
Catalytic selective reduction of NO with ethylene over a series of copper catalysts on amorphous...
11
Applied Catalysis B: Environmental 28 (2000) 175–185 Catalytic selective reduction of NO with ethylene over a series of copper catalysts on amorphous silicas Paolo Carniti a , Antonella Gervasini a,* , Vittorio H. Modica a , Nicoletta Ravasio b a Dipartimento di Chimica Fisica ed Elettrochimica, Università degli Studi di Milano, via Golgi 19, I-20133 Milan, Italy b CNR Centro CSSCMTBSO, c/o Dipartimento di Chimica Inorganica, Metallorganica ed Analitica, Università degli Studi di Milano, via Venezian 21, I-20133 Milan, Italy Received 13 February 2000; received in revised form 23 April 2000; accepted 11 May 2000 Abstract Catalytic selective reduction of NO to N 2 was studied comparing a series of Cu-based catalysts (ca. 8 wt.%) supported over amorphous pure and modified silicas: SiO 2 , SiO 2 -Al 2 O 3 , SiO 2 -TiO 2 , SiO 2 -ZrO 2 . The catalysts were prepared by the chemisorption-hydrolysis method which ensured the formation of a unique copper phase well dispersed over all supports, as confirmed by scanning electron micrographs (SEMs). Temperature-programmed reduction (TPR) analyses confirmed the presence of dispersed copper species which underwent complete reduction at a temperature of about 220 ◦ C, independently of the support. It was found that the support affects the extent of NO reduction as well as the selectivity to N 2 formation. Maximum N 2 yield was found in the range 275–300 ◦ C. The catalyst prepared over SiO 2 -Al 2 O 3 was the most active and selective with respect to the other silicas. Competitiveness factors (c.f.’s) as high as 13–20% in the temperature range 200–250 ◦ C could be calculated. For all catalysts, the temperature of the N 2 peak maximum did not correspond to that of the maximum C 2 H 4 oxidation to CO 2 , suggesting the presence of two different sites for the oxidation and the reduction activity. On the catalyst prepared on SiO 2 -Al 2 O 3 , a kinetic interpretation of catalytic data collected at different contact times and temperatures permitted evaluating the ratio between kinetic coefficients as well as the difference between activation energies of NO reduction by C 2 H 4 and C 2 H 4 oxidation by O 2 . © 2000 Elsevier Science B.V. All rights reserved. Keywords: Catalyst preparation; Cu/silica catalysts; Selective catalytic reduction; Nitrogen oxide; Hydrocarbon; Kinetics of reaction 1. Introduction Copper-containing catalysts are being extensively studied since the first reports of Iwamoto et al. [1–3] that proposed Cu/ZSM-5 as an active catalyst for se- lective lean NO x reduction to N 2 by hydrocarbons. From that moment, in relation to the different pro- cesses for removing NO x in exhaust gases, the selec- tive catalytic reduction of NO x by hydrocarbons in the * Corresponding author. Tel.: +39-02-26603293; fax: +39-02-70638129. E-mail address: [email protected] (A. Gervasini). presence of excess oxygen (denoted as NO-HC-O 2 ) has attracted much interest [4–6]. Poor thermal and hy- drothermal stability of copper-based zeolite catalysts [7–9] led the research towards the study of amorphous copper catalysts [10,11]. The hydrothermal stability of supports such as alumina, silica and zirconia is higher than that of zeolites. Therefore, such oxides would be very desirable catalyst supports. A large number of amorphous copper catalytic systems have been inves- tigated, paying particular attention to the preparation method [12], type and purity of oxide support [13,14], and copper loading [12,15,16]. Most of the results ap- pearing in the literature reporting screening tests show 0926-3373/00/$ – see front matter © 2000 Elsevier Science B.V. All rights reserved. PII:S0926-3373(00)00172-7
Transcript of Catalytic selective reduction of NO with ethylene over a series of copper catalysts on amorphous...

Applied Catalysis B: Environmental 28 (2000) 175–185
Catalytic selective reduction of NO with ethylene overa series of copper catalysts on amorphous silicas
Paolo Carnitia, Antonella Gervasinia,∗, Vittorio H. Modicaa, Nicoletta Ravasioba Dipartimento di Chimica Fisica ed Elettrochimica, Università degli Studi di Milano, via Golgi 19, I-20133 Milan, Italy
b CNR Centro CSSCMTBSO, c/o Dipartimento di Chimica Inorganica, Metallorganica ed Analitica,Università degli Studi di Milano, via Venezian 21, I-20133 Milan, Italy
Received 13 February 2000; received in revised form 23 April 2000; accepted 11 May 2000
Abstract
Catalytic selective reduction of NO to N2 was studied comparing a series of Cu-based catalysts (ca. 8 wt.%) supportedover amorphous pure and modified silicas: SiO2, SiO2-Al2O3, SiO2-TiO2, SiO2-ZrO2. The catalysts were prepared by thechemisorption-hydrolysis method which ensured the formation of a unique copper phase well dispersed over all supports,as confirmed by scanning electron micrographs (SEMs). Temperature-programmed reduction (TPR) analyses confirmed thepresence of dispersed copper species which underwent complete reduction at a temperature of about 220◦C, independentlyof the support. It was found that the support affects the extent of NO reduction as well as the selectivity to N2 formation.Maximum N2 yield was found in the range 275–300◦C. The catalyst prepared over SiO2-Al2O3 was the most active andselective with respect to the other silicas. Competitiveness factors (c.f.’s) as high as 13–20% in the temperature range200–250◦C could be calculated. For all catalysts, the temperature of the N2 peak maximum did not correspond to that of themaximum C2H4 oxidation to CO2, suggesting the presence of two different sites for the oxidation and the reduction activity.On the catalyst prepared on SiO2-Al2O3, a kinetic interpretation of catalytic data collected at different contact times andtemperatures permitted evaluating the ratio between kinetic coefficients as well as the difference between activation energiesof NO reduction by C2H4 and C2H4 oxidation by O2. © 2000 Elsevier Science B.V. All rights reserved.
Keywords:Catalyst preparation; Cu/silica catalysts; Selective catalytic reduction; Nitrogen oxide; Hydrocarbon; Kinetics of reaction
1. Introduction
Copper-containing catalysts are being extensivelystudied since the first reports of Iwamoto et al. [1–3]that proposed Cu/ZSM-5 as an active catalyst for se-lective lean NOx reduction to N2 by hydrocarbons.From that moment, in relation to the different pro-cesses for removing NOx in exhaust gases, the selec-tive catalytic reduction of NOx by hydrocarbons in the
∗ Corresponding author. Tel.:+39-02-26603293;fax: +39-02-70638129.E-mail address:[email protected] (A. Gervasini).
presence of excess oxygen (denoted as NO-HC-O2)has attracted much interest [4–6]. Poor thermal and hy-drothermal stability of copper-based zeolite catalysts[7–9] led the research towards the study of amorphouscopper catalysts [10,11]. The hydrothermal stability ofsupports such as alumina, silica and zirconia is higherthan that of zeolites. Therefore, such oxides would bevery desirable catalyst supports. A large number ofamorphous copper catalytic systems have been inves-tigated, paying particular attention to the preparationmethod [12], type and purity of oxide support [13,14],and copper loading [12,15,16]. Most of the results ap-pearing in the literature reporting screening tests show
0926-3373/00/$ – see front matter © 2000 Elsevier Science B.V. All rights reserved.PII: S0926-3373(00)00172-7

176 P. Carniti et al. / Applied Catalysis B: Environmental 28 (2000) 175–185
that amorphous copper-based catalysts are inferior toZSM-5-based ones [11,17,18]. However, zeolitic sup-ports do not play an essential role by themselves, butonly favour copper dispersion which has been shownto be the key factor for catalytic activity [18–20].Therefore, the preparation method is a critical factorfor the activity of copper-based oxides for NO catalyticreduction by hydrocarbons. Methods that favour theformation of isolated CuO-like species (i.e. adsorptionof Cu2+ ions from basic solution), which are believedto be the active sites for NO reduction [12,15,21,22],lead to a higher activity than those recognized as moreconventional (i.e. impregnation) that, on the contrary,lead to large CuO-like aggregates. The latter ones aremore capable of oxidizing hydrocarbons, and there-fore, the overall NO-HC-O2 process follows the di-rection towards combustion reaction (HC by O2) [15]rather than that towards NO reduction (HC by NO).Because O2 is present in a much higher concentrationthan NO, a selective catalyst must be able to effectivelydirect the reaction towards the desired reduction ratherthan towards the undesired combustion [5,23–26].
The presence of a very well dispersed copperphase has been evidenced on the surface of cat-alysts prepared by the chemisorption-hydrolysismethod, thoroughly characterized in previous pa-pers through transmission electron microscopy(TEM), temperature-programmed reduction (TPR),DR-UV–VIS-NIR and FT-IR of adsorbed CO[27,28]. Specific Cu(0) surface areas as high as130 m2 g−1
Cu were measured by N2O titration ex-periments on Cu/SiO2 and Cu/SiO2-TiO2 samples[28]. Moreover, Cu/TiO2 catalysts prepared by thechemisorption-hydrolysis method exhibited a Cu(0)surface area much higher than those prepared bythe conventional incipient wetness impregnationmethod, also for an 8 wt.% copper loading (55 ver-sus 9 m2 g−1
Cu). Therefore, these catalysts appear to begood candidates for the reaction of selective catalyticreduction of nitrogen oxides.
The present study aimed at determining the perfor-mances of copper catalysts prepared on pure and mod-ified silicas, i.e. SiO2, SiO2-Al2O3, SiO2-TiO2, andSiO2-ZrO2, by the chemisorption-hydrolysis methodtowards the selective reduction of NO with ethylene(denoted as NO-C2H4-O2). The copper weight load-ing was chosen to be as high as 8 wt.% in any case. It isknown that, at low copper loading, isolated or weakly
interacting Cu2+ species can be easily stabilized overoxide supports, but CuO-like aggregates are formedabove a certain threshold loading [16,29]. However,the preparation method utilized ensured high disper-sion of the copper phase, avoiding formation of largeCuO aggregates and spinel compounds with the oxidesupport independently of the support itself [28].
A kinetic model was applied to the catalytic data,allowing the determination of rate coefficient ratios aswell as activation energy differences of NO reductionby C2H4 and C2H4 oxidation by O2, related to theactivity of the catalytic sites.
2. Experimental
2.1. Catalyst preparation
The catalysts Cu/SiO2, Cu/SiO2-Al2O3, Cu/SiO2-TiO2, and Cu/SiO2-ZrO2, hereafter referred to asCu/Si, Cu/SiAl, Cu/SiTi, and Cu/SiZr, respectively(Table 1), were prepared by what was called in pre-vious papers [27,28] the chemisorption-hydrolysismethod. The supports, all from Grace Davison, wereadded to a [Cu(NH3)4]2+ solution prepared by addingNH4OH to a Cu(NO3)2·3H2O solution until pH 9.After 20 min under stirring, the slurry, held in an icebath at 0◦C, was slowly diluted in order to allow hy-drolysis of the copper complex and deposition of thefinely dispersed product to occur. The solid was sepa-rated by filtration, washed with water, dried overnightat 110◦C, and calcined in air at 350◦C for 4 h. In allcases, the Cu content was ca. 8 wt.% (Table 1).
2.2. Catalyst characterization
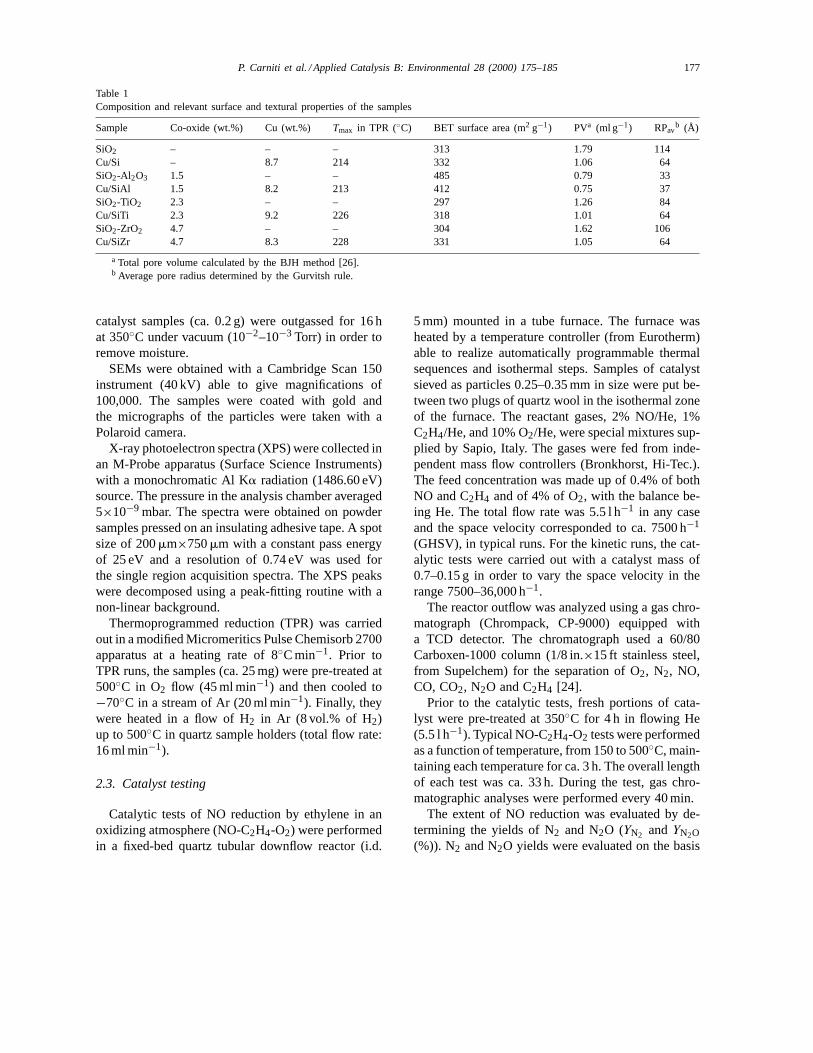
The copper content of all samples was establishedby atomic absorption spectrometry (AAS) analysis af-ter dissolution in HNO3. Table 1 shows the results ex-pressed as wt.%.
BET surface area and pore volume were obtainedfrom N2 adsorption isotherms measured at liquidnitrogen temperature (−196◦C) using a Sorptomatic1900 instrument (CE Instruments) working with thestatic volumetric technique. The analysis was con-trolled by microcomputer processing usingmiles-200and mileadp software for computations. Nitrogenwas of 99.9995% purity. Prior to the analysis, the
P. Carniti et al. / Applied Catalysis B: Environmental 28 (2000) 175–185 177
Table 1Composition and relevant surface and textural properties of the samples
Sample Co-oxide (wt.%) Cu (wt.%) Tmax in TPR (◦C) BET surface area (m2 g−1) PVa (ml g−1) RPavb (Å)
SiO2 – – – 313 1.79 114Cu/Si – 8.7 214 332 1.06 64SiO2-Al2O3 1.5 – – 485 0.79 33Cu/SiAl 1.5 8.2 213 412 0.75 37SiO2-TiO2 2.3 – – 297 1.26 84Cu/SiTi 2.3 9.2 226 318 1.01 64SiO2-ZrO2 4.7 – – 304 1.62 106Cu/SiZr 4.7 8.3 228 331 1.05 64
a Total pore volume calculated by the BJH method [26].b Average pore radius determined by the Gurvitsh rule.
catalyst samples (ca. 0.2 g) were outgassed for 16 hat 350◦C under vacuum (10−2–10−3 Torr) in order toremove moisture.
SEMs were obtained with a Cambridge Scan 150instrument (40 kV) able to give magnifications of100,000. The samples were coated with gold andthe micrographs of the particles were taken with aPolaroid camera.
X-ray photoelectron spectra (XPS) were collected inan M-Probe apparatus (Surface Science Instruments)with a monochromatic Al Ka radiation (1486.60 eV)source. The pressure in the analysis chamber averaged5×10−9 mbar. The spectra were obtained on powdersamples pressed on an insulating adhesive tape. A spotsize of 200mm×750mm with a constant pass energyof 25 eV and a resolution of 0.74 eV was used forthe single region acquisition spectra. The XPS peakswere decomposed using a peak-fitting routine with anon-linear background.
Thermoprogrammed reduction (TPR) was carriedout in a modified Micromeritics Pulse Chemisorb 2700apparatus at a heating rate of 8◦C min−1. Prior toTPR runs, the samples (ca. 25 mg) were pre-treated at500◦C in O2 flow (45 ml min−1) and then cooled to−70◦C in a stream of Ar (20 ml min−1). Finally, theywere heated in a flow of H2 in Ar (8 vol.% of H2)up to 500◦C in quartz sample holders (total flow rate:16 ml min−1).
2.3. Catalyst testing
Catalytic tests of NO reduction by ethylene in anoxidizing atmosphere (NO-C2H4-O2) were performedin a fixed-bed quartz tubular downflow reactor (i.d.
5 mm) mounted in a tube furnace. The furnace washeated by a temperature controller (from Eurotherm)able to realize automatically programmable thermalsequences and isothermal steps. Samples of catalystsieved as particles 0.25–0.35 mm in size were put be-tween two plugs of quartz wool in the isothermal zoneof the furnace. The reactant gases, 2% NO/He, 1%C2H4/He, and 10% O2/He, were special mixtures sup-plied by Sapio, Italy. The gases were fed from inde-pendent mass flow controllers (Bronkhorst, Hi-Tec.).The feed concentration was made up of 0.4% of bothNO and C2H4 and of 4% of O2, with the balance be-ing He. The total flow rate was 5.5 l h−1 in any caseand the space velocity corresponded to ca. 7500 h−1
(GHSV), in typical runs. For the kinetic runs, the cat-alytic tests were carried out with a catalyst mass of0.7–0.15 g in order to vary the space velocity in therange 7500–36,000 h−1.
The reactor outflow was analyzed using a gas chro-matograph (Chrompack, CP-9000) equipped witha TCD detector. The chromatograph used a 60/80Carboxen-1000 column (1/8 in.×15 ft stainless steel,from Supelchem) for the separation of O2, N2, NO,CO, CO2, N2O and C2H4 [24].
Prior to the catalytic tests, fresh portions of cata-lyst were pre-treated at 350◦C for 4 h in flowing He(5.5 l h−1). Typical NO-C2H4-O2 tests were performedas a function of temperature, from 150 to 500◦C, main-taining each temperature for ca. 3 h. The overall lengthof each test was ca. 33 h. During the test, gas chro-matographic analyses were performed every 40 min.
The extent of NO reduction was evaluated by de-termining the yields of N2 and N2O (YN2 and YN2O(%)). N2 and N2O yields were evaluated on the basis

178 P. Carniti et al. / Applied Catalysis B: Environmental 28 (2000) 175–185
of the NO fed. As concerns C2H4, both its conversion(XC2H4 (%)) and the yields of CO2 and CO (YCO2 andYCO (%)) were determined. CO2 and CO yields wereevaluated on the basis of the C2H4 fed. The selectiv-ity to N2 was evaluated by the competitiveness factor(c.f. (%)), calculating the ratio between the C2H4 con-sumed to reduce NO to N2 and the total amount ofC2H4 consumed [11,26].
3. Results and discussion
3.1. Catalyst characterization
Despite the high copper loading (ca. 8 wt.%), SEMsdid not show any significant difference between themorphological features of the catalysts and those of thebare supports. The micrographs of SiO2-TiO2 and therelevant catalyst are shown in Fig. 1, as an example.
Previous findings on Cu/Si and Cu/SiTi samples byTEM analysis [28] showed that the dimensions of cop-per particles after calcination are distributed over the2.0–5.5 nm range with a mean diameterdm=3.0 nm.
TPR profiles of all catalysts examined showed aunique narrow reduction peak centred at ca. 220◦C(Fig. 2 and Table 1). The lack of reduction peaks attemperatures lower than 200◦C indicated the absenceof oxocations (Cu–O–Cu)2+, while the lack of peaksat temperatures higher than 250◦C indicated the ab-sence of copper crystalline phases [12,16]. Accord-ing to what has already been observed on Cu/TiO2and Cu/ZrO2 samples [27,30], the unique peak ob-served can be attributed to a single reduction step:Cu2+ → Cu0, that is to the presence of well dispersedCu2+ species. Quantitative evaluations of hydrogenconsumption are in full agreement with this hypoth-esis. This is true independently of the support used,as already shown for some of these catalysts [27,28],and therefore, the behaviour has to be ascribed to thepreparation method. The presence of a unique reduc-tion peak for Cu loading as high as 8% has also al-ready been observed by Shimokawabe et al. [30]. Theyshowed, by monitoring the TPR peak of catalysts at220◦C, that highly dispersed CuO was the only specieson Cu/ZrO2 (8 wt.%) calcined below 300◦C.
The surface of fresh catalysts, without any thermaltreatment, was examined for the determination of cop-per oxidation state. Unique copper species were ob-
Fig. 1. Scanning electron micrographs of SiO2-TiO2 (A) supportand the relevant Cu/SiTi (B) catalyst at 10,000 magnifications.
served with binding energy values of Cu 2p3/2 around933 eV, very similar to that of CuO species (933.7 eV).No important differences were observed between freshand used (after NO-C2H4-O2 reaction) catalysts.
The N2 adsorption isotherms on all oxide supportsbelonged to type IV (mesoporous solids in whichcapillary condensation takes place at higher pressuresof adsorbate in addition to multilayer adsorption atlower pressures), according to IUPAC classification[31]. In agreement with the porous character of theoxides, the desorption branch of the isotherms showeda marked hysteresis whose position depended on thetype of oxide. The corresponding Cu catalysts showedanalogous behaviour, suggesting that metal deposition

P. Carniti et al. / Applied Catalysis B: Environmental 28 (2000) 175–185 179
Fig. 2. TPR profiles for the copper catalysts prepared on differentsilica supports by 8% H2/Ar. Heating rate: 8◦C min−1.
did not alter the textural properties of supports. In thecase of Cu/Si and Cu/SiZr, the hysteresis was smallerand shifted to lowerP/P0 values than that of the cor-responding supports, indicating an important decreaseboth in total pore volume and in pore diameter.Table 1 reports the values of total pore volume (PV)and average pore radius (RPav) for all supports andcatalysts. From these data, it is apparent that copperdeposition affected the porosity of the supports. As ageneral trend, it was observed that total PV and RPavdecreased with copper deposition, particularly forCu/Si and Cu/SiZr, as already mentioned. As regardsthe surface of the samples examined, BET surfacevalues in the range 300–500 m2 g−1 were obtained forthe supports and these values were maintained for allcopper catalysts. A peculiar behaviour was observedfor Cu/SiAl which exhibited a lower BET surfacearea than the relevant support. In this case, constantvalues of PV and RPav for both catalyst and supportwere found, indicating the predominant presence ofCu on the external surface of SiAl.
3.2. Catalytic studies
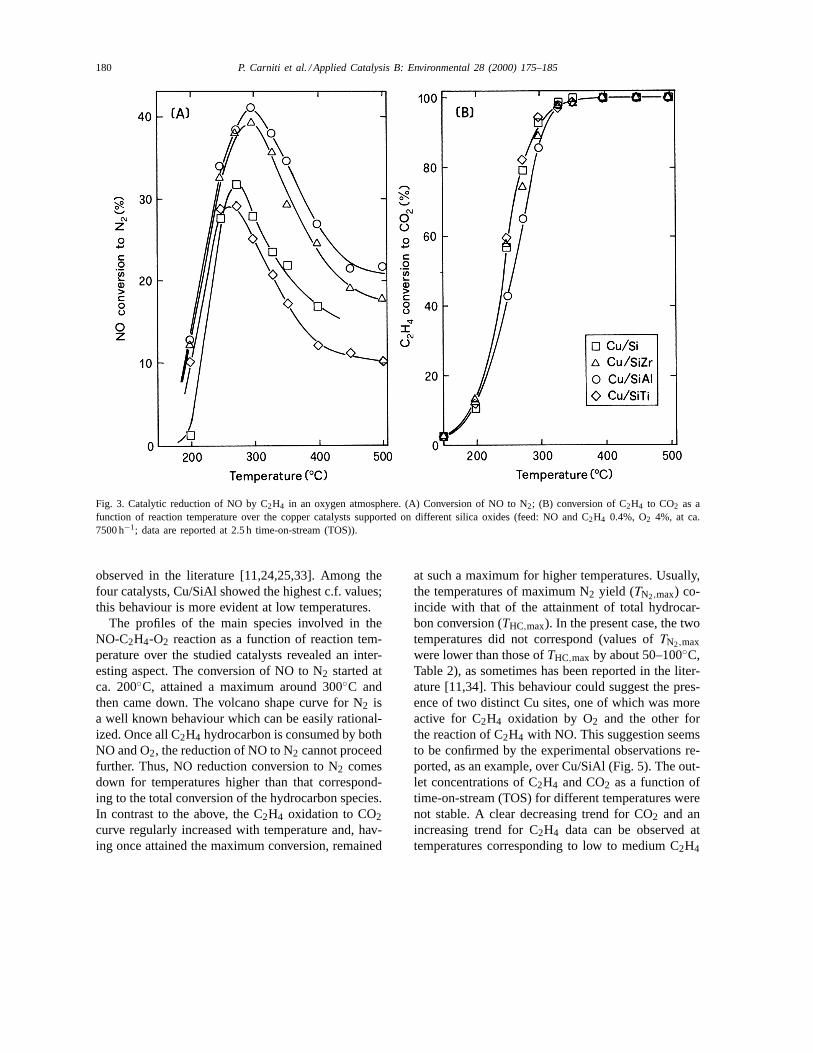
Fig. 3 shows the NO reduction conversions to N2and the C2H4 oxidation conversions to CO2 overthe four copper catalysts prepared on the different
silica oxide supports. Similar trends could be ob-served regardless of the support. The N2 formationcurves as a function of temperature showed a vol-cano shape with a detectable maximum in the range275–300◦C. The maximum NO reduction conver-sion to N2 ranged from 29 to 41% in the followingorder: Cu/SiTi<Cu/Si<Cu/SiZr≈Cu/SiAl. For themore active catalysts, the temperature of maximumNO conversion shifted to higher values. On the otherhand, the curves of C2H4 oxidation conversion toCO2 as a function of temperature were very slightlyaffected by the support. Quite unique curves could beobserved, except for the Cu/SiAl catalyst whose curvewas shifted to higher temperatures. Maximum C2H4conversions to CO2 were obtained at a temperatureof around 350◦C. The temperatures of maximum NOreduction and of attainment of total C2H4 oxidationdid not correspond, conversely to what is usually ob-served in most catalytic systems (i.e. ion-exchangedzeolites, supported metal catalysts) [21,24,32].
As concerns the catalyst selectivity, among theN-products from the reactor, N2O besides N2 was de-tected in any case. For all catalysts, the yield of N2Owas about 15–20% without a clear dependence ontemperature. Among the C-products from the reactor,CO was observed only on Cu/SiAl at low temperatures(200–325◦C) and in little amount (maximum CO for-mation of 5% at 275◦C). Carbon balance showed thatthere were no other C-compounds other than C2H4,CO, and CO2 at any time and temperature of reaction.
The c.f. (%) has been used as an index to controla particular type of selectivity of catalysts. It givesa measure of the ability of the C2H4 hydrocarbonspecies to be oxidized by NO rather than by O2. De-tails on its computation can be found in [24]. Fig. 4shows the trends of the calculated c.f. values as a func-tion of reaction temperature, for all catalysts. As ex-pected, c.f. values decreased as temperature increased,because C2H4 oxidation by O2 prevailed on C2H4 ox-idation by NO (that is, reduction of NO to N2). At alow temperature, 200–250◦C, high c.f. values (in therange 15–20%) were obtained, suggesting that C2H4was oxidized to a large extent by NO rather than byO2. Starting from 300◦C, a temperature at which max-imum NO reduction conversion to N2 was achievedin any case, the c.f. values fell down to 2–4%. In thistemperature range, the combustion process (C2H4 ox-idation by O2) prevailed. Analogous evidences were
180 P. Carniti et al. / Applied Catalysis B: Environmental 28 (2000) 175–185
Fig. 3. Catalytic reduction of NO by C2H4 in an oxygen atmosphere. (A) Conversion of NO to N2; (B) conversion of C2H4 to CO2 as afunction of reaction temperature over the copper catalysts supported on different silica oxides (feed: NO and C2H4 0.4%, O2 4%, at ca.7500 h−1; data are reported at 2.5 h time-on-stream (TOS)).
observed in the literature [11,24,25,33]. Among thefour catalysts, Cu/SiAl showed the highest c.f. values;this behaviour is more evident at low temperatures.
The profiles of the main species involved in theNO-C2H4-O2 reaction as a function of reaction tem-perature over the studied catalysts revealed an inter-esting aspect. The conversion of NO to N2 started atca. 200◦C, attained a maximum around 300◦C andthen came down. The volcano shape curve for N2 isa well known behaviour which can be easily rational-ized. Once all C2H4 hydrocarbon is consumed by bothNO and O2, the reduction of NO to N2 cannot proceedfurther. Thus, NO reduction conversion to N2 comesdown for temperatures higher than that correspond-ing to the total conversion of the hydrocarbon species.In contrast to the above, the C2H4 oxidation to CO2curve regularly increased with temperature and, hav-ing once attained the maximum conversion, remained
at such a maximum for higher temperatures. Usually,the temperatures of maximum N2 yield (TN2,max) co-incide with that of the attainment of total hydrocar-bon conversion (THC,max). In the present case, the twotemperatures did not correspond (values ofTN2,maxwere lower than those ofTHC,max by about 50–100◦C,Table 2), as sometimes has been reported in the liter-ature [11,34]. This behaviour could suggest the pres-ence of two distinct Cu sites, one of which was moreactive for C2H4 oxidation by O2 and the other forthe reaction of C2H4 with NO. This suggestion seemsto be confirmed by the experimental observations re-ported, as an example, over Cu/SiAl (Fig. 5). The out-let concentrations of C2H4 and CO2 as a function oftime-on-stream (TOS) for different temperatures werenot stable. A clear decreasing trend for CO2 and anincreasing trend for C2H4 data can be observed attemperatures corresponding to low to medium C2H4

P. Carniti et al. / Applied Catalysis B: Environmental 28 (2000) 175–185 181
Fig. 4. Trends of the competitiveness factor (c.f.) in theNO-C2H4-O2 reaction as a function of reaction temperature forthe copper catalysts supported on different silica oxides (feed: NOand C2H4 0.4%, O2 4%, at ca. 7500 h−1; data are reported at 2.5 htime-on-stream (TOS)).
conversion and CO2 formation. Moreover, the valuesof N2 concentration as a function of TOS did not showclear decreasing or increasing trends. From the aboveevidences, one can argue that two different kinds of
Table 2Summary of the catalytic activities in the NO-C2H4-O2 reactionof the copper catalystsa
Sample TN2,max
(◦C)THC,max
(◦C)YN2
b
(%)YCO2
b
(%)105 TOFc
(s−1)
Cu/Si 275 350 31.7 79.1 9.27Cu/SiAl 300 350 41.0 85.6 12.06Cu/SiTi 275 350 29.0 82.1 7.80Cu/SiZr 300 350 39.3 89.4 12.57
a Obtained at ca. 7500 h−1 (GHSV). Feed concentration: NOand C2H4, 0.4%; and O2, 4%.
b Calculated atTN2,max.c Expressed as molN2 mol−1
Cu s−1 and calculated atTN2,max.
Fig. 5. Outlet concentrations of C2H4 (s) and CO2 (h) in theNO-C2H4-O2 reaction as a function of time-on-stream (TOS) fordifferent reaction temperatures over the Cu/SiAl catalyst (feed:NO and C2H4, 0.4%; O2, 4%; at ca. 7500 h−1).
copper sites could be active, one with predominant ac-tivity towards the reaction of C2H4 with O2 and theother with predominant activity towards the reactionwith NO, the former being subjected to deactivation.
3.3. Kinetic interpretation
Cu/SiAl, the most active and selective catalystamong the series of copper-on-silica catalysts studied,was selected for a kinetic study aimed at quantify-ing the ability of the active copper sites to oxidizeC2H4 by NO rather than by O2. For the kinetic study,NO-C2H4-O2 runs were performed at different con-tact times (from 7500 to 36,000 h−1, expressed asGHSV) and temperatures from 200 to 400◦C, main-taining the feed concentration at a constant level.Most of the significant experimental results have beencollected in Table 3. As expected, at a given temper-ature, N2 yield as well as CO2 yield decreased, as

182 P. Carniti et al. / Applied Catalysis B: Environmental 28 (2000) 175–185
Table 3Catalytic activity in the NO-C2H4-O2 reaction of Cu/SiAl for runsat different contact times and selected reaction temperatures (TR)a
Contacttime (s)b
T (◦C) YN2
(%)YN2O
(%)YCO2
(%)YCO
(%)
0.48 (7500) 250 34 17 43 4.7300 41 23 86 4.0350 35 21 100 –
0.24 (15,000) 250 17 17 20 4.2300 30 21 55 8.5350 26 22 93 3.8
0.14 (25,000) 250 14 16 15 2.9300 24 20 41 7.4350 25 21 76 9.4
0.10 (36,000) 250 – 17 7.8 2.4300 20 19 25 6.2350 24 19 58 11
a Feed concentration: NO and C2H4, 0.4%; and O2, 4%.b Values in parentheses are those of the space velocity (h−1,
GHSV).
contact time decreased. A careful inspection of thedata revealed that the decrease was more pronouncedfor the CO2 yield than for that of N2. Therefore, lowcontact times did not favour hydrocarbon combustion(C2H4 by O2). This behaviour was expected as smallCuO aggregates, that are present on the Cu/SiAlcatalyst, do not possess enhanced oxidation activity[15,21].
The overall NO-C2H4-O2 reaction can be viewed,for a simplified kinetic interpretation, as a combi-nation of simultaneous competitive reactions whichrun at different rates and possess different activationparameters [23,24]. The following reaction schemethat provides equations leading to the reaction prod-ucts N2, N2O, CO2, and CO has been taken intoaccount:
6NO+ C2H4k1→3N2 + 2CO2 + 2H2O (1)
3O2 + C2H4k2→2CO2 + 2H2O (2)
8NO+ C2H4k3→4N2O + 2CO+ 2H2O (3)
2O2 + C2H4k4→2CO+ 2H2O (4)
wherek1 andk3 are the rate constants of the reactionsassociated with the NO reduction activity and wherek2 and k4 are those associated with the combustion
activity of the catalyst. In the reaction scheme, the con-version of NO to NO2 has been ruled out. Accordingto reported data [35,36], copper-based catalysts haveonly minor activity in NO2 formation.
Considering altogether the NO reduction productsN2 and N2O as well as the oxidation products CO2and CO, the following rate equations can be written,indicating the rates of C2H4 consumption in reactions(1)–(4) byr1, r2, r3, andr4, respectively:
d(PN2 + PN2O)
dt= 3r1 + 4r3 (5)
d(PCO2 + PCO)
dt= 2(r1 + r2 + r3 + r4) (6)
where Pi indicates the partial pressure of theithspecies.
On the basis of the procedure described in [23,24],in which reactions (1) and (2) were only taken intoaccount, and including in the procedure reactions (3)and (4) also, it is possible to write the variation in theamount of N2 and N2O, in terms of partial pressures,with respect to CO2 and CO:
d(PN2 + PN2O)
d(PCO2 + PCO)
= 3
2
(k1 + (4/3)k3
k1 + k3
)
× P inNO − 2(PN2 + PN2O)
[P inNO − 2(PN2 + PN2O)] + [P in
O2+ (PN2 + PN2O)
−(3/2)(PCO2 + (4/3)PCO)][(k2 + k4)/(k1 + k3)]
(7)
where P ini indicates the partial pressure of theith
species in the feed.If one can assume that
k1 + (4/3)k3
k1 + k3
∼= 1 and (PCO2 + (4/3)PCO)
' (PCO2 + PCO)
Eq. (7) can be rewritten as
d(PN2 + PN2O)
d(PCO2 + PCO)
=3
2
P inNO − 2(PN2 + PN2O)
[P inNO − 2(PN2 + PN2O)] + [P in
O2+ (PN2 + PN2O)
−(3/2)(PCO2 + PCO)][(k2 + k4)/(k1 + k3)]
(8)
P. Carniti et al. / Applied Catalysis B: Environmental 28 (2000) 175–185 183
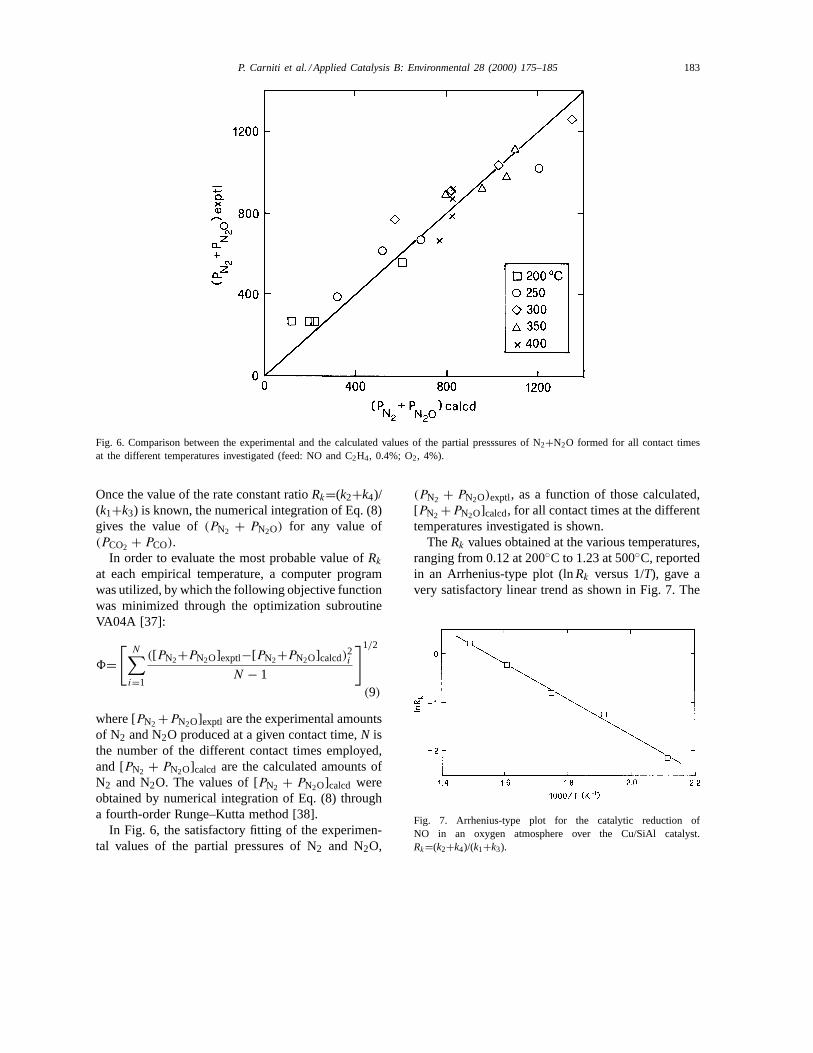
Fig. 6. Comparison between the experimental and the calculated values of the partial presssures of N2+N2O formed for all contact timesat the different temperatures investigated (feed: NO and C2H4, 0.4%; O2, 4%).
Once the value of the rate constant ratioRk=(k2+k4)/(k1+k3) is known, the numerical integration of Eq. (8)gives the value of(PN2 + PN2O) for any value of(PCO2 + PCO).
In order to evaluate the most probable value ofRk
at each empirical temperature, a computer programwas utilized, by which the following objective functionwas minimized through the optimization subroutineVA04A [37]:
8=[
N∑i=1
([PN2+PN2O]exptl−[PN2+PN2O]calcd)2i
N − 1
]1/2
(9)
where [PN2 +PN2O]exptl are the experimental amountsof N2 and N2O produced at a given contact time,N isthe number of the different contact times employed,and [PN2 + PN2O]calcd are the calculated amounts ofN2 and N2O. The values of [PN2 + PN2O]calcd wereobtained by numerical integration of Eq. (8) througha fourth-order Runge–Kutta method [38].
In Fig. 6, the satisfactory fitting of the experimen-tal values of the partial pressures of N2 and N2O,
(PN2 + PN2O)exptl, as a function of those calculated,[PN2 +PN2O]calcd, for all contact times at the differenttemperatures investigated is shown.
TheRk values obtained at the various temperatures,ranging from 0.12 at 200◦C to 1.23 at 500◦C, reportedin an Arrhenius-type plot (lnRk versus 1/T), gave avery satisfactory linear trend as shown in Fig. 7. The
Fig. 7. Arrhenius-type plot for the catalytic reduction ofNO in an oxygen atmosphere over the Cu/SiAl catalyst.Rk=(k2+k4)/(k1+k3).

184 P. Carniti et al. / Applied Catalysis B: Environmental 28 (2000) 175–185
slope of the calculated line (1Ea/R) permits obtaininga rough estimate of the difference between the acti-vation energy of the oxidation of C2H4 by O2 (con-sidering reactions (2) and (4) together) and that of thereduction of NO by C2H4 (considering reactions (1)and (3) together):
1Ea = Ea,O2 − Ea,NO (10)
The numerical value of this difference was found to be30.5±1.3 kJ mol−1. Thus, the reaction of C2H4 oxida-tion by O2 showed an activation energy much higherthan those of C2H4 with NO. This is in agreementwith the above-reported results; in particular, the ox-idation of C2H4 is mainly due to NO for tempera-tures up to 250◦C, while at higher temperatures, thereactions in which the oxidation reagent is O2 wereabruptly started.
4. Conclusions
Results reported in Table 2, in particular, the activityin the NO-C2H4-O2 reaction in terms of TOF, indicatethat the copper systems investigated here are a goodstarting point for improving the preparation of selec-tive catalysts for the above-mentioned reaction. Thus,although selectivity is not yet as good as one would de-sire, the high amount of the N2+N2O products showsa good reducing activity of the catalytic site. A betterdistribution between N2 and N2O requires optimiza-tion of the catalytic system preparation, particularlyas far as the metal loading is concerned.
As regards the nature of active sites, evidences thathave appeared in the recent literature show that iso-lated Cu2+ species are desirable as active sites inthe catalytic reduction of NO. Thus, XANES analysison Cu/Al2O3 and Cu/SiO2 systems [21] conclusivelyshowed that Cu(II) centres, either exchanged and/orsmall size particles of CuO-like species, catalyze thereduction of NO with C3H6. The initial existence ofan isolated CuO-like phase is, in turn, a direct con-sequence of the preparation method, it being stronglyfavoured by basic solution preparations [22,39]. Theoperative methodology for the preparation of the cata-lyst studied, that is the use of a [Cu(NH3)4]2+ at pH 9,well above the support’s zero-point charge, ensures theformation of a highly dispersed CuO-like phase. Therelevance of the preparation method is also enlight-
ened by our direct observation that all four catalystsprepared on four different silica oxides exhibit similaractivity. As already reported [11], the NO-C2H4-O2 re-action is primarily affected by the nature of active cen-tres, the support playing only a secondary role. Amongthe four silicas used, only a small increase in activityis observed with increase in the co-ion Lewis acidity.
On the other hand, a very strong influence of thesize of the copper particles has already been recog-nized in the recent literature [12,21,39]. Aggregatedcopper species were found to be more active for C3H8oxidation by O2 than for the reaction of C3H8 withNO. A similar behaviour can be suggested for theNO-C2H4-O2 reaction. As mentioned above, a statis-tical analysis of the dimension of the metallic particlespresent on the surfaces of Cu/Si and Cu/SiTi catalystsafter calcination resulted in a narrow size distributionover the 2.0–5.5 nm range [28]. This may justify thehigh activity observed.
However, from the reaction profiles for N2 and CO2formation as a function of reaction temperature (Ta-ble 2), it appears that the temperature of maximumN2 formation (TN2,max) and that of maximum C2H4oxidation (THC,max) do not coincide. The existence oftwo different copper sites, that is of copper aggregatesof different nuclearity, could account for this unusualbehaviour. DifferentTmax’s for the two reactions havealready been reported [11,34], but no interpretationhas been proposed till now.
Although the preparation method adopted leadsto a unique CuO-like well dispersed phase, one ofus has already reported on the presence of plate-liketwo-dimensional (2D) and three-dimensional (3D)metal particles [27,28]. HRTEM and FT-IR character-ization of a Cu/Si sample before and after having beenused in the polymerization of 2,6-dimethylphenolin the presence of molecular O2 allowed identi-fying the active sites for this particular reactionas two-dimensional metallic particles [40], whilethree-dimensional stepped ones are more involved inmolecular H2 activation [41].
The analysis of the results reported here prompt usto further investigate the activity of these catalytic sys-tems, particularly as far as the copper loading is con-cerned. Moreover, work is already in progress to eluci-date the nature of the copper active sites. The tailoringof metallic particles would allow us to design a cata-lyst with optimum properties towards NO reduction.

P. Carniti et al. / Applied Catalysis B: Environmental 28 (2000) 175–185 185
Acknowledgements
N.R. wishes to acknowledge Grace Davison GmbH,Worms, Germany, for financial and technical support.
References
[1] M. Iwamoto, H. Hamada, Catal. Today 10 (1991) 57.[2] M. Iwamoto, H. Yahiro, S. Shundo, Y. Yu-u, N. Mizuno,
Appl. Catal. 69 (1991) L15.[3] S. Sato, Y. Yu-u, H. Yahiro, N. Mizuno, M. Iwamoto, Appl.
Catal. 70 (1991) L1.[4] M. Shelef, Chem. Rev. 95 (1995) 209.[5] M. Misono, CATTECH 2 (1998) 183.[6] Y. Traa, B. Burger, J. Weitkamp, Micropor. Mesopor. Mater.
30 (1999) 3.[7] R. Grinsted, H. Zhen, C. Montreuil, M. Rokosz, M. Shelef,
Zeolite 13 (1993) 602.[8] J. Yan, G.-D. Lei, W.M.H. Sachtler, H.H. Kung, J. Catal. 161
(1996) 43.[9] K.C.C. Kharas, H.J. Robota, D.J. Liu, Appl. Catal. B 2 (1993)
225.[10] H. Hamada, Catal. Today 22 (1994) 21.[11] K.A. Bethke, M.C. Kung, B. Yang, M. Shah, D. Alt, C. Li,
H.H. Kung, Catal. Today 26 (1995) 169.[12] L. Chen, T. Horiuchi, T. Osaki, T. Mori, Appl. Catal. B 23
(1999) 259.[13] N. Okazaki, Y. Shiina, H. Itoh, A. Tada, M. Iwamoto, Catal.
Lett. 49 (1997) 169.[14] M. Ozawa, H. Toda, O. Kato, S. Suzuki, Appl. Catal. B 8
(1996) 123.[15] Z. Chajar, M. Primet, H. Praliaud, M. Chevrier, C. Gauthier,
F. Mathis, in: A. Frennet, J.-M. Bastin (Eds.), Catalysis andAutomotive Pollution Control III, Stud. Surf. Sci. Catal., Vol.96, Elsevier, Amsterdam, 1995, p. 591.
[16] J.A. Anderson, C. Márquez-Alvarez, M.J. López-Muñoz,I. Rodrıguez-Ramos, A. Guerrero-Ruiz, Appl. Catal. B 14(1997) 189.
[17] H. Praliaud, S. Mikhailenko, Z. Chajar, M. Primet, Appl.Catal. B 6 (1998) 359.
[18] Z. Chajar, V.C.L. Chanu, M. Primet, H. Praliaud, Catal. Lett.52 (1998) 97.
[19] D. Pietrogiacomi, D. Sannino, S. Tuti, P. Ciambelli, V.Indovina, M. Occhiuzzi, F. Pepe, Appl. Catal. B 21 (1999)141.
[20] V. Indovina, M. Occhiuzzi, D. Pietrogiacomi, S. Tuti, J. Phys.Chem. B 103 (1999) 9967.
[21] C. Márquez-Alvarez, I. Rodrıguez-Ramos, A. Guerrero-Ruiz,G.L. Haller, M. Fernández-Garcıa, J. Am. Chem. Soc. 119(1997) 2905.
[22] M. Fernández-Garcıa, I. Rodrıguez-Ramos, P. Ferreira-Aparicio, A. Guerrero-Ruiz, J. Catal. 178 (1998) 253.
[23] P. Carniti, A. Gervasini, React. Kinet. Catal. Lett. 67 (1999)233.
[24] A. Gervasini, P. Carniti, V. Ragaini, Appl. Catal. B 22 (1999)201.
[25] H.H. Kung, M.C. Kung, Catal. Today 30 (1996) 5.[26] M.C. Kung, K.A. Bethke, J.-H. Lee, H.H. Kung, Appl. Surf.
Sci. 121/122 (1997) 261.[27] F. Boccuzzi, A. Chiorino, G. Martra, M. Gargano, N. Ravasio,
B. Carrozzini, J. Catal. 165 (1997) 129.[28] F. Boccuzzi, S. Coluccia, G. Martra, N. Ravasio, J. Catal.
184 (1999) 316.[29] F. Radtke, R.A. Koeppel, E.G. Minardi, A. Baiker, J. Catal.
167 (1997) 127.[30] M. Shimokawabe, H. Asakawa, N. Takezawa, Appl. Catal.
59 (1990) 45.[31] P. Schneider, Appl. Catal. A 129 (1995) 157.[32] R. Gopalakrishnan, P.R. Stafford, J.E. Davidson, W.C. Hecker,
C.H. Bartholomew, Appl. Catal. B 2 (1993) 165.[33] J.Y. Yan, W.M.H. Sachtler, H.H. Kung, Catal. Today 22 (1997)
279.[34] K. Bethke, D. Alt, M.C. Kung, Catal. Lett. 25 (1994) 37.[35] M. Shelef, C.N. Montreuil, H.W. Jen, Catal. Lett. 26 (1994)
277.[36] V. Tomašic, Z. Gomzi, S. Zrncevic, Appl. Catal. B 18 (1998)
233.[37] M.J.D. Powell, Comput. J. 7 (1965) 303.[38] B. Carnahan, H.A. Luther, J.O. Wilkes, Applied Numerical
Methods, Wiley, New York, 1969, p. 361.[39] Z. Chajar, M. Primet, H. Praliaud, J. Catal. 180 (1998)
279.[40] F. Boccuzzi, G. Martra, G. Partipilo Papalia, N. Ravasio, J.
Catal. 184 (1999) 327.[41] F. Boccuzzi, A. Chiorino, M. Gargano, N. Ravasio, J. Catal.
165 (1997) 140.




















