
Chronic Obstructive Pulmonary Disease Is Associated with an Increase in Urinary Levels of...
11
Journal of Pathology J Pathol 2005; 206: 28–38 Published online 16 March 2005 in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/path.1748 Original Paper Chronic obstructive pulmonary disease is associated with enhanced bronchial expression of FGF-1, FGF-2, and FGFR-1 Andor R Kranenburg, 1 Anna Willems-Widyastuti, 1 Wolter J Mooi, 2 Pramod R Saxena, 1 Peter J Sterk, 3 Willem I de Boer 4 and Hari S Sharma 1 * 1 Department of Pharmacology, Erasmus MC, University Medical Center, Rotterdam, The Netherlands 2 Department of Pathology, The Netherlands Cancer Institute, Amsterdam, The Netherlands 3 Department of Pulmonology, Leiden University Medical Center, Leiden, The Netherlands 4 Department of Pulmonary Medicine, Erasmus MC, University Medical Center, Rotterdam, The Netherlands *Correspondence to: Hari S Sharma, Mphil, PhD, Institute of Pharmacology, Erasmus MC, University Medical Center, Dr Molewaterplein 50, 3015 GE Rotterdam, The Netherlands. E-mail: [email protected] Received: 20 February 2004 Revised: 20 October 2004 Accepted: 26 October 2004 Abstract An important feature of chronic obstructive pulmonary disease (COPD) is airway remod- elling, the molecular mechanisms of which are poorly understood. In this study, the role of fibroblast growth factors (FGF-1 and FGF-2) and their receptor, FGFR-1, was assessed in bronchial airway wall remodelling in patients with COPD (FEV 1 < 75%; n = 15) and with- out COPD (FEV 1 > 85%; n = 16). FGF-1 and FGFR-1 were immunolocalized in bronchial epithelium, airway smooth muscle (ASM), submucosal glandular epithelium, and vascular smooth muscle. Quantitative digital image analysis revealed increased cytoplasmic expres- sion of FGF-2 in bronchial epithelium (0.35 ± 0.03 vs 0.20 ± 0.04, p < 0.008) and nuclear localization in ASM (p < 0.0001) in COPD patients compared with controls. Elevated lev- els of FGFR-1 in ASM (p < 0.005) and of FGF-1 (p < 0.04) and FGFR-1 (p < 0.001) in bronchial epithelium were observed. In cultured human ASM cells, FGF-1 and/or FGF-2 (10 ng/ml) induced cellular proliferation, as shown by [ 3 H]thymidine incorporation and by cell number counts. Steady-state mRNA levels of FGFR-1 were elevated in human ASM cells treated with either FGF-1 or FGF-2. The increased bronchial expression of fibroblast growth factors and their receptor in patients with COPD, and the mitogenic response of human ASM cells to FGFs in vitro suggest a potential role for the FGF/FGFR-1 system in the remodelling of bronchial airways in COPD. Copyright 2005 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd. Keywords: chronic obstructive pulmonary disease; fibroblast growth factor; airway remodelling; airway smooth muscle; gene expression; human Introduction Chronic obstructive pulmonary disease (COPD) is a global health problem with increasing morbidity and mortality [1]. One of the major determining factors is tobacco smoking [2]. However, only 10% of all smokers develop COPD. One of the key pathological features of COPD is thickening of the airway walls, which is thought to be a result of a chronic smoul- dering inflammatory process, in which neutrophils, macrophages, and T-lymphocytes play a role, and which is associated with hyperplasia of airway smooth muscle cells and (myo-) fibroblasts, and increased deposition of extracellular matrix [3]. The bronchial epithelium and airway smooth muscle are two major cellular structures involved in airway remodelling [3]. A variety of growth factors and cytokines, including platelet-derived growth factor-B (PDGF-B), epider- mal growth factor (EGF), and transforming growth factor-β (TGF-β ) that are released from these sites of the airway wall, have the potential to contribute to the pathogenesis of airway remodelling [4–6]. Sup- porting in vitro evidence for a relationship between epithelial injury and enhanced airway remodelling is provided by studies of co-cultures of bronchial epithelial cells and myo-fibroblasts [7,8]: these studies revealed enhanced cellular proliferation and increased collagen expression resulting from the interaction of these cells with several growth factors, including basic FGF (FGF-2), insulin-like growth factor-1, PDGF-B, TGF-β , endothelin-1, and EGF. Fibroblast growth factors (FGFs) may well play a pivotal role in regulating airway wall remodelling. A number of studies have demonstrated that members of the EGF and FGF family contribute to chronic inflammatory and tissue repair processes as well as to fibrosis in chronic airway diseases such asthma [9,10]. Fibroblast growth factors bind to four high-affinity, transmembrane tyrosine-kinase receptors (FGFR1-4). Distinct FGF subtypes bind with different affinities Copyright 2005 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Chronic Obstructive Pulmonary Disease Is Associated with an Increase in Urinary Levels of...

Journal of PathologyJ Pathol 2005; 206: 28–38Published online 16 March 2005 in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/path.1748
Original Paper
Chronic obstructive pulmonary disease is associatedwith enhanced bronchial expression of FGF-1, FGF-2,and FGFR-1Andor R Kranenburg,1 Anna Willems-Widyastuti,1 Wolter J Mooi,2 Pramod R Saxena,1 Peter J Sterk,3
Willem I de Boer4 and Hari S Sharma1*1Department of Pharmacology, Erasmus MC, University Medical Center, Rotterdam, The Netherlands2Department of Pathology, The Netherlands Cancer Institute, Amsterdam, The Netherlands3Department of Pulmonology, Leiden University Medical Center, Leiden, The Netherlands4Department of Pulmonary Medicine, Erasmus MC, University Medical Center, Rotterdam, The Netherlands
*Correspondence to:Hari S Sharma, Mphil, PhD,Institute of Pharmacology,Erasmus MC, University MedicalCenter, Dr Molewaterplein 50,3015 GE Rotterdam, TheNetherlands.E-mail: [email protected]
Received: 20 February 2004Revised: 20 October 2004Accepted: 26 October 2004
AbstractAn important feature of chronic obstructive pulmonary disease (COPD) is airway remod-elling, the molecular mechanisms of which are poorly understood. In this study, the role offibroblast growth factors (FGF-1 and FGF-2) and their receptor, FGFR-1, was assessed inbronchial airway wall remodelling in patients with COPD (FEV1 < 75%; n = 15) and with-out COPD (FEV1 > 85%; n = 16). FGF-1 and FGFR-1 were immunolocalized in bronchialepithelium, airway smooth muscle (ASM), submucosal glandular epithelium, and vascularsmooth muscle. Quantitative digital image analysis revealed increased cytoplasmic expres-sion of FGF-2 in bronchial epithelium (0.35 ± 0.03 vs 0.20 ± 0.04, p < 0.008) and nuclearlocalization in ASM (p < 0.0001) in COPD patients compared with controls. Elevated lev-els of FGFR-1 in ASM (p < 0.005) and of FGF-1 (p < 0.04) and FGFR-1 (p < 0.001) inbronchial epithelium were observed. In cultured human ASM cells, FGF-1 and/or FGF-2(10 ng/ml) induced cellular proliferation, as shown by [3H]thymidine incorporation and bycell number counts. Steady-state mRNA levels of FGFR-1 were elevated in human ASMcells treated with either FGF-1 or FGF-2. The increased bronchial expression of fibroblastgrowth factors and their receptor in patients with COPD, and the mitogenic response ofhuman ASM cells to FGFs in vitro suggest a potential role for the FGF/FGFR-1 system inthe remodelling of bronchial airways in COPD.Copyright 2005 Pathological Society of Great Britain and Ireland. Published by JohnWiley & Sons, Ltd.
Keywords: chronic obstructive pulmonary disease; fibroblast growth factor; airwayremodelling; airway smooth muscle; gene expression; human
Introduction
Chronic obstructive pulmonary disease (COPD) is aglobal health problem with increasing morbidity andmortality [1]. One of the major determining factorsis tobacco smoking [2]. However, only 10% of allsmokers develop COPD. One of the key pathologicalfeatures of COPD is thickening of the airway walls,which is thought to be a result of a chronic smoul-dering inflammatory process, in which neutrophils,macrophages, and T-lymphocytes play a role, andwhich is associated with hyperplasia of airway smoothmuscle cells and (myo-) fibroblasts, and increaseddeposition of extracellular matrix [3]. The bronchialepithelium and airway smooth muscle are two majorcellular structures involved in airway remodelling [3].A variety of growth factors and cytokines, includingplatelet-derived growth factor-B (PDGF-B), epider-mal growth factor (EGF), and transforming growthfactor-β (TGF-β) that are released from these sites
of the airway wall, have the potential to contribute tothe pathogenesis of airway remodelling [4–6]. Sup-porting in vitro evidence for a relationship betweenepithelial injury and enhanced airway remodellingis provided by studies of co-cultures of bronchialepithelial cells and myo-fibroblasts [7,8]: these studiesrevealed enhanced cellular proliferation and increasedcollagen expression resulting from the interaction ofthese cells with several growth factors, including basicFGF (FGF-2), insulin-like growth factor-1, PDGF-B,TGF-β, endothelin-1, and EGF.
Fibroblast growth factors (FGFs) may well play apivotal role in regulating airway wall remodelling. Anumber of studies have demonstrated that membersof the EGF and FGF family contribute to chronicinflammatory and tissue repair processes as well as tofibrosis in chronic airway diseases such asthma [9,10].Fibroblast growth factors bind to four high-affinity,transmembrane tyrosine-kinase receptors (FGFR1-4).Distinct FGF subtypes bind with different affinities
Copyright 2005 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.

Airway remodelling in COPD 29
to the various FGF receptors. Alternative splicingand regulated protein trafficking further modulate theintra-cellular events initiated by FGF ligand–receptorinteraction [11]. Increased expression of FGF-1 andFGFR-1 has been shown during the development oflung fibrosis [12], and FGF-2 has been implicated inthe pathogenesis of obliterative bronchiolitis in lungtransplants [13].
We postulate that the FGF–FGFR system is invol-ved in the pathogenesis of COPD. We investigatedthe expression patterns of FGF-1, FGF-2 and FGFR-1in bronchial airways of (ex-) smokers with or withoutCOPD. In addition, we examined cell proliferation andthe expression of FGFR-1 in cultured human ASMcells stimulated with FGF-1 and FGF-2.
Materials and methods
Selection of specimens
The Medical Ethics Committees of the Leiden Uni-versity Medical Center and Southern Hospital Rot-terdam, The Netherlands approved the study. Lungtissue from the hospitals’ pathology archives wasobtained from patients who underwent lobectomyor pneumonectomy. Based on lung function data,patients were assigned [6,14] to the COPD group(n = 15) consisting of 15 subjects with a forced 1-s expiratory volume (FEV1) < 75% of the predictedvalue [15] before bronchodilatation, an FEV1/FVCratio < 75%, a reversibility in FEV1 � 12% of thepredicted value after 400 µg of inhaled salbutamol,and a carbon monoxide diffusion capacity (Kco) �80% of the predicted value, or to the non-COPDgroup (n = 16) consisting of 16 subjects with anFEV1 > 85% before bronchodilation, an FEV1/FVCratio > 85%, and a total lung capacity (TLC) of over80% [15]. The patients in these two groups partic-ipated in a larger research project, part of whichhas been published previously [16,17]. Clinical datafrom all patients were examined for possible co-morbidity and medication usage. All pulmonary func-tion tests were performed within 3 months priorto surgery as described earlier [16]. Lung functiondata and other patient characteristics are shown inTable1.
Immunohistochemistry
Serial sections of 4 µm were dewaxed, rehydrated, andimmunostained using a Multilink labelling system(Biogenex, San Ramon, USA) and specific anti-humanmouse monoclonal antibodies against α-smooth mus-cle actin (α-SMA; NeoMarkers, Fremont, USA), Ki-67 (Biogenex, San Ramon, USA), FGF-2 (Transduc-tion Laboratories, Lexington, USA), and FGF-1 andFGFR-1 (kind gift from Dr J Walters) as describedpreviously [18,19]. Colour was developed using NewFuchsin or 3,3-diaminobenzidine as the chromogen.
Table 1. Subject characteristics and clinical parameters
Non-COPD COPD
FEV1 97 ± 1.6 54 ± 3.3∗dFEV1 3 ± 0.6 4 ± 0.9FEV1/FVC 100 ± 2.1 58 ± 2.3∗TLC 104 ± 1.9 103 ± 3.6RV 117 ± 5.4 141 ± 10∗Kco 94 ± 2.0 55 ± 5.4∗Sex (male/female) 13/3 14/1Age (years) 59 ± 3.5 64 ± 2.6Smokers/ex-smokers/non-smokers 11/3/2 12/3/0Pack-years 44 ± 8.6 31 ± 0.3Steroid use (yes/no/unknown) 0/15/1 3/10/2
FEV1 = forced expiratory volume in 1 s; FVC = forced vital capacity;TLC = total lung capacity; RV = residual volume. Reversibility of FEV1after 400 µg of salbutamol (dFEV1) and the carbon monoxide diffusionconstant (Kco) are given as a percentage of the predicted value.∗ p < 0.005 vs non-COPD. The patients in these two groupsparticipated in a larger project, part of which has been publishedpreviously [16,17].
Slides were counter-stained with Mayer’s haema-toxylin. Positive controls consisted of human breastcarcinoma and placental tissue. The optimal dilutionsfor all antibodies were identified by examining theintensity of staining obtained with a series of dilu-tions: the optimum concentration resulted in a specificand easily visible signal on control specimens. Neg-ative controls consisted of omission of the primaryantibody.
Quantitative analyses of immunostaining
Digital images (pixel size 736 × 574) from each sub-ject were analysed using the Leica Qwin image anal-ysis system (Leica BV, Rijswijk, The Netherlands).Staining patterns of FGF-1, FGF-2, FGFR-1, and α-SMA were analysed by drawing areas interactivelyand assessing the area of positive staining dividedby the total measured cellular area of the respectiveepithelial or ASM layer. The nuclear localization ofFGF-2 in ASM was assessed by computerized count-ing of individual nuclei and the data are expressedas the number of positive nuclei divided by the totalnuclei (labelling index, LI). For vascular expressionof FGFs and FGFR-1, quantitative analysis was per-formed using an arbitrary visual scale with gradingscores of 0, 1, 2, and 3 (Figure 1) representing no(panel A), weak (panel B), moderate (panel C), andintense (panel D) staining, respectively [6,14].
Isolation and culture of human ASM cells
Human airway smooth muscle cells were from threedifferent non-asthmatic, non-COPD, and (ex-) smokerdonors who underwent lobectomy or pneumonectomyas described previously [20,21]. ASM cells were char-acterized immunocytochemically (α-SMA and smoothmuscle myosin heavy chain staining) and used forexperiments at passage 4–5.
J Pathol 2005; 206: 28–38

30 AR Kranenburg et al
Figure 1. Immunohistochemical localization of FGF-2 in bronchial vessels. Representative examples of staining intensity patternused for visual scoring. Photomicrographs depict lung tissue sections from patients without COPD (A and B) and with COPD (Cand D) showing nuclear staining of FGF-2 in vascular smooth muscle cells. Panels A–D show representative examples of stainingintensities used for visual scoring, 0–3 respectively. Original magnification × 100
Figure 2. Immunohistochemical localization of FGF-1 and FGF-2 in central airways. Photomicrographs showing central bronchialtissue sections from patients without COPD (A, C, E, G) and with COPD (B, D, F, H). Panels A and B show representativeexamples of FGF-1 protein staining (red, New Fuchsin) in bronchial epithelium. Panels C and D show representative staining inairway smooth muscle (ASM) cells. Original magnification × 200. Panels E and F show representative examples of FGF-2 proteinstaining (brown, 3,3-diaminobenzidine) in bronchial epithelium. Panels G and H show representative nuclear staining (shownby solid arrows) and no nuclear staining (blue nuclei) shown by open arrows in airway smooth muscle (ASM) cells. Originalmagnification × 400. Scale bar = 50 µm
J Pathol 2005; 206: 28–38
Airway remodelling in COPD 31
ASM cell proliferation assaysCells were seeded at a density 1 × 104 cells per wellin 96-well plates, cultured until confluence, subse-quently serum-deprived to synchronize the growth,and incubated with either 0.1, 1.0, 10, or 50 ng/mlhuman recombinant FGF-1 (Promega, Madison, USA)and/or FGF-2 (Sigma-Aldrich, St Louis, USA) for 8,24, and 48 h. Control cells received FBS-free DMEMalone. Five hours prior to the end of the treatment,1 µCi/well of [3H]thymidine (Amersham, Roosendaal,The Netherlands) was added. The cells were harvestedon glass fibre filters and radioactivity was assessedusing a Microplate Scintillation β-counter (Topcount,Packard, Meridan, USA). The mean counts per minuteof quadruple wells and subsequently from three dif-ferent cell batches was expressed as fold change com-pared with controls. In a parallel series of experiments,cells in quadruple were stimulated for 24 and 48 hand processed for cell counting in the Casy1 system(Scharfe System GmbH, Reutingen, Germany) [20].
RNA isolation and RT-PCR
Growth-arrested ASM cells were incubated with eitherFGF-1 or FGF-2 (10 ng/ml) for 1, 2, 4, 8, 24, and
48 h. Total RNA was extracted, treated with Rnase-free DNase to eliminate contaminating genomic DNA,and processed for the synthesis of cDNA and PCR[20,21]. Human-specific forward and reverse primersspanning over a 497 bp fragment encoding FGFR-1 and a 625 bp fragment of β-actin cDNAs wereemployed [22,23]. The PCR products were separatedon 1.5% agarose gels, digitally photographed, andthe intensity of the bands was quantified in relationto the β-actin band using the Molecular Analyst (V1.5) image analysis program (Biorad Laboratories,Hercules, USA); values were expressed as a ratio tothe controls.
Statistical analysis
Data were analysed for statistical significance usingthe unpaired, two-tailed Student’s t-test as well as thenon-parametric Mann–Whitney test, where appropri-ate. The data were expressed as mean ± SEM. Stainingfor different compartments was correlated with FEV1and Kco using Pearson’s correlation analysis. Differ-ences with p � 0.05 were considered to be statisticallysignificant.
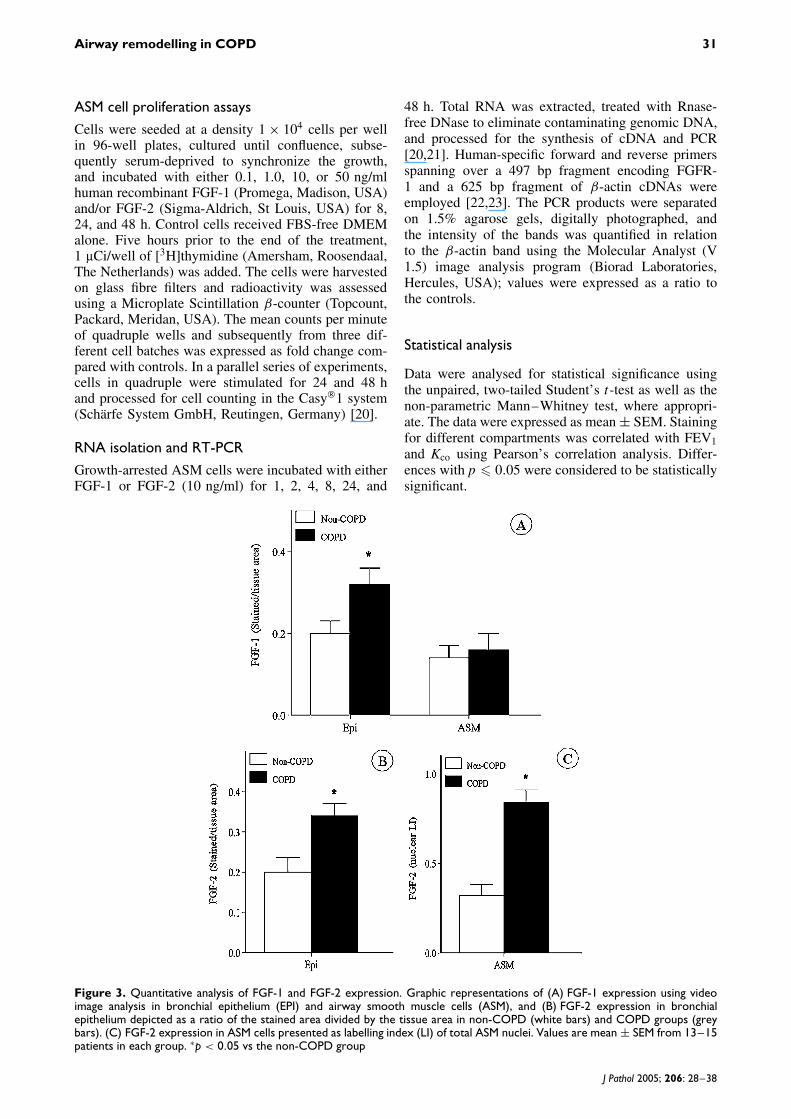
Figure 3. Quantitative analysis of FGF-1 and FGF-2 expression. Graphic representations of (A) FGF-1 expression using videoimage analysis in bronchial epithelium (EPI) and airway smooth muscle cells (ASM), and (B) FGF-2 expression in bronchialepithelium depicted as a ratio of the stained area divided by the tissue area in non-COPD (white bars) and COPD groups (greybars). (C) FGF-2 expression in ASM cells presented as labelling index (LI) of total ASM nuclei. Values are mean ± SEM from 13–15patients in each group. ∗p < 0.05 vs the non-COPD group
J Pathol 2005; 206: 28–38
32 AR Kranenburg et al
Results
Clinical parameters
The clinical and lung function characteristics of allsubjects included in the study are listed in Table 1[16]. The COPD group demonstrated an elevatedresidual volume (RV), whereas the CO diffusion (Kco)was reduced compared with controls (p < 0.005). Thesubjects in the two groups did not differ significantlyin age, total lung capacity (TLC), reversibility inFEV1, smoking status (pack-years) or steroid use(Table 1).
Localization and quantification of FGF-1 and FGF-2
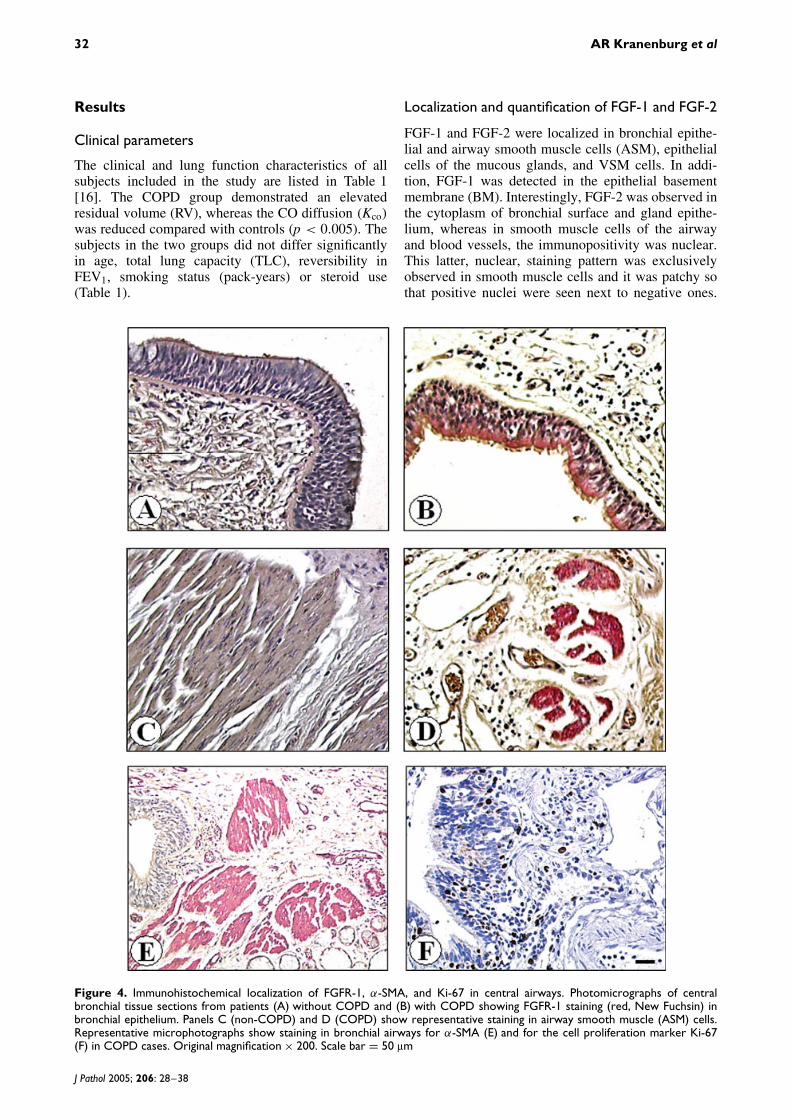
FGF-1 and FGF-2 were localized in bronchial epithe-lial and airway smooth muscle cells (ASM), epithelialcells of the mucous glands, and VSM cells. In addi-tion, FGF-1 was detected in the epithelial basementmembrane (BM). Interestingly, FGF-2 was observed inthe cytoplasm of bronchial surface and gland epithe-lium, whereas in smooth muscle cells of the airwayand blood vessels, the immunopositivity was nuclear.This latter, nuclear, staining pattern was exclusivelyobserved in smooth muscle cells and it was patchy sothat positive nuclei were seen next to negative ones.
Figure 4. Immunohistochemical localization of FGFR-1, α-SMA, and Ki-67 in central airways. Photomicrographs of centralbronchial tissue sections from patients (A) without COPD and (B) with COPD showing FGFR-1 staining (red, New Fuchsin) inbronchial epithelium. Panels C (non-COPD) and D (COPD) show representative staining in airway smooth muscle (ASM) cells.Representative microphotographs show staining in bronchial airways for α-SMA (E) and for the cell proliferation marker Ki-67(F) in COPD cases. Original magnification × 200. Scale bar = 50 µm
J Pathol 2005; 206: 28–38
Airway remodelling in COPD 33
Microphotographs showing the expression patterns ofFGF-1 and FGF-2 are presented in Figures 2A, 2C and2E, 2G (non-COPD), and 2B, 2D and 2F, 2H (COPD),respectively.
Video image analysis revealed that the expres-sion levels for FGF-1 in the bronchial epithelium(Figure 3A) were increased significantly (stained/totalepithelial area: 0.32 ± 0.04 vs 0.20 ± 0.03, p < 0.04)in COPD cases compared with non-COPD. In ASMcells, no difference was found for FGF-1 (0.16 ± 0.04vs 0.14 ± 0.03, p = 0.77). FGF-2 expression, how-ever, was clearly up-regulated in the bronchial epithe-lium of COPD cases (0.35 ± 0.03 vs 0.20 ± 0.04, p <
0.008, Figure 3B) and ASM nuclei (LI ASM nuclei,0.84 ± 0.07 vs 0.32 ± 0.06, p < 0.0001, Figure 3C).The distribution of total nuclei/total ASM tissue arearemained unchanged in both groups, indicating thatthe number of nuclei as well as the ASM areaincreased simultaneously, keeping the ratio equal inboth groups (data not shown). Furthermore, it appearedthat COPD was associated with an increase in FGF-2 expression in ASM cells, with perhaps an increasein their size but without their apparent prolifera-tion.
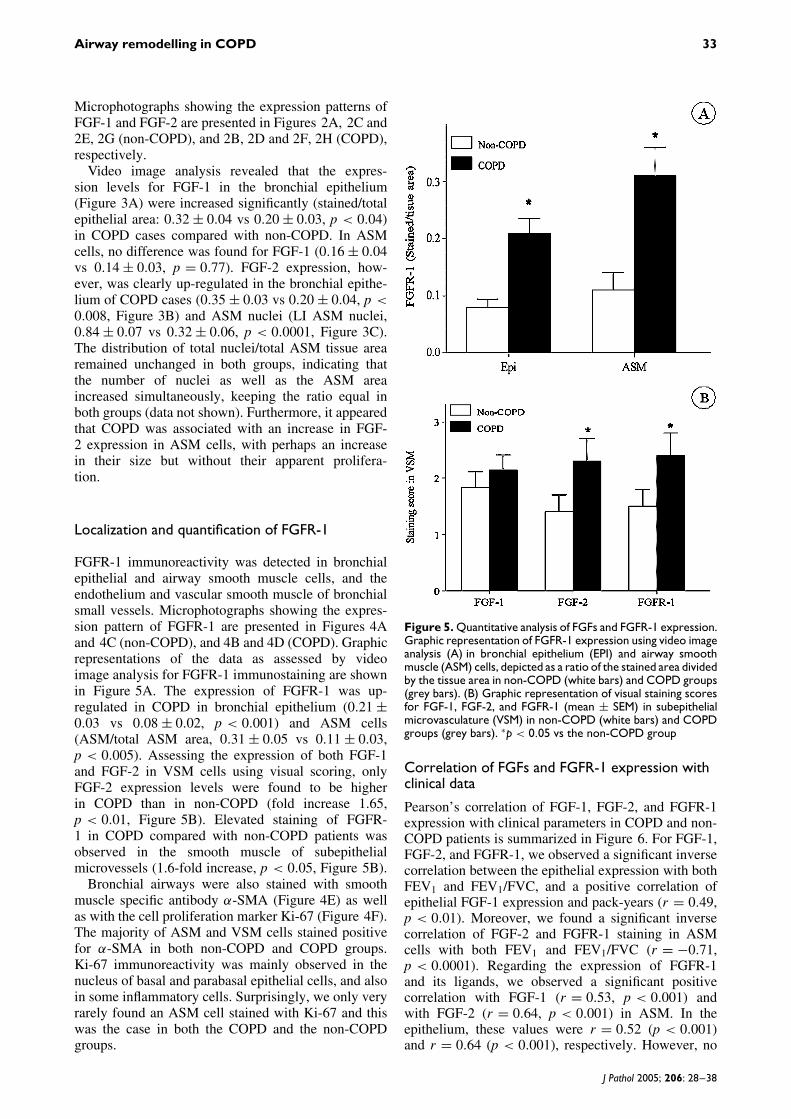
Localization and quantification of FGFR-1
FGFR-1 immunoreactivity was detected in bronchialepithelial and airway smooth muscle cells, and theendothelium and vascular smooth muscle of bronchialsmall vessels. Microphotographs showing the expres-sion pattern of FGFR-1 are presented in Figures 4Aand 4C (non-COPD), and 4B and 4D (COPD). Graphicrepresentations of the data as assessed by videoimage analysis for FGFR-1 immunostaining are shownin Figure 5A. The expression of FGFR-1 was up-regulated in COPD in bronchial epithelium (0.21 ±0.03 vs 0.08 ± 0.02, p < 0.001) and ASM cells(ASM/total ASM area, 0.31 ± 0.05 vs 0.11 ± 0.03,p < 0.005). Assessing the expression of both FGF-1and FGF-2 in VSM cells using visual scoring, onlyFGF-2 expression levels were found to be higherin COPD than in non-COPD (fold increase 1.65,p < 0.01, Figure 5B). Elevated staining of FGFR-1 in COPD compared with non-COPD patients wasobserved in the smooth muscle of subepithelialmicrovessels (1.6-fold increase, p < 0.05, Figure 5B).
Bronchial airways were also stained with smoothmuscle specific antibody α-SMA (Figure 4E) as wellas with the cell proliferation marker Ki-67 (Figure 4F).The majority of ASM and VSM cells stained positivefor α-SMA in both non-COPD and COPD groups.Ki-67 immunoreactivity was mainly observed in thenucleus of basal and parabasal epithelial cells, and alsoin some inflammatory cells. Surprisingly, we only veryrarely found an ASM cell stained with Ki-67 and thiswas the case in both the COPD and the non-COPDgroups.
Figure 5. Quantitative analysis of FGFs and FGFR-1 expression.Graphic representation of FGFR-1 expression using video imageanalysis (A) in bronchial epithelium (EPI) and airway smoothmuscle (ASM) cells, depicted as a ratio of the stained area dividedby the tissue area in non-COPD (white bars) and COPD groups(grey bars). (B) Graphic representation of visual staining scoresfor FGF-1, FGF-2, and FGFR-1 (mean ± SEM) in subepithelialmicrovasculature (VSM) in non-COPD (white bars) and COPDgroups (grey bars). ∗p < 0.05 vs the non-COPD group
Correlation of FGFs and FGFR-1 expression withclinical data
Pearson’s correlation of FGF-1, FGF-2, and FGFR-1expression with clinical parameters in COPD and non-COPD patients is summarized in Figure 6. For FGF-1,FGF-2, and FGFR-1, we observed a significant inversecorrelation between the epithelial expression with bothFEV1 and FEV1/FVC, and a positive correlation ofepithelial FGF-1 expression and pack-years (r = 0.49,p < 0.01). Moreover, we found a significant inversecorrelation of FGF-2 and FGFR-1 staining in ASMcells with both FEV1 and FEV1/FVC (r = −0.71,p < 0.0001). Regarding the expression of FGFR-1and its ligands, we observed a significant positivecorrelation with FGF-1 (r = 0.53, p < 0.001) andwith FGF-2 (r = 0.64, p < 0.001) in ASM. In theepithelium, these values were r = 0.52 (p < 0.001)and r = 0.64 (p < 0.001), respectively. However, no
J Pathol 2005; 206: 28–38

34 AR Kranenburg et al
Figure 6. Correlation analysis of FGFs and FGFR-1 expression. Correlation analysis was performed for FGF-1 expression inbronchial epithelium with pack-years (A); and FGF-2 (B) and FGFR-1 (C) in ASM with forced vital capacity. Panel D showsthe staining correlation for FGF-2 with FGFR-1 in ASM cells. Correlation coefficients (r) were obtained using linear regression(Pearson’s) analysis and significance level (p value, p < 0.05) was considered at absolute values of r > 0.37. FEV1 = forcedexpiratory volume in 1 s; FVC = forced vital capacity; Kco = carbon monoxide diffusion constant
significant correlation was found between FGF-1 andFGF-2 localization.
Mitogenic effects of FGFs in cultured human ASMcells
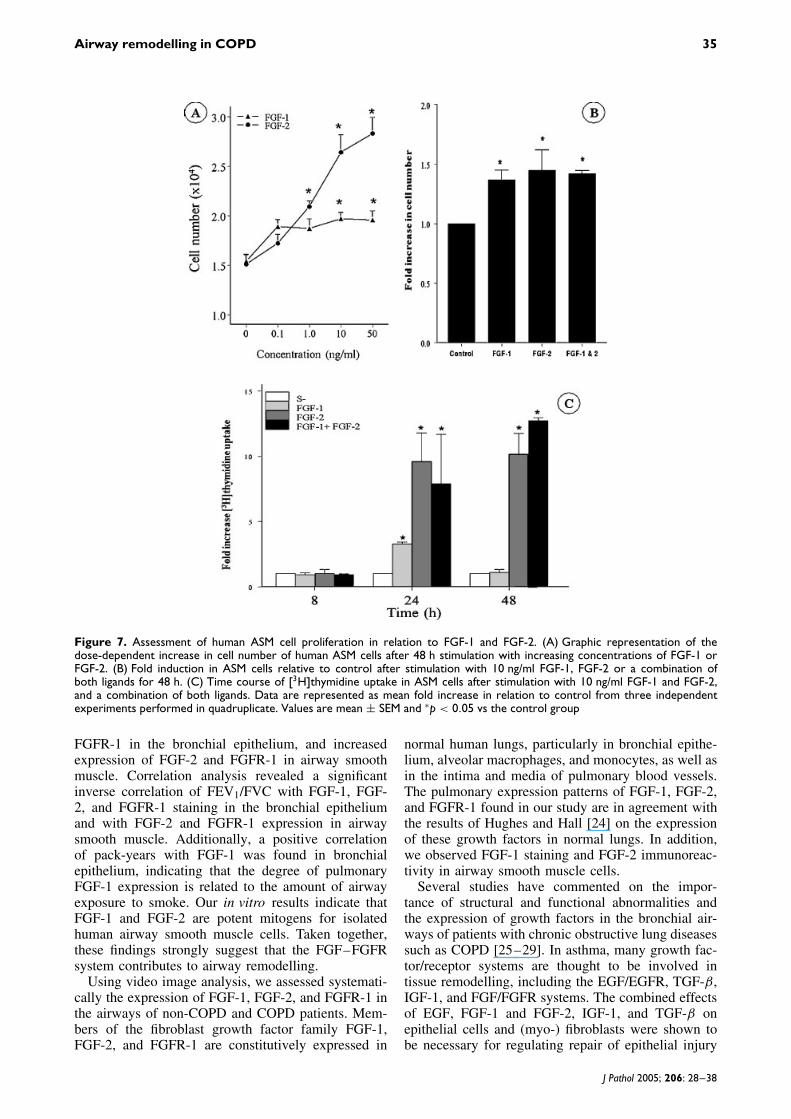
In order to investigate the role of FGFs on airwaysmooth muscle remodelling further, isolated humanairway smooth muscle cells were stimulated in vitrowith increasing concentrations of FGF-1 or FGF-2.Graphic representation of the dose-dependent increasein cell number for FGF-1 and FGF-2 is shownin Figure 7A. Both FGF-1 and FGF-2 resulted insignificantly increased cell numbers at a concentrationof 10 ng/ml after 48 h of incubation. We thereforeopted for this concentration for both growth factorsin our further experiments. Figure 7B shows the foldincrease in cell number after 48 h of stimulation with10 ng/ml FGF-1, FGF-2, and the combination of thetwo over the control. A significant increase in ASMcell numbers (fold increase) after 48 h of incubationwith FGF-1 (1.37 ± 0.08, p < 0.01) or FGF-2 (1.45 ±0.17, p = 0.05) or both ligands (1.42 ± 0.14, p <
0.03) was observed.A graphic representation of the time-dependent
[3H]TdR uptake at a concentration of 10 ng/ml ofFGF-1 or FGF-2 is presented in Figure 7C. After 24 ofstimulation, we found significantly increased [3H]TdRuptake with FGF-1 and FGF-2, but only after 48 hwith FGF-2. The combined incubation with 10 ng/mleach of FGF-1 and FGF-2 resulted in significantlyincreased thymidine uptake that was comparable to
10 ng/ml of FGF-2 alone. Eight hours of stimulationwith either FGF-1 or FGF-2 did not result in a markedincrease in [3H]TdR uptake (Figure 7C).
Expression of FGFR-1 mRNA in human ASM cells
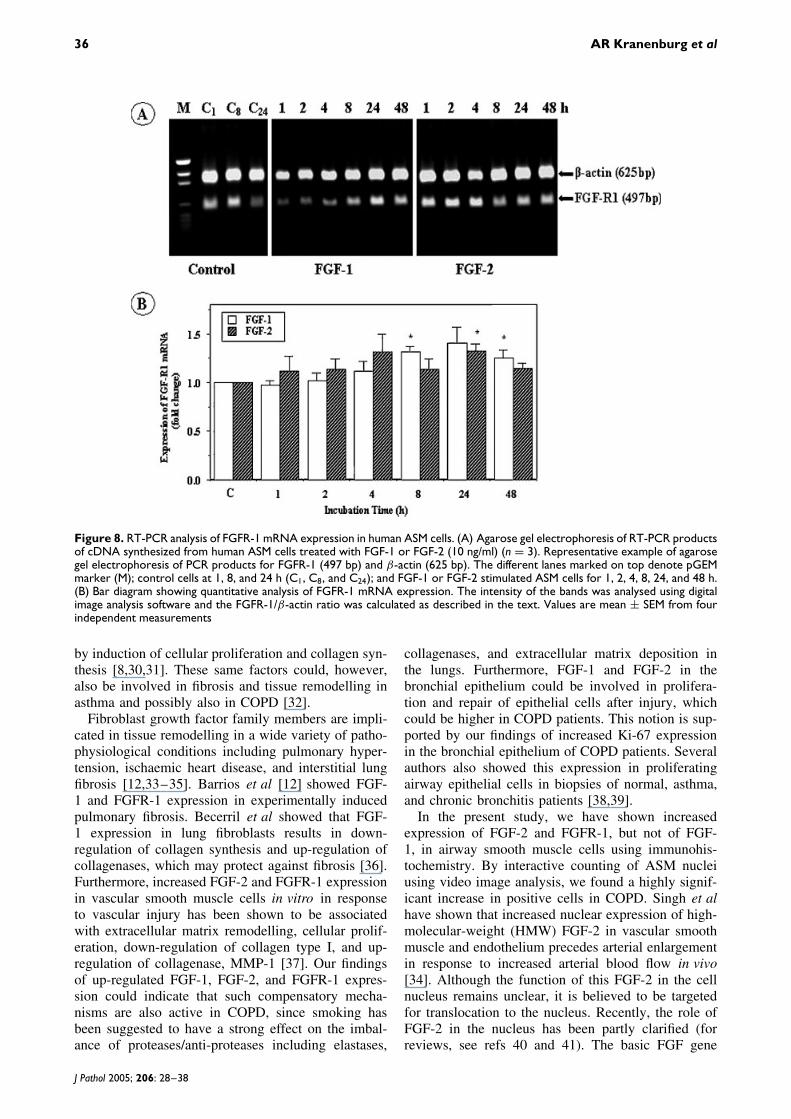
To examine whether human ASM cells express FGFR-1 and whether this expression is regulated by FGF-1and/or FGF-2, we performed RT-PCR on cDNA tem-plates derived from cells treated with 10 ng/ml FGF-1or FGF-2 for various time periods and compared theexpression pattern with controls. A photograph show-ing a representative example, after agarose gel elec-trophoresis with PCR products for FGFR-1 (497 bp)and β-actin (625 bp), is shown in Figure 8A. Bothbands were analysed using appropriate image anal-ysis software and FGFR-1/β-actin values of FGF-1-or FGF-2-treated ASM cells at different time pointswere assessed in relation to controls (Figure 8B).Stimulation with 10 ng/ml FGF-1 increased FGFR-1 mRNA expression by 1.31 ± 0.11 fold at 8 h andby 1.23 ± 0.12 fold at 48 h of incubation comparedwith controls (p < 0.05), whereas FGF-2 stimulationresulted in elevated levels for FGFR-1 mRNA at 24 h(1.32 ± 0.14 fold, p < 0.05) and at 48 h (1.21 ± 0.13,ns) of incubation.
Discussion
In this study, we have shown that COPD is associatedwith increased expression of FGF-1, FGF-2, and
J Pathol 2005; 206: 28–38
Airway remodelling in COPD 35
Figure 7. Assessment of human ASM cell proliferation in relation to FGF-1 and FGF-2. (A) Graphic representation of thedose-dependent increase in cell number of human ASM cells after 48 h stimulation with increasing concentrations of FGF-1 orFGF-2. (B) Fold induction in ASM cells relative to control after stimulation with 10 ng/ml FGF-1, FGF-2 or a combination ofboth ligands for 48 h. (C) Time course of [3H]thymidine uptake in ASM cells after stimulation with 10 ng/ml FGF-1 and FGF-2,and a combination of both ligands. Data are represented as mean fold increase in relation to control from three independentexperiments performed in quadruplicate. Values are mean ± SEM and ∗p < 0.05 vs the control group
FGFR-1 in the bronchial epithelium, and increasedexpression of FGF-2 and FGFR-1 in airway smoothmuscle. Correlation analysis revealed a significantinverse correlation of FEV1/FVC with FGF-1, FGF-2, and FGFR-1 staining in the bronchial epitheliumand with FGF-2 and FGFR-1 expression in airwaysmooth muscle. Additionally, a positive correlationof pack-years with FGF-1 was found in bronchialepithelium, indicating that the degree of pulmonaryFGF-1 expression is related to the amount of airwayexposure to smoke. Our in vitro results indicate thatFGF-1 and FGF-2 are potent mitogens for isolatedhuman airway smooth muscle cells. Taken together,these findings strongly suggest that the FGF–FGFRsystem contributes to airway remodelling.
Using video image analysis, we assessed systemati-cally the expression of FGF-1, FGF-2, and FGFR-1 inthe airways of non-COPD and COPD patients. Mem-bers of the fibroblast growth factor family FGF-1,FGF-2, and FGFR-1 are constitutively expressed in
normal human lungs, particularly in bronchial epithe-lium, alveolar macrophages, and monocytes, as well asin the intima and media of pulmonary blood vessels.The pulmonary expression patterns of FGF-1, FGF-2,and FGFR-1 found in our study are in agreement withthe results of Hughes and Hall [24] on the expressionof these growth factors in normal lungs. In addition,we observed FGF-1 staining and FGF-2 immunoreac-tivity in airway smooth muscle cells.
Several studies have commented on the impor-tance of structural and functional abnormalities andthe expression of growth factors in the bronchial air-ways of patients with chronic obstructive lung diseasessuch as COPD [25–29]. In asthma, many growth fac-tor/receptor systems are thought to be involved intissue remodelling, including the EGF/EGFR, TGF-β,IGF-1, and FGF/FGFR systems. The combined effectsof EGF, FGF-1 and FGF-2, IGF-1, and TGF-β onepithelial cells and (myo-) fibroblasts were shown tobe necessary for regulating repair of epithelial injury
J Pathol 2005; 206: 28–38
36 AR Kranenburg et al
Figure 8. RT-PCR analysis of FGFR-1 mRNA expression in human ASM cells. (A) Agarose gel electrophoresis of RT-PCR productsof cDNA synthesized from human ASM cells treated with FGF-1 or FGF-2 (10 ng/ml) (n = 3). Representative example of agarosegel electrophoresis of PCR products for FGFR-1 (497 bp) and β-actin (625 bp). The different lanes marked on top denote pGEMmarker (M); control cells at 1, 8, and 24 h (C1, C8, and C24); and FGF-1 or FGF-2 stimulated ASM cells for 1, 2, 4, 8, 24, and 48 h.(B) Bar diagram showing quantitative analysis of FGFR-1 mRNA expression. The intensity of the bands was analysed using digitalimage analysis software and the FGFR-1/β-actin ratio was calculated as described in the text. Values are mean ± SEM from fourindependent measurements
by induction of cellular proliferation and collagen syn-thesis [8,30,31]. These same factors could, however,also be involved in fibrosis and tissue remodelling inasthma and possibly also in COPD [32].
Fibroblast growth factor family members are impli-cated in tissue remodelling in a wide variety of patho-physiological conditions including pulmonary hyper-tension, ischaemic heart disease, and interstitial lungfibrosis [12,33–35]. Barrios et al [12] showed FGF-1 and FGFR-1 expression in experimentally inducedpulmonary fibrosis. Becerril et al showed that FGF-1 expression in lung fibroblasts results in down-regulation of collagen synthesis and up-regulation ofcollagenases, which may protect against fibrosis [36].Furthermore, increased FGF-2 and FGFR-1 expressionin vascular smooth muscle cells in vitro in responseto vascular injury has been shown to be associatedwith extracellular matrix remodelling, cellular prolif-eration, down-regulation of collagen type I, and up-regulation of collagenase, MMP-1 [37]. Our findingsof up-regulated FGF-1, FGF-2, and FGFR-1 expres-sion could indicate that such compensatory mecha-nisms are also active in COPD, since smoking hasbeen suggested to have a strong effect on the imbal-ance of proteases/anti-proteases including elastases,
collagenases, and extracellular matrix deposition inthe lungs. Furthermore, FGF-1 and FGF-2 in thebronchial epithelium could be involved in prolifera-tion and repair of epithelial cells after injury, whichcould be higher in COPD patients. This notion is sup-ported by our findings of increased Ki-67 expressionin the bronchial epithelium of COPD patients. Severalauthors also showed this expression in proliferatingairway epithelial cells in biopsies of normal, asthma,and chronic bronchitis patients [38,39].
In the present study, we have shown increasedexpression of FGF-2 and FGFR-1, but not of FGF-1, in airway smooth muscle cells using immunohis-tochemistry. By interactive counting of ASM nucleiusing video image analysis, we found a highly signif-icant increase in positive cells in COPD. Singh et alhave shown that increased nuclear expression of high-molecular-weight (HMW) FGF-2 in vascular smoothmuscle and endothelium precedes arterial enlargementin response to increased arterial blood flow in vivo[34]. Although the function of this FGF-2 in the cellnucleus remains unclear, it is believed to be targetedfor translocation to the nucleus. Recently, the role ofFGF-2 in the nucleus has been partly clarified (forreviews, see refs 40 and 41). The basic FGF gene
J Pathol 2005; 206: 28–38

Airway remodelling in COPD 37
can produce at least five different isotypes: the con-ventional 18 kD extracellular bFGF, as well as fouradditional HMW forms that are predominantly nuclearin localization. All five isoforms are able to translo-cate to the nucleus upon activation of different cells.In the nucleus, FGF-2 can act as a modulator of ribo-somal gene transcription via direct interaction of theregulatory subunit of the protein kinase CKII. Also,the FGF receptors can be translocated to the nucleus,as was demonstrated by a study by Stachowiak et alshowing co-localization of the receptor FGFR-1 andFGF-2 in the nucleus, which could indicate a novelFGFR-1 and FGF-2 functional mechanism [42]. Fromthe pattern that we observed, we assume that the pos-itivity in the nuclei was not due to an artefact but wasrepresentative of specific localization of the appropri-ate antigen by the antibody used. In the same section,some nuclei were distinctly positive, whereas adjoin-ing nuclei were clearly negative. Taken together, therole of FGF-2 isoforms in the nucleus is very com-plex, but may well represent an important feature offunctional regulation.
Our ASM cell culture experiments in vitro indicatethat FGF-2 and to a lesser extent FGF-1 are potentmitogens for airway smooth muscle cells, as shownby increased [3H]thymidine incorporation. However,scarce Ki-67-positive ASM cells in COPD despiteenhanced FGF expression indicate a low cell turnoverand untimely proliferation due to tissue damage. Ourresults are in accordance with previous studies onthe mitogenic activity of these molecules [43,44] andfurther strengthen the role of FGFs in COPD.
Pearson’s correlation analysis revealed a significantinverse correlation of FEV1 on the one hand withexpression of FGF-1, FGF-2, and receptor FGFR-1 inbronchial epithelium, and on the other hand with FGF-2 and FGFR-1 in ASM. These findings may indicatethat the expression of these molecules is related to air-flow limitation. Additionally, we observed a positivecorrelation of epithelial FGF-1 expression and pack-years in all patients, although no significant differencewas observed when comparing pack-years betweennon-COPD and COPD patients. This suggests thatresponses to cigarette smoke exposure are involved inepithelial cell function. We also observed a highly sig-nificant correlation of FGF-1/FGFR-1 co-localizationin bronchial epithelium and FGF-2/FGFR-1 in ASMcells. These findings indicate that FGF-1 and FGF-2are differentially expressed and may regulate locallydifferent events in the corresponding tissues.
In vivo and in vitro data indicate that smooth mus-cle cells, and their cross-talk with myo-fibroblasts andinflammatory cells via growth factors and cytokines,are major factors in airway remodelling due to a vari-ety of pathophysiological conditions [36,45–47]. Inline with this general picture, our findings suggest thatthe FGF–FGFR system contributes to airway remod-elling in COPD. Taken together, our results supportthe notion that increased bronchial expression of FGF-1, FGF-2, and FGFR-1 in patients with COPD could
participate in regulating the process of pulmonary air-way remodelling. Blockade of these pathways shouldbe considered in the development of therapeutic inter-ventions aimed to prevent or reverse chronic airflowlimitation in COPD.
Acknowledgements
We thank Drs J Stolk (Leiden University Medical Center)and R Slingerland (Southern Hospital, Rotterdam) for theirhelp in analysing the clinical data. Financial support from TheNetherlands Asthma Foundation (grant No NAF 32.97.73) isgratefully acknowledged.
References
1. Pauwels RA, Buist AS, Ma P, Jenkins CR, Hurd SS. Globalstrategy for the diagnosis, management, and prevention of chronicobstructive pulmonary disease: National Heart, Lung, and BloodInstitute and World Health Organization Global Initiative forChronic Obstructive Lung Disease (GOLD): executive summary.Respir Care 2001; 46: 798–825.
2. Madison JM, Irwin RS. Chronic obstructive pulmonary disease.Lancet 1998; 352: 467–473.
3. Jeffery PK. Remodeling in asthma and chronic obstructive lungdisease. Am J Respir Crit Care Med 2001; 164: S28–S38.
4. Aubert JD, Dalal BI, Bai TR, Roberts CR, Hayashi S, Hogg JC.Transforming growth factor beta 1 gene expression in humanairways. Thorax 1994; 49: 225–232.
5. Aubert JD, Hayashi S, Hards J, Bai TR, Pare PD, Hogg JC.Platelet-derived growth factor and its receptor in lungs frompatients with asthma and chronic airflow obstruction. Am J Physiol1994; 266: L655–L663.
6. de Boer WI, van Schadewijk A, Sont JK, et al. Transforminggrowth factor beta1 and recruitment of macrophages and mast cellsin airways in chronic obstructive pulmonary disease. Am J RespirCrit Care Med 1998; 158: 1951–1957.
7. Zhang L, Rice AB, Adler K, et al. Vanadium stimulates humanbronchial epithelial cells to produce heparin-binding epidermalgrowth factor-like growth factor. A mitogen for lung fibroblasts.Am J Respir Cell Mol Biol 2001; 24: 123–131.
8. Zhang S, Smartt H, Holgate ST, Roche WR. Growth factorssecreted by bronchial epithelial cells control myofibroblastproliferation: an in vitro co-culture model of airway remodelingin asthma. Lab Invest 1999; 79: 395–405.
9. Holgate ST. Asthma: a dynamic disease of inflammation andrepair. Ciba Found Symp 1997; 206: 5–28; discussion 28–34,106–110.
10. Chung KF, Sterk PJ. The airway smooth muscle cell: a majorcontributor to asthma? Eur Respir J 2000; 15: 438–439.
11. Szebenyi G, Fallon JF. Fibroblast growth factors as multifunc-tional signaling factors. Int Rev Cytol 1999; 185: 45–106.
12. Barrios R, Pardo A, Ramos C, et al. Upregulation of acidicfibroblast growth factor during development of experimental lungfibrosis. Am J Physiol 1997; 273: L451–L458.
13. al-Dossari GA, Jessurun J, Bolman RM, et al. Pathogenesis ofobliterative bronchiolitis. Possible roles of platelet-derived growthfactor and basic fibroblast growth factor. Transplantation 1995;59: 143–145.
14. Grashoff WF, Sont JK, Sterk PJ, et al. Chronic obstructivepulmonary disease: role of bronchiolar mast cells andmacrophages. Am J Pathol 1997; 151: 1785–1790.
15. Quanjer PH, Tammeling GJ, Cotes JE, et al. Lung volumes andforced ventilatory flows. Report Working Party Standardizationof Lung Function Tests, European Community for Steel and Coal.Official Statement of the European Respiratory Society. Eur RespirJ Suppl 1993; 16: 5–40.
16. Kranenburg AR, De Boer WI, Van Krieken JH, et al. Enhancedexpression of fibroblast growth factors and receptor FGFR-1
J Pathol 2005; 206: 28–38

38 AR Kranenburg et al
during vascular remodeling in chronic obstructive pulmonarydisease. Am J Respir Cell Mol Biol 2002; 27: 517–525.
17. Kranenburg AR, Willems-Widyastuti A, Mooi WJ, et al. COPDis associated with increased bronchial deposition of extracellularmatrix proteins. Eur Respir J (in press).
18. Coope RC, Browne PJ, Yiangou C, et al. The location of acidicfibroblast growth factor in the breast is dependent on the activityof proteases present in breast cancer tissue. Br J Cancer 1997; 75:1621–1630.
19. Yiangou C, Cox H, Bansal GS, et al. Down-regulation of a novelform of fibroblast growth factor receptor 1 in human breast cancer.Br J Cancer 1997; 76: 1419–1427.
20. McKay S, de Jongste JC, Saxena PR, Sharma HS. AngiotensinII induces hypertrophy of human airway smooth muscle cells:expression of transcription factors and transforming growth factor-beta1. Am J Respir Cell Mol Biol 1998; 18: 823–833.
21. McKay S, Hirst SJ, Haas MB, et al. Tumor necrosis factor-alphaenhances mRNA expression and secretion of interleukin-6 incultured human airway smooth muscle cells. Am J Respir CellMol Biol 2000; 23: 103–111.
22. Yamaguchi T, Iwano M, Kubo A, et al. IL-6 mRNA synthesisby peripheral blood mononuclear cells (PBMC) in patients withchronic renal failure. Clin Exp Immunol 1996; 103: 279–284.
23. Isacchi A, Bergonzoni L, Sarmientos P. Complete sequence of ahuman receptor for acidic and basic fibroblast growth factors.Nucleic Acids Res 1990; 18: 1906.
24. Hughes SE, Hall PA. Immunolocalization of fibroblast growthfactor receptor 1 and its ligands in human tissues. Lab Invest 1993;69: 173–182.
25. Lams BE, Sousa AR, Rees PJ, Lee TH. Subepithelialimmunopathology of the large airways in smokers with and withoutchronic obstructive pulmonary disease. Eur Respir J 2000; 15:512–516.
26. Di Stefano A, Capelli A, Lusuardi M, et al. Decreased Tlymphocyte infiltration in bronchial biopsies of subjects withsevere chronic obstructive pulmonary disease. Clin Exp Allergy2001; 31: 893–902.
27. Mitchell RS, Stanford RE, Johnson JM, Silvers GW, Dart G,George MS. The morphologic features of the bronchi, bronchioles,and alveoli in chronic airway obstruction: a clinicopathologicstudy. Am Rev Respir Dis 1976; 114: 137–145.
28. Nagai A. Pathology and pathophysiology of chronic obstructivepulmonary disease. Intern Med 2002; 41: 265–269.
29. Tiddens HA, Pare PD, Hogg JC, Hop WC, Lambert R, de Jong-ste JC. Cartilaginous airway dimensions and airflow obstruction inhuman lungs. Am J Respir Crit Care Med 1995; 152: 260–266.
30. Dube J, Chakir J, Dube C, Grimard Y, Laviolette M, Boulet LP.Synergistic action of endothelin (ET)-1 on the activation ofbronchial fibroblast isolated from normal and asthmatic subjects.Int J Exp Pathol 2000; 81: 429–437.
31. Morishima Y, Nomura A, Uchida Y, et al. Triggering theinduction of myofibroblast and fibrogenesis by airway epithelialshedding. Am J Respir Cell Mol Biol 2001; 24: 1–11.
32. Aubry MC, Wright JL, Myers JL. The pathology of smoking-related lung diseases. Clin Chest Med 2000; 21: 11–35, vii.
33. Liebler JM, Picou MA, Qu Z, Powers MR, Rosenbaum JT.Altered immunohistochemical localization of basic fibroblastgrowth factor after bleomycin-induced lung injury. Growth Factors1997; 14: 25–38.
34. Singh TM, Abe KY, Sasaki T, Zhuang YJ, Masuda H, Zarins CK.Basic fibroblast growth factor expression precedes flow-inducedarterial enlargement. J Surg Res 1998; 77: 165–173.
35. Scheinowitz M, Abramov D, Eldar M. The role of insulin-likeand basic fibroblast growth factors on ischemic and infarctedmyocardium: a mini review. Int J Cardiol 1997; 59: 1–5.
36. Becerril C, Pardo A, Montano M, Ramos C, Ramirez R, Sel-man M. Acidic fibroblast growth factor induces an antifibrogenicphenotype in human lung fibroblasts. Am J Respir Cell Mol Biol1999; 20: 1020–1027.
37. Pickering JG, Uniyal S, Ford CM, et al. Fibroblast growth factor-2 potentiates vascular smooth muscle cell migration to platelet-derived growth factor: upregulation of alpha2beta1 integrin anddisassembly of actin filaments. Circ Res 1997; 80: 627–637.
38. Boers JE, Ambergen AW, Thunnissen FB. Number and prolifera-tion of basal and parabasal cells in normal human airway epithe-lium. Am J Respir Crit Care Med 1998; 157: 2000–2006.
39. Demoly P, Simony-Lafontaine J, Chanez P, et al. Cell prolifera-tion in the bronchial mucosa of asthmatics and chronic bronchitics.Am J Respir Crit Care Med 1994; 150: 214–217.
40. Delrieu I. The high molecular weight isoforms of basic fibroblastgrowth factor (FGF-2): an insight into an intracrine mechanism.FEBS Lett 2000; 468: 6–10.
41. Boilly B, Vercoutter-Edouart AS, Hondermarck H, Nurcombe V,Le Bourhis X. FGF signals for cell proliferation and migrationthrough different pathways. Cytokine Growth Factor Rev 2000;11: 295–302.
42. Stachowiak MK, Maher PA, Joy A, Mordechai E, Stachowiak EK.Nuclear localization of functional FGF receptor 1 in human astro-cytes suggests a novel mechanism for growth factor action. MolBrain Res 1996; 38: 161–165.
43. Hawker KM, Johnson PR, Hughes JM, Black JL. Interleukin-4inhibits mitogen-induced proliferation of human airway smoothmuscle cells in culture. Am J Physiol 1998; 275: L469–L477.
44. Hirst SJ, Twort CH, Lee TH. Differential effects of extracellularmatrix proteins on human airway smooth muscle cell proliferationand phenotype. Am J Respir Cell Mol Biol 2000; 23: 335–344.
45. Chen CH, Henry PD. Atherosclerosis as a microvascular disease:impaired angiogenesis mediated by suppressed basic fibroblastgrowth factor expression. Proc Assoc Am Physicians 1997; 109:351–361.
46. Jones R, Jacobson M, Steudel W. Alpha-smooth-muscle actinand microvascular precursor smooth-muscle cells in pulmonaryhypertension. Am J Respir Cell Mol Biol 1999; 20: 582–594.
47. Ambalavanan N, Bulger A, Philips IJ. Hypoxia-induced release ofpeptide growth factors from neonatal porcine pulmonary arterysmooth muscle cells. Biol Neonate 1999; 76: 311–319.
J Pathol 2005; 206: 28–38




















