
Balanced biosynthesis of major membrane components through regulated degradation of the committed...
12
Balanced biosynthesis of major membrane components through regulated degradation of the committed enzyme of lipid A biosynthesis by the AAA protease FtsH (HflB) in Escherichia coli Teru Ogura, 1 * Koichi Inoue, 2² Takashi Tatsuta, 1 Toshinobu Suzaki, 3 Kiyonobu Karata, 1 Katherine Young, 4 Lin-Hui Su, 5 Carol A. Fierke, 5 Jane E. Jackman, 5 Christian R. H. Raetz, 5 Jack Coleman, 6‡ Toshifumi Tomoyasu 1§ and Hiroshi Matsuzawa 7 1 Department of Molecular Cell Biology, Institute of Molecular Embryology and Genetics, Kumamoto University School of Medicine, Kumamoto 862-0976, Japan. 2 Department of Molecular Biology, Faculty of Science, Saitama University, 255 Shimo-Ohkubo, Urawa 338-8570 Japan. 3 Faculty of Science, Kobe University, 1-1 Rokkodai-cho, Nada-ku, Kobe 657-8501, Japan. 4 Department of Microbiology, Merck Research Laboratories, Rahway, NJ 07065, USA. 5 Department of Biochemistry, Duke University Medical Center, Durham, NC 27710, USA. 6 Department of Biochemistry and Molecular Biology, Louisiana State University Medical Center, New Orleans, LA 70112, USA. 7 Department of Biotechnology, The University of Tokyo, 1-1-1 Yayoi, Bunkyo-ku, Tokyo 113-8657, Japan. Summary The suppressor mutation, named sfhC21, that allows Escherichia coli ftsH null mutant cells to survive was found to be an allele of fabZ encoding R-3-hydroxy- acyl-ACP dehydrase, involved in a key step of fatty acid biosynthesis, and appears to upregulate the dehy- drase. The ftsH1(Ts) mutation increased the amount of lipopolysaccharide at 428C. This was accompanied by a dramatic increase in the amount of UDP-3-O-(R-3- hydroxymyristoyl)-N-acetylglucosamine deacetylase [the lpxC (envA) gene product] involved in the com- mitted step of lipid A biosynthesis. Pulse-chase experi- ments and in vitro assays with purified components showed that FtsH, the AAA-type membrane-bound metalloprotease, degrades the deacetylase. Genetic evidence also indicated that the FtsH protease activity for the deacetylase might be affected when acyl-ACP pools were altered. The biosynthesis of phospholipids and the lipid A moiety of lipopolysaccharide, both of which derive their fatty acyl chains from the same R-3-hydroxyacyl-ACP pool, is regulated by FtsH. Introduction The product of the Escherichia coli ftsH gene is a mem- brane-bound ATP-dependent metalloprotease (Tomoyasu et al ., 1995). It degrades the heat shock transcription factor s 32 (Herman et al ., 1995; Tomoyasu et al ., 1995), l lysis- lysogeny determination proteins CII (Herman et al ., 1993; Kihara et al ., 1997; Shotland et al ., 1997) and CIII (Herman et al ., 1997), and uncomplexed membrane proteins such as the secretory machinery subunit SecY (Kihara et al ., 1995; Akiyama et al ., 1996a) and subunit a of the F 1 F o ATPase complex (Akiyama et al ., 1996b). FtsH belongs to a novel ATPase family, called AAA ( ATPases associated with diverse cellular activities), involved in a variety of cellu- lar processes (Kunau et al ., 1993; Tomoyasu et al ., 1993a; Confalonieri and Duguet, 1995; Patel and Latterich, 1998). However, the common function of the AAA family members has not yet been elucidated. FtsH forms a homo-oligomer mediated by the direct interaction between the N-terminal regions of FtsH molecules (Akiyama et al ., 1994a, 1995, 1998). Electron microscopy revealed that purified FtsH molecules form ring-like structures (Shotland et al ., 1997). There is evidence that FtsH also associates with the mem- brane proteins HflK/HflC that modulate the proteolytic activity of FtsH (Kihara et al ., 1996, 1997). Several lines of evidence have also suggested that FtsH has a chaperone-like activity. It has been reported that ftsH mutants show defects in membrane functions, such as anchoring of integral membrane proteins (Akiyama et al ., 1994b), export of secretory proteins (Akiyama et al ., 1994a, 1994b), colicin tolerance (Matsuzawa et al ., 1984; Molecular Microbiology (1999) 31(3), 833–844 Q 1999 Blackwell Science Ltd Received 31 August, 1998; revised 14 October, 1998; accepted 19 October, 1998. Present addresses: ²Department of Biochemistry, Robert Wood Johnson Medical School, Piscataway, NJ 08854, USA. ‡Enzo Biochemicals, 60 Executive Boulevard, Farmingdale, NY 11735, USA. §Institut fu ¨r Biochemie und Molekularbiologie, Hermann- Herder-Strasse, 79104 Freiburg, Germany. *For correspondence. E-mail [email protected]; Tel. (þ81) 96 373 5335; Fax (þ81) 96 371 2408.
Transcript of Balanced biosynthesis of major membrane components through regulated degradation of the committed...

Balanced biosynthesis of major membrane componentsthrough regulated degradation of the committed enzymeof lipid A biosynthesis by the AAA protease FtsH (HflB)in Escherichia coli
Teru Ogura, 1* Koichi Inoue, 2† Takashi Tatsuta, 1
Toshinobu Suzaki, 3 Kiyonobu Karata, 1 KatherineYoung, 4 Lin-Hui Su, 5 Carol A. Fierke, 5 Jane E.Jackman, 5 Christian R. H. Raetz, 5 Jack Coleman, 6‡
Toshifumi Tomoyasu 1§ and Hiroshi Matsuzawa 7
1Department of Molecular Cell Biology, Institute ofMolecular Embryology and Genetics, KumamotoUniversity School of Medicine, Kumamoto 862-0976,Japan.2Department of Molecular Biology, Faculty of Science,Saitama University, 255 Shimo-Ohkubo, Urawa 338-8570Japan.3Faculty of Science, Kobe University, 1-1 Rokkodai-cho,Nada-ku, Kobe 657-8501, Japan.4Department of Microbiology, Merck ResearchLaboratories, Rahway, NJ 07065, USA.5Department of Biochemistry, Duke University MedicalCenter, Durham, NC 27710, USA.6Department of Biochemistry and Molecular Biology,Louisiana State University Medical Center, New Orleans,LA 70112, USA.7Department of Biotechnology, The University of Tokyo,1-1-1 Yayoi, Bunkyo-ku, Tokyo 113-8657, Japan.
Summary
The suppressor mutation, named sfhC21 , that allowsEscherichia coli ftsH null mutant cells to survive wasfound to be an allele of fabZ encoding R-3-hydroxy-acyl-ACP dehydrase, involved in a key step of fattyacid biosynthesis, and appears to upregulate the dehy-drase. The ftsH1 (Ts) mutation increased the amountof lipopolysaccharide at 42 8C. This was accompaniedby a dramatic increase in the amount of UDP-3- O-(R-3-hydroxymyristoyl)- N-acetylglucosamine deacetylase
[the lpxC (envA ) gene product] involved in the com-mitted step of lipid A biosynthesis. Pulse-chase experi-ments and in vitro assays with purified componentsshowed that FtsH, the AAA-type membrane-boundmetalloprotease, degrades the deacetylase. Geneticevidence also indicated that the FtsH protease activityfor the deacetylase might be affected when acyl-ACPpools were altered. The biosynthesis of phospholipidsand the lipid A moiety of lipopolysaccharide, both ofwhich derive their fatty acyl chains from the sameR-3-hydroxyacyl-ACP pool, is regulated by FtsH.
Introduction
The product of the Escherichia coli ftsH gene is a mem-brane-bound ATP-dependent metalloprotease (Tomoyasuet al., 1995). It degrades the heat shock transcription factors32 (Herman et al., 1995; Tomoyasu et al., 1995), l lysis-lysogeny determination proteins CII (Herman et al., 1993;Kihara et al., 1997; Shotland et al., 1997) and CIII (Hermanet al., 1997), and uncomplexed membrane proteins suchas the secretory machinery subunit SecY (Kihara et al.,1995; Akiyama et al., 1996a) and subunit a of the F1Fo
ATPase complex (Akiyama et al., 1996b). FtsH belongs toa novel ATPase family, called AAA (ATPases associatedwith diverse cellular activities), involved in a variety of cellu-lar processes (Kunau et al., 1993; Tomoyasu et al., 1993a;Confalonieri and Duguet, 1995; Patel and Latterich, 1998).However, the common function of the AAA family membershas not yet been elucidated. FtsH forms a homo-oligomermediated by the direct interaction between the N-terminalregions of FtsH molecules (Akiyama et al., 1994a, 1995,1998). Electron microscopy revealed that purified FtsHmolecules form ring-like structures (Shotland et al., 1997).There is evidence that FtsH also associates with the mem-brane proteins HflK/HflC that modulate the proteolyticactivity of FtsH (Kihara et al., 1996, 1997).
Several lines of evidence have also suggested that FtsHhas a chaperone-like activity. It has been reported thatftsH mutants show defects in membrane functions, suchas anchoring of integral membrane proteins (Akiyama etal., 1994b), export of secretory proteins (Akiyama et al.,1994a, 1994b), colicin tolerance (Matsuzawa et al., 1984;
Molecular Microbiology (1999) 31(3), 833–844
Q 1999 Blackwell Science Ltd
Received 31 August, 1998; revised 14 October, 1998; accepted 19October, 1998. Present addresses: †Department of Biochemistry,Robert Wood Johnson Medical School, Piscataway, NJ 08854, USA.‡Enzo Biochemicals, 60 Executive Boulevard, Farmingdale, NY11735, USA. §Institut fur Biochemie und Molekularbiologie, Hermann-Herder-Strasse, 79104 Freiburg, Germany. *For correspondence.E-mail [email protected]; Tel. (þ81) 96 373 5335; Fax(þ81) 96 371 2408.

Qu et al., 1996) and the membrane potential (DC) (Mat-suzawa et al., 1984). Some of these defects are partiallysuppressed by overproduction of the molecular chaper-ones GroEL/ES (Hsp60/10) or HtpG (Hsp90) (Shirai etal., 1996). However, the molecular basis of these defectsin membrane functions in ftsH mutants remains to be solved.Eukaryotic mitochondrial homologues of FtsH are involvedin both protein degradation (Pajic et al., 1994; Nakai et al.,1995; Guelin et al., 1996; Leonhard et al., 1996; Weber etal., 1996) and assembly of membrane proteins (Paul andTzagoloff, 1995; Arlt et al., 1996; Rep et al., 1996), similarto FtsH. Genetic evidence has indicated that the latterfunction, most probably chaperone-like, is independentof the proteolytic activity (Arlt et al., 1996; Rep et al.,1997).
The ftsH gene is essential for cell viability (Tomoyasu etal., 1993a; Akiyama et al., 1994b). However, the essentialfunction of FtsH has not been elucidated. We have foundthat one particular ftsH mutant, tolZ21, has a mutationat a His residue within a putative zinc-binding motif (Quet al., 1996). This substitution of His-418 by Tyr mustgreatly reduce the protease activity of FtsH. The tolZ21mutant is viable, although it is temperature sensitive (Mat-suzawa et al., 1984; Qu et al., 1996). These findings sug-gest that the proteolytic function of FtsH is not essential forviability and, therefore, another function such as the pro-posed chaperone activity must be essential in wild-typecells. To identify the essential function of FtsH, we havestudied conditionally lethal ftsH1(Ts) and tolZ21 mutantsand found that the latter contains a secondary mutationwhich suppresses the lethality of the tolZ21 mutation. Inthis paper, we report that the sfhC mutation (for suppres-sor of ftsH ) suppresses the lethality of the ftsH mutation,and that the sfhC21 mutation is a fabZ allele.
The fabZ gene codes for R-3-hydroxyacyl-ACP dehy-drase (Mohan et al., 1994; Heath and Rock, 1996). Oneof its substrates, R-3-hydroxymyristoyl-ACP, is situated atan important biosynthetic branch point in the synthesis ofphospholipids and the lipid A moiety of lipopolysaccharide(LPS) (Mohan et al., 1994). R-3-Hydroxymyristoyl-ACP canbe elongated by the enzymes of fatty acid synthesis to formpalmitoyl-ACP, which then serves as a key acyl donor in theformation of the membrane phospholipids (Magnuson etal., 1993; Heath and Rock, 1996). R-3-Hydroxymyristoyl-ACP can also serve directly as the initial acyl donor forlipid A/LPS biosynthesis (Anderson et al., 1985, 1988,1993; Anderson and Raetz, 1987; Galloway and Raetz,1990; Kelly et al., 1993). Presumably, the synthesis oflipid A and phospholipids must be properly balanced, butthe mechanisms by which this is achieved are unknown.
Recent studies have revealed that the envA (lpxC ) geneproduct UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucos-amine deacetylase (the commited step of lipid A biosynth-esis) is an importantant point of regulation (Young et al.,
1995; Sorensen et al., 1996). Deacetylase levels areelevated when any of the early enzymes of lipid A biosyn-thesis are inhibited by pharmacological (Sorensen et al.,1996; Onishi et al., 1996) or genetic methods (Andersonet al., 1993; Sorensen et al., 1996). The mechanisms thatregulate UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucos-amine deacetylase remain to be elucidated (Sorensen etal., 1996). We report here that ftsH mutations cause a 5-to 10-fold accumulation of UDP-3-O-(R-3-hydroxymyris-toyl)-N-acetylglucosamine deacetylase under non-permis-sive conditions, comparable to what is seen when lipid Abiosynthesis is blocked (Sorensen et al., 1996), and thatthe deacetylase is a proteolytic substrate of FtsH pro-tease. Our findings show that FtsH is an important regula-tor of the production of membrane lipid components inE. coli. In addition, our findings strongly suggest that thesfhC21 mutation elevates the activity of R-3-hydroxy-myristoyl-ACP dehydrase, thereby compensating for thehigh levels of deacetylase when FtsH is missing.
Results
Identification of the sfhC mutation that suppressesthe lethality of the ftsH mutations
The tolZ21 mutation, which causes tolerance to colicins E2,E3 and D, has been identified as a mutation in the ftsH genein which His-418 is replaced by Tyr in the FtsH protein(Qu et al., 1996). The substitution is within a putative zinc-binding motif (His–Glu–Ala–Gly–His-418), which mustbe essential for the proteolytic activity of FtsH. This sug-gests either that the proteolytic activity of FtsH is notessential for cell viability or that the original tolZ21 mutantUM21 has a suppressor mutation which allows cells to sur-vive. Recently, our studies revealed that the latter possi-bility was the case (Qu et al., 1996; Makino et al., 1997).We named the suppressor sfhC21 (for suppressor offtsH ) (Makino et al., 1997).
We noticed that the ftsHþ (tolZþ) derivative of UM21,AR3099, was hypersensitive to rifampicin. Genetic map-ping indicated that the mutation was located at the 4-minregion of the E. coli chromosome. We transduced the muta-tion responsible for the rifampicin hypersensitivity into wild-type cells and examined whether these clones survivedwithout ftsH. The DftsH mutation could be introduced intoall rifampicin-hypersensitive clones tested. Thus, the muta-tion mapped to the 4-min region is referred to as sfhC21.A set of strains, AR3307 (wild type), AR3289 (sfhC ) andAR3291 (sfhC DftsH ) which are isogenic derivatives ofW3110, were constructed and examined by Westernblotting for both FtsH and s32. Sigma-32 is a proteolyticsubstrate of FtsH protease (Tomoyasu et al., 1995). Asexpected, AR3291 lacking FtsH showed a very highlevel (<20-fold compared with the wild type) of s32,
Q 1999 Blackwell Science Ltd, Molecular Microbiology, 31, 833–844
834 T. Ogura et al.

whereas the sfhC21 mutation did not affect levels of eitherFtsH or s32 (Fig. 1A; Tatsuta et al., 1998).
To map the sfhC21 mutation more precisely, we exam-ined several plasmids carrying various segments of the4-min region, i.e. the hlpA-firA (lpxD ) and fabZ-lpxA-lpxBoperons, for their ability to complement sfhC. However,the sfhC21 suppressor mutation was not complementedby any of these clones. We sequenced the entire segmentof this region. A single-base substitution was found in thecoding sequence of fabZ at codon CTG for Leu-85 beingchanged to CCG for Pro. There was no mutation in thecoding sequences of the other genes. Because the plas-mid harbouring the wild-type fabZ gene does not com-plement the sfhC21 mutation, the mutation found in fabZmight increase the activity of the fabZ gene product, lead-ing to the observed suppressor activity. The DftsH muta-tion could be introduced into the strain carrying thefabZþ plasmid, but not into the strain carrying the vector.In addition, the sfhC21 mutant showed a 30–50% increase
in the specific activity of R-3-hydroxymyristoyl-ACP dehy-drase compared with the wild type. These results indicatethat higher levels of fabZ-encoded activity suppress thelethality of DftsH, and that the mutation Leu-85 to Pro infabZ is the sfhC21 suppressor mutation. It is likely thatthe sfhC21 mutation is a gain-of-function allele, althoughwe cannot exclude other possibilities, e.g. a decrease inthe turnover rate of this mutant protein.
The sfhC21 mutation also suppresses thetemperature sensitivity of the ftsH1(Ts) mutant
To examine whether the sfhC21 mutation can also suppressthe lethality of the ftsH1(Ts) mutation at 428C, we introducedthe sfhC21 mutation into the ftsH1(Ts) mutant. The clonesshowing hypersensitivity to rifampicin were able to grow at428C, whereas the clones resistant to the low concentrationof rifampicin remained temperature sensitive. Thus, thesfhC21 mutation suppresses the lethal effect of ftsH1(Ts)as well as DftsH, indicating that the ftsH1(Ts) mutationsimply reduces the activity of FtsH at the non-permissivetemperature.
Inactivation of FtsH results in overproduction of LPS
To understand the mechanism of the suppression by thesfhC21 mutation and the lethal effect caused by ftsH muta-tions, we analysed mutant cells for phospholipid and LPScontent. The amount and composition of phospholipidswere not significantly affected by the sfhC, ftsH1(Ts) orDftsH mutation (data not shown). In contrast, the amountof LPS, measured as the amount of the 2-keto-3-deoxyoc-tonate (KDO), increased in the ftsH1(Ts) mutant at 428C(Table 1), but was not markedly affected by the sfhC21mutation. This increase in LPS might result from the defec-tive release of LPS from the cell. We determined the ratioof LPS/phospholipids in cells and the whole culture andfound that it was essentially the same (data not shown).These results suggest that the lack of ftsH causes overpro-duction of LPS, and that this may be related to the lethality
Q 1999 Blackwell Science Ltd, Molecular Microbiology, 31, 833–844
Fig. 1. Western blots of mutants for FtsH, s32 and LpxC (EnvA).A. Western blots of sfhC and sfhC DftsH mutants for FtsH and s32.Cells were grown in L medium at 378C, collected and suspendedwith the SDS sample buffer. Samples of equal amounts of totalproteins were analysed by SDS–PAGE followed by Westernblotting with anti-FtsH or anti-s32 serum. Lane 1, AR3307 (wildtype); lane 2, AR3289 (sfhC ); lane 3, (sfhC DftsH ).B. Western blots of ftsH1(Ts) mutants for LpxC (EnvA). Cells weregrown continuously at 308C (left) or grown at 308C, transferred to428C and incubated for 2.5 h (right). Samples were analysed as in(A) with anti-FtsH, anti-s32 or anti-LpxC serum. Lane 1, AR3307(wild type); lane 2, AR3317 (ftsH1 ); lane 3, AR3289 (sfhC ); lane 4,AR3319 (sfhC ftsH1 ).
Table 1. Amounts of lipopolysaccharide.
Genotype mg of KDO per mg of protein
Strain ftsH sfhC 308C 378C 428C
AR3307 þ þ 9.2 6 0.1 8.6 6 0.5 9.0 6 0.2AR3317 ftsH1 þ 9.8 6 0.4 NT 13.2 6 0.1AR3319 ftsH1 sfhC21 8.1 6 0.2 NT 7.3 6 0.1AR3289 þ sfhC21 NT 7.8 6 0.2 NTAR3291 DftsH sfhC21 NT 10.3 6 0.4 NTAR753 þ þ 10.1 6 0.2 NT 8.3 6 0.1AR754 ftsH1 þ 9.3 6 0.7 NT 14.0 6 0.5
NT, not tested.
FtsH controls biosynthesis of membrane components 835

of ftsH mutations. This is fully consistent with the fact thathigher levels of fabZ-encoded activity suppresses the leth-ality of the ftsH mutations because phospholipids and LPSare synthesized from the same precursor R-3-hydroxy-myristoyl-ACP in pathways outlined in Fig. 2. In this respect,it should be noted that Kloser et al. (1998) have reportedthat the assembly defect of a mutant outer membrane pro-tein, OmpF315, which is corrected by mutations in eitherlocus and leads to reduced LPS levels and an elevatedratio of phospholipid/LPS, can be suppressed by eitherintroducing a fabZ-inducible plasmid or transducing thesfhC21 mutation. These results are consistent with ourresults and thus support our hypothesis.
Formation of abnormal membrane structures in theperiplasm of the ftsH1(Ts) cells at non-permissivetemperature
Overproduction of LPS may alter cell morphology becauseoverproduced LPS is accumulated in the cell (see above).We observed that ftsH1(Ts) cells grown at 428C containedabnormal new membranes that were layered and distinctfrom the inner and outer membranes, whereas they werenot observed in wild-type or sfhC mutant cells (Fig. 3). For-mation of these membrane structures was suppressed bythe presence of the sfhC21 mutation (data not shown). Todetermine the exact location of these aberrant membranes,we observed plasmolysed cells. It is clear that the newmembranes were located, at least predominantly, in theperiplasmic space (Fig. 3C and D). We have not yet iso-lated these membranes to analyse their composition. It isreasonable to expect, however, that they contain LPS asa major component.
Overproduction of LPS in ftsH mutants results fromstabilization of UDP-3-O-( R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase
It has recently been indicated that the level of UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylaseencoded by the lpxC (envA) gene (Young et al., 1995),the committed reaction in the biosynthetic pathway ofLPS (Fig. 2), is post-transcriptionally regulated (Sorensenet al., 1996). We found that the amount of the deacetylasewas dramatically increased in the ftsH1(Ts) mutant at428C (Fig. 1B). Results of enzymatic assays of extractsof these strains were fully consistent with the results ofWestern blotting (Table 2).
Then, we examined the stability of the deacetylase bypulse-chase experiments. It was clearly shown that thedeacetylase is unstable in wild-type cells (half-life of<5 min), and that it was stabilized in ftsH1(Ts) cells at428C (Fig. 4A). Because FtsH has been demonstrated tobe an ATP-dependent protease, we examined in vitro pro-teolysis of the purified deacetylase by FtsH. As shownin Fig. 4C, the deacetylase was degraded by FtsH in the
Q 1999 Blackwell Science Ltd, Molecular Microbiology, 31, 833–844
Fig. 2. Schematic representation of biosyntheticpathways of major membrane components.Functions of FtsH in the regulation ofbiosynthesis of LPS and phospholipids are drawn.T-bars represent negative control (degradation,inhibition or suppression). FtsH degrades LpxC,and this proteolytic activity is modulated byperturbation of the acyl-ACP pools (see the textfor details). ACP, acyl carrier protein; UDP-GlcNAc, UDP-N-acetylglucosamine; G3P,glycerol-3-phosphate; LPA, lysophosphatidicacid; PA, phosphatidic acid; PGP,phosphatidylglycerolphosphate; PS,phosphatidylserine; PG, phosphatidylglycerol;PE, phosphatidylethanolamine; CL, cardiolipin.
Table 2. Specific activity of the LpxC (EnvA) deacetylase in extracts.
Genotype pmol min¹1 mg¹1
Strain ftsH sfhC 308C 428C
AR3307 þ þ 75.5 104AR3317 ftsH1 þ 101 484AR3289 þ sfhC21 617 750AR3319 ftsH1 sfhC21 524 991AR3291 DftsH sfhC21 305 NT
NT, not tested.
836 T. Ogura et al.
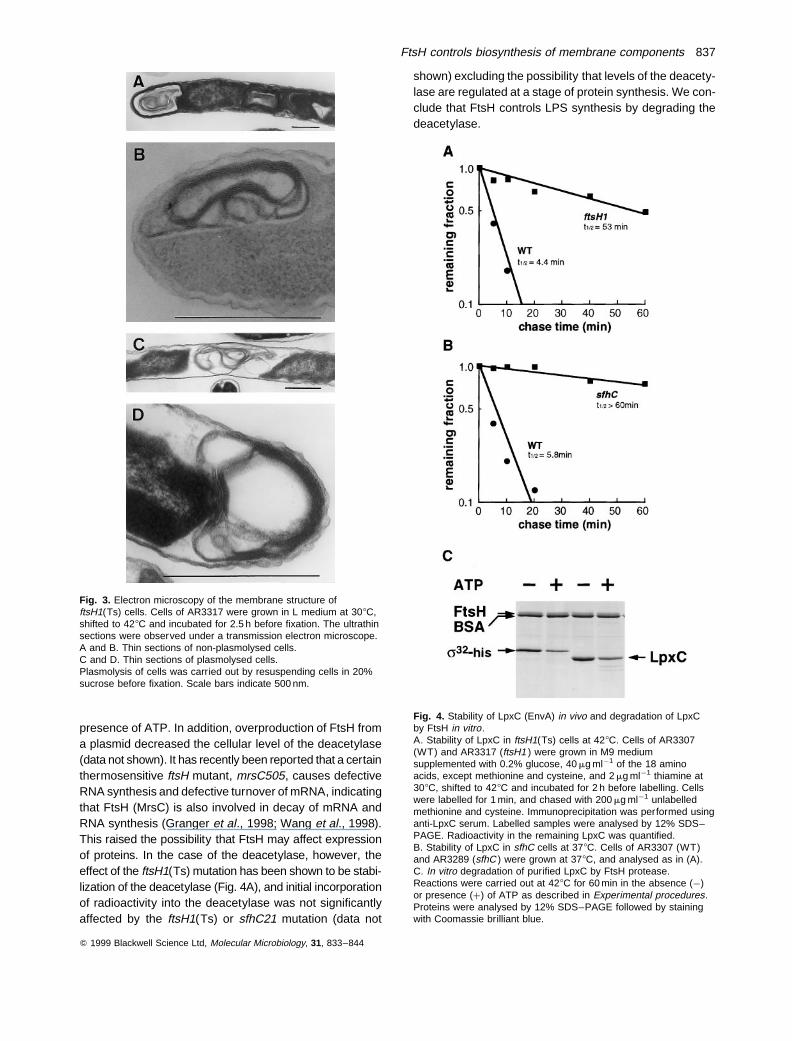
presence of ATP. In addition, overproduction of FtsH froma plasmid decreased the cellular level of the deacetylase(data not shown). It has recently been reported that a certainthermosensitive ftsH mutant, mrsC505, causes defectiveRNA synthesis and defective turnover of mRNA, indicatingthat FtsH (MrsC) is also involved in decay of mRNA andRNA synthesis (Granger et al., 1998; Wang et al., 1998).This raised the possibility that FtsH may affect expressionof proteins. In the case of the deacetylase, however, theeffect of the ftsH1(Ts) mutation has been shown to be stabi-lization of the deacetylase (Fig. 4A), and initial incorporationof radioactivity into the deacetylase was not significantlyaffected by the ftsH1(Ts) or sfhC21 mutation (data not
shown) excluding the possibility that levels of the deacety-lase are regulated at a stage of protein synthesis. We con-clude that FtsH controls LPS synthesis by degrading thedeacetylase.
Q 1999 Blackwell Science Ltd, Molecular Microbiology, 31, 833–844
Fig. 3. Electron microscopy of the membrane structure offtsH1(Ts) cells. Cells of AR3317 were grown in L medium at 308C,shifted to 428C and incubated for 2.5 h before fixation. The ultrathinsections were observed under a transmission electron microscope.A and B. Thin sections of non-plasmolysed cells.C and D. Thin sections of plasmolysed cells.Plasmolysis of cells was carried out by resuspending cells in 20%sucrose before fixation. Scale bars indicate 500 nm.
Fig. 4. Stability of LpxC (EnvA) in vivo and degradation of LpxCby FtsH in vitro.A. Stability of LpxC in ftsH1(Ts) cells at 428C. Cells of AR3307(WT) and AR3317 (ftsH1 ) were grown in M9 mediumsupplemented with 0.2% glucose, 40 mg ml¹1 of the 18 aminoacids, except methionine and cysteine, and 2 mg ml¹1 thiamine at308C, shifted to 428C and incubated for 2 h before labelling. Cellswere labelled for 1 min, and chased with 200 mg ml¹1 unlabelledmethionine and cysteine. Immunoprecipitation was performed usinganti-LpxC serum. Labelled samples were analysed by 12% SDS–PAGE. Radioactivity in the remaining LpxC was quantified.B. Stability of LpxC in sfhC cells at 378C. Cells of AR3307 (WT)and AR3289 (sfhC ) were grown at 378C, and analysed as in (A).C. In vitro degradation of purified LpxC by FtsH protease.Reactions were carried out at 428C for 60 min in the absence (¹)or presence (þ) of ATP as described in Experimental procedures.Proteins were analysed by 12% SDS–PAGE followed by stainingwith Coomassie brilliant blue.
FtsH controls biosynthesis of membrane components 837

Interestingly, the sfhC21 mutation also caused anincrease in the amount of the deacetylase (Fig. 1B andTable 2) through its stabilization (Fig. 4B), similar to theftsH mutation. This is consistent with the fact that thesfhC21 mutation does not significantly affect the ratio ofphospholipid/LPS. Stabilization of the deacetylase in thesfhC21 mutant may be explained by a feedback mechan-ism. Phospholipid synthesis and LPS synthesis competefor R-3-hydroxymyristoyl-ACP (Fig. 2). Because fabI isalso involved in the pathway of phospholipid synthesis(Fig. 2), we examined the fabI(Ts) mutants for levels ofthe deacetylase, and found that the fabI(Ts) mutants alsoincreased amounts of the deacetylase at 428C (Fig. 5).Taken together, we propose a novel control mechanismused to maintain the appropriate balance between phos-pholipids and LPS in E. coli. FtsH protease regulates thedeacetylase level through direct degradation and theFtsH protease activity for the deacetylase is modulatedby perturbation of the acyl-ACP pools (Fig. 2). It shouldbe noted that the sfhC21 and fabI(Ts) mutations affectthe deacetylase level but not the s32 level, indicating thatthis modulation is substrate specific.
Reduction of LPS synthesis suppresses the lethalityof the ftsH mutations
Many genes are involved in LPS synthesis. Most muta-tions in these genes have been isolated as conditionallylethal mutations. Activities of some mutant genes mightbe lowered, even under permissive conditions. Such muta-tions might suppress the lethality of DftsH if reduction ofLPS synthesis is generally sufficient for the suppression.In support of the hypothesis, the DftsH mutation could beintroduced into lpxA2, firA66 and firA200 mutants at308C, whereas it could not be introduced into the wild-type strain. It is likely that lpxA2, firA66 and firA200 muta-tions reduce enzymatic activities to some extent even at308C. This is supported by the observation that thelpxA2 mutant still shows hypersensitivity to rifampicin at308C (Vuorio and Vaara, 1992). These results strongly
indicate that reduction of LPS synthesis is sufficient to sup-press the lethality of the ftsH null mutation.
Growth of ftsH1(Ts) cells is severely inhibited at hightemperature. If the observed increase of the deacetylaselevel in ftsH1(Ts) cells at high temperature is responsiblefor this growth defect, partial inhibition of the deacetylasemight also suppress the lethality of ftsH1(Ts). The R-iso-mer of the compound L-573 655 is a competitive inhibitorof the deacetylase (Onishi et al., 1996). We found that alow concentration (25 mg ml¹1) of L-573 655, which did notsignificantly affect the growth of wild-type cells, partiallysuppressed the growth defect of the ftsH1(Ts) mutantat 408C (data not shown). We have also found that thedeacetylase is toxic to the wild-type cell when overpro-duced, and that this toxicity is suppressed by the sfhC21mutation (data not shown) excluding the possibility thatftsH mutations exhibit the lethal effect in some other waydespite the overproduction of the deacetylase and LPS.
Further characterization of the DftsH strain
Several phenotypes, for example stop-transfer defect(Std¹) (Akiyama et al., 1994b), high frequency of l lysogen-ization (Hfl¹) (Herman et al., 1993), tolerance to colicins(Tol¹) (Qu et al., 1996), and failure to grow on non-fermen-table carbon sources (Nfc¹) (Qu et al., 1996), have beenobserved in ftsH mutants. Among these, Std¹ and Hfl¹
phenotypes are common to ftsH1(Ts) ftsH101, hflB29and tolZ21, but the other two phenotypes, Tol¹ and Nfc¹,are specific to the tolZ21 mutant (Qu et al., 1996). Weexamined the constructed DftsH sfhC strain for thesephenotypes. As shown in Table 3, the DftsH sfhC strainAR3291 showed Tol¹ and Nfc¹ phenotypes as well asStd¹ and Hfl¹ phenotypes, comparable to the tolZ21mutant. These results indicate that the complete loss ofthe FtsH protease activity causes all of the four pheno-types described above.
Previously, we demonstrated that l CIII protein inhibitedFtsH (Tomoyasu et al., 1995). Because the sfhC21 muta-tion suppresses the lethality of DftsH, the sfhC21 mutantshould become resistant to overproduction of CIII. Strains
Q 1999 Blackwell Science Ltd, Molecular Microbiology, 31, 833–844
Fig. 5. Western blots of fabI(Ts) mutants for LpxC. Cells of twofabI(Ts) mutant strains were continuously grown at 308C or grownat 308C, transferred to 428C and incubated for 2.5 h. Samples wereanalysed as in Fig. 1 with anti-LpxC or anti-s32 serum.
Table 3. Phenotypes of mutants.
Genotype Phenotype
Strain ftsH sfhC Hfl Std Tol Nfc CIIIa
W2252 þ þ þ þ þ þ SUM21 tolZ21 sfhC21 – – – – RAR3099 þ sfhC21 þ þ þ þ RAR3307 þ þ þ þ þ þ SAR3289 þ sfhC21 þ þ þ þ RAR3291 DftsH sfhC21 – – – – R
a. S and R indicate sensitivity and resistance to l CIII respectively.
838 T. Ogura et al.

carrying the sfhC21 mutation, but not wild-type strains,were resistant to overproduction of CIII (Table 3), whichis consistent with the notion that CIII directly interactswith FtsH to inhibit its function.
The tolZ21 mutant UM21 shows temperature-sensitivegrowth (Qu et al., 1996). However, the DftsH mutantAR3291, derived from W3110, did not show Ts¹ pheno-type. It grew at all temperatures between 258C and 448C,although it showed slow growth (<twofold slower thanthat of the wild-type or sfhC mutant) at all of these tem-peratures (Tatsuta et al., 1998). The tolZ21 derivativeof AR3289 also did not show a Ts¹ phenotype, unlikeUM21. We have noticed that the parental strain of UM21,W2252, grows poorly at 428C, suggesting that the Ts¹
phenotype of UM21 is due to combined effects of thetolZ (ftsH ) mutation and an unidentified mutation presentin W2252.
Discussion
In this paper, we have shown that the inactivation of theftsH gene elevates the cellular amount of LPS by increas-ing the amount of UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase, involved in the committedreaction in lipid A biosynthesis (Anderson et al., 1993;Sorenson et al., 1996), due to stabilization of the deacety-lase, and that FtsH protease degrades the deacetylase.Increasing the amount of LPS results in formation of abnor-mal membrane structures in the periplasm. The lethality offtsH mutations is clearly related to the overproduction ofLPS, given that it is suppressed under conditions in whichLPS synthesis is reduced. Because the lipid A moiety ofLPS and phospholipids both derive a large portion oftheir fatty acyl chains from the same precursor molecule,R-3-hydroxymyristoyl-ACP (Mohan et al., 1994; Heathand Rock, 1996), it is reasonable that the biosynthesis ofthese two major membrane lipids is mutually competitive.Accordingly, stimulation of phospholipid synthesis can alsosuppress the lethality of ftsH mutations. These findingslead us to conclude that the essential function of FtsH isthe control of biosynthesis of phospholipids and LPS, assur-ing a proper balance of their amounts. Recently, it wasreported that, in contrast to E. coli, the ftsH gene was dis-pensable in Bacillus subtilis (Deuerling et al., 1997). Thisseems reasonable because the lethality of the E. coli ftsHmutations is caused by the overproduction of LPS, whichB. subtilis, a Gram-positive bacterium lacking the outermembrane, does not produce.
The inhibition of lipid A biosynthesis by the lpxA or lpxD(firA) mutations elevates the specific activity of the deacety-lase (Anderson et al., 1993; Sorenson et al., 1996), asdoes treatment of wild-type cells with L-573 655 (Onishiet al., 1996). The inhibition of lipid A synthesis by thesfhC21 mutation also elevates deacetylase levels to the
same extent as is seen in ftsH mutations under non-permis-sive conditions (Fig. 1B and Table 2). The effect of sfhC21on the deacetylase is clearly stabilization (Fig. 4B). It couldbe explained by the inhibition of FtsH due to a low transientpool of newly made lipid A in the inner membrane. How-ever, the stabilization of the deacetylase by fabI(Ts) muta-tions does not support this mechanism. From the pathwayof phospholipid synthesis depicted in Fig. 2, 2(E )-tetrade-cenoyl-ACP seems to be a candidate for the inhibitor of thedeacetylase degradation because both higher activity ofthe FabZ dehydrase (caused by the sfhC21 mutation orthe presence of a fabZ plasmid) and inactivation of theFabI activity at 428C [caused by fabI(Ts) mutations] stabil-ize the deacetylase (Figs 1B and 5). However, it seemsunlikely that fabI(Ts) mutations cause accumulation of2(E )-tetradecenoyl-ACP (Heath and Rock, 1995). Never-theless, it is also evident that fabI(Ts) mutations affectthe acyl-ACP pools (Heath and Rock, 1995). We speculatethat the cellular concentration of one such acyl-ACP isaffected by the sfhC21 mutation similarly. Because theproteolytic activity of FtsH for s32 is not affected by thesfhC21 mutation (Fig. 1), the modulation of the proteolyticactivity of FtsH by altered acyl-ACP pools seems to be sub-strate specific. The responsible acyl-ACP might modulateFtsH activity by interacting with either FtsH or the deacety-lase. Further studies are required to identify the respons-ible acyl-ACP and to elucidate the molecular mechanismof this substrate-specific inhibition of proteolysis by FtsH.
As discussed above, the sfhC21 mutation does not affectthe level of s32. This implies that an excess of s32 itself isnot deleterious to the cell. In this respect, we have foundthat accumulated s32 in ftsH mutants is not fully active fortranscription of the heat shock genes, indicating that func-tional inactivation of s32, at least, occurs when it is accumu-lated in ftsH mutant cells (Tatsuta et al., 1998; Tomoyasu etal., 1998). This undoubtedly provides us with new insightinto the control mechanism of heat shock regulation.
It is still uncertain how excess LPS causes lethality. Theformation of abnormal layered membrane structures in theperiplasm might directly be deleterious to the cell. Alterna-tively, export of excess LPS may alter membrane structuresby disturbing membrane protein assembly, which mayalso cause defects in membrane functions observed inftsH mutants. Further studies are needed to determinehow membrane structure is affected by overproductionof LPS. Isolation of three different membranes, i.e. inner,outer and abnormal periplasmic membranes, from theftsH1(Ts) mutant cell and biochemical analysis of thesemembranes should provide some clues.
The formation of abnormal membrane structures inftsH1(Ts) cells suggests that export of LPS through theinner membrane is not diminished by the mutation. Itseems, however, that the integration of LPS into theouter membrane is more strictly regulated. This limited
Q 1999 Blackwell Science Ltd, Molecular Microbiology, 31, 833–844
FtsH controls biosynthesis of membrane components 839

integration of excess LPS might be due to a defectivematuration of the overproduced LPS. We examined thechain length of LPS overproduced in ftsH1(Ts) cells andfound no detectable difference (data not shown), indicatingthat its maturation is completed. Thus, ftsH mutants willalso be useful in investigating the mechanism of LPS inte-gration into the outer membrane.
As discussed above, ftsH mutations affect integrationand assembly of membrane proteins, and the FtsH pro-tease degrades some unassembled membrane proteins.Here, we have shown that FtsH is involved in balancedsynthesis of LPS and phospholipids. There is evidenceindicating that FtsH seems to have multiple functions inthe regulation of biosynthesis of membrane components.Another possible regulatory function of FtsH in phospholi-pid/LPS synthesis is suggested by the fact that the cdsSmutation, which suppresses the pH-sensitive cds-8 muta-tion, is an allele of ftsH (K. Karata and T. Ogura, unpub-lished). The cds gene product CDP-diglyceride synthaseis an integral membrane enzyme involved in the biosyn-thetic pathway of phospholipids (Fig. 2). In addition, theactivity of another integral membrane enzyme involvedin the phospholipid pathway, the plsC gene product, is alsoaffected by ftsH mutations (J. Coleman and T. Ogura,unpublished). In conclusion, FtsH must be one of themost important regulatory proteins in membrane biogen-esis. It controls biosynthesis of major membrane com-ponents, phospholipids and LPS, and also controls thequality of membrane proteins by both assisting theirassembly and degrading abnormal forms.
Experimental procedures
Bacterial, plasmid and phage strains
The strains of E. coli and plasmids used are listed in Table 4.l c17 and l c17 cII were used to examine the Hfl phenotype.The P1vir phage was used for P1 transduction.
Media and growth conditions
Bacteria were usually grown in L medium (10 g of tryptone, 5 gof yeast extract, 5 g of NaCl l¹1) or LB medium (10 g of tryp-tone, 5 g of yeast extract, 10 g of NaCl l¹1). M9 medium (Miller,1972) was used as a minimal medium. G-56 medium [45 mMMES, 0.3 mM KH2PO4, 10 mM KCl, 10 mM MgCl2, 15 mM(NH4)2SO4, 5 mg ml¹1 thiamine, 0.2% glucose, 0.4% Casa-mino acid (vitamin free)] was used for labelling with 32Pi.Media were solidified with 1.5% agar for plates and 0.7%agar for top agar. Antibiotics, when required, were added atthe following concentrations: tetracycline, 15 mg ml¹1; kana-mycin, 20 mg ml¹1; chloramphenicol, 20 mg ml¹1; ampicillin,50 mg ml¹1; and rifampicin, 1 or 5 mg ml¹1.
Phenotype tests
The hypersensitivity to rifampicin was defined as a strain’s
inability to form colonies on an L plate containing 1 mg ml¹1
of rifampicin at 308C.The Hfl phenotype was examined as described previously
(Herman et al., 1993). The Std phenotype was examinedaccording to the method described previously (Akiyama etal., 1994b), using pKY251 instead of pKY221. The Tol andNfc phenotypes were defined as a strain’s sensitivity to colicinsE2 and E3 (Matsuzawa et al., 1984; Qu et al., 1996) and itsability to grow on an M9 plate containing 30 mM succinate at378C (Qu et al., 1996). To test the sensitivity to CIII, ptaccIIIwas introduced into the strains for test, and the strains carryingptaccIII was grown on an L plate containing 1 mM IPTG toinduce expression of cIII under the tac promoter.
Western blots
Gel electrophoresis was carried out according to Laemmli(1970) using 10% or 12% SDS-polyacrylamide gels. Westernblotting was as described previously (Towbin et al., 1979),except that immunoblots were developed by an ECL Westernblotting detection kit (Amersham) according to the manufac-turer’s instructions. Antisera used were anti-FtsH (Tomoyasuet al., 1993b), anti-s32 (Gamer et al., 1992), anti-LpxC (EnvA)(Sorensen et al., 1996) and anti-PhoA (a gift from K. Yoda).
Pulse-chase labelling and immunoprecipitation
Labelling and immunoprecipitation were carried out asdescribed previously (Ito et al., 1981). Cells were grown inM9 medium (supplemented with 0.2% glucose, 40 mg ml¹1
of the 18 amino acids except methionine and cysteine, and2 mg ml¹1 thiamine at 308C), shifted to 428C and incubatedfor 2 h before labelling. Cells were labelled with RedivueTM
PRO-MIXTM L-[35S] in vitro Cell Labelling Mix (>37 TBqmmol¹1; Amersham) for 1 min, and chased with 200 mg ml¹1
unlabelled methionine and cysteine. Immunoprecipitation wasperformed using anti-LpxC serum. Labelled samples wereanalysed by 12% SDS–PAGE. Radioactivity in the remainingLpxC was quantified with an image analyser BAS2000 (FujiFilm).
In vitro degradation assay with FtsH
In vitro proteolysis by FtsH was carried out as described pre-viously (Tomoyasu et al., 1995). The complete reaction mixture(25 ml) consisted of the following: 50 mM Tris-acetate (pH 8.0),5 mM magnesium acetate, 12 mM zinc acetate, 80 mM NaCl,1.4 mM b-mercaptoethanol, 4 mM ATP, 0.04 mg ml¹1 bovineserum albumin (BSA), 100 mg ml¹1 purified s32-C-his or LpxC(EnvA) and 40 mg ml¹1 purified FtsH. Reactions were perfor-med at 428C for 60 min. The reaction mixtures were analysedby 12% SDS–PAGE followed by staining with Coomassie bril-liant blue.
Electron microscopy
Cells were grown at 308C in L medium, shifted to 428C andincubated for 2 h. Cells were fixed with glutaraldehyde to afinal concentration of 3%, followed by post-fixation withbuffered 1% OsO4. Plasmolysis of cells was carried out by
Q 1999 Blackwell Science Ltd, Molecular Microbiology, 31, 833–844
840 T. Ogura et al.

resuspending cells into 20% sucrose before fixation. The fixedcells were dehydrated through a graded ethanol series andembedded in Spurr’s low viscosity embedding medium(Spurr, 1969). Ultrathin sections were prepared using an ultra-microtome (LKB 2088 Ultratome V) equipped with glass knivesand stained with 2% aqueous uranyl acetate and Reynold’slead citrate stain (Reynolds, 1963). The ultrathin sectionswere observed under a transmission electron microscope(Phillips CM120) operating at 100 kV.
Analysis of phospholipids
Cells were grown in L medium to early log phase, shifted to428C and incubated for 3 h. Phospholipids were extractedessentially by the method of Ames (1968), with minor modifi-cations, and separated by Silica Gel 60 (no. 5721, E. MerckAG, Darmstadt, Germany) thin-layer chromatography withchloroform/methanol/acetic acid [65: 25: 10, (v/v/v) (Nicols,
1963)]. Spots of phospholipids were visualized by uniformlyspraying a Dittmer-Lester reagent (Dittmer and Lester, 1964)and analysed with a high-speed TLC scanner (model CS-920, Shimadzu) at a wavelength of 628 nm. The molar per-centage of each component was calculated.
Measurement of the amount of KDO
KDO was estimated as described by Karkhanis et al. (1978).Cells were grown in L medium and harvested. The pellet wasresuspended in 10 mM HEPES buffer (pH 7.4) and disruptedby sonication at 48C. The cell lysate was used for the KDOassay. Protein concentrations were determined by the methodof Peterson (1977) with crystalline BSA as the standard.
Measurement of the content of lipid A by 32Pi labelling
Cells were grown to mid-log phase at 308C in G-56 medium,
Q 1999 Blackwell Science Ltd, Molecular Microbiology, 31, 833–844
Table 4. Strains and plasmids used in thisstudy. Strain Relevant genotype Source or reference
E. coli strainsW2252 HfrC metB1 relA1 spoT1 Matsuzawa et al. (1984)UM21 Like W2252, but tolZ21 sfhC21 Qu et al. (1996)SJ16 F¹ panD2 metB1 relA1 gyrA216
zad220 ::Tn10 l¹ lrA. Nishimura
AR3099 Like UM21, but ftsHþ(tolZþ) zha-3168 ::Tn10kan
This study
W3110 F¹ IN(rrnD-rrnE )1 Laboratory stockAR3307 Like W3110, but zad220 ::Tn10 Tatsuta et al. (1998)AR3289 Like W3110, but sfhC21
zad220 ::Tn10Tatsuta et al. (1998)
AR3291 Like W3110, but DftsH3 ::kan sfhC21zad220 ::Tn10
Tatsuta et al. (1998)
AR453 Like W3110, but ftsH1(Ts)zgj-3198 ::Tn10kan
This study
AR3317 Like W3110, but ftsH1(Ts)zgj-3198 ::Tn10kan zad220 ::Tn10
This study
AR3319 Like W3110, but ftsH1(Ts)zgj-3198 ::Tn10kan sfhC21zad220 ::Tn10
This study
AR753 thr-1 leu-6 thi-1 supE44 lacY1 tonA21zha-6 ::Tn10
Begg et al. (1992)
AR754 Like AR753, but ftsH1(Ts) Begg et al. (1992)MF6 thr-1 ara-14 dapD4 D(gal-att l)99
hisG4 rpsL136 xyl-5 mtl-1 lacY1tsx-78 eda-50 rfbD1 thi-1
Ganong and Raetz (1982)
AR3377 Like MF6, but dapDþ This studyAR3379 Like MF6, but dapDþ sfhC21 This studySM101 Like MF6, but dapDþ lpxA2(Ts) Galloway and Raetz (1990)SM66 Like MF6, but dapDþ firA66(Ts)
zae::Tn10Galloway and Raetz (1990)
AR3381 Like MF6, but dapDþ firA200(Ts) This studyJP1111 galE45 relA1 spoT1 thi-1 fabI392
(envM ) (Ts)Egan and Russel (1973)
TKG49 F¹ thr leu thi pyrF codA thyA argGilvA his lacY fhuA tsx deoC supEsus uvrB vtr qmeA (fabI ) (Ts)
Wijsman (1972);Kleerebezem et al.(1996)
AR423 met gal supE hsdR sfiC D(srl-recA)306 ::Tn10 DftsH3 ::kan(pAR171)
Akiyama et al. (1994b)
PlasmidsptaccIII l cIII Kornitzer et al. (1989)pHSG575 Takeshita et al. (1987)pKY251 secY 8-8phoA Y. Akiyama
FtsH controls biosynthesis of membrane components 841

were shifted to 428C and further grown for 3 h. The method ofGalloway and Raetz (1990) was used to estimate the lipid A/glycerophospholipid molar ratio before and, at various times,after the temperature shift. Cells were usually labelled with 5–10 mCi ml¹1 32Pi.
LPS analysis by Tricine SDS–PAGE
LPS was prepared (Kido et al., 1990) and analysed by TricineSDS–PAGE (Pradel and Schnaitman, 1991). Harvested cellsgrown in L medium were suspended in 10 mM HEPES buffer(pH 7.4). LPS was detected using a silver-staining procedure.
UDP-3-O-Acyl-GlcNAc deacetylase assay
The deacetylase assays were performed essentially as des-cribed by Sorensen et al. (1996). The reaction was carriedout in 600 ml microcentrifuge tubes at 308C in a final volumeof 20 ml. Briefly, the reaction mixtures typically contained:3 mM [a-32P]UDP-3-O-(R-3-hydroxymyristoyl)-GlcNAc (2.6 ×105 c.p.m. nmol¹1), 40 mM bis-tris pH 5.5, 1 mg ml¹1 fatty-acid-free BSA, 0.5 mM AMP, and cell extract (0.02–1.0mg ml¹1 of protein depending on the strain). The cell extractswere prepared from cells grown to mid-log phase either at308C, or after a 3 h shift during mid-log phase to 428C. Thereaction was initiated by adding the extract and placing thetubes at 308C. After 2, 4, 6 and 8 min to assure a linear rateof product formation, 5 ml samples were removed from thereaction and mixed with 1 ml of 1.25 M NaOH in a secondmicrocentrifuge tube to stop the reaction. The tubes werethen incubated at 308C for 10 min to remove ester-linkedR-3-hydroxymyristate from both the remaining substrateand product. The samples were then neutralized with 1 ml of1.25 M acetic acid and 1 ml of 5% trichloroacetic acid. Thetubes were placed on ice for 5 min, and then centrifugedfor 2 min. A 2 ml portion of supernatant was spotted onto aflexible PEI-cellulose plate (E. Merck). The plate was dried,washed in methanol for 5 min, dried again, and developed in0.2 M guanidine-HCl. The developed plate was dried andexposed to a PhosphorImager screen overnight. The percen-tage deacetylation was calculated from the relative amountsof [a-32P]UDP-GlcNAc and [a-32P]UDP-GlcN detected by aPhosphorImager (Molecular Dynamics).
R-3-Hydroxymyristoyl-ACP dehydrase activity assay
The dehydrase was assayed using 10 mM R-3-hydroxymyris-toyl-ACP as the substrate, prepared as described previously(Mohan et al., 1994). The dehydration product [2(E )-tetrade-cenoyl-ACP] was separated by electrophoresis from the R-3-hydroxymyristoyl-ACP substrate on a urea-containing nativegel (Post-Beittenmiller et al., 1991), as described by Mohanet al. (1994). The percentage dehydration was determined bygel densitometry and used to calculate the specific activityof the dehydrase in cell extracts.
Acknowledgements
We are grateful to A. Nishimura and J. Tommassen for strains,Y. Akiyama for a plasmid, and K. Yoda for an antiserum. We
also thank M. Nishijima, I. Shibuya, M. Anderson and S. Hiragafor stimulating discussions, helpful advice and warm support.This work was supported in part by Grants-in-Aid for ScientificResearch from the Ministry of Education, Science, Sports andCulture, Japan to T. Ogura, NIH grants GM-51310 to C. R. H.Raetz and GM-40602 to C. A. Fierke, and by a NationalScience Foundation Predoctoral Fellowship to J. E. Jackman.
References
Akiyama, Y., Shirai, Y., and Ito, K. (1994a) Involvement ofFtsH in protein assembly into and through the membrane.II. Dominant mutations affecting FtsH functions. J BiolChem 269: 5225–5229.
Akiyama, Y., Ogura, T., and Ito, K. (1994b) Involvement ofFtsH in protein assembly into and through the membrane.I. Mutations that reduce retention efficiency of a cytoplas-mic reporter. J Biol Chem 269: 5218–5224.
Akiyama, Y., Yoshihisa, T., and Ito, K. (1995) FtsH, a mem-brane-bound ATPase, forms a complex in the cytoplasmicmembrane of Escherichia coli. J Biol Chem 270: 23485–23490.
Akiyama, Y., Kihara, A., Tokuda, H., and Ito, K. (1996a) FtsH(HflB) is an ATP-dependent protease selectively acting onSecY and some other membrane proteins. J Biol Chem271: 31196–31201.
Akiyama, Y., Kihara, A., and Ito, K. (1996b) Subunit a of pro-ton ATPase Fo sector is a substrate of the FtsH protease inEscherichia coli. FEBS Lett 399: 26–28.
Akiyama, Y., Kihara, A., Mori, H., Ogura, T., and Ito, K. (1998)Roles of the periplasmic domain of Escherichia coli FtsH(HflB), in protein interactions and activity modulation.J Biol Chem 273: 22326–22333.
Ames, G.F. (1968) Lipids of Salmonella typhimurium andEscherichia coli : structure and metabolism. J Bacteriol 95:833–843.
Anderson, M.S., and Raetz, C.R.H. (1987) Biosynthesis oflipid A precursors in Escherichia coli : a cytoplasmic acyl-transferase that converts UDP-N-acetylglucosamine toUDP-3-O-(R-3-hydroxymyristoyl) -N-acetylglucosamine. JBiol Chem 262: 5159–5169.
Anderson, M.S., Bulawa, C.E., and Raetz, C.R.H. (1985) Thebiosynthesis of Gram-negative endotoxin: formation of lipidA precursors from UDP-GlcNAc in extracts of Escherichiacoli. J Biol Chem 260: 15536–15541.
Anderson, M.S., Robertson, A.D., Macher, I., and Raetz,C.R.H. (1988) Biosynthesis of lipid A in Escherichia coli :identification of UDP-3-O-(R-3-hydroxymyristoyl)-a-D-glu-cosamine as a precursor of UDP-N2-O3-bis-(R-3-hydroxy-myristolyl)-a-D-glucosamine. Biochemistry 27: 1908–1917.
Anderson, M.S., Bull, H.S., Galloway, S.M., Kelly, T.M.,Mohan, S., Radika, K., and Raetz, C.R.H. (1993) UDP-N-acetylglucosamine acyltransferase of Escherichia coli :the first step of endotoxin biosynthesis is thermodynami-cally unfavorable. J Biol Chem 268: 19858–19865.
Arlt, H., Tauer, R., Feldmann, H., Neupert, W., and Langer,T. (1996) The YTA10–12 complex, an AAA proteasewith chaperone-like activity in the inner membrane of mito-chondria. Cell 85: 875–885.
Begg, K.J., Tomoyasu, T., Donachie, W.D., Khattar, M.,Niki, H., Yamanaka, K., Hiraga, S., and Ogura, T. (1992)
Q 1999 Blackwell Science Ltd, Molecular Microbiology, 31, 833–844
842 T. Ogura et al.

Escherichia coli mutant Y16 is a double mutant carryingthermosensitive ftsH and ftsI mutations. J Bacteriol 174:2416–2417.
Confalonieri, F., and Duguet, M. (1995) A 200-amino acidATPase module in search of a basic function. Bioessays17: 639–665.
Deuerling, E., Mogk, A., Richter, C., Purucker, M., and Schu-mann, W. (1997) The ftsH gene of Bacillus subtilis isinvolved in major cellular processes such as sporulation,stress adaptation and secretion. Mol Microbiol 23: 921–933.
Dittmer, J.C., and Lester, R.L. (1964) A simple specific sprayfor the detection of phospholipids on thin layer chromato-grams. J Lipid Res 5: 126–127.
Egan, A.F., and Russel, R.R.B. (1973) Conditional mutantsaffecting the cell envelope of E. coli K12. Genet Res 21:139–152.
Galloway, S.M., and Raetz, C.R.H. (1990) A mutant ofEscherichia coli defective in the first step of endotoxin bio-synthesis. J Biol Chem 265: 6394–6402.
Gamer, J., Bujard, H., and Bukau, B. (1992) Physical interac-tion between heat shock proteins DnaK, DnaJ, and GrpEand the bacterial heat shock transcription factor s32. Cell69: 833–842.
Ganong, B.R., and Raetz, C.R.H. (1982) Massive accumula-tion of phophatidic acid in conditionally lethal CDP-diglycer-ide synthetase mutants. J Biol Chem 257: 389–394.
Granger, L.L., O’Hara, E.B., Wang, R.-F., Meffen, F.V., Arm-strong, K., Yancy, S.D., Babitzke, P., and Kushner, S.R.(1998) The Escherichia coli mrsC gene is required forcell growth and mRNA decay. J Bacteriol 180: 1920–1928.
Guelin, E., Rep, M., and Grivell, L.A. (1996) Afg3p, a mito-chondrial ATP-dependent metalloprotease, is involved indegradation of mitochondrially encoded Cox1, Cox3, Cob,Su6, Su8 and Su9 subunits of the inner membrane com-plexes III, IV and V. FEBS Lett 381: 42–46.
Heath, R.J., and Rock, C.O. (1995) Enoyl-acyl carrier proteinreductase (fabI ) plays a determinant role in completingcycles of fatty acid elongation in Escherichia coli. J BiolChem 270: 26538–26542.
Heath, R.J., and Rock, C.O. (1996) Roles of the FabA andFabZ b-hydroxyacyl-acyl carrier protein dehydratases inEscherichia coli fatty acid biosynthesis. J Biol Chem 271:27795–27801.
Herman, C., Ogura, T., Tomoyasu, T., Hiraga, S., Akiyama,Y., Ito, K., Thomas, R., D’Ari, R., and Bouloc, P. (1993)Cell growth and l phage development controlled by thesame essential Escherichia coli gene, ftsH/hflB. ProcNatl Acad Sci USA 90: 10861–10865.
Herman, C., Thevenet, D., D’Ari, R., and Bouloc, P. (1995)Degradation of s32, the heat shock regulator in E. coli isgoverned by HflB. Proc Natl Acad Sci USA 92: 3516–3520.
Herman, C., Thevenet, D., D’Ari, R., and Bouloc, P. (1997)The HflB protease of Escherichia coli degrades its inhibitorlcIII. J Bacteriol 179: 358–363.
Ito, K., Bassford Jr, K.P.J., and Beckwith, J. (1981) Proteinlocalization in E. coli: is there a common step in the secre-tion of periplasmic and outer-membrane proteins? Cell 24:707–717.
Karkhanis, Y.D., Zeltner, J.Y., Jackson, J.J., and Carlo, D.J.
(1978) A new and improved microassay to determine2-keto-3-deoxyoctonate in lipopolysaccharide of Gram-negative bacteria. Anal Biochem 85: 595–601.
Kelly, T.M., Stachula, S.A., Raetz, C.R.H., and Anderson,M.S. (1993) The firA gene of Escherichia coli encodesUDP-3-O-(R-3-hydroxymyristoyl)-a-D-glucosamine N-acyl-transferase: the third step of endotoxin biosynthesis. J BiolChem 268: 19866–19874.
Kido, N., Ohta, M., and Kato, N. (1990) Detection of lipopoly-saccharides by ethidium bromide staining after sodiumdodecyl sulfate-polyacrylamide gel electrophoresis. J Bac-teriol 172: 1145–1147.
Kihara, A., Akiyama, Y., and Ito, K. (1995) FtsH is requiredfor proteolytic elimination of uncomplexed forms of SecY,an essential protein translocase subunit. Proc Natl AcadSci USA 92: 4532–4536.
Kihara, A., Akiyama, Y., and Ito, K. (1996) A protease com-plex in the Escherichia coli plasma membrane: HflKC(HflA) forms a complex with FtsH (HflB), regulating its pro-teolytic activity against SecY. EMBO J 15: 6122–6131.
Kihara, A., Akiyama, Y., and Ito, K. (1997) Host regulation oflysogenic decision in bacteriophage l: transmembranemodulation of FtsH (HflB), the cII degrading protease, byHflKC (HflA). Proc Natl Acad Sci USA 94: 5544–5549.
Kleerebezem, M., Heutink, M., de Cock, H., and Tommassen,J. (1996) The qmeA (ts) mutation of Escherichia coli islocalized in the fabI gene, which encodes enoyl-ACPreductase. Res Microbiol 147: 609–613.
Kloser, A., Laird, M., Deng, M., and Misra, R. (1998) Modula-tions in lipid A and phospholipid biosynthesis pathwaysinfluence outer membrane protein assembly in Escherichiacoli K-12. Mol Microbiol 27: 1003–1008.
Kornitzer, D., Teff, D., Altuvia, S., and Oppenheim, A.B.(1989) Genetic analysis of bacteriophage l cIII gene:mRNA structural requirements for translation initiation. JBacteriol 171: 2563–2572.
Kunau, W.H., Beyer, A., Franken, T., Gotte, K., Marzioch, M.,Saidowsky, J., Skaletz-Rorowski, A., and Wiebel, F.F.(1993) Two complementary approaches to study peroxi-some biogenesis in Saccharomyces cerevisiae: forwardand reversed genetics. Biochimie 75: 209–224.
Laemmli, U.K. (1970) Cleavage of structural proteins duringthe assembly of the head of bacteriophage T4. Nature227: 680–685.
Leonhard, K., Herrmann, J.M., Stuart, R.A., Mannhaupt, G.,Neupert, W., and Langer, T. (1996) AAA proteases withcatalytic sites on opposite membrane surfaces comprisea proteolytic system for the ATP-dependent degradationof inner membrane proteins in mitochondria. EMBO J 15:4218–4229.
Magnuson, K., Jackowski, S., Rock, C.O., and Cronan Jr,J.E. (1993) Regulation of fatty acid biosynthesis in Escheri-chia coli. Microbiol Rev 57: 522–542.
Makino, S., Qu, J.-N., Uemori, K., Ichikawa, H., Ogura, T.,and Matsuzawa, H. (1997) A silent mutation in the ftsHgene of Escherichia coli that affects FtsH protein produc-tion and colicin tolerance. Mol Gen Genet 254: 578–583.
Matsuzawa, H., Ushiyama, S., Koyama, Y., and Ohta, T.(1984) Escherichia coli K-12 tolZ mutants tolerant tocolicins E2, E3, D, Ia, and Ib: defect in generation of theelectrochemical proton gradient. J Bacteriol 160: 733–739.
Q 1999 Blackwell Science Ltd, Molecular Microbiology, 31, 833–844
FtsH controls biosynthesis of membrane components 843

Miller, J.H. (1972) Experiments in Molecular Genetics ColdSpring Harbor, NY: Cold Spring Harbor Laboratory Press.
Mohan, S., Kelly, T.M., Eveland, S.S., Raetz, C.R.H., andAnderson, M.S. (1994) An Escherichia coli gene (fabZ )encoding R-3-hydroxymyristoyl acyl carrier protein dehy-drase: relation to fabA and suppression of mutations inlipid A biosynthesis. J Biol Chem 269: 32896–32903.
Nakai, T., Yasuhara, T., Fujiki, T., and Ohashi, A. (1995)Multiple genes, including a member of the AAA family,are essential for degradation of unassembled subunit 2 ofcytochrome c oxidase in yeast mitochondria. Mol Cell Biol15: 4441–4452.
Nichols, B.W. (1963) Separation of the lipids of photosyn-thetic tissues: improvement in analysis by thin layer chroma-tography. Biochim Biophys Acta 70: 417–422.
Onishi, H.R., Pelak, B.A., Gerckens, L.S., Silver, L.L., Kahan,F.M., Chen, M.-H., Patchett, A.A., Galloway, S.M., Hyland,S.A., Anderson, M.S., and Raetz, C.R.H. (1996) Antibac-terial agents that inhibit lipid A biosynthesis. Science274: 980–982.
Pajic, A., Tauer, R., Feldmann, H., Neupert, W., and Langer,T. (1994) Yta10p, a member of a novel ATPase family inyeast, is essential for mitochondrial function. FEBS Lett353: 197–200.
Patel, S., and Latterich, M. (1998) The AAA team: relatedATPases with diverse functions. Trends Cell Biol 8: 65–71.
Paul, M.-F., and Tzagoloff, A. (1995) Mutations in RCA1 andAFG3 inhibit F1-ATPase assembly in Saccharomyces cer-evisiae. FEBS Lett 373: 66–70.
Peterson, G.L. (1977) A simplification of the protein assaymethod of Lowry et al. which is more genetically applic-able. Anal Biochem 83: 346–356.
Post-Beittenmiller, D., Jaworski, J.G., and Ohlrogge, J.B.(1991) In vivo pools of free and acylated acyl carrier pro-teins in spinach. J Biol Chem 266: 1858–1865.
Pradel, E., and Schnaitman, C.R. (1991) Effect of rfaH (sfrB )and temperature on expression of rfa genes of Escherichiacoli K-12. J Bacteriol 173: 6428–6431.
Qu, J.-N., Makino, S., Adachi, H., Koyama, Y., Akiyama, Y.,Ito, K., Tomayasu, T., Ogura, T., and Matsuzawa, H.(1996) The tolZ gene of Escherichia coli is identified asthe ftsH gene. J Bacteriol 178: 3457–3461.
Rep, M., van Dijl, J.M., Suda, K., Schatz, G., Grivell, L.A.,and Suzuki, C.K. (1996) Promotion of mitochondrial mem-brane complex assembly by a proteolytically inactive yeastLon. Science 274: 103–106.
Reynolds, E.S. (1963) The use of lead citrate at high pH asan electron-opaque stain in electron microscopy. J CellBiol 17: 208–212.
Shirai, Y., Akiyama, Y., and Ito, K. (1996) Suppression of ftsHmutant phenotypes by overproduction of molecular chaper-ones. J Bacteriol 178: 1141–1145.
Shotland, Y., Koby, S., Teff, D., Mansur, N., Oren, D.A.,Tetematsu, K., Tomoyasu, T., Kessel, M., Bukau, B.,Ogura, T., and Oppenheim, A.B. (1997) Proteolysis of thephage l CII regulatory protein by FtsH (HflB) of Escheri-chia coli. Mol Microbiol 24: 1303–1310.
Sorensen, P.G., Lutkenhaus, J., Young, K., Eveland, S., Ander-son, M.A., and Raetz, C.R.H. (1996) Regulation of UDP-3-O-[R-3-hydroxymyristoly]-N-acetylglucosamine deacetylase
in Escherichia coli : the second enzymatic step of lipid Abiosynthesis. J Biol Chem 271: 25898–25905.
Spurr, A.R. (1969) A low-viscosity epoxy resin embeddingmedium for electron microscopy. J Ultrastruct Res 26: 31–43.
Takeshita, S., Sato, M., Toba, M., Masahashi, W., and Hashi-moto-Gotoh, T. (1987) High-copy-number and low-copy-number plasmid vectors for lacZ a-complementation andchloramphenicol- or kanamycin-resistance selection. Gene61: 63–74.
Tatsuta, T., Tomoyasu, T., Bukau, B., Kitagawa, M., Mori, H.,Karata, K., and Ogura, T. (1998) Heat shock regulationin the ftsH null mutant of Escherichia coli : dissection ofstability and activity control mechanisms of s32 in vivo.Mol Microbiol. 30: 583–594.
Tomoyasu, T., Yuki, T., Morimura, S., Mori, H., Yamanaka,K., Niki, H., Hiraga, S., and Ogura, T. (1993a) The Escheri-chia coli FtsH protein is a prokaryotic member of a proteinfamily of putative ATPases involved in membrane func-tions, cell cycle control, and gene expression. J Bacteriol175: 1344–1351.
Tomoyasu, T., Yamanaka, K., Murata, K., Suzaki, T., Bouloc,P., Kato, A., Niki, H., Hiraga, S., and Ogura, T. (1993b)Topology and subcellular localization of FtsH in Escheri-chia coli. J Bacteriol 175: 1352–1357.
Tomoyasu, T., Gamer, J., Bukau, B., Kanemori, M., Mori, H.,Rutman, A.J., Oppenheim, A.B., Yura, T., Yamanaka, K.,Niki, H., Hiraga, S., and Ogura, T. (1995) Escherichiacoli FtsH is a membrane-bound, ATP-dependent proteasewhich degrades the heat shock transcription factor s32.EMBO J 14: 2551–2560.
Tomoyasu, T., Ogura, T., Tatsuta, T., and Bukau, B. (1998)Levels of DnaK and DnaJ provide tight control of heatshock gene expression and protein repair in Escherichiacoli. Mol Microbiol 30: 567–582.
Towbin, H., Staehelin, T., and Gordon, J. (1979) Electro-phoretic transfer of proteins from polyacrylamide gels tonitrocellulose sheets: procedure and some applications.Proc Natl Acad Sci USA 76: 4350–4354.
Vuorio, R., and Vaara, M. (1992) The lipid A biosynthesismutation lpxA2 of Escherichia coli results in drastic anti-biotic supersusceptibility. Antimicrobiol Agents Chemother36: 826–829.
Wang, R.-F., O’Hara, E.B., Aldea, M., Bargmann, C.I.,Gromley, H., and Kushner, S.R. (1998) Escherichia colimrsC is an allele of hflB, encoding a membrane-associatedATPase and protease that is required for mRNA decay.J Bacteriol 180: 1929–1939.
Weber, E.R., Hanekamp, T., and Thorsness, P.E. (1996) Bio-chemical and functional analysis of the YME1 gene pro-duct, an ATP and zinc-dependent mitochondrial proteasefrom S. cerevisiae. Mol Biol Cell 7: 307–317.
Wijsman, H.J.W. (1972) Mutation affecting plasmolysis inEscherichia coli. J Bacteriol 110: 789–790.
Young, K., Silver, L.L., Bramhill, D., Cameron, P., Eveland, S.,Raetz, C.R.H., Hyland, S.A., and Anderson, M.S. (1995)The envA permeability/cell division gene of Escherichia coliencodes the second enzyme of lipid A biosyntheis. UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacety-lase. J Biol Chem 270: 30384–30391.
Q 1999 Blackwell Science Ltd, Molecular Microbiology, 31, 833–844
844 T. Ogura et al.




















