
Research and Clinical Landscape of Bispecific Antibodies for ...
Upload
khangminh22Category
view
3download
0
∗To whom correspondence should addressed. Jijie Gu or Siwei Nie. Email: [email protected]; [email protected].
© The Author(s) 2020. Published by Oxford University Press on behalf of Antibody Therapeutics.
Antibody Therapeutics, 2020, Vol. 3, No. 1 18–62doi:10.1093/abt/tbaa003
Advance Access Publication on 17 February 2020
Review Article
Biology drives the discovery of bispecificantibodies as innovative therapeuticsSiwei Nie1,*, Zhuozhi Wang1, Maria Moscoso-Castro2, Paul D’Souza2,Can Lei2, Jianqing Xu1 and Jijie Gu1,*1WuXi Biologics, 299 Fute Zhong Road, Waigaoqiao Free Trade Zone, Shanghai 200131, China and 2ClarivateAnalytics, Friars House, 160 Blackfriars Road, London SE1 8EZ, UK
Received: December 10, 2019; Revised: February 7, 2020; Accepted: Month 0, 2000
ABSTRACT
A bispecific antibody (bsAb) is able to bind two different targets or two distinct epitopes on the sametarget. Broadly speaking, bsAbs can include any single molecule entity containing dual specificities withat least one being antigen-binding antibody domain. Besides additive effect or synergistic effect, the mostfascinating applications of bsAbs are to enable novel and often therapeutically important concepts otherwiseimpossible by using monoclonal antibodies alone or their combination. This so-called obligate bsAbs couldopen up completely new avenue for developing novel therapeutics. With evolving understanding of structuralarchitecture of various natural or engineered antigen-binding immunoglobulin domains and the connectionof different domains of an immunoglobulin molecule, and with greatly improved understanding of molecularmechanisms of many biological processes, the landscape of therapeutic bsAbs has significantly changedin recent years. As of September 2019, over 110 bsAbs are under active clinical development, and near180 in preclinical development. In this review article, we introduce a system that classifies bsAb formatsinto 30 categories based on their antigen-binding domains and the presence or absence of Fc domain.We further review the biology applications of approximately 290 bsAbs currently in preclinical and clinicaldevelopment, with the attempt to illustrate the principle of selecting a bispecific format to meet biologyneeds and selecting a bispecific molecule as a clinical development candidate by 6 critical criteria. Given thenovel mechanisms of many bsAbs, the potential unknown safety risk and risk/benefit should be evaluatedcarefully during preclinical and clinical development stages. Nevertheless we are optimistic that next decadewill witness clinical success of bsAbs or multispecific antibodies employing some novel mechanisms ofaction and deliver the promise as next wave of antibody-based therapeutics.
Statement of Significance: This article comprehensively reviewed various bispecific antibody formats andthe biology driving the design and selection of a right bispecific antibody to enable novel therapeuticconcept and match intended therapeutic applications. The principles and the examples discussed couldprovide a general guidance for people interested in exploring bispecific antibody therapeutics.
KEYWORDS: bispecific antibody; bsAb; multispecific antibody; msAb
A BRIEF HISTORICAL VIEW OF BISPECIFICANTIBODIES
The invention of hybridoma technology in 1975 markedthe arrival of new era of monoclonal antibody (mAb)-based therapy [1]. However, the first wave of clinicalattempts with mouse antihuman mAb therapeutics during1975–86 largely failed, due to immunogenicity of mouse
sequences, with only one mAb (anti-CD3 muromonab)being approved. It took another decade for the field tosolve the immunogenicity issues, and the lessons learnedfrom the first wave of clinical trials of antibody therapeuticsis the key driver leading to invention of innovative anti-body humanization technologies represented by antibodychimerization, CDR graft, in vitro display of human
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

Antibody Therapeutics, 2020 19
antibody repertoire, and human immunoglobulin trans-genic rodents. The approval of rituximab by the USFDA in 1997 marked the field entering into the boomingstage. About 100 antibody-based therapeutics have beenapproved by the regulatory agencies worldwide, since then,antibody therapeutics have now become one of the main-stays for developing new medicines. The history of devel-opment of bispecific antibodies (bsAbs) almost followedthe footprint of the development of mAb therapeutics.As illustrated in a recent review article [2], starting in the1960s, scientists explored generation of antigen-bindingfragments (Fabs) from two different polyclonal sera andreassociated them into bispecific F(ab’)2 molecules. Afterhybridoma technology was established in 1975, chemicalconjugation of two rodent mAbs or fusion of two antibody-producing hybridomas (so-called quodroma) was exploredimmediately to make bsAbs with defined specificities.The first therapeutic bsAb catumaxomab (Removab
®)
approved by the EMA in 2009 was made by this earlytechnology. The bsAbs made by these two methods prior toestablishing antibody humanization technologies, however,suffered from the same issue of immunogenicity in additionto stability, solubility, and manufacturability challenges.The development of methods to produce recombinantantibodies in the 1980s enabled the rapid generation ofvarious bsAbs with defined structure, composition, andbiochemical, functional, and pharmacological properties,but it still took scientists more than 2 decades to reallyunderstand the unique structural features of variousantigen-binding building blocks such as Fab, Fv, scFv,SDA, etc., to develop various innovative engineeringsolutions to generate homo- and heterodimerizationbuilding blocks necessary for making various bispecificformats and most importantly understand the structuralbiology of how to connect them together to enablevarious biology concepts while maintaining favorabledevelopability. In the later paragraphs, we will review theevolution of some of those landmark solutions for bsAbconstruction. But before we get into detailed discussion ofhow to make various recombinant bsAbs, we will discussthe principles governing how to define and identify a goodbsAb therapeutics first.
THE PRINCIPLES GOVERNING A GOODTHERAPEUTIC BISPECIFIC ANTIBODY
Though mAbs have demonstrated definitive therapeuticbenefits in multiple disease areas, it is believed that bsAbscan further advance the success of therapeutic antibodiesby enabling the molecules with new mechanisms of action(MOAs) and by providing new functional advantages thatcannot be achieved by mAbs. We believe that identificationof a good bsAb should be based on three principles (Fig. 1):(1) the molecule should be able to provide unique biologicalfunction to achieve desired efficacy with appropriate safetyprofile, driven by unique biology; (2) the format chosenshould enable the molecule to fulfill its proposed function,match biology with an optimal format; and (3) the moleculeselected as a clinical development candidate should satisfythe six criteria critical for clinical development and
commercial manufacturing, i.e., desired clinical efficacy,appropriate safety profile, favorable pharmacokinetic/pharmacodynamic (PK/PD) properties, appropriate physic-ochemical properties, scalable manufacturability, and min-imal or no immunogenicity risk—select a right molecule.Unfortunately, these six criteria, particularly those crit-ical for biological function (efficacy, safety, PK/PD,immunogenicity) and those critical for developability(expression, homogeneity, solubility, stability, viscosity,formulation ability, etc.), often are not correlated witheach other, sometimes even counterbalance each other thatrequires balancing when selecting a therapeutic molecule.Identification of a good therapeutic bispecific moleculetherefore usually requires starting with good therapeuticmolecular design defined by molecular product profile(MPP) that is developed based on target product profile(TPP), followed by rigorous molecular and functionalscreen, selection and characterization using pharmacologi-cal assays, mechanistic and/or disease models, and otherpreclinical translational systems relevant to the humandisease one intends to treat.
THE MAKING OF RECOMBINANT BISPECIFICANTIBODIES
In a recent review article, Brinkmann and Kontermannthoroughly reviewed many experimentally verified formatsthat had been described in the literature as of September2016 [3]. We concur with their opinion that besides thefreedom-to-operate (FTO) and the desire to generate pro-prietary intellectual properties (IP) for competitive reason,one of the critical drivers for explosive diversity of so manybsAb formats is the plethora of desired functionalities andapplications of bsAbs. Format variability is essential toserve diverse bsAb applications defined by different TPP.These formats may vary in size, domain composition andarrangement, binding kinetics and valencies, flexibility andgeometry of their binding modules, as well as in their bio-distribution and pharmacokinetic properties to fulfill a par-ticular clinical application. Small variations, such as minorchanges in linker length or composition of domains, canbe crucial determinants for functionality. Some designedparameters may be deduced from structural modeling ofdrug-target interaction. In many cases, however, a suitablemolecule must be identified by generating and compar-ing the functionalities of different formats and differentmolecules in the systems relevant to clinical settings.
Here we review various bsAb formats and classify theminto 30 categories: (1) what are the building blocks ofantigen-binding and their combination, and (2) whetherthey contain fragment of crystallizable region (Fc) domain.From published reports and our practice, most bispecificformats contain the antigen-binding sites derived fromimmunoglobulin domain of native antibodies. We identifysingle-domain antibody (SDA or VHH), variable fragment(Fv), single-chain variable fragment (scFv), Fab, and single-chain antigen-binding fragment (scFab) as the five keybuilding blocks of bispecific formats. As shown in Fig. 2,most of bsAb formats can be classified into 30 groups basedon the above classification. As there are more than 200
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

20 Antibody Therapeutics, 2020
Figure 1. The principles, criteria and screening funnel in discovering a good therapeutic bsAb. (A) Three principles of governing the discovery of a goodbsAb, (B) Six criteria of defining a bsAb as a clinical development candidate. (C) Detailed function and developability screenings to identify a goodtherapeutic bsAb molecule.
bispecific formats from published data and our practice,we do not intend to list all these formats in Fig. 2. Instead,we have just listed an example of each category to illustratethe concept.
In each category, the bispecific formats can be furtherclassified by their geometry (such as homodimer vs. het-erodimer) and valency (number of antigen-binding sites). AbsAb with one binding site to target A and one binding siteto target B is called 1 + 1 format. Similarly there are 1 + 2,1 + 3, and 2 + 2 formats. The formats with more than fourantigen-binding sites are uncommon but growing, so theyare just mentioned as examples in this review.
In addition to the building blocks, absence or presenceof Fc, and different valency, multiple fusion sites of Fc-containing formats increase the complexity of bispecificformats. As shown in Fig. 3A, an antigen-binding build-ing block can be fused to N-terminus or C-terminus ofan Fc fragment or inserted between CH2 domain andCH3 domain. On a heterodimeric Fc-containing bispe-cific format, there are at least six fusion sites. If an Fc-containing format also comprises of CL, the fusion sitesincrease to 12 (Fig. 3A). Moreover, theoretically all theloops of each immunoglobulin domain (CL, CH1, CH2,and CH3) can be used as fusion sites to integrate an antigen-
binding building block. It becomes obvious to employ thesefusion sites to make a bispecific format with desired bindingactivity.
Bispecific molecules containing non-antibody-bindingdomains such as peptides, ligands, receptors, or alternativescaffolds may not fall into this classification system.However, depending on how many polypeptide chainsof the antigen-binding sites are used, the non-antibodybispecific molecules can be constructed using similarapproaches as the above bsAbs.
Bispecific antibody fragments without Fc
In this category, all antigen-binding sites are from the afore-mentioned building blocks (SDA, Fv, scFv, Fab, and scFab)and the bsAbs do not contain Fc. Many different bispecificformats, including 1 + 1, 1 + 2, 1 + 3, and 2 + 2 formats,and trispecific formats have been used for preclinical andclinical development (Tables 1–6). BsAb fragments usuallyare smaller than IgG and lack of Fc-related functions suchas Fcγ R-, FcRn-, and complement-binding and relatedactivities. Due to large number of the bsAb fragment for-mats, only some examples of bsAbs fragments are brieflydescripted below.
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

Antibody Therapeutics, 2020 21
Tab
le1.
Pro
gram
sin
clin
ical
and
prec
linic
alst
ages
tobl
ock
the
angi
ogen
esis
and/
ortu
mor
igen
esis
for
canc
ertr
eatm
ent
Ant
ibod
yna
me
Org
aniz
atio
nT
arge
tsH
ighe
stph
ase
Bio
logi
calf
unct
ion
Typ
eof
mec
hani
smF
orm
atC
linic
alst
udie
s
Dilp
acim
ab,
AB
T-1
65A
bbV
ieV
EG
F×
DL
L4
Pha
seII
Ant
i-an
giog
enes
isC
ombi
nato
rial
effe
ctF
ab+
Fv
wit
hF
c,2
+2
NC
T01
9460
74,
NC
T01
9460
74,
NC
T03
3688
59,
NC
T03
3688
59M
P02
50M
olec
ular
Par
tner
sA
GV
EG
F×
HG
F×
albu
min
Pha
seII
Ant
i-an
giog
enes
isC
ombi
nato
rial
effe
ctSc
affo
ld1
+1
+1
NC
T02
1944
26,
NC
T03
1366
53,
NC
T03
4185
32A
BL
-001
,N
OV
-150
1,T
R-0
09A
BL
Bio
,TR
IGR
The
rape
utic
sV
EG
F×
DL
L4
Pha
seI
Ant
i-an
giog
enes
isC
ombi
nato
rial
effe
ctF
ab+
scF
vw
ith
Fc,
2+
2N
CT
0329
2783
Van
uciz
umab
,R
G-7
221
Roc
he,H
arva
rdM
edic
alSc
hool
,N
atio
nalC
ance
rC
entr
eof
Sing
apor
e
AN
GP
T2
×V
EG
FP
hase
IA
nti-
angi
ogen
esis
Com
bina
tori
alef
fect
Fab
+F
abw
ith
Fc,
1+
1N
CT
0168
8206
,N
CT
0214
1295
,N
CT
0266
5416
BI-
8368
80B
oehr
inge
rIn
gelh
eim
,Sa
nofi
AN
GP
T2
×V
EG
F,al
bum
inP
hase
IA
nti-
angi
ogen
esis
Com
bina
tori
alef
fect
VH
+V
H,1
+1
+1
NC
T02
6741
52,
NC
T02
6895
05,
NC
T03
4684
26,
NC
T03
8612
34,
NC
T03
9721
50N
avic
ixiz
umab
,O
MP
-305
B83
Onc
oMed
Pha
rmac
euti
cals
VE
GF
×D
LL
4P
hase
IA
nti-
angi
ogen
esis
Com
bina
tori
alef
fect
Fab
+F
abw
ith
Fc,
1+
1N
CT
0229
8387
,N
CT
0303
0287
,N
CT
0303
5253
KN
-026
Jian
gsu
Alp
ham
abB
ioph
arm
aceu
tica
lsH
ER
2×
HE
R2
Pha
seII
Ant
i-tu
mor
igen
esis
Bip
arat
opic
Fab
+F
abw
ith
Fc,
1+
1N
CT
0361
9681
,N
CT
0384
7168
,N
CT
0392
5974
,N
CT
0404
0699
ZW
-25
Zym
ewor
ks,B
eiG
ene
HE
R2
×H
ER
2P
hase
IIA
nti-
tum
orig
enes
isB
ipar
atop
icF
ab+
scF
vw
ith
Fc,
1+
1N
CT
0289
2123
,N
CT
0392
9666
MC
LA
-128
Mer
usH
ER
3×
HE
R2
Pha
seII
Ant
i-tu
mor
igen
esis
Com
bina
tori
alef
fect
Fab
+F
abw
ith
Fc,
1+
1N
CT
0291
2949
,N
CT
0332
1981
EM
B-0
1,F
IT-0
13a
Epi
mA
bB
ioth
erap
euti
csE
GF
R×
cME
TP
hase
I/II
Ant
i-tu
mor
igen
esis
Com
bina
tori
alef
fect
Fab
+F
abw
ith
Fc,
2+
2N
CT
0379
7391
JNJ-
6118
6372
,JN
J-63
72Ja
nsse
nE
GF
R×
cME
TP
hase
IA
nti-
tum
orig
enes
isC
ombi
nato
rial
effe
ctF
ab+
Fab
wit
hF
c,1
+1
NC
T02
6097
76,
NC
T04
0774
63B
CD
-147
Bio
cad
HE
R2
×H
ER
2P
hase
IA
nti-
tum
orig
enes
isB
ipar
atop
icF
ab+
scF
vw
ith
Fc,
1+
2N
CT
0391
2441
MB
S-30
1B
eijin
gM
abw
orks
Bio
tech
HE
R2
×H
ER
2P
hase
IA
nti-
tum
orig
enes
isB
ipar
atop
icF
ab+
Fab
wit
hF
c,1
+1
NC
T03
8420
85
Con
tinu
ed
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022
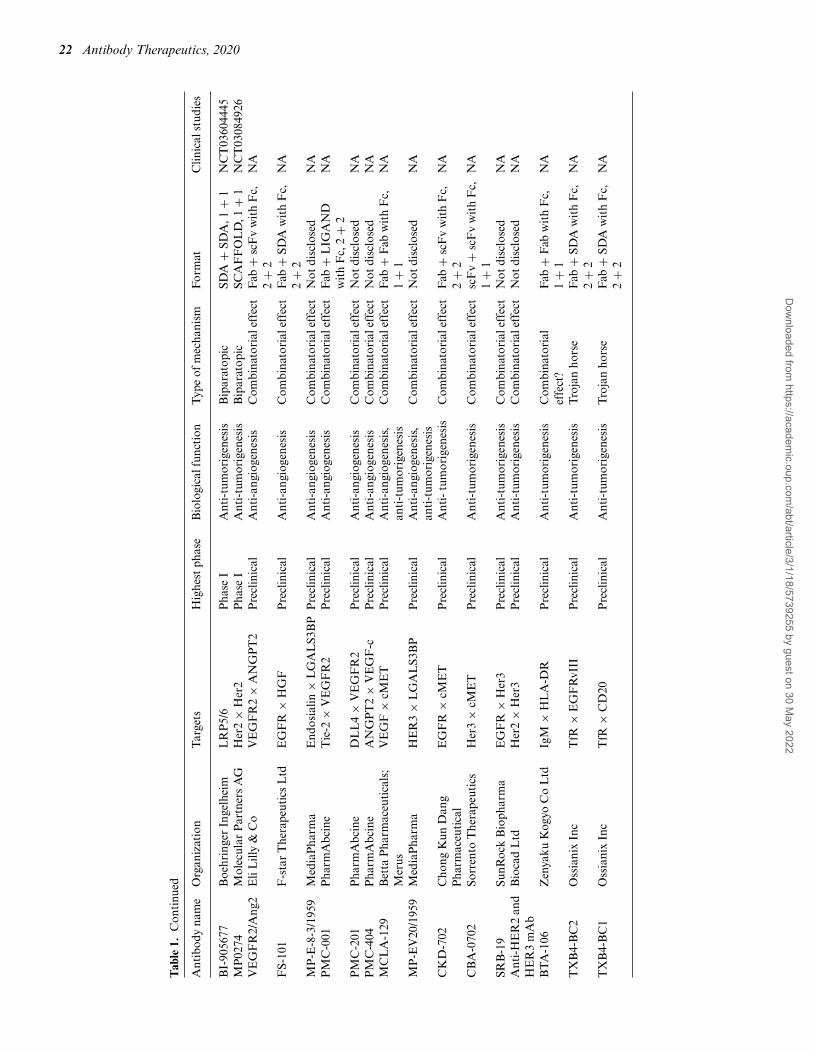
22 Antibody Therapeutics, 2020
Tab
le1.
Con
tinu
ed
Ant
ibod
yna
me
Org
aniz
atio
nT
arge
tsH
ighe
stph
ase
Bio
logi
calf
unct
ion
Typ
eof
mec
hani
smF
orm
atC
linic
alst
udie
s
BI-
9056
77B
oehr
inge
rIn
gelh
eim
LR
P5/
6P
hase
IA
nti-
tum
orig
enes
isB
ipar
atop
icSD
A+
SDA
,1+
1N
CT
0360
4445
MP
0274
Mol
ecul
arP
artn
ers
AG
Her
2×
Her
2P
hase
IA
nti-
tum
orig
enes
isB
ipar
atop
icSC
AF
FO
LD
,1+
1N
CT
0308
4926
VE
GF
R2/
Ang
2E
liL
illy
&C
oV
EG
FR
2×
AN
GP
T2
Pre
clin
ical
Ant
i-an
giog
enes
isC
ombi
nato
rial
effe
ctF
ab+
scF
vw
ith
Fc,
2+
2N
A
FS-
101
F-s
tar
The
rape
utic
sL
tdE
GF
R×
HG
FP
recl
inic
alA
nti-
angi
ogen
esis
Com
bina
tori
alef
fect
Fab
+SD
Aw
ith
Fc,
2+
2N
A
MP
-E-8
-3/1
959
Med
iaP
harm
aE
ndos
ialin
×L
GA
LS3
BP
Pre
clin
ical
Ant
i-an
giog
enes
isC
ombi
nato
rial
effe
ctN
otdi
sclo
sed
NA
PM
C-0
01P
harm
Abc
ine
Tie
-2×
VE
GF
R2
Pre
clin
ical
Ant
i-an
giog
enes
isC
ombi
nato
rial
effe
ctF
ab+
LIG
AN
Dw
ith
Fc,
2+
2N
A
PM
C-2
01P
harm
Abc
ine
DL
L4
×V
EG
FR
2P
recl
inic
alA
nti-
angi
ogen
esis
Com
bina
tori
alef
fect
Not
disc
lose
dN
AP
MC
-404
Pha
rmA
bcin
eA
NG
PT
2×
VE
GF
-cP
recl
inic
alA
nti-
angi
ogen
esis
Com
bina
tori
alef
fect
Not
disc
lose
dN
AM
CL
A-1
29B
etta
Pha
rmac
euti
cals
;M
erus
VE
GF
×cM
ET
Pre
clin
ical
Ant
i-an
giog
enes
is,
anti
-tum
orig
enes
isC
ombi
nato
rial
effe
ctF
ab+
Fab
wit
hF
c,1
+1
NA
MP
-EV
20/1
959
Med
iaP
harm
aH
ER
3×
LG
AL
S3B
PP
recl
inic
alA
nti-
angi
ogen
esis
,an
ti-t
umor
igen
esis
Com
bina
tori
alef
fect
Not
disc
lose
dN
A
CK
D-7
02C
hong
Kun
Dan
gP
harm
aceu
tica
lE
GF
R×
cME
TP
recl
inic
alA
nti-
tum
orig
enes
isC
ombi
nato
rial
effe
ctF
ab+
scF
vw
ith
Fc,
2+
2N
A
CB
A-0
702
Sorr
ento
The
rape
utic
sH
er3
×cM
ET
Pre
clin
ical
Ant
i-tu
mor
igen
esis
Com
bina
tori
alef
fect
scF
v+
scF
vw
ith
Fc,
1+
1N
A
SRB
-19
SunR
ock
Bio
phar
ma
EG
FR
×H
er3
Pre
clin
ical
Ant
i-tu
mor
igen
esis
Com
bina
tori
alef
fect
Not
disc
lose
dN
AA
nti-
HE
R2
and
HE
R3
mA
bB
ioca
dL
tdH
er2
×H
er3
Pre
clin
ical
Ant
i-tu
mor
igen
esis
Com
bina
tori
alef
fect
Not
disc
lose
dN
A
BT
A-1
06Z
enya
kuK
ogyo
Co
Ltd
IgM
×H
LA
-DR
Pre
clin
ical
Ant
i-tu
mor
igen
esis
Com
bina
tori
alef
fect
?F
ab+
Fab
wit
hF
c,1
+1
NA
TX
B4-
BC
2O
ssia
nix
Inc
TfR
×E
GF
RvI
IIP
recl
inic
alA
nti-
tum
orig
enes
isT
roja
nho
rse
Fab
+SD
Aw
ith
Fc,
2+
2N
A
TX
B4-
BC
1O
ssia
nix
Inc
TfR
×C
D20
Pre
clin
ical
Ant
i-tu
mor
igen
esis
Tro
jan
hors
eF
ab+
SDA
wit
hF
c,2
+2
NA
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

Antibody Therapeutics, 2020 23
Tab
le2.
Pro
gram
sin
clin
ical
and
prec
linic
alst
ages
toen
hanc
etu
mor
imm
unit
yfo
rca
ncer
trea
tmen
t
Ant
ibod
yna
me
Org
aniz
atio
nT
arge
tsH
ighe
stph
ase
Bio
logi
calf
unct
ion
Typ
eof
mec
hani
smF
orm
atC
linic
alst
udie
s
KN
-046
Jian
gsu
Alp
ham
abB
ioph
arm
aceu
tica
lsP
D-L
1×
CT
LA
-4P
hase
IIE
nhan
cetu
mor
imm
unit
yT
umor
orti
ssue
loca
lizat
ion
SDA
+SD
Aw
ith
Fc,
2+
2N
CT
0352
9526
,N
CT
0373
3951
,N
CT
0383
8848
,N
CT
0387
2791
,N
CT
0392
5870
,N
CT
0392
7495
,N
CT
0404
0699
AK
-104
Ake
soB
ioph
arm
aP
D-1
×C
TL
A-4
Pha
seI/
IIE
nhan
cetu
mor
imm
unit
yC
ombi
nato
rial
effe
ctF
ab+
scF
vw
ith
Fc,
2+
2N
CT
0326
1011
,N
CT
0385
2251
Duo
Bod
y-P
D-L
1x4-
1BB
,G
EN
-104
6
Bio
NT
ech,
Gen
mab
PD
-L1
×4-
1BB
Pha
seI/
IIE
nhan
cetu
mor
imm
unit
yT
umor
orti
ssue
loca
lizat
ion
Fab
+F
abw
ith
Fc,
1+
1N
CT
0391
7381
RE
GN
-567
8R
egen
eron
PSM
A×
CD
28P
hase
I/II
Enh
ance
tum
orim
mun
ity
Tum
oror
tiss
uelo
caliz
atio
nF
ab+
Fab
wit
hF
c,1
+1
NC
T03
9726
57
FS1
18m
Ab2
,F
S-11
8,L
AG
-3/P
D-L
1m
Ab2
F-s
tar
PD
-L1
×L
AG
-3P
hase
IE
nhan
cetu
mor
imm
unit
yT
umor
orti
ssue
loca
lizat
ion
Fab
+SD
A,2
+2
NC
T03
4404
37
IBI-
318
Inno
vent
Bio
logi
cs,L
illy
PD
-1×
PD
-L1
Pha
seI
Enh
ance
tum
orim
mun
ity
Pro
mot
edo
wnr
egul
atio
nN
otdi
sclo
sed
NC
T03
8751
57
LY-3
4341
72E
liL
illy
PD
-1×
PD
-L1
Pha
seI
Enh
ance
tum
orim
mun
ity
Pro
mot
edo
wnr
egul
atio
nF
ab+
Fab
wit
hF
c,1
+1
NC
T03
9369
59
MG
D-0
13M
acro
Gen
ics,
ZA
IL
abP
D-1
×L
AG
-3P
hase
IE
nhan
cetu
mor
imm
unit
yC
ombi
nato
rial
effe
ctF
v+
Fv
wit
hF
c,2
+2
NC
T03
2192
68,
NC
T04
0823
64X
mA
b-23
104
Xen
cor
PD
-1×
ICO
SP
hase
IE
nhan
cetu
mor
imm
unit
yC
ombi
nato
rial
effe
ctF
ab+
scF
vw
ith
Fc,
1+
1N
CT
0375
2398
AB
BV
-428
Abb
Vie
MSL
N×
CD
40P
hase
IE
nhan
cetu
mor
imm
unit
yT
umor
orti
ssue
loca
lizat
ion
scF
v+
scF
vw
ith
Fc,
2+
2N
CT
0295
5251
AD
C-1
015,
AT
OR
-101
5A
lliga
tor
Bio
scie
nce
OX
40×
CT
LA
-4P
hase
IE
nhan
cetu
mor
imm
unit
yC
ombi
nato
rial
effe
ctF
ab+
LIG
AN
Dw
ith
Fc,
2+
2N
CT
0378
2467
INB
RX
-105
-1,
INB
RX
-105
,ES-
101
Inhi
brx,
Elp
isci
ence
Bio
Pha
rma
PD
-L1
×4-
1BB
Pha
seI
Enh
ance
tum
orim
mun
ity
Tum
oror
tiss
uelo
caliz
atio
nSD
A+
SDA
wit
hF
c,2
+2
NC
T03
8096
24
MC
LA
-145
Mer
us,I
ncyt
eP
D-L
1×
4-1B
BP
hase
IE
nhan
cetu
mor
imm
unit
yT
umor
orti
ssue
loca
lizat
ion
Fab
+F
abw
ith
Fc,
1+
1N
CT
0392
2204
ME
DI-
5752
Med
Imm
une
PD
-1×
CT
LA
-4P
hase
IE
nhan
cetu
mor
imm
unit
yC
ombi
nato
rial
effe
ctF
ab+
Fab
wit
hF
c,1
+1
NC
T03
5303
97
MG
D-0
19M
acro
Gen
ics
PD
-1×
CT
LA
-4P
hase
IE
nhan
cetu
mor
imm
unit
yC
ombi
nato
rial
effe
ctF
v+
Fv
wit
hF
c,2
+2
NC
T03
7610
17
PR
S-34
3P
ieri
sH
ER
2×
4-1B
BP
hase
IE
nhan
cetu
mor
imm
unit
yT
umor
orti
ssue
loca
lizat
ion
Fab
+SC
AF
FO
LD
wit
hF
c,2
+2
NC
T03
3305
61,
NC
T03
6503
48R
G-7
769,
RO
-712
1661
Roc
heP
D-1
×T
IM-3
Pha
seI
Enh
ance
tum
orim
mun
ity
Com
bina
tori
alef
fect
Fab
+F
abw
ith
Fc,
1+
1N
CT
0370
8328
Con
tinu
ed
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022
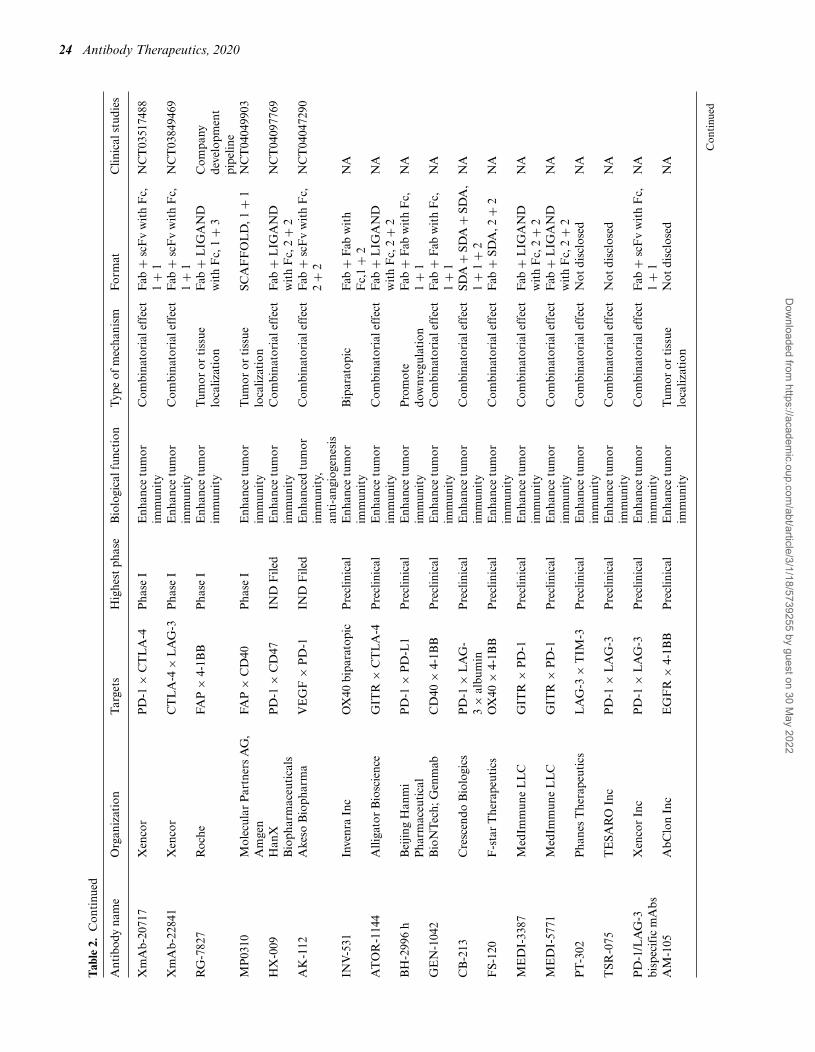
24 Antibody Therapeutics, 2020
Tab
le2.
Con
tinu
ed
Ant
ibod
yna
me
Org
aniz
atio
nT
arge
tsH
ighe
stph
ase
Bio
logi
calf
unct
ion
Typ
eof
mec
hani
smF
orm
atC
linic
alst
udie
s
Xm
Ab-
2071
7X
enco
rP
D-1
×C
TL
A-4
Pha
seI
Enh
ance
tum
orim
mun
ity
Com
bina
tori
alef
fect
Fab
+sc
Fv
wit
hF
c,1
+1
NC
T03
5174
88
Xm
Ab-
2284
1X
enco
rC
TL
A-4
×L
AG
-3P
hase
IE
nhan
cetu
mor
imm
unit
yC
ombi
nato
rial
effe
ctF
ab+
scF
vw
ith
Fc,
1+
1N
CT
0384
9469
RG
-782
7R
oche
FAP
×4-
1BB
Pha
seI
Enh
ance
tum
orim
mun
ity
Tum
oror
tiss
uelo
caliz
atio
nF
ab+
LIG
AN
Dw
ith
Fc,
1+
3C
ompa
nyde
velo
pmen
tpi
pelin
eM
P03
10M
olec
ular
Par
tner
sA
G,
Am
gen
FAP
×C
D40
Pha
seI
Enh
ance
tum
orim
mun
ity
Tum
oror
tiss
uelo
caliz
atio
nSC
AF
FO
LD
,1+
1N
CT
0404
9903
HX
-009
Han
XB
ioph
arm
aceu
tica
lsP
D-1
×C
D47
IND
File
dE
nhan
cetu
mor
imm
unit
yC
ombi
nato
rial
effe
ctF
ab+
LIG
AN
Dw
ith
Fc,
2+
2N
CT
0409
7769
AK
-112
Ake
soB
ioph
arm
aV
EG
F×
PD
-1IN
DF
iled
Enh
ance
dtu
mor
imm
unit
y,an
ti-a
ngio
gene
sis
Com
bina
tori
alef
fect
Fab
+sc
Fv
wit
hF
c,2
+2
NC
T04
0472
90
INV
-531
Inve
nra
Inc
OX
40bi
para
topi
cP
recl
inic
alE
nhan
cetu
mor
imm
unit
yB
ipar
atop
icF
ab+
Fab
wit
hF
c,1
+2
NA
AT
OR
-114
4A
lliga
tor
Bio
scie
nce
GIT
R×
CT
LA
-4P
recl
inic
alE
nhan
cetu
mor
imm
unit
yC
ombi
nato
rial
effe
ctF
ab+
LIG
AN
Dw
ith
Fc,
2+
2N
A
BH
-299
6h
Bei
jing
Han
mi
Pha
rmac
euti
cal
PD
-1×
PD
-L1
Pre
clin
ical
Enh
ance
tum
orim
mun
ity
Pro
mot
edo
wnr
egul
atio
nF
ab+
Fab
wit
hF
c,1
+1
NA
GE
N-1
042
Bio
NT
ech;
Gen
mab
CD
40×
4-1B
BP
recl
inic
alE
nhan
cetu
mor
imm
unit
yC
ombi
nato
rial
effe
ctF
ab+
Fab
wit
hF
c,1
+1
NA
CB
-213
Cre
scen
doB
iolo
gics
PD
-1×
LA
G-
3×
albu
min
Pre
clin
ical
Enh
ance
tum
orim
mun
ity
Com
bina
tori
alef
fect
SDA
+SD
A+
SDA
,1
+1
+2
NA
FS-
120
F-s
tar
The
rape
utic
sO
X40
×4-
1BB
Pre
clin
ical
Enh
ance
tum
orim
mun
ity
Com
bina
tori
alef
fect
Fab
+SD
A,2
+2
NA
ME
DI-
3387
Med
Imm
une
LL
CG
ITR
×P
D-1
Pre
clin
ical
Enh
ance
tum
orim
mun
ity
Com
bina
tori
alef
fect
Fab
+L
IGA
ND
wit
hF
c,2
+2
NA
ME
DI-
5771
Med
Imm
une
LL
CG
ITR
×P
D-1
Pre
clin
ical
Enh
ance
tum
orim
mun
ity
Com
bina
tori
alef
fect
Fab
+L
IGA
ND
wit
hF
c,2
+2
NA
PT
-302
Pha
nes
The
rape
utic
sL
AG
-3×
TIM
-3P
recl
inic
alE
nhan
cetu
mor
imm
unit
yC
ombi
nato
rial
effe
ctN
otdi
sclo
sed
NA
TSR
-075
TE
SAR
OIn
cP
D-1
×L
AG
-3P
recl
inic
alE
nhan
cetu
mor
imm
unit
yC
ombi
nato
rial
effe
ctN
otdi
sclo
sed
NA
PD
-1/L
AG
-3bi
spec
ific
mA
bsX
enco
rIn
cP
D-1
×L
AG
-3P
recl
inic
alE
nhan
cetu
mor
imm
unit
yC
ombi
nato
rial
effe
ctF
ab+
scF
vw
ith
Fc,
1+
1N
A
AM
-105
AbC
lon
Inc
EG
FR
×4-
1BB
Pre
clin
ical
Enh
ance
tum
orim
mun
ity
Tum
oror
tiss
uelo
caliz
atio
nN
otdi
sclo
sed
NA
Con
tinu
ed
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022
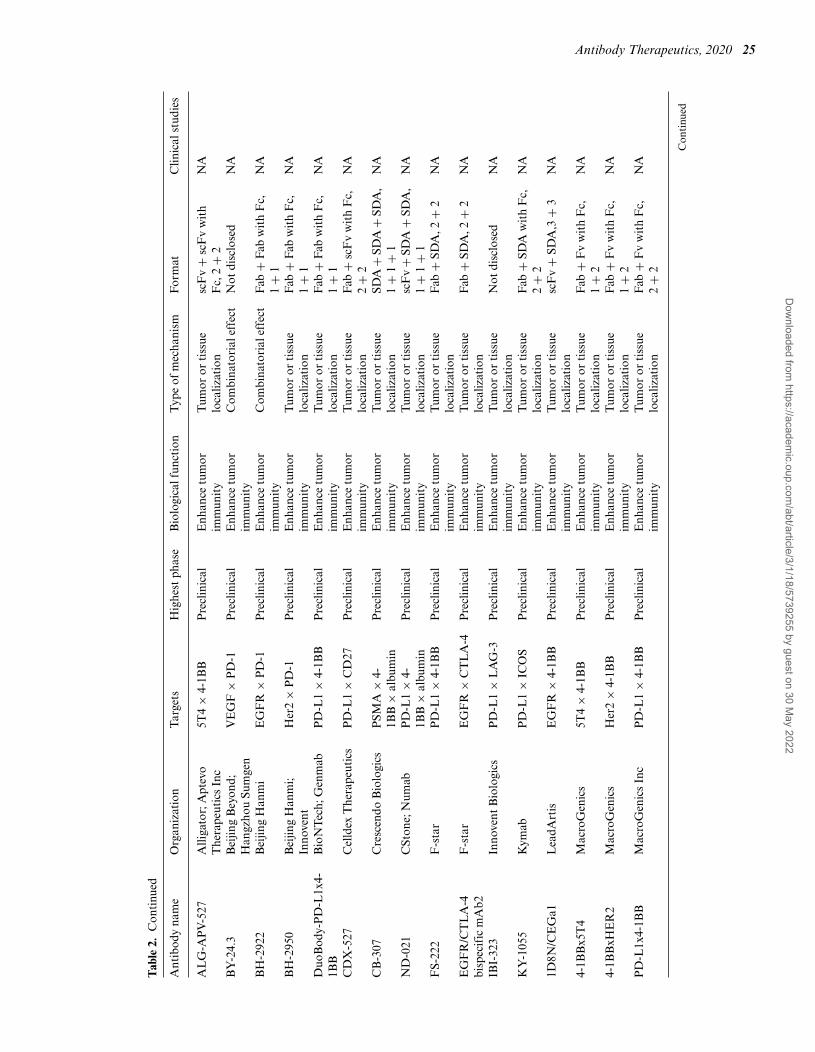
Antibody Therapeutics, 2020 25
Tab
le2.
Con
tinu
ed
Ant
ibod
yna
me
Org
aniz
atio
nT
arge
tsH
ighe
stph
ase
Bio
logi
calf
unct
ion
Typ
eof
mec
hani
smF
orm
atC
linic
alst
udie
s
AL
G-A
PV
-527
Alli
gato
r;A
ptev
oT
hera
peut
ics
Inc
5T4
×4-
1BB
Pre
clin
ical
Enh
ance
tum
orim
mun
ity
Tum
oror
tiss
uelo
caliz
atio
nsc
Fv
+sc
Fv
wit
hF
c,2
+2
NA
BY
-24.
3B
eijin
gB
eyon
d;H
angz
hou
Sum
gen
VE
GF
×P
D-1
Pre
clin
ical
Enh
ance
tum
orim
mun
ity
Com
bina
tori
alef
fect
Not
disc
lose
dN
A
BH
-292
2B
eijin
gH
anm
iE
GF
R×
PD
-1P
recl
inic
alE
nhan
cetu
mor
imm
unit
yC
ombi
nato
rial
effe
ctF
ab+
Fab
wit
hF
c,1
+1
NA
BH
-295
0B
eijin
gH
anm
i;In
nove
ntH
er2
×P
D-1
Pre
clin
ical
Enh
ance
tum
orim
mun
ity
Tum
oror
tiss
uelo
caliz
atio
nF
ab+
Fab
wit
hF
c,1
+1
NA
Duo
Bod
y-P
D-L
1x4-
1BB
Bio
NT
ech;
Gen
mab
PD
-L1
×4-
1BB
Pre
clin
ical
Enh
ance
tum
orim
mun
ity
Tum
oror
tiss
uelo
caliz
atio
nF
ab+
Fab
wit
hF
c,1
+1
NA
CD
X-5
27C
elld
exT
hera
peut
ics
PD
-L1
×C
D27
Pre
clin
ical
Enh
ance
tum
orim
mun
ity
Tum
oror
tiss
uelo
caliz
atio
nF
ab+
scF
vw
ith
Fc,
2+
2N
A
CB
-307
Cre
scen
doB
iolo
gics
PSM
A×
4-1B
B×
albu
min
Pre
clin
ical
Enh
ance
tum
orim
mun
ity
Tum
oror
tiss
uelo
caliz
atio
nSD
A+
SDA
+SD
A,
1+
1+
1N
A
ND
-021
CSt
one;
Num
abP
D-L
1×
4-1B
B×
albu
min
Pre
clin
ical
Enh
ance
tum
orim
mun
ity
Tum
oror
tiss
uelo
caliz
atio
nsc
Fv
+SD
A+
SDA
,1
+1
+1
NA
FS-
222
F-s
tar
PD
-L1
×4-
1BB
Pre
clin
ical
Enh
ance
tum
orim
mun
ity
Tum
oror
tiss
uelo
caliz
atio
nF
ab+
SDA
,2+
2N
A
EG
FR
/CT
LA
-4bi
spec
ific
mA
b2F
-sta
rE
GF
R×
CT
LA
-4P
recl
inic
alE
nhan
cetu
mor
imm
unit
yT
umor
orti
ssue
loca
lizat
ion
Fab
+SD
A,2
+2
NA
IBI-
323
Inno
vent
Bio
logi
csP
D-L
1×
LA
G-3
Pre
clin
ical
Enh
ance
tum
orim
mun
ity
Tum
oror
tiss
uelo
caliz
atio
nN
otdi
sclo
sed
NA
KY
-105
5K
ymab
PD
-L1
×IC
OS
Pre
clin
ical
Enh
ance
tum
orim
mun
ity
Tum
oror
tiss
uelo
caliz
atio
nF
ab+
SDA
wit
hF
c,2
+2
NA
1D8N
/CE
Ga1
Lea
dArt
isE
GF
R×
4-1B
BP
recl
inic
alE
nhan
cetu
mor
imm
unit
yT
umor
orti
ssue
loca
lizat
ion
scF
v+
SDA
,3+
3N
A
4-1B
Bx5
T4
Mac
roG
enic
s5T
4×
4-1B
BP
recl
inic
alE
nhan
cetu
mor
imm
unit
yT
umor
orti
ssue
loca
lizat
ion
Fab
+F
vw
ith
Fc,
1+
2N
A
4-1B
BxH
ER
2M
acro
Gen
ics
Her
2×
4-1B
BP
recl
inic
alE
nhan
cetu
mor
imm
unit
yT
umor
orti
ssue
loca
lizat
ion
Fab
+F
vw
ith
Fc,
1+
2N
A
PD
-L1x
4-1B
BM
acro
Gen
ics
Inc
PD
-L1
×4-
1BB
Pre
clin
ical
Enh
ance
tum
orim
mun
ity
Tum
oror
tiss
uelo
caliz
atio
nF
ab+
Fv
wit
hF
c,2
+2
NA
Con
tinu
ed
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

26 Antibody Therapeutics, 2020
Tab
le2.
Con
tinu
ed
Ant
ibod
yna
me
Org
aniz
atio
nT
arge
tsH
ighe
stph
ase
Bio
logi
calf
unct
ion
Typ
eof
mec
hani
smF
orm
atC
linic
alst
udie
s
ME
DI-
1109
Med
Imm
une
PD
-L1
×O
X40
Pre
clin
ical
Enh
ance
tum
orim
mun
ity
Tum
oror
tiss
uelo
caliz
atio
nF
ab+
LIG
AN
Dw
ith
Fc,
2+
2N
A
PR
S-30
0se
ries
AP
ieri
sH
er2
×C
TL
A-4
Pre
clin
ical
Enh
ance
tum
orim
mun
ity
Tum
oror
tiss
uelo
caliz
atio
nN
otdi
sclo
sed
NA
PR
S-34
2P
ieri
sG
PC
3×
4-1B
BP
recl
inic
alE
nhan
cetu
mor
imm
unit
yT
umor
orti
ssue
loca
lizat
ion
SCA
FF
OL
D+
SCA
F-
FO
LD
wit
hF
c,2
+2
NA
PR
S-34
4P
ieri
s;Se
rvie
rP
D-L
1×
4-1B
BP
recl
inic
alE
nhan
cetu
mor
imm
unit
yT
umor
orti
ssue
loca
lizat
ion
Fab
+SC
AF
FO
LD
wit
hF
c,2
+2
NA
PD
-1×
BT
LA
Xen
cor
BT
LA
×P
D-1
Pre
clin
ical
Enh
ance
tum
orim
mun
ity
Com
bina
tori
alef
fect
Fab
+sc
Fv
wit
hF
c,1
+1
NA
TX
B4-
BC
3O
ssia
nix
Inc
TfR
×P
D-L
1P
recl
inic
alE
nhan
cetu
mor
imm
unit
yT
roja
nho
rse
Fab
+SD
Aw
ith
Fc,
2+
2N
A
CB
A-0
710
Sorr
ento
cME
T×
PD
-L1
Pre
clin
ical
Enh
ance
tum
orim
mun
ity,
anti
-tum
orig
enes
is
Com
bina
tori
alef
fect
Fab
+F
abw
ith
Fc,
1+
1N
A
Tab
le3.
Pro
gram
sin
clin
ical
and
prec
linic
alst
ages
tom
odul
ate
TM
Efo
rca
ncer
trea
tmen
t
Ant
ibod
yna
me
Org
aniz
atio
nT
arge
tsH
ighe
stph
ase
Bio
logi
cal
func
tion
Typ
eof
mec
hani
smF
orm
atC
linic
alst
udie
s
Bin
traf
usp
alfa
Gla
xoSm
ithK
line,
Mer
ckK
GaA
PD
-L1
×T
GF
beta
Pha
seII
IM
odul
ate
TM
ET
umor
orti
ssue
loca
lizat
ion
Fab
+R
EC
EP
TO
Rw
ith
Fc,
2+
2N
CT
0406
6491
,N
CT
0384
0902
,N
CT
0383
3661
,N
CT
0363
1706
,N
CT
0384
0915
,N
CT
0269
9515
,N
CT
0251
7398
AG
EN
-142
3,G
S-14
23A
genu
s,G
ilead
CD
73×
TG
Fbe
taP
hase
IM
odul
ate
TM
EC
ombi
nato
rial
effe
ctN
otdi
sclo
sed
NC
T03
9547
04
SHR
-170
1Ji
angs
uH
engr
uiP
D-L
1×
TG
Fbe
taP
hase
IM
odul
ate
TM
ET
umor
orti
ssue
loca
lizat
ion
Fab
+R
EC
EP
TO
Rw
ith
Fc,
2+
2N
CT
0371
0265
,N
CT
0377
4979
AK
-123
Ake
soB
ioph
arm
aP
D-1
×C
D73
Pre
clin
ical
Enh
ance
tum
orim
mun
ity,
mod
ulat
eT
ME
Tum
oror
tiss
uelo
caliz
atio
nN
otdi
sclo
sed
NA
Uni
TI-
101
Els
tar
The
rape
utic
sC
CR
2×
CSF
1RP
recl
inic
alM
odul
ate
TM
EC
ombi
nato
rial
effe
ctF
ab+
Fab
wit
hF
c,1
+1
NA
Fm
Ab-
2B
ioco
n;IA
TR
ICa
EG
FR
×T
GF
beta
Pre
clin
ical
Mod
ulat
eT
ME
Tum
oror
tiss
uelo
caliz
atio
nF
ab+
RE
CE
PT
OR
wit
hF
c,2
+2
NA
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

Antibody Therapeutics, 2020 27
Tab
le4.
Pro
gram
sin
clin
ical
and
prec
linic
alst
ages
topr
omot
eta
rget
cell
depl
etio
nfo
rca
ncer
trea
tmen
t
Ant
ibod
yna
me
Org
aniz
atio
nT
arge
tsH
ighe
stph
ase
Bio
logi
cal
func
tion
Typ
eof
mec
hani
smF
orm
atC
linic
alst
udie
s
Teb
enta
fusp
Imm
unoc
ore
gp10
0/H
LA
-A
∗ 020
1×
CD
3P
hase
III
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
TC
R+
scF
v,1
+1
NC
T03
0703
92,
NC
T02
8898
61,
NC
T02
5703
08,
NC
T02
5350
78,
NC
T01
2112
62,
NC
T01
2096
76O
XS-
1550
,DT
-221
9G
TB
ioph
arm
aC
D19
×C
D22
Pha
seII
Tar
get
cell
depl
etio
nA
DC
scF
v+
scF
v,1
+1
NC
T00
8894
08,
NC
T02
3701
60A
FM
-13
Aff
imed
CD
16×
CD
30P
hase
IIT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
v+
Fv,
2+
2N
CT
0122
1571
,N
CT
0232
1592
,N
CT
0266
5650
,N
CT
0319
2202
,N
CT
0407
4746
Odr
onex
tam
ab,
RE
GN
-197
9R
egen
eron
CD
3×
CD
20P
hase
IIT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
Fab
wit
hF
c,1
+1
NC
T02
6516
62,
NC
T03
8881
05IM
C-C
103C
Gen
ente
ch;
Imm
unoc
ore
MA
GE
-A
4/H
LA
∗ A02
01×
CD
3P
hase
IIT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tT
CR
+sc
Fv,
1+
1N
CT
0397
3333
IMC
nyes
oG
laxo
Smit
hKlin
e;Im
mun
ocor
eN
Y-E
SO-
1/H
LA
∗ A02
01×
CD
3P
hase
IIT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tT
CR
+sc
Fv,
1+
1N
CT
0351
5551
Mos
unet
uzum
ab,
RG
-782
8G
enen
tech
,Roc
he,
Chu
gai
CD
3×
CD
20P
hase
I/II
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+F
abw
ith
Fc,
1+
1N
CT
0250
0407
,N
CT
0367
1018
,N
CT
0367
7141
,N
CT
0367
7154
OX
S-35
50,
CD
1615
33T
riK
EG
TB
ioph
arm
a,A
ltor
Bio
Scie
nce,
U.
Min
neso
ta
CD
16×
CD
33,I
L-1
5P
hase
I/II
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
scF
v+
scF
v+
LIG
AN
D,
1+
1+
1N
CT
0321
4666
GE
N-3
013
Gen
mab
CD
3×
CD
20P
hase
I/II
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+F
abw
ith
Fc,
1+
1N
CT
0362
5037
MC
LA
-117
Mer
usC
D3
×C
LE
C12
Pha
seI/
IIT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
Fab
wit
hF
c,1
+1
NC
T03
0382
30
Flo
tetu
zum
ab,
MG
D-0
06M
acro
Gen
ics,
Serv
ier
CD
3×
CD
123
Pha
seI/
IIT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
v+
Fv,
1+
1N
CT
0215
2956
,N
CT
0373
9606
MG
D-0
07M
acro
Gen
ics
CD
3×
GPA
33P
hase
I/II
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fv
+F
vw
ith
Fc,
1+
1N
CT
0224
8805
,N
CT
0353
1632
RE
GN
-401
8R
egen
eron
,San
ofi
CD
3×
MU
C16
Pha
seI/
IIT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
Fab
wit
hF
c,1
+1
NC
T03
5643
40
Cib
isat
amab
,R
O-6
9586
88,
RG
-780
2
Gen
ente
ch,R
oche
,C
huga
iC
D3
×C
EA
Pha
seI/
IIT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
Fab
wit
hF
c,1
+1
NC
T02
3242
57,
NC
T02
6507
13,
NC
T03
3376
98,
NC
T03
8662
39
Con
tinu
ed
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

28 Antibody Therapeutics, 2020
Tab
le4.
Con
tinu
ed
Ant
ibod
yna
me
Org
aniz
atio
nT
arge
tsH
ighe
stph
ase
Bio
logi
cal
func
tion
Typ
eof
mec
hani
smF
orm
atC
linic
alst
udie
s
huG
D2-
BsA
bY
-mA
bsC
D3
×G
D2
Pha
seI/
IIT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
scF
vw
ith
Fc,
2+
2N
A
AM
G-7
01A
mge
nC
D3
×B
CM
AP
hase
IT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tsc
Fv
+sc
Fv,
1+
1N
CT
0328
7908
A−3
37G
ener
on(S
hang
hai)
CD
3×
EpC
AM
Pha
seI
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+sc
Fv,
1+
2C
ompa
nyde
velo
pmen
tpi
pelin
eA
MG
-160
Am
gen
CD
3×
PSM
AP
hase
IT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tsc
Fv
+sc
Fv,
1+
1N
CT
0379
2841
AM
G-3
30,
MT
-114
Am
gen
CD
3×
CD
33P
hase
IT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tsc
Fv
+sc
Fv,
1+
1N
CT
0252
0427
AM
G-4
24A
mge
nC
D3
×C
D38
Pha
seI
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+sc
Fv
wit
hF
c,1
+1
NC
T03
4456
63
AM
G-4
27A
mge
nC
D3
×F
LT3
Pha
seI
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
scF
v+
scF
v,1
+1
NC
T03
5413
69
AM
G-5
62A
mge
nC
D3
×C
D19
Pha
seI
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
scF
v+
scF
v,1
+1
NC
T03
5718
28
AM
G-5
96A
mge
nC
D3
×E
GF
RvI
IIP
hase
IT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tsc
Fv
+sc
Fv,
1+
1N
CT
0329
6696
AM
G-6
73A
mge
nC
D3
×C
D33
Pha
seI
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
scF
v+
scF
v,1
+1
NC
T03
2248
19
AM
G-7
57A
mge
nC
D3
×D
LL
3P
hase
IT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tsc
Fv
+sc
Fv,
1+
1N
CT
0331
9940
AM
V-5
64,
Tan
dAb
T56
4A
ffim
ed,F
red
Hut
ch,A
mph
iven
aC
D3
×C
D33
Pha
seI
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fv
+F
v,2
+2
NC
T03
1442
45,
NC
T03
5165
91A
PV
O-4
36A
ptev
oC
D3
×C
D12
3P
hase
IT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tsc
Fv
+sc
Fv
wit
hF
c,2
+2
NC
T03
6478
00
BI-
8369
09,
AM
G-4
20A
mge
n,B
oehr
inge
rIn
gelh
eim
CD
3×
BC
MA
Pha
seI
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
scF
v+
scF
v,1
+1
NC
T02
5142
39,
NC
T03
8360
53R
G-6
026,
RO
-708
2859
Roc
heC
D3
×C
D20
Pha
seI
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+F
abw
ith
Fc,
1+
2C
ompa
nyde
velo
pmen
tpi
pelin
eE
M-9
01,
CC
-932
69C
elge
neC
D3
×B
CM
AP
hase
IT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
Fab
wit
hF
c,1
+2
NC
T03
4860
67
ER
Y-9
74C
huga
iC
D3
×G
PC
3P
hase
IT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
Fab
wit
hF
c,1
+1
NC
T02
7488
37
GB
R-1
302
Gle
nmar
k,H
arbo
urB
ioM
edC
D3
×H
ER
2P
hase
IT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
scF
vw
ith
Fc,
1+
1N
CT
0282
9372
,N
CT
0398
3395
GB
R-1
342
Gle
nmar
kC
D3
×C
D38
Pha
seI
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+sc
Fv
wit
hF
c,1
+1
NC
T03
3091
11
Con
tinu
ed
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

Antibody Therapeutics, 2020 29
Tab
le4.
Con
tinu
ed
Ant
ibod
yna
me
Org
aniz
atio
nT
arge
tsH
ighe
stph
ase
Bio
logi
cal
func
tion
Typ
eof
mec
hani
smF
orm
atC
linic
alst
udie
s
GE
M-3
33G
EM
oaB
,Cel
gene
CD
3×
CD
33P
hase
IT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tsc
Fv
+sc
Fv,
1+
1N
CT
0351
6760
GE
M-3
PSC
A,
GE
M3P
SCA
GE
Moa
B,C
elge
neC
D3
×P
SCA
Pha
seI
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
scF
v+
scF
v,1
+1
NC
T03
9275
73
IGM
-232
3IG
MB
iosc
ienc
esC
D3
×C
D20
Pha
seI
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+sc
Fv
wit
hF
c,1
+10
NC
T04
0829
36
JNJ-
6757
1244
,JN
J-12
44Ja
nsse
nR
esea
rch
&D
evel
opm
ent
CD
3×
CD
33P
hase
IT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
Fab
wit
hF
c,1
+1
NC
T03
9153
79
JNJ-
6370
9178
,JN
J-91
78Ja
nsse
nR
esea
rch
&D
evel
opm
ent
CD
3×
CD
123
Pha
seI
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+F
abw
ith
Fc,
1+
1N
CT
0271
5011
JNJ-
6400
7957
,JN
J-79
57Ja
nsse
nR
esea
rch
&D
evel
opm
ent
CD
3×
BC
MA
Pha
seI
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+F
abw
ith
Fc,
1+
1N
CT
0314
5181
JNJ-
6389
8081
,JN
J-80
81Ja
nsse
nR
esea
rch
&D
evel
opm
ent
CD
3×
PSM
AP
hase
IT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
Fab
wit
hF
c,1
+1
NC
T03
9260
13
Orl
otam
ab,
MG
D-0
09M
acro
Gen
ics
CD
3×
B7-
H3
Pha
seI
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fv
+F
vw
ith
Fc,
1+
1N
CT
0262
8535
,N
CT
0340
6949
Pas
otux
izum
ab,
AM
G-2
12,
Am
gen,
Bay
erC
D3
×P
SMA
Pha
seI
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
scF
v+
scF
v,1
+1
NC
T01
7234
75,
NC
T01
7234
75P
F-0
6671
008
Pfi
zer
CD
3×
CD
H3
Pha
seI
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fv
+F
vw
ith
Fc,
1+
1N
CT
0265
9631
PF
-068
6313
5,P
F-3
135
Pfi
zer
CD
3×
BC
MA
Pha
seI
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+F
abw
ith
Fc,
1+
1N
CT
0326
9136
RE
GN
-545
8R
egen
eron
,San
ofi
CD
3×
BC
MA
Pha
seI
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+F
abw
ith
Fc,
1+
1N
CT
0376
1108
RG
-619
4,B
TR
C-4
017A
Gen
ente
chC
D3
×H
ER
2P
hase
IT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tN
otdi
sclo
sed
NC
T03
4480
42
TN
B-3
83B
Ten
eoB
io,A
bbV
ieC
D3
×B
CM
AP
hase
IT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
SDA
wit
hF
c,1
+2
NC
T03
9337
35
Xm
Ab-
1367
6,T
HG
-338
Xen
cor
CD
3×
CD
20P
hase
IT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
scF
vw
ith
Fc,
1+
1N
CT
0292
4402
Xm
Ab-
1404
5,SQ
Z-6
22X
enco
r,N
ovar
tis
CD
3×
CD
123
Pha
seI
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+sc
Fv
wit
hF
c,1
+1
NC
T02
7303
12
Xm
Ab-
1808
7,X
EN
P-1
8087
Xen
cor
CD
3×
SST
R2
Pha
seI
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+sc
Fv
wit
hF
c,1
+1
NC
T03
4119
15
HP
N-4
24H
arpo
onC
D3
×P
SMA
×al
bu-
min
Pha
seI
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
SDA
-SD
A-s
cFv,
1+
1+
1N
CT
0357
7028
M-8
02W
uhan
YZ
YB
ioph
arm
aC
D3
×H
ER
2P
hase
IT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
scF
vw
ith
Fc,
1+
1N
A
Con
tinu
ed
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

30 Antibody Therapeutics, 2020
Tab
le4.
Con
tinu
ed
Ant
ibod
yna
me
Org
aniz
atio
nT
arge
tsH
ighe
stph
ase
Bio
logi
cal
func
tion
Typ
eof
mec
hani
smF
orm
atC
linic
alst
udie
s
JNJ-
6440
7564
Jans
sen
CD
3×
GP
RC
5DP
hase
IT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
Fab
wit
hF
c,1
+1
NC
T04
1081
95,
NC
T03
3997
99R
G-6
160
Gen
ente
chC
D3
×F
cRH
5P
hase
IT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
Fab
wit
hF
c,1
+1
NC
T03
2751
03
NI-
1701
,TG
-180
1N
ovIm
mun
e,T
GT
hera
peut
ics
CD
19×
CD
47P
hase
IT
arge
tce
llde
plet
ion
Enh
ance
phag
ocyt
osis
Fab
+F
abw
ith
Fc,
1+
1N
CT
0380
4996
MC
LA
-158
Mer
usE
GF
R×
LG
R5
Pha
seI
Tar
get
cell
depl
etio
nF
cef
fect
orF
ab+
Fab
wit
hF
c,1
+1
NC
T03
5268
35
ZW
-49
Zym
ewor
ksH
ER
2×
HE
R2
Pha
seI
Tar
get
cell
depl
etio
nA
DC
Fab
+sc
Fv
wit
hF
c,1
+1
NC
T03
8212
33
A-3
19G
ener
on(S
hang
hai)
CD
3×
CD
19IN
DF
iled
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+sc
Fv,
1+
2N
CT
0405
6975
SAR
-440
234
Sano
fiC
D3
×C
D12
3Su
spen
ded
(1/2
)T
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
Fv
wit
hF
c,1
+1
NC
T03
5949
55
AF
M-1
1A
ffim
edC
D3
×C
D19
Susp
ende
d(1
)T
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
v+
Fv,
2+
2N
CT
0210
6091
,N
CT
0284
8911
cMet
×E
GF
RA
DC
Sorr
ento
EG
FR
×cM
ET
Pre
clin
ical
Tar
get
cell
depl
etio
nA
DC
Not
disc
lose
dN
A
AP
LP
2×
HE
R2
AD
CR
egen
eron
AP
LP
2×
HE
R2
Pre
clin
ical
Tar
get
cell
depl
etio
nA
DC
Fab
+F
abw
ith
Fc,
1+
1N
A
AB
P-1
50A
bpro
Cla
udin
18.2
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+sc
Fv
wit
hF
c,2
+2
NA
AB
P-1
10A
bpro
GP
C3
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+sc
Fv
wit
hF
c,2
+2
NA
AB
P-1
40A
bpro
;Luy
eC
EA
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+sc
Fv
wit
hF
c,2
+2
NA
AB
P-1
30A
bpro
;Luy
eC
D38
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+sc
Fv
wit
hF
c,2
+2
NA
AB
P-1
00A
bpro
;MSK
Can
cer
Cen
ter
Her
2×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
scF
vw
ith
Fc,
2+
2N
A
CD
16×
BC
MA
×C
D20
0A
ffim
edB
CM
A×
CD
16×
CD
200
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fv
+F
v+
Fv,
1+
2+
1N
A
AF
M-2
6A
ffim
edB
CM
A×
CD
16P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
v+
Fv,
2+
2N
A
AF
M-2
4A
ffim
edE
GF
R×
CD
16P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
v+
Fv,
2+
2N
A
AF
M-2
1A
ffim
edE
GF
RvI
II×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
v+
Fv,
2+
2N
A
B05
/CD
3A
ffim
ed;I
mm
atic
sM
MP
1-00
3/H
LA
-A
∗ 02
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fv
+F
v,2
+2
NA
Con
tinu
ed
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

Antibody Therapeutics, 2020 31
Tab
le4.
Con
tinu
ed
Ant
ibod
yna
me
Org
aniz
atio
nT
arge
tsH
ighe
stph
ase
Bio
logi
cal
func
tion
Typ
eof
mec
hani
smF
orm
atC
linic
alst
udie
s
CD
3×
FLT
3A
lloge
ne;M
aver
ick;
Pfi
zer
FLT
3×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
Fab
wit
hF
c,1
+1
NA
Fol
-aC
D3
Am
brx
Fol
Ra
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+L
IGA
ND
wit
hF
cN
A
CD
3×
MSL
NA
mge
nM
SLN
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
scF
v+
scF
v,1
+1
NA
CD
H19
×C
D3
HL
EB
iTE
Am
gen
Cad
heri
n19
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
scF
v+
scF
vw
ith
Fc,
1+
1N
A
CD
3×
EG
FR
Pb-
TC
BA
mge
n;C
ytom
XT
hera
peut
ics
EG
FR
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+sc
Fv
wit
hF
c,2
+2
NA
AM
X-1
68A
mun
ixE
pCA
M×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tsc
Fv
+sc
Fv,
1+
1N
A
AP
VO
-425
Apt
evo
The
rape
utic
sIn
cR
OR
1×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tsc
Fv
+sc
Fv
wit
hF
c,2
+2
NA
AR
B-2
01A
rbel
eC
adhe
rin-
17×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tsc
Fv
+sc
Fv
wit
hF
c,1
+1
NA
AVA
-012
Ava
cta
CD
22×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tN
otdi
sclo
sed
NA
CD
3×
CD
19A
vact
aC
D19
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Not
disc
lose
dN
A
CD
3×
CD
123
Bay
lor
Scot
t&
Whi
teR
esea
rch
Inst
itut
e
CD
123
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
scF
v+
scF
vw
ith
Fc,
2+
2N
A
CD
3×
HE
R2
Bei
jing
Han
mi
Her
2×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tN
otdi
sclo
sed
NA
CD
3×
DL
L3
Boe
hrin
ger
Inge
lhei
mD
LL
3×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tsc
Fv
+sc
Fv
wit
hF
c,1
+1
NA
CC
W-7
02C
IBR
∗ ;Sc
ripp
sP
SMA
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+SM
OL
∗N
A
CB
A-1
535
Chi
ome
Bio
scie
nce
5T4
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+sc
Fv,
1+
2N
A
CT
X-4
419
Com
pass
The
rape
utic
sB
CM
A×
NK
p30
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+F
abw
ith
Fc,
2+
2N
A
CO
VA-4
231
Cov
agen
;Fre
dH
utch
CD
33×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
SCA
FF
OL
Dw
ith
Fc,
2+
2N
A
CD
3×
EG
FR
vIII
Duk
eU
nive
rsit
yE
GF
RvI
II×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tsc
Fv
+sc
Fv,
1+
1N
A
Con
tinu
ed
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022
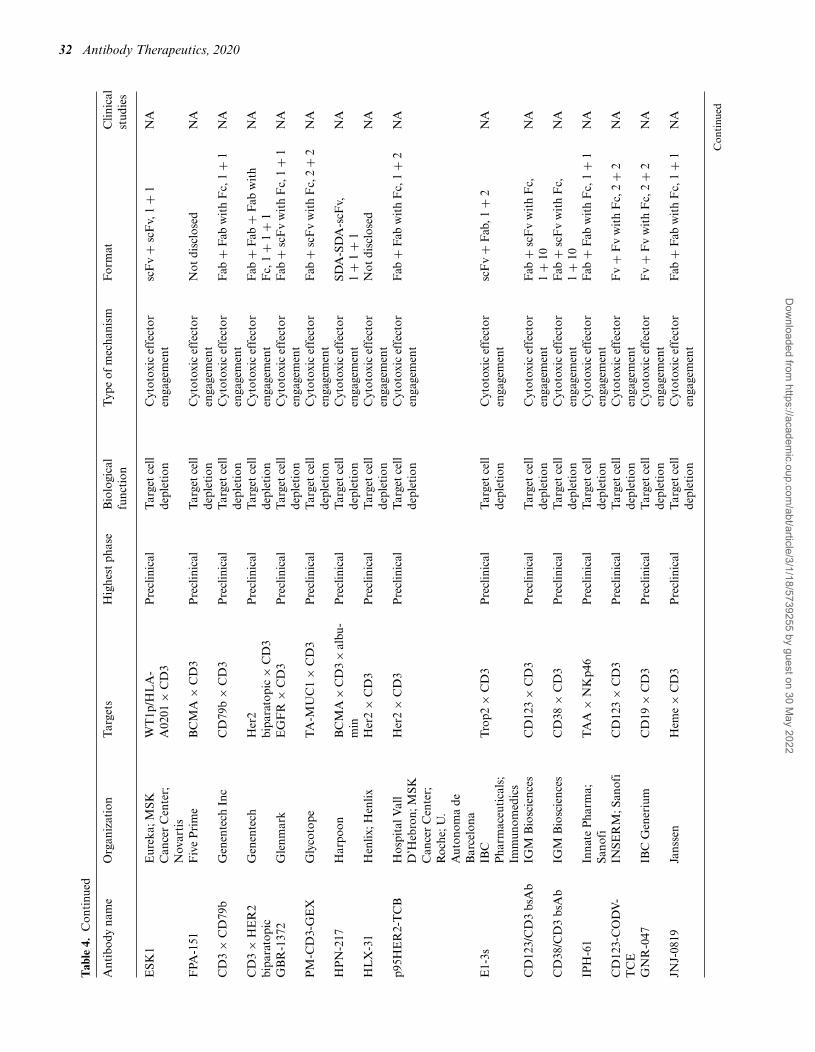
32 Antibody Therapeutics, 2020
Tab
le4.
Con
tinu
ed
Ant
ibod
yna
me
Org
aniz
atio
nT
arge
tsH
ighe
stph
ase
Bio
logi
cal
func
tion
Typ
eof
mec
hani
smF
orm
atC
linic
alst
udie
s
ESK
1E
urek
a;M
SKC
ance
rC
ente
r;N
ovar
tis
WT
1p/H
LA
-A
0201
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
scF
v+
scF
v,1
+1
NA
FPA
-151
Fiv
eP
rim
eB
CM
A×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tN
otdi
sclo
sed
NA
CD
3×
CD
79b
Gen
ente
chIn
cC
D79
b×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
Fab
wit
hF
c,1
+1
NA
CD
3×
HE
R2
bipa
rato
pic
Gen
ente
chH
er2
bipa
rato
pic
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+F
ab+
Fab
wit
hF
c,1
+1
+1
NA
GB
R-1
372
Gle
nmar
kE
GF
R×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
scF
vw
ith
Fc,
1+
1N
A
PM
-CD
3-G
EX
Gly
coto
peT
A-M
UC
1×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
scF
vw
ith
Fc,
2+
2N
A
HP
N-2
17H
arpo
onB
CM
A×
CD
3×
albu
-m
inP
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tSD
A-S
DA
-scF
v,1
+1
+1
NA
HL
X-3
1H
enlix
;Hen
lixH
er2
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Not
disc
lose
dN
A
p95H
ER
2-T
CB
Hos
pita
lVal
lD
’Heb
ron;
MSK
Can
cer
Cen
ter;
Roc
he;U
.A
uton
oma
deB
arce
lona
Her
2×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
Fab
wit
hF
c,1
+2
NA
E1-
3sIB
CP
harm
aceu
tica
ls;
Imm
unom
edic
s
Tro
p2×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tsc
Fv
+F
ab,1
+2
NA
CD
123/
CD
3bs
Ab
IGM
Bio
scie
nces
CD
123
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+sc
Fv
wit
hF
c,1
+10
NA
CD
38/C
D3
bsA
bIG
MB
iosc
ienc
esC
D38
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+sc
Fv
wit
hF
c,1
+10
NA
IPH
-61
Inna
teP
harm
a;Sa
nofi
TA
A×
NK
p46
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+F
abw
ith
Fc,
1+
1N
A
CD
123-
CO
DV
-T
CE
INSE
RM
;San
ofi
CD
123
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fv
+F
vw
ith
Fc,
2+
2N
A
GN
R-0
47IB
CG
ener
ium
CD
19×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
v+
Fv
wit
hF
c,2
+2
NA
JNJ-
0819
Jans
sen
Hem
e×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
Fab
wit
hF
c,1
+1
NA
Con
tinu
ed
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

Antibody Therapeutics, 2020 33
Tab
le4.
Con
tinu
ed
Ant
ibod
yna
me
Org
aniz
atio
nT
arge
tsH
ighe
stph
ase
Bio
logi
cal
func
tion
Typ
eof
mec
hani
smF
orm
atC
linic
alst
udie
s
Vγ
9/V
δ2
TC
R×
EG
FR
Lav
aE
GF
R×
g9/d
2T
CR
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
SDA
+SD
A,1
+1
NA
CD
3×
5T4
Mac
roG
enic
s5T
4×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
v+
Fv
wit
hF
c,1
+1
NA
Nex
t-ge
nera
tion
CD
19×
CD
3D
AR
T
Mac
roG
enic
sC
D19
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fv
+F
vw
ith
Fc,
1+
1N
A
CD
123
×C
D3
DA
RT
Mac
roG
enic
sC
D12
3×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
v+
Fv
wit
hF
c,1
+1
NA
Eph
A2x
CD
3D
AR
TM
acro
Gen
ics
Eph
a2×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
v+
Fv
wit
hF
c,1
+1
NA
CD
3×
IL13
Ra2
Mac
roG
enic
sIL
-13R
a2×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
v+
Fv
wit
hF
c,1
+1
NA
CD
3×
RO
R1
Mac
roG
enic
sR
OR
1×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
v+
Fv
wit
hF
c,1
+1
NA
CD
3×
CD
133
McM
aste
rU
nive
rsit
y;U
nive
rsit
yof
Toro
nto
CD
133
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+sc
Fv,
1+
1N
A
h8F
4-B
iTE
MD
And
erso
nC
ance
rC
ente
rP
R1/
HL
A-A
2×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tsc
Fv
+sc
Fv,
1+
1N
A
ZW
-38
Mer
ck;Z
ymew
orks
CD
19×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
scF
vw
ith
Fc,
1+
1N
A
CD
3×
HE
R2
Mol
ecul
arP
artn
ers
Her
2×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tSC
AL
FF
OL
D,1
+1
NA
CD
3×
PSM
AR
egen
eron
PSM
A×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
Fab
wit
hF
c,1
+1
NA
CD
3×
CD
20R
inat
-Pfi
zer
CD
20×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
Fab
wit
hF
c,1
+1
NA
CD
3×
RO
R1
Scri
pps
Res
earc
hIn
stit
ute
RO
R1
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
scF
v+
scF
vw
ith
Fc,
1+
1N
A
B-1
93Sh
ando
ngD
anho
ng;
Shan
ghai
Yan
yiC
D19
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Not
disc
lose
dN
A
CD
3×
Sial
yl-T
nSi
amab
The
rape
utic
sSi
alyl
-Tn
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+sc
Fv
wit
hF
c,2
+2
NA
19-3
-19
Spec
traM
abC
D19
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
scF
v+
scF
v,1
+2
NA
SV-2
02SY
SVA
XC
D19
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
SDA
+SD
A,1
+1
NA
Con
tinu
ed
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

34 Antibody Therapeutics, 2020
Tab
le4.
Con
tinu
ed
Ant
ibod
yna
me
Org
aniz
atio
nT
arge
tsH
ighe
stph
ase
Bio
logi
cal
func
tion
Typ
eof
mec
hani
smF
orm
atC
linic
alst
udie
s
SV-2
01SY
SVA
XH
er2
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
SDA
+SD
A,1
+1
NA
TN
B-5
85T
eneo
Bio
PSM
A×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
SDA
wit
hF
c,1
+1
or1
+1
+1
NA
TN
B-4
86T
eneo
Bio
CD
19×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
SDA
wit
hF
c,1
+1
NA
TN
B-3
81M
Ten
eoB
ioB
CM
A×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
SDA
wit
hF
c,1
+1
NA
CD
3×
CD
19T
ianj
inC
hase
Sun
Jinb
oda
CD
19×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tN
otdi
sclo
sed
NA
CD
3×
MO
SPD
2V
BL
The
rape
utic
sM
OSP
D2
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
scF
v+
scF
v,1
+1
NA
M-7
01W
uhan
YZ
YE
pCA
M×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
scF
vw
ith
Fc,
1+
1N
A
Y-1
50W
uhan
YZ
YC
D38
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+sc
Fv
wit
hF
c,1
+1
NA
CD
3×
EM
P2
Wuh
anY
ZY
EM
P2
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+sc
Fv
wit
hF
c,1
+1
NA
CD
3×
EG
FR
Wuh
anY
ZY
EG
FR
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+sc
Fv
wit
hF
c,1
+1
NA
CD
3×
CD
19W
uhan
YZ
YC
D19
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+sc
Fv
wit
hF
c,1
+1
NA
CD
3×
CD
20W
uhan
YZ
YC
D20
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+sc
Fv
wit
hF
c,1
+1
NA
MS-
133
Wuh
anY
ZY
CD
133
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+sc
Fv
wit
hF
c,1
+1
NA
Xm
Ab-
1448
4X
enco
rP
SMA
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+sc
Fv
wit
hF
c,1
+1
NA
YB
L-0
13Y
-Bio
logi
csP
D-L
1×
CD
3P
recl
inic
alT
arge
tce
llde
plet
ion
Cyt
otox
icef
fect
oren
gage
men
tF
ab+
Fv,
1+
2N
A
huC
D33
-BsA
bY
-mA
bsC
D33
×C
D3
Pre
clin
ical
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fab
+sc
Fv
wit
hF
c,2
+2
NA
BI-
9057
11B
oehr
inge
rIn
gelh
eim
Cad
heri
n-17
×T
RA
IL-R
2P
recl
inic
alT
arge
tce
llde
plet
ion
Enh
ance
apop
tosi
sF
ab+
scF
vw
ith
Fc,
2+
2N
A
Nov
otar
gP
rom
ethe
raC
D20
×C
D95
Pre
clin
ical
Tar
get
cell
depl
etio
nE
nhan
ceap
opto
sis
Fab
+sc
Fv,
1+
1N
A
Con
tinu
ed
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

Antibody Therapeutics, 2020 35
Tab
le4.
Con
tinu
ed
Ant
ibod
yna
me
Org
aniz
atio
nT
arge
tsH
ighe
stph
ase
Bio
logi
cal
func
tion
Typ
eof
mec
hani
smF
orm
atC
linic
alst
udie
s
AB
P-1
60A
bpro
PD
-L1
×C
D47
Pre
clin
ical
Tar
get
cell
depl
etio
nE
nhan
ceph
agoc
ytos
isN
otdi
sclo
sed
NA
BH
-29x
xB
eijin
gH
anm
iP
D-L
1×
CD
47P
recl
inic
alT
arge
tce
llde
plet
ion
Enh
ance
phag
ocyt
osis
Fab
+F
abw
ith
Fc,
1+
1N
A
IMM
-030
6G
atew
ayB
iolo
gics
;Im
mun
eOnc
oC
D20
×C
D47
Pre
clin
ical
Tar
get
cell
depl
etio
nE
nhan
ceph
agoc
ytos
isF
ab+
LIG
AN
Dw
ith
Fc,
2+
2N
A
HM
BD
-004
AH
umm
ingb
ird
CD
33×
CD
47P
recl
inic
alT
arge
tce
llde
plet
ion
Enh
ance
phag
ocyt
osis
Fab
+sc
Fv,
1+
1N
A
HM
BD
-004
BH
umm
ingb
ird
BC
MA
×C
D47
Pre
clin
ical
Tar
get
cell
depl
etio
nE
nhan
ceph
agoc
ytos
isF
ab+
Fab
wit
hF
c,1
+1
NA
IMM
-250
5Im
mun
eOnc
oP
D-L
1×
CD
47P
recl
inic
alT
arge
tce
llde
plet
ion
Enh
ance
phag
ocyt
osis
Fab
+L
IGA
ND
wit
hF
c,2
+2
NA
IMM
-260
11Im
mun
eOnc
oF
LT-3
×C
D47
Pre
clin
ical
Tar
get
cell
depl
etio
nE
nhan
ceph
agoc
ytos
isF
ab+
LIG
AN
Dw
ith
Fc,
2+
2N
A
IMM
-020
7Im
mun
eOnc
oV
EG
F×
CD
47P
recl
inic
alT
arge
tce
llde
plet
ion
Enh
ance
phag
ocyt
osis
RE
CE
PT
OR
+L
IGA
ND
wit
hF
c,2
+2
NA
IMM
-290
2Im
mun
eOnc
oH
er2
×C
D47
Pre
clin
ical
Tar
get
cell
depl
etio
nE
nhan
ceph
agoc
ytos
isF
ab+
LIG
AN
Dw
ith
Fc,
2+
2N
A
IBI-
322
Inno
vent
PD
-L1
×C
D47
Pre
clin
ical
Tar
get
cell
depl
etio
nE
nhan
ceph
agoc
ytos
isN
otdi
sclo
sed
NA
NI-
1801
Nov
imm
une
MSL
N×
CD
47P
recl
inic
alT
arge
tce
llde
plet
ion
Enh
ance
phag
ocyt
osis
Fab
+F
abw
ith
Fc,
1+
1N
A
PT
-886
Pha
nes
The
rape
utic
sC
laud
in18
.2×
CD
47P
recl
inic
alT
arge
tce
llde
plet
ion
Enh
ance
phag
ocyt
osis
Not
disc
lose
dN
A
PT
-217
Pha
nes
The
rape
utic
sD
LL
3×
CD
47P
recl
inic
alT
arge
tce
llde
plet
ion
Enh
ance
phag
ocyt
osis
Not
disc
lose
dN
A
PM
C-1
22P
harm
Abc
ine
PD
-L1
×C
D47
Pre
clin
ical
Tar
get
cell
depl
etio
nE
nhan
ceph
agoc
ytos
isN
otdi
sclo
sed
NA
Duo
Hex
aBod
y-C
D37
Gen
mab
CD
37bi
para
topi
cP
recl
inic
alT
arge
tce
llde
plet
ion
Fc
effe
ctor
Fab
+F
abw
ith
Fc,
1+
1N
A
PM
-PD
L-G
EX
Gly
coto
peT
A-M
UC
1×
PD
-L1
Pre
clin
ical
Tar
get
cell
depl
etio
nF
cef
fect
orF
ab+
scF
vw
ith
Fc,
2+
2N
A
CD
38×
IGF
-1R
I’ro
mG
roup
CD
38×
IGF
-1R
Pre
clin
ical
Tar
get
cell
depl
etio
nF
cef
fect
orsc
Fv
+sc
Fv
wit
hF
c,1
+1
NA
∗ CIB
R,C
alif
orni
aIn
stit
ute
for
Bio
med
ical
Res
earc
h;SM
OL
,sm
allm
olec
ule.
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

36 Antibody Therapeutics, 2020
Tab
le5.
Pro
gram
sin
clin
ical
and
prec
linic
alst
ages
for
infl
amm
ator
yco
ndit
ions
Ant
ibod
yna
me
Org
aniz
atio
nT
arge
tsH
ighe
stph
ase
Bio
logi
calf
unct
ion
Typ
eof
mec
hani
smF
orm
atC
linic
alst
udie
s
Ozo
raliz
umab
,T
S-15
2,P
F-5
2308
96,
AT
N-1
03
Sano
fi,T
aish
o,E
ddin
gpha
rmT
NF
×al
bum
inP
hase
III
Hal
f-lif
eex
tens
ion
Hal
f-lif
eex
tens
ion
SDA
+SD
A,1
+2
NC
T00
9590
36,
NC
T01
0071
75,
NC
T01
0638
03,
NC
T04
0775
67V
obar
ilizu
mab
Abb
Vie
;Abl
ynx
IL-6
R×
albu
min
Pha
seII
Hal
f-lif
eex
tens
ion
Hal
f-lif
eex
tens
ion
SDA
+SD
A,1
+1
NC
T02
5186
20,
NC
T02
4378
90,
NC
T02
3093
59,
NC
T02
2879
22R
omilk
imab
,SA
R-1
5659
7Sa
nofi
IL-4
×IL
-13
Pha
seII
Com
bina
tori
alef
fect
Com
bina
tori
alef
fect
Fab
+F
vw
ith
Fc,
2+
2N
CT
0152
9853
,N
CT
0234
5070
,N
CT
0292
1971
M-1
095,
AL
X-0
761
Avi
llion
;Mer
ckSe
rono
IL-1
7A×
albu
min
×IL
-17F
Pha
seII
Com
bina
tori
alef
fect
Com
bina
tori
alef
fect
SDA
+SD
A,1
+1
+1
NC
T03
3847
45,
NC
T02
1564
66M
GD
-010
,P
RV
-327
9M
acro
Gen
ics,
Pro
vent
ion
CD
32B
×C
D79
BP
hase
I/II
Dom
inan
tne
gati
veD
omin
ant
nega
tive
Fv
+F
vw
ith
Fc,
1+
1N
CT
0237
6036
AM
G-5
70,
ME
DI-
0700
Am
gen,
Vie
laB
io,
Ast
raZ
enec
aB
AF
F×
ICO
SLP
hase
IC
ombi
nato
rial
effe
ctC
ombi
nato
rial
effe
ctF
ab+
PE
PT
IDE
wit
hF
c,2
+2
NC
T02
6189
67,
NC
T03
1560
23,
NC
T04
0580
28T
ibul
izum
abE
liL
illy
BA
FF
×IL
-17A
Pha
seI
Com
bina
tori
alef
fect
Com
bina
tori
alef
fect
Fab
+sc
Fv
wit
hF
c,2
+2
Com
pany
deve
lopm
ent
pipe
line
JNJ-
6117
8104
Jans
sen
Res
earc
h&
Dev
elop
men
tT
NF
×IL
-17A
Pha
seI
Com
bina
tori
alef
fect
Com
bina
tori
alef
fect
Fab
+F
abw
ith
Fc,
1+
1N
CT
0275
8392
ON
O-4
685
Ono
CD
3×
PD
-1P
hase
ID
omin
ant
nega
tive
Dom
inan
tne
gati
veF
ab+
Fab
wit
hF
c,1
+1
NC
T04
0790
62
ES-
210,
AP
VO
-210
Apt
evo
The
rape
utic
sC
D86
-IL
10P
hase
IT
issu
esp
ecif
icit
yT
umor
orti
ssue
loca
lizat
ion
scF
v+
scF
vw
ith
Fc,
2+
2N
CT
0376
8219
CD
19×
CD
11c
Nat
iona
lJew
ish
Hea
lth
CD
19×
CD
11c
Pre
clin
ical
Tar
get
cell
depl
etio
nF
cef
fect
orF
ab+
Fab
wit
hF
c,1
+1
NA
AM
-201
AbC
lon
IL-6
R×
TN
FP
recl
inic
alA
nti-
infl
amm
atio
nC
ombi
nato
rial
effe
ctF
ab+
SCA
FF
OL
Dw
ith
Fc,
2+
2N
A
IL4R
alph
a/IL
-5bs
Ab
arG
EN
-XIL
-4R
a×
IL-5
Pre
clin
ical
Ant
i-in
flam
mat
ion
Com
bina
tori
alef
fect
Fab
+sc
Fv
wit
hF
c,2
+2
NA
Con
tinu
ed
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022
Antibody Therapeutics, 2020 37
Tab
le5.
Con
tinu
ed
Ant
ibod
yna
me
Org
aniz
atio
nT
arge
tsH
ighe
stph
ase
Bio
logi
calf
unct
ion
Typ
eof
mec
hani
smF
orm
atC
linic
alst
udie
s
BH
-165
7B
eijin
gH
anm
iT
NF
×IL
-17A
Pre
clin
ical
Ant
i-in
flam
mat
ion
Com
bina
tori
alef
fect
Fab
+F
abw
ith
Fc,
1+
1N
A
IL-4
×IL
-13
Bei
jing
VD
JBio
IL-4
×IL
-13
Pre
clin
ical
Ant
i-in
flam
mat
ion
Com
bina
tori
alef
fect
Not
disc
lose
dN
AIL
-1×
TN
Fα
Bei
jing
VD
JBio
IL-1
×T
NF
Pre
clin
ical
Ant
i-in
flam
mat
ion
Com
bina
tori
alef
fect
Not
disc
lose
dN
AC
MX
-02
Com
plix
TN
F×
IL-2
3P
recl
inic
alA
nti-
infl
amm
atio
nC
ombi
nato
rial
effe
ctF
ab+
SCA
FF
OL
Dw
ith
Fc,
2+
2N
A
ND
-016
Inta
rcia
TN
F×
IL-
17A
×al
bum
inP
recl
inic
alA
nti-
infl
amm
atio
nC
ombi
nato
rial
effe
ctF
v+
Fv
+F
v,1
+1
+1
NA
MT
-619
4M
itsu
bish
iTan
abe
Pha
rma
IL-6
R×
IL-1
7AP
recl
inic
alA
nti-
infl
amm
atio
nC
ombi
nato
rial
effe
ctF
ab+
SCA
FF
OL
Dw
ith
Fc,
2+
2N
A
YB
L-0
04Y
-Bio
logi
csT
NF
×IL
-17A
Pre
clin
ical
Ant
i-in
flam
mat
ion
Com
bina
tori
alef
fect
Fab
+sc
Fv
wit
hF
c,2
+2
NA
PT
-001
Pan
dion
MA
dCA
M×
PD
-1P
recl
inic
alA
nti-
infl
amm
atio
nT
umor
orti
ssue
loca
lizat
ion
Fab
+sc
Fv
wit
hF
c,2
+2
NA
AL
XN
-172
0A
lexi
onC
5×
albu
min
Pre
clin
ical
Hal
f-lif
eex
tens
ion
Ext
ende
dha
lf-l
ife
scF
v+
scF
v,1
+1
NA
SDA-based. Two different VHHs can be fused to forma bsAb [4]. This format may be the smallest bsAb formatwith molecular weight approximately 25 KD. It has beenreported that two different VHHs can be fused to coiled-coil peptide to form Combody. The peptide facilitates theoligomerization of the antibody and renders the antibodyavidity effect [5]. Two different VHH can also be engineeredon the N-terminus of CH1 and CL to form a Fab-like 1 + 1bsAb fragment [6].
ScFv-based. Bispecific T cell engager (BiTE), one ofthe formats used to redirect T cells to tumor cells, com-prises two tandem linked scFvs: one scFv against a tumor-associated antigen and another binding to CD3 on T cells.The structure and mechanism of BiTE was well reviewed byWolf [7]. Two scFvs can also be indirectly linked, such asvia a CL, to form a bsAb in scFv-CL-scFv format [8]. Dueto aggregative tendency of scFv, various techniques wereemployed to stabilize scFv. Brolucizumab (Beovu) was engi-neered using scFv-λcap platform [9]. Similar technologywas also used to build multispecific antibody (msAb)-basedtherapeutics by cognate heterodimerization (MATCH) [10],where up to four distinct binding sites can be integrated intoa multispecific Fv- or scFv-based molecule.
Fv-based. A diabody molecule is formed by two polypep-tides: one polypeptide contains VHa and VLb; anotherpolypeptide contains VHb and VLa. Due to the shortlinker, VHa associates with VLa on another polypeptideand similarly VLb associates with VHb to form 1 + 1 bsAbfragment. A diabody-based bispecific format is called dual-affinity retargeting antibody or DART [11–13]. DARTmolecules may contain Fc domain to extend in vivo half-life and grand effect functions. TandAb is another Fv-basedbispecific fragment: two polypeptides are forced to fold ina head-to-tail fashion to form 2 + 2 bsAb fragment [14].
Combination. In a native antibody, VH and VL are onthe N-terminus of Fab region and CH1 and CL on C-terminus. It was found that CH1 and CL can also facilitatethe association of VH and VL on C-terminus of a Fab-Fv fusion protein. This Fab directed VH-VL associationcan be further improved by introducing a disulfide bondbetween the VH and VL on C-terminus [15]. A VH on theC-terminus of a Fab-Fv may associate with a C-terminalVL on another Fab-Fv to form 2 + 2 tetramer Fab-Fv[16]. Similarly, a scFv can be fused on the C-terminus of aFab to form Fab-scFv fusion proteins. The so-called bibodyhas one Fab with one scFv, and “tribody” has one Fabwith two scFvs [17]. A tribody can be either bispecific ortrispecific, depending on the specificity of the two attachedscFvs. A VHH can be fused to a light chain C-terminus ofa Fab to form 1 + 1 bispecific antibody fragment [18]. Itwas reported that three tandem linked VHHs can be fusedwith a scFv to form 1 + 3 bispecific fragment [19]. A bsAbfragment containing a VHH or scFv specific to humanserum albumin is a common strategy to extend serum half-life of such molecules.
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

38 Antibody Therapeutics, 2020
Tab
le6.
Pro
gram
sin
clin
ical
and
prec
linic
alst
ages
for
othe
rco
ndit
ions
Ant
ibod
yna
me
Org
aniz
atio
nT
arge
tsH
ighe
stph
ase
Bio
logi
calf
unct
ion
Typ
eof
mec
hani
smF
orm
atC
ondi
tion
sC
linic
alst
udie
s
Far
icim
ab,
RG
-771
6,R
O-6
8674
61
Roc
he,C
huga
iP
harm
aceu
tica
lV
EG
F×
AN
GP
T2
Pha
seII
IA
nti-
angi
ogen
esis
Com
bina
tori
alef
fect
Fab
+F
abw
ith
Fc,
1+
1O
cula
r,di
abet
icre
tino
path
yN
CT
0194
1082
,N
CT
0248
4690
,N
CT
0269
9450
,N
CT
0303
8880
,N
CT
0362
2580
,N
CT
0362
2593
,N
CT
0382
3287
,N
CT
0382
3300
IBI-
302
Inno
vent
VE
GF
×co
mpl
e-m
ent
Pha
seI
Ant
i-an
giog
enes
is;
anti
-inf
lam
mat
ion
Com
bina
tori
alef
fect
Not
disc
lose
dO
cula
rN
CT
0381
4291
Gre
mub
amab
,M
ED
I390
2M
edIm
mun
eP
crV
×P
sIP
hase
IIC
ombi
nato
rial
effe
ctC
ombi
nato
rial
effe
ctF
ab+
scF
vw
ith
Fc,
2+
2A
ntib
acte
rial
NC
T02
2557
60,
NC
T02
6969
02M
ED
I-73
52A
stra
Zen
eca
NG
F×
TN
FP
hase
IIC
ombi
nato
rial
effe
ctC
ombi
nato
rial
effe
ctsc
Fv
+R
EC
EP
-T
OR
wit
hF
c,2
+2
Ana
lges
icdr
ugs
NC
T02
5081
55,
NC
T03
7559
34
10E
8.4/
iMab
Tai
Med
,Aar
onD
iam
ond
AID
SR
esea
rch
Cen
ter
HIV
-1E
nv×
CD
4P
hase
IB
road
enpr
otec
tion
Com
bina
tori
alef
fect
Fab
+F
abw
ith
Fc,
1+
1H
IV-1
NC
T03
8752
09
SAR
-441
236
Sano
fi,N
IHH
IV-1
Env
trip
arat
opic
Pha
seI
Com
bina
tori
alef
fect
Com
bina
tori
alef
fect
Fab
+F
vw
ith
Fc,
1+
1+
1H
IV-1
NC
T03
7051
69
MG
D-0
14M
acro
Gen
ics,
NIA
IDC
D3
×H
IV-1
Env
prot
ein
Pha
seI
Tar
get
cell
depl
etio
nC
ytot
oxic
effe
ctor
enga
gem
ent
Fv
+F
vw
ith
Fc,
1+
1H
IV-1
NC
T03
5709
18
BF
KB
-848
8A,
RG
-799
2G
enen
tech
FG
FR
1×
beta
-K
loth
oP
hase
IT
issu
esp
ecif
icit
yT
umor
orti
ssue
loca
lizat
ion
scF
v+
scF
vw
ith
Fc,
1+
1D
iabe
tes
NC
T02
5933
31,
NC
T03
0605
38A
BP
-201
AbM
edV
EG
F×
AN
GP
T2
Pre
clin
ical
Ant
i-an
giog
enes
isC
ombi
nato
rial
effe
ctF
ab+
scF
vw
ith
Fc,
2+
2O
cula
rN
A
SL-6
34U
nive
rsit
yof
Col
orad
oSy
stem
VE
GF
×A
NG
PT
2P
recl
inic
alA
nti-
angi
ogen
esis
Com
bina
tori
alef
fect
Not
disc
lose
dO
cula
rN
A
KSI
-501
Kod
iak
Scie
nces
VE
GF
×IL
-6P
recl
inic
alA
nti-
angi
ogen
esis
,an
ti-i
nfla
mm
atio
nC
ombi
nato
rial
effe
ctF
ab+
RE
CE
P-
TO
Rw
ith
Fc,
2+
2
DM
E,U
veit
isN
A
FIT
-1H
umab
sB
ioM
edZ
ika
Epr
otei
nbi
para
topi
cP
recl
inic
alB
road
enpr
otec
tion
Bip
arat
opic
Fab
+F
abw
ith
Fc,
2+
2In
fect
ion
NA
TM
B-b
ispe
cifi
cA
aron
Dia
mon
dA
IDS
Res
earc
hC
ente
r;T
aiM
ed
HIV
gp12
0×
CD
4P
recl
inic
alB
road
enpr
otec
tion
Com
bina
tori
alef
fect
Fab
+F
abw
ith
Fc,
1+
1In
fect
ion
NA
VIS
-RSV
Vis
terr
a,V
irB
iote
chno
logy
RSV
F×
RSV
GP
recl
inic
alB
road
enpr
otec
tion
Com
bina
tori
alef
fect
Fab
+sc
Fv
wit
hF
c,1
+1
Infe
ctio
nN
A
Con
tinu
ed
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

Antibody Therapeutics, 2020 39
Tab
le6.
Con
tinu
ed
Ant
ibod
yna
me
Org
aniz
atio
nT
arge
tsH
ighe
stph
ase
Bio
logi
calf
unct
ion
Typ
eof
mec
hani
smF
orm
atC
ondi
tion
sC
linic
alst
udie
s
Dua
lFZ
Dan
dL
RP
5/6
agon
ist
Ant
lerA
The
rape
utic
sF
ZD
×L
RP
5/6
bipa
rato
pic
Pre
clin
ical
Cof
acto
rm
imet
icC
ofac
tor
mim
etic
Fv
+F
vw
ith
Fc,
2+
1+
1T
issu
ere
pair
NA
AB
L-3
01A
BL
Bio
?×
alph
a-sy
nucl
ein
Pre
clin
ical
Neu
tral
izin
gpa
thog
enic
targ
etT
roja
nho
rse
Fab
+sc
Fv
wit
hF
c,2
+2
Neu
rolo
gy/P
sych
iatr
icN
A
AT
V:B
AC
E1/
Tau
Den
ali
TfR
×B
AC
E1
orT
fR×
Tau
Pre
clin
ical
Neu
tral
izin
gpa
thog
enic
targ
etT
roja
nho
rse
Fab
+F
ab+
SDA
wit
hF
c,1
+1
+1
Neu
rolo
gy/P
sych
iatr
icN
A
KY
-104
9K
ymab
Ltd
FIX
×F
XP
recl
inic
alC
ofac
tor
mim
etic
Cof
acto
rm
imet
icF
ab+
Fab
wit
hF
c,1
+1
Hem
atol
ogic
NA
Bis
peci
fic
fully
hum
anIg
GSh
ire
plc
FIX
×F
XP
recl
inic
alC
ofac
tor
mim
etic
Cof
acto
rm
imet
icF
ab+
Fab
wit
hF
c,1
+1
Hem
atol
ogic
NA
Fc-containing bispecific antibodies
The Fc region, part of natural antibody, is homodimerof two polypeptide chains. Depending on the isotype ofthe antibody, each comprising two to three heavy chainconstant domains (CH2, CH3, and CH4). The Fc regionnot only imparts an antibody effector functions due toFcγ R binding and complement-binding but also extendsits in vivo half-life via FcRn binding.
When two different heavy chains and two differentlight chains of IgG antibodies are expressed in onecell, these different heavy chains and light chains mayscramble randomly, possibly to form 10 different IgGantibody molecules. Among these, statistically only 12.5%would be desired bsAb. For symmetric Fc-containingbispecific formats, a challenge is to avoid heavy chain/lightchain mispairing. For asymmetric Fc-containing bispecificformats, an additional challenge is to force heterodimericheavy chain formation.
Forming heterodimeric Fc can be achieved by engineer-ing the interface of two CH3 (for IgG) or CH4 (for IgMor IgE) domains, by changing size (knob-into-hole) [20–22] and electrostatic steering [23]. Computational methodshave been used to identify the mutations that facilitate het-erodimeric association [24, 25]. Several groups also used theinterface of different Ig proteins to design the heterodimericFc. Davis et al. developed derivatives of human IgG andIgA CH3 domains composed of alternating segments ofhuman IgA and IgG CH3 sequences [26]. Skegro et al.grafted some residues from T cell receptor (TCR) constantregion to CH3 of IgG1 or CH4 of IgM [27]. An alter-native strategy is to purify heterodimer from unwantedhomodimers. With the modification on CH3 domain, theheterodimer and homodimer have different affinity bindingon Protein A, and the bsAb with heterodimeric Fc can beisolated [28].
In order to ensure cognate heavy chain and light chainpairing, several strategies have been reported. The firststrategy is to use SDA, scFv, or scFab as antigen-bindingbuilding blocks. In addition, single-chain IgG has beenreported [29], where two heavy chains, two light chains,and three linker sequences were expressed from one gene.Protease cleavage sites were integrated into these linkers,allowing protease digestion to cleave the linkers. The secondstrategy is using common light chain or common heavychain. In the case of common light chain, an identical lightchain is used as the partner for two different heavy chains[30–33]. Common heavy chain was also reported in a κλ-body case [34]. The third strategy is to modify the interfaceof VH-CH1 and VL-CL, including developing orthogonalFab interface [35, 36], altering the location of interchaindisulfide bond [37–39], and addition of charged pairs andknob-into-hole [40, 41]. These strategies usually involvechanges on both VH-VL interface and CH1/CL interface.Yet, Bonish et al. reported that the preferential associationcan be achieved by only engineering the CH1/CL interface[42]. To associate cognate VH and VL, the CH2 domainfrom IgM or IgE have been engineered to form heterodimerto replace CH1/CL [43] [Dong, WO2017011342].
There are additional strategies to avoid heavy chain/lightchain mispairing. Schaefer et al. described a CrossMab
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

40 Antibody Therapeutics, 2020
Figure 2. The classification of bsAb formats based on assembly of antibody fragments as building blocks. The first row and column list the five basicbuilding blocks (SDA, Fv, scFv, Fab, scFab). The different color and shape of VH and VL represent their origins from different parental antibodies. Theassembly of different building blocks creates various bsAb formats classified into 30 groups. An exemplary format and its molecular weight of each groupare listed. The diagonal line divides the formats into bispecific formats without Fc (top right with number 1-15) and bispecific formats with Fc (bottomleft with number 16-30): 1, tandemly linked SDAs; 2, a SDA tandemly linked on the VH of a Fv; 3, a SDA tandemly linked on the VH of a scFv; 4,two SDAs are separately linked on the carboxyl-terminus of constant domain of a Fab; 5, a SDA tandemly linked on the VL of a scFab; 6, the VHs andVLs of two Fvs cross pair to each other to form diabody; 7, a scFv tandemly linked on the VH of a Fv; 8, the VH and VL of a Fv each linked on thecarboxyl-terminus of CH1 and CL of a Fab; 9, the VH of a Fv linked to the CH1 of a scFab; 10, two tandemly linked scFvs; 11, a scFv linked on the VHof a Fab; 12, a scFab
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

Antibody Therapeutics, 2020 41
approach: exchange of heavy chain and light chain domainswithin the Fab of one half of the bsAb [44]. Moore et al.described a Mab-Fv approach: the first pair of variableregions was present on the regular position of an IgG. Thesecond pair variable region VL and VH were fused to theC-terminus of the heterodimeric heavy chain, respectively,to form a 1 + 2 bsAb [24]. A similar 1 + 2 bsAb formatwith protease-inducible activity was also reported [45].
Recently, a versatile bispecific platform namedWuXiBody
®has been developed [Xu, WO2019057122A1].
The CH1/CL domains of one of the two parental antibodiesare replaced by stabilized TCR α and β constant regions(Cα/Cβ) to ensure the cognate light chain-heavy chainpairing. This approach has been tested on more than100 pairs of antibodies and compatible with most of theantibody pairs (data not shown).
Below are some representative examples of Fc-containingbispecific formats with SDA, Fv, scFv, Fab, and scFab.There are more examples that use the combination of thesefive building blocks.
SDA-based. As a small, flexible, and modular antigen-binding site, it is obvious that SDA or VHH can be fusedto N-terminus or C-terminus of heavy chain or light chainof an IgG antibody. As shown in Fig. 3, SDA can alsobe inserted in to other fusion sites. Shen et al. reportedthat a SDA antibody can be fused with VL to form 2 + 2bispecific format [46, 47]. Shi et al. used two different SDAsto replace VH and VL of an IgG, respectively, to form 2 + 2biparatopic antibody [48]. In addition to variable domain,CH3 domain on Fc has been engineered as binding site[49]. Broadly speaking, the engineered CH3 is a SDA thatcan be integrated into a bispecific format, such as IgG-likeBsAb [50].
Fv-based. VH and VL domains are tandemly linked withVH and VL of another IgG antibody, to form a 2 + 2bispecific IgG format, named as “dual variable domainimmunoglobulin” or DVD-Ig [51]. Fab + Fv-based 1 + 2format called mAb-Fv [24] was mentioned above. Fv canalso replace CH2 domain to form a 1 + 2 bispecific formatcalled TriFab [52]. Seifert et al. employed diabody formatcombined with heterodimeric CH2 from IgM or IgE toconstruct 2 + 2 bispecific format [43]. AforementionedWuXiBody
®formats are also Fv-based formats, including
1 + 1, 1 + 2, and 2 + 2 formats.
ScFv-based. There are many Fc-containing bispecificformats comprising of scFv, although scFv is prone to
aggregate. A scFv can be fused with heavy chain of IgG toform symmetric 2 + 2 bsAb [53], called Morrison format.Morrison format is one of the earliest bispecific formatsthat have still been widely used. ScFv can also be fusedwith light chain [54]. To form 1 + 1 format, several groupsused scFv to replace one of the Fabs on an IgG and usedengineered Fc heterodimer [55, 56]. Two different scFvs canbe used to replace both Fabs on an IgG antibody to form1 + 1 bispecific format [57, 58]. ScFv can also be placedin the hinge region or CH3 domain to form 2 + 2 formats[59, 60]. Kim et al. fused scFv with CH1-CH2-CH3 and co-expressed LC domain, potentially masking the hydropho-bic part of scFv to make the molecule more stable [61].
Fab/scFab-based. Several Fab- or scFab-based bispecificformats have been reported. Fab-based CrossMab, a 1 + 1bispecific IgG format [44], was mentioned above. Fabs-in-tandem immunoglobulin (FIT-Ig) is a format where a Fab isfused to the N-terminus of another IgG antibody: the lightchain of the Fab is fused with the heavy chain of the IgG,and FD chain of the Fab and light chain of the IgG areseparate polypeptides. These three different polypeptidescan be co-expressed from single cells and be assembled toform the bsAb [62]. In theory, the FD of antibody A andthe light chain of antibody B can associate to each otherto form a hybrid Fab, but this hybrid Fab can be removedin Protein A purification step. Bostrom et al. described atwo-in-one bsAb, a bsAb in regular IgG form, and eacharm can bind two distinct antigens [63–65]. Hu et al. evendeveloped a four-in-one antibody [66]. Strop et al. showedthat making mutations in hinge region and CH3 domain ofhuman IgG1 and IgG2 could facilitate heterodimerizationof heavy chain. Two parental antibodies can be expressedand purified separately and mixed together under appro-priate redox conditions, resulting in formation of a stablebsAb [67]. Labrijn et al. reported a similar platform, latercalled DuoBody [68]. scFab can be used to construct 1 + 1format [69], and it can also be used as one of the buildingblocks to construct tetraspecific antibody [70].
Other binding modalities
As mentioned earlier, peptides, ligands, receptors, anddifferent protein scaffolds, either native form or engineeredform, can be used as antigen-binding sites. The non-antibody scaffolds include Adnectin, DARPin, Affilins,alpha helix scaffold, avimer, Centyrin, Duocalin, Ecallan-tide, Fynomer, microprotein, peptide, Protein A domain,trimeric, TCR, etc. There are numerous possibilities to
tandemly linked with a scFv; 13, two tandemly linked Fabs: the light chain of one Fab linked with the heavy chain of another Fab and vice versa; 14, ascFab linked on the VH of a Fab; 15, tandemly linked two scFab; 16, two tandemly linked SDA on Fc to form homodimer; 17, a SDA and the VH of a Fvlinked to the FcA to pair with the VL of a Fv linked to another FcB to form heterodimer; 18, a diabody on FcA to pair with FcB to form heterodimer;19, a SDA on FcA to pair with a scFv on FcB to form heterodimer; 20, a scFv and the VH of a Fv linked to FcA to pair with the VL of the Fv linked withFcB to form heterodimer; 21, two scFv tandemly linked to the amino- and carboxyl-terminus of a Fc to form homodimer; 22, a SDA tandemly link to thelight chain of a IgG to form homodimer; 23, a TCR constant domain anchored Fv linked to FcA to pair with a half IgG with FcB to form heterodimer(WuXiBodyTM); 24, a scFv linked FcA to pair with a half IgG with FcB to form heterodimer; 25, two tandemly linked Fabs (the light chain of a Fab linkedon the heavy chain of another Fab) on Fc to form homodimer (FIT-Ig); 26, a SDA on FcA to pair with a scFab on FcB to form heterodimer; 27, a scFaband the VH of a Fv linked on FcA to pair with the VL linked on FcB to form heterodimer; 28, a scFv linked on FcA to pair with a scFab linked on FcBto form heterodimer; 29, a half IgG with FcA paired with scFv linked on FcB to form heterodimer; 30, two scFab each linked on FcA and FcB to formheterodimer. Above mentioned FcA and FcB are engineered Fc pair to facilitate Fc heterodimerization.
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

42 Antibody Therapeutics, 2020
Figure 3. Fusion sites for antigen-binding building blocks. A) A heterodimeric Fc fragment has at least six fusion sites: amino-terminus (1 and 4), carboxyl-terminus (3 and 6) of Fc and between CH2 and CH3 (2 and 5). B) The fragment made of heterodimeric Fc and two differently heterodimerized CL-CH1domains provides at least twelve fusion sites: amino-terminus of CL (1 and 7) and CH1 (3 and 9), carboxyl-terminus of CL (2 and 8) and CH3 (6 and 12),hinge region (4 and 10), between CH2 and CH3 (5 and 11).
generate bispecific formats using these binding modalities.Recent examples include peptide [71], VEGF receptor [72],TCR [73], and single-chain TRAIL [74].
In the new paradigm of bsAbs, many novel formatshave been designed and tested. The general goal is todesign a molecule to enable novel therapeutic mechanismsand to make homogeneous product in large scale to meetthe need of clinical development and commercial manu-facturing. Additionally, more multispecific formats havebeen reported in the recent years, including trispecific [75],tetraspecific [70], and pentaspecific [71].
THE RESURGENCE OF BISPECIFIC ANTIBODIES
During the last few years, the number of clinical stud-ies using bsAbs has increased exponentially (Fig. 4A). Infact, this increase in 2014 matched with the launch ofBlincyto (Amgen), the first commercialized BiTE for thetreatment of acute lymphoblastic leukemia. However, it wasnot until 2017 that another bsAb, Hemlibra (Roche), waslaunched for the treatment of hemophilia A. Currently,most bsAbs in clinical development are in early studies(67% in Phase I, 25% in Phase II) with only five productsin Phase III studies (Fig. 4B). The majority of clinical stagebsAbs (∼84%) are designed to treat cancer, especially solidtumors, breast cancer, and acute myeloid leukemia. Nev-ertheless, there are some products designed to treat otherconditions such as rheumatoid arthritis or autoimmunediseases (Fig. 4C). The company with more bsAbs underactive development is Amgen, followed by MacroGenicsand then Lilly, Janssen, Roche, Sanofi, and Xencor. Thestrategy in nearly half of the developing bispecific productsis to deplete the malignant cells by engaging cytotoxic effec-tor cells including T or natural killer (NK) cells using CD3or FcGR3A (CD16) targeting arms. Another commonlyused strategy is to identify tumor or tissue-specific markersto act only in the affected areas. For that, many companieshave designed their own technology to manufacture bsAbs,including Amgen’s BiTE, MacroGenics’ DART, or Roche’sCrossMab platforms.
Accordingly, with the increase in bsAb development, thenumber of deals (excluding mergers and acquisitions) havefocused on clinical stage bsAbs resulting in an increaseof 140% in the last 3 years, while the total disclosed dealvalue decreased by 50%, from $3.2 billion to $1.6 billion;$6.8 billion is recorded across the whole period (Fig. 5A).From these deals, nine were worth more than $100 million,and most were structured with milestones, signifying thebalancing of risk and reward between the partners. Sanofiand Regeneron’s 2015 co-development agreement focusedon various antibodies, including CD3 × MUC16 (REGN-401) and CD3 × BCMA (REGN-5458) (Fig. 5B). Fromthese deals, $3.8 billion were spent on oncology followedby $1 billion on infection diseases from a total of $6.8billion, probably due to the clinical and commercial poten-tial of treating patients in these disease areas with bsAbs(Fig. 5C).
The global market of bsAb was worth $0.46 billion in2018, which was dominated almost equally by Blincyto($230 million) and Hemlibra ($229 million). As predicted,the market for Hemlibra will boom in the next few yearsand grow up to $3.96 billion by 2024. Instead, Blincytowill only have a moderate increase. With the massive salesgrowth of Hemlibra and the potential approval of newentrants, for instance, faricimab, gremubamab, MCLA-117, and XmAb-14045, the bsAb market will surpass $5.43billion in 2024 (Fig. 5D).
BIOLOGY DRIVES DEVELOPMENT OF VARIOUSBISPECIFIC ANTIBODIES
Most human diseases are complex, often driven by multipleredundant or distinct mechanisms; thereby single-targetapproach such as mAb may not be sufficient to achieveoptimal therapeutic efficacy. Especially, many therapeuticconcepts need physical linkage of two or more targets. Inthis case, bsAbs or msAbs targeting two or more targetsmay offer novel therapeutic applications that are difficultto achieve by mAbs. In a recent comprehensive reviewarticle, Aran Labrijn, Maarten Janmaat, Janice Reichert,
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022
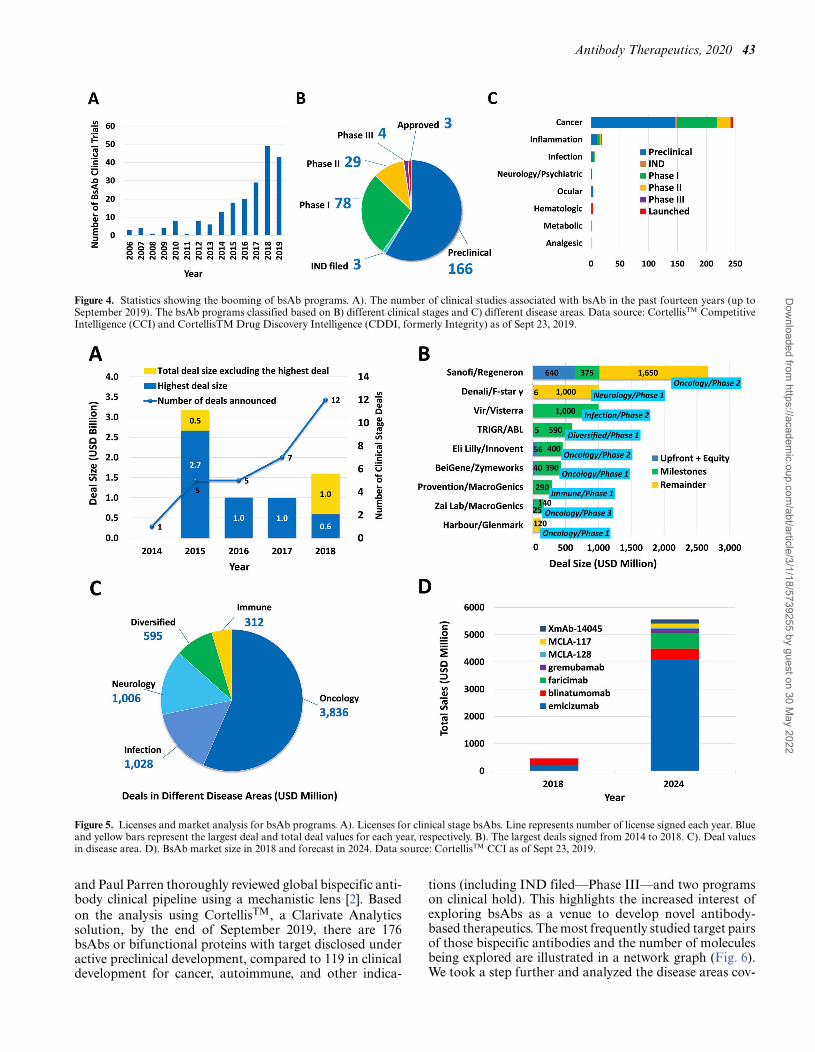
Antibody Therapeutics, 2020 43
Figure 4. Statistics showing the booming of bsAb programs. A). The number of clinical studies associated with bsAb in the past fourteen years (up toSeptember 2019). The bsAb programs classified based on B) different clinical stages and C) different disease areas. Data source: CortellisTM CompetitiveIntelligence (CCI) and CortellisTM Drug Discovery Intelligence (CDDI, formerly Integrity) as of Sept 23, 2019.
Figure 5. Licenses and market analysis for bsAb programs. A). Licenses for clinical stage bsAbs. Line represents number of license signed each year. Blueand yellow bars represent the largest deal and total deal values for each year, respectively. B). The largest deals signed from 2014 to 2018. C). Deal valuesin disease area. D). BsAb market size in 2018 and forecast in 2024. Data source: CortellisTM CCI as of Sept 23, 2019.
and Paul Parren thoroughly reviewed global bispecific anti-body clinical pipeline using a mechanistic lens [2]. Basedon the analysis using CortellisTM, a Clarivate Analyticssolution, by the end of September 2019, there are 176bsAbs or bifunctional proteins with target disclosed underactive preclinical development, compared to 119 in clinicaldevelopment for cancer, autoimmune, and other indica-
tions (including IND filed—Phase III—and two programson clinical hold). This highlights the increased interest ofexploring bsAbs as a venue to develop novel antibody-based therapeutics. The most frequently studied target pairsof those bispecific antibodies and the number of moleculesbeing explored are illustrated in a network graph (Fig. 6).We took a step further and analyzed the disease areas cov-
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

44 Antibody Therapeutics, 2020
Figure 6. A network graph characterizing the target pairs of most bispecific programs in both preclinical and clinical investigations. Each node in thenetwork is one target, and each edge connecting two nodes represents one bispecific program. The circular edges are biparatopic programs. The node sizeshows the degree of a particular target being paired with other different targets. The colors of the edges are marked in black if only one program is availablefor that particular pair, otherwise in red if more than one are being explored. The popularity of that bispecific program is reflected from the thickness ofthe red edges. Source data are from CortellisTM (Table 1-4). Tri-specific and albumin-relevant bispecific programs are not included. The albumin-relevanttri-specific are analyzed as bispecific projects.
ered and mechanisms employed by these clinical moleculesand preclinical drug development candidates as well, withthe attempt to illustrate the principle of selecting a bispe-cific format to meet biology needs and selecting a bispecificmolecule as a clinical development candidate by six criticalcriteria.
Bispecific antibodies for cancer treatment
According to the CortellisTM’ analysis, the bsAb pipelineis composed predominantly by programs for cancertreatment, with 99/119 programs in clinical stages and153/176 preclinical programs (Fig. 4C). As reviewed byHanahan D. and Weinberg RA., there are eight hallmarksof cancer, and targeting these biological capabilities ofcancer cells may lead to new therapeutic options for cancer[76]. Therefore, based on the biological functions, wecategorize the bsAb programs into the following groups:anti-angiogenesis, anti-tumorigenesis, enhancing tumorimmunity, modulating tumor microenvironment (TME),and depletion of target cells.
Anti-angiogenesis. As angiogenesis plays an essentialrole in promoting tumor progression and metastasis, anti-angiogenesis for cancer treatments have been extensivelyexplored. Though several therapies, such as bevacizumab(anti-VEGF) and ramucirumab (anti-VEGFR2), havebeen approved to treat several types of tumors, onlymoderate levels of antitumor activity were observed.Along with the booming of bispecific programs and betterunderstanding of the angiogenesis process, new generationof anti-angiogenesis treatments are emerging. As shown inSupplementary Table S1 available online at ABT Online,12 programs are under active development. Majority of theprograms are focusing on improving the anti-angiogenesiseffect by combinatory targeting two or even three moleculesthat are involved in angiogenesis, such as VEGF, VEGFR2,DLL4, and ANGPT2.
Dilpacimab (AbbVie) targeting DLL4 and VEGF isone of the most advanced programs in this category.DLL4-Notch signaling plays a critical role in angiogenicsprouting, and DLL4 blockade alone has shown inhibition
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

Antibody Therapeutics, 2020 45
in tumor growth [77, 78]. Dilpacimab was designed toco-inhibit both DLL4 and VEGF signaling to achievemore prominent antitumor efficacy [79]. Dilpacimab wasgenerated using DVD-Ig platform with the variable domain(VD) of anti-DLL4 located at the outer position and anti-VEGF VD located at the inner position. Interestingly, in thepresence of VEGF, dilpacimab showed 20–50× enhancedcapability of blocking DLL4 signaling, which may be dueto the VEGF homodimerization-mediated cross-linking ofdilpacimab, then enhanced its binding avidity to DLL4on the cell surface, and promoted the downregulationof DLL4 [79]. Considering higher levels of VEGF attumor sites than in peripheral circulation, this uniquecharacteristic of dilpacimab may conditionally enhanceDLL4 neutralization activity only at tumor sites. Currently,the treatment of dilpacimab along or in combinationwith chemotherapy in patients with advanced solid tumorhave shown acceptable safety profile and demonstratedpreliminary antitumor efficacy [80, 81].
Anti-tumorigenesis. Anti-tumorigenesis by targetingoncogenic receptors is another well-validated anticancertreatment. Trastuzumab targeting Her2 was approved totreat Her2-overexpressing breast cancer in 1999. Later,pertuzumab recognizing a different epitope of Her2 wasapproved in 2012 for treatment of patients with Her2-positive metastatic breast cancer in combination withtrastuzumab and chemotherapy. To develop an ideal com-binatorial treatment with trastuzumab and pertuzumab, ahandful of biparatopic Her2 bsAb programs are under earlyclinical testing. ZW25, a biparatopic Her2 bsAb designedbased on Azymetric platform, able to bind domains 2 and 4of extracellular region of HER2 simultaneously to promoteinternalization of HER2 and to inhibit HER2/HER3heterodimer formation, demonstrated promising clinicalefficacy in a Phase I study (ESMO-Asia 2019). Addi-tionally, several other oncogenic targets are also underevaluation, such as EGFR, Her3, cMET, and lipoproteinreceptor-related proteins (LRP) 5/6. Leading players in thisarea are Merus (Her3 × Her2), Jiangsu Alphamab (Her2biparatopic), Zymeworks (Her2 biparatopic), Janssen(EGFR × cMET), and EpimAb (EGFR × cMET),followed by Beijing Mabworks, Boehringer Ingelheim (BI),Molecular Partners, etc. (Table 1).
In collaboration with Genmab, Janssen has developedJNJ-61186372 to concurrently block both EGFR andcMET pathways for treatment of patients who are resistantto EGFR tyrosine kinase inhibitors (TKIs). It has beenshown that JNJ-61186372 not only effectively blocksthe ligand binding-induced EGFR and cMET activationbut also promotes the downregulation of both EGFRand cMET. To further enhance its antitumor potency,the antibody-dependent cellular cytotoxicity (ADCC)function of JNJ-61186372 is augmented by production ina fucosylation defective CHO cell line [82]. In the first-in-human (FIH) Phase I study, JNJ-61186372 has been testedon 142 patients who were progressed after EGFR TKItherapies and has shown promising antitumor activity with∼30% partial response rate.
Though most of the companies are focusing on mod-ulating the signaling of the well-validated ErBB family
members, Boehringer Ingelheim is developing a first-in-class biparatopic antibody to block the function ofLRP5/LRP6 and Wnt/β-catenin pathway. LRP5/LRP6forms trimeric complex with the serpentine receptorFrizzled and Wnt and mediates the stabilization of β-catenin, the transcriptional activator of the Wnt targetinggenes. Aberrant Wnt/β-catenin pathway activation cancontribute to the carcinogenesis and has been observedin many types of tumors. It has been suggested thatLRP5/LRP6 can interact with different Wnt ligands atdifferent domains, and mAbs blocking different domainsshowed different profile [83]. Therefore, BI generatedthe LRP5/LRP6 biparatopic nanobody, BI 905677, withhigh affinity and complete blockage of the binding ofWnt ligands to LRP5/LRP6, thereby inhibiting the Wnt-mediated cancer cell proliferation and survival [84]. Thismolecule is currently at Phase I in patients with differenttypes of solid tumors.
To further expand the antitumor activity, strategiesin combining the blockage of both angiogenic andtumorigenic pathways have been exploited, such astargeting VEGF and cMET (Merus), as well as Her3 andLGALS3BP (MediaPharma). Both programs are still atpreclinical stage (Table 1).
Enhance tumor immunity. Though the idea of immunother-apy dates back to the 1890s, it was not until the 2010s whenit had significant breakthrough with ipilimumab launchedin 2011 and Keytruda and Opdivo in 2014. The aim ofthe immunotherapy is to boost the patients’ own immunesystem to generate antitumor T cell responses. This canbe achieved by either blocking the inhibitory signals, suchas CTLA-4 and PD-1, or enhancing the co-stimulatorysignals, such as 4-1BB and OX40. Till today, anti-CTLA-4and anti-PD(L)1 treatments have shown promising efficacyand revolutionized cancer treatment. Nevertheless, only10–30% of the patients benefit from the treatment [85, 86].
The immune system is a fine-tuned system, withredundancy in most of the regulatory pathways to avoiddamage to the host while it remains effective to clearinfection and tumor cells. To further improve the antitumorefficacy of anti-CTLA-4 and anti-PD(L)1 therapies, severalstrategies are being evaluated, including combining theanti-angiogenesis treatment with anti-PD-1 treatment(VEGF + PD-1) and combining the blockage of multipleimmune checkpoints (CTLA-4 + PD-1 [87], PD-1 + LAG-3, etc.). Though the additive effect can be achieved bycombining two mAbs, bsAbs have the advantages indevelopment as a single molecule entity, sometimes mayeven have synergistic efficacy. As of September 2019,there are 11 bsAbs targeting multiple inhibitory immunemodulatory pathways such as PD-1, CTLA-4, LAG-3,TIM-3, etc. under active development at clinical and 13 atpreclinical stage (Table 2). For example, the co-expressionof PD-1 and LAG-3 on tumor-infiltrating lymphocytesidentifies the tumor-specific T cells [88], which are mostlydysfunctional [89]. The co-treatment of anti-PD-1 andanti-LAG-3 can effectively restore the T cell function [90]and have showed antitumor activity in PD-1-resistantpatients [91]. Based on this fact, several companies areevaluating PD-1 × LAG-3 bsAbs. Some of the molecules
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

46 Antibody Therapeutics, 2020
represent preferential binding on the double-positive Tcells and are more effective in upregulating the T effectorcell function, as compared to the combination of thetwo parental antibodies [WO2017019846; WO2018134279;WO2018185043; WO2019158942].
Despite the success achieved by the immune checkpointinhibitors, the development of co-stimulatory signal ago-nists was hindered by the intriguing balance between safetyand efficacy. For instance, the co-stimulatory molecule4-1BB is a promising target for cancer immunotherapy,as it can activate T cells, NK cells, and other immune cellsand has been clinically validated in CAR-T therapies tosustain T cell activation. However, the clinical developmentof anti-4-1BB monoclonal antibodies was stagnated byeither liver toxicity or lack of efficacy [92]. To minimizethe safety issue associated with the systemic activationof 4-1BB, strategies have been employed to localize theactivation of 4-1BB at tumor site. Roche developed a4-1BBL fusion protein targeting fibroblast activationprotein (FAP) with Fc region mutations to abrogate Fcγ Rbinding but maintain favorable PK profile. The in vitrofunctional tests suggested that, only in the co-presenceof anti-CD3 signal and FAP-expressing cells, FAP-4-1BBL can increase T cell activation and proliferation.Furthermore, preclinical studies showed that FAP-4-1BBLcannot inhibit tumor growth by itself. The combined treat-ment with relevant T cell-redirecting bispecific antibodies(TRBAs) or immune checkpoint inhibitor, such as anti-PD-L1, can efficiently inhibit tumor growth without prominentliver toxicity [93]. Recently, at the 34th SITC annualmeeting, Pieris Pharmaceuticals reported the preliminaryresults of the Phase I study of PRS-343 (Her2 × 4-1BB) inpatients with Her2+ malignancies. Among the 18 patientswho received active doses of PRS-343, 2 patients reachedpartial response, and 8 patients had stable diseases. This isthe first 4-1BB agonistic treatment reported with promisingefficacy as well as good safety profile.
The tumor site localization strategy has also beenexplored to selectively activate other co-stimulatoryreceptors, such as OX40, CD27, CD28, CD40, and ICOS,as well as to selectively inhibit co-inhibitory receptors,including CTLA-4 and PD-1. There are 10 bsAbs utilizingthis strategy under active development at clinical and 19at preclinical stages, which reflects the growing interests inthis area (Table 2).
Modulate TME. To evade the immune surveillance,tumor cells can commonly influence the microenviron-ment around them by expressing immunosuppressivemolecules, such as TGFβ and CD73, and by recruitingor promoting the differentiation of immunosuppressivecells, such as myeloid-derived suppressor cells (MDSCs),tumor-associated macrophages (TAMs), and regulatory Tcells (Tregs). A few bsAbs and bifunctional proteins aredeveloped to overcome immunosuppressive TME, suchas bintrafusp alfa, an anti-PD-L1, and TGFβRII fusionprotein. TGFβ is a pleiotropic cytokine and plays dualfunctions in cancer progression. Though TGFβ suppressestumor progression at tumor initiation stage, at later stages,TGFβ facilitates tumor progression and metastasis andhas been suggested to contribute to resistance to anti-PD-1
and chemotherapy. Bintrafusp alfa (aka M7824) has theTGFβRII extracellular domain fused to the C-terminalof avelumab [94]. In preclinical studies, M7824 exhibitedstrong antitumor activity and significantly decreasedthe incidence of metastasis in mouse tumor models. Inclinical tests, M7824 displayed acceptable safety profileand encouraging clinical efficacy in patients with heavilypretreated advanced solid tumors [95]. Other strategies tar-geting TME, including CD73 × TGFβ, EGFR × TGFβ,and CCR2 × CSF1R, are also under development at earlyclinical stage or preclinical stage (Table 3).
Target cell depletion. The last group represents themajority of the bsAb programs (clinical, 60/99; preclinical,99/153) to promote the target cell depletion by differentmechanisms (Table 4). According to the MOAs, this groupcan be further divided into subgroups, including cytotoxiceffector engagement, Fc effector function (ADCC, ADCP,complement-dependent cytotoxicity [CDC]), enhancedphagocytosis, enhanced apoptosis, and drug conjugation.
Cytotoxic effector engagement is the largest subgroupin this category. Two out of the three launched bsAbs,catumaxomab, and blinatumomab are in this subgroup.Catumaxomab contains the antigen-binding sites for CD3on the T cells and EpCAM on the cancer cells [96]. It wasfirst authorized for market by the EMA in 2009 for thetreatment of malignant ascites [97], but was withdrawn in2017 due to commercial reasons. On the other hand, blina-tumomab targeting CD3 and CD19 has shown impressiveclinical results since launched in 2014 [98, 99].
T cells identify the target cells by recognizing the peptidespresented by the major histocompatibility complex (MHC)through TCR. Based on the dynamic segregation model,the interaction of TCR with cognate peptide/MHC com-plex (pMHCs) brings the T cells and target cells in closeproximity (∼14 nm) and results in TCR clustering at thecenter of immune synapse (IS) and exclusion of the largeinhibitory tyrosine phosphatases from this region [100].Following TCR clustering, cytolytic granules move towardthe center supramolecular activation cluster (cSMAC) andrelease perforin and granzymes into the target cells. Oncethe target cells undergo cell death, the T cells quicklydetach from the dying cells and move to the next targetcell [100].
TRBAs are a group of bsAbs that can simultaneouslytarget a component of the TCR complex (most commonlyCD3ε) on a T cell and a target on the tumor cell surface[101–103]. By this approach, TRBAs promote IS formationbetween T cells and cancer cells independent of ligationof TCR with pMHCs, leading to T cell activation andkilling of the tumor cells [104–107]. Due to the clinicalsuccess of blinatumomab, the development of TRBAshas gained substantial attention with 51 programs are atclinical and 66 at preclinical stages. Molecules in differentformats are under evaluation to prove whether they candeliver the proposed biological function or have therapeuticwindow. For examples, BiTE and half-life extended BiTEare used by Amgen; DART and DART-Fc format areevaluated by MacroGenics; common light chain formatis under investigation by Regeneron; and DuoBody format
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

Antibody Therapeutics, 2020 47
is under development for multiple projects by Genmaband Janssen. Glenmark’s BEAT platform, Xencor’s XmAbplatform, Aptevo’s ADAPTIR platform, and Teneo-Bio’s unique anti-CD3 platform are also under activeexploration. Recently, IGM Biosciences announced theinitiation of FIH Phase I clinical trial of IGM-2323, anIgM-based CD20 × CD3 TRBA. Unlike other formats,containing only 1 or 2 binding units for the tumor-associated antigen (TAA), the IGM-2323 contains 10binding units for CD20. It is hypothesized that the higheravidity to CD20 of IGM-2323 may provide an advantage totreat CD20low tumor cells over other formats. Moreover, theIgM-based TRBAs can more efficiently mediate CDC thanIgG antibody. However, whether the IgM-based TRBAscan deliver superior efficacy to other formats still needs tobe confirmed in the clinical studies.
Approaches to engage effector cell populations otherthan conventional αβ T cells, such as CD8 T cells, γ δ Tcells, NK cells, and iNKT cells, have also been exploredand were reviewed by Ellerman recently [108]. The γ δ Tcells represent 10% of the total T lymphocyte populationin circulating blood. Unlike conventional T cells, γ δ Tcells recognize stressed and malignant cells independent ofMHC molecules, and their activation does not require co-stimulatory signals [109]. Besides strong cytotoxic activity,one unique property of activated γ δ T cells is that theycan cross-present tumor antigens to enhance CD8 T cellresponse [110]. A few strategies to improve the antitumoractivity of the γ δ T cells have been explored at preclini-cal and early clinical stages [111]. A selective Vγ 9Vδ2 Tcell engager (Her2 × Vγ 9) showed superior activity ininducing Vγ 9Vδ2 T cell-mediated tumor cell lysis thanHer2 × CD3 TRBAs in vitro and exhibited antitumor activ-ity in combination with IL-2 and activated γ δ T cells adop-tive transfer treatments in the PancTu-1 xenograft mousemodel [112].
It has been argued that NK cell engagement may havebetter safety profile over T cell engagement therapies,while providing similar levels of clinical efficacy. CD16is the most commonly used target for engaging NKcells. Results published by Affimed have suggested thatNK engagers may induce efficient target cell killing withlower cytokine release risk, when compared to CD3 Tcell engagers [113]. Early clinical results reported at 60thASH meeting and 15th ICML meeting had shown thatAFM-13 (CD16 × CD30) was well tolerated and effica-cious when administrated alone or in combination withpembrolizumab [114, 115]. The definitive clinical benefitof NK cell engagement still remains to be demonstratedin ongoing clinical studies. Other NK cell-activatingreceptors that are considered to have distinct advantagesto overcome certain deficiencies in TME, such as NKG2D,NKp30, and NKp46, are under preclinical evaluation [116].Additionally, strategies that redirecting iNKT cells by usingCD1d extracellular domain fusion protein is also at earlyresearch stage [117].
Along with the growing depth of knowledge in Fc effec-tor function, several approaches have been adopted forbsAbs, including mutations in the Fc region to enhancethe Fcγ R binding, and afucosylation. Genmab has estab-lished a HexaBody platform, which contains mutations
(E430G) in the IgG1 Fc region to enhance hexameriationupon antigen engagement and thereby enhance the CDCeffect. By combining the HexaBody and DuoBody plat-forms, Genmab has developed a DuoHexaBody anti-CD37biparatopic antibody. In ex vivo CDC assays using samplesisolated from lymphoma patients, the DuoHexaBody anti-CD37 biparatopic antibody showed more potent tumor celllysis, as compared to the control anti-CD20 antibodies,rituximab, ofatumumab, and obinutuzumab [118].
CD47-SIRPα signaling plays an inhibitory effect onthe phagocytosis by phagocytes, such as macrophages.Tumor cells overexpress CD47 on the cell surface toescape the elimination by phagocytes. Antibodies againstCD47 and SIRPα have been developed to interruptCD47-SIRPα signaling. The combined treatment ofa CD47 antagonist, Hu5F9-G4, with rituximab showedpromising therapeutic efficacy in patients with non-Hodgkin lymphoma (NHL) [119]. By taking the advantageof the CD47 antagonist, a series of bsAbs using anti-CD47as one moiety to enhance the phagocytosis to the cancercells have been developed. Though there is only 1 programat clinical Phase I (NI-1701, CD19 × CD47), 14 programsare undergoing active development at preclinical phase withCD47 coupled with different tumor-associated antigens(Table 4), suggesting there is substantial growing interestsin this area. Recently, the results published by Hattereret al. have suggested that in addition to the enhancementof phagocytosis, the co-engagement of CD47 and CD19by NI-1701 can prevent the colocalization of CD19 toBCR cluster during B cell activation, therefore inhibitingactivated B cell proliferation [120].
Unlike previously mentioned bispecific programs, whichall rely on the cytotoxic function of the effector cells or thecomplement system, two preclinical programs are focusingon actuating the apoptotic process of the cancer cells byactivating the apoptotic receptors. BI and Promethera gen-erated bsAbs targeting CDH17 × TRAILR2 (BI-905711)and CD20 × CD95 (Novotarg), respectively. According tothe report published by BI on 2019 AACR Annual Meeting,BI-905711 induced TRAILR2 clustering on a CDH17-dependent manner and selectively triggered the apoptosisof CDH17 expressing tumor cells. BI-905711 also demon-strated significant antitumor activity in multiple colorectalcancer xenograft models [121].
Lastly, there are a couple of bsAbs that are being usedto deliver toxin into cells that are positive for either or bothtargets based on the particular design of each molecule. Forexample, Regeneron is working on APLP2 × Her2 bispe-cific antibody-drug conjugate (ADC). Amyloid precursor-like protein 2 (APLP2) has been suggested to be involvedin increased tumor cell proliferation and migration, andaberrant APLP2 expression was observed in multiple typesof cancers, such as breast cancer [122]. Though APLP2 is aninternalizing receptor, due to its ubiquitous expression andthe presence of secreted form, APLP2 is not an ideal targetfor ADC. Trastuzumab emtansine (T-DM1) has shownpotent efficacy in cancer cells with high level of Her2expression, but has little effect on cells with mid to low lev-els of Her2 expression. To improve the therapeutic efficacyof Her2 ADC, Regeneron developed the Her2 × APLP2-DM1. The bsAb binds to Her2-positive cells with the high-
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

48 Antibody Therapeutics, 2020
affinity Her2 binding arm and then bridges to the cellsurface APLP2 with the low-affinity APLP2 binding arm,which promotes rapid antibody internalization, lysosomaltrafficking, and tumor cell killing [123].
Bispecific antibodies for inflammatory conditions
Autoimmune disease is the second largest area for bsAbs’applications, with 10 clinical programs and 12 preclinicalprograms ongoing. Most of these programs are aiming toblock the function of multiple pro-inflammatory cytokinesby combining the neutralizing antibodies into one moleculeentity, such as IL-1α × IL-1β, IL-17 × IL-13, IL-4 × IL-13,and BAFF × IL-17 (Table 5).
In immune cells, when it is in-cis coupled with an activat-ing receptor, the inhibitory receptor can play a dominantnegative role by diminishing the transduction of the activesignal [124]. It has been shown that MGD-010 targetingCD79B, one component of the BCR complex and theinhibitory receptor CD32B, can decrease B cell responsewithout depleting the B cells in healthy donors [125]. Onois developing ONO-4685 (CD3 × PD-1) to turn down theT cell responses in autoimmune diseases. However, it is stillnot clear whether the effect of ONO-4685 is dependent onthe in-cis engagement of CD3 and PD-1 to block the Tcell activation or by in-trans interaction to deplete PD-1expressing activated T cells.
Bispecific antibodies for other conditions
Hemophilia A. Hemophilia A is another successfulexample in the application of bsAbs, with the approvalof emicizumab in 2017 as a landmark event. Emicizumabbridges factor IXa and X in spatially appropriate positionsto facilitate the factor IXa-catalyzed factor X activation,which is usually mediated by factor VIII in healthyindividuals, but is deficient in patients with hemophiliaA. Though the etiology of hemophilia A has been wellunderstood for a long period of time, the treatment optionsare still limited. Recombinant factor VIII and humanplasma-derived factor concentrates are the commonly usedpractices for hemophilia A. However, the short half-lifeof factor VIII and development of anti-drug antibody(ADA) remains the major challenges for factor replacementtherapy [126]. Emicizumab was intentionally designed tofunction as factor VIII with prolonged plasma half-life[127, 128]. Clinical results in hemophilia A patients withfactor VIII inhibitor showed that the weekly subcutaneous(SC) treatment of emicizumab significantly reduced thefrequency of bleeding episodes with no detectable anti-drug antibody [129]. Based on its promising efficacyand superior regimen schedule, emicizumab was initiallylaunched in the USA for hemophilia A patients with factorVIII inhibitor in 2017; and then its usage was quicklyexpanded to patient without factor VIII inhibitor and waslaunched in EU and Japan in 2018. Similar programs areunder preclinical development by Kymab and Shire.
Ocular. Excessive neovascularization, bleed, and fluidleakage from the abnormal blood vessels result in rapidvision loss or even blindness in patients with wet form
age-related macular degeneration (AMD). Anti-angiogenesistreatment, such as Lucentis, Eylea, and Beovu, has beenapproved for treatment of this condition and has shownsignificant improvement in visual acuity and prevention ofvision loss. Though over 90% of the patients can benefitfrom the treatment (i.e., maintain vision), eventually thesepatients become resistant to the treatment. New therapiesare needed to further improve the therapeutic efficacy. Aswe mentioned above, several bsAbs have been developedto block the process of angiogenesis for cancer indications.Similar strategy has also been exploited for the treatmentof wet AMD and diabetic macular edema (DME). Roche’sfaricimab is an Ig-like bsAb targeting VEGF and ANGPT2using CrossMab technology. During clinical tests, faricimabhas shown superior efficacy and safety in patients withDME, as compared to Lucentis [130]. Phase III studiesto evaluate faricimab’s therapeutic efficacy are initiated inearly 2019; and the filing for BLA is expected in 2021.
Neurology. Despite the tremendous efforts in developingbiological therapeutics for neurodegeneration diseases,effective treatment remains elusive. One of the obstaclesin developing biological drugs for neurological disease is toeffectively deliver the large molecule into the central neuronsystem. “Trojan horse” bsAb has one binding specificityresponsible for the transportation of the antibody tothe location that otherwise cannot be reached naturally,whereas the other binding specificity fulfills its function. Byusing this approach, a group of bsAbs have been developedto cross the blood-brain barrier (BBB). These antibodiesusually have one binding arm recognizing the receptorsin the receptor-mediated transcytosis system, such asinsulin receptor, transferrin receptor, and lipoproteintransport receptors [131], and the other arm targetingthe pathogenic molecules (Table 6). Bifunctional fusionproteins or antibody-drug conjugates are also under activedevelopment as therapeutic drugs or diagnostic reagentsfor central nervous system diseases, but they are not underthe scope of this review.
Infectious diseases. Due to the high frequency of escapemutations and development of drug resistance to single-agent treatment, combinatory treatment with mixture ofmAbs or by bsAbs to broaden the protection spectrum andto decrease the chance to establish drug resistance is beingdeveloped to fight against infections. MEDI3902 was orig-inally designed to achieve broader protection against Pseu-domonas aeruginosa by combining two clinically provenanti-PcrV and anti-Psl antibodies into one molecule. Psland PcrV are present in ∼90% of the P. aeruginosa clinicalisolates, respectively. Theoretically, the bsAb targeting Psland PcrV simultaneously can protect the host from theinfection of 97–100% of the isolates, which express eitheror both targets. Surprisingly, when compared to the mixtureof the parental antibodies in preclinical studies, MEDI3902showed enhanced efficacy. By further dissecting the MOA,it was found that the format of MEDI3902 rendered a high-avidity low-affinity binding to Psl, which led to the accu-mulation of MEDI3902 around the bacterium and moreefficient blocking of PcrV-mediated cytotoxicity [132].
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

Antibody Therapeutics, 2020 49
The “Trojan horse” strategy is also employed by somebsAbs with elegant design for infectious diseases treatment.During filoviruses (e.g., Ebola virus) infection, the mem-brane envelope glycoprotein (GP) is responsible for the cellattachment and membrane fusion. A unique feature aboutthe GP of filoviruses is that it first binds to the receptoron the cell surface which induces the internalization ofthe virus particle, and then in the late endosome, GP iscleaved to expose the highly conserved receptor-bindingsite (RBS) for Niemann-Pick C1 (NPC1), which mediatesthe membrane fusion and cell entry [133]. Therefore, toprovide broad protection against filoviruses, bsAbs weredesigned to block the intracellular GPCL-NPC1 interac-tion by coupling the GPCL-NPC1 blocking arm with adelivering arm targeting a broadly conserved epitope inuncleaved GP. The delivering arm binds to the virus par-ticles and goes into the endosome together with the virus,where the blocking arm functions to abrogate the GPCL-NPC1 interaction when it is exposed and prevents theviral entry [134].
Diabetes. Fibroblast growth factor 21 (FGF21) playskey roles in stimulating metabolism and has shown somepreliminary clinical benefits in obese patients with diabetes.However, the poor PK profile and potential adverse effectsassociated with long-term usage of recombinant FGF21limit its usage. RG7992 (FGFR1 × KLB) was thereforedesigned to mimic the function of FGF21 but selectivelyactivate FGFR1/KLB complex in the liver, adipose, andpancreas tissues, where KLB is present, to avoid safetyconcern associated with broad FGFRs activation, but stillbe able to provide clinical benefit in obesity and diabetes[135].
MATCH BIOLOGY WITH AN OPTIMAL BISPECIFICFORMAT
As discussed in the early section, format diversity is essen-tial to serve the plethora of applications of bsAbs defined byTPPs. Variances in affinity, valency, epitope, and geometryof their binding domains, linkers, as well as in size- and Fc-mediated distribution and pharmacokinetic properties tofulfill a particular clinical application define a bsAb format.In practice, many variances or attributes for selecting anoptimal format are intertwined and must be addressed forselecting the right molecule. Therefore, we will discuss theseattributes below.
Antigen-binding affinity and valency
Affinity. Even though one of the advantages of usingantibody-based therapeutics is that they may interactwith their antigens with substantially high affinities,higher affinity does not always translate into betterefficacy. Unlike antagonistic molecule, whose potency isusually associated with its affinity, agonistic molecule ismore difficult to predict and to optimize its potency byincreasing the binding affinity. Based on different modesthe receptor uses for activation, different binding kineticsof the agonistic bsAb to reach optimal receptor activation
are required. For receptors depending on clustering toactivate, fast-on fast-off binding kinetics is preferred toensure efficient recruitment of receptors [136, 137]. Onthe contrary, for receptors activated by ligand binding-induced conformational change, the slow off bindingkinetics would endorse more durable activating efficacy[138]. Furthermore, there are evidences that the affinityto CD3 may significantly affect the function and safetyprofile for TRBAs. It has been suggested that T cells requirelower threshold for mediating cytotoxic killing than forcytokine production perhaps due to different number ofITAM motifs of TCR complex being phosphorylated,it may be possible to dissociate TRBAs’ potency fromtoxicity by modulating the CD3 affinity of the bsAbs.As shown by Leong et al., by lowering the affinity toCD3, the CD3 × CLL1 bsAb with low affinity to CD3exhibited better safety profile and retained equivalent invivo efficacy, as compared to the ones with high affinity toCD3 [139] when net impact on T cell activation, receptorinternalization, and PK all combined. Similar results werealso shown by Zuch de Zafra et al. By comparing a series ofCD38 × CD3 bsAbs with different affinities to CD3, theyfound that lowering the affinity to CD3 can dramaticallydecrease the cytokine release, but still maintain potencyin mediating cytotoxic killing [140]. In November 2019,AMG-424, the final lead from the aforementioned study,was granted with orphan drug designation for multiplemyeloma by the FDA.
As for the affinities of TRBAs to TAAs, due to the differ-ent expression profile of the TAAs in normal tissues versusin tumors, and the tolerability and the ability of regener-ation of TAA-positive cell populations in normal tissues,the TRBAs targeting different TAAs may require differentbinding kinetics. For low-expression, tumor-specific anti-gens, a TRBA with high affinity to the antigen wouldbe required to elicit efficient tumor cell killing. However,for TAA with low expression on essential normal tissues/organs, to spare the normal cells and avoid on-target off-tumor toxicity, low-affinity high-avidity TRBAs would bepreferred, which can be achieved by modulating the valency(see below).
Moreover, for a bsAb, difference in affinities of twodifferent antigen-binding specificities may determine whicharm drives tissue distribution, tissue penetration, and reten-tion of a therapeutic molecule at the site of MOA. Forexamples, high affinity to TAA and low affinity to CD3may enable the preferential binding of TRBAs to the targetcells and implement serial killing of the target cells by asingle T cell [141]; and as mentioned above, APLP2 × Her2bispecific ADC with high affinity to Her2 and low affinityto APLP2 preferentially binds to Her2-positive cells andthen bridges APLP2 on the cell surface to mediate efficientendocytosis to avoid the toxicity associated with the panexpression of APLP2.
For BBB crossing bsAbs, along with other consider-ations, careful selection of the transport receptor andselection of a molecule with appropriate binding kinetics tothe transport receptor is critical for success of this strategy.As reported by the scientists from Genentech, to ensurethe effectiveness of the transcytosis, the “Trojan horse”antibody using the TfR pathway needs to have low affinity
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

50 Antibody Therapeutics, 2020
to TfR [142]. Later, another study by the University ofWisconsin-Madison showed that TfR bsAb with highbinding affinity to TfR at pH 7.4 but low affinity atpH 5.5 can effectually release the bsAbs from BBB into thebrain and avoid the degradation of bsAb in the endosome[143].
Valency. The valency for each target can dramaticallyaffect the function of the bsAbs. For the TRBAs, mono-valency of anti-CD3 arms may help to avoid non-specificactivation of the T cells without engagement of tumor cells,as shown by Bardwell et al. [144]. Interestingly, Y-mAband Abpro have CD3 scFv fused to the C-terminus ofthe light chain. Even though the format ends up with twobinding sites for CD3, both companies claimed that thisformat was actually functional monovalent toward CD3.Additionally, Aptevo and Affimed also developed TRBAsusing bivalency to CD3. Preclinical evidence has suggestedthat the adoption of the ADAPTIR format can inducepotent T cell activation and target cell killing, but low levelsof cytokine release [145]. AFM-11 (CD19 × CD3) alsoshowed more potent T cell activation than BiTE controland strict CD19-dependent T cell activation preclinically[146]. However, due to one death and two life-threateningevents in clinical trial, AFM-11 was placed on clinicalhold.
The valence for the TAA may vary based on the prop-erties of the TAA, such as tumor specificity, antigen size,expression level on the tumor versus normal tissue, and thetolerance of complete elimination of TAA-positive cells. Inthe case of some types of hematopoietic tumors, the deple-tion of both normal and malignant cells expressing TAAs,such as CD19 and/or CD20, can be tolerated. However, formost of the other TAAs, the expression levels may be lowon normal tissues, but the killing of these low-expressionnormal cells can lead to deleterious consequences. To dis-tinguish the targethigh tumor cells from the targetlow normaltissue, RG7802 (CEA × CD3) was optimized to have low-affinity high-avidity 2 + 1 format in appropriate geometryto facilitate the selection of CEAhigh cells with a threshold of∼10 000 CEA-binding sites/cell [105].
Based on the lessons learned from the initial mAb devel-opment for cMET treatment, bivalency to cMET mayelicit agonistic, instead of antagonistic, effect resulting fromthe mAb-mediated dimerization of cMET. Though mono-valent binding to cMET can function as an antagonist,it can only block the HGF-mediated cMET activation.Later, Wang et al. demonstrated that ABT-700, a trulyantagonistic mAb against cMET, can bind to a uniqueepitope on cMET. The bivalency to cMET of ABT-700 andstringent hinge region was essential to inhibit both HGF-dependent and HGF-independent activation of cMET andinduce cMET downregulation [147]. Interestingly, half ofthe cMET bsAb programs are using monovalency againstcMET to avoid agonistic effect, while the other half choosebivalency. EMB-01 (EGFR × cMET) has two binding sitesfor cMET, and has no obvious cMET activation in theabsence of ligands. Furthermore, it can effectively induceEGFR and cMET degradation, therefore preventing thecMET activation [62].
Epitope, geometry, and distance between differentantigen-binding domains
Epitope. In respect of antagonistic bsAb, the bindingepitope of the corresponding binding units are requiredto prevent the receptor/ligand engagement, or the receptorsignal complex formation, or any step that is crucial for theinitiation or passage of signaling cascade into the cells toplay its biological function.
In general, the receptor-binding epitope for agonisticmolecules is not as predictive as it is for antagonisticmolecules. However, there is evidence to suggest thatthe binding epitopes do contribute significantly to thebsAb efficacy. It was found that anti-CD3 binding armsrecognizing different epitopes on CD3-activated T celldifferently. TeneoBio, therefore, identified a dozen ofanti-CD3 antibodies with different binding epitopes anddifferent binding kinetics to CD3 molecules to disassociatethe capabilities of TRBAs in inducing cytotoxic killingfrom promoting cytokine production post T cell activation.They identified a clone (F2B) that recognizes a uniqueepitope on CD3δε, but not CD3 γ ε, at a low affinity(34 nM). By comparing to another clone (F1F), whichbinds to both CD3δε and CD3 γ ε with high affinity(<1 pM), they found that BCMA × CD3 bsAb usingF2B arm (CD3_F2BxBCMA) can induce moderate levelsof cell killing but very weak cytokine production, ascompared to the one using F1F arm (CD3_F1FxBCMA)in vitro. Moreover, the in vivo efficacy study showedthat CD3_F2BxBCMA exhibited antitumor activity in awide dose range (0.01–10 μg), while CD3_F1FxBCMAcompletely lost its therapeutic efficacy at the high dose(10 μg) [148].
As we mentioned above, to effectively redirect T cellkilling, the TRBAs must be able to induce the IS formationbetween the T cells and target tumor cells. Besides theformat of the TRBAs, the tumor antigen selection, the sizeof the antigen, antigen surface density, as well as the dis-tance between the TRBAs binding epitope to target cellmembrane, all can influence the formation of the IS. Com-paring to large antigens or antigens with protruding struc-ture, the small antigens or antigens with structure closeto the cell membrane can more effectively promote the ISformation [106]. When the selected tumor antigen is largein size, such as melanoma chondroitin sulfate proteoglycan(MCSP) [149] and FcRH5 [150], the membrane-proximalepitope is desired. For cell surface targets that can be shedinto the bloodstream, to avoid antigen sink, the bsAbsshould recognize the membrane-bound but not the solubleform of the antigen [151].
Geometry. Besides the distance between the epitope tothe target cell membrane, the distance between the twotargets engaged by TRBAs also plays a crucial role in deter-mining whether it can effectively promote IS formation andT cell activation. Considering the distances between theTAA and CD3 epitopes to target cell and T cell, respec-tively, the format of the TRBAs needs to bring TAA andCD3 to a close proximity much less than 14 nm. Moreover,the whole molecule has to be able to physically fit into thesmall junction between the two cells in a density to effec-
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

Antibody Therapeutics, 2020 51
tively form a cluster with several engaged target pairs toinitiate TCR signaling. Despite its short serum half-life, thesmall size of BiTE format with two binding units locatingin opposite sides is extremely potent in redirecting T cellcytotoxicity by inducing serial killing of tumor cells at aneffector-to-target ratio as low as 1:5 [141]. In another case,Aptevo fused the scFvs against the TAA and CD3 at the N-and C-terminus of Fc (scFv + scFv with Fc, 2 + 2), whichended up with longer distances between the two bindingdomains. The in vitro studies showed that this molecule hadmore potent target cell killing, but less cytokine release,as compared to the BiTE format [145]. Unfortunately, theclinical development for this molecule was discontinueddue to high frequency of anti-drug antibody development.
The same situation also applies to T cell co-stimulatoryand co-inhibitory receptors, which co-cluster with TCRduring IS formation and regulate T cell activation. PD-1and PD-L1 interaction leads to the accumulation of PD-1microclusters at cSMAC and destabilizes the IS. When theextracellular domain of PD-1 was elongated by insertingextra Ig domains, the inhibitory role of PD-1 decreasedalong with the increase of the number of Ig domainsinserted [152]. Though current anti-PD-1 molecules allblock the PD-1 signaling by inhibiting the PD-1/PD-L1interaction, in theory, the designs that can prevent the PD-1 colocalization to cSMAC should also be able to diminishthe inhibitory role of PD-1 in T cell responses. On thecontrary, bsAbs to activate the co-stimulatory receptorsuch as 4-1BB must exert its function at the site of IS[93]; therefore, a format that can meet these criteria isnecessary. As reported by Pieris, the geometry of the 4-1BBanticalin attachment significantly affected the function ofthe Her2 × 4-1BB bispecific anticalins. PRS-343 with 4-1BB anticalin attached at the C-terminus of the heavy chainshowed the most effective T cell activation, as compared toother formats. One possible explanation is that the bindingsites for Her2 and 4-1BB are approximately 15 nm apart,which is close to the distance of the IS. However, aftermeasuring the distances from the binding epitopes to thecell membrane, the distance between the target cell and theeffector cell might be much longer than 15 nm. On the otherhand, ND-021 (PD-L1 × 4-1BB × HSA) is an Fc-lackingscFv-VHH-based molecule. With its small size and flexiblestructure, it may have better potential in colocalizing atcSMAC and enhance TCR signaling. It will be interestingto see how it will perform in the clinical trials.
Cases are also shown in bsAbs programs developed forother conditions. For example, when the scientists at Med-Immune tested their Psl × PcrV bsAbs, they examined sev-eral different formats with varying intramolecular distancesbetween the two binding components. After comparisonof these formats in both in vitro and in vivo efficacy stud-ies, BiS4aPa, with an intermediate distance, exhibited themost effective protection against P. aeruginosa infectionand therefore was selected as the final therapeutic candidateformat [132].
Linker design
As reviewed by Brinkmann and Kontermann, various con-necting linkers have been explored [3]. Similar to the hinge
region of the IgG subclass, the length, flexibility, and aminoacid composition of the linkers used to connect the buildingblocks (scFv, Fab, etc.) may determine the correct for-mation, functionality, and developability of the resultingbispecific molecules, as shown by Le Gall et al. [153] andDiGiammarino et al. [154].
Size
The bsAbs have made significant impact on hematologicmalignancy treatments. However, the therapeutic benefitsdelivered by bsAb for solid tumor are still waiting to beunveiled. One of the concerns using bsAbs for solid tumortreatment is how to increase the drug tumor penetrationand accumulation. Though molecules with smaller sizewould have better chance entering the tumor site byincreased tumor penetration, the molecules with sizesmaller than the threshold of renal clearance of proteins arerapidly cleared from the blood and therefore have decreasedflux into the tumor [155]. Using a compartmental model,Schmidt and Wittrup predicted that molecules with thesize of 150 kDa would have the best tumor localization,whereas molecules with the size of 25 kDa would havethe worst tumor uptake [156]. However, due to their largesize, molecules at the size of ∼150 kDa have decreasedextravasation and normally take days to reach maximumtumor uptake. On the other hand, molecules of smaller sizereach the maximum tumor uptake within a short period oftime. The fast tumor penetration and systemic clearanceof small-sized molecule therefore lead to high tumor/bloodlocalization ratio, which is preferred for some applications,such as imaging [157], as well as safety management todecrease the systemic drug exposure-associated toxicity.
To improve the serum half-life, while still retaining thefast extravasation property, Harpoon developed the Tri-TAC platform, which targets TAA, CD3, as well as humanalbumin for extended half-life with a total size of ∼50 kDa.It is believed that with its improved drug exposure andsmall size, TriTAC would enable faster and better tumorpenetration, compared to large-sized bsAbs.
Fc region
The Fc region can substantially influence the bsAbs’ func-tion. It was found that the properties of IgG subclass hingeregion, such as length, sequences, flexibility, and disulfidebond structures, can influence the variable region presen-tation and thereby affect the functionality of an antibody[158]. While it is not always desired, the format with Fc canprolong the bsAb serum half-life through FcRn-mediatedrecycling and may provide Fc effector function through theinteraction with Fcγ Rs.
IgG subclass. Recently, Kapelski et al. reported theinfluence of the IgG subclass on TRBAs. They found thatdue to its short and rigid hinge region, IgG2 cannot effec-tively promote the IS formation. However, by replacing thehinge region of the IgG2 with the hinge region of IgG4 orIgG1, the function of IgG2 chimeric bsAb can effectivelyinduce IS formation and redirect T cell killing [159].
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

52 Antibody Therapeutics, 2020
Similarly, the Fc region also showed significant influenceon the factor VIII-mimetic activity of emicizumab. Aftercomparison between different IgG subclasses, interchaindisulfide bonds, and mutations in hinge region and CH2domain, IgG4 was selected as it presented with the mostpotent factor VIII-mimetic activity [160].
Fc effector function. As mentioned above, severalstrategies have been developed to enhance the bindingbetween Fc and Fcγ Rs to increase the Fc effector function.This could be important for bsAbs against TAAs foreffective killing tumor cells or for bsAbs against infectiousagents for pathogen uptake and clearance. However, the Fceffector function and Fcγ R binding are usually abrogatedfor TRBAs and some other agonistic bsAbs to avoidthe Fcγ R-mediated cross-linking, which may cause non-specific activation of T cells and the targeted receptors,respectively. Advances in Fc engineering allow tailoredmodification of Fc effector functions for specific need. Forexample, Xencor developed a series of TRBAs using theXmAb platforms, including AMG-424 (CD38 × CD3,Fab + scFv with Fc, 1 + 1), and used a combinationof mutations (E233P/L234V/L235A/G236del/S267K) tocompletely eliminate the binding of IgG1 Fc to Fcγ Rs [55].
Because IgG4 only binds to Fcγ R1 with high affin-ity and mediates weaker effector function than IgG1,it is commonly used for antagonistic antibodies tar-geting immune cells, to avoid Fc effector function-mediated cell elimination. However, the research by Zhanget al. showed that the anti-PD-1/IgG4 antibody caninduce the phagocytosis of PD-1+ T cells by activatingFcγ RI+ macrophages. By introducing five additionalmutations (E233P/F234VL235A/D265A/R409K), BGB-A317 showed no binding to Fcγ R1, and more efficientpreclinical antitumor activity, as compared to the anti-PD-1/IgG4 control [161]. The recently reported results ofthe pivotal study of BGB-A317 also exhibited its superiorantitumor efficacy in patients with relapsed/refractoryclassical Hodgkin lymphoma, with an overall response rate(ORR) of 87% and 63% complete response rate (CRR).
As we discussed above, the format contains many com-ponents that can be tweaked, their final impact on pharma-cological properties of a bsAb is intertwined, and here weonly mentioned some of them. The fine-tuned parts work inconcert with each other to determine the success of bsAbs.To obtain the optimal therapeutic candidate, the selectionof any component in the final format should be carefullyevaluated for specific target pairs; and the matched formatwill not only facilitate the bsAbs to elicit biological functionbut also may enable a molecule for further product devel-opment, which otherwise may not be suitable for clinicalapplication.
SELECT A RIGHT MOLECULE TO MEET BOTHFUNCTION AND DEVELOPABILITY REQUIREMENT
As illustrated in the early section, the six criteria criticalfor clinical development and commercial manufacturing(Fig. 1) define a good bsAb. Identification of a goodtherapeutic bispecific molecule usually requires starting
with good therapeutic and molecular design definedby MPP that is developed based on TPP, followed byvigorous in vitro and in vivo screening and characterizations.Below we will discuss these six criteria and how they canhave significant impact on the outcome of the resultingbsAbs: physiochemical properties, manufacturability,immunogenicity, PK/PD property, and, most importantly,efficacy and safety.
Physiochemical properties and manufacturability. Asaforementioned, many strategies have been exploredto solve the CMC quality issues, such as mispairing,stability, aggregation, segmentation, solubility, viscosity,purification, etc. A good bispecific clinical candidateshould (1) be easily expressed with high percentage ofcorrectly assembled product in manufacturing scale; (2)display no significant aggregation or low percentage ofaggregation that can be easily removed, as aggregation mayaffect the therapeutic efficacy and increase immunogenicityrisk; (3) have good solubility, high stability, and lowviscosity to meet drug product formulation needs forintended clinical dosage and route of administration; and(4) have low manufacturing cost for economical reason.The stringency of those requirements may differ based onvarious clinical applications. For instance, reconstitutedlyophilized formulation for intravenous infusion (IV) isgenerally acceptable for oncology applications, while liquidformulation developed for SC administration may bepreferred for most of autoimmune indications. For oculardisease, the high solubility, high stability, and low viscosityare imperative for a competitive product. With the advanceof the bsAb technology, more and more reported bsAbformats can be expressed and purified with reasonableyield and meet the reasonable physiochemical propertiesfor a given clinical application and are scalable for large-scale manufacture, although some of the formats do requiresignificant CMC optimization and longer developmenttimeline than the others.
Immunogenicity. Immunogenicity is one of the criticalfactors limiting clinical use of biological therapeutics, as thegeneration of ADA may lead to fast drug clearance, neutral-ization of therapeutic effect, and even severe adverse eventsin clinic. The duration of the ADA response can be cate-gorized into transient and persistent ADAs. The persistentADA requires the T cell help and commonly leads to moredeleterious consequences. The nature and the levels of theADA generated are influenced by both the patients’ physi-cal conditions (autoimmune-prone vs. immunosuppressive,pre-existing ADAs, etc.) and the intrinsic properties of anantibody (i.e., sequences, impurities, format, MOAs, dos-ing regimens, etc.) [162]. For example, the cancer patientsare usually immunosuppressive, while the patients withautoimmune diseases are prone to develop auto-reactiveantibodies and ADAs. The antibodies that contain strongT cell epitopes have high risk to induce T cell-dependentpersistent ADA. Compared to bsAbs that deplete B cells,the bsAbs that enhance immune system response may havea higher chance to induce ADA. And the bsAbs dosed bySC and intramuscular (IM) administration may be easier
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

Antibody Therapeutics, 2020 53
to be picked up by dendritic cells (DCs) and present bsAb-derived peptides to T cells, as compared to the ones givenby IV infusion.
As reviewed by Davda et al. [163], using the approvedmAb clinical results, they found that although both ate-zolizumab and durvalumab were Fc-engineered anti-PD-L1 mAbs, only atezolizumab showed higher rate of ADA,as compared to the other anti-PD(L)1 mAbs. The combi-national treatment of the anti-PD(L)1 and anti-CTLA-4could increase the rate of ADA: the ADA rates againstnivolumab were increased from ∼12% (monotherapy) to24–38% (combo therapy with ipilimumab). Furthermore,the antibodies mediating B cell depletion usually hadlow ADA rates. As most of the reported bsAb formatsare heavily engineered and with non-native Ig sequencesintroduced, it is very likely that the bsAbs have higherimmunogenicity risk than regular mAbs. However, mostof the bsAb programs are still at early clinical stages, andonly very limited information and results are available toevaluate the immunogenicity issue for bsAbs. As mentionedabove, Aptevo developed APVO-414 (PSMA × CD3)using the ADAPTIR platform. In the initial Phase Idose escalation study, 58% of the patients developedADA with the titers as high as 1:250 000, leading to fastdrug clearance from the blood. By modifying the doseregimen from weekly IV to continuous IV infusion, theADA titers were decreased dramatically to the range of1:160–1:320. However, there were still 50% of the patientsdeveloped ADAs. Later, this program was discontinued asno therapeutic benefit was observed.
Due to its critical impact on the clinical outcomes,methods to minimize the immunogenicity risk have beenexploited at early discovery phase. Firstly, more and moreantibody therapeutics are utilizing humanized or evenfully human antibodies or fragments to decrease the non-human sequences, thereby decreasing the immunogenicityrisk. Secondly, in silico approaches are being employedto identify the immunogenic epitopes, especially T cellepitopes can be removed to prevent the generation of Tcell-dependent persistent ADAs. Though these approachesstill need to be validated in clinical practice, various in silicoalgorithms have been developed to predict the presenceof potential T cell epitopes. To compliment the in silicoprediction, in vitro assays are utilized, which include HLAbinding assays, primary peripheral blood mononuclear cell(PBMC) assays, mixed lymphocyte reaction assays, and 3Dmodels to mimic the conditions in specific tissues [164].The integrated results from in silico algorithm and in vitroassays can provide some suggestive information and helpthe bsAb development. For example, during rounds ofengineering and optimization, emicizumab adopted a de-immunization strategy that the effects of each mutation onimmunogenicity were evaluated by using algorithm, andany mutation that may increase immunogenicity risk wasavoided [127]. The clinical data suggested that there werelow level or no ADA observed in treated patients.
Pharmacokinetic and pharmacodynamic properties. PK,described as what the body does to a drug, refers to thedrug absorption, distribution, metabolism, and excretion.
PD, described as what the drug does to the body, involvesthe target binding and the following effect. The PK/PDprofiles play an important role in effecting the drug efficacyand safety and therefore are critical for the developmentof bsAbs. Many factors of the bsAbs can influence the PKprofiles, including molecule format, size, physicochemicalproperties, Fcγ R binding, as well as target binding affin-ity. For example, Harpoon is developing a novel protease-activated T cell engager platform, ProTriTAC based on theaforementioned TriTAC platform. By modifying the non-CDR region, the anti-albumin SDA can bind and mask theanti-CD3 arm while maintaining its binding to albumin.Furthermore, a tumor-associated protease cleavage site isintroduced to the linker between the anti-CD3 bindingdomain and anti-albumin SDA. In the circulation, theanti-albumin keeps anti-CD3 arm inactive and imparts themolecule long serum half-life. Once it enters into the TME,ProTriTACs are cleaved by tumor-associated proteases tolose the anti-albumin SDA and expose the anti-CD3 bind-ing site to function. If the cleaved molecules enter intothe circulation again, they will be rapidly cleared from thesystem due to its small size. By using this strategy, theydeveloped a ProTriTAC targeting EGFR, which was noteasy to be targeted by TRBAs due to its wide expression inthe normal tissue.
As long serum half-life may increase tissue penetrationand therapeutic efficacy, as well as require lower dosageand less frequent drug administration, sometimes, a longerserum half-life is preferred. IgGs and albumin are bothabundant in plasma with long half-life due to the bindingto FcRn, which rescues them from degradation in theendo/lysosomal compartment. Therefore, enhancing the Fcbinding to FcRn, or by adding a HSA binding domaininto the format (without Fc), is commonly used by bsAbsto improve the serum PK. Many mutations in the CH2-CH3 region have been tested to increase the Fc binding toFcRn, yet only the YTE and LS mutation combinations(YTE = M252Y/S254T/T256E; LS = M428L/N434S) havebeen clinically validated [165]. YTE mutations can increasethe antibody serum half-life ∼4-fold in human, but alsodecrease the ADCC activity of the antibody. VRC01LScontaining the LS mutations also showed more than 4-foldincrease in serum half-life in human [166]. Unlike YTEmutations, LS mutations have no impact on antibody’sADCC activity. On the other hand, in some applications,when the prolonged half-life is undesired, mutations todecrease the Fc to FcRn binding can also be applied.Detailed methods in modulating FcRn binding to modifyPK were reviewed by Leipold [167]. Though the effect ofFcRn on influencing serum half-life has been well studied,it still remains controversial on how it affects the drugmetabolism in other tissues, such as the brain and eyes. Asmatter of fact, Lucentis (Fab) and Eylea (Fc fusion protein)only showed slightly different ocular half-life in humans,suggesting FcRn binding may not play a major role indetermining ocular half-life, while the molecular size mayplay some, but not determining roles on PK properties ofmolecules in retina.
Due to the high binding affinity to the target, target-mediated drug disposition (TMDD) is common forantibody-based therapeutics, especially for those targeting
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

54 Antibody Therapeutics, 2020
surface antigens. As mentioned in the previous section,decreasing the target binding affinity, in some cases, canprolong the drug half-life and therefore improve the thera-peutic efficacy. As shown by Leong et al., the relationshipbetween CD3 affinity of the CD3 × CLL1 TRBA to itsactivity, PK and safety are quite complicated. During invitro characterization, they found that the one with lowaffinity to CD3 (CLL1/CD3L) showed decreased potency,but had more favorable safety profiles, as compared tothe one with high affinity to CD3 (CLL1/CD3H). Moreimportantly, when they tested these molecules in vivo, theyfound that CLL1/CD3L had slower drug clearance (50%)and increased drug exposure, which led to more durableantitumor responses, as compared to CLL1/CD3H [139].A similar case was also observed in an IL-15/Rα × PD-1 bifunctional protein. The fusion protein was engineeredto decrease the IL-15/Rα potency, thereby decrease theantigen sink, and increase half-life. Several variants withdecreased potency were generated and compared in vivo. Asthey predicted, the low potency variants showed dramatichalf-life extension from 0.5 day (wild type) to 9 days[US20180118828].
Besides affecting the serum PK, target binding affinitymay also influence the tumor/tissue distribution of thebsAbs. For example, the affinity of the bsAb to the tumorantigen can influence the tumor penetration. BsAbs withextremely high affinity to tumor antigen get stuck at theentrance and therefore have poor tumor penetration [168,169]. While low-affinity bsAbs distribute further into thetumor, but bsAbs with small sizes may have decreasedretention time in the tumor. The distribution of TRBAsfor solid tumors, as predicted by Friedrich et al., may besignificantly affected by the distribution of T cells, andmodifying the affinity to CD3 or TAA may not be sufficientto accumulate TRBAs and T cells into the tumor [170].Other methods may be used to affect the bsAb distributioninside the tumor tissue and include target selection, Fc, andutilizing of transcytosis [155].
The ultimate goal of all previously discussed strategies tomodulate PK was to enhance the overall clinical efficacyand/or to minimize the toxicity of the therapeutic bsAbs.Similarly, improving the PD profiles can also be achieved bymodifying the antigen-binding activity and by modifyingthe Fc-mediated effector function to further increase clini-cal potential of the bsAbs. Thus, the PK/PD profiles can bemodified by adjusting multiple factors, while most of thesefactors are interdependent, which highlight the inherentchallenges in therapeutic antibody design, and improvingone property can sometimes affect the others. Therefore,we should bear in mind that due to the complexity of theMOAs of bsAbs, the PK/PD profiles may not be the same aswe expected (hoped). Robust technologies and tools (bothexperimental and in silico) are critically needed to advancethe understanding of structural determinants of the bsAbsthat can impact the PK/PD properties and to guide theoptimization of bsAbs.
Efficacy and safety. A reasonable efficacy/safety win-dow is fundamental for a good clinical candidate; andPK/PD profiles, efficacy, and safety profiles commonly
influence each other. It is common that the drug showinghigh potency in discovery stage tends to be selected as thetherapeutic candidate. However, highly potent drug thatinduces toxicity at low dose leaves no or very limited ther-apeutic window, which may significantly hinder its clinicalapplication. On the other hand, the drug with a reasonablepotency but better safety profile may have wide therapeuticwindow, and the therapeutic efficacy may be improved byreadily increasing the dose without inducing significanttoxicity. Increasing drug exposure may be another wayto enhance the efficacy and prolong the response dura-tion, as we discussed above. However, increased systemicexposure may also increase the chance and the severityof adverse event. It is hard to predict which compositionof the MPP can translate into an optimal TPP in clinicalapplication.
For example, despite its extreme potency in eliminatingthe tumor cells, the life-threatening adverse effect associ-ated with the treatment of blinatumomab, as well as shortserum half-life, both significantly limit the application ofblinatumomab [171]. To improve the therapeutic efficacyand prolong the serum half-life, Affimed developed AFM-11 (Fv + Fv, 2 + 2), a tetravalent CD19 × CD3 bsAb [146].In vitro characterization studies showed that AFM-11 wasmore potent than BiTE molecule to elicit target cell killing.Though bivalent for CD3, AFM-11 showed stringenttarget-dependent activation of T cells. Using a NOD/SCIDxenograft model, AFM-11 showed favorable PK profileswith preferential tumor localization over normal tissue anda half-life of ∼20 h. In Phase I dose escalation study, AFM-11 was dosed by continuous infusion (Week 1, 0.7 ng/kg/wkto 130 ng/kg/wk; Week 2+, 2 ng/kg/wk to 400 ng/kg/wk).During the study, among the 14 patients who completedthe dose limiting toxicity observation period, 3 patientsshowed complete response (CR), but 2 were transient andpatients relapsed after cycle 2. Serum half-life was rangedfrom 7.14 to 10.6 h in four evaluable patients. Although nocytokine release syndrome (CRS) was observed, two Grade3 neurotoxicity and one fatal event were recorded in the twohighest dose groups. AFM-11 was placed on clinical hold,due to the severe adverse events.
TRBAs in formats containing Fc may have improvedstability and manufacture profile, as well as prolongedserum half-life. The long-term drug exposure may provideimproved efficacy and more flexible dosing strategy, butmay be more difficult to handle if undesired effect is expe-rienced. Regeneron developed REGN-1979 (Fab + Fabwith Fc, 1 + 1), a CD20 × CD3 bsAb. In vitro assaysshowed that REGN-1979 can effectively and specificallymediate the killing of CD20+ cells. The preclinical phar-macology studies using cynomolgus monkeys showed thatREGN-1979 can cause durable and deep B cell depletionwith a serum half-life of ∼14 days [31]. In June 2019,Regeneron reported the early-stage dose escalation trialresults of REGN-1979: 93% ORR and 71% CRR in 14patients with follicular lymphoma treated with REGN-1979 (5–320 mg); and 57% ORR in 7 patients with diffuselarge B cell lymphoma (DLBCL) treated with REGN-1979(80–160 mg), which were all CR. Among the total of 81evaluable patients, 7% experienced Grade 3 or higher CRS,and at least 10% of patients experienced Grade 3 or higher
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

Antibody Therapeutics, 2020 55
adverse event. The incidence and severity of CRS can bemitigated by optimized premedication. Recently, in 2019ASH annual meeting, similar results were also reportedfor mosunetuzumab (CD20 × CD3, Roche) with ORRand CRR of 62.7 and 43.3%, respectively, in patients withslow-growing non-Hodgkin lymphoma. Both REGN-1979and mosunetuzumab showed benefit to patients who haddisease progressed post CAR-T therapies.
The comprehensive review regarding TRBAs publishedby Ellerman made a perfect case of how complex it canbe to optimize a TRBA, and the change of a factor of thebsAb may influence multiple profiles of the molecule, anda molecule profile can be modulated by multiple factors.For example, to uncouple the capabilities of TRBAs toinduce cytotoxic killing and cytokine production by theT cells, the TRBAs can be modified by (1) decreasing theaffinity to CD3, as T cell cytotoxic killing requires a loweractivation threshold; (2) using a different CD3 bindingepitope, as based on the “permissive geometry” model,different binding epitope may lead to different CD3 confor-mational change and T cell signaling; and (3) switching toanother format. In another case, to distinguish the antigen-overexpressing tumor cells and the low-expression normalcells, one can (1) decrease the binding affinity and usemultivalency to the antigen and (2) increase the distance ofthe IS, by either choosing a membrane distal epitope on theantigen or using a format with longer distance between thetwo binding domain. From another aspect, decreasing theaffinity of CD3 may diminish the target cell killing potencyin vitro; it may also increase the PK profile and tumor accu-mulation which ends up with comparable or even improvedin vivo efficacy and therapeutic window [108].
Another group of bsAbs that represents with challengesin leveraging the safety and efficacy is the agonistic bsAbtargeting co-stimulatory receptors, such as 4-1BB. Recently,the results reported for PRS-343 showed first sign of hopefor development anti-4-1BB treatment (see above). Numabdeveloped ND-021, a monovalent trispecific antibody tar-geting PD-L1, 4-1BB, and HSA. The in vitro efficacy testssuggested that the ultrahigh affinity (2 × 10−12 M) to PD-L1determined the potency of the molecule; binding to a distalepitope on 4-1BB can promote the 4-1BB clustering moreeffectively; and when the affinity to 4-1BB was way lowerthan the affinity to PD-L1, the effective dose range can besignificantly extended. As compared to the combinationsof mAbs, ND-021 showed superior activity in enhancingactivated T cell responses. Due to the monovalency and lackof Fc region, ND-021 displayed strictly PD-L1-dependent4-1BB activation and spared antigen-presenting cells fromdepletion. In in vivo efficacy tests, ND-021 showed higherantitumor activity than combined treatment with mAbs inmice. Most importantly, ND-021 did not induce liver toxi-city, and systemic T cell activation in cynomolgus monkeyposts a single-dose IV injection [172] although it remainselusive how this may translate into safety in humans. Cur-rently, this program is at IND-enabling study stage, and weare looking forward to see its clinical results.
On the basis of the strong biological rationale, empow-ered by the carefully harmonized format, and with themeticulously selected binding units, bispecific moleculesjust finish the first step to its final success. A good bispecific
clinical candidate not only needs to show promising thera-peutic potential but also needs to have good physiochemicalproperties and scalable manufacturability. Furthermore,favorable PK properties and low immunogenicity are alsocritical to assure the success of the candidate. Besides allthe above mentioned factors, the efficacy/safety ratio isone of the major determinants whether a bsAb moves intodevelopment stages in the end.
KEY CHALLENGES THE FIELD STILL FACING
Though bsAbs development has made significant progressand several strategies have been exploited to solve some ofthe challenges, many still remain. We would like to reviewthese challenges in two categories: technical challenges andmechanistic or biology challenges.
Technical challenges
Discovery. Compared to mAbs, bsAbs display signifi-cant complexity in the research and development stages.Special testing systems are needed to characterize thepotential therapeutic efficacy, toxicity, and PK/PD profilesof the bsAb therapeutic candidates, and many of thesesystems may be quite complicated, as compared to thesystems used to evaluate mAbs.
For example, artificial cell line used to evaluate bsAbfunction needs to overexpress both targets and include bothsignaling pathways, and the generation of such cell line mayhave huge technical challenges. Also, the expression leveland temporal order of the two targets on the artificial cellline may not reflect the disease situation in human. Forprimary cell-based efficacy tests, a specific population ofcells may need to be isolated and cultured ex vivo to inducethe expression of both targets, which makes the assaysextremely time- and cost-consuming and low throughput.Furthermore, even though researchers try to mimic thereal situation under which the bsAb plays its functionalroles, the in vitro assay system cannot completely reflect theimmune system, and therefore the effect of the bsAb cannotbe accurately evaluated in vitro.
The selection of species and relevant disease model forefficacy, pharmacology, and toxicology studies can be com-plicated, with considerations for the properties of both tar-gets, such as the cross-species specificity, the functionalityof the bsAb, as well as the expression and function of thetargets. Although, transgenic animals and animals graftedwith human immune systems are developed for bsAbs with-out cross-species binding, it is still doubtful how closelythese models can reflect the actual clinical conditions andhow accurately they predict the therapeutic efficacy, safetyrisk, and PK/PD profiles of a bsAb.
CMC. With the advanced protein engineering technol-ogy and elegantly designed bispecific formats, the physio-chemical properties and manufacturability are no longersignificant hurdles in developing bispecific clinical candi-dates. However, different formats do vary in the degreesof difficulty in Chemistry, Manufacturing and Controls(CMC) development, and the ones that fulfill developabil-
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

56 Antibody Therapeutics, 2020
ity criteria no doubt would significantly lower developmentrisk and shorten development timeline.
Preclinical pharmacology and toxicology. The preclinicalpharmacology and toxicology studies are very critical forthe development of bsAbs, as the results from these studiesnot only support the scientific rationale of the bsAbs butalso provide valuable information for selecting the FIHdose. Though the scope of the bsAb preclinical studies maybe similar to that for mAbs, the selection of the relevantspecies may be more challenging for bsAbs due to theadditional target. The relevant species should be selectedbased on the following: (1) both targets should have similarexpression profiles and biological functions as the targetsin human, respectively, and (2) the bsAb should bind toboth targets with similar properties as it binds to the humantargets. In the case that Fc effector function is required,especially the ones with modified binding to Fcγ Rs, theselected species should also be able to predict the Fc func-tion in human. If such a species is available, the FIHdose may be selected based on the no-observed-adverse-effect level (NOAEL). If a relevant species is not available,in vitro pharmacology studies and in vivo pharmacologystudies using surrogate bsAb or transgenic animals maybe required to provide supporting information. The FIHdose may be selected by using the minimum anticipatedbiological effect level (MABEL) approach if no relevanttoxicity species are available, especially for molecules withagonistic activities. Several case studies of the bsAb preclin-ical studies were reviewed by Prell et al. [173] and by Trivediet al. [174] to illustrate the complexity and challenge duringbsAb preclinical development.
Clinical development. Based on the draft guidance forbsAb development programs published by the FDA inApril 2019, several factors should be considered duringbsAb clinical development: (1) scientific rationale (e.g.,MOA, therapeutic advantages over standard of care); (2)mode of action (e.g., bridge two target cells, simultaneousor sequential binding); (3) binding kinetics to each target;(4) special pharmacology studies (e.g., PK/PD assessmentfor active form of the bsAb, immunogenicity assessment foreach domain of the bsAb); and (5) in certain cases, factorialdesign of clinical trials to inform risk/benefit ratio.
TGN1412, an anti-CD28 agonistic antibody case, alertedus that cautions must be taken in regard to clinical develop-ment of bsAbs with novel MOAs, especially for agonisticmolecules. Therefore, it is recommended that for bsAbsplaying agonistic function, especially for unprecedentedtarget pairs, the selection of the initial dose of the FIHtrial should use MABEL approach. Additionally, agonis-tic bsAbs may have a bell-shaped dose-response that thetherapeutic efficacy peaks at a dose that receptor occu-pancy is not saturated and then decreases along with theincreased drug dose [175]. Therefore, an agonistic bsAbwith a narrowed bell-shaped dose-response curve may besignificantly difficult for researchers to select the optimaldoses for different patients.
Comprehensive examinations for anti-drug antibodiesmay be required to evaluate the immunogenicity risk of
different domains of the bsAbs, as some of the bsAbsare heavily engineered with potential immunogenic epitopeintroduced. Special attention needs to be taken on ADAagainst TRBAs and agonistic bsAbs using the tumor/tissuelocalization strategy, as the presence of ADA may break theTAA dependency of these bsAbs and lead to non-specificactivation of immune cells and unpredictable severe adverseevent. One should always follow FDA outlined and rec-ommended adoption of a risk-based approach to evaluateand mitigate immune responses or adverse immunologi-cally related responses associated with therapeutic proteinproducts that affect their safety and efficacy during clinicaldevelopment of a bsAb.
Furthermore, in some instances, combinational therapyprovides the flexibility in adjusting the dosing regimen,which cannot be achieved by bsAbs. Although variousbispecific formats can provide some degree of flexibility inadjusting affinity and valency of a binding specificity tosuit different needs, once the format is determined, the ratioagainst two targets is fixed, and it cannot be adjusted basedon the clinical results, which may pose clinical developmentchallenge for a drug. Moreover, an optimal treatment mayrequire sequential target intervention. For example, theconcurrent treatment of anti-PD-1 with anti-OX40 treat-ment leads to substantial increase in serum cytokines andthe expression of inhibitory receptors on T cells, as well asdecreased T cell proliferation, thereby attenuating the anti-tumor efficacy of anti-OX40 treatment. However, delayingthe PD-1 treatment can increase the antitumor activity ofanti-OX40 treatment [176]. In another case, NK cells canbe activated and upregulate 4-1BB expression by exposingto rituximab-coated CD20+ tumor cells or trastuzumab-coated Her2-overexpressing breast cancer cells. The anti-4-1BB treatment following the treatment of rituximab ortrastuzumab can enhance the ADCC effect of NK cellsto antibody-coated tumor cells [177, 178]. In such cases,the combinational therapy with mAbs offers the flexibilitywhich cannot be accomplished by current bsAb strategies.
Mechanistic or biology challenges
The most fascinating applications of bsAbs are to enablenovel biological function and therapeutic MOA otherwiseimpossible by using mAbs alone or in combination. How-ever, the novel MOA may also impose unknown safety riskon bsAbs, which cannot be readily predicted or evaluatedin preclinical studies, and possibly result in severe or evenlife-threatening adverse events during the clinical stage.Therefore, the uncertainty in function and safety of thesebsAbs represents a major challenge for development ofbsAb therapeutics.
When selecting the target pair, researchers shouldconsider the spatial and temporal presence of both targets.Whether both targets are expressed at the same location atthe same time? Whether their levels are within a reasonablerange that can be effectively treated by a bsAb with fixedstoichiometry? Whether the two targets expressed on differ-ent cells or on the same cells? Whether the bsAb will medi-ate in-cis or in-trans engagement of the two targets? Willdifferent engagement models result in different outcomes
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

Antibody Therapeutics, 2020 57
in efficacy and safety? Those are all important questionsone need to think through when embarking a bsAb project.
Bispecific antibodies engaging CD32B and FcεR weredesigned to employ the dominant negative role of CD32Band inhibit the activation of FcεR to alleviate IgE-mediateddiseases. The bsAb 9202.1/5411 with IgG1 format wasproduced using Escherichia coli cell line and therefore hadno Fc effector function due to lack of glycosylation. In vitroanalysis showed that this bsAb can inhibit IgE-mediatedactivation of mast cells and basophils. As mentioned by theauthors, several formats that were bivalent for FcεR mightcross-link FcεR in the absence of CD32B, thereby activat-ing rather than inhibiting FcεR [179]. One may speculatein the worst scenario in vivo, sometime may be inevitable,if such a molecule formed aggregates, it may function toactivate rather than inhibit FcεR as one initially designed.
This becomes even more complicated for agonistic bsAbsto activate receptors. As a receptor is co-evolved with itscognate ligand, the signaling upon ligand-receptor engage-ment is evolved to be tightly controlled under physiologicalconditions. Due to the plasticity of receptors, polygamywidely exists for ligand-receptor interaction. When usingantibody-based therapeutics to mimic the function of aligand, the antibody may bind to the site on the receptor dif-ferent from its cognate ligand binding site, which may elicitdifferent signals. The deviation from the cognate activationmay result in unexpected consequence, and their potentialsafety risk is unknown.
For example, as reported by Gu et al., a panel ofbiparatopic anti-Her2 antibodies in DVD-Ig formatgenerated from the same parental mAbs only differedby VD orientations or linker length. Surprisingly, DVD-Ig molecules with one VD orientation showed agonisticeffect and increased tumor cell proliferation, whereasmolecules with the opposite VD orientation remainedantagonistic. Further studies revealed that a particularVD orientation interrupted Her2/EGFR and Her2/Her3interaction, resulting in increased Her2 homodimerizationand activation [180]. Similarly, a biparatopic anti-CTLA-4 bsAb unexpectedly changed the signalosome assemblyon the cytoplasmic domain of CTLA-4 and completelyconverted the inhibitory receptor into a stimulatoryreceptor [181].
The preclinical and clinical development path havelargely paved for bsAbs with precedent mechanisms.However, the development of bsAbs with novel biologicalmechanisms still faces a few challenges and pitfalls. It mayrequire more preclinical studies and early discussion withregulatory agencies for clinical development plans. Webelieve that, in the future, biology will be the key driver fordesign and selection of a bsAb and the key considerationfor clinical development of bsAb drugs.
PERSPECTIVE
A growing number of recombinant bsAbs are now in clini-cal development. These bsAbs represent quite different for-mats. The number of the formats may reflect the diversityin desired features of therapeutic applications and may alsoreflect the different understanding of biology. For instance,
for T cell-redirected cytotoxicity, a variety of formats, withdifference in affinity, valency, domain geometry, Fc proper-ties, and pharmacokinetic properties, have progressed intoclinical development. It will be interesting to see clinicalvalidation of various preclinical rationales behind thedesign of those molecules in the coming years.
With the advent of gene therapy, RNA therapy, celltherapy, and various other new therapies, we should alwayscompare those different therapeutic options and pay closeattention to those new therapeutic modalities that mayhave disruptive potentials, for instance, both chimericantigen receptors T cell (CAR-T) therapy and TRBAs havedemonstrated dramatic effects in patients with hematologictumors. One TRBA and two CAR-T cell products havebeen approved by major regulatory agencies within thelast 10 years for the treatment of hematological cancers,and an additional approximately 60 TRBAs and 300 CARcell constructs are in clinical trials today. CAR-Ts aredesigned to activate T cells via intracellular T cell co-stimulatory signaling modules in tandem and to form acytolytic synapse with target cells that is very differentfrom the classical immune synapse both physically andmechanistically, whereas the TRBA-induced synapse issimilar to the classic immune synapse by bringing T cellsclose proximity to tumor cells via a bispecific molecule.As published in 2018 ASH annual meeting, in patientswith relapsed and refractory multiple myeloma (r/r MM),AMG-420 (BCMA × CD3, BiTE) showed 70% ORRand 40% CRR. Similarly, bb-2121 (BCMA CAR-T) andJCARH125 also demonstrated ∼80% ORR and ∼30%CRR. On the other hand, both TRBAs and CAR-Ttherapies showed similar adverse effect, which may bedue to their MOA in redirecting T cell cytotoxicity totumor cells. Blincyto and CAR-T therapies, Kymriah andYescarta, are all targeting CD19+ tumor cells and provedfor treatment of B cell lymphomas, and all of them have theblock box warnings for CRS and neurological toxicities.From the manufacturing aspect, due to the characteristicsof BiTE molecules, the manufacture of Blincyto still hasquite a few challenges, but this has been solved by thenext generation of TRBAs in the clinical development.For autologous CAR-T therapies, a complicated and time-consuming (3–4 weeks) manufacturing process is requiredfor each patient. Additionally, as the CAR-T therapies arelive cells, the regulatory requirements for CAR-T therapiesare more complicated and stringent than regular biologicaltherapeutics. Most CAR-T cells today are autologous,although significant strides are being made to developoff-the-shelf allogeneic CAR-based products. Therefore,in general comparing these two therapeutic platforms,TRBAs are the off-the-shelf products and may be moreconvenient and affordable to patients in the near futurewhen more TRBAs are available, while CAR-T therapymay be tedious but may have advantage to mobilize theentire T cell machinery in a very different mechanism tofight cancer cells. Both platforms currently are facing thesame moderate anticancer effects in solid tumor settings,probably due to inaccessibility of immune effector cells tosolid tumors and complex immunosuppressive mechanismsat TME. The knowledge learned from clinical trials foreither one will definitely help to improve the design of both
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

58 Antibody Therapeutics, 2020
therapies with additional immunomodulatory features toovercome the key challenges they are still facing.
Nevertheless, bsAbs and msAbs open up tremendousopportunities to explore previously unexplored therapeuticoptions. We believe that the next decade will witnessthe clinical success of bsAbs or msAbs employing somenovel MOAs in the applications in cancer and infectious,metabolic, ocular, and other diseases with significant unmetmedical needs.
DECLARATIONS
Siwei Nie, Zhuozhi Wang, Jianqing Xu, and Jijie Gu arecurrent employees of WuXi Biologics and may hold WuXiBiologics’ stocks.
CONFLICT OF INTEREST STATEMENT
Siwei Nie, Zhuozhi Wang, Jianqing Xu and Jijie Gu arecurrent employees of WuXi Biologics, and may hold WuXiBiologics’ stocks.
ABBREVIATIONS:bsAb bispecific antibodymAb monoclonal antibodyMPP molecular product profileTPP target product profileMOA mechanism of actionUMN unmet medical needsTMDD target-mediated drug dispositionSDA single-domain antibodyFv variable fragmentscFv single-chain variable fragmentFab antigen-binding fragmentscFab single-chain antigen-binding fragmentVH heavy chain variable domainVL light chain variable domainCH1 heavy chain constant domain 1CH2 heavy chain constant domain 2CH3 heavy chain constant domain 3CH4 heavy chain constant domain 4Fc fragment of crystallizable regionFD the heavy chain of a Fab, i.e. VH domain plus
CH1 domainPK/PD pharmacokinetic/pharmacodynamicsTRBA T cell-redirecting bispecific antibodyTAA tumor-associated antigen
REFERENCES1. Husain, B, Ellerman, D. Expanding the boundaries of
biotherapeutics with Bispecific antibodies. BioDrugs 2018; 32:441–64.
2. Labrijn, AF, Janmaat, ML, Reichert, JM et al. Bispecificantibodies: a mechanistic review of the pipeline. Nat Rev DrugDiscov 2019; 18: 585–608.
3. Brinkmann, U, Kontermann, RE. The making of bispecificantibodies. MAbs 2017; 9: 182–212.
4. Harmsen, MM, Van Solt, CB, Fijten, HPD et al. Prolonged in vivoresidence times of llama single-domain antibody fragments in pigs
by binding to porcine immunoglobulins. Vaccine 2005; 23:4926–34.
5. Zhu, X, Wang, L, Liu, R et al. COMBODY: one-domain antibodymultimer with improved avidity. Immunol Cell Biol 2010; 88:667–75.
6. Del Bano, J, Florès-Florès, R, Josselin, E et al. A bispecificantibody-based approach for targeting Mesothelin in triple negativebreast cancer. Front Immunol 2019; 10: 1593.
7. Wolf, E, Hofmeister, R, Kufer, P et al. BiTEs: bispecific antibodyconstructs with unique anti-tumor activity. Drug Discov Today2005; 10: 1237–44.
8. Kim, S, Kim, H, Jo, DH et al. Bispecific anti-mPDGFRβ ×cotinine scFv-Cκ-scFv fusion protein and cotinine-duocarmycincan form antibody-drug conjugate-like complexes that exertcytotoxicity against mPDGFRβ expressing cells. Methods 2019;154: 125–35.
9. Tietz, J, Spohn, G, Schmid, G et al. Affinity and potency ofRTH258 (ESBA1008), a novel inhibitor of vascular endothelialgrowth factor a for the treatment of retinal disorders. InvestOphthalmol Vis Sci 2015; 56: 1501.
10. Egan, TJ, Diem, D, Weldon, R et al. Novel multispecificheterodimeric antibody format allowing modular assembly ofvariable domain fragments. MAbs 2017; 9: 68–84.
11. Moore, PA, Zhang, W, Rainey, GJ et al. Application of dualaffinity retargeting molecules to achieve optimal redirected T-cellkilling of B-cell lymphoma. Blood 2011; 117: 4542–51.
12. Veri, M-C, Burke, S, Huang, L et al. Therapeutic control of B cellactivation via recruitment of Fcγ receptor IIb (CD32B) inhibitoryfunction with a novel bispecific antibody scaffold. Arthritis Rheum2010; 62: 1933–43.
13. Johnson, S, Burke, S, Huang, L et al. Effector cell recruitment withnovel Fv-based dual-affinity re-targeting protein leads to potenttumor cytolysis and in vivo B-cell depletion. J Mol Biol 2010; 399:436–49.
14. McAleese, F, Eser, M. RECRUIT-TandAbs®
: harnessing theimmune system to kill cancer cells. Futur Oncol 2012; 8:687–95.
15. Davé, E, Adams, R, Zaccheo, O et al. Fab-dsFv: a bispecificantibody format with extended serum half-life through albuminbinding. MAbs 2016; 8: 1319–35.
16. Bhatta, P, Humphreys, DP. Relative contribution of frameworkand CDR regions in antibody variable domains to multimerisationof Fv- and scFv-containing bispecific antibodies. Antibodies (Basel,Switzerland) 2018; 7: 35.
17. Schoonjans, R, Willems, A, Schoonooghe, S et al. Fab chains as anefficient heterodimerization scaffold for the production ofrecombinant bispecific and trispecific antibody derivatives. JImmunol 2000; 165: 7050–7.
18. Lin, L, Li, L, Zhou, C et al. A HER2 bispecific antibody can beefficiently expressed in Escherichia coli with potent cytotoxicity.Oncol Lett 2018; 16: 1259–66.
19. Harwood, SL, Alvarez-Cienfuegos, A, Nuñez-Prado, N et al.ATTACK, a novel bispecific T cell-recruiting antibody withtrivalent EGFR binding and monovalent CD3 binding for cancerimmunotherapy. Oncoimmunology 2017; 7: e1377874–4.
20. Ridgway, JBB, Presta, LG, Carter, P. ‘Knobs-into-holes’engineering of antibody CH3 domains for heavy chainheterodimerization. Protein Eng Des Sel 1996; 9: 617–21.
21. Atwell, S, Ridgway, JBB, Wells, JA et al. Stable heterodimers fromremodeling the domain interface of a homodimer using a phagedisplay library. J Mol Biol 1997; 270: 26–35.
22. Spiess, C, Bevers, J 3rd, Jackman, J et al. Development of a humanIgG4 bispecific antibody for dual targeting of interleukin-4 (IL-4)and interleukin-13 (IL-13) cytokines. J Biol Chem 2013; 288:26583–93.
23. Gunasekaran, K, Pentony, M, Shen, M et al. Enhancing antibodyFc heterodimer formation through electrostatic steering effects:applications to bispecific molecules and monovalent IgG. J BiolChem 2010; 285: 19637–46.
24. Moore, GL, Bautista, C, Pong, E et al. A novel bispecific antibodyformat enables simultaneous bivalent and monovalentco-engagement of distinct target antigens. MAbs 2011; 3: 546–57.
25. Von Kreudenstein, TS, Escobar-Carbrera, E, Lario, PI et al.Improving biophysical properties of a bispecific antibody scaffold
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

Antibody Therapeutics, 2020 59
to aid developability: quality by molecular design. MAbs 2013; 5:646–54.
26. Davis, JH, Aperlo, C, Li, Y et al. SEEDbodies: fusion proteinsbased on strand-exchange engineered domain (SEED) CH3heterodimers in an Fc analogue platform for asymmetric binders orimmunofusions and bispecific antibodies. Protein Eng Des Sel2010; 23: 195–202.
27. Skegro, D, Stutz, C, Ollier, R et al. Immunoglobulin domaininterface exchange as a platform technology for the generation ofFc heterodimers and bispecific antibodies. J Biol Chem 2017; 292:9745–59.
28. Tustian, AD, Endicott, C, Adams, B et al. Development ofpurification processes for fully human bispecific antibodies basedupon modification of protein A binding avidity. MAbs 2016; 8:828–38.
29. Dimasi, N, Fleming, R, Sachsenmeier, KF et al. Guiding bispecificmonovalent antibody formation through proteolysis of IgG1single-chain. MAbs 2017; 9: 438–54.
30. Merchant, AM, Zhu, Z, Yuan, JQ et al. An efficient route tohuman bispecific IgG. Nat Biotechnol 1998; 16: 677–81.
31. Smith, EJ, Olson, K, Haber, LJ et al. A novel, native-formatbispecific antibody triggering T-cell killing of B-cells is robustlyactive in mouse tumor models and cynomolgus monkeys. Sci Rep2015; 5: 17943.
32. De, Nardis, C, Hendriks, LJA, Poirier, E et al. A new approach forgenerating bispecific antibodies based on a common light chainformat and the stable architecture of human immunoglobulin G(1).J Biol Chem 2017; 292: 14706–17.
33. Shiraiwa, H, Narita, A, Kamata-Sakurai, M et al. Engineering abispecific antibody with a common light chain: identification andoptimization of an anti-CD3 epsilon and anti-GPC3 bispecificantibody, ERY974. Methods 2019; 154: 10–20.
34. Fischer, N, Elson, G, Magistrelli, G et al. Exploiting light chainsfor the scalable generation and platform purification of nativehuman bispecific IgG. Nat Commun 2015; 6: 6113.
35. Lewis, SM, Wu, X, Pustilnik, A et al. Generation of bispecific IgGantibodies by structure-based design of an orthogonal Fabinterface. Nat Biotechnol 2014; 32: 191.
36. Froning, KJ, Leaver-Fay, A, Wu, X et al. Computational design ofa specific heavy chain/κ light chain interface for expressing fullyIgG bispecific antibodies. Protein Sci 2017; 26: 2021–38.
37. Mazor, Y, Oganesyan, V, Yang, C et al. Improving target cellspecificity using a novel monovalent bispecific IgG design. MAbs2015; 7: 377–89.
38. Vaks, L, Litvak-Greenfeld, D, Dror, S et al. Design principles forbispecific IgGs, opportunities and pitfalls of artificial disulfidebonds. Antibodies (Basel, Switzerland) 2018; 7: 27.
39. Litvak-Greenfeld, D, Vaks, L, Dror, S et al. “BIClonals”:production of bispecific antibodies in IgG format in transientlytransfected mammalian cells. In: Steinitz, M (ed). Methods inMolecular Biology. New York, NY: Springer, 2019, 431–54
40. Liu, Z, Leng, EC, Gunasekaran, K et al. A novel antibodyengineering strategy for making monovalent bispecificheterodimeric IgG antibodies by electrostatic steering mechanism. JBiol Chem 2015; 290: 7535–62.
41. Dillon, M, Yin, Y, Zhou, J et al. Efficient production of bispecificIgG of different isotypes and species of origin in single mammaliancells. MAbs 2017; 9: 213–30.
42. Bönisch, M, Sellmann, C, Maresch, D et al. Novel CH1:CLinterfaces that enhance correct light chain pairing in heterodimericbispecific antibodies. Protein Eng Des Sel 2017; 30: 685–96.
43. Seifert, O, Rau, A, Beha, N et al. Diabody-Ig: a novel platform forthe generation of multivalent and multispecific antibody molecules.MAbs 2019; 11: 919–29.
44. Schaefer, W, Regula, JT, Bähner, M et al. Immunoglobulin domaincrossover as a generic approach for the production of bispecific IgGantibodies. Proc Natl Acad Sci USA 2011; 108: 11187–92.
45. Metz, S, Panke, C, Haas, AK et al. Bispecific antibody derivativeswith restricted binding functionalities that are activated byproteolytic processing. Protein Eng Des Sel 2012; 25:571–80.
46. Shen, J, Vil, MD, Jimenez, X et al. Single variable domain-IgGfusion: a novel recombinant approach to Fc domain-containingbispecific antibodies. J Biol Chem 2006; 281: 10706–14.
47. Shen, J, Vil, MD, Jimenez, X et al. Single variable domain antibodyas a versatile building block for the construction of IgG-likebispecific antibodies. J Immunol Methods 2007; 318: 65–74.
48. Shi, SY, Lu, Y-W, Liu, Z et al. A biparatopic agonistic antibodythat mimics fibroblast growth factor 21 ligand activity. J Biol Chem2018; 293: 5909–19.
49. Wozniak-Knopp, G, Bartl, S, Bauer, A et al. Introducingantigen-binding sites in structural loops of immunoglobulinconstant domains: Fc fragments with engineeredHER2/neu-binding sites and antibody properties. Protein Eng DesSel 2010; 23: 289–97.
50. Everett, KL, Kraman, M, Wollerton, FPG et al. Generation ofFcabs targeting human and murine LAG-3 as building blocks fornovel bispecific antibody therapeutics. Methods 2019; 154:60–9.
51. Wu, C, Ying, H, Grinnell, C et al. Simultaneous targeting ofmultiple disease mediators by a dual-variable-domainimmunoglobulin. Nat Biotechnol 2007; 25: 1290–7.
52. Dickopf, S, Lauer, ME, Ringler, P et al. Highly flexible,IgG-shaped, trivalent antibodies effectively target tumor cells andinduce T cell-mediated killing. Biol Chem 2019; 400: 343.
53. Coloma, MJ, Morrison, SL. Design and production of noveltetravalent bispecific antibodies. Nat Biotechnol 1997; 15: 159–63.
54. Hoseini, SS, Guo, H, Wu, Z et al. A potent tetravalentT-cell-engaging bispecific antibody against CD33 in acute myeloidleukemia. Blood Adv 2018; 2: 1250–8.
55. Moore, GL, Bernett, MJ, Rashid, R et al. A robust heterodimericFc platform engineered for efficient development of bispecificantibodies of multiple formats. Methods 2019; 154: 38–50.
56. Yu, S, Zhang, J, Yan, Y et al. A novel asymmetricalanti-HER2/CD3 bispecific antibody exhibits potent cytotoxicity forHER2-positive tumor cells. J Exp Clin Cancer Res 2019; 38: 355.
57. Qi, J, Li, X, Peng, H et al. Potent and selective antitumor activity ofa T cell-engaging bispecific antibody targeting amembrane-proximal epitope of ROR1. Proc Natl Acad Sci USA2018; 115: E5467–76.
58. Robinson, HR, Qi, J, Cook, EM et al. A CD19/CD3 bispecificantibody for effective immunotherapy of chronic lymphocyticleukemia in the ibrutinib era. Blood 2018; 132: 521–32.
59. Bezabeh, B, Fleming, R, Fazenbaker, C et al. Insertion of scFv intothe hinge domain of full-length IgG1 monoclonal antibody resultsin tetravalent bispecific molecule with robust properties. MAbs2017; 9: 240–56.
60. Manikwar, P, Mulagapati, SHR, Kasturirangan, S et al.Characterization of a novel bispecific antibody with improvedconformational and chemical stability. J Pharm Sci 2019. doi:10.1016/j.xphs.2019.06.025.
61. Kim, HS, Dunshee, DR, Yee, A et al. Tethered-variable CLbispecific IgG: an antibody platform for rapid bispecific antibodyscreening. Protein Eng Des Sel 2017; 30: 627–37.
62. Gong, S, Ren, F, Wu, D et al. Fabs-in-tandem immunoglobulin is anovel and versatile bispecific design for engaging multipletherapeutic targets. MAbs 2017; 9: 1118–28.
63. Bostrom, J, Yu, S-F, Kan, D et al. Variants of the antibodyHerceptin that interact with HER2 and VEGF at the antigenbinding site. Science (80-) 2009; 323: 1610–4.
64. Schaefer, G, Haber, L, Crocker, LM et al. A two-in-one antibodyagainst HER3 and EGFR has superior inhibitory activitycompared with monospecific antibodies. Cancer Cell 2011; 20:472–86.
65. Lee, CV, Koenig, P, Fuh, G. A two-in-one antibody engineeredfrom a humanized interleukin 4 antibody through mutation inheavy chain complementarity-determining regions. MAbs 2014; 6:622–7.
66. Hu, S, Fu, W, Xu, W et al. Four-in-one antibodies have superiorcancer inhibitory activity against EGFR, HER2, HER3, andVEGF through disruption of HER/MET crosstalk. Cancer Res2015; 75: 159–70.
67. Strop, P, Ho, W-H, Boustany, LM et al. Generating bispecifichuman IgG1 and IgG2 antibodies from any antibody pair. J MolBiol 2012; 420: 204–19.
68. Labrijn, AF, Meesters, JI, de Goeij, BECG et al. Efficientgeneration of stable bispecific IgG1 by controlled fab-armexchange. Proc Natl Acad Sci USA 2013; 110: 5145–50.
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

60 Antibody Therapeutics, 2020
69. Schanzer, JM, Wartha, K, Croasdale, R et al. A novelglycoengineered bispecific antibody format for targeted inhibitionof epidermal growth factor receptor (EGFR) and insulin-likegrowth factor receptor type I (IGF-1R) demonstrating uniquemolecular properties. J Biol Chem 2014; 289: 18693–706.
70. Castoldi, R, Schanzer, J, Panke, C et al. TetraMabs: simultaneoustargeting of four oncogenic receptor tyrosine kinases for tumorgrowth inhibition in heterogeneous tumor cell populations. ProteinEng Des Sel 2016; 29: 467–75.
71. LaFleur, DW, Abramyan, D, Kanakaraj, P et al. Monoclonalantibody therapeutics with up to five specificities: functionalenhancement through fusion of target-specific peptides. MAbs2013; 5: 208–18.
72. Yin, W, Zhu, J, Gonzalez-Rivas, D et al. Construction of a novelbispecific antibody to enhance antitumor activity against lungcancer. Adv Mater 2018; 30: 1805437.
73. Harper, J, Adams, KJ, Bossi, G et al. An approved in vitroapproach to preclinical safety and efficacy evaluation of engineeredT cell receptor anti-CD3 bispecific (ImmTAC) molecules. PLoSOne 2018; 13: e0205491–1.
74. Siegemund, M, Schneider, F, Hutt, M et al. IgG-single-chainTRAIL fusion proteins for tumour therapy. Sci Rep 2018; 8: 7808.
75. Gantke, T, Weichel, M, Herbrecht, C et al. Trispecific antibodiesfor CD16A-directed NK cell engagement and dual-targeting oftumor cells. Protein Eng Des Sel 2017; 30: 673–84.
76. Hanahan, D, Weinberg, RA. Hallmarks of cancer: the nextgeneration. Cell 2011; 144: 646–74.
77. Ridgway, J, Zhang, G, Wu, Y et al. Inhibition of Dll4 signallinginhibits tumour growth by deregulating angiogenesis. Nature 2006;444: 1083–7.
78. Yan, M. Therapeutic promise and challenges of targetingDLL4/NOTCH1. Vasc Cell 2011; 3: 17.
79. Li, Y, Hickson, JA, Ambrosi, DJ et al. Abt-165, a dual variabledomain immunoglobulin (dvd-ig) targeting dll4 and vegf,demonstrates superior efficacy and favorable safety profiles inpreclinical models. Mol Cancer Ther 2018; 17: 1039–50.
80. Gordon, MS, Nemunaitis, JJ, Ramanathan, RK et al. Phase 1,open-label, dose-escalation and expansion study of ABT-165, adual variable domain immunoglobulin (DVD-Ig) targeting bothDLL4 and VEGF, in patients (pts) with advanced solid tumors. JClin Oncol 2016; 34: 2507.
81. Wainberg, Z, Strickler, J, Gordon, M et al. P-234 Phase 1bopen-label study evaluating the safety, pharmacokinetics, andpreliminary efficacy of ABT-165 plus FOLFIRI in patients withsecond-line (2L) colorectal cancer (CRC). Ann Oncol 2018; 29. doi:10.1093/annonc/mdy151.233.
82. Moores, SL, Chiu, ML, Bushey, BS et al. A novel bispecificantibody targeting EGFR and cMet is effective against EGFRinhibitor-resistant lung tumors. Cancer Res 2016; 76: 3942–53.
83. MacDonald, BT, He, X. Frizzled and LRP5/6 receptors forWnt//b-catenin signaling. Cold Spring Harb Perspect Biol 2012; 4:a007880.
84. Zinzalla, V, Drobits-Handl, B, Savchenko, A et al. AbstractDDT01-01: BI 905677: a first-in-class LRP5/6 antagonist targetingWnt-driven proliferation and immune escape. Cancer Res 2019; 79:DDT01-01.
85. Schadendorf, D, Hodi, FS, Robert, C et al. Pooled analysis oflong-term survival data from phase II and phase III trials ofIpilimumab in unresectable or metastatic melanoma. J Clin Oncol2015; 33: 1889–94.
86. Kwok, G, Yau, TCC, Chiu, JW et al. Pembrolizumab (Keytruda).Hum Vaccin Immunother 2016; 12: 2777–89.
87. Rotte, A. Combination of CTLA-4 and PD-1 blockers fortreatment of cancer. J Exp Clin Cancer Res 2019; 38: 255.
88. Gros, A, Robbins, PF, Yao, X et al. PD-1 identifies thepatient-specific CD8+ tumor-reactive repertoire infiltrating humantumors. J Clin Invest 2014; 124: 2246–59.
89. Yang, Z-Z, Kim, HJ, Villasboas, JC et al. Expression of LAG-3defines exhaustion of intratumoral PD-1(+) T cells and correlateswith poor outcome in follicular lymphoma. Oncotarget 2017; 8:61425–39.
90. Woo, S-R, Turnis, ME, Goldberg, MV et al. Immune inhibitorymolecules LAG-3 and PD-1 synergistically regulate T-cell function
to promote tumoral immune escape. Cancer Res 2012; 72:917–27.
91. Ascierto, PA, Melero, I, Bhatia, S et al. Initial efficacy ofanti-lymphocyte activation gene-3 (anti-LAG-3; BMS-986016) incombination with nivolumab (nivo) in pts with melanoma (MEL)previously treated with anti-PD-1/PD-L1 therapy. J Clin Oncol2017; 35: 9520.
92. Chester, C, Sanmamed, MF, Wang, J et al. Immunotherapytargeting 4-1BB: mechanistic rationale, clinical results, and futurestrategies. Blood 2018; 131: 49–57.
93. Claus, C, Ferrara, C, Xu, W et al. Tumor-targeted 4-1BB agonistsfor combination with T cell bispecific antibodies as off-the-shelftherapy. Sci Transl Med 2019; 11: eaav5989.
94. Lan, Y, Zhang, D, Xu, C et al. Enhanced preclinical antitumoractivity of M7824, a bifunctional fusion protein simultaneouslytargeting PD-L1 and TGF-β. Sci Transl Med 2018; 10: eaan5488.
95. Strauss, J, Heery, CR, Schlom, J et al. Phase I trial of M7824(MSB0011359C), a bifunctional fusion protein targeting PD-L1and TGFβ, in advanced solid tumors. Clin Cancer Res 2018; 24:1287–95.
96. Chelius, D, Ruf, P, Gruber, P et al. Structural and functionalcharacterization of the trifunctional antibody catumaxomab. MAbs2010; 2: 309–19.
97. Linke, R, Klein, A, Seimetz, D. Catumaxomab: clinicaldevelopment and future directions. MAbs 2010; 2: 129–36.
98. Dufner, V, Sayehli, CM, Chatterjee, M et al. Long-term outcomeof patients with relapsed/refractory B-cell non-Hodgkin lymphomatreated with blinatumomab. Blood Adv 2019; 3: 2491–8.
99. Kantarjian, H, Stein, A, Gökbuget, N et al. Blinatumomab versuschemotherapy for advanced acute lymphoblastic leukemia. N EnglJ Med 2017; 376: 836–47.
100. Davis, SJ, van der Merwe, PA. The kinetic-segregation model: TCRtriggering and beyond. Nat Immunol 2006; 7: 803–9.
101. Strohl, WR, Naso, M. Bispecific T-cell redirection versus chimericantigen receptor (CAR)-T cells as approaches to kill cancer cells.Antibodies 2019; 8: 41.
102. Clynes, RA, Desjarlais, JR. Redirected T cell cytotoxicity in cancertherapy. Annu Rev Med 2019; 70: 437–50.
103. Trabolsi, A, Arumov, A, Schatz, JH. T cell–activating bispecificantibodies in cancer therapy. J Immunol 2019; 203: 585–92.
104. Wong, R, Pepper, C, Brennan, P et al. Blinatumomab inducesautologous T-cell killing of chronic lymphocytic leukemia cells.Haematologica 2013; 98: 1930–8.
105. Bacac, M, Fauti, T, Sam, J et al. A novel carcinoembryonic antigenT-cell bispecific antibody (CEA TCB) for the treatment of solidtumors. Clin Cancer Res 2016; 22: 3286–97.
106. Rossi, DL, Rossi, EA, Cardillo, TM et al. A new class of bispecificantibodies to redirect T cells for cancer immunotherapy. MAbs2014; 6: 381–91.
107. Offner, S, Hofmeister, R, Romaniuk, A et al. Induction of regularcytolytic T cell synapses by bispecific single-chain antibodyconstructs on MHC class I-negative tumor cells. Mol Immunol2006; 43: 763–71.
108. Ellerman, D. Bispecific T-cell engagers: towards understandingvariables influencing the in vitro potency and tumor selectivity andtheir modulation to enhance their efficacy and safety. Methods2019; 154: 102–17.
109. Castella, B, Melaccio, A, Foglietta, M et al. Vγ 9Vδ2 T cells asstrategic weapons to improve the potency of immune checkpointblockade and immune interventions in human myeloma. FrontOncol 2018; 8: 508.
110. Brandes, M, Willimann, K, Bioley, G et al. Cross-presentinghuman gammadelta T cells induce robust CD8+ alphabeta T cellresponses. Proc Natl Acad Sci USA 2009; 106: 2307–12.
111. Hoeres, T, Smetak, M, Pretscher, D et al. Improving the efficiencyof Vγ 9Vδ2 T-cell immunotherapy in cancer. Front Immunol 2018;9: 800.
112. Oberg, H-H, Peipp, M, Kellner, C et al. Novel Bispecific antibodiesincrease γ δ T-cell cytotoxicity against pancreatic cancer cells.Cancer Res 2014; 74: 1349–60.
113. Reusch, U, Burkhardt, C, Fucek, I et al. A novel tetravalentbispecific TandAb (CD30/CD16A) efficiently recruits NK cells forthe lysis of CD30+ tumor cells. MAbs 2014; 6: 728–39.
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

Antibody Therapeutics, 2020 61
114. Bartlett, NL, Chen, RW, Domingo-Domenech, E et al. A phase 1bstudy investigating the combination of the tetravalent Bispecific NKcell engager AFM13 and pembrolizumab in patients withrelapsed/refractory Hodgkin lymphoma after brentuximab vedotinfailure: updated safety and efficacy data. Blood 2018; 132: 1620.
115. Sawas, A, Chen, P, Vlad, G et al. Clinical and biological evaluationof the novel Cd30/Cd16a tetravalent bispecific antibody (Afm13) inrelapsed or refractory Cd30-positive lymphoma with cutaneouspresentation: a biomarker phase Ib/Iia study (Nct03192202).Hematol Oncol 2019; 37: 314–6.
116. Chan, WK, Kang, S, Youssef, Y et al. A CS1-NKG2D bispecificantibody collectively activates cytolytic immune cells againstmultiple myeloma. Cancer Immunol Res 2018; 6: 776–87.
117. Corgnac, S, Perret, R, Derré, L et al. CD1d-antibody fusionproteins target iNKT cells to the tumor and trigger long-termtherapeutic responses. Cancer Immunol Immunother 2013; 62:747–60.
118. Van Der Horst, HJ, Oostindie, SC, Cillessen, SAGM et al.Targeting CD37 in B-cell malignancies using the novel therapeuticDuoHexaBody-CD37 results in efficient killing of tumor B-cells exvivo via complement-dependent cytotoxicity, even in relapsedand/or refractory patient samples. Blood 2018; 132: 4179.
119. Advani, R, Flinn, I, Popplewell, L et al. CD47 blockade byHu5F9-G4 and rituximab in non-Hodgkin’s lymphoma. N Engl JMed 2018; 379: 1711–21.
120. Hatterer, E, Barba, L, Noraz, N et al. Co-engaging CD47 andCD19 with a bispecific antibody abrogates B-cell receptor/CD19association leading to impaired B-cell proliferation. MAbs 2019; 11:322–34.
121. Garcia-Martinez, JM, Wernitznig, A, Rinnenthal, J et al. Abstract2051: BI 905711, a novel CDH17-targeting TRAILR2 agonist,effectively triggers tumor cell apoptosis and tumor regressionsselectively in CDH17-positive colorectal cancer models. Cancer Res2019; 79: 2051–1.
122. Pandey, P, Sliker, B, Peters, HL et al. Amyloid precursor proteinand amyloid precursor-like protein 2 in cancer. Oncotarget 2016; 7:19430–44.
123. Bay, AP, Kalsy, A, Tiwari, S et al. Abstract 233: bispecific HER2ADC: making more potent HER2 ADC by improving targetinternalization. Cancer Res 2019; 79: 233–3.
124. Ravetch, JV, Lanier, LL. Immune inhibitory receptors. Science(80-) 2000; 290: 84–9.
125. Chen, W, Shankar, S, Lohr, J et al. SAT0027 Immunomodulatory
effects of MGD010, a dart®
molecule targeting human B-CELLCD32B and CD79B. Ann Rheum Dis 2017; 76: 777–8.
126. Knight, T, Callaghan, MU. The role of emicizumab, a bispecificfactor IXa- and factor X-directed antibody, for the prevention ofbleeding episodes in patients with hemophilia A. Ther Adv Hematol2018; 9: 319–34.
127. Kitazawa, T, Igawa, T, Sampei, Z et al. A bispecific antibody tofactors IXa and X restores factor VIII hemostatic activity in ahemophilia A model. Nat Med 2012; 18: 1570–4.
128. Sampei, Z, Igawa, T, Soeda, T et al. Identification andmultidimensional optimization of an asymmetric bispecific IgGantibody mimicking the function of factor VIII cofactor activity.PLoS One 2013; 8: e57479.
129. Oldenburg, J, Mahlangu, JN, Kim, B et al. Emicizumab prophylaxisin hemophilia A with inhibitors. N Engl J Med 2017; 377: 809–18.
130. Sahni, J, Patel, SS, Dugel, PU et al. Simultaneous inhibition ofangiopoietin-2 and vascular endothelial growth factor-a withFaricimab in diabetic macular Edema BOULEVARD phase 2randomized trial. Ophthalmology 2019; 126: 1155–70.
131. Pulgar, VM. Transcytosis to cross the blood brain barrier. NewAdvancements and Challenges. Front Neurosci 2019; 12: 1019.
132. DiGiandomenico, A, Keller, AE, Gao, C et al. A multifunctionalbispecific antibody protects against Pseudomonas aeruginosa. SciTransl Med. 2014; 6: 262ra155.
133. Takada, A. Filovirus tropism: cellular molecules for viral entry.Front Microbiol 2012; 3: 1–9.
134. Holtsberg, W, Bakken, RR, Mittler, E et al. A “Trojan horse”bispecific-antibody strategy for broad protection againstebolaviruses. Science 2017; 354: 350–4.
135. Kolumam, G, Chen, MZ, Tong, R et al. Sustained brown fatstimulation and insulin sensitization by a humanized bispecific
antibody agonist for fibroblast growth factor receptor 1/βKlothocomplex. EBioMedicine 2015; 2: 730–43.
136. Chodorge, M, Züger, S, Stirnimann, C et al. A series of Fasreceptor agonist antibodies that demonstrate an inverse correlationbetween affinity and potency. Cell Death Differ 2012; 19: 1187–95.
137. Liu, Z, Stoll, VS, DeVries, PJ et al. A potenterythropoietin-mimicking human antibody interacts through anovel binding site. Blood 2007; 110: 2408–13.
138. Hothersall, JD, Guo, D, Sarda, S et al. Structure-activityrelationships of the sustained effects of adenosine A2A receptoragonists driven by slow dissociation kinetics. Mol Pharmacol 2017;91: 25–38.
139. Leong, SR, Sukumaran, S, Hristopoulos, M et al. Ananti-CD3/anti-CLL-1 bispecific antibody for the treatment of acutemyeloid leukemia. Blood 2017; 129: 609–18.
140. Zuch de Zafra, CL, Fajardo, F, Zhong, W et al. Targeting multiplemyeloma with AMG 424, a novel anti-CD38/CD3 bispecificT-cell–recruiting antibody optimized for cytotoxicity and cytokinerelease. Clin Cancer Res 2019; 25: 3921–33.
141. Hoffmann, P, Hofmeister, R, Brischwein, K et al. Serial killing oftumor cells by cytotoxic T cells redirected with aCD19-/CD3-bispecific single-chain antibody construct. Int JCancer 2005; 115: 98–104.
142. Yu, YJ, Zhang, Y, Kenrick, M et al. Boosting brain uptake of atherapeutic antibody by reducing its affinity for a transcytosistarget. Sci Transl Med 2011; 3: 84ra44.
143. Tillotson, BJ, Goulatis, LI, Parenti, I et al. Engineering ananti-transferrin receptor ScFv for pH-sensitive binding leads toincreased intracellular accumulation. PLoS One 2015; 10:e0145820–0.
144. Bardwell, PD, Staron, MM, Liu, J et al. Potent and conditionalredirected T cell killing of tumor cells using half DVD-Ig. ProteinCell 2018; 9: 121–9.
145. Hernandez-Hoyos, G, Sewell, T, Bader, R et al. MOR209/ES414, anovel bispecific antibody targeting PSMA for the treatment ofmetastatic castration-resistant prostate cancer. Mol Cancer Ther2016; 15: 2155–65.
146. Reusch, U, Duell, J, Ellwanger, K et al. A tetravalent bispecificTandAb (CD19/CD3), AFM11, efficiently recruits T cells for thepotent lysis of CD19(+) tumor cells. MAbs 2015; 7: 584–604.
147. Wang, J, Goetsch, L, Tucker, L et al. Anti-c-Met monoclonalantibody ABT-700 breaks oncogene addiction in tumors with METamplification. BMC Cancer 2016; 16: 105.
148. Trinklein, ND, Pham, D, Schellenberger, U et al. Efficient tumorkilling and minimal cytokine release with novel T-cell agonistbispecific antibodies. MAbs 2019; 11: 639–52.
149. Bluemel, C, Hausmann, S, Fluhr, P et al. Epitope distance to thetarget cell membrane and antigen size determine the potency of Tcell-mediated lysis by BiTE antibodies specific for a largemelanoma surface antigen. Cancer Immunol Immunother 2010; 59:1197–209.
150. Li, J, Stagg, NJ, Johnston, J et al. Membrane-proximal epitopefacilitates efficient T cell synapse formation by anti-FcRH5/CD3and is a requirement for myeloma cell killing. Cancer Cell 2017; 31:383–95.
151. Crawford, A, Haber, L, Kelly, MP et al. A Mucin 16 bispecific Tcell-engaging antibody for the treatment of ovarian cancer. SciTransl Med 2019; 11: eaau7534.
152. Yokosuka, T, Takamatsu, M, Kobayashi-Imanishi, W et al.Programmed cell death 1 forms negative costimulatorymicroclusters that directly inhibit T cell receptor signaling byrecruiting phosphatase SHP2. J Exp Med 2012; 209:1201–17.
153. Le Gall, F, Reusch, U, Little, M et al. Effect of linker sequencesbetween the antibody variable domains on the formation, stabilityand biological activity of a bispecific tandem diabody. Protein EngDes Sel 2004; 17: 357–66.
154. DiGiammarino, EL, Harlan, JE, Walter, KA et al. Ligandassociation rates to the inner-variable-domain of adual-variable-domain immunoglobulin are significantly impactedby linker design. MAbs 2011; 3: 487–94.
155. Thurber, GM, Schmidt, MM, Wittrup, KD. Antibody tumorpenetration: transport opposed by systemic and antigen-mediatedclearance. Adv Drug Deliv Rev 2008; 60: 1421–34.
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022

62 Antibody Therapeutics, 2020
156. Schmidt, MM, Wittrup, KD. A modeling analysis of the effects ofmolecular size and binding affinity on tumor targeting. Mol CancerTher 2009; 8: 2861–71.
157. Goldenberg, DM, Chatal, J-F, Barbet, J et al. Cancer imaging andtherapy with bispecific antibody pretargeting. Update Cancer Ther2007; 2: 19–31.
158. Roux, KH, Strelets, L, Michaelsen, TE. Flexibility of human IgGsubclasses. J Immunol 1997; 159: 3372–82.
159. Kapelski, S, Cleiren, E, Attar, RM et al. Influence of the bispecificantibody IgG subclass on T cell redirection. MAbs 2019; 11:1012–24.
160. Sampei, Z, Igawa, T, Soeda, T et al. Non-antigen-contacting regionof an asymmetric bispecific antibody to factors IXa/X significantlyaffects factor VIII-mimetic activity. MAbs 2015; 7: 120–8.
161. Zhang, T, Song, X, Xu, L et al. The binding of an anti-PD-1antibody to Fcγ RI has a profound impact on its biologicalfunctions. Cancer Immunol Immunother 2018; 67: 1079–90.
162. Singh, SK. Impact of product-related factors on immunogenicity ofbiotherapeutics. J Pharm Sci 2011; 100: 354–87.
163. Davda, J, Declerck, P, Hu-Lieskovan, S et al. Immunogenicity ofimmunomodulatory, antibody-based, oncology therapeutics. JImmunother Cancer 2019; 7: 105.
164. Groell, F, Jordan, O, Borchard, G. In vitro models forimmunogenicity prediction of therapeutic proteins. Eur J PharmBiopharm 2018; 130: 128–42.
165. Saunders, KO. Conceptual approaches to modulating antibodyeffector functions and circulation half-life. Front Immunol 2019; 10:1296.
166. Gaudinski, MR, Coates, EE, Houser, KV et al. Safety andpharmacokinetics of the Fc-modified HIV-1 human monoclonalantibody VRC01LS: a phase 1 open-label clinical trial in healthyadults. PLoS Med 2018; 15: e1002493.
167. Leipold, D, Prabhu, S. Pharmacokinetic and pharmacodynamicconsiderations in the design of therapeutic antibodies. Clin TranslSci 2019; 12: 130–9.
168. Adams, GP, Schier, R, Mccall, AM et al. High affinity restricts thelocalization and tumor penetration of single-chain Fv antibodymolecules. Cancer Res 2001; 61: 4750–5.
169. Rudnick, SI, Lou, J, Shaller, CC et al. Influence of affinity andantigen internalization on the uptake and penetration of anti-HER2antibodies in solid tumors. Cancer Res 2011; 71: 2250–9.
170. Friedrich, SW, Lin, SC, Stoll, BR et al. Antibody-directed effectorcell therapy of tumors: analysis and optimization using aphysiologically based pharmacokinetic model. Neoplasia 2002; 4:449–63.
171. Goebeler, M-E, Knop, S, Viardot, A et al. Bispecific T-cell engager(BiTE) antibody construct blinatumomab for the treatment ofpatients with relapsed/refractory non-Hodgkin lymphoma: finalresults from a phase I study. J Clin Oncol 2016; 34:1104–11.
172. Gunde, T, Brock, M, Warmuth, S et al. Abstract 1532: a novel,monovalent tri-specific antibody-based molecule thatsimultaneously modulates PD-L1 and 4-1BB exhibits potentanti-tumoral activity in vivo. Cancer Res 2019; 79: 1532–2.
173. Prell, RA, Lee, DW, Halpern, WG et al. Chapter 14. Nonclinicaldevelopment of multi-targeting biopharmaceuticals. In: Plitnick,LM, Biosimilars, H (eds). Vaccines and Specialty BiologicsDJBT-ND of NB. San Diego: Academic Press, 2013, 343–71
174. Trivedi, A, Stienen, S, Zhu, M et al. Clinical pharmacology andtranslational aspects of bispecific antibodies. Clin Transl Sci 2017;10: 147–62.
175. Mayes, PA, Hance, KW, Hoos, A. The promise and challenges ofimmune agonist antibody development in cancer. Nat Rev DrugDiscov 2018; 17: 509–27.
176. Messenheimer, DJ, Jensen, SM, Afentoulis, ME et al. Timing ofPD-1 blockade is critical to effective combination immunotherapywith anti-OX40. Clin Cancer Res 2017; 23: 6165–77.
177. Kohrt, HE, Houot, R, Goldstein, MJ et al. CD137 stimulationenhances the antilymphoma activity of anti-CD20 antibodies.Blood 2011; 117: 2423–32.
178. Kohrt, HE, Houot, R, Weiskopf, K et al. Stimulation of naturalkiller cells with a 4-1BB-specific antibody enhances trastuzumabefficacy in xenotransplant models of breast cancer. J Clin Invest2012; 122: 1066–75.
179. Jackman, J, Chen, Y, Huang, A et al. Development of a two-partstrategy to identify a therapeutic human bispecific antibody thatinhibits IgE receptor signaling. J Biol Chem 2010; 285: 20850–9.
180. Gu, J, Yang, J, Chang, Q et al. Identification of anti-ErbB2 dualvariable domain immunoglobulin (DVD-IgTM) proteins withunique activities. PLoS One 2014; 9: e97292–2.
181. Madrenas, J, Chau, LA, Teft, WA et al. Conversion of CTLA-4from inhibitor to activator of T cells with a bispecific tandemsingle-chain Fv ligand. J Immunol 2004; 172: 5948–56.
Dow
nloaded from https://academ
ic.oup.com/abt/article/3/1/18/5739255 by guest on 30 M
ay 2022
Copyright © 2022 FDOKUMEN




















