
Calponin1 inhibits dilated cardiomyopathy development in mice through the εPKC pathway
9
This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues. Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited. In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/authorsrights
Transcript of Calponin1 inhibits dilated cardiomyopathy development in mice through the εPKC pathway

This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/authorsrights

Author's personal copy
Calponin1 inhibits dilated cardiomyopathy development inmice throughthe εPKC pathway☆
Dan Lu a, Li Zhang a, Dan Bao a, Yingdong Lu a, Xu Zhang a, Ning Liu a, Wenping Ge a, Xiang Gao a,Hongliang Li b, Lianfeng Zhang a,⁎a Key Laboratory of Human Disease Comparative Medicine, Ministry of Health, Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences & Comparative Medical Center,Peking Union Medical College, Chinab Department of Cardiology, Renmin Hospital of Wuhan University, Cardiovascular Research Institute of Wuhan University, China
a b s t r a c ta r t i c l e i n f o
Article history:Received 22 October 2013Received in revised form 24 January 2014Accepted 8 February 2014Available online 25 February 2014
Keywords:Calponin1Dilated cardiomyopathy (DCM)Heart failure (HF)Transgenic miceEpsilon isoform of protein kinase C (εPKC)
Background: Calponin1 (CNN1) is involved in the regulation of smooth muscle contraction in physiological situ-ation and it also expresses abnormally in a variety of pathological situations. We found that the expression ofCNN1 decreased significantly in the heart tissue of a cTnTR141W transgenic dilated cardiomyopathy (DCM)mouse model and an adriamycin (ADR)-induced DCM mouse model, suggesting that CNN1 is involved in thepathogenesis of DCM. However, the role of CNN1 on cardiac function, especially on pathogenesis of DCM, hasnot been clarified. In this study, we tested whether rescued expression of CNN1 could prevent the developmentof DCM and investigated its possible mechanisms.Methods and results: TheDCMphenotypeswere significantly improvedwith the transgenic expressionof CNN1 inthe cTnTR141W×CNN1double transgenic (DTG)mice,whichwas demonstrated by the survival, cardiac geometryand function analyses, as well as microstructural and ultrastructural observations based on echocardiographyand histology examination. The expression of CNN1 could also resist the cardiac geometry breakage and dysfunc-tion in the ADR-induced DCM mice model. Meanwhile, the epsilon isoform of protein kinase C (εPKC) activatorand inhibitor could reverse the activation of εPKC/ERK/mTOR pathway and DCM phenotypes in the cTnTR141W
and cTnTR141W × CNN1 double transgenic (DTG) mice.Conclusions: εPKC/ERK/mTOR pathway activation induced by the rescued expression of CNN1 contributed to theimprovement of cardiac dysfunction and pathological changes observed in the DTG mice. CNN1 could be a ther-apeutic target to prevent the development of DCM and heart failure (HF).
© 2014 Elsevier Ireland Ltd. All rights reserved.
1. Introduction
Calponin1 (CNN1, also called basic calponin or calponin h1) is a34-kDa troponin-like molecule that was originally isolated from thesmoothmuscles of bovine aortas and chicken gizzards and was indicat-ed later to be present in most vertebrate smooth muscles. CNN1 bindsto actin, tropomyosin, and calmodulin and is involved in the regulationof smoothmuscle contraction in physiological situation [1–6]. CNN1 ex-presses abnormally in a variety of pathological situations, including ab-normal gastrointestinal motility and multiple tumors [7–13]. The boneformation is increased which associated with enhancement of bonemorphogenetic protein responses in mice lacking smooth muscle
CNN1 [14]. CNN1 is not only involved in smooth muscle contraction,but also function as a integrating molecule in signal transduction thatinteracts with the proteins including ERK1/2, p160Rho kinase, proteinkinase C (PKC) α and PKC ε [6,15–19]. CNN1 plays a significant role inthe regulation of vascular smooth muscle contraction [6,20–23] andmay also be a useful target for therapies aiming to disrupt tumor vascu-lature [12,24–29].
It has been shown that CNN1 is expressed in embryonic cardiacmus-cle and in smooth muscle cells (SMCs) [30–32]. We found that the ex-pression of CNN1 decreased significantly in the heart tissue of acTnTR141W transgenic dilated cardiomyopathy (DCM) mouse modeland an adriamycin (ADR)-inducedDCMmousemodel [33–35], suggest-ing that CNN1 is involved in the pathogenesis of DCM. However,the effect of CNN1 on cardiac function, especially on pathogenesis ofDCM, remains largely unknown. In the present study, we generated acardiac-specific overexpression of CNN1 transgenic mouse to rescuethe expression of CNN1 in heart tissue of cTnTR141W transgenic micein vivo to determine whether and by what mechanism this would pre-vent the development of DCM.
International Journal of Cardiology 173 (2014) 146–153
☆ All authors take responsibility for all aspects of the reliability and freedom from bias ofthe data presented and their discussed interpretation.⁎ Corresponding author at: Building 5, Panjiayuan Nanli, Chaoyang District, Beijing
100021, China. Tel.: +86 10 87778442; fax: +86 10 67776394.E-mail address: [email protected] (L. Zhang).
http://dx.doi.org/10.1016/j.ijcard.2014.02.0320167-5273/© 2014 Elsevier Ireland Ltd. All rights reserved.
Contents lists available at ScienceDirect
International Journal of Cardiology
j ourna l homepage: www.e lsev ie r .com/ locate / i j ca rd

Author's personal copy
2. Materials and methods
2.1. Generation of the transgenic mice
Full-length human CNN1 cDNA was cloned into an expression plasmid under theα-MHC promoter. The heart-specific CNN1 transgenic mouse was generated by microin-jection andwas genotyped by PCRwith the primers 5′AAGGGCGGAACATCATTGGGCT and5′CTCGAAGATCTGCCGCTTGGT. For genotyping, a 215-bp fragment of the transgenic genewas amplified with 35 PCR cycles consisting of 94 °C for 30 s, 62 °C for 30 s and 72 °C for30 s. The expression of CNN1 was screened by western blot analysis using a rabbit mono-clonal antibody (Abcam). The α-MHC-cTnTR141W (referred to as cTnTR141W) dilated car-diomyopathy (DCM) transgenic mice were previously generated in our laboratory [33].The α-MHC-cTnTR141W × CNN1 double transgenic (referred to as DTG) mice were gener-ated throughmating of theα-MHC-CNN1 (referred to as CNN1)with the cTnTR141W trans-genicmice. Allmice used in this studyweremaintained on a C57BL/6J genetic backgroundand were bred in an AAALAC-accredited facility. The use of animals was approved by theAnimal Care and Use Committees of the Institute of Laboratory Animal Science of PekingUnion Medical College (ILAS-GC-2012-001).
2.2. ADR-induced DCM mice model
The CNN1 mice and non-transgenic (NTG) littermates at 2 months of age were usedfor adriamycin (ADR) treatment. In the groups of NTG + ADR and CNN1 + ADR, ADRwas injected intraperitoneally at a constant volume of saline at 4 mg/kg every other dayfor a total of 2 weeks [34,35]. The groups designated as NTG+ saline and CNN1+ salinereceived the same amount of saline. Echocardiographywas performed on all mice at 0 day(the day before the treatment of ADR). All of the survivingmicewere chosen for follow-upechocardiography at 3 months of age (2 weeks after the cessation of ADR treatment) andwere used to analyze the expression of CNN1 in the heart tissues.
2.3. Survival analysis
The cumulative percent mortality in each group of mice was calculated every month,and the data from 1 to 8 months of agewere summarized. Upon the death of eachmouse,the body was autopsied by a pathologist and the morphological and pathological changesof the heart were recorded. Kaplan–Meier curves for the survival analysis were comparedby the log-rank test (SPSS 16.0 software).
2.4. Echocardiography
M-mode echocardiographywasperformed at 1, 3, 6 and 8 months of age on the trans-genic mice and their littermates with the small animal echocardiography analysis system(Vevo770, Canada) as previously described [34,36].
2.5. Histological analysis
For light microscopy, mice were euthanized by cervical dislocation at 6 months of ageand cardiac tissue was fixed in 4% formaldehyde and mounted in paraffin blocks, and thesections were stained with H&E or Masson trichrome as previously described [34,36] andanalyzed using Aperio ImageScope v8.2.5 software. For transmission electron microscopy(TEM), cardiac tissue was prepared as previously described [34,36]. Myocytes were ana-lyzed by an observer who was blinded to the genotypes of the mice.
2.6. RNA extraction, quantification and RT-PCR
Mice were euthanized by cervical dislocation at 6 months of age, and total RNA wasisolated from the heart tissues using TRIzol Reagent (Invitrogen). First-strand cDNA wassynthesized from 2 μg of total RNA using random hexamer primers and Superscript III re-verse transcriptase according to the manufacturer's protocol (Invitrogen). Procollagentype III α1 (Col3α1) mRNA was detected by RT-PCR using GAPDH for normalizationunder standard conditions (primers: for Col3α1, forward 5′CTCAAGAGCGGAGAATACTGG and reverse 5′CAATGTCATAGGGTGCGATA; for GAPDH, forward 5′CAAGGTCATCCATGACAACTTTG and reverse 5′GTCCACCACCCTGTTGCTGTAG).
2.7. Protein extraction and immunoblotting
Mice were euthanized by cervical dislocation, and total protein lysates frommouse heart tissues were prepared as previously described [34,36]. After performingSDS-PAGE and transferring the bands to nitrocellulose (Millipore), the membranes wereincubated overnight with antibodies against CNN1 (Abcam), εPKC, phospho-p44/42MAPK (Erk1/2) (Thr202/Tyr204), phospho-mTOR (Ser2448), p44/42 MAPK (Erk1/2)andmTOR. After incubationwith the appropriate secondary antibody for 1 h at room tem-perature, the antibody binding was detected with an HRP-conjugated immunoglobulinG (Santa Cruz) using a chemiluminescence detection system (Santa Cruz). The quantita-tive analysis of the level of phosphorylated proteins uses corresponding total protein fornormalization, others use GAPDH for normalization, and bands were quantified usingthe ImageJ software.
2.8. Immunohistochemical staining
Mice were euthanized by cervical dislocation, and the sections of the hearts were pre-pared by a standard pathological procedure. For paraffin-embedded heart tissues, 6-μmsections were dewaxed, rehydrated, unmasked, blocked and then incubated with ananti-CNN1 monoclonal antibody (Abcam) overnight at 4 °C. The sections were washedwith PBS and incubatedwith an anti-rabbit horseradish peroxidase conjugated secondaryantibody for 1 h at room temperature, and all slides were counterstained with hematoxy-lin. The slides were then dehydrated, coverslips were placed on top, and the specimenswere photographed. Images of the sections were collected and analyzed using AperioImageScope v8.2.5 software.
2.9. Drug treatment
The cTnTR141W mice were treated with an εPKC activator (TOCRIS, FR 236924,CAS Number: 28399-31-7, IUPAC Name: 2-[(2-pentylcyclopropyl)methyl]cyclopropaneoctanoic acid, 0.60 mol/kg weight) [37], and the DTG mice were treatedwith an εPKC inhibitor (Sigma, PKC isoenzyme inhibitor PKC Y translocation peptide,0.15 mol/kg) [38] from 6 months of age. In the group of cTnTR141W + activator andDTG + inhibitor mice, the activator or inhibitor was injected intraperitoneally everyother day for a total of 3 weeks. The groups of cTnTR141W + saline and DTG + saline re-ceived the same amount of saline. Echocardiography was performed on all mice at0 day (the day before the treatment with the εPKC activator/inhibitor). All survivingmice were chosen for follow-up echocardiography analysis, histological analysis and ex-pression analysis of signaling molecules in the heart tissues at 7 months of age (1 weekafter cessation of drug treatment).
2.10. Statistical analysis
The data were analyzed by one-way ANOVA for multiple groups followed by Tukey'spost-hoc analysis. The data are expressed as themeans± SD from individual experiments.The differences were considered significant at P b 0.05.
3. Results
3.1. Rescue of CNN1 expression in cTnTR141W mice by heart-specifictransgenic expression of CNN1
We tested the expression of CNN1 in the NTG and DCM hearts fromcTnTR141W mice and the ADR-treated mice, and the results indicatedthat CNN1 was expressed in the adult hearts, and its expressionwas decreased significantly by 54.0% and 59.1%, respectively, in theDCM hearts from cTnTR141W mice and ADR-treated mice (Fig. 1A and B,n = 3, P b 0.01). To verify whether the rescue of CNN1 expression inthe heart tissues of cTnTR141Wmice could improve the DCM phenotypes,we generated CNN1 transgenic mice overexpressing CNN1 in the hearttissues (Fig. 1C–E, n = 3, P b 0.01), which were indistinguishable fromtheir non-transgenic (NTG) littermates at birth and in young mice. TheCNN1 mice were then crossed with the cTnTR141W mice to generate thecTnTR141W × CNN1 double transgenic (DTG) mice, which compensatedfor the loss of CNN1 expression in the heart tissues of the cTnTR141W
mice (Fig. 1F).
3.2. Rescue of CNN1 expression increased the survival rate of cTnTR141W
mice
Cumulative mouse mortality data of the NTG, CNN1, cTnTR141W andDTG mice were recorded between 1 and 8 months of age. Dilationand mural thrombi were observed in the hearts of mice during thepost-mortem examinations. The survival rate was 100% in the NTGgroup (n = 67) and CNN1 group (n = 34), while the survival rate wasonly 81.4% in the cTnTR141W group (Fig. 2, n = 59, P b 0.001 versusNTG group) until 8 months of age. However, the survival rate in theDTG group (n = 25) was increased by 14% compared with that of thecTnTR141Wgroup (Fig. 2, P b 0.01) due to the rescued expression of CNN1.
3.3. Rescue of CNN1 expression improves cardiacmorphology breakage anddysfunction in DCM mice models
Rescue of CNN1 expression increased the survival rate of cTnTR141W
mice. Furthermore, we analyzed the cardiac geometry and function in
147D. Lu et al. / International Journal of Cardiology 173 (2014) 146–153

Author's personal copy
DTG mice and ADR-treated CNN mice by echocardiography examina-tion to verify whether rescue of CNN1 expression could also improvethe phenotypes of the DCMmodels.
The CNN1mice (n=20)presented thick-walled ventricles and small-er left ventricular diameters compared with the NTG mice (n = 25) at6 months of age (Fig. 3A and B, left ventricular diameter at end-diastole(LVEDD) decreased by 8.1% (P b 0.01); left ventricular posterior wallthickness at end-diastole (LVPWD) increased by 15.5% (P b 0.05)).Mean-while, transgenic expression of CNN1 in the DTG mice (n = 28) signifi-cantly improved the DCM phenotypes, which was demonstrated by the6.2% reduced LVEDD (P b 0.05), 26.0% increased LVPWD (P b 0.001)and 28.7% increased left ventricular percent fractional shortening(LVFS) (P b 0.001) compared with that of the cTnTR141W mice (n = 22)at 6 months of age (Fig. 3A–C). The data for the echocardiographic char-acteristics of the NTG, CNN1, cTnTR141W and DTG mice were shown inTable S1–S4 at 1, 3, 6 and 8 months of age.
Furthermore, the expression of CNN1 could also reduce the cardiacgeometry breakage and dysfunction in the ADR-induced DCM models.The DCM phenotypes were obviously induced by ADR treatment in theNTG mice (n = 10), with increased LVEDD (5.8%, P b 0.05), decreasedLVPWD (21.0% P b 0.001) and FS% (16.5%, P b 0.01), compared withthat of the shammice (n=10) 2 weeks after the cessation of ADR treat-ment (Fig. 3D–F). However, the DCM phenotypes induced by ADR were
Fig. 1. Determination of the CNN1 expression and generation of the transgenic mice. (A) The expression of CNN1 in the heart tissues of cTnTR141W transgenic mice and adriamycin (ADR)induced DCMmicemodels. (B) The quantitative analysis of the expression of CNN1 using GAPDH for normalization (n=3, ⁎⁎P b 0.01 versusNTGmice). (C) Theα-MHC-CNN1 transgenicconstruct was generated by inserting the target gene under the control of the α-MHC cardiac-specific promoter and the transgenic mice were created following microinjection. (D) Theexpression of CNN1 in the heart tissues of CNN1 transgenic mice at 2 months of age weremeasured bywestern blot. (E) The quantitative analysis of the expression of CNN1 using GAPDHfor normalization (n = 3, ⁎⁎P b 0.01 versus NTG mice). (F) Immunodectection of CNN1 in the heart tissues of NTG, CNN1, cTnTR141W and DTG transgenic mice at 6 months of age.
Fig. 2. Rescue of CNN1 expression increased the survival rate of cTnTR141W transgenicmice. Cumulative percent mortality for NTG (n= 67), CNN1 (n= 34), cTnTR141W (n= 59,⁎⁎⁎P b 0.001 versus NTG group) and DTG (n = 25, ##P b 0.01 versus cTnTR141W group)transgenic mice was calculated every month from 1 to 8 months of age.
148 D. Lu et al. / International Journal of Cardiology 173 (2014) 146–153

Author's personal copy
improved in the CNN1mice, with no significance between CNN1+ADRmice (n = 7) and CNN1 + saline mice (n = 8) (Fig. 3D–F, the otherechocardiographic characteristics were supplied in Table S5).
3.4. Rescue of CNN1 expression ameliorates cardiac pathological changes incTnTR141W mice
The morphological changes of themicrostructure and ultrastructureof the ventricles and myocytes from NTG, CNN1, cTnTR141W and DTGmice were observed by light and electron microscopy.
We did not find any obvious changes in the microstructure and ul-trastructure in theCNN1mice. However, themalalignment and collagenaccumulation in the interstitial space were significantly reduced in theDTGmice compared with the cTnTR141W mice due to the transgenic ex-pression of CNN1 (Fig. 4A–E). Both the prominence of swollen mito-chondria and poorly organized myofibrils were also clearly lessenedby rescuing the expression of CNN1 in the DTG mice compared withthat of the cTnTR141W mice (Fig. 4F).
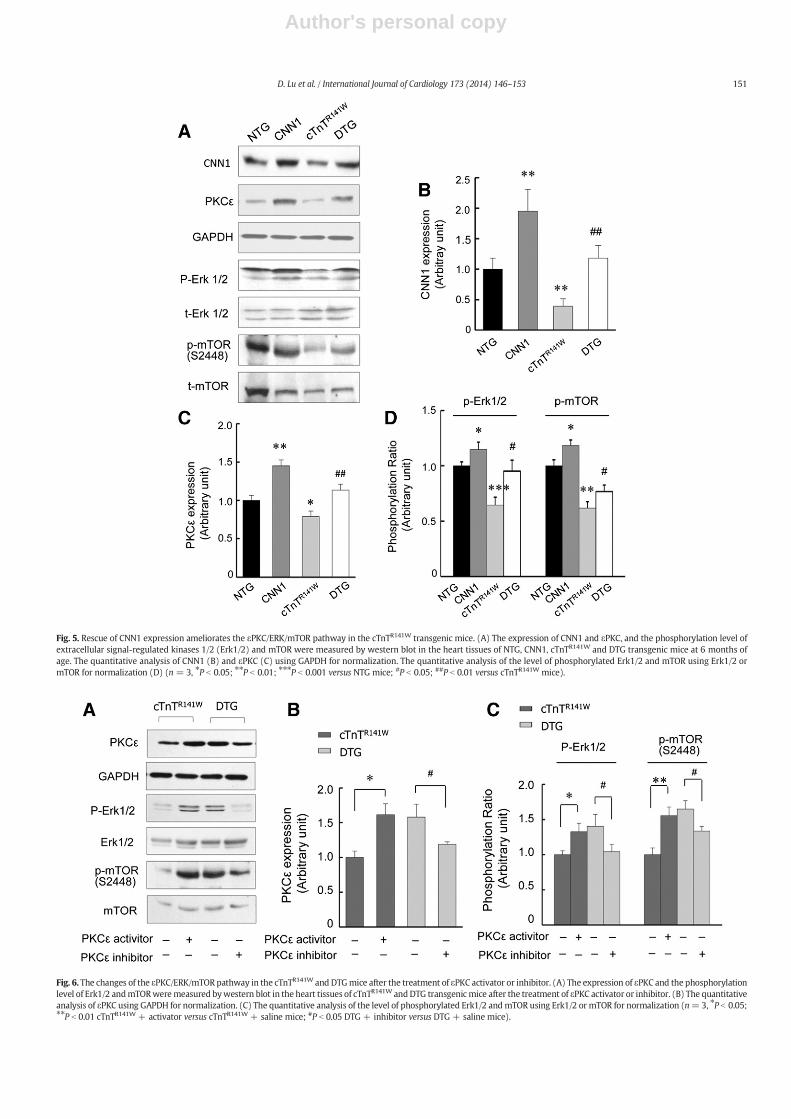
3.5. Rescue of CNN1 expression ameliorates the εPKC/ERK/mTOR pathwayin the cTnTR141W mice
CNN1 interacts with εPKC, Erk1/2 and mTOR (mammalian target ofrapamycin), which are all involved in the pathogenesis of DCM andHF. Therefore, we measured the expression of CNN1 and εPKC and thephosphorylation level of extracellular signal-regulated kinases 1/2(Erk1/2) andmTOR in the heart tissues from the NTG, CNN1, cTnTR141W
and DTG mice. Corresponding with the decrease of CNN1, the level ofεPKC expressionwas also reduced in the cTnTR141Wmice, and the phos-phorylation of Erk1/2 and mTOR was subsequently inhibited comparedwith the NTG mice. CNN1 can increase the εPKC expression and phos-phorylation level of Erk1/2 and mTOR in the CNN1 mice comparedwith the NTG mice. When the decreased expression of CNN1 was res-cued, the εPKC expression and the phosphorylation of Erk1/2 andmTOR were reversed in the DTG mice compared with the cTnTR141W
mice (Fig. 5A–D).
3.6. The εPKC is essential for the prevention of CNN1 on cardiomyopathydevelopment
To verify whether the εPKC is essential for the prevention of CNN1on cardiomyopathy development, the cTnTR141W mice were treatedwith the εPKC activator (FR236924, 0.62 mol/kg), and the DTG micewere treated with the εPKC inhibitor (PKC isoenzyme inhibitor PKC Ytranslocation peptide, 0.15 mol/kg). The treatment with the εPKC acti-vator increased the phosphorylation level of Erk1/2 (n = 3, P b 0.05)and mTOR (n = 3, P b 0.01) in the cTnTR141W mice (Fig. 6A–C). Mean-while, the treatment with the εPKC inhibitor decreased the phosphory-lation level of Erk1/2 (n=3, P b 0.05) andmTOR (n=3, P b 0.05) in theDTG mice (Fig. 6A–C).
The cardiac geometry and morphology were improved significantlyby the treatment with the εPKC activator in the cTnTR141W mice, andshowed a similar phenotype to the DTGmice, which was demonstratedby a 5.8% reduction in the LVEDD (P b 0.05), a 13.5% increase in the
Fig. 3.Rescue of CNN1 expression improves cardiac geometry and dysfunction in transgenic and chemical inducedDCMmicemodels. Echocardiographic parameters of left ventricular (LV)diameter at end-diastole (LVEDD) (A), LV posterior wall at end-diastole (LVPWD) (B) and LV fractional shortening (LVFS) (C)were analyzed inNTG, CNN1, cTnTR141W and DTG transgenicmice at 1, 3, 6 and 8 months of age. Echocardiographic parameters of LVEDD (D), LVPWD (E) and LVFS (F) were analyzed in NTG and CNN1mice after the treatment of ADR (⁎P b 0.05;⁎⁎P b 0.01; ⁎⁎⁎P b 0.001 NTG + ADR versus NTG + Saline mice, CNN1 + ADR versus CNN1 + Saline mice given no significance (NS)).
149D. Lu et al. / International Journal of Cardiology 173 (2014) 146–153

Author's personal copy
LVPWD (P b 0.05) and an improved malalignment of the myofibrils(Fig. 7A–E, cTnTR141W + activator (n = 7) versus cTnTR141W + saline(n = 15)). The improved cardiac geometry and morphology causedby CNN1 expression in the DTG mice were eliminated by treatmentwith the εPKC inhibitor, which was demonstrated by a 4.3% increasein the LVEDD (P b 0.05) and a 14.4% decrease in the LVPWD (P b 0.01)and deteriorated malalignment of the myofibrils (Fig. 7A–E, DTG +inhibitor (n = 9) versus DTG + saline (n = 13)). The other echocar-diographic characteristics induced by the εPKC activator or inhibitortreatment also showed a tendency of reversion (Table S6).
4. Discussion
CNN1 expresses abnormally in abnormal gastrointestinal motilityand multiple tumors [7–13]. A study of gene expression fingerprint ofhuman heart failure indicated that the expression of CNN1 was downregulated in failure heart [39]. Our result showed that CNN1 wasexpressed in the myocytes and decreased obviously in DCM heartsfrom the cTnTR141W transgenic mice and ADR-induced DCM mice(Fig. 1). When the decrease of CNN1 was rescued by the transgenic ex-pression of CNN1, the DCM phenotypes of the cTnTR141W transgenicmice were obviously improved in the DTG mice. The amelioration wasdemonstrated as the increased survival rate (Fig. 2), the improved car-diac geometry and the function parameters of echocardiography(Fig. 3), the reduced collagen accumulation in the interstitial space
and the inhibition of the damages of mitochondria (Fig. 4) due to therescued expression of CNN1 in the DTG mice.
CNN1 interacts with εPKC and connects PKC cascade to the ERK cas-cade as an adaptor protein [6,15–19]. The ERK1/2 activation appears tobe advantageous for a failing or dilatedmyocardium, and ERK1/2 activa-tion has been shown to confer cardioprotection in vivo against ischemiareperfusion injury [40–43], while the inhibition of the εPKC with a se-lective inhibitor can result in DCM and HF in mice [44], suggestingthat εPKC was important for the functional maintenance of heart. Ourresult showed that the εPKC expressionwas reduced and the phosphor-ylation of Erk1/2 and mTOR was subsequently inhibited in thecTnTR141W mice. When the decreased expression of CNN1 was rescued,the εPKC expression and the phosphorylation of Erk1/2 andmTORwerereversed in the DTG mice (Fig. 5). The εPKC activator increased thephosphorylation level of Erk1/2 and mTOR and could also improve sig-nificantly the DCM phenotype of cTnTR141W transgenic mice, mean-while, the improved cardiac function and geometry caused bytransgenic expression of CNN1 in the DTG mice were eliminatedby treatment with the εPKC inhibitor (Figs. 6–7), which suggestedthat εPKC was essential for the prevention of the CNN1 on DCMdevelopment.
DCM is a primary heart muscle disease characterized by the dilationof the heart chambers and the attendant impaired systolic functions[45]. Despite recent advances in pharmacological and surgical therapies,disability and morbidity due to DCM remain common [46–48]. Heart
Fig. 4. Rescue of CNN1 expression ameliorates cardiac microstructure and ultrastructure in cTnTR141W transgenic mice. (A) H&E staining patterns of the whole-heart longitudinal sectionsfrom mice at 6 months of age. (B) Magnification of H&E-stained sections of left ventricle (magnification ×400), showing disparate pathological changes. (C) Magnification of Massontrichrome-stained sections of left ventricle (magnification ×400); myocytes, stained red; collagenous tissue, stained green. (D) The expression of Col3α1 was detected by RT-PCR.(E) The quantitative analysis of the expression of Col3α1 using GAPDH for normalization (n=3, ⁎P b 0.05; ⁎⁎P b 0.01versusNTGmice; #P b 0.05 versus cTnTR141Wmice). (F) TEM analysisof sarcomeres (blue hollow arrow) and mitochondria (blue star) of left ventricular free walls from mice at 6 months of age, showing disparate ultrastructural characteristics of hearttissues.
150 D. Lu et al. / International Journal of Cardiology 173 (2014) 146–153
Author's personal copy
Fig. 5. Rescue of CNN1 expression ameliorates the εPKC/ERK/mTOR pathway in the cTnTR141W transgenic mice. (A) The expression of CNN1 and εPKC, and the phosphorylation level ofextracellular signal-regulated kinases 1/2 (Erk1/2) and mTOR were measured by western blot in the heart tissues of NTG, CNN1, cTnTR141W and DTG transgenic mice at 6 months ofage. The quantitative analysis of CNN1 (B) and εPKC (C) using GAPDH for normalization. The quantitative analysis of the level of phosphorylated Erk1/2 and mTOR using Erk1/2 ormTOR for normalization (D) (n = 3, ⁎P b 0.05; ⁎⁎P b 0.01; ⁎⁎⁎P b 0.001 versus NTG mice; #P b 0.05; ##P b 0.01 versus cTnTR141W mice).
Fig. 6. The changes of the εPKC/ERK/mTOR pathway in the cTnTR141W and DTGmice after the treatment of εPKC activator or inhibitor. (A) The expression of εPKC and the phosphorylationlevel of Erk1/2 andmTORweremeasured bywestern blot in the heart tissues of cTnTR141W andDTG transgenicmice after the treatment of εPKC activator or inhibitor. (B) The quantitativeanalysis of εPKC using GAPDH for normalization. (C) The quantitative analysis of the level of phosphorylated Erk1/2 andmTOR using Erk1/2 or mTOR for normalization (n=3, ⁎P b 0.05;⁎⁎P b 0.01 cTnTR141W + activator versus cTnTR141W + saline mice; #P b 0.05 DTG + inhibitor versus DTG + saline mice).
151D. Lu et al. / International Journal of Cardiology 173 (2014) 146–153

Author's personal copy
transplants, though considered to be themost effective therapy for end-stage DCM, cannot be used for all DCM patients. Therefore, screeningand investigation of important modifier genes involved in the patho-genesis of cardiomyopathy are still the major concerns of the heat dis-ease field, which might lead to the discovery of new therapeuticstrategies. The abnormal expression of CNN1 may play a significantrole in a variety of diseases, including heart failure [7–13,39]. Modula-tors of PKC isozymes are currently in clinical trials for a variety of indi-cations, and recent clinical trials suggest that the systemic delivery ofinhibitors and activators of PKC isozymes is well-tolerated [49–57].
Our major funding was that CNN1 could improve the DCM pheno-types through the εPKC/ERK/mTOR pathway in mice, which suggestedthat CNN1 could be a therapeutic target to control the development ofDCM and HF, as the CNN1 is a potential target for therapies aiming todisrupt tumor vasculature [12,24–29].
Acknowledgments
This research was supported in part by the National Key TechnologyResearch and Development Program of the Ministry of Science andTechnology (2012BA139B02); National Science and Technology MajorProject of the Ministry of Science and Technology (2012ZX09301001-006); and PUMC Youth Fund (3332013140). The authors thankDr. Harold James for patiently reading and correcting the Englishwriting of this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.ijcard.2014.02.032.
References
[1] Takahashi K, Hiwada K, Kokubu T. Vascular smooth muscle calponin. A noveltroponin T-like protein. Hypertension 1988;11:620–6.
[2] Abe M, Takahashi K, Hiwada K. Effect of calponin on actin-activated myosin ATPaseactivity. J Biochem 1990;108:835–8.
[3] Winder SJ, Walsh MP. Smooth muscle calponin. Inhibition of actomyosin MgATPaseand regulation by phosphorylation. J Biol Chem 1990;265:10148–55.
[4] LehmanW. Calponin and the composition of smooth muscle thin filaments. J MuscleRes Cell Motil 1991;12:221–4.
[5] Takahashi K, Nadal-Ginard B. Molecular cloning and sequence analysis of smoothmuscle calponin. J Biol Chem 1991;266:13284–8.
[6] Winder SJ, Allen BG, Clement-Chomienne O, Walsh MP. Regulation of smooth mus-cle actin–myosin interaction and force by calponin. Acta Physiol Scand 1998;164:415–26.
[7] Wang X, Wu K, Zhang Z, Lan M, Jin J, Fan D. The effect of calponin and caldesmon inregulation of the gastrointestinal motility during pathophysiological adaptation.Zhonghua Nei Ke Za Zhi 2001;40:459–62.
[8] Islam AH, Ehara T, Kato H, et al. Calponin h1 expression in renal tumor vessels:correlations with multiple pathological factors of renal cell carcinoma. J Urol2004;171:1319–23.
[9] Yanagisawa Y, Takeoka M, Ehara T, Itano N, Miyagawa S, Taniguchi S. Reduction ofcalponin h1 expression in human colon cancer blood vessels. Eur J Surg Oncol2008;34:531–7.
[10] Koganehira Y, Takeoka M, Ehara T, et al. Reduced expression of actin-bindingproteins, h-caldesmon and calponin h1, in the vascular smooth muscle inside
Fig. 7. CNN1 inhibits cardiomyopathy development in the cTnTR141W transgenic mice through εPKC pathway. Echocardiographic parameters of LVEDD (A), LVPWD (B) and LVFS (C)were analyzed in cTnTR141W and DTG transgenic mice after the treatment of εPKC activator or inhibitor (⁎P b 0.05 cTnTR141W + activator (n = 7) versus cTnTR141W + saline (n = 15)mice; #P b 0.05; ##P b 0.01DTG+ inhibitor (n=9) versusDTG+ saline (n=13)mice). (D) H&E staining patterns of thewhole-heart longitudinal sections frommice after the treatmentof εPKC activator or inhibitor. (E) Magnification of H&E-stained sections of left ventricle (magnification ×400), showing disparate pathological changes.
152 D. Lu et al. / International Journal of Cardiology 173 (2014) 146–153

Author's personal copy
melanoma lesions: an adverse prognostic factor for malignant melanoma. BrJ Dermatol 2003;148:971–80.
[11] Sasaki Y, Yamamura H, Kawakami Y, et al. Expression of smooth muscle calponin intumor vessels of human hepatocellular carcinoma and its possible association withprognosis. Cancer 2002;94:1777–86.
[12] Yamamura H, Yoshikawa H, Tatsuta M, Akedo H, Takahashi K. Expression of thesmooth muscle calponin gene in human osteosarcoma and its possible associationwith prognosis. Int J Cancer 1998;79:245–50.
[13] Sugenoya Y, Yoshimura A, Yamamura H, et al. Smooth-muscle calponin inmesangialcells: regulation of expression and a role in suppressing glomerulonephritis. J AmSoc Nephrol 2002;13:322–31.
[14] Yoshikawa H, Taniguchi SI, Yamamura H, et al. Mice lacking smoothmuscle calponindisplay increased bone formation that is associated with enhancement of bonemorphogenetic protein responses. Genes Cells 1998;3:685–95.
[15] Leinweber B, Parissenti AM, Gallant C, et al. Regulation of protein kinase C by thecytoskeletal protein calponin. J Biol Chem 2000;275:40329–36.
[16] Nakamura F, Mino T, Yamamoto J, Naka M, Tanaka T. Identification of the regulatorysite in smooth muscle calponin that is phosphorylated by protein kinase C. J BiolChem 1993;268:6194–201.
[17] Menice CB, Hulvershorn J, Adam LP, Wang CA, Morgan KG. Calponin and mitogen-activated protein kinase signaling in differentiated vascular smooth muscle. J BiolChem 1997;272:25157–61.
[18] Leinweber BD, Leavis PC, Grabarek Z, Wang CL, Morgan KG. Extracellular regulatedkinase (ERK) interaction with actin and the calponin homology (CH) domain ofactin-binding proteins. Biochem J 1999;344(Pt 1):117–23.
[19] Ping P, Zhang J, Huang S, et al. PKC-dependent activation of p46/p54 JNKs duringischemic preconditioning in conscious rabbits. Am J Physiol 1999;277:H1771–85.
[20] Gerthoffer WT, Pohl J. Caldesmon and calponin phosphorylation in regulation ofsmooth muscle contraction. Can J Physiol Pharmacol 1994;72:1410–4.
[21] Takahashi K, Yoshimoto R, Fuchibe K, et al. Regulation of shortening velocity bycalponin in intact contracting smooth muscles. Biochem Biophys Res Commun2000;279:150–7.
[22] Matthew JD, Khromov AS, McDuffie MJ, et al. Contractile properties and proteins ofsmooth muscles of a calponin knockout mouse. J Physiol 2000;529(Pt 3):811–24.
[23] Facemire C, Brozovich FV, Jin JP. The maximal velocity of vascular smooth muscleshortening is independent of the expression of calponin. J Muscle Res Cell Motil2000;21:367–73.
[24] Yamamura H, Hirano N, Koyama H, Nishizawa Y, Takahashi K. Loss of smoothmusclecalponin results in impaired blood vessel maturation in the tumor-host microenvi-ronment. Cancer Sci 2007;98:757–63.
[25] Horiuchi A, Nikaido T, Taniguchi S, Fujii S. Possible role of calponin h1 as a tumorsuppressor in human uterine leiomyosarcoma. J Natl Cancer Inst 1999;91:790–6.
[26] KanekoM, TakeokaM, OguchiM, et al. Calponin h1 suppresses tumor growth of Src-induced transformed 3Y1 cells in association with a decrease in angiogenesis. Jpn JCancer Res 2002;93:935–43.
[27] Ogura T, Kobayashi H, Ueoka Y, et al. Adenovirus-mediated calponin h1 gene therapydirected against peritoneal dissemination of ovarian cancer: bifunctional therapeuticeffects on peritoneal cell layer and cancer cells. Clin Cancer Res 2006;12:5216–23.
[28] Taniguchi S. Suppression of cancer phenotypes through a multifunctional actin-binding protein, calponin, that attacks cancer cells and simultaneously protectsthe host from invasion. Cancer Sci 2005;96:738–46.
[29] Takahashi K, Yamamura H. Studies and perspectives of calponin in smooth muscleregulation and cancer gene therapy. Adv Biophys 2003;37:91–111.
[30] Hossain MM, Hwang DY, Huang QQ, Sasaki Y, Jin JP. Developmentally regulatedexpression of calponin isoforms and the effect of h2-calponin on cell proliferation.Am J Physiol Cell Physiol 2003;284:C156–67.
[31] Draeger A, Gimona M, Stuckert A, Celis JE, Small JV. Calponin. Developmentalisoforms and a low molecular weight variant. FEBS Lett 1991;291:24–8.
[32] Duband JL, Gimona M, Scatena M, Sartore S, Small JV. Calponin and SM 22 as differ-entiation markers of smooth muscle: spatiotemporal distribution during avianembryonic development. Differentiation 1993;55:1–11.
[33] Juan F, Wei D, Xiongzhi Q, et al. The changes of the cardiac structure and function incTnTR141W transgenic mice. Int J Cardiol 2008;128:83–90.
[34] Lu D, Ma Y, Zhang W, et al. Knockdown of cytochrome P450 2E1 inhibits oxidativestress and apoptosis in the cTnT(R141W) dilated cardiomyopathy transgenic mice.Hypertension 2012;60:81–9.
[35] Smith HJ, Nuttall A. Experimental models of heart failure. Cardiovasc Res1985;19:181–6.
[36] Lu D, Lian H, Zhang X, et al. LMNA E82K mutation activates FAS and mitochondrialpathways of apoptosis in heart tissue specific transgenic mice. PLoS One 2010;5:e15167.
[37] Yamamoto S, Kanno T, Nagata T, Yaguchi T, Tanaka A, Nishizaki T. The linoleic acidderivative FR236924 facilitates hippocampal synaptic transmission by enhancingactivity of presynaptic alpha7 acetylcholine receptors on the glutamatergic termi-nals. Neuroscience 2005;130:207–13.
[38] Inagaki K, Koyanagi T, Berry NC, Sun L, Mochly-Rosen D. Pharmacological inhibitionof epsilon-protein kinase C attenuates cardiac fibrosis and dysfunction inhypertension-induced heart failure. Hypertension 2008;51:1565–9.
[39] Tan FL, Moravec CS, Li J, et al. The gene expression fingerprint of human heart failure.Proc Natl Acad Sci U S A Aug 20 2002;99(17):11387–92.
[40] Bueno OF, Molkentin JD. Involvement of extracellular signal-regulated kinases 1/2 incardiac hypertrophy and cell death. Circ Res 2002;91:776–81.
[41] Wang Y. Mitogen-activated protein kinases in heart development and diseases.Circulation 2007;116:1413–23.
[42] Lips DJ, Bueno OF, Wilkins BJ, et al. MEK1–ERK2 signaling pathway protects myocar-dium from ischemic injury in vivo. Circulation 2004;109:1938–41.
[43] Bueno OF, De Windt LJ, Tymitz KM, et al. The MEK1–ERK1/2 signaling pathwaypromotes compensated cardiac hypertrophy in transgenic mice. EMBO J 2000;19:6341–50.
[44] Palaniyandi SS, Sun L, Ferreira JC, Mochly-Rosen D. Protein kinase C in heart failure: atherapeutic target? Cardiovasc Res May 1 2009;82(2):229–39.
[45] Richardson P, McKenna W, Bristow M, et al. Report of the 1995 World HealthOrganization/International Society and Federation of Cardiology Task Force on thedefinition and classification of cardiomyopathies. Circulation 1996;93:841–2.
[46] Jessup M, Brozena S. Heart failure. N Engl J Med 2003;348:2007–18.[47] Ho KK, Anderson KM, Kannel WB, Grossman W, Levy D. Survival after the onset of
congestive heart failure in Framingham Heart Study subjects. Circulation 1993;88:107–15.
[48] Rakar S, Sinagra G, Di Lenarda A, et al. Epidemiology of dilated cardiomyopathy.A prospective post-mortem study of 5252 necropsies. The Heart Muscle DiseaseStudy Group. Eur Heart J 1997;18:117–23.
[49] Advani R, Peethambaram P, Lum BL, et al. A phase II trial of aprinocarsen, an anti-sense oligonucleotide inhibitor of protein kinase C alpha, administered as a 21-dayinfusion to patients with advanced ovarian carcinoma. Cancer 2004;100:321–6.
[50] Bates E, Bode C, Costa M, et al. Intracoronary KAI-9803 as an adjunct to primarypercutaneous coronary intervention for acute ST-segment elevation myocardial in-farction. Circulation 2008;117:886–96.
[51] The effect of ruboxistaurin on visual loss in patients with moderately severe to verysevere nonproliferative diabetic retinopathy: initial results of the Protein KinaseC beta Inhibitor Diabetic Retinopathy Study (PKC-DRS) multicenter randomizedclinical trial. Diabetes 2005;54:2188–97.
[52] Effect of ruboxistaurin in patients with diabetic macular edema: thirty-monthresults of the randomized PKC-DMES clinical trial. Arch Ophthalmol 2007;125:318–24.
[53] Ajani JA, Jiang Y, Faust J, et al. A multi-center phase II study of sequential paclitaxeland bryostatin-1 (NSC 339555) in patients with untreated, advanced gastric or gas-troesophageal junction adenocarcinoma. Invest New Drugs 2006;24:353–7.
[54] Casellini CM, Barlow PM, Rice AL, et al. A 6-month, randomized, double-masked,placebo-controlled study evaluating the effects of the protein kinase C-beta inhibitorruboxistaurin on skin microvascular blood flow and other measures of diabeticperipheral neuropathy. Diabetes Care 2007;30:896–902.
[55] Grossman SA, Alavi JB, Supko JG, et al. Efficacy and toxicity of the antisense oligonu-cleotide aprinocarsen directed against protein kinase C-alpha delivered as a 21-daycontinuous intravenous infusion in patients with recurrent high-grade astrocyto-mas. Neuro Oncol 2005;7:32–40.
[56] Herbst RS, Oh Y, Wagle A, Lahn M. Enzastaurin, a protein kinase Cbeta-selectiveinhibitor, and its potential application as an anticancer agent in lung cancer. ClinCancer Res 2007;13:s4641–6.
[57] Ritch P, Rudin CM, Bitran JD, et al. Phase II study of PKC-alpha antisense oligonucle-otide aprinocarsen in combination with gemcitabine and carboplatin in patientswith advanced non-small cell lung cancer. Lung Cancer 2006;52:173–80.
153D. Lu et al. / International Journal of Cardiology 173 (2014) 146–153




















