
Children with frequent infections: A proposal for a stepwise assessment and investigation of the...
11
Review Article Children with frequent infections: A proposal for a stepwise assessment and investigation of the immune system ÔThe immune defense to foreign invadersÕ Symphony. Which instrument is out of tune? The immune system (IS) is characterized by spontaneity and ÔeducatedÕ function regarding specificity, diversity, and self-recognition. Its structure, organization and ÔharmoniousÕ func- tion resemble a symphonic orchestra. The integrity of the human organism relies on the prompt and adequate production of various cells and proteins. The inability of the organism to respond satisfactorily by the production of all the appropriate substances leads to an ineffective immune response. This resembles an orchestra in which the various instruments are not ÔtunedÕ together with final result cacophony and turmoil. Cassimos DC, Liatsis M, Stogiannidou A, Kanariou MG. Children with frequent infections: A proposal for a stepwise assessment and investigation of the immune system. ÔThe immune defense to foreign invadersÕ Symphony. Which instrument is out of tune? Pediatr Allergy Immunol 2010: 21: 463–473. Ó 2009 John Wiley & Sons A/S Although many children develop frequent infections, only a few have an underlying immune disorder. Children with dysfunction of the immune system develop frequent infections and/or recurrent, persistent, severe, and rare infections. The aim of this review is to provide to the clinician a valuable tool for recognizing any ÔdiscordsÕ of the Ôimmune-system symphonic orchestraÕ. By following a reverse route, it will be possible to brighten up the dark and winding road of immunodeficiencies and identify the exact point of immune dysfunction. This is fundamental and crucial to perceive etiologic management and subsequently achieve the best for these young patients and their families. Dimitrios C. Cassimos 1,2 , Manolis Liatsis 1 , Anastasia Stogiannidou 2 and Maria G. Kanariou 1 1 Department of Immunology-Histocompatibility, Center for Primary Immunodeficiencies – Paediatric Immunology, Aghia Sophia ChildrenÕs Hospital, Athens, Greece, 2 Department of Pediatrics, Democritus University of Thrace, Alexandroupolis, Greece Key words: infections; immune system; investigation; algorithm; immunodeficiencies Maria G. Kanariou, Department of Immunology- Histocompatibility, Center for Primary Immunodeficiencies – Paediatric Immunology, Aghia Sophia ChildrenÕs Hospital, Athens, Greece E-mail: [email protected] Accepted 15 September 2009 Abbreviations: AFP, a-Fetoprotein; AH50, alternative hemolytic 50; AIDS, acquired immunodeficiency syndrome; ALPS, autoimmune lymphoproliferative syndrome; APC, antigen presenting cells; APECED, autoimmune polyendocrinopathy candidiasis ectodermal dystrophy; AT, ataxia telangiectasia; BMT, bone marrow transplantation; CD, cluster differentiation; CEA, carcino- embryonic antigen; CGD, chronic granulomatous disease; CH50, classical hemolytic 50; CHS, Chediak-Higashi Syndrome; CID, combined immunodeficiencies; CMCC, chronic mucocataneous candidiasis; CMV, cytomegalovirus; CVID, common variable immunodeficiency; DGS, Di George Syndrome; DHR, dihydrorhodamine; EBV, Epstein–Barr virus; FBC, full blood count; FcR, Fc portion of immunoglobulin receptor; GI, gastrointestinal system; GS, Griscelli Syndrome; GVHD, graft vs. host disease; HIES, hyper IgE syndrome; HSV, herpes simplex virus; IBD, inflammatory bowel disease; ICOS, inducible co-stimulation; ID, immuno- deficiency; IFN, interferon; Ig(s), immunoglobulin(s); IL-12R, interleukin-12 receptor; IPEX, immune dysregulation, polyendo- crinopathy, enteropathy X-Linked inheritance; IS, immune system; IVIG, intravenous immunoglobulin; LAD, leukocyte adhesion deficiency; MHC major histocompatibility complex; NBT nitroblue tetrazolium test; NK natural killer cell; PID primary immun- odeficiencies; PRP pattern recognition receptors; SCID severe combined immunodeficiency; SCIG subcutaneous immunoglobulins; SLE systemic lupus erythematosus; TCR T-cell receptor; TLR Toll-like receptor; VZV Varicella Zoster Virus; WAS Wiskott Aldrich Syndrome; WHIM warts, hypogammaglobulinemia, infections, myelokathexis; XLA, X linked agammaglobulinemia; XLP X linked proliferative disease. Pediatr Allergy Immunol 2010: 21: 463–473 DOI: 10.1111/j.1399-3038.2009.00964.x Ó 2009 John Wiley & Sons A/S PEDIATRIC ALLERGY AND IMMUNOLOGY 463
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Children with frequent infections: A proposal for a stepwise assessment and investigation of the...

Review Article
Children with frequent infections: A proposalfor a stepwise assessment and investigation ofthe immune system�The immune defense to foreign invaders�Symphony. Which instrument is out of tune?
The immune system (IS) is characterized byspontaneity and �educated� function regardingspecificity, diversity, and self-recognition. Itsstructure, organization and �harmonious� func-tion resemble a symphonic orchestra.The integrity of the human organism relies on
the prompt and adequate production of various
cells and proteins. The inability of the organismto respond satisfactorily by the production of allthe appropriate substances leads to an ineffectiveimmune response. This resembles an orchestra inwhich the various instruments are not�tuned� together with final result cacophony andturmoil.
Cassimos DC, Liatsis M, Stogiannidou A, Kanariou MG. Childrenwith frequent infections: A proposal for a stepwise assessment andinvestigation of the immune system. �The immune defense to foreigninvaders� Symphony. Which instrument is out of tune?Pediatr Allergy Immunol 2010: 21: 463–473.� 2009 John Wiley & Sons A/S
Although many children develop frequent infections, only a few have anunderlying immune disorder. Children with dysfunction of the immunesystem develop frequent infections and/or recurrent, persistent, severe,and rare infections. The aim of this review is to provide to the clinician avaluable tool for recognizing any �discords� of the �immune-systemsymphonic orchestra�. By following a reverse route, it will be possible tobrighten up the dark and winding road of immunodeficiencies andidentify the exact point of immune dysfunction. This is fundamentaland crucial to perceive etiologic management and subsequently achievethe best for these young patients and their families.
Dimitrios C. Cassimos1,2, ManolisLiatsis1, Anastasia Stogiannidou2 andMaria G. Kanariou1
1Department of Immunology-Histocompatibility,Center for Primary Immunodeficiencies – PaediatricImmunology, Aghia Sophia Children�s Hospital,Athens, Greece, 2Department of Pediatrics,Democritus University of Thrace, Alexandroupolis,GreeceKey words: infections; immune system; investigation;algorithm; immunodeficiencies
Maria G. Kanariou, Department of Immunology-Histocompatibility, Center for PrimaryImmunodeficiencies – Paediatric Immunology,Aghia Sophia Children�s Hospital, Athens, GreeceE-mail: [email protected]
Accepted 15 September 2009
Abbreviations: AFP, a-Fetoprotein; AH50, alternative hemolytic 50; AIDS, acquired immunodeficiency syndrome; ALPS,autoimmune lymphoproliferative syndrome; APC, antigen presenting cells; APECED, autoimmune polyendocrinopathy candidiasisectodermal dystrophy; AT, ataxia telangiectasia; BMT, bone marrow transplantation; CD, cluster differentiation; CEA, carcino-embryonic antigen; CGD, chronic granulomatous disease; CH50, classical hemolytic 50; CHS, Chediak-Higashi Syndrome; CID,combined immunodeficiencies; CMCC, chronic mucocataneous candidiasis; CMV, cytomegalovirus; CVID, common variableimmunodeficiency; DGS, Di George Syndrome; DHR, dihydrorhodamine; EBV, Epstein–Barr virus; FBC, full blood count; FcR, Fcportion of immunoglobulin receptor; GI, gastrointestinal system; GS, Griscelli Syndrome; GVHD, graft vs. host disease; HIES,hyper IgE syndrome; HSV, herpes simplex virus; IBD, inflammatory bowel disease; ICOS, inducible co-stimulation; ID, immuno-deficiency; IFN, interferon; Ig(s), immunoglobulin(s); IL-12R, interleukin-12 receptor; IPEX, immune dysregulation, polyendo-crinopathy, enteropathy X-Linked inheritance; IS, immune system; IVIG, intravenous immunoglobulin; LAD, leukocyte adhesiondeficiency; MHC major histocompatibility complex; NBT nitroblue tetrazolium test; NK natural killer cell; PID primary immun-odeficiencies; PRP pattern recognition receptors; SCID severe combined immunodeficiency; SCIG subcutaneous immunoglobulins;SLE systemic lupus erythematosus; TCR T-cell receptor; TLR Toll-like receptor; VZV Varicella Zoster Virus; WAS Wiskott AldrichSyndrome; WHIM warts, hypogammaglobulinemia, infections, myelokathexis; XLA, X linked agammaglobulinemia; XLP X linkedproliferative disease.
Pediatr Allergy Immunol 2010: 21: 463–473
DOI: 10.1111/j.1399-3038.2009.00964.x
� 2009 John Wiley & Sons A/S
PEDIATRIC ALLERGY AND
IMMUNOLOGY
463

Normally, all children have the experience ofinfections. The question that the clinician isfacing in his everyday practice is in which casesshould he suspect an underlying immune disor-der and how should he stratify the investigation.To answer these questions, it is important first ofall to review the structure and the function of theIS (1–8).Immune defense commences within hours with
an early (innate) non-specific response to foreigninvaders, continues with a slower specificresponse to certain molecules, and a non-specificaugmentation �crescendo� of this response. Thememory of the IS for specific antigens providesitself with the ability for a more immediateand vigorous response to a future challenge bythe same microorganism (adaptive-specificimmunity).The efficient immune response is completed in
two phases; the recognition and the effectorphases. The recognition of the foreign sub-stances is performed by PRP or TLR and thesubsequent binding to APC. Thereafter, thecascade of the immune response will follow.APC will process and present the antigentogether with MHC molecules to T cells viatheir receptors (TCR).Innate immunity, which is the first line defense
of the organism, is �performed� by cells (e.g., thephagocytes) and soluble components (e.g.,the complement) and in our metaphore is thebasic �tempo� of the orchestra. Specific immunity,the delicate complete immune response, derivedfrom the lymphocytes, that is of the �solo�instruments, which are mostly responsible forthe melody itself.
Phagocytic compartment
The phagocytic compartment of the immunesystem includes neutrophils, eosinophils, andmonocytes–macrophages. The latest migrate tothe tissues where they differentiate into specifictissue macrophages. The response of phagocytesagainst bacteria is integrated through phago-cytosis, which is accomplished in a stepwiseprocess. The first step in phagocytosis is chemo-taxis by which the phagocytic cells get attractedto the infected site moving towards bacteria andvarious substances that are produced in animmune response. The next step is adherence ofthe antigen to the phagocytic cell membrane. Thephagocytes have specific receptors at their cellu-lar membrane, thus enhancing the ability toattract antigens and bind to them more readily.By this process (opsonization), some antigens arerendered more susceptible to phagocytosis.
Complement system
The complement system participates, activates,and reinforces various immune functions likechemotaxis and opsonization. The complementactivates neutrophils and macrophages that sub-sequently induce lysis of bacteria and increasethe vascular permeability. Activation of eitherthe classical or the alternative pathway of thecomplement leads to C3 activation, consequentlyconverging to the same final route, ending to theproduction of the same biologic particles. Thecomplement functions as an autoregulated, auto-restricted system. In humans, deficiencies ofcomplement components are related to charac-teristic clinical syndromes. Patients with C1, C4,or C2 deficiency develop symptoms that resembleSLE and less frequently recurrent pyogenicinfections. Deficiencies of C3 and componentsof the final part of the complement (C5–9) arerelated with recurrent bacterial infections (9, 10).Nevertheless, hypocomplementemia can be the
final result of complement consumption, as mayoccur in autoimmune diseases or during aninfection (2, 11–13).
Lymphocytic system (LS)
The basic component of the LS is the lympho-cyte. The major populations of the lymphocytesare T, B, and Natural Killer (NK) cells. T and Bcells are further divided into subpopulations withdiscrete functions. The hallmark for differentiat-ing them is their immunophenotype that isdetermined in relation to specific markers (gly-coproteins) that they express on their surface andin the cytoplasm.T lymphocytes are divided into two main cell
types: CD4+ (T-helper – Th) and CD8+
(T-suppressor/cytotoxic-Ts/c). Th cells have acentral role in the immune response, as the�conductor� of the orchestra of the IS. Theyrecognize antigens, which are presented to themby APC in conjunction with MHC class IImolecules, and consequently they activate otherimmune cells inducing an immune response(Fig. 1). This process is enhanced by otherfunctioning molecules that are lymphocyte sur-face proteins, which increase binding, signaling,and homing of antigens. Th cells that are derivedfrom naı̈ve Th0 are divided into three distinctsubgroups depending on the cytokines theyproduce (Th1, Th2, and Th17). Th1 cells secreteIL-2 and IFN-c, which are favoring thedevelopment of cellular immunity. Th2 cellssecrete IL-4, IL-5, and IL-13 that regulate B-cellresponses, immunity against parasites, and are
Cassimos et al.
464

crucial for allergic diseases by the induction ofIgE production. Th17 cells play a key role inimmunoregulation (14). Ts/c cells restrict theimmune response and induce cytotoxicity totarget cells. They recognize viral antigens inconjunction with MHC class I molecules.B lymphocytes constitute the other major
lymphocytic population. After an antigenic stim-ulation, most of the B cells differentiate intoplasma cells that produce and secrete specificantibodies. Initially, these are IgM and later IgMand IgD and finally IgG, IgA, or IgE. Thisgradual process of Igs production that is knownas �isotype switching� requires the interaction ofseveral proteins, mainly CD40 on B-cell surfacewith its ligand (CD40L) on activated T cells. Bcells also function as APC in a secondaryimmune response.Some of the T and B cells differentiate to
memory cells that act as a reservoir of immuneresponse in a future antigenic challenge (Fig. 1).T cells are mostly responsible for the defenseagainst viruses, fungi, and parasites, as well astumors and foreign grafts while B cells towardsbacteria. There is also a great deal of interactionof these two populations to accomplish a satis-factory immune response. The communication ofthese and other cells of the IS is achieved by:(i) soluble mediators known as cytokines, mono-kines, or lymphokines, secreted by macrophages,monocytes, or lymphocytes, respectively. (Cyto-
kines that induce chemotaxis of leucocytes arereferred to as chemokines), (ii) specific cytokinereceptors on the surface of these cells (IL-12R,IFN-cR), and (iii) adhesion molecules.The NK cells form another part of the
lymphocytic population. They do not havememory and have limited specificity. Their actionagainst tumor cells and cells infected with virusesdoes not require the presence of antibodies orantigenic stimulation and therefore belongs tothe innate immunity.
Immunodeficiencies and time of onset
All children have the experience of infections.Children in playgroups – nurseries can develop8–10 upper respiratory tract infections and 1–2infections of the GI tract annually. Thereare some characteristics from the personaland family history that are more indicative ofan underlying immune disorder (Table 1) (15–18).The suspicion about an immune deficiency
should emerge in various clinical symptoms andsigns that the clinician comes across, and shouldbe able to identify (Table 2) (15, 16). It is alsoimportant to determine the time of onset ofthe disease (19). Inherited defective productionof antibodies become evident after the age of6 months, when antibodies of maternal originhave dissipated. On the other hand, SCID
ThB
memory
B
Teffector
NK
Tmemory
APC
CD80/CD86
CD28
CD40L
CD40Plasma
cell
MHC II
Cytokines
Antigen
TCR
CD80/CD86
CD28
Fig. 1. Schematic illustration ofthe immune response after anantigenic stimulation.
An immune investigation algorithm for children with frequent infections
465

presents soon after birth, while LAD causesdelay in the separation of the umbilical cordbeyond the age of 2 wk. Finally, CVID has noage preference and can present from late infancyto young adulthood with a broad spectrum ofsymptoms. The combination of various symp-toms and signs is pathognomonic of certainsyndromes or specific immunodeficiencies(Table 3) (19–40). Apart from the detailedmedical history, a thorough clinical examinationis a prerequisite for the objective evaluation ofthe problem.
Disease entity – epidemiology
Immunodeficiencies (IDs) are divided into pri-mary and secondary. In children, primary im-munodeficiencies (PID) are more common; whilein adults predominate the secondary. PID arevery rare, with an overall incidence one in 2000live births (41). In a recent publication, theprevalence of PID is estimated to be one in 1200persons. (42). Nevertheless, there is evidence thatPID are more common than what was previouslythought (43, 44).There is a lot of heterogeneity in the etiology
and the underlying genetic disorder of more than200 (45) known PID. Antibody deficiency is themost common type of PID, accounting for about2/3 of them, while complement deficiency com-prises <1%. Cellular and phagocytic defectsaccount for 20–30% of PID. There can also be agreat deal of variability in the clinical presenta-tion and the severity of the disease making thelong and winding pathway to the correct diag-nosis more difficult and more obscure (24, 41). Inregards to the immunologic pathway that isimpaired, PID are divided into eight groups ofsyndromes (Table 4) (46).
Table 1. Signals from history in children with possible immunodeficiency
Personal historyInfections Severe, recurrent, in multiple sites,
with prolonged duration, withpoor response to
treatment, with major complications,abscesses
Infective agents OpportunisticImmunizations With complication or disseminated
infection (e.g., BCG-itis)Dermatitis/eczema Severe and/or persistentBronchial asthma Severe, resistant, or corticoid dependentChronic lung diseaseAutoimmune manifestations Cytopenias, vasculitisDysmorphism, MicrocephalyDevelopmental delayChronic diarrheaDelayed separation of the
umbilical cord (>2 wk)Family history
Unexplained death in infancyKnown or possible immunodeficiencyParental consanguinityInfections at the home environment (HIV)Autoimmune diseases, SLELymphoma
Table 2. Clinical signs and symptoms in children with possible immunodefi-ciency
Failure to thriveDysmorphism, microcephalyExtremities, nails Digital clubbing, palenessMouth Periodonditis, gingivostomatitis,
delayed shedding of primary teethCardiovascular Congenital heart diseaseRespiratory Chronic lung disease,
bronchiectasis, pneumatocelesGastrointestinal Hepatosplenomegaly, IBDLymphatic tissue Absent or excessive proliferation
with lymphadenopathy, thymomaSkin, hair Eczema, petechiae, scars,
granullomas, angioedema, albinismPigmentary changes, severe
skin infections, abnormal hairEyes Retinal abnormalities, telangiectasiaSkeletal abnormalitiesNervous system Ataxia
Table 3. Characteristic clinical signs and/or laboratory findings in certainimmunodeficiencies
AT Cerebellar ataxia, oculocutaneous telangiectasia,elevated AFP and CEA
CGD Deep seated infections, abscesseswith granulomatous formation
CHS Partial oculocutaneous albinism, neurologic symptoms,and giant azurophil granules in neutrophils
CMCC Recurrent candidal infections of nails, skin, andmucous membranes, defective T-cell response
Complementdeficiency
Neisserial infection orautoimmune disorders
DGS Hypocalcaemic seizures, cardiacdisease, abnormal facies
GS Pigmentary dilution, neurologic abnormalities,pyogenic infections, and a hemophagocytic syndrome
HIES Failure to shed primary teeth, thickened skin,skin and lung infections, skeletal abnormalities
IFN-cR, IL-12R Atypical mycobacterial orSalmonella infections
LAD Delayed separation of the umbilical cord,chronic severe periodontitis, lack of pus
SCID Viral or fungal infections, chronic diarrhea,failure to thrive in early infancy
XLA Absent lymph nodes and tonsils,agammaglobulinaemia, and absence of B-cells
XLP Fulminant infectious mononucleosis,lymphoma and dysgammaglobulineamia
WAS Thrombocytopenia with small in size platelets,eczema, and recurrent infections by
encapsulated bacteriaWHIMS Warts, hypogammaglobulinemias,
immunodeficiency, and myelocathexis
Cassimos et al.
466

Diagnostic approach – stepwise evaluation andassessment
Although in the majority of children withfrequent infections, there is no underlyingimmune disorder, a delay in recognizing animmune dysfunction could be devastating forthese young patients. The earliest diagnosis iscrucial to pursue an etiologic management of thedisease and, subsequently, avoid the conse-quences of severe and/or recurrent infections.The recognition of the �damaged� immune path-way is substantial in identifying the possible�uninvited visitors�-microorganisms (Table 5)(2, 47–49). By following the reverse route, it ispossible to orientate to the possible implicatedimmune pathway. Functioning as a compass tothe clinician, are the presenting symptoms, thetype of infection(s), the severity of the diseaseand the microorganisms involved. Certainly, it isimportant to exclude secondary immunodeficien-cies and other causes of frequent infections(Table 6).The development of abscesses and deep sited
infections from Staphylococcus, Serratia, orAspergillus is consistent with neutrophils defi-ciency/dysfunction, as in CGD (50–53). Recur-rent infections from Neisseria (meningitidis orgonorrhoeae) emerge in patients with deficiencyof the final part of the complement (C5–C9) (12,54). Pyogenic bacteria (Haemophilus influenzaetype b, Pneumococcus, Pseudomonas) develop inpatients with deficient antibody response (55),whilst infections by intracellular organisms(Mycobacterium, Salmonella) and viruses aremore indicative of an underlying disorder of thecellular part of immunity and, more specifically,of the T cells and type 1 cytokine production (25,56). Recurrent and/or severe Herpes or Papil-loma virus infections may be seen in NKdysfunction. Patients with combined immunode-ficiencies (CID) can experience infections fromthe entire range of pathogens (2, 41).Although there are some outstanding proto-
cols for the diagnostic screening of PID, our
effort is to present a stepwise rational approachto the child with frequent infections, for a �handy�use in the �bed side� medicine (41, 47, 48, 57, 58).The initial investigation screening includes:
FBC, Igs (IgG, IgA, IgM, IgE), complement(CH50, C3, C4), and isohemmaglutinins(Table 7). From the FBC, it is possible to excludeneutropenia or lymphopenia. Neutropenia war-rants further investigation as well as lymphope-nia that can be the �prelude� of SCID.Nevertheless, marked leukocytosis and neutro-philia is almost always present in cases of LAD(41). White cells morphology can attribute to thediagnosis, like in CHS, where neutrophils havecharacteristic giant azurophil granules. Plateletsthat reflect the bone marrow function, can bediminished in WAS and in autoimmune diseases.Hb, Hct, and red cells morphology are goodmarkers for an underlying chronic disease.Cytopenias that are very alarming should bemonitored and investigated thoroughly (41, 46,59). CVID, one of the most common PID, exceptfrom autoimmune cytopenias and infections, canalso develop IBD and lymphomas (19). Erythro-phagocytosis can be the presenting feature ofsome PID (60). Evaluation of B-cell function, inthe first place, is assessed by measurement of Igs.The screening is extended in accordance withthe clinical picture and in conjunction withthe pathogenic microorganisms (Tables 6–8).The antibody function is further evaluated bymeasurement of specific antibodies, isohemmag-glutinins (anti-A, anti-B IgM antibodies), andIgG subclasses. (8, 61). However, infants 6–8 months old often develop transient hypo-
Table 4. Classification of PID (IUIS, 2007)
Combined T-cell and B-cell immunodeficienciesPredominantly antibody deficienciesOther well-defined immunodeficiency syndromesDiseases of immune dysregulationCongenital defects of phagocyte number, function, or bothDefects in innate immunityAutoinflammatory disordersComplement deficiencies
Table 5. Common pathogens related with infections in specific types of im-munodeficiencies
Immunodeficiencies Common pathogens
Antibody deficiency Bacteria: Staphylococcus aureus, StreptococcusHaemophilus influenza bEnteroviruses: polio, echo
T-cell deficiency Intracellular bacteria: Mycobacteriumspecies, listeria
VirusesFungi: Candida, Pneumocystis jiroveci
Combined T- andB-cell deficiency
BacteriaFungi: Pneumocystis jiroveci, Candida
Viruses: CMV, HSV, MeaslesParasites: Cryptococcus, Cryptosporidium
Phagocytes deficiency Bacteria: Staphylococcus aureus, Salmonella,Nocardia, Mycobacterium species-(atypical)
Fungi: AspergillusInnate immunity deficiency Bacteria: Staphylococcus aureus
Mycobacterium speciesHuman pappiloma virus (HPV)
Complement deficiency Bacteria: Streptococcus, Haemophilus influenza b,Neisseria meningitidis, Neisseria gonorrhoeae
An immune investigation algorithm for children with frequent infections
467
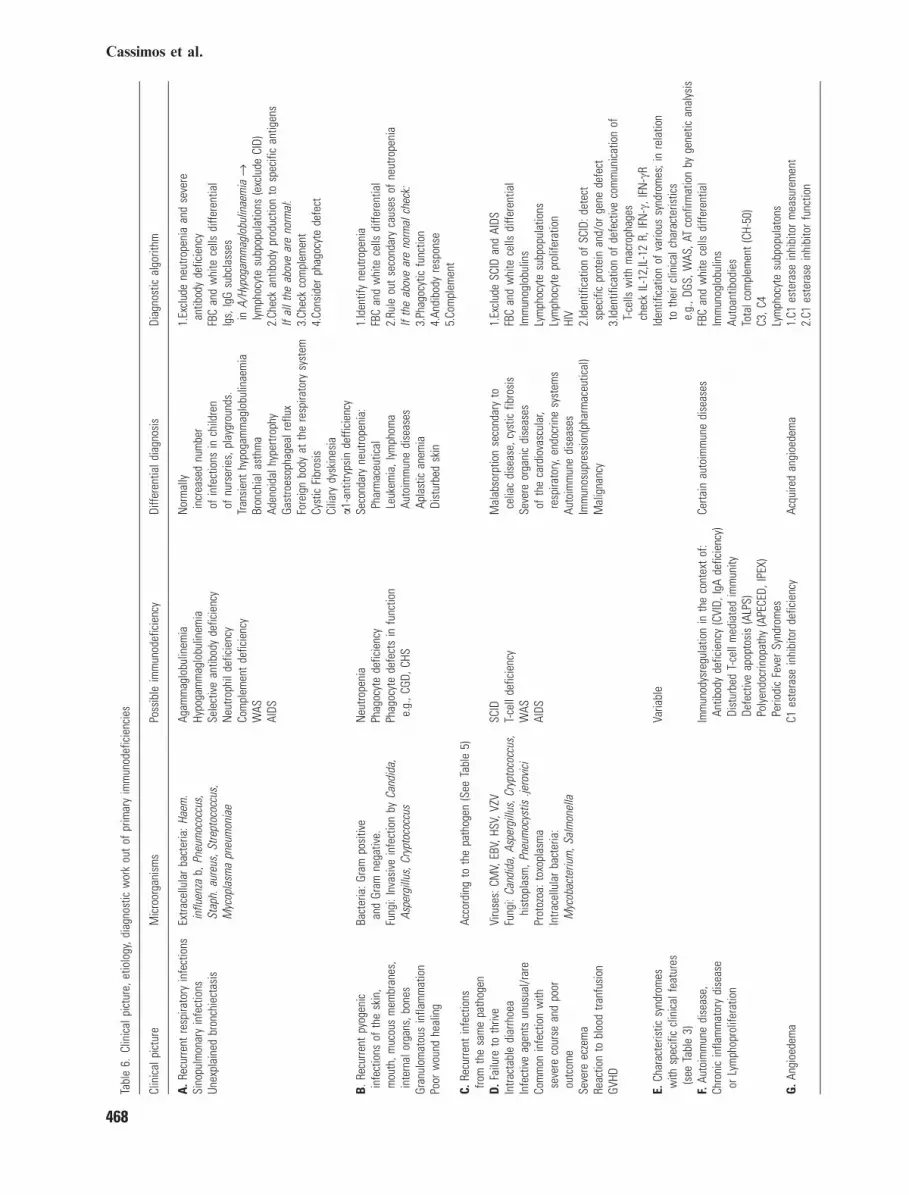
Tabl
e6.
Clin
ical
pict
ure,
etio
logy
,dia
gnos
ticw
ork
out
ofpr
imar
yim
mun
odef
icie
ncie
s
Clin
ical
pict
ure
Mic
roor
gani
sms
Poss
ible
imm
unod
efic
ienc
yDi
ffere
ntia
ldia
gnos
isDi
agno
stic
algo
rithm
A.R
ecur
rent
resp
irato
ryin
fect
ions
Sino
pulm
onar
yin
fect
ions
Unex
plai
ned
bron
chie
ctas
is
Extra
cellu
lar
bact
eria
:Hae
m.
influ
enza
b,Pn
eum
ococ
cus,
Stap
h.au
reus
,Stre
ptoc
occu
s,M
ycop
lasm
apn
eum
onia
e
Agam
mag
lobu
linem
iaHy
poga
mm
aglo
bulin
emia
Sele
ctiv
ean
tibod
yde
ficie
ncy
Neu
troph
ilde
ficie
ncy
Com
plem
ent
defic
ienc
yW
ASAI
DS
Nor
mal
lyin
crea
sed
num
ber
ofin
fect
ions
inch
ildre
nof
nurs
erie
s,pl
aygr
ound
s.Tr
ansi
ent
hypo
gam
mag
lobu
linae
mia
Bron
chia
last
hma
Aden
oida
lhyp
ertro
phy
Gast
roes
opha
geal
reflu
xFo
reig
nbo
dyat
the
resp
irato
rysy
stem
Cyst
icFi
bros
isCi
liary
dysk
ines
iaa1
-ant
itryp
sin
deffi
cien
cy
1.Ex
clud
ene
utro
peni
aan
dse
vere
antib
ody
defic
ienc
yFB
Can
dw
hite
cells
diffe
rent
ial
Igs,
IgG
subc
lass
esin
A/Hy
poga
mm
aglo
bulin
aem
iafi
lym
phoc
yte
subp
opul
atio
ns(e
xclu
deCI
D)2.
Chec
kan
tibod
ypr
oduc
tion
tosp
ecifi
can
tigen
sIf
allt
heab
ove
are
norm
al:
3.Ch
eck
com
plem
ent
4.Co
nsid
erph
agoc
yte
defe
ct
B.R
ecur
rent
pyog
enic
infe
ctio
nsof
the
skin
,m
outh
,muc
ous
mem
bran
es,
inte
rnal
orga
ns,b
ones
Gran
ulom
atou
sin
flam
mat
ion
Poor
wou
ndhe
alin
g
Bact
eria
:Gra
mpo
sitiv
ean
dGr
amne
gativ
e.Fu
ngi:
Inva
sive
infe
ctio
nby
Cand
ida,
Aspe
rgill
us,C
rypt
ococ
cus
Neu
trope
nia
Phag
ocyt
ede
ficie
ncy
Phag
ocyt
ede
fect
sin
func
tion
e.g.
,CGD
,CHS
Seco
ndar
yne
utro
peni
a:Ph
arm
aceu
tical
Leuk
emia
,lym
phom
aAu
toim
mun
edi
seas
esAp
last
ican
emia
Dist
urbe
dsk
in
1.Id
entif
yne
utro
peni
aFB
Can
dw
hite
cells
diffe
rent
ial
2.Ru
leou
tse
cond
ary
caus
esof
neut
rope
nia
Ifth
eab
ove
are
norm
alch
eck:
3.Ph
agoc
ytic
func
tion
4.An
dibo
dyre
spon
se5.
Com
plem
ent
C.Re
curre
ntin
fect
ions
from
the
sam
epa
thog
enAc
cord
ing
toth
epa
thog
en(S
eeTa
ble
5)
D.F
ailu
reto
thriv
eIn
tract
able
diar
rhoe
aIn
fect
ive
agen
tsun
usua
l/rar
eCo
mm
onin
fect
ion
with
seve
reco
urse
and
poor
outc
ome
Seve
reec
zem
aRe
actio
nto
bloo
dtra
nfus
ion
GVHD
Viru
ses:
CMV,
EBV,
HSV,
VZV
Fung
i:Ca
ndid
a,As
perg
illus
,Cry
ptoc
occu
s,hi
stop
lasm
,Pne
umoc
ystis
.jero
vici
Prot
ozoa
:tox
opla
sma
Intra
cellu
lar
bact
eria
:M
ycob
acte
rium
,Sal
mon
ella
SCID
T-ce
llde
ficie
ncy
WAS
AIDS
Mal
abso
rptio
nse
cond
ary
toce
liac
dise
ase,
cyst
icfib
rosi
sSe
vere
orga
nic
dise
ases
ofth
eca
rdio
vasc
ular
,re
spira
tory
,end
ocrin
esy
stem
sAu
toim
mun
edi
seas
esIm
mun
osup
ress
ion(
phar
mac
eutic
al)
Mal
igna
ncy
1.Ex
clud
eSC
IDan
dAI
DSFB
Can
dw
hite
cells
diffe
rent
ial
Imm
unog
lobu
lins
Lym
phoc
yte
subp
opul
atio
nsLy
mph
ocyt
epr
olife
ratio
nHI
V2.
Iden
tific
atio
nof
SCID
:det
ect
spec
ific
prot
ein
and/
orge
nede
fect
3.Id
entif
icat
ion
ofde
fect
ive
com
mun
icat
ion
ofT-
cells
with
mac
roph
ages
chec
kIL
-12,
IL-1
2R,
IFN
-c,I
FN-c
RE.
Char
acte
ristic
synd
rom
esw
ithsp
ecifi
ccl
inic
alfe
atur
es(s
eeTa
ble
3)
Varia
ble
Iden
tific
atio
nof
vario
ussy
ndro
mes
;in
rela
tion
toth
eir
clin
ical
char
acte
ristic
se.
g.,D
GS,W
AS,A
Tco
nfirm
atio
nby
gene
tican
alys
isF.
Auto
imm
une
dise
ase,
Chro
nic
infla
mm
ator
ydi
seas
eor
Lym
phop
rolif
erat
ion
Imm
unod
ysre
gula
tion
inth
eco
ntex
tof
:An
tibod
yde
ficie
ncy
(CVI
D,Ig
Ade
ficie
ncy)
Dist
urbe
dT-
cell
med
iate
dim
mun
ityDe
fect
ive
apop
tosi
s(A
LPS)
Poly
endo
crin
opat
hy(A
PECE
D,IP
EX)
Perio
dic
Feve
rSy
ndro
mes
Certa
inau
toim
mun
edi
seas
esFB
Can
dw
hite
cells
diffe
rent
ial
Imm
unog
lobu
lins
Auto
antib
odie
sTo
talc
ompl
emen
t(C
H-50
)C3
,C4
Lym
phoc
yte
subp
opul
aton
sG
.Ang
ioed
ema
C1es
tera
sein
hibi
tor
defic
ienc
yAc
quire
dan
gioe
dem
a1.
C1es
tera
sein
hibi
tor
mea
sure
men
t2.
C1es
tera
sein
hibi
tor
func
tion
Cassimos et al.
468

gammaglobulinemia and toddlers are notexpected to produce antibodies against polysac-charide antigens (e.g., Haemophilus influenzaetype b and/or Pneumonococcus). Responses topure polysaccharide antigens can be evaluated inpatients older than 2 yr of age (55, 62). Specificantibody levels are preferably interpreted 3 wkpost-vaccination and are expected to rise four-fold for protein antigens and two-fold for poly-saccharide antigens (41, 63). Specific antibodyresponses may be impaired as a result of a B-celldefect or a T-cell failure to help for antibodyproduction (2, 41, 62, 64). In cases with normallevels of Igs, impaired polysaccharide responsescould be due to IgG2 subclass deficiency orspecific antibody deficiency. (62, 64–67). Elevatedserum IgE levels matched with staphylococcus-binding IgE, and eosinophilia are characteristicof HIES.Cellular immunity is assessed by measurement
of the lymphocytes and their subpopulations.Also, with skin prick tests to antigens already�introduced� to the patient by vaccination(tetanus, mumps, diphtheria), or environmentally(Trichopyton, Candida, Proteus) cutaneousdelayed hypersensitivity, an in vivo T-cell specificantigenic response is evaluated (63, 68). A normalresponse is 2–5 mm of induration after 48–72 hof intracutaneous injection (12).The phagocytic part of immunity is assessed
primarily by white cells number and morphol-ogy. To investigate the phagocytic function, it isimportant to evaluate chemotaxis, phagocytosis,and neutrophil oxidative burst. The latter ismeasured by the nitroblue tetrazolium test(NBT) and by the dihydrorhodamine test(DHR), a flow cytometric assay (69, 70).The complement is evaluated initially by mea-
surement of CH50, C3, C4. In the case that thelevels of CH50 are low, the investigation has to
be extended to the other Complement compo-nents. If they are normal and the suspicion aboutcomplement deficiency remains strong, functionof the alternative pathway is further evaluatedwith measurement of AH50. This pathway canbe influenced in properdin or factor D deficiency(12, 41).If an allergic disease is considered as an
underlying cause for the clinical picture, beyondtotal IgE, specific IgE should be measured andin vivo allergic responsehas tobe acknowledgedbyskin prick and/or patch tests for certain allergens.If any of these first line investigations are
abnormal, more specific studies have to beorganized: immunophenotype of T and B cells,evaluation of the response to specific antigens,lymphocyte cultures, immunohistochemistry ofgranule contents, and, finally, measurement of allthe fractions of the complement (Table 8). Invery specific cases, it is required to pursue moreextended investigations with evaluation of theimmune cells communication, measurement ofspecific enzymes, and analysis of white cellglycoproteins. In cases in which certain syn-dromes are suspected, specific investigations areperformed (Table 3). Diagnosis at molecularlevel is mostly desired for the establishment ofunequivocal diagnosis, the identification of can-didates for gene specific therapy, and detection ofcarriers (69, 71–77). An online directory ofspecialized laboratory testing for immunodefi-ciencies has recently become available (78).
Prognosis
The prognosis of children with PID is consideredto be much better in the last two decades,something that is expected to improve even morewith the valuable input, assistance, and collabo-ration of the clinicians and scientists.Nevertheless, the key point for a favorable
prognosis is early diagnosis (15, 16, 48). With thesubsequent prompt etiologic and aggressivetreatment, it is possible to avoid permanent andlife-threatening complications and achieve longerand better quality of life (21, 41). There are threemajor categories of management:
1 Preventive: antimicrobial chemoprophylaxis,immunizations
2 Supportive: (i) replacement (IVIG, SCIG, etc),(ii) treatment of infections and complications
3 Curative: with the inclusion of hematopoieticstem cell transplantation and gene therapy inthe therapeutic repertoire of PID, it has beenachieved complete cure in cases of SCID andother PID (79–81)
Table 7. Investigation for possible immunodeficiency in children with frequentinfections
1st line diagnostic work outFull blood countImmunoglobulins: IgG, IgA, IgM, IgEIsohemagglutininsCH50, C3,C4
2nd line diagnostic work outImmunoglobulin subclasses In frequent bacterial infectionsComplement investigation In recurrent infections
by MeningococcusPhacocytic investigation In infections by Staphylococcus
aureus, gram negative bacteria, fungiLymphocytes subpopulations In severe viral or opportunistic
infectionsHIV investigation
3rd line or specialized diagnosticinvestigation
In opportunistic infections, TBC
An immune investigation algorithm for children with frequent infections
469

Finally, family counseling is an important issue,which can be extremely facilitated with geneticdiagnosis.
Conclusions
PID are a challenge for the clinical pediatrician(4, 43, 81, 82). The immune system resembles anorchestra that comprises many instruments andcan produce �melodies� balsam for the earsequivalent to – complete and effective immuneresponse(s), balsam for the �wounded� organism.A trained pediatrician enjoys the melody ofa �natural normal immune response�, but hasalso the trained �musical ear� that enables him/herto recognize any �discords� equivalent to aninconclusive immune response. The isolation ofthese �discords� will give him/her the hint aboutthe �instrument� – immune pathway, that is �outof tune� – dysfunctioning. The appropriateinvestigation – screening for a possible PID willthereafter be organized, brightening up theobscure pathways of immunodeficiencies (47,48, 83).
In conclusion, this review is intended to be avaluable tool for the clinical pediatrician. Theidentification of signs of PID will be the crucialpoint for the ignition of the diagnostic �mara-thon�. In collaboration with immunologic cen-ters, it will facilitate the revelation of the hiddenimmunopathogenetic route and identify the ID.With the diagnosis of a certain PID, the mostappropriate treatment is organized at specializedcenters and genetic counseling is obtainedaccomplishing the best for these young trouble-some patients and their families.
AcknowledgmentsWe would like to thank Mrs. Kelly Chorafopoulou,Mrs. Penio Kassari, Mr. Sotirios Livas & Mrs. Efi Symeo-nov for their support and contribution.
References
1. Bonilla FA, Geha RS. 12. Primary immunodeficiencydiseases. J Allergy Clin Immunol 2003: 111: S571–81.
2. Chapel H, Haeney M, Misbah S, Snowden N. Basiccomponents: structure and function. In: Chapel H,Haeney M, Misbah S, Snowden N, eds. Essentials of
Table 8. Microorganisms, infections & immunological investigation of Children
Cassimos et al.
470

Clinical Immunology. Oxford: Blackwell Publishing,2006: 1–32.
3. Conley ME, Broides A, Hernandez-Trujillo V,et al. Genetic analysis of patients with defects in earlyB-cell development. Immunol Rev 2005: 203: 216–34.
4. Fischer A. Human primary immunodeficiency diseases:a perspective. Nat Immunol 2004: 5: 23–30.
5. Notarangelo LD, Giliani S, Mazzolari E, Gulino
AV. Primary immune deficiencies unravel the molecularbasis of immune response. Rev Clin Exp Hematol 2003:7: 84–111.
6. Rosen F, Wedgwood R, Eibl M, et al. Primaryimmunodeficiency diseases. Report of a WHO scientificGroup. Clin Exp Immunol 1997: 109: 1–28.
7. Smith CIE, Ochs HD, Puck JM. Genetically deter-mined immunodeficiency diseases: a perspective. In:Ochs HD, Smith CIE, Puck JM, eds. Primary Immu-nodeficiency Diseases, a Molecular and GeneticApproach, 2nd edn.Oxford:UniversityPress, 2007: 3–15.
8. Wood P, Stanworth S, Burton J, et al. Recognition,clinical diagnosis and management of patients withprimary antibody deficiencies: a systematic review. ClinExp Immunol 2007: 149: 410–23.
9. Frank MM. Complement deficiencies. Pediatr ClinNorth Am 2000: 47: 1339–54.
10. Walport MJ. Complement. Second of two parts.N Engl J Med 2001; 344: 1140–4.
11. Fleisher TA. Immune function. Pediatr Rev 1997: 18:351–6.
12. Wen L, Atkinson JP, Giclas PC. Clinical and labo-ratory evaluation of complement deficiency. J AllergyClin Immunol 2004: 113: 585–93; quiz 94.
13. Wood RA, Sampson HA. The child with frequentinfections. Curr Probl Pediatr 1989: 19: 229–84.
14. Dong C. TH17 cells in development: an updated view oftheir molecular identity and genetic programming. NatRev Immunol 2008: 8: 337–48.
15. Ballow M. Primary immunodeficiency disorders: anti-body deficiency. J Allergy Clin Immunol 2002: 109:581–91.
16. Buckley RH. Primary cellular immunodeficiencies.J Allergy Clin Immunol 2002: 109: 747–57.
17. Stiehm RE. The four most common pediatric immun-odeficiencies. Adv Exp Med Biol 2007: 601: 15–26.
18. de Martino M, Ballotti S. The child with recurrentrespiratory infections: normal or not? Pediatr AllergyImmunol 2007: 18 (Suppl. 18): 13–8.
19. Paul ME, Shearer WT. Approach to the evaluation ofthe immunodeficient patient. In: Rich RR, Fleisher
TA, Shearer WT, Schroeder H, Kotzin B, eds.Clinical Immunology: Principles and Practice. London:Mosby International, 2001: 33.1–33.11.
20. Bartsch O, Nemeckova M, Kocarek E, et al.DiGeorge/velocardiofacial syndrome: FISH studies ofchromosomes 22q11 and 10p14, and clinical reports onthe proximal 22q11 deletion. Am J Med Genet A 2003:117A: 1–5.
21. Buckley RH. Primary immunodeficiency or not?Making the correct diagnosis. J Allergy Clin Immunol2006: 117: 756–8.
22. Buckley RH, Schiff RI, Schiff SE, et al. Humansevere combined immunodeficiency: genetic, pheno-typic, and functional diversity in one hundred eightinfants. J Pediatr 1997: 130: 378–87.
23. Cabana MD, Crawford TO, Winkelstein JA,Christensen JR, Lederman HM. Consequences of the
delayed diagnosis of ataxia-telangiectasia. Pediatrics1998: 102: 98–100.
24. Conley ME, Notarangelo LD, Etzioni A. Diagnosticcriteria for primary immunodeficiencies. RepresentingPAGID (Pan-American Group for Immunodeficiency)and ESID (European Society for Immunodeficiencies).Clin Immunol 1999: 93: 190–7.
25. Conley ME, Rohrer J, Minegishi Y. X-linked agam-maglobulinemia. Clin Rev Allergy Immunol 2000: 19:183–204.
26. de Moraes-Vasconcelos D, Orii NM, Romano CC,Iqueoka RY, Duarte AJ. Characterization of the cel-lular immune function of patients with chronic muco-cutaneous candidiasis. Clin Exp Immunol 2001: 123:247–53.
27. Etzioni A, Tonetti M. Leukocyte adhesion deficiencyII-from A to almost Z. Immunol Rev 2000: 178: 138–47.
28. Grimbacher B, Holland SM, Gallin JI, et al. Hyper-IgE syndrome with recurrent infections – an autosomaldominant multisystem disorder. N Engl J Med 1999:340: 692–702.
29. Hamilton JK, Paquin LA, Sullivan JL, et al.X-linked lymphoproliferative syndrome registry report.J Pediatr 1980: 96: 669–73.
30. Hord JD, Whitlock JA, Gay JC, Lukens JN. Clinicalfeatures of myelokathexis and treatment with hemato-poietic cytokines: a case report of two patients andreview of the literature. J Pediatr Hematol Oncol 1997:19: 443–8.
31. Mancini AJ, Chan LS, Paller AS. Partial albinismwith immunodeficiency: Griscelli syndrome: report of acase and review of the literature. J Am Acad Dermatol1998: 38: 295–300.
32. Nowak-Wegrzyn A, Crawford TO, Winkelstein JA,Carson KA, Lederman HM. Immunodeficiency andinfections in ataxia-telangiectasia. J Pediatr 2004: 144:505–11.
33. OchsHD,ThrasherAJ.TheWiskott-Aldrichsyndrome.J Allergy Clin Immunol 2006: 117: 725–38; quiz 39
34. Rosenzweig SD, Holland SM. Phagocyte immun-odeficiencies and their infections. J Allergy ClinImmunol 2004: 113: 620–6.
35. Sanal O, Ersoy F, Tezcan I, et al. Griscelli disease:genotype-phenotype correlation in an array of clinicalheterogeneity. J Clin Immunol 2002: 22: 237–43.
36. Schuster V, Kreth HW. X-linked lymphoproliferativedisease is caused by deficiency of a novel SH2 domain-containing signal transduction adaptor protein. Immu-nol Rev 2000: 178: 21–8.
37. Sugimoto T, Sawada T, Tozawa M, Kidowaki T,Kusunoki T, Yamaguchi N. Plasma levels of carcino-embryonic antigen in patients with ataxia-telangiectasia.J Pediatr 1978: 92: 436–9.
38. Waldmann TA, McIntire KR. Serum-alpha-fetopro-tein levels in patients with ataxia-telangiectasia. Lancet1972: 2: 1112–5.
39. Wetzler M, Talpaz M, Kleinerman ES, et al. A newfamilial immunodeficiency disorder characterized bysevere neutropenia, a defective marrow release mecha-nism, and hypogammaglobulinemia. Am J Med 1990:89: 663–72.
40. Kutukculer N, Karaca NE, Demircioglu O, Aksu
G. Increases in serum immunoglobulins to age-relatednormal levels in children with IgA and/or IgG sub-class deficiency. Pediatr Allergy Immunol 2007: 18:167–73.
An immune investigation algorithm for children with frequent infections
471

41. Bonilla FA, Bernstein IL, Khan DA, et al. Practiceparameter for the diagnosis and management of pri-mary immunodeficiency. Ann Allergy Asthma Immunol2005: 94: S1–63.
42. Boyle JM, Buckley RH. Population prevalence ofdiagnosed primary immunodeficiency diseases in theUnited States. J Clin Immunol 2007: 27: 497–502.
43. Casanova JL, Abel L. Primary immunodeficiencies:a field in its infancy. Science 2007: 317: 617–9.
44. Puck JM. Neonatal screening for severe combined im-mune deficiency. Curr Opin Allergy Clin Immunol 2007:7: 522–7.
45. Eades-Perner AM, Gathmann B, Knerr V, et al. TheEuropean internet-based patient and research databasefor primary immunodeficiencies: results 2004–06. ClinExp Immunol 2007: 147: 306–12.
46. Geha RS, Notarangelo LD, Casanova JL, et al.Primary immunodeficiency diseases: an update fromthe International Union of Immunological SocietiesPrimary Immunodeficiency Diseases ClassificationCommittee. J Allergy Clin Immunol 2007: 120: 776–94.
47. de Vries E. Patient-centred screening for primaryimmunodeficiency: a multi-stage diagnostic protocoldesigned for non-immunologists. Clin Exp Immunol2006: 145: 204–14.
48. Slatter MA, Gennery AR. Clinical immunologyreview series: an approach to the patient with recurrentinfections in childhood. Clin Exp Immunol 2008: 152:389–96.
49. Wahn U. Evaluation of the child with suspected pri-mary immunodeficiency. Pediatr Allergy Immunol 1995:6: 71–9.
50. Roos D, Kuijpers TW, Curnutte JT. Chronic granu-lomatous disease. In: Ochs HD, Smith CIE, Puck JM,eds. Primary Immunodeficiency Diseases. A Molecularand Genetic Approach. 2nd edn. Oxford: Oxford Uni-versity Press, 2007; 525–49.
51. Martire B, Rondelli R, Soresina A, et al. Clinicalfeatures, long-term follow-up and outcome of a largecohort of patients with Chronic Granulomatous Dis-ease: an Italian multicenter study. Clin Immunol 2008:126: 155–64.
52. Seger RA. Modern management of chronic granu-lomatous disease. Br J Haematol 2008: 140: 255–66.
53. Carnide EG, Jacob CA, Castro AM, Pastorino AC.Clinical and laboratory aspects of chronic granuloma-tous disease in description of eighteen patients. PediatrAllergy Immunol 2005: 16: 5–9.
54. Fasano MB, Sullivan K, Ibsen L, Winkelstein JA.Chronic meningococcemia in a child with a deficiency ofthe sixth component of complement. Pediatr AllergyImmunol 1993: 4: 214–6.
55. Boyle RJ, Le C, Balloch A, Tang ML. The clinicalsyndrome of specific antibody deficiency in children.Clin Exp Immunol 2006: 146: 486–92.
56. Casanova JL, Abel L. Genetic dissection of immunityto mycobacteria: the human model. Annu Rev Immunol2002: 20: 581–620.
57. Sewell WA, Khan S, Dore PC. Early indicators ofimmunodeficiency in adults and children: protocols forscreening for primary immunological defects. Clin ExpImmunol 2006: 145: 201–3.
58. Fleisher TA. Evaluation of suspected immunodefi-ciency. Adv Exp Med Biol 2007: 601: 291–300.
59. Cham B, Bonilla MA, Winkelstein J. Neutropeniaassociated with primary immunodeficiency syndromes.Semin Hematol 2002: 39: 107–12.
60. Filipovich AH. Hemophagocytic lymphohistiocytosisand related disorders. Curr Opin Allergy Clin Immunol2006: 6: 410–5.
61. Pachman LM, Lynch PA, Silver RK, Ozog DL,Poznanski AK. Primary immunodeficiency disease inchildren: an update. Curr Probl Pediatr 1989: 19: 1–64.
62. Sorensen RU, Leiva LE, Javier FC III, et al. Influenceof age on the response to Streptococcus pneumoniaevaccine in patients with recurrent infections and normalimmunoglobulin concentrations. J Allergy Clin Immu-nol 1998: 102: 215–21.
63. Noroski LM, Shearer WT. Screening for primaryimmunodeficiencies in the clinical immunology labo-ratory. Clin Immunol Immunopathol 1998: 86: 237–45.
64. UmetsuDT,AmbrosinoDM,Quinti I,SiberGR,Geha
RS. Recurrent sinopulmonary infection and impairedantibody response to bacterial capsular polysaccharideantigen in children with selective IgG-subclass deficiency.N Engl J Med 1985: 313: 1247–51.
65. Ambrosino DM, Siber GR, Chilmonczyk BA,Jernberg JB, Finberg RW. An immunodeficiencycharacterized by impaired antibody responses to poly-saccharides. N Engl J Med 1987: 316: 790–3.
66. Ambrosino DM, Umetsu DT, Siber GR, et al. Selec-tive defect in the antibody response to Haemophilusinfluenzae type b in children with recurrent infectionsand normal serum IgG subclass levels. J Allergy ClinImmunol 1988: 81: 1175–9.
67. Epstein MM, Gruskay F. Selective deficiency inpneumococcal antibody response in children withrecurrent infections. Ann Allergy Asthma Immunol1995: 75: 125–31.
68. Folds JD, Schmitz JL. 24. Clinical and laboratoryassessment of immunity. J Allergy Clin Immunol 2003:111: S702–11.
69. Crockard AD, Thompson JM, Boyd NA, Haughton
DJ, McCluskey DR, Turner CP. Diagnosis and car-rier detection of chronic granulomatous disease in fivefamilies by flow cytometry. Int Arch Allergy Immunol1997: 114: 144–52.
70. Vowells SJ, Sekhsaria S, Malech HL, Shalit M,Fleisher TA. Flow cytometric analysis of the granu-locyte respiratory burst: a comparison study of fluo-rescent probes. J Immunol Methods 1995: 178: 89–97.
71. Futatani T, Watanabe C, Baba Y, Tsukada S, Ochs
HD. Bruton�s tyrosine kinase is present in normalplatelets and its absence identifies patients with X-linkedagammaglobulinaemia and carrier females. Br J Hae-matol 2001: 114: 141–9.
72. Grimbacher B, Hutloff A, Schlesier M, et al.Homozygous loss of ICOS is associated with adult-on-set common variable immunodeficiency. Nat Immunol2003: 4: 261–8.
73. Kanegane H, Tsukada S, Iwata T, et al. Detection ofBruton�s tyrosine kinase mutations in hypogamma-globulinaemic males registered as common variableimmunodeficiency (CVID) in the Japanese Immunode-ficiency Registry. Clin Exp Immunol 2000: 120: 512–7.
74. KawaiS,MinegishiM,OhashiY, et al. Flowcytometricdetermination of intracytoplasmic Wiskott-Aldrich syn-drome protein in peripheral blood lymphocyte subpop-ulations. J Immunol Methods 2002: 260: 195–205.
Cassimos et al.
472

75. Morra M, Silander O, Calpe S, et al. Alterations ofthe X-linked lymphoproliferative disease gene SH2D1Ain common variable immunodeficiency syndrome.Blood 2001: 98: 1321–5.
76. Weston SA, Prasad ML, Mullighan CG, Chapel H,Benson EM. Assessment of male CVID patients formutations in the Btk gene: how many have beenmisdiagnosed? Clin Exp Immunol 2001: 124: 465–9.
77. Yamada M, Ariga T, Kawamura N, et al. Determi-nation of carrier status for the Wiskott-Aldrich syn-drome by flow cytometric analysis of Wiskott-Aldrichsyndrome protein expression in peripheral bloodmononuclear cells. J Immunol 2000: 165: 1119–22.
78. Samarghitean C, Valiaho J, Vihinen M. Online reg-istry of genetic and clinical immunodeficiency diagnosticlaboratories, IDdiagnostics. J Clin Immunol 2004: 24:53–61.
79. Amrolia P, Gaspar HB, Hassan A, et al. Non-myeloablative stem cell transplantation for congenitalimmunodeficiencies. Blood 2000: 96: 1239–46.
80. Notarangelo LD, Forino C, Mazzolari E. Stem celltransplantation in primary immunodeficiencies. CurrOpin Allergy Clin Immunol 2006: 6: 443–8.
81. Fischer A, Le Deist F, Hacein-Bey-Abina S, et al.Severe combined immunodeficiency. A model diseasefor molecular immunology and therapy. Immunol Rev2005: 203: 98–109.
82. Shearer WT, Fischer A. The last 80 years in primaryimmunodeficiency: how far have we come, how far needwe go? J Allergy Clin Immunol 2006: 117: 748–52.
83. Chapel H, Misbah S, Webster AD. Assessment of theimmune system. In: Ochs HD, Smith CIE, Puck JM,eds. Primary Immunodeficiency Diseases, 2nd edn, NY,Oxford University Press, 2007: 611–32.
An immune investigation algorithm for children with frequent infections
473




















