
Porous Alginate Hydrogels: Synthetic Methods for Tailoring the Porous Texture

Cite this: RSC Advances, 2013, 3, 5547
Blood triggered rapid release porous nanocapsules3
Received 30th October 2012,Accepted 23rd January 2013
DOI: 10.1039/c3ra22693j
www.rsc.org/advances
Tiffany P. Gustafson,{a Sergey A. Dergunov,b Walter J. Akers,a Qian Cao,a
Selena Magalotti,a Samuel Achilefu,a Eugene Pinkhassik*b and Mikhail Y. Berezin*a
Rapid-release drug delivery systems present a new paradigm in emergency care treatments. Such systems
combine a long shelf life with the ability to provide a significant dose of the drug to the bloodstream in
the shortest period of time. Until now, development of delivery formulations has concentrated on slow
release systems to ensure a steady concentration of the drug. To address the need for a quick release
system, we created hollow polyacrylate nanocapsules with nanometer-thin porous walls. Burst release
occurs upon interaction with blood components that leads to escape of the cargo. The likely mechanism of
release involves a conformational change of the polymer shell caused by binding albumin. To demonstrate
this concept, a near-infrared fluorescent dye indocyanine green (ICG) was incorporated inside the
nanocapsules (NCs). ICG-loaded nanocapsules demonstrated a remarkable shelf life in aqueous buffers
with no release of ICG for twelve months. Rapid release of the dye was demonstrated first in vitro using
albumin solution and serum. SEM and light scattering analysis demonstrated the retention of the
nanocapsule architecture after the release of the dye upon contact with albumin. In vivo studies using
fluorescence lifetime imaging confirmed quick discharge of ICG from the nanocapsules following
intravenous injection.
Introduction
A general trend in drug formulations centers on advanceddelivery systems where the in vivo release of a drug is carefullycontrolled to improve the effectiveness of therapy.1,2 Themajority of drug release systems developed to date rely uponthe sustained release of an active ingredient from the deliverymatrix to establish and maintain a therapeutically effectiveconsistent concentration within the body. Many systems havebeen developed and approved for the treatment of cancer,glaucoma, diabetes, and chronic pain.3–5 While sustained-release formulations are essential for the treatment of patientswith prolonged illnesses, there is an unmet need for a drugdelivery system where the active component is releasedquickly. Rapid release systems could find an immediateapplication in emergency medicine where a fast rise in drugconcentration is critical to saving lives. Therefore, a small butgrowing number of rapid release formulations have recentlybeen proposed as first aid or emergency treatments in life-threatening conditions, including thrombosis, stroke, acutepoisoning and sharp pain.6–9
Intensive care treatments are based on administering drugsthrough parenteral routes, such as an intravenous (IV)infusion. Although direct injection of the drugs in thebloodstream is currently standard for emergency applications,release systems provide the advantage to formulate therapeu-tic agents that would be too unstable for practical use intraditional intravenous administration. The factors affectingthe stability of intravenous medications are well documentedand include drug precipitation when added to intravenous (IV)solutions, incompatibility with other drugs, buffering agentsand preservative in the diluents.10–12 Formulation of the drugsthrough protective encapsulation-type drug release systems isexpected to minimize these mitigating phenomena and reducethe risk of potentially fatal side effects.
The current generation of controlled release systems basedon passive diffuse transport of the active ingredient underphysiological conditions cannot be adapted for emergencytreatment because of its relatively long diffusion time. The newgeneration of drug release systems utilizes the release ofentrapped cargo triggered by endogenous factors,13,14 e.g.,biodegradation of certain polymers,15 enzymatic reactions,16
change in pH17 or external factors such as light.18 While agreat number of triggered nanosystems have been proposed,none of these devices are suitable for rapid release.
Porous polymeric nanoparticles are frequently utilized incontrolled drug delivery for the encapsulation of activeingredients.19,20 The regulated pore size nanoconstructsaccommodate a variety of pharmaceuticals from small
aDepartment of Radiology, Washington University School of Medicine, St. Louis, MO
63110 E-mail: [email protected] of Chemistry, Saint Louis University, St. Louis, MO 63103
E-mail: [email protected]
3 Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra22693j{ Present address: Department of Chemistry, Texas A&M University, CollegeStation, TX 77842
RSC Advances
PAPER
This journal is � The Royal Society of Chemistry 2013 RSC Adv., 2013, 3, 5547–5555 | 5547

molecules to relatively large polypeptides.20,21 We recentlyreported the synthesis of thin wall hollow polymeric nanocap-sules with controlled pores sizes as promising candidates forthe cellular delivery of therapeutic or diagnostic agents.22–28
Spherical nanocapsules were formed by the polymerization ofstyrene derivatives or acrylates in the hydrophobic interior oflipid bilayers. Due to the nanometer-thin walls, these hollownanocapsules have a high payload capacity. They demonstratedexcellent cellular biocompatibility with no inhibition of cellularproliferation, induced apoptosis or generation of reactiveoxygen species.28
We have also shown that the confinement of molecules innanocapsules dramatically increases the molecules’ stability.27
High water solubility, a long shelf life, low cellular toxicity28
and the tunable pore size of the nanocapsules23–25 encouragedus to investigate this promising material as an emergency drugdelivery vehicle. To visualize the release in vitro and in livemice, an imaging reporter, the dye ICG, was used as a modelcargo. ICG is a well validated imaging agent fluorescent inNIR, it is often used in evaluating new drug delivery systems invivo.29,30 ICG was also chosen because of its unique opticalproperties that are dependent on the environment, thus theblood triggered release can be monitored by optical means.The size of ICG is similar to the size of most commonly useddrug molecules. The utilization of imaging agents in the NIRrange (700–950 nm) extends imaging capabilities up to severalcentimeters in depth due to attenuated scattering and a lack ofendogenous fluorophores within this range.31–35 Because ofthe high penetration of NIR photons through tissue, ICGrelease can be easily observed in small animals with standardoptical imaging techniques.
Herein, we demonstrate a novel type of drug delivery systemthat releases their cargo upon contact with blood. The deliverysystem is based on porous nanocapsules that retain the drugduring formulation and storage. After in vivo administration,rapid release of the capsules’ contents (represented here by anear-infrared dye) upon contact with blood componentsoccurs. This liberation of the encapsulated content is triggeredby the interaction of the nanocapsule’s shell with plasmaproteins. The mechanism of such a triggered release and thefirst demonstration of the rapid release system in smallanimals are given below.
Results and discussion
Nanocapsules feature a hydrophobic interior
The synthesis of the nanocapsules with encapsulated ICG asan imaging reporter was conducted in a fashion similar to thepreviously reported vesicle-templated synthesis of nanocap-sules.24,25,27 Monomers were loaded into the lipid bilayerduring the preparation of liposomes. Liposomes were pre-pared by hydration of the lipid–monomer mixture with anaqueous solution of ICG. Unilamellar liposomes were formedby extrusion and non-entrapped ICG was removed by size-exclusion chromatography. Polymerization was initiated with
UV irradiation. To maximize the efficiency of polymerizationin the presence of a large amount of dye, the pathlength oflight in the quartz vessel holding the solution was kept short,approximately 3 mm, using a glass insert. The use of the UVlight to crosslink polymer shell nanocapsules could bedetrimental to ICG, resulting in permanent bleaching of thedye. Addition of ascorbic acid, a known antioxidant recentlyshown to protect NIR dyes from degradation by radiation,36
alleviated this problem. The presence of ascorbic acid in theaqueous solution had no negative effect on the rate ofpolymerization and successful formation of nanocapsules.The absorbance and fluorescence spectra of free ICG remainedunchanged under these conditions.
Successful incorporation of the dye in nanocapsules wasconfirmed by steady state (Fig. 2A,B) and dynamic spectro-scopy (Fig. 2C). ICG encapsulation led to a substantial y20nm bathochromic shift of ICG absorption/emission maxima,accompanied by a substantial increase in the quantum yieldand a longer fluorescence lifetime (Fig. 2A,B and Table 1).Such trends generally indicate a change from a hydrophilic toa more hydrophobic environment around the fluorophore.37,38
Indeed, a polarity function, known as the solvent orientationpolarizability (Dfs)
39 of the nanocapsule interior, determinedby the fluorescence lifetime, was found to be 0.305, which issimilar to the polarity of methanol.38 This information wascritical to establish the initial state of ICG in nanocapsules.
Table 1 Optical properties of ICG and ICG–NCs in water
lab, nm lem, nm e, M21 cm21 W t, ns r, (st. dev)
ICG 779 802 183 00038 0.03038 ,0.2 0.31 (0.037)ICG–NCs 801 821 167 000 0.062 0.55 0.22 (0.022)
Fig. 1 Injection of the cargo carrying nanocapsules (large green spheres) intothe blood stream is followed by the attachment of serum proteins, such asalbumin (small white spheres) to the surface of the nanocapsule. That leads tothe release of the cargo (small green spheres) leaving the empty nanocapsules(large grey spheres) intact.
5548 | RSC Adv., 2013, 3, 5547–5555 This journal is � The Royal Society of Chemistry 2013
Paper RSC Advances

Below we demonstrate that the release of the dye can be simplymonitored by the dye’s spectral characteristic, resulting fromthe change in the environment.
ICG–NCs demonstrate a long-term shelf-life stability
For a drug delivery system the shelf-life should be sufficientlylong to endure storage and shipment before use of theformulation.40 Following preparation, ICG–NCs dispersed inwater were stable for more 18 months at 4 uC in the dark withno change in the optical properties, absorption, emission (Fig.S1, ESI3), lifetime (0.56 ns) and overall appearance. Thermaltreatment also showed no sign of nanocapsule deterioration.The accelerated thermal stability study of ICG–NCs wasevaluated by heating their aqueous suspension up to 85 uCfor 5 min (Fig. 1D). The intensity at the monitored wavelength(820 nm) linearly decreased with heating and returned to theprevious state after cooling (see also Fig. S2, ESI,3 showing thatno change in the spectrum shape occurred). This behavior iscommon for polymethine dyes because of their temperaturedependent conformational flexibility in the excited state.32
Complete reversibility of the fluorescence intensity and otherfluorescent parameters such as the shape of the spectra andthe fluorescence lifetime indicated that ICG remained insidethe nanocapsules during a heating–cooling cycle, renderingsufficient thermal stability of the ICG–NC system.
Albumin triggers the release of nanocapsule’s imaging cargoin vitro
The release of the dye from the nanocapsules upon contactwith blood was demonstrated using fluorescence lifetimemeasurements and gel electrophoresis. The kinetics of therelease was measured with high speed steady state spectralanalysis.
We have previously shown that the fluorescence lifetime ofpolymethine dyes is highly sensitive to solvent polarity and canbe used to evaluate polarity inside local microenvironments.38
We have also shown that the binding pockets of albumin areessentially hydrophobic with a solvent orientation polariz-ability higher than that of methanol.38,41 Therefore, wehypothesized that if ICG, with its known strong bindingaffinity to albumin (Kd = 554 000 M21)33 is released from thenanocapsules, albumin would bind the dye, thereby increasingits fluorescence lifetime. Alternatively, if the ICG–NCs areintact, then no change in the fluorescence lifetime of the dyeshould be observed.
A control experiment with 4% BSA (to mimic the concentra-tion of albumin in serum and blood) revealed a rapid increaseof the fluorescence lifetime of ICG (Table 2). The time decaysof the intact ICG–NCs and with albumin illustrate this increase(Fig. 2C). The initial fluorescence lifetime of ICG in NCs was0.56 ns. The resulting fluorescence lifetime of ICG–NCs treatedwith BSA (0.83 ns) was close to the lifetime value of neat ICGbound to albumin (y0.88 ns).41 Similar to BSA, treatment withfetal bovine serum (FBS) led to a rapid increase of the lifetime
Fig. 2 Optical characteristics of ICG and ICG–NCs and effect of BSA. Absorption (A) and emission (B) spectra of ICG and ICG–NCs in water at room temp. Ex/em.: 720/735–900 nm. C: fluorescent decays, ex/em.: 773/820 nm. D: thermal stability of ICG/NCs in water. Ex/em.: 720/820 nm, forward arrow (solid circles) – heating from20 to 85 uC, backward arrow (empty circles) – return from 85 to 20 uC.
This journal is � The Royal Society of Chemistry 2013 RSC Adv., 2013, 3, 5547–5555 | 5549
RSC Advances Paper

from initially 0.56 ns to 0.68 ns within a few minutes aftermixing, suggesting essentially the same mechanism of release.
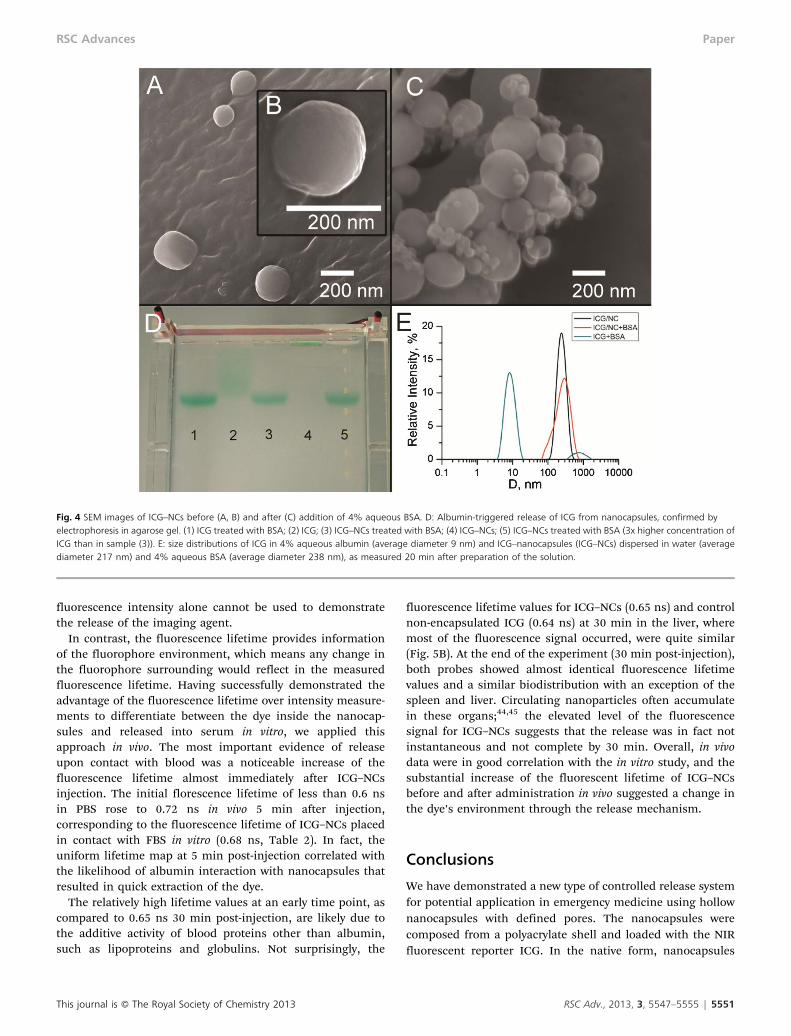
Albumin-triggered release of ICG was independently con-firmed by gel electrophoresis (Fig. 4D). A control pre-madeICG–albumin complex moved quickly and appeared in onespot in agarose gel (well #1). Another control, free ICG,exhibited significant tailing (well #2). When the dye-loadednanocapsules ICG–NCs were treated with albumin (wells #3and 5), the single spot matched to that in well #1, indicatingthat almost the entire content of the nanocapsules wasreleased. In contrast, ICG–NCs with no albumin treatment(well #4) did not move under the applied electric field,apparently because of the low charge of the nanocapsules(zeta potential y0 mV) and/or low mobility of relatively largenanocapsules in the agarose gel, indicating that all the contentretained in nanocapsules.
The intensity of the emission from ICG–NCs placed in thesolution of 4% albumin measured with a high speed CCDdiode array detector was shown to decrease with time,reflecting the release of ICG (Fig. 3A). The decrease of thesignal was also accompanied with a hypsochromic shift of theemission of y14 nm (Fig. S3A, ESI3). Based on the wavelengthmaxima data a release profile was constructed. (Fig. 3B andFig. S3B, ESI3). The data illustrate the rapid release of ICG fromthe nanocapsules: the half-life of ICG in NCs is y6 s.Importantly, the release was not complete; ca. 9% ICGremained in the NCs 30 min after their contact with analbumin solution.
DLS and SEM data indicate expansion of the nanocapsules as apotential mechanism of the release
Scanning electron microscopy (SEM) images of the nanocap-sules indicate that ICG–NCs preserved their shape aftertreatment with excess albumin (Fig. 4A–C). Small sphericalobjects in the albumin-treated sample (Fig. 4C) are character-istic of albumin aggregates. SEM images revealed no broken ordeformed nanocapsules, supporting the proposed mechanismof release rather than collapse of the nanocapsules. Dynamiclight scattering (DLS) data correlated well with the average sizeof nanocapsules measured by SEM. The broadened sizedistribution observed in DLS data of albumin-treated sampleswere in agreement with the presence of albumin aggregates(Fig. 4E). DLS performed in water and albumin solutionsrevealed a slight size increase of ICG-loaded nanocapsules inthe albumin solution. This increase of the average diameterfrom 217 nm to 238 nm was apparently critical for dye release.Such expansion is likely to be a result of a conformationalchange in the polymer structure, and not solely due to theabsorption of proteins to the surface of the nanoparticle.Albumin adsorbs on the polymer surface as a monolayer notexceeding 3 nanometers.42 We suggest that the adsorption ofalbumin favors the exposure of hydrophobic regions of thepolymer network accompanied with pore expansion leading tocargo release. The detailed mechanism of such notablebehavior is currently under investigation.
Fluorescence lifetime imaging confirmed the quick release ofICG in vivo
Fluorescence intensity imaging in vivo provides information ofthe fluorophore distribution. Indeed, longitudinal fluores-cence intensity mapping showed high fluorescence from theliver, gall bladder and intestines 30 min post-injection,indicating clearance via a hepatobiliary excretion pathway(Fig. 5A). This pathway is the primary route of elimination forICG due to its strong binding affinity to albumin,43 and is alsothe principal clearance pathway for the majority of knownnanoparticles due to their relatively large size. Since both thenanocapsule and the dye have similar clearance pathways,
Table 2 Serum stability of ICG–NCs measured by the fluorescence lifetimea
Solvent system
5 min 60 min
Average lifetime,t ns x2b
Average lifetime,t ns x2b
PBS 0.56 1.17 0.59 1.214% albumin/water 0.83 1.34 0.84 1.32FBS 0.68 1.28 0.74 1.32
a Ex/em. 773/820 nm, room temperature, from three exponential fits.b Goodness of fit parameter.
Fig. 3 (A) Change on the emission spectra of ICG–NCs upon interaction with 4% BSA in water. Ex/em: 720/735–900 nm. A bathochromic shift of the ICG–NC contactwith BSA in water is observed. (B) Cumulative release of ICG from ICG–NCs upon contact with BSA.
5550 | RSC Adv., 2013, 3, 5547–5555 This journal is � The Royal Society of Chemistry 2013
Paper RSC Advances
fluorescence intensity alone cannot be used to demonstratethe release of the imaging agent.
In contrast, the fluorescence lifetime provides informationof the fluorophore environment, which means any change inthe fluorophore surrounding would reflect in the measuredfluorescence lifetime. Having successfully demonstrated theadvantage of the fluorescence lifetime over intensity measure-ments to differentiate between the dye inside the nanocap-sules and released into serum in vitro, we applied thisapproach in vivo. The most important evidence of releaseupon contact with blood was a noticeable increase of thefluorescence lifetime almost immediately after ICG–NCsinjection. The initial florescence lifetime of less than 0.6 nsin PBS rose to 0.72 ns in vivo 5 min after injection,corresponding to the fluorescence lifetime of ICG–NCs placedin contact with FBS in vitro (0.68 ns, Table 2). In fact, theuniform lifetime map at 5 min post-injection correlated withthe likelihood of albumin interaction with nanocapsules thatresulted in quick extraction of the dye.
The relatively high lifetime values at an early time point, ascompared to 0.65 ns 30 min post-injection, are likely due tothe additive activity of blood proteins other than albumin,such as lipoproteins and globulins. Not surprisingly, the
fluorescence lifetime values for ICG–NCs (0.65 ns) and controlnon-encapsulated ICG (0.64 ns) at 30 min in the liver, wheremost of the fluorescence signal occurred, were quite similar(Fig. 5B). At the end of the experiment (30 min post-injection),both probes showed almost identical fluorescence lifetimevalues and a similar biodistribution with an exception of thespleen and liver. Circulating nanoparticles often accumulatein these organs;44,45 the elevated level of the fluorescencesignal for ICG–NCs suggests that the release was in fact notinstantaneous and not complete by 30 min. Overall, in vivodata were in good correlation with the in vitro study, and thesubstantial increase of the fluorescent lifetime of ICG–NCsbefore and after administration in vivo suggested a change inthe dye’s environment through the release mechanism.
Conclusions
We have demonstrated a new type of controlled release systemfor potential application in emergency medicine using hollownanocapsules with defined pores. The nanocapsules werecomposed from a polyacrylate shell and loaded with the NIRfluorescent reporter ICG. In the native form, nanocapsules
Fig. 4 SEM images of ICG–NCs before (A, B) and after (C) addition of 4% aqueous BSA. D: Albumin-triggered release of ICG from nanocapsules, confirmed byelectrophoresis in agarose gel. (1) ICG treated with BSA; (2) ICG; (3) ICG–NCs treated with BSA; (4) ICG–NCs; (5) ICG–NCs treated with BSA (3x higher concentration ofICG than in sample (3)). E: size distributions of ICG in 4% aqueous albumin (average diameter 9 nm) and ICG–nanocapsules (ICG–NCs) dispersed in water (averagediameter 217 nm) and 4% aqueous BSA (average diameter 238 nm), as measured 20 min after preparation of the solution.
This journal is � The Royal Society of Chemistry 2013 RSC Adv., 2013, 3, 5547–5555 | 5551
RSC Advances Paper

retain ICG over an extended time. The composition was stablein aqueous buffers, indicating no dye leakage within 1.5 years.Upon contact with serum proteins or blood a rapid release ofthe cargo into the media or blood stream was observed in vitroand in vivo. The release was attributed to a potentialconformational change in the polymer frame upon contactwith albumin, resulting in the expansion of the nanocapsuleand therefore the pore size. This expansion facilitateddiffusion of the cargo into the surrounding environment.
The limitation of the current system includes optimizationof the nanocapsules design for every type of cargo to reach themaximum efficiency in drug delivery. Other limitationsinclude potential side effects due to the non-degradablenature of the nanocapsules. Creating biodegradable nanocap-sules capable of pore expansion caused by interactions withserum proteins is an attractive future extension of this work.
The general design of the nanocapsules fits well to theirpotential applications in medicine as rapid release nanocar-riers and sets the stage for the development of other rapiddrug release systems for emergency treatments and medicaldiagnostics. If necessary, the nanocapsules can be freeze-driedfor prolonged storage and re-suspended in aqueous bufferswithout the loss of content. The diameter of the pores as wellas the size of the nanoparticles can potentially be tuned for aspecific drug molecule in order to achieve the highest rate ofrelease and guide the required clearance pathway after thecargo is released. Such next generation systems will provide
higher synthetic flexibility, facilitating the development of newtherapeutic and imaging systems for emergency applications.
Materials and methods
Materials
Solvents: DMSO, chloroform, buffers and high purity water(18.2 MV) were used throughout the study. Bovine serumalbumin (BSA, grade agarose gel electrophoresis, 99%), ICG(Sigma-Aldrich), fetal bovine serum (FBS), agarose (BiolineUSA Inc.) were used without purification. 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) was purchased from AvantiPolar Lipids, Inc. as a dry powder. Tert-butyl methacrylate(t-BMA), butyl methacrylate (BMA), used as monomers, andethylene glycol dimethacrylate (EGDMA), used as a cross-linking agent, were purchased from Sigma-Aldrich and werepassed through an alumina column to remove the inhibitorshortly before polymerization. The photoinitiator 2,2-dimethoxy-2-phenyl-acetophenone (DPA), from Sigma-Aldrich, was used without purification.
Synthesis of nanocapsules
DMPC (160 mg) was dissolved in 0.4 mL chloroform in a testtube, then t-BMA (32 mL, 0.193 mmol), BMA (32 mL, 0.2 mmol),EGDMA (32 mL, 0.17 mmol) and initiator 2,2-dimethoxy-2-phenyl-acetophenone (3 mg, 0.01 mmol) were added.Chloroform was evaporated using a stream of purified argonto form a lipid–monomer mixture. The mixture was further
Fig. 5 Time-domain diffuse optical imaging of mice injected with ICG and ICG–NCs. The results showing fluorescence intensity (A) (30 min) and fluorescence lifetime(B) maps of mice at about 5 and 30 min after injection of either ICG (upper) or ICG–NCs (lower). Fluorescence lifetime of ICG–NCs was initially uniformly distributed inthe body with a high value of 0.72 ns, and then lowered to that observed with free ICG within 30 min after injection. Ex/em.: 775/820 nm. (C): ex vivo fluorescencedistribution imaging at 30 min post-injection of softgel–ICG (black) or ICG (grey). The biodistribution profile of ICG–NCs is similar to free ICG with the exception ofhigher fluorescence from the spleen and liver.
5552 | RSC Adv., 2013, 3, 5547–5555 This journal is � The Royal Society of Chemistry 2013
Paper RSC Advances

dried in a vacuum to remove traces of solvent. The solution ofICG (150 mM, 8 mL) in Tris buffer (pH 7.4) was added to thetest tube with the lipid–monomer mixture and incubated at 35uC for 30 min. During this period, the mixture was brieflyvortexed every 5 min. ICG was not stable in high intensity UVlight. To minimize the degradation of dye during thepolymerization process, ascorbic acid was added to thereaction mixture (5 mg mL21). The suspension was extruded20 times at 35 uC through a track-etched polyester nucleoporemembrane (Sterlytech) with 100 nm pore size using a Lipexstainless steel extruder (Northern Lipids).
The sample was irradiated for 1.5 h with UV light (l = 254nm) in a photochemical reactor (10 lamps, 32 W each; thedistance between the lamps and the sample was 10 cm) usinga quartz tube with a path length of light of approximately 3mm. A short path length is important for efficient polymeriza-tion in the presence of the dye. After polymerization, non-entrapped dye was removed using size exclusion chromato-graphy. The sample was passed through a Sephadex G-25column (10 mL) twice to ensure complete removal of free dye.The faster-eluting nanocapsule fraction was collected.
Optical measurements
UV/Vis spectra of samples were recorded on a spectrophot-ometer equipped with an integrating cavity absorption meterto minimize the effect of scattering (OLIS Inc.) or DU 640 UV-visible spectrophotometer (Beckman Coulter). Samples withnanocapsules were diluted with water to four differentconcentrations, so that their absorptions lay between 0.1 and1. Using Beer–Lambert’s law and known molar absorptivitiesof ICG, the concentration of the samples are calculated.
Steady state fluorescence spectra and the fluorescencelifetime were recorded on a Fluorolog-3 spectrofluorometer(Horiba Jobin Yvon). The photophysical data of ICG, ICG–nanocapsules (steady-state absorption, fluorescence) and life-time were obtained in DMSO, water, PBS buffer, 4% (mass)BSA solution and undiluted FBS as indicated. Fluorescencequantum yields of the samples were measured using acomparative method with ICG in DMSO as a standard.46 Thefluorescence lifetime of the nanocapsules was determinedusing a time-correlated single photon counting (TCSPC)technique with a NanoLed 773 nm excitation source asdescribed previously.38 Fast kinetics studies were performedwith a Synapse CCD diode array as a detector. Fluorescenceanisotropy was conducted in L-format with automated Glan-Thompson polarizing prisms controlled by FluorEssencesoftware (Horiba) as described earlier.47
Measurement of NCs interior solvent polarity
Solvent orientation polarizability (Df) was utilized as a solventpolarity function. This function is defined as following39:
Df ~e{1
2ez1{
n2{1
2n2z1(1)
e – solvent dielectric constant, n – solvent refractive indexSolvent orientation polarizabilities of typical solvents are:
water – 0.320, methanol – 0.309, ethanol – 0.289, DMSO –0.263, methylene chloride – 0.217.
The polarity inside the nanocapsules was defined as theinterior solvent orientation polarizability and was evaluatedfrom the measured fluorescence lifetimes using the followingequation and parameters previously determined in ref. 38:
Dfs~Df0z1
Kln 1{
ts
tV
� �~0:33z
1
Kln 1{
ts
tV
� �(2)
ts – measured fluorescence lifetime in NCs, ns, tv – lifetime ofa probe in vacuum, for ICG, tv = 1.11 ¡ 0.11 ns, Df0 – solventorientation polarizability in a hypothetical solvent where thefluorescence lifetime is approaching 0, Df0 = 0.33, K – dye-specific lifetime-solvatochromic coefficient, for ICG, K = 27.5¡ 6.7.
Kinetics of the release in vitro
The experiments were conducted using a CCD diode arraycamera Synapse (Horiba) integrated in a spectrophotometerFluorolog-3. The camera allows a full spectrum (735–900 nm)to be acquired in 6 s. A bolus of 20 ml of the ICG–NCs wasadded through an injection port into a cuvette containing 2mL 4% BSA in water with constant stirring. The firstacquisition was obtained 6 s after the injection, the shortestpossible time. The acquisitions were conducted continuouslyup to 2000 s. The cumulative release was calculated from theequation:
%Release~la{ls
la{lb
|100% (3)
la – emission maxima of ICG–NCs in water, nm; lb emissionmaximum of ICG in 4% BSA, nm; ls – emission maxima ofICG–NCs at certain time point in 4% albumin, nm. At time t =0, ls = la (%Release = 0%, no release), at t A ‘, ls = lb
(%Release = 100%, complete release)
Thermal stability
ICG–NCs were suspended in PBS buffer and their temperaturestability evaluated by heating from 20 to 85 uC with 10 degreeincrements within 1 h. The sample was then cooled down withapproximately the same rate (1 h). The sample was allowed toequilibrate for 5 min at each temperature point and thefluorescence spectra of the sample were recorded. The detailsof the setup and temperature ramp profile were publishedpreviously.48
Serum stability
The stability of the ICG–NCs was tested by measuring thefluorescence lifetime of the nanocapsules in PBS, 4% aqueousBSA solution, and FBS at 5 min and 1 h after the solutions wereprepared. Dynamic light scattering (DLS) of the selectedsamples was performed on a Malvern Zetasizer Nano 20 minafter preparation.
Electrophoresis experiments were carried out using an OwlA5 Large Gel System with Owl EC-105 apparatus(ThermoScientific) with an upright positioned separationchamber. All separations were performed in agarose gel witha chamber temperature of 25 uC, and an electric field of 130 V.A sample (each sample contained 10% v/v glycerol, total
This journal is � The Royal Society of Chemistry 2013 RSC Adv., 2013, 3, 5547–5555 | 5553
RSC Advances Paper

volume 100 mL) was injected near the anode into the flowingseparation buffer (TAE buffer).
In vivo imaging
Animal studies were performed according to protocolsapproved by Washington University School of MedicineAnimal Studies Committee for humane care and use oflaboratory animals. Mice were anesthetized with ketamine(85 mg kg21) and xylazine (15 mg kg21), IP for depilation,intravenous injections and initial post-injection imaging.Imaging agents ICG in water (25 mL, conc 60 mM) and ICG–NCs (20 mL of aqueous solutions) were administered via thelateral tail vein of 6–8 week-old female Balb/c mice. The micewere imaged with a Pearl Imager (LI-Cor Biosciences) forfluorescence intensity in the 700 nm (685 lex/710 lem) and 800nm (785 lex/810 lem) emission channels at 5 min, 1 h, 5 h and24 h after injection. Time-domain diffuse optical imaging ofliving mice was performed using the Optix MX2 system(Advanced Research Technologies) as reported previously.49
Briefly, the animals were positioned supine on the heatedimaging platform. Pre-injection scans were performed toassess background and autofluorescence signals, then againat 5 and 30 min post-injection. The regions of interest wereraster-scanned at 780 nm excitation with emission detectioncentered at 830 nm in 1.5 mm steps. Fluorescence intensityand lifetime values were determined by integration or singleexponential fitting of the acquired temporal point spreadfunction (TPSF) fluorescence decay curves for each measure-ment using Optiview software (Advanced ResearchTechnologies). Fluorescence intensity and lifetime maps werecreated by assigning each respective value to the correspond-ing measurement location on a white light reference image ofthe mouse. The mean fluorescence intensity and lifetimevalues for tumor and non-tumor tissue regions of interest(ROI) for each mouse were manually selected and reported foranalysis.
Biodistribution of the probes was assessed using themethod described previously.50,51 Aliquots of blood and piecesof major organs (tumor, heart, kidney, lung, spleen, stomach,intestine, muscle, liver, skin and brain) were harvested andplaced on a clear plastic petri dish. Fluorescence images wereacquired with the Pearl Imager as described above. The meanfluorescence intensity was determined for each tissue by ROIanalysis and combined for each group for statistical analysis.
Acknowledgements
We gratefully acknowledge financial support from the NCI/NIH R21CA149814 (MB), NHLBI/NIH as a Program ofExcellence in Nanotechnology HHSN268201000046C (MB),Washington University Optical Spectroscopy Core NCRR/NIH1S10RR031621 (MB), ORIP/NIH KO1RR026095 (WA),Mallinckrodt Institute of Radiology Summer ResearchProgram (SM, QC), NSF CHE-1012951 (EP), NIAMS/NIH R21,AR060408-01A1 (EP), Saint Louis University PresidentialResearch Fund (EP) and FedEx Institute of Technology (EP).
References
1 F. Gu, L. Zhang, B. A. Teply, N. Mann, A. Wang, A.F. Radovic-Moreno, R. Langer and O. C. Farokhzad, Proc.Natl. Acad. Sci. U. S. A., 2008, 105, 2586–2591.
2 K. E. Uhrich, S. M. Cannizzaro, R. S. Langer and K.M. Shakesheff, Chem. Rev., 1999, 99, 3181–3198.
3 R. Singh and J. W. Lillard, Jr., Exp. Mol. Pathol., 2009, 86,215–223.
4 T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818–1822.5 G. H. Hsiue, S. H. Hsu, C. C. Yang, S. H. Lee and I. K. Yang,
Biomaterials, 2002, 23, 457–462.6 J. Myerson, L. He, G. Lanza, D. Tollefsen and S. Wickline,
JTH, 2011, 9, 1292–1300.7 T. W. Chung, S. S. Wang and W. J. Tsai, Biomaterials, 2008,
29, 228–237.8 J. E. Frampton, Drugs, 2010, 70, 1719–1743.9 K. Cai, J. Li, Z. Luo, Y. Hu, Y. Hou and X. Ding, Chem.
Commun., 2011, 47, 7719–7721.10 S. Weinstein andA. L. Plumer, Plumer’s principles & practice
of intravenous therapy, Lippincott Williams & Wilkins,Philadelphia, 2007.
11 A. Foinard, B. Decaudin, C. Barthelemy, B. Debaene andP. Odou, Ann. Intensive Care, 2012, 2, 28.
12 S. Kanji, J. Lam, C. Johanson, A. Singh, R. Goddard,J. Fairbairn, T. Lloyd, D. Monsour and J. Kakal, Crit. CareMed., 2010, 38, 1890–1898.
13 K. Raemdonck, J. Demeester and S. De Smedt, Soft Matter,2009, 5, 707–715.
14 L. Zha, B. Banik and F. Alexis, Soft Matter, 2011, 7,5908–5916.
15 K. S. Soppimath, T. M. Aminabhavi, A. R. Kulkarni and W.E. Rudzinski, J. Controlled Release, 2001, 70, 1–20.
16 Z. Ma and J.-S. Taylor, Proc. Natl. Acad. Sci. U. S. A., 2000,97, 11159–11163.
17 S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2008, 4, 435–449.18 N. Fomina, J. Sankaranarayanan and A. Almutairi, Adv.
Drug Delivery Rev., 2012, 64, 1005–1020.19 S. Hyuk Im, U. Jeong and Y. Xia, Nat. Mater., 2005, 4,
671–675.20 P. L. Lu, Y. C. Chen, T. W. Ou, H. H. Chen, H. C. Tsai, C.
J. Wen, C. L. Lo, S. P. Wey, K. J. Lin, T. C. Yen and G.H. Hsiue, Biomaterials, 2011, 32, 2213–2221.
21 S. Hyuk Im, U. Jeong and Y. Xia, Nat. Mater., 2005, 4,671–675.
22 A. G. Richter, S. A. Dergunov, B. Ganus, Z. Thomas, S.V. Pingali, V. Urban, Y. Liu, L. Porcar and E. Pinkhassik,Langmuir, 2011, 27, 3792–3797.
23 S. A. Dergunov and E. Pinkhassik, Angew. Chem., Int. Ed.,2008, 47, 8264–8267.
24 S. A. Dergunov, K. Kesterson, W. Li, Z. Wang andE. Pinkhassik, Macromolecules, 2010, 43, 7785–7792.
25 D. C. Danila, L. T. Banner, E. J. Karimova, L. Tsurkan,X. Wang and E. Pinkhassik, Angew. Chem., Int. Ed., 2008,47, 7036–7039.
26 L. T. Banner, D. C. Danila, K. Sharpe, M. Durkin,B. Clayton, B. Anderson, A. Richter and E. Pinkhassik,Langmuir, 2008, 24, 11464–11473.
27 M. D. Kim, S. A. Dergunov, E. Lindner and E. Pinkhassik,Anal. Chem., 2012, 84, 2695–2701.
5554 | RSC Adv., 2013, 3, 5547–5555 This journal is � The Royal Society of Chemistry 2013
Paper RSC Advances

28 Q. Zhou, S. A. Dergunov, Y. Zhang, X. Li, Q. Mu, Q. Zhang,G. Jiang, E. Pinkhassik and B. Yan, Nanoscale, 2011, 3,2576–2582.
29 D. Turner, D. Moshkelani, C. Shemesh, D. Luc andH. Zhang, Pharm. Res., 2012, 29, 2092–2103.
30 J. R. Rajian, M. L. Fabiilli, J. B. Fowlkes, P. L. Carson andX. Wang, Opt. Express, 2011, 19, 14335–14347.
31 S. Achilefu, Technology in cancer research & treatment, 2004,3, 393–409.
32 W. J. Akers, Z. Zhang, M. Berezin, Y. Ye, A. Agee, K. Guo, R.W. Fuhrhop, S. A. Wickline, G. M. Lanza and S. Achilefu,Nanomedicine, 2010, 5, 715–726.
33 M. Solomon, K. Guo, G. P. Sudlow, M. Y. Berezin, W.B. Edwards, S. Achilefu and W. J. Akers, J. Biomed. Opt.,2011, 16, 066019.
34 Z. Zhang, J. Fan, P. P. Cheney, M. Y. Berezin, W.B. Edwards, W. J. Akers, D. Shen, K. Liang, J. P. Culverand S. Achilefu, Mol. Pharm., 2009, 6, 416–427.
35 J. Lee and M. Bogyo, ACS Chem. Biol., 2010, 5, 233–243.36 M. Y. Berezin, K. Guo, B. Teng, W. B. Edwards, C.
J. Anderson, O. Vasalatiy, A. Gandjbakhche, G.L. Griffiths and S. Achilefu, J. Am. Chem. Soc., 2009, 131,9198–9200.
37 I. Texier, M. Goutayer, A. Da Silva, L. Guyon, N. Djaker,V. Josserand, E. Neumann, J. Bibette and F. Vinet, J.Biomed. Opt., 2009, 14, 054005.
38 M. Y. Berezin, H. Lee, W. Akers and S. Achilefu, Biophys. J.,2007, 93, 2892–2899.
39 J. R. Lakowicz, Principles of fluorescence spectroscopy,Springer, New York, 2006.
40 FDA, 2003, vol. revision 2.41 M. Y. Berezin, H. Lee, W. Akers, G. Nikiforovich and
S. Achilefu, Photochem. Photobiol., 2007, 83, 1371–1378.42 H. M. Kowalczynska, M. Nowak-Wyrzykowska, A.
A. Szczepankiewicz, J. Dobkowski, M. Dyda, J. Kaminskiand R. Kołos, Colloids Surf., B, 2011, 84, 536–544.
43 C. M. Leevy, C. L. Mendenhall, W. Lesko and M.M. Howard, J. Clin. Invest., 1962, 41, 1169–1179.
44 H. Soo Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer,B. Itty Ipe, M. G. Bawendi and J. V. Frangioni, Nat.Biotechnol., 2007, 25, 1165–1170.
45 S. M. Moghimi, A. C. Hunter and J. C. Murray, Pharmacol.Rev., 2001, 53, 283–318.
46 R. C. Benson and H. A. Kues, Phys. Med. Biol., 1978, 23,159–163.
47 T. P. Gustafson, Q. Cao, S. Achilefu and M. Y. Berezin,ChemPhysChem, 2012, 13, 716–723.
48 T. P. Gustafson, Q. Cao, S. T. Wang and M. Y. Berezin,Chem. Commun., 2013, 49, 680–682.
49 S. Achilefu, S. Bloch, M. A. Markiewicz, T. Zhong, Y. Ye, R.B. Dorshow, B. Chance and K. Liang, Proc. Natl. Acad. Sci.U. S. A., 2005, 102, 7976–7981.
50 S. Achilefu, R. B. Dorshow, J. E. Bugaj and R. Rajagopalan,Invest. Radiol., 2000, 35, 479–485.
51 J. E. Bugaj, S. Achilefu, R. B. Dorshow and R. Rajagopalan,J. Biomed. Opt., 2001, 6, 122–133.
This journal is � The Royal Society of Chemistry 2013 RSC Adv., 2013, 3, 5547–5555 | 5555
RSC Advances Paper
Copyright © 2022 FDOKUMEN




















