
Bile acids reduce SR-BI expression in hepatocytes by a pathway involving FXR/RXR, SHP, and LRH-1
10
Bile acids reduce SR-BI expression in hepatocytes by a pathway involving FXR/RXR, SHP, and LRH-1 q Lene Malerød a,1 , Marita Sporstøl a, * ,1 , Lene K. Juvet b , Seyed Ali Mousavi a , Tor Gjøen c , Trond Berg a , Norbert Roos a , Winnie Eskild a a Programme for Cell Biology, Department of Molecular Biosciences, University of Oslo, Norway b Institute of Nutrition Research, University of Oslo, Norway c Division of Pharmacy, Institute of Pharmacy, University of Oslo, Norway Received 8 August 2005 Available online 9 September 2005 Abstract Hepatic SR-BI mediates uptake of circulating cholesterol into liver hepatocytes where a part of the cholesterol is metabolised to bile acids. In the hepatocytes, bile acids reduce their own synthesis by a negative feedback loop to prevent toxic high levels of bile acids. Bile acid- activated FXR/RXR represses expression of CYP7A1, the rate-limiting enzyme during bile acid synthesis, by inducing the expression of SHP, which inhibits LXR/RXR and LRH-1-transactivation of CYP7A1. The present paper presents data indicating that CDCA suppresses SR-BI expression by the same pathway. As previously reported, LRH-1 induces SR-BI promoter activity. Here we show that CDCA or over- expression of SHP inhibit this transactivation. No FXR-response element was identified in the bile acid-responsive region of the SR-BI pro- moter (1200 bp/937 bp). However, a binding site for LRH-1 was characterised and shown to specifically bind LRH-1. The present study shows that also the SR-BI-mediated supply of cholesterol, the substrate for bile acid synthesis, is feedback regulated by bile acids. Ó 2005 Elsevier Inc. All rights reserved. Keywords: SR-BI; Liver; Bile acids; FXR; LRH-1; SHP Excess cellular cholesterol in extrahepatic tissues is transferred to plasma HDL and transported to the liver in a process called reverse cholesterol transport (reviewed in [1]). A crucial factor in this pathway is scavenger recep- tor class B, type I (SR-BI). SR-BI was cloned in 1994 as the receptor mediating selective uptake of HDL-cholesterol into liver, adrenals, testes, and ovaries [2–4]. In steroido- genic cells, SR-BI supplies cholesterol required for steroid hormone synthesis, and the receptor is regulated in parallel with steroidogenesis by adrenocorticotropic hormone [5–7], human chorionic gonadotropin [4,8], and estradiol [9]. In the liver, SR-BI is responsible for hepatic uptake of HDL-cholesterol in the reverse cholesterol transport [10]. HDL also mediates transport of cholesterol from non-parenchymal liver cells (liver endothelial cells and Kupffer cells) to the hepatocytes, the main site for catabo- lism of cholesterol [11–14]. Estradiol has been shown to de- crease rat hepatocytic SR-BI levels, and at the same time upregulate SR-BI levels in Kupffer cells in vivo [4,13,15]. Hepatic SR-BI expression is regulated by fatty acids [16,17], cholesterol, estradiol [13], vitamin E [18], and li- gands of liver X receptor (LXR) (oxysterols) [19]. Further- more, it has been shown that activators of peroxisome proliferator-activated receptor a (PPARa) reduce [20], whereas PPARc stimulates hepatic SR-BI expression [21]. In hepatocytes, cholesterol delivered to the liver is used for synthesis of new lipoproteins or conversion to bile acids 0006-291X/$ - see front matter Ó 2005 Elsevier Inc. All rights reserved. doi:10.1016/j.bbrc.2005.08.237 q Abbreviations: CDCA, chenodeoxycholic acid; CETP, cholesteryl ester transfer protein; CYP7A1, cholesterol 7a-hydroxylase; CYP27, sterol 27- hydroxylase; FXR, farnesoid X receptor; HMG-CoA, 3-hydroxy-3- methylglutaryl coenzyme A; LRH-1, liver receptor homologue-1; LXR, liver X receptor; RXR, 9-cis-retinoic acid receptor; SHP, small heterodi- mer partner; SR-BI, scavenger receptor class B, type I; TCA, taurocholate. * Corresponding author. Fax: +47 22854605. E-mail address: [email protected] (M. Sporstøl). 1 These authors contributed equally to the present work. www.elsevier.com/locate/ybbrc Biochemical and Biophysical Research Communications 336 (2005) 1096–1105 BBRC
Transcript of Bile acids reduce SR-BI expression in hepatocytes by a pathway involving FXR/RXR, SHP, and LRH-1

www.elsevier.com/locate/ybbrc
Biochemical and Biophysical Research Communications 336 (2005) 1096–1105
BBRC
Bile acids reduce SR-BI expression in hepatocytes by a pathwayinvolving FXR/RXR, SHP, and LRH-1q
Lene Malerød a,1, Marita Sporstøl a,*,1, Lene K. Juvet b, Seyed Ali Mousavi a, Tor Gjøen c,Trond Berg a, Norbert Roos a, Winnie Eskild a
a Programme for Cell Biology, Department of Molecular Biosciences, University of Oslo, Norwayb Institute of Nutrition Research, University of Oslo, Norway
c Division of Pharmacy, Institute of Pharmacy, University of Oslo, Norway
Received 8 August 2005Available online 9 September 2005
Abstract
Hepatic SR-BImediates uptake of circulating cholesterol into liver hepatocyteswhere a part of the cholesterol ismetabolised to bile acids.In the hepatocytes, bile acids reduce their own synthesis by a negative feedback loop to prevent toxic high levels of bile acids. Bile acid-activated FXR/RXR represses expression of CYP7A1, the rate-limiting enzyme during bile acid synthesis, by inducing the expression ofSHP,which inhibits LXR/RXRandLRH-1-transactivation ofCYP7A1. The present paper presents data indicating that CDCA suppressesSR-BI expression by the samepathway.As previously reported, LRH-1 induces SR-BI promoter activity.Herewe show thatCDCAorover-expression of SHP inhibit this transactivation. No FXR-response element was identified in the bile acid-responsive region of the SR-BI pro-moter (�1200 bp/�937 bp). However, a binding site for LRH-1was characterised and shown to specifically bind LRH-1. The present studyshows that also the SR-BI-mediated supply of cholesterol, the substrate for bile acid synthesis, is feedback regulated by bile acids.� 2005 Elsevier Inc. All rights reserved.
Keywords: SR-BI; Liver; Bile acids; FXR; LRH-1; SHP
Excess cellular cholesterol in extrahepatic tissues istransferred to plasma HDL and transported to the liverin a process called reverse cholesterol transport (reviewedin [1]). A crucial factor in this pathway is scavenger recep-tor class B, type I (SR-BI). SR-BI was cloned in 1994 as thereceptor mediating selective uptake of HDL-cholesterolinto liver, adrenals, testes, and ovaries [2–4]. In steroido-genic cells, SR-BI supplies cholesterol required for steroidhormone synthesis, and the receptor is regulated in parallel
0006-291X/$ - see front matter � 2005 Elsevier Inc. All rights reserved.
doi:10.1016/j.bbrc.2005.08.237
q Abbreviations: CDCA, chenodeoxycholic acid; CETP, cholesteryl estertransfer protein; CYP7A1, cholesterol 7a-hydroxylase; CYP27, sterol 27-hydroxylase; FXR, farnesoid X receptor; HMG-CoA, 3-hydroxy-3-methylglutaryl coenzyme A; LRH-1, liver receptor homologue-1; LXR,liver X receptor; RXR, 9-cis-retinoic acid receptor; SHP, small heterodi-mer partner; SR-BI, scavenger receptor class B, type I; TCA, taurocholate.* Corresponding author. Fax: +47 22854605.E-mail address: [email protected] (M. Sporstøl).
1 These authors contributed equally to the present work.
with steroidogenesis by adrenocorticotropic hormone[5–7], human chorionic gonadotropin [4,8], and estradiol[9]. In the liver, SR-BI is responsible for hepatic uptakeof HDL-cholesterol in the reverse cholesterol transport[10]. HDL also mediates transport of cholesterol fromnon-parenchymal liver cells (liver endothelial cells andKupffer cells) to the hepatocytes, the main site for catabo-lism of cholesterol [11–14]. Estradiol has been shown to de-crease rat hepatocytic SR-BI levels, and at the same timeupregulate SR-BI levels in Kupffer cells in vivo [4,13,15].Hepatic SR-BI expression is regulated by fatty acids[16,17], cholesterol, estradiol [13], vitamin E [18], and li-gands of liver X receptor (LXR) (oxysterols) [19]. Further-more, it has been shown that activators of peroxisomeproliferator-activated receptor a (PPARa) reduce [20],whereas PPARc stimulates hepatic SR-BI expression [21].
In hepatocytes, cholesterol delivered to the liver is usedfor synthesis of new lipoproteins or conversion to bile acids

L. Malerød et al. / Biochemical and Biophysical Research Communications 336 (2005) 1096–1105 1097
by two alternative pathways, the classical/neutral pathwaycatalysed by the rate-limiting enzyme cholesterol 7a-hy-droxylase (CYP7A1), or the alternative/acidic pathwaycatalysed by sterol 27-hydroxylase (CYP27) [22,23].CYP7A1 is positively regulated by oxysterols [24–27]whereas both CYP7A1 and CYP27 are negatively feedbackregulated by bile acids [28–34]. Bile acids repress their ownsynthesis by activating FXR/RXR [27,35–37], which indi-rectly suppresses CYP7A1 via induction of SHP expression[31,38]. The nuclear SHP inhibits CYP7A1 expression byinactivating liver receptor homologue-1 (LRH-1) [39] andLXR/RXR [26,40], which synergistically stimulate transac-tivation of CYP7A1 [32]. SHP has also been shown todirectly inhibit LXR/RXR [41]. HDL-cholesterol is thepreferred substrate for bile acid synthesis [42], and theHDL receptor, SR-BI, is regulated in parallel with bile acidsynthesis by LXR/RXR [19]. In the present study, wetherefore asked whether the negative feedback regulationof bile acid synthesis via the FXR/SHP pathway also af-fects SR-BI expression in hepatocytes. We found that bileacids as well as the synthetic FXR-agonist GW404764 re-duced SR-BI expression in rodent and human hepatocytesby initiating a nuclear cascade similar to that operating inthe regulation of CYP7A1 transcription [29]. The FXR/RXR-heterodimer is activated by bile acids and inducesthe expression of SHP. This repressor of transcriptionbinds to LRH-1 and inhibits its activation of SR-BI expres-sion in the hepatocytes. This means that not only conver-sion of cholesterol to bile acids (by CYP7A1 and CYP27)but also the uptake of HDL-cholesterol by SR-BI is con-trolled by FXR/RXR in hepatocytes.
Materials and methods
Plasmids. Expression plasmids for human RXRa (RXRa-pCMV),SHP-pSG5, and b-galactosidase-pSV were kindly provided by Dr. HildeNebb (Institute of Nutrition Research, University of Oslo). Human LRH-1-pSG5 and FXR-pCMX were from Dr. Bryan Goodwin (NuclearReceptor Sysmtem Research, GlaxoSmithKline Research & Development,USA) and Dr. Heidi R. Kast (Department of Biological Chemistry andMedicine, University of California, Los Angeles, USA) [43], respectively.A luciferase reporter construct containing the human SR-BI promoter waskindly provided by Dr. Helen H. Hobbs (Department of MolecularGenetics, University of Texas, USA) [44]. The 1200 bp 5 0-flanking se-quence was subcloned into pGL3-Basic luciferase vector (Promega).Deletion promoter constructs were made by restriction enzyme digestion.The plasmids were sequenced to verify DNA sequence fidelity by Cyber-
Table 1Primers used for real-time RT-PCR
Gene GenBankAccession No.
Forward primer Reverse
hSR-BI NM005505 CTGTGGGTGAGATCATGTGG GCCAGmSR-BI BC004656 CAGGCTGTGGGAACTCTAGC GAAAArSR-BI NM031541 CAAGAAGCCAAGCTGTAGGG CCCAAhSHP BC030207 GCTGTCTGGAGTCCTTCTGG GAAAGmSHPa BC019540 CCCAAGATGCTGTGACCTTT TCCAGhGADPH AF261085 GGCCTCCAAGGAGTAAGACC AGGGGmGADPHa NM023964 ACCCAGAAGACTGTGGATGG CACATT
a The primer is recognizing both the murine and rat sequence.
gene AB (Sweden). The pcDNA3.1/myc-His vector (Invitrogen) was usedto prepare myc-tagged LRH-1-expression plasmid.
Animals. The rats were treated according to established guidelines forexperimental animals. Male Wistar rats (approximately 200 g) were fastedfor 24 h before they were fed standard laboratory chow (control animals)or standard diet supplemented with 1% (w/w) taurocholate (TCA) (Sigma)or 2% (w/w) cholestyramine (Sigma) for 6 days ad libitum. The animals (4per group) were anaesthetised with pentobarbital before livers were ex-cised and stored at �80 �C.
Isolation of rat primary liver cells. Primary rat hepatocytes were iso-lated as described elsewhere [45]. Cells were stimulated with the indicatedconcentrations of the primary bile acid chenodeoxycholic acid (CDCA)(Sigma), the synthetic FXR-agonist GW404764 (gift from Glaxo-SmithKline Research & Development, USA) or vehicle (96% ethanol) intheir respective media containing 0.5% charcoal-treated foetal bovineserum. Lipoprotein-depleted serum was prepared as described in [46].
Cell cultures and transient transfection. The murine hepatoma cell lineHepa 1c1c-7 and COS-7 cells were grown in DMEM supplemented with2 mM L-glutamine, 100 U penicillin, 100 lg/ml streptomycin, and 10%foetal bovine serum (medium A). HepG2 cells (a human hepatoma cellline) were cultured in medium A containing 1· non-essential amino acids.200,000 Hepa 1c1c-7 cells or COS-7 cells were cultured in six-well platesthe day before transfection. The transfection mixture contained 0.3 lgreporter construct and 0.1 lg of each nuclear receptor-expression plasmid.The amount of DNA was adjusted with carrier DNA (pcDNA3.1, Stra-tagene) to a total of 1 lg. In each assay, 0.5 lg b-galactosidase-pSV wasco-transfected as an internal standard to assess transfection efficiency.Hepa 1c1c-7 and COS-7 cells were transfected using FuGENE 6 Trans-fection Reagent according to the manufacturer�s instructions (Roche).Twenty-four hours after transfection, medium A was replaced withDMEM supplemented with 2 mM L-glutamine, 100 U penicillin, 100 lg/ml streptomycin, and 0.5% charcoal-stripped fetal bovine serum, free oflipoproteins (medium B). Indicated concentrations of CDCA, GW404764or vehicle (ethanol or DMSO) were included in the medium during thefinal 24 h. The cells were washed twice with phosphate-buffered saline andlysed with Reporter lysis buffer (Promega). Luciferase activity and b-ga-lactosidase activity were measured according to manufacturer�s instruc-tions. Luciferase activities were normalised to b-galactosidase activities toaccount for any variation in transfection efficiency. At least three separateexperiments carried out in duplicate were performed.
Quantitative real time RT-PCR. Total RNA was isolated using TriZolreagent (Invitrogen) from liver tissue samples and liver cells treated withligands or vehicle according to the manufacturer. Five micrograms of totalRNA was reverse transcribed using SuperScript II Reverse RNase HTranscriptase (Invitrogen) and oligo(dT) primers. The cDNA was dilutedfivefold and 1 ll was used as template for quantitative real time RT-PCRon a Light Cycler machine (Roche) using SYBR Green technology.Primer3 program was used to design the primers, which are listed inTable 1 in addition to the PCR conditions used. The primers were de-signed such that they do not amplify from genomic DNA. This was testedusing RNA reverse transcribed without the enzyme which was more than10 crossing point-values different from RNA samples reversed transcribedusing the enzyme. SR-BI and SHP expression was measured relative to thehousekeeping gene GADPH expression.
primer Product size (bp) Tm (�C) [MgCl2] (mM)
AAGTCAACCTTGCTC 216 62 3AGCGCCAGATACAGC 248 62 3CAGGCTCTACTCAGC 230 62 3AAGAGGTCCCCAAGC 261 62 3GACTTCACACAGCAC 310 62 3TCTACATGGCAACTG 147 62 3GGGGGTAGGAACAC 171 62 3
100
75
42
1Fold regulation 0.67 2.5
kDa con
con
TCA cholestyramine
SR-BI
β-actin
1%
1% TCA
2%
2% cholestyramine
rela
tive
SR-B
I m
RN
A le
vels 3,5
2,5
1,5
0,5
3
2
1
0
A
B
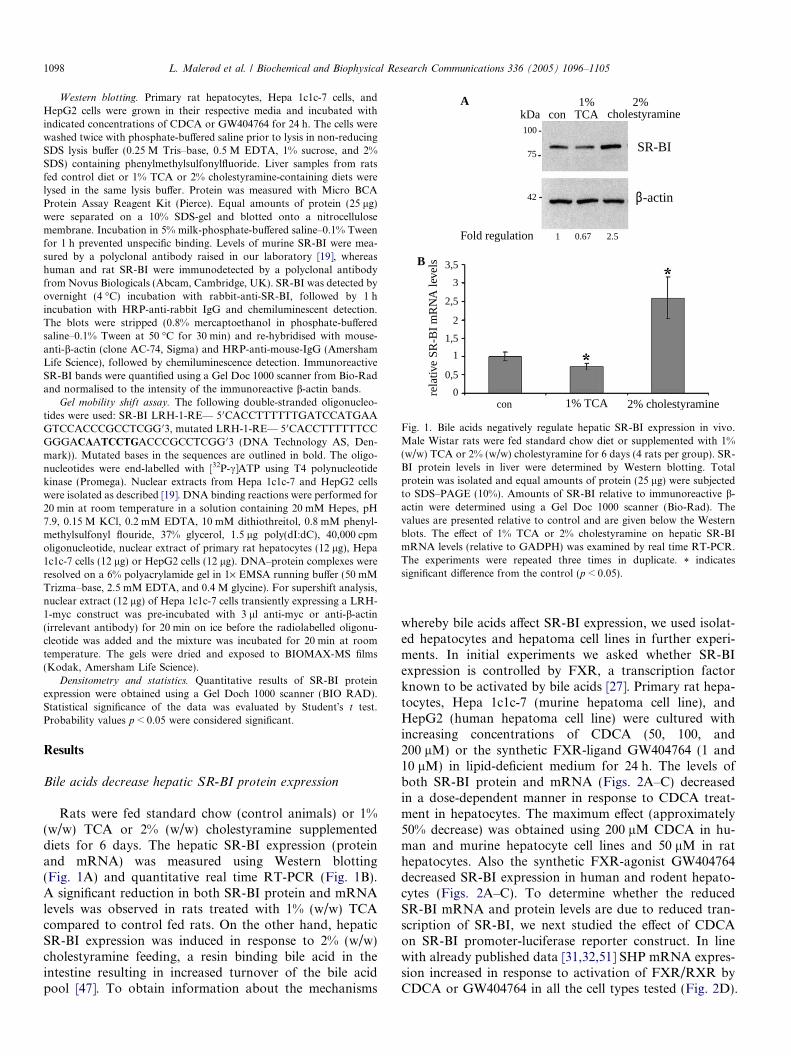
Fig. 1. Bile acids negatively regulate hepatic SR-BI expression in vivo.Male Wistar rats were fed standard chow diet or supplemented with 1%(w/w) TCA or 2% (w/w) cholestyramine for 6 days (4 rats per group). SR-BI protein levels in liver were determined by Western blotting. Totalprotein was isolated and equal amounts of protein (25 lg) were subjectedto SDS–PAGE (10%). Amounts of SR-BI relative to immunoreactive b-actin were determined using a Gel Doc 1000 scanner (Bio-Rad). Thevalues are presented relative to control and are given below the Westernblots. The effect of 1% TCA or 2% cholestyramine on hepatic SR-BImRNA levels (relative to GADPH) was examined by real time RT-PCR.The experiments were repeated three times in duplicate. * indicatessignificant difference from the control (p < 0.05).
1098 L. Malerød et al. / Biochemical and Biophysical Research Communications 336 (2005) 1096–1105
Western blotting. Primary rat hepatocytes, Hepa 1c1c-7 cells, andHepG2 cells were grown in their respective media and incubated withindicated concentrations of CDCA or GW404764 for 24 h. The cells werewashed twice with phosphate-buffered saline prior to lysis in non-reducingSDS lysis buffer (0.25 M Tris–base, 0.5 M EDTA, 1% sucrose, and 2%SDS) containing phenylmethylsulfonylfluoride. Liver samples from ratsfed control diet or 1% TCA or 2% cholestyramine-containing diets werelysed in the same lysis buffer. Protein was measured with Micro BCAProtein Assay Reagent Kit (Pierce). Equal amounts of protein (25 lg)were separated on a 10% SDS-gel and blotted onto a nitrocellulosemembrane. Incubation in 5% milk-phosphate-buffered saline–0.1% Tweenfor 1 h prevented unspecific binding. Levels of murine SR-BI were mea-sured by a polyclonal antibody raised in our laboratory [19], whereashuman and rat SR-BI were immunodetected by a polyclonal antibodyfrom Novus Biologicals (Abcam, Cambridge, UK). SR-BI was detected byovernight (4 �C) incubation with rabbit-anti-SR-BI, followed by 1 hincubation with HRP-anti-rabbit IgG and chemiluminescent detection.The blots were stripped (0.8% mercaptoethanol in phosphate-bufferedsaline–0.1% Tween at 50 �C for 30 min) and re-hybridised with mouse-anti-b-actin (clone AC-74, Sigma) and HRP-anti-mouse-IgG (AmershamLife Science), followed by chemiluminescence detection. ImmunoreactiveSR-BI bands were quantified using a Gel Doc 1000 scanner from Bio-Radand normalised to the intensity of the immunoreactive b-actin bands.
Gel mobility shift assay. The following double-stranded oligonucleo-tides were used: SR-BI LRH-1-RE— 5 0CACCTTTTTTGATCCATGAAGTCCACCCGCCTCGG 03, mutated LRH-1-RE— 5 0CACCTTTTTTCCGGGACAATCCTGACCCGCCTCGG 03 (DNA Technology AS, Den-mark)). Mutated bases in the sequences are outlined in bold. The oligo-nucleotides were end-labelled with [32P-c]ATP using T4 polynucleotidekinase (Promega). Nuclear extracts from Hepa 1c1c-7 and HepG2 cellswere isolated as described [19]. DNA binding reactions were performed for20 min at room temperature in a solution containing 20 mM Hepes, pH7.9, 0.15 M KCl, 0.2 mM EDTA, 10 mM dithiothreitol, 0.8 mM phenyl-methylsulfonyl flouride, 37% glycerol, 1.5 lg poly(dI:dC), 40,000 cpmoligonucleotide, nuclear extract of primary rat hepatocytes (12 lg), Hepa1c1c-7 cells (12 lg) or HepG2 cells (12 lg). DNA–protein complexes wereresolved on a 6% polyacrylamide gel in 1· EMSA running buffer (50 mMTrizma–base, 2.5 mM EDTA, and 0.4 M glycine). For supershift analysis,nuclear extract (12 lg) of Hepa 1c1c-7 cells transiently expressing a LRH-1-myc construct was pre-incubated with 3 ll anti-myc or anti-b-actin(irrelevant antibody) for 20 min on ice before the radiolabelled oligonu-cleotide was added and the mixture was incubated for 20 min at roomtemperature. The gels were dried and exposed to BIOMAX-MS films(Kodak, Amersham Life Science).
Densitometry and statistics. Quantitative results of SR-BI proteinexpression were obtained using a Gel Doch 1000 scanner (BIO RAD).Statistical significance of the data was evaluated by Student�s t test.Probability values p < 0.05 were considered significant.
Results
Bile acids decrease hepatic SR-BI protein expression
Rats were fed standard chow (control animals) or 1%(w/w) TCA or 2% (w/w) cholestyramine supplementeddiets for 6 days. The hepatic SR-BI expression (proteinand mRNA) was measured using Western blotting(Fig. 1A) and quantitative real time RT-PCR (Fig. 1B).A significant reduction in both SR-BI protein and mRNAlevels was observed in rats treated with 1% (w/w) TCAcompared to control fed rats. On the other hand, hepaticSR-BI expression was induced in response to 2% (w/w)cholestyramine feeding, a resin binding bile acid in theintestine resulting in increased turnover of the bile acidpool [47]. To obtain information about the mechanisms
whereby bile acids affect SR-BI expression, we used isolat-ed hepatocytes and hepatoma cell lines in further experi-ments. In initial experiments we asked whether SR-BIexpression is controlled by FXR, a transcription factorknown to be activated by bile acids [27]. Primary rat hepa-tocytes, Hepa 1c1c-7 (murine hepatoma cell line), andHepG2 (human hepatoma cell line) were cultured withincreasing concentrations of CDCA (50, 100, and200 lM) or the synthetic FXR-ligand GW404764 (1 and10 lM) in lipid-deficient medium for 24 h. The levels ofboth SR-BI protein and mRNA (Figs. 2A–C) decreasedin a dose-dependent manner in response to CDCA treat-ment in hepatocytes. The maximum effect (approximately50% decrease) was obtained using 200 lM CDCA in hu-man and murine hepatocyte cell lines and 50 lM in rathepatocytes. Also the synthetic FXR-agonist GW404764decreased SR-BI expression in human and rodent hepato-cytes (Figs. 2A–C). To determine whether the reducedSR-BI mRNA and protein levels are due to reduced tran-scription of SR-BI, we next studied the effect of CDCAon SR-BI promoter-luciferase reporter construct. In linewith already published data [31,32,51] SHP mRNA expres-sion increased in response to activation of FXR/RXR byCDCA or GW404764 in all the cell types tested (Fig. 2D).
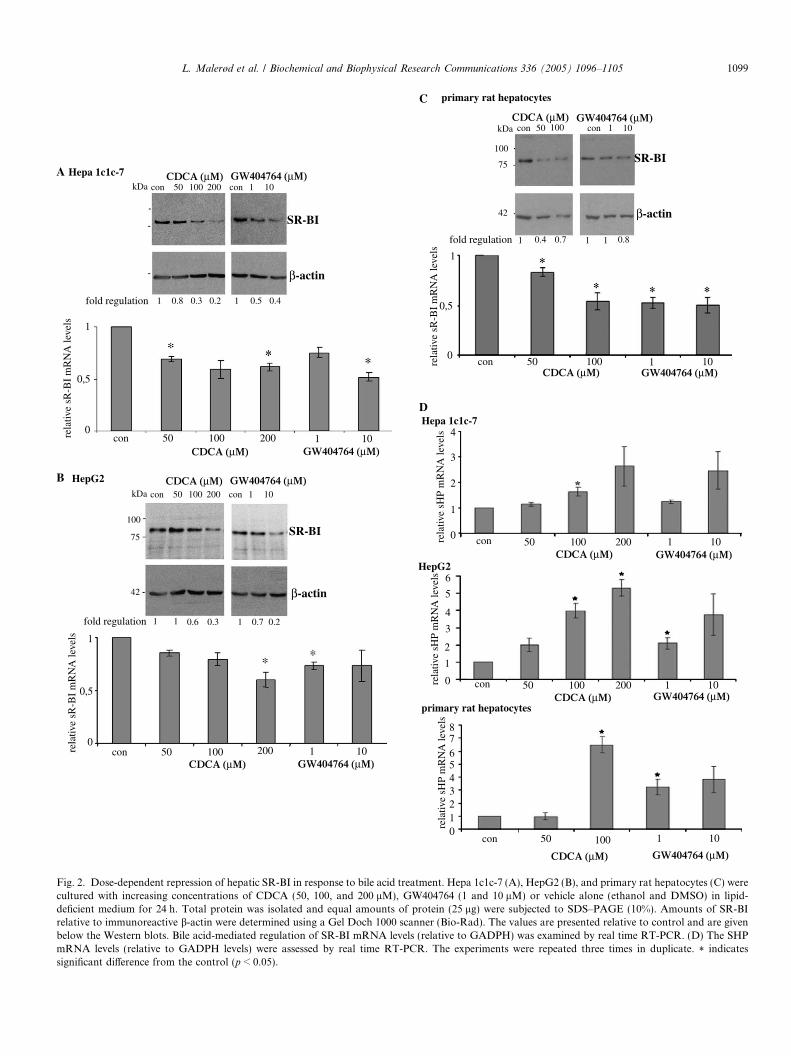
Fig. 2. Dose-dependent repression of hepatic SR-BI in response to bile acid treatment. Hepa 1c1c-7 (A), HepG2 (B), and primary rat hepatocytes (C) werecultured with increasing concentrations of CDCA (50, 100, and 200 lM), GW404764 (1 and 10 lM) or vehicle alone (ethanol and DMSO) in lipid-deficient medium for 24 h. Total protein was isolated and equal amounts of protein (25 lg) were subjected to SDS–PAGE (10%). Amounts of SR-BIrelative to immunoreactive b-actin were determined using a Gel Doch 1000 scanner (Bio-Rad). The values are presented relative to control and are givenbelow the Western blots. Bile acid-mediated regulation of SR-BI mRNA levels (relative to GADPH) was examined by real time RT-PCR. (D) The SHPmRNA levels (relative to GADPH levels) were assessed by real time RT-PCR. The experiments were repeated three times in duplicate. * indicatessignificant difference from the control (p < 0.05).
L. Malerød et al. / Biochemical and Biophysical Research Communications 336 (2005) 1096–1105 1099

1100 L. Malerød et al. / Biochemical and Biophysical Research Communications 336 (2005) 1096–1105
CDCA activation of FXR/RXR represses SR-BI promoter
activity
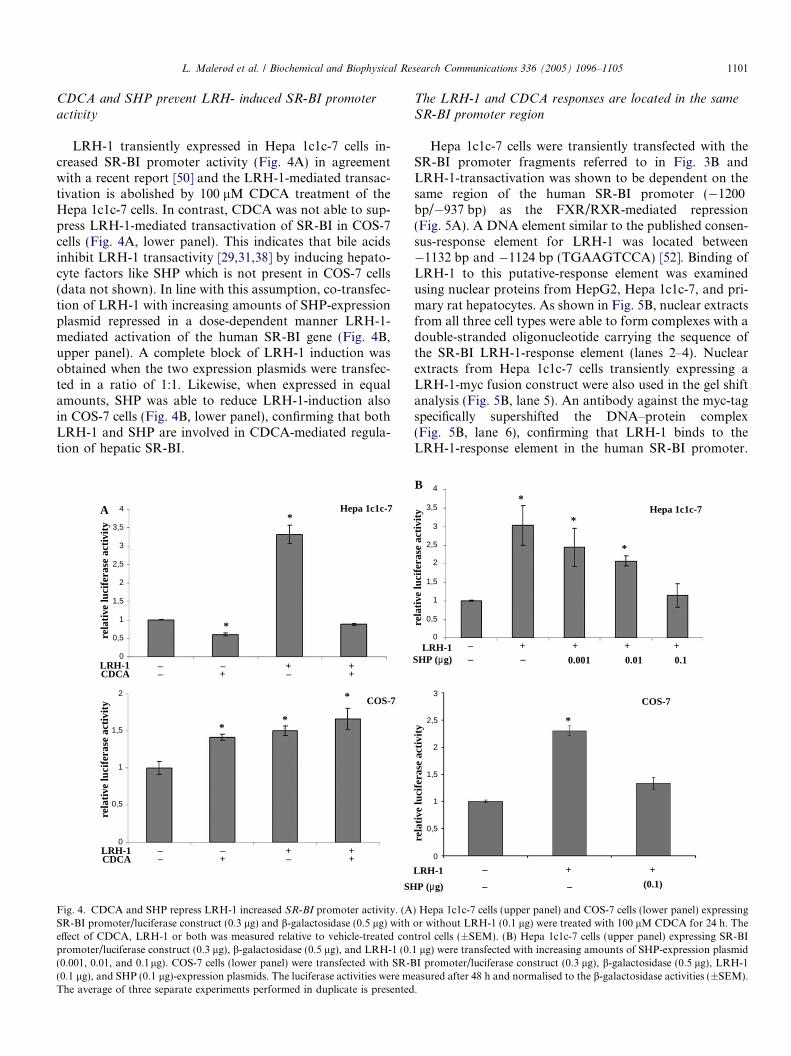
Hepa 1c1c-7 cells were transiently transfected with avector expressing the reporter gene luciferase under thecontrol of the human SR-BI promoter fragment. Fig. 3Ashows that treatment of the cells with 100 lM CDCAresulted in a repression of promoter activity in both the ab-sence and presence of co-expressed FXR/RXR, whereastransfection of FXR/RXR alone only modestly reducedpromoter activity. The negative CDCA-effect on SR-BIpromoter activity was not observed using a non-hepatocytecell line; COS-7 cells (Fig. 3A). In contrast to what was ob-served in Hepa 1c1c-7 cells, bile acid treatment slightlystimulated SR-BI promoter activity in transiently transfec-ted COS-7 cells. Transient co-expression of FXR/RXR inCOS-7 cells decreased SR-BI promoter activity. However,promoter activity was not reduced below control levels inCOS-7 cells in contrast to what was found in transfectedHepa 1c1c-7 cells. This indicates that additional factors(e.g., SHP) are involved in the bile acid suppression ofSR-BI seen in hepatocytes.
In order to map regulatory elements in the SR-BI pro-moter, progressive 5 0-deletion fragments of the promoterwere generated by endonuclease digestion and cloned up-stream of the luciferase reporter gene in pGL3 Basic Vec-
0
0
0.5
1.5
1
2
0.2
0.4
0.6
0.8
1
1.2
FXR/RXRF
F
F
rela
tive
luci
fera
se a
ctiv
ity
rela
tive
luci
fera
se a
ctiv
ity
CDCA
FXR/RXRCDCA
COS-7
Hepa 1c1c-7A
Fig. 3. FXR/RXR represses SR-BI promoter activity. (A) Hepa 1c1c-7 cellspromoter/luciferase reporter construct (0.3 lg) together with an expression plasefficiency. Twenty-four hours after transfection, the cells were treated with 1presence or absence of co-transfected expression plasmids for FXR (0.1 lg)galactosidase activities. The results represent experiments performed in duplicvehicle (ethanol + DMSO)-treated cells (±SEM). (B) Various 5 0 deleted fragm1c1c-7 cells together with b-galactosidase (0.5 lg), FXR (0.1 lg), and RXR (0.The effect of CDCA (dark grey columns) was expressed relative to the vehiperformed three times, in duplicate. * indicates significantly different from con
tor (Promega). Three promoter/luciferase reporterconstructs were transiently transfected into Hepa 1c1c-7cells; �1200 bp (full-length promoter), �937 bp, and�574 bp. Fig. 3B shows that CDCA activation of FXR/RXR decreased promoter activity in Hepa 1c1c-7 cellstransfected with the full-length promoter construct(�1200 bp). In contrast, bile acid-activated FXR/RXRdid not affect promoter activity when sequences down-stream of �937 bp were removed. Therefore, the deletionanalysis of the SR-BI promoter revealed a bile acid-re-sponding region between �1200 and �937 bp. Upon li-gand-binding FXR forms an obligate heterodimer withRXR and binds to response elements arranged as invertedrepeats spaced by 1 nucleotide (IR-1) [48,49]. No sequenc-es similar to the FXR consensus sequence (IR-1) wereidentified in the bile acid-responsive region. However, bileacids have been shown to repress their own synthesis ina negative feedback loop where FXR/RXR stimulatesthe expression of SHP [29], which inactivates LRH-1[31,32,39] and LXR/RXR-transactivation [41] of CYP7A1.It is therefore conceivable that bile acids suppress cellularuptake of cholesterol via SR-BI by the pathway used to re-duce bile acid synthesis via CYP7A1. We therefore exam-ined the effect of CDCA on LRH-1-mediatedtransactivation of the SR-BI promoter activity in transfec-ted Hepa 1c1c-7 cells.
XR/RXR/CDCA
XR/RXR/CDCA
XR/RXR/CDCA
relative luciferase activity
pGL3
0 0,2 0,4 0,6 0,8 1 1,2
-1200
-937
-574
-574+
-937+
-1200+
B
(upper panel) or COS-7 cells (lower panel) were transfected with SR-BImid for b-galactosidase (0.5 lg) used as an internal control for transfection00 lM CDCA for additional 24 h. The CDCA-effect was assessed in theand RXR (0.1 lg). The luciferase activity was normalised against the b-ate and repeated three times. The change in promoter activity relative toents of the SR-BI promoter (0.3 lg) were transiently expressed in Hepa
1 lg), and cultured with 100 lM CDCA or vehicle (ethanol) the final 24 h.cle-treated controls (light grey columns) (±SEM). The experiments weretrol (p < 0.05).
L. Malerød et al. / Biochemical and Biophysical Research Communications 336 (2005) 1096–1105 1101
CDCA and SHP prevent LRH- induced SR-BI promoter
activity
LRH-1 transiently expressed in Hepa 1c1c-7 cells in-creased SR-BI promoter activity (Fig. 4A) in agreementwith a recent report [50] and the LRH-1-mediated transac-tivation is abolished by 100 lM CDCA treatment of theHepa 1c1c-7 cells. In contrast, CDCA was not able to sup-press LRH-1-mediated transactivation of SR-BI in COS-7cells (Fig. 4A, lower panel). This indicates that bile acidsinhibit LRH-1 transactivity [29,31,38] by inducing hepato-cyte factors like SHP which is not present in COS-7 cells(data not shown). In line with this assumption, co-transfec-tion of LRH-1 with increasing amounts of SHP-expressionplasmid repressed in a dose-dependent manner LRH-1-mediated activation of the human SR-BI gene (Fig. 4B,upper panel). A complete block of LRH-1 induction wasobtained when the two expression plasmids were transfec-ted in a ratio of 1:1. Likewise, when expressed in equalamounts, SHP was able to reduce LRH-1-induction alsoin COS-7 cells (Fig. 4B, lower panel), confirming that bothLRH-1 and SHP are involved in CDCA-mediated regula-tion of hepatic SR-BI.
4
3,5
2
1,5
1
0,5
0
3
2,5
2
1,5
1
0,5
0LRH-1CDCA
LRH-1CDCA
SH
––
––
+–
++
–+
+–
++
–+
*
**
*
*Hepa 1c1c-7
COS-7
rela
tive
luci
fera
se a
ctiv
ity
rela
tive
luci
fera
se a
ctiv
ity
A
Fig. 4. CDCA and SHP repress LRH-1 increased SR-BI promoter activity. (ASR-BI promoter/luciferase construct (0.3 lg) and b-galactosidase (0.5 lg) witheffect of CDCA, LRH-1 or both was measured relative to vehicle-treated conpromoter/luciferase construct (0.3 lg), b-galactosidase (0.5 lg), and LRH-1 (0.(0.001, 0.01, and 0.1lg). COS-7 cells (lower panel) were transfected with SR-B(0.1 lg), and SHP (0.1 lg)-expression plasmids. The luciferase activities were meThe average of three separate experiments performed in duplicate is presented
The LRH-1 and CDCA responses are located in the same
SR-BI promoter region
Hepa 1c1c-7 cells were transiently transfected with theSR-BI promoter fragments referred to in Fig. 3B andLRH-1-transactivation was shown to be dependent on thesame region of the human SR-BI promoter (�1200bp/�937 bp) as the FXR/RXR-mediated repression(Fig. 5A). A DNA element similar to the published consen-sus-response element for LRH-1 was located between�1132 bp and �1124 bp (TGAAGTCCA) [52]. Binding ofLRH-1 to this putative-response element was examinedusing nuclear proteins from HepG2, Hepa 1c1c-7, and pri-mary rat hepatocytes. As shown in Fig. 5B, nuclear extractsfrom all three cell types were able to form complexes with adouble-stranded oligonucleotide carrying the sequence ofthe SR-BI LRH-1-response element (lanes 2–4). Nuclearextracts from Hepa 1c1c-7 cells transiently expressing aLRH-1-myc fusion construct were also used in the gel shiftanalysis (Fig. 5B, lane 5). An antibody against the myc-tagspecifically supershifted the DNA–protein complex(Fig. 5B, lane 6), confirming that LRH-1 binds to theLRH-1-response element in the human SR-BI promoter.
4
3,5
3
2,5
2
1,5
1
0,5
0
3
2,5
2
1,5
1
0,5
0
LRH-1SHP (µg)
LRH-1
P (µg)
––
–
– –
+ +
(0.1)
+–
+
0.001
+
0.01
+
0.1
*
*
*
*
COS-7
Hepa 1c1c-7
rela
tive
luci
fera
se a
ctiv
ity
rela
tive
luci
fera
se a
ctiv
ity
B
) Hepa 1c1c-7 cells (upper panel) and COS-7 cells (lower panel) expressingor without LRH-1 (0.1 lg) were treated with 100 lM CDCA for 24 h. Thetrol cells (±SEM). (B) Hepa 1c1c-7 cells (upper panel) expressing SR-BI1 lg) were transfected with increasing amounts of SHP-expression plasmidI promoter/luciferase construct (0.3 lg), b-galactosidase (0.5 lg), LRH-1asured after 48 h and normalised to the b-galactosidase activities (±SEM)..
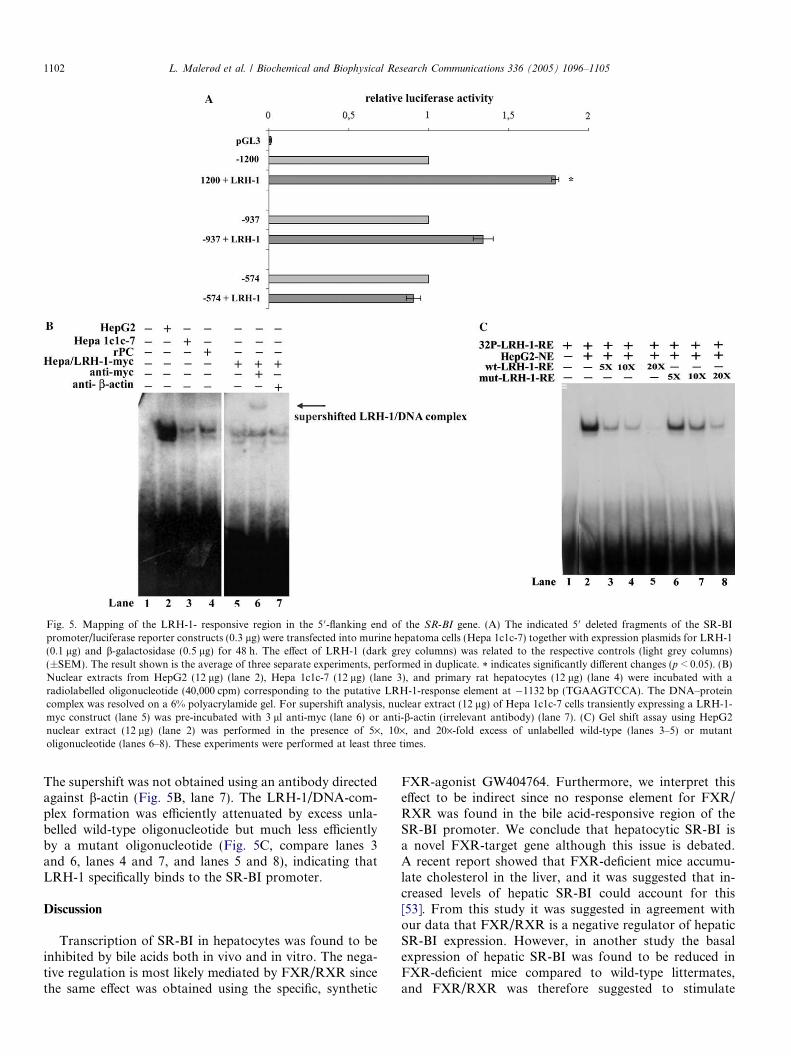
Fig. 5. Mapping of the LRH-1- responsive region in the 5 0-flanking end of the SR-BI gene. (A) The indicated 5 0 deleted fragments of the SR-BIpromoter/luciferase reporter constructs (0.3 lg) were transfected into murine hepatoma cells (Hepa 1c1c-7) together with expression plasmids for LRH-1(0.1 lg) and b-galactosidase (0.5 lg) for 48 h. The effect of LRH-1 (dark grey columns) was related to the respective controls (light grey columns)(±SEM). The result shown is the average of three separate experiments, performed in duplicate. * indicates significantly different changes (p < 0.05). (B)Nuclear extracts from HepG2 (12 lg) (lane 2), Hepa 1c1c-7 (12 lg) (lane 3), and primary rat hepatocytes (12 lg) (lane 4) were incubated with aradiolabelled oligonucleotide (40,000 cpm) corresponding to the putative LRH-1-response element at �1132 bp (TGAAGTCCA). The DNA–proteincomplex was resolved on a 6% polyacrylamide gel. For supershift analysis, nuclear extract (12 lg) of Hepa 1c1c-7 cells transiently expressing a LRH-1-myc construct (lane 5) was pre-incubated with 3 ll anti-myc (lane 6) or anti-b-actin (irrelevant antibody) (lane 7). (C) Gel shift assay using HepG2nuclear extract (12 lg) (lane 2) was performed in the presence of 5·, 10·, and 20·-fold excess of unlabelled wild-type (lanes 3–5) or mutantoligonucleotide (lanes 6–8). These experiments were performed at least three times.
1102 L. Malerød et al. / Biochemical and Biophysical Research Communications 336 (2005) 1096–1105
The supershift was not obtained using an antibody directedagainst b-actin (Fig. 5B, lane 7). The LRH-1/DNA-com-plex formation was efficiently attenuated by excess unla-belled wild-type oligonucleotide but much less efficientlyby a mutant oligonucleotide (Fig. 5C, compare lanes 3and 6, lanes 4 and 7, and lanes 5 and 8), indicating thatLRH-1 specifically binds to the SR-BI promoter.
Discussion
Transcription of SR-BI in hepatocytes was found to beinhibited by bile acids both in vivo and in vitro. The nega-tive regulation is most likely mediated by FXR/RXR sincethe same effect was obtained using the specific, synthetic
FXR-agonist GW404764. Furthermore, we interpret thiseffect to be indirect since no response element for FXR/RXR was found in the bile acid-responsive region of theSR-BI promoter. We conclude that hepatocytic SR-BI isa novel FXR-target gene although this issue is debated.A recent report showed that FXR-deficient mice accumu-late cholesterol in the liver, and it was suggested that in-creased levels of hepatic SR-BI could account for this[53]. From this study it was suggested in agreement withour data that FXR/RXR is a negative regulator of hepaticSR-BI expression. However, in another study the basalexpression of hepatic SR-BI was found to be reduced inFXR-deficient mice compared to wild-type littermates,and FXR/RXR was therefore suggested to stimulate

L. Malerød et al. / Biochemical and Biophysical Research Communications 336 (2005) 1096–1105 1103
SR-BI expression [54]. This result is in contrast to thenegative regulation demonstrated in the present studyand the data presented by Sinal et al. [53]. Disruption ofthe hepatic FXR-gene was shown to be accompanied by al-tered expression of several other molecules in lipid metab-olism, e.g., HMG-CoA reductase [54]. The reduced levelsof HMG-CoA reductase that was reported result in de-creased de novo synthesis of cholesterol. Furthermore, de-creased levels of cholesterol are also associated withreduced levels of different modified forms of cholesterolsuch as oxysterols, which are cognate ligands for the nucle-ar receptor LXR [55]. The LXR/RXR-heterodimer hasbeen shown to be a positive regulator of hepatic SR-BIexpression [19] and this may therefore explain the lowlevels of SR-BI reported in FXR-deficient mice. That thepositive LXR-effect dominates over the negative FXR-effect on SR-BI expression has also been observed in ourlaboratory (preliminary data).
Schoonjans et al. [50] identified a LRH-1-response ele-ment in the human SR-BI promoter located between �77and �69 bp. Likewise binding of steroidogenic factor-1(SF-1), the twin-brother of LRH-1, to the same responseelement was already described by Cao et al. [44]. In thepresent study, we identify a second response element forLRH-1 in the human SR-BI promoter, which is not in con-flict with the previous data. Schoonjans et al. [50] observedonly partly reduction in the LRH-1-induction in responseto mutation of the response element at �77 to �69 bp. Inaddition, the deletion analysis shows that LRH-1 stimu-lated SR-BI promoter activity less when the region between�2913 and �258 bp was removed. These results stronglyindicate that additional response elements for LRH-1 existin the humanSR-BI promoter, as shown in the present study.
Hepatic SR-BI mediates selective uptake of HDL-cho-lesteryl esters [3,10]. Within the hepatocytes cholesterol isconverted to bile acids, secreted directly into the bile can-aliculus or incorporated into lipoproteins [56]. Bile acidsrepress their own synthesis by initiating a nuclear cascadeinvolving activation of FXR/RXR, which increasesexpression of SHP [29]. This repressor inhibits LRH-1and LXR/RXR-transactivation of CYP7A1 in rats andmice [39,41]. According to previous studies, it appears thatcholesterol metabolism in hepatocytes is strictly regulatedby LXR/RXR and FXR/RXR. LXR/RXR regulates cho-lesterol metabolism in hepatocytes by stimulating the selec-tive uptake of HDL-cholesterol (via SR-BI) [19], byconverting cholesterol to bile acids (via CYP7A1) [24],and by promoting the direct secretion of cholesterol intothe bile (via ABCG5, ABCG8 [57–60] and possibly viaSR-BI) [61–63]. On the other hand, FXR/RXR preventsaccumulation of toxic high levels of bile acids, whichmay cause liver damage by decreasing bile acid synthesis(via CYP7A1) [32], and as shown in the current paper,HDL-cholesterol uptake (via SR-BI). Furthermore, bileacids have been shown to reduce hepatic HMG-CoAreductase mRNA levels [64] and consequently the de novocholesterol synthesis.
In summary, we have shown that bile acids downregu-late SR-BI in a similar manner as observed for CYP7A1,i.e., bile acid-activated FXR/RXR induces the expressionof SHP, which binds to and inhibits LRH-1 resulting in re-duced expression of CYP7A1 [34]. We show that LRH-1binds specifically to an element at �1132 bp/�1124 bp inthe human SR-BI promoter and therefore identifies a newLRH-1-response element in addition to the one (�77 bp/�69 bp) previously described [50]. We conclude that hepat-ic SR-BI expression is regulated by FXR/RXR in concertwith other molecules involved in cholesterol metabolismin the liver such as CYP7A1, CYP27, and HMG-CoAreductase.
Acknowledgments
We thank Drs. Hilde Nebb, Bryan Goodwin, Helen H.Hobbs, and Heidi R. Kast for providing us with the neces-sary expression plasmids. A special thank to German Tapiaand Folkert Kuipers for valuable help with the in vivoexperiments. The National Research Council and Norwe-gian Cancer Society funded this work.
References
[1] D. Sviridov, P. Nestel, Dynamics of reverse cholesterol transport:protection against atherosclerosis, Atherosclerosis 161 (2002) 245–254.
[2] S.L. Acton, P.E. Scherer, H.F. Lodish, M. Krieger, Expressioncloning of SR-BI, a CD36-related class B scavenger receptor, J. Biol.Chem. 269 (1994) 21003–21009.
[3] S. Acton, A. Rigotti, K.T. Landschulz, S. Xu, H.H. Hobbs, M.Krieger, Identification of scavenger receptor SR-BI as a high densitylipoprotein receptor, Science 271 (1996) 518–520.
[4] K.T. Landschulz, R.K. Pathak, A. Rigotti, M. Krieger, H.H. Hobbs,Regulation of scavenger receptor, class B, type I, a high densitylipoprotein receptor, in liver and steroidogenic tissues of the rat, J.Clin. Invest. 98 (1996) 984–995.
[5] S. Azhar, A. Nomoto, E. Reaven, Hormonal regulation of adrenalmicrovillar channel formation, J. Lipid Res. 43 (2002) 861–871.
[6] A. Rigotti, E.R. Edelman, P. Seifert, S.N. Iqbal, R.B. DeMattos,R.E. Temel, M. Krieger, D.L. Williams, Regulation by adrenocor-ticotropic hormone of the in vivo expression of scavenger receptorclass B type I (SR-BI), a high density lipoprotein receptor, insteroidogenic cells of the murine adrenal gland, J. Biol. Chem. 271(1996) 33545–33549.
[7] Y. Sun, N. Wang, A.R. Tall, Regulation of adrenal scavengerreceptor-BI expression by ACTH and cellular cholesterol pools, J.Lipid Res. 40 (1999) 1799–1805.
[8] X. Li, H. Peegel, K.M. Menon, Regulation of high density lipoproteinreceptor messenger ribonucleic acid expression and cholesteroltransport in theca-interstitial cells by insulin and human chorionicgonadotropin, Endocrinology 142 (2001) 174–181.
[9] D. Lopez, M.D. Sanchez, W. Shea-Eaton, M.P. McLean, Estrogenactivates the high- density lipoprotein receptor gene via binding toestrogen response elements and interaction with sterol regulatoryelement binding protein-1A, Endocrinology 143 (2002) 2155–2168.
[10] M. Krieger, Charting the fate of the ‘‘good cholesterol’’: identificationand characterization of the high-density lipoprotein receptor SR-BI,Annu. Rev. Biochem. 68 (1999) 523–558.
[11] R. Blomhoff, C.A. Drevon, W. Eskild, P. Helgerud, K.R. Norum, T.Berg, Clearance of acetyl low density lipoprotein by rat liverendothelial cells. Implications for hepatic cholesterol metabolism, J.Biol. Chem. 259 (1984) 8898–8903.

1104 L. Malerød et al. / Biochemical and Biophysical Research Communications 336 (2005) 1096–1105
[12] R. Blomhoff, W. Eskild, T. Berg, Endocytosis of formaldehyde-treated serum albumin via scavenger pathway in liver endothelialcells, Biochem. J. 218 (1984) 81–86.
[13] K. Fluiter, D.R. van der Westhuijzen, T.J. van Berkel, In vivoregulation of scavenger receptor BI and the selective uptake of highdensity lipoprotein cholesteryl esters in rat liver parenchymal andKupffer cells, J. Biol. Chem. 273 (1998) 8434–8438.
[14] T.J. van Berkel, G.J. Ziere, M.K. Bijsterbosch, J. Kuiper, Lipoproteinreceptors and atherogenic receptor-mediated mechanisms, Curr.Opin. Lipidol. 5 (1994) 331–338.
[15] G.A. Graf, K.L. Roswell, E.J. Smart, 17beta-Estradiol promotes theup-regulation of SR-BII in HepG2 cells and in rat livers, J. Lipid Res.42 (2001) 1444–1449.
[16] C. Loison, F. Mendy, C. Serougne, C. Lutton, Dietary myristic acidmodifies the HDL-cholesterol concentration and liver scavengerreceptor BI expression in the hamster, Br. J. Nutr. 87 (2002) 199–210.
[17] D.K. Spady, D.M. Kearney, H.H. Hobbs, Polyunsaturated fattyacids up-regulate hepatic scavenger receptor B1 (SR-BI) expressionand HDL cholesteryl ester uptake in the hamster, J. Lipid Res. 40(1999) 1384–1394.
[18] W. Witt, I. Kolleck, H. Fechner, P. Sinha, B. Rustow, Regulation byvitamin E of the scavenger receptor BI in rat liver and HepG2 cells, J.Lipid Res. 41 (2000) 2009–2016.
[19] L. Malerod, L.K. Juvet, A. Hanssen-Bauer, W. Eskild, T. Berg,Oxysterol-activated LXRalpha/RXR induces hSR-BI-promoteractivity in hepatoma cells and preadipocytes, Biochem. Biophys.Res. Commun. 299 (2002) 916–923.
[20] P. Mardones, A. Pilon, M. Bouly, D. Duran, T. Nishimoto, H. Arai,K.F.Kozarsky,M.Altayo, J.F.Miquel,G.Luc,V.Clavey,B. Staels,A.Rigotti, Fibrates down-regulate hepatic scavenger receptor class B typeI protein expression in mice, J. Biol. Chem. 278 (2003) 7884–7890.
[21] L. Malerod, M. Sporstol, L.K. Juvet, A. Mousavi, T. Gjoen, T. Berg,Hepatic scavenger receptor class B, type I is stimulated by peroxisomeproliferator-activated receptor gamma and hepatocyte nuclear factor4alpha, Biochem. Biophys. Res. Commun. 305 (2003) 557–565.
[22] B. Khan, H.G. Wilcox, M. Heimberg, Cholesterol is required forsecretion of very-low-density lipoprotein by rat liver, Biochem. J. 258(1989) 807–816.
[23] Z.R. Vlahcevic, W.M. Pandak, R.T. Stravitz, Regulation of bile acidbiosynthesis, Gastroenterol. Clin. North Am. 28 (1999) 1–25, v.
[24] J.Y. Chiang, R. Kimmel, D. Stroup, Regulation of cholesterol7alpha-hydroxylase gene (CYP7A1) transcription by the liver orphanreceptor (LXRalpha), Gene 262 (2001) 257–265.
[25] S. Gupta, W.M. Pandak, P.B. Hylemon, LXR alpha is the dominantregulator of CYP7A1 transcription, Biochem. Biophys. Res. Com-mun. 293 (2002) 338–343.
[26] J.M. Lehmann, S.A. Kliewer, L.B. Moore, T.A. Smith-Oliver, B.B.Oliver, J.L. Su, S.S. Sundseth, D.A. Winegar, D.E. Blanchard, T.A.Spencer, T.M. Willson, Activation of the nuclear receptor LXR byoxysterols defines a new hormone response pathway, J. Biol. Chem.272 (1997) 3137–3140.
[27] M. Makishima, A.Y. Okamoto, J.J. Repa, H. Tu, R.M. Learned, A.Luk, M.V. Hull, K.D. Lustig, D.J. Mangelsdorf, B. Shan, Identifi-cation of a nuclear receptor for bile acids, Science 284 (1999) 1362–1365.
[28] J.Y. Chiang, R. Kimmel, C. Weinberger, D. Stroup, Farnesoid Xreceptor responds to bile acids and represses cholesterol 7alpha-hydroxylase gene (CYP7A1) transcription, J. Biol. Chem. 275 (2000)10918–10924.
[29] R.A. Davis, J.H. Miyake, T.Y. Hui, N.J. Spann, Regulation ofcholesterol-7alpha-hydroxylase: BAREly missing a SHP, J. LipidRes. 43 (2002) 533–543.
[30] A. del Castillo-Olivares, G. Gil, Role of FXR and FTF in bile acid-mediated suppression of cholesterol 7alpha-hydroxylase transcrip-tion, Nucleic Acids Res. 28 (2000) 3587–3593.
[31] B. Goodwin, S.A. Jones, R.R. Price, M.A. Watson, D.D. McKee,L.B. Moore, C. Galardi, J.G. Wilson, M.C. Lewis, M.E. Roth, P.R.Maloney, T.M. Willson, S.A. Kliewer, A regulatory cascade of the
nuclear receptors FXR, SHP-1, and LRH-1 represses bile acidbiosynthesis, Mol. Cell 6 (2000) 517–526.
[32] T.T. Lu, M. Makishima, J.J. Repa, K. Schoonjans, T.A. Kerr, J.Auwerx, D.J. Mangelsdorf, Molecular basis for feedback regulationof bile acid synthesis by nuclear receptors, Mol. Cell 6 (2000) 507–515.
[33] J. Twisk, E.C. de Wit, H.M. Princen, Suppression of sterol 27-hydroxylase mRNA and transcriptional activity by bile acids incultured rat hepatocytes, Biochem. J. 305 (Pt. 2) (1995) 505–511.
[34] J. Twisk, E.M. Lehmann, H.M. Princen, Differential feedbackregulation of cholesterol 7 alpha-hydroxylase mRNA and transcrip-tional activity by rat bile acids in primary monolayer cultures of rathepatocytes, Biochem. J. 290 (Pt. 3) (1993) 685–691.
[35] J.A. Gustafsson, Seeking ligands for lonely orphan receptors, Science284 (1999) 1285–1286.
[36] D.J. Parks, S.G. Blanchard, R.K. Bledsoe, G. Chandra, T.G. Consler,S.A. Kliewer, J.B. Stimmel, T.M. Willson, A.M. Zavacki, D.D.Moore, J.M. Lehmann, Bile acids: natural ligands for an orphannuclear receptor, Science 284 (1999) 1365–1368.
[37] H. Wang, J. Chen, K. Hollister, L.C. Sowers, B.M. Forman,Endogenous bile acids are ligands for the nuclear receptor FXR/BAR, Mol. Cell 3 (1999) 543–553.
[38] W. Chen, E. Owsley, Y. Yang, D. Stroup, J.Y. Chiang, Nuclearreceptor-mediated repression of human cholesterol 7alpha-hydroxy-lase gene transcription by bile acids, J. Lipid Res. 42 (2001) 1402–1412.
[39] Y.K. Lee, D.D. Moore, Dual mechanisms for repression of themonomeric orphan receptor liver receptor homologous protein-1 bythe orphan small heterodimer partner, J. Biol. Chem. 277 (2002)2463–2467.
[40] M. Nitta, S. Ku, C. Brown, A.Y. Okamoto, B. Shan, CPF: an orphannuclear receptor that regulates liver-specific expression of the humancholesterol 7alpha- hydroxylase gene, Proc. Natl. Acad. Sci. USA 96(1999) 6660–6665.
[41] C. Brendel, K. Schoonjans, O.A. Botrugno, E. Treuter, J. Auwerx,The small heterodimer partner interacts with the liver X receptoralpha and represses its transcriptional activity, Mol. Endocrinol. 16(2002) 2065–2076.
[42] C.C. Schwartz, L.G. Halloran, Z.R. Vlahcevic, D.H. Gregory, L.Swell, Preferential utilization of free cholesterol from high-densitylipoproteins for biliary cholesterol secretion in man, Science 200(1978) 62–64.
[43] H.R. Kast, C.M. Nguyen, C.J. Sinal, S.A. Jones, B.A. Laffitte, K.Reue, F.J. Gonzalez, T.M. Willson, P.A. Edwards, Farnesoid X-activated receptor induces apolipoprotein C-II transcription: amolecular mechanism linking plasma triglyceride levels to bile acids,Mol. Endocrinol. 15 (2001) 1720–1728.
[44] G. Cao, C.K. Garcia, K.L. Wyne, R.A. Schultz, K.L. Parker, H.H.Hobbs, Structure and localization of the human gene encoding SR-BI/CLA-1. Evidence for transcriptional control by steroidogenicfactor 1, J. Biol. Chem. 272 (1997) 33068–33076.
[45] L. Malerod, K. Juvet, T. Gjoen, T. Berg, The expression of scavengerreceptor class B, type I (SR-BI) and caveolin-1 in parenchymal andnonparenchymal liver cells, Cell Tissue Res. 307 (2002) 173–180.
[46] K. Fronsdal, N. Engedal, F. Saatcioglu, Efficient DNA-mediatedgene transfer into prostate cancer cell line LNCaP, Prostate 43 (2000)111–117.
[47] B.E. Gustafsson, B. Angelin, K. Einarsson, J.A. Gustafsson, Influ-ence of cholestyramine on synthesis of cholesterol and bile acids ingermfree rats, J. Lipid Res. 19 (1978) 972–977.
[48] B.M. Forman, E. Goode, J. Chen, A.E. Oro, D.J. Bradley, T.Perlmann, D.J. Noonan, L.T. Burka, T. McMorris, W.W. Lamph,R.M. Evans, C. Weinberger, Identification of a nuclear receptor thatis activated by farnesol metabolites, Cell 81 (1995) 687–693.
[49] B.A. Laffitte, H.R. Kast, C.M. Nguyen, A.M. Zavacki, D.D. Moore,P.A. Edwards, Identification of the DNA binding specificity andpotential target genes for the farnesoid X-activated receptor, J. Biol.Chem. 275 (2000) 10638–10647.
[50] K. Schoonjans, J.S. Annicotte, T. Huby, O.A. Botrugno, E. Fayard,Y. Ueda, J. Chapman, J. Auwerx, Liver receptor homolog 1 controls

L. Malerød et al. / Biochemical and Biophysical Research Communications 336 (2005) 1096–1105 1105
the expression of the scavenger receptor class B type I, EMBO Rep. 3(2002) 1181–1187.
[51] L. Wang, Y.K. Lee, D. Bundman, Y. Han, S. Thevananther, C.S.Kim, S.S. Chua, P. Wei, R.A. Heyman, M. Karin, D.D. Moore,Redundant pathways for negative feedback regulation of bile acidproduction, Dev. Cell 2 (2002) 721–731.
[52] J.J. Repa, D.J. Mangelsdorf, Nuclear receptor regulation of choles-terol and bile acid metabolism, Curr. Opin. Biotechnol. 10 (1999)557–563.
[53] C.J. Sinal, M. Tohkin, M. Miyata, J.M. Ward, G. Lambert, F.J.Gonzalez, Targeted disruption of the nuclear receptor FXR/BARimpairs bile acid and lipid homeostasis, Cell 102 (2000) 731–744.
[54] G. Lambert, M.J. Amar, G. Guo, H.B. Brewer Jr., F.J. Gonzalez,C.J. Sinal, The farnesoid X-receptor is an essential regulator ofcholesterol homeostasis, J. Biol. Chem. 278 (2003) 2563–2570.
[55] B.A. Janowski, P.J. Willy, T.R. Devi, J.R. Falck, D.J. Mangelsdorf,An oxysterol signalling pathway mediated by the nuclear receptorLXR alpha, Nature 383 (1996) 728–731.
[56] S. Kang, R.A. Davis, Cholesterol and hepatic lipoprotein assemblyand secretion, Biochim. Biophys. Acta 1529 (2000) 223–230.
[57] K.E. Berge, H. Tian, G.A. Graf, L. Yu, N.V. Grishin, J. Schultz, P.Kwiterovich, B. Shan, R. Barnes, H.H. Hobbs, Accumulation ofdietary cholesterol in sitosterolemia caused by mutations in adjacentABC transporters, Science 290 (2000) 1771–1775.
[58] T. Sudhop, Y. Sahin, B. Lindenthal, C. Hahn, C. Luers, H.K.Berthold, K. von Bergmann, Comparison of the hepatic clearances ofcampesterol, sitosterol, and cholesterol in healthy subjects suggests
that efflux transporters controlling intestinal sterol absorption alsoregulate biliary secretion, Gut 51 (2002) 860–863.
[59] L. Yu, R.E. Hammer, J. Li-Hawkins, K. Von Bergmann, D.Lutjohann, J.C. Cohen, H.H. Hobbs, Disruption of Abcg5 andAbcg8 in mice reveals their crucial role in biliary cholesterol secretion,Proc. Natl. Acad. Sci. USA 99 (2002) 16237–16242.
[60] L. Yu, J. Li-Hawkins, R.E. Hammer, K.E. Berge, J.D. Horton, J.C.Cohen, H.H. Hobbs, Overexpression of ABCG5 and ABCG8promotes biliary cholesterol secretion and reduces fractional absorp-tion of dietary cholesterol, J. Clin. Invest. 110 (2002) 671–680.
[61] Y. Ji, N. Wang, R. Ramakrishnan, E. Sehayek, D. Huszar, J.L.Breslow, A.R. Tall, Hepatic scavenger receptor BI promotes rapidclearance of high density lipoprotein free cholesterol and its transportinto bile, J. Biol. Chem. 274 (1999) 33398–33402.
[62] K.F. Kozarsky,M.H. Donahee, A. Rigotti, S.N. Iqbal, E.R. Edelman,M. Krieger, Overexpression of the HDL receptor SR-BI alters plasmaHDL and bile cholesterol levels, Nature 387 (1997) 414–417.
[63] E. Sehayek, J.G. Ono, S. Shefer, L.B. Nguyen, N. Wang, A.K.Batta, G. Salen, J.D. Smith, A.R. Tall, J.L. Breslow, Biliarycholesterol excretion: a novel mechanism that regulates dietarycholesterol absorption, Proc. Natl. Acad. Sci. USA 95 (1998)10194–10199.
[64] M. Rudling, B. Angelin, L. Stahle, E. Reihner, S. Sahlin, H.Olivecrona, I. Bjorkhem, C. Einarsson, Regulation of hepatic low-density lipoprotein receptor, 3-hydroxy-3- methylglutaryl coenzymeA reductase, and cholesterol 7alpha-hydroxylase mRNAs in humanliver, J. Clin. Endocrinol. Metab. 87 (2002) 4307–4313.




















