
Why Unemployment rate vary, Finnish Unemployment in a comparative international context
Upload
independentCategory
view
1download
0
Acta Neuropathol (2006) 112: 95–105 DOI 10.1007/s00401-006-0078-4
ORIGINAL PAPER
Ana S. Falcão · Adelaide Fernandes · Maria A. Brito Rui F. M. Silva · Dora Brites
Bilirubin-induced immunostimulant effects and toxicity vary with neural cell type and maturation state
Received: 30 January 2006 / Revised: 24 April 2006 / Accepted: 24 April 2006 / Published online: 30 May 2006© Springer-Verlag 2006
Abstract Hyperbilirubinemia remains one of the mostfrequent clinical diagnoses in the neonatal period. Theincreased vulnerability of premature infants to unconju-gated bilirubin (UCB)-induced brain damage may bedue to a proneness of immature nerve cells to UCB-toxicstimulus. Thus, in this study, we evaluated UCB-inducedcell death, glutamate release and cytokine production, inastrocytes and neurons cultured for diVerent days, inorder to relate the diVerentiation state with cell vulnera-bility to UCB. The age-dependent activation of thenuclear factor-�B (NF-�B), an important transcriptionfactor involved in inXammation, was also investigated.Furthermore, responsiveness of neurons and astrocytesto UCB were compared in order to identify the most sus-ceptible to each induced eVect, as an approach to whathappens in vivo. The results clearly showed that imma-ture nerve cells are more vulnerable than the most diVer-entiated ones to UCB-induced cell death, glutamaterelease and tumour necrosis factor (TNF)-� secretion.Moreover, astrocytes seem to be more competent cells inreleasing glutamate and in producing an inXammatoryresponse when injured by UCB. Activation of NF-�B byUCB also presents a cell-age-dependent pattern, and val-ues vary with neural cell type. Again, astrocytes have thehighest activation levels, which are correlated with thegreater amount of cytokine production observed in thesecells. These results contribute to a better knowledge ofthe mechanisms leading to UCB encephalopathy by elu-cidation of age- and type-related diVerences in neural cellresponses to UCB.
Keywords Astrocytes · Cell maturation · Cell reactivity · Neurons · Unconjugated bilirubin
Introduction
Hyperbilirubinemia is probably the most common clini-cal diagnosis during neonatal life [37, 63], and reXects theinterplay between developmentally modulated changesin bilirubin production and metabolism [77]. This neona-tal jaundice may be physiological, without any clinicalconsequence, or it can lead to an acute form of bilirubinencephalopathy with minimal neurological damage, orto a more severe condition called kernicterus, character-ized by chronic and permanent neurological sequelae, asa result of unconjugated bilirubin (UCB) neurotoxicity[2, 54, 68]. The evidence that children with a moderateneonatal jaundice can display minor neurologic dysfunc-tions during the Wrst year of life [72] and that prematu-rity is considered a signiWcant factor for adverseneurodevelopmental sequelae [76], led clinicians to con-cern about the infants who are more vulnerable to UCB-induced neurological damage.
Preterm newborns have an increased susceptibility toUCB-toxic eVects, during moderate to severe neonataljaundice [17, 26, 37] because several conditions of thepremature newborn contribute to a poor clearance ofUCB, such as a decreased hepatic UDP-glucuronosyl-transferase activity [39], a reduced uptake and secretionof UCB [76] and a shorter life span of their red bloodcells [73], which further increases UCB production. Inaddition, these infants normally present hypoalbumin-emia, leading to a greater amount of free UCB, that cancross the more immature and permeable blood–brainbarrier. As a result of all these factors, the neurotoxiceVect will be more pronounced in premature than in terminfants, even at relatively low levels of UCB [52]. More-over, prematurity is often associated with sepsis, alsoconsidered to be a risk factor for bilirubin encephalopa-thy [32, 37, 41], probably by altering the blood–brainbarrier permeability [56, 59].
In order to further explain the higher propensity ofpreterm infants to UCB neurotoxicity, we have recentlyhypothesized that this could be due to a proneness ofimmature nerve cells to neurotoxic injury by UCB. The
A. S. Falcão · A. Fernandes · M. A. Brito R. F. M. Silva · D. Brites (&)Centro de Patogénese Molecular-UBMBE, Faculdade de Farmácia, University of Lisbon, Av. Forças Armadas, 1600-083 Lisbon, PortugalE-mail: [email protected].: +351-21-7946450Fax: +351-21-7946491

96
results obtained in our studies showed that youngerastrocytes are more prone to UCB-induced cell death,glutamate eZux and inXammatory response than olderones [18]. We also demonstrated that incubation ofimmature glial cells with lipopolysaccharide (LPS), anendotoxin used to mimic infection, exacerbates some ofthe deleterious eVects of UCB, namely cell death bynecrosis, pointing out that hyperbilirubinemia should bemore cautiously monitored in premature infants wheninfection by bacteria is anticipated.
Since neurons are considered to be the main targetsfor UCB neurotoxicity [50], we extended our previousstudies in order to establish how these neural cells atdiVerent stages of diVerentiation behave when exposedto clinically relevant concentrations of UCB. Compari-son of the results obtained in astrocytes and neuronsallow us to ascertain whether these neural cells responddiVerently to UCB-toxic stimulus. Thus, in the presentstudy, we evaluate and compare UCB-induced cell death,glutamate release and cytokine production in neuronsand astrocytes, according to cell age in culture. We alsoinvestigate the activation of nuclear factor-�B (NF-�B),an important transcription factor that positively regu-lates the induction of some inXammatory genes [15, 74],and was shown to be activated by UCB [20]. Moreover,we evaluate if co-incubation with LPS further exacer-bates the UCB-induced eVects in neuronal cells, as previ-ously demonstrated in astrocytes [18].
The results clearly show that neurons are mainly vul-nerable to UCB-induced cell death, displaying a lowinXammatory response, whereas astrocytes are morecompetent in releasing glutamate and the pro-inXamma-tory cytokine tumour necrosis factor (TNF)-� whenexposed to UCB. Moreover, both immature neural celltypes show an increased susceptibility to UCB-inducedcell death, glutamate release and TNF-� secretion whencompared with the most diVerentiated ones. The UCB-induced activation of NF-�B has also a cell-age-depen-dent proWle, with astrocytes showing the highest activa-tion levels, an eVect that can be related with the greateramount of cytokine secretion by these cells. Further-more, and in contrast to what happens in astrocytes, LPSdid not signiWcantly exacerbate the UCB-induced immu-nostimulant eVects and toxicity in neuronal cells.
These Wndings contribute to a better knowledge of themechanisms underlying UCB neurotoxicity by showing adiVerent behavior of the two main nerve-cell types whenexposed to UCB. Furthermore, this study provides anadditional supportive evidence for the higher susceptibil-ity of premature babies to UCB brain damage.
Materials and methods
Materials
Dulbecco’s modiWed Eagle’s medium (DMEM) and fetalcalf serum (FCS) were purchased from Biochrom AG
(Berlin, Germany). Neurobasal medium, B-27 Supple-ment 50£, Hanks’ balanced salt solution (HBSS-1),Hanks’ balanced salt solution without Ca2+ and Mg2+
(HBSS-2), gentamicin (50 mg/ml) and trypsin (0.025%)were acquired from Invitrogen (Carlsbad, CA, USA).Antibiotic antimycotic solution (20£), bilirubin, humanserum albumin (HSA), fraction V, fatty acid free, andHoechst dye 33258 were purchased from Sigma Chemi-cal Co. (St Louis, MO, USA). Escherichia coli O111:B4LPS was purchased from Calbiochem (La Jolla, CA,USA). Rabbit antibody anti-p65 NF-�B subunit wasfrom Santa Cruz Biotechnology (Santa Cruz, CA, USA),FITC-labeled goat antibody anti-rabbit was from Sigmaand TRITC-labeled goat antibody anti-rabbit was fromSanta Cruz Biotechnology. Lactate dehydrogenase(LDH) cytotoxicity detection kit and L-glutamic acid kitwere obtained from Roche Molecular Biochemicals(Manheim, Germany). DuoSet ELISA kits for TNF-�,interleukin (IL)-1� and IL-6 were from R&D SystemsInc. (Minneapolis, MN, USA). All other chemicals wereof analytical grade and were purchased from Merck(Darmstadt, Germany).
Cell cultures
The Institutional Animal Ethics Committee approvedthe study protocol and all procedures complied withinternational standards of humane care in animal experi-mentation. Thus, Wistar rats were maintained on a 12-hlight/dark cycle, under conditions of constant tempera-ture and humidity. Animals were supplied with standardlaboratory chow and water ad libitum.
Neurons were isolated from fetuses of 17–18 daypregnant Wistar rats, as previously described [70]. Inshort, pregnant rats were anesthetized and decapitated.The fetuses were collected in HBSS-1 and rapidly decap-itated. After removal of meninges and white matter, thebrain cortex was collected in HBSS-2 and mechanicallyfragmented. The cortex fragments were transferred to a0.025% trypsin in HBSS-2 solution and incubated for15 min at 37°C. Following trypsinization, the cells werewashed twice in HBSS-2 containing 10% FCS, and resus-pended in Neurobasal medium supplemented with0.5 mM L-glutamine, 25 �M L-glutamic acid, 2% B-27Supplement, and 0.12 mg/ml gentamicin. Aliquots of1.0 £ 105 cells/cm2 were plated on 12-well tissue cultureplates and maintained at 37°C in a humidiWed atmo-sphere of 5% CO2. Every 3 days, 0.5 ml of the oldmedium was removed by aspiration and replaced by thesame volume of fresh medium without L-glutamic acid.Cells were used after 4, 8 and 18 days in vitro (DIV) andwere morphologically characterized by phase-contrastmicroscopy.
Astrocytes were isolated from 2-day-old rats as previ-ously described [8], with minor modiWcations [69].BrieXy, rats were decapitated and the brains collected inDMEM containing 11 mM sodium bicarbonate,38.9 mM glucose and 1% antibiotic antimycotic solution.The cortical fraction was homogenized by mechanical

97
fragmentation, and the cells collected after centrifuga-tion (10 min at 700g) were resuspended in culturemedium supplemented with 10% FCS. Finally,2.0 £ 105 cells/cm2 were plated on 12-well tissue-cultureplates and maintained at 37°C in a humidiWed atmo-sphere of 5% CO2. Experimental incubations were per-formed at 5, 10 and 20 DIV.
To exclude the interference of contaminant astrocytesand microglia in our primary cultures of neurons andastrocytes, respectively, that could be responsible for glu-tamate release and cytokine production [28, 49] we eval-uated the purity of our nerve-cell cultures. We concludedthat in neuronal primary cultures, the contaminant glialcells (GFAP-positive) increased with age in culture, buteven at 18 DIV the percentage of these cells was nothigher than 5.6%. Likewise, in astrocyte cultures, thecontaminant microglial cells (OX-42-positive) wereinsigniWcant, ranging from 0.9% at 5 DIV to 3.6% at 20DIV.
This model of aging in vitro is based on the fact thatthe timing of development in glial cell cultures is likelyrepresentative of maturation in vivo [1, 78] and that full-term newborns correspond best to 12 day-old rats [61].This way, in our cultures, 10 DIV astrocytes will matchup to a full-term neonate and cells used in a less-diVeren-tiated stage to a premature condition [71]. In addition,diVerentiation of neurons is obtained by the eighth dayin culture, while comparable diVerentiation and conXu-ence of astrocytes is reached only by the 10th to 11th dayin culture, as was observed by morphological analysisand immunolabeling of speciWc neuronal and astrocyticmarkers [10, 43].
Cortical neurons and astrocytes were isolated fromrat cortices based on the fact that, despite classicalkernicterus has been characterized by an intense color-ing of the basal ganglia and the medulla oblongata,diVuse yellow spots were observed in most of the jaun-diced infants autopsied by Schmorl [31], and that nodiVerences were found regarding the acute entry ofUCB between those regions and the cortical region[30].
Bilirubin puriWcation
Unconjugated bilirubin was puriWed by a modiWcation ofthe method described by McDonagh and Assisi [44], asfollows: commercial UCB was Wrst dissolved (1 mg/ml)in spectroscopy grade chloroform (Uvasol®), under heat(65°C) and agitation. After complete dissolution, thissolution was Wltered by a grade 40 Whatman paper(Whatman International Ltd, Maidstone, Kent,England), previously soaked in chloroform, and washedwith 0.1 M NaHCO3 under vigorous shaking. The aque-ous phase, containing the UCB contaminants, wasremoved and the organic phase was washed Wve moretimes. Afterwards, a wash with 10% NaCl (w/v), followedby three washes with distilled water were performed. Theobtained puriWed chloroformic phase was Wlteredthrough a grade 40 Whatman paper, in the presence of
anidrous Na2SO4, and evaporated under a nitrogenstream, with heating (70–80°C) and shaking till half ofthe initial volume. Spectroscopy grade methanol (Uva-sol) was then added drop wise until the UCB crystalsstart to form. Heating was maintained until completechloroform evaporation and a suspension of UCB inmethanol was obtained. This suspension was centrifugedby 5 min at 2,500 rpm and the resulting crystals werewashed twice with methanol and dried under vacuum.The puriWed UCB was then sealed under a nitrogenatmosphere and preserved at ¡20°C. All the proceduresand storage were performed under light protection.
Treatment of nerve cells
All UCB solutions were prepared from a 10-mM stocksolution in 0.1 N NaOH, and used brieXy after prepara-tion, in diminished light conditions. HCl 0.1 N was usedto restore the pH value to 7.4. LPS was dissolved in phos-phate buVered saline (PBS, pH 7.4) at 1 mg/ml.
Neurons at 4, 8 and 18 DIV and astrocytes at 5, 10and 20 DIV were incubated with 50 or 100 �M UCB, inthe presence of 100 �M HSA (UCB/HSA molar ratios of0.5 and 1.0, respectively), for 4 h at 37°C. For co-incuba-tion studies, neurons were exposed for 4 h to UCB(molar ratio of 0.5) and 1 ng/ml LPS. Controls were per-formed in the absence of UCB and LPS.
At the end of the incubation period, the cell-freemedium was collected for LDH, glutamate and cytokinedeterminations, while attached cells were Wxed for30 min with freshly prepared 4% paraformaldehydein PBS for both apoptosis and immunocytochemicalstudies.
Cell death
Cell death by apoptosis was evaluated by assessment ofnuclear morphology. In brief, cells were incubated withHoechst dye 33258 at 5 �g/ml in PBS, for 2 min at roomtemperature, washed with PBS and mounted using PBS/glycerol (3:1, v/v). Fluorescent nuclei were visualizedusing an Axioscop® microscope (Zeiss, Germany), cate-gorized according to condensation and staining charac-teristics of chromatin, and scored by investigatorsblinded as to the treatment received by cells. Apoptoticnuclei were identiWed by condensed chromatin, contigu-ous to the nuclear membrane, as well as nuclear fragmen-tation of the condensed chromatin. At least Wve randommicroscopic Welds were counted per sample, and meanvalues were expressed as the percentage of apoptoticnuclei.
The assay used to estimate loss of cell viability wasbased on the release of LDH by nonviable cells. LDHwas determined in the incubation medium using thecytotoxicity detection kit, LDH. All readings were cor-rected for the possible interference of UCB absorptionand the results were expressed as percent of total LDHrelease, obtained by treating non-incubated cells with 2%Triton X-100 in incubation medium for 30 min.
98
Measurement of glutamate
Release of glutamate to the culture medium was deter-mined by an adaptation of the L-glutamic acid kit, usinga ten-fold reduction in kit reagents. Samples were pip-etted to a 96-well plate and read at 490 nm, using a refer-ence Wlter of 620 nm, in a microplate reader PR 2100(Bio-Rad Laboratories, Hemel, Hempstead, UK). A newcalibration curve was used in each assay and the resultsnormalized to 105 cells.
Cytokine determinations
Culture supernatants, free from cellular debris, wereassessed in duplicate for TNF-�, IL-1� and IL-6 withspeciWc Quantikine ELISA kits, according to the manu-facturer’s instructions, using a microplate reader. Theresults were expressed in picograms per milliliter andnormalized to 105 cells for each culture.
Assessment of NF-�B activation
Translocation of NF-�B from the cytoplasm to thenucleus was visualized by staining with a primary anti-body raised against the p65 subunit of NF-�B (1:200),followed by a goat anti-rabbit secondary antibodylabeled with FITC (1:160) or TRITC (1:100) for astro-cytes and neurons, respectively. Nuclei were stained withHoechst dye 33258 as previously described and pairs ofUV and Xuorescence images of Wve random microscopicWelds (original magniWcation 516£) were acquired persample. NF-�B-positive nuclei and total cells werecounted to determine the percentage of NF-�B-positivenuclei.
Statistical analysis
Results of, at least, three diVerent experiments, per-formed in duplicate, were expressed as mean § SEM.DiVerences between groups were determined by one-wayanalysis of variance with Dunnett’s or Bonferroni’s mul-tiple comparisons post tests, using the Prism Program(GraphPad Software Inc.). We considered P < 0.05 to bestatistically signiWcant.
Results
Cell death depends on neural cell type and diVerentiation state and is enhanced by bilirubin
We reported in one of our most recent works [18], thatimmature astrocytes (5 DIV) were more prone to UCB-induced cell death than older ones (20 DIV). In the workpresented here, we extended these previous studies, incu-bating not only astrocytes, but also neurons, withdiVerent UCB concentrations in order to clarify if thiscell-age-dependent toxicity is also extensive to other
nerve cells, like neurons and if it occurs in a concentra-tion-dependent way.
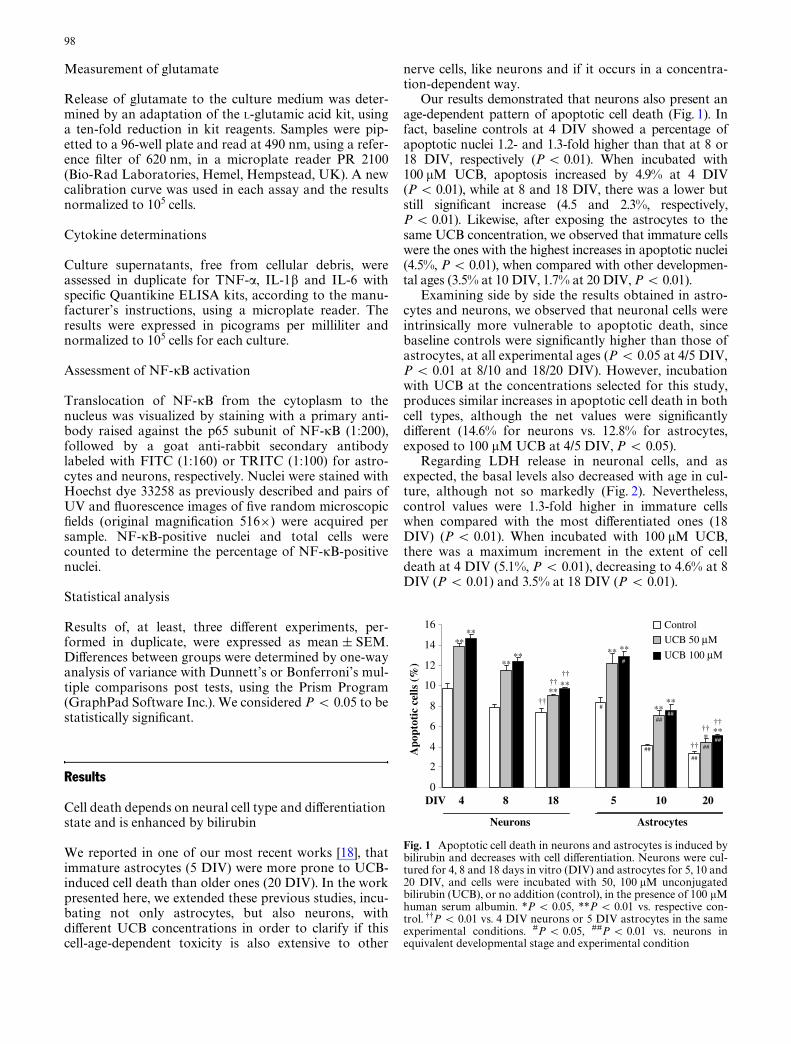
Our results demonstrated that neurons also present anage-dependent pattern of apoptotic cell death (Fig. 1). Infact, baseline controls at 4 DIV showed a percentage ofapoptotic nuclei 1.2- and 1.3-fold higher than that at 8 or18 DIV, respectively (P < 0.01). When incubated with100 �M UCB, apoptosis increased by 4.9% at 4 DIV(P < 0.01), while at 8 and 18 DIV, there was a lower butstill signiWcant increase (4.5 and 2.3%, respectively,P < 0.01). Likewise, after exposing the astrocytes to thesame UCB concentration, we observed that immature cellswere the ones with the highest increases in apoptotic nuclei(4.5%, P < 0.01), when compared with other developmen-tal ages (3.5% at 10 DIV, 1.7% at 20 DIV, P < 0.01).
Examining side by side the results obtained in astro-cytes and neurons, we observed that neuronal cells wereintrinsically more vulnerable to apoptotic death, sincebaseline controls were signiWcantly higher than those ofastrocytes, at all experimental ages (P < 0.05 at 4/5 DIV,P < 0.01 at 8/10 and 18/20 DIV). However, incubationwith UCB at the concentrations selected for this study,produces similar increases in apoptotic cell death in bothcell types, although the net values were signiWcantlydiVerent (14.6% for neurons vs. 12.8% for astrocytes,exposed to 100 �M UCB at 4/5 DIV, P < 0.05).
Regarding LDH release in neuronal cells, and asexpected, the basal levels also decreased with age in cul-ture, although not so markedly (Fig. 2). Nevertheless,control values were 1.3-fold higher in immature cellswhen compared with the most diVerentiated ones (18DIV) (P < 0.01). When incubated with 100 �M UCB,there was a maximum increment in the extent of celldeath at 4 DIV (5.1%, P < 0.01), decreasing to 4.6% at 8DIV (P < 0.01) and 3.5% at 18 DIV (P < 0.01).
Fig. 1 Apoptotic cell death in neurons and astrocytes is induced bybilirubin and decreases with cell diVerentiation. Neurons were cul-tured for 4, 8 and 18 days in vitro (DIV) and astrocytes for 5, 10 and20 DIV, and cells were incubated with 50, 100 �M unconjugatedbilirubin (UCB), or no addition (control), in the presence of 100 �Mhuman serum albumin. *P < 0.05, **P < 0.01 vs. respective con-trol. 99P < 0.01 vs. 4 DIV neurons or 5 DIV astrocytes in the sameexperimental conditions. #P < 0.05, ##P < 0.01 vs. neurons inequivalent developmental stage and experimental condition
0
2
4
6
8
10
12
14
16
Apo
ptot
ic c
ells
(%
)
††
**
††
**
****
****
** **
****
***
††
††
††††
##
####
##
####
#
#
Control
UCB 50 µM
UCB 100 µM
DIV 4 8 18 5 10 20
Neurons Astrocytes

99
The same pattern of vulnerability was observed forastrocytes, where LDH release induced by 100 �M UCBwas maximum at 5 DIV (7.0%, P < 0.01), decreasing to4.0 and 3.0% at 10 and 20 DIV, respectively (P < 0.01).A similar response in cell death was obtained for thelower UCB concentration (50 �M), but with smaller,although signiWcant, increases. Again, when comparingthe two cell types, UCB produced equivalent levels inLDH release, despite the marked diVerences observed inabsolute values (11.3% for neurons vs. 18.4% for astro-cytes, exposed to 100 �M UCB at 4/5 DIV, P < 0.01).
Glutamate secretion depends on neural cell type and diVerentiation state and is enhanced by bilirubin
As shown in Fig. 3, control levels of extracellular gluta-mate in neuronal cultures decreased from 1.2 nmol/105
cells at 4 DIV to 0.3 nmol/105 cells at 18 DIV (P < 0.05).UCB markedly increased glutamate release, particularlyin the immature cells, achieving in the extracellularmedium the highest value of 19.8 nmol/105 cells(P < 0.01), by incubation with 100 �M UCB.
Astrocytes present a similar age-dependent decrease inglutamate release, with baseline values that fall from9.2 nmol/105 cells at 5 DIV to 1.4 nmol/105 cells at 20 DIV(P < 0.01). However, it deserves to be noted that afterUCB incubation, immature glial cells are the ones with thehighest increases in the extracellular glutamate, mainlywith 100 �M UCB, a condition producing 49 nmol/105
cells (P < 0.01). Although less pronounced, the valuesobtained for glutamate release in all conditions, using50 �M UCB were also highly signiWcant (P < 0.01).
Comparing the results obtained in both cell types, andcontrasting with cell death, the glial cells were muchmore reactive to UCB, as demonstrated by the increased
levels of glutamate release (»two-fold higher values thanthose observed in neurons, P < 0.01).
TNF-� secretion depends on neural cell type and diVerentiation state and is enhanced by bilirubin, while IL-1� is not produced by neurons
Having previously demonstrated that UCB is immuno-stimulant to diVerentiated astrocytes [19], we proceededto evaluate if this UCB eVect is extensive to neurons andif it is also dependent on the diVerentiation state.
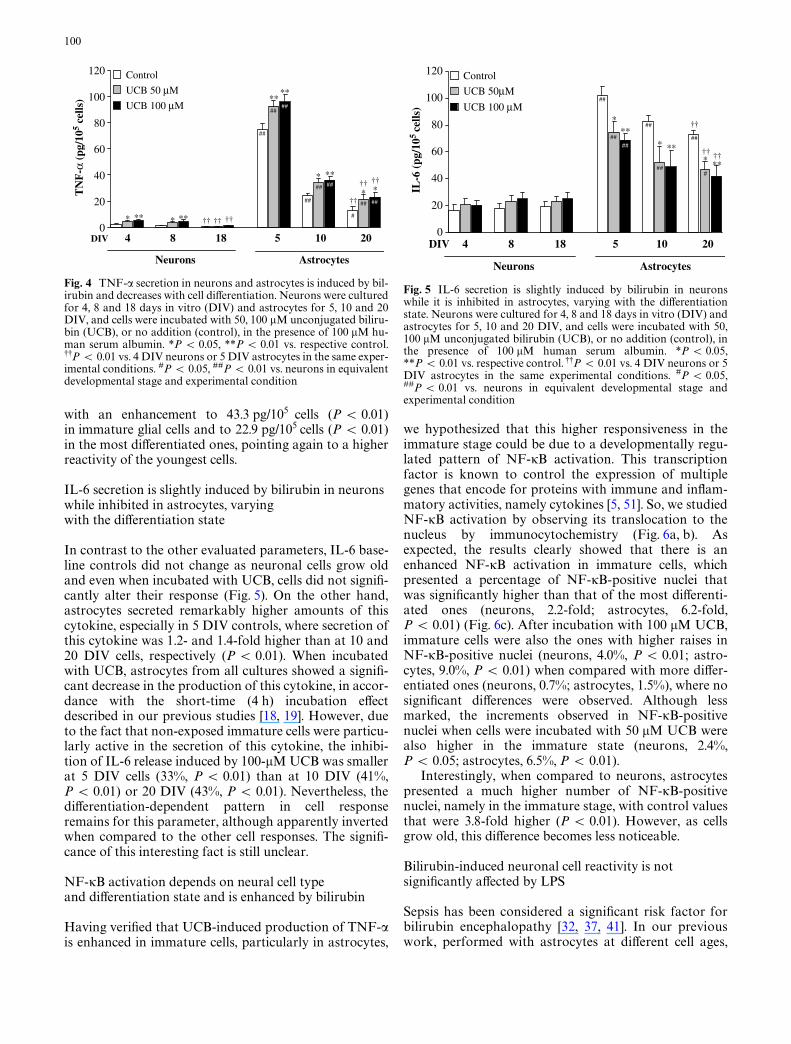
As observed for glutamate release, TNF-� basal levelsdecreased with cell maturation in both neural cell types(Fig. 4), but now with even greater expression, particu-larly in astrocytes. While in neuronal cells, there is areduction in baseline controls from 2.3 pg/105 cells at 4DIV to 0.5 pg/105 cells at 18 DIV (P < 0.01), in astro-cytes these values decrease from 74.9 pg/105 cells at 5DIV to 12.6 pg/105 cells at 20 DIV (P < 0.01).
Although neurons were capable to develop an inXam-matory response when incubated with UCB, particularlyat 4 and 8 DIV (P < 0.05 and 0.01, UCB 50 and 100 �MUCB, respectively), astrocytes are much more reactive.Once again, the UCB-induced production of TNF-� washigher at 5 DIV astrocytes, with a signiWcant raise of 17.6and 20.9 pg/105 cells by exposure to 50 and 100 �MUCB, respectively (P < 0.01), whereas in the oldest cells,this increase was only of 8.9 and 10.4 pg/105 cells (50 and100 �M UCB, respectively, P < 0.05).
Interestingly, we were not able to detect the secretionof IL-1� by neurons either in the absence or in the pres-ence of UCB (data not shown). Conversely, basal levels ofIL-1� in astrocyte cultures were detected and decreasedfrom 24.7 pg/105 cells at 5 DIV to 11.6 pg/105 cells at 20DIV (P < 0.01). After incubation with 100 �M UCB, theproduction of this cytokine was increased at all cell ages
Fig. 2 Lactate dehydrogenase release by neurons and astrocytes isinduced by bilirubin and decreases with cell diVerentiation. Neuronswere cultured for 4, 8 and 18 days in vitro (DIV) and astrocytes for5, 10 and 20 DIV, and cells were incubated with 50, 100 �M uncon-jugated bilirubin (UCB), or no addition (control), in the presence of100 �M human serum albumin. *P < 0.05, **P < 0.01 vs. respec-tive control. 9P < 0.05, 99P < 0.01 vs. 4 DIV neurons or 5 DIV as-trocytes in the same experimental conditions. #P < 0.05, ##P < 0.01vs. neurons in equivalent developmental stage and experimentalcondition
0
5
10
15
20
25L
DH
rel
ease
(%
)
DIV 4 8 18 5 10 20
Neurons Astrocytes
#
**
††
**** **
***
****
****
*
**
†††
††
††
††
##
####
###
##
#
Control
UCB 50 µM
UCB 100 µM
Fig. 3 Glutamate release in neurons and astrocytes is induced bybilirubin and decreases with cell diVerentiation. Neurons were cul-tured for 4, 8 and 18 days in vitro (DIV) and astrocytes for 5, 10 and20 DIV, and cells were incubated with 50, 100 �M unconjugated bil-irubin (UCB), or no addition (control), in the presence of 100 �Mhuman serum albumin. **P < 0.01 vs. respective control. 9P < 0.05,99P < 0.01 vs. 4 DIV neurons or 5 DIV astrocytes in the same exper-imental conditions. #P < 0.05, ##P < 0.01 vs. neurons in equivalentdevelopmental stage and experimental condition
0
10
20
30
40
50
60
Glu
tam
ate
(nm
ol/1
05 ce
lls)
DIV 4 8 18 5 10 20
Neurons Astrocytes
**
†
****
**
**
**
**
**
**
**
**
**
††
†† ††
††
††
##
##
##
#
##
##
##
##
Control
UCB 50 µM
UCB 100 µM
100
with an enhancement to 43.3 pg/105 cells (P < 0.01)in immature glial cells and to 22.9 pg/105 cells (P < 0.01)in the most diVerentiated ones, pointing again to a higherreactivity of the youngest cells.
IL-6 secretion is slightly induced by bilirubin in neurons while inhibited in astrocytes, varying with the diVerentiation state
In contrast to the other evaluated parameters, IL-6 base-line controls did not change as neuronal cells grow oldand even when incubated with UCB, cells did not signiW-cantly alter their response (Fig. 5). On the other hand,astrocytes secreted remarkably higher amounts of thiscytokine, especially in 5 DIV controls, where secretion ofthis cytokine was 1.2- and 1.4-fold higher than at 10 and20 DIV cells, respectively (P < 0.01). When incubatedwith UCB, astrocytes from all cultures showed a signiW-cant decrease in the production of this cytokine, in accor-dance with the short-time (4 h) incubation eVectdescribed in our previous studies [18, 19]. However, dueto the fact that non-exposed immature cells were particu-larly active in the secretion of this cytokine, the inhibi-tion of IL-6 release induced by 100-�M UCB was smallerat 5 DIV cells (33%, P < 0.01) than at 10 DIV (41%,P < 0.01) or 20 DIV (43%, P < 0.01). Nevertheless, thediVerentiation-dependent pattern in cell responseremains for this parameter, although apparently invertedwhen compared to the other cell responses. The signiW-cance of this interesting fact is still unclear.
NF-�B activation depends on neural cell type and diVerentiation state and is enhanced by bilirubin
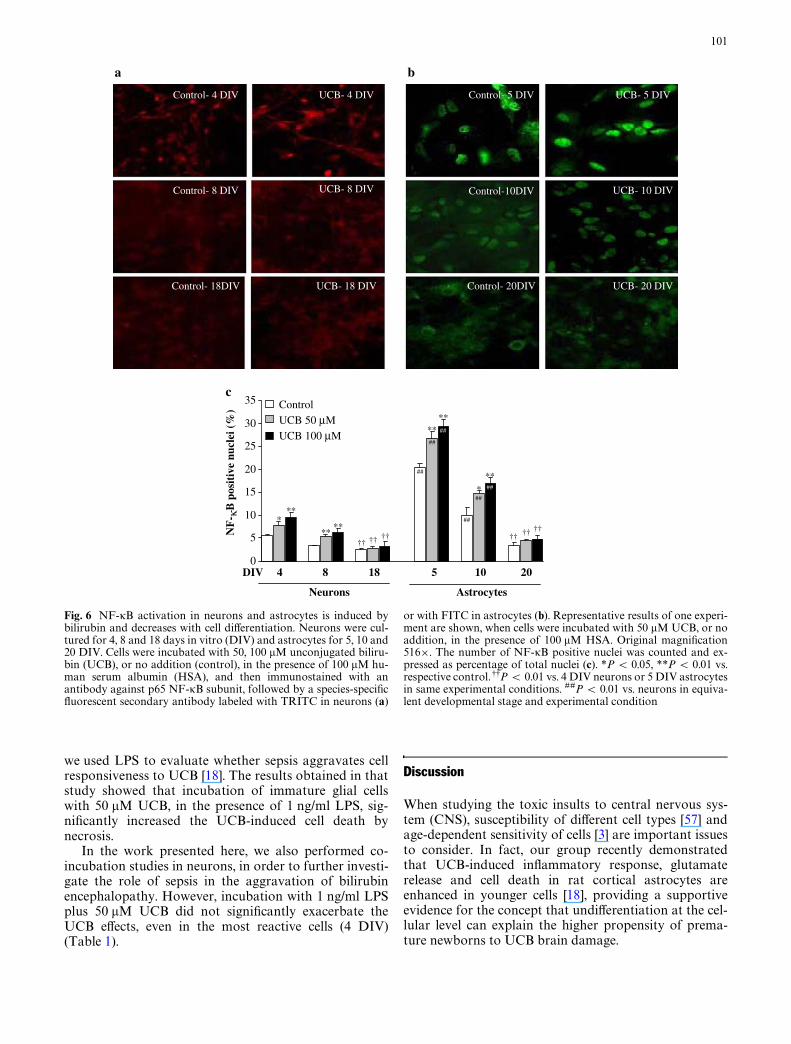
Having veriWed that UCB-induced production of TNF-�is enhanced in immature cells, particularly in astrocytes,
we hypothesized that this higher responsiveness in theimmature stage could be due to a developmentally regu-lated pattern of NF-�B activation. This transcriptionfactor is known to control the expression of multiplegenes that encode for proteins with immune and inXam-matory activities, namely cytokines [5, 51]. So, we studiedNF-�B activation by observing its translocation to thenucleus by immunocytochemistry (Fig. 6a, b). Asexpected, the results clearly showed that there is anenhanced NF-�B activation in immature cells, whichpresented a percentage of NF-�B-positive nuclei thatwas signiWcantly higher than that of the most diVerenti-ated ones (neurons, 2.2-fold; astrocytes, 6.2-fold,P < 0.01) (Fig. 6c). After incubation with 100 �M UCB,immature cells were also the ones with higher raises inNF-�B-positive nuclei (neurons, 4.0%, P < 0.01; astro-cytes, 9.0%, P < 0.01) when compared with more diVer-entiated ones (neurons, 0.7%; astrocytes, 1.5%), where nosigniWcant diVerences were observed. Although lessmarked, the increments observed in NF-�B-positivenuclei when cells were incubated with 50 �M UCB werealso higher in the immature state (neurons, 2.4%,P < 0.05; astrocytes, 6.5%, P < 0.01).
Interestingly, when compared to neurons, astrocytespresented a much higher number of NF-�B-positivenuclei, namely in the immature stage, with control valuesthat were 3.8-fold higher (P < 0.01). However, as cellsgrow old, this diVerence becomes less noticeable.
Bilirubin-induced neuronal cell reactivity is not signiWcantly aVected by LPS
Sepsis has been considered a signiWcant risk factor forbilirubin encephalopathy [32, 37, 41]. In our previouswork, performed with astrocytes at diVerent cell ages,
Fig. 4 TNF-� secretion in neurons and astrocytes is induced by bil-irubin and decreases with cell diVerentiation. Neurons were culturedfor 4, 8 and 18 days in vitro (DIV) and astrocytes for 5, 10 and 20DIV, and cells were incubated with 50, 100 �M unconjugated biliru-bin (UCB), or no addition (control), in the presence of 100 �M hu-man serum albumin. *P < 0.05, **P < 0.01 vs. respective control.99P < 0.01 vs. 4 DIV neurons or 5 DIV astrocytes in the same exper-imental conditions. #P < 0.05, ##P < 0.01 vs. neurons in equivalentdevelopmental stage and experimental condition
0
20
40
60
80
100
120T
NF
-α (p
g/10
5 ce
lls)
DIV 4 8 18 5 10 20
Neurons Astrocytes
* * **
****
* **
* *
†† ††
†† ††
** ††
††
##
####
##
####
#
####
Control
UCB 50 µM
UCB 100 µM
Fig. 5 IL-6 secretion is slightly induced by bilirubin in neuronswhile it is inhibited in astrocytes, varying with the diVerentiationstate. Neurons were cultured for 4, 8 and 18 days in vitro (DIV) andastrocytes for 5, 10 and 20 DIV, and cells were incubated with 50,100 �M unconjugated bilirubin (UCB), or no addition (control), inthe presence of 100 �M human serum albumin. *P < 0.05,**P < 0.01 vs. respective control. 99P < 0.01 vs. 4 DIV neurons or 5DIV astrocytes in the same experimental conditions. #P < 0.05,##P < 0.01 vs. neurons in equivalent developmental stage andexperimental condition
0
20
40
60
80
100
120
IL-6
(pg
/105
cells
)
DIV 4 8 18 5 10 20
Neurons Astrocytes
***
* *** **
†† ††
††
##
####
##
##
###
Control
UCB 50µM
UCB 100 µM
101
we used LPS to evaluate whether sepsis aggravates cellresponsiveness to UCB [18]. The results obtained in thatstudy showed that incubation of immature glial cellswith 50 �M UCB, in the presence of 1 ng/ml LPS, sig-niWcantly increased the UCB-induced cell death bynecrosis.
In the work presented here, we also performed co-incubation studies in neurons, in order to further investi-gate the role of sepsis in the aggravation of bilirubinencephalopathy. However, incubation with 1 ng/ml LPSplus 50 �M UCB did not signiWcantly exacerbate theUCB eVects, even in the most reactive cells (4 DIV)(Table 1).
Discussion
When studying the toxic insults to central nervous sys-tem (CNS), susceptibility of diVerent cell types [57] andage-dependent sensitivity of cells [3] are important issuesto consider. In fact, our group recently demonstratedthat UCB-induced inXammatory response, glutamaterelease and cell death in rat cortical astrocytes areenhanced in younger cells [18], providing a supportiveevidence for the concept that undiVerentiation at the cel-lular level can explain the higher propensity of prema-ture newborns to UCB brain damage.
Fig. 6 NF-�B activation in neurons and astrocytes is induced bybilirubin and decreases with cell diVerentiation. Neurons were cul-tured for 4, 8 and 18 days in vitro (DIV) and astrocytes for 5, 10 and20 DIV. Cells were incubated with 50, 100 �M unconjugated biliru-bin (UCB), or no addition (control), in the presence of 100 �M hu-man serum albumin (HSA), and then immunostained with anantibody against p65 NF-�B subunit, followed by a species-speciWcXuorescent secondary antibody labeled with TRITC in neurons (a)
or with FITC in astrocytes (b). Representative results of one experi-ment are shown, when cells were incubated with 50 �M UCB, or noaddition, in the presence of 100 �M HSA. Original magniWcation516£. The number of NF-�B positive nuclei was counted and ex-pressed as percentage of total nuclei (c). *P < 0.05, **P < 0.01 vs.respective control. 99P < 0.01 vs. 4 DIV neurons or 5 DIV astrocytesin same experimental conditions. ##P < 0.01 vs. neurons in equiva-lent developmental stage and experimental condition
a
Control- 4 DIV
Control- 8 DIV
Control- 18DIV
UCB- 4 DIV
UCB- 8 DIV
UCB- 18 DIV
b
Control-10DIV
Control- 20DIV
UCB- 10 DIV
UCB- 20 DIV
Control- 5 DIV UCB- 5 DIV
c
0
5
10
15
20
25
30
35
NF
- κB
pos
itiv
e nu
clei
(%
)
DIV 4 8 18 5 10 20
Neurons Astrocytes
***
**
****
*
†† †† ††
**
††††
##
##
##
**
††
##
##
##
Control UCB 50 µMUCB 100 µM

102
In the present study, we evaluated how neuronsbehave when exposed to UCB, and used neuron culturesat diVerent developmental stages to establish the role ofcell diVerentiation in neuronal sensitivity to the insult.Astrocytes were also used and the results compared tothose obtained in neurons in order to assess the varia-tions in cell reactivity to the same toxic stimulus.
Regarding cell death, our results showed that imma-ture neurons and astrocytes are more vulnerable to bothapoptosis and necrosis, when compared with the olderones. In fact, the importance of cell death during devel-opment, as a process of neurogenesis regulation is welldocumented [33, 62]. This phenomenon may beexplained, in part, by an age-dependent caspase-3 activa-tion [35, 78, 79], which may be implicated in the higherlevels of apoptosis observed in controls of immaturecells. Additionally, these cells are also exposed to othermolecules that are developmentally released and mayinjure cells, such as the excitotoxic glutamate, which mayaccount for the increase in necrotic cell death [36] inimmature neurons. This greater sensitivity of immaturecells was maintained when cells were exposed to UCB,which further contribute to an increase in apoptotic celldeath and necrosis [19, 29]. Studies concerning the UCB-induced apoptosis have demonstrated that this processalso involves caspase-3 activation [60] as well as hyper-phosphorylation of p38 MAP kinase [40]. Consequently,and as expected, immature cells were the ones most vul-nerable to this insult, with the highest increases in celldeath. Curiously, while less-diVerentiated neurons seemto degenerate more by an apoptotic-like pathway,“young” astrocytes are destructed mostly by a necrotic-similar one. This is in agreement with the concept thatUCB toxicity to astrocytes and neurons occurs by diVer-ent pathways, as stressed in more diVerentiated cells andhigher UCB concentrations [70]. It should be mentioned,however, that the incubation conditions used weredesigned to avoid high levels of cell death, in order toalso evaluate alterations in cell functionality.
Concerning glutamate, there are relatively few studiesabout the relationship between this excitotoxic moleculeand development or aging, and these are mainly per-formed in astrocytes, the cells primarily involved in glu-tamate uptake and neuronal metabolic support in CNS[34]. Our results clearly show that UCB-induced
glutamate release is markedly enhanced in immaturecells, resulting in higher levels of this molecule in theextracellular space. This fact may also explain the highervulnerability of undiVerentiated cells to UCB toxicity,namely in neurons, since the exposure to this excitotoxicstimulus may lead to higher rates of cell death [27] andthe consequent brain damage. Our group already showedthat the UCB-induced enhancement of glutamate in theextracellular space may be due, at least in part, to aninhibition of glutamate uptake [70]. This uptake reduc-tion was observed not only in astrocytes, the main cellsperforming this function, but also in neurons, althoughless markedly. This fact is in line with the results pre-sented here, where UCB-exposed astrocytes presenthigher concentrations of glutamate in the extracellularmedium, when compared with neurons. To this fact, shallwe account the higher capacity of astrocytes to toleratethis excitotoxic molecule, since they can easily metabo-lize glutamate to glutamine that has no transmitter activ-ity [34]. It is interesting to notice that while secretion ofglutamate by exposure of astrocytes and neurons toUCB occurs by a concentration-dependent mechanism,other UCB eVects, like apoptosis, were not clearly dose-dependent, which points to distinct mechanisms of inter-action.
Cytokines play a signiWcant role in several importantprocesses during brain development, including neuronalsurvival, cellular diVerentiation, synaptic regulation andneuritic growth [45]. Recently, we have described theimmunostimulant role of UCB in cultured astrocytes[19]. This Wnding assumes a special relevance since feverepisodes during acute bilirubin encephalopathy havebeen observed [38], and the presence of extracellular andintracellular brain edema has been identiWed in kernic-teric patients at autopsy [55, 75]. Although the neuropa-thology of kernicterus has not been associated with thepresence of inXammation, there are other subtle neuro-logical abnormalities associated with hyperbilirubinemiaand a great number of autopsied jaundiced newbornsonly reveal diVusely yellow spots in the brain [31], inwhich the underlying molecular mechanisms of UCBinjury are unknown. In addition, some papers connectneonatal hyperbilirubinemia and Gilbert’s syndromewith the development of mental illness, namely schizo-phrenia [16, 46]. Moreover, prenatal exposure to infec-tion has been associated with the risk for schizophrenia[24] and reduction in dendrite development by cytokinesis consistent with the neuropathology of schizophrenia[25]. Interestingly, we found that an early exposure toUCB leads to reduction in dendrite extension and ramiW-cation (personal communication). Thus, it is becomingapparent that the release of cytokines due to UCBimmunostimulation may trigger long-term neuropatho-logical eVects. Therefore, we were also interested ininvestigating if there is a cell-age-dependent pattern ofcytokine secretion and whether it is aVected by UCBexposure.
Several reports indicate that neurons produce TNF-�[9, 23] and IL-6 [58, 66] in low levels, suggesting that
Table 1 EVect of co-incubation of unconjugated bilirubin (UCB)and lipopolysaccharide (LPS) vs. UCB alone (fold change). Reactiv-ity of neurons to bilirubin does not increase by co-incubation withLPS
Neurons at 4 days in vitro were incubated with 50 �M UCB and1 ng/ml LPS, in the presence of 100 �M human serum albumin, asdescribed in Sect. ”Materials and methods”
Apoptosis 1.02Necrosis 1.01Glutamate 1.02TNF-� 1.03IL-6 1.10NF-�B 1.07

103
these cells can also participate in some inXammatoryresponse activities in the brain. TNF-� is known to betransiently expressed in high levels in the immatureembryonic stage [23, 45], being involved in the develop-ment of the CNS. In neuronal cells, this cytokine canpromote diVerentiation and neuritic growth [48], while inastrocytes, it also induces proliferation [7, 67] and stimu-lates the synthesis of some neurotrophic factors, such asnerve-growth factor [22]. Accordingly, we observedhigher levels of TNF-� secretion in less-diVerentiatedcells, where proliferation is still occurring, both in astro-cytes and neurons, although astrocytes were much morereactive.
We also observed that both cells participate in UCB-induced inXammatory response, although with diVerentcytokine levels. However, it remains to be clariWed if thisresponse involves the same signaling pathways for bothcells. These quantitative diVerences observed in cytokineproduction between astrocytes and neurons were just aswe expected, since astrocytes are considered to be themost important cells in the CNS cytokine network, alongwith microglia, providing structural and environmentalsupport for neurons [65]. This UCB-induced release ofcytokines by astrocytes appears to involve the activationof MAP kinases [20], which ultimately leads to apoptosis[53]. In addition, we have observed that speciWc inhibi-tors of MAP kinases reduce UCB-induced apoptosis(unpublished observations).
IL-6 is also implicated in neuronal development,inducing diVerentiation of immature human and mouseneuroblastomas [47, 48], and acting as a developmentalneurotrophic factor in astrocytes [21]. Interestingly, ourresults did not show an increase on IL-6 production inimmature neurons, even after UCB incubation. Substan-tiating these observations, it is described that neuronsare capable of producing IL-6, but at lower basal levels[9], which only signiWcantly increase when neurons arestimulated with high doses of IL-1� and/or TNF-� [58].However, in neuronal extracellular medium and in allexperimental conditions, the levels of TNF-� were verylow and IL-1� was undetectable, and therefore, insuY-cient to induce IL-6 secretion by neurons. In opposite,basal levels of IL-6 in astrocytes are much higher thanthose of neurons and are reduced following UCB expo-sure at all cell-diVerentiation states, being the young cellsthe most responsive ones. We can speculate that IL-6production by astrocytes in an early diVerentiation statecan participate in the induction of neuronal diVerentia-tion, acting as a neurotrophic growth factor [65]. If so,the interaction of UCB in early life could aVect the neu-ronal normal development, a feature that may have animportant impact in the development of the CNS.
The distinct cytokine-secretion proWle observed inastrocytes and neurons led us to hypothesize that possi-bly there are also some diVerences in the UCB-inducedand developmental activation of some transcription fac-tors, like NF-�B, that positively regulate the induction ofinXammatory genes [15, 74]. It is described that thistranscription factor plays an important role in the
diVerentiation of the CNS, presenting a developmentallyregulated pattern [4, 14]. Accordingly, our results haveshown that in baseline controls of astrocytes and neu-rons, the activation of this transcription factor washighly enhanced in immature cells, as compared with themore diVerentiated ones.
In our most recent report, we demonstrated that stim-ulation of astrocytes with UCB activates NF-�B [20],and inhibition of NF-�B pathway prevents UCB-stimu-lated cytokine release by astrocytes (unpublished obser-vations). Here, we extended those studies to evaluatewhether this UCB-induced NF-�B activation also pre-sents a cell-age-dependent proWle.
The action of NF-�B depends from the nerve-celltype. In fact, NF-�B activation in neurons has been gen-erally involved in synaptic plasticity, neuronal function,development and survival [42, 51], whereas in astrocytesit has been mainly related to the inXammatory response,inducing the production of TNF-�, Il-1� and IL-6 [6, 64].Interestingly, the quantitative diVerences observed in theNF-�B-activation proWle between astrocytes and neu-rons are similar to the ones observed in TNF-� produc-tion. This observation reinforces the fact that astrocytesplay an important role in the UCB-induced inXamma-tory response, mainly in the less-diVerentiated cells, andmay explain the higher susceptibility of premature new-borns to UCB encephalopathy. In neurons, NF-�B acti-vation is probably related to UCB-induced cell injury. Infact, in our ongoing studies, we already observed thatUCB toxicity is associated with neuronal oxidative dam-age as well as disruption of glutathione metabolism [11],events that are known to be important stimuli for NF-�Bactivation [51]. However, more studies have to be per-formed in order to clarify the roles of NF-�B in theUCB-induced cell- injury and/or -recovery process.
In our previous report [18], we have observed that inimmature astrocytes, LPS signiWcantly increased theUCB-induced cell death by necrosis, pointing out thathyperbilirubinemia should be monitored more carefullyin premature newborns with infection by bacteria. Con-versely, LPS is not capable of aggravating the neuronalresponse induced by UCB, probably because LPS exertsits eVects by producing an inXammatory response [12,13], which is not particularly relevant in neurons, inaccordance with the present results.
In conclusion, with this work we demonstrate thatimmature neurons and astrocytes are more vulnerable toUCB-induced cell death, glutamate release and TNF-�secretion than the older ones. Moreover, the diVerentia-tion-dependent proWle of cytokine secretion is reXectedby the pattern of NF-�B activation following UCB expo-sure. Accordingly, the greater amount of cytokine secre-tion by astrocytes is correlated with the highestactivation levels observed in this cell type. Hence, we canspeculate that UCB toxicity in neurons is accomplishedmainly through an increase in cell death both by necrosisand apoptosis, whereas the mechanisms that underlieastrocyte injury, are mainly related to excitotoxicity andto inXammatory signaling pathways.

104
Altogether, these results shed some light on the mech-anisms underlying UCB neurotoxicity by discriminatingthe eVects induced by this molecule in diVerent nerve-celltypes and provide an additional supportive evidence forthe higher susceptibility of premature babies to UCBneurotoxicity.
Acknowledgments This work was supported by grants POCI/39906/FCB/2001 and POCI/SAU-MMO/55955/2004, from Fun-dação para a Ciência e a Tecnologia (FCT), Lisbon, Portugal, andFEDER (to D.B.), and Ph.D. Fellowships SFRH/BD/8436/2002 andSFRH/BD/9204/2002 from FCT (to A.S.F. and A.F.)
References
1. Abney ER, Bartlett PP, RaV MC (1981) Astrocytes, ependymalcells, and oligodendrocytes develop on schedule in dissociatedcell cultures of embryonic rat brain. Dev Biol 83:301–310
2. American Academy of Pediatrics (2004) Management of hyper-bilirubinemia in the newborn infant 35 or more weeks of gesta-tion. Pediatrics 114:297–316
3. Amit Y, Brenner T (1993) Age-dependent sensitivity of culturedrat glial cells to bilirubin toxicity. Exp Neurol 121:248–255
4. Bakalkin GY, Yakovleva T, Terenius L (1993) NF-�B-like fac-tors in the murine brain. Developmentally-regulated and tissue-speciWc expression. Brain Res Mol Brain Res 20:137–146
5. Baldwin AS Jr (1996) The NF-�B and I�B proteins: new discov-eries and insights. Annu Rev Immunol 14:649–683
6. Bales KR, Du Y, Dodel RC, Yan GM, Hamilton-Byrd E, PaulSM (1998) The NF-�B/Rel family of proteins mediates A�-in-duced neurotoxicity and glial activation. Brain Res Mol BrainRes 57:63–72
7. Barna BP, Estes ML, Jacobs BS, Hudson S, RansohoV RM(1990) Human astrocytes proliferate in response to tumor necro-sis factor �. J Neuroimmunol 30:239–243
8. Blondeau JP, Beslin A, Chantoux F, Francon J (1993) Triiodo-thyronine is a high-aYnity inhibitor of amino acid transport sys-tem L1 in cultured astrocytes. J Neurochem 60:1407–1413
9. Breder CD, Tsujimoto M, Terano Y, Scott DW, Saper CB(1993) Distribution and characterization of tumor necrosis fac-tor-alpha-like immunoreactivity in the murine central nervoussystem. J Comp Neurol 337:543–567
10. Brewer GJ (1997) Isolation and culture of adult rat hippocam-pal neurons. J Neurosci Methods 71:143–155
11. Brito MA, Rosa AI, Fernandes A, Falcão AS, Silva RFM, BritesD (2005) Hyperbilirubinemia induces oxidative stress in ratbrain primary neuronal cultures. Free Radic Res 39(Suppl.1):S52
12. Cai Z, Pan ZL, Pang Y, Evans OB, Rhodes PG (2000) Cytokineinduction in fetal rat brains and brain injury in neonatal rats af-ter maternal lipopolysaccharide administration. Pediatr Res47:64–72
13. Cai Z, Pang Y, Lin S, Rhodes PG (2003) DiVerential roles of tu-mor necrosis factor-� and interleukin-1� in lipopolysaccharide-induced brain injury in the neonatal rat. Brain Res 975:37–47
14. Cauley K, Verma IM (1994) �B enhancer-binding complexesthat do not contain NF-�B are developmentally regulated inmammalian brain. Proc Natl Acad Sci USA 91:390–394
15. Da SJ, Pierrat B, Mary JL, Lesslauer W (1997) Blockade of p38mitogen-activated protein kinase pathway inhibits inducible ni-tric-oxide synthase expression in mouse astrocytes. J Biol Chem272:28373–28380
16. Dalman C, Cullberg J (1999) Neonatal hyperbilirubinaemia—avulnerability factor for mental disorder? Acta Psychiatr Scand100:469–471
17. Dennery PA, Seidman DS, Stevenson DK (2001) Neonatal hyp-erbilirubinemia. N Engl J Med 344:581–590
18. Falcão AS, Fernandes A, Brito MA, Silva RFM, Brites D (2005)Bilirubin-induced inXammatory response, glutamate release,and cell death in rat cortical astrocytes are enhanced in youngercells. Neurobiol Dis 20:199–206
19. Fernandes A, Silva RFM, Falcão AS, Brito MA, Brites D (2004)Cytokine production, glutamate release and cell death in rat cul-tured astrocytes treated with unconjugated bilirubin and LPS. JNeuroimmunol 153:64–75
20. Fernandes A, Falcão AS, Silva RFM, Gordo AC, Gama MJ,Brito MA, Brites D (2006) InXammatory signaling pathways in-volved in astroglial activation by unconjugated bilirubin. J Neu-rochem 96:1667–1679
21. Gadient RA, Otten U (1994) Expression of interleukin-6 (IL-6)and interleukin-6 receptor (IL-6R) mRNAs in rat brain duringpostnatal development. Brain Res 637:10–14
22. Gadient RA, Cron KC, Otten U (1990) Interleukin-1� and tu-mor necrosis factor-� synergistically stimulate nerve growth fac-tor (NGF) release from cultured rat astrocytes. Neurosci Lett117:335–340
23. Gendron RL, Nestel FP, Lapp WS, Baines MG (1991) Expres-sion of tumor necrosis factor-� in the developing nervous sys-tem. Int J Neurosci 60:129–136
24. Gilmore JH, Jarskog LF, Vadlamudi S, Lauder JM (2004) Pre-natal infection and risk for schizophrenia: IL-1�, IL-6 and TNF-� inhibit cortical neuron dendrite development. Neuropsycho-pharmacology 29:1221–1229
25. Grima G, Benz B, Parpura V, Cuenod M, Do KQ (2003) Dopa-mine-induced oxidative stress in neurons with glutathione deW-cit: implication for schizophrenia. Schizophr Res 62:213–224
26. Gourley GR (1997) Bilirubin metabolism and kernicterus. AdvPediatr 44:173–229
27. Grojean S, Koziel V, Vert P, Daval JL (2000) Bilirubin inducesapoptosis via activation of NMDA receptors in developing ratbrain neurons. Exp Neurol 166:334–341
28. Hanisch UK (2002) Microglia as a source and target of cyto-kines. Glia 40:140–155
29. Hankø E, Hansen TWR, Almaas R, Lindstad J, Rootwelt T(2005) Bilirubin induces apoptosis and necrosis in human NT2-N neurons. Pediatr Res 57:179–184
30. Hansen TWR (1995) Acute entry into rat brain regions. BiolNeonate 67:203–207
31. Hansen TWR (2000) Pioneers in the scientiWc study of neonataljaundice and kernicterus. Pediatrics 106:e15
32. Hansen TWR, Maynard EC, Cashore WJ, Oh W (1993) Endo-toxemia and brain bilirubin in the rat. Biol Neonate 63:171–176
33. Haydar TF, Kuan CY, Flavell RA, Rakic P (1999) The role ofcell death in regulating the size and shape of the mammalianforebrain. Cereb Cortex 9:621–626
34. Hertz L, Dringen R, Schousboe A, Robinson SR (1999) Astro-cytes: glutamate producers for neurons. J Neurosci Res 57:417–428
35. Hong HN, Yoon SY, Suh J, Lee JH, Kim D (2002) DiVerentialactivation of caspase-3 at two maturational stages during oka-daic acid-induced rat neuronal death. Neurosci Lett 334:63–67
36. Hutchins JB, Barger SW (1998) Why neurons die: cell death inthe nervous system. Anat Rec 253:79–90
37. Kaplan M, Hammerman C (2004) Understanding and prevent-ing severe neonatal hyperbilirubinemia: is bilirubin neurotoxityreally a concern in the developed world? Clin Perinatol 31:555–575
38. Kaplan M, Hammerman C (2005) Understanding severe neona-tal hyperbilirubinemia and preventing kernicterus: adjuncts inthe interpretation of neonatal serum bilirubin. Clin Chim Acta356:9–21
39. Kawade N, Onishi S (1981) The prenatal and postnatal develop-ment of UDP-glucuronyltransferase activity towards bilirubinand the eVect of premature birth on this activity in the humanliver. Biochem J 196:257–260
40. Lin S, Yan C, Wei X, Paul SM, Du Y (2003) p38 MAP kinasemediates bilirubin-induced neuronal death of cultured rat cere-bellar granule neurons. Neurosci Lett 353:209–212

105
41. Lucey JF (1972) The unsolved problem of kernicterus in the sus-ceptible low birth weight infant. Pediatrics 49:646–647
42. Mattson MP, Culmsee C, Yu Z, Camandola S (2000) Roles ofnuclear factor �B in neuronal survival and plasticity. J Neuro-chem 74:443–456
43. McCarthy KD, de Vellis J (1980) Preparation of separate as-troglial and oligodendroglial cell cultures from rat cerebral tis-sue. J Cell Biol 85:890–902
44. McDonagh AF, Assisi F (1972) The ready isomerization of bili-rubin IX-� in aqueous solution. Biochem J 129:797–800
45. Mehler MF, Kessler JA (1997) Hematolymphopoietic andinXammatory cytokines in neural development. Trends Neuro-sci 20:357–365
46. Miyaoka T, Seno H, Itoga M, Iijima M, Inagaki T, Horiguchi J(2000) Schizophrenia-associated idiopathic unconjugated hyper-bilirubinemia (Gilbert’s syndrome). J Clin Psychiatry 61:868–871
47. Muñoz-Fernandez MA, Armas-Portela R, Diaz-Nido J, AlonsoJL, Fresno M, Avila J (1991) DiVerential eVects of tumor necro-sis factor on the growth and diVerentiation of neuroblastomaand glioma cells. Exp Cell Res 194:161–164
48. Muñoz-Fernandez MA, Cano E, O’Donnell CA, Doyle J, LiewFY, Fresno M (1994) Tumor necrosis factor-� (TNF-�), inter-feron-�, and interleukin-6 but not TNF-� induce diVerentiationof neuroblastoma cells: the role of nitric oxide. J Neurochem62:1330–1336
49. Nakamura Y, Ohmaki M, Murakami K, Yoneda Y (2003)Involvement of protein kinase C in glutamate release from cul-tured microglia. Brain Res 962:122–128
50. Notter MF, Kendig JW (1986) DiVerential sensitivity of neuralcells to bilirubin toxicity. Exp Neurol 94:670–682
51. O’Neill LA, Kaltschmidt C (1997) NF-�B: a crucial transcrip-tion factor for glial and neuronal cell function. Trends Neurosci20:252–258
52. Oh W, Tyson JE, FanaroV AA, Vohr BR, Perritt R, Stoll BJ, Eh-renkranz RA, Carlo WA, Shankaran S, Poole K, Wright LL(2003) Association between peak serum bilirubin and neurode-velopmental outcomes in extremely low birth weight infants.Pediatrics 112:773–779
53. Oh HL, Seok JY, Kwon CH, Kang SK, Kim YK (2006) Role ofMAPK in ceramide-induced cell death in primary cultured astro-cytes from mouse embryonic brain. Neurotoxicology 27:31–38
54. Ostrow JD, Pascolo L, Brites D, Tiribelli C (2004) Molecular basisof bilirubin-induced neurotoxicity. Trends Mol Med 10:65–70
55. Perlman JM, Rogers BB, Burns D (1997) Kernicteric Wndings atautopsy in two sick near term infants. Pediatrics 99:612–615
56. Porter ML, Dennis BL (2002) Hyperbilirubinemia in the termnewborn. Am Fam Physician 65:599–606
57. Pulsinelli WA (1985) Selective neuronal vulnerability: morpho-logical and molecular characteristics. Prog Brain Res 63:29–37
58. Ringheim GE, Burgher KL, Heroux JA (1995) Interleukin-6mRNA expression by cortical neurons in culture: evidence forneuronal sources of interleukin-6 production in the brain. JNeuroimmunol 63:113–123
59. Ritter DA, Kenny JD, Norton HJ, Rudolph AJ (1982) A pro-spective study of free bilirubin and other risk factors in thedevelopment of kernicterus in premature infants. Pediatrics69:260–266
60. Rodrigues CMP, Solá S, Brites D (2002) Bilirubin induces apop-tosis via the mitochondrial pathway in developing rat brain neu-rons. Hepatology 35:1186–1195
61. Romijn HJ, Hofman MA, Gramsbergen A (1991) At what age isthe developing cerebral cortex of the rat comparable to that ofthe full-term newborn human baby? Early Hum Dev 26:61–67
62. Roth KA, D’Sa C (2001) Apoptosis and brain development.Ment Retard Dev Disabil Res Rev 7:261–266
63. Rubaltelli FF, GriYth PF (1992) Management of neonatal hyp-erbilirubinaemia and prevention of kernicterus. Drugs 43:864–872
64. Sanz O, Acarin L, Gonzalez B, Castellano B (2002) NF-�B andI�B� expression following traumatic brain injury to the imma-ture rat brain. J Neurosci Res 67:772–780
65. Sawada M, Suzumura A, Marunouchi T (1995) Cytokine net-work in the central nervous system and its roles in growth anddiVerentiation of glial and neuronal cells. Int J Dev Neurosci13:253–264
66. Schobitz B, Voorhuis DA, De Kloet ER (1992) Localization ofinterleukin 6 mRNA and interleukin 6 receptor mRNA in ratbrain. Neurosci Lett 136:189–192
67. Selmaj KW, Farooq M, Norton WT, Raine CS, Brosnan CF(1990) Proliferation of astrocytes in vitro in response to cyto-kines. A primary role for tumor necrosis factor. J Immunol144:129–135
68. Shapiro SM (2005) DeWnition of the clinical spectrum of kern-icterus and bilirubin-induced neurologic dysfunction (BIND). JPerinatol 25:54–59
69. Silva R, Mata LR, Gulbenkian S, Brito MA, Tiribelli C, BritesD (1999) Inhibition of glutamate uptake by unconjugated biliru-bin in cultured cortical rat astrocytes: role of concentration andpH. Biochem Biophys Res Commun 265:67–72
70. Silva RFM, Rodrigues CMP, Brites D (2002) Rat cultured neu-ronal and glial cells respond diVerently to toxicity of unconju-gated bilirubin. Pediatr Res 51:535–541
71. Silva RFM, Falcão AS, Fernandes A, Gordo AC, Brito MA,Brites D (2006) Cell cultures as models for assessment of neuro-toxicity. Toxicol Lett 163:1–9
72. Soorani-Lunsing I, Woltil HA, Hadders-Algra M (2001) Aremoderate degrees of hyperbilirubinemia in healthy term neo-nates really safe for the brain? Pediatr Res 50:701–705
73. Stevenson DK, Dennery PA, Hintz SR (2001) Understandingnewborn jaundice. J Perinatol 21(Suppl. 1):S21–S24
74. Tak PP, Firestein GS (2001) NF-�B: a key role in inXammatorydiseases. J Clin Invest 107:7–11
75. Turkel SB, Miller CA, Guttenberg ME, Moynes DR, GodgmanJE (1982) A clinical pathologic reappraisal of kernicterus. Pedi-atrics 69:267–272
76. Watchko JF, Maisels MJ (2003) Jaundice in low birthweight in-fants: pathobiology and outcome. Arch Dis Child Fetal Neona-tal Ed 88:F455–F458
77. Watchko JF, Daood MJ, Biniwale M (2002) Understandingneonatal hyperbilirubinaemia in the era of genomics. SeminNeonatol 7:143–152
78. Xu L, Chock VY, Yang EY, GiVard RG (2004) Susceptibility toapoptosis varies with time in culture for murine neurons and as-trocytes: changes in gene expression and activity. Neurol Res26:632–643
79. Yakovlev AG, Ota K, Wang G, Movsesyan V, Bao WL, Yoshi-hara K, Faden AI (2001) DiVerential expression of apoptoticprotease-activating factor-1 and caspase-3 genes and suscepti-bility to apoptosis during brain development and after trau-matic brain injury. J Neurosci 21:7439–7446
Copyright © 2022 FDOKUMEN




















