
Centrosomal Chk2 in DNA damage responses and cell cycle progession
10
www.landesbioscience.com Cell Cycle 2647 Cell Cycle 9:13, 2647-2656; July 1, 2010; © 2010 Landes Bioscience REPORT REPORT *Correspondence to: David F. Stern; Email: [email protected] Submitted: 04/02/10; Accepted: 04/19/10 Previously published online: www.landesbioscience.com/journals/cc/article/12121 DOI: 10.4161/cc.9.13.12121 Introduction Cell cycle checkpoint controls maintain the temporal order of biochemically unlinked processes. It is increasingly appar- ent that checkpoint systems integrate activities of multiple cel- lular systems. Recent findings suggest, also, communication between DNA damage signaling systems and mitotic regulators. For example, polo-like kinases Plk1 and Plk3 are important for progression into and through mitosis, and they are regulated by DNA damage. Both Plks interact with Chk2. 1-3 A number of DNA checkpoint proteins, including ATM, ATR, BRCA1, Chk1, Chk2 and p53 have now been localized to centrosomes, the microtubule organizing centers, suggest- ing a direct connection with mitotic regulation. 1,4-7 In principle, checkpoint proteins at the centrosome could serve as points of cross-regulation for DNA damage response and mitotic spindle processes. Alternatively, the centrosome could serve as a site of sequestration for inhibition of signaling 8,9 or a meeting place for interacting proteins independent of spindle functions. For example, Chk1 tonically inhibits Cyclin B-Cdk1 activity that drives mitotic entry and prevents premature centrosome separa- tion and activation of centrosome-localized Cdk1. Activation of Cdk1 by dephosphorylation coincides with loss of Chk1 from the centrosomes, so that Chk1 seems to be an important regulator that forestalls premature M phase transit. 4 Here, the centrosomal Two major control systems regulate early stages of mitosis: activation of Cdk1 and anaphase control through assembly and disassembly of the mitotic spindle. In parallel to cell cycle progression, centrosomal duplication is regulated through proteins including Nek2. Recent studies suggest that centrosome-localized Chk1 forestalls premature activation of centrosomal Cdc25b and Cdk1 for mitotic entry, whereas Chk2 binds centrosomes and arrests mitosis only after activation by ATM and ATR in response to DNA damage. Here, we show that Chk2 centrosomal binding does not require DNA damage, but varies according to cell cycle progression. These and other data suggest a model in which binding of Chk2 to the centrosome at multiple cell cycle junctures controls co-localization of Chk2 with other cell cycle and centrosomal regulators. Centrosomal Chk2 in DNA damage responses and cell cycle progession Amnon Golan, 1,† Elah Pick, 2,‡ Lyuben Tsvetkov, 1,¥ Yasmine Nadler, 3 Harriet Kluger 3 and David F. Stern 1, * 1 Department of Pathology; Yale School of Medicine; New Haven, CT USA; 2 Department of Molecular, Cellular and Developmental Biology; Yale University; New Haven, CT USA; 3 Section of Oncology; Department of Internal Medicine; Yale School of Medicine; New Haven, CT USA Current addresses: † Wolfson Advanced Research Center for Protein Turnover; The Bruce Rappaport Faculty of Medicine; Technion-Israel Institute of Technology; Haifa, Israel; ‡ Department of Biology; Haifa University at Oranim; Tivon, Israel; ¥ Drug Discovery; SRB-3; Moffitt Research Institute; Tampa, FL USA Key words: Chk2, centrosome, checkpoint, DNA damage, wild type, kinase-defective Abbreviations: IF, immunofluorescence; GFP, green fluorescent protein; WT, wild type; KD, kinase-defective localization of Chk1 facilitates coordination of Cdk1 activation with centrosomal processes. One of the earliest associations of a checkpoint protein with centrosomes was our finding that phosphorylated Chk2 co-local- izes with Plk1 at centrosomes. 1 Chk2 threonine 68 is one of the sites in the Chk2 serine-glutamine/threonine-glutamine cluster domain (SCD) that is phosphorylated in response to DNA dam- age. Phosphorylation at this site is mediated by the PIKK ATM, and by Chk2 itself. We found in immunofluorescence (IF) experi- ments that immunoreactive T68-phosphorylated Chk2 (PT68- Chk2) was detectable at centrosomes, even in the absence of DNA damage, as was a small proportion of HA-tagged Chk2 (and GFP- tagged Chk2, unpublished data). Despite the extensive purifica- tion and characterization of the PT68-Chk2 antibody, and our finding that epitope-tagged Chk2 showed some centrosomal local- ization, concerns about cross-reactivity of the phospho-antibody remained, especially since some commercial anti-PT68 antibodies cross-react. 10 Another report documented the presence of centro- somal Chk2, but only after DNA damage, 4 and another labora- tory reported that Chk2 is localized to centrosomes in interphase and mitosis, and phosphorylated at sites associated with activation. In a cell fusion-based model for induction of mitotic arrest, Chk2 co-localized with the centrosomal component γ-tubulin. 11 These disparate results raised a number of questions regard- ing Chk2, including the verity of centrosomal localization, the
Transcript of Centrosomal Chk2 in DNA damage responses and cell cycle progession

www.landesbioscience.com Cell Cycle 2647
Cell Cycle 9:13, 2647-2656; July 1, 2010; © 2010 Landes Bioscience
RepoRt RepoRt
*Correspondence to: David F. Stern; Email: [email protected]: 04/02/10; Accepted: 04/19/10Previously published online: www.landesbioscience.com/journals/cc/article/12121DOI: 10.4161/cc.9.13.12121
Introduction
Cell cycle checkpoint controls maintain the temporal order of biochemically unlinked processes. It is increasingly appar-ent that checkpoint systems integrate activities of multiple cel-lular systems. Recent findings suggest, also, communication between DNA damage signaling systems and mitotic regulators. For example, polo-like kinases Plk1 and Plk3 are important for progression into and through mitosis, and they are regulated by DNA damage. Both Plks interact with Chk2.1-3
A number of DNA checkpoint proteins, including ATM, ATR, BRCA1, Chk1, Chk2 and p53 have now been localized to centrosomes, the microtubule organizing centers, suggest-ing a direct connection with mitotic regulation.1,4-7 In principle, checkpoint proteins at the centrosome could serve as points of cross-regulation for DNA damage response and mitotic spindle processes. Alternatively, the centrosome could serve as a site of sequestration for inhibition of signaling8,9 or a meeting place for interacting proteins independent of spindle functions. For example, Chk1 tonically inhibits Cyclin B-Cdk1 activity that drives mitotic entry and prevents premature centrosome separa-tion and activation of centrosome-localized Cdk1. Activation of Cdk1 by dephosphorylation coincides with loss of Chk1 from the centrosomes, so that Chk1 seems to be an important regulator that forestalls premature M phase transit.4 Here, the centrosomal
two major control systems regulate early stages of mitosis: activation of Cdk1 and anaphase control through assembly and disassembly of the mitotic spindle. In parallel to cell cycle progression, centrosomal duplication is regulated through proteins including Nek2. Recent studies suggest that centrosome-localized Chk1 forestalls premature activation of centrosomal Cdc25b and Cdk1 for mitotic entry, whereas Chk2 binds centrosomes and arrests mitosis only after activation by AtM and AtR in response to DNA damage. Here, we show that Chk2 centrosomal binding does not require DNA damage, but varies according to cell cycle progression. these and other data suggest a model in which binding of Chk2 to the centrosome at multiple cell cycle junctures controls co-localization of Chk2 with other cell cycle and centrosomal regulators.
Centrosomal Chk2 in DNA damage responses and cell cycle progession
Amnon Golan,1,† elah pick,2,‡ Lyuben tsvetkov,1,¥ Yasmine Nadler,3 Harriet Kluger3 and David F. Stern1,*
1Department of pathology; Yale School of Medicine; New Haven, Ct USA; 2Department of Molecular, Cellular and Developmental Biology; Yale University; New Haven, Ct USA; 3Section of oncology; Department of Internal Medicine; Yale School of Medicine; New Haven, Ct USA
Current addresses: †Wolfson Advanced Research Center for protein turnover; the Bruce Rappaport Faculty of Medicine; technion-Israel Institute of technology; Haifa, Israel; ‡Department of Biology; Haifa University at oranim; tivon, Israel; ¥Drug Discovery; SRB-3; Moffitt Research Institute; tampa, FL USA
Key words: Chk2, centrosome, checkpoint, DNA damage, wild type, kinase-defective
Abbreviations: IF, immunofluorescence; GFP, green fluorescent protein; WT, wild type; KD, kinase-defective
localization of Chk1 facilitates coordination of Cdk1 activation with centrosomal processes.
One of the earliest associations of a checkpoint protein with centrosomes was our finding that phosphorylated Chk2 co-local-izes with Plk1 at centrosomes.1 Chk2 threonine 68 is one of the sites in the Chk2 serine-glutamine/threonine-glutamine cluster domain (SCD) that is phosphorylated in response to DNA dam-age. Phosphorylation at this site is mediated by the PIKK ATM, and by Chk2 itself. We found in immunofluorescence (IF) experi-ments that immunoreactive T68-phosphorylated Chk2 (PT68-Chk2) was detectable at centrosomes, even in the absence of DNA damage, as was a small proportion of HA-tagged Chk2 (and GFP-tagged Chk2, unpublished data). Despite the extensive purifica-tion and characterization of the PT68-Chk2 antibody, and our finding that epitope-tagged Chk2 showed some centrosomal local-ization, concerns about cross-reactivity of the phospho-antibody remained, especially since some commercial anti-PT68 antibodies cross-react.10 Another report documented the presence of centro-somal Chk2, but only after DNA damage,4 and another labora-tory reported that Chk2 is localized to centrosomes in interphase and mitosis, and phosphorylated at sites associated with activation. In a cell fusion-based model for induction of mitotic arrest, Chk2 co-localized with the centrosomal component γ-tubulin.11
These disparate results raised a number of questions regard-ing Chk2, including the verity of centrosomal localization, the

2648 Cell Cycle Volume 9 Issue 13
Sedimentation of the core centrosomal protein γ-tubulin sepa-rated a “free” component, that co-sedimented with the cytoplas-mic non-centrosomal protein Grb2 (Fig. 1D, fractions 1–3), and a component that sedimented rapidly (Fig. 1D, fractions 10 and beyond). Other proteins known to associate with centrosomes, including Mps1, p53 and CDK1 also showed this dual sedimen-tation pattern (Fig. 1D).
Chk2 co-purifies with centrosomes. In pilot experiments with Gradient II (Fig. 2A), soluble Grb2 sedimented slowly, but centrosomal proteins including γ-tubulin and CDK1 showed the bimodal distribution. The second peak was centered in the range of 40 to 45% sucrose (Fig. 2A, fractions 10–12). Other proteins associated with centrosomes, including Mps1, cosedimented with γ-tubulin in this range, whereas p53 showed a broader distribu-tion (Fig. 1D, fractions 11–13). Immunoblotting gradient-puri-fied centrosome preparations from HEK 293 cells with anti-Chk2 identified bands with approximate molecular weights of 62 kD
cell cycle-dependency of such localization, and the impact of Chk2 on centrosomal functions and co-localization of other proteins. Moreover, the dependency of Chk2 centrosomal local-ization on induced DNA damage and on Chk2 activity and phosphorylation was uncertain. For this reason, we decided to use a complementary approach to IF, biochemical purification of centrosomes.
Results
Centrosome purification. Centrosomes were purified by consec-utive sedimentation steps (Fig. 1A). Typical profiles of proteins in gradients II and III are shown shown in Figure 1B and C, Gradients I and II only were used for most experiments described herein. Separation of proteins mid-gradient (gradient II, fractions 6 and 7) from the 70% sucrose cushion avoided collection of aggregates that sediment to the cushion (Fig. 1B, fraction 12).
Figure 1. Gradient purification of centrosomes. (A) Centrosome purification from HeK293 cells by three successive sucrose gradients. (B and C) Sucrose gradient fractions were analyzed by 5–20% gradient gel SDS-pAGe with proteins detected by silver staining. (B) Centrosome purification through gra-dients I and II only. Lanes 1–4 include mainly soluble proteins, lanes 6 and 7 are enriched for centrosomes, and lane 12 contains high molecular weight aggregates. (C) Centrosome purification through gradients I, II and III. StI, soy trypsin inhibitor carrier. (D) Gradient fractions (gradient II) analyzed by immunoblotting. Grb2 was used to mark cytosolic proteins, γ-tubulin marks centrosomes, p53, Mps1 have soluble and centrosomal components.

www.landesbioscience.com Cell Cycle 2649
absence of DNA damage, under conditions in which PT68 is not detectable in the bulk population of Chk2.1 Despite extensive efforts, we have been unable to reliably detect immunoreactive endogenous Chk2 phosphorylated at T68 in partially purified centrosome preparations (data not shown). Also, although Chk2 electrophoretic mobility is retarded by DNA damage-dependent phosphorylation (Fig. 2D, compare lanes 2 and 4), the major form of Chk2 in centrosome preparations comigrated with the form of Chk2 found in basal (Fig. 2D, lane 4), but not irradiated cells (Fig. 2D, lane 2; see also, B). (We were able to detect DNA damage-dependent mobility shift of transiently overexpressed GFP-Chk2 as discussed below, Fig. 5). Nonetheless, we can-not rule out dephosphorylation of Chk2 during the many hours required for these preparations. The association of kinase-defec-tive Chk2 with centrosomes (Fig. 2C) means that Chk2 kinase activity is not required for binding.
Multiple forms of Chk2. Chk2 typically migrates with an approximate molecular weight of 62 kDa. However, anti-Chk2
and 52 kD, (Fig. 2B, lanes 7–9). The larger band comigrates with Chk2 from non-fractionated extracts of non-irradiated cells (Fig. 2B, lane 2). Similarly, in HEK 293 cells expressing green fluorescent-protein (GFP)-tagged kinase-defective (KD) Chk2, full-length GFP-Chk2 KD co-purified with centrosomes as well as endogenous truncated Chk2 bands (Fig. 2C). In some experi-ments (Fig. 2C), but not others, Chk2 sedimented rapidly, but with a distribution slightly slower than the center of γ-tubulin distribution. This may mean that Chk2 associates with a subset of centrosomes, which include many loosely- and tightly-associ-ated proteins.12 Taken together, these data provide biochemical evidence supporting the conclusion that a component of Chk2 associates with centrosomes in the absence of DNA damage or Chk2 kinase activity.
Kinase activity and phosphorylation of centrosomal Chk2. Phosphorylation of Chk2 at the PIKK target site T68 contributes to and coincides with Chk2 activation.13 Chk2 was originally detected in centrosomes with anti-Chk2 PT68 antibodies in the
Figure 2. Chk2 localizes to centrosomes without DNA damage. (A) Western blot analysis of endogenous Chk2 in fractions (gradient II) from non-irra-diated HeK293 cells. (B) extracts from cells without (lane 2) or with (lane1) prior γ-irradiation. Lanes 3 through 12, gradient fractions encompassing the centrosome peak from non-irradiated HeK293 cells. (C) GFp-Chk2 (KD) stably expressed in HeK293 stable cells, endogenous Chk2, and endogenous γ-tubulin were detected by immunoblotting. Lane 1, total extract before application to gradient I. Fractions 2 through 9 are from a portion of gradient II including the centrosome peak. (D) Anti-Chk2 immunoblot of extracts of mock-treated HeK293 (lane 4), HeK293 cells exposed to ionizing radiation (10 Gy, lane 2) or (lanes 1 and 3) two different centrosome preparations concentrated twenty-fold with Strataclean resin (Stratagene). “total” extracts are supernatants from gradient I.

2650 Cell Cycle Volume 9 Issue 13
kDa form than parallel extracts pre-pared with centrosome buffer (Fig. 3A). Incubation at room tempera-ture for 30 min. with DNase only slightly enhanced recovery of the 52 kd form, so only small amounts of in vitro cleavage occur during the DNase step in centrosome puri-fication (Fig. 3B). The presence or absence of either Nocodazole or the proteasome inhibitor MG132 had little impact on the recovery of the smaller form (data not shown).
We used amino and carboxyl ter-minal epitope tags to map the major endogenous Chk2 cleavage sites (Fig. 3C and D). HEK 293 cells were engineered to express GFP-Chk2, with a 27 kD N-terminal GFP tag, or GFP-Chk2-AKAP, with an additional 8 kd carboxyl-terminal AKAP tag. Full length and clipped endogenous Chk2 (bands III and IV), GFP-Chk2 (band II) and GFP-Chk2-AKAP (band I) are detectable with anti-Chk2 in lanes 2–4, respectively. Blotting with anti-GFP marks the full length bands, and also a major approximately 37 KD cleavage product (band V, lanes 7 and 8) that is approximately 10 kDa larger than the 27 kDa GFP tag (band VI). The simplest interpretation is that the major cleavage site removes a 10 kDa amino terminal fragment of Chk2 that, with GFP fusions, is fused to GFP. Then, the larger of the two small GFP-containing products (band V) is the reciprocal cleavage product complementing band IV, and containing between 2 and 10 kd of Chk2 sequence that does not react with the Chk2 anti-
body. Since the shortened form of Chk2 (band IV) is associated with centrosomal material, it is likely that the major 52 kd cleav-age product including the carboxyl terminus retains sequences sufficient for binding to centrosomes or centrosome-associated macromolecules. This portion would still retain the full kinase catalytic domain, but not the FHA domain, which binds phos-phopeptides and is involved in DNA damage-dependent localiza-tion and activation of Chk2.
Cell cycle. We next determined the distribution of full-length and truncated Chk2 during the cell cycle. HeLa cells were syn-chronized at the beginning of S phase using a double thymi-dine block, and released for various periods of time (Fig. 4A).
immunoblots of centrosome preparations revealed a smaller 52 kDa form of immunoreactive Chk2 (Fig. 2B). In cells stably expressing GFP-Chk2 and endogenous Chk2 (Figs. 2C, lane 1; 3C, lane 7) bands include endogenous full-length 62 kDa Chk2, 52 kDa form, full-length GFP-Chk2, and shorter forms associ-ated with expression of GFP-Chk2. Alternative splicing of Chk2 has been reported.14 However, since smaller forms are produced with Chk2 expressed from tagged cDNAs (Fig. 2C and Figures following), it is likely that some result from cleavage of Chk2, rather than expression of Chk2 mRNA variants.
Protein extracts produced with the more stringent TSD solu-bilization buffer contained significantly lower quantities of the 52
Figure 3. Cleavage of centrosomal Chk2. (A–C) Centrosome preparations from exponentially growing HeK293 cells were analyzed by western blot with anti-Chk2 or anti-GFp antibodies. (A) extracts contain-ing endogenous Chk2 in centrosome isolation buffer (left) or tSD buffer (right). (B) Lane 1, full-length endogenous Chk2 marker. Recovery of full-length and cleaved forms of endogenous Chk2 and GFp-Chk2 (Wt) in extracts that were kept on ice (lane 2) or incubated thirty min Rt in 10 mM Hepes pH 7.2 and 2 units/ml DNase. (C) protein extracts from HeK293 cells (lanes 1, 2, 5 and 6); HeK293 cells stably expressing GFp-Chk2 (lanes 3 and 7); or GFp-Chk2-pACt [AKAp] (lanes 4 and 8); were analyzed by immunoblotting with anti-Chk2 and anti-GFp. (D) Schematic interpretation of Chk2 forms marked with Roman numerals as in (C), with proposed amino-terminal Chk2 cleavage site marked by arrow.

www.landesbioscience.com Cell Cycle 2651
of Chk2 relative to γ-tubulin occurred in the preparations nine and eleven hours post-release that were enriched for cells in mito-sis and G
1 (Fig. 4A). Interestingly, the centrosomal preparations
during these time points are enriched for the smaller form, even with the greater prevalence of full-length Chk2 at those times (Fig. 4B) taken into account.
Chk2 is associated with centrosomes after DNA damage. It has been reported that Chk2 associates with centrosomes only in response to DNA damage, which activates Chk2.4 This associa-tion does not require Chk2 kinase activity, since kinase-defective GFP-Chk2-KD stably expressed in HEK293 cells co-sedimented with centrosomal proteins (Fig. 4D). However, it was possible that endogenous wild-type (WT) Chk2 in these cells phosphorylated the GFP-Chk2-KD, since Chk2 molecules cross-phosphorylate in vivo.13 To determine the effects of kinase domain mutations
Total (non-sedimented) preparations prepared with centrosome extraction buffer were analyzed. By nine hours post-release, a substantial proportion of cells had G
2 DNA content (Fig. 4A),
and M phase was marked at 11 hours by predominance of hyper-phosphorylated CDC25b, and loss of Nek2a (Fig. 4B). Chk2 cleavage was apparent first at nine hours, and highest at times with predominantly fast-migrating Nek2 and hyperphosphory-lated CDC25B, when cells are predominantly in mitosis and/or have returned to return to G
1 (Fig. 4B),15 suggesting mitotic
cleavage.We attempted to determine the relationship of the varying
recovery of full-length and cleaved Chk2 to centrosome-asso-ciated Chk2 (Fig. 4C). (This centrosomal isolation experiment was only performed once because of the large number of cells required for each of these preparations). The greatest recovery
Figure 4. Binding of Chk2 to centrosomes during cell cycle progression. (A–C) HeLa cells were synchronized at S phase entry by double thymidine block, released and incubated for various periods of time. (A) Flow microfluorimetry analysis of cell samples. Red line marks cells that had been synchronized and released from thymidine block, and black line marks non-synchronized cells analyzed in parallel. (B) Analysis of non-fractionated ex-tracts (after Gradient I) from cells chased for periods of time between two and eighteen hours, as marked, post-synchronization, experiment repeated three times. (C) Immunoblots of centrosome fractions (stage II, 40–45% sucrose fractions). Grb2 is a cytoplasmic marker; Nek2 a cell cycle marker; γ-tubulin a centrosome marker, Cdk1 and Mps1 are centrosome binding proteins. “total” lanes are extracts prior to gradient purification. (D) Gradient II fractions enriched for centrosomes from HeK293 cells stably expressing GFp-Chk2 KD and γ-irradiated (10 Gy) one hour prior to harvest. GFp-Chk2 KD and γ-tubulin were detected by immunoblotting.
2652 Cell Cycle Volume 9 Issue 13
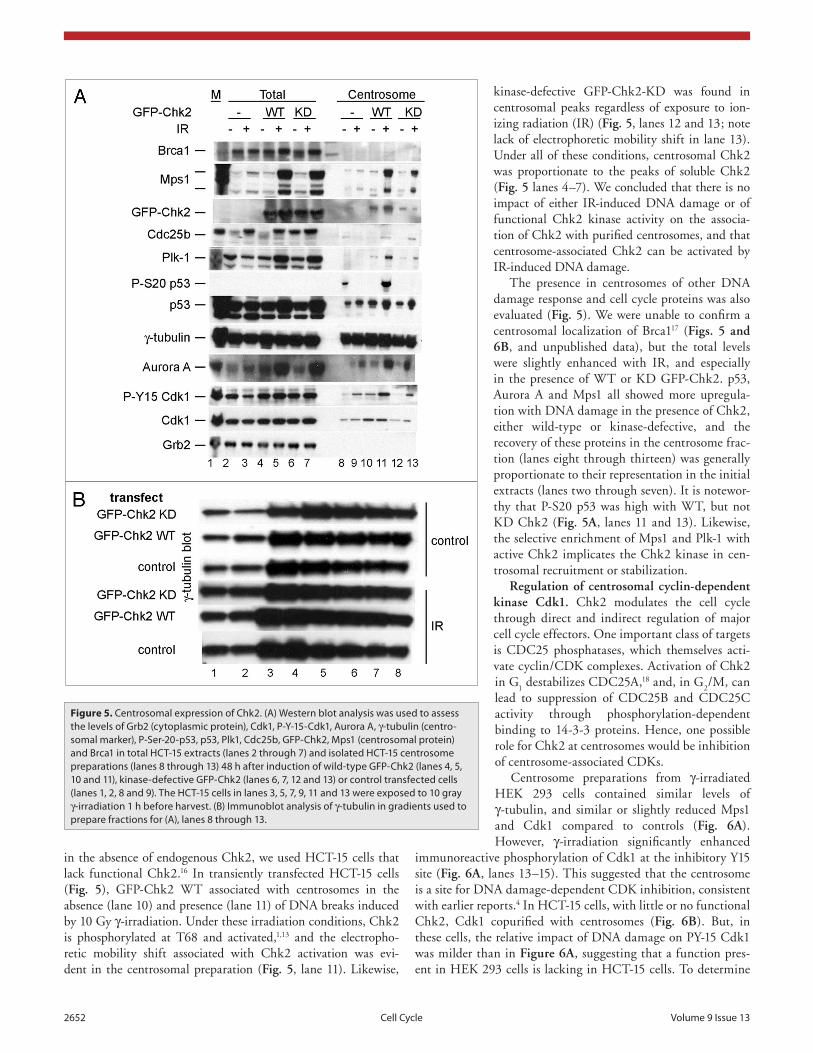
kinase-defective GFP-Chk2-KD was found in centrosomal peaks regardless of exposure to ion-izing radiation (IR) (Fig. 5, lanes 12 and 13; note lack of electrophoretic mobility shift in lane 13). Under all of these conditions, centrosomal Chk2 was proportionate to the peaks of soluble Chk2 (Fig. 5 lanes 4–7). We concluded that there is no impact of either IR-induced DNA damage or of functional Chk2 kinase activity on the associa-tion of Chk2 with purified centrosomes, and that centrosome-associated Chk2 can be activated by IR-induced DNA damage.
The presence in centrosomes of other DNA damage response and cell cycle proteins was also evaluated (Fig. 5). We were unable to confirm a centrosomal localization of Brca117 (Figs. 5 and 6B, and unpublished data), but the total levels were slightly enhanced with IR, and especially in the presence of WT or KD GFP-Chk2. p53, Aurora A and Mps1 all showed more upregula-tion with DNA damage in the presence of Chk2, either wild-type or kinase-defective, and the recovery of these proteins in the centrosome frac-tion (lanes eight through thirteen) was generally proportionate to their representation in the initial extracts (lanes two through seven). It is notewor-thy that P-S20 p53 was high with WT, but not KD Chk2 (Fig. 5A, lanes 11 and 13). Likewise, the selective enrichment of Mps1 and Plk-1 with active Chk2 implicates the Chk2 kinase in cen-trosomal recruitment or stabilization.
Regulation of centrosomal cyclin-dependent kinase Cdk1. Chk2 modulates the cell cycle through direct and indirect regulation of major cell cycle effectors. One important class of targets is CDC25 phosphatases, which themselves acti-vate cyclin/CDK complexes. Activation of Chk2 in G
1 destabilizes CDC25A,18 and, in G
2/M, can
lead to suppression of CDC25B and CDC25C activity through phosphorylation-dependent binding to 14-3-3 proteins. Hence, one possible role for Chk2 at centrosomes would be inhibition of centrosome-associated CDKs.
Centrosome preparations from γ-irradiated HEK 293 cells contained similar levels of γ-tubulin, and similar or slightly reduced Mps1 and Cdk1 compared to controls (Fig. 6A). However, γ-irradiation significantly enhanced
immunoreactive phosphorylation of Cdk1 at the inhibitory Y15 site (Fig. 6A, lanes 13–15). This suggested that the centrosome is a site for DNA damage-dependent CDK inhibition, consistent with earlier reports.4 In HCT-15 cells, with little or no functional Chk2, Cdk1 copurified with centrosomes (Fig. 6B). But, in these cells, the relative impact of DNA damage on PY-15 Cdk1 was milder than in Figure 6A, suggesting that a function pres-ent in HEK 293 cells is lacking in HCT-15 cells. To determine
in the absence of endogenous Chk2, we used HCT-15 cells that lack functional Chk2.16 In transiently transfected HCT-15 cells (Fig. 5), GFP-Chk2 WT associated with centrosomes in the absence (lane 10) and presence (lane 11) of DNA breaks induced by 10 Gy γ-irradiation. Under these irradiation conditions, Chk2 is phosphorylated at T68 and activated,1,13 and the electropho-retic mobility shift associated with Chk2 activation was evi-dent in the centrosomal preparation (Fig. 5, lane 11). Likewise,
Figure 5. Centrosomal expression of Chk2. (A) Western blot analysis was used to assess the levels of Grb2 (cytoplasmic protein), Cdk1, p-Y-15-Cdk1, Aurora A, γ-tubulin (centro-somal marker), p-Ser-20-p53, p53, plk1, Cdc25b, GFp-Chk2, Mps1 (centrosomal protein) and Brca1 in total HCt-15 extracts (lanes 2 through 7) and isolated HCt-15 centrosome preparations (lanes 8 through 13) 48 h after induction of wild-type GFp-Chk2 (lanes 4, 5, 10 and 11), kinase-defective GFp-Chk2 (lanes 6, 7, 12 and 13) or control transfected cells (lanes 1, 2, 8 and 9). the HCt-15 cells in lanes 3, 5, 7, 9, 11 and 13 were exposed to 10 gray γ-irradiation 1 h before harvest. (B) Immunoblot analysis of γ-tubulin in gradients used to prepare fractions for (A), lanes 8 through 13.

www.landesbioscience.com Cell Cycle 2653
lane 3). Interestingly, preliminary data (Fig. 6D, lane 5) show this did not occur in nocadozole-synchronized cells. Overexpression of GFP-Chk2 (Fig. 6D, lanes 6–9) indicates that Chk2 reduces the impact of γ-irradiation on total Cdk1 and PY-15 Cdk1 in asynchronous cells (Fig. 6D, lanes 7 and 9). Therefore, the centro-somal Chk2 may mediate communication between γ-irradiation and the centrosomal Cdk1 or phosphorylation of centrosomal P-Y-15-Cdk1 in asynchronous or nocadazole-synchronized cells. This may be connected with earlier studies of Chk2 in mitotic catastrophe.6,19 Under these conditions, preliminary experiments did not identify changes in centrosomal p53 associated with sta-ble expression of GFP-Chk2 (Fig. 6D).
whether Chk2 is involved in regulation of Cdk1 in HCT-15 cells, we induced GFP-Chk2 expression by gene transfer, and measured the effects on Cdk1 phosphorylation. Although there was only slightly more Cdk1 in centrosomes from GFP-Chk2 expressing cells (Fig. 6C lanes 7–9) than controls (lanes 15–17), this Cdk1 was phosphorylated to substantially higher levels. This difference was not evident in Cdk1 phosphorylation in the total extracts (lanes 3 and 4). This implicated Chk2 in DNA damage-dependent phosphorylation of Cdk1 at the centrosomes.
In asynchronous HEK 293 cells stably expressing GFP-Chk2, γ-irradiation promoted phosphorylation of centrosomal GFP-Chk2 293 cells and Y15-phosphorylated Cdk1 (Fig. 6D,
Figure 6. Impact of Chk2 on responses of centrosomal cell cycle proteins to γ-irradiation. Immunoblots were performed for total extracts and for isolated centrosome preparations from exponentially growing HeK293 (A and D) or from transiently transfected HCt-15 cells expressing GFp-Chk2 (B and C), using antibodies against Grb2 (cytosolic), CenpF, Mps1, Nek2, Cdk1, p-Y-15-Cdk1, p53 (centrosomal proteins), γ-tubulin (centrosomal marker) and Chk2. (A) Centrosome preparations from control (lanes 3–10) or γ-irradiated (lanes 11–17) HeK293 cells. (B) Centrosome preparations from control (lanes 3–10) or γ-irradiated (lanes 11–17) HCt-15 cells. (C) Centrosome preparations from irradiated HCt-15 cells transiently transfected to express GFp-Chk2 (lanes 5 through 11) or non-transfected controls (lanes 12 through 18). Markers are extracts of HeLa (lane 1), HCt-15 cells (lane 2), HCt-15 cells with GFp-Chk2 (lane 3). (D) Centrosome preparations from control HeK293 cells (lanes 2–5) or HeK293 cells stably expressing GFp-Chk2 (lanes 6–9). Cells were irradiated (“IR”, 10 Gy) or synchronized with Nocodazole (“Noc”) as marked.

2654 Cell Cycle Volume 9 Issue 13
late in mitosis or after return to G1 with cytokinesis. This may
reflect a regulated cleavage, changes in masking of Chk2 through binding to other proteins, or differential regulation of a protease that targets Chk2, and it will be of interest to determine whether the cleavage is associated with phosphorylation of Chk2.
Elucidation of centrosome-specific functions for DNA dam-age checkpoint proteins linked to centrosomal sites, including Chk1, ATM, ATR, p53 and BRCA1 is a great challenge, since bulk populations of these proteins are major DNA damage and cell cycle regulators, and only a small proportion of the total are localized to centrosomes. Centrosomes have been proposed as targets for functions of DDR proteins, as sites for sequestration of DDR proteins, and as sites for organization and initiation of DDR functions that are then carried to other subcellular loca-tions. Since centrosomal amplification that can lead to multipo-lar spindle formation is a common finding in cancer cells with DDR mutations, altered centrosomal dysregulation may be a fre-quent mechanism promoting chromosomal instability that can lead to genomic rearrangement.
Our results shed light on the importance of Chk2 for associa-tion of other proteins with centrosomes, either as a consequence of scaffolding, or through action of the Chk2 kinase. One trend was that expression of Chk2 or GFP-Chk2 enhanced co-purification of the spindle checkpoint protein kinase Mps1. Figure 5 displays centrosome buffer extracts (lanes two through seven) and centro-somal preparations (lanes eight through thirteen) from HCT-15 cells expressing GFP-Chk2 having or lacking kinase activity. The presence of GFP-Chk2 correlated with an enhancement of the total extractable Mps1, regardless of Chk2 kinase activity, and the increase was markedly enhanced by DNA damage through γ-irradiation. Expression of GFP-Chk2 also enhanced recovery of the mitotic regulators Plk1 and Aurora A, especially with DNA damage (Fig. 5). There was only a modest impact of the kinase mutation in this experiment indicating either that kinase activity was unimportant, or that there may be trans-complementation between FHA-defective molecules expressed endogenously in HCT-15, and kinase-defective GFP-Chk2 brought in by gene transfer. The correlation with p53 levels is consistent with GFP-Chk2 enhancing a damage-dependent process, that may have indirectly affected MPS1 levels, or alternatively enriched for cell cycle stages with higher levels of MPS1, Plk1 and Aurora A. In this experiment (also, Fig. 6C), centrosomal associated compo-nents of MPS1, Plk1 and Aurora A were all increased with DNA damage, and were all more evident in the cells with WT or KD GFP-Chk2. In this experiment, also, phosphorylation of p53 at S20 was specifically enhanced at centrosomes with wild-type Chk2. Since S20 is a known target for Chk1 and Chk2, this is evidence for the importance of Chk2 in on-site p53 phosphoryla-tion at the centrosome. This suggests a possible role for Chk2 in stabilizing Mps1 throughout the cell, and at the centrosomes, which could most easily occur through direct protein interaction. There is a possibility, though, that partial cell cycle synchroni-zation from expression of Chk2 contributes to this result, since MPS1 levels are regulated within the cell cycle (Fig. 4B).
Previous work suggested that Chk1-dependent inhibition of Cdc25b in the G
2/M transition takes place in centrosomes and
Discussion
There is increasing evidence that centrosomes are important for protective regulation of the cell cycle. A number of pro-teins involved in DNA damage checkpoint responses and cell cycle regulation have now been localized to centrosomes, along with cell cycle regulatory kinases including Cdk1, Mps1, polo-like kinases and Aurora kinases.2,19-21 We show here that Chk2 does co-purify with centrosomal preparations. This association does not require kinase activity of Chk2, and the centrosome-associated Chk2 is not phosphorylated at T68 upon completion of the purification. A portion of centrosome-associated Chk2 is truncated at the amino terminus. Although the major functions of Chk2 at centrosomes remain elusive, our data support the pos-sibility that Chk2 modulates recruitment and phosphorylation of other centrosomal proteins.
Our main goal for the present study was to use a method inde-pendent from IF to validate the presence of Chk2 at centrosomes, and to investigate the major dependencies for centrosomal local-ization. The most stringent purifications used employed con-secutive centrifugation steps to avoid combining high molecular weight aggregates as in Figure 1B, fraction 12, with centrosomal proteins. Chk2 co-purified with centrosomes through multiple sedimentation steps in several independent purifications, which confirmed the apparent association of Chk2 with centrosomes originally found by immunofluorescence. We had originally found Chk2 in centrosomes using a PT-68 antibody1 and other labs have suggested that Chk2 associates with centrosomes only after DNA damage.4 The forms of Chk2 that co-purified with centrosomes were not apparently phosphorylated at T68, and their electrophoretic mobility matched that of the basal form, not that of the form induced by DNA damage. Finally, the FHA domain is required for activation of Chk2 by DNA damage, but this domain is lacking in the truncated forms associated with centrosomes. In our hands, Chk2 readily co-purified with cen-trosomes from non-treated cells and cells with DNA damage induced by ionizing radiation. Hence, neither phosphorylation, catalytic activity, nor catalytic activation through DNA damage are required for association of Chk2 with centrosomes.
Targeted proteolysis is an important mechanism of cell cycle regulation. Chk2 associated with centrosomes was often trun-cated. This truncation would eliminate a portion of the FHA domain, which is normally employed in recruitment of Chk2 to phospho-epitopes on Chk2 interacting proteins that are cre-ated by PIKKs, and in recruitment of Chk2 into homodimers in which inactive Chk2 molecules are activated in a cross-phospho-rylation reaction. Hence it is unlikely that these truncated forms would be subject to normal modes of DNA damage-dependent activation. However, an open conformation promoted by this truncation may permit other forms of interaction with other pro-teins, or even greater baseline kinase activity. We cannot rule out the possibility that the truncation is simply an artefact of the extended duration of these sedimentation conditions. It is note-worthy, though, that the relative recovery of cleaved Chk2 in cell extracts (Fig. 4B) and centrosomal preparations (Fig. 4C) varied within the cell cycle, with the cleavage activity most prevalent

www.landesbioscience.com Cell Cycle 2655
2 mM thymidine (Sigma Chemical Co.,) for 18 h., washed with phosphate-buffered saline, and grown in fresh medium without thymidine for 8 h. Thymidine was added again for 18 h. to block cells at the G
1/S boundary. The medium was replaced with fresh
thymidine-free medium and cells were harvested at the indicated times thereafter.
Plasmids. Xba1/EcoR1 fragments containing WT CHK2 and KD CHK2 (CHK2 D368A) were subcloned into pEBFP-C1 con-taining an amino-terminal GFP tag then subcloned into a GFP-Chk1-PACT AKAP plasmid (accession number BC004202.2),4 replacing CHK1 with CHK2.
Antibodies. Affinity-purified polyclonal anti-PT68 Chk2 was produced as described.1 Antibodies that recognize Aurora A/AIK (3092), Cdc25B (9525), Cdc2 (9112), Phospho-Chk2 T68 (80F5), phospho-p53 S15 (16G8) (9286), Phospho-p53 S20 (9287), PLK1 (4535) were obtained from Cell Signaling Technology; BRCA1 (BL319), CHK2 (BL2882), were from Bethyl laboratories; antibodies against Cdc25B (Ab-1), Cdk1 PY15 (C219440) were from Calbiochem; Cdk1/Cdc2 (C610038), GRB2 (C610112), were from BD Transduction lab-oratories; CHK2, clone 7 (C05-649), Mps1 NT Clone 3-472-1 (C05-682), were from Upstate Biotechnologies; Chk2 (A-12): sc-5278, GFP (FL): (sc-8334), p53 (FL-393); sc-6243, p53 Ab-3 (Clone BP53-12) were from Santa Cruz Biotechnology; NEK2 (C629402), were from BioLegend; p53 Ab3 (Clone BP53-12), and antibodies against γ-Tubulin (T6557) were from Sigma.
Immunoblotting. Cell lysates were harvested in TSD buf-fer (20 mM Tris [pH 7.5], 100 mM NaCl, 0.1% sodium deoxy-cholate, 0.1% Triton x-100), and protease inhibitor cocktail [Roche Applied Science] or as described below.
Isolation and analysis of human centrosomes. Between 1 x 107 and 2.5 x 107 cells total were harvested from ten 150 mm tissue culture plates or twenty-five 100 mm culture dishes (HeLa cell preparations). Centrosomes were isolated from cells as described, with some modifications,27-29 but Nocodazole was omitted from HeLa cell preparations. Extracts from HEK293 cells or HCT-15 cells arrested in mitosis (nocodazole treatment for 24 h) were prepared as described below. Cells were grown to 80% confluence. Before harvest, Nocodazole (330 ng/ml) and Cytochalasin (1 ug/ml; Sigma Chemical Co.,) was added in 10 ml and the plates incubated at 37°C for 1 h. Cells were transferred to centrifuge tubes, collected by centrifugation at 2,000 xg, washed twice with TBS (pH 7.4) and then 8% sucrose in 0.1X TBS. Cells were shaken slowly in Lysis buffer (1 mM dithiothreitol [DTT], 1 mM phenylmethanesulphonylfluoride, 10 mM β-glycerophosphate, 10 mM sodium fluoride, and 1:50 protease inhibitor mix [Roche complete EDTA-free tablets]) and triturated with a 1 ml pipette until chromatin aggregates were evident, with yellow coloring and cords. The extracts were cleared by centrifugation for 10 min, then through 70 µm Nylon filters (Falcon 08-771-1). Preparations were incubated for thirty minutes at RT in 2 units/ml Deoxyribonuclease (DNase; Sigma Chemical Co.,) and 10 mM HEPES pH 7.2. Lysis buf-fer was added to a final volume of nine ml and samples were layered on 1.1 ml of 42% sucrose in gradient buffer (PIPES pH 7.2 10 mM, Triton X-100 0.1%, β-mercaptoethanol 0.1%,
results in inhibition of Cdk1 through stabilization of phosphory-lation at PY-15.4 Also, Chk1 facilitates the phosphorylation of P-Y-15-Cdk1 by AuroraA.22,23 Additionally, the phosphorylation of Cdc25b by Aurora A in centrosomes is influenced by DNA damage,23 while its inhibition by centrosomal Chk1 is not.24
Our finding that DNA damage enhances PY-15 phosphory-lation of centrosomal Cdk1, presumably reducing Cdk1 activ-ity, is consistent with several earlier studies.19,25 The effect of γ-irradiation on the centrosomal accumulation of P-Y-15-Cdk1 was not influenced in 293 cells by expression of GFP-Chk2 (KD or WT) (Fig. 6A, lanes 2, 4 and 6). The lack of synergistic effect between centrosomal fractions in 293 cells that were treated by Chk2 enrichment, and DNA damage diverges from the results in HCT-15 cells (Fig. 5, lanes 9, 11 and 13), but this may not be surprising since 293 cells express functional Chk2, and the Chk2 forms expressed by gene transfer only added incrementally to the total pool of centrosomal Chk2.
Restoration of Chk2 expression in HCT-15 cells generally enhanced damage-dependent PY15-phosphorylation of CDK1 at centrosomes (Figs. 5B, lanes 10 versus 11; 6C peak lanes seven through ten versus fifteen through eighteen). Hence, Chk2 activity does promote DNA damage-dependent inhibition of CDK1 at centrosomes. The CDC25 phosphatases are major CDK regulators that promote CDK activation through removal of the inhibitory phosphorylations at the ATP binding pocket, including PY15. Since they are negatively regulated by Chk1 and Chk2, they are logical candidate regulators for changes in CDK1 phosphorylation. Our analysis of Chk2 dependencies of CDC25B in centrosomes (Fig. 5 and unpublished data) yielded some inconsistencies, since CDK1 PY15 assorted independently from Cdc25B abundance and mobility shift in centrosomes. This issue will warrant further analysis to quantify activating and inhibitory phosphorylations. Overall, the impact of expression of Chk2 on centrosomal P-Y-15-Cdk1 following DNA damage may be more complex than the current working model in which Chk2 acts mainly as an amplifier of the signal induced by DNA dam-age.4,26 Our data confirm the localization of Chk2 to centrosomes and demonstrate that this localization does not require Chk2 kinase activity or DNA damage. The centrosomal localization of a short, previously unknown, 52 kDa Chk2 form, a product of proteolytic cleavage, raise interesting questions about its function and regulation. Ectopic expression of Chk2 in cells with little or no functional endogenous Chk2 affects centrosomal levels of Plk1 and Mps1 as well as the phosphorylation status of cen-trosomal Cdk1 and p53, indicating broad functional significance for Chk2 at centrosomes.
Materials and Methods
Cell culture. HeLa cells, HCT-15 cells and HEK 293 cells were cultured in Dulbecco’s modified Eagle’s medium contain-ing 10% fetal bovine serum in a CO
2 (5%) incubator at 37°C.
Transfected derivatives of HCT-15 cells and HEK293 cells were generated using Lipofectamine 2000 (Invitrogen Corp.,) accord-ing to the manufacturer’s instructions. For cell cycle synchro-nization experiments, HeLa cells were grown in the presence of

2656 Cell Cycle Volume 9 Issue 13
20. Tsvetkov LM, Tsekova RT, Xu X, Stern DF. The Plk1 Polo box domain mediates a cell cycle and DNA dam-age regulated interaction with Chk2. Cell Cycle 2005; 4:609-17.
21. Mohammad DH, Yaffe MB. 14-3-3 proteins, FHA domains and BRCT domains in the DNA damage response. DNA Repair (Amst) 2009; 8:1009-17.
22. Dutertre S, Cazales M, Quaranta M, Froment C, Trabut V, Dozier C, et al. Phosphorylation of CDC25B by Aurora-A at the centrosome contributes to the G
2-M
transition. J Cell Sci 2004; 117:2523-31.23. Cazales M, Schmitt E, Montembault E, Dozier C,
Prigent C, Ducommun B. CDC25B phosphorylation by Aurora-A occurs at the G
2/M transition and is inhib-
ited by DNA damage. Cell Cycle 2005; 4:1233-8.24. Schmitt E, Boutros R, Froment C, Monsarrat B,
Ducommun B, Dozier C. CHK1 phosphorylates CDC25B during the cell cycle in the absence of DNA damage. J Cell Sci 2006; 119:4269-75.
25. Loffler H, Rebacz B, Ho AD, Lukas J, Bartek J, Kramer A. Chk1-dependent regulation of Cdc25B functions to coordinate mitotic events. Cell Cycle 2006; 5:2543-7.
26. Loffler H, Lukas J, Bartek J, Kramer A. Structure meets function—centrosomes, genome maintenance and the DNA damage response. Exp Cell Res 2006; 312:2633-40.
27. Bornens M, Moudjou M. In: Celis J, ed. Cell Biology: A laboratory handbook. San Diego: Academic Press 1994; 595-604.
28. do Carmo Avides M, Glover DM. Abnormal spindle protein, Asp, and the integrity of mitotic centro-somal microtubule organizing centers. Science 1999; 283:1733-5.
29. Fabunmi RP, Wigley WC, Thomas PJ, DeMartino GN. Activity and regulation of the centrosome-associated proteasome. J Biol Chem 2000; 275:409-13.
10. Lukas C, Falck J, Bartkova J, Bartek J, Lukas J. Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat Cell Biol 2003; 5:255-60.
11. Castedo M, Perfettini JL, Roumier T, Yakushijin K, Horne D, Medema R, et al. The cell cycle checkpoint kinase Chk2 is a negative regulator of mitotic catastro-phe. Oncogene 2004; 23:4353-61.
12. Andersen JS, Wilkinson CJ, Mayor T, Mortensen P, Nigg EA, Mann M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature 2003; 426:570-4.
13. Xu X, Tsvetkov LM, Stern DF. Chk2 activation and phosphorylation-dependent oligomerization. Mol Cell Biol 2002; 22:4419-32.
14. Staalesen V, Falck J, Geisler S, Bartkova J, Borresen-Dale AL, Lukas J, et al. Alternative splicing and mutation status of CHEK2 in stage III breast cancer. Oncogene 2004; 23:8535-44.
15. Hames RS, Wattam SL, Yamano H, Bacchieri R, Fry AM. APC/C-mediated destruction of the centrosomal kinase Nek2A occurs in early mitosis and depends upon a cyclin A-type D-box. EMBO J 2001; 20:7117-27.
16. Falck J, Lukas C, Protopopova M, Lukas J, Selivanova G, Bartek J. Functional impact of concomitant versus alternative defects in the Chk2-p53 tumour suppressor pathway. Oncogene 2001; 20:5503-10.
17. Hsu LC, White RL. BRCA1 is associated with the centrosome during mitosis. Proc Natl Acad Sci USA 1998; 95:12983-8.
18. Falck J, Mailand N, Syljuasen RG, Bartek J, Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 2001; 410:842-7.
19. Doxsey S, McCollum D, Theurkauf W. Centrosomes in cellular regulation. Annu Rev Cell Dev Biol 2005; 21:411-34.
recovered and stored at -80°C. Fractions containing centro-somes were identified by immunoblotting for γ-tubulin. For gradient III, the peak fraction at 40–45% sucrose were dialyzed and re-run on step gradients.
Acknowledgements
This work was supported by United States Army Medical Research and Materiel Command grant DAMD17-03-1-0331, and US PHS R01CA82257 to D.F.S.
1 mM DTT and 200 µg of Soybean Trypsin Inhibitor (STI) as carrier). Following centrifugation at 2,000 xg (7,500 rpm) for 1 h at 4°C, the 1.1 ml cushion was dialyzed (nominal 10 kDa cutoff, Pierce 69570) against gradient buffer overnight at 4°C, and was layered on a discontinuous sucrose gradient with 750 µl steps (bottom to top) of 70%, 65%, 60%, 55%, 50%, 45%, 42%, 40%, 35%, 30%, 25% and 20% sucrose solution in gradient buffer, containing 1 mM DTT and STI 200 µg ml. Gradients were centrifuged in a SW41 rotor at 40,000 xg for 12 h at 4°C. Twenty fractions of 0.5 ml each were manually
References1. Tsvetkov L, Xu X, Li J, Stern DF. Polo-like kinase 1 and
Chk2 interact and co-localize to centrosomes and the midbody. J Biol Chem 2003; 278:8468-75.
2. Tsvetkov L, Stern DF. Phosphorylation of Plk1 at S137 and T210 is inhibited in response to DNA damage. Cell Cycle 2005; 4:166-71.
3. Bahassi el M, Myer DL, McKenney RJ, Hennigan RF, Stambrook PJ. Priming phosphorylation of Chk2 by polo-like kinase 3 (Plk3) mediates its full activation by ATM and a downstream checkpoint in response to DNA damage. Mutat Res 2006; 596:166-76.
4. Kramer A, Mailand N, Lukas C, Syljuasen RG, Wilkinson CJ, Nigg EA, et al. Centrosome-associated Chk1 prevents premature activation of cyclin-B-Cdk1 kinase. Nat Cell Biol 2004; 6:884-91.
5. Zhang S, Hemmerich P, Grosse F. Centrosomal local-ization of DNA damage checkpoint proteins. J Cell Biochem 2007; 101:451-65.
6. Takada S, Kelkar A, Theurkauf WE. Drosophila check-point kinase 2 couples centrosome function and spindle assembly to genomic integrity. Cell 2003; 113:87-99.
7. van Vugt MA, Gardino AK, Linding R, Ostheimer GJ, Reinhardt HC, Ong SE, et al. A mitotic phosphoryla-tion feedback network connects Cdk1, Plk1, 53BP1 and Chk2 to inactivate the G(2)/M DNA damage checkpoint. PLoS Biol 2010; 8:1000287.
8. Hong Y, Cervantes RB, Tichy E, Tischfield JA, Stambrook PJ. Protecting genomic integrity in somatic cells and embryonic stem cells. Mutat Res 2007; 614:48-55.
9. Hong Y, Stambrook PJ. Restoration of an absent G1
arrest and protection from apoptosis in embryonic stem cells after ionizing radiation. Proc Natl Acad Sci USA 2004; 101:14443-8.




















