
Charge-remote fragmentations: Analytical applications and fundamental studies
46
Charge-remote fragmentations: Analytical applications and fundamental studies Jeanette Adams Department of Chemistry, Emory University, Atlanta, Georgia 30322 I. INTRODUCTION Charge-remote fragmentations of closed-shell ions are a new class of gas-phase ion decompositions that are both analytically useful and fundamentally interest- ing. The ion fragmentations are called ”charge-remote” because bond cleavage reactions take place at a site in the ion that is removed from the charge site, and because the mechanisms of fragmentation do not involve any significant inter- vention of the site of charge. The cleavage reactions thus are not “charge-me- diated.” For instance, some charge-remotefragmentations involve losses of C,H2, +2r CnH2n+l, H20, and other neutral species from locations in ionized molecules that are spatially separated from the charge site by many atoms. The only structural requirement for inducing charge-remote fragmentations is that the site of charge be stable so as to not migrate into, or be involved in, the fragmenting portion of the ion. A stable negative site of charge is present in some carboxylate anions, sulfates, and sulfonates; a stable positive site of charge is present in some pro- tonated amines and other polar molecules cationized with alkali and alkaline earth ions. Many charge-remote fragmentations require keV collisional activation (CA), but some can arise from low-eV CA, from ion source reactions following fast atom bombardment desorption, or from metastable ion decompositions. Charge-remote fragmentations of ions that contain a stable site of charge are mechanistic analogies to gas-phase thermal decompositions of neutral molecules. It is this thermochemical analogy that makes the ion fragmentations so structurally informative: bonds distant from the charge-site are cleaved via simple rearrange- ments and homolytic bond dissociations that are governed by classic thermolytic- like chemistry. For instance, the thermal analogy predicts that charge-remote fragmentations of ions containing a double bond would disfavor energy-intensive thermal-like rearrangements of the double bond, and instead favor more ener- getically accessible allylic and other cleavages that arise via simpler, lower-energy reactions. Indeed, this minimization of double-bond migration and hydrogen scrambling, which is characteristic of classic charge-mediated fragmentations, Mass Spectrometry Reviews 1990, 9, 141-186 0 1990 John Wiley & Sons, Inc. CCC 0277-70371 901020141-46$4.00
Transcript of Charge-remote fragmentations: Analytical applications and fundamental studies

Charge-remote fragmentations: Analytical applications and fundamental studies
Jeanette Adams Department of Chemistry, Emory University, Atlanta, Georgia 30322
I. INTRODUCTION
Charge-remote fragmentations of closed-shell ions are a new class of gas-phase ion decompositions that are both analytically useful and fundamentally interest- ing. The ion fragmentations are called ”charge-remote” because bond cleavage reactions take place at a site in the ion that is removed from the charge site, and because the mechanisms of fragmentation do not involve any significant inter- vention of the site of charge. The cleavage reactions thus are not “charge-me- diated.” For instance, some charge-remote fragmentations involve losses of C,H2, +2r
CnH2n+l, H20, and other neutral species from locations in ionized molecules that are spatially separated from the charge site by many atoms. The only structural requirement for inducing charge-remote fragmentations is that the site of charge be stable so as to not migrate into, or be involved in, the fragmenting portion of the ion. A stable negative site of charge is present in some carboxylate anions, sulfates, and sulfonates; a stable positive site of charge is present in some pro- tonated amines and other polar molecules cationized with alkali and alkaline earth ions. Many charge-remote fragmentations require keV collisional activation (CA), but some can arise from low-eV CA, from ion source reactions following fast atom bombardment desorption, or from metastable ion decompositions.
Charge-remote fragmentations of ions that contain a stable site of charge are mechanistic analogies to gas-phase thermal decompositions of neutral molecules. It is this thermochemical analogy that makes the ion fragmentations so structurally informative: bonds distant from the charge-site are cleaved via simple rearrange- ments and homolytic bond dissociations that are governed by classic thermolytic- like chemistry. For instance, the thermal analogy predicts that charge-remote fragmentations of ions containing a double bond would disfavor energy-intensive thermal-like rearrangements of the double bond, and instead favor more ener- getically accessible allylic and other cleavages that arise via simpler, lower-energy reactions. Indeed, this minimization of double-bond migration and hydrogen scrambling, which is characteristic of classic charge-mediated fragmentations,
Mass Spectrometry Reviews 1990, 9, 141-186 0 1990 John Wiley & Sons, Inc. CCC 0277-70371 901 020141-46$4.00
142 ADAMS
mlz
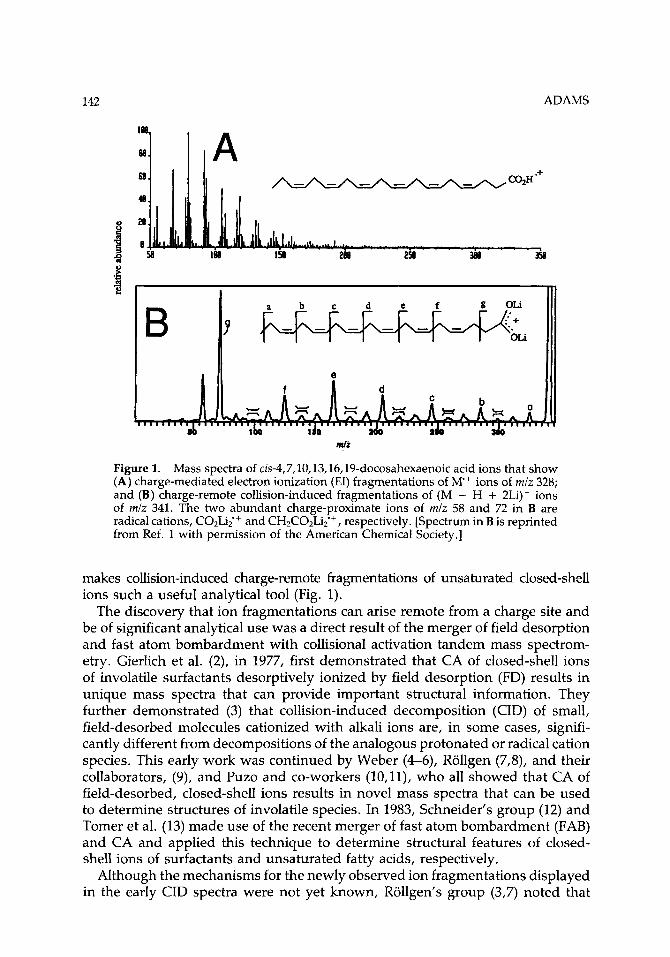
Figure 1. Mass spectra of cis-4,7,10,13,16,19-docosahexaenoic acid ions that show (A) charge-mediated electron ionization (EI) fragmentations of M + ions of m/z 328; and (B) charge-remote collision-induced fragmentations of (M - H + 2Li)+ ions of mlz 341. The two abundant charge-proximate ions of mlz 58 and 72 in B are radical cations, C02Li;+ and CH2C02Li;+, respectively. [Spectrum in B is reprinted from Ref. 1 with permission of the American Chemical Society.]
makes collision-induced charge-remote fragmentations of unsaturated closed-shell ions such a useful analytical tool (Fig. 1).
The discovery that ion fragmentations can arise remote from a charge site and be of significant analytical use was a direct result of the merger of field desorption and fast atom bombardment with collisional activation tandem mass spectrom- etry. Gierlich et al. (2), in 1977, first demonstrated that CA of closed-shell ions of involatile surfactants desorptively ionized by field desorption (FD) results in unique mass spectra that can provide important structural information. They further demonstrated (3) that collision-induced decomposition (CID) of small, field-desorbed molecules cationized with alkali ions are, in some cases, signifi- cantly different from decompositions of the analogous protonated or radical cation species. This early work was continued by Weber (4-6), Rollgen (7,8), and their collaborators, (9), and Puzo and co-workers (lO,ll), who all showed that CA of field-desorbed, closed-shell ions results in novel mass spectra that can be used to determine structures of involatile species. In 1983, Schneider’s group (12) and Tomer et al. (13) made use of the recent merger of fast atom bombardment (FAB) and CA and applied this technique to determine structural features of closed- shell ions of surfactants and unsaturated fatty acids, respectively.
Although the mechanisms for the newly observed ion fragmentations displayed in the early CID spectra were not yet known, Rollgen’s group (3,7) noted that

CHARGE-REMOTE FRAGMENTATIONS 143
the alkali ion contained in cationized molecules appears to serve only as a "spec- tator" ion, primarily to carry the charge. They likened the collision-induced ion decompositions to decompositions of excited neutrals. Tomer et al. (13) hypoth- esized that the structurally informative fragmentations that arise by CA of closed- shell fatty acid anions occur independent of and remote from the site of charge. In 1985, Liehr et al. (14) studied fragmentations of bile salts and also proposed that fragmentations of these species arise independent of the site of charge. They also hypothesized that cleavages of C-C bonds in alkyl chains arise via 1,2- eliminations of CnHZn+z. It was Jensen et al. (15), however, who formally presented evidence for a mechanism for fragmentations of alkyl chains that involves a 1,4- elimination of CnH2n+2 that occurs remote from the charge site. They also observed that the reactions are analogous to thermal fragmentations of fatty acid esters (16). Later mechanistic work by Adams and Gross (17,18) confirmed the earlier, intuitive ideas of Rollgen (3,7) that the alkali ion contained in certain cationized molecules is not directly involved in the mechanisms of fragmentation. They also presented further evidence that charge-remote fragmentations are indeed direct mechanistic analogies to thermal decompositions of neutrals.
The purpose of this review is to show the utility of charge-remote fragmen- tations for solving structural problems and to describe research that has addressed mechanisms and energetics of the ion decornpositions. Many analytical studies have shown charge-remote fragmentations to be a useful tool for determining structures of a large variety of biologically and otherwise important molecules. Research on mechanisms and energetics of the ion fragmentations, the latter to which Wysocki et al. (19-22) have contributed, has provided insights into the fundamental chemical characteristics of charge-remote fragmentations.
11. ANALYTICAL APPLICATIONS
Charge-remote fragmentations have been implicated as being responsible for producing structurally informative collision-induced decomposition (CID) spectra of a large variety of biological and other molecules. Some of the applications described here are a result of directly applying the ion fragmentations to structural problems. Other research represents new discoveries of structural applications, and some was performed before the charge-remote nature of the ion fragmen- tations was understood.
A. Compounds containing an alkyl chain
The majority of analytical or structural applications of charge-remote fragmen- tations has involved studies of closed-shell ions derived from molecules that contain a long hydrocarbon chain. In these studies, the stable site of charge is either anionic, as in carboxylate, sulfate, and sulfonate anions, or cationic, as in protonated and quaternary amines, sulfonium and phosphonium cations, and molecules cationized with alkali and alkaline earth metal ions. Characteristic charge- remote fragmentations of ions that contain a saturated hydrocarbon chain involve losses of the elements of CnH2n+z that originate from the alkyl terminus, distant

144 ADAMS
from the site of charge. Other types of charge-remote fragmentations arise in cases in which there are substituents on the alkyl chain. Fast atom bombardment (FAB) is now typically used to desorptively ionize the molecules, but field de- sorption (FD) and chemical ionization (CI) are also viable alternatives in some cases.
1. Fatty acids, esters, and prostaglandins
The discovery (13) that collision-induced decompositions of unsaturated fatty acid (M - H)- ions provide for direct determination of double-bond location was the first of many applications of charge-remote fragmentations to structural elu- cidation of fatty acids. The earliest, and some of the most comprehensive, studies of CID of fatty acid anions are found in the papers of Jensen, Tomer, Gross, et al. (13,15,23-29). Others also have made important contributions in studies of the anions. Deterding and Gross (35,36), Adams et al. (1,18,37-40), Crockett and Gross (41), and Davoli et al. (42) have further demonstrated that positive closed- shell fatty acid ions also can be collisionally activated to give structurally infor- mative charge-remote fragmentations. The positive ions are (M + H)+ ions of fatty acid picoliriyl ester derivatives (35,36,38); fatty acid (M + Cat)+ and (M - H + 2Cat)+ ions, in which Cat is an alkali metal ion (1,18,37-40); and fatty acid (M - H + Cat)+ ions, in which Cat is an alkaline earth metal ion (41,42).
In contrast to fatty acid anions and the other ionic species described above, fatty acid (M + H)+ ions do not undergo charge-remote fragmentations because the site of charge does not remain localized on the carboxylate terminus (1). Instead, the (M + H)+ ions fragment by collisional activation (CA) either by eliminating H20 to give an acylium ion or by losing two molecules of H20 to give a carbocation [Fig. 2(A)]. The acylium ion and carbocation decompose to give a variety of fragment ions that do not give information either about location or about type of substituent. A localized and stable site of charge on the carboxylate terminus is provided, however, in resonance-stabilized (M - H)- anions, (M - H + 2Cat)+ ions, and (M - H + Cat)+ ions, as described above. The site of charge in (M + H)+ ions of fatty acid picolinyl esters is stabilized because of the high proton affinity of the pyridine nitrogen. As a result of the stable site of charge, and the resultant stability of the charged molecule as a whole, CA results in fragmentations that arise via thermal-like cleavages of C-C bonds [Fig. 2(B,C)]. Saturated long-chain fatty acid ions fragment to give a pattern of peaks that proceeds uninterrupted, which are evenly spaced by 14 amu [Fig. 2(B)]. The highest-mass fragment ion results from loss of CH4, and the pattern continues with losses of CnH2n+2, at least through the ion of mlz 99. As shown in Figure 2(C), however, the presence of the double bond perturbs the fragmentation pat- tern so that a "gap" appears. The "gap" is comprised of ions of low abundances that would arise by energetically unfavorable cleavages vinylic to and through the double bond. Abundant ions are formed, however, via more energetically favorable allylic cleavages.
The use of charge-remote fragmentations of closed-shell ions of fatty acids is not only limited to determining the location of a single double bond, but extends

CHARGE-REMOTE FRAGMENTATIONS 145
50 100 150 20 0 250
5’0 I00 I50 200 2 i O
50 Id0 I50 200 250 m / Z
Figure 2. CID spectra of (A) (M + H)+ ions of cis-9-octadecenoic (oleic) acid of mlz 283; (B) (M - H)- ions of octadecanoic (stearic) acid of mlz 283; and (C) (M - H)- ions of oleic acid of mlz 281. Fragment ions labeled with squares, triangles, and circles in A arise from charge-mediated processes. (Spectrum in A is reprinted from Ref. 1, and B and C from Ref. 25, with permission of the American Chemistry Society.)
to determining the location and identity of a variety of substituents. Jensen and Gross (43) reviewed mass spectrometric methods, generally through the year 1986, for determining structures of fatty acids. Their review describes many applications of FAB-MS-MS and charge-remote fragmentations for elucidating structures of fatty acid anions. There is a particularly striking example in which CIDs of anions can differentiate is0 from anfeiso methyl-branched acids. Other examples reveal important differences in CID spectra of hydroxy-, epoxy-, cyclopropane-, and cyclopropene-substituted fatty acid anions. The CID spectra not only indicate location of substituent, but the fragmentation patterns are unique “fingerprints” and can serve to identify the type of substituent present.
The chemistry of carboxylate anions was applied by Chauhan et al. (44) to structural confirmation of V-[~-L-( + )-glutamy1]-4-~arboxyphenylhydrazine (1). Some hydrazine derivatives induce tumors in laboratory animals, and this one (1) was isolated from Agaricus bisporus, the common mushroom commercially

146 ADAMS
available in food stores. The most stable site of anionic charge is the resonance- stabilized deprotonated benzoic acid terminus, and the (M - H)- ions undergo charge-remote fragmentations to produce four important ions of mlz 205, 177, 151, and 136, that verify the structure of the unusual alkyl chain. Only one abundant, structurally significant charge-remote fragment ion arises from decom- position of an ion in which the other carboxylic acid terminus is deprotonated.
1
Adams et al. (38) reviewed differences between CID spectra of fatty acid (M - H)-, (M - H + 2Li)+, and picolinyl ester (M + H)+ ions. Characteristically, (M - H)- anions decompose to give lower relative abundances of low-mass fragment ions [see Fig. 2(B,C)]. The (M - H + 2Li)+ ions decompose to give two charge- proximate abundant low-mass radical cations of mlz 58 (C02Li$+ and of m/z 72 (C2H202Li2)’+ [see Fig. l(B)]. The (M + H)+ ions of picolinyl ester derivatives fragment to give an abundant low-mass doublet, seen in B/E scans (acquired by the author) as m/z 92 and 93, and a triplet, seen in B/E scans as m/z 108, 109, and 110. The low-mass fragment ion of m/z 93 is (CH-C5H4N-H>’+ and the ion of m/z 109 is (O-CH2-C5H4N-H)’+. Spectra of the three types of precursor ions are usually complementary. There are some cases, however, in which the anions undergo charge-driven decompositions so that structurally informative charge- remote fragmentations are either weakly abundant or missing. Structures of short- chain acids, of acids in which a double bond is in close proximity to the site of charge, of some polyunsaturated acids, and of 2-hydroxy acids are best deter- mined by CA of either (M - H + 2Li)+ or picolinyl ester (M + H)+ ions. For cis-4,7,10,13,16,19-docosahexaenoic acid, only CIDs of the (M - H + 2Li)+ ions give structural information [see Fig. l(B)].
A recent study by Crockett and Gross (41) directly addresses the effect of double- bond location on CIDs of (M - H)-, (M - H + 2Li)+, and (M - H + Ba)+ ions of a series of homoconjugated octadecadienoic acids. Fragmentations of the anions are most affected in that charge-driven losses of C02 and sometimes H 2 0 dom- inate the spectra in cases in which the double bonds are within a distance of five carbon atoms from the carboxylate terminus. Structurally informative charge- remote fragmentations of the bariated ions are suppressed by charge-proximate reactions, although structural determination is possible. It is only in the case of a double bond in the 3-position that the bariated ions undergo a charge-driven loss of C02. The lithiated species, however, give the most uniformly consistent spectra and provide reliable structural information independent of double-bond location.
The problems that are introduced in CID spectra of anions and bariated species by double bonds in proximity to the carboxylate terminus can be overcome by deuterating the double bonds. Jensen et al. (25) used this technique for studying

CHARGE-REMOTE FRAGMENTATIONS 147
anions of homoconjugated polyunsaturated acids. CID spectra of the mass-se- lected, fully deuterated saturated acid anions can be interpreted to determine original locations of the double bonds.
The different types of fatty acid closed-shell ions can be formed a variety of ways. The (M - H)- anions can be formed either by dissolving the free fatty acid in a FAB matrix and desorbing the anionic species (13,15,23-27), or by preparing pentafluorobenzyl ester derivatives and creating the anions by dissociative elec- tron capture (30-33). Savagnac et al. (33) applied both approaches for determining structures of unknown mycolic acids. Closely related is the approach of Bam- bagiotti and co-workers (34), who determined structures of fatty acid esters by preparing the (M - CH& carboxylate anions by negative ion CI. Deterding and Gross (35) prepared fatty acid picolinyl esters by treating fatty acids with thionyl chloride and 3-(hydroxy-methyl)pyridine7 and the (M + H)+ ions are formed by FAB. The picolinyl esters, however, can also be determined after GC-MS by positive ion, proton-transfer CI. The (M - H + 2Li)+ ions can be formed in situ on the FAB probe tip by dissolving the fatty acid in a FAB matrix that is saturated with either LiI or LiOH (1,37). The (M - H + Ba)+ and other alkaline earth- cationized species are most easily formed in situ on the FAB probe tip by dissolving the fatty acid in a FAB matrix saturated with an alkaline earth hydroxide (41,42).
The work of Prom6 et al. (30) and others (31-33) and Bambagiotti et al. (34), in which fatty acid anions are formed by using negative ion CI, has important analytical utility in analysis of complex mixtures. Although FAB-MS-MS some- times can provide for direct mixture analysis of free fatty acids (25,27,35), in practice FAB desorption of fatty acid mixtures can result in preferential desorption of some ions (37). The nonuniform responses make accurate quantification dif- ficult; some fatty acids are not detected at all; detection limits may be hampered by the presence of chemical noise; and structural isomers may be difficult to characterize. It may be possible to eliminate FAB matrix discrimination by using n-dodecanol as a matrix for fatty acids (45), although chemical noise may still be a problem. A CID spectrum of a FAB-desorbed isomeric mixture of two fatty acid anions, which appear at the same mlz in the FAB spectrum and thus are mass- selected together, can indicate the presence of the two structural isomers as long as the concentration dynamic range is <10:1 (27,43). The approach of Prom6 and co-workers, however, in which complex mixtures of mycobacterial monounsa- turated and polyunsaturated fatty acids are derivatized to their pentafluorobenzyl esters, separated by capillary GC, and structurally determined by CA of the (M - H)- anions formed by dissociative electron capture, can avoid the problems of FAB matrix-induced discrimination, chemical noise, low sensitivity, and inter- ferences of structural isomers. The alternative approach of Bambagiotti et al., in which fatty acid methyl esters are separated by capillary gas chromatography (GC) and (M - CH3)- anions are formed by OH- and NHF negative ion CI, can also alleviate the problems inherent in FAB.
Methods that use GC-MS and negative ion CI also have potential for reducing detection limits for structural determination of trace levels of fatty acids. Methods that use FAB-MS-MS of picolinyl ester (M + H)+ ions can provide a detection limit of 50 ng at a signal-to-noise ratio (S/N) of 35 (35). FAB-desorbed (M - H +

148 ADAMS
2Li)+ ions can be similarly determined at a level of 10 ng at a S/N of 4 (l), and (M - H)- anions can be structurally determined at levels of 25-50 ng (no S/N reported) (25,43). Collisionally activating the (M - H + Ba)+ ions may improve the FAB detection limits because of increased efficiency of desorption and the ability to mass-select the ions with less interference from chemical noise (41,42). The use of pentafluorobenzyl ester derivatives to form anions by dissociative electron capture, however, provides a detection limit of 10 ng for pentafluoro- benzyl oleate introduced by capillary GC (32). The CID spectrum was acquired by using a single, 1.5-s scan, and the spectrum is easily interpretable. Thus, the ultimate detection limit for this approach may be significantly lower.
With basis in the studies of charge-remote fragmentations of fatty acids, meth- ods also were developed to determine structures of fatty acid esters. As discussed above, Bambagiotti and co-workers (34) used negative ion CI and collisionally activated the (M - CH3)- anions. Although they do not report detection limits, this approach is useful for GC-MS and should provide high sensitivity. An al- ternative method is to collisionally activate the (M + Li)+ ions produced by FAB (39). The latter technique can achieve detection limits of 15 ng.
Studies performed to determine structures of prostaglandins, which are highly substituted derivatives of arachidonic acid and are biologically active, have also grown from the earlier work on charge-remote fragmentations of fatty acids. CA of either (M - H + 2Li)+ ions (39,40) or (M - H + Ba)+ ions, as shown by Davoli et al. (42), provides information about the alkyl chain in proximity to the carboxylate terminus, about the structure of the 5-membered ring, and about the number of hydroxyl groups present. CA of either the dilithiated or the bariated ions results in more structural information than can be attained by CA of either the (M - H)- or the (M + Li)+ ions.
2. Fatty Alcohols
Adams and Gross (17) found that neither fatty alcohol (M - H)- ions nor (M + H)+ ions undergo structurally specific fragmentations. The (M - H)- anions of saturated fatty alcohols decompose by eliminating H2 to give a stable enolate; unsaturated alcohol (M - H)- anions fragment either by eliminating H2 or by eliminating formaldehyde to give a stable monounsaturated carbanion. The (M + H) + ions decompose primarily by losing H20 to give a carbocation that further fragments to produce CnHln+l+ and CnHZn-l+ ions, but no useful structural in- formation.
Attaching an alkali ion to the fatty alcohol molecule, however, produces a stable site of charge. Thus, (M + Li)+ ions, in sharp contrast to (M - H)- and (M + H)+ ions for this class of substances, decompose to give structurally informative charge-remote fragmentations that are analogous to those of the fatty acid ions described above. Other characteristic ions are two charge-proximate radical cations of m/z 52 (C2H50Li)’+ and of mlz 66 (C3H70Li)’+. The (M + Li)+ ions are produced in situ by using FAB desorption from a FAB matrix saturated with LiI.
Polyunsaturated fatty alcohols cationized with Li+ also fragment to give struc- tural information, but here the Li+ ion also interacts with the double bonds. This

CHARGE-REMOTE FRAGMENTATIONS 149
interaction results in CID spectra that are more difficult to interpret because now some of the charge-remote fragmentations arise from species that contain Li+ attached to the alkyl chain. Thus, there are fragment ions that result from loss of the hydroxy terminal of the alkyl chain.
The most significant limitation of FAB-MS-MS of (M + Li)+ ions of fatty alcohols is detection limit. For oleyl alcohol (M + Li) + ions, the detection limit for structure determination is approximately 30 pg at a SIN of 4. The poor detection limit is most likely because the FAB matrix effectively competes against the alcohol for Li+ and because FAB desorption may produce (M + Li)+ ions that have enough internal energy in excess of the ROH-Li bond strength (-2 eV) so that Li+ is released in decompositions in the ion source.
3. Surfactants
Surfactants are commercially important compounds that usually contain long alkyl chains, and sometimes polar head groups, and are particularly amenable to charge-remote cleavage reactions. Most surfactants occur as complex mixtures, and the use of FAB- or FD-MS-MS also has been particularly relevant in structural determination. There are several types of surfactants that undergo charge-remote fragmentations as a result of CA. These include various types of cationic, anionic, and nonionic surfactants that either are present as preformed ions or are either protonated or cationized in situ in the ion source.
Although the first report of CID spectra of surfactants did not clearly indicate the analytical utility of charge-remote fragmentations, it is still appropriate to discuss those spectra here. These early CID spectra, first published in 1977 by Gierlich et al. (2), reveal characteristic charge-remote losses of the elements of CnHZn+2. These early reports present a link between the development of FD and FAB, in conjunction with MS-MS, that led to the later, formalized documentation of charge-remo te fragmentations.
Cu tionic. Cationic surfactants, present as preformed ions, are comprised of qua- ternary ammonium salts, %N+. The first CID spectra of alkyl quaternary am- monium ions were acquired by using FD-MS-MS (2). Because the alkyl chains are relatively short, only some charge-remote losses of the elements of CnHZn+2 can be seen in the spectra. The most structurally informative, and highly abundant, fragment ions arise by charge-mediated as opposed to charge-remote decompo- sitions. The charge-mediated fragmentations involve losses of the R groups via three processes: (a) losses of R’ to produce higher-mass abundant trialkyl amine fragment ions (NR,’+); (b) losses of RH to produce abundant imminium fragment ions (R2N+=CHR’), which are adjacent in mass to the ions formed via (a); and (c) loss of two R groups to produce a low-mass abundant fragment ion, also an imminium ion, (R2N+=CH2).
It is in CID spectra of alkyl ammonium ions that contain longer alkyl chains that charge-remote losses of CnH2n+z become of greater analytical utility: the charge-mediated fragmentations produce less abundant ions. Veith (9) used FD- and B/E-linked scans to acquire CID spectra of longer-chain (C4-C8) quaternary ammonium ions, and Weber and co-workers (5,6) studied even larger alkyl am-

150 ADAMS
monium ions (C8-CI8). The charge-mediated fragmentations, a s described above, immediately give the masses of the R groups. It is the less facile, charge-remote losses of CnH2n+2, however, that allow the length of the alkyl chains to be con- firmed, and that show that there are no modifications in the alkyl chains. An isomeric mixture of alkylammonium ions, of the same mlz and thus mass-selected together, can be readily identified as such from the CID spectrum (5). FAB-MS- MS can be substituted for FD-MS-MS to analyze complex mixtures, and FAB-MS- MS spectra are similar to those from FD-MS-MS (12). In addition, pg levels of the surfactants serve to suppress completely all chemical noise that otherwise can arise from the FAB matrix.
Lyon et al. (46) also demonstrated that mixtures and structures of alkyl qua- ternary and other types of quaternary amine surfactants can be determined by FAB-MS-MS. In addition to quaternary alkylamines, fragmentations of cetylpyr- idinium bromide and polyethoxylated quaternary amines were investigated. Here, CID spectra of all the alkyl quaternary amines clearly show structurally diagnostic charge-remote losses of CnHzn+z, which show the presence of n-alkyl chains, The ethoxylated quaternary amines, CH3(CH&-(CH3)N + (CHZCH~OH)~, also undergo structurally important charge-driven cleavages to expel either ethylene oxide or an alkene.
Anionic. Anionic surfactants are comprised partially of alkyl and alkylarylsul- fonates, and alkyl and alkylether sulfates. As opposed to the cationic quaternary ammonium surfactants discussed above, charge-remote fragmentations have played a more important role in structure elucidation of anionic surfactants. Both positive- and negative-ion MS-MS have been used to determine anionic surfactants. The positive-ion CID spectra of (M + H)+ and (M + Na)+ ions of sodium alkyl- and alkylbenzenesulfonates have been acquired by using FD-MS-MS (5,6), whereas the negative ion CID spectra of alkyl- and alkylarylsulfonates, perfluoroalkane- sulfonates, alkylether sulfates, N-acylated amino acids, and sulfopolycarboxylates have been evaluated by using FAB-MS-MS (47,48).
CID spectra of either the (M + H)+, the (M + Na)+, or the (M - Na)- ions of sodium alkylsulfonates, CH3(CH2),S03Na, and sodium alkylarylsulfonates, CH3(CH2)nC6H4S03Na, give complementary structural information (5,6,47). In analogy to spectra of carboxylate anions [see Fig. 2(B)], charge-remote losses of CnHZn+2 from closed-shell ions of long-chain sulfonates indicate length of the n- alkyl chain. Alterations in the fragmentation patterns indicate the presence of methyl branch-points. It should be noted that the original CID spectra of Weber et al. (5), acquired by using energy scans, indicate that the ions normally formed by losses of CnH2n+2 are instead radical cations formed by losses of CnH2n+l. CID spectra acquired by using B/E-linked scans (by the author), however, show that the most abundant ions are indeed formed by the typical losses of CnH2n+2. In contrast, (M - H)- anions of perfluoroalkanesulfonates undergo charge-remote CIDs to lose primarily CnF2n+l radicals, which give a series of radical anions: loss of CnF2n+2 is a comparatively minor process (48).
Various n-alkyl, branched alkyl, unsaturated alkyl, and alkyl ether sulfates were studied by Lyon et al. (47) who used FAB-MS-MS of (M - H)- anions. Charge- remote fragmentations of the various alkyl sulfates are analogous to fragmenta-

CHARGE-REMOTE FRAGMENTATIONS 151
50 100 150 200 250
.. .;;o.. ..... . .......,...“ ““,,. I . 1 1 . . m , , , . .,,.. ~
200 250 300 350
Figure 3. CID spectra of anions of (A) tetradecyl sulfate of m/z 293 and (B) tetra- decyl-diethoxy-sulfate of m/z 381. The ions of m/z 96 and 80, SO<, are charge- induced. (Spectra are reprinted from Ref. 47 with permission of the American Chemical Society.)
tions of carboxylate anions, as shown in Figure 3(A) for the anions of n-tetradecyl sulfate. Charge-remote cleavages of anions of alkyl ether sulfates, CH3(CH2),- (OCH2CH2),0S0<, however, produce a uniquely characteristic fragmentation pattern that immediately indicates the presence and number of the ethylene oxide units [Fig. 3(B)]. The ethylene oxide groups can be discerned by a pattern of three peaks, which repeats every 44 amu. Each peak is a result of a cleavage through one of the three (-0-CHrCH2) bonds.
Other anionic surfactants, N-acylated amino acids and sulfosuccinates, also can be determined using FAB-MS-MS (47). Although charge-remote losses of C,H2, +
from the N-acylated amino acids are useful for indicating the structure of the alkyl chain, the CID spectra are dominated by charge-mediated fragmentations. Spectra of the sulfosuccinates lack characteristic charge-remote fragmentations that can provide information about structure of the alkyl chains.
Nonionic. Nonionic surfactants do not exist as preformed ions. They include alkyl amines, amine oxides, and ethoxylated alkyl amines and phenols. They have been studied by both FD- (5,6) and FAB- (46) MS-MS as protonated (M + H)+ ions.
Long-chain aliphatic amine (M + H)+ ions (46) fragment to give CID spectra in which the higher-mass ends of the spectra are dominated by structurally in-

152 ADAMS
formative charge-remote cleavages of the alkyl chain. These cleavages provide information concerning length of the alkyl chain and sites of unsaturation anal- ogous to charge-remote cleavages of carboxylate anions. The low-mass ends of the spectra, however, are dominated by charge-induced fragmentations. The lower- mass fragment ions comprise a series of alkyl and alkenyl cations that arise as a result of loss of the protonated ammonium terminus. That is, loss of NH3 from the (M + H)+ ions produces a carbocation that undergoes a series of charge- mediated reactions. The charge-induced fragmentations are analogous to those that arise from (M + H)+ ions of fatty alcohols and acids [see Fig. 2(A)]. Protonated amine oxides, (CH3(CH2),-(CH3)2N0 + H) + , undergo both charge-remote frag- mentations and charge-mediated loss of H20 (46). Loss of H20 also results in formation of a low-mass fragment ion that arises from concomitant loss of the alkyl chain.
Protonated ethoxylated amines, [CH3(CH2)xN(CH2CH20)yH + HI +, and ethox- ylated phenols, [CH3(CH2),C6H40(CH2CH20)nH + HI + , fragment analogously to ethoxylated sulfates described above (5,6). Charge-remote fragmentations pro- vide information about the length of the hydrocarbon chain and about the number of ethylene oxide groups. Here, as for the ethoxylated sulfates, the presence of ethylene oxide groups is indicated by a series of three fragment ions that repeats every 44 amu.
4 . Phosphonium salts
Phosphonium [(C6H5)3P+ R] salts are synthetic precursors to phosphorus ylides, which are important intermediates that undergo nucleophilic addition of the R group to either aldehyde or ketone carbonyl carbons to produce olefins (the Wittig reaction) (49). The R group of phosphonium salts may be unsaturated, saturated, or contain various functional groups. Confirmation of the structure of the salts can be performed via FD or FAB and collision-induced charge-remote fragmen- tations.
The first study of CIDs of phosphonium salts was done by Weber et al. (4) who used FD to form gas-phase ions. The salts contained short alkyl-chains, and thus charge-remote cleavages of the R groups are of minor importance. Later investigations by Veith (9) and particularly by McCrery et al. (50), however, show the analytical utility of charge-remote fragmentations for determining the struc- ture of the R group. In addition to delineating isomeric structures of pentyl- and butyltriphenylphosphonium salts (Fig. 4), charge-remote cleavages of the C-C bonds of the R group also readily identify the structures of salts containing longer alkyl chains.
5. Vitamins
Such vitamins as E (2) and K (3) contain a polar functional group to which is attached a long alkyl, or isoprenoid, chain. Collision-induced charge-remote frag- mentations of the (M - H)- phenolate anions of vitamin E (2) were demonstrated by Tomer and Gross (51) to be an effective way to locate the branch points in the isoprenoid chain. This chemistry is in contrast to the chemistry of alkoxy anions

CHARGE-REMOTE FRAGMENTATIONS 153
b
A 1 ) a
(GHshF''
Q 4 f - l! e (c6HS)3p + 50 100 150 200 250 300
mlz
Figure 4. CID spectra of three structural isomers of the butyltriphenylphosphon- ium cation of mlz 319. The location of the methyl branch point in B and Cis indicated by a "gap" of 28 amu between the peaks labelled a and b. The ions of mlz 108 and 183, and of 199 in A and B, are formed by charge-mediated processes. (Spectra are reprinted from Ref. 50 with permission of the American Chemical Society.)
of fatty alcohols, as described in an earlier section, because phenolate anions contain a stable site of charge as a result of charge delocalization. Complementary results can be obtained from collisionally activating (M + Li)+ ions of vitamin E (39). Similarly, vitamin K (3) can be cationized with Li+, desorptively ionized by using FAB, and collisionally activated to give charge-remote fragmentations that indicate the structure of the isoprenoid chain.
0
3
B. Complex lipids
Some of the most productive analytical applications of charge-remote frag- mentations have been in the determination of unknown structures of complex

154 ADAMS
lipids. These biological molecules are vital components of cell membranes, and many are involved in cellular transport, cell-cell recognition, and other biological activity. The use of FAB-MS-MS has been particularly useful in structure eluci- dation because complex lipids are involatile molecules that frequently contain charged functional groups.
0 H~~-O-;-R~ II
H C - 0 - C - ~ 2
HzC-0-1-0-R3 l o I OH 4
One class of complex lipids are phospholipids, which are derivatives of struc- ture 4, in which R1 and R2 are alkyl chains and R3 can be choline, -CH2CH2N+ (CH3)3; serine, -CH2CH(NH2)C0,H; ethylamine, -CH2CH2NH2; glycerol, -CH2CH(OH)CH20H; or other species. Jensen et al. (28,29) and Miinster and Budzikiewicz (52) studied various types of phospholipids by using negative ion FAB-MS-MS and demonstrated the types of structural information obtainable. In addition, Jensen and Gross (53) reviewed, through the year 1986, mass spectro- metric methods for determination of phospholipids, and examples of CID spectra that summarize their approach to structure determination are provided. They elucidate structure by using information obtained both from negative ion FAB and from CID spectra of selected ions.
0 H2C-O-C-R3' I 1
:: HI-O-C-R4' I:: -CHz-CH-CHz-O-C-O- Hz
I I OH OH
5
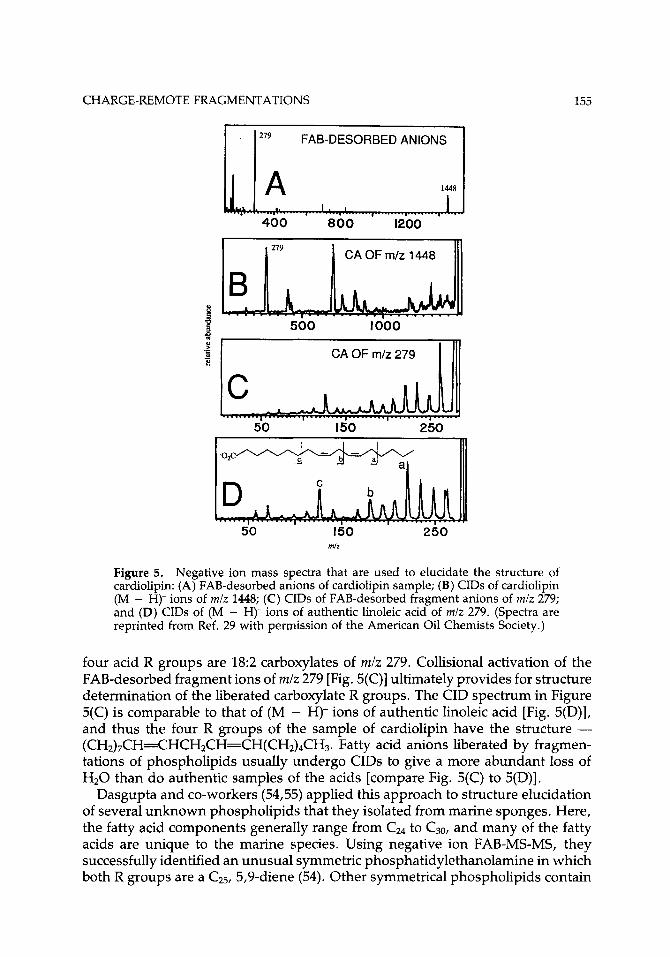
An example of their method is shown in Figure 5 in the negative ion FAB and MS-MS spectra that are used to elucidate the structure of a sample of cardiolipin (29). This phospholipid has the general structure shown by 4 except that here R3 is of the structure 5. FAB desorption results in an abundant (M - H)- ion of m/z 1448 and a lower-mass, more highly abundant ion of m/z 279 [Fig. 5(A)]. Collisional activation of the (M - H)- ion [Fig. 5(B)] confirms that the low-mass ion of m/z 279 indeed arises by fragmentation of the precursor (M - H)- ion. Complex phospholipids fragment by both negative ion FAB and CA to liberate carboxylate anions that contain the R groups. Lipids of higher organisms liberate carboxylates that are generally in the range of m/z 255 to 339 (CI6-Cz). Thus, both the FAB and the CID spectra imply that the cardiolipin is symmetric in that all
CHARGE-REMOTE FRAGMENTATIONS 155
400 800 1200
CA OF m/z 1448 219
1000
- t CA OF m/z 279 2
50 I50 250
50 I50 250
Figure 5. Negative ion mass spectra that are used to elucidate the structure of cardiolipin: (A) FAB-desorbed anions of cardiolipin sample; (B) CIDs of cardiolipin (M - H)- ions of m/z 1448; (C) CIDs of FAB-desorbed fragment anions of m/z 279; and (D) CIDs of (M - H)- ions of authentic linoleic acid of m/z 279. (Spectra are reprinted from Ref. 29 with permission of the American Oil Chemists Society.)
four acid R groups are 18:2 carboxylates of mlz 279. Collisional activation of the FAB-desorbed fragment ions of mlz 279 [Fig. 5(C)] ultimately provides for structure determination of the liberated carboxylate R groups. The CID spectrum in Figure 5(C) is comparable to that of (M - H)- ions of authentic linoleic acid [Fig. 5(D)], and thus the four R groups of the sample of cardiolipin have the structure - (CH2)7CH=CHCH2CH=CH(CH2)4CH3. Fatty acid anions liberated by fragmen- tations of phospholipids usually undergo CIDs to give a more abundant loss of H20 than do authentic samples of the acids [compare Fig. 5(C) to 5(D)].
Dasgupta and co-workers (54,55) applied this approach to structure elucidation of several unknown phospholipids that they isolated from marine sponges. Here, the fatty acid components generally range from C24 to C30, and many of the fatty acids are unique to the marine species. Using negative ion FAB-MS-MS, they successfully identified an unusual symmetric phosphatidylethanolamine in which both R groups are a CZ5, 5,9-diene (54). Other symmetrical phospholipids contain

156 ADAMS
either a phytyl chain or R groups that contain various sites of unsaturation (55). The confirmation of polyunsaturated arachidonic acid as a component of one symmetrical phospholipid, however, required alternative methods.
6
An analogous application provides for structural confirmation of two homol- ogous ornithine-containing lipids ( 6 ) that were isolated as a mixture from Thiob- acillus fhiooxidans (56,57). The negative ion FAB spectrum reveals two high-mass (M - H)- ions of mlz 679 and 665 along with lower-mass ions of mlz 311 and 297 (56). CA of the two respective (M - H)- ions results in formation of the two respective low-mass ions of mlz 311 and 297, which arise by release of the re- spective R groups as carboxylate anions. From these first data, the higher-mass lipid was proposed to contain a cyclopropane-substituted R group, -CH(OH)(CH2)8(CHCH2CH)(CH2)5CH3, and the lower-mass homologue to con- tain -CH(OH)C16H31. Later (57), the FAB-desorbed low-mass carboxylate anions were collisionally activated, and the structure of the higher-mass R group was confirmed as proposed, while the lower-mass R group was shown to be -CH( OH)( CH&CH=CH(CH2)5CH3.
0 II
H,C-C-O-H[ CH,]
I H,C-N+-(CH&
I
Carnitines and acylcarnitines, 7, represent another class of complex lipids that are biologically important. Abnormal carnitinelacylcarnitine ratios are clinical in- dicators of metabolic disorders. Gaskell and co-workers (58) distinguished be- tween isomeric short-chain acylcarnitines and identified them in urine of children with metabolic diseases. CID spectra of (M + H)+ ions of authentic samples of isomeric acylcarnitines, in which R is either -C4H9 or -C5Hll, are compared to CID spectra of acylcarnitines contained in patients’ urine. The CID spectra show charge-remote fragmentations similar to those demonstrated in Figure 4, frdm which the structures of n-alkyl versus other branched acylcarnitines are easily differentiated. One set of results reveals that the structure of one acylcarnitine, which is contained in one urine sample, is clearly that of isovaleryl carnitine. The data are important in that they ccnfirm previous diagnoses of ethylmalonic-adipic aciduria and isovaleric acidemia. In addition, the structural studies provide evi- dence for a new variant of the ethylmalonicadipic aciduria disorder in children.

CHARGE-REMOTE FRAGMENTATIONS 157
0 8
The structures of other complex lipids, glycosphingolipids and gangliosides, can be probed by using FAB-MS-MS. Domon and Costello (59) acquired CID spectra of (M - H)-ions of glycosphingolipids that contain galactose (8) or lactose, and weakly abundant ions that arise by charge-remote cleavages of the long alkyl ceramide R groups are formed. These charge-remote fragmentations may be less useful, however, than cleavages that involve the oligosaccharide moieties that are contained in the larger ganglioside homologues. The latter cleavages, which may be charge-remote, are discussed further below.
C. Carbohydrates and glycosides
In most studies of CIDs of carbohydrate ions, charge-remote fragmentation mechanisms have not yet been implicated (7,8,10,11,60). By comparing CID spec- tra of (M + H)+ and (M + Na)+ ions of sucrose, it is obvious that the (M + Na) + ions undergo a significantly larger number of structurally informative frag- mentations (57). The (M + H)+ ions of many flavonoid and steroidal glycosides, such as digitoxin and digoxin, also undergo structurally informative CIDs [Fig. 6(A)] (61). Assuming that the preferred site of proton attachment is the carbonyl oxygen of the steroid, structurally diagnostic cleavages of the glycosidic bonds would be a result of charge-remote fragmentations. Evidence in favor of a charge- remote mechanism for cleavages of glycosidic bonds in digoxin was recently presented by Light et al. (62). In an analogous manner, (M - H)- anions of gangliosides, which are the same as structure 8 except that there is an oligosac- charide chain, fragment upon collision via cleavages of the glycosidic bonds (59). Although there are no abundant charge-remote cleavages of the ceramide R- groups (refer to structure 8), cleavages of the glycosidic bonds and through the pyran rings instead provide information about the structure of the oligosaccharide moiety. Here, it is less clear whether the fragmentations arise via charge-remote or via charge-induced cleavages.
There is one example of CIDs of carbohydrates that is most likely an example of charge-remote fragmentations. Carr et al. (63) made 2-aminopyridine deriva- tives of oligosaccharides, and the (M + H) + ions undergo CIDs to give structurally informative fragment ions that contain the protonated 2-aminopyridine moiety [Fig. 6(B)]. Two series of fragment ions arise that are similar to those described by Domon and Costello (59): one via cleavages of glycosidic bonds and another via cleavages of the pyran rings. Here, the 2-aminopyridine functionality may be analogous to the picolinyl group that is used to localize the site of charge for derivatized fatty acids, as discussed above.

158 ADAMS
A 0 +-0 I II I
Y
digitose digitose
100 200 300 400 500 600 700 800
1 ~ 5 9
287
300 500 700 900
Figure 6 . CID spectra of (A) (M + H)+ ions of digitoxin of miz 781; and (B) (M + H)' ions of a derivatized hexasaccharide of mlz 1069. (Spectrum in A is reprinted from ref. 61 with permission of Academic Press; and B from Ref. 63 with permission of John Wiley & Sons.)
D. Steroids, bile salts, and sterols
The application of FAB and tandem mass spectrometry to the study of biological compounds containing the cholestane ring system was introduced in 1983 by Gaskell and co-workers (64). Using normal- and reverse-geometry mass spec- trometers, they investigated metastable ion decompositions of steroid sulfate an- ions in order to qualitatively and quantitatively determine isomers in blood sam- ples. Ions studied include dehydroepiandrosterone sulfate (9) and testosterone sulfate (10). Although the fragmentations were not known to be charge-remote, the two isomers were easily differentiated from the metastable ion decomposi- tions. Species 9 undergoes a predominant charge-remote cleavage through the D

CHARGE-REMOTE FRAGMENTATIONS 159
ring to give an ion of m/z 310 that contains the -0SOy site of charge and the A, B, and C rings. In contrast, 10 fragments across the B ring to give an ion of mlz 243 that contains the anionic site of charge and the C and D rings.
0 OSO3'
9 10
FAB-desorbed (M + Na)+ and (M + H)+ ions of bile salts, the sodium gly- cocholates (11) and sodium taurocholates (12), were later studied by Liehr et al. (14). Here, the R groups are either H or OH, and the (M + Na) + ions are analogous to the (M - H + 2Cat)+ ions discussed above for carboxylates and sulfonates. Both types of sodiated or protonated bile salts undergo unique and structurally informative CIDs [Fig. (7A)]. Liehr et al. (14) specifically point out that the positive charge resides on the sulfonate or carboxylate moieties and does not migrate to other functional groups. Thus, fragmentations both of the side chains and of the steroid ring system are charge-remote cleavages. Cleavages of the cholestane ring system reflect structural differences such as location of OH groups, which can be determined by shifts in masses of the fragment ions. Some of the ring-cleavage reactions arise with and without hydrogen migration to the neutral fragment. Tomer et al. (65,66) document fragmentations of the corresponding anions of similar bile salts, and the CID spectra reflect analogous, complementary frag- mentations except that the carboxylates fragment to give an abundant loss of H20, and the structurally important charge-remote fragmentations are suppressed. # -CQzNa &m&s03Na 19 7 R3
HO R' HO RZ R'
11 12
The earlier investigation by Tomer and Gross et al. (65) was later extended to a more extensive series of steroid conjugates and bile salts (66). The latter research specifically addresses structural utility of both charge-remote and charge-proxi- mate fragmentations, and mechanisms for various ring-cleavage reactions are proposed. Among the compounds studied are C(17) side-chain sulfates and bile salts; C(3) sulfates and one C(19) sulfate; and C(3) and C(17) side-chain glucu- ronides (13 and 14, respectively). All the anions, except for estrogen glucuronides, undergo abundant charge-remote fragmentations to give cleavages of the cho-

160 ADAMS
d 9
100 200 300 400 500
200 300 400
50 150 250 350 d 2
Figure 7. CID spectra of (A) (M + Na)+ ions of sodium tauroursodeoxycholic acid of m/z 544; (B) (M - H)- anions of dihydrocholesterol sulfate of m/z 467; and (C) (M + Li)' ions of 5a-cholest-7-en-3pl-01 of mlz 393. [Spectrum in A is reprinted from Ref. 14 and B is from Ref. 66 with permission of John Wiley & Sons.]
COf I
& OH OH
0 & 13
lestane rings. The estrogen glucuronides primarily fragment by charge-mediated decompositions to give abundant ions from losses of the glucuronide and H20.

CHARGE-REMOTE FRAGMENTATIONS 161
Several general trends characterize all structurally diagnostic charge-remote fragmentations of various cholestane derivatives (66). The ring-cleavage reactions preferentially involve the ring that is most distant from the site of charge; locations of ring substituents can be determined from shifts in mass of the fragment ions; and stereoisomers, such as 3-a versus 3-p or 5-a versus 5-p, can be determined from relative abundances of the fragment ions. Locations of double bonds in the ring system also can be determined. For instance, a 5,6-double bond suppresses the typical ring-cleavage reaction, which would cleave the 5,6-bond, and instead, a retro-Diels-Alder reaction occurs to give allylic cleavage between the 7,8-bond. Another fragmentation produces a stable allylic radical anion, which also indicates double bond location.
Other charge-remote cleavages involve angular -CH3 groups, alkyl chains, and the glucuronide moiety (66). Loss of CH4 is a predominant charge-remote reaction, and this fragmentation is believed to arise via loss of an angular CH3 with concerted transfer of H. Charge-remote fragmentations of C(3) sterol sulfates that contain a substituted alkyl chain on C(17) can be used to elucidate the struc- ture of the chain [Fig. 7(B)]. The C(3) glucuronides (13) fragment between the oxygen and the cholestane A-ring to give a diagnostic ion of m/z 191 that can be explained by a 1,Ctransfer of H from one of the glucuronide -OH groups to the C(3) of the ring. Conversely, the C(17) side-chain glucuronides (14) fragment between the oxygen and the alkyl carbon that is a to the carbonyl carbon to give an ion of m/z 193 that can arise by a 1,4-transfer of H from the cholestane D ring to the oxygen.
Tomer and Gross (66) also discussed charge-mediated reactions that are useful in structure determination of sulfated cholestane derivatives. The C(3) sulfates fragment to give HS04, whereas the C(17) sulfates decompose to give both HS04 and SO4-. The C(17) glucuronides that also contain -OH on the C(17) of the D ring lose 206 amu possibly through an ion-dipole complex. In contrast, other glucuronides that do not have an -OH group on C(17), which include other C(3) and C(17) side-chain glucuronides, undergo loss of 176 amu that may be via an internal displacement reaction.
There have been some preliminary investigations of CIDs of positive ions of sterols (39). Here, the cholestane alcohols are FAB desorbed as (M + Li)+ ions, and the fragmentations are analogous to those of the sulfate anions [Fig. 7(C)]. Potentially, this technique can be applied to many sterols and some steroids that do not contain functional groups that can compete for the Li+ ion. Competition for the Li+ ion can result in CID spectra that are more difficult to interpret, analogously to those of polyunsaturated fatty alcohols described in an earlier section, because charge-remote fragmentations would arise from different ends of the ions.
E. Peptides
Correct sequencing of polypeptides that contain leucine (15) or isoleucine (16) by CA of (M + H)+ ions was difficult until recently. Johnson et al. (67), however, have differentiated these two structural isomers by using a structure-specific charge-
162 ADAMS
p 100 300 500 700 900
"I" a71 ae
100 300 $0 700 900 1100 1300 m l 2
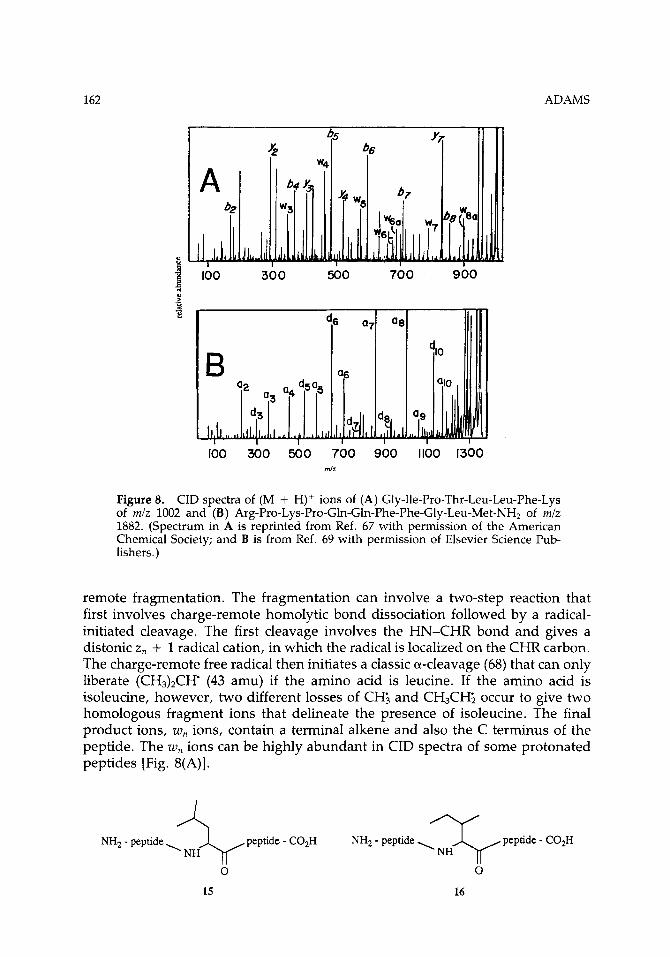
Figure 8. CID spectra of (M + H)+ ions of (A) Gly-Ile-Pro-Thr-Leu-Leu-Phe-Lys of m/z 1002 and (B) Arg-Pro-Lys-Pro-Gln-Gln-Phe-Phe-Gly-Leu-Met-NH2 of m/z 1882. (Spectrum in A is reprinted from Ref. 67 with permission of the American Chemical Society; and B is from Ref. 69 with permission of Elsevier Science Pub- lishers.)
remote fragmentation. The fragmentation can involve a two-step reaction that first involves charge-remote homolytic bond dissociation followed by a radical- initiated cleavage. The first cleavage involves the HN-CHR bond and gives a distonic z , + 1 radical cation, in which the radical is localized on the CHR carbon. The charge-remote free radical then initiates a classic a-cleavage (68) that can only liberate (CH&CH' (43 amu) if the amino acid is leucine. If the amino acid is isoleucine, however, two different losses of CH; and CH3CHz occur to give two homologous fragment ions that delineate the presence of isoleucine. The final product ions, w, ions, contain a terminal alkene and also the C terminus of the peptide. The w, ions can be highly abundant in CID spectra of some protonated peptides [Fig. 8(A)].
peptide - C02H NH2 - peptide peptide - C02H ' NH 0 0
16
NJ3, - peptide
15

CHARGE-REMOTE FRAGMENTATIONS 163
Other structurally informative ions are formed by charge-remote cleavage re- actions of peptide (M + H)+ ions. Johnson and co-workers (69) presented other evidence that other distonic radical cations, a, + 1 and x, + 1 ions, can be formed by charge-remote homolytic bond cleavages. The a, + 1 ions then fragment by charge-remote mechanisms to give d, and a, ions; the x,, + 1 ions fragment to give v, ions. The a, fragment ions are important sequence ions that contain the N terminus. The d, ions can be formed analogously to the w,, ions described above, but they contain the N terminus instead of the C terminus. The v, ions contain the C terminus but are related to the structure of the N-terminal amino acid contained in the fragment ion. As shown in Figure 8(B), these other newly described fragmentations also give rise to structurally significant and sometimes highly abundant ions that are indicative of peptide structure.
Details of the mechanisms for some of these reactions are covered in a later section that discusses Schemes 6 and 7.
F. Others
The analytical applications discussed above are ones in which charge-remote fragmentations have been implicated either by the referenced investigators or by the author. There are, however, other examples of structurally informative col- lision-induced fragmentations of closed-shell ions that may not be true charge- remote fragmentations. Bambagiotti and co-workers (70,71) described a series of unusual CIDs of (M - H)- anions of fatty acid esters that result in CID spectra that resemble those that are clear examples of charge-remote fragmentations. Chang, Siege1 et al. (72,73) studied CIDs of (M + K)+ and (M + K - CH203)+ ions of polyether antibiotics (17) and propose schemes to account for cleavages of the (M + K - CH203)+ ions. Some fragmentations involve cleavages of the rings, whereas others arise from cleavages of the bonds between the rings. Be- cause the cleavages produce fragment ions that arise from both ends of the pre- cursor, and the K f ion may be at various locations in the species, the ultimate utility of the fragmentations for structural determination of unknowns may be limited. \ lo--
R' 0' I HO O P 0 -
HO
17
Zwitterionic compounds were recently studied by Hand and Cooks (74) by using secondary ion mass spectrometry (SIMS). The SIMS spectrum of C12H2+5N(CH3)2-(CH2)5C02H shows a series of ions that are assigned to originate via charge-remote losses of 16 amu and other masses, whch correspond to CnH2,+2, from the CI2 alkyl chain of the quaternary ammonium ion. Interestingly, the SIMS spectrum of (H3C);N(CH2)5C02H also shows a similar series of ions that include losses of 16,30, and 44 amu. For the latter species, however, charge-remote losses

164 ADAMS
of CnH2n+2 should be precluded. That is, charge-remote fragmentations of the latter ion would be expected to involve losses of neutrals that contain the car- boxylic acid moiety.
111. FUNDAMENTAL STUDIES
Investigations of the fundamental aspects of the chemistry of charge-remote fragmentations have involved studies of the mechanisms and of the energetics of fragmentation. Experiments performed to elucidate the mechanisms of frag- mentation have included deuterium labeling, MS-MS-MS, B/E-linked scan, and B2/E-linked scan experiments. The energetics of the fragmentations have been investigated by evaluating competitive fragmentations, by studying both high- and low-energy (laboratory frame of reference) collisions, and by performing angle-resolved experiments. Some theoretical calculations have been performed to address both the mechanisms and energetics of some of the fragmentations.
A. Mechanisms of fragmentation
Fragmentations to be discussed here are those that specifically occur several bonds distant from the putative site of charge. This specification is invoked so as to not include fragmentations that may in actuality be charge mediated because of delocalization of the charge site onto adjacent atoms. In addition, the frag- mentations can be explained by mechanisms that are analogous to thermolytic decompositions and result from either simple rearrangements or homolytic dis- sociations that lead to bond cleavages. Thus, fragmentations such as the one recently proposed by Renner and Spiteller (75) and Grese et al. (76) for loss of the C-terminal amino acid of (M + Na)+ and (M + Li)+ ions of FAB-desorbed polypeptides are not included. Although the mechanism for this particular frag- mentation does not involve the putative site of charge, which may be on the amino terminus of the peptide chain, the rearrangement reaction is induced by a carbonyl that is highly polarized as a result of a (-C02-Li+) bond.
The mechanisms discussed here are also organized into two general types. More detail is given to mechanisms that have been studied at least by using either deuterium labeling, MS-MS-MS, or B2/E parent ion scan experiments. The B2/E scans indicate precursors of fragment ions, and can confirm some mechanisms. A general section describes other fragmentations that have been proposed to arise via charge-remote mechanisms.
1. 1,4-Elirnination of H2 and C,H2,
Jensen et al. (15) published the first evidence in support of a mechanism for the structurally informative CIDs of saturated fatty acid (M - H)- anions that does not require intervention of the site of charge. Results from various experiments indicate that coiling in the gas phase most likely does not bring the charge site into contact with the alkyl end of the ions. One piece of supportive evidence is that cholesteryl

CHARGE-REMOTE FRAGMENTATIONS 165
hemisuccinate anions (18) also undergo losses of C,H2, + 2 that arise from the alkyl chain distant from the charge-site [also see spectra in Fig. 7(B) and (C)]. The choles- tane-containing compounds, many of which are discussed in an earlier section, are large, rigid molecules in which the charge site is physically unable to contact most sites of reaction. The collision-induced fragmentations of the choiestane derivatives also are unlike the ion-molecule-dipole-complex-induced fragmentations that are described by Longevialle and Botter (77,78).
18
Consistent with the evidence above and with deuterium-labeling studies, cleav- ages of C-C bonds in the alkyl chain of saturated fatty acid anions are proposed to arise via charge-remote, thermally allowed 1,4-eliminations of H2 and C,H2, (Scheme 1). Although the structures of the neutral fragments are unknown, Wy- socki et al. (22) and Adams and Gross (18) verified by MS-MS-MS experiments that the ionic products are indeed terminally unsaturated fatty acid ions.
The mechanism and its thermal analogy is strengthened by early evidence from the pyrolysis of deuterated fatty acid esters (16). That is, pyrolysis of deuterated fatty acid esters produces a homologous series of alkenes and monounsaturated fatty acid esters, all of which contain the correct number of deuterium atoms as would be predicted by the mechanism shown in Scheme 1. Thus, a 1,2-elimination of alkane seems unreasonable with basis both in the results of Sun et al. (16) and on energetic considerations. Furthermore, 1,2-eliminations also would be ex- pected to form terminally unsaturated alkenes and saturated fatty acid anions: saturated fatty acid fragment anions are not observed in the spectra. It should be noted that concerted elimination reactions, which may not be synchronous, in which three groups are involved in the bond breaking and bond making processes are not unusual in thermolysis: chloroformates and carbonates (79), and mag- nesium, zinc, and aluminum alkoxides (80,81) are believed to decompose ther- mally via 1,4-eliminations to produce three neutral products.
D 0
D
Scheme 1
CIDs of 7,7,8,8-hexadecanol-d4 (M + Li)+ ions, and CIDs of 7,7,8,8-hexade- canoic-d4 acid (M - H + 2Li)+ ions and (M + H)+ ions of the picolinyl ester,

166 ADAMS
also are explained by the same mechanism involving 1,4-eliminations of H2 and C,H2, (1,17,35). These results imply that the fragmentations are not charge me- diated as a result of ion-molecule-dipole complexes (77,78) because the same fragmentations occur both for the anions and for the various types of cations. In addition, the CID spectrum of protonated octadecyl-N,N,N-d3-amine displays no mass shifts that would correspond to transfer of deuterium from the amino ter- minus to the alkyl chain as part of the losses of CnH2n+2 (15).
With basis in studies of the energetics of the losses of CnH2n+2, to be discussed in detail in a later section, Wysocki et al. (19-22) have raised some questions concerning the 1,4-elimination mechanism. They have proposed that alternative mechanisms should be considered, such as intervention of the charge-site either through coiling of the alkyl chain or through an intermediate charge-mediated ion-molecule-dipole complex. Another proposed alternative is that losses of C,H2, + 2 occur via successive radical eliminations (22). As discussed above, the two former alternative mecha- nisms were seriously considered (15,18), and neither coiling nor an intermediate ion-molecule-dipole complex seems to explain the experimental results. Until the neutral products of the fragmentations are determined, however, the possibility that losses of C,H2, + 2 arise as a result of successive radical eliminations cannot be entirely eliminated from consideration. If such radical eliminations occurred, they would most likely involve loss of an element of CnH2n+I', which then would be followed by a radical-induced a-cleavage to lose H . Indeed, radical ions, adjacent in mass to the closed-shell fragment ions, can be detected in quadrupole CID spectra of some species that contain a long alkyl chain, and they can undergo further fragmentations to give closed-shell ions (22). This alternative mechanism also would be a thermal analogy, and the fragmentations would still arise remote from the site of charge. Losses of CnHZn+z via a radical mechanism seem unlikely as the major mechanism on energetic grounds, however, as will be discussed further in a later section.
2. 0-Hydro-C-ally1 and hydro-hydroxy eliminations
Some new mechanisms for charge-remote fragmentations recently were de- scribed by Adams and Gross (18). These fragmentations do not require collisional activation and also occur as ion source reactions in FAB and as decompositions of metastable ions. One fragmentation is an 0-hydro-C-ally1 elimination (Scheme 2) and the other is a hydro-hydroxy elimination (Scheme 3) that occur upon decomposition of (M - H)-, (M + Li)+, and (M - H + 2Li)+ ions of ricinoleic
OLi
Scheme 2

CHARGE-REMOTE FRAGMENTATIONS 167
OLi
Scheme 3
and ricinelaidic acids, and upon decomposition of (M + Li)+ ions of methyl ricinoleate and ricinoleyl alcohol. These mechanisms were elucidated by using deuterium labeling and MS-MS-MS experiments.
This investigation (18) provides strong evidence in support of a direct analogy between charge-remote fragmentations and thermolytic decompositions of neu- trals. Results from MS-MS-MS experiments indicate that the transferred deuterium atom shown in Scheme 2 is in the allylic position in the fragment ion: this same result was shown by Arnold and Smolinsky (82) in their earlier studies of the O-hydro-C- ally1 elimination that arises by thermolysis of the neutral molecules. Other MS-MS- MS experiments show that the structure of the product ion from the hydro-hydroxy elimination is that shown in Scheme 3, although the structure of the deuterium- containing neutral lost is not confirmed. Because the same elimination reactions occur both for anionic and for cationic precursors, it is unlikely that these fragmen- tations arise via charge-mediated ion-molecule-dipole complexes.
3 . Other mechanisms for fragmentations of alkyl chains
Other thermal-like mechanisms for charge-remote fragmentations of alkyl chains were reported by Contado et al. (83). Results from CIDs of (M - H)-, (M + Li)+, and (M - H + 2Li)+ ions of deuterium-labeled cis-7-dodecenoic acids provide evi- dence for mechanisms responsible for fragment ions that arise via allylic cleavages in monounsaturated fatty acid ions. A thermally analogous retro-ene reaction (19) explains allylic cleavage on the carboxylate terminus side of the double bond, whereas a thermal-like 1’4-conjugate elimination (20) accounts for allyliccleavage on the alkyl terminus side of the double bond. These mechanisms contrast with those previously suggested by Bambagiotti and co-workers (34), who proposed that the allylic cleav- ages arise via complex mechanisms involving transfers of vinylic hydrogens. It is interesting that one reaction preferentially occurs on the carboxylate side of the dou- ble bond, whereas the other reaction preferentially occurs on the alkyl side. A con- sistent feature of both rearrangement mechanisms, however, is that the product ion always contains one or more double bonds. This suggests a second-order interaction between the charge site and the reaction site whereby the transition state energy
19 20

168 ADAMS
may be lowered by involvement with the charge site. The mechanisms for reaction at allylic bonds are still under investigation.
An abundant radical anion fragment ion also is observed in CID spectra of anionic precursor ions that contain a site of unsaturation in the alkyl chain (34,54,55,83). As mentioned in an earlier section, an abundant radical anion also is produced by allylic cleavage of an unsaturated cholestane ring (66). The radical anions can be explained (34,66,83) as arising from homolytic bond dissociations, and the fragment ions may be stable, distonic, allylic radicals such as that shown by 21. Distonic ions are ones that have the charge and radical sites either on different atoms or at least in different molecular orbitals. Although radical anions are only weakly abundant in CID spectra of anionic species that contain a saturated hydrocarbon chain, they are of significant abundance in spectra of saturated perfluoroalkanesulfonate anions (48). Here, losses of C,F2, + 2 are suppressed by energetically more favorable fragmentations that give rise to losses of CnF2n+l. This is a result of two factors. One is that fluorine is more electronegative than hydrogen, and this raises the electron affinity so that the distonic perfluorocarbon anion radical products are significantly more stable than the analogous hydro- carbon radicals. Another factor is that a 1,4-elimination of F2 and C,F2, from perfluorocarbons can be calculated (84) to be -5 eV more endothermic than the analogous elimination of H2 and C,H2, from hydrocarbons.
21
A few highly abundant radical cations also have been observed in CID spectra of cationic precursor ions that contain a hydrocarbon chain (1,17,18,37). Abundant low-mass radical cations, which are important in structure elucidation, are seen in CID spectra of (M + Li)+ ions of fatty alcohols, and in (M - H + 2Li)+ and (M - H + Ba)+ ions of fatty acid derivatives [see Fig. l(B)] (1,17,18,37,41,42). Homolytic bond dissociations may be responsible for formation of these radical cations, and the species also may be distonic ions (Scheme 4). It should be noted, however, that the site of charge is in fairly close proximity to the putative radical site in these low-mass fragment ions.
Li+ - OH ;
D
Scheme 4
Some weakly abundant, longer-chain radical cations can be seen in CID spectra of some types of closed-shell precursor ions, and there is evidence that these ions also are distonic species. Wysocki et al. (22) performed MS-MS-MS (MS3) exper-

CHARGE-REMOTE FRAGMENTATIONS 169
iments to compare structures of radical cations that are formed by losses of CnHzn+ to structures of adjacent, more abundant closed-shell ions that are formed by losses of CnH2n+2. The MS3 spectrum of a first-generation radical cation that arises by CA of protonated 2-pentadecylpyridine is similar to the MS3 spectrum of the adjacent first-generation closed-shell ion. That is, series of second-generation fragment ions are formed from both types of first-generation ions, and the second- generation fragment ions are identical in mass. These results are exactly to be expected if the radical cations are indeed distonic ions. For instance, a first- generation closed-shell ion of miz 218, which is formed by loss of C5H12, would undergo allylic cleavage via a retro-ene reaction (83) to give a second-generation fragment ion of mlz 176 that contains a terminal alkene (Scheme 5a). The adjacent first-generation radical cation of mlz 219 instead, as suggested here, would undergo a classic, thermally analogous radical-induced hydrogen rearrangement reaction followed by an a-cleavage (85,86), also to give an ion of m/z 176 that also would be a terminal alkene (Scheme 5b). Other second-generation closed-shell fragment ions that arise from the first-generation radical cation can be envisioned as oc- curring via other analogous radical mechanisms, each of which results in the formation of a double bond. Other radical cations are formed by FAB-induced decompositions of protonated peptides, and these also are proposed to be distonic ions (67,69). These fragmentations are discussed immediately below.
cH3 (cH2) &&.@+H
a b
Scheme 5
4. Fragmentations of protonated peptides
Mechanisms proposed by Johnson and co-workers (67,69) for fragmentations involving structurally specific losses of side chains of (M + H)+ ions of peptides involve two different types of charge-remote fragmentations. One type is induced

170 ADAMS
by distonic radical sites and the other type involves charge-remote thermolytic- like eliminations of neutrals. The mechanisms that involve radical cations have basis in results from CIDs of radical cation fragment ions that are produced in FAB spectra: the radical cations are generally missing in CID spectra of the pre- cursor-protonated peptides. Because the radical cation fragments, and others, are produced by FAB, B2/E precursor ion scans give further evidence for the frag- mentation mechanisms.
Distonic radical cations are believed to be formed first by homolytic bond dis- sociations that arise remote from the site of charge. The distonic radical site then induces either a classic, thermally analogous cleavage reaction, an a-cleavage (68), or a ring-closure reaction, both of which involve the peptide side chain. One example of this type of fragmentation is shown in Scheme 6 (67). Here, the (M + H)+ ions undergo collision-induced homolytic bond cleavages to give z , + 1 distonic radical cations. The radical site in the 2, + 1 ion then induces an a- cleavage to give wn ions [also see Fig. 8A]. Charge-remote a-cleavages similar to the one shown in Scheme 6 account for formation of d, and u, ions that arise from distonic u, + 1 radical cations (69). It should be noted that the product ions shown in Scheme 6 also could be formed directly by a 1,2-elimination of (CH&CH-NH peptide from the (M + H)+ precursor ions.
0
peptide
- NH-peptide. I 0
0
I H+
Scheme 6
There is also evidence that a charge-remote 1,2-elimination of R,-H from y, fragment ions is one route for formation of P, ions (Scheme 7) (69). The alternative route is a charge-remote fragmentation of distonic x , + 1 radical cations. Johnson et al. (69) further discuss (M + H)+ ions of peptides that contain a basic amino acid, and they suggest that the proton is somewhat fixed at the basic amino acid so that the ions tend to undergo fragmentations remote from the site of charge.

CHARGE-REMOTE FRAGMENTATIONS 171
Their conclusions are supported by evidence obtained from CIDs of peptides that have been derivatized to contain the 2,4,6-trimethylpyridinium cation.
0
Y .
Scheme 7
5. Other mechanisms
Other mechanisms that involve fragmentations that arise remote from the site of charge have been proposed by Tomer and Gross (66), Siege1 et al. (73), and Cerny et al. (87) to account for product ions that arise from fragmentations of steroid conjugates, polyether antibiotics, and dinucleotides, respectively. Al- though these fragmentation mechanisms have not yet been investigated by either deuterium labeling or other techniques, they are worthy of mention.
Several charge-remote mechanisms explain fragment ions that are formed by decompositions of steroid sulfates and glucuronides (66). One, shown in Scheme 8, describes formation of abundant ions that arise by concomitant loss of an angular methyl group and the C(17) substituent from C(3) steroid sulfates. Other charge-remote mechanisms explain important cleavage reactions that involve the glucuronide moiety in steroidal glucuronides.
SO;
so, L P Scheme 8
+ CH, + C H + k O
Two charge-remote fragmentation mechanisms describe decompositions of pol- yether antibiotics cationized with Kf (73). One fragmentation, shown in Scheme 9, is similar to the mechanism for the thermolytic decomposition of P-r unsatu- rated carboxylic acids (79). Another mechanism explains loss of C2H602 via charge- remote eliminations of vicinal dimethoxy groups.

172 ADAMS
Scheme 9
The most structurally important ions that arise from cleavages of (M - H)- anions of dinucleotides are charge mediated (87). Ring-cleavages of the sugar groups, however, may involve charge-remote fragmentations, such as that shown in Scheme 10. Here, R' is OH for ribonucleotides and H for 2'-deoxy ribonucle- otides, and the precursor in the reaction is the (M - H - BH)- fragment ion, in which B is the nucleotide base.
kH Ho3 0
I 0 __c
0 R' - 0 - p : o -0- P =o
I I
R ' 0 R/
/o R
-0. p :o I
/o R
I -0- P =o
0
Scheme 10
B. Energetics
Studies of the energetics of charge-remote fragmentations have involved the characterization of two important and distinguishable quantities: the energy re- quired for the reactions and the energy required to produce detectable fragment ions. The fragmentations that have been primarily evaluated are the collision- induced losses of CnH2n+2. The energy required for the reactions, which is the activation energy, has been approximated by relating the charge-remote frag- mentations to competitive reactions for which activation energies are known or can be estimated (17,36). The energy required to produce detectable fragment ions has been studied both by using keV and low-eV (laboratory-frame) collisional activation (19-22,34,50,88-91) and by measuring inelastic energy loss (Q) that occurs upon keV CA (92,93). Angle-resolved mass spectrometry (22,94) also has been used to investigate the energetics of the reactions. To provide some back- ground, a review of general concepts that are related to energetics of ion frag- mentations is first presented.
The reaction coordinate diagram presented in Figure 9(A) shows simplified relationships between potential energy and two exemplary competitive fragmen- tation routes that are available upon collision-induced decompositions of (M + Na)+ ions of fatty alcohols (17). The precursor (ROH-Na)+ ions predominantly lose Na+, but some charge-remote losses of CnH2n+2 also occur. For heterolytic

CHARGE-REMOTE FRAGMENTATIONS 173
log (sec
E
4 4 4 1
E ::
A (C,H2,.,0H-Na)+
n H,,,.-2.2 eV A Hd.l .35 eV
( E O d V V V
(ROH-Na)+ Reaction Coordinate
k
-' 1 (CnH2,., OH-Cat)+
Energy - Figure 9. Diagrams that show (A) potential energy in relation to the reaction coordinate for loss of Na+ vs. loss of CnH2n+2 from (ROH-Na)+ ions; and (B) hypothetical log k vs. ion internal energy curves for losses of Na+, Li+, or CnHzn+2 from (ROH-Cat)+ ions. The numbers shown in A are only approximate values.
loss of Na+, AH,,, is approximated from literature values, as described previously (17), and is considered to be equal to the forward activation energy (Eofor) of the cleavage reaction. The value of EOfor for competitive loss of CnHZn+2 is not known, but AH,,, can be approximated from additivity rules (84). For loss of CnHZncz, AH,,, is not equal to the forward activation energy because the rearrangement reaction has a reverse activation energy (Earev). The average internal energy of FAB-desorbed precursor (M + Na)+ ions that do not decompose unimolecularly in the field-free region of the mass spectrometer is represented by IEinit. Collision between the (M + Na)+ ions and neutral target atoms results in transfer of some of the ion translational energy into internal energy, and this average inelastic kinetic energy loss is Q (95). Because the ions contain extra energy above their ground vibrational levels both before and after collision, and because extra energy above Eofor may be needed to produce enough fragment ions to be detectable, the transition state complexes contain some amount of excess energy E*. Some of this

174 ADAMS
excess energy, plus some of the reverse activation energy for the rearrangement reaction, is released as translational energy (KER) of the product fragments (96).
The extra energy, above the activation energy, that is needed to produce enough product ions for detection is related to the change in the rate of the reaction with respect to ion internal energy. Although the RRKM theory of unimolecular de- compositions gives a more accurate evaluation of changes in rate (k) with energy (E), the older and simpler RRK formulation [Eq. (l)] is useful to show
general effects of transition state energy (E*) , activation energy ( Eofor), transition state geometry ( A ) , and "effective" degrees of freedom (s) on rates of reactions (79). The activation energy is related to the enthalpy of the transition state, whereas the A factor, which is sometimes called a frequency factor v and is the same as the Arrhenius A parameter, is related to the entropy of the transition state. Ex- perimentally, it has been established that direct homolytic C-C cleavage reactions ("loose") are Gnthalpically disfavored (high activation energies, -4-5 eV), but are entropically favored (high A factors, -1015-1018 s-') (79). Conversely, C-C bond cleavages via rearrangement reactions ("tight") are enthalpically favored (low activation energies, -1-4 eV depending upon transition state geometry), but are entropically disfavored (lower A factors, - 1013-10'4 s-* for a four-membered tran- sition state, and even lower A factors, -1011-1013 s-', for a six-membered transition state) (79). The effects of these relationships on rate constants is presented in Figure 9(B), which illustrates hypothetical log k( E ) curves for reactions such as those shown in Figure 9(A) in which either Cat+ or CnH2n+2 are lost from (M + Cat)+ ions of a fatty alcohol. The intercept of a log k ( E ) curve with the energy axis ideally represents the activation energy of the reaction, whereas both the slope of the curve and the maximum rate constant that the reaction can achieve at high energies are related both to the activation energy and to the A factor. That is, direct cleavages, such as heterolytic formation of Na+ and Lif in Figure 9(B), have more steeply-sloped k( E ) curves that level at higher values of log k, whereas rearrangement reactions, such as loss of H2 and C,H2, in Figure 9(B), have k ( E ) curves that have more shallow slopes and that level at lower values of log k . The heterolytic bond cleavages that give rise to Na+ and Li+ are different from ho- molytic C-C bond cleavages in that the heterolytic cleavage reactions have much lower activation energies.
Changes in size, or the degrees of freedom [dof, or s in Eq. (l)], of the precursor ion have dramatic effects on k ( E ) curves. In general, increasing the dof causes slopes of k( E ) curves to become more shallow, and they level at lower values of log k. These changes, however, are not the same for direct cleavages and rear- rangements. Bente and co-workers (97) studied the "degrees of freedom effect" by comparing experimental changes in fragment ion abundances to theoretical log k( E) curves for a series of 2-alkanone molecular ions ranging from C6-C3'. An important observation was that the effect of differences in activation energy for a C-C bond cleavage either via a rearrangement or via a homolytic cleavage is magnified as size of the molecular ion increases. As a result, the probability of

CHARGE-REMOTE FRAGMENTATIONS 175
the rearrangement reaction, which has a lower activation energy, becomes greater with increasing size of the molecular ion, except, of course, at very high energies. That is, for large ions, a very large excess energy is needed to localize statistically enough internal energy into a higher-energy homolytic cleavage reaction coor- dinate in order for the homolytic cleavage to be competitive with a lower energy rearrangement reaction.
The dashed-line in Figure 9(B) represents a hypothetical mass spectrometric time window in relation to a particular value of precursor ion internal energy. For example, for CA in a sector mass spectrometer in a cm-size collision cell, reaction products can only be detected if their rate constants are above the hor- izontal dashed-line. Although the time window varies with precursor ion velocity, in general, the window for observing collision-induced product ions in sector mass spectrometers corresponds to a period of time shorter than a ps (log k - 6). At the particular value of ion internal energy shown in Figure 9(B), relative abundances of ions in a CID spectrum would reflect the areas under the log k( E ) curves that are contained in the time window. Thus, Na+ would be a highly abundant ion in CID spectra of (M + Na)+ ions, and loss of CnH2n+2 would be of minor abundance. In contrast, (M + Li)+ ions would decompose to give a greater abundance of fragment ions from loss of C,H2, +2, but very little Li+. It can be seen from Figure 9(B) that by increasing the ion internal energy, the vertical dashed-line will be shifted to the right. Thus, for (M + Li)+ ions, increasing the ion internal energy would result in increased detection of Li+. Eventually, Li+ would become the most abundant fragment ion in the CID spectrum. The "kinetic shift" is the difference between the activation energies for the reactions vs. the excess energies that are needed to detect the ions in the time window (98). It can be seen in Figure 9(B) that there is a kinetic shift associated with all three frag- mentation routes, and it is greatest for the rearrangement reaction. That is, E' for the rearrangement reaction is greater than E" for either of the heterolytic cleavages.
The three curves shown in Figure 9(B) are hypothetical, but they show curves for loss of Li+ and for loss of CnH2,+2 that cross: the curve-crossing has basis in experimental results reported by Adams and Gross (17). That is, the relative abundances of Naf and Li+ versus (C,H2,-10H-Cat)+ in CID spectra of fatty alcohol (M + Cat)+ ions are roughly described by the areas under the curves in the time window shown in Figure 9(B). From these considerations, the energy required, or the activation energy, for a charge-remote loss of CnH2n+2 was esti- mated to be between the bond enthalpies for loss of Na+ and for loss of Li+: 1.3- 1.9 eV (17). From Figure 9(B), if the activation energy of the rearrangement reaction were larger than for loss of Li+, the log k ( E ) curve for loss of CnH2n+2 would be shifted much farther to the right, and loss of Li+ would be the predominant pathway of reaction. Wysocki et al. (21) do report a more abundant loss of Li+ when they perform MS-MS experiments by using a VG-ZAB. The Li+ ion abun- dance in their spectrum is still less than the abundances of any losses of C,H2, +2,
and their results are consistent with the k ( E ) curves shown in Figure 9(B). The low energy required for the reaction that leads to loss of CnH2n+2 has been
further substantiated by other results. Deterding and Gross (36) compared losses of H 2 0 and NH3 from protonated fatty alcohols and amines, respectively, to

176 ADAMS
competitive losses of CnH2n+2, and they estimated the activation energy of the rearrangement reaction to be 1.4-2.7 eV. From the calculated AH,,, of 2.2 eV shown in Figure 9(A), this estimate of the activation energy may be more appro- priate than the previously estimated value of 1.3-1.9 eV (17).
These original estimates of the energy required for the loss of CnHZn+2 are supported by results both of Cody (91) and of Wysocki et al. (22). Cody studied charge-remote fragmentations of (M + H) + ions of 4-pentadecylpyridine by using Fourier transform ion cyclotron resonance (FTICR) MS. Here, the time window is increased to 100-500 ms (log k - 23) , which allows slower reactions to be observed at energies closer to threshold. In the MS time window observable in FTICR, charge-remote losses of C,H2, + 2 are detected at a laboratory collision energy of 2.5 eV (center-of-mass energy, E,, of 1.4-2.5 eV, discussed below). Wysocki et al. used low-eV CA and a triple quadrupole and found that the fragmentations are detectable both from protonated pentadecylpyridine and from lithiated oleyl alcohol at laboratory energies of 5 eV (€cm of 1.6 eV). Furthermore, Siege1 and Colthup (99) recently approximated the activation energy for a 1,4- elimination of C,H2, + 2 by using semiempirical molecular orbital calculations. Their theoretical calculations give an energy requirement of 2.7 eV.
The relatively low activation energy thus is consistent with a pericyclic elimination reaction such as a 1,4-elimination of H2 and C,H2,. An alternative mechanism (22) that would involve a two-step, charge-remote homolytic C-C bond cleavage can be evaluated with respect to the activation energy, to the RRKM theory [Fig. 9(B)], and to the experimental results of Adams and Gross (17). The activation energy for a homolytic, two-step radical reaction can be approximated from known activation energies for similar thermolytic radical eliminations from alkanes. For instance, homolytic cleavage of n-butane to give CH3 and n-C3H7 has an activation energy of 3.7 eV (84). This first step would then be followed by a-cleavage of n-C2H7 to eliminate H to give the final n- CH,CH=CH product. The radical-induced a-cleavage has an activation en- ergy of 1.6 eV (85), and therefore, the overall activation energy for this two- step homolytic cleavage would be 5.3 eV. The A factor for this reaction would be approximately the same as that for heterolytic loss of Na+ or Li+, or -1017 s-l, and thus the k ( E ) curve for the C-C homolytic bond cleavage reaction should be similarly shaped in that it also rises more steeply with increasing ion internal energy, although it would level at lower values of log k. The en- tire k ( € ) curve for the homolytic radical mechanism, however, would be moved to higher energies as a result of the larger activation energy for the process. As a result, to access this fragmentation route in the time window shown in Figure 9(B), the vertical dashed-line would have to be moved to much higher energies. It should be evident from Figure 9(B), and from the above discus- sion about the effects of increasing ion energy on spectra of (M + Li)+ ions, that this would result in CID spectra that would show highly abundant Li+ ions and weakly abundant (C,H2,-10H-Li)+ ions. This is not the case experi- mentally (17,21), however, and this, coupled with all the experimental data that indicate an activation energy less than 3 eV, implies that a two-step homolytic radical mechanism is unlikely to be the major mechanism for loss of CnH2n+2. It is unquestionable, however, that the weakly abundant radical cations that

CHARGE-REMOTE FRAGMENTATIONS 177
are formed by initial homolytic loss of CnH2n+l can further react to form some of the closed-shell ions, as shown by Wysocki et al. (22) and further sug- gested here in Scheme 5.
It is clear from Figure 9(A) and (B) that the energy required for the 1,4-elimi- nation of H2 is not necessarily the same as the energy required to detect the product ions. This would be the case only if there were neither a kinetic shift nor a competitive shift, the latter which also can reduce the detectability of product ions. A competitive shift is a result of lower-energy or faster fragmentation path- ways that effectively compete against the fragmentation path of interest. For instance, in the case of (M + Na)+ ions of fatty alcohols, many of the precursor ions fragment via the alternative reaction that results in formation of Na+ . Thus, the ions that instead fragment via charge-remote fragmentations are fewer, and the product ions from the charge-remote fragmentations are more difficult to detect.
Investigations by Wysocki et al. (19-22) and others (34,50,88,89) reveal the importance of competitive shifts and their effects on the energy required to detect charge-remote losses of CnH2n+2 from different types of precursor ions. These studies reconcile the low activation energy for the reactions with earlier, seemingly conflicting, data of Fraley and Lawrence (88), in which losses of CnHZn+2 from carboxylate anions cannot be detected at a laboratory collision energy of 300 eV (88), but analogous fragmentations from alkyltriphenylphosphonium ions, as re- ported by McCrery et al. (50), can be detected at a laboratory energy of 175 eV (50). The failure of CA to produce detectable charge-remote fragmentations of carboxylate anions at low laboratory energies was later confirmed by Bambagiotti and co-workers (34). Wysocki et al. (19-22) studied a greater variety of com- pounds, including sulfates, carboxylates, protonated amines, and lithiated oleyl alcohol, and found that the detectability of losses of CnHZn+2 was highly compound dependent. Gaskell and Reilly (89) investigated fragmentations of several other different types of biological ions, such as protonated isovaleryl carnitine, choles- tanol sulfate, and protonated androsterone glucuronides, and reported that struc- turally informative charge-remote fragmentations were missing at low laboratory energies. In addition, although Hunt et al. (100) and Brinegar et al. (101) find that many types of fragmentations of peptides can be induced at low eV collision energies, Alexander et al. (90) recently demonstrate that charge-remote fragmen- tations proposed by Johnson and co-workers (67) do not occur at laboratory collision energies <200 eV.
Results from these various studies of the energy required to produce detectable losses of CnH2n+2, which have been conducted by using sector and quadrupole mass spectrometers, are summarized in Table I. The data are arranged in de- creasing order of center-of-mass energy (E,,), which is calculated for the purpose of this review by using Eq. (2), in which Eo is initial kinetic
energy (laboratory collision energy) of the projectile ion; mo is mass of the projectile ion; and in2 is mass of the neutral target (collision) gas. Values of Ec, give the

1 78 ADAMS
Table I. quadrupole mass spectrometers
Losses of C,jHZn+2 under different collision conditions in sector and
Laboratory Detectable Mass Collision Collision E,, loss of
Ion spectrometer energy (eV) gas (ev). C,,H2,,+, Ref. Sector Sector Sector Quad. Sector Quad. Quad. Quad. Sector Quad. Quad. Quad. Quad. Quad. Quad. Quad. Quad. Quad. Quad. Quad. Quad.
8000 8000 8000 200
8000 400 250 300
2000 50 50 50
300 100 200 50 47 30 20 5 5
He He He Xe He Airb Ar Airb He Xe Xe Xe Airb Ar Airb Xe Ar Ar Ar Xe Xe
128 111 68 65 53 37 31 28 17 16 16 16 16 12 10 10 3.7 3.1 2.8 1.6 1.6
Yes 89 Yes 25 Yes 89 Yes 22 Yes 48 Yes 88 Yes 90 No 88 Yes 50 Yes 20 Yes 20 Yes 20 Yes 88 No 90 No 88 Yes 20 No 89 No 20 No 89 Yes 22 Yes 22
CI&ZJ" (C6H5)3 Sector 100 He 0.86 No 50 "The values calculated here for E,, assume single collisions. bThe mass of air used in the E,, calculations is assumed to be 28.964 (102).
maximum theoretical amount of the projectile ion's translational energy that can be converted into internal energy as a result of collision.
There are several features about the data presented in Table I that are worthy of note. One is that there is no relationship between laboratory collision energies and the detectability of charge-remote fragmentations: the only meaningful energy relationship involves the center-of-mass energy. Thus, charge-remote losses of CnHZn+2 can be detected at both keV and at eV laboratory collision energies as long as the target gas is massive enough in the eV collisions. This relationship reveals that charge-remote losses of CnHZn+2 generally are undetected in either sector or quadrupole mass spectrometers at center-of-mass collision energies that are a 1 2 eV, except for the three notable exceptions that occur at E,, of 10 and of 1.6 eV. Another feature is that the data are generally consistent among a large variety of different types of ions at least at the higher energies: compare data acquired at values of E,, of 16-17 eV. At higher E,,, the only major variant from this general pattern of consistency is carboxylate anions. At lower E,,, however, the situation changes in that some precursor ions fragment to give detectable losses of CnHZn+2, whereas others do not.
These changes in the energy required to detect collision-induced charge-remote fragmentations can be explained by the presence of competitive shifts, as sug- gested by Wysocki et al. (20). The anomalous, and striking, chemistry of carbox- ylate anions is thus understood from results of Giblin and Gross (92) and Tomer

CHARGE-REMOTE FRAGMENTATIONS 179
Table 11. and fragment ion abundances as a result of charge-reversal
Ratio between fragment ion abundances as a result of charge-remote fragmentations
~
Ratio
Oleate 5.5
Anion (c-remotek-reversal)
Octadecyl sulfonate 33 Octadecyl sulfate 43
Source: From Ref. 103.
(103). By performing CA and changing the polarity of the analyzer that follows the collision cell, products from collision-induced charge-reversal reactions can be observed. These experiments reveal that there is a significant competitive shift that is associated with CIDs of carboxylate anions that results from a highly favorable charge-reversal reaction (Table 11). Similarly, sulfonate and sulfate an- ions also undergo a charge-reversal reaction that offers important alternate frag- mentation routes to the charge-remote reactions (also in Table 11).
Competition against more favorable reactions also determines the detectability of charge-remote fragmentations at lower values of E,, for other types of ions. In cases in which the charge can be transferred to the alkyl chain, competitive fragmentations to give alkyl and alkenyl ions result in a competitive shift that reduces the detectability of losses of CnHznf2. Protonated alkyl amines lose NH3 to give a carbocation that undergoes charge-mediated fragmentations to give alkyl and alkenyl ions, as discussed in an earlier section. Inspection of CID spectra shown in Figures 3, 4, 6, and 7 reveals that CIDs of many ions are comprised of both charge-remote and charge-mediated fragmentations, in which the latter com- pete against the charge-remote fragmentations and serve to reduce their detect- ability.
Table 111. conditions in quadrupole and Fourier transform ICR mass spectrometers
Formation of w2 ions from peptides and losses of C,H2,1+2 under different collision
Laboratory collision Charge-remote
Mass energy Collision E,, fragmentation Ion spectrometer (eV) gas (eV). observed Ref.
lys-lys-H' Quad. 300 Ar 38 w2 90 tyr-gly-gly-phe-leu-arg-H+ Quad. 300 Ar 16 w2 90 tyr-gly-gly-phe-leu-lys-H+ Quad. 400 Ar 22 ~ ~ ( 5 0 % RA)b 90 tyr-gly-gly-phe-leu-lys-H' Quad. 300 Ar 16 ~ ~ ( 2 0 % RA)b 90 tyr-gly-gly-phe-leu-lys-H' Quad. 200 Ar 11 ~ ~ ( 4 % RA)b 90 CSHIIP+ (C&)3 FT 145 Ar 16' Loss of CnH2n+2 50 C,HI~P+(C&;)~ FT 145 He 1.7' None detected 50 (CisHdCsH2J' H FT 860 Ar 104' LOSS of CnH2,1+2d 91 (CEH&J%N 'H FT 2.5 Kr 1.4-2.5' LOSS of CnHZn+Zd 91
"The values calculated here for E,, assume single collisions unless otherwise noted. bThe relative abundance (RA) calculated here is relative to the B2 ions of miz 221 shown in the
'Here, E,, was calculated assuming single collisions, although multiple collisions most likely are
dLosses of CnH2n+2 do not include CH,. 'Calculated by Cody (91) who considered multiple collisions.
CID spectra in Ref. 90.
occurring.

180 ADAMS
More insight about the energetics of charge-remote fragmentations can be gained from data presented in Table 111, which summarizes other results obtained by using quadrupole and FTICR mass spectrometers. The data presented in Table I11 are grouped according to the type of charge-remote fragmentations that were investigated.
The data of Alexander's group (90) indicate that other types of charge-remote fragmentations require similar energies to detect product ions. They investigated formation of w2 ions that arise by charge-remote cleavages of (M + H)+ ions of leucine enkephalin-lysine. The energy required to detect w2 ions, as shown in Table 111, is similar to results presented in Table I for charge-remote losses of CnH2n+2. It can be seen that abundances of w2 ions decrease with decreasing values of E,,, and that the onset for observation of the w2 ions is at an E,, of approximately 11 eV.
Other data presented in Table I11 refer to studies performed by using FTICR MS to study charge-remote losses of C,H2, + 2 from closed-shell ions containing long alkyl chains. These results indicate the importance of observation time in detecting charge-remote fragmentations of some types of precursor ions. McCrery et al. (50) used a 1.3-ms collision period following ion cyclotron excitation and observed detectable losses of CnH2n+2 from alkyl triphenyl phosphonium ions at an E,, of at least 16 eV. None were detected, however, at an E,, of approximately 1.7 eV. Cody (91), however, used a 100-500-ms collision period following ion cyclotron excitation and was able to detect losses of CnH2n+2 (n > 1) from pro- tonated pentadecylpyridine at a reported E,, of 1.4-2.5 eV. The main difference between the two sets of FTICR data appears to be the collision and observation time periods: prolonging these times allows both for increased number of colli- sions and for longer times in which to observe slower reactions.
Another type of experiment has been performed to investigate the energy required to produce detectable charge-remote losses of CnH2n+2. Giblin and Gross (92,93) measured the translational energy loss, AE, in a series of charge-remote fragmentations at keV laboratory energies. From AE, the inelastic energy loss, Q, is calculated from Eq. (3), in which mo and m2 are masses of the
projectile ion and neutral target, respectively; Eo and El are kinetic energies of the projectile ion before and after collision, respectively; and 9 is the angle through which the projectile ion is deflected upon collision (95). The collision deflection angle 8 is normally assumed to be O", and the maximum value for Q is Ec,. The physical meaning of Q is discussed further above in relation to Figure 9(A): in theory, Q gives an estimate of the activation energy of a reaction plus excess energy of the transition state. The preliminary investigations (92,93) indicate that Qs of approximately 12-13 eV at Eo = 8 keV when He is used as collision gas are needed for losses of CnH2,+3 from carboxylate and dodecyl sulfate anions. These numbers imply that collision-induced losses of C,H2, + 2 from carboxylates and sulfates in sector spectrometers are associated with either a kinetic or a competitive

CHARGE-REMOTE FRAGMENTATIONS 181
shift of -10-11 eV. It should be noted, however, that the measured values of Q increase at lower precursor translational energies, and this may partially be a result of collisional scattering. That is, 8 > 0". If this is the case, then the real values for inelastic energy loss are smaller.
Angle-resolved mass spectrometry (ARMS) also has been used to study the energetics of charge-remote losses of CnHZn+2 (21,22,94). The basis for ARMS is that inelastic energy loss, as shown in Eq. (3), is directly related to the angle 8 through which the projectile ion is inelastically scattered by a collision. It follows that a fragment ion that arises from a higher internal energy precursor, which is inelastically scattered, is also characterized by the scattering angle of the precursor. Thus, an ARMS experiment entails selecting the angle at which collision-induced fragment ions are allowed to exit the collision cell, and their relative abundances theoretically should give an indication of the relative internal energies of their precursors. At higher collision deflection angles, fragment ions that arise from higher energy precursors should appear in higher abundances, and vice versa. The ideal case in which ions are energetically selected by collision deflection angle, however, must be tempered with the knowledge that the deflection angle includes contributions not only from inelastic scattering but from KER (104,105). Indeed, some early interpretations of ARMS results were later found to be incorrect be- cause scattering as a result of KER was not taken into account (105). Preliminary results of Giblin (94) and Wysocki et al. (21,22) indicate that there are changes in relative ion abundances of charge-remote losses of CnHZn+* with collision deflec- tion angle. As the laboratory angle is increased, fragment ions that arise by cleav- ages in closer proximity to the site of charge increase in abundance. One inter- pretation of these results (21,22) implies that the energy required for either producing or detecting the high-mass ions is smaller than the energy required for either producing or detecting the low-mass ions. Whether this is indeed the case, or alternatively, whether the differences in ion abundances with changes in collection angle are simply a result of KER or a result of differences in ion collection effi- ciency, will have to await further experimentation.
IV. CONCLUSIONS
This reviewer has attempted to describe the historical development of, and the contributions of a number of research groups to, the current understanding of charge-remote fragmentations. Retrospection reveals that the observation of charge- remote fragmentations was directly linked to the timely development of soft ionization techniques and collisional activation tandem mass spectrometry. In perspective, it can be seen that the nature of the ion fragmentations is quite reasonable: with available internal energy, if bond dissociation routes that re- quired either an unoccupied or a half-occupied (charged) molecular orbital were inaccessible, then alternate routes that involved other molecular orbitals (un- charged) would be accessed instead. That is, with available energy, an ion that contains a strongly localized, stable and unreactive site of charge undergoes ther- mal-like simple bond dissociations and rearrangement reactions that are charac- teristic of uncharged species.

182 ADAMS
Analytically important implications may be drawn from results presented here. Data from the analytical applications imply that any large molecule that contains a stable site of charge may be collisionally activated to undergo charge-remote fragmentations that give useful structural information. Thus, molecules of interest can contain either inherent ionic sites, or polar functional groups that associate strongly with alkali or alkaline-earth metal ions, or a moiety that can be chemically modified to produce a structural derivative that would serve to localize the site of charge. Results from the studies of the energetics of the ion decompositions imply that to detect collision-induced charge-remote fragmentations by using either sector or quadrupole mass spectrometers, center-of-mass collision energies greater than 16-17 eV are optimal, assuming that no significant competitive shifts occur. Energy loss measurements acquired at keV energies (laboratory frame) with He imply that at low collision energies, collisional scattering may be im- portant. This phenomenon might reduce the detectability of charge-remote frag- ment ions at low collision energies in sector mass spectrometers that have limited acceptance angles. Collisional scattering, however, should not significantly affect detectability-of fragment ions that arise from collisions occurring in quadrupoles in which the ions are constrained by rf fields.
There are still incompletely answered questions about the fundamental aspects of charge-remote fragmentations. One involves the mechanism surrounding losses of C,H2, + 2 from closed-shell ions containing long alkyl chains. Experimental evi- dence does not seem to support coiling of the alkyl chain or an ion-molecule- dipole complex, and energetics does not seem to support a primary mechanism that involves successive losses of CnH2,+< and H . The ultimate proof of the mechanism for loss of C,H2, + 2 requires determination of the neutral products by other mass spectrometric techniques (106).
Some aspects of the energetics of the different types of charge-remote frag- mentations are still unresolved. Some reactions occur as metastable ion decom- positions whereas some only occur as a result of CA. Evidence summarized in this review indicates that losses of CnH2n+2 definitely occur in the time frames that are needed for observation in sector and quadrupole mass spectrometers at an E,, of 16-17 eV (except for carboxylate anions, as discussed), although the onset for the fragmentations may be closer to an E,, of 10-12 eV. It is yet not known whether this range of E,, of 10-12 eV is only fortuitously similar to the range of 12-13 eV measured for energy loss Q in 8-keV collisions with He: the latter values are believed to be enhanced as a result of collisional scattering. In addition, it is necessary to consider that in the ms and longer time frame of FTICR, losses of CnH2,+2 are reported to occur at an E,, of 1.4-2.5 eV, which is virtually identical with estimates of the activation energy of the reaction. These results together imply that there may be a significant kinetic shift associated with the reactions in the p s time frame.
V. ACKNOWLEDGEMENTS
The author of this review acknowledges the help of M. J. Contado and R. L. Woodlee, and the many researchers who responded to requests for reprints and

CHARGE-REMOTE FRAGMENTATIONS 183
preprints of their work: A. M. Bambagiotti, R. K. Boyd, R. B. Cody, R. G. Cooks, S. J. Gaskell, M. L. Gross, J. L. Holmes, D. F. Hunt, J.-C. Prome, V. H. Reinhold, M. M. Siegel, K. B. Tomer, and V. H. Wysocki. Efforts to prepare this review were supported in part by the Emory University Research Fund and by the Pe- troleum Research Fund, which is administered by the American Chemical Society.
This work is dedicated in memory of Charles E. Lindsey and Myron C. A. Anderson.
1. 2.
3.
4. 5.
6.
7.
8. 9.
10. 11.
12.
13. 14. 15. 16. 17. 18. 19.
20. 21.
22.
23.
24.
25. 26. 27. 28. 29. 30.
VI. REFERENCES
Adams, 1.; Gross, M. L. Anal. Chem. 1987,59, 1576-1582. Gierlich,-H. H.; Rollgen, F. W.: Borchers, F.; Levsen, K. Org. Muss Spectrom. 1977, 22, 387-390. Rollgen, F. W.K; Borchers, F.; Giessmann, U.; Levsen, K. Org. Muss Spectrom. 1977, 22, 541-543. Weber, R.; Borchers, F.; Levsen, K.; Rollgen, F. W. Z. Nuturforsch. 1978, 33u, 540-548. Weber, R.; Levsen, K.; Louter, G. J.; Boerboom, A. J. H.; Haverkamp, J. Anal. Chem.
Weber, R.; Levsen, K.; Boerboom, A. J . H.; Haverkamp, J . Int. J. Muss Spectrom. Ion Phys. 1983,46, 305-308. Rollgen, F. W.; Giessmann, U.; Borchers, F.; Levsen, K. Org. Muss Spectrom. 1978, 23, 459-461. Rollgen, F. W.; Giessmann, U.; Levsen, K. Adv. Muss Spectrom. 1980, 8, 997-1003. Veith, H. J. Org. Muss Spectrom. 1978, 13, 280-283. Puzo, G.; Prome, J.-C.; Maxime, 8. Adu. Muss Spectrom. 1980, 8, 1003-1011. Puzo, G.; Maxime, B.; Prome, J. C. Int. J. Muss Spectrom. Ion Phys. 1983, 48, 169- 172. Schneider, E.; Levsen, K.; Dahling, P.; Rollgen, F. W. F . Z . Anal. Chem. 1983,326, 277- 285. Tomer, K. B.; Crow, F. W.; Gross, M. L. J. Am. Chem. SOC. 1983, 205, 5487-5488. Liehr, J. G.; Kingston, E. E.; Beynon, J. H. Biomed. Muss Spectrom. 1985, 12, 95-99. Jensen, N. J.; Tomer, K. B.; Gross, M. L. 1. A m . Chem. SOC. 1985, 207, 1863-1868. Sun, K. K.; Hayes, H. W.; Holman, R. T. Org. Muss Spectrom. 1970, 3, 1035-1042. Adams, J.; Gross, M. L. J. Am. Chem. SOC. 1986, 208, 6915-6921. Adams, J.; Gross, M. L. 1. A m . Chem. SOC. 1989, 222 , 435-440. Wysocki, V. H.; Bier, M. E.; Cooks, R. G. Proc. 35th A S M S Conf. Muss Spectrom. Allied Top. 1987, 223-224. Wysocki, V. H.; Bier, M. E.; Cooks, R. G. Org. Muss Spectrom. 1988, 23, 627-633. Wysocki, V. H.; Ross, M. M.; Cooks, R. G. Proc. 36th A S M S Conf. Muss Spectrom. Allied
Wysocki, V. H.; Ross, M. M.; Horning, S. R.; Cooks, R. G. Rapid Commun. Muss Spectrom.
Gross, M. L.; Jensen, N. J.; Lippstreu-Fisher, D. L.; Tomer, K. B. In: “Mass Spectrometry in the Health and Life Sciences”; Burlingame, A. L., Castagnoli, N., Jr., Eds., Elsevier: Amsterdam, 1985; pp. 209-238. Jensen, N. J.; Tomer, K. B.; Gross, M. L.; Lyon, P. A. In: “Desorption Mass Spectrom- etry”; ACS Symp. Ser. No. 291; Lyon, P. A., Ed., American Chemical Society: Wash- ington, DC, 1985; pp. 194-208. Jensen, N. J.; Tomer, K. B.; Gross, M. L. Anal. Chem. 1985, 57, 2018-2021. Tomer, K. B.; Jensen, N. J.; Gross, M. L. Anal. Chem. 1986, 58, 2429-2433. Jensen, N. J.; Gross, M. L. Lipids 1986, 22, 362-365. Jensen, N. J.; Tomer, K. B.; Gross, M. L. Lipids, 1986, 22, 580-588. Jensen, N. J.; Tomer, K. B.; Gross, M. L. Lipids, 1987, 22, 480-489. Prome, J.-C.; Aurelle, H.; Couderc, F.; Savagnac, A. Rapid Commun. Muss Spectrom. 1987,
1982,54, 1458-1466.
TOP. 1988, 381-382.
1988, 2, 214-216.
2, 50-52.

184 ADAMS
31. Aurelle, H.; Treilhou, M.; Prome, D.; Savagnac, A.; Prome, J. C. Rapid Commun. Muss Spectrom. 1987, 1, 65-66.
32. Couderc, F.; Aurelle, H.; Prome, D.; Savagnac, A.; Prome, J. C. Biomed. Enuiron. Mass Spectrom. 1988, 16, 317-321.
33. Savagnac, A.; Aurelle, H.; Casas, C.; Couderc, F.; Gavard, P.; Prome, D.; Prome, J . C. Chem. Phys. Lipids, in press.
34. Bambagiotti, A. M.; Coran, S. A.; Vincieri, F. F.; Petrucciani, T.; Traldi, P. Org. Mass Spectrom. 1986, 21, 485-488.
35. Deterding, L. J.; Gross, M. L. Anal. Chim. Acta 1987, 200, 431-445. 36. Deterding, L. J.; Gross, M. L. Org. Mass Spectrom. 1988, 23, 169-177. 37. Adams, J.; Gross, M. L. Org. Mass Spectrom. 1988, 23, 307-316. 38. Adams, J.; Deterding, L. J.; Gross, M. L. Spectros. Int. I . 1987, 5, 199-228. 39. Adams, J. Proc. 35th ASMS Conf. Mass Spectrom. Allied Top. 1987, 227-228. 40. Contado, J. C.; Adams, J.; Gross, M. L. Adv. Mass Spectrorn., in press. 41. Crockett, J . S.; Gross, M. L. Proc. 37th ASMS Conf. Mass Spectrom. Allied Top. 1989, 240-
241. 42. Davoli, E.; Bettazzoli, L.; Gross, M. L.; Murphy, R. C. Proc. 37th ASMS Conf. Mass
Spectrom. Allied Top. 1989, 754-755. 43. Jensen, N. J.; Gross, M. L. Mass Spectrom. Rev. 1987, 6 , 497-536. 44. Chauhan, Y.; Nagel, D.; Gross, M.; Cerny, R.; Toth, B. 1. Agric. Food Chem. 1985, 33,
45. Caldwell, K.; Gross, M. L. Anal. Chem. 1989, 61, 494-496. 46. Lyon, P. A.; Crow, F. W.; Tomer, K. B.; Gross, M. L. Anal. Chern. 1984, 56, 2278-
2284. 47. Lyon, P. A.; Stebbings, W. L.; Crow, F. W.; Tomer, K. B.; Lippstreu, D. L.; Gross, M.
L. Anal. Chem. 1984, 56, 8-13. 48. Lyon, P. A.; Tomer, K. 8.; Gross, M. L. Anal. Chem. 1985, 57, 2984-2989. 49. March, J. ”Advanced Organic Chemistry”, 3rd ed.; John Wiley & Sons: New York, 1985;
50. McCrery, D. A.; Peake, D. A.; Gross, M. L. Anal. Chem. 1985,57, 1181-1186. 51. Tomer, K. B.; Gross, M. L. In: “Ion Formation from Organic Solids (IFOS 111)”; Ben-
ninghoven, A. Ed., Springer-Verlag: Berlin, 1986; pp. 134-139. 52. Miinster, H.; Budzikiewicz, H. Rapid Commun. Mass Spectrom. 1987, 1, 126-
128. 53. Jensen, N. J.; Gross, M. L. Mass Spectrom. Rev. 1988, 7, 41-69. 54. Dasgupta, A.; Ayanoglu, E.; Wegmann-Szente, A.; Tomer, K. B.; Djerassi, C. Chem.
Phys. Lipids 1986, 41, 335-347. 55. Dasgupta, A.; Ayanoglu, E.; Tomer, K. B.; Djerassi, C. Chem. Phys. Lipids 1987,43, 101-
111. 56. Tomer, K. B.; Crow, F. W.; Knoche, H. W.; Gross, M. L. Anal. Chem. 1983, 55, 1033-
1036. 57. Cerny, R. L.; Tomer, K. B.; Gross, M. L. Org. Mass Spectrom. 1986, 21, 655-660. 58. Gaskell, S. 1.; Guenat, C.; Millington, D. S.; Maltby, D. A.; Roe, C. R. Anal. Chem. 1986,
58, 2801-2805. 59. Domon, B.; Costello, C. E. Biochemistry 1988, 27, 1534-1543. 60. Wright, L. G.; Cooks, R. G.; Wood, K. V. Biomed. Mass Spectrom. 1985, 12, 159-
162. 61. Crow, F. W.; Tomer, K. B.; Looker, J. H.; Gross, M. L. Anal. Biochem. 1986, 155, 286-
307. 62. Light, K.; Schultz, G.; Allision, J. Proc. 37th ASMS Conf. Mass Spectrom. Allied Top. 1989,
256-257. 63. Carr, S. A.; Reinhold, V. N.; Green, B. N.; Hass, J. R. Biomed. Enuiron. Mass Spectrom.
64. Gaskell, S . J,; Brownsey, B. G.; Brooks, P. W.; Green, B. N. Biomed. Enuiron. Mass Spectrom. 1983, 10, 215-219.
65. Tomer, K. B.; Jensen, N. J.; Gross, M. L.; Whitney, J. Biomed. Envivon. Muss Spectrom.
81 7-820.
pp. 845-854.
1985, 12, 288-295.
1986, 13, 265-272.

CHARGE-REMOTE FRAGMENTATIONS 185
66. 67.
68.
69.
70.
71.
72. 73.
74. 75. 76. 77. 78. 79.
80. 81
82. 83.
84.
85.
86. 87. 88.
89. 90.
91. 92.
93.
94. 95.
96. 97.
98. 99.
100.
101.
102.
Tomer, K. B.; Gross, M. L. Biomed. Environ. Mass Spectrom. 1988, 15, 89-98. Johnson, R. S.; Martin, S. A.; Biemann, K.; Stults, J. T.; Watson, J. T. Anal. Chem. 1987, 59, 2621-2625. McLafferty, F. W. "Interpretation of Mass Spectra", 3rd ed.; University Science Books: Mill Valley, CA, 1980. Johnson, R. S.; Martin, S. A.; Biemann, K. lnt. J. Mass Spectrom. lon Proc. 1988, 86, 137- 154. Bambagiotti, A. M.; Coran, S. A.; Giannellini, V.; Vincieri, F. F.; Daolio, S.; Traldi, P. OrR. Mass Spectrom. 1983, 18, 133-134. Bambagiotti, A. M.; Coran, S. A.; Giannellini, V.; Vincieri, F. F.; Daolio, S.; Traldi, P. Org. Mass Spectrom. 1984, 19, 577-580. Chang, T. T.; Tsou, H.-R.; Siegel, M. M. Anal. Chem. 1987, 59, 614-617. Siegel, M. M.; McGahren, W. J.; Tomer, K. B.; Chang, T. T. Biomed. Environ. Mass Spectrorn. 1987, 14, 29-38. Hand, 0. W.; Cooks, R. G. Int. J. Mass Spectrom. Zon Proc., in press. Renner, D.; Spiteller, G. Biomed. Environ. Mass Spectrom. 1988, 15, 75-77. Grese, R. P.; Cerny, R. L.; Gross, M. L. J. A m . Chem. SOC. 1989, 111, 2835-2842. Longevialle, P.; Botter, R. Org. Mass Spectrom. 1983, 18, 1-8. Longevialle, P. Org. Mass Spectrom. 1985, 20, 644-645. Robinson, P. J.; Holbrook, K. A. Unimolecular Reactions; Wiley-Interscience: London, 1972. Ashby, E. C.; Willard, G. F.; Goel, A. B. J. Org. Chem. 1979,44, 1221-1232. Brieger, G.; Watson, S. W.; Barar, D. G.; Shene, A. L. J. Org. Ckem. 1979, 44, 1340- 1342. Arnold, R. T.; Smolinksy, G. J. Org. Chem. 1960, 25, 129-131. Contado, J. C.; Adams, J.; Jensen, N. J.; Gross, M. L. Proc. 37thASMS Conf. Mass Spectrom. Allied Top. 1989, 654-655. Benson, S. W.; ONeal, H. E. Kinetic Data on Gas Phase Unimolecular Reactions; NSRDS- NBS 21; U.S. Government Printing Office: Washington, D.C., 1970. Kerr, J. A. In: "Free Radicals"; Kocki, J. K. ed., John Wiley & Sons: New York, 1973; Vol. 1, pp. 1-36. Kossiakoff, A.; Rice, F. 0. J. Am. Chem. SOC. 1943, 65, 590-595. Cerny, R. L.; Gross, M. L.; Grotjahn, L. Anal. Biockem. 1986, 156, 424-435. Fraley, D. F.; Lawrence, D. L. Proc. 33rd ASMS Conf. Mass Spectrom. Allied Top. 1985, 144-145. Gaskell, S. J.; Reilly, M. H. Rapid Commun. Mass Spectrom. 1988, 2, 139-142. Alexander, A. J.; Thibault, P.; Boyd, R. K. Rapid Commun. Mass Spectrom., 1989, 3, 30- 34. Cody, R. B. Rapid Commun. Mass Spectrom. 1988, 2, 260-261. Giblin, D. E.; Gross, M. L. Proc. 36th ASMS Conf. Mass Spectrom. Allied Top. 1988, 83- 84. Giblin, D. E.; Gross, M. L. Proc. 37th ASMS Conf. Mass Spectrom. Allied Top. 1989, 238- 239. Giblin, D. E. Proc. 33rd ASMS Conf. Mass Spectrom. Allied Top. 1985, 921-922. Kessell, Q. C.; Pollack, E.; Smith, W. W. In Collision Spectroscopy; Cooks, R. G. (ed.); Plenum Press: New York, 1978; pp. 147-225. Williams, D. H. Acc. Chem. Res. 1977, 10, 280-286. Bente, P. F., 111; McLafferty, F. W.; McAdoo, D. J.; Lifshitz, C. J. Phys. Ckem. 1975, 79, 713-721. Rosenstock, H. M. Znt. J. Mass Spectrom. Ion Proc. 1976, 20, 139-190. Siegel, M. M.; Colthup, N. B. Appl. Spectrosc. 1988, 42, 1214-1221. Hunt, D. F.; Yates, J . R., 111; Shabanowitz, J.; Winston, S.; Hauer, C. R. Proc. Natl. Acad. Sci. U S A 1986, 83, 6233-6237. Brinegar, A. C.; Cooper, G.; Stevens, A.; Hauer, C. R.; Shabanowitz, J.; Hunt, D. F.; Fox, J. E. Proc. Natl. Acad. Sci. U S A 1988, 1988, 85, 5927-5931. Weast, R. C., Ed., "CRC Handbook of Chemistry and Physics", 58th ed.; CRC Press: West Palm Beach, FL, 1977.

186 ADAMS
103. 104.
105.
106.
Tomer, K. B. Org. Mass Spectrom., 1989, 24, 969-972. Sigh, S.; Thatker, M. S.; Harris, F. M.; Beynon, J. H. Org. Mass Spectrom. 1985, 20, 156- 157. Waddell, D. S.; Boyd, R. K.; Brenton, A. G.; Beynon, J. H. Int. J. Mass Spectrom. Ion Proc. 1986, 68, 71-90. Clair, R.; Holmes, J. L.; Mommers, A. A.; Burgers, P. C. Org. Mass Spectrorn. 1985, 20, 207-212.




















