
catalyst studies of vapor phase esteripication
173
CATALYST STUDIES OF VAPOR PHASE ESTERIPICATION OF n-OCTYL ALCOHOL AND ACETIC ACID DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By YERRAM VENKATESHAM, H. So. r' «1,';1 ■, The Ohio State University 1953 Approved byt
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of catalyst studies of vapor phase esteripication

CATALYST STUDIES OF VAPOR PHASE ESTERIPICATIONOF n-OCTYL ALCOHOL AND ACETIC ACID
DISSERTATION
Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the
Graduate School of The Ohio State University
By
YERRAM VENKATESHAM, H. So.r'
«1, ';1 ■,
The Ohio State University 1953
Approved byt

ACKMOVUtDaMIKT
The following work wee done under the guidance of Dr. Aldrich Syreraon of the Chemical Engineering Department of The Ohio State University. The author is extremely grateful for his interest in and guidance of the project.
lerraa Tenkatesham

TABLE OF CONTENTSfagft,
SUMMARY............................................ 1STATEMENT OF THE PROBLEM............ ............ 6INTRODUCTION ..................................... 7RELATED LITERATURE
E s ter if lea t ion...... 8Equilibrium Constant...................... ISCatalytic Vapor Phase Esterification..... 25Catalytic Reaction.............. ............ • 36Longitudinal Diffusion.......... 52Porosity of the Catalyst............. 55
EXPERIMENTAL...................................... 57Equipment for Esterificatlon.............. 57Materials Used in the Investigation...... 67Calibration of the Feed System......... ...» 69Operating Procedure...................... 71
DATA A HD OBSERVATIONS............................ 83
STATEMENT OF RESULTS............................. 88
CORRELATION OF DATA AND PROFOSED MECHANISM.... 102Evaluation of Constants by Method ofLeast Squares................. 112
REE DEa 1 S DA TA..................................... 133BACK CALCULATION OF DATA ......................... 1*0CONCLUSIONS....................................... 152RECOMMENDATIONS................................... 157APPENDIX........................................... 158BIBLIOGRAPHY...................................... 163AUTOBIOGRAPHY 169

CATALYST STUDIES OF VAPOR PHASE BSTKRIFICATIOHOF n-OCTYL ALCOHOL AND ACETIC ACID
SUMMARYLow iron low silica bauxite was found to be an
excellent catalyst for the vapor phase esterifIcatlon of n—octyl alcohol and acetic acid. Side reactions and decomposition of the products were negligible between 218°C. and 244*5°C. It was found that a & fold change in the particle size of the catalyst had no measurable effect on the activity, other conditions being equal catalyst from 6/14 mesh to 40/60 mesh gave the same results. The experiments were carried out at two pressures, 0.974 atm. and at about 0.470 atm., using a feed containing from 50 mole % to 30 mole % octanol.The feed rates were varied from 8 g./or to 40 g./ir. and the catalyst weight from 4 to 10 grams. Z?o effect on conversion was noticed when the mass velocity of the feed was doubled as long as the ratio of feed rate to catalyst mass was constant.
The catalyst was found to be very permeable and had a value of 0*558 for the fraction of internal voids. A volumenometer was used to find the true density of the catalyst. Since it was found that air was adsorbed by the catalyst, helium was used in the volumenometer. The feed system for runs made at 0.470 atmospheres had to be modified in order to prevent the suction of feed

from "the pump into the reactor. The modification consisted in introducing a capillary tube of the right length between the pump and the reactor, to offer a resistance to the flow of the feed, that was greater than the value that could be overcome by pressure drop betwee the pump and the reactor. The vacuum pump was connected to a coarse and a fine bleed nozzles which were controlled by a "Thermocap Kelay" attached to a mercury manometer. This arrangement proved to be very effective in maintaining the pressure In the reactor constant within one m.m. of mercury.
The degree to which the eaterificatlon took place was determined by titrating the product containing the unreacted acid with standard sodium hydroxide solution. The amount of acid consumed enabled the calculation of octanol reacted. During each run the conversion of octanol was high to start with and slowly decreased until it reached a steady state condition. The experimental data were plotted as x. g. moles octanol con— verted/g. of feed Xft* W/F, the ratio of weight of the catalyst to the feed rate g/hr. From these curves values of x, W/F and were obtained. Valxms of xwere plotted against Q d/X, and a smooth curve was drawnn xthrough the points to get values of rates directly for any value of x. For a particular value of x values of pA , pg and p^ and r were obtained and tabulated. It

could be shown by qualitative considerations, In the light of the data obtained, that neither the adsorption of any one of the products nor the adsorption of acid was the rate controlling step in the vapor phase esteri- fication. The effect of diffusion from the bulk of the gas phase to the gross surface of the catalyst particle was evaluated and the partial pressure gradient due to diffusion was estimated to be 2—3% of the total. In the range of mass velocity used in this investigation it appears that longitudinal diffusion may not have been a negligible factor, although it is not a predominant factor.
Two other possible rate controlling mechanisms to be considered are: adsorption of octanol controllingand surface reaction controlling. In order to determine if one of the two mechanisms is rate controlling, evaluation of the adsorption equilibrium constants In the appropriate rate equation by the method of least squares was employed. In conjunction with the equation for adsorption of octanol controlling, the experimental data yielded negative values for equilibrium adsorption constants. Thus the possibility of adsorption of octanol being the rate controlling step, was ruled out. The rate equation for surface reaction controlling:
ksLKAKB (aA «B - SS£S. )r « ■ - — .i ...... . ----
(l+aAKA +aBKB +aRKR','aS K8 )

was found to fit the data very well. The term kaLK4,K£ le a constant, for a given temperature and for simplicitywill be called Q. The activities were expressed as partial pressures. Values of aB and ag being always equal were factored out resulting in a term SpCK^+Kg) in the denominator. The sum of constants Kp+Kg will be called Kp. The form of the final equation that fitted the data was:
Q (PA*PB “r m ---
(1+Pjl A "*■ Pbk B+PR*R The values of the constants were:
T°C. Q K*. KB Kp K218 2.06 1.42 11.94 12.38 16.A244.5 2.34 1.30 8.49 8.95 11.4
By using the above values for the constants in backcalculation^ values of r corresponding to given values ofx were obtained. From these a plot of x Y6. l/r wasobtained. In view of the equation W/F ■ The
Jo rvalues of W/F for any given x were obtained by the graphical integration of the curve x ifi. l/r* The calculated values were in close conformity with the experimental curves. Some deviation of the calculated values from the experimental curves was observed in the case of runs made with the feeds containing 50 mole % octanol. Hence it was concluded that the mechanism of surface

reaction controlling vaa followed more closely when the feed contained an excess of octanol.

STATEMENT OF THE PROBLEMThe work reported in the following pages was under
taken to throw more light on these questions:1. Is the catalyst used, low silica low iron
bauxite, porous and if so how porous?2. Does the size of the catalyst affect the acti
vity? What happens if the catalyst used is in the form of a powder?
3. To what extent does the phenomenon of diffusion enter the esterlfication reaction, under the conditions in which it is carried out?
4. How important is back diffusion of products in the reactor?
5• Does reduced pressure alter the mechanism of the vapor phase catalytic esterification?
6. Can the mechanism of vapor phase catalytic esterification of n—octyl alcohol and acetic acid, suggested by Clyde Reeder, Jr», be arrived at independently and thus be confirmed?
Some related problems were:1* To find an easy and fairly accurate method of
measuring the true volume of the catalyst*2, To devise an effective and easy way of pumping
the feed at accurate rates into the reactor kept at reduced pressure,
3. To devise a system to maintain a constant reduced pressure in the reactor.

7
INTRODUCTIONEsterification is a very Important unit process in
our technology. The most common method Tor the preparation oT esters is the reaction of a carboxylic acid and an alcohol with elimination of water. The vapor phase catalytic esterification was selected for this investigation for two reasons. The side reactions in the esterification of n-octyl alcohol and acetic acid in the vapor phase were negligible between the 213 and 245°C. The analytical procedure necessary to follow the reaction was simple.
With a view to obtain kinetic data for the esteri— fication reaction, Clyde Reeder, Jr. studied the catalytic, vapor phase esterification of n—octyl alcohol and acetic acid using bauxite as a catalyst. In his study of the catalyst many questions regarding the behavior of the catalyst were left unanswered.
He obtained a great deal of data by conducting the reactions at 218° and 24l°C. The boiling points of octyl alcohol and octyl ester are 19-4° and 210°C. respectively. In his experiments the temperature of the reactor was close to the boiling points of the alcohol and the ester. It was quite possible that the vapors of alcohol and ester were being absorbed on the catalyst and thus the mechanism arrived at might not have been

completely a true one. If the reaction was carried out at higher temperatures, there was a tendency for the ester to decompose.
In order to obtain a greater difference between the. boiling points of ester and octanol and the reaction temperature, it was considered necessary to study the esterification reaction under reduced pressure. These reactions carried out under reduced pressure would also provide additional information to verify the mechanism proposed by Clyde Reeder, Jr.
fia.tarlXiCLfl.tijaaThe most common method for the preparation of esters
is the reaction of a carboxylic acid and an alcohol with elimination of water. Esters are also formed by a number of other reactions including the use of acid anhydrides, acid chlorides, amides, nitriles, unsaturated hydrocarbons, ethers, aldehydes, and ketones and by dehydrogenation of alcohols.
The making of esters la big business. Approximate figures for the production of esters in 194-8 in the United States in millions of pounds are:
(l) Reid, E. E., "Esterification", Ind. Eng. Chera., 1596 (1948),

Ethyl acetate Butyl acetate
125 (1945) 100Dibutyl phthalate Cellulose acetate
45 (1945)Alkyd resins Rosin estersCellulose xanthate Plastic i zers
28019083
700170
Alkyd resins are mixed esters from phthalic and other acids with penta srythritol, glycerol and glycols, Plastioiaers are largely phthalates and include dibutyl phthalates,
Esterification as a chemical reaction has been well known for many years, but it reached its commercial significance only during the first World War, A short history of the use of ester solvents is very interesting from the standpoint of the growth of the ester manufacture.
(2) Keyes, D, B,, nEsterification Processes and Equipment", Ind, Eng, Chem., 2L . 1096 (1932),
Organic solvents with the exception of ethyl alcohol and methanol, had little commercial use until 1914,Amyl acetate in the form of fusel oil acetate was the best-known ester solvent at that time. It was not,however, used in large quantities. The demand for airplane dope and the corresponding need for cellulose nitrate and cellulose acetate solvents increased rapidly during the war period. The only ester that was developed on a large scale during the war to replace amyl acetate was

-10-methyl acetate.
Immediately after the var there vas available an enormous quantity of nitrocellulose and a correspondingly large potential source of this product in the United States. The manufacture of ethyl alcohol increased enormously during the war, and the stocks of this solvent were unusually great at that time. Fermentation process which produced acetone also produced enormous quantities of n—butyl alcohol. This product had little or no peacetime use. Furthermore, in order to obtain a satisfactory production of acetone, various processes for the manufacture of acetic acid had been developed, notably the fermentation of ethyl alcohol by acetic acid. The acetone was produced by the heat treatment of calcium acetate. The situation in the U.S.A. at the close of the war was such that there was an over-production of nitrocellulose, ethyl alcohol, butyl alcohol and acetic acid.
It was natural that the course of development should be to produce acetic acid esters of both amyl and butyl alcohol and to mix these solvents with nitrocellulose in the hope of utilising the final solution as a covering material. Another factor in this development was the formation of a low viscosity nitrocellulose which would permit the dissolving of an unusually large amount of nitrocellulose in these solvents without the resulting

-11-solution becoming too viscous for practical use* Nitrocellulose recovered from smokeless povder vas largely of this variety, and special methods to produce the original product with this low viscosity property were also developed at this time*
The resulting solution containing (in addition to the constituents mentioned above) natural resins, diluents such as the hydrocarbons and plasticizing agentB, and pigments, constituted the beginning of our modern Jsequer. At that time the varnishes and enamels used on automobiles were far from satisfactory, in that the time of application was enormous if the resulting product were to show any durability* Manufacturers of automobiles in U.S.A. were anxious to obtain quick—drying enamel that would be durable, and could be readily washed and polished without removing an appreciable amount of the surface. The solution of esters and nitrocellulose together with the other constituents seemed to answer this particular problem. The film was formed by evaporation and the pigmented nitrocellulose was apparently quite resistant toward light and general atmospheric conditions. The application could be carried out in a matter of minutes, whereas the old finish, consisting of many layers of varnish, required several months. The economic advantage of this covering from the standpoint of the automobile and of the furniture manufacture was

-12-088117 appreciated.
Today the commercial quantities of moat of the simple esters are of synthetic origin, although a number of them do occur in nature. Some of the naturally occurring esters other than fats and waxes, and some of their sources are :
Ethyl acetate - in many wines, brandy, wine vinegar and some fruits such as pineapple; amyl acetate — In apples, bananas, and other fruits; geranyl formate and citronellyl formate and acetate - In geranium oil; terpinyl acetate - in cypress oil; methyl acetate — in peppermint oil; benzyl acetate — in jasmine, hyacinth and gardenias; methyl benzoate - in clove oil. Host of these naturally occurring esters have very pleasant odors and either they or their synthetic counterparts find uses in the confectionery, beverage, perfume, cosmetic and soap industries.
Uses _uf esters in general.Solvents and Plasticizersi — The largest use of
esters are in the solvent and plasticizer fields. The lower-molecular esters enter the laequer, paint and varnish field, while the higher ones are used primarily as plasticizers. Alkyl acetates are used in solvent field and those of the phthalates and tricreeyl—phosphate for the plasticizer field.
R e s i n s P l a s t i c s and Coatings: - Certain esters

-13-form polymers useful as resins and plastics, and many of these resins are used as coatings.
Perfumes. Flavors- Cosmetics and Soaoi - Compared to solvents and plasticizers, the tonnage of esters used in improving odors and flavors is small hut nevertheless the quantity is important economically and esthetically.
Medicinelai — Esters of aromatic acids find extensive use in medicine. Aspirin, the acetate of hydroxy acid (salicylic acid) is one of the more commonly used compounds.
An ester is usually defined as a compound formed by substituting a group such as ethyl (—C2H^) for the ionlzable H of an acid. In the older chemical literature esters were commonly called "ethereal salts" ahd ethyl acetate was regarded as the analogue of sodium acetate. Doubt was thrown on this theory by a study of the esterifIcation of mercaptan by acetic acid and of alcohol by thioacetic acid which react as follows:
CH3CO OH + H SC2H 5 ---- CH3COSC2H 5CH3CO SH + H OC2H5 --- CH3COOC2H 5 + H2S
According to this the esterificatlon reaction should bewritten the other way,
(3 ) Reid, E. E., "EsterIfication of thiobenzoic Acid and of Benzoic A d d by Mercaptan", Am. Chem. J., A3 r 489-504 (1910).

This view has been cosfirmed by a study of the esterl— fioatlon of methanol containing heavy oxygen all of which went into the ester.
(4) Roberts, I.and Urey, H. C., "A Study of the Esteri- fication of Benzoic Acid with Methyl Alcohol Using Isotoplc Oxygen", J. Am. Chem. Soc., ££, 2391 (1938)*
The ester should be regarded as having the acyl group substituted for the active hydrogen of the alcohol, ethyl acetate is acetyl—ethanol. however, the name ethyl acetate is too well established to be changed.
The rate at which different alcohols and acids are esterlfied, as well as the extent of the reaction, is dependent upon the structure of the molecules and types of radicals present.
The general effects of structure on the esterifi— cation of a number of acids and alcohols has been studied
(5) Kirk, R. E., and Qthmer, D. F., "Esterification" (editors) "Encyclopedia of Chemical Technology", Vol. V. The Interscience Encyclopedia, Inc., Hew York, 1950.
With acetic acid at 155°C* the primary alcohols were found to esterify most rapidly and completely with methanol giving the highest yield and most rapid reaction

Ethyl, n-propyl and n—butyl alcohols reacted with about equal velocities and limits. Under the same conditions the secondary alcohols were much slower reacting and had lower limits of esterification; however, wide variations were noted among the different members of this series.The tertiary alcohols were very slow in reacting and the limits were generally low, one to ten per cent conversion at equilibrium. Testa with isobutyl alcohol at 155°C, and various acids showed that those containing a straight chain, e,g., acetic, propranic and butyric, and phenyl acetic and 0—phenylproplonic acids were esterl fled readily. Formic acid had the fastest initial rate of esterification, but the esterification limits of the acids were noted to increase with increasing molecular weight of the acid. The introduction of a branched chain in the acid decreased the rate of esterification and two branches caused a still greater retarding effect. Double bonds also had a retarding influence. However, the limits of esterification of these substituted acids were higher than for the normal straight chain acids.
Michael and Wolgast% determinations show some devia—
(6) Michael A .f and Wolgast K,, "Nature of Steric Hindrance III, Relationship Between Structure of Aliphatic Alcohols and the Velocity of Their Esterification" Ber., 3157 (1909),

-16-tlon from earlier studies. The esterifications vlth trlchloro acetic acid showed that for normal aliphatic alcohols the speed of esterifleation was Increased with Increasing length of the carbon chain of the alcohol, thus n— octyl alcohol reacted with this acid at two to four times the speed of ethyl alcohol at 25 and 50°C. Tert—butyl and tert-amyl alcohols reacted faster with trichloro-acetic acid than secondary alcohols such as isopropyl, see-butyl, sec-amyl, and eec-octyl alcohols.It la evident that under certain ccndltions the secondary and tertiary alcohols may be very reactive. The comparative behavior Is not determined by a single component but rather by all of the components in the system) the alcohol, the acid, the solvent and sometimes the catalyst.
Caquil studied the esterification of cyclohexanol
(7) Caquil, M . , "The Esterification of Cyclohexanol and Some of Its Homologs", Compt. rend., 178. 323—6 (1924)j "The Esterification of Homologs of Cyclohexanol", Corapt. rend., 178. 1536-40 (1924).
and various methyl— and ethyl—substituted derivatives with acetic acid at 95°C. The initial velocity of the reaction was practically the same in each system, but the completeness of the esterification was influenced

by the position of the substituent group and was less than with the corresponding open—chain secondary alcohols.
Some workers have studied the Influence of substituents on the rate of esterification of aliphatic acids. The nitrile group has a pronounced inhibiting effect. With the chloroacetic acids the velocity decreases with Increased substitution. The rate of esteri- fication of the straight chain fatty acids from propionic through stearic is substantially constant. Branching of the chain causes retardation, especially with acids below valeric. In the saturated dibasic acids the rate of esterification increases to a maximum with glutarlc acid and then falls. The ease of esterification of the cycloparaffin monocarboxylic acids Increases in the orders C7 , C^, and C^ rings; with the exceptionof cyclopropane carboxylic acid, they are esterified more rapidly than the corresponding open chain acids.
The effect of branching on the rate of esterifica— tion of aliphatic acids has been investigated by Smith.
(3) Smith, H. A., "Kinetics of the Catalyzed Ksterifica— tion of Normal Aliphatic Acid in Methyl Alcohol", J. Am. Chem, Soc., J2JL, 254 (1939); "The Acid Catalyzed Esteri— flcation of Aliphatic Acids", 1136 (1940).
Ordinarily, substitutions must take place in the a—

-18-or p-positlon to affect the reaction velocity. The greater the number of alkyl substituents, the greater Is the effect on the retardation of the rate of reaction and the greater Is the Increase In the activation energy for the reaction. Acids with more than four substituents in the a— and p—positions should be practically unesterifiable under ordinary conditions.
The effect of substitutions in the benzene ring on the rate of esterification of aromatic acids has been studied by Hartman and co—workers.
(9) Hartman, E. J . , Storms, L. B., and Gassman, A. G., "Effect of Polar Groups Upon Esterification Velocities of Substituted Benzoic Acids with Cyclohexane” , J. Am.Chem. Soc., 2167 (1939).
Substitutions that displace electrons towards the carboxyl group diminish the rate of reaction. The substitution of fluoro, methoxy, or ethoxy groups in the 0- posltion has an accelerating action, whereas iodo, bromo, nitro, or methyl groups produce retardation. The influence of groups in the m— and p—positions is not nearly so marked.
Equilibrium Constants.The reaction between an organic acid and an alcohol
to produce an ester and water according to the equation*

-19-R« COOH + ROH - R* COOR + HgO
Is an example of a reversible equilibrium. This was first demonstrated in 1362 by Berthelot and F'ean de St. Gilles,
■
(10) Berthelot and F'ean de St. Glllesf Ann. Chim. (3)385 (1862).
who found that when equimolal quantities of ethyl alcohol and acetic acid were heated together, the esterification stopped when about 2/3 of the acid had been reacted. Similarly, when equimolal proportions of ethyl acetate and water were heated together, hydrolysis of the ester stopped when approximately l/3 of the ester was hydrolyzed. By varying the molal ratios of alcohol to acid, yields of ester above 66j6 were obtained by displacement of the equilibrium. The results of these tests were reported to be in accordance with the mass—action law:
(ester)(water)* (acid)(alcohol)
The accuracy of these early studies were questionedby Poznanskl
(ll) Poznanski, S., "Equilibrium of the Esterification Reaction in the Liquid Phase", Rocznlkl Chera. , 377(1928). C. A., 23, 1559.

-20-who pointed out that the equilibrium "constant" for the reaction of ethyl alcohol and acetic acid varied from 1,0 to 6 .8 .
Swletoslawaki
(12) Swletoslawaki, W. J., "Determining the Equilibrium Constant of Esterification", J. Phys. Chen., XL, 701(1933).
used an ebullioscopic method to study this reaction and confirmed the effect of proportions of reactants on the equilibrium constant for the system.
The reported results are:AlcoholsAcid 3:1 1:1 1:3K (Poznanski) 2.45 3.79 4.73K(Swletoslawski) 2.47 3.82 4.74
Corla
(13) Coria, P. E., "Equilibrium Constant of the System Ethanol-Acetic Acid," Rev. facultad Cienc. quim. (Univ. nacl. Laplata), 10, 67, (1935); C. A., 7427.
studied this reaction and obtained the relation:K • 1.31 + 4-*86 C, where C is the ratio of number of moles of acetic acid to the total moles of acid and ethyl alcohol in the starting mixture.
Similar variation in the calculated equilibrium

-21-constant with proportions of reactants have been reported)
(14-) Leyes, C. E., and Othmer, D. F. , "E sterif Ication of Butyl Alcohol and Acetic Acid,” Ind. Eng. Chem., 37,968 (1945).
with n—butyl alcohol and acetic acid.The catalyst may also affect the value of the equili
brium constant. With hydrogen chloride, It was found that the equilibrium constant for the esterification of ethyl alcohol and acetic acid increases in an approximately linear manner with increase in the initial catalyst concentration.
(15) Durruty, C. A., "The Catalytic Action of Hydrogen Chloride on the System Acetic Acid Ethanol, Ethyl Acetate, Water," Anales. Asoc. quim. Argentina, JL2, 227 (1931);C. A., 2£, 3721.
Using perchloric acid as a catalyst for the same reaction, a linear relation between the equilibrium constant and the mole per cent of catalyst is reported.
(16) Trimble, H. M., and Richardson, E. L., J. Am. Chem.S oc • , £ 2 , 1018 (1940). "Equilibrium in an EsterificationReaction with Perchloric Acid as Catalyst".
Temperature and the presence of salts may also have

-22-an influence on the equilibrium constant.
Poznanski
Loc, cit.
hat studied the effect of water on the equilibrium constant for the reaction of 1 mole of ethyl alcohol, 1 mole of acetic acid and 23 moles of water; he obtained a value of 3.56 for this mixture, compared with 3.79 for the reaction with anhydrous materials.
The numerical value of the equilibrium constant is dependent upon the particular acid and alcohol involved in the esterification and must be determined experimentally. Using the esterification data for acetic acid and various alcohols at 155°C. values for the respective equilibrium constants have been calculated and vary between 5.24 for methyl acetate and 1.0 for allyl carbinol acetate (3-butenyl acetate) for various primary and secondary alcohols.
(17) Reid, E. E., "Esterification", in Groggins1 "Unit Processes in Organic Chemistry Synthesis", 3rd. ed. McGrav Hill, Hew York, 1947.
With tertiary alcohols, the equilibrium constants are much lower and range from 0.0192 for thymyl acetate to 0.000070 for dimethyl propyl carbinol acetate (1,1—

dimethyl butyl acetate).Similar studies by Menschutkin using isobutyl al
cohol and various acids at 155°C. give equilibrium constants varying from 3*22 for isobutyl formate to 8.63 for isobutyl—clnnamate, the general increase being parallel to the increase in molecular weight of the acid. With aromatic acids, equilibrium constants of 7.0 and 10.62 were calculated for isobutyl benzoate and p—toluate respectively.
In a technical process, the numerical value of the equilibrium constant is very Important as it determines the yield of ester attainable at equilibrium and also indicates what the effect of increasing the molar ratio of reactants will have on the yield. For a general esteri— fication reaction, assuming equilibrium constant does not vary with molar proportions of reactants and temperature, a general equatloh may be written relating the yield and equilibrium constant. With no water or ester present at the start, for every mole of these products formed,1 mole of acid and 1 mole of alcohol must have reacted.In the general case, with n moles of alcohol per mole of acid (or vice versa) these concentration terms may be substituted in the equation for the equilibrium constant:

where x is moles of water or ester formed at equilibrium.In these idealized calculations, assuming K to be
Independent of temperature and feed composition the acid or alcohol may be taken in excess with no difference in the yield of ester. However, in the esterification of ethyl alcohol and acetic acid there was found a distinct advantage in employing an excess of the acid rather than of the alcohol, as the numerical value of the equilibrium constant is Increased by using an excess of acid, and decreased with excess of alcohol. Thus for ethyl acetate with 3 moles of alcohol per mole of acetic acid the equilibrium constant is 2 .4,5, which is equivalent to B6% yield of ester. With 3 moles of acetic acid per mole of alcohol, the equilibrium constant is 4-#73 and the corresponding yield of ester is 92%m Industrially the relative costs of the reactants and ease of recovery are factors that must be considered along with the equili brium constants in determining which reactant should be used in excess*
In general, esters having equilibrium constants below unity are not prepared by direct interaction of alcohol and acid; in these cases the acid anhydrides or acid chlorides are used. Reactions with latter agents do not involve reversible equilibriums and hence will give high yields of esters.
Another important use of the equilibrium constant

-2 5-is to determine, in a given mixture of alcohol, ester, acid and water, whether eaterification or hydrolysis will take place.
At equilibrium the rate of esterification and hydrolysis are equal and opposite. When the value calculated for an apparent equilibrium constant for a given four component system is numerically less than the value for the true equilibrium cohstant, ester if ica tion is still possible and is occurring at a more rapid rate than hydrolysis. If the numerical value is greater than the actual value of the equilibrium constant, then hydrolysis must predominate.
Catalytic Vapor lhase ^sterification: - The catalytic esterification of alcohols and acid in the vapor phase has received considerable attention because the conversions obtained are generally higher thin in the corresponding liquid phase reactions. However, there have been few investigations made to obtain engineering data for the rea c ti on. W o e ommerc ia1 a ppllcations of vapor pha se esteri- fication methods have been developed.
fclsterification of ethanol and acetic acid has been the subject of a number of investigations. Over the temperature range of 40-300°C. the values of equilibrium constant range from 6 to 559 (Table I) with 71—95$ ester as the equilibrium concentration from an equimolar mixture of ethyl alcohol, and acetic acid, depending upon the technique used.
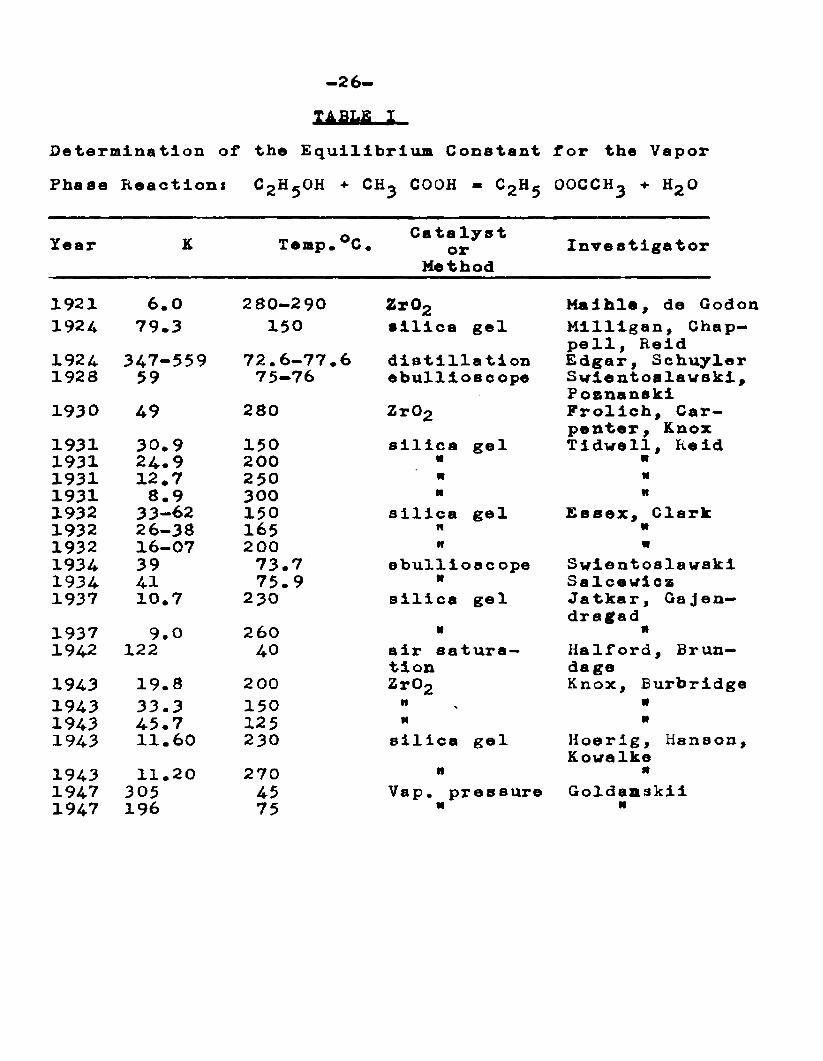
XABhfi 1Determination of the Equilibrium Constant for the VaporPhase Reaction* CgHjOH + CH^ COOH - C2H5 OOCCH^ + H2Q
Year K Temp.°C. Catelyst or
MethodInvestigator
1921 6.0 280-290 Zr°2 Maihie, de Godon1924 79.3 150 silica gel Milligan, Chap
pell, Reid1924 347-559 72.6-77.6 distillation Edgar, Schuyler1928 59 75-76 ebullioscope Swlentoslavski ,
Posnanski1930 49 280 Zr02 Frolich, Car
penter, Knox1931 30.9 150 silica gel Tidwell, Reid1931 24.9 200 « m1931 12.7 250 n N1931 8.9 300 M it1932 33-62 150 silica gel Essex, Clark1932 26-38 165 n M1932 16-07 2 00 n N1934 39 73.7 ebulliosc ope Swlentoslawski1934 41 75.9 N Salcewicz1937 10.7 230 silica gel Jatkar, Gajen—
dragad1937 9.0 260 H n1942 122 40 air satura
tionHalford, Brun- da ge
1943 19.8 2 00 Zr02 Knox, Burbridge1943 33.3 150 It a1943 45.7 125 M N1943 11.60 230 silica gel Hoerig, Hanson,
Kowalke1943 11.20 270 n it1947 305 45 Vap. pressure Goldaaskii1947 196 75 « M

-27-(18) Mai hie, A . , and de Godon, F., "Preparation of Esters by Catalysis," Bull. Soc. Chem., 101 (1921).(19) Milligan, C. II., Chappell, J. T., and Reid, E. E., "Esterification in the Presence of Silica Gel", J. Phys. Chem., 2£, 872 (1924).(20) Edgar, G. , and Schuyler, W. II., "Esterification Equilibrium in the Gaseous Phase", J. Am. Chem. Soc.,A£, 64 (1924).(21) Svletoslawski, irf., and Poznanski, S., "The Equilibrium Constant of Esterification Reaction in the Gaseous Phase", Roezniki Chem., £, 527-41 (541 French) (1928).(22) Frolich, K., Carpenter, G. B., and Knox, W. J., Jr., "Vapor Phase Esterification of Acetic Acid by Ethyl Alcohol", J. Am. Chen, ^oc., 1565-70 (1930).(23) Tidwell, II. C., and Reid, E. E., "Vapor Phase Esterification in the Presence of Silica Gel", J. Am. Chem. Soc., 4353 (1931).(24) Essex, H., and Clark, J. D . , "Free Energy ofFormation of Lthyl Acetate—Equilibrium in the Gaseous Phase", J. Am. Chen. r,oc, , 5A r 1290—1306 (1932).(25) Swietosl awski, W . , and Galcewiez, J., "The Equilibrium Constant of Esterification in the Gaseous, Coexisting with the Liquid Phase", Conpt. rend., 199. 1308—10(1934).(26) Jatkar, 3. L. K., and Gajendragad,, K. G., "VaporPhase Esterification Equilibrium", J. a m. Chem. Joe.,£2, 798 (1937).

-28-(27) Halford, J. 0. , and Brundage, P., "Vapor Phase Esterif ication Equilibrium", J# Am, Chen* Soc • }36-40 (1942).(28) Knox, W. J., Jr., and Burbrldge, T. N. ,"Vapor Phase Esterification over Zlrconclum Oxide*, J. Am. Chem. Soc., ££, 999-1001 (1943)*(29) Hoerig, H. F,, Hansoa, P., and Kowalke, 0. L., "Vapor Phase Esterification Hates", Ind. Eng. Chem.,22, 575 (1943).(30) Goldanslcii, V. I., "Heterogeneous Catalysis in Multi Component Adsorption Layers Esterification, Equilibrium In Two—Phase Systems", J. Phys. Chem. (U.S.S.R.) 2X, 431 (1947); C. A., £1, 6801.
Tidwell and Held have pointed out that adsorption of material on the catalyst may be responsible for some of the discrepancies noted in these experiments. With fresh silica gel, a yield of 90*6% ester was obtained, but on continuous running over a 6 day period this dropped to 83.5%, at which value it remained constant for five days.
Hoerig, Hanson and Kowalke have studied the rate of esterification of acetic acid and ethyl alcohol in equimolar quantities in a dynamic system using silica gel catalyst at 150—270°C.
(29) Loc. cit. Hoerig, Hanson and Kowalke.

Experimental evidence Indicates that the rate of the vapor phase reaction Is controlled by the rate of mass transfer or diffusion through a condensed phase present In the capillaries of the catalyst rather than by actual chemical rate of esterification.
Goldanskii and Chirkov
(31) Gollanskii and Chirkov, N. M. , J. Fhys. Chem. (U.3.S.E.) g&» 1333 (1946)5 C. A., 41, 2973.
Investigated the reaction of ethyl alcohol and acetic acid vapors at 75°C. in a glass vessel at reduced pressures* Vi thout a catalyst the reaction velocity is immeasurably slow, but upon addition of hydrogen chloride (1—3656 by weight of the acetic acid) an equilibrium is reached at 92 mole 56 ester. The rate of esterification up to 40^ conversion was independent of time (indicating a aero order reaction); this was confirmed by inserting glass tubing to increase the surface area and the reaction rate was increased proportionally. The velocity was increased exponentially with increase in the ratio of total gas pressure to the pressure at which droplets appear on the walls of the reactor. The temperature coefficient of the reaction was constant and corresponded to an activation energy of 15*000 Cal. From these factors it was concluded that the reaction takes place in

-30-the liquid adsorption layer and Is an example of heterogeneous catalysis,
Vernon and Brown
(32) Vernon, A. A,, and Brown, B, M . , "Vapor—Phase Esterification of Benzole Acid with Ethyl Alcohol", Ind. Eng. Chem., 22., 534 (1940); "Vapor-Phase Esterification of Benzoic Acid with Ethyl Alcohol. Effect of Oxides on the Catalytic Activity of Silicon Carbide and Alundum", ibid., 22, 12$9 (1941).
have studied the vapor phase esterification of benzoic aeid and ethyl alcohol using a number of difficultly reducible oxides suspended on silica gel, alumina, and silicon carbide, and found that the activity of the catalyst mass varies with the carrier for the same oxide. A ratio of 5 moles of alcohol per mole of benzoic acid was used with a temperature range of 370—450°C. Data are given in Table XI for esterifications with titanium dioxide and the different carriers.
A 6 hour run made with titanium dioxide on alumina at 370°C. ahd a space velocity of 276 litres of benzoic acid vapor per hour per litre of catalyst gave 85% conversion of the acid to ester and 7% to side reactions at the start. After 4*5 hours, the side reactions were nil and the conversion to ester increased to 99%*

-31- I ABIS,.U
Esterification with Titanium Dioxide and Different Carriers
Carrier Temp.° c .
Space*Velocity
Acid to Ester, %
Acid to sii Reaction, :
Silica gel 3 9 0 2 7 8 8 4 01 3 9 85 6
7 0 8 7 1 0
Silicon Carbide 3 9 0 2 7 8 2 8 21 3 9 36 0
7 0 4 9 2
Alumina 3 7 0 2 7 0 98 01 3 5 97 0
68 88 3
* Space velocity in liners of benzoic acid vapor per hour per liter of catalyst at the reaction temperature.

at which value It remained.Most of the oxides tested indicated increasing
extent of esterification with decreased space velocity. However, titania or alumina at 370°C. and manganoua oxide on silicon carbide at 410°C. showed increasing yields of ester with increased space velocity over the range investigated (up to 360 litres of acid vapor per hour per litre of catalyst). In general as the space velocity is decreased, a large amount of the acid was diverted to side reactions.
Spangenberg
(33) Spangenberg, J. F. , Industrie y Quimica Buenos Aires, Z, 393 (1945)j C. A., 41, 4028.
has studied the esterification of acetic acid with various alcohols in the vapor phase using several catalysts precipitated on pumice. The maximum yields with thorla and titania are given in Table III.
Buckley and Altpeter
(34) Buckley, R. A., and Altpeter, E. J., "Vapor-Phaae Catalytic Esterification Rates", Chem. Engr. Progress, 42, 243-50 (1951).
conducted Investigation of reaction rates for the vapor phase esterification of acetic acid and ethyl alcohol with a silica gel catalyst, and showed that the reaction
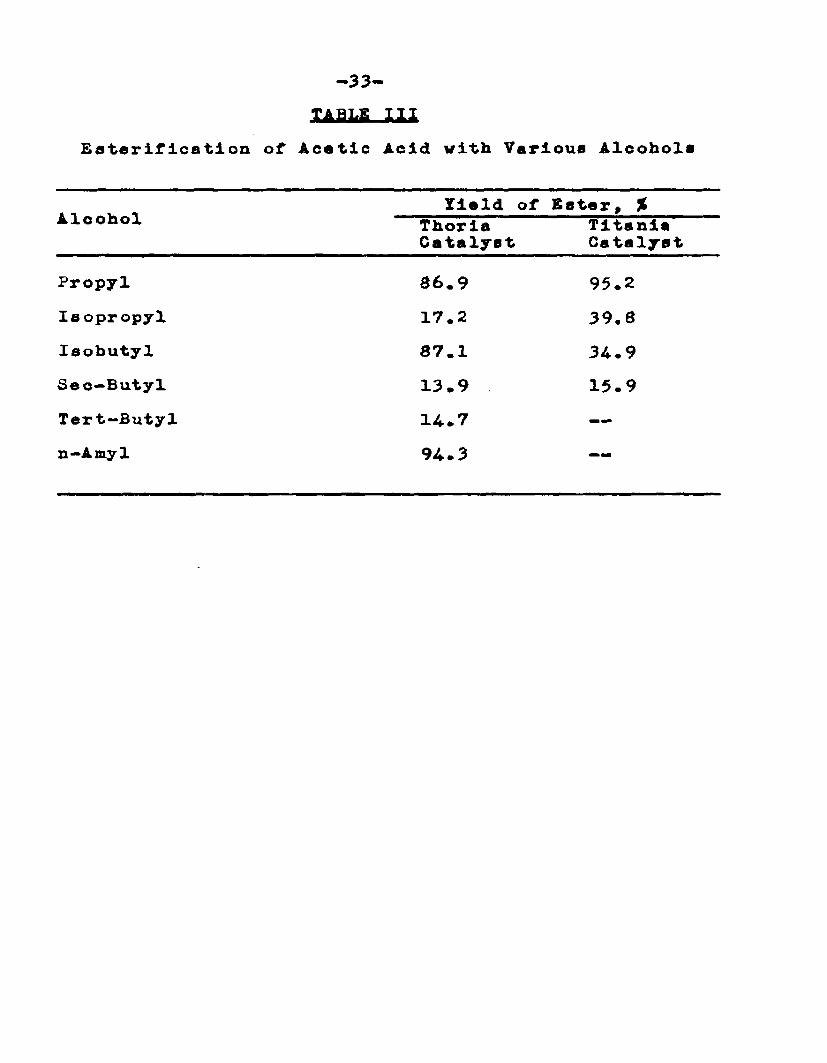
lABfcfi I HEaterification of Acetic Acid with Various Alcohols
Yield of Ester, %Alcohol Thorla Titania
Catalyst Catalyst
Propyl 86.9 95.2Isopropyl 17.2 39,8Iaobutyl 87.1 34-.9Seo—Butyl 13.9 15.9

rate was limited by the rate at which acetic acid was activatedly adsorbed where the mole fraction of acetic acid in the feed was less than 0.7 at temperatures be— tween 200—260°C. and under total pressure of 1 to 2,33 atm. The rate of adsorption of acetic acid was found to be dependent on the water content of the silica gel. A reaction rate equation was established based on a function having the form of an adsorption isotherm relating the partial pressure of water vapor in the reacting stream to the total number of active sites available for reaction, The integrated rate equation correlated data taken over a wide range of feed compositions, at 3 temps and under 3 pressures. The apparent overall reaction velocity constant formed from the product of effectiveness factor, reaction velocity constant for the activated a sorption'of acetic acid, and an equilibrium constant for the effect of water on the number of active sites, was found to be an approximately linear function of the reciprocal absolute temperature,
Clyde Reeder, Jr. (a)
(35) Reeder, Clyde Jr., "Studies of vapor Phase Catalytic Esterification Using Acetic Acid". Ph.D. Dissertation The Ohio State University, 1951*
found that low iron, low silica bauxite is an excellent

- 35-catalyst for "the vapor phase esterification of n—octylalcohol and acetic acid. The equilibrium constant andthe standard free energy change ware determined overthe temperature range 218*l°-309.0°C.
Temp. K. The Standard freeC. equilibrium energy change
constant cal./g. mole
218.1 16.4 1 0.6 -2,730 1 40241-1 11.7 1 0.4 -2,510 1 40283-0 6.961 0.20 - 2,140 1 40309.0 4.951 0.14 -1,850 1 40
(b) The variation of equilibrium constant, standard free .energy change, and entropy with temperature are expressed by means of the following equationsi
tog K « 1672/T -2.174 6 F ° - -7,652 + 9.949T A S ° - 7,287/T - 9.949
(c) The activated adsorption of the alcohol is the rate determining step in the vapor phase catalytic esterifl—cation using acetic acid and n-octyl alcohol in thepresence of bauxite.(d) The relation between weight of catalyst, feed rate, and conversion, at a given temperature are expressed by:
w /f -•J* r
r - E ««/*»*)l+aBKB+aRKp

where :r — reaction rate, g. moles of alcohol
converted per hour per g. of feed W * grams of catalystP » Feed rate, grams of feed/hourx * Conversion g. moles of alcohol per g. of feed
Activity of Compound A in the vapor stream k, Kg, Kp, K * Constants which vary with temperature
f ifl .taIytis .-& oa.sA l.a& a,
Catalysis: — Catalysis is a process in which the rate of a chemical reaction is influenced by a substance which itself remains unchanged chemically at the end. The power of a suitable catalyst lies not in its ability to modify the free energies of the reactants or products, but rather in its ability to change the rate at which a reaction determined by the free energy relationscan take place. For any reaction to be thermodynamicallyfeasible the change in free energy must be negative. Nevertheless, even with a negative A f the rate of transformation may be so slow as to make the reactants appear to be inert. In such cases the purpose of a catalyst is to speed the reaction, and to permit a more rapid approach to equilibrium. From a thermodynamic standpoint, therefore, catalysis introduces no complications into the energy relations of the system.

-37-Since the catalyst cannot change the F of a reac
tion neither can It modify the equilibrium constant. Consequently the forward and the reverse reactions must be affected to the same extent. The equilibrium constant is the ratio of the rates of the competing reactions, that Is, it is the velocity constant of the forward reaction divided by the velocity constant of the reverse reaction. The rate of the reactions
A B = K + S la the difference between the rates of the forward and reverse reactions.
r • kiaAaB - k2aRas
where "a" * activities, k « the velocity constant.At equilibrium when the rate is zero, this becomes
aR as/aA aB * kl/k2 “ K
Solid Catalysts: — It is believed that solid catalysts function through the occurrence of intermediate reactions on their surfaces. As a result the extent and character of the surface are of primary importance in determining catalytic effectiveness. In general it is desirable that the catalyst shall have a large surface area per unit mass or volume and that this surface be relatively accessible to the fluid—reactant mixture through inter-connected pores and openings.
It was proposed by Taylor

-38-
(36) Taylor, H. S., National Research Council "Twelfth Report of Committee on Catalysis", John Wiley & Sons, (1940).
that reactions which are catalyzed by solids actually occur on the surfaces of the solids at points of high chemical activity, which are termed active centers. On this basis the activity of a catalytic surface is proportional to the number of active centers per unit area. There is evidence that in many cases this concentration of active centers is relatively low, as Indicated by the extremely small quantities of "poisons" which are sufficient to destroy the activity of a catalyst. The exact nature of an active center and the conditions which must be fulfilled in order that a point on the surface may become an active center remains the subject of much speculation. There is evidence that the inter atomic spacing of the solid structure is important as well as its chemical constitution and lattice structure.
Adsorption: - There are two types of adsorption, one referred to as Van der Waals or physical adsorption and the other as activated adsorption or Chemisorption*
Van der Waals AdsorptionAll molecules both like and unlike are subject to
Van der Waals forces of attraction, which bring about the normal condensation of vapor at its d»w point as well as

the adsorption of gases by solids above the dew point.On a smooth surface Van der Waals adsorption is restricted to a layer of one or a few molecules in thickness. However, on a solid possessing a minute capillary structure surface adsorption is supplemented by capillary condensation which is also brought about by the Van der Waals forces of attraction.
In Van der Waals adsorption the union between the surface of the solid and the adsorbed molecule is not permanent. Adsorbed molecules which have acquired sufficient energy to overcome the surface forces continually evaporate while other molecules are being adsorbed. When a gas is brought into contact with an adsorbent surface, adsorption takes place until the rate at which gas molecules strike the surface and are adsorbed is equal to the rate of evaporation of adsorbed molecules. No further change will then take place in the concentration of the gas in either the gaseous or adsorbed phases and a condition of dynamic equilibrium will exist. The amount of gas which is adsorbed at equilibrium always increases with Increase in partial pressure and decreases with increase in temperature.
£hsmiaorption or Activated Adsorption Chemisorption is believed to involve definite
electron bonds corresponding to the formation of a chemical compound between the adsorbate and the surface. Like

-40-a chemical reaction, chemisorptlon is a highly specific phenomenon depending upon the chemical natures of ad— sorbets and adsorbent.
It is believed that activated adsorption occurs only on specific active centers which may represent only a small fraction of the total surface. On this basis the maximum capacity of a surface for a specific chemi— sorption is frequently much less than the amount of ad— sorbate required to form a monomolecular layer. By Van der Waals adsorption it is possible to adsorb much larger quantities which form layers several molecules in thickness or result in capillary condensation.
Because of the larger energy changes involved in the formation of valence bonds the enthalpy changes accompanying chemlsorption are generally highly negative, in the range of —10 to —100 kilo calories per g. mole.The enthalpy change of Von der Waals adsorption is of the same order as the heat of condensation, in the range of —5 to —10 kilo calories per g* mole. It follows that the effect of temperature In diminishing the quantity of adsorbate at equilibrium Is greater for activated than for Van der Waals adsorption.
The most significant difference between Von der Waals and activated adsorption is In the rate at which equilibrium is approached. Activated adsorption, as the name implies, requires a definite energy of activation

which corresponds to relatively slow rates of adsorption. These rates are greatly affected by changes in temperature, just as is the case of any chemical reaction.
Langmuir
(37) Langmuir, Irving, "Theory of Adsorption", rhys. Lev., £, 79-80 (1915).
has suggested that the forces Involved in chemisorption are exerted over distances of the order of 2 — 3A°. Only a unimolecular layer of adsorbed gas results under these conditions. The surface is to be regarded as one of the reactants. This is the same as saying that the activated complex involves the reactants and an atom or a group of atoms of the catalyst. In other words in the reaction of vapors in the presence of a solid material which acts as a catalyst the reactants are probably absorbed on the adjacent points of the surface. Then if the molecules possess the required energy, an activated complex may be formed involving the catalyst atom or atoms. This activated complex is in equilibrium with the adjacent adsorbed molecules. The rate determining step for this process is either the activated adsorption of reactants or the decomposition of the activated complex.
In order that a reactant in the main fluid phase may be converted catalytically to a product in the main

fluid phase, it is necessary that the reactant be transferred from Its position in the fluid to the catalytic interface, be activatedly adsorbed on the surface, and undergo reaction to form the adsorbed product. The product must then be desorbed and transferred from the inter* face to a position in the fluid phase. The rate at which each of these steps occurs determines the overall rate.
According to Hougen and Watson
(38) Hougen, 0. A., and Watson, K. M., "Chemical Process Principles," Fart III, New Yorks John Wiley and ->ons, 1947.
these rate determining steps are;1. The transfer of reactant molecules and product
molecules to and from the gross exterior surface of the catalyst particle and the main body of the fluid.
2. The flow of reactant molecules and the product molecules through the pore structure of the catalyst, if reaction takes place on interior surfaces.
3. The activated adsorption of reactants and the activated desorption of products at the catalytic Interface.
4. The surface reaction of adsorbed reactants to form chemically adsorbed products.
Now consider the various factors affecting the rates

-4.3-of the four steps listed above. The movement of mole— cules to end from the catalyst surface is determined by the mass velocity of the fluid stream past the catalyst particle, the size of the particles, and the diffusional characteristics of the fluid* These rates are important in reactions having very rapid surface reaction and sorption velocities, or if flow conditions are not favorable.
Since the rate of this operation is little affected by temperature, this step is negligible at low temperatures, in determining the rate. At high temperatures, this step is not very different, while the other steps are much faster, it may become very important.
Step 2, is determined by the degree of porosity of the catalyst, the dimensions of the pores, the extent to which they are inter-connected. The size of the catalyst particles, the diffusional characteristics of the fluid, and the rate at which the reaction occurs at the interface. This step is frequently negligible for catalysts of low activity in small particles with large inter-connected pores and ceases to be a factor for non-porous catalyst in moderately large particles having large internal surfaces. However, in the general case of an active catalyst in moderately large particles having large internal surfaces with restricted capillaries it may be of great importance*

- 44-Step 3 ia determined by the character and extent
of the catalyatic surface, and by the specific activation energies required for the adsorption and desorption of each of the components of the fluid*
Step 4 is determined by the nature and extent of the catalytic surface and by the activation energies required for the reaction oh the surface.
Steps 3 and 4 are chemical phenomena usually associated with relatively large e thalpies of activation.Hence they are highly sensitive to temperature. Chemical reaction rates vary over wide ranges, so it is very unlikely that the rates of activated adsorption or desorption (step 3) and surface reaction (step 4) will be equal for any one system. For this reason it is assumed that all the chemical steps are fast in comparison to one step, which becomes the rate determining step, and the overall rate is determined by this slow step in combination with steps 1 and 2. The other chemical steps are then considered to be in a state of equilibrium.
Taylor
(36) Loc. cit.
maintains that the reactions which are catalyzed by solids take place on parts of the surface which are called active centers. There is a great deal of discussion about the

exact nature of an active center. Some investigators consider an active center to be the most unsaturated section of a crystal such as an edge or corner. Others consider the surface of the catalyst as being like a checker-board, different parts of which can be active for the adsorption of different parts of a molecule.
If the theory of active centers is accepted, and if the active centers are all considered to be alike, expressions can be derived for the rate of reactions when different steps in the process are controlling.
An equation will be derived showing the relationship between weight of catalyst, feed rate, composition of feed and conversion, for a reaction blmolecular in both directions, for two different rate controlling steps (slowest steps) (1) Activated adsorption of one of the reactants is the slowest step.
(2) Surface reaction is the slowest step.A + B R + S
Assume that a unit mass of catalyst has L molal active centers (moles/weight) on which adsorption may occur. The rate of adsorption per unit area of a component A from a fluid in contact with the surface is then proportional to Its activity a^^ in the fluid at the interface and to the concentration of vacant active centers per unit weight of catalyst.

(a) Since the adsorption of A is controlling the reaction rate, surface reaction and other adsorption steps are assumed to be in equilbrium. The rate of adsorption of component A becomes:
*AA- activity of A in the fluid at the Interface moles of A per mole of vacant active centers
* moles of vacant active centers/weight of catalyst Since chemisorption Is a reversible phenomenon,component A 1b also desorbed from the surface at a rate proportional to the concentration of adsorbed molecules on the surface. Thus
vrhen the adsorption equilibrium is reached the net rate of adsorption is aero i,e., the rate of adsorption is equal to the rate of desorption
moles of A ads orbed/(time)(weight of catalyst) adsorption velocity constant of A in reciprocaltime
(2)where
« moles of A adsorbed/moles of catalyst The net rate of adsorption is the difference betweenthe rate of adsorption and desorption:
r - kA aAi Cx - k*A CA (3)
(4 )

where ■ cdsorption equilibrium constant of A .If (3) and (5) are combined an expression is ob
tained for the net rate of adsorption where all sites are equally accessible,
r - kA (aA1 Ojl - 2*) (6 )*A
In the application of above equation all surface concentrations may be expressed in terms of the equilibrium of equation (5) with the exception of C^. This surface concentration must be arrived at from the condition of equilibrium in the surface reaction.
°R CS. . K* (7)CA CB
°A ” °H CSCB K' ( 8 )
where K* * surface equilibrium constant « £k'
When component A is in admixture with other components, B, h, and S which are also adsorbed on active centers of the same type, rate and equilibrium equationssimilar to (3) and (5) may be written for each componentThen
3 ^R aRi an< CS * Kg ag^ Cx etc. (9)Substituting for CB , Cs and Cg in (8)
C* . KR aRiclKS aSi C1 (1Q)KB aBl CX K*

at equilibrium the net rate becomes sero
£jk_£s . k < - k (11)k r .k s
£b_£s » (12)k b k * k '
Substituting In 10,CA - ■hi. r a t h ° i (13)
aBi KThe number of vacant molal active centers per unit
weight of cataljst is the total number of active centers per unit weight,less the number of molal active centers occupied by the components of the fluid mixture.
* L — (CA + Cg + CR + Cg ) (14-)Substitute in (14) the value of CA as in (13) and values of CB , Cr , Cg as in (9)
C. = L — C / aRi aSi KA . _ v v , _ v \! --- ------- + aBi Kg + aRi Kr + aSi Kg )B1 (15)
CX + BX * a k b ♦ aBi K* + • Kg) = LBi
°1 f1 + K±aB^ — “ + aBi KB + aRi Si + °Si KsJ “ L
Cx *---------- -------------- — ---- (16)i + aRi aSi KA . 0 kBi KB aEi KR aSI KSAaBiK
from equation (13)
aB±K

-49—Substitute for in equation (6)
*A
(18)
••• r * k (19)
Substitute for in (19)
1 4.aRiaSiKA aBiK +aBiKB+aRiKR +aSikS
[s. aKlaSi 7I aBik J <2°)
Equation (20) is the expression for the rate of reaction in a differential section of catalyst bed. By a consideration of this equation it is obvious that when the adsorption of a reactant, A, is controlling,
by a higher concentration of the other reactant, B, in the feed.(b) Surface Reaction Controlling
When a reaction is catalyzed by a solid it is presumed that the actual combination of the reactants occurs on the surface of the solid. Such surface reactions may take place between adsorbed molecules on adjacently situated active centers. In such a case a reaction proceeds at rates proportional to the concentrations of adjacently adsorbed reactants. Thus, if adsorbed molecule A reacts with adsorbed molecule B, the rate of the

reaction is proportional to the number of pairs of adjacently adsorbed A and B molecules per unit area of surface.
A + B ^ss R + S For a surface reaction between adsorbed A and B molecules
Loc. clt* (38) pp. 916—917. Hougen and Watson.
r - k CAB * CA°B (21)where a number of equidistant active centers surrounding each active center. If products R and S are formed, thenet rate of the forward reaction on the surface is expressed by
r . Jfe (CACB- £$£S) (22)
where K* * the equilibrium constant of the surface reaction ■ CRCs/C^Cg at equilibrium.The number of vacant molal centers per unit weight of catalyst is the total number of active centers, per unit weight, less the number of molal active centers occupied by the components of the fluid mixture:
= L — + Cg + Cg + Cg) (14)at adsorption equilibrium
°A ' *A ®A± C1 (5>At equiltdum conditions each of the adsorbate terms in(14) 1 e * g • f Cji r CB» CR and Cg may be replaced byexpressions similar to (5).

ci * L~cl(aAiKA + aBiKB + aRikB + aSiKS^ (23)
" ■■ ■■■■■— 1 ■ ■ (24-)1+aAiKA +aBiKB+aRiKR +aSikS
Substituting for the value from equation (3)
CA ------ *>1. kA..L . . ... , (25)^ +aAiKA +aBikB*aRllcB +aSilcS
Similar expressions can be written for Cg, Cg and Cg. Substituting the values of CA , Cg, Cr and Cg in equation (22)
“ L|(1+aAiKA +aBikBL ) (agjlcRL) (asjKgL)♦aBikB+aRikR +aSiKS )'* K» (l+aAiKA + ... )2J (26)
p A i kA*aBikB- aRlkK-a3iksj(1+aAikA +aBikB+aRlkR +aSiKS^
(27)
r. IcaL KAKB T aAl*aBl— “1 ^ — g f - 1 (28)
at equilibrium net rate ** O
a5^ a ^ " Kr~^-K- “ K (29)aAi aBi KftKs
*JLL. * & - .a RjL.°£J. (3 0 )kA k BK *

Substituting (30) In (28)
fcfllr KA.&R____________________(1+aAiKA +aBiKB+aRiKK +aSikS)2
Longitudinal Diffusioni - Diffusion of this typeresults from the concentration gradients established by the conversion of the reactants. Thus, products tend to diffuse back against the stream while reactants tend to diffuse forward. The relative importance of these effects depends on the magnitudes of the concentration gradients which in turn are determined by the depth of the catalyst—bed and by the velocity of the fluid stream. In a reactor operated at a fixed space-velocity the effects of longitudinal diffusion are negligible with a deep catalyst bed having a small cross-sectional area, but they may be of considerable importance in a shallow bed of large cross-section.
The general problem of longitudinal diffusion has been analyzed by Hulburt
(39) Hulburt, H. M. , Ind. Eng. Chem. , Xl, 1012 (194-4); XL, 1063 (1945).
who developed integrated expressions for simplified cases Even where these expressions are applicable their usefulness is limited by uncertainty as to the proper value

-53-of the diffusion coefficient. It is evident that the effective or apparent coefficient for diffusion in a stream flowing through a granular bed is quite different from that determined from static conditions in the absence of a granular solid.
In general, the mass velocities in commercial scale reactors with fixed catalyst beds are such that longitudinal diffusion is negligible. In the corresponding pilot—plant operations it may be a serious factor which should be carefully considered. Since the effects are difficult to analyze or to translate to large-scale operations it is desirable if possible to design the pilot plants so that they are negligible. The problem is not serious in an experimental differential reactor in which only small Incremental conversions are produced. According to Hougen and Watson
(38) Loc. cit. Hougen and Watson.
for a small experimental reactor of the Integral type where large overall conversions are produced the design may be verified by applying the following equation to a small section at the Inlet of the reactor.
-FdnA «rAfill-A A B RT(ir+PA >A )nt A Zwhere: F • feed rate mass per unit time per unit cross
sectional area.n^a moles of A per unit mass of feed

« total number of moles per unit mass of feedrA ■ rate of reaction of A, moles/Cmass of cata
lyst )(time)DAm * mean diffusion coefficient of A in the
stream at position ZS a = „(rig a-fr)
It ia first assumed that diffusion is negligible, and is calculated by omitting the last term of theabove equation. The last term is then evaluated with this value o f T h e diffusion coefficients are calculated from Gilliland's equation
T * temperature degrees Kelvin Ma ,Mg » Molecular weights of A and B
"TI" = total pressure, atmospheres VA t VB “ molecular volumes of A and B
If the last term is negligible in comparison with the second, longitudinal diffusion may be neglected. This calculation should be made for the reactant or product having the largest coefficient of diffusion.
a■ bulk density of catalyst bed
dAB * 0.0043
where:®AB “ diffusion coefficient cm^/sec

-5 5-PoroaitT of the Catalyst: - In chemical reactiona
catalyzed by solid surfaces the reaction rate per unit mass of catalyst is Influenced by the size and shape of the catalyst particle. In general an increase in gross external surface area or decrease in particle size for given surface conditions of temperature and component activities increases the rate. For a completely impervious catalyst the reaction is confined to the external surface, and the rate is hence directly proportional to the external surface area. In permeable catalysts the reaction extends to the Interior surfaces, and the gross external area is generally a negligible fraction of the total effective Interfacial area.
The availability of the interior of the pellet for catalysis depends upon size, shape and permeability of pore structure. For high effectiveness it is required that the pores and capillaries be of large and reasonably uniform c d o s s section and be inter-connected with the external surface of the pellet. The effectiveness of the interior surface also depends upon the rate and nature of the reaction.
Hougen and Watson
Loc. cit.
state that in considering the properties of a catalyst bed the external void space which surrounds the pellets

should be distinguished from the Internal voids within the particles. This distinction is established from the measurement of three densities, bulk density expressed as mass per unit volume of bed, particle density /p, mass per unit volume of particle, and solid density^,, mass per unit volume of solid free from all voids; external and internal. For many catalysts the particle density is conveniently determined by displacement of mercury while the solid density is measured by helium displacement. The external void fraction of the bed is given by the relation:
Fe - 1 - £|_
and the internal voil fraction by*
Fi - 1
Francis and Oxnard- f t
(40) Francis, A. W., and Oxnard, E. P., "An Improved Volumenometer”, Ind. Eng. Chem. Anal. Ed., 3-70 (1923)*
describe a relatively simple apparatus to measure the true volume of a porous solid; knowing the true volume the true density or particle density can be easily obtained.

— 57—
BXPgBIMENTALSouiDBflDt
a . £ .cr Sfl G r i l l e a t i o n
The equipment used for the study of the esterlfi- cation reaction consisted of the following main unite:
1. Reactor (Fig* 1)2. Feed system (Fig* 2)3. Pressure regulating system (Fig. 3)4-. Condenser5. Nitrogen purge system6* Temperature indicator
1. Reactor (Fig- 1): - The reactor was constructed from pyrex glass* It consisted of a 2*5 cm* i.d. 20 cm* long, catalyst tube, R, wound around by the preheater tube, S. The preheater tube and the catalyst tube were enclosed in a long jacket, J, wound with nichrorae wire and well lagged with asbestos* A liter flask was connected at the bottom of the jacket by means of a tapered joint. The flask containing a suitable heating fluid was heated by an electric mantle. The jacket was also heated electrically. The condensing vapors in the jacket maintained the catalyst tube at a constant temperature. These vapors were prevented from escaping by using a condenser at the top of the Jacket*
The catalyst tube was closed at the top by means of
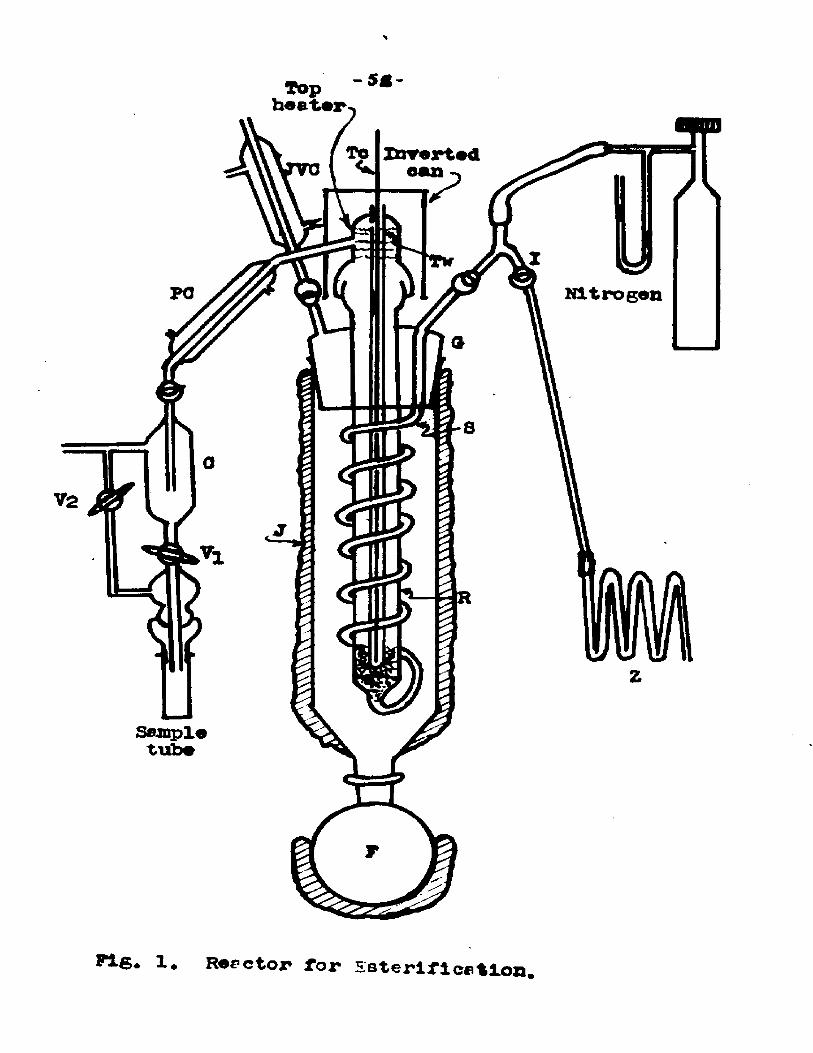
Fi6* 1* Reactor for Esterlficctlon.

2.5 cm. l.d. tube containing ball and socket joint.This lid for the catalyst tube contained a side arm, to serve as an outlet for the product, and a thermo-wellTw reaching up to the bottom of the catalyst tube.
The necks of catalyst tube and the preheater tubeswere joined to a big taper joint G, which fitted intothe female joint at the top of the jacket. The top of the catalyst tube was heated by means of an electric heating tape and an inverted can lagged with asbestos served to minimize the heat losses from the top.
The preheater tube was connected to the feed inlet tube I, containing a side arm, by means of ball and socket joints. The side arm was connected to the nitrogen supply cylinder. When the feed entered the preheater tube, it vaporized and entered the catalyst tube at the bottom and came out at the top. The feed inlet tube I, contained a small plug of glass wool which helped to make the feed continuous rather than dropwise.
The jacket heater, the mantle of the flask and the heater at the top of the catalyst tube were all individually controlled by separate variacs.
2. Fsa<j IJaiV-Cfflg«.,2) I - The feed syst em consisted of a carriage holding a hypodermic syringe L, the plunger of which was moved by a screw device M. The screw was driven by a synchronous motor K, connected to the screw through a set of gears. The plunger moved down and forced
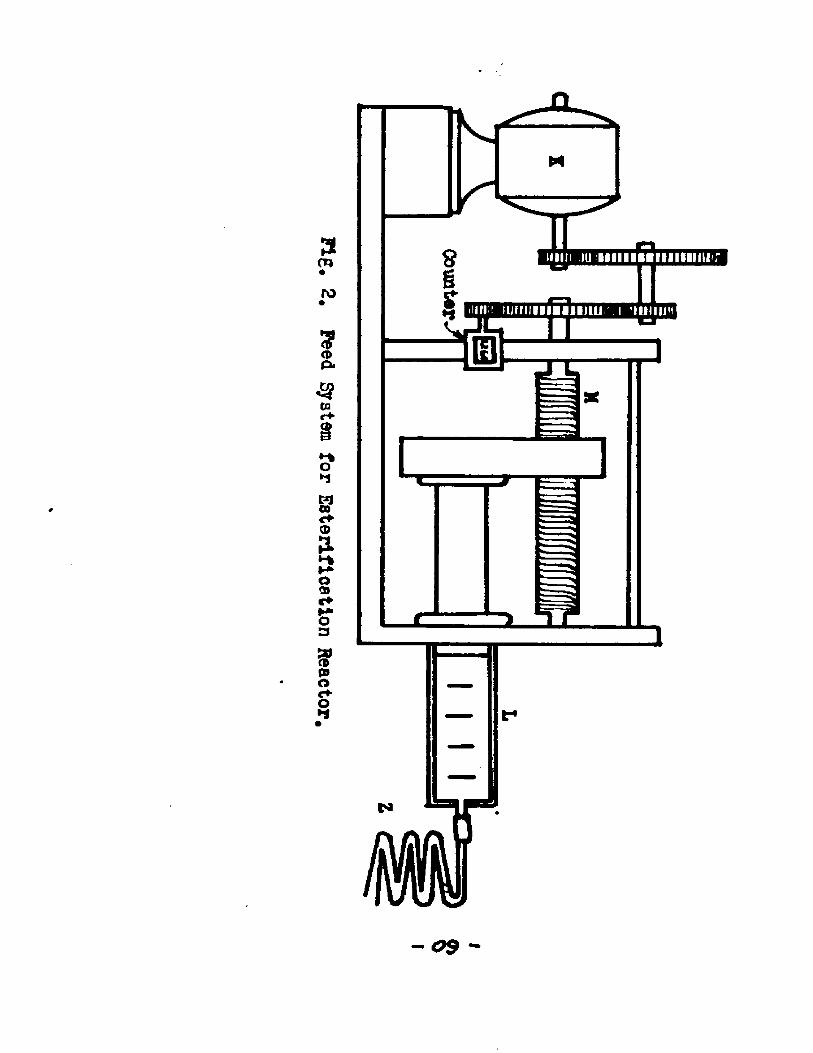
Peed System
for Esterlflcetlon
Reactor
m
to♦8§tH
turn hi iiiiiiiaitiiniiu
iin i.niiiiiaijiiiii i nmu"
-09 -

-61-the feed mixture into the reactor through a capillary tube Z, connected to the inlet tube I. The capillary tube Z was connected to the nozzle of the syringe by means of a abort piece of tygon tube* Tygon tube was found to be inert to the chemicals used and thus proved to be a convenient and flexible material to be used for joining the tubes.
The feed system was fixed in such a way that the hypodermic syringe was vertical and the plunger moved down* This position for the syringe was arrived at after encountering considerable difficulty in other positions of the syringe. When the syringe was not vertical the plunger tended to stick and thus failed to deliver a constant feed* By fixing the syringe in a vertical position and by keeping the plunger wet with the feed mixture the above difficulty was overcome*
3* Pressure regulating- unit (Fig. 3)s - When it was desired to carry on the reaction at reduced pressure of about half an atmosphere, two difficulties were encountered*(a) Delivering a constant feed into the reactor maintained at a reduced pressure and (b) maintaining the reduced pressure in the system at a fairly constant value*
(a) As soon as the reactor was evacuated the feed mixture in the syringe was sucked into the reactor, even without starting the feed system. In order to correct this situation, various methods were tried, including a

"Thennocap" Relay
Fl$. 3* Pressure Regulating System for Esterlfication Reactor.

-63-check valve. The simplest and the easiest method vras found to be the introduction of a suitable capillary tube Z, between the feed system and the reactor. The capillary tube used was such that the pressure drop across was slightly greater than the pressure drop between the syringe and the reactor. The plunger had to overcome this slight excess of pressure drop and thus the feed system always had to work against a positive pressure.
For a particular feed rate the length of the capillary required was determined by trial and error.
(b) A vacuum pump was used to evacuate the reactor.A surge bottle B, of about 5 gallon capacity was included in the system in order to minimize the fluctuations in pressure. The system was connected to a manometer P. "Thermo cap relay"was connected by means of a clip to one leg of the manometer at a position giving the desired amount of reduced pressure. An electro magnet £, was plugged into the"Thermocap relay1!. The system was also connected to a fine nozzle D, with a plug H, and to the base of a burner, serving as a coarse adjustable nozzle Q. When the pressure inside the system was lower than desired the mercury column in the leg of the manometer rose high and passed through the clip of the thermocap, and caused the relay to turn the electro magnet on. This in turn pulled back the plug H and allowed some air into the system thus raising the pressure a little. By ad—

—justing the nozzle Q the unit could be made to maintain the pressure in the system constant within one millimeter *
4. The product condenser (Fig- 3 ) : - The product outlet arm was connected to a condenser and then to a device C containing a two-way valve and a three-way valve V2 . A sample tube S, can be connected to the device by a ball and socket joint. If it is desired to take a sample when the system is under reduced pressure, sample tube S is connected with the system by means of V2 * Then the pressure in sample tube S becomes the same as in the eystem, and the product from C could be transferred into sample tube S by opening valve V^. After the sample is taken is closed and valve V2 is turned so the space between S and V2 is open to the atmosphere. Sample tube 3 can now be detached,
5. Nitrogen puree unit; - This unit consisted of a nitrogen cylinder N, to which was connected a needle valve for the release of nitrogen. A mercury manometer was included in the nitrogen line, which indicated the nitrogen pressure in the reaction,
6. Temperature Indicator; - Thi3 unit consisted of a movable thermocouple TQ which could be moved anywhere along the axis of the catalyst tube, and a thermocouple extending down the vapor condenser into the vapor— space surrounding the catalyst tube. These thermocouples were

-65-connected “to a Brown electronic temperature Indicator and the temperatures directly read on the dial.
b . f P e A e r a J L B a t i v aThe equipment used Tor determining the true density
of the catalyst is as follows:1. Volumenometer2. Helium cylinder3. Vacuum pumpU• PytooaeterThe volumenometer consisted of a sample bulb S
(Fig. 4.) made of two short pieces of 2.5 cm. i.d. pyrexglass tubing, closed at one end and joined to ball and socket joint on the other end. The two pieces could be clamped together forming an air tight bulb. The upper piece of the bulb had two side arms, one had a valve VT,in it and the other was connected to a short piece ofcapillary tube. A pipette p, of about 30 cc. capacity was joined to the end of the capillary. The lower leg of the pipette had a side arm, which was connected to a levelling bulb D, full of mercury, by means of a pressure tubing. The open end of the pipette was connected to a tube T, about two meters long by means of pressure tubing. The sample bulb S was connected to a helium cylinder, a manometer IT, and a vacuum pump. The valve W served to isolate the sample bulb from vacuum pump, manometer and helium cylinder.
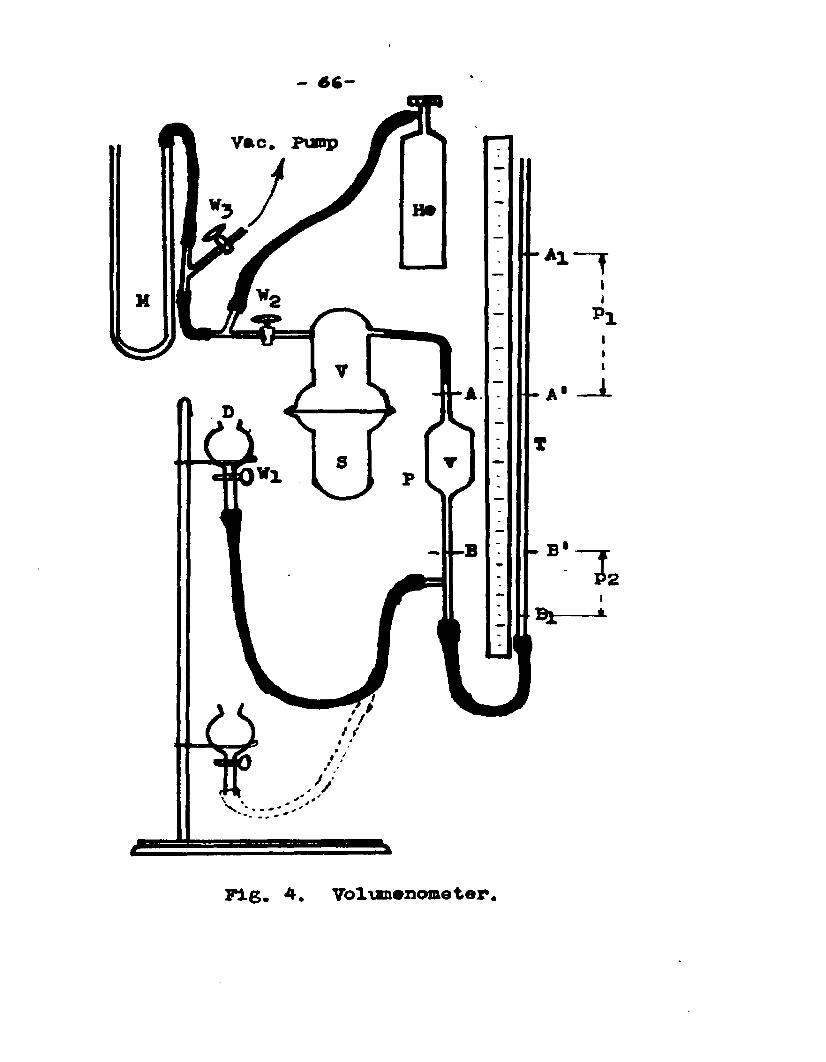
Fig. 4. Volumenometer

-67-Materials Used in the InvestigationThe following chemicals and catalysts were used in
the investigation.1. Normal octyl alcohol of molecular wt. 130.23
and with formulach3 (ch2 )6 ch2oh
was used. It was obtained from Mallinkrodt ChemicalWorks and was redistilled before being used. The initialand bottom cuts were rejected and the material distilledover between 195 and 195*4°C. was used. This materialhad a specific gravity of .8246/^Q°C- (and a refractive
4index of 1.4-27.0 to 1.4273.)
2. Acetic acid, molecular weight 60.05 having the following description was used.
Keagent Acid Acetic Glacial MerckAssay 99.5% CH3COOHFreezing point 15.8°C.
Maximum impuritiesNon volatile 0.001%Substances precipitated by water 0.000%Chloride (Cl) 0.0001%Sulfate (SO^) 0.0002,2Heavy metals(as p ^ 0.0004%Iron (Fe) 0.0002%
This product was manufactured by Merck and Co., Inc.,

68-3. The water used in the analyses was distilled*4* The standard sodium hydroxide used in the analy
tical work was standardized against potassium acid phthalate, with phenophthalein as indicator. It was prepared from a saturated solution of carbonate free sodium hydroxide. All volumes were corrected to 20°C*The base was kept in wax—lined bottles and was protected from being contaminated by atmospheric CO^*
5* The indicator used was a solution of phenol— phthalein In alcohol.
6. The nitrogen used to purge the reactor was a Linde product.
7. Helium cylinder was obtained from Ohio Chemical and Surgical Equipment Co., Madison, Wisconsin.
8 . The catalyst used in this'investigation was the product of the Attapulgus Glay Company (Porocel)210 West Washington Square, Philadelphia 5, Pennsylvania. It was 6/lA mesh low Iron, low silica bauxite having the following properties determined in the course of this investigation.
A Bulk density ■ 0.856 gr/ccParticle density = 1.56a gr/cc Solid density ** 3*52 gr/cc Color = light red
External void fraction:

-69-Internal void fraction:
1 ■ stfj4, ’ 1- * ^ - *£56
Calibration of the Feed SystemThe synchronous motor - hypodermic syringe feed
system was calibrated by weighing a rather dense, non— hygroscopic, non volatile liquid delivered from the hypodermic syringe during a measured number of counts on the counter* Ethyl phthalate of density 1.116 g/cc was considered to be a good liquid to use.
The hypodermic syringe was filled with ethyl phthalate and the synchronous motor started, using the set of gears which gave the slowest movement of the syringe plunger. A counter was used to check the constancy of the revolutions per unit time. Below are some of the data taken using a 50 cc. hypodermic syringe:
TABLE IVCalibration of Feed System
Counter Reading Wt. of Ethylphthalatecollected
Wt. of de-Ko,
from tolivered per count
123A
2540 3160 3680 42 00
31003610A130A740
2 *192 5 1.7514 1.7505 2.102 5
Ave .
3.9152x10"^ 3 *8921x10“' 3 *8900x10“' 3.902 8x10“:; 3 • 9000x10"-*
The volume delivered per count of the counter is
3'*1Q116° 3 = 3.505x10-3 cc.

The gears used in the calibration had the following numbers of teeth, starting with the gear nearest to the motor: 14* 192, 18, 192
The counter gear had 18 teeth.The volume delivered per one revolution of the
screw device may be calculated knowing the number ofteeth on the gears. The teeth of the gear of the countermesh with the teeth of the fourth gear from the motor.The latter is the gear that drives the screw. Therefore, the volume delivered per revolution of screw is:
x 3«505x10“^ * 0.0374 cc. per revolution.18The calibration of the hypodermic syringe was re
peated over the whole length of the syringe and the volume delivered per revolution was found to be constant.
From Table IV it is seen that the feed mechanism used in this investigation delivered a constant feed.Any variation in the feed rate due to changes in temperature would be exti'emely small.
To obtain faster feed rates another set of gears were used. The numbers of xeeth on these gears is as follows, the gear nearest to the motor being first.
14 , 192, 66, 144 The rate using these gears is greater than the rate with

-71-The number of counts per minute for the counter
was measured during a period of several hours. For the slow gear ratio, that Is gears with 14, 192, 18, 192 teeth, the counter was turning at the rate of 4.3*82 counts per mlhute*
In order to get a greater range in the feed rates, a hypodermic syringe of 20 cc. capacity was also used.In the case of this syringe the weight of ethyl phthalate delivered per count was found to be 1*996x10 gr*Hence the volume delivered per count is
1.996xl0“^/l.116 = 1.789x10—3 cc./count, how the calculations for the deternination of the
weight rate of feed will be shown. The density of feed containing 50 mole % n—octyl alcohol is 0.895 g/cc.For the 50 cc. syringe:3.505xlO“*3 cc/count x 43.82 cts/min. x 60 min/hr.
x 0.895 g/cc. = 8.249 g./hr.for the 20 cc. syringe It Is x 1.789 - 4.21 g./hr.
3 • 505
For the same mixture using the fast gear ratio, the feed rate for 20 cc. syringe will be
4.21 x 4.889 * 20.6 g./hr.
Operating froceflnrQA. Ssterification Keactlon: - The following se
quence of operations was carried out for each experiment on vapor-phase catalytic esterification.

72-1. The heating tape and the inverted can from the
top of the catalyst tube were removed and the thermocouple pulled out of the thermovell.
2. The inlet tube I, the jacket vapor condenser JVC, and the product condenser PC, were all disengaged from the catalyst tube R.
3. Then the catalyst tube was c arefully taken out of the jacket, the lid of the catalyst tube along with the thermowell was removed and the used catalyst from the previous experiment was dumped out of the catalyst tube.
U* The catalyst tube R, and the preheater vaporizer tube S, were cleaned with acetone and any catalyst dust in the tubes was flushed out with compressed air.
5* The desired amount of bauxite catalyst was carefully and quickly weighed on a filter paper, and was transferred into the catalyst tube, without exposing the catalyst too much, to the atmosphere. The catalyst rested on a cushion of glass wool which in turn rested on a perforated plate. A loose cushion of glass wool was inserted on top of the catalyst, leaving enough space between the glass wool and the catalyst. This served to prevent the catalyst dust and particles flying up, when being purged with nitrogen. The lid of the catalyst tube containing the thermowell was carefully placed on the catalyst tube.

6. The big taper joint G, was veil greased with high vacuum grease and the catalyst tube was inserted back into the jacket* Then the product condenser PC, the jacket vapor condenser JVC and the inlet tube I are all connected to the catalyst tube. The thermocouple was inserted in the thermowell,
7. The mantle heating the flask F, the jacket heater, and the top heater were all turned on, the top heater was kept at low voltage to start with.
8 . The amounts of octanol and acetic acid for the desired feed mixture, were calculated, and each reactant was accurately weighed separately. The weighed reactants were then carefully mixed together in a bottle,
9. Proper gears were put on the feed device,10. The hypodermic syringe and the plunger were
well cleaned with acetone and ether and dried. If the plunger were not clean it tended to stick, and interrupt the feed. The feed mixture was then pulled into the syringe. The plunger was held in position with the help of a rubber band. If the experiment was to be run at a reduced pressure, the predetermined length of capil lary tube Z, acting as resistance to the flow of feed, was connected to the syringe and the other end of the capillary was connected to the inlet tube I, The synchronous motor K was always run at 110 volts,
11. For a reduced pressure run, the vacuum pump, an

-74-the surge bottle B along with the pressure regulating device, were connected to the condensing bulb C. Valve V^ was used to make or break the connection between the evacuating unit and the reactor*
12. For the whole reactor to attain the temperature of the vapors from flask F, it took about an hour and a half. When this temperature was attained, the vacuum pump was started and the "Thermocap Relay" clip was adjusted to give the desired pressure. Then nozzle Q was adjusted so that the pressure could be maintained just by the "Thermocap Relay" turning the electromagnet off and letting air through nozzle D.
13. The feed device was started by turning the synchronous motor on, and then by opening the valve V^ the reactor was connected to the evacuating unit maintained at the desired reduced pressure.
14. As the reaction took place; the product condensed in the product condenser and then was collected in bulb C. To collect a sample a tared sample tube was attached to C by means of a ball and socket joint. Then by means of V2 the sample tube was connected with the evacuating unit and the reactor. Thus sample tube was brought down to the same pressure as the rest of the system. Then valve V^ was opened till the desired amount of the product was collected in the tube. Valve Vland then V2 were turned so that the sample tube was open

to the atmosphere. The sample tube was then taken out and weighed,
15# During the course of a run following data were recorded.
(a) Temperature of the catalyst(b) Temperature of the vapor surrounding the
reactor tube(c) Barometric pressure(d) The reduced pressure in the system.16* A run was considered complete when three
samples analyzed approximately the same. When the run was completed the feed was stopped, all the heaters were turned off, and the reactor was allowed to cool.The system was purged with nitrogen while cooling.
B. Analytical Procedure: - The reaction proceeds according to the equation
CH3 COOH + C8 H1 7 0H = CH3 COOCgH1 7 + H20
for every mole of acid reacted; there is one mole of ester and one mole of water formed. It was found that at the temperatures between 218°C* and 24-5°C., side reactions and decomposition of the ester were very negligible. 00, the analysis of the sample collected during the run gave the amount of acid used up for esterifi— cation; which was the only reaction taking place to any great extent. The amount of acetic acid in the feed was known. The amount of acetic acid present in the sample

-76-was given by tirration of the sample with standard sodium hydroxide solution. The difference gave the amount of acid reacted, from which per cent acid reacted was calculated. The procedure then was — the sample was weighedand was transferred from the sample tube into a 250 cc,beaker. The sample tube was carefully rinsed with distilled water. The contents of the beaker were diluted with distilled water, and a few drops of phenolphthalein solution were added. The whole mixture was titratedwith 0.102 IT. sodium hydroxide solution to pink endpoint.
Sample Data Sheet and Analysis Hun Ho. 4.3 Barometer m 28.730" hg.Pressure in the reactor “ Barometric pressure — 3B0 mm*Feed composition: 50 mole % octanolOctanol taken: 65*704-5 g*Mol. wt. of octanol: 130.23Hoi. wt. of acid: 60.05Acid required * 6 9 *.05 x 65.704,5 g*
130.23Correcting for the purity of acid (99*5^ pure)
Acid *= 60. 05 _ 65*704.5x100 ~ -1 3 0 . 2 3 9 9 . 5 " 3 0 - ^ s B.
Feed rate = 8.24-9 g./hr.Wt. of catalyst =* 4-.0 g.Heating liquid ■* diethylene , glycolBoiling point = 24.4 .5°C.

-77-Wt* of tube X + sample 1 14.9235Wt, of tube 1 14.4473Y/t. of sample 1 0.4762Wt, oW tube 2 + sample 2 15.8120Wt. of tube 2 15.2472Wt. of sample 2 0.5648 gTitrations with 0.102 Li. NaOHSample 1 required 16.02 c.c.Sample 2 required 13.21 c.c.Wt. of acid in the samples:
Sample 1 16.02 x (0.06005) x (.102) a .0811Sample 2 13.21 x (0.06005) x (.102) a .0981
Wt. of acid/gram of product Sample 1 QM Q&41 = .1732
Wt. of acid per gram of feed60.05/(60.05+130.23) - 0.3156
per cent conversion of the acidSample 1 (.3156 - .1732) x 100/0.3156 = 45.05&Sample 2 (.3156 - .1735) x 100/0.3156 = 45.12
Average conversion ■ 45.052C. Porosity Determination: - Porosity is fraction
of internal voids expressed as per cent of voids in a sample. In order to determine the fraction of internal voids Fi
0.4762 ~Sample 2 Q.0981 .1735
0.5648
Fi « 1

-78-Values of /*p, pellet density and solid density were needed* To find the pellet density a 25 cc, pyknometer and some mercury were used* The following data were taken to calculate the pellet density:
Wt. of pyknometer: 30.6185 g.Wt* of pyknometer and
mercury 339.3640 g.Wt. of mercury 308*7455 g.Wt. of bauxite catalyst
sample 4.7325 g.Pyknometer+mercury+cata —
lyst sample 303.1100Wt. of mercury « (303.11 - 4.7325 - 30.6185) g.
« 267.7590 g.Wt. of displaced mercury « 308.7455 — 267.7590
« 40.9865 g.Density of mercury * 13.546 g./cc.Volume of displaced mercury - Z.0- 9865 _ 3^026 cc.
13.546
Volume of catalyst sample = 3*026 cc.Wt. of catalyst sample = 4*7325 g.Pallet density f v = = 1.564. g./co/
3 • 02 ©
To find the solid density or true density, f* , of the catalyst sample, the true volume of the sample was measured by means of the volumenometer. To enable the measurement of true volume of the sample it was necessary to measure the volumes v, and V of the pipette

-79-P and sample tube S respectively (Fig, A) • To determine the volume of the pipette P between the marks A and B the amount of water filling the pipette up to mark A was weighed. From this weight was subtracted the weight of water filling the pipette up to B. Thus the weight of the water in the pipette between A and B was found to be 32.660 gr. The volume of the pipette between A and B was taken to be 32.660 cc*
The volume V of the sample tube S was found as follows:
The mercury funnel D, was adjusted so that the mercury in the pipette was somewhere between A and B. The valve W, was then closed, and the levelling bulb D was raised till the mercury in the pipette reached mark A. The level of mercury in tube T, rose to A. The height of mercury column A*A^ in tube T was measured. Then the levelling bulb D was lowered and adjusted in such a way that mercury in the pipette was at B. The level of mercury in tube T fell down to B^. The distance B'B^ was measured.
Since p*V is constantPXV = P2 (v+V) (1)
where p^ * upper reading A*A^ p2 m lower reading B'B^
when volume was V, the pressure P^ equalled Barometric pressure B plus A*A-^

—8 0—P1 * B + p^ (2)
when vo 1 lime was (v+V) the pressure equalled barometricpressure B minus B '.V P2 = B-p2Substituting the values of P^ and Pg in (l)
(3)
(B + p1 )v = (B — P2 ) (v + V) (4)Expanding
pxV - Bv - vp2 - Vp2 PlV + P2V * v(B “ P2 )= V(p! + p2 ) - v(B - p2 )V - V (B ~ B2.)
Pi + P2v > B* Pi and p2 were known and V could be solved
for.For values of* p _ and P2 average of* three readings
were taken:Average upper reading p^ = 20.9 cm.Average lower reading p2 » 8.6 cm.Barometer 749 mm.
An accurately weighed amount of bauxite catalystwas transferred into the sample tube and the above procedure was repeated in order to find the new volume ofthe sample tube. The difference between the volume ofthe sample tube and the volume of the same with known weight of catalyst in it gave the volume of the known weight of the sample. Knowing the weight and true
V 73.4 c.c.

-61-volume of the sample the true density of the sample could be calculated.
When the catalyst sample was Introduced Into the sample tube S and the procedure to obtain the new volume of the sample tube S was conducted; it was experienced that the catalyst adsorbed some of the air enclosed in the system, when the air was compressed and desorbed lot of air when the air was expanded. This lead to a much lower value for the true volume of th# catalyst sample, and consequently the value for true density came out to be as high as 9*12 gr./cc.
In order to correct the above situation helium gas was used instead of air. The helium cylinder, a vacuum pump and a mercury manometer M, were connected to the sample tube o as shown in Fig. 4. Keeping the valve W^ of the levelling bulb D closed and the valve W opened; the system was evacuated till the manometer showed a complete vacuum in the system. Then the vacuum pump was disconnected from the system by closing the valve W^jand helium was admitted into the system. The valve Wwas closed and the procedure to find the volume V ofthe sample tube was repeated in order to measure the newvolume of the sample tube. The procedure was repeated twice. For values of p^ and pg average of three readings were taken.

-82-(1) Wt. of catalyst sample ■ 25.133 gr.
Average upper reading: p1 » 18.1 cm.Average lower reading: ■ 12.3 cm*Barometric pressure « 740 mm.v = 2Z*.t>$.LZAQ-l2 2 ) » - 66.4 c.c.
181+123 304
(2) Wt. of the catalyst sample * 25.133 g.Average of upper reading p^= 8.35 cm.Average lower reeding p2 » 18.9 cm.Barometric pressure * 740 mm.
v . ?3.f6g f7$g-189) , 6 6 . 1 cc.8 3 5 + 1 8 9 2 7 2 . 5
The new volume of the sample tube was taken to be theaverage value of 66.25 c.c. The true volume of the sample373.4 - 66.25 * 7.15 c.c.The true density was 2 5.133 m 3,52 g./c.c.
7.15
F± - 1 - /%/fa
” 1 - 52 = 1 - - 0.556Fraction internal voids » 0.556 Porosity - 55.6$.

-83-
PATA.-AJiiP OfigB&VATIQflSThe data on which the correlation of results and
proposed mechanism of the reaction were based are given in Tables V and VI. ^ome comments about certain runs will be made now. Nearly half the runs were made at a reduced pressure of about half an atmosphere. The boiling point of the ester produced in the reaction is 210°C. The temperatures at which the runs were made were 218°C and 244.5°C. It was thought desirable tomake some runs in which the difference between the boiling point of ester and the temperature of the reactor was much greater. The temperature of the reactor could not be raised higher than 244.5°C. since at higher temperatures decomposition of the product took place. Hence some of the runs were made at reduced pressure. Kuna (l) to (5l) were all made for obtaining kinetic data which could be used to find the mechanism of the reaction ta king place.
huns (52) through (57) and (59) were conducted to study the effect of particle size of the catalyst on conversion. hun (58) was intended to find the effect of increasing the mass velocity on conversion.
Runs (60) through (63) were intended to give anidea of the extent to which the catalyst adsorbed eachof the components of the system — octanol, acetic acid, octyl acetate and water.
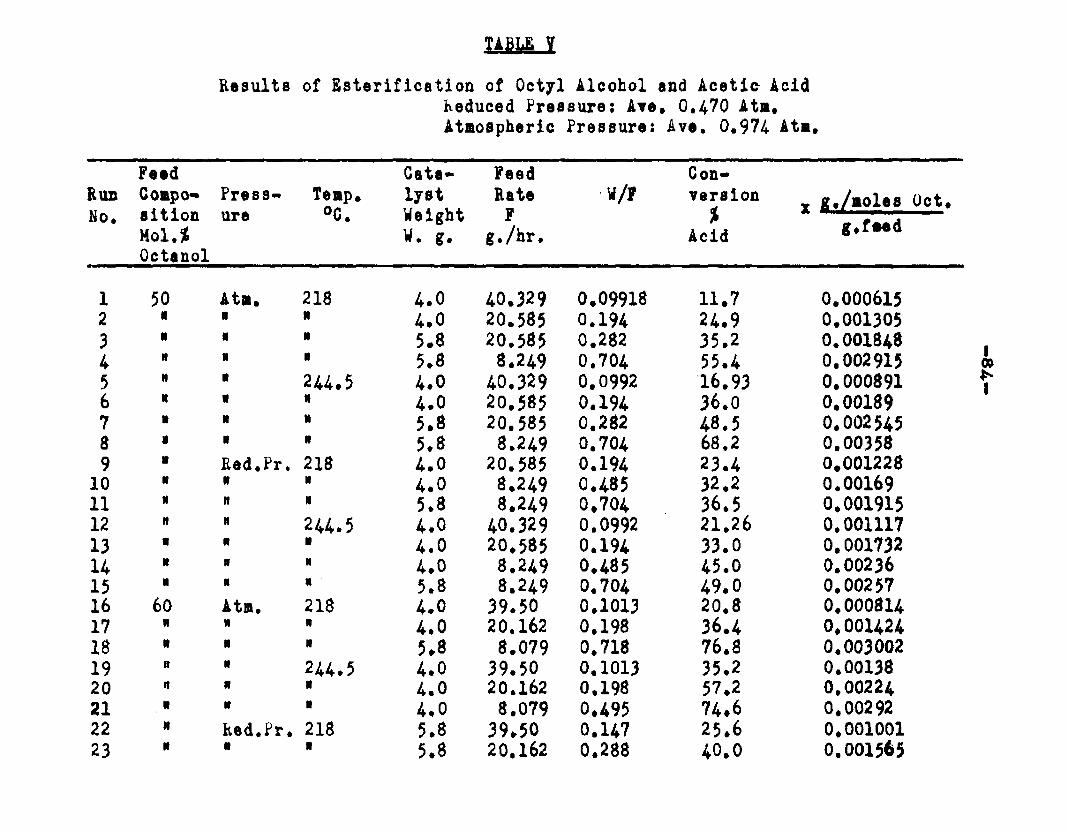
RunNo*
123456789
1011121314151617181920212223
u m i
Results of Esterification of Octyl Alcohol and Acetic AcidReduced Pressure; Are* 0.470 Atm. Atmospheric Pressure: Ave. 0.974 Atn.
Feed
siMo0c
.56anol
50
60R
RRIt
NRH
Pressure
Temp.° c .Catalyst Weight W. g.
FeedKateF
g./hr.tf/F
Conversion
56Acid
v g,/moles g.feed
Atm. 218 4.0 40.329 0.09918 11.7 0.000615R R 4.0 20.585 0.194 24.9 0.001305R R 5.8 20.585 0.282 35.2 0.001848R R 5.8 8.249 0.704 55.4 0.002915R 244.5 4.0 40.329 0.0992 16.93 0.000891R R 4.0 20.585 0.194 36.0 0.00189R R 5.8 20.585 0.282 48.5 0.002545R R 5.8 8.249 0.704 68.2 0.00358Red.Pr. 218 4.0 20.585 0.194 23.4 0.001228R R 4.0 8.249 0.485 32 .2 0.00169
R 5.8 8.249 0*704 36.5 0.001915N 244.5 4.0 40.329 0.0992 21.26 0.001117R R 4.0 20.585 0.194 33.0 0.001732R R 4.0 8.249 0.485 45.0 0.00236R 5.8 8.249 0.704 49.0 0.00257Atm. 218 4.0 39.50 0.1013 20.8 0.000814R R 4.0 20,162 0.198 36.4 0.001424H R 5.8 8.079 0.718 76.8 0.003002R 244.5 4.0 39.50 0.1013 35.2 0.00138R R 4.0 20.162 0.198 57.2 0 ,00224R R 4.0 8.079 0.495 74.6 0.00292Red.Pr. 218 5.8 39.50 0.147 25.6 0.001001R R 5.8 20,162 0.288 4 0 .0 0.001565
ICOI
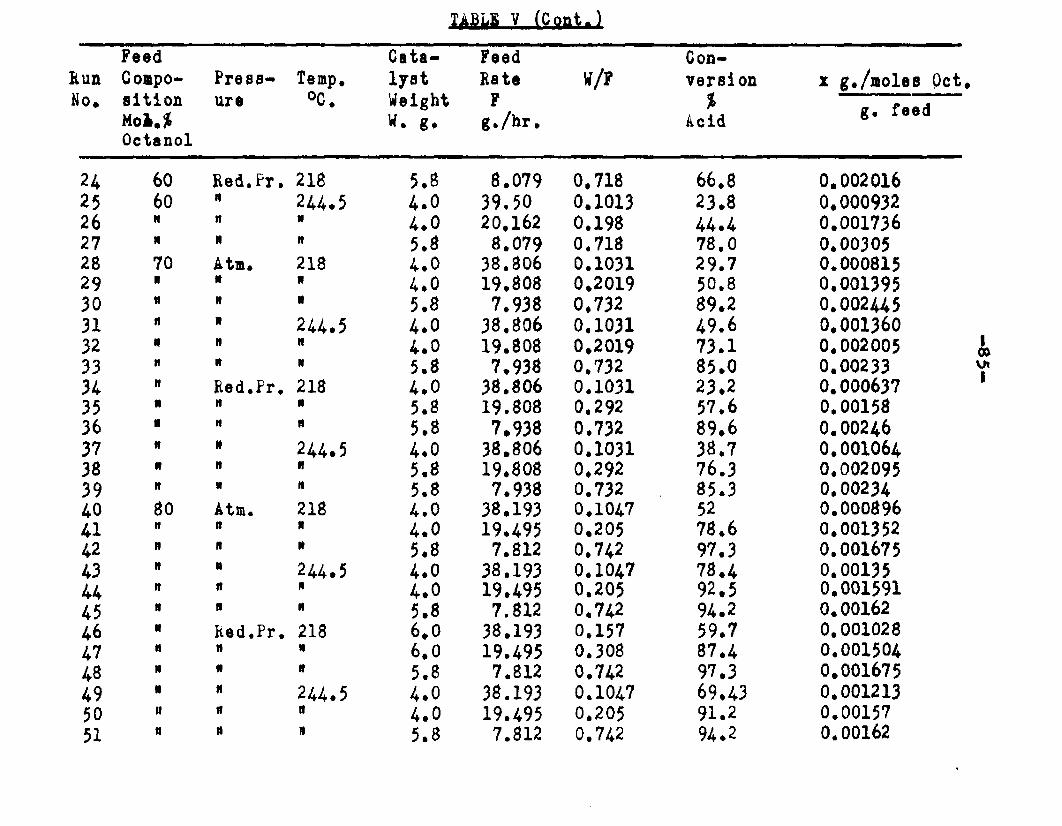
u m ,v
lunJo*
Feed Composition Mol. 4 Octanol
Pressure
Temp.° c .
Catalyst Weight W. g*
FeedRateFg./hr.
24 60 Red.Pr. 218 5.8 8.07925 60 n 244.5 4.0 39.5026 H n R 4.0 20.16227 R R R 5.8 8.07928 70 Atm. 218 4.0 38.80629 R R R 4.0 19.80830 N H R 5.8 7.93831 It R 244.5 4.0 38.80632 N R R 4.0 19.80833 R R R 5.8 7.93834 H Red.Pr. 218 4.0 38.80635 R H R 5.8 19.80836 R R H 5.8 7.93837 H R 244.5 4.0 38.80638 R R H 5.8 19.80839 R R R 5.8 7.93840 80 Atm. 218 4.0 38.19341 H R R 4.0 19.49542 R R R 5.8 7.81243 R R 244.5 4.0 38.19344 R R R 4.0 19.49545 R R R 5.8 7.81246 H Red.Pr. 218 6.0 38.19347 It H R 6,0 19.49548 R R R 5.8 7.81249 R R 244.5 4.0 38.19350 R R R 4.0 19.49551 n n n 5.8 7.812
ont>)Con-
W/F verBion x g./moles Oct,^ e, feedAcid
0.718 66.8 0.0020160.1013 23.8 0,0009320.198 44.4 0.0017360.718 78.0 0.003050.1031 29.7 0.0008150.2019 50.8 0.0013950.732 89.2 0.0024450.1031 49.6 0.0013600,2019 73.1 0.0020050.732 85.0 0.002330.1031 23.2 0.0006370.292 57.6 0.001580.732 89.6 0.002460.1031 38.7 0,0010640.292 76.3 0.0020950.732 85.3 0.002340.1047 52 0.0008960.205 78.6 0.0013520.742 97.3 0.0016750.1047 78.4 0.001350.205 92.5 0.0015910.742 94.2 0.001620.157 59.7 0.0010280.308 87.4 0.0015040.742 97.3 0.0016750.1047 69.43 0.0012130.205 91.2 0.001570.742 94.2 0.00162
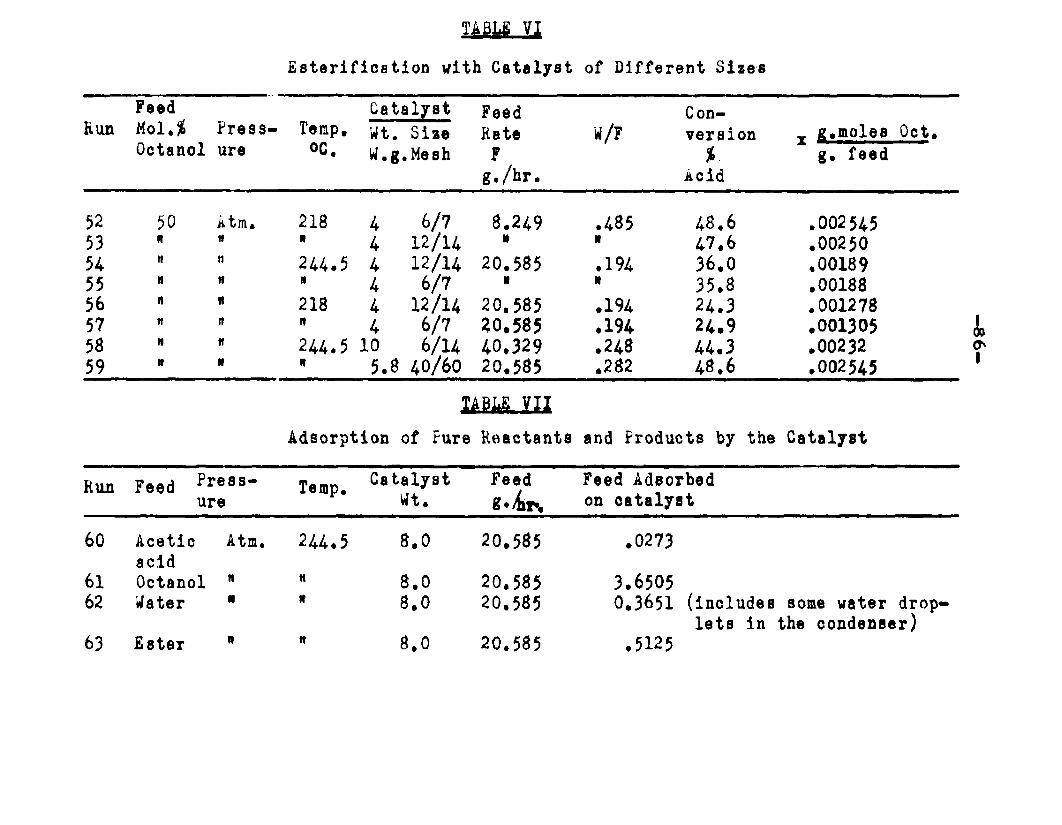
5253545556575859
Ru
606162
63
TABLE VIEsterification with Catalyst of Different Sizes
FeedMol*# Press- Octanol ure
Catalyst Temp, Size°®* W.g.Mesh
FeedRateF
g.Ar.
tf/FConversionSt.
Acid
x g.moles Oct, g. feed
50 Atm. 218 4 6 /7 8 .2 4 9 .485 48.6 .002 545It 11 ■ 4 12/ H N ■ 47.6 .00250I! » 244.5 4 12 /14 20.585 .194 36.0 .00189N n n 4 6 /7 * " 35.8 .00188It 11 218 4 12 /14 20.585 .194 24.3 .001278tt it n 4 6 /7 20.585 .194 24.9 .001305II « 244.5 10 6 /1 4 40,329 .248 44.3 .00232II H H 5.8 4 0 / 6 0 20.585 .282 4 8 .6 .002545
i a m . mAdsorption of Pure Reactants and Products by the Catalyst
Feed Pressure Temp, Catalyst Feed Feed Adsorbed
Wt. g.Ar*, on catalystAcetic Atm. 244.5acidOctanol n nWater " *Ester " ■
8 . 0
8.08.08.0
20.58520.58520.58520.585
.0273 3.65050,3651 (includes some water drop*
lets in the condenser)• 5125

-87-Palj_Qii_ the. Catalyst
Low Iron, Low silica Bauxite. 6/14 mesh Bulk density of the catalysts 0.856 gr./c.c.
* particle density or pellet density 1.564 g./c.c /* * Solid density or true density 3.52 g./e.e.
External Void Fraction
1 “ 7^ - 1 “ - .453r\p 1.564
Internal Void Fraction
1 - 4 - 1 - l.SfrA _ .558 /i 3.52
Porosity » 55.856

—88—
STA-T£M£J?T OF BESULTSThe experiments were made at two temperatures
218°C. and 244.5°C. and at two pressures, 0.974 atm. and 0.470 atm. The value x, g. moles of octanol converted per g. of feed is plotted against W/F.^"the ratio of catalyst weight to feed rate.
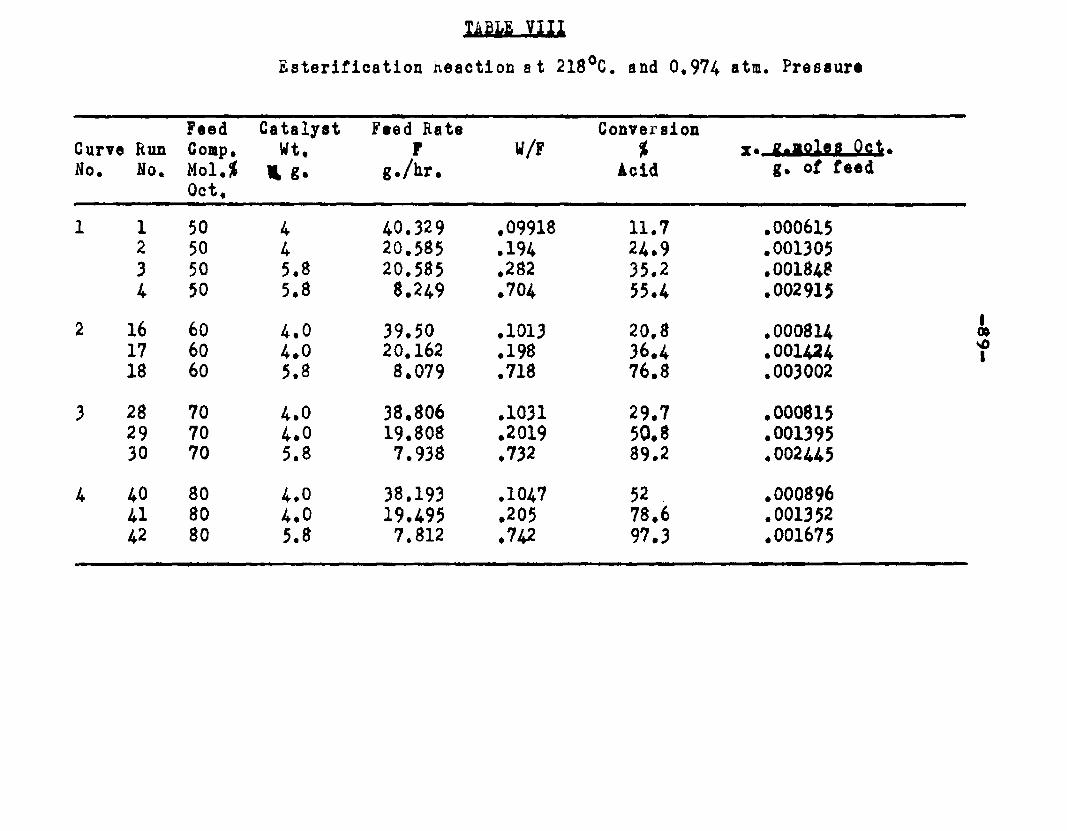
1ABMS1111Esterification neaction at 218°C. and 0.974 atm. Preaaur.
CurveNo.
i Run No.
FeedComp.Mol.*Oct.
CatalystWt,
* g.
Feed Rata Fg./hr.
W/FConversion
*Acid g. of feed
1 1 50 4 40.329 .09918 11.7 .0006152 50 4 20,585 .194 24.9 .0013053 50 5.8 20.585 .282 35.2 .0018484 50 5.8 8.249 .704 55.4 .002915
2 16 60 4,0 39.50 .1013 20,8 .00081417 60 4*0 20.162 .198 36.4 •00142418 60 5.8 8.079 .718 76.8 .003002
3 28 70 4.0 38.806 .1031 29.7 .00081529 70 4.0 19.808 .2019 50.8 .00139530 70 5.8 7.938 .732 89.2 .002445
4 40 80 4.0 38.193 .1047 52 . .00089641 80 4.0 19.495 .205 78.6 .00135242 80 5.8 7.812 .742 97.3 .001675
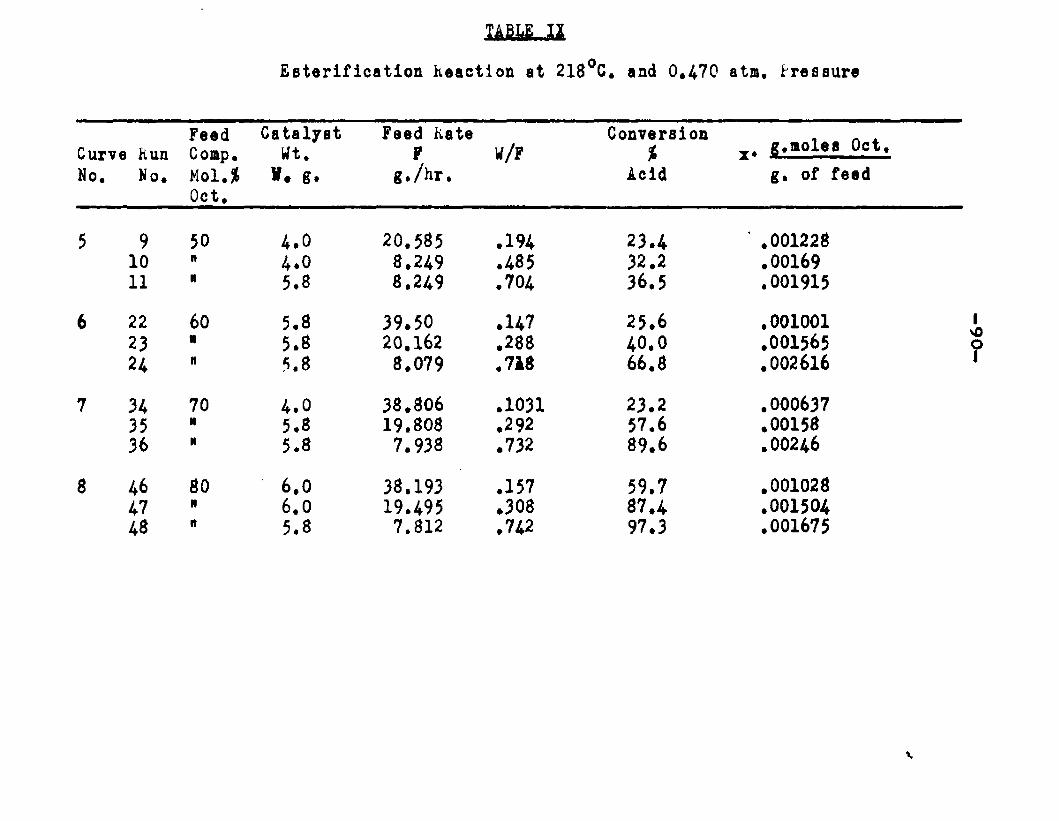
I M L U I
Esterificatioa heaction at 218°C. and 0*470 atm. Pressure
Curve ftun No. No.
FeedComp.Mol.#Oct.
CatalystWt.
V* g.
Feed F
g./hr.hate
W/FConversion
%Acid
x. g.aol<a Oct, g. of feed
5 9 50 4.0 20.585 .194 23.4 .00122810 n 4.0 8.249 .485 32.2 .0016911 N 5.8 8.249 .704 36.5 .001915
6 22 60 5.8 39.50 .147 25.6 .00100123 ■ 5.8 20.162 .288 4 0 .0 .00156524 n 5.8 8.079 .718 66.8 .002616
7 34 70 4.0 38.806 .1031 23.2 .00063735 N 5.8 19.808 .292 57.6 ,0015836 n 5.8 7.938 .732 89.6 .00246
8 46 80 6.0 38.193 .157 59.7 .00102847 N 6.0 19.495 .308 87.4 .00150448 It 5.8 7.812 .742 97.3 .001675
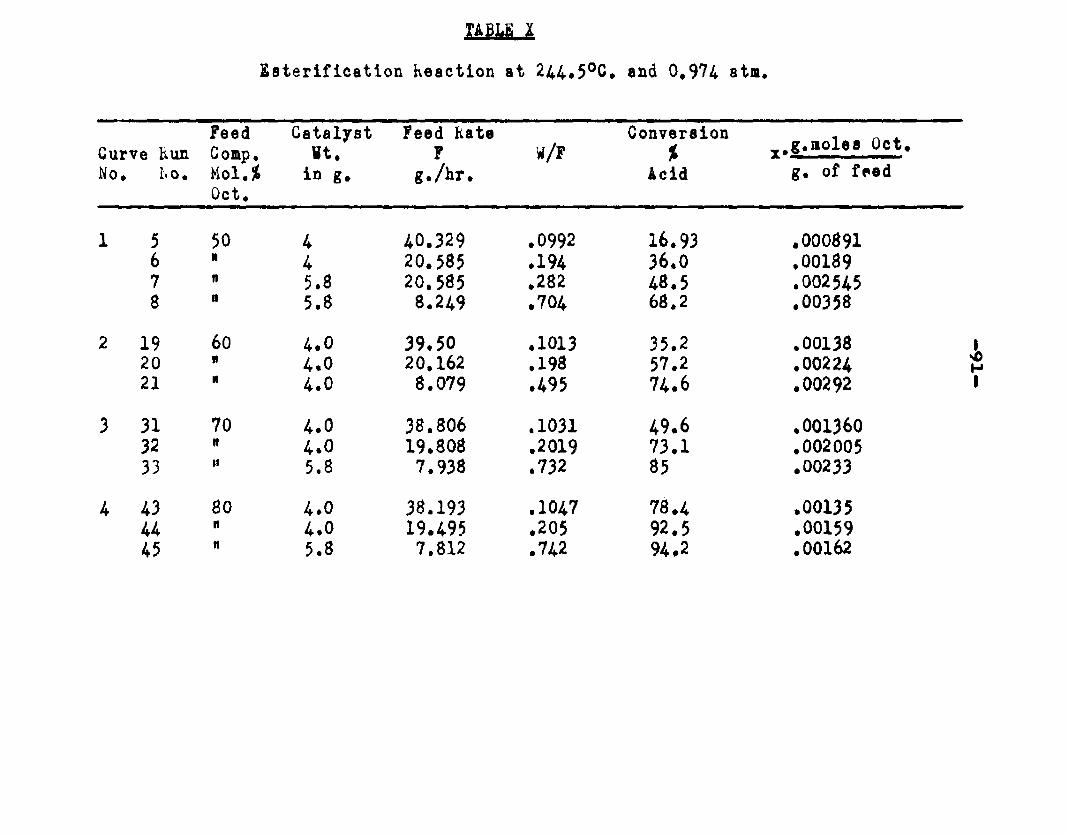
TA3M iEsterification heaction at 244,5°C,
Curve hun No* No.
Feed Comp. Mol.56 Oct,
Catalyst it. in g.
Feed hate F
g./hr.W/F
1 5 50 4 40 .329 .09926 N 4 20.585 .1947 n 5.8 20 .585 .2828 0 5 .8 8 .2 4 9 .704
2 19 60 4 .0 3 9 .5 0 .101320 R 4 .0 20 .162 .19821 N 4 .0 8 ,079 .495
3 31 70 4 .0 38 .806 .103132 n 4 .0 19 .808 .201933 i) 5.8 7 .9 3 8 .732
4 43 80 4 .0 38 .193 .104744 n 4 .0 19.495 .20545 it 5 .8 7 .812 .742
Conversion , .% ^.g.moles Oct,
Acid i• of fwd
16 .93 .0008913 6 .0 .001894 8 .5 .00254568.2 .00358
35.2 .0013857.2 .002247 4 .6 .00292
4 9 .6 .0013607 3 .1 .00200585 .00233
7 8 .4 .001359 2 .5 .0015994.2 .00162

TABLE II
Esterification heaction at 244.
Curve RunNo. No.
Feed Comp. Mol j Oct.
Catalyst Wt. in g.
Feed hate F
g./hr.W/F
5 12 50 4.0 40.329 .099213 R 4.0 20.585 .19414 It 4.0 8.249 .48515 tl 5.8 8.249 .704
6 25 60 4.0 39.50 .101326 n 4.0 20.162 .19827 R 5.8 8.079 .718
7 37 70 4.0 38.806 .103138 R 5.8 19.808 .29239 R 5.8 7.938 .732
8 49 80 4.0 38.193 .104750 R 4.0 19.495 .20551 H 5.8 7,812 .742
5°C. and 0.470 atm.
Conversion _ „ ,% x#g^moles_0ct.
Acid g. of feed
21.26 .00111733.0 .00173245.0 .0023649.0 .0025723.8 .00093244* A .00173678.0 .0030538.7 .00106476.3 .00209585.3 .0023469.43 .00121391.2 ,0015794.2 .00162
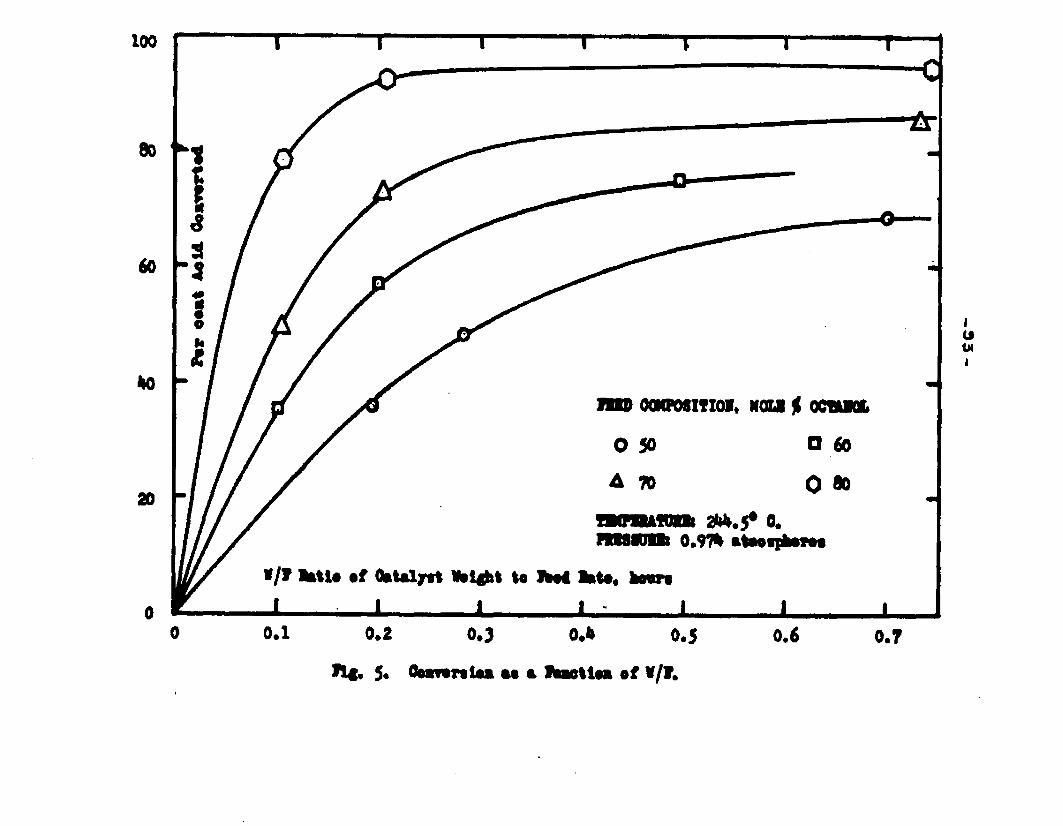
m OWFOSIVIOV, NOLI $ OCHMO 50 o 60A 70 0 90
2WK5* 0. FUSfOlU 0.97% atMipksntV/y M U if QiUlyrt *«l*ht to J M feto, hovi-I I_________I_________I *0.1 0,2 0.3 0.* 0.5
ft*. 5. OosTortlom «• o taetloa of W/F.0.6 o.r
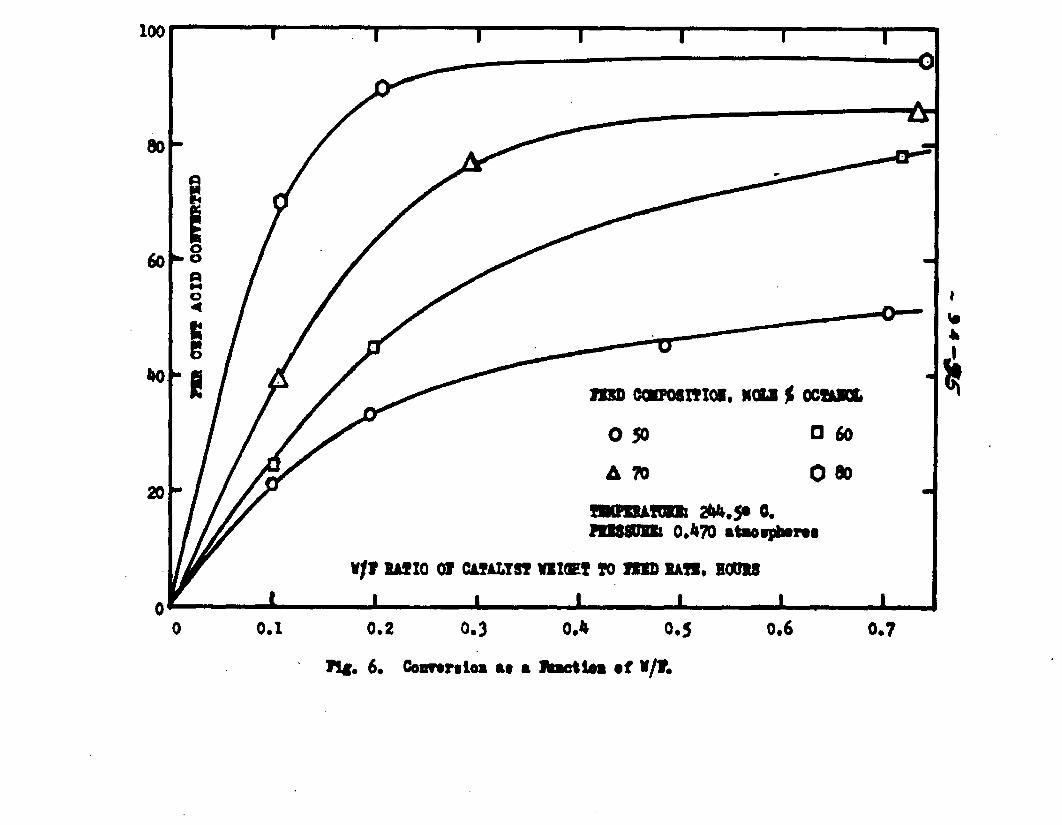
IBRD CCDOSmoi. MOU % OCn X L
O 50 □ 60A 70 o 80
TmmxvxMi 244.5a o.nSSSOSXi 0*470 ataoapterM
¥/r RATIO or CATALYST WIIOE? TO ]RO BATS, BOORS_i ________i________i i________i_______
0.2 0.3 0.4 0.5
flf. 6. Convaraloa at a Ibacttoi af V/T.0.6 0.7
H
+ €
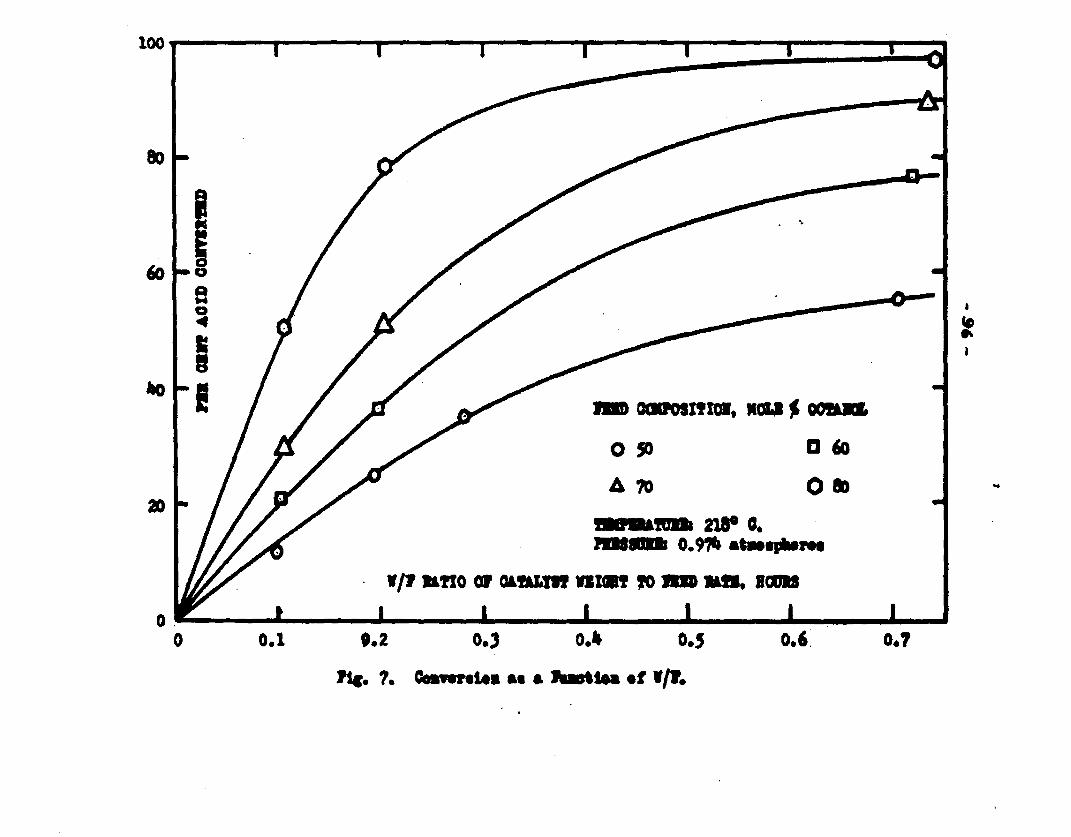
100
80
60 • o
*0
20
HID QCUOSXf I0lt NOLI $ OOfUGILo 50 □ 60A 70 0 80
tmsi 218° 0. flOSSUBl 0.97*> atSMpftwrM
v /r i& no or oitaith vxxon to m o xtn, boobs
i L i 10.1 8*2 0.3 0.* 0.5H|, 7. Co»**r«l#» m * favtUa of V/V*
0.6 0.7

1___________0 0.1
Hf.
m oaooimoi, rou $ otmn op aA 70 0
fMRBAIQUt 218° 0. nsflm 0.470 *ta»«]iNTM
v/r uTio of a m i m num to m us, 10mJ______I_______ I_____ » » 10.2 0.3 0.4 0*5 0*6 0.7(!. Coavoroioft at a Ibaetloa of V/T.
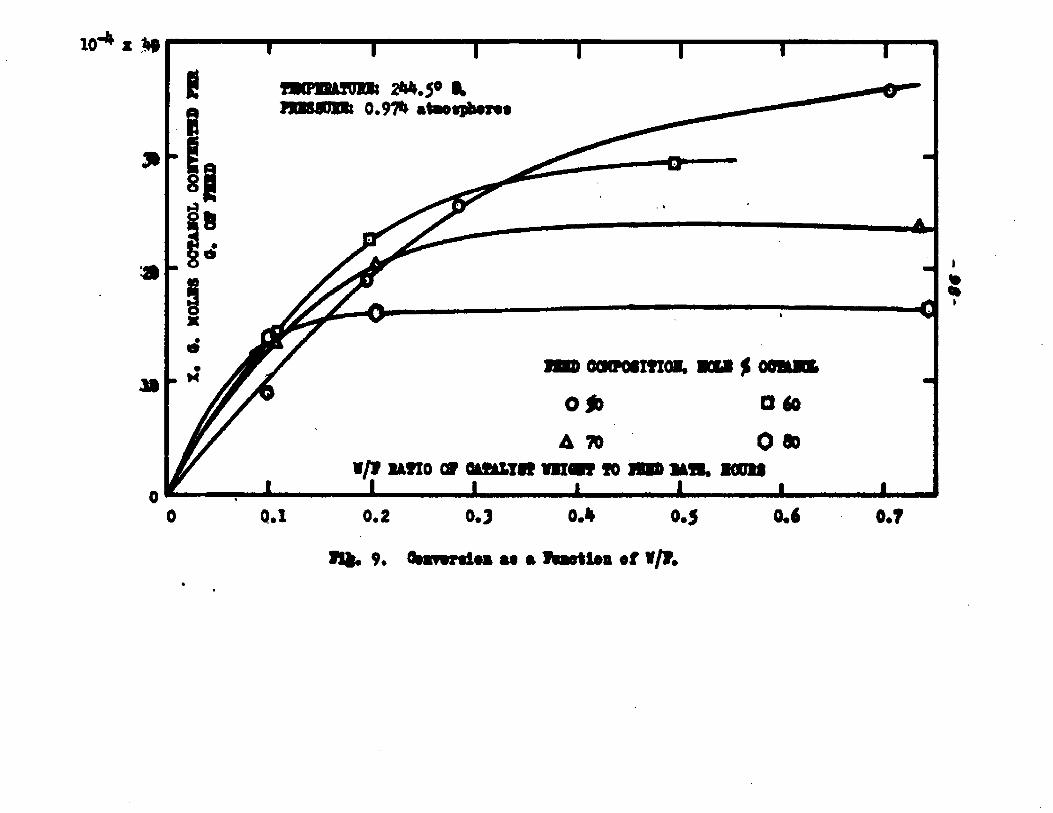
: 498
I
3» *! *S B
#a -
X
U K*
T Tm m u m 204.5° 9. m s f lo n 0.979
HD) CCMPOOlflOX. KXI £ 0 0 * »0|6 090a 70 0 a
v/v x&no ov amiitt vnan vo m o m s , warn1 JL X X X
0.1 0.2 0 .3 0.4 0.5 0.9life. 9. flbmnlii u » Mnatios of V/F.
0.7
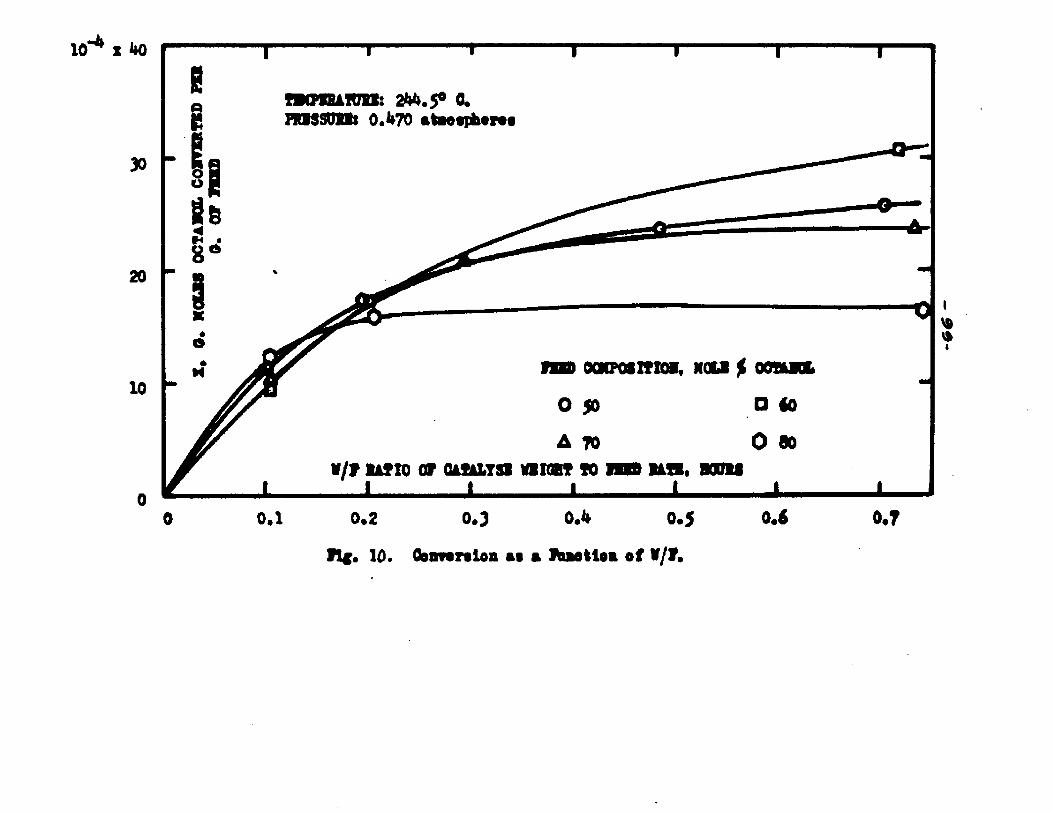
NO
US
OCT
aVQ
L CO
ITXB
TKI)
FD
l------- 1------- 1------- 1------- 1------- 1------- rm p m r n 2*4.5° c.FRISSOn: 0.470 ataoopborot
0 0,1 0.2 0.3 0.4 0*5 0.6 0.Tfix. 10. Contortion at a Itanotlom of W/I,
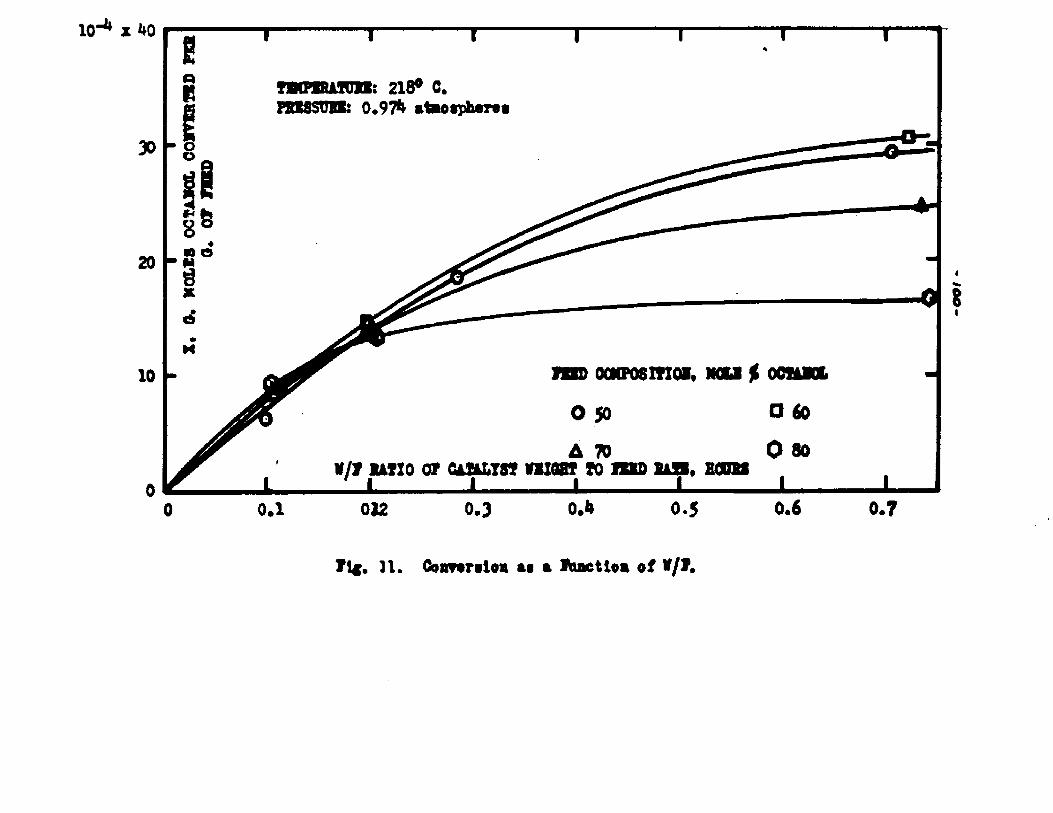
10" x 40epK
30 “
20 -
I 6n o H
10 -
TWFKUTOH: 218° c. mSSUHB: 0.97*1 ataoapharaa
IBD OCNFOSmoi, MOU i OCHMJLo 50 a toA 70
V/J IATI0 or GlStiiTS? VUOBI TO 1B0 U S , HOBBS' J________ I------------ J________ L
0.3 0.4 0.5 0.6
%
0.7
?1*. 31. Convaraloa at a Itaetloa of V/f.
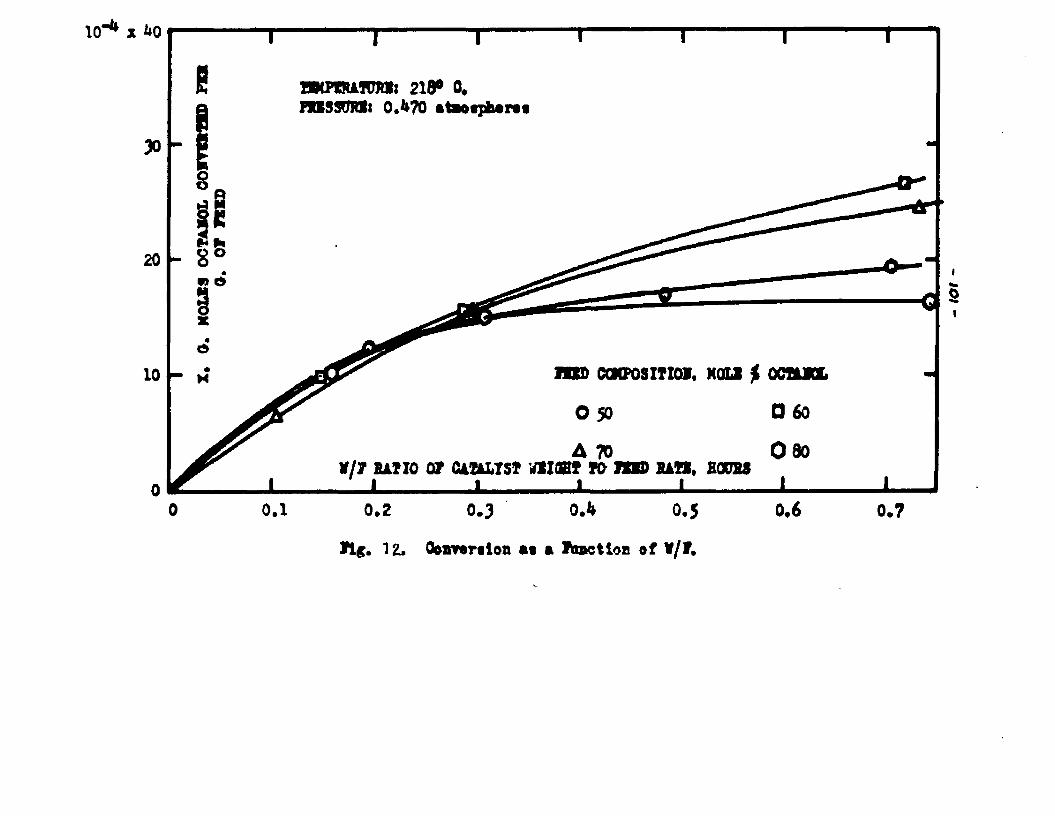
10** x fco
30
10
g
g
20 -
8
£ * 8 °
*
BHPIRA1TO*: 210» 0. FRSSSOBBt 0.470 a ta o tp h a ro s
0.1
K D COMPOS ITIOV* MC3L1 $ OCTAJOLO 50 0 60A 70 0 80
ir/7 sap io or capu ts* v tm r to mo b a h . boobs J________ I________ I________ I________ L0.2 0.3 0.4 0.5 0.6
fig. 12. Obnvartlon at a function of V/7.0.7

CQftfrgLATIOK OF SATA AliBEKO POSED MECHANISMFor a completely impervious catalyst the reaction
is confined to the external surface and the rate is hence directly proportional to the external surface area. In permeable catalysts the reaction extends to the interior surfaces, and the gross external area is generally a negligible fraction of the total effective interfacial area. This was found true with the bauxite catalyst used in this investigation. The runs (52) through (57) show that there was practically no difference in the conversion when the catalyst used had two different particle sizes 6/7 mesh and 12/14. mesh (Tylor). The surface area of 6/7 mesh catalyst is four times that of the 12/14. mesh particles, and this change in surface area did not affect the conversion. This proved that the catalyst used was porous and the external surface was but a small fraction of the total interfacial area.
To confirm the above observation run (59) was made. In this run powdered catalyst, £0/60 mesh was used.The conversion per cent acid was 4-8.6. Kun i'io. 7, under the same conditions, but using catalyst of 4./1& mesh gave the same conversion of 48.5 per cent acid.This confirmed that the conversion was completely independent of particle size of the catalyst; and that the catalyst is highly porous. In order to arrive at the high porosity of the catalyst by an Independent method,

moasuremeats of pellet density and , solid densityof the catalyst were conducted. The porosity was evaluated to be 55.85t, the fraction of internal voids being 0.558.
The possibility of diffusion being the controlling factor inthe reaction was studied by run (58), in which the mass velocity was nearly doubled. Increase in mass velocity increases the rate of diffusion. If diffusion were the slowest process in the reaction, the increased mass velocity would increase the diffusion, which in turn would lead to higher conversion. But it was found that the conversion of run (58) closely conformed to the conversions obtained at lower mass velocities as shown in the curve representing runs 5, 6, 7 and 8.This proved that the process of diffusion was not the rate controlling one in the vapor phase, esterification reaction.
The above conclusions were also arrived at independently by calculations along the lines indicated by lang and Hougen
(41) Yang, K. H,, and Hougen, 0. A., "Determination of Mechanism of Catalyzed Gaseous Reactions", Chem, Eng. Progress, No. 3. 146 (1950).
According to them, the fraction of pressure drop across

-104-tho gas film contributed by diffusion is:
r **m Pf \ f ^ /J e T ^ G \ n_1pa o' p ' b^irK C '
vbere:«p ■ surface area per single catalyst pelletr ■ reaction rate moles/(unit mass of catalyst)
(t i me )Mm ■ mean molecular weightPf ■ film pressure factor,for equimolal diffusion
Pf - 7ram ■ surface area of pellets per unit mass
* density of gas G ■ mass velocity mass/(area)(time)
D^m " Average diffusivity of component A
for values of P ^ less than 620/*■
a 3 2,44 and n = 0,51.D. oct. acetic acid was taken to be the average diffu— sivity of octanol. The results of calculations using the above equation are shown in Table III. It is seen that for runs conducted at atmospheric pressure diffusion accounts for only 2% of the p and at reduced pressure diffusion is responsible for 3% of the pressure drop. These calculations confirm that diffusion is not the rate controlling step in our reaction.
In the attempt to find the controlling step ih the esterification reaction, the effects of diffusion and

TABLE XII
Fraction of Ap Acroaa the Gas Film on the Catalyst Contributed by Diffusion
Curve Temp°c. Pr.
Atm*Feedrateg./hr.
r PAAtmG
#/hr.ft2 D°ath p/pA
la 245 Atm 40.329 .01031 .394 16.34 6.0 0*328 .0152lb n Atm 20.585 .00746 .3015 8.34 3.06 N .0201lc H Atm 8.249 .00323 .209 3.34 1.22 M .0199lb 218 Atm 20.585 .00431 .209 8.34 3.26 .302 .01716b 245 .470 20.585 .00588 .1680 8.34 3 .0 6 .68 .0307
105

-106-particle size have been eliminated from the picture, thus far. Mow the possible steps that could prove to be the controlling factors in the reaction are the followingiFor a reaction of the type:
A + B 5P=S R + S
(1) Adsorption of K controlling*___________kRLK_______________________ /aAaB - ^R
1 ’ T T k - a K ■ aA»B^RK A < as1 aAKA B B --- 5^----- SKS
(2) Adsorption of A controlling:
(»A - T l ) r « — aUa"q"RA----- B* *BKB ♦ *r kb * aSKS
(3) Surface reaction controlling:
kBLKAKB (»A«R - ^ )r . ------------------------------- K.(l+aA^A * aBKB + ahKR + aSKS^
In order to analyze the data qualitatively with respect to the above three possible mechanisms, the equations for the initial rate rQ, when the value of conversion r is zero, will be obtained.
when x = 0 a^ ■ a^ assuming the activities to be equal to partial pre s sures.

-107-(A) Adsorption of k controlling:
ro - ---------- — P*- o)A A + aB B *
ro - --------- — --------- (-•- o)1+aAKA + aBKB +*°
This shows that in this case initial rate is independent of reactant activities.(5) Adsorption of A controlling:
kAL » aA_____ m k 1 aA° l+aBKB + o — 1+aBKB
In this case the initial rate is directly proportional to the activity of A. An increase in the activity of the other component has an adverse affecton the values of r Q .(6) Surface reaction controlling:
ksu Ka Kb (a^ag ) k' aA^r° (l+aA^A + aBkB ^ ^ + aAKA + aBKB ^
In this case, the numerator contains the activities of both A and B. As the activity of A is Increased, activity of B decreases and the product a^aB decreases slowly. In the denominator the term is squared, this makes the denominator decrease faster as the activity of A Is Increased. The net result is that the value of r Q increases as the activity of A increases, but

-108-lesa than In proportion to the increased activity of A.It is possible that the values of and Kg be such that the increase in initial rate could be nearly proportional to the increased activity of A.
The following table gives the values of initial rates for rQ for different curves.
TABLS XIIIValues of initial rates ro at different values of and PgTemp. Curve
No. PA PB ro
244.5°C. 1 • 487 .487 .016692 .584 .390 .020753 .682 .292 ,024484 .778 .196 .026455 .235 .235 .011876 .282 .188 .014147 .329 .141 .015778 .376 .094 .01582
218°C. 1 .487 .487 .008862 .584 .390 .011153 .682 .292 .013784 .778 .196 .015895 .235 .235 .006636 .282 .188 .008227 .329 .141 .009628 .376 .094 .01031
Wow consider the case of adsorption of E or S i.e.,one of the products controlling. In this case as was shown previously the initial rate is independent of reactant activities. That is the values of rQ should not change with increase of p^ or decrease of pg. From the above table it is clear that values of rQ do depend on

-109-the values of and pg, Thus it is obvious that neither the adsorption of K, nor that of S is the controlling factor in the reaction.
This leaves us with two other possible mechanisms to choose from; adsorption of one of the reactants A or B controlling and surface reaction controlling.In both cases as was siiown before, initial rates rQ increase with increasing activity of A, Since asshown in Table XIII, values of r^ Increase with in—' ocrease in p^ , and decrease with increase in pg; it is again obvious that adsorption of B, i.e., acid, cannot be the controlling factor, uence it should be either adsorption of A or surface reaction that should be the slowest step in the reaction.
The detailed calculations to choose the best possible mechanism from the above two, are based on the lines followed by Hougen and Watson.
(33) Loct. cit., Chapt, XIX

-110-In a reaction bimolecular in both directions:
A + Bm b R + S
When the slowest step is the surface reaction, the mechanism of the reaction can be expressed by the equation:
_ _______ ksL KAKm___________ (aA ’aB—(l+aAKA +«b KB *aRKR +*SKsJ2

-111-let ksL * Q
aA a B - aR >aS . G K
the equation can be written ass* G
r « (l+a^K^ + afiKB + a^K. +
_______________ f ^ G ___________________1+aA KA 4 aBKB + aKKK + a3liS
rearranging *J~G 1+ aA KA + aBKB + ahKR + aSKSfr~ Q
This can be written as:
KaTt + hk aA + aR + £& an + Ks‘A B lS
a.Since equal moles of ester and water a^j taking activities to b.e equal to
ures, the above equation can be written as
£5. . - l . + Eh. PA + £fi PB ♦ LKii + KS ) pufr Q Q <4 Q
let Kp + Kc = K„
••• G L =
LR
JL_ + 2L fa + p b + p r r Q Q Q g
The above equation is of the form:R • a + b pA + o pfi + d pR
This involves four constants.
are produced partial press—

-112-Svaluatioa of Constants by Least Squares In developing the constants of an equation to re
present experimentally observed data it is desired to arrive at the most probable or "best" relationship which will represent all of the data with a minimum of average error of deviation. The principle of least squares helps the calculation of the "best" values for the constants. According to this principle, to evaluate all the four constants of the above equation, following four equations in summation form are obtained:
na + b 5 pA + c ^ p B + d £ p R » £ R (1)
a iPA + b i PA2 + c I P A ’Pb + d 2 PA*PR s J P a 'H (2)a Z pb + b lPA*PB + c Z P b2 + d 2 p b *pr =Xp b *r (3)a 2 ph + b ZPA*PR + cj.pg-p^ + d f p R 2 - I P r -R U )Where n represents the total number of readings.
In order to obtain the above equations in terms of the data collected, following procedure was adopted: Thevalues of conversion x, g. moles of octanol/g. of feed
were plotted against W/F. Thus eight curves were obtained for each of the temperatures, 244-»5°C. and 218°C. For each curve three values of x were picked and the corresponding values of , pg and pR were calculat— ed. (Refer to Appendix for sample calculation of partial pressures).
Calculation of Kates from Data:Starting from an elementary section of reactor

-113-containlng a mass of catalyst dW in which a conversion dx is produced, then
F dx = rdWwhere
F = feed rate, mass per unit timeW * mass of catalyst in reactorr » reaction rate, moles/(raass of catalyst)(time)x = conversion, moles per unit mass of feed*Integrating the above equation:
s ■ / d zr
A ItL F r
X « * K/f,r A x
from the curves of x xfi* W/F the values of x, tf/F and A ^/^X were obtained. The above values were used to Plot curves of x vs.A x that is x XSL» From these curves
the corresponding value of r for any value of x could be easily obtained. Curve 2, (60 mole % octanol feed,24-4 *5°C.) will be taken to illustrate the above calculations. Fig. 13 is the curve where x Is plotted vs.W/F*the values of x, W/F andA W/F are given on the
* A xfollowing page*
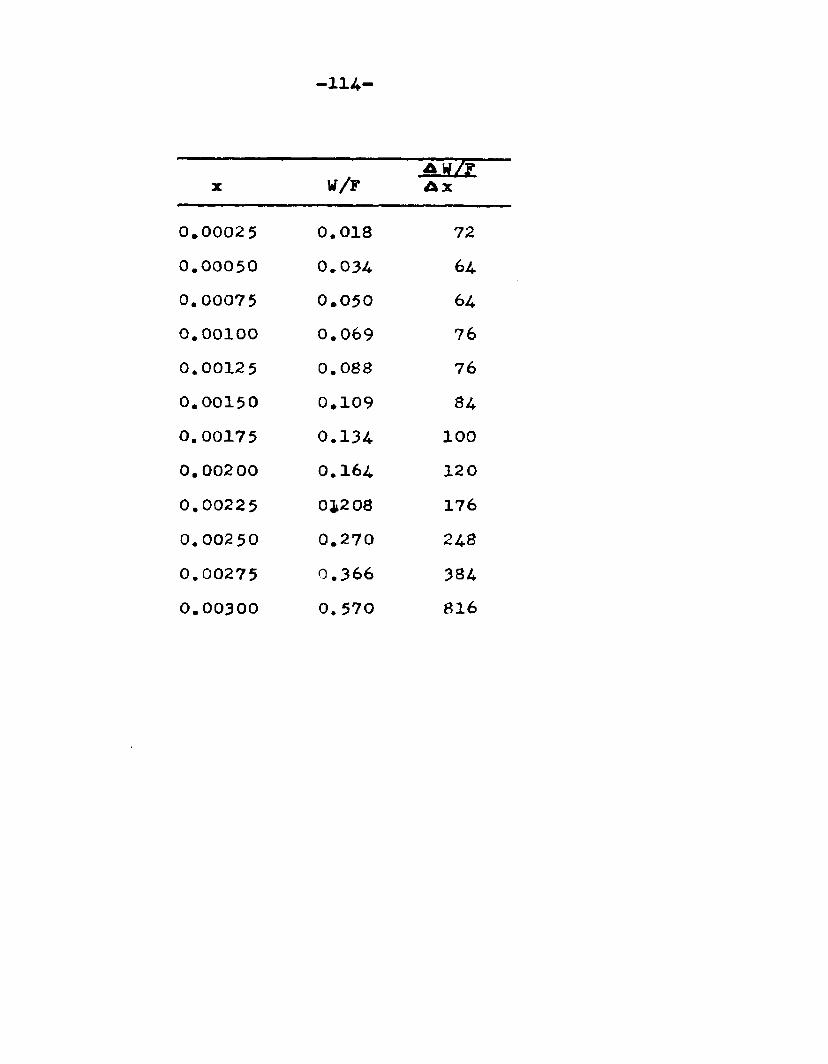
-114-
' AW/F.x tf/F A x
0*00025 0.018 720.00050 0.034 640.00075 0.050 640.00100 0.069 760.00125 0.088 760.00150 0.109 840.00175 0.134 1000.00200 0.164 1200.00225 0*2 08 1760.00250 0.270 2480.00275 0.366 3840.00300 0. 570 816

X. 0 . MOUSS OCTAIOL COIVXRHO FIRa. or r a o
OC * ^0
1

J<4
800
700
JBBDt 60 NOLI % OOZ4K3L T R Fl l l TUHJi 2W*,5° 0.nxSOTBi 0.97t f tta o a p h a f ii60 oc -
500
300
200
100
5 io .X. 0 . M0L18 OCTAFOL OOJ TBHBM) F I* O. OF R ID
t i a A u c tio n o f O o sro ro lo iu
20 30 *

-117-The values of x and^ W/F were plotted as in Fig. 14.
AxThe Y coordinate of each point was drawn and the x coordinate of each point was then drawn to meet the Y coordinate of the previous point K making a rectangle. This gave a step-wise appearance to the plot.A smooth curve was drawn through the steps in such a way that the area excluded from each rectangle was equal to the area included in the rectangle. That is the area under the curve remained the same as the area of all the rectangles. For any value of x, thissmooth curve gave the corresponding value of Aw/F
A xwhich is reciprocal of the rate.
Thus for a particular value of x correspondingvalues of r, p^ , pg and p^ can be obtained. With the
2help of these figures all the terms; , ^.Pb ’PRJ2L PgR etc., required to evaluate the constants from the simultaneous equations, could be calculated. These various terms are tabulated in Table 14, along with their summations.
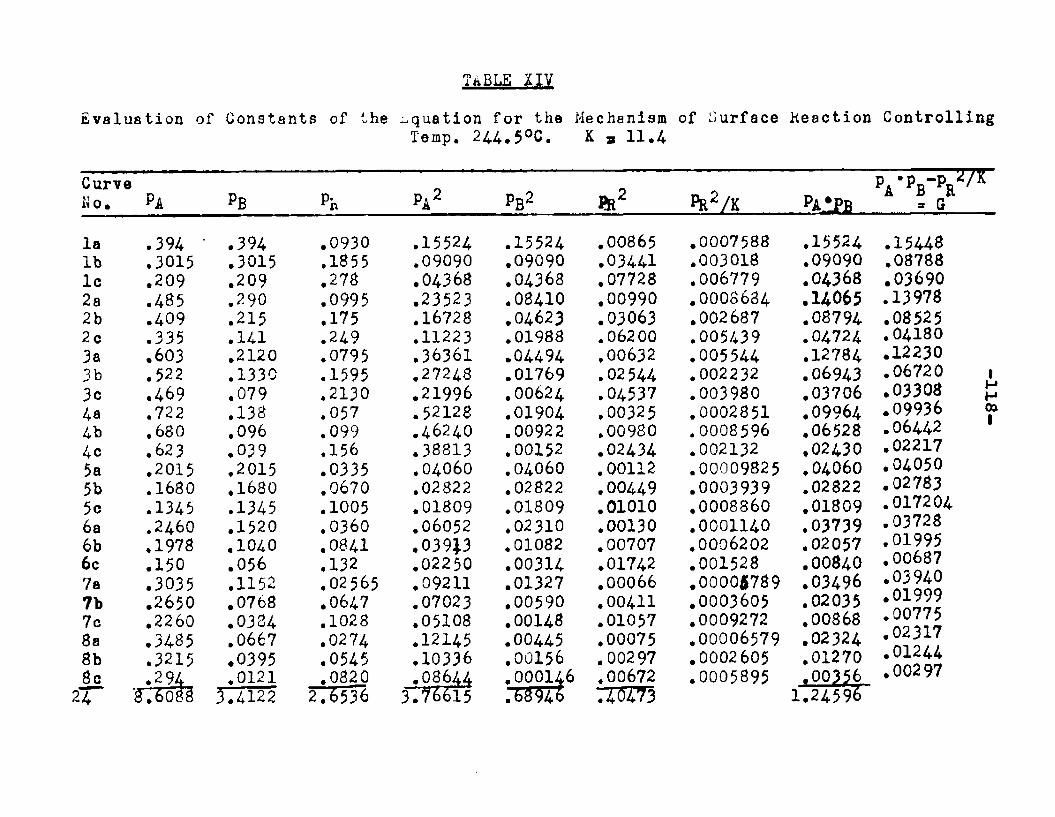
TABLE XIVEvaluation of Constants of the equation for the
Temp. 244.5°C.
CurveHo. pa PB P- pa2 PB2
la .394 ■ .394 .0930 .15524 .15524lb .3015 .3015 ,1855 .09090 .090901c .209 .209 .278 .04368 .043682a .485 .290 .0995 .23523 .084102b .409 .215 .175 .16728 .046232c .335 .141 .249 .11223 .019883a .603 .2120 .0795 .36361 .044943b .522 .1330 .1595 .27248 .017693c .469 .079 .2130 .21996 .006244a .722 .138 .057 .52128 .019044b .680 .096 .099 .46240 .009224c .623 .039 .156 .38813 .001525a .2015 .2015 .0335 .04060 .040605b .1680 .1680 .0670 .02822 .028225c .1345 .1345 .1005 .01809 .018096a .2460 .1520 .0360 .06052 .023106b .1978 .1040 .0841 • 0394.3 .010826c .150 .056 .132 .02250 .003147a .3035 .1152 .02565 .09211 .013277b .2650 .0768 .0647 .07023 .005907c .2260 .03 34 .1028 .05108 .001488a .3485 .0667 .0274 .12145 .004458b .3215 .0395 .0545 .10336 .001568c .294 .0121 .0820 .08644 .000146
24 S.608§ 3.4122" 2.6536 3.76615 .TS946
ylechanism of Burface heaction Controlling K a 11.4
Pr2/k Pa *Pb A rB "R = G
.00865
.03441
.07728
.00990
.03063
.06200
.00632
.02 544
.04537
.00325
.00980
.02434
.00112
.00449
.01010
.00130
.00707
.01742 .00066
.00411
.01057
.00075
.00297
.00672
.0007588
.003018
.006779
.0008684
.002687
.005439
.005544
.002232
.003980
.0002851
.0008596
.002132
.00009825
.0003939
.0008860
.0001140
.0006202
.001528
.00005789
.0003605
.0009272
.00006579
.0002 605
.0005895
.15524
.09090
.04368
.14065
.08794
.04724
.12784
.06943,03706.09964.06528.02430.04060.02822.01809.03739.02057.00840.03496.02035.00868.02324.01270.00356
1.24596
15448 08788 03690 13978 08525 04180 12230 06720 03 3 08 09936 06442 02217 04050 02783 017204 03728 01995 00687 03940 01999 00775 02317 01244 00297
1HHQ»I
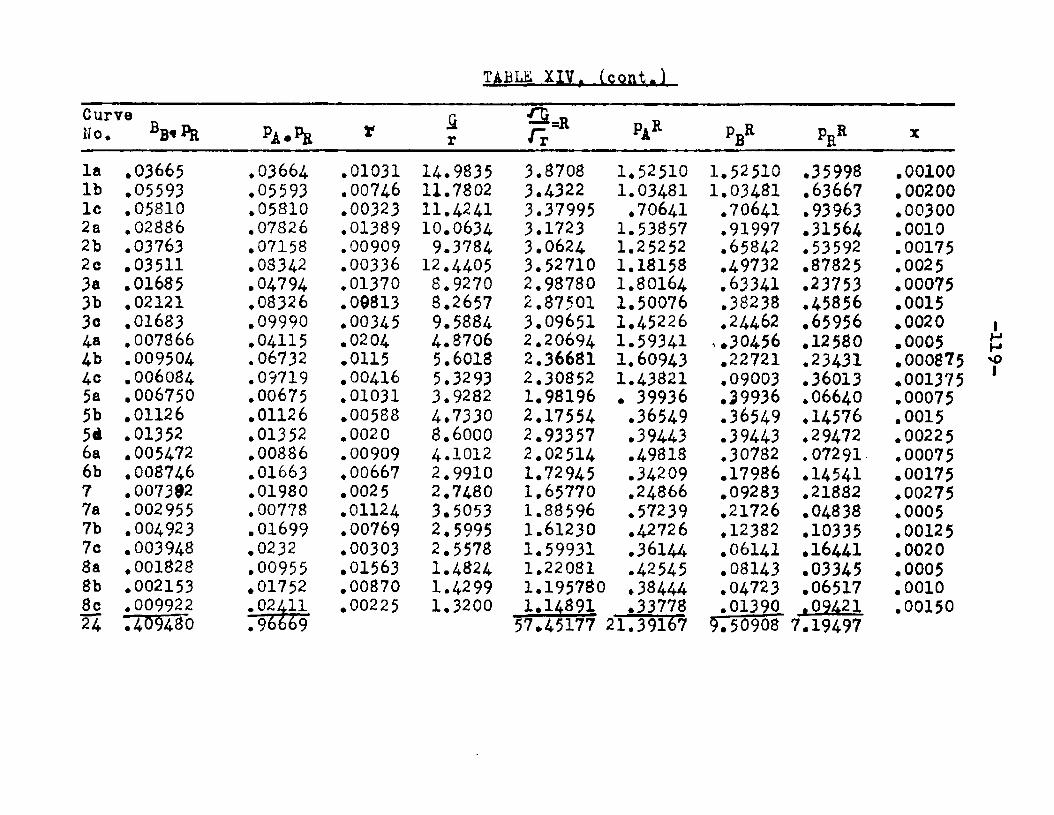
TABLE XIV. (coat.)CurveNo* BB « % pa .Pr V £r
a *• r pAR pBB pRE X
la .03665 .03664 .01031 14.9835 3.8708 1.52510 1.52510 .35998 .00100lb .05593 .05593 .00746 11.7802 3.4322 1.03481 1.03481 .63667 .00200lc .05810 .05810 .00323 11.4241 3.37995 .70641 .70641 .93963 .003002a .02866 .07826 .01389 10.0634 3.1723 1.53857 .91997 .31564 .00102b .03763 .07158 .00909 9.3784 3.0624 1.25252 .65842 .53592 .001752c .03511 .03342 .00336 12.4405 3.52710 1.18158 .49732 .87825 .00253a .01685 .04794 .01370 8.9270 2.98780 1.80164 .63341 .23753 .000753b .02121 .08326 .00813 8.2657 2.87501 1.50076 .38238 .45856 .00153c .01683 .09990 .00345 9.5884 3.09651 1.45226 .24462 .65956 .00204a .007866 .04115 .0204 4.8706 2.20694 1.59341 ».30456 .12580 .00054b .009504 .06732 .0115 5.6013 2 . 3 6 6 8 1 1.60943 .22721 .23431 .0008754c .006084 .09719 .00416 5.3293 2.30852 1.43821 .09003 .36013 .0013755a .006750 .00675 .01031 3.9282 1.98196 . 39936 .39936 .06640 .000755b .01126 .01126 .00588 4.7330 2.17554 .36549 .36549 .14576 .00155* .01352 .01352 .0020 8.6000 2.93357 .39443 .39443 .29472 .002256a .005472 .00886 .00909 4.1012 2.02514 .49818 .30782 .07291 .000756b .008746 .01663 .00667 2.9910 1.72945 .34209 .17986 .14541 .001757 .0073*2 .01980 .002 5 2.7480 1.65770 .24866 .09283 .21882 .002757a .002955 .00778 .01124 3.5053 1.88596 .57239 .21726 .04838 .00057b .004923 .01699 .00769 2.5995 1.61230 .42726 .12382 .10335 .001257c .003948 .0232 .00303 2.5578 1.59931 .36144 .06141 .16441 .00206a .001828 .00955 .01563 1.4824 1.22081 .42545 .08143 .03345 .00056b .002153 .01752 .00870 1.4299 1.195780 .38444 .04723 .06517 .00106c24
.009922
.409430.02411.966^9
.00225 1.3200 1.1489157.45177
.3377821.39167
.013909.50908
,,094217.19497
.00150

-120-Subs tituting the values of summations of the terms
in the simultaneous equations, the following four equations were obtained:2^a + 8.6088b + 3.4122c + 2.6536d = 57.45177 8.6088a + 3.76615b + 1.24596c + 0.96669d « 21.391673.4122a + 1.24596b + 0.68946c + 0.40948d * 9.509082.6536a + 0.96669b + 0.40948c + 0.40473d = 7.19497from the above equations a, was eliminated to obtainthe following three equations:16.27616b + .52809c + .35625d = 18.80928
.52809b + 4.90393c + .77291d = 32.18099
.35625b + .77291c + 2.67193d = 20.22526The above three equations were solved by determinants and the values obtained for b, c and d were:
b = 0.84752 c a 5.54877 d - 5.85144
By substituting the above values in one of the four equations ;he value of a was found to be
a = 0.65394From the values of a, b, c and d, the values of (*,A* ant* were calculated.
= a = .65394 Q = 2.33842k a - - t f s i H ■ i-29602
KB - o/* ' ■£^53 9A = « ^ 8513

-121-KP * d/* - *^5394 ' 8*94797
Q - k«LKA KB = 2.3384-2The equation that described the mechanism of the reaction at 244.5°C. was:
2.33842 fA *(l+1.29602pA + 8.48513pB + 8.94797
E-Ps]11.4
Same treatment as above was given to the data obtained at 218°C. The constants were evaluated by the method of least squares. The values of different terms are given in Table XV.
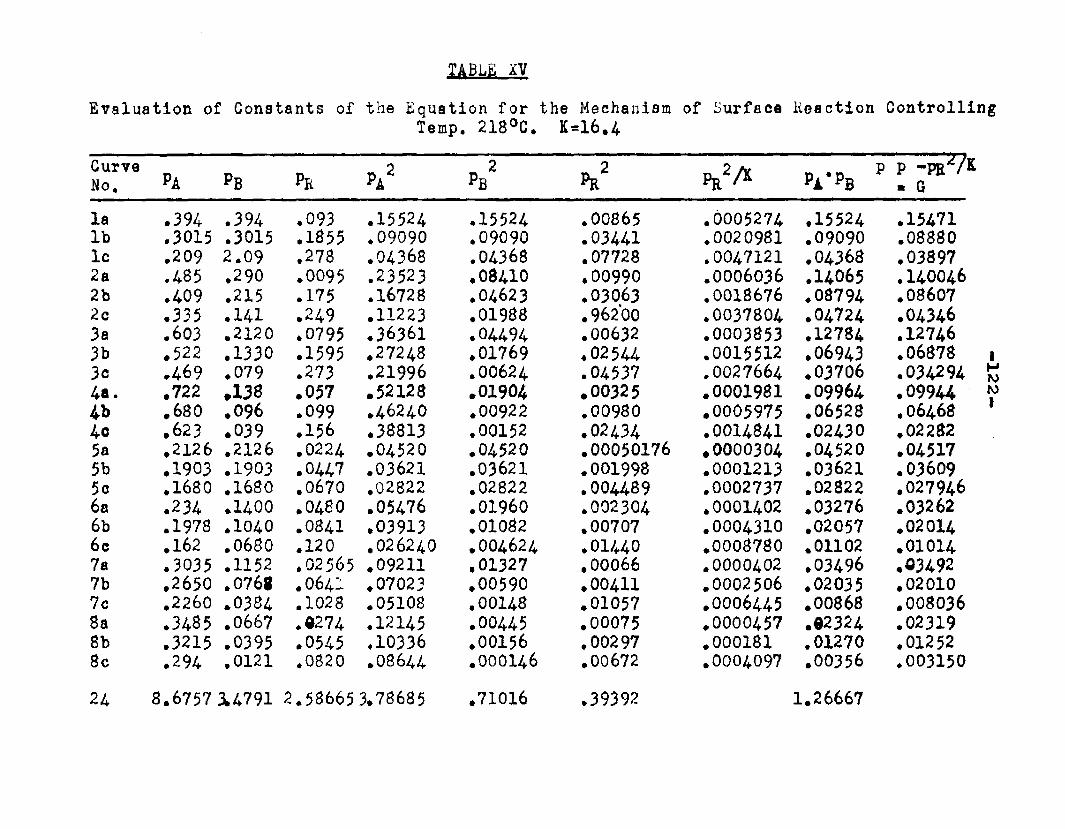
TABLE XVEvaluation of Constants of the Equation for the
Temp, 218°C. KCurveNo, Pa PB PR pa2 Pb2la .394 .394 .093 .15524 .15524lb .3015 .3015 .1855 .09090 .09090lc .209 2.09 .278 .04368 .043682a .485 .290 .0095 .23523 .084102b .409 .215 .175 .16728 .046232c .335 .141 .249 .11223 .019883a .603 .2120 .0795 .36361 .044943b .522 .1330 .1595 .27248 .017693c .469 .079 .273 .21996 .006244a. .722 .138 .057 .52128 .019044b .680 .0 9 6 .099 .46240 .009224c .623 .039 .156 .38813 .001525a .2126 .2126 .0224 .04520 .045205b .1903 .1903 .0447 .03621 .036215c .1680 .1680 .0670 .02822 .028226a .234 .1400 .0480 .05476 .019606b .1978 .1040 .0841 .03913 .010826c .162 .0680 .120 .026240 .0046247a .3035 .1152 .02565 .09211 .013277b .2650 .0768 .0641 *07023 .005907c .2260 .0384 .1028 .05108 .001488a .3485 .0667 .8274 .12145 .004458b .3215 .0395 .0545 .10336 .001568c .294 .0121 .0820 .08644 .000146
24 8.6757 14791 2.58665 3.78685 .71016
Mechanism of Surface Reaction Controlling 16.4
h 2 ^ pa -pb p !
00865 .000527403441 .002098107728 .004712100990 .000603603063 .001867696200 .003780400632 .000385302 544 .001551204537 .002766400325 .000198100980 .000597502434 .001484100050176 .0000304001998 .0001213004489 .0002737002304 .000140200707 .000431001440 .000878000066 .000040200411 .0002 50601057 .000644500075 .000045700297 .00018100672 .000409739392
.15524 .15471
.09090 .08880
.04368 .03897
.14065 .140046
.08794 .08607
.04724 .04346
.12784 .12746
.06943 .06878
.03706 .034294
.09964 .09944
.06528 .O6468
.02430 .02282
.04520 .04517
.03621 .03609
.02822 .027946
.03276 .03262
.02057 .02014
.01102 .01014
.03496 .03492
.02035 .02010
.00868 .008036
.02324 .02319
.01270 .01252
.00356 .0031501.26667
122

TABLE XV. (Coat.)CurveNo. PB'PR PA-PK r £
r Jr* R pa *r PB’* p r*r X
la .03664 .03664 .006660 23.22973 4.81972 1.89897 1.89897 .44823 .0010lb .05595 .05593 .004310 20.60325 4.53898 1.36850 1.36850 .84198 .0020lc .05810 ,05810 .001041 37.43516 6.11843 1.27875 1.27875 1.70092 .00302a .02886 .04826 .006710 20.87124 4.56851 2.21573 1.32487 .45457 .00102b .03763 .07158 .004608 18.67839 4.32185 1.76764 ..92920 .75632 .001752c .03111 .08342 .002695 16.12616 4.01579 1.34529 .57130 1.00890 .00253a .01686 .04794 .006849 18.61002 4.31393 2.60130 .91455 .34296 .000753b .02121 .08326 .006060 11.20099 3.34688 1.74707 .44514 .53383 .00153c .01683 .09990 .002315 14.81382 3.84887 1.80512 .30406 .8198k .00204a .007866 .04115 .009090 10.93949 3.30749 2.38801 .45643 .18852 .000504b .009504 .06732 .007042 9.18489 3.03065 2.06084 .2 9094 .30003 .0008754c .006084 .09719 .002857 7.98740 2.82620 1.76072 .11022 .44089 .0013755a .004762 .004762 .008065 5.60074 2.36659 .50314 .50314 .05301 .00055b .00851 .00851 .004739 7.61553 2.75962 .52516 .52516 .12336 .00105c .01126 .01126 .001776 15.73536 3.96678 .66642 .66642 .26577 .00156a .006720 .01123 .00500 6.52400 2.55421 .59769 .35759 .1226 .00106b .008746 .01663 .003030 6.64686 2.57815 .50996 .26813 .21682 .001756c .00816 .0^944 .001942 5.22142 2.28504 .37018 .15538 .2742 .002507a .002955 .00778 .006250 5.58720 2.36373 .71739 .27230 .06063 .00057b .004923 .01699 .004587 4.38195 2.09331 .55473 .16077 .13418 .001257c .003043 .02323 .002380 3.37647 1.83752 .41523 .07056 .18390 .002008 a .001828 .00955 .006757 3.43200 1.85257 .64562 .12357 .05076 .00058b .002153 .01752 .004878 2.56b63 1.60207 .51507 .06328 .08731 .0010Sc .009922 .02411 .002041 1.54336 1.24232 .36524 .01503 .10187 .0015
24 .40448 .96170 76.55921 28.62382 13.07426 9.51638

-124-By substituting the values of various terms from
the Table XV in the equations the following four equations were obtained:24a + 8.6757b + 3.4791c + 2.58665d - 76.55921 8.6757a + 3.78685b + 1.26667c + 0.96l70d = 28.623823.4791a + 1.26667b + 0.71016c + 0.40443d = 13.074262.58665a + 0.96170b + 0.40448c + 0.39392d = 9.51638
Eliminating a, from the above equations, the following equations were obtained:15.61663b + 0.21645c + 0.6398d » 22.766940.21645b + 4.93970c + 0.70831d - 47.425090.63980b + 0.70831c + 2.76332d * 30.36124
The constants b, c and d were evaluated by determinants , to b e :
b - 0.98916 c « 6.32065 d - 8.62 549a was evaluated by substituting values of b, c
and d in the first of the four equations, a = 0.69661 Q = ksLKAKB= 2.06072
K a * b/a = = 1.41996A 0.69661Kfi - c/a = « 11.94449
kp - d/a - o'A U i l ■ 1 2 - 3 8 2 0 9

Using the rounded figures the equation describing the mechanism of the reaction is:
r « 2.06 ( pa * PB " tS’P| )l+1.4.2pA + 11.95pg + 1 2 . 3 8 ^
Adsorption of Qctanol Controlling.The data obtained at 218°C. and 2A4.5°C. were
treated as for the surface reaction controlling. The equation for a reaction where adsorption of a reactant is controlling is: s
"aBKStj a o
kA L (*1* artK )r = ^ a
* aBKB + “*** ' 8SKSID
asLET ajas . 8R2 . M
aBK
l e t P a - * G
let KaL = QThe above equation can be written as
QGr = ' '1+ MKa + agKg + agKp
Taking activities to be equal to partial pressures, a s __________________1+MX.a + pBKB + Pk K]

-126-Rearranging —
* • r Q Q Q “ Q RThe above equation is of the form £ = a + bM + cpg + dpjAccording to the method of least squares the four
equations required to evaluate the four constants a, b, c and d are:
na + b ^ M + c ^-Pb + d Pr * 5-R alM + b 2. + c Z M p B + d X MpR = 2_M.R ®2Pb + b i M . p B + c i p B d £ P B . p R “i P g •£a 2 PR + b ZM.pr + c^pg.pR + djpj^2 = 2_PR*R The values of above terms for the data obtained at
244*5°C. are all tabulated in the Table XVI. The values of * P A# £ M, ZPr* etc. are obtained from the above table* By substituting the appropriate values for all the terms in the equations the following four equations were obtained.24a + 0.36306b + 3.4122c + 2.6536d = 1267.17355 0.30306a + 0.01257b + 0.03549c + 0.05838d = 31.03941 3.4122a + .03549b + .68946c + .40948d = 152.60969 2.6536a + ,05838b + .40948c + ,40473d = 167.44281
By eliminating a, the following three equations were obtained:

-127-0.16987b - 0.38707c + ,43770d * 284.88582 0.38707b - 4.90393c- 0.77291d - 661.21702 0.10502b - .43232c + ,29442d * 166.38329 The constants b, c and d were evaluated by deter
minant sb * 1901.72012 c = 24.22335 d *-94.7 5 592One of* the constants had a negative value, hence
It was unlikely that this equation could describe the mechanism of the reaction. The possibility of adsorption of octanol controlling was thus ruled out
ofor the runs made at 244*5 C.Similar treatment for the data obtained at 218°C.
gave the Table XVII, The table yielded the following four equations:
24a + .24264b + 3.4791c + 2.58665d * 2103.45535 0.24264a + 0.005860b + 0.02402c + .04626d = 32.583563.4791a * 0.02402b + 0.71016c + 0.40448d * 277.902342.58665a + 0.04626b + 0.40448c + 0.39392d * 279.88300 eliminating a, following equations were obtained: 0.08177b - ,26769c + .482628 * 271.62303 0.26769b - 4.9397c - 0.708318 * 648.475350.48262b + ,70831c + 2.76332d * 1276.28922The consta nts b f c and d were evaluated by deter—
mina nts
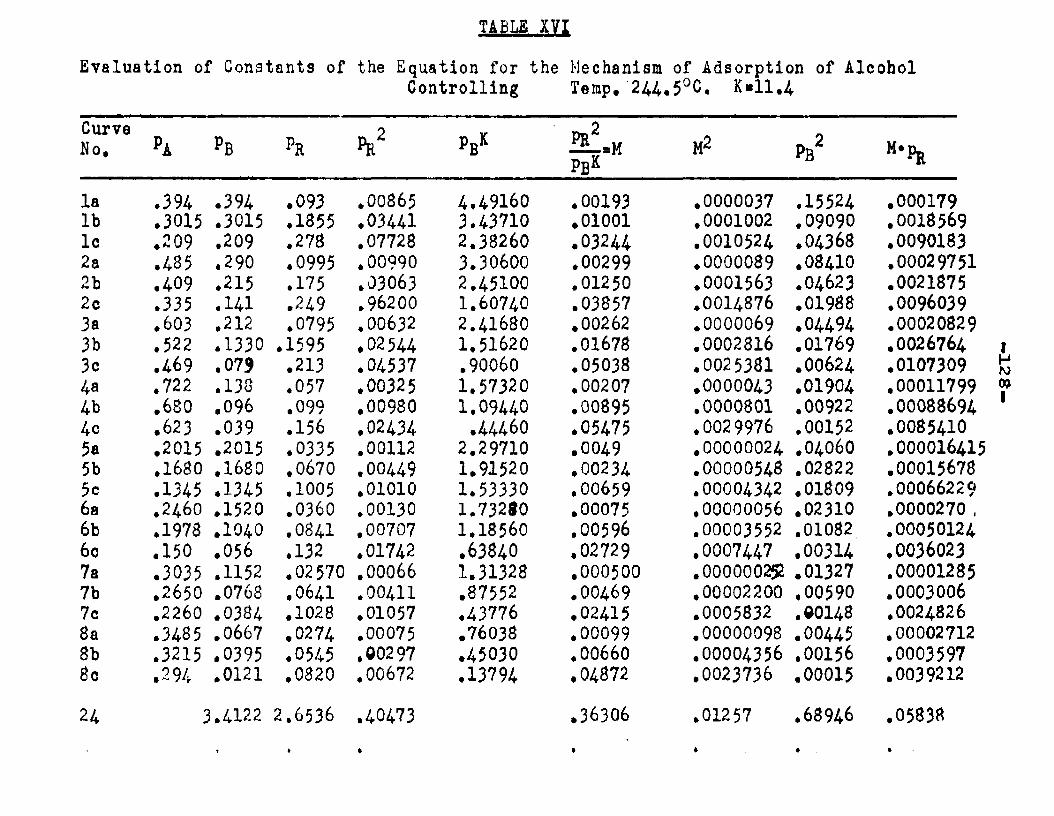
Evaluation of Constants of the Equation for the Mechanism of Adsorption of AlcoholControlling Temp* 244.5°C. K«ll,4
CurveNo, PA Pb PR
CM pbk m2 D.2pb m ,Pr
la .394 .394 .093 .00865 4.49160 .00193 .0000037 .15524 .000179lh .3015 .3015 .1855 .03441 3.43710 .01001 .0001002 .09090 .0018569lc .209 .209 .278 .07728 2.38260 .03244 .0010524 .04368 .00901832a .435 .290 .0995 .00990 3.30600 .00299 .0000089 .08410 .000297512b .409 .215 .175 .03063 2.45100 .01250 .0001563 .04623 .00218752c .335 .141 .249 .96200 1.60740 .03857 .0014876 .01988 .00960393a .603 .212 .0795 .00632 2.41680 .00262 .0000069 .04494 .000208293b .522 .1330 .1595 .02544 1,51620 .01678 .0002816 .01769 .00267643c .469 .073 .213 .04537 .90060 .05038 .0025381 .00624 .01073094a .722 .138 .057 .00325 1.57320 .00207 .0000043 .01904 .000117994b .680 .096 .099 .00980 1.09440 .00895 .0000801 .00922 .000886944c .623 .039 .156 .02434 .44460 .05475 .002 9976 .00152 ,00854105a .2015 .2015 .0335 .00112 2.29710 .0049 .00000024 .04060 .0000164155b .1680 .1680 .0670 .00449 1.91520 .00234 .00000548 .02822 .000156785c .1345 .1345 .1005 .01010 1.53330 .00659 .00004342 .01809 ,000662296a .2460 .1520 .0360 .00130 1.73200 .00075 ,00000056 .02310 .0000270 ,6b .1978 .1040 .0841 .00707 1.18560 .00596 .00003552 .01082 .000501246c .150 .056 .132 .01742 .63840 ,02729 .0007447 ,003H ,00360237a .3035 .1152 .02570 .00066 1.31328 .000500 .000000252 .01327 .000012857b .2650 .0768 .0641 .00411 .87552 .00469 .00002200 .00590 .00030067c .2260 .0384 .1028 .01057 .43776 .02415 .0005832 .©0148 .00248268a .3485 .0667 ,0274 .00075 .76038 .00099 .00000098 .00445 .000027128b .3215 .0395 .0545 .00297 .45030 .00660 ,00004356 .00156 .00035978c .294 .0121 .0820 ,00672 .13794 ,04872 ,0023736 .00015 .0039212
24 3.4122 2.6536 .40473 .36306 .01257 .68946 .05838

TABU XVI. (Cont.)CurveNo. M*pB P * P B R pA-M=G r f * M*R Pr *r Pb-R
la .000760 .03664 .39207 .01031 38.02812 .07339 3.53662 14.98308lb .0030180 .05593 .29149 .00746 39.07372 .39113 7.24818 11.78073lc .0067799 .05810 .17656 .00323 54.66253 1.77325 15.19518 11.424472a .00086710 .02886 .48201 .01389 34.70194 .10376 3.45284 10.063562b .0026875 .03763 .39650 .00909 43.61936 .54524 7.63339 9.378162c .0054384 .03511 .29643 .00336 88.22321 3.40277 21.96757 12.439473a .0005554 .01685 .60038 .01370 43.82336 .11482 3.48396 9.290553b .0022317 .02121 .50522 .00813 62.14268 1.04275 9.91176 8.264983c .0039800 .01683 .41862 .00345 121.33913 6.11307 25.84523 9.585794a .00028566 .007866 .71993 .0204 35.29069 .07305 2.01157 4.870124b .00085920 .009504 .67105 .01150 58.35217 .52225 5.77686 5.601814c .0021353 .006084 .56825 .00416 136.59856 7.47877 21.30938 5.327345a .00009874 .006750 .20101 .01031 19.49661 .00955 .65314 3.928575b .00039312 .01126 .16566 .00588 28.17347 .06593 1.88762 4.733145c .00088635 .01352 .12791 .0020 63.95500 .42146 6.42748 8.601956a .0001140 .005472 .24525 .00909 26.98020 .02024 .97129 4.101006b .00061984 .008746 .19184 .00667 28.76162 .17142 2.41885 2.991216c .0015282 .007392 .12271 .002 5 49.98400 1.33950 6.47909 2.748707a .00005760 .002955 .30300 .01124 26.95730 .01348 .69280 3.105487b .0003602 .004923 .26031 .00769 33.85046 .15876 2.16981 2.599727c .00092736 .003948 .20185 .00303 66.61716 1.63279 6 .84824 2.558108a .00006603 .001828 .34751 .01563 22.23353 .02201 .60920 1.482988b .0002607 .002153 .31490 .00870 36.19540 .23889 1.97265 11.429728c .00057862 .009922 .24528 .00225 109.01333 5.31113 8.93910 1.31906
24 .035490 .40948 1267.17355 31.03941 167.44281 152.6069
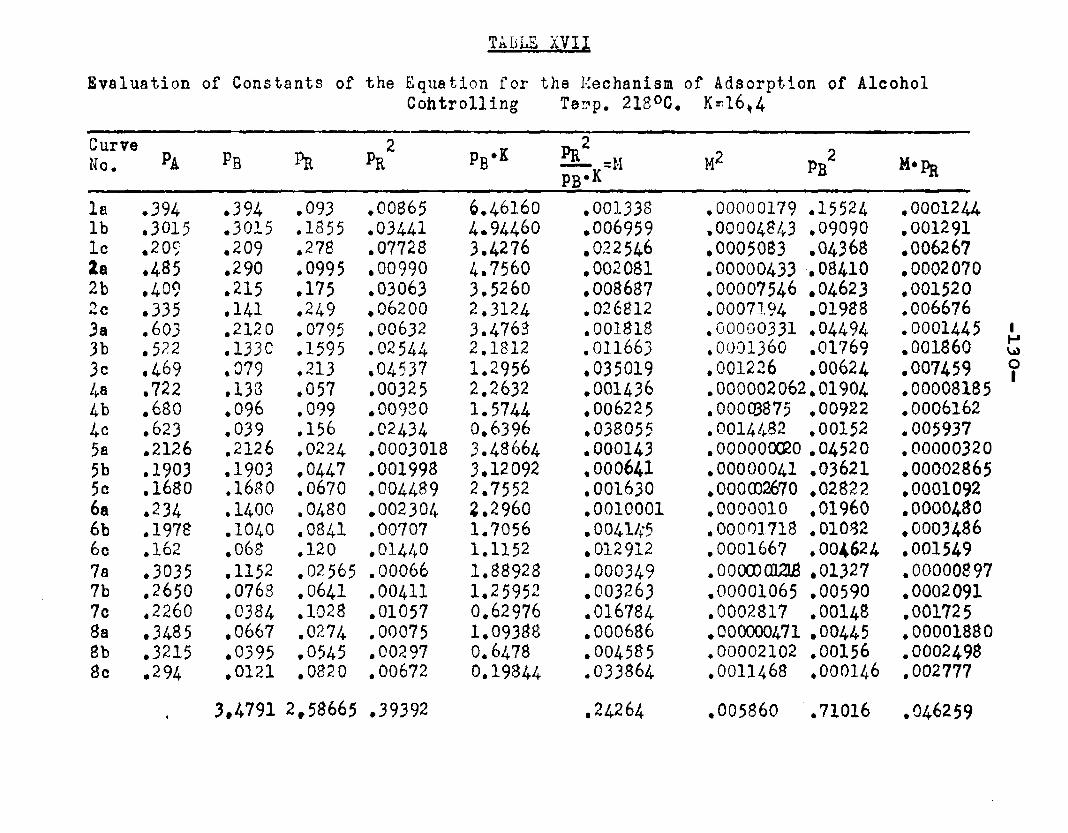
TABLEEvaluation of Constants of the Equation for
CohtrollingCurveNo. pa Pb %
2PR pb‘K
la .394 .394 .093 .00865 6 .46160lb .3015 .3015 .1855 .03441 4 .9 4 4 6 0lc .209 .209 .278 .07728 3.42762a .485 .290 .0995 .00990 4.75602b .409 .215 .175 .03063 3.52602c .335 .141 .249 .06200 2.31243a .603 .2120 .0795 .00632 3.47633b • 522 .1330 .1595 .02 544 2,13123c .469 .079 .213 .04537 1.29564a .722 .138 .057 .00325 2.26324b .680 .096 .099 .00930 1.57444c .623 .039 .156 .02434 0.63965a .2126 .2126 .0224 ,0003018 3.486645b .1903 .1903 .0447 .001998 3.12 0925c .1680 .1680 .0670 .004489 2.75526a .234 .1400 .0480 .002304 2.29606b .1978 .1040 .0841 .00707 1.70566c .162 .063 .120 .01440 1.11527a .3035 .1152 .02565 .00066 1.889287b .2650 .0763 .0641 .00411 1.259527c .2260 .0384 .1028 .01057 0.629768a .3485 ,0667 .0274 .00075 1.093888b .3215 .0395 .0545 .00297 0.64788c .294 .0121 .0820 .00672 0.19844
3.4791 2*58665 .39392
e Kechanism of Adsorption of Alcohol Ten'p. 218°C, K-l6*4
2- aM i*K m2 _ 2
pb M*Pr
.001338 .00000179 .15524 .0001244
.006959 ,00004343 .09090 .001291
.022546 ,0005083 .04368 .006267,002081 .00000433 .08410 .0002070.008687 .00007546 .04623 .001520.026812 .0007194 .01988 .006676.001818 .00000331 .04494 ,0001445.011663 .0001360 .01769 .001860.035019 .001226 .00624 .007459.001436 .000002062 .01904 .00008185.00622 5 .000Q38 75 ,00922 .0006162.038055 .0014482 .00152 .005937.000143 .000000020 .04520 .00000320,000641 .00000041 .03621 .00002865.001630 ,000002670 .02822 .0001092.0010001 .0000010 .01960 .0000480.00414*5 .00001713 .01032 .0003486.012912 .0001667 .004624 .001549.000349 .00000(2218 .01327 .00000897.003263 .00001065 .00590 .0002091.016784 .0002817 .00148 .001725.000686 .000000471 .00445 .00001880.004585 .00002102 .00156 .0002498.033864 .0011468 .000146 .002777.24264 .005860 .71016 .046259
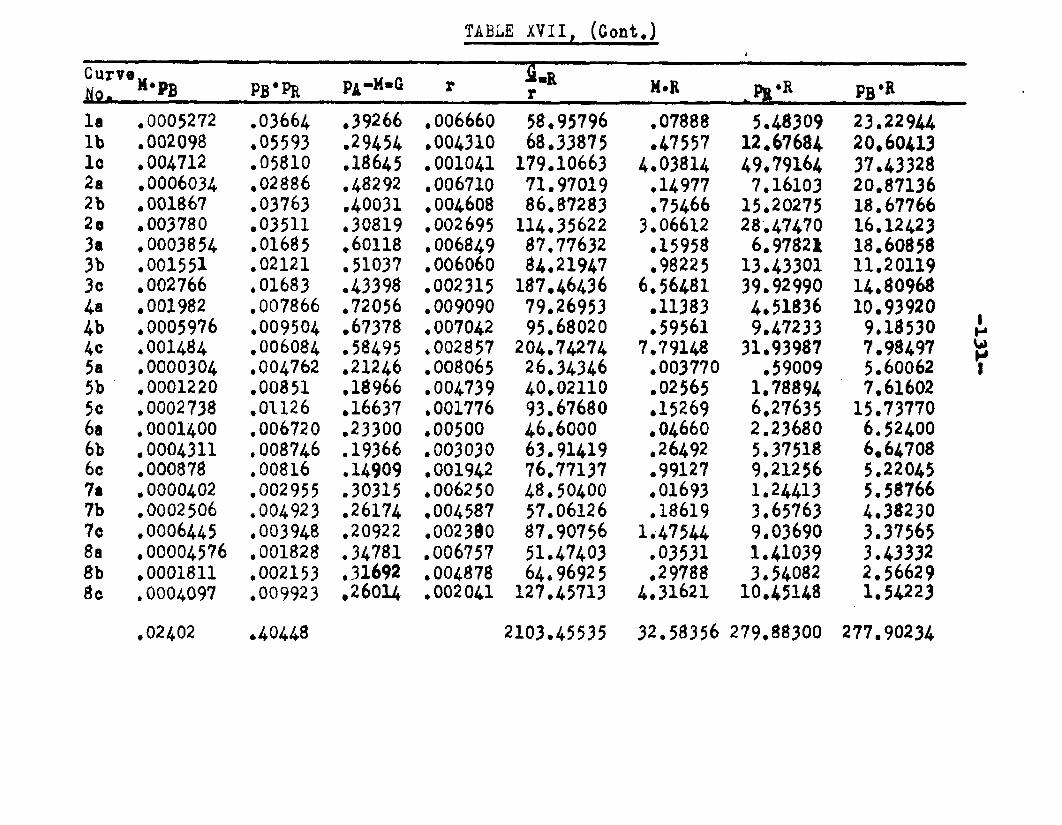
TABLE XVII, (Cont.)i
CurveW9«
>w ^H«pb p b*pr p^-M«G r ■ c 'r M.R .PRtR p b*rla .0005272 .03664 .39266 .006660 58.95796 .07888 5.48309 23.22944lb .002098 .05593 .29454 .004310 68.33875 .47557 12.67684 20.60413lc .004712 .05810 .18645 .001041 179.10663 4.03814 49.79164 37.433282a .0006034 .02886 .48292 .006710 71.97019 .14977 7.16103 20.871362b .001867 .03763 .40031 .004608 86.87283 .75466 15.20275 18.677662o .003780 .03511 .30819 .002695 114.35622 3.06612 28.47470 16.124233a .0003854 .01685 .60118 .006849 87.77632 .15958 6.97821 18.608583b .001551 .02121 .51037 .006060 84.21947 .98225 13.43301 11.201193c .002766 .01683 .43398 .002315 187.46436 6.56481 39.92990 14.809684a .001982 .007866 .72056 .009090 79.26953 .11383 4.51836 10.939204b .0005976 .009504 .67378 .007042 95.68020 .59561 9.47233 9.18530Ac .OOI484 ,006084 .58495 .002857 204.74274 7.79148 31.93987 7.984975a .0000304 .004762 .21246 .008065 26.34346 .003770 .59009 5.600625b .0001220 .00851 .18966 .004739 40.02110 .02565 1.78894 7.616025c .0002738 .01126 .16637 .001776 93.67680 .15269 6.27635 15.737706a .0001400 .006720 .23300 .00500 46.6000 ,04660 2.23680 6.524006b .0004311 .008746 .19366 .003030 63.91419 .26492 5.37518 6.647086c .000878 .00816 .14909 .001942 76.77137 .99127 9.21256 5.220457a .0000402 .002955 .30315 .006250 48.50400 .01693 1.24413 5.587667b .0002506 .004923 .26174 .004587 57.06126 .18619 3.65763 4.382307c .0006445 .003948 .20922 .002380 87.90756 1.47544 9.03690 3.375658a .00004576 .001828 .34781 .006757 51.47403 .03531 1.41039 3.433328b .0001811 .002153 .31692 .004878 64.96925 .29788 3.54082 2.566298c .0004097 .009923 .26014 .002041 127.45713 4.31621 10.45148 1.54223
.02402 .40448 2103.45535 32.58356 279.88300 277.90234

-132-b ■ 40^.397c » -130.700d « 336.609Again 11 is seen that, one of the constants is
negative, proving that adsorption of octanol is not a controlling step at 218°C. also.
(

-133
EKSBBP^s d a t a
Reeder
L o c . c i t . (35)
used a lovr iron low silica bauxite catalyst, supplied by the same concern, then operating under a different name, for his study of the esterification reaction.The equilibrium constants for the reaction with his catalyst and with the catalyst used in this investigation were proved to be the same by Lindsey.
(4.2) Lindsey, Rolland, "Chemical Equilibrium in the System Acetic Acid, n— Octanol, n-Octyl Acetate and Water", M. Sc. Thesis, The Ohio State University, 1953.
It has been noticed however, that in this investigation the catalyst used gave smaller conversions than those obtained by Reeder. This could mean that the present catalyst has a different activity which is less compared to the activity of Reeder*s catalyst.
Reeder evaluated the constants kg and for hisdata and the values were very much higher than those found in this investigation (Table XVIIJ). He assumed
to be negligible in comparison with Kg and K^. He also neglected 1 In the denominator of the rate e quation for adsorption of alcohol controlling.
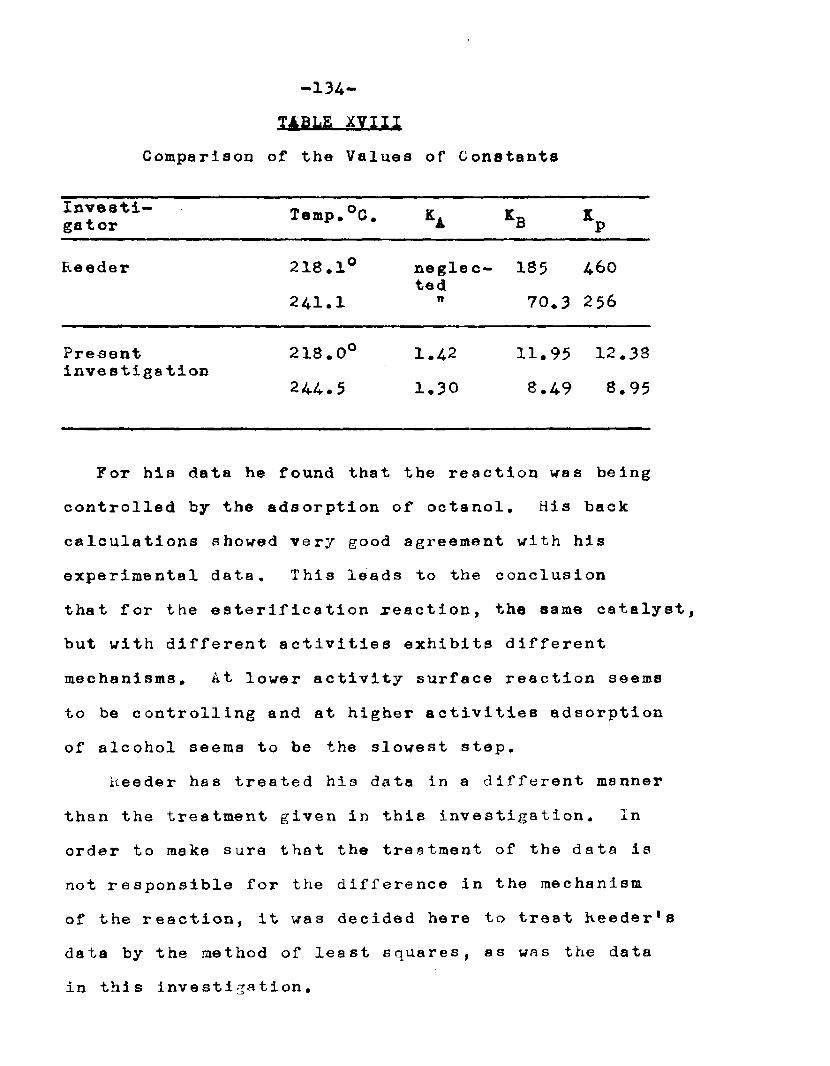
-134- TABLE XVIII
Comparison of the Values of Constants
Investigator Temp.°C. ka *B k p
Reeder 218.1°241.1
negle c—tedn
18570.3
460256
Present 218.0° investigation
244.51.421.30
11.958.49
12.388.95
For his data he found that the reaction was beingcontrolled by the adsorption of octanol. His backcalculations showed very good agreement with hisexperimental data. This leads to the conclusion that for the esterification reaction, the same catalyst, but with different activities exhibits different mechanisms. At lower activity surface reaction seems to be controlling and at higher activities adsorption of alcohol seems to be the slowest step,
heeder has treated his data in a different manner than the treatment given in this investigation. In order to make sure that the treatment of the data is not responsible for the difference in the mechanism of the reaction, it was decided here to treat heeder1s data by the method of least squares, as was the data in this investigation.

-135-heeder's data was treated exactly the way described
previously and the values of different terras of the simultaneous equations are tabulated in Table XVIX.For surface reaction controlling, following four equations were obtained:
12a + 2.4624b + 4.6054c + 2.3103d * 37.76032 2.4624a + 0.66284b + 0.79394c + 0.47078d = 7.327034.6054a + 0.793946 + 2.37982c + 0.65604d = 16.691782.3103a + 0.47078b + 0.65604c + 0.56173d = 6.38064Eliminating a from the above equations, following
three equations were obtained-1.89067b + 1.81306c + 0.03952d = 5.05665-1.81306b + 7.34813c - 2.76733d = 26.399980.03952b + 2.76738c - 1.40327d = 10.66999The constants b, c and d were evaluated by deter
minants .The denominator D = 5.01599 - 4.8111 - .20977
» 0.00488The difference between the positive and the nega
tive terras was very small.dolving for b:b*D = -13.41540 + 70.0544-6 - 56.63449 = +.00457Again the difference between positive and nagative
terms is very small.
b * - z2PJt!?J = -.93647 -.00488

Solving for c.c.D - 14.21478 - 13.62973 - .59426 = -.00921
'• c ■ : :8 8 g I ■ i-8872*value of d was foutfd to be:
+»Q1911 = -3.91598-.00488Some of the constants came out to be negative
but it was noticed that in case of evaluation of the denominator D, b and c the differences between negative and positive values has been very small, and the \alues became negative by a very narrow margin.This shows that the data does not perfectly fit this equation for surface reaction controlling, but indications are that the data are quite close to fit this equation.
The data were then tried to fit into the equation for adsorption of octanol controlling. Differentterms required to evaluate the constants by the
«
method of least squares, have been given in Table XX.From this table four equations given below were obtained .
12a + .34436b + 4.6054c + 2.3103d « 381.44135 ,34436a + 0.031221b 4 0.048010c + 0.10683d = 14.61769 4.6054a + 0.048010b + 2.37982c + 0.65604 = 127.27985 2.3103a + 0.10683b + 0.65604c + 0.56173d * 79.5668
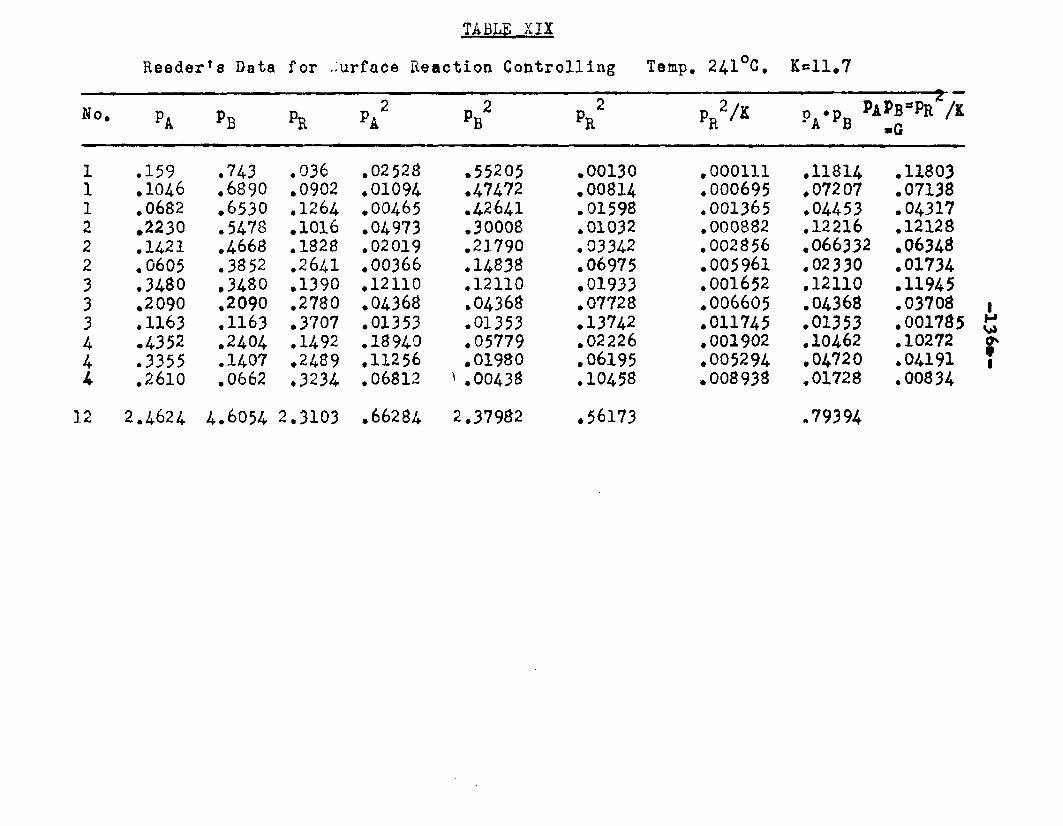
TABLE XIXReeder's Data for -Surface Reaction Controlling Temp. 241°C. Kell.7
No. Pa Pb PR_ 2 A « 2B
n 2 PR ph 2/k
. ......... .. go *p PAPBSPR /£ ‘A B ,G
1 .159 .7A3 .036 .02528 .55205 .00130 .000111 .11814 .118031 .1046 .6890 .0902 .0109A .A7A72 .0081A .000695 ,07207 .071381 .0682 .6530 .126A .00A65 .A26A1 .01598 .001365 .0AA53 .043172 .2230 .5A7S .1016 •0A973 .30008 .01032 .000882 .12216 .121282 .1A21 .A668 .1328 .02019 .21790 .033A2 .002856 .066332 .063482 .0605 .3852 .26A1 .00366 .H838 .06975 .005961 .02330 .017343 .3480 .3A80 .1390 .12110 .12110 .01933 .001652 .12110 .119453 .2090 .2090 .2780 .0A368 .0A368 .07728 .006605 .0A368 .03708 t3 .1163 .1163 .3707 .01353 .01353 .137A2 .0117A5 .01353 .001785 HVjJA .A3 52 .2A0A • 1A92 .139A0 .05779 .02226 ,001902 .10A62 .10272 SA .33 55 .H07 .2A89 .112 56 .01980 .06195 .00529A .04720 .04191 ¥1A .2610 .0662 .323A .06812 ' .00A38 .10A58 .008938 ,01728 .00834
12 2.A62A 4.6054 2.3103 .6628A 2.37982 .56173 .79394
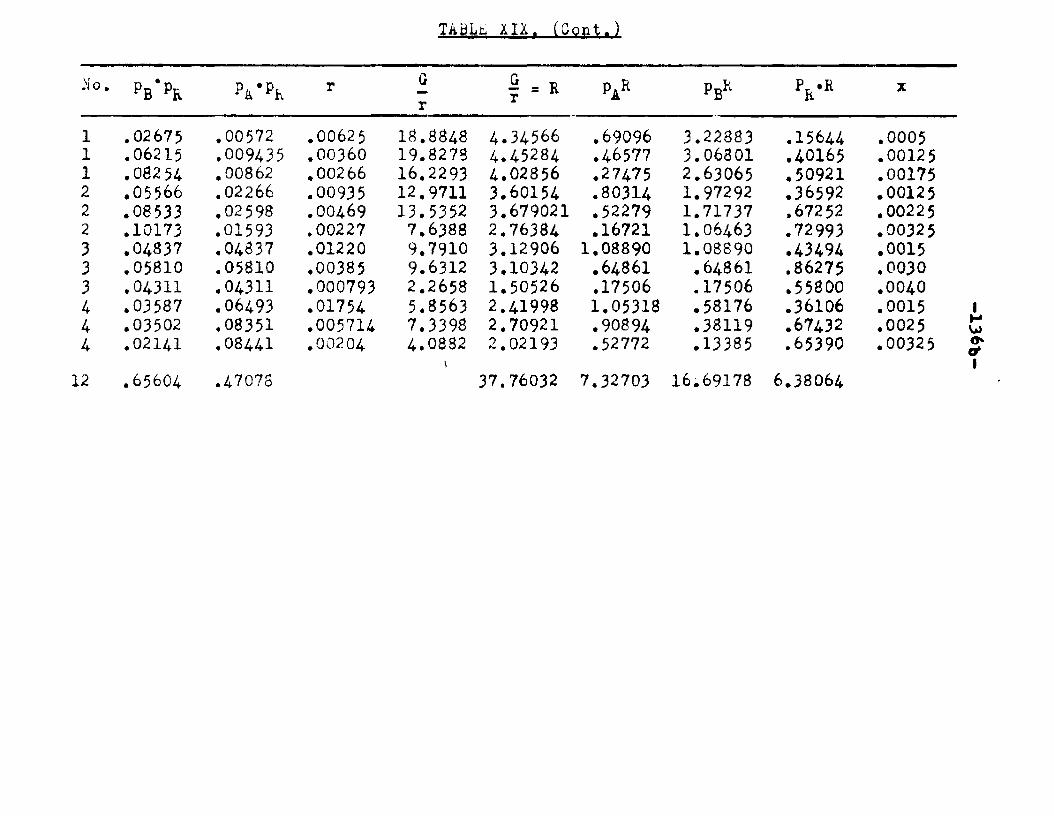
TABLL 111. (Cont.)
I'l 0 . pb ‘Pr V PK r Gr 1 * * pAR PBh V R X
1 .02675 .00572 .00625 18.8848 4.34566 .69096 3.22383 .15644 .00051 .06215 .009435 .00360 19.8273 4.45284 .46577 3.06801 .40165 .001251 .082 54 .00862 .00266 16.2293 4.02856 .27475 2.63065 .50921 .001752 .05566 .02266 .00935 12.9711 3.60154 .80314 1.97292 .36592 .0012 52 .08533 .02 598 .00469 13.5352 3.679021 .52279 1.71737 .672 52 .002252 .10173 .01593 .00227 7.6388 2.76384 .16721 1.06463 .72 993 .003253 .04837 .04337 .01220 9.7910 3.12906 1.08890 1.08890 .43494 .00153 .05810 .05810 .00385 9.6312 3.10342 .64861 .64861 .86275 .00303 .04311 .04311 .000793 2.2658 1.50526 .17506 .17506 .55800 .00404 .03587 .06493 .01754 5.8563 2.41998 1.05318 .58176 .36106 .00154 .03502 .08351 .005714 7.3398 2.70921 .90894 .38119 .67432 .00254 .02141 .08441 .002 04 4.0832 2.02193 .52772 .13385 .65390 .0032512 .65604 .47078
\37.76032 7.32703 16.69178 6.38064
-136b-

Reeder fs Data for AdsorptionTABLE
of Octanoli XXControlling Temp. 241°C. K-11.7
No. pa pb PR PR2 pb *k Pp2iiL =m Pb*k
M2 pb2 M*pr
1 .159 .743 .036 .00130 8.6931 .000149 .0000000222 .55205 .000005361 .104.6 .6890 .0902 .00814 8.0613 .001009 .000001018 .47472 .000091011 .0682 .6530 .1264 .01598 7.6401 .002091 .000004372 .42641 .00026432 .2230 .5478 .1016 .01032 6.40926 .001610 .00000259 .30008 .00016362 .1421 .4668 .1828 .03342 5.46156 .006119 .00003744 .21790 .00111862 .0605 .3852 .2641 .06975 4.50684 .015476 .0002395 .14838 .0040873 .3480 .3480 .1390 .01933 4.0716 .004747 .00002253 .12110 .00065983 .2090 .2090 .2780 .07728 2.4453 .031603 .0009987 .04368 .0087863 .1163 .1163 .3707 .13742 1.36071 .100991 .0101992 .01353 .0374374 .4352 .2404 .1492 .02226 2.81268 .007914 .00006263 .05779 .0011807A .3355 .1407 .2489 .06195 ' 2.64619 .037632 .0014162 .01980 .0093660A ,2610 . 0662 .3234 .10458 0.77454 .135022 .018231 ,00438 .043666
12 2.4624 4.6054 2.3103 .56173 .34436 .031215 2.37982 .106826
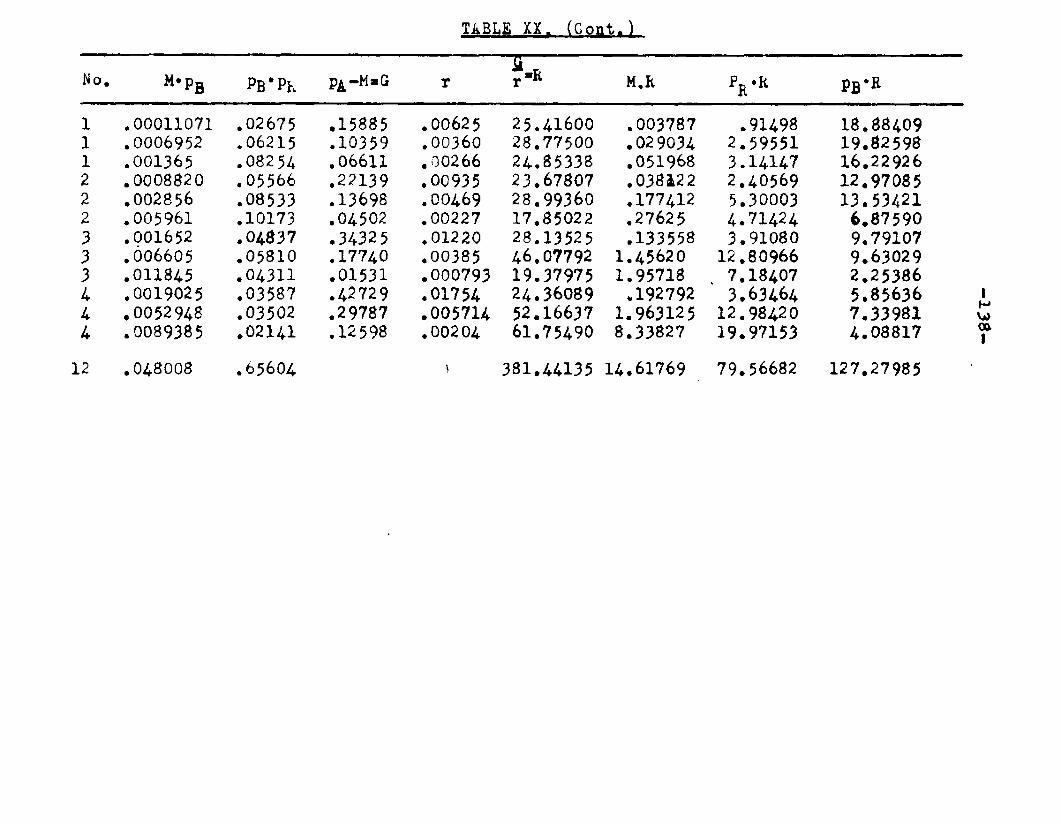
TABLE XX. (Coat.)
No. M.pB PB'PK PA-M»G r r-B M.R V R pB -R
1 .00011071 .02675 .15885 .0062 5 25.41600 .003787 .91498 18.884091 .0006952 .06215 .10359 .00360 28.77500 .02 9034 2.59551 19.825981 .001365 .082 54 .06611 .00266 24.85338 .051968 3.14147 16.229262 .0008820 .05566 .22139 .00935 23.67807 .038122 2.40569 12.970852 .002856 .08533 .13698 .00469 28.99360 .177412 5.30003 13.534212 .005961 .10173 .04502 .00227 17.85022 .27625 4.71424 6.875903 .001652 .04837 .3432 5 .01220 28.13525 .133558 3.91080 9.791073 .006605 .05810 .17740 .00385 46.07792 1.45620 12.80966 9.630293 .01184-5 .04311 .01531 .000793 19.37975 1.95718 7.18407 2.253864 .0019025 .03587 .42729 .01754 24.36089 .192792 3.63464 5.856364 .0052948 .03502 .29787 .005714 52.16637 1.963125 12.98420 7.339814 .0089385 .02141 .12 598 .00204 61.75490 8.33827 19.97153 4.08817
12 .048008 .65604 381.44135 14.61769 79.56682 127.27985

-139-Eliminating a, the three equations obtained were: 0.25606b - 1.00980c + 0.4S639d = 44.059141.00980b - 7.34813c + 2.76733d - 229.331790.48639b - 2.76738c + 1.40327d * 73.55789b, c and d were evaluated by determinants.
The denominator of the determinants was found to be:D » -.67933 + .07169 + 3.7707 * -0.23057
Solving for b:b*D = -116.88978 + 16.28026 + 57.34279 = -43.26673
b = - A ? 2 3 0 5 7 3' “ 1 8 7 *6 5 1 1 7
Solving for c:D*c « 30.27955 - 26.30433 + 5.05052 - 9.02574
c « -*23057 ** -39.14533
The values of the constants in this case were bigger and the differences between negative and positive terms were much bigger. The big negative values for the constant c proves that the data does not fit the equation for adsorption of octanol controlling and that the data seems to be closer to fitting the mechanism of surface reaction controlling than the adsorption of octanol controlling.

-140-
BACK CALCULATIONS OF DATA USING THE DETERMINED VALUED OF CONSTANTS
The equation for surface reaction controlling is:
P . teL ka*b <papb- hdffi )(1+PAKA + pbk b + PRKp)
letksL K^Kg “ Q
PA#PB “ ZSL'HS. . GK.The equation can now be written as:
r * ______ Sl£___________*_(i +Pa k a *p b k b +Pr V
The values of Q, , Kg and K^ have been obtained from the experimental data by using the method of least squares, G c^n be evaluated. By picking a value of x, corresponding values of p^, pg and pg can be calculated (Appendix). Thus all the terms in the right- hand side of the rate equation are known and by substituting for these terms in the equation, values of r could be evaluated. Tables XXI and XXII show the values of different terms of the equation for corresponding value of x f for the experiments made at 244.5°C. and 218°C. The reciprocals of the rates were plotted against x to give the curve in Fig. 15.
SinceFdx = rdW
W/Fr

-141-the value of W/F for a particular value of x could be obtained from graphical integration of the curve, x l/r. In this way three points on each curvewere back calculated. It is seen from the figures (Figs. 16, 17, 18, 19) that the calculated points closely follow the curves obtained experimentally.

TABLE XXIBack Calculation Data -Surface heaction Controlling 244.5°C.g = 2.33842, Ka = 1*29602, KB = 8.48513, Kp * 3.94797, Z = 1 + pAKA +pgKB +p^Kp
No, X PA PB Pfi G QG kapa1 .00100 .394 .394 .093 .15448 .36124 .510631 .00200 .3015 .3015 .1855 .08788 .20550 .390751 .00350 .1628 .1628 .3242 .01728 .04408 .210992 .0010 .485 .290 .0995 .13978 .32686 .628572 .00175 .409 .215 .175 .08525 .19935 530072 .00300 .2855 .0912 .2985 .01822 .04261 .370013 .00075 .603 .212 .0795 . 1 2 2 3 0 .28599 .781503 .0015 .522 .133 .1595 .06720 .15714 .676523 .00225 .4425 .0527 .2394 .01829 .04277 .573494 .0005 .722 .138 .057 .09936 .23235 .935734 .000875 .680 . 0 9 6 .099 .06442 .15064 .88129A .00160 .5980 .0136 .1812 .005253 .01228 .775025 .00075 .2015 .2015 .03355 .04050 .09471 .261155 .0015 .1680 .1680 .0670 .20783 .06508 .217735 .00265 .1165 .1165 .1185 .01234 .02886 .150996 .0075 .2460 .1520 .0360 .03728 .08718 .138826 .00175 .1978 .1040 .0841 .01995 .04665 .256356 .00310 .1332 .0393 .1490 .003288 .007693 .172637 .0005 .3035 .1152 .6257 .03940 .09213 .393347 .00125 .2560 .0768 .0641 .01999 .04675 .343457 .00232 5 .2095 .0216 .1194 .003274 .00765 .271528 .0005 .3485 .0067 .0274 .02317 .05418 .451668 .0010 .3215 .0395 .0545 .01244 .02909 .416678 .00160 .2884 .00658 .0875 .001226 .00288 .37377
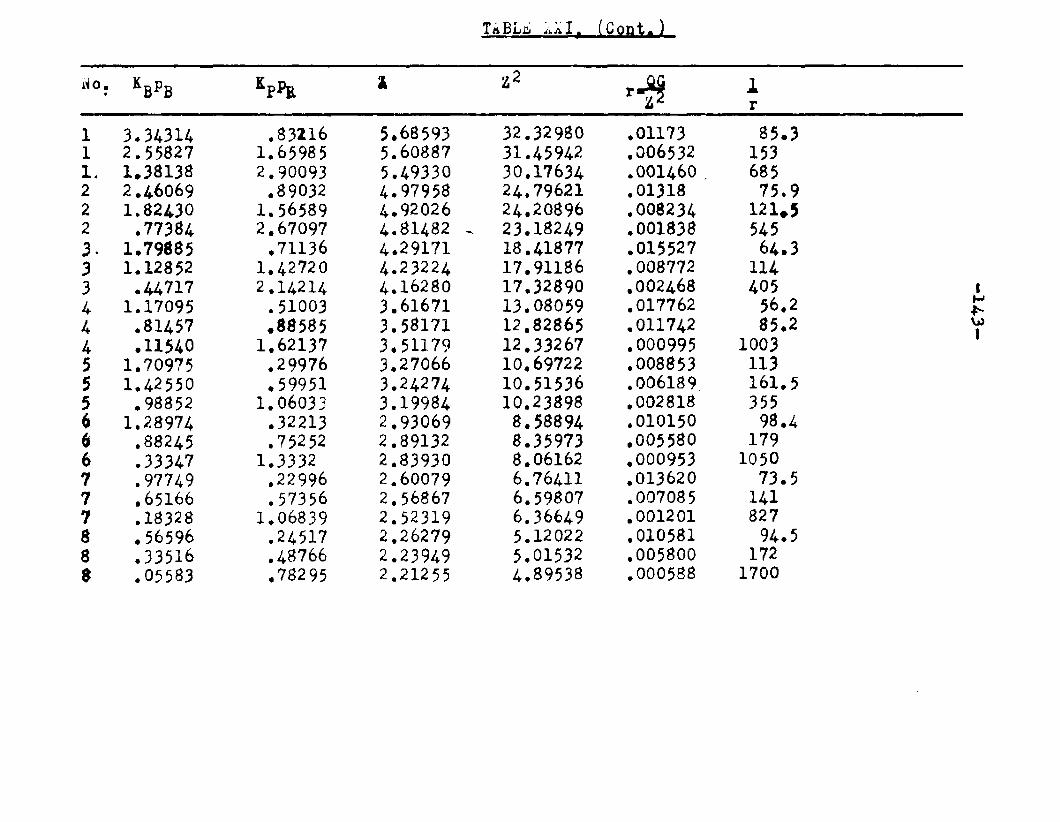
TABLa AXIr (Cont.)
Ao.* k bpb k p*e 1 Z2 OlcM 1
T
1 3.34314 .83116 5.68593 32.32980 .01173 85.31 2.55827 1.65985 5.60887 31.45942 .006532 1531 . 1.38138 2.90093 5.49330 30.17634 .001460 6852 2.46069 .89032 4.97958 24.79621 .01318 75.92 1.82430 1.56589 4.92026 24.20896 .008234 121*52 .77384 2.67097 4.81482 23.18249 .001838 5453- 1.79885 .71136 4.29171 18.41877 .015527 64.33 1.12852 1.42720 4.23224 17.91186 .008772 1143 .44717 2.14214 4.16280 17.32890 .002468 405A 1.17095 .51003 3.61671 13.08059 .017762 56.24 .81457 .88585 3.58171 12.82865 ,011742 85.24 .11540 1.62137 3.51179 12.33267 .000995 10035 1.70975 .29976 3.27066 10.69722 .008853 1135 1.42550 .59951 3.24274 10.51536 .006189 161.55 .98852 1.06033 3.19984 10.23898 .002818 3556 1.28974 .32213 2.93069 8.58894 .010150 98.46 .88245 .75252 2 .89132 8.35973 .005580 1796 .33347 1.3332 2.83930 8.06162 .000953 10507 .97749 .22996 2.60079 6.76411 .013620 73.57 .65166 .57356 2.56867 6.59807 .007085 1417 .18328 1.06839 2.52319 6.36649 .001201 8278 .56596 .24517 2.26279 5.12022 .010581 94.58 .33516 .48766 2.23949 5.01532 .005800 1728 .05583 .78295 2.21255 4.89538 .000588 1700
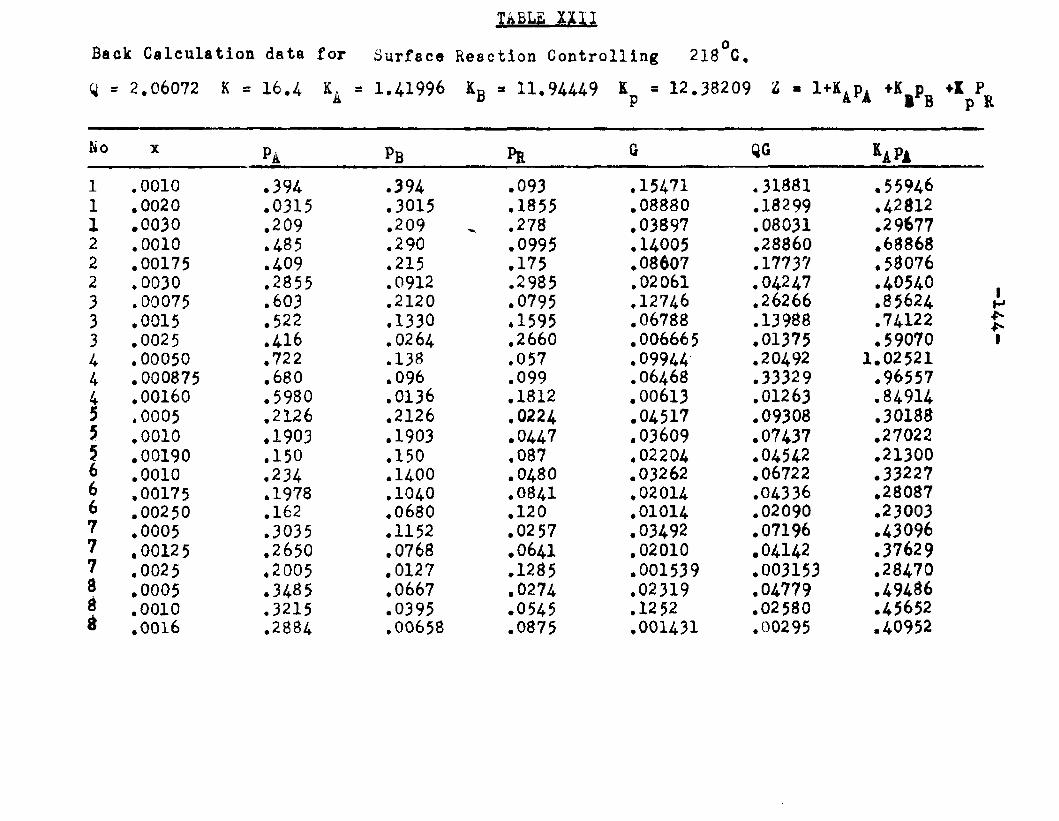
TABLE XXIIBack Calculation data for Surface Reaction Controlling 218°C.Q = 2.06072 K = 16.4 K, * 1.41996 Kg » 11.94U9 K = 12.38209 Z » 1+kaPa +K«P +X p" P A * 1 8 p R
No X Pa PB PH G QG KAPA1 .0010 .394 .394 .093 .15471 .31881 .559461 ,0020 .0315 .3015 .1855 ,08880 .18299 .428121 *0030 .209 .209 .278 .03897 .08031 .296772 .0010 .485 .290 .0995 .14005 .28860 .688682 ,00175 .409 .215 .175 .08607 .17737 .580762 .0030 .2855 .0912 .2985 .02061 .04247 .405403 .00075 .603 .2120 .0795 .12746 ,26266 .856243 .0015 .522 .1330 .1595 .06788 .13988 .741223 .0025 .416 .0264 .2660 .006665 .01375 .590704 .00050 .722 .138 .057 .09944 .20492 1.025214 .000875 .680 .096 .099 .06468 .33329 .965574 .00160 .5980 .0136 .1812 .00613 .01263 .849145 .0005 .2126 .2126 .0224 .04517 .09308 .301885 .0010 .1903 .1903 .0447 .03609 .07437 .270225 .00190 .150 .150 .087 .02204 .04542 .213006 .0010 .234 .1400 .0480 .03262 .06722 .332276 .00175 .1978 ,1040 .0841 .02014 .04336 .280876 .00250 .162 .0680 .120 .01014 .02090 .230037 .0005 .3035 .1152 .02 57 .03492 .07196 .430967 .0012 5 .2650 .0768 .0641 .02010 .04142 .376297 .0025 .2005 .0127 .1285 .001539 .003153 .284708 .0005 .3485 .0667 .0274 .02319 .04779 .494868 .0010 .3215 .0395 .0545 .12 5 2 .02580 .456528 .0016 .2884 .00658 .0875 .001431 .00295 .40952
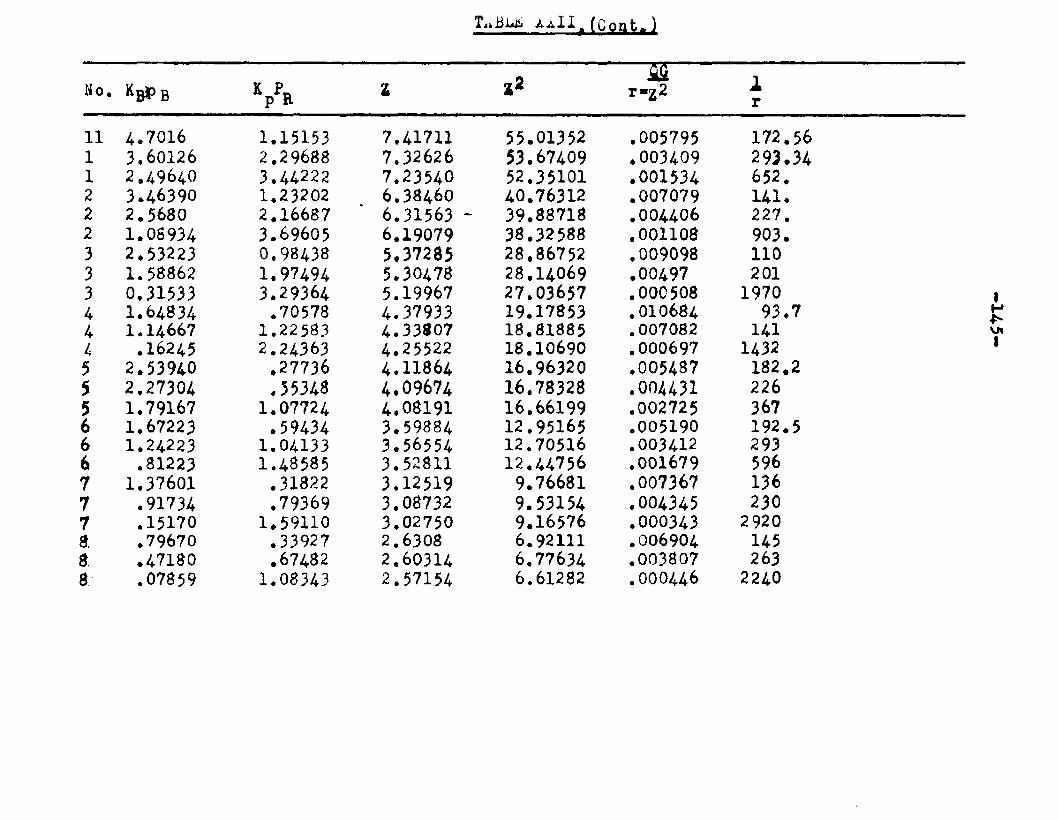
HBUC j a a II^(Coat.)
No. kb$ b K P_ P ft 2 22 r-^2 1r11 a .7016 1.15153 7.41711 55.01352 .005795 172.561 3.60126 2.29688 7.32626 53.67409 .003409 293.341 2.49640 3.44222 7.23540 52.35101 .001534 652.2 3.46390 1.23202 6,38460 40.76312 .007079 HI.2 2.5680 2.16687 6.31563 - 39.88718 .004406 227.2 1.OS934 3.69605 6.19079 38.32588 .001108 903.3 2.53223 0.98438 5.37285 28.86752 .009098 1103 1.58862 1.97494 5.30478 28.14069 .00497 2013 0.31533 3.29364 5.19967 27.03657 .000508 19704 1.64834 .70578 4.37933 19.17853 .010684 93.74 1.14667 1.22583 4.33807 18.81885 .007082 1414 .16245 2.24363 4.25522 18.10690 .000697 14325 2.53940 .27736 4.11864 16.96320 .005487 182.25 2.27304 .55348 4.09674 16.78328 .004431 2265 1,79167 1.07724 4.08191 16.66199 .002725 3676 1.67223 .59434 3.59884 12.95165 .005190 192.56 1.24223 1.04133 3.56554 12.70516 .003412 2936 .31223 1.48585 3.52811 12.44756 .001679 5967 1.37601 .31822 3.12519 9.76681 .007367 1367 .91734 .79369 3.08732 9.53154 .004345 2307 .15170 1.59110 3.02750 9.16576 .000343 2 920a .79670 .33927 2.6308 6.92111 .006904 145a .47180 .67482 2.60314 6.77634 .003807 2638. .07859 1.08343 2.57154 6.61282 ,000446 2240
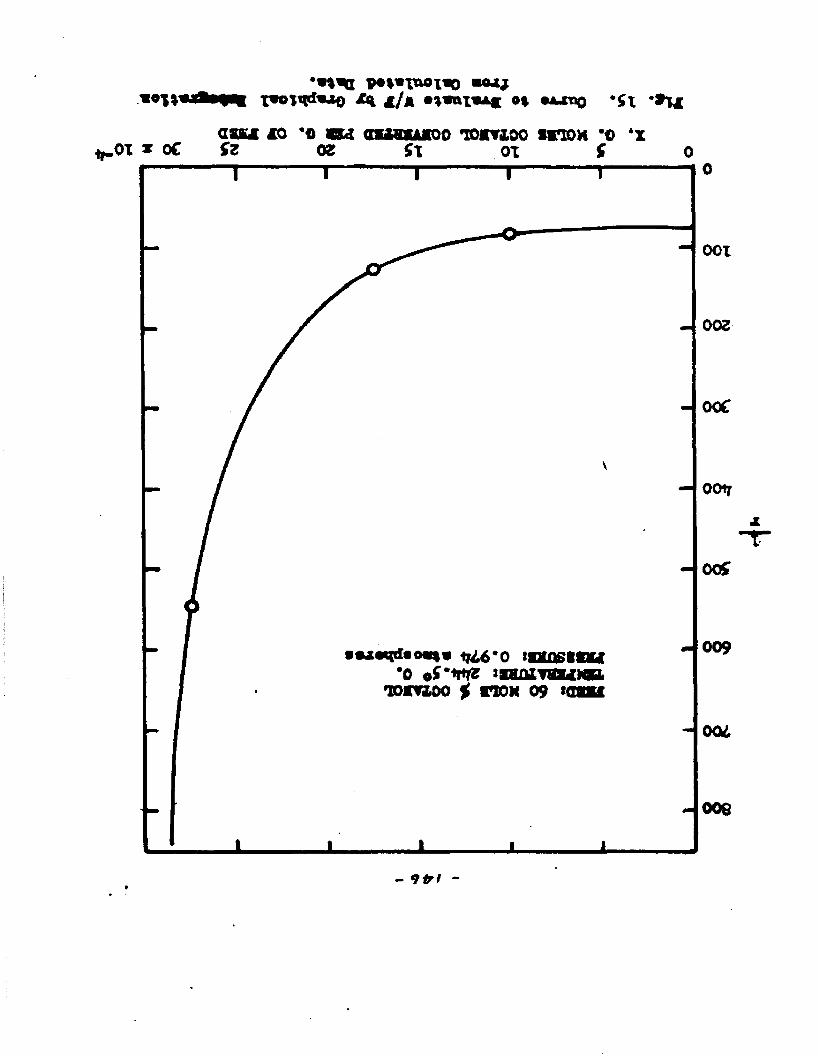
*-01
PM*T*oi*D aooj t*OfqcI«Jo A, i/a o% lunQ *£x *>U
a x u 10 *o w ausxAioo tomyioo sroox •© *xsz* oc OS 01 0
oox
002
OOC
009S M w p te o v iv ©46*0 tntQSKCU‘0 Ofmmz T H U M B
to x tio o % noN 09 ><nou
008

10“* X *9
t HWIRASUHBs 2**.5° >• IBS80KB: 0.97* itaoiph«niBET P0I2TTS: Calculated
H D CCMP08TYI0V, KXS 9 OOBUKXi 0*> O 60A 70 0 BO
w/r iA.no or o t t u m v n o r to i*b» b a s , booiiI_________ I_________ u ..— ..I *0.2 o o O'* 0.5
Hfc.j6. O D m n l M m a function of Y/l.
a#-/
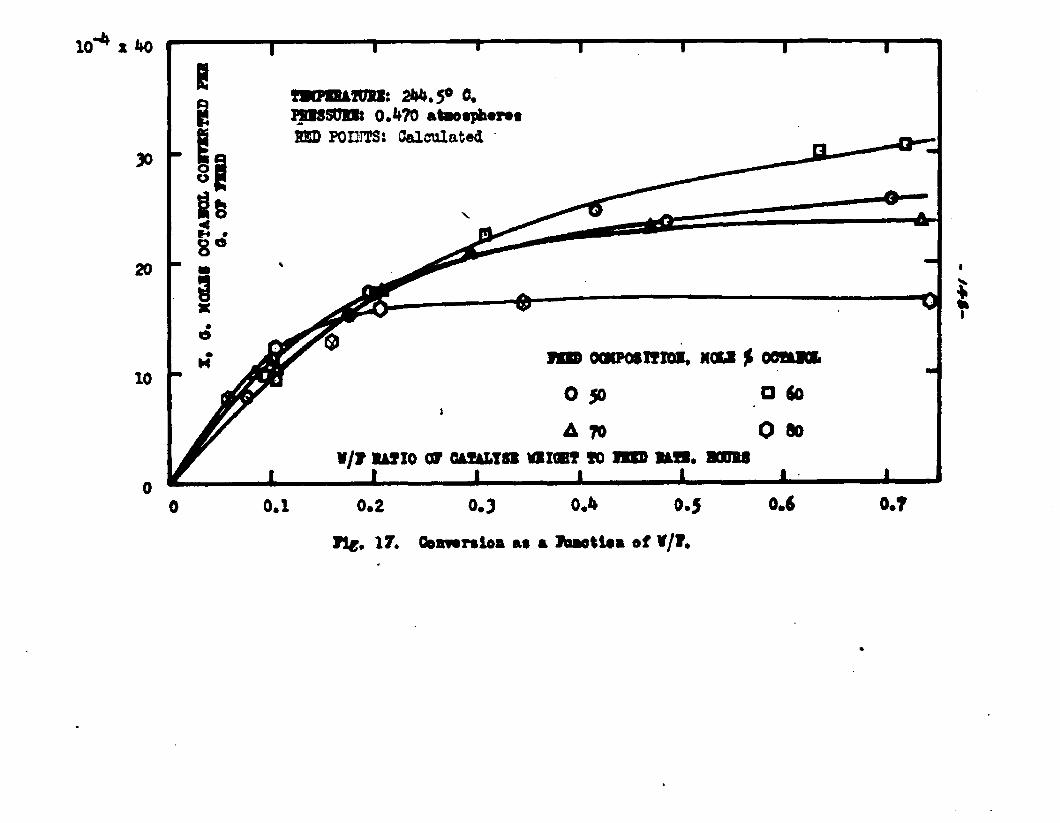
10+ x bO
30
20
10
S mnilOXS: 2W».5° 0. nOSSOKIt 0.*»70 ataoepherotBSD PODITS: Calculated '
S s
o
0.1 0.2 0.3 0.4 0.5
Tig* 17. Ooarerelon a* a tootle* of V/T*0.6 0.T
-#*v
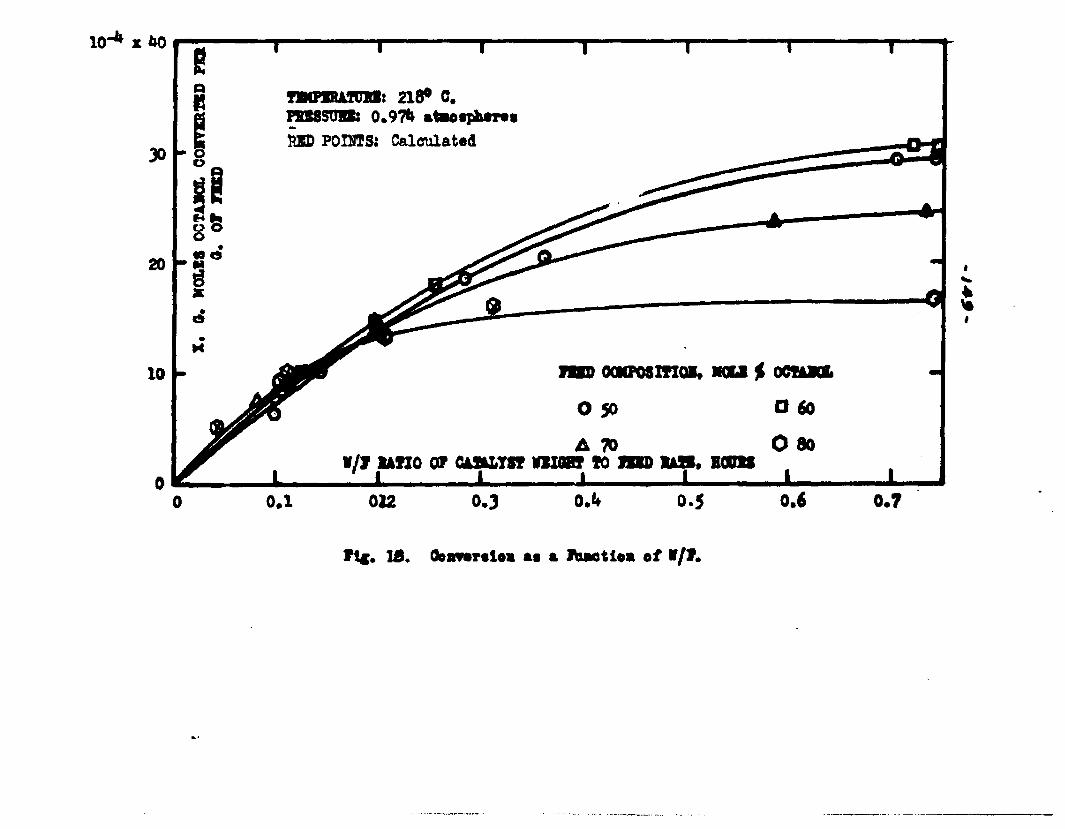
10-* x 60
30
20
10
0 0 0*1 012 0.3 0.4 0.5 0.6 0.7
Bn m u m i i 218° c.FBS8S0BB: 0.976 ataoophmaiz s POIKTS: Calculated
§
t o d ooMPosmoB. mqu % o c t r a
o 50 o 60
A 70 O 80V/J 1A9I0 07 CiULISt WIKJH9 90 HID UB. BCOtt I I I_________ I_________ I
71*. IB. Oenvereloa at a Ibactloa of V/7.

10"* x fco
BMFERATGKI: 218° 0. mSSJKSt 0.U7O ataomharasEED POIHTS; Calculated
8
3
IBS OMKSITKa. KOU $ OOtUOL0 50 0 60A n Oso v/i uiio or ottum w o n » m o u b , boom1
0.2 0.3 0.4 0.5 0.6fig, 19. Ooarar*lon a* a Inaction of V/T.
0.7

-151-Summarv of Kate Equations and Constants
The esterificatlon reaction was found to be having the mechanism of surface reaction controlling whose equations could be written as:
W/F - p XdxI r
r „ Q* ( PA*PB —(1+PAkA +PBkE +pRp )2
Where Q. - lrs.L.K^. KBThe values of the constants were:
t°C. Q Ka Kb Ip K218 2.06 1.42 11.95 12.38 16.4244.5 2.34 1.30 8.49 8.95 11.4

CONCLUSIONSThe work reported on the previous pages had led
to the following conclusions:1. The low Iron, low silica bauxite catalyst
used in this investigation has relatively high fraction of Internal voids, the value of the fraction of internal voids being 0.558. The same value expressed as porosity is 55.8%.
2. The activity of the catalyst used in this investigation is less than that of the catalyst used by Heeder; but the equilibrium constants for theesterification of n-octyl alcohol by acetic acid are same for both catalysts. The present catalyst has been taken out of a second shipment of the sam pie from the same company, now operating under a different name.
Previous name: Attapulgus Clay Co.210 W. Washington Square Philadelphia 5, Is.
Present name: Porocel Co.same address
3. The volumenometer used proved to be a simple and easy device to measure the porosity of catalysts, provided helum gas was used.
U* The low iron, low silica bauxite is proved to be excellent catalyst for the vapor phase esteri- fication of n.octyl alcohol with acetic acid. Side

-153-re a ct i on a and decomposition of the products was very insignificant between the temperatures of 218° and 24.4*5°C, By proper selection of the temperature and ratio of feed rate to catalyst weight, it is possible to obtain conversions approaching equilibrium values.
5. The rate determining step in the vapor phase esterification reaction is the surface reaction taking place between adjacently adsorbed molecules.By the back calculation of the data from the values of the constants determined by the method of least squares, it is seen that the mechanism of the surface reaction controlling very well fits the experimental data, between the temperatures of 218 and 24,4. 5°C.It is seen that the deviation of the calculated points from the experimental curves is greatest In case of the runs made with feeds containing 50 mole % octyl alcohol and for the runs made with feeds having excess alcohol the deviations are very small. Hence this mechanism seems to apply very well for the reaction when the feed contained an excess of alcohol.
6. To make the feed mechanism work under reduced pressure, the simplest device that can be used is to Introduce a capillary tube between the feed mechanism and the reactor. The size of the capillary selected should be such that the pressure drop created by it is slightly greater than the pressure

-154-drop existing between the pump and the reactor. The feed pump when operating has to overcome the excess pressure drop and thus gets a chance to operate against a positive pressure, avoiding the suction of the feed into the reactor,
7. It was found that for the reactor used longitudinal diffusion was not negligible. The calculations of longitudinal diffusion for different runs are given in the Appendix. For the runs made with faster feed rates of about 40 g./nr. and 20 g./hr. the longitudinal diffusion was found to be 10 to 16%. For slower feed rates the back diffusion given by the equation was much larger. This seems to Indicate that back diffusion is not negligible. However, the equation which is used to calculate longitudinal diffusion seems to be more suitable for incremental height of the catalyst bed. The conversions obtained at slow feed rates were close to the equilibrium conversions and hence increasing the amount of catalyst could not have increased the conversion to any extent. The high values of back aiffusion at low feed rates, given by the equation makes it appear that the equation used in calculating longitudinal diffusion is not the right one to be used for the data obtained in this investigation*
8. The catalyst adsorbs moisture at room

-155-temperature but at high temperatures i.e., 218°—244.5°C. no moisture is adsorbed.
9. The magnitudes of adsorption of the pure reactants and products, by the catalyst are in the following order of descendency* octyl aloohol, octyl acetate, water and acetic acid.
10. Keeder's data, when given a similar mathematical treatment as data in this investigation seems to point more towards the mechanism of surface reaction than that of adsorption of octyl alcohol.
11. A catalyst bed surrounded by a vapor bathof a pure chemical compound simplifies the maintenance of a nearly constant temperature:
Naphthalene 218°C.Diethylene glycol 2.44,5°C.12. The relation between weight of catalyst,
feed rote, and conversion are given by:
______ ksL ka k b (pa -pb- ^ S r s )(1+ Pa Ka + PbkB + pr k p )
where: r = reaction rate, g. moles of alcohol per(hour)(g. of feed)
\f * g. of catalyst F =* feed rate g./hr.

- 1 5 6 -x = conversion, g. moles of alcohol per g. of feed
* partial pressure of octanol in vapor stream^A * ~ a(lsorption equilibrium constants of
octanol and acid
Kp “ % + KSA, B, R and S - represent, n—octyl alcohol,
acetic acid, n-octyl acetate and water, respectively.

-157-Re commendations
1, It would be desirable to study the reaotion with feeds containing excess qf acid,
2, Additional runs at lower pressures than,4-70 atm. should be carried out to see if the mechanism holds for wide ranges of temperatures and pressures.
3, The same reaction should be carried out usingacids of higher molecular weights to study the effectof acid on the reaction. Similarly with a particular acid the reaction should be carried out using alcohols of different molecular weight,
4* Other catalysts like silica gel should be triedand the mechanism established. This will tell moreabout the reaction itself,
5. It is very desirable to find ways to directly measure the adsorption equilibrium constants of various components of the reaction for a particular catalyst,
6. In order to avoid the possibility of back diffusion, it is recommended that greater amounts of catalyst be used along with increased feed rates.

AFiPfiflPIX
Longitudinal diffusionThe following equations from Hougen and Watson,
are used here*
- f l n 4 * rA /»B kZm — EabJT &J±_A A ' B KT U + pA * A )Qt A Zwhere
F « feed rate lb./hr. ft.^nA = moles of A per unit mass of feedn-t = total number of moles per unit mass of feedrA * rate of reaction of A moles/lb.hr.
DAm » mean diffusion coefficient of A in thestrean at point Z
i k - (*2S=}=S)= bulk density of catalyst bed
Diffusivities were calculated using Gilliland'sequation:--------------------------- ----------
T 3/2 I 1 i 1dab . .0043 r(Vii/3 + vBV5)i V "* "bThe mean diffusivities were obtained from the
equation:
O-Na ) °Am “ nB °AB + NR °AR + NS °A5where NA , Ng - - - - = average mole fractions of
components A, B - - - in the diffusional film.

-159-Calculation of D oct_©Bter
Temp - 245°C. - 518°K.Pressure ■ 0.974 atm. v octanol ■ c8h g0 C * 8 x 14*8
H - 18 x 3.7 0 = 1 x 11.0
v ester = ^10^20^2 C = 10 x 14.8 H = 20 x 3.7 0 - 2 x 11.0
(.0043)(518)3/2
118.4 66. 5 11.0
195.9 148. 074.022.0
244.0
1 1 T30 * T72
oct—ester 974 (19 5 . 91>/3+2 44. 01/3 )2
(.0043X11780) .974 x 145 x .1161
» 0.0417 cm2/sec.. (0.0417)(3.87) = 0.161 ft2/hr.
imilarlyDoct-water was f°und be 0.707 ft2/hr. and Doct-acid to be °*328 ft /hr.Now longtiduinal diffusion will be calculated for
K un No. 7:feed ■ 20.585 g./hr. 50 mole % octanol F * 8.34 lb./hr. ft2

-160-Catalyst used « 5*8 gr./b m ^ g./in.3 a 0.856 g./c.c. = 53*height of catalyst ■ 5.8 x 1 1
14. .785 12
x - .002545Partial pressures for this value of x
Poet ■ °'252pacid “ °'252P + 0.235*esterp + * 0.235*wete r
(1-NA ) DAm = (.252)(.328) + (.235)(.161) .748 DAm = .2867
DAm “ 0.383 ft.2/hr.
Average rate for an x * .002545 Is (.00505 + .0106/2 = .00782
rearranging the equation for longitudinal noting that £ A = 0
_ A nA (-nA a^y _ . -F). r / B d z
= r A*>Am» - FRTnt^2
R = 1.3145 atm. ft.3/lb. mol°K.= 0.974 atm.
ntB .0105
4 lb./ft.3
- .044 ft.
.235 (.707)
diffusion

- 1 6 1 -- A n * « « . Q f l 732 x 52*L , k *QLL
A («383)(.974)_______________(518)(1.3145)(.044)7*0105)
“JnA M *orW$2- %°Lk . .002 567,155
Second terst in the equation: r/| Az.00782 x 53.4 x .044 = 0.1835
Last term » (,3$3H.974) x «°Q25fr = 0 00306(518) (l.3145TTT0T05) x .044
Last term is 16.7/6 of the second term./. Back diffusion is 16.7/6.
Similarly for hun No. 5, with feed rate of 40.329 g./hr. back diffusion according to the above equations was found to be 10.4%.
For a value of x calculation of corresponding values of partial preasurea;_______
given: Feed = 60 mole % octanolx = .00250 g. mole oct/g.feedp = .974 atm.
3 moles oct = 3 x 130.23 = 390.692 moles acid 2 x 60.05 = 120.10
510.79 g.= .002 5 g. moles oc-tanol . 510.79 e.
g. feed 5 moles feed= 1.278 g. moles of octanol converted per 5 moles
of feed.

-162-mol.fr. part.pr.
Octanol loft - 3-1.278 - 1.722 .345 .335Acid left » 2-1.278 - 0.722 .145 .141
Ester in reaction * 1.278 .256 .249Water H " * 1.278 .256 .249
mols 5.00 1.000 .974
♦• * PA - .335
PB = .141
PR - .249
ps * .249

- 1 6 3
BIBLIOGRAPHY(10) Berthelot and P*eau de St. Gilles, A m . Chlm.,(3) & l, 385 (1862).(34) Buckley and Altpeter, R. J., "Vapor Phase Catalytic Esterification Rates", Chem. Engr. Progress,AZ, 243-50 (1951).( 7) Caquil, M., "The Esterification of Cyclohexanol and some of its Homologs", Conpt. rend., 178. 323—6 (1924)J "The Esterification of Homologs of Cyclohexanol" , Compt. rend., 178. 1538-40 (1924).(13) Coria, P. E., "Equilibrium Constant of the System Ethanol-Acetic Acid", R^v, facultad Ciene. quim.(Univ. nacl. Laplata), 67, (1935); C. A., 7427.(4-5) Durruty, C. A,, "The Catalytic Action of Hydrogen CHoride on the System Acetic Acid, Ethanol, Lthyl Acetate, Water", Anales. asoc. quim. Argentina, i.2, 227 (1931); C. A., 26, 3721.(20) Edgar, G., and Schuyler, W. H., "Esterification Equilibrium in the Gaseous Phase", J. Am. Chem. Soc.,A£, 64 (1924).(24) Essex, H., and Clark, J. D., "Free Energy of Formation of Ethyl Acetate Equilibrium in the Gaseous Phase", J. Am. Chem. Soc., ^A, 1290—1306 (1932).(40) Francis, A. W., and Oxnard, E. P., "An Improved Volumenometer", Ind. Eng. Chem. Anal. Ed., A, 170 (1923).

-164-(22) Frolich, K., Carpeter, G. B., and Knox, tf. J.,Jr. "Vapor Phase Eaterification of Acetic Acid by Ethyl Alcohol", J. Am. Chem. Soc., 52 f 1565—70 (1930),(30) Goldanskii, V. I., "Heterogeneous Catalysis in Multicomponent Adsorption Layers Esterification Equilibrium in Two-Phase Systems", J. Phys. Chem., (U.S.S.ft), 2JL, 431 (1947); C. A. £1, 6801.(31)' Goldanskii and Chirkov, N. M., J. Phys. Chem., (O.S.S.R.), 242, 1333 (1946); C. A., Al, 2973.(27) Halford, J. 0., and Brundage, D., "Vapor Phase Esterification Equilibrium", J. Am. Chem. Soc., 2A, 36-40 (1942).( 9) Hartman, K. J., Storms, L. B. and Gassman, A. G., "Effect of Polar Groups Upon Esterification Velocities of Substituted Benzoic Acids with Cyclohexanol", J.Am. Chem. Soc., £1, 2167 (1939).(29) Hoe rig, H. F., Hanson, D., and Kowalke, j . j-.., "Vapor Phase Esterification Hates", Ind. Eng. Chem.,22, 575 (1943).(38) Hougen, 0. A., and Watson, K. M., "Chemical Process rrinciples", Part III. John Wiley and Hons,New York, 1947.(39) Hulburt, H. M., Ind. Eng. Chem., 22, 1012 (1944); 22, 1063 (1945).(26) Jatkar, 3. K. u. and Gajendragad, N. G., "Vapor Phase Esterification Equilibrium", J. Am. Chem. Hoc.,
59, 798 (1937).

-165-( 2) Keys, D. B., "Esterification Process and Equipment" , Ind. Eng. Chem., 1096 (1932).(/ 5) Kirk, h. E., and Othmer, D. F., "Esterifica — tion" (editors). "Encyclopedia of Chemical Technology" Vol. V. The Interscience Encyclopedia Inc., New York,1950.(28) Knox, W. J., Jr., and Burbrldge, J. N., "Vapor Phase Esterification over Zirconium Oxide", J. Am.Chem. Soc., ££, 999-1001 (194-3).(37) Langmuir, Irivng, "Theory of Adsorption",. Phys. Rev., £, 79-80 (1915).(14) Leyes, C. E., and Othmmr, D. F., "Esterification of Butyl Alcohol and Acetic Acid", Ind. Eng. Chem.,22, 968 (1945).(42) Lindsey, holland, "Chemical equilibrium in the System Acetic Acid, n—octyl Alcohol, n-octyl acetate and Water", M. Sc. Thesis, The Ohio State University, 1953.(18) Maihle, A . , and de Godon, F., "Preparation of Esters by Catalysis", Bull. ~oc. Chem., 2^, 101 (1921).( 6) Michael, A., a n d Jolg&st, K., "Nature of Steric Hindrance III. r- ela ti ons hip Between Structure of Aliphatic Alcohols and the Velocity of Their Esterifi— cation", Ber. , 2., 3157 (1909).

-166-(19) Milligan, C. K., Chappell, J. T., and Reid, E. E., "Esterification in the Presence of Silica Gel", J.Phys. ohera., 872 (1924).(ll) Foznanski, S., "Equilibrium of the Esterifica- tion Reaction inthe Liquid Phase", Roczniki Chem.,377 (I928)j C. A., 2^, 1559.(35) Reeder, Clyde, Jr., "Studies of Vapor Phase Catalytic Esterification Using Acetic Acid", Ph.D. Dissertation, The Ohio State University, 1951.( 1) Reid, E. E., "Esterification", Ind. Eng. Chem.,
1596 (1943).( 3) heid, E. E., "Egterification of Thio benzoic Acid and of Benzoic Kcid by Hercaptan", ^m. ohera.J., 489-504 (1910).(17) Reid, E. E ., "Esterification" in Groggins*"Unit Processes in Organic Chemistry Synthesis",3rd ed. Me Grew Hill, New l'ork, 1947.( 4) Roberts, I., and Urey, Pi. C., "A Study of the Esterification of Benzoic Acid with Methyl Alcohol Using Isotopic Oxygen", J. Am. ohem. Soc., 60. 2391 (1938).( 8) Smith, II. A., "Kinetics of the Catalyzed Esteri- ficstion of Normal Aliphatic Acids in Methyl Alcohol", J. Am. Chem. Soc., 6^, 254 (1939)} "The Acid Catalyzed Esterification of Aliphatic Acids", 62. 1136 (1940).

-167-(33) Spangenberg, J. P., Industrai y Quimica Buenos Aires, 2, 393 (1945); C. A., £1, 4028.(12) Swietoslawski, W. J . , "Determining the Equilibrium Constant of Laterification", J. Phys. Chem.,21, 701 (1933).(21) Swietoslawski, J . , and Poznanski, S., "TheEquilibrium Constant of ^sterification Reaction in the Gaseous Phase", r.oczni ki Chera., £, 527-41 (541 French).(25) Swietoslawki, W. J., and Salcewicz, Jr., "The uquilibrium Constant of Esterification in the Gaseous, Coexisting with the liquid Phase", Compt. rend., 199 1308-10 (1934).(36) Taylor, H. S., Rational iiesearch Council "Twelfth Report of Committee on Catalysis", John Jiley and Cons (1940).(2 3) Tidwell, A. o., and i.e id, E. l, . , "Vapor Phase Esterification in the rresence of oilica Gel", J. Am. Chem. Soc., $2, 4353 (1931).(16) Trimble, . S., and Richardson, E. L. , J. Am.ohem. ooc., j 2, 1018 (1940); "Equilibrium in an Esteri-fieation reaction with Perchloric Acid as Catalyst.(32) Vernon, A. A., and brown, B. M., "Vapor Phase Esterification of Benzoic Acid with Ethyl Alcohol", Ind. Eng. Chem., 534 (1940); "Vapor Phase Esterif icationof Benzoic Acid with Ethyl Alcohol. Effect of Oxides

-168-on the Catalytic Activity of Silicon Carbide and Alundum", JJstlA. , 21, 1289 (194-1).(41) Yang, K. H., and Hougen, 0. A., "Determination of Mechanism of Catalyzed Gaseous Reactions", Chem. Eng. Progress, ,46, Ho. 3. 146 (1950)*

-169-
AUTOBIQGhAEILI__I, Yerram Venkaksham, was born in Bhongir, Hydera
bad, India, September 14, 1923. I received my secondary school education in the Government School in Singareni Collieries and in Secunderabad. My undergraduate training was obtained at Nizam College Madras University for which I received the degree Bachelor of Science in 194.2, having majored in Chemistry, From Osmania University I received the degree Master of Science in 1944*
I served the Hyderabad Government as a research assistant for four years. In 1948 I received a scholarship to study Chemical Engineering in the U.S.A. In 1950 I finished my undergraduate training In the Illinois Institute of Technology for which I received B .Ch.£. •
I entered the graduate school of The Ohio S^ate University from where I received the degree M.Sc. in1951. I had a university scholarship for six months.I held the position of research assistant for the Re
search Foundation, while completing the requirements
for the degree Doctor of Philosophy.




















