
Inducible mouse models illuminate parameters influencing epigenetic inheritance
Upload
independentCategory
view
1download
0
Molecular and Cellular Pathobiology
ALK-Dependent Control of Hypoxia-Inducible FactorsMediates Tumor Growth and Metastasis
Cinzia Martinengo1,2, Teresa Poggio1,2, Matteo Menotti1,2, Maria Stella Scalzo2, Cristina Mastini1,2,Chiara Ambrogio3, Elisa Pellegrino1,2, Ludovica Riera1,2, Roberto Piva1,2, Domenico Ribatti4,5,Fabio Pastorino6, Patrizia Perri6, Mirco Ponzoni6, Qi Wang7, Claudia Voena1,2, and Roberto Chiarle1,2,7
AbstractRearrangements involving the anaplastic lymphoma kinase (ALK) gene are defining events in several
tumors, including anaplastic large-cell lymphoma (ALCL) and non–small cell lung carcinoma (NSCLC). Insuch cancers, the oncogenic activity of ALK stimulates signaling pathways that induce cell transformation andpromote tumor growth. In search for common pathways activated by oncogenic ALK across different tumorstypes, we found that hypoxia pathways were significantly enriched in ALK-rearranged ALCL and NSCLC, ascompared with other types of T-cell lymphoma or EGFR- and K-RAS–mutated NSCLC, respectively.Consistently, in both ALCL and NSCLC, we found that under hypoxic conditions, ALK directly regulatedthe abundance of hypoxia-inducible factors (HIF), which are key players of the hypoxia response in normaltissues and cancers. In ALCL, the upregulation of HIF1a and HIF2a in hypoxic conditions required ALKactivity and its downstream signaling proteins STAT3 and C/EBPb. In vivo, ALK regulated VEGFA productionand tumor angiogenesis in ALCL and NSCLC, and the treatment with the anti-VEGFA antibody bevacizumabstrongly impaired ALCL growth in mouse xenografts. Finally, HIF2a, but not HIF1a, was required for ALCLgrowth in vivo whereas the growth and metastasis potential of ALK-rearranged NSCLC required both HIF1aand HIF2a. In conclusion, we uncovered an ALK-specific regulation of the hypoxia response across differentALKþ tumor types and propose HIFs as a powerful specific therapeutic target in ALK-rearranged ALCL andNSCLC. Cancer Res; 74(21); 6094–106. �2014 AACR.
IntroductionThe anaplastic lymphoma kinase (ALK) is a receptor tyro-
sine kinase involved in chromosomal rearrangements in ana-plastic large-cell lymphoma (ALCL), non–small cell lung car-cinoma (NSCLC; ref. 1), and other solid cancers (2). Frequently,in ALCL the Nucleophosmin 1 (NPM1) gene and ALK generatethe NPM–ALK fusion, but more than 20 other partners of
ALK have been cloned in ALCL (2). In NSCLC, ALK rearrange-ments occur in 6% to 7% of the cases and mainly involve theechinoderm microtubule–associated protein-like 4 (EML4)gene as a partner (1) but additional ALK rearrangementshave also been described previously (3, 4).
The cell-transforming potential of ALK in tumors largelydepends on its deregulated tyrosine kinase activity that resultsfrom spontaneous dimerization (2). Several downstream path-ways are activated by NPM–ALK, with a broad range of signalsthat lead to increased cell proliferation, survival, motility, andcytoskeletal rearrangements (2, 5). In ALCL, ALK oncogenicsignals aremediated by a series of keymolecules and pathways,including STAT3, PI3K, RAS/MAPK/ERK, SHP2, p130CAS,PLCg , and Src (2, 5). In NSCLC, conversely, downstream path-ways are less extensively characterized (6).
Early clinical trials have shown that ALK-rearrangedtumors respond quite dramatically to the inhibition of theALK activity by specific inhibitors such as crizotinib (5). Highrate of responses were observed in patients with ALK-rear-ranged NSCLC (7) and ALCL (8). Unfortunately, the benefit ofALK inhibition is only transient because tumors relapsealmost invariably in patients due to the development ofresistance to crizotinib (9, 10). Therefore, novel therapeutictargets are needed for potential combination therapies withALK inhibitors.
Our group and others previously described a link betweenALK and angiogenesis in lymphoma (11) and neuroblastoma
1Department ofMolecular Biotechnology andHealth Sciences, University ofTorino, Torino, Italy. 2Center for Experimental Research andMedical Studies(CERMS), Torino, Italy. 3Molecular Oncology Program, Centro Nacional deInvestigaciones Oncol�ogicas, Madrid, Spain. 4Department of Basic MedicalSciences, Neurosciences and Sensory Organs, University of Bari MedicalSchool, Bari, Italy. 5National Cancer Institute "Giovanni Paolo II," Bari, Italy.6Experimental Therapy Unit, Laboratory of Oncology, G. Gaslini Children'sHospital, Genoa, Italy. 7Department of Pathology, Children's Hospital Bos-ton and Harvard Medical School, Boston, Massachusetts.
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
C.Martinengo, T. Poggio, andM.Menotti contributed equally to this article.
CorrespondingAuthors:Claudia Voena, Department ofMolecular Biotech-nologyandHealthSciences,UniversityofTorino,Torino, Italy,10126.Phone:39-0116336861; Fax: 39-0116336887; E-mail: [email protected]; andRoberto Chiarle, Department of Pathology, Children's Hospital Boston;Associate Professor in Pathology, Harvard Medical School, Enders1116.1, 300 Longwood Avenue, Boston, MA 02115. Phone: 617-919-2662; Fax: 617-730-0148; E-mail: [email protected]
doi: 10.1158/0008-5472.CAN-14-0268
�2014 American Association for Cancer Research.
CancerResearch
Cancer Res; 74(21) November 1, 20146094
on January 8, 2015. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 5, 2014; DOI: 10.1158/0008-5472.CAN-14-0268

(12). In ALCL, oncogenic ALK was shown to control VEGFexpression in vitro in murine cells and human ALCL (11, 13).Furthermore, ALK-dependent regulation of platelet-derivedgrowth factor receptor-b (PDGFRb) could contribute to tumorangiogenesis in a mouse model of ALK-rearranged lymphoma(14). In neuroblastoma, ALK-deregulated activity controlledboth tumor growth and tumor vessel formation (12).Tumor angiogenesis provides oxygen and nutrients to the
growing mass (15), and hypoxic conditions are critical tostimulate new vessel formation in tumors (16). The regulatorsof hypoxic response in the cells are transcription factors calledhypoxia-inducible factors (HIF). HIFs are heterodimers com-posed of an O2-labile a subunit (HIF1a, HIF2a, and the lesscharacterized HIF3a) and a stable b subunit (HIF1b). WhereasHIF1a is ubiquitously expressed, HIF2a and HIF3a have amore restricted pattern of expression, including vascularendothelial cells and type II pneumocytes (17). The role ofHIFs in different cancer types is variable and sometimescontradictory, sharing redundant functions but also opposingeffects on angiogenesis, metabolism, and other processes thatcan influence tumor growth (18).In the present study, we show that the hypoxia response is
specifically enriched in a large series of human ALK-rear-ranged lymphoma and NSCLC cases, and we provide evidencethat ALK specifically regulates HIF1a and HIF2a expressionunder hypoxia conditions in both ALCL and NSCLC. In vivo,ALK activity induced VEGF production and tumor angiogen-esis and we demonstrated that HIF2a, but not HIF1a, wasrequired for ALCL growth, whereas both HIF1a and HIF2awere essential for NSCLC growth and metastasis. Thus, wehave identified an ALK-dependent hypoxic response sharedby ALK-rearranged ALCL and NSCLC, and suggest VEGFAand HIFa proteins as powerful therapeutic targets for ALK-rearranged tumors.
Materials and MethodsGene-expression profiling and GSEAGene-expression profiling (GEP) data from T-cell lympho-
mas were generated by our group (19). Expression data forNSCLC are publicly available (20, 21).
Cell lines and reagentsALK-rearranged (TS, SU-DHL1, JB6, and Karpas-299) and
ALK� (CEM, FePD, JURKAT, and MAC-1) ALCL cell lineswere obtained from the DSMZ (German collection of Micro-organisms and Cell Cultures) collection and were previouslydescribed (22). Methods of characterization from the cellbank include karyotyping and DNA fingerprinting. Cells werepassaged for fewer than 6 months after receipt and resus-citation. Cells were internally tested by ALK and p53 muta-tional status within 6 months after receipt and resuscitation.EML4–ALK-rearranged H2228 (variant 2, EML4/ex6–ALK/ex20) and H3122 (variant 1, EML4/ex13–ALK/ex20), B-RAF–mutated H1395 (p.G469A), EGFR-mutated H1975 (p.L858R,T790M) and H3255 (p. L858R), and K-RAS–mutated A549(p.G12S), H441 (p.G12V), and H460 (p.Q61H) lung carcinomacell lines were obtained from the ATCC and DSMZ collec-
tions and were passaged for fewer than 6 months afterreceipt and resuscitation. These cell lines were furtherinternally tested for the mutational status of their respectivecharacterizing oncogene mutation (EML4-ALK, B-RAF,EGFR, and K-RAS). Cell were cultivated in presence ofNVP-TAE684 (synthesized by Axon Medchem), crizotinib(PF-02341066), deferoxamine mesylate (DFX; Sigma), doxy-cicline hyciclate (Sigma), MG-132 (Sigma), and bevacizumab(Genentech/Roche) for the indicated times and concentra-tions. For experiments in hypoxia, cells were cultured inHeracell 150i CO2 incubator (Thermo Scientific) at 1% O2.ELISA for hVEGFA was performed on supernatants collectedafter 8 hours of culture by the Quantikine human VEGFELISA Kit (R&D Systems) according to the manufacturer'sinstructions.
Lentiviral-mediated shRNA targetingLentiviral shRNA clones targeting HIF1a and HIF2a were
obtained from Sigma. Inducible shRNAs cells were obtainedby transduction of pLVtTRKRAB vector followed by pLVTHMvectors containing the cloned shRNA cassettes (23). ALK-,C/EBPb-, and Stat3-specific shRNAs have been previouslydescribed (23, 24). Retrovirus expressing NPM–ALK (25),STAT3C (26) or lentivirus expressing C/EBPb (23) have beenpreviously described.
ImmunoblottingTotal cellular proteins were extracted as previously
described (22). Primary antibodies used were: anti-HIF1a(1:800; Cell Signaling Technology), anti-HIF2a (1:800; NovusBiologicals), anti-ALK (1:2,000; Invitrogen), anti–phospho-ALK(1:1,000; Cell Signaling Technology), anti-actin (1:2,000; Sigma),anti–phospho-IKB (1:1,000; Cell Signaling Technology), anti-STAT3 (1:1,000; Cell Signaling Technology), anti-C/EBPb(1:1,000; Cell Signaling Technology), anti-EGFR (1:1,000; CellSignaling Technology), anti–phospho-EGFR (Y1068; 1:1,000;Cell Signaling Technology), anti-ERBB2 (1:1,000; Cell SignalingTechnology), and anti–phospho-ERBB2 (Y1221/1222; 1:1,000;Cell Signaling Technology).
Histology, immunohistochemistry, andimmunofluorescence
The following primary antibodies were used: CD34 (cloneMEC14.7; Santa Cruz Biotechnology), a-smooth muscle actin(clone 1A4; DakoCytomation), desmin (clone D33; DakoCyto-mation), VEGFA (clone 26503; R&D Systems), and Ki-67 (cloneMIB-1; DakoCytomation). Positive cells were evaluated onrandomly selected high power field (HPF; �100 objective)using an Image Analysis software (Olympus Italia).
Quantitative real-time PCRTotal cDNA was synthesized as previously described (22).
Normalization was performed against the housekeeping genesmurine actin for mouse samples and human RPLP0 for humansamples according to the formula 2�DDCt , where the DCt ¼ Ct(threshold cycle) gene of interest—Ct internal control, as indi-cated by the manufacturer. Primers used in quantitative real-time PCR (qRT-PCR) as indicated in Supplementary Table S1.
HIFs in ALK-Rearranged Lymphoma and Lung Carcinoma
www.aacrjournals.org Cancer Res; 74(21) November 1, 2014 6095
on January 8, 2015. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 5, 2014; DOI: 10.1158/0008-5472.CAN-14-0268

Mice strains and in vivo xenograft and metastasis assaysStrains of mice used in this study include K-RasLSLG12V (27),
TgEGFRL858R (28), B-RafLmLV600E (29), and EML4–ALK trans-genic mice (C. Voena; submitted for publication).
For in vivo induction of inducible lentiviral constructs, wedissolved doxycicline hydrochloride (MPBiomedicals, 1 g/L) inthe drinkable water. For ALK knockdown, samples were col-lected at 96 hours after induction to achieve the maximumbefore the induction of apoptosis (24).
Statistical analysisFor GEP, we used false discovery rate (FDR) to calculate the
statistical significance of enrichment scores. For the otherexperiments, statistical significance was calculated with theStudent t test, and only values lower than 0.05 were consideredsignificant.
ResultsHypoxia pathways are significantly enriched in ALK-rearranged ALCL and NSCLC
In the search for pathways significantly enriched in ALK-rearranged ALCL, we performed gene set enrichment analysis(GSEA) of GEP data in a series of 169 T-cell lymphoma cases,which included54ALCLsamples (30ALK-rearrangedALCLand24 ALK� ALCL), 74 peripheral T-cell lymphoma NOS (PTCL-NOS), and 41 angioimmunoblastic T-cell lymphoma. We foundthat several GSEA hypoxia datasets were significantly enrichedin ALCL, as compared with other T-cell lymphoma types(Supplementary Fig. S1A–S1C). Furthermore, when we furtherseparated ALCL into ALKþ and ALK� cases, GSEA indicatedthat hypoxia pathways enrichment was significantly associatedwith ALKþ ALCL cases (Fig. 1 and Supplementary Fig. S2), thussuggesting that the deregulated ALK activity in ALCL could bedirectly related to a hypoxia signature. We next asked whetherhypoxia pathways were enriched also in other tumors expres-sing ALK translocations. We compared the GEP dataset fromEML4–ALK-rearranged NSCLC with EGFR-mutated NSCLC, inwhich EGFR-L858R or EGFR-Del19 mutations aberrantly acti-vate the EGFR tyrosine kinase receptor. Hypoxia pathwaysweresignificantly enriched in ALK-rearranged NSCLC as comparedwith EGFR-mutated cases in two independent GEP datasets(Supplementary Fig. S3; refs. 20, 21). Thus, we concluded thatALK rearrangements are associated with an enriched hypoxiasignature in both ALCL and NSCLC.
ALK regulates HIFs expression in ALCL via STAT3- andC/EBPb-dependent transcription
Because hypoxia pathways are largely dependent on theactivity of HIFs, we asked whether ALK activity could controlHIFs expression in human cell lines derived from patientswith ALK-rearranged ALCL. In all NPM–ALK-rearrangedALCL cell lines tested (SU-DHL-1, TS, JB6, and KARPAS-299), levels of HIF1a and HIF2a were low in standard 21%oxygen conditions, but were strongly upregulated in hypoxiccondition (1% oxygen; Fig. 2A) or after treatment with thechemical compound that mimics hypoxia DFX (Supplemen-tary Fig. S4). Conversely, in hypoxic condition ALK� ALCL
cell lines (FeDP and MAC-1) as well as other T-cell Lym-phoma lines (Jurkat and CEM) showed upregulation onlyof HIF1a and no changes in HIF2a expression (Fig. 2B).When we treated ALK-rearranged ALCL with the specificALK inhibitors TAE684 or crizotinib to block the kinaseactivity of the NPM–ALK fusion, ALK phosphorylation wasstrongly reduced and HIF1a and HIF2a protein levels werestrongly reduced in both normoxic and hypoxic conditions(Fig. 2A and Supplementary Fig. S4). In contrast, HIF1aexpression was modestly and inconsistently reduced inALK� lymphoma cell lines, possibly due to some nonspecificeffects of ALK inhibitors (Fig. 2B). To further confirm therole of ALK in HIFs expression, we also performed a geneticknockdown of ALK by shRNA. Again, efficient knockdown ofALK induced a marked reduction of both HIF1a and HIF2aprotein levels (Supplementary Fig. S5), thus supportingour conclusions that in ALK-rearranged ALCL the expres-sion of both HIF1a and HIF2a in hypoxic conditions isspecifically regulated by the activity of ALK. Furthermore,although HIF1a upregulation in hypoxic conditions wasdetectable in all lymphoma cell lines, HIF2a upregulationappeared to be a specific feature of ALKþ ALCL. To furthersupport the ALK specificity of HIF2a regulation, we analyz-ed the expression levels of HIF2a in ALKþ ALCL comparedwith ALK� ALCL and other T-cell lymphoma. Indeed, HIF2aexpression was significantly higher in ALK-rearranged ALCLas compared with the other T-cell lymphoma analyzed(Fig. 2C).
HIFa proteins are regulated in normoxic and hypoxiccells through various mechanisms that include protea-some-dependent degradation and transcription (16, 18, 30).In well-oxygenated conditions, hydroxylation of HIFa sub-units induces their proteosomal degradation by an E3 ubi-quitin ligase, the von Hippel–Lindau protein (pVHL) complex.In contrast, in hypoxic conditions, HIFa proteins are stabi-lized by a decreased proteasome-dependent degradation.Therefore, we asked whether the low levels of HIFa proteinsduring ALK inhibition could be explained by an increasedproteasome-dependent degradation. To test this hypothesis,we treated ALCL with the proteasome inhibitor MG132. Theinhibition of the proteasome-dependent degradation byMG132 clearly stabilized the known degradation target phos-pho-IKBa but it was unable to restore high levels of HIFaproteins in ALCL treated with the ALK inhibitor, thus rulingout proteasome-dependent degradation as a major mecha-nism of ALK-dependent HIFa regulation (SupplementaryFig. S6).
Next, we asked whether ALK regulated HIFa proteins bya transcription-dependent mechanism. By qRT-PCR, ALCLcells treated with an ALK inhibitor showed a significantreduction of both HIF1a and HIF2a mRNA levels (Fig. 2D),thus indicating that ALK controls HIFa expression at atranscriptional level. Because we and others previouslyshowed that both STAT3 and C/EBPb are key transcriptionfactors in the oncogenic ALK signaling (2, 23, 31–33), andSTAT3 has been shown to regulate HIF1a transcription (13),we tested whether they could be involved in ALK-dependentHIFa protein regulation. In ALCL cells, shRNA-mediated
Martinengo et al.
Cancer Res; 74(21) November 1, 2014 Cancer Research6096
on January 8, 2015. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 5, 2014; DOI: 10.1158/0008-5472.CAN-14-0268

knockdown of either STAT3 or C/EBPb resulted in a markedreduction of both HIF1a and HIF2a protein levels (Sup-plementary Fig. S7). Consistently, ectopic overexpressionof C/EBPb and/or a constitutively active form of STAT3(STAT3C; ref. 34) partially rescued HIF2a upregulationunder hypoxic conditions even in the presence of the ALKinhibitor (Supplementary Fig. S8). In contrast, in ALK� celllines, basal levels of STAT3 phosphorylation and C/EBPb
were low but increased after ectopic expression of NPM–ALK. Still, HIF2a expression remained undetectable evenunder hypoxic conditions, possibly suggesting a block ofHIF2a expression in ALK� lymphoma cells mediated byother mechanisms such as epigenetic silencing (Supplemen-tary Fig. S9). Therefore, we concluded that in ALK-rearrang-ed ALCL cells both STAT3 and C/EBPb are key transcriptionfactors in the ALK-mediated regulation of HIFa proteins.
ALK+ ALK–
ALCL
PYGLANLNPVRLDHARHOC
PPP4R1S100A10SLC2A1TPD52L2GAPDHTUBB2CMNAT1TEAD4BNIP3PGK1TANC2CA12TPI1C200RF20MTX1CNIH4ANGPTL4PMSD2TNS4
ALDOA
PSMA7CDCA4LDLRRUVBL2NDUFA4L2VEZTGMFBSIP1PSMB7TRMT5DPM2ANKRD9TPBGRANIGF2BP2PDZD11VAPBPTGFRNMIFC140RF156NDRG1C160RF74SLC16A1S100A3BCAR1PFKFB4C160FR68B4GALT2RNPS1NUDT15GSSPPARDEIF2S1AK3L1HES2NME1SNX24RNF24TMEM30BECE2MRPL14CA9CORO1CADORA2BP4HA1KCTD11TMTC3KRT17IL8HOMER1ANKRD37TFAP2CSLC6A8PLEKHG3XPO5COL4A5SLCO1B3PGFPLAU
A
B
Enrichment plot: WINTER_HYPOXIA_UP
0.6
0.5
0.4
0.3
0.2
0.1
0.0
1.5
1.0
0.5
0.0
–0.5
–1.0
0 2,500 5,000 7,500 10,000Rank in ordered dataset
Zero cross at 11,964
‘0’ (negatively correlated)
‘1’ (positively correlated)
Enrichment profile Hits Ranking metric scores
Ran
ked
list m
etric
(si
gnal
2Noi
se)
Enr
ichm
ent s
core
12,500 15,000 17,500 20,000
Figure 1. ALK-rearranged ALCLs show enriched expression of hypoxia pathway–associated genes. A, the GSEA plot of Winter_hypoxia_uppathway based on ALK-rearranged ALCL versus ALK� ALCL GEP. B, heatmap of the genes enriched in the indicated hypoxia pathway. Hypoxia FDRq value ¼ 0.18, P < 0.005.
HIFs in ALK-Rearranged Lymphoma and Lung Carcinoma
www.aacrjournals.org Cancer Res; 74(21) November 1, 2014 6097
on January 8, 2015. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 5, 2014; DOI: 10.1158/0008-5472.CAN-14-0268

A
HIF1a
p-ALK
ALK
Actin
HIF2a
TAE684
Crizotinib
TS
– –++– –– +––– +
Hy HyHyNN N
SU-DHL-1
– –++– –– +––– +
Hy HyHyNN N
KARPAS-299
– –++– –– +––– +
Hy HyHyNN N
JB6
– –++– –– +––– +
Hy HyHyNN N
HIF1a
p-ALK
ALK
Actin
HIF2a
TAE684
Crizotinib
JURKAT
– –++– –– +––– +
Hy HyHyNN N
MAC-1
– –++– –– +––– +
Hy HyHyNN N
FePD
– –++– –– +––– +
Hy HyHyNN N
CEM
– –++– –– +––– +
Hy HyHyNN N
B
0.181.000.561.00 0.291.000.060.01 0.181.000.17 0.2
0.011.00 1.001.00 0.210.381.000.130.070.020.07 0.02
1.00 0.27 1.00 0.91 51.100.153.116.0 0.78 1.00 0.730.73
C
SU-DHL1TS
DMSO
TAE684
Fo
ld c
han
ge
HIF1a
***
00.20.40.60.81.01.2
Fo
ld c
han
ge
HIF2a
*
00.20.40.60.81.01.2
Fo
ld c
han
ge
HIF1a
**
00.20.40.60.81.01.2
Fo
ld c
han
ge
HIF2a
***
00.20.40.60.81.01.2
D
ALK+ALK–
Exp
ress
ion
ALK (P = 0.0001)
ALK+ALK–
Exp
ress
ion
TNFRSF8/CD30 (P = 0.39)
ALK+ALK–
Exp
ress
ion
HIF2α (P = 0.005)
ALK+ALK–
Exp
ress
ion
HIF1α (P = 0.055)
6,000
4,000
2,000
0
6,0005,000
4,000
3,000
2,000
1,000
0
4,000
2,000
50
100
150
200
0
Martinengo et al.
Cancer Res; 74(21) November 1, 2014 Cancer Research6098
on January 8, 2015. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 5, 2014; DOI: 10.1158/0008-5472.CAN-14-0268

HIF2a, but not HIF1a, is essential for tumor growth inALCLAswe showed thatHIF1a andHIF2a are regulated by theALK
activity in ALK-rearranged ALCL, we next seek to establish theirbiologic roles inALCL growth. In a previous in vitro study inALK-rearranged ALCL cell lines, HIF1a depletion has been shown toincrease cell proliferation but to decrease VEGF synthesis (13),thus suggesting potentially contrasting effects for ALCL growthin vivo. Indeed, both HIF1a and HIF2a have been shown topossess contradicting roles in vitro and in vivo, mostly due to thedifferent types of tumor analyzed (18, 30). Therefore, we decidedto test directly in vivo the role of HIF1a and HIF2a in ALK-rearranged ALCL tumor growth by xenograft assays. We gener-
ated inducible lentiviral vectors to specifically knockdownHIF1aor HIF2a proteins upon treatment with doxycycline (Supple-mentary Fig. S10). When HIF1a was knocked down by treatingmice with doxycycline, we did not observe any impairment oftumor growth in vivo as compared with control untreated mice(Fig. 3A and C), consistent with previous in vitro results (13). Incontrast, HIF2a knockdown strongly impaired the growth ofALCL tumors in vivo (Fig. 3B andD and Supplementary Fig. S11).Remarkably, the knockdown of HIF1a or HIF2a did not reducetumor growth in ALK� lymphomas (Supplementary Fig. S12).Finally, when we induced the HIF2a knockdown in establishedtumors, they arrested their growth and partially regressed (Fig.3B and D, arrows). In sum, our data show that ALK specifically
Figure 2. Oncogenic ALK controls HIF1a and HIF2a expression by transcription-dependent mechanisms. A, NPM–ALK-rearranged ALCLs wereincubated for 8 hours in normoxic (N, 21% O2) or hypoxic (Hy, 1% O2) conditions with or without the ALK inhibitors TAE684 (300 nmol/L) andcrizotinib (300 nmol/L), as indicated. Western Blot analyses were performed with the indicated antibodies. HIF1a and HIF2a levels in cells in hypoxicconditions and treated with ALK inhibitors were quantified by densitometry, normalized to actin, and represented as fold changes compared withthe corresponding expression levels in cells without ALK inhibitors. B, ALK� lymphoma cell lines were treated and analyzed as in A. C, ALKþ
ALCL cases show higher expression levels of HIF2a as compared with ALK� ALCL. Box-plot showing the expression of ALK, TNFRSF8/CD30, HIF1a,and HIF2a, in 30 ALKþ and 24 ALK� ALCL, calculated on the basis of GEP data. The paired t test P value is indicated for each gene transcript.D, qRT-PCR for HIF1a and HIF2amRNA was performed in ALK-rearranged ALCL cell lines treated with TAE684 (300 nmol/L) for 8 hours; error bars, theSD of the mean; �, P < 0. 05; ��, P < 0.005; ���, P < 0.0001.
shHIF1a #1 shHIF2a #1
0 5 10 15 200
2
4
6
8
10
12
14
Days
Tum
or
dia
met
er(m
m) – Doxy
+ DoxyDoxy
0 5 10 15 20 25 30 35 400
2
4
6
8
10
12
14
Days
Tum
or
dia
met
er(m
m)
*
– Doxy + Doxy
TS
SU-DHL-1
shHIF1a #1
0 5 10 15 20 250
2
4
6
8
10
12
14
Days
Tu
mo
r d
iam
eter
(m
m) – Doxy
+ Doxy
shHIF2a #1
**
0
2
4
6
8
10
12
14
Days
Tu
mo
r d
iam
eter
(m
m)
Doxy
– Doxy+ Doxy
4035302520151050
A B
C D
Figure 3. HIF2a is essential for the growth of ALK-rearranged ALCL in vivo. SU-DHL-1 (A and B) and TS (C and D) ALCL cell lines were transduced with adoxycycline-inducible shRNA against HIF1a (A–C) or HIF2a (B–D) and injected s.c. in the flank of NOD/SCID mice (n ¼ 6 for each group). For treated mice(þDoxy), treatment with doxycycline started the same day of injection. Tumor diameter was measured over time. To study the role of HIF2a in alreadyestablished tumors,weadministereddoxycycline to theuntreatedcontrol groupwhen the tumor size reached8 to10mm indiameter, as indicatedby anarrow;error bars, the SEM; �, P < 0.05; ��, P < 0.0001.
HIFs in ALK-Rearranged Lymphoma and Lung Carcinoma
www.aacrjournals.org Cancer Res; 74(21) November 1, 2014 6099
on January 8, 2015. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 5, 2014; DOI: 10.1158/0008-5472.CAN-14-0268

regulated HIF2a levels in lymphoma cells and that HIF2a isessential for ALCL growth and maintenance in vivo.
ALK-mediated VEGFA production is a therapeutic targetin ALCL
The induction of hypoxia pathways mediated by ALK couldcontribute to the regulation of angiogenesis and metabolismof ALCL cells. Indeed, previous reports have shown thatpatients with ALK-rearranged ALCL have higher VEGFA levelsthan ALK� ALCL (11), that ALK regulates VEGFA secretion infibroblasts in vitro (11), and that HIF1a contributes at leastpartially to VEGFA secretion (13). However, it is not knownwhether ALK-dependent VEGFA secretion has a role on tumor
angiogenesis in vivo and whether the therapeutic blockade ofVEGFA could be an effective therapeutic approach in ALCL. Toaddress these points, we first confirmed in vitro that VEGFAmRNA and protein levels are regulated by ALK also in ALCL(Supplementary Fig. S13). Next, we showed that expression ofVEGFA was downregulated in ALCL when ALK was knockeddown by specific shRNA in vivo (Fig. 4A–C). Because VEGFAregulates tumor vessels formation (35), we expected that areduced production of VEGFA would decrease the formationand the number of tumor vessels. Indeed, ALCL tumors inwhich ALK was knocked down, and therefore VEGFA wasreduced, showed a significant decrease in tumor vessels, asmeasured by CD34, SMA, and Desmin stainings (Fig. 4D).
C
TS
SU-DHL-1
shALKshCtrl
VE
GFA
TS
020
40
60
80
100
VE
GFA
+ ce
lls (
%) *
SU-DHL1
VE
GFA
+ ce
lls (
%)
0
20
40
60
80
100 *
shCtrl
shALK
A BshALKshCtrl
SU-DHL-1
+–+–
shALKshCtrl
ALK
Actin
Doxy +–+–
TS
ED
CtrlBevacizumab
SU-DHL-1
0 5 10 15 20 25 30 350
10
20
30
40
Days
Tum
or
dia
met
er(m
m)
**
TS
0 5 10 15 20 250
5
10
15
DaysTum
or
dia
met
er(m
m)
**
CtrlBevacizumab
shCtrlshALK
020
60
100
Des
min
+ ce
lls *
*
Des
min
shALK shCtrl
CD
34S
MA
0
20
40
60
CD
34+
cells
*
010
30
50
SM
A+
cells
Figure 4. VEGFA production is regulated by ALK and is a target for therapy in ALCL. A, TS and SU-DHL-1 ALCL cells were transduced with lentivirusexpressing doxycycline-inducible shRNA against ALK (shALK) or a mutated control sequence (shCtrl), as we previously published (24). B and C, tumorxenografts were obtained by s.c. injection in NOD/SCID mice of 1 � 107 ALCL cells. Immunohistochemistry for VEGFA was performed in TS or SU-DHL-1 tumor xenografts 96 hours after in vivo treatment of mice with doxycycline. C, histograms, counts of VEGFA-positive cells/HFP in at least threeindependent areas for each tumor. Data were collected from at least four tumors in each group; error bars, SD; �, P < 0.001. D, in the sametumor xenografts as in C from SU-DHL-1 cells immunofluorescence stainings for CD34, SMA, and Desmin were performed to measure tumor vesselformation; error bars, SD; �, P < 0.001. E, a total of 1 � 107 TS and SU-DHL-1 ALCL cells were injected s.c. in NOD/SCID mice. Treatment withbevacizumab started at day 3 and continued twice a week for 3 weeks. Data are from at least six tumors for each group; ��, P < 0.005.
Martinengo et al.
Cancer Res; 74(21) November 1, 2014 Cancer Research6100
on January 8, 2015. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 5, 2014; DOI: 10.1158/0008-5472.CAN-14-0268

To exploit ALK-induced VEGFA production as a target fortherapeutic strategy, we treated mice that received ALCLxenografts with the VEGFA blocking antibody bevacizumabthat is currently used in several clinical trials for differentcancer types (36). Remarkably, ALCL tumors in mice treatedwith bevacizumab grew significantly slower than in controlmice, thus indicating an important role for the ALK-dependentVEGFA production in ALCL growth (Fig. 4E). The growth ofALK� lymphoma xenografts was also impaired by bevacizu-mab (Supplementary Fig. S14), thus indicating that in ALK�
lymphoma VEGFA secretion could be regulated by differentmechanisms but still be important for tumor growth. We thusconcluded that VEGFA production induced by oncogenic ALKregulates tumor vessels formation in ALCL and can be apotential target for ALCL therapy.
Oncogenic ALK regulates HIF1a and HIF2a and VEGFAexpression in NSCLC
We showed that hypoxia pathways are enriched also inother ALK-rearranged tumor such as EML4–ALK-rearrangedNSCLC (Supplementary Fig. S3). Therefore, we asked whetheroncogenic ALK regulated HIFa proteins and VEGF expressionalso in NSCLC. Treatment with DFX to mimic hypoxic condi-tions induced increased levels of both HIF1a or HIF2a inseveral NSCLC cell lines, including EML4–ALK-rearrangedH2228 and H3122 (Fig. 5A), K-Ras–mutated A549, H460, andH441 (Fig. 5B), and EGFR-mutated H1975 and H3255 (Fig.5C), whereas the B-RAF–mutated cell line H1395 showed onlyHIF1a upregulation (Fig. 5D). However, treatment of NSCLCcell lines with the ALK inhibitors TAE684 or crizotinib-impaired HIF1a and HIF2a protein upregulation only in
A B
C D
A549 (KRASG12S)
– –++– –– +––– +
+ ++–– –
HIF1a
p-ALK
ALK
Actin
HIF2a
TAE684Crizotinib
DFX
H441 (KRASG12V)
– –++– –– +––– +
+ ++–– –
H460 (KRASQ61H)
– –++– –– +––– +
+ ++–– –
1.021.001.531.00 0.731.000.89 0.61.56
1.001.001.00 0.541.181.010.81 0.83 0.6
H3122 (EML4-ALK)
– –++– –– +––– +
+ ++–– –
H2228 (EML4-ALK)
– –++– –– +––– +
+ ++–– –
HIF1a
p-ALK
ALK
Actin
HIF2a
TAE684Crizotinib
DFX
0.461.00 1.00 0.490.09 0.35
1.00 1.00 0.110.22 0.090.01
HIF1a
p-ALK
ALK
Actin
HIF2a
TAE684Crizotinib
DFX
1.00 0.75 1.00 0.511.030.79
H3255 (EGFRL858R)
– –++– –– +––– +
+ ++–– –
1.00 1.331.66
H1975 (EGFRL858R)
– –++– –– +––– +
+ ++–– –
1.00 0.660.73
H1395 (B-RAF G469A)
– –++– –– +––– +
+ ++–– –
1.00 1.660.55
HIF1a
p-ALK
ALK
Actin
HIF2a
TAE684Crizotinib
DFX
0
0.05
0.10
0.15
0.20
HIF1a
*
**
2-DC
t
2-DC
t
E
0
0.20
0.40
0.60
0.80
HIF2a
*
**
F
Figure 5. ALK kinase activity regulates HIF1a and HIF2a in NSCLC cells. EML4–ALK-rearranged H2228 and H3122 (A), K-RAS–mutated A549, H460,and H441 (B), EGFR-mutated H1975 and H3255 (C), B-RAF–mutated H1395 (D) NSCLC cell lines were incubated for 16 hours with DFX (100 mmol/L) tomimic hypoxic conditions and treated with TAE684 (300 nmol/L) or crizotinib (300 nmol/L) as indicated. Western blot analyses were performed withthe indicated antibodies. HIF1a and HIF2a levels were normalized to actin and represented as fold changes compared with the correspondingexpression levels in cells without ALK inhibitors. E and F, HIF1a and HIF2a mRNAs are higher in tumors from EML4–ALK transgenic mice than fromother oncogenic mutations as demonstrated by qRT-PCR analysis for HIF1a (E) and HIF2a (F) mRNA levels performed on cDNA from fiveindependent tumors dissected for each GEM; �, P < 0.05; ��, P < 0.005.
HIFs in ALK-Rearranged Lymphoma and Lung Carcinoma
www.aacrjournals.org Cancer Res; 74(21) November 1, 2014 6101
on January 8, 2015. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 5, 2014; DOI: 10.1158/0008-5472.CAN-14-0268

ALK-rearranged H2228 and H3122 (Fig. 5A). Consistently, ALKknockdown by specific shRNA reduced HIF1a or HIF2a upre-gulation under DFX treatment (Supplementary Fig. S15).
Given that in ALK-rearranged NSCLC, the EGFR familymembers are active and share common signaling pathwayswith ALK that can compensate for an inhibited ALK signaling(6, 37), we next investigated whether inhibition of EGFRsignaling would affect HIF1a or HIF2a expression. In contrastwith ALK inhibitors, HIF1a and HIF2a protein levels wereminimally affected by treatment with gefitinib, an EGFR-specific inhibitor (Supplementary Fig. S16A), or with lapatinib,a broader EGFR and ERBB2 inhibitor (Supplementary Fig.S16B). Overall, these data indicate a specific role for ALK inthe regulation of HIF1a and HIF2a in ALK-rearranged NSCLC.
To further prove the specific role of oncogenic ALK in HIFaregulation, we studied HIFa expression in genetically engi-neered mice (GEM) harboring the most common mutationsfound in NSCLC.We compared HIF1a andHIF2amRNA levelsin tumors isolated from GEM carrying the EML4–ALK trans-location or the K-RasV12G, EGFRL858R, or B-RafV600E mutations(Supplementary Fig. S17A). We observed high HIF1a expres-sion in K-Ras–mutated tumors that confirmed previous obser-vations that oncogenic K-Ras increases HIF1a expressionlikely by reactiveoxygen species generation (Fig. 5E; refs. 30, 38).Strikingly, HIF1a transcripts in EML4–ALK tumors werealmost as high as K-Ras–mutated tumors, and significantly
higher than HIF1a transcripts in normal lung, EGFRL858R- orB-RafV600E–mutated tumors (Fig. 5E). When we analyzedHIF2a mRNA levels, as expected, we found high expressionin normal lung given that HIF2a is highly expressed invessels and alveolar type II pneumocytes (17). Remarkably,EML4–ALK mice showed significantly higher HIF2a tran-scripts than all the other lung cancer genotypes (Fig. 5F).
Consistent with these data on HIFa regulation mediated byALK inNSCLC, inGEMmodels VEGFA levels were significantlyhigher in EML4–ALK than in K-RasV12G–, EGFRL858R-, orB-RafV600E–mutated tumors (Supplementary Fig. S17B) andthe inhibition of ALK decreased VEGFA expression in humanNSCLC (Supplementary Fig. S17C), thus indicating a directcontrol of VEGFA by oncogenic ALK also in NSCLC, as weshowed in ALCL.
HIF1a and HIF2a regulate tumor growth, vesselformation, and metastasis in ALK-rearranged NSCLC
We next wished to examine the functions of HIFa pro-teins in ALK-rearranged NSCLC growth and metastasisformation. Knockdown of both HIF1a and HIF2a did nothave significant effect on cell growth in vitro (data notshown) but significantly impaired the growth of ALK-rear-ranged NSCLC xenografts in both H2228 and H3122 EML4–ALK NSCLC cell lines (Fig. 6A, C, E and G). Reduced growthwas associated with reduced proliferation, as determined
H2228 shHIF1a H2228 shHIF2aA
H3122 shHIF1a
*
0 5 10 15 20 25 30 350
2
4
6
8
Days
Tum
or
dia
met
er (
mm
) - Doxy
+ Doxy
**
0 5 10 15 20 25 300
2
4
6
8
10
12
14
16
Days
Tum
or
dia
met
er (
mm
) – Doxy
+ Doxy
–Doxy +Doxy0
10
20
30
40
50
Ki-
67+
cells
(%)
***
–Doxy +Doxy0
10
20
30
40
50
Ki-
67+
cells
(%
)
*
–Doxy +Doxy0
10
20
30
40
50
Ki-
67+
cells
(%
)
**
–Doxy +Doxy0
10
20
30
40
50
Ki-
67+
cells
(%
)
*
H3122 shHIF2a
**
0 5 10 15 20 25 30 35 400
2
4
– Doxy
+ Doxy6
8
Days
Tum
or
dia
met
er(m
m)
**
0 5 10 15 20 25 300
2
4
8
10
12
Days
Tum
or
dia
met
er(m
m)
6
– Doxy
+ Doxy
DCB
HGFE
Figure 6. HIF1a and HIF2a are essential for the growth of ALK-rearranged NSCLC tumors. H2228 and H3122 EML4–ALK NSCLC cell lines were transducedwith lentiviral vectors expressing doxycycline-inducible shRNAagainst HIF1a (A andE) andHIF2a (C andG) andwere injected s.c. (1�107 cells) inNOD/SCIDmice. Treatment with doxycycline started the same day of injection. Tumor growth was measured over time; error bars, SEM from six independenttumors for eachgroup.B,D, F, andH, histograms, thepercentagesofKi-67–positive cells in tumors isolated from thecorrespondinggroupsofmice.Mean andSDs are calculated on the basis of six independent areas from at least six independent mice for the group; �, P < 0.05; ��, P < 0.005; ���, P < 0.001.
Martinengo et al.
Cancer Res; 74(21) November 1, 2014 Cancer Research6102
on January 8, 2015. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 5, 2014; DOI: 10.1158/0008-5472.CAN-14-0268

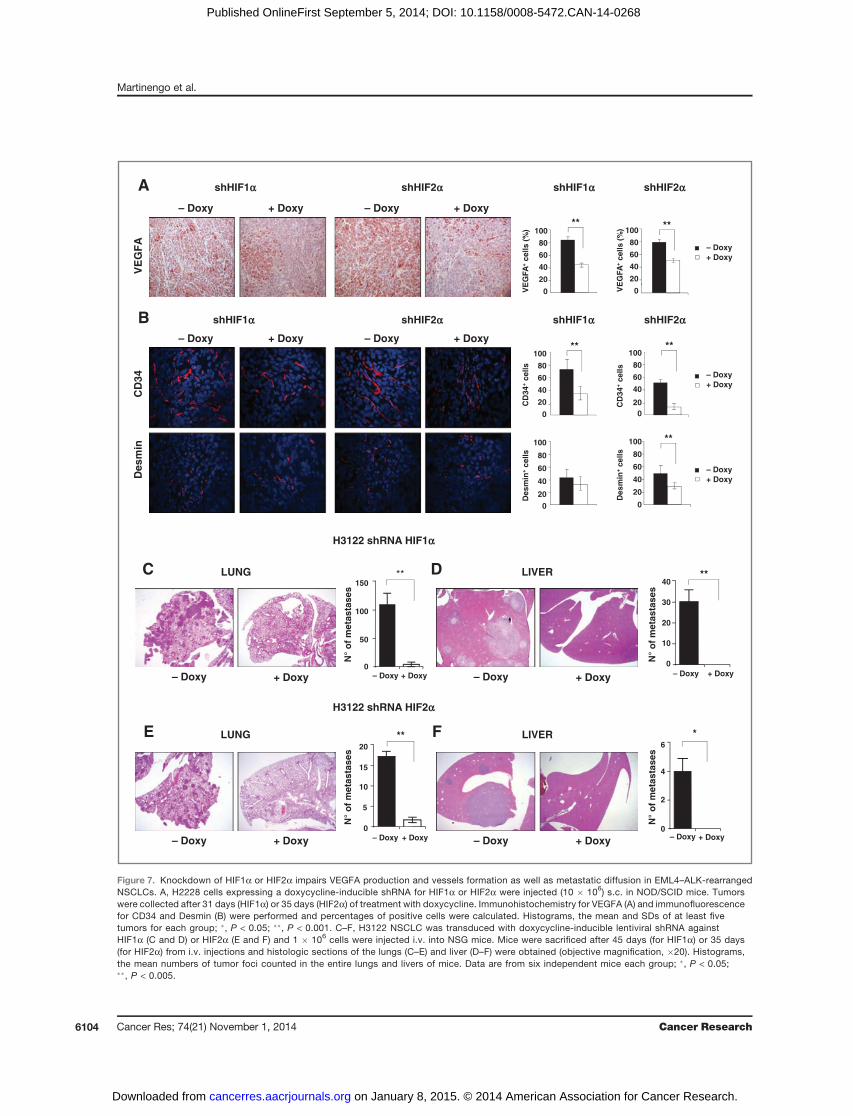
by Ki-67 stainings (Fig. 6B, D, F and H). In contrast, in theK-RasG12S–mutated NSCLC cell line HIF1a knockdown didnot affect tumor growth or proliferation, whereas HIF2aknockdown slightly increased tumor growth (SupplementaryFig. S18), in accordance with previously published observa-tions in K-Ras–mutated GEM (39). In ALK-rearrangedNSCLC xenografts, HIF1a or HIF2a knockdown induced adecrease in VEGFA production (Fig. 7A) and tumor vesselformation (Fig. 7B).Finally, because ALK-rearranged NSCLCs are typically asso-
ciated with advanced disease (stage IV; ref. 40) and metastaticspread, in particular liver metastasis (41), and because HIFshave been described to regulate tumormetastasis formation insolid cancers (18), we tested the roles of HIFa proteins inmetastatic assays in vivo. Strikingly, either HIF1a or HIF2aknockdown almost completely abolished the metastaticpotential of ALK-rearranged NSCLC in the lungs and in theliver (Fig. 7C–F and Supplementary Fig. S19), thus indicatingnonredundant functions ofHIF1a orHIF2a in ALK-rearrangedNSCLC metastasis formation.
DiscussionIn the present study, we showed that the ALK activity
specifically controls hypoxia pathways and tumor vesselsformation in ALK-rearranged ALCL andNSCLC. Together withthe evidences on the role of ALK in neuroblastoma tumorangiogenesis (12), collectively these data support the conceptthat controlling the hypoxia response could be one majorfunction of oncogenic ALK in various tumor types. Both HIF1aand HIF2a were potently regulated by ALK in hypoxic condi-tions inALKþALCL.Of note, we propose thatHIF2a regulationis a specific feature of ALK-rearranged ALCL, as ALK� ALCLand cell lines showed significantly lower levels of HIF2a and alack of HIF2a regulation under hypoxic conditions. Remark-ably, we showed that both STAT3 and C/EBPb transcriptionfactors are required to sustain hypoxia-induced HIF2a upre-gulation in ALK-rearranged ALCL.Because HIF1a and HIF2a have unique and sometimes
opposing functions in tumor biology (18), their precise roleas oncogenes or tumor suppressor cannot be predicted andneeds validation in each tumor in vivo. For example, in amouse model of K-Ras–mutated NSCLC, HIF2a deletionincreased tumor progression, thus acting as a tumor sup-pressor rather than an oncogene (39). In contrast, HIF2asupports tumor growth in renal cell carcinoma (42). Weshowed that HIF1a was dispensable for ALCL growth, thusconfirming previous results in vitro (13), whereas HIF2a wasrequired not only for ALCL growth but also for its mainte-nance. Because it has been shown that ALK regulateslymphoma proliferation by sustaining c-myc levels in ALCL(43) and that HIF2a regulates c-myc expression (44), oneintriguing possibility is that ALK could promote lymphomaproliferation by an HIF2a-mediated c-myc regulation.Importantly, in ALCL oncogenic ALK not only specificallyregulated HIF2a but lymphoma cells were addicted to itsfunctions, suggesting that HIF2a targeting could work as anadditional therapeutic option for ALCL.
With striking similarities to ALK-rearranged ALCL,EML4–ALK rearranged NSCLC showed enriched hypoxiapathways when compared with NSCLC carrying other com-mon driver mutations, such as EGFR and K-RAS mutation.Regulation of HIF1a and HIF2a in ALK-rearranged NSCLCwas less potent but consistent with that observed in ALK-rearranged ALCL. One possible explanation for such differ-ence could be that rearranged ALK is expressed at higherlevels in ALCL than in NSCLC (2, 45). As a result, oncogenicALK activity is more dominant in ALCL than in NSCLCin terms of signaling pathway activation. In contrast withALCL, in which oncogenic ALK activity dominates thesignaling, the ALK blockade results in a milder inhibitionof the same downstream pathways and in milder biologiceffects in NSCLC (46). Indeed, STAT3 phosphorylation isonly minimally impaired by ALK inhibitor treatment (10),whereas C/EBPb is not expressed in ALK-rearranged NSCLC(data not shown), indicating that other pathways are likelyinvolved in HIFs regulation in NSCLC. In this context, it isknown that several additional tyrosine kinases can contrib-ute to the biology of ALK-rearranged NSCLC (9). We exclud-ed a role for EGFR family receptors by gefitinib or lapatinibtreatment as well as for c-MET because the selective ALKinhibitor TAE684 or the specific ALK shRNA knockdown hadsimilar effects to the dual ALK–MET inhibitor crizotinib butit is possible that other tyrosine kinases, such as PDGFR orc-KIT, or other signaling pathways active in NSCLC wouldpartially compensate for ALK inhibition. This "by pass"compensation of the ALK activity is also a recurrent mech-anism that explains resistance to ALK inhibitors in NSCLCcells (9). Of note, knockdown of either HIF1a or HIF2ainhibited not only tumor growth but also metastasis forma-tion in EML4–ALK-rearranged NSCLC, therefore indicatingthat, in contrast with ALCL, in ALK-rearranged NSCLC bothHIFa proteins act as oncogenes and could serve as potentialtargets for therapy.
Oncogenic ALK activity is also required for VEGFA produc-tion by ALCL and NSCLC cells. ALK knockdown significantlyreduced VEGFA production and vessel formation in tumors invivo. EML4–ALK lung tumors in GEM had significantly higherlevels of VEGFA as compared with EGFR-, K-Ras–, and B-Raf–mutated tumors, thus reinforcing the specificity of the ALK-hypoxia-angiogenesis axis. In ALCL, the mechanisms of ALK-mediated VEGF regulation could be quite complex becauseALK controls at least four factors that regulate VEGF expres-sion, such as HIF1a, HIF2a, STAT3, and C/EBPb as well asmiRNA-16 (11). Single knockdown of each of these four factorsdid not significantly change VEGF expression (data notshown). To this end, more complex combinatorial knockdownexperiments are needed to precisely unveil the mechanisms ofALK-mediated VEGF regulation.
Blockade of VEGFA by the VEGFA-specific antibody bev-acizumab in ALCL reduced vessels formation and tumorgrowth, thus indicating a potential efficacy of antiangiogenicdrugs in ALK-rearranged lymphoma. The use of bevacizumabin lymphoma patients is still under debate due to the limitedefficacy and associated toxicities (47) and should be restrictedto lymphoma types with proven efficacy (48). In this context,
HIFs in ALK-Rearranged Lymphoma and Lung Carcinoma
www.aacrjournals.org Cancer Res; 74(21) November 1, 2014 6103
on January 8, 2015. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 5, 2014; DOI: 10.1158/0008-5472.CAN-14-0268
CD
34V
EG
FA
A
B shHIF1a shHIF2a
Des
min
+ ce
lls
shHIF1a shHIF2a
Des
min
020
40
60
80
100
shHIF1a shHIF2a
+ Doxy– Doxy+ Doxy– Doxy
+ Doxy– Doxy+ Doxy– Doxy
shHIF1a shHIF2a
– Doxy+ Doxy
– Doxy+ Doxy
– Doxy+ Doxy
VE
GFA
+ ce
lls (
%)
0
20
40
60
80
100**
CD
34+
cells
0
20
40
60
80
100**
0
20
40
60
80
100
Des
min
+ ce
lls
**
VE
GFA
+ ce
lls (
%) **
0
20
40
60
80
100
CD
34+
cells
**
020
40
60
80
100
H3122 shRNA HIF2a
– Doxy + Doxy
LUNG
LIVER
H3122 shRNA HIF1a
– Doxy + Doxy
– Doxy + Doxy
C D
– Doxy + Doxy
LUNG
LIVER
N°
of
met
asta
ses
N°
of
met
asta
ses
N°
of
met
asta
ses
N°
of
met
asta
ses
– Doxy
20
10
15
0
5
+ Doxy
**
– Doxy + Doxy
6
4
2
0
*
– Doxy
150
100
50
0+ Doxy
**
+ Doxy– Doxy
40
30
20
10
0
**
E F
Figure 7. Knockdown of HIF1a or HIF2a impairs VEGFA production and vessels formation as well as metastatic diffusion in EML4–ALK-rearrangedNSCLCs. A, H2228 cells expressing a doxycycline-inducible shRNA for HIF1a or HIF2a were injected (10 � 106) s.c. in NOD/SCID mice. Tumorswere collected after 31 days (HIF1a) or 35 days (HIF2a) of treatment with doxycycline. Immunohistochemistry for VEGFA (A) and immunofluorescencefor CD34 and Desmin (B) were performed and percentages of positive cells were calculated. Histograms, the mean and SDs of at least fivetumors for each group; �, P < 0.05; ��, P < 0.001. C–F, H3122 NSCLC was transduced with doxycycline-inducible lentiviral shRNA againstHIF1a (C and D) or HIF2a (E and F) and 1 � 106 cells were injected i.v. into NSG mice. Mice were sacrificed after 45 days (for HIF1a) or 35 days(for HIF2a) from i.v. injections and histologic sections of the lungs (C–E) and liver (D–F) were obtained (objective magnification, �20). Histograms,the mean numbers of tumor foci counted in the entire lungs and livers of mice. Data are from six independent mice each group; �, P < 0.05;��, P < 0.005.
Martinengo et al.
Cancer Res; 74(21) November 1, 2014 Cancer Research6104
on January 8, 2015. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 5, 2014; DOI: 10.1158/0008-5472.CAN-14-0268

our data indicate that patientswithALK-rearranged ALCLmayindeed benefit with treatment with bevacizumab. In solidcancers, bevacizumab has been approved as treatment forseveral late-stage advanced metastatic cancers (35). However,the rate of response to bevacizumab in NSCLC is still unpre-dictable for unclear reasons. It has been suggested that astratification based on the molecular genetics of NSCLC couldimprove the efficacy of bevacizumab, possibly by restricting itsuse inNSCLCwith selected genetic lesions (36, 49). On the basisof the results of the present study, ALK-rearranged NSCLCcould represent a subset of tumors that could mostly benefitfrom such antiangiogenic therapy.In summary, in this work, we present evidences for an ALK-
dependent hypoxia-angiogenesis regulation in both ALCL andNSCLC. ALK regulates HIF1a and HIF2a, which in turnbecome essential for tumor growth. Thus, treatments aimedat blocking the ALK-driven VEGFA production in ALCL andNSCLC, or directly at targeting HIF2a in ALCL and both HIF1aand HIF2a in NSCLC, have potentials to become additionaltherapeutic strategies in ALK-rearranged tumors.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: C. Martinengo, M. Menotti, R. Piva, R. ChiarleDevelopment of methodology: C. Martinengo, T. Poggio, M. Menotti,C. Ambrogio, C. Mastini, L. Riera, C. Voena, R. ChiarleAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): C. Martinengo, M. Menotti, E. Pellegrino, R. Piva,D. Ribatti, F. Pastorino, R. ChiarleAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): C. Martinengo, E. Pellegrino, R. Piva, D. Ribatti,F. Pastorino, P. Perri, M. Ponzoni, Q. Wang, R. ChiarleWriting, review, and/or revision of the manuscript: M. Menotti, F. Pastor-ino, P. Perri, M. Ponzoni, C. Voena, R. ChiarleAdministrative, technical, or material support (i.e., reporting ororganizing data, constructing databases): M.S. Scalzo, C. Ambrogio,R. ChiarleStudy supervision: R. Chiarle
Grant SupportThe work was supported by grants FP7 ERC-2009-StG (proposal no.
242965—"Lunely"), Associazione Italiana per la Ricerca sul Cancro (AIRC)grant IG-12023, and International Association for Cancer Research (AICR)grant 12-0216.
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicate thisfact.
Received January 28, 2014; revised August 5, 2014; accepted August 13, 2014;published OnlineFirst September 5, 2014.
References1. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S,
et al. Identification of the transforming EML4–ALK fusion gene in non–small cell lung cancer. Nature 2007;448:561–6.
2. Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplasticlymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer2008;8:11–23.
3. Takeuchi K, Choi YL, Togashi Y, Soda M, Hatano S, Inamura K, et al.KIF5B-ALK, a novel fusion oncokinase identified by an immunohis-tochemistry-based diagnostic system for ALK-positive lung cancer.Clin Cancer Res 2009;15:3143–9.
4. Togashi Y, Soda M, Sakata S, Sugawara E, Hatano S, Asaka R,et al. KLC1-ALK: a novel fusion in lung cancer identified using aformalin-fixed paraffin-embedded tissue only. PLoS ONE 2012;7:e31323.
5. Hallberg B, Palmer RH. Mechanistic insight into ALK receptor tyrosinekinase in human cancer biology. Nat Rev Cancer 2013;13:685–700.
6. Voena C, Di Giacomo F, Panizza E, D'Amico L, Boccalatte FE, Pelle-grino E, et al. The EGFR family members sustain the neoplasticphenotype of ALKþ lung adenocarcinoma via EGR1. Oncogenesis2013;2:e43.
7. Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, et al.Crizotinib versus chemotherapy in advanced ALK-positive lung can-cer. N Engl J Med 2013;368:2385–94.
8. Gambacorti-Passerini C, Messa C, Pogliani EM. Crizotinib in anaplas-tic large-cell lymphoma. N Engl J Med 2011;364:775–6.
9. Shaw AT, Engelman JA. ALK in lung cancer: past, present, and future.J Clin Oncol 2013;31:1105–11.
10. Voena C, Chiarle R. The battle against ALK resistance: successes andsetbacks. Expert Opin Investig Drugs 2012;21:1751–4.
11. Dejean E, Renalier MH, Foisseau M, Agirre X, Joseph N, de Paiva GR,et al. Hypoxia-microRNA-16 downregulation induces VEGF expres-sion in anaplastic lymphoma kinase (ALK)-positive anaplastic large-cell lymphomas. Leukemia 2011;25:1882–90.
12. Di Paolo D, Ambrogio C, Pastorino F, Brignole C, Martinengo C,Carosio R, et al. Selective therapeutic targeting of the ana-plastic lymphoma kinase with liposomal siRNA induces apoptosisand inhibits angiogenesis in neuroblastoma. Mol Ther 2011;19:2201–12.
13. Marzec M, Liu X, Wong W, Yang Y, Pasha T, Kantekure K, et al.Oncogenic kinase NPM/ALK induces expression of HIF1alpha mRNA.Oncogene 2011;30:1372–8.
14. Laimer D, Dolznig H, Kollmann K, Vesely PW, Schlederer M, Merkel O,et al. PDGFR blockade is a rational and effective therapy for NPM–
ALK-driven lymphomas. Nat Med 2012;18:1699–704.15. Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev
Cancer 2011;11:393–410.16. Semenza GL. Oxygen sensing, homeostasis, and disease. N Engl
J Med 2011;365:537–47.17. Bertout JA, Patel SA, Simon MC. The impact of O2 availability on
human cancer. Nat Rev Cancer 2008;8:967–75.18. Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling
rivalry in hypoxic tumour growth and progression. Nat Rev Cancer2012;12:9–22.
19. Agnelli L, Mereu E, Pellegrino E, Limongi T, Kwee I, Bergaggio E, et al.Identification of a 3-gene model as a powerful diagnostic tool for therecognition of ALK-negative anaplastic large-cell lymphoma. Blood2012;120:1274–81.
20. ChenZ, Sasaki T, TanX,Carretero J, Shimamura T, Li D, et al. Inhibitionof ALK, PI3K/MEK, and HSP90 in murine lung adenocarcinomainduced by EML4–ALK fusion oncogene. Cancer Res 2010;70:9827–36.
21. Okayama H, Kohno T, Ishii Y, Shimada Y, Shiraishi K, Iwakawa R,et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res 2012;72:100–11.
22. Ambrogio C, Martinengo C, Voena C, Tondat F, Riera L, di Celle PF,et al. NPM–ALK oncogenic tyrosine kinase controls T-cell identity bytranscriptional regulation and epigenetic silencing in lymphoma cells.Cancer Res 2009;69:8611–9.
23. Piva R, Pellegrino E, Mattioli M, Agnelli L, Lombardi L, Boccalatte F,et al. Functional validation of the anaplastic lymphoma kinase signa-ture identifies CEBPB and BCL2A1 as critical target genes. J ClinInvest 2006;116:3171–82.
24. PivaR,Chiarle R,ManazzaAD, Taulli R, SimmonsW,AmbrogioC, et al.Ablation of oncogenic ALK is a viable therapeutic approach for ana-plastic large-cell lymphomas. Blood 2006;107:689–97.
HIFs in ALK-Rearranged Lymphoma and Lung Carcinoma
www.aacrjournals.org Cancer Res; 74(21) November 1, 2014 6105
on January 8, 2015. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 5, 2014; DOI: 10.1158/0008-5472.CAN-14-0268

25. Ambrogio C, Voena C, Manazza AD, Piva R, Riera L, Barberis L, et al.p130Cas mediates the transforming properties of the anaplastic lym-phoma kinase. Blood 2005;106:3907–16.
26. Camporeale A,Marino F, Papageorgiou A, Carai P, Fornero S, FletcherS, et al. STAT3 activity is necessary and sufficient for the developmentof immune-mediatedmyocarditis inmice andpromotes progression todilated cardiomyopathy. EMBO Mol Med 2013;5:572–90.
27. Guerra C, Mijimolle N, Dhawahir A, Dubus P, Barradas M, Serrano M,et al. Tumor induction by an endogenous K-ras oncogene is highlydependent on cellular context. Cancer Cell 2003;4:111–20.
28. Politi K, ZakowskiMF, FanPD, Schonfeld EA, PaoW,VarmusHE. Lungadenocarcinomas induced in mice by mutant EGF receptors found inhuman lung cancers respond to a tyrosine kinase inhibitor or todownregulation of the receptors. Genes Dev 2006;20:1496–510.
29. Dankort D, Filenova E, ColladoM, SerranoM, Jones K,McMahonM. Anewmousemodel to explore the initiation, progression, and therapy ofBRAFV600E-induced lung tumors. Genes Dev 2007;21:379–84.
30. Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors andthe response to hypoxic stress. Mol Cell 2010;40:294–309.
31. Zamo A, Chiarle R, Piva R, Howes J, Fan Y, Chilosi M, et al. Anaplasticlymphoma kinase (ALK) activates Stat3 and protects hematopoieticcells from cell death. Oncogene 2002;21:1038–47.
32. Zhang Q, Raghunath PN, Xue L, Majewski M, Carpentieri DF, OdumN,et al. Multilevel dysregulation of STAT3 activation in anaplastic lym-phoma kinase-positive T/null-cell lymphoma. J Immunol 2002;168:466–74.
33. Quintanilla-Martinez L, Pittaluga S, Miething C, Klier M, Rudelius M,Davies-Hill T, et al. NPM-ALK-dependent expression of the transcrip-tion factor CCAAT/enhancer binding protein beta in ALK-positiveanaplastic large cell lymphoma. Blood 2006;108:2029–36.
34. Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG,Albanese C, et al. Stat3 as an oncogene. Cell 1999;98:295–303.
35. Carmeliet P, Jain RK. Principles and mechanisms of vessel normal-ization for cancer and other angiogenic diseases. Nat Rev Drug Discov2011;10:417–27.
36. Amit L, Ben-Aharon I, Vidal L, Leibovici L, Stemmer S. The impact ofbevacizumab (avastin) on survival in metastatic solid tumors - a meta-analysis and systematic review. PLoS ONE 2013;8:e51780.
37. Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ,Halmos B, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med 2012;4:120ra17.
38. Gerald D, Berra E, Frapart YM, Chan DA, Giaccia AJ, Mansuy D, et al.JunD reduces tumor angiogenesis by protecting cells from oxidativestress. Cell 2004;118:781–94.
39. Mazumdar J, Hickey MM, Pant DK, Durham AC, Sweet-Cordero A,Vachani A, et al. HIF-2alpha deletion promotes Kras-driven lung tumordevelopment. Proc Natl Acad Sci U S A 2010;107:14182–7.
40. Shaw AT, Yeap BY, Mino-Kenudson M, Digumarthy SR, Costa DB,Heist RS, et al. Clinical features and outcome of patients with non–small cell lung cancer who harbor EML4–ALK. J Clin Oncol 2009;27:4247–53.
41. Doebele RC, Lu X, Sumey C, Maxson DA, Weickhardt AJ, Oton AB,et al. Oncogene status predicts patterns of metastatic spreadin treatment-naive non–small cell lung cancer. Cancer 2012;118:4502–11.
42. KondoK,Klco J,Nakamura E, LechpammerM,KaelinWGJr. Inhibitionof HIF is necessary for tumor suppression by the von Hippel–Lindauprotein. Cancer Cell 2002;1:237–46.
43. Raetz EA, Perkins SL, Carlson MA, Schooler KP, Carroll WL, VirshupDM. The nucleophosmin-anaplastic lymphoma kinase fusion proteininduces c-Myc expression in pediatric anaplastic large-cell lympho-mas. Am J Pathol 2002;161:875–83.
44. Gordan JD, ThompsonCB,SimonMC.HIF and c-Myc: sibling rivals forcontrol of cancer cell metabolism and proliferation. Cancer Cell 2007;12:108–13.
45. Inamura K, Takeuchi K, Togashi Y, Nomura K, Ninomiya H, Okui M,et al. EML4-ALK fusion is linked to histological characteristics in asubset of lung cancers. J Thorac Oncol 2008;3:13–7.
46. Settleman J. Cell culture modeling of genotype-directed sensitivity toselective kinase inhibitors: targeting the anaplastic lymphoma kinase(ALK). Semin Oncol 2009;36(2 Suppl 1):S36–41.
47. Stopeck AT, Unger JM, Rimsza LM, LeBlanc M, Farnsworth B,Iannone M, et al. A phase 2 trial of standard-dose cyclophospha-mide, doxorubicin, vincristine, prednisone (CHOP) and rituximabplus bevacizumab for patients with newly diagnosed diffuse largeB-cell non-Hodgkin lymphoma: SWOG 0515. Blood 2012;120:1210–7.
48. Dupire S,CoiffierB. Targeted treatment andnewagents in diffuse largeB-cell lymphoma. Int J Hematol 2010;92:12–24.
49. Metro G, Crino L. Novel molecular trends in the management ofadvanced non–small cell lung cancer. Expert Rev Anticancer Ther2012;12:729–32.
Cancer Res; 74(21) November 1, 2014 Cancer Research6106
Martinengo et al.
on January 8, 2015. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 5, 2014; DOI: 10.1158/0008-5472.CAN-14-0268

2014;74:6094-6106. Published OnlineFirst September 5, 2014.Cancer Res Cinzia Martinengo, Teresa Poggio, Matteo Menotti, et al. Tumor Growth and MetastasisALK-Dependent Control of Hypoxia-Inducible Factors Mediates
Updated version
10.1158/0008-5472.CAN-14-0268doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2014/09/05/0008-5472.CAN-14-0268.DC1.html
Access the most recent supplemental material at:
Cited Articles
http://cancerres.aacrjournals.org/content/74/21/6094.full.html#ref-list-1
This article cites by 49 articles, 17 of which you can access for free at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
To request permission to re-use all or part of this article, contact the AACR Publications Department at
on January 8, 2015. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 5, 2014; DOI: 10.1158/0008-5472.CAN-14-0268
Copyright © 2022 FDOKUMEN




















