
Assessment of 115 Candidate Genes for Diabetic Nephropathy by Transmission/Disequilibrium Test
14
Original Article Assessment of 115 Candidate Genes for Diabetic Nephropathy by Transmission / Disequilibrium Test Kathryn Gogolin Ewens, 1 Roberta Ann George, 1 Kumar Sharma, 2 Fuad N. Ziyadeh, 3 and Richard S. Spielman 1 Several lines of evidence, including familial aggregation, suggest that allelic variation contributes to risk of diabetic nephropathy. To assess the evidence for specific suscepti- bility genes, we used the transmission/disequilibrium test (TDT) to analyze 115 candidate genes for linkage and association with diabetic nephropathy. A comprehensive survey of this sort has not been undertaken before. Single nucleotide polymorphisms and simple tandem repeat poly- morphisms located within 10 kb of the candidate genes were genotyped in a total of 72 type 1 diabetic families of European descent. All families had at least one offspring with diabetes and end-stage renal disease or proteinuria. As a consequence of the large number of statistical tests and modest P values, findings for some genes may be false-positives. Furthermore, the small sample size re- sulted in limited power, so the effects of some tested genes may not be detectable, even if they contribute to suscepti- bility. Nevertheless, nominally significant TDT results (P < 0.05) were obtained with polymorphisms in 20 genes, in- cluding 12 that have not been studied previously: aqua- porin 1; B-cell leukemia/lymphoma 2 (bcl-2) proto- oncogene; catalase; glutathione peroxidase 1; IGF1; laminin alpha 4; laminin, gamma 1; SMAD, mothers against DPP homolog 3; transforming growth factor, beta receptor II; transforming growth factor, beta receptor III; tissue inhibitor of metalloproteinase 3; and upstream transcrip- tion factor 1. In addition, our results provide modest support for a number of candidate genes previously studied by others. Diabetes 54:3305–3318, 2005 D iabetic nephropathy is the most serious long- term complication of diabetes, accounting for 40% of new cases of end-stage renal disease (ESRD) in the U.S. (1). Two lines of evidence suggest a strong genetic component in susceptibility to diabetic kidney disease. 1) Epidemiological studies indi- cate that the prevalence of diabetic nephropathy increases during the first 15–20 years after onset of diabetes and then reaches a plateau, suggesting that only a subset of patients is susceptible to the development of kidney disease (2). 2) Family studies show clustering of diabetic nephropathy in both type 1 and type 2 diabetes; diabetic siblings of probands with diabetic nephropathy have a significantly greater risk for developing kidney complica- tions than diabetic siblings of probands without diabetic nephropathy (3– 6). In addition, segregation analyses of diabetic nephropathy in both Caucasians and Pima Indians with type 2 diabetes provide evidence for the presence of a major locus, with a possible role for several minor loci (7,8). Numerous metabolic pathways and associated groups of genes have been proposed as candidates to play a role in the genetic susceptibility to diabetic nephropathy (9 –12). Before onset of overt proteinuria, functional changes are observed in the kidney (altered glomerular filtration rates and increasing albumin excretion rates), which are thought to result from the underlying pathological changes that occur. These changes include thickening of the glo- merular basement membrane and expansion of the mes- angium due to accumulation of extracellular matrix proteins. Products of a wide range of genes might mediate these renal changes. Examples include 1) the synthesis and degradation of glomerular basement membrane and mesangial matrix components; 2) components of meta- bolic pathways involving glucose metabolism and trans- port; 3) blood pressure regulation and the renin- angiotensin system; 4) cytokines, growth factors, signaling molecules, and transcription factors; and 5) advanced glycation processes. Many of these candidate genes have been tested for association with diabetic nephropathy, typically in case-control studies of only one or a few genes (Table 1). In many instances, initial reports were not confirmed in follow-up studies. We have carried out family-based studies with simple tandem repeat polymorphisms (STRPs) and single nucle- otide polymorphisms (SNPs) in 83 candidate genes that have not been studied previously and 32 genes or gene regions that have been reported as having significant association or linkage with diabetic nephropathy (Table From the 1 Department of Genetics, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania; the 2 Department of Medicine, Center for Diabetic Kidney Disease, Division of Nephrology, Thomas Jefferson University, Philadelphia, Pennsylvania; and the 3 Renal-Electrolyte and Hyper- tension Division and Penn Center for Molecular Studies of Kidney Diseases, Department of Medicine, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania Address correspondence and reprint requests to Dr. Richard S. Spielman, Department of Genetics, University of Pennsylvania School of Medicine, Philadelphia, PA 19104-6145. E-mail: [email protected]. Received for publication 1 April 2005 and accepted in revised form 5 August 2005. Additional information for this article can be found in an online appendix at http://diabetes.diabetesjournals.org. For a complete list of gene abbreviations, see the APPENDIX. CEPH, Centre d’Etude du Polymorphisme Humain; ESRD, end-stage renal disease; HBDI, Human Biological Data Interchange; SNP, single nucleotide polymorphism; STRP, simple tandem repeat polymorphism; TDT, transmis- sion/disequilibrium test; UTR, untranslated region. © 2005 by the American Diabetes Association. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. DIABETES, VOL. 54, NOVEMBER 2005 3305
Transcript of Assessment of 115 Candidate Genes for Diabetic Nephropathy by Transmission/Disequilibrium Test

Original Article
Assessment of 115 Candidate Genes for DiabeticNephropathy by Transmission/Disequilibrium TestKathryn Gogolin Ewens,
1Roberta Ann George,
1Kumar Sharma,
2Fuad N. Ziyadeh,
3and
Richard S. Spielman1
Several lines of evidence, including familial aggregation,
suggest that allelic variation contributes to risk of diabetic
nephropathy. To assess the evidence for specific suscepti-
bility genes, we used the transmission/disequilibrium test
(TDT) to analyze 115 candidate genes for linkage and
association with diabetic nephropathy. A comprehensive
survey of this sort has not been undertaken before. Single
nucleotide polymorphisms and simple tandem repeat poly-
morphisms located within 10 kb of the candidate genes
were genotyped in a total of 72 type 1 diabetic families of
European descent. All families had at least one offspring
with diabetes and end-stage renal disease or proteinuria.
As a consequence of the large number of statistical tests
and modest P values, findings for some genes may be
false-positives. Furthermore, the small sample size re-
sulted in limited power, so the effects of some tested genes
may not be detectable, even if they contribute to suscepti-
bility. Nevertheless, nominally significant TDT results (P <0.05) were obtained with polymorphisms in 20 genes, in-
cluding 12 that have not been studied previously: aqua-
porin 1; B-cell leukemia/lymphoma 2 (bcl-2) proto-
oncogene; catalase; glutathione peroxidase 1; IGF1;
laminin alpha 4; laminin, gamma 1; SMAD, mothers against
DPP homolog 3; transforming growth factor, beta receptor
II; transforming growth factor, beta receptor III; tissue
inhibitor of metalloproteinase 3; and upstream transcrip-
tion factor 1. In addition, our results provide modest
support for a number of candidate genes previously studied
by others. Diabetes 54:3305–3318, 2005
Diabetic nephropathy is the most serious long-term complication of diabetes, accounting for�40% of new cases of end-stage renal disease(ESRD) in the U.S. (1). Two lines of evidence
suggest a strong genetic component in susceptibility todiabetic kidney disease. 1) Epidemiological studies indi-cate that the prevalence of diabetic nephropathy increasesduring the first 15–20 years after onset of diabetes andthen reaches a plateau, suggesting that only a subset ofpatients is susceptible to the development of kidneydisease (2). 2) Family studies show clustering of diabeticnephropathy in both type 1 and type 2 diabetes; diabeticsiblings of probands with diabetic nephropathy have asignificantly greater risk for developing kidney complica-tions than diabetic siblings of probands without diabeticnephropathy (3–6). In addition, segregation analyses ofdiabetic nephropathy in both Caucasians and Pima Indianswith type 2 diabetes provide evidence for the presence ofa major locus, with a possible role for several minor loci(7,8).
Numerous metabolic pathways and associated groups ofgenes have been proposed as candidates to play a role inthe genetic susceptibility to diabetic nephropathy (9–12).Before onset of overt proteinuria, functional changes areobserved in the kidney (altered glomerular filtration ratesand increasing albumin excretion rates), which arethought to result from the underlying pathological changesthat occur. These changes include thickening of the glo-merular basement membrane and expansion of the mes-angium due to accumulation of extracellular matrixproteins. Products of a wide range of genes might mediatethese renal changes. Examples include 1) the synthesisand degradation of glomerular basement membrane andmesangial matrix components; 2) components of meta-bolic pathways involving glucose metabolism and trans-port; 3) blood pressure regulation and the renin-angiotensin system; 4) cytokines, growth factors, signalingmolecules, and transcription factors; and 5) advancedglycation processes. Many of these candidate genes havebeen tested for association with diabetic nephropathy,typically in case-control studies of only one or a few genes(Table 1). In many instances, initial reports were notconfirmed in follow-up studies.
We have carried out family-based studies with simpletandem repeat polymorphisms (STRPs) and single nucle-otide polymorphisms (SNPs) in 83 candidate genes thathave not been studied previously and 32 genes or generegions that have been reported as having significantassociation or linkage with diabetic nephropathy (Table
From the 1Department of Genetics, University of Pennsylvania School ofMedicine, Philadelphia, Pennsylvania; the 2Department of Medicine, Centerfor Diabetic Kidney Disease, Division of Nephrology, Thomas JeffersonUniversity, Philadelphia, Pennsylvania; and the 3Renal-Electrolyte and Hyper-tension Division and Penn Center for Molecular Studies of Kidney Diseases,Department of Medicine, University of Pennsylvania School of Medicine,Philadelphia, Pennsylvania
Address correspondence and reprint requests to Dr. Richard S. Spielman,Department of Genetics, University of Pennsylvania School of Medicine,Philadelphia, PA 19104-6145. E-mail: [email protected].
Received for publication 1 April 2005 and accepted in revised form 5 August2005.
Additional information for this article can be found in an online appendix athttp://diabetes.diabetesjournals.org.
For a complete list of gene abbreviations, see the APPENDIX.CEPH, Centre d’Etude du Polymorphisme Humain; ESRD, end-stage renal
disease; HBDI, Human Biological Data Interchange; SNP, single nucleotidepolymorphism; STRP, simple tandem repeat polymorphism; TDT, transmis-sion/disequilibrium test; UTR, untranslated region.
© 2005 by the American Diabetes Association.The costs of publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby marked “advertisement” in accordance
with 18 U.S.C. Section 1734 solely to indicate this fact.
DIABETES, VOL. 54, NOVEMBER 2005 3305

1). No previous studies have undertaken a comprehensiveassessment of the evidence for many candidate genes atonce, applying the same approaches and using a singlesample of patient material. We therefore had two relatedgoals: review briefly all relevant published studies, andcarry out a thorough assessment ourselves. All our resultswere obtained from patients who have both diabeticnephropathy and type 1 diabetes. Consequently, it isformally possible that positive findings are due to diabetesrather than diabetic nephropathy. All of the candidategenes were chosen for a possible role in kidney disease,not in diabetes. Positive results would be of interest ineither case, and the possibilities can be resolved bystudying patients who have long-standing diabetes withoutdiabetic nephropathy.
For analysis of our own data, we used the transmission/disequilibrium test (TDT) in its original form (13). TheTDT tests for the simultaneous presence of linkage andallelic association between a genetic marker and a puta-tive disease susceptibility locus. Because linkage andassociation, when present together, define linkage disequi-librium, we refer to the TDT as a test for linkage disequi-librium. If there is only loose (or no) linkage, or if allelicassociation is only weak or absent, linkage disequilibriumwill not be strong, and the TDT will not detect an effect.
RESEARCH DESIGN AND METHODS
Forty-three families of European descent were ascertained through an indexcase subject with type 1 diabetes and diabetic nephropathy through the Penn
Transplant Center of the University of Pennsylvania Health System. Diabeticindividuals were considered to have diabetic nephropathy if they had ESRD orif their albumin-to-creatinine ratio was �300 �g/mg in two of three randomurine samples collected at least 6 weeks apart. When available, diabeticsiblings of the index case subject were phenotyped using the same criteria.Twenty-nine additional families with type 1 diabetes from the Human Biolog-ical Data Interchange (HBDI) collection (14) were also included in this study.These families were contacted in collaboration with HBDI to obtain updatedmedical information, including the presence of ESRD and information onrelevant medications. In the absence of ESRD, diabetic nephropathy statuswas determined as described above. The total family material consisted of 72families with type 1 diabetes: 68 parent-child trios and 4 multiplex families.Among the 77 diabetic offspring in these families, 73 had received a kidneytransplant. The mean � SD age at diagnosis of diabetes was 11.1 � 6.1 years(range, 1–30), and the mean duration of diabetes before transplant was 23.9 �5.9 years (range, 12–42). At the time of enrollment into this study, the meanduration of diabetes was 29.7 � 8.6 years (range, 17–53). The mean timeelapsed between transplant and enrollment (or until death 8 years aftertransplant in one case subject) was 6.5 � 5.5 years (range, �1–30). This studywas carried out in accordance with the protocol and informed consent formsapproved by the Institutional Review Board of the University of Pennsylvania.
Thirty-six Centre d’Etude du Polymorphisme Humain (CEPH) families(two parents and three offspring in each family) were studied for transmissiondistortion in nondiabetic control subjects. In these families, we genotyped 29SNP markers that showed nominally significant evidence for linkage disequi-librium with diabetic nephropathy.DNA preparation. For individuals ascertained through the University ofPennsylvania, total genomic DNA was prepared from peripheral blood leuko-cytes using the PureGene protocol (Gentra Systems). DNA for the HBDI andCEPH families was obtained from the Coriell Cell Repositories (CoriellInstitute for Medical Research).Candidate genes and genotyping. Candidate genes were chosen because oftheir role in normal or pathological kidney function and from published
TABLE 1Candidate genes (n � 115) for diabetic nephropathy (DN) tested for linkage disequilibrium (LD)
Functional categoryGenes (n � 83) not tested previously
for LD with DNGenes (n � 32) tested by others for
association or linkage with DN
Glomerular basement membrane andmesangial matrix components andtheir metabolism; cell adhesion
CD36 (58); COL1A1 (59–61); COL4A2 (60,61); COL4A3(60,61); COL4A4 (60,61); FBLN1 (11); FBN1*; FN1(62); HSPG1/SDC2 (62); ICAM1 (63,64); ITGA1 (65);ITGA3 (65); LAMA4*; LAMB1*; LAMC1*; LAMC2*;MMP1 (11,66); MMP2 (11); MMP3 (11,66); NID*;OPN/SPP1 (58); SELE (67); TIMP2 (62); TIMP3 (62)
COL4A1 (18,19); HSPG2 (68,69); MMP9(35,36); NPHS1 (27,70,71); SELL (72)
Glucose metabolism and transport GLUT2/SLC2A2* AKR1B1 (45–51); GFPT2 (73);GLUT1/SLC2A1 (74–76)
Blood pressure regulation and therenin-angiotensin system
EDN1 (77,78); EDN2 (79); EDN3 (79); REN*; SAH (80);UTS2 (81)
ACE (37–44); AGT (40–42,82,83);AGTR1 (20–24); NPPA (84–86)
Cytokines, growth factors, andreceptors
ACVR2 (11); BMP2 (11,71); BMP7 (11,87,88); CCL2(89); CTGF (11,62,90,91); EGF (11); GH1 (62,92);IGF1 (62,92–94); IGF1R (95); IL10*; LTBP1 (96);PDGFB (62,97); PDGFRB (97); TGFB2*; TGFB3*;TGFBR2 (98); TGFBR3*; TNFRSF1A*;TNFSF6/FASLG (11,99); VEGF (11,71,100,101)
CCR5 (102); IL1A (103–105); IL1B(103–105); IL1R1 (103–105); IL1RN(103–106); NRP1 (27); TGFB1 (93,107–109)
Lipid metabolism APOC2*; APOC4* APOE (25,52–56); LPL (25,55,110)
Protein and amino acid metabolism CTSD (111); CTSL*; ECE1*; SGK (112); UBA52 (113) MTHFR (114–116); NOS3 (117–121)
Nucleic acid metabolism ANG (10,62,122) ENPP/PC-1 (123–126)
Transcription factors and regulatoryand signaling molecules
AXL (127); EDNRA (128,129); EDNRB*; FOS*; GAS6(127); MIG6 (130); NFKB1 (89); PRKCA (131,132);SMAD3 (133); UNC13B (134); USF1*; USF2*;VEGFR/KDR (11)
BDKRB2 (135–137); CNOT4/D7S500(8); HNF1B/TCF2 (28–32); PPARG(138,139); PRKCB1 (26,93); TSC22(140)
Electron transport CAT (141); NOX4 (142) p22phox/CYBA (33,34,142)
Transport function AQP1 (71,143); SLC9A1 (93,144,145); SLC12A3 (146);TCN2 (147)
Miscellaneous BCL2 (11,148–150); GPX1 (141,151); GREM1/CKTSF1B1(11,152); HSD3B1 (58); LGALS3 (11)
CALD1 (153)
Underline indicates nominally significant results in this study. *To our knowledge, not previously proposed as candidate gene for diabeticnephropathy. For a complete list of gene abbreviations, see the APPENDIX.
CANDIDATE GENES FOR DIABETIC NEPHROPATHY
3306 DIABETES, VOL. 54, NOVEMBER 2005

reports of candidate gene or expression studies. In the initial phase of thisstudy, linkage disequilibrium with diabetic nephropathy was assessed usingSTRPs mapping in or close to the candidate gene. These markers wereselected from the UCSC Genome Bioinformatics site (http://genome.cse.ucsc.edu/). PCR primers were designed from the surrounding sequence, and PCRamplification was carried out by standard methods using fluorescently labeledprimers (15). PCR products were electrophoresed on an Applied Biosystems377 DNA Sequencer, and the genotypes were analyzed using Genescan andGenotyper software.
SNPs in candidate genes were identified using either dbSNP at NationalCenter for Biotechnology Information (http://www.ncbi.nlm.nih.gov/SNP/) orApplied Biosystems/Celera Discovery System (http://www.appliedbiosystems.com and http://www.celeradiscoverysystem.com). Polymorphic markers re-ported by others to be associated with diabetic nephropathy (Table 1) werealso genotyped. (In most cases, the restriction digest assays described in theliterature were converted to Applied Biosystems Taqman Genotyping Assays.)The goal was to genotype one SNP approximately every 20 kb. (Mean spacingof SNPs was 17.3 kb; range, 1.2–88.4 kb; median, 13.4 kb). For genes �20 kbin genomic extent, typically one SNP was typed. When available, SNPs locatedin exons were genotyped in preference to those in introns if the minor allelefrequency exceeded �0.2. Some of the SNP genotyping was carried out byrestriction enzyme digestion, sequencing, or fluorescent polarizatation withAcycloPrime-FP SNP detection assays read on a Victor multilabel reader(Perkin Elmer Life Sciences). For most SNPs, we used Applied BiosystemsTaqman SNP Genotyping Assays and read results on an Applied Biosystems7900HT Sequence Detection System. For specific PCR primer information andinformation on individual SNP locations, see supplemental Tables 1 and 2,respectively, which are presented in the online appendix (available at http://diabetes.diabetesjournals.org).Statistical analysis. To assess linkage disequilibrium, differential transmis-sion of polymorphic variants from heterozygous parent to affected child wastested by the TDT (13). TDT for haplotypes was carried out with Genehunter(16). In multiplex families, the TDT is not strictly valid as a test of association.However, in view of the small number of multiplex families (4 of 72), we didnot correct for the small effect of this departure from the assumptions. Themaximum number of transmissions in our sample was 83, and some rareralleles provided samples of fewer than 30. To avoid compromising statisticalpower excessively, we restricted analysis to alleles for which the sum oftransmissions and nontransmissions from informative parents was 40 orgreater. For this minimum sample size of 40, we calculated the power todetect departures from the null hypothesis of 50% transmission in a two-sidedtest with � � 0.05. We used the normal approximation to the binomialdistribution as implemented in SISA (Simple Interactive Statistical Analysis)(17). For a transmission rate of 0.6, power is 0.24; for transmission rate 0.7,power is 0.73. These values are lower limits for the anticipated power. We alsocalculated the corresponding values of power for 60 transmissions: 0.34 and0.89 for transmission rates of 0.6 and 0.7, respectively. For most markers, thesample size was larger than 40, providing greater power to detect the stateddegree of differential transmission.
Nominal P values for significance of the TDT �2 are reported withoutcorrection for multiple testing, but we indicate here what minimal P valueswould be required if Bonferroni correction were used. The number ofstatistical tests for markers at one candidate gene was typically three to four;for four tests, Bonferroni correction would require a nominal P of 0.0125 foradjusted P � 0.05 and 0.0025 for adjusted P � 0.01. The total number ofstatistical tests was �380. Bonferroni correction would require a nominal P of1.3 104 for an adjusted P of 0.05 and 2.6 105 for an adjusted P of 0.01.
RESULTS
Diabetic nephropathy candidate gene polymorphismsnot previously tested (83 genes). Of the total of 115genes with results reported here, 83 have not been testedpreviously, to our knowledge. Among these 83 genes, theTDT was nominally significant (P � 0.05) for 12 (summa-rized individually below and in Table 2). The nonsignifi-cant results for the remaining 71 genes are summarized inTable 3.B-cell leukemia/lymphoma 2 (bcl-2) proto-oncogene.Ten SNPs in B-cell leukemia/lymphoma 2 (bcl-2) proto-oncogene (BCL2) were genotyped in the 72 diabetic ne-phropathy families. Three gave nominally significantevidence for linkage disequilibrium with diabetic nephrop-athy: rs2062011 (P � 0.001), rs12457700 (P � 0.006), and
rs1481031 (P � 0.009). All three SNPs lie in a 24-kb regionin intron 1 (192 kb) of BCL2.Catalase. We genotyped two SNPs in catalase (CAT).Both were nominally significant: rs1049982, located in the5�-untranslated region (UTR) (P � 0.006); and rs560807,located in intron 1 (P � 0.044).Laminin, alpha 4. Eight SNPs and one STRP weregenotyped in laminin, alpha 4 (LAMA4). One SNP,rs3734287, located in an intron, gave a nominally signifi-cant result (P � 0.016).Transforming growth factor, beta receptor II andtransforming growth factor, beta receptor III. SevenSNPs were genotyped in transforming growth factor, betareceptor II (TGFBR2) and 10 in transforming growthfactor, beta receptor III (TGFBR3). One SNP in each ofthese unlinked genes gave a nominally significant result:rs6792117, located in an intron of TGFBR2 (P � 0.024); andrs12756024, located in an intron of TGFBR3 (P � 0.018).Glutathione peroxidase 1. The single SNP we tested inglutathione peroxidase 1 (GPX1), rs1800668, was nomi-nally significant (P � 0.022).Laminin, gamma 1. We tested 12 SNPs in laminin,gamma 1 (LAMC1). Significant TDT results were foundacross the entire gene, suggesting strong linkage disequi-librium. We found that the linkage disequilibrium param-eter D� for the mostly widely spaced markers (separatedby 125 kb) ranged from 0.7 to 0.9 (P �� 0.01). Thestrongest evidence for linkage disequilibrium with diabeticnephropathy was found with a synonymous SNP, rs20557(Asn837Asn, P � 0.026). There is thus modest evidence forassociation of diabetic nephropathy with LAMC1; how-ever, the strong linkage disequilibrium across the gene willmake it difficult to narrow the critical region using geneticmeans.SMAD, mothers against DPP homolog 3. We testedseven SNPs in SMAD, mothers against DPP homolog 3(SMAD3). Linkage disequilibrium with two intronic SNPs,rs12594610 and rs4776890, located 2.9 kb apart, was nom-inally significant (P � 0.033 and 0.046, respectively).Upstream transcription factor 1. Four SNPs weregenotyped in upstream transcription factor 1 (USF1). Oneof these, rs2516839, located in the 3�-UTR, gave a nomi-nally significant result (P � 0.047).Aquaporin 1, IGF1, and tissue inhibitor of metallo-proteinase 3. Nominally significant results were foundfor STRP markers near three genes: aquaporin 1 (AQP1),IGF1, and tissue inhibitor of metalloproteinase 3 (TIMP3).The markers were D7S526 located 2.7 kb 5� of AQP1(125-bp allele, P � 0.027), MFD1 (GDB: 171128) located 0.7kb 5� of IGF1 (209-bp allele, P � 0.047), and D22S280 in the3�-UTR region of TIMP3 (214-bp allele, P � 0.048). Foreach of these genes, we followed up by testing two orthree SNPs in or near the gene and found no evidence tosupport the result from the STRP. We have not pursuedthese genes further.
Table 3 presents the results for SNPs and STRPs in 71additional “new” candidate genes (not previously tested)that showed no significant linkage disequilibrium withdiabetic nephropathy. In view of the marker spacing(mean of 17.2 kb) and the modest power of the sample, weconsider the absence of significant linkage disequilibriumto be inconclusive evidence concerning a role for thesegenes.Follow-up of previously reported diabetic nephropa-thy associations (32 genes). We genotyped SNPs in 32candidate genes that have been studied previously by
K.G. EWENS AND ASSOCIATES
DIABETES, VOL. 54, NOVEMBER 2005 3307

TABLE 2Candidate genes (n � 12) for diabetic nephropathy not previously tested; nominal P � 0.05 for at least one marker
Genesymbol Locus Assay ID dbSNP ID Location Alleles T
NotT Total %T �2 P
AQP1 7p14.3 hCV2973378 rs763422 5.1 kb 5� T/C T 33 31 64 0.52 0.1D7S526 2.6 kb 5� 125 bp 38 21 59 0.64 4.9 0.027hCV2973385 rs1049305 3�-UTR G/C G 27 24 51 0.53 0.2
BCL2 18q21.33 hCV7905447 rs1564483 3�-UTR C/T C 31 29 60 0.52 0.1hCV7905342 rs3943258 Intron T/C T 36 30 66 0.55 0.5hCV8685764 rs1481031 Intron C/T C 39 19 58 0.67 6.9 0.009hCV1408500 rs12457700 Intron C/T C 36 16 52 0.69 7.7 0.006hCV1408502 rs2062011 Intron A/T T 42 17 59 0.71 10.6 0.001hCV1408482 rs8083946 Intron G/A G 40 27 67 0.60 2.5hCV1728132 rs8084922 Intron G/C G 46 31 77 0.60 2.9hCV8687299 rs1381548 Intron G/A G 33 25 58 0.57 1.1hCV2855833 rs11152377 Intron C/T C 32 27 59 0.54 0.4hCV2855835 rs2551402 4.1 kb 5� C/A C 38 30 68 0.56 0.9
CAT 11p13 hCV1883211 rs1049982 5�-UTR C/T C 43 21 64 0.67 7.6 0.006hCV3102895 rs560807 Intron A/T A 44 27 71 0.62 4.1 0.044
GPX1 3p21.3 hCV7912052 rs1800668 5�-UTR A/G G 29 14 43 0.67 5.2 0.022IGF1 12q23.2 hCV2801121 rs2946834 1.9 kb 3� A/G A 24 22 46 0.52 0.1
hCV2801103 rs972936 Intron T/C C 28 28 56 0.50 0.0hCV346219 rs10735380 Intron A/G G 30 27 57 0.53 0.2MFD1 0.7 kb 5� 209 bp 15 28 43 0.35 3.9 0.047
LAMA4 6q21 hCV2462170 rs1050353 Val(A)1713Val(T) A/T A 30 29 59 0.51 0.0hCV2462178 rs969139 Intron C/T T 44 32 76 0.58 1.9hCV2462186 rs3734287 Intron C/T C 37 19 56 0.66 5.8 0.016hCV2462219 rs11153344 Intron A/G G 35 31 66 0.53 0.2LAMA4-STRP1 Intron 119 bp 27 15 42 0.64 3.4hCV2462251 rs1050348 His(C)491Tyr(T) A/G A 28 24 52 0.54 0.3hCV2462280 rs3777928 Intron A/C A 33 30 63 0.52 0.1hCV2462319 rs2157547 Intron C/G G 20 18 38 0.53 0.1hCV11903282 rs1894682 Intron A/G A 33 23 56 0.59 1.8
LAMC1 1q25.3 hCV505167 rs10737236 4 kb 5� C/T T 45 30 75 0.60 3.0hCV26124236 rs10911194 Ala(C)58Ala(T) A/G G 46 31 77 0.60 2.9hCV9066112 rs10797819 Intron G/A A 46 28 74 0.62 4.4 0.036hCV1770066 rs4652775 Intron A/T A 45 29 74 0.61 3.5hCV3127531 rs2296288 Cys(C)182Cys(T) T/C T 46 29 75 0.61 3.9 0.050hCV11632431 rs7556132 Ile(A)458Val(G) A/G A 47 29 76 0.62 4.3 0.039hCV3127590 rs2296292 Ala(C)592Ala(A) A/C A 45 28 73 0.62 4.0 0.047hCV3127518 rs20557 Asn(C)837Asn(T) T/C T 46 27 73 0.63 4.9 0.026hCV3127512 rs7410919 Leu888Pro T/C T 47 29 76 0.62 4.3 0.039LAMC1-STRP1 Intron 215 bp 31 20 51 0.61 2.4hCV3127470 rs4651146 Arg(C)1376Arg(T) T/C C 42 28 70 0.60 2.8hCV3127469 rs3818419 Ala(A)1433Ala(G) G/A G 33 32 65 0.51 0.0hCV3127459 rs1547715 3�-UTR A/G A 47 30 77 0.61 3.8
SMAD3 15q22.33 hCV9707890 rs1498506 Intron A/C A 28 18 46 0.61 2.2hCV2113018 rs4776890 Intron C/G T 40 24 64 0.63 4.0 0.046hCV11306173 rs12594610 Intron G/A G 36 20 56 0.64 4.6 0.033hCV2112975 rs11631380 Intron C/T T 32 19 51 0.63 3.3hCV2112965 rs745103 Intron A/G A 29 29 58 0.50 0.0hCV1044749 rs731874 Intron A/G G 31 23 54 0.57 1.2hCV2112907 rs2289791 Intron G/T T 29 19 48 0.60 2.1
TGFBR2 3p24.1 hCV3158972 rs13081419 Intron A/C C 41 31 72 0.57 1.4hCV11565979 rs1431131 Intron A/T T 34 30 64 0.53 0.3hCV1612549 rs1155705 Intron A/G G 34 32 66 0.52 0.1hCV972343 rs1078985 Intron A/G G 24 22 46 0.52 0.1hCV8778179 rs995435 Intron A/G G 27 21 48 0.56 0.8hCV1612506 rs6792117 Intron A/G G 41 23 64 0.64 5.1 0.024hCV1612480 rs744751 2.8 kb 3� A/G A 29 25 54 0.54 0.3
TGFBR3 1p22.1 hCV945103 rs284878 Thr(C)746Thr(T) A/G A 10 5 15 0.67 1.7hCV1931721 rs1805113 Phe(C)673Phe(T) A/G G 38 30 68 0.56 0.9hCV3130156 rs284180 Intron A/C A 38 32 70 0.54 0.5hCV3130147 rs284190 Intron A/T T 37 29 66 0.56 1.0hCV3130125 rs12756024 Intron A/C C 42 23 65 0.65 5.6 0.018hCV11643684 rs5019497 Intron A/C A 38 34 72 0.53 0.2
Continued on following page
CANDIDATE GENES FOR DIABETIC NEPHROPATHY
3308 DIABETES, VOL. 54, NOVEMBER 2005

others. Table 4 shows results from our TDT studies for 11of these genes. In eight of these, we found nominallysignificant results. Table 4 also includes results for SNPs inthree genes (ACE, aldose reductase [AKR1B1], and apoli-poprotein E [APOE]) that deserve attention because theyhave been the subject of numerous diabetic nephropathyassociation studies. For these genes, we found a trend thatsupports published results, although our results were notsignificant, perhaps because of the small sample size. Thenonsignificant results for the remaining 21 genes aresummarized in Table 5.Collagen, type IV, alpha 1. Nine SNPs and one STRPwere genotyped in collagen, type IV, alpha 1 (COL4A1).Two SNPs in intron 1 showed significant association withdiabetic nephropathy: rs614282 (P � 0.002) and rs679062(P � 0.0002). Because of the strong evidence with thelatter SNP, we looked for nearby coding SNPs. We se-quenced a 700-bp region that included all of exon 2(located �4 kb from rs614282) in two sets of pooled DNAsamples: 16 diabetic nephropathy and 42 CEPH individu-als. No sequence variants were found, suggesting that nocommon disease-associated variant is located in thisnearby exon.
Two studies of COL4A1 by others (18,19) led to contra-dictory conclusions that have not been followed up since.The region of association we found in intron 1 lies �100 kb5� to a polymorphic HindIII restriction site found byKrolewski et al. (19) to be associated with increased riskfor progression to overt nephropathy. Chen et al. (18)failed to confirm this finding with a larger sample (n � 116diabetic nephropathy and 91 individuals with long-stand-ing diabetes but no evidence of kidney disease [diabeticnephropathy negative]). In our studies, SNP rs1133219,located only 8 kb from the site first tested by Krolewski etal. (19), provided no significant evidence (55 transmis-sions, P � 0.53).Angiotensin II receptor, type 1 region. Moczulski et al.(20) reported linkage and association studies in discordantsibpairs and parent-offspring trios with a diabetic nephrop-athy or diabetic nephropathy–negative offspring. Theyfound linkage with the STRPs ATCA (located near theangiotensin II receptor, type 1 [AGTR1 gene]) andD3S1308 (located 575 kb telomeric to AGTR1), but noassociation was found with six SNPs in AGTR1 or with anyalleles of ATCA. (No association results were reported forD3S1308.) We tested these two STRPs, plus three addi-tional SNPs in AGTR1. These included the A1166C SNP
reported previously (21–24). We also tested 11 SNPslocated in the 1-Mb region telomeric to AGTR1 (summa-rized in Table 4). The only significant evidence for linkagedisequilibrium with diabetic nephropathy is seen atD3S1308 itself (allele 2 [106 bp], P � 0.001; and allele 3[108 bp], P � 0.009; alleles named as in GDB allele set:63031, http://gdbwww.gdb.org).Lipoprotein lipase. Five SNPs in lipoprotein lipase(LPL) were tested. Three of the SNPs, located in a 5.4-kbregion near the 3� end of the gene, had nominally signifi-cant TDT results: rs320 (P � 0.005), rs326 (P � 0.011), andrs13702 (P � 0.004). In a study of Caucasian type 1diabetic patients, Orchard et al. (25) reported an associa-tion between rs320 (a HindIII restriction site) and in-creased albumin-to-creatinine ratio.Protein kinase C, beta 1. Eleven SNPs and one STRP inor near protein kinase C, beta 1 (PRKCB1) were geno-typed. Only SNP rs1015408, located in intron 4, wasnominally significant (P � 0.025). Two of the SNPs wegenotyped were previously found to be associated withdiabetic nephropathy (26): rs3760106 (C-1504T) andrs2575390 (G-546C). However, in our families, there wasno significant evidence for linkage disequilibrium witheither of these SNPs.Neuropilin 1. Iyengar et al. (27) found linkage betweenD10S1654 and diabetic nephropathy in Caucasian sibpairswith type 2 diabetes. Because this marker maps in anintron of neuropilin 1 (NRP1), we tested seven SNPs in thisgene. Two of these, rs869636 and rs2804495, located 40 kbapart in intron 2, were nominally significant (P � 0.047 and0.027, respectively).HNF1B1/transcription factor 2, hepatic (MODY5).Several studies have reported that rare mutations inHNF1B1 are associated with renal dysfunction in Japaneseand Caucasian maturity-onset diabetes of the young fam-ilies (28–31). However, no HNF1B1 mutations were foundamong 63 German and Czech type 2 diabetic patients withdiabetic nephropathy (32). In our type 1 diabetic familieswith diabetic nephropathy, we found nominally significantevidence with an SNP located in the 3�-UTR (rs2688, P �0.029), but three SNPs in introns of HNF1B1 and onelocated 2.2 kb 3� of the gene failed to support this finding.p22phox/cytochrome b-245, �-polypeptide. ThreeSNPs were genotyped in p22phox, including rs4673(C242T, His72Tyr) previously studied for association withdiabetic nephropathy in Caucasians with type 1 diabetes(33) and Japanese with type 2 diabetes (34). In our type 1
TABLE 2—Continued
Genesymbol Locus Assay ID dbSNP ID Location Alleles T
NotT Total %T �2 P
hCV11643667 rs10783040 Intron A/G G 38 28 66 0.58 1.5hCV1931638 rs11165595 Intron A/G A 34 30 64 0.53 0.3hCV3130092 rs1192524 Intron A/G A 32 32 64 0.50 0.0hCV3181378 rs7550034 Intron A/G A 37 35 72 0.51 0.1D1S1588 Intron 132 bp 17 28 45 0.38 2.7
TIMP3 22q12.3 hCV8712827 rs135025 Intron A/G A 38 26 64 0.59 2.3D22S280 Intron 214 bp 32 18 50 0.64 3.9 0.048hCV3294872 rs242075 Intron A/G G 39 37 76 0.51 0.1hCV8712964 rs1065314 3�-UTR T/C C 26 25 51 0.51 0.0
USF1 1q23.3 hCV1459759 rs3737787 3�-UTR A/G G 25 24 49 0.51 0.0rs2073658 rs2073658 Intron C/T C 22 19 41 0.54 0.2hCV15949520 rs2073656 Intron C/G G 22 21 43 0.51 0.0hCV1839183 rs2516839 5�-UTR C/T T 45 28 73 0.62 4.0 0.047
T, number of transmissions in the TDT analysis. For a complete list of gene abbreviations, see the APPENDIX.
K.G. EWENS AND ASSOCIATES
DIABETES, VOL. 54, NOVEMBER 2005 3309
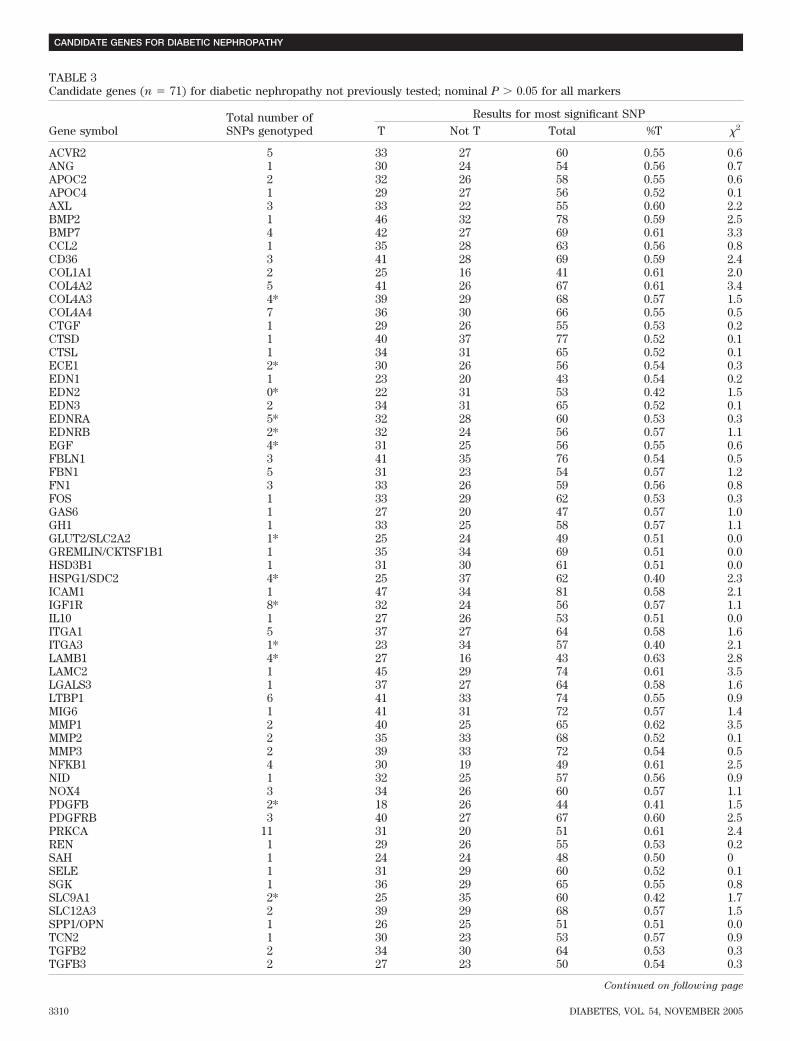
TABLE 3Candidate genes (n � 71) for diabetic nephropathy not previously tested; nominal P � 0.05 for all markers
Gene symbolTotal number ofSNPs genotyped
Results for most significant SNPT Not T Total %T �2
ACVR2 5 33 27 60 0.55 0.6ANG 1 30 24 54 0.56 0.7APOC2 2 32 26 58 0.55 0.6APOC4 1 29 27 56 0.52 0.1AXL 3 33 22 55 0.60 2.2BMP2 1 46 32 78 0.59 2.5BMP7 4 42 27 69 0.61 3.3CCL2 1 35 28 63 0.56 0.8CD36 3 41 28 69 0.59 2.4COL1A1 2 25 16 41 0.61 2.0COL4A2 5 41 26 67 0.61 3.4COL4A3 4* 39 29 68 0.57 1.5COL4A4 7 36 30 66 0.55 0.5CTGF 1 29 26 55 0.53 0.2CTSD 1 40 37 77 0.52 0.1CTSL 1 34 31 65 0.52 0.1ECE1 2* 30 26 56 0.54 0.3EDN1 1 23 20 43 0.54 0.2EDN2 0* 22 31 53 0.42 1.5EDN3 2 34 31 65 0.52 0.1EDNRA 5* 32 28 60 0.53 0.3EDNRB 2* 32 24 56 0.57 1.1EGF 4* 31 25 56 0.55 0.6FBLN1 3 41 35 76 0.54 0.5FBN1 5 31 23 54 0.57 1.2FN1 3 33 26 59 0.56 0.8FOS 1 33 29 62 0.53 0.3GAS6 1 27 20 47 0.57 1.0GH1 1 33 25 58 0.57 1.1GLUT2/SLC2A2 1* 25 24 49 0.51 0.0GREMLIN/CKTSF1B1 1 35 34 69 0.51 0.0HSD3B1 1 31 30 61 0.51 0.0HSPG1/SDC2 4* 25 37 62 0.40 2.3ICAM1 1 47 34 81 0.58 2.1IGF1R 8* 32 24 56 0.57 1.1IL10 1 27 26 53 0.51 0.0ITGA1 5 37 27 64 0.58 1.6ITGA3 1* 23 34 57 0.40 2.1LAMB1 4* 27 16 43 0.63 2.8LAMC2 1 45 29 74 0.61 3.5LGALS3 1 37 27 64 0.58 1.6LTBP1 6 41 33 74 0.55 0.9MIG6 1 41 31 72 0.57 1.4MMP1 2 40 25 65 0.62 3.5MMP2 2 35 33 68 0.52 0.1MMP3 2 39 33 72 0.54 0.5NFKB1 4 30 19 49 0.61 2.5NID 1 32 25 57 0.56 0.9NOX4 3 34 26 60 0.57 1.1PDGFB 2* 18 26 44 0.41 1.5PDGFRB 3 40 27 67 0.60 2.5PRKCA 11 31 20 51 0.61 2.4REN 1 29 26 55 0.53 0.2SAH 1 24 24 48 0.50 0SELE 1 31 29 60 0.52 0.1SGK 1 36 29 65 0.55 0.8SLC9A1 2* 25 35 60 0.42 1.7SLC12A3 2 39 29 68 0.57 1.5SPP1/OPN 1 26 25 51 0.51 0.0TCN2 1 30 23 53 0.57 0.9TGFB2 2 34 30 64 0.53 0.3TGFB3 2 27 23 50 0.54 0.3
Continued on following page
CANDIDATE GENES FOR DIABETIC NEPHROPATHY
3310 DIABETES, VOL. 54, NOVEMBER 2005

diabetic families, the 242C-allele was significantly over-transmitted (P � 0.032). This result supports the findingsof Matsunaga-Irie et al. (34), but is not consistent withthose of Hodgkinson et al. (33), in which the TT genotypewas significantly more frequent in diabetic patients withnephropathy than in the control group.Matrix metalloproteinase 9. Maeda et al. (35) andHirakawa et al. (36) found evidence for association inJapanese and Caucasian type 2 diabetic patients, respec-tively, between diabetic nephropathy and D20S838, anSTRP located in the promoter region of matrix metallo-proteinase 9 (MMP9). In contrast, we found no evidencefor an association with any allele of D20S838. Our resultsdid provide nominally significant evidence for linkagedisequilibrium between diabetic nephropathy andrs11697325, an SNP located 8.2 kb 5� of MMP9 (P � 0.029),but this was not supported by results from rs2664538, anonsynonymous SNP (Gln279Arg) in exon 6 of MMP9.Other previously tested genes. Table 5 gives the resultsfor the 21 genes with previously reported diabetic ne-phropathy associations for which we found no significantlinkage disequilibrium with diabetic nephropathy. Asnoted above, three genes for which our results are nega-tive (ACE, AKR1B1, and APOE) have been the subject ofmany studies of association in diabetic nephropathy, sowe comment further here. The variants tested were asfollows: 1) the 287-bp insertion/deletion (in/del) polymor-phism in intron 16 of ACE (37–44), 2) the CA-repeat STRPat AKR1B1 (45–51), and 3) the APOE polymorphism(25,52–56). In our results (Table 4), we see a trend thatsupports these findings, but our sample size is small, andresults are mostly not significant: ACE in/del (deletionallele, 38:31 transmissions:nontransmissions, 55.1% trans-missions in the TDT analysis, P � 0.5); AKR1B1 5�CA-repeat polymorphism (Z2 allele, 27:22, 55.1% transmissions,P � 0.5; Z�2 allele, 8:15, 34.8% transmissions, P � 0.5); andAPOE (e2 “risk” allele, 12:2, 85.7% transmissions, P �0.008).
For all of the genes in Table 5 in which we tested morethan one marker, we also examined results of the TDTwith the corresponding haplotypes. Among 12 genestested, we found nominally significant results with several(smallest P � 0.009). However, in this analysis, all possiblehaplotypes were tested, and the results in all cases arebased on fewer than 40 transmissions, reducing our con-fidence that these are true positives.TDT in CEPH control families. We were concerned thatan SNP allele that appeared to be associated with diabeticnephropathy might be preferentially transmitted, for rea-
sons unrelated to diabetes or diabetic nephropathy. Toaddress this possibility of transmission distortion, wefocused on genes in which at least one SNP was significantat P � 0.05 in the TDT analysis. (There were 29 such SNPsin 16 genes; in 4 additional genes, the only markers withP � 0.05 were STRPs, and these were not tested in controlsubjects.) We genotyped the 29 SNPs in 36 CEPH families,considered as unselected control subjects (detailed resultsnot shown). For most transmissions, the sample size wassomewhat larger (maximum, 114) than in the diabeticnephropathy families.
Only three SNPs had transmission distortion with nom-inal P � 0.05 in the CEPH families. For rs560807 in CAT(P � 0.022) and rs11697325 in MMP9 (P � 0.035), the allelethat was over-transmitted in the diabetic nephropathyfamilies was significantly under-transmitted in the CEPHfamilies, slightly strengthening the evidence from thediabetic nephropathy families. At the third SNP, rs6792117in TGFBR2, the same allele was over-transmitted in bothsets of families, but the effect was barely significant in theCEPH families (P � 0.048). For a more global view, welooked at the whole set of 29 SNPs in 16 genes. In thediabetic nephropathy data, the P values range from 0.0002to 0.05, and almost 50% (13 of 29) have P � 0.025. Incontrast, in the CEPH families, there is only one SNP withP � 0.025 (rs560807 in CAT), and as noted above, thisresult is “in the direction” opposite to that seen in thediabetic nephropathy families.
DISCUSSION
Our principal goal was to assess the evidence for acontribution to diabetic nephropathy susceptibility at 115candidate genes. By carrying out a comprehensive analysisof all of the genes on the same family material, we haveprovided a large set of comparable findings, a featurelacking in the results from very heterogeneous existingstudies. One of our findings is significant beyond thenominal P � 0.001 level (0.0002, for COL4A1), but inter-pretation of this and all of our findings is complicated bythe multiple testing problem. For interpretation of Pvalues, we suggest the following approach, which is basedon genes, not on individual markers. Markers within agene tend to be correlated to varying degrees. For this andother reasons (57), adjustment for the full number ofmarkers tested (e.g., by Bonferroni correction) is likely tobe too stringent. Instead of considering individual P val-ues, we identified the genes with at least one P value�0.05. Among the 83 “new” genes, we would expect 0.05
TABLE 3—Continued
Gene symbolTotal number ofSNPs genotyped
Results for most significant SNPT Not T Total %T �2
TIMP2 2 34 28 62 0.55 0.6TNFRSF1A 1 37 29 66 0.56 1.0TNFSF6/FASLG 1 39 35 74 0.53 0.2UBA52 1 36 35 71 0.51 0.0UNC13B 6 33 27 60 0.55 0.6USF2 1 20 16 36 0.56 0.4UTS2 2 37 29 66 0.56 1.0VEGF 1* 38 32 70 0.54 0.5VEGFR2/KDR 3 31 23 54 0.57 1.2
For detailed results, see supplemental Table 2 in the online appendix. T, number of transmissions in the TDT analysis. *One STRP or variablenumber tandem repeat was genotyped in addition to the number of SNPs indicated. For a complete list of gene abbreviations, see the APPENDIX.
K.G. EWENS AND ASSOCIATES
DIABETES, VOL. 54, NOVEMBER 2005 3311

TABLE 4Candidate genes (n � 11) for diabetic nephropathy previously studied by others
Genesymbol Assay ID dbSNP ID Location Alleles T
NotT Total %T �2 P Reference
ACE hCV1247701 rs4293 Intron A/G A 39 33 72 0.54 0.5 (37–43)hCV1247713 rs4329 Intron A/G A 37 32 69 0.54 0.4in/del Intron 16-in/del del 38 31 69 0.55 0.7 (22,37–44)hCV1247681 rs4267385 Intron C/T C 31 30 61 0.51 0.0
AGTR1region
rs1492103 rs1492103 AGTR1-intron C/T C 30 30 60 0.50 0.0rs5182 rs5182 AGTR1-Leu(C)191Leu(T) C/T C 32 26 58 0.55 0.6rs5186 rs5186 AGTR1-A1166C A/C C 29 19 48 0.60 2.1 (20–24)rs427832 rs427832 Intergenic C/T C 23 20 43 0.54 0.2ATCA 9.3kb 5� of AGTR1 9 38 34 72 0.53 0.2hCV9146233 rs1845413 CPA3-intron G/A A 28 23 51 0.55 0.5hCV8759101 rs812249 SMARCA3-Thr(A)303Thr(G) C/T C 23 21 44 0.52 0.1hCV1732626 rs6440589 HPS3-Gln(A)498Gln(G) G/A A 31 19 50 0.62 2.9D3S1308 573 kb 5� of AGTR1 106 bp 17 42 59 0.29 10.6 0.001 (20)
108 bp 47 25 72 0.65 6.7 0.009hCV2041187 rs2293418 Intergenic A/G A 43 28 71 0.61 3.2hCV3201872 N/A Intergenic G/A A 22 20 42 0.52 0.1hCV265602 N/A TM4SF4-intron G/A A 35 25 60 0.58 1.7hCV2726141 N/A TAZ-intron T/G G 27 19 46 0.59 1.4rs1344816 rs1344816 TAZ-intron T/G G 31 21 52 0.60 1.9hCV9148272 rs6807742 TAZ-intron A/T A 24 24 48 0.50 0.0hCV1794446 rs1002896 Intergenic A/G G 27 23 50 0.540 0.3
AKR1B1 rs759853 rs759853 C-106T in 5�-UTR C/T C 24 24 48 0.50 0.0 (48,49)STRP1-AKR1B1 1.9 kb 5� Z2 27 22 49 0.55 0.5 (45–51)
Z 27 28 55 0.49 0.0Z�2 8 15 23 0.35 2.1
APOE APOE RFLP* rs429358 Arg(C)112Cys(T) 2 12 2 14 0.86 7.1 (25,52–56)rs7412 Arg(C)158Cys(T) 3 20 30 50 0.40 2.0
4 19 19 38 0.50 0.0COL4A1 hCV1964948 rs1133219 Ala(T)1490Ala(C) G/A A 29 26 55 0.53 0.2 (18,19)
hCV3147619 rs2305080 Intron T/C C 43 30 73 0.59 2.3afm073we5 Intron 175 bp 30 36 66 0.46 0.5hCV3147628 rs532625 Ala(A)144Ala(T) A/T A 33 25 58 0.57 1.1hCV3147652 rs639562 Intron T/C T 28 24 52 0.54 0.3hCV3147669 rs614282 Intron T/C C 40 17 57 0.72 9.3 0.002hCV3147671 rs679062 Intron C/T T 43 15 58 0.74 13.5 0.0002hCV3147675 rs9559749 Intron G/A G 29 18 47 0.62 2.6hCV3147696 rs627527 Intron G/A A 44 29 73 0.60 3.1hCV1433329 rs12431029 Intron C/T C 34 27 61 0.56 0.8
HNF1B1/TCF2
hCV2559950 rs739753 2.2 kb 3� T/A T 24 16 40 0.60 1.6 (28–32)hCV11415601 rs2688 3�-UTR C/A C 43 25 68 0.63 4.8 0.029hCV2559930 rs2269843 Intron G/A A 28 18 46 0.61 2.1hCV2559920 rs2285740 Intron C/T T 33 32 65 0.51 0.0hCV2559889 rs4430796 Intron C/T T 26 25 51 0.51 0.0
LPL hCV9642885 rs10104051 Intron C/T C 28 26 54 0.52 0.1rs285 rs285 Intron C/T T 35 33 68 0.52 0.1rs320 rs320 Intron G/T T 41 19 60 0.68 8.1 0.005 (25,110)hCV1843005 rs326 Intron A/G A 41 21 62 0.66 6.5 0.011hCV9639448 rs13702 3�-UTR C/T T 40 18 58 0.69 8.3 0.004
MMP9 hCV1414746 rs11697325 8.2 kb 5� A/G A 31 16 47 0.66 4.8 0.029D20S838 5�-UTR A14 30 26 56 0.54 0.3 (35,36)
A21 24 22 46 0.52 0.1hCV11655953 rs2664538 Gln(A)279Arg(G) A/G A 30 20 50 0.60 2.0
NRP1 hCV347431 rs2247015 Intron T/G T 36 35 71 0.51 0 (27)hCV346947 rs2474714 Intron G/A A 37 35 72 0.51 0.1hCV2738770 rs927099 Intron C/T T 39 30 69 0.57 1.2hCV7467750 rs1319013 Intron T/G T 35 33 68 0.51 0.1hCV7467760 rs869636 Intron T/C C 36 21 57 0.63 4.0 0.047hCV2738721 rs1331326 Intron T/C C 41 30 71 0.58 1.7hCV11659809 rs2804495 Intron T/G G 42 24 66 0.64 4.9 0.027
p22phox/CYBA
rs1049255 rs1049255 Ala(C)174Val(T) C/T T 33 31 64 0.52 0.1hCV11291909 rs3794622 Intron C/T C 35 34 69 0.51 0.0hCV2038 rs4673 His(C)72Tyr(T) C/T C 32 17 49 0.65 4.6 0.032 (33,34)
Continued on following page
CANDIDATE GENES FOR DIABETIC NEPHROPATHY
3312 DIABETES, VOL. 54, NOVEMBER 2005
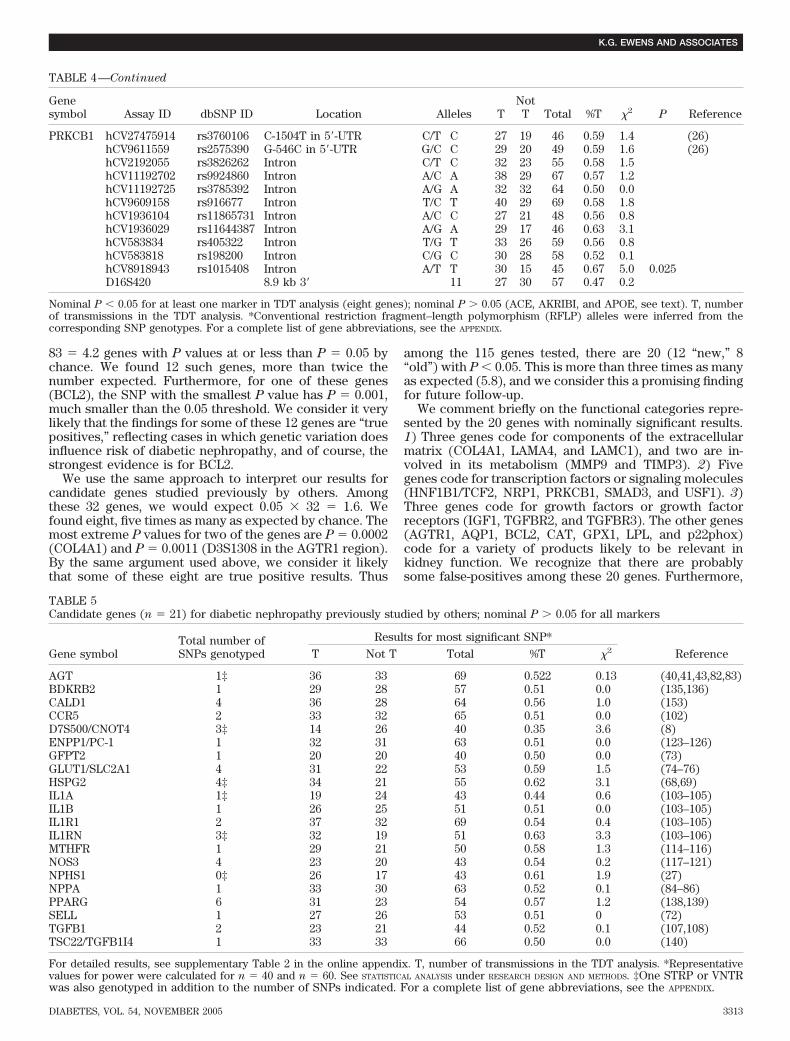
83 � 4.2 genes with P values at or less than P � 0.05 bychance. We found 12 such genes, more than twice thenumber expected. Furthermore, for one of these genes(BCL2), the SNP with the smallest P value has P � 0.001,much smaller than the 0.05 threshold. We consider it verylikely that the findings for some of these 12 genes are “truepositives,” reflecting cases in which genetic variation doesinfluence risk of diabetic nephropathy, and of course, thestrongest evidence is for BCL2.
We use the same approach to interpret our results forcandidate genes studied previously by others. Amongthese 32 genes, we would expect 0.05 32 � 1.6. Wefound eight, five times as many as expected by chance. Themost extreme P values for two of the genes are P � 0.0002(COL4A1) and P � 0.0011 (D3S1308 in the AGTR1 region).By the same argument used above, we consider it likelythat some of these eight are true positive results. Thus
among the 115 genes tested, there are 20 (12 “new,” 8“old”) with P � 0.05. This is more than three times as manyas expected (5.8), and we consider this a promising findingfor future follow-up.
We comment briefly on the functional categories repre-sented by the 20 genes with nominally significant results.1) Three genes code for components of the extracellularmatrix (COL4A1, LAMA4, and LAMC1), and two are in-volved in its metabolism (MMP9 and TIMP3). 2) Fivegenes code for transcription factors or signaling molecules(HNF1B1/TCF2, NRP1, PRKCB1, SMAD3, and USF1). 3)Three genes code for growth factors or growth factorreceptors (IGF1, TGFBR2, and TGFBR3). The other genes(AGTR1, AQP1, BCL2, CAT, GPX1, LPL, and p22phox)code for a variety of products likely to be relevant inkidney function. We recognize that there are probablysome false-positives among these 20 genes. Furthermore,
TABLE 5Candidate genes (n � 21) for diabetic nephropathy previously studied by others; nominal P � 0.05 for all markers
Gene symbolTotal number ofSNPs genotyped
Results for most significant SNP*ReferenceT Not T Total %T �2
AGT 1‡ 36 33 69 0.522 0.13 (40,41,43,82,83)BDKRB2 1 29 28 57 0.51 0.0 (135,136)CALD1 4 36 28 64 0.56 1.0 (153)CCR5 2 33 32 65 0.51 0.0 (102)D7S500/CNOT4 3‡ 14 26 40 0.35 3.6 (8)ENPP1/PC-1 1 32 31 63 0.51 0.0 (123–126)GFPT2 1 20 20 40 0.50 0.0 (73)GLUT1/SLC2A1 4 31 22 53 0.59 1.5 (74–76)HSPG2 4‡ 34 21 55 0.62 3.1 (68,69)IL1A 1‡ 19 24 43 0.44 0.6 (103–105)IL1B 1 26 25 51 0.51 0.0 (103–105)IL1R1 2 37 32 69 0.54 0.4 (103–105)IL1RN 3‡ 32 19 51 0.63 3.3 (103–106)MTHFR 1 29 21 50 0.58 1.3 (114–116)NOS3 4 23 20 43 0.54 0.2 (117–121)NPHS1 0‡ 26 17 43 0.61 1.9 (27)NPPA 1 33 30 63 0.52 0.1 (84–86)PPARG 6 31 23 54 0.57 1.2 (138,139)SELL 1 27 26 53 0.51 0 (72)TGFB1 2 23 21 44 0.52 0.1 (107,108)TSC22/TGFB1I4 1 33 33 66 0.50 0.0 (140)
For detailed results, see supplementary Table 2 in the online appendix. T, number of transmissions in the TDT analysis. *Representativevalues for power were calculated for n � 40 and n � 60. See STATISTICAL ANALYSIS under RESEARCH DESIGN AND METHODS. ‡One STRP or VNTRwas also genotyped in addition to the number of SNPs indicated. For a complete list of gene abbreviations, see the APPENDIX.
TABLE 4—Continued
Genesymbol Assay ID dbSNP ID Location Alleles T
NotT Total %T �2 P Reference
PRKCB1 hCV27475914 rs3760106 C-1504T in 5�-UTR C/T C 27 19 46 0.59 1.4 (26)hCV9611559 rs2575390 G-546C in 5�-UTR G/C C 29 20 49 0.59 1.6 (26)hCV2192055 rs3826262 Intron C/T C 32 23 55 0.58 1.5hCV11192702 rs9924860 Intron A/C A 38 29 67 0.57 1.2hCV11192725 rs3785392 Intron A/G A 32 32 64 0.50 0.0hCV9609158 rs916677 Intron T/C T 40 29 69 0.58 1.8hCV1936104 rs11865731 Intron A/C C 27 21 48 0.56 0.8hCV1936029 rs11644387 Intron A/G A 29 17 46 0.63 3.1hCV583834 rs405322 Intron T/G T 33 26 59 0.56 0.8hCV583818 rs198200 Intron C/G C 30 28 58 0.52 0.1hCV8918943 rs1015408 Intron A/T T 30 15 45 0.67 5.0 0.025D16S420 8.9 kb 3� 11 27 30 57 0.47 0.2
Nominal P � 0.05 for at least one marker in TDT analysis (eight genes); nominal P � 0.05 (ACE, AKRIBI, and APOE, see text). T, numberof transmissions in the TDT analysis. *Conventional restriction fragment–length polymorphism (RFLP) alleles were inferred from thecorresponding SNP genotypes. For a complete list of gene abbreviations, see the APPENDIX.
K.G. EWENS AND ASSOCIATES
DIABETES, VOL. 54, NOVEMBER 2005 3313

as noted above, the results could in principle be due totype 1 diabetes instead of diabetic nephropathy, but inview of the known functions of these genes, this possibil-ity seems unlikely.
Our many negative results call for some comment. Forseveral very large genes (for example, latent transforminggrowth factor beta binding protein 1 [LTBP1] and IGF1receptor) the small number of SNPs we tested led to verylarge spacing between SNPs, so a negative result does notconstitute strong evidence against a contribution by thegene. In addition, we note that our study is based entirelyon type 1 diabetic patients of European ancestry. Ourresults might not be directly comparable with those forcandidate genes studied previously in other ethnic groupsor in type 2 diabetes. Finally, in any study, including thepresent one, both positive and negative results must beinterpreted with awareness of the limitations imposed bysample size and multiple testing. In particular, nonsignifi-cant results must be viewed against the background ofanticipated effect size and likely statistical power. Withour modest sample size throughout, it is likely that someeffects of candidate genes have not been detected, or notbeen confirmed, even though they are “real.”
ACKNOWLEDGMENTS
R.S.S. has received support from National Institutes ofHealth Grant DK-55227 and U.S. Army Medical ResearchGrant DAMD17-01-1-0009).
We are grateful to HBDI for recontacting families andto the families who volunteered to participate in thisstudy through HBDI and the Hospital of the University ofPennsylvania.
APPENDIX
Gene abbreviations. ACE, angiotensin I converting en-zyme; ACVR2, activin A receptor, type IIA; AGT, angio-tensinogen; AGTR1, angiotensin II receptor, type 1;AKR1B1(AR), aldose reductase; ANG, angiogenin, ribonu-clease, RNase A family, 5; APOC2, apolipoprotein C2;APOC4, apolipoprotein C4; APOE, apolipoprotein E;AQP1, aquaporin 1; AXL, AXL receptor tyrosine kinase;BCL2, B-cell leukemia/lymphoma 2 (bcl-2) proto-onco-gene; BDKRB2, bradykinin receptor B2; BMP2, bonemorphogenetic protein 2 precursor; BMP7, bone morpho-genetic protein 7; CALD1, caldesmon 1; CAT, catalase;CCL2, chemokine (C-C motif) ligand 2; CCR5, chemokine(C-C motif) receptor 5; CD36, CD36 antigen; CNOT4,CCR4-NOT transcription complex, subunit 4; COL1A1,collagen, type I, alpha 1; COL4A1, collagen, type IV, alpha1; COL4A2, collagen, type IV, alpha 2; COL4A3, collagen,type IV, alpha 3; COL4A4, collagen, type IV, alpha 4; CPA3,carboxypeptidase A3 ; CTGF, connective tissue growthfactor; CTSD, cathepsin D; CTSL, cathepsin L; ECE1,endothelin converting enzyme 1; EDN1, endothelin 1;EDN2, endothelin 2; EDN3, endothelin 3; EDNRA, endo-thelin receptor type A; EDNRB, endothelin receptor typeB; EGF, epidemal growth factor; ENPP1 (PC-1), ectonucle-otide pyrophosphatase/phosphodiesterase 1; FBLN1, fibu-lin 1; FBN1, fibrillin; FN1, fibronectin 1; FOS, v-fos FBJmurine osteosarcoma viral oncogene homolog; GAS6,growth arrest–specific 6; GFPT2, glutamine-fructose-6-phosphate transaminase 2; GH1, growth hormone; GLUT1(SLC2A1), glucose transporter-1, solute carrier family 2,member 1; GLUT2 (SLC2A2), glucose transporter-2, solutecarrier family 2, member 2; GPX1, glutathione peroxidase
1; GREM (CKTSF1B1), gremlin 1 homolog, cysteine knotsuperfamily; HNF1B1 (TCF2), transcription factor 2, he-patic; HPS3, Hermansky-Pudlak syndrome 3; HSD3B1,hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroiddelta-isomerase 1; HSPG1 (SDC2), heparan sulfate proteo-glycan 1 (syndecan 2); HSPG2, heparan sulfate proteogly-can 2 (perlecan); ICAM1, intercellular adhesion molecule1; IGF1R, IGF1 receptor; IL10, interleukin 10; IL1A, inter-leukin-1, alpha; IL1B, interleukin-1, beta; IL1R1, interleu-kin-1 receptor type 1; IL1RN, interleukin-1 receptorantoginist; ITGA1, integrin, alpha 1; ITGA3, integrin, alpha3; LAMA4, laminin, alpha 4; LAMB1, laminin, beta 1;LAMC1, laminin, gamma 1; LAMC2, laminin, gamma 2 ;LGALS3, lectin, galactoside-binding, soluble, 3; LPL, lipo-protein lipase; LTBP1, latent transforming growth factorbeta binding protein 1; MIG6, mitogen-inducible gene 6protein; MMP1, matrix metalloproteinase 1; MMP2, matrixmetalloproteinase 2; MMP3, matrix metalloproteinase 3;MMP9, matrix metalloproteinase 9; MTHFR, 5,10-methyl-enetetrahydrofolate reductase (NADPH); NFKB1, nuclearfactor of kappa light polypeptide gene enhancer in B-cells1; NID, nidogen (enactin); NOS3, nitric acid synthetase 3(endothelial); NOX4, NADPH oxidase 4; NPHS1, nephrin;NPPA, natriuretic peptide precursor A; NRP1, neuropilin 1;OPN (SPP1), osteopontin (secreted phosphoprotein 1);p22phox, (CYBA), cytochrome b-245, alpha polypeptide;PDGFB, platelet-derived growth factor beta polypeptide;PDGFRB, platelet-derived growth factor receptor, beta;PPARG, peroxisome proliferative–activated receptor,gamma; PRKCA, protein kinase C, alpha; PRKCB1, proteinkinase C, beta 1; REN, renin; SAH, SA hypertension-associated homolog (rat); SELE, selectin E; SELL, selectinL; SGK, serum/glucocorticoid regulated kinase; SLC12A3,solute carrier family 12 (sodium/chloride transporters),member 3; SLC9A1, solute carrier family 9 (Na�/H�antiporter); SMAD3, SMAD, mothers against DPP homolog3 (Drosophila); SMARCA3, SWI/SNF-related, matrix-asso-ciated, actin-dependent regulator of chromatin, subfamilya, member 3; TAZ, tafazzin; TCF2, transcription factor 2,hepatic; TCN2, transcobalamin II; TGFB1, transforminggrowth factor, beta 1; TGFB2, transforming growth factor,beta 2; TGFB3, transforming growth factor, beta 3;TGFBR2, transforming growth factor, beta receptor II;TGFBR3, transforming growth factor, beta receptor III;TIMP2, tissue inhibitor of metalloproteinase 2; TIMP3,tissue inhibitor of metalloproteinase 3; TM4SF4, trans-membrane 4 superfamily member 4; TNFRSF1A, tumornecrosis factor receptor 1 precursor; TNFSF6/FASLG,tumor necrosis factor (ligand) superfamily, member 6;TSC22 (TGFB1I4), transforming growth factor beta 1–in-duced transcript 4; UBA52, ubiquitin A-52 residue ribo-somal protein fusion product 1; UNC13B, unc-13 homologB (C. elegans); USF1, upstream transcription factor 1;USF2, upstream transcription factor 2; UTS2, urotensin 2;VEGF, vascular endothelial growth factor; VEGFR2(KDR), kinase insert domain receptor.
REFERENCES
1. American Diabetes Association: Diabetic nephropathy (Position State-ment). Diabetes Care 26 (Suppl. 1):S94–S98, 2003
2. Krolewski AS, Warram JH, Christlieb AR, Busick EJ, Kahn CR: Thechanging natural history of nephropathy in type I diabetes. Am J Med
78:785–794, 19853. Seaquist ER, Goetz FC, Rich S, Barbosa J: Familial clustering of diabetic
kidney disease: evidence for genetic susceptibility to diabetic nephropa-thy. N Engl J Med 320:1161–1165, 1989
4. Borch-Johnsen K, Norgaard K, Hommel E, Mathiesen ER, Jensen JS,
CANDIDATE GENES FOR DIABETIC NEPHROPATHY
3314 DIABETES, VOL. 54, NOVEMBER 2005

Deckert T, Parving HH: Is diabetic nephropathy an inherited complica-tion? Kidney Int 41:719–722, 1992
5. Quinn M, Angelico MC, Warram JH, Krolewski AS: Familial factorsdetermine the development of diabetic nephropathy in patients withIDDM. Diabetologia 39:940–945, 1996
6. Satko SG, Langefeld CD, Daeihagh P, Bowden DW, Rich SS, Freedman BI:Nephropathy in siblings of African Americans with overt type 2 diabeticnephropathy. Am J Kidney Dis 40:489–494, 2002
7. Fogarty DG, Hanna LS, Wantman M, Warram JH, Krolewski AS, Rich SS:Segregation analysis of urinary albumin excretion in families with type 2diabetes. Diabetes 49:1057–1063, 2000
8. Imperatore G, Knowler WC, Pettitt DJ, Kobes S, Bennett PH, Hanson RL:Segregation analysis of diabetic nephropathy in Pima Indians. Diabetes
49:1049–1056, 20009. Doria A, Warram JH, Krolewski AS: Genetic susceptibility to nephropathy
in insulin-dependent diabetes: from epidemiology to molecular genetics.Diabete Metab Rev 11:287–314, 1995
10. Caramori ML, Mauer M: Diabetes and nephropathy. Curr Opin Nephrol
Hypertens 12:273–282, 200311. Dolan V, Hensey C, Brady HR: Diabetic nephropathy: renal development
gone awry? Pediatr Nephrol 18:75–84, 200312. Lindner TH, Monks D, Wanner C, Berger M: Genetic aspects of diabetic
nephropathy. Kidney Int 84 (Suppl.):S186–S191, 200313. Spielman RS, McGinnis RE, Ewens WJ: Transmission test for linkage
disequilibrium: the insulin gene region and insulin-dependent diabetesmellitus (IDDM). Am J Hum Genet 52:506–516, 1993
14. Lernmark A, Ducat L, Eisenbarth G, Ott J, Permutt MA, Rubenstein P,Spielman R: Family cell lines available for research. Am J Hum Genet
47:1028–1030, 199015. Urbanek M, Legro RS, Driscoll DA, Azziz R, Ehrmann DA, Norman RJ,
Strauss JF, III, Spielman RS, Dunaif A: Thirty-seven candidate genes forpolycystic ovary syndrome: strongest evidence for linkage is with follista-tin. Proc Natl Acad Sci U S A 96:8573–8578, 1999
16. Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES: Parametric and non-parametric linkage analysis: a unified multipoint approach. Am J Hum
Genet 58:1347–1363, 199617. Uitenbroek DG: SISA-Binomial [article online], 1997. Available from
http://home.clara.net/sisa/binomial.htm. Accessed 3 January 200518. Chen JW, Hansen PM, Tarnow L, Hellgren A, Deckert T, Pociot F: Genetic
variation of a collagen IV alpha 1-chain gene polymorphism in Danishinsulin-dependent diabetes mellitus (IDDM) patients: lack of associationto nephropathy and proliferative retinopathy. Diabet Med 14:143–147,1997
19. Krolewski AS, Tryggvason K, Warram J, Laffel L, Housman D: Diabeticnephropathy and polymorphism in the gene coding for the alpha 1 chainof collagen IV. Kidney Int 37: 510, 1990
20. Moczulski DK, Rogus JJ, Antonellis A, Warram JH, Krolewski AS: Majorsusceptibility locus for nephropathy in type 1 diabetes on chromosome3q: results of novel discordant sib-pair analysis. Diabetes 47:1164–1169,1998
21. Chowdhury TA, Dyer PH, Kumar S, Gough SC, Gibson SP, Rowe BR,Smith PR, Dronsfield MJ, Marshall SM, Mackin P, Dean JD, Morris PJ,Davies S, Dunger DB, Boulton AJ, Barnett AH, Bain SC: Lack ofassociation of angiotensin II type 1 receptor gene polymorphism withdiabetic nephropathy in insulin-dependent diabetes mellitus. Diabet Med
14:837–840, 199722. Doria A, Onuma T, Warram JH, Krolewski AS: Synergistic effect of
angiotensin II type 1 receptor genotype and poor glycaemic control onrisk of nephropathy in IDDM. Diabetologia 40:1293–1299, 1997
23. Miller JA, Thai K, Scholey JW: Angiotensin II type 1 receptor genepolymorphism and the response to hyperglycemia in early type 1 diabe-tes. Diabetes 49:1585–1589, 2000
24. Savage DA, Feeney SA, Fogarty DG, Maxwell AP: Risk of developingdiabetic nephropathy is not associated with synergism between theangiotensin II (type 1) receptor C1166 allele and poor glycaemic control.Nephrol Dial Transplant 14:891–894, 1999
25. Orchard TJ, Chang YF, Ferrell RE, Petro N, Ellis DE: Nephropathy in type1 diabetes: a manifestation of insulin resistance and multiple geneticsusceptibilities? Further evidence from the Pittsburgh Epidemiology ofDiabetes Complication Study. Kidney Int 62:963–970, 2002
26. Araki S, Ng DP, Krolewski B, Wyrwicz L, Rogus JJ, Canani L, Makita Y,Haneda M, Warram JH, Krolewski AS: Identification of a common riskhaplotype for diabetic nephropathy at the protein kinase C-beta1(PRKCB1) gene locus. J Am Soc Nephrol 14:2015–2024, 2003
27. Iyengar SK, Fox KA, Schachere M, Manzoor F, Slaughter ME, Covic AM,Orloff SM, Hayden PS, Olson JM, Schelling JR, Sedor JR: Linkage analysis
of candidate loci for end-stage renal disease due to diabetic nephropathy.J Am Soc Nephrol 14:S195–S201, 2003
28. Horikawa Y, Iwasaki N, Hara M, Furuta H, Hinokio Y, Cockburn BN,Lindner T, Yamagata K, Ogata M, Tomonaga O, Kuroki H, Kasahara T,Iwamoto Y, Bell GI: Mutation in hepatocyte nuclear factor-1 beta gene(TCF2) associated with MODY. Nat Genet 17:384–385, 1997
29. Lindner T, Njolstad P, Horikawa Y, Bostad L, Bell G, Sovik O: A novelsyndrome of diabetes mellitus, renal dysfunction and genital malforma-tion associated with a partial deletion of the pseudo-POU domain ofhepatocyte nuclear factor-1beta. Hum Mol Genet 8:2001–2008, 1999
30. Carbone I, Cotellessa M, Barella C, Minetti C, Ghiggeri GM, Caridi G,Perfumo F, Lorini R: A novel hepatocyte nuclear factor-1beta (MODY-5)gene mutation in an Italian family with renal dysfunctions and early-onsetdiabetes. Diabetologia 45:153–154, 2002
31. Nishigori H, Yamada S, Kohama T, Tomura H, Sho K, Horikawa Y, Bell GI,Takeuchi T, Takeda J: Frameshift mutation, A263fsinsGG, in the hepato-cyte nuclear factor-1beta gene associated with diabetes and renal dys-function. Diabetes 47:1354–1355, 1998
32. Selisko T, Vcelak J, Bendlova B, Graessler J, Schwarz PE, Schulze J:Mutations and intronic variants in the HNF-1 beta gene in a group ofGerman and Czech Caucasians with type 2 diabetes mellitus and progres-sive diabetic nephropathy. Exp Clin Endocrinol Diabetes 110:145–147,2002
33. Hodgkinson AD, Millward BA, Demaine AG: Association of the p22phoxcomponent of NAD(P)H oxidase with susceptibility to diabetic nephrop-athy in patients with type 1 diabetes. Diabetes Care 26:3111–3115, 2003
34. Matsunaga-Irie S, Maruyama T, Yamamoto Y, Motohashi Y, Hirose H,Shimada A, Murata M, Saruta T: Relation between development ofnephropathy and the p22phox C242T and receptor for advanced glycationend product G1704T gene polymorphisms in type 2 diabetic patients.Diabetes Care 27:303–307, 2004
35. Maeda S, Haneda M, Guo B, Koya D, Hayashi K, Sugimoto T, Isshiki K,Yasuda H, Kashiwagi A, Kikkawa R: Dinucleotide repeat polymorphism ofmatrix metalloproteinase-9 gene is associated with diabetic nephropathy.Kidney Int 60:1428–1434, 2001
36. Hirakawa S, Lange EM, Colicigno CJ, Freedman BI, Rich SS, Bowden DW:Evaluation of genetic variation and association in the matrix metallopro-teinase 9 (MMP9) gene in ESRD patients. Am J Kidney Dis 42:133–142,2003
37. Doria A, Warram JH, Krolewski AS: Genetic predisposition to diabeticnephropathy: evidence for a role of the angiotensin I–converting enzymegene. Diabetes 43:690–695, 1994
38. Jacobsen P, Tarnow L, Carstensen B, Hovind P, Poirier O, Parving HH:Genetic variation in the renin-angiotensin system and progression ofdiabetic nephropathy. J Am Soc Nephrol 14:2843–2850, 2003
39. Tarnow L, Gluud C, Parving HH, Nielsen FS: Diabetic nephropathy andthe insertion/deletion polymorphism of the angiotensin-converting en-zyme gene. Nephrol Dial Transplant 13:1125–1130, 1998
40. Lovati E, Richard A, Frey BM, Frey FJ, Ferrari P: Genetic polymorphismsof the renin-angiotensin-aldosterone system in end-stage renal disease.Kidney Int 60:46–54, 2001
41. Marre M, Jeunemaitre X, Gallois Y, Rodier M, Chatellier G, Sert C,Dusselier L, Kahal Z, Chaillous L, Halimi S, Muller A, Sackmann H,Bauduceau B, Bled F, Passa P, Alhenc-Gelas F: Contribution of geneticpolymorphism in the renin-angiotensin system to the development ofrenal complications in insulin-dependent diabetes: Genetique de la Ne-phropathie Diabetique (GENEDIAB) study group. J Clin Invest 99:1585–1595, 1997
42. Gilbert RE, Krum H, Wilkinson-Berka J, Kelly DJ: The renin-angiotensinsystem and the long-term complications of diabetes: pathophysiologicaland therapeutic considerations. Diabet Med 20:607–621, 2003
43. Hadjadj S, Belloum R, Bouhanick B, Gallois Y, Guilloteau G, Chatellier G,Alhenc-Gelas F, Marre M: Prognostic value of angiotensin-I convertingenzyme I/D polymorphism for nephropathy in type 1 diabetes mellitus: aprospective study. J Am Soc Nephrol 12:541–549, 2001
44. Boright AP, Paterson AD, Mirea L, Bull SB, Mowjoodi A, Scherer SW,Zinman B: Genetic variation at the ACE gene is associated with persistentmicroalbuminuria and severe nephropathy in type 1 diabetes: the DCCT/EDIC Genetics Study. Diabetes 54:1238–1244, 2005
45. Heesom AE, Hibberd ML, Millward A, Demaine AG: Polymorphism in the5�-end of the aldose reductase gene is strongly associated with thedevelopment of diabetic nephropathy in type I diabetes. Diabetes 46:287–291, 1997
46. Hodgkinson AD, Sondergaard KL, Yang B, Cross DF, Millward BA,Demaine AG: Aldose reductase expression is induced by hyperglycemiain diabetic nephropathy. Kidney Int 60:211–218, 2001
47. Dyer PH, Chowdhury TA, Dronsfield MJ, Dunger D, Barnett AH, Bain SC:
K.G. EWENS AND ASSOCIATES
DIABETES, VOL. 54, NOVEMBER 2005 3315

The 5�-end polymorphism of the aldose reductase gene is not associatedwith diabetic nephropathy in Caucasian type I diabetic patients. Diabe-
tologia 42:1030–1031, 199948. Moczulski DK, Scott L, Antonellis A, Rogus JJ, Rich SS, Warram JH,
Krolewski AS: Aldose reductase gene polymorphisms and susceptibilityto diabetic nephropathy in type 1 diabetes mellitus. Diabet Med 17:111–118, 2000
49. Neamat-Allah M, Feeney SA, Savage DA, Maxwell AP, Hanson RL,Knowler WC, El Nahas AM, Plater ME, Shaw J, Boulton AJ, Duff GW, CoxA: Analysis of the association between diabetic nephropathy and poly-morphisms in the aldose reductase gene in type 1 and type 2 diabetesmellitus. Diabet Med 18:906–914, 2001
50. Ng DP, Conn J, Chung SS, Larkins RG: Aldose reductase (AC)(n)microsatellite polymorphism and diabetic microvascular complications inCaucasian type 1 diabetes mellitus. Diabetes Res Clin Pract 52:21–27,2001
51. Lajer M, Tarnow L, Fleckner J, Hansen BV, Edwards DG, Parving HH,Boel E: Association of aldose reductase gene Z�2 polymorphism withreduced susceptibility to diabetic nephropathy in Caucasian type 1diabetic patients. Diabet Med 21:867–873, 2004
52. Onuma T, Laffel LM, Angelico MC, Krolewski AS: Apolipoprotein Egenotypes and risk of diabetic nephropathy. J Am Soc Nephrol 7:1075–1078, 1996
53. Chowdhury TA, Dyer PH, Kumar S, Gibson SP, Rowe BR, Davies SJ,Marshall SM, Morris PJ, Gill GV, Feeney S, Maxwell P, Savage D, BoultonAJ, Todd JA, Dunger D, Barnett AH, Bain SC: Association of apolipopro-tein epsilon2 allele with diabetic nephropathy in Caucasian subjects withIDDM. Diabetes 47:278–280, 1998
54. Araki S, Moczulski DK, Hanna L, Scott LJ, Warram JH, Krolewski AS:APOE polymorphisms and the development of diabetic nephropathy intype 1 diabetes: results of case-control and family-based studies. Diabetes
49:2190–2195, 200055. Hadjadj S, Gallois Y, Simard G, Bouhanick B, Passa P, Grimaldi A, Drouin
P, Tichet J, Marre M: Lack of relationship in long-term type 1 diabeticpatients between diabetic nephropathy and polymorphisms in apoli-poprotein epsilon, lipoprotein lipase and cholesteryl ester transfer pro-tein. Nephrol Dial Transplant 15:1971–1976, 2000
56. Tarnow L, Stehouwer CD, Emeis JJ, Poirier O, Cambien F, Hansen BV,Parving HH: Plasminogen activator inhibitor-1 and apolipoprotein E genepolymorphisms and diabetic angiopathy. Nephrol Dial Transplant 15:625–630, 2000
57. Westfall P, Young S: Resampling-Based Multiple Testing: Examples and
Methods for P-Value Adjustment. New York, John Wiley & Sons, 199358. Susztak K, Bottinger E, Novetsky A, Liang D, Zhu Y, Ciccone E, Wu D,
Dunn S, McCue P, Sharma K: Molecular profiling of diabetic mousekidney reveals novel genes linked to glomerular disease. Diabetes 53:784–794, 2004
59. Fukui M, Nakamura T, Ebihara I, Shirato I, Tomino Y, Koide H: ECM geneexpression and its modulation by insulin in diabetic rats. Diabetes
41:1520–1527, 199260. Kim Y, Kleppel MM, Butkowski R, Mauer SM, Wieslander J, Michael AF:
Differential expression of basement membrane collagen chains in dia-betic nephropathy. Am J Pathol 138:413–420, 1991
61. Yagame M, Kim Y, Zhu D, Suzuki D, Eguchi K, Nomoto Y, Sakai H,Groppoli T, Steffes MW, Mauer SM: Differential distribution of type IVcollagen chains in patients with diabetic nephropathy in non-insulin-dependent diabetes mellitus. Nephron 70:42–48, 1995
62. Mason RM, Wahab NA: Extracellular matrix metabolism in diabeticnephropathy. J Am Soc Nephrol 14:1358–1373, 2003
63. Okada S, Shikata K, Matsuda M, Ogawa D, Usui H, Kido Y, Nagase R,Wada J, Shikata Y, Makino H: Intercellular adhesion molecule-1–deficientmice are resistant against renal injury after induction of diabetes.Diabetes 52:2586–2593, 2003
64. Guler S, Cakir B, Demirbas B, Yonem A, Odabasi E, Onde U, Aykut O,Gursoy G: Plasma soluble intercellular adhesion molecule 1 levels areincreased in type 2 diabetic patients with nephropathy. Horm Res
58:67–70, 200265. Jin DK, Fish AJ, Wayner EA, Mauer M, Setty S, Tsilibary E, Kim Y,
Anderson SS: Distribution of integrin subunits in human diabetic kidneys.J Am Soc Nephrol 7:2636–2645, 1996
66. Nakamura T, Fukui M, Ebihara I, Osada S, Tomino Y, Koide H: Abnormalgene expression of matrix metalloproteinases and their inhibitor inglomeruli from diabetic rats. Ren Physiol Biochem 17:316–325, 1994
67. Narumi S, Onozato ML, Tojo A, Sakamoto S, Tamatani T: Tissue-specificinduction of E-selectin in glomeruli is augmented following diabetesmellitus. Nephron 89:161–171, 2001
68. Hansen PM, Chowdhury T, Deckert T, Hellgren A, Bain SC, Pociot F:
Genetic variation of the heparan sulfate proteoglycan gene (perlecangene): association with urinary albumin excretion in IDDM patients.Diabetes 46:1658–1659, 1997
69. Liu L, Xiang K, Zheng T, Zhang R, Li M, Wang Y, Lu H, Li J: The heparansulfate proteoglycan gene polymorphism: association with type 2 diabeticnephropathy in Chinese. Mol Cell Biochem 245:121–126, 2003
70. Doublier S, Salvidio G, Lupia E, Ruotsalainen V, Verzola D, Deferrari G,Camussi G: Nephrin expression is reduced in human diabetic nephropa-thy: evidence for a distinct role for glycated albumin and angiotensin II.Diabetes 52:1023–1030, 2003
71. Baelde HJ, Eikmans M, Doran PP, Lappin DW, de Heer E, Bruijn JA: Geneexpression profiling in glomeruli from human kidneys with diabeticnephropathy. Am J Kidney Dis 43:636–650, 2004
72. Kamiuchi K, Hasegawa G, Obayashi H, Kitamura A, Ishii M, Yano M,Kanatsuna T, Yoshikawa T, Nakamura N: Leukocyte-endothelial celladhesion molecule 1 (LECAM-1) polymorphism is associated with dia-betic nephropathy in type 2 diabetes mellitus. J Diabetes Complicat
16:333–337, 200273. Zhang H, Jia Y, Cooper JJ, Hale T, Zhang Z, Elbein SC: Common variants
in glutamine:fructose-6-phosphate amidotransferase 2 (GFPT2) gene areassociated with type 2 diabetes, diabetic nephropathy, and increasedGFPT2 mRNA levels. J Clin Endocrinol Metab 89:748–755, 2004
74. Tarnow L, Grarup N, Hansen T, Parving HH, Pedersen O: Diabeticmicrovascular complications are not associated with two polymorphismsin the GLUT-1 and PC-1 genes regulating glucose metabolism in Cauca-sian type 1 diabetic patients. Nephrol Dial Transplant 16:1653–1656, 2001
75. Ng DP, Canani L, Araki S, Smiles A, Moczulski D, Warram JH, KrolewskiAS: Minor effect of GLUT1 polymorphisms on susceptibility to diabeticnephropathy in type 1 diabetes. Diabetes 51:2264–2269, 2002
76. Hodgkinson AD, Millward BA, Demaine AG: Polymorphisms of theglucose transporter (GLUT1) gene are associated with diabetic nephrop-athy. Kidney Int 59:985–989, 2001
77. Itoh Y, Imamura S, Yamamoto K, Ono Y, Nagata M, Kobayashi T, Kato T,Tomita M, Nakai A, Itoh M, Nagasaka A: Changes of endothelin instreptozotocin-induced diabetic rats: effects of an angiotensin convertingenzyme inhibitor, enalapril maleate. J Endocrinol 175:233–239, 2002
78. Minchenko AG, Stevens MJ, White L, Abatan OI, Komjati K, Pacher P,Szabo C, Obrosova IG: Diabetes-induced overexpression of endothelin-1and endothelin receptors in the rat renal cortex is mediated via poly(ADP-ribose) polymerase activation. FASEB J 17:1514–1516, 2003
79. Kohan DE: Endothelins in the normal and diseased kidney. Am J Kidney
Dis 29:2–26, 199780. Nabika T, Bonnardeaux A, James M, Julier C, Jeunemaitre X, Corvol P,
Lathrop M, Soubrier F: Evaluation of the SA locus in human hypertension.Hypertension 25:6–13, 1995
81. Langham RG, Kelly DJ, Gow RM, Zhang Y, Dowling JK, Thomson NM,Gilbert RE: Increased expression of urotensin II and urotensin II receptorin human diabetic nephropathy. Am J Kidney Dis 44:826–831, 2004
82. Rogus JJ, Moczulski D, Freire MB, Yang Y, Warram JH, Krolewski AS:Diabetic nephropathy is associated with AGT polymorphism T235: resultsof a family-based study. Hypertension 31:627–631, 1998
83. Doria A, Onuma T, Gearin G, Freire MBS, Warram JH, Krolewski AS:Angiotensinogen polymorphism M235T, hypertension, and nephropathyin insulin-dependent diabetes. Hypertension 27:1134–1139, 1996
84. Nannipieri M, Penno G, Pucci L, Colhoun H, Motti C, Bertacca A, Rizzo L,De Giorgio L, Zerbini G, Mangili R, Navalesi R: Pronatriodilatin genepolymorphisms, microvascular permeability, and diabetic nephropathy intype 1 diabetes mellitus. J Am Soc Nephrol 10:1530–1541, 1999
85. Schmidt S, Bluthner M, Giessel R, Strojek K, Bergis KH, Grzeszczak W,Ritz E: A polymorphism in the gene for the atrial natriuretic peptide anddiabetic nephropathy. Diabetic Nephropathy Study Group. Nephrol Dial
Transplant 13:1807–1810, 199886. Roussel R, Tregouet DA, Hadjadj S, Jeunemaitre X, Marre M: Investigation
of the human ANP gene in type 1 diabetic nephropathy: case-control andfollow-up studies. Diabetes 53:1394–1398, 2004
87. Wang S, Hirschberg R: BMP7 antagonizes TGF-beta–dependent fibrogen-esis in mesangial cells. Am J Physiol Renal Physiol 284:F1006–F1013,2003
88. Wang SN, Lapage J, Hirschberg R: Loss of tubular bone morphogeneticprotein-7 in diabetic nephropathy. J Am Soc Nephrol 12:2392–2399, 2001
89. Mezzano S, Aros C, Droguett A, Burgos ME, Ardiles L, Flores C, SchneiderH, Ruiz-Ortega M, Egido J: NF-kappaB activation and overexpression ofregulated genes in human diabetic nephropathy. Nephrol Dial Transplant
19:2505–2512, 200490. Wang S, Denichilo M, Brubaker C, Hirschberg R: Connective tissue
growth factor in tubulointerstitial injury of diabetic nephropathy. Kidney
Int 60:96–105, 2001
CANDIDATE GENES FOR DIABETIC NEPHROPATHY
3316 DIABETES, VOL. 54, NOVEMBER 2005

91. Weston BS, Wahab NA, Mason RM: CTGF mediates TGF-beta–inducedfibronectin matrix deposition by upregulating active alpha5beta1 integrinin human mesangial cells. J Am Soc Nephrol 14:601–610, 2003
92. Cummings EA, Sochett EB, Dekker MG, Lawson ML, Daneman D:Contribution of growth hormone and IGF-I to early diabetic nephropathyin type 1 diabetes. Diabetes 47:1341–1346, 1998
93. Mogyorosi A, Ziyadeh FN: Update on pathogenesis, markers and manage-ment of diabetic nephropathy. Curr Opin Nephrol Hypertens 5:243–253,1996
94. Lupia E, Elliot SJ, Lenz O, Zheng F, Hattori M, Striker GE, Striker LJ:IGF-1 decreases collagen degradation in diabetic NOD mesangial cells:implications for diabetic nephropathy. Diabetes 48:1638–1644, 1999
95. Sugimoto H, Shikata K, Makino H, Ota K, Ota Z: Upregulation ofinsulin-like growth factor receptor gene in experimental diabetic ratglomeruli. J Diabetes Complicat 9:262–264, 1995
96. Huang C, Kim Y, Caramori ML, Fish AJ, Rich SS, Miller ME, Russell GB,Mauer M: Cellular basis of diabetic nephropathy: II. The transforminggrowth factor-� system and diabetic nephropathy lesions in type 1diabetes. Diabetes 51:3577–3581, 2002
97. Nakagawa H, Sasahara M, Haneda M, Koya D, Hazama F, Kikkawa R:Immunohistochemical characterization of glomerular PDGF B-chain andPDGF [beta]-receptor expression in diabetic rats. Diabetes Res Clin Prac
48:87–98, 200098. Kim HW, Kim BC, Song CY, Kim JH, Hong HK, Lee HS: Heterozygous mice
for TGF-betaIIR gene are resistant to the progression of streptozotocin-induced diabetic nephropathy. Kidney Int 66:1859–1865, 2004
99. Kelly DJ, Stein-Oakley A, Zhang Y, Wassef L, Maguire J, Koji T, ThomsonN, Wilkinson-Berka JL, Gilbert RE: Fas-induced apoptosis is a feature ofprogressive diabetic nephropathy in transgenic (mRen-2)27 rats: attenu-ation with renin-angiotensin blockade. Nephrology 9:7–13, 2004
100. Hovind P, Tarnow L, Oestergaard PB, Parving HH: Elevated vascularendothelial growth factor in type 1 diabetic patients with diabeticnephropathy. Kidney Int 75 (Suppl.)S56–S61, 2000
101. Onozaki A, Midorikawa S, Sanada H, Hayashi Y, Baba T, Katoh T,Watanabe T: Rapid change of glucose concentration promotes mesangialcell proliferation via VEGF: inhibitory effects of thiazolidinedione. Bio-
chem Biophys Res Commun 317:24–29, 2004102. Nakajima K, Tanaka Y, Nomiyama T, Ogihara T, Ikeda F, Kanno R,
Iwashita N, Sakai K, Watada H, Onuma T, Kawamori R: RANTESpromoter genotype is associated with diabetic nephropathy in type 2diabetic subjects. Diabetes Care 26:892–898, 2003
103. Tarnow L, Pociot F, Hansen PM, Rossing P, Nielsen FS, Hansen BV,Parving HH: Polymorphisms in the interleukin-1 gene cluster do notcontribute to the genetic susceptibility of diabetic nephropathy in Cau-casian patients with IDDM. Diabetes 46:1075–1076, 1997
104. Loughrey BV, Maxwell AP, Fogarty DG, Middleton D, Harron JC, Patter-son CC, Darke C, Savage DA: An interluekin 1B allele, which correlateswith a high secretor phenotype, is associated with diabetic nephropathy.Cytokine 10:984–988, 1998
105. Bensen JT, Langefeld CD, Hawkins GA, Green LE, Mychaleckyj JC,Brewer CS, Kiger DS, Binford SM, Colicigno CJ, Allred DC, Freedman BI,Bowden DW: Nucleotide variation, haplotype structure, and associationwith end-stage renal disease of the human interleukin-1 gene cluster.Genomics 82:194–217, 2003
106. Blakemore AI, Cox A, Gonzalez AM, Maskil JK, Hughes ME, Wilson RM,Ward JD, Duff GW: Interleukin-1 receptor antagonist allele (IL1RN*2)associated with nephropathy in diabetes mellitus. Hum Genet 97:369–374, 1996
107. Ng DP, Warram JH, Krolewski AS: TGF-beta 1 as a genetic susceptibilitylocus for advanced diabetic nephropathy in type 1 diabetes mellitus: aninvestigation of multiple known DNA sequence variants. Am J Kidney
Dis 41:22–28, 2003108. Wong TY, Poon P, Chow KM, Szeto CC, Cheung MK, Li PK: Association of
transforming growth factor-beta (TGF-beta) T869C (Leu 10Pro) genepolymorphisms with type 2 diabetic nephropathy in Chinese. Kidney Int
63:1831–1835, 2003109. Sharma K, Ziyadeh FN: Hyperglycemia and diabetic kidney disease: the
case for transforming growth factor-beta as a key mediator. Diabetes
44:1139–1146, 1995110. Mattu RK, Trevelyan J, Needham EW, Khan M, Adiseshiah MA, Richter D,
Murray RG, Betteridge DJ: Lipoprotein lipase gene variants relate topresence and degree of microalbuminuria in type II diabetes. Diabetolo-
gia 45:905–913, 2002111. Leehey DJ, Song RH, Alavi N, Singh AK: Decreased degradative enzymes
in mesangial cells cultured in high glucose media. Diabetes 44:929–935,1995
112. Lang F, Klingel K, Wagner CA, Stegen C, Warntges S, Friedrich B,
Lanzendorfer M, Melzig J, Moschen I, Steuer S, Waldegger S, Sauter M,Paulmichl M, Gerke V, Risler T, Gamba G, Capasso G, Kandolf R, HebertSC, Massry SG, Broer S: Deranged transcriptional regulation of cell-volume-sensitive kinase hSGK in diabetic nephropathy. Proc Natl Acad
Sci U S A 97:8157–8162, 2000113. Sun L, Pan X, Wada J, Haas CS, Wuthrich RP, Danesh FR, Chugh SS,
Kanwar YS: Isolation and functional analysis of mouse UbA52 gene andits relevance to diabetic nephropathy. J Biol Chem 277:29953–29962, 2002
114. Bluthner M, Bruntgens A, Schmidt S, Strojek K, Grzeszczak W, Ritz E:Association of methylenetetrahydrofolate reductase gene polymorphismand diabetic nephropathy in type 2 diabetes? Nephrol Dial Transplant
14:56–57, 1999115. Makita Y, Moczulski DK, Bochenski J, Smiles AM, Warram JH, Krolewski
AS: Methylenetetrahydrofolate reductase gene polymorphism and sus-ceptibility to diabetic nephropathy in type 1 diabetes. Am J Kidney Dis
41:1189–1194, 2003116. Smyth JS, Savage DA, Maxwell AP: MTHFR gene polymorphism and
diabetic nephropathy in type 1 diabetes. Lancet 353:1156–1157, 1999117. Weekers L, Hadjadj S, Belloum R, YG, Bouhanick B, MM: Lack of
contribution of two endothelial nitric-oxide synthase (eNOS) gene poly-morphisms to diabetic nephropathy in type 1 diabetes. Diabetologia 42(Suppl. 1):A266, 1999
118. Zanchi A, Moczulski DK, Hanna LS, Wantman M, Warram JH, KrolewskiAS: Risk of advanced diabetic nephropathy in type 1 diabetes is associ-ated with endothelial nitric oxide synthase gene polymorphism. Kidney
Int 57:405–413, 2000119. Degen B, Schmidt S, Ritz E: A polymorphism in the gene for the
endothelial nitric oxide synthase and diabetic nephropathy. Nephrol Dial
Transplant 16:185, 2001120. Ng DP, Warram JH, Krolewski AS: To: Rippin JD, Patel A, Belyaev ND,
Gill GV, Barnett AH, Bain SC (2003) Nitric oxide synthase gene polymor-phisms and diabetic nephropathy. Diabetologia 46:426–428. Diabetologia
46:1706, 2003121. Rippin JD, Patel A, Belyaev ND, Gill GV, Barnett AH, Bain SC: Nitric oxide
synthase gene polymorphisms and diabetic nephropathy. Diabetologia
46:426–428, 2003122. Chiarelli F, Pomilio M, Mohn A, Tumini S, Verrotti A, Mezzetti A,
Cipollone F, Wasniewska M, Morgese G, Spagnoli A: Serum angiogeninconcentrations in young patients with diabetes mellitus. Eur J Clin Invest
32:110–114, 2002123. De Cosmo S, Argiolas A, Miscio G, Thomas S, Piras GP, Trevisan R, Perin
PC, Bacci S, Zucaro L, Margaglione M, Frittitta L, Pizzuti A, Tassi V,Viberti GC, Trischitta V: A PC-1 amino acid variant (K121Q) is associatedwith faster progression of renal disease in patients with type 1 diabetesand albuminuria. Diabetes 49:521–524, 2000
124. De Cosmo S, Miscio G, Zucaro L, Margaglione M, Argiolas A, Thomas S,Piras G, Trevisan R, Perin PC, Bacci S, Frittitta L, Pizzuti A, Tassi V, DiMinno G, Viberti G, Trischitta V: The role of PC-1 and ACE genes indiabetic nephropathy in type 1 diabetic patients: evidence for a polygeniccontrol of kidney disease progression. Nephrol Dial Transplant 17:1402–1407, 2002
125. Jacobsen P, Grarup N, Tarnow L, Parving HH, Pedersen O: PC-1 aminoacid variant (K121Q) has no impact on progression of diabetic nephrop-athy in type 1 diabetic patients. Nephrol Dial Transplant 17:1408–1412,2002
126. Canani LH, Ng DP, Smiles A, Rogus JJ, Warram JH, Krolewski AS:Polymorphism in ecto-nucleotide pyrophosphatase/phosphodiesterase 1gene (ENPP1/PC-1) and early development of advanced diabetic nephrop-athy in type 1 diabetes. Diabetes 51:1188–1193, 2002
127. Nagai K, Arai H, Yanagita M, Matsubara T, Kanamori H, Nakano T, IeharaN, Fukatsu A, Kita T, Doi T: Growth arrest-specific gene 6 is involved inglomerular hypertrophy in the early stage of diabetic nephropathy. J Biol
Chem 278:18229–18234, 2003128. Nakamura T, Ebihara I, Tomino Y, Koide H: Effect of a specific endothelin
A receptor antagonist on murine lupus nephritis. Kidney Int 47:481–489,1995
129. Khan MA, Dashwood MR, Mumtaz FH, Thompson CS, Mikhailidis DP,Morgan RJ: Upregulation of endothelin A receptor sites in the rabbitdiabetic kidney: potential relevance to the early pathogenesis of diabeticnephropathy. Nephron 83:261–267, 1999
130. Makkinje A, Quinn DA, Chen A, Cadilla CL, Force T, Bonventre JV,Kyriakis JM: Gene 33/Mig-6, a transcriptionally inducible adapter proteinthat binds GTP-Cdc42 and activates SAPK/JNK: a potential markertranscript for chronic pathologic conditions, such as diabetic nephropa-thy: possible role in the response to persistent stress. J Biol Chem
275:17838–17847, 2000131. Kang N, Alexander G, Park JK, Maasch C, Buchwalow I, Luft FC, Haller
K.G. EWENS AND ASSOCIATES
DIABETES, VOL. 54, NOVEMBER 2005 3317

H: Differential expression of protein kinase C isoforms in streptozotocin-induced diabetic rats. Kidney Int 56:1737–1750, 1999
132. Menne J, Park JK, Boehne M, Elger M, Lindschau C, Kirsch T, Meier M,Gueler F, Fiebeler A, Bahlmann FH, Leitges M, Haller H: Diminished lossof proteoglycans and lack of albuminuria in protein kinase C-alpha-deficient diabetic mice. Diabetes 53:2101–2109, 2004
133. Isono M, Chen S, Hong SW, Iglesias-de la Cruz MC, Ziyadeh FN: Smadpathway is activated in the diabetic mouse kidney and Smad3 mediatesTGF-beta-induced fibronectin in mesangial cells. Biochem Biophys Res
Commun 296:1356–1365, 2002134. Song Y, Ailenberg M, Silverman M: Cloning of a novel gene in the human
kidney homologous to rat munc13s: its potential role in diabetic nephrop-athy. Kidney Int 53:1689–1695, 1998
135. Maltais I, Bachvarova M, Maheux P, Perron P, Marceau F, Bachvarov D:Bradykinin B2 receptor gene polymorphism is associated with alteredurinary albumin/creatinine values in diabetic patients. Can J Physiol
Pharmacol 80:323–327, 2002136. Zychma MJ, Gumprecht J, Zukowska-Szczechowska E, Grzeszczak W:
Polymorphisms in the genes encoding for human kinin receptors and therisk of end-stage renal failure: results of transmission/disequilibrium test:the End-Stage Renal Disease Study Group. J Am Soc Nephrol 10:2120–2124, 1999
137. Kakoki M, Takahashi N, Jennette JC, Smithies O: Diabetic nephropathy ismarkedly enhanced in mice lacking the bradykinin B2 receptor. Proc Natl
Acad Sci U S A 101:13302–13305, 2004138. Herrmann SM, Ringel J, Wang JG, Staessen JA, Brand E: Peroxisome
proliferator–activated receptor-�2 polymorphism Pro12Ala is associatedwith nephropathy in type 2 diabetes: the Berlin Diabetes Mellitus (Be-DiaM) study. Diabetes 51:2653–2657, 2002
139. Caramori ML, Canani LH, Costa LA, Gross JL: The human peroxisomeproliferator-activated receptor gamma2 (PPARgamma2) Pro12Ala poly-morphism is associated with decreased risk of diabetic nephropathy inpatients with type 2 diabetes. Diabetes 52:3010–3013, 2003
140. Sugawara F, Yamada Y, Watanabe R, Ban N, Miyawaki K, Kuroe A,Hamasaki A, Ikeda H, Kurose T, Usami M, Ikeda M, Seino Y: The role ofthe TSC-22 (-396) A/G variant in the development of diabetic nephropa-thy. Diabetes Res Clin Pract 60:191–197, 2003
141. Hodgkinson AD, Bartlett T, Oates PJ, Millward BA, Demaine AG: Theresponse of antioxidant genes to hyperglycemia is abnormal in patientswith type 1 diabetes and diabetic nephropathy. Diabetes 52:846–851, 2003
142. Etoh T, Inoguchi T, Kakimoto M, Sonoda N, Kobayashi K, Kuroda J,Sumimoto H, Nawata H: Increased expression of NAD(P)H oxidasesubunits, NOX4 and p22phox, in the kidney of streptozotocin-induced
diabetic rats and its reversibility by interventive insulin treatment.Diabetologia 46:1428–1437, 2003
143. Agre P, Kozono D: Aquaporin water channels: molecular mechanisms forhuman diseases. FEBS Lett 555:72–78, 2003
144. Trevisan R, Viberti G: Sodium-hydrogen antiporter: its possible role in thegenesis of diabetic nephropathy. Nephrol Dial Transplant 12:643–645,1997
145. Zerbini G, Gabellini D, Ruggieri D, Maestroni A: Increased sodium-lithiumcountertransport activity: a cellular dysfunction common to essentialhypertension and diabetic nephropathy. J Am Soc Nephrol 15 (Suppl.1):S81–S84, 2004
146. Tanaka N, Babazono T, Saito S, Sekine A, Tsunoda T, Haneda M, TanakaY, Fujioka T, Kaku K, Kawamori R, Kikkawa R, Iwamoto Y, Nakamura Y,Maeda S: Association of solute carrier family 12 (sodium/chloride)member 3 with diabetic nephropathy, identified by genome-wide analysesof single nucleotide polymorphisms. Diabetes 52:2848–2853, 2003
147. Anwar W, Gerard P, Moutabarrek A, Namour F, Gueant J-L: End-stagerenal disease increases plasma transcobalamin and neutralizes influenceof TCN 776C�G polymorphism. Kidney Int 66:2092–2093, 2004
148. Murata I, Takemura G, Asano K, Sano H, Fujisawa K, Kagawa T, Baba K,Maruyama R, Minatoguchi S, Fujiwara T, Fujiwara H: Apoptotic cell lossfollowing cell proliferation in renal glomeruli of Otsuka Long-EvansTokushima Fatty rats, a model of human type 2 diabetes. Am J Nephrol
22:587–595, 2002149. Kang BP, Urbonas A, Baddoo A, Baskin S, Malhotra A, Meggs LG: IGF-1
inhibits the mitochondrial apoptosis program in mesangial cells exposedto high glucose. Am J Physiol Renal Physiol 285:F1013–F1024, 2003
150. Li H, Telemaque S, Miller RE, Marsh JD: High glucose inhibits apoptosisinduced by serum deprivation in vascular smooth muscle cells viaupregulation of Bcl-2 and Bcl-xl. Diabetes 54:540–545, 2005
151. Ceriello A, Morocutti A, Mercuri F, Quagliaro L, Moro M, Damante G,Viberti GC: Defective intracellular antioxidant enzyme production in type1 diabetic patients with nephropathy. Diabetes 49:2170–2177, 2000
152. McMahon R, Murphy M, Clarkson M, Taal M, Mackenzie HS, Godson C,Martin F, Brady HR: IHG-2, a mesangial cell gene induced by high glucose,is human gremlin: regulation by extracellular glucose concentration,cyclic mechanical strain, and transforming growth factor-beta1. J Biol
Chem 275:9901–9904, 2000153. Conway BR, Maxwell AP, Savage DA, Patterson CC, Doran PP, Murphy M,
Brady HR, Fogarty DG: Association between variation in the actin-bindinggene caldesmon and diabetic nephropathy in type 1 diabetes. Diabetes
53:1162–1165, 2004
CANDIDATE GENES FOR DIABETIC NEPHROPATHY
3318 DIABETES, VOL. 54, NOVEMBER 2005




















