
Differential Detergent Fractionation for Non-electrophoretic Eukaryote Cell Proteomics
Upload
independentCategory
view
0download
0
Analysis of detergent-resistant membranes ofHelicobacter pylori infected gastric adenocarcinomacells reveals a role for MARK2/Par1b in CagA-mediateddisruption of cellular polarity
Zaher Zeaiter,1 David Cohen,2 Anne Müsch,2
Fabio Bagnoli,3 Antonello Covacci3 andMarkus Stein1*1Department of Medical Microbiology and Immunology,University of Alberta, Edmonton, Alberta, Canada,T6G 2H7.2Dyson Vision Research Institute, Cornell UniversityMedical College, New York, NY 10021, USA.3Novartis, Via Fiorentina 1, Siena, Italy.
Summary
Detergent-resistant membranes of eukaryotic cellsare enriched in many important cellular signallingmolecules and frequently targeted by bacterialpathogens. To learn more about pathogenic mecha-nisms of Helicobacter pylori and to elucidate noveleffects on host epithelial cells, we investigated howbacterial co-cultivation changes the protein composi-tion of detergent-resistant membranes of gastricadenocarcinoma (AGS) tissue culture cells. UsingiTRAQ (isobaric tags for relative and absolute quan-tification) analysis we identified several cellularproteins, which are potentially related to H. pylorivirulence. One of the proteins, which showed a sig-nificant infection-dependent increase in detergentresistance, was the polarity-associated serine/threonine kinase MARK2 (EMK1/Par-1b). We demon-strate that H. pylori causes the recruitment of MARK2from the cytosol to the plasma membrane, where itcolocalizes with the bacteria and interacts with CagA.Using Mardin Darby Canine Kidney (MDCK) monolay-ers and a three-dimensional MDCK tissue culturemodel we showed that association of CagA withMARK2 not only causes disruption of apical junc-tions, but also inhibition of tubulogenesis and celldifferentiation.
Introduction
Bacterial pathogens use an arsenal of sophisticated strat-egies to exploit host cell proteins and signalling pathwaysfor their own benefit to achieve adherence, invasion,replication and survival in the respective host. A growingnumber of these pathogens have been described toexploit specialized regions of the eukaryotic cell mem-brane called lipid rafts for cell entry, intracellular survivalor toxin delivery (Rosenberger et al., 2000; Lafont andvan der Goot, 2005). Lipid rafts are micro domains, whichare enriched in cholesterol and sphingolipid causinglateral segregation of lipids and proteins. This segregationis functional relevant, and acts as a concentration andselection platform for many important signalling molecules(Brown and London, 1998; Simons and Toomre, 2000;Hancock, 2006; Jacobson et al., 2007). Originally, lipidraft proteins have been identified by their insolubility inTriton X-100 and for this reason the term detergent-resistant membranes (DRMs) had frequently been usedsynonymously in the literature. However, in recent years ithas been recognized that detergent resistance is not asufficient criterion to decide, whether a protein is localizedin membrane rafts or not. Nevertheless, insolubility inTriton X-100 is an accepted method for carrying out pro-teomic screens to identify putative raft-associated pro-teins, which than would need to be confirmed as lipid raftcomponents using complementary methods. Since thetwo Helicobacter pylori (Hp) toxins VacA (Schraw et al.,2002; Nakayama et al., 2006) and CagA (Asahi et al.,2003) were both previously shown to associate withDRMs we reasoned that a proteomic analysis of thesemembranes would identify new CagA and VacA interact-ing proteins and associated signalling pathways.
Hp is a human pathogen that colonizes the gastricepithelium and in some cases triggers the development ofsevere gastric diseases including peptic ulcer disease,MALT-lymphoma and adenocarcinoma (Peek and Blaser,2002). The virulence protein that is thought to be a majorfactor for cellular transformation and cancer developmentis the CagA toxin (Covacci et al., 1993; Tummuru et al.,1993), although the vacuolating toxin VacA and other viru-lence factors may also contribute to disease development
Received 12 July, 2007; revised 22 October, 2007; accepted 23October, 2007. *For correspondence. E-mail [email protected];Tel. (+1) 780 492 5495; Fax (+1) 780 492 7521.
Cellular Microbiology (2008) 10(3), 781–794 doi:10.1111/j.1462-5822.2007.01084.xFirst published online 27 November 2007
© 2007 The AuthorsJournal compilation © 2007 Blackwell Publishing Ltd

(Peek and Blaser, 2002). Tissue culture experiments dem-onstrated that CagA positive Hp strains disrupt cellularpolarity by targeting tight junctions (Amieva et al., 2003)(Lai et al., 2006) and adherence junctions (Suzuki et al.,2005; Murata-Kamiya et al., 2007), induce morphologicalchanges known as the hummingbird phenotype (Segalet al., 1999), and cause an increase in motility (Churinet al., 2001) and cell proliferation (Peek et al., 1997).Following adherence CagA is translocated into epithelialcells by the pathogenicity island-encoded type IV secretionsystem (Segal et al., 1999; Asahi et al., 2000; Backertet al., 2000; Stein et al., 2000; Odenbreit et al., 2001) andphosphorylated on a variable number of tyrosine residuesby members of the Src-kinase family (Asahi et al., 2000;Stein et al., 2002) and the Abl tyrosine kinase (Tammeret al., 2007). CagA mediates its various effects by recruit-ing several cellular proteins in a phosphorylation-dependent (Shp-2, c-Met, Crk2) (Higashi et al., 2002;Churin et al., 2003; Suzuki et al., 2005) or independent(Grb-2 and ZO-1) (Mimuro et al., 2002; Amieva et al.,2003) manner. Although ZO-1 colocalization with CagAhas been described to coincide with disruption of tightjunctions leading to loss of apical-basolateral cell polarity adeeper understanding of the contributing molecularmechanisms remains elusive (Amieva et al., 2003).
Studies in Drosophila and in cultured mammalianepithelial cells have revealed a role for PAR proteins inepithelial polarization. Initially identified as Partitioningdeterminants in the C. elegans embryo (Kemphues et al.,1988), Par proteins mark distinct cortical domains (see(Macara, 2004) (Suzuki and Ohno, 2006) for review). Thus,the scaffolding proteins Par3 and Par6 form a ternarycomplex with aPKC at the apical cell surface, while theserine/threonine kinase Par1 is restricted to the lateraldomain of epithelial cells. In mammalian kidney epithelial(MDCK) cells the Par3/Par6/aPKC complex is part of anapical junctional complex that contributes to the develop-ment of zonula adherens, and is critical for the initialformation and maintenance of apical-basolateral cell polar-ity (Suzuki et al., 2001; 2004). Drosophila Par1 and themammalian Par1 isoform Par1b/MARK2/EMK1 are essen-tial for cell-cell adhesion, compaction and integrity of theepithelial monolayer (Bohm et al., 1997; Cox et al., 2001;Cohen et al., 2004, Suzuki et al., 2004, Benton and StJohnston, 2003; Elbert et al., 2006). Par1 isoforms wereindependently identified as Microtubule-Affinity-Regulat-ing Kinases (Drewes et al., 1997), hence the acronymMARK and have been shown to regulate the epithelialMT-cytoskeleton (Doerflinger et al., 2003, Cohen et al.,2004). aPKC is central for the mutual exclusive activities ofthe Par3/Par6/aPKC complex and Par1 along the apical-basolateral polarity axis: Cdc42, a cofactor for aPKC activ-ity is restricted to the apical surface of epithelial cells(Martin-Belmonte et al., 2007), effectively preventing
aPKC activity at the basolateral surface. aPKC-mediatedphosphorylation of MARK2 on Thr595, on the other hand,causes its membrane dissociation, thereby preventingMARK2 accumulation and activity at the apical cortex(Hurov et al., 2004, Suzuki et al., 2004). Here we per-formed iTRAQ proteomic analysis to determine modifica-tions of DRMs of AGS cells during infection. We identified7 proteins that became enriched in DRMs upon Hp infec-tion, and further characterized one of them, MARK2. Weverified that Hp causes a CagA-dependent shift of MARK2from a soluble to a detergent-resistant compartment anddetermined that CagA and MARK2 interact and colocalizein Hp infected AGS cells. Furthermore, we show thatCagA-expression in MDCK cells mimics the effect ofMARK2 depletion on epithelial morphogenesis and dem-onstrate that overexpression of MARK2 attenuates theCagA-induced polarity phenotypes in this polarized epithe-lial cells line. Our data suggest that CagA-mediated inhibi-tion of MARK2 function represents an important molecularmechanism for Hp virulence.
Results
iTRAQ analysis of detergent resistance membranes ofHp infected and uninfected AGS tissue culture cells
To quantify relative differences in the proteome of DRMsof AGS tissue culture cells following infection with Hpwild-type we used the isobaric tagging reagents (iTRAQ;Applied Biosystems) followed by multidimensional liquidchromatography (LC) and tandem mass spectrometry(MS/MS) analysis. Cells were infected for four hours andDRMs were isolated by cell lysis in 1% Triton X-100 for 1 hand flotation on a sucrose step gradient as described inExperimental procedures. Successful isolation of DRMswas confirmed by immunoblotting using antibodiesagainst proteins that are known to be present or absent inlipid rafts. Flotillin 1 and Lyn kinase were used as positivemarkers and the transferrin receptor (CD71) was used asa negative control (Fig. 1a–c).
When analysing the results from the iTRAQ analysis atleast 7 proteins were enriched by a factor of 1.8-fold ormore in the DRMs of Hp infected cells compared withuninfected cells. Four proteins were identified to be sig-nificant decreased in the DRMs during infection and fourrepresentatives are shown for proteins that were notmodified. Table 1 shows a representative list of theseproteins that were all identified with a confidence > 99%and a P-value < 0.05 for quantification.
Hp induces increased expression of the serine/threoninekinases MARK2
The two proteins that showed the strongest increase inthe DRMs of infected cells compared with uninfected cells
782 Z. Zeaiter et al.
© 2007 The AuthorsJournal compilation © 2007 Blackwell Publishing Ltd, Cellular Microbiology, 10, 781–794

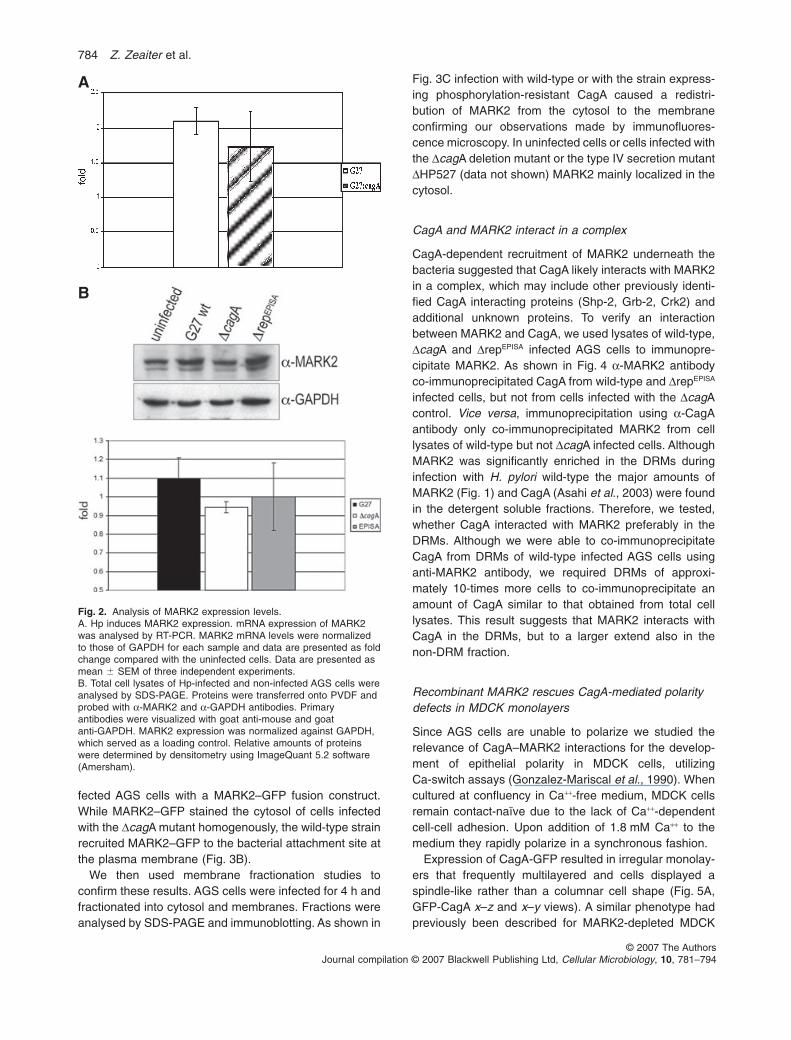
were the serine-threonine kinases MARK2 and MARK3,which were enriched approximately fourfold. SinceMARK2 is a well-studied mediator for the development ofcellular polarity and since Hp CagA is known to disrupttight and adherence junctions we decided to further inves-tigate the role of MARK2 in this process. We first con-firmed the presence of MARK2 in the DRMs of infectedcells by immunoblotting. Figure 1d and e show thatMARK2 was indeed present in raft-fractions of infected,but was not detected in corresponding fractions of unin-fected cells. To further test, whether this enrichmentwas due to increased expression of MARK2 or rather dueto redistribution from the Triton X-100 soluble to theinsoluble fraction, we performed quantitative RT-PCR.The results showed that in Hp infected cells MARK2 tran-scription was increased approximately twofold (Fig. 2A).Increased expression, although at slightly lower levels,was also observed for infection with the DcagA mutant.However, when total cell lysates of uninfected cells or
cells infected with G27, DcagA or DrepEPISA were analysedby immunoblotting no significant differences for MARK2protein expression were observed (Fig. 2B). The DrepEPISA
mutant expressed a modified version of CagA, whichwas lacking the repeat-region and the tyrosine-phosphorylation motifs.
CagA causes the redistribution of MARK2 from thecytosol to the cell membrane in non-polarized AGS cells
Since MARK2 was fourfold enriched in the DRMs ofinfected cells, a modest to no increase in protein expres-sion could hardly account for our finding, especially sinceit was suggested that protein ratios identified by iTRAQanalyses often suggest smaller increases/decreases thanwould be detected with alternative methods. This phe-nomenon is also supported by our immunoblotting data inFig. 1d and e, which suggest a more drastic increase forMARK2 in the DRM fractions of infected versus unin-fected cells. We thus tested, whether co-culture of AGScells with Hp affects the cellular distribution of MARK2.Following infection for 4 h with wild-type or an isogenicDcagA mutant the cells were washed, fixed and analysedby immunofluorescence microscopy (Fig. 3A). Uninfectedcells and the DcagA deletion mutant showed an evendistribution of MARK2 throughout the cytosol. In contrast,Hp wild-type and the DrepEPISA mutant caused the recruit-ment of MARK2 from the cytosol to the plasma membrane(Fig. 3A). Here MARK2 mainly colocalized in foci under-neath the attaching bacteria. This demonstrates thatMARK2 recruitment to the bacterial attachment site isdependent on CagA translocation, but not on CagAtyrosine phosphorylation of the EPIYA motif. Analysis ofwild-type infected AGS cells at different time points indi-cated that MARK2 recruitment to the membrane wasdetectable as early as 30 min after infection (Fig. 3A),which coincides with the time required for CagA translo-cation to the host cell (Asahi et al., 2000). We also trans-
Fig. 1. Infection with Hp induces significant increase of MARK2 inDRMs. AGS cells were infected with Hp wild-type (a–d) or a DcagA(e) mutant for 4 h. DRMs were isolated by cell-lysis in 1% TritonX-100 followed by flotation on a sucrose step-gradient. One millilitrefractions (1–12) were collected from the top of the gradient andanalysed by immunoblotting with antibodies against a-Flotillin,a-Lyn, a-CD71 (transferring receptor) and a-MARK2 as indicated.
Table 1. Representative list of differentially partitioned DRM proteins following infection of AGS cells with H. pylori.
Accession Name Fold
LSR_HUMAN (Q86 ¥ 29) Lipolysis-stimulated lipoprotein receptor -2.27CAPON_HUMAN (O75052) Carboxyl-terminal PDZ ligand of neuronal nitric oxide synthase protein -2.22K2C6E_HUMAN (P48668) Keratin, type II cytoskeletal 6E -2.17MYH14_HUMAN (Q7Z406) Myosin-14 (Myosin heavy chain, non-muscle IIc) -1.64HM13_HUMAN (Q8TCT9) Minor histocompatibility antigen H13 1.08ITB4_HUMAN (P16144) Integrin beta-4 precursor (GP150) (CD104 antigen) 1.11APMAP_HUMAN (Q9HDC9) Adipocyte plasma membrane-associated protein 1.12AT1A1_HUMAN (P05023) Sodium/potassium-transporting ATPase alpha-1 chain precursor 1.12KU70_HUMAN (P12956) ATP-dependent DNA helicase 2 subunit 1 1.82UQCR1_HUMAN (P31930) Ubiquinol-cytochrome-c reductase complex core protein I, mitochondrial precursor 2.02SA7L1_HUMAN (Q86SG5) Protein S100-A7-like 1 2.75UPAR_HUMAN (Q03405) Urokinase plasminogen activator surface receptor precursor 2.90YBOX1_HUMAN (P67809) Nuclease sensitive element-binding protein 1 (YB1) 3.40MARK3_HUMAN (P27448) MAP/microtubule affinity-regulating kinase 3 3.90MARK2_HUMAN (Q7KZI7) Serine/threonine-protein kinase MARK2 4.03
iTraq analysis of Helicobacter pylori infected cells 783
© 2007 The AuthorsJournal compilation © 2007 Blackwell Publishing Ltd, Cellular Microbiology, 10, 781–794
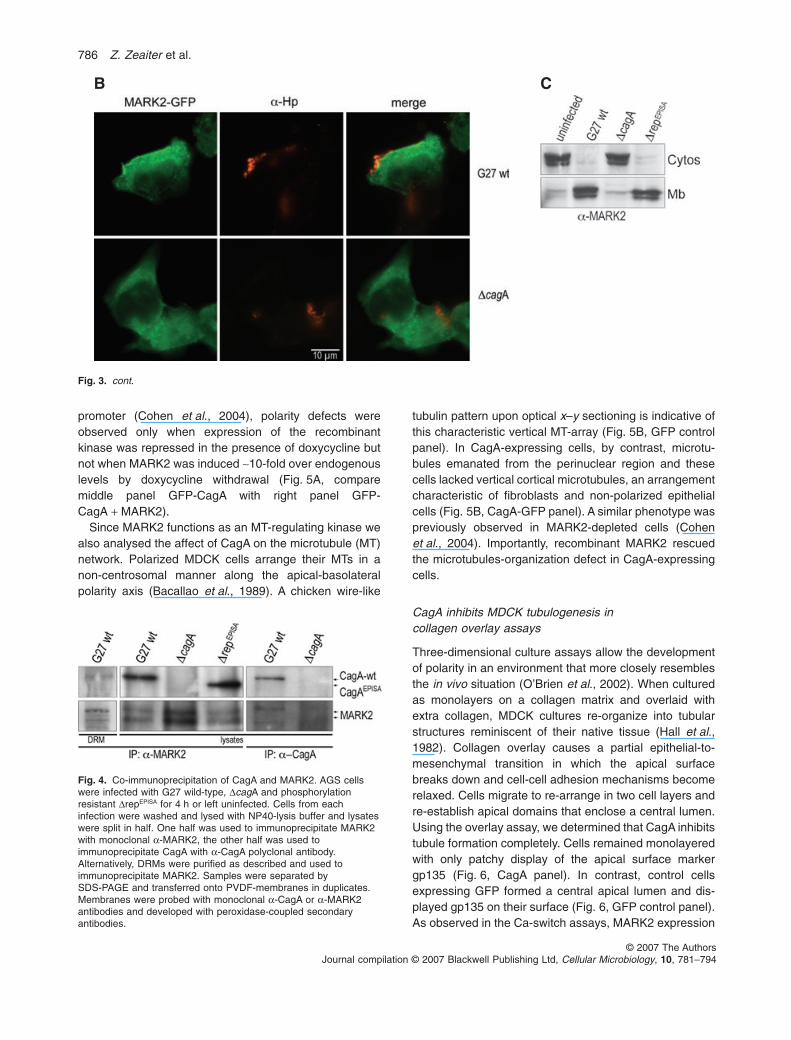
fected AGS cells with a MARK2–GFP fusion construct.While MARK2–GFP stained the cytosol of cells infectedwith the DcagA mutant homogenously, the wild-type strainrecruited MARK2–GFP to the bacterial attachment site atthe plasma membrane (Fig. 3B).
We then used membrane fractionation studies toconfirm these results. AGS cells were infected for 4 h andfractionated into cytosol and membranes. Fractions wereanalysed by SDS-PAGE and immunoblotting. As shown in
Fig. 3C infection with wild-type or with the strain express-ing phosphorylation-resistant CagA caused a redistri-bution of MARK2 from the cytosol to the membraneconfirming our observations made by immunofluores-cence microscopy. In uninfected cells or cells infected withthe DcagA deletion mutant or the type IV secretion mutantDHP527 (data not shown) MARK2 mainly localized in thecytosol.
CagA and MARK2 interact in a complex
CagA-dependent recruitment of MARK2 underneath thebacteria suggested that CagA likely interacts with MARK2in a complex, which may include other previously identi-fied CagA interacting proteins (Shp-2, Grb-2, Crk2) andadditional unknown proteins. To verify an interactionbetween MARK2 and CagA, we used lysates of wild-type,DcagA and DrepEPISA infected AGS cells to immunopre-cipitate MARK2. As shown in Fig. 4 a-MARK2 antibodyco-immunoprecipitated CagA from wild-type and DrepEPISA
infected cells, but not from cells infected with the DcagAcontrol. Vice versa, immunoprecipitation using a-CagAantibody only co-immunoprecipitated MARK2 from celllysates of wild-type but not DcagA infected cells. AlthoughMARK2 was significantly enriched in the DRMs duringinfection with H. pylori wild-type the major amounts ofMARK2 (Fig. 1) and CagA (Asahi et al., 2003) were foundin the detergent soluble fractions. Therefore, we tested,whether CagA interacted with MARK2 preferably in theDRMs. Although we were able to co-immunoprecipitateCagA from DRMs of wild-type infected AGS cells usinganti-MARK2 antibody, we required DRMs of approxi-mately 10-times more cells to co-immunoprecipitate anamount of CagA similar to that obtained from total celllysates. This result suggests that MARK2 interacts withCagA in the DRMs, but to a larger extend also in thenon-DRM fraction.
Recombinant MARK2 rescues CagA-mediated polaritydefects in MDCK monolayers
Since AGS cells are unable to polarize we studied therelevance of CagA–MARK2 interactions for the develop-ment of epithelial polarity in MDCK cells, utilizingCa-switch assays (Gonzalez-Mariscal et al., 1990). Whencultured at confluency in Ca++-free medium, MDCK cellsremain contact-naïve due to the lack of Ca++-dependentcell-cell adhesion. Upon addition of 1.8 mM Ca++ to themedium they rapidly polarize in a synchronous fashion.
Expression of CagA-GFP resulted in irregular monolay-ers that frequently multilayered and cells displayed aspindle-like rather than a columnar cell shape (Fig. 5A,GFP-CagA x–z and x–y views). A similar phenotype hadpreviously been described for MARK2-depleted MDCK
A
B
Fig. 2. Analysis of MARK2 expression levels.A. Hp induces MARK2 expression. mRNA expression of MARK2was analysed by RT-PCR. MARK2 mRNA levels were normalizedto those of GAPDH for each sample and data are presented as foldchange compared with the uninfected cells. Data are presented asmean � SEM of three independent experiments.B. Total cell lysates of Hp-infected and non-infected AGS cells wereanalysed by SDS-PAGE. Proteins were transferred onto PVDF andprobed with a-MARK2 and a-GAPDH antibodies. Primaryantibodies were visualized with goat anti-mouse and goatanti-GAPDH. MARK2 expression was normalized against GAPDH,which served as a loading control. Relative amounts of proteinswere determined by densitometry using ImageQuant 5.2 software(Amersham).
784 Z. Zeaiter et al.
© 2007 The AuthorsJournal compilation © 2007 Blackwell Publishing Ltd, Cellular Microbiology, 10, 781–794

cells (Bohm et al., 1997; Cohen et al., 2004; Suzuki et al.,2004). In addition, CagA disrupted the localization of thetight junction marker ZO1, which distributed along theentire length of the lateral cortex (Fig. 5A, arrow in GFP-CagA panel) with additional intracellular staining. To
evaluate whether the morphological defects were dueto CagA-mediated MARK2 inhibition, we attempted torescue the CagA-phenotype by overexpressing MARK2.When CagA-GFP was transfected into MDCK–MARK2cells that express MARK2 under an doxycycline-inducible
A Fig. 3. CagA causes MARK2-recruitment tothe plasma membrane of AGS cells.A AGS cells were infected with various Hpstrains for different time points as indicatedand analysed by immunofluorescencemicroscopy. MARK2 was labelled withanti-MARK2 monoclonal antibody andbacteria with polyclonal anti-Hp serum.Primary antibodies were visualized withanti-mouse Alexa 488 and anti-chicken Alexa594 secondary antibodies respectively.B. AGS cells were transiently transfected withMARK2–GFP and infected with Hp wild-typeor the DcagA isogenic mutant for 4 h. Bacteriawas stained and visualized as describedabove and GFP-fluorescence is shown ingreen.C. AGS cells were infected with various Hpstrains for 4 h as indicated. Then the cellswere washed, scraped and fractionated intocytosol (Cyt) and membranes (Mb). Fractionswere analysed by SDS-PAGE. Proteins weretransferred onto PVDF-membrane andmembranes were probed a-MARK2monoclonal antibody. The blot was developedwith peroxidase-coupled secondary antibody.
iTraq analysis of Helicobacter pylori infected cells 785
© 2007 The AuthorsJournal compilation © 2007 Blackwell Publishing Ltd, Cellular Microbiology, 10, 781–794
promoter (Cohen et al., 2004), polarity defects wereobserved only when expression of the recombinantkinase was repressed in the presence of doxycycline butnot when MARK2 was induced ~10-fold over endogenouslevels by doxycycline withdrawal (Fig. 5A, comparemiddle panel GFP-CagA with right panel GFP-CagA + MARK2).
Since MARK2 functions as an MT-regulating kinase wealso analysed the affect of CagA on the microtubule (MT)network. Polarized MDCK cells arrange their MTs in anon-centrosomal manner along the apical-basolateralpolarity axis (Bacallao et al., 1989). A chicken wire-like
tubulin pattern upon optical x–y sectioning is indicative ofthis characteristic vertical MT-array (Fig. 5B, GFP controlpanel). In CagA-expressing cells, by contrast, microtu-bules emanated from the perinuclear region and thesecells lacked vertical cortical microtubules, an arrangementcharacteristic of fibroblasts and non-polarized epithelialcells (Fig. 5B, CagA-GFP panel). A similar phenotype waspreviously observed in MARK2-depleted cells (Cohenet al., 2004). Importantly, recombinant MARK2 rescuedthe microtubules-organization defect in CagA-expressingcells.
CagA inhibits MDCK tubulogenesis incollagen overlay assays
Three-dimensional culture assays allow the developmentof polarity in an environment that more closely resemblesthe in vivo situation (O’Brien et al., 2002). When culturedas monolayers on a collagen matrix and overlaid withextra collagen, MDCK cultures re-organize into tubularstructures reminiscent of their native tissue (Hall et al.,1982). Collagen overlay causes a partial epithelial-to-mesenchymal transition in which the apical surfacebreaks down and cell-cell adhesion mechanisms becomerelaxed. Cells migrate to re-arrange in two cell layers andre-establish apical domains that enclose a central lumen.Using the overlay assay, we determined that CagA inhibitstubule formation completely. Cells remained monolayeredwith only patchy display of the apical surface markergp135 (Fig. 6, CagA panel). In contrast, control cellsexpressing GFP formed a central apical lumen and dis-played gp135 on their surface (Fig. 6, GFP control panel).As observed in the Ca-switch assays, MARK2 expression
B C
Fig. 3. cont.
Fig. 4. Co-immunoprecipitation of CagA and MARK2. AGS cellswere infected with G27 wild-type, DcagA and phosphorylationresistant DrepEPISA for 4 h or left uninfected. Cells from eachinfection were washed and lysed with NP40-lysis buffer and lysateswere split in half. One half was used to immunoprecipitate MARK2with monoclonal a-MARK2, the other half was used toimmunoprecipitate CagA with a-CagA polyclonal antibody.Alternatively, DRMs were purified as described and used toimmunoprecipitate MARK2. Samples were separated bySDS-PAGE and transferred onto PVDF-membranes in duplicates.Membranes were probed with monoclonal a-CagA or a-MARK2antibodies and developed with peroxidase-coupled secondaryantibodies.
786 Z. Zeaiter et al.
© 2007 The AuthorsJournal compilation © 2007 Blackwell Publishing Ltd, Cellular Microbiology, 10, 781–794

alleviated the CagA-induced morphogenetic defects.Although the kinase did not allow for a complete rescue oftubulogenesis, MARK2 promoted the formation of laterallumen between neighbouring cells in CagA-expressingcells (Fig. 6, panel CagA + MARK2). These lumina likelyrepresent intermediates in apical lumen formation thathave been observed during the course of tubulogenesis in
control cells and in MARK2 overexpressing cells (Ojakianet al., 1997; Cohen and Musch, 2003).
Discussion
Proteomic approaches offer powerful screening methodsto compare the abundance of a large number of cellular
Fig. 5. Recombinant Par1b rescues CagA-mediated polarity defects in MDCK cells. MARK2-MDCK cells were cultured in the presence (‘GFPcontrol’ and ‘GFP-CagA′ panels) or absence (‘GFP-CagA + Par1b’ panels) of doxycycline to repress or induce recombinant Par1b respectively.Cells were transfected with GFP or CagA-GFP constructs and analysed in Ca-switch assays.A. Confocal x–z and x–y views of monolayers labelled with antibodies to the apical marker gp135 (red) and the tight junction marker ZO1(blue). GFP-fluorescence is shown in green. ‘max’ indicates a maximal x–y projection.B. Confocal x–y sections through the cell centre of monolayers extracted with Tx100 before methanol fixation and labelled for tubulin.
Fig. 6. CagA inhibits MDCK tubulogenesis in collagen overlay assays. Par1b-MDCK cells were cultured and transfected as above andanalysed in collagen overlay assays. Shown are confocal x–z views of representative fields of the collagen sandwich labelled for the apicalmarker gp135 (red) and with DAPI for the nucleus.
iTraq analysis of Helicobacter pylori infected cells 787
© 2007 The AuthorsJournal compilation © 2007 Blackwell Publishing Ltd, Cellular Microbiology, 10, 781–794

proteins using different stimulatory conditions and further-more allow limiting the analysis to specific cellularfractions of interest. Investigating redistribution andmodification of proteins during cell stimulation can oftenprovide more insights into protein function than the soledetermination of changing protein expression levels.Rapid improvements of mass spectrometry techniquesenable us to study the cellular proteome in steadilyincreasing detail. Several techniques have become avail-able to quantitatively compare proteomes includingFluorescence 2-D Difference Gel Electrophoresis (DIGE)(Marouga et al., 2005), SILAC (Ong et al., 2002), iCAT(von Haller et al., 2003) and more recently iTRAQ (Zieske,2006). Each of these complementary methods has advan-tages and allows the identification of novel sets of proteinswhen compared with the others, but iTRAQ probablyallows studying proteomes most broadly.
In recent years, it has been recognized that a variety ofpathogens and toxins interact with microdomains in theplasma membrane known as DRMs or lipid rafts (Maneset al., 2003). Lipid rafts are membrane microdomains thatare enriched in cholesterol, sphingolipids and glyco-sylphosphatidylinositol GPI-anchored proteins (Keller andSimons, 1997). These membrane microdomains havebeen shown to mediate internalization of bacteria, virusesand parasites into the host cell (Manes et al., 2003;Lafont et al., 2004). Recently, it was reported that Hpassociates with and affects raft-like membrane domains(Wunder et al., 2006). Others have demonstrated that themajor Hp cytotoxins VacA (Schraw et al., 2002;Nakayama et al., 2006) and CagA (Asahi et al., 2003)become partially Triton X-100 insoluble during the infec-tion process. Thus, we assumed that a proteomic study ofthe DRMs would potentially identify CagA or VacA inter-acting host cell proteins that should logically be part of thedetergent-resistant complexes as well. Additionally, weaimed to identify membrane associated signalling compo-nents that are recruited to or excluded from the DRMswithout directly interacting with the bacterial cytotoxins.Therefore, the aim in this study was not to identify newcomponents of lipid rafts, but rather use detergent resis-tance simply as a method to limit the number of totalmembrane proteins analysed, while enriching for proteinswith possible functional importance for CagA and VacA-mediated signalling events.
Results from iTRAQ analysis presented in this studyrevealed that the serine/threonine kinases MARK2 andMARK3 were strongly enriched in the DRMs of AGS cellsinfected with Hp wild-type, but were not detectable inDRMs isolated from uninfected cells. Since CagA waspreviously reported to disrupt cellular junctions (Amievaet al., 2003) and since MARK2 is an important regulator ofepithelial cell polarity we investigated a possible connec-tion between the CagA-mediated effects and MARK2.
MARK2 is a family member of four Par-1/MARK kinases(MARK1–4) that are conserved from yeast to human(Drewes, 2004; Tassan and Le Goff, 2004). Although allMARK-kinases are able to phosphorylate microcotubule-associated proteins in vitro, only MARK 2 has been impli-cated directly in the development of polarized mammalianepithelial cells. Inhibition of MARK2 function disruptedseveral hallmarks of epithelial differentiation, notably cell-cell adhesion, maintenance of a well organized mono-layer, cell shape changes that lead to cell columnarizationand compaction, development of the luminal domain andepithelial-specific MT- and actin-organization (Bohmet al., 1997; Cohen et al., 2004).
When we studied the distribution of MARK2 in wild-typeinfected AGS cells using immunofluorescence microscopyand membrane fractionation we found that MARK2 redis-tributed from the cytosol to the membrane, where it local-ized underneath the attaching bacteria. Colocalization wasobserved only if cells were infected with Hp strains thatwere able to translocate CagA into the host cell. CagA andMARK2 furthermore co-immunoprecipitated with eachother indicating that both molecules were part of the samecomplex and this effect was independent of CagA tyrosine-phosphorylation. CagA/MARK2 interaction was not limitedto the DRMs and in fact, the majority of the interactionappeared to be DRM-independent. At present we do notknow, whether CagA dependent recruitment of MARK2 tothe DRMs is crucial for interference with MARK2 signallingpathways or rather reflects a partial association of CagAand/or MARK2 with other detergent-insoluble molecules.In polarized MDCK cells, CagA localized to the lateralmembrane (Fig. 5A) where MARK2 is also enriched.Intriguingly, CagA expression in polarizing MDCK cellscaused polarity defects reminiscent of those previouslyreported for MARK2 inhibition. These findings are in agree-ment with earlier observations on CagA toxicity in epithelialcells (Amieva et al., 2003; Bagnoli et al., 2005). Impor-tantly, we determined that overexpression of MARK2antagonizes CagA-induced polarization defects. Together,our data support a scenario in which Hp infection causesCagA to interact with and inhibit MARK2 (and potentiallyMARK3) at the lateral cell cortex. Inhibition of MARK2activity is likely to contribute to the disease phenotype ofinfected gastric epithelia. CagA is only the fourth proteinimplicated in MARK2 distribution and activity. The kinasesLKB1/Par4 and MAKK/Tao-1 have been characterized asupstream activators (Spicer et al., 2003; Timm et al., 2003;Lizcano et al., 2004) while aPKC-mediated phosphoryla-tion leads to inhibition of MARK2 kinase activity andits dissociation from the membrane (Hurov et al., 2004)Suzuki (2004). The mechanism by which CagA inhibitsMARK2 remains to be elucidated.
While this manuscript was in preparation, Saadat et al.published findings similar to ours (Saadat et al., 2007).
788 Z. Zeaiter et al.
© 2007 The AuthorsJournal compilation © 2007 Blackwell Publishing Ltd, Cellular Microbiology, 10, 781–794

The authors identified MARK2 as a CagA interactionpartner in co-immunoprecitation experiments and alsoreported rescue of CagA-induced tight junction defectsand MDCK monolayer organization by MARK2overexpression. They furthermore demonstrated an inhi-bition of MARK2 kinase-activity by CagA in vitro. Thus, ourstudy confirms these important insights into the molecularmechanisms of Hp pathology. In addition we haveextended the findings by Saadat (Saadat et al., 2007) bydemonstrating that the MARK2–CagA interaction not onlydisrupts junctional integrity of MDCK cells, but also pre-vents lumen formation and tubulogenesis, which areimportant hallmarks of epithelial differentiation. Further-more, we determined that CagA recruits MARK2 intothe DRM fraction, suggesting that the kinase becomesassociated with the lipid microdomains that function asplatforms for signalling complexes that mediate Hppathogenesis. It is tempting to speculate that in addition tobeing target of CagA-signalling the kinase itself partici-pates in downstream signalling events mediated by theCagA complex.
Finally, our proteomic analysis of DRMs not only iden-tified MARK2, but additional proteins that may be impor-tant factors for Hp-mediated pathogenesis. Although thereis some evidence that MARK3 may act redundantly withMARK2, MARK3 has mainly been described in connec-tion with unrelated signalling events including the activa-tion of the Ras/MAPK/ERK pathway (Muller et al., 2001)and cell cycle control via phosphorylation of cdc25c(Sebastian et al., 1993). Since CagA is a known activatorof ERK1/2 activation and cell proliferation (Keates et al.,1999; Meyer-ter-Vehn et al., 2000) investigations into thepossible involvement of CagA/MARK3 in these pathwaysare currently ongoing in our laboratory.
Another protein that was enriched in the DRMs followinginfection was the nuclease sensitive element-bindingprotein 1 (YB-1), which was recognized as an importantprognostic factor in ovarian carcinoma. This protein wasidentified as an effector of transcription and also as atranslational repressor (Evdokimova et al., 2001; 2006;Basaki et al., 2007). Furthermore, YB-1 expression isincreased in melanoma cells in vitro and in vivo, andfollowing translocation into the nucleus stimulates prolif-eration, migration and tumour invasion (Schittek et al.,2007). Since Hp induces related signalling pathways, YB-1may be an important regulator of these events. Whetherincrease of YB-1 in the DRMs reflects either increasedexpression levels or alternatively reflects nuclear import orassociation with the capped mRNA remains to be tested.As well it would be interesting to see, whether YB-1 expres-sion is induced in gastric adenocarcinomas.
Urokinase plasminogen activator receptor (uPAR/CD87) is a GPI-anchored protein, whose activity wasaccelerated following partial localization to the DRMs
(Cunningham et al., 2003; Sitrin et al., 2004). uPARexpression, which is known to closely correlate withgastric cancer invasion and involved in changes in cellmorphology, migration and adhesion, was shown to beupregulated by Hp previously (Kim et al., 2005) (Kimet al., 2007). The Hp-induced increase of uPAR in theDRMs may account for these changes.
Studies using recombinant S100A7 indicated thatS100A7 adheres to and reduces E. coli survival (Lee andEckert, 2007). Additionally, S100A7 expression wasupregulated in human epithelial skin tumours (Moubayedet al., 2007). The function of the S100A7-like protein is stillunknown but we may speculate that the S100A7-likeprotein, which showed increase in detergent-resistanceduring infection may have similar functions and play a rolein the host response to Hp infection.
In conclusion, proteomic analysis of DRMs proofed tobe a successful tool in the search for new Hp biomarkersand pathogen-associated signalling pathways. Deter-mination of proteins differentially recruited to certainmembrane domains during infection, in addition to theidentification of the responsible bacterial factor(s), willwithout doubt provide new clues in the dissection of Hppathogenesis mechanisms.
Experimental procedures
Bacterial strains and growth conditions
Hp strain G27 and its isogenic mutants DcagA, DHP527, andDrepEPISA were used in this study (Covacci et al., 1993; Steinet al., 2002). Strain were cultured on brucella broth agar platessupplemented with selective antibiotics and 10% fetal bovineserum (FBS) and incubated at 37°C for 48 h in an anaerobic jarcontaining 5% CO2, 5% H2 and 90% N2. Liquid cultures weregrown in brucella broth supplemented with 10% FBS in ananaerobic jar containing a Campygen gas pack (5% O2, 10% CO2
and 85% N2) (Oxoid). Cultures were incubated with 165 r.p.m.rotation at 37°C overnight.
Cell culture
The human gastric adenocarcinoma cell line AGS was obtainedfrom ATCC (ATCC# CRL-1739). The canine kidney tubular epi-thelial line MDCK II was kindly provided by Dr W. James Nelson(Stanford University). MDCK II cells were cultured in Dulbecco’smodified Eagle’s medium (DMEM, Invitrogen) and AGS cellswere cultured in RPMI 1640 medium (Invitrogen). Both mediawere supplemented with 10% fetal bovine serum (FBS,Invitrogen). Cell lines were maintained at 37°C in 5% CO2 atmo-sphere and 95% humidity.
MDCK-tet-off cells stably expressing myc-tagged caninePAR1b were used as described previously (Cohen et al., 2004).Recombinant PAR1b is expressed 10-fold above endogenouslevels in the absence of doxycycline. For Ca2+-switch experi-ments, cells were cultured for 24 h in DMEM at subconfluency,transfected with recombinant cDNAs and plated at confluency in
iTraq analysis of Helicobacter pylori infected cells 789
© 2007 The AuthorsJournal compilation © 2007 Blackwell Publishing Ltd, Cellular Microbiology, 10, 781–794

S-MEM (low Ca2+ medium, Invitrogen) with 10% FBS dialysedagainst PBS and switched back to DMEM (1.8 mM Ca2+) after8 h. Cells were analysed 24 h after the Ca-switch.
Collagen overlay assay
Collagen I overlay of subconfluent monolayers grown oncollagen-coated coverslips was done as described (Cohen andMusch, 2003) with the following modification: Monolayers wereoverlaid with collagen 8 h after transfection and plating oncollagen-coated coverslips and analysed 24 h after overlay. Theshortening of the assay time was necessary because CagA-expression deceased sharply 48 h after transfection.
Plasmids and transfection
P-CagA-GFP (pEGFP-C3-1216) was previously described(Bagnoli et al., 2005). pEGFP-HA-MARK2 was a generous giftfrom Dr D.R. Alessi (University of Dundee).
AGS cells were tranfected with Lipofectamin 2000 (Invitrogen).MDCK cells were transfected using AMAXA nucleofection tech-nology (Amaxa) (Cohen et al., 2004). Ten micrograms of cDNAand 4 ¥ 106 cells were used per transfection.
Antibodies and Immunoblotting
Anti-CD71 (3B82A1) (Santa Cruz), anti-Lyn (H-6) (Santa Crusz),anti-Flotillin 1 (BD Transduction Laboratories), anti-Par1b(Upstate), anti-CagA (Austral Biological), anti-gp135 (mouse,clone 3F21D8, provided by G. Ojakian, SUNY DownstateMedical Center, Brooklyn, NY), anti-ZO1 (rat, Chemicon), ratantitubulin antibody (clone YL1/2, Abcam), anti-GST (Oncogene)and Alexa-Fluor 488, 594 or 305 conjugated secondary antibod-ies (Invitrogen) were used for immunostaining. Chicken anti-Helicobacter pylori serum was a generous gift from Dr M.R.Amieva (Stanford University). For immunoblotting proteins wereseparated by SDS-PAGE and transferred to PVDF membranes.PVDF membranes were blocked for 1 h with protein-free T20blocking buffer (Pierce), washed, and incubated with primaryantibodies overnight at 4°C. Horseradish peroxidase-conjugatedsecondary antibodies (Amersham) were used together with theSuper Sgnal kit (Pierce) and Hyperfilm-ECL (Amersham Bio-sciences) to visualize proteins.
Immunofluorescent and confocal microscopy
Tissue culture cells were seeded on sterile 12 mm glass cover-slips in a 24-well plate at a density of 3 ¥ 105 cells ml-1. After 18 hconfluent monolayers were washed three times with infectionmedium (RPMI plus 5% FBS, pH 6.5). Hp was infected at amultiplicity of infection of 100:1 in all assays except immunofluo-rescence microscopy (10:1). Following infection cells werewashed briefly in ice-cold wash buffer (PBS plus 1.5% FCS) andfixed in 4% (v/v) paraformaldehyde in PBS for 1 h on ice. Thencells were washed and permeabelized with 0.2% NP-40 in PBSfor 10 min at room temperature. Coverslips were stained with theappropriate primary antibodies diluted in wash buffer for 1 h atroom temperature, washed and stained with Alexa-Fluor conju-gated secondary antibodies for 1 h at room temperature. All
secondary antibodies were used in a 1/500 dilution. Coverslipswere mounted on glass slides using ProLong® Gold antifadereagent (invitrogen). All immunofluorescence microscopy wasperformed on a Leica DMI6000 B-inverted fluorescence micro-scope (Leica Microsystems, Wetzlar, Germany). Fluorescentdyes were detected with an L5 or TX2 filter set. Images wereprocessed in OpenLab 5.0.2 for contrast and Adobe Photoshopfor formatting. Confocal microscopy was performed with a modelSP2 (Leica) using a 63 ¥ oil objective. Presented are individualconfocal x–y and x–z sections. Images were processed withAdobe Photoshop.
Immunoprecipitation
9 ¥ 106 AGS cells were infected for 3 h with Hp strains G27,G27DcagA and DrepEPISA or left uninfected. Cells were washedthree times with 5 ml ice-cold TBS buffer and scraped in 1 ml ofTBS. Then cells were precipitated by centrifugation (7000 g) andresuspended in 600 ml of lysis buffer [50 mM Tris/CL pH 7.5, 1 mMEGTA, 1 mM EDTA, 1%NP40, 0.27 M sucrose, 1 mM DTT, 1 mMPMSF and 1 mM Na3VO4, 1 mM NaF, plus a cocktail of proteaseinhibitors (complete, EDTA-free, Roche)]. After solubilization onice for 1 h, the insoluble fraction was precipitated (16 100 g,15 min) and the lysates transferred to new tubes.Alternatively, 109
infected cells were used to purify DRMs as described below. TheDRMs were lysed in TBS/Triton X-100 lysis buffer (describedbelow) supplemented with 1% NP-40 for 1 h at 12°C. Total celllysates and alternatively lysates of the purified DRMs were treatedas follows. To preclear the lysate 50 ml of Protein G magneticbeads (Invitrogen) were added to the lysates and the samplesrotated for 1 h at 4°C. Then the magnetic beads were removed bya magnet (Promega) and the lysate transferred to a new tube andincubated for 1 h on ice with 5 mg of MARK2 or CagA antibody.Then 50 ml Protein G magnetic beads were added and the samplerotated at 4°C overnight. The beads were then washed three timesin 1 ml of lysis buffer, resuspended in 60 ml of 2 ¥ Laemmli samplebuffer (5% b-merceptoethanol) and boiled for 10 min
Cell fractionation
AGS cells were grown, infected, washed, scraped and precipitatedas described above. Approximately 9 ¥ 106 AGS cells were resus-pended in 100 ml of saponin buffer [50 mM Tris/CL, pH 7.5, 1 mMPMSF, 1 mM Na3VO4, 1 mM NaF, plus a cocktail of proteaseinhibitors (complete, EDTA-free, Roche), 1% (w/v) saponin] andincubated on ice for 10 min. Samples were pelleted (16 100 g for5 min) and the supernatant transferred to a new tube (cytosolfractions). The cells were resuspended a second time with 800 mlof saponin buffer and the pelleted again. The supernatants werediscarded and 100 ml of lysis buffer [saponin buffer plus 1% TritonX-100 (v/v)] was added to solubelize the cells. Lysates wereincubated on ice for 10 min, pelleted to remove the Triton X-100insoluble fraction and supernatants were transferred to new tubes(membrane fraction). Twenty-five microlitres of 5 ¥ Laemmlisample buffer (5% b-merceptoethanol) were added to each of thecytosol and membrane fractions and samples were boiled for10 min. For total cell lysates, 9 ¥ 106 AGS cells were washed,scraped and precipitated as described. Precipitates were resus-pended in 400 ml of PBS, 100 ml of 5 ¥ Laemmli sample buffer wasadded, and cells lysates were boiled for 10 min.
790 Z. Zeaiter et al.
© 2007 The AuthorsJournal compilation © 2007 Blackwell Publishing Ltd, Cellular Microbiology, 10, 781–794

Real-time reverse-transcription polymerase chainreaction (RT-PCR)
Total RNA was isolated from AGS cells using the RNeasy Minikit (Qiagen) following the manufacturer’s instructions. RNAwas tested for quality, quantity and genomic DNA contaminationusing the Agilent 2100 bioanalyser. Two mg of total RNA werereverse transcribed using Oligo-dt (invitrogen) and Omniscriptreverse transcription kit (Qiagen) following the manufacturer’sinstructions. The resulting cDNA was stored at -80°C. MARK2and GAPDH mRNA levels from different samples were deter-mined using real-time PCR with an ABI Prism 7900HT SequenceDetection System (Applied Biosystems). Serial dilutions of thecDNA were denatured for 3 min at 95°C, and then subjected to 40cycles of denaturation for 15 s at 95°C, annealing for 15 s at 55°Cand extension for 1 min at 72°C, followed by denaturation for 15 sat 95°C. The gene-specific primers used were purchased fromSuperArray (Frederick, USA). MARK2 levels in each samplewere normalized to GAPDH expression, and the relative changein mRNA level was expressed as fold induction compared withuntreated AGS cells using the -DD cycle threshold method.
AGS infection and DRM preparation
AGS cells were washed twice using RPMI medium at 37°C andincubated for 30 min at 37°C in RPMI medium containing 5%FBS, pH 6.5. One part of AGS cells (6–8 ¥ 107) was infected withHp strain G27 for 4 h at 37°C. The humming bird phenotype wasverified by microscopy. AGS cells were washed 5 times in ice-cold Tris-buffered saline (TBS: 25 mM Tris/HCl, 140 mM NaCl,pH 7.5), scrapped on ice in TBS, and pelleted at 250 g for 10 minat 4°C. The pellet was treated on ice with 3 ml of lysis bufferconsisting of [1% Triton X-100 (w/v in TBS, Sigma) 1 mM EDTA,1 mM PMSF and 1 mM Na3VO4, 1 mM NaF, plus a cocktail ofprotease inhibitors (complete, EDTA-free, Roche)], homogenizedby 10 strokes of a loose fit Dounce Homogenizer and incubatedon ice for 1 h. The lysate was passed 10 times through a22-gauge needle and adjusted to 40% (w/v) sucrose by mixing itwith an equal volume of 80% sucrose in TBS. This sample wasadded on top of 1 ml of 80% sucrose in a 13 ml ultracentrifugetube, overlaid with 6 ml of 30% sucrose and topped by 5%sucrose. Samples were centrifuged at 39 000 r.p.m. for 18 h at4°C using a SW40Ti rotor (Beckman). Fractions of 1 ml werecollected from the top of the gradient and immunoblot analysiswas performed to identify and confirm the fractions containingDRMs. Gradient fractions containing DRMs (4 and 5) werepooled and diluted 3 times with TBS (to remove sucrose) andprecipitated at 59 000 r.p.m. for 3 h at 4°C using an Ti70 rotor(Beckman). Pellets from 4 preparations were dissolved in 100 mlof iTRAQ buffer [50 mM TEAB (triethylammonium bicarbonate),4.75 M Urea (1/2 Saturated) and 0.2% SDS]. Protein concentra-tions were estimated by the BCA assay (Pierce) and 100 mg wereused in further steps. All the following steps were carried out atthe University of Victoria Genome BC Proteomics Centre (Victo-ria, BC, Canada).
Sample preparation for mass spectrometry
Reducing of proteins, blocking the cysteine and digesting withtrypsin were performed using standard protocols. iTRAQ labelling
was performed using iTRAQ ReagentsMultiplex kit accordingto the manufacturer’s instructions (Applied Biosystems, FosterCity, CA).
Strong cation exchange (SCX) HPLC and LC-MS/MS
A Vision Workstation (AB, Foster City, USA) was equipped witha Polysulfoethyl A (Poly LC, Columbia, MD) 100 mm ¥ 4.6 mm,5 mm, 300 A SCX column. Buffer A was 10 mM KPO4 (pH 2.7),25% ACN. Buffer B was 10 mM KH2PO4, 25% ACN, 0.5 M KCl.The flow rate was set to 0.5 ml min-1. Samples were brought upto 2 ml with buffer A and injected onto the column. The columnwas allowed to equilibrate for 20 min in buffer A before a gra-dient was applied; 0–35% B in 30 min. Fractions were collectedevery minute after injection. The collected fractions were thenreduced in volume in a Speed-Vac (Savant Instruments, Hol-brook, NY) and transferred to autosampler vials (LC Packings,Amsterdam).
LC-MS/MS analysis was performed using an integrated Famosautosampler, SwitchosII switching pump and UltiMate micropump (LC Packings, Amsterdam) system with an HybridQuadrupole-TOF LC/MS/MS Mass Spectrometer (QStar Pulsar i)equipped with a nano-electrospray ionization source (Proxeon,Odense, Denmark) and fitted with a 10 mm fused-silica emitter tip(New Objective, Woburn, MA). Chromatographic separation wasachieved on a 75 mm ¥ 15 cm C18 PepMap Nano LC column(3 mm, 100 Å, LC Packings, Amsterdam) and a 300 mm ¥ 5 mmC18 PepMap guard column (5 mm, 100 Å, LC Packings, Amster-dam) was in place before switching inline with the analyticalcolumn and the MS. The mobile phase (solvent A) consisted ofwater/acetonitrile [98:2 (v/v)] with 0.05% formic acid for sampleinjection and equilibration on the guard column at a flow rate of100 ml min-1. A linear gradient was created upon switching thetrapping column inline by mixing with solvent B which consistedof acetonitrile/water [98:2 (v/v)] with 0.05% formic acid and theflow rate was reduced to 200 nl min-1 for high-resolution chroma-tography and introduction into the mass spectrometer.
Samples were brought up to 20 ml with 5% ACN and 3% FAand transferred to autosampler vials (LC Packings, Amsterdam).Ten microlitres of sample were injected in 95% solvent A andallowed to equilibrate on the trapping column for 10 min to washaway any contaminants. Upon switching inline with the MS, alinear gradient from 95% to 40% solvent A developed for 40 minand in the following 5 min the composition of mobile phase wasincreased to 20% A before decreasing to 95% A for a 15 minuteequilibration before the next sample injection. MS data wasacquired automatically using Analyst QS 1.0 software ServicePack 8 (ABI MDS SCIEX, Concord, Canada). An informationdependent acquisition method consisting of a 1 second TOFMSsurvey scan of mass range 400–1200 amu and two 2.5 secondproduct ion scans of mass range 100–1500 amu. The two mostintense peaks over 20 counts, with charge state 2–5 wereselected for fragmentation and a 6 amu window was used toprevent the peaks from the same isotopic cluster from beingfragmented again. Once an ion was selected for MS/MS frag-mentation it was put on an exclude list for 180 s. Curtain gas wasset at 23, nitrogen was used as the collision gas and the ioniza-tion tip voltage used was 2700 V. If the observed A215 wasgreater than 0.1 for any fraction collected during the SCX a 2.5 hgradient (95–50% solvent A) was used to compensate for thehigher peptide concentration in that fraction.
iTraq analysis of Helicobacter pylori infected cells 791
© 2007 The AuthorsJournal compilation © 2007 Blackwell Publishing Ltd, Cellular Microbiology, 10, 781–794

Data analysis
Pro Quant software v1.1 (Applied Biosystems MDS SCIEX) wasused to identify and quantify proteins resolved by LC/MS/MS.Briefly, Pro Quant calculated peak areas and iTRAQ ratios foriTRAQ-labelled peptides using a threshold iTRAQ cluster valueof 40 counts. iTRAQ peptide data were used to search the CeleraDiscovery System (CDS; v. Rev C) FASTA Files (Applied Biosys-tems), the International Protein Index (v. 3.04) (IPI) (http://www.ebi.ac.uk/IPI/IPIhuman.html) and the Swiss-Prot database(v. March 05; ftp://ftp.ncbi.nih.gov/blast/db/FASTA/). Pro Quant-generated results were analysed using the Protein Pilot software(Ver. 1.0) (AB, Foster City, USA).
Acknowledgements
We thank Dr W. James Nelson (Stanford University) for provid-ing us with MDCK II cells, Dr Manuel R. Amieva (Stanford Uni-versity) for chicken anti-Helicobacter pylori serum, and Dr G.Ojakian (SUNY Downstate Medical Center) for anti-gp135 anti-body. We would also like to thank Derek Smith for his help withthe iTRAQ analysis. This work was supported by a CanadianInstitutes of Health Research (CIHR) operating Grant (MOP-62779) and an Alberta Heritage Foundation for MedicalResearch (AHFMR) establishment Grant (200200583) to M.S.We are also thankful for the support received by the CanadaFoundation of Innovation (CFI) and the Alberta Science andResearch Investments Program (ASRIP). M.S. is an AHFMRResearch Scholar.
References
Amieva, M.R., Vogelmann, R., Covacci, A., Tompkins, L.S.,Nelson, W.J., and Falkow, S. (2003) Disruption of the epi-thelial apical-junctional complex by Helicobacter pyloriCagA. Science 300: 1430–1434.
Asahi, M., Azuma, T., Ito, S., Ito, Y., Suto, H., Nagai, Y., et al.(2000) Helicobacter pylori CagA protein can be tyrosinephosphorylated in gastric epithelial cells. J Exp Med 191:593–602.
Asahi, M., Tanaka, Y., Izumi, T., Ito, Y., Naiki, H., Kersulyte,D., et al. (2003) Helicobacter pylori CagA containing ITAM-like sequences localized to lipid rafts negatively regulatesVacA-induced signaling in vivo. Helicobacter 8: 1–14.
Bacallao, R., Antony, C., Dotti, C., Karsenti, E., Stelzer, E.H.,and Simons, K. (1989) The subcellular organization ofMadin-Darby canine kidney cells during the formation of apolarized epithelium. J Cell Biol 109: 2817–2832.
Backert, S., Ziska, E., Brinkmann, V., Zimny-Arndt, U., Fau-connier, A., Jungblut, P.R., et al. (2000) Translocation ofthe Helicobacter pylori CagA protein in gastric epithelialcells by a type IV secretion apparatus. Cell Microbiol 2:155–164.
Bagnoli, F., Buti, L., Tompkins, L., Covacci, A., and Amieva,M.R. (2005) Helicobacter pylori CagA induces a transitionfrom polarized to invasive phenotypes in MDCK cells. ProcNatl Acad Sci USA 102: 16339–16344.
Basaki, Y., Hosoi, F., Oda, Y., Fotovati, A., Maruyama, Y.,Oie, S., et al. (2007) Akt-dependent nuclear localization ofY-box-binding protein 1 in acquisition of malignant charac-
teristics by human ovarian cancer cells. Oncogene 26:2736–2746.
Benton, R., and St Johnston, D. (2003) Drosophila PAR-1and 14-3-3 inhibit Bazooka/PAR-3 to establish complemen-tary cortical domains in polarized cells. Cell 115: 691–704.
Bohm, H., Brinkmann, V., Drab, M., Henske, A., andKurzchalia, T.V. (1997) Mammalian homologues of C.elegans PAR-1 are asymmetrically localized in epithelialcells and may influence their polarity. Curr Biol 7: 603–606.
Brown, D.A., and London, E. (1998) Functions of lipid rafts inbiological membranes. Annu Rev Cell Dev Biol 14: 111–136.
Churin, Y., Kardalinou, E., Meyer, T.F., and Naumann, M.(2001) Pathogenicity island-dependent activation of RhoGTPases Rac1 and Cdc42 in Helicobacter pylori infection.Mol Microbiol 40: 815–823.
Churin, Y., Al-Ghoul, L., Kepp, O., Meyer, T.F., Birchmeier,W., and Naumann, M. (2003) Helicobacter pylori CagAprotein targets the c-Met receptor and enhances the moto-genic response. J Cell Biol 161: 249–255.
Cohen, D., and Musch, A. (2003) Apical surface formation inMDCK cells: regulation by the serine/threonine kinaseEMK1. Methods 30: 269–276.
Cohen, D., Brennwald, P.J., Rodriguez-Boulan, E., andMusch, A. (2004) Mammalian PAR-1 determines epitheliallumen polarity by organizing the microtubule cytoskeleton.J Cell Biol 164: 717–727.
Covacci, A., Censini, S., Bugnoli, M., Petracca, R., Burroni,D., Macchia, G., et al. (1993) Molecular characterization ofthe 128-kDa immunodominant antigen of Helicobacterpylori associated with cytotoxicity and duodenal ulcer. ProcNatl Acad Sci USA 90: 5791–5795.
Cox, D.N., Lu, B., Sun, T.Q., Williams, L.T., and Jan, Y.N.(2001) Drosophila par-1 is required for oocyte differentia-tion and microtubule organization. Curr Biol 11: 75–87.
Cunningham, O., Andolfo, A., Santovito, M.L., Iuzzolino, L.,Blasi, F., and Sidenius, N. (2003) Dimerization controls thelipid raft partitioning of uPAR/CD87 and regulates its bio-logical functions. Embo J 22: 5994–6003.
Doerflinger, H., Benton, R., Shulman, J.M., and St Johnston,D. (2003) The role of PAR-1 in regulating the polarisedmicrotubule cytoskeleton in the Drosophila follicularepithelium. Development 130: 3965–3975.
Drewes, G. (2004) MARKing tau for tangles and toxicity.Trends Biochem Sci 29: 548–555.
Drewes, G., Ebneth, A., Preuss, U., Mandelkow, E.M., andMandelkow, E. (1997) MARK, a novel family of proteinkinases that phosphorylate microtubule-associated pro-teins and trigger microtubule disruption. Cell 89: 297–308.
Elbert, M., Cohen, D., and Musch, A. (2006) PAR1b promotescell-cell adhesion and inhibits dishevelled-mediated trans-formation of Madin-Darby canine kidney cells. Mol Biol Cell17: 3345–3355.
Evdokimova, V., Ruzanov, P., Imataka, H., Raught, B., Svitkin,Y., Ovchinnikov, L.P., and Sonenberg, N. (2001) The majormRNA-associated protein YB-1 is a potent 5′-cap-dependent mRNA stabilizer. EMBO J 20: 5491–5502.
Evdokimova, V., Ovchinnikov, L.P., and Sorensen, P.H.(2006) Y-box binding protein 1: providing a new angle ontranslational regulation. Cell Cycle 5: 1143–1147.
Gonzalez-Mariscal, L., Contreras, R.G., Bolivar, J.J., Ponce,A., Chavez De Ramirez, B., and Cereijido, M. (1990) Role
792 Z. Zeaiter et al.
© 2007 The AuthorsJournal compilation © 2007 Blackwell Publishing Ltd, Cellular Microbiology, 10, 781–794

of calcium in tight junction formation between epithelialcells. Am J Physiol 259: C978–C986.
Hall, H.G., Farson, D.A., and Bissell, M.J. (1982) Lumenformation by epithelial cell lines in response to collagenoverlay: a morphogenetic model in culture. Proc Natl AcadSci USA 79: 4672–4676.
von Haller, P.D., Yi, E., Donohoe, S., Vaughn, K., Keller, A.,Nesvizhskii, A.I., et al. (2003) The application of new soft-ware tools to quantitative protein profiling via isotope-coded affinity tag (ICAT) and tandem mass spectrometry. I.Statistically annotated datasets for peptide sequences andproteins identified via the application of ICAT and tandemmass spectrometry to proteins copurifying with T cell lipidrafts. Mol Cell Proteomics 2: 426–427.
Hancock, J.F. (2006) Lipid rafts: contentious only from sim-plistic standpoints. Nat Rev Mol Cell Biol 7: 456–462.
Higashi, H., Tsutsumi, R., Muto, S., Sugiyama, T., Azuma, T.,Asaka, M., and Hatakeyama, M. (2002) SHP-2 tyrosinephosphatase as an intracellular target of Helicobacterpylori CagA protein. Science 295: 683–686.
Hurov, J.B., Watkins, J.L., and Piwnica-Worms, H. (2004)Atypical PKC phosphorylates PAR-1 kinases to regulatelocalization and activity. Curr Biol 14: 736–741.
Jacobson, K., Mouritsen, O.G., and Anderson, R.G. (2007)Lipid rafts: at a crossroad between cell biology andphysics. Nat Cell Biol 9: 7–14.
Keates, S., Keates, A.C., Warny, M., Peek, R.M., Jr, Murray,P.G., and Kelly, C.P. (1999) Differential activation ofmitogen-activated protein kinases in AGS gastric epithelialcells by cag+ and cag-Helicobacter pylori. J Immunol 163:5552–5559.
Keller, P., and Simons, K. (1997) Post-Golgi biosynthetictrafficking. J Cell Sci 110 (Part 24): 3001–3009.
Kemphues, K.J., Priess, J.R., Morton, D.G., and Cheng, N.S.(1988) Identification of genes required for cytoplasmiclocalization in early C. elegans embryos. Cell 52: 311–320.
Kim, M.H., Yoo, H.S., Chang, H.J., Hong, M.H., Kim, H.D.,Chung, I.J., et al. (2005) Urokinase plasminogen activatorreceptor is upregulated by Helicobacter pylori in humangastric cancer AGS cells via ERK, JNK, and AP-1. BiochemBiophys Res Commun 333: 874–880.
Kim, M.H., Yoo, H.S., Kim, M.Y., Jang, H.J., Baek, M.K., Kim,H.R., et al. (2007) Helicobacter pylori stimulates urokinaseplasminogen activator receptor expression and cell inva-siveness through reactive oxygen species and NF-kappaBsignaling in human gastric carcinoma cells. Int J Mol Med19: 689–697.
Lafont, F., Abrami, L., and van der Goot, F.G. (2004) Bacterialsubversion of lipid rafts. Curr Opin Microbiol 7: 4–10.
Lafont, F., and van der Goot, F.G. (2005) Bacterial invasionvia lipid rafts. Cell Microbiol 7: 613–620.
Lai, Y.P., Yang, J.C., Lin, T.Z., Lin, J.T., and Wang, J.T.(2006) Helicobacter pylori infection and CagA proteintranslocation in human primary gastric epithelial cellculture. Helicobacter 11: 451–459.
Lee, K.C., and Eckert, R.L. (2007) S100A7 (Psoriasin) –mechanism of antibacterial action in wounds. J Invest Der-matol 127: 945–957.
Lizcano, J.M., Goransson, O., Toth, R., Deak, M., Morrice,N.A., Boudeau, J., et al. (2004) LKB1 is a master kinase
that activates 13 kinases of the AMPK subfamily, includingMARK/PAR-1. EMBO J 23: 833–843.
Macara, I.G. (2004) Parsing the polarity code. Nat Rev MolCell Biol 5: 220–231.
Manes, S., del Real, G., and Martinez, A.C. (2003) Patho-gens: raft hijackers. Nat Rev Immunol 3: 557–568.
Marouga, R., David, S., and Hawkins, E. (2005) The devel-opment of the DIGE system: 2D fluorescence differencegel analysis technology. Anal Bioanal Chem 382: 669–678.
Martin-Belmonte, F., Gassama, A., Datta, A., Yu, W.,Rescher, U., Gerke, V., and Mostov, K. (2007) PTEN-mediated apical segregation of phosphoinositides controlsepithelial morphogenesis through Cdc42. Cell 128: 383–397.
Meyer-ter-Vehn, T., Covacci, A., Kist, M., and Pahl, H.L.(2000) Helicobacter pylori activates mitogen-activatedprotein kinase cascades and induces expression of theproto-oncogenes c-fos and c-jun. J Biol Chem 275: 16064–16072.
Mimuro, H., Suzuki, T., Tanaka, J., Asahi, M., Haas, R., andSasakawa, C. (2002) Grb2 is a key mediator of helico-bacter pylori CagA protein activities. Mol Cell 10: 745–755.
Moubayed, N., Weichenthal, M., Harder, J., Wandel, E.,Sticherling, M., and Glaser, R. (2007) Psoriasin (S100A7)is significantly up-regulated in human epithelial skintumours. J Cancer Res Clin Oncol 133: 253–261.
Muller, J., Ory, S., Copeland, T., Piwnica-Worms, H., andMorrison, D.K. (2001) C-TAK1 regulates Ras signaling byphosphorylating the MAPK scaffold, KSR1. Mol Cell 8:983–993.
Murata-Kamiya, N., Kurashima, Y., Teishikata, Y., Yama-hashi, Y., Saito, Y., Higashi, H., et al. (2007) Helicobacterpylori CagA interacts with E-cadherin and deregulates thebeta-catenin signal that promotes intestinal transdifferen-tiation in gastric epithelial cells. Oncogene 26: 4617–4626.
Nakayama, M., Hisatsune, J., Yamasaki, E., Nishi, Y., Wada,A., Kurazono, H., et al. (2006) Clustering of Helicobacterpylori VacA in lipid rafts, mediated by its receptor, receptor-like protein tyrosine phosphatase beta, is required forintoxication in AZ-521 Cells. Infect Immun 74: 6571–6580.
O’Brien, L.E., Zegers, M.M., and Mostov, K.E. (2002)Opinion: Building epithelial architecture: insights fromthree-dimensional culture models. Nat Rev Mol Cell Biol 3:531–537.
Odenbreit, S., Gebert, B., Puls, J., Fischer, W., and Haas, R.(2001) Interaction of Helicobacter pylori with professionalphagocytes: role of the cag pathogenicity island and trans-location, phosphorylation and processing of CagA. CellMicrobiol 3: 21–31.
Ojakian, G.K., Nelson, W.J., and Beck, K.A. (1997) Mecha-nisms for de novo biogenesis of an apical membrane com-partment in groups of simple epithelial cells surrounded byextracellular matrix. J Cell Sci 110 (Part 22): 2781–2794.
Ong, S.E., Blagoev, B., Kratchmarova, I., Kristensen, D.B.,Steen, H., Pandey, A., and Mann, M. (2002) Stable isotopelabeling by amino acids in cell culture, SILAC, as a simpleand accurate approach to expression proteomics. Mol CellProteomics 1: 376–386.
Peek, R.M., Jr, and Blaser, M.J. (2002) Helicobacter pyloriand gastrointestinal tract adenocarcinomas. Nat RevCancer 2: 28–37.
iTraq analysis of Helicobacter pylori infected cells 793
© 2007 The AuthorsJournal compilation © 2007 Blackwell Publishing Ltd, Cellular Microbiology, 10, 781–794

Peek, R.M., Jr, Moss, S.F., Tham, K.T., Perez-Perez, G.I.,Wang, S., Miller, G.G., et al. (1997) Helicobacter pyloricagA+ strains and dissociation of gastric epithelial cell pro-liferation from apoptosis. J Natl Cancer Inst 89: 863–868.
Rosenberger, C.M., Brumell, J.H., and Finlay, B.B. (2000)Microbial pathogenesis: lipid rafts as pathogen portals.Curr Biol 10: R823–R825.
Saadat, I., Higashi, H., Obuse, C., Umeda, M., Murata-Kamiya, N., Saito, Y., et al. (2007) Helicobacter pyloriCagA targets PAR1/MARK kinase to disrupt epithelial cellpolarity. Nature 447: 330–333.
Schittek, B., Psenner, K., Sauer, B., Meier, F., Iftner, T., andGarbe, C. (2007) The increased expression of Y box-binding protein 1 in melanoma stimulates proliferation andtumor invasion, antagonizes apoptosis and enhanceschemoresistance. Int J Cancer 120: 2110–2118.
Schraw, W., Li, Y., McClain, M.S., van der Goot, F.G., andCover, T.L. (2002) Association of Helicobacter pylori vacu-olating toxin (VacA) with lipid rafts. J Biol Chem 277:34642–34650.
Sebastian, B., Kakizuka, A., and Hunter, T. (1993) Cdc25M2activation of cyclin-dependent kinases by dephosphoryla-tion of threonine-14 and tyrosine-15. Proc Natl Acad SciUSA 90: 3521–3524.
Segal, E.D., Cha, J., Lo, J., Falkow, S., and Tompkins, L.S.(1999) Altered states: involvement of phosphorylatedCagA in the induction of host cellular growth changes byHelicobacter pylori. Proc Natl Acad Sci USA 96: 14559–14564.
Simons, K., and Toomre, D. (2000) Lipid rafts and signaltransduction. Nat Rev Mol Cell Biol 1: 31–39.
Sitrin, R.G., Johnson, D.R., Pan, P.M., Harsh, D.M., Huang,J., Petty, H.R., and Blackwood, R.A. (2004) Lipid raft com-partmentalization of urokinase receptor signaling in humanneutrophils. Am J Respir Cell Mol Biol 30: 233–241.
Spicer, J., Rayter, S., Young, N., Elliott, R., Ashworth, A., andSmith, D. (2003) Regulation of the Wnt signalling compo-nent PAR1A by the Peutz–Jeghers syndrome kinase LKB1.Oncogene 22: 4752–4756.
Stein, M., Rappuoli, R., and Covacci, A. (2000) Tyrosinephosphorylation of the Helicobacter pylori CagA antigenafter cag-driven host cell translocation. Proc Natl Acad SciUSA 97: 1263–1268.
Stein, M., Bagnoli, F., Halenbeck, R., Rappuoli, R., Fantl,W.J., and Covacci, A. (2002) c-Src/Lyn kinases activateHelicobacter pylori CagA through tyrosine phosphorylationof the EPIYA motifs. Mol Microbiol 43: 971–980.
Suzuki, A., and Ohno, S. (2006) The PAR-aPKC system:lessons in polarity. J Cell Sci 119: 979–987.
Suzuki, A., Yamanaka, T., Hirose, T., Manabe, N., Mizuno,K., Shimizu, M., et al. (2001) Atypical protein kinase C isinvolved in the evolutionarily conserved par proteincomplex and plays a critical role in establishing epithelia-specific junctional structures. J Cell Biol 152: 1183–1196.
Suzuki, A., Hirata, M., Kamimura, K., Maniwa, R., Yamanaka,T., Mizuno, K., et al. (2004) aPKC acts upstream of PAR-1bin both the establishment and maintenance of mammalianepithelial polarity. Curr Biol 14: 1425–1435.
Suzuki, M., Mimuro, H., Suzuki, T., Park, M., Yamamoto, T.,and Sasakawa, C. (2005) Interaction of CagA with Crkplays an important role in Helicobacter pylori-induced lossof gastric epithelial cell adhesion. J Exp Med 202: 1235–1247.
Tammer, I., Brandt, S., Hartig, R., Konig, W., and Backert, S.(2007) Activation of Abl by Helicobacter pylori: a novelkinase for CagA and crucial mediator of host cell scattering.Gastroenterology 132: 1309–1319.
Tassan, J.P., and Le Goff, X. (2004) An overview of theKIN1/PAR-1/MARK kinase family. Biol Cell 96: 193–199.
Timm, T., Li, X.Y., Biernat, J., Jiao, J., Mandelkow, E., Vande-kerckhove, J., and Mandelkow, E.M. (2003) MARKK, aSte20-like kinase, activates the polarity-inducing kinaseMARK/PAR-1. EMBO J 22: 5090–5101.
Tummuru, M.K., Cover, T.L., and Blaser, M.J. (1993) Cloningand expression of a high-molecular-mass major antigen ofHelicobacter pylori: evidence of linkage to cytotoxinproduction. Infect Immun 61: 1799–1809.
Wunder, C., Churin, Y., Winau, F., Warnecke, D., Vieth, M.,Lindner, B., et al. (2006) Cholesterol glucosylation pro-motes immune evasion by Helicobacter pylori. Nat Med 12:1030–1038.
Zieske, L.R. (2006) A perspective on the use of iTRAQreagent technology for protein complex and profilingstudies. J Exp Bot 57: 1501–1508.
794 Z. Zeaiter et al.
© 2007 The AuthorsJournal compilation © 2007 Blackwell Publishing Ltd, Cellular Microbiology, 10, 781–794
Copyright © 2022 FDOKUMEN




















