
Morphological Characteristics of Brain Tumors Causing Seizures

Structural and Functional Effects of Disease-CausingAmino Acid Substitutions Affecting Residues Ala72 andGlu76 of the Protein Tyrosine Phosphatase SHP-2
Gianfranco Bocchinfuso,1y Lorenzo Stella,1y Simone Martinelli,2 Elisabetta Flex,2 Claudio Carta,2 FrancescaPantaleoni,2 Basilio Pispisa,1 Mariano Venanzi,1 Marco Tartaglia,2 and Antonio Palleschi1*1Dipartimento di Scienze e Tecnologie Chimiche, Universita di Roma Tor Vergata, Rome, Italy2Dipartimento di Biologia Cellulare e Neuroscienze, Istituto Superiore di Sanita, Rome, Italy
ABSTRACT Mutations of the protein tyrosinephosphatase SHP-2 are implicated in human dis-eases, causing Noonan syndrome (NS) and relateddevelopmental disorders or contributing to leuke-mogenesis depending on the specific amino acidsubstitution involved. SHP-2 is composed by a cat-alytic (PTP) and two regulatory (N-SH2 andC-SH2) domains that bind to signaling partnersand control the enzymatic activity by limiting theaccessibility of the catalytic site. Wild type SHP-2and four disease-associated mutants recurring inhematologic malignancies (Glu76Lys and Ala72Val)or causing NS (Glu76Asp and Ala72Ser), withaffected residues located in the PTP-interactingregion of the N-SH2 domain, were analyzed bymolecular dynamics simulations and in vitro bio-chemical assays. Simulations demonstrate thatmutations do not affect significantly the confor-mation of the N-SH2 domain. Rather they desta-bilize the interaction of this domain with thecatalytic site, with more evident effects in thetwo leukemia associated mutants. Consistentwith this structural evidence, mutants exhibitan increased level of basal phosphatase activityin the order Glu76Lys > Ala72Val > Glu76Asp >
Ala72Ser > WT. The experimental data also showthat the mutants with higher basal activity aremore responsive to an activating phosphopep-tide. A thermodynamic analysis demonstratesthat an increase in the overall phosphopeptideaffinity of mutants can be explained by a shiftin the equilibrium between the inactive andactive SHP-2 structure. These data support theview that an increase in the affinity of SHP-2 forits binding partners, caused by destabilization ofthe closed, inactive conformation, rather thanprotein basal activation per se, would representthe molecular mechanism, leading to pathogene-sis in these mutants. Proteins 2007;66:963–974. VVC 2006 Wiley-Liss, Inc.
Key words: molecular dynamics simulation; phos-phatase activity; SHP-2 mutants;Noonan syndrome; leukemia; interdo-main interaction
INTRODUCTION
SHP-2 is a member of a small subfamily of cytoplasmicSrc homology 2 (SH2) domain-containing protein tyrosinephosphatases (PTPs) that control cellular proliferation anddifferentiation.1 SHP-2 is a key molecule in intracellularsignaling and is necessary for activation of the RAS/MAPKcascade in response to a variety of growth factors, hor-mones, and cytokines.2–4 SHP-2 is expressed widely and isrequired in development5–9 and hematopoiesis.8,10,11
Increasing interest on SHP-2 has emerged since the dis-covery that mutations in PTPN11, the gene encoding forthis phosphatase, are involved in different human dis-eases.12 Among these, a first group of molecular lesionsrecur as germinally transmitted defects causing Noonansyndrome (NS),13,14 a developmental disorder character-ized by facial dysmorphisms, short stature, as well as skel-etal and congenital heart defects.15 A second group oflesions is somatically acquired and contributes to leuke-mogenesis, being documented with variable prevalence inmyeloproliferative/myelodysplastic disorders16–18 and acuteleukemias.16,17,19–21 Studies from our group and others pro-vided evidence that, except a few in-frame small deletions,these molecular lesions are missense mutations. Remark-ably, the individual amino acid substitutions identified inNS (germline origin) and leukemia (somatic origin) rarelyoverlap, suggesting different effects of the two classes ofmutations on protein structure and function.22,23
SHP-2 is composed of two tandemly arranged amino-terminal SH2 domains (N-SH2 and C-SH2; residues 3–104 and 112–216, respectively), a single catalytic domain(PTP; residues 221–524), and a carboxy-terminal tail con-taining two tyrosyl phosphorylation sites and a proline-rich stretch (Fig. 1). Both the SH2 domains selectivelybind to short amino acid motifs containing a phosphotyro-syl residue, and promote SHP-2’s association with cell
*Correspondence to: Antonio Palleschi, Dipartimento di Scienze eTecnologie Chimiche, Universita di Roma Tor Vergata, Via dellaRicerca Scientifica s.n.c., 00133 Roma, Italy.E-mail: [email protected]
yThese authors contributed equally to this work.
Received 27 January 2006; Revised 24 March 2006; Accepted 19April 2006
Published online 18 December 2006 in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/prot.21050
VVC 2006 WILEY-LISS, INC.
PROTEINS: Structure, Function, and Bioinformatics 66:963–974 (2007)

surface receptors, cell adhesion molecules, and scaffoldingadapters. Crystallographic data24 suggest a mechanism forSHP-2 regulation: in the absence of a tyrosin-phosphoryl-ated binding partner, a loop of the N-SH2 domain (residues58–62, blocking loop) blocks the catalytic site of PTP do-main. This interaction alters the structure of the N-SH2domain, disrupting its phosphopetide binding cleft. Con-versely, in the crystal structure of the phosphopeptidebound N-SH2 domain, the conformation of the blockingloop is perturbed, loosing its complementarity for the PTPactive site.25 Therefore, N-SH2 binding to phosphotyrosylligands weakens the auto-inhibiting interdomain interac-tion,26 making the catalytic site available to substrates,thereby activating the phosphatase.NS-causing and leukemia-associated SHP-2 mutations
affect different protein regions, but most frequently involveresidues located at the interface between the PTP and N-SH2 domains.23 Genetic and biochemical studies supportthe view that most of these interface lesions promote again of function of the protein by perturbing the autoinhi-bitory interaction between the two domains.13,16,27,28
Recent studies also indicate that the leukemia-associatedPTPN11 mutations have a stronger activating effect thanthe NS-causing ones.16,28–32
To test this hypothesis further, we investigated thestructural and functional consequences of two pairs ofSHP-2 mutants involving residues 72 and 76, which arelocated in the surface of the N-SH2 domain interactingwith the PTP domain (Fig. 1). Ala72Val and Glu76Lys areamong the most common lesions associated with leukemiaand have never been observed in individuals with NS. Con-versely, Ala72Ser and Glu76Asp specifically recur amongsubjects with NS, and were selected for direct comparison
with the leukemia-associated lesions. To explore the conse-quences of these selected mutations on SHP-2 structureand function, molecular dynamics (MD) simulations (up to10 ns long) were performed on the whole protein. A prelim-inary account of these simulations has already been pre-sented,23 but the thorough analysis of the trajectories, pre-sented here, provides a clear picture of the molecularmechanism leading to SHP-2 upregulation. In vitro phos-phatase activities were also determined, basally and afterstimulation with different concentrations of synthetic SH2domain-binding phosphopeptide, and a thermodynamicmodel interpreting these activation profiles is presented.Overall, this study provides a picture of the molecularmechanisms leading to SHP-2 upregulation.
MATERIALS AND METHODSMD Simulations
Initial coordinates of human SHP-2 were taken from theX-ray crystal structure24 (chain A of protein data bankentry 2shp). The recombinant protein used in the crystallo-graphic study lacked the 66-residue C-terminal tail, which,therefore, was not considered in the simulations. Further-more, the coordinates of residues 1, 156–160, 235–245,294–301, 313–323, and 526–527 were not determined.These regions, belonging to flexible loops, are totally sol-vent exposed and are not involved in catalytic or bindingfunctions of the protein, or in the interaction between theN-SH2 and PTP domains. Finally, the crystallized proteindiffers from the reference sequence (Swiss-Prot entryQ06124, WT SHP-2 hereinafter) at three sites (Thr2Lys,Phe41Leu, and Phe513Ser). The conformation of the miss-ing loops was determined by homology modeling by em-ploying the program DeepView 3.7.33 A database of knownloops was scanned to find the best loops connecting the twoamino acid anchor points. These possible loops were eval-uated by sequence homology, and by the number of clashes,putative H-bonds that they can make, and GROMOS96energy in the final model. The same program was used tocorrect residues 2, 41, and 513 as well as to introduce thepathologic mutations, after an analysis of all possiblerotamers of the side chains, and to add polar and aromatichydrogen atoms, providing the initial coordinates of thecomplete chain from residue 2 to 525.
Molecular dynamics (MD) simulations were performedusing GROMACS 3.1.4, with the ffgmx force field.34 Thesimulations were performed according to previously de-scribed procedures.35,36 Briefly, after energy minimiza-tion in vacuo, the protein was centered in a triclinic box(6.4 3 8.5 3 8.7 nm3) and hydrated using the SPC watermodel,37 maintaining the water molecules included in thecrystallographic structure and located within 0.5 nmfrom the protein. The final fully hydrated system con-tained more than 13,000 water molecules. Initial strainsin the system were released by a two-step energy minimi-zation, first restraining all protein atoms to their posi-tions with a harmonic constant of 1000 kJ mol�1 nm�2,and then removing the restraints. Initial atomic veloc-
Fig. 1. Crystallographic structure of SHP-2 (residues 2–525) in its cat-alytically inactive conformation (pdb entry 2shp, chain A). The N-SH2, C-SH2, and PTP domains are shown in blue, green, and red, respectively.The signature motif of the PTP active site (residue 457–467) is colored inyellow and the N-SH2 blocking loop (residue 58–62) in cyan. The sidechains of residues Ala72 and Glu76 (affected in the four mutants ana-lyzed in this study), and Cys459 (the catalytic nucleophile) are reported inlight blue, white, and orange, respectively. [Color figure can be viewed inthe online issue, which is available at www.interscience.wiley.com.]
964 G. BOCCHINFUSO ET AL.
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot

ities were randomly assigned from a Maxwell distributioncorresponding to 300 K. The system was kept at constanttemperature (300 K) and pressure (1 bar) by the Berend-sen weak-coupling method,38 using separate temperaturebaths for protein and solvent, with a relaxation time of0.1 ps for temperature, and 1 ps for pressure. Nonbondedinteractions were treated according to the twin-rangemethod (cut-off radii of 1 and 1.5 nm). A time-step of 2 fswas employed.The initial equilibrations were attained by performing
MD simulations of 100 ps, in which protein atoms (exceptthose belonging to mutated or modeled residues) wereposition restrained. The final files resulting from thesecalculations were used as inputs for subsequent long-scale MD runs. The simulated trajectory was 5.36 ns longin the case of mutant Glu76Lys where the effects of themutation were more marked, while they were prolongedup to 9.86 ns for mutants Glu76Asp, Ala72Val, andAla72Ser simulations. Two simulations were performedfor the WT protein, differing for the random initial veloc-ities: one trajectory was 5.98 ns long (indicated as WT_s)and the other 9.86 ns (WT_l). These calculations wereperformed on the computer cluster of the E. FermiResearch Center of Rome (32 Pentium IV nodes intercon-nected through Myrinet 2000).Root mean square positional fluctuations (RMSF) and
root mean square deviations (RMSD) were calculatedaccording to standard definitions. The following equationwas employed to convert experimentally determined crys-tallographic Debye–Waller B factors to RMSF values:
RMSFðiÞ ¼ffiffiffiffiffiffiffiffiffiffiffiffi3BðiÞ8p2
r:
H-bonds were assigned according to standard GROMACScriteria.Poisson–Boltzmann electrostatic potential calculations
were performed with the program APBS.39 The input filewas prepared by using PDB2PQR,40 and a dielectric con-stant of 2 and 78.54 were employed for the protein andwater, respectively. Molecular graphics and solvent-accessi-ble surface calculations were performed with MOLMOL.41
Figure 9 was produced using the UCSF Chimera package(http://www.cgl.ucsf.edu/chimera/) from the Resource for
Biocomputing, Visualization, and Informatics at the Uni-versity of California, San Francisco.42
Phosphatase Assay
Full-length human His-tagged PTPN11 cDNA cloned inpET-26b (Novagen) was a kind gift from Antonio Pizzuti(Department of Experimental Medicine and Pathology,University ‘‘La Sapienza’’, Rome, Italy). The single nucleo-tide changes resulting in the Ala72Ser, Ala72Val,Glu76Asp, and Glu76Lys were introduced by site-directedmutagenesis (QuikChange Site-Directed Mutagenesis Kit,Stratagene). Recombinant SHP-2 proteins were expressedin E. coli Rosetta 2 (DE3) competent cells (Novagen). Fol-lowing induction, harvesting, and cell lysis, proteins werepurified by chromatography, using Ni-NTA magnetic aga-rose beads (Qiagen). In vitro phosphatase assays were per-formed using 20 pmol of purified recombinant SHP-2 pro-teins in 200 lL of PTP buffer (25 mM Hepes pH 7,4; 50mM NaCl; 2.5 mM EDTA; 60 lg/mL BSA; 5 mM DTT) sup-plemented with 20 mM of para-nitrophenyl phosphate(pNPP) (Sigma) as substrate, either in basal condition orwith the activating SHPS1 (DITpYADLNLPKGKKPAP-
Fig. 2. Root mean square positional fluctuations (RMSF) of SHP-2 Ca atoms calculated between 4.36and 5.36 ns. WT_l (light gray), WT_s (dark gray), Ala72Ser (orange), Ala72Val (red), Glu76Asp (violet), andGlu76Lys (blue). The black curve represents RMSF calculated from the crystallographic B factors (pdb entry2shp, chain A). The line is interrupted in regions whose structure was not determined in the pdb file. The sec-ondary structure of the protein backbone is also shown: green and cyan segments indicate a-helices andb-strands, respectively. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Fig. 3. (A) Root mean square deviations (RMSD) of the N-SH2 do-main Ca atoms from their starting position. Rototranslational motions ofthis domain were removed before RMSD calculations. (B) RMSD fromthe initial structure of the N-SH2 residues forming the phosphopeptidebinding site. Only residues that are in contact with phosphopeptideatoms in the X-ray structure of the N-SH2-peptide complex structure(1aya pdb entry) were considered (residues 13, 14, 17, 32, 34–36, 42,45, 51–55, 65–68, 81, 87–93, and 96). Rototranslational motions of thisregion were removed before RMSD calculations. The color code is asreported in the Figure 2 legend. [Color figure can be viewed in the onlineissue, which is available at www.interscience.wiley.com.]
965SHP-2 MUTANTS: A STRUCTURAL AND FUNCTIONAL STUDY
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot

QAAEPNNHTEpYASIQTS-NH2) (Primm) BTAM peptide,and incubated for 30 min at 293 K. Reaction were stoppedby adding 800 lL of 0.1NNaOH. pNPP dephosphorylationwas evaluated by measuring absorbance at 410 nm.Amount, purity, and integrity of recombinant SHP-2 pro-teins were evaluated using the Protein Assay Kit (Bio-Rad), coomassie staining, and by immunoblot analysiswith anti-SHP-2 monoclonal antibody (Santa Cruz Bio-technology).
RESULTSBackbone Fluctuations
The RMSF of the protein Ca atoms provides a quantita-tive measurement of backbone flexibility. To compare alltrajectories at the same time, this quantity was calculatedbetween 4.36 and 5.36 ns (Fig. 2), corresponding to the lastnanosecond of the shortest simulation (Glu76Lys). Thesame RMSF profile was obtained at different times alongthe trajectories (data not shown). The extent of flexibilityalong the protein sequence was comparable in mutantsand WT protein, and reproduced the mobility profile de-rived from the crystallographic Debye–Waller factors, vali-dating the simulation results. Observed discrepanciesbetween the calculated and experimental values are justi-fied by the different temperature and physicochemicalstate in the simulation and in the protein crystal, and bythe contribution of crystal disorder to the temperature fac-tors.35 Of note, the four loops whose structure was not
determined by X-ray crystallography (residues 156–160,236–245, 295–301, and 313–323) were among the mostmobile regions of the protein in all simulations performed.
N-SH2 Domain Structure
The N-SH2 domain has a crucial role in SHP-2 function:it acts as a conformational switch, regulating the catalyticactivation of the enzyme through its interaction with thePTP domain, and it promotes the protein association withcell surface receptors and scaffolding adapters.24 The fourinvestigated mutations involve residues located in this do-main at the interface with the catalytic site. Therefore,they might perturb both the interdomain interaction andthe conformation of the N-SH2 phosphopeptide bindingpocket, thereby affecting SHP-2 affinity for binding part-ners. To analyze this possible effect, the RMSD from thecrystal structure of the N-SH2 Ca atoms in the WT proteinand in the four mutants was calculated [Fig. 3(A)]. No sub-stantial difference was observed between the differentsimulations, indicating that these mutations did not in-duce any structural perturbation in this protein region.
An analysis of the contribution of each residue to theoverall RMSD showed that the slight deviation from thecrystallographic structural data was localized in the flexi-ble loop regions formed by residues 34–39 and 84–96.These deviations took place within the first 2 ns of simu-lations, after which the structures remained approxi-mately stable. The conformational transition of theseregions with respect to the crystallographic structure wassimilar in all simulations: in the case of residues 34–39deviations were caused by the formation of a salt bridgebetween Lys35 and Glu249, while in the case of residues84–96 they resulted from the formation of a short-betasheet structure (Fig. 4). Interestingly, also the RMSD of
Fig. 4. Comparison between the starting conformation of the N-SH2domain (red) and the structure attained after 2 ns of the WT_l simulation(blue). The regions of maximal deviation (residues 34–39 and 84–96)are evidenced in yellow and light blue on the first and second structure,respectively. [Color figure can be viewed in the online issue, which isavailable at www.interscience.wiley.com.]
Fig. 5. Root mean square deviations (RMSD) of the N-SH2 loop (res-idues 58–62) from its starting position. Rototranslational motions of theprotein were removed by fitting the positions of the PTP signature motifatoms to their coordinates in the initial structure. The color code is asreported in the Figure 2 legend. [Color figure can be viewed in the onlineissue, which is available at www.interscience.wiley.com.]
966 G. BOCCHINFUSO ET AL.
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot

all atoms of the N-SH2 residues in contact with the phos-phopeptide in the X-ray structure of the complex (pdbentry 1aya)25 was comparable in all the different simula-tions [Fig. 3(B)], suggesting that the mutations shouldhave no major direct effect on the affinity of this domainfor its binding partners.An analysis of intradomain interactions of the mutated
residues indicated that no unfavorable energies were in-troduced by the amino acid substitutions (data not shown),providing a rationale for the observed lack of structuraldifferences between the various mutants. Surprisingly,this was the case even for the Glu76Lys mutant, in whichthe side-chain charge sign changed. In the WT protein, theelectrostatic interaction of residue 76 with the rest ofthe domain was attractive. However, as a consequence ofthe different position of the charged atoms in the side-chains of Glu and Lys, no repulsion arose in the Glu76Lysmutant.
Interdomain Motions
Behavior of SHP-2 mutants was markedly differentwhen considering the N-SH2/PTP interdomain interaction.The simulations of the leukemia-related mutants demon-strated a distinct perturbation of the interface between thePTP domain and the N-SH2 loop (residues 58–62), whichwas displaced significantly from its starting position whereit blocks the active site. This relative motion was quanti-fied by the RMSD of the blocking loop, calculated after therigid roto-translational motions of the protein were re-moved by fitting the PTP signature motif (residues 457–467) to its starting position (Fig. 5). Remarkably, theleukemia-associated Ala72Val and Glu76Lys mutants dis-played a much larger deviation from the starting confor-mation than the other simulations. On the other hand, theRMSD of the NS-causing Glu76Asp and Ala72Ser mu-tants were not significantly different from those of the twoWT trajectories.This displacement of the N-SH2 loop was mainly due to
a collective motion of the regulatory domain with respect
to the active site rather than to a local rearrangement ofthe N-SH2 structure, as shown by the lack of structuraldifferences between the various mutants in this domain.
Interactions Stabilizing the Inactive Conformationin the WT Protein
To individuate the causes leading to the perturbation ofthe interface between N-SH2 and PTP domains in thepathogenic mutants, the interdomain interactions stabiliz-ing the inactive conformation in the WT protein were ana-lyzed. In the crystal structure of SHP-224 and the structur-ally and functionally related SHP-143 the interfacebetween the N-SH2 and PTP domains is stabilized by foursalt bridges (formed by Arg4-Glu252, Arg4-Glu258, Asp61-Lys366, Glu76-Arg265 in SHP-2) and by several hydrogenbonds involving residues Asn58 and Gln506, Gly60 andGln510, Glu69 and Asn281, Ala72 and Gln506, and Glu76and Ser502 (Fig. 6). All these interactions were essentiallymaintained during both simulations of the WT protein(Table I and Fig. 7). Three additional interdomain bondswith a comparable stability were identified. Lys35 andGlu249 formed a salt bridge within the first 100 ps of thesimulations, taking advantage of the flexibility of the N-SH2 loop comprising residues 34–39 (see earlier). Further-more, Asp61 formed two extra interdomain H-bonds in
Fig. 6. Crystallographic structure of the N-SH2/PTP interdomain region, evidencing the salt bridges (Panel A) and H-bonds (Panel B) stabilizing thecatalytically inactive conformation in the WT SHP-2 protein. The PTP domain is represented by partially transparent red ribbons, with the signaturemotif (residues 457–467) evidenced in yellow. The N-SH2 domain is colored in blue, with the blocking loop (residues 58–62) evidenced in cyan. Inthe side chain of residues involved in salt bridges or H-bonds, C atoms (gray), N atoms (blue), O atoms (red), and H atoms (white) are evidenced. Forthe sake of clarity, only H atoms involved in H-bonds are shown. The interactions previously documented in the crystallographic structure24 arereported as black dashed lines, while those formed during the present simulations of the WT protein are reported in green. [Color figure can be viewedin the online issue, which is available at www.interscience.wiley.com.]
TABLE I. Distance Between the Centers ofMass of Side-Chain Charged Atoms of ResiduesInvolved in Interdomain Salt Bridges in the
WT SHP-2 Simulations
Salt bridge WT_l (nm) WT_s (nm)
Arg4-Glu252 0.23 � 0.03 0.4 � 0.1Arg4-Glu258 0.35 � 0.07 0.38 � 0.09Lys35-Glu249 0.4 � 0.1 0.4 � 0.1Asp61-Lys366 0.27 � 0.06 0.26 � 0.03Glu76-Arg265 0.29 � 0.06 0.33 � 0.08
The distances were computed from molecular dynamics trajectories;average values and standard deviations are reported.
967SHP-2 MUTANTS: A STRUCTURAL AND FUNCTIONAL STUDY
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot

both simulations of the WT protein. One, involving Ser460,was already present after the initial energy minimizationprocedure; the second, involving Gln506, formed afterapproximately 50 ps. Both remained stable during the sim-ulations of the WT protein (Fig. 7).
Glu76Lys
The simulation of the Glu76Lys mutant displayed themost drastic perturbation of the interface between N-SH2and PTP domains. In this case, the relative motion be-tween these two protein regions was clearly due to anelectrostatic repulsion. Figure 8(A) shows the Coulombpotential energy between residue 76 and the PTP domain,calculated for the WT (simulation WT_s) and the mutant.In the latter, a repulsive strain occurred. This was mainlydue to the interaction of residue 76 with Arg265, involvedin a salt bridge in the WT protein, and with Arg498,although to a lesser extent. In the initial conformation,the electrostatic interaction of residue 76 with Arg265 andArg498 accounted respectively for �67 and 27% of theenergy difference between WT and the Glu76Lys mutantshown in Figure 8(A). The effects of the Glu76Lys substi-tution on the electrostatic interaction between the N-SH2and PTP domains are shown pictorially in Figure 9.Figure 9(A) indicates the position of charged side-chainsin the region surrounding Glu76, in the starting structureof the MD simulation of WT SHP-2, evidencing the salt-bridges between residues Glu76 and Arg265, Arg4 andGlu252, and Arg4 and Glu258. Figure 9(B) shows thevalue of the electrostatic potential generated by the N-SH2 domain on the PTP surface, resulting from Poisson–Boltzmann electrostatic calculations performed on thesame structure. Positively and negatively PTP chargedresidues are located in regions where the N-SH2 gener-ated potential is negative and positive, respectively, ac-counting for the favorable electrostatic energy. By con-
Fig. 7. Existence plot of interdomain H-bonds between PTP and N-SH2 for the WT_l (gray) and WT_s (black) simulations. The presence ofa specific H-bond at a given time is indicated by a vertical bar. The resi-dues involved in H-bond formation are reported on the vertical axis.
Fig. 8. Glu76Lys simulation. (A) Electrostatic energy between residue76 and the PTP domain, during the MD trajectories Glu76Lys (black) andWT_s (gray). (B) Distance between the centers of mass of the N-SH2blocking loop (residues 58–62) and of the PTP signature motif (residues457–467). Glu76Lys (black), WT_s (gray). (C) Comparison between theinitial (light gray) and final (dark gray) conformations attained during thesimulation of the Glu76Lys SHP-2 mutant. The N-SH2 domain and the sig-nature motif of the PTP domain active site (residues 457–467) aredepicted with a ribbon representation. To evidence interdomain motions,rototraslation of the whole protein was removed by fitting the coordinatesof the PTP signature motif atoms to their initial positions.
968 G. BOCCHINFUSO ET AL.
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot
trast, the Glu76Lys substitution generates a region of pos-itive potential in correspondence of basic residues Lys265and Arg498, determining a strong repulsion between thetwo domains [Fig. 9(C)]. During the MD simulation thisrepulsion caused a slow drift between the N-SH2 loop andPTP active site [Fig. 8(B)], leading to a relaxation of theelectrostatic stress [Fig. 8(A)], even though the simulationtime was not sufficient to attain equilibrium.The structural effects caused by this mutation during
the simulation are shown in Figure 8(C), where the ini-tial structure is compared with that attained at the endof the trajectory.
Fig. 9. Electrostatic effects of the Glu76Lys mutation. (A) Position ofcharged residues surrounding Glu76 in WT SHP-2, in the starting con-formation of MD simulations. The position of charged PTP residues isindicated by a red (negative charge) or blue (positive charge) color onthe PTP surface. The side chain of N-SH2 charged residues Glu76 andArg4 are shown in a ball and stick representation, with oxygen and nitro-gen atoms colored in red and blue, respectively. (B) Electrostatic poten-tial generated by the N-SH2 domain on the PTP surface in the startingconformation of WT SHP-2. Red and blue colors indicate potential val-ues going from �40 kT/e to þ40 kT/e. (C) Electrostatic potential gener-ated by the N-SH2 domain on the PTP surface in the starting conforma-tion of the E76K mutant. Red and blue colors indicate potential valuesgoing from �40 kT/e to þ40 kT/e. Fig. 10. Ala72Val simulation. (A) van der Waals energy between res-
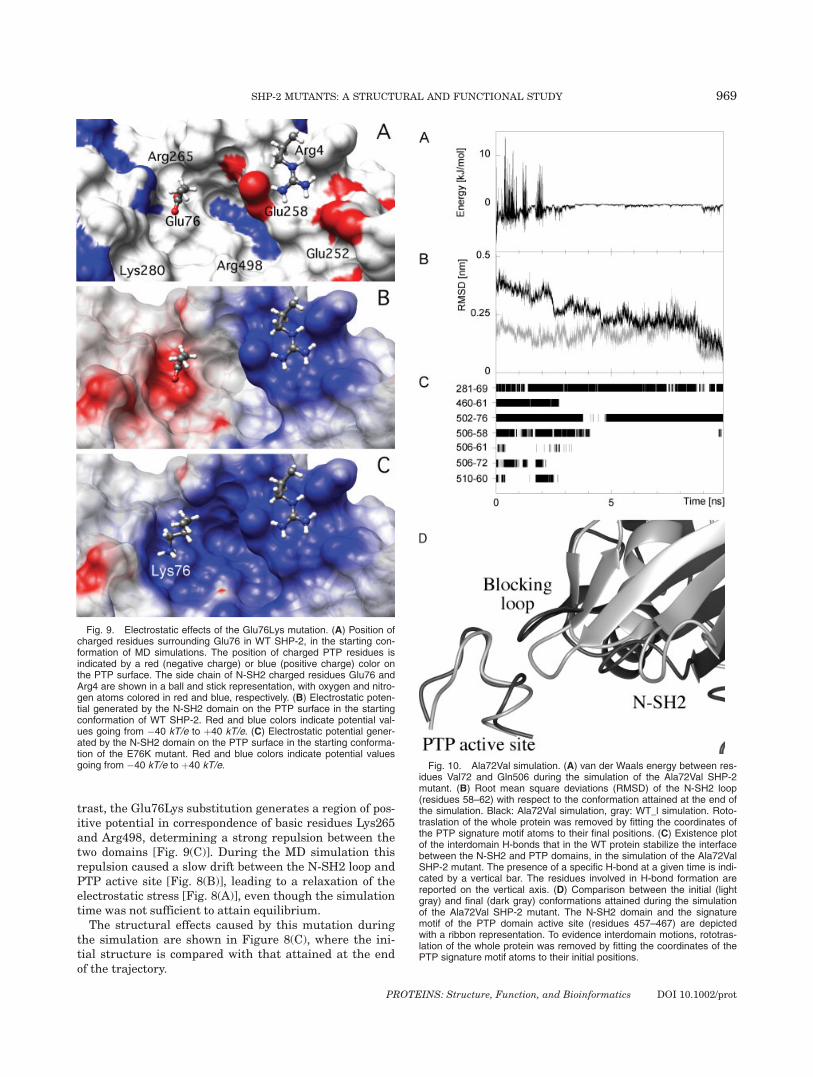
idues Val72 and Gln506 during the simulation of the Ala72Val SHP-2mutant. (B) Root mean square deviations (RMSD) of the N-SH2 loop(residues 58–62) with respect to the conformation attained at the end ofthe simulation. Black: Ala72Val simulation, gray: WT_l simulation. Roto-traslation of the whole protein was removed by fitting the coordinates ofthe PTP signature motif atoms to their final positions. (C) Existence plotof the interdomain H-bonds that in the WT protein stabilize the interfacebetween the N-SH2 and PTP domains, in the simulation of the Ala72ValSHP-2 mutant. The presence of a specific H-bond at a given time is indi-cated by a vertical bar. The residues involved in H-bond formation arereported on the vertical axis. (D) Comparison between the initial (lightgray) and final (dark gray) conformations attained during the simulationof the Ala72Val SHP-2 mutant. The N-SH2 domain and the signaturemotif of the PTP domain active site (residues 457–467) are depictedwith a ribbon representation. To evidence interdomain motions, rototras-lation of the whole protein was removed by fitting the coordinates of thePTP signature motif atoms to their initial positions.
969SHP-2 MUTANTS: A STRUCTURAL AND FUNCTIONAL STUDY
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot

Ala72Val
The Ala72Val mutant is the other instance in which asignificant perturbation of the interdomain interface wasobserved. In this case the origin of this effect is more puz-zling, since this amino acid substitution is rather conserv-ative. According to the crystal structure of SHP-2 in itscatalytically inactive conformation, Ala72 is in close con-tact with the PTP domain surface, in particular with resi-dues Asn281, Ile282, Ile463, Ser502, Gly503, and Gln506.This area of the PTP surface plays a very delicate role inthe interaction with the N-SH2 domain since a number ofdisease-causing mutations of the PTP domain are found inthis region,23 and residues 281, 502, and 506 contribute tothe H-bond network stabilizing the inactive conformation(Fig. 7). The mutation to Val introduces steric clashes withthe PTP surface. Most of these strains relaxed very rap-idly during the simulation, but an unfavorable van derWaals interaction between Val72 and Gln506 (which arehydrogen bonded) was still present from time to time upto approximately 2 ns [Fig. 10(A)]. At that time a confor-mational rearrangement of the interdomain interfaceoccurred, driving residues 72 and 506 apart.The RMSD should be a useful parameter to evidence
this structural transition. However, when the startingstructure was taken as a reference, effects related to theequilibration of the simulation obscured deviations causedby sampling of the conformational space. These two contri-butions can be separated by calculating the RMSD withrespect to the final structure.44 Indeed, a clear stepbetween 2 and 3 ns was visible in the RMSD of the N-SH2loop atoms, when calculated taking the final conformationas a reference [Fig. 10(B)], while it was less evident in the
RMSD calculated from the initial structure (Fig. 5). Theinteractions of the N-SH2 loop with the PTP domainchanged markedly after this transition. Most of the H-bonds occurring in the interdomain interface of the WTprotein were lost in the simulation of the Ala72Val mu-tant, including the one involving the mutated residue andGln506 [Fig. 10(C)]. Furthermore, at the same time saltbridges between Arg4 and Glu252, and Asp61 and Lys366increased their distance from approximately 0.25 to 0.45nm. The structural effects of this conformational transi-tion at the interface between PTP active site and N-SH2domain are shown in Figure 10(D), where the initial struc-ture is compared with that attained at the end of the tra-jectory.
NS-Causing Mutants: Glu76Asp and Ala72Ser
In contrast to the leukemia-associated SHP-2 mutants,the NS-causing Glu76Asp and Ala72Ser mutants did notdisplay any significant displacement of the N-SH2 loopfrom the PTP active site during the simulated trajecto-ries. However, the Glu76Asp substitution caused a desta-bilization of the interdomain interface. By shortening theside-chain of residue 76, this mutation weakened itsinterdomain H-bond with Ser502. This apparently smalleffect generated a cascade of events, leading to a subtlerearrangement of the interface between the N-SH2 andPTP domains and determining the loss of several stabiliz-ing interactions present in the WT protein. After approxi-mately 1 ns the H-bond between Asp76 and Ser502 waspermanently lost and Asp76 formed a new H-bond withAsn281, which, in turn, lost its H-bond with Glu69 [Fig.11(A)]. Furthermore, after approximately 3 ns, loss of the
Fig. 11. Glu76Asp simulation. (A) Existence plot of the interdomain H-bonds that in the WT protein stabi-lize the interface between the N-SH2 and PTP domains, in the simulation of the Glu76Asp SHP-2 mutant.The presence of a specific H-bond at a given time is indicated by a vertical bar. The residues involved in H-bond formation are reported on the vertical axis. The H-bond formed by residues 281 and 76 (which is notstably present in the simulations of the WT protein) is shown in gray. (B) Histogram of the relative population(P) of the dihedral angle values for w of Gly503 and / of Met504. The Glu76Asp simulation (black) is com-pared with the WT_s (dark gray) and WT_l (light gray) trajectories. (C) Distance between the centers of massof side-chain charged atoms of residues Asp61 and Lys366 during the simulation of the Glu76Asp SHP-2mutant.
970 G. BOCCHINFUSO ET AL.
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot

H-bond between Asp76 and Ser502 caused a conforma-tional rearrangement of the PTP loop formed by residues500–508, in correspondence of Gly503 and Met504, asevidenced by a transition in the w angle of Gly503 and inthe / angle of Met504 [Fig. 11(B)]. This region is crucialin the interdomain interface, and its structural rear-rangement led to loss of Asp61 interaction with Ser460(H-bond) and Lys366 (salt bridge) [Fig. 11(C)].No destabilization of the interdomain interface was
observed during the simulation of the Ala72Ser mutant,and only the hydrogen bond between residues Asp61 andSer460 was lost after 6.75 ns (data not shown). However,it is interesting to note that, during our simulation, theSer72 side chain did not form any additional interdomainH-bonds, but rather bound to the side chain of Glu76.
Enzymatic Activity
To explore the functional consequences of the mutationsinvestigated, WT and mutant SHP-2 proteins were ex-pressed in bacteria and their phosphatase activities weredetermined in vitro, basally and after stimulation withvarious concentrations of the BTAM phosphotyrosyl pep-tide (Fig. 12). As expected, all the mutants increased theirbasal catalytic activity with respect to the WT protein,with highest activation observed in the two leukemia-as-sociated mutants. The order of basal activity was Glu76Lys> Ala72Val > Glu76Asp > Ala72Ser > WT. Addition of theBTAM phosphopeptide caused a further activation of theproteins, with the exception of Glu76Lys that appeared
completely activated already in basal conditions. Of note,the experimental data also showed a differential behaviorof the phosphatase activity of the other mutants as a func-tion of BTAM peptide concentration, indicating that ahigher basal activity is correlated with an increased re-sponsiveness to the activating phosphotyrosyl peptide.
No substantial structural effect of mutations on thephosphopeptide-binding pocket was documented by ourMD analyses. Consistently, the increased responsivenessof mutants to the BTAM peptide does not necessarilyimply a direct effect of the mutations on the N-SH2 do-main, as a simple thermodynamic analysis can demon-strate. In the crystallographic structure, with the PTPactive site blocked by the N-SH2 loop, no phosphataseactivity is possible.24 Therefore, the presence of a basalenzymatic activity indicates that, even in absence ofBTAM peptide, an equilibrium is present between thisclosed, inactive conformation (indicated by I, in SchemeI) and an open, active conformation (A). Since all muta-tions analyzed in this study are located in the N-SH2 do-main, we assume that the phosphatase activity of the Aconformation (a) is not affected by the aminoacidic sub-stitutions. Therefore, the higher basal activity (A0)observed in the mutants would be due to an increase inthe equilibrium constant (Kact) for the I$A conforma-tional transition. Under these hypotheses, the basal ac-tivity is given by
A0 ¼ aKact
1þ Kactð1Þ
Let KA and KI be the association constants of BTAM peptidewith the A and I conformations, respectively (Scheme I).According to this simple scheme, the phosphatase activity(A) as a function of BTAM peptide concentration (C) isgiven by
A ¼ A0 þ ðA1 � A0Þ KC
1þ KCð2Þ
where
A1 ¼ A01þ Kact
KI
KAþ Kact
ð3Þ
Fig. 12. In vitro phosphatase assay of WT (circles) and mutatedSHP-2 proteins (Ala72Ser: squares, Glu76Asp: triangles, Ala72Val: dia-monds, Glu76Lys: inverted triangles). Activity was measured as pmolesof phosphate released using para-nitrophenyl phosphate (pNPP) as sub-strate, as a function of SH2 domain-binding peptide (phosphorylatedSHPS1 BTAM motif45). Values are means � SD of at least four inde-pendent experiments, and are normalized to unstimulated WT SHP-2.Continuous lines represent a global fit performed according to Eq. (2),with a common A? value for all mutants (A? ¼ 8.92).
Scheme 1
971SHP-2 MUTANTS: A STRUCTURAL AND FUNCTIONAL STUDY
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot

is the activity at saturating BTAM peptide concentra-tions, and
K ¼ KI1
1þ Kactþ KA
1
1þ 1Kact
ð4Þ
is the apparent constant describing the hyperbolic behav-ior of the activity as a function of phosphopeptide concen-tration.BTAM binding leads to an increase in activity, and
therefore A? > A0: this implies that KA > KI [see Eq. (3)].The fact that BTAM binding shifts the conformationalequilibrium towards A indicates that this conformationhas a higher affinity than I for the peptide. This finding isconfirmed by the closed conformation observed for the N-SH2 phosphotyrosyl peptide binding site in the crystalstructure.24
This model provides a simple explanation for the ob-served correlation between basal activity A0 and the de-pendence of activation on BTAM peptide concentration.When a mutation increases Kact (and A0), the equilibriumshifts towards the A conformation, which has a higher af-finity for the phosphopeptide. As a result, according toEq. (4), the apparent constant K increases (as qualita-tively observed in our experimental data) even if theamino acidic substitution does not have a significantdirect effect on the N-SH2 domain affinity for the activat-ing peptide (KI and KA). This is in agreement with ourMD simulations, showing the absence of structural per-turbations in the phosphopeptide binding cleft.This model can be further simplified by assuming KA �
KI, an approximation justified by the crystallographicstructure of the inactive conformation. In this case, Eq. (3)becomes
A1 ¼ a ð5Þ
indicating that the plateau activity value is the same forWT protein and SHP-2 mutants.This approximation, employed in the fits of Figure 12,
accounts satisfactorily for all the experimental data. Byintroducing this value in Eq. (1) it was possible to deriveKact from the basal activity A0, obtaining an estimate of
the standard free energy of the I–A equilibrium (DG0 ¼�RT ln Kact, Table II).
DISCUSSION
Germinally transmitted PTPN11 mutations representa major molecular event underlying NS, while somaticmutations in the same gene contribute to a wide spec-trum of leukemic disorders.22 An analysis of the NS-caus-ative and leukemia-associated PTPN11 mutations indicatesa clear-cut genotype/phenotype correlation, supporting amodel in which distinct gain-of-function thresholds forSHP-2 are required to induce cell-, tissue-, or developmental-specific phenotypes.16,23
In this study, MD simulations were employed to ana-lyze the structural and functional consequences of fourdisease-associated PTPN11 missense mutations affectingresidues Ala72 (Ala72Val and Ala72Ser) and Glu76(Glu76Lys and Glu76Asp) that are located within the N-SH2 portion of the protein interacting with the PTP do-main and participate to the interdomain bonding networkthat stabilizes SHP-2 in its closed, catalytically inactiveconformation. Our data indicate that these amino acidsubstitutions do not have any significant effect on theconformation of the regulatory N-SH2 domain, wherethey are located, but rather affect the interaction of thisdomain with the catalytic site. This observation providesa rationale for the increased basal activity of thesemutants and supports the hypothesis that these NS-caus-ing and leukemia-associated mutations destabilize theclosed conformation observed in the crystallographicstructure, where the PTP active site is masked by the N-SH2 loop, favoring a more open, active conformation.
Proper interpretation of the computational data re-quires a careful consideration of the time-scale involved inthese interdomain motions. At present, MD simulations ofa relatively large protein, such as SHP-2, are limited toapproximately 10 ns, but interdomain motions generallytake place on a much longer time-scale, probably of theorder of several ls.46 Consequently, our MD trajectorieswere not able to attain complete equilibration, and do notprovide information on the open, active protein conforma-tion. On the other hand, the present study illustrates indetail the molecular origin of the destabilization of the N-SH2/PTP interdomain interaction.
In the leukemia-related Ala72Val and Glu76Lys mu-tants a significant displacement of the N-SH2 loop fromthe PTP site was observed. Such perturbation probablyrepresents the beginning of the conformational transitionleading to the open state. Therefore, we can reasonablyassume that in these two mutants the driving force forthis process is comparatively higher, as confirmed by theanalysis of interdomain interactions, and by the experi-mental data on phosphatase activities. Since no effect ofthe mutations on the overall N-SH2 domain structurewas observed during simulations, the destabilization ofthe interdomain association must arise from a perturba-tion of specific interactions. For this reason, salt-bridgesand H-bonds formed between the PTP and N-SH2
TABLE II. Experimental Standard FreeEnergy for the Equilibrium Betweenthe Inactive and Active Conformation
Protein DG8 (kJ mol�1)a D(DG8) (kJ mol�1)b
WT 5.1 � 0.2 —Ala72Ser 3.0 � 0.1 2.1 � 0.2Ala72Val �0.2 � 0.5 5.3 � 0.5Glu76Asp 1.6 � 0.2 3.6 � 0.3Glu76Lys �5.2 � 2 10 � 2
Standard deviations are also reported.aStandard free energy for the equilibrium between the inactive andactive conformation, derived from the basal activity data reported inFigure 12.bRelative destabilization of the inactive conformation induced by themutation. D(DG8) ¼ DG8(WT) � DG8(mutant).
972 G. BOCCHINFUSO ET AL.
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot

domains were analyzed in detail. Our analyses indicatethat the Glu76Lys substitution introduces a strong elec-trostatic repulsion with Arg265, while Ala72Val gener-ated a steric stress with the PTP surface, leading to lossof several stabilizing interactions. No appreciable dis-placement of the N-SH2 loop from the PTP active sitewas observed in the Ala72Ser and Glu76Asp simulations.However, in the latter, the side chain shortening led toloss of hydrogen bond with Ser502, which, in turn, causeda subtle rearrangement of the interdomain interface. Oursimulation did not exhibit any destabilization of theclosed protein conformation in the case of the Ala72Sermutant, while the experimental data showed a small butsignificant activation of this protein with respect to theWT enzyme. It should be considered that a shift towardsthe active conformation of the protein can be obtained ei-ther by a destabilization of the closed structure, or by astabilization of the open conformation. The latter effectwas not taken into account in our simulations, and isprobably relevant in the case of Ala72Ser substitution,which introduces a hydrophilic side chain. A statisticalanalysis of globular protein supports this hypothesis,since alanine residues are on average only 12% solventexposed, while serine residues are solvent exposed for28%.47 More quantitatively, the solvation free energiesfor these residues differ by approximately 2 kJ mol�1.48
It is interesting to note that this value agrees perfectlywith the relative destabilization free energy of the inac-tive conformation in Ala72Ser. The change in side-chainhydrophobicity introduced by the other mutations isminor, and probably negligible because of the largereffects observed in the closed conformation during thesimulations.Overall, our simulations suggest a much higher destabili-
zation of the interdomain interface in the leukemia relatedSHP-2 mutants, with the highest effect associated with theGlu76Lys change. A significant destabilization was ob-served for the NS-causing Glu76Asp mutant, while no di-rectly observable effects where present in the Ala72Ser mu-tant. This ranking nicely parallels the experimentallydetermined basal phosphatase activities, strongly support-ing the reliability of the simulations.The experimental data also indicate that the mutants
with higher basal activity are more responsive to activat-ing phosphopeptide. Our simulations suggest that this isnot due to a conformational change in the N-SH2 domain,but mainly to a shift in the equilibrium between the inac-tive and active conformations of SHP-2. The latter form,more populated in the disease-associated SHP-2 mutantsthan in the WT protein, has a higher affinity for the phos-phopeptides. Therefore, the mutations investigated in thisstudy have a twofold effect: they both destabilize the cata-lytically inactive, closed conformation, and favor SHP-2association with its signaling partners. This finding is rel-evant with respect to the functional consequences of theSHP-2 mutants in perturbing intracellular signal flow. Ofnote, no developmental defect was observed in mice heter-ozygous for a catalytically activated SHP-2 mutant lack-ing the N-SH2 domain,6 demonstrating that a simple
increase in basal activity caused by the removal of the N-SH2 blocking loop from the PTP active site is not sufficientto affect signaling cascades regulated by SHP-2. On theother hand, mice homozygous for the same mutant alleledie in utero at mid gestation. These findings suggest thatproper binding of SHP-2 to its intracellular signaling part-ners is required for function of this phosphatase. Consist-ent with this experimental evidence, the present data sup-port the view that the mutations affecting the N-SH2stretch interacting with the PTP domain increase theoverall protein affinity for phosphopeptides by shifting theconformational equilibrium towards the open conforma-tion. According to this model, the augmented basal phos-phatase activity documented for the NS-causative and leu-kemia-associated mutants should be considered solely asan indirect measure of the degree of destabilization of theinterdomain interaction. On the other hand, the presentmodel implicitly suggests that local structural changes ofthe N-SH2 domain not involving the PTP-interacting sur-face would represent an alternative molecular mechanismpromoting SHP-2 gain of function. Indeed, this is the caseof the recurrent pathogenic Thr42Ala and Glu139Asp sub-stitutions affecting the phosphopeptide binding clefts ofthe SH2 domains recently documented to promote higheractivation of the protein in response to phosphopeptides,without altering the basal SHP-2 catalytic activity.23,28
REFERENCES
1. Neel BG, Gu H, Pao L. The ‘Shp’ing news: SH2 domain-contain-ing tyrosine phosphatases in cell signaling. Trends Biochem Sci2003;28:284–293.
2. Maroun CR, Naujokas MA, Holgado-Madruga M, Wong AJ, ParkM. The tyrosine phosphatase SHP-2 is required for sustainedactivation of extracellular signal-regulated kinase and epithelialmorphogenesis downstream from the met receptor tyrosine kinase.Mol Cell Biol 2000;20:8513–8525.
3. Shi ZQ, Yu DH, Park M, Marshall M, Feng GS. Molecular mech-anism for the Shp-2 tyrosine phosphatase function in promotinggrowth factor stimulation of Erk activity. Mol Cell Biol 2000;20:1526–1536.
4. Cunnick JM, Meng S, Ren Y, Desponts C, Wang HG, Djeu JY,Wu J. Regulation of the mitogen-activated protein kinase signal-ing pathway by SHP2. J Biol Chem 2002;277:9498–9504.
5. Tang TL, Freeman RM Jr, O’Reilly AM, Neel BG, Sokol SY. TheSH2-containing protein-tyrosine phosphatase SH-PTP2 is requiredupstream of MAP kinase for early Xenopus development. Cell1995;80:473–483.
6. Saxton TM, Henkemeyer M, Gasca S, Shen R, Rossi DJ, Shalaby F,Feng G-S, Pawson T. Abnormal mesoderm pattering in mouse em-bryos mutant for the SH2 tyrosine phosphatase Shp-2. EMBO J1997;16:2352–2364.
7. Saxton TM, Ciruna BG, Holmyard D, Kulkarni S, Harpal K,Rossant J, Pawson T. The SH2 tyrosine phosphatase Shp2 is re-quired for mammalian limb development. Nat Genet 2000;24:420–423.
8. Qu C-K, Yu W-M, Azzarelli B, Cooper S, Broxmeyer HE, Feng G-S.Biased suppression of hematopoiesis and multiple developmentaldefects in chimeric mice containing Shp-2 mutant cells. Mol CellBiol 1998;18:6075–6082.
9. Chen B, Bronson RT, Klaman LD, Hampton TG, Wang J-F,Green PJ, Magnuson T, Douglas PS, Morgan JP, Neel BG. Micemutants for Egfr and Shp2 have defective cardiac semilunar val-vulogenesis. Nat Genet 2000;24:296–299.
10. Qu CK, Shi ZQ, Shen R, Tsai FY, Orkin SH, Feng GS. A deletionmutation in the SH2-N domain of Shp-2 severely suppresses he-matopoietic cell development. Mol Cell Biol 1997;17:5499–5507.
973SHP-2 MUTANTS: A STRUCTURAL AND FUNCTIONAL STUDY
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot

11. Qu CK, Nguyen S, Chen J, Feng GS. Requirement of Shp-2 tyro-sine phosphatase in lymphoid and hematopoietic cell development.Blood 2001;97:911–914.
12. Tartaglia M, Gelb BD. Noonan syndrome and related disorders:genetics and pathogenesis. Ann Rev Genomics Hum Genet 2005;6:45–68.
13. Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG,Kremer H, van der Burgt I, Crosby AH, Ion A, Jeffery S, KalidasK, Patton MA, Kucherlapati RS, Gelb BD. Mutations in PTPN11,encoding the protein tyrosine phosphatase SHP-2, cause Noonansyndrome. Nat Genet 2001;29:465–468.
14. Tartaglia M, Kalidas K, Shaw A, Song X, Musat DL, van derBurgt I, Brunner HG, Bertola DR, Crosby A, Ion A, Kucherlapati RS,Jeffery S, Patton MA, Gelb BD. PTPN11 mutations in Noonan syn-drome: molecular spectrum, genotype–phenotype correlation, andphenotypic heterogeneity. Am J Hum Genet 2002;70:1555–1563.
15. Noonan JA. Noonan syndrome. An update and review for the pri-mary pediatrician. Clin Pediatr (Phila) 1994;33:548–555.
16. Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J,Jung A, Hahlen K, Hasle H, Licht JD, Gelb BD. Somatic muta-tions in PTPN11 in juvenile myelomonocytic leukemia, myelodys-plastic syndromes and acute myeloid leukemia. Nat Genet 2003;34:148–150.
17. Loh M, Vattikuti S, Schubbert S, Reynolds MG, Carlson E,Lieuw KH, Cheng JW, Lee CM, Stokoe D, Bonifas JM, Curtiss NP,Gotlib J, Meshinchi S, Le Beau MM, Emanuel PD, Shannon KM.Mutations in PTPN11 implicate the SHP-2 phosphatase in leuke-mogenesis. Blood 2004;103:2325–2331.
18. Loh ML, Martinelli S, Cordeddu V, Reynolds MG, Vattikuti S,Lee CM, Wulfert M, Germing U, Haas P, Niemeyer C, Beran ME,Strom S, Lubbert M, Sorcini M, Estey EH, Gattermann N,Tartaglia M. Acquired PTPN11 mutations occur rarely in adultpatients with myelodysplastic syndromes and chronic myelo-monocytic leukemia. Leuk Res 2005;29:459–462.
19. Tartaglia M, Martinelli S, Cazzaniga G, Cordeddu V, Iavarone I,Spinelli M, Palmi C, Carta C, Pession A, Arico M, Masera G,Basso G, Sorcini M, Gelb BD, Biondi A. Genetic evidence for line-age- and differentiation stage-related contribution of somaticPTPN11 mutations to leukemogenesis in childhood acute leuke-mia. Blood 2004;104:307–313.
20. Tartaglia M, Martinelli S, Iavarone I, Cazzaniga G, Spinelli M,Giarin E, Petrangeli V, Carta C, Masetti R, Arico M, Locatelli F,Basso G, Sorcini M, Pession A, Biondi A. Somatic PTPN11mutationsin childhood acute myeloid leukaemia. Br J Haematol 2005;129:333–339.
21. Loh ML, Reynolds MG, Vattikuti S, Gerbing RB, Alonzo TA,Carlson E, Cheng JW, Lee CM, Lange BJ, Meshinchi S. PTPN11mutations in pediatric patients with acute myeloid leukemia:results from the Children’s Cancer Group. Leukemia 2004;18:1831–1834.
22. Tartaglia M, Gelb BD. Germ-line and somatic PTPN11 mutationsin human disease. Eur J Med Genet 2005;48:81–96.
23. Tartaglia M, Martinelli S, Stella L, Bocchinfuso G, Flex E,Cordeddu V, Bale S, Zampino G, van der Burgt I, Palleschi A,Petrucci TC, Sorcini M, Schoch C, Foa R, Emanuel PE, Gelb BD.Diversity and functional consequences of germline and somaticPTPN11 mutations in human disease. Am J Hum Genet 2006;78:279–290.
24. Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE. Crystalstructure of the tyrosine phosphatase SHP-2. Cell 1998;92:441–450.
25. Lee CH, Kominos D, Jacques S, Margolis B, Schlessinger J,Shoelson SE, Kuriyan J. Crystal structures of peptide complexesof the amino-terminal SH2 domain of the Syp tyrosine phospha-tase. Structure 1994;2:423–438.
26. Dechert U, Adam M, Harder KW, Clark-Lewis I, Jirik F. Charac-terization of protein tyrosine phosphatase SH-PTP2. Study ofphosphopeptide substrates and possible regulatory role of SH2domains. J Biol Chem 1994;269:5602–5611.
27. Fragale A, Tartaglia M, Wu J, Gelb BD. Noonan syndrome-asso-ciated SHP2/PTPN11 mutants cause EGF-dependent prolongedGAB1 binding and sustained ERK2/MAPK1 activation. HumMutat 2004;23:267–277.
28. Keilhack H, David FS, McGregor M, Cantley LC, Neel BG.Diverse biochemical properties of Shp2 mutants: implications fordisease phenotypes. J Biol Chem 2005;280:30984–30993.
29. Chan RJ, Leedy MB, Munugalavadla V, Voorhorst CS, Li Y, Yu M,Kapur R. Human somatic PTPN11 mutations induce hematopoi-etic cell hypersensitivity to granulocyte-macrophage colony stimu-lating factor. Blood 2005;105:3737–3742.
30. Mohi MG, Williams IR, Dearolf CR, Chan G, Kutok JL, Cohen S,Morgan K, Boulton C, Shigematsu H, Keilhack H, Akashi K,Gilliland DG, Neel BG. Prognostic, therapeutic, and mechanisticimplications of a mouse model of leukemia evoked by Shp2(PTPN11) mutations. Cancer Cell 2005;7:179–191.
31. Niihori T, Aoki Y, Ohashi H, Kurosawa K, Kondoh T,Ishikiriyama S, Kawame H, Kamasaki H, Yamanaka T, TakadaF, Nishio K, Sakurai M, Tamai H, Nagashima T, Suzuki Y, KureS, Fujii K, Imaizumi M, Matsubara Y. Functional analysis ofPTPN11/SHP-2 mutants identified in Noonan syndrome andchildhood leukemia. J Hum Genet 2005;50:192–202.
32. Shubbert S, Lieuw K, Rowe SL, Lee CM, Li X, Loh ML, Clapp DW,Shannon KM. Functional analysis of leukemia-associated PTPN11mutations in primary hematopoietic cells. Blood 2005;106:311–317.
33. Guex N, Peitsch MC. Swiss-model and the Swiss-PdbViewer: anenvironment for comparative protein modeling. Electrophoresis1997;18:2714–2723.
34. Lindhal E, Hess B, van der Spoel D. GROMACS 3.0: a package formolecular simulation and trajectory analysis. J Mol Mod 2001;7:306–317.
35. Stella L, Nicotra M, Ricci G, Rosato N, Di Iorio EE. Moleculardynamic simulations of human glutathione transferase P1-1:analysis of the induced-fit mechanism by GSH binding. Proteins1999;37:1–9.
36. Palleschi A, Bocchinfuso G, Coviello T, Alhaique F. Molecular dy-namics investigations for the polysaccharide scleroglucan: firststudy on the triple helix structure. Carbohydr Res 2005;340:2154–2162.
37. Berendsen HJC, Postma JM, van Gunsteren WF, Hermans J.Interaction models for water in relation to protein hydration. In:Pullman B, editor. Intermolecular forces. Dordrecht: Reidel;1981. pp 331–342.
38. Berendsen HJC, Postma JPM, van Gunsteren WF, Di Nola A,Haak JR. Molecular dynamics with coupling to an external bath.J Chem Phys 1984;81:3684–3690.
39. Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electro-statics of nanosystems: application to microtubules and the ribo-some. Proc Natl Acad Sci USA 2001;98:10037–10041.
40. Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR:an automated pipeline for the setup, execution, and analysis ofPoisson–Boltzmann electrostatics calculations. Nucleic Acids Res2004;32:W665–W667.
41. Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for dis-play and analysis of macromolecular structures. J Mol Graph 1996;14:51–55.
42. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM,Meng EC, Ferrin TE. UCSF chimera—a visualization system forexploratory research and analysis. J Comput Chem 2004;25:1605–1612.
43. Yang J, Liu L, He D, Song X, Liang X, Zhao ZJ, Zhou GW. Crystalstructure of human protein-tyrosine phosphatase SHP-1. J BiolChem 2003;278:6516–6520.
44. Stella L, Melchionna S. Equilibration and sampling in moleculardynamics simulations of biomolecules. J Chem Phys 1998;109:10115–10117.
45. O’Reilly AM, Pluskey S, Shoelson SE, Neel BG. Activated mu-tants of SHP-2 preferentially induce elongation of Xenopus ani-mal caps. Mol Cell Biol 2000;20:299–311.
46. Derreumaux P, Schlick T. The loop opening/closing motion ofthe enzyme triosephosphate isomerase. Biophys J 1998;74:72–81.
47. Lins L, Thomas A, Brasseur R. Analysis of accessible surface ofresidues in proteins. Prot Sci 2003;12:1406–1417.
48. Eisenberg D, McLachan A. Solvation energy in protein foldingand binding. Nature 1986;319:199–203.
974 G. BOCCHINFUSO ET AL.
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot
Copyright © 2022 FDOKUMEN




















