
Ap120207 tap
7
Eur. Phys. J. Appl. Phys. (2013) 61: 10103 DOI: 10.1051/epjap/2012120207 Density functional theory study of the structural and electronic properties of Mg 3 Bi 2 in hexagonal and cubic phases Matin Sedighi, Borhan Arghavani Nia, Hanif Zarringhalam, and Rostam Moradian
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Ap120207 tap

Eur. Phys. J. Appl. Phys. (2013) 61: 10103 DOI: 10.1051/epjap/2012120207
Density functional theory study of the structural andelectronic properties of Mg3Bi2 in hexagonal and cubic phases
Matin Sedighi, Borhan Arghavani Nia, Hanif Zarringhalam, and Rostam Moradian

Eur. Phys. J. Appl. Phys. (2013) 61: 10103DOI: 10.1051/epjap/2012120207
THE EUROPEANPHYSICAL JOURNAL
APPLIED PHYSICS
Regular Article
Density functional theory study of the structural and electronicproperties of Mg3Bi2 in hexagonal and cubic phases
Matin Sedighi1, Borhan Arghavani Nia1,a, Hanif Zarringhalam1, and Rostam Moradian2,3
1 Department of Physics, Kermanshah Branch, Islamic Azad University, Kermanshah, Iran2 Nano-Science and Nano-Technology Research Center, Razi University, Kermanshah, Iran3 Computational Physics Science Research Laboratory, Department of Nano-Science,
Institute for Studies in Theoretical Physics and Mathematics (IPM), P.O. Box 19395-1795, Tehran, Iran
Received: 31 May 2012 / Received in final form: 23 September 2012 / Accepted: 23 November 2012Published online: 25 January 2013 – c© EDP Sciences 2013
Abstract. We study the structural and electronic properties of Mg3Bi2 in both hexagonal and body centercubic (bcc) phases by using first-principles calculations based on the density functional theory (DFT).We solve the Kohn-Sham equations using the full potential linearized augmented plane wave (FP-LAPW)method. The generalized gradient approximations as proposed by Perdew-Burke-Ernzerhof (PBE sol GGA)and Engel-Vosko (EV-GGA) are employed to calculate the exchange correlation potential. Using GGA, wecompute the optimized lattice parameters of the bcc. This is the first instance of such application of GGA.In close agreement with experimental results, our electronic calculations show that the hexagonal phase isa direct gap semiconductor with energy gap of 0.2520 eV. The bcc phase calculated band structure showsa direct gap semiconductor with band gap of 1.0306 eV. We further study the effects of applying pressureon the band structure of the system.
1 Introduction
Wide band gap semiconductors have gained considerableattention due to their numerous applications in electronicand optoelectronic devices. Semiconductors are widelyused in lasers, detectors and integrated circuits, while thesmall gap semiconductors such as Mg3Bi2 have applica-tions in thermoelectric element [1], Li batteries [2] andphotoconduction [3]. It can also be used as a superionicmaterial [4]. Combination of Mg with the fifth group ele-ments (pnicogens (Pn): N, P, As, Sb and Bi) of periodictable produces semiconductor compounds with the generalformula Mg3Pn2 (e.g., magnesium bismuthide Mg3Bi2).There are two crystallographic structures for Mg3Bi2, thebody center cubic (bcc) structure and the hexagonal struc-ture. Mg3Bi2 has (Fe, Mn)2O3 type structure with thespace group Ia3 (2 0 6) in the cubic phase and La2O3 typestructure with the space group of P3m1 in the hexagonalphase [5,6]. To the best of our knowledge there are veryfew reports on physical properties of this compound. Themeasured value of the hexagonal phase is 0.15 eV [7]. In [8]the lattice parameters and elastic properties of the hexag-onal phase were investigated. In reference [9] the latticeparameters and band structure of Mg3Bi2 were calculatedby using pseudo-potential density functional method. Inreference [9] it was predicted that Mg3Bi2 should have asemimetal feature, which is not in agreement with exper-
a e-mail: [email protected]
imental results [7]. Kajikawa et al. measured the thermo-electric properties of Mg3Bi2 and Mg3Sb2 in the hexagonalphase at different temperatures [1]. It was shown that theelectrical conductivity decreases by increasing the temper-ature. The electrical conductivity of Mg3Bi2 under pres-sure in both liquid and in crystalline states was studied inreference [10]. Its conductivity and band structure were in-vestigated by Xu et al. [11]. The band structure of Mg3Bi2was calculated using the augmented spherical wave (ASW)method and a metallic feature was predicted [11]. First-principles molecular dynamics simulation of liquid Mg3Bi2was performed by de Wijs et al. [12]. In our previous workwe investigated the physical properties of Mg3Sb2 fromthe alkaline earth pnictides series [13]. Unlike the major-ity of Mg3Pn2 compounds, which are more stable in thebcc phase, the Mg3Bi2 is more stable in the hexagonalphase. Experimental works performed on Mg3Bi2 in thebcc phase are rare as Mg3Bi2 exists at high temperatureand high pressure. Just the lattice parameter was mea-sured to be 25.7951 a.u. in bcc phase [5]. Therefore ourresults can be considered as a physical parameter’s pre-diction for this phase. The disagreement between experi-mental and theoretical lattice parameters, band gap andelectronic properties of Mg3Bi2 in the hexagonal phasemotivated us to reexamine the calculations by ab initiofull potential density functional theory. Interestingly ourresults show that the hexagonal Mg3Bi2 is a semiconduc-tor with energy gap of 0.2520 eV and lattice constants of
10103-p1

The European Physical Journal Applied Physics
a = 8.8133 and c = 13.8587 a.u. We also investigated theeffects of pressure on the electronic properties of hexag-onal and bcc Mg3Bi2. The paper is organized as follows:in Section 2 we described our computational details, inSection 3 we present our results and discussions. Conclud-ing remarks are given in last section.
2 Calculation method
In the present work, we employ FP-LAPW density func-tional method as embodied in Wien2k package [14] tosolve the single particle Kohn-Sham equations [15] forMg3Bi2 compound in the cubic and hexagonal phases. Theexchange correlation potential is computed within bothgeneralized gradient approximation as introduced byPerdew-Burke-Ernerhof (PBE sol GGA) [16] andEngel-vosko (EV-GGA) [17]. The unit cell is divided intotwo distinct regions: one around the ionic core, i.e., themuffin tin region, and the other on the remaining spaceoutside the muffin tin region called the interstitial region.The wave functions were expanded in spherical harmon-ics in the muffin tin region. In the interstitial region thewave functions were expanded in plane waves. The max-imum value of angular momentum was set to lmax = 10.The muffin tin radii were set to 2.4 and 2.3 a.u. for Mgatom in the cubic and the hexagonal phases respectively.In the case of Bi atom, the muffin tin radii were selectedto be 2.7 a.u. in the cubic phase and 2.6 a.u. in the hexag-onal phase. 250 and 300 k-points were sampled in the ir-reducible wedge of the first Brillouin zone for body centercubic and hexagonal phases respectively which correspondto 6×6×6 and 8×8×4 grids in Monkhorst-Park schemerespectively [18].
3 Results and discussion
3.1 Structural properties
Magnesium bismuthide crystallizes in both hexagonal andcubic phases. There are five atoms in the primitive cell ofMg3Bi2 in its hexagonal phase, i.e., 1 formula unit perprimitive cell. Two kinds of Mg atoms with different sym-metries exist in the primitive cell, labeled by Mg1 andMg2. In the Mg1 type there are six Bi neighbors all withthe same bond lengths. In the Mg2 type there are four Bineighbors, three of which have the same bond lengths andthe fourth with a different bond length. In the hexagonalphase, the Bi, Mg1 and Mg2 atoms are in ±(1/3, 2/3, u),(0, 0, 0) and ±(1/3, 2/3, v) positions respectively [19]. Theprimitive cell of Mg3Bi2 in its cubic phase has 40 atoms.This primitive cell contains eight formula units of Mg3Bi2.There are 24 identical Mg atoms in this primitive cell. TheBi atoms are divided into two different groups. We refer tothese groups as Bi1 and Bi2. Each group has six Mg neigh-bors. In the Bi1 group, the six bonds between the Bi withMg atoms are all of the same length. In the Bi2 group, thesix bonds between the Bi with Mg atoms are divided into
Fig. 1. (Color online) The unit cell of Mg3Bi2 and its atomspositions in hexagonal structure.
three subgroups. Within each subgroup, the bond lengthsare identical, but they vary slightly across the subgroups.The Mg, Bi1 and Bi2 atoms in this phase are at (x, y,z, etc.), (1/4, 1/4, 1/4, etc.) and (u, 0, 1/4, etc.) posi-tions respectively [19]. Figure 1 illustrates the positions ofMg3Bi2 unit cell atoms in the hexagonal phase.
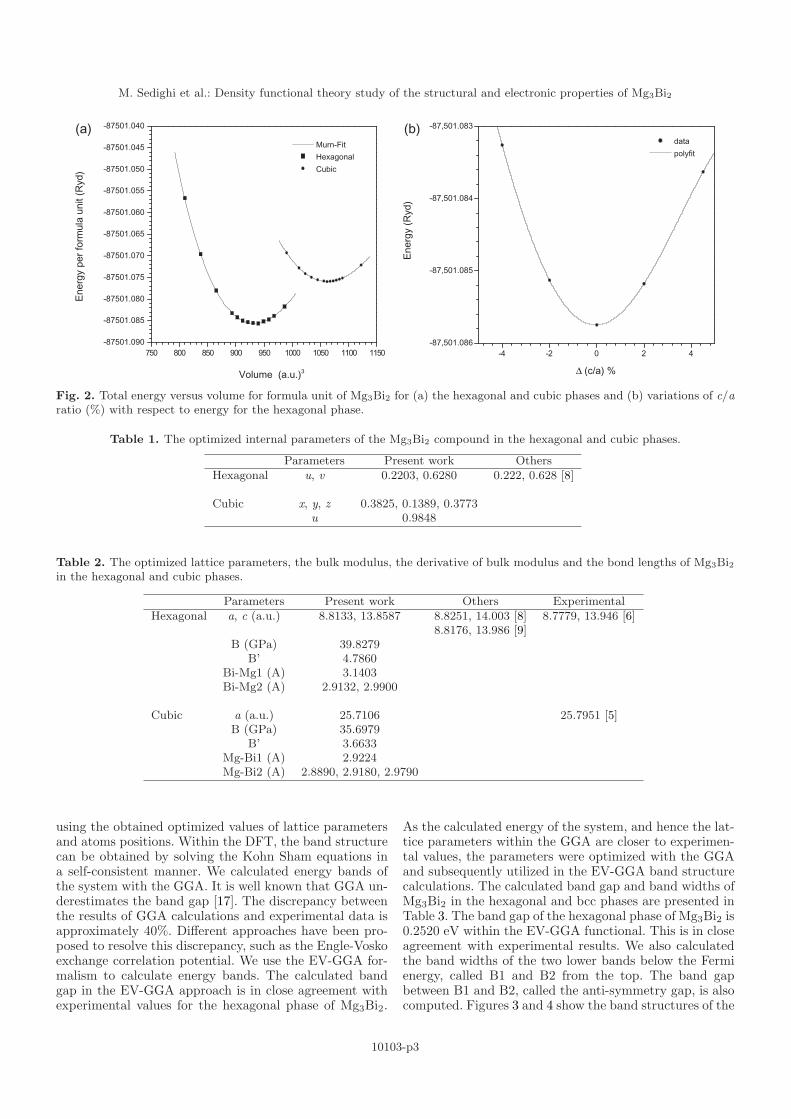
To investigate the structural and electronic propertiesof magnesium bismuthide, we first calculated the opti-mized values of the lattice and internal parameters forboth phases. The Birch-Murnaghan equation of state [20]was used to obtain the energy curve in terms of volume. Itis worth mentioning that in the hexagonal phase the valueof c/a ratio should be optimized first. Figure 2 shows thediagram of energy in terms of volume per formula unit forthe hexagonal and cubic phases. The energy volume curveobtained by Wien2k package is energy of a unit cell. In thehexagonal phase of Mg3Bi2 we have five atoms (or 1 for-mula unit) while in the bcc phase there are 40 atoms (or8 formula unit). In order to have the comparable energyvolume curves of both structures in the same figure, wenormalized the energy of the 40 atoms of the bcc phaseunit cell to Mg3Bi2 formula unit number. Therefore forthis phase both energy and volume are divided by 8.
The internal and lattice parameters were computedand optimized by relaxing the atomic positions in the forcedirections within the scheme of Hellmann-Feynman [21].Tables 1 and 2 list the results. As can be seen, our cal-culated lattice parameters are in good agreement withexperimental values.
3.2 Electronic properties
Detailed description of energy bands provides valuable in-sight into the electronic and optoelectronic properties ofsemiconductors. In this section we study the electronicproperties of Mg3Bi2 in the hexagonal and cubic phases
10103-p2
M. Sedighi et al.: Density functional theory study of the structural and electronic properties of Mg3Bi2
750 800 850 900 950 1000 1050 1100 1150-87501.090
-87501.085
-87501.080
-87501.075
-87501.070
-87501.065
-87501.060
-87501.055
-87501.050
-87501.045
-87501.040
Murn-Fit Hexagonal Cubic
Ener
gy p
er fo
rmul
a un
it (R
yd)
Volume (a.u.)3
-4 -2 0 2 4-87,501.086
-87,501.085
-87,501.084
-87,501.083
data polyfit
Ener
gy (R
yd)
Δ (c/a) %
(a) (b)
Fig. 2. Total energy versus volume for formula unit of Mg3Bi2 for (a) the hexagonal and cubic phases and (b) variations of c/aratio (%) with respect to energy for the hexagonal phase.
Table 1. The optimized internal parameters of the Mg3Bi2 compound in the hexagonal and cubic phases.
Parameters Present work OthersHexagonal u, v 0.2203, 0.6280 0.222, 0.628 [8]
Cubic x, y, z 0.3825, 0.1389, 0.3773u 0.9848
Table 2. The optimized lattice parameters, the bulk modulus, the derivative of bulk modulus and the bond lengths of Mg3Bi2in the hexagonal and cubic phases.
Parameters Present work Others ExperimentalHexagonal a, c (a.u.) 8.8133, 13.8587 8.8251, 14.003 [8] 8.7779, 13.946 [6]
8.8176, 13.986 [9]B (GPa) 39.8279
B’ 4.7860Bi-Mg1 (A) 3.1403Bi-Mg2 (A) 2.9132, 2.9900
Cubic a (a.u.) 25.7106 25.7951 [5]B (GPa) 35.6979
B’ 3.6633Mg-Bi1 (A) 2.9224Mg-Bi2 (A) 2.8890, 2.9180, 2.9790
using the obtained optimized values of lattice parametersand atoms positions. Within the DFT, the band structurecan be obtained by solving the Kohn Sham equations ina self-consistent manner. We calculated energy bands ofthe system with the GGA. It is well known that GGA un-derestimates the band gap [17]. The discrepancy betweenthe results of GGA calculations and experimental data isapproximately 40%. Different approaches have been pro-posed to resolve this discrepancy, such as the Engle-Voskoexchange correlation potential. We use the EV-GGA for-malism to calculate energy bands. The calculated bandgap in the EV-GGA approach is in close agreement withexperimental values for the hexagonal phase of Mg3Bi2.
As the calculated energy of the system, and hence the lat-tice parameters within the GGA are closer to experimen-tal values, the parameters were optimized with the GGAand subsequently utilized in the EV-GGA band structurecalculations. The calculated band gap and band widths ofMg3Bi2 in the hexagonal and bcc phases are presented inTable 3. The band gap of the hexagonal phase of Mg3Bi2 is0.2520 eV within the EV-GGA functional. This is in closeagreement with experimental results. We also calculatedthe band widths of the two lower bands below the Fermienergy, called B1 and B2 from the top. The band gapbetween B1 and B2, called the anti-symmetry gap, is alsocomputed. Figures 3 and 4 show the band structures of the
10103-p3

The European Physical Journal Applied Physics
Table 3. Calculated values of the band gap, bandwidths (B1 and B2) and the anti-symmetry gap of Mg3Bi2 in the hexagonaland bcc phases. For comparison experimental values of the band gap and one of the other DFT calculation methods for thehexagonal phase are also presented.
Phase Method Band gap (eV) Bandwidths (eV) Anti-Sym-Gap (eV)
B1 B2Cubic DFT(GGA) 0.4064 4.1471 0.8195 5.8590
DFT(EV-GGA) 1.0306 4.0029 0.8435 5.8152
Hexagonal DFT(GGA) 0.0 4.9438 1.1345 5.4997DFT(EV-GGA) 0.2520 4.8243 1.1248 5.5174
DFT pseudo 0.0 [9]Experimental 0.15 [7]
(a) (b)
Fig. 3. Band structures of Mg3Bi2 for the hexagonal phase within the (a) GGA and (b) EV-GGA.
(a) (b)
Fig. 4. Band structures of Mg3Bi2 for the bcc phase within the (a) GGA and (b) EV-GGA.
10103-p4

M. Sedighi et al.: Density functional theory study of the structural and electronic properties of Mg3Bi2
0 1 2 3 4 5 6 7 8 9 10 11 12 13 140
1
2
3
4
5
6
0 1 2 3 4 5 6 7 8 9 10 11 12 13 140
1
2
3
4
5
6
Ener
gy (e
v)
Ener
gy (e
v)Pressure (GPa) Pressure (GPa)
GGA-Hex EV-GGA-Hex(a) (b)
Fig. 5. The variations of the band gap, the bandwidths (B1 and B2) and the anti-symmetry gap versus pressure for thehexagonal phase within the (a) GGA and (b) EV-GGA.
0 1 2 3 4 5 6 7 8 9 10 11 12 13 140
1
2
3
4
5
6
0 1 2 3 4 5 6 7 8 9 10 11 12 13 140
1
2
3
4
5
6
Ener
gy (e
v)
Ener
gy (e
v)
Pressure (GPa)
GGA-BCC EV-GGA-BCC
Pressure (GPa)
(a) (b)
Fig. 6. The variations of the band gap, the bandwidths (B1 and B2) and the anti-symmetry gap versus pressure for the bccphase within the (a) GGA and (b) EV-GGA.
hexagonal and bcc phases of Mg3Bi2 respectively. As canbe seen, the GGA method predicts a semimetal feature forMg3Bi2 in the hexagonal phase which is not in agreementwith experimental results. However, the EV-GGA methodshows that this system is a direct gap semiconductor witha band gap of 0.2502 eV. For the bcc phase both GGAand EV-GGA methods show that system is a direct gapsemiconductor with band gaps of 0.4064 and 1.0306 eVrespectively.
Finally, we use the relation P (V )= B0B′
0
[(V0V
)B′0− 1
][22]
to study effects of applying pressure on the band gap, anti-symmetry gap and band widths. The results are presentedin Figures 5 and 6. In the bcc phase both GGA and EV-GGA predict a direct gap at ambient pressure and underpressure. In the bcc phase, by increasing the pressure upto 5.8 GPa (with the GGA method) and 8 GPa (with theEV-GGA method) the value of the band gap increases.
10103-p5

The European Physical Journal Applied Physics
Applying further pressure results in a decrease in the bandgap with both the GGA and EV-GGA methods. By ex-trapolating the variations of the band gap versus pressure,for the bcc phase the metallization pressure was calculatedto be 59.3 GPa with the GGA and 102.894 GPa with theEV-GGA. In the hexagonal phase, the semiconducting be-havior cannot be seen at ambient condition using the GGAmethod. As can be seen in Figure 5 by increasing thepressure from 2 to 6 GPa Mg3Bi2 behaves as a semicon-ductor. Within the GGA method a direct to indirect gaptransition takes place for the semiconductor at 3.1 GPapressure. Within the EV-GGA method the band gap iscloser to its experimental value. The band gap is direct inthe pressure range of 0 to 2.3 GPa but for pressure valuesbigger than 2.3 GPa, the band gap is predicted to be in-direct. In this phase, the metallization pressure is extrap-olated to be 6 GPa with the GGA and 27.986 GPa withthe EV-GGA. Note that for the hexagonal phase applica-tion of pressure results in an increase and a subsequentdecrease in the band gap in both methods.
4 Conclusions
In this paper, we investigated the structural and electronicproperties of Mg3Bi2 in both hexagonal and bcc phaseswithin the framework of density functional theory. Sincethe calculation of energy within the GGA is better thanEV-GGA, the optimized structural parameters are firstcalculated using GGA and then utilized in the EV-GGAto calculate the band structure. For the hexagonal phasethe obtained lattice constants are a = 8.8133 a.u. andc = 13.8587 a.u. For the bcc phase our calculated latticeconstant is 25.7951 a.u. In contrast to previous work wherea semimetal feature was predicted for the hexagonal phase,our results show that Mg3Bi2 is a semiconductor in thisphase. We used both the GGA and EV-GGA formalismsto calculate the band structures and the band gaps. Theresults of the EV-GGA calculations show that the hexag-onal phase of magnesium bismuthide is a direct gap semi-conductor with energy gap of 0.2520 eV while the GGAcalculations show that the system is a semimetal. As ex-pected the results of the band structure and the band gapwithin the EV-GGA are in good agreement with experi-mental results. Finally, we investigated effects of pressureon the band structure and band gap. We found by increas-ing pressure for both phases the gap initially increases andthen decreases. As a result of applying pressure a semicon-ductor phase transition from direct to indirect occurs forthe hexagonal phase.
Authors would like to thank Dr. Kourosh Zarringhalam fromthe department of mathematics of University of MassachusettsBoston for proofreading the manuscript.
References
1. T. Kajikawa, N. Kimura, T. Yokoyama, 22nd InternationalConference on Thermo Electrics, 2003
2. H. Honda, H. Sakaguchi, I. Tanaka, T. Esaka, J. PowerSources 123, 216 (2003)
3. P. Singh, K.K. Sarkar, Solid State Commun. 55, 439 (1985)4. A.C. Bames, C. Guo, W.S. Howells, J. Phys.: Condens.
Matter 6, L467 (1994)5. G.V. Samsonov, M.N. Abdusalyamova, V.B.
Chernogorenko, Vismutidy (Bismuthides) (NaukovaDumka, Kiev, 1977)
6. L.G. Sevast’yanova, O.V. Kravchenko, O.K. Gulish, V.A.Stupnikov, M.E. Leonova, M.G. Zhizhin, NeorganicheskieMaterialy (Inorganic Materials) 42, 863 (2006)
7. L.M. Watson, C.A.W. Marshall, C.P. Cardoso, J. Phys. F:Met. Phys. 14, 113 (1984)
8. D.W. Zhou, J.S. Liu, S.H. Xu, P. Peng, Physica B 405,2863 (2010)
9. Y. Imai, A. Watanabe, J. Mater. Sci. 41, 2435 (2006)10. V.V. Brazhkin, D.R. Dmitriev, R.N. Voloshin, Phys. Lett.
A 193, 102 (1994)11. R. Xu, R.A. de Groot, W. van der Lugt, J. Phys.: Condens.
Matter 5, 7551 (1993)12. G.A. de Wijs, G. Pastore, A. Selloni, W. van der Lugt, J.
Phys.: Condens. Matter 8, 1879 (1996)13. M. Sedighi, B. Arghavani Nia, H. Zarringhalam, R.
Moradian, Physica B 406, 3149 (2011)14. P. Blaha, K. Schwarz, G.K.H. Madsen, D.K. Vasnicka,
J. Luitz, WIEN2K, An Augmented Plane Wave + LocalOrbitals Program for calculating Crystal Properties(Karlheinz Schwarz, Techn. Universitat Wien, Austria,ISBN 3-9501031-1-2, 2001)
15. W. Kohn, L.J. Sham, Phys. Rev. 140, A1133 (1965)16. J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77,
3865 (1996)17. E. Engel, S.H. Vosko, Phys. Rev. 47, 13164 (1993)18. H.J. Monkhorst, J.D. Park, Phys. Rev. B 13, 5188
(1976)19. R.W.G. Wyckoff, Crystal Structures, 2nd edn. (Krieger,
Malabar, FL, 1986)20. F.D. Murnaghan, Proc. Natl. Acad. Sci. USA 30, 244
(1944)21. P. Pulay, Mol. Phys. 17, 197 (1969)22. V.G. Tyuterev, N. Vast, Comput. Mater. Sci. 38, 350
(2006)
10103-p6




















