
Factors influencing the use of antibiotics and knowledge about ...
Upload
khangminh22Category
view
1download
0
Bacterial response to
membrane‐active peptide antibiotics
Dissertation to obtain the degree Doctor rerum naturalium (Dr. rer. nat.)
at the Faculty of Biology and Biotechnology, International Graduate School Biosciences,
Ruhr University Bochum
at the department
Microbial Biology
submitted by
Michaela Wenzel
from Essen
Bochum, April 2013
Advisor: Jun. Prof. Dr. Julia E. Bandow Second Advisor: Prof. Dr. Dr. Dr. med. habil. Hanns Hatt

Bakterielle Antwort auf
membranaktive Peptid‐Antibiotika
Dissertation zur Erlangung des Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.)
der Fakultät für Biologie und Biotechnologie an der internationalen Graduiertenschule Biowissenschaften,
Ruhr‐Universität Bochum
angefertigt am
Lehrstuhl für Biologie der Mikroorganismen
vorgelegt von
Michaela Wenzel
aus Essen
Bochum, April 2013 Referent: Jun. Prof. Dr. Julia E. Bandow Korreferent: Prof. Dr. Dr. Dr. med. habil. Hanns Hatt

Vielen Dank…
all denen, die zum erfolgreichen Gelingen dieser Arbeit beigetragen haben, insbesondere
Frau Jun. Prof. Dr. Julia Bandow für die Ermöglichung dieser Arbeit, die Bereitstellung des
interessanten und ergiebigen Themas und ihr wissenschaftliches Engagement.
Herrn Prof. Dr. Dr. Dr. med. habil. Hanns Hatt für die freundliche Übernahme des
Korreferats.
meinen Kooperationspartnern Nils Metzler‐Nolte, Hans‐Georg Sahl, Dagmar Zweytick, Heike
Brötz‐Oesterhelt, Ute Krämer, Ralf Erdmann, Dörte Becher, Caroline May, Ronald Gust, Ingo
Ott und Suzana Straus. Ganz besonderer Dank gilt Iulia Chiriac, Bauke Albada und Malay
Patra.
allen kollaborierenden TAs, insbesondere Petra Düchting, Monika Bürger, Stephanie Tautges
und Pascal Prochnow.
meinen Studenten Bastian Kohl, Jennifer Stepanek, Christoph Senges und Patrick Schriek für
ihr erfolgreiches Mitwirken an dieser Arbeit und all den Unfug im Labor.
allen Mitarbeitern des Zentralen Isotopenlabors, insbesondere Stephan Spöllmann, Thomas
Lenders und Michael Siewert, für die exzellente technische Unterstützung und amüsanten
Flur‐Gespräche.
allen Mitarbeitern des Lehrstuhls Biologie der Mikroorganismen, insbesondere der
Mikrobiellen Antibiotikaforschung, für das angenehme Arbeitsklima und die
freundschaftliche Atmosphäre. Besonders danken möchte ich Jan Lackmann und Nadja
Raatschen für all die amüsanten Episoden im Labor und darüber hinaus.
sowie all meinen Freunden und meiner Familie für ihren ständigen Rückhalt.
I

The most exciting phrase to hear in science, the one that heralds new discoveries,
is not 'Eureka!' but 'That's funny ...'
‐ Isaac Asimov ‐
II

Contents
Abbreviations 1
A Introduction 3
1 End of an era ‐ dawn of a new age of antibiotics 3
2 Antimicrobial peptides ‐ ancient molecules to combat modern superbugs 4
2.1 Structural diversity of natural antimicrobial peptides 5
2.2 Synthetic antimicrobial peptides 6
2.3 Organometal‐substituted peptidomimetics 8
3 Mechanisms of action of membrane‐targeting antimicrobial peptides 9
3.1 Specific ion transport 9
3.2 Membrane interaction of RW‐rich antimicrobial peptides 11
4 The Gram‐positive cell envelope 15
4.1 The cell wall and its biosynthesis 15
4.2 The cytoplasmic membrane and membrane biogenesis 18
4.3 The physiological role of the bacterial membrane 20
4.4 Membrane stress response 23
5 Proteomics in antibiotic research 23
6 Objectives 25
B Proteomic signature of fatty acid biosynthesis inhibition available for in vivo
mechanism of action studies 27
C Proteomic response of Bacillus subtilis to lantibiotics reflects differences in
interaction with the cytoplasmic membrane 35
D Modulating the activity of short arginine‐tryptophan containing antibacterial
peptides with N‐terminal metallocenoyl groups 45
E An antimicrobial peptide delocalizes peripheral membrane proteins 58
F Influence of lipidation on the mechanism of action of an RW‐rich antimicrobial
peptide 121
G Quantitative tracing of ruthenocene derivatives for subcellular localization of
antimicrobial peptides in bacteria 149
H Ferrocene‐ and ruthenocene‐specific modulation of the mechanism of action of
metal‐substituted short antimicrobial peptides 167
I Analysis of the mechanism of action of potent antibacterial hetero‐tri‐organometallic
compounds ‐ a structurally new class of antibiotics 201
Contents
III

J Structural optimization of an antibacterial hetero‐tri‐organometallic compound:
Identification of the redundant organometallic moiety required for antibacterial
activity 243
K Discussion 288
1 Proteomic signatures 288
2 Mechanism of action of MP196, a short cationic antimicrobial peptide 293
3 Bacterial stress adaptation to short cationic antimicrobial peptides 298
4 MP196 derivatives: novel implications for antibacterial drug discovery 301
5 Organometallic PNA derivatives: a novel antibiotic class 303
6 Metal complexes in antibiotic drug discovery 307
7 Antimicrobial peptides as therapeutics: possibilities and limitations 309
L Summary 311
M Zusammenfassung 312
N References 314
O Publications 330
P Appendix 334
1 Catalog of proteomic response profiles to antibiotics 335
2 Curriculum vitae 402
3 Contributions to the integrated publications and manuscripts 403
4 Erklärung 405
Contents
IV

Abbreviations
Abbreviations used in the introduction and discussion are listed here. Specific abbreviations
used in the individual publications and manuscripts are explained therein. Standard units
according to the International System of Units (SI) and chemical formulas are not listed
separately. The single or triple letter codes are used for designation of standard amino acids.
Abu 2‐aminoisobutyric acid
ACP acyl carrier protein
ATP adenosine triphosphate
CoA coenzyme A
CWB cell wall biosynthesis
Cyt c cytochrome c
Dha 2,3‐dehydroalanine
Dhb (Z)‐2,3‐dehydrobutyrine
DNA deoxyribonucleic acid
ECF extracytoplasmic function
e.g. exempli gratia
et al. et alii
i.e. id est
FAB fatty acid biosynthesis
Fc ferrocene
FcPNA ferrocene peptide nucleic acid
GFP green‐fluorescing protein
LTA lipoteichoic acid
MRSA methicillin‐resistant Staphylococcus aureus
NAD(P)+ nicotinamide adenine dinucleotide (phosphate)
PBP penicillin‐binding protein
PLB phospholipid biosynthesis
PNA peptide nucleic acid
Q ubiquinone
Rc ruthenocene
RcPNA ruthenocene peptide nucleic acid
Abbreviations
1

RNA ribonucleic acid
ROS reactive oxygen species
RSC respiratory chain
RW‐rich arginine‐tryptophan‐rich
SAR structure‐activity relationship
UDP uridine diphosphate
UDP‐GlcNAc uridine diphosphate N‐acetyl glucosamine
UDP‐MurNAc uridine diphosphate N‐acetyl muramic acid
WTA wall teichoic acid
Abbreviations
2

A Introduction
1 End of an era ‐ dawn of a new age of antibiotics
Selman Waksman, the discoverer of streptomycin, coined the term antibiotic for a low‐
molecular‐weight compound that inhibits growth of microorganisms at low concentrations
but does not inhibit growth of its producer [Waksman, 1947]. Already before, the concept of
antibacterial therapeutic agents was established by Paul Ehrlich in the late 19th century. In
search for a “magic bullet” that kills pathogenic bacteria in the human body without harming
the patient, Ehrlich developed the synthetic arsenic drug arsphenamin. Being successfully
applied in the treatment of syphilis, arsphenamin sold under the trademark salvarsan was
the first antibiotic agent in clinical use. It was followed by a variety of antibiotics produced
by organisms from all domains of life as well as of synthetic origin. The discovery and clinical
application of the sulfonamides and penicillins heralded the “golden age of antibiotics”,
revolutionizing treatment of infectious diseases [Amyes, 2001].
The biosynthesis and integrity of DNA, RNA, proteins, and the cell wall constitute the
antibacterial targets most heavily exploited so far. Although functionality of these structures
is essential for viability, the remarkable genetic adaptability of bacteria has led to the
development of antibiotic resistances. Accumulation and spread of resistance genes gave
rise to multi‐resistant bacteria, so‐called superbugs. The most prominent superbug is the
methicillin‐resistant Staphylococcus aureus (MRSA), which is insensitive against a variety of
antibiotic classes including β‐lactam antibiotics [Waters et al., 2011]. Infections with such
multi‐resistant strains are highly difficult to treat and constitute a serious challenge to public
health. The number of newly approved antibiotic agents steadily decreases since the
1990ies, marking the end of the golden age of antibiotics [Wenzel and Bandow, 2011].
Together, increasing bacterial resistance and the decreasing number of antibiotic approvals
have founded an urgent need for novel, resistance‐breaking antibacterial agents.
Addressing the bacterial resistance problem, structurally novel antibiotic classes as well as
compounds with novel or multiple targets are highly desirable [Brötz‐Oesterhelt and
Brunner, 2008; Patra et al., 2012a]. Development of completely novel antibacterial agents,
re‐evaluation of so far neglected compound classes, and alternative therapeutic strategies
might open up new avenues in antibiotic treatment and herald a new era of antibacterial
therapy.
A Introduction
3

2 Antimicrobial peptides ‐ ancient molecules to combat modern superbugs
The antibiotic class of antimicrobial peptides often exhibits broad‐spectrum activity against
both Gram‐positive and Gram‐negative bacteria. Some peptides are further known to
inactivate viruses, fungi, protozoa, and even cancer cells [Guiliani et al., 2007]. Antimicrobial
peptides naturally occur in all domains of life and constitute one of the evolutional oldest
host defense strategies. In vertebrates they are part of innate immunity. They inactivate
pathogenic microorganisms and modulate the immune response [Ganz, 2003]. In
invertebrates and plants, both of which lack adaptive immunity, antimicrobial peptides are
the major players in host defense [Fritig et al., 1998; Meister et al., 1997]. Bacteria mainly
secrete antimicrobial peptides in order to inhibit growth of competing microorganisms.
Moreover, bacterial peptides have recently been shown to trigger signaling cascades in
plants and to induce plant resistance against virus infections [Henry et al., 2011, Desoignies
et al., 2012]. Although antimicrobial peptides have been widely distributed in nature for
millions of years, only very few resistances are known today [Huang et al., 2010]. Thus, they
hold great promise as potential therapeutic agents.
The first antimicrobial peptides attracting interest as antibiotics were the gramicidins, which
were isolated from the Gram‐positive soil bacterium Bacillus brevis [Hotchkiss and Dubos,
1940a; Hotchkiss and Dubos, 1940b]. They were the first antimicrobial peptides
manufactured commercially and were predominantly administered in wound treatment
[Gause and Brazhnikova, 1944]. Clinical application of gramicidin was followed by several
peptide‐based antibiotics. One of these was the glycopeptide vancomycin, which has been
the first‐line treatment for multidrug‐resistant S. aureus infections for almost 30 years [Tan
et al., 2000; Moellering RC Jr, 2006]. However, vancomycin‐resistant enterococci as well as
vancomycin‐tolerant and ‐resistant S. aureus strains started to spread in the late 1980ies
[Lütticken and Kunstmann, 1988; Smith et al., 1999]. Today, the cyclic lipopeptide
daptomycin is one of the last resort antibiotics against infections with vancomycin‐resistant
Gram‐positive bacteria [Boucher and Sakoulas, 2007]. However, even tolerance against
daptomycin has been described for S. aureus rendering bacteria also cross‐resistant to
vancomycin and telavancin [Bertsche et al., 2011, Rose et al., 2012]. Further peptide‐based
antibiotics are currently evaluated in clinical studies hopefully giving rise to the resistance‐
breaking therapeutics of tomorrow [Butler and Cooper, 2011].
A Introduction
4

2.1 Structural diversity of natural antimicrobial peptides
Antimicrobial peptides are structurally diverse. Their length might vary from 4 to over 100
amino acids and their secondary structures can be characterized by linear, α‐helical, β‐sheet,
or complex structures [http://aps.unmc.edu/AP/main.php]. Organisms might genetically
encode antimicrobial peptide sequences and use the transcription and translation
machineries for their synthesis. Several microorganisms are known to synthesize peptides
independently from the ribosome complex. Such peptides are referred to as non‐ribosomal.
Their production requires complex biosynthesis machineries as non‐ribosomal peptides are
often heavily modified containing e.g. cyclic structures, glycosylations, or lipid modifications
[Samel et al., 2008]. Vancomycin, daptomycin, and the gramicidins belong to this class
[Caboche et al., 2008; http://bioinfo.lifl.fr/norine/]. However, ribosomally synthesized
peptides might undergo heavy posttranslational modifications. The lantibiotics, which hold
great promise for clinical and biotechnological applications, are characterized by complex
secondary structures. They contain the amino acid lanthionine, which often forms sulfur
bridges resulting in polycyclic peptide structures [Bierbaum and Sahl, 2009]. Lantibiotics can
be further subdivided into class A, which comprises long flexible molecules tending to
interact with the bacterial membrane, and class B, comprising more globular molecules
[Chatterjee et al., 2005]. The probably most prominent lantibiotic is the food preservative
nisin, a typical class A member (Figure 1).
Figure 1: Structures of selected cell wall biosynthesis‐inhibiting lantibiotics. Nisin and
gallidermin belong to the lantibiotic class A and integrate into the membrane. Mersacidin,
more globular in structure, belongs to class B and does not insert into the lipid bilayer.
Structures were freely adapted according to Chatterjee et al., 2005, and Bonelli et al., 2006.
Dha: 2,3‐dehydroalanine; Dhb: (Z)‐2,3‐dehydrobutyrine; Abu: 2‐aminoisobutyric acid.
A Introduction
5

Nisin binds to the cell wall precursor lipid II using it as docking molecule for forming an
unselective pore [Reisinger et al., 1980; Brötz and Sahl, 2000; Hasper et al., 2004].
Gallidermin and mersacidin (Figure 1) are examples for lantibiotics, which are currently
examined for their potential applications. Similar to nisin, they inhibit cell wall biosynthesis
by binding the peptidoglycan precursor lipid II [Schneider and Sahl, 2010]. Gallidermin
integrates into the membrane and, depending on the phospholipid composition, forms nisin‐
like pores [Christ et al., 2008]. Class B lantibiotic mersacidin does not interact with the lipid
bilayer [Brötz et al., 1998].
Modifications of antimicrobial peptides such as the sulfide bonds or non‐proteinogenic
amino acids within the lantibiotic structure are known to confer higher resistance against
proteolysis [Bierbaum et al., 1996; Chatterjee et al., 2005]. In fact, proteolysis prevents oral
administration of several peptide antibiotics, strictly limiting their applicability
[Schellenberger et al., 1994]. Additionally, several peptides possess cytotoxic and hemolytic
activity or might trigger immune responses, possibly leading to anaphylactic shock [Polk et
al., 1991]. Toxicity and immunogenicity thus additionally restrict several antimicrobial
peptides to topical administration.
2.2 Synthetic antimicrobial peptides
To address these problems, a variety of synthetic peptides and peptide derivatives has been
developed. Synthetic approaches often feature the naturally widely distributed amphiphilic
peptide archetype, characterized by hydrophilic amino acids such as tryptophan and
phenylalanine and cationic side chains like arginine and lysine. Such peptides are often
simply referred to as cationic antimicrobial peptides. The amphiphilic amino acid pattern is
thought to facilitate interaction with bacterial membranes, where many antimicrobial
peptides act [Yeaman and Yount, 2003].
Especially, arginine‐tryptophan‐rich (RW‐rich) peptides have been extensively investigated
and a variety of both fully and semi‐synthetic peptides with excellent antibacterial activities
has been developed [Rathinakumar et al., 2008; Rathinakumar et al., 2009; Sharma et al.,
2010]. In an attempt to reveal the minimal pharmacophore of short cationic antimicrobial
peptides, the hexapeptide RWRWRW‐NH2 (MP196, Figure 2) was developed. It represents
the shortest alternating RW sequence possessing potent antibacterial activity [Strøm et al.,
2003].
A Introduction
6

Figure 2: MP196 and organometallic derivatives. Peptides of the MP series are typically N‐
terminally amidated or modified. Both all‐L and all‐D versions of the peptides have been
synthesized, all of them displaying potent activity against Gram‐positive bacteria. Structures
were kindly supplied by Bauke Albada. Rc: ruthenocene, Fc: ferrocene.
Its short and simple sequence together with its high antibacterial and low cytotoxic activities
[Albada et al., 2012b] rendered MP196 an ideal lead structure for chemical derivatization
approaches. Earlier reports on chemical modification of other RW‐rich peptide sequences
revealed potent strategies to overcome some of the typical weaknesses of antimicrobial
peptides. Thus, both acetylation and amidation of the peptide termini as well as introduction
of D‐ and β‐amino acids have been shown to efficiently enhance resistance against
proteolysis [Raguse et al., 2002; Hunter et al., 2005; Nguyen et al., 2010].
MP196 and its derivatives usually carry a C‐terminal amide group or other C‐terminal
modifications to provide higher stability. In MP196‐based derivatization series, aiming at
improved activity against resistant bacteria, lipid chains and organometallic moieties were
introduced to the hexapeptide structure. Lipidation of the termini with acyl chains of
different lengths, attached by an additional lysine side chain, was shown to enhance
antibacterial activity, especially against Gram‐negative bacteria. However, at the same time
it featured hemolysis [Albada et al., 2012a]. Systematic L‐ to D‐amino acid substitutions of
such lipidated peptides have recently been shown to fully eliminate the hemolytic potential,
while completely retaining antibacterial activity [Albada et al., submitted].
Further, MP196‐based peptides conjugated with organometallics, precisely metallocene
residues featuring cobalt, iron, and ruthenium, were developed, two of which are displayed
in Figure 2 [Chantson et al., 2005; Chantson et al., 2006; Albada et al., 2012b]. Especially
ferrocene has been suggested to award additional metal‐specific mechanistic features due
to its potential to participate in electron transfer. Despite possessing the same structural
features as ferrocene, ruthenocene does not seem to exhibit considerable redox properties
A Introduction
7

[Dubar et al., 2011; Biot et al., 2012]. Substitution of the N‐terminal arginine of MP196 for
the respective metallocene moieties resulted in modified activities ranging from
considerably lowered to significantly improved antibacterial potency, altered antibacterial
activity spectra, and modified kinetic properties [Chantson et al., 2005; Chantson et al.,
2006; Albada et al., 2012b].
2.3 Organometal‐substituted peptidomimetics
Another approach to developing peptide‐based antibiotics is the substitution of amino acid
side chains with diverse non‐peptidic structures. The resulting compounds are referred to as
peptidomimetics and often exhibit higher serum stabilities [Haug et al., 2008; Liskamp et al.,
2011]. Diverse peptidomimetic classes have been developed, typically displaying a
membrane‐related mode of action following the example of natural antimicrobial peptides.
The peptide nucleic acid (PNA) compounds were designed as peptidic DNA analogs for
antisense approaches and have recently been employed as antibiotic agents [Hatamoto et
al., 2010; Bai et al., 2012]. Derivatizing the PNA backbone structure with different
organometallic moieties yielded tri‐organometallic PNA compounds (Figure 3) [Patra et al.,
2010].
Figure 3: Structures of tri‐organometallic compounds with PNA backbones. A PNA
backbone was combined with an alkyne side chain, a cymantrene, a (di‐picolyl)Re(CO)3
moiety, and a ferrocene or ruthenocene, respectively. Structures were kindly supplied by
Malay Patra.
The PNA backbone was complemented with an alkyne side chain, a manganese‐containing
residue (cymantrene), a rhenium complex (di‐picolyl)Re(CO)3), and either a ferrocene or a
ruthenocene. The resulting compounds, FcPNA and RcPNA, are structurally completely new.
This might be a favorable feature in terms of resistance development. Such novel structures
might circumvent already established resistances [Patra et al., 2012a]. However, such heavily
A Introduction
8

modified peptidomimetics constitute a so far unprecedented structural class and could
exhibit completely different modes of action.
3 Mechanisms of action of membrane‐targeting antimicrobial peptides
Although antimicrobial peptides are described that target intracellular structures, such as
the DNA gyrase‐targeting microcin B17 [Parks et al., 2007], most peptides affect the
bacterial cell envelope, which consists of the cytoplasmic membrane, the cell wall, and, in
Gram‐negative bacteria, the outer membrane. Within the mechanistic subclass of envelope‐
targeting peptides different cell wall and membrane structures as well as their biosynthetic
routes might be affected [Yeaman and Yount, 2003].
3.1 Specific ion transport
The most prominent mechanism of action of antimicrobial peptides is based on changing
permeability of the cytoplasmic membrane for ions. Antibiotic‐mediated ion transport might
be highly selective for one or few ions or rather unspecific depending on the individual
molecular translocation mechanism. Potassium as the smallest metal ion is most often
described to be transported. Translocation of potassium ions across the bilayer leads to
break‐down of the membrane potential, a widely distributed feature of peptide antibiotics.
The mechanisms of action of the peptide antibiotics valinomycin, gramicidin A, nisin, and
gramicidin S differ considerably and represent four typical ways of ion translocation across
biological membranes (Figure 4).
Ionophores transport ions across the membrane and consequently disrupt the
electrochemical gradient. They are often secreted by soil bacteria to outcompete other
microorganisms. The divalent cation ionophores ionomycin and calcimycin have been
supposed to extract metal micronutrients from competing microorganisms making them
available for uptake by the producer [Raatschen et al., 2013]. They might be peptides
(valinomycin, gramicidin A) but non‐peptidic ionophores are common (calcimycin,
ionomycin). Ionophores are subdivided into carriers, such as the highly selective potassium‐
transpoting valinomycin, and channels, such as the potassium/sodium‐transporting
gramicidin A [Duax et al., 1996]. Carriers are mobile in the lipid phase. They bind ions on one
side of the bilayer, diffuse through the membrane, and release them on the other side [Duax
A Introduction
9

et al., 1996]. Channels might reach transport rates up to four orders of magnitude higher
than carrier ionophores [Haynes et al., 1974].
While ionophores display different grades of ion selectivity, pore formers facilitate
membrane transport of any molecule large enough to pass the pore. A well‐characterized
example is the lantibiotic nisin, which forms unspecific membrane pores after initial binding
to the cell wall precursor lipid II. After reaching a certain concentration threshold, a nisin‐
lipid II pore is formed large enough to allow passing of ions and small amino acids [Reisinger
et al., 1980; Hasper et al., 2004]. Thus, nisin displays a dual mechanism compromising cell
wall biosynthesis and forming membrane pores [Brötz and Sahl, 2000], a feature
contributing to low resistance development [Brötz‐Oesterhelt and Brunner, 2008]. A similar
mechanistic duality is known for gallidermin. While nisin needs lipid II as a docking molecule
and is able to form pores in different membranes, gallidermin might integrate independently
from lipid II, but its ability to form pores depends on membrane thickness and fatty acid
branching [Christ et al., 2008].
Figure 4: Mechanisms of action of potassium transport. Valinomycin is a potassium‐specific
carrier, gramicidin A a channel ionophore transporting potassium and sodium, and nisin
forms large unselective pores. Gramicidin S probably induces clusters of negatively charged
phospholipids. The exact mechanism of potassium translocation by gramicidin S is yet
unknown.
Gramicidin S is a cationic peptide that structurally differs from the channel‐forming
gramicidins A, B, and C. The mechanism of membrane interaction of gramicidin S still
remains to be fully elucidated. However, it was reported to induce formation of
A Introduction
10

phospholipid domains. It has been proposed that its two positive charges trigger
accumulation of negatively charged phospholipids around the peptide molecule. This lipid
de‐mixing is believed to result in “frozen” membrane areas impairing functionality of the
membrane [Kaprel’iants et al., 1977; Vostroknutova et al., 1981]. Gramicidin S facilitates
transport of potassium ions [Katsu et al., 1989]. This might be attributed to the distance
between its two positive charges, which are thought to force apart the phospholipid head
groups. It was further described to inhibit components of the respiratory chain (RSC) [Mogi
et al., 2008].
The mechanisms of cationic antimicrobial peptides in general, but especially of RW‐rich
peptides, are less well characterized. Forming pores is most frequently discussed as their
mode of action, however different models of membrane interaction are proposed.
3.2 Membrane interaction of RW‐rich antimicrobial peptides
RW‐rich short cationic peptides, such as cathelicidins and lactoferricins, are consistently
described to interact with and typically integrate into the bacterial membrane. Several
peptides were shown to enhance membrane permeability for ions and/or larger molecules
[Chan et al., 2006]. Different models for membrane disruption, i.e. influencing the
membrane structure in a way leading to rupture of the bilayer, are currently discussed. Most
models were established to describe the membrane interaction of α‐helical amphiphilic
peptide structures resulting in forming of pores or membrane holes [Sato and Feix, 2006;
Teixeira et al., 2012]. The prevailing models are the barrel‐stave, toroidal pore, and carpet
mechanism models (Figure 5A‐D) [Costa et al., 2011].
The barrel‐stave model proposes initial attachment of amphiphilic peptides to the surface of
the outer membrane leaflet (Figure 5A). Perpendicular membrane integration leads to
organized, barrel‐like formation of a stable pore (Figure 5B). Peptides are packed parallel to
the phospholipid chains resulting in a pore fully lined by the peptides themselves [Melo et
al., 2009].
In contrast, the toroidal pore model suggests less organized but still perpendicular
membrane insertion after initial surface contact (Figure 5C). Thereby, phospholipids are
forced into an altered alignment promoting the forming of a pore consisting of both peptide
bundles and lipids [Melo et al., 2009]. This undefined conformation allows the peptides to
disassociate from the pore and cross the membrane. Thus, the toroidal pore model provides
A Introduction
11

explanation for cell penetration and leaves room for the binding of antimicrobial peptides to
intracellular structures [van’t Hof et al., 2001].
Figure 5: Models of the interaction of antimicrobial peptides with membranes. (A) Binding
of amphipathic α‐helical peptides to the membrane surface (red: hydrophilic, blue:
hydrophobic). (B) Barrel‐stave model (Melo et al., 2009), (C) toroidal pore model (van’t Hof
et al., 2001), (D) carpet mechanism model (Steiner et al., 1988), (E) molecular
electroporation model (Miteva et al., 1999), (F) sinking raft model (Pkorny et al., 2002;
Pkorny and Almeida, 2004), (G) interfacial activity model (Wimley, 2010).
A Introduction
12

Recently, a modified toroidal pore model was established. According to this concept, a less
rigid peptide and lipid conformation results in pores lined mainly by phospholipid head
groups instead of the peptide helices [Leontiadou et al., 2006].
The carpet mechanism resembles detergent behavior. Peptides bind to the membrane
surface and cause micellation of the lipid bilayer (Figure 5D). As a result, lipid portions are
removed from the membrane enhancing permeability up to the point of cell lysis [Steiner et
al., 1988]. Although the carpet model has been established for α‐helical peptides, it is also
applicable for small unstructured peptides.
In addition to these “classical” models, several new approaches to explain antimicrobial
peptide action have been developed (Figure 5E‐F). They are more applicable to smaller
unstructured peptides, which do not adapt α‐helical conformations. However, all of them
propose membrane disruption and leakiness.
In the molecular electroporation model it is assumed that densely charged molecules
binding to the membrane surface result in an electrostatic potential across the bilayer.
When this potential is strong enough (>0.2 V), molecular electroporation occurs resulting in
membrane pores. Established for the α‐helix‐rich antimicrobial protein NK‐lysine, the
peptides’ secondary structures are irrelevant in this model as long as the molecule is densely
charged. Moreover, the molecular electroporation model provides an explanation for cell
penetration by peptides, which are small enough to enter the induced pores [Miteva et al.,
1999].
The sinking raft model likewise works for small unstructured peptides. Here, a change of
membrane curvature is believed to occur upon peptide binding to the outer membrane
surface. Possibly accompanied by sinking of peptides into the bilayer, a transient membrane
pore is formed. The sinking of peptides into the bilayer furthermore explains the presence of
small cationic peptides in the inner membrane leaflet [Pkorny et al., 2002; Pkorny and
Almeida, 2004].
The interfacial activity model is based on the tumultuous chemical heterogeneity between
the hydrocarbon (lipid) part of the membrane bilayer and the hydrophilic (head group) part
[Wiener and White, 1992]. By accumulation in this interfacial zone, antimicrobial peptides
are believed to deform the lipid bilayer. They promote a deeper, funnel‐like incursion of the
phospholipid head groups into the hydrocarbon layer, which perturbs the normally strict
separation of hydrophilic and hydrophobic membrane parts. This intermixture allows
A Introduction
13

shuttling of polar molecules through the bilayer along the peptide‐phospholipid structure
[Wimley, 2010]. Thus, the interfacial activity model does not propose an aqueous channel
but rather a permeation pathway consisting of both peptides and lipids.
The interfacial activity model ideally suits RW‐rich peptides due to the nature of tryptophan.
Owing to its uncharged side chain, tryptophan is typically described as hydrophobic residue.
However, the π‐electron system of the indole ring displays a significant quadrupole moment.
Such a quadrupole can be imagined as two negatively charged electron clouds extending
perpendicular from each surface of the positively charged ring structure, forming two
dipoles with the ring plane. It confers a certain polarity to the molecule, which manifests in
the observation that tryptophan residues do not always co‐localize with the hydrocarbon
region of lipid bilayers [Dougherty, 1996; Chan et al., 2006]. Thus, interfacial localization of
RW‐rich peptides is a coherent deduction of their structural properties, especially as short
peptides often do not exhibit distinct secondary structures.
Another recently discussed hypothesis proposes that the occurrence of holes in the
membrane is an inherent feature of the dynamic bilayer structure itself [Fuertes et al.,
2011]. The interaction of peptides with membranes fosters curvature changes, promoting
transient holes in the bilayer structure. The most important difference to the other models is
that the peptide structures themselves do not form the pore or contribute to its structure
but rather induce transient lipid‐lined pores. This mechanism could be best described as
“lipocentric pore model”.
Antimicrobial peptides might also perturb the phospholipid bilayer, i.e. interfering with its
architecture without membrane rupture. So far, no model describing a non‐disruptive but
phospholipid bilayer‐disturbing interaction between antimicrobial peptides and biological
membranes has been established.
In contrast to the huge efforts that have been undertaken to describe the molecular
interaction of antimicrobial peptides with lipid bilayers, their influence on bacterial
physiology is not well understood yet. The cell envelope in general and the cytoplasmic
membrane in particular are essential cellular structures. Disturbing their functionality has
manifold different consequences on cellular physiology. In the following, the bacterial cell
envelope, its biosynthesis, and its potential as antibiotic target will be described in detail for
the soil bacterium Bacillus subtilis, a model organism for Gram‐positive bacteria.
A Introduction
14

4 The Gram‐positive cell envelope
Gram‐positive pathogens constitute a growing threat to public health. MRSA is the best‐
known Gram‐positive superbug but also other Staphylococci, Streptococci, and Enterococci
are worth mentioning. Gram‐positive pathogenic bacteria obtain their perilousness from
their remarkable ability to develop and accumulate multiple resistances. Thus, they have at
least as much impact on public health as Gram‐negative pathogens. Those, in contrast,
possess a notably high inherent antibiotic tolerance. This difference in inherent resistance to
chemical agents is based on the different composition of their cell envelopes. Gram‐negative
bacteria possess a cytoplasmic membrane, a thin peptidoglycan cell wall, and an outer
membrane. Especially the outer membrane serves as very efficient barrier for larger
molecules. It prevents numerous chemicals from approaching the cell wall, the membrane,
and the cytosol. Thus, it considerably restricts access to any structure that might be targeted
by antibiotics. Gram‐positive bacteria do not possess an outer membrane, resulting in a
higher susceptibility to chemical stress. Their cell wall is much thicker than that of Gram‐
negative bacteria conferring higher resistance to physical stress like osmotic pressure
[Madigan and Martinenko, 2006]. Gram‐positive bacteria are highly susceptible to cell wall‐
targeting antibiotics but rapidly develop β‐lactam resistance.
4.1 The cell wall and its biosynthesis
The bacterial cell wall serves as protective shield, molecular filter, and outer barrier limiting
turgor‐dependent cell expansion. Figure 6 shows the cell envelope of Gram‐positive model
organism B. subtilis, a close relative of important pathogens like S. aureus. Gram‐positive
bacteria possess a thick peptidoglycan cell wall composed of multiple layers of N‐
acetylmuramic acid (MurNAc) and N‐acetylglucosamine (GlcNAc). Peptide bridges between
the sugar chains confer high stability to the cell wall structure. Their amino acid composition
varies between different organisms, though typically D‐amino acids are prevalent [Madigan
and Martinenko, 2006].
The Gram‐positive cell envelope further contains teichoic acids. B. subtilis possesses one
species of lipid‐anchored teichoic acids (lipoteichoic acids, LTA) and two wall‐anchored
species (wall teichoic acids, WTA). Lipoteichoic acids of B. subtilis consist of a
glycerolphosphate chain, which is attached to a diaglycerol membrane lipid. Wall teichoic
acid chains are mostly composed of sugar units and glycosidically bound to MurNAc. Teichoic
A Introduction
15

acids are involved in cell division and cation homoeostasis and play an important role in cell
envelope protection as they serve as mechanical barrier. Moreover, their modification with
positively charged D‐alanine restricts accessibility of the cytoplasmic membrane for
positively charged antimicrobial peptides [Weidenmaier and Peschel, 2008].
Figure 6: The cell wall of B. subtilis. The peptidoglycan layer consists of alternating MurNAc
and GlcNAc subunits, peptide chains, and teichoic acids (modified according to Weidenmaier
and Peschel, 2008). LTA: lipoteichoic acid; WTA: wall teichoic acid.
Cell wall biosynthesis takes place in the cytosol and at the cytoplasmic membrane. The
peptidoglycan precursor molecule uridine diphosphate (UDP) MurNAc pentapeptide is
synthesized in the cytosol in five steps from UDP acetylglucosamine. The successive cell wall
biosynthesis steps are located at the membrane. These membrane‐bound steps are also
referred to as the lipid II cycle (Figure 7). UDP MurNAc pentapeptide is attached to the
carrier lipid bactoprenol phosphate by the MraY enzyme forming lipid I. The peripheral
membrane protein MurG converts lipid I to lipid II by attaching a GlcNAc moiety.
Successively, the bactoprenol carrier flips across the membrane bilayer transporting the cell
A Introduction
16

wall precursor to the outer membrane surface. Incorporation of the cell wall building block
into the peptidoglycan layer is performed by penicillin‐binding proteins (PBPs).
Figure 7: The lipid II cycle in B. subtilis. Intracellularly synthesized UDP‐MurNAc
pentapeptide is converted to lipid II and transported across the membrane by flipping of the
bactoprenolphosphate. Penicillin‐binding proteins (PBPs) incorporate the precursor into the
peptidoglycan layer and crosslink the peptide chains (modified according to Schneider and
Sahl, 2010).
Penicillin‐binding proteins, which constitute one of the oldest and most successful targets in
antibiotic history, possess transglycosylase function, connecting the sugar subunits, and
transpeptidase function, building the interpeptide bridges. In B. subtilis, the pentapeptide
chain is composed of L‐alanine1, D‐glutamic acid2, meso‐diaminopimelic acid3, and two D‐
alanine4,5 units. Direct peptide bridges are built between the D‐alanine4 and the meso‐
diaminopimelic acid3 of another pentapeptide chain, thereby cleaving the terminal D‐
alanine5 [Warth and Strominger, 1971; Scheffers, 2012]. In S. aureus, the pentapeptide
chains are cross‐linked by a pentaglycine chain, which is attached to the immature lipid II
molecule by the additional membrane‐bound enzyme complex FemXAB located at the
cytosolic side of the lipid bilayer. The mature lipid II is then flipped across the membrane by
A Introduction
17

the bactoprenol carrier and incorporated in the cell wall by penicillin‐binding proteins
[Schneider et al., 2010].
Following lipid II delivery, bactoprenol pyrophosphate is converted into its mono‐phosphate
form, which is able to flip back across the membrane, where it is available again for another
lipid II cycle [Schneider and Sahl, 2010]. The same carrier is also involved in wall teichoic acid
biosynthesis while lipoteichoic acids are synthesized by direct addition of the sugar and
glycerol components to diaglycerol lipids [Weidenmaier and Peschel, 2008].
Several antimicrobial peptides are known to inhibit distinct steps of cell wall biosynthesis,
some of which are displayed in Figure 7. Lysozyme, an antibacterial protein, enzymatically
cleaves glycosidic bonds, thus digesting the cell wall [Rupley, 1967]. The β‐lactam antibiotics,
comprising the penicillins and cephalosporins, are structurally based on a tripeptide and are,
therefore, peptide antibiotics in a broader sense. They prevent cross‐linking of the cell wall
peptidoglycan [Fisher et al., 2005]. Several compounds like glycopeptide vancomycin or the
lantibiotics nisin, gallidermin, and mersacidin bind to the precursor molecule lipid II,
inhibiting its incorporation into the cell wall. The cyclic peptides bacitracin and friulimicin B
prevent recycling of the bactoprenol carrier, thus inhibiting both lipid II and wall teichoic acid
synthesis. Non‐peptidic tunicamycin competitively inhibits MraY. Cytosolic steps of cell wall
precursor biosynthesis are inhibited by D‐cycloserine and the non‐peptide antibiotic
fosfomycin B [Schneider and Sahl, 2010]. Several of these compounds are currently
employed as therapeutics demonstrating the still attractive clinical potential of cell wall
biosynthesis inhibitors.
4.2 The cytoplasmic membrane and membrane biogenesis
The cytoplasmic membrane constitutes the main antibiotic target of most antimicrobial
peptides. Membrane composition might differ considerably even between closely related
bacterial species. Additionally, it crucially depends on the growth conditions. The charge of
the phospholipid head groups as well as the length and branching of the fatty acyl chains
critically determines bacterial susceptibility to membrane‐active compounds [Christ et al.,
2008; Cheng et al., 2011]. Although different B. subtilis membrane lipid compositions have
been reported, neutral to polar lipid ratios in the range of 30:70 are most frequently
described in the literature. The main polar phospholipid species is consistently reported to
be phosphatidylglycerol followed by phosphatidylethanolamine. Diaglycerol lipids are most
A Introduction
18

abundant among the neutral lipid portion [Bishop et al., 1967; den Kamp et al., 1969; Clejan
et al., 1986; López et al., 1998; Seydlová et al., 2008].
Membrane biogenesis consists of a repeated cycle of precursor condensation, reduction,
dehydration, and a second reduction step (Figure 8). In contrast to e.g. Escherichia coli, B.
subtilis is able to synthesize both straight and branched‐chain fatty acids by use of different
precursors for the initial condensation step [Fujita et al., 2007]. With over 90% of the total
fatty acids being branched, B. subtilis displays high membrane fluidity under standard
growth conditions [Clejan et al., 1986]. After completing the condensation and elongation
cycles, fatty acids are attached to the phospholipid head groups and incorporated into the
cytoplasmic membrane.
Figure 8: Composition and biogenesis of the cytoplasmic membrane of B. subtilis. The
amount of polar phospholipids (dark grey) predominates in the B. subtilis membrane while
fewer lipids feature neutral head groups (light grey). Branched fatty acyl chains clearly
predominate. Fatty acyl branching is adjusted by initial condensation of different precursors
during fatty acid biosynthesis (modified according to Fujita et al., 2007). CoA: coenzyme A.
Fatty acid biosynthesis (FAB) has attracted interest as antibiotic target with the discovery of
platensimycin from the actinomycete Streptomyces platensis [Wang et al., 2006]. Both
platensimycin and long known cerulenin target the FabF enzyme, which initiates fatty acid
elongation. A triple inhibitor of FabF, FabHA, and FabHB, which perform the initial
condensing steps utilizing different precursors, was found with platencin from another S.
A Introduction
19

platensis strain [Wang et al., 2007]. The enoyl‐acyl carrier protein (ACP) reductase FabI is
inhibited by triclosan, which is clinically applied in skin infections and is further used in
disinfectants and personal care products [von der Ohe et al., 2012]. So far, the only peptide
antibiotic described to target fatty acid biosynthesis is andrimid, which was already
discovered in the late 1980ies [Fredenhagen et al., 1987]. Almost two decades later, it was
found to inhibit the acetyl‐coenzyme A (CoA) carboxylases (AccABCD) [Pohlmann et al.,
2005], which recently advanced as novel antibiotic targets [Freiberg et al., 2004; Freiberg et
al., 2005].
Fatty acid biosynthesis is considered an attractive antibiotic target pathway. Platensimycin
and platencin display excellent antibacterial activity but are non‐toxic for mammalian cells
[Martens and Demain, 2011]. However, their in vivo activities are limited. Mainly attributed
to poor pharmacokinetics, this effect started a controversy about the essentiality of fatty
acid biosynthesis in bacteria during infection. Brinster et al. provided evidence that several
Gram‐positive pathogens might use fatty acids from the blood stream rendering fatty acid
biosynthesis inhibitors ineffective [Brinster et al., 2009; Zlitni and Brown, 2009]. Balemans et
al. indeed could show that this is not true for S. aureus [Balemans et al., 2010], which could
recently be correlated with the absence of a feedback regulation mechanism [Parsons et al.,
2011] suggesting application of fatty acid biosynthesis inhibitors in the treatment of S.
aureus infections. Recently, three novel FabI inhibitors, two of which based on the triclosan
structure, entered phase I clinical trials [Butler and Cooper, 2011].
While fatty acid biosynthesis inhibitors are limited to specific application in the treatment of
S. aureus infections, the bacterial membrane itself is an attractive broad‐spectrum target
important for different cellular processes.
4.3 The physiological role of the bacterial membrane
The cytoplasmic membrane acts as a permeability barrier protecting the cytosol from
environmental influences. While the cell wall essentially constitutes a mechanical barrier,
the membrane maintains the chemical environment in the cytosol, restricts access for
harmful molecules, and maintains the cellular milieu. The membrane controls the
distribution of ions between the cytosol and the surrounding medium, which contributes to
the membrane potential and trans‐membrane transport [Lodish et al., 2000]. It harbors a
A Introduction
20
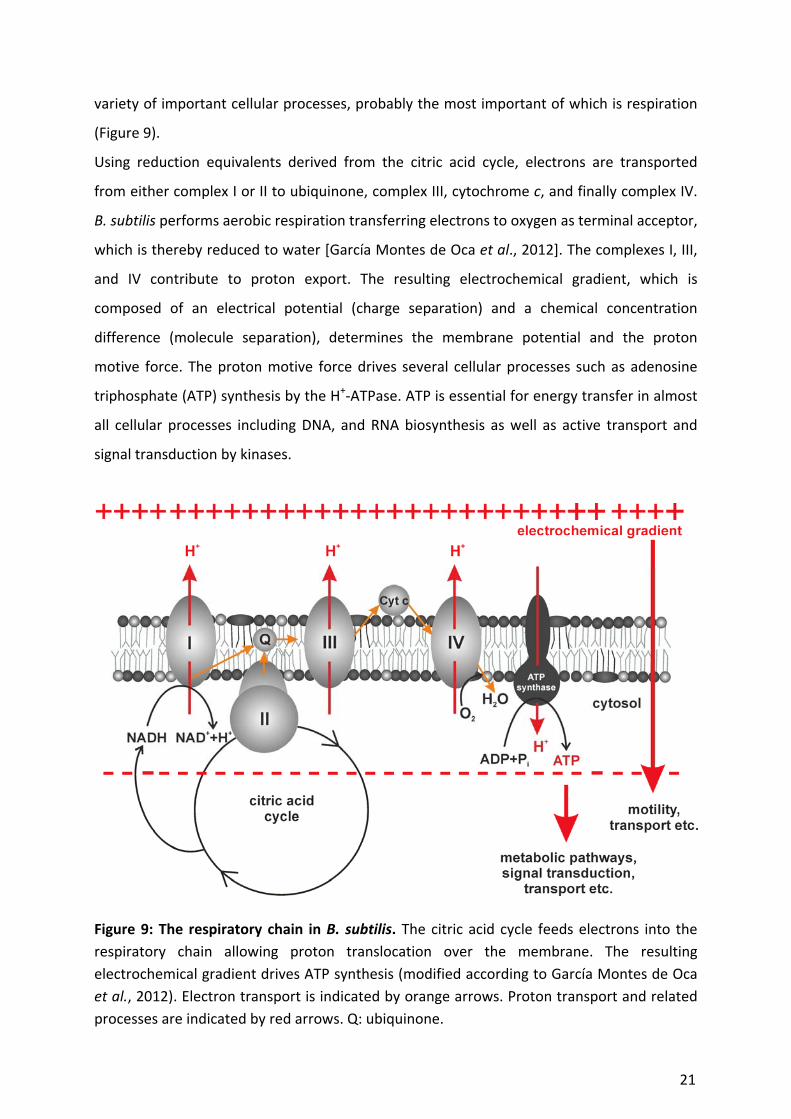
variety of important cellular processes, probably the most important of which is respiration
(Figure 9).
Using reduction equivalents derived from the citric acid cycle, electrons are transported
from either complex I or II to ubiquinone, complex III, cytochrome c, and finally complex IV.
B. subtilis performs aerobic respiration transferring electrons to oxygen as terminal acceptor,
which is thereby reduced to water [García Montes de Oca et al., 2012]. The complexes I, III,
and IV contribute to proton export. The resulting electrochemical gradient, which is
composed of an electrical potential (charge separation) and a chemical concentration
difference (molecule separation), determines the membrane potential and the proton
motive force. The proton motive force drives several cellular processes such as adenosine
triphosphate (ATP) synthesis by the H+‐ATPase. ATP is essential for energy transfer in almost
all cellular processes including DNA, and RNA biosynthesis as well as active transport and
signal transduction by kinases.
Figure 9: The respiratory chain in B. subtilis. The citric acid cycle feeds electrons into the
respiratory chain allowing proton translocation over the membrane. The resulting
electrochemical gradient drives ATP synthesis (modified according to García Montes de Oca
et al., 2012). Electron transport is indicated by orange arrows. Proton transport and related
processes are indicated by red arrows. Q: ubiquinone.
21

The proton motive force further directly drives motility and several transport processes.
Therefore, disruption of the respiratory chain is likely to result in impairment of multiple
cellular processes. Such mechanistic multiplicity is favorable in terms of antibiotic resistance
development [Brötz‐Oesterhelt and Brunner, 2008].
Loss of the proton gradient and in turn of the proton motive force is not the only effect of
respiratory chain inhibition. Impaired electron transport might also lead to improper delivery
of electrons to oxygen, resulting in emergence of reactive oxygen species (ROS) like e.g.
superoxide (O2∙‐), the hydroxyl radical (HO∙), or hydrogen peroxide (H2O2). Such are highly
reactive molecules causing DNA, RNA, and protein damage [Mols and Abee, 2011]. Oxidative
stress might constitute an additional mechanistic component of membrane‐targeting
compounds affecting the respiratory chain and has been proposed to occur after treatment
with bactericidal antibiotics [Hassett and Imlay, 2007; Kohanski et al., 2007; Foti et al., 2012].
However, newer studies could not prove a correlation between bactericidy and ROS
formation [Keren et al., 2013; Liu and Imlay, 2013].
Apart from respiration, further important cellular processes are located at the cytoplasmic
membrane, which might also be affected by membrane‐active antimicrobial peptides. Thus,
cell wall biosynthesis seems not only to be inhibited by direct interaction of a compound
with involved enzymes or precursor molecules. As shown for daptomycin, membrane‐
targeting compounds might also induce strong cell wall stress responses, although direct
interaction of daptomycin with the cell wall biosynthesis machinery was excluded [Wecke et
al., 2009].
Cell division is another important membrane‐associated process in bacteria. Inhibition of cell
separation is not lethal as was demonstrated by highly elongated but viable B. subtilis
mutants [Beall and Lutkenhaus, 1992; Rabenau, 2010]. However, several cell division
proteins seem essential for viability [Kobayashi et al., 2003] and bactericidal antibiotics
targeting cell division have been found. The cephalosporin cephalexin, which is successfully
employed in treating several bacterial infections including pharyngitis, tonsillitis, respiratory,
and urinary tract infections [http://www.drugs.com/monograph/cephalexin.html], inhibits
peptidoglycan biosynthesis during septum formation and causes rapid cell lysis [Chung et al.,
2009].
Transport processes might also be affected by a disturbed phospholipid bilayer. Especially,
mechanosensitive channels, which react to alterations of membrane tension [Hoffmann et
A Introduction
22

al., 2008], are likely influenced by compounds that perturb the membrane structure.
Further, uptake and export systems might be affected.
4.4 Membrane stress response
Bacteria have developed several adaptation and compensation strategies in order to
overcome membrane stress. They e.g. can adapt their phospholipid composition. Under heat
shock conditions membrane structures tend to melt, while under cold shock conditions they
become very rigid impeding membrane‐located processes. Bacteria adjust membrane
fluidity towards higher rigidity under heat shock and towards higher fluidity under cold
shock conditions. Similarly, low osmolarity requires a more rigid membrane structure,
whereas high osmolarity is antagonized by adaptation towards lower membrane viscosity
[Šajbidor, 1997].
Membrane integrity is also stabilized by protective membrane‐binding proteins, such as the
B. subtilis phage shock protein A (PspA). PspA was shown to protect membrane integrity
under stress conditions and to prevent proton leakage [Kobayashi et al., 2007]. The PspA
homolog LiaH displays similar membrane‐stabilizing properties. It is part of the LiaRS two‐
component system, which is mainly responsive to membrane‐mediated cell wall biosynthesis
inhibition [Wolf et al., 2010].
In B. subtilis, the membrane and cell wall stress responses are regulated by alternative sigma
factors, more precisely by the extracytoplasmic function (ECF) sigma factors σM, adjusting
cell wall synthesis and cell shape, σW, responsible for detoxification, bacteriocin expression,
and adaptation of the membrane composition, and σX, adjusting the cell envelope surface
charge, but also by the general stress response‐controlling σB. Together with the LiaRS two‐
component system, the ECF sigma factors constitute the first line of defense against cell
envelope damage [Hecker and Völker, 2002; Helmann, 2002; Cao et al., 2002; Cao and
Helmann, 2004; Jordan et al., 2006; Eiamphungporn and Helmann, 2008].
5 Proteomics in antibiotic research
B. subtilis is a model organism frequently employed for studying bacterial stress adaptation
[Wolf et al., 2010; Winter et al., 2011]. The bacterial stress response provides insight into
bacterial adaptation to different environments and thus of basic physiological processes.
Physical and chemical stresses such as heat and cold shock, salt, and ethanol stress have
A Introduction
23

been studied using comparative proteomics [Petersohn et al., 2001; Budde et al., 2006;
Hahne et al., 2010]. Similarly, the acute B. subtilis stress responses to various antibiotic
agents with different modes of action has been extensively investigated by 2D gel‐based
proteomics (Figure 10) [Bandow et al., 2003].
Radioactive pulse labeling of proteins newly synthesized under stress allows selective
monitoring of the acute stress reaction. Studying bacterial adaptation to antibiotic stress
provides insight into how microorganisms cope with antibacterial compounds secreted by
competing organisms in a natural habitat like soil [Raatschen et al., 2013]. It also allows
insight into how microbes might evade immune responses in the host and how they resist
antibiotic treatment. Moreover, the stress response to an antibiotic agent is typically
indicative of the antibacterial mechanism of action [Raatschen and Bandow, 2012]. Thus,
proteomic response profiles can efficiently aid mode of action elucidation [Brötz‐Oesterhelt
et al., 2005]. This is particularly facilitated by an antibiotic reference compendium, which
was established by Bandow et al. and is constantly expanded [Bandow et al., 2003;
Raatschen et al., 2013]. This library currently comprises proteome response profiles to more
than 50 antibiotic agents, toxins, and genetic down‐regulation of target genes. It can be used
to compare the response patterns of novel compounds with that of well‐known antibiotics.
An overlapping stress response is indicative of a similar mechanism, allowing fast mode of
action diagnosis.
Figure 10: 2D gel‐based proteomics. Cytosolic protein extracts are separated in a first
dimension by isoelectric focusing and in a second dimension by SDS‐PAGE. Protein synthesis
patterns of the untreated controls are false colored in green and those of the antibiotic‐
treated cultures in red. Gel images of antibiotic‐treated cultures are overlaid with their
untreated controls. Proteome response patterns are then compared with reference profiles
allowing mode of action diagnosis.
A Introduction
24

Marker proteins overlapping in the response profiles between compounds targeting the
same pathway, allow establishing of so‐called proteomic signatures. Such signatures are
indicative of the inhibition of a target pathway or structure [van Bogelen et al., 1999]. They
allow fast designation of novel compounds to mechanistic classes. In particular, they are
useful for mode of action validation or falsification of compounds derived from known
antibiotic structures or from molecular modeling approaches. Certainly, they also aid mode
of action analysis of completely novel compounds.
6 Objectives
Two main objectives were pursued in this work. On the one hand, the proteome response
library was complemented with proteomic signatures for different aspects of cell envelope
stress. On the other hand, the mechanisms of action of novel peptide antibiotics were
investigated.
A proteomic signature for inhibition of fatty acid biosynthesis was established by use of
triclosan, cerulenin, platensimycin, and platencin. The newly established signature was
employed for mode of action analysis of the chromium organometallic‐substituted
platensimycin derivative PM47.
The lantibiotics mersacidin, gallidermin, and nisin, each of which binds to cell wall precursor
lipid II but displays different stages of interaction with the cytoplasmic membrane, were
employed for setting up signatures for cell envelope stress. In combination with further
reference profiles this subset of lantibiotics allowed definition of distinct signatures for
general cell envelope stress, membrane stress, and inhibition of the membrane‐bound cell
wall biosynthesis machinery.
The newly established signatures served as basis for mechanistic analyses of the
antimicrobial hexapeptide MP196 (RWRWRW‐NH2). Its antibacterial mode of action should
be elucidated with special emphasis on its effects on bacterial physiology. Further, insight
into bacterial stress adaptation to membrane‐targeting antibiotics should be obtained.
The antibiotic mechanism of lipidated MP196 derivatives was investigated and compared to
that of MP196 by comparative proteome analysis. To this end, the most active peptides,
carrying lipid chains of 8 C‐atoms in length at either the N‐ or C‐terminus (H15C7(O)C‐
KRWRWRW‐NH2 (N‐C8) and RWRWRWK‐C(O)C7H15 (C‐C8)) were chosen. To evaluate the
effects of the additional lysine residues employed for attaching the lipid tails, the
A Introduction
25

correspondent non‐lipidated peptides (KRWRWRW‐NH2 (N‐C0) and RWRWRWK‐NH2 (C‐C0))
were included in the study (see Appendix 1 for structures).
The ruthenocene‐modified peptide MP276 (RcCO‐WRWRW‐NH2, Figure 1) was employed for
studying peptide localization in vivo by electron microscopy and atomic absorption
spectrometry‐based metal tracing.
In order to monitor metal‐specific mechanistic differences, organometallic peptides carrying
either ferrocene or ruthenocene were analyzed. Both all‐L and all‐D amino acid versions of
MP276 and the ferrocene‐substituted MP66 (FcwRWRWRW‐NH2, Figure 1) as well as the all‐
L cyclic MP66 derivative MP159 (FcCO‐G‐cCWRWRWRWC, see Appendix 1 for structure)
were investigated by proteomic profiling and compared to all‐L and all‐D MP196. The MP276
mode of action was compared with that of MP196 in detail in order to evaluate the influence
of metallocene derivatization on the molecular mechanism.
The completely novel antibiotic class of organometallic PNA backbone derivatives,
represented by the tri‐organometallic compounds FcPNA and RcPNA, were analyzed
regarding their antibacterial potency, mechanism of action, and metal‐based mechanistic
differences. A systematic structure‐activity relationship (SAR) study was conducted in order
to identify the essential organometallic moiety required for antibacterial activity.
A Introduction
26

B
Proteomic signature of fatty acid biosynthesis
inhibition available for in vivo mechanism of action
studies
Michaela Wenzel, Malay Patra, Dirk Albrecht, David Y.‐K. Chen, Kyriakos
C. Nicolaou, Nils Metzler‐Nolte, Julia E. Bandow
Antimicrobial Agents and Chemotherapy, 2011
B Fatty acid biosynthesis inhibition signature
27

ANTIMICROBIAL AGENTS AND CHEMOTHERAPY, June 2011, p. 2590–2596 Vol. 55, No. 60066-4804/11/$12.00 doi:10.1128/AAC.00078-11Copyright © 2011, American Society for Microbiology. All Rights Reserved.
Proteomic Signature of Fatty Acid Biosynthesis Inhibition Availablefor In Vivo Mechanism-of-Action Studies�
Michaela Wenzel,1 Malay Patra,2 Dirk Albrecht,3 David Y.-K. Chen,4 K. C. Nicolaou,5Nils Metzler-Nolte,2 and Julia E. Bandow1*
Ruhr University Bochum, Biology of Microorganisms, Universitatsstraße 150, 44801 Bochum, Germany1; Ruhr University Bochum,Bioinorganic Chemistry, Universitatsstraße 150, 44801 Bochum, Germany2; Ernst Moritz Arndt University Greifswald, Institute for
Microbiology, Friedrich-Ludwig-Jahn-Straße 15a, 17489 Greifswald, Germany3; Chemical Synthesis Laboratory@Biopolis,Institute of Chemical and Engineering Sciences (ICES), Agency for Science, Technology and Research (ASTAR), 11 Biopolis Way,The Helios Block, #03-08, Singapore 138667, Singapore4; and Department of Chemistry and The Skaggs Institute for
Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037,and Department of Chemistry and Biochemistry, University of California,
San Diego, 9500 Gilman Drive, La Jolla, California 920935
Received 19 January 2011/Returned for modification 14 February 2011/Accepted 24 February 2011
Fatty acid biosynthesis is a promising novel antibiotic target. Two inhibitors of fatty acid biosynthesis,platencin and platensimycin, were recently discovered and their molecular targets identified. Numerousstructure-activity relationship studies for both platencin and platensimycin are currently being undertaken.We established a proteomic signature for fatty acid biosynthesis inhibition in Bacillus subtilis using platencin,platensimycin, cerulenin, and triclosan. The induced proteins, FabHA, FabHB, FabF, FabI, PlsX, and PanB,are enzymes involved in fatty acid biosynthesis and thus linked directly to the target pathway. The proteomicsignature can now be used to assess the in vivo mechanisms of action of compounds derived from structure-activity relationship programs, as demonstrated for the platensimycin-inspired chromium bioorganometallicPM47. It will further serve as a reference signature for structurally novel natural and synthetic antimicrobialcompounds with unknown mechanisms of action. In summary, we described a proteomic signature in B. subtilisconsisting of six upregulated proteins that is diagnostic of fatty acid biosynthesis inhibition and thus can beapplied to advance antibacterial drug discovery programs.
Bacterial infections continue to be a challenge throughoutthe world, especially in light of increasing development anddissemination of multiresistant pathogens that are more andmore difficult to treat (33). Therefore, prudent use of approvedantibiotics and, more importantly, new antibiotics, preferablywith new mechanisms of action and low resistance develop-ment rates, are urgently required to restrain infectious dis-eases. Two main strategies for antibiotic development are be-ing pursued today: (i) identification of structurally novelantibiotics using screening approaches based on either naturalor synthetic compounds and (ii) chemical modification ofknown antibiotics aiming at improving their antibacterial orpharmacological properties or at circumventing existing resis-tance mechanisms.
Since the recent discovery of platencin and platensimycin,two potent natural inhibitors of bacterial growth from Strepto-myces platensis (15, 24, 31, 32), there is renewed interest in fattyacid biosynthesis as an antibacterial target. Platensimycin aswell as cerulenin, discovered in the 1960s, inhibit the 3-oxoacyl-acyl carrier protein (ACP) synthase II FabF (21, 32),whereas platencin inhibits both FabF and 3-oxoacyl-ACP syn-thases III FabHA and FabHB (15, 31). These enzymes catalyzethe initial condensation of acyl-ACPs and existing fatty acid
chains with malonyl-ACP, respectively (9). In contrast, tri-closan, another fatty acid biosynthesis inhibitor, discovered inthe 1970s, targets the second reduction step in the fatty acidchain elongating biosynthesis cycle, inhibiting the enoyl-ACPreductase FabI (11) (see Fig. 1 for an overview). Neither tri-closan nor cerulenin is used clinically as an antimicrobial agent.However, triclosan is used in consumer products, such astoothpaste (17, 29, 30), while cerulenin is currently being eval-uated as an antitumor therapeutic in combination therapies(10). Platensimycin and platencin showed efficacy in a Staphy-lococcus aureus mouse infection model (31). A number ofstructure-activity relationship (SAR) studies are being per-formed to identify lead structures for further development (14,16, 18, 20). In addition, the search for effective natural ana-logues continues (12, 35, 36).
Proteomic profiling can support antibacterial drug discoveryby contributing to target identification and mechanism-of-ac-tion studies (4, 7). We have previously established a compre-hensive proteomic response reference compendium using theGram-positive model organism Bacillus subtilis. It containsproteomic response patterns for over 40 antibacterial com-pounds (2). As the bacterial response to antibiotic treatmentmirrors the inflicted damage, it is highly specific and closelylinked to the antibiotic mechanism. Proteomic signatures in-dicative of the antibiotic mechanism of action can be estab-lished, if structurally different inhibitors of the same pathwayare available (28). Once they are established, these signaturescan aid in mechanism-of-action identification of structurallynovel compounds. For instance, the proteomic signature for
* Corresponding author. Mailing address: Ruhr-Universitat Bo-chum, Mikrobielle Antibiotikaforschung, Biologie der Mikroorganis-men, Universitatsstraße 150, Bochum 44801, Germany. Phone: 49-234-32-23102. Fax: 49-234-32-14620. E-mail: [email protected].
� Published ahead of print on 7 March 2011.
2590
B Fatty acid biosynthesis inhibition signature
28

inhibition of translation identified peptidyltransferase inhibi-tion as the mechanism of action of the structurally novel com-pound Bay 50-2369 (2). The signatures can also serve asreferences to confirm the in vivo mechanism of action of SAR-derived compounds, provided the modified compound still hasthe same mechanism as the lead structure.
In this study we investigated the bacterial response to fattyacid biosynthesis inhibitors triclosan, cerulenin, platensimycin,and platencin. On the basis of the proteomic response profilesof B. subtilis, we were able to establish a proteomic signaturefor fatty acid biosynthesis inhibition. We then applied thenewly established signature to investigate the mechanism ofaction of the chromium bioorganometallic PM47, a compoundinspired by platensimycin, which displayed low activity againstGram-positive bacteria (19). On the basis of the findings ofproteome analysis, we could rule out fatty acid biosynthesis asits primary mechanism of action.
MATERIALS AND METHODS
Bacterial strains and growth conditions. Bacillus subtilis 168 (trpC2) (1) wasgrown at 37°C under steady agitation in a defined medium previously described(25). Cerulenin and triclosan were purchased from Merck KGaA, Darmstadt,Germany; platencin was synthesized by D. Chen; and platensimycin was providedby Merck & Co., Inc., Rahway, NJ. All antibiotic stock solutions were preparedin dimethyl sulfoxide (DMSO). MICs were determined in a test tube assay asdescribed previously (2). Two milliliters of defined medium was inoculated with
105 bacteria per ml, and the mixture was incubated at 37°C under agitation for18 h. The MIC was defined as the lowest concentration inhibiting visible growth.
In growth experiments, bacterial cultures were exposed to antibiotics at dif-ferent concentrations during early exponential growth phase after they reachedan optical density at 500 nm (OD500) of 0.35. An antibiotic concentration leadingto a reduction in growth rate of approximately 50 to 70% was chosen forproteomic profiling experiments.
Preparation of cytoplasmic L-[35S]methionine-labeled protein fractions. Forpulse-labeling experiments, 5 ml of a bacterial culture in early exponentialgrowth phase was exposed to 0.5 �g/ml triclosan, 5 �g/ml cerulenin, 5 �g/mlplatensimycin, 0.2 �g/ml platencin, or 25 �g/ml PM47 or was left untreated as acontrol. After 10 min of antibiotic treatment, cells were pulse-labeled radioac-tively with 1.8 MBq L-[35S]methionine (Hartmann Analytic, Braunschweig, Ger-many) for 5 min. Methionine incorporation was stopped by adding 1 mg/mlchloramphenicol and an excess of nonradioactive L-methionine (10 mM) and byimmediately transferring samples onto ice. Cells were harvested by centrifuga-tion and washed three times with 100 mM Tris–1 mM EDTA buffer, beforedisruption by ultrasonication in a VialTweeter instrument (Hielscher, Teltow,Germany) in 10 mM Tris buffer containing 1.39 mM phenylmethylsulfonyl flu-oride. The soluble protein fraction was separated from cell debris by centrifu-gation at 16.1 � g for 20 min. Protein concentrations were estimated using aBradford-based Roti NanoQuant assay (Roth, Karlsruhe, Germany).
2D-PAGE. Unless otherwise noted, chemicals for two-dimensional (2D) gelelectrophoresis were ordered from Sigma-Aldrich or Roth in electrophoresis-grade quality. Cytosolic proteins were solubilized in 400 �l buffer containing 7 Murea, 2 M thiourea, 6.5 mM 3-[(3-cholamidopropyl)-dimethylammonio]-1-pro-panesulfonate, 0.5% Triton X-100, 1.04% Pharmalyte 3-10 (GE Healthcare,Uppsala, Sweden), and 50 mM dithiothreitol (DTT). Fifty or 300 �g of proteinfor radioactive analytical gels and for nonradioactive preparative gels (for pro-tein identification by mass spectrometry), respectively, was loaded onto 24-cmimmobilized pH gradient (IPG) strips, pH 4 to 7 (GE Healthcare), by passive
FIG. 1. Fatty acid biosynthesis in B. subtilis. Cerulenin, platensimycin, and platencin inhibit the 3-oxoacyl-ACP synthase FabF. Platencin targetsFabF and FabHA/HB. Triclosan inhibits the enoyl-ACP-reductase FabI. Proteins belonging to the FapR regulon are circled. Proteins induced byfatty acid biosynthesis inhibitors are placed inside boxes. Modified according to Fujita et al. (9) with permission of the publisher.
VOL. 55, 2011 FATTY ACID BIOSYNTHESIS PROTEOMIC SIGNATURE 2591
B Fatty acid biosynthesis inhibition signature
29

rehydration for 18 h. Isoelectric focusing was carried out using a Multiphor IIapparatus (GE Healthcare), applying the following gradient: 0 to 500 V for 1kVh, 500 V for 0.02 kVh, 500 to 3,500 V for 3 kVh, and 3,500 V for 57 kVh at20°C. Prior to SDS-PAGE, proteins were reduced and subsequently alkylated for20 min each in equilibration buffer (50 mM Tris, pH 8.8, 6 M urea, 30% glycerol,2% SDS) supplemented with 1% DTT and 2.5% iodoacetamide, respectively.IPG strips were placed onto 12.5% SDS-polyacrylamide gels (acrylamide, bisa-crylamide, 0.375 M Tris, 01.% SDS, 0.05% ammonium persulfate, 0.0138%N,N,N�,N�-tetramethylethylenediamine) and covered with 0.5% agarose in run-ning buffer (25 mM tris, 192 mM glycine, 0.1% SDS). Electrophoresis was carriedout using an Ettan DALTtwelve system from GE Healthcare at 0.5 W/gel for 1 hto allow transfer of proteins from the IPG strip into the polyacrylamide gel,followed by 10 W/gel for protein separation using the above-described runningbuffer (2� buffer in the upper chamber, 1� buffer in the lower chamber).Proteins were stained with 0.003% ruthenium(II) Tris(4,7-diphenyl-1,10-phenan-trolin disulfonate) (RuBPs) (22) and imaged using a Typhoon Trio� variable-mode imager (GE Healthcare) set at an excitation wavelength of 532 nm andusing a 610-nm emission filter. Analytical gels were dried on Whatman paper andexposed to storage phosphor screens (GE Healthcare). Screens were scannedwith the Typhoon Trio� imager at a 633-nm excitation wavelength and using a390-nm emission filter. Images were analyzed as described by Bandow et al. (3)using Decodon Delta 2D image analysis software (Decodon, Greifswald, Ger-many). Proteins found to be induced more than 2-fold in three independentbiological replicates are reported as marker proteins.
Protein identification. Protein spots were excised from preparative 2D gelsand transferred into 96-well microtiter plates. Tryptic digestion with subsequentspotting on a matrix-assisted laser desorption ionization (MALDI) target wascarried out automatically with an Ettan spot handling workstation (AmershamBiosciences, Uppsala, Sweden) as described by Eymann et al. (8).
MALDI-time of flight (TOF) measurements were carried out on a 4800MALDI TOF/TOF analyzer (Applied Biosystems, Foster City, CA) designed forhigh-throughput measurements. The instrument allows automatic measurementof the samples, calibration of the spectra, and analysis of the data using 4000Explorer software, version 3.5.3. Spectra were recorded in a mass range from 900to 3,700 Da with a focus mass of 2,000 Da. For one main spectrum, 25 subspectrawith 100 shots per subspectrum were accumulated using a random-search pat-tern. If the autolytical fragment of trypsin with the monoisotopic (M�H)� m/zat 2211.104 reached a signal-to-noise (S/N) ratio of at least 10, internal calibra-tion was automatically performed as one-point calibration using this peak. Thestandard mass deviation was less then 0.15 Da. If the automatic mode failed (forless than 1% of samples), the calibration was carried out manually. After cali-bration, the peak lists were created by using the “peak-to-mascot” script of the4000 Explorer software, version 3.5.3, using the following settings: mass rangefrom 900 to 3,700 Da, peak density of 50 peaks per 200 Da, minimal area of 100,and a maximum of 200 peaks per spot. The peak list was created for an S/Nratio of 6.
The MALDI-TOF-TOF measurements were also carried out on the 4800MALDI TOF/TOF analyzer. The three strongest peaks of the TOF spectra weremeasured. For one main spectrum, 20 subspectra with 125 shots per subspectrumwere accumulated using a random-search pattern. Internal calibration was au-tomatically performed as one-point calibration, with the monoisotopic arginine(M�H)� m/z at 175.119 or lysine (M�H)� m/z at 147.107 reaching an S/N ratioof at least 5. The peak lists were created by using the script of the 4000 ExplorerSoftware, version 3.5.3, with the following settings: mass range from 60 to pre-cursor mass plus 20 Da, peak density of 5 peaks per 200 Da, minimal area of 100,and a maximum of 20 peaks per precursor. The peak list was created for an S/Nratio of 5. For database search, the Mascot search engine, version 2.1.04 (MatrixScience Ltd., London, United Kingdom), with a specific B. subtilis sequencedatabase was used.
RESULTS
Proteomic signature of fatty acid biosynthesis inhibition.Bacterial cells react quickly to subinhibitory concentrations ofantibiotics by adjusting their protein synthesis (2). In manycases, translation capacity is allocated to production of pro-teins that counteract the loss of function or the damage in-flicted by the antibiotic action. The cellular response is highlyspecific for each antibiotic compound, with some of the re-sponder proteins reflecting the antibiotic mechanism of action
and others being structure specific. Structurally different anti-biotics that cause the same physiological condition in the cellinduce the same responder proteins. Those responder proteinsindicative of the antibiotic mechanism constitute the antibioticproteomic signature.
To establish an antibiotic proteomic signature for fatty acidbiosynthesis inhibition, the protein synthesis patterns of tri-closan, cerulenin, platensimycin, and platencin were comparedto identify common responder proteins. Reproducibility of theproteome response patterns for each of the antibiotics is ab-solutely critical and highly dependent on reproducibility of thecellular growth and antibiotic treatment conditions. Differ-ences in antibiotic concentration, cell density, or incubationtime may have a significant impact on protein synthesis pro-files. This is of particular concern, when an antibiotic mode ofaction is known to shift in a concentration-dependent manner.Triclosan, e.g., inhibits fatty acid biosynthesis at low concen-trations but at higher doses also damages multiple cytoplasmicand membrane targets (23, 26, 27).
We first determined the MICs of the four antibiotics againstB. subtilis under growth conditions similar to those used forproteomic sample generation. The MICs in defined mediumwith cells aerated during overnight incubation were 0.1 �g/mlfor triclosan, 0.2 �g/ml for platencin, 1 �g/ml for platensimy-cin, and 5 �g/ml for cerulenin. Growth experiments with dif-ferent antibiotic concentrations added to the cultures in earlyexponential growth phase were then performed to identify aconcentration that inhibited bacterial growth visibly withoutkilling the cells. A growth rate reduction of 50 to 70% in theexponential growth phase compared to the untreated controlgrowth had previously proven useful for proteome analysis (2).In order to generate the desired growth inhibition, concentra-tions of up to five times the MIC were used to treat thecultures. Representative growth curves of B. subtilis culturesexposed to fatty acid biosynthesis inhibitors at concentrationsselected for proteome analyses are shown in Fig. 2.
Once a suitable antibiotic concentration was found, pulse-labeling experiments with L-[35S]methionine were per-formed to specifically label those proteins newly synthesizedduring the 5-min pulse either after treatment with a fattyacid biosynthesis inhibitor or under control conditions.Highly reproducible protein expression profiles were ob-tained from three biological replicate experiments for eachantibiotic following a standardized protocol for protein sep-aration and gel image analysis. Marker proteins reproduc-ibly induced at least 2-fold for each antibiotic are listed inTable 1. All fatty acid biosynthesis inhibitors tested inducedsix common marker proteins (Fig. 3). These six markersmake up the proteomic signature for fatty acid biosynthesisinhibition. All of them are involved in fatty acid biosynthe-sis. Indirectly contributing to fatty acid or phospholipid bio-synthesis is PanB, an enzyme of the pantothenate biosyn-thesis pathway. Pantothenate is a precursor of the cofactorof ACP and a precursor of coenzyme A (CoA). Coenzyme Ais essential for fatty acid biosynthesis, as the condensationcycle always starts with acetyl-CoA or branched-chain CoAs(Fig. 1) and chain elongation requires malonyl-CoA. Theother five marker proteins are involved directly in fattyacid biosynthesis. The 3-oxoacyl-ACP synthases FabHA,FabHB, and FabF initiate the condensation reaction with
2592 WENZEL ET AL. ANTIMICROB. AGENTS CHEMOTHER.
B Fatty acid biosynthesis inhibition signature
30

different substrate preferences, whereas enoyl-ACP reduc-tase FabI catalyzes the second reduction step of the con-densation cycle. PlsX starts the conversion of fatty acids tophospholipids. On the transcriptional level in B. subtilis, allfive enzymes are negatively controlled by the FapR repres-sor and are therefore part of the same regulon (Fig. 1).
Platensimycin exclusively induced the marker proteins of theproteomic signature for fatty acid biosynthesis inhibition. Tri-closan, cerulenin, and platencin each induced some additionalmarker proteins specific for each compound (Table 1). Some
FIG. 3. Details of 2D gels depict the six marker proteins (markedby arrowheads) making up the proteomic signature of fatty acid bio-synthesis inhibition under control conditions as well as triclosan,cerulenin, platensimycin, and platencin treatment. Induction factorsdisplayed in the lower right reflect averages over three biologicalreplicates.
FIG. 2. Antibiotic concentrations leading to approximately 50%growth reduction. B. subtilis was grown in defined medium to expo-nential phase and treated with platensimycin (5 �g/ml) (A), platencin(0.2 �g/ml) (B), cerulenin (5 �g/ml) (C), and triclosan (0.5 �g/ml) (D).The time point of antibiotic addition is marked by arrowheads.
TABLE 1. Marker proteins induced by fatty acid biosynthesis inhibitors
Marker proteinaInduction factor
Protein function PathwayPlatensimycin Platencin Cerulenin Triclosan
FabHA 5.1 5.2 4.3 7.2 3-Oxoacyl-ACP synthase III Fatty acid biosynthesisFabHB 17.6 18.6 14.2 20.0 3-Oxoacyl-ACP synthase III Fatty acid biosynthesisFabF 2.6 3.7 4.2 3.7 3-Oxoacyl-ACP synthase II Fatty acid biosynthesisFabI 12.7 3.7 6.7 6.2 Enoyl-ACP reductase Fatty acid biosynthesisPlsX 3.1 4.0 3.2 2.6 Putative glycerol-3-phosphate
acyltransferase PlsXPhospholipid biosynthesis
PanB 2.8 4.7 3.0 3.9 3-Methyl-2-oxobutanoatehydroxymethyltransferase
Pantothenate biosynthesis
CP_1 7.0 5.4 Protein not identifiedYkrS 2.6 Methylthioribose-1-phosphate isomerase Amino acid metabolismSerA 3.5 D-3-Phosphoglycerate dehydrogenase Amino acid metabolismPlc_3 3.1 Protein not identifiedPlc_4 4.0 Protein not identifiedAld 2.6 Putative alanine dehydrogenase Amino acid metabolismCer_1 4.4 Protein not identifiedYurP 2.8 Similar to glutamine–fructose-6-phosphate
transaminaseAmino acid metabolism
Cer_4 2.3 Protein not identifiedPdhC 4.3 Pyruvate dehydrogenase subunit E2b Acetyl-CoA, branched-chain
CoA metabolismTrc_2 3.9 Protein not identifiedSucD 2.3 Succinyl-CoA synthetase subunit alpha Citric acid cycleTrc_4 2.6 Protein not identified
a Cer, unidentified protein induced by cerulenin; CP, unidentified protein induced by cerulenin and platencin; Plc, unidentified protein induced by platencin; Trc,unidentified protein induced by triclosan.
b This subunit is also part of branched-chain alpha 2-oxoacid dehydrogenase.
VOL. 55, 2011 FATTY ACID BIOSYNTHESIS PROTEOMIC SIGNATURE 2593
B Fatty acid biosynthesis inhibition signature
31

of these proteins are related indirectly to fatty acid biosynthe-sis, as they are involved in acetyl-CoA production. Cerulenin-induced Ald is a putative dehydrogenase forming pyruvatefrom L-alanine, while triclosan-induced PdhC is a subunitof pyruvate dehydrogenase as well as branched-chain alpha2-oxoacid dehydrogenase, which generate acetyl-CoA andbranched-chain CoAs, respectively.
Some of the marker proteins, such as platencin-inducedYkrS involved in the methionine salvage pathway, could not beconnected directly to fatty acid biosynthesis. Others could notbe identified by mass spectrometry due to a lack of proteinaccumulation. However, these protein signals defined by theircoordinates on the 2D gel map still serve as antibiotic-specificmarkers, as they are reproducibly induced after antibiotictreatment.
Proteomic analysis of platensimycin analogue PM47. Patraet al. synthesized different organometallic platensimycin deriv-atives aiming to preserve the steric properties while at thesame time simplifying synthesis and potentially improving an-tibacterial properties (19). One of these compounds is thechromium bioorganometallic PM47 (Fig. 4), which shows lowactivity against Gram-positive bacteria and has a MIC of 80�g/ml against B. subtilis (19).
The proteomic response of B. subtilis to 25 �g/ml PM47 wasinvestigated to test whether PM47 still inhibits fatty acid bio-synthesis or has another mechanism of action. Induction of the
marker proteins belonging to the proteomic signature for fattyacid biosynthesis inhibition would be expected if the mecha-nism of action were preserved.
The proteomic responses to PM47 and platensimycin werecompared, as the latter induced exclusively the proteins be-longing to the fatty acid biosynthesis inhibition signature (Fig.5). None of the six signature markers nor any of the com-pound-specific marker proteins were induced by PM47, indi-cating that PM47 does not inhibit fatty acid biosynthesis. Sev-enteen proteins were significantly upregulated upon PM47treatment. The comparison of the proteomic response toPM47 with the proteomic profiles in the reference compen-dium (2) did not yield a close match, although some of thePM47 marker proteins are also induced by membrane-activeantibiotics (data not shown). There was further evidence thatplatensimycin derivative PM47 has another mechanism of ac-tion than its lead compound. In contrast to bacteriostatic plat-ensimycin, PM47 was shown to be bacteriolytic at higher doses.
DISCUSSION
Bacteria respond to antibiotic treatment in a highly specificway. The proteome mirrors the physiological impairmentcaused by the antibiotic. Due to this tight connection betweenmechanism of action and proteomic response, we can use pro-teomics to support mechanism-of-action studies in antibacte-rial drug discovery programs (2, 5, 6). To this end, we estab-lished a reference compendium of proteome response profilesfor over 40 antibiotics, including almost all clinically relevantantibiotic classes. Recently, the discovery of novel inhibitorswith antibacterial activity sparked renewed interest in fattyacid biosynthesis as an antibiotic target (34). The comparisonof proteomic responses to four different inhibitors of fatty acidbiosynthesis allowed the description of a proteomic signaturediagnostic of fatty acid biosynthesis inhibition consisting of sixupregulated proteins connected to the target pathway. Thisproteomic signature, together with the signatures already avail-able in the reference compendium, serves to classify structur-
FIG. 4. Structures of platensimycin (structure 1) and PM47 (struc-ture 2). The tetracyclic cage of platensimycin was replaced by anorganometallic core. Me, methyl.
FIG. 5. The protein expression profiles of B. subtilis in response to treatment with 5 �g/ml platensimycin (A) and 25 �g/ml PM47 (B) do notshare marker proteins. In panel B, unidentified marker proteins of PM47 are indicated by arrowheads, while boxes indicate the locations of fattyacid biosynthesis marker proteins. Pulse-labeled cytosolic proteins were separated by 2D PAGE. Red false-color images representing proteinbiosynthesis after antibiotic treatment were warped onto green false-color images showing protein synthesis under control conditions. Antibiotic-induced proteins appear red, repressed proteins appear green, and proteins synthesized at equal rates under both conditions appear yellow.
2594 WENZEL ET AL. ANTIMICROB. AGENTS CHEMOTHER.
B Fatty acid biosynthesis inhibition signature
32

ally novel synthetic or natural compounds with regard to theirmechanisms of action. Proteome analyses of the bacterial re-sponse to different fatty acid biosynthesis inhibitors revealedthat five enzymes regulated by fatty acid biosynthesis repressorFapR are induced regardless of the molecular target in thepathway. They are directly involved in fatty acid or phospho-lipid biosynthesis (Fig. 1). For the four enzymes directly in-volved in fatty acid biosynthesis, FabI, FabF, FabHA, andFabHB, Hutter et al. previously reported induction of tran-scription by fatty acid biosynthesis inhibitors cerulenin andtriclosan (13). Malonyl-CoA sterically inhibits FapR (9). It canbe expected that this precursor of fatty acid biosynthesis accu-mulates upon fatty acid biosynthesis inhibition, causing dere-pression of the fatty acid biosynthesis enzymes. The sixthmarker protein of the proteomic signature, PanB, is involved inthe biosynthesis of coenzyme A, an essential cofactor for fattyacid biosynthesis, as the condensation reaction always startswith acetyl-CoA or branched-chain CoAs. None of the sixsignature proteins were induced by any of the antibiotics withmechanisms other than fatty acid biosynthesis inhibition testedso far.
Beside the six proteins induced by all inhibitors tested, someadditional proteins are upregulated in response to one or twoof the antibiotics. Some of these are connected to fatty acidbiosynthesis as they contribute to the acetyl-CoA metabolism.Others may be induced as a result of compound-specific reac-tions.
The newly established proteomic signature for fatty acidbiosynthesis inhibition was used to evaluate the mechanism ofaction of a compound derived from an SAR program aroundthe lead structure of platensimycin. Chromium-containing or-ganometallic analogue PM47 showed low antibacterial activity(19) against Gram-positive pathogens. PM47 treatment did notinduce fatty acid biosynthesis enzymes, proving that the in vivomechanism of action is not inhibition of fatty acid biosynthesis.This finding is interesting, as in silico docking studies indicatedthat the compound fits well into the same binding pocket asplatensimycin (19). However, a different mechanism of actionis in concordance with the observation that, unlike fatty acidbiosynthesis inhibitors, PM47 is bacteriolytic at higher doses.Interestingly, the entire reference compendium does not con-tain a proteomic signature or protein response profile match-ing that of PM47, pointing to an unprecedented mechanism ofaction. There is some overlap in marker proteins with mem-brane-active antibiotics. Further studies would be necessary toelucidate the mechanism of action. However, PM47 showscytotoxicity against mammalian cells at sub-MICs (19), sug-gesting a nonspecific, toxic mechanism.
ACKNOWLEDGMENTS
We thank Merck & Co., Inc., Rahway, NJ, for providing platensi-mycin. We further thank Elmar Langenfeld, formerly of the Medizinis-ches Proteom-Center, Ruhr University Bochum, for helping us withRuBP synthesis and Lars I. O. Leichert from the same institution forcritically reading the manuscript. We are also grateful for the superbtechnical support by our colleagues at Rubion.
This work was financially supported by a startup grant from theRuhr University Bochum and a grant from the state of North Rhine-Westphalia (NRW), Germany, and the European Union, EuropeanRegional Development Fund, Investing in your future, to J.E.B.
REFERENCES
1. Agnostopoulos, C., and J. Spizizen. 1961. Requirements for transformationin Bacillus subtilis. J. Bacteriol. 81:741–746.
2. Bandow, J. E., H. Brotz, L. I. Leichert, H. Labischinski, and M. Hecker.2003. Proteomic approach to understanding antibiotic action. Antimicrob.Agents Chemother. 47:948–955.
3. Bandow, J. E., et al. 2008. Improved image analysis workflow for 2D gelsenables large-scale 2D gel-based proteomics studies—COPD biomarker dis-covery study. Proteomics 8:3030–3041.
4. Bandow, J. E., and M. Hecker. 2007. Proteomic profiling of cellular stressesin Bacillus subtilis reveals cellular networks and assists in elucidating antibi-otic mechanisms of action. Prog. Drug Res. 64:79, 81-101.
5. Beyer, D., et al. 2004. New classes of bacterial phenylalanyl-tRNA synthetaseinhibitors with high potency and broad-spectrum activity. Antimicrob.Agents Chemother. 48:525–532.
6. Brotz-Oesterhelt, H., et al. 2005. Dysregulation of bacterial proteolytic ma-chinery by a new class of antibiotics. Nat. Med. 11:1082–1087.
7. Brotz-Oesterhelt, H., J. E. Bandow, and H. Labischinski. 2005. Bacterialproteomics and its role in antibacterial drug discovery. Mass Spectrom. Rev.24:549–565.
8. Eymann, C., et al. 2004. A comprehensive proteome map of growing Bacillussubtilis cells. Proteomics 4:2849–2876.
9. Fujita, Y., H. Matsuoka, and K. Hirooka. 2007. Regulation of fatty acidmetabolism in bacteria. Mol. Microbiol. 66:829–839.
10. Haase, D., et al. 2010. Fatty acid synthase as a novel target for meningiomatherapy. Neuro. Oncol. 12:844–854.
11. Heath, R. J., Y. T. Yu, M. A. Shapiro, E. Olson, and C. O. Rock. 1998. Broadspectrum antimicrobial biocides target the FabI component of fatty acidsynthesis. J. Biol. Chem. 273:30316–30320.
12. Herath, K. B., et al. 2008. Structure and semisynthesis of platensimide A,produced by Streptomyces platensis. Org. Lett. 10:1699–1702.
13. Hutter, B., et al. 2004. Prediction of mechanisms of action of antibacterialcompounds by gene expression profiling. Antimicrob. Agents Chemother.48:2838–2844.
14. Jang, K. P., et al. 2009. Isoplatensimycin: synthesis and biological evaluation.Bioorg. Med. Chem. Lett. 19:4601–4602.
15. Jayasuriya, H., et al. 2007. Isolation and structure of platencin: a FabH andFabF dual inhibitor with potent broad-spectrum antibiotic activity. Angew.Chem. Int. ed. Engl. 46:4684–4688.
16. Krauss, J., V. Knorr, V. Manhardt, S. Scheffels, and F. Bracher. 2008.Synthesis of platensimycin analogues and their antibiotic potency. Arch.Pharm. (Weinheim) 341:386–392.
17. Moran, J., M. Addy, R. G. Newcombe, and I. Marlow. 2001. A study to assessthe plaque inhibitory action of a newly formulated triclosan toothpaste.J. Clin. Periodontol. 28:86–89.
18. Nicolaou, K. C., G. S. Tria, D. J. Edmonds, and M. Kar. 2009. Totalsyntheses of (�/�)-platencin and (�)-platencin. J. Am. Chem. Soc. 131:15909–15917.
19. Patra, M., et al. 2009. Synthesis and biological evaluation of chromiumbioorganometallics based on the antibiotic platensimycin lead structure.ChemMedChem. 4:1930–1938.
20. Patra, M., et al. 2010. Synthesis and biological evaluation of ferrocene-containing bioorganometallics inspired by the platensimycin lead structure.Organometallics 29:4312–4319.
21. Price, A. C., et al. 2001. Inhibition of beta-ketoacyl-acyl carrier proteinsynthases by thiolactomycin and cerulenin. Structure and mechanism. J. Biol.Chem. 276:6551–6559.
22. Rabilloud, T., J. M. Strub, S. Luche, A. van Dorsselaer, and J. Lunardi.2001. A comparison between SyproRuby and ruthenium II Tris (batho-phenanthroline disulfonate) as fluorescent stains for protein detection ingels. Proteomics 1:699–704.
23. Russell, A. D. 2004. Whither triclosan? J. Antimicrob. Chemother. 53:693–695.
24. Singh, S. B., et al. 2006. Isolation, structure, and absolute stereochemistry ofplatensimycin, a broad spectrum antibiotic discovered using an antisensedifferential sensitivity strategy. J. Am. Chem. Soc. 128:11916–11920.
25. Stulke, J., R. Hanschke, and M. Hecker. 1993. Temporal activation of beta-glucanase synthesis in Bacillus subtilis is mediated by the GTP pool. J. Gen.Microbiol. 139:2041–2045.
26. Suller, M. T. E., and A. D. Russell. 1999. Antibiotic and biocide resistance inmethicillin-resistant Staphylococcus aureus and vancomycin-resistant entero-coccus. J. Hosp. Infect. 43:281–291.
27. Suller, M. T. E., and A. D. Russell. 2000. Triclosan and antibiotic resistancein Staphylococcus aureus. J. Antimicrob. Chemother. 46:11–18.
28. vanBogelen, R. A., E. E. Schiller, J. D. Thomas, and F. C. Neidhardt. 1999.Diagnosis of cellular states of microbial organisms using proteomics. Elec-trophoresis 20:2149–2159.
29. Van Loveren, C., J. F. Bujis, and J. M. ten Cate. 1999. Protection of dentinby triclosan toothpaste in a bacterial demineralization model. Eur. J. OralSci. 107:114–120.
30. van Loveren, C., J. F. Bujis, and J. M. ten Cate. 2000. The effect of triclosan
VOL. 55, 2011 FATTY ACID BIOSYNTHESIS PROTEOMIC SIGNATURE 2595
B Fatty acid biosynthesis inhibition signature
33

toothpaste on enamel demineralization in a bacterial demineralizationmodel. J. Antimicrob. Chemother. 45:153–158.
31. Wang, J., et al. 2007. Discovery of platencin, a dual FabF and FabH inhibitorwith in vivo antibiotic properties. Proc. Natl. Acad. Sci. U. S. A. 104:7612–7616.
32. Wang, J., et al. 2006. Platensimycin is a selective FabF inhibitor with potentantibiotic properties. Nature 441:358–361.
33. World Health Organization. 2005. Containing antimicrobial resistance.World Health Organization, Geneva, Switzerland.
34. Wright, H. T., and K. A. Reynolds. 2007. Antibacterial targets in fatty acidbiosynthesis. Curr. Opin. Microbiol. 10:447–453.
35. Zhang, C., et al. 2010. Isolation, structure and biological activities of platen-cin A(2)-A(4) from Streptomyces platensis. Bioorg. Med. Chem. 18:2602–2610.
36. Zhang, C., et al. 2008. Isolation, structure and fatty acid synthesis inhibitoryactivities of platensimycin B1-B3 from Streptomyces platensis. Chem. Com-mun. (Camb.) 5034-5036.
2596 WENZEL ET AL. ANTIMICROB. AGENTS CHEMOTHER.
B Fatty acid biosynthesis inhibition signature
34

C
Proteomic response of Bacillus subtilis to
lantibiotics reflects differences in interaction with
the cytoplasmic membrane
Michaela Wenzel, Bastian Kohl, Daniela Münch, Nadja Raatschen, H.
Bauke Albada, Leendert Hamoen, Nils Metzler‐Nolte, Hans‐Georg Sahl,
Julia E. Bandow
Antimicrobial Agents and Chemotherapy, 2012
C Lantibiotic stress response
35

Proteomic Response of Bacillus subtilis to Lantibiotics ReflectsDifferences in Interaction with the Cytoplasmic Membrane
Michaela Wenzel,a Bastian Kohl,a* Daniela Münch,b Nadja Raatschen,a H. Bauke Albada,c Leendert Hamoen,d Nils Metzler-Nolte,c
Hans-Georg Sahl,b and Julia E. Bandowa
Biology of Microorganisms, Ruhr University Bochum, Bochum, Germanya; Medical Microbiology, Immunology and Parasitology, Pharmaceutical Microbiology, Universityof Bonn, Bonn, Germanyb; Bioinorganic Chemistry, Ruhr University Bochum, Bochum, Germanyc; and Centre for Bacterial Cell Biology, Institute for Cell and MolecularBiosciences, Newcastle University, Newcastle-upon-Tyne, United Kingdomd
Mersacidin, gallidermin, and nisin are lantibiotics, antimicrobial peptides containing lanthionine. They show potent antibacte-rial activity. All three interfere with cell wall biosynthesis by binding lipid II, but they display different levels of interaction withthe cytoplasmic membrane. On one end of the spectrum, mersacidin interferes with cell wall biosynthesis by binding lipid IIwithout integrating into bacterial membranes. On the other end of the spectrum, nisin readily integrates into membranes, whereit forms large pores. It destroys the membrane potential and causes leakage of nutrients and ions. Gallidermin, in an intermedi-ate position, also readily integrates into membranes. However, pore formation occurs only in some bacteria and depends onmembrane composition. In this study, we investigated the impact of nisin, gallidermin, and mersacidin on cell wall integrity,membrane pore formation, and membrane depolarization in Bacillus subtilis. The impact of the lantibiotics on the cell envelopewas correlated to the proteomic response they elicit in B. subtilis. By drawing on a proteomic response library, including otherenvelope-targeting antibiotics such as bacitracin, vancomycin, gramicidin S, or valinomycin, YtrE could be identified as the mostreliable marker protein for interfering with membrane-bound steps of cell wall biosynthesis. NadE and PspA were identified asmarkers for antibiotics interacting with the cytoplasmic membrane.
Over the last decades, bacteria have demonstrated their im-pressive ability to adapt to changing environmental condi-
tions by rapidly developing and accumulating antibiotic resis-tances. Helped by an extensive use of antibiotics in health care andagriculture, multidrug-resistant strains, so-called superbugs, arespreading with a remarkable impact on human health. Recentstatistics of the World Health Organization show that even indeveloped countries, infectious diseases are back among the topfive causes of death, with the majority of deaths being attributed tobacterial infections (49). At the same time, approval rates of newantibiotic agents have been steadily decreasing since their apex inthe 1980s (5). In view of these developments, in addition to im-proving hospital hygiene and to devising and implementing strat-egies to preserve effectiveness of available antimicrobial agents, areinvigoration of antibiotic research and development is urgentlyneeded to meet the superbug challenge.
One of the promising antibiotic classes for treatment of infec-tions caused by multidrug-resistant pathogens are antimicrobialpeptides, which occur naturally as components of the host defenseand typically target the bacterial cell envelope. The peptide sub-class of lantibiotics, which are produced by Gram-positive bacte-ria, is characterized by containing the nonproteinogenic aminoacid lanthionine and often also other unusual amino acids. How-ever, the group of lantibiotics is structurally and mechanisticallydiverse and can be further divided into class A, comprisingstretched, flexible peptides, and class B, with more globular struc-tures. The majority of peptides in both classes have been shown toinhibit cell wall biosynthesis. In contrast to class B members, someclass A lantibiotics are also capable of integrating into the bacterialmembrane and of pore formation, a process facilitated by bindingof cell wall precursor lipid II (7, 13). This mechanistic dualitydepicts an interesting advantage of lantibiotics over conventionalsingle-mechanism antibiotics. It was shown for various antibacte-
rial compounds that inhibit targets encoded by multiple genesthat resistance development is much slower than for antibioticsinhibiting a single target of a specific metabolic pathway (11, 32).As two essential complex targets, the cell wall biosynthesis ma-chinery and the cell membrane, need to be altered to preventcellular damage by dual-mechanism lantibiotics, resistance de-velopment by target mutation and stress adaptation is effec-tively deferred (10, 32). Such properties highlight antimicro-bial peptides as attractive antibacterial agents.
Here, we investigate three different lantibiotics. Nisin, the firstlantibiotic ever described, belongs to class A. The bactericidal ef-fects of nisin involve two distinct mechanisms. At lower concen-trations, nisin forms aggregates with lipid II, thereby preventingthem from participating in cell wall biosynthesis (23, 35). Afterreaching a certain concentration threshold, complexes of 8 nisinand 4 lipid II molecules are formed that integrate into the bacterialmembrane. They form pores large enough for ions and smallamino acids to pass through (9, 23). Structurally similar gallider-min also belongs to class A and inhibits cell wall biosynthesis bylipid II binding. Potassium leakage indicating pore formation wasobserved in some but not all bacteria (8); however, B. subtilis wasnot tested. Christ et al. were able to correlate pore formation with
Received 6 July 2012 Returned for modification 28 July 2012Accepted 16 August 2012
Published ahead of print 27 August 2012
Address correspondence to Julia Bandow, [email protected].
* Present address: Bastian Kohl, Biochemistry II—Biomolecular Spectroscopy, RuhrUniversity Bochum, Bochum, Germany.
Copyright © 2012, American Society for Microbiology. All Rights Reserved.
doi:10.1128/AAC.01380-12
on October 31, 2012 by U
NIV
ER
SIT
AT
BO
CH
UM
http://aac.asm.org/
Dow
nloaded from
C Lantibiotic stress response
36

membrane thickness and fatty acid branching, suggesting that thissecond mechanism of action of gallidermin is dependent on mem-brane composition (16). In contrast to nisin, which needs to dockto lipid II for membrane integration and pore formation, gallider-min shows very high association and low dissociation constantsfor binding phospholipid bilayers, indicating that it readily inte-grates into the membrane independently of lipid II binding. Theinsertion of gallidermin into the bilayer already influences mem-brane properties without a necessity for pore formation (16).Class B lantibiotic mersacidin is structurally and mechanisticallydifferent from nisin or gallidermin. More globular in structure, itis not able to integrate into the membrane or to form pores, so itsantibacterial activity is based solely on inhibition of cell wall bio-synthesis by binding to lipid II. In contrast to vancomycin, a gly-copeptide which does not integrate into the membrane either,mersacidin does not seem to bind the amino acid tail of lipid II.Rather, it binds to the disaccharide headgroup of the lipid II mol-ecule and additionally interacts with the pyrophosphate, suggest-ing that lipid II-bound lantibiotic molecules are localized near theouter layer of the cell membrane (6, 14).
These three lantibiotics were chosen to investigate the pro-teomic response of B. subtilis to cell wall biosynthesis inhibition bylipid II binding coupled with different levels of interference withmembrane integrity. The physiological response of B. subtilismanifest in the proteome after antibiotic stress was previouslyshown to correlate with the antibacterial mechanism (2, 4, 47) andcontributed to elucidating the antibacterial mechanisms of novelantibiotic compounds (12, 48). Proteomic profiles of B. subtilistreated with several cell wall biosynthesis inhibitors have beenreported (4, 39, 46), but proteomic signatures indicative of spe-cific aspects of cell wall biosynthesis inhibition have not yet beendescribed. Utilizing lantibiotics, which range in mechanism fromjust binding lipid II to binding lipid II and forming large pores,and by further drawing on an extensive library of proteomic re-sponse profiles previously described (4), we identified responderproteins indicative of the different lantibiotic mechanisms. Todirectly correlate the proteome profiles with the influence of mer-sacidin, gallidermin, and nisin on B. subtilis, the impact of thelantibiotics on membrane integrity was studied in this model or-ganism. Lantibiotic interference with cell wall biosynthesis wasexamined microscopically, looking at cell shape after methanol-acetic acid fixation. Potassium efflux measurements served to in-vestigate pore formation. And finally, the influence of lantibioticson the membrane potential was studied microscopically with thehelp of a green fluorescent protein (GFP)-MinD fusion protein.MinD is a cell division protein requiring an intact membrane po-tential for localization at the cell poles and the cell division plane(41). Based on the correlation of these physiological experimentswith the proteomic response, marker proteins for different mech-anisms of membrane interference could be proposed.
MATERIALS AND METHODSAntibiotics. All antibiotic stock solutions were prepared at 10 mg/ml.Nisin was dissolved in 0.01 M HCl, gallidermin in double-distilled water(both were purified according to Bonelli et al. [8]), and mersacidin(Sanofi-Aventis, Paris, France) in methanol. Valinomycin and gramicidinA were dissolved in dimethyl sulfoxide (DMSO), and bacitracin was dis-solved in double-distilled water (all from Sigma-Aldrich, St. Louis, MO).Vancomycin (Applichem, Darmstadt, Germany) was dissolved in DMSO.Gramicidin S was synthetized and purified according to Wadhwani et al.(45) and dissolved in DMSO.
Bacterial strains and growth conditions. Bacillus subtilis 168 (trpC2)(1) was grown at 37°C under steady agitation in Belitzky minimal medium(BMM) (42). MICs were determined in a modified MIC test describedpreviously to match the growth conditions of the proteome experiment,specifically using chemically defined medium and supplying sufficientoxygen (48). Briefly, in a test tube, 2 ml of defined medium was inoculatedwith 5 � 105 bacteria per ml and incubated with different lantibioticconcentrations at 37°C under steady agitation for 18 h. The MIC wasdefined as the lowest concentration inhibiting visible growth. In growthexperiments, bacterial cultures were treated with different antibiotic con-centrations in mid-exponential phase after reaching an optical density at500 nm (OD500) of 0.35. For physiological stress experiments, includingproteomic studies, antibiotic concentrations were chosen that reducedgrowth rates to approximately 50 to 70% compared to that of the un-treated control culture.
Light microscopy. B. subtilis 168 cultures were grown in BMM to anOD500 of 0.35 and subsequently treated with 0.75 �g/ml nisin, 6 �g/mlgallidermin, 30 �g/ml mersacidin, 10 �g/ml valinomycin, 0.025�g/ml gramicidin A, 1.5 �g/ml vancomycin, 12 �g/ml bacitracin, or 1�g/ml gramicidin S. After 15 min of antibiotic exposure, 200 �l of bacte-rial culture were immediately fixed in 1 ml of a 1:3 mixture of acetic acidand methanol. Five microliters of fixed cells were immobilized in 5 �lBMM containing 0.5% low-melting agarose at 40°C. Cells were observedwith an Olympus BX51 microscope using a U-UCD8 condenser and aUPlanSApo 100XO objective. Pictures were taken using a CC12 digitalcolor camera and cell imaging software (all components by Olympus,Hamburg, Germany).
GFP-MinD localization. B. subtilis 1981 GFP-MinD (41) was grownovernight in BMM. Cells were then inoculated to an OD500 of 0.1 inmodified BMM containing xylose instead of glucose to induce expressionof the GFP-MinD fusion protein. After reaching an OD500 of 0.35, cellswere exposed to antibiotics at concentrations described above. After 15min of antibiotic stress, 5 �l of the bacterial culture was withdrawn andthe nonfixed, nonimmobilized samples were imaged immediately in fluo-rescent mode using the described equipment with a U-LH100HGAPOburner and a U-RFL-T power supply (Olympus, Hamburg, Germany).
Potassium release. Potassium efflux experiments were performed us-ing a microprocessor pH meter (pH 213; Hanna Instruments, Kehl, Ger-many) with an MI-442 potassium electrode and MI-409F reference elec-trode. The electrodes were preconditioned by immersing both thepotassium selective and the reference electrodes in choline buffer (300mM choline chloride, 30 mM MES, 20 mM Tris, pH 6.5) for at least 1 hbefore starting calibration or measurements. Calibration was carried outprior to each determination by immersing the electrodes in fresh standardsolutions containing 0.01, 0.1, or 1 mM KCl in choline buffer. B. subtilis168 was grown in BMM and harvested at an OD500 of 1.0 to 1.5 (3,300 �g, 4°C, 3 min), washed with 50 ml cold choline buffer, and resuspended inthe same buffer to an OD500 of 30. The concentrated cell suspension waskept on ice and used within 30 min. For each measurement, the cells werediluted in choline buffer (25°C) to an OD500 of about 3. Antibiotics wereapplied at the concentrations described above. Calculations of potassiumefflux were performed according to the equations established by Orlov etal. (31). Antibiotic-induced leakage was monitored for 3 min with read-ings taken every 10 s and expressed relative to the total potassium releaseinduced by nisin.
Proteome analysis. Protein extracts for 2D-PAGE were prepared asdescribed previously (48). In short, 5 ml of a bacterial culture in earlyexponential growth phase were exposed to 6 �g/ml gallidermin, 30 �g/mlmersacidin, 10 �g/ml valinomycin, or 1 �g/ml gramicidin S or left un-treated as the control. When B. subtilis cultures were treated during expo-nential growth, nisin exhibited a very narrow window of concentrationsthat solicit a stress response without causing massive cell lysis. Addressingthis problem, concentrations from 0.25 to 1.25 �g/ml of nisin were usedin parallel for each of the replicate labeling experiments. For the pro-teomic analysis, only those cultures were processed that showed growth
on October 31, 2012 by U
NIV
ER
SIT
AT
BO
CH
UM
http://aac.asm.org/
Dow
nloaded from
C Lantibiotic stress response
37
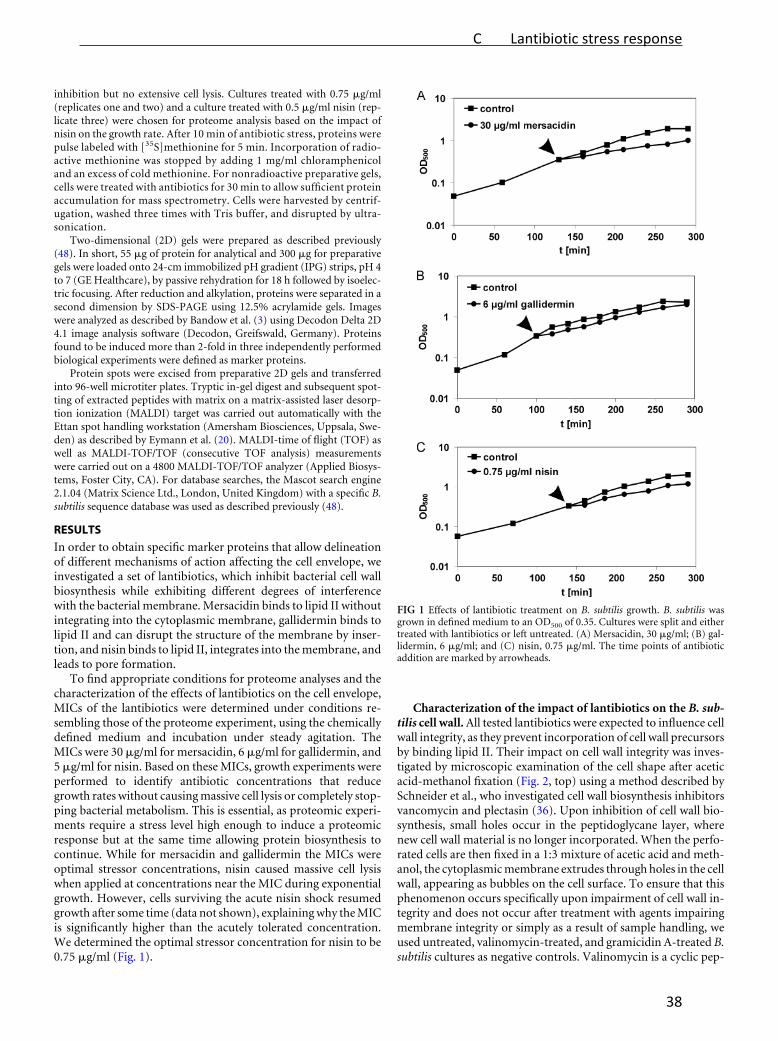
inhibition but no extensive cell lysis. Cultures treated with 0.75 �g/ml(replicates one and two) and a culture treated with 0.5 �g/ml nisin (rep-licate three) were chosen for proteome analysis based on the impact ofnisin on the growth rate. After 10 min of antibiotic stress, proteins werepulse labeled with [35S]methionine for 5 min. Incorporation of radio-active methionine was stopped by adding 1 mg/ml chloramphenicoland an excess of cold methionine. For nonradioactive preparative gels,cells were treated with antibiotics for 30 min to allow sufficient proteinaccumulation for mass spectrometry. Cells were harvested by centrif-ugation, washed three times with Tris buffer, and disrupted by ultra-sonication.
Two-dimensional (2D) gels were prepared as described previously(48). In short, 55 �g of protein for analytical and 300 �g for preparativegels were loaded onto 24-cm immobilized pH gradient (IPG) strips, pH 4to 7 (GE Healthcare), by passive rehydration for 18 h followed by isoelec-tric focusing. After reduction and alkylation, proteins were separated in asecond dimension by SDS-PAGE using 12.5% acrylamide gels. Imageswere analyzed as described by Bandow et al. (3) using Decodon Delta 2D4.1 image analysis software (Decodon, Greifswald, Germany). Proteinsfound to be induced more than 2-fold in three independently performedbiological experiments were defined as marker proteins.
Protein spots were excised from preparative 2D gels and transferredinto 96-well microtiter plates. Tryptic in-gel digest and subsequent spot-ting of extracted peptides with matrix on a matrix-assisted laser desorp-tion ionization (MALDI) target was carried out automatically with theEttan spot handling workstation (Amersham Biosciences, Uppsala, Swe-den) as described by Eymann et al. (20). MALDI-time of flight (TOF) aswell as MALDI-TOF/TOF (consecutive TOF analysis) measurementswere carried out on a 4800 MALDI-TOF/TOF analyzer (Applied Biosys-tems, Foster City, CA). For database searches, the Mascot search engine2.1.04 (Matrix Science Ltd., London, United Kingdom) with a specific B.subtilis sequence database was used as described previously (48).
RESULTS
In order to obtain specific marker proteins that allow delineationof different mechanisms of action affecting the cell envelope, weinvestigated a set of lantibiotics, which inhibit bacterial cell wallbiosynthesis while exhibiting different degrees of interferencewith the bacterial membrane. Mersacidin binds to lipid II withoutintegrating into the cytoplasmic membrane, gallidermin binds tolipid II and can disrupt the structure of the membrane by inser-tion, and nisin binds to lipid II, integrates into the membrane, andleads to pore formation.
To find appropriate conditions for proteome analyses and thecharacterization of the effects of lantibiotics on the cell envelope,MICs of the lantibiotics were determined under conditions re-sembling those of the proteome experiment, using the chemicallydefined medium and incubation under steady agitation. TheMICs were 30 �g/ml for mersacidin, 6 �g/ml for gallidermin, and5 �g/ml for nisin. Based on these MICs, growth experiments wereperformed to identify antibiotic concentrations that reducegrowth rates without causing massive cell lysis or completely stop-ping bacterial metabolism. This is essential, as proteomic experi-ments require a stress level high enough to induce a proteomicresponse but at the same time allowing protein biosynthesis tocontinue. While for mersacidin and gallidermin the MICs wereoptimal stressor concentrations, nisin caused massive cell lysiswhen applied at concentrations near the MIC during exponentialgrowth. However, cells surviving the acute nisin shock resumedgrowth after some time (data not shown), explaining why the MICis significantly higher than the acutely tolerated concentration.We determined the optimal stressor concentration for nisin to be0.75 �g/ml (Fig. 1).
Characterization of the impact of lantibiotics on the B. sub-tilis cell wall. All tested lantibiotics were expected to influence cellwall integrity, as they prevent incorporation of cell wall precursorsby binding lipid II. Their impact on cell wall integrity was inves-tigated by microscopic examination of the cell shape after aceticacid-methanol fixation (Fig. 2, top) using a method described bySchneider et al., who investigated cell wall biosynthesis inhibitorsvancomycin and plectasin (36). Upon inhibition of cell wall bio-synthesis, small holes occur in the peptidoglycane layer, wherenew cell wall material is no longer incorporated. When the perfo-rated cells are then fixed in a 1:3 mixture of acetic acid and meth-anol, the cytoplasmic membrane extrudes through holes in the cellwall, appearing as bubbles on the cell surface. To ensure that thisphenomenon occurs specifically upon impairment of cell wall in-tegrity and does not occur after treatment with agents impairingmembrane integrity or simply as a result of sample handling, weused untreated, valinomycin-treated, and gramicidin A-treated B.subtilis cultures as negative controls. Valinomycin is a cyclic pep-
FIG 1 Effects of lantibiotic treatment on B. subtilis growth. B. subtilis wasgrown in defined medium to an OD500 of 0.35. Cultures were split and eithertreated with lantibiotics or left untreated. (A) Mersacidin, 30 �g/ml; (B) gal-lidermin, 6 �g/ml; and (C) nisin, 0.75 �g/ml. The time points of antibioticaddition are marked by arrowheads.
on October 31, 2012 by U
NIV
ER
SIT
AT
BO
CH
UM
http://aac.asm.org/
Dow
nloaded from
C Lantibiotic stress response
38

tide carrier ionophore that selectively transports potassium ionsacross the bacterial membrane, while peptide channel ionophoregramicidin A transports monovalent cations (18). Neither the un-treated controls (Fig. 2A) nor the valinomycin or gramicidin A-treated cultures (Fig. 2B or C, respectively) displayed any mem-brane extrusions. As expected, all lantibiotics led to membraneextrusions (Fig. 2E, F, G), demonstrating their influence on cellwall biosynthesis in B. subtilis. Further, we tested the cyclic peptideantibiotic gramicidin S (Fig. 2D), which served as a positive con-trol for depolarization experiments. Gramicidin S had been sug-gested to affect membrane-bound processes indirectly by causingdelocalization of membrane-associated enzymes (25, 26, 44, 55).Indeed, after gramicidin S treatment, cells also showed membraneextrusions, corroborating an effect of gramicidin S on cell wallbiosynthesis.
Lantibiotics’ influence on B. subtilis membrane potential. Inaddition to cell wall biosynthesis inhibition, gallidermin and nisinare expected to influence membrane integrity due to integrationinto the membrane and pore formation, respectively. Therefore,we investigated the influence of lantibiotics on the membranepotential monitoring microscopically the localization of a GFP-MinD fusion protein (Fig. 2, central and bottom panels). MinD ispart of the cell division regulation machinery of B. subtilis. As longas the membrane potential remains intact, MinD is attached to themembrane and localizes at the cell poles and in the cell divisionplain. It was shown that MinD localization is altered by mem-brane-depolarizing agents like valinomycin or proton ionophorecarbonyl cyanide m-chlorophenylhydrazone (CCCP), due to theinterruption of electrostatic interactions between MinD and themembrane surface (41). This phenomenon was employed here toinvestigate depolarizing properties of antibiotic agents. As de-scribed by Strahl and Hamoen (41), valinomycin was used as apositive control, as it causes rapid depolarization (Fig. 2B). Dis-ruption of the membrane potential results in delocalization ofGFP-MinD, which appears in fluorescing spots irregularly distrib-
uted throughout the cells. Nisin and gallidermin (Fig. 2E and F,respectively) exhibited the same GFP-MinD distribution patternas valinomycin, indicating the collapse of the membrane potential(Fig. 2). In contrast, mersacidin did not disturb the polar andseptal localization of GFP-MinD (Fig. 2G). These findings suggestthat nisin and gallidermin but not mersacidin cause membranedepolarization in B. subtilis. Delocalization of GFP-MinD was alsocaused by the membrane-integrating antimicrobial peptide gram-icidin S (Fig. 2D), which is neither an ionophore nor a pore former(25, 44).
Lantibiotics’ ability to form pores in the B. subtilis mem-brane. In order to investigate the ability of mersacidin, gallider-min, and nisin to form pores in B. subtilis, we measured the lan-tibiotic-induced potassium release using a potassium-sensitiveelectrode (Fig. 3). Consistent with the literature, pore-forminglantibiotic nisin led to strong, immediate potassium release,
FIG 2 Influence of mersacidin, gallidermin, and nisin on B. subtilis cell wall integrity and membrane potential. (Top) Light microscopy images of B. subtilis fixedwith acetic acid-methanol and immobilized with low-melting agarose show the influence of lantibiotics and comparator compounds on cell wall integrity.(Center) Fluorescence microscopy images of nonfixed cells show GFP-MinD localization after treatment with envelope-targeting agents. Cultures of B. subtilis168 or B. subtilis 1981 GFP-MinD were grown until early exponential phase, divided into aliquots, and stressed with antibiotics for 15 min: (A) untreated control;(B) valinomycin (10 �g/ml); (C) gramicidin A (0.025 �g/ml); (D) gramicidin S (1 �g/ml); (E) nisin (0.75 �g/ml); (F) gallidermin (6 �g/ml); (G) mersacidin (30�g/ml); (H) bacitracin (12 �g/ml); (I) vancomycin (1.5 �g/ml). (Bottom) Corresponding light microscopy pictures of the B. subtilis GFP-MinD cells shownabove. Note that B. subtilis GFP-MinD mutants typically grow longer than wild-type B. subtilis due to MinD overexpression.
FIG 3 Potassium release from B. subtilis induced by mersacidin, gallidermin,nisin, and valinomycin. Potassium efflux from living cells was monitored witha potassium-sensitive electrode. Ion leakage is expressed relative to the totalamount of potassium released after addition of the pore-forming lantibioticnisin (100% value). Antibiotics were added at 50 s and applied at the sameconcentrations as for proteome analysis: 0.75 �g/ml nisin, 6 �g/ml gallider-min, 30 �g/ml mersacidin, and 10 �g/ml valinomycin. The symbol-free solidline represents the baseline without antibiotics.
on October 31, 2012 by U
NIV
ER
SIT
AT
BO
CH
UM
http://aac.asm.org/
Dow
nloaded from
C Lantibiotic stress response
39

whereas the potassium ionophore valinomycin (positive control)led to release of the same amount of potassium, following a slowerkinetic characteristic for carrier ionophores. In contrast, as de-scribed previously, mersacidin did not cause an increase in extra-cellular potassium levels (6, 9, 14, 23). Gallidermin, which had notyet been tested regarding its ability to form pores in B. subtilis,caused only a minor potassium release of up to 20% compared tonisin, suggesting that it does not effectively form pores in B. sub-tilis under the tested conditions.
Proteomic response to lantibiotic treatment. Comparativeproteome analyses were carried out to investigate whether or notthe proteomic response would reveal marker proteins for the lan-tibiotic mechanism of action of cell wall biosynthesis inhibition bylipid II binding and how far it could provide insights into addi-tional membrane-impairing properties of lantibiotics. To thisend, B. subtilis was grown to early exponential phase and stressedwith the appropriate concentrations of lantibiotics. [35S]-L-methi-onine pulse labeling was used to label selectively those proteinsnewly synthesized under antibiotic stress. This procedure allowsmonitoring of the acute bacterial stress response with exquisitesensitivity. The soluble intracellular protein fraction was then sep-arated by 2D-PAGE. Representative proteome expression profilesof mersacidin, gallidermin, and nisin are shown in Fig. 4. Proteinsinduced at least 2-fold are referred to as marker proteins. Theymirror the physiological stress conditions the cells are facing andproved valuable for studying antibiotic mechanisms of action.
Twenty-five (25) marker protein spots were induced by treat-ment with gallidermin, 13 by mersacidin, and 8 by nisin (Table 1).Using MALDI-TOF/TOF, we were able to identify 13, 7, and 6protein spots, respectively. The remaining protein spots wereclearly visible on autoradiographs reflecting protein synthesis af-ter antibiotic treatment but did not accumulate to any appreciableextent. Although these unidentified protein spots do not allowrational insights into the antibiotic mechanism, they still serve asantibiotic-specific markers, as they are reproducibly induced aftertreatment.
DISCUSSION
In this study, we set out to characterize the stress response of B.subtilis to three different lantibiotics with gradually differentmechanisms of action regarding interactions with the cytoplasmicmembrane but each inhibiting cell wall biosynthesis by binding toprecursor lipid II. It was previously described that mersacidin actssolely on cell wall biosynthesis, whereas gallidermin is able to in-tegrate into the membrane, although pores are formed only insome bacterial species and not others (8, 16). Nisin was shown tointegrate into the membrane upon lipid II binding, forming poresof 2 to 2.5 nm in diameter (50), if concentrations exceed a certainthreshold (8, 38). In preparation of the proteomic study, we testednisin, gallidermin, and mersacidin for their effects on the B. sub-tilis cell envelope under growth conditions matching those of theproteomic profiling analysis. As expected, all lantibiotics signifi-cantly affected cell wall integrity as shown microscopically (Fig. 2).Protein localization studies using GFP-labeled MinD showed pro-tein delocalization after treatment with nisin and gallidermin, in-dicating disruption of the membrane potential. Mersacidin testednegative for MinD-GFP delocalization. Significant potassium re-lease demonstrating pore formation was observed only for nisinbut not gallidermin or mersacidin (Fig. 2 and 3). Taken together,these results suggest that in B. subtilis, both nisin and gallidermin
FIG 4 Differential proteome analysis of B. subtilis 168 in response to mersaci-din (A), gallidermin (B), and nisin (C). 2D gel-based protein synthesis profilesof the controls false colored in green were overlaid with those of the antibiotic-treated samples false colored in red. In the overlays, downregulated proteinsappear green, upregulated proteins appear red, and proteins expressed at equalrates appear yellow. Unidentified proteins are labeled as follows: M, inducedby mersacidin; G, induced by gallidermin; N, induced by nisin; NGM, inducedby nisin, gallidermin, and mersacidin.
on October 31, 2012 by U
NIV
ER
SIT
AT
BO
CH
UM
http://aac.asm.org/
Dow
nloaded from
C Lantibiotic stress response
40

effectively depolarize the membrane, with only nisin formingpores. Our results indicate that the GFP-MinD delocalization ob-served for gallidermin may be due to membrane depolarizationcaused by the lantibiotic integrating into the membrane ratherthan to pore formation. As Christ et al. demonstrated, galliderminleads to effective pore formation in lipid bilayers containingmainly acyl chains up to 15 C atoms in length. Limited pore for-mation is observed in membranes composed predominantly of 17C lipid tails (16). Consistently, Bonelli et al. reported pore forma-tion for Staphylococcus simulans and Micrococcus flavus (both 15C) but not for Lactococcus lactis (17 C) (8). Additionally to themembrane thickness, a large amount of branched-chain fatty ac-ids in the membrane seems to negatively affect pore formation bygallidermin (16). The B. subtilis membrane consists mostly of 15 Clipid chains but at the same time possesses about 80% branched-chain fatty acids (17). Therefore, it is likely that gallidermin is notable to form pores in this organism due to membrane densityrather than thickness. However, integration of gallidermin intothe membrane might affect the physicochemical properties of themembrane, resulting in depolarization and consequently MinD-GFP delocalization. Similar effects have already been described forcationic antimicrobial peptide gramicidin S, a depolarizing agent.At sublytical concentrations, gramicidin S seems to disturb themembrane structure leading to dysfunction of membrane proteincomplexes. It uncouples electron transport by inhibiting cyto-chrome bd quinol oxidase and consequently disrupts the electro-chemical gradient (25, 30, 44, 55). Gramicidin S also affects cell
wall integrity (Fig. 2), potentially also due to disturbing the mem-brane-bound cell wall biosynthesis machinery by integrating intothe membrane. There are also reports that gramicidin S increasesmembrane permeability for potassium ions (26), which might bethe effect rather than the cause of membrane depolarization (30).
Based on their common mechanism of inhibiting cell wall bio-synthesis by binding lipid II, the three lantibiotics were ideallysuited for comparative proteomic studies aimed at describing pro-teomic signatures for cell wall biosynthesis inhibition. At the sametime, this collection of lantibiotics in combination with the exist-ing library of proteomic response profiles could be used to corre-late the differences in the proteome according to the differences inmembrane interaction. Many of the identified marker proteins areknown to be upregulated in response to cell envelope stress (Table1). YvlB, for instance, belongs to the SigW regulon induced by cellenvelope stress. Taking into account the lantibiotic proteome pro-files as well as previously published reference patterns of an exten-sive antibiotic proteome response library (4), we delineated threegroups of marker proteins (Table 2, Fig. 5): those indicative of cellwall biosynthesis inhibition, those indicating membrane stress,and those induced in response to general envelope stress.
Markers for interference with cell wall biosynthesis by lipidII binding. Four proteins, namely, LiaH, YtrE, and TrmB, as wellas unidentified protein NGM1, were induced by all tested lantibi-otics. LiaH belongs to the LiaRS regulon. It forms multimericringlike structures consisting of 36 identical subunits that protectcell envelope integrity probably by binding to the membrane upon
TABLE 1 Responder proteins induced by nisin, gallidermin, and mersacidin
Responderprotein
Induction factora
Protein name Function RegulatorNisin(25% inhibition)
Nisin(50% inhibition) Gallidermin Mersacidin
LiaH 6.6 19 86 92 Modulator of liaIHGFSR operonexpression
TrmB 3.4 2.8 9.1 8.6 tRNA [guanin-N(7)]methyltransferaseYtrE 1.6 2.1 21 14 Similar to ABC transporterNGM1 1.6 2.5 13 8.5 Not identifiedPspA 1.8 5.5 6.4 Phage shock protein A Protection against cell envelope
stressSigW
NadE 1.6 2.3 6.7 NAD synthetase Energy limitation-mediated stressresponse
SigB
YceC 13 4.2 Similar to tellurium resistance protein Stress response SigW/SigBYceH 5.8 2.4 Similar to toxic anion resistance protein Stress response SigW/SigBYtrB 14 9.8 Similar to ABC transporter Putative ABC transporter ATPase
subunitYtrA
GM1 7.9 8.3 Not identifiedGM2 8.6 6.7 Not identifiedSerA 1.2 2.6 D-3-Phosphoglycerate dehydrogenase Serine biosynthesis/oxidative
stress/NADH regenerationN1 1 13 Not identifiedDltA 4.2 D-Alanyl:D-alanine carrier protein ligase Modification of lipoteichoic acids SigXYvlBa 3.6 Unknown Stress response SigWYvlBb 7 Unknown Stress responsePbpC 27 Penicillin binding protein 3 Cell wall biosynthesisRpsB 4.1 Ribosomal protein S2 TranslationG1 7.2 Not identifiedG2 3 Not identifiedG3 3.9 Not identifiedG4 2.4 Not identifiedG5 3.3 Not identifiedG6 47 Not identifiedG7 5.8 Not identifiedG8 2.8 Not identifiedG9 11 Not identifiedYqiG 5 NADH-dependent flavin oxidoreductase Electron transportM1 7.3 Not identifiedM2 7.2 Not identifiedM3 3.2 Not identified
a Bold numbers indicate reliable marker proteins, more than 2-fold induced in three independent biological replicates.
on October 31, 2012 by U
NIV
ER
SIT
AT
BO
CH
UM
http://aac.asm.org/
Dow
nloaded from
C Lantibiotic stress response
41

cell wall stress (51). LiaRS has been shown before to be induced byvarious cell wall biosynthesis inhibitors, such as bacitracin, nisin,ramoplanin, and to a lesser extent vancomycin, measuring liaI-lacZ reporter gene activation (29). Further, the LiaRS system was
induced after severe secretion stress, suggesting that protein accu-mulation in the cell envelope may also trigger LiaH induction(24). Besides the three lantibiotics investigated here, two inhibi-tors targeting membrane-bound steps of cell wall precursor syn-thesis were previously analyzed using proteomic profiling. Vanco-mycin prevents transglycosylation by binding to the amino acidside chain of lipid II, while bacitracin prevents recycling of thebactoprenol carrier that transports cell wall precursor moleculesthrough the membrane (15). LiaH was upregulated in response toall lantibiotics, as well as bacitracin and gramicidin S, but notvancomycin. A possible explanation might be that the lipid IIbinding site of vancomycin is the amino acid chain of lipid II,which is remote from the membrane (15), while for all other com-pounds, the lipid II binding sites are in close proximity to themembrane.
YtrE and YtrB, which are upregulated by all investigated cellwall biosynthesis inhibitors with the exception of nisin, are en-coded in the ytrABCDEF operon and form two cytoplasmicATPase subunits of a putative ABC transporter (52). Earlier stud-ies by Yoshida et al. demonstrated impaired acetoin import inytr-deficient knockout mutants (53). However, it should be notedthat the YtrF substrate recognition subunit shares homology withboth antimicrobial peptide efflux systems and the FtsX permeaseinvolved in cell division. Delayed spore formation was observed inthe absence of FtsX (21) as well as in ytr-deficient mutants (53),possibly indicating similar functions of both proteins. Although itis yet unclear how YtrE/YtrB induction might be connected to theantibiotic mechanism, they serve as specific markers for cell wallbiosynthesis inhibition. YtrE is the only marker protein shared byall lantibiotics, vancomycin, and bacitracin. It is not induced byother antibiotics tested and is, therefore, the most reliable markerfor inhibition of a membrane-bound step of cell wall biosynthesis.
TrmB is a tRNA-modifying [guanine-N(7)-]-methyltrans-ferase involved in tRNA maturation (54). What role it might playin the response to lantibiotic treatment remains unclear. LikeLiaH, unidentified NGM1 is induced by all lantibiotics and baci-tracin but not vancomycin, demonstrating a different cell re-sponse to vancomycin.
Likewise, the response pattern induced by the lipopeptide fri-ulimicin B differs from that of cell wall biosynthesis-targeting an-tibiotics that bind proximal to the membrane. In contrast to thelipid II-binding lantibiotics tested here, friulimicin B binds to bac-
TABLE 2 Marker proteins for antibiotics targeting membrane integrity and/or cell wall biosynthesis correlated with influence on cell envelope integrityc
Compound Mode of actionCell wallintegrity
MinDdelocalization
Potassiumleakage NadE PspA YceC YceH TrmB LiaH YtrE YtrB NGM1
Valinomycinb Carrier ionophore � � � x x x xGramicidin Aa Channel ionophore � � � x x xGramicidin Sb Membrane integration � � � x x x x x xNisin Pore formation and cell wall
biosynthesis� � � x x x x x x
Gallidermin Membrane integration and cellwall biosynthesis
� � � x x x x x x x x x
Mersacidin Cell wall biosynthesis � � � x x x x x x xBacitracina Cell wall biosynthesis � � � x x x x xVancomycina Cell wall biosynthesis � � � x x xa Previously published (4).b 2D gel images not shown.c �, envelope functionality as measured in cell wall integrity assay, MinD delocalization assay, or potassium release assay was impaired; �, control-like envelope functionality; x,marker protein upregulated at least twofold in response to antibiotic treatment.
FIG 5 Details of autoradiographs of 2D gels depicting the specific markerproteins for cell wall biosynthesis, membrane stress, and general cell envelopestress. Induction factors are displayed in the lower right corner as the averageover three independent biological replicates.
on October 31, 2012 by U
NIV
ER
SIT
AT
BO
CH
UM
http://aac.asm.org/
Dow
nloaded from
C Lantibiotic stress response
42

toprenol monophosphate, thus inhibiting carrier recycling. As aresult, cell wall biosynthesis and biosynthesis of wall teichoic acidsand carbohydrate-containing capsular material are impaired, andcell envelope modification reactions such as glycosylations are in-hibited. All these pathways depend on bactoprenol-mediatedbuilding block shuffling to the outside (37). As expected for anantibiotic interfering with cell wall biosynthesis, friulimicin Bleads to membrane extrusions in the methanol-acetic acid assay. Itdoes not depolarize the bacterial membrane in the GFP-MinDassay (data not shown) and does not cause potassium leakage (37).Friulimicin B shares a high mode of action similarity with bacitra-cin, which binds to bactroprenol pyrophosphate. However, whilebacitracin induces all cell wall biosynthesis marker proteins, fri-ulimicin B induces only the unidentified marker proteins NGM1and GM1 but not LiaH or YtrE (data not shown). In a previousstudy, Wecke et al. further showed induction of YceC and YceH(46), which in the present study were upregulated in response toseveral of the cell envelope-targeting antibiotics but were not cor-related strictly with either interference with the membrane or withcell wall biosynthesis.
A clear distinction based on the proteome response can bemade between inhibition of membrane-bound steps of cell wallbiosynthesis and inhibition of either intracellular or extracellularsteps. This could be shown using D-cycloserine or methicillin, re-spectively. D-Cycloserine, which inhibits alanine racemase, andD-alanine:D-alanine ligase, two intracellular enzymes of the path-way (28, 43), elicit a completely different proteomic response withno marker proteins overlapping with any of the other investigatedcell wall biosynthesis inhibitors (4). Methicillin inhibits penicil-lin-binding protein 2 (PBP-2), a membrane-anchored transpep-tidase and transglycosylase (33); however, the methicillin bindingsite is not located near the membrane anchor, and a direct inter-action of methicillin with the cytoplasmic membrane has not beenreported. For this compound in B. subtilis, no acute stress re-sponse at all was observed on the proteome level (4), suggestingthat B. subtilis cells do either not sense the consequences of meth-icillin action and/or are unable to react to them. Based on theseresults, differences in the proteomic response to cell wall biosyn-thesis inhibition relating to the localization of the steps inhibitedand potentially even the proximity of the antibiotic binding sitesto the bacterial membrane can be observed.
Marker proteins for interference with membrane integrity.PspA and NadE are both induced by nisin and gallidermin as wellas gramicidin A, gramicidin S, and valinomycin. Among all agentstested so far, only compounds acting on the bacterial membraneled to induction of these two proteins, supporting that they arespecific marker proteins for membrane stress.
Interestingly, upregulation of PspA and NadE was observedonly at higher nisin concentrations, leading to approximately 50%growth inhibition, while the cell wall biosynthesis-specific mark-ers LiaH and TrmB were upregulated already under moderatestress conditions, causing approximately 25% growth inhibition(Table 1). This is in line with a two-staged mechanism of action ofnisin, which is characterized by lipid II complexation at lowerconcentrations and pore formation above a threshold concentra-tion (9, 23). PspA, like LiaH, forms homomultimeric ringlikestructures (22). Although Standar et al. showed that those 36-mersare able to form large scaffolds (40), more recent studies demon-strated localization of PspA at midcell and at the cell poles by useof a GFP fusion (19). It was shown that PspA is able to bind to
membrane phospholipids and prevent proton leakage. It isthought to counteract external membrane damage and to stabilizethe membrane during membrane transport (27). Structural ho-mology between PspA and LiaH suggests similar functions in sta-bilizing the cell envelope by binding to the membrane surface.Our results indicate that PspA is responsive to membrane damage,while LiaH is induced by lipid II-mediated cell wall stress. How-ever, the different triggers inducing both stress proteins remain tobe identified (51).
NAD synthase NadE, which catalyzes the final step in NADsynthesis, is regulated on the transcriptional level by the house-keeping sigma factor SigA and by the alternative sigma factor SigBcoordinating the general stress response in B. subtilis in responseto environmental stress and energy limitation. As a consequenceof antibiotic-related impairment of membrane integrity evi-denced by the disturbed membrane potential, energy limitation isa possible explanation for NadE upregulation.
Differentiation between pore formation by nisin and mem-brane integration by gallidermin could not be securely establishedbased on specific marker proteins. This may be due to rapid killingof cells once nisin concentrations reach the necessary thresholdfor pore formation, hardly leaving a chance for a coordinatedstress response. For this reason, pore formation might be difficultto distinguish from other forms of membrane stress by proteomeanalysis alone. However, monitoring concentration-dependentkilling kinetics already allows differentiation between compoundsthat integrate into the membrane and those that form pores.Rapid bacteriolytic pore formers leave only a very narrow concen-tration window for proteomic profiling experiments and shouldinduce the membrane stress-specific marker proteins PspA andNadE at the appropriate antibiotic concentrations.
Marker proteins for envelope stress. Putative tellurium resis-tance protein YceC, induced by gallidermin and mersacidin, wasalso induced by several of the envelope-targeting compounds inour proteomic response library, namely, valinomycin, gramicidinS, bacitracin, and vancomycin, and can therefore serve as a generalmarker for envelope stress. Putative anion resistance proteinYceH was also upregulated in response to gallidermin, mersacidin,valinomycin, gramicidin S, and gramicidin A. YceC and YceH areunder the dual control of SigW and SigB, but to our knowledgetheir functions are yet unknown.
In conclusion, using lantibiotics and a number of other antibi-otics with different targets related to the cellular envelope, wedescribed marker proteins correlating with cell wall biosynthesisinhibition, membrane stress, and general cell envelope stress. Theestablished marker proteins can now be used to characterize newantibiotic agents, particularly lantibiotics, with regard to their an-tibacterial mechanisms.
ACKNOWLEDGMENTS
Mersacidin was provided by Sanofi-Aventis (formerly Hoechst AG). Wegratefully acknowledge Jia Yii Airina Lee from Pennsylvania State Univer-sity as well as Christoph H. R. Senges (Ruhr University Bochum) forassistance with the bubble and MinD-GFP assays. We are also grateful forexcellent support by the technical staff at Rubion, Ruhr University Bo-chum, and to Michaele Josten, University of Bonn, for purifying nisin.
This work was financially supported by “Innovative Antibiotics fromNRW,” a grant from the state of North Rhine-Westphalia (NRW), Ger-many, and the European Union, European Regional Development Fund,“Investing in Your Future,” to J.E.B. and H.G.S.
on October 31, 2012 by U
NIV
ER
SIT
AT
BO
CH
UM
http://aac.asm.org/
Dow
nloaded from
C Lantibiotic stress response
43

REFERENCES1. Anagnostopoulos C, Spizizen J. 1961. Requirements for transformation
in Bacillus subtilis. J. Bacteriol. 81:741–746.2. Bandow JE. 2005. Proteomic approaches to antibiotic drug discovery.
Curr. Protoc. Microbiol. 1:1F.2.3. Bandow JE, et al. 2008. Improved image analysis workflow for 2D gels
enables large-scale 2D gel-based proteomics studies—COPD biomarkerdiscovery study. Proteomics 8:3030 –3041.
4. Bandow JE, Brötz H, Leichert LI, Labischinski H, Hecker M. 2003.Proteomic approach to understanding antibiotic action. Antimicrob.Agents Chemother. 47:948 –955.
5. Bandow JE, Metzler-Nolte N. 2009. New ways of killing the beast: pros-pects for inorganic-organic hybrid nanomaterials as antibacterial agents.Chembiochem 10:2847–2850.
6. Barrett MS, Wenzel RP, Jones RN. 1992. In vitro activity of mersacidin(M87-1551), an investigational peptide antibiotic tested against Gram-positive bloodstream isolates. Diagn. Microbiol. Infect. Dis. 15:641– 644.
7. Bierbaum G, Sahl HG. 2009. Lantibiotics: mode of action, biosynthesisand bioengineering. Curr. Pharm. Biotechnol. 10:2–18.
8. Bonelli RR, Schneider T, Sahl HG, Wiedemann I. 2006. Insights into invivo activities of lantibiotics from gallidermin and epidermin mode-of-action studies. Antimicrob. Agents Chemother. 50:1449 –1457.
9. Breukink E, et al. 2003. Lipid II is an intrinsic component of the pore inducedby nisin in bacterial membranes. J. Biol. Chem. 278:19898–19903.
10. Brötz-Osterhelt H, Sass P. 2010. Postgenomic strategies in antibacterialdrug discovery. Future Microbiol. 5:1553–1579.
11. Brötz-Osterhelt H, Brunner NA. 2008. How many modes of actionshould an antibiotic have? Curr. Opin. Pharmacol. 8:564 –573.
12. Brötz-Oesterhelt H, et al. 2005. Dysregulation of bacterial proteolyticmachinery by a new class of antibiotics. Nat. Med. 11:1082–1087.
13. Brötz H, Sahl HG. 2000. New insights into the mechanism of action oflantibiotics— diverse biological effects by binding to the same moleculartarget. J. Antimicrob. Chemother. 46:1– 6.
14. Brötz H, Bierbaum G, Leopold K, Reynolds PE, Sahl HG. 1998. Thelantibiotic mersacidin inhibits peptidoglycan synthesis by targeting lipidII. Antimicrob. Agents Chemother. 42:154 –160.
15. Bugg TDH, Braddick D, Dowson CG, Roper DI. 2011. Bacterial cell wallassembly: still an attractive antibacterial target. Trends Biotechnol. 29:167–173.
16. Christ K, et al. 2008. Membrane lipids determine the antibiotic activity ofthe lantibiotic gallidermin. J. Membr. Biol. 226:9 –16.
17. Clejan S, Krulwich TA, Mondrus KR, Seto-Young D. 1986. Membranelipid composition of obligately and fakultatively alkalophilic strains ofBacillus spp. J. Bacteriol. 168:334 –340.
18. Duax WL, et al. 1996. Molecular structure and mechanisms of action ofcyclic and linear ion transport antibiotics. Biopolymers 40:141–155.
19. Engl C, et al. 2009. In vivo localizations of membrane stress controllersPspA and PspG in Escherichia coli. Mol. Microbiol. 73:382–396.
20. Eymann C, et al. 2004. A comprehensive proteome map of growingBacillus subtilis cells. Proteomics 4:2849 –2876.
21. Garti-Levi S, Hazan R, Kain J, Fujita M, Ben-Yehuda S. 2008. The FtsEXABC transporter directs cellular differentiation in Bacillus subtilis. Mol.Microbiol. 69:1018 –1028.
22. Hankamer BD, Elderkin SL, Buck M, Nield J. 2004. Organization of theAAA� adaptor protein PspA is an oligomeric ring. J. Biol. Chem. 279:8862– 8866.
23. Hasper HE, de Kruijff B, Breukink E. 2004. Assembly and stability ofnisin-lipid II pores. Biochemistry 43:11567–11575.
24. Hyyryläinen HL, Sarvay M, Kontinen VP. 2005. Transcriptome analysisof the secretion stress response of Bacillus subtilis. Appl. Microbiol. Bio-technol. 67:389 –396.
25. Kaprel’iants AS, et al. 1977. Membranes of bacteria and mechanism ofaction of the antibiotic gramicidin S. Biokhimiia. 42:329 –337.
26. Katsu T, et al. 1989. Mechanism of membrane damage induced by theamphiphilic peptides gramicidin S and melittin. Biochim. Biophys. Acta983:135–141.
27. Kobayashi R, Suzuki T, Yoshida M. 2007. Escherichia coli phage-shockprotein A (PspA) binds to membrane phospholipids and repairs protonleakage of the damaged membranes. Mol. Microbiol. 66:100 –109.
28. Lambert MP, Neuhaus FC. 1972. Mechanism of D-cycloserine action:alanine racemase from Escherichia coli W. J. Bacteriol. 109:1156 –1161.
29. Mascher T, Zimmer SL, Smith TA, Helmann JD. 2004. Antibiotic-
inducible promotor regulated by the cell envelope stress-sensing two-component system LiaRS of Bacillus subtilis. Antimicrob. Agents Che-mother. 48:2888 –2896.
30. Mogi T, Ui H, Shiomi K, Omura S, Kita K. 2008. Gramicidin S identifiedas a potent inhibitor for cytochrome bd-type quinol oxidase. FEBS Lett.582:2299 –2302.
31. Orlov DS, Nguyen T, Lehrer RI. 2002. Potassium release, a useful tool forstudying antimicrobial peptides. J. Microbiol. Methods 49:325–328.
32. Peschel A, Sahl HG. 2006. The co-evolution of host cationic antimicro-bial peptides and microbial resistance. Nat. Rev. Microbiol. 4:529 –536.
33. Pinho MG, de Lencastre H, Tomasz A. 2001. An acquired and a nativepenicillin-binding protein cooperate in building the cell wall of drug-resistant staphylococci. Proc. Natl. Acad. Sci. U. S. A. 98:10886 –10891.
34. Rabilloud T, Strub JM, Luche S, van Dorsselaer A, Lunardi J. 2001. Acomparison between SyproRuby and ruthenium II Tris (bathophenanth-roline disulfonate) as fluorescent stains for protein detection in gels. Pro-teomics 1:699 –704.
35. Reisinger P, Seidel H, Tschesche H, Hammes WP. 1980. The effect ofnisin on murein synthesis. Arch. Microbiol. 127:187–193.
36. Schneider T, et al. 2010. Plectasin, a fungal defensin, targets the bacterialcell wall precursor lipid II. Science 328:1168 –1172.
37. Schneider T, et al. 2009. The lipopeptide antibiotic friulimicin B inhibitscell wall biosynthesis through complex formation with bactoprenol phos-phate. Antimicrob. Agents Chemother. 53:1610 –1618.
38. Schneider T, Sahl HG. 2010. An oldie but a goodie— cell wall biosynthe-sis as antibiotic target pathway. Int. J. Med. Microbiol. 300:161–169.
39. Singh VK, Jayaswal RK, Wilkinson BJ. 2001. Cell wall-active antibioticinduced proteins of Staphylococcus aureus identified using a proteomicapproach. FEMS Microbiol. Lett. 199:79 – 84.
40. Standar K, et al. 2008. PspA forms large scaffolds in Escherichia coli. FEBSLett. 582:3585–3589.
41. Strahl H, Hamoen LW. 2010. Membrane potential is important for bac-terial cell division. Proc. Natl. Acad. Sci. U. S. A. 27:12281–12286.
42. Stülke J, Hanschke R, Hecker M. 1993. Temporal activation of betaglu-canase synthesis in Bacillus subtilis is mediated by the GTP pool. J. Gen.Microbiol. 139:2041–2045.
43. Vicario PP, Green BG, Katzen HM. 1987. A single assay for simultane-ously testing effectors of alanine racemase and/or D-alanine:D-alanine li-gase. J. Antibiot. (Tokyo) 40:209 –216.
44. Vostroknutova GN, Bulgakova VG, Udalova TP, Sepetov NF, Sibel’dinaLA. 1981. Localization of gramicidin S on the cytoplasmic membrane ofBacillus brevis and its effect on the activity of membrane enzymes. Biokh-imiia 46:657– 666.
45. Wadhwani P, Afonin S, Ieronimo M, Buerck J, Ulrich AS. 2006. Opti-mized protocol for synthesis of cyclic gramicidin S: starting amino acid iskey to high yield. J. Org. Chem. 71:55– 61.
46. Wecke T, et al. 2009. Daptomycin versus friulimicn B: in-depth profilingof Bacillus subtilis cell envelope stress responses. Antimicrob. Agents Che-mother. 53:1619 –1623.
47. Wenzel M, Bandow JE. 2011. Proteomic signatures in antibiotic research.Proteomics 11:3256 –3268.
48. Wenzel M, et al. 2011. Proteomic signature of fatty acid biosynthesisinhibition available for in vivo mechanism-of-action studies. Antimicrob.Agents Chemother. 55:2590 –2596.
49. WHO. 2012. The evolving threat of antimicrobial resistance—options foraction. World Health Organization, Geneva, Switzerland. http://whqlibdoc.who.int/publications/2012/9789241503181_eng.pdf.
50. Wiedemann I, Benz R, Sahl HG. 2004. Lipid II-mediated pore formationby the peptide antibiotic nisin—a black lipid membrane study. J. Bacte-riol. 186:3259 –3261.
51. Wolf D, et al. 2010. In-depth profiling of LiaR response in Bacillus subtilis.J. Bacteriol. 192:4680 – 4693.
52. Xiao Z, Xu P. 2007. Acetoin metabolism in bacteria. Crit. Rev. Microbiol.33:127–140.
53. Yoshida KI, Fujita Y, Ehrlich SD. 2000. An operon for a putative ATP-binding cassette transport system involved in acetoin utilization of Bacil-lus subtilis. J. Bacteriol. 182:5454 –5461.
54. Zegers I, et al. 2006. Crystal structure of Bacillus subtilis TrmB, the tRNA(m7G46) methyltransferase. Nucleic Acids Res. 34:1925–1934.
55. Zhang L, Dhillon P, Yan H, Farmer S, Hancock REW. 2000. Interactionsof bacterial cationic peptide antibiotics with outer and cytoplasmic mem-branes of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 44:3317–3321.
on October 31, 2012 by U
NIV
ER
SIT
AT
BO
CH
UM
http://aac.asm.org/
Dow
nloaded from
C Lantibiotic stress response
44

D
Modulating the activity of short arginine‐
tryptophan containing antibacterial peptides with
N‐terminal metallocenoyl groups
H. Bauke Albada, Alina Iulia Chiriac, Michaela Wenzel, Maya Penkova,
Julia E. Bandow, Hans‐Georg Sahl, Nils Metzler‐Nolte
Beilstein Journal of Organic Chemistry, 2012
D Modulating peptide activity with metallocenoyl groups
45

1753
Modulating the activity of short arginine-tryptophancontaining antibacterial peptides with N-terminal
metallocenoyl groupsH. Bauke Albada1, Alina-Iulia Chiriac2, Michaela Wenzel3, Maya Penkova1,
Julia E. Bandow3, Hans-Georg Sahl2 and Nils Metzler-Nolte*1
Full Research Paper Open Access
Address:1Inorganic Chemistry I – Bioinorganic Chemistry, Faculty of Chemistryand Biochemistry, Ruhr University Bochum, Universitätsstraße 150,44801 Bochum, Germany, 2Institute for Medical Microbiology,Immunology, and Parasitology, Pharmaceutical Microbiology Section,University of Bonn, Meckenheimer Allee 168, 53115 Bonn, Germanyand 3Microbial Biology, Faculty of Biology and Biotechnology, RuhrUniversity Bochum, Universitätsstraße 150, 44801 Bochum, Germany
Email:Nils Metzler-Nolte* - [email protected].
* Corresponding author
Keywords:antimicrobial peptides; arginine; medicinal organometallic chemistry;metallocenoyl; peptides; tryptophan
Beilstein J. Org. Chem. 2012, 8, 1753–1764.doi:10.3762/bjoc.8.200
Received: 14 July 2012Accepted: 06 September 2012Published: 15 October 2012
This article is part of the Thematic Series "Antibiotic and cytotoxicpeptides".
Guest Editor: N. Sewald
© 2012 Albada et al; licensee Beilstein-Institut.License and terms: see end of document.
AbstractA series of small synthetic arginine and tryptophan containing peptides was prepared and analyzed for their antibacterial activity.
The effect of N-terminal substitution with metallocenoyl groups such as ferrocene (FcCO) and ruthenocene (RcCO) was investi-
gated. Antibacterial activity in different media, growth inhibition, and killing kinetics of the most active peptides were determined.
The toxicity of selected derivatives was determined against erythrocytes and three human cancer cell lines. It was shown that the
replacement of an N-terminal arginine residue with a metallocenoyl moiety modulates the activity of WRWRW-peptides against
Gram-positive and Gram-negative bacteria. MIC values of 2–6 µM for RcCO-W(RW)2 and 1–11 µM for (RW)3 were determined.
Interestingly, W(RW)2-peptides derivatized with ferrocene were significantly less active than those derivatized with ruthenocene
which have similar structural but different electronic properties, suggesting a major influence of the latter. The high activities
observed for the RcCO-W(RW)2- and (RW)3-peptides led to an investigation of the origin of activity of these peptides using several
important activity-related parameters. Firstly, killing kinetics of the RcCO-W(RW)2-peptide versus killing kinetics of the (RW)3
derivative showed faster reduction of the colony forming units for the RcCO-W(RW)2-peptide, although MIC values indicated
higher activity for the (RW)3-peptide. This was confirmed by growth inhibition studies. Secondly, hemolysis studies revealed that
both peptides did not lead to significant destruction of erythrocytes, even up to 500 µg/mL for (RW)3 and 250 µg/mL for RcCO-
D Modulating peptide activity with metallocenoyl groups
46

Beilstein J. Org. Chem. 2012, 8, 1753–1764.
1754
W(RW)2. In addition, toxicity against three human cancer cell lines (HepG2, HT29, MCF7) showed that the (RW)3-peptide had an
IC50 value of ~140 µM and the RcW(RW)2 one of ~90 µM, indicating a potentially interesting therapeutic window. Both the killing
kinetics and growth inhibition studies presented in this work point to a membrane-based mode of action for these two peptides, each
having different kinetic parameters.
Beilstein J. Org. Chem. 2012, 8, 1753–1764.
1754
IntroductionNew antibacterial agents need to be discovered since estab-
lished antibiotics are increasingly losing ground against resis-
tant bacteria and at the same time the pipeline that is supposed
to produce new antibiotics is running dry [1]. For example, the
number of methicillin-resistant Staphylococcus aureus (MRSA)
infections in hospitals are still very high and new infectious
agents like Acinetobacter baumannii are on the rise, both
leading to increased numbers of mortality. In view of this, the
discovery of host-defense and antimicrobial peptides with
bacteria-specific membrane targeting modes of action (MOA)
to which resistance cannot easily develop has led to high expec-
tations in the treatment of bacterial infections [2-4]. Whereas
host-defense peptides are found in many multicellular organ-
isms as part of their innate immune system, the name “anti-
microbial peptides” (abbreviated as AMPs) defines a larger
group of peptides that also encompasses synthetic peptides, and
peptidomimetics, for example. Among these, synthetic peptide-
based antimicrobial agents are especially interesting because
isolation and/or synthesis of traditional organic molecules is
often time-consuming and costly [3,5]. In fact, already in World
War II, peptides belonging to a certain group of antimicrobial
peptides, i.e., the gramicidins, found application in the treat-
ment of gunshot wounds [6]. Unfortunately, their general
toxicity prevented widespread systemic administration in the
clinic. However, during the past couple of decades a large
number of peptides with very potent antimicrobial activity and
lower general toxicity were discovered [7].
Inspired by these antimicrobial peptides, many synthetic deriva-
tives of naturally occurring antimicrobial peptides have been
studied [5]. In addition, chemical syntheses of a large number of
peptides that do not have natural counterparts have furnished
promising synthetic antimicrobial peptides (synAMPs) [8]. For
example, peptide-dendrimers [9-13], lipidated short peptides
[14], trivalent lipidated short peptides with antifungal activity
[15], peptoids [16], peptides containing D-amino acids [17], and
foldamers based on β-amino acid residues with antibacterial
activity [18] have been described. Whereas nature has to stick
to products compatible with biosynthetic pathways, the syn-
thetic chemist is free to apply all available compounds and tech-
niques, thereby introducing even the most exotic molecular
entities. The most recent and exotic additions are conjugates of
metallocenes with short synthetic antimicrobial peptides [19-22]
and organometallic derivatives of platensimycin [23-28].
Among the synAMPs known to date, those based on arginine
(Arg or R) and tryptophan (Trp or W) residues are amongst the
smallest peptides that still possess significant antibacterial
activity. For example, Strøm et al. [29] described short
RW-based synAMPs with different N- and C-terminal
substituents, which showed low micromolar antibacterial
activity against various strains of Gram-positive bacteria and
moderate activity against Gram-negative bacteria. Interestingly,
head-to-tail cyclized RW-based synAMPs with clustered func-
tionalities increased the activity against Gram-negative
Escherichia coli much more than against Gram-positive
Bacillus subtilis [30,31], and only slightly increased the
hemolytic activity [32]. Moreover, the alkylation of tryptophan
residues by tert-butyl groups resulted in increased activity and
low hemolytic activity of the constructs [33]. Our group has
previously shown that the covalent attachment of metal
complexes to RW-based synAMPs yields more active deriva-
tives with a changed activity profile for Gram-positive and
Gram-negative bacteria. In this work, the attachment of the
neutral ferrocenoyl group (ferrocene: dicyclopentadienyl iron,
Cp2Fe; ferrocenoyl: FcCO) was beneficial over the presence of
the monocationic cobaltocenium (Cc+CO) fragment [20].
To gain a better understanding of the origin of the activity of
these RW-based synAMPs and of the effect exerted on it by a
metallocene moiety, a set of peptides was synthesized and
tested for antimicrobial activity. In this paper we add another
metal to the spectrum of existing organometallic synAMPs and
we provide a detailed assessment of the kinetic parameters of
this peptide. Specifically, we describe the effect of the introduc-
tion of ruthenocenoyl (ruthenocene: dicyclopentadienyl ruthe-
nium, Cp2Ru; ruthenocenoyl: RcCO), an organometallic moiety
that is almost isostructural to ferrocenoyl (FcCO) but has
different electro- and physicochemical properties [34]. For
example, the more extended d-orbitals of Rc form stronger
hydrogen bonds with OH or NH groups than Fc [35]. The
activities of the synAMPs (MIC values) were compared to those
of GS(K2Y2) (Y = D-tyrosine), a gramicidin S analogue, and
vancomycin, one of the last lines of defense against Staphylo-
coccus infections. From the antibacterial activity screening, the
two most active peptides were selected for further analysis, i.e.,
H-Arg-Trp-Arg-Trp-Arg-Trp-NH2 (referred to as (RW)3), and
RcCO-Trp-Arg-Trp-Arg-Trp-NH2 (referred to as RcCO-
W(RW)2). For these peptides, toxicity against three human
D Modulating peptide activity with metallocenoyl groups
47

Beilstein J. Org. Chem. 2012, 8, 1753–1764.
1755
Figure 1: Structures of the most active peptides that have been used in this study. The top row shows two representative structures of the Arg-Trpbased peptides (left) and their metallocene-derivatives (right); the lower row shows the structure of pore-forming gramicidin S derivative GS(K2Y2)(left) and lipid II-binding cell wall biosynthesis inhibitor vancomycin (right).
cancer cell lines was assessed, followed by determination of
their killing kinetics and growth inhibition potential. Please note
that underlined one-letter codes of the amino acid residues
represent D-amino acids, not underlined one-letter codes of the
residues are L-amino acids.
ResultsSynthesis of the synAMPsAll peptides described in this study were prepared according to
established or recently published procedures [36]. In short,
Fmoc-protected amino acids were coupled in a solid-state syn-
thesis scheme using HOBt, TBTU, and DiPEA under
microwave irradiation. Using suitably protected amino acids,
i.e., Fmoc-Arg(Pbf)-OH, Fmoc-Trp(Boc)-OH, and Fmoc-
Lys(Boc)-OH, and polystyrene-based resin decorated with
Fmoc-protected Rink linkers, a set of peptides were prepared
(Table 1 and Figure 1). These peptides were obtained after
acidic cleavage of the resin-bound protected precursors, puri-
fied by preparative HPLC, and the fractions containing the
desired compound in high purity were lyophilized from the
prep-HPLC buffers. All these peptides were obtained in high
yields and close to 100% HPLC purity.
Table 1: Overview of the studied sequences and analysis thereof(retention times and m/z values). Underlined amino acids are D-enan-tiomers, not underlined residues are L-enantiomers; FcCO refers toferrocenoyl and RcCO to ruthenocenoyl (Figure 1).
entry sequence tR(min)
m/z found(calcd for [M + H]+)
1 H-RWRWRW-NH2 17.2 1044.25 (1044.58)2 H-RWRWRW-NH2 17.2 1044.27 (1044.58)3 RcCO-WRWRW-NH2 20.1 1146.27 (1146.44)4 RcCO-WRWRW-NH2 20.1 1146.11 (1146.44)5 FcCO-WRWRW-NH2 20.2 1100.36 (1100.47)6 Ac-RWRWRW-NH2 17.6 1086.45 (1086.59)7 Ac-RWRWRW-NH2 17.6 1086.37 (1086.59)8 FcCO-RWRWRW-NH2 19.0 1256.48 (1256.57)9 FcCO-RWRWRW-NH2 19.0 1256.46 (1256.59)10 H-KWKWKW-NH2 16.7 959.43 (959.55)11 vancomycina 11.7 1448.56 (1448.44)12 GS(K2Y2)b 25.0 1201.46 (1201.73)
aVancomycin was obtained from Sigma-Aldrich Fluka and purified bypreparative HPLC using a C18-reversered phase column; bGS(K2Y2) =cyclo([Pro-Val-Lys-Leu-D-Tyr]2) was prepared according to [37].
D Modulating peptide activity with metallocenoyl groups
48
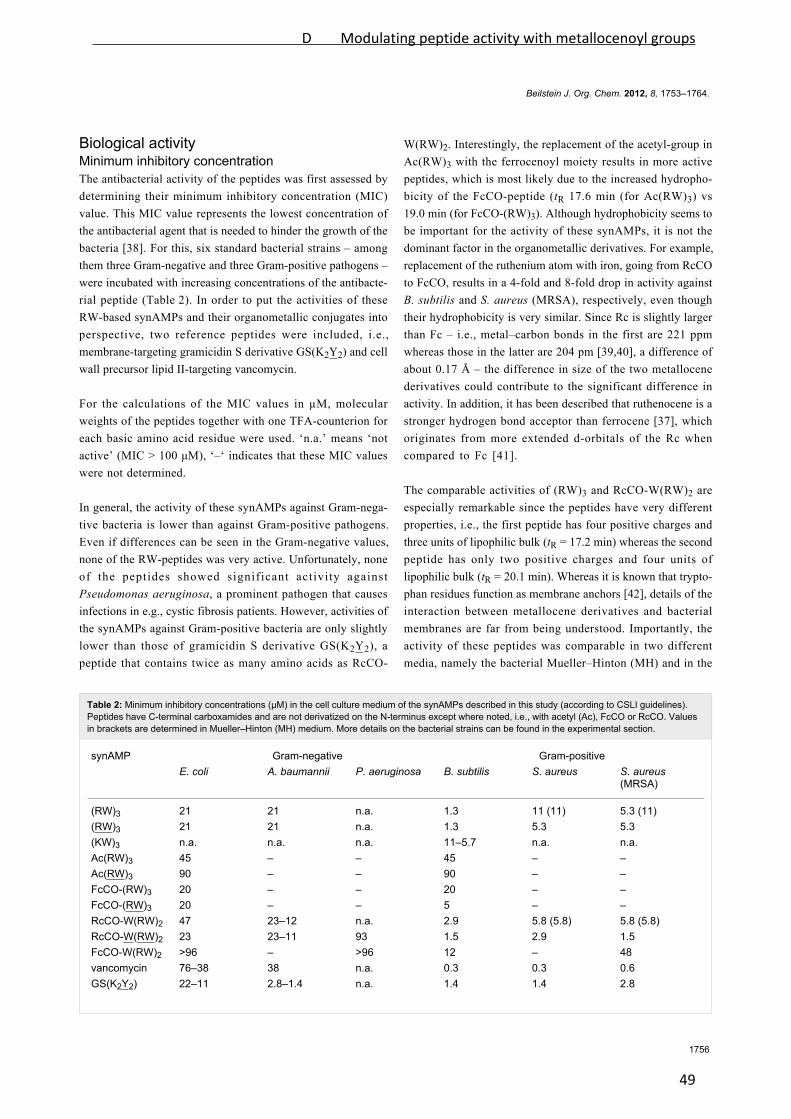
Beilstein J. Org. Chem. 2012, 8, 1753–1764.
1756
Table 2: Minimum inhibitory concentrations (µM) in the cell culture medium of the synAMPs described in this study (according to CSLI guidelines).Peptides have C-terminal carboxamides and are not derivatized on the N-terminus except where noted, i.e., with acetyl (Ac), FcCO or RcCO. Valuesin brackets are determined in Mueller–Hinton (MH) medium. More details on the bacterial strains can be found in the experimental section.
synAMP Gram-negative Gram-positiveE. coli A. baumannii P. aeruginosa B. subtilis S. aureus S. aureus
(MRSA)
(RW)3 21 21 n.a. 1.3 11 (11) 5.3 (11)(RW)3 21 21 n.a. 1.3 5.3 5.3(KW)3 n.a. n.a. n.a. 11–5.7 n.a. n.a.Ac(RW)3 45 – – 45 – –Ac(RW)3 90 – – 90 – –FcCO-(RW)3 20 – – 20 – –FcCO-(RW)3 20 – – 5 – –RcCO-W(RW)2 47 23–12 n.a. 2.9 5.8 (5.8) 5.8 (5.8)RcCO-W(RW)2 23 23–11 93 1.5 2.9 1.5FcCO-W(RW)2 >96 – >96 12 – 48vancomycin 76–38 38 n.a. 0.3 0.3 0.6GS(K2Y2) 22–11 2.8–1.4 n.a. 1.4 1.4 2.8
Biological activityMinimum inhibitory concentrationThe antibacterial activity of the peptides was first assessed by
determining their minimum inhibitory concentration (MIC)
value. This MIC value represents the lowest concentration of
the antibacterial agent that is needed to hinder the growth of the
bacteria [38]. For this, six standard bacterial strains – among
them three Gram-negative and three Gram-positive pathogens –
were incubated with increasing concentrations of the antibacte-
rial peptide (Table 2). In order to put the activities of these
RW-based synAMPs and their organometallic conjugates into
perspective, two reference peptides were included, i.e.,
membrane-targeting gramicidin S derivative GS(K2Y2) and cell
wall precursor lipid II-targeting vancomycin.
For the calculations of the MIC values in µM, molecular
weights of the peptides together with one TFA-counterion for
each basic amino acid residue were used. ‘n.a.’ means ‘not
active’ (MIC > 100 μM), ‘–‘ indicates that these MIC values
were not determined.
In general, the activity of these synAMPs against Gram-nega-
tive bacteria is lower than against Gram-positive pathogens.
Even if differences can be seen in the Gram-negative values,
none of the RW-peptides was very active. Unfortunately, none
of the peptides showed significant activity against
Pseudomonas aeruginosa, a prominent pathogen that causes
infections in e.g., cystic fibrosis patients. However, activities of
the synAMPs against Gram-positive bacteria are only slightly
lower than those of gramicidin S derivative GS(K2Y2), a
peptide that contains twice as many amino acids as RcCO-
W(RW)2. Interestingly, the replacement of the acetyl-group in
Ac(RW)3 with the ferrocenoyl moiety results in more active
peptides, which is most likely due to the increased hydropho-
bicity of the FcCO-peptide (tR 17.6 min (for Ac(RW)3) vs
19.0 min (for FcCO-(RW)3). Although hydrophobicity seems to
be important for the activity of these synAMPs, it is not the
dominant factor in the organometallic derivatives. For example,
replacement of the ruthenium atom with iron, going from RcCO
to FcCO, results in a 4-fold and 8-fold drop in activity against
B. subtilis and S. aureus (MRSA), respectively, even though
their hydrophobicity is very similar. Since Rc is slightly larger
than Fc – i.e., metal–carbon bonds in the first are 221 ppm
whereas those in the latter are 204 pm [39,40], a difference of
about 0.17 Å – the difference in size of the two metallocene
derivatives could contribute to the significant difference in
activity. In addition, it has been described that ruthenocene is a
stronger hydrogen bond acceptor than ferrocene [37], which
originates from more extended d-orbitals of the Rc when
compared to Fc [41].
The comparable activities of (RW)3 and RcCO-W(RW)2 are
especially remarkable since the peptides have very different
properties, i.e., the first peptide has four positive charges and
three units of lipophilic bulk (tR = 17.2 min) whereas the second
peptide has only two positive charges and four units of
lipophilic bulk (tR = 20.1 min). Whereas it is known that trypto-
phan residues function as membrane anchors [42], details of the
interaction between metallocene derivatives and bacterial
membranes are far from being understood. Importantly, the
activity of these peptides was comparable in two different
media, namely the bacterial Mueller–Hinton (MH) and in the
D Modulating peptide activity with metallocenoyl groups
49

Beilstein J. Org. Chem. 2012, 8, 1753–1764.
1757
Table 3: Detailed assessment of the MIC values (in µg/mL) of both L- and D-versions of the (RW)3 and RcCO-W(RW)2 synAMPs against severalGram-positive bacterial strains. More details on the bacterial strains can be found in the experimental section.
strain (RW)3 (RW)3 RcCO-W(RW)2 RcCO-W(RW)2
S. aureus (133) 2.1 ± 0.7 2.1 ± 0.7 3.3 ± 1.4 6.7 ± 2.9S. simulans (22) 5.0 ± 0.0 3.3 ± 1.4 5.0 ± 0.0 6.7 ± 2.9S. aureus (SG511) 6.7 ± 2.9 4.2 ± 1.4 4.2 ± 1.4 8.3 ± 2.9B. subtilis 3.5 ± 1.8 3.8 ± 1.8 3.8 ± 1.8 3.8 ± 1.8B. megaterium 0.8 ± 0.7 0.5 ± 0.2 1.9 ± 0.9 2.5 ± 0.0M. luteus 0.6 ± 0.0 0.6 ± 0.0 0.9 ± 0.4 1.9 ± 0.9
richer cell culture medium. Interestingly, replacement of the
arginine residues with lysine residues resulted in an almost
completely inactive (KW)3 peptide [43]. Although the center of
the positive charge in both residues is found at five atoms from
the backbone, the different structures of the functional groups
and the hydrophobicity of the side chain seem to cause a signifi-
cant difference in activity [44,45].
From the initial screening, four peptides were selected for
further biological characterization. In view of the mentioned
differences in structure, including the addition of a novel metal
core in one of them, but rather similar activity the (RW)3 and
RcCO-W(RW)2 peptides were chosen. Since both L- and
D-amino acid versions of the (RW)3 and RcCO-W(RW)2
peptides are comparable in activity and represent the most
promising synAMPs (Table 2), we next determined the MIC
values of these four peptides against other Gram-positive
bacteria in order to expand the panel of test strains (Table 3).
As can be inferred from this table, the L- and D-amino acid
versions of these two synAMPs show comparable activity
although small differences can be observed. For example,
RcCO-W(RW)2 is almost twice as active against S. aureus
(SG511) as the D-amino acid isomer RcCO-W(RW)2, a differ-
ence that can be seen as a tendency against all but one of the
bacterial strains. Since the biological world is chiral, it is not
surprising to see some small differences between both chiral
forms of the synAMPs. Similar differences in the interaction
with chiral molecules and biological membranes have been
described before [46-48], although only in a few examples and
not without exceptions [49]. An opposite trend is seen for the
(RW)3-peptides, where the D-peptides show higher MIC values
than the L-peptides. These values could have been corroborated,
however, by preferential proteolytic degradation of the
N-terminally unprotected (RW)3-peptides [50].
In order to obtain more information on the antibacterial prop-
erties of these four peptides, we performed killing kinetics and
growth inhibition studies. Finally, we assessed the selectivity of
the RcCO-W(RW)2 and (RW)3 peptides towards bacteria by
determining their activity against human red blood cells and
several human cancer cell lines.
Killing kineticsKilling kinetics experiments show the rate at which bacteria are
killed over time and indicate whether an antibacterial agent has
a bacteriostatic or bactericidal activity. The killing kinetics of
the L-amino acid containing (RW)3- and RcCO-W(RW)2-
peptides were determined against S. aureus and B. megaterium
(Figure 2).
For this, peptides were added in various concentrations to bacte-
rial cultures at the optical densities of 0.1 at OD600. Aliquots of
the mixture were taken at given time points, plated in duplicate
on MH agar and incubated at 37 °C. Then, the number of
colony forming units (CFU) was counted (see Experimental
section for details).
The addition of the (RW)3 peptide to the bacterial culture
resulted in a strong inhibition and in an immediate reduction of
CFUs by a factor of 103 after 1 min, for both S. aureus and
B. megaterium. Similarly, treatment with RcCO-W(RW)2 also
decreased the number of CFUs and has shown increased
potency since only one dose at the MIC value was needed to
decrease the CFUs by 2–3 log units, whereas 5 × MIC of (RW)3
was required for a similar drop of CFUs. The immediate drop in
CFUs highlights the bactericidal nature of these synAMPs and
typically occurs with membrane acting compounds [51,52].
Growth inhibitionWhereas the killing kinetics studies determine the number of
viable cells as a function of time and thereby classify a com-
pound as bacteriostatic or bactericidal, monitoring the optical
density of a treated culture may give hints as to the lytic activity
of a compound.
In this work, the growth inhibition of Bacillus megaterium was
determined under the influence of the same four peptides used
D Modulating peptide activity with metallocenoyl groups
50
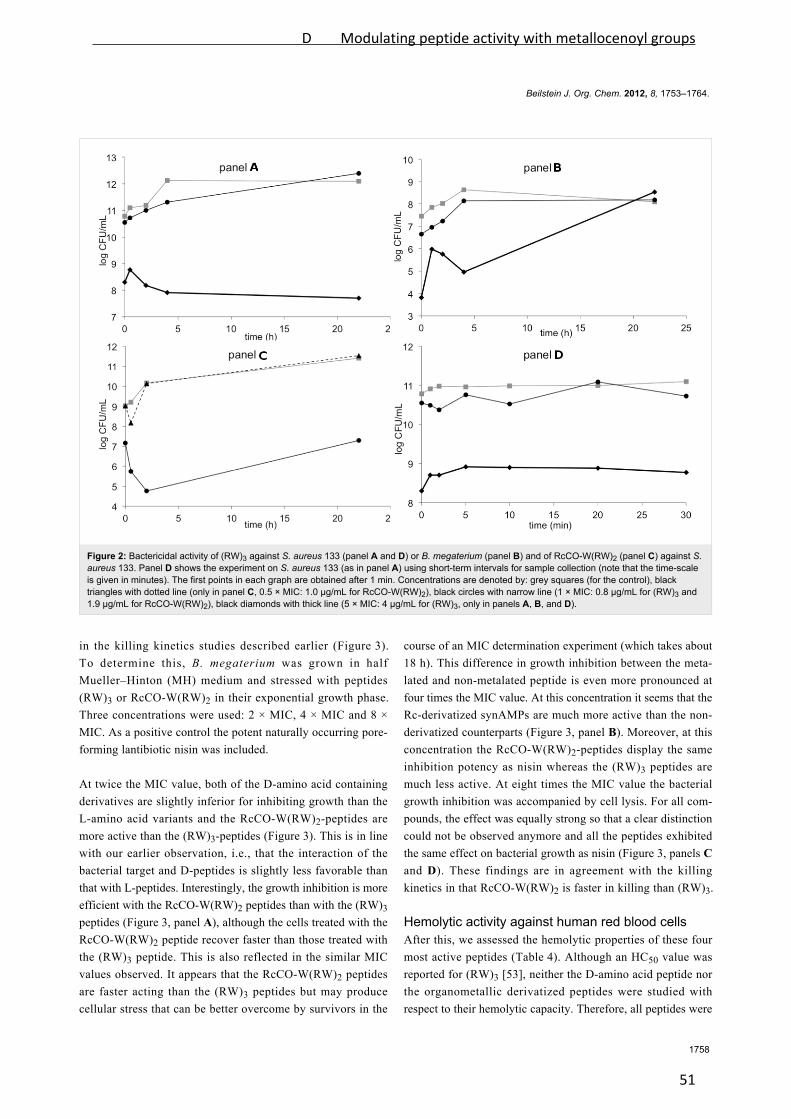
Beilstein J. Org. Chem. 2012, 8, 1753–1764.
1758
Figure 2: Bactericidal activity of (RW)3 against S. aureus 133 (panel A and D) or B. megaterium (panel B) and of RcCO-W(RW)2 (panel C) against S.aureus 133. Panel D shows the experiment on S. aureus 133 (as in panel A) using short-term intervals for sample collection (note that the time-scaleis given in minutes). The first points in each graph are obtained after 1 min. Concentrations are denoted by: grey squares (for the control), blacktriangles with dotted line (only in panel C, 0.5 × MIC: 1.0 µg/mL for RcCO-W(RW)2), black circles with narrow line (1 × MIC: 0.8 µg/mL for (RW)3 and1.9 µg/mL for RcCO-W(RW)2), black diamonds with thick line (5 × MIC: 4 µg/mL for (RW)3, only in panels A, B, and D).
in the killing kinetics studies described earlier (Figure 3).
To determine this, B. megaterium was grown in half
Mueller–Hinton (MH) medium and stressed with peptides
(RW)3 or RcCO-W(RW)2 in their exponential growth phase.
Three concentrations were used: 2 × MIC, 4 × MIC and 8 ×
MIC. As a positive control the potent naturally occurring pore-
forming lantibiotic nisin was included.
At twice the MIC value, both of the D-amino acid containing
derivatives are slightly inferior for inhibiting growth than the
L-amino acid variants and the RcCO-W(RW)2-peptides are
more active than the (RW)3-peptides (Figure 3). This is in line
with our earlier observation, i.e., that the interaction of the
bacterial target and D-peptides is slightly less favorable than
that with L-peptides. Interestingly, the growth inhibition is more
efficient with the RcCO-W(RW)2 peptides than with the (RW)3
peptides (Figure 3, panel A), although the cells treated with the
RcCO-W(RW)2 peptide recover faster than those treated with
the (RW)3 peptide. This is also reflected in the similar MIC
values observed. It appears that the RcCO-W(RW)2 peptides
are faster acting than the (RW)3 peptides but may produce
cellular stress that can be better overcome by survivors in the
course of an MIC determination experiment (which takes about
18 h). This difference in growth inhibition between the meta-
lated and non-metalated peptide is even more pronounced at
four times the MIC value. At this concentration it seems that the
Rc-derivatized synAMPs are much more active than the non-
derivatized counterparts (Figure 3, panel B). Moreover, at this
concentration the RcCO-W(RW)2-peptides display the same
inhibition potency as nisin whereas the (RW)3 peptides are
much less active. At eight times the MIC value the bacterial
growth inhibition was accompanied by cell lysis. For all com-
pounds, the effect was equally strong so that a clear distinction
could not be observed anymore and all the peptides exhibited
the same effect on bacterial growth as nisin (Figure 3, panels C
and D). These findings are in agreement with the killing
kinetics in that RcCO-W(RW)2 is faster in killing than (RW)3.
Hemolytic activity against human red blood cellsAfter this, we assessed the hemolytic properties of these four
most active peptides (Table 4). Although an HC50 value was
reported for (RW)3 [53], neither the D-amino acid peptide nor
the organometallic derivatized peptides were studied with
respect to their hemolytic capacity. Therefore, all peptides were
D Modulating peptide activity with metallocenoyl groups
51
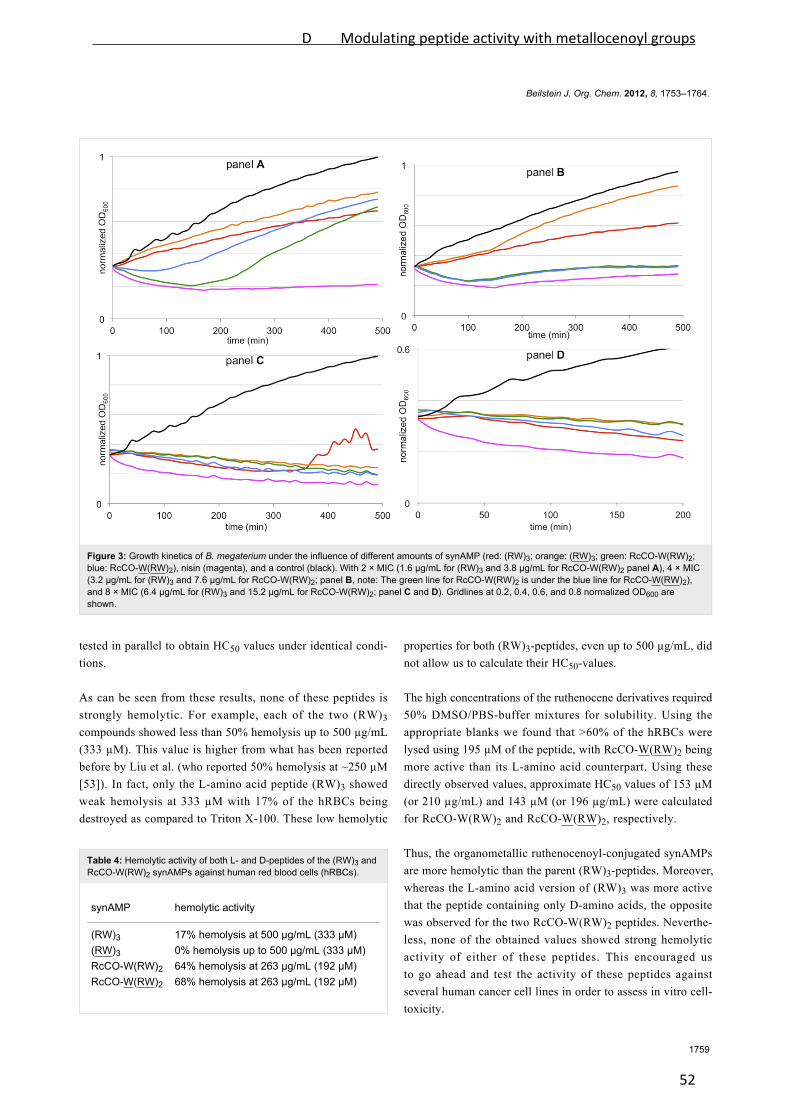
Beilstein J. Org. Chem. 2012, 8, 1753–1764.
1759
Figure 3: Growth kinetics of B. megaterium under the influence of different amounts of synAMP (red: (RW)3; orange: (RW)3; green: RcCO-W(RW)2;blue: RcCO-W(RW)2), nisin (magenta), and a control (black). With 2 × MIC (1.6 µg/mL for (RW)3 and 3.8 µg/mL for RcCO-W(RW)2 panel A), 4 × MIC(3.2 µg/mL for (RW)3 and 7.6 µg/mL for RcCO-W(RW)2; panel B, note: The green line for RcCO-W(RW)2 is under the blue line for RcCO-W(RW)2),and 8 × MIC (6.4 µg/mL for (RW)3 and 15.2 µg/mL for RcCO-W(RW)2; panel C and D). Gridlines at 0.2, 0.4, 0.6, and 0.8 normalized OD600 areshown.
Table 4: Hemolytic activity of both L- and D-peptides of the (RW)3 andRcCO-W(RW)2 synAMPs against human red blood cells (hRBCs).
synAMP hemolytic activity
(RW)3 17% hemolysis at 500 µg/mL (333 µM)(RW)3 0% hemolysis up to 500 µg/mL (333 µM)RcCO-W(RW)2 64% hemolysis at 263 µg/mL (192 µM)RcCO-W(RW)2 68% hemolysis at 263 µg/mL (192 µM)
tested in parallel to obtain HC50 values under identical condi-
tions.
As can be seen from these results, none of these peptides is
strongly hemolytic. For example, each of the two (RW)3
compounds showed less than 50% hemolysis up to 500 µg/mL
(333 µM). This value is higher from what has been reported
before by Liu et al. (who reported 50% hemolysis at ~250 µM
[53]). In fact, only the L-amino acid peptide (RW)3 showed
weak hemolysis at 333 µM with 17% of the hRBCs being
destroyed as compared to Triton X-100. These low hemolytic
properties for both (RW)3-peptides, even up to 500 µg/mL, did
not allow us to calculate their HC50-values.
The high concentrations of the ruthenocene derivatives required
50% DMSO/PBS-buffer mixtures for solubility. Using the
appropriate blanks we found that >60% of the hRBCs were
lysed using 195 µM of the peptide, with RcCO-W(RW)2 being
more active than its L-amino acid counterpart. Using these
directly observed values, approximate HC50 values of 153 µM
(or 210 µg/mL) and 143 µM (or 196 µg/mL) were calculated
for RcCO-W(RW)2 and RcCO-W(RW)2, respectively.
Thus, the organometallic ruthenocenoyl-conjugated synAMPs
are more hemolytic than the parent (RW)3-peptides. Moreover,
whereas the L-amino acid version of (RW)3 was more active
that the peptide containing only D-amino acids, the opposite
was observed for the two RcCO-W(RW)2 peptides. Neverthe-
less, none of the obtained values showed strong hemolytic
activity of either of these peptides. This encouraged us
to go ahead and test the activity of these peptides against
several human cancer cell lines in order to assess in vitro cell-
toxicity.
D Modulating peptide activity with metallocenoyl groups
52
Beilstein J. Org. Chem. 2012, 8, 1753–1764.
1760
Toxicity against human cancer cell linesFinally, to determine whether the peptides are selective for
bacterial cells, we tested the toxicity against mammalian cells
using three malignant cell lines: human liver carcinoma
(HepG2), human colon adenocarcinoma grade II (HT29) and
human breast adenocarcinoma (MCF7) cell lines (Table 5).
Table 5: IC50 values (in µM) of both (RW)3 and RcCO-W(RW)2 againsthuman liver carcinoma (HepG2), human colon cancer (HT29) andhuman breast cancer (MCF7) cell lines.
synAMP HepG2 HT29 MCF7
(RW)3 143 ± 21 132 ± 12 159 ± 7RcCO-W(RW)2 92 ± 5 94 ± 6 90 ± 1
In general, we consider a peptide with an IC50 value higher than
100 µM to be inactive. As can be seen from Table 5, the
peptides with the highest activity against Gram-positive bacteria
are not toxic against the three selected human cancer cell lines.
Based on average values from these cell lines, i.e., 142 μM for
(RW)3 and 92 μM for RcCO-W(RW)2, a potential therapeutic
window of about 7 and 4 can be calculated for Gram-negative
pathogens using (RW)3 and RcCO-W(RW)2, respectively.
Concerning the threatening Gram-positive S. aureus strains an
even better window of >13 is calculated for (RW)3 and RcCO-
W(RW)2. Interestingly, again the ruthenocenoyl-derivatized
synAMP is more active than the (RW)3 model peptide, as was
seen in both the antibacterial and hemolysis studies.
DiscussionRuthenium is one of the most promising metals in anticancer
drug candidates [54-57], with two Ru-compounds even in clin-
ical trials [58-62]. Surprisingly however, its potential in antibac-
terial research has not been explored so far. In this paper, we
present the effects of the attachment of the organometallic
ruthenocene (Rc) moiety to RW-based synthetic antimicrobial
peptides (synAMPs). A comparison of the MIC values from a
first screening of peptides that were N-terminally derivatized
with a ruthenocenoyl (RcCO) group with that of the ferro-
cenoyl (FcCO)-derivatized peptides showed superior properties
of the Rc-conjugated synAMPs (Table 2). Although both metal-
locenes have very similar hydrophobic properties, as confirmed
herein again by their almost identical retention times on a C18-
column during HPLC-analysis, they have slightly different
dimensions and very different physicochemical and electro-
chemical properties. Firstly, ferrocene derivatives have redox
potentials that are within the realm of biological systems, but
ruthenocene derivatives do not [41]. Secondly, while most
ferrocene derivatives exhibit a reversible one-electron oxi-
dation, ruthenocene and its derivatives typically undergo irre-
versible two-electron redox chemistry. Whether this difference
in redox chemistry of the two metallocenes could interplay with
the piezoelectric properties of phospholipid membranes [63]
remains to be determined.
Ruthenocene is known to have more extended d-orbitals and is
a stronger hydrogen-bond acceptor than ferrocene. In addition,
ruthenocene is slightly larger than ferrocene, which might result
in a possibly more disruptive interaction with bacterial
membranes.
Small differences between the L- and D-amino acid versions of
the peptides could be observed in their MIC values (Table 3)
and within growth inhibition studies (Figure 3). Examples for
this difference were found in other systems (see above) and
points to a delicate contribution of the chirality of the peptides
used. This effect was not observed in the first MIC values deter-
mined (Table 2), which indicates that this is a very subtle factor.
Indeed, the quantification requires a more sophisticated
analysis. This can be done using sensitive biophysical model
systems like those used in quartz crystal microbalance (QCM)
studies. This information can then be used to further optimize
an active synAMP.
Fortunately, while retaining antibacterial activity in cell culture
medium, the cellular toxicity of both the (RW)3 and the RcCO-
W(RW)2 peptides is low, and only high peptide concentrations
cause significant hemolysis. Apparently, these peptides have a
strong preference for prokaryotic membranes over eukaryotic
membranes, e.g., erythrocytes (Table 4 and Table 5). Neverthe-
less, it remains to be seen to what extent these short synAMPs
can be used in vivo.
Concerning the antibacterial effect of these peptides, the killing
kinetics showed rapid bactericidal properties of both the (RW)3
and RcCO-W(RW)2 peptides, and the growth kinetics showed
growth arrest and also indicated bacteriolytic properties.
Naturally occurring AMPs such as nisin and magainin typically
have >20 amino acids and often have a specific target, like
nisin, or are long enough to penetrate the membrane, like maga-
inin. For these long peptides descriptions of their action mode
with the “carpet-model”, the “toroidal pore model” or the
“barrel stave model” [64] are quite suitable. Considering the
rapid upon-contact killing and bacteriolytic properties, it
appears that the small synAMPs studied herein interact with the
bacterial membrane as well. The monomers of these peptides
are, however, too short to penetrate a bacterial membrane in
order to form pores, and therefore, probably act slightly
different from the more or less well-established mechanisms for
longer AMPs. We are currently undertaking efforts to uncover
more details on the mode of action. Specifically, proteomic
D Modulating peptide activity with metallocenoyl groups
53

Beilstein J. Org. Chem. 2012, 8, 1753–1764.
1761
analysis of the changes in the bacterial proteome as result of
exposure to these synAMPs, and prokaryotic and eukaryotic
membrane model systems will be used to precisely determine if
it is simply a membrane-based mechanism or if there are more
factors. While we attempt to elucidate the mode of action of
these synAMPs, we are also interested in a detailed under-
standing of the effects of the organometallic fragment on the
activity – for example by determining the contributions of
hydrogen-bond forming processes in membrane environments –
and the effect of the redox potential on the activity. We assume
that the application of model systems will help us to determine
the extent in which differences in chirality of the amino acids
used to construct the peptides result in more or less favorable
interactions.
ConclusionWe have shown that the replacement of the N-terminal arginine
residue in non-toxic and non-hemolytic (RW)3 peptides can
modulate the kinetics of the peptide’s antibacterial activity.
Acetylation completely suppresses this activity. In comparison,
replacement of the N-terminal arginine residue with the
organometallic ferrocenoyl moiety reduces the activity only
5- to 10-fold, whereas the replacement with ruthenocene
completely restores the level of activity. In summary, the data
supports a metal-specific activity-enhancing effect of the added
organometallic moiety. This effect is most likely due to the
added lipophilic bulk together with intricate contributions from
the electro- and physicochemical properties of the
organometallic fragment. None of these peptides is hemolytic
and both are hardly toxic against human cancer cell lines.
Thereby, they represent an interesting group of synthetic anti-
microbial peptides to be used in a therapeutic setting. Analysis
of the antibacterial properties of these peptides showed that they
are rapidly bactericidal and also bacteriolytic. Even though both
peptides have similar MIC values, RcCO-W(RW)2 is acting
faster than (RW)3, but is losing activity after 100–200 min,
which is significantly faster compared to (RW)3. Future studies
on these peptides are directed towards a better understanding of
their mode of action and attempts are being made for the
improvement of their activity to increase the therapeutic
window of these compounds.
ExperimentalMinimal inhibitory concentration (results areshown in Table 2)The minimal inhibitory concentrations (MIC) were tested
against Escherichia coli DSM 30083, Acinetobacter baumannii
DSM 30007, Pseudomonas aeruginosa DSM 50071, Bacillus
subtilis DSM 402, Staphylococcus aureus DSM 20231 (type
strain), and Staphylococcus aureus ATCC 43300 (MRSA) in a
microtiter plate assay according to CLSI guidelines [65].
E. coli, A. baumannii, S. aureus, and B. subtilis were grown in
Mueller–Hinton (MH) broth, P. aeruginosa in cation adjusted
Mueller–Hinton II. Peptides were dissolved in DMSO to give
10 mg/mL stock solutions. Serial dilution in culture media was
carried out automatically with the Tecan Freedom Evo 75 liquid
handling workstation (Tecan, Männedorf, Switzerland) from
512 to 0.5 µg/mL. Peptide dilutions were inoculated with
105 bacteria/mL taken from late exponential cultures grown in
the same media in a total volume of 200 µL per well. Cells were
incubated for 16 h at 37 °C. The lowest peptide concentration
inhibiting visible bacterial growth was taken as MIC.
For MIC determination in cell culture broth, the peptides were
diluted manually in DMEM high glucose (with 4.5 g/L glucose,
no penicillin). Only S. aureus DSM 20231 and ATCC 43300
were capable of growing in cell culture broth and were used for
MIC determination. Cells were grown in DMEM until late
exponential phase before using them for inoculation. Peptide
concentrations, inoculation and incubation were performed as
described above.
Minimum inhibitory concentration (results areshown in Table 3)Determination of MIC values was performed in 96-well
polypropylene microtiter plates (Life Technologies) in order to
reduce the AMP binding [66]. A series of 2-fold dilutions of the
peptides was prepared directly in the plate in half-concentrated
MH broth. The tested strains were grown to an optical density
(600 nm) of 0.5 in half-concentrated MH broth and diluted
1:105 using the same medium. Then, 100 µL of this suspension
was mixed with 100 µL of the peptide solution already prepared
in the wells of the microtiter plate as mentioned earlier. After
incubation for 18 h at 37 °C, the MIC value was read as the
lowest concentration of antimicrobial agent that resulted in
complete inhibition of visible growth. The results given are
mean values of three or more independent determinations.
Killing kineticsThe cells were grown in half-concentrated MH broth up to an
optical density of 0.5 and diluted in fresh medium to an optical
density of 0.1. Peptides were added in concentrations corres-
ponding to 0.5 to 5 × MIC. The viable count was monitored up
to 18 h. Aliquots were taken at defined time intervals, diluted in
10 mM potassium phosphate buffer, and 100 µL of several
decimal dilutions were plated in duplicate on MH agar. The
plates were incubated at 37 °C and the plates containing 30–300
colony forming units (CFU) were counted after 24 h.
Kinetic growth inhibitionGrowth kinetic experiments were performed in microtiter plates
using 200 µL half concentrated MH broth. The cells were
D Modulating peptide activity with metallocenoyl groups
54

Beilstein J. Org. Chem. 2012, 8, 1753–1764.
1762
grown to an optical density of up to 0.5 and diluted in fresh
medium to an optical density of 0.25. After this, peptides were
added in concentrations corresponding to 2 × MIC, 4 × MIC,
and 8 × MIC and the optical density was registered for 8 h using
a multichannel absorbance plate reader (SunriseTM, Tecan).
Hemolysis and in vitro cell toxicity studiesAfter drawing whole blood into anticoagulant containing tubes
(BD Vacutainer®, K2 EDTA 3.6 mg, Ref 368841, Lot
1248213), its fractionation was executed with one volume
whole blood added to nine volumes sterile 0.9% NaCl and
centrifugation (800g, 10 min, 4 °C). Subsequently, the lowest
fraction containing all hRBCs was washed twice with nine
volumes 1 × PBS (PAA), triturating carefully. The concen-
trated hRBCs were re-suspended with 1 × PBS to an erythro-
cyte concentration of 5% (v/v). Wells of a 96-well plate were
filled with 100 µL of the appropriate peptide solutions: The
peptides were dissolved in 1 × PBS and DMSO (5% for (RW)3
and 50% for RcCO-W(RW)2). These were mixed with 100 µL
of the 5% hRBCs solution and incubated under agitation on a
flat shaker (170 rpm, 30 min, 37 °C). After sedimenting all
probes under centrifugation (800g, 10 min, 4 °C), all super-
natants were transferred into a clean 96-well plate. The release
of hemoglobin was monitored by measuring the absorbance of
the supernatant at 550 nm using an automated 96-well plate
reader. Controls for 0 and 100% hemolysis consisted of hRBC
5% (v/v) suspended in PBS containing DMSO in appropriate
concentrations and 1% Triton X-100, respectively. Toxicity on
human cancer cell lines was determined according to previ-
ously described procedures [67,68].
AcknowledgementsWe thank Annegret Knüfer for determining the IC50 values of
the RcW(RW)2 and (RW)3 peptides. We also thank Vera
Eßmann for preparing GS10(K2Y2), and Hung Bahn and Damla
Yaprak for determining the MIC values of Ac(RW)3 and
Fc(RW)3.
References1. Wenzel, M.; Bandow, J. E. Proteomics 2011, 11, 3256–3268.
doi:10.1002/pmic.2011000462. Hancock, R. E. W.; Lehrer, R. Trends Biotechnol. 1998, 16, 82–88.
doi:10.1016/S0167-7799(97)01156-63. Hancock, R. E. W.; Chapple, D. S. Antimicrob. Agents Chemother.
1999, 43, 1317–1323.4. Hancock, R. E. W.; Sahl, H.-G. Nat. Biotechnol. 2006, 24, 1551–1557.
doi:10.1038/nbt12675. Findlay, B.; Zhanel, G. G.; Schweizer, F.
Antimicrob. Agents Chemother. 2010, 54, 4049–4058.doi:10.1128/AAC.00530-10
6. Gause, G. F.; Brazhnikova, M. G. Nature 1944, 154, 703.doi:10.1038/154703a0
7. Zasloff, M. Nature 2002, 415, 389–395. doi:10.1038/415389a
8. Fjell, C. D.; Hiss, J. A.; Hancock, R. E. W.; Schneider, G.Nat. Rev. Drug Discovery 2012, 11, 37–51. doi:10.1038/nrd3591
9. Kadam, R. U.; Bergmann, M.; Hurley, M.; Garg, D.; Cacciarini, M.;Swiderska, M. A.; Natici, C.; Sattler, M.; Smyth, A. R.; Williams, P.;Cámara, M.; Stocker, A.; Darbre, T.; Reymond, J.-L.Angew. Chem., Int. Ed. 2011, 50, 10631–10635.doi:10.1002/anie.201104342
10. Appelt, C.; Schrey, A. K.; Söderhäll, J. A.; Schmieder, P.Bioorg. Med. Chem. Lett. 2007, 17, 2334–2337.doi:10.1016/j.bmcl.2007.01.075
11. Young, A. W.; Liu, Z.; Zhou, C.; Totsingan, F.; Jiwrajka, N.; Shi, Z.;Kallenbach, N. R. Med. Chem. Commun. 2011, 2, 308–314.doi:10.1039/c0md00247j
12. Johansson, E. M. V.; Kadam, R. U.; Rispoli, G.; Crusz, S. A.;Bartels, K.-M.; Diggle, S. P.; Cámara, M.; Williams, P.; Jaeger, K.-E.;Darbre, T.; Reymond, J.-L. Med. Chem. Commun. 2011, 2, 418–420.doi:10.1039/c0md00270d
13. Stach, M.; Maillard, N.; Kadam, R. U.; Kalbermatter, D.; Meury, M.;Page, M. G. P.; Fotiadis, D.; Darbre, T.; Reymond, J.-L.Med. Chem. Commun. 2012, 3, 86–89. doi:10.1039/c1md00272d
14. Makovitzki, A.; Avrahami, D.; Shai, Y. Proc. Natl. Acad. Sci. U. S. A.2006, 103, 15997–16002. doi:10.1073/pnas.0606129103
15. Arnusch, C. J.; Albada, H. B.; van Vaardegem, M.; Liskamp, R. M. J.;Sahl, H.-G.; Shadkchan, Y.; Osherov, N.; Shai, Y. J. Med. Chem. 2012,55, 1296–1302. doi:10.1021/jm2014474
16. Chongsiriwatana, N. P.; Patch, J. A.; Czyzewski, A. M.; Dohm, M. T.;Ivankin, A.; Gidalevitz, D.; Zuckermann, R. N.; Barron, A. E.Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 2794–2799.doi:10.1073/pnas.0708254105
17. Oren, Z.; Shai, Y. Biochemistry 1997, 36, 1826–1835.doi:10.1021/bi962507l
18. Tew, G. N.; Scott, R. W.; Klein, M. L.; DeGrado, W. F. Acc. Chem. Res.2010, 43, 30–39. doi:10.1021/ar900036b
19. Chantson, J. T.; Verga Falzacappa, M. V.; Crovella, S.;Metzler-Nolte, N. J. Organomet. Chem. 2005, 690, 4564–4572.doi:10.1016/j.jorganchem.2005.07.007
20. Chantson, J. T.; Verga Falzacappa, M. V.; Crovella, S.;Metzler-Nolte, N. ChemMedChem 2006, 1, 1268–1274.doi:10.1002/cmdc.200600117
21. Fränzel, B.; Frese, C.; Penkova, M.; Metzler-Nolte, N.; Bandow, J. E.;Wolters, D. A. J. Biol. Inorg. Chem. 2010, 15, 1293–1303.doi:10.1007/s00775-010-0689-z
22. Patra, M.; Gasser, G.; Metzler-Nolte, N. Dalton Trans. 2012, 41,6350–6358. doi:10.1039/c2dt12460b
23. Patra, M.; Gasser, G.; Pinto, A.; Merz, K.; Ott, I.; Bandow, J. E.;Metzler-Nolte, N. ChemMedChem 2009, 4, 1930–1938.doi:10.1002/cmdc.200900347
24. Patra, M.; Gasser, G.; Wenzel, M.; Merz, K.; Bandow, J. E.;Metzler-Nolte, N. Eur. J. Inorg. Chem. 2011, 3295–3302.doi:10.1002/ejic.201100497
25. Patra, M.; Gasser, G.; Wenzel, M.; Merz, K.; Bandow, J. E.;Metzler-Nolte, N. Organometallics 2010, 29, 4312–4319.doi:10.1021/om100614c
26. Wenzel, M.; Patra, M.; Albrecht, D.; Chen, D. Y.-K.; Nicolau, K. C.;Metzler-Nolte, N.; Bandow, J. E. Antimicrob. Agents Chemother. 2011,55, 2590–2596. doi:10.1128/AAC.00078-11
27. Patra, M.; Gasser, G.; Wenzel, M.; Merz, K.; Bandow, J. E.;Metzler-Nolte, N. Organometallics 2012, 31, 5760–5771.doi:10.1021/om201146c
D Modulating peptide activity with metallocenoyl groups
55

Beilstein J. Org. Chem. 2012, 8, 1753–1764.
1763
28. Patra, M.; Merz, K.; Metzler-Nolte, N. Dalton Trans. 2012, 41, 112–117.doi:10.1039/c1dt10918a
29. Strøm, M. B.; Haug, B. E.; Skar, M. L.; Stensen, W.; Stiberg, T.;Svendsen, J. S. J. Med. Chem. 2003, 46, 1567–1570.doi:10.1021/jm0340039
30. Junkes, C.; Wessolowski, A.; Farnaud, S.; Evans, R. W.; Good, L.;Bienert, M.; Dathe, M. J. Pept. Sci. 2008, 14, 535–543.doi:10.1002/psc.940
31. Wessolowski, A.; Bienert, M.; Dante, M. J. Pept. Res. 2004, 64,159–169. doi:10.1111/j.1399-3011.2004.00182.x
32. Dathe, M.; Nikolenko, H.; Klose, J.; Bienert, M. Biochemistry 2004, 43,9140–9150. doi:10.1021/bi035948v
33. Haug, B. E.; Stensen, W.; Kalaaji, M.; Rekdal, Ø.; Svendsen, J. S.J. Med. Chem. 2008, 51, 4306–4314. doi:10.1021/jm701600a
34. Schatzschneider, U.; Metzler-Nolte, N. Angew. Chem., Int. Ed. 2006,45, 1504–1507. doi:10.1002/anie.200504604
35. Shubina, E. S.; Krylov, A. N.; Kreindlin, A. Z.; Rybinskaya, M. I.;Epstein, L. M. J. Mol. Struct. 1993, 301, 1–5.doi:10.1016/0022-2860(93)80225-K
36. Kirin, S. I.; Noor, F.; Mier, W.; Metzler-Nolte, N. J. Chem. Educ. 2007,84, 108–111. doi:10.1021/ed084p108
37. Wadhwani, P.; Afonin, S.; Ieronimo, M.; Buerck, J.; Ulrich, A. S.J. Org. Chem. 2006, 71, 55–61. doi:10.1021/jo051519m
38. Mosby’s Medical Dictionary, 8th ed.; Elsevier: 2009.39. Zanello, P. Inorganic Electrochemistry, theory, practise and
applications; Royal Society of Chemistry: Cambridge, UK, 2003;pp 159–203.
40. Elschenbroich, C. Organometallchemie; B. G. Teubner Verlag:Wiesbaden, Germany, 2008; pp 457–458.
41. Mutoh, H.; Masuda, S. J. Chem. Soc., Dalton Trans. 2002, 1875–1881.doi:10.1039/B111486G
42. Schibli, D. J.; Epand, R. F.; Vogel, H. J.; Epand, R. M.Biochem. Cell Biol. 2002, 80, 667–677. doi:10.1139/o02-147
43. Gopal, R.; Kim, Y. J.; Seo, C. H.; Hahm, K.-S.; Park, Y. J. Pept. Sci.2011, 17, 329–334. doi:10.1002/psc.1369
44. Su, Y.; Doherty, T.; Waring, A. J.; Ruchala, P.; Hong, M. Biochemistry2009, 48, 4587–4595. doi:10.1021/bi900080d
45. Kyte, J.; Doolittle, R. F. J. Mol. Biol. 1982, 157, 105–132.doi:10.1016/0022-2836(82)90515-0
46. Nandi, N. J. Phys. Chem. A 2003, 107, 4588–4591.doi:10.1021/jp030076s
47. Pathirana, S.; Neely, W. C.; Myers, L. J.; Vodyanoy, V.J. Am. Chem. Soc. 1992, 114, 1404–1405. doi:10.1021/ja00030a041
48. Alakoskela, J.-M.; Sabatini, K.; Jiang, X.; Laitala, V.; Covey, D. F.;Kinnunen, P. K. J. Langmuir 2008, 24, 830–836.doi:10.1021/la702909q
49. Alakoskela, J.-M.; Covey, D. F.; Kinnunen, P. K. J.Biochim. Biophys. Acta 2007, 1768, 131–145.
50. Nguyen, L. T.; Chau, J. K.; Perry, N. A.; de Boer, L.; Zaat, S. A. J.;Vogel, H. J. PLoS One 2010, 5, e12684.doi:10.1371/journal.pone.0012684
51. Pag, U.; Oedenkoven, M.; Sass, V.; Shai, Y.; Shamova, O.;Antcheva, N.; Tossi, A.; Sahl, H.-G. J. Antimicrob. Chemother. 2008,61, 341–352. doi:10.1093/jac/dkm479
52. Sass, V.; Pag, U.; Tossi, A.; Bierbaum, G.; Sahl, H.-G.Int. J. Med. Microbiol. 2008, 298, 619–633.doi:10.1016/j.ijmm.2008.01.011
53. Lui, Z.; Brady, A.; Young, A.; Rasimick, B.; Chen, K.; Zhou, C.;Kallenbach, N. R. Antimicrob. Agents Chemother. 2007, 51, 597–603.doi:10.1128/AAC.00828-06
54. Page, S. M.; Ross, S. R.; Barker, P. D. Future Med. Chem. 2009, 1,541–559. doi:10.4155/fmc.09.25
55. Schluga, P.; Hartinger, C. G.; Egger, A.; Reisner, E.; Galanski, M.;Jakupec, M. A.; Keppler, B. K. Dalton Trans. 2006, 1796–1802.doi:10.1039/b511792e
56. Therrien, B.; Süss-Fink, G.; Govindaswamy, P.; Renfrew, A. K.;Dyson, P. J. Angew. Chem., Int. Ed. 2008, 47, 3773–3776.doi:10.1002/anie.200800186
57. Gasser, G.; Ott, I.; Metzler-Nolte, N. J. Med. Chem. 2011, 54, 3–25.doi:10.1021/jm100020w
58. Rademaker-Lakhai, J. M.; van den Bongard, D.; Pluim, D.;Beijnen, J. H.; Schellens, J. H. M. Clin. Cancer Res. 2004, 10,3717–3727. doi:10.1158/1078-0432.CCR-03-0746
59. Lentz, F.; Drescher, A.; Lindauer, A.; Henke, M.; Hilger, R. A.;Hartinger, C. G.; Scheulen, M. E.; Dittrich, C.; Keppler, B. K.;Jaehde, U. Anti-Cancer Drugs 2009, 20, 97–103.doi:10.1097/CAD.0b013e328322fbc5
60. Hartinger, C. G.; Zorbas-Seifried, S.; Jakupec, M. A.; Kynast, B.;Zorbas, H.; Keppler, B. K. J. Inorg. Biochem. 2006, 100, 891–904.doi:10.1016/j.jinorgbio.2006.02.013
61. Hartinger, C. G.; Jakupec, M. A.; Zorbas-Seifried, S.; Groessl, M.;Egger, A.; Berger, W.; Zorbas, H.; Dyson, P. J.; Keppler, B. K.Chem. Biodiversity 2008, 5, 2140–2155. doi:10.1002/cbdv.200890195
62. Groessl, M.; Reisner, E.; Hartinger, C. G.; Eichinger, R.;Semenova, O.; Timerbaev, A. R.; Jakupec, M. A.; Arion, V. B.;Keppler, B. K. J. Med. Chem. 2007, 50, 2185–2193.doi:10.1021/jm061081y
63. Harden, J.; Diorio, N.; Petrov, A. G.; Jakli, A. Phys. Rev. E 2009, 79,011701. doi:10.1103/PhysRevE.79.011701
64. Brogden, K. A. Nat. Rev. Microbiol. 2005, 3, 238–250.doi:10.1038/nrmicro1098
65. M07-A8, Vol. 29, No. 2; Methods for Dilution AntimicrobialSusceptibility Tests for Bacteria That Grow Aerobically; ApprovedStandard – 8th edition, Clinical and Laboratory Standards Institute,Wayne, Pennsylvania, USA, 2009.
66. Giacometti, A.; Cirioni, O.; Barchiesi, F.; Del Prete, M. S.; Fortuna, M.;Caselli, F.; Scalise, G. Antimicrob. Agents Chemother. 2000, 44,1694–1696. doi:10.1128/AAC.44.6.1694-1696.2000
67. Bernhardt, G.; Reile, H.; Birnböck, H.; Spruß, T.; Schönenberger, H.J. Cancer Res. Clin. Oncol. 1992, 118, 35–43.doi:10.1007/BF01192309
68. Neukamm, M. J.; Pinto, A.; Metzler-Nolte, N. Chem. Commun. 2008,232–234. doi:10.1039/b712886j
D Modulating peptide activity with metallocenoyl groups
56

Beilstein J. Org. Chem. 2012, 8, 1753–1764.
1764
License and TermsThis is an Open Access article under the terms of the
Creative Commons Attribution License
(http://creativecommons.org/licenses/by/2.0), which
permits unrestricted use, distribution, and reproduction in
any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic
Chemistry terms and conditions:
(http://www.beilstein-journals.org/bjoc)
The definitive version of this article is the electronic one
which can be found at:
doi:10.3762/bjoc.8.200
D Modulating peptide activity with metallocenoyl groups
57

E
An antimicrobial peptide delocalizes peripheral
membrane proteins
Michaela Wenzel, Alina Iulia Chiriac, Andreas Otto, Dagmar Zweytick,
Caroline May, Catherine Schumacher, H. Bauke Albada, Maya
Penkova, Ute Krämer, Ralf Erdmann, Nils Metzler‐Nolte, Heike Brötz‐
Oesterhelt, Dörte Becher, Hans‐Georg Sahl, Julia E. Bandow
submitted
E An antimicrobial peptide delocalizes peripheral membrane proteins
58

An antimicrobial peptide delocalizes peripheral membrane proteins
Michaela Wenzel1, Alina Iulia Chiriac2, Andreas Otto3, Dagmar Zweytick4, Caroline May5,
Catherine Schumacher6, H. Bauke Albada7, Maya Penkova7, Ute Krämer8, Ralf Erdmann9, Nils
Metzler‐Nolte7, Heike Brötz‐Oesterhelt6, Dörte Becher3, Hans‐Georg Sahl2, Julia E. Bandow1*
1Biology of Microorganisms, Ruhr University Bochum, Germany 2Institute for Medical Microbiology, Immunology, and Parasitology, Pharmaceutical
Microbiology Section, University of Bonn, Germany 3Microbial Physiology and Molecular Biology, Ernst Moritz Arndt University, Greifswald,
Germany 4Institute of Molecular Biosciences, Biophysics Division, University of Graz, Austria 5Immune Proteomics, Medical Proteome Center, Ruhr University Bochum, Germany 6Institute for Pharmaceutical Biology and Biotechnology, Heinrich Heine University,
Düsseldorf, Germany 7Bioinorganic Chemistry, Ruhr University Bochum, Germany 8Department of Plant Physiology, Ruhr University Bochum, Germany 9Institute of Physiological Chemistry, Ruhr University Bochum, Germany
*Corresponding author. Mailing address: Ruhr‐Universität Bochum, Biologie der
Mikroorganismen, Universitätsstraße 150, 44801 Bochum, Germany. Phone: +49‐234‐32‐
23102. Fax: +49‐234‐32‐14620. E‐mail: [email protected]
ABSTRACT
Short arginine‐ and tryptophan‐rich (RW‐rich) peptides target the bacterial membrane. We
characterized the effects on bacterial physiology of the synthetic hexapeptide NH2‐
RWRWRW‐CONH2. Peptide integration into the membrane is dependent on the phospholipid
composition, occurring readily in mixtures of dipalmitoyl‐glycero‐phosphatidylglycerol
(DPPG) and dipalmitoyl‐glycero‐phosphatidylethanolamine (DPPE), less well in membranes
composed of DPPG, and not at all in DPPE membranes. Integration of the peptide alters lipid
bilayer architecture, resulting in delocalization of membrane‐associated proteins involved in
cell wall biosynthesis, respiration, and cell division. Consequently, energy within the cells is
limited and cell wall integrity is undermined. The bacteria respond to this peptide stress with
an integrated strategy, including the adjustment of membrane lipid composition,
modification of the cell wall, and release of osmoprotective amino acids.
E An antimicrobial peptide delocalizes peripheral membrane proteins
59

INTRODUCTION
Antimicrobial peptides are potent antibiotic agents that occur in all domains of life. They act
against bacteria but also against viruses, fungi, protozoa, and cancer cells1,2. Ranging in
length from four to over one hundred amino acids, they fall into a number of different
structural classes, including α‐helical amphiphiles, lantibiotics, lipopeptides, glycopeptides,
and short cationic peptides.
Short cationic antimicrobial peptides are characterized by positively charged amino acids like
arginine and lysine, and hydrophobic amino acids like tryptophan and phenylalanine3‐4. Such
peptides typically target the bacterial membrane interfering with ion transport, membrane
architecture, specific interactions between membrane components, or forming pores5.
Previous mechanistic studies in vitro have examined the interactions of the peptides with
membranes or membrane extracts6‐7 but their effects on bacterial physiology have gone
underexplored.
To generate a more complete understanding of the impact of short cationic antimicrobial
peptides on bacterial physiology, we selected the synthetic hexapeptide NH2‐RWRWRW‐
CONH2 (referred to in the following as MP196). MP196 represents the minimal
pharmacophore of positively charged and hydrophobic amino acids8‐9. It is a potent
alternative to longer natural antimicrobial peptides, which can be difficult to isolate and
complicated to synthesize chemically. Short peptides are readily generated by solid‐phase
peptide synthesis and are easily accessible for chemical derivatization10‐11. MP196 is
effective against gram‐positive bacteria including MRSA strains, moderately effective against
Gram‐negative bacteria and has low toxicity against human cell lines and low hemolytic
activity12. It is therefore a promising lead structure for derivatization, and has already yielded
peptides with improved activities and altered pharmacological properties13.
Proteomic profiling of Bacillus subtilis exposed to MP196 provided a starting point for
mechanistic studies. Previously, the proteomic response to antibiotics was found to reflect
the general mode of action, in some cases even correlating with the antibiotic’s molecular
binding site14‐15. We monitored the acute bacterial stress response to MP196 using
metabolic pulse‐labeling of proteins newly synthesized in response to antibiotic stress,
followed by separation of the cytosolic proteome by 2D‐PAGE. In parallel, the membrane
proteome was investigated by LC‐ESI‐MS/MS to identify proteome changes at the site of
binding of MP196 16. Proteome‐based hypotheses were challenged and corroborated by a
E An antimicrobial peptide delocalizes peripheral membrane proteins
60

diverse set of further methods including differential scanning calorimetry, ionomics, and
amino acid analysis, providing insights into both the mechanism of action of cationic
antimicrobial peptides and bacterial adaptation to peptide stress.
RESULTS
Inhibition of macromolecule biosynthesis
To explore inhibition of macromolecule biosynthesis in vivo, we studied incorporation of
radioactive precursors. B. subtilis cells were exposed to MP196 concentrations that reduced
growth rates by approximately 50 percent unless noted otherwise (Supplementary Fig. 1).
MP196 moderately inhibited incorporation of DNA, RNA, protein, and cell wall precursors
(Supplementary Fig. 2), findings typical for membrane‐targeting antibiotics. The activation of
reporter genes previously shown to respond to inhibition of metabolic pathways and
different types of stress in B. subtilis17 was also studied (Supplementary Fig. 3). MP196 did
not activate transcription from promoters of yorB, helD, or bmrC , which are indicative of
DNA damage, RNA synthesis inhibition, and translation inhibition, respectively. The liaI
promoter, responsive to inhibition of membrane‐bound cell wall biosynthesis steps, was
activated weakly, while ypuA, which is responsive to membrane‐associated and membrane‐
independent cell wall stress, was strongly activated.
Proteomic profiling
Proteomic analyses were performed using B. subtilis 168, for which a library of proteome
response profiles to over 40 antibiotic agents exists19‐21. Cultures were stressed with
sublethal peptide concentrations (Supplementary Fig. 1) prior to analysis of the cytosolic
and membrane proteome. After 10 minutes of exposure to the peptide, newly synthesized
proteins were pulse labeled with L‐[35S]‐methionine for five minutes and analyzed by 2D
gelelectrophoresis. Thirty‐six proteins were selectively upregulated in response to MP196
treatment, 32 of which were identified by mass spectrometry (Fig. 1, Supplementary Table 1
and 2). In parallel, the membrane proteome response was analyzed by LC‐ESI‐MS/MS.
Differential 14N/15N labeling was used for relative quantitation of proteins in the membrane
fractions of untreated and peptide‐treated cultures. After 65 minutes of peptide treatment,
178 proteins were upregulated at least two‐fold (Supplementary Table 3 and 4). Most
differentially regulated proteins are predicted to localize in the cytosol yet are associated
with membrane functions, including general cell envelope stress response, membrane stress
response, cell wall biosynthesis, lipid biosynthesis, and energy metabolism.
E An antimicrobial peptide delocalizes peripheral membrane proteins
61

The tellurium resistance protein, YceC, and toxic anion resistance protein, YceH, previously
described as marker proteins for both cell wall and membrane stress21, were upregulated,
along with six other proteins regulated by the cell envelope‐responsive alternative sigma
factors W (σW) and M (σM). Other notable proteins upregulated included LiaH, which
protects cell envelope integrity by binding to the cytoplasmic membrane, the ABC
transporter protein, YtrE, and the tRNA‐modifying enzyme, TrmB, which are specific markers
for interference with the membrane‐bound part of the cell wall biosynthesis machinery21.
Upregulation of MurAA, MurAB, and MurG, which are involved in the synthesis of the cell
wall precursor lipid II, indicates inhibition of cell wall biosynthesis, as does upregulation of
MreB, which is important for localization of the cell wall biosynthesis and cell division
machineries, and of the MreB‐like protein Mbl. The amino acid racemase RacX and proteins
belonging to the dlt operon were upregulated. They are involved in cell envelope
modification by synthesis of D‐amino acids, probably feeding into D‐alanylation reactions
and lipoteichoic acid biosynthesis, respectively.
Figure 1. Cytosolic protein biosynthesis profile. The protein synthesis pattern of untreated
B. subtilis cells was false‐colored in green and overlayed with that of the MP196‐treated
culture, false‐colored in red. Down‐regulated proteins appear green, upregulated proteins
red, and proteins expressed at equal rates yellow. Unidentified proteins are marked by
circles. Blue labels indicate marker proteins for general cell envelope stress, green labels
identify cell wall biosynthesis inhibition, and red labels are markers for membrane stress.
E An antimicrobial peptide delocalizes peripheral membrane proteins
62

PspA, an LiaH homologue, is specifically upregulated following membrane stress, as opposed
to peptidoglycan stress21. Other proteins upregulated in response to MP196 are involved in
restructuring of the membrane lipid composition. Among these are proteins involved in fatty
acid biosynthesis (FabG and AcpA), alternative fatty acid biosynthesis (YjdA and YoxD),
phospholipid biosynthesis (PlsX), lipid‐degradation (FadE), and membrane fluidity control
(FloT). NAD synthase (NadE) is crucial for energy metabolism and, together with PspA,
constitutes a proteomic signature for membrane‐mediated stress21. Other proteins, whose
upregulation indicates substantial energy limitation, are involved in oxidative
phosphorylation, NAD metabolism, the TCA cycle, glycolysis, and ATP synthesis. Similarly,
several ABC transporter and protease ATP‐binding proteins were upregulated. Although
biosynthesis of housekeeping proteins was largely down‐regulated after MP196 treatment,
elongation factors Tu and G, which require GTP for activity, were upregulated. Also, many
proteins involved in amino acid biosynthesis were upregulated, particularly those involved in
glutamate and aspartate anabolism.
To investigate the importance of peptide stereochemistry for antibacterial activity and
MP196‐target interaction, the proteomic response to the all‐D‐amino acid peptide D‐MP196
was studied using 2D gel electrophoresis. D‐MP196 was as active against B. subtilis as
MP19612 and both shared many marker proteins (Supplementary Fig. 4). Thus,
stereochemistry does not seem to be crucial for target interaction, making it unlikely that
MP196 binds a specific site on a protein target.
The findings above prompted the following questions about MP196: How does it affect cell
wall biosynthesis? Does it interact with the bacterial membrane and, if so, how? How does it
influence energy metabolism? What is the significance of the strong upregulation of amino
acid biosynthesis proteins? And what is the primary event that leads to cell death?
To address these questions, we compared MP196 with a set of compounds with different
cell envelope‐related modes of action. Selection of reference compounds was aided by
comparative proteome analysis using the existing library of B. subtilis antibiotic response
profiles19‐21 (Supplementary Table 5). The strongest overlap was observed with gramicidin S,
a cyclic peptide that disrupts membrane function by integrating into the lipid bilayer, and
with the potassium carrier ionophore valinomycin. We also chose vancomycin, a lipid II‐
binding cell wall biosynthesis inhibitor, and nisin, which, in addition to inhibiting cell wall
biosynthesis by binding lipid II, forms heteromultimeric membrane pores with lipid II.
E An antimicrobial peptide delocalizes peripheral membrane proteins
63

Effects on the cell wall
Cell wall integrity was investigated by examining the shape of B. subtilis cells after acetic
acid/methanol fixation: where the incorporation of cell wall material is inhibited at a
membrane‐bound biosynthesis step, fixation leads to extrusion of the cell membrane
through holes in the cell wall22.
Figure 2. Effects on the cell wall. (a) Light microscopy images showing cell wall integrity of B.
subtilis after treatment with MP196 and nisin. Acetic acid‐methanol fixation visualizes cell
wall damage by extrusions of the membrane through cell wall holes. (b) Thin layer
chromatography (TLC) of radioactively labeled lipid II showing influence of MP196 and nisin
on in vitro lipid II synthesis. (c) TLC probing for in vitro lipid II binding capability by MP196.
Nisin, which retains lipid II by binding to it, serves as positive control. (d) HPLC
chromatographs showing accumulation of the cell wall precursor UDP‐MurNac pentapeptide
by S. aureus cultures treated with MP196 and vancomycin. (e) Overview of bacterial cell wall
biosynthesis (according to Schneider et al.22), illustrating how MurG delocalization might
interfere with the cell wall biosynthesis cycle in vivo. In contrast to integral membrane
protein MraY, the peripheral membrane protein MurG attaches to the membrane surface
through electrostatic interactions. (f) Western blot detection of the cell wall biosynthesis
protein, MurG, in membrane fractions of B. subtilis five minutes after antibiotic addition.
Membranes were stained with Ponceau S as sample loading control. (g) Transmission
electron microscopy images of B. subtilis treated with MP196 and gramicidin S showing the
influence of the peptides on the cell shape and cell wall structure.
E An antimicrobial peptide delocalizes peripheral membrane proteins
64

Untreated cells and cells treated with valinomycin did not display membrane extrusions (Fig.
2a) while nisin and MP196 did induce membrane extrusions, as did gramicidin S, although it
does not interfere directly with cell wall biosynthesis. Membrane extrusions are suggestive
of inhibition of cell wall biosynthesis at the level of lipid II22. However, MP196 did not impair
lipid II synthesis in an in vitro assay using concentrated cell lysate (Fig. 2b). Furthermore,
while nisin bound lipid II in vitro, preventing it from migrating in the thin layer
chromatography matrix, MP196 did not affect lipid II migration (Fig. 2c). We therefore
investigated the influence of MP196 on early and late steps of cell wall biosynthesis in intact
cells. As described above, glucosamine incorporation was partially inhibited by MP196
(Supplementary Fig. 2). HPLC analysis showed that MP196 exposure led to an accumulation
of UDP‐MurNAc pentapeptide in the cell (Fig. 2d), albeit not to the same extent as
vancomycin. Both assays suggest that lipid II is not synthesized efficiently in vivo. We next
investigated the localization of MurG, a membrane‐associated protein that converts lipid I to
lipid II, by attaching N‐acetyl‐glucosamine (GlcNAc) to the bactoprenol carrier‐conjugated
UDP‐MurNAc pentapeptide molecule (Fig. 2e).
Western Blot analysis of B. subtilis cells showed that both MP196 and gramicidin S treatment
caused almost complete loss of MurG from the membrane fraction within five minutes (Fig.
2f). The removal of MurG from the cytosolic membrane surface may explain, in combination
with energy limitation (see below), the reduced glucosamine incorporation, UDP‐MurNAc
pentapeptide accumulation, loss of cell wall integrity, and induction of a cell wall‐specific
stress response. The morphology of B. subtilis cells treated with non‐lytic concentrations of
MP196 or gramicidin S was examined by transmission electron microscopy (TEM, Fig. 2g).
MP196‐treated cells had cell wall lesions lined with characteristic structures. The cell wall at
these lesion sites appeared to be thinner, corroborating the observed loss of integrity. In
contrast, gramicidin S‐treated cells did not retain their shape, culminating in the leakage of
cell contents and cell collapse. Thus, while the peptides induce similar proteome response
patterns and cause delocalization of the MurG protein, B. subtilis counteracts MP196‐
induced lesions more efficiently than those caused by gramicidin S.
Interaction with the membrane
Interaction of MP196 with the membrane was investigated by differential scanning
calorimetry (DSC) using model membranes consisting of the two most abundant
E An antimicrobial peptide delocalizes peripheral membrane proteins
65

phospholipids in the B. subtilis membrane23, 1,2‐dipalmitoyl‐sn‐glycero‐3‐
phosphatidylglycerol (DPPG) and 1,2‐dipalmitoyl‐sn‐glycero‐3‐phosphatidylethanolamine
(DPPE), in an 88:12 ratio (Fig. 3a). DSC monitors the thermotropic phase behaviour of the
lipid bilayer. Changes in pattern are indicative of perturbation of the membrane bilayer and
packaging of the phospholipids’ fatty acyl chains and, therefore, of peptide integration.
Deconvolution of the thermograms indicates that MP196 induces peptide‐affected
domains6, reflected in a broadening of the existing transition and the appearance of a small
transition at lower temperatures. This causes rearrangement of the lipid mixture, and,
consequently, alterations in packaging of the lipids at the emerging borderlines.
When bilayers composed of a single type of phospholipid were used, the influence of MP196
on a DPPG bilayer was far less pronounced, although broadening of the shoulder at lower
temperatures indicated the formation of some peptide‐affected domains. No effect was
seen using pure DPPE or using 1,2‐dipalmitoyl‐sn‐glycero‐3‐phosphatidylcholine (DPPC)
(Supplementary Fig. 5), which is typically used to model erythrocyte membranes24. The fact
that MP196 affected DPPG but not DPPE implies a preference for negatively charged over
neutral lipids.
We used a fluorescence stain to test whether peptide integration influences membrane
permeability in B. subtilis in vivo. BacLight staining combines a green‐fluorescent dye that
crosses intact membranes with a red‐fluorescent dye that enters bacterial cells through
membrane pores (Fig. 3b). Pore‐forming nisin allowed both dyes to enter B. subtilis cells,
resulting in orange cells. MP196 treatment, as well as treatment with non‐pore‐forming
gramicidin S and valinomycin, resulted in green‐fluorescent cells like the untreated control,
suggesting that MP196 does not act by forming large pores. Similar conclusions were drawn
from measuring peptide‐induced potassium leakage from Bacillus megaterium cells into
choline buffer (Supplementary Fig. 6a). In contrast to the positive control, nisin, MP196
released little potassium even at concentrations equivalent to 50 times the minimal
inhibitory concentration (MIC).
The effect of MP196 on membrane permeability was further studied using a global ionomics
approach. Total cellular metal concentrations were determined after treating B. subtilis with
peptide in salt‐free Tris buffer, which, in contrast to the choline buffer used for the
potassium leakage assay, is not osmotically stabilizing (Fig. 3c). While no effect was seen on
sodium, calcium, and zinc levels, potassium concentrations decreased by 96 percent,
E An antimicrobial peptide delocalizes peripheral membrane proteins
66

magnesium by 86 percent and manganese by 59 percent. This was also the case for
gramicidin S, in contrast to potassium carrier ionophore valinomycin, which selectively
caused potassium efflux.
Figure 3. Interaction with the membrane. (a) DSC thermograms of lipid bilayers consisting
of 88:12 DPPG/DPPE incubated with MP196. Changes in thermotropic phase behaviour due
to perturbation of fatty acyl packing indicate peptide integration. (b) Overlayed fluorescence
microscopy images of BacLight‐stained B. subtilis treated with valinomycin, nisin, gramicidin
S, and MP196. A red‐fluorescing dye selectively stains cells with large membrane holes
whereas a green‐fluorescing dye stains all cells independently of membrane damage. Cells
with intact membranes are green and cells with perforated membranes are orange. (c) Total
ion profiles of B. subtilis stressed with valinomycin, gramicidin S, and MP196 in Tris buffer.
(d) Total ion profiles of B. subtilis stressed with valinomycin, gramicidin S, and MP196 in
chemically defined medium under conditions resembling those of the proteome analysis.
To investigate whether ion efflux mediated by MP196 is relevant in Tris‐buffered chemically
defined culture broth with regular salt content, total ion concentrations were determined
after peptide treatment under the same conditions used for proteomic profiling (Fig. 3d). In
E An antimicrobial peptide delocalizes peripheral membrane proteins
67

agreement with the observed lack of potassium efflux in choline buffer, no significant
decrease of any metal ion was observed after MP196 treatment under these conditions,
suggesting that ion efflux either does not occur or is compensated for in chemically defined
medium. In contrast, gramicidin S treatment led to a decrease of potassium, magnesium,
and manganese levels comparable to the effect in Tris buffer, consistent with the disruptive
effect of gramicidin S seen by TEM. Valinomycin‐treated cells accumulated potassium (an
eight‐fold increase), probably due to overcompensation. This fits well with the observation
that, despite provoking a strong proteome response, valinomycin does not inhibit B. subtilis
growth19.
Effects on membrane potential and respiration
Membrane‐targeting antibiotics typically depolarize the bacterial membrane. Depolarization
was investigated using a B. subtilis strain carrying a GFP fusion to the cell division‐regulating
protein MinD. Normally localized at the cell poles and in the cell division plane, MinD
delocalizes upon depolarization, resulting in a spotty GFP‐MinD distribution pattern25. In
contrast to the normal MinD pattern in the untreated control, MinD delocalization was seen
in MP196‐ and gramicidin S‐treated cells, indicating membrane depolarization (Fig. 4a).
Depolarization upon MP196 treatment was confirmed in Bacillus megaterium employing the
voltage‐sensitive fluorescent probe DiSC35 (Supplementary Fig. 6b).
Sudden depolarization should result in energy limitation, consistent with the proteome
response. Intracellular ATP levels of B. subtilis, determined with a luciferase assay, showed a
60 percent reduction in cellular ATP content in MP196‐treated and a 70 percent reduction in
gramicidin S‐treated cells (Fig. 4b), while valinomycin and nisin caused 30 percent drops.
Thus, MP196 has a considerable impact on energy metabolism. No significant ion efflux was
observed under the test conditions, indicating that depolarization and energy limitation are
not due solely to ion transfer. We therefore tested, whether components of the respiratory
chain are inhibited by MP196. H+‐ATPase inhibition would lead directly to ATP limitation.
Proton pumping activity was monitored in Micrococcus flavus inverted vesicles employing
the pH‐sensitive probe acridine orange (Fig. 4c). The H+‐ATPase inhibitor lactoferrin26 served
as a positive control. MP196 did not inhibit H+‐ATPase pumping activity at a peptide
concentration corresponding to that used for proteome analysis.
E An antimicrobial peptide delocalizes peripheral membrane proteins
68

Figure 4. Effects on membrane potential and respiration. (a) Fluorescence microscopy
images (top row) and corresponding bright field images (bottom row) show GFP‐MinD
localization in untreated B. subtilis cells and cells incubated with gramicidin S or MP196,
respectively. GFP‐MinD delocalization indicates membrane depolarization. (b) ATP levels of
B. subtilis cells treated with valinomycin, nisin, gramicidin S, or MP196. In a luciferase assay,
ATP concentrations were determined under conditions resembling those of the proteome
experiment. (c) H+‐ATPase activity of M. flavus inverted vesicles treated with lactoferrin or
MP196. Proton pumping activity was measured by the pH‐dependent absorbance change of
acridine orange at 495 nm. (d) Activity of the respiratory chain of inverted M. flavus vesicles
treated with rotenone, antimycin A, or MP196. Electron transport efficiency was monitored
by reduction of iodonitrotetrazolium chloride (INT) leading to decreased absorbance at 485
nm. (e) Western blot analysis of cytochrome c in membrane fractions of B. subtilis after
treatment with gramicidin S and MP196 for five minutes. Ponceau S is displayed as loading
control. (f) Overview of the bacterial respiratory chain (according to Vonck and Schäfer24).
Proton movements are indicated by green arrows, electron movements by orange arrows.
ATP limitation might also stem from inhibition of the respiratory chain, which would
contribute to the breakdown of the membrane potential and a subsequent loss of ATP
synthesis. Using M. flavus inverted vesicles, inhibition of the respiratory chain was
69

monitored with iodonitrotetrazolium chloride, a reduction‐sensitive dye (Fig. 4d). Rotenone
and antimycin A, specific inhibitors of complexes I and III of the respiratory chain,
respectively, were used as comparator compounds. Rotenone reduced electron transport
activity by 20 percent, antimycin A by 50 percent. MP196 displayed similar inhibition
efficiency, reducing electron transport by 30 percent. MP196 inhibition was additive with
either rotenone or antimycin A, suggesting that MP196 inhibits a different component of the
respiratory chain. An alteration in membrane architecture, as suggested by DSC, might affect
proteins attached to the membrane surface, as was shown for cell wall biosynthesis enzyme
MurG. Cytochrome c is located on the outer membrane surface and transfers electrons from
complex III to complex IV. Its localization after MP196 treatment was investigated by
Western blot analysis of membrane fractions of antibiotic‐treated B. subtilis cells (Fig. 4e).
The amount of cytochrome c in the membrane was drastically reduced after five minutes
treatment with MP196, suggesting dissociation of the protein from the extracellular
membrane surface. A similar effect was observed using gramicidin S, which was shown
earlier to inhibit components of the respiratory chain27. Detachment of cytochrome c from
the outer membrane leaflet explains the inhibition of the respiratory chain and subsequent
energy limitation (Fig. 4f), and probably contributes to the breakdown of membrane
potential.
Cellular and extracellular amino acid composition
Proteome analysis revealed upregulation of proteins involved in amino acid anabolism,
especially of branched‐chain amino acid but also of glutamate and aspartate. To further
investigate this, intracellular and extracellular free amino acid levels were measured by HPLC
after 15 minutes of antibiotic treatment. MP196‐treated cells accumulated free arginine and
valine intracellularly (Fig. 5a, Supplementary Fig. 7a), consistent with the upregulation of
proteins involved in valine biosynthesis (Tab. 1, Supplementary Table 3).
Intracellular glutamine/glutamate, asparagine/aspartate (the assay employed here does
distinguish between glutamate and glutamine or between aspartate and asparagine), lysine,
and proline levels were substantially reduced, with concomitant dramatic increases in
extracellular glutamine/glutamate and asparagine/aspartate and moderate increases in
extracellular arginine, lysine, and proline (Fig. 5b, Supplementary Fig. 7b).
E An antimicrobial peptide delocalizes peripheral membrane proteins
70

Figure 5. Amino acid composition. (a) HPLC analysis of the intracellular amino acid
composition of B. subtilis treated with MP196. (b) Extracellular amino acid composition of
the same cultures. Only those amino acids, whose concentrations changed significantly after
peptide treatment, are displayed here (see Supplementary Fig. 7 for full amino acid
profiles). Amino acids are written in one letter code in the order of elution time from the
column. Glutamate and glutamine as well as aspartate and asparagine are not
distinguishable by this method, and appear as one peak. Tryptophan was not quantified
here. (c‐e) Minimal inhibitory concentrations (MICs) of MP196 against B. subtilis in defined
medium supplemented with rising glutamate (c), NaCl (d), or KCl (e) concentrations. MICs
were independently determined twice. (f) Intra‐ and extracellular glutamate concentrations
in B. subtilis cells under different antibiotic and osmotic stress conditions determined by
amino acid analysis.
The absolute amounts of released glutamine/glutamate exceeded by far the sum of
intracellular and extracellular levels of untreated cells, indicating dedicated biosynthesis and
export (Supplementary Fig. 8a). Feeding experiments using radioactive glutamate
performed in Staphylococcus simulans showed that glutamate uptake is reduced
immediately upon MP196 treatment (Supplementary Fig. 8b). Supplementing exogenous
glutamate to defined medium increased the minimal inhibitory concentration (MIC) of
MP196 against B. subtilis up to eight‐fold (Fig. 5c), establishing a protective effect of
glutamate against MP196 stress. Similar results were obtained with exogenously
supplemented sodium chloride (Fig. 5d) and potassium chloride (Fig. 5e), suggesting that the
protection against MP196 is not specific for glutamate but rather due to osmotic
E An antimicrobial peptide delocalizes peripheral membrane proteins
71

stabilization. Glutamate release was also observed in response to osmotic downshift or
treatment with gramicidin S or nisin, but not to osmotic upshift or valinomycin treatment
(Fig. 5f). Mechanosensitive channels appear to contribute to glutamate release after MP196
treatment. A B. subtilis mutant that lacks the four known mechanosensitive channels29
accumulated high levels of glutamate intracellularly (Fig. 5f) and showed heightened
sensitivity to MP196 (Fig. 5c‐e). However, these channels are not the only route for
glutamate release, as extracellular glutamate levels did increase when the triple mutant was
treated with MP196 (Fig. 5f).
DISCUSSION
The cationic hexapeptide MP196, which is the minimal pharmacophore of RW‐rich peptides,
was used here to study the mechanism of action of short cationic RW‐rich antimicrobial
peptides and subsequent bacterial adaptation strategies. MP196 acts on the bacterial
cytoplasmic membrane, where essential physiological processes, including cell wall
biosynthesis (Fig. 6a.II), respiration (Fig. 6a.III) and membrane transport (Fig. 6a.IV), take
place. At the molecular level (Fig. 6b.I), MP196 readily integrates into the lipid bilayer and
disturbs membrane architecture, as revealed by perturbation of fatty acyl packing measured
using DSC. This membrane integration is likely promoted by interaction of the cationic
arginine residues with negatively charged phospholipid head groups, and facilitated by
lipophilic tryptophan residues. This membrane‐integration mechanism is typical of
amphipathic peptides30 and predicts that MP196 accumulates in the membrane near the
interface with the cytosol, making contact with lipid head groups and fatty acyl chains.
We found no evidence for pore formation in model membranes or in vivo, yet MP196 was
able to induce the release of potassium, magnesium, and manganese into a salt‐free buffer
solution, but not into chemically defined bacterial culture medium. For other peptides, it has
been suggested that integration leads to local lipid reorganization, causing transient
membrane leakiness that allows metal ions to cross the membrane31 but the lack of ion
release in chemically defined medium suggests that ion efflux is not a major effect of MP196
under the growth conditions used for the proteome experiment. Osmotic membrane
stabilization may prevent ion leakage in culture media with high ion concentrations, a
possibility supported by the observation that MP196 triggered only weak potassium efflux in
osmotically stabilizing choline buffer.
E An antimicrobial peptide delocalizes peripheral membrane proteins
72

The integration of MP196 into the membrane affects multiple cellular processes (Fig. 6b.II‐
IV). One is cell wall biosynthesis, as evidenced by a reduction in precursor incorporation, loss
of cell wall integrity, promoter activation of cell wall stress‐responsive genes, and
upregulation of proteins indicative of cell wall biosynthesis stress. Similar observations have
been reported for gramicidin S, which is thought to change membrane architecture and
induces markers specific for cell wall biosynthesis inhibition21. Here, both MP196 and
gramicidin S treatment lead to detachment of the lipid II biosynthesis protein MurG from the
intracellular surface of the membrane within five minutes, probably due to peptide
integration causing alterations in membrane architecture. MurG biosynthesis is strongly
upregulated after 65 minutes, suggesting a compensations strategy by B. subtilis. This
mechanism also provides a potential explanation for the induction of the LiaRS cell wall
stress response by membrane‐targeting peptides such as daptomycin32, which has also been
suggested to alter membrane architecture33‐34.
Integration of MP196 into the membrane has a profound impact on the bacterial respiratory
chain (Fig. 6b.III). The observed inhibition can be attributed to detachment of cytochrome c
from the membrane, resulting in disruption of the electron transfer chain and subsequent
breakdown of the proton gradient. Thus, MP196 impairs ATP synthesis, causing energy
limitation that impacts cell wall biosynthesis and all other macromolecule biosynthesis
pathways.
A further consequence of membrane depolarization is delocalization of the cell division
regulation protein, MinD. Unlike cytochrome c variants in B. subtilis, which attach to the
outer leaflet of the membrane by a lipid anchor or transmembrane helices35‐36, MinD and
MurG attach to the intracellular surface of the membrane through electrostatic interactions
using an amphipathic helix motif25,37.
Like MurG, two additional membrane‐associated proteins, SecA and FtsA, were strongly
upregulated in the membrane proteome after 65 minutes. And like MurG, FtsA docks to the
membrane via an amphipathic helix 25. To summarize, the strongest effects of MP196 are on
cell wall biosynthesis and energy metabolism. We propose that substantial energy limitation,
together with impairment of cell wall biosynthesis, due to detachment of cytochrome c and
MurG, respectively, are the major factors contributing to MP196‐mediated bacterial cell
death.
E An antimicrobial peptide delocalizes peripheral membrane proteins
73

Figure 6. Model of the mechanism of action of MP196 and the bacterial response to
exposure. Model of changes to membrane structure (I). Membrane harboring the cell wall
biosynthesis machinery (II), the respiratory chain (III), and transport channels (IV). (a) Intact
B. subtilis membrane, (b) influence of MP196 on membrane architecture and membrane‐
associated components, (c) stress adaptation of B. subtilis to MP196‐mediated membrane
stress. MSC: mechanosensitive channel
74

This report represents the first detailed physiological study of the mechanism of action of a
short RW‐rich antimicrobial peptide. It provides insights into the biological action of peptides
beyond the lipid bilayer itself. The dissociation of peripheral membrane proteins in inner and
outer leaflets of the cytoplasmic membrane, reported here for MurG and cyochrome c,
respectively, have not previously been proposed as antibacterial mechanisms. They provide
new perspectives on the action of cationic antimicrobial peptides that are complementary to
the lipid‐focused pore formation and carpet mechanism models. Based on the data
presented here, we suggest that MP196 action follows the interfacial activity model
described by Wimley38, whereby at 50 percent growth rate reduction, phospholipid
perturbation predominates over membrane disruption.
While the main functional impact of MP196 is on proteins located in the membrane, its
molecular target is the lipid bilayer itself. This means that MP196 interferes with a broad
range of targets, complicating the development of bacterial resistance. DSC experiments
showed that the interaction of MP196 with the membrane critically depends on lipid
composition, offering the opportunity to design peptides selective for Gram‐positive and
Gram‐negative organisms.
This study also uncovered mechanisms by which B. subtilis counteracts peptide‐mediated
membrane stress (Fig. 6c). One survival strategy is to adapt membrane lipid composition
(Fig. 6c.I). Following exposure to MP196, the branched‐chain amino acid valine, which is
needed as precursor molecule for branched‐chain fatty acids, accumulates in the cytosol.
Changing the membrane lipid composition by modulating branched‐chain fatty acids may
interfere with the peptide‐membrane interaction. Previous experiments have suggested this:
in response to a ferrocene‐conjugated MP196 derivative, Corynebacterium glutamicum
altered the phospholipid headgroups by reducing negatively charged phosphatidylglycerol
lipids and increasing uncharged diaglycerol lipids. This impeded attachment of the peptide to
the membrane. In addition, cardiolipin levels were increased, giving the membrane a more
rigid structure, which further obstructed peptide integration39.
We also observed the induction of FloT, a protein controlling membrane fluidity that is
involved in orchestrating physiological processes in lipid microdomains40. Upregulation of
this protein underscores the importance of membrane properties and indicates that B.
subtilis alters membrane architecture to impair membrane‐associated physiological
processes.
E An antimicrobial peptide delocalizes peripheral membrane proteins
75

Additional bacterial adaptation strategies were observed (Fig. 6c.II‐IV). First, induction of the
dlt operon and the RacX protein suggest that there is adjustment of the bacterial cell wall by
enhancing wall teichoic acid D‐alanylation, to restrict access of the peptide to the
membrane. This is a common survival strategy, observed in daptomycin and vancomycin‐
resistant S. aureus strains41‐42. Second, the LiaRS cell wall biosynthesis stress response and
the σW‐mediated membrane stress response were strongly upregulated. The LiaH protein
forms ring‐like multimeric structures and binds to the inner surface of the cytoplasmic
membrane upon lipid II‐mediated cell wall biosynthesis inhibition43. It is thought that this
stabilizes the membrane. The σW‐controlled PspA protein is a homologue of LiaH, which
fulfils the same stabilizing function when the membrane is damaged44. Upregulation of both
PspA and LiaH reflects the duality of the MP196 mode of action: it impairs membrane
function and cell wall biosynthesis.
Adaptation of the membrane and cell wall structure and LiaH and PspA induction are known
bacterial strategies in response to cell envelope‐targeting antibiotics5,45. Another survival
strategy, namely the release of selected amino acids, is described here for the first time.
Glutamine/glutamate, asparagine/aspartate, arginine, proline, and lysine were all released
into the medium and biosynthesis of these amino acids was reflected in the proteome
response. Glutamate in particular is released in large quantities. It was already known as an
osmoprotectant that accumulates intracellularly upon heat and salt stress46‐47. We show that
B. subtilis releases glutamate in response to hypoosmotic stress conditions (Fig. 5c). Such
amino acid release is mediated by mechanosensitive channels, which measure membrane
tension and are activated by pressure on the lipid bilayer48. It is likely that membrane
structure alterations induced by MP196 activate mechanosensitive channels, resulting in the
amino acid release (Fig. 5c). The addition of exogenous glutamate to the growth medium
protected against MP196 (Fig. 5 d), suggesting that glutamate release is a deliberate survival
strategy rather than a nonspecific side effect. Protection is probably based on osmotic
stabilization, a suggestion supported by the finding that sodium chloride and potassium
chloride similarly protect against MP196 (Fig. 5e,f). Salts in the culture medium effectively
protect against peptide‐induced ion leakage, explaining why the MICs of membrane‐
targeting antimicrobial peptides show considerable medium dependency49. Release of
glutamate was also observed for other bacteriolytic peptides (Fig. 5c), suggesting that
osmoprotective glutamate release is a general reaction to bacteriolytic peptides.
E An antimicrobial peptide delocalizes peripheral membrane proteins
76

Amino acid analysis also revealed arginine accumulation in MP196‐treated cells. As
described for Bacillus licheniformis, arginine catabolism is negatively affected by low
glutamate levels50. Thus, arginine accumulation may be due to the low intracellular
glutamate concentration.
This report constitutes a comprehensive mechanistic study of the antibacterial mechanism
of action of a RW‐rich antimicrobial peptide in vivo. It shows that a peptide causes
dissociation of the essential proteins MurG and cytochrome c from the bacterial membrane,
providing new insights into how membrane‐targeting antibiotics inhibit cell wall biosynthesis
and respiration without interfering directly with components of these pathways. And it
confirms several bacterial adaptation strategies and describes a novel strategy that relies on
osmotic stabilization through release of selected amino acids into the medium.
METHODS
Experimental details and citations for all methods are provided as Supplementary
Information.
Radioactive precursor incorporation
The influence of MP196 on the major macromolecular synthesis routes was studied by
incorporation of radioactively labeled precursor molecules by Staphylococcus simulans as
described by Schneider et al.22.
Reporter gene activation
Damage to the main cellular macromolecules was monitored by promoter activation of
selected marker genes fused to the firefly luciferase reporter gene in the genetic background
of B. subtilis IS34. Serial two‐fold dilutions of each peptide (0.031 to 64 µg/ml) were
inoculated with bacterial cell suspensions and incubated at 37 °C for a time period
depending on the induction kinetics of the reporter strain. 2 mM luciferin was added and
flash luminescence measured.
Proteomics
Treatment of B. subtilis 168, metabolic labeling of newly synthesized proteins with [35S]‐L‐
methionine, and subsequent protein separation by 2D PAGE were performed as described
previously21. For gel‐free proteome analysis of membrane proteins, B. subtilis 168 was grown
aerobically at 37 °C in Belitzky minimal medium (BMM) supplemented with 14N‐ammonium
sulfate and 14N‐L‐tryptophane. Cells were stressed at an OD500 of 0.4 with 22.5 µg/mL MP196
for 65 minutes or left untreated as control. Untreated cultures grown on 15N‐ammonium
E An antimicrobial peptide delocalizes peripheral membrane proteins
77

sulfate and 15N‐L‐tryptophane were used for relative quantification. Mixing of cell extracts
prior to subcellular fractionation steps for relative quantification was carried out according
to Otto et al.51. The enriched membrane protein fraction was prepared according to the
workflow published by Eymann et al.52, omitting the n‐dodecyl‐β‐D‐maltoside treatment
step. The preparation of the integral membrane peptides was carried out as described by
Wolff et al.35. Sample preparation, mass spectrometric measurement, and subsequent data
analysis were carried out as described by Otto et al.51. The mass spectrometry proteomics
data have been deposited to the ProteomeXchange Consortium
(http://proteomecentral.proteomexchange.org) via the PRIDE partner repository53 with the
dataset identifier PXD000181.
Cell wall integrity and biosynthesis inhibition
Sample preparation and bright field microscopic inspection of B. subtilis 168 was performed
as described previously21. Inhibition of in vitro lipid II synthesis was performed using
Micrococcus flavus DSM 1790 membrane preparations as described by Schneider et al.22. For
in vitro lipid II binding, peptides were incubated with 2 nmol lipid II in molar ratios 1:1.
Reaction mixtures were incubated at 30 °C for 30 min and applied onto TLC plates (TLC Silica
Gel 60 F254, Merck, Darmstadt, Germany). Chromatography was performed in chloroform‐
methanol‐water‐ammonia (88:48:10:1) and stained with phosphomolybdic acid stain at
140°C (PMA). Accumulation of UPP‐MurNac pentapeptide was analyzed by HPLC as
described by Schmitt et al.54.
For detection of MurG in membrane fractions of B. subtilis 168, cells were grown in BMM
until early logarithmic growth phase, treated with antibiotics for 5 minutes, and harvested
by centrifugation. Membrane fractions were separated by differential centrifugation,
subjected to SDS PAGE, and blotted onto a nitrocellulose membrane. MurG was detected
with an E. coli MurG‐specific antibody produced in rabbit and a secondary goat anti‐rabbit
IgG‐HRP conjugate (BioRad, Berkeley, CA, USA).
Transmission electron microscopy
Cells were grown in BMM to an OD500 of 0.35. The main culture was then subdivided into 50
mL aliquots and subcultures were treated with the respective antibiotics for 15 minutes or
left untreated as control. Cells were harvested by centrifugation and washed twice in 100
mM Tris/1 mM EDTA, pH 7.5 and subsequently washed once in the same buffer without
EDTA. Preparation of bacterial samples for TEM was performed according to Santhana Raj et
E An antimicrobial peptide delocalizes peripheral membrane proteins
78

al.55 with some modifications (see Supplementary Methods). Sections were stained with 0.2
% lead citrate in 0.1 M NaOH for 3 seconds. Samples were examined at 23,000 ‐ 230,000
magnification with a Philips EM410 transmission electron microscope (Philips, Eindhoven,
Netherlands) equipped with a Gatan (Pleasanton, CA, USA) digital camera system at an
accelerating voltage of 80 kV.
Differential scanning calorimetry
Membrane integration was investigated by DSC‐based determination of peptide‐induced
changes in the thermotropic phase behaviour of liposomes consisting of pure DPPG, pure
DPPE, pure DPPC, and a mixture of 88% DPPG and 12% DPPE, respectively.
Permeability assays
In vivo pore formation in B. subtilis 168 was monitored using the LIVE/DEAD BacLight
bacterial viability kit (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s
instructions. Potassium release from B. megaterium was performed as described by
previously21. Ionomics experiments were performed with B. subtilis 168 either in Tris buffer
or in minimal medium under the same conditions as the proteomics experiments. Ion
concentrations were determined by inductively coupled plasma atomic emission
spectroscopy.
Membrane potential measurements
GFP‐MinD localization assays were performed with B. subtilis 1981 GFP‐MinD23 as described
previously21. Determination of membrane potential changes by use of DiSC35 was performed
as described by Andrés and Fierro26 in B. megaterium.
Influence on energy metabolism
ATP concentrations were determined from B. subtilis cytosolic extracts using the Perkin
Elmer ATPlite 1step assay kit (Waltham, MA, USA) according to the manufacturer’s
instructions. M. flavus inverted vesicles for H+‐ATPase and respiratory chain activity
measurements were prepared according to Burstein et al.56. Proton pumping into M. flavus
inverted vesicles by H+‐ATPase was measured in a microtiter plate assay based on the pH‐
sensitive probe acridine orange as described by Palmgren et al.57. Antibiotic Influence on the
bacterial electron transport chain was monitored by reduction of iodonitrotetrazolium
chloride using M. flavus inverted vesicles as described by Smith and McFeters58. Cytochrome
c localization was determined by Western Blot detection of cytochrome c on the same blots
used for MurG detection.
E An antimicrobial peptide delocalizes peripheral membrane proteins
79

Amino acid analysis
Amino acid analysis of B. subtilis 168 cellular extracts and culture supernatants was
performed by HPLC using the Acquity HPLC and AccQ•Tag Ultra system (Waters GmbH,
Milford, MA, USA) according to the manufacturer's instructions. Prior to HPLC, proteins were
removed by acetone precipitation.
Uptake and efflux of radioactively labeled glutamate was performed with S. simulans as
described by Ruhr and Sahl59. MIC values against B. subtilis 168 were determined with 5x105
cells/mL in BMM supplemented with rising glutamate concentrations. The lowest
concentration inhibiting visible growth was taken as MIC.
ACKNOWLEDGEMENTS
We thank our collaborating technicians Michaele Josten, University of Bonn, for purifying
nisin, Petra Düchting, Ruhr University Bochum (RUB) Plant Physiology for helping us establish
AES, Monika Bürger and Beate Menzel, RUB Physiological Chemistry, for their help with TEM,
and Stephanie Tautges, Kathrin Barlog, and Jale Stoutjesdijk, Medical Proteome Center, RUB,
for assisting with amino acid analysis. We further acknowledge Christoph H. R. Senges, RUB,
for his help with B. subtilis sample preparation. We thank Helmut Meyer, Medical Proteome
Center, RUB, for providing amino acid analysis technology, and Suzana K. Straus, Department
of Chemistry, University of British Columbia, for fruitful discussion. We further thank Klaus
Funke and Ellen Kloosterboer, RUB Neurophysiology, for providing the cytochrome c
antibody as well as Tanneke de Blaauwen, Swammerdam Institute for Life Sciences,
University of Amsterdam, and Franz Naberhaus, RUB Microbial Biology for providing the
MurG antibody. AiCuris, Wuppertal, is acknowledged for providing the B. subtilis reporter
strains. Further, we thank Erhard Bremer, Philipps University Marburg, for providing the B.
subtilis mechanosensitive channel quadruple mutant. We thank Richard Gallagher for
critically reading the manuscript and for his many helpful suggestions.
This work was financially supported by a grant from the German federal state of North
Rhine‐Westphalia and the European Union (N.M.N, H.B.O, H.G.S, J.E.B), and the RUB
Research Department Interfacial Systems Chemistry (N.M.N, J.E.B). CM was supported by
the Protein Unit for Research in Europe (PURE), a project of North Rhine‐Westfalia.
E An antimicrobial peptide delocalizes peripheral membrane proteins
80

Author contributions
M.W. designed and performed experiments, designed the mode of action model, and wrote
the paper; A.I.C., A.O., D.Z., and C.S. designed and performed experiments; H.B.A., M.P., and
N.M.N. designed and synthesized MP196 and derivatives; C.M., U.K., R.E., H.B.O., D.B., and
H.G.S. contributed tools, instrumentation and intellectual input; J.E.B. designed experiments
and wrote the paper.
Competing financial interest statement
The authors declare no competing financial interests.
REFERENCES
1. Giuliani, A., Pirri, G. & Nicoletto, S.F. Antimicrobial peptides: an overview of a
promising class of therapeutics. Cent. Eur. J. Biol. 2, 1‐33 (2007).
2. Riedl, S., Zweytick, D. & Lohner, K. Membrane‐active host defense peptides –
Challenges and perspectives fort the development of novel anticancer drugs. Chem. Phys.
Lipids 164, 766‐81 (2011).
3. Rathinakumar, R., Walkenhorst, W.F. & Wimley, W.C. Broad‐spectrum antimicrobial
peptides by rational combinatory design and high‐throughput screening: the importance of
interfacial activity. J. Am. Chem. Soc. 131, 7609‐17 (2009).
4. Sharma, R.K., Sundriyal, S., Wangoo, N., Tegge, W. & Jain, R. New antimicrobial
hexapeptides: synthesis, antimicrobial activities, cytotoxicity, and mechanistic studies.
ChemMedChem 5, 86‐95 (2010).
5. Yeaman, M.R. & Yount, N.Y. Mechanisms of antimicrobial peptide action and
resistance. Pharmacol. Rev. 55, 27‐55 (2003).
6. Zweytick, D. et al. Studies on lactoferricin‐derived Escherichia coli membrane‐active
peptides reveal differences in the mechanism of N‐acylated versus nonacylated peptides. J.
Biol. Chem. 286, 21266‐76 (2011).
7. Zweytick, D., Tumer, S., Blondelle, S.E. & Lohner, K. Membrane curvature stress and
antibacterial activity of lactoferricin derivatives. Biochem. Biophys. Res. Commun. 369, 395‐
400 (2008).
8. Chantson, J.T., Varga Falzacappa, M.V., Crovella, S. & Metzler‐Nolte, N. Solid‐phase
synthesis, characterization, and antibacterial activities of metallocene‐peptide
bioconjugates. ChemMedChem. 1,1268‐74 (2006).
9. Strøm, M.B. et al. The pharmacophore of short cationic antibacterial pepides. J. Med.
Chem. 46, 1567‐70 (2003).
10. Patra, M., Gasser, G. & Metzler‐Nolte, N. Small organometallic compounds as
antibacterial agents. Dalton Trans. 41, 6350‐8 (2012).
E An antimicrobial peptide delocalizes peripheral membrane proteins
81

11. Zelezetsky, I. et al. Controlled alteration and conformational stability of alpha‐helical
cell‐lytic peptides: effect on mode of action and cell specificity. Biochem. J. 390, 177‐88
(2005).
12. Albada, H.B. et al. Modulating the activity of short arginine‐tryptophan containing
antibacterial peptides with N‐terminal metallocenoyl groups. Beilstein J. Org. Chem. 8,
1765‐70 (2012).
13. Albada, H. B. et al. Tuning the activity of short Arg‐Trp antimicrobial peptides by
lipidation of C‐ or N‐terminal lysine side‐chain. ACS Med. Chem. Lett. DOI:
10.1021/ml300148v (2012).
14. Bandow, J. E. Proteomic approaches to antibiotic drug discovery. Curr. Protoc.
Microbiol. 1, 1F.2 (2005).
15. Wenzel, M. & Bandow, J.E. Proteomic signatures in antibiotic research. Proteomics
11, 3256‐68 (2011).
16. Chan., D.I., Prenner, E.J. & Vogel, H.J. Tryptophan‐ and arginine‐rich antimicrobial
peptides: structures and mechanisms of action. Biochim. Biophys. Acta 1758, 1184‐202
(2006).
17. Urban A. et al. Novel whole‐cell antibiotic biosensors for compound discovery. Appl.
Environ. Microbiol. 73, 6436‐6443 (2007).
18. Brötz‐Oesterhelt, H. et al. Dysregulation of bacterial proteolytic machinery by a new
class of antibiotics. Nat. Med. 11, 1082‐7 (2005).
19. Bandow, J.E., Brötz, H, Leichert, L.I., Labischinski, H. & Hecker, M. Proteomic
approach to understanding antibiotic action. Antimicrob. Agents Chemother. 47, 948–55
(2003).
20. Wenzel, M. et al. Proteomic signature of fatty acid biosynthesis inhibition available
for in vivo mechanism‐of‐action studies. Antimicrob. Agents Chemother. 55, 2590‐6 (2011).
21. Wenzel, M. et al. Proteomic response of Bacillus subtilis to lantibiotics reflects
differences in interaction with the cytoplasmic membrane. Antimicrob. Agents Chemother.
56, 5749‐57 (2012).
22. Schneider, T. et al. In vitro assembly of a complete, pentaglycine interpeptide bridge
containing cell wall precursor (lipid II‐Gly5) of Staphylococcus aureus. Mol. Microbiol. 53,
675–85 (2004).
23. Clejan, S., Krulwich, T.A., Mondrus, K.R. & Seto‐Young, D. Membrane lipid
composition of obligately and facultatively alkalophilic strains of Bacillus spp. J. Bacteriol.
168, 334‐40 (1986).
24. Brisebois, P.P., Arnold, A.A., Chabre, Y.M., Roy, R. & Marcotte, I. Comparative study
of the interaction of fullerenol nanoparticles with eukaryotic and bacterial model
membranes using solid‐state NMR and FTIR spectroscopy. Eur. Biophys. J. 41, 535‐44
(2012).
25. Strahl, H. & Hamoen, L.W. Membrane potential is important for bacterial cell division.
Proc. Natl. Acad. Sci. USA 27, 12281‐6 (2010).
26. Andrés, M.T. & Fierro, J.F. Antimicrobial mechanism of action of transferrins:
Selective inhibition of H+‐ATPase. Antimicrob. Agents Chemother. 54, 4335–42 (2010).
E An antimicrobial peptide delocalizes peripheral membrane proteins
82

27. Mogi, T., Ui, H., Shiomi, K., Omura, S. & Kita K. Gramicidin S identified as a potent
inhibitor for cytochrome bd‐type quinol oxidase. FEBS Lett. 291, 157‐61 (2008).
28. Vonck, J. & Schäfer, E. Supramolecular organization of protein complexes in the
mitochondrial inner membrane. Biochim. Biophys. Acta 1793, 117‐24 (2009).
29. Hoffmann, T. Boiangu, C., Moses, S. & Bremer, E. Responses of Bacillus subtilis to
hypotonic challenges: physiological contributions of mechanosensitive channels to cellular
survival. Appl. Environ. Microbiol. 74, 2454‐60 (2008).
30. Sitaram, N. and Nagaraj, R. Interaction of antimicrobial peptides with biological and
model membranes: structural and charge requirements for activity. Biochim. Biophys. Acta
15, 29‐54 (1999).
31. Fuertes, G., Giménez, D., Esteban‐Martín, S., Sánchez‐Muñoz, O.L. & Salgado, J. A
lipocentric view of peptide‐induced pores. Eur. Biophys. J. 40, 399‐415 (2011).
32. Wecke, T. et al. Daptomycin versus Friulimicn B: in‐depth profiling of Bacillus subtilis
cell envelope stress responses. Antimicrob. Agents Chemother. 53, 1619‐23 (2009).
33. Pogliano, J., Pogliano, N. & Silverman, J.A. Daptomycin‐mediated reorganization of
membrane architecture causes mislocalization of essential cell division proteins. J.
Bacteriol. 194, 4494‐504 (2012).
34. Bayer, A.S., Schneider, T. & Sahl, H.G. Mechanisms of daptomycin resistance in
Staphylococcus aureus: role of the cell membrane and cell wall. Ann. N. Y. Acad. Sci. 1277,
139‐58 (2013).
35. Bengtsson, J., Tjalsma, H., Rivolta, C. & Hederstedt, L. Subunit II of Bacillus subtilis
cytochrome c oxidase is a lipoprotein. J. Bacteriol. 181, 685‐8 (1999).
36. Bengtsson, J., Rivolta, C., Hederstedt, L. & Karamata, D. Bacillus subtilis contains two
small c‐type cytochromes with homologous heme domains but different types of
membrane anchors. J. Biol. Chem. 274, 26179‐84 (1999).
37. Ha, S., Walker, D., Shi, Y., Walker, S. The 1.9 Å crystal structure of Escherichia coli
MurG, a membrane‐associated glycosyltransferase involved in peptidoglycan biosynthesis.
Protein Sci. 9, 1045‐52 (2000).
38. Wimley, W.C. Describing the mechanism of antimicrobial peptide action with the
interfacial activity model. ACS Chem. Biol. 5, 905‐17 (2010).
39. Fränzel, B. et al. Corynebacterium glutamicum exhibits a membrane‐related response
to a small ferrocene‐conjugated antimicrobial peptide. J. Biol. Iorg. Chem. 15, 1293‐303
(2010).
40. López, D. & Kolter, R. Functional microdomains in bacterial membranes. Genes Dev.
24,1893‐902 (2012).
41. Bertsche, U. et al. Correlation of daptomycin resistance in a clinical Staphylococcus
aureus strain with increased cell wall teichoic acid production and D‐alanylation.
Antimicrob. Agents Chemother. 55, 3922‐8 (2011).
42. Rose, W.E., Fallon, M., Moran, J.J. & Vanderloo, J.P. Vancomycin tolerance in
methicillin‐resistant Staphylococcus aureus: influence of vancomycin, daptomycin, and
telavancin on differential resistance gene expression. Antimicrob. Agents Chemother. 56,
4422‐7 (2012).
E An antimicrobial peptide delocalizes peripheral membrane proteins
83

43. Wolf, D. et al. In‐depth profiling of LiaR response in Bacillus subtilis. J. Bacteriol. 192,
4680‐93 (2010).
44. Kobayashi, R., Suzuki, T. & Yoshida, M. Escherichia coli phage‐shock protein A (PspA)
binds to membrane phospholipids and repairs proton leakage of the damaged membranes.
Mol. Microbiol. 66, 100‐9 (2007).
45. Kingston, A.W., Subramanian, C., Rock, C.O. & Helmann, J.D. A σW‐dependent stress
response in Bacillus subtilis that reduces membrane fluidity. Mol. Microbiol. 81, 69‐79
(2011).
46. Hoffmann, T. & Bremer, E. Protection of Bacillus subtilis against cold stress via
compatible solute acquisition. J. Bacteriol. 193:1552‐62 (2011).
47. Kempf, B., & Bremer, E. Uptake and synthesis of compatible solutes as microbial
stress response to high‐osmolality environments. Arch. Microbiol. 170, 319‐30 (1998).
48. Sukharev, S. Purification of the small mechanosensitive channel of Escherichia coli
(MscS): the subunit structure, conduction, and gating characteristics in liposomes. Biophys.
J. 83, 290‐8 (2002).
49. Schwab, U., Gilligan, P., Jaynes, J. & Henke, D. In vitro activities of designed
antimicrobial peptides against multidrug‐resistant cystic fibrosis pathogens. Antimicrob.
Agents Chemother. 43, 1435‐40 (1999).
50. Laishley, E.J. & Bernlohr, R.W. Regulation of arginine and proline catabolism in
Bacillus licheniformis. J. Bacteriol. 96, 322‐9 (1968).
51. Otto, A., et al. Systems‐wide temporal proteomic profiling in glucose‐starved Bacillus
subtilis. Nat Commun. 1, 137. doi: 10.1038/ncomms1137 (2010).
52. Eymann, C. et al. A comprehensive proteome map of growing Bacillus subtilis cells.
Proteomics 4, 2849–76 (2004).
53. Vizcaino, J.A. et al. The Proteomics Identifications (PRIDE) database and associated
tools: status in 2013. Nucleic Acids Res. 41, D1063‐9 (2013).
54. Schmitt, P. et al. Insight into invertebrate defensin mechanism of action: oyster
defensins inhibit peptidoglycan biosynthesis by binding to lipid II. J. Biol. Chem. 285, 29208‐
16 (2010).
55. Santhana Raj, L. et al. Rapid Method for Transmission Electron Microscopy Study of
Staphylococcus aureus ATCC 25923. Annals of Microscopy 7, 102‐8 (2007).
56. Burstein, C., Tiankova, L. & Kepes, A. Respiratory control in Escherichia coli K 12. Eur.
J. Biochem. 94, 387‐92 (1979).
57. Palmgren, M.G, Sommarin, M., Ulvskov, P. & Larsson, C. Effect of detergents on the
H(+)‐ATPase activity of inside‐out and right‐side‐out plant plasma membrane vesicles.
Biochim. Biophys. Acta 1021, 133‐40 (1990).
58. Smith, J.J. & McFeters, G.A. Mechanisms of INT (2‐(4‐iodophenyl)‐3‐(4‐nitrophenyl)‐
5‐phenyl tetrazolium chloride), and CTC (5‐cyano‐2,3‐ditolyl tetrazolium chloride) reduction
in Escherichia coli K‐12, J. Microbiol. Methods 29, 161‐75 (1997).
59. Ruhr, E. & Sahl, H.G. Mode of action of the peptide antibiotic nisin and influence on
the membrane potential of whole cells and on cytoplasmic and artificial membrane
vesicles. Antimicrob. Agents Chemother. 27, 841‐5 (1985).
E An antimicrobial peptide delocalizes peripheral membrane proteins
84

Supplementary Information
Content
i) Experimental Details
ii) Supplementary Tables
iii) Supplementary Figures
iv) References
i) Experimental details
Antibiotics
Antibiotic stock solutions of 10 mg/ml were prepared in sterile DMSO (MP196, D‐MP196,
valinomycin, gramicidin S, vancomycin) or 0.01 M HCl (nisin). MP276, L‐ and D‐MP196 were
synthesized by solid‐phase synthesis as described previously1. Nisin was purified according to
Bonelli et al.2. Valinomycin and vancomycin were purchased from Sigma‐Aldrich (St Louis, MO,
USA). Gramicidin S was synthesized according to Wadwhani et al.3. Lactoferrin, rotenone, and
antimycin A were purchased from Sigma‐Aldrich. Stock solutions were prepared in water
(lactoferrin, 1 mM), acetone (rotenone, 100 mM), or ethanol (antimycin A, 10 mM). Sublethal
antibiotic concentrations applied in the single experiments were adjusted to the respective
bacterial strains and experimental conditions aiming at adequate bacterial growth inhibition
(reduction of the growth rate by 50‐70%) without killing the cells or abolishing their metabolic
activity completely (see Supplementary Figure 3).
Radioactive precursor incorporation
Influence of MP196 on major metabolic pathways was studied by incorporation of radioactively
labeled precursor molecules into Staphylococcus simulans 22 as described by Schneider et al.4.
[14C]‐thymidine was used for monitoring DNA, 5‐[3H]‐uridine for RNA, L‐[14C]‐isoleucine for
E An antimicrobial peptide delocalizes peripheral membrane proteins
85

protein and [3H]‐glucosamine‐hydrochloride for cell wall biosynthesis inhibition, respectively.
Cells were grown in half‐concentrated Mueller Hinton broth (henceforth MH, Oxoid,
Basingstoke, United Kingdom) at 37 °C until reaching an optical density at 600 nm (OD600) of 0.5
and diluted to an OD600 of 0.1 to 0.2. Newly synthesized DNA, RNA, protein or peptidoglycane
was radioactively labeled by addition of the respective precursors to a final concentration of
0.15 µCi/mL [14C]‐thymidine, 1 μCi/mL 5‐[3H]‐uridine, 0.15 µCi/mL L‐[14C]‐isoleucine, and 1
µCi/mL [3H]‐glucosamine‐hydrochloride. After 15 ([14C]‐thymidine, 5‐[3H]‐uridine, L‐[14C]‐
isoleucine) or 30 minutes ([3H]‐glucosamine‐hydrochloride) of radioactive incorporation,
subcultures of the differentially labeled samples were treated with 2 µg/ml MP196, 0.7 µg/ml
ciprofloxacin, 0.07 µg/ml rifampicin, 0.7 µg/ml tetracycline, and 0.7 µg/ml vancomycin,
respectively, or left untreated as negative controls. Samples of 200 μl were collected at 0, 10,
20, and 30 minutes after antibiotic addition, mixed with ice‐cold trichloroacetic acid (TCA)
precipitation buffer (10% TCA, 1M NaCl), incubated on ice for 30 minutes, and filtered through
glass microfibre filters (Whatman, GE Healthcare, Uppsala, Sweden). Filters were subsequently
washed with 5 ml washing buffer (2.5% TCA, 1M NaCl), dried, and transferred to counting vials.
5 ml of scintillation fluid (Filtersafe, Zinsser, Frankfurt, Germany) were added and radioactivity
was measured in counts per minute (CPM) using a TriCarb 3110TR liquid scintillation analyzer
(Perkin Elmer, Waltham, MA, USA).
Reporter gene fusions
For quantitative expression analysis of selected marker genes, five bacterial reporter strains
were used, carrying the promotor of either yorB, bmrC (synonym yheI), helD (synonym yvgS),
ypuA and liaI (synonym yvqI) fused to the firefly luciferase reporter gene in the genetic
background of B. subtilis IS345. Reporter strains were cultured overnight in lysogeny broth with
5 µg/ml erythromycin (resistance marker on the luciferase plasmid) at 37 °C and 200 rpm.
Cultures were diluted to OD600 of 0.05 in 20 ml of either Belitzky minimal medium (BMM)6
(bmrC strain) or lysogeny broth (all other strains) with 5 µg/ml erythromycin, grown to an OD600
of 0.4 (bmrC strain) or 0.9 (all other strains) and diluted to an OD600 of 0.02. Serial twofold
dilutions of MP196 (0.031 to 64 µg/ml) in 60 µl lysogeny broth or BMM were prepared in white
96‐well flat bottom polystyrol microtiter plates (Greiner, Frickenhusen, Germany) and were
inoculated with 60 µl of the adjusted bacterial suspension. Plates were incubated at 37 °C for a
E An antimicrobial peptide delocalizes peripheral membrane proteins
86

predetermined time period depending on the induction kinetics of the reporter strain: 1 hour
for the liaI and ypuA strains, 1.5 hours for the helD strain, 3.5 hours for the yorB strain and 4
hours for the bmrC strain. Then 60 µl citrate buffer (0.1 M, pH 5) containing 2 mM luciferin
(Serva, Heidelberg, Germany) was added and flash luminescence was measured using a
microtiter plate reader (infinite M200, Tecan, Männedorf, Switzerland).
Proteomics
Bacterial strains and growth conditions
Bacillus subtilis 168 (trpC2)7 was grown at 37 °C under steady agitation in BMM. Minimal
inhibitory concentrations (MICs) were determined under proteomics conditions as described
previously8. In short, two ml of antibiotic‐supplemented BMM were inoculated with 5x105
bacteria per ml and incubated at 37 °C under steady agitation for 18 hours. The MIC was defined
as the lowest antibiotic concentration inhibiting visible growth. In growth experiments, bacterial
cultures were treated with different MIC‐based antibiotic concentrations after growing to an
OD500 of 0.35. For proteomic experiments those antibiotic concentrations were chosen that led
to a reduced growth rate compared to the untreated control.
Preparation of cytoplasmic [35S]‐L‐methionine‐labeled protein fractions
Radioactive labeling was performed as described previously8. In short, 5 ml of a bacterial culture
in early exponential growth phase were exposed to 22.5 µg/ml MP196 or 50 µg/ml D‐MP196,
respectively, or left untreated as control. After 10 minutes of antibiotic stress cells were pulse‐
labeled with [35S]‐L‐methionine for additional 5 minutes. Radioactive incorporation was stopped
by inhibition of protein biosynthesis by chloramphenicol and an excess of cold methionine. Cells
were harvested by centrifugation, washed three times in Tris buffer and disrupted by
ultrasonication.
2D‐PAGE
2D gels were prepared as described previously8. In short, 55 µg of protein for analytical and 300
µg for preparative gels were loaded onto 24 cm immobilized pH gradient (IPG) strips pH 4‐7 (GE
Healthcare) by passive rehydration for 18 hours. Proteins were separated in a first dimension by
isoelectric focusing and in a second dimension by SDS‐PAGE using 12.5% acrylamide gels.
Analytical gel images were analyzed as described by Bandow et al.9 using Decodon Delta 2D 4.1
image analysis software (Decodon, Greifswald, Germany). Proteins found to be induced more
E An antimicrobial peptide delocalizes peripheral membrane proteins
87

than two‐fold in three independent biological replicates were defined as marker proteins.
Protein spots were manually excised from preparative 2D gels and transferred into 96 well
microtiter plates. Tryptic digest with subsequent spotting on a MALDI target was carried out
automatically with the Ettan Spot Handling Workstation (Amersham Biosciences, Uppsala,
Sweden) as described by Eymann et al.10. MALDI‐ToF as well as MALDI‐ToF‐ToF measurements
were carried out on a 4800 MALDI‐ToF/ToF Analyzer (Applied Biosystems, Foster City, CA, USA)
as described previously8. For the five most intense peaks in the MS spectra, MS/MS data was
obtained. Peak lists were generated using the “peak to mascot” script of the 4000 Explorer
Software V3.5.3 and searched against an in house strain‐specific B. subtilis 168 sequence
database with 4767 entries downloaded from UniProt (May 2nd, 2008) using the Mascot search
engine Version 2.1.04 (Matrix Science Ltd, London, UK). Search parameters allowed for one
missed cleavage. Carbamidomethylation of cysteine was defined as fixed modification, oxidation
of methionine as variable modification. Mass tolerance of precursor ions was set to 50 ppm, and
known contaminants were excluded. Protein scores, ‐10*Log(P), where P is the probability that
the observed match is a random event, are derived from ion scores as a non‐probabilistic basis
for ranking protein hits. Protein scores higher than 49 were considered significant (significance
threshold p<0.05).
Protein spots, which could not be identified by MALDI‐ToF/ToF (indicated by asterisks in the
identification table) were identified using a Synapt G2S high definition mass spectrometer
equipped with a lock spray source for electrospray ionization and a ToF detector (Waters,
Milford, MA, USA). Manually excised protein spots were destained with 20 mM
ammoniumbicarbonate/30% acetonitrile, dried completely, and subsequently tryptically
digested (6.25 ng/µL, Promega, Fitchburg, WI, USA). Peptides were eluted into ultrapure water
by 15 minutes of ultrasonication. Eluates were loaded on a trap column (C18, pore size 100 Å,
particle diameter 5 µm, inner diameter 180 µm, length 20 mm) and were then eluted using
gradients of acetonitrile with 0.1% formic acid (350 µL/min, linear gradient 2‐60% in 40 min)
from an analytical column at 50 °C (C18, pore size 130 Å, particle diameter 1.7 µm, inner
diameter 75 µm, length 150 mm) to be subjected to mass spectrometry. Spectra were recorded
in positive resolution mode over a mass range of 50 to 1800 m/z with 1 s/scan. The following
parameters were used for the NanoLockSpray source: capillary voltage, 3.3 kV; sampling cone
E An antimicrobial peptide delocalizes peripheral membrane proteins
88

voltage, 30 V; source temperature, 80 °C; desolvation temperature, 200 °C; cone gas flow; 50
L/h; desolvation gas flow, 600 L/h. [Glu1]‐Fibrinopeptide B serving as lock mass analyte was fed
through the lock spray channel (lock mass capillary voltage, 3.2 V). Analysis of the spectra was
performed using MassLynx V4.1 SCN813.
Preparation of 15N/14N‐labeled membrane proteome fractions
For gel‐free proteome analysis of membrane proteins B. subtilis 168 was grown aerobically at 37
°C in BMM supplemented with either 15N‐ammonium sulfate/15N‐ L‐ tryptophan (0.078 mM, 98
atom % excess, Cambridge Isotope Laboratories, Andover, USA) or 14N‐ ammonium sulfate and
14N‐ L‐ tryptophane. Cells were harvested by centrifugation either with antibiotic stress imposed
at an optical density at 500 nm of 0.4 for 65 minutes or as a control directly before setting of the
stress. Mixing of cell extracts prior to subcellular fractionation steps for relative quantification
was carried out according to Otto et al.11. The enriched membrane protein fraction was
prepared according to the workflow published in Eymann et al.10 omitting the n‐dodecyl‐β‐D‐
maltoside treatment step. Preparation of integral membrane peptides (membrane shaving
fraction) was carried out as described by Wolff et al.12.
ESI‐MS measurement
Sample preparation, mass spectrometric measurement for both the enriched membrane
protein and membrane shaving fractions, and subsequent data analysis (protein identification
and relative quantitation) were carried out as described by Otto et al.11. Logarithmic peak
intensities (ratio to the 15N‐labelled standard culture) were normalized to the median of the
respective datasets. The cutoff for significant protein upregulation was set to a lognorm change
of 0.35, representing the maximal biological variation between different control cultures in
>99% of the data points.
Light microscopy
Sample preparation and microscopic inspection of the cell shape was performed as described
previously13. Briefly, B. subtilis 168 was grown in BMM until early exponential growth phase and
subsequently treated with 22.5 µg/ml MP196, 0.75 µg/mL nisin, or left untreated as control.
After 15 minutes of antibiotic stress cells were fixed in a 1:3 mixture of acetic acid and
methanol, immobilized in low melting agarose, and subjected to bright field microscopy.
In vitro lipid II synthesis
E An antimicrobial peptide delocalizes peripheral membrane proteins
89

In vitro lipid II synthesis was performed using Micrococcus flavus DSM 1790 membrane
preparations as described by Schneider et al.4, adding [14C]‐UDP‐N‐acetyl‐D‐glucosamine
(GlcNAc) to label newly synthesized lipid II. Membranes were isolated from lysozyme‐treated
cells by centrifugation at 40,000 x g, washed twice in washing buffer (50 mM Tris‐HCl, 10 mM
MgCl2, pH 7.5) and frozen in liquid nitrogen. In vitro lipid II synthesis was performed in a total
volume of 75 µL using 400 µg of membrane protein, 5 nmol C55‐P, 50 nmol UDP‐N‐
acetylmuramylpentapeptide (UDP‐MurNAc‐PP, purified according to Kohlrausch and Höltje14),
and 50 nmol [14C]‐UDP‐GlcNAc in reaction buffer (60 mM Tris‐HCl, 5 mM MgCl2, 0.5% (w/v)
triton X‐100, pH 8.0). Peptides were added in a 2:1 molar ratio to C55‐P and incubated with the
reaction mixture for 1 hour at 37 °C. Subsequently, lipid extraction was performed by addition
of 75 µL 2:1 n‐butanol/6 M pyridine‐acetate, pH 4.2. Lipids were separated by thin layer
chromatography using 60F254 silica plates (Merck, Darmstadt, Germany) and
chloroform/methanol/water/ammonia (88:48:10:1) as solvent as described by Rick et al.15.
Radiolabeled spots were visualized using a Storm 820 Phosphor Imager (Amersham Biosciences,
Uppsala, Sweden). Image analysis was performed using the ImageQuant TL v2005 software
(Nonlinear Dynamics Ltd, Newcastle Upon Tyne, UK).
Lipid II‐binding
For in vitro lipid II binding of peptides were incubated with 2 nmol lipid II in molar ratios 1:1.
Reaction mixtures were incubated at 30 °C for 30 min and applied onto TLC plates (TLC Silica Gel
60 F254, Merck, Darmstadt, Germany). Chromatography was performed in chloroform‐methanol‐
water‐ammonia (88:48:10:1)15 and stained with phosphomolybdic acid stain and 140°C (PMA).
Accumulation of UDP‐MurNAC‐pentapeptide
Accumulation of UPP‐MurNac was analysed by HPLC as described by Schmitt et al.16.
S. simulans 22 was grown in MH broth to an OD600 of 0.5. 130 µglmL chloramphenicol were
added in order to prevent induction of autolysis and de novo synthesis of enzymes hydrolyzing
the nucleotide‐activated sugars interfering with determination of the soluble precursor17 (Dai
and Ishiguro, 1988). After 15 min, 5 µg/mL MP196 and 0.7 µg/mL vancomycin were added. After
30 min of incubation with the respective peptides cells were harvested and boiled in water.
Insoluble components were removed by centrifugation at 13,000 × g for 5 min. Supernatants
were adjusted to pH 2 with H3PO4, filtered through cellulose acetate filters (pore size 0.2 μm)
E An antimicrobial peptide delocalizes peripheral membrane proteins
90

and analyzed by RP‐HPLC in 50 mM sodium phosphate buffer, pH 5.2, developed in isocratic
mode over 30 min at a flow rate 1 ml/min on a Nucleosil 100‐C18 column (Schambeck SFD
GmbH, Bad Honnef, Germany).
SDS‐PAGE and Western blot
B. subtilis 168 was grown in BMM until early logarithmic growth phase and subsequently
treated with 22.5 µg/mL MP196, 1 µg/mL gramicidin S, or left untreated as control. Cells were
harvested by centrifugation, washed three times in washing buffer (100 mM tris/1 mM EDTA,
pH 7.5), and resuspended in disruption buffer (10 mM tris, pH 7.5). Cells were disrupted by
ultrasonication, cell debris was removed by centrifugation, and cell extracts were subjected to
ultracentrifugation at 150,000 x g for 4 h. The resulting membrane pellet was dissolved in
washing buffer. Protein concentrations were determined by a Bradford‐based assay (Roti‐
Nanoquant, Carl Roth, Karlsruhe, Germany) according to the manufacturer’s instructions. 30 µg
of protein were loaded onto a 15% SDS polyacrylamide gel and proteins were separated at 120
V. Proteins were then blotted onto a nitrocellulose membrane (Hybond, GE Healthcare,
Uppsala, Sweden) at 250 mA and 25 V per blot for 2 h. Membranes were stained with Ponceau S
prior to antibody detection to control equal sample loading. Subsequently, membranes were
destained in TBST (100 mM tris, 150 mM NaCl, 0.1% Tween 20, pH 7.5), blocked with 3% non‐fat
dried milk powder in TBST for 2 h, and subsequently incubated with a rabbit anti‐(E.coli‐)MurG‐
His antibody18 in a 1:10,000 dilution in TBST with 1% milk powder for 18 h. Membranes were
then washed three times in TBST with 3% milk powder, incubated with a goat anti‐rabbit IgG‐
HRP secondary antibody (Bio‐Rad, Berkeley, CA, USA) in a 1:3,000 dilution in TBST with 3% milk
powder for 1 h, washed three times with TBST and were then covered with chemiluminescence
reagent (Luminata™ Forte Western HRP Substrate, Merck Millipore, Darmstadt, Germanay).
Signals were detected with a FluorChem® Imaging Workstation (Alpha Innotech, now part of
Proteinsimple, Santa Clara, CA, USA). Quantification of band intensity was performed with the
ImageQuant TL 1D analysis software (GE Healthcare, Uppsala, Sweden). The same membranes
were analogously used for cytochrome c detection using a mouse anti‐(pigeon‐)cytochrome c
antibody (7H8.2C12, abcam, Cambridge, UK) at a final concentration of 2.5 µg/mL and a goat
anti‐mouse IgG‐HRP secondary antibody (Bio‐Rad, Berkeley, CA, USA).
Transmission electron microscopy
E An antimicrobial peptide delocalizes peripheral membrane proteins
91

Cells were grown in BMM to an OD500 of 0.35. The main culture was then subdivided into 50 mL
aliquots and subcultures were treated with the respective antibiotics for 15 minutes or left
untreated as control. Cells were harvested by centrifugation and washed twice in 100 mM Tris/1
mM EDTA, pH 7.5 and subsequently washed once in the same buffer without EDTA. Preparation
of bacterial samples for TEM was performed according to Santhana Raj et al.19 with some
modifications. Cells were fixed in 2% glutaraldehyde for 20 min, washed twice with double‐
distilled water, and subsequently incubated with 2% uranylacetate for 5 min Samples were then
washed twice with double‐distilled water and stained with 2% osmium tetroxide (OT). Cells
were centrifuged down and excess of OT solution was discarded. Samples were then
dehydrated by an incubation series with 50%, 70%, 90%, and 100% acetone for 5 min each. Cells
were subsequently incubated with 1:1 acetone/epoxy resin for 15 min and then embedded in
pure epoxy resin. For preparation of the epoxy resin, solution A (38% Epon 812/62% dodecyl
succinic anhydride) was mixed with solution B (47% epon 812/53% methylnadic anhydride) at a
ratio of 3:7. To this mixture tris‐2,3,6‐(dimethylaminomethyl)phenol (DMP30) was added to a
final concentration of 1.5% to accelerate polymerization. Polymerization was performed at 75 °C
for 18 hours. Blocks were trimmed, cut to 50 nm ultrathin sections and mounted on Formvar‐
coated (R1201, Plano, Wetzlar, Germany), 75 mesh thin bar copper grids (Stork Veco, Eerbeek,
Nertherlands). Samples were either stained with 0.2 % lead citrate in 0.1 M NaOH for 3 seconds
or remained unstained. Samples were examined at 23,000 ‐ 230,000 magnification with a Philips
EM410 transmission electron microscope (Philips, Eindhoven, Netherlands) equipped with a
Gatan (Pleasanton, CA, USA) digital camera system at an accelerating voltage of 80 kV.
Differential scanning calorimetry (DSC)
1,2‐Dipalmitoyl‐sn‐glycero‐3‐phosphoglycerol (Na‐salt) (DPPG), 1,2‐Dipalmitoyl‐sn‐glycero‐3‐
phosphoethanolamine (DPPE), and 1,2‐Dipalmitoyl‐sn‐glycero‐3‐phosphocholine were
purchased from Avanti Polar Lipids Inc. (Alabaster, AL, USA). Peptides were dissolved in
phosphate buffered saline (PBS, 20 mM NaPi, 130 mM NaCl, pH 7.4) at a concentration of 3
mg/ml before each experiment. Aqueous dispersions of lipids of 0.1% (w/w) in PBS buffer were
prepared before measurement in the presence (lipid‐to‐peptide molar ratio of 25:1 and 120:1)
and absence of peptides. The respective liposomes were always prepared 10‐15 °C above the
phase transition temperature by vigorous mixing at certain time points. For the preparation of
E An antimicrobial peptide delocalizes peripheral membrane proteins
92

DPPG liposomes we followed a protocol described by Zweytick et al.20. Lipid films of DPPE and
DPPG/DPPE 88:12 (w/w) were hydrated at 70 °C for 2 hours. For the mixture a freeze thaw
protocol was used to increase homogenous mixing. DSC experiments were performed with a
differential scanning calorimeter (VP‐DSC) from MicroCal, Inc. (Northampton, MA, USA). Heating
scans were performed at a scan rate of 30 °C/hour with a final temperature approximately 10 °C
above the main transition temperature (Tm) and cooling scans at the same scan rate with a final
temperature about 20 °C below Tm. The heating/cooling cycle was repeated twice, pre‐scan
thermostating was allowed for 15 minutes for the heating scans and 1 minute for the cooling
scans. Enthalpies were calculated by integrating the peak areas after normalization to
phospholipid concentration and baseline adjustment using the MicroCal Origin software (VP‐
DSC version).
Live/Dead staining
Pore formation was monitored using the LIVE/DEAD BacLight bacterial viability kit (Invitrogen,
Carlsbad, CA, USA) following the manufacturer’s instructions. B. subtilis 168 was grown at 37 °C
in BMM under steady agitation until early exponential phase. The main culture was split and
aliquots were treated with 22.5 µg/ml MP196, 0.75 µg/ml nisin, 1 µg/ml gramicidin S 10 µg/mL
valinomycin, or left untreated as control. After 15 minutes of antibiotic stress, 2 µl of a 1:1
mixture of the green and red‐fluorescing dyes were added per ml of culture and incubated for
15 minutes in the dark under steady agitation. Cells were washed with 100 mM Tris/1mM EDTA,
pH 7.5 and resuspended in the same buffer. 5 µl of the cell suspension were imaged without
fixation or immobilization in fluorescent mode as described before13. Single channel pictures
were combined using the Cell* software (Olympus, Hamburg, Germany). Background correction
was performed applying the same constant (25) for all images.
Potassium release
Potassium efflux experiments were performed as described previously13 with minor
modifications. Briefly, B. megaterium ATCC 13632 was grown in half MH to an OD500 of 1.0 to
1.5, washed with 50 ml cold choline buffer, and resuspended in the same buffer to an OD500 of
30. For each measurement, cells were diluted in choline buffer (25 °C) to an OD500 of about 3.
Nisin was applied at 3,35 µg/ml (1 µM). MP196 at 125 µg/ml (80 µM), which is equivalent to 50x
MIC. No effect was seen with lower MP196 concentrations (2x, 10x MIC). A control culture was
E An antimicrobial peptide delocalizes peripheral membrane proteins
93

left untreated. Potassium efflux was monitored using a pH‐meter pH 213 (Hanna Instruments,
Kehl am Rhein, Germany) with an MI‐442 potassium electrode and an MI‐409F reference
electrode. Before each experiment, the electrodes were calibrated with standard solutions
containing 0.01, 0.1, or 1 mM KCl. Calculations of potassium efflux in percent were performed
according to Orlov et al.21. Antibiotic‐induced leakage was monitored for 5 minutes with values
taken every 10 seconds and expressed relatively to the total amount of potassium release
induced by nisin.
Ionomics
For ionomics only metal‐free plastic ware and ultrapure water (Bernd Kraft, Duisburg, Germany)
were used. All centrifugation steps were performed for 2 minutes to reduce sample handling
time. B. subtilis 168 was grown in BMM until early exponential growth phase. For determination
of ion concentrations of cells stressed in medium, subcultures were directly treated with 22.5
µg/ml MP196, 1 µg/ml gramicidin S, and 10 µg/mL valinomycin, respectively, or left untreated
as a negative control. After 15 minutes of antibiotic treatment (proteomic conditions), cells
were harvested by centrifugation, washed twice in 100 mM Tris/1mM EDTA, pH 7.5 and washed
once in 100 mM Tris, pH 7.5. For determination of ion concentrations of cells stressed in buffer,
exponentially growing cultures were harvested by centrifugation, washed twice in 100 mM
Tris/1 mM EDTA, pH 7.5, washed once in 100 mM Tris, pH 7.5, and subsequently resuspended in
the same buffer. The OD500 was readjusted to 0.4 and cells were stressed as described above.
After 5 minutes (due to rapid ion release kinetics) of antibiotic stress cells were harvested by
centrifugation without further washing steps.
Cell pellets were completely dissolved in 2.5 ml 65% nitric acid (Bernd Kraft, Duisburg, Germany)
and incubated at 80 °C for 16 hours. Dissolved samples were filled up with ultrapure water
(Bernd Kraft, Duisburg, Germany) to 10 ml. Elemental concentrations were determined by
inductively coupled plasma atomic emission spectroscopy using an iCAP* 6300 Duo View ICP
Spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). Liquid calibration standards from
10 µg/L to 10 mg/L of each element of interest (Bernd Kraft, Duisburg, Germany) were run
before each series of measurement and selected standards were additionally run every 20
samples as a quality control. Resulting element concentrations were converted into intracellular
ion concentrations based on calculation of the cytosolic volume of B. subtilis. This volume was
E An antimicrobial peptide delocalizes peripheral membrane proteins
94

taken as 3.09x10‐9 µl based on average rod size and B. subtilis cell wall and membrane thickness
determined by cryo‐electron microscopy by Matias and Beveridge22‐23.
GFP‐MinD localization
Localization assays were performed with B. subtilis 1981 GFP‐MinD24 as described previously13.
In short, cells were grown over night in BMM and then inoculated in modified BMM containing
xylose instead of glucose to induce GFP‐MinD expression. Cultures were grown until early
exponential growth phase, and subsequently stressed with 22.5 µg/mL MP196, 1 µg/mL
gramicidin S or left untreated. After 15 minutes of antibiotic exposure, cells were imaged in
fluorescent mode without fixation or immobilization.
DiSC35‐based membrane potential measurement
Determination of membrane potential changes was performed as described by Andrés and
Fierro25 using the membrane potential‐sensitive fluorescent dye 3,3′‐dipropylthiadicarbocyanine
iodide (DiSC35, Sigma‐Aldrich, St. Louis, MO, USA). In short, Bacillus megaterium ATCC 13632
was grown in half‐concentrated MH to an OD600 of 0.5 and subsequently incubated for 5 min
with 3 µM DiSC35. Samples were then treated with 3.3 µg/ml nisin or 25 µg/ml MP196,
respectively, or left untreated as control. Fluorescence was measured continuously for 5
minutes at 651 nm excitation and 675 nm emission wavelength using a Shimadzu RF‐5301PC
Spectrofluorometer (Shimaduzu, Duisburg, Germany).
Determination of cellular ATP levels
B. subtilis 168 was grown in BMM until reaching an OD500 of 0.35 and subsequently exposed to
22.5 µg/ml MP196, 0.75 µg/ml nisin, 10 µg/ml valinomycin, 1 µg/ml gramicidin S, and 0.5 µg/ml
erythromycin (negative control) for 15 minutes or left untreated. After stress, cells were
harvested by centrifugation, resuspended in 500 µl 10 mM Tris, pH 7.5, and disrupted by
ultrasonication. ATP determination was performed using the Perkin Elmer ATPlite 1step assay
kit (Waltham, MA, USA) following the manufacturer's instructions. ATP standards were
prepared from 1 x 10‐7 to 8.5 x 10‐7 mol ATP in the provided buffer. For determination of cellular
ATP levels 100 µl of cytosolic cell extracts were used. All measurements were performed in
quintuple biological and double technical replicates using the Tecan Infinite® 200 PRO
multimode reader (Tecan, Männedorf, Switzerland).
Preparation of inverted vesicles from M. flavus
E An antimicrobial peptide delocalizes peripheral membrane proteins
95

Inverted vesicles were prepared from M. flavus according to Burstein et al.26 with some
modifications. Briefly, cells were grown in tryptic soy broth until exponential growth phase. 1 L
of culture was harvested and washed with 100 mL Tris buffer (50 mM Tris‐HCl, 2 mM MgCl2, 0.5
mM dithiothreitol, 0.5 mM EDTA, pH 8.0). The cell pellet was resuspended in 5 mL Tris buffer,
followed by pH readjustment to 8.0 and addition of each 10 µg/ml DNase and RNase. Cells were
disrupted mechanically in a Precellys 24 homogenizer using 0.1 mm glass beads (Bertin
Technologies, Montigny‐le‐Bretonneux, France). The crude homogenate was centrifuged at
30,000 x g for 20 minutes. The resulting supernatant was subsequently centrifuged at 175,000 x
g for 120 minutes. The resulting vesicle‐containing pellet was resuspended in 2 mL Tris buffer
supplemented with 10% glycerol and stored in liquid nitrogen until usage.
H+‐ATPase inhibition
Proton uptake into inverted M. flavus vesicles was measured in a microtiter plate assay in a final
volume of 200 µL based on the pH‐sensitive probe acridine orange as described by Palmgren et
al.27. The reaction mixture (20 µM acridine orange, 4 mM MgCl2, 10 mM MOPS‐BTP (pH 7.0),
140 mM KCI, 1 mM EDTA, 1 mM DTT, 1 mg/ml BSA (essentially fatty acid free), 2.5 µg/ml
valinomycin, 75 µg/ml vesicle protein) was preincubated with 20 µM MP196, 20 µM nisin, 20
µM lactoferrin or left untreated as control, respectively, at 25 °C for 30 minutes. The reaction
was initiated by addition of 2mM ATP‐BTP and absorbance was measured at 495 nm for 6h
using a SunriseTM multichannel plate reader (Tecan, Männedorf, Switzerland).
INT reduction assay
Antibiotic influence on the bacterial electron transport chain was monitored by reduction of
iodonitrotetarzolium chloride (INT) using M. flavus inverted vesicles as described by Smith and
McFeters28. 20 µg of vesicle protein were preincubated for 30 minutes in pure phosphate buffer
(10 mM potassium phosphate, 5 mM magnesium acetate, pH 6.5) or buffer with 40 µM of
MP196, 40 µM nisin, 100 µM antimycin A, or 5 mM rotenone, respectively. Subsequently, 1 mM
INT and 0.6 mM NADH or 10 mM succinate were added as substrate and the solution was
incubated for 1 hour at 25 °C in the dark. INT reduction was stopped by addition of 5% TCA.
Insoluble formazan was pelleted for 5 minutes at 13,000xg, extracted with 1 mL ethanol and
centrifuged again to remove insoluble particles. Absorbance of the supernatant was measured
at 485 nm.
E An antimicrobial peptide delocalizes peripheral membrane proteins
96

Amino acid analysis (AAA)
B. subtilis 168, JH642, and SMB8029 were grown in BMM until an OD500 of 0.35, harvested by
centrifugation (3,220 x g, 10 minutes, 37 °C), and washed three times in pre‐warmed AAA buffer
(10.2 mM Na2HPO4, 1.8 mM KH2PO4, 25.8 mM KCl, 4.3 mM NaCl). Cells were then resuspended
in AAA buffer, readjusted to an OD500 of 0.35, and stressed with 22.5 µg/ml MP196, 1 µg/mL
gramicidin S, 0.75 µg/mL nisin, or 10 µg/mL valinomycin for 15 minutes or left untreated as
control, respectively. Hypoosmotic stress was set by resuspending the cells in double distilled
water, hyperosmotic stress by resuspending in AAA buffer containing 6% NaCl, followed by 15
minutes incubation at 37 °C. Subsequently, cells were harvested by centrifugation (16,100xg, 10
minutes, 4 °C). The supernatant was kept for measurement of amino acid leakage whereas the
cells were washed three times in AAA buffer (16,100 x g, 10 minutes, 4 °C), resuspended in the
same buffer and disrupted by ultrasonication as described above. In order to analyze only free
amino acids, proteins were precipitated with acetone in both intra‐ and extracellular fractions.
Therefore, 100 µl of cell extract and 200 µl of culture supernatant, respectively, were mixed
with the five‐fold volume of ice‐cold 80% acetone and incubated over night at ‐20 °C.
Precipitated proteins were pelleted (16,100 x g, 20 minutes, 4 °C) and the resulting, protein‐free
supernatant was used for HPLC. 500 µl of the protein‐free cell extract and 1 ml of protein‐free
culture supernatant samples, respectively, were dried completely in AAA glass tubes and
subsequently dissolved in 10 µl of 20 mM HCl. Amino acid analysis was performed using the
Acquity HPLC and AccQ•Tag Ultra system (Waters GmbH, Milford, MA, USA) according to the
manufacturer's instructions. In short, 10 µl AccQTag reagent and 30 µl internal norvalin
standard (final concentration 10 pmol/µl) was added to the samples for derivatization. Those
samples were then incubated for 10 minutes to allow conversion of primary and secondary
amines into stabile derivatives. Amino acid derivatives were separated using an AccQ•Tag Ultra
RP column and detected by an Acquity UPLC‐TUV detector (Waters GmbH, Eschborn, Germany).
Amino acids were quantified using 10 pmol/µl amino acid standards. Intracellular amino acid
concentrations were calculated based on the B. subtilis cellular volume as described above.
Glutamate uptake and efflux
Glutamate uptake and efflux was performed as described previously30. In short, S. simulans 22
was cultured at 37 °C to an OD600 of 0.3 and subsequently pre‐incubated for 15 minutes with
E An antimicrobial peptide delocalizes peripheral membrane proteins
97

100 µg/ml chloramphenicol to prevent glutamate incorporation into proteins. Afterwards,
radioactively labeled [3H]‐L‐glutamate was added to a final concentration of 1 μCi/ml and the
culture was immediately subdivided. To determine [3H]‐glutamate uptake, aliquots were treated
with 5 µg/ml MP196 or 5 µg/ml nisin, respectively or left untreated as control. After 30 minutes
of incubation, the control was further subdivided into aliquots. To determine efflux of
previously ingested [3H]‐glutamate, such aliquots were then treated with 5 µg/ml MP196 or 5
µg/ml nisin, respectively or left untreated as control.
Samples of 100 μl were collected every 10 minutes, filtered through cellulose acetate filters
(pore size 0.2 μm; Schleicher & Schüll, Dassel, Germany) and washed twice with 5 ml potassium
phosphate buffer (200 mM potassium phosphate, 100 µM non‐radioactive glutamate, pH 7.0)
Filters were dried, transferred to counting vials and filled with 5 ml of scintillation fluid
(Filtersafe, Zinsser, Frankfurt, Germany). Radioactivity was measured in a 1900CA beta‐counter
(Packard/Perkin Elmer, Waltham, MA, USA).
Minimal inhibitory concentration (MIC) dependence on glutamate and salt concentration
MIC determination of MP196 under different glutamate and salt concentrations was performed
in a microtiter plate assay in modified BMM containing rising concentrations of glutamate (0,
4.5, 45, 450, 3000 mM), NaCl (0, 10, 100, 500, 1000 mM), or KCl (0, 100, 500 mM, cells did not
survive 1000 mM KCl). BMM without glutamate was additionally supplemented with 4.5 mM
ammonium nitrate as alternative nitrogen source. 200 µl of medium were inoculated with
5x105 cells/mL of B. subtilis 168 and incubated with different antibiotic concentrations for 18
hours at 37 °C. The MIC was defined as the lowest antibiotic concentration inhibiting visible
growth.
E An antimicrobial peptide delocalizes peripheral membrane proteins
98

ii) Supplementary Tables
Supplementary Table 1: Marker proteins induced in the cytosolic proteome
protein ID protein function functional category regulator
SpoVG negative effector of asymetric septation at the onset of sporulation sporulation σH Spo0M sporulation‐control gene sporulation σW/σH
YdaG general stress protein general stress σB
YtxH general stress protein general stress σB/σH Dps DNA‐protecting protein, ferritin general stress σB ClpP ATP‐dependent Clp protease proteolytic subunit general stress σB
YdbD manganese‐containing catalase oxidative stress unknown AzoR2 azoreductase oxidative stress σG YceC similar to tellurium resistance protein cell envelope stress σB/σW/σM YceH similar to toxic anion resistance protein cell envelope stress σB/σW/σM
YthP similar to ABC transporter cell envelope stress σW YfhM similar to epoxide hydrolase cell envelope stress σB/σW FosB bacillithiol‐S‐transferase cell envelope stress σW
YtrE similar to ABC transporter cell envelope stress YtrA LiaH modulator of LiaIHGFSR operon expression cell wall LiaRS DltA D‐alanyl‐D‐alanine carrier protein ligase cell wall σD/σX/σM RacX amino acid racemase cell wall σW
PspA phage shock protein A homolog membrane σW YjdA similar to 3‐ketoacyl‐acyl‐carrier protein reductase membrane σE YoxD similar to 3‐oxoacyl‐acyl‐carrier protein reductase membrane unknownYuaI involved in reducing membrane fluidity membrane σW NadE NAD synthetase energy σB BglH phospho‐beta‐glucosidase energy CcpA CitZ citrate synthase energy CcpAYwrO similar to NAD(P)H oxidoreductase energy unknown IolS also/keto reductase energy IolR YhdN aldo/keto reductase specific for NADPH energy σB
E An antimicrobial peptide delocalizes peripheral membrane proteins
99

protein ID protein function functional category regulator
NfrA FMN‐containing NADPH‐linked nitro/flavin reductase energy Spx RocA 3‐hydroxy‐1‐pyrroline‐5‐carboxylate dehydrogenase amino acids RocRTrmB tRNA (m7G46) methyltransferase tRNA modification unknown RpsB ribosomal protein S2 translation unknown YvlB unknown unknown unknown
Supplementary Table 2: MALDI‐ToF‐MS data of identified cytosolic marker proteins (2D gel‐based approach)
protein ID
protein name mass weight
pI peptide count
protein score
protein score C.I. %
AzoR2 azoreductase 23257 5.26 7 117 100 BglH phospho‐beta‐glucosidase 53255 5.13 13 287 100 CitZ citrate synthase 41702 5.55 16 444 100 ClpP ATP‐dependent Clp protease proteolytic subunit 21668 5.19 12 353 100 DltA D‐alanyl‐D‐alanine carrier protein ligase 55773 5.10 13 276 100 Dps DNA‐protecting protein, ferritin 16583 4.64 8 168 100 FosB bacillithiol‐S‐transferase 17161 6.14 4 754 * ‐ IolS aldo/keto reductase 35146 5.50 16 280 100 LiaH modulator of LiaIHGFSR operon expression 25682 6.20 13 112 100 NadE NAD synthetase 30376 5.07 16 459 100 NfrA FMN‐containing NADPH‐linked nitro/flavin reductase 28302 5.73 20 9602 * ‐ PspA phage shock protein A 25125 5.87 11 91 100 RacX amino acid racemase 25270 5.46 3 73 100 RocX 3‐hydroxy‐1‐pyrroline‐5‐carboxylate dehydrogenase 56284 5.58 52 12601 * ‐ RpsB ribosomal protein S2 27950 6.27 16 279 100 Spo0M sporulation‐control gene 29714 4.26 63 25783 * ‐ SpoVG negative effector of sporulation 10886 5.25 10 348 100 TrmB tRNA (guanine‐N(7)‐)‐methyltransferase 24488 6.32 7 53 98
E An antimicrobial peptide delocalizes peripheral membrane proteins
100

protein ID
protein name mass weight
pI peptide count
protein score
protein score C.I. %
YceC similar to tellurium resistance protein 21810 5.46 14 224 100 YceH unknown 41646 5.90 13 263 100 YdaG general stress protein 15867 5.33 5 100 100 YdbD manganese‐containing catalase 30238 5.06 10 103 100 YfhM similar to epoxide hydrolase 32737 6.07 20 7499 * ‐ YhdN aldo/keto reductase specific for NADPH 37289 4.96 10 132 100 YjdA similar to 3‐ketoacyl‐ acyl‐carrier protein reductase 27432 5.74 5 55 99 YoxD similar to 3‐oxoacyl‐ acyl‐carrier protein reductase 25283 5.48 20 434 100 YqiG similar to NADH‐dependent flavin oxidoreductase 40780 5.34 19 325 100 YthP similar to ABC transporter 26490 5.39 5 195 100 YtrE similar to ABC transporter 25444 5.99 11 241 100 YtxH general stress protein 16675 5.30 5 131 100 YuaI involved in reducing membrane fluidity 19830 5.31 7 167 100 YvlB unknown 41056 5.50 15 121 100 YwrO similar to NAD(P)H oxidoreductase 19942 5.33 7 166 100
* Identification was carried out using a Synapt G2S HDMS mass spectrometer (see supplementary methods)
E An antimicrobial peptide delocalizes peripheral membrane proteins
101

Supplementary Table 3: Marker proteins induced in the membrane proteome
ID protein function pathway category repressor activator
PtkA protein tyrosine kinase involved in biofilm formation antibiotic stress stress response AbrB
YaaN similar to toxic cation resistance protein detoxication stress response σW
YfhM similar to epoxide hydrolase detoxication, sigma b dependent, phosphate starvation
stress response σW/σ
B
RsbW anti‐sigma factor general stress response sigma b stress response σB
PcrA ATP‐dependent DNA helicase mismatch repair, nucleotide excision repair stress response LexA
ClpX ATP‐dependent Clp protease ATP‐binding subunit heat shock stress response CtsR
HslU ATP‐dependent protease ATP‐binding subunit heat shock stress response CodY
DnaK class I heat‐shock protein heat shock stress response HrcA
LonA class III heat‐shock ATP‐dependent Lon protease heat shock stress response CtsR
YciC putative metallochaperone with NTPase activity/probably part of a low‐affinity pathway of zinc transport
chaperone/zinc transport stress response Zur
EngD GTP‐dependent nucleic acid‐binding protein competence stress response ComK
NucA endonuclease competence stress response ComK
DegS two‐component sensor histidine kinase competence stress response
KinD two‐component sensor histidine kinase sporulation stress response
SpoIVA required for proper spore cortex formation and coat assembly sporulation stress response σE
YlbC unknown sporulation stress response
McsB protein arginine kinase protein modification stress response
SrfAA surfactin synthetase antibiotic production stress response Abh, CodY, Spx
ComA, PerR
SrfAB surfactin synthetase antibiotic production stress response Abh, CodY, Spx
ComA, PerR
SrfAC surfactin synthetase antibiotic production stress response Abh, CodY, Spx
ComA, PerR
YsdB unknown control of SigW activity, survival of heat stress
cell envelope σB/σ
W
YacL unknown survival of salt and ethanol stresses cell envelope σB/σ
M
YpuA unknown unknown cell envelope σM
YobJ unknown unknown cell envelope σW
YteJ unknown unknown cell envelope σW
BioD dethiobiotin synthetase biotin metabolism membrane BirA
BioI cytochrome P450 enzyme biotin metabolism, detoxication membrane BirA
AcpA acyl carrier protein fatty acid biosynthesis membrane FapR
FabG beta‐ketoacyl‐acyl carrier protein reductase fatty acid biosynthesis membrane FapR
E An antimicrobial peptide delocalizes peripheral membrane proteins
102
ID protein function pathway category repressor activator
PlsX involved in fatty acid/phospholipid synthesis fatty acid biosynthesis membrane FapR
YhfT similar to long‐chain fatty‐acid‐CoA ligase fatty acid biosynthesis membrane
FadE acyl‐CoA dehydrogenase fatty acid biosynthesis membrane CcpA, FadE SdpR
GlpD glycerol‐3‐phosphate dehydrogenase glycerophospholipid metabolism membrane CcpA AbrB, GlpP
FloT similar to flotillin 1, orchestration of physiological processes in lipid microdomains
control of membrane fluidity membrane
DynA dynamin‐like protein membrane fusion membrane
DltB D‐alanine esterification of lipoteichoic acid and wall teichoic acid/D‐alanyl transfer from Dcp to undecaprenol‐phosphate
lipoteichoic acid biosynthesis cell wall Spo0A, stringent response
σM/σ
X/σ
D,
YvrHb
DltD D‐alanine transfer from undecaprenol‐phosphate to the poly(glycerophosphate) chain of LTA
lipoteichoic acid biosynthesis cell wall Spo0A, stringent response
σM/σ
X/σ
D,
YvrHb
Ddl D‐alanyl‐D‐alanine ligase A peptidoglycan biosynthesis cell wall σM
MreB cell shape‐determining protein peptidoglycan biosynthesis cell wall σM
Mbl MreB‐like protein peptidoglycan biosynthesis cell wall stringent response
σE
MurAB UDP‐N‐acetylglucosamine 1‐carboxyvinyltransferase peptidoglycan biosynthesis cell wall
MurAA UDP‐N‐acetylglucosamine 1‐carboxyvinyltransferase peptidoglycan biosynthesis cell wall
MurG UDP‐N‐acetylglucosamine‐N‐acetylmuramyl‐(pentapeptide)pyrophosphoryl‐undecaprenol N‐acetylglucosamine transferase
peptidoglycan biosynthesis cell wall
LytA involved in the secretion of major autolysin LytC (amidase) lysis and remodeling of peptidoglycan cell wall SlrR σD, YvrHb
YycH negative effector of WalK control of cell wall metabolism cell wall
GdpP cyclic di‐AMP phosphodiesterase cell wall homeostasis cell wall
YbbP maybe involved in the synthesis of c‐di‐AMP in vegetative cells cell wall homeostasis cell wall
FtsA membrane anchor of FtsZ septum formation cell division σH, WalR
YydI ABC transporter (ATP‐binding protein) antibiotic transport system transporter AbrB, Rok
YybJ similar to ABC transporter (ATP‐binding protein) antibiotic transport system, iorganic ion transport
transporter
YdbJ similar to ABC transporter (ATP‐binding protein) antibiotic transport system, multidrug transporter
transporter
YutK similar to Na+/nucleoside cotransporter concentrative nucleoside transporter transporter
OpuAA glycine betaine ABC transporter osmoprotectant transport system, choline transport
transporter
OpuCC glycine betaine/carnitine/choline ABC transporter osmoprotectant transport system, choline transport
transporter
OpuBA choline ABC transporter (ATP‐binding protein) osmoprotectant transport system, choline transport
transporter
DppE dipeptide ABC transporter peptide transport transporter CodY
OppD oligopeptide ABC transporter (ATP‐binding protein) peptide transport transporter ScoC TnrA
E An antimicrobial peptide delocalizes peripheral membrane proteins
103

ID protein function pathway category repressor activator
SufC similar to ABC transporter, iron metabolism (ATP‐binding protein) transport transporter
ThiV putative HMP/thiamine permease amino acid/sugar metabolism transporter Thi‐box Thi‐box
YlaG GTPase signal transduction protein secretion stringent response
SecA translocase binding subunit protein secretion protein secretion
AtpC ATP synthase oxidative phosphorylation energy metabolism stringent response
QoxC cytochrome aa3 quinol oxidase (subunit III) oxidative phosphorylation energy metabolism stringent response
HemL glutamate‐1‐semialdehyde 2,1‐aminotransferase porphyrin biosynthesis energy metabolism PerR
SufB synthesis of Fe‐S‐clusters iron metabolism energy metabolism
ResE two‐component sensor histidine kinase regulation of electron transport energy metabolism CcpA PhoP, ResD
NadA quinolinate synthetase nicotineamide and nicotinate metabolism energy metabolism NadR
NadB L‐aspartate oxidase nicotineamide and nicotinate metabolism energy metabolism NadR
NadF probable inorganic polyphosphate/ATP‐NAD kinase 2 nicotineamide and nicotinate metabolism energy metabolism
PncB nicotinate phosphoribosyltransferase nicotineamide and nicotinate metabolism energy metabolism
ThiC biosynthesis of the pyrimidine moiety of thiamin amino acid/sugar metabolism energy metabolism Thi‐box Thi‐box
SucC succinyl‐CoA synthetase (beta subunit) TCA cycle energy metabolism CcpA
CitB aconitate hydratase TCA cycle energy metabolism CcpC, CodY, FrsA
Icd isocitrate dehydrogenase TCA cycle energy metabolism CcpA, CcpC
PycA pyruvate carboxylase TCA cycle energy metabolism stringent response
AckA acetate kinase pyruvate metabolism energy metabolism CcpA, CodY
Tkt transketolase glycolysis energy metabolism Spo0A
Pgk phosphoglycerate kinase glycolysis energy metabolism CggR
Pyk pyruvate kinase glycolysis energy metabolism
GapA glyceraldehyde‐3‐phosphate dehydrogenase glycolysis energy metabolism CggR
Tal putative transaldolase pentose phosphate pathway energy metabolism
PtsI phosphotransferase system (PTS) enzyme I PTS energy metabolism stringent response
GlcT
FrlB similar to glutamine‐fructose‐6‐phosphate transaminase carbohydrate & nitrogen metabolism energy metabolism CodY, FlrR
FrlO similar to multiple sugar‐binding protein carbohydrate & nitrogen metabolism energy metabolism CodY, FrlR
YoaC similar to xylulokinase carbohydrate metabolism energy metabolism S‐box S‐box
BdhA acetoin reductase/2,3‐butanediol dehydrogenase carbohydrate metabolism energy metabolism AbrB
YqfL modulator of CcpN activity carbohydrate metabolism energy metabolism
GndA NADP‐dependent phosphogluconate dehydrogenase carbohydrate metabolism energy metabolism
LutB lactate catabolic enzyme carbohydrate metabolism energy metabolism LutR
E An antimicrobial peptide delocalizes peripheral membrane proteins
104

ID protein function pathway category repressor activator
YitJ probable 5,10‐methylenetetrahydrofolate reductase (NADP) unknown energy metabolism S‐box S‐box
PurA adenylosuccinate synthetase purine nucleotide synthesis PurR, G‐box G‐box
PurB adenylosuccinate lyase purine nucleotide synthesis PurR, G‐box G‐box
PurC phosphoribosylaminoimidazole succinocarboxamide synthetase purine nucleotide synthesis PurR, G‐box G‐box
PurD phosphoribosylglycinamide synthetase purine nucleotide synthesis PurR, G‐box G‐box
PurF glutamine phosphoribosylpyrophosphate amidotransferase purine nucleotide synthesis PurR, G‐box G‐box
PurH phosphoribosylaminoimidazole carboxy formyl formyltransferase and inosine‐monophosphate cyclohydrolase
purine nucleotide synthesis PurR, G‐box G‐box
PurK phosphoribosylaminoimidazole carboxylase II purine nucleotide synthesis PurR, G‐box G‐box
PurL phosphoribosylformylglycinamidine synthetase II purine nucleotide synthesis PurR, G‐box G‐box
Xpt xanthine phosphoribosyltransferase purine nucleotide synthesis PurR, G‐box G‐box
MtnN methylthioadenosine/S‐adenosylhomocysteine nucleosidase purine nucleotide synthesis Spx CymR
PyrAB carbamoyl‐phosphate synthetase pyrimidine nucleotide synthesis PyrR
PyrE orotate phosphoribosyltransferase pyrimidine nucleotide synthesis PyrR
PyrH uridylate kinase pyrimidine nucleotide synthesis PyrR
PyrR transcriptional attenuator and uracil phosphoribosyltransferase activity pyrimidine nucleotide synthesis PyrR
CarA carbamoyl‐phosphate transferase‐arginine Ala, Asp, Glu amino acids AhrC
CarB carbamoyl‐phosphate transferase‐arginine Ala, Asp, Glu amino acids AhrC
GltA glutamate synthase (large subunit) Ala, Asp, Glu amino acids GltC FsrA, TnrA
AsnB asparagine synthetase Ala, Asp, Glu amino acids
GudB glutamate dehydrogenase Ala, Asp, Glu, Arg, Pro amino acids
ArgH argininosuccinate lyase Ala, Asp, Glu, Arg, Pro amino acids AhrC
ArgJ ornithine acetyltransferase and amino‐acid acetyltransferase Ala, Asp, Glu, Arg, Pro amino acids AhrC
YhdR similar to aspartate aminotransferase Ala, Asp, Glu, Arg, Pro, Cys, Met, Phe, Tyr, Trp
amino acids
AlaT alanine transaminase Arg, Pro, Lys amino acids
MetC similar to cystathionine beta‐lyase Cys, Met amino acids S‐box S‐box
MetE cobalamin‐independent methionine synthase Cys, Met amino acids S‐box S‐box
MetK S‐adenosylmethionine synthetase Met, SAM amino acids S‐box S‐box
MtnK methylthioribose kinase Cys, Met amino acids S‐box S‐box
YxjG putative methionine synthase met amino acids S‐box S‐box
CysJ similar to sulfite reductase sulfur metabolism amino acids CysL
Sat sulfate adenylyltransferase sulfur metabolism amino acids S‐box S‐box
ThiO glycine oxidase Gly, Ala, Val, Pro amino acids Thi‐box Thi‐box
HisA phosphoribosylformimino‐5‐aminoimidazole carboxamide ribotide isomerase His amino acids T‐box T‐box
E An antimicrobial peptide delocalizes peripheral membrane proteins
105

ID protein function pathway category repressor activator
LysC aspartokinase II alpha subunit (aa 1‐408) and beta subunit (aa 246‐408) Lys, Cys, Met, Gly, Ser, Thr amino acids
AroA 3‐deoxy‐D‐arabino‐heptulosonate 7‐phosphate synthase and chorismate mutase‐isozyme 3
Phe, Tyr, Trp amino acids
AroB 3‐dehydroquinate synthase Phe, Tyr, Trp amino acids
AroE 5‐enolpyruvoylshikimate‐3‐phosphate synthase Phe, Tyr, Trp amino acids
AroF chorismate synthase Phe, Tyr, Trp amino acids
ProA gamma‐glutamyl phosphate reductase Arg, Pro amino acids T‐box T‐box
LeuA 2‐isopropylmalate synthase Val, Leu, Ile amino acids T‐box, CodY, CcpA, TnrA
T‐box
LeuC 3‐isopropylmalate dehydratase (large subunit) Val, Leu, Ile amino acids T‐box, CodY, CcpA, TnrA
T‐box
LeuD 3‐isopropylmalate dehydratase (small subunit) Val, Leu, Ile amino acids T‐box, CodY, CcpA, TnrA
T‐box
IlvB acetolactate synthase catalytic subunit Val, Leu, Ile amino acids T‐box, CodY, CcpA, TnrA
T‐box
IlvD dihydroxy‐acid dehydratase Val, Leu, Ile amino acids T‐box, CodY, CcpA, TnrA
T‐box
YwaA similar to branched‐chain amino acid aminotransferase Val, Leu, Ile amino acids
RelA ppGpp hydrolase/synthase stringent response, amino acid starvation amino acids
YdbR similar to ATP‐dependent RNA helicase transcription regulation transcription
GlpP transcription antiterminator transcription regulation transcription GlpP
Rho transcriptional terminator Rho transcription regulation transcription
TufA elongation factor TU elongation translation stringent response
FusA elongation factor G elongation translation stringent response
RplA ribosomal protein L1 ribosomal proteins translation stringent response
YydA similar to pseudouridine methyltransferase rRNA maturation translation DnaA
YmcB tRNA methylthiotransferase tRNA maturation translation
ProS prolyl‐tRNA synthetase tRNA synthetase (Pro) translation
ThrS threonyl‐tRNA synthetase tRNA synthetase (Thr) translation T‐box T‐box
ThrZ threonyl‐tRNA synthetase tRNA synthetase (Thr) translation T‐box T‐box
LysS lysyl‐tRNA synthetase tRNA synthetase (Lys) translation
HisS histidyl‐tRNA synthetase tRNA synthetase (His) translation T‐box T‐box
GatA glutamyl‐tRNA(Gln) amidotransferase tRNA transferase (Glu) translation
GatB glutamyl‐tRNA(Gln) amidotransferase tRNA transferase (Glu) translation
YebA probable membrane protein unknown unknown
YfhO putative membrane protein unknown unknown
E An antimicrobial peptide delocalizes peripheral membrane proteins
106

ID protein function pathway category repressor activator
YddK unknown unknown unknown
YgaO unknown unknown unknown
YydB unknown unknown unknown
YufK unknown unknown unknown
YjcH unknown unknown unknown
YjjA unknown unknown unknown
YjlC unknown unknown unknown
YqeG unknown unknown unknown
E An antimicrobial peptide delocalizes peripheral membrane proteins
107

Supplementary Table 4: ESI‐MS data of the marker proteins identified in the membrane
proteome
control (replicate 1) control (replicate 2) MP196 (replicate 1) MP196 (replicate 2)
protein ID ratio SD peptides ratio SD peptides ratio SD peptides ratio SD peptides
O05249|yufK 1.71 0 2 2.21 0.15 2
O06491|gatA 1.4 0.14 4 14.35 8.69 9 10.78 14.49 4
O06745|yitJ 0.52 0.03 2 0.55 0.03 7 4.4 0.54 12 9.53 1.23 9
O07021|yvfW 1.71 0.18 2 10.03 1.31 5 16.02 5.6 4
O07587|yhdR 25.25 8.15 5 52.68 71.85 2
O07619|yhfT 23.06 2.96 2 38.66 12.71 4
O07631|typA 1.3 0.13 4 6.73 0.48 8 18.58 2.12 4
O30509|gatB 1.54 0.32 3 16.72 9.55 4 40.15 27.31 3
O31581|yfhM 21.9 1.78 3 30.97 2.39 2
O31582|yfhO 1.39 0.07 3 3.67 0.34 5 4.77 0.5 3
O31630|yjcH 5.66 0.59 3 5.2 0.97 3
O31632|yjcJ 10.16 1.13 2 24.1 26.22 2
O31663|mtnK 1.33 0.08 4 1.41 0.07 7 18.27 1.81 12 53.31 6.45 6
O31671|kinD 2.4 0.49 4 3.07 0.45 2
O31749|pyrH 0.82 0.03 2 0.93 0.04 2 8.06 1 5 12.27 0.4 2
O31755|proS 9.17 9.34 3 39.81 30.51 2
O31778|ymcB 27.39 20.03 3 68.55 4.23 2
O32028|mtnN 1.64 0.12 2 16.5 12.38 3 39.83 9.41 2
O32039|hisS 1.13 0.03 4 6.51 0.28 4 11.98 1.95 2
O32076|yuaG 1.4 0.16 15 1.41 0.09 13 13.26 1.29 28 13.47 1.56 18
O32090|yueK 3.13 0.32 4 5.68 0.81 5
O32115|yutK 0.7 0.05 2 1.31 0.01 2 1.73 0.36 6
O32156|yurO 0.79 0.07 2 2.45 0.2 8 4.33 0.06 3
O32157|yurP 0.81 0.1 3 19.31 9.97 4 45.29 4.68 2
O32162|yurU 1.19 0.08 2 23.31 1.96 6 62.73 3.09 3
O32176|yusJ 18.81 12.36 2 24.44 14.4 2
O32213|yvgQ 16.21 6.06 4 24.38 2.68 3
O32243|opuCC 1.27 0.07 6 2.93 0.05 3 3.54 0.07 3
O34394|yjjA 17.96 0.17 2 29.44 5.56 2
O34424|yteJ 3.7 0.46 2 4.06 0.55 4
O34580|pcrA 1.24 0.09 2 1.28 0.29 5 7.69 1.42 3 31.05 4.62 2
O34586|ylbC 2.18 0.31 2 2.63 0.35 3
O34633|yjlC 1.68 0.06 2 2.76 0.04 2
O34738|ykoE 1.34 0.12 4 1.8 0.13 2
O34764|sat 2.46 2.48 2 12.92 2.1 3
O34774|yobJ 0.81 0.12 4 0.86 0.1 7 5.78 0.47 7 6.44 0.79 5
O34788|ydjL 1.29 0.05 2 3.74 0.23 6 11.06 0.58 4
O34858|argH 7.99 0.94 4 28.52 5.81 4
O34861|yoaC 4.69 0.26 6 6.4 5.3 3
O34934|ppnK2 21.31 2.49 2 24.87 0.89 2
O35006|hisA 5.9 0.71 4 10.07 0.74 3
O54408|relA 1.02 0.06 9 4.64 0.53 8 12.12 1.05 3
P00497|purF 1.31 0.14 3 10.58 2.1 4 24.59 1.07 2
P08495|lysC 2.18 1.13 2 18.32 5.18 4 87.52 8.66 2
P08838|ptsI 0.97 0.09 7 2.45 0.1 4 10.28 0.93 5
E An antimicrobial peptide delocalizes peripheral membrane proteins
108
control (replicate 1) control (replicate 2) MP196 (replicate 1) MP196 (replicate 2)
protein ID ratio SD peptides ratio SD peptides ratio SD peptides ratio SD peptides
P09339|citB 0.79 0.19 3 3.36 0.63 3 7.72 1.22 9
P09124|gapA 1.28 0.12 7 1.21 0.09 6 31.91 5.15 5 71.4 40.82 6
P12039|purD 1.4 0.1 4 1.7 0.04 2 19.18 5.95 4 30.92 2.22 8
P12042|purL 1.29 0.05 3 1.27 0.02 8 26.81 2.12 11 40.4 1.84 9
P12045|purK 7.39 1.57 6 11.22 0.81 3
P12046|purC 1.62 0.13 2 1.38 0.26 3 20.33 9.8 7 43.25 8.5 3
P12047|purB 1.07 0.09 3 5.86 0.11 6 14.41 3.35 7
P12048|purH 1.82 0.25 3 1.59 0.16 5 20.81 4.22 6 73.17 48.09 5
P12667|nucA 0.72 0.06 3 1.11 0.41 2
P13799|degS 1.11 0.04 2 11.66 0.92 6 18.85 1.21 3
P17820|dnaK 1.7 0.04 7 40.41 4.5 6 58.99 31.1 10
P17904|rsbW 20.96 4.87 2 40.23 1.16 2
P18185|carB 1.28 0.18 2 1.19 0.04 4 9.55 0.88 5 28.6 3.26 10
P18255|thrS 0.66 0.11 6 4.11 0.26 9 21.44 9.09 7
P18256|thrZ 0.91 0.03 7 10.98 1.1 7 24.32 3.32 7
P19669|tal 1.06 0.29 2 2.07 0.17 4 4.28 0.28 3
P19670|murAB 18.25 2.28 2 14.27 18.88 2
P20691|aroE 28.16 2.08 4 35.76 38.25 2
P24136|oppD 3.15 0.33 2 4.01 0.06 3
P25972|pyrE 1.09 0.19 2 9.46 0.82 2 11.65 1.12 2
P25994|pyrAB 0.66 0.03 10 3.28 0.41 13 7.01 0.57 17
P26906|dppE 0.78 0.03 3 4.83 0.01 2 5.38 0.75 4
P27206|srfAA 0.37 0.07 12 0.38 0.05 31 1.99 0.49 37 3.86 0.98 37
P28264|ftsA 10.64 1.52 2 15.15 0.48 2
P28366|secA 1.07 0.11 6 1.04 0.04 17 10.56 0.89 18 16.05 0.77 15
P29726|purA 0.84 0.09 6 0.92 0.12 4 6.25 1.57 13 8.39 2.31 9
P30300|glpP 3.25 0.15 2 4.66 1.19 2
P30949|hemL 7.88 0.33 5 8.33 5.49 4
P31102|aroB 1.7 0.12 4 1.81 0.23 7 20.1 8.99 8 92.54 21.03 2
P31104|aroF 2.12 0.11 2 16.64 0.7 3 45.26 42.15 2
P31847|ypuA 0.86 0.1 4 8.65 1.54 3 10.55 1.32 2
P33166|tuf 1.33 0.06 11 1.35 0.07 12 14.29 2.68 14 32.7 20.2 11
P34958|qoxC 0.72 0.2 5 1.95 0.13 4 2.14 0.05 2
P35149|spoIVA 1.77 0.11 2 0.87 0.13 3
P35164|resE 0.86 0.06 4 2.86 0.66 7 4.04 1.09 5
P36838|carA 1.12 0 2 4.93 0.34 5 13.23 3.95 6
P36843|argJ 61.37 6.39 2 118.98 14.3 2
P37484|yybT 0.96 0.08 2 2.53 0.26 5 3.3 0.29 6
P37494|yybJ 0.67 0.02 4 3.32 0.18 4 3.42 1.04 2
P37518|engD 3.22 0.29 4 13.05 5.67 6 21.23 13.14 4
P37535|yaaN 1.29 0.04 2 19.02 3 6 30.73 9.04 4
P37570|yacI 7.15 0.55 2 12.29 2.4 2
P37585|murG 0.8 0.05 4 4.98 1.05 2 8.78 1.66 2
P37812|atpC 2.78 0.07 2 2.85 0.26 3
P37877|ackA 65.81 37.52 4 102.55 95.95 3
P37945|lonA 0.92 0.16 2 0.97 0.07 7 5.21 0.35 8 12.03 1.84 6
P38032|nadB 6.57 2.71 6 14.48 1.14 6
P39580|dltB 0.7 0.01 2 4.48 0.19 3 5.36 0.04 2
E An antimicrobial peptide delocalizes peripheral membrane proteins
109
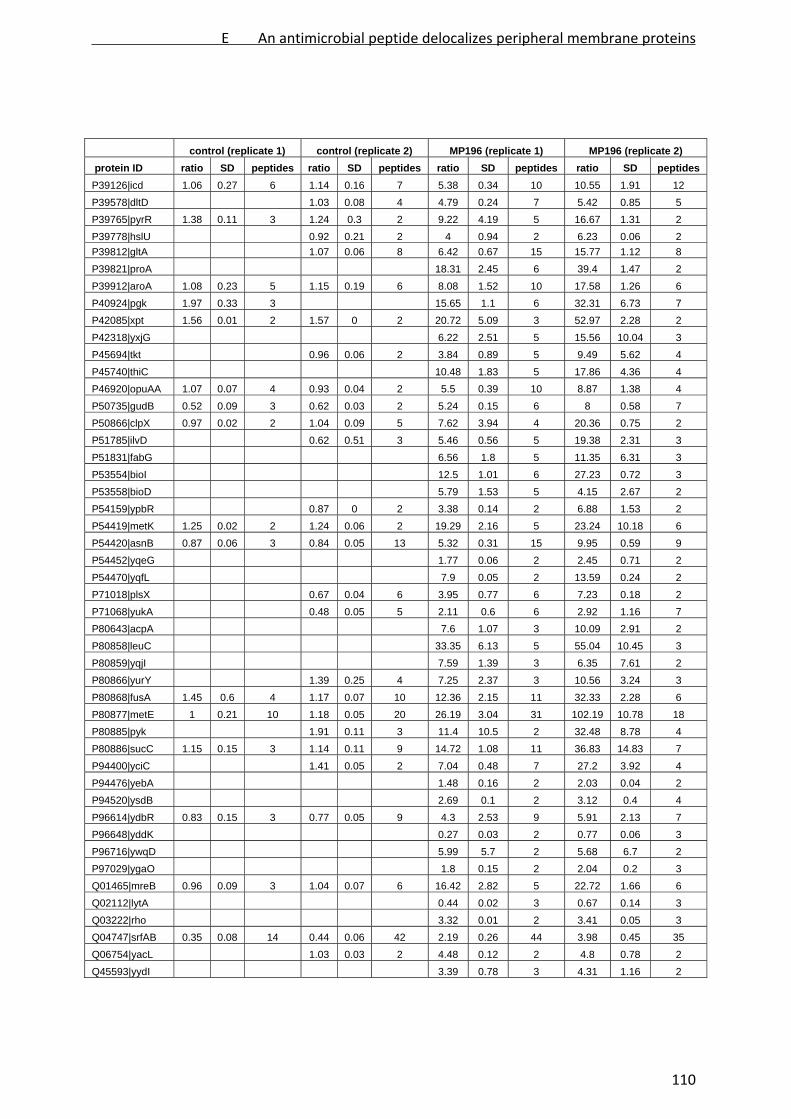
control (replicate 1) control (replicate 2) MP196 (replicate 1) MP196 (replicate 2)
protein ID ratio SD peptides ratio SD peptides ratio SD peptides ratio SD peptides
P39126|icd 1.06 0.27 6 1.14 0.16 7 5.38 0.34 10 10.55 1.91 12
P39578|dltD 1.03 0.08 4 4.79 0.24 7 5.42 0.85 5
P39765|pyrR 1.38 0.11 3 1.24 0.3 2 9.22 4.19 5 16.67 1.31 2
P39778|hslU 0.92 0.21 2 4 0.94 2 6.23 0.06 2
P39812|gltA 1.07 0.06 8 6.42 0.67 15 15.77 1.12 8
P39821|proA 18.31 2.45 6 39.4 1.47 2
P39912|aroA 1.08 0.23 5 1.15 0.19 6 8.08 1.52 10 17.58 1.26 6
P40924|pgk 1.97 0.33 3 15.65 1.1 6 32.31 6.73 7
P42085|xpt 1.56 0.01 2 1.57 0 2 20.72 5.09 3 52.97 2.28 2
P42318|yxjG 6.22 2.51 5 15.56 10.04 3
P45694|tkt 0.96 0.06 2 3.84 0.89 5 9.49 5.62 4
P45740|thiC 10.48 1.83 5 17.86 4.36 4
P46920|opuAA 1.07 0.07 4 0.93 0.04 2 5.5 0.39 10 8.87 1.38 4
P50735|gudB 0.52 0.09 3 0.62 0.03 2 5.24 0.15 6 8 0.58 7
P50866|clpX 0.97 0.02 2 1.04 0.09 5 7.62 3.94 4 20.36 0.75 2
P51785|ilvD 0.62 0.51 3 5.46 0.56 5 19.38 2.31 3
P51831|fabG 6.56 1.8 5 11.35 6.31 3
P53554|bioI 12.5 1.01 6 27.23 0.72 3
P53558|bioD 5.79 1.53 5 4.15 2.67 2
P54159|ypbR 0.87 0 2 3.38 0.14 2 6.88 1.53 2
P54419|metK 1.25 0.02 2 1.24 0.06 2 19.29 2.16 5 23.24 10.18 6
P54420|asnB 0.87 0.06 3 0.84 0.05 13 5.32 0.31 15 9.95 0.59 9
P54452|yqeG 1.77 0.06 2 2.45 0.71 2
P54470|yqfL 7.9 0.05 2 13.59 0.24 2
P71018|plsX 0.67 0.04 6 3.95 0.77 6 7.23 0.18 2
P71068|yukA 0.48 0.05 5 2.11 0.6 6 2.92 1.16 7
P80643|acpA 7.6 1.07 3 10.09 2.91 2
P80858|leuC 33.35 6.13 5 55.04 10.45 3
P80859|yqjI 7.59 1.39 3 6.35 7.61 2
P80866|yurY 1.39 0.25 4 7.25 2.37 3 10.56 3.24 3
P80868|fusA 1.45 0.6 4 1.17 0.07 10 12.36 2.15 11 32.33 2.28 6
P80877|metE 1 0.21 10 1.18 0.05 20 26.19 3.04 31 102.19 10.78 18
P80885|pyk 1.91 0.11 3 11.4 10.5 2 32.48 8.78 4
P80886|sucC 1.15 0.15 3 1.14 0.11 9 14.72 1.08 11 36.83 14.83 7
P94400|yciC 1.41 0.05 2 7.04 0.48 7 27.2 3.92 4
P94476|yebA 1.48 0.16 2 2.03 0.04 2
P94520|ysdB 2.69 0.1 2 3.12 0.4 4
P96614|ydbR 0.83 0.15 3 0.77 0.05 9 4.3 2.53 9 5.91 2.13 7
P96648|yddK 0.27 0.03 2 0.77 0.06 3
P96716|ywqD 5.99 5.7 2 5.68 6.7 2
P97029|ygaO 1.8 0.15 2 2.04 0.2 3
Q01465|mreB 0.96 0.09 3 1.04 0.07 6 16.42 2.82 5 22.72 1.66 6
Q02112|lytA 0.44 0.02 3 0.67 0.14 3
Q03222|rho 3.32 0.01 2 3.41 0.05 3
Q04747|srfAB 0.35 0.08 14 0.44 0.06 42 2.19 0.26 44 3.98 0.45 35
Q06754|yacL 1.03 0.03 2 4.48 0.12 2 4.8 0.78 2
Q45593|yydI 3.39 0.78 3 4.31 1.16 2
E An antimicrobial peptide delocalizes peripheral membrane proteins
110

control (replicate 1) control (replicate 2) MP196 (replicate 1) MP196 (replicate 2)
protein ID ratio SD peptides ratio SD peptides ratio SD peptides ratio SD peptides
Q06797|rplA 0.98 0.36 2 2.41 0.52 2 4.12 0.2 2
Q45589|ybbP 0.71 0.15 2 2.26 0.37 5 2.78 0.62 4
Q45600|yydB 19.62 5.97 6 38.72 50.53 2
Q45601|yydA 1.26 0.05 2 2.65 0.69 2
Q794W0|yycH 0.94 0.1 3 2.29 0.26 7 3.12 0.27 7
Q795M6|alaT 4.35 1.08 4 9.29 0.37 2
Q9KWU4|pyc 1.17 0.01 2 1.08 0.05 9 10.42 1.04 17 23.43 1.85 16
Q9KWZ1|nadA 44.25 8.8 4 49.76 43.2 3
Supplementary Table 5: Marker proteins of MP196 overlapping with markers of other cell
envelope‐targeting antibiotics ‐ a comparison with the B. subtilis proteome response library13,31.
MP
196
van
com
ycin
ba
citr
acin
me
rsac
idin
nis
in
gal
lider
min
gra
mic
idin
S
da
pto
myc
in
gra
mic
idin
A
valin
om
yci
n
general stress YtxH x sporulation Spo0M x x x x x
cell envelope stress
YceC x x x x x x
YceH x x x x
LiaH x x x x x x
YtrE x x x x x
DltA x RacX x
PspA x x x x x x YthP YuaI x YjdA x x YoxD x x x x
YfhM x FosB x
energy
NadE x x x x BglH x CitZ x NfrA x x x YwrO x
tRNA modification TrmB x x x translation RpsB x x unknown YvlB x x x
total overlap 35/35 3/5 4/11 5/13 5/8 10/25 12/21 2/8 4/5 19/22
E An antimicrobial peptide delocalizes peripheral membrane proteins
111

iii) Supplementary Figures
Supplementary Figure 1: Growth of B. subtilis in chemically defined medium after treatment
with MP196 (a) and D‐MP196 (b). Arrowheads indicate the time point of antibiotic addition
during logarithmic growth. Peptide concentrations leading to 50‐70% growth inhibition in
relation to the untreated controls were chosen for radioactive labeling and follow up
experiments (chosen concentrations are underlined).
E An antimicrobial peptide delocalizes peripheral membrane proteins
112
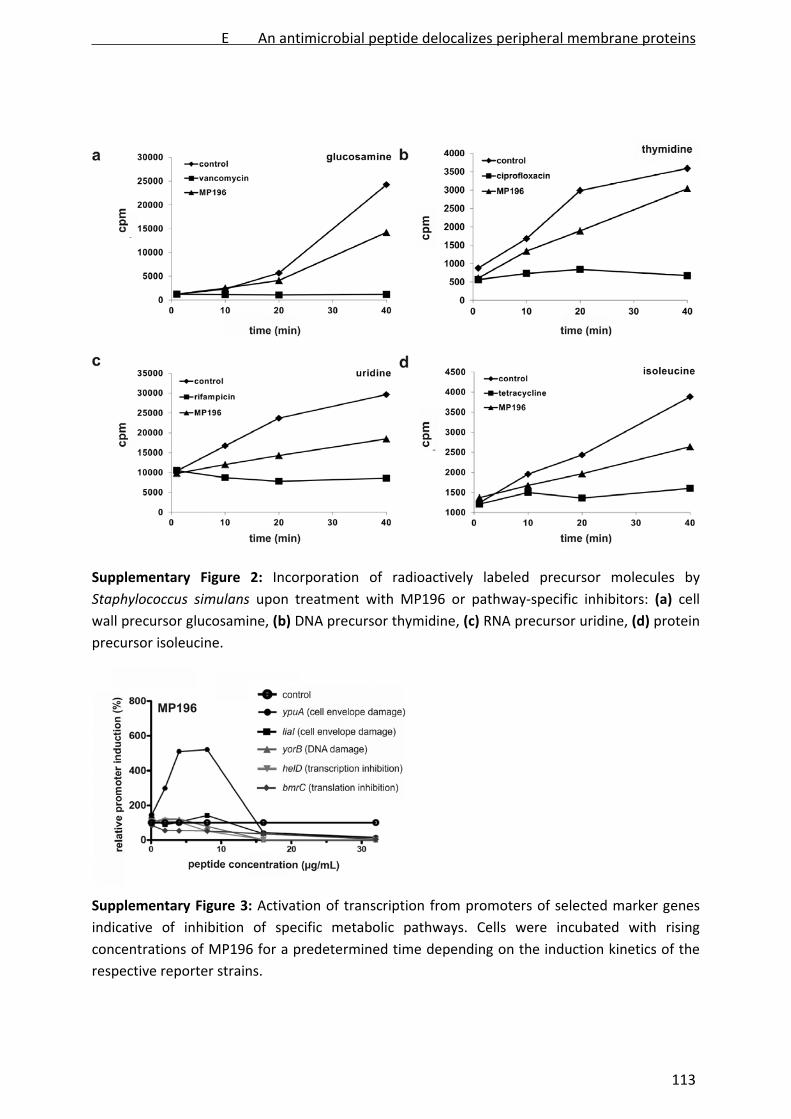
Supplementary Figure 2: Incorporation of radioactively labeled precursor molecules by
Staphylococcus simulans upon treatment with MP196 or pathway‐specific inhibitors: (a) cell
wall precursor glucosamine, (b) DNA precursor thymidine, (c) RNA precursor uridine, (d) protein
precursor isoleucine.
Supplementary Figure 3: Activation of transcription from promoters of selected marker genes
indicative of inhibition of specific metabolic pathways. Cells were incubated with rising
concentrations of MP196 for a predetermined time depending on the induction kinetics of the
respective reporter strains.
E An antimicrobial peptide delocalizes peripheral membrane proteins
113

Supplementary Figure 4: 2D gel‐based proteome response patterns of B. subtilis after
treatment with MP196 (a) and D‐MP196 (b). Marker proteins upregulated in response to both
peptides (c). Overlapping proteins are labeled in orange. Unidentified proteins are circled.
E An antimicrobial peptide delocalizes peripheral membrane proteins
114

Supplementary Figure 5: DSC thermograms of pure DPPG (a), DPPE (b), and DPPC lipids (c)
incubated with MP196.
E An antimicrobial peptide delocalizes peripheral membrane proteins
115

Supplementary Figure 6: (a) Potassium release from whole B. megaterium cells. Values are
given relative to positive control nisin, which forms pores. (b) DiSC35‐based depolarization
measurements of B. megaterium treated with MP196 and nisin, a pore‐forming lantibiotic
serving as positive control.
E An antimicrobial peptide delocalizes peripheral membrane proteins
116

Supplementary Figure 7: Free amino acid profiles of B. subtilis after MP196‐treatment
determined by HPLC: (a) intracellular amino acid concentrations (b) extracellular amino acid
concentrations. Amino acids are written in one letter code in the order of elution time from the
column. Glutamate and glutamine as well as aspartate and asparagine are not separated and
appear as one peak. Tryptophan was not quantified here.
E An antimicrobial peptide delocalizes peripheral membrane proteins
117

Supplementary Figure 8: (a) Total amount of glutamate in the B. subtilis cytosol and in the
supernatant of MP196‐treated cells and untreated control cultures. (b) Uptake and efflux of
radioactively labeled [3H]‐L‐glutamate upon treatment with MP196 and pore former nisin.
iv) References
1. Chantson, J.T., Verga Falzacappa, M.V., Crovella, S. & Metzler‐Nolte, N. Solid‐phase
synthesis, characterization, and antibacterial activities of metallocene‐peptide
bioconjugates. ChemMedChem. 1,1268‐74 (2006).
2. Bonelli, R.R., Schneider, T., Sahl, H.G. & Wiedemann, I. Insights into in vivo activities from
gallidermin and epidermin mode‐of‐action studies. Antimicrob. Agents Chemother. 50,
1449‐57 (2006).
3. Wadhwani, P., Afonin, S., Ieronimo, M., Buerck, J. & Ulrich A.S. Optimized protocol for
synthesis of cyclic gramicidin S: Starting amino acid is key to high yield. J. Org. Chem. 71,
55‐61 (2006).
E An antimicrobial peptide delocalizes peripheral membrane proteins
118

4. Schneider, T. et al. In vitro assembly of a complete, pentaglycine interpeptide bridge
containing cell wall precursor (lipid II‐Gly5) of Staphylococcus aureus. Mol. Microbiol. 53,
675–85 (2004).
5. Urban, A. et al. Novel whole‐cell antibiotic biosensors for compound discovery. Appl.
Environ. Microbiol. 73, 6436‐43 (2007).
6. Stülke, J., Hanschke, R. & Hecker, M. Temporal activation of betaglucanase synthesis in
Bacillus subtilis is mediated by the GTP pool. J. Gen. Microbiol. 139, 2041–45 (1993).
7. Agnostopoulos, C. & Spizizen, J. Requirements for transformation in Bacillus subtilis. J.
Bacteriol. 81, 741–46 (1961).
8. Wenzel, M. et al. Proteomic signature of fatty acid biosynthesis inhibition available for in
vivo mechanism‐of‐action studies. Antimicrob. Agents Chemother. 55, 2590‐6. (2011).
9. Bandow, J.E. et al. Improved image analysis workflow for 2D gels enables large‐scale 2D
gel‐based proteomics studies ‐ COPD biomarker discovery study. Proteomics 8, 3030–41
(2008).
10. Eymann, C. et al. A comprehensive proteome map of growing Bacillus subtilis cells.
Proteomics 4, 2849–76 (2004).
11. Otto, A., et al. Systems‐wide temporal proteomic profiling in glucose‐starved Bacillus
subtilis. Nat Commun. 1, 137. doi: 10.1038/ncomms1137 (2010).
12. Wolff, S., Hahne, H., Hecker, M. & Becher, D. Complementary analysis of the vegetative
membrane proteome of the human pathogen Staphylococcus aureus. Mol Cell
Proteomics 7, 1460‐8 (2008).
13. Wenzel, M. et al. Proteomic response of Bacillus subtilis to lantibiotics reflects
differences in interaction with the cytoplasmic membrane. Antimicrob. Agents
Chemother. 56, 5749‐57 (2012).
14. Kohlrausch, U. & Höltje, J.V. One‐step purification procedure for UDP‐N‐acetylmuramyl‐
peptide murein precursors from Bacillus cereus. FEMS Microbiol. Lett. 62, 253–7 (1991).
15. Rick, P.D. et al. Characterization of the lipid carrier involved in the synthesis of
enterobacterial common antigen (ECA) and identification of a novel phosphoglyceride in
a mutant of Salmonella typhimurium defective in ECA synthesis. Glycobiology 8, 557–67
(1998).
16. Schmitt, P. et al. Insight into invertebrate defensin mechanism of action: oyster
defensins inhibit peptidoglycan biosynthesis by binding to lipid II. J. Biol. Chem. 285,
29208‐16 (2010).
17. Dai, D. & Ishiguro, E.E. MurH, a new genetic locus in Escherichia coli involved in cell wall
peptidoglycan biosynthesis. J. Bacteriol. 170, 2197‐201 (1988).
18. Mohammadi, T. et al. The essential peptidoglycan glycosyltransferase MurG forms a
complex with proteins involved in lateral envelope growth as well as with proteins
involved in cell division in Escherichia coli. Mol. Microbiol. 65, 1106‐21 (2007).
E An antimicrobial peptide delocalizes peripheral membrane proteins
119

19. Santhana Raj, L. et al. Rapid Method for Transmission Electron Microscopy Study of
Staphylococcus aureus ATCC 25923. Annals of Microscopy 7, 102‐8 (2007).
20. Zweytick, D. et al. Influence of N‐acylation of a peptide derived from human lactoferricin
on membrane selectivity. Biochim. Biophys. Acta 1758, 1426‐143 (2006).
21. Orlov, D.S., Nguyen, T. & Lehrer, R.I. Potassium release, a useful tool for studying
antimicrobial peptides. J. Microbiol. Methods 49, 325‐8 (2002).
22. Matias, V.R. & Beveridge, T.J. Cryo‐electron microscopy reveals native polymeric cell wall
structure in Bacillus subtilis 168 and the existence of a periplasmic space. Mol. Microbiol.
56, 240‐51 (2005).
23. Matias, V.R. & Beveridge, T.J. Lipoteichoic acid is a major component of the Bacillus
subtilis periplasm. J. Bacteriol. 190, 7414‐8 (2008).
24. Strahl, H. & Hamoen, L.W. Membrane potential is important for bacterial cell division.
Proc. Natl. Acad. Sci. USA 27, 12281‐86 (2010).
25. Andrés, M.T. & Fierro, J.F. Antimicrobial mechanism of action of transferrins: Selective
inhibition of H+‐ATPase. Antimicrob. Agents Chemother. 54, 4335–42 (2010).
26. Burstein, C., Tiankova, L. & Kepes, A. Respiratory control in Escherichia coli K 12. Eur. J.
Biochem. 94, 387‐92 (1979).
27. Palmgren, M.G, Sommarin, M., Ulvskov, P. & Larsson, C. Effect of detergents on the H(+)‐
ATPase activity of inside‐out and right‐side‐out plant plasma membrane vesicles.
Biochim. Biophys. Acta 1021, 133‐40 (1990).
28. Smith, J.J. & McFeters, G.A. Mechanisms of INT (2‐(4‐iodophenyl)‐3‐(4‐nitrophenyl)‐5‐
phenyl tetrazolium chloride), and CTC (5‐cyano‐2,3‐ditolyl tetrazolium chloride)
reduction in Escherichia coli K‐12, J. Microbiol. Methods 29, 161‐75 (1997).
29. Hoffmann, T. Boiangu, C., Moses, S. & Bremer, S. Responses of Bacillus subtilis to
hypotonic challenges: physiological contributions of mechanosensitive channels to
cellular survival. Appl. Environ. Microbiol. 74, 2454‐60 (2008).
30. Ruhr, E. & Sahl, H.G. Mode of action of the peptide antibiotic nisin and influence on the
membrane potential of whole cells and on cytoplasmic and artificial membrane vesicles.
Antimicrob. Agents Chemother. 27, 841‐5 (1985).
31. Bandow, J.E., Brötz, H, Leichert, L.I., Labischinski, H. & Hecker, M. Proteomic approach to
understanding antibiotic action. Antimicrob. Agents Chemother. 47, 948–55 (2003).
E An antimicrobial peptide delocalizes peripheral membrane proteins
120

F
Influence of lipidation on the mechanism of action
of an RW‐rich antimicrobial peptide
Michaela Wenzel, Patrick Schriek, Pascal Prochnow, H. Bauke Albada,
Nils Metzler‐Nolte, Julia E. Bandow
In preparation
F Influence of lipidation on peptide mode of action
121

Influence of lipidation on the mechanism of action of an RW‐rich antimicrobial peptide
Michaela Wenzel1, Patrick Schriek1, Pascal Prochnow1, H. Bauke Albada2, Nils Metzler‐Nolte2,
Julia E. Bandow1*
1Biology of Microorganisms, Ruhr University Bochum, Germany 2Bioinorganic Chemistry, Ruhr University Bochum, Germany
*Corresponding author. Mailing address: Ruhr‐Universität Bochum, Biologie der
Mikroorganismen, Universitätsstraße 150, 44801 Bochum, Germany. Phone: +49‐234‐32‐23102.
Fax: +49‐234‐32‐14620. E‐mail: [email protected]
Abstract
The synthetic antimicrobial peptide RWRWRW‐NH2 integrates into the bacterial membrane and
delocalizes essential peripheral membrane proteins involved in cell wall biosynthesis and
respiration. Lipidated derivatives showed significantly improved antibacterial activity against
both Gram‐positive and Gram‐negative bacteria. C8 acyl chains were coupled to the hexapeptide
by means of a C‐ or N‐terminally positioned lysine residue. Here, we report a comparative study
on the mechanism of action of the lipidated and non‐lipidated heptapeptides. All derivatives
depolarized the bacterial membrane without forming pores and affected cell wall integrity.
Proteomic profiling of the bacterial stress response to the small RW‐rich antimicrobial peptides
revealed few alterations compared to the parent peptide and indicated perturbation of
membrane phospholipids due to peptide integration. No specific markers were found to
correlate with lipidation while five proteins correlated with strict alteration of positively charged
and hydrophobic residues. We conclude that lipidation of the antimicrobial peptide enhances
antibacterial activity without significantly altering the mechanism of action.
Introduction
Antimicrobial peptides are often characterized by a high number of positively charged and
hydrophobic amino acids, resulting in a high affinity to bacterial membranes [1]. The cationic
hexapeptide RWRWRW‐NH2 (MP196) was designed with the objective to develop very short
antimicrobially active peptide sequences [2], [3]. Being neither cytotoxic nor hemolytic, MP196
displayed good Gram‐positive but only moderate Gram‐negative activity [4]. The peptide was
F Influence of lipidation on peptide mode of action
122
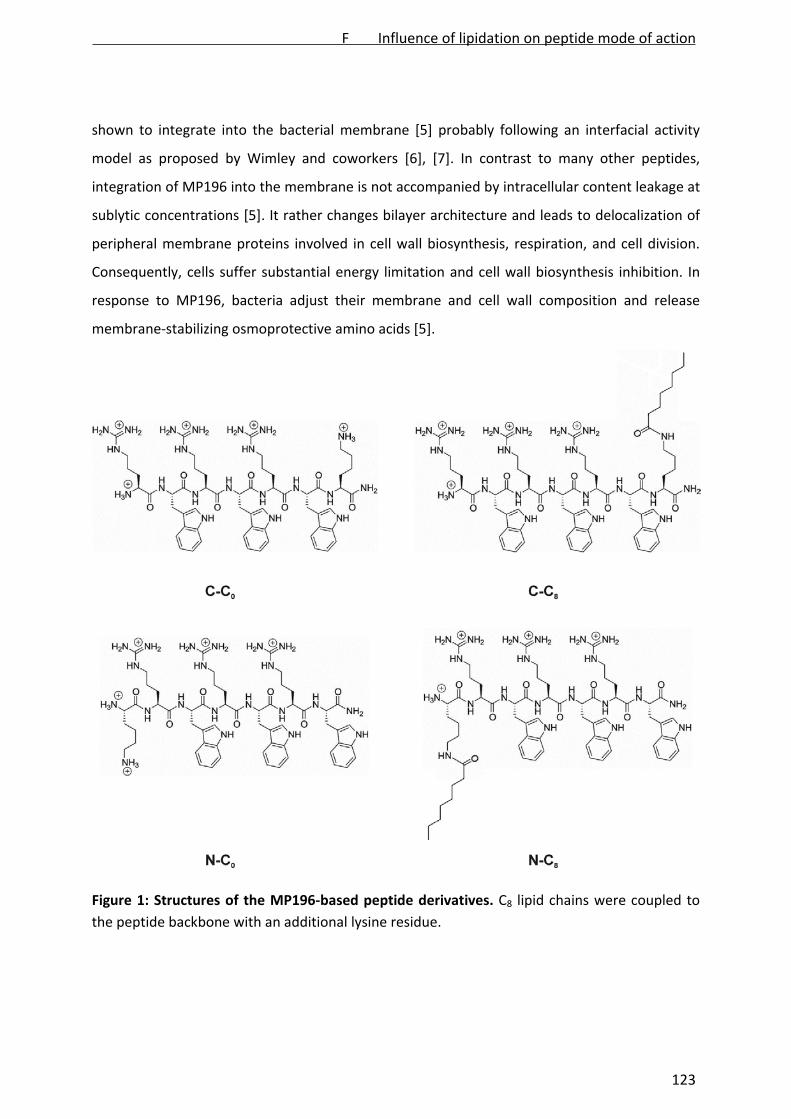
shown to integrate into the bacterial membrane [5] probably following an interfacial activity
model as proposed by Wimley and coworkers [6], [7]. In contrast to many other peptides,
integration of MP196 into the membrane is not accompanied by intracellular content leakage at
sublytic concentrations [5]. It rather changes bilayer architecture and leads to delocalization of
peripheral membrane proteins involved in cell wall biosynthesis, respiration, and cell division.
Consequently, cells suffer substantial energy limitation and cell wall biosynthesis inhibition. In
response to MP196, bacteria adjust their membrane and cell wall composition and release
membrane‐stabilizing osmoprotective amino acids [5].
Figure 1: Structures of the MP196‐based peptide derivatives. C8 lipid chains were coupled to
the peptide backbone with an additional lysine residue.
F Influence of lipidation on peptide mode of action
123

Short synthetic peptides like MP196 constitute a potent alternative to the typically much longer
natural peptides, as they are both easy to synthesize and to derivatize [6], [8], [9]. Being one of
the shortest antimicrobially active peptides known so far, MP196 is thought to represent the
minimal pharmacophore of positively charged and hydrophobic amino acids required for
antibacterial activity [10]. It is therefore regarded a lead structure for derivatization approaches.
Albada et al. described a set of heptapeptide derivatives amended with an either N‐ or C‐
terminally added lysine residue that facilitates introduction of acyl chains to the peptide
backbone. Additional lipophilic tails were thought to improve interaction with the lipid bilayer.
Among derivatives with C0 to C14 acyl chains, both the C‐ and N‐terminally C8‐substituted
peptides C‐C8 (H15C7(O)C‐KRWRWRW‐NH2) and N‐C8 (RWRWRWK‐C(O)C7H15) (Fig. 1) were the
most potent in terms of antibacterial activity. They displayed minimal inhibitory concentrations
in the low micro molar range even against highly resistant bacteria like Pseudomonas
aeruginosa and methicillin‐resistant Staphylococcus aureus. However, lipidation also
significantly increased cytotoxic and hemolytic activity suggesting alterations in interaction with
the membrane [11].
To investigate whether or not lipidation affects the mechanism of action of the peptide, the
lipidated derivatives C‐C8 and N‐C8 were analyzed in comparison with the hexapeptide lead
structure MP196 with regard to depolarization, pore formation, cell wall integrity, and their
impact on bacterial physiology. The latter one was investigated using proteomic profiling of
peptide‐stressed Bacillus subtilis, a method that supported previous structure‐activity
relationship and mechanism of action studies [5], [12], [13]. To distinguish between lipid‐
dependent and lysine‐dependent mechanistic properties, the non‐lipidated derivatives C‐C0
(KRWRWRW‐NH2) and N‐C0 (RWRWRWK‐NH2) (Fig. 1) carrying only the additional lysine residue
were also included in the study.
Materials and Methods
Antibiotics
Peptides were synthesized by solid‐phase synthesis as described by Albada et al. [11]. Antibiotic
stock solutions of 10 mg/ mL were prepared in sterile DMSO (MP196, C‐C0, C‐C8, N‐C0, N‐C8) or
0.01 M HCl (nisin). Nisin was a gift from H.G. Sahl, University of Bonn.
Bacterial strains and growth conditions
F Influence of lipidation on peptide mode of action
124

Bacillus subtilis 168 (trpC2) [14] was aerobically grown in Belitzky minimal medium (BMM) [15]
at 37°C. Minimal inhibitory concentrations (MICs) were determined as described previously. In
subsequent growth experiments, exponentially growing cultures were challenged with different
multiples of MIC [13]. For further analyses, antibiotic concentrations leading to a 50‐70%
reduced growth rate compared to an untreated control were chosen (Fig. S1): 15 µg/ mL C‐C0, 3
µg/ mL C‐C8, 15.75 µg/ mL N‐C0, 4.5 µg/ mL N‐C8, and 0.75 µg/ mL nisin.
Microscopy
Depolarization assays were performed with B. subtilis 1981 [16] as reported recently [17].
Briefly, cells were grown overnight in BMM. Those cells were then inoculated in modified BMM
containing xylose instead of glucose to induce GFP‐MinD expression. Cultures were grown to
early exponential phase and subsequently stressed with antibiotics. After 15 min of antibiotic
exposure, cells were imaged in fluorescent mode without fixation or immobilization.
Pore formation was monitored in B. subtilis 168 using the live/dead BacLight bacterial viability
kit (Invitrogen, Carlsbad, CA, USA) as described previously [5]. In short, exponentially growing
cells were treated with antibiotics for 15 min. Subsequently, 2 mL of culture were incubated
with 4 µL of a 1:1 mixture of the fluorescent dyes for another 15 min in the dark. Cells were
washed and resuspended in TE buffer (100 mM Tris/ 1mM EDTA, pH 7.5). Five (5) µL of the cell
suspension were imaged without fixation or immobilization in fluorescent mode.
Sample preparation and bright field microscopy was performed as described by Schneider et al.
[18] with minor modifications [17]. Briefly, B. subtilis 168 was grown in BMM and treated with
antibiotics in early exponential growth phase. After 15 min of antibiotic stress cells were fixed in
a 1:3 mixture of acetic acid and methanol, immobilized in low melting agarose, and
microscopically examined.
Radioactive 2D PAGE
Radioactive labeling of newly synthesized proteins and subsequent separation of the cytosolic
proteome by two‐dimensional polyacrylamide gel electrophoresis (2D PAGE) was performed as
described previously [13]. In short, 5 mL of a B. subtilis culture were exposed to antibiotics in
early exponential growth phase. After 10 min cells were pulse‐labeled with [35S]‐L‐methionine
for 5 min. Radioactive incorporation was stopped by inhibition of protein biosynthesis by adding
F Influence of lipidation on peptide mode of action
125

1 mg/mL chloramphenicol and an excess of non‐radioactive methionine. Cells were harvested
by centrifugation, washed three times with TE buffer, and disrupted by ultrasonication.
Fifty‐five (55) µg of protein for analytical and 300 µg for preparative gels were loaded onto 24
cm immobilized pH gradient (IPG) strips pH 4‐7 (GE Healthcare, Uppsala, Sweden) by passive
rehydration for 18 h. Proteins were separated in a first dimension by isoelectric focusing and in
a second dimension by SDS‐PAGE using 12.5% acrylamide gels. Analytical gel images were
analyzed as described by Bandow et al. [19] using Decodon Delta 2D 4.1 image analysis software
(Decodon, Greifswald, Germany). Proteins more than two‐fold upregulated in three
independent biological replicates were defined as marker proteins. Protein spots were
identified by nano HPLC‐ESI‐MS/MS as previously reported [20] using a Synapt G2S high
definition mass spectrometer equipped with an electrospray ionization source and an additional
ToF detector (Waters, Milford, MA, USA).
Results and Discussion
Influence on membrane and cell wall integrity
The influence of the peptide on the B. subtilis cell envelope was investigated using a set of
microscopic assays previously applied to MP196 [5]. The lantibiotic nisin, which is both a cell
wall biosynthesis inhibitor and a membrane pore former, was chosen as comparator compound.
Membrane depolarization was monitored employing a B. subtilis strain carrying a GFP fusion to
the cell division protein MinD, which localizes at the cell poles and in the cell division plane only
when the membrane potential is intact [16]. In a depolarized cell MinD delocalizes resulting in
irregular GFP distribution throughout the cell. While the untreated control showed distinct GPF‐
MinD localization, all four heptapeptides as well as lead structure MP196 and positive control
nisin led to similarly delocalized GFP‐MinD clusters (Fig. 2A,B) suggesting efficient membrane
depolarization.
MP196 differed from many other small peptides in its ability to disrupt membrane integrity at
sublytic concentrations. In contrast to e.g. nisin or daptomycin [21], it did not lead to
intracellular content leakage into defined medium [5]. To investigate, whether lipidation leads
to a higher membrane‐disruptive potential, pore formation was monitored with the live/dead
BacLight staining kit, which is based on two different fluorescent dyes. The green‐fluorescing
dye SYTO 9 is able to cross intact bacterial membranes. Propidium iodide, fluorescing red, may
F Influence of lipidation on peptide mode of action
126

only enter bacterial cells through membrane pores, thus indicating membrane disruption. In a
fluorescence overlay, cells with intact membranes appear green while porous cells appear
orange. In contrast to nisin, neither MP196 nor the heptapeptide derivatives significantly
facilitated penetration of the red‐fluorescing dye into B. subtilis (Fig. 2C). Apparently, neither
addition of the lysine side chain nor of the lipid tail enhanced the membrane‐disruptive
potential of the MP196 core structure.
Figure 2: Influence of the peptides on the bacterial cell envelope. (A) Fluorescence microscopy
images of B. subtilis 1981. Delocalization of MinD from the cell poles and the cell division plane
indicates membrane depolarization. (B) Bright field microscopy images of the same cells
displayed above. (C) Dual‐channel fluorescence microscopy images of B. subtilis stained with
live/dead BacLight. Green fluorescence indicates an intact, red fluorescence a porous
membrane. (D) Bright field microscopy images of B. subtilis fixed with acetic acid and methanol.
Loss of cell wall integrity is indicated by extrusion of the cell membrane through holes in the cell
wall.
MP196 was shown to disturb cell wall integrity, probably at least in part due to delocalization of
a peripheral membrane protein, MurG, involved in cell wall biosynthesis, and by causing energy
limitation [5]. The B. subtilis cell shape after acetic acid/methanol fixation was examined by
F Influence of lipidation on peptide mode of action
127

bright field microscopy (Fig. 2D). This treatment leads to excrescence of the cytoplasmic
membrane through holes in the cells wall, which typically occurs when cell wall biosynthesis is
inhibited at a membrane‐bound step [17], [18] as shown for MP196 and nisin. While untreated
control cells retained their shape after fixation, all four heptapeptide derivatives led to
membrane extrusions.
Influence on bacterial physiology
In order to characterize the physiological impact of the different derivatives, proteome analyses
of the bacterial stress response to antibiotic treatment were performed. Radioactive pulse
labeling with [35S]‐L‐methionine of proteins newly synthesized upon antibiotic exposure allowed
monitoring of the acute bacterial stress reaction reflective of the antibacterial mode of action.
Proteins were subsequently separated by 2D PAGE. Autoradiographs of the antibiotic‐treated
cultures were overlayed with those of the untreated controls. Proteins upregulated more than
two‐fold compared to the untreated control in three independent biological replicate
experiments were defined as marker proteins for treatment with a particular compound [17],
[19], [23].
Full protein expression patterns of B. subtilis in response to the different derivatives are
displayed in Figure S2. Details of protein function and regulation are compiled in Table 1, mass
spectrometric identification data in Table S1. Several marker proteins were regulated by the
extracytoplasmic function sigma factor W (σW), which is activated upon cell envelope stress by
antibiotics [24]. The proteome response patterns were dominated by proteins involved in the
cell envelope, cell wall, and membrane stress responses, as well as in energy and amino acid
metabolism. The proteins upregulated in response to all five peptides illustrate this particularly
well and are shown in Figure 3.
Some markers have been reported to be specific for different aspects of cell envelope stress.
YceC and YceH constitute a signature for cell envelope stress [17]. YceC, sharing similarity with a
tellurium resistance protein, was upregulated in response to all derivatives. YceH, annotated as
toxic anion resistance protein, was identified as a marker for C‐C0, N‐C0, and N‐C8, only closely
missing the cutoff of two‐fold induction in one out of three replicates of C‐C8.
F Influence of lipidation on peptide mode of action
128

Table 1: Induction factors and protein function of the identified marker proteins upregulated by the four peptide derivatives.
induction factor
protein C‐C0 C‐C8 N‐C0 N‐C8 function regulator stress category
YceC* 19.8 8.5 11.5 16.7 tellurium resistance protein σB, σW, σM cell envelope
YceH* 10.5 8.7 10.1 16.0 toxic anion resistance protein σB, σM, σW cell envelope
YjbC/BacB 3.7 1.9 8.7 8.2 general stress protein/involved in L‐anticapsin synthesis PerR, σB, σM, σW, σX/ unknown
cell envelope
YfhM 7.7 3.5 9.5 7.3 similar to epoxide hydrolase σB, σW cell envelope
FosB 16.6 4.9 28.1 18.6 bacillithiol‐S‐transferase σW cell envelope
YvlB 11.8 7.1 6.6 8.2 unknown σW cell envelope
YvlB 12.7 8.5 13.4 20.6 unknown σW cell envelope
YpuA 5.1 4.6 8.6 4.7 unknown σM cell envelope
YknY/LiaH# 5.7 5.1 5.8 3.7 ABC transporter (ATP‐binding protein) for the export of the SdpC toxin/similar to phage shock protein
σW/LiaRS cell envelope
LiaH# 48.1 26.7 18.3 4.0 similar to phage shock protein LiaRS cell wall
LiaH# 18.2 9.1 8.1 2.9 similar to phage shock protein LiaRS cell wall
RacX 21.7 13.6 18.8 6.6 amino acid racemase, production of D‐ animo acids, control of biofilm formation
σW cell wall
PbpE 7.7 6.6 13.5 7.6 penicillin‐binding protein σW cell wall
PbpE 3.9 0.8 2.3 11.8 penicillin‐binding protein σW cell wall
MurB 4.9 5.8 5.1 2.8 UDP‐N‐acetylenolpyruvoylglucosamine reductase σE, σM, SpoIIID cell wall
PspA° 16.2 6.6 13.1 9.7 phage shock protein A homolog σW membrane
AcsA 12.3 9.0 3.2 14.7 acetyl‐CoA synthetase unknown membrane
AcsA 12.0 9.4 0.7 1.1 acetyl‐CoA synthetase unknown membrane
FabF 2.6 1.9 2.8 2.6 beta‐ketoacyl‐acyl carrier protein synthase II, involved in control of membrane fluidity
FapR, σW membrane
YoxD 3.0 5.3 8.6 2.7 similar to 3‐oxoacyl‐ acyl‐carrier protein reductase unknown membrane
YjdA 8.2 3.7 13.3 10.0 similar to 3‐oxoacyl‐acyl‐carrier protein reductase unknown membrane
YuaI 8.3 3.9 7.4 8.9 involved in reducing membrane fluidity σW membrane
YkpA 4.5 6.0 32.7 35.0 similar to ABC transporter (ATP‐binding protein) unknown membrane
F Influence of lipidation on peptide mode of action
129
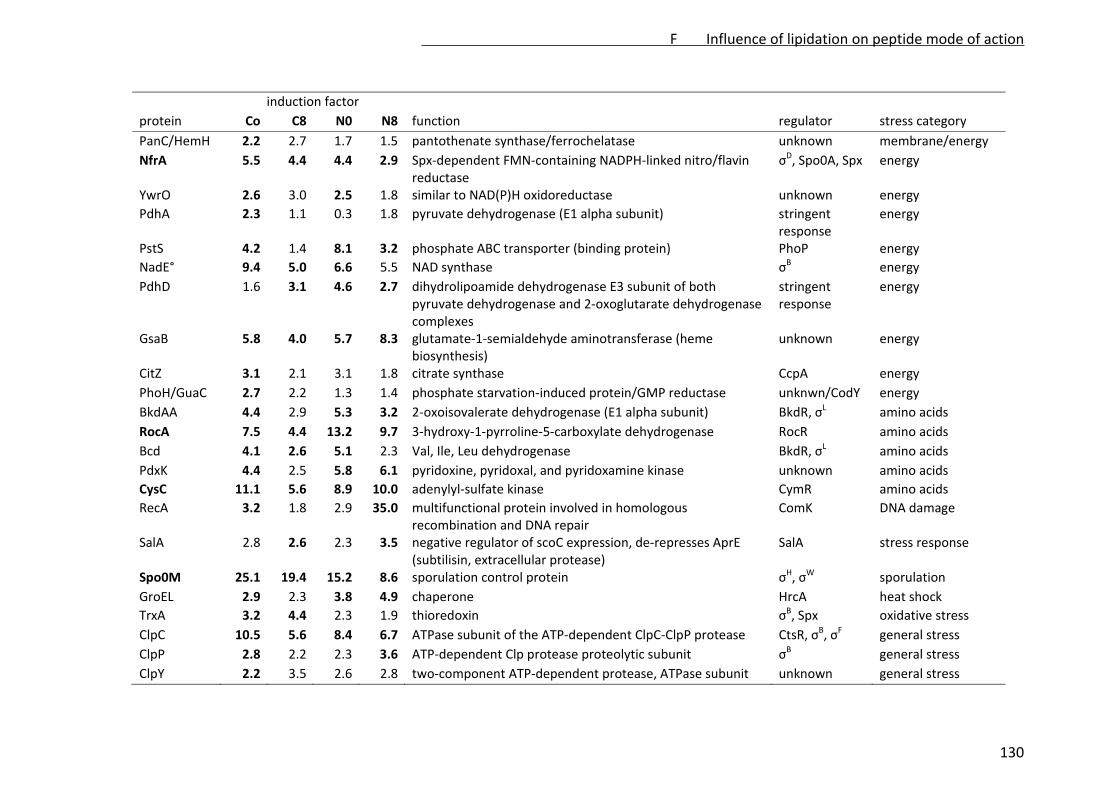
induction factor
protein Co C8 N0 N8 function regulator stress category
PanC/HemH 2.2 2.7 1.7 1.5 pantothenate synthase/ferrochelatase unknown membrane/energy
NfrA 5.5 4.4 4.4 2.9 Spx‐dependent FMN‐containing NADPH‐linked nitro/flavin reductase
σD, Spo0A, Spx energy
YwrO 2.6 3.0 2.5 1.8 similar to NAD(P)H oxidoreductase unknown energy
PdhA 2.3 1.1 0.3 1.8 pyruvate dehydrogenase (E1 alpha subunit) stringent response
energy
PstS 4.2 1.4 8.1 3.2 phosphate ABC transporter (binding protein) PhoP energy
NadE° 9.4 5.0 6.6 5.5 NAD synthase σB energy
PdhD 1.6 3.1 4.6 2.7 dihydrolipoamide dehydrogenase E3 subunit of both pyruvate dehydrogenase and 2‐oxoglutarate dehydrogenase complexes
stringent response
energy
GsaB 5.8 4.0 5.7 8.3 glutamate‐1‐semialdehyde aminotransferase (heme biosynthesis)
unknown energy
CitZ 3.1 2.1 3.1 1.8 citrate synthase CcpA energy
PhoH/GuaC 2.7 2.2 1.3 1.4 phosphate starvation‐induced protein/GMP reductase unknwn/CodY energy
BkdAA 4.4 2.9 5.3 3.2 2‐oxoisovalerate dehydrogenase (E1 alpha subunit) BkdR, σL amino acids
RocA 7.5 4.4 13.2 9.7 3‐hydroxy‐1‐pyrroline‐5‐carboxylate dehydrogenase RocR amino acids
Bcd 4.1 2.6 5.1 2.3 Val, Ile, Leu dehydrogenase BkdR, σL amino acids
PdxK 4.4 2.5 5.8 6.1 pyridoxine, pyridoxal, and pyridoxamine kinase unknown amino acids
CysC 11.1 5.6 8.9 10.0 adenylyl‐sulfate kinase CymR amino acids
RecA 3.2 1.8 2.9 35.0 multifunctional protein involved in homologous recombination and DNA repair
ComK DNA damage
SalA 2.8 2.6 2.3 3.5 negative regulator of scoC expression, de‐represses AprE (subtilisin, extracellular protease)
SalA stress response
Spo0M 25.1 19.4 15.2 8.6 sporulation control protein σH, σW sporulation
GroEL 2.9 2.3 3.8 4.9 chaperone HrcA heat shock
TrxA 3.2 4.4 2.3 1.9 thioredoxin σB, Spx oxidative stress
ClpC 10.5 5.6 8.4 6.7 ATPase subunit of the ATP‐dependent ClpC‐ClpP protease CtsR, σB, σF general stress
ClpP 2.8 2.2 2.3 3.6 ATP‐dependent Clp protease proteolytic subunit σB general stress
ClpY 2.2 3.5 2.6 2.8 two‐component ATP‐dependent protease, ATPase subunit unknown general stress
F Influence of lipidation on peptide mode of action
130
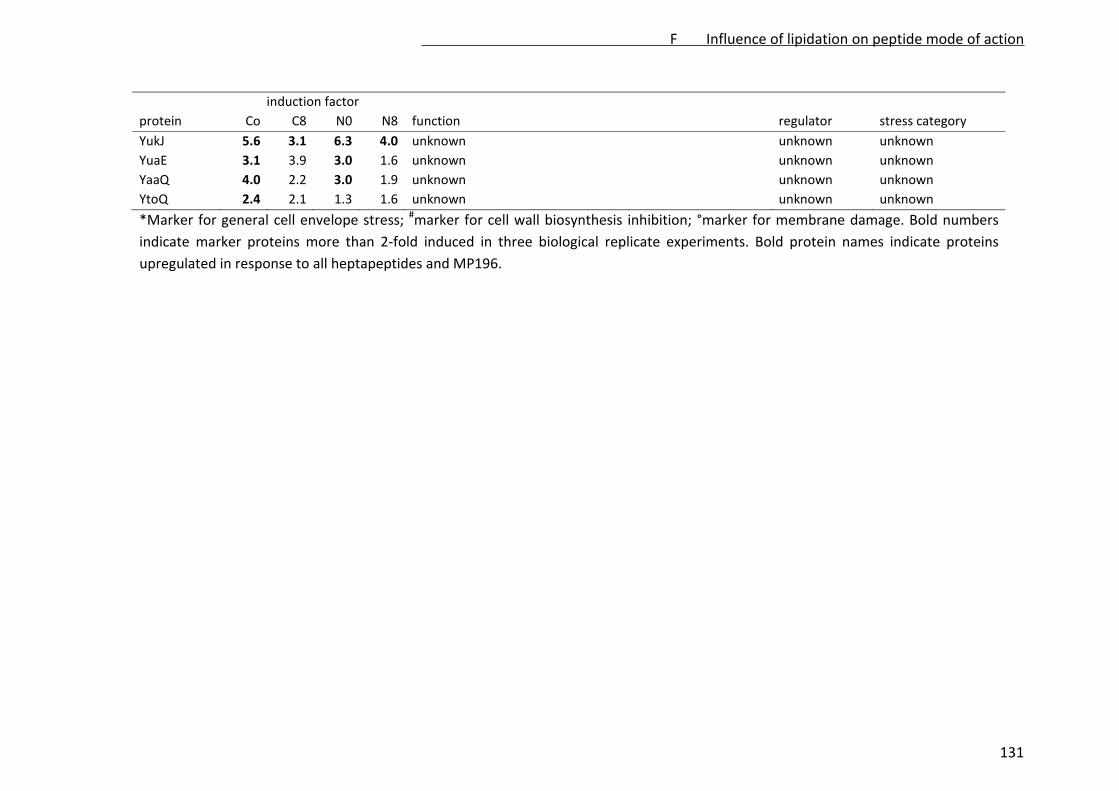
induction factor
protein Co C8 N0 N8 function regulator stress category
YukJ 5.6 3.1 6.3 4.0 unknown unknown unknown
YuaE 3.1 3.9 3.0 1.6 unknown unknown unknown
YaaQ 4.0 2.2 3.0 1.9 unknown unknown unknown
YtoQ 2.4 2.1 1.3 1.6 unknown unknown unknown
*Marker for general cell envelope stress; #marker for cell wall biosynthesis inhibition; °marker for membrane damage. Bold numbers
indicate marker proteins more than 2‐fold induced in three biological replicate experiments. Bold protein names indicate proteins
upregulated in response to all heptapeptides and MP196.
F Influence of lipidation on peptide mode of action
131
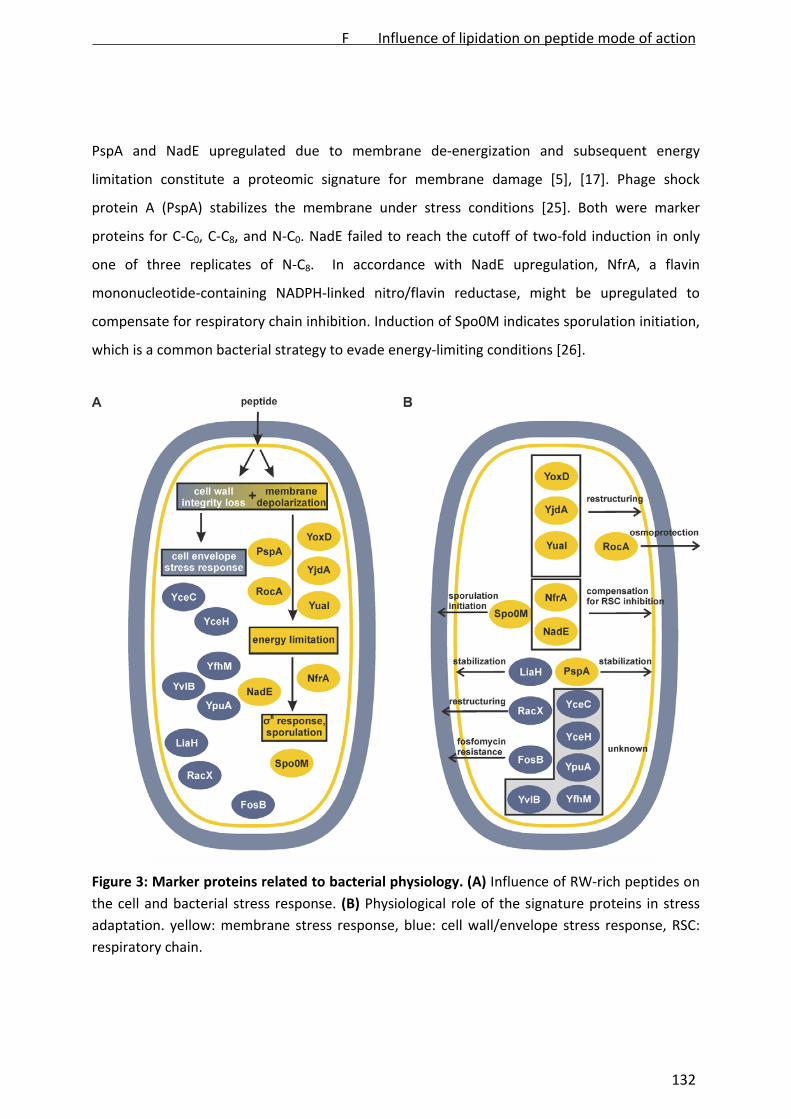
PspA and NadE upregulated due to membrane de‐energization and subsequent energy
limitation constitute a proteomic signature for membrane damage [5], [17]. Phage shock
protein A (PspA) stabilizes the membrane under stress conditions [25]. Both were marker
proteins for C‐C0, C‐C8, and N‐C0. NadE failed to reach the cutoff of two‐fold induction in only
one of three replicates of N‐C8. In accordance with NadE upregulation, NfrA, a flavin
mononucleotide‐containing NADPH‐linked nitro/flavin reductase, might be upregulated to
compensate for respiratory chain inhibition. Induction of Spo0M indicates sporulation initiation,
which is a common bacterial strategy to evade energy‐limiting conditions [26].
Figure 3: Marker proteins related to bacterial physiology. (A) Influence of RW‐rich peptides on
the cell and bacterial stress response. (B) Physiological role of the signature proteins in stress
adaptation. yellow: membrane stress response, blue: cell wall/envelope stress response, RSC:
respiratory chain.
F Influence of lipidation on peptide mode of action
132

Several other markers reflect attempts of B. subtilis to adapt to membrane stress. YoxD and
YjdA share similarity with fatty acid biosynthesis enzymes but are not part of the standard
biosynthetic pathway. YuaI belongs to the yuaF‐floT‐yuaI operon. Together with FabF (marker
for C‐C0, N‐C0, and N‐C8, closely missing the cutoff for C‐C8) (Fig. 4, Table 1), YuaI was shown to
reduce membrane fluidity [27], [28]. Together, these proteins might be involved in phospholipid
adaptation in order to prevent peptide binding and integration into the bilayer. Adjustment of
the lipid composition has been demonstrated before in Corynebacterium glutamicum in
response to a metal‐substituted MP196 derivative [29]. Similarly, the amino acid racemase RacX
may be involved in enhancing cell wall lipoteichoic acid D‐alanylation, another strategy to adapt
to peptide stress [5], [30]. Upregulation of RocA, a dehydrogenase converting (S)‐1‐pyrroline‐5‐
carboxylate into L‐glutamate might reflect glutamate release, which has been shown to play a
role in bacterial survival under peptide stress, probably due to osmoprotective effects of
glutamate [5].
The PspA homologue LiaH, which was strongly induced by all derivatives, has been described as
specific marker for inhibition of membrane‐bound cell wall biosynthesis steps. Both PspA and
LiaH bind to the membrane conferring higher stability under stress conditions [25], [31].
Another cell wall biosynthesis inhibition marker upregulated by all derivatives is the unknown
protein YpuA (formerly referred to as NGM1 [17]), which has also been demonstrated to be
upregulated by different kinds of cell wall stress [32]. Together, LiaH and YpuA indicate
disturbance of cell wall biosynthesis by all heptapeptides. In agreement with the microscopy‐
based assays, the proteome analysis data reflect high mechanistic similarity between MP196
and its heptapeptide derivatives.
Differences to the lead structure
Nine marker proteins, 6 of which were identified, were upregulated by all heptapeptide
derivatives but not by MP196 (Table S2). They were involved in cell wall modification or
connected to amino acid and energy metabolism, thus belonging to protein categories similarly
upregulated in response to MP196 treatment and probably not reflecting a major difference in
mode of action. However, by comparing the proteome response patterns to an antibiotic
reference compendium, distinct differences between the heptapeptides and MP196 were
revealed. The library comprises B. subtilis proteome response profiles to over 50 antibiotic
F Influence of lipidation on peptide mode of action
133

agents [13], [17], [22], [33]. The highest degree of overlap with MP196 and its heptapeptide
derivatives was found for membrane‐integrating gallidermin, depolarizing gramicidin S, and
potassium ionophore valinomycin (Table 2, see Table S2 for details) [5].
Table 2: Marker proteins overlapping between lead structure MP196, its four derivatives, and
comparator compounds
total
MP196
C‐C
0
C‐C
8
N‐C
0
N‐C
8
gram
icidin S
galliderm
in
valin
omycin
MP196 36 21 16 19 17 17 10 19
C‐C0 43 21 26 34 32 16 13 10
C‐C8 28 16 26 26 24 14 12 10
N‐C0 36 19 34 26 30 15 10 10
N‐C8 35 17 32 24 30 14 10 10
gramicidin S 20 17 16 14 15 14 7 8
gallidermin 25 10 13 15 10 10 7 4
valinomycin 22 19 10 14 10 10 8 4
While gallidermin and gramicidin S, which both disturb the phospholipid bilayer structure,
shared almost the same subset of markers with MP196 and the derivatives, only half of the
marker proteins common to valinomycin and MP196 were also shared with the derivatives.
Valinomycin does not perturb the phospholipid bilayer structure but affects ion homoeostasis
and energy metabolism [5]. The proteins upregulated by valinomycin and MP196 are involved in
energy metabolism and the general stress response. In contrast, a number of the proteins
upregulated by the derivatives ‐ but not MP196 or the comparators ‐ are involved in membrane
and cell wall modification (Table 1). B. subtilis seems to intensify envelope‐related peptide
defense strategies in response to the lipidated and non‐lipidated derivatives, set off by
perturbations of the phospholipid bilayer, and compensation for downstream effects related to
energy limitation in response to MP196.
The cell wall biosynthesis inhibition signature reported previously comprises LiaH, the ABC
transporter subunits YtrE and YtrB, and the tRNA‐modifying enzyme TrmB, all of which are
upregulated in response to MP196. In response to the heptapeptides only LiaH was
F Influence of lipidation on peptide mode of action
134

upregulated, which could point to a reduced impact on cell wall biosynthesis by the derivatives
compared to MP196.
Both energy limitation and cell wall biosynthesis inhibition result from delocalization of
peripheral membrane proteins due to integration of MP196 into the membrane (cytochrome c
and MurG) [5]. The additional lipid tails and/or positive charges could alter peptide‐
phospholipid interaction, amplifying impact on the architecture of the phospholipid bilayer.
MP196 itself showed a similar effect at higher concentrations (Fig. S3). The stress response of B.
subtilis to moderate MP196 concentrations showed a high overlap with lipid II‐binding
antibiotics, affecting cell wall biosynthesis, while the overlap was stronger with membrane‐
integrating antibiotics at high MP196 concentrations.
Differences between the heptapeptide derivatives
Some differences between the derivatives were observed (Fig. 4A, S2). Keeping in mind the
higher antibacterial and hemolytic activity of the lipidated peptides compared to the lead
structure or the non‐lipidated heptapeptides, we looked for markers that correlate with
lipidation. The lipidated peptides C‐C8 and N‐C8 shared only one marker protein that was not
upregulated by the other peptides. SalA derepresses the extracellular protease subtilisin E,
which is secreted to acquire nitrogen from extracellular protein sources [34]. As the non‐
lipidated heptapeptides C‐C0 and N‐C0 showed the same trend of SalA upregulation, albeit
missing the stringent two‐fold cutoff, SalA cannot be considered to be a reliable indicator for
lipid‐derived mechanistic differences. The uncharacterized protein YuaE was a marker for the
two non‐lipidated heptapeptides C‐C0 and N‐C0, but was also upregulated in response to the
lipidated peptides without meeting the cutoff of two‐fold upregulation. The site of modification
seems to be of some importance however. A total of five proteins were differentially expressed
depending on the position of the lysine residue. Unknown protein YtoQ, phosphate starvation‐
induced protein PhoH (identified in one spot with the GMP reductase GuaC), and three
unidentified proteins were upregulated in response to C‐C0 and by C‐C8, closely missing the
cutoff in the latter, but not by the N‐terminally modified peptides.
The oxidative stress‐protective thioredoxin was upregulated by both C‐terminally modified
heptapeptides C‐C0 and C‐C8 but showed the same trend in response the other peptides.
F Influence of lipidation on peptide mode of action
135

Figure 4: Marker proteins of all MP196 derivatives. (A) Proteome response. purple N‐terminal
modification (alternating pattern disrupted), pink C‐terminal modification (alternating pattern
retained); CCNN_X shared markers of C‐C0, C‐C8, N‐C0, and N‐C8, CCN0_X shared markers of C‐C0,
C‐C8, and N‐C0, CCN8_X shared markers of C‐C0, C‐C8, and N‐C8, C0NN_X shared markers of C‐C0,
N‐C0, and N‐C8, C8NN_X shared markers of C‐C8, N‐C0, and N‐C8, CC_X shared markers of C‐C0
and C‐C8, NN_X shared markers of N‐C0 and N‐C8, C0N0_X shared markers of C‐C0 and NC‐0,
C8N8_X shared markers of C‐C8 and N‐C8, C0N8_X shared markers of C‐C0 and N‐C8, C0_X specific
markers of C‐C0, C8_X specific markers of C‐C8, N0_X specific markers of N‐C0, N8_X specific
markers of N‐C8. Boxes highlight signature proteins: orange short RW‐rich peptides, purple
addition of a lysine side chain, red lipidation, pink C‐terminal lysine modification. Dotted lines
indicate that in one of the peptide derivatives the protein was upregulated but did not meet the
two‐fold cutoff. (B) Relation of specific marker protein upregulation and peptide sequence. +
positively charged residue, o hydrophobic residue.
F Influence of lipidation on peptide mode of action
136

Introduction of the lysine residue at the C‐terminus retains the pattern of alternating positively
charged and hydrophobic amino acids. In contrast, N‐terminal derivatization results in two
successive positive charges at the N‐terminus (Fig. 4B). According to the difference in marker
proteins, the charge distribution in the RW‐backbone structure seems to affect the bacterial
response more than the introduction of a lipid tail. However, lipidation of the N‐terminal lysine
(N‐C8) produced 9 markers, 5 of which showed no trend of upregulation by the other peptides.
N‐terminal lipidation seems to affect the mechanistic properties of the peptide structure in a
unique way, which was also reflected in the elevated hemolysis.
Concluding Remarks
Lipidation has recently been shown to enhance antibacterial activity of a small cationic
antimicrobial peptide RWRWRW‐NH2. Hemolytic activity was increased as well. A systematic
exchange of L‐ to D‐amino acids in the lipidated N‐C8 and C‐C8 structures identified peptides
without hemolytic activity but fully retained antibacterial potency [35] making lipidated
peptides attractive for antibacterial drug discovery. Here, we report the influence of positive
charge and a lipidation on the mechanism of action of a small cationic antimicrobial peptide.
Validating the cytoplasmic membrane as their antibiotic target, pore‐independent membrane
depolarization and corruption of cell wall integrity were demonstrated for all derivatives.
Proteome analysis confirmed a highly similar membrane‐related mode of action for all
derivatives and the lead structure MP196, which has recently been investigated in depth for its
impact on bacterial physiology. These results indicate that antimicrobial peptides can be
derivatized with lipid chains without considerably changing the mode of action. Upregulation of
derivative‐specific marker proteins suggests that interaction of the peptides with the
phospholipid bilayer can be modulated by altering the distribution of positive charges and
lipophilic residues in the peptide.
Acknowledgements
The authors thank H.‐G. Sahl, University of Bonn, for supplying nisin. This work was financially
supported by a start‐up grant from the Ruhr University Bochum (JEB), and a grant from the state
of North Rhine‐Westphalia (NRW), Germany and the European Union, European Regional
Development Fund, "Investing in your future" (JEB, NMN). JEB acknowledges financial support
F Influence of lipidation on peptide mode of action
137

for the mass spectrometer by the state of North Rhine Westphalia (Forschungsgroßgerät der
Länder).
The authors declare no financial/commercial conflicts of interest.
References
1. Yeaman MR, Yount NY. (2003) Mechanisms of Antimicrobial Peptide Action and Resistance.
Pharmacol Rev 55: 27‐55.
2. Chantson JT, Verga Falzacappa MV, Crovella S, Metzler‐Nolte N. (2006) Solid‐phase
synthesis, characterization, and antibacterial activities of metallocene‐peptide
bioconjugates. ChemMedChem 1: 1268‐1274.
3. Chantson JT, Varga Falzacappa MV, Crovella S, Metzler‐Nolte N. (2005) Antibacterial
activities of ferrocenoyl‐ and cobaltocenium‐peptide bioconjugates. J Organomet Chem 690:
4564‐4574.
4. Albada HB, Chiriac AI, Wenzel M, Penkova M, Bandow JE, et al. (2012) Modulating the
activity of short arginine‐tryptophan containing antibacterial peptides with N‐terminal
metallocenoyl groups. Beilstein J. Org. Chem 8: 1753‐1764.
5. Wenzel M, Chiriac AI, Otto A, Zweytick D, May C, et al. An antimicrobial peptide delocalizes
peripheral membrane proteins. submitted.
6. Rathinakumar R, Walkenhorst WF, Wimley WC. (2009) Broad‐spectrum antimicrobial
peptides by rational combinatory design and high‐throughput screening: the importance of
interfacial activity. J Am Chem Soc 131: 7609‐7617.
7. Wimley WC. (2010) Describing the Mechanism of Antimicrobial Peptides with the Interfacial
Activity Model. ACS Chem Biol 5: 905‐917.
8. Patra M, Gasser G, Metzler‐Nolte N. (2012) Small organometallic compounds as antibacterial
agents. Dalton Trans 41: 6350‐6358.
9. Sharma RK, Sundriyal S, Wangoo N, Tegge W, Jain R. (2010) New antimicrobial hexapeptides:
synthesis, antimicrobial activities, cytotoxicity, and mechanistic studies. ChemMedChem 5:
86‐95.
10. Strøm MB, Haug BE, Skar ML, Stensen W, Stiberg T, et al. (2003) The pharmacophore of
short cationic antibacterial peptides. J Med Chem 46: 1567‐1570.
11. Albada HB, Prochnow P, Bobersky S, Langklotz S, Schriek P, et al. (2012) Tuning the activity
of short Arg‐Trp antimicrobial peptides by lipidation of C‐ or N‐terminal lysine side‐chain.
ACS Med Chem Lett 3: 980‐984.
12. Brötz‐Oesterhelt H, Beyer D, Kroll HP, Endermann R, Ladel C, et al. (2005) Dysregulation of
bacterial proteolytic machinery by a new class of antibiotics. Nat Med 11: 1082‐1087.
F Influence of lipidation on peptide mode of action
138

13. Wenzel M, Patra M, Albrecht D, Chen DYK, Nicolaou KC, et al. (2011) Proteomic signature of
fatty acid biosynthesis inhibition available for in vivo mechanism‐of‐action studies.
Antimicrob Agents Chemother 55: 2590‐2596.
14. Agnostopoulos C, Spizizen J. (1961) Requirements for transformation in Bacillus subtilis. J
Bacteriol 81: 741‐746.
15. Stülke J, Hanschke R, Hecker M. (1993) Temporal activation of betaglucanase synthesis in
Bacillus subtilis is mediated by the GTP pool. J Gen Microbiol 139: 2041‐2045.
16. Strahl H, Hamoen LW. (2010) Membrane potential is important for bacterial cell division.
Proc Natl Acad Sci USA 27: 12281‐12286.
17. Wenzel M, Kohl B, Münch D, Raatschen N, Albada HB, et al. Proteomic response of Bacillus
subtilis to lantibiotics reflects differences in interaction with the cytoplasmic membrane.
Antimicrob Agents Chemother 56: 5749‐5757.
18. Schneider T, Kruse T, Wimmer R, Wiedemann I, Sass V, et al. (2010) Plectasin, a fungal
defensin, targets the bacterial cell wall precursor lipid II. Science 328: 1168‐1172.
19. Bandow JE, Baker JD, Berth M, Painter C, Sepulveda OJ et al. (2008) Improved image analysis
workflow for 2D gels enables large‐scale 2D gel‐based proteomics studies ‐ COPD biomarker
discovery study. Proteomics 8: 3030‐3041.
20. Wenzel M, Patra M, Senges CHR, Ott I, Stepanek JJ, et al. Analysis of the mechanism of
action of potent antibacterial hetero‐tri‐organometallic compounds – a structurally new
class of antibiotics. ACS Chem Biol. doi: 10.1021/cb4000844.
21. Hobbs JK, Miller K, O’Neill AJ, Chopra I. (2008) Consequences of daptomycin‐mediated
membrane damage in Staphylococcus aureus. J Antimicrob Chermother 62: 1003‐1008.
22. Bandow JE, Brötz H, Leichert LI, Labischinski H, Hecker M. (2003) Proteomic approach to
understanding antibiotic action. Antimicrob Agents Chemother 47: 948‐955.
23. Wenzel M, Bandow JE. (2012) Proteomic signatures in antibiotic research. Proteomics 11:
3256‐3268.
24. Butcher BG, Helmann JD. (2006) Identification of Bacillus subtilis sigma‐dependent genes
that provide intrinsic resistance to antimicrobial compounds produced by Bacilli. Mol
Microbiol 60: 765‐782.
25. Kobayashi R, Suzuki T, Yoshida M. (2007) Escherichia coli phage‐shock protein A (PspA) binds
to membrane phospholipids and repairs proton leakage of the damaged membranes. Mol
Microbiol 66: 100‐109.
26. Voelker U, Voelker A, Haldenwang WG. (1996) Reactivation of the Bacillus subtilis anti‐sigma
B antagonist, RsbV, by stress‐ or starvation‐induced phosphatase activities. J Bacteriol 178:
5456‐5463.
27. Kingston AW, Subramanian C, Rock CO, Helmann JD. (2011) A σW‐dependent stress response
in Bacillus subtilis that reduces membrane fluidity. Mol Microbiol 81: 69‐79.
28. Lee YH, Kingston AW, Helmann JD. (2012) Glutamate dehydrogenase affects resistance to
cell wall antibiotics in Bacillus subtilis. J Bacteriol 194: 993‐1001.
F Influence of lipidation on peptide mode of action
139

29. Fränzel B, Frese C, Penkova M, Metzler‐Nolte N, Bandow JE, et al. (2010) Corynebacterium
glutamicum exhibits a membrane‐related response to a small ferrocene‐conjugated
antimicrobial peptide. J Biol Inorg Chem 15: 1293‐1303.
30. Bertsche U, Weidenmaier C, Kuehner D, Yang SJ, Baur S, et al. (2011) Correlation of
daptomycin resistance in a clinical Staphylococcus aureus strain with increased cell wall
teichoic acid production and D‐alanylation. Antimicrob Agents Chemother 55: 3922‐3928.
31. Wolf D, Kalamorz F, Wecke T, Juszczak A, Mäder U, et al. In‐depth profiling of the LiaR
response of Bacillus subtilis. J Bacteriol 192: 4680‐4693.
32. Urban A, Eckermann S, Fast B, Metzger S, Gehling M, et al. (2007) Novel whole‐cell antibiotic
biosensors for compound discovery. Appl Environ Microbiol 73: 6436‐6443.
33. Raatschen N, Bandow JE. 2‐D gel‐based proteomic approaches to antibiotic drug discovery.
Curr Protoc Microbiol Chapter 1:Unit1F.2
34. Ogura M, Matsuzawa A, Yoshikawa H, Tanaka T. (2004) Bacillus subtilis SalA (YbaL)
negatively regulates expression of scoC, which encodes the repressor for the alkaline
exoprotease gene, aprE. J Bacteriol 184: 3056‐3064.
35. Albada HB, Prochnow P, Bobersky S, Langklotz S, Bandow JE, et al. Short antibacterial
peptides with significantly reduced hemolytic activity can be identified by an L‐to‐D
exchange scan of their amino acid residues. submitted.
Supplementary Information
Contents
i. Supplementary Tables
ii. Supplementary Figures
iii. References
F Influence of lipidation on peptide mode of action
140

i. Supplementary Tables
Supplementary Table 1: nano UPLC‐ESI‐MS/MS data of spot identification
protein MW [Da]
pI PLGS score
peptides coverage (%) precursor mass error (ppm)
number of peptides with fragment data
product mass error (ppm)
AcsAl 64850 5.55 4947 80 45 1.6169 75 3.9868
AcsAr 64850 5.54 4431 53 36 1.3795 48 4.0771
BacB 26822 5.19 6750 17 41 2.8160 13 4.5349
Bcd 39966 4.94 10682 56 55 2.6251 50 4.0140
BkdAA 36310 4.78 5404 4 48 3.1813 18 8.4847
ClpC 90063 5.75 22605 182 60 1.5483 159 4.8319
ClpP 21668 5.00 7705 36 28 1.2729 32 2.9824
ClpY 52553 5.31 10546 90 56 1.3788 82 3.9932
CysC 22530 5.08 23981 51 60 1.7163 44 4.2176
FabF 43977 4.76 4091 69 46 1.5398 60 4.3794
FosB 17161 6.14 754 4 17 1.5744 4 2.1651
GroEL 57388 4.53 17614 121 77 2.4981 110 5.6355
GsaB 46154 5.65 3403 4 50 5.1234 21 8.8421
GuaC 35827 6.08 4667 41 56 1.5061 36 4.4410
HemH 35325 4.62 1418 15 52 5.8581 20 10.2544
LiaHl 25682 6.17 4794 14 46 1.5921 13 4.6401
MurB 32787 6.08 17578 57 67 1.9137 52 4.0263
NfrA 28302 5.73 9602 24 33 1.5843 20 4.4483
PanC 31938 4.63 5324 15 65 6.1887 21 9.3234
PbpEl 51404 4.84 8109 63 41 1.8203 58 4.8336
PbpEr 51404 4.84 12871 7 55 3.7195 43 8.2619
PdhA 41522 5.83 12509 80 59 3.1268 71 3.9294
PdhD 49701 4.76 7868 17 66 3.225 52 9.1011
PdxK 28998 4.92 2498 20 42 1.0566 15 4.1123
PhoH 35518 6.12 11735 41 54 1.4392 35 4.2039
F Influence of lipidation on peptide mode of action
141

protein MW [Da]
pI PLGS score
peptides coverage (%) precursor mass error (ppm)
number of peptides with fragment data
product mass error (ppm)
PstS 31664 4.84 12665 48 48 1.5004 44 4.0909
RecA 38035 4.82 23208 58 63 1.8056 50 3.5429
RocA 56284 5.58 12601 56 57 2.2908 52 4.2925
SalA 38614 5.23 6829 37 44 1.4237 33 3.6666
Spo0M 29714 4.26 25783 71 60 1.5721 63 3.9270
TrxA 11385 4.30 11205 14 62 1.0655 11 2.5525
YaaQ 11959 6.10 11749 18 39 1.8629 14 0.0125
YceC 21809 5.31 8639 46 66 2.0471 40 4.0478
YfhM 32737 6.07 7499 22 44 1.2305 20 3.8769
YjbC 23105 5.14 6233 38 54 2.2088 32 3.3452
YknY 25255 5.81 6242 26 38 1.3441 25 3.0882
YkpA 61017 4.98 5582 101 53 1.7916 89 4.5519
YoxD 25283 5.34 12493 50 66 1.6707 43 3.9615
YpuA 31275 4.51 20679 38 53 1.7477 30 4.3916
YtoQ 16780 5.77 9248 14 40 0.8658 14 3.3474
YuaE 19098 6.20 5138 18 39 1.7526 15 3.6214
YuaI 19830 5.15 6665 19 49 1.7138 18 3.5182
YukJ 25538 4.42 3593 24 26 1.9362 21 4.2716
YwrO 19942 5.20 13172 50 63 2.2595 46 4.4720
CitZ* 41702 5.55 444° 1 ‐ ‐ 5/16# ‐
LiaHr* 25682 6.20 112° 1 ‐ ‐ 5/13# ‐
NadE* 30376 5.07 459° 1 ‐ ‐ 5/16# ‐
YjdA* 27432 5.74 55° 1 ‐ ‐ 5/5# ‐
YvlBl* 41056 5.50 121° 1 ‐ ‐ 5/15# ‐
YvlBr* 41055 5.50 303° 1 ‐ ‐ 5/23# ‐
* Protein was identified by comparison of the spot pattern to a proteome response library [1‐3]. Mass spectrometric identification was
carried out on a 4800 MALDI‐ToF/ToF Analyzer. Database search was performed with the Mascot search engine [3]. ° Mascot score, # 16
peptides found, 5 of them fragmented by MS/MS. l left of two spots on the gel, r right of two spots on the gel.
F Influence of lipidation on peptide mode of action
142

Supplementary Table 2: Overlapping marker proteins of lead structure MP196, its four
derivatives, and comparator compounds
protein
MP196
C‐C
0
C‐C
8
N‐C
0
N‐C
8
gram
icidin S
galliderm
in
valin
omycin
stress
category
YceC* x x x x x x x x cell envelope
YceH x x x x x x x cell envelope
YjbC/BacB x x x cell envelope
YfhM* x x x x x x cell envelope
FosB* x x x x x x cell envelope
YvlB* x x x x x x x cell envelope
YknY/LiaH* x x x x x x x cell envelope
YpuA x x x x x cell wall
LiaH* x x x x x x x x cell wall
RacX* x x x x x x x cell wall
PbpE x x x x cell wall
MurB x x x cell wall
PspA* x x x x x x x membrane
AcsA x x x membrane
FabF x x x membrane
YoxD* x x x x x x x x membrane
YjdA* x x x x x x x x membrane
YuaI* x x x x x x membrane
YkpA x x x x membrane
PanC/HemH x membrane/energy
NfrA* x x x x x x x x energy
YwrO x x x x x energy
PdhA x energy
PdhD x x x energy
PstS x x x x energy
NadE x x x x x x x energy
GsaB x x x x energy
PhoH/GuaC x energy
CitZ x x x energy
BkdAA x x x amino acids
RocA* x x x x x amino acids
Bcd x x x amino acids
PdxK x x x amino acids
CysC* x x x x x energy
F Influence of lipidation on peptide mode of action
143
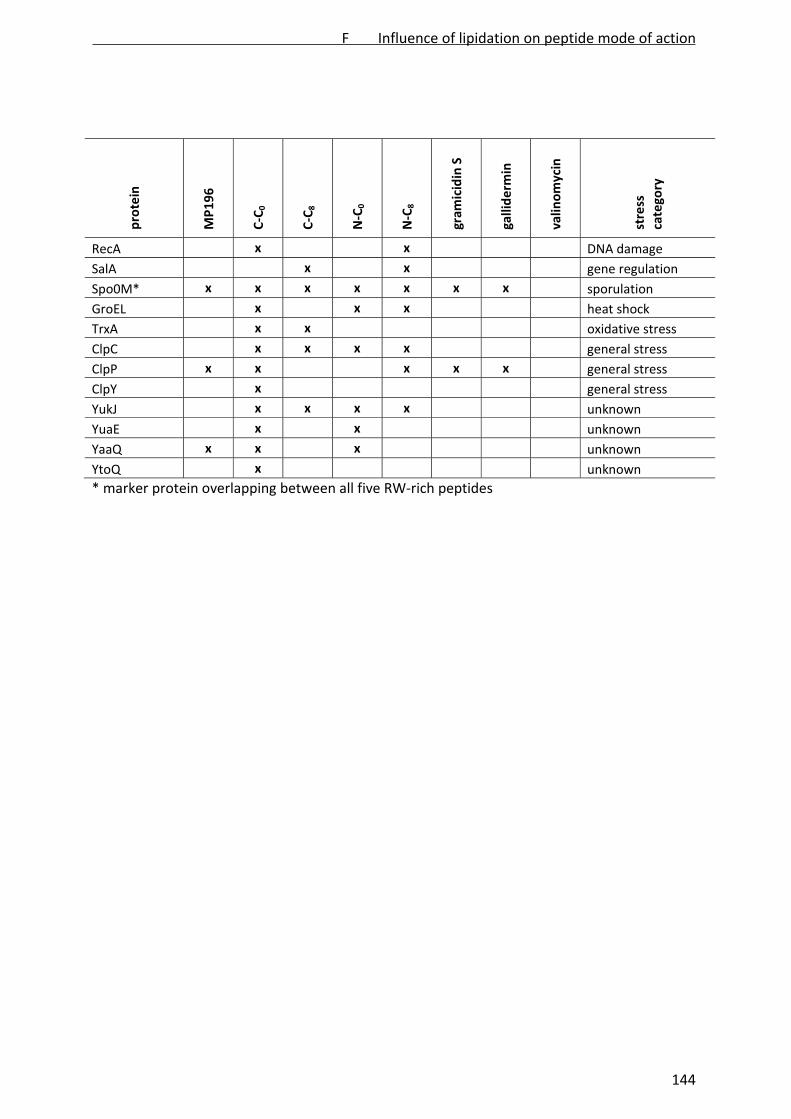
protein
MP196
C‐C
0
C‐C
8
N‐C
0
N‐C
8
gram
icidin S
galliderm
in
valin
omycin
stress
category
RecA x x DNA damage
SalA x x gene regulation
Spo0M* x x x x x x x sporulation
GroEL x x x heat shock
TrxA x x oxidative stress
ClpC x x x x general stress
ClpP x x x x x general stress
ClpY x general stress
YukJ x x x x unknown
YuaE x x unknown
YaaQ x x x unknown
YtoQ x unknown
* marker protein overlapping between all five RW‐rich peptides
F Influence of lipidation on peptide mode of action
144

ii. Supplementary Figures
Supplementary Figure 1: Growth inhibition of B. subtilis aerobically grown in BMM: (A) C‐C0, (B)
C‐C8, (C) N‐C0, (D) N‐C8. The time point of antibiotic addition is marked by arrows. Peptide
concentrations used for proteomic profiling are indicated by boxes.
F Influence of lipidation on peptide mode of action
145

Supplementary Figure 2: Cytosolic protein biosynthesis profiles of peptide‐treated B. subtilis.
Autoradiographs of the untreated controls were false‐colored in green and overlayed with the
antibiotic‐treated cultures false colored in red. Down‐regulated proteins appear green,
upregulated proteins red, and proteins synthesized at equal rates yellow. Orange protein labels
indicate marker proteins shared by all four heptapeptides. Unidentified proteins are marked
with circles.
F Influence of lipidation on peptide mode of action
146

Supporting Figure 3: Proteome response profile of B. subtilis in response to 9 µg/mL (A) and
22.5 µg/mL MP196 (B).
F Influence of lipidation on peptide mode of action
147

Supplementary Figure 3: Marker proteins overlapping between the MP196 derivatives. Orange
protein names indicate proteins, which are also induced by MP196 [3].
III) References
1. Bandow JE, Brötz H, Leichert LI, Labischinski H, Hecker M. (2003) Proteomic approach to
understanding antibiotic action. Antimicrob Agents Chemother 47: 948–955.
2. Wenzel M, Kohl B, Münch D, Raatschen N, Albada HB, et al. Proteomic response of Bacillus
subtilis to lantibiotics reflects differences in interaction with the cytoplasmic membrane.
Antimicrob Agents Chemother 56: 5749‐5757.
3. Wenzel M, Chiriac AI, Otto A, Zweytick D, May C, et al. An antimicrobial peptide delocalizes
peripheral membrane proteins. submitted.
F Influence of lipidation on peptide mode of action
148

G
Quantitative tracing of ruthenocene derivatives for
subcellular localization of antimicrobial peptides in
bacteria
Michaela Wenzel, Ronald Gust, Monika Bürger, H. Bauke Albada, Maya
Penkova, Nils Metzler‐Nolte, Ralf Erdmann, Julia E. Bandow
In preparation
G Peptide quantitation by ruthenocene modification
149

Quantitative tracing of ruthenocene derivatives for subcellular localization of antimicrobial
peptides in bacteria
Michaela Wenzel1, Ronald Gust2, Monika Bürger3, H. Bauke Albada4, Maya Penkova4, Nils
Metzler‐Nolte4, Ralf Erdmann3, Julia E. Bandow1*
1Biology of Microorganisms, Ruhr University Bochum, Germany 2Department of Pharmaceutical Chemistry, University of Innsbruck, Austria 3Physiological Chemistry, Ruhr University Bochum, Germany 4Bioinorganic Chemistry, Ruhr University Bochum, Germany
*Corresponding author. Mailing address: Ruhr‐Universität Bochum, Biologie der
Mikroorganismen, Universitätsstraße 150, 44801 Bochum, Germany. Phone: +49‐234‐32‐23102.
Fax: +49‐234‐32‐14620. E‐mail: [email protected]
ABSTRACT
Antimicrobial peptides are a potent class of antibiotics that can target the cell envelope or
intracellular structures. For mode of action analysis, the subcellular localization is of crucial
importance. In a proof‐of‐concept study, the localization of the antimicrobial hexapeptide NH2‐
RWRWRW‐CONH2 was analyzed visually by transmission electron microscopy and quantitatively
by graphite furnace atomic absorption spectroscopy employing a ruthenocene‐substituted
derivative (Rc‐WRWRW‐CONH2). Proteomic profiling of the Bacillus subtilis stress response
confirmed a similar membrane‐related mechanism of action for NH2‐RWRWRW‐CONH2 and RC‐
WRWRW‐CONH2. Transmission electron microscopy revealed a similar impact on the cell
envelope. Omitting the lead‐staining step in the preparation of ultrathin slices, the modified
peptide was visualized by electron microscopy on the basis of its electron‐dense ruthenium
label. Atomic absorption spectrometry allowed absolute quantitation of the ruthenium‐labeled
peptide in subcellular fractions. Both methods independently showed ruthenium accumulation
in the bacterial membrane. Our results indicate that ruthenocene is suitable for quantitative
monitoring of antimicrobial peptide localization in bacteria.
G Peptide quantitation by ruthenocene modification
150

INTRODUCTION
For the development of novel antibacterial agents the identification of the molecular target and
the elucidation of the mechanism of action are indispensable. The localization of antibiotics in
the bacterial cell is crucial for their mode of action. The question of antibiotic localization has
become more important as membrane‐targeting compounds have come into focus in antibiotic
research (1). This development was prompted by lipopeptide daptomycin, which validated the
bacterial membrane as a clinically attractive antibacterial target and showed that membrane‐
targeting compounds may display high selectivity for bacterial over mammalian membranes.
The antibacterial mechanism of action of daptomycin is still not completely understood (2, 3),
demonstrating that the identification of the mode of action of membrane‐targeting compounds
can be rather complicated. This is true in particular for demonstrating interactions with the
cytoplasmic membrane in vivo. Typically, studies focus on compound interaction with model
lipid layers or isolated biological membranes (4, 5). Several studies have shown that peptide‐
lipid interaction can differ considerably depending on the phospholipid composition or
membrane type, making it uncertain, how in vitro studies translate to the living system (6‐9).
Methods that allow studying different aspects of the interaction of antimicrobial compounds
with bacterial membranes in vivo are highly desirable.
We have recently characterized the mechanism of action of a small cationic arginine‐
tryptophan‐rich antimicrobial peptide (MP196, Fig. 1A) (10). We could demonstrate peptide
integration into model phospholipid bilayers. We conclude based on the bacterial response, that
it integrates into the bacterial membrane, probably at the interface of phospholipid head
groups and fatty acyl chains, and disturbs bilayer architecture. Consequently, it causes
delocalization of the peripheral membrane proteins MurG and cytochrome c involved in cell
wall biosynthesis and respiration, respectively. However, direct confirmation of MP196
localization in the cytoplasmic membrane in vivo would further corroborate the hypothesis on
its mechanism of action.
Monitoring antibiotic localization has repeatedly been approached by directly fusing fluorescent
labels to a molecule (11‐13). Most commonly used fluorophores have much higher molecular
weights than common antibiotics. Direct labeling with such large moieties may critically
influence compound activity, uptake, and mechanism of action (14). Direct fluorescence labeling
G Peptide quantitation by ruthenocene modification
151

approaches can therefore be restricted to in vitro techniques (11) or to use for larger molecules,
which are not affected by the addition of a fluorophore (12, 13). Recently, fluorescent amino
acids have been developed as small labels applicable to proteins and peptides (14).
Fluorescence labeling can only be relatively quantified. For in vivo analysis direct labeling of
antibiotic molecules allowing absolute compound quantitation is desirable.
Chantson et al. reported on the derivatization of very short antimicrobial peptides based on the
MP196 core structure NH2‐RWRWRW‐CONH2 (Fig.1A) with cobaltocene and ferrocene as an
approach to modulating antibacterial activity (15). Such organometallic substitutions also open
up new perspectives for studying peptide localization. Electron‐dense metals can be visualized
by transmission electron microscopy (TEM) (16, 17) and graphite furnace atomic absorption
spectrometry allows absolute quantitation of metal concentrations (18‐21).
Figure 1: Structures of the hexapeptide MP196 (A) and the ruthenocene‐substituted MP276 (B).
Ruthenocene was substituted for the N‐terminal arginine residue resulting in higher
hydrophobicity compared to the hexapeptide (22).
Regarding the MP196 derivatives, cobaltocene was shown to compromise antibacterial activity
(15) and ferrocene quantitation might be hampered by a high cellular iron background,
motivating development of alternative organometallic peptide derivatives. Ruthenocene has
G Peptide quantitation by ruthenocene modification
152

previously been employed for studying cellular uptake of cytotoxic compounds into eukaryotic
cells by graphite furnace atomic absorption spectrometry (21). It was further used to study
localization of antimalarial chloroquine derivatives by both metal analysis and TEM (17).
Inspired by these successful metal‐tracing approaches, the antimicrobial peptide derivative
MP276 (Rc‐WRWRW‐CONH2, Fig. 1B) was synthesized. Ruthenocene substitution did not
negatively affect antibacterial activity (22). As ruthenium is not toxic by itself, MP276 is an ideal
candidate for a proof‐of‐concept study, aiming at evaluating the potential of organometallic
derivatization of antimicrobial peptides for in vivo localization studies in bacteria.
In a first step the impact of the organometallic label on the peptide mechanism of action was
investigated by profiling the proteomic stress response in Bacillus subtilis. Proteome analysis
has been used previously to compare modes of action of antibiotics and derivatives (23). In a
second step, ruthenium distribution was visualized by TEM, providing insight into peptide
localization in a whole cell assay. Finally, the ruthenium concentration was quantified in
subcellular fractions of MP276‐treated cells by element analysis using graphite furnace atomic
absorption spectrometry.
METHODS
Peptides and bacterial growth conditions
MP196 and MP276 were synthesized by solid‐phase synthesis as described by Albada et al. (22).
Gramicidin S was synthesized according to Wadhwani et al. (24). Daptomycin was purchased
from Enzo Life Sciences (Farmingdale, NY, USA). Peptides were dissolved to 10 mg/ml stock
solutions in sterile DMSO. Bacillus subtilis 168 (25) was grown aerobically in Belitzky minimal
medium (BMM) (26) at 37 °C. For an optimal stress response, antibiotics were applied at
concentrations inhibiting bacterial growth in exponential phase by approximately 50% (22.5
µg/ml MP196, 1 µg/ml MP276, 1 µg/ml gramicidin S, and 3.5 µg/ml daptomycin).
Proteomics
Radioactive labeling of proteins newly synthesized under antibiotic stress, 2D gel‐based
proteome analysis of cytosolic protein fractions, and protein identification by MALDI‐ToF/ToF
were performed as previously reported (23).
G Peptide quantitation by ruthenocene modification
153

Transmission electron microscopy
Cells were cultured and prepared for TEM as described earlier (10) based on a method
described by Santhana Raj et al. (27). Briefly, B. subtilis 168 was cultured in BMM and stressed
with antibiotics in early logarithmic growth phase. Cells were washed twice in 100 mM Tris/1
mM EDTA, pH 7.5 and once in the same buffer without EDTA. Cells were fixed with 2%
glutaraldehyde, treated with 2% uranylacetate, stained with 0.2% osmium tetroxide,
dehydrated with a series of solutions containing rising acetone concentrations, and embedded
in epoxy resin. Ultrathin slices for the examination of cellular damage were stained with 0.2%
lead citrate in 0.1 M NaOH for 3 seconds. Cuts used for ruthenium localization remained
unstained. Samples were examined at 23,000 x and 153,000 x magnification.
Graphite furnace atomic absorption spectrometry
Cells were grown in BMM until early exponential growth phase. Cultures were divided and 50 ml
aliquots were either stressed with MP276 or left untreated as controls. After 15 min of
antibiotic stress, cells were harvested at 3,320 x g, washed five times in 100 mM Tris/1 mM
EDTA, pH 7.5, resuspended in 10 mM Tris, pH 7.5 and disrupted by ultrasonication in a Vial
Tweeter instrument (Hielscher, Teltow, Germany). All washing steps were collected and
subjected to ruthenium measurement. Cell debris was separated from the cytosolic/membrane‐
containing fraction by centrifugation at 16,100 x g. The resulting cell wall pellet was dissolved in
10 mM Tris, pH 7.5. The membrane fraction was separated from the cytosolic fraction by
ultracentrifugation at 150,000 x g for 4 h. The cytosolic fraction was collected and the
membrane pellet was dissolved in 10 mM Tris, pH 7.5. All fractions were lyophilized at ‐20 °C for
24 h in a Freeze Dryer BETA I instrument (Martin Christ, Osterode, Germany). Ruthenium
contents were quantified using graphite furnace atomic absorption spectroscopy following a
previously described procedure (28‐30). Briefly, samples were dissolved in 2 ml of double‐
distilled water. For ruthenium determination, samples of 160 µl were stabilized by addition of
20 µl of Triton X‐100 (final concentration 1%) and 40 µl of 1 N HCl and measured immediately.
Standards for calibration purposes were prepared as aqueous dilutions from a MP276 stock (1
mg/ml in DMSO). A Vario 6 graphite furnace atomic absorption spectrometer (Analytik Jena,
Jena, Germany) was used for ruthenium quantitation. Ruthenium was detected at a wavelength
of 349.9 nm with a bandpass of 0.5 nm. A deuterium lamp was used for background correction.
G Peptide quantitation by ruthenocene modification
154

A volume of 25 µl was injected into the graphite tubes. Drying, pyrolysis, and atomization in the
graphite furnace were performed according to published procedures (29, 30). The mean areas
under the curve (AUC) of peaks in the absorption spectra of duplicate injections were used
throughout the study.
RESULTS
Proteome response of B. subtilis
To confirm that the mechanism of action of MP276 is not substantially altered compared to
MP196, the acute bacterial stress response to the peptide was investigated by proteomic
profiling. To this end, B. subtilis was exposed to MP276 in early logarithmic growth phase and
proteins newly synthesized in response to antibiotic stress were radioactively pulse‐labeled with
L‐[35S]‐methionine. The cytosolic proteins were then separated by 2D‐PAGE and protein spots
on autoradiographs of 2D gels were measured densitometrically, normalized, and quantified
relative to an untreated control. Proteins upregulated more than 2‐fold in three independent
experiments were defined as marker proteins for treatment with MP276.
The proteomic response to MP276 was compared to that of the unmodified hexapeptide
MP196 (10). MP196 inhibits respiration and cell wall biosynthesis manifesting in the proteome
response by specific membrane and cell wall stress responses, initiation of cell envelope
modification, and upregulation of energy metabolism (10). Protein biosynthesis patterns in
response to both peptides are displayed in figure 2, details of protein function are given in table
1. In response to MP196 and MP276, 36 and 43 marker proteins were upregulated, respectively.
Of these, 20 markers were shared by both antimicrobial peptides including the most strongly
upregulated proteins (Table 1).
A number of shared marker proteins have previously been described as specific markers for
different aspects of cell envelope stress (31). Phage shock protein PspA and NAD synthase NadE
are specific marker proteins for membrane stress. Upregulation of the PspA homolog LiaH
suggests impairment of a membrane‐bound step of cell wall biosynthesis. YceC, annotated as
tellurium resistance protein, is a more general marker for cell envelope stress (31). Other shared
marker proteins were also related all to membrane and cell wall stress and suggest a
mechanistic duality as previously described for MP196. YjdA and YoxD share similarity with fatty
acid biosynthesis enzymes but do not belong to the standard biosynthesis pathway. They are
G Peptide quantitation by ruthenocene modification
155

probably involved in modifying the membrane lipid composition. Similarly, the amino acid
racemase RacX probably supplies D‐amino acids for cell wall modifications like D‐alanylation of
lipoteichoic acids. FosB, a bacillithiol‐S‐transferase, is known to confer resistance against
antimicrobial compounds targeting cell wall biosynthesis (32).
Figure 2: Protein synthesis profiles of B. subtilis in response to the peptides. Autoradiographs of
proteins separated by 2D gels of the untreated controls false‐colored in green were overlayed
with those of the MP196‐treated (A) or MP276‐treated (B) samples false‐colored in red. Down‐
regulated proteins appear green, upregulated proteins red, and proteins synthesized at equal
rates yellow. Unidentified proteins are indicated by circles. Orange labels indicate marker
proteins upregulated by both MP196 and MP276, white labels indicate non‐overlapping marker
proteins. (C) Functional categories and overlap of the marker proteins upregulated in response
to MP196 and MP276.
Proteins involved in energy metabolism were upregulated in response to both peptides. YwrO
shows similarity to an NAD(P)H oxidoreductase and NfrA is an FMN‐containing NADPH‐linked
nitro/flavin reductase. Their upregulation suggests energy limitation due to impaired membrane
G Peptide quantitation by ruthenocene modification
156

function as was proposed for the membrane stress marker NadE. Limitation of ATP might also
trigger secondary defense strategies. Further marker proteins shared by both peptides belonged
to the B. subtilis general stress response (YdaG, YtxH, Dps, GsiB), which is regulated by the
alternative sigma factor B (σB). Sporulation was indicated by upregulation of SpoVG and Spo0M.
The σB‐dependent general stress response and sporulation are two alternative strategies
upregulated in response to energy limitation and contribute to outliving the stress conditions
(33).
Marker proteins, which were unique to either of the peptides, belonged to the same functional
categories of membrane and cell wall stress, energy metabolism, and general stress, supporting
high mechanistic similarity between both peptides. Most of the proteins described as marker
proteins for only one of the peptides showed the same trend of upregulation in response to the
other peptide, but failed to meet the twofold cut‐off in all three the biological replicates.
Table 1: Marker proteins upregulated in response to MP196 and MP276
protein ID
induction factor protein function functional category regulator
MP196 MP276
LiaH 44.8 43.8 similar to phage shock protein cell envelope stress LiaRS
PspA 20.1 4.2 phage shock protein A homolog cell envelope stress σW
YceC 29.7 2.5 similar to tellurium resistance protein cell envelope stress σB/σW/σM
FosB 5.0 20.8 bacillithiol‐S‐transferase cell envelope stress σW
RacX 8.6 6.8 amino acid racemase cell envelope stress σW
YthP 2.9 3.6 similar to ABC transporter cell envelope stress σW
YtrE 2.7 0.6 similar to ABC transporter cell envelope stress YtrA
DltA 9.4 0.8 D‐alanyl‐D‐alanine carrier protein ligase cell envelope stress σD/σX/σM
YceH 13.1 1.9 similar to toxic anion resistance protein cell envelope stress σB/σW/σM
YuaI 5.7 3.3 unknown cell envelope stress σW
YfhM 7.0 2.9 similar to epoxide hydrolase cell envelope stress σB/σW
RocA 1.2 14.1 3‐hydroxy‐1‐pyrroline‐5‐carboxylate dehydrogenase cell envelope stress σL
YjdA 14.3 3.5 similar to 3‐ketoacyl‐ acyl‐carrier protein reductase fatty acid bioynthesis σE
YoxD 5.1 3.1 similar to 3‐oxoacyl‐ acyl‐carrier protein reductase fatty acid bioynthesis unknown
NadE 12.2 3.7 NAD synthetase energy σB
NfrA 10 6.8 FMN‐containing NADPH‐linked nitro/flavin reductase energy Spx
YwrO 3.8 2.9 similar to NAD(P)H oxidoreductase energy unknown
YqkF 2.6 2.9 NADPH‐dependent aldo‐keto reductase energy unknown
BglH 3.3 1.6 phospho‐beta‐glucosidase energy CcpA
CitZ 4.2 1.0 citrate synthase energy CcpA
IolS 4.0 2.2 aldo/keto reductase energy IolR
G Peptide quantitation by ruthenocene modification
157

protein ID
induction factor protein function functional category regulator
MP196 MP276
YhdN 2.0 1.3 aldo/keto reductase specific for NADPH energy σB
Mdh 0.8 2.2 malate dehydrogenase energy CcpA
ArgC 2.3 3.3 N‐acetyl‐gamma‐glutamyl‐phosphate reductase energy unknown
YtxH 7.8 4.6 general stress protein general stress σB/σH
Dps 5.8 2.9 DNA‐protecting protein, ferritin general stress σB
GsiB 2.5 4.4 general stress protein general stress σB
PpiB 0.8 5.6 peptidyl‐prolyl isomerase (chaperone) general stress unknown
YdaG 4.8 7.9 general stress protein general stress σB
ClpP 4.2 1.8 ATP‐dependent Clp protease proteolytic subunit general stress σB
YtkL 3.6 2.7 general stress protein general stress σB
Spo0M 6.1 9.3 sporulation control gene sporulation σW/σH
SpoVG 5.8 3.8 negative effector of asymetric septation at the onset of sporulation sporulation σH
AzoR2 5.4 2.3 azoreductase oxidative stress σG
MrgA 1.7 3.5 metalloregulation DNA‐binding stress protein oxidative stress PerR
YwbC 1.0 4.1 putative methylglyoxylase oxidative stress unknown
YphP 3.5 2.7 disulfide isomerase, bacilliredoxin oxidative stress Spx
RpsB 3.4 15.3 ribosomal protein S2 translation unknown
Frr 1.1 2.9 ribosome recycling factor translation unknown
TrmB 2.1 1.4 tRNA (m7G46) methyltransferase translation unknown
YvlB 8.9 2.1 unknown unknown unknown
Induction factors are displayed as average over three independent biological replicates. Bold
numbers indicate marker proteins induced more than 2‐fold in all replicate experiments.
* marker for general cell envelope stress, # specific marker for cell wall biosynthesis inhibition, °
specific marker for membrane damage.
A difference in markers responsive to oxidative stress was observed, which were upregulated in
response to the ruthenocene‐substituted peptide but not to MP196 (Fig. 2C). Using CellROX, a
dye fluorescing red in the presence of reactive oxygen species, including peroxide and
superoxide, revealed no fluorescence in the peptide‐treated cells. Thus, the observed
upregulation of oxidative stress response proteins might be effected by σB, which controls a
large stress response regulon as preventive measure against future stress. Taken together, the
protein expression profiles in response to MP196 and ruthenocene‐substituted MP276 were
highly similar.
G Peptide quantitation by ruthenocene modification
158

Influence on cell envelope morphology
Morphology of the B. subtilis cell envelope after peptide treatment was investigated by electron
microscopy (Fig. 3). Ultrathin slices were stained with lead citrate to provide better contrast of
the cell envelope structures. Two antimicrobial peptides were chosen for comparison.
Gramicidin S integrates into the bacterial membrane but in contrast to MP196 it strongly
facilitates leakage of intracellular contents (10). The lipopeptide daptomycin is thought to dock
to the bacterial membrane bilayer by inserting its lipid tail (2).
Figure 3: TEM images of B. subtilis showing cellular damage by antimicrobial peptides: (A)
control; (B) MP196; (C) MP276; (D) gramicidin S; (E) daptomycin. Cells were fixed with 2%
uranylacetate and slices were stained with 0.2% lead citrate in 0.1 M NaOH.
MP196‐treated cells showed cell wall lesions lined with characteristic structures and thinner cell
wall domains as described previously (10). Gramicidin S likewise induced lesions but additionally
caused intracellular content leakage culminating in cell lysis. The applied concentrations do not
G Peptide quantitation by ruthenocene modification
159

cause significant cell lysis under normal growth conditions suggesting that dehydration during
sample preparation causes already perforated cells to rupture. Cells treated with daptomycin
were altered in their cell shape showing bulky excrescences. Concomitantly, thinner cell wall
domains were observed. Although MP196, gramicidin S, and daptomycin all target the bacterial
membrane, they apparently affect the cell shape in distinct ways. MP276‐treated cells resemble
those treated with MP196 suggesting that the membrane‐related mechanism of action is not
considerably altered by the ruthenocene substitution.
Ruthenocene‐peptide visualization in whole cells
Ruthenocene‐peptide localization was monitored in B. subtilis by TEM (Fig. 4). Ruthenium as an
electron‐dense metal can be directly employed for visualizing the modified peptide without the
need for metal‐labeled antibodies. To avoid interference of lead citrate with the ruthenium
signal, ultrathin slices were left unstained. The cytosol of untreated control cells appeared dark
indicating high electron density.
Figure 4: TEM images of B. subtilis showing ruthenocene peptide localization: (A) control; (B)
MP276. Cells were fixed with 2% uranylacetate. Slices remained unstained to avoid interference
of lead with the ruthenium signal.
As typical for unstained cells, the cell envelope structures were of a light grey. In MP276‐treated
cells the cell envelope was stained by the ruthenium peptide. The membrane structure
G Peptide quantitation by ruthenocene modification
160

appeared darker than the cell wall suggesting that the peptide primarily localizes in the lipid
bilayer. Both the membrane and the cell wall were equally stained indicating an even
distribution of the peptide between both compartments. No difference was observed between
the cytosol of untreated and MP276‐treated cells suggesting that no significant amounts of
localize intracellularly.
Absolute quantitation of the ruthenocene‐labeled peptide
Ruthenium was absolutely quantified using graphite furnace atomic absorption spectrometry.
To this end, antibiotic‐treated and untreated B. subtilis cells were fractionated into cytosolic,
membrane, and cell wall fractions. An MP276 stock solution was used as calibration standard. In
untreated control cells only traces of ruthenium, amounts close to the detection limit, were
measured (Table 2). In MP276‐treated samples higher ruthenium quantities were detected.
Taken together, the ruthenium measured in the individual fractions of MP276‐treated cells
exceeded the ruthenium measured in whole cells, which might be due to more efficient
atomization of pre‐fractionated samples.
Table 2: Ruthenium concentration in B. subtilis subcellular fractions
ruthenium per cell [pmol] ruthenium per compartment volume* [pmol/µL] control MP276 control MP276
cells 1.57 x 10‐9 1.45 x 10‐7 ‐ ‐ cytosol 2.59 x 10‐9 1.85 x 10‐7 0.65 46.29 membrane 1.85 x 10‐9 6.37 x 10‐8 20.61 709.41 cell wall 0 3.38 x 10‐8 0 41.93
* cytosolic volume 3.09 x 10‐9 µL, membrane volume 8.99 x 10‐11 µL, cell wall volume 8.08 x 10‐10
µL; calculated based on cyro‐electron microscopy results by Matias and Beveridge (34, 35).
Aiming at revealing the peptide concentrations in the respective cellular compartments, it has
to be considered that the volumes of the cell wall and the membrane differ from that of the
cytosol by one and two orders of magnitude, respectively. Hence, absolute values measured in
the individual fractions are not representative of the actual peptide concentration in the
different cellular compartments. Addressing this problem, the measured ruthenium amounts
were related to the respective compartment volumes to give compartment concentrations.
Volumes were calculated based on cryo‐electron microscopy studies by Matias and Beverige,
who measured the B. subtilis membrane and cell wall structures in high resolution (34, 35).
G Peptide quantitation by ruthenocene modification
161

Adjusted to the calculated compartment volumes (Table 2), the ruthenium concentration in the
membrane fraction exceeded the concentrations measured in the cytosolic and cell wall
fractions by a factor of 15 and 17, respectively. In agreement with our TEM results, the principal
localization of MP276 seems to be the membrane. Cells were washed with EDTA several times
during sample preparation without eluting significant amounts of ruthenium (~10‐21 mol/cell)
suggesting very strong binding of the peptide to the lipid bilayer. This is consistent with
membrane integration, which was shown in model phospholipid bilayers for the unmodified
peptide MP196 (10). Significant amounts of peptide were also detected in the cell wall. This
might be due to the barrier function of the cell wall structure detaining antibiotic compounds
and preventing them from reaching the membrane structure (36). Above background ruthenium
concentrations were detected in the cytosolic fraction, suggesting that the peptide is able to
cross the lipid bilayer to some extent.
DISCUSSION
Hexapeptide MP196 has recently been characterized with regard to its mechanism of action
(10). Substitution of the N‐terminal arginine residue of MP196 against a ruthenocene moiety
yielded the metallo‐peptide MP276 (22), which was deliberately designed for metal‐based in
vivo localization studies. In this proof‐of‐concept study we demonstrated that an organometallic
derivatization facilitates studying peptide localization. The antibiotic mechanism of the
ruthenocene‐substituted peptide was validated by proteomic profiling showing essentially
consistent proteome response patterns to both the unmodified MP196 and the ruthenocene‐
substituted MP276. Marker protein upregulation corresponded well to the mode of action
described for MP196, which integrates into the bacterial membrane and delocalizes essential
components of the cell wall biosynthesis machinery and the respiratory chain. Consequently,
cells suffer strong energy limitation and cell wall integrity is corrupted (10). TEM of peptide‐
treated cells further demonstrated that the cell envelope damage caused by MP196 and MP276
is indistinguishable. Our data suggest that the mechanism of action of MP276 was not
substantially altered by the introduced ruthenocene moiety.
The high mechanistic similarity of such metallocene‐substituted peptides to their lead structures
ideally predisposes them as tools in mode of action studies. Inspired by previous studies using
ruthenium as tracer (17, 21), MP276 was employed for metal‐based peptide localization in vivo.
G Peptide quantitation by ruthenocene modification
162

The electron‐dense ruthenium was exploited to localize the ruthenocene peptide in the
bacterial cell by TEM. The bulk of MP276 was detected in the cytoplasmic membrane supporting
our current model on the mode of action of MP196. This was confirmed by quantifying
ruthenium with graphite furnace atomic absorption spectrometry of subcellular fractions.
Based on these data we conclude that organometallic substitutions are useful tools for metal‐
based localization studies on antimicrobial peptides. Compared to alternative strategies to
follow compound localization, metallocene substitutions have several advantages. Metal‐labels
can be small, in case of ruthenocene not significantly bulkier than a tryptophan residue. They
can be designed to optimally fit into a specific compound structure minimizing any effects on
target interaction. Alternatively, antibodies, e.g. labeled with immunogold, may be applied to
bacterial ultrathin slices allowing indirect antibiotic detection. Successful immunogold detection
of a 15 amino acid long antimicrobial peptide has recently been reported (16). Yet, antibody‐
based compound detection depends on immunogenicity and antibody specificity. In the future
organometallic labels may also prove useful in studying compound distribution in infection
models.
ACKNOWLEDGEMENTS
This work was financially supported by “Innovative Antibiotics from NRW”, a grant from the
state of North Rhine‐Westphalia (NRW), Germany and the European Union, European Regional
Development Fund, "Investing in your future" to JEB and NMN.
REFERENCES
1. Hurdle JG, O'Neill AJ, Chopra I, Lee RE. 2011. Targeting bacterial membrane function: an
underexploited mechanism for treating persistent infections. Nat. Rev. Microbiol. 9:62‐75.
2. Pogliano J, Pogliano N, Silverman JA. 2012. Daptomycin‐mediated reorganization of
membrane architecture causes mislocalization of essential cell division proteins. J. Bacteriol.
194:4494‐4504.
3. Bayer AS, Schneider T, Sahl HG. 2013. Mechanisms of daptomycin resistance in
Staphylococcus aureus: role of the cell membrane and cell wall. Ann. N. Y. Acad. Sci.
1277:139‐158.
4. Zweytick D, Deutsch G, Andrä J, Blondelle SE, Vollmer E, Jerala R, Lohner K. 2011. Studies
on lactoferricin‐derived Escherichia coli membrane‐active peptides reveal differences in the
mechanism of N‐acylated versus nonacylated peptides. J. Biol. Chem. 286:21266‐21276.
G Peptide quantitation by ruthenocene modification
163

5. Zweytick D, Tumer S, Blondelle SE, Lohner K. 2008. Membrane curvature stress and
antibacterial activity of lactoferricin derivatives. Biochem. Biophys. Res. Commun. 369:395‐
400.
6. Cheng JT, Hale JD, Elliot M, Hancock RE, Straus SK. 201 The importance of bacterial
membrane composition in the structure and function of aurein 2.2 and selected variants.
Biochim. Biophys. Acta. 1808:622‐633.
7. Cheng JT, Hale JD, Elliot M, Hancock RE, Straus SK. 2009. Effect of membrane composition
on antimicrobial peptides aurein 2.2 and 2.3 from Australian southern bell frogs. Biophys. J.
96:552‐565.
8. Gonçalves S, Abade J, Teixeira A, Santos NC. 2012. Lipid composition is a determinant for
human defensing HNP1 selectivity. Biopolymers. 98:313‐321.
9. dos Santos Cabrera MP, Arcisio‐Miranda M, Gorjão R, Leite NB, de Souza BM, Curi R,
Procopio J, Ruggiero Neto J, Palma MS. Influence of the bilayer composition on the binding
and membrane disrupting effect of Polybia‐MP1, an antimicrobial mastoparan peptide with
leukemic T‐lymphocyte cell selectivity. 2012. Biochemistry. 51:4898‐4908.
10. Wenzel M, Chiriac AI, Otto A, Zweytick D, May C, Schumacher C, Albada HB, Penkova M,
Krämer U, Erdmann R, Metzler‐Nolte N, Brötz‐Oesterhelt H, Becher D, Sahl HG, Bandow JE.
An antimicrobial peptide delocalizes peripheral membrane proteins. submitted.
11. März A, TruppS, Rösch P, Mohr GJ, Popp J. 2011. fluorescence dye as novel label molecule
for quantitative SERS investigations of an antibiotic. Anal. Bioanal. Chem. 402:2625‐2631.
12. Eliassen LT, Berge G, Leknessund A, Wikman M, Lindin I, Løkke C, Ponthan F, Johnson JI,
Sveinjørnsson B, Kogner P, Flaegstad T, Rekdal Ø. 2006. The antimicrobial peptide.
lactoferricin B, is cytotoxic to neuroblastoma cells in vitro and inhibits xenograft growth in
vivo. Int. J. Cancer 119:493‐500.
13. Knappe D, Piantavigna S, Hansen A, Mechler A, Binas A, Nolte O, Martin LL, Hoffmann R.
2012. Oncocin (VDKPPLYLPRPRPPRRIYNR‐NH2): A novel antibacterial peptide optimized
against Gram‐negative human pathogens. J. Med. Chem. 53:5240‐5247.
14. Katritzky AR, Narindoshvili T. 2009. Fluorescent amino acids: advances in protein‐extrinsic
fluorophores. Org. Biomol. Chem. 7:627‐634.
15. Chantson JT, Verga Falzacappa MV, Crovella S, Metzler‐Nolte N. 2006. Solid‐phase
synthesis, characterization, and antibacterial activities of metallocene‐peptide
bioconjugates. ChemMedChem. 1:1268‐1274.
16. Azad MA, Huttunen‐Hennelly HE, Ross Friedman C. 2011. Bioactivity and the first
transmission electron microscopy immunogold studies of short de novo‐designed
antimicrobial peptides. Antimicrob. Agents Chemother. 55:2137‐2145.
17. Biot C, Dubar F, Khalife J, Slomianny C. 2012. Opening up the advantages of the
ruthenocenic bioprobes of ferroquine: distribution and localization in Plasmodium
falciparum‐infected erythrocytes. Metallomics. 4:780‐783.
G Peptide quantitation by ruthenocene modification
164

18. Kirin SI, Ott I, Gust R, Mier W, Weyhermüller T, Metzler‐Nolte N. 2008. Cellular uptake
quantification of metalated peptide and peptide nucleic acid bioconjugates by atomic
absorption spectroscopy. Angew. Chem. Int. Ed. Engl. 47:955‐999.
19. Wenzel M, Patra M, Senges CHR, Ott I, Stepanek JJ, Pinto A, Prochnow P, Vuong C,
Langklotz S, Metzler‐Nolte N, Bandow JE. Analysis on the mechanism of action of potent
antibacterial hetero‐tri‐organometallic compounds – a structurally new class of antibiotics.
ACS Chem. Biol. doi: 10.1021/cb4000844.
20. Patra M, Gasser G, Pinto A, Merz K, Ott I, Bandow JE, Metzler‐Nolte N. 2009. Synthesis and
biological evaluation of chromium bioorganometallics based on the antibiotic platensimycin
lead structure. ChemMedChem. 4:1930‐1938.
21. Gross A, Hüsken N, Schur J, Raszeja Ł, Ott I, Metzler‐Nolte N. 2012. A ruthenocene‐PNA
bioconjugate‐‐synthesis, characterization, cytotoxicity, and AAS‐detected cellular uptake.
Bioconjug. Chem. 23:1764‐1774.
22. Albada HBA, Chiriac AI, Wenzel M, Penkova M, Bandow JE, Sahl HG, Metzler‐Nolte N.
2012. Modulating the activity of short arginine‐tryptophan containing antibacterial peptides
with N‐terminal metallocenoyl groups. Beilstein J. Org. Chem. 8:1753‐1764.
23. Wenzel M, Patra M, Albrecht D, Chen DY, Nicolaou KC, Metzler‐Nolte N, Bandow JE. 2011.
Proteomic signature of fatty acid biosynthesis inhibition available for in vivo mechanism‐of‐
action studies. Antimicrob. Agents Chemother. 55:2590‐2596.
24. Wadhwani P, Afonin S, Ieronimo M, Buerck J, Ulrich AS. 2006. Optimized protocol for
synthesis of cyclic gramicidin S: starting amino acid is key to high yield. J. Org. Chem. 71:55‐
61.
25. Agnostopoulos C, Spizizen J. 1961. Requirements for transformation in Bacillus subtilis. J.
Bacteriol. 81:741‐746.
26. Stülke J, Hanschke R, Hecker M. 1993. Temporal activation of betaglucanase synthesis in
Bacillus subtilis is mediated by the GTP pool. J. Gen. Microbiol. 139:2041‐2045.
27. Santhana Raj L, Hing HL, Baharudin O, Aida Suhana R, Nor Asiha CP, Paramsarvaran S,
Sumarni G, Hanjeet K. 2007. Rapid method for transmission electron microscopy study of
Staphylococcus aureus ATCC 25923. Ann. Microscopy 7:102‐108.
28. Gust R, Schnurr B, Krauser R, Bernhardt G, Koch M, Schmid B, Hummel E, Schönenberger
H. 1998. Stability and cellular studies of [rac‐1,2‐bis(4‐fluorophenyl)ethylenediamine][cyclo‐
butane‐1,1‐dicarboxylato]platinum(II), a novel, highly active carboplatin derivative. J. Cancer
Res. Clin. Oncol. 124:585‐597.
29. Schäfer S, Ott I, Gust R , Sheldrick WS. 2007. Influence of the polypyridyl (pp) ligand size on
the DNA binding properties, cytotoxicity and cellular uptake of organoruthenium(II)
complexes of the type [(6‐C6Me6)Ru(L)(pp)]n+ [L = Cl, n = 1; L = (NH2)2CS, n = 2]. Eur. J. Inorg.
Chem. 19:3034‐3046.
30. Schatzschneider U, Niesel J, Ott I, Gust R, Alborzinia H, Wölfl S. 2008. Cellular uptake,
cytotoxicity, and metabolic profiling of human cancer cells treated with coordinatively
G Peptide quantitation by ruthenocene modification
165

saturated octahedral ruthenium(II) complexes [Ru(bpy)2(N‐N)]Cl2 with N‐N = bpy, phen, dpq,
dppz, and dppn. ChemMedChem. 3:1104‐1109.
31. Wenzel M, Kohl B, Münch D, Raatschen N, Albada HB, Hamoen LW, Metzler‐Nolte N, Sahl
HG, Bandow JE. 2012. Proteomic response of Bacillus subtilis to lantibiotics reflects
differences in interaction with the cytoplasmic membrane. Antimicrob. Agents Chemother.
56:5749‐5757.
32. Butcher BG, Helmann JD. 2006. Identification of Bacillus subtilis sigma‐dependent genes
that provide intrinsic resistance to antimicrobial compounds produced by Bacilli. Mil.
Microbiol. 60:765‐782.
33. Voelker U, Voelker A, Haldenwang WG. 1996. Reactivation of the Bacillus subtilis antisigma
B antagonist, RsbV, by stress‐ or starvation‐induced phosphatase activities. J. Bacteriol.
178:5456‐5463.
34. Matias VR, Beveridge TJ. 2005. Cryo‐electron microscopy reveals native polymeric cell wall
structure in Bacillus subtilis 168 and the existence of a periplasmic space. Mol. Microbiol.
56:240‐251.
35. Matias VR, Beveridge TJ. 2008. Lipoteichoic acid is a major component of the Bacillus
subtilis periplasm. J. Bacteriol. 190:7414‐7418.
36. Weidenmeier C, Peschel A. 2008. Teichoic acids and related cell‐wall glycopolymers in
Gram‐positive physiology and host interactions. Nature Rev. Microbiol. 6:276‐287.
G Peptide quantitation by ruthenocene modification
166

H
Ferrocene‐ and ruthenocene‐specific modulation of
the mechanism of action of metal‐substituted
short antimicrobial peptides
Alina Iulia Chiriac*, Michaela Wenzel*, Dagmar Zweytick, Catherine
Schumacher, H. Bauke Albada, Jonas Krämer, Maya Penkova, Nils
Metzler‐Nolte, Heike Brötz‐Oesterhelt, Hans‐Georg Sahl, Julia E.
Bandow
In preparation
* equally contributed
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
167

Ferrocene‐ and ruthenocene‐specific modulation of the mechanism of action of metal‐
substituted short antimicrobial peptides
Alina Iulia Chiriac1§, Michaela Wenzel2§, Dagmar Zweytick3, Catherine Schumacher4, H. Bauke
Albada5, Jonas Krämer1, Maya Penkova5, Nils Metzler‐Nolte5, Heike Brötz‐Oesterhelt4, Hans‐
Georg Sahl1, Julia E. Bandow2*
1Institute for Medical Microbiology, Immunology, and Parasitology, Pharmaceutical
Microbiology Section, University of Bonn, Germany 2Biology of Microorganisms, Ruhr University Bochum, Germany 3Institute of Molecular Biosciences, Biophysics Division, University of Graz, Austria 4Pharmaceutical Biology and Biotechnology, Heinrich Heine University Düsseldorf, Germany 5Bioinorganic Chemistry, Ruhr University Bochum, Germany
§ These authors contributed equally to this work.
*to whom correspondence should be addressed: Jun.‐Prof. Dr. Julia E. Bandow, Ruhr‐Universität
Bochum, Biologie der Mikroorganismen, Universitätsstr. 150, 44801 Bochum, Germany, Phone:
+49‐234‐3223102, email: [email protected]
Abstract
Derivatization with moieties not occurring in natural products constitutes a promising approach
to developing novel resistance‐breaking antibiotics. Organometallic substitutions may offer a
metal‐specific modulation of the antibiotic mode of action. Here we report a comparative study
on the mechanism of action of ferrocene‐ and ruthenocene‐substituted antimicrobial peptides
based on the core structure RWRWRW‐NH2. This small peptide has been shown to integrate
into the bacterial membrane, changing the phospholipid bilayer architecture and delocalizing
essential membrane proteins involved in respiration and cell wall biosynthesis. The bacterial
stress response to the metallocene‐substituted peptides shows that peptide‐membrane
interaction results in energy limitation and cell wall biosynthesis inhibition. The ruthenocene‐
substituted peptides induce leakage of potassium from the cytosol. Ferrocene‐substituted
peptides cause formation of reactive oxygen species.
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
168

Introduction
Bacterial infections continue to challenge public health worldwide. This is owing to the rapid
development, dissemination, and accumulation of antibiotic resistances resulting in multi‐
resistant pathogens [1]. Introducing structurally novel moieties to antibiotic lead structures is
one approach to overcome bacterial resistance. Not occurring naturally, organometallic
complexes are promising building blocks for antibiotic derivatization [2]. The antimalarial drug
candidate ferroquine, a ferrocene‐substituted chloroquine derivative, did overcome microbial
chloroquine resistance. It also successfully passed phase II clinical trials [3], [4] demonstrating
the potential of organometallics in antimicrobial therapy. So far, organometallic compounds
have been developed predominantly as anti‐cancer and anti‐malarial drugs [3], [5], while less
attention was paid to antibacterial drug development [2]. Since the arsenic‐containing
salvarsan, marketed for treatment of syphilis in 1910 [6], organometallic derivatives of e.g.
quinolones, β‐lactams, and platensimycin have been reported, which contain iron, tungsten,
gold, silver, or ruthenium [2].
Recently, we reported on a hetero‐tri‐organometallic peptide nucleic acid (PNA) derivative
containing a manganese complex (cymantrene), a rhenium moiety (di‐picolyl‐Re(CO)3), and
either a ferrocene or a ruthenocene [7]. Only the rhenium moiety was shown to be essential for
antibacterial activity [8]. The ferrocene‐containing derivative provoked oxidative stress in
bacteria, the ruthenocene‐analog did not [7]. Formation of reactive oxygen species was
correlated with increased antibacterial activity. These data suggested the employment of
ferrocene as an activity‐enhancing building block in drug design. Adding an oxidative component
to the mode of action may prove useful in limiting resistance development as resistance
development is lower for compounds with multiple targets or mechanisms [9].
The ruthenocene moiety did not influence the mechanism of action of the PNA derivative [7],
[8]. It proved useful as a label for metal‐based compound tracing of the antimicrobial peptide
RWRWRW‐NH2 (MP196, Fig. 1) [10], which was originally developed in an attempt to identify
the minimal pharmacophore of short cationic antimicrobial peptides [11]. MP196 was shown to
perturb phospholipid bilayer architecture thereby delocalizing peripheral membrane proteins
involved in respiration and cell wall biosynthesis [10].
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
169

Figure 1: Structures of the investigated metallo‐peptides. (A) MP196, RWRWRW‐NH2, (B)
MP276, RcCO‐WRWRW‐NH2, (C) MP66, FcCO‐WRWRW‐NH2, (D) MP159, FcCO‐G‐cCWRWRWC.
Substitution of the N‐terminal arginine residue for ruthenocene yielded the peptide RcCO‐
WRWRW‐NH2 (MP276, Fig. 1). Proteome analysis of the bacterial stress response to MP276
suggested that the mode of action of the lead peptide was retained. The ruthenium label
allowed locating the peptide with electron microscopy and quantifying cellular peptide
distribution by atomic absorption spectrometry confirming peptide accumulation in the
bacterial membrane in vivo [12].
Exploring the antibacterial potential of such organometallic‐conjugated peptides, cobaltocene‐
and ferrocene‐containing derivatives were synthesized [13], [14]. While cobaltocene negatively
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
170

affected antibacterial activity, ferrocene‐ and ruthenocene‐substituted peptides were as active
as the lead compound opening up new opportunities in drug discovery.
Here, we present a comparative analysis of the mode of action of different ruthenocene‐ and
ferrocene‐substituted peptides based on the MP196 lead structure. All‐L and all‐D‐amino acid
versions of each MP196, ruthenocene‐substituted MP276, and ferrocene‐substituted MP66, as
well as L‐MP159, a cyclic variant of MP66 (Fig. 1), were analyzed.
Materials and Methods
Antibiotics
Peptides were synthesized by solid‐phase synthesis as described previously [15]. Antibiotic stock
solutions were prepared in sterile dimethylsulfoxide (L‐MP196, D‐MP196, L‐MP276, D‐MP276, L‐
MP66, D‐MP66, L‐MP159, vancomycin, rifampicin), 0.01 M HCl (nisin, ciprofloxacin), water
(lactoferrin), acetone (rotenone), or ethanol (antimycin A, tetracycline). Nisin was purified
according to Bonelli et al. [16]. Vancomycin, lactoferrin, rotenone, antimycin A, ciprofloxacin,
rifampicin, and tetracycline were purchased from Sigma Aldrich (St Lous, MO, USA). For
mechanistic studies, antibiotic concentrations were adjusted to inhibit growth by approximately
50% compared to an untreated control, to elicit stress responses without causing significant cell
lysis (see Fig. S1 for example).
Proteomics
Minimal inhibitory concentrations (MIC), growth experiments, radioactive labeling, and
separation of cytosolic protein extracts by two‐dimensional polyacrylamide gel electrophoresis
was performed as described previously [17]. For radioactive labeling Bacillus subtilis 168 (trpC2)
[18] was grown aerobically in Belitzky minimal medium (BMM) [19] at 37°C until early
exponential growth phase. Culture aliquots of 5 mL were exposed to 22.5 µg/ mL L‐MP196, 50
µg/ mL D‐MP196, 1 µg/ mL L‐MP276, 5 µg/ mL D‐MP276, 5.25 µg/ mL L‐MP66, 3.75 µg/ mL D‐
MP66, and 5 µg/ mL L‐MP159, respectively. After 10 min of antibiotic stress cells were pulse‐
labeled with [35S]‐L‐methionine for additional 5 minutes. Cells were harvested, washed three
times in 100 mM Tris/ 1 mM EDTA (TE), and disrupted by ultrasonication. Fifty‐five (55) µg of
protein for analytical and 300 µg for preparative gels were loaded onto 24 cm immobilized pH
gradient strips pH 4‐7 (GE Healthcare, Uppsala, Sweden) by passive rehydration for 18 h.
Proteins were separated in a first dimension by isoelectric focusing and in a second dimension
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
171

by SDS‐PAGE using 12.5% acrylamide gels. Autoradiographs of gel images were analyzed as
described by Bandow et al. [20] using Decodon Delta 2D 4.1 image analysis software (Decodon,
Greifswald, Germany). Proteins found to be induced more than two‐fold in three independent
biological replicates were defined as marker proteins. Protein spots were identified by matrix‐
associated laser desorption/ ionization‐time of flight‐time of flight (MALDI‐ToF/ ToF) mass
spectrometry (MS) as described previously [17]. Proteins that could not be identified by MALDI‐
MS were subjected to nano ultra‐performance liquid chromatography (nanoUPLC)‐electrospray
ionization (ESI)‐MS/ MS [7] using a Synapt G2S high definition mass spectrometer equipped with
an ESI source and an additional ToF detector (Waters, Milford, MA, USA).
Microscopy
Microscopy‐based experiments were performed under proteomic profiling conditions. B. subtilis
1981 [21] and B. subtilis 168 were grown aerobically in BMM until early exponential phase and
treated with 22.5 µg/ mL L‐MP196, 50 µg/ mL D‐MP196, 1 µg/ mL L‐MP276, 5 µg/ mL D‐MP276,
or 5.25 µg/ mL L‐MP66. Samples were taken after 15 min of treatment.
CellROX fluorescence staining was performed as recently reported [7]. Samples treated with 57
µM hydrogen peroxide, respectively, were used as positive controls. Cells were stained with the
CellROX Deep Red reagent (Invitrogen, Carlsbad, CA, USA) for 30 min, washed in TE buffer, and
imaged without fixation.
MinD localization assays were performed as described previously [22]. Cells were grown
overnight in BMM. Overnight cultures were used to inoculate modified BMM containing xylose
instead of glucose for induction of GFP‐MinD expression. Antibiotic‐treated samples were
directly imaged without fixation or immobilization.
Pore formation was monitored with the live/dead BacLight bacterial viability kit (Invitrogen,
Carlsbad, CA, USA) as described previously [7]. Samples were stained with the fluorescent dyes
according to the manufacturer’s instructions, washed with TE buffer, and imaged without
fixation or immobilization.
Sample preparation for bright field microscopy was performed according to Schneider et al. [23]
with minor modifications [22]. Samples were fixed in a 1:3 mixture of acetic acid and methanol,
immobilized in low melting agarose, and microscopically examined.
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
172

Reporter gene fusions
Reporter gene activation assays were performed as previously reported [10] using strains
carrying fusions of the firefly luciferase gene to the promoters of either yorB, bmrC, helD, ypuA,
or liaI in the genetic background of B. subtilis IS34 [24]. Cultures were incubated with serial
twofold dilutions of L‐MP196, D‐MP196, L‐MP276, and D‐MP276, luciferin‐containing citrate
buffer was added, and luminescence was measured.
Radioactive precursor incorporation
Incorporation of radioactively labeled precursor molecules into macromolecules was
determined in Staphylococcus simulans 22 as described recently [10]. Newly synthesized DNA,
RNA, protein, or peptidoglycane was radioactively labeled by incorporation of [14C]‐thymidine,
5‐[3H]‐uridine, L‐[14C]‐isoleucine, and [3H]‐glucosamine‐hydrochloride, respectively. Subcultures
of the differently labeled samples were treated with 2 µg/ mL L‐MP196, 2 µg/ mL D‐MP196,
0.75 µg/ mL L‐MP276, 0.75 µg/ mL D‐MP276, 0.7 µg/ mL ciprofloxacin, 0.07 µg/ mL rifampicin,
0.7 µg/ mL tetracycline, and 0.7 µg/ mL vancomycin, respectively.
In vitro lipid II synthesis
In vitro lipid II synthesis was performed using Micrococcus flavus DSM 1790 membrane
preparations as described by Schneider et al. [25] with some modifications [10]. Briefly, 400 µg
of membrane protein, 5 nmol undecaprenylphosphate, 50 nmol uridine diphosphate ‐N‐
acetylmuramylpentapeptide, and 50 nmol [14C]‐uridine diphosphate‐glucosamine were mixed
with reaction buffer (60 mM Tris‐HCl, 5 mM MgCl2, 0.5% (w/ v) triton X‐100, pH 8.0, total
volume 75 µL). Peptides were added in a 2:1 molar ratio to undecaprenylphosphate and the
mixture was incubated for 1 h at 37°C. Lipids were extracted with 75 µL 2:1 n‐butanol/ 6 M
pyridine‐acetate, pH 4.2, and separated by thin layer chromatography with chloroform/
methanol/ water/ ammonia (88:48:10:1) as solvent [26].
In vitro lipid II‐binding
Peptides were incubated with 2 nmol lipid II in molar ratios of 1:1. Reaction mixtures were
incubated at 30°C for 30 min and applied to thin layer chromatography plates. Chromatography
was performed in chloroform‐methanol‐water‐ammonium (88:48:10:1) and stained with
phosphomolybdic acid.
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
173

Potassium efflux
Potassium efflux experiments were performed with Bacillus megaterium ATCC 13632 as
described previously [10]. Nisin was applied at 7 µg/ ml, L‐MP196 at 125 µg/ ml, D‐ MP196 at
125 µg/ mL, L‐MP276 at 12.5 µg/ mL, and D‐MP276 at 12.5 µg/ mL. Calculations of potassium
efflux in percent were performed according to Orlov et al. [27]. Antibiotic‐induced leakage was
monitored for 5 min with values taken every 10 seconds and expressed relative to the total
amount of potassium released by nisin.
H+‐ATPase proton pumping activity
Inverted vesicles were prepared from M. flavus as described previously [10]. Proton uptake into
vesicles was measured based on the pH‐sensitive probe acridine orange [10]. The reaction
mixture (20 µM acridine orange, 4 mM MgCl2, 10 mM MOPS‐bis‐tris propane (pH 7.0), 140 mM
KCI, 1 mM EDTA, 1 mM dithiothreitol, 1 mg/ mL bovine serum albumine (essentially fatty acid
free), 2.5 µg/ mL valinomycin, 75 µg/ mL vesicle protein) was preincubated for 30 min with 20
µM L‐MP196, 20 µM L‐MP276, 20 µM nisin, or 20 µM lactoferrin. The reaction was then
initiated by addition of 2 mM ATP‐bis‐tris propane, and absorbance was measured at 495 nm.
Respiratory chain activity
Antibiotic influence on the bacterial electron transport chain was monitored by reduction of
iodonitrotetrazolium chloride (INT) using M. flavus inverted vesicles as described before [10].
Briefly, 20 µg of vesicle protein were preincubated for 30 min in pure phosphate buffer (10 mM
potassium phosphate, 5 mM magnesium acetate, pH 6.5) or buffer with 40 µM of L‐MP196, 40
µM D‐MP196, 40 µM L‐MP276, 40 µM D‐MP276, 40 µM nisin, 100 µM antimycin A, or 5 mM
rotenone, respectively. Samples were incubated with 1 mM INT and 0.6 mM NADH as substrate
for 1 h. INT reduction was stopped by addition of 5% trichloroacetic acid. Insoluble formazan
was removed and fluorescence was measured at 485 nm.
Differential scanning calorimetry (DSC)
1,2‐Dipalmitoyl‐sn‐glycero‐3‐phosphoglycerol (Na‐salt) (DPPG) and 1,2‐Dipalmitoyl‐sn‐glycero‐3‐
phosphoethanolamine (DPPE) were purchased from Avanti Polar Lipids Inc. (Alabaster, AL, USA).
Peptides were dissolved in phosphate‐buffered saline (20 mM sodium phosphate, 130 mM
sodium chloride, pH 7.4) (PBS) at a concentration of 3 mg/ mL before each experiment. Aqueous
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
174

dispersions of lipids of 0.1% (w/ w) in PBS buffer were prepared before measurement in the
presence (lipid‐to‐peptide molar ratios of 25:1 and 120:1) or absence of peptides. Liposomes
were prepared 10‐15°C above the phase transition temperature by vigorous mixing. DPPG
liposomes were prepared as described by Zweytick et al. [28]. Lipid films of DPPE and DPPG/
DPPE 88:12 (w/ w) were hydrated at 70°C for 2 h. A freeze thaw protocol was used to increase
homogeneity of the lipid mixture. DSC experiments were performed as described previously
[10].
Results
Proteomic stress response to ferrocene‐ and ruthenocene‐conjugated peptides
Mechanistic differences between the individual antimicrobial peptides were investigated by
proteomic profiling of the B. subtilis antibiotic stress response to sublethal peptide
concentrations (Supplementary Fig. 1). To this end, proteins newly synthesized under antibiotic
stress were radioactively pulse‐labeled with [35S]‐L‐methionine. Proteins more than two‐fold
upregulated in three independent replicate experiments are referred to as marker proteins for
the compound under investigation [29], [30]. Both all‐L and all‐D versions of the unmodified
MP196, the ruthenocene‐substituted MP276, the ferrocene‐substituted MP66, and the all‐L
cyclic MP66 derivative MP159 were investigated. A number of marker proteins, which were
described previously as specific indicators for different aspects of cell envelope damage, were
upregulated in response to the peptides (Fig. 1, Table 1, Supplementary Table 1, Supplementary
Fig. 2‐5). YceC and YceH, marker proteins shared by the peptides, are upregulated in response
to several cell wall and membrane‐targeting antibiotics but their functions are unknown [22].
Several other marker proteins are regulated by the extracytoplasmic function sigma factors M
(σM), X (σX), and W (σW), which are responsive to cell envelope stress [31, [32], [33], [34].
PspA and NadE, specific markers for membrane stress [22], were upregulated in response to all
peptides. The phage shock protein A (PspA) is known to stabilize the cytoplasmic membrane
under stress conditions [35]. NAD synthase (NadE) is indicative of energy limitation and its
upregulation may compensate for respiratory chain inhibition. Other proteins were involved in
compensating for energy limitation such as NfrA and YwrO were upregulated. There were also
marker proteins that indicate an adaptation of the membrane composition, such as YjdA, YoxD,
YuaI, and FabF.
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
175

Table 1: Marker proteins induced by the different peptide conjugates
protein ID L‐MP196 D‐MP196 L‐MP276 D‐MP276 L‐MP66 D‐MP66 L‐MP159 protein function stress category regulator
SpoVG x x x
x negative effector of asymmetric
septation at the onset of sporulation sporulation σH
Spo0M x x x x x sporulation‐control gene sporulation σW/ σH
YdaG x x x x x general stress protein general stress σB
YtxH x x general stress protein general stress σB/ σH
GsiB x x x x x general stress protein general stress σB
Dps x x x x DNA‐protecting protein, ferritin general stress σB
ClpP x x
x
x x ATP‐dependent Clp protease
proteolytic subunit general stress σB
YtkL x x general stress protein general stress σB
PpiB x peptidyl‐prolyl isomerase chaperone unknown
YdbD x x x x x manganese‐containing catalase oxidative stress unknown
AzoR2 x x x x x x azoreductase oxidative stress σG
MrgA
x
x mini‐ferritin, DNA‐binding stress
protein oxidative stress PerR
YwbC x putative methylglyoxalase oxidative stress unknown
YphP
x
disulfide isomerase, putative
bacilliredoxin oxidative stress unknown
Tpx x probable thiol peroxidase oxidative stress Spx
TrxA
x
x
thioredoxin A oxidative stress CtsR, Spx,
σB
SodA x x x x superoxide dismutase oxidative stress σB
YceC* x x x x x x x similar to tellurium resistance protein cell envelope stress σB/ σW/ σM
YceH* x x
x
x similar to toxic anion resistance
protein cell envelope stress σB/ σW/ σM
YthP x x x similar to ABC transporter cell envelope stress σW
YfhM x x similar to epoxide hydrolase cell envelope stress σB/ σW
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
176
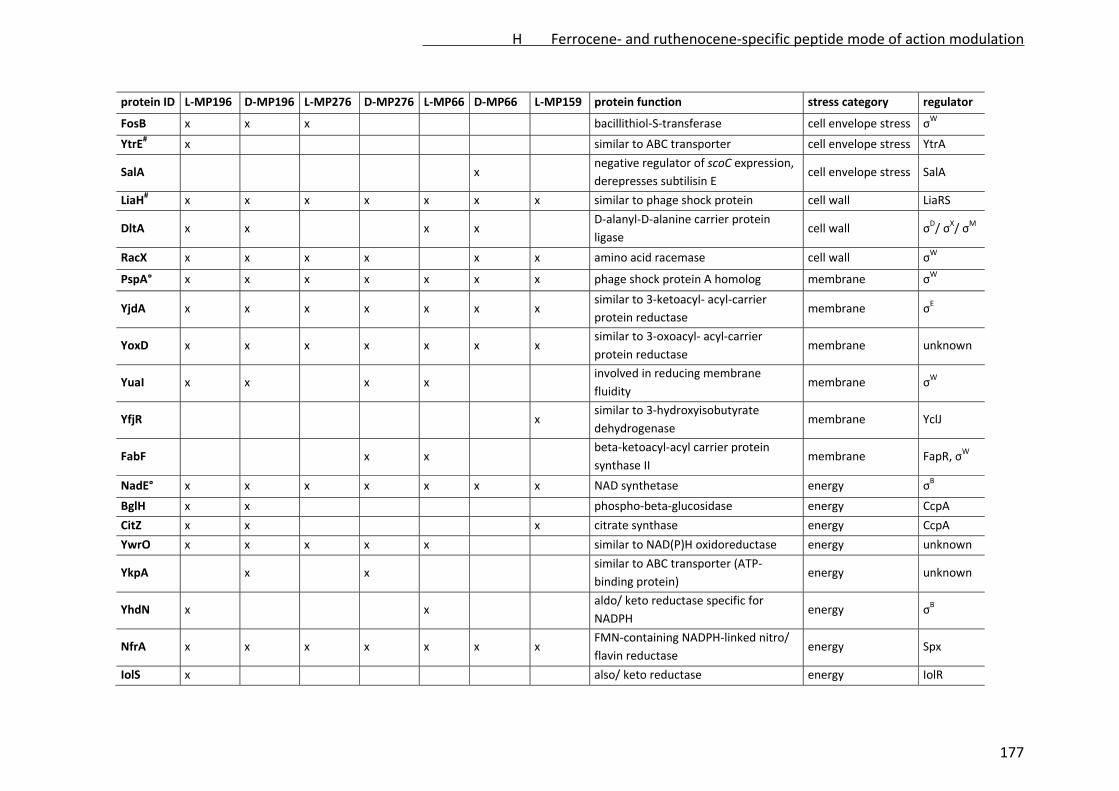
protein ID L‐MP196 D‐MP196 L‐MP276 D‐MP276 L‐MP66 D‐MP66 L‐MP159 protein function stress category regulator
FosB x x x bacillithiol‐S‐transferase cell envelope stress σW
YtrE# x similar to ABC transporter cell envelope stress YtrA
SalA
x
negative regulator of scoC expression,
derepresses subtilisin E cell envelope stress SalA
LiaH# x x x x x x x similar to phage shock protein cell wall LiaRS
DltA x x
x x
D‐alanyl‐D‐alanine carrier protein
ligase cell wall σD/ σX/ σM
RacX x x x x x x amino acid racemase cell wall σW
PspA° x x x x x x x phage shock protein A homolog membrane σW
YjdA x x x x x x x similar to 3‐ketoacyl‐ acyl‐carrier
protein reductase membrane σE
YoxD x x x x x x x similar to 3‐oxoacyl‐ acyl‐carrier
protein reductase membrane unknown
YuaI x x
x x
involved in reducing membrane
fluidity membrane σW
YfjR
x similar to 3‐hydroxyisobutyrate
dehydrogenase membrane YclJ
FabF
x x
beta‐ketoacyl‐acyl carrier protein
synthase II membrane FapR, σW
NadE° x x x x x x x NAD synthetase energy σB
BglH x x phospho‐beta‐glucosidase energy CcpA
CitZ x x x citrate synthase energy CcpA
YwrO x x x x x similar to NAD(P)H oxidoreductase energy unknown
YkpA x x similar to ABC transporter (ATP‐
binding protein) energy unknown
YhdN x
x
aldo/ keto reductase specific for
NADPH energy σB
NfrA x x x x x x x FMN‐containing NADPH‐linked nitro/
flavin reductase energy Spx
IolS x also/ keto reductase energy IolR
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
177

protein ID L‐MP196 D‐MP196 L‐MP276 D‐MP276 L‐MP66 D‐MP66 L‐MP159 protein function stress category regulator
YqkF
x x
x x
NADPH‐dependent aldo‐keto
reductase energy unknown
RocA x
3‐hydroxy‐1‐pyrroline‐5‐carboxylate
dehydrogenase amino acids RocR
ArgC
x
N‐acetyl‐g‐glutamyl‐phosphate
reductase amino acids AhrC
TrmB# x tRNA (m7G46) methyltransferase tRNA modification unknown
GreA x x transcription elongation factor transcription unknown
EfTU
x
elongation factor TU translation stringent
response
Frr
x
ribosome recycling factor translation stringent
response
RpsB x x ribosomal protein S2 translation unknown
YvlB x x x x unknown unknown unknown
YtoQ x unknown unknown unknown
YqiW x x x unknown unknown unknown
*marker for general cell envelope stress, #specific marker for cell wall biosynthesis inhibition, °specific marker for membrane damage
[21]
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
178

The PspA homolog LiaH, a specific proteomic marker for the inhibition of membrane‐bound
steps of cell wall biosynthesis [22], was strongly upregulated. The amino acid racemase RacX
might feed D‐amino acids into cell wall biosynthesis or lipoteichoic acid D‐alanylation, a
bacterial strategy to restrict access to the cytoplasmic membrane [36]. DltA, which is involved in
lipoteichoic acid D‐alanylation, was upregulated by MP196 and MP66.
Overall, the proteomic responses to the different peptides were in good agreement suggesting
that the peptides act by the same mechanism. The ruthenium‐substituted peptide L‐MP276 led
to upregulation of a unique subset of proteins regulated by the general stress sigma factor B
(σB) and the regulators Spx and PerR, which are responsive to oxidative stress and to
membrane‐related stress [37], [38], [39].
Metal‐dependent formation of reactive oxygen species
To examine oxidative stress in vivo, the formation of reactive oxygen species in response to the
metallated and umetallated peptides was investigated using the CellROX fluorescent dye. In the
presence of different reactive oxygen species, including superoxide and hydroxyl radicals, the
dye fluoresces red. Hydrogen peroxide‐treated cells served as positive control and showed
strong fluorescence, while no signal was detected in untreated control cells (Fig. 2).
Figure 2: Formation of reactive oxygen species in B. subtilis. Red fluorescence indicates
presence of reactive oxygen species in the cells. Hydrogen peroxide was used as a positive
control.
The unmodified peptide L‐MP196 and the ruthenocene‐containing L‐MP276 did not give a
fluorescence signal, while cells treated with the ferrocene‐peptide L‐MP66 strongly fluoresced
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
179

red suggesting high abundance of reactive oxygen species. The fact that no oxidative stress was
observed after treatment with the ruthenocene‐peptides suggested a difference in peptide‐
membrane interaction rather than ruthenium‐specific formation of reactive oxygen species.
Ruthenocene‐specific aspects of the in vivo mechanism of action
Proteomic profiling indicated that interactions of any of the peptides with the membrane do not
involve pore formation and that cell wall biosynthesis is inhibited at a membrane‐bound step.
Both aspects were validated using a set of microscopic assays performed under proteomic
profiling conditions in the same chemically defined medium (Supplementary Fig. 2).
Delocalization of a GFP‐labeled cell division protein MinD indicated depolarization by all
peptides (Supplementary Fig. 6A). No pore formation was observed when probing membrane
permeabilization for the red‐fluorescing propidium iodide (Supplementary Fig. 6B). Corruption
of cell wall integrity due to impaired lipid II synthesis was confirmed by extrusion of the
cytoplasmic membrane through cell wall holes after acetic acid/ methanol fixation
(Supplementary Fig. 6C).
None of the three assays revealed any difference in mechanism between the different peptides.
In the chemically defined medium the ruthenocene‐derivatives were 10‐times more potent than
the non‐metallated peptides. Minimal inhibitory concentrations (MICs) were 9 µg/ mL L‐MP196,
10 µg/ mL D‐MP196, 0.9 µg/ mL MP276, and 1 µg/ mL D‐MP276. The further characterization of
the peptides’ mechanisms of action was performed in complex culture broth, where peptides
could be applied at similar molarity.
The influence of the peptides on the main metabolic routes was investigated by promoter
activity assays. To this end, B. subtilis strains with the firefly luciferase gene fused to the
promoters of selected marker genes responsive to DNA damage (yorB), transcription inhibition
(helD), translation inhibition (bmrC), and cell envelope damage (ypuA, liaI) [10], [24] were
treated with the peptides. None of the peptides activated transcription from the promoters of
yorB, helD, or bmrC. The promoter of the cell envelope stress gene ypuA (Fig. 3) was activated
by all peptides, supporting their action on the cell envelope. The liaI promoter was strongly
activated by all peptides except for L‐MP196, which led to only marginal activation. The
ruthenocene‐peptides activated both the liaI and the ypuA promoter at a lower concentration
and a very narrow concentration range compared to the unmodified peptides.
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
180

Figure 3: Activation of transcription from promoters indicative of different types of stress. (A) L‐
MP196, (B) D‐MP196, (C) L‐MP276, (D) D‐MP276. Promoter regions are fused to the firefly
luciferase gene allowing chemiluminescence‐based detection of transcription activation.
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
181

Incorporation of radioactive precursor molecules into all macromolecules was moderately
inhibited by both L‐MP196 and L‐MP276 (Supplementary Fig. 7) indicating no significant
difference between the unmodified and the ruthenocene‐peptide. Such effects on multiple
macromolecule synthesis pathways are frequently observed for membrane‐targeting antibiotics
due to energy limitation. In a previous study no direct interaction of MP196 with components of
the membrane‐bound cell wall biosynthesis machinery has been observed in vitro [10]. L‐MP196
was shown to influence cell wall biosynthesis indirectly by causing a peripheral membrane
protein involved in lipid II synthesis, MurG, to dissociate from the membrane. As in case of
MP196, no influence on in vitro lipid II synthesis was observed for the ruthenocene peptides and
none of them bound to lipid II in vitro (Supplementary Fig. 8). In contrast to L‐ and D‐MP196, the
ruthenocene‐peptides L‐ and D‐MP276 moved with the mobile phase in the thin layer
chromatography, probably due to increased hydrophobicity due to the hydrophobic metal
complex and reduced charge (two instead of four positive charges).
Figure 4: Influence on membrane function. (A) Respiratory chain activity in Micrococcus flavus
inverted vesicles was monitored with the formazan dye iodonitrotetrazolium chloride changing
fluorescence upon reduction. (B) Potassium release from Bacillus megaterium whole cells was
measured with a K+‐sensitive electrode and expressed relative to positive control nisin.
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
182

Energy limitation after L‐MP196 treatment was due to inhibition of the respiratory chain, which
was accompanied by delocalization of the peripheral membrane protein cytochrome c involved
in electron transport [10]. An even stronger effect on the respiratory chain was observed for
both L‐ and D‐MP276 compared to L‐ and D‐MP196 (Fig. 4A). All peptides were more effective in
inhibiting electron transfer than rotenone and antimycin A, specific inhibitors of complex I and
III, respectively. As for L‐MP196, no direct effect of L‐MP276 on the H+‐ATPase was observed
(Supplementary Fig. 9).
In case of L‐MP196, membrane depolarization and inhibition of the respiration was not
accompanied by ion leakage. It has been shown not to facilitate any ion fluxes in regular, salt‐
containing medium or in osmotically stabilizing choline buffer [10]. Here, both L‐ and D‐MP276
led to immediate potassium release into choline buffer, while no significant effect was observed
for L‐ and D‐MP196 even at 50x MIC (Fig. 4B).
Discussion
The influence of ferrocene and ruthenocene substitutions on the mechanism of action of the
short antimicrobial hexapeptide MP196 was investigated. Proteomic profiling of the bacterial
stress responses to the individual peptides revealed no major differences in their physiological
impact on B. subtilis. The stress responses to all peptides similarly reflected a membrane‐related
mechanism of action consistent with peptide integration into the phospholipid bilayer. Further,
energy limitation and cell wall biosynthesis inhibition were apparent, a dual stress response
described previously for L‐MP196. Due to a change of the membrane architecture upon peptide
integration into the phospholipid bilayer, peripheral membrane proteins are delocalized, an
effect observed for cytochrome c involved in respiration and MurG involved in cell wall
biosynthesis. Consequently, the respiratory chain is inhibited resulting in ATP limitation and
integrity of the cell wall is corrupted [10].
Although the ruthenocene‐substituted L‐MP276 did not induce accumulation of reactive oxygen
species, upregulation of a number of oxidative stress‐related proteins regulated by σB, Spx, and
PerR was observed. The σB‐dependent general stress response is typically activated by
membrane‐targeting compounds through energy limitation [37], [38]. PerR is activated upon ion
limitation [39] and has been shown to be responsive to ionophores [40]. Spx is also connected
with thermo tolerance [41], a process also strongly dependent on the membrane composition
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
183

[42]. Thus, such proteins might be rather upregulated in response to peptide‐membrane
interaction than to formation of reactive oxygen species.
The ferrocene‐peptide L‐MP66 strongly induced formation of reactive oxygen species. This is in
line with the results obtained for ferrocene‐ and ruthenocene‐conjugated PNA derivatives [7] as
well as with observations made for the antimalarial ferroquine [43], [44]. The ferrocenoyl‐PNA
elicited a strong oxidative stress response on the proteome level and the ferrocene moiety
positively influenced antibacterial activity [7]. MP66, while causing formation of reactive oxygen
species, did not induce a dedicated oxidative stress response and did not considerably differ
from MP196 in its antibacterial activity in a standardized minimal inhibitory concentration test
[13], [15]. Apparently, ferrocene confers reactive oxygen species‐producing properties to
different compound classes but does not necessarily significantly affect the antibiotic activity.
The mode of action of the ruthenocenoyl‐peptides, which were designed originally for
intracellular peptide tracing [12], did not differ from L‐MP196 with regard to depolarization,
pore formation, cell wall biosynthesis inhibition, and incorporation of precursors into
macromolecules. Minor differences were observed in promoter activation studies. While all
peptides similarly activated transcription from the ypuA promoter indicating cell wall stress, L‐
MP196 was the only peptide not activating transcription from the liaI promoter. Such a
difference was not observed on the proteome level, where LiaH (encoded by liaH in one operon
with liaI) was the most strongly upregulated protein in response to all of the peptides including
L‐MP196. Promoter induction was measured after 30 minutes at different antibiotic
concentrations [24]. On the proteome level the highest LiaH synthesis rates were observed after
15 minutes, while no synthesis was observed after 30 or 60 minutes (unpublished results),
suggesting that the lia operon is expressed intermittently in response to L‐MP196.
The ruthenocene‐substituted peptides differed from the unmodified peptides in that they
increase membrane permeability, leading to a fast release of potassium ions into choline buffer.
This was not observed for L‐ or D‐MP196 at concentrations up to 50x MIC. It was shown that
MP196 is able to provoke ion leakage in salt‐free buffer, probably due to membrane
destabilization under low‐osmolarity conditions [10]. The minimal inhibitory concentrations of
L‐ and D‐MP196 were higher in the chemically defined comparably salt‐rich medium used for
proteomics than in Mueller‐Hinton broth. This effect was not observed for L‐ and D‐MP276,
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
184

which displayed slightly lower minimal inhibitory concentrations in chemically defined medium.
Apparently, osmolarity does not protect equally well against membrane permeabilization by
MP196 and MP276. Since osmoprotection was identified as a bacterial defense strategy against
bacteriolytic peptides, it will be interesting to investigate the reasons underlying the difference
in membrane permeabilization on molecular level in future studies. A first hint is constituted by
the finding that the ferrocene‐conjugated MP66, which displays membrane‐disruptive
properties (Supplementary Fig. 10), exhibits less phospholipid selectivity than MP196 in a
differential scanning calorimetry experiment monitoring changes in fatty acyl chain packing of
different model membranes after peptide addition (Supplementary Fig. 11), suggesting different
interaction of metallocene‐substituted and unmodified peptides with phospholipid bilayers.
Our results show that derivatization of an antimicrobial peptide with ferrocene or ruthenocene
does not change the principle mode of action yet allows metal‐specific modulations. In contrast
to ruthenocene, ferrocene leads to formation of reactive oxygen species in the cytosol. In some
instances oxidative stress generation may be an attractive additional mechanistic component to
build into existing molecules as shown for ferroquine [44]. Ruthenocene might be used to
enhance bacteriolytic properties of peptides.
Acknowledgements
This work was financially supported by a RUB start‐up grant (J.E.B), the RUB Research
Department Interfacial Systems Chemistry (N.M.N, J.E.B), and a grant from the German federal
state of North Rhine‐Westphalia and the European Union, European Regional Development
Fund, "Investing in your future" (N.M.N, J.E.B, H.G.S). JEB acknowledges financial support for the
mass spectrometer by the state of North Rhine Westphalia (Forschungsgroßgerät der Länder).
References
1. Wenzel M, Bandow, JE. (2012) Proteomic signatures in antibiotic research. Proteomics 11:
3256‐3268.
2. Patra M, Gasser G, Metzler‐Nolte N. (2012) Small organometallic compounds as antibacterial
agents. Dalton Trans 41: 6350‐6358.
3. Biot C, Nosten F, Fraisse L, Ter‐Minassian D, Khalife J, et al. (2011) The antimalarial
ferroquine: from bench to clinic. Parasite 18: 207‐214.
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
185

4. Dubar F, Egan TJ, Pradines B, Kuter D, Ncokazi KK, et al. (2011) The antimalarial ferroquine:
role of the metal and intramolecular hydrogen bond in activity and resistance. ACS Chem
Biol 6: 275‐287.
5. Gasser G, Ott I, Metzler‐Nolte N. (2011) Organometallic anticancer compounds. J Med Chem
54: 3‐25.
6. Amyes SGB. (2001) Magic bullets lost horizons: The rise and fall of antibiotics. Taylor &
Francis Inc, London.
7. Wenzel M, Patra M, Senges CHR, Ott I, Stepanek JJ, et al. (2013) Mechanism of action
analysis of potent antibacterial hetero‐tri‐organometallic compounds ‐ a structurally new
class of antibiotics. ACS Chem Biol. doi: 10.1021/cb4000844.
8. Patra P, Wenzel M, Prochnow P, Pierroz V, Gasser G, et al. (2012) Structural optimization of
an antibacterial hetero‐tri‐organometallic compound: identification of the essential
organometallic moiety required for antibacterial activity. in preparation.
9. Brötz‐Oesterhelt H, Brunner NA. (2008) How many modes of action should an antibiotic
have? Curr Opin Pharmacol 8: 564‐573.
10. Strøm MB, Haug BE, Skar ML, Stensen W, Stiberg T, et al. (2003) The pharmacophore of
short cationic antibacterial peptides. J Med Chem 46: 1567‐1570.
11. Wenzel M, Chiriac AI, Otto A, Zweytick D, May C, et al. An antimicrobial peptide delocalizes
peripheral membrane proteins. submitted.
12. Wenzel M, Gust R, Bürger M, Albada HB, Penkova M, et al. Quantitative tracing of
ruthenocene derivatives for subcellular localization of antimicrobial peptides in bacteria. in
preparation.
13. Chantson JT, Verga Falzacappa MV, Crovella S, Metzler‐Nolte N. (2006) Solid‐phase
synthesis, characterization, and antibacterial activities of metallocene‐peptide
bioconjugates. ChemMedChem 1: 1268‐1274.
14. Chantson JT, Varga Falzacappa MV, Crovella S, Metzler‐Nolte N. (2005) Antibacterial
activities of ferrocenoyl‐ and cobaltocenium‐peptide bioconjugates. J Organomet Chem 690:
4564‐4574.
15. Albada HB, Chiriac AI, Wenzel M, Penkova M, Bandow JE, et al. (2012) Modulating the
activity of short arginine‐tryptophan containing antibacterial peptides with N‐terminal
metallocenoyl groups. Beilstein J Org Chem 8: 1753‐1764.
16. Bonelli RR, Schneider T, Sahl HG, Wiedemann I. (2006) Insights into in vivo activities of
lantibiotics from gallidermin and epidermin mode‐of‐action studies. Antimicrob Agents
Chemother 50: 1449‐57.
17. Wenzel M, Patra M, Albrecht D, Chen DY, Nicolaou KC, et al. (2011) Proteomic signature of
fatty acid biosynthesis inhibition available for in vivo mechanism‐of‐action studies.
Antimicrob Agents Chemother 55: 2590‐2596.
18. Agnostopoulos C, Spizizen J. (1961) Requirements for transformation in Bacillus subtilis. J
Bacteriol 81: 741‐746.
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
186

19. Stülke J, Hanschke R, Hecker M. (1993) Temporal activation of betaglucanase synthesis in
Bacillus subtilis is mediated by the GTP pool. J Gen Microbiol 139: 2041‐2045.
20. Bandow JE, Baker JD, Berth M, Painter C, Sepulveda OJ, et al. (2008) Improved image
analysis workflow for 2D gels enables large‐scale 2D gel‐based proteomics studies ‐ COPD
biomarker discovery study. Proteomics 8: 3030‐3041.
21. Strahl H, Hamoen LW. (2010) Membrane potential is important for bacterial cell division.
Proc Natl Acad Sci USA 27: 12281‐12286.
22. Wenzel M, Kohl B, Münch D, Raatschen N, Albada HB, et al. (2012) Proteomic response of
Bacillus subtilis to lantibiotics reflects differences in interaction with the cytoplasmic
membrane. Antimicrob Agents Chemother 56: 5749‐5757.
23. Schneider, T, Kruse T, Wimmer R, Wiedemann I, Sass V, et al. (2010) Plectasin, a fungal
defensin, targets the bacterial cell wall precursor lipid II. Science 328: 1168‐1172.
24. Urban A, Eckermann S, Fast B, Metzger S, Gehling M, et al. (2007) Novel whole‐cell antibiotic
biosensors for compound discovery. Appl Environ Microbiol 73: 6436‐6443.
25. Schneider T, Kruse T, Wimmer R, Wiedemann I, Sass V, et al. (2004) In vitro assembly of a
complete, pentaglycine interpeptide bridge containing cell wall precursor (lipid II‐Gly5) of
Staphylococcus aureus. Mol Microbiol 53: 675‐685.
26. Rick PD, Hubbard GL, Kitaoka M, Nagaki H, Kinoshita T, et al. (1998) Characterization of the
lipid carrier involved in the synthesis of enterobacterial common antigen (ECA) and
identification of a novel phosphoglyceride in a mutant of Salmonella typhimurium defective
in ECA synthesis. Glycobiology 8: 557‐567.
27. Orlov DS, Nguyen T, Lehrer RI. (2002) Potassium release, a useful tool for studying
antimicrobial peptides. J Microbiol Methods 49: 325‐328.
28. Zweytick D, Pabst G, Abuja PM, Jilek A, Blondelle SE, et al. (2006) Influence of N‐acylation of
a peptide derived from human lactoferricin on membrane selectivity. Biochim Biophys Acta
1758: 1426‐1143.
29. Bandow JE, Brötz H, Leichert LI, Labischinski H, Hecker M. (2003) Proteomic approach to
understanding antibiotic action. Antimicrob Agents Chemother. 47: 948‐955.
30. Brötz‐Oesterhelt H, Beyer D, Kroll HP, Endermann R, Ladel C, et al. (2005) Dysregulation of
bacterial proteolytic machinery by a new class of antibiotics. Nat Med 11: 1082‐1087.
31. Cao M, Helmann JD. (2004) The Bacillus subtilis ectracytoplasmic‐function sigmaX factor
regulates modification of the cell envelope and resistance to cationic antimicrobial peptides.
J Bacteriol. 186: 1136‐1146.
32. Cao M, Kobel PA, Morshedi MM, Wu MFW, Paddon C, et al. ( 2002) Defining the Bacillus
subtilis σW regulon: a comparative analysis of promoter consensus search, run‐off
transcription/microarray analysis (ROMA), and transcriptional profiling approaches 1. J Mol
Biol 316: 443‐457.
33. Eiamphungporn W, Helmann JD. (2008) The Bacillus subtilis σM regulon and its contribution
to cell envelope stress responses. Mol Microbiol. 67, 830‐48.
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
187

34. Helmann JD. (2002) The extracytoplasmic function (ECF) sigma factors. Adv Microb Physiol
46: 47‐110.
35. Kobayashi R, Suzuki T, Yoshida M. (2007) Escherichia coli phage‐shock protein A (PspA) binds
to membrane phospholipids and repairs proton leakage of the damaged membranes. Mol
Microbiol 66: 100‐109.
36. Weidenmaier C, Peschel A. (2008) Teichoic acids and related cell‐wall glycopolymers in
Gram‐positive physiology and host interactions. Nat Rev Microbiol 6: 276‐267.
37. Hecker M, Völker U. (2001) General stress response in Bacillus subtilis and other bacteria.
Adv Microb Physiol 44: 35‐91.
38. Turgay K. (2013) Protein arginine phosphorylation in Bacillus subtilis. Annual Conference of
the German Society for General and Applied Microbiology (VAAM) 2013. abstr RSV2‐FG.
39. Chen L, Keramati L, Helmann JD. (1995) Coordinate regulation of Bacillus subtilis peroxide
stress genes by hydrogen peroxide and metal ions. Proc Natl Acad Sci USA 92: 8190‐8194.
40. Voelker U, Voelker A, Haldenwang WG. (1996) Reactivation of the Bacillus subtilis anti‐sigma
B antagonist, RsbV, by stress‐ or starvation‐induced phosphatase activities. J Bacteriol 178:
5456‐5463.
41. Raatschen N, Wenzel M, Leichert LI, Düchting P, Krämer U, et al. Extracting iron and
manganese from bacteria with ionophores ‐ a mechanism against competitors characterized
by increased potency in environments low in micronutrients. Proteomics. doi:
10.1002/pmic.201200556.
42. Šajbidor J. (1997) Effect of Some Environmental Factors on the Content and Compositions of
Microbial Membrane Lipids. Crit Rev Biotechnol 17: 87‐103.
43. Biot C, Castro W, Botté CY, Navarro M. (2012) The therapeutic potential of metal‐based
antimalarial agents: implications for the mechanism of action. Dalton Trans 41: 6335‐6349.
44. Dubar F, Bohic S, Dive D, Guérardel Y, Cloetens P, et al. (2012) Deciphering the Resistance‐
Counteracting Functions of Ferroquine in Plasmodium falciparum‐Infected Erythrocytes. ACS
Med Chem Lett 3: 480‐483.
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
188

Supplementary Information
Contents
i. Supplementary Tables
ii. Supplementary Figures
i. Supplementary Tables
Table S1: Mass spectrometric identification of upregulated marker proteins
protein ID molecular weight pI peptide count Mascot score Mascot % score
ArgC 38,049 5.30 14 342 100
AzoR2 23,257 5.26 7 117 100
BglH 53,255 5.13 13 287 100
CitZ 41,702 5.55 16 444 100
ClpP 21,668 5.19 12 353 100
DltA 55,773 5.10 13 276 100
Dps 16,583 4.64 8 168 100
EfTU 44,565 4.92 6 458 100
FabF 43,977 4.76 60 4,091 *
FosB 17,161 6.14 4 754 * ‐
Frr 20,754 5.52 9 31 100
GreA 17,261 4.68 11 339 100
GsiB 13,789 5.31 5 122 100
IolS 35,146 5.50 16 280 100
LiaH 25,682 6.20 13 112 100
MrgA 17,322 4.79 3 184 100
NadE 30,376 5.07 16 459 100
NfrA 28,302 5.73 20 9,602 * ‐
PpiB 15,246 5.53 3 149 100
PspA 25,125 5.87 11 91 100
RacX 25,270 5.46 3 73 100
RocA 56,284 5.58 52 12,601 * ‐
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
189

protein ID molecular weight pI peptide count Mascot score Mascot % score
RpsB 27,950 6.27 16 279 100
SalA 38,614 5.23 33 6,829 * ‐
SodA 22,561 5.33 6 450 100
Spo0M 29,714 4.26 63 25,783 * ‐
SpoVG 10,886 5.25 10 348 100
Tpx 18,204 4.89 15 403 100
TrmB 24,488 6.32 7 53 98
TrxA 11,385 4.30 11 11,205 * ‐
YceC 21,810 5.46 14 224 100
YceH 41,646 5.90 13 263 100
YdaG 15,867 5.33 5 100 100
YdbD 30,238 5.06 10 103 100
YfhM 32,737 6.07 20 7,499 * ‐
YfjR 27,848 4.97 4 109 100
YhdN 37,289 4.96 10 132 100
YjdA 27,432 5.74 5 55 99
YkpA 61,017 4.98 89 5,582 * ‐
YoxD 25,283 5.48 20 434 100
YphP 15,865 4.81 5 94 100
YqiG 40,780 5.34 19 325 100
YqiW 16,186 5.00 7 137 100
YqkF 34,695 5.30 10 126 100
YthP 26,490 5.39 5 195 100
YtkL 24,816 5.14 8 75 100
YtoQ 16,780 5.77 14 9,248 * ‐
YtrE 25,444 5.99 11 241 100
YtxH 16,675 5.30 5 131 100
YuaI 19,830 5.31 7 167 100
YvlB 41,056 5.50 15 121 100
YwbC 14,424 4.62 7 231 100
YwrO 19,942 5.33 7 166 100
* Identification was carried out using a Synapt G2S HDMS mass spectrometer. PLGS scores are displayed.
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
190

ii. Supplementary Figures
Figure S1: Growth of B. subtilis in chemically defined medium under acute peptide stress.
Arrowheads mark the time points of antibiotic addition. Concentrations chosen for proteomic
profiling are underlined.
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
191

Figure S2: Proteomic response of B. subtilis after treatment with D‐MP276. Protein synthesis
profiles of the untreated control were false colored in green, those of the antibiotic‐treated
cultures in red. Images of autoradiographs were overlayed and protein spots quantified. Marker
proteins more than twofold upregulated in each of three independent replicates are labeled
with the protein names, unidentified markers are circled.
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
192

Figure S3: Figure S1: Proteomic profile of B. subtilis in response to L‐MP66.
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
193

Figure S4: Proteomic profile of B. subtilis in response to D‐MP66
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
194

Figure S5: Proteomic profile of B. subtilis in response to L‐MP159
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
195
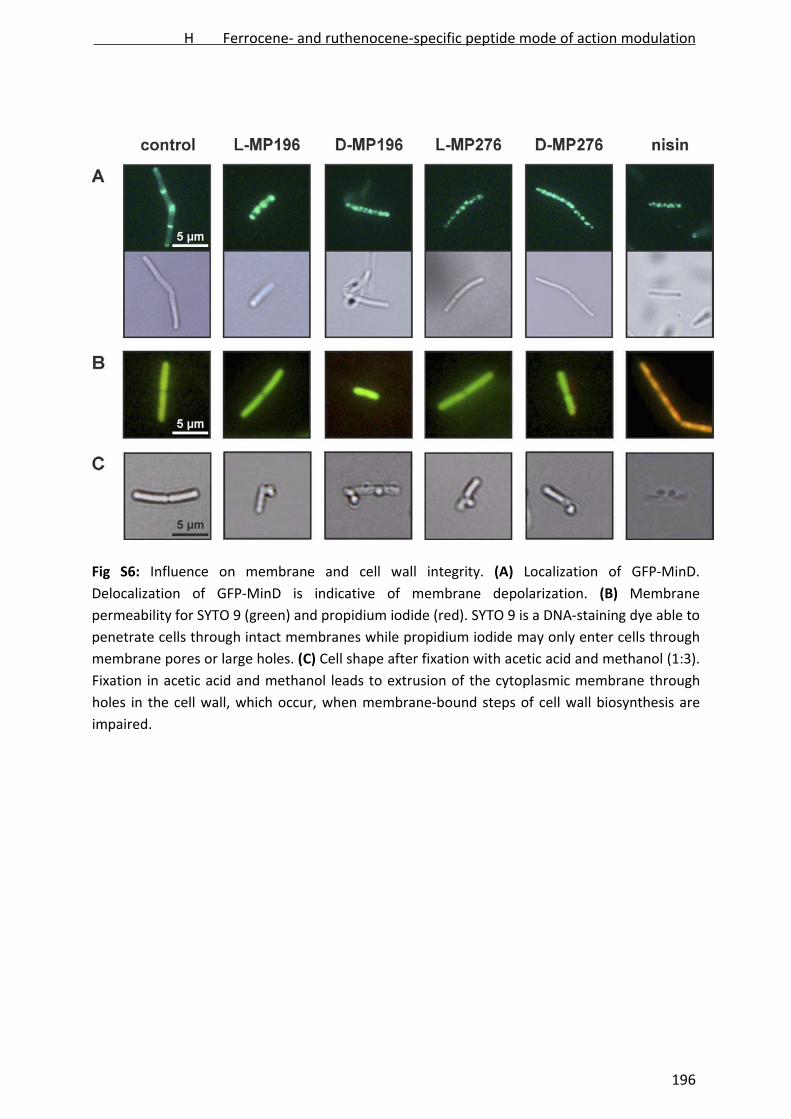
Fig S6: Influence on membrane and cell wall integrity. (A) Localization of GFP‐MinD.
Delocalization of GFP‐MinD is indicative of membrane depolarization. (B) Membrane
permeability for SYTO 9 (green) and propidium iodide (red). SYTO 9 is a DNA‐staining dye able to
penetrate cells through intact membranes while propidium iodide may only enter cells through
membrane pores or large holes. (C) Cell shape after fixation with acetic acid and methanol (1:3).
Fixation in acetic acid and methanol leads to extrusion of the cytoplasmic membrane through
holes in the cell wall, which occur, when membrane‐bound steps of cell wall biosynthesis are
impaired.
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
196
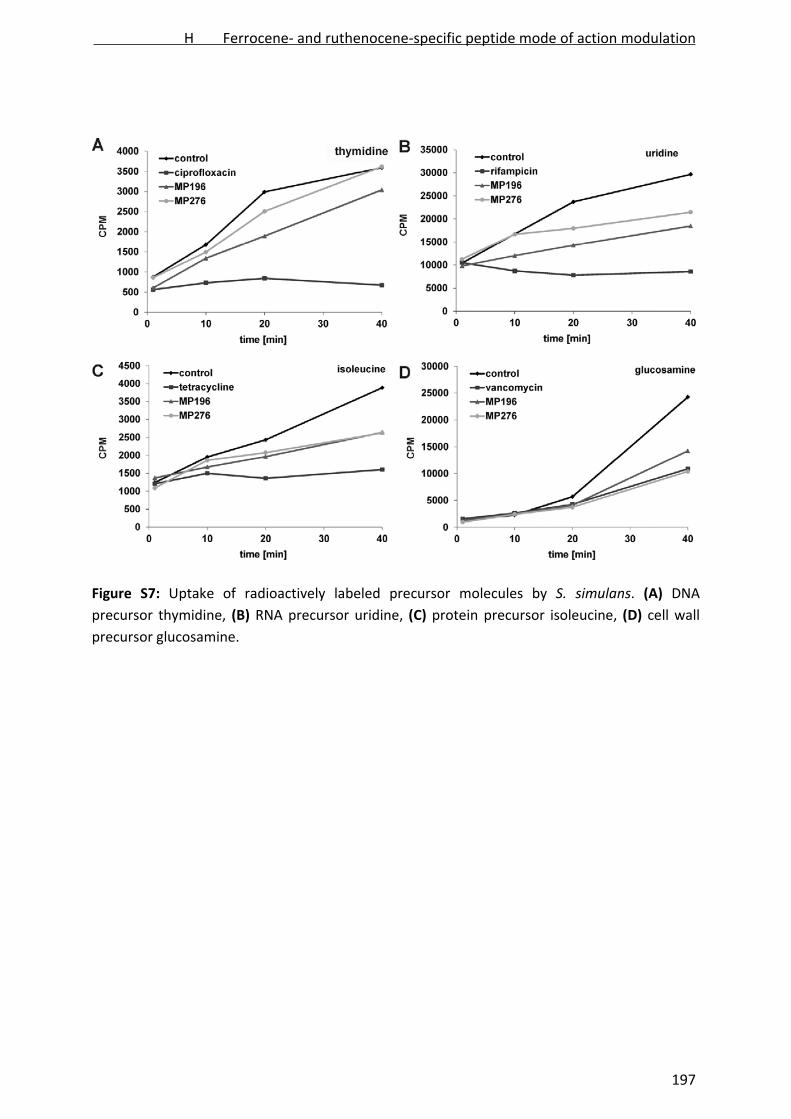
Figure S7: Uptake of radioactively labeled precursor molecules by S. simulans. (A) DNA
precursor thymidine, (B) RNA precursor uridine, (C) protein precursor isoleucine, (D) cell wall
precursor glucosamine.
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
197

Figure S8: Influence on the cell wall biosynthesis machinery. (A) Influence on in vitro lipid II
synthesis, (B) binding to lipid II in vitro, (C) thin layer chromatography of peptides without lipid
II.
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
198

Figure S9: Influence on H+‐ATPase activity in M. flavus inverted vesicles. Proton pumping activity
was monitored with acridine orange, a dye changing absorption in a pH‐dependent manner.
Figure S10: Leakage of ANTS/DPX from unilamellar POPG vesicles induced by MP196 and MP66.
Leakage is expressed relative to positive control triton X‐100 causing vesicle lysis.
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
199

Figure S11: Influence of MP196 and MP66 on model membranes. (A) DPPG, (B) DPPE, (C) 88:12
DPPG/DPPE.
H Ferrocene- and ruthenocene-specific peptide mode of action modulation
200

I
Analysis of the mechanism of action of potent
antibacterial hetero‐tri‐organometallic compounds
‐ a structurally new class of antibiotics
Michaela Wenzel*, Malay Patra*, Christoph H. R. Senges, Ingo Ott,
Jennifer J. Stepanek, Antonio Pinto, Pascal Prochnow, Cuong Vuong,
Sina Langklotz, Nils Metzler‐Nolte, Julia E. Bandow
ACS Chemical Biology, in press
* equally contributed
I Hetero-tri-organometallic PNA backbone derivatives
201

Analysis of the mechanism of action of potent antibacterial hetero‐tri‐organometallic
compounds ‐ a structurally new class of antibiotics
Michaela Wenzel1#, Malay Patra2#+, Christoph H. R. Senges1, Ingo Ott4, Jennifer J. Stepanek1,
Antonio Pinto2++, Pascal Prochnow1, Cuong Vuong3, Sina Langklotz1, Nils Metzler‐Nolte2*, and
Julia E. Bandow1†
1Biology of Microorganisms, Ruhr University Bochum, Germany 2Bioinorganic Chemistry, Ruhr University Bochum, Germany 3AiCuris GmbH & Co. KG, Wuppertal, Germany 4Institute of Medicinal and Pharmaceutical Chemistry, Technische Universität Braunschweig,
Germany +Current address: Department of Bioinorganic Chemistry, University of Zurich, Switzerland ++Current address: Clinic of Cardiovascular Surgery, Heinrich Heine University Düsseldorf,
Germany
#These authors contributed equally to this work.
*Corresponding author for reported chemistry. Mailing address: Ruhr‐Universität Bochum,
Anorganische Chemie I ‐ Bioanorganische Chemie, Universitätsstraße 150, 44801 Bochum,
Germany. Phone: +49‐234‐32‐24153. Fax: +49‐234‐32‐14378. E‐mail: nils.metzler‐[email protected]
†Corresponding author concerning biological findings. Mailing address: Ruhr‐Universität
Bochum, Biologie der Mikroorganismen, Mikrobielle Antibiotikaforschung, Universitätsstraße
150, 44801 Bochum, Germany. Phone: +49‐234‐32‐23102. Fax: +49‐234‐32‐14620. E‐mail:
ABSTRACT
Two hetero‐tri‐organometallic compounds with potent activity against Gram‐positive bacteria
including multi‐resistant Staphylococcus aureus (MRSA) were identified. The compounds consist
of a peptide nucleic acid backbone with an alkyne side chain, substituted with a cymantrene, a
(di‐picolyl)Re(CO)3 moiety, and either a ferrocene (FcPNA) or a ruthenocene (RcPNA).
Comparative proteomic analysis indicates the bacterial membrane as antibiotic target structure.
FcPNA accumulation in the membrane was confirmed by manganese tracing with atomic
absorption spectroscopy. Both organometallics disturbed several essential cellular processes
taking place at the membrane such as respiration and cell wall biosynthesis, suggesting that the
I Hetero-tri-organometallic PNA backbone derivatives
202

compounds affect membrane architecture. Correlating with enhanced antibacterial activity,
oxidative stress was induced only by the ferrocene‐substituted compound. The organometallics
described here target the cytoplasmic membrane, a clinically proven antibacterial target
structure, feature a bactericidal but non‐bacteriolytic mode of action, and limited cytotoxicity
within the limits of solubility. Thus, FcPNA represents a promising lead structure for the
development of a new synthetic class of antibiotics.
INTRODUCTION
Bacterial antibiotic resistance has become a major public health problem of our time. This is
owed to the enormous bacterial adaptability causing rapid resistance development, acquisition,
and spread. With a prevalence of methicillin‐resistant S. aureus (MRSA) in hospitals of up to 50%
and the prevalence of multi‐resistant Gram‐negative pathogens steadily rising, it is clear that
new antibiotics with full activity against multi‐resistant pathogens are urgently needed.
However, the number of newly approved antibiotics steadily declined since the 1980ies.
Currently, on average less than one antibiotic reaches the market each year [1]. Most of these
newly approved antibiotics belong to long‐established compound classes and are likely affected
by cross‐resistance. It is therefore thought that the development of new compounds from old
classes is not going to be sufficient to counteract bacterial antibiotic resistance, and the
identification of structurally completely new classes is highly desirable.
During the last decade, only three new antibiotic classes were approved for clinical use: cyclic
lipopeptides (daptomycin), pleuromutilines (ratapamuline), and glycylcyclines (tigecycline). The
glycylcylines are synthetic tetracycline derivatives, thus structurally and mechanistically related
to a long established antibiotic class. The cyclic lipopeptides and pleuromutilines are structurally
and mechanistically new compounds of microbial origin. Such naturally occurring structures
bear the risk of triggering the clinical manifestation of bacterial adaptation strategies long‐
established in the natural environment of the producer. This has already been observed for
daptomycin‐resistant strains, which show protective modifications of the cell envelope due to
enhanced lipoteichoic acid production [2,3]. In the natural environment, this adaptation of the
cell envelope occurs in response to exposure to antimicrobial compounds secreted by other
species rendering bacteria cross‐resistant to vancomycin and telavancin [4,5]. Fully synthetic
I Hetero-tri-organometallic PNA backbone derivatives
203

structurally novel antibiotic classes might give us an edge in the fight against multi‐resistant
pathogens by avoiding pre‐existing resistance mechanisms.
Recently, we reported a proof‐of‐principle study on the synthesis of the hetero‐tri‐
organometallic compound FcPNA (Figure 1), attaching three distinct organometallic residues to
a building block containing both a peptide nucleic acid (PNA) backbone and an alkyne side chain
[6].
Figure 1. Structures of the hetero‐tri‐organometallic compounds FcPNA and RcPNA.
Characterized by a pseudo‐peptide backbone, a positive charge, and three lipophilic
organometallic residues, namely ferrocene, cymantrene, and a (di‐picolyl)Re(CO)3 moiety,
FcPNA possesses distinct structural features not occurring in nature. At the same time, it shares
chemical properties with membrane‐active antimicrobial peptides, which often are both
positively charged and lipophilic [7‐9]. Here we report on the antibiotic properties of FcPNA and
the identification of the bacterial membrane as its antibiotic target structure. We observed an
elevation in reactive oxygen species (ROS) in vivo in response to FcPNA treatment and were able
to link ROS formation to the ferrocene moiety using a ruthenocene‐substituted reference
compound (RcPNA).
RESULTS AND DISCUSSION
Antibacterial potency and cytotoxicity
Antibacterial activity of FcPNA was tested against both Gram‐positive and Gram‐negative strains
according to the Clinical and Laboratory Standards Institute (CLSI) guidelines (Table 1) [10]. In
water and growth media the limit of solubility was 25 µg mL‐1. While within the limit of solubility
the organometallic compound was inactive against Gram‐negative pathogens (Pseudomonas
I Hetero-tri-organometallic PNA backbone derivatives
204

aeruginosa, Escherichia coli, Acinetobacter baumannii), it exhibited activity against Gram‐
positive bacteria (Staphylococcus aureus, Bacillus subtilis) in the low micro molar range
comparable to clinically used antibiotics.
Table 1: Minimal inhibitory concentrations in µg mL‐1 and µM
FcPNA RcPNA amoxicillin norfloxacin vancomycin
[µg mL‐1]
[µM]
[µg mL‐1]
[µM]
[µg mL‐1]
[µM]
[µg mL‐1]
[µM]
[µg mL‐1]
[µM]
B. subtilis (DSM 402) 2 1.4 32 21 3 8.2 1 3.1 0.5 0.3
S. aureus (DSM 20231) 2 1.4 4 2.7 2 5.5 0.5 1.6 0.5 0.3
S. aureus (ATCC43300)(MRSA) 2 1.4 6 4 48 131 0.5 1.6 1 0.7
S. aureus (COL)(MRSA) 2 1.4 n.d. n.d. n.d. n.d. n.d. n.d. n.d. n.d.
S. aureus (Mu50)(VISA) 6 4 n.d. n.d. n.d. n.d. n.d. n.d. n.d. n.d.
E. coli (DSM 30083) n.d. n.d. n.d. n.d. 64 175 0.5 1.6 96 66
A. baumannii (DSM 30007) n.d. n.d. n.d. n.d. >256 >700 0.75 2.3 64 44
P. aeruginosa (DSM 50071) n.d. n.d. n.d. n.d. >256 >700 4 13 >512 >353
MIC testing was performed according to Clinical and Laboratory Standards Institute (CLSI)
guidelines. Results are reported in the CLSI standard unit for MICs (µg mL‐1) and in µM. n.d.: MIC
was not reached up to the limit of solubility (25 µg ml‐1 for FcPNA and RcPNA).
In contrast to amoxicillin, it exhibited full activity against MRSA strains ATCC 43300 and COL at 2
µg mL‐1. FcPNA further showed good activity against the vancomycin‐intermediate S. aureus
(VISA) strain Mu50 at 6 µg mL‐1. Survival assays indicated that FcPNA is a bactericidal
compound. For B. subtilis the minimal inhibitory concentration of FcPNA equals the minimal
bactericidal concentration, achieving more than 5‐log reduction in colony forming units after 24
h. Survival rates even 15 min after treatment with 4 µg mL‐1 FcPNA were reduced to 5%
compared to untreated controls. Growth experiments showed that neither compound is
bacteriolytic within the limits of solubility (data not shown).
The cytotoxicity of FcPNA against several mammalian cell lines was tested in three different
assays (Table 2). Moderate cytotoxicity close to the limit of solubility (around 25 µg ml‐1) was
I Hetero-tri-organometallic PNA backbone derivatives
205
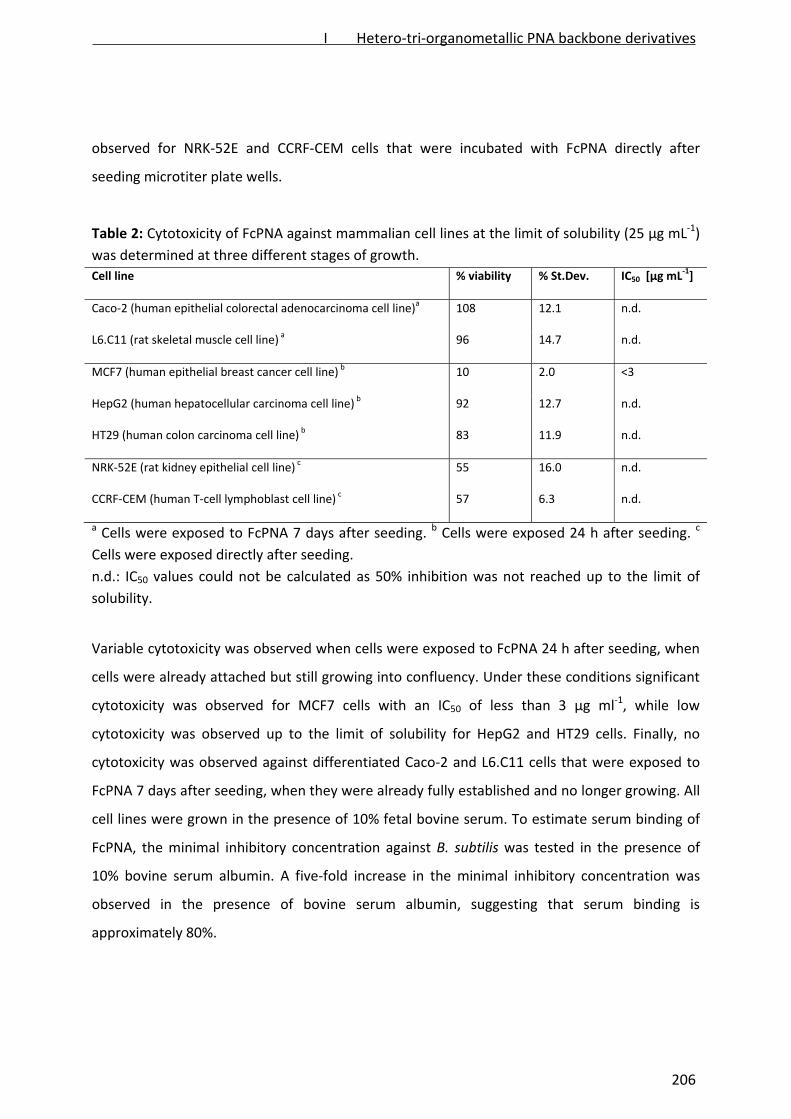
observed for NRK‐52E and CCRF‐CEM cells that were incubated with FcPNA directly after
seeding microtiter plate wells.
Table 2: Cytotoxicity of FcPNA against mammalian cell lines at the limit of solubility (25 µg mL‐1)
was determined at three different stages of growth.
Cell line % viability % St.Dev. IC50 [µg mL‐1]
Caco‐2 (human epithelial colorectal adenocarcinoma cell line)a 108 12.1 n.d.
L6.C11 (rat skeletal muscle cell line) a 96 14.7 n.d.
MCF7 (human epithelial breast cancer cell line) b 10 2.0 <3
HepG2 (human hepatocellular carcinoma cell line) b 92 12.7 n.d.
HT29 (human colon carcinoma cell line) b 83 11.9 n.d.
NRK‐52E (rat kidney epithelial cell line) c 55 16.0 n.d.
CCRF‐CEM (human T‐cell lymphoblast cell line) c 57 6.3 n.d.
a Cells were exposed to FcPNA 7 days after seeding. b Cells were exposed 24 h after seeding. c
Cells were exposed directly after seeding.
n.d.: IC50 values could not be calculated as 50% inhibition was not reached up to the limit of
solubility.
Variable cytotoxicity was observed when cells were exposed to FcPNA 24 h after seeding, when
cells were already attached but still growing into confluency. Under these conditions significant
cytotoxicity was observed for MCF7 cells with an IC50 of less than 3 µg ml‐1, while low
cytotoxicity was observed up to the limit of solubility for HepG2 and HT29 cells. Finally, no
cytotoxicity was observed against differentiated Caco‐2 and L6.C11 cells that were exposed to
FcPNA 7 days after seeding, when they were already fully established and no longer growing. All
cell lines were grown in the presence of 10% fetal bovine serum. To estimate serum binding of
FcPNA, the minimal inhibitory concentration against B. subtilis was tested in the presence of
10% bovine serum albumin. A five‐fold increase in the minimal inhibitory concentration was
observed in the presence of bovine serum albumin, suggesting that serum binding is
approximately 80%.
I Hetero-tri-organometallic PNA backbone derivatives
206

A ruthenocene analogue, RcPNA (Figure 1), was designed to investigate metal‐specific
mechanistic differences related to the different redox potentials of ferrocene and ruthenocene
(see Supplementary Information for synthesis, Supplementary Figures 1‐3). Compared to FcPNA,
RcPNA displayed lower antibacterial activity (MICs against B. subtilis were 2 and 32 µg mL‐1,
respectively) (Table 1). The small organometallic building blocks of FcPNA and RcPNA by
themselves containing a (di‐picolyl)Re(CO)3 or cymantrene as well as the ferrocene or
ruthenocene carboxylic acids (Supplementary Figure 4) showed no antibacterial activity up to
512 µg mL‐1.
Proteomic profiling
To gain first insights into the antibacterial mode of action of this new class of hetero‐tri‐
organometallics, the response of Bacillus subtilis to treatment with FcPNA and RcPNA was
investigated by global proteome analyses. Changes in protein synthesis triggered by antibiotic
stress often reveal cellular mechanisms of damage control and compensation for loss of
function. Over the years, a proteomic response library for the Gram‐positive model organism B.
subtilis has been built that allows to compare proteome profiles of structurally new compounds
to proteomic profiles of antibacterial agents of known antibiotic classes. Sets of target‐ or
mechanism‐specific marker proteins, so‐called proteomic signatures, provide a diagnostic tool
that can aid in narrowing down the antibiotic target area [11]. As the proteome is highly
sensitive to changes in growth conditions, all mechanistic studies were carried out under
standardized growth conditions in a chemically defined medium as previously described [12].
Cells were stressed with antibiotic concentrations leading to a significant reduction in growth
rates without inflicting lethal damage (Supplementary Figure 5). The FcPNA and RcPNA
concentrations required to reduce growth rates of a B. subtilis culture growing exponentially in
chemically defined medium were similar (2 and 1 µg ml‐1, respectively (Supplementary Figure
5)). Proteins newly synthesized in response to the organometallics were then pulse‐labelled
with L‐[35S]‐methionine. Cytosolic proteins were subsequently separated by two‐dimensional
SDS‐polyacrylamide gel electrophoresis and protein synthesis patterns of antibiotic‐treated cells
were compared to those of untreated controls. The proteomic responses to FcPNA and RcPNA
treatment were generally very similar (Figure 2).
I Hetero-tri-organometallic PNA backbone derivatives
207

Figure 2. Differential proteome analysis of B. subtilis in response to FcPNA and RcPNA. a) FcPNA,
b) RcPNA. 2D gel‐based protein biosynthesis profiles of the controls false‐colored in green were
overlayed with those of the antibiotic‐treated samples false‐colored in red. Down‐regulated
proteins appear green, upregulated proteins red, and proteins expressed at pre‐stress rates
yellow. Unidentified upregulated proteins are circled. Orange labels indicate proteins induced
by both FcPNA and RcPNA, white labels indicate non‐overlapping upregulated proteins.
Biosynthesis of many housekeeping proteins was substantially diminished in both cases,
evidenced by the majority of protein spots on the 2D gel overlays appearing green representing
proteins no longer synthesized after treatment with the organometallics. FcPNA and RcPNA
shared 23 marker proteins (proteins induced more than twofold), while 32 markers were unique
to FcPNA, and 12 to RcPNA treatment (details on protein function, regulation, and induction
factors are provided in Supplementary Tables 1 and 2). Many of the induced marker proteins
I Hetero-tri-organometallic PNA backbone derivatives
208

can be grouped into three related functional categories: cell envelope stress, energy
metabolism, and general stress response (Figure 3). Two marker proteins indicative of general
cell envelope stress, YceC and YceH [13], were induced, classifying FcPNA and RcPNA as cell
envelope‐targeting agents. Moreover, induction of the phage shock protein A (PspA), which
stabilizes the membrane by binding to its inner surface, allows narrowing down the antibiotic
target to the cell membrane. Upregulation of both PspA and NAD synthase (NadE) suggests that
the compounds integrate into the membrane [13]. YoxD, YjdA, and IspH, are proteins involved
in fatty acid biosynthesis. However, none of the three proteins is part of the standard
biosynthetic pathway suggesting that compound treatment causes a restructuring of the
membrane lipid composition. The proteome responses to the organometallics were compared
to the antibiotic response library (Supplementary Table 3). Proteins upregulated in response to
FcPNA and RcPNA most strongly overlap with marker proteins of potassium carrier ionophore
valinomycin and of depolarizing agent gramicidin S (Supplementary Figure 6).
Figure 3. Functional categories of the FcPNA and RcPNA‐induced marker proteins. a)
Overlapping marker proteins of FcPNA and RcPNA. Proteins previously described as markers for
general cell envelope and membrane stress are indicated in bold letters. b) Scheme of the
bacterial stress response to the PNA organometallics.
I Hetero-tri-organometallic PNA backbone derivatives
209

In addition to the cell envelope stress response, some of the overlapping proteins were involved
in energy metabolism, a further common characteristic for membrane‐targeting antibiotics.
NadE falls into this category of indicators for energy limitation [13]. Other proteins involved in
energy metabolism were also upregulated particularly by FcPNA, most of which, however, are
poorly characterized with regard to their functions. Many of the upregulated proteins are
known to participate in the σB‐dependent general stress response or in sporulation, two
alternative strategies of B. subtilis for coping with energy limitation [14]. Comparing the
proteome responses to FcPNA and RcPNA treatment, several marker proteins of oxidative stress
were upregulated only by the ferrocene but not the ruthenocene‐substituted compound,
among them metalloregulation DNA‐binding stress protein MrgA and thiol peroxidase Tpx. Both
of these stress proteins are regulated in a σB‐independent fashion. Oxidative stress is also
known to trigger the σB‐dependent general stress response [15] and we observed that more
proteins of the σB‐regulon were upregulated in response to FcPNA treatment.
Oxidative stress
CellROX, a fluorescent probe sensitive to reactive oxygen species, was employed to test for
intracellular oxidative stress (Figure 4a,b).
Paraquat, which causes superoxide formation, served as positive control and led to B. subtilis
cells fluorescing red. Similarly, red fluorescence was observed after treatment with FcPNA
indicating oxidative stress. In contrast, RcPNA‐treated cells and untreated controls did not give a
fluorescence signal. We conclude that the iron in the ferrocene moiety is redox‐active under
physiological conditions, probably resulting in the generation of a ferrocene/ferrocenium redox
pair that in vivo generates reactive oxygen species as part of its redox cycle.
However, ferrocene by itself does not seem to be sufficient for antibacterial activity as the
ferrocene carboxylic acid building block used to synthesize FcPNA did not show any antibacterial
activity (Supplementary Figure 4). As in MIC tests FcPNA showed higher antibacterial potency
than RcPNA, it is tempting to speculate that, while it is not essential for bactericidal activity, the
oxidative stress component can enhance antibiotic potency.
I Hetero-tri-organometallic PNA backbone derivatives
210
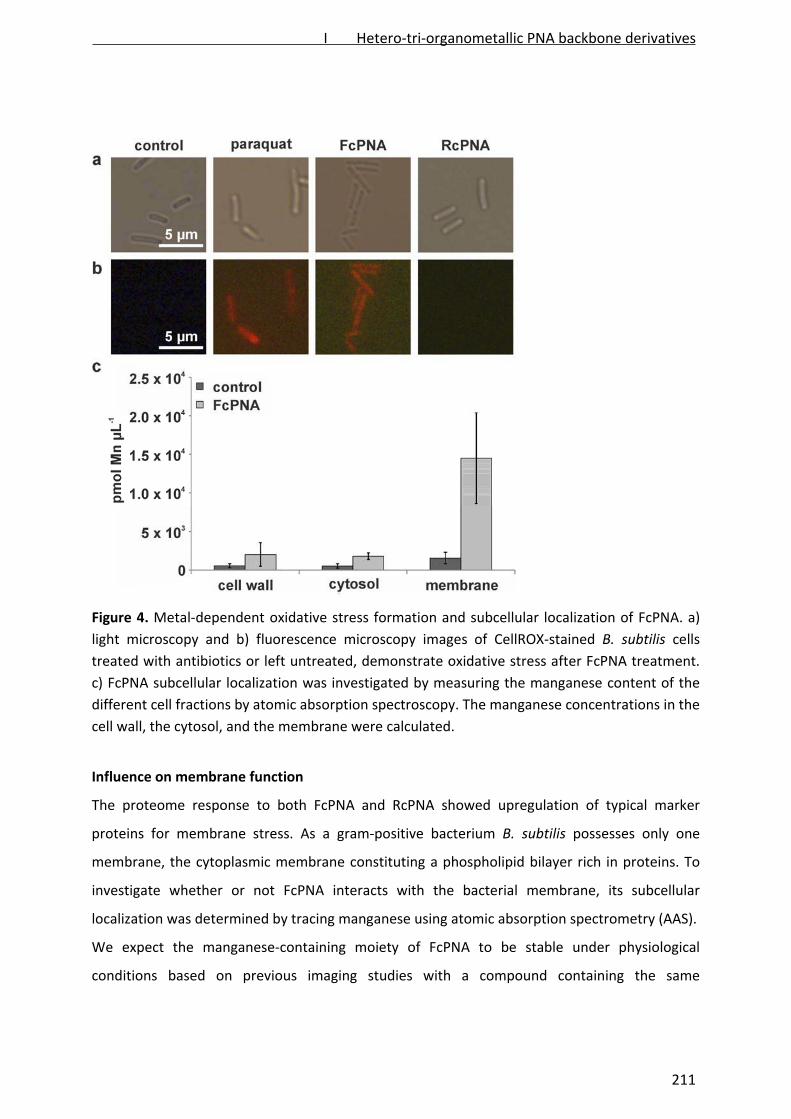
Figure 4. Metal‐dependent oxidative stress formation and subcellular localization of FcPNA. a)
light microscopy and b) fluorescence microscopy images of CellROX‐stained B. subtilis cells
treated with antibiotics or left untreated, demonstrate oxidative stress after FcPNA treatment.
c) FcPNA subcellular localization was investigated by measuring the manganese content of the
different cell fractions by atomic absorption spectroscopy. The manganese concentrations in the
cell wall, the cytosol, and the membrane were calculated.
Influence on membrane function
The proteome response to both FcPNA and RcPNA showed upregulation of typical marker
proteins for membrane stress. As a gram‐positive bacterium B. subtilis possesses only one
membrane, the cytoplasmic membrane constituting a phospholipid bilayer rich in proteins. To
investigate whether or not FcPNA interacts with the bacterial membrane, its subcellular
localization was determined by tracing manganese using atomic absorption spectrometry (AAS).
We expect the manganese‐containing moiety of FcPNA to be stable under physiological
conditions based on previous imaging studies with a compound containing the same
I Hetero-tri-organometallic PNA backbone derivatives
211

manganese‐carbonyl building block [16]6 Antibiotic‐treated cells were fractionated by
differential centrifugation and the manganese content was determined in the cytosolic, the
membrane, and the cell wall fractions (Figure 4c). After treatment with FcPNA, the
concentration of manganese in the membrane fraction was about 10‐fold higher than in
untreated controls and about 6‐fold higher than in the cytosolic and cell wall fractions,
demonstrating that a majority of FcPNA localizes in or at the B. subtilis membrane
(Supplementary Figure 7). During sample preparation, cells were repeatedly washed with
Tris/EDTA buffer to remove loosely bound manganese. FcPNA seems to bind to the bacterial
membrane very tightly as little manganese was removed by the washing steps. At the same
time, as manganese levels in the cytosol and cell wall fractions increased only slightly after
FcPNA treatment, it is unlikely that FcPNA targets cell wall or cytosolic components.
Figure 5. Influence of FcPNA and RcPNA on the B. subtilis membrane and cell wall integrity. a)
GFP‐MinD localization after antibiotic treatment. b) Light microscopy images corresponding to
panel a. c) Light microscopy images of antibiotic‐treated cells stained with BacLight green dye
passing through intact membranes and red dye passing through membrane pores. d) Antibiotic‐
stressed cells fixed with acetic acid/methanol.
I Hetero-tri-organometallic PNA backbone derivatives
212

The overlap of marker proteins between the organometallics and valinomycin and gramicidin S
suggests that the organometallics might also cause disruption of the cellular membrane
potential. Membrane depolarization was monitored using an assay that utilizes the membrane
potential‐dependent localization of cell division protein MinD. If the membrane potential is
intact, MinD localizes at the cell poles and the cell division plane, while upon depolarization
MinD delocalizes and distributes irregularly throughout the cell [13,17]. Using a GFP‐MinD
fusion strain, MinD localization was monitored by fluorescence microscopy (Figure 5a,b). The
untreated control as well as vancomycin, an inhibitor of cell wall biosynthesis that does not
affect the membrane potential, displayed normal MinD localization. Like valinomycin and
gramicidin S, FcPNA and RcPNA led to MinD delocalization, demonstrating membrane
depolarization by both organometallics.
Membrane depolarization can be the result of a number of different challenges ranging from
inhibition of the respiratory chain to an enhanced bilayer permeability, e.g. due to pore
formation or potassium or proton translocation. Using a pair of fluorescent dyes we investigated
the potential of FcPNA and RcPNA to form pores. The commercially available BacLight staining
kit combines a green‐fluorescent dye that readily crosses intact membranes and a red‐
fluorescent dye, which can only enter bacterial cells through pores in the membrane (Figure 5c).
Only nisin, a pore‐forming lantibiotic serving as positive control, permeabilized B. subtilis for the
red dye, resulting in cells fluorescing both red and green. Cells not treated with antibacterial
agents as well as cells treated with non‐pore‐forming comparator compounds fluoresced only
green. Since the organometallics fluoresced green, formation of large pores can be excluded as
a mechanism of both FcPNA and RcPNA.
Energy limitation
To examine, if the interaction of FcPNA and RcPNA with the membrane causes energy limitation
in the cell as suggested by the proteome response, we determined cytosolic ATP levels after
treatment with the organometallics (Figure 6). Compared to the untreated control, valinomycin,
a potassium ionophore, and gramicidin S, which was previously shown to permeabilize the
membrane for potassium [18], reduced the ATP content by 40% and 80%, respectively. In
contrast, protein biosynthesis inhibitor erythromycin did not influence intracellular ATP levels.
I Hetero-tri-organometallic PNA backbone derivatives
213
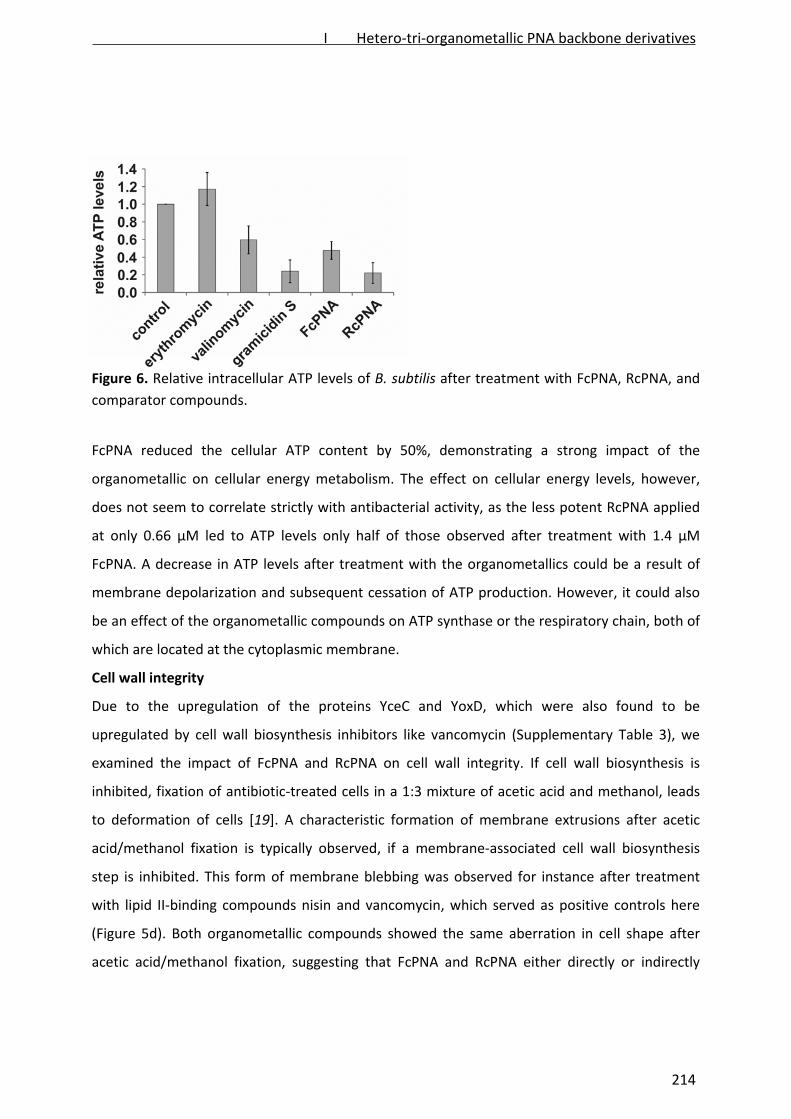
Figure 6. Relative intracellular ATP levels of B. subtilis after treatment with FcPNA, RcPNA, and
comparator compounds.
FcPNA reduced the cellular ATP content by 50%, demonstrating a strong impact of the
organometallic on cellular energy metabolism. The effect on cellular energy levels, however,
does not seem to correlate strictly with antibacterial activity, as the less potent RcPNA applied
at only 0.66 µM led to ATP levels only half of those observed after treatment with 1.4 µM
FcPNA. A decrease in ATP levels after treatment with the organometallics could be a result of
membrane depolarization and subsequent cessation of ATP production. However, it could also
be an effect of the organometallic compounds on ATP synthase or the respiratory chain, both of
which are located at the cytoplasmic membrane.
Cell wall integrity
Due to the upregulation of the proteins YceC and YoxD, which were also found to be
upregulated by cell wall biosynthesis inhibitors like vancomycin (Supplementary Table 3), we
examined the impact of FcPNA and RcPNA on cell wall integrity. If cell wall biosynthesis is
inhibited, fixation of antibiotic‐treated cells in a 1:3 mixture of acetic acid and methanol, leads
to deformation of cells [19]. A characteristic formation of membrane extrusions after acetic
acid/methanol fixation is typically observed, if a membrane‐associated cell wall biosynthesis
step is inhibited. This form of membrane blebbing was observed for instance after treatment
with lipid II‐binding compounds nisin and vancomycin, which served as positive controls here
(Figure 5d). Both organometallic compounds showed the same aberration in cell shape after
acetic acid/methanol fixation, suggesting that FcPNA and RcPNA either directly or indirectly
I Hetero-tri-organometallic PNA backbone derivatives
214

interfere with the cell wall biosynthesis machinery. A direct inhibition of cell wall biosynthesis
components by the organometallics is unlikely as the proteome responses to both compounds
lack the typical marker proteins for cell wall biosynthesis inhibition LiaH, YtrE, and YtrB [13]. A
similar effect was observed for gramicidin S, which integrates into the bacterial membrane and
acts rapidly depolarizing [13,16]. Both gramicidin S and the organometallics might indirectly
affect cell wall biosynthesis by disturbing membrane architecture. Despite FcPNA and RcPNA
having gramicidin S‐like effects on the membrane potential, ATP levels, cell wall integrity, and
the proteome, the organometallics differ from gramicidin S in that they are not bacteriolytic.
CONCLUSION
The recently reported hetero‐tri‐organometallic compound FcPNA showed potent antibacterial
activity against Gram‐positive bacteria. It was active against S. aureus strains including MRSA
and VISA strains in the low micro molar range, comparable to antibiotics currently used
clinically. FcPNA represents a fully synthetic novel class of antibiotics with structures completely
new to nature. Generally, aside from antiseptics, organometallics have been underexplored in
antibacterial research and development [20‐23] and to our knowledge this work represents the
first study on a metal‐based antibacterial agent that addresses both the antimicrobial potency
and the antibiotic target.
We initiated global proteome‐based mechanistic studies to identify the general target area for
FcPNA and designed a ruthenocene‐substituted derivative, RcPNA, to study metal‐specific
effects. The proteomic response of B. subtilis to the organometallics showed upregulation of
marker proteins for cell envelope stress in general and membrane stress in particular. A strong
interaction of FcPNA with the cytoplasmic membrane was demonstrated by tracing manganese
as a surrogate for the manganese‐containing FcPNA using AAS. A direct interaction of FcPNA
with the bacterial membrane is consistent with an induction of alternative fatty acid
biosynthesis enzymes involved in adjusting the composition of the membrane to antagonize
interactions with the organometallics. As shown by fluorescence microscopy, FcPNA and RcPNA
efficiently depolarized B. subtilis cells. While the cause of depolarization remains to be
elucidated, we were able to exclude formation of large pores as a mechanism. Consistent with
membrane depolarization, the proteome and a luciferase‐based ATP assay revealed severe
energy limitation.
I Hetero-tri-organometallic PNA backbone derivatives
215

An elevation in reactive oxygen species was observed after treatment with FcPNA, but not
RcPNA. To our knowledge, we could show here for the first time that ferrocene can induce
oxidative stress in a living system. Similar results were previously obtained in vitro for the
ferrocene‐substituted chloroquine‐based antimalarial drug candidate ferroquine [24]. Further,
Dunbar et al. provided evidence for co‐localization of ferroquine with a sulfur‐containing
compound, presumably the antioxidant glutathione [25]. Ferroquine provides a rare example of
an organometallic compound in clinical development for antimicrobial therapy targeting a
unicellular parasite Plasmodium falciparum. It has been shown to overcome chloroquine
resistance and is about to complete phase II clinical trials [26]. Ferroquine passing phase 1
clinical trials suggests that there are no major safety issues connected to the ferrocene moiety
itself, clearing the way for future ferrocene‐containing organometallic drug candidates in the
antimicrobial field.
While the molecular mechanism of action of FcPNA remains to be studied further, we were able
to identify the cytoplasmic membrane as the target structure. Based on the effects observed we
postulate that FcPNA integrates into the phospholipid bilayer causing a change in membrane
architecture that affects the function of one or more membrane proteins. However, it is also
possible that FcPNA interacts directly with different membrane proteins. The bacterial
membrane has been validated as an antibacterial target only in the past decade. As
demonstrated by daptomycin, membrane‐targeting antibiotics can be successfully applied in the
clinic using systemic administration [27]. Membrane‐targeting antibiotics were shown to be less
prone to bacterial resistance development than antibiotics inhibiting a single enzyme target
[28], making the bacterial membrane a particularly attractive target for future antibiotic
development. Based on its potent antibacterial activity and the limited cytotoxicity, FcPNA,
representing a novel class of antibiotics, is a promising lead structure that warrants further
investigation. The solubility of FcPNA is rather poor and currently prevents systemic
administration, thereby limiting evaluation of acute toxicity in animal models. The main goal of
future structure‐activity‐relationship studies will be to increase solubility while retaining
antibacterial activity and retaining or increasing selectivity for bacteria over mammalian cells.
I Hetero-tri-organometallic PNA backbone derivatives
216

METHODS
Antibiotics. FcPNA was synthesized as described previously [5]. RcPNA synthesis largely parallels
FcPNA synthesis and is described in the supplementary methods. All antibiotic stock solutions
were prepared at 10 mg mL‐1 in DMSO. Valinomycin and bacitracin were purchased from Sigma‐
Aldrich. Vancomycin was purchased from Applichem. Gramicidin S was synthesized and purified
according to Wadhwani et al. [29] and nisin was obtained from H.‐G. Sahl (University of Bonn,
Germany).
Minimal inhibitory concentration. Minimal inhibitory concentrations (MIC) were tested against
Escherichia coli DSM 30083, Acinetobacter baumannii DSM 30007, Pseudomonas aeruginosa
DSM 50071, Bacillus subtilis DSM 402, Staphylococcus aureus DSM 20231 (type strain),
Staphylococcus aureus ATCC 43300 and COL (MRSA), and Staphylococcus aureus Mu50 (VISA) in
a microtiter plate assay according to CLSI guidelines as described previously [10,30]. To estimate
serum binding, MIC tests were analogously performed in the presence of 10% bovine serum
albumin.
Minimal bactericidal concentration. Minimal bactericidal concentrations (MBCs) against B.
subtilis 168 were determined in MH medium in a microtiter plate assay. 5 x 105 cells per mL
were treated with 1, 2, 5, and 10‐fold MIC (2, 4, 10, 20 µg/mL FcPNA and 32, 64, 160, 320 µg/mL
RcPNA). An untreated control was directly plated on MH agar plates and incubated at 37 °C for
16 h to determine the amount of applied colony‐forming units (CFUs). Antibiotic‐treated
cultures were incubated at 37 °C for 24 h. Samples were then plated on MH agar plates and
incubated at 37 °C for 16 h. CFUs were counted and related to those of the untreated control.
The antibiotic concentration that led to a CFU reduction of 5 log units was taken as MBC.
Cytotoxicity. Cytotoxiciy against human cancer cell lines was tested in three different assays (for
detailed experimental procedures see Supplementary Information). Caco‐2 and L6.C11 cells
were exposed to FcPNA 7 days after seeding when cells had grown into confluency and were
fully established and no longer growing. Metabolic activity was determined in a resazurin‐based
assay. MCF7, HepG2, and HT29 cells were exposed to FcPNA 24 h after seeding, when cells had
attached but were still growing into confluency. Viability was determined using a crystal violet‐
based assay. NRK‐52E and CCRF‐CEM cells were exposed to FcPNA directly after seeding and
metabolic activity determined in an Alamar Blue‐based assay.Bacterial strains and growth
I Hetero-tri-organometallic PNA backbone derivatives
217

conditions. For mechanistic studies, B. subtilis 168 (trpC2) [31] was grown at 37 °C under steady
agitation in chemically defined Belitzky Minimal Medium (BMM) [32]. In growth experiments,
bacterial cultures were treated with different antibiotic concentrations in mid exponential
growth phase after reaching an optical density at 500 nm (OD500) of 0.35. For physiological
stress experiments, including proteomic studies, antibiotic concentrations were chosen that led
to reduced growth rates compared to the untreated control (Supplementary Figure 5).
Determination of survival rates. B. subtilis was grown in Luria Bertani (LB) medium at 37 °C
under steady agitation to an OD500 of 0.35. 5 x 105 cells per mL were exposed to 4 µg mL‐1 FcPNA
for 15 min or left untreated. Cells were then plated on LB agar plates and incubated overnight
before colony forming units were counted. The experiment was performed 4 times
independently.
Proteome analysis. Preparation of radioactively labeled protein extracts and two‐dimensional
polyacrylamide gelelectrophoresis were performed as described previously [12]. FcPNA was
applied at a concentration of 2 µg mL‐1 (2.9 µM) and RcPNA of 1 µg mL‐1 (1.5 µM). Proteins were
identified by MALDI‐ToF/ToF as described previously [12]. Protein spots that could not be
identified with this method were identified by nLC‐ESI‐MS/MS as described in the
Supplementary Methods.
Atomic absorption spectroscopy. Cells were grown in BMM until early logarithmic growth
phase and stressed with 2 µg mL‐1 (2.9 µM) FcPNA as in the proteomic experiments. After 15
min of antibiotic stress, cells were harvested by centrifugation, washed three times with
Tris/EDTA buffer (100 mM Tris/1 mM EDTA, pH 7.5), and disrupted by ultrasonication. Cell
debris was kept for measuring the cell wall fraction. The crude cell extract was subjected to
ultracentrifugation at 80,000 x g for 4 h. The resulting supernatant yielded the cytosolic fraction.
The membrane pellet was dissolved in EDTA‐free Tris buffer (100 mM Tris, pH 7.5). Samples
were lyophilized using a freeze dryer BETA I instrument (Martin Christ) at ‐20 °C overnight. For
determination of the manganese content, samples were resuspended in distilled H2O. Triton X‐
100 and HNO3 were added to 200 µL of resuspended samples to final concentrations of 1% and
13%, respectively. Manganese measurements were performed using a ContrAA 700 graphite
furnace high resolution continuum source atomic absorption spectrometer (Analytik Jena).
Samples were injected at a volume of 20 µL into regular graphite tubes and processed in the
I Hetero-tri-organometallic PNA backbone derivatives
218

furnace according to a recently described protocol [33]. Manganese was detected at a
wavelength of 279.4817 nm. The mean absorbance of duplicate injections was used throughout
the study. A pure sample of FcPNA was used for calibration. Sensitivities and detection limits of
the AAS method depend on the sample matrix and instrument performance. For biological
samples measured in this study sensitivities ranged between 0.35–1.12 A µM‐1 and detection
limits reported by the instrument software as characteristic concentrations were in the range of
0.0039–0.0096 µM (1%A)‐1. Measured manganese concentrations were related to the B. subtilis
cytosolic, cell wall, and membrane volumes. Volumes of these cell fractions were calculated
based on cryo‐electron micrographs by Matias and Beveridge [34‐35]. The volumes of the
cellular cytosolic fraction, the cell wall fraction, and the cell membrane fraction were estimated
at 3.09 x 10‐9 µl, 8.08 x 10‐10 µl, and 8.99 x 10‐11 µl, respectively. The experiment was performed
three times independently.
Microscopy. All microscopy experiments were performed in three biological replicates. For
fluorescence and light microscopy experiments, the following antibiotic concentrations were
used to treat B. subtilis: 8 µg mL‐1 (11.6 µM) FcPNA, 6 µg mL‐1 (8.8 µM) RcPNA, 10 µg mL ‐1 (11.1
µM) valinomycin, 1 µg mL ‐1 (1.1 µM) gramicidin S, 0.75 µg mL ‐1 (2.5 µM) nisin, and 1.5 µg mL ‐1
(2.2 µM) vancomycin. MinD‐GFP localization and cell wall integrity were examined as described
previously13 using the B. subtilis 1981 GFP‐MinD strain [15] obtained from L. Hamoen
(Newcastle University, England) or B. subtilis 168, respectively.
For the BacLight assay, B. subtilis 168 was grown in BMM until reaching mid logarithmic growth
phase. The culture was split and the subcultures were treated with the same antibiotic
concentrations described above. After 15 min of antibiotic stress, 2 µl of a 1:1 mixture of the
green and red‐fluorescing dyes were added per mL culture volume and incubated for 15 min in
the dark under steady agitation. Cells were washed with 100 mM Tris/1 mM EDTA, pH 7.5 and
resuspended in the same buffer. Five (5) µl of the cell suspension were imaged in fluorescent
mode as described previously [13]. The Cell Imaging Software was used to merge the green and
red channel pictures. The same background value (factor = 25) was subtracted from each single
picture.
For the CellROX assay, B. subtilis was grown in BMM until early logarithmic growth phase and
subsequently divided into subcultures, which were stressed with 25.7 µg mL‐1 (100 µM)
I Hetero-tri-organometallic PNA backbone derivatives
219

paraquat, 8 µg mL‐1 (11.6 µM) FcPNA, 6 µg mL‐1 (8.8 µM) RcPNA, or left untreated as control.
After 15 min of antibiotic stress, CellROX Deep Red reagent (Invitrogen) was added and the cells
were incubated for another 30 min. Subsequently, cells were washed in 100 mM Tris/1 mM
EDTA, and resuspended in the same buffer. Five (5) µL of cells were imaged without fixation in
fluorescence mode using an Olympus BX51 microscope with a U‐UCD8 condenser, an
UPlanSApo 100XO objective, a U‐LH100HGAPO burner, and a U‐RFL‐T power supply. Pictures
were taken using a CC12 digital color camera and the Cell Imaging Software (all components by
Olympus).
Determination of cellular ATP levels. B. subtilis 168 was grown in BMM to an OD500 of 0.35 and
subsequently exposed to the antibiotics at the same concentrations described above.
Erythromycin was applied at 0.5 µg mL‐1 (0.37 µM). Cells were harvested after 15 min of
antibiotic stress by centrifugation, resuspended in 500 µL 10 mM Tris, pH 7.5, and disrupted by
ultrasonication as described for proteome analysis. ATP levels were determined using the Perkin
Elmer ATPlite 1step assay kit following the manufacturer's instructions. ATP standards were
prepared from 1 x 10‐7 to 8.5 x 10‐7 M ATP in the provided buffer. For determination of cellular
ATP levels, 100 µL of cytosolic cell extracts were used. All measurements were performed in five
independent biological experiments with two technical replicates each using the Tecan Infinite®
200 PRO multimode reader (Tecan).
ACKNOWLEDGEMENTS
This work was financially supported by a fellowship of the International Max Planck Research
School for Chemical Biology (MP), the Research Department Interfacial Systems Chemistry at
Ruhr‐University Bochum (NMN, JEB), the DFG (NMN) and the European Regional Development
Fund "Investing in your future" (JEB). The authors thank H.‐G. Sahl for providing S. aureus COL
and nisin, Prof. K. Hiramatsu for providing S. aureus Mu50, L. Hamoen for providing B. subtilis
1981, A. Knüfer and C. Dilk for technical assistance with cytotoxicity testing, M. Strack for his
help during RcPNA synthesis, and T. Bracht for sharing the Tecan plate reader.
The authors declare no competing financial interests.
I Hetero-tri-organometallic PNA backbone derivatives
220

REFERENCES
1. Taubes, G. (2008) The bacteria fight back. Science 321, 360.
2. Cafiso, V., Bertuccio, T., Spina, D., Purrello, S., Campanile, F., Di Pietro, C., Purrello, M., and
Stefani, S. (2012) Modulating activity of vancomycin and daptomycin on the expression of
autolysis cell‐wall turnover and membrane charge genes in hVISA and VISA strains. PLoS One
7, e29573. doi: 10.1371/journal.pone.0029573.
3. Bertsche, U., Weidenmaier, C., Kuehner, D., Yang, S. J., Baur, S., Wanner, S., Francois, P.,
Schrenzel, J., Yeaman, M. R., and Bayer, A. S. (2011) Correlation of daptomycin resistance in
a clinical Staphylococcus aureus strain with increased cell wall teichoic acid production and
D‐alanylation. Antimicrob. Agents Chemother. 55, 3922–8.
4. López, D., Vlamakis, H., Losick, R, and Kolter, R. (2009) Cannibalism enhances biofilm
development in Bacillus subtilis. Mol. Microbiol. 74, 609–18.
5. Rose, W. E., Fallon, M., Moran, J. J. & Vanderloo, J. P. (2012) Vancomycin tolerance in
methicillin‐resistant Staphylococcus aureus: influence of vancomycin, daptomycin, and
telavancin on differential resistance gene expression. Antimicrob. Agents Chemother. 56,
4422–7.
6. Patra, M., Gasser, G., Bobukhov, D., Merz, K., Shtemenko, A. V., and Metzler‐Nolte, N. (2012)
Sequential insertion of three different organometallics into a versatile building block
containing a PNA backbone. Dalton Trans. 39, 5617–9.
7. Bremner, J. B., Keller, P. A., Pyne, S. G., Boyle, T. P., Brkic, Z., David, D. M., Garas, A., Morgan,
J., Robertson, M., Somphol, K., Miller, M. H., Howe, A. S., Ambrose, P., Bhavnani, S., Fritsche,
T. R., Biedenbach, D. J., Jones, R. N., Buckheit, R. W. Jr., Watson, K. M., Baylis, D., Coates, J.
A., Deadman, J., Jeevarajah, D., McCracken, A., and Rhodes, D. I. (2010) Binaphthyl‐based
dicationic peptoids with therapeutic potential. Angew. Chem. Int. Ed. 49, 537–40.
8. Haug, B. E., Stense, W., Kalaaji, M., Rekdal, Ø., and Svendsen, J. S. (2008) Synthetic
antimicrobial peptidomimetics with therapeutic potential. J. Med. Chem. 51, 4306–14.
9. Thaker, H. D., Som, A., Ayaz, F., Lui, D., Pan, W., Scott, R. W., Anguita, J., and Tew, G. N.
(2011) Synthetic mimics of antimicrobial peptides with immunomodulatory responses. J.
Am. Chem. Soc. 11, 11088–91.
10. Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility
Tests for Bacteria That Grow Aerobically. Approved Standard – Eight Edition, Mo7‐A8. Vol.29
No.2
11. Wenzel, M., and Bandow, J. E. (2011) Proteomic signatures in antibiotic research. Proteomics
11, 3256–68.
12. Wenzel, M., Patra, M., Albrecht, D., Chen, Y. C., Nicolaou, K. C., Metzler‐Nolte, N., and
Bandow, J. E. (2011) Proteomic signature of fatty acid biosynthesis inhibition available for in
vivo mechanism‐of‐action studies. Antimicrob. Agents Chemother. 55, 2590–6.
13. Wenzel, M., Kohl, B., Münch, D., Raatschen, N., Albada, H. B., Hamoen, L., Metzler‐Nolte, N.,
Sahl, H.‐G., and Bandow, J. E. (2012) Proteomic response of Bacillus subtilis to lantibiotics
I Hetero-tri-organometallic PNA backbone derivatives
221

reflects differences in interaction with the cytoplasmic membrane. Antimicrob. Agents
Chemother. 56, 5749–57.
14. Völker, U., Völker, A., and Haldenwang, W. G. (1996) Reactivation of the Bacillus subtilis anti‐
sigma B antagonist, RsbV, by stress‐ or starvation‐induced phosphatase activities. J.
Bacteriol. 178, 5456–63.
15. Hecker, M, and Völker, U. (2001) General Stress Response of Bacillus subtilis and Other
Bacteria. Adv. Microb. Physiol. 44, 35–91.
16. Meister, K., Niesel, J., Schatzschneider, U., Metzler‐Nolte, N., Schmidt, D. A., and Havenith,
M. (2010) Label‐free imaging of metal‐carbonyl complexes in live cells by Raman
microspectroscopy. Angew. Che. Int. Ed. Engl. 49, 3310–2.
17. Strahl, H., and Hamoen, L. W. (2012) Membrane potential is important for bacterial cell
division. Proc. Natl. Acad. Sci. USA 27, 12281–6.
18. Katsu, T., Kuroko, M., Morikawa, T., Sanchika, K., Fujita, Y., Yamamura, H., and Uda, M.
(1989) Mechanism of membrane damage induced by the amphipathic peptides gramicidin S
and melittin. Biochim. Biophys. Acta 939, 135–41.
19. Schneider, T., Kruse, T., Wimmer, R., Wiedemann, I., Sass, V., Pag, U., Jansen, A., Nielsen, A.
K., Mygind, P. H., Raventós, D. S., Neve, S., Ravn, B., Bonvin, A. M., De Maria, L., Andersen, A.
S., Gammelgaard, L. K., Sahl, H.‐G., Kristensen, H. H. (2010) Plectasin, a Fungal Defensin,
Targets the Bacterial Cell Wall Precursor Lipid II. Science 328, 1168–72.
20. Patra, M., Gasser, G., Pinto, A., Merz, K., Ott, I., Bandow, J. E., and Metzler‐Nolte, N. (2009)
Synthesis and biological evaluation of chromium bioorganometallics based on the antibiotic
platensimycin lead structure. ChemMedChem, 4, 1930–8.
21. Patra, M., Gasser, G., and Metzler‐Nolte, N. (2012) Small organometallic compounds as
antibacterial agents. Dalton Trans. 41, 6350–8.
22. Chantson, J. T., Verga Falzacappa, M. V., Crovella, S. & Metzler‐Nolte, N. (2006) Solid‐phase
synthesis, characterization, and antibacterial activities of metallocene‐peptide
bioconjugates. ChemMedChem 1,1268–74.
23. Chantson, J. T., Varga Falzacappa, M. V., Crovella, S. & Metzler‐Nolte, M. (2005) Antibacterial
activities of ferrocenoyl‐ and cobaltocenium‐peptide bioconjugates. J. Organomet. Chem.
690, 4564–74.
24. Dubar, F., Egan, T.J., Pradines, B., Kuter, D., Ncokazi, K. K., Forge, D., Paul, J. F., Pierrot, C.,
Kalamou, H., Khalife, J., Buisine, E., Rogier, C., Vezin, H., Forfar, I., Slomianny, C., Trivelli, X.,
Kapishnikov, S., Leiserowitz, L., Dive, D., and Biot, C. (2011) The antimalarial ferroquine: role
of the metal and intramolecular hydrogen bond in activity and resistance. ACS Chem. Biol. 6,
275–87.
25. Dubar, F., Bohic, S., Dive, D., Guérardel, Y. Cloetens, P., Khalife, J., and Biot, C. (2012)
Deciphering the Resistance‐Counteracting Functions of Ferroquine in Plasmodium
falciparum‐Infected Erythrocytes. ACS Med. Chem. Lett. 3, 480‐483.
I Hetero-tri-organometallic PNA backbone derivatives
222

26. Biot, C., Nosten, F., Fraisse, L., Ter‐Minassian, D., Khalife, J., and Dive, D. (2011) The
antimalarial ferroquine: from bench to clinic. Parasite 18, 207–14.
27. Brötz‐Oesterhelt, H., and Sass, P. (2010) Postgenomic strategies in antibacterial drug
discovery. Future Microbiol. 5, 1553–79.
28. Brötz‐Oesterhelt, H., and Brunner, N. A. (2008) How many modes of action should an
antibiotic have? Curr. Opin. Pharmacol. 8, 564–73.
29. Wadhwani, P., Afonin, S., Ieronimo, M., Buerck, J., and Ulrich A. S. (2006) Optimized protocol
for synthesis of cyclic gramicidin S: Starting amino acid is key to high yield. J. Org. Chem. 71,
55–61.
30. Albada, H. B., Chiriac, A.I., Wenzel, M., Penkova, M., Bandow, J. E., Sahl, H.G., and Metzler‐
Nolte, N. (2012) Modulating the activity of short arginine‐tryptophan containing
antibacterial peptides with N‐terminal metallocenoyl groups. Beilstein J. Org. Chem. 8,
1753–64.
31. Agnostopoulos, C., and Spizizen, J. (1961) Requirements for transformation in Bacillus
subtilis. J. Bacteriol. 81, 741–6.
32. Stülke, J., Hanschke, R., and Hecker, M. (1993) Temporal activation of betaglucanase
synthesis in Bacillus subtilis is mediated by the GTP pool. J. Gen. Microbiol. 139, 2041–5.
33. Niesel, J., Pinto, A., Peindy N'Dongo, H. W., Merz, K., Ott, I., Gust, R., and Schatzschneider, U.
(2008) Photoinduced CO release, cellular uptake and cytotoxicity of a tris(pyrazolyl)methane
(tpm) manganese tricarbonyl complex. Chem. Commun. 15, 1798–800.
34. Matias, V. R., and Beveridge, T. J. (2008) Lipoteichoic acid is a major component of the
Bacillus subtilis periplasm. J. Bacteriol. 190, 7414–8.
35. Matias, V. R., and Beveridge, T. J. (2005) Cryo‐electron microscopy reveals native polymeric
cell wall structure in Bacillus subtilis 168 and the existence of a periplasmic space. Mol.
Microbiol. 56, 240–51.
I Hetero-tri-organometallic PNA backbone derivatives
223

Supplementary Information
Contents
i. Supplementary Methods
ii. Supplementary Tables
iii. Supplementary Figures
iv. References
i. Supplementary Methods
General remarks
All chemicals were of reagent grade quality or better, obtained from commercial suppliers and
used without further purification. Solvents were used as received or dried over 3/4 Å molecular
sieves. All preparations were carried out using standard Schlenk techniques. The reactions of all
the compounds containing CpMn(CO)3 or ruthenocene residues were carried out in the dark.
Azidomethyl‐ruthenocene,[1] carboxylic acid 1,[2] β‐cymantrenoyl‐propionic acid (4),[3] and the
rhenium tricarbonyl complex (6)[4] were prepared following literature procedures. FcPNA was
synthesized following the procedure as published previously except for purification was done
twice by column chromatography on silica gel.[4] RcPNA was synthesized following a similar
synthetic sequence. 1H and 13C NMR spectra were recorded in deuterated solvents on Bruker
DRX 200 or 400 spectrometers at room temperature. The chemical shifts, δ, are reported in
ppm (parts per million). The residual solvent peaks have been used as an internal reference. The
abbreviations for the peak multiplicities are as follows: s (singlet), d (doublet), dd (doublet of
doublets), t (triplet), q (quartet), m (multiplet) and br (broad). Infrared spectra were recorded
either on an ATR unit using a Bruker Tensor 27 FT‐IR spectrophotometer. Signal intensity is
abbreviated br (broad), s (strong), m (medium), and w (weak). ESI mass spectra were recorded
I Hetero-tri-organometallic PNA backbone derivatives
224

on a Bruker Esquire 6000. Analytical HPLC was performed using a Nucleosil 100‐5 C18 column
(250 × 3 mm) on a Hitachi chromaster HPLC system at 30 °C. The flow rate was 0.5 mL/min and
UV‐absorption was measured at 250 nm. The runs were performed with a linear gradient of A
(acetonitrile (Sigma Aldrich HPLC‐grade) and B (distilled water containing 0.1% v/v TFA): t = 0‐
3 min, 5% A; t = 17‐22 min, 100% A; t = 22‐25 min, 5% A.
Synthesis of RcPNA
Carboxylic acid 2. A solution of 1 (980 mg, 2.3 mmol) and azidomethyl‐ruthenocene (1.3 g, 4.67
mmol) in 36 mL THF was degassed by two ‘freeze‐pump‐thaw’ cycles. Aqueous solutions of
CuSO4∙5H2O (114 mg, 0.46 mmol in 2 mL water) and sodium ascorbate (182 mg, 0.92 mmol in 2
mL water) were added sequentially. The resulting mixture was degassed by one more ‘freeze‐
pump‐thaw’ cycle and allowed to stir at 35 °C for 48 hours under argon atmosphere. The THF
was then removed and CH2Cl2 (100 mL) was added. The organic phase was washed with 0.1 M
aqueous Na2EDTA solution (until the aqueous phase became colorless), brine, dried over
anhydrous Na2SO4, filtered and concentrated. Column chromatography (silica gel,
CH2Cl2/MeOH/AcOH 5/1/0.01) yielded 2 as white sticky solid (yield: 800 mg, 49%).
NH
NOH
OO
CuSO4,Sodium ascorbate
HATU, DIPEA
i) 20% piperidine in DMF
ii) HATU, DIPEA
1
N
N
N
Re
COCO
CO
PF6 OCOOH
Mn(CO)3
Ru
N3
NH
NOH
OO
NNN
O
O
2
Ru
IH2N
NH
NNH
OO
NNN
O
O
3
Ru
I
NH
NNH
OO
NNN
O
OI
Ru
Mn
COOC
OC
5
NH
NNH
OO
NNN
Ru
O
+
Re
N
N
N
CO
CO
CO
MnOC
OC CO O
Rc-PNA
Pd(PPh3)2Cl2, CuBr
+
4
6
O
O
PF6
I Hetero-tri-organometallic PNA backbone derivatives
225

Data for 2: Rf = 0.36 (silica gel, CH2Cl2/MeOH/AcOH 5/1/0.01). 1H NMR (400 MHz, CD2Cl2): δ
(ppm) 2.55 (min) and 2.68 (maj) (rotamers, m, 2H, CH2‐CH2‐triazole ring), 2.86 (m, 2H, CH2‐CH2‐
triazole ring), 3.19 (m, 2H, NH‐CH2‐CH2), 3.37 (m, 2H, CH2‐CH2‐N), 3.84‐4.21 (rotamers, m, 5H, N‐
CH2‐COOH, CH Fmoc and Fmoc‐CH2O), 4.32‐4.56 (m, 9H, Rc), 4.82 and 4.87 (rotamers, s, 2H, Rc‐
CH2‐triazole ring), 5.62 (min) and 6.09 (maj) (rotamers, s, br, 1H, NH), 7.13‐7.28 (m, 5H, CH
triazole ring and CH Fmoc arom), 7.48 (m, 2H, CH Fmoc arom), 7.68 (m, 2H, CH Fmoc arom),
10.46 (s, br, 1H, COOH). 13C NMR (100.6 MHz, CD2Cl2): δ (ppm) 19.8 (maj) and 20.1(min)
(rotamers, CH2‐CH2‐triazole ring), 31.3 (maj) and 31.5 (min) (rotamers, CH2‐CH2‐triazole ring ),
38.5 (NH‐CH2‐CH2 ), 46.4 (maj) and 46.5 (min) (rotamers, CH Fmoc), 47.1 (NH‐CH2‐CH2), 48.3 (Rc‐
CH2‐triazole ring), 48.8 (maj) and 48.9 (min) (rotamers, N‐CH2‐COOH), 66.1 (Fmoc‐CH2O), 69.8
(CH Rc), 70.1 (min) and 70.2 (maj) (rotamers, CH Rc), 70.3 (maj) and 70.4 (min) (rotamers, CH
Rc), 84.1 (min) and 84.2 (maj) (rotamers, C Rc), 119.1 (CH Fmoc arom), 120.6 (CH triazole ring),
124.4 (maj) and 124.5 (min) (rotamers, CH Fmoc arom), 126.3 (CH Fmoc arom), 126.9 (CH Fmoc
arom), 140.3 (CH Fmoc arom), 143.2 (min) and 143.4 (maj) (rotamers, CH Fmoc arom), 145.5 (C
triazole ring), 155.7 (NHCOO), 172.5 (COOH), 174.3 (CH2CON). IR bands(ν): 2962w, 1710s, 1636s,
1522m, 1448w, 1409w, 1258m (br), 1100m, 1021m (br), 807m, 759s, 739s cm−1. ESI‐MS
(negative detection mode): m/z (%): 819.92 (30) [M+TFA–H]–, 803.92 (100) [M+TFA–OH]–.
Compound 3: To a stirred solution of the carboxylic acid 2 (800 mg, 1.13 mmol) in 25 mL of
DMF, HATU (850 mg, 2.26 mmol) and DIPEA (291 mg, 2.26 mmol) were added. The mixture was
allowed to stir for 30 minutes under argon atmosphere. P‐iodo‐aniline (495 mg, 2.26 mmol) was
NH
NOH
OO
NNN
O
O
2
Ru
NH
NNH
OO
NNN
O
O
3
Ru
I
I Hetero-tri-organometallic PNA backbone derivatives
226

then added and the mixture was stirred for 36 h at room temperature. The DMF was removed
under reduced pressure and the resulting residue was dissolved in 100 mL of DCM, washed with
distilled water and brine. The organic phase was dried over anhydrous Na2SO4, filtered and
concentrated. Flash column chromatography (silica gel, EtOAc/MeOH 25/1 → 20/1 → 15/1)
gave compound 3 as white solid (yield: 667 mg, 52%).
Data for 3: Rf = 0.31 (silica gel, EtOAc/MeOH 25/1). 1H NMR (200 MHz, CD2Cl2): δ (ppm) 2.60 (m,
2H, CH2‐CH2‐triazole ring), 2.85 (m, 2H, CH2‐CH2‐triazole ring), 3.19 (m, 2H, NH‐CH2‐CH2), 3.41
(m, 2H, CH2‐CH2‐N), 3.84 (min) and 3.90 (maj) (rotamers, s, 2H, N‐CH2‐CONH), 3.99‐4.24 (m, 3H,
CH Fmoc and Fmoc‐CH2O), 4.30‐4.51 (m, 9H, Rc), 4.79 (maj) and 4.83, 4.88 (min) (rotamers, s,
2H, Rc‐CH2‐triazole ring), 5.94, 6.37 (min) and 6.23 (maj) (rotamers, s, br, 1H, NH), 7.11‐7.47 (m,
11H, 8×CH Fmoc arom, 2×CH benzene ring and CH triazole ring), 7.64 (m, 2H, 2×CH benzene
ring), 9.18, 9.48 (min) and 9.27 (maj) (rotamers, s, br, 1H, NH). 13C NMR (50 MHz, CD2Cl2): δ
(ppm) 21.1 (min) and 21.7 (maj) (rotamers, CH2‐CH2‐triazole ring), 32.1 (CH2‐CH2‐triazole ring),
39.8 (NH‐CH2‐CH2), 47.5 (rotamers, CH Fmoc and NH‐CH2‐CH2), 49.9 (Fc‐CH2‐triazole ring), 52.3
(N‐CH2‐CONH), 67.1 (Fmoc‐CH2O), 70.9 (min) and 71.1 (maj) (rotamers, CH Rc), 71.2 (min) and
71.4 (maj) (rotamers, CH Rc), 71.6 (maj) and 71.7 (min) (rotamers, CH Rc), 85.3 (maj) and 85.5
(min) (rotamers, CI benzene ring), 87.5 (maj) and 87.7 (min) (rotamers, C Rc), 120.3 (CH Fmoc
arom), 121.3 (maj) and 121.8 (min) (rotamers, CH triazole ring), 122.4 (maj) and 122.6 (min)
(rotamers, CNH benzene ring), 125.5 (CH Fmoc arom), 127.4 (CH Fmoc arom), 128.1 (CH Fmoc
arom), 137.9 (maj) and 138.1 (min) (rotamers, CH benzene ring), 138.3 (min) and 138.6 (maj)
(rotamers, CH benzene ring), 141.5 (CH Fmoc arom), 144.4 (CH Fmoc arom), 146.6 (maj) and
146.7 (min) (rotamers, C triazole ring), 156.9 (NHCOO), 168.6 (CONH), 173.9 (CH2CON). IR
bands(ν): 3271w, 2926w, 1700s, 1630s, 1586m, 1527s, 1485s, 1449m, 1371m, 1005w, 1242s,
1044w, 843s, 818s, 758s, 740s, 621w cm−1. ESI‐MS (positive detection mode): m/z (%): 930.84
(100) [M+Na]+, 908.87 (20) [M+Na]+.
NH
NNH
OO
NNN
O
OI
Ru
Mn
COOC
OC
5
I Hetero-tri-organometallic PNA backbone derivatives
227

Hetero‐bimetallic compound 5: To a stirred solution of 3 (250 mg, 0.28 mmol) in 2 mL DMF, 5
mL of 20% piperidine in DMF was added at 0 °C and stirred for 10 minutes. Then the ice bath
was removed and the mixture was stirred for 2 hours at room temperature. DMF and piperidine
was removed using a high vacuum pump. The resulting residue was washed several times with
pentane. The solid residue thus obtained was dissolved in 3 mL DMF. The solution was added to
a solution of cymantrene keto carboxylic acid 4 (146 mg, 0.48 mmol), HATU (183 mg, 0.48
mmol) and DIPEA (62 mg, 0.48 mmol) in DMF (2 mL), which has been initially stirred for 30
minutes at room temperature. The reaction mixture was stirred for 48 hours before DMF was
removed under reduced pressure. Column chromatography (silica gel, EtOAc/MeOH 20/1 →
THF) yielded 5 as a light yellow solid (yield: 89.1 mg, 34 %).
Data for 5: Rf = 0.25 (silica gel, EtOAc/MeOH 20/1). 1H NMR (400 MHz, CD2Cl2): δ (ppm) 2.33 (m,
2H, cymantrene‐CO‐CH2‐CH2), 2.68‐2.97 (m, 6H, CH2‐CH2‐triazole ring, cymantrene‐CO‐CH2‐CH2
and CH2‐CH2‐triazole ring), 3.24‐3.50 (m, 4H, NH‐CH2‐CH2 and CH2‐CH2‐N), 4.44 (s, br, 9H, N‐CH2‐
CONH, CH Rc), 4.57 (s, br, 2H, CH Rc), 4.77 (s, br, 2H, CH cymantrene), 4.95 (m, 2H, rotamers, Rc‐
CH2‐triazole ring), 5.36 (s, br, 2H, CH cymantrene), 7.04‐7.53 (m, 6H, 4×CH benzene ring, CH
triazole ring and CO‐NH). 13C NMR (62.9 MHz, CD2Cl2): δ (ppm) 21.1 (min) and 22.5 (maj)
(rotamers, CH2‐CH2‐triazole ring), 29.2 (min) and 29.7 (maj) (rotamers, cymantrene‐CO‐CH2‐
CH2), 31.6 (min) and 31.8 (maj) (rotamers, CH2‐CH2‐triazole ring), 33.7 (min) and 33.9 (maj)
(rotamers, cymantrene‐CO‐CH2‐CH2), 37.9 (maj) and 38.3 (min) (rotamers, NH‐CH2‐CH2), 47.3
(maj) and 47.9 (min) (rotamers, NH‐CH2‐CH2), 49.4 (min) and 49.7 (maj) (rotamers, Rc‐CH2‐
triazole ring), 51.9 (N‐CH2‐CONH), 70.6 (min) and 70.7 (maj) (rotamers, CH Rc), 71.1 (CH Rc),
71.2 (maj) and 71.4 (min) (rotamers, CH Rc), 83.9 (cymantrene‐CH), 85.1 (C Rc), 86.8
(cymantrene‐CH), 87.1 (C‐I benzene ring), 91.4 (cymantrene‐C), 120.9 (min) and 121.3 (maj) (CH
triazole ring), 122.1 (CNH benzene ring), 137.5 (CH benzene ring), 138.1 (CH benzene ring),
146.2 (C triazole ring), 168.1 (CONH), 171.9 (CONH), 173.6 (CH2CON), 196.4 (maj) and 196.9
(min) (rotamers, cymantrene‐CO‐CH2‐CH2), 223.1 (maj) and 224.4 (min) (rotamers, Mn‐C≡O). IR
bands(ν): 2924w, 2022s and 1928s (M‐C≡O), 1670m (br), 1529m, 1484w, 1461w, 1305w, 1263w,
1181w, 1035w, 948w, 875m, 667m, 627s cm−1. ESI‐MS (positive detection mode): m/z (%):
994.79 (100) [M+Na]+, 972.79 (10) [M+H]+.
I Hetero-tri-organometallic PNA backbone derivatives
228

Hetero‐trimetallic compound RcPNA: A solution of 5 (89 mg, 0.092 mmol) and 6 (60 mg, 0.092
mmol) in 3 mL DMF/NEt3 mixture (2/1, v/v) was degassed by three ‘freeze‐pump‐thaw’ cycles.
Then CuBr (1.63 mg, 0.01 mmol) and cis‐dichlorobis (triphenylphosphine) palladium(II) (1.94 mg,
0.003 mmol) were added under nitrogen atmosphere and the mixture was degassed again by
two ‘freeze‐pump‐thaw’ cycles. The deep brown‐colored solution thus obtained was allowed to
stir for 22 hours at room temperature in the dark. The DMF and NEt3 were removed using high
vacuum pump. The residue was diluted with 50 mL of CH2Cl2 and washed with distilled water
and brine. The CH2Cl2 layer was dried over anhydrous Na2SO4, filtered and evaporated to
dryness. The residue was washed with pentane to remove the remaining triethylamine and
finally dried in vacuum. Flash column chromatography (two times, silica gel, CH2Cl2:MeOH 5:1)
afforded a light brown sticky solid which was re‐dissolved in DCM and filtered. The filtrate was
dried and washed with pentane to give the desired compound RcPNA as a light brown power
(yield: 23 mg, 17 %).
Data for RcPNA: Rf = 0.51 (silica gel, CH2Cl2/MeOH 5/1). Retention time (tR) in RP‐HPLC = 17.9
min. ESI‐MS (positive detection mode): m/z (%): 1351.28 (100) [M‐PF6]+. IR bands (ν): 2923w,
2027s and 1909s (M‐C≡O), 1655m (br), 1514m, 1446m, 1245w, 1060w, 833s, 765m, 733w,
667w, 628m cm−1.
Toxicity against mammalian cell lines MCF7, HepG2, and HT29
Cells were cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS), 2 mM
L‐glutamine, 200 U ml‐1 penicillin, and 200 µg ml‐1 streptomycin in 10% CO2 atmosphere.
Absolute cell numbers were determined by use of crystal violet. Cells were seeded in 96‐well
cell culture‐treated microtiter plates and grown for 24 hours under standard conditions. FcPNA
was dissolved in DMSO (10 mg ml‐1 stock solution) and cell cultures were then exposed to 3, 10,
30, 100, and 300 µg ml‐1 for 65 hours. Each concentration was tested in six replicates each in
two independent experiments. Before crystal violet was added to the cells, they were washed
NH
NNH
OO
NNN
Ru
O
+
Re
N
N
N
CO
CO
CO
MnOC
OC CO O
RcPNA
PF6
I Hetero-tri-organometallic PNA backbone derivatives
229

three times with phosphate buffered saline (PBS), and fixed with 2% glutardialdehyde for 20
minutes at 37 °C. Glutardialdehyde was discarded and membranes permeabilized with Triton X‐
100 (0.1% in PBS). Afterwards, cells were incubated in crystal violet solution (0.04% in 4%
ethanol) on a shaker for 1 h. Cells were washed six times with H2O before crystal violet was
eluted with ethanol for 3.5 h. Absorbance was measured at 570 nm. Cell biomass was calculated
by subtracting the absorbance values read prior to substance addition at 24 h. For
determination of the half maximal inhibitory concentrations (IC50), viability in percent was
plotted against the compound concentration on a half‐logarithmical scale. Sigmoidal function fit
was performed using Origin 7 (Originlab, Northampton, USA). IC50 values were directly
calculated from the fit function.
Toxicity against mammalian cell lines NRK‐52E and CCRF‐CEM
NRK‐52E rat kidney epithelial cells were cultured in DMEM PAN (PAN Biotech, Cat.‐Nr. P04‐
03500) supplemented with 10% fetal calf serum (FCS), 100 U mL‐1 penicillin, 100 µg mL‐1
streptomycin, and 1% L‐glutamine at 37 °C in 5% CO2 atmosphere. CCRF‐CEM human T‐cell
lymphoblast cells were cultured in RPMI 1640 medium supplemented with 10% FCS, 1%
penicillin/ streptomycin, and 1% L‐glutamine, at 37 °C in a 5% CO2 atmosphere.
FcPNA (20 mg ml‐1 stock solution in 100% DMSO) was diluted into culture medium without FCS
so that 10 µl of the dilution gave final compound concentrations of 200, 66, 22.2, 7.4, 2.4, 0.82,
2.7, and 0.09 µg ml‐1 in the cell culture. Ten µl of the dilutions were transferred into 96‐well
tissue‐culture microtiter plate wells to which 90 µl of cells cultured in DMEM PAN or RPMI 1640
medium were added (final concentrations: 1% DMSO, 10% FCS, 2.5x1003 NRK‐52E cells and
6.2x1004 CCTF‐CEM cells). Plates were incubated at 37 °C for 72 h in 5% CO2 atmosphere. Ten
(10) µl of Alamar Blue (Invitrogen) were added to the plates. After 3 h at 37 °C in 5% CO2
atmosphere, fluorescence was detected with a Tecan Spectra Fluor Plus instrument (exitation at
550 nm, emission at 595 nm as a readout for cell viability. Each FcPNA concentration was tested
in duplicate in two independent experiments.
Toxicity against Caco‐2 and L6.C11 cell lines. Caco‐2 cells were cultured and differentiated in
DMEM (Dulbecco’s modified eagle medium) supplemented with 20% fetal calf serum (FCS), 100
U mL‐1 penicillin and 100 µg mL‐1 streptomycin (PAA, Austria, Pasching), 2 mM Glutamax and
non‐essential amino acids (Invitrogen, Carlsbad, California, USA). L6.C11 cells were proliferated
I Hetero-tri-organometallic PNA backbone derivatives
230

in DMEM supplemented with 10% FCS, 100 U mL‐1 penicillin and 100 µg mL‐1 streptomycin and 2
mM glutamax. Caco‐2 (6000 cells per well) or L6.C11 (5000 cells per well) were seeded in a
microtiter plate. Caco‐2 cells were differentiated for 7 days in the culture medium described
above while L6.C11 cells were differentiated in DMEM supplemented with 2% FCS,
penicillin/streptomycin and glutamax.
Fc‐PNA was dissolved in DMSO and diluted from 0 to 700 µM in cell culture medium, resulting in
a final amount of DMSO of 1%. Cells were incubated with antibiotic for 48 hours. Every
concentration was tested 8‐fold. Afterwards, resazurin (10% in DMEM) was added to the cells
and absorbance decrease at 690 nm was measured over 120 minutes in an Optima Microwell
Reader (BMG Labtech, Ortenberg, Germany).
Protein identification by nLC‐ESI‐MS/MS. Protein spots were identified using a Synapt G2‐S
high definition mass spectrometer equipped with a lock spray source for electrospray ionization
and a ToF detector (Waters, Milford, MA, USA). Manually excised protein spots were destained
with 20 mM ammoniumbicarbonate in 30% acetonitrile, dried completely, and subsequently
tryptically digested (6.25 ng µL‐1, 16 h, Promega, Fitchburg, WI, USA). Peptides were eluted into
ultrapure water by 15 minutes of ultrasonication. Eluates were loaded on a trap column (C18,
pore size 100 Å, particle diameter 5 µm, inner diameter 180 µm, length 20 mm) and were then
eluted using gradients of acetonitrile with 0.1% formic acid (350 nL min‐1, linear gradient 2‐60%
in 40 min) from an analytical column at 50 °C (C18, pore size 130 Å, particle diameter 1.7 µm,
inner diameter 75 µm, length 150 mm) to be subjected to mass spectrometry. Spectra were
recorded in positive resolution mode over a mass range of 50 to 1800 m z‐1 with 1 s per scan.
The following parameters were used for the NanoLockSpray source: capillary voltage, 3.3 kV;
sampling cone voltage, 30 V; source temperature, 80 °C; desolvation temperature, 200 °C; cone
gas flow; 50 L h‐1; desolvation gas flow, 600 L h‐1. [Glu1]‐Fibrinopeptide B serving as lock mass
analyte was fed through the lock spray channel (lock mass capillary voltage, 3.2 V). Analysis of
the spectra was performed using MassLynx V4.1 SCN813.
I Hetero-tri-organometallic PNA backbone derivatives
231

ii. Supplementary Tables
Supplementary Table 1: Details on mass spectrometric identification of proteins excised from preparative 2D gels
protein ID protein molecular mass
[Da] protein pI peptide count Mascot protein score Mascot protein score C.I. %
sequence coverage [%]
SpoVG 10886 5.25 9 426 100 77
SpoVC 20859 5.68 11 222 100 55
SkfG 14467 5.33 3 87 100 19
GsiB 13789 5.31 5 122 100 n.d.
YqxA 34375 5.83 7 83 100 21
Nin 14987 5.09 9 392 100 49
ClpP 21668 5.19 12 353 100 67
GroEL 57388 4.53 11 146 100 29
PpiB 15246 5.53 3 149 100 36
CheV 34612 4.82 13 378 100 46
Dps 16583 4.64 7 86 100 60
YtkL 24816 5.14 8 75 100 38
YdaG 15867 5.33 5 111 100 52
YtxH 16675 5.30 5 131 100 28
SodA 22561 5.33 6 450 100 36
AzoR2 23257 5.26 7 117 100 38
YdbD 30238 5.06 10 103 100 39
YphP 15865 4.81 5 94 100 40
YwbC 14424 4.62 7 231 100 50
Tpx 18204 4.89 15 403 100 91
MrgA 17322 4.79 3 184 100 26
YqiW 16186 5.00 7 137 100 66
YceC 21810 5.46 12 215 100 43 YceH 41646 5.90 13 263 100 35PspA 25125 5.87 11 91 100 41
RacX 25270 5.46 3 73 100 12
IspH 34902 5.68 2 91 100 10
YoxD 25283 5.48 16 570 100 65
YjdA 27432 5.74 5 55 100 18
I Hetero-tri-organometallic PNA backbone derivatives
232

protein ID
protein molecular mass
[Da]
protein pI
peptide count
Mascot protein score
Mascot protein score C.I. %
sequence coverage
NadE 30376 5.07 16 459 100 55
Adk 24104 4.65 11 205 100 65
NudF 20967 4.78 7 109 100 42
YqiG 40780 5.34 19 325 100 45
YhdN 37289 4.96 10 132 100 41
YwrO 19942 5.33 7 166 100 44
IolS 35146 5.50 16 280 100 62
ArgC 38049 5.3 14 342 100 39
YfjR 27848 4.97 4 109 100 21
IolS 35146 5.50 16 280 100 62
YqkF 34695 5.30 10 126 100 33
LuxS 17703 5.29 5 301 100 47
GreA 17261 4.68 11 339 100 75
Frr 20754 5.52 9 361 100 53
YmaE 26560 9.11 4 60 100 15
YqeY 16755 5.71 6 184 100 26
protein ID protein molecular mass
[Da] protein pI
peptide count
PLGS score coverage
[%] precursor mass error
[ppm] product mass error
[ppm]
NfrA* 28302 5.73 20 9602 33 1.5843 4.4483
PstS* 31664 4.84 44 12665 48 1.5004 4.0909
* Protein was identified by nLC‐ESI‐MS/MS.
I Hetero-tri-organometallic PNA backbone derivatives
233

Supplemental Table 2: Marker proteins induced after FcPNA and RcPNA treatment. Induction
factors are displayed as averages over three independently performed biological experiments.
Protein ID F
cPN
A
RcP
NA
protein function category regulator
SpoVG 6.0 n.u. regulatory protein SpoVG stress response SigH
SpoVC 3.1 n.u. peptidyl-tRNA hydrolase stress response SigB
SkfG 3.7 n.u.
unknown stress response PhoP, Spo0A
GsiB general stress protein stress response SigB
YqxA 4.2 n.u. unknown stress response SigE
Nin 5.3 n.u. inhibitor of the DNA degrading activity of NucA stress response ComK
ClpP 3.5 3.8 ATP-dependent Clp protease proteolytic subunit stress response SigB
GroEL 3.7 n.u. chaperonin stress response unknown
PpiB 2.5 n.u. peptidyl-prolyl isomerase (chaperone) stress response unknown
CheV 3.5 2.9 CheA modulator stress response SigD
Dps 7.3 5.0 DNA-protecting protein, ferritin stress response SigB
YtkL 3.6 n.u. metal-dependent hydrolase stress response SigB
YdaG 12.1
90.7 general stress protein stress response SigB
YtxH 4.5 3.6 unknown stress response SigB/H
SodA 5.6 n.u. superoxide dismutase oxidative stress SigB
AzoR2 3.4 n.u. azoreductase oxidative stress SigG
YdbD 27.5
9.3 manganese-containing catalase oxidative stress unknown
YphP 2.4 n.u. disulfide isomerase, putative bacilliredoxin oxidative stress unknown
YwbC 3.8 n.u. putative methylglyoxylase oxidative stress unknown
Tpx 6.4 n.u. thiol peroxidase oxidative stress Spx
MrgA 4.1 n.u. metalloregulation DNA-binding stress protein oxidative stress PerR
YqiW 5.3 n.u. unknown, disulfide isomerase family oxidative stress unknown
YceC 5.9 117.4 similar to tellurium resistance protein cell envelope stress
SigB/M/W
YceH n.u. 18.5 similar to toxic anion resistance protein cell envelope stress
SigB/M/W
PspA 12.5
6.0 phage shock protein A cell envelope stress
SigW
RacX 18.0
24.7 amino acid racemase cell envelope stress
unknown
IspH 6.0 n.u. isopentenyl diphosphate biosynthesis cell envelope stress
unknown
YoxD 3.5 117.4 similar to 3-oxoacyl- acyl-carrier protein reductase cell envelope stress
unknown
YjdA 17.7
27.8 similar to 3-ketoacyl-acyl-carrier protein reductase cell envelope stress
unknown
NadE 2.7 13.9 NAD synthetase energy SigB
NfrA 11.7
7.2 Spx-dependent FMN-containing NADPH-linked nitro/flavin reductase
energy SigD, Spx
PstS n.u. 30.9 phosphate ABC transporter (binding protein) energy PhoP
Adk 6.4 47.8 adenylate kinase energy unknown
NudF 2.5 n.u. ADP-ribose pyrophosphatase energy unknown
YqiG 5.0 n.u. NADH-dependent flavin oxidoreductase energy unknown
YhdN 4.6 n.u. aldo/keto reductase specific for NAD(P)H energy SigB
YwrO 5.4 n.u. similar to NAD(P)H oxidoreductase energy unknown
IolS 3.4 n.u. aldo-keto reductase energy IolR
ArgC 3.0 n.u. N-acetyl-gamma-glutamyl-phosphate reductase energy unknown
YfjR 2.9 n.u. beta-hydroxyacid dehydrogenase energy YclJ
I Hetero-tri-organometallic PNA backbone derivatives
234

Protein ID
FcP
NA
RcP
NA
protein function category regulator
YqkF 3.3 3.3 similar to oxidoreductase energy unknown
LuxS 2.4 n.u. S-ribosylhomocysteinase energy unknown
GreA 4.0 5.0 transcription elongation factor GreA transcription unknown
Frr 2.8 n.u. ribosome recycling factor translation unknown
YmaE 3.5 n.u. unknown unknown unknown
YqeY 7.1 n.u. unknown unknown unknown
n.u. not upregulated
I Hetero-tri-organometallic PNA backbone derivatives
235

Supplemental Table 3: Marker proteins of FcPNA and RcPNA overlapping with those of
comparator compounds
fun
cti
on
al c
ate
go
ry
pro
tein
ID
mem
bra
ne
mem
bra
ne
and
cel
l w
all
bio
syn
thes
is
cell
wal
l b
iosy
nth
esis
pro
tein
m
od
ific
ati
on
tra
nsl
ati
on
tra
nsc
rip
tio
n
valin
om
ycin
gra
mic
idin
S
gra
mic
idin
A
nis
in
gal
lide
rmin
me
rsac
idin
van
com
ycin
dia
mid
e
pu
rom
ycin
rifa
mp
icin
stress response
SpoVG x SpoVC SkfG GsiB x x YqxA Nin
ClpP x x x GroEL x x PpiB CheV Dps x YtkL
YdaG YtxH x
oxidative stress
SodA x x MrgA x Tpx x
YdbD x x YphP YwbC YqiW
AzoR2
cell envelope stress
YceC x x x x YceH x x x x PspA x x x x x RacX x IspH YoxD x x x YjdA x x
energy metabolism
NadE x x x x x NfrA x x x Adk x
NudF YqiG x x YhdN x YwrO x IolS
ArgC YfjR LuxS
YqkF PstS x
transcription/ translation
GreA x Frr
unknown YmaE YqeY
total markers 57/35 24 20 5 8 25 13 5 29 28 32
I Hetero-tri-organometallic PNA backbone derivatives
236

iii. Supplementary Figures
Supplementary Figure 1: Analytical HPLC chromatogram (a) and ESI‐MS spectrum (b) of RcPNA.
I Hetero-tri-organometallic PNA backbone derivatives
237
Supplementary Figure 2: IR spectrum of RcPNA
I Hetero-tri-organometallic PNA backbone derivatives
238

Supplementary Figure 3: Analytical HPLC chromatogram (a)nand ESI‐MS spectrum (b) of FcPNA.
I Hetero-tri-organometallic PNA backbone derivatives
239

Supplementary Figure 4: Structures of the individual organometallic complexes that were
tested for antibacterial activity.
Supplementary Figure 5: Growth inhibition of B. subtilis 168 by FcPNA and RcPNA. Arrowheads
indicate antibiotic addition. Radioactive labelling was performed 10 min after treatment with
the underlined sublethal concentrations.
I Hetero-tri-organometallic PNA backbone derivatives
240

Supplementary Figure 6: Proteome response to valinomycin (a) and gramicidin S (b)
I Hetero-tri-organometallic PNA backbone derivatives
241

Supplemental Figure 7: Manganese content in the cell fractions: Concentrations were
calculated to starting culture volume. Whole cells and supernatant add up to the total amount
of manganese measured in the culture. Similarly, manganese contents of the subcellular
fractions almost add up to the amount measured in whole cells.
iv. References
1. Patra, M., and Metzler-Nolte, N. (2011) Azidomethyl-ruthenocene: facile synthesis of a useful metallocene derivative and its application in the ‘click’ labelling of biomolecules. Chem. Commun. (Camb.) 47, 11444-11446.
2. Gasser, G., Hüsken, N., Köster, S. D., and Metzler-Nolte, N. (2008) Synthesis of organometallic PNA oligomers by click chemistry. Chem. Commun. (Camb.) 3675-3677.
3. N'Dongo, H. W. P., Neundorf, I., Merz, K., and Schatzschneider, U. (2008) Synthesis, characterization, X-ray crystallography, and cytotoxicity of a cymantrene keto carboxylic acid for IR labelling of bioactive peptides on a solid support J. Inorg. Biochem. 102, 2114-2119.
4. Patra, M., Gasser, G., Bobukhov, D., Merz, K., Shtemenko, A. V., and Metzler-Nolte, N. (2010) Sequential insertion of three different organometallics into a versatile building block containing a PNA backbone. Dalton Trans. 39, 5617-5619.
I Hetero-tri-organometallic PNA backbone derivatives
242

J
Structural optimization of an antibacterial
hetero‐tri‐organometallic compound:
Identification of the redundant organometallic
moiety required for antibacterial activity
Malay Patra, Michaela Wenzel, Pascal Prochnow, Vanessa Pierroz,
Gilles Gasser, Julia E. Bandow, Nils Metzler‐Nolte
in preparation
J Organometallic PNA backbone derivatives SAR
243

Structural optimization of an antibacterial hetero‐tri‐organometallic compound:
Identification of the redundant organometallic moiety required for antibacterial activity
Malay Patra,†, # Michaela Wenzel,‡ Pascal Prochnow,‡ Vanessa Pierroz,# Gilles Gasser,# Julia
E. Bandow,‡ and Nils Metzler‐Nolte†,*
†Lehrstuhl für Anorganische Chemie I ‐ Bioanorganische Chemie, Fakultät für Chemie und
Biochemie, Ruhr‐Universität Bochum, Gebäude NC3 Nord, Universitätsstraße 150, D‐44801
Bochum, Germany, Fax: 0234‐32‐14378, e‐mail: nils.metzler‐[email protected] ‡Lehrstuhl für Biologie der Mikroorganismen, Fakultät für Biologie und Biotechnologie, Ruhr‐
Universität Bochum. Universitätsstraße 150, D‐44801 Bochum, Germany #Institute of Inorganic Chemistry, University of Zurich, Winterthurerstraße 190, CH‐8057
Zurich, Switzerland
*to whom correspondence should be addressed.
ABSTRACT
A systematic structure‐activity relationship (SAR) study on an antibacterial hetero‐tri‐
metallic compound (FcPNA) containing a ferrocenyl (Fc), a CpMn(CO)3 (cymantrene) and a
[(Dpa)Re(CO)3, Dpa = di‐(2‐picolyl)amine] residue is presented in this report. In order to
understand the impact of each organometallic moiety on the antibacterial activity, a series
of mono‐ and bi‐metallic compounds was synthesized and the biological activities were
evaluated. The [(Dpa)Re(CO)3] moiety was discovered to be the essential unit for the
observed antibacterial activity of FcPNA. The ferrocenyl and CpMn(CO)3 units can be
replaced one by one or both together by organic moieties such as a phenyl ring without
alteration of antibacterial activity. The mono‐metallic (8c′) as well as the two active bi‐
metallic (8a and 8b) compounds were shown to target the same bacterial structure as the
lead compound: the cytoplasmic membrane. The mono‐metallic derivative 8c′ containing the
[(Dpa)Re(CO)3] moiety as only organometallic complex, displayed the highest water solubility
and lowest cytotoxicity of all organometallic‐containing compounds while retaining potent
antibacterial activity making 8c′ a promising new lead structure for developing
organometallic antibiotics.
J Organometallic PNA backbone derivatives SAR
244

INTRODUCTION
The steady increase of microbial resistance to commercial drugs is a serious health issue
worldwide. One pathogen of high clinical importance is methicillin‐resistant Staphylococcus
aureus (MRSA), which is not only resistant to methicillin but also to other clinically used
antibiotics such as gentamycin, erythromycin, and neomycin.1, 2 The treatment of bacterial
infections with such multi‐resistant germs is highly difficult. Even in developed countries like
the US, over 100’000 life‐threatening MRSA infections were reported in 2005.3 According to
a report by the European Centre of Disease Control and Prevention, MRSA has reached an
average prevalence of 50% in Europe.4 Microbial resistance to conventional organic
antibiotics often develops and spreads amazingly fast.1 In case of several antibiotics like
linezolid or some β‐lactams, resistance was observed already before the compound was
commercialized for human use.5‐7 Since the increasing occurrence of antimicrobial resistance
in the 1990s, the annual approval rate of antibiotic therapeutics steadily decreased.
Currently, on average only one antibiotic per year reaches the market.1, 8 During the last two
decades, the number of new antibiotics that have been discovered or commercialized is
insufficient to control infections with multi‐resistant pathogens. These facts emphasize the
need for new classes of antimicrobial compounds. Compounds, which possess completely
new modes of action, target more than one bacterial structure, or attack essential structures
that are less prone to target mutation such as the bacterial cell envelope, are promising
candidates for resistance‐breaking antibacterials.9
Fe
HN N
NCl
Ferroquine
As
As
As
R
RRAs As
AsAs
As RR
R R
R
OH
NH2
R =Salvarsan®
N
N N
NAg
O
O
O
O
Silvamist®
Figure 1: Structures of organometallic antimalarial and antibacterial drug candidates.10
J Organometallic PNA backbone derivatives SAR
245

Derivatization of bioactive organic molecules with organometallic fragments was shown to
be an efficient approach to solve the resistance problem of conventional organic drug
candidates.11 The rationale behind this idea is that the metal‐based compounds might offer
metal‐specific modes of action not available for organic drugs.12, 13 This strategy was shown
to be successful for the antimalarial drug candidate chloroquine (CQ). Ferroquine (FQ, Figure
1), a ferrocene derivative of CQ, is not only capable of overcoming CQ resistance but also
showed enhanced antimalarial activity.14 Mechanistic studies suggested that the activity of
FQ against the CQ‐resistant P. falciparum is related to increased lipophilicity, change in
basicity, and the redox‐properties by ferrocene.15, 16
Despite promising results in anticancer and antimalerial research,11, 13, 14, 17 use of
organometallic compounds in antibacterial drug development is still limited.18 However,
their potential for treatment of infectious diseases was already proven in the early 1900s
with the discovery of Salvarsan (Figure 1), an organoarsenical antimicrobial agent.19, 20 Silver
complexes with N‐heterocyclic carbene (NHC) ligands are another emerging class of
promising compounds.21, 22 For example, Silvamist (Figure 1), a caffeine derived Ag‐NHC
complex exhibits high activity against tobramycin‐resistant pathogenic bacteria both in vitro
and in vivo.21, 23
For a few years, our groups are involved in the development of organometallic‐containing
antibacterial agents.18, 24‐27 Very recently, we disclosed the antibacterial activity of the
hetero‐tri‐organometallic compound FcPNA (see Table 1 for structure).28 FcPNA inhibits the
growth of Gram‐positive bacteria, including MRSA, at a minimal inhibitory concentration
(MIC) of 2 µg/mL (Table 1). A ruthenocene derivative, RcPNA (see Table 1 for structure), was
found to be slightly less active. A mechanism of action study revealed that both compounds
interact with the bacterial membrane and interfere with membrane‐associated processes
such as cell wall biosynthesis and the respiratory chain.28 Additionally, FcPNA induces
oxidative stress in bacteria, which is not the case for RcPNA. Solubility of both compounds
was around 25 µg/mL in aqueous solution limiting pharmacological evaluation of the
compounds. However, due to its promising antibacterial activity, we decided to optimize the
structure of FcPNA. We aimed at obtaining derivatives with higher solubility while retaining
or improving antibacterial potency and selectivity over mammalian cells, which are at the
J Organometallic PNA backbone derivatives SAR
246

same time easier to synthesize. With this in mind, understanding of the importance of each
organometallic moiety present in the FcPNA molecule, namely ferrocene, CpMn(CO)3, and
[(Dpa)Re(CO)3] and their contributions to antibacterial activity is crucial. As a preliminary SAR
study, we have already demonstrated that the redox property of ferrocene is not essential
for the antibacterial activity of FcPNA28 as the ruthenocene‐containing analogue RcPNA still
exhibited activity, although it was lacking the mechanistic feature of provoking oxidative
stress in bacteria. Herein, we report on the synthesis and biological activity of a series of
non‐, mono‐, and bi‐metallic compounds designed based on the structure of FcPNA. This
study allowed us to identify the essential organometallic moiety required for the
antibacterial activity of this class of molecules.
RESULTS AND DISCUSSION
Synthesis and spectroscopic characterization
Compounds 8a‐c were synthesized following a similar synthetic route to that previously
reported for FcPNA (Scheme 1).29 As a representative example, the synthesis of 8c was
initiated with the CuI‐catalysed [3+2] cycloaddition reaction30 of azidomethyl benzene to the
side chain of the alkyne‐containing peptide nucleic acid (PNA) building block 1.31 Compound
2 was isolated in 82% yield. Formation of 2 was confirmed by the presence of the
characteristic singlet from the triazole ring at 5.30 (maj) and 5.39 (min) (rotamers) ppm in its
proton NMR spectrum. Treatment of 2 with TFA, followed by amide coupling with p‐iodo‐
aniline gave 4b in 52% yield. The Fmoc‐group of 4b was then cleaved with 20% piperidine in
DMF. HATU‐mediated peptide coupling of the resulting residue with carboxylic acid 5a
yielded compound 6c. A signal at 199 ppm for the keto carbonyl group present in the 13C
NMR spectrum of 6c confirmed the presence of the expected compound. The last synthetic
step consisted of the insertion of the alkyne‐substituted (Dpa)Re(CO)3 complex (7)29 into the
iodo‐substituted aromatic ring of 6c by Pd‐catalyzed Sonogashira coupling to give compound
8c. The 13C NMR spectrum of 8c showed a signal at 195 ppm (rotamers) corresponding to the
Re(CO)3 moiety present. Furthermore, a peak at m/z = 1071.9 for the [M‐PF6]+ species
confirmed the formation of 8c. After silica column chromatography, 8c was further purified
by semi‐preparative RP‐HPLC to remove trace amount of impurity. The counter anion of 8c
was therefore changed to CF3COO‐ and the compound was designated as 8c′.
J Organometallic PNA backbone derivatives SAR
247

FmocHNN
OtBu
OO
FmocHNN
OtBu
OO
NNN
COOH
ONH
NNH
OO
NNN
R2
O
OI
FmocHNN
OH
OO
NNN
TFA
FmocHNN
NH
OO
NNN
I
R2
CuSO4,sodium ascorbate
CH2Cl2
N3
HATU
IH2N
R1
PhPh
i) 20% piperidine in DMF
ii) HATU
R1
1 2 3
R1 = Fc (4a)R1 = Ph (4b)
R1 = Fc, R2 = Ph (6a)R1 = Ph, R2 = Cy (6b)R1 = Ph, R2 = Ph (6c)
NH
NNH
OO
NNN
O
ON
N
N
Re
COCO
COR2
R1
7
Pd(PPh3)2Cl2, CuBr
R1 = Fc, R2 = Ph (8a)R1 = Ph, R2 = Cy (8b)R1 = Ph, R2 = Ph (8c)
R1 = Ph (5a)R1 = Cy (5b)
N
N
N
Re
COCO
CO
PF6+
7
PF6+
Fe
Fc =
MnOC CO
CO
Cy =
NH
NNH
OO
NNN
O
ON
N
N6c
N
N
N
Pd(PPh3)2Cl2, CuBr
9
Scheme 1: Synthesis of non‐, mono‐, and bi‐metallic compounds.
All new compounds were characterized by nuclear magnetic resonance (NMR) spectroscopy,
electrospray ionization (ESI) mass spectrometry, and infrared (IR) spectroscopy. It is worth
mentioning that all intermediates as well as final compounds showed the presence of
J Organometallic PNA backbone derivatives SAR
248

rotamers in solution leading to a sometimes tedious assignment of 1H and 13C NMR signals.
Compounds 4a, 5b, 6d were synthesized following literature procedures.29 32 Purity of the
compounds used for biological activity test was ≥95% as determined by reverse phase
analytical high performance liquid chromatography (HPLC).
Antimicrobial activity
The antimicrobial activities of all newly synthesized non‐, mono‐, and bi‐metallic derivatives
were tested against Gram‐positive Bacillus subtilis and S. aureus strains including one MRSA
strain (Table 1). As reported recently, the tri‐metallic FcPNA inhibits Gram‐positive bacterial
growth at an MIC of 2 µg/mL.28 The analogous tri‐metallic compound RcPNA, in which the
ferrocene moiety is replaced by a ruthenocene, was found to be slightly less active (4‐32
µg/mL).28 Thus, oxidative stress by ferrocene seems to enhance antibacterial potency but is
not the only component determining antibiotic activity.28 This is consistent with the similar
modes of action of both tri‐metallic compounds targeting the bacterial membrane.28
Among the three bi‐metallic derivatives, 6d lacks the [(Dpa)Re(CO)3] moiety but contains the
ferrocenyl and CpMn(CO)3 moieties. Compounds 8a and 8b have the [(Dpa)Re(CO)3] moiety
in common. However, 8a contains a ferrocenyl moiety while 8b contains the CpMn(CO)3.
When tested for antibacterial activity, 6d was found to be inactive up to 512 µg/mL. Both 8a
and 8b exhibited potent antibacterial activity with MIC values in a range from 2 to 4 µg/mL.
The mono‐metallic compounds lacking the [(Dpa)Re(CO)3] moiety (4a, 6a, 6b) were inactive.
The mono‐metallic derivative 8c′, which contains the [(Dpa)Re(CO)3] as the only
organometallic residue while both ferrocenyl and CpMn(CO)3 were replaced by phenyl
groups, was found to retain the antibacterial activity. It inhibited the growth of MRSA at a
concentration of 2 µg/mL. Its activity is comparable to that of the parent compound FcPNA
and the bimetallic derivatives 8a and 8b. These results suggest that neither the ferrocene
nor the CpMn(CO)3 moiety is essential but presence of the [(Dpa)Re(CO)3] moiety is
necessary for antibacterial activity. However, the alkyne‐substituted [(Dpa)Re(CO)3] complex
7 was found to be inactive up to 512 µg/mL,28 demonstrating that the organometallic moiety
alone is not sufficient for antibiotic activity but needs the pseudo‐peptide backbone. The
purely organic intermediate compounds 4b and 6c were also inactive. We then synthesized
compound 9, the purely organic ligand of the active mono‐metallic derivative 8c′. Compound
9 was found to have no antibacterial activity.
J Organometallic PNA backbone derivatives SAR
249
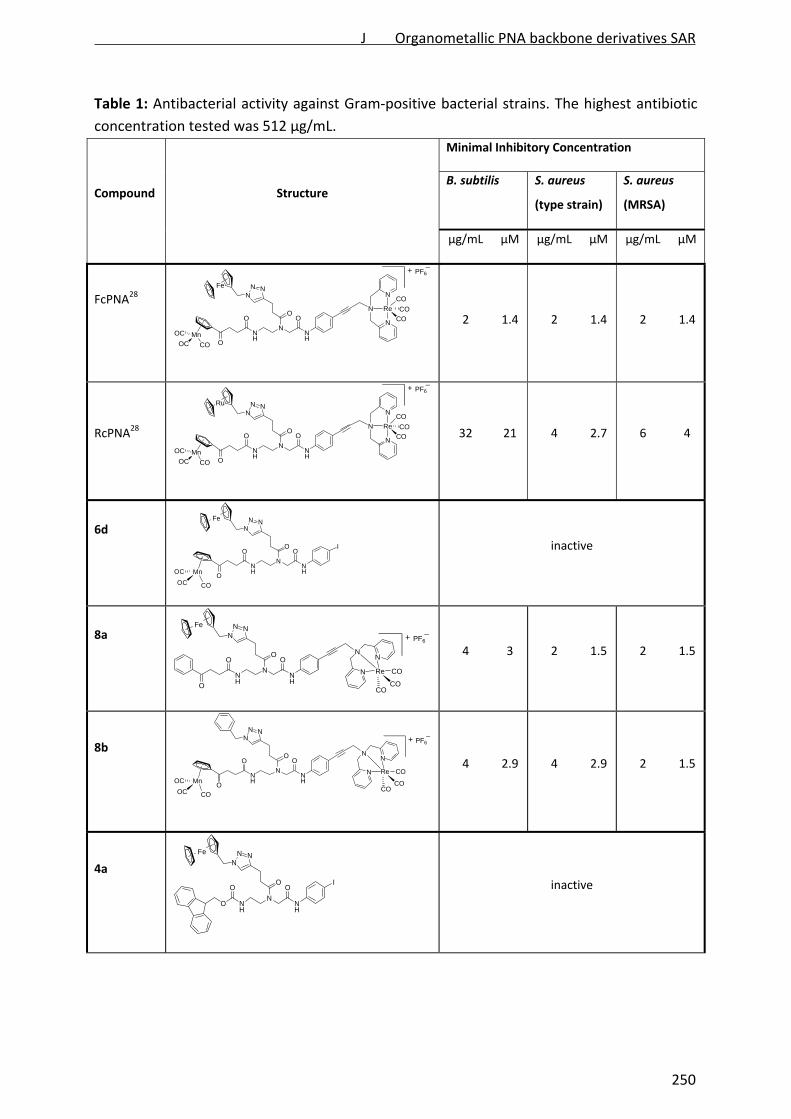
Table 1: Antibacterial activity against Gram‐positive bacterial strains. The highest antibiotic
concentration tested was 512 µg/mL.
Compound
Structure
Minimal Inhibitory Concentration
B. subtilis S. aureus
(type strain)
S. aureus
(MRSA)
μg/mL μM μg/mL μM μg/mL μM
FcPNA28
NH
NNH
OO
NNN
Fe
ORe
N
N
N
CO
CO
CO
MnOC
OC CO O
PF6+
2 1.4 2 1.4 2 1.4
RcPNA28 NH
NNH
OO
NNN
Ru
ORe
N
N
N
CO
CO
CO
MnOC
OC CO O
PF6+
32 21 4 2.7 6 4
6d
NH
NNH
OO
NNN
O
OI
Fe
Mn
COOC
OC
inactive
8a
NH
NNH
OO
NNN
O
O
Fe
N
N
N
Re
COCO
CO
PF6+
4 3 2 1.5 2 1.5
8b
NH
NNH
OO
NNN
O
ON
N
N
Re
COCO
COMn
COOC
OC
PF6+
4 2.9 4 2.9 2 1.5
4a
NH
NNH
OO
NNN
O
OI
Fe
inactive
J Organometallic PNA backbone derivatives SAR
250
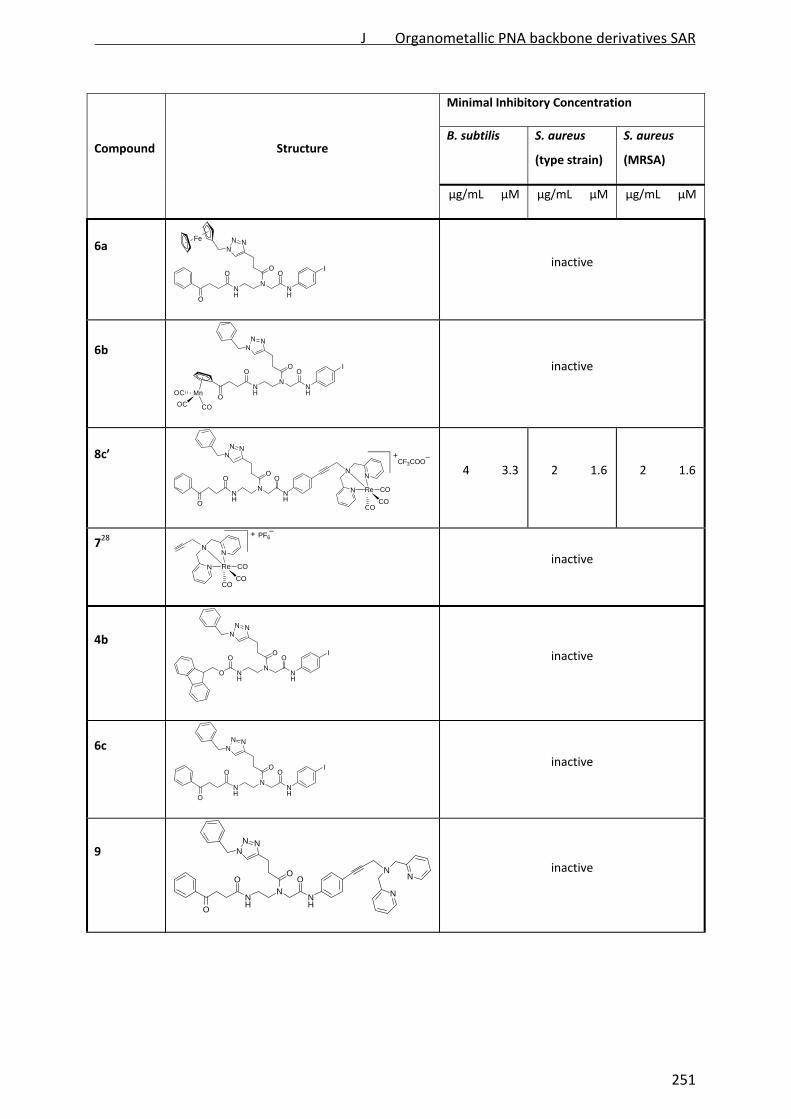
Compound
Structure
Minimal Inhibitory Concentration
B. subtilis S. aureus
(type strain)
S. aureus
(MRSA)
μg/mL μM μg/mL μM μg/mL μM
6a
NH
NNH
OO
NNN
O
OI
Fe
inactive
6b
NH
NNH
OO
NNN
IO
OMn
COOC
OC
inactive
8c′
+
NH
NNH
OO
NNN
O
ON
N
N
Re
COCO
CO
CF3COO
4 3.3 2 1.6 2 1.6
728
PF6+
N
N
N
Re
COCO
CO
inactive
4b
NH
NNH
OO
NNN
I
O
O
inactive
6c
NH
NNH
OO
NNN
IO
O
inactive
9
NH
NNH
OO
NNN
O
ON
N
N
inactive
J Organometallic PNA backbone derivatives SAR
251

This observation emphasizes that the organometallic fragment {Re(CO)3} but not ferrocene
and CpMn(CO)3 is essential for antibacterial activity of the PNA backbone derivatives.
It is worth mentioning that none of the active compounds, FcPNA, RcPNA, 8a, 8b, and 8c′,
showed activity against Gram‐negative Pseudomonas aeruginosa, Escherichia coli and
Acinetobacter baumannii. This might be due to the outer membrane of Gram‐negative
bacteria, which limits access of many compounds to the cytoplasmic membrane, where
FcPNA exerts its activity.28
Solubility in culture broth
Maximum solubility of the active derivatives was determined in Belitzky minimal medium
(BMM), a bacterial culture medium, and in Dulbecco’s modified Eagle’s medium (DMEM), a
standard growth medium for mammalian cells (Table 2).
Table 2: Maximum solubility of active compounds in bacterial (BMM) and mammalian
(DMEM) cell culture media.
Maximum solubility (µg/mL)
BMM DMEM
FcPNA 25 25
8a 100 50
8b 100 50
8c‘ 300 50
Compared to the tri‐metallic FcPNA, the di‐ and mono‐metallic derivatives displayed two‐
fold higher solubility in DMEM. In BMM, solubility of the di‐metallic compounds 8a and 8b
was 4‐fold increased. Solubility of the mono‐metallic compound 8c‘ was even 12‐fold higher
compared to that of FcPNA.
Toxicity against mammalian cell lines
Antibacterial compounds should be selective for bacterial over mammalian cells. To obtain
preliminary insight into cytotoxicity the active compounds (8a, 8b, and 8c′) were tested
against different mammalian cancerous (HeLa and HepG2) and non‐cancerous (MRC‐5) cell
lines. The metal‐based cytotoxic anticancer drug cisplatin was used as positive control (Table
3).
J Organometallic PNA backbone derivatives SAR
252

Table 3: Cytotoxicity of active compounds against mammalian cell lines.
IC50 values
HeLa HepG2 MRC‐5
μg/mL μM μg/mL μM μg/mL μM
FcPNA 12.7 ± 1 7.8 ± 1.9 > 145 > 100 > 145 100
8a 13.5 ± 2.6 10.2 ± 2.0 > 133 > 100 30.2 ± 4.1 22.8 ± 3.1
8b 10.7 ± 1.3 8.0 ± 1.0 > 134 > 100 not determined
8c′ 31.2 ± 0.9 26.3 ± 0.8 62.5 ± 11.5 52.7 ± 9.7 43.7 ± 5.9 36.9 ± 5.0
cisplatin 3.5 ± 0.9 11.5 ± 2.9 1.6 ± 0.01 5.5 ± 0.5 2.4 ± 0.4 7.9 ± 1.2
As reported for FcPNA,28 cytotoxicity of the derivatives was highly dependent on the cell
lines. For the tri‐metallic FcPNA as well as the bi‐metallic 8a and 8b, IC50 values comparable
to cisplatin were determined against HeLa cells. They showed moderate toxicity against
MRC‐5 and were non‐toxic to HepG2 cells up to 100 μM.
The mono‐metallic derivative 8c′ exhibited moderate toxicity towards all tested mammalian
cell lines with IC50 values 15‐30 times higher than the MIC against MRSA. Thus, the mono‐
metallic compound 8c′ is not only highly selective for Gram‐positive over Gram‐negative
bacteria but also by at least one order of magnitude less toxic to mammalian than to
bacterial cells.
Influence on the bacterial cell envelope
FcPNA targets the bacterial membrane.28 Binding of FcPNA to the lipid bilayer leads to loss of
the membrane potential resulting in substantial energy limitation. Depolarization is thereby
thought to be caused by interference with membrane‐bound processes like the respiratory
chain. FcPNA has also been shown to affect membrane‐bound steps of cell wall biosynthesis,
which is often observed after treatment with membrane‐integrating compounds.12 In order
to determine whether the active derivatives 8a, 8b, and 8c′ still possess the same
antibacterial mechanism, we tested their influence on membrane potential, membrane
permeabilization, and cell wall integrity. The antimicrobial peptide nisin, which binds to cell
wall precursor lipid II and forms membrane pores, was used as a positive control.
J Organometallic PNA backbone derivatives SAR
253
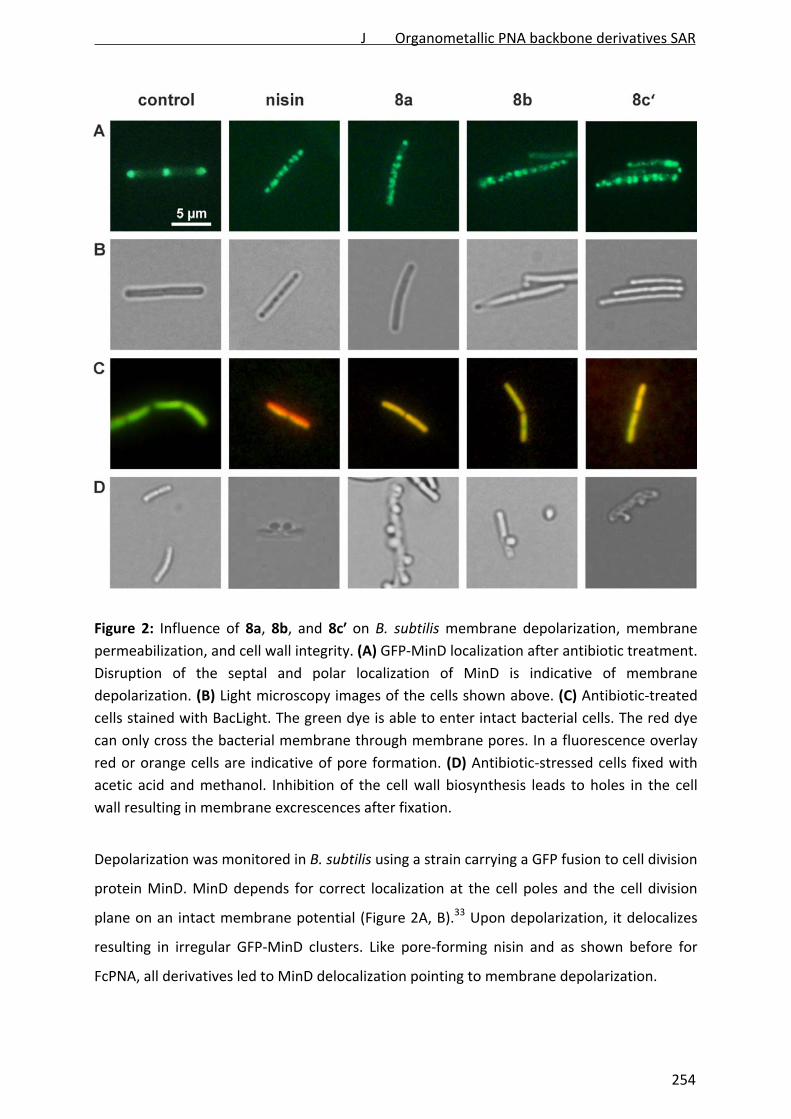
Figure 2: Influence of 8a, 8b, and 8c′ on B. subtilis membrane depolarization, membrane
permeabilization, and cell wall integrity. (A) GFP‐MinD localization after antibiotic treatment.
Disruption of the septal and polar localization of MinD is indicative of membrane
depolarization. (B) Light microscopy images of the cells shown above. (C) Antibiotic‐treated
cells stained with BacLight. The green dye is able to enter intact bacterial cells. The red dye
can only cross the bacterial membrane through membrane pores. In a fluorescence overlay
red or orange cells are indicative of pore formation. (D) Antibiotic‐stressed cells fixed with
acetic acid and methanol. Inhibition of the cell wall biosynthesis leads to holes in the cell
wall resulting in membrane excrescences after fixation.
Depolarization was monitored in B. subtilis using a strain carrying a GFP fusion to cell division
protein MinD. MinD depends for correct localization at the cell poles and the cell division
plane on an intact membrane potential (Figure 2A, B).33 Upon depolarization, it delocalizes
resulting in irregular GFP‐MinD clusters. Like pore‐forming nisin and as shown before for
FcPNA, all derivatives led to MinD delocalization pointing to membrane depolarization.
254
J Organometallic PNA backbone derivatives SAR

The BacLight assay was employed to investigate, if depolarization is accompanied by
disruption of the membrane structure. This staining method combines a green‐fluorescent
dye that crosses intact membranes and a red‐fluorescent dye, which may only enter
bacterial cells through membrane pores or larger holes. In fluorescence overlays,
permeabilized cells appear orange. While pore‐forming nisin led to red‐fluorescencing cells,
the FcPNA derivatives 8a, 8b, and 8c′ led to yellow‐fluorescence (Figure 2C). This provides
evidence for slight membrane permeabilization by 8a, 8b, and 8c’ rather than for organized
pore formation. Permeabilization could be due to a detergent‐like carpet mechanism of
action as was proposed for some antimicrobial peptides.34 However, FcPNA was not shown
to permeabilize the bacterial membrane in the BacLight assay 28 suggesting slight differences
in membrane interaction between the tri‐metallic compound and the di‐ and mono‐metallic
derivatives.
FcPNA has been shown to affect cell wall integrity, probably by impairing membrane‐bound
cell wall biosynthesis steps.28 The effects of the derivatives 8a, 8b, and 8c‘on B. subtilis cell
wall integrity were monitored by microscopic examination of the cell shape after fixation
with acetic acid and methanol. This treatment reliably leads to membrane extrusions
through cell wall holes, which occur upon inhibition of membrane‐bound cell wall
biosynthesis steps.35 As positive control nisin, inhibiting cell wall biosynthesis by binding to
precursor lipid II, all tested PNA derivatives led to membrane extrusions (Figure 2D). This
demonstrates the same, probably indirect effect on cell wall biosynthesis by all derivatives.
Taken together, the derivatives 8a, 8b, and 8c‘ target the bacterial membrane and influence
cell wall integrity but show more efficient membrane permeabilization abilities than the tri‐
metallic FcPNA.
CONCLUSION
In order to identify the essential organometallic moiety responsible for antibacterial activity,
a series of non‐, mono‐, and bi‐metallic derivatives was synthesized based on the lead
compound FcPNA. All compounds were tested against different Gram‐positive bacterial
strains including MRSA. The study suggests that the [(Dpa)Re(CO)3] moiety is essential for
the antibacterial activity of FcPNA. The ferrocene and CpMn(CO)3 residues can be replaced
by hydrophobic moieties like phenyl groups without affecting antibacterial activity. Hence,
J Organometallic PNA backbone derivatives SAR
255

the bi‐metallic derivatives 8a and 8b, containing the [(Dpa)Re(CO)3] and either a ferrocenyl
or CpMn(CO)3 moiety, and the mono‐metallic derivative 8c′ that contains the [(Dpa)Re(CO)3]
as sole organometallic residue exhibited potent activity against Gram‐positive bacteria
comparable to that of the tri‐metallic FcPNA. Similarly to FcPNA, all three active compounds
8a, 8b, and 8c′ target the bacterial membrane and influence cell wall integrity. However, the
derivatives show more efficient membrane permeabilization abilities than the original
FcPNA. All active derivatives displayed higher solubility in culture media than the tri‐metallic
FcPNA, with 8c′ being most soluble. The monometallic derivative 8c′ is 15‐30 times more
toxic to Gram‐positive bacteria than to mammalian cell lines. The main goal of future studies
will now be to develop compounds based on the new 8c’ lead structure that display even
higher selectivity for bacteria over mammalian cells.
So far, ReI tricarbonyl compounds are well‐known for their luminescent properties and for
potential as anticancer therapeutics.13, 36‐39 To best of our knowledge, this is the first study
reporting that a {ReI(CO)3} core with an appropriate organic ligand could be attractive for the
development of antibacterial compounds. We believe that this discovery will open up new
avenues in the preparation of novel organometallic antimicrobial drug candidates.
EXPERIMENTAL SECTION
Materials. All chemicals were of reagent grade quality or better, obtained from commercial
suppliers, and used without further purification. Solvents were used as received or dried
over molecular sieves. All preparations were carried out using standard Schlenk techniques.
Reactions involving CpMn(CO)3‐containing compounds were carried out in the dark. N,N‐
bis(pyridine‐2‐ylmethyl)prop‐2‐yn‐1‐amine, compound 1, 4a, 5b, 6d, and 7 were synthesized
following the literature procedure.29, 31, 32, 40 The analytical data matched what was
previously reported.
Instrumentation and methods. 1H and 13C NMR spectra were recorded in deuterated
solvents on Bruker DRX 200, 250, 400, or 600 spectrometers at 30 °C. The chemical shifts, δ,
are reported in ppm (parts per million). The residual solvent peaks have been used as an
internal reference. The abbreviations for the peak multiplicities are as follows: s (singlet), d
(doublet), dd (doublet of doublets), t (triplet), q (quartet), m (multiplet), and br (broad).
Infrared spectra were recorded on an ATR unit using a Bruker Tensor 27 FTIR
J Organometallic PNA backbone derivatives SAR
256

spectrophotometer at 4 cm‐1 resolution. Signal intensity is abbreviated br (broad), s (strong),
m (medium), and w (weak). ESI mass spectra were recorded on a Bruker Esquire 6000.
Analytical HPLC was performed using a Nucleosil 100‐5 C18 column (250 × 3 mm) on a
Hitachi chromaster HPLC system at 25 °C. The flow rate was 0.5 mL min‐1 and UV‐absorption
was measured at 250 nm. The runs were performed with a linear gradient of A (acetonitrile
(Sigma‐Aldrich HPLC‐grade) and B (distilled water containing 0.1% v/v TFA): t = 0‐3 min, 5%
A; t = 17‐22 min, 100% A; t = 22‐25 min, 5% A. Preparative HPLC was performed using a
Nucleosil 100‐7 C18 column (250 × 21 mm) on a Varian Prostar HPLC system at 25 °C. The
flow rate was 15 mL min‐1 and UV‐absorption was measured at 250 nm. The runs were
performed with a linear gradient of A (acetonitrile (Sigma‐Aldrich HPLC‐grade) and B
(distilled water containing 0.1% v/v TFA): t = 0‐3 min, 10% A; t = 3‐6 min, 50% A; t = 6‐23 min,
100% A; t = 23‐27 min, 100% A, t = 30 min, 100% A.
Minimal inhibitory concentration (MIC)
The minimal inhibitory concentrations (MIC) were tested against Escherichia coli DSM 30083,
Acinetobacter baumannii DSM 30007, Pseudomonas aeruginosa DSM 50071, Bacillus subtilis
DSM 402, Staphylococcus aureus DSM 20231 (type strain), and Staphylococcus aereus ATCC
43300 (MRSA) in a microtiter plate assay according to the guidelines of the Clinical and
Laboratory Standards Institute (CSLI).41 E. coli, A. baumannii, S. aureus, and B. subtilis were
grown in Mueller Hinton broth, P. aeruginosa in cation‐adjusted Mueller Hinton II. Peptides
were dissolved in DMSO to give 10 mg/mL stock solutions. Serial dilution in culture media
was carried out automatically with the Tecan Freedom Evo 75 liquid handling workstation
(Tecan, Männedorf, Switzerland) from 512 to 0.5 µg/mL. Peptide dilutions were inoculated
with 105 bacteria/mL taken from late exponential cultures grown in the same media in a
total volume of 200 µL per well. Cells were incubated for 16 hours at 37 °C. The lowest
compound concentration inhibiting visible bacterial growth is reported as MIC.
Mammalian cell culture and cytotoxicity test. Cytotoxicity studies were performed on three
different cell lines by a fluorometric cell viability assay using resazurin (Promocell
GmbH). Human cervical carcinoma cells (HeLa) were cultured in DMEM (Gibco)
supplemented with 5% fetal calf serum (FCS, Gibco), 100 U/mL penicillin, 100 mg/mL
streptomycin at 37 °C and 5% CO2. The normal human lung fibroblast MRC‐5 cell line was
maintained in F‐10 medium (Gibco) supplemented with 10% FCS (Gibco), penicillin (100
J Organometallic PNA backbone derivatives SAR
257

U/mL), and streptomycin (100 mg/mL). The human hepatomacarcinoma (HepG2) cells were
cultured in DMEM (Gibco) supplemented with 10% fetal calf serum (FCS, Gibco), 100 U/mL
penicillin, 100 mg/mL streptomycin at 37 °C and 5% CO2. One day before treatment, cells
were seeded in triplicate in 96‐well plates at a density of 4 x 103 cells/well for HeLa and
HepG2, and 7 x 103 for MRC‐5 in 100 mL growth medium. Upon treating cells with increasing
concentrations of respective compounds for 48 h, the medium was removed, and 100 mL
complete medium containing resazurin (0.2 mg/mL final concentration) was added. After 4 h
of incubation at 37 °C, fluorescence of the highly red fluorescent product resorufin was
quantified at 590 nm emission with 540 nm excitation wavelength in a SpectraMax M5
microplate reader.
GFP‐MinD depolarization assay. B. subtilis 1981 GFP‐MinD33 was grown in Belitzky Minimal
Medium (BMM)42 until early logarithmic phase. The main culture was subdivided and
aliquots were treated with 8 µg/mL (2x MIC) 8a, 8b, and 8c′, respectively, or left untreated
as control. Nisin was used as positive control at a concentration of 0.75 µg/mL. After 15
minutes of antibiotic stress, cells were microscopically examined in fluorescent mode as
described previously.43
BacLigh membrane disruption assay. BacLight staining (live/dead BacLight Bacterial Viability
Kit, Invitrogen, Carlsbad, CA, USA) was performed following the manufacturer’s instructions.
B. subtilis 16844 was grown and stressed with antibiotics as described above. After 15
minutes of antibiotic exposure, cells were stained with a 1:1 mixture of SYTO 9 and
propidium iodide (1 µL per mL cells) for 15 minutes, washed twice in 100 mM Tris/1 mM
EDTA, pH 7.5, resuspended in the same buffer, and subjected to fluorescence microscopy43.
Cell wall integrity assay. B. subtilis 168 was grown and stressed with antibiotics as described
above. After 15 minutes of antibiotic treatment, 200 µL of culture were withdrawn and
immediately fixed in 1 mL of a 1:3 mixture of acetic acid and methanol. 5 µL of fixed bacteria
were immobilized in agarose and subjected to light microscopy as described previously.43
ACKNOWLEDGMENTS
The financial support from The International Max Planck Research School for Chemical
Biology (fellowship to MP), the Research Department Interfacial Systems Chemistry at Ruhr
University Bochum (NMN, JEB), the DFG (NMN), the State of North Rhine‐Westphalia (NRW),
J Organometallic PNA backbone derivatives SAR
258

Germany, the European Union, European Regional Development Fund, "Investing in your
future" (JEB), the Swiss National Science Foundation (Professorship to GG, grant number
PP00P2_133568) and the University of Zurich are gratefully acknowledged. The authors
thank PD Dr. Stefano Ferrari for access to cell culture laboratories.
REFERENCES
1. Taubes, G., Science 2008, 321, 356‐361.
2. Saxena, S.; Gomber, C., Cent. Eur. J. Med. 2010, 5, 12‐29.
3. Klevens, R. M.; Morrison, M. A.; Nadle, J.; Petit, S.; Gershman, K.; Ray, S.; Harrison, L. H.;
Lynfield, R.; Dumyati, G.; Townes, J. M.; Craig, A. S.; Zell, E. R.; Fosheim, G. E.; McDougal,
L. K.; Carey, R. B.; Fridkin, S. K., J. Am. Med. Assoc. 2007, 298, 1763‐1771.
4. ECDC/EMEA Joint Working Group; The Bacterial Challenge, The Publications Office of the
European Union 2009, Stockholm.
5. Bandow, J. E.; Metzler‐Nolte, N., ChemBioChem 2009, 10, 2847‐2850.
6. Kloss, P.; Xiong, L.; Shinabarger, D. L.; Mankin, A. S., J. Mol. Biol. 1999, 294, 93‐101.
7. Abraham, E. P.; Chain, E.; Fletcher, C. M.; Gardner, A. D.; Heatley, N. G.; Jennings, M. A.;
Florey, H. W., Lancet 1941, 238, 177‐189.
8. Wenzel, M.; Bandow, J. E., Proteomics 2011, 11, 3256‐3268.
9. Brötz‐Oesterhelt, H.; Brunner, N. A., Curr. Opin. Pharmacol. 2008, 8, 564‐573.
10. Lloyd, N. C.; Morgan, H. W.; Nicholson, B. K.; Ronimus, R. S., Angew. Chem. Int. Ed. 2005,
44, 941 –944.
11. Jaouen, G.; Metzler‐Nolte, N., Medicinal Organometallic Chemistry. In Topics in
Organometallic Chemistry, 1st ed.; Springer: Heidelberg, Germany, 2010; Vol. 32; and
references therein.
12. Gasser, G.; Metzler‐Nolte, N., Curr. Opin. Chem. Biol. 2012, 16, 84‐91.
13. Gasser, G.; Ott, I.; Metzler‐Nolte, N., J. Med. Chem. 2011, 54, 3‐25.
14. Biot, C.; Dive, D., Bioorganometallic Chemistry and Malaria. In Medicinal Organometallic
Chemistry (Topics in Organometallic Chemistry), Jaouen, G.; Metzler‐Nolte, N., Eds.
Srringer: Heidelberg, Germany, 2010; Vol. 32, pp 155‐193.
15. Biot, C.; Castro, W.; Botte, C. Y.; Navarro, M., Dalton Trans. 2012, 41, 6321‐6580.
16. Dubar, F.; Egan, T. J.; Pradines, B.; Kuter, D.; Ncokazi, K. K.; Forge, D.; Paul, J.‐F.; Pierrot,
C.; Kalamou, H.; Khalife, J.; Buisine, E.; Rogier, C.; Vezin, H.; Forfar, I.; Slomianny, C.;
Trivelli, X.; Kapishnikov, S.; Leiserowitz, L.; Dive, D.; Biot, C., ACS Chem. Biol. 2011, 6,
275‐287.
17. Hartinger, C. G.; Dyson, P. J., Chem. Soc. Rev. 2009, 38, 391‐401.
18. Patra, M.; Gasser, G.; Metzler‐Nolte, N., Dalton Trans. 2012, 41, 6350‐6358.
19. Strebhard, K.; Ullrich, A., Nat. Rev. Cancer 2008, 8, 473‐480
J Organometallic PNA backbone derivatives SAR
259

20. Gibaud, S.; Jaouen, G., Arsenic‐Based Drugs: From Fowler's Solution to Modern
Anticancer Chemotherapy. In Medicinal Organometallic Chemistry, Jaouen, G.; Metzler‐
Nolte, N., Eds. Springer‐Verlag: Berlin Heidelberg, 2010; pp 1‐20, and references therein.
21. Youngs, W. J.; Knapp, A. R.; Wagersa, P. O.; Tessiera, C. A., Dalton Trans. 2012, 41, 327‐
336; and references therein.
22. Hindi, K. M.; Panzner, M. J.; Tessier, C. A.; Cannon, C. L.; Youngs, W. J., Chem. Rev. 2009,
109, 3859‐3884; and references therein.
23. Cannon, C. L.; Hogue, L. A.; Vajravelu, R. K.; Capps, G. H.; Ibricevic, A.; Hindi, K. M.;
Kascatan‐Nebioglu, A.; Walter, M. J.; Brody, S. L.; Youngs, W. J., Antimicrob. Agents
Chemother. 2009, 53, 3285‐3293.
24. Patra, M.; Gasser, G.; Pinto, A.; Merz, K.; Ott, I.; Bandow, J. E.; Metzler‐Nolte, N.,
ChemMedChem 2009, 4, 1930‐1938.
25. Patra, M.; Gasser, G.; Wenzel, M.; Merz, K.; Bandow, J. E.; Metzler‐Nolte, N.,
Organometallics 2012, 31, 5760‐5771.
26. Patra, M.; Gasser, G.; Wenzel, M.; Merz, K.; Bandow, J. E.; Metzler‐Nolte, N., Eur. J.
Inorg. Chem. 2011, 3295‐3302.
27. Chantson, J. T.; Falzacappa, M. V. V.; Crovella, S.; Metzler‐Nolte, N., ChemMedChem
2006, 1, 1268‐1274.
28. Wenzel, M.; Patra, M.; Senges, C. H. R.; Stepanek, J. J.; Pinto, A.; Prochnow, P.; Ott, I.;
Metzler‐Nolte, N.; Bandow, J. E., ACS Chem. Biol. doi: 10.1021/cb4000844.
29. Patra, M.; Gasser, G.; Bobukhov, D.; Merz, K.; Shtemenko, A. V.; Metzler‐Nolte, N.,
Dalton Trans. 2010, 39, 5617‐5619.
30. Moses, J. E.; Moorhouse, A. D., Chem. Soc. Rev. 2007, 36, 1249–1262.
31. Gasser, G.; Hüsken, N.; Köster, S. D.; Metzler‐Nolte, N., Chem. Commun. 2008, 3675‐
3677.
32. N'Dongo, H. W. P.; Neundorf, I.; Merz, K.; Schatzschneider, U., J. Inorg. Biochem. 2008,
102, 2114‐2119.
33. Strahl, H.; Hamoen, L. W., Proc. Natl. Acad. Sci. U.S.A 2010, 107, 12281‐12286.
34. Teixeira, V.; Feio, M. J.; Bastos, M., Progress in Lipid Research 2012, 51, 149‐177.
35. Schneider, T.; Kruse, T.; Wimmer, R.; Wiedemann, I.; Sass, V.; Pag, U.; Jansen, A.;
Nielsen, A. K.; Mygind, P. H.; Raventós, D. S.; Neve, S.; Ravn, B.; Bonvin, A. M. J. J.; Maria,
L. D.; Andersen, A. S.; Gammelgaard, L. K.; Sahl, H.‐G.; Kristensen, H.‐H., Science 2010,
328, 1168‐1172.
36. Fernandez‐Moreira, V.; Thorp‐Greenwood, F. L.; Coogan, M. P., Chem. Commun. 2010,
46, 186‐202, and references therein.
37. Thorp‐Greenwood, F. L.; Balasingham, R. G.; Coogan, M. P., J. Organomet. Chem. 2012,
714, 12–21 and references therein.
38. Lo, K. K.‐W.; Choi, A. W.‐T.; Law, W. H.‐T., Dalton Trans. 2012, 41, 6021‐6047, and
references therein.
39. Patra, M.; Gasser, G., ChemBioChem 2012, 13, 1232 – 1252.
J Organometallic PNA backbone derivatives SAR
260

40. Cabrera, D. G.; Koivisto, B. D.; Leigh, D. A., Chem. Commun. 2007, 4218‐4220.
41. Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial
Susceptibility Tests for Bacteria That Grow Aerobically. Approved Standard – Eight
Edition, Mo7‐A8. Vol.29 No.2.
42. Stülke, J.; Hanschke, R.; Hecker, M., J. Gen. Microbiol. 1993, 139, 2041‐2045.
43. Wenzel, M.; Kohl, B.; Münch, D.; Raatschen, N.; Albada, H. B.; Hamoen, L. W.; Metzler‐
Nolte, N.; Sahl, H. G.; Bandow, J. E., Antimicrob. Agents Chemother. 2012, submitted.
44. Agnostopoulos, C.; Spizizen, J., J. Bacteriol. 1961, 81, 741‐746.
J Organometallic PNA backbone derivatives SAR
261

Supplementary Information
Table of Contents
i. Synthesis and characterization of compounds
ii. RP‐HPLC traces and ESI mass spectra of 8a‐b, 8c′, and 9
iii. 1H and 13C NMR spectra of compounds
iv. References
i. Synthesis and characterization of compounds
NH
NO
OO
NNN
O
O
2
Compound 2. To a stirred solution of 1 (390 mg, 0.82 mmol) and benzyl azide (164 mg, 1.23
mmol) in degassed acetone (15 mL), aqueous solution of CuSO4∙5H2O (41 mg, 0.16 mmol in
3.5 mL degassed water) and sodium ascorbate (66 mg, 0.32 mmol in 3.5 mL degassed water).
The resulting mixture was allowed to stir at room temperature for 30 h under nitrogen
atmosphere and the progress of the reaction was monitored by TLC (silica gel, EtOAc). After
completion of the reaction, acetone was removed under vacuum, brine (100 mL) was added
and extracted with EtOAc (3×75 mL). The combined organic phase was washed with 50 mL of
0.1M aqueous Na2EDTA solution (to remove the Cu), brine, dried over anhydrous Na2SO4,
J Organometallic PNA backbone derivatives SAR
262

filtered and concentrated. Flash column chromatography (silica gel, EtOAc:MeOH 20:0 →
20:1) yielded pure 2 as colorless oil (yield: 410 mg, 82%).
Data for 2. Rf = 0.57 (silica gel, EtOAc). 1H NMR (400 MHz, CDCl3): δ (ppm) 1.43 (maj) and
1.47 (min) (rotamers, s, 9H, OC(CH3)3), 2.54 (maj) and 2.60 (min) (rotamers, m, 1H, CH2‐CH2‐
triazole ring), 2.75 (m, 1H, CH2‐CH2‐triazole ring), 2.98‐3.04 (m, 2H, CH2‐CH2‐triazole ring),
3.31 (m, 2H, NH‐CH2‐CH2‐N), 3.45 (maj) and 3.51(min) (rotamers, m, 2H, NH‐CH2‐CH2‐N),
3.90 (maj) and 3.94 (min) (rotamers, s, 2H, N‐CH2‐COOtBu), 4.13 (maj) and 4.20 (min)
(rotamers, m, 1H, CH Fmoc), 4.31(maj) and 4.50 (min) (rotamers, m, 2H, Fmoc‐CH2O), 5.30
(maj) and 5.39 (min) (rotamers, s, 2H, triazole‐CH2‐Ph), 5.79 (min) and 6.18 (maj) (rotamers,
s, br, 1H, NH), 7.12‐7.30 (m, 8H, triazole ring, CH Fmoc arom and benzene ring protons), 7.36
(m, 2H, CH Fmoc arom), 7.58 (m, 2H, CH Fmoc arom), 7.73 (d, 2H, benzene ring protons). 13C
NMR (62.9 MHz, CDCl3): δ (ppm) 21.0 (min) and 21.1(maj) (rotamers, CH2‐CH2‐triazole ring),
27.9 (min) and 28.1 (maj) (rotamers, OC(CH3)3), 32.0 (maj and 32.3 (min) (rotamers, CH2‐CH2‐
triazole ring), 39.4 (NH‐CH2‐CH2‐N), 47.1 (maj) and 47.2 (min) (rotamers, CH Fmoc), 48.1
(min) and 49.1 (maj) (rotamers, NH‐CH2‐CH2‐N), 49.7 (maj) and 51.5 (min) (rotamers, N‐CH2‐
COOtBu), 53.8 (maj) 53.9 (min) (rotamers, Ph‐CH2‐triazole ring), 66.6 (min) and 66.8 (maj)
(rotamers, Fmoc‐CH2O), 82.0 (min) and 82.9 (maj) (rotamers, OC(CH3)3), 119.9 (CH Fmoc
arom), 121.8 (CH triazole ring), 125.2 (CH Fmoc arom), 127.0 (maj) and 127.1 (min)
(rotamers, CH Fmoc arom), 127.6 (maj) and 127.7 (min) (benzene ring C), 127.8 (maj) and
127.9 (min) (benzene ring C), 128.4 (maj) and 128.5 (min) (rotamers, CH Fmoc arom), 128.9
(maj) and 129 (min) (benzene ring C), 134.8 (benzene ring C), 141.1 (br, CH Fmoc arom),
143.9 (CH Fmoc arom), 147.1 (rotamers, C triazole ring), 156.6 (NHCOO), 168.7(min) and
169.5 (maj) (rotamers, COOtBu), 172.4 (maj) and 173.2 (min) (rotamers, CH2CON). IR
bands(ν): 3136w, 1715s, 1644s, 1517w, 1449m, 1367w, 1231s, 1151s, 1077w, 943w, 846w,
728s (br) cm−1. ESI‐MS (pos. detection mode): m/z (%): 610.29 (100) [M+H]+.
NH
NNH
OO
NNN
O
OI
4b
NH
NOH
OO
NNN
O
O
3
J Organometallic PNA backbone derivatives SAR
263

Compound 3 and 4b. To a stirred solution of 2 (1 g, 1.64 mmol) in 20 mL of DCM, TFA (6 mL)
and triethylsilane (2.5 mL) was added at 0 °C. After 30 minutes, the mixture was allowed to
warm to room temperature and stirred for another 4 h. The solvent was then removed
under reduced pressure to give colorless oil. Addition of cold hexane gave 3 as white
precipitate. The hexane was decanted off and the precipitate was washed several times with
hexane to give 3 as white powder which was used for the next step without further
purification (yield: 830 mg, 91%).
To a stirred solution of the carboxylic acid 3 (553 mg, 1.0 mmol) in 20 mL of DMF, HATU (760
mg, 2.0 mmol) and N,N‐diisopropylethylamine (DIPEA) (155 mg, 1.2 mmol) were added. The
mixture was allowed to stir for 30 min under nitrogen atmosphere. P‐iodo‐aniline (328.5 mg,
1.5 mmol) in 10 mL of DCM was then added and the mixture was stirred for 40 h at room
temperature. The reaction mixture was then evaporated under reduced pressure and the
residue was diluted with 100 mL of DCM. The organic layer was washed with 0.5M aqueous
HCl, brine, dried over anhydrous Na2SO4, filtered and concentrated. Flash column
chromatography (silica gel, EtOAc:MeOH 15:0 → 15:1) was performed to obtain pure 4b as
light yellowish solid (yield: 392 mg, 52%).
Data for 4b. Rf = 0.45 (silica gel, EtOAc:MeOH 15:1). 1H NMR (400 MHz, CDCl3): δ (ppm) 2.78
(m, 2H, CH2‐CH2‐triazole ring), 3.01 (m, 2H, CH2‐CH2‐triazole ring), 3.38 (m, 2H, NH‐CH2‐CH2‐
N), 3.49 (m, 2H, NH‐CH2‐CH2‐N), 4.02 (s, 2H, N‐CH2‐CO), 4.16 (m, 1H, CH Fmoc), 4.32 (m, 2H,
Fmoc‐CH2O), 5.29 (maj) and 5.38 (min) (rotamers, s, 2H, triazole‐CH2‐Ph), 6.11 (min) and 6.50
(maj) (rotamers, s, br, 1H, NH), 7.10‐7.15 (m, 2H, triazole ring and CH Fmoc arom), 7.22‐7.60
(m, 14H, CH Fmoc arom and benzene ring protons ), 7.73 (d, 2H, benzene ring protons), 9.47
(maj) and 9.60 (min) (rotamers, NHCO). 13C NMR (100.6 MHz, CDCl3): δ (ppm) 20.9 (min) and
21.2 (maj) (rotamers, CH2‐CH2‐triazole ring), 31.6 (min) and 31.9 (maj) (rotamers, CH2‐CH2‐
triazole ring), 38.6 (min) and 39.5 (maj) (NH‐CH2‐CH2‐N), 47.1 (CH Fmoc), 48.1 (min) and 49.5
(maj) (rotamers, NH‐CH2‐CH2‐N), 51.9 (maj) and 52.7 (min) (rotamers, N‐CH2‐CO), 53.8 (maj)
and 53.9 (min) (rotamers, Ph‐CH2‐triazole ring), 66.6 (Fmoc‐CH2O), 87.3 (maj) and 87.6 (min)
(rotamers, C‐I benzene ring), 119.9 (CH Fmoc arom), 121.3 (maj) and 121.6 (min) (rotamers,
CH triazole ring), 122.1 (min) and 122.2 (maj) (rotamers, CONH‐C benzene ring), 125.2 (CH
Fmoc arom), 126.9 (CH Fmoc arom), 127.5 (maj) and 127.6 (min) (rotamers, benzene ring C),
127.8 (maj) and 127.9 (min) (rotamers, benzene ring C), 128.5 (maj) and 128.6 (min)
J Organometallic PNA backbone derivatives SAR
264

(rotamers, CH Fmoc arom), 128.8 (maj) and 128.9 (min) (rotamers, benzene ring C), 134.5
(benzene ring C), 137.3 (maj) and 137.5 (min) (rotamers, CONH‐benzene ring CH), 137.6
(min) and 137.7 (maj) (rotamers, CONH‐benzene ring CH), 141.1 (br, CH Fmoc arom), 143.9
(CH Fmoc arom), 147.1 (rotamers, C triazole ring), 156.6 (maj) and 156.7 (min) (rotamers,
CONH), 167.2 (min) and 168.5 (maj) (rotamers, CONH), 173.4 (CH2CON). IR bands(ν): 3309w,
2923m, 2853w, 1677s, 1632s, 1586w, 1545m, 1526s, 1494m, 1302w, 1256s, 1107w, 1012w,
819m, 757w, 730s cm−1. ESI‐MS (pos. detection mode): m/z (%): 755.05 (100) [M+H]+, 776.93
(25) [M+Na]+.
NH
NNH
OO
NNN
O
O
Fe
I
6a
Compound 6a. To a stirred solution of 4a1 (600 mg, 0.70 mmol) in 4 mL DMF, 10 mL of 20%
piperidine in DMF was added at 0 °C and stirred for 10 min. Then the ice bath was removed
and the mixture was stirred 2 h at room temperature. The DMF and piperidine was removed
using high vacuum pump. The resulting residue was washed several times with pentane. The
yellow color solid thus obtained was dissolved in 8 mL DMF. The solution was added to a
solution of 4‐oxo‐4‐phenylbutanoic acid (5a) (278 mg, 1.5 mmol), HATU (570 mg, 1.5 mmol)
and DIPEA (193 mg, 1.5 mmol) in DMF (3 mL) which has been initially stirred for 30 minutes
at room temperature. The reaction mixture was stirred for 48 h before being diluted with
150 mL of EtOAc. The organic phase was washed with 1M HCl (50 mL), sat. NaHCO3 solution
(50 mL), water (2×75 mL), brine (2×75 mL), dried over anhydrous Na2SO4, filtered and
concentrated. Flash column chromatography on silica gel (EtOAc:MeOH 20:1 → 15:1 → 10:1)
yielded compound 6a as yellow solid (yield: 228 mg, 42%).
Data for 6a. Rf = 0.07 (silica gel, EtOAc:MeOH 20:1). 1H NMR (400 MHz, CD2Cl2): δ (ppm) 2.29
(m, 2H, Ph‐CO‐CH2‐CH2), 2.63 (min) and 2.71 (maj) (rotamers, m, 2H, CH2‐CH2‐triazole ring),
2.92 (m, 2H, CH2‐CH2‐triazole ring), 3.15 (m, 2H, Ph‐CO‐CH2‐CH2), 3.30 (m, 2H, NH‐CH2‐CH2‐
N), 3.38 (m, 2H, NH‐CH2‐CH2‐N), 3.95 (maj) and 3.99 (min) (rotamers, s, 2H, N‐CH2‐CO), 4.07‐
4.17 (m, 9H, CH Fc), 5.03 (maj), 5.08, 5.14 (min) (rotamers, s, 2H, triazole‐CH2‐Fc), 6.82 (min)
and 7.17 (maj) (rotamers, 1 NH‐CO), 7.19‐7.49 (m, 8H, 1×triazole ring proton, 7×benzene ring
J Organometallic PNA backbone derivatives SAR
265

protons), 7.84 (m, 2H, benzene ring protons), 9.20 (maj) and 9.31 (min) (rotamers, 1H,
NHCO). 13C NMR (100.6 MHz, CD2Cl2): δ (ppm) 21.2 (min) and 21.6 (maj) (rotamers, CH2‐CH2‐
triazole ring), 30.1 (maj) and 30.2 (min) (rotamers, Ph‐CO‐CH2‐CH2), 31.8 (maj) and 32.1 (min)
(rotamers, CH2‐CH2‐triazole ring), 33.9 (maj) and 34.1 (min) (rotamers, Ph‐CO‐CH2‐CH2), 38.4
(maj) and 38.6 (min) (rotamers, NH‐CH2‐CH2‐N), 47.8 (maj) and 48.2 (min) (rotamers, NH‐
CH2‐CH2‐N), 49.7 (min) and 50.2 (maj) (rotamers, N‐CH2‐CO), 52.3 (Fc‐CH2‐triazole ring), 69.2
(CH Fc), 69.3 (CH Fc), 69.4 (CH Fc), 81.5 (maj) and 81.7 (min) (rotamers, C Fc), 87.2 (maj) and
87.6 (min) (rotamers, C‐I), 121.3 (maj) and 121.4 (min) (rotamers, CH triazole ring), 122.4
(maj) and 122.6 (min) (rotamers, benzene ring carbon), 128.2 (benzene ring carbon), 128.9
(benzene ring carbon), 133.4 (maj) and 133.5 (min) (rotamers, benzene ring carbon), 136.9
(min) and 137.1 (maj) (rotamers, benzene ring carbon), 137.9 (maj) and 138.1 (min)
(rotamers, benzene ring carbon), 138.3 (min) and 138.5 (maj) (rotamers, benzene ring
carbon), 146.8 (C triazole ring), 167.8 (min) and 168.7 (maj) (rotamers, CONH), 172.9,
(CONH), 173.9 (maj) and 174.1 (min) (rotamers, CH2CON), 199.1 (maj) and 199.6 (min)
(rotamers, Ph‐CO). IR bands(ν): 3266w, 3090w, 2930w, 2360m, 2342w, 1731w, 1682s, 1637s,
1530s, 1484s, 1447w, 1392w, 1370w, 1239s, 1180m, 1001m, 818s, 668m, 606s cm−1. ESI‐MS
(pos. detection mode): m/z (%): 799.95 (100) [M]+, 822.90 (70) [M+Na]+, 838.85 (25) [M+K]+.
NH
NNH
OO
NNN
IO
OMn
COOC
OC6b
Compound 6b: To a stirred solution of 4b (1 g, 1.33 mmol) in 5 mL DMF, 18 mL of 20%
piperidine in DMF was added at 0 °C and stirred for 10 min. Then the ice bath was removed
and the mixture was stirred 2 h at room temperature. The DMF and piperidine was removed
using high vacuum pump. The resulting residue was washed several times with pentane. The
light yellow color solid thus obtained was dissolved in 10 mL DMF. The solution was added to
a solution of 4‐oxo‐4‐cymantrenylbutanoic acid (5b)2 (340 mg, 1.12 mmol), HATU (855 mg,
2.25 mmol) and DIPEA (290 mg, 2.25 mmol) in DMF (5 mL), which has been initially stirred
for 30 minutes at room temperature. The reaction mixture was stirred for 48 h before being
J Organometallic PNA backbone derivatives SAR
266

diluted with 150 mL of ethyl acetate. The organic phase was washed with 1M HCl (50 mL),
sat. NaHCO3 solution (50 mL), water (2×75 mL) and brine (2×75 mL), dried over anhydrous
Na2SO4, filtered and concentrated. After flash column chromatography on silica gel
(EtOAc:MeOH 15:1 → 10:1 → 5:1), the solu on was concentrated and pentane was added to
get a light yellow precipitate. Pentane was decanted off to yield the desired compound 6b as
light yellow powder (yield: 620 mg, 57%).
Data for 6b. Rf = 0.14 (silica gel, EtOAc:MeOH 10:1). 1H NMR (250 MHz, CD2Cl2): δ (ppm) 2.47
(m, 2H, cymantrene‐CO‐CH2‐CH2), 2.76 (min) and 2.83 (maj) (rotamers, m, 2H, CH2‐CH2‐
triazole ring), 2.92 (m, 2H, CH2‐CH2‐triazole ring), 3.07 (m, 2H, cymantrene‐CO‐CH2‐CH2), 3.45
(m, 2H, NH‐CH2‐CH2‐N), 3.51 (m, 2H, NH‐CH2‐CH2‐N), 4.09 (maj) and 4.22 (min) (rotamers, s,
2H, N‐CH2‐CO), 4.88 (m, 2H, CH cymantrene), 5.45 (s, 2H, triazole‐CH2‐Ph), 5.48 (m, 2H, CH
cymantrene), 6.87 (min) and 7.01 (maj) (rotamers, 1H, NH‐CO), 7.25‐7.44 (m, 8H, 1×triazole
ring proton, 7×benzene ring protons), 7.51‐7.69 (m, 2H, benzene ring protons), 9.49 (maj)
and 9.61 (min) (rotamers, 1H, NHCO). 13C NMR (62.9 MHz, CD2Cl2): δ (ppm) 21.1 (min) and
21.2 (maj) (rotamers, CH2‐CH2‐triazole ring), 29.2 (maj) and 29.4 (min) (rotamers,
cymantrene‐CO‐CH2‐CH2), 31.5 (maj) and 31.8 (min) (rotamers, CH2‐CH2‐triazole ring), 33.8
(maj) and 33.9 (min) (rotamers, cymantrene‐CO‐CH2‐CH2), 37.9 (maj) and 38.1 (min)
(rotamers, NH‐CH2‐CH2‐N), 47.4 (NH‐CH2‐CH2‐N), 49.1 (N‐CH2‐CO), 51.8 (Ph‐CH2‐triazole
ring), 83.9 (maj) and 84.9 (min) (rotamers, CH cymantrene), 86.8 (CH cymantrene), 87.1
(maj) and 87.4 (min) (rotamers, C‐I), 91.1 (min) and 91.2 (maj) (rotamers, C cymantrene),
121.6 (CH triazole ring), 121.9 (maj) and 122.1 (min) (rotamers, benzene ring carbon), 127.9
(benzene ring carbon), 128.5 (benzene ring carbon), 129.1 (benzene ring carbon), 135.1
(benzene ring carbon), 137.5 (maj) and 137.7 (min) (rotamers, benzene ring carbon), 137.9
(min) and 138.1 (maj) (rotamers, benzene ring carbon), 147.1 (C triazole ring), 167.8 (min)
and 168.5 (maj) (rotamers, CONH), 171.9, (CONH), 173.4 (maj) and 173.8 (min) (rotamers,
CH2CON), 196.5 (maj) and 196.3, 197.5 (min) (rotamers, cymantrene‐CO), 223.2 (maj) and
224.3 (min) (rotamers, Mn‐CO). IR bands(ν): 2023s, 1931s, 1646m (br), 1532m, 1462w,
1392w, 1305w, 1245w, 1181w, 1059w, 1003w, 822w, 730w, 630s cm−1. ESI‐MS (pos.
detection mode): m/z (%): 818.87 (100) [M+H]+, 840.84 (35) [M+Na]+, 856.79 (15) [M+K]+.
J Organometallic PNA backbone derivatives SAR
267

NH
NNH
OO
NNN
O
OI
6c
Compound 6c. To a stirred solution of 4b (600 mg, 0.79 mmol) in 4 mL DMF, 10 mL of 20%
piperidine in DMF was added at 0 °C and stirred for 10 min. Then the ice bath was removed
and the mixture was stirred 2 h at room temperature. The DMF and piperidine was removed
using high vacuum pump. The resulting residue was washed several times with pentane. The
white color solid thus obtained was dissolved in 9 mL DMF. The solution was added to a
solution of 4‐oxo‐4‐phenylbutanoic acid (5a) (255 mg, 1.43 mmol), HATU (544 mg, 1.43
mmol) and DIPEA (185 mg, 1.43 mmol) in DMF (4 mL), which has been initially stirred for 30
minutes at room temperature. The reaction mixture was stirred for 40 h before the solvent
was removed. Flash column chromatography on silica gel (THF:MeOH 10:0 → 10:1) yielded
the desired compound 6c as white solid (yield: 310 mg, 56%). For analytical purpose the
compound was washed with little amount of DCM.
Data for 6c. Rf = 0.45 (silica gel, EtOAc:MeOH 12:1). 1H NMR (400 MHz, DMSO‐d6): δ (ppm)
2.47 (maj) and 2.59 (min) (rotamers, m, 2H, PhCO‐CH2‐CH2), 2.76 (m, 2H, CH2‐CH2‐triazole
ring), 2.87 (m, 2H, CH2‐CH2‐triazole ring), 3.22 (m, 4H, NH‐CH2‐CH2‐N and PhCO‐CH2‐CH2),
3.41 (m, 2H, NH‐CH2‐CH2‐N), 4.11(maj) and 4.16, 4.29 (min) (rotamers, s, 2H, N‐CH2‐CO), 5.52
(maj) and 5.51, 5.55 (min) (rotamers, s, 2H, triazole‐CH2‐Ph), 7.25‐7.54 (m, 7H, triazole ring
and benzene ring protons), 7.59‐7.65 (m, 4H, benzene ring protons), 7.84‐8.12 (m, 4H,
benzene ring protons), 10.1 (min) and 10.2 (maj) (rotamers, 1H, NHCO), 10.5 (min), 10.6
(maj) (rotamers, 1H NHCO). 13C NMR (100.6 MHz, DMSO‐d6): δ (ppm) 21.2 (min) and 21.3,
21.4 (maj) (rotamers, CH2‐CH2‐triazole ring), 29.4, 29.5 (min) and 29.6, 29.8 (maj) (rotamers,
PhCO‐CH2‐CH2), 31.5, 32.1 (min) and 31.8, 32.5 (maj) (rotamers, CH2‐CH2‐triazole ring), 33.8
(maj) and 33.9 (min) (rotamers, PhCO‐CH2‐CH2) 37.1, 37.8 (maj) and 37.5, 38.6 (min) (NH‐
CH2‐CH2‐N ), 46.4, 47.4 (min) and 46.9, 48.3 (maj) (rotamers, NH‐CH2‐CH2‐N), 50.1, 51.9 (maj)
and 50.3, 51.7 (min) (rotamers, N‐CH2‐CO), 53.1 (min) and 53.2 (maj) (rotamers, Ph‐CH2‐
triazole ring), 86.9, 92.1 (maj) and 87.3, 92.2 (min) (rotamers, C‐I benzene ring), 115.7 (maj)
and 119.1 (min) (rotamers, benzene ring C), 121.8 (maj) and 121.9 (min) (rotamers, CH
J Organometallic PNA backbone derivatives SAR
268

triazole ring), 122.6 (min) and 122.7 (maj) (rotamers, CONH‐C benzene ring), 126.0 (min) and
126.1 (maj) (rotamers, benzene ring C), 128.2 (maj) and 128.3 (min) (rotamers, benzene ring
C), 128.4 (maj) and 128.6 (min) (rotamers, benzene ring C), 129.1, 129.7 (min) and 129.2
(maj) (rotamers, benzene ring C), 133.5 (benzene ring C), 136.6 (min) and 136.9 (maj)
(rotamers, benzene ring C), 139.1, 139.3 (maj) and 139.2 (min) (rotamers, benzene ring C),
143.9 (min) and 144.1 (maj) (rotamers, benzene ring C), 146.8 (min) and 147.1 (rotamers, C
triazole ring), 167.8, 168.1 (min) and 168.4 (maj) (rotamers, CONH), 171.8, 171.9 (min) and
172.0, 172.2 (maj) (rotamers, CONH), 174.6 (min) and 174.7 (maj) (rotamers, CH2CON), 199.1
(Ph‐CO). IR bands(ν): 3236m, 1680m, 1654m, 1625m, 1589m, 1438s, 1400s, 1351w, 1207s,
1135s, 1114s, 935w, 769s, 640m cm−1. ESI‐MS (pos. detection mode): m/z (%): 715.02 (100)
[M+Na]+.
8a
NH
NNH
OO
NNN
Fe
ORe
N
N
N
CO
CO
CO
O
PF6+
Compound 8a. A solution of 6a (240 mg, 0.35 mmol) and 7 (163 mg, 0.25 mmol) in 9 mL
DMF/NEt3 mixture (2/1, v/v) was degassed by two ‘freeze‐pump‐thaw’ cycles. Then CuBr (4.4
mg, 0.029 mmol) and cis‐dichlorobis (triphenylphosphine) palladium(II) (5.3 mg, 0.0075
mmol) were added under nitrogen atmosphere and the mixture was degassed again by one
‘freeze‐pump‐thaw’ cycles. The wine color solution thus obtained was allowed to stir 22 h at
room temperature. The reaction mixture was then diluted with 100 mL of CH2Cl2 and
washed with distilled water (4×60 mL) and brine (2×60 mL). The CH2Cl2 layer was dried over
Na2SO4, filtered and concentrated. Addition of 40 mL of diethyl ether gave a brown color
solid which was collected and further washed with 15 mL of cold ethyl acetate. The solid was
then loaded on a silica column and eluted immediately with CH2Cl2:MeOH 5:1. The combined
fractions was evaporated and redissolved in minimum volume CH2Cl2 and filtered. Filtrate
was dried to give the desired compound 6.23 as light brown solid (yield: 144 mg, 41%).
(Note: If the compound stacks in the column, CH2Cl2:MeOH 5:1 saturated with KPF6 should
be used as eluent).
J Organometallic PNA backbone derivatives SAR
269

Data for 8a. Rf = 0.66 (silica gel, DCM:MeOH 5:1). tR (RP‐HPLC) = 17.3 min. 1H NMR (400 MHz,
CD2Cl2): δ (ppm) 2.33‐2.79 (m, 4H, PhCO‐CH2‐CH2 and CH2‐CH2‐triazole ring), 2.84‐3.01 (m,
2H, CH2‐CH2‐triazole ring), 3.23 (m, 2H, PhCO‐CH2‐CH2), 3.30‐3.45 (m, 4H, NH‐CH2‐CH2‐N and
NH‐CH2‐CH2‐N), 4.11 (s, br, 2H, N‐CH2‐CO), 4.15‐4.30 (m, 9H, CH Fc), 4.66 (s, br, 2H, N‐CH2‐
C≡C), 4.83 (s, 4H, 2×N‐CH2‐Py), 5.15 (maj) and 5.20 (min) (rotamers, s, 2H, triazole‐CH2‐Fc),
6.89 (min) and 6.98 (maj) (rotamers, s, br, 1H, NHCO), 7.29‐7.96 (m, 16H, 1×triazole ring
proton and 15×aromatic protons), 8.76 (m, 2H, aromatic protons), 9.04, 9.28, 9.55 (min) and
9.19 (maj) (rotamers, s, br, 1H, NHCO). 13C NMR (100.6 MHz, CD2Cl2): δ (ppm) 20.9 (min) and
21.2 (maj) (rotamers, CH2‐CH2‐triazole ring), 29.6 (min) and 30.2 (maj) (rotamers, PhCO‐CH2‐
CH2), 31.7 (maj) and 32.3 (min) (rotamers, CH2‐CH2‐triazole ring), 33.9 (PhCO‐CH2‐CH2), 37.7
(min) and 38.3 (maj) (rotamers, NH‐CH2‐CH2‐N), 48.4 (NH‐CH2‐CH2‐N), 50.2 (N‐CH2‐CO), 52.1
(Fc‐CH2‐triazole ring), 59.6 (N‐CH2‐C≡C), 68.4 (2×N‐CH2‐Py), 69.2 (CH Fc), 69.3 (CH Fc), 69.4
(CH Fc), 81.1 (C≡C‐CH2‐N), 81.5 (C Fc), 90.6 (N‐CH2‐C≡C), 116.7 (maj) and 117.1 (min)
(rotamers, aromatic carbon), 120.1 (maj) and 120.3 (min) (rotamers, aromatic carbon), 121.6
(min) and 121.9 (maj) (rotamers, CH triazole ring), 124.2 (aromatic carbon), 125.7 (min) and
126.3 (maj) (rotamers, aromatic carbon), 128.4 (aromatic carbon), 129.1 (aromatic carbon),
133.2 (aromatic carbon), 133.6 (aromatic carbon), 136.7 (min) and 136.8 (maj) (rotamers,
aromatic carbon), 139.3 (min) and 139.6 (maj) (rotamers, aromatic carbon), 140.9 (aromatic
carbon), 146.7 (maj) and 147.1 (min) (rotamers, C triazole ring), 152.1 (aromatic carbon),
160.1 (aromatic carbon), 168.1, 168.3 (min) and 169.0, 169.1 (maj) (rotamers, CONH), 173.3
(maj) and 173.5 (min) (CONH), 174.5 (min) and 174.7 (maj) (rotamers, CH2CON), 195.7 (Re‐
CO), 199.1 (maj) and 200.4 (min) (rotamers, Ph‐CO). IR bands(ν): 2030s, 1910s, 1648m (br),
1610w, 1514m, 1365w, 1293w, 1105w, 1000w, 834s, 733m, 622w cm−1. ESI‐MS (pos.
detection mode): m/z (%): 1179.93 (100) [M‐PF6]+.
8b
NH
NNH
OO
NNN
O
Re
N
N
N
CO
CO
CO
MnOC
OC CO O
PF6+
J Organometallic PNA backbone derivatives SAR
270

Compounds 8b. A stirred solution of 6b (300 mg, 0.37 mmol) and 7 (199 mg, 0.31 mmol) in 9
mL DMF/NEt3 mixture (2/1, v/v) was degassed by two ‘freeze‐pump‐thaw’ cycles. Then CuBr
(5.4 mg, 0.03 mmol) and cis‐dichlorobis (triphenylphosphine) palladium(II) (6.4 mg, 0.0091
mmol) were added under nitrogen atmosphere and the mixture was degassed again by one
‘freeze‐pump‐thaw’ cycle. The wine color solution thus obtained was allowed to stir 22 h at
room temperature. The reaction mixture was then diluted with 100 mL of CH2Cl2 and
washed with distilled water (4×60 mL) and brine (2×60 mL). The CH2Cl2 layer was dried over
anhydrous Na2SO4, filtered and concentrated. Addition of 40 mL of pentane gave a brown
color solid. Flash column chromatography on silica gel (eluent: EtOAc:MeOH 5:1 and then
DCM:MeOH 3:1 saturated with KPF6 to elute the 8b) was done. The fractions containing the
desired compound was combined and evaporated. The residue was dissolved in 10 mL of
CH2Cl2 and filtered. The residue obtained after evaporation was loaded again on a small filter
silica column and eluted immediately with DCM:MeOH 5:1. The desired fractions are
combined and dried, redissolved in minimum volume CH2Cl2 and filtered. Filtrate was dried
to give the desired compound 8b as light brown solid (yield: 180 mg, 44%).
Data for 8b. Rf = 0.27 (silica gel, CH2Cl2:MeOH 5:1). tR (RP‐HPLC) =17.1 min. 1H NMR (250
MHz, DMSO‐d6): δ (ppm) 2.37 (m, 2H, cymantrene‐CO‐CH2‐CH2), 2.50 (maj) and 2.57 (min)
(rotamers, m, 2H, CH2‐CH2‐triazole ring), 2.71‐2.95 (m, 2H, CH2‐CH2‐triazole ring), 3.15‐3.48
(m, 6H, cymantrene‐CO‐CH2‐CH2, NH‐CH2‐CH2‐N and NH‐CH2‐CH2‐N), 4.11 (maj) and 4.25
(min) (rotamers, s, 2H, N‐CH2‐CO), 4.83 (s, 2H, N‐CH2‐C≡C), 4.85 (min) and 4.92 (maj)
(rotamers, s, 2H, N‐CH2‐Py), 5.03 (maj) and 5.10 (min) (rotamers, s, 2H, N‐CH2‐Py), 5.18 (maj)
and 5.25 (min) (rotamers, m, 2H, CH cymantrene), 5.54 (s, 2H, triazole‐CH2‐Ph), 5.78
(rotamers, m, 2H, CH cymantrene), 6.43 (min) and 6.50 (maj) (rotamers, 1H, NHCO), 7.25‐
7.54 (m, 8H, 1×triazole ring proton and 7×aromatic protons), 7.66 (m, 4H, aromatic protons),
7.82‐8.04 (m, 4H, aromatic protons), 8.85 (d, 2H, aromatic protons), 10.21 (maj) and 10.35
(min) (rotamers, s, 1H, NHCO). 13C NMR (62.9 MHz, DMSO‐d6): δ (ppm) 21.1, 21.2 (min) and
21.3 (maj) (rotamers, CH2‐CH2‐triazole ring), 29.2, 29.5 (min) and 29.4 (maj) (rotamers,
cymantrene‐CO‐CH2‐CH2), 31.8 (maj) and 32.3 (min) (rotamers, CH2‐CH2‐triazole ring), 34.2
(cymantrene‐CO‐CH2‐CH2), 37.1 (min) and 37.7 (maj) (rotamers, NH‐CH2‐CH2‐N), 48.3 (min)
and 50.2 (maj) (rotamers, NH‐CH2‐CH2‐N), 51.3 (min) and 53.1 (maj) (rotamers, N‐CH2‐CO),
55.1 (Ph‐CH2‐triazole ring), 58.4 (maj) and 59.8 (min) (rotamers, N‐CH2‐C≡C), 68.2 (maj) and
J Organometallic PNA backbone derivatives SAR
271

70.1 (min) (rotamers, 2×N‐CH2‐Py), 82.9 (maj) and 83.1 (min) (rotamers, C≡C‐CH2‐N), 85.3
(CH cymantrene), 87.9 (CH cymantrene), 89.1 (N‐CH2‐C≡C), 92.3 (C cymantrene), 115.9 (maj)
and 116.2 (min) (rotamers, aromatic carbon), 119.3 (maj) and 119.4 (min) (rotamers,
aromatic carbon), 122.7 (CH triazole ring), 124.1 (aromatic carbon), 126.1 (aromatic carbon),
128.2 (aromatic carbon), 128.4 (aromatic carbon), 129.1 (aromatic carbon), 132.9 (aromatic
carbon), 136.5 (aromatic carbon), 139.9 (min) and 140.2 (maj) (rotamers, aromatic carbon),
140.9 (aromatic carbon), 146.9 (C triazole ring), 152.4 (aromatic carbon), 160.8 (aromatic
carbon), 168.0, 168.1 (min) and 168.4 (maj) (rotamers, CONH), 171.4, 171.9 (min) and 171.6
(maj) (CONH), 172.3 (maj) and 172.4, 171.5 (min) (rotamers, CH2CON), 195.6 (min) 196.1
(maj) (rotamers, Re‐C≡O), 197.1 (maj) and 197.2 (min) (rotamers, cymantrene‐CO‐), 224.1
(maj) and 225.4, 225.6 (min) (rotamers, Mn‐CO). IR bands(ν): 2027s, 1910s (br), 1670m (br),
1518m, 1445w, 1311w, 1245w, 1060w, 832s, 765m, 667w, 629m cm−1. ESI‐MS (pos.
detection mode): m/z (%): 610.6 (50) [M+Na‐PF6]2+, 1197.91 (100) [M‐PF6]
+.
X = PF6 (8c), CF3COO (8c')
NH
NNH
OO
NNN
ORe
N
N
N
CO
CO
CO
O
X+
Compound 8c′. A solution of 6c (371 mg, 0.53 mmol) and 7 (350 mg, 0.53 mmol) in 12 mL
DMF/NEt3 mixture (2/1, v/v) was degassed by two ‘freeze‐pump‐thaw’ cycles. Then CuBr
(9.86 mg, 0.07 mmol) and cis‐dichlorobis (triphenylphosphine) palladium(II) (7.7 mg, 0.01
mmol) were added under nitrogen atmosphere and the mixture was degassed again by one
‘freeze‐pump‐thaw’ cycle. The wine color solution thus obtained was allowed to stir 20 h at
room temperature. The solvent was removed and the residue was diluted with 100 mL of
CH2Cl2, washed with distilled water (4×60 mL) and brine (2×60 mL). The CH2Cl2 layer was
dried over Na2SO4, filtered and concentrated. The solid was then loaded on a silica column
and eluted immediately with CH2Cl2:MeOH 5:1 → 3:1. The combined frac ons was
evaporated and redissolved in CH2Cl2 and filtered. Filtrate was dried to give 8c as light brown
solid (yield: 256 mg, 39%). For biological applications purpose 8c was further purified by RP‐
J Organometallic PNA backbone derivatives SAR
272

HPLC to remove a trace of impurity present after purification by silica column
chromatography to give 8c′.
Data for 8c. Rf = 0.51 (silica gel, CH2Cl2:MeOH 4:1). tR (RP‐HPLC) = 16.8 min. 1H NMR (250
MHz, CD2Cl2): δ (ppm) 2.54 (m, 2H, Ph‐CO‐CH2‐CH2), 2.75 (min) and 2.82 (maj) (rotamers, m,
2H, CH2‐CH2‐triazole ring), 3.03 (m, 2H, CH2‐CH2‐triazole ring), 3.27 (m, 2H, PhCO‐CH2‐CH2),
3.34‐3.56 (m, 4H, NH‐CH2‐CH2‐N and NH‐CH2‐CH2‐N), 4.15 (maj) and 4.32 (min) (rotamers, s,
2H, N‐CH2‐CO), 4.68 (min) and 4.71 (maj) (rotamers, s, 2H, N‐CH2‐C≡C), 4.87‐5.18 (rotamers,
m, 4H, 2×N‐CH2‐Py), 5.42 (maj) and 5.45, 5.20 (min) (rotamers, s, 2H, triazole‐CH2‐Ph), 6.52,
6.78 (min) and 6.99 (maj) (rotamers, 1H, NHCO), 7.19‐7.50 (m, 13H, 1×triazole ring proton
and 12×aromatic protons), 7.56‐7.69 (m, 4H, aromatic protons), 7.82‐7.95 (m, 4H, aromatic
protons), 8.76 (m, 2H, aromatic protons), 9.43 (maj) and 9.50, 9.60, 10.13 (min) (rotamers,
NHCO). 13C NMR (62.9 MHz, CD2Cl2): δ (ppm) 20.9 (min) and 21.1 (maj) (rotamers, CH2‐CH2‐
triazole ring), 29.6 (maj) and 39.9 (min) (rotamers, PhCO‐CH2‐CH2), 31.6 (maj) and 31.9 (min)
(rotamers, CH2‐CH2‐triazole ring), 33.6 (maj) and 33.9 (min) (rotamers, PhCO‐CH2‐CH2), 37.9
(maj) and 38.3 (min) (rotamers, NH‐CH2‐CH2‐N), 49.1 (NH‐CH2‐CH2‐N), 51.8 (N‐CH2‐CO), 52.1
(Ph‐CH2‐triazole ring, overlaps with solvent residual signal), 59.1 (maj) and 59.2 (min)
(rotamers, N‐CH2‐C≡C), 68.1, 68.3 (maj) and 72.2, 72.4 (min) (rotamers, 2×N‐CH2‐Py), 80.8
(C≡C‐CH2‐N), 90.3 (N‐CH2‐C≡C), 116.1 (maj) and 116.3 (min) (rotamers, aromatic carbon),
119.5, 120.1 (min) and 119.7 (maj) (rotamers, aromatic carbon), 121.6 (min) and 121.9 (maj)
(rotamers, CH triazole ring), 123.9 (aromatic carbon), 124.2 (aromatic carbon), 125.4, 125.6
(min) and 125.8, 125.9 (maj) (rotamers, 2×aromatic carbons), 127.8 (min) and 127.9 (maj)
(rotamers, aromatic carbon), 128.4 (min) and 128.5 (maj) (rotamers, aromatic carbon), 128.9
(aromatic carbon), 132.7 (aromatic carbon), 133.1 (aromatic carbon), 135.2 (aromatic
carbon), 136.6 (aromatic carbon), 139.3 (min) and 139.6 (maj) (rotamers, aromatic carbon),
140.5 (aromatic carbon), 146.7 (maj) and 147.1 (min) (rotamers, C triazole ring), 151.4 (min)
and 151.6 (maj) (rotamers, aromatic carbon), 159.3 (min) and 159.6 (maj) (rotamers,
aromatic carbon), 167.1, 167.9, 168.1 (min) and 168.4 (maj) (rotamers, CONH), 172.4
(CONH), 173.4 (maj) and 173.6 (min) (rotamers, CH2CON), 194.9 (min) and 195.3 (maj)
(rotamers, Re‐CO), 198.8 (min) and 199.2 (maj) (rotamers, Ph‐CO). IR bands(ν): 2032s, 1913s,
1670m (br) cm−1. ESI‐MS (pos. detection mode): m/z (%): 1071.98 (100) [M‐PF6]+.
J Organometallic PNA backbone derivatives SAR
273

NH
NNH
OO
NNN
ON
N
N
O9
Compound 9. A solution of 6c (180 mg, 0.26 mmol) and N,N‐bis(pyridine‐2‐ylmethyl)prop‐2‐
yn‐1‐amine3 (62 mg, 0.26 mmol) in 9 mL DMF/NEt3 mixture (2/1, v/v) was degassed by two
‘freeze‐pump‐thaw’ cycles. Then CuBr (60 mg, 0.31 mmol) and cis‐dichlorobis
(triphenylphosphine) palladium (II) (21 mg, 0.03 mmol) were added under nitrogen
atmosphere and the mixture was degassed again by one ‘freeze‐pump‐thaw’ cycle. The wine
color solution thus obtained was allowed to stir 20 h at room temperature. The DMF and
NEt3 were removed and the residue was diluted with 125 mL of CH2Cl2/isopropanol mixture
(5/1, v/v). The organic phase was washed with aqueous sat. Na2EDTA, brine, dried over
anhydrous Na2SO4, filtered and concentrated. Flash column chromatography (two times)
(silica gel, CH2Cl2:CH3CN:MeOH:NH4OH 50:50:25:2) with the residue was performed. The
resulting solution was dried and the residue was redissolved in 15 mL DCM and filtered to
remove the silica. Evaporation of the CH2Cl2 solution afforded the desired compound 9 as
brown sticky solid (yield: 69 mg, 35%). For biological applications purpose 9 was further
purified by RP‐HPLC.
Data for 9. tR (RP‐HPLC) = 14.1 min. 1H NMR (400 MHz, CDCl3): δ (ppm) 2.42 (m, 2H, PhCO‐
CH2‐CH2), 2.65 (min) and 2.73 (maj) (rotamers, m, 2H, CH2‐CH2‐triazole ring), 2.97 (m, 2H,
CH2‐CH2‐triazole ring), 3.16 (m, 2H, PhCO‐CH2‐CH2), 3.33 (m, 2H, NH‐CH2‐CH2‐N), 3.41 (m, 2H,
NH‐CH2‐CH2‐N), 3.53 (s, 2H, N‐CH2‐C≡C), 3.89 (s, 4H, 2×N‐CH2‐Py), 4.08 (maj) and 4.21 (min)
(rotamers, s, br, 2H, N‐CH2‐CO), 5.28 (maj) and 5.33 (min) (rotamers, s, 2H, triazole‐CH2‐Ph),
7.07‐7.56 (m, 20H, 1×triazole ring proton, 1×NH‐CO, 18×aromatic protons), 7.82 (d, 2H,
aromatic protons), 8.46 (m, 2H, aromatic protons), 9.43 (maj) and 10.01 (min) (rotamers, s,
1H, NHCO). 13C NMR (100.6 MHz, CDCl3): δ (ppm) 21.1 (min) and 21.2 (maj) (rotamers, CH2‐
CH2‐triazole ring), 29.6, 29.9 (min) and 29.8 (maj) (rotamers, PhCO‐CH2‐CH2), 31.7 (maj) and
32.1 (min) (rotamers, CH2‐CH2‐triazole ring), 33.5 (maj) and 33.8 (min) (rotamers, PhCO‐CH2‐
CH2), 37.9 (maj) and 38.1, 38.3 (min) (rotamers, NH‐CH2‐CH2‐N), 43.6 (N‐CH2‐C≡C), 47.7 (min)
and 49.1 (maj) (rotamers, NH‐CH2‐CH2‐N), 51.7 (maj) and 51.2 (min) (rotamers, N‐CH2‐CO),
J Organometallic PNA backbone derivatives SAR
274

54.1 (maj) and 54.2 (min) (rotamers, Ph‐CH2‐triazole ring), 59.5 (N‐CH2‐Py), 83.6 (N‐CH2‐C≡C),
85.6 (C≡C‐CH2‐N), 118.4 (CH triazole ring), 119.7 (aromatic carbon), 121.7 (aromatic carbon),
122.2 (aromatic carbon), 123.3 (aromatic carbon), 125.3 (aromatic carbon), 128.1 (aromatic
carbon), 128.6 (aromatic carbon), 128.9 (aromatic carbon), 131.9 (aromatic carbon), 132.2
(aromatic carbon), 133.1 (aromatic carbon), 134.7 (aromatic carbon), 136.5 (aromatic
carbon), 136.7 (aromatic carbon), 138.1 (aromatic carbon), 147.1 (C triazole ring), 149.1
(aromatic carbon), 158.6 (aromatic carbon), 167.6, 167.8 (min) and 168.2 (maj) (rotamers,
CONH), 172.5 (CONH), 173.4 (maj) and 173.7 (min) (rotamers, CH2CON), 199.1 (Ph‐CO). IR
bands(ν): 2926w, 2360w, 1648s (br), 1594s, 1529s, 1435s, 1308m, 1179m, 1118m, 999w,
839m, 722s, 693s cm−1. ESI‐MS (pos. detection mode): m/z (%): 802.35 (100) [M+H]+, 824.19
(25) [M+Na]+.
J Organometallic PNA backbone derivatives SAR
275

ii. RP‐HPLC traces and ESI mass spectra of 8a, 8b, 8c,′ and 9
Compound 8a.
8a
NH
NNH
OO
NNN
Fe
O
Re
N
N
N
CO
CO
CO
O
PF6+
Compound 8b.
8b
NH
NNH
OO
NNN
O
Re
N
N
N
CO
CO
CO
MnOC
OC CO O
PF6+
J Organometallic PNA backbone derivatives SAR
276

Compound 8c′ (after RP‐HPLC purification).
8c'
NH
NNH
OO
NNN
O
Re
N
N
N
CO
CO
CO
O
CF3COO+
Compound 9 (after RP‐HPLC purification).
NH
NNH
OO
NNN
ON
N
N
O9
J Organometallic PNA backbone derivatives SAR
277

iii. 1H and 13C NMR spectra of compounds
NH
NO
OO
NNN
O
O
2 1H, CDCl3, 400 MHz
13C, CDCl3, 62.9 MHz
J Organometallic PNA backbone derivatives SAR
278

NH
NNH
OO
NNN
O
OI
4b
1H, CDCl3, 100.6 MHz
13C, CDCl3, 250 MHz
J Organometallic PNA backbone derivatives SAR
279

NH
NNH
OO
NNN
O
O
Fe
I
6a 1H, CD2Cl2, 400 MHz
13C, CD2Cl2, 100.6 MHz
J Organometallic PNA backbone derivatives SAR
280

NH
NNH
OO
NNN
IO
OMn
COOC
OC6b
1H, CD2Cl2, 250 MHz
13C, CD2Cl2, 62.9 MHz
J Organometallic PNA backbone derivatives SAR
281

NH
NNH
OO
NNN
O
OI
6c
1H, DMSO‐d6, 400 MHz
13C, DMSO‐d6, 100.6 MHz
0306090130170210ppm
J Organometallic PNA backbone derivatives SAR
282
8a
NH
NNH
OO
NNN
Fe
O
Re
N
N
N
CO
CO
CO
O
PF6+
1H, CD2Cl2, 400 MHz
13C, CD2Cl2, 100.6 MHz
J Organometallic PNA backbone derivatives SAR
283
8b
NH
NNH
OO
NNN
O
Re
N
N
N
CO
CO
CO
MnOC
OC CO O
PF6+
1H, DMSO‐d6, 250 MHz
13C, DMSO‐d6, 62.9 MHz
-2-1012345678910111213ppm
Triethyl amine
J Organometallic PNA backbone derivatives SAR
284
8c
NH
NNH
OO
NNN
O
Re
N
N
N
CO
CO
CO
O
PF6+
1H, CD2Cl2, 250 MHz
13C, CD2Cl2, 62.9 MHz
J Organometallic PNA backbone derivatives SAR
285

NH
NNH
OO
NNN
ON
N
N
O9
1H, CDCl3, 400 MHz
13C, CDCl3, 100.6 MHz
205080120160200ppm
J Organometallic PNA backbone derivatives SAR
286

iv. References
1. Patra, M.; Gasser, G.; Bobukhov, D.; Merz, K.; Shtemenko, A. V.; Metzler‐Nolte, N., Dalton
Trans. 2010, 39, 5617‐5619.
2. N'Dongo, H. W. P.; Neundorf, I.; Merz, K.; Schatzschneider, U., J. Inorg. Biochem. 2008,
102, 2114‐2119.
3. Cabrera, D. G.; Koivisto, B. D.; Leigh, D. A., Chem. Commun. 2007, 4218‐4220.
J Organometallic PNA backbone derivatives SAR
287

K Discussion
The bacterial cell envelope is an attractive antibiotic target. The penicillins and
cephalosporins targeting peptidoglycan cross‐linking are probably the most successful
antibiotic agents in history. Different steps of cell wall biosynthesis as well as membrane
biogenesis and integrity are further promising antibiotic targets that have been proposed to
be less prone to bacterial resistance [Brötz and Brunner, 2006; Peschel and Sahl, 2006]. In
this work, reference antibiotics targeting membrane biogenesis and structure as well as cell
wall biosynthesis were investigated. Targeting the cell wall structure itself was already
shown not to elicit a proteome response in B. subtilis [Bandow et al., 2003, Rabenau, 2010].
Findings obtained for the reference compounds were then employed in mode of action
analysis of novel peptide‐based antibiotics. Two groups of compounds were investigated:
short cationic peptides based on the MP196 lead structure RWRWRW‐NH2 and the
structurally novel class of organometallic PNA derivatives.
1 Proteomic signatures
Proteomic profiles of the acute bacterial stress response to treatment with an antibiotic
compound can be indicative of its mechanism of action. Comparison of the proteome
response to novel antibiotics with established reference patterns allows compound
classification into mechanistic categories. Thus, proteome analysis serves as an informative
starting point for mode of action elucidation [Wenzel and Bandow, 2011]. In order to
complement the existing antibiotic reference library [Bandow et al., 2003], proteomic
signatures for different aspects of cell envelope‐related stress were established. A proteomic
signature is a set of specific marker proteins indicative of a defined physiological condition.
Such signatures are typically established by comparing the proteomic response of different
compounds targeting the same pathway or structure [VanBogelen et al., 1999].
A signature for fatty acid biosynthesis inhibition was established employing the FabI inhibitor
triclosan, the FabF inhibitors cerulenin and platensimycin, and the triple inhibitor platencin,
binding to FabF, FabHA, and FabHB. The proteomic signature consists of six proteins, all of
which involved in membrane lipid biosynthesis (Figure 11). PanB is involved in the synthesis
of coenzyme A, an essential cofactor for fatty acid biosynthesis. FabF, FabHA, and FabHB
start the fatty acid condensation/elongation cycle. FabI performs the last reduction step.
PlsX is the first enzyme in the successive phospholipid biosynthesis. Upregulation of the
K Discussion
288

metabolic bottlenecks of an inhibited pathway is a common stress response strategy and
reflects the cellular attempt to compensate for the antibiotic‐mediated loss of function.
Figure 11: Proteomic signature for fatty acid biosynthesis inhibition. Proteins upregulated
in response to triclosan, cerulenin, platensimycin, and platencin are highlighted in red. B.
subtilis responds by upregulation of enzymes catalyzing the initial and last steps of the fatty
acid biosynthesis cycle as well as proteins involved in coenzyme A and phospholipid
biosynthesis.
The newly established signature was employed to investigate the target area of the
chromium organometallic‐substituted platensimycin derivative PM47. Although molecular
modeling suggested that the derivative fits into the platensimycin binding site of the FabF
enzyme [Patra et al., 2009], none of the fatty acid biosynthesis signature proteins was
upregulated in response to the chromium bioorganometallic. Thus, membrane biogenesis is
not its antibiotic target. Moreover, none of the proteome response profiles of the B. subtilis
reference compendium shared similarity with the PM47 stress response pattern indicating a
novel mechanism of action. Its low selectivity for bacterial cells however suggests a rather
unspecific toxic mechanism.
PM47 target falsification demonstrates how proteomic signatures can aid narrowing down
the target area. Since the development of platensimycin and platencin, huge efforts have
been undertaken to modify their structures and to develop analogs or novel FAB inhibitors
K Discussion
289

by rational design in order to overcome the low in vivo activity of these otherwise promising
antibiotics [Shen et al., 2009; Patra et al., 2010b; Patra et al., 2011; Patra et al., 2012b;
Plesch et al., 2012]. The proteomic signature for inhibition of fatty acid biosynthesis can now
aid mode of action validation of promising derivatives.
A subset of lantibiotics with distinct mechanisms of action was employed to generate
individual proteomic signatures for general cell envelope stress, membrane damage, and
inhibition of membrane‐bound cell wall biosynthesis (Figure 12).
The lantibiotics nisin, gallidermin, and mersacidin target cell wall biosynthesis by binding to
the precursor lipid II [Schneider and Sahl, 2010]. Additionally, gallidermin and nisin insert
into the bacterial membrane, but only nisin forms pores in B. subtilis. Although gallidermin
permeates the membrane of Stapyhlococci, the B. subtilis membrane composition, rich in
branched chain fatty acids, prevents it from efficiently forming pores [Christ et al., 2008].
This set of lantibiotics together with further profiles from the reference compendium
allowed definition of a proteomic signature for general cell envelope stress, constituted by
the detoxifying proteins YceC and YceH. They share similarity with a tellurium and a toxic
anion resistance protein, respectively. However, their particular roles in the cell envelope
stress response are not understood so far.
The proteome responses to the respective lantibiotics revealed a specific signature for
membrane damage, constituted by PspA and NadE. PspA binds to the inner membrane
surface, stabilizing the bilayer structure and preventing proton leakage [Kobayashi et al.,
2007]. NAD synthase is probably upregulated because of energy limitation due to impaired
membrane function. It reflects a cellular attempt to compensate for respiratory chain
inhibition.
The stress responses to the individual lantibiotics provide unique insight into the bacterial
reaction to different grades of membrane interaction. Mersacidin‐treatment provokes
upregulation of very few compound‐specific marker proteins in addition to the cell wall
biosynthesis signature. This is consistent with its single mechanism of binding lipid II.
Gallidermin elicits a more complex stress response reflective of the multiple consequences
of binding lipid II and integrating into the membrane. B. subtilis responds to integration of
gallidermin into the membrane by upregulation of cell envelope stress proteins, such as the
penicillin‐binding protein PBP 4 or DltA, the latter of which is involved in lipiteichoic acid
synthesis and indicates cell wall adaptation. In contrast, the stress response to nisin revealed
K Discussion
290

almost no specific markers apart from the signature proteins. This could be due to its pore‐
forming mechanism. As soon as a nisin concentration sufficient for pore formation is reached
[Huang et al., 2002], it is likely that the membrane potential immediately breaks down,
strictly limiting energy for the biosynthesis of stress proteins. Owing to this fast kinetic, nisin‐
treated cells have only very little chance to appropriately react to antibiotic stress. Thus, no
specific proteomic markers for pore formation can be observed.
Figure 12: Proteomic signatures for general cell envelope stress, cell wall biosynthesis
inhibition, and membrane damage. Both PspA and LiaH bind to the membrane and stabilize
its structure. While PspA is specifically upregulated by membrane damage, LiaH is responsive
to inhibition of membrane‐bound cell wall biosynthesis. NadE is probably upregulated due to
energy limitation. The particular roles of the other marker proteins in the cell envelope
stress response remain to be fully elucidated. CWB: cell wall biosynthesis; RSC: respiratory
chain.
A signature for inhibition of membrane‐bound cell wall biosynthesis is constituted by LiaH,
YtrB, YtrE, TrmB, and YpuA (formerly referred to as NGM1 [Wenzel et al., 2012]). What
particular roles the ATP transporter subunits YtrB and YtrE, the tRNA‐modifying TrmB, and
K Discussion
291

the unknown protein YpuA play in the stress response remains unclear so far. The PspA
homolog LiaH forms ring structures consisting of 36 LiaH proteins, which bind to the
cytoplasmic membrane surface. It is thought to stabilize the lipid bilayer under stress
conditions [Wolf et al., 2010]. Upregulation of LiaH correlates with the proximity of the
antibiotic binding site to the cytoplasmic membrane. The investigated lantibiotics bind to the
membrane‐proximal sugar part of the lipid II molecule. Bacitracin binds to undecaprenyl
pyrophosphate. They all strongly induce liaH expression. In contrast, LiaH was not
upregulated in response to vancomycin, which binds to the membrane‐distant amino acid
side chain of lipid II. Similar results were obtained by Mascher et al., who measured
activation of a liaH‐lacZ reporter gene fusion by different antibiotics. Although there was an
activation of liaH transcription after vancomycin treatment, it was far less pronounced than
in response to membrane‐proximally lipid II‐binding compounds like bacitracin and nisin
[Mascher et al., 2004].
LiaH was further upregulated in the proteome response to gramicidin S, which does not
directly interact with cell wall biosynthesis but does affect cell wall integrity. Gramicidin S is
described to disorganize lipid mixing in the membrane, gathering negatively charged
phospholipids around its positive charges [Kaprel’iants et al., 1977; Vostroknutova et al.,
1981]. Localization of the cell wall biosynthesis machinery was proposed to take place in
membrane regions rich in negatively charged phospholipids [Matsumoto et al., 2006;
Muchová et al., 2011]. Thus, domain formation in the membrane bilayer by gramicidin S
could contribute to impairment of cell wall biosynthesis, explaining upregulation of LiaH in
the proteome response.
The differential regulation of liaH expression depending on membrane proximity
demonstrates how sophisticated proteomic profiling can distinguish between mechanisms of
action. This specificity becomes even more apparent upon comparison of the novel signature
with compounds targeting intracellular steps of cell wall biosynthesis. D‐cycloserine, limiting
the supply of D‐alanine for peptidoglycan synthesis [Lambert and Neuhaus, 1972; Vicario et
al., 1987], induced a distinct stress response, which did not overlap with that to lantibiotic
treatment [Bandow et al., 2003]. Compounds inhibiting membrane‐distal extracellular steps
do not elicit a stress response in B. subtilis at all. This was true for methicillin, binding to PBP
2, as well as for cephalexin, binding to PBP 4 [Bandow et al., 2003, Rabenau, 2010]. B. subtilis
168 possesses a functional β‐lactamase encoded by penP [Imanaka et al., 1983], which is
K Discussion
292

expressed and secreted after long‐term treatment with ampicillin [unpublished results].
Thus, B. subtilis does react to treatment with β‐lactam antibiotics although no stress
response could be detected in the 2D gel‐based proteomics approach in the investigated
time frame.
The new signature for cell wall biosynthesis inhibition is highly specific to inhibition on a
membrane‐bound step. It further allows prediction of the proximity of the antibiotic’s
binding site to the bilayer. Together with the signatures for membrane and general cell
envelope stress it complements the existing proteome reference compendium. The new
signatures place the already established proteome profiles of cell envelope‐targeting
compounds into a broader context and refine the reference compendium in terms of
signatures for cell envelope‐related stress, recently complemented by a signature for
treatment with divalent cation ionophores [Raatschen and Bandow, 2012; Raatschen et al.,
2013]. They are now available for mode of action analysis of further cell envelope‐targeting
antibiotics and will prove useful especially for investigating antimicrobial peptides, which
might affect further cellular processes in addition to interacting with membranes [Nicolas,
2009; Wilmes et al., 2011; Wimley, 2011]. The newly established signatures were employed
here for mode of action analysis of typical short cationic antimicrobial peptides and novel
organometallic PNA derivatives.
2 Mechanism of action of MP196, a short cationic antimicrobial peptide
The synthetic hexapeptide MP196 consists of the amino acid sequence RWRWRW‐NH2. It
derived from an approach to reveal the minimal pharmacophore of short cationic peptides
[Strøm et al., 2003]. It is therefore ideally suited to study the mechanism of action of this
peptide class and its influence on bacterial physiology.
Interaction of MP196 with bacterial membranes is best described with the interfacial activity
model [Wimley, 2011]. Interfacial activity is defined as “the ability to bind to a membrane,
partition in the membrane‐water interface and to alter the packing and organization of
lipids” [Rathinakumar and Wimley, 2008; Rathinakumar et al., 2009]. The positively charged
arginines and the more hydrophobic tryptophans, which at the same time display a
significant quadrupole moment, favor accumulation of the peptide at the interface between
the hydrophilic phospholipid head groups and the hydrocarbon layer [Chan et al., 2006].
K Discussion
293

In fact, integration of MP196 into model lipid bilayers leads to perturbation of fatty acyl
chain packing. This is consistent with the interfacial activity model proposing perturbed
separation of hydrophilic and hydrophobic membrane parts. Insertion of the polar peptide
into the bilayer fosters incursion of polar lipid head groups deeper into the bilayer as was
demonstrated for the bee venom peptide melittin (Figure 13).
Figure 13: Molecular dynamics simulation of peptide‐induced pore formation according to
the interfacial activity model. The antimicrobial peptide melittin perturbs the segregation of
polar and nonpolar moieties of the lipid bilayer and forms a permeation pathway shuttling
small ions along the peptide‐lipid structure [modified according to Wimley, 2010; based on
results from Sengupta et al., 2008].
According to molecular dynamics simulations, melittin forms a permeation pathway
consisting of the peptide and lipid head groups that allows ions to cross the bilayer. In
contrast, MP196 treatment did not foster ion efflux. Probably, MP196 disturbs bilayer
architecture by inducing the same funnel‐like incursion of phospholipid head groups and
destabilizes the membrane structure but does not accomplish a membrane‐spanning
permeation pathway. This could be due to its short and compact structure. It could also be
attributed to the alternating RW sequence. Interfacial activity requires an imperfect
amphipathicity as a molecule with ideal separation of hydrophilic and hydrophobic residues
would exhibit less potential to deform the lipid bilayer [Rathinakumar et al., 2009]. However,
under hypoosmotic conditions, MP196 facilitates ion fluxes, especially potassium efflux,
across the B. subtilis membrane. Hypoosmosis destabilizes the membrane structure. It is
possible that higher lipid mobility and increased turgor pressure facilitate formation of a
functional permeation pathway by MP196.
The interaction of several antimicrobial peptides with model membranes or isolated
membrane extracts has been extensively studied [Zweytick et al., 2011; Zweytick et al.,
K Discussion
294

2012]. However, only very little is known about their in vivo mechanism of action. Even less
information is available on their influence on bacterial cells apart from their influence on
membrane lipids. MP196 was employed to shed light on the influence of short cationic
antimicrobial peptides on bacterial physiology. Based on proteome analysis of both the
cytosolic and the membrane proteome, hypotheses on the mechanism of action of MP196
were generated. The cytosolic proteome response revealed upregulation of almost all
signature proteins for cell envelope stress, cell wall biosynthesis inhibition (except for YpuA
and YtrB), and membrane stress. The proteome response was further indicative of strong
energy limitation. Such mechanistic duality was similarly observed for gallidermin,
suggesting membrane integration and inhibition of cell wall biosynthesis by MP196.
Comparison with the proteome reference compendium further revealed similarities of
MP196 with the membrane‐integrating peptide gramicidin S and the potassium ionophore
valinomycin. Extensive comparative follow up experiments emphasizing inhibition of cell
wall biosynthesis, membrane interaction, and influence on energy metabolism allowed
developing a comprehensive model for the in vivo mechanism of action of MP196 (Figure
14).
Figure 14: Mechanism of action of MP196. MP196 binds to the interfacial membrane
regions and disturbs membrane architecture. Consequently, peripheral membrane proteins,
such as MurG and cytochrome c, are detached from the membrane surface resulting in
inhibition of cell wall biosynthesis and respiration.
Integration of MP196 into the B. subtilis membrane perturbs phospholipid packing and leads
to detachment of the peripheral membrane proteins MurG involved in cell wall biosynthesis
295

and cytochrome c involved in respiration. Consequently, both processes are impaired
leading to loss of cell wall integrity and substantial ATP limitation.
MurG binds to the cytoplasmic membrane with an amphipathic α‐helix, which is horizontally
attached to the bilayer [Hu et al., 2003]. It is probably detached due to membrane
deformation. This phenomenon might explain an observation repeatedly made for
membrane‐interacting peptides like daptomycin. Similar to MP196, no interaction of
daptomycin with components of the cell wall biosynthesis machinery was observed
[Schneider et al., 2009]. However, both compounds strongly induce a LiaRS response [Wecke
et al., 2009]. Daptomycin has also been shown to influence membrane architecture and to
delocalize the cell division protein DivIVA [Pogliano et al., 2012]. It is likely that it similarly
delocalizes MurG, which would explain the upregulation of LiaH and daptomycin influence
on cell wall integrity [unpublished results].
MurG detachment also plays a role in the effects on cell wall biosynthesis observed for
gramicidin S. The protein almost completely disappears from the membrane fractions of
gramicidin S‐treated cultures. This might be due to gramicidin S‐membrane interaction.
MurG has been shown to preferentially co‐localize with negatively charged phospholipids
[Matsumoto et al., 2006], which are disturbed upon integration of gramicidin S into the
bilayer [Kaprel’iants et al., 1977; Vostroknutova et al., 1981].
MurG seems to be the major player in cell wall biosynthesis impairment by membrane‐active
antibiotics. Its delocalization explains how such compounds elicit strong cell wall stress
responses and impair cell wall integrity without interacting with a particular component of
the biosynthesis pathway. The induction mechanism of the LiaRS two‐component system is
still not completely understood [Wolf et al., 2010]. Based on the observations that (i)
perturbation of membrane architecture causes delocalization of MurG and (ii) compounds
interfering with membrane architecture lead to LiaH upregulation, it is plausible that
induction of the LiaRS system is strongly connected to a disturbed membrane architecture
and subsequent protein delocalization. This would be in contrast to the LiaH homolog PspA,
which is induced by membrane‐targeting compounds independent of their influence on
membrane architecture.
Detachment of cytochrome c explains the substantial energy limitation after MP196
treatment. Cytochrome c is also released from the mitochondrial membrane upon treatment
with valinomycin and from liposomes by some cationic agents [Jutila et al., 1998; Yamamoto
K Discussion
296

et al., 2012]. B. subtilis possesses four c‐type cytochromes [Hodson et al., 2008]. Cytochrome
c550 (CccA) is attached to the membrane with a transmembrane peptide helix, while
cytochrome c551 (CccB) anchors in the lipid bilayer with a lipid tail [von Wachenfeldt and
Hederstedt, 1990; Bengtsson et al., 1999; García Montes de Oca et al., 2012]. QcrC and CtaC
are subunits of the cytochrome bc complex and the cytochrome c oxidase caa3, respectively
[van der Oost et al., 1991; Yu and Le Brun, 1998]. Whether all four c‐type cytochromes are
detached by MP196 could not be determined so far, although it appears likely that only the
free variants are affected. Consistently, about 40% of total cytochrome c disappears from
the membrane fraction after treatment resulting in a strong impact on the electron
transport chain and cellular ATP levels.
MinD is another example for MP196‐dependent protein delocalization. It possesses an
amphipathic α‐helix, which is horizontally attached to the inner membrane surface by
electrostatic interactions. The primary cause of MinD delocalization is the loss of the
membrane potential [Pichoff et al., 2005; Strahl and Hamoen, 2010]. Consistently, MinD was
also delocalized by valinomycin, which should not affect membrane architecture.
FtsA and SecA, both peripheral membrane proteins, were found to be upregulated in the
membrane proteome after 65 minutes of antibiotic treatment. As suggested for MurG,
upregulation of FtsA and SecA could point to a cellular attempt to compensate for their
detachment from the membrane surface. They might be further candidates for MP196‐
dependent membrane protein delocalization. FtsA is attached to the membrane with an
amphipathic α‐helix similar to that of MinD. Its localization has therefore been discussed to
similarly depend on the membrane potential [Szeto et al., 2003; Adams and Errington, 2009;
Strahl and Hamoen, 2010]. SecA is located at the inner membrane surface and is bound to
the SecYEG intramembrane complex [Vrontau and Economou, 2004]. However, the B.
subtilis stress response to MP196 centered on compensation for energy limitation and
impairment of cell wall biosynthesis, not on inhibition of cell division and protein secretion,
and no cell elongation or disordered septation could be observed microscopically.
Apparently, cell wall biosynthesis inhibition and energy limitation have the strongest impact
on B. subtilis physiology. They are, therefore, probably the main factors contributing to
bacterial death.
As proteins located at both the outer and inner membrane surfaces are delocalized, it is
probable that MP196 accumulates in both interfacial zones, facing the cytosol and the
K Discussion
297

extracellular space. This was supported by metal tracing of the ruthenocene‐conjugated
derivative MP276, which accumulated in the membrane but also penetrated into the
cytosol.
The multidisciplinary in vivo mechanism study on MP196 allows so far unique insight into
antimicrobial peptide action in the whole biological system and supplements in vitro studies
on peptide‐membrane interaction [Joshi et al., 2010; Andrushchenko et al., 2008; Appelt et
al., 2005]. Several peptides have been developed that do not follow the mode of action
models described so far [Scheinpflug and Dathe, personal communication1], which usually
propose formation of pores or ulterior membrane disruption [Teixeira et al., 2012]. The
MP196 mechanism of action model might be useful for exploring the mechanism of action of
further antimicrobial peptides, especially of those, whose antibacterial activity is not
primarily based on increasing membrane permeability.
3 Bacterial stress adaptation to short cationic antimicrobial peptides
The proteome response pattern to MP196 did not only reflect its mechanism of action but
also allowed deeper insight into strategies of B. subtilis to adapt to peptide stress. Basically,
B. subtilis pursues three strategies to encounter membrane stress by MP196: stabilization of
the membrane structure, restriction of membrane access for cationic compounds, and
metabolic compensation for the physiological consequences of peptide integration into the
membrane (Figure 14).
B. subtilis compensates for inhibition of cell wall biosynthesis and respiration by
upregulation of proteins involved in peptidoglycan synthesis and energy metabolism, e.g.
MurG or H+‐ATPase subunits (AhpC). Energy limitation in turn leads to activation of the σB‐
dependent general stress response and of sporulation, two alternative strategies to evade
resource‐limiting conditions [Hecker and Völker, 2001].
In order to restrict peptide access to the cytoplasmic membrane, B. subtilis adapts its cell
envelope structure. Enhanced D‐alanylation of lipoteichoic acids, indicated by upregulation
of the dlt operon and the amino acid racemase RacX, is a known strategy of protecting
against antimicrobial peptides. Such cell wall modifications are e.g. involved in vancomycin
and daptomycin resistance [Weidenmaier and Peschel, 2008; Bertsche et al., 2011; Rose et
al., 2012]. Several upregulated proteins point to adaptation of the phospholipid
composition. FloT and YuaI e.g. are known to be involved in reducing membrane fluidity 1 Chemical Biology, Leipniz‐Institut für Molekulare Pharmakologie, Robert‐Roessle‐Str. 10, 13125 Berlin
K Discussion
298

[Kingston et al., 2011; López and Kolter, 2012;]. Lipidomics of cells treated with the
ferrocene‐substituted peptide MP66 revealed an exchange of negatively charged
phospholipid head groups against neutral lipids. Apparently, cells attempt a more neutral
surface charge in order to prevent attachment of MP196 to the bilayer. Further, fatty acid
adaptation towards a more rigid membrane structure was observed, complicating peptide
integration while at the same time stabilizing the membrane structure [Fränzel et al., 2010].
Figure 14: Stress response of B. subtilis to MP196: Three strategies, membrane stabilization,
restriction of compound access, and metabolic compensation, are pursued by MP196‐
treated cells. Representative stress proteins are displayed. Cell wall‐related processes are
depicted in blue, membrane‐related processes in yellow.
Stabilization of the membrane is further achieved by the membrane‐binding proteins PspA
and LiaH, which are known to stabilize the bilayer under stress conditions [Kobayashi et al.,
2007; Wolf et al., 2010]. Moreover, the proteome response showed upregulation of a
number of proteins involved in amino acid metabolism. Analysis of the free amino acids after
peptide treatment revealed highly elevated biosynthesis and release of the osmoprotective
amino acids aspartate, proline, lysine, and, especially, glutamate. Amino acid release is a
novel adaptation strategy. Exogenously supplied glutamate was shown to be effectively
protective against treatment with MP196. Probably, the local increase of compatible solutes
at the outer membrane surface stabilizes bilayer structure, a strategy similarly pursued
under hypoosmotic stress conditions. Membrane stabilization by glutamate is most probably
K Discussion
299

an osmoprotective effect as high salt concentrations similarly protect against peptide stress.
It was further shown that glutamate release is a common reaction to several membrane‐
integrating compounds such as gramicidin S and nisin. It did not occur after treatment with
the potassium ionophore valinomycin. Such carrier ionophores diffuse through the lipid
bilayer and do not disturb membrane architecture. The different reaction to valinomycin and
the other investigated compounds point to glutamate release being a specific response to
membrane‐integrating compounds that interfere with the membrane structure.
It was further shown that the observed amino acid release is partly mediated by the B.
subtilis mechanosensitive channels MscL, YkuT, YhyY, and YfkC. Those are activated upon
changes in osmolarity, which are sensed by pressure on the lipid bilayer [Sukharev, 2002,
Hoffmann et al., 2012]. Such pressure might be applied to the mechanosensitive channel
proteins by the structural changes induced by peptide integration. It is likely that activation
of amino acid‐transporting mechanosensitive channels is a direct effect of peptide‐
membrane interaction. This is supported by the recent finding that antimicrobial peptides
trigger signal cascades in plants by similar membrane‐curvature‐dependent activation of
mechanosensors [Henry et al., 2011, Desoignies et al., 2012].
Lee et al. were able to show that the glutamate dehydrogenase RocG plays a crucial role in
resistance of B. subtilis against cell wall antibiotics. This was supposed to be due to a yet
unknown regulatory influence of RocG on induction of σW. σW regulates the expression of
the yuaFGI operon, which is involved in the control of membrane fluidity in response to
membrane stress [Lee et al., 2012]. Keeping in mind the osmoprotective role of glutamate,
its secretion by mechanosensitive channels, and the role of antimicrobial peptides in plant
signaling, the supposed RocG‐related regulatory mechanism on σW could also involve direct
mechanosensing of disturbed bacterial membrane architecture.
All these recent findings on the interaction of antimicrobial peptides with living cells
demonstrate that the cellular response to membrane stress, its regulation, and the role of
antimicrobial peptides, not only as antibiotics but also as natural signaling molecules, are far
more complex than presumed so far.
Strong upregulation of amino acid metabolism was predominantly detected in the
membrane and not in the cytosolic proteome response, although amino acid biosynthesis
takes place in the cytosol. The same was observed for proteins involved in translation and
fatty acid biosynthesis. It is tempting to speculate that processes, such as biosynthesis of
K Discussion
300

osmoprotective amino acids, generation of fatty acids for adjusting phospholipid
composition, and biosynthesis of membrane stress proteins like PspA and LiaH, proceed near
to the membrane, where their products are needed.
4 MP196 derivatives: novel implications for antibacterial drug discovery
Representing the minimal pharmacophore of RW‐rich peptides, MP196 has been employed
as a lead structure for several derivatization approaches [Chantson et al., 2005; Chantson et
al., 2006]. An either N‐ or C‐terminally added lysine residue has been exploited to introduce
lipid chains of different length to the peptide structure [Albada et al., 2012a]. The most
active peptides C‐C8 and N‐C8 as well as their corresponding non‐lipidated derivatives C‐C0
and N‐C0 have been mechanistically investigated. Their mode of action was validated by
comparative proteomics. No major mechanistic differences to MP196 were revealed
suggesting a similar interfacial activity‐based mechanism of action. All four heptapeptides
elicited a more membrane‐focused stress response, while MP196‐treated cells rather
compensated for energy limitation. This could be a hint for more efficient membrane
integration of the derivatives compared to MP196. Ruthenocene‐ and ferrocene‐substituted
peptides similarly retained the MP196 mechanism of action. The cyclic ferrocene derivative
MP159 did neither differ in activity nor in its mechanism of action. Either, the cyclization
does not affect membrane interaction or the disulfide bridge is unstable under bacterial
culture conditions resulting in linearization of the peptide.
Conjugation of MP196 with metallocene moieties resulted in a higher disruptive potential on
both model membranes and B. subtilis in vivo. In fact, the low membrane‐disruptive
potential of MP196 is an important difference to other antimicrobial peptides and to the
prevailing models of peptide‐membrane interaction. The MP196 sequence seems to be
sufficient for membrane integration but not for permeation. This might be a matter of length
or of its probably perfect amphipathic sequence. The latter is supported by the membrane‐
disruptive potential of the metallocene peptides, which interact with different model lipids,
another prediction of the interfacial activity model [Wimley, 2011]. Their alternating
sequence is disrupted by the metal complex and they possess higher hydrophobicity. An RW‐
rich cyclic peptide with an ideal amphiphilic amino acid distribution has also been shown not
to enhance membrane permeability [Scheinpflug, personal communication2]. Consistently,
2 Chemical Biology, Leipniz‐Institut für Molekulare Pharmakologie, Robert‐Roessle‐Str. 10, 13125 Berlin
K Discussion
301

differences between the lipidated derivatives were observed depending on disruption or
conservation of the alternating pattern of positively charged and hydrophobic residues. This
further supports that the grade of amhipathicity determines membrane interaction. It is
possible that changing the MP196 sequence towards higher hydrophobicity or imperfect
amphipathicity results in more efficient formation of a permeation pathway. It would now
be interesting to evaluate, whether the lipid and/or lysine‐conjugated heptapeptides might
also facilitate ion leakage. It is further interesting, if a longer alternating RW sequence would
be sufficient for translocating ions or if the limited ability of MP196 to promote ion transport
solely depends on its perfectly alternating sequence.
Apart from unprecedented insight into the relationship between structure and membrane
interaction, the MP196 derivatization series provide novel implications for drug discovery.
MP196 displayed potent activity against Gram‐positive and moderate activity against Gram‐
negative bacteria. At the same time, it was neither significantly cytotoxic nor hemolytic. The
ferrocene‐conjugated MP66 displayed slightly lower activity against Gram‐positives but
reduced activity against Gram‐negatives [Chantson et al., 2006]. The ruthenocene‐
substituted MP276 was comparably active but also slightly more cytotoxic and more
hemolytic than MP196. All‐L and all‐D peptides did not differ considerably in their activity or
mechanism suggesting a minor role of stereochemistry.
While metallocene substitution did not significantly enhance the antibiotic properties, it
might be employed as potent tool in drug development. Formation of ROS by ferrocene
might prove useful in terms of bacterial resistance as compounds with multiple mechanisms
are less prone to fast resistance development [Brötz‐Oesterhelt and Brunner, 2008].
Ruthenocene has been employed for quantitative ruthenium tracing, which allowed studying
peptide localization in vivo. Although MP276 differed mechanistically from MP196 in its
ability to induce ion leakage, it still most probably acts by the same interfacial mechanism.
Thus, ruthenocene derivatization might be employed as localization tool in antibiotic
research. So far, mainly fluorescence labels but also antibody detection have been employed
to follow compound distribution. The mostly very large fluorescence labels are often not
well applicable to small compounds like MP196 as they are prone to influence antibiotic
target interaction. Development of fluorescent amino acids has recently been reported as
promising alternative for protein and peptide labeling [Katritzky and Narindoshvili, 2009].
Antibodies, which can also be immunogold labeled allowing detection by TEM, might not
K Discussion
302

detect molecules as small as MP196 [Scheinpflug, personal communication3]. Metal tracers
like ruthenocene indeed can be individually incorporated into the compound structure.
Ruthenocene might e.g. replace bulky amino acid side chains like tryptophan or
phenylalanine without much effect on the molecule’s properties. Another important
advantage of metal‐based detection compared to alternative methods is the possibility of
absolute compound quantitation by spectroscopy‐based metal analysis. Further, its
detectability by electron microscopy additionally facilitates a graphic visualization of
compound localization.
The lipidated MP derivatization series yielded peptides with significantly higher antibacterial
activity against both Gram‐positive and Gram‐negative bacteria. However, the most active
derivatives were considerably hemolytic and highly cytotoxic [Albada et al., 2012a]. The C8
and C10 series have been subjected to extensive structural optimization focused on
systematic L to D exchange of C‐ and N‐terminally lipidated peptides [Albada et al.,
submitted]. In course of that study, peptides were developed, which fully retained
antibacterial activity, e.g. in the nanomolar range against MRSA, while completely
eliminating their hemolytic potential. Such derivatives hold great promise to undergo further
drug development.
However, the lead structure MP196 exhibited high acute toxicity in mice [Vuong, personal
communication4]. As shown for further positively charged peptides [Graham et al., 2006;
Peng et al., 2010], this could be due to histamine release and an anaphylactic shock reaction.
Histamine release by activation of CD63‐positive basophils, diagnostic of the IgE‐mediated
allergic reaction [Amin, 2012], could be excluded [Höxtermann, personal communication5].
The high immunogenicity of MP196 limits intravenous application and restricts its potential
as a systemic drug. Lowering immunogenicity will be one major aim in optimizing promising
derivatives.
5 Organometallic PNA derivatives: a novel antibiotic class
A structurally completely novel antibiotic class was developed in course of a chemical proof‐
of‐principle study [Patra et al., 2010a]. The resulting tri‐metallic compound FcPNA,
containing a manganese moiety (cymantrene), a rhenium complex (di‐picolyl)Re(CO)3), and a
3 Chemical Biology, Leipniz‐Institut für Molekulare Pharmakologie, Robert‐Roessle‐Str. 10, 13125 Berlin 4 AiCuris GmbH & Co. KG, Friedrich‐Ebert‐Str. 475, 42117 Wuppertal 5 Klinik für Dermatologie, Venerologie und Allergologie, St. Josef‐Hospital, Gudrunstr. 56, 44791 Bochum
K Discussion
303

ferrocene, exhibited potent activity against Gram‐positive bacteria including MRSA, being
even more efficient than the clinically applied antibiotics amoxicillin and norfloxacin. Dubar
et al. suggested that ferrocene in contrast to ruthenocene promotes formation of ROS
contributing to antimicrobial activity [Dubar et al., 2011; Dubar et al., 2012]. To evaluate, if
this is true in bacteria in vivo, the derivative RcPNA containing a ruthenocene instead of a
ferrocene, was developed. The mechanism of action of FcPNA and RcPNA was investigated
by proteomic profiling. The bacterial response to the PNA derivatives is shown in Figure 15.
Figure 15: Cellular response to FcPNA and RcPNA. Both derivatives insert into the bacterial
membrane. Depolarization activates the membrane stress response. Energy limitation
activates the general stress response and sporulation. Cell wall biosynthesis is probably
impaired due to altered membrane architecture and energy limitation. FcPNA additionally
leads to formation of ROS.
Both compounds share a similar mechanism of action. They integrate into the bacterial
membrane bilayer but do not form pores. Both elicit a membrane stress response and cause
break‐down of the membrane potential. Consequently, cells suffer substantial energy
limitation. Consistently, σB activation and sporulation initiation were observed in the
proteome pattern, two strategies activated upon energy‐limiting conditions [Hecker and
Völker, 2001]. Although no reproducible markers for inhibition of cell wall biosynthesis were
upregulated, cell wall integrity was corrupted by both FcPNA and RcPNA. The acetic
acid/methanol fixation that was employed to visualize cell wall damage allows distinction
between inhibition of the lipid II cycle, of transglycosylation, and of cell wall digestion
K Discussion
304

[Schneider et al., 2010]. The membrane excrescences observed after FcPNA and RcPNA
treatment point to inhibition of the lipid II cycle. Membrane‐targeting compounds can impair
the lipid II cycle by changing membrane architecture, a mechanism that might also apply to
FcPNA and RcPNA. However, so far such membrane‐associated inhibition of cell wall
biosynthesis has always been accompanied by upregulation of the cell wall stress marker
LiaH. The missing upregulation of LiaH by FcPNA and RcPNA could point to a modified
mechanism, which is based less on protein detachment due to interference with membrane
architecture and instead more on interference with the respiratory chain and energy
limitation.
However, depolarization and energy limitation without membrane disruption does point
towards inhibition of the respiratory chain by altered membrane architecture. This could be
similarly achieved as described for MP196: by delocalization of cytochrome c. The data on
hand do not provide conclusive evidence of the nature of the membrane interaction but
suggest that the main antibacterial effects are based on impairment of the respiratory chain
rather than cell wall biosynthesis. The molecular basis underlying this effect is worth further
investigation and might provide essential insights into how membrane interaction
determines physiological effects.
An important mechanistic difference between the ferrocene and the ruthenocene
derivatives is constituted by their ability to induce oxidative stress. In contrast to RcPNA,
FcPNA causes formation of ROS in vivo. These results concur with those obtained for the
ruthenocene and ferrocene‐substituted MP196 derivatives MP276 and MP66. However,
FcPNA was much more active than RcPNA, while no such difference was seen with the
MP196 derivatives. By a mechanism not understood so far, ferrocene bound to the PNA
backbone structure seems to contribute to bacterial killing, while it does not in the peptide
conjugates. This could be due to a difference in membrane interaction. It is tempting to
speculate that the bulky PNA derivative positions the redox‐active ferrocene moiety near
components of the respiratory chain, where it may capture and mislead electrons more
efficiently. This would also fit to the idea that the PNA derivatives primarily target the
respiratory chain, probably accompanied by interference with membrane architecture.
Both the structure and the mechanism of action of FcPNA are not exploited in antibacterial
drug discovery so far. In terms of resistance development, the bacterial membrane is a
favorable target structure as resistances are less common [Peschel and Sahl, 2006; Brötz‐
K Discussion
305

Oesterhelt and Brunner, 2008]. Especially the highly active FcPNA is a promising antibiotic
candidate belonging to a completely novel class of antimicrobial agents.
While it is active against all tested Gram‐positive bacteria, its cytotoxicity strongly depends
on the cell lines and the growth conditions. A trend towards increased cytotoxicity
depending on the growth rate of the mammalian cells became apparent. While fast‐growing
tumor cell lines were efficiently killed, cells that were already growing into confluence for a
few days were less affected. The tested cells that were differentiated and fully confluent
were not affected at all. This behavior could be due to higher metabolic activity of fast‐
growing cells. Moreover, it has been shown for some tumor cell lines that they differ from
normal cells in their membrane composition [Liu et al., 2010; Hilvo et al., 2011, Harris et al.,
2012]. It is possible that the cell selectivity of FcPNA depends on more or less efficient
membrane interaction as similarly supposed for MP196. Based on the data available to date,
the molecular basis underlying the cell line selectivity of FcPNA could not be determined.
The limited number of tested cell lines does further not allow a definite conclusion on the
cytotoxicity for cancerous and differentiated cell lines. A detailed analysis of the underlying
mechanisms of toxicity and selectivity could provide novel insight into both toxicity of
antibiotics and anti‐tumor drugs and could advance targeted drug design.
One substantial limitation of both FcPNA and RcPNA is their low solubility in aqueous
solution, which might be due to the organometallic complexes rendering the molecule highly
hydrophobic. In a systematic SAR study, the essential organometallic residue for
antibacterial activity was found to be the rhenium moiety, a metal complex not exploited in
antibacterial development so far. Interestingly, (di‐picolyl)Re(CO)3 alone does not inhibit
growth of bacteria, suggesting that its combination with the PNA backbone structure is
essential for antibacterial activity. Three derivatives, the PNA backbone with the rhenium
moiety alone or in combination with either cymantrene or ferrocene, were as active as
FcPNA and essentially retained its mechanism of action. The Re‐only derivative 8c’
additionally displayed the lowest cytotoxicity and the highest solubility in culture medium,
thus advancing to a new lead structure for further compound optimization. Structural
optimization should focus on lowering cytotoxicity, which still is difficult to predict, and on
further improvement of solubility in order to yield drug candidates for systemic application.
However, even a moderately soluble, hydrophobic compound like 8c’ might already find
topical application, e.g. as ointment for treatment of skin infections. In face of the rising
K Discussion
306

number of epidermal MRSA infections, the clinical importance of such topically applicable
antibiotic agents should not be underestimated.
6 Metal complexes in antibiotic drug discovery
Organometallic complexes, i.e. containing a metal directly bound to a carbon atom, are
unusual in nature. They constitute an antibiotic compound family bacteria have not been
extensively challenged with before. Thus, they might open up new avenues for antibacterial
drug design. Especially ferrocene attracted much interest over the last decades. It has been
employed in the development of both antimalarial (ferroquine) and anticancer (ferrocifen)
drug candidates [Dive and Biot, 2008; Hillard et al., 2005]. Ferroquine was shown to
overcome plasmodial resistance against its lead structure chloroquine and has recently
completed phase II clinical trials for the treatment of uncomplicated malaria [Biot et al.,
2012]. The success of ferroquine demonstrates the clinical potential of organometallic drugs.
It further suggests that no toxic effects are inherent to ferrocene itself encouraging its
employment in drug development.
Despite this inspiring success, the potential of organometallics in antibacterial drug
development has been rather underexplored. Approaches that have been undertaken so far
employed iron, ruthenium, tungsten, gold, and silver complexes [Patra et al., 2012a]. With
the PNA derivatives, the first antibiotic class featuring rhenium was developed. Moreover,
most organometallic antibacterial compounds are based on an active organic lead structure.
The metal is mostly employed to enhance antibacterial properties but is essentially
dispensable [Patra et al., 2012a]. Not so the PNA derivatives, where rhenium critically
determines antibacterial activity. Apart from the recently developed silver‐containing
antibiotic SCC1 [Panzner et al., 2009], they constitute the first class of organometallic
antibacterial agents, whose activity is based on a metal complex, since the development of
arsphenamin.
Comparing different metallocene substitutions, it becomes apparent that different metals
incorporated into the same organic structure might distinctly influence antibacterial
properties. While cobalt negatively affected antibacterial activity, iron and ruthenium did
not significantly influence the antibacterial potential of metallocene‐substituted MP196
derivatives. However, the ferrocene PNA derivative was much more active than the
ruthenocence PNA, probably due to oxidative stress caused by ferrocene. The ferrocene‐
K Discussion
307

substituted MP66 induced formation of ROS but did not elicit an oxidative stress response.
Apparently, the PNA structure advances efficient ROS formation by ferrocene while this
mechanism plays a minor role within the MP structure. This interesting finding suggests a
strong correlation between the action of a metal complex and the organic structure it is
conjugated with.
The dependency of ROS formation on the presence of ferrocene provides insight into the
recent “oxidative stress controversy”. While some groups proposed the occurrence of
oxidative stress after treatment with bactericidal antibiotics [Hassett and Imlay, 2007;
Kohanski et al., 2007] and provided experimental evidence for oxidative damage to
nucleotides by β‐lactams and quinolones [Foti et al., 2012], others could not show a
correlation between ROS formation and bacterial death [Keren et al., 2013; Liu and Imlay,
2013]. The ferrocene‐ and ruthenocene‐substituted MP196 derivatives, all of which are
bactericidal, share a highly similar mechanism of action and equally influence bacterial
physiology. The same was observed for FcPNA and RcPNA. Only the ferrocene‐substituted
compounds induced formation of ROS in bacteria, probably due to the ferrocene redox
potential, suggesting that oxidative stress is not a general mechanistic component of
bactericidal antibiotics.
Ferrocene and ruthenocene have been considered “fancy bulky moieties” that do not elicit
considerable effects on the mechanism of action of peptide antibiotics [Albada, conference
contribution6]. Here it was shown that both complexes shifted the molecular mechanism of
MP196 towards a higher membrane‐disruptive potential. Ferrocene was additionally shown
to reduce peptide selectivity for the phospholipid composition. At the same time, both
ferrocene‐ and ruthenocene‐substituted compounds retained the general mode of
membrane interaction as well as their physiological impact on B. subtilis in vivo. While the
effect of ferrocene on the mode of action of MP196 derivatives was rather subtle, it
considerably modified the action of FcPNA compared with RcPNA, demonstrating that
ferrocene is not simply another bulky residue but might efficiently tune a compound’s
antibacterial properties. It constitutes a potent tool, which confers the ability to induce
formation of ROS and might enhance antibacterial activity, depending on the structure of
the individual molecule. Thus, it could contribute to overcoming bacterial resistance by
adding another mechanistic component. Ruthenocene had a more subtle influence on the
6 Albada HB. Metallocenoyl derivatives of antibacterial peptides. 11th ferrocene colloquium, Feb 6th‐8th, 2013. Hannover, Germany
K Discussion
308

mechanism of action. It is therefore very well suited as a subtle labeling tool allowing metal‐
based peptide tracing. Recently, an osmocene‐conjugated MP196 derivative was developed
[Albada, personal communication7]. As osmium is more electron‐dense than ruthenium, the
osmocene‐conjugate could improve the contrast in electron microscopy. Employing the
ruthenium derivative, the membrane and cell wall structures were well “stained”. However,
the peptide did not accumulate in the cytosol to an extent that could give a significant signal.
A more electron‐dense metal could improve detection of compounds located in the cytosol
or at distinct subcellular binding sites.
Taken together, organometallic substitutions open up new avenues in drug development,
both as novel resistance‐breaking structures and as efficient research tools. Metal
complexes are a rich repository of bioactive building blocks. Exploring the properties of
further metallocene complexes might yield new interesting properties and feature novel
applications.
7 Antimicrobial peptides as therapeutics: possibilities and limitations
Antimicrobial peptides have been discussed as possible new generation of antibiotic agents
mostly due to their broad‐spectrum activity and low resistance development [Giuliani et al.,
2007; Brogden and Brogden, 2011]. Clinically successful peptides like vancomycin and
daptomycin greatly nourished these hopes. Currently, several peptide‐based antibiotics
derived from different structural subclasses, e.g. glycopeptides, lipopeptides, and defensins,
are in the drug discovery and development pipeline [Butler and Cooper, 2011]. Many
peptides are highly potent in terms of bacterial killing and advantageous in resistance
development. However, cationic peptides might also display high cytotoxic or hemolytic
activity such as gramicidin S [Semrau et al., 2010] or trigger allergic shock reactions [Graham
et al., 2006; Peng et al., 2010]. This behavior is not extraordinary keeping in mind that
cationic peptides are widely distributed immunomodulators in vertebrates [Steinsträßer et
al. 2011; Choi et al., 2012]. Thus, it remains doubtful, if cationic peptides, such as the RW‐
rich MP196, will ever be suitable for systemic application, even after chemical compound
optimization. Rather, they are suited for topical application, whose importance should not
be underestimated in face of the rising number of complicated skin infections caused by
MRSA. Surface coating of medical catheters or prostheses but also of food wrappings might
be another possible application of cationic peptides [Cooksey, 2005]. Furthermore, the 7 Bioinorganic Chemistry, Ruhr‐Universität Bochum, Universitätsstr. 150, 44801 Bochum
K Discussion
309

extensive research currently conducted on immunomodulation by antimicrobial peptides
might open up novel therapeutic approaches shifting the focus from killing bacteria towards
stimulating the patients’ own immune response.
Anionic peptides like daptomycin or glycopeptides like vancomycin do not suffer from the
same limitations as the cationic RW‐rich peptides. They constitute alternative lead structures
for development of novel systemic drugs. The semi‐synthetic vancomycin derivative
telavancin is currently in clinical use. However, resistance against daptomycin, vancomycin,
and televancin has already been described [Lütticken and Kunstmann, 1988; Smith et al.,
1999; Bertsche et al., 2011, Rose et al., 2012] and it is only a matter of time until bacterial
resistance demands novel therapeutics. Peptidomimetics and peptide derivatives with
structurally novel features might constitute a next generation of antibacterial agents.
Introducing non‐natural moieties to peptide‐based core structures might contribute to
overcoming bacterial resistance as well as the application‐limiting issues of the natural
compounds.
K Discussion
310

L Summary
Increasing antibiotic resistance in hand with decreasing antibiotic approvals has founded an
urgent need for novel antimicrobial agents. Antibiotics acting on the bacterial cell envelope
have been highly successful in terms of inhibition of peptidoglycan crosslinking. However,
the cell envelope offers further interesting antibiotic targets such as membrane biogenesis
and integrity. Specific proteomic signatures for fatty acid biosynthesis inhibition, membrane
damage, and inhibition of membrane‐bound cell wall biosynthesis steps formed a basis for
mode of action diagnosis of novel cell envelope‐targeting antibiotics. The established
signatures were successfully employed in target falsification of a potential fatty acid
biosynthesis inhibitor and in mode of action elucidation of antimicrobial peptides and
organometallic peptidomimetics. The hexapeptide MP196, a small but yet typical
representative of short cationic antimicrobial peptides, was found to integrate into the
bacterial membrane. Its antibacterial action is mainly based on delocalization of peripheral
membrane proteins like cytochrome c and MurG, resulting in inhibition of respiration and
cell wall biosynthesis. Consequently, cells suffer substantial energy limitation and cell wall
integrity is corrupted. Bacteria adapt to peptide stress by adjusting their membrane and cell
wall composition, upregulation of impaired cellular processes, synthesis of membrane‐
stabilizing proteins, and release of osmoprotective amino acids. Lipidated derivatives of
MP196 have been shown to essentially retain the same mechanism of action. So did
organometallic‐substituted variants. Metallocene derivatization slightly alters membrane
interaction leading to enhanced disruptive potential. Ruthenocene modification has
additionally been shown to be a powerful tool for metal‐based peptide tracing while at the
same time not considerably influencing the peptides’ mechanism.
The organometallic PNA backbone derivatives constitute a completely novel class of
antibiotics. Displaying excellent antibacterial activity and limited toxicity for mammalian
cells, the hetero‐tri‐metallic compound FcPNA targets the cytoplasmic membrane leading to
depolarization and energy limitation. The ferrocene moiety induced formation of ROS in
bacterial cells, whereas ruthenocene did not display a metal‐specific mode of action. In a
systematic SAR study, the essential organometallic moiety for antibacterial activity was
found to be the rhenium complex. A mono‐metallic Re‐containing compound was
established as new lead structure displaying good antibacterial activity and increased water
solubility compared the tri‐metallic compound.
L Summary
311

M Zusammenfassung
Der Anstieg bakterieller Antibiotika‐Resistenzen zusammen mit sinkenden Neu‐
Zulassungsraten verantwortet einen dringenden Bedarf an neuen antimikrobiellen
Substanzen. Antibiotika, die die bakterielle Zellhülle angreifen, waren bisher äußerst
erfolgreich, insbesondere in Hinblick auf die Hemmung der Vernetzung von Peptidoglykan.
Sie bietet jedoch noch weitere, interessante Angriffsorte für Antibiotika wie die Biogenese
und Integrität der Zytoplasma‐Membran. Die Etablierung spezifischer Proteom‐Signaturen
für Inhibition der Fettsäure‐Biosynthese, Membran‐Schäden und Inhibition Membran‐
gebundener Zellwand‐Biosynthese‐Schritte lieferten die Basis für die Analyse des
Wirkmechanismus neuer Zellhüllen‐angreifender Antibiotika. Diese Signaturen wurden
erfolgreich in der Angriffsort‐Falsifizierung eines potenziellen Fettsäure‐Biosynthese‐
Inhibitors und in der Wirkmechanismus‐Analyse antimikrobieller Peptide und
metallorganischer Peptidomimetika eingesetzt.
Für das Hexapeptid MP196, einen kleinen, aber typischen Vertreter kurzer, kationischer,
antimikrobieller Peptide, konnte gezeigt werden, dass es in bakterielle Membranen
integriert. Seine antibakterielle Wirkung basiert hauptsächlich auf der Delokalisation
peripherer Membranproteine wie Cytochrom c und MurG, was in einer Inhibierung der
Zellatmung und Zellwand‐Biosynthese resultiert. Infolgedessen tritt eine kritische Energie‐
Limitation ein und die Zellwand‐Integrität wird gestört. Bakterien passen sich an diesen
Peptid‐Stress durch Anpassung der Membran‐ und Zellwand‐Zusammensetzung,
Hochregulation betroffener zellulärer Prozesse, Synthese Membran‐stabilisierender Proteine
und Freisetzung osmoprotektiver Aminosäuren an.
Acylierte sowie metallorganisch‐substituierte MP196‐Derivate behielten diesen
Wirkmechanismus grundsätzlich bei. Metallocen‐Substituierungen haben einen geringen
Einfluss auf die Interaktion mit der Membran, was in einer erhöhten Permeabilisierung
resultiert. Modifikation der Peptid‐Struktur mit Ruthenocen ist zudem eine interessante
Möglichkeit, die Lokalisation antimikrobieller Peptide anhand des Metalles nachzuverfolgen
ohne den Wirkmechanismus durch die Derivatisierung grundlegend zu ändern.
Mit den metallorganischen PNA‐Derivaten wurde eine völlig neue Antibiotika‐Klasse
beschrieben. Die hetero‐tri‐metallische Verbindung FcPNA besitzt eine hohe antibakterielle
Aktivität und nur eingeschränkte Zytotoxizität. Sie greift die bakterielle Membran an und
führt zu Depolarisation und Energie‐Limitation. Der Ferrocen‐Rest führt zu Bildung reaktiver
M Zusammenfassung
312

Sauerstoff‐Spezies, während Ruthenocen keinen Metall‐spezifischen Einfluss auf den
Wirkmechanismus zu haben scheint. In einer systematischen SAR‐Studie stellte sich der
Rhenium‐Komplex als essentiell für die antibakterielle Aktivität heraus. Ein mono‐
metallisches Rhenium‐Derivat mit guter Aktivität und verbesserter Wasserlöslichkeit konnte
als neue Leitstruktur etabliert werden.
M Zusammenfassung
313

N References
Adams DW, Errington J. Bacterial cell division: Assembly, maintenance and disassembly of
the Z ring. Nat Rev Microbiol 7, 642‐653 (2009).
Albada HB, Prochnow P, Bobersky S, Langklotz S, Bandow JE, Metzler‐Nolte N. Short
antibacterial peptides with significantly reduced hemolytic activity can be identified by an L‐
to‐D exchange scan of their amino acid residues. submitted.
Albada HB, Prochnow P, Bobersky S, Langklotz S, Schriek P, Bandow JE, Metzler‐Nolte N.
Tuning the activity of a short Arg‐Trp antimicrobial peptide by lipidation of a C‐ or N‐terminal
lysine side‐chain. ACS Med Chem Lett 3, 980‐4 (2012a).
Albada HB, Chiriac AI, Wenzel M, Penkova M, Bandow JE, Sahl HG, Metzler‐Nolte N.
Modulating the activity of short arginine‐tryptophan containing antibacterial peptides with
N‐terminal metallocenoyl groups. Beilstein J Org Chem 8, 1753‐64 (2012b).
Amin K. The role of mast cells in allergic inflammation. Respir Med 106, 9‐14 (2012).
Amyes SGB. Magic bullets lost horizons: The rise and fall of antibiotics. Taylor & Francis Inc,
London (2001).
Andrushchenko VV, Aarabi MH, Nguyen LT, Prenner EJ, Vogel HJ. Thermodynamics of the
interactions of tryptophan‐rich cathelicidin antimicrobial peptides with model and natural
membranes. Biochim Biophys Acta 1778, 1004‐14 (2008).
Appelt C, Eisenmenger F, Kühne R, Schmieder P, Söderhäll JA. Interaction of the
antimicrobial peptide cyclo(RRWWRF) with membranes by molecular dynamics simulations.
Biophys J 89, 2296‐306 (2995).
Bai H, You Y, Yan H, Meng J, Xue X, Hou Z, Zhou Y, Ma X, Sang G, Luo X. Antisense inhibition
of gene expression and growth in Gram‐negative bacteria by cell‐penetrating peptide
conjugates of peptide nucleic acids targeted to rpoD gene. Biomaterials 33, 659‐67 (2012).
Balemans W, Lounis N, Gilissen R, Guillemont J, Simmen K, Andries K, Koul A. Essentiality of
the FASII pathway for Staphylococcus aureus. Nature 463, E3; discussion E4 (2010).
Beall B, Lutkenhaus J. Impaired cell division and sporulation of a Bacillus subtilis strain with
the ftsA gene deleted. J Bacteriol 174, 2398‐403 (1992).
Bengtsson J, Rivolta C, Hederstedt L, Karamata D. Bacillus subtilis contains two small c‐type
cytochromes with homologous heme domains but different types of membrane anchors. J
Biol Chem 274, 26179‐84 (1999).
Bertsche U, Weidenmaier C, Kuehner D, Yang SJ, Baur S, Wanner S, Francois P, Schrenzel J,
Yeaman MR, Bayer AS. Correlation of daptomycin resistance in a clinical Staphylococcus
N References
314

aureus strain with increased cell wall teichoic acid production and D‐alanylation. Antimicrob
Agents Chemother 55:3922‐8 (2011).
Bierbaum G, Sahl HG. Lantibiotics: mode of action, biosynthesis and bioengineering. Curr
Pharm Biotechnol 10, 2‐18 (2009).
Bierbaum G, Szekat C, Josten M, Heidrich C, Kempter C, Jung G, Sahl HG. Engineering of a
novel thioether bridge and role of modified residues in the lantibiotic Pep5. Appl Environ
Microbiol 62, 385‐92 (1996).
Biot C, Castro W, Botte CY, Navarro M. The therapeutic potential of metal‐based antimalarial
agents: Implications for the mechanism of action. Dalton Trans 41, 6335‐49 (2012).
Bishop DG, Rutberg L, Samuelsson B. The chemical composition of the cytoplasmic
membrane of Bacillus subtilis. Eur J Biochem 2, 448‐53 (1967).
Bonelli RR, Schneider T, Sahl HG, Wiedemann I. Insights into in vivo Activities of lantibiotics
from gallidermin and epidermin mode‐of‐action studies. Antimicrob Agents Chemother 50,
1449‐57 (2006).
Brinster S, Lamberet G, Staels B, Trieu‐Cuot P, Gruss A, Poyart C. Type II fatty acid synthesis is
not a suitable target for Gram‐positive pathogens. Nature 458, 83‐6 (2009).
Brogden NK, Brogden KA. Will new generations of modified antimicrobial peptides improve
their potential as pharmaceuticals? Int J Antimicrob Agents 38, 217‐25 (2011).
Brötz‐Oesterhelt H, Beyer D, Kroll HP, Endermann R, Ladel C, Schroeder W, Hinzen B,
Raddatz S, Paulsen H, Henninger K, Bandow JE, Sahl HG, Labischinski H. Dysregulation of
bacterial proteolytic machinery by a new class of antibiotics. Nat Med 11, 1082‐7 (2005).
Brötz‐Oesterhelt H, Brunner NA. How many modes of action should an antibiotic have? Curr
Opin Pharmacol 8, 564‐73 (2008).
Brötz H, Sahl HG. New insights into the mechanism of action of lantibiotics‐‐diverse
biological effects by binding to the same molecular target. J Antimicrob Chemother 46, 1‐6
(2000).
Brötz H, Bierbaum G, Leopold K, Reynolds PE, Sahl HG. The lantibiotic mersacidin inhibits
peptidoglycan synthesis by targeting lipid II. Antimicrob Agents Chemother 42, 154‐60
(1998).
Brötz H, Bierbaum G, Markus A, Molitor E, Sahl HG. Mode of action of the lantibiotic
mersacidin: inhibition of peptidoglycan biosynthesis via a novel mechanism? Antimicrob
Agents Chemother 714‐9 (1995).
N References
315

Budde I, Steil L, Scharf C, Völker U, Bremer E. Adaptation of Bacillus subtilis to growth at low
temperature: a combined transcriptomic and proteomic appraisal. Microbiology 152, 831‐53
(2006).
Burchall JJ. Mechanism of action of trimethoprim‐sulfamethoxazole. II. J Infect Dis 128, 437‐
41 (1973).
Butler MS, Cooper MA. Antibiotics in the clinical pipeline in 2011. J Antibiot (Tokyo) 64, 413‐
25 (2011).
Caboche S, Pupin M, Leclère V, Fontaine A, Jacques P, Kucherov G. NORINE: a database of
nonribosomal peptides. Nucleic Acids Res 36, D326‐31 (2008).
Cao M, Helmann JD. The Bacillus subtilis ectracytoplasmic‐function sigmaX factor regulates
modification of the cell envelope and resistance to cationic antimicrobial peptides. J
Bacteriol 186, 1136‐46 (2004).
Cao M, Kobel PA, Morshedi MM, Wu MFW, Paddon C, Helmann JD. Defining the Bacillus
subtilis σW regulon: a comparative analysis of promoter consensus search, run‐off
transcription/microarray analysis (ROMA), and transcriptional profiling approaches 1. J Mol
Biol 316, 443‐57 (2002).
Casadio S. General characteristics and solvent activity of triton X 100. Boll Chim Farm 91, 3‐7
(1952).
Chan DI, Prenner EJ, Vogel HJ. Tryptophan‐ and arginine‐rich antimicrobial peptides:
structures and mechanisms of action. Biochim Biophys Acta 1758, 1184‐202 (2006).
Chantson JT, Verga Falzacappa MV, Crovella S, Metzler‐Nolte N. Solid‐phase synthesis,
characterization, and antibacterial activities of metallocene‐peptide bioconjugates.
ChemMedChem 1, 1268‐74 (2006).
Chantson JT, Verga Falzacappa MV, Crovella S, Metzler‐Nolte N. Antibacterial activities of
ferrocenoyl‐ and cobaltocenium‐peptide bioconjugates. J Organomet Chem 690, 4564‐74
(2005).
Chatterjee C, Paul M, Xie L, van der Donk WA. Biosynthesis and mode of action of
lantibiotics. Chem Rev 105, 633‐84 (2005).
Chatterjee S, Chatterjee S, Lad SJ, Phansalkar MS, Rupp RH, Ganguli BN, Fehlhaber HW,
Kogler H. Mersacidin, a new antibiotic from Bacillus. Fermentation, isolation, purification
and chemical characterization. J Antibiot (Tokyo) 45, 832‐8 (1992).
Cheng JT, Hale JD, Elliott M, Hancock RE, Straus SK. The importance of bacterial membrane
composition in the structure and function of aurein 2.2 and selected variants. Biochim
Biophys Acta 1808, 622‐33 (2011).
N References
316

Cheng JT, Hale JD, Kindrachuk J, Jenssen H, Elliott M, Hancock RE, Straus SK. Importance of
residue 13 and the C‐terminus for the structure and activity of the antimicrobial peptide
aurein 2.2. Biophys J 99, 2926‐35 (2010).
Chiriac AI, Wenzel M, Zweytick D, Schumacher C, Albada HB, Krämer J, Penkova M, Brötz‐
Oesterhelt H, Metzler‐Nolte N, Brötz‐Oesterhelt H, Sahl HG, Bandow JE. Influence of
ferrocene and ruthenocene modification on the mechanism of action of a small
antimicrobial peptide. in preparation.
Choi KY, Chow LN, Mookherjee N. Cationic host defence peptides: multifaceted role in
immune modulation and inflammation. J Innate Immun 4, 361‐70 (2012).
Christ K, Al‐Kaddah S, Wiedemann I, Rattay B, Sahl HG, Bendas G. Membrane lipids
determine the antibiotic activity of the lantibiotic gallidermin. J Membr Biol 226, 9‐16 (2008).
Chung HS, Yao Z, Goehring NW, Kishony R, Beckwith J, Kahne D. Rapid beta‐lactam‐induced
lysis requires successful assembly of the cell division machinery. Proc Natl Acad Sci USA 106,
21872‐7 (2009).
Clejan S, Krulwich TA, Mondrus KR, Seto‐Young D. Membrane lipid composition of obligately
and facultatively alkalophilic strains of Bacillus spp. J Bacteriol 168, 334‐40 (1986).
Cooksey K. Effectiveness of antimicrobial food packaging materials. Food Addit Contam 22,
980‐7 (2005).
Cooper RG, Wald M. Successful treatment of Proteus septicaemia with a new drug
trimethoprim. Med J Aust 2, 93‐6 (1964).
Costa F, Carvalho IF, Montelaro RC, Gomes P, Martins MCL. Covalent immobilization of
antimicrobial peptides (APMs) onto biomaterial surfaces. Acta Biomater 7, 1431‐40 (2011).
Day RV. Sulfanilamide in the treatment of gonorrhea. Cal West Med 47, 79‐80 (1937).
den Kamp JA, Redai I, van Deenen LL. Phospholipid composition of Bacillus subtilis. J
Bacteriol 99, 298‐303 (1969).
Desoignies N, Schramme F, Ongena M, Legrève A. Systemic resistance induced by Bacillus
lipopeptides in Beta vulgaris reduces infection by the rhizomania disease vector Polymyxa
betae. Mol Plant Pathol doi: 10.1111/mpp.12008 (2012).
Dive D, Biot C. Ferrocene conjugates of chloroquine and other antimalarials: the
development of ferroquine, a new antimalarial. ChemMedChem 3, 383‐91 (2008).
Dougherty DA. Cation‐pi interactions in chemistry and biology: a new view on benzene Phe,
Tyr, and Trp. Science 271, 163‐168 (1996).
N References
317

Dubar F, Bohic S, Dive D, Guérardel Y, Cloetens P, Khalife J, Biot C. Deciphering the
resistance‐counteracting functions of ferroquine in Plasmodium falciparum‐infected
erythrocytes. ACS Med Chem Lett 3:480‐3 (2012).
Dubar F, Egan TJ, Pradines B, Kuter D, Ncokazi KK, Forge D, Paul JF, Pierrot C, Kalamou H,
Khalife J, Buisine E, Rogier C, Vezin H, Forfar I, Slomianny C, Trivelli X, Kapishnikov S,
Leiserowitz L, Dive D, Biot C. The antimalarial ferroquine: role of the metal and
intramolecular hydrogen bond in activity and resistance. ACS Chem Biol 6, 275‐87 (2011).
Duax WL, Griffin JF, Langs DA, Smith GD, Grochulski P, Pletnev V, Ivanov V. Molecular
structure and mechanisms of action of cyclic and linear ion transport antibiotics.
Biopolymers, 40, 141‐55 (1996).
Eiamphungporn W, Helmann JD. The Bacillus subtilis σM regulon and its contribution to cell
envelope stress responses. Mol Microbiol 67, 830‐48 (2008).
Eliopoulos GM, Willey S, Reiszner E, Spitzer PG, Caputo G, Moellering RC Jr. In vitro and in
vivo activity of LY 146032, a new cyclic lipopeptide antibiotic. Antimicrob Agents Chemother
30, 532‐5 (1986).
Fisher JF, Meroueh SO, Mobashery S. Bacterial resistance to β‐lactam antibiotics: compelling
opportunism, compelling opportunity. Chem Rev 105, 395‐424 (2005).
Foti JJ, Devadoss B, Winkler JA, Collins JJ, Walker GC. Oxidation of the guanine nucleotide
pool underlies cell death by bactericidal antibiotics. Science 336, 315‐9 (2012).
Fränzel B, Frese C, Penkova M, Metzler‐Nolte N, Bandow JE, Wolters DA. Corynebacterium
glutamicum exhibits a membrane‐related stress response to a small ferrocene‐conjugated
antimicrobial peptide. J Biol Iorg Chem. 15, 1293‐303 (2010).
Fredenhagen A, Tamura SY, Kenny PTM, Komura H, Naya Y, Nakanishi K, Nishiyama K,
Sugiura M, Kita H. 1987. Andrimid, a new peptide antibiotic produced by an intracellular
bacterial symbiont isolated from a brown planthopper. J Am Chem Soc 109, 4409‐11 (1987).
Freiberg C, Fischer HP, Brunner NA. Discovering the mechanism of action of novel
antibacterial agents through transcriptional profiling of conditional mutants. Antimicrob
Agents Chemother 49, 749‐759 (2005).
Freiberg C, Schiffer G, Brunner N, Lampe T, Pohlmann J, Brands M, Haebich D, Ziegelbauer K.
Identification and characterization of the first class of potent bacterial acetyl‐CoA
carboxylase inhibitors with antibacterial activity. J Biol Chem 279, 26066‐73 (2004).
Fritig B, Heitz T, Legrand M. Antimicrobial proteins in induced plant defense. Curr Opin
Immunol 10, 16‐22 (1998).
Fuertes G, Giménez D, Esteban‐Martín S, Sánchez‐Muñoz OL, Salgado J. A lipocentric view of
peptide‐induced pores. Eur Biophys J 40, 399‐415 (2011).
N References
318

Ganz T. The role of antimicrobial peptides in innate immunity. Integr Comp Biol 43, 300‐4
(2003).
García Montes de Oca LY, Chagolla‐López A, González de la Vara L, Cabellos‐Avelar T, Gómez‐
Lojero C, Gutiérrez Cirlos EB. The composition of the Bacillus subtilis aerobic respiratory
chain supercomplexes. J Bioenerg Biomembr 44, 473‐86 (2012).
Gause GF, Brazhnikova MG. Gramicidin S and its use in the treatment of infected wounds.
Nature 154, 703 (1944).
Giuliani A, Pirri G, Nicoletto SF. Antimicrobial peptides: an overview of a promising class of
therapeutics. Cent Eur J Biol 2, 1‐33 (2007).
Graham C, Richter SC, McClean S, O'Kane E, Flatt PR, Shaw C. Histamine‐releasing and
antimicrobial peptides from the skin secretions of the dusky gopher frog, Rana sevosa.
Peptides 27, 1313‐9 (2006).
Hahne H, Mäder U, Otto A, Bonn F, Steil L, Bremer E, Hecker M, Becher D. A comprehensive
proteomics and transcriptomics analysis of Bacillus subtilis salt stress adaptation. J Bacteriol
192, 870‐82 (2010).
Harris F, Dennison SR, Singh J, Phoenix DA. On the selectivity and efficacy of defense
peptides with respect to cancer cells. Med Res Rev 33, 190‐234 (2013).
Hasper HE, de Kruiff B, Breukink E. Assembly and stability of nisin‐lipid II pores. Biochemistry
43, 11567‐75 (2004).
Hassett DJ, Imlay JA. Bactericidal antibiotics and oxidative stress: a radical proposal. ACS
Chem Biol 2, 708‐10.
Hatamoto M, Ohashi A, Imachi H. Peptide nucleic acids (PNAs) antisense effect to bacterial
growth and their application potentiality in biotechnology. Appl Microbiol Biotechnol 86,
397‐402 (2010).
Haug BE, Stensen W, Kalaaji M, Rekdal O, Svendsen JS. Synthetic antimicrobial
peptidomimetics with therapeutic potential. J Med Chem 51, 4306‐14 (2008).
Haynes DH, Wiens T, Pressman BC. Turnover numbers for ionophore‐catalyzed cation
transport across the mitochondrial membrane. J Membr Biol 18, 23‐38 (1974).
Heath RJ, Rubin JR, Holland DR, Zhang E, Snow ME, Rock CO. Mechanism of triclosan
inhibition of bacterial fatty acid synthesis. J Biol Chem 274, 11110‐4 (1999).
Hecker M, Völker U. General stress response in Bacillus subtilis and other bacteria. Adv
Microb Physiol 44, 35‐91 (2001).
Helmann JD. The extracytoplasmic function (ECF) sigma factors. Adv Microb Physiol 46, 47‐
110 (2002).
N References
319

Henry G, Deleu M, Jourdan E, Thonart P, Ongena M. The bacterial lipopeptide surfactin
targets the lipid fraction of the plant plasma membrane to trigger immune‐related defence
responses. Cell Microbiol 13, 1824‐37 (2011).
Hillard E, Vessières A, Thouin L, Jaouen G, Amatore C. Ferrocene‐mediated proton‐coupled
electron transfer in a series of ferrocifen‐type breast‐cancer drug candidates. Angew Chem
Int Ed Engl 45, 285‐90 (2005).
Hilvo M, Denkert C, Lehtinen L, Müller B, Brockmöller S, Seppänen‐Laakso T, Budczies J,
Bucher E, Yetukuri L, Castillo S, Berg E, Nygren H, Sysi‐Aho M, Griffin JL, Fiehn O, Loibl S,
Richter‐Ehrenstein C, Radke C, Hyötyläinen T, Kallioniemi O, Iljin K, Oresic M. Novel
theranostic opportunities offered by characterization of altered membrane lipid metabolism
in breast cancer progression. Cancer Res 71, 3236‐45 (2011).
Hoffmann T, von Blohn C, Stanek A, Moses S, Barzantny H, Bremer E. Synthesis, release, and
recapture of compatible solute proline by osmotically stressed Bacillus subtilis cells. Appl
Environ Microbiol 78, 5753‐62 (2012).
Hoffmann T, Boiangu C, Moses C, Bremer S. Responses of Bacillus subtilis to hypotonic
challenges: physiological contributions of mechanosensitive channels to cellular survival.
Appl Environ Microbiol 74, 2454‐60 (2008).
Hotchkiss RD, Dubos RJ. Fractionation of the bactericidal agent from cultures of a soil
bacillus. J Biol Chem 132, 791‐2 (1940a).
Hotchkiss RD, Dubos RJ. Chemical properties of bactericidal substances isolated from
cultures of a soil bacillus. J Biol Chem 132, 793‐4 (1940b).
Hu Y, Chen L, Ha S, Gross B, Falcone B, Walker D, Mokhtarzadeh M, Walker S. Crystal
structure of the MurG:UDP‐GlcNAc complex reveals common structural principles of a
superfamily of glycosyltransferases. Proc Natl Acad Sci USA 100, 845‐9 (2003).
Huang Y, Huang J, Chen Y. Alpha‐helical cationic antimicrobial peptides: relationships of
structure and function. Protein Cell 1, 143‐52 (2010).
Huang W, Zhang Z, Han X, Wang J, Tang J, Dong S, Wang E. Concentration‐dependent
behavior of nisin interaction with supported bilayer lipid membrane. Biophys Chem 99, 271‐
9 (2002).
Hunter HN, Jing W, Schibli DJ, Trinh T, Park IY, Kim SC, Vogel HJ. The interactions of
antimicrobial peptides derived from lysozyme with model membrane systems. Biochim
Biophys Acta 1668: 175‐89 (2005).
Imanaka T Oshihara W, Himeno T, Aiba S. Comparative studies on extracellular penicillinase
of the same structural gene, penP, expressed in Bacillus licheniformis and Bacillus subtilis. J
Gen Microbiol 129, 2621‐8 (1983).
N References
320

Johnson BA, Anker H, Meleney FL. Bacitracin: a new antibiotic produced by a member of the
B. subtilis group. Science 102, 376‐7 (1945).
Jordan S, Junker A, Helmann JD, Mascher T. Regulation of LiaRS‐Dependent Gene Expression
in Bacillus subtilis: Identification of inhibitor proteins, regulator binding sites, and target
genes of a conserved cell envelope stress‐sensisng two‐component system J Bacteriol 188,
5153‐66 (2006).
Joshi S, Bisht GS, Rawat DS, Kumar A, Kumar R, Maiti S, Pasha S. Interaction studies of novel
cell selective antimicrobial peptides with model membranes and E. coli ATCC 11775. Biochim
Biophys Acta 1798, 1864‐75 (2010).
Jutila A, Rytömaa M, Kinnunen PK. Detachment of cytochrome c by cationic drugs from
membranes containing acidic phospholipids: comparison of lidocaine, propranolol, and
gentamycin. Mol Pharmacol 54, 722‐32 (1998).
Kaprel'iants AS, Nikiforov VV, Miroshnikov AI, Snezhkova LG, Eremin VA, Ostrovskiĭ DN.
Membranes of bacteria and mechanism of action of the antibiotic gramicidin S. Biokhimiia
42, 329‐37 (1977).
Katritzky AR, Narindoshvili T. Fluorescent amino acids: advances in protein‐extrinsic
fluorophores. Org Biomol Chem 7, 627‐34 (2009).
Katsu T, Kuroko M, Morikawa T, Sanchika K, Fujita Y, Yamamura H, Uda M. Mechanism of
membrane damage induced by the amphipathic peptides gramicidin S and melittin. Biochim
Biophys Acta 983, 135‐41 (1989).
Kellner R, Jung G, Hörner T, Zähner H, Schnell N, Entian KD, Götz F. Gallidermin: a new
lanthionine‐containing polypeptide antibiotic. Eur J Biochem 177, 53‐9 (1988).
Keren I, Wu Y, Inocencio J, Mulcahy LR, Lewis K. Killing by bactericidal antibiotics does not
depend on reactive oxygen species. Science 339, 1213‐6 (2013).
Kim SH, Shon HK, Ngo HH. Adsorption characteristics of antibiotics trimethoprim on
powdered granular activated carbon. J Ind Eng Chem 16, 344‐9 (2007).
Kingston AW, Subramanian C, Rock CO, Helmann JD. A σW‐dependent stress response in
Bacillus subtilis that reduces membrane fluidity. Mol Microbiol 81, 69‐79 (2011).
Kobayashi R, Suzuki T, Yoshida M. Escherichia coli phage‐shock protein A (PspA) binds to
membrane phospholipids and repairs proton leakage of the damaged membranes. Mol
Microbiol 66, 100‐9 (2007).
Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. A common mechanism of cellular
death induced by bactericidal antibiotics. Cell 130, 797‐810.
N References
321

Kroteń MA, Bartoszewicz M, Swiecicka I. Cereulide and valinomycin, two important natural
dodecadepsipeptides with ionophoretic activities. Pol J Microbiol 59, 3‐10 (2010).
Lambert MP, Neuhaus FC. Mechanism of D.cycloserine action: alanine racemase from
Escherichia coli W. J Bacteriol 109, 1156‐1161 (1972).
Lee YH, Kingston AW, Helmann JD. Glutamate dehydrogenase affects resistance to cell wall
antibiotics in Bacillus subtilis. J Bacteriol 194, 993‐1001 (2012).
Leontiadou H, Mark AE, Marrink S J. Antimicrobial peptides in action. J Am Chem Soc 128,
12156–61 (2006).
Liskamp RM, Rijkers DT, Kruijtzer JA, Kemmink J. Peptides and proteins as a continuing
exciting source of inspiration for peptidomimetics. Chembiochem 12, 1626‐53 (2011).
Liu X, Tao X, Zou A, Yang S, Zhang L, Mu B. Effect of the microbial lipopeptide on tumor cell
lines: apoptosis induced by disturbing the fatty acid composition of cell membrane. Protein
Cell 1, 584‐94 (2010).
Liu Y, Imlay JA. Cell death from antibiotics without the involvement of reactive oxygen
species. Science 339, 1210‐3 (2013).
Lodish H, Berk A, Zipursky SL, Matsudaira P, Darnell J. Molecular cell biology (4. Auflage), Kap
16.2. WH Freeman, New York. (2000).
López D, Kolter R. Functional microdomains in bacterial membranes. Genes Dev 24, 1893‐
902 (2012).
López CS, Heras H, Ruzal SM, Sánchez‐Rivas C, Rivas EA. Variations of the envelope
composition of Bacillus subtilis during growth in hyperosmotic medium. Curr Microbiol 36,
55‐61 (1998).
Lütticken R, Kunstmann G. Vancomycin‐resistant Streptococcaceae from clinical material.
Zentralbl Bakteriol Mikrobiol Hyg A 267, 379‐82 (1988).
MacDonald JC. Biosynthesis of valinomycin. Can J Microbiol 6, 27‐34 (1960).
Madigan MT, Martinenko JM. Brock Mikrobiologie (11. Auflage), Addison‐Wesley Verlag,
München. pp 81‐87 (2006).
Martens E, Demain AL. Platensimycin and platencin: promising antibiotics for future
application in human medicine. J Antibiot (Tokyo) 64, 705‐10 (2011).
Mascher T, Zimmer SL, Smith TA, Helmann JD. Antibiotic‐inducible promoter regulated by
the cell envelope stress‐sensing two‐component system LiaRS of Bacillus subtilis. Antimicrob
Agents Chemother 48, 2888‐96 (2004).
N References
322

Matsumae A, Kamio Y, Hata T. Studies on cerulenin. I. Studies on cerulenin producing strain.
J Antibiot (Tokyo) 16, 236‐8 (1963).
Matsumoto K, Kusaka J, Nishibori A, Hara H. Lipid domains in bacterial membranes. Mol
Microbiol 61, 1110‐7 (2006).
Mattik AT, Hirsch A. Further observations on an inhibitory substance (nisin) from lactic
streptococci. Lancet 2, 5‐8 (1947).
Meister B, Lemaitre B, Hoffmann JA. Antimicrobial peptide defense in Drosophila. Bioessays
19, 1019‐26 (1997).
Melo MN, Ferre R, Castanho MARB. Antimicrobial peptides: linking partition, activity and
high membrane‐bound concentrations. Nature Rev Microbiol 7, 245‐50 (2009).
Miteva M, Andersson M, Karshikoff A, Otting G. Molecular electroporation: a unifying
concept for the description of membrane pore formation by antibacterial peptides,
exemplified with NK‐lysin. FEBS Lett 462, 155‐158 (1999).
Moellering RC Jr. Vancomycin: a 50‐year reassessment. Clin Infect Dis 42 Suppl 1, S3‐4
(2006).
Mogi T, Ui H, Shiomi K, Omura S, Kita K. Gramicidin S identified as a potent inhibitor for
cytochrome bd‐type quinol oxidase. FEBS Lett 582, 2299‐302 (2008).
Mols M, Abee T. Primary and secondary oxidative stress in Bacillus. Environ Microbiol 13,
1387‐94 (2011).
Muchová K, Wilkinson AJ, Barák I. Changes of lipid domains in Bacillus subtilis cells with
disrupted cell wall peptidoglycan. FEMS Microbiol Lett 325, 92‐8 (2011).
Nguyen LT, Chau JK, Perry NA, de Boer L, Zaat SA, Vogel HJ. Serum stabilities of short
tryptophan‐ and arginine‐rich antimicrobial peptide analogs. PLoS One 5, e12684 (2010).
Nicolas P. Multifunctional host defense peptides: intracellular‐targeting antimicrobial
peptides. FEBS J 276, 6483‐96 (2009).
Oleson FB, Berman CL, Li AP. An evaluation of the P450 inhibition and induction potential of
daptomycin in primary human hepatocytes. Chem Biol Interact 150, 137‐47 (2004).
Parks WM, Bottrill AR, Pierrat OA, Durrant MC, Maxwell A. The action of the bacterial toxin,
microcin B17, on DNA gyrase. Biochimie 89, 500‐7 (2007).
Panzner MJ, Hindi KM, Wright BD, Taylor JB, Han DS, Youngs WJ, Cannon CL. A theobromine
derived silver N‐heterocyclic carbine: synthesis, characterization, and antimicrobial efficacy
studies on cystic fibrosis relevant pathogens. Dalton Trans 21, 7308‐13 (2009).
N References
323

Parsons JB, Frank MW, Subramanian C, Saenkham P, Rock CO. Metabolic basis for the
differential susceptibility of Gram‐positive pathogens to fatty acid synthesis inhibitors. Proc
Natl Acad Sci USA 108, 15378‐83 (2011).
Patra M, Gasser G, Bobukhov D, Merz K, Shtemenko AV, Metzler‐Nolte N. Sequential
insertion of three different organometallics into a versatile building block containing a PNA
backbone. Dalton Trans 39, 5617‐9 (2010a).
Patra M, Gasser G, Metzler‐Nolte N. Small organometallic compounds as antibacterial
agents. Dalton Trans 41, 6350‐8 (2012a).
Patra M, Gasser G, Pinto A, Merz K, Ott I, Bandow JE, Metzler‐Nolte N. Synthesis and
biological evaluation of chromium bioorganometallics based on the antibiotic platensimycin
lead structure. ChemMedChem 4, 1930‐8 (2009).
Peng K, Kong Y, Zhai L, Wu X, Jia P, Liu J, Yu H. Two novel antimicrobial peptides from
centipede venoms. Toxicon 55, 274‐9 (2010).
Penkova M. Arginine and tryptophan rich antimicrobial peptides (AMPs): modifications,
application and mode of action. PhD thesis. Ruhr‐Universität Bochum (2010).
Peschel A, Sahl HG. The co‐evolution of host cationic antimicrobial peptides and microbial
resistance. Nat Rev Microbiol 4, 529‐36 (2006).
Petersohn A, Brigulla M, Haas S, Hoheisel JD, Völker U, Hecker M. Global analysis of the
general stress response of Bacillus subtilis. J Bacteriol 183, 5617‐31 (2001).
Pichoff S, Lutkenhaus J. Tethering the Z ring to the membrane through a conserved
membrane targeting sequence in FtsA. Mol Microbiol 55, 1722‐34 (2005).
Plesch E, Bracher F, Krauss J. Synthesis and antimicrobial evaluation of novel platensimycin
analogues. Arch Pharm (Weinheim) 345, 657‐62 (2012).
Pkorny A, Almeida PFF. Kinetics of dye efflux and lipid flip‐flop induced by δ‐lysin in
phosphatidylcholine vesicles and the mechanism of graded release by amphipathic, α‐helical
peptides. Biochemistry 43, 8846‐57 (2004).
Pkorny, A, Birkbeck TH, Almeida PFF. Mechanism and kinetics of δ‐lysin interaction with
phospholipid vesicles. Biochemistry 41, 11044‐56 (2002).
Pogliano J, Pogliano N, Silverman JA. Daptomycin‐mediated reorganization of membrane
architecture causes mislocalization of essential cell division proteins. J Bacteriol 194, 4494‐
504 (2012).
Polk RE. Anaphylactoid reactions to glycopeptide antibiotics. J Antimicrob Chermother, Suppl
B, 17‐29 (1991).
N References
324

Price AC, Choi KH, Heath RJ, Li Z, White SW, Rock CO. Inhibition of beta‐ketoacyl‐acyl carrier
protein synthases by thiolactomycin and cerulenin. Structure and mechanism. J Biol Chem
276, 6551‐9 (2001).
Raatschen N, Wenzel M, Leichert LI, Düchting P, Krämer U, Bandow JE. Extracting iron and
manganese from bacteria with ionophores ‐ a mechanism against competitors characterized
by increased potency in environments low in micronutrients. Proteomics, doi:
10.1002/pmic.201200556.
Raatschen N, Bandow JE. 2‐D gel‐based proteomic approaches to antibiotic drug discovery.
Curr Protoc Microbiol Chapter 1:Unit1F.2 (2012).
Rabenau A. Auswirkungen von Zellteilungshemmung auf Morphologie und Proteom von
Bacillus subtilis. Diploma thesis, Ruhr‐Universität Bochum (2010).
Raguse TL, Porter EA, Weisblum B, Gellman SH. Structure‐activity studies of 14‐helical
antimicrobial beta‐peptides: probing the relationship between conformational stability and
antimicrobial potency. J Am Chem Soc 124, 12774‐85 (2002).
Rathinakumar R, Walkenhorst WF, Wimley WC. Broad‐spectrum antimicrobial peptides by
rational combinatory design and high‐throughput screening: the importance of interfacial
activity. J Am Chem Soc 131, 7609‐17 (2009).
Rathinakumar R, Wimley WC. Biomolecular engineering by combinatorial design and high‐
throughput screening: small, soluble peptides that permeabilize membranes. J Am Chem Soc
130, 9849‐58 (2008).
Reisinger P, Seidel H, Tschesche H, Hammes WP. The effect of nisin on murein synthesis.
Arch Microbiol 127, 187‐93 (1980).
Rose WE, Fallon M, Moran JJ, Vanderloo JP. Vancomycin tolerance in methicillin‐resistant
Staphylococcus aureus: influence of vancomycin, daptomycin, and telavancin on differential
resistance gene expression. Antimicrob Agents Chemother 56: 4422‐7 (2012).
Rozek T, Wegener KL, Bowie JH, Olver IN, Carver JA, Wallace JC, Tyler MJ. The antibiotic and
anticancer active aurein peptides from the Australian Bell Frogs Litoria aurea and Litoria
raniformis the solution structure of aurein 1.2. Eur J Biochem 267, 5330‐41 (2000).
Rupley JA. The binding and cleavage by lysozyme of N‐acetylglucosamine oligosaccharides.
Proc Roy Soc B167, 416‐428 (1967).
Šajbidor J. Effect of some environmental factors on the content and compositions of
microbial membrane lipids. Crit Rev Biotechnol 17, 87‐103 (1997).
Samel SA, Marahiel MA, Essen LO. How to tailor non‐ribosomal peptide products ‐ new clues
about the structures and mechanisms of modifying enzymes. Mol Biosyst 4, 387‐93 (2008).
N References
325

Sato H, Feix JB. Peptide‐membrane interactions and mechanisms of membrane destruction
by amphipathic α‐helical antimicrobial peptides. Biochim Biophys Acta 1758, 1245‐56 (2006).
Scheffers DJ. The cell wall of Bacillus subtilis. in: Bacillus: cellular and molecular biology (2nd
Edition), Caister Academic Press, Norfolk. pp 285‐318 (2012).
Schellenberger V, Turck C, Rutter WJ. Role of the S' subsites in serine protease catalysis.
Active‐site mapping of rat chymotrypsin, rat trypsin, alpha‐lytic protease, and cercarial
protease from Schistosoma mansoni. Biochemistry 33, 4251‐7 (1994).
Schneider T, Sahl HG. An oldie but a goodie ‐ cell wall biosynthesis as antibiotic target
pathway. Int J Med Microbiol 300, 161‐9 (2010).
Schneider T, Kruse T, Wimmer R, Wiedemann I, Sass V, Pag U, Jansen A, Nielsen AK, Mygind
PH, Raventós DS, Neve S, Ravn B, Bonvin AM, De Maria L, Andersen AS, Gammelgaard LK,
Sahl HG, Kristensen HH. Plectasin, a fungal defensin targets the bacterial cell wall precursor
lipid II. Science 328, 1168‐72 (2010).
Schneider T, Gries K, Josten M, Wiedemann I, Pelzer S, Labischinski H, Sahl HG. The
lipopeptide friulimicin B inhibits cell wall biosynthesis by complex formation with
bactoprenol phosphate. Antimicrob Agents Chemother 53, 1610‐8 (2009).
Semrau S, Monster MW, van der Knaap M, Florea BI, Schmidt T, Overhand M. Membrane
lysis by gramicidin S visualized in red blood cells and giant vesicles. Biochim Biophys Acta
1798, 2033‐9 (2010).
Seydlová G, Svobodová J. Development of membrane lipids in the surfactin producer Bacillus
subtilis. Folia Microbiol (Praha) 53, 303‐7 (2008).
Sharma RK, Sundriyal S, Wangoo N, Tegge W, Jain R. New antimicrobial hexapeptides:
synthesis, antimicrobial activities, cytotoxicity, and mechanistic studies. ChemMedChem 5,
86‐95 (2010).
Shen Y, Liu Y, Estiu G, Isin B, Ahn YY, Lee DS, Barabási AL, Kapatral V, Wiest O, Oltvai ZN.
Blueprint for antimicrobial hit discovery targeting metabolic networks. Proc Natl Acad Sci
USA 107, 1082‐7.
Smith TL, Pearson ML, Wilcox KR, Cruz C, Lancaster MV, Robinson‐Dunn B, Tenover FC,
Zervos MJ, Band JD, White E, Jarvis WR. Emergence of vancomycin resistance in
Staphylococcus aureus. Glycopeptide‐Intermediate Staphylococcus aureus Working Group. N
Engl J Med 340, 493‐501 (1999).
Steiner H, Andreu D, Merrifield RB. Binding and action of cecropin and cecropin analogues:
antibacterial peptides from insects. Biochim Biophys Acta 939, 260‐6 (1988).
Steinsträßer L, Kraneburg U, Jacobsen F, Al‐Benna S. Host defense peptides and their
antimicrobial‐immunomodulatory duality. Immunobiology 216, 322‐33 (2011).
N References
326

Strøm MB, Haug BE, Skar ML, Stensen W, Stiberg T, Stiberg T, Svendsen JS. The
pharmacophore of short cationic antibacterial peptides. J Med Chem 46, 1567‐70 (2003).
Sukharev S. Purification of the small mechanosensitive channel of Escherichia coli (MscS):
the subunit structure, conduction, and gating characteristics in liposomes. Biophys J 83, 290‐
8 (2002).
Szeto TH, Rowland SL, Habrukowich CL, King GF. The MinD membrane targeting sequence is
a transplantable lipid‐binding helix. J Biol Chem 278, 40050‐6 (2003).
Tan YT, Tillet DJ, McKay IA. Molecular strategies for overcoming antibiotic resistance in
bacteria. Mol Med Today 6, 309‐14 (2000).
Teixeira V, Feio MJ, Bastos M. Role of membrane lipids in the interaction of antimicrobial
peptides with membranes. Prog Lipid Res 51, 149‐77 (2012).
VanBogelen RA, Schiller EE, Thomas JD, Neidhardt FC. Diagnosis of cellular states of microbial
organisms using proteomics. Electrophoresis 20, 2149‐59 (1999).
von der Ohe PC, Schmitt‐Jansen M, Slobodnik J, Brack W. Triclosan ‐ the forgotten priority
substance? Environ Sci Pollut Res Int 19, 585‐91 (2012).
van der Oost J, von Wachenfeld C, Hederstedt L, Saraste M. Bacillus subtilis cytochrome
oxidase mutants: biochemical analysis and genetic evidence for two aa3‐type oxidases. Mol
Microbiol 5, 2063‐72 (1991).
van Heusden HE, de Kruijff B, Breukink E. Lipid II induces a transmembrane orientation of the
pore‐forming peptide lantibiotic nisin. Biochemistry 41, 12171‐8 (2002).
van‘t Hof W, Veerman EC, Helmerhorst EJ, Amerongen AV. Antimicrobial peptides:
properties and applicability. Biol Chem 382, 597–619 (2001).
Vértesy L, Ehlers E, Kogler H, Kurz M, Meiwes J, Seibert G, Vogel M, Hammann P. Friulimicins:
novel lipopeptide antibiotics with peptidoglycan synthesis inhibiting activity from
Actinoplanes friuliensis sp. nov. II. Isolation and structural characterization. J Antibiot (Tokyo)
53, 816‐27 (2000).
Vicario PP, Green BG, Katzen HM. A single assay for simultaneously testing effectors of
alanine racemase and/or d‐alanine:D‐alanine ligase. J Antibiot (Tokyo) 40, 209‐16 (1987).
Vischer WA, Regös J. Antimicrobial spectrum of triclosan, a broad‐spectrum antimicrobial
agent for topical application. Zentralbl Bakteriol Orig A 226, 376‐89 (1974).
von Wachenfeldt C, Hederstedt L. Bacillus subtilis 13‐kilodalton cytochrome c‐550 encoded
by cccA consists of a membrane‐anchor and a heme domain. J Biol Chem 265, 13939‐48
(1990).
N References
327

Vostroknutova GN, Bulgakova VG, Udalova TP, Sepetov NF, Sibel'dina LA. Localization of
gramicidin S on the cytoplasmic membrane of Bacillus brevis and its effect on the activity of
membrane enzymes. Biokhimiia 46, 657‐66 (1981).
Vrontou E, Economou A. Structure and function of SecA, the preprotein translocase
nanomotor. Biochim Biophys Acta 1694, 67‐80 (2004).
Wang J, Kodali S, Lee SH, Galgoci A, Painter R, Dorso K, Racine F, Motyl M, Hernandez L,
Tinney E, Colletti SL, Herath K, Cummings R, Salazar O, González I, Basilio A, Vicente F,
Genilloud O, Pelaez F, Jayasuriya H, Young K, Cully DF, Singh SB. Discovery of platencin, a
dual FabF and FabH inhibitor with in vivo antibiotic properties. Proc Natl Acad Sci U S A 104,
7612‐6 (2007).
Wang J, Soisson SM, Young K, Shoop W, Kodali S, Galgoci A, Painter R, Parthasarathy G, Tang
YS, Cummings R, Ha S, Dorso K, Motyl M, Jayasuriya H, Ondeyka J, Herath K, Zhang C,
Hernandez L, Allocco J, Basilio A, Tormo JR, Genilloud O, Vicente F, Pelaez F, Colwell L, Lee
SH, Michael B, Felcetto T, Gill C, Silver LL, Hermes JD, Bartizal K, Barrett J, Schmatz D, Becker
JW, Cully D, Singh SB. Platensimycin is a selective FabF inhibitor with potent antibiotic
properties. Nature 441, 358‐61 (2006).
Waksman SA. What is an antibiotic or an antibiotic substance? Mycologia 39, 565‐9 (1947).
Warth AD, Strominger JL. Structure of the peptidoglycan from vegetative cell walls of Bacillus
subtilis. Biochemistry 10, 4349‐58 (1971).
Waters AE, Contente‐Cuomo T, Buchhagen J, Liu CM, Watson L, Pearce K, Foster JT, Bowers
J, Driebe EM, Engelthaler DM, Keim PS, Price LB. Multidrug‐resistant Staphylococcus aureus
in US meat and poultry. Clin Infect Dis 52, 1227‐30 (2011).
Wecke T, Zühlke D, Mäder U, Jordan S, Voigt B, Pelzer S, Labischinski H, Homuth G, Hecker
M, Mascher T. Daptomycin versus friulimicin B: in‐depth profiling of Bacillus subtilis cell
envelope stress responses. Antimicrob Agents Chemother 53, 1619‐23 (2009).
Weidenmaier C, Peschel A. Teichoic acids and related cell‐wall glycopolymers in Gram‐
positive physiology and host interactions. Nat Rev Microbiol 6, 276‐87 (2008).
Wenzel M, Schriek P, Prochnow P, Albada HB, Metzler‐Nolte N, Bandow JE. Influence of
lipidation on the mechanism of action of an RW‐rich antimicrobial peptide. in preparation.
Wenzel M, Patra M, Senges CHR, Ott I, Stepanek JJ, Pinto A, Prochnow P, Vuong C, Langklotz
S, Metzler‐Nolte N, Bandow JE. Analysis pf the mechanism of action of potent antibacterial
hetero‐tri‐organometallic compounds ‐ a structurally new class of antibiotics. ACS Chem Biol.
doi: 10.1021/cb4000844.
Wenzel M, Kohl B, Münch D, Raatschen N, Albada HB, Hamoen L, Metzler‐Nolte N, Sahl HG,
Bandow JE. Proteomic response of Bacillus subtilis to lantibiotics reflects differences in
N References
328

interaction with the cytoplasmic membrane. Antimicrob Agents Chemother 56, 5749‐57
(2012).
Wenzel M, Bandow JE. Proteomic signatures in antibiotic research. Proteomics 11, 3256‐68
(2011).
Wiener MC, White SH. Structure of a fluid doileoylphosphatidylcholine bilayer determined
by joint refinement of x‐ray and neutron diffraction data. III. Complete structure. Biophys J
61, 434‐47 (1992).
Wilmes M, Cammue BP, Sahl HG, Thevissen K. Antibiotic activities of host defense peptides:
more to it than lipid bilayer perturbation. Nat Prod Rep 28, 1350‐8 (2011).
Wimley WC. Describing the mechanism of action of antimicrobial peptide action with the
interfacial activity model. ACS Chem Biol 5, 905‐17 (2010).
Winter T, Winter J, Polak M, Kusch K, Mäder U, Sietmann R, Ehlbeck J, van Hijum S,
Weltmann KD, Hecker M, Kusch H. Characterization of the global impact of low temperature
gas plasma on vegetative microorganisms. Proteomics 11, 3518‐30 (2011).
Wolf D, Kalamorz F, Wecke T, Juszczak A, Mäder U, Homuth G, Jordan S, Kirstein J, Hoppert
M, Voigt B, Hecker M, Mascher T. In‐depth profiling of the LiaR response of Bacillus subtilis. J
Bacteriol 192, 4680‐93 (2010).
Yamamoto T, Yamada A, Yoshimura Y, Terada H, Shinohara Y. The mechanisms of the release
of cytochrome c from mitochondria revealed by proteomics analysis. Yakugaku Zasshi 132,
1099‐104 (2012).
Yeaman MR, Yount NY. Mechanisms of antimicrobial peptide action and resistance.
Pharmacol Rev 55, 27‐55 (2003).
Yu J, Le Brun NE. Studies of the cytochrome subunits of menaquinone:cytochrome c
reductase (bc complex) of Bacillus subtilis. Evidence for the covalent attachment of heme to
the cytochrome b subunit. J Biol Chem 273, 8860‐6 (1998).
Zlitni S, Brown ED. Drug discovery: Not as fab as we thought. Nature 458, 39‐40 (2009).
Zweytick D, Tumer S, Blondelle SE, Lohner K. Membrane curvature stress and antibacterial
activity of lactoferricin derivatives. Biochem Biophys Res Commun 369, 395‐400 (2008).
Zweytick D, Deutsch G, Andrä J, Blondelle SE, Vollmer E, Jerala R, Lohner K. Studies on
lactoferricin‐derived Escherichia coli membrane‐active peptides reveal differences in the
mechanism of N‐acylated versus nonacylated peptides. J Biol Chem 286, 21266‐76 (2011).
N References
329

O Publications
Research articles
Wenzel M, Chiriac AI, Otto A, Zweytick D, May C, Schumacher C, Albada HB, Penkova
M, Krämer U, Erdmann R, Metzler‐Nolte N, Brötz‐Oesterhelt H, Becher D, Sahl HG,
Bandow JE. An antimicrobial peptide delocalizes peripheral membrane proteins.
submitted.
Wenzel M*, Patra M*, Senges CHR, Ott I, Stepanek JJ, Pinto A, Prochnow P, Vuong C,
Langklotz S, Metzler‐Nolte N, Bandow JE. Analysis of the mechanism of action of
potent antibacterial hetero‐tri‐organometallic compounds ‐ a structurally new class
of antibiotics. ACS Chem Biol. doi: 10.1021/cb4000844.
Raatschen N, Wenzel M, Düchting P, Leichert LI, Krämer U, Bandow JE. Extracting iron
and manganese from bacteria with ionophores – a mechanism against competitors
characterized by increased potency in environments low in micronutrients.
Proteomics. doi: 10.1002/pmic.201200556.
Albada HB, Chiriac AI, Wenzel M, Penkova M, Bandow JE, Sahl HG, Metzler‐Nolte N.
Modulating the activity of short arginine‐tryptophan containing antibacterial
peptides with N‐terminal metallocenoyl groups. Beilstein J Org Chem 8, 1753‐64
(2012).
Wenzel M, Kohl B, Münch D, Raatschen N, Albada HB, Hamoen L, Metzler‐Nolte N, H
Sahl HG, Bandow JE. Proteomic response of Bacillus subtilis to lantibiotics reflects
differences in interaction with the cytoplasmic membrane. Antimicrob Agents
Chemother 56, 5749‐57 (2012).
Patra M, Gasser G, Wenzel M, Merz K, Bandow JE, Metzler‐Nolte N. Sandwich and
half‐sandwich derivatives of platensimycin: Synthesis and biological evaluation.
Organometallics 31, 5760‐71 (2012).
Patra M, Gasser G, Wenzel M, Merz K, Bandow JE, Metzler‐Nolte N. Synthesis of
optically active ferrocene‐containing platensimycin derivatives with a C6‐C7
substitution pattern. Eur J Inorg Chem 22, 3295‐302 (2011).
Wenzel M, Patra M, Albrecht D, Chen DYK, Nicolaou KC, Metzler‐Nolte N, Bandow JE.
Proteomic signature of fatty acid biosynthesis inhibition available for in vivo
mechanism of action studies. Antimicrob Agents Chemother 55, 2590‐6 (2011).
* equally contributed
O Publications
330

Patra M, Gasser G, Wenzel M, Merz K, Bandow JE, Metzler‐Nolte N. Synthesis and
biological evaluation of ferrocene‐containing bioorganometallics inspired by the
platensimycin lead structure. Organometallics 29, 4312‐9 (2010).
Review articles
Wenzel M, Bandow JE. Proteomic signatures in antibiotic research. Proteomics. 11,
3256‐68 (2011).
Conference contributions
Talks
Wenzel M*, Chiriac AI, Otto A, Zweytick D, May C, Schumacher C, Albada HB, Penkova
M, Krämer U, Erdmann R, Metzler‐Nolte N, Brötz‐Oesterhelt H, Becher D, Sahl HG,
Bandow JE. Short cationic antimicrobial peptide RWRWRW‐NH2 inhibits cell wall
biosynthesis and bacterial respiration by delocalizing essential membrane proteins.
2nd Innovative Antibiotics from NRW Progress Meeting, Bochum, Germany. January
30th, 2013.
Wenzel M. Hunting the Red Queen – Proteomic Profiling in der Entwicklung neuer
Antibiotika. 19th Arbeitstagung Micromethods in Protein Chemistry. Bochum,
Germany. June 25th‐27th, 2012.
Wenzel M, Chiriac AI, Albada HB, Knüfer A, Hamoen L, Sahl HG, Metzler‐Nolte N,
Bandow JE. Antimicrobial hexapeptide MP196 disturbs membrane integrity without
pore formation. 3rd International Symposium on Antimicrobial Peptides, Lille, France.
June 13th‐15th, 2012.
Wenzel M, Chiriac AI, Albada HB, Otto A, Knüfer A, Becher D, Hamoen L, Sahl HG,
Metzler‐Nolte N, Bandow JE. The cell envelope as target of a novel antimicrobial
peptide. Annual Conference of the Association for General and Applied Microbiology
(VAAM), Tübingen, Germany. March 18th‐21st, 2012.
Wenzel M, Chiriac AI, Otto A, Gust R, May C, Sahl HG, Bandow JE. Antibiotic
mechanism of a small cationic hexapeptide. 1st Innovative Antibiotics from NRW
Progress Meeting, Bochum, Germany. February 8th, 2012.
Wenzel M, Bandow JE. 2D gel‐based proteomics. Innovative Antibiotics from NRW
Kickoff Meeting, Wuppertal, Germany. February 14th, 2011.
* presenting author underlined
O Publications
331

Bandow JE, Wenzel M, Raatschen N, Rabenau A, Kutscher B, Penkova M, Patra M,
Metzler‐Nolte N. Investigating antibiotic targets and mechanisms of action using
proteome analyses. 3rd joint conference of the Association for General and Applied
Microbiology (VAAM) and the German Society for Hygiene and Microbiology (DGHM),
Hannover, Germany. March 28th‐31st, 2010.
Wenzel M, Bandow JE. MP66 and derivatives: Biological activity and proteome
response of Bacillus subtilis. 1st IFSC Mini‐Symposium, Bochum, Germany. August
28th, 2009.
Posters
Wenzel M*, Chiriac AI, Otto A, Albada HB, May C, Zweytick D, Krämer U, Becher D,
Metzler‐Nolte N, Sahl HG, Bandow JE. In depth analysis of the mechanism of action of
the short RW‐rich peptide MP196. Gordon Research Conference on Antimicrobial
Peptides, Ventura, California, USA. February 23rd ‐ March 1st, 2013.
Chiriac AI, Wenzel M, Albada HB, Josten M, Krämer J, Sahl HG, Metzler‐Nolte N,
Bandow JE. Effect of synthetic short cationic antimicrobial peptide on bacterial cell
envelope. Gordon Research Conference on New Antimicrobial Discovery and
Development, Lucca, Italy. April 15th‐20th, 2012.
Wenzel M, Penkova M, Josten M, Chiriac AI, Sahl HG, Metzler‐Nolte N, Bandow JE.
Cell envelope‐targeting hexapeptide exhibits a concentration‐dependent shift in
mode of action. Gordon Research Conference on Antimicrobial Peptides, Lucca, Italy.
May 15th‐20th, 2011.
Wenzel M, Penkova M, Otto A, Becher D, Metzler‐Nolte N, Bandow JE. On a highway
to the antibiotic mechanism: proteomic investigation of small, cationic peptides.
Proteomic Forum, Berlin, Germany. April 3rd‐7th, 2011.
Kohl B, Wenzel M, Bandow JE. Comparative proteomic study of cell envelope stress
caused by lipopeptide antibiotics. Proteomic Forum, Berlin, Germany. April 3rd‐7th,
2011.
Wenzel M, Penkova M, Otto A, Becher D, Metzler‐Nolte N, Bandow JE. What can we
learn from the cytosolic and the membrane proteome about the mechanism of
action of short cationic peptides? 9th Human Proteome Organisation (HUPO) World
Congress, Sydney, Australia. September 19th‐23rd, 2010.
Wenzel M, Penkova M, Raatschen N, Metzler‐Nolte N, Bandow JE. Antibiotic
mechanism of action studies in Bacillus subtilis: Proteomic investigation of short
* presenting author underlined
O Publications
332

cationic peptides and fatty acid biosynthesis inhibitors. 3rd joint conference of the
Association for General and Applied Microbiology (VAAM) and the German Society
for Hygiene and Microbiology (DGHM),, Hannover, Germany. March 28th‐31st, 2010.
Wenzel M, Penkova M, Raatschen N, Metzler‐Nolte N, Bandow JE. Short cationic
ferrocene‐peptide bioconjugate targets the cytoplasmic membrane. 49th Interscience
Conference on Antimicrobial Agents and Chemotherapy (ICAAC), San Francisco,
California, USA. September 12th‐15th, 2009.
O Publications
333

P Appendix
1 Catalog of proteomic response profiles to antibiotics
2 Curriculum vitae
3 Contributions to the integrated publications and manuscripts
4 Erklärung
P Appendix
334

Treatment parameters
Minimal inhibitory concentration 14 µg/mL
Optimal stressor concentration 8.4 µg/mL
Bacteriolytic concentration 14 µg/mL
Bacterial response analyzed after 15 min
Aurein 2.2 (GLFDIVKKVVGALGSL‐CONH2)
Mechanistic properties
Mode of action ion transport
Molecular target phospholipids
Depolarizationa yes
Permeabilizationb no
Cell wall integrityc not disturbed
General information
Source Litoria aurea
MW 1613 g/mol
Ref. Rozek et al., 2000
control/aurein 2.2
Wen
zel andSenges, unpublished
a determined by MinD localization; b determined by acetic acid/methanol fixation; c determined by BacLight
P Appendix
335
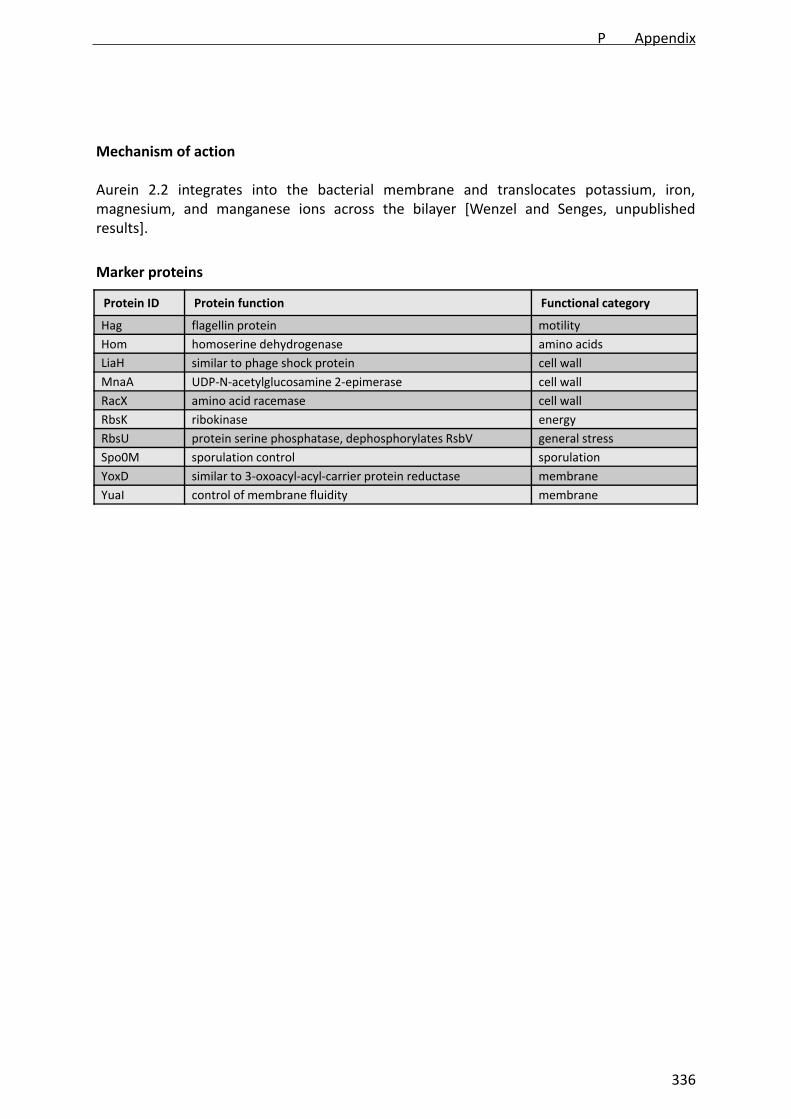
Mechanism of action
Aurein 2.2 integrates into the bacterial membrane and translocates potassium, iron,magnesium, and manganese ions across the bilayer [Wenzel and Senges, unpublishedresults].
Protein ID Protein function Functional category
Hag flagellin protein motility
Hom homoserine dehydrogenase amino acids
LiaH similar to phage shock protein cell wall
MnaA UDP‐N‐acetylglucosamine 2‐epimerase cell wall
RacX amino acid racemase cell wall
RbsK ribokinase energy
RbsU protein serine phosphatase, dephosphorylates RsbV general stress
Spo0M sporulation control sporulation
YoxD similar to 3‐oxoacyl‐acyl‐carrier protein reductase membrane
YuaI control of membrane fluidity membrane
Marker proteins
P Appendix
336

Treatment parameters
Minimal inhibitory concentration 14 µg/mL
Optimal stressor concentration 10.5 µg/mL
Bacteriolytic concentration 14 µg/mL
Bacterial response analyzed after 15 min
Aurein 2.3 (GLFDIVKKVGAIGSL‐CONH2)
Mechanistic properties
Mode of action ion transport
Molecular target phospholipids
Depolarization yes
Permeabilization no
Cell wall integrity not disturbed
General information
Source Litoria aurea
MW 1613 g/mol
Ref. Rozek et al., 2000
control/aurein 2.3
Wen
zel andSenges, unpublished
P Appendix
337

Mechanism of action
Aurein 2.3 integrates into the bacterial membrane and translocates potassium, iron,magnesium, and manganese across the bilayer [Wenzel and Senges, unpublished results].
Protein ID Protein function Functional category
AccD acetyl‐CoA carboxylase (beta subunit) membrane
Adk adenylate kinase energy
AroA3‐deoxy‐D‐arabino‐heptulosonate 7‐phosphate synthase /
chorismate mutase‐isozyme 3 energy
CotEouter spore coat morphogenetic protein, controls the
assembly of the outer spore coat layercell envelope
CysC adenylyl‐sulfate kinase sulfur metabolism
GlcR transcriptional repressor (DeoR family) energy
Hag flagellin protein motility
Hom homoserine dehydrogenase (NADPH) amino acids
LiaH similar to phage shock protein cell wall
NadE NAD synthase energy
Ndk nucleoside diphosphate kinase energy
PpaC inorganic pyrophosphatase phosphate metabolism
PspA phage shock protein A energy
RbsK ribokinase energy
RbsU protein serine phosphatase, dephosphorylates RsbV general stress
RacX amino acid racemase cell wall
Spo0M sporulation control protein sporulation
SufD synthesis of Fe‐S‐clusters iron metabolism
TrxA thioredoxin A oxidative stress
YceC similar to tellurium resistance protein cell envelope
YceH similar to toxic anion resistance protein cell envelope
YezE similar to transcriptional regulator (TetR family) unknown
YhcG similar to ABC transporter (ATP‐binding protein) transport
YkaA unknown unknown
YoxD similar to 3‐oxoacyl‐acyl‐carrier protein reductase membrane
YpuA unknown cell envelope
YpxDtwo‐component response regulator, regulation of aerobic
and anaerobic respiration energy
YqiG similar to NADH‐dependent flavin oxidoreductase energy
YuaI control of membrane fluidity membrane
YugJ similar to NADH‐dependent butanol dehydrogenase energy
YvyE unknown unknown
YxxG unknown unknown
Marker proteins
P Appendix
338

Treatment parameters
Minimal inhibitory concentration 20 µg/mL
Optimal stressor concentration 13.3 µg/mL
Bacteriolytic concentration 20 µg/mL
Bacterial response analyzed after 15 min
Aurein 2.2Δ‐3 (GLFDIVKKVVGAL‐CONH2)
Mechanistic properties
Mode of action ion transport
Molecular target phospholipids
Depolarization yes
Permeabilization no
Cell wall integrity not disturbed
General information
Source Litoria aurea
MW 1301 g/mol
Ref. Cheng et al., 2010
control/aurein 2.2Δ‐3
Wen
zel andSenges, unpublished
P Appendix
339
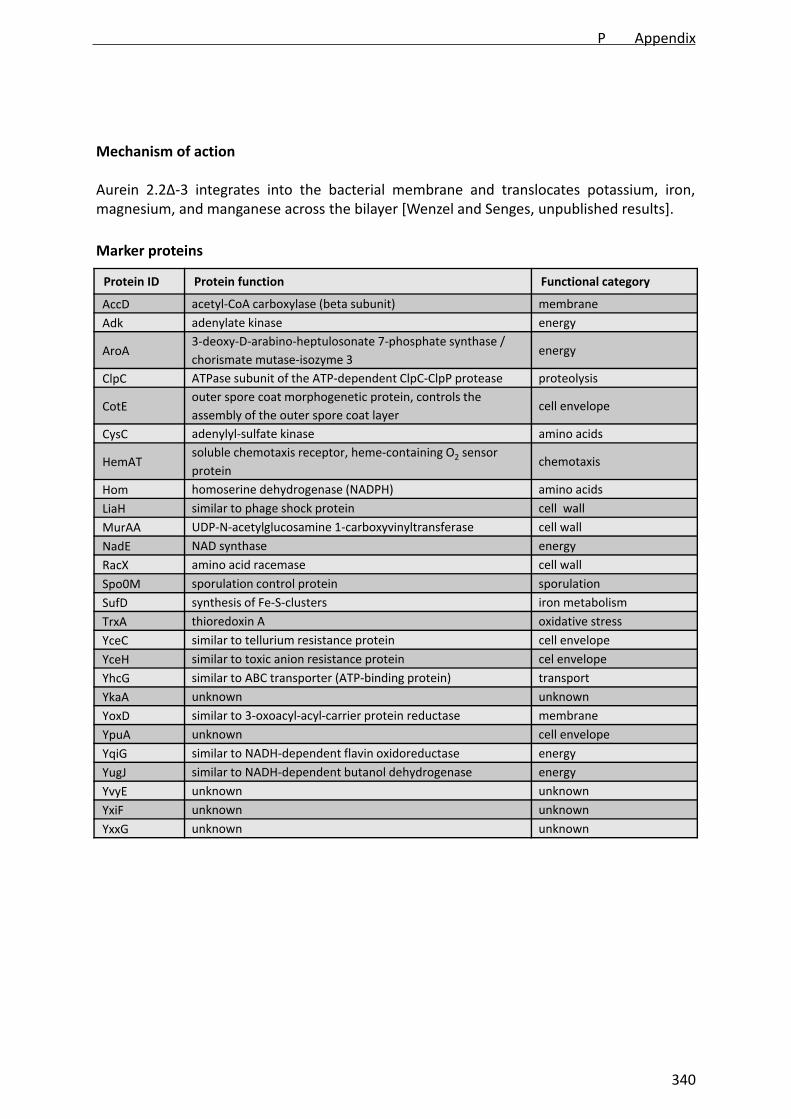
Mechanism of action
Aurein 2.2Δ‐3 integrates into the bacterial membrane and translocates potassium, iron,magnesium, and manganese across the bilayer [Wenzel and Senges, unpublished results].
Protein ID Protein function Functional category
AccD acetyl‐CoA carboxylase (beta subunit) membrane
Adk adenylate kinase energy
AroA3‐deoxy‐D‐arabino‐heptulosonate 7‐phosphate synthase /
chorismate mutase‐isozyme 3 energy
ClpC ATPase subunit of the ATP‐dependent ClpC‐ClpP protease proteolysis
CotEouter spore coat morphogenetic protein, controls the
assembly of the outer spore coat layercell envelope
CysC adenylyl‐sulfate kinase amino acids
HemATsoluble chemotaxis receptor, heme‐containing O2 sensor
protein chemotaxis
Hom homoserine dehydrogenase (NADPH) amino acids
LiaH similar to phage shock protein cell wall
MurAA UDP‐N‐acetylglucosamine 1‐carboxyvinyltransferase cell wall
NadE NAD synthase energy
RacX amino acid racemase cell wall
Spo0M sporulation control protein sporulation
SufD synthesis of Fe‐S‐clusters iron metabolism
TrxA thioredoxin A oxidative stress
YceC similar to tellurium resistance protein cell envelope
YceH similar to toxic anion resistance protein cel envelope
YhcG similar to ABC transporter (ATP‐binding protein) transport
YkaA unknown unknown
YoxD similar to 3‐oxoacyl‐acyl‐carrier protein reductase membrane
YpuA unknown cell envelope
YqiG similar to NADH‐dependent flavin oxidoreductase energy
YugJ similar to NADH‐dependent butanol dehydrogenase energy
YvyE unknown unknown
YxiF unknown unknown
YxxG unknown unknown
Marker proteins
P Appendix
340
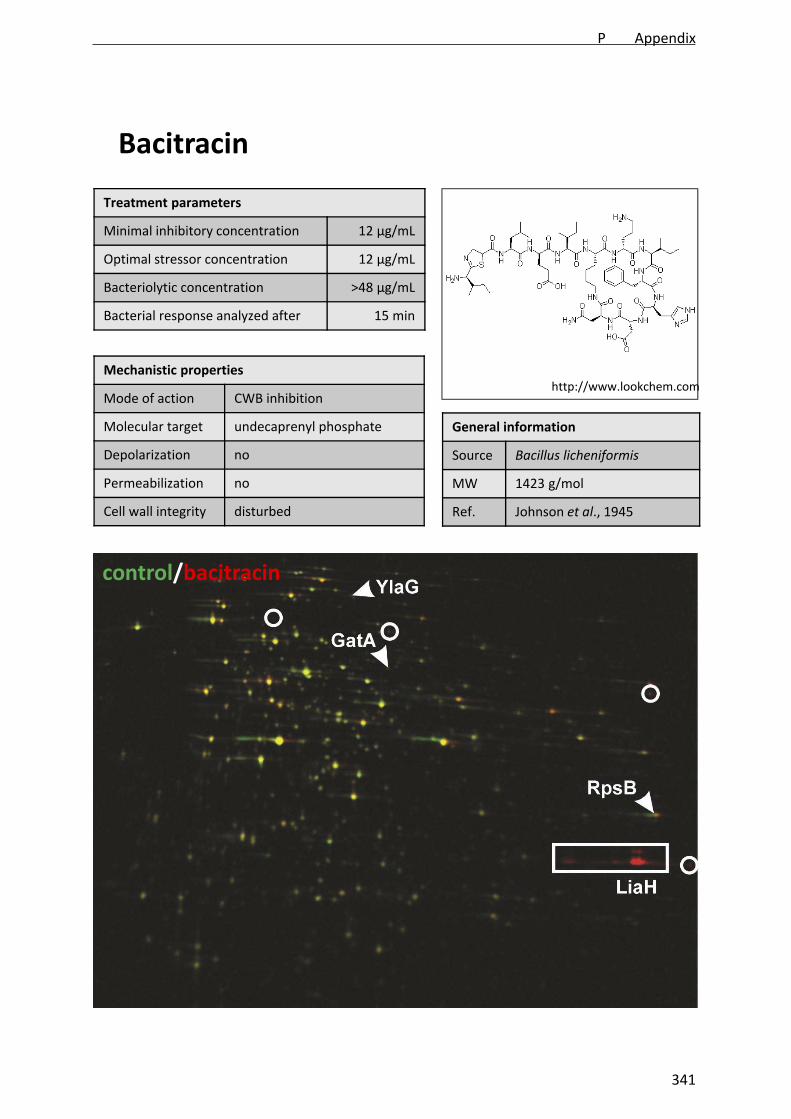
Treatment parameters
Minimal inhibitory concentration 12 µg/mL
Optimal stressor concentration 12 µg/mL
Bacteriolytic concentration >48 µg/mL
Bacterial response analyzed after 15 min
Bacitracin
Mechanistic properties
Mode of action CWB inhibition
Molecular target undecaprenyl phosphate
Depolarization no
Permeabilization no
Cell wall integrity disturbed
General information
Source Bacillus licheniformis
MW 1423 g/mol
Ref. Johnson et al., 1945
control/bacitracin
http://www.lookchem.com
P Appendix
341

Mechanism of action
Bacitracin binds to undecaprenylpyrophosphate and prevents recycling of the bactoprenolcarrier. Consequently, lipid II and WTA synthesis are inhibited [Schneider and Sahl, 2010].
Protein ID Protein function Functional category
GatA production of glutamyl‐tRNA translation
LiaH similar to phage shock protein cell wall
RpsB ribosomal protein translation
YlaG similar to GTP‐binding elongation factor translation
Marker proteins
Protein ID Protein function Functional category
DltA D‐alanyl‐D‐alanine carrier protein ligase cell wall
Hom homoserine dehydrogenase (NADPH) amino acids
IolS unknown, may be involved in myo‐inositol catabolism energy
LiaH similar to phage shock protein cell wall
PtsHHPr, general component of the sugar phosphotransferase
system (PTS). energy
Sat sulfate adenylyltransferase sulfate metabolism
YceC similar to tellurium resistance protein cell envelope
YtrB ABC transporter (ATP‐binding protein) transport
YtrE ABC transporter (ATP‐binding protein) transport
Marker proteins described by Bandow et al., 2003
P Appendix
342

Treatment parameters
Minimal inhibitory concentration 12 µg/mL
Optimal stressor concentration 15 µg/mL
Bacteriolytic concentration 50 µg/mL
Bacterial response analyzed after 15 min
C‐C0 (RWRWRWK‐NH2)
Mechanistic properties
Mode of action interfacial activity
Molecular target phospholipids
Depolarization yes
Permeabilization no
Cell wall integrity disturbed
General information
Source synthetic
MW 1176 g/mol
Ref. Albada et al., 2012a
control/C‐C0
Wenzel et al., in preparation a
P Appendix
343

Mechanism of action
C‐C0 integrates into the bacterial membrane and disturbs membrane architecture accordingto the interfacial activity model. It impairs respiration and cell wall biosynthesis [Wenzel etal., in preparation a].
Protein ID Protein function Functional category
AcsA acetyl‐CoA synthetase membrane
Bcd valine, isoleucine, leucine dehydrogenase amino acids
ClpPATP‐dependent Clp protease proteolytic subunit (class III heat‐shock protein)
proteolysis
ClpY two‐component ATP‐dependent protease, ATPase subunit proteolysis
CysC adenylyl‐sulfate kinase amino acids
FabF beta‐ketoacyl‐acyl carrier protein synthase II membrane
FosB bacillithiol‐S‐transferase cell envelope
GroEL chaperonin and co‐repressor for HrcA chaperone
IlvC ketol‐acid reductoisomerase amino acids
LiaH similar to phage shock protein A cell wall
MurB UDP‐N‐acetylenolpyruvoylglucosamine reductase cell wall
NfrASpx‐dependent FMN‐containing NADPH‐linked nitro/flavin reductase
energy
PbpE penicillin‐binding protein PBP 4 cell wall
PdhA pyruvate dehydrogenase (E1 alpha subunit) energy
PdxK pyridoxine, pyridoxal, pyridoxamine kinase energy/amino acids
PspA phage shock protein A membrane
PstS phosphate ABC transporter (binding protein) transport
RacX amino acid racemase cell wall
RecAmultifunctional protein involved in homologous recombination and DNA repair
DNA repair
RocA 3‐hydroxy‐1‐pyrroline‐5‐carboxylate dehydrogenase amino acids
SalA negative regulator of scoC expression, subtilisin derepression proteolysis
Spo0M sporulation control protein sporulation
TrxA thioredoxin A oxidative stress
YaaQ unknown unknown
YceC similar to tellurium resistance protein cell envelope
YceH similar to toxic anion resistance protein cell envelope
YdhMglucomannan‐specific phosphotransferase system, EIIB component of the PTS
energy
Ydjl acetoine/butanediol dehydrogenase energy
YjbC general stress protein, required for survival of salt stress general stress
YkpA similar to ABC transporter (ATP‐binding protein) energy
YoxD similar to 3‐oxoacyl‐acyl‐carrier protein reductase membrane
YpuA unknown cell envelope
YqiG similar to NADH‐dependent flavin oxidoreductase energy
YtoQ unknown unknown
Marker proteins
P Appendix
344

Protein ID Protein function Functional category
YuaE unknown unknown
YuaI control of membrane fluidity membrane
YugJ similar to NADH‐dependent butanol dehydrogenase energy
YukJ unknown unknown
YvIB unknown cell envelope
YvyDgeneral stress protein, required for ribosome dimerization in the stationary phase
general stress
YwrO similar to NAD(P)H oxidoreductase energy
P Appendix
345

Treatment parameters
Minimal inhibitory concentration 3 µg/mL
Optimal stressor concentration 3 µg/mL
Bacteriolytic concentration 7.5 µg/mL
Bacterial response analyzed after 15 min
C‐C8 (RWRWRWK‐C7H15)
Mechanistic properties
Mode of action interfacial activity
Molecular target phospholipids
Depolarization yes
Permeabilization no
Cell wall integrity disturbed
General information
Source synthetic
MW 1300 g/mol
Ref. Albada et al., 2012a
control/C‐C8
Wenzel et al., in preparation a
P Appendix
346

Mechanism of action
C‐C8 integrates into the bacterial membrane and disturbs membrane architecture accordingto the interfacial activity model. It impairs respiration and cell wall biosynthesis [Wenzel etal., in preparation a].
Protein ID Protein function Functional category
AcsA acetyl‐CoA synthetase membrane
Bcd valine, isoleucine, leucine dehydrogenase amino acids
ClpPATP‐dependent Clp protease proteolytic subunit (class III heat‐shock protein)
proteolysis
ClpY two‐component ATP‐dependent protease, ATPase subunit proteolysis
CysC adenylyl‐sulfate kinase amino acids
FosB bacillithiol‐S‐transferase cell envelope
GroEL chaperonin and co‐repressor for HrcA chaperone
LiaH similar to phage shock protein A cell wall
MurB UDP‐N‐acetylenolpyruvoylglucosamine reductase cell wall
NfrASpx‐dependent FMN‐containing NADPH‐linked nitro/flavinreductase
energy
PbpE penicillin‐binding protein PBP 4 cell wall
PdxK pyridoxine, pyridoxal, pyridoxamine kinase energy/amino acids
PspA phage shock protein A membrane
RacX amino acid racemase cell wall
RocA 3‐hydroxy‐1‐pyrroline‐5‐carboxylate dehydrogenase amino acids
SalA negative regulator of scoC expression, subtilisin derepression proteolysis
Spo0M sporulation control protein sporulation
TrxA thioredoxin A oxidative stress
YaaQ unknown unknown
YceC similar to tellurium resistance protein cell envelope
YceH similar to toxic anion resistance protein cell envelope
YdhMglucomannan‐specific phosphotransferase system, EIIB component of the PTS
energy
Ydjl acetoine/butanediol dehydrogenase energy
YkpA similar to ABC transporter (ATP‐binding protein) transport
YoxD similar to 3‐oxoacyl‐acyl‐carrier protein reductase membrane
YpuA unknown cell envelope
YqiG similar to NADH‐dependent flavin oxidoreductase energy
YtoQ unknown unknown
YuaE unknown unknown
YuaI control of membrane fluidity membrane
YugJ similar to NADH‐dependent butanol dehydrogenase energy
YukJ unknown unknown
YvIB unknown cell envelope
YvyDgeneral stress protein, required for ribosome dimerization in the stationary phase
general stress
YwrO similar to NAD(P)H oxidoreductase energy
Marker proteins
P Appendix
347

Treatment parameters
Minimal inhibitory concentration 5 µg/mL
Optimal stressor concentration 5 µg/mL
Bacteriolytic concentration n.d.
Bacterial response analyzed after 15 min
Cerulenin
Mechanistic properties
Mode of action FAB inhibition
Molecular target FabF
Depolarization n.d.
Permeabilization n.d.
Cell wall integrity n.d.
General information
Source Cephalosporium caerulens
MW 223.27 g/mol
Ref. Matsumae et al., 1964
control/cerulenin
http://www.lookchem.com
P Appendix
348

Mechanism of action
Cerulenin inhibits fatty acid biosynthesis by binding to the 3‐oxoacyl‐ACP synthase II (FabF)[Price et al., 2001].
Protein ID Protein function Functional category
Ald L‐alanine dehydrogenase amino acids
FabHA 3‐oxoacyl‐ACP synthase III membrane (FAB)
FabHB 3‐oxoacyl‐ACP synthase III membrane (FAB)
FabI enoyl‐ACP reductase membrane (FAB)
FabF 3‐oxoacyl‐ACP synthase II membrane (FAB)
PlsXputative glycerol‐3‐phosphateacyltransferase
membrane (PLB)
PanB3‐Methyl‐2‐oxobutanoatehydroxymethyltransferase
CoA synthesis
YurPsimilar to glutamine–fructose‐6‐phosphatetransaminase
energy
Marker proteins
P Appendix
349

Treatment parameters
Minimal inhibitory concentration 2 µg/mL
Optimal stressor concentration 3.5 µg/mL
Bacteriolytic concentration 10 µg/mL
Bacterial response analyzed after 15 min
Daptomycin (LY146032)
Mechanistic properties
Mode of action membrane perturbation
Molecular target probably phospholipids
Depolarization yes
Permeabilization no
Cell wall integrity disturbed
General information
Source Streptomyces roseosporus
MW 1621 g/mol
Ref. Eliopulos et al., 1986
control/daptomycin
Oleson et al., 2004
P Appendix
350
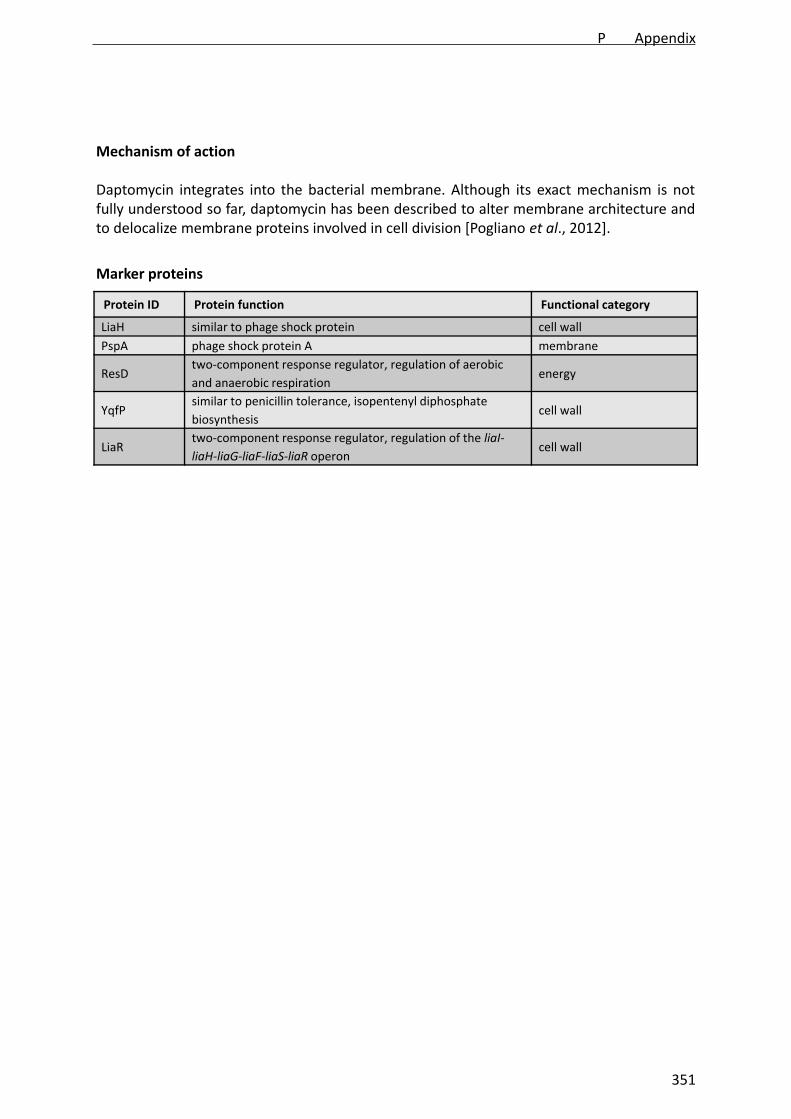
Mechanism of action
Daptomycin integrates into the bacterial membrane. Although its exact mechanism is notfully understood so far, daptomycin has been described to alter membrane architecture andto delocalize membrane proteins involved in cell division [Pogliano et al., 2012].
Protein ID Protein function Functional category
LiaH similar to phage shock protein cell wall
PspA phage shock protein A membrane
ResDtwo‐component response regulator, regulation of aerobic
and anaerobic respiration energy
YqfPsimilar to penicillin tolerance, isopentenyl diphosphate
biosynthesis cell wall
LiaRtwo‐component response regulator, regulation of the liaI‐
liaH‐liaG‐liaF‐liaS‐liaR operoncell wall
Marker proteins
P Appendix
351

Treatment parameters
Minimal inhibitory concentration 1:1000
Optimal stressor concentration 1:1000
Bacteriolytic concentration 1:100
Bacterial response analyzed after 15 min
Dekont
Mechanistic properties
Mode of action detergent
Molecular target phospholipids
Depolarization n.d.
Permeabilization n.d.
Cell wall integrity n.d.
General information
Source unknown
MW unknown
Ref. unknown
control/dekont
unknown
P Appendix
352

Mechanism of action
Dekont is a detergent solution, which is applied to remove radioactive liquids from surfaces.
Protein ID Protein function Functional category
no marker proteins determined
Marker proteins
P Appendix
353
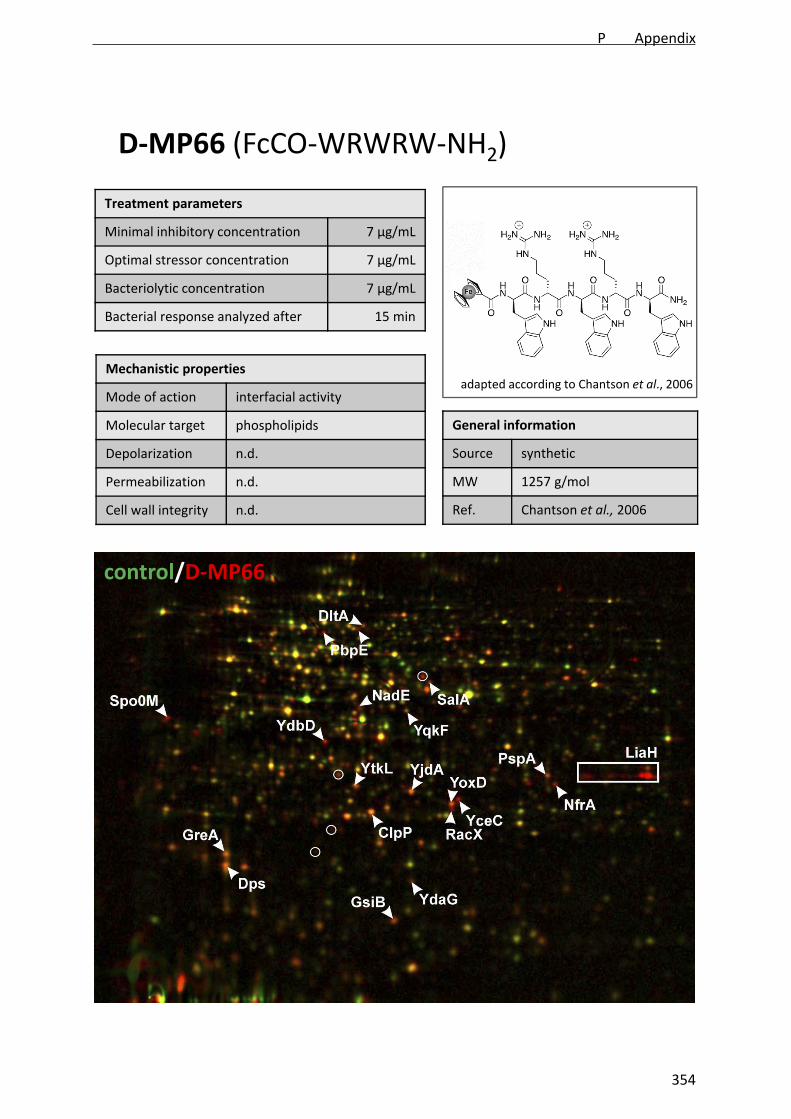
Treatment parameters
Minimal inhibitory concentration 7 µg/mL
Optimal stressor concentration 7 µg/mL
Bacteriolytic concentration 7 µg/mL
Bacterial response analyzed after 15 min
D‐MP66 (FcCO‐WRWRW‐NH2)
Mechanistic properties
Mode of action interfacial activity
Molecular target phospholipids
Depolarization n.d.
Permeabilization n.d.
Cell wall integrity n.d.
General information
Source synthetic
MW 1257 g/mol
Ref. Chantson et al., 2006
control/D‐MP66
adapted according to Chantson et al., 2006
P Appendix
354

Mechanism of action
D‐MP66 integrates into the bacterial membrane and disturbs membrane architectureaccording to the interfacial activity model. It impairs respiration and cell wall biosynthesis[Wenzel et al., in preparation b.
Protein ID Protein function Functional category
ClpPATP‐dependent Clp protease proteolytic subunit (class III
heat‐shock protein) proteolysis
DltA D‐alanyl‐D‐alanine carrier protein ligas cell wall
Dpsiron storage protein, general stress protein, resistance
against ethanol stress and survival at low temperatures general stress
GreAtranscription elongation factor, resolves stalled RNA
polymerase at promoter or promoter‐proximal regionstranscription
GsiB general stress protein general stress
LiaH similar to phage shock protein cell wall
NadE NAD synthase energy
NfrA
Spx‐dependent FMN‐containing NADPH‐linked nitro/flavin
reductase energy
PbpE penicillin‐binding protein 4 cell wall
PspA phage shock protein A membrane
RacX amino acid racemase cell wall
SalA negative regulator of scoC expression, subtilisin derepression proteolysis
Spo0M sporulation control protein sporulation
YceC similar to tellurium resistance protein cell envelope
YdaG general stress protein general stress
YdbD similar to manganese‐containing catalase oxidative stress
YjdA similar to 3‐oxoacyl‐acyl‐carrier protein reductase membrane
YoxD similar to 3‐oxoacyl‐acyl‐carrier protein reductase membrane
YqkF similar to oxidoreductase energy
YtkL general stress protein general stress
Marker proteins
P Appendix
355

Treatment parameters
Minimal inhibitory concentration 10 µg/mL
Optimal stressor concentration 50 µg/mL
Bacteriolytic concentration >50 µg/mL
Bacterial response analyzed after 15 min
D‐MP196 (RWRWRW‐NH2)
Mechanistic properties
Mode of action interfacial activity
Molecular target phospholipids
Depolarization yes
Permeabilization no
Cell wall integrity disturbed
General information
Source synthetic
MW 1044 g/mol
Ref. Albada et al., 2012b
control/D‐MP196
adapted according to Chantson et al., 2006
P Appendix
356
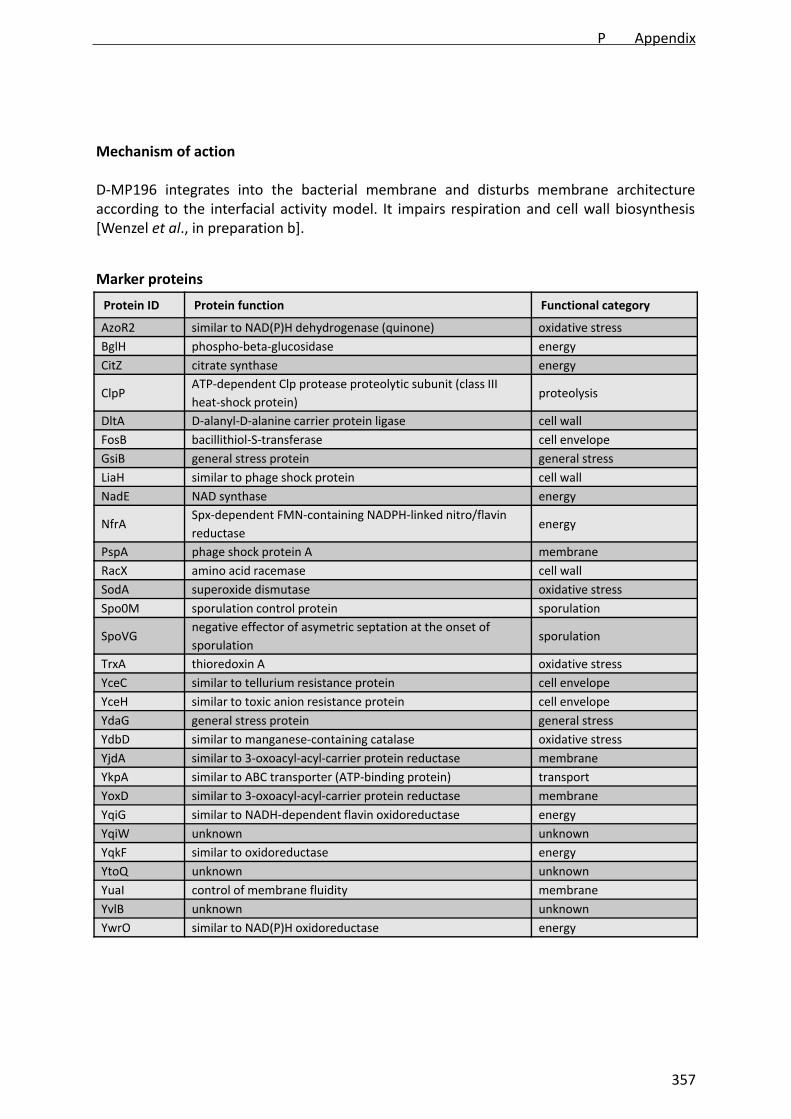
Mechanism of action
D‐MP196 integrates into the bacterial membrane and disturbs membrane architectureaccording to the interfacial activity model. It impairs respiration and cell wall biosynthesis[Wenzel et al., in preparation b].
Protein ID Protein function Functional category
AzoR2 similar to NAD(P)H dehydrogenase (quinone) oxidative stress
BglH phospho‐beta‐glucosidase energy
CitZ citrate synthase energy
ClpPATP‐dependent Clp protease proteolytic subunit (class III
heat‐shock protein) proteolysis
DltA D‐alanyl‐D‐alanine carrier protein ligase cell wall
FosB bacillithiol‐S‐transferase cell envelope
GsiB general stress protein general stress
LiaH similar to phage shock protein cell wall
NadE NAD synthase energy
NfrASpx‐dependent FMN‐containing NADPH‐linked nitro/flavin
reductaseenergy
PspA phage shock protein A membrane
RacX amino acid racemase cell wall
SodA superoxide dismutase oxidative stress
Spo0M sporulation control protein sporulation
SpoVGnegative effector of asymetric septation at the onset of
sporulation sporulation
TrxA thioredoxin A oxidative stress
YceC similar to tellurium resistance protein cell envelope
YceH similar to toxic anion resistance protein cell envelope
YdaG general stress protein general stress
YdbD similar to manganese‐containing catalase oxidative stress
YjdA similar to 3‐oxoacyl‐acyl‐carrier protein reductase membrane
YkpA similar to ABC transporter (ATP‐binding protein) transport
YoxD similar to 3‐oxoacyl‐acyl‐carrier protein reductase membrane
YqiG similar to NADH‐dependent flavin oxidoreductase energy
YqiW unknown unknown
YqkF similar to oxidoreductase energy
YtoQ unknown unknown
YuaI control of membrane fluidity membrane
YvlB unknown unknown
YwrO similar to NAD(P)H oxidoreductase energy
Marker proteins
P Appendix
357

Treatment parameters
Minimal inhibitory concentration 5 µg/mL
Optimal stressor concentration 25 µg/mL
Bacteriolytic concentration n.d.
Bacterial response analyzed after 15 min
D‐MP276 (RcCO‐WRWRW‐NH2)
Mechanistic properties
Mode of action interfacial activity
Molecular target phospholipids
Depolarization yes
Permeabilization no
Cell wall integrity disturbed
General information
Source synthetic
MW 1146 g/mol
Ref. Albada et al., 2012b
control/D‐MP276
adapted according to Albada et al., 2012
P Appendix
358

Mechanism of action
D‐MP276 integrates into the bacterial membrane and disturbs membrane architectureaccording to the interfacial activity model. It impairs respiration and cell wall biosynthesis[Wenzel et al., in preparation b].
Protein ID Protein function Functional category
AzoR2 similar to NAD(P)H dehydrogenase (quinone) oxidative stress
ClpPATP‐dependent Clp protease proteolytic subunit (class III
heat‐shock protein) proteolysis
FabF beta‐ketoacyl‐acyl carrier protein synthase II membrane
GsiB general stress protein general stress
LiaH similar to phage shock protein cell wall
NadE NAD synthase energy
NfrASpx‐dependent FMN‐containing NADPH‐linked nitro/flavin
reductaseenergy
PspA phage shock protein A membrane
RacX amino acid racemase cell wall
SodA superoxide dismutase oxidative stress
TrxA thioredoxin A oxidative stress
YceC similar to tellurium resistance protein cell envelope
YceH similar to toxic anion resistance protein cell envelope
YdaG general stress protein general stress
YdbD similar to manganese‐containing catalase oxidative stress
YjdA similar to 3‐oxoacyl‐acyl‐carrier protein reductase membrane
YkpA similar to ABC transporter (ATP‐binding protein) energy
YoxD similar to 3‐oxoacyl‐acyl‐carrier protein reductase membrane
YqiG similar to NADH‐dependent flavin oxidoreductase energy
YqiW unknown unknown
YvlB unknown cell envelope
YuaI control of membrane fluidity membrane
YwrO similar to NAD(P)H oxidoreductase energy
Marker proteins
P Appendix
359

Treatment parameters
Minimal inhibitory concentration 2 µg/mL
Optimal stressor concentration 2 µg/mL
Bacteriolytic concentration >25 µg/mL
Bacterial response analyzed after 15 min
FcPNA
Mechanistic properties
Mode of action membrane integration
Molecular target phospholipids
Depolarization yes
Permeabilization no
Cell wall integrity disturbed
General information
Source synthetic
MW 1451 g/mol
Ref. Patra et al., 2010
control/FcPNA
adapted according to Patra et al., 2010
P Appendix
360

Mechanism of action
FcPNA integrates into the bacterial membrane and probably affects membrane architecture.It strongly inhibits respiration, impairs cell wall biosynthesis, and induces formation of ROS[Wenzel et al., 2013].
Protein ID Protein function Functional category
Adk adenylate kinase energy
ArgC N‐acetyl‐gamma‐glutamyl‐phosphate reductase energy
AzoR2 similar to NAD(P)H dehydrogenase (quinone) oxidative stress
CheV CheA modulator general stress
ClpP ATP‐dependent Clp protease proteolytic subunit proteolysis
Dps DNA‐protecting protein, ferritin general stress
Frr ribosome recycling factor miscellaneous
GreA transcription elongation factor GreA miscellaneous
GroEL chaperonin chaperone
IolS aldo‐keto reductase energy
IspH isopentenyl diphosphate biosynthesis cell envelope
LuxS S‐ribosylhomocysteinase energy
MrgA metalloregulation DNA‐binding stress protein oxidative stress
NadE NAD synthase energy
Nin inhibitor of the DNA degrading activity of NucA general stress
PpiB peptidyl‐prolyl isomerase (chaperone) chaperone
PspA phage shock protein A cell envelope
RacX amino acid racemase cell envelope
SodA superoxide dismutase oxidative
SpoVC peptidyl‐tRNA hydrolase sporulation
SpoVG regulatory protein SpoVG sporulation
Tpx thiol peroxidase oxidative stress
YceC similar to tellurium resistance protein cell envelope
YceH similar to toxic anion resistance protein cell envelope
YdaG general stress general stress
YdbD manganese‐containing catalase oxidative stress
YfjR beta‐hydroxyacid dehydrogenase energy
YhdN aldo/keto reductase specific for NAD(P)H energy
YjdA similar to 3‐ketoacyl‐acyl‐carrier protein reductase cell envelope
YmaE unknown unknown
YoxD similar to 3‐oxoacyl‐acyl‐carrier protein reductase cell envelope
YphP disulfide isomerase, putative bacilliredoxin oxidative stress
YqiG NADH‐dependent flavin oxidoreductase energy
YqiW unknown, disulfide isomerase family unknown
YqxA unknown general stress
YtkL metal‐dependent hydrolase general stress
YwbC putative methylglyoxylase oxidative stress
YwrO similar to NAD(P)H oxidoreductase energy
Marker proteins
P Appendix
361

Treatment parameters
Minimal inhibitory concentration 4 µg/mL
Optimal stressor concentration 1 µg/mL
Bacteriolytic concentration 20 µg/mL
Bacterial response analyzed after 15 min
Friulimicin B
Mechanistic properties
Mode of action CWB inhibition
Molecular target undecaprenyl pyrophosphate
Depolarization no
Permeabilization n.d.
Cell wall integrity disturbed
General information
Source Actinoplanes friuliensis
MW 1303 g/mol
Ref. Vértesy et al., 2000
control/friulimicin B
Schneider et al., 2009
P Appendix
362

Mechanism of action
Friulimicin B binds to undecaprenyl phosphate, prevents recycling of the bactoprenol carrier,and inhibits the lipid II cycle [Schneider et al., 2009].
Protein ID Protein function Functional category
no marker proteins identified
Marker proteins
P Appendix
363

Treatment parameters
Minimal inhibitory concentration 6 µg/mL
Optimal stressor concentration 6 µg/mL
Bacteriolytic concentration 30 µg/mL
Bacterial response analyzed after 15 min
Gallidermin
Mechanistic properties
Mode of action CWB inhibition, membrane
Molecular target lipid II, phospholipids
Depolarization yes
Permeabilization no
Cell wall integrity disturbed
General information
Source Staphylococcus gallinarium
MW 2166 g/mol
Ref. Kellner et al., 1988
control/gallidermin
adapted according to Bonelli et al., 2006
P Appendix
364
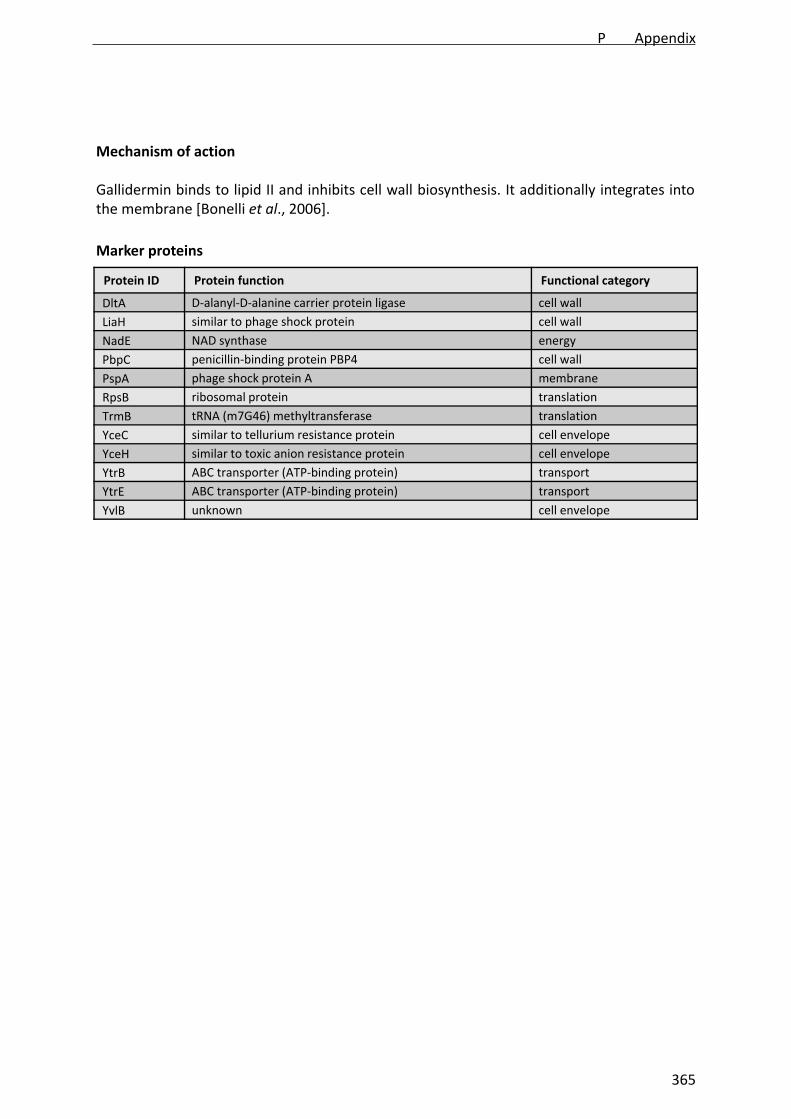
Mechanism of action
Gallidermin binds to lipid II and inhibits cell wall biosynthesis. It additionally integrates intothe membrane [Bonelli et al., 2006].
Protein ID Protein function Functional category
DltA D‐alanyl‐D‐alanine carrier protein ligase cell wall
LiaH similar to phage shock protein cell wall
NadE NAD synthase energy
PbpC penicillin‐binding protein PBP4 cell wall
PspA phage shock protein A membrane
RpsB ribosomal protein translation
TrmB tRNA (m7G46) methyltransferase translation
YceC similar to tellurium resistance protein cell envelope
YceH similar to toxic anion resistance protein cell envelope
YtrB ABC transporter (ATP‐binding protein) transport
YtrE ABC transporter (ATP‐binding protein) transport
YvlB unknown cell envelope
Marker proteins
P Appendix
365

Treatment parameters
Minimal inhibitory concentration 2 µg/mL
Optimal stressor concentration 1 µg/mL
Bacteriolytic concentration 2 µg/mL
Bacterial response analyzed after 15 min
Gramicidin S
Mechanistic properties
Mode of action membrane integration
Molecular target phospholipids
Depolarization yes
Permeabilization no
Cell wall integrity disturbed
General information
Source Bacillus brevis
MW 1141 g/mol
Ref. Gause & Brazhnikova, 1944
control/gramicidin S
academic.ru
P Appendix
366

Mechanism of action
Gramicidin S integrates into the bacterial membrane and leads to rearrangement ofnegatively charged phospholipids [Kaprel’iants et al., 1977; Vostroknutova et al., 1981]. Ittriggers potassium release [Katsu et al., 1989] and delocalizes cytochrome c and MurG.
Protein ID Protein function Functional category
AroA3‐deoxy‐D‐arabino‐heptulosonate 7‐phosphate synthase /
chorismate mutase‐isozyme 3amino acids
ClpPATP‐dependent Clp protease proteolytic subunit (class III
heat‐shock protein) proteolysis
LiaH similar to phage shock protein cell wall
NadE NAD synthase energy
PspA phage shock protein A membrane
RacX amino acid racemase cell wall
ThrC threonine synthase amino acids
YceC similar to tellurium resistance protein cell envelope
YceH similar to toxic anion resistance protein cell envelope
YjdA similar to 3‐oxoacyl‐acyl‐carrier protein reductase membrane
YoxD similar to 3‐oxoacyl‐acyl‐carrier protein reductase membrane
YuaE unknown unknown
YugJ similar to NADH‐dependent butanol dehydrogenase energy
YvIB unknown cell envelope
YwrO similar to NAD(P)H oxidoreductase energy
Marker proteins
P Appendix
367

Treatment parameters
Minimal inhibitory concentration 1:10000
Optimal stressor concentration 1:100000
Bacteriolytic concentration 1:10000
Bacterial response analyzed after 15 min
Helipur
Mechanistic properties
Mode of action detergent
Molecular target phospholipids
Depolarization n.d.
Permeabilization n.d.
Cell wall integrity disturbed
General information
Source synthetic
MW unknown
Ref. B. Braun Melsungen AG
control/helipur
unknown
P Appendix
368

Mechanism of action
Helipur is a toxic detergent used for disinfection of glass ware.
Protein ID Protein function Functional category
no marker proteins determined
Marker proteins
P Appendix
369

Treatment parameters
Minimal inhibitory concentration 9 µg/mL
Optimal stressor concentration 0.9 µg/mL
Bacteriolytic concentration 6.75 µg/mL
Bacterial response analyzed after 15 min
Lanti X
Mechanistic properties
Mode of action CWB inhibition
Molecular target lipid II
Depolarization n.d.
Permeabilization n.d.
Cell wall integrity n.d.
General information
Source unknown
MW unknown
Ref. unknown
control/lanti X
unknown
P Appendix
370

Mechanism of action
According to the proteome pattern, lanti X acts similar to lipid II‐binding antibiotics likegallidermin and nisin. It induces a pronounced cell wall stress response but no specificmembrane stress markers (Wenzel and Kohl, unpublished results).
Protein ID Protein function Functional category
CysC adenylyl‐sulfate kinase sulfur metabolism
GamA glucosamine‐6‐phosphate deaminase cell wall
LiaH similar to phage shock protein cell wall
NrfASpx‐dependent FMN‐containing NADPH‐linked nitro/flavin
reductaseenergy
PbpC penicillin‐binding protein cell wall
RpsB ribosomal protein translation
YceC similar to tellurium resistance protein cell envelope
YceH similar to toxic anion resistance protein cell envelope
YqiG similar to NADH‐dependent flavin oxidoreductase energy
YtrE ABC transporter (ATP‐binding protein) transport
YvcRABC transporter (ATP‐binding protein) for the export of lipid
II‐binding lantibioticstransport
YvIB unknown cell envelope
LiaRtwo‐component response regulator, regulation of the liaI‐
liaH‐liaG‐liaF‐liaS‐liaR operoncell wall
Marker proteins
P Appendix
371

Treatment parameters
Minimal inhibitory concentration 7 µg/mL
Optimal stressor concentration 5.75 µg/mL
Bacteriolytic concentration 7 µg/mL
Bacterial response analyzed after 15 min
L‐MP66 (FcCO‐RWRWRW‐NH2)
Mechanistic properties
Mode of action interfacial activity
Molecular target phospholipids
Depolarization n.d.
Permeabilization n.d.
Cell wall integrity n.d.
General information
Source synthetic
MW 1257 g/mol
Ref. Chantson et al., 2006
control/L‐MP66
adapted according to Chantson et al., 2006
P Appendix
372

Mechanism of action
L‐MP66 integrates into the bacterial membrane and disturbs membrane architectureaccording to the interfacial activity model. It impairs respiration and cell wall biosynthesis[Wenzel et al., in preparation b].
Protein ID Protein function Functional category
AzoR2 similar to NAD(P)H dehydrogenase (quinone) oxidative stress
DltA D‐alanyl‐D‐alanine carrier protein ligase cell wall
EfTU elongation factor TU translation
FabF beta‐ketoacyl‐acyl carrier protein synthase II membrane
GsiB general stress protein general stress
LiaH similar to phage shock protein cell wall
LuxSS‐ribosylhomocysteine lyase, autoinducer‐2 production
protein, required for swarming motility and biofilm formation
amino acids, chemotaxis,
biofilm formation
NadE NAD synthase energy
NfrASpx‐dependent FMN‐containing NADPH‐linked nitro/flavin
reductaseenergy
PspA phage shock protein A membrane
Spo0M sporulation control protein sporulation
YceC similar to tellurium resistance protein cell envelope
YhdN aldo/keto reductase general stress
YjdA similar to 3‐oxoacyl‐acyl‐carrier protein reductase membrane
YoxD similar to 3‐oxoacyl‐acyl‐carrier protein reductase membrane
YqiW unknown oxidative stress
YqkF similar to oxidoreductase energy
YuaI control of membrane fluidity membrane
YwrO similar to NAD(P)H oxidoreductase energy
Marker proteins
P Appendix
373

Treatment parameters
Minimal inhibitory concentration 5 µg/mL
Optimal stressor concentration 5 µg/mL
Bacteriolytic concentration >25 µg/mL
Bacterial response analyzed after 15 min
L‐MP159 (FcCO‐G‐cCRWRWRWC)
Mechanistic properties
Mode of action interfacial activity
Molecular target phospholipids
Depolarization n.d.
Permeabilization n.d.
Cell wall integrity n.d.
General information
Source synthetic
MW 1361 g/mol
Ref. Penkova, 2009
control/L‐MP159
Chiriac et al., in preparation
P Appendix
374

Mechanism of action
L‐MP159 integrates into the bacterial membrane and disturbs membrane architectureaccording to the interfacial activity model. It impairs respiration and cell wall biosynthesis[Wenzel et al., in preparation b].
Protein ID Protein function Functional category
AzoR2 similar to NAD(P)H dehydrogenase (quinone) oxidative stress
CitZ citrate synthase energy
ClpPATP‐dependent Clp protease proteolytic subunit (class III
heat‐shock protein) proteolysis
Dpsiron storage protein, general stress protein, resistance
against ethanol stress and survival at low temperatures general stress
IolS unknown, may be involved in myo‐inositol catabolism energy
LiaH similar to phage shock potein cell wall
LuxSS‐ribosylhomocysteine lyase, autoinducer‐2 production
protein, required for swarming motility and biofilm formation
amino acids, chemotaxis,
biofilm formation
MrgA iron storage protein, DNA‐binding stress protein oxidative stress
NadE NAD synthase energy
NfrASpx‐dependent FMN‐containing NAD(P)H‐linked nitro/flavin
reductaseenergy
PbpE penicillin‐binding protein PBP4 cell wall
PspA phage shock protein A membrane
RacX amino acid racemase cell wall
SodA superoxide dismutase oxidative stress
SpoVGnegative effector of asymetric septation at the onset of
sporulation sporulation
YceC similar to tellurium resistance protein cell envelope
YceH similar to toxic anion resistance protein cell envelope
YdbD similar to manganese‐containing catalase oxidative stress
YjdA similar to 3‐oxoacyl‐acyl‐carrier protein reductase membrane
YfhM similar to epoxide hydrolase cell envelope
YfjR similar to 3‐hydroxyisobutyrate dehydrogenase memrane
YoxD similar to 3‐oxoacyl‐acyl‐carrier protein reductase membrane
YthP similar to ABC transporter (ATP‐binding protein) transport
YvlB unknown cell envelope
Marker proteins
P Appendix
375

Treatment parameters
Minimal inhibitory concentration 9 µg/mL
Optimal stressor concentration 22.5 µg/mL
Bacteriolytic concentration 45 µg/mL
Bacterial response analyzed after 15 min
L‐MP196 (H‐RWRWRW‐NH2)
Mechanistic properties
Mode of action interfacial activity
Molecular target phospholipids
Depolarization yes
Permeabilization no
Cell wall integrity disturbed
General information
Source synthetic
MW 1043 g/mol
Ref. Chantson et al., 2006
control/MP196
adapted according to Chantson et al., 2006
P Appendix
376

Mechanism of action
L‐MP196 integrates into the bacterial membrane and disturbs membrane architectureaccording to the interfacial activity model. It impairs respiration and cell wall biosynthesis[Wenzel et al., submitted].
Protein ID Protein function Functional category
BglH phospho‐beta‐glucosidase energy
CitZ citrate synthase energy
DltA D‐alanyl‐D‐alanine carrier protein ligase cell wall
LiaH modulator of LiaIHGFSR operon expression cell wall
NadE NAD synthetase energy
PspA phage shock protein A homolog membrane
RacX amino acid racemase cell wall
RpsB ribosomal protein S2 translation
TrmB tRNA (m7G46) methyltransferase translation
YceC similar to tellurium resistance protein cell envelope
YceH similar to toxic anion resistance protein cell envelope
YjdA similar to 3‐ketoacyl‐acyl‐carrier protein reductase membrane
YoxD similar to 3‐oxoacyl‐acyl‐carrier protein reductase membrane
YthP similar to ABC transporter (ATP‐binding protein) transport
YtrE similar to ABC transporter (ATP‐binding protein) transport
YtxH general stress protein general stress
YuaI control of membrane fluidity membrane
YvlB unknown cell envelope
YwrO similar to NAD(P)H oxidoreductase energy
Marker proteins
P Appendix
377

Treatment parameters
Minimal inhibitory concentration 1 µg/mL
Optimal stressor concentration 1 µg/mL
Bacteriolytic concentration n.d.
Bacterial response analyzed after 15 min
L‐MP276 (RcCO‐RWRWRW‐NH2)
Mechanistic properties
Mode of action interfacial activity
Molecular target phospholipids
Depolarization yes
Permeabilization no
Cell wall integrity disturbed
General information
Source synthetic
MW 1146 g/mol
Ref. Albada et al., 2012b
control/L‐MP276
adapted according to Albada et al., 2012
P Appendix
378

Mechanism of action
L‐MP276 integrates into the bacterial membrane and disturbs membrane architectureaccording to the interfacial activity model. It impairs respiration and cell wall biosynthesis[Wenzel et al., in preparation b].
Protein ID Protein function Functional category
ArgC N‐acetyl‐g‐glutamyl‐phosphate reductase amino acids
Dpsiron storage protein, general stress protein, resistance against ethanol stress and survival at low temperatures
general stress
Frr ribosome recycling factor translation
GreAtranscription elongation factor, resolves stalled RNA polymerase at promoter or promoter‐proximal regions
translation
LiaH similar to phage shock protein cell wall
Mdh malate dehydrogenase energy
MrgA iron storage protein, DNA‐binding stress protein oxidative stress
NadE NAD synthase energy
PpiB peptidyl‐prolyl isomerase chaperone
PspA phage shock protein A membrane
RacX amino acid racemase cell wall
RpsB ribosomal protein translation
SodA superoxide dismutase oxidative stress
SpoVGnegative effector of asymetric septation at the onset of sporulation
sporulation
Tpx thiol peroxidase Oxidative stress
YceC similar to tellurium resistance protein cell envelope
YdaG general stress protein general stress
YjdA similar to 3‐oxoacyl‐acyl‐carrier protein reductase membrane
YoxD similar to 3‐oxoacyl‐acyl‐carrier protein reductase membrane
YphP disulfide isomerase, putative bacilliredoxin oxidative stress
YqkF similar to oxidoreductase energy
YthP similar to ABC transporter (ATP‐binding protein) transport
YtkL general stress protein general stress
YtxH general stress protein general stress
YwbC putative methylglyoxalase oxidative stress
YwrO similar to NAD(P)H oxidoreductase energy
Marker proteins
P Appendix
379

Treatment parameters
Minimal inhibitory concentration 30 µg/mL
Optimal stressor concentration 30 µg/mL
Bacteriolytic concentration n.d.
Bacterial response analyzed after 15 min
Mersacidin
Mechanistic properties
Mode of action CWB inhibition
Molecular target lipid II
Depolarization no
Permeabilization n.d.
Cell wall integrity disturbed
General information
Source Bacillus sp. HIL Y‐85,54728
MW 1834 g/mol
Ref. Chatterjee et al., 1992
control/mersacidin
adapted according to Chatterjee et al., 2005
P Appendix
380

Mechanism of action
Mersacidin binds to lipid II and inhibits cell wall biosynthesis [Brötz et al., 1995].
Protein ID Protein function Functional category
LiaH similar to phage shock protein cell wall
TrmB tRNA (m7G46) methyltransferase translation
YceC similar to tellurium resistance protein cell envelope
YceH similar to toxic anion resistance protein cell envelope
YqiQ methylisocitrate lyase sporulation
YtrB ABC transporter (ATP‐binding protein) transport
YtrE ABC transporter (ATP‐binding protein) transport
Marker proteins
P Appendix
381

Treatment parameters
Minimal inhibitory concentration 10 µg/mL
Optimal stressor concentration 15.75 µg/mL
Bacteriolytic concentration 22.5 µg/mL
Bacterial response analyzed after 15 min
N‐C0 (KRWRWRW‐NH2)
Mechanistic properties
Mode of action interfacial activity
Molecular target phospholipids
Depolarization yes
Permeabilization no
Cell wall integrity disturbed
General information
Source synthetic
MW 1176 g/mol
Ref. Albada et al. 2012a
control/N‐C0
Wenzel et al., in preparation a
P Appendix
382

Mechanism of action
N‐C0 integrates into the bacterial membrane and disturbs membrane architecture accordingto the interfacial activity model. It impairs respiration and cell wall biosynthesis [Wenzel etal., in preparation a].
Protein ID Protein function Functional category
Bcd valine, isoleucine, leucine dehydrogenase amino acids
ClpY two‐component ATP‐dependent protease, ATPase subunit proteolysis
CysC adenylyl‐sulfate kinase amino acids
FabF beta‐ketoacyl‐acyl carrier protein synthase II membrane
FosB bacillithiol‐S‐transferase cell envelope
GroEL chaperonin and co‐repressor for HrcA chaperone
IlvC ketol‐acid reductoisomerase amino acids
LiaH similar to phage shock protein A cell wall
MurB UDP‐N‐acetylenolpyruvoylglucosamine reductase cell wall
NfrASpx‐dependent FMN‐containing NADPH‐linked nitro/flavin reductase
energy
PbpE penicillin‐binding protein PBP 4 cell wall
PdxK pyridoxine, pyridoxal, pyridoxamine kinase energy/amino acids
PspA phage shock protein A membrane
PstS phosphate ABC transporter (binding protein) energy
RacX amino acid racemase cell wall
RocA 3‐hydroxy‐1‐pyrroline‐5‐carboxylate dehydrogenase amino acids
SalA negative regulator of scoC expression, subtilisin derepression proteolysis
Spo0M sporulation control protein sporulation
TrxA thioredoxin A oxidative stress
YaaQ unknown unknown
YceC similar to tellurium resistance protein cell envelope
YceH similar to toxic anion resistance protein cell envelope
YdhMglucomannan‐specific phosphotransferase system, EIIB component of the PTS
energy
YjbC general stress protein, required for survival of salt stress general stress
YkpA similar to ABC transporter (ATP‐binding protein) energy
YoxD similar to 3‐oxoacyl‐acyl‐carrier protein reductase fatty acid biosynthesis
YpuA unknown cell envelope
YqiG similar to NADH‐dependent flavin oxidoreductase energy
YuaE unknown unknown
YuaI control of membrane fluidity membrane
YugJ similar to NADH‐dependent butanol dehydrogenase energy
YukJ unknown unknown
YvIB unknown cell envelope
YvyDgeneral stress protein, required for ribosome dimerization in the stationary phase
general stress
YwrO similar to NAD(P)H oxidoreductase energy
Marker proteins
P Appendix
383

Treatment parameters
Minimal inhibitory concentration 6 µg/mL
Optimal stressor concentration 4.5 µg/mL
Bacteriolytic concentration 6 µg/mL
Bacterial response analyzed after 15 min
N‐C8 (H15C7‐KRWRWRW‐NH2)
Mechanistic properties
Mode of action interfacial activity
Molecular target phospholipids
Depolarization yes
Permeabilization no
Cell wall integrity disturbed
General information
Source synthetic
MW 1300 g/mol
Ref. Albada et al. 2012a
control/N‐C8
Wenzel et al., in preparation a
P Appendix
384
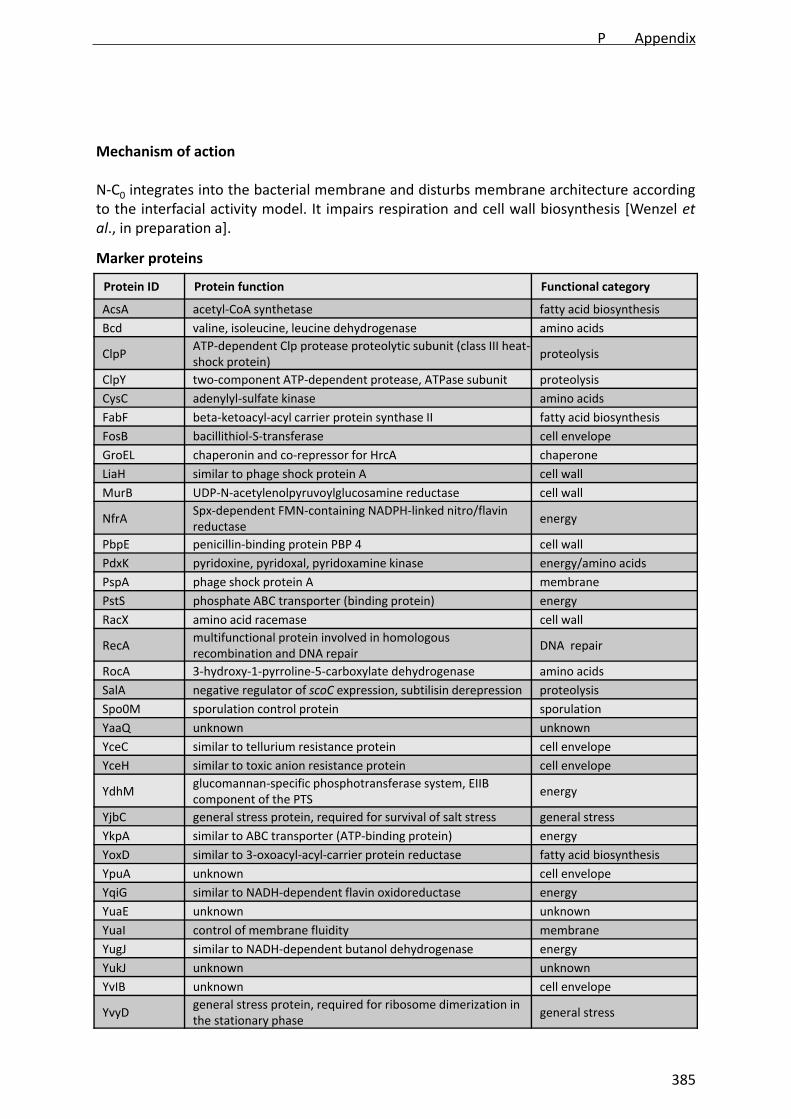
Mechanism of action
N‐C0 integrates into the bacterial membrane and disturbs membrane architecture accordingto the interfacial activity model. It impairs respiration and cell wall biosynthesis [Wenzel etal., in preparation a].
Protein ID Protein function Functional category
AcsA acetyl‐CoA synthetase fatty acid biosynthesis
Bcd valine, isoleucine, leucine dehydrogenase amino acids
ClpPATP‐dependent Clp protease proteolytic subunit (class III heat‐shock protein)
proteolysis
ClpY two‐component ATP‐dependent protease, ATPase subunit proteolysis
CysC adenylyl‐sulfate kinase amino acids
FabF beta‐ketoacyl‐acyl carrier protein synthase II fatty acid biosynthesis
FosB bacillithiol‐S‐transferase cell envelope
GroEL chaperonin and co‐repressor for HrcA chaperone
LiaH similar to phage shock protein A cell wall
MurB UDP‐N‐acetylenolpyruvoylglucosamine reductase cell wall
NfrASpx‐dependent FMN‐containing NADPH‐linked nitro/flavin reductase
energy
PbpE penicillin‐binding protein PBP 4 cell wall
PdxK pyridoxine, pyridoxal, pyridoxamine kinase energy/amino acids
PspA phage shock protein A membrane
PstS phosphate ABC transporter (binding protein) energy
RacX amino acid racemase cell wall
RecAmultifunctional protein involved in homologous recombination and DNA repair
DNA repair
RocA 3‐hydroxy‐1‐pyrroline‐5‐carboxylate dehydrogenase amino acids
SalA negative regulator of scoC expression, subtilisin derepression proteolysis
Spo0M sporulation control protein sporulation
YaaQ unknown unknown
YceC similar to tellurium resistance protein cell envelope
YceH similar to toxic anion resistance protein cell envelope
YdhMglucomannan‐specific phosphotransferase system, EIIB component of the PTS
energy
YjbC general stress protein, required for survival of salt stress general stress
YkpA similar to ABC transporter (ATP‐binding protein) energy
YoxD similar to 3‐oxoacyl‐acyl‐carrier protein reductase fatty acid biosynthesis
YpuA unknown cell envelope
YqiG similar to NADH‐dependent flavin oxidoreductase energy
YuaE unknown unknown
YuaI control of membrane fluidity membrane
YugJ similar to NADH‐dependent butanol dehydrogenase energy
YukJ unknown unknown
YvIB unknown cell envelope
YvyDgeneral stress protein, required for ribosome dimerization in the stationary phase
general stress
Marker proteins
P Appendix
385
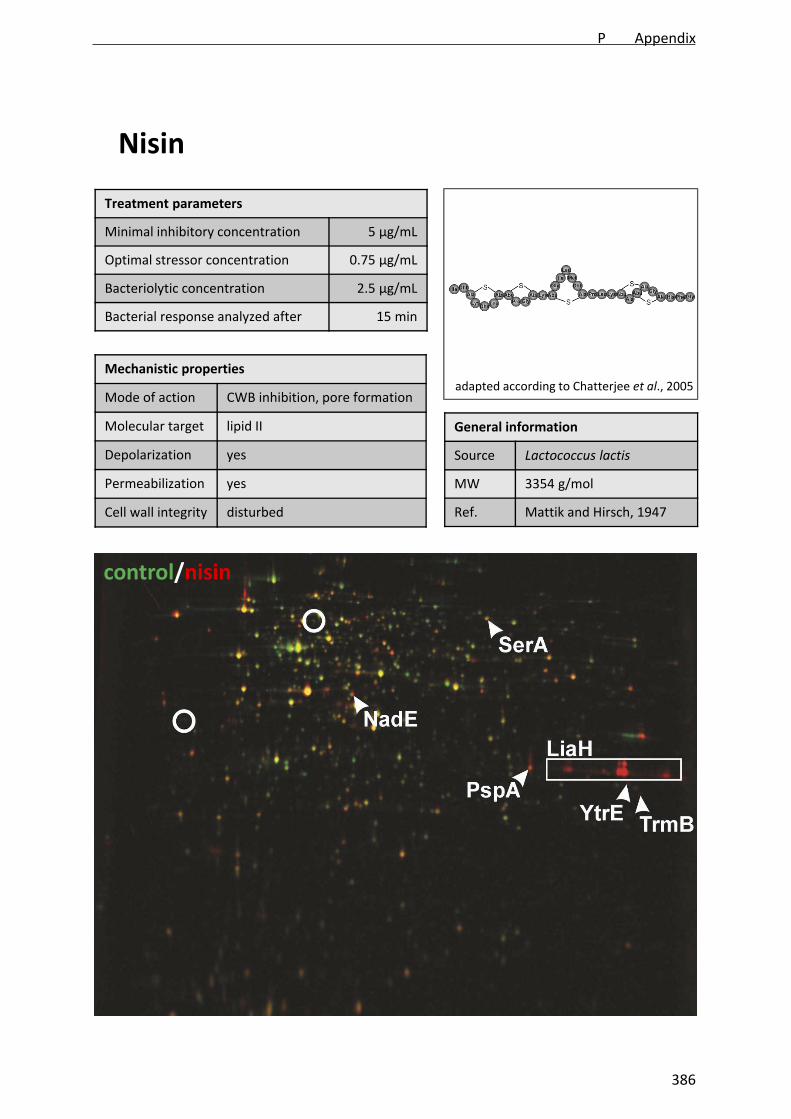
Treatment parameters
Minimal inhibitory concentration 5 µg/mL
Optimal stressor concentration 0.75 µg/mL
Bacteriolytic concentration 2.5 µg/mL
Bacterial response analyzed after 15 min
Nisin
Mechanistic properties
Mode of action CWB inhibition, pore formation
Molecular target lipid II
Depolarization yes
Permeabilization yes
Cell wall integrity disturbed
General information
Source Lactococcus lactis
MW 3354 g/mol
Ref. Mattik and Hirsch, 1947
control/nisin
adapted according to Chatterjee et al., 2005
P Appendix
386

Mechanism of action
Nisin binds to lipid II and inserts into the membrane. After reaching a certain concentrationthreshold a nisin‐lipid II pore is formed [van Heudsen et al., 2002].
Protein ID Protein function Functional category
LiaH similar to phage shock protein cell wall
NadE NAD synthase energy
PspA phage shock protein A membrane
SerA phosphoglycerate dehydrogenase amino acids
TrmB tRNA (m7G46) methyltransferase translation
YtrE ABC transporter (ATP‐binding protein) transport
Marker proteins
P Appendix
387
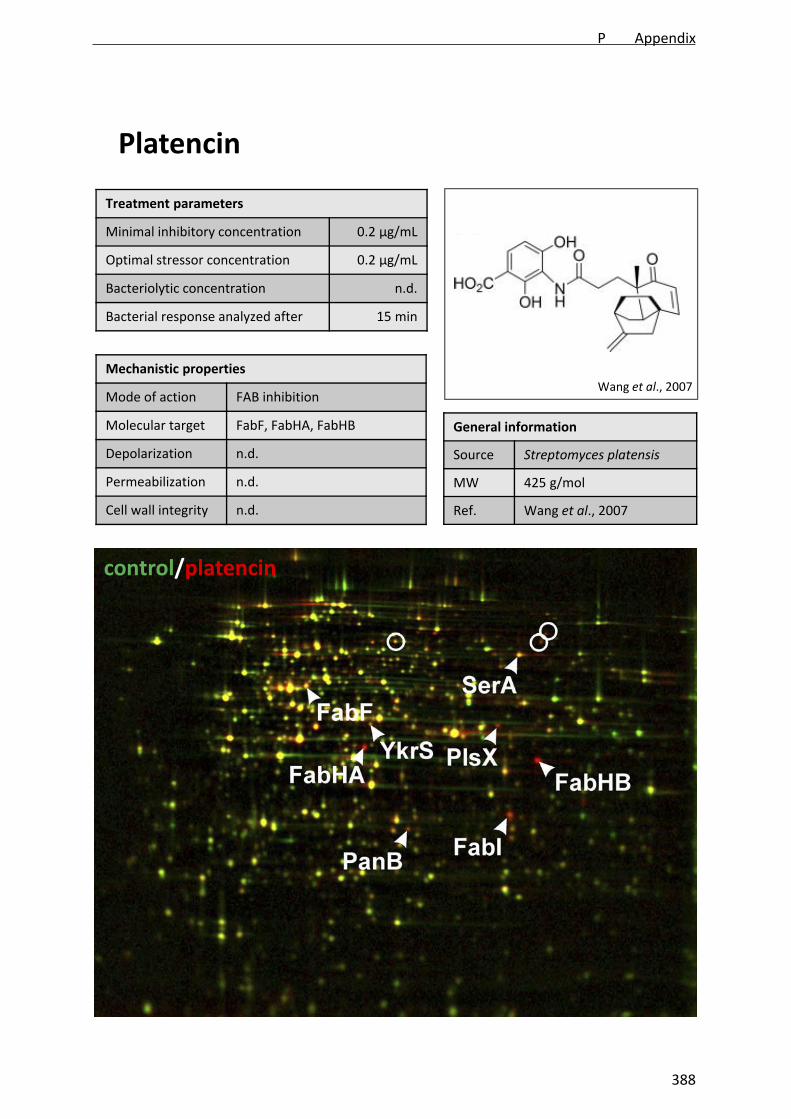
Treatment parameters
Minimal inhibitory concentration 0.2 µg/mL
Optimal stressor concentration 0.2 µg/mL
Bacteriolytic concentration n.d.
Bacterial response analyzed after 15 min
Platencin
Mechanistic properties
Mode of action FAB inhibition
Molecular target FabF, FabHA, FabHB
Depolarization n.d.
Permeabilization n.d.
Cell wall integrity n.d.
General information
Source Streptomyces platensis
MW 425 g/mol
Ref. Wang et al., 2007
control/platencin
Wang et al., 2007
P Appendix
388

Mechanism of action
Platencin is a triple inhibitor of the 3‐oxoacyl ACP synthases FabF, FabHA, and FabHB andinhibits fatty acid biosynthesis [Wang et al., 2007].
Protein ID Protein function Functional category
FabHA 3‐oxoacyl‐ACP synthase III membrane (FAB)
FabHB 3‐oxoacyl‐ACP synthase III membrane (FAB)
FabF 3‐oxoacyl‐ACP synthase II membrane (FAB)
FabI enoyl‐ACP reductase membrane (FAB)
PlsX putative glycerol‐3‐phosphate acyltransferase membrane (PLB)
PanB 3‐methyl‐2‐oxobutanoate hydroxymethyltransferase CoA synthesis
SerA D‐3‐phosphoglycerate dehydrogenase amino acids
YkrS methylthioribose‐1‐phosphate isomerase amino acids
Marker proteins
P Appendix
389

Treatment parameters
Minimal inhibitory concentration 1 µg/mL
Optimal stressor concentration 5 µg/mL
Bacteriolytic concentration n.d.
Bacterial response analyzed after 15 min
Platensimycin
Mechanistic properties
Mode of action FAB inhibition
Molecular target FabF
Depolarization n.d.
Permeabilization n.d.
Cell wall integrity n.d.
General information
Source Streptomyces platensis
MW 441 g/mol
Ref. Wang et al., 2006
control/platensimycin
Wang et al., 2007
P Appendix
390
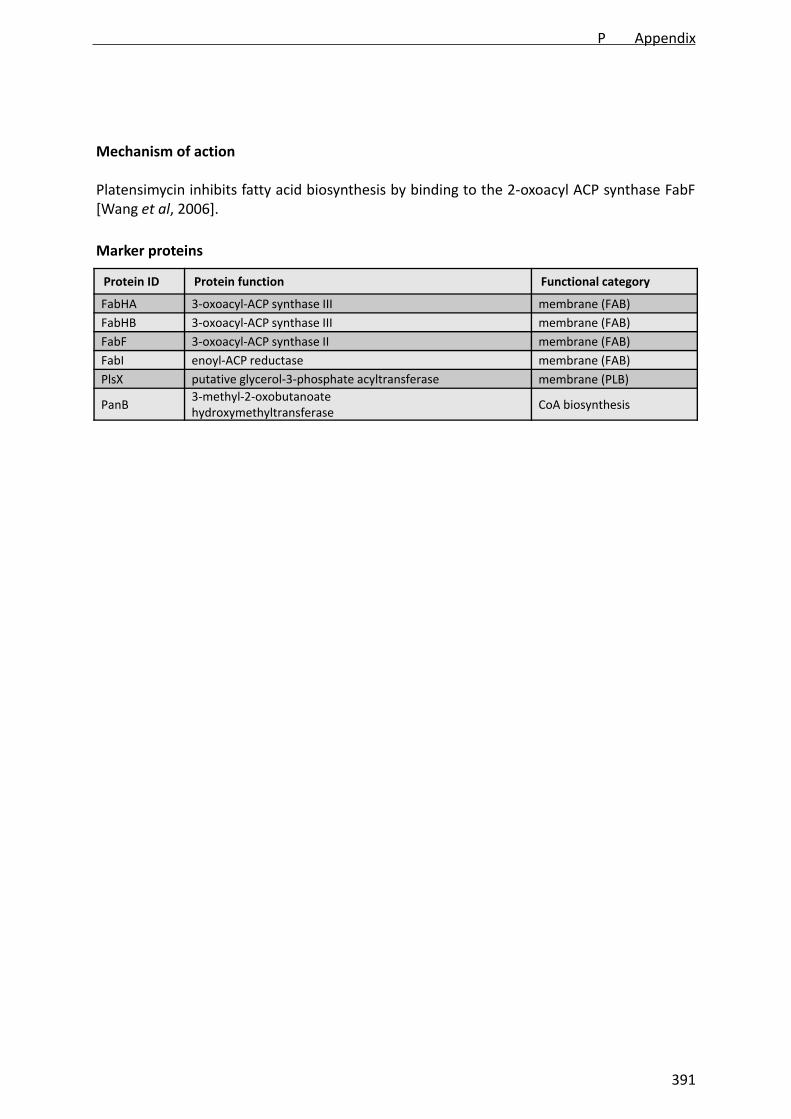
Mechanism of action
Platensimycin inhibits fatty acid biosynthesis by binding to the 2‐oxoacyl ACP synthase FabF[Wang et al, 2006].
Protein ID Protein function Functional category
FabHA 3‐oxoacyl‐ACP synthase III membrane (FAB)
FabHB 3‐oxoacyl‐ACP synthase III membrane (FAB)
FabF 3‐oxoacyl‐ACP synthase II membrane (FAB)
FabI enoyl‐ACP reductase membrane (FAB)
PlsX putative glycerol‐3‐phosphate acyltransferase membrane (PLB)
PanB3‐methyl‐2‐oxobutanoatehydroxymethyltransferase
CoA biosynthesis
Marker proteins
P Appendix
391
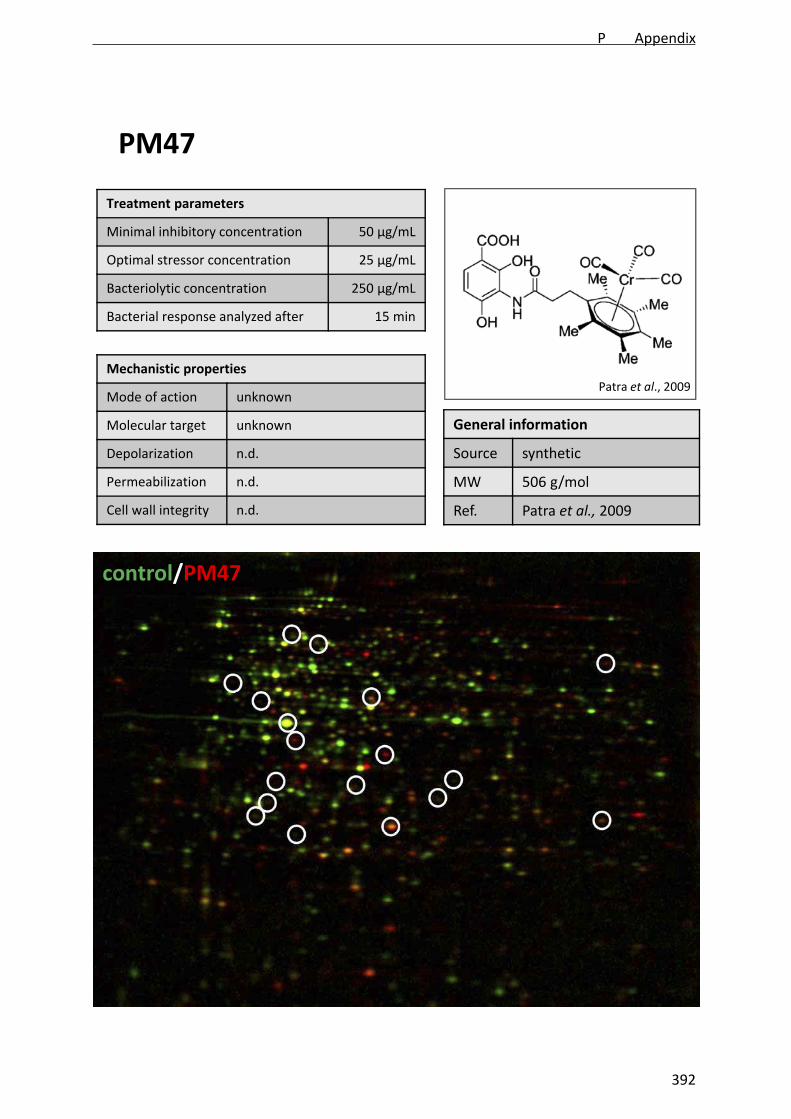
Treatment parameters
Minimal inhibitory concentration 50 µg/mL
Optimal stressor concentration 25 µg/mL
Bacteriolytic concentration 250 µg/mL
Bacterial response analyzed after 15 min
PM47
Mechanistic properties
Mode of action unknown
Molecular target unknown
Depolarization n.d.
Permeabilization n.d.
Cell wall integrity n.d.
General information
Source synthetic
MW 506 g/mol
Ref. Patra et al., 2009
control/PM47
Patra et al., 2009
P Appendix
392

Mechanism of action
PM47 is an organometallic derivative of platensimycin with unknown mechanism of action.Fatty acid biosynthesis was excluded as target [Wenzel et al., 2011.
Protein ID Protein function Functional category
no marker proteins identified
Marker proteins
P Appendix
393

Treatment parameters
Minimal inhibitory concentration 1 µg/mL
Optimal stressor concentration 1 µg/mL
Bacteriolytic concentration >25 µg/mL
Bacterial response analyzed after 15 min
RcPNA
Mechanistic properties
Mode of action membrane integration
Molecular target phospholipids
Depolarization yes
Permeabilization no
Cell wall integrity disturbed
General information
Source synthetic
MW 1351 g/mol
Ref. Wenzel et al., 2013
control/RcPNA
Wenzel et al., in press
P Appendix
394
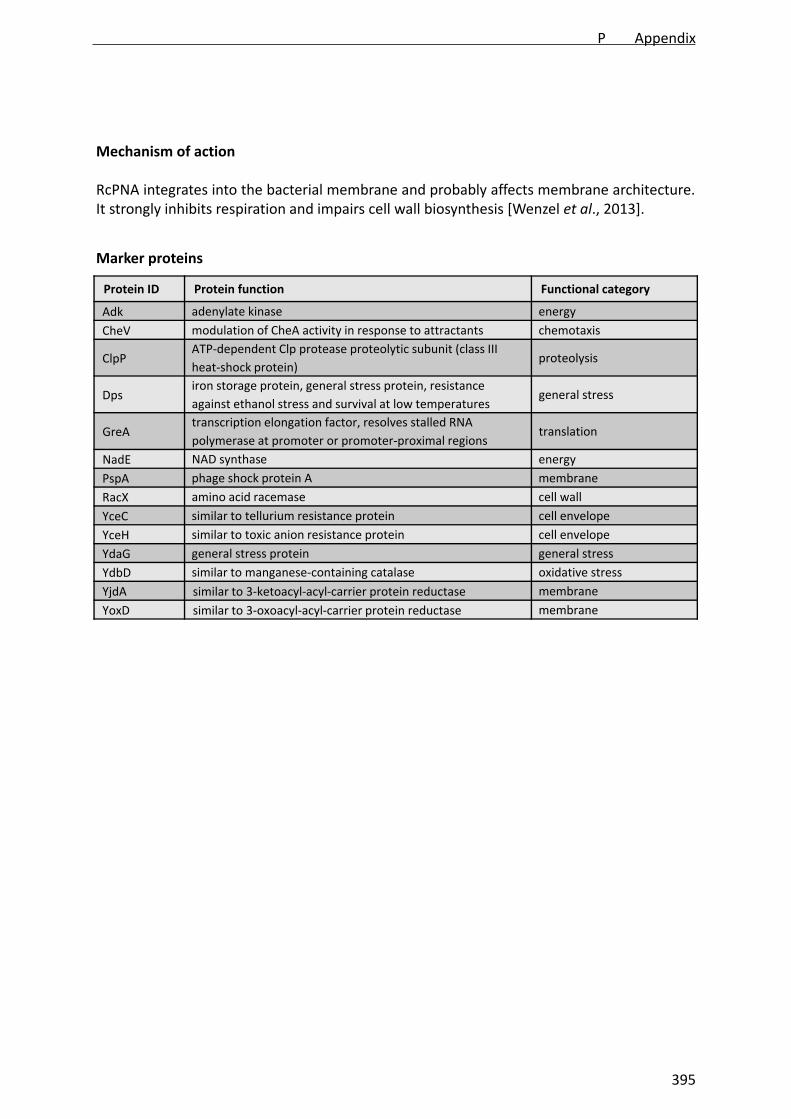
Mechanism of action
RcPNA integrates into the bacterial membrane and probably affects membrane architecture.It strongly inhibits respiration and impairs cell wall biosynthesis [Wenzel et al., 2013].
Protein ID Protein function Functional category
Adk adenylate kinase energy
CheV modulation of CheA activity in response to attractants chemotaxis
ClpPATP‐dependent Clp protease proteolytic subunit (class III
heat‐shock protein) proteolysis
Dpsiron storage protein, general stress protein, resistance
against ethanol stress and survival at low temperatures general stress
GreAtranscription elongation factor, resolves stalled RNA
polymerase at promoter or promoter‐proximal regionstranslation
NadE NAD synthase energy
PspA phage shock protein A membrane
RacX amino acid racemase cell wall
YceC similar to tellurium resistance protein cell envelope
YceH similar to toxic anion resistance protein cell envelope
YdaG general stress protein general stress
YdbD similar to manganese‐containing catalase oxidative stress
YjdA similar to 3‐ketoacyl‐acyl‐carrier protein reductase membrane
YoxD similar to 3‐oxoacyl‐acyl‐carrier protein reductase membrane
Marker proteins
P Appendix
395

Treatment parameters
Minimal inhibitory concentration 0.1 µg/mL
Optimal stressor concentration 0.5 µg/mL
Bacteriolytic concentration n.d.
Bacterial response analyzed after 15 min
Triclosan
Mechanistic properties
Mode of action FAB inhibition
Molecular target FabI
Depolarization n.d.
Permeabilization n.d.
Cell wall integrity n.d.
General information
Source synthetic
MW 289.53 g/mol
Ref. Vischer & Regös, 1974
control/triclosan
chemicalbook.com
P Appendix
396

Mechanism of action
Triclosan interferes with fatty acid biosynthesis by competetively inhibiting the enoyl‐ACPreductase FabI [Heath et al., 1999].
Protein ID Protein function Functional category
FabHA 3‐oxoacyl‐ACP synthase III membrane (FAB)
FabHB 3‐oxoacyl‐ACP synthase III membrane (FAB)
FabF 3‐oxoacyl‐ACP synthase II membrane (FAB)
FabI enoyl‐ACP reductase membrane (FAB)
PlsXputative glycerol‐3‐phosphateacyltransferase
membrane (PLB)
PanB3‐methyl‐2‐oxobutanoatehydroxymethyltransferase
CoA biosynthesis
PdhC pyruvate dehydrogenase subunit E2b energy
SucD succinyl‐CoA synthetase subunit alpha energy
Marker proteins
P Appendix
397

Treatment parameters
Minimal inhibitory concentration 0.09 µg/mL
Optimal stressor concentration 0.225 µg/mL
Bacteriolytic concentration n.d.
Bacterial response analyzed after 15 min
Trimethoprim
Mechanistic properties
Mode of action folate biosynthesis inhibition
Molecular target DfrA
Depolarization n.d.
Permeabilization n.d.
Cell wall integrity n.d.
General information
Source synthetic
MW 290.32 g/mol
Ref. Cooper & Wald, 1964
control/trimethoprim
Kim et al., 2007
P Appendix
398

Mechanism of action
Trimethoprim inhibits folate biosynthesis by binding to the dihydrofolate reductase DfrA[Burchall, 1973]. Impairment of folate biosynthesis affects nucleotide synthesis andsubsequently limits ATP levels.
Protein ID Protein function Functional category
DhaS aldehyde dehydrogenase (NAD) energy
GamA glucosamine‐6‐phosphate deaminase cell wall
GsiB general stress protein general stress
GuaC GMP reductase purine metabolism
HisB imidazoleglycerol‐phosphate dehydratase histidine metabolism
HisD histidinol dehydrogenase histidine metabolism
HisF cyclase‐like protein histidine metabolism
HisH tyrosine transaminase histidine metabolism
LeuA 2‐isopropylmalate synthase amino acids
NfrASpx‐dependent FMN‐containing NADPH‐linked nitro/flavin reductase, stress protein
energy
PurA adenylosuccinate synthetase purine metabolism
PurE phosphoribosylaminoimidazole carboxylase (ATP‐dependent) purine metabolism
PurF glutamine phosphoribosyldiphosphate amidotransferase purine metabolism
PurHphosphoribosylaminoimidazole carboxamide formyltransferase
purine metabolism
PurM phosphoribosylaminoimidazole synthetase purine metabolism
PurQ phosphoribosylformylglycinamidine synthase purine metabolism
PurR transcription repressor of the pur operon purine metabolism
PyrE orotate phosphoribosyltransferase pyrimidine metabolism
RocA 3‐hydroxy‐1‐pyrroline‐5‐carboxylate dehydrogenase amino acids
Spo0F phosphotransferase of the sporulation initiation phosphorelay sprulation
Xpt xanthine phosphoribosyltransferase unknwon
YaaQ unknown purine metabolism
YdaG general stress protein general stress
YdbD similar to manganese‐containing catalase oxidative stress
YhfE similar to glucanase energy
YhxAsimilar to adenosylmethionine‐8‐amino‐7‐oxononanoate aminotransferase
unknown
YkzA general stress protein general stress
YsnF general stress protein, survivsl of ethanol stress general stress
YtxH general stress protein general stress
YukJ unknown unknown
YvfW actate catabolic enzyme energy
YvyDgeneral stress protein, required for ribosome dimerization in the stationary phase
general stress
Marker proteins
P Appendix
399

Treatment parameters
Minimal inhibitory concentration > 50 µg/mL
Optimal stressor concentration 10 µg/mL
Bacteriolytic concentration >50 µg/mL
Bacterial response analyzed after 15 min
Valinomycin
Mechanistic properties
Mode of action potassium carrier ionophore
Molecular target phospholipids
Depolarization yes
Permeabilization no
Cell wall integrity not disturbed
General information
Source Streptomyces spp.
MW 1111 g/mol
Ref. MacDonald, 1960
control/valinomycin
Kroterí et al., 2010
P Appendix
400

Mechanism of action
Valinomycin is a carrier ionophore specific for potassium ions [Kroterí et al., 2010].
Protein ID Protein function Functional category
BglH phospho‐beta‐glucosidase energy
CitZ citrate synthase energy
Dps iron storage protein, general stress protein general stress
MelA alpha‐galactosidase energy
NadE NAD synthase energy
PspA phage shock protein A membrane
RacX amino acid racemase cell wall
SdhA succinate dehydrogenase (flavoprotein subunit) energy
SodA superoxide dismutase oxidative stress
SpoVGnegative effector of asymetric septation at the onset of
sporulation sporulation
YceC similar to tellurium resistance protein cell envelope
YceH similar to toxic anion resistance protein cell envelope
YdbD similar to manganese‐containing catalase oxidative stress
YjdA similar to 3‐oxoacyl‐acyl‐carrier protein reductase membrane
YtxH general stress protein general stress
YuaI control of membrane fluidity membrane
YvaB similar to NAD(P)H dehydrogenase (quinone) membrane
YvIB unknown cell envelope
Marker proteins
P Appendix
401

2 Curriculum vitae
Personal information
Name Michaela Wenzel
Born November 28th, 1986 in Essen
Family status unmarried
Nationality German
Education
12/2009 ‐ presence PhD studies in Biology (Fast Track), Ruhr‐Universität Bochum
10/2009 ‐ 10/2010 Master of Science‐equivalent studies, Ruhr‐Universität Bochum
Average grade 1.1
10/2006 ‐ 08/2009 Bachelor of Science studies in Biology, Ruhr‐Universität Bochum
Average grade 1.4
08/1997 – 06/2006 Viktoriaschule, Essen
08/1993 – 07/1997 Grundschule am Wasserturm, Essen
Work experience
12/2009 ‐ presence Research scientist, Ruhr‐Universität Bochum
04/2009 ‐ 10/2009 Student assistant, Ruhr‐Universität Bochum
Awards
06/2012 1st Price DGPF Young Talent Award,
19th Arbeitstagung “Micromethods in Protein Chemistry”
Service
02/2013 ‐ 03/2015 Appointed Chair of the Gordon Research Seminar “Antimicrobial
Peptides”, Lucca, Italy. to be held in spring 2015.
06/2011 ‐ 02/2013 Associate Chair of the Gordon Research Seminar “Antimicrobial
Peptides”, Ventura, CA, USA. February 23rd ‐ 24th, 2013.
P Appendix
402

3 Contributions to the integrated publications and manuscripts
[1] Wenzel M, Patra M, Albrecht D, Chen DYK, Nicolaou KC, Metzler‐Nolte N, Bandow JE.
Proteomic signature of fatty acid biosynthesis inhibition available for in vivo mechanism
of action studies. Antimicrob Agents Chemother 55, 2590‐6 (2011).
90% design, execution, and evaluation of experiments and writing of the manuscript
[2] Wenzel M, Kohl B, Münch D, Raatschen N, Albada HB, Hamoen L, Metzler‐Nolte N, H
Sahl HG, Bandow JE. Proteomic response of Bacillus subtilis to lantibiotics reflects
differences in interaction with the cytoplasmic membrane. Antimicrob Agents
Chemother 56, 5749‐57 (2012).
75% design, 30% execution and evaluation of experiments, 75% writing of the
manuscript
[3] Albada HB, Chiriac AI, Wenzel M, Penkova M, Bandow JE, Sahl HG, Metzler‐Nolte N.
Modulating the activity of short arginine‐tryptophan containing antibacterial peptides
with N‐terminal metallocenoyl groups. Beilstein J Org Chem 8, 1753‐64 (2012).
20% design, execution, and evaluation of experiments and writing of the manuscript
[4] Wenzel M, Chiriac AI, Otto A, Zweytick D, May C, Schumacher C, Albada HB, Penkova M,
Krämer U, Erdmann R, Metzler‐Nolte N, Brötz‐Oesterhelt H, Becher D, Sahl HG, E.
Bandow JE. An antimicrobial peptide delocalizes peripheral membrane proteins.
submitted.
75% design, 60% execution, and 75% evaluation of experiments, 75% writing of the
manuscript
[5] Wenzel M, Schriek P, Prochnow P, Albada HB, Metzler‐Nolte N, Bandow JE. Influence of
lipidation on the mechanism of action of an RW‐rich antimicrobial peptide. in
preparation.
75% design, 25% execution, 50% evaluation of experiments, 90% writing of the
manuscript
P Appendix
403

[6] Wenzel M, Gust R, Bürger M, H Albada HB, Penkova M, Metzler‐Nolte N, Erdmann R,
Bandow JE. Quantitative tracing of ruthenocene derivatives for subcellular localization of
antimicrobial peptides in bacteria. in preparation
90% design, 75% execution, 90% evaluation of experiments, 90% writing of the
manuscript
[7] Chiriac AI*, Wenzel M*, Zweytick D, Schumacher C, Albada HB, Krämer J, Penkova M,
Brötz‐Oesterhelt H, Metzler‐Nolte N, Sahl HG, Bandow JE. Ferrocene‐ and ruthenocene‐
specific modulation of the mechanism of action of metal‐substituted short antimicrobial
peptides. in preparation
40% design, execution, and evaluation of experiments and writing of the manuscript
[8] Wenzel M*, Patra M*, Senges CHR, Ott I, Stepanek JJ, Pinto A, Prochnow P, Vuong C,
Langklotz S, Metzler‐Nolte N, Bandow JE. Analysis of the mechanism of action of potent
antibacterial hetero‐tri‐organometallic compounds ‐ a structurally new class of
antibiotics. ACS Chem Biol. doi: 10.1021/cb4000844.
40% design, 70% execution, and evaluation of experiments and 80% writing of the
manuscript
[9] Patra M, Wenzel M, Prochnow P, Gasser G, Bandow JE, Metzler‐Nolte N. Structural
optimization of an antibacterial hetero‐tri‐organometallic compound: Identification of
the redundant organometallic moiety required for antibacterial activity. in preparation.
25% design and execution, 50% evaluation of experiments, 30% writing of the
manuscript
* equally contributed
404

4 Erklärung
Hiermit erkläre ich, dass ich die Arbeit selbstständig verfasst und bei keiner anderen Fakultät
eingereicht und dass ich keine anderen als die angegebenen Hilfsmittel verwendet habe. Es
handelt sich bei der heute von mir eingereichten Dissertation um sechs in Wort und Bild völlig
übereinstimmende Exemplare.
Weiterhin erkläre ich, dass digitale Abbildungen nur die originalen Daten enthalten und in
keinem Fall inhaltsverändernde Bildbearbeitung vorgenommen wurde.
Bochum, den 15.04.2013
(Unterschrift)
P Appendix
405
Copyright © 2022 FDOKUMEN




















