
Lymphokines Enhance the Capacity of Human Monocytes to Secrete Reactive Oxygen Intermediates

Archives of Biochemistry and Biophysics xxx (2006) xxx–xxx
www.elsevier.com/locate/yabbi
ARTICLE IN PRESS
Allosteric modulation of the human P-glycoprotein involves conformational changes mimicking catalytic transition intermediates �
Pratiti Ghosh, Karobi Moitra, Nazli Maki, Saibal Dey ¤
Department of Biochemistry and Molecular Biology, Uniformed Services University of the Health Sciences, F. Edward Hébert School of Medicine, 4301 Jones Bridge Road, Bethesda, MD 20814-4799, USA
Received 25 January 2006, and in revised form 21 February 2006
Abstract
The drug transport function of human P-glycoprotein (Pgp, ABCB1) can be inhibited by a number of pharmacological agents collec-tively referred to as modulators or reversing agents. In this study, we demonstrate that certain thioxanthene-based Pgp modulators withan allosteric mode of action induce a distinct conformational change in the cytosolic domain of Pgp, which alters susceptibility to proteo-lytic digestion. Both cis and trans-isomers of the Pgp modulator Xupentixol confer considerable protection of an 80 kDa Pgp fragmentagainst trypsin digestion, that is recognized by a polyclonal antibody speciWc for the NH2-terminal half to Pgp. The protection by Xupen-tixol is abolished in the Pgp F983A mutant that is impaired in modulation by Xupentixols, indicating involvement of the allosteric site ingenerating the conformational change. A similar protection to an 80 kDa fragment is conferred by ATP, its nonhydrolyzable analogATP�S, and by trapping of ADP-vanadate at the catalytic domain, but not by transport substrate vinblastine or by the competitive mod-ulator cyclosporin A, suggesting diVerent outcomes from modulator interaction at the allosteric site and at the substrate site. In summary,we demonstrate that allosteric interaction of Xupentixols with Pgp generates conformational changes that mimic catalytic transition inter-mediates induced by nucleotide binding and hydrolysis, which may play a crucial role in allosteric inhibition of Pgp-mediated drug trans-port.Published by Elsevier Inc.
Keywords: Trypsin digestion; Thioxanthene derivatives; Flupentixol; N-�-Benzoylarginine-L-p-nitroanilide (BAPNA); Vanadate-trapping; ABCB1
The human P-glycoprotein (Pgp),1 encoded by theMDR1 gene, confers multidrug resistance in cancer cellsand restricts bioavailability of many therapeutic agentsdirected against pathological conditions and microbialpathogens [1,2]. Structurally, Pgp is a 1280 amino acidplasma membrane protein with two highly homologous
� This work was supported by United States Public Health ServicesGrant GM067926 and Uniformed Services University of the Health Sci-ences Intramural Grant C071FU. The costs of the publication of this arti-cle were defrayed in part by the payment of page charges. This article musttherefore hereby be marked “advertisement” in accordance with 18 U.S.C.section 1734 solely to indicate this fact.
* Corresponding author. Fax: +1 301 295 3512.E-mail address: [email protected] (S. Dey).
1 Abbreviations used: Pgp, P-glycoprotein; BAPNA, N-�-benzoylargi-nine-L-P-nitroanilide; PBS, phosphate-buVered saline.
0003-9861/$ - see front matter Published by Elsevier Inc.doi:10.1016/j.abb.2006.02.025
halves connected by an 80 amino acid linker region [3].Each half of the protein contains six putative transmem-brane helices and a large cytosolic domain. The transmem-brane regions from the two halves associate with each otherto form the drug-translocating pathway capable of recog-nizing a large spectrum of structurally unrelated hydropho-bic compounds. The cytosolic domains interact intimatelyto form two ATP binding/hydrolysis sites, together knownas the catalytic domain [3], involved in generating the driv-ing force for transport. The linker region between the twohalves has multiple sites for phosphorylation, of which thefunctional signiWcance is yet to be clearly understood [4,5].
A number of compounds have been identiWed that inter-act with Pgp and inhibit its ability to transport chemothera-peutic drugs [6,7]. These compounds are collectively knownas Pgp modulators or reversing agents. Several of these

2 P. Ghosh et al. / Archives of Biochemistry and Biophysics xxx (2006) xxx–xxx
ARTICLE IN PRESS
agents are now being tested for their clinical eVectiveness[8]. However, there remains a growing need for Pgp inhibi-tors with improved eYcacy [9]. It is apparent that a strate-gic approach towards developing such compounds requiresa clear understanding of Pgp function and the possiblemechanisms by which its function can be inhibited. Thisincludes identiWcation of the modulator interaction sites,their molecular characterization, and understanding theirinXuence on the diVerent functional aspects of Pgp. Some ofthe Pgp modulators, such as cyclosporin A [10] and verapa-mil, are themselves high aYnity substrates of the pump, andthey modulate drug transport in a competitive manner [11–15]. These compounds are broadly referred to as competi-tive modulators. On the other hand, several of the newlydiscovered compounds are believed to inhibit Pgp-medi-ated drug transport in an allosteric manner [16–22]. Thesites of interaction with Pgp of these allosteric modulators,and their mechanisms of inactivation remain to be eluci-dated.
In recent studies, we have demonstrated that certainthioxanthene-based Pgp modulators, such as the cis andtrans-isomers of Xupentixol and their closely related ana-logs, act through interaction at a site functionally distinctfrom the site of substrate recognition [20–22]. Using ala-nine-scanning mutagenesis, we identiWed a single phenyl-alanine residue at position 983 (F983) that is activelyinvolved in the modulatory mechanism [21]. Availablestructural information on Pgp from molecular modeling[23,24] and cysteine-scanning mutagenesis [25,26] placesTM12 outside the catalytic domain of Pgp, and residueF983 lies opposite from the drug binding helical face ofTM12. Because the Xupentixol interaction site (the residueF983) is spatially distinct from the catalytic domain (theATP sites) and functionally distinct from the substrate-binding site, it raises the possibility that an interdomainsignaling mechanism is involved in mediating modulatorychanges. Consistent with this idea, binding of monoclonalantibody UIC2, which is speciWc for a conformation-sen-sitive extracellular epitope of Pgp, was markedly reducedupon interaction with Xupentixol [22], whereas antibodybinding was increased by the Pgp substrate vinblastineand the competitive modulator cyclosporin A. This diVer-ential reactivity to UIC2 indicated the possibility of afunctionally relevant conformational change beinginduced by the thioxanthene derivatives that may extendto the cytosolic catalytic domain.
Susceptibility to protease digestion has been found tobe a useful technique in understanding global conforma-tional changes associated with ligand binding to solubleas well as to membrane proteins [27–30]. In Pgp, by study-ing susceptibility to limited trypsin digestion in inside-outmembrane vesicles and in puriWed and reconstituted pro-teins, distinct conformational changes have been detectedin response to interaction with nucleotides as well as drugsubstrates [31–33].
In the present study, we used trypsin digestion to probefor possible conformational changes in Pgp in response to
allosteric modulator interaction. The results show experi-mentally detectable conformational changes induced by thethioxanthene-based Pgp modulators cis-(Z)-Xupentixol andtrans-(E)-Xupentixol that mimic catalytic transition inter-mediates of Pgp during ATP binding and hydrolysis. Nosuch conformational changes were observed in the presenceof drug substrate or competitive modulator of Pgp.
Materials and methods
Materials
cis-(Z)-Flupentixol and trans-(E)-Xupentixol were fromResearch Biochemicals International. Cyclosporin A waspurchased from Calbiochem. [125I]Iodoarylazidoprazosin([125I]IAAP) (2200 Ci/mmol) was purchased from NENPerkin-Elmer Life Science. TPCK treated trypsin frombovine pancreas and N-�-benzoylarginine-L-P-nitroanilide(BAPNA) were purchased from Sigma. All other chemicalswere obtained from either Sigma or Bio-Rad. Previously,characterized mouse NIH3T3 Wbroblasts (drug-sensitivecell line) and the drug-resistant wild-type human Pgpexpressing NIHMDR1 cells [12] were used for drug trans-port studies, and Pgp-speciWc polyclonal antibodiesPEPG12 and PEPG13 were obtained from the laboratoryof Dr. Michael M. Gottesman, National Cancer Institute,NIH. The Goat anti-rabbit IgG conjugated with horserad-ish peroxidase and High Five insect cells were obtainedfrom Invitrogen.
Baculovirus mediated expression of human Pgp
A recombinant baculovirus harboring either the wild-type or the F983A mutant human MDR1 cDNA, with6£His-tag at the C-terminal end, BV-MDR1-(H6) [34] wasused to infect High Five insect cells grown in serum freeExcell 400 medium as described [35]. Cells were grown to80% conXuency at 27 °C in monolayer and infected with therecombinant baculovirus with multiplicity of infection of10, and harvested after 72 h of infection.
Isolation of crude membranes from high Wve insect cells
Crude membranes were prepared according to Dey et al.[20] with slight modiWcations. BrieXy, infected cells wereharvested and, washed twice in phosphate-buVered saline(PBS) containing 1% Aprotinin. Washed cells were incu-bated on ice for 45 min in homogenization buVer (50 mMTris–HCl, pH 7.5, 50 mM mannitol, 2 mM EGTA, 2 mMDTT, 1 mM AEBSF, and 1% aprotinin) and disrupted byrepeated strokes of a Dounce homogenizer. Followinghomogenization, undisrupted cells and nuclei wereremoved by centrifugation at 500g for 20 min. The superna-tant was collected and diluted with resuspension buVer(containing 50 mM Tris, pH 7.5, 300 mM mannitol, 1 mMEGTA, 1 mM DTT, 1 mM AEBSF, and 1% aprotinin) andcentrifuged at 100,000g for 1 h. The pellet was washed once

P. Ghosh et al. / Archives of Biochemistry and Biophysics xxx (2006) xxx–xxx 3
ARTICLE IN PRESS
with the same buVer and resuspended in resuspensionbuVer containing 10% glycerol by passing through a hypo-dermic needle (gauge size 19 and then 23). Membranes werestored at-¡70 °C in aliquots. Protein concentration wasmeasured by a modiWed Lowry method [36] using BSA as astandard.
Limited trypsin digestion of Pgp in isolated membranes
Pgp in isolated insect cell membranes (10–15�g) was pre-incubated with diVerent ligands in labeling buVer (50 mMTris–HCl, pH 7.5, 50 mM sodium chloride and 300 mM man-nitol), in a Wnal volume of 100�l. Trypsin at the indicatedWnal concentrations was added to the incubation mix toachieve the desired trypsin–protein ratio and incubated at37 °C for varying times. The reactions were stopped by addi-tion of 25�l of 5£SDS–PAGE sample buVer (5% SDS, 25%glycerol, 0.125 mM Tris–HCl, pH 6.8, 40 mM DTT, 0.01%pyronin Y) [37]. The samples were incubated at room tem-perate for 30min, mixed well by vortexing, and the peptidefragments of Pgp were resolved by SDS–PAGE in a 4–20%Tris–glycine gradient gel. Resolved Pgp fragments weretransferred to a nitrocellulose membrane, and probed withpolyclonal antisera PEPG13 or PEPG12, speciWc for theNH2- and COOH-terminal halves of Pgp, respectively [38].
PhotoaYnity labeling of Pgp with [125I]IAAP
PhotoaYnity labeling of crude membranes with the Pgpsubstrate [125I]IAAP was carried out according to Dey et al.[20]. BrieXy, insect cell membranes (10 �g protein) express-ing human Pgp were incubated at room temperature for15 min under subdued light with 5 nM [125I]IAAP in 25 mMTris–HCl, pH 7.5, and 1% aprotinin (labeling buVer). Fol-lowing incubation, membranes were exposed to UV illumi-nation at 365 nm (General Electric F15T8-BLB) for 15 min,at room temperature. Following UV crosslinking 5£ SDS–PAGE sample buVer was added to the reaction mixture andthe samples were held at room temperature (21–23 °C) foranother 30 min and mixed well by vortexing before analyz-ing by SDS–PAGE. Where indicated, membranes were pre-incubated for 5 min with Pgp substrates or modulatorsprior to the addition of [125I]IAAP and photocrosslinking.For determining the eVect of ATP and ADP-vanadate trap-ping, membranes were preincubated for 5 min at 37 °C witheither 5 mM MgATP or 5 mM MgATP and 250 �M sodiumorthovanadate, prior to photocrosslinking with [125I]IAAP.
Vanadate trapping and trypsin digestion
Isolated insect cell membranes (10�g protein) were prein-cubated in 100�l of labeling buVer (same as trypsin digestionbuVer) supplemented with 5mM ATP, 10 mM MgCl2, and0.25 mM sodium orthovanadate at 37°C for 10min, prior toincubation with trypsin at 37°C for 15min. The reaction wasstopped by addition of 25�l of 5X SDS–PAGE sample buVer.Samples were incubated at room temperature for 30min,
mixed thoroughly, and resolved by SDS–PAGE in a 4–20%Tris–glycine gel. Tryptic fragments of Pgp were detected byimmunoblotting with the Pgp-speciWc antibody PEPG13.
Radioactive drug accumulation in intact cells
Radioactive drug accumulation was studied accordingto [22]. 0.5£106 cells/well were grown in monolayers in a24-well tissue culture plate at 37 °C in the presence of 5%CO2 in DMEM supplemented with 10% FBS(DMEM + 10% FBS). Cells were washed once with 1 ml/well DMEM + 10% FBS, 15 min prior to the initiation ofthe assay. The assay was initiated by incubating cells witheither 0.5�M [3H]cis-(Z)-Xupentixol, 0.5 �M [3H]trans-(E)-Xupentixol, 15 nM [3H] vinblastine, or 1 �M [3H]cyclo-sporin A, in 0.3 ml DMEM + 10 % FBS at 37 °C in the pres-ence of 5% CO2. After incubation for varying time periods,cells were washed twice with 1 ml/well of ice-cold PBS.Washed cells were harvested by treatment with 0.5 ml/wellof Trypsin–EDTA at 37 °C for 15 min. Harvested cells werediluted in 5 ml of Biosafe II scintillation Xuid and mixedwell by vortexing. Radioactivity associated with the cellswas measured in a scintillation counter. Cells washed withice-cold PBS immediately after addition of the assay mixwere used as the “0” minute time point and the value foraccumulated radioactive drug was subtracted from eachdata point as nonspeciWc binding to the cells. The CPM val-ues were converted to nmol/million cells.
SDS–PAGE and immunoblot analysis
Electrophoresis and immunoblot analysis were per-formed as previously described [4].
Colorimetric assay for trypsin activity
The eVects of the ligands on trypsin activity were mea-sured using N-�-benzoylarginine-L-p-nitroanilide (BAPNA)as a chromogenic substrate [39,40]. The activity of trypsinwas tested at room temperature in 200 �l of assay buVercontaining 10 mM Hepes, pH 7.4, 0.5 mM BAPNA, and10�g trypsin, with or without the ligands. The activity ofthe enzyme was determined by measuring the absorbanceof p-nitroanilide at 450 nm generated from the reaction.Measurements were recorded using a Pharmacia BiotechUltrospec 3000 spectrophotometer at 1-min intervalsimmediately after mixing, and continued up to 10 min.Ligands were added 3 min prior to initiation of the assay.
Results
Modulator interaction at the allosteric site induces a distinct conformational change in Pgp detectable by trypsin susceptibility
Limited trypsin digestion of Pgp in inside-out membranevesicles has been a useful tool for detecting global
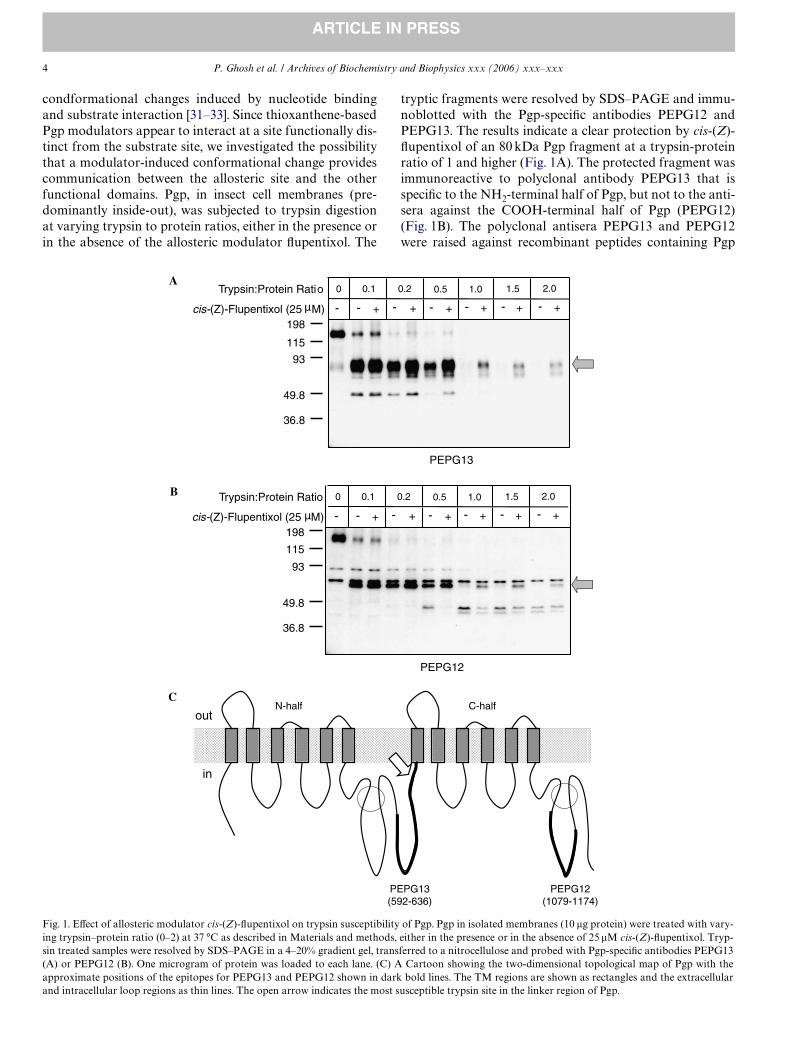
4 P. Ghosh et al. / Archives of Biochemistry and Biophysics xxx (2006) xxx–xxx
ARTICLE IN PRESS
condformational changes induced by nucleotide bindingand substrate interaction [31–33]. Since thioxanthene-basedPgp modulators appear to interact at a site functionally dis-tinct from the substrate site, we investigated the possibilitythat a modulator-induced conformational change providescommunication between the allosteric site and the otherfunctional domains. Pgp, in insect cell membranes (pre-dominantly inside-out), was subjected to trypsin digestionat varying trypsin to protein ratios, either in the presence orin the absence of the allosteric modulator Xupentixol. The
tryptic fragments were resolved by SDS–PAGE and immu-noblotted with the Pgp-speciWc antibodies PEPG12 andPEPG13. The results indicate a clear protection by cis-(Z)-Xupentixol of an 80 kDa Pgp fragment at a trypsin-proteinratio of 1 and higher (Fig. 1A). The protected fragment wasimmunoreactive to polyclonal antibody PEPG13 that isspeciWc to the NH2-terminal half of Pgp, but not to the anti-sera against the COOH-terminal half of Pgp (PEPG12)(Fig. 1B). The polyclonal antisera PEPG13 and PEPG12were raised against recombinant peptides containing Pgp
Fig. 1. EVect of allosteric modulator cis-(Z)-Xupentixol on trypsin susceptibility of Pgp. Pgp in isolated membranes (10 �g protein) were treated with vary-ing trypsin–protein ratio (0–2) at 37 °C as described in Materials and methods, either in the presence or in the absence of 25 �M cis-(Z)-Xupentixol. Tryp-sin treated samples were resolved by SDS–PAGE in a 4–20% gradient gel, transferred to a nitrocellulose and probed with Pgp-speciWc antibodies PEPG13(A) or PEPG12 (B). One microgram of protein was loaded to each lane. (C) A Cartoon showing the two-dimensional topological map of Pgp with the
198
49.8
36.8
115
93
+- +- +- +- +- +-cis-(Z)-Flupentixol (25 M) -
Trypsin:Protein Ratio 0 0.1 0.2 0.5 1.0 1.5 2.0
PEPG13
+- +- +- +- +- +-cis-(Z)-Flupentixol (25
µ
µM) -
Trypsin:Protein Ratio 0 0.1 0.2 0.5 1.0 1.5 2.0
198
49.8
36.8
115
93
PEPG12
A
B
in
out
PEPG13(592-636)
PEPG12(1079-1174)
N-half C-halfC
approximate positions of the epitopes for PEPG13 and PEPG12 shown in dark bold lines. The TM regions are shown as rectangles and the extracellularand intracellular loop regions as thin lines. The open arrow indicates the most susceptible trypsin site in the linker region of Pgp.

P. Ghosh et al. / Archives of Biochemistry and Biophysics xxx (2006) xxx–xxx 5
ARTICLE IN PRESS
fragments spanning amino acid residues 592–636, and1079–1174, respectively (see cartoon in Fig. 1C), as COOH-terminal fusions with Pseudomonas endotoxin [38].
Immunoblotting with PEPG12 revealed moderate pro-tection of a lower molecular weight Pgp fragment (Fig. 1B).This suggested that the initial cleavage was in the linkerregion of Pgp separating the two homologous halves, andthat the conformational change selectively restricts suscep-tibility of the NH2-terminal domain to trypsin. The protec-tion was concentration dependent, reaching its maximallevel at a cis-(Z)-Xupentixol concentration of 25�M(Fig. 2A). At the higher concentrations, the modulator alsorendered protection to an additional Pgp fragment with anapproximate molecular weight of 60 kDa, reactive to anti-sera PEPG13. As reported earlier, cis-(Z)-Xupentixol stimu-lates [125I]IAAP (substrate) binding to Pgp in aconcentration dependent manner [21] with maximal stimu-lation observed at a concentration (25 �M) similar to thatobserved in the trypsin protection experiment. SigniWcantly,no detectable protection from trypsin digestion wasobserved for the Pgp F983A mutant (Fig. 2C) that is
impaired in modulation by Xupentixol [21]. These resultssuggested that the change in trypsin susceptibility repre-sents a functionally relevant conformational change thatoriginates from Xupentixol interaction at the allosteric site.
The observed conformational change occurs independent of the functional state of the substrate site
The trans-isomer of Xupentixol allosterically modulatesPgp function through the same site of interaction as the cisisomer [21]. However, unlike cis-(Z)-Xupentixol, it inhibitssubstrate binding to Pgp [21]. To distinguish whether theconformational change observed is a mediator of modula-tory signal, or whether it originates as a downstream conse-quence of the modulatory changes occurring in the substratesite we investigated the eVect of trans-(E)-Xupentixol on tryp-sin susceptibility of Pgp. As indicated in Fig. 2B, trans-(E)-Xupentixol eVectively protected the same 80 kDa and the60kDa fragments from trypsin digestion in a concentrationdependent manner. Therefore, irrespective of the nature ofthe modulatory eVects (stimulatory or inhibitory) on the sub-
Fig. 2. Protection to limited trypsin digestion is induced by both isomers of Xupentixol, which is aVected in Pgp F983A mutant. Pgp in isolated membranes(10 �g protein) was subjected to trypsin digestion at a Wxed trypsin–protein ration of 1, in the presence of varying concentrations (0–25 �M) of either thecis (A) or the trans (B) isomers of Xupentixol. The trypsin treated membranes were resolved by SDS–PAGE in a 4–20% Tris–glycine gradient gel, andimmunoblotted with the Pgp-speciWc antibody PEGP13. Each lane contains 1 �g of total protein. (C) Similar experiments were carried out with the PgpF983A mutant in the presence of varying concentrations (0–25 �M) of cis-(Z)-Xupentixol and resolved as stated above.
198
49.8
115
93
+-Trypsin
0 1 2.5 5 10cis-(Z)-Flupentixol (µM) 250
Wild Type
198
49.8
115
93
+-Trypsin
0 1 2.5 5 10trans-(E)-Flupentixol (µM) 250
Wild Type
198
49.8
115
93
+-Trypsin
0 1 2.5 5 10cis-(Z)-Flupentixol (µM) 250
F983A
A
B
C
6 P. Ghosh et al. / Archives of Biochemistry and Biophysics xxx (2006) xxx–xxx
ARTICLE IN PRESS
strate site, both isomers of Xupentixol induced similar con-formational changes in Pgp. These results indicated that theconformational change observed in the presence of Xupen-tixol was not dependent on the functional state of the sub-strate site especially on the accessibility to substrate binding.
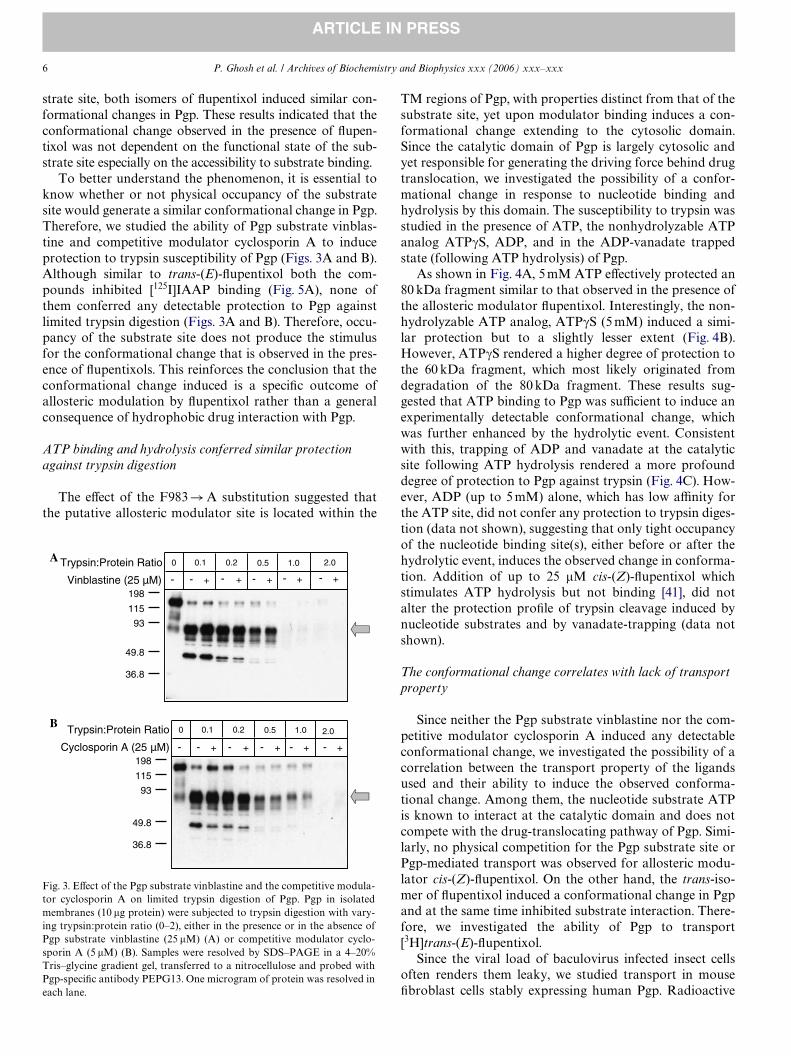
To better understand the phenomenon, it is essential toknow whether or not physical occupancy of the substratesite would generate a similar conformational change in Pgp.Therefore, we studied the ability of Pgp substrate vinblas-tine and competitive modulator cyclosporin A to induceprotection to trypsin susceptibility of Pgp (Figs. 3A and B).Although similar to trans-(E)-Xupentixol both the com-pounds inhibited [125I]IAAP binding (Fig. 5A), none ofthem conferred any detectable protection to Pgp againstlimited trypsin digestion (Figs. 3A and B). Therefore, occu-pancy of the substrate site does not produce the stimulusfor the conformational change that is observed in the pres-ence of Xupentixols. This reinforces the conclusion that theconformational change induced is a speciWc outcome ofallosteric modulation by Xupentixol rather than a generalconsequence of hydrophobic drug interaction with Pgp.
ATP binding and hydrolysis conferred similar protection against trypsin digestion
The eVect of the F983!A substitution suggested thatthe putative allosteric modulator site is located within the
Fig. 3. EVect of the Pgp substrate vinblastine and the competitive modula-tor cyclosporin A on limited trypsin digestion of Pgp. Pgp in isolatedmembranes (10 �g protein) were subjected to trypsin digestion with vary-ing trypsin:protein ratio (0–2), either in the presence or in the absence ofPgp substrate vinblastine (25 �M) (A) or competitive modulator cyclo-sporin A (5 �M) (B). Samples were resolved by SDS–PAGE in a 4–20%Tris–glycine gradient gel, transferred to a nitrocellulose and probed withPgp-speciWc antibody PEPG13. One microgram of protein was resolved ineach lane.
198
49.8
36.8
115
93
+- +- +- +-Vinblastine (25 µM) -
Trypsin:Protein Ratio 0 0.1 0.2 0.5 1.0
+-
2.0
198
49.8
36.8
115
93
+- +- +- +-Cyclosporin A (25 µM) -
Trypsin:Protein Ratio 0 0.1 0.2 0.5 1.0
+-
2.0
A
B
TM regions of Pgp, with properties distinct from that of thesubstrate site, yet upon modulator binding induces a con-formational change extending to the cytosolic domain.Since the catalytic domain of Pgp is largely cytosolic andyet responsible for generating the driving force behind drugtranslocation, we investigated the possibility of a confor-mational change in response to nucleotide binding andhydrolysis by this domain. The susceptibility to trypsin wasstudied in the presence of ATP, the nonhydrolyzable ATPanalog ATP�S, ADP, and in the ADP-vanadate trappedstate (following ATP hydrolysis) of Pgp.
As shown in Fig. 4A, 5 mM ATP eVectively protected an80 kDa fragment similar to that observed in the presence ofthe allosteric modulator Xupentixol. Interestingly, the non-hydrolyzable ATP analog, ATP�S (5 mM) induced a simi-lar protection but to a slightly lesser extent (Fig. 4B).However, ATP�S rendered a higher degree of protection tothe 60 kDa fragment, which most likely originated fromdegradation of the 80 kDa fragment. These results sug-gested that ATP binding to Pgp was suYcient to induce anexperimentally detectable conformational change, whichwas further enhanced by the hydrolytic event. Consistentwith this, trapping of ADP and vanadate at the catalyticsite following ATP hydrolysis rendered a more profounddegree of protection to Pgp against trypsin (Fig. 4C). How-ever, ADP (up to 5 mM) alone, which has low aYnity forthe ATP site, did not confer any protection to trypsin diges-tion (data not shown), suggesting that only tight occupancyof the nucleotide binding site(s), either before or after thehydrolytic event, induces the observed change in conforma-tion. Addition of up to 25 �M cis-(Z)-Xupentixol whichstimulates ATP hydrolysis but not binding [41], did notalter the protection proWle of trypsin cleavage induced bynucleotide substrates and by vanadate-trapping (data notshown).
The conformational change correlates with lack of transport property
Since neither the Pgp substrate vinblastine nor the com-petitive modulator cyclosporin A induced any detectableconformational change, we investigated the possibility of acorrelation between the transport property of the ligandsused and their ability to induce the observed conforma-tional change. Among them, the nucleotide substrate ATPis known to interact at the catalytic domain and does notcompete with the drug-translocating pathway of Pgp. Simi-larly, no physical competition for the Pgp substrate site orPgp-mediated transport was observed for allosteric modu-lator cis-(Z)-Xupentixol. On the other hand, the trans-iso-mer of Xupentixol induced a conformational change in Pgpand at the same time inhibited substrate interaction. There-fore, we investigated the ability of Pgp to transport[3H]trans-(E)-Xupentixol.
Since the viral load of baculovirus infected insect cellsoften renders them leaky, we studied transport in mouseWbroblast cells stably expressing human Pgp. Radioactive

P. Ghosh et al. / Archives of Biochemistry and Biophysics xxx (2006) xxx–xxx 7
ARTICLE IN PRESS
drug accumulation in drug sensitive NIH3T3 cells wascompared with that of drug resistant NIHMDR1 cells(NIH3T3 transfected with the human MDR1 cDNA). Asshown in Fig. 5B, a 5- to 10-fold less accumulation of[3H]vinblastine (lower left) as well as [3H]cyclosporin A(lower right) was detected in the Pgp-expressingNIHMDR1 cells compared to the untransfected NIH3T3cells, suggesting eYcient transport of the these compoundsfrom the Pgp-expressing cells. The transport of [3H]vinblas-tine as well as of [3H]cyclosporin A from NIHMDR1 cellscould be inhibited by inclusion of the Pgp modulator rapa-mycin, resulting in increased intracellular accumulation ofthe radioactive compounds (data not shown, see [22]),which conWrmed that the observed transport was Pgp med-iated. In contrast, no diVerence in intracellular accumula-tion of either [3H]cis-(Z)-Xupentixol (top left) or [3H]trans-(E)-Xupentixol (top right) was observed between the twocell lines (Fig. 5B), indicating no Pgp-mediated eZux of
radioactive Xupentixol from the cells. Therefore, althoughtrans-(E)-Xupentixol inhibits substrate binding, the inhibi-tion is not through direct competition for the substrate site.The data also suggests that the observed conformationalchange in Pgp correlates with modulator interactions thatare allosteric in nature.
None of the ligands aVect the enzymatic activity of trypsin
To determine whether any of the ligands used in thestudy directly inXuence the enzymatic activity of trypsin,the eVects of the ligands on the catalytic activity of trypsinwas measured using N-�-benzoylarginine-L-p-nitroanilide(BAPNA) as a chromogenic substrate, as described [39,40].None of the compounds used had any appreciable eVect onthe ability of trypsin to cleave the ester bond of BAPNA,which conWrmed no alteration in the enzymatic activity oftrypsin by any of the compounds (data not shown).
Fig. 4. EVect of ATP, nonhydrolyzable ATP analog ATP�S, and ADP-vanadate trapping on trypsin susceptibility of Pgp. Trypsin digestion of Pgp in iso-lated insect cell membranes was carried out with varying trypsin–protein ratio (0–2) either in the presence or in the absence of 5 mM ATP (A) or 5 mMATP�S (B). Trypsin treated samples were resolved by SDS–PAGE in 4–20% Tris–glycine gradient gel, transferred to nitrocellulose and probed with PgpspeciWc antibody PEPG13. One microgram of protein was added to each lane. (C) The eVect of vanadate-trapping on trypsin susceptibility of Pgp. Pgp inisolated membranes (10 �g) was vanadate-trapped with 5 mM ATP and 0.25 mM sodium orthovanadate, as described, and then subjected to trypsin diges-tion with varying trypsin–protein ratio (0–2.5). Control samples were treated the same way except no ATP was added to the reaction. The samples wereresolved by SDS–PAGE in a 4–20% Tris-glycine gradient gel and immunoblotted with Pgp-speciWc antibody PEPG13. Each lane contains 1 �g protein.
198
49.8
36.8
115
93
+- +- +- +- +- +--
Trypsin:Protein Ratio 0 0.1 0.2 0.5 1.0
0.1 0.2 0.5 1.0
1.5 2.0
ATP (5 mM)
+- +- +- +- +- +-ATPγS (5 mM) -
Trypsin:Protein Ratio 0 1.5 2.0
198
49.8
36.8
115
930 0.
1
0.2
0.5
1 1.5
2
198
49.8
36.8
115
93
Trypsin:Protein Ratio
2.5
0 0.1
0.2
0.5
1 1.5
2 2.5
ATP + VanadateVanadate
A
B
C

8 P. Ghosh et al. / Archives of Biochemistry and Biophysics xxx (2006) xxx–xxx
ARTICLE IN PRESS
Therefore, the observed eVects on the trypsin digestionwere due to changes induced on Pgp by the respectiveligands.
Discussion
Several of the Pgp modulators have been predicted tohave an allosteric mode of action on Pgp-mediated drugtransport [16–22,42], however, their sites of interaction with
Pgp and the exact mechanisms of action are not yet fullyunderstood. Our prior studies have demonstrated that thethioxanthene-based Pgp modulator Xupentixols and theirclosely related analogs interact with Pgp at a site function-ally distinct from the site of substrate recognition and mod-ulate substrate binding and ATP hydrolysis in an allostericmanner [20,22]. In modulation by these compounds, a sin-gle phenylalanine residue in TM12 at position 983 (F983)plays a crucial role [21,22]. According to the helical wheel
Fig. 5. (A) EVect of Pgp substrates, modulators, ATP and ADP-vanadate trapping on substrate binding to Pgp. Pgp in isolated insect cell membranes(15 �g protein) were photoaYnity labeled with 5 nM [125I]IAAP, as described, either in the absence (None) or in the presence of, 25 �M cis-(Z)-Xupentixol[Cis(Z)] or trans-(E)-Xupentixol [Trans(E)], 1 �l dimethylsulfoxide (DMSO), 25 �M vinblastine (Vinb), 5 �M cyclosporin A (CsA), 5 mM ATP (ATP), or5 mM MgATP + 0.25 mM sodium orthovanadate (ATP·Vi). Following labeling, the samples (4 �g protein/well) were resolved by SDS–PAGE in a 8%Tris–glycine gel, and the radioactivity associated was captured on an X-ray Wlm and quantiWed in a PhosphorImager. (B) Transport properties of allostericmodulators cis- and trans-Xupentixol, competitive modulator cyclosporine A and Pgp substrate vinblastine. NIH3T3 (3T3) cells or NIHMDR1 (MDR1)cells (5 £ 106 cells) were incubated at 37 °C with either 0.5 �M of [3H]cis-(Z)-Xupentixol (top left), 0.5 �M [3H]trans-(E)-Xupentixol (top right), 15 nM[3H]vinblastine (bottom left), or 1 �M [3H]cyclosporin A (bottom right), for 60 min, and intracellular accumulation of the radioactive drugs was deter-mined as described. The data are plotted as nanomoles of respective drugs accumulated per million cells. The data represents average of two independentexperiments (A and B).

P. Ghosh et al. / Archives of Biochemistry and Biophysics xxx (2006) xxx–xxx 9
ARTICLE IN PRESS
projection map of the Pgp TM regions, describing thearrangement of the transmembrane �-helices relative toeach other, as well as to the major drug-binding (substrate-binding) site, residue F983 is located opposite to the drug-binding face of TM12 [25,26]. This indicates that the keyinteraction site for Xupentixol lies outside the conventionaldrug recognition site of Pgp. Consistent with this, theF983!A substitution selectively altered the modulatoryproperties of the Xupentixols without any appreciable eVecton the interaction with Pgp substrates or competitive mod-ulators [21,22]. However, the mechanism by which residueF983, upon interaction with Xupentixols, communicateswith the substrate- or the ATP-sites is not clearly under-stood. In this report, by studying susceptibility to limitedtrypsin digestion of Pgp in inside-out membrane vesicles,we demonstrate a modulator-induced conformationalchange as a possible mode of communication between theputative allosteric modulator site and the other functionaldomains.
In the absence of high-resolution structural informationof Pgp, understanding of ligand-induced conformationalchange largely depends on biochemical and biophysicaltechniques. Of these methods, change in susceptibility tolimited trypsin digestion has been found to be particularlyinformative [31–33,43]. These studies have demonstratedthat during its catalytic turnover, Pgp undergoes a consid-erable degree of conformational change that can be experi-mentally detected. SpeciWc conformational changes areinduced by interaction with nucleotides [31], as well as,drug substrates [32,33], and the data are consistent with thechanges observed using other experimental approaches[44–48]. The eVect of Xupentixol on binding of monoclonalantibody UIC2 to a conformation-sensitive external epi-tope of Pgp (in intact cells) suggested a distinct conforma-tional change induced by these modulators [22]. Toinvestigate whether the conformational change observedextends to the cytosolic domain, and to understand the cor-relation of such a change to the modulatory eVects inducedon the substrate site, we performed a more detailed analysisof the phenomenon.
The data indicate that Xupentixol interaction at the allo-steric site induces a conformational change in the cytosolicdomain of Pgp that confers protection to a unique 80 kDaPgp fragment against limited trypsin digestion, detectableby the polyclonal antiserum PEPG13 (Fig. 1A) speciWc forthe NH2-terminal half of Pgp. Antibody speciWc to theCOOH-terminal half of Pgp (PEPG12) did not recognizethe protected fragment (Fig. 1B) suggesting that the cleav-age site lies in the linker region between the two halves.Since the Pgp F983A mutant that is impaired in modula-tion by Xupentixol [21,22] displays no such protection(Fig. 2C) the functional signiWcance of the conformationalchange in the mechanism of modulation is underscored.This further suggests that the change in conformation isinduced through modulator interaction at the allosteric siteand not elsewhere in the protein. For instance, the possibil-ity of Xupentixols rendering protection to the 80 kDa frag-
ment by interacting directly to the cytosolic domain of Pgpis ruled out. It is conceivable that a direct interaction of cis-(Z)-Xupentixol as well as ATP with the second site of tryp-sin cleavage could result in a similar protection. However,the fact that cis-(Z)-Xupentixol does not block ATP bind-ing to Pgp [41], argues against a common site of interactionfor the two ligands. Furthermore, for cleavage, trypsin sitesneed to be solvent accessible, a condition that is not favor-able for interaction with hydrophobic molecules like Xup-entixols. Instead, the transport substrates and modulatorsof Pgp are known to interact with the transmembraneregions of the protein, either by hydrogen bonding orthrough hydrophobic interaction. Consistent with thatF983 is located in the hydrophobic environment of TM12corresponding to the upper leaXet of the lipid bilayer,which lies outside the cytosolic domain of the protein. Thisindicates that, despite being spatially separated, the alloste-ric site is capable of communicating with the catalyticdomain. It is possible that the conformational change inresponse to Xupentixol interaction may mediate the Xow ofinformation from the allosteric site to the substrate, and theATP, sites. In addition Xupentixol-induced conformationalchange has been demonstrated using monoclonal antibodyUIC2 binding to an external epitope of Pgp [22].
The most susceptible trypsin cleavage site in Pgp hasbeen found to be at the linker region [49–51], which is con-sistent with the data obtained in our current experiments.Studies with proteolytic fragments of Pgp have demon-strated retention of the substrate binding and ATP hydro-lytic activity even after the initial cleavage at the linkerregion. The association between the NH2- and COOH-ter-minal fragments is tight and virtually impossible to sepa-rate without denaturing the protein totally [43]. Along thisline, we demonstrated the ability of cis-(Z)-Xupentixol tostimulate [125I]IAAP binding to Pgp that is cleaved at thelinker region [52], which suggested that the modulatoryproperties of the Xupentixol site is preserved even when theprotein is cleaved into two halves. Consistent with this, ourpresent study shows the ability of cis-(Z)-Xupentixol toeVectively protect the 80 kDa and the 60 kDa fragmentsfrom further digestion (Figs. 1A and 2A), despite having noeVect on the initial cleavage at the linker region of the fulllength protein. It is interesting to observe that residue F983,which plays an indispensable role in the conformationalchange, is located at the COOH-terminal half of Pgp yetmediates protection of the NH2-terminal fragment fromfurther trypsin digestion.
Interestingly, in our study, the Pgp substrate vinblastine,as well as the competitive modulator cyclosporin A do notrender any detectable protection to Pgp against trypsindigestion (Figs. 3A and B), which further reinforces thefunctional distinctness of the Xupentixol interaction sitefrom that of the substrate site. Furthermore, both [3H]vin-blastine and [3H]cyclosporin A are eVectively transportedout from the cells by Pgp (Fig. 5B, bottom), whereas notransport activity is detected for [3H]cis-(Z)-Xupentixol and[3H]trans-(E)-Xupentixol (Fig. 5B, top). This underscores

10 P. Ghosh et al. / Archives of Biochemistry and Biophysics xxx (2006) xxx–xxx
ARTICLE IN PRESS
the fact that ability to inhibit substrate binding does notnecessarily indicate that the modulator is a transport sub-strate of the pump. Although substrate-induced alterationin the susceptibility to proteolytic digestion has beenobserved in prior studies [32,33], the identity of the pro-tected fragments diVer from that protected by Xupentixols.In the said study, substrate molecule vinblastine or compet-itive modulator verapamil conferred protection to two Pgpfragments from the COOH-terminal half of the protein,and the major conformational change was predicted to bein the loop region between the TM8 and TM9 [33]. SincePEPG13 does not detect Pgp fragments from the COOH-terminal half, our results do not rule out the possibility of asubstrate-induced conformational change in Pgp, insteadthey highlight the fact that the conformational changeinduced by the allosteric modulator Xupentixol is distinctfrom that induced by substrates or competitive modulators.
On the other hand, the nucleotide substrate ATP andnonhydrolyzable nucleotide analog ATP�S those binddirectly to the cytosolic domain induced a similar kind ofprotection to trypsin digestion (Figs. 4A and B). Theseresults are in close agreement with studies on the mousePgp homolog Mdr3 [31]. As revealed by structural analysis,Pgp and other ABC transporters, in their functional formundergo dimerization of the two nucleotide-bindingdomains, with nucleotides bound at the interface betweenthe two [23]. It is possible that the conformational changethat brings the two ATP sites together also causes inaccessi-bility of a trypsin site that otherwise remains exposedallowing further degradation of the protein. Since the pro-Wle of protected Pgp fragments induced by Xupentixol(Figs. 1A and 2A) was indistinguishable from that inducedby nucleotide binding (Figs. 4A and B) or vanadate-trap-ping (Fig. 4C), it suggests that the allosteric modulator-induced conformational change may mimic a catalytictransition state generated during ATP binding and hydro-lysis.
The functional signiWcance of the conformationalchange remains open to interpretation. Interestingly, the cisand trans-isomers of Xupentixol, although protect the samePgp fragment from trypsin (Figs. 2A and B), have oppositeeVects on substrate binding to Pgp (Fig. 5A). One possibil-ity is that the conformational changes induced by the twoisomers are not identical but close enough to render similarprotections to Pgp against limited trypsin digestion. Thisperhaps is analogous to the eVect of ATP (or ATP�S) bind-ing and ADP-vanadate trapping on trypsin digestion. ATPbinding, which induces a catalytic intermediate stateachieved prior to hydrolysis, do not aVect substrate bindingby Pgp, whereas ADP-vanadate-trapping, which stabilizesa catalytic transition state conformation following ATPhydrolysis, almost completely prevent Pgp-substrate inter-action (Fig. 5A), yet both events render similar protectionagainst trypsin digestion.
A less likely explanation is that the observed conforma-tional change occurs early in the communication process,and although act as a mediator of allosteric modulation,
does not appear to be the determinant of the nature of allo-steric changes (stimulatory or inhibitory) induced on thesubstrate- and the ATP-sites. Nevertheless, our data sug-gests that the conformational change occurs independent ofthe substrate-site occupancy, which is further supported bythe fact that both trans-(E)-Xupentixol and cyclosporin Ainhibit substrate recognition by Pgp (Fig. 5A), but confor-mational change is induced only by the former (Fig. 2B)and not by the latter (Fig. 3B). The data provide furthersupport for discrete modes of action by the two modulators(trans-(E)-Xupentixol and cyclosporin A); while trans-(E)-Xupentixol interacts at a site functionally distinct from thesite of substrate (or competitive inhibitor) recognition andcommunicates with the substrate site via a conformationalchange, the competitive modulator cyclosporin A blockssubstrate binding through a direct physical competitionwithout any need for signaling. Consistent with that, in thePgp F983A mutant, the loss of cis-(Z)-Xupentixol-inducedconformational change coincides with impaired modula-tion of [125I]IAAP binding, whereas inhibition of [125I]IAAPbinding by cyclosporin A remained unaltered in the mutant[21].
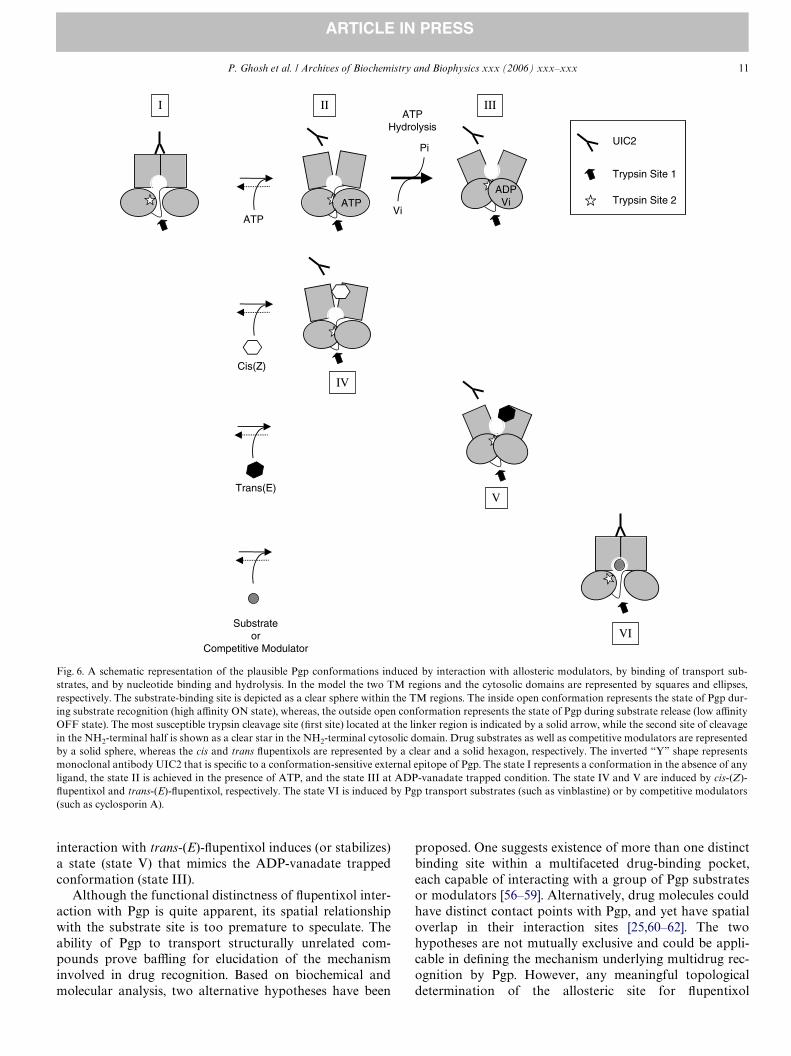
Based on our present study and on the data from mono-clonal antibody UIC2 binding to a conformation-sensitiveexternal epitope of Pgp [22,53,54], we propose a workingmodel describing the functional signiWcance of the confor-mational changes induced by the allosteric modulators cis-and trans- Xupentixol (Fig. 6). According to this model, cis-(Z)-Xupentixol-induced conformation (state IV) is similarto that generated by ATP binding (state II), which involvesmovement of the ATP sites without aVecting substrateinteraction, but results in a protection of the second site oftrypsin cleavage, presumably located within the NH2-termi-nal half of the protein. On the other hand, the trans-(E)-Xupentixol-induced state (state V) mimics a post-hydrolyticconformation of Pgp that is induced by ADP-vanadatetrapping (state III). Consistent with that, modulation bytrans-(E)-Xupentixol and vanadate-trapping, both inhibitedsubstrate interaction with Pgp (Fig. 5A) and at the sametime rendered an identical protection to Pgp from cleavageat the second trypsin site (Figs. 2B and 4C). We also pro-pose that, as reported earlier [33], substrate binding mightinduce a conformational change in Pgp (state VI), but thechange does not result in protection from the second tryp-sin cleavage. The proposed model is in good agreementwith the data obtained from studies in intact cells usingmonoclonal antibody UIC2 [22,53,54]. UIC2 binding toPgp expressing cells is maximal under metabolic starvationthat leads to a low intracellular ATP pool, or in the pres-ence of Pgp substrate [54,55], suggesting similar conforma-tional states under those two conditions. Whereas, anabundance of intracellular ATP [54,55], sodium orthovana-date treatment [22], or incubation with Xupentixols [22], allresult in reduced levels of UIC2 binding to Pgp expressingcells. In summary, we propose that cis-(Z)-Xupentixol stabi-lizes (or induces) a Pgp conformation (state IV) similar tothat generated by ATP binding (state II), whereas
P. Ghosh et al. / Archives of Biochemistry and Biophysics xxx (2006) xxx–xxx 11
ARTICLE IN PRESS
interaction with trans-(E)-Xupentixol induces (or stabilizes)a state (state V) that mimics the ADP-vanadate trappedconformation (state III).
Although the functional distinctness of Xupentixol inter-action with Pgp is quite apparent, its spatial relationshipwith the substrate site is too premature to speculate. Theability of Pgp to transport structurally unrelated com-pounds prove baZing for elucidation of the mechanisminvolved in drug recognition. Based on biochemical andmolecular analysis, two alternative hypotheses have been
proposed. One suggests existence of more than one distinctbinding site within a multifaceted drug-binding pocket,each capable of interacting with a group of Pgp substratesor modulators [56–59]. Alternatively, drug molecules couldhave distinct contact points with Pgp, and yet have spatialoverlap in their interaction sites [25,60–62]. The twohypotheses are not mutually exclusive and could be appli-cable in deWning the mechanism underlying multidrug rec-ognition by Pgp. However, any meaningful topologicaldetermination of the allosteric site for Xupentixol
Fig. 6. A schematic representation of the plausible Pgp conformations induced by interaction with allosteric modulators, by binding of transport sub-strates, and by nucleotide binding and hydrolysis. In the model the two TM regions and the cytosolic domains are represented by squares and ellipses,respectively. The substrate-binding site is depicted as a clear sphere within the TM regions. The inside open conformation represents the state of Pgp dur-ing substrate recognition (high aYnity ON state), whereas, the outside open conformation represents the state of Pgp during substrate release (low aYnityOFF state). The most susceptible trypsin cleavage site (Wrst site) located at the linker region is indicated by a solid arrow, while the second site of cleavagein the NH2-terminal half is shown as a clear star in the NH2-terminal cytosolic domain. Drug substrates as well as competitive modulators are representedby a solid sphere, whereas the cis and trans Xupentixols are represented by a clear and a solid hexagon, respectively. The inverted “Y” shape representsmonoclonal antibody UIC2 that is speciWc to a conformation-sensitive external epitope of Pgp. The state I represents a conformation in the absence of anyligand, the state II is achieved in the presence of ATP, and the state III at ADP-vanadate trapped condition. The state IV and V are induced by cis-(Z)-Xupentixol and trans-(E)-Xupentixol, respectively. The state VI is induced by Pgp transport substrates (such as vinblastine) or by competitive modulators(such as cyclosporin A).
Substrateor
Competitive ModulatorVI
ATPHydrolysis
I
V
ATP
ATP
II III
IVCis(Z)
Trans(E)
ADPVi
UIC2
Trypsin Site 1
Trypsin Site 2Vi
Pi

12 P. Ghosh et al. / Archives of Biochemistry and Biophysics xxx (2006) xxx–xxx
ARTICLE IN PRESS
interaction and its spatial relationship with the substrateinteraction site have to wait till the high resolution three-dimensional structure of the protein becomes available. Atpresent, the location of residue F983 (which plays a crucialrole in modulation by Xupentixol) away from the majordrug-binding pocket encourages us to predict a certaindegree of spatial distinctness for the allosteric site. This,however, does not mean that all the modulators with allo-steric mode of action share the same site for interaction.
In summary, our data demonstrate the ability of boththe cis and the trans-isomers of Xupentixol to generate aconformational change in Pgp that can be detected througha change in accessibility of trypsin sites. This change is nota part of a general response to hydrophobic drug interac-tion with Pgp at the TM region. The conformationalchanges induced by the allosteric modulators mimic Pgpcatalytic transitions states with respect to trypsin suscepti-bility as well as interaction with substrate molecules, andmay play an important role in the mechanism by whichXupentixols inhibit Pgp-mediated drug transport. To fur-ther investigate the molecular details of the phenomenon,we have now generated four diVerent versions of trypto-phan-less Pgp mutants with diVerent degrees of tryptophansubstitution (by phenylalanine), which will be used to mea-sure change in intrinsic tryptophan Xuorescence of Pgp asan indicator of modulator-induced conformational change.
Acknowledgments
We thank Dr. Michael Gottesman for his encourage-ment and support, and Dr. Teresa Dunn for her criticalassessment of the manuscript.
References
[1] M.M. Gottesman, I. Pastan, Annu. Rev. Biochem. 62 (1993) 385–427.[2] S.V. Ambudkar, S. Dey, C.A. Hrycyna, M. Ramachandra, I. Pastan,
M.M. Gottesman, Annu. Rev. Pharmacol. Toxicol. 39 (1999) 361–398.[3] C.-J. Chen, J.E. Chin, K. Ueda, D.P. Clark, I. Pastan, M.M. Gottes-
man, I.B. Roninson, Cell 47 (1986) 381–389.[4] U.A. Germann, T.C. Chambers, S.V. Ambudkar, T. Licht, C.O. Car-
darelli, I. Pastan, M.M. Gottesman, J. Biol. Chem. 271 (1996) 1708–1716.
[5] C.A. Hrycyna, L.E. Airan, U.A. Germann, S.V. Ambudkar, I. Pastan,M.M. Gottesman, Biochemistry 37 (1998) 13660–13673.
[6] J.M. Ford, W.N. Hait, Pharmacol. Rev. 42 (1990) 155–199.[7] J.A. Kellen, J. Exp. Ther. Oncol. 3 (2003) 5–13.[8] B.I. Sikic, Oncology (Huntingt) 13 (1999) 183–187.[9] M.M. Gottesman, T. Fojo, S.E. Bates, Nat. Rev. Cancer 2 (2002) 48–58.
[10] M. Bohme, M. Buchler, M. Muller, D. Keppler, FEBS Lett. 333 (1993)193–196.
[11] J.M. Ford, Eur. J. Cancer 32A (1996) 991–1001.[12] A. Tsuji, I. Tamai, A. Sakata, Y. Tenda, T. Terasaki, Biochem. Phar-
macol. 46 (1993) 1096–1099.[13] T. Saeki, K. Ueda, Y. Tanigawara, R. Hori, T. Komano, J. Biol. Chem.
268 (1993) 6077–6080.[14] E. Crivellato, L. Candussio, A.M. Rosati, F. Bartoli-Klugmann, F.
Mallardi, G. Decorti, J. Histochem. Cytochem. 50 (2002) 731–734.[15] G. Luurtsema, C.F. MolthoV, A.D. Windhorst, J.W. Smit, H. Keizer,
R. Boellaard, A.A. Lammertsma, E.J. Franssen, Nucl. Med. Biol. 30(2003) 747–751.
[16] C. Martin, G. Berridge, P. Mistry, C. Higgins, P. Charlton, R. Calla-ghan, Br. J. Pharmacol. 128 (1999) 403–411.
[17] C. Martin, G. Berridge, C.F. Higgins, R. Callaghan, Br. J. Pharmacol.122 (1997) 765–771.
[18] D.R. Ferry, P.J. Malkhandi, M.A. Russell, D.J. Kerr, Biochem. Phar-macol. 49 (1995) 1851–1861.
[19] R. Boer, M. Dichtl, C. Borchers, W.R. Ulrich, J.F. Marecek, G.D. Prest-wich, H. Glossmann, J. Striessnig, Biochemistry 35 (1996) 1387–1396.
[20] S. Dey, M. Ramachandra, I. Pastan, M.M. Gottesman, S.V. Ambud-kar, Proc. Natl. Acad. Sci. USA 94 (1997) 10594–10599.
[21] S. Dey, P. Hafkemeyer, I. Pastan, M.M. Gottesman, Biochemistry 38(1999) 6630–6639.
[22] N. Maki, P. Hafkemeyer, S. Dey, J. Biol. Chem. 278 (2003) 18132–18139.
[23] C.F. Higgins, K.J. Linton, Nat. Struct. Mol. Biol. 11 (2004) 918–926.[24] K. Pleban, S. Kopp, E. Csaszar, M. Peer, T. Hrebicek, A. Rizzi, G.F.
Ecker, P. Chiba, Mol. Pharmacol. 67 (2005) 365–374.[25] T.W. Loo, D.M. Clarke, J. Biol. Chem. 276 (2001) 14972–14979.[26] T.W. Loo, D.M. Clarke, Biochem. Biophys. Res. Commun. 329 (2005)
419–422.[27] X.V. Gomes, L.A. Henricksen, M.S. Wold, Biochemistry 35 (1996)
5586–5595.[28] J.G. Stout, Q. Zhou, T. Wiedmer, P.J. Sims, Biochemistry 37 (1998)
14860–14866.[29] A. Rothman, Y. Gerchman, E. Padan, S. Schuldiner, Biochemistry 36
(1997) 14572–14576.[30] P. Morsomme, S. Dambly, O. Maudoux, M. Boutry, J. Biol. Chem.
273 (1998) 34837–34842.[31] M. Julien, P. Gros, Biochemistry 39 (2000) 4559–4568.[32] G. Wang, R. Pincheira, M. Zhang, J.T. Zhang, Biochem. J. 328 (1997)
897–904.[33] G. Wang, R. Pincheira, J.T. Zhang, Eur. J. Biochem. 255 (1998) 383–
390.[34] M. Ramachandra, S.V. Ambudkar, D. Chen, C.A. Hrycyna, S. Dey,
M.M. Gottesman, I. Pastan, Biochemistry 37 (1998) 5010–5019.[35] U.A. Germann, M.C. Willingham, I. Pastan, M.M. Gottesman, Bio-
chemistry 29 (1990) 2295–2303.[36] J.L. Bailey, in, Elsevier Publishing Co., New York, 1967, pp. 340-341.[37] U.K. Laemmli, Nature 227 (1970) 680–685.[38] E.P. Bruggemann, V. Chaudhary, M.M. Gottesman, I. Pastan, Bio-
techniques 10 (1991) 202–209.[39] K. Hjelmeland, J. Raa, Comp. Biochem. Physiol. B 71 (1982) 557–562.[40] Y. Jacquot-Armand, G. Krebs, FEBS Lett. 4 (1969) 21–24.[41] N. Maki, K. Moitra, C. Silver, P. Ghosh, A. Chattopadhyay, S. Dey,
Biochemistry 45 (2006) 2739–2751.[42] F.J. Sharom, X. Yu, G. DiDiodato, J.W. Chu, Biochem. J. 320 (1996)
421–428.[43] U.S. Rao, S.L. Nuti, J. Biol. Chem. 278 (2003) 46576–46582.[44] N. Sonveaux, A.B. Shapiro, E. Goormaghtigh, V. Ling, J.M. Ruyss-
chaert, J. Biol. Chem. 271 (1996) 24617–24624.[45] N. Sonveaux, C. Vigano, A.B. Shapiro, V. Ling, J.M. Ruysschaert, J.
Biol. Chem. 274 (1999) 17649–17654.[46] T.W. Loo, M.C. Bartlett, D.M. Clarke, J. Biol. Chem. 278 (2003) 1575–
1578.[47] T.W. Loo, M.C. Bartlett, D.M. Clarke, J. Biol. Chem. 278 (2003)
13603–13606.[48] M.F. Rosenberg, G. Velarde, R.C. Ford, C. Martin, G. Berridge, I.D.
Kerr, R. Callaghan, A. Schmidlin, C. Wooding, K.J. Linton, C.F. Hig-gins, EMBO J. 20 (2001) 5615–5625.
[49] E.P. Bruggemann, U.A. Germann, M.M. Gottesman, I. Pastan, J. Biol.Chem. 264 (1989) 15483–15488.
[50] E. Georges, J.T. Zhang, V. Ling, J. Cell Physiol. 148 (1991) 479–484.[51] A. Yoshimura, Y. Kuwazuru, T. Sumizawa, M. Ichikawa, S. Ikeda, T.
Ueda, S.-I. Akiyama, J. Biol. Chem. 264 (1989) 16282–16291.[52] S. Dey, M. Ramachandra, I. Pastan, M.M. Gottesman, S.V. Ambud-
kar, Methods Enzymol. 292 (1998) 318–328.[53] E.B. Mechetner, I.B. Roninson, Proc. Natl. Acad. Sci. USA 89 (1992)
5824–5828.

P. Ghosh et al. / Archives of Biochemistry and Biophysics xxx (2006) xxx–xxx 13
ARTICLE IN PRESS
[54] E.B. Mechetner, B. Schott, B.S. Morse, W.D. Stein, T. Druley, K.A.Davis, T. Tsuruo, I.B. Roninson, Proc. Natl. Acad. Sci. USA 94 (1997)12908–12913.
[55] T.E. Druley, W.D. Stein, I.B. Roninson, Biochemistry 40 (2001) 4312–4322.
[56] A.B. Shapiro, V. Ling, Eur. J. Biochem. 250 (1997) 130–137.[57] A.B. Shapiro, K. Fox, P. Lam, V. Ling, Eur. J. Biochem. 259 (1999)
841–850.
[58] C. Martin, G. Berridge, C.F. Higgins, P. Mistry, P. Charlton, R. Calla-ghan, Mol. Pharmacol. 58 (2000) 624–632.
[59] T.W. Loo, M.C. Bartlett, D.M. Clarke, J. Biol. Chem. 278 (2003)39706–39710.
[60] T.W. Loo, M.C. Bartlett, D.M. Clarke, J. Biol. Chem. 278 (2003)50136–50141.
[61] T.W. Loo, D.M. Clarke, J. Biol. Chem. 277 (2002) 44332–44338.[62] T.W. Loo, D.M. Clarke, J. Biol. Chem. 275 (2000) 39272–39278.
Copyright © 2022 FDOKUMEN




















