
Arp2/3 Complex from Acanthamoeba Binds Profilin and Cross-links Actin Filaments
Upload
independentCategory
view
0download
0
A R T I C L E S
Amyloid-like filaments and water-filled nanotubes formedby SOD1 mutant proteins linked to familial ALSJennifer Stine Elam1, Alexander B Taylor1, Richard Strange2, Svetlana Antonyuk2, Peter A Doucette3, Jorge A Rodriguez3, S Samar Hasnain2, Lawrence J Hayward4, Joan Selverstone Valentine3, Todd O Yeates3
& P John Hart1
Mutations in the SOD1 gene cause the autosomal dominant, neurodegenerative disorder familial amyotrophic lateral sclerosis(FALS). In spinal cord neurons of human FALS patients and in transgenic mice expressing these mutant proteins, aggregatescontaining FALS SOD1 are observed. Accumulation of SOD1 aggregates is believed to interfere with axonal transport, proteindegradation and anti-apoptotic functions of the neuronal cellular machinery. Here we show that metal-deficient, pathogenicSOD1 mutant proteins crystallize in three different crystal forms, all of which reveal higher-order assemblies of aligned �-sheets.Amyloid-like filaments and water-filled nanotubes arise through extensive interactions between loop and �-barrel elements ofneighboring mutant SOD1 molecules. In all cases, non-native conformational changes permit a gain of interaction betweendimers that leads to higher-order arrays. Normal �-sheet–containing proteins avoid such self-association by preventing their edgestrands from making intermolecular interactions. Loss of this protection through conformational rearrangement in the metal-deficient enzyme could be a toxic property common to mutants of SOD1 linked to FALS.
NATURE STRUCTURAL BIOLOGY VOLUME 10 NUMBER 6 JUNE 2003 461
has remained undefined. Here we show that two members of a largerclass of pathogenic SOD1 mutants termed ‘metal-binding regionmutants’22 undergo conformational changes that facilitate their poly-merization, via extensive non-native protein–protein interactions,into higher order filamentous assemblies.
RESULTSMetal-deficient SOD1 and FALSTo better understand how mutations in SOD1 can alter the wild-typeprotein and potentially lead to aggregation, we determined and refinedthe X-ray crystal structures of the human pathogenic SOD1 mutantproteins S134N, apo H46R and zinc-bound H46R (Zn–H46R) to resolutions of 1.3, 2.5 and 2.15 Å, respectively (Table 1). Both muta-tions result in defective metal binding22–24, a feature postulated tounderlie the toxicity of many pathogenic SOD1 mutants14,23,25–27. TheS134N and H46R mutant proteins studied here are all metal deficientto varying degrees when they are isolated from their respective yeast orbaculovirus Sf21 expression systems (see Methods), and their low levels of metallation in these studies are likely representative of theirstates in vivo. The relevance of metal-deficient FALS SOD1 proteins todisease etiology is underscored by recent studies showing that theycause motor neuron disease. For example, transgenic mice over-expressing the FALS SOD1 double mutant H46R/H48Q, a protein thatis incapable of binding copper ion, show disease onset and progression
Amyotrophic lateral sclerosis (ALS), known also as Lou Gehrig’s dis-ease, is a neurodegenerative disorder characterized by the destructionof large motor neurons in the spinal cord and brain. The diseaseresults in progressive paralysis, usually culminating in death within2–5 years after the onset of symptoms1. Of all ALS cases, ∼ 10% arefamilial, and ∼ 20% of these familial ALS (FALS) cases are associatedwith dominantly inherited mutations in copper-zinc superoxide dismutase (SOD1)2,3, a 32-kDa homodimeric antioxidant enzyme4.SOD1-linked FALS was initially believed to result from oxidativedamage caused by diminished SOD1 activity2,5, but Sod1-null micedevelop normally and live to adulthood without developing motorneuron disease6. However, transgenic mice expressing human FALSSOD1 mutant proteins become paralyzed, despite possessing normal7,8 or elevated9,10 levels of SOD1 activity . Together, theseobservations indicate that pathogenic SOD1 molecules act throughthe gain of a cytotoxic property and not a loss of enzymatic function.The precise nature of this toxicity is unknown, with explanatoryhypotheses ranging from aberrant copper-mediated catalysis11–14 tomutant SOD1 misfolding and aggregation8,15,16. Recent reports sug-gest that aggregates, composed in part of FALS SOD1, play a role inpathogenesis either by sequestering heat-shock proteins and mole-cular chaperones17,18 or by interfering with the neuronal axonaltransport19,20 and protein degradation21 machineries. However, themolecular basis underlying the formation of these SOD1 aggregates
1Department of Biochemistry and the Center for Biomolecular Structure Analysis, The University of Texas, Health Science Center at San Antonio, 7703 Floyd CurlDrive, San Antonio, Texas 78229-3900, USA. 2Molecular Biophysics Group, Department of Synchrotron Radiation, CCLRC Daresbury Laboratory, Warrington,Cheshire, WA44AD, UK. 3Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, USA. 4Department of Neurology,University of Massachusetts Medical School, 55 Lake Avenue North, Worcester, Massachusetts 01655, USA. Correspondence should be addressed to P.J.H.([email protected]).
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
rest
ruct
ura
lbio
log
y

A R T I C L E S
profiles nearly identical to those of mice expressing other FALS SOD1proteins that have mutations remote from the metal-binding sites16.These mice demonstrate pathology in their motor neurons dominatedby fibrillar (thioflavin-S–positive) inclusions similar to those observedin mouse models that express other variants of human SOD1 protein16. Other transgenic mice that lack the copper chaperone forSOD1 (CCS), a protein that inserts copper specifically into nascentSOD1, also become paralyzed when they express a variety of differentFALS SOD1 proteins that are thus forced to be copper deficient. Thepathology observed in these mice also includes aggregates composedin part of SOD1 (ref. 28). Thus, the metal-deficient pathogenic SOD1proteins are not only relevant to FALS, they could provide key clues tohow the aggregation of pathogenic SOD1 variants might occur (see below).
Gain of interaction in pathogenic SOD1Although the eight-stranded Greek key β-barrel components of thesemetal-deficient, pathogenic S134N and H46R SOD1 structures arepreserved relative to the wild-type protein, they exhibit significant dis-order and conformational changes in their zinc and electrostatic loopelements (Fig. 1). In all three structures, the disorder in these loop
regions deprotects the edges of β-strands 5 and6, which together form a cleft located betweenthe two β-sheets of the SOD1 β-barrel in theseproteins. This deprotected depression serves asthe molecular interface for non-native SOD1protein–protein contacts. We refer to thesenew contacts as ‘gain-of-interaction’ (GOI)contacts to distinguish them from the natu-rally occurring interfaces between subunits ofthe native SOD1 dimer. When the GOI con-tacts are combined with the native dimericinteractions, the pathogenic SOD1 dimersassemble into either linear or helical filaments.
S134N and apo H46R linear amyloid-likefilamentsAmyloid-like filaments of pathogenic SOD1dimers are linear and occur in two differentcrystalline environments: one in the S134Northorhombic crystal form (S134N filament)and one in the apo H46R monoclinic crystalform (apo H46R filament 1). The two fila-ments are nearly identical, and their backboneatoms align with an r.m.s. deviation of 1.6 Å.The GOI interfaces are symmetric, and the∼ 180° symmetry axes they produce are parallelto the symmetry axes of the natural dimers(Fig. 2a). Residues 125–131 of the electrostaticloop adopt an extended, non-native confor-mation and participate in extensive hydrogenbonding and apolar interactions with thedeprotected surface depression on the β-barrelof a neighboring SOD1 dimer (Fig. 2b). TheseGOI interfaces are extensive, burying up to∼ 640 Å2 of solvent-accessible surface area perpolypeptide, which corresponds to approxi-mately the same amount of surface area buriedin the highly stable native SOD1 homodimericinterface (∼ 660 Å2) (Fig. 2a). A smallhydrophobic core is formed around the
pseudo dyad, with Leu42 and Leu126 from each subunit participating(Fig. 2c). The overall arrangement of SOD1 dimeric building blocks issimilar to the arrangement recently proposed for transthyretin (TTR)amyloid fibrils29, in which the β-sheets of the protein molecules lieparallel to the long axis of the fiber while their constituent β-strandsrun perpendicular to this axis. An additional parallel to SOD1 andFALS is that ∼ 73 distinct point mutations of TTR have been identifiedthat enhance the amyloidogenicity of the protein and cause anotherautosomal dominant neurodegenerative disease, familial amyloidpolyneuropathy (FAP)30.
The close packing of the β-barrels observed in these linear SOD1 filaments cannot occur when the proteins possess zinc and electrostaticloops in their well-ordered, wild-type conformations. Superposition offully metallated native SOD1 dimers onto mutant dimers comprisingthe linear filaments show that steric and electrostatic repulsion fromzinc loop residues Asp76 and Glu77 occur at the mutant GOI interface(data not shown). Well-ordered SOD1 electrostatic and zinc loops arethus critical to prevent spontaneous aggregation (data not shown), amechanism consistent with the various strategies that natural β-sheet–containing proteins use to avoid unintended edge-to-edge, β-sheet–to–β-sheet association (reviewed in ref. 31).
462 VOLUME 10 NUMBER 6 JUNE 2003 NATURE STRUCTURAL BIOLOGY
a
b
Figure 1 Disorder and conformational changes in pathogenic SOD1 molecules leading to GOIinterfaces and filamentous assembly. (a) Stereo view of structures of S134N and apo H46R SOD1monomers (green) superimposed on a monomer of human wild-type SOD1 (gray). The disorder in themetal-deficient S134N and apo H46R proteins is nearly the same, so they are both represented by thegreen structure. Amino acid residues that are N- and C-terminal to the disordered regions are indicatedby red and black dots for the zinc and electrostatic loop elements, respectively. The electrostatic loop(blue) interacts with the exposed cleft (red) of an adjacent molecule in the crystal lattice. Yellow dotsrepresent acidic residues in the wild-type protein that clash and prevent SOD1–SOD1 interactions fromoccurring at the cleft (see text). (b) Structure of a Zn–H46R SOD1 monomer (black) superimposed on amonomer of human wild-type SOD1 (gray). Amino acid residues are colored as in a. The zinc ion isshown as a green sphere. The zinc loop (blue) interacts with deprotected β-strands 5 and 6 (red) ofadjacent molecules in the crystal lattice. The yellow dots are as in a.©
2003
Nat
ure
Pu
blis
hin
g G
rou
p
htt
p:/
/ww
w.n
atu
re.c
om
/nat
ure
stru
ctu
ralb
iolo
gy

A R T I C L E S
NATURE STRUCTURAL BIOLOGY VOLUME 10 NUMBER 6 JUNE 2003 463
a
Figure 2 GOI interfaces in pathogenic SOD1 giverise to cross-β fibrils in two different crystalsystems. (a) Orthogonal views of the linear,amyloid-like filaments represented by threedimers shown from top to bottom in green, goldand blue, respectively. Both the S134N and apoH46R linear filaments are represented by thesingle filament shown in panels i–iv. The GOIinterface is red in the filament in i and boxed inii–iv. In iv, which is rotated 90° relative to ii andiii, β-strands 1, 2, 3 and 6, comprising one-halfof each SOD1 β-barrel, are shown in red. The‘cross-β’ structure observed in amyloid fibrils isshown schematically in v. (b) Stereo view of theGOI interface in the S134N filament. Residues125–131 of the electrostatic loop from oneS134N dimer (orange) interact with a depression
in the β-barrel of a neighboring S134N dimer (green) in the crystal lattice. Water molecules are represented as black spheres. The 1.3 Å σA55-weighted
electron density, with coefficients 2mFo – DFc, is contoured at 1.0 σ. (c) Small hydrophobic core formed at the GOI interface in the linear, pathogenic SOD1filaments (see text). The image is an enlargement of the region boxed in image iii of panel a.
c
b
These crystal structures provide a first glimpse of how pathogenicSOD1 mutant proteins might assemble into amyloid filaments. In theabsence of constraints imposed by the crystal repeat, SOD1 filamentsmay adopt arrangements different in detail from the arrangementsreported here. For example, X-ray diffraction from most amyloidfibers suggest that the β-stands tend to run roughly perpendicular tothe filament axis and seem to have a spacing of 4.7 Å over a length ofmany strands. The linear models of SOD1 reported here are consistentwith currently accepted ideas of amyloid structure in the broad sense.At a finer level of detail, however, some structural rearrangementsbetween crystalline and noncrystalline forms of SOD1 filamentswould be necessary to bring the observed filaments in line with modelsproposed for other amyloid fibrils on the basis of their X-ray fiber dif-fraction. The nature of the aggregated mutant SOD1 protein in vivo iscomplex and unlikely to represent only the ‘clean’ associations weobserve in our in vitro studies with purified recombinant pathogenicSOD1 proteins. For example, the SOD1-containing protein aggregatesare ubiquitinated in murine models of the disease, and other modifi-cations also occur, such as crosslinking, protein oxidation and theassociation of heat-shock proteins16,32. Further study of the aggregatesformed in vivo is required to address this issue.
Apo H46R zigzag filamentThe second filament in the monoclinic crystal system, apo H46R fila-ment 2, shares some features with the two amyloid-like linear fila-ments described above, but it also shows some significant differences.
Here, the rotation axes at the GOI interfaces are nearly parallel to thedimeric symmetry axes, but the rotation angles are 145° rather than180°. Thus, the H46R filament 2 ‘zigzags’ (Fig. 3) compared with thelinear filaments (Fig. 2). This zigzag arrangement occurs because oneof the dimers possesses fully ordered electrostatic and zinc loops,thereby preventing the close approach of β-barrels from neighboringmolecules observed in the more robust, linear amyloid-like filaments.Although the amino acids participating in the interdimer contacts arethe same as in the linear fibrils, they interact in a different orientation,burying only 270 Å2 of solvent-accessible surface area per polypeptide.However, this interaction is not reciprocal across the GOI interface asis observed for the linear filaments. The significance of the zigzag fila-ment to FALS is uncertain because of the tenuous nature of the con-tacts between SOD1 dimers and also because a nearly identical zigzagfilament has recently been observed for the human wild-type apoSOD1 protein33. Together, these structures suggest that an equilibriumexists in solution between the metal-bound loop conformations anddisordered conformations of the apo proteins. Because the wild-typeenzyme has presumably evolved to avoid nonproductive assemblywhen it is metal free, this equilibrium may be shifted to favor the dis-ordered conformations in pathogenic SOD1 proteins known to bedefective in metal binding.
Zn–H46R water-filled nanotubesIn contrast to the linear amyloid-like filaments described above for theS134N and apo H46R SOD1 proteins, a distinct but related GOI inter-
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
rest
ruct
ura
lbio
log
y

A R T I C L E S
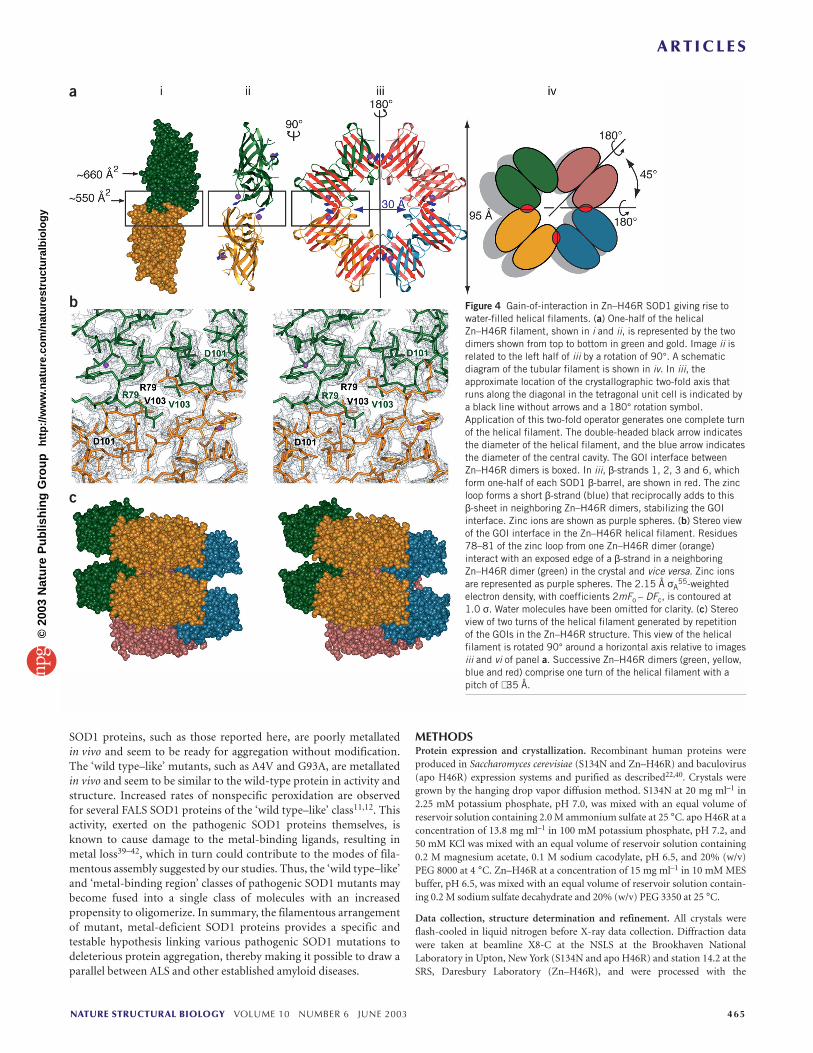
face between Zn–H46R dimers results in the assembly of helical fila-ments consisting of four Zn–H46R dimers per turn. A helical filamentrather than a linear filament arises because the 180° symmetrical GOIis offset 45° from the 180° symmetrical interaction between SOD1subunits in the homodimer (Fig. 4a). The GOI interface in this tetra-gonal crystal system also occurs between β-strands 5 and 6 of theSOD1 β-barrel, but the intermolecular contact surface is shifted byseveral amino acids relative to that of the linear filaments and, in thiscase, residues of the zinc loop rather than the electrostatic loop partic-ipate (Fig. 1b). Zinc loop residues 78–81 adopt a non-native confor-mation and hydrogen bond to the deprotected edge of β-strand 6,extending the β-sheet by one strand (Fig. 4a). Repetition of this GOIin the Zn–H46R crystal results in the formation of hollow ‘nanotubes’with an overall diameter of ∼ 95 Å and an inner water-filled cavity witha diameter of ∼ 30 Å. The reciprocal addition of β-strands to the β-sheet from the neighboring SOD1 molecules around the GOI dyadgives rise to continuously hydrogen-bonded β-sheets that spiralaround the long axis of the nanotube. This architecture represents avariation on a theme recently proposed for polyglutamine amyloidfibrils34, except that in the case of Zn–H46R, the plane of the continu-ous β-sheets is perpendicular to the long axis of the nanotube ratherthan parallel, as proposed for the polyglutamine case. The GOI inter-face in Zn–H46R buries ∼ 550 Å2 per polypeptide. As with the GOIinterfaces in the linear filaments, the extensive contacts are a combi-nation of apolar and hydrogen-bonding interactions (Fig. 4b) that areprevented in the normally folded and metallated wild-type protein.
The observation of the hollow, pore-like structure formed byZn–H46R is particularly intriguing because ‘amyloid pores’ areobserved in mutants of α-synuclein and β-amyloid, proteins thatcause Parkinson’s and Alzheimer’s disease, respectively35. For α-synuclein, electron microscopy reveals annular α-synucleinoligomeric species with an external diameter of 80–120 Å and an innerdiameter of 20–25 Å. For β-amyloid protein, electron microscopyreveals annular oligomeric species with an external diameter of70–100 Å and an inner diameter of 15–20 Å (ref. 35). Thus, both the
α-synuclein and β-amyloid pores exhibit dimensions similar to thoseobserved here for the Zn–H46R nanotubes (external diameter = 95 Å;internal diameter = 30 Å). There is growing support for the hypothesisthat the toxic species in neurodegenerative diseases such asAlzheimer’s and Parkinson’s diseases are not the long, insoluble fibrils(which may in fact be cytoprotective) but rather the ‘amyloidoligomers’ or ‘protofibrils’ formed by their soluble precursors36.
DISCUSSIONSignificance of GOI interfacesSeveral lines of evidence point to the significance of the GOI inter-faces between pathogenic SOD1 molecules. First, they bury approxi-mately the same amount of solvent-accessible surface area as thestable, naturally occurring homodimer interface. Second, the FALSSOD1 filaments are not always generated by crystallographic symme-try operators alone, but instead involve noncrystallographic symme-try operations in the monoclinic and tetragonal crystal forms. Thismeans that the formation of filaments is not simply a necessary consequence of crystallization. Third, the GOI that forms the linear,amyloid-like FALS SOD1 filaments are observed multiple times independently—with two different pathogenic SOD1 mutants in twodifferent crystal systems and in crystallographically distinct environ-ments within and between the asymmetric units of a single crystalform of the protein. Similarly, the GOI forming the helical FALSSOD1 filament appears independently in crystallographically distinctenvironments within and between the asymmetric units of the tetrag-onal crystal form. Fourth, each GOI interface arises through subunitsof SOD1 that are metal deficient and possess disorder in both the zincand electrostatic loop elements. Defective metal binding is a well-characterized feature common to multiple pathogenic SOD1mutants14,16,22–27 that could all exhibit similar disorder. Fifth, high-molecular-weight FALS SOD1 aggregates are observed in humans37
and mouse models of the disease8,15,32,38. The pathology of many ofthese transgenic mice is dominated by fibrillar inclusions of patho-genic SOD1 protein16. Thus, the crystallographic observation of fibrillar, amyloid-like SOD1 filaments is generally consistent within vivo observations. Sixth, a survey of molecular packing interactionsin crystal structures of fully metallated wild-type SOD1 from a varietyof organisms, including human, bovine, yeast and spinach, revealsthat the structurally intact zinc and electrostatic loop elements pro-tect against the formation of the GOI interfaces and filamentousassemblies reported here (data not shown). Finally, preliminaryanalyses of two additional metal-deficient pathogenic SOD1 mole-cules, G85R and E133∆, reveal that they crystallize isomorphouslywith the orthorhombic S134N crystal form and possess a linear fila-mentous arrangement of SOD1 dimers similar to that describedabove (X. Cao, L.J. Whitson and P.J.H., unpublished data).
Implications for familial ALSNeuronal damage mediated by toxic, aggregation-prone proteins is acommon theme emerging from studies on a broad range of neuro-degenerative disorders, including Alzheimer’s disease, Parkinson’sdisease, prion disease and polyglutamine disease39. It has becomeclear that a major effect of mutations in the genes associated withthese disorders is the abnormal assembly of misfolded mutant proteininto neuronal inclusions, plaques and, more recently, protofibrilpores. Our studies suggest that FALS-causing mutant SOD1 may bethe latest addition to an ever increasing list39. Finally, we suggest thatthe two most prominent theories of FALS etiology—aberrant copperchemistry and mutant protein aggregation—are not necessarilymutually exclusive. The ‘metal-binding region’ mutant pathogenic
464 VOLUME 10 NUMBER 6 JUNE 2003 NATURE STRUCTURAL BIOLOGY
Figure 3 Two orthogonal views of apo H46R filament 2, represented by threeSOD1 dimers in the same orientation as in Fig. 2a. Although the residuesinvolved in this GOI interface are nearly identical to those of the other linearfilaments, they touch each other in a different way, resulting in asubstantially less extensive interface and a zigzag arrangement of subunits.
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
rest
ruct
ura
lbio
log
y
A R T I C L E S
SOD1 proteins, such as those reported here, are poorly metallatedin vivo and seem to be ready for aggregation without modification.The ‘wild type–like’ mutants, such as A4V and G93A, are metallatedin vivo and seem to be similar to the wild-type protein in activity andstructure. Increased rates of nonspecific peroxidation are observedfor several FALS SOD1 proteins of the ‘wild type–like’ class11,12. Thisactivity, exerted on the pathogenic SOD1 proteins themselves, isknown to cause damage to the metal-binding ligands, resulting inmetal loss39–42, which in turn could contribute to the modes of fila-mentous assembly suggested by our studies. Thus, the ‘wild type–like’and ‘metal-binding region’ classes of pathogenic SOD1 mutants maybecome fused into a single class of molecules with an increasedpropensity to oligomerize. In summary, the filamentous arrangementof mutant, metal-deficient SOD1 proteins provides a specific andtestable hypothesis linking various pathogenic SOD1 mutations todeleterious protein aggregation, thereby making it possible to draw aparallel between ALS and other established amyloid diseases.
METHODSProtein expression and crystallization. Recombinant human proteins wereproduced in Saccharomyces cerevisiae (S134N and Zn–H46R) and baculovirus(apo H46R) expression systems and purified as described22,40. Crystals weregrown by the hanging drop vapor diffusion method. S134N at 20 mg ml–1 in2.25 mM potassium phosphate, pH 7.0, was mixed with an equal volume ofreservoir solution containing 2.0 M ammonium sulfate at 25 °C. apo H46R at aconcentration of 13.8 mg ml–1 in 100 mM potassium phosphate, pH 7.2, and50 mM KCl was mixed with an equal volume of reservoir solution containing0.2 M magnesium acetate, 0.1 M sodium cacodylate, pH 6.5, and 20% (w/v)PEG 8000 at 4 °C. Zn–H46R at a concentration of 15 mg ml–1 in 10 mM MESbuffer, pH 6.5, was mixed with an equal volume of reservoir solution contain-ing 0.2 M sodium sulfate decahydrate and 20% (w/v) PEG 3350 at 25 °C.
Data collection, structure determination and refinement. All crystals wereflash-cooled in liquid nitrogen before X-ray data collection. Diffraction datawere taken at beamline X8-C at the NSLS at the Brookhaven NationalLaboratory in Upton, New York (S134N and apo H46R) and station 14.2 at theSRS, Daresbury Laboratory (Zn–H46R), and were processed with the
NATURE STRUCTURAL BIOLOGY VOLUME 10 NUMBER 6 JUNE 2003 465
Figure 4 Gain-of-interaction in Zn–H46R SOD1 giving rise towater-filled helical filaments. (a) One-half of the helicalZn–H46R filament, shown in i and ii, is represented by the twodimers shown from top to bottom in green and gold. Image ii isrelated to the left half of iii by a rotation of 90°. A schematicdiagram of the tubular filament is shown in iv. In iii, theapproximate location of the crystallographic two-fold axis thatruns along the diagonal in the tetragonal unit cell is indicated bya black line without arrows and a 180° rotation symbol.Application of this two-fold operator generates one complete turnof the helical filament. The double-headed black arrow indicatesthe diameter of the helical filament, and the blue arrow indicatesthe diameter of the central cavity. The GOI interface betweenZn–H46R dimers is boxed. In iii, β-strands 1, 2, 3 and 6, whichform one-half of each SOD1 β-barrel, are shown in red. The zincloop forms a short β-strand (blue) that reciprocally adds to this β-sheet in neighboring Zn–H46R dimers, stabilizing the GOIinterface. Zinc ions are shown as purple spheres. (b) Stereo viewof the GOI interface in the Zn–H46R helical filament. Residues78–81 of the zinc loop from one Zn–H46R dimer (orange)interact with an exposed edge of a β-strand in a neighboringZn–H46R dimer (green) in the crystal and vice versa. Zinc ionsare represented as purple spheres. The 2.15 Å σA
55-weightedelectron density, with coefficients 2mFo – DFc, is contoured at1.0 σ. Water molecules have been omitted for clarity. (c) Stereoview of two turns of the helical filament generated by repetitionof the GOIs in the Zn–H46R structure. This view of the helicalfilament is rotated 90° around a horizontal axis relative to imagesiii and vi of panel a. Successive Zn–H46R dimers (green, yellow,blue and red) comprise one turn of the helical filament with apitch of ∼ 35 Å.
a
b
c
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
rest
ruct
ura
lbio
log
y

A R T I C L E S
DENZO/SCALEPACK suite41. The structure of SOD1 mutant G37R (PDBentry 1AZV)42 was used as the search model for molecular replacement inAMoRe43 (S134N) and EPMR44 (apo H46R), whereas the structure of nativehuman SOD1 (ref. 33) was used as the search model for molecular replacementin MOLREP45 (Zn–H46R). The structures were iteratively refined using REF-MAC5 (ref. 46) (S134N and Zn–H46R) and CNS47 (apo H46R). The modelswere manually adjusted in O48. For S134N, anisotropic atomic displacementthermal parameters were refined late in the process49. For Zn–H46R, TLSrefinement was implemented in the end stages. The final models were evaluatedusing WHATCHECK50 and PROCHECK51. No residues of the S134N andH46R structures fall outside of the allowable regions of their respectiveRamachandran plots.
Metal deficiency. All three pathogenic SOD1 proteins are metal deficient, asindicated by inductively coupled plasma mass spectroscopy, when purifiedfrom their respective expression systems22,24. The S134N subunits are desig-nated as A–B, where the (–) symbol represents the naturally occurring homo-dimer interface. X-ray data collected at the copper and zinc absorption edgescoupled with anomalous difference Fourier calculations reveal that in S134N
subunit A, there is a mixture of copper and zinc ionsin the Cu-binding site and zinc alone is bound in theZn-binding site. S134N subunit B contains almostexclusively zinc in the Cu-binding site and no metalin the Zn-binding site. The apo H46R subunits aredesignated as G–H, I–J, K–L and M–N in the fourdimers, and all are completely devoid of both copperand zinc ions in their binding sites. The apo H46Rfilaments 1 and 2 are packed in layers that alternateand are approximately normal to the ac plane of themonoclinic crystal. The Zn–H46R subunits are des-ignated as W–X and Y–Z, respectively, and all havezinc bound in their Zn-binding sites but no metal intheir Cu-binding sites.
Structure analysis and figure preparation. Solvent-accessible surface areas were calculated in CNS47
using a probe radius of 1.4 Å. Structural alignmentswere performed using ALIGN52. Figures were pre-pared using MolScript53, BobScript54 and POV-Ray(http://www.povray.org).
Coordinates. Coordinates and structure factors havebeen deposited in the Protein Data Bank (accessioncodes 1OZU, 1OZT and 1OEZ for S134N, apo H46Rand Zn–H46R SOD1 structures, respectively).
ACKNOWLEDGMENTSWe thank L. Flaks, and J. Berendzen for support atbeamline X8-C at the NSLS, Brookhaven NationalLaboratory; D. Cascio and M. Hough for their interestand valuable discussions; S. Holloway for assistancewith the illustrations and our colleagues who haveoffered comments during the preparation of thismanuscript. This work was supported by the NationalInstitutes of Health (L.J.H., J.S.V. and P.J.H.), theRobert A. Welch Foundation (P.J.H.), the ALSAssociation (L.J.H., J.S.V and P.J.H.), the MNDAssociation (S.S.H.) and a predoctoral fellowship fromthe Association for the Advancement of AgingResearch (J.S.E.). Funding from CCLRC and resourcesat Daresbury are also gratefully acknowledged.
COMPETING INTERESTS STATEMENTThe authors declare that they have no competingfinancial interests.
Received 6 December 2002; accepted 22 April 2003Published online 19 May 2003; doi:10.1038/nsb935
1. Haverkamp, L.J., Appel, V. & Appel, S.H. Natural history of amyotrophic lateralsclerosis in a database population. Validation of a scoring system and a model forsurvival prediction. Brain 118, 707–719 (1995).
2. Deng, H.X. et al. Amyotrophic lateral sclerosis and structural defects in Cu,Znsuperoxide dismutase. Science 261, 1047–1051 (1993).
3. Rosen, D.R. et al. Mutations in Cu/Zn superoxide dismutase gene are associatedwith familial amyotrophic lateral sclerosis. Nature 362, 59–62 (1993).
4. Fridovich, I. Superoxide dismutases. An adaptation to a paramagnetic gas. J. Biol.Chem. 264, 7761–7764 (1989).
5. Bowling, A.C., Schulz, J.B., Brown, R.H. Jr. & Beal, M.F. Superoxide dismutaseactivity, oxidative damage, and mitochondrial energy metabolism in familial and sporadic amyotrophic lateral sclerosis. J. Neurochem. 61, 2322–2325 (1993).
6. Reaume, A.G. et al. Motor neurons in Cu/Zn superoxide dismutase-deficient micedevelop normally but exhibit enhanced cell death after axonal injury. Nat. Genet. 13,43–47 (1996).
7. Ripps, M.E., Huntley, G.W., Hof, P.R., Morrison, J.H. & Gordon, J.W. Transgenic miceexpressing an altered murine superoxide dismutase gene provide an animal model ofamyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 92, 689–693 (1995).
8. Bruijn, L.I. et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytesand promotes rapidly progressive disease with SOD1-containing inclusions. Neuron18, 327–338 (1997).
9. Gurney, M.E. et al. Motor neuron degeneration in mice that express a humanCu,Zn superoxide dismutase mutation. Science 264, 1772–1775 (1994).
466 VOLUME 10 NUMBER 6 JUNE 2003 NATURE STRUCTURAL BIOLOGY
Table 1 Crystallographic data and refinement statistics
S134N apo H46R Zn–H46R
Data collection
Space group P212121 P21 P412121
Unit cell
a (Å) 40.1 79.6 190.8
b (Å) 56.5 70.0 190.8
c (Å) 105.4 113.8 34.6
β (°) 110.4
X-ray data
λ (Å) 1.100 0.989 0.978
Number of observations
Total 270,095 167,752 652,196
Unique 58,507 40,758 35,432
Resolution range (Å)a 50–1.3 (1.38–1.30) 100–2.5 (2.56–2.50) 20–2.15 (2.21–2.15)
Completeness (%)a 97.9 (87.1) 99.8 (100.0) 99.3 (98.6)
Rsym (on I) (%)a,b 5.0 (49.8) 8.6 (40.8) 9.0 (48.0)
Refinement
Number of dimers 1 4 2
per asymmetric unit
Resolution range (Å) 21.1–1.3 42.4–2.5 20–2.15
Rcryst (%)c 18.8 22.2 20.6
Rfree (%)d 21.1 27.0 23.6
Raniso (%)e 17.7
Rfree_aniso (%)e 19.3
F / σ F >0 >0 >0
R.m.s. deviations
Bonds (Å) 0.010 0.011 0.016
Angles (°) 1.5 1.7 1.7
Number of atoms
Protein 2,005 7,966 3,657
Water 247 235 277
Metal ions 3 0 4
Sulfate anions 2 0 4
aThe number in parentheses is for the last shell. bRsym = Σ|I – <I>|/ΣI, where I is the observed intensity and <I> is theaverage intensity of multiple symmetry-related observations of that reflection. cRcryst = Σ||Fop| – |Fcp|| / Σ|Fop|. dRfree =Σ||Fop| – |Fcp|| / Σ|Fop| where |Fop| is from a test set not used in the structural refinement. eRaniso and Rfree_aniso are asRcryst and Rfree, except with anisotropic atomic displacement parameter refinement.
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
rest
ruct
ura
lbio
log
y

A R T I C L E S
10. Wong, P.C. et al. An adverse property of a familial ALS-linked SOD1 mutationcauses motor neuron disease characterized by vacuolar degeneration of mitochon-dria. Neuron 14, 1105–1116 (1995).
11. Wiedau-Pazos, M. et al. Altered reactivity of superoxide dismutase in familialamyotrophic lateral sclerosis. Science 271, 515–518 (1996).
12. Yim, M.B. et al. A gain-of-function of an amyotrophic lateral sclerosis-associatedCu,Zn-superoxide dismutase mutant: an enhancement of free radical formation dueto a decrease in Km for hydrogen peroxide. Proc. Natl. Acad. Sci. USA 93,5709–5714 (1996).
13. Beckman, J.S., Chen, J., Crow, J.P. & Ye, Y.Z. Reactions of nitric oxide, superoxideand peroxynitrite with superoxide dismutase in neurodegeneration. Prog. Brain Res.103, 371–380 (1994).
14. Estevez, A.G. et al. Induction of nitric oxide-dependent apoptosis in motorneurons by zinc-deficient superoxide dismutase. Science 286, 2498–2500 (1999).
15. Bruijn, L.I. et al. Aggregation and motor neuron toxicity of an ALS-linked SOD1mutant independent from wild-type SOD1. Science 281, 1851–1854 (1998).
16. Wang, J. et al. Fibrillar inclusions and motor neuron degeneration in transgenicmice expressing superoxide dismutase 1 with a disrupted copper-binding site.Neurobiol. Dis. 10, 128–138 (2002).
17. Bruening, W. et al. Up-regulation of protein chaperones preserves viability ofcells expressing toxic Cu/Zn-superoxide dismutase mutants associated with amy-otrophic lateral sclerosis. J. Neurochem. 72, 693–699 (1999).
18. Okado-Matsumoto, A. & Fridovich, I. Amyotrophic lateral sclerosis: a proposedmechanism. Proc. Natl. Acad. Sci. USA 99, 9010–9014 (2002).
19. Borchelt, D.R. et al. Axonal transport of mutant superoxide dismutase 1 and focalaxonal abnormalities in the proximal axons of transgenic mice. Neurobiol. Dis. 5,27–35 (1998).
20. Williamson, T.L. & Cleveland, D.W. Slowing of axonal transport is a very earlyevent in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat. Neurosci. 2,50–56 (1999).
21. Johnston, J.A., Dalton, M.J., Gurney, M.E. & Kopito, R.R. Formation of highmolecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mousemodel for familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 97,12571–12576 (2000).
22. Hayward, L.J. et al. Decreased metallation and activity in subsets of mutantsuperoxide dismutases associated with familial amyotrophic lateral sclerosis. J. Biol.Chem. 277, 15923–15931 (2002).
23. Liu, H. et al. Copper2+ binding to the surface residue cysteine 111 of His46Arghuman copper-zinc superoxide dismutase, a familial amyotrophic lateral sclerosismutant. Biochemistry 39, 8125–8132 (2000).
24. Rodriguez, J.A. et al. Familial ALS-associated mutations decrease the thermalstability of distinctly metallated species of human copper-zinc superoxide dismutase.J. Biol. Chem. 277, 15932–15937 (2002).
25. Lyons, T.J. et al. Mutations in copper-zinc superoxide dismutase that causeamyotrophic lateral sclerosis alter the zinc binding site and the redox behavior of theprotein. Proc. Natl. Acad. Sci. USA 93, 12240–12244 (1996).
26. Crow, J.P., Sampson, J.B., Zhuang, Y., Thompson, J.A. & Beckman, J.S.Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismu-tase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J.Neurochem. 69, 1936–1944 (1997).
27. Lyons, T.J. et al. The metal binding properties of the zinc site of yeast copper-zinc superoxide dismutase: implications for amyotrophic lateral sclerosis. J. Biol.Inorg. Chem. 5, 189–203 (2000).
28. Subramaniam, J.R. et al. Mutant SOD1 causes motor neuron disease independentof copper chaperone-mediated copper loading. Nat. Neurosci. 5, 301–307 (2002).
29. Serag, A.A., Altenbach, C., Gingery, M., Hubbell, W.L. & Yeates, T.O.Arrangement of subunits and ordering of β-strands in an amyloid sheet. Nat. Struct.Biol. 9, 734–739 (2002).
30. Connors, L.H., Richardson, A.M., Theberge, R. & Costello, C.E. Tabulation oftransthyretin (TTR) variants as of 1/1/2000. Amyloid 7, 54–69 (2000).
31. Richardson, J.S. & Richardson, D.C. Natural β-sheet proteins use negative
design to avoid edge-to-edge aggregation. Proc. Natl. Acad. Sci. USA 99,2754–2759 (2002).
32. Watanabe, M. et al. Histological evidence of protein aggregation in mutant SOD1transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol. Dis. 8,933–941 (2001).
33. Strange, R.W. et al. The structure of metal deficient wild type human Cu,Znsuperoxide dismutase and its relevance to familial amyotrophic lateral sclerosis. J. Mol. Biol. 328, 877–891 (2003).
34. Perutz, M.F., Finch, J.T., Berriman, J. & Lesk, A. Amyloid fibers are water-fillednanotubes. Proc. Natl. Acad. Sci. USA 99, 5591–5595 (2002).
35. Lashuel, H.A., Hartley, D., Petre, B.M., Walz, T. & Lansbury, P.T. Jr.Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature 418,291 (2002).
36. Sherman, M.Y. & Goldberg, A.L. Cellular defenses against unfolded proteins: acell biologist thinks about neurodegenerative diseases. Neuron 29, 15–32 (2001).
37. Shibata, N. et al. Intense superoxide dismutase-1 immunoreactivity inintracytoplasmic hyaline inclusions of familial amyotrophic lateral sclerosis with pos-terior column involvement. J. Neuropathol. Exp. Neurol. 55, 481–490 (1996).
38. Wang, J., Xu, G. & Borchelt, D.R. High molecular weight complexes of mutantsuperoxide dismutase 1: age-dependent and tissue-specific accumulation.Neurobiol. Dis. 9, 139–148 (2002).
39. Taylor, J.P., Hardy, J. & Fischbeck, K.H. Toxic proteins in neurodegenerativedisease. Science 296, 1991–1995 (2002).
40. Goto, J.J. et al. Loss of in vitro metal ion binding specificity in mutant copper-zinc superoxide dismutases associated with familial amyotrophic lateral sclerosis.J. Biol. Chem. 275, 1007–1014 (2000).
41. Otwinowski, Z. & Minor, W. Processing of X-ray diffraction data collected inoscillation mode. Methods Enzymol. 276, 306–326 (1997).
42. Hart, P.J. et al. Subunit asymmetry in the three-dimensional structure of a humanCuZnSOD mutant found in familial amyotrophic lateral sclerosis. Protein Sci. 7,545–555 (1998).
43. Navaza, J. & Saludjian, P. AMoRe: an automated molecular replacement programpackage. Methods Enzymol. 276, 581–594 (1997).
44. Kissinger, C.R., Gehlhaar, D.K. & Fogel, D.B. Rapid automated molecularreplacement by evolutionary search. Acta Crystallogr. D 55, 484–491 (1999).
45. Vagin, A.A. & Teplyakov, A. MOLREP: an automated program for molecularreplacement. J. Appl. Crystallogr. 30, 1022–1025 (1997).
46. Murshudov, G.N., Vagin, A.A., Lebedev, A., Wilson, K.S. & Dodson, E.J.Efficient anisotropic refinement of macromolecular structures using FFT. ActaCrystallogr. D 55, 247–255 (1999).
47. Brunger, A.T. et al. Crystallography & NMR system: a new software suite formacromolecular structure determination. Acta Crystallogr. D 54, 905–921 (1998).
48. Jones, T.A., Zou, J.Y., Cowan, S.W. & Kjeldgaard, M. Improved methods forbuilding protein models in electron density maps and the location of errors in thesemodels. Acta Crystallogr. A 47, 110–119 (1991).
49. Winn, M.D., Isupov, M.N. & Murshudov, G.N. Use of TLS parameters to modelanisotropic displacements in macromolecular refinement. Acta Crystallogr. D 57,122–133 (2001).
50. Hooft, R.W., Vriend, G., Sander, C. & Abola, E.E. Errors in protein structures.Nature 381, 272 (1996).
51. Laskowski, R.A., McArthur, M.W., Moss, D.S. & Thornton, J.M. PROCHECK: a pro-gram to check the stereochemical quality of protein structures. J. Appl. Crystallogr.26, 283–291 (1993).
52. Cohen, G.E. ALIGN: a program to superimpose protein coordinates, accounting forinsertions and deletions. J. Appl. Crystallogr. 30, 1160–1161 (1997).
53. Kraulis, P.J. MOLSCRIPT: a program to produce both detailed and schematicplots of protein structures. J. Appl. Crystallogr. 24, 946–950 (1991).
54. Esnouf, R.M. Further additions to MolScript version 1.4, including reading andcontouring of electron-density maps. Acta Crystallogr. D 55, 938–940 (1999).
55. Read, R.J. Improved Fourier coefficients for maps using phases from partial struc-tures with errors. Acta Crystallogr. A 42, 140–149 (1986).
NATURE STRUCTURAL BIOLOGY VOLUME 10 NUMBER 6 JUNE 2003 467
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
rest
ruct
ura
lbio
log
y
Copyright © 2022 FDOKUMEN




















