
Attenuated, flow-induced ATP release contributes to absence of flow-sensitive, purinergic Cai2+...
14
doi: 10.1152/ajprenal.90542.2008 296:F1464-F1476, 2009. First published 25 February 2009; Am J Physiol Renal Physiol Rossetti, Peter C. Harris, Angela Wandinger-Ness, Robert L. Bacallao and Seth L. Alper Chang Xu, Boris E. Shmukler, Katherine Nishimura, Elzbieta Kaczmarek, Sandro human ADPKD cyst epithelial cells signaling in 2+ i absence of flow-sensitive, purinergic Ca Attenuated, flow-induced ATP release contributes to You might find this additional info useful... for this article can be found at: Supplementary material DC1.html http://ajprenal.physiology.org/http://ajprenal.physiology.org/content/suppl/2009/03/30/90542.2008. 64 articles, 39 of which you can access for free at: This article cites http://ajprenal.physiology.org/content/296/6/F1464.full#ref-list-1 6 other HighWire-hosted articles: This article has been cited by http://ajprenal.physiology.org/content/296/6/F1464#cited-by including high resolution figures, can be found at: Updated information and services http://ajprenal.physiology.org/content/296/6/F1464.full found at: can be American Journal of Physiology - Renal Physiology about Additional material and information http://www.the-aps.org/publications/ajprenal This information is current as of November 22, 2012. 0363-6127, ESSN: 1522-1466. Visit our website at http://www.the-aps.org/. Rockville Pike, Bethesda MD 20814-3991. Copyright © 2009 the American Physiological Society. ISSN: volume and composition. It is published 12 times a year (monthly) by the American Physiological Society, 9650 relating to the kidney, urinary tract, and their respective cells and vasculature, as well as to the control of body fluid publishes original manuscripts on a broad range of subjects American Journal of Physiology - Renal Physiology by guest on November 22, 2012 http://ajprenal.physiology.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Attenuated, flow-induced ATP release contributes to absence of flow-sensitive, purinergic Cai2+...

doi: 10.1152/ajprenal.90542.2008296:F1464-F1476, 2009. First published 25 February 2009;Am J Physiol Renal Physiol
Rossetti, Peter C. Harris, Angela Wandinger-Ness, Robert L. Bacallao and Seth L. AlperChang Xu, Boris E. Shmukler, Katherine Nishimura, Elzbieta Kaczmarek, Sandrohuman ADPKD cyst epithelial cells
signaling in2+iabsence of flow-sensitive, purinergic Ca
Attenuated, flow-induced ATP release contributes to
You might find this additional info useful...
for this article can be found at: Supplementary material
DC1.htmlhttp://ajprenal.physiology.org/http://ajprenal.physiology.org/content/suppl/2009/03/30/90542.2008.
64 articles, 39 of which you can access for free at: This article citeshttp://ajprenal.physiology.org/content/296/6/F1464.full#ref-list-1
6 other HighWire-hosted articles: This article has been cited by http://ajprenal.physiology.org/content/296/6/F1464#cited-by
including high resolution figures, can be found at: Updated information and serviceshttp://ajprenal.physiology.org/content/296/6/F1464.full
found at: can beAmerican Journal of Physiology - Renal Physiology about Additional material and information
http://www.the-aps.org/publications/ajprenal
This information is current as of November 22, 2012.
0363-6127, ESSN: 1522-1466. Visit our website at http://www.the-aps.org/. Rockville Pike, Bethesda MD 20814-3991. Copyright © 2009 the American Physiological Society. ISSN: volume and composition. It is published 12 times a year (monthly) by the American Physiological Society, 9650relating to the kidney, urinary tract, and their respective cells and vasculature, as well as to the control of body fluid
publishes original manuscripts on a broad range of subjectsAmerican Journal of Physiology - Renal Physiology
by guest on Novem
ber 22, 2012http://ajprenal.physiology.org/
Dow
nloaded from

Attenuated, flow-induced ATP release contributes to absenceof flow-sensitive, purinergic Cai
2� signaling in human ADPKDcyst epithelial cells
Chang Xu,1,2 Boris E. Shmukler,1,2 Katherine Nishimura,1,2 Elzbieta Kaczmarek,3 Sandro Rossetti,4
Peter C. Harris,4 Angela Wandinger-Ness,5 Robert L. Bacallao,6 and Seth L. Alper1,2
1Molecular and Vascular Medicine and Renal Divisions, Beth Israel Deaconess Medical Center and Departmentsof 2Medicine and 3Surgery, Harvard Medical School, Boston, Massachusetts; 4Departments of Medicine and Biochemistry,Mayo Medical School, Rochester, Minnesota; 5Department of Pathology, University of New Mexico School of Medicine,Albuquerque, New Mexico; and 6Department of Medicine, University of Indiana School of Medicine, Indianapolis, Indiana
Submitted 11 September 2008; accepted in final form 24 February 2009
Xu C, Shmukler BE, Nishimura K, Kaczmarek E, Rossetti S,Harris PC, Wandinger-Ness A, Bacallao RL, Alper SL. Attenuated,flow-induced ATP release contributes to absence of flow-sensitive, puri-nergic Cai
2� signaling in human ADPKD cyst epithelial cells. Am JPhysiol Renal Physiol 296: F1464–F1476, 2009. First published Febru-ary 25, 2009; doi:10.1152/ajprenal.90542.2008.—Flow-induced cytoso-lic Ca2� Cai
2� signaling in renal tubular epithelial cells is mediated inpart through P2 receptor (P2R) activation by locally released ATP.The ability of P2R to regulate salt and water reabsorption hassuggested a possible contribution of ATP release and paracrine P2Ractivation to cystogenesis and/or enlargement in autosomal dominantpolycystic kidney disease (ADPKD). We and others have demon-strated in human ADPKD cyst cells the absence of flow-induced Cai
2�
signaling exhibited by normal renal epithelial cells. We now extendthese findings to primary and telomerase-immortalized normal andADPKD epithelial cells of different genotype and of both proximaland distal origins. Flow-induced elevation of Cai
2� concentration([Ca2�]i) was absent from ADPKD cyst cells, but in normal cells wasmediated by flow-sensitive ATP release and paracrine P2R activation,modulated by ecto-nucleotidase activity, and abrogated by P2R inhi-bition or extracellular ATP hydrolysis. In contrast to the elevated ATPrelease from ADPKD cells in static isotonic conditions or in hypo-tonic conditions, flow-induced ATP release from cyst cells was lowerthan from normal cells. Extracellular ATP rapidly reduced thapsigar-gin-elevated [Ca2�]i in both ADPKD cyst and normal cells, but cystcells lacked the subsequent, slow, oxidized ATP-sensitive [Ca2�]i
recovery present in normal cells. Telomerase-immortalized cyst cellsalso exhibited altered CD39 and P2X7 mRNA levels. Thus the loss offlow-induced, P2R-mediated Cai
2� signaling in human ADPKD cystepithelial cells was accompanied by reduced flow-sensitive ATPrelease, altered purinergic regulation of store-operated Ca2� entry,and altered expression of gene products controlling extracellularnucleotide signaling.
autosomal dominant polycystic kidney disease; telomerase; monoci-lium; shear stress; luciferase; fura 2
AUTOSOMAL DOMINANT POLYCYSTIC kidney disease (ADPKD) ischaracterized by progressive enlargement of fluid-filled cystsoriginating from only 1–5% of nephrons. The disease affectsbetween 1:400 and 1:1,000 people, and leads to end-stage renaldisease in half of those affected. Between 70 and 85% of casesare caused by mutations in the PKD1 gene encoding the 4,302
amino acid (aa) protein polycystin-1 (PC1). PC1 includes a�3,000 aa NH2-terminal extracellular domain, � 11 trans-membrane spans, and a �200 aa COOH-terminal cytoplasmictail. Most or all other cases of ADPKD are caused by mutationsin the 968 aa polycystin-2 (PC2) (52), a cation channel inter-acting with and regulated by PC1, and forming heteromerswith the TRPV4 and TRPC1 cation channels (1, 24, 53).
ATP release is a property shared by most cell types sub-jected to mechanical deformation (10, 41, 55). Epithelialmonolayers of renal, respiratory, and gastrointestinal originrelease ATP from both apical and basolateral surfaces (14, 20,22, 28). The released nucleotides achieve concentrations suf-ficient to activate purinergic receptors, in turn regulating se-cretion and reabsorption of ions and water (26, 27, 35, 55).These effects of ATP on solute and water transport haveencouraged the hypothesis that ATP release from cyst epithe-lial cells regulates cyst enlargement and possibly also cysto-genesis in ADPKD (48, 60).
ADPKD cyst epithelial cells grown on permeable supportsin primary culture released ATP at higher rates at rest andunder hypotonic stress than did cells isolated from normalkidneys (60). ATP release can be detected in both apical andbasolateral compartments. Cyst epithelial cells express P2Xand P2Y receptors, which, upon activation, elevated cytosolicCa2� concentration ([Ca2�]i) and stimulated Cl� secretion byconfluent cyst cell monolayers. Cysts microdissected fromADPKD kidneys also released ATP, and cyst fluid contained asmuch as 10 �M ATP, suggesting that ATP release from cystepithelial cells might regulate cyst enlargement in ADPKD(48, 60). However, purinergic signaling may produce opposingeffects on models of dominant and recessive cystic kidneydisease (18, 54), and the role of purinergic signaling in earlycystogenesis remains unclear.
Flow stimulates Cai2� signaling in epithelial monolayers
(42) by mechanisms proposed to include bending of the pri-mary cilium (40, 43). Flow also regulates [Ca2�]i in intactrenal tubules (14, 22, 27), contributing to modulation of ionand water transport in response to changes in luminal soluteload and/or concentration (47, 55). The flow-induced Cai
2�
signal evident in primary renal cortical epithelial cell mono-layers isolated from mouse (33) and human kidney (34, 63)was absent from Pkd1�/� mouse embryonic collecting ductepithelial cells or cells isolated from human ADPKD cystsexpressing lectin markers of nominal distal or proximal origin(33, 34, 63). Isolated, perfused collecting ducts from the
Address for reprint requests and other correspondence: S. L. Alper, Molec-ular and Vascular Medicine and Renal Divs., Beth Israel Deaconess MedicalCenter, 330 Brookline Ave., E/RW763, Boston, MA 02215 (e-mail: [email protected]).
Am J Physiol Renal Physiol 296: F1464–F1476, 2009.First published February 25, 2009; doi:10.1152/ajprenal.90542.2008.
0363-6127/09 $8.00 Copyright © 2009 the American Physiological Society http://www.ajprenal.orgF1464
by guest on Novem
ber 22, 2012http://ajprenal.physiology.org/
Dow
nloaded from

orpk/orpk mouse model of recessive polycystic kidney dis-ease exhibited attenuated flow-induced [Ca2�]i signalingapparent by postnatal day 14 (29). Reduced or absentflow-induced [Ca2�]i signaling also characterized primaryembryonic orpk/orpk collecting duct epithelial monolayers,albeit accompanied by increased resting rates of apical Ca2�
entry (21, 50). In contrast, primary human ARPKD cystepithelial cell monolayers exhibited enhanced flow-induced[Ca2�]i signaling (44).
Since flow stimulates both ATP release and elevation of[Ca2�]i, the relationship between these two events is of inter-est. Flow-stimulated elevation of [Ca2�]i in isolated, perfusedmouse medullary thick ascending limb (MTAL) was attenuatedby P2R blockade and by extracellular ATP hydrolysis (22), andwas reduced in MTAL from P2y2�/� mice (22). P2R inhibi-tion and extracellular ATP hydrolysis reduced spontaneous[Ca2�]i oscillations in resting Madin-Darby canine kidney(MDCK) monolayers and in perfused mouse MTAL. Sponta-neous [Ca2�]i oscillation amplitude was also reduced in am-plitude in a P2R-deficient cell line and in the perfused P2y2�/�
MTAL (14). P2R inhibition and ATP hydrolysis nearly abol-ished flow-induced elevation of [Ca2� ]i in established humanand rat cholangiocyte cell lines (62).
We therefore tested the hypotheses that the normal flow-induced elevation of [Ca2�]i that is deficient in primary humanADPKD cyst epithelial cells is mediated by paracrine puriner-gic signaling, and that defective purinergic signaling mightexplain in part the loss of flow-sensitive Cai
2� elevation inADPKD cells.
We found that flow-induced Cai2� signaling of primary
human epithelial cells is preserved in telomerase-immortalizednormal kidney epithelial (NK cell lines), and its absence inADPKD cyst cells is preserved in telomerase-immortalized celllines of defined germline PKD1 genotype. ADPKD cyst cellsexhibited enhanced resting and hypotonicity-induced ATP re-lease. In contrast, flow-induced ATP release from ADPKD cystcells was markedly reduced compared with that of normal renalepithelial cells, despite near-normal nucleotide-induced peak[Ca2�]i elevations in cyst cells. Purinergic control of [Ca2�]i inthapsigargin-pretreated cells was also altered in ADPKD cystcells, and thapsigargin-sensitive stores were reduced. Varia-tions in P2R and ecto-nucleotidase gene expression by cyst celllines were also detected. We conclude that loss of flow-sensitive Cai
2� signaling in human ADPKD cyst epithelial cellsis due in part to reduced flow-sensitive ATP release and isaccompanied by altered purinergic responsiveness in the set-ting of nominal store depletion.
METHODS
Cell culture. Discarded portions of human kidney cortex wereharvested according to CCI/IRB protocols reviewed and approved atthe Indiana University School of Medicine and Beth Israel DeaconessMedical Center. NK cortical tubular epithelial cells from individualNK57M03 and ADPKD cyst cells from an anonymized ADPKDpatient undergoing nephrectomy were grown in primary culture asdescribed (63). Aliquots of NK cells from the same individual(NK57M03) and aliquots of ADPKD cyst cells from a different,anonymized ADPKD patient kidney were similarly grown in primaryculture. These pools of primary cells were referred to as NK and PKDcells (Supplemental Table 1; all supplementary material for this articleare accessible on the journal web site).
Primary NK and PKD cells were subjected to fluorescence-activatedcell sorting (FACS). Cells were separated according to intensity of thebound fluorophore-conjugated lectins Lotus tetragonolobus agglutinin(LTA; a marker of proximal nephron origin) and Dolichos biflorusagglutinin (DBA; a marker of distal nephron origin; Vector Laboratories,Burlingame, CA) (6, 49, 63). Marker-enriched, sorted cell pools weretransformed with human telomerase (hTERT) cDNA as previously de-scribed (5, 65). The EcoRI fragment from pGRN145 encoding hTERTcDNA was subcloned into retroviral vector pBABEneo. Recombinantviruses were generated by electroporation-transfection of the ecotropicpackaging line PE501 followed by selection with 400 �g/ml G418.Supernatants containing amphotropic virus released from confluent cul-tures were filtered through 0.45-�m filters. This virus preparation wasused to infect NK and PKD cells, which were clonally selected in G418and expanded.
Cloned cells were maintained in Clonetics REGM Renal EpithelialCell Medium (Lonza, Portsmouth, NH) in the presence of penicillin/streptomycin and G418. After seven passages, G418 was removedfrom the growth medium. Cells for experiments were transferred toglass coverslips coated with Vitrogen collagen (Conhesion Technol-ogies, Palo Alto, CA), fed every other day with REGM, and grown toconfluence 5–6 days after plating, at which time they were used forexperiments. Primary cells were used through passage 6 only. ThehTERT-transformed cells were passaged weekly and maintained astable growth rate and phenotype through passage 34 and beyond(Bacallao R, unpublished observations). These hTERT-immortalizedcell lines were referred to as LTA�-NKTERT, DBA�-NKTERT,LTA�-PKDTERT, and DBA�-PKDTERT (Supplemental Table 1).
Genomic DNA analysis. Genomic DNA was extracted from LTA�-PKDTERT cells by the salting-out method. All coding exons and splicejunctions of the PKD1 and PKD2 genes were amplified by PCR fromgenomic DNA (46, 63) and directly sequenced on both strands. Theduplicated region of the PKD1 gene was amplified with rTth DNApolymerase (PerkinElmer Applied Biosystems, Foster City, CA) inthe supplied DMSO-containing buffer. Primers were either anchoredin the single-copy region of the gene or mismatched with the homol-ogous gene sequences to generate five, locus-specific Long RangePCR fragments of 3–9 kb in length. The DNA sequencing allowedidentification of heterozygous variation in gene sequence. Any muta-tion found was confirmed with a second, independent amplification.
RNA preparation and RT-PCR. Total RNA from NK and PKD cystcells was isolated with an RNeasy kit (Qiagen, Valencia, CA). Humankidney total RNA was purchased from Ambion (Woodlands, TX).Reverse transcription was performed with a Retroscript first-strandcDNA synthesis kit (Ambion) using 1 �g of total RNA. Of thereaction volume, 5% was used for PCR with HotStart DNA polymer-ase (Qiagen) in a total reaction volume of 50 �l in the supplier’srecommended buffer. cDNA fragments were amplified using specificprimer pairs and annealing conditions for P2Y and P2X receptorproducts (Supplemental Table 2) and for CD39 family ecto-nucleo-side triphosphate diphosphohydrolase products (NTPDases; Supple-mental Table 3). Enzyme activation and initial template denaturationwere at 95°C for 15 min, followed by 35–38 cycles of 45 s at 94°C,2 min at 52–60°C, and 2 min at 72°C, with a 7-min final extensionstep at 72°C. PCR products were separated on 1% agarose gels,visualized with ethidium bromide, purified with a Qiagen Gel Extrac-tion kit, and validated by DNA sequence analysis. PCR productabundance was documented (GelDoc, Bio-Rad) and semiquantitatedwith ImageJ (National Institutes of Health). PCR cycle number wasadjusted to ensure detection of low-abundance transcripts and tomaintain higher abundance transcripts within the log-linear range ofamplification.
Confocal immunofluorescence microscopy. Cell monolayers grownon glass coverslips were fixed for 30 min at room temperature with140 mM NaCl and 10 mM sodium phosphate, pH 7.4 (PBS) contain-ing 3% (wt/vol) paraformaldehyde. Fixed cells were extensivelyrinsed with PBS, quenched with 50 mM lysine HCl, pH 8.0, exposed
F1465PARACRINE PURINERGIC MEDIATION OF FLOW-INDUCED Ca SIGNALING
AJP-Renal Physiol • VOL 296 • JUNE 2009 • www.ajprenal.org
by guest on Novem
ber 22, 2012http://ajprenal.physiology.org/
Dow
nloaded from

to 1% SDS in PBS for 15 min for permeabilization and epitopeunmasking, and blocked for 15 min in PBS with 1% bovine serumalbumin and 0.05% saponin. For lectin staining, cells were incubatedfor 2 h with FITC-conjugated LTA or with FITC-conjugated DBA,then washed. For PC1 and PC2 staining, after 4°C overnight incuba-tion with 1:100 dilutions of rabbit polyclonal antibody NM005 againsthuman PC1 COOH-terminal aa 4070–4302 or rabbit polyclonalantibody against GST fusion protein with human PC2 C-terminal aa687–968, coverslips were incubated with Cy3-conjugated donkeyanti-rabbit Ig secondary antibody (1:500, Jackson ImmunoResearch,West Grove, PA) for 2 h at room temperature as described (63). Somecoverslips immunostained and fixed as above for PC1 or PC2 weresubsequently stained with mouse monoclonal antibody to the ciliarymarker N-acetylated �-tubulin (1:500, Sigma, St. Louis, MO). P2X7immunostaining was performed by overnight incubation (1:100) withpolyclonal antibody to rat P2X7 intracellular COOH-terminal tail aa576–595 KIRKEFPKTQGQYSGFKYPY (Alomone, Jerusalem, Is-rael). Anti-CD39 immunostaining with mAb clone BU-61 (Bayport,MN) was performed by overnight incubation of fixed cells, followedby PBS wash and visualization with Cy3-conjugated donkey anti-mouse Ig secondary antibody (1:500). In some experiments as indi-cated, antibodies were pre- and coincubated with excess specificpeptide antigen. Immunostained cells on coverslips were imaged witha Zeiss LSM 510 Meta confocal laser scanning microscope.
Measurement of [Ca2�]i. NK and PKD cells cultured to confluenceon collagen-coated 35-mm glass coverslips were loaded with 5 �Mfura 2-AM (Molecular Probes, Eugene, OR) in HEPES-bufferedHBSS (pH 7.4) at 37°C for 30 min. Extracellular fura 2-AM wasremoved by washing twice with HEPES-HBSS. Coverslips were thenmounted into the base of a parallel-plate flow chamber 0.5 cm in widthand 0.0254 cm in depth (GlycoTech, Gaithersburg, MD) and perfusedwith a Harvard Syringe pump at 37°C using a calibrated, in-line heater(WPI, Sarasota, FL), with monitoring of inflow and outflow temper-atures. Perfusion medium (Hanks’ solution) composition was (in mM)127 NaCl, 5.4 KCl, 1.27 CaCl2, 1 MgCl2, 5.6 glucose, and 11.6HEPES, final pH 7.4. [Ca2�]i was measured by fluorescence ratioimaging with a Metafluor digital imaging system (Universal Imaging,West Chester, PA) equipped with an Olympus IMT-2 inverted micro-scope, and a CoolSNAP CCD camera (Photometrics, Tucson, AZ).Fura 2 emission images were recorded at 510 nm during alternatingexcitation at 340 and 380 nm. Fura 2 fluorescence ratio valuesdetermined by in situ calibration in immortalized epithelial cells didnot differ from values determined by in vitro calibration for [Ca2�]i,
and so were calibrated in vitro with the same experimental settings forthe imaging system, using a Ca2� Calibration Kit No. 2 (MolecularProbes) with concentrations between 36 nM and 4 �M (32). Theminimal fluorescence ratio (Rmin) was determined at “zero Ca2�”(�10 nM) and the maximal fluorescence ratio (Rmax) at 4 �M totalCa2�. The equilibrium constant (Kb) was determined by fitting ex-perimental fluorescence ratio R values at various free [Ca2�] with theequation [Ca2�]free � Kb (Sf2/Sb2)[(R � Rmin)/(Rmax � R)], wherethe factor Sf2/Sb2 corrects for fura 2 ion selectivity at 380 nm. Foreach coverslip, one visual field was selected as a region of interest,recorded before and during imposition of a uniform rate of fluid flow.Shear stress � was calculated as � � 6�Q/a2b, where � � apparentviscosity of superfusate (1.00 for H2O at 20°C, 0.70 at 37°C), Q �volumetric flow rate (ml/s), and a and b � flow chamber depth andwidth, respectively.
Measurement of ATP release. To measure flow-induced ATPrelease, confluent monolayers of 5 104 NK or PKD cells mountedin the Glycotech chamber were subjected to defined laminar flow ratesat 37°C as described above. Perfusate volumes of at least 200 �l werecollected at a single time for each flow rate. At 0.1 dyne/cm2 (flow of50 �l/min), the collection period was 6 min (including 2 min to fill the100 �l of initially empty postchamber dead space at the start of flow,followed by 4 min to collect 200 �l containing ATP released underflow, in addition to that accumulated in the 25-�l chamber volume
during the 15-min preincubation period before the initiation of flow).At both 0.75 dyne/cm2 (flow of 320 �l/min) and 10 dyne/cm2 (flow of5,000 �l/min), the collection period was 1 min.
Tonicity-regulated ATP release was measured after 30-min expo-sure of 5 105 confluent cells in 35-cm culture dishes to isotonicHanks’ solution or to a hypotonic 4:6 vol/vol mixture of Hanks’solution and distilled water, with a relative osmolarity of 40%. ATPconcentration was measured by luciferin-luciferase assay (Sigma).Duplicate 100-�l aliquots of perfusate or incubation medium wereadded to glass tubes containing 100 �l ATP assay mix dilution buffer(25-fold dilution), and luminescence intensity was measured (LumatLB 9507, Berthold, Oak Ridge, TN). Flow-induced ATP release from5 104 cells was expressed as picomoles per minute. Tonicity-regulated ATP release from 5 105 cells during a 30-min period wasexpressed as picomoles. Identity of the ATP-associated luciferasesignal was confirmed by its complete abolition after apyrase treat-ment.
ATP concentrations of samples were extrapolated from a calibra-tion curve constructed with ATP standards in isotonic Hanks’ solu-tion. Although chloride can inhibit luciferase (8, 19), several subse-quent studies of ATP release by renal and respiratory epithelial cellsexposed to low [Cl�] hypotonic solutions have shown luciferase lightemission across this range of [Cl�] to not differ significantly (21, 45,51, 60) or to differ by �10% (35).
Statistics. Data are presented as means � SE for n measurements.Means of the data were compared by Student’s two-tailed unpairedt-test, with P � 0.05 defined as the threshold for significance.
RESULTS
Shear stress elevates [Ca2�]i in both primary and telomer-ase-immortalized NK epithelial cells. We showed previouslythat confluent primary human NK cells expressing predom-inantly the proximal nephron marker LTA exhibit flow-sensitive Cai
2� signaling, whereas predominantly LTA�
primary ADPKD cyst cells harboring the germline mutation�L2433 lack this response (63). Previous reports havesuggested that shear sensitivity of human and mouse NKcells expressing the distal marker DBA (33, 34) is higherthan that of human primary LTA�-NK cells (63). Wetherefore generated and studied hTERT-immortalized clonalisolates of NK and ADPKD cyst cells of nominal proximalorigin (LTA�) and distal origin (DBA�). The latter areshown in Supplemental Fig. 1.
LTA�-PKDTERT cells harbor the heterozygous C-to-T transi-tion at nucleotide 12010 of PKD1 cDNA (counting A of the ATGimitator codon as �1), converting amino acid residue Q4004 ofpolycystin-1 (PC1) to a nonsense codon (Q4004X; SupplementalFig. 2). The previously reported PKD1 Q4004X mutation (39)encodes a polypeptide lacking the COOH-terminal 300 aa of PC1,a region encompassing terminal transmembrane spans and theCOOH-terminal cytoplasmic tail. No mutation was found in thecoding region of the PKD2 gene of LTA�-PKDTERT cells.
LTA�-NKTERT cells subjected to superfusion at low (Fig. 1A,0.75 dyne/cm2) and high (Fig. 1C, 10 dyne/cm2) levels of shearstress transiently elevated [Ca2�]i, whereas LTA�-PKDTERT
cyst cells lines failed to elevate [Ca2�]i in response to superfusionat either low (Fig. 1B) or high (Fig. 1D) shear stress. Similardifferences between LTA�-NKTERT and LTA�-PKDTERT cellswere observed at shear stress levels of 0.32 and 2.3 dyne/cm2
(not shown). [Ca2�]i elevation in NK cells remained at asubmaximal plateau value in the presence of higher shear stress(Fig. 1C). Thus hTERT immortalization and cell cloning did
F1466 PARACRINE PURINERGIC MEDIATION OF FLOW-INDUCED Ca SIGNALING
AJP-Renal Physiol • VOL 296 • JUNE 2009 • www.ajprenal.org
by guest on Novem
ber 22, 2012http://ajprenal.physiology.org/
Dow
nloaded from

not change the flow-sensitivity phenotypes of either LTA�-NKor LTA�-PKD cells.
DBA�-NKTERT cells also elevated [Ca2�]i in response tolow levels of shear stress (Fig. 2A), with a peak response atlower shear stress (2.3 dyne/cm2) than that exhibited by pri-mary (63) and by LTA�-NKTERT cells at 10 dyne/cm2 (Fig. 1).In contrast, [Ca2�]i in DBA�-PKDTERT cells was unresponsiveto either low (Fig. 2B) or high levels (Fig. 2D) of shear stress.As was true for primary NK and �L2433 PKD cells (63),resting [Ca2�]i did not differ between LTA�-NKTERT andQ4004X LTA�-PKDTERT cyst cells (not shown). Thus the lossof flow-induced [Ca2�]i elevation characterizes primary andimmortalized ADPKD cyst cells of distinct nephron segmentorigin, and bearing distinct heterozygous germline mutations.
LTA�-PKDTERT cells form primary cilia lacking PC1.Among their multiple sites of subcellular localization (25),PC1 and PC2 are expressed along with many other cystickidney disease gene products in the primary cilium (33, 34,63). Figure 3 shows PC1 (Fig. 3A, left) and PC2 (Fig. 3A, right)in the cilium of LTA�-NKTERT cells. However, PC1 wasundetectable (Fig. 3B, left) and PC2 nearly so (Fig. 3B, right)in LTA�-PKDTERT cell cilia 5.0 � 0.2 �m in length (n � 11),indistinguishable from the 4.8 � 0.3-�m (n � 19) length ofLTA�-NKTERT cilia. Ciliary PC2 localization in the x-y planeis shown in Supplemental Fig. 3. DBA�-NKTERT cell cilia of8.7�0.4 �M in length (n � 21), and the slightly shorterDBA�-PKDTERT cilia of 7.4 � 0.3 �M in length (n � 25; P �0.05 compared with NK) each were significantly longer than
Fig. 1. Human telomerase (hTERT)-immortalized, clonal Lotus tetragonolobus agglutinin (LTA�) autosomal dominant polycystic kidney disease (ADPKD) cystcells (LTA�-PKDTERT) lack the flow-sensitive elevation in cytosolic Ca2� concentration ([Ca2�]i) exhibited by LTA�-NKTERT cells, where NK refers to normalkidney. Imposition of laminar flow at t � 0 increases [Ca2�]i in LTA� NKTERT cells at low (0.75 dyne/cm2; A) and high levels of shear stress (10 dyne/cm2;C). In contrast, laminar flow fails to increase [Ca2�]i in LTA�-PKDTERT cells in response to shear stress at low (B) or high levels (D). Values are means � SEfor (n) coverslips, with fura 2 ratio recorded from a single 20 visual field encompassing 40–60 confluent cells on each coverslip.
F1467PARACRINE PURINERGIC MEDIATION OF FLOW-INDUCED Ca SIGNALING
AJP-Renal Physiol • VOL 296 • JUNE 2009 • www.ajprenal.org
by guest on Novem
ber 22, 2012http://ajprenal.physiology.org/
Dow
nloaded from

LTA�-TERT cells (P � 0.01). Thus clonal PKDTERT cellsheterozygous for the PC1 germline mutation Q4004X resemblemixed primary PKD cyst cells with the PC1 germline mutation�L2433 in their absence of detectable ciliary PC1, but they aremore severely impaired in ciliary PC2 localization than were�L2433 primary cyst cells, 30% of which expressed apparentlynormal ciliary levels of PC2 (63). Extraciliary localization ofPC1 and PC2 in NKTERT and ADPKDTERT cells did not differ(Supplemental Fig. 4).
Shear stress-induced net ATP release is selectively reducedin PKD cyst cells compared with NK cells. NKTERT cellsreleased ATP in response to various uncontrolled types ofmechanical stimulation (not shown), including complete re-
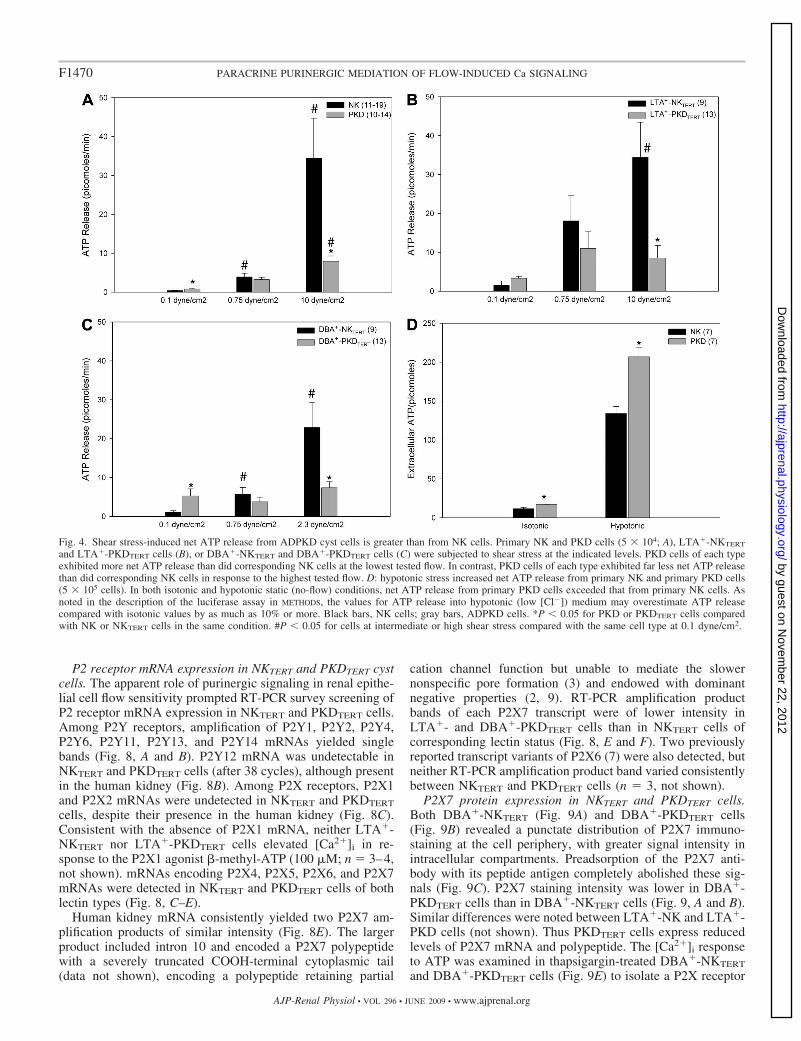
placement of medium, tilting of the monolayer, and touching ofthe cell monolayer surface with a glass pipette, as reported withother cell types (16, 59). Although slow superfusion at 0.1dyne/cm2 produced little net ATP release into the medium, thenet ATP release at this low flow rate from primary �L2433cells (Fig. 4A) and from Q4004X PKDTERT cells (Fig. 4, B andC) exceeded that from NK (Fig. 4A) or NKTERT cells (Fig. 4,B and C). This elevated net ATP release was especially markedin DBA�-PKDTERT cells (Fig. 4C), consistent with previousreports of elevated resting net ATP release by primary ADPKDcyst cells (48). Low (0.75 dyne/cm2) and high flow (10 dyne/cm2) elicited higher magnitudes of net ATP release fromprimary NK and PKD cells (Fig. 4A) and from LTA� (Fig.
Fig. 2. hTERT-immortalized, clonal Dolichos biflorus agglutinin (DBA�) ADPKD cyst cells (DBA�-PKDTERT) lack the flow-sensitive elevation in [Ca2�]i
exhibited by DBA�-NKTERT cells. Imposition of laminar flow at t � 0 increases [Ca2�]i in DBA�-NKTERT cells at low levels of shear stress (0.75 or 2.3dyne/cm2; A). Higher shear stress (10 dyne/cm2; C) slightly reduces the magnitude of peak [Ca2�]i, but prolongs peak duration. In contrast, laminar flow failsto increase [Ca2�]i in DBA�-PKDTERT cyst cells in response to shear stress at low (B) or high levels (D). Values are means � SE for (n) coverslips, with 40–60confluent cells/20 visual field on each coverslip.
F1468 PARACRINE PURINERGIC MEDIATION OF FLOW-INDUCED Ca SIGNALING
AJP-Renal Physiol • VOL 296 • JUNE 2009 • www.ajprenal.org
by guest on Novem
ber 22, 2012http://ajprenal.physiology.org/
Dow
nloaded from

4B)-or DBA� (Fig. 4C)-NKTERT and PKDTERT cells. Primary�L2433 ADPKD cyst cells (Fig. 4A) and Q4004X LTA�-PKDcells (Fig. 4B) released substantially less net ATP in responseto 10 dyne/cm2 shear stress than did the corresponding NKcells (P � 0.05). Q4004X DBA�-PKD cells (Fig. 4C) releasedless net ATP in response to 2.3 dyne/cm2 shear stress than didNK cells. In contrast, primary �L2433 PKD cyst cells at rest inisotonic and hypotonic conditions released significantly morenet ATP than did primary NK cells (Fig. 4D; P � 0.05).Apyrase treatment of the collected perfusates abolished theluciferase signal, confirming its source as released ATP (n � 3,data not shown). The applied shear stresses and times did notdetach, loosen, or morphologically alter the cells under study,and cessation of flow returned [Ca2�]i to preflow levels in allcases. The results demonstrate a selective defect in shearstress-induced net ATP release from both primary �L2433primary PKD cells and Q4004X PKDTERT cells.
Shear stress-induced ATP release mediates flow-inducedCai
2� in NK cells. We hypothesized that flow-induced ATPrelease might mediate shear stress-induced [Ca2�]i increase inNK cells. Thus primary NK cells were pretreated with P2receptor blockers or with the ATP hydrolase apyrase for 10min before the onset of flow at 10 dyne/cm2 shear stress in thecontinued presence of the drugs. As shown in Fig. 5A, flow-induced elevation in [Ca2�]i was abolished by apyrase (5U/ml) and by the semiselective P2Y receptor antagonistsuramin (300 �M). Figure 5B shows that the semiselectiveP2X receptor antagonist PPADS (100 �M) also abolished theflow-induced [Ca2�]i increase. Suramin also greatly reducedflow-induced [Ca2�]i elevation in LTA�-NKTERT cells (Fig.5C) and abolished it in DBA�-NKTERT cells (Fig. 5D). Con-versely, inhibition of ATP hydrolysis with the 5 -ecto-nucle-otidase inhibitor ARL67156 (300 �M) prolonged the plateau-phase duration of [Ca2�]i elevation in response to both highand low shear stress (Fig. 6, A and B). This potentiationsuggests that released ATP is actively degraded by cellularnucleotidase activities and that the values of net ATP released
in response to hypotonic swelling (Fig. 4D) and in response toshear stress (Fig. 4, A–C) are underestimates. The data indicatethat released ATP is a crucial intermediate in the transductionof a flow signal leading to elevation of [Ca2�]i in NK cells.Defective flow-sensitive autocrine/paracrine ATP release byPKD cyst cells likely contributes to their inability to elevate[Ca2�]i in response to flow.
PKDTERT cyst cells exhibit intact ATP-induced [Ca2�]i ele-vation but modestly reduced [Ca2�]i elevation in response toother nucleotide P2 receptor ligands. Since PKDTERT cellslack flow-responsive Cai
2� signaling, and exhibit decreasedflow-responsive net ATP release, we tested the possibility thatATP responsiveness was also deficient. Exogenous extracellu-lar ATP triggered similar [Ca2�]i responses in primary NK and�L2433 PKD cells (63). Exposure to extracellular ATP elevated[Ca2�]i in both LTA�-NKTERT and Q4004X LTA�-PKDTERT
cells, and differed only in the more prolonged and stable plateauphase of the PKDTERT cells (Fig. 7; P � 0.03 at 90 s, 0.01 at 100 scompared with NKTERT). Nominally Ca2�-free medium did notchange peak ATP-induced [Ca2�]i in PKDTERT cells but re-duced the peak by �30% in NKTERT cells. Ca2�-free mediumalso accelerated the [Ca2�]i decline from peak ATP-stimulatedvalues in both NKTERT and PKDTERT cells (Fig. 7). Similar[Ca2�]i differences between cell lines and conditions wereevident at 20°C (not shown). The nucleotide-induced Cai
2�
increase in primary NK cells was elicited by nucleotides in aconcentration-dependent manner with the rank order of po-tency UTP � ATP � ADP (Supplemental Fig. 5A) and in�L2433 PKD cells UTP � ATP � ADP (Supplemental Fig.5B). Similar peak [Ca2�]i elevations and rank orders ofpotency in response to 10 �M UTP, ADP, and ATP weremeasured in LTA� (Suppl. Fig. 5C) and DBA� (Suppl Fig.5D) clones of NKTERT and Q4004X PKDTERT cells. Thesefindings are consistent with the previously reported expres-sion of multiple functional P2X and P2Y receptor subtypesin human primary cultures of normal and ADPKD renalepithelia (48).
Fig. 3. Ciliary localization of polycystins PC1and PC2 is reduced or absent in PKDTERT
cells. Confocal x-z plane reconstructions ofLTA�-PKDTERT cells show undetectable cili-ary PC1 (B, left) and reduced levels of ciliaryPC2 (B, right), as judged by colocalizationwith N-acetylated �-tubulin in contrast tothe presence of PC1 (A, left) and PC2 (A,right) in cilia of LTA�-NKTERT cells.Scale bar � 10 �m.
F1469PARACRINE PURINERGIC MEDIATION OF FLOW-INDUCED Ca SIGNALING
AJP-Renal Physiol • VOL 296 • JUNE 2009 • www.ajprenal.org
by guest on Novem
ber 22, 2012http://ajprenal.physiology.org/
Dow
nloaded from
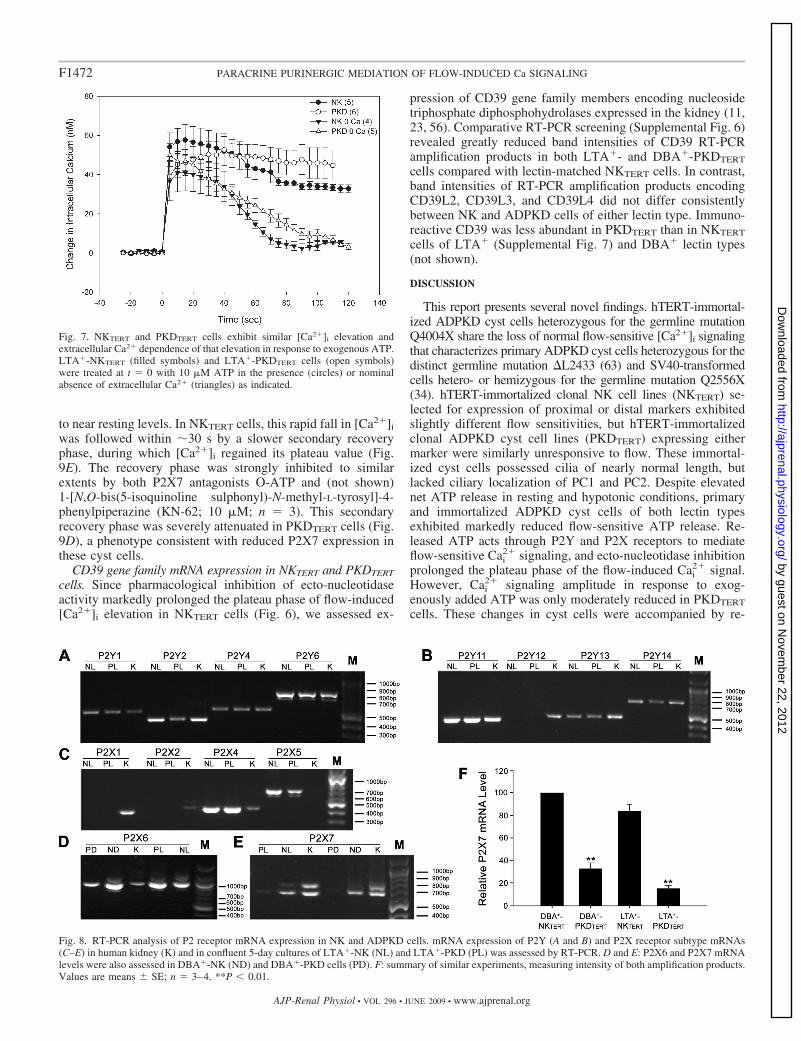
P2 receptor mRNA expression in NKTERT and PKDTERT cystcells. The apparent role of purinergic signaling in renal epithe-lial cell flow sensitivity prompted RT-PCR survey screening ofP2 receptor mRNA expression in NKTERT and PKDTERT cells.Among P2Y receptors, amplification of P2Y1, P2Y2, P2Y4,P2Y6, P2Y11, P2Y13, and P2Y14 mRNAs yielded singlebands (Fig. 8, A and B). P2Y12 mRNA was undetectable inNKTERT and PKDTERT cells (after 38 cycles), although presentin the human kidney (Fig. 8B). Among P2X receptors, P2X1and P2X2 mRNAs were undetected in NKTERT and PKDTERT
cells, despite their presence in the human kidney (Fig. 8C).Consistent with the absence of P2X1 mRNA, neither LTA�-NKTERT nor LTA�-PKDTERT cells elevated [Ca2�]i in re-sponse to the P2X1 agonist �-methyl-ATP (100 �M; n � 3–4,not shown). mRNAs encoding P2X4, P2X5, P2X6, and P2X7mRNAs were detected in NKTERT and PKDTERT cells of bothlectin types (Fig. 8, C–E).
Human kidney mRNA consistently yielded two P2X7 am-plification products of similar intensity (Fig. 8E). The largerproduct included intron 10 and encoded a P2X7 polypeptidewith a severely truncated COOH-terminal cytoplasmic tail(data not shown), encoding a polypeptide retaining partial
cation channel function but unable to mediate the slowernonspecific pore formation (3) and endowed with dominantnegative properties (2, 9). RT-PCR amplification productbands of each P2X7 transcript were of lower intensity inLTA�- and DBA�-PKDTERT cells than in NKTERT cells ofcorresponding lectin status (Fig. 8, E and F). Two previouslyreported transcript variants of P2X6 (7) were also detected, butneither RT-PCR amplification product band varied consistentlybetween NKTERT and PKDTERT cells (n � 3, not shown).
P2X7 protein expression in NKTERT and PKDTERT cells.Both DBA�-NKTERT (Fig. 9A) and DBA�-PKDTERT cells(Fig. 9B) revealed a punctate distribution of P2X7 immuno-staining at the cell periphery, with greater signal intensity inintracellular compartments. Preadsorption of the P2X7 anti-body with its peptide antigen completely abolished these sig-nals (Fig. 9C). P2X7 staining intensity was lower in DBA�-PKDTERT cells than in DBA�-NKTERT cells (Fig. 9, A and B).Similar differences were noted between LTA�-NK and LTA�-PKD cells (not shown). Thus PKDTERT cells express reducedlevels of P2X7 mRNA and polypeptide. The [Ca2�]i responseto ATP was examined in thapsigargin-treated DBA�-NKTERT
and DBA�-PKDTERT cells (Fig. 9E) to isolate a P2X receptor
Fig. 4. Shear stress-induced net ATP release from ADPKD cyst cells is greater than from NK cells. Primary NK and PKD cells (5 104; A), LTA�-NKTERT
and LTA�-PKDTERT cells (B), or DBA�-NKTERT and DBA�-PKDTERT cells (C) were subjected to shear stress at the indicated levels. PKD cells of each typeexhibited more net ATP release than did corresponding NK cells at the lowest tested flow. In contrast, PKD cells of each type exhibited far less net ATP releasethan did corresponding NK cells in response to the highest tested flow. D: hypotonic stress increased net ATP release from primary NK and primary PKD cells(5 105 cells). In both isotonic and hypotonic static (no-flow) conditions, net ATP release from primary PKD cells exceeded that from primary NK cells. Asnoted in the description of the luciferase assay in METHODS, the values for ATP release into hypotonic (low [Cl�]) medium may overestimate ATP releasecompared with isotonic values by as much as 10% or more. Black bars, NK cells; gray bars, ADPKD cells. *P � 0.05 for PKD or PKDTERT cells comparedwith NK or NKTERT cells in the same condition. #P � 0.05 for cells at intermediate or high shear stress compared with the same cell type at 0.1 dyne/cm2.
F1470 PARACRINE PURINERGIC MEDIATION OF FLOW-INDUCED Ca SIGNALING
AJP-Renal Physiol • VOL 296 • JUNE 2009 • www.ajprenal.org
by guest on Novem
ber 22, 2012http://ajprenal.physiology.org/
Dow
nloaded from

component of Ca2� signaling in these cells (64). The thapsi-gargin-induced elevation of [Ca2�]i in NK cells (E) wasattenuated in PKDTERT cells (F), as observed previously inprimary PKD cells (63). NKTERT cell pretreatment with theP2X7 antagonist oxidized ATP (O-ATP; 300 �M, ‚) enhanced
the rate of [Ca2�]i rise elicited by thapsigargin, but attenuatedthe magnitude of the plateau phase. Subsequent exposure to 1mM ATP, a concentration sufficient for near-maximal P2X7activation, produced a biphasic effect on [Ca2�]i. In the firstphase, ATP acutely reduced the thapsigargin-elevated [Ca2�]i
Fig. 5. ATP released in response to flow mediates flow-induced Cai2� signaling by NK cells. A: primary NK cell [Ca2�]i increase in response to 10 dyne/cm2
shear stress was monitored in absence or continued presence of the nucleotidase apyrase (5 U/ml) or the P2Y receptor antagonist suramin (300 �M). B: primaryNK cell [Ca2�]i increase in response to 10 dyne/cm2 shear stress was monitored in absence or continued presence of the P2X receptor antagonist PPADS (100�M). C: LTA�-NKTERT cell [Ca2�]i increase in response to 10 dyne/cm2 shear stress was monitored in the absence or continued presence of suramin. D:DBA�-NKTERT cell [Ca2�]i increase in response to 2.3 dyne/cm2 shear stress was monitored in the absence or continued presence of suramin.
Fig. 6. Ecto-nucleotidase inhibition prolongs the flow-induced elevation of [Ca2�]i. NK cell [Ca2�]i increase in response to 10 (A) or 0.75 dyne/cm2 (B) shearstress was monitored in absence (E) or continued presence of the 5 -ectonucleotidase inhibitor ARL67156 (300 �M, F).
F1471PARACRINE PURINERGIC MEDIATION OF FLOW-INDUCED Ca SIGNALING
AJP-Renal Physiol • VOL 296 • JUNE 2009 • www.ajprenal.org
by guest on Novem
ber 22, 2012http://ajprenal.physiology.org/
Dow
nloaded from
to near resting levels. In NKTERT cells, this rapid fall in [Ca2�]i
was followed within �30 s by a slower secondary recoveryphase, during which [Ca2�]i regained its plateau value (Fig.9E). The recovery phase was strongly inhibited to similarextents by both P2X7 antagonists O-ATP and (not shown)1-[N,O-bis(5-isoquinoline sulphonyl)-N-methyl-L-tyrosyl]-4-phenylpiperazine (KN-62; 10 �M; n � 3). This secondaryrecovery phase was severely attenuated in PKDTERT cells (Fig.9D), a phenotype consistent with reduced P2X7 expression inthese cyst cells.
CD39 gene family mRNA expression in NKTERT and PKDTERT
cells. Since pharmacological inhibition of ecto-nucleotidaseactivity markedly prolonged the plateau phase of flow-induced[Ca2�]i elevation in NKTERT cells (Fig. 6), we assessed ex-
pression of CD39 gene family members encoding nucleosidetriphosphate diphosphohydrolases expressed in the kidney (11,23, 56). Comparative RT-PCR screening (Supplemental Fig. 6)revealed greatly reduced band intensities of CD39 RT-PCRamplification products in both LTA�- and DBA�-PKDTERT
cells compared with lectin-matched NKTERT cells. In contrast,band intensities of RT-PCR amplification products encodingCD39L2, CD39L3, and CD39L4 did not differ consistentlybetween NK and ADPKD cells of either lectin type. Immuno-reactive CD39 was less abundant in PKDTERT than in NKTERT
cells of LTA� (Supplemental Fig. 7) and DBA� lectin types(not shown).
DISCUSSION
This report presents several novel findings. hTERT-immortal-ized ADPKD cyst cells heterozygous for the germline mutationQ4004X share the loss of normal flow-sensitive [Ca2�]i signalingthat characterizes primary ADPKD cyst cells heterozygous for thedistinct germline mutation �L2433 (63) and SV40-transformedcells hetero- or hemizygous for the germline mutation Q2556X(34). hTERT-immortalized clonal NK cell lines (NKTERT) se-lected for expression of proximal or distal markers exhibitedslightly different flow sensitivities, but hTERT-immortalizedclonal ADPKD cyst cell lines (PKDTERT) expressing eithermarker were similarly unresponsive to flow. These immortal-ized cyst cells possessed cilia of nearly normal length, butlacked ciliary localization of PC1 and PC2. Despite elevatednet ATP release in resting and hypotonic conditions, primaryand immortalized ADPKD cyst cells of both lectin typesexhibited markedly reduced flow-sensitive ATP release. Re-leased ATP acts through P2Y and P2X receptors to mediateflow-sensitive Cai
2� signaling, and ecto-nucleotidase inhibitionprolonged the plateau phase of the flow-induced Cai
2� signal.However, Cai
2� signaling amplitude in response to exog-enously added ATP was only moderately reduced in PKDTERT
cells. These changes in cyst cells were accompanied by re-
Fig. 7. NKTERT and PKDTERT cells exhibit similar [Ca2�]i elevation andextracellular Ca2� dependence of that elevation in response to exogenous ATP.LTA�-NKTERT (filled symbols) and LTA�-PKDTERT cells (open symbols)were treated at t � 0 with 10 �M ATP in the presence (circles) or nominalabsence of extracellular Ca2� (triangles) as indicated.
Fig. 8. RT-PCR analysis of P2 receptor mRNA expression in NK and ADPKD cells. mRNA expression of P2Y (A and B) and P2X receptor subtype mRNAs(C–E) in human kidney (K) and in confluent 5-day cultures of LTA�-NK (NL) and LTA�-PKD (PL) was assessed by RT-PCR. D and E: P2X6 and P2X7 mRNAlevels were also assessed in DBA�-NK (ND) and DBA�-PKD cells (PD). F: summary of similar experiments, measuring intensity of both amplification products.Values are means � SE; n � 3–4. **P � 0.01.
F1472 PARACRINE PURINERGIC MEDIATION OF FLOW-INDUCED Ca SIGNALING
AJP-Renal Physiol • VOL 296 • JUNE 2009 • www.ajprenal.org
by guest on Novem
ber 22, 2012http://ajprenal.physiology.org/
Dow
nloaded from

duced mRNA and protein levels of P2X7 and CD39 and by areduction in a pharmacological index of P2X7 activity. Theseobservations are the first to demonstrate paracrine purinergicmediation of flow-sensitive Ca2� signaling in human NK cellsand to show deficient flow-sensitive net ATP release fromADPKD cells.
Role of genotype and nephron segment in flow-sensitiveCai
2� signaling. The present work adds to the range of definedPKD1 genotypes associated with loss of flow-sensitive Cai
2�
signaling to include heterozygosity for the mutation Q4004X inaddition to the previously reported heterozygosity for themutation �L2433 (63) and heterozygosity and likely hemizy-gosity for the germline mutation Q2556X (34). These germlinemutations together encompass a range of total PC1 polypeptideexpression that is normal or elevated (this work and Ref. 63),or reduced, or absent (34). They also now extend across thegamut of primary cells, SV40-transformed cell lines, andhTERT-immortalized cell lines. The defective flow phenotypeis present in cells of both proximal and distal origin, as judgedby lectin expression. Thus loss of flow-sensitive [Ca2�]i sig-naling has proven to be a robust phenotype of ADPKD cyst
cells in culture, thus far regardless of germline genotype or ofnephron segment of apparent origin.
The emerging picture is more complicated in recessivecystic disease. Embryonic orpk/orpk collecting duct cells withstunted cilia exhibited a moderate reduction of the flow-induced [Ca2�]i elevation observed in Tg737/polaris-rescuedcells. This reduction was noted in the context of increased“basal” Ca2� permeability and elevated apical PC2 expression(50). Similar reduction in flow-triggered [Ca2�]i elevation wasevident in isolated, perfused orpk/orpk collecting ducts from2-wk-old, but not 1-wk-old mice (29). Small interference RNAknockdown (�90%) of the ARPKD gene fibrocystin producedreductions of comparable magnitude in flow-induced [Ca2�]i
elevation in mIMCD3 and murine embryonic collecting ductcells (58). In contrast, both clonal and pooled SV40-immortal-ized human ARPKD cyst cells responded to flow with [Ca2�]i
elevations twofold higher than those of immortalized collectingduct cells from age-matched normal kidneys (44).
The reported requirements of flow-sensitive Cai2� signaling
for extracellular Ca2� entry and for Ca2� release from intra-cellular stores have also varied. In human ADPKD cyst cells
Fig. 9. Reduced P2X7 protein and a reduced index of P2X7 function in ADPKD cyst cells. P2X7 immunostaining in DBA�-NK cells (A), DBA�-PKD cells(B), and DBA�-NK cells in the added presence of peptide antigen (C). Scale bar � 10 �m. D: biphasic effect of 1 mM extracellular ATP on [Ca2�]i in 500nM thapsigargin-treated NKTERT cells (E) and PKDTERT cells (F) and in thapsigargin-treated NKTERT cells pretreated with and in the continued presence of theP2X7 antagonist oxidized ATP (O-ATP; 300 �M, ‚).
F1473PARACRINE PURINERGIC MEDIATION OF FLOW-INDUCED Ca SIGNALING
AJP-Renal Physiol • VOL 296 • JUNE 2009 • www.ajprenal.org
by guest on Novem
ber 22, 2012http://ajprenal.physiology.org/
Dow
nloaded from

and in murine Pkd1�/� embryonic kidney cells, Ca2� entrywas required, and stores were ryanodine-sensitive (33, 34, 63).MDCK cells (42) and intact or split-open rabbit collecting duct(30) required Ca2� entry, but the stores were inositol triphos-phate (IP3)-sensitive. orpk/orpk tubules also required Ca2�
entry (29), but flow-induced elevation of [Ca2�]i in cell mono-layers was bath Ca2�-independent (21). The pharmacosensi-tivity of flow-induced Cai
2� elevation reported in the orpk/orpktubule experiments did not define the nature of the Ca2� stores(29, 50). Flow-induced [Ca2�]i increase in the isolated, per-fused mouse MTAL did not require luminal Ca2� but didrequire basolateral extracellular Ca2� to sustain the post-peakplateau response (22). These differences likely reflect species,developmental stage, and differentiation state, with differingcontributions of shear stress, mechanical stretch, and hydro-static pressure according to experimental system and geometry.
Role of ATP release in flow-sensitive Cai2� signaling. The
selective reduction in flow-sensitive net ATP release observedin ADPKD cyst cells (Fig. 4) suggests that it contributes to theloss of flow-sensitive Cai
2� signaling in ADPKD cells. The roleof paracrine ATP signaling in flow-induced Ca2� signaling hasbeen investigated in several cell types. Flow-induced [Ca2�]i
increase in perfused mouse MTAL was inhibited by apyraseand suramin, mediated by P2y2 receptors on both apical andbasolateral membranes, and substantially reduced in P2y2�/�
tubules (22). P2y2 activation was also found to underlie tem-perature-dependent, spontaneous [Ca2�]i oscillations in MDCKcells at rest (14). In orpk/orpk mouse collecting duct cellsrescued with the Tg737/polaris transgene, flow-induced eleva-tion of [Ca2�]i was blocked by apyrase and suramin. Incontrast to the selective defect in flow-induced net ATP releaseof ADPKD cyst cells, the unrescued orpk/orpk cells withstubby cilia exhibited attenuated net ATP release in response tothree different stimuli: hypotonic shock, harsh pipetting, andionomycin (21).
Flow elevates [Ca2�]i in isolated rat bile duct segments andin cholangiocytes by a cilium-dependent mechanism (31).Flow also triggers cholangiocytes to release ATP, which me-diates flow-induced Cai
2� elevation by paracrine P2 receptorstimulation (62). P2R activation in cholangiocytes activatesanion channels in parallel with increased bicarbonate secretionbile flow. However, flow-induced ATP release and subsequentpurinergic elevation of [Ca2�]i in some human cholangiocytecell lines was not cilium dependent (62). Thus the properties offlow-stimulated net ATP release appear to differ among celltypes, may reflect predominant utilization of distinct ATPrelease pathways, and may be differentially affected by muta-tions in different cystic disease genes.
Purinergic receptors and nucleotidases in flow sensing.Intraluminal ATP concentrations in superficial proximal tu-bules of anesthetized rats have been estimated at 100–300 nM,several-fold higher than in Bowman’s space, and several-foldhigher than in superficial distal convoluted tubule (57). Theseconcentrations likely underestimate true juxtamembrane con-centrations, in view of the high activities of both secreted andapical membrane ecto-nucleotidases. Thus released luminalATP is believed to reach concentrations sufficient for acti-vation of apical purinergic receptors (27, 55). A subset ofADPKD cyst fluids contained ATP at the higher concentrationsof 0.5–10 �M (60).
Normal and ADPKD kidneys express a wide range ofpurinergic receptor and ecto-nucleotidase mRNAs and immu-noreactive polypeptides (27, 48, 55). The absence inLTA�-NK and PKD cyst cells of some P2R subtype mRNAsexpressed in the kidney (Fig. 8) may reflect either axialheterogeneity of P2R expression or alterations in gene expressionaccompanying immortalization. Although ATP-responsive[Ca2�]i elevation was normal or modestly reduced in NK andADPKD cyst cells, cyst cell responses to UTP and ADP werereduced to greater extents (Supplemental Fig. 5). PKDTERT cellsdisplayed lower mRNA levels and immunostaining intensityfor P2X7 and CD39 than detected in NKTERT cells of eitherlectin type (Figs. 8 and 9; Supplemental Figs. 6 and 7).ADPKD cyst cells also exhibited a delayed and attenuated[Ca2�]i recovery following the profound reduction in [Ca2�]i
induced by 1 mM ATP, a recovery sensitive to the P2X7antagonists O-ATP and KN-62 (Fig. 9). Thus reductions inflow-induced net ATP release and flow-induced, extracellularATP-dependent Cai
2� signaling in PKDTERT cells were asso-ciated with apparent decreases in P2X7 expression and in Cai
2�
signaling sensitive to P2X7 inhibitors.The ATP-induced rapid fall in thapsigargin-elevated
[Ca2�]i observed in NKTERT cells has been observed inother cell types. In thapsigargin-pretreated CFPAC-1 cells,10 �M ATP rapidly reduced [Ca2�]i by 40% without de-creasing plasmalemmal Ca2� entry (as measured by Mn2�
quench of Cai2� signal) and without increasing Cai
2� extru-sion by either Na�/Ca2� exchange or vanadate-sensitive Ca2�-ATPase (61). Similar inhibitory effects of ATP were noted inHT-29 cells. In rat brown adipocytes, 10 �M ATP also rapidlydepressed thapsigargin-elevated [Ca2�]i by 92% (37) by amechanism insensitive to phorbol ester but sensitive to highconcentrations of suramin and of PPADS (36). The inhibitoryeffect of ATP on thapsigargin-elevated [Ca2�]i correlated withincreased peripheral actin polymerization and was blocked bypharmacological disruption of the actin cytoskeleton (38). Themechanism of this inhibitory effect of ATP on [Ca2�]i inNKTERT and PKDTERT cells, as well as in brown adipocytes,CFPAC-1, and HT-29 cells, remains unclear. The secondaryelevation of depressed [Ca2�]i might be mediated by slower P2receptor activation (or recovery from thapsigargin-associatedinactivation), with pharmacological properties suggestive ofP2X7.
Reduced expression of both P2X7 and CD39 in PKDTERT
cells suggests important roles for these proteins in normalflow-sensitive Cai
2� signaling, but would be predicted to exertopposing effects. Generation of adenosine by CD39 and otherecto-nucleotidases may play an additional, potentially impor-tant role in terminating or otherwise modulating the flowsignal. However, suramin is a poor P2X7 antagonist (13), andpreliminary experiments suggest that neither P2X7 antagonistO-ATP nor KN-62 can reproduce the inhibitory effects ofsuramin and of PPADS on flow-induced Cai
2� signaling inLTA� or DBA� NKTERT cells (Xu C and Alper SL, unpub-lished observations). In addition, primary ADPKD cyst cellsbearing the heterozygous germline mutation �L2433 exhibitedincreased levels of mRNA encoding P2X7 (n � 4) and CD39(n � 2; not shown), in contrast to the decreased levels inimmortalized Q4004X ADPKD cyst cells (Fig. 8 and Supple-mental Fig. 6). Thus levels of these mRNAs may vary as afunction of either genotype or immortalization state. Taken
F1474 PARACRINE PURINERGIC MEDIATION OF FLOW-INDUCED Ca SIGNALING
AJP-Renal Physiol • VOL 296 • JUNE 2009 • www.ajprenal.org
by guest on Novem
ber 22, 2012http://ajprenal.physiology.org/
Dow
nloaded from

together, the data suggest that the identity of the P2 receptor(s)mediating flow-sensitive Cai
2� signaling in primary NK cellsand in NKTERT cells remains to be determined. P2X7 is apotentiator of apoptosis and inflammatory cytokine release inepithelial (4) as well as in other cells (12). Alteration of P2X7expression in ADPKD (17) may be related to the diseasestage-specific, variable increases in apoptosis detected inADPKD cyst epithelial cells (15). This variation, in turn,may be reflected in the differentiation state-dependence ofP2R and ecto-nucleotidase mRNA levels.
Additional work is needed to define the pathway(s) offlow-sensitive ATP release that are selectively impaired inADPKD cyst cells while ATP release in basal and hypotonicswelling conditions is elevated. The central role of vectorialfluid flow-induced paracrine signaling in the embryonic nodein the developmental establishment of left-right asymmetrystrongly suggests that deficient flow-induced ATP release byADPKD cyst cells likely has consequences for the control ofgene expression. However, the roles of flow-sensitive, puriner-gically mediated Cai
2� signaling by renal epithelial cells in thegeneration and/or maintenance of normal axial tubular struc-ture and in the prevention of cystogenesis remain to be estab-lished.
ACKNOWLEDGMENTS
We thank Drs. W. Junger and D. H. Friedman (Harvard) and O. Ibraghi-mova-Beskrovnaya (Genzyme) for antibodies and D. H. Friedman, W. Junger,and S. A. Ness (University of New Mexico) for helpful discussions.
GRANTS
C. Xu was supported by postdoctoral fellowship F32 DK69049 from theNational Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)and by a postdoctoral fellowship from the Polycystic Kidney Disease Foun-dation. This work was additionally supported by NIDDK Grants R01-DK57662 to S. L. Alper, R01 DK58816 to P. C. Harris, R01 DK50141 to A.Wandinger-Ness, by a Polycystic Kidney Disease Foundation research grant toR. Bacallao, and by Shared Instrument Grant S10-RR017927 to Beth IsraelDeaconess Medical Center.
REFERENCES
1. Bai CX, Giamarchi A, Rodat-Despoix L, Padilla F, Downs T, TsiokasL, Delmas P. Formation of a new receptor-operated channel by hetero-meric assembly of TRPP2 and TRPC1 subunits. EMBO Rep 9: 472–479,2008.
2. Becker D, Woltersdorf R, Boldt W, Schmitz S, Braam U, SchmalzingG, Markwardt F. The P2X7 carboxyl tail is a regulatory module of P2X7receptor channel activity. J Biol Chem 2008.
3. Cheewatrakoolpong B, Gilchrest H, Anthes JC, Greenfeder S. Identi-fication and characterization of splice variants of the human P2X7 ATPchannel. Biochem Biophys Res Commun 332: 17–27, 2005.
4. Chen L, Brosnan CF. Regulation of immune response by P2X7 receptor.Crit Rev Immunol 26: 499–513, 2006.
5. Condon J, Yin S, Mayhew B, Word RA, Wright WE, Shay JW,Rainey WE. Telomerase immortalization of human myometrial cells. BiolReprod 67: 506–514, 2002.
6. D’Agati V, Trudel M. Lectin characterization of cystogenesis in the SBMtransgenic model of polycystic kidney disease. J Am Soc Nephrol 3:975–983, 1992.
7. da Silva RL, Resende RR, Ulrich H. Alternative splicing of P2X6receptors in developing mouse brain and during in vitro neuronal differ-entiation. Exp Physiol 92: 139–145, 2007.
8. Denburg JL, McElroy WD. Anion inhibition of firefly luciferase. ArchBiochem Biophys 141: 668–675, 1970.
9. Feng YH, Li X, Wang L, Zhou L, Gorodeski GI. A truncated P2X7receptor variant (P2X7-j) endogenously expressed in cervical cancer cellsantagonizes the full-length P2X7 receptor through hetero-oligomerization.J Biol Chem 281: 17228–17237, 2006.
10. Fitz JG. Regulation of cellular ATP release. Trans Am Clin ClimatolAssoc 118: 199–208, 2007.
11. Friedman DJ, Rennke HG, Csizmadia E, Enjyoji K, Robson SC. Thevascular ectonucleotidase ENTPD1 is a novel renoprotective factor indiabetic nephropathy. Diabetes 56: 2371–2379, 2007.
12. Garcia-Marcos M, Pochet S, Marino A, Dehaye JP. P2X7 and phos-pholipid signalling: the search of the “missing link” in epithelial cells. CellSignal 18: 2098–2104, 2006.
13. Gever JR, Cockayne DA, Dillon MP, Burnstock G, Ford AP. Pharma-cology of P2X channels. Pflugers Arch 452: 513–537, 2006.
14. Geyti CS, Odgaard E, Overgaard MT, Jensen ME, Leipziger J,Praetorius HA. Slow spontaneous [Ca2�]i oscillations reflect nucleotiderelease from renal epithelia. Pflugers Arch 455: 1105–1117, 2008.
15. Goilav B, Satlin LM, Wilson PD. Pathways of apoptosis in humanautosomal recessive and autosomal dominant polycystic kidney diseases.Pediatr Nephrol 23: 1473–1482, 2008.
16. Grygorczyk R, Hanrahan JW. CFTR-independent ATP release fromepithelial cells triggered by mechanical stimuli. Am J Physiol Cell Physiol272: C1058–C1066, 1997.
17. Hillman KA, Burnstock G, Unwin RJ. The P2X7 ATP receptor in thekidney: a matter of life or death? Nephron Exp Nephrol 101: e24–30,2005.
18. Hillman KA, Johnson TM, Winyard PJ, Burnstock G, Unwin RJ,Woolf AS. P2X(7) receptors are expressed during mouse nephrogenesisand in collecting duct cysts of the cpk/cpk mouse. Exp Nephrol 10: 34–42,2002.
19. Holmsen H, Holmsen I, Bernhardsen A. Microdetermination of adeno-sine diphosphate and adenosine triphosphate in plasma with firefly lucif-erase system. Anal Biochem 17: 456–473, 1966.
20. Homolya L, Steinberg TH, Boucher RC. Cell to cell communication inresponse to mechanical stress via bilateral release of ATP and UTP inpolarized epithelia. J Cell Biol 150: 1349–1360, 2000.
21. Hovater MB, Olteanu D, Hanson EL, Cheng NL, Siroky B, Fintha A,Komlosi P, Liu W, Satlin LM, Bell PD, Yoder BK, Schwiebert EM.Loss of apical monocilia on collecting duct principal cells impairs ATPsecretion across the apical cell surface and ATP-dependent and flow-induced calcium signals. Purinergic Signal 4: 155–170, 2008.
22. Jensen ME, Odgaard E, Christensen MH, Praetorius HA, Leipziger J.Flow-induced [Ca2�]i increase depends on nucleotide release and subse-quent purinergic signaling in the intact nephron. J Am Soc Nephrol 18:2062–2070, 2007.
23. Kishore BK, Isaac J, Fausther M, Tripp SR, Shi H, Gill PS, Braun N,Zimmermann H, Sevigny J, Robson SC. Expression of NTPDase1 andNTPDase2 in murine kidney: relevance to regulation of P2 receptorsignaling. Am J Physiol Renal Physiol 288: F1032–F1043, 2005.
24. Kottgen M, Buchholz B, Garcia-Gonzalez MA, Kotsis F, Fu X,Doerken M, Boehlke C, Steffl D, Tauber R, Wegierski T, Nitschke R,Suzuki M, Kramer-Zucker A, Germino GG, Watnick T, Prenen J,Nilius B, Kuehn EW, Walz G. TRPP2 and TRPV4 form a polymodalsensory channel complex. J Cell Biol 182: 437–447, 2008.
25. Kottgen M, Walz G. Subcellular localization and trafficking of poly-cystins. Pflugers Arch 451: 286–293, 2005.
26. Lazarowski ER, Tarran R, Grubb BR, van Heusden CA, Okada S,Boucher RC. Nucleotide release provides a mechanism for airway surfaceliquid homeostasis. J Biol Chem 279: 36855–36864, 2004.
27. Leipziger J. Control of epithelial transport via luminal P2 receptors. Am JPhysiol Renal Physiol 284: F419–F432, 2003.
28. Lewis SA, Lewis JR. Kinetics of urothelial ATP release. Am J PhysiolRenal Physiol 291: F332–F340, 2006.
29. Liu W, Murcia NS, Duan Y, Weinbaum S, Yoder BK, Schwiebert E,Satlin LM. Mechanoregulation of intracellular Ca2� concentration isattenuated in collecting duct of monocilium-impaired orpk mice. Am JPhysiol Renal Physiol 289: F978–F988, 2005.
30. Liu W, Xu S, Woda C, Kim P, Weinbaum S, Satlin LM. Effect of flowand stretch on the [Ca2�]i response of principal and intercalated cells incortical collecting duct. Am J Physiol Renal Physiol 285: F998–F1012,2003.
31. Masyuk AI, Masyuk TV, Splinter PL, Huang BQ, Stroope AJ, La-Russo NF. Cholangiocyte cilia detect changes in luminal fluid flow andtransmit them into intracellular Ca2� and cAMP signaling. Gastroenter-ology 131: 911–920, 2006.
32. Merlin D, Jiang L, Strohmeier GR, Nusrat A, Alper SL, Lencer WI,Madara JL. Distinct Ca2�- and cAMP-dependent anion conductances in
F1475PARACRINE PURINERGIC MEDIATION OF FLOW-INDUCED Ca SIGNALING
AJP-Renal Physiol • VOL 296 • JUNE 2009 • www.ajprenal.org
by guest on Novem
ber 22, 2012http://ajprenal.physiology.org/
Dow
nloaded from

the apical membrane of polarized T84 cells. Am J Physiol Cell Physiol275: C484–C495, 1998.
33. Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE,Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J. Polycystins 1 and 2mediate mechanosensation in the primary cilium of kidney cells. NatGenet 33: 129–137, 2003.
34. Nauli SM, Rossetti S, Kolb RJ, Alenghat FJ, Consugar MB, HarrisPC, Ingber DE, Loghman-Adham M, Zhou J. Loss of polycystin-1 inhuman cyst-lining epithelia leads to ciliary dysfunction. J Am Soc Nephrol17: 1015–1025, 2006.
35. Okada SF, Nicholas RA, Kreda SM, Lazarowski ER, Boucher RC.Physiological regulation of ATP release at the apical surface of humanairway epithelia. J Biol Chem 281: 22992–23002, 2006.
36. Omatsu-Kanbe M, Isono T, Matsuura H. Multiple P2 receptors con-tribute to a transient increase in intracellular Ca2� concentration inATP-stimulated rat brown adipocytes. Exp Physiol 87: 643–652, 2002.
37. Omatsu-Kanbe M, Matsuura H. Inhibition of store-operated Ca2� entryby extracellular ATP in rat brown adipocytes. J Physiol 521: 601–615,1999.
38. Omatsu-Kanbe M, Shibata M, Yamamoto T, Isono T, Matsuura H.Actin filaments play a permissive role in the inhibition of store-operatedCa2� entry by extracellular ATP in rat brown adipocytes. Biochem J 381:389–396, 2004.
39. Peltola P, Lumiaho A, Miettinen R, Pihlajamaki J, Sandford R,Laakso M. Genetics and phenotypic characteristics of autosomal domi-nant polycystic kidney disease in Finns. J Mol Med 83: 638–646, 2005.
40. Praetorius HA, Frokiaer J, Nielsen S, Spring KR. Bending the primarycilium opens Ca2�-sensitive intermediate-conductance K� channels inMDCK cells. J Membr Biol 191: 193–200, 2003.
41. Praetorius HA, Leipziger J. Fluid flow sensing and triggered nucleotiderelease in epithelia. J Physiol 586: 2669, 2008.
42. Praetorius HA, Spring KR. Bending the MDCK cell primary ciliumincreases intracellular calcium. J Membr Biol 184: 71–79, 2001.
43. Praetorius HA, Spring KR. Removal of the MDCK cell primary ciliumabolishes flow sensing. J Membr Biol 191: 69–76, 2003.
44. Rohatgi R, Battini L, Kim P, Israeli S, Wilson PD, Gusella GL, SatlinLM. Mechanoregulation of intracellular Ca2� in human autosomal reces-sive polycystic kidney disease cyst-lining renal epithelial cells. Am JPhysiol Renal Physiol 294: F890–F899, 2008.
45. Roman RM, Feranchak AP, Davison AK, Schwiebert EM, Fitz JG.Evidence for Gd3� inhibition of membrane ATP permeability and puri-nergic signaling. Am J Physiol Gastrointest Liver Physiol 277: G1222–G1230, 1999.
46. Rossetti S, Consugar MB, Chapman AB, Torres VE, Guay-WoodfordLM, Grantham JJ, Bennett WM, Meyers CM, Walker DL, Bae K,Zhang QJ, Thompson PA, Miller JP, Harris PC. Comprehensivemolecular diagnostics in autosomal dominant polycystic kidney disease.J Am Soc Nephrol 18: 2143–2160, 2007.
47. Satlin LM, Carattino MD, Liu W, Kleyman TR. Regulation of cationtransport in the distal nephron by mechanical forces. Am J Physiol RenalPhysiol 291: F923–F931, 2006.
48. Schwiebert EM, Wallace DP, Braunstein GM, King SR, Peti-PeterdiJ, Hanaoka K, Guggino WB, Guay-Woodford LM, Bell PD, SullivanLP, Grantham JJ, Taylor AL. Autocrine extracellular purinergic signal-ing in epithelial cells derived from polycystic kidneys. Am J Physiol RenalPhysiol 282: F763–F775, 2002.
49. Silva FG, Nadasdy T, Laszik Z. Immunohistochemical and lectin dis-section of the human nephron in health and disease. Arch Pathol Lab Med117: 1233–1239, 1993.
50. Siroky BJ, Ferguson WB, Fuson AL, Xie Y, Fintha A, Komlosi P,Yoder BK, Schwiebert EM, Guay-Woodford LM, Bell PD. Loss ofprimary cilia results in deregulated and unabated apical calcium entry inARPKD collecting duct cells. Am J Physiol Renal Physiol 290: F1320–F1328, 2006.
51. Taylor AL, Kudlow BA, Marrs KL, Gruenert DC, Guggino WB,Schwiebert EM. Bioluminescence detection of ATP release mechanismsin epithelia. Am J Physiol Cell Physiol 275: C1391–C1406, 1998.
52. Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidneydisease. Lancet 369: 1287–1301, 2007.
53. Tsiokas L, Kim S, Ong EC. Cell biology of polycystin-2. Cell Signal 19:444–453, 2007.
54. Turner CM, King BF, Srai KS, Unwin RJ. Antagonism of endogenousputative P2Y receptors reduces the growth of MDCK-derived cysts cul-tured in vitro. Am J Physiol Renal Physiol 292: F15–F25, 2007.
55. Vallon V. P2 receptors in the regulation of renal transport mechanisms.Am J Physiol Renal Physiol 294: F10–F27, 2008.
56. Vekaria RM, Shirley DG, Sevigny J, Unwin RJ. Immunolocalization ofectonucleotidases along the rat nephron. Am J Physiol Renal Physiol 290:F550–F560, 2006.
57. Vekaria RM, Unwin RJ, Shirley DG. Intraluminal ATP concentrationsin rat renal tubules. J Am Soc Nephrol 17: 1841–1847, 2006.
58. Wang S, Zhang J, Nauli SM, Li X, Starremans PG, Luo Y, RobertsKA, Zhou J. Fibrocystin/polyductin, found in the same protein complexwith polycystin-2, regulates calcium responses in kidney epithelia. MolCell Biol 27: 3241–3252, 2007.
59. Watt WC, Lazarowski ER, Boucher RC. Cystic fibrosis transmembraneregulator-independent release of ATP. Its implications for the regulationof P2Y2 receptors in airway epithelia. J Biol Chem 273: 14053–14058,1998.
60. Wilson PD, Hovater JS, Casey CC, Fortenberry JA, Schwiebert EM.ATP release mechanisms in primary cultures of epithelia derived from thecysts of polycystic kidneys. J Am Soc Nephrol 10: 218–229, 1999.
61. Wolff T, Leipziger J, Fischer KG, Klar B, Nitschke R, Greger R.Evidence for agonist-induced export of intracellular Ca2� in epithelialcells. Pflugers Arch 424: 423–430, 1993.
62. Woo K, Dutta AK, Patel V, Kresge C, Feranchak AP. Fluid flowinduces mechanosensitive ATP release, calcium signalling and Cl� trans-port in biliary epithelial cells through a PKCzeta-dependent pathway.J Physiol 586: 2779–2798, 2008.
63. Xu C, Rossetti S, Jiang L, Harris PC, Brown-Glaberman U, Wand-inger-Ness A, Bacallao R, Alper SL. Human ADPKD primary cystepithelial cells with a novel, single codon deletion in the PKD1 geneexhibit defective ciliary polycystin localization and loss of flow-inducedCa2� signaling. Am J Physiol Renal Physiol 292: F930–F945, 2007.
64. Yeung D, Zablocki K, Lien CF, Jiang T, Arkle S, Brutkowski W,Brown J, Lochmuller H, Simon J, Barnard EA, Gorecki DC. Increasedsusceptibility to ATP via alteration of P2X receptor function in dystrophicmdx mouse muscle cells. FASEB J 20: 610–620, 2006.
65. Yi X, Tesmer VM, Savre-Train I, Shay JW, Wright WE. Bothtranscriptional and posttranscriptional mechanisms regulate human telom-erase template RNA levels. Mol Cell Biol 19: 3989–3997, 1999.
F1476 PARACRINE PURINERGIC MEDIATION OF FLOW-INDUCED Ca SIGNALING
AJP-Renal Physiol • VOL 296 • JUNE 2009 • www.ajprenal.org
by guest on Novem
ber 22, 2012http://ajprenal.physiology.org/
Dow
nloaded from




















