
Administration of Agonistic Anti4-1BB Monoclonal Antibody Leads to the Amelioration of Experimental...
10
of October 23, 2013. This information is current as Encephalomyelitis Amelioration of Experimental Autoimmune Monoclonal Antibody Leads to the Administration of Agonistic Anti-4-1BB K. Subudhi, Lieping Chen and Yang-Xin Fu Yonglian Sun, Xiaoqi Lin, Helen M. Chen, Qiang Wu, Sumit http://www.jimmunol.org/content/168/3/1457 2002; 168:1457-1465; ; J Immunol References http://www.jimmunol.org/content/168/3/1457.full#ref-list-1 , 20 of which you can access for free at: cites 38 articles This article Subscriptions http://jimmunol.org/subscriptions is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/ji/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/cgi/alerts/etoc Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved. Copyright © 2002 by The American Association of 9650 Rockville Pike, Bethesda, MD 20814-3994. The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on October 23, 2013 http://www.jimmunol.org/ Downloaded from by guest on October 23, 2013 http://www.jimmunol.org/ Downloaded from by guest on October 23, 2013 http://www.jimmunol.org/ Downloaded from by guest on October 23, 2013 http://www.jimmunol.org/ Downloaded from by guest on October 23, 2013 http://www.jimmunol.org/ Downloaded from by guest on October 23, 2013 http://www.jimmunol.org/ Downloaded from by guest on October 23, 2013 http://www.jimmunol.org/ Downloaded from by guest on October 23, 2013 http://www.jimmunol.org/ Downloaded from by guest on October 23, 2013 http://www.jimmunol.org/ Downloaded from by guest on October 23, 2013 http://www.jimmunol.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Administration of Agonistic Anti4-1BB Monoclonal Antibody Leads to the Amelioration of Experimental...

of October 23, 2013.This information is current as
EncephalomyelitisAmelioration of Experimental AutoimmuneMonoclonal Antibody Leads to the Administration of Agonistic Anti-4-1BB
K. Subudhi, Lieping Chen and Yang-Xin FuYonglian Sun, Xiaoqi Lin, Helen M. Chen, Qiang Wu, Sumit
http://www.jimmunol.org/content/168/3/14572002; 168:1457-1465; ;J Immunol
Referenceshttp://www.jimmunol.org/content/168/3/1457.full#ref-list-1
, 20 of which you can access for free at: cites 38 articlesThis article
Subscriptionshttp://jimmunol.org/subscriptions
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/ji/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/cgi/alerts/etocReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2002 by The American Association of9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on October 23, 2013
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on October 23, 2013
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on October 23, 2013
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on October 23, 2013
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on October 23, 2013
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on October 23, 2013
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on October 23, 2013
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on October 23, 2013
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on October 23, 2013
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on October 23, 2013
http://ww
w.jim
munol.org/
Dow
nloaded from

Administration of Agonistic Anti-4-1BB Monoclonal AntibodyLeads to the Amelioration of Experimental AutoimmuneEncephalomyelitis1
Yonglian Sun,*‡ Xiaoqi Lin, † Helen M. Chen,* Qiang Wu,* Sumit K. Subudhi,* ‡
Lieping Chen,§ Yang-Xin Fu2*‡
4-1BB, a member of the TNFR superfamily, is a costimulatory receptor primarily expressed on activated T cells. It has been shownthat the administration of agonistic anti-4-1BB Abs enhances tumor immunity and allogenic immune responses. Paradoxically, wefound that the administration of an agonistic anti-4-1BB mAb (2A) dramatically reduced the incidence and severity of experi-mental autoimmune encephalomyelitis (EAE). Adoptive transfer of T cells from such treated mice failed to induce EAE, whereasanti-4-1BB treatment following adoptive transfer of encephalitogenic T cells did not prevent EAE pathogenesis. These resultssuggest that anti-4-1BB treatment during the induction phase inhibits autoreactive T cell immune responses rather than pre-venting T cell trafficking into the CNS. This was substantiated by the observations that draining lymph node cells from anti-4-1BB-treated mice failed to respond to Ag stimulation in vitro. In addition, we found that such treatment initially promotes theactivation and proliferation of Ag-specific CD4� T cells but subsequently increases their probability of undergoing activation-induced cell death, thereby inhibiting effector T cell responses. More importantly, 2A treatment also inhibits the relapse of EAEin a clinically relevant murine model of multiple sclerosis. This study indicates that the agonistic Ab against 4-1BB can potentiallybe used as a novel immunotherapeutic agent for treating autoimmune diseases.The Journal of Immunology, 2002, 168: 1457–1465.
E fficient activation and differentiation of T cells requirestwo signals: a primary signal initiated by the engagementof a TCR with a MHC/peptide complex and a secondary
signal mediated through one of several accessory molecules ex-pressed on the surface of the T cell. CD28 is a well-characterizedcostimulatory molecule that is constitutively expressed on the sur-face of T cells, and it provides the primary costimulatory signal forT cell activation through interaction with its ligands, B7-1 andB7-2, expressed on APCs (1). However, studies of immune re-sponses in CD28-deficient mice showed that CD28-independentimmune responses exist (2–4). An example of a CD28-indepen-dent response involves 4-1BB (CDw137), a member of the TNFRsuperfamily, which is primarily expressed on activated T cells (5)and NK cells (6). Its natural ligand, the 4-1BB ligand (4-1BBL),3
is a member of TNF superfamily, and it has been detected on
activated B and T cells, macrophages, dendritic cells, mouse lym-phomas, and human carcinoma lines of epithelial origin (7–11).
In vitro studies have shown that 4-1BB signaling augments Tcell proliferation and cytokine production through both CD28-de-pendent and -independent mechanisms (4, 5, 7, 12–14). Further-more, agonistic anti-4-1BB mAbs preferentially stimulate prolif-eration of and IFN-� production by CD8� T cells as comparedwith CD4� T cells (15), suggesting that 4-1BB is a costimulatorymolecule primarily for CD8� T cells. In vivo experiments usingagonistic 4-1BB mAbs or 4-1BBL-transfected tumor cells haveshown that signaling through 4-1BB can induce the preferentialexpansion of CD8� CTLs that recognize and reject tumors andallograft transplants (15–17). Agonistic anti-4-1BB mAb also pre-vents superantigen-induced T cell death, especially of CD8� Tcells (18). In addition, the study of immune responses in 4-1BBL-deficient mice showed that CD8� T cell responses to virus infec-tions were reduced (19, 20). It appears that activation via 4-1BB isrequired for optimal CD8� T cell-mediated immune responsesin vivo.
The function of 4-1BB in CD4� T cell-mediated immunity hasbeen explored, with most studies suggesting that it plays a co-stimulatory role (10, 12, 21–23). A previous study showed that4-1BBL could provide a costimulatory signal for T cell activationon APCs lacking B7 molecules by using a MHC class II-restrictedautoreactive T cell hybridoma and purified CD4� T cells (10).Another study used purified CD4� T cells responding to alloge-neic 4-1BBL-expressing stimulator cells to demonstrate that4-1BB ligation could augment CD28-independent cytokine pro-duction by T cells (12). Pigeon cytochrome c-presenting fibroblastAPCs that were transfected with 4-1BBL were used to suggest that4-1BB engagement on naive CD4� T cells promotes proliferation,cell cycle progression, and IL-2 secretion (21). In addition, bothCD4� and CD8� T cell-mediated in vivo alloresponses were
Departments of *Pathology and †Neurology, and ‡Committee in Immunology, Uni-versity of Chicago, Chicago, IL 60637; and §Department of Immunology, MayoClinic, Rochester, MN 55905
Received for publication April 20, 2001. Accepted for publication November 2, 2001.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported in part by the National Multiple Sclerosis Society (GrantsRG3068 and RG3127-A-1), by the National Institutes of Health (Grant HD73104),and by the Juvenile Diabetes Foundation (Grant 1-2000-875). Y.-X.F. is a recipient ofa clinical investigator award (no. AI01431) from the National Institutes of Health.2 Address correspondence and reprint requests to Dr. Yang-Xin Fu, Department ofPathology, University of Chicago, MC3083, Chicago, IL 60637. E-mail address:[email protected] Abbreviations used in this paper: 4-1BBL, 4-1BB ligand; GVHD, graft-vs-host dis-ease; EAE, experimental autoimmune encephalomyelitis; MOG, myelin oligodendro-cyte glycoprotein; AICD, activation-induced cell death; DLN, draining lymph node;DTH, delayed-type hypersensitivity; 7-AAD, 7-aminoactinomycin D; PLP,proteolipoprotein.
Copyright © 2002 by The American Association of Immunologists 0022-1767/02/$02.00

regulated by 4-1BB/4-1BBL interactions to approximately thesame extent (22). 4-1BBL induced cell division and enhancedCD4� and CD8� T cell effector functions with similar efficacywhen using purified TCR-transgenic T cells responding to a specificpeptide and allogeneic stimulator cells that express 4-1BBL (23).
However, the introduction of the same agonistic anti-4-1BBmAb (1D8 and 3E1) clones that were used to promote tumor re-jection and enhancing graft-vs-host disease (GVHD) abrogated Tcell-dependent humoral immune responses in vivo, possibly byinducing Th cell anergy (24). To further investigate the role of4-1BB signaling in CD4� T cell-mediated immune responses andits effects on autoimmune diseases, we studied its function in thedevelopment of experimental autoimmune encephalomyelitis(EAE). EAE is a Th type 1 cell-mediated demyelinating disease ofthe CNS that is often used as an animal model for human multiplesclerosis. It can be induced in several strains of animals by immu-nization with various myelin proteins or immunodominant peptideepitopes derived from myelin basic protein, proteolipoprotein(PLP), or myelin oligodendrocyte glycoprotein (MOG) (25, 26).Th1-type responses are believed to be responsible for EAE patho-genesis, whereas Th2 responses appear to be protective (27, 28).Because the immunodominant autoreactive Ag in this disease hasbeen identified, EAE serves as an excellent model for studying thefunction of costimulatory molecules in an Ag-specific system.
Interestingly, we found that the administration of an agonisticanti-4-1BB mAb reduced the incidence and severity of EAE bypreventing autoreactive T cell immune responses. Taking advan-tage of DO11.10 TCR-transgenic mice, we were able to monitorAg-specific CD4� T cells, and we found that the administration ofanti-4-1BB mAb initially promotes CD4� T cell proliferation butsubsequently accelerates their activation-induced cell death(AICD). Therefore, the engagement of 4-1BB by an agonistic Abmay provide a novel approach to effectively depleting autoreactiveT cells.
Materials and MethodsMice
Six- to 10-wk-old C57BL/6, SJL, and BALB/c female mice were pur-chased from the National Cancer Institute (Frederick, MD). C57BL/6RAG-1�/� male mice were purchased from The Jackson Laboratory (BarHarbor, ME). DO11.10 BALB/c mice were kindly provided by Dr. A.Sperling (University of Chicago, Chicago, IL). All mice were housed in theUniversity of Chicago Animal Care Facilities. In each experiment, age-matched mice were used.
Peptides
MOG35–55 peptide (MEVGWYRSPFSRVVHLYRNGK) and PLP139–151
(HSLGKWLGHPDKF) were synthesized by Alpha Diagnostic Interna-tional (San Antonio, TX). The peptides were �76% pure as determined byHPLC. Chicken OVA323–339 (ISQAVHAAHAEINEAGR) peptide wassynthesized by the Divisional Protein-Peptide Core Facility of the Univer-sity of Chicago. The peptides were �86% pure as determined by HPLC.
Antibodies
Agonistic anti-4-1BB Ab (2A and IgG2a) was generated by immunizing aLewis rat (Harlan Sprague-Dawley, Indianapolis, IN) with mouse4-1BBIg.4 Hybridomas were produced by fusing rat spleen cells withmouse Sp2/0 myeloma cells. The culture supernatants were screened byELISA, and the hybridoma secreting the mAb 2A was selected for addi-tional experiments. To prepare 4-1BBIg fusion protein, the extracellulardomain of mouse 4-1BB was amplified from activated spleen cell cDNAusing sequence-specific primers and was then fused to the CH2-CH3 do-main of mouse IgG2a in an expression plasmid pmIgV, which was trans-
fected into Chinese hamster ovary cells. The protein in the culture super-natants was purified using a HiTrap Protein G-Sepharose column(Amersham Pharmacia Biotech, Piscataway, NJ) and was dialyzed inLPS-free PBS.
mAb 2A stained �80% of purified T cells that had been activated for24 h by anti-CD3 and anti-CD28 mAbs. Binding is specific, as Ab bindingcould be competitively inhibited by inclusion of 4-1BB-Ig, whereas inclu-sion of a control rat IgG Ab did not inhibit binding. Furthermore, mAb 2Abinds specifically to the mouse T cell lymphoma S49.1 that constitutivelyexpresses 4-1BB, as demonstrated by another anti-4-1BB mAb, clone1AH2. Similar to previously reported agonistic anti-4-1BB mAb (1D8 and3E1; Ref. 15), mAb 2A enhanced T cell proliferation in a dose-dependentfashion in the presence of a suboptimal dose of anti-CD3 mAb in vitro, andit induced rejection of well-established EL4E7 tumor in vivo.4
Rat IgG was purchased from Sigma-Aldrich (St. Louis, MO) and servedas a control Ab. The KJ1-26 mAb, which binds only to the particulartransgenic TCR heterodimer in DO11.10 TCR-transgenic mice, was kindlyprovided by Dr. A. Sperling.
Induction of EAE and mAb treatment
To induce EAE by active immunization, female C57BL/6 mice were im-munized s.c. at four sites on the flank, with a total of 100 �g of MOG35–55
peptide emulsified in an equal volume of CFA (Life Technologies, Gaith-ersburg, MD) containing 1 mg/ml Mycobacterium tuberculosis H37 RA(Difco, Detroit, MI) on days 0 and 7. Mice also received i.v. injections of200 ng of pertussis toxin (List Biological Laboratories, Campbell, CA) ondays 0 and 2 after immunization. Mice were administered with anti-4-1BBor control rat IgG (or PBS) i.p. on day 0. Because the doses of anti-4-1BBfrom 100 to 300 �g/mouse showed similar effects, in most experiments, thedose of anti-4-1BB and control rat IgG was used at 150 �g/mouse. Foradoptively transferred EAE, draining lymph node (DLN) cells were har-vested from female C57BL/6 mice on day 10 post-s.c. immunization with100 �g of MOG35–55 emulsified in CFA. The cells (5 � 106 cells/ml) werecultured with MOG35–55 peptide (25 �g/ml) for 4 days and were then i.v.transferred into sublethally (600 rad) irradiated female C57BL/6 mice (2 �107 cells per mouse). The recipients received 200 ng of pertussis toxin byi.v. injection immediately after cell transfer and 2 days later. Recipientswere also treated i.p. with anti-4-1BB or control rat IgG on the day oftransfer. In some experiments, C57BL/6 mice were immunized s.c. with100 �g of MOG35–55 emulsified in CFA twice at a 1-wk interval andtreated with anti-4-1BB or control IgG on the day of the first immunization.One week after the second immunization, DLN cells and splenocytes wereisolated, pooled, and directly transferred into male RAG-1�/� mice.
For induction of the relapsing-remitting EAE model, female SJL micewere immunized s.c. once with 100 �g of PLP139–151 emulsified in CFAcontaining 500 �g of M. tuberculosis H37 RA at three sites on the flank.On the same day or after the onset of disease, mice were administered 200�g of anti-4-1BB or control rat IgG i.p once or once a week for a total of3 wk. Mice were scored daily for signs of EAE according to the followingclinical scoring system: 0, no clinical signs; 0.5, partial loss of tail tonicity;1, complete loss of tail tonicity; 2, flaccid tail and abnormal gait; 3, hind legparalysis; 4, hind leg paralysis with hind body paresis; 5, hind and fore legparalysis; and 6, death.
DTH assay
Female C57BL/6 mice were immunized s.c. at four sites on the flank, witha total of 100 �g of MOG35–55 peptide emulsified in an equal volume ofCFA on days 0 and 7. Mice also received an i.v. injection of 200 ng ofpertussis toxin on days 0 and 2 after immunization. Mice were adminis-tered 150 �g of anti-4-1BB or control rat IgG i.p. on day 0. Six weeks afterthe first immunization, mice were injected in the footpad with 15 �g ofMOG in 15 �l of PBS. The thickness of the footpad was measured 24 hafter injection. Naive mice were used as negative controls.
Preparation of tissue for histologic examination
Mice were anesthetized with pentobarbital and were perfused by intracar-diac puncture with 50 ml of Trump’s fixative (phosphate-buffered 4% para-formaldehyde and 1% glutaraldehyde, pH 7.2). Spinal cords were removed,paraffin-embedded, sectioned coronally, and stained with H&E for visual-ization of inflammatory infiltrates (29).
4 R. A. Wilcox, D. B. Flies, G. Zhu, A. J. Johnson, K. Tamada, A. I. Chapoval, S. E.Strome, L. R. Pease, and L. Chen. Treatment of poorly immunogenic establishedtumors by harnessing ignorant cytotoxic T cells with epitope peptide and anti-4-1BBcostimulatory monoclonal antibody. Submitted for publication.
1458 REGULATION OF AUTOIMMUNE DISEASES BY ANTI-4-1BB Ab

In vitro proliferation assay
Ten days after immunization, DLN cells were isolated and cultured in96-well flat-bottom plates at a concentration of 5 � 105 cells/well in com-plete RPMI 1640 medium (Life Technologies) that contained 10% heat-inactivated FCS, 1 mM glutamine, 1% penicillin-streptavidin, 1 mM non-essential amino acids, and 5 � 10�5 M 2-ME with various concentrationsof MOG35–55 peptide. Plates were pulsed with [3H]thymidine (AmershamPharmacia Biotech) at 0.5 �Ci/well on day 4 of culture for the final 18 h.Incorporation of thymidine into DNA was measured by liquid scintillationcounting, and the mean was calculated from triplicate wells.
Detection of cytokine production by ELISA
Four and 10 days after immunization, DLN cells were isolated and culturedwith or without MOG35–55 peptide (25 �g/ml) in complete RPMI 1640medium for 2 or 3 days. Supernatants were collected for the detection ofIFN-�, GM-CSF, IL-10, and IL-4 by ELISA according to the manufactur-er’s instructions. All anti-mouse cytokine mAbs and recombinant cytokinestandards were purchased from BD PharMingen (San Diego, CA). Allcytokine levels were calculated using standard curves with known amountsof recombinant cytokines.
Real-time PCR
Ten days after immunization, DLNs were removed and total RNA wasisolated using TRI reagent (Life Technologies) and was then treated withDNase I (Amersham Pharmacia Biotech). Total RNA (5 �g) was reversetranscribed using the First Strand cDNA Synthesis kit (Amersham Phar-macia Biotech). The mRNA of cytokines and GAPDH were detected byreal-time RT-PCR using the ABI Prism 7700-sequence detection system(Applied Biosystems, Foster City, CA) as described previously (30). Theprimer and probe sequences have previously been reported (31). Theprobes for cytokines or GAPDH were labeled with 6-carboxy-fluoresceinor tetrachloro-6-carboxy-fluorescein, respectively, as a reporter dye. Allreactions were conducted in triplicate 20-�l reaction volumes containingTaqMan Universal PCR master mixture (Applied Biosystems). PCR am-plification conditions were as follows: 95°C for 10 min, and 40 cycles at95°C for 15 s and 60°C for 1 min. PCR results were analyzed using therelative standard curve method according to the manufacturer’s instruc-tions. The amount of cytokine mRNA in each sample was corrected forGAPDH. Final results are displayed as the ratio of the amount of cytokinemRNA over that of control group.
5,6-Carboxy-succinimidyl-fluoresceine-ester labeling of DO11.10cells
Before fluorescence labeling, TCR-transgenic lymph node and spleen cellsfrom DO11.10 transgenic mice were isolated, and CD3� T cells were pu-rified by immunomagnetic column by negative depletion method (Stem-Cell Technologies, Vancouver, British Columbia, Canada). The purified Tcells containing �90% of CD4�KJ126� cells were resuspended in PBS at2 � 107 cells/ml and they were incubated with 10 �M 5,6-carboxy-suc-cinimidyl-fluoresceine-ester (Molecular Probes, Eugene, OR) at 37°C for30 min.
Tracing adoptively transferred cells
To trace Ag-specific T cells in vivo, we took advantage of DO11.10 TCR-transgenic mice (32, 33). CFSE-labeled and unlabeled TCR-transgeniclymph node and spleen cells from DO11.10 transgenic mice were isolated,and the percentage of CD4�KJ1-26� cells was measured by flow cyto-metric analysis. Three to five million CD4�KJ1-26� cells were i.v. in-jected into nonirradiated BALB/c mice. On the same day, recipient micewere s.c. injected with OVA323–339 (300 �g) emulsified in CFA in a vol-ume of 0.1 ml distributed among three sites on the hind back and treatedwith anti-4-1BB or control IgG (150 �g/mouse) i.p. DLN cells fromBALB/c recipients of CFSE-labeled DO11.10 cells were isolated 54 h afterimmunization and analyzed by flow cytometry. DLN cells from BALB/crecipients of nonlabeled cells were isolated 4, 9, and 19 days after immu-nization and incubated with anti-FcR mAb (2.4G2; American Type CultureCollection, Manassas, VA) in staining buffer (PBS containing 1% FCS and0.1% sodium azide) on ice for 10 min to block FcR. FITC-labeled anti-CD4 (BD PharMingen) and biotinylated KJ1-26 mAb were then incubatedwith the cells at 4°C for 20 min. After a wash with staining buffer, the cellswere incubated with PE-labeled streptavidin (Immunotech, Luminy,France) at 4°C for 20 min. Following one wash, cells were collected on aFACScan (BD Biosciences, Mountain View, CA) and were analyzed usingCellQuest software (BD Immunocytometry Systems, San Jose, CA).
Detection of apoptosis
Four to 5 days after immunization, DLN cells from immunized BALB/crecipients were isolated and stained with biotin-labeled KJ1-26 mAb fol-lowed by PE-labeled streptavidin. Following two washes, the cells werestained with either FITC-labeled annexin V (BD PharMingen) or 7-ami-noactinomycin D (7-AAD; Sigma-Aldrich) and then analyzed by flow cy-tometry. For staining with 7-AAD, cells were incubated with FACS buffercontaining 20 �g/ml 7-AAD at 4°C for 20 min. The fluorescence of 7-AADwas detected by red channel FL-3 of FACScan. KJ1-26-positive cells weregated to analyze annexin V and 7-AAD staining.
Statistical analysis
Comparison of the mean peak disease severity between two groups of micewas analyzed by the Student’s t test.
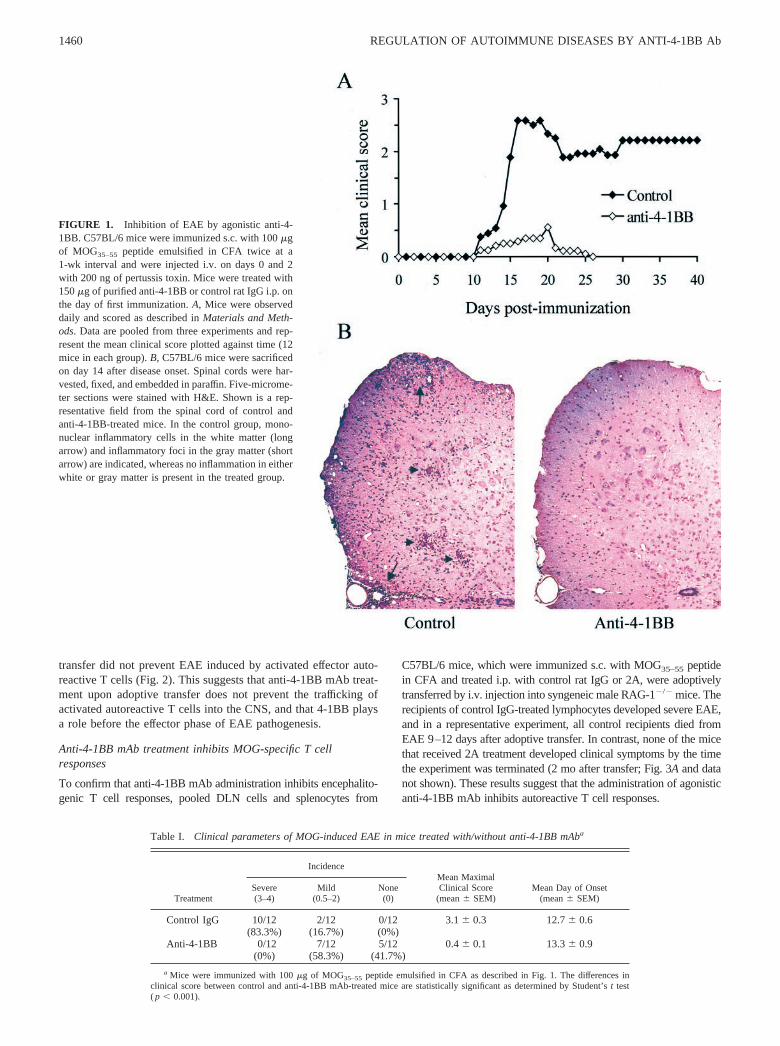
ResultsAnti-4-1BB mAb treatment inhibits the development ofactive EAE
To address the role of 4-1BB in the development of EAE,C57BL/6 mice were treated with either agonistic anti-4-1BB mAb(2A) or rat IgG control i.p. on the day of the first s.c. immunizationwith MOG35–55 peptide emulsified in CFA. Interestingly, the re-sults showed that 2A treatment greatly inhibited the developmentof EAE and reduced the disease incidence and severity (Fig. 1A;Table I). All of the control mice developed moderate (2 of 12;clinical score, 0.5–2) to severe (10 of 12; 83.3%; clinical score,3–4) EAE. Recovery from the disease was not observed in any ofthe control mice up to 30 days after onset, and similar results wereobtained when the mice were treated with PBS (data not shown).In contrast, in the 2A-treated group, 5 of 12 mice (41.7%) wereentirely asymptomatic, whereas the rest (7 of 12; 58.3%) onlydisplayed moderate symptoms (clinical score, 0.5–2). More im-portantly, complete recovery was observed in virtually all themildly symptomatic mice 10–15 days after disease onset. Themean maximal clinical score of 2A-treated mice was significantlylower than that of the control group (0.4 for the 2A-treated groupvs 3.1 for the control group; p � 0.001).
The presence/absence and degree of EAE were further evaluatedby histological examination of spinal cord sections from both con-trol and 2A-treated mice. Fourteen days postimmunization in con-trol mice, dense lymphocytic infiltrate was observed in the menin-ges and the white matter and gray matter of the spinal cord (Fig.1B). In contrast, no or only minimal perivascular lymphocytic in-filtrate was observed in the sections obtained from mice treatedwith 2A (Fig. 1B). These results suggest that 4-1BB engagementduring the priming stage of an immune response strongly inhibitsthe infiltration of autoreactive T cells in the CNS and EAEdevelopment.
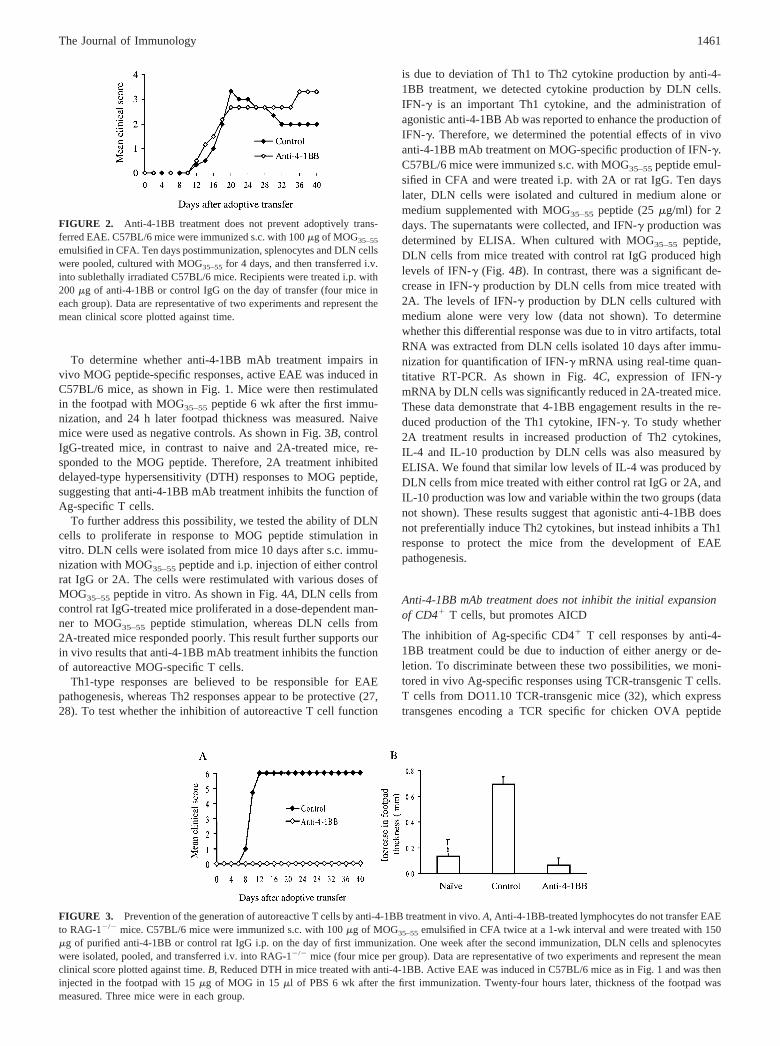
Anti-4-1BB mAb treatment does not prevent adoptivelytransferred EAE
The mechanism by which anti-4-1BB mAb administration inhibitsEAE development may be due to the inhibition of encephalito-genic T cell responses and/or their migration into the CNS. Todissect this issue, we tested the effect of 2A treatment in an adop-tive transfer model of EAE. Activated MOG-specific T cells wereobtained by coculture for 4 days of MOG35–55 peptide with DLNcells from C57BL/6 mice immunized with MOG35–55 peptideemulsified in CFA. EAE was established by transferring these ac-tivated MOG-specific T cells by i.v. injection into sublethally ir-radiated C57BL/6 mice that were then treated i.p. with either 2A orcontrol rat IgG at 200 or 500 �g/mouse. The results showed thatthe administration of anti-4-1BB mAb at the time of adoptive
1459The Journal of Immunology
transfer did not prevent EAE induced by activated effector auto-reactive T cells (Fig. 2). This suggests that anti-4-1BB mAb treat-ment upon adoptive transfer does not prevent the trafficking ofactivated autoreactive T cells into the CNS, and that 4-1BB playsa role before the effector phase of EAE pathogenesis.
Anti-4-1BB mAb treatment inhibits MOG-specific T cellresponses
To confirm that anti-4-1BB mAb administration inhibits encephalito-genic T cell responses, pooled DLN cells and splenocytes from
C57BL/6 mice, which were immunized s.c. with MOG35–55 peptidein CFA and treated i.p. with control rat IgG or 2A, were adoptivelytransferred by i.v. injection into syngeneic male RAG-1�/� mice. Therecipients of control IgG-treated lymphocytes developed severe EAE,and in a representative experiment, all control recipients died fromEAE 9–12 days after adoptive transfer. In contrast, none of the micethat received 2A treatment developed clinical symptoms by the timethe experiment was terminated (2 mo after transfer; Fig. 3A and datanot shown). These results suggest that the administration of agonisticanti-4-1BB mAb inhibits autoreactive T cell responses.
FIGURE 1. Inhibition of EAE by agonistic anti-4-1BB. C57BL/6 mice were immunized s.c. with 100 �gof MOG35–55 peptide emulsified in CFA twice at a1-wk interval and were injected i.v. on days 0 and 2with 200 ng of pertussis toxin. Mice were treated with150 �g of purified anti-4-1BB or control rat IgG i.p. onthe day of first immunization. A, Mice were observeddaily and scored as described in Materials and Meth-ods. Data are pooled from three experiments and rep-resent the mean clinical score plotted against time (12mice in each group). B, C57BL/6 mice were sacrificedon day 14 after disease onset. Spinal cords were har-vested, fixed, and embedded in paraffin. Five-microme-ter sections were stained with H&E. Shown is a rep-resentative field from the spinal cord of control andanti-4-1BB-treated mice. In the control group, mono-nuclear inflammatory cells in the white matter (longarrow) and inflammatory foci in the gray matter (shortarrow) are indicated, whereas no inflammation in eitherwhite or gray matter is present in the treated group.
Table I. Clinical parameters of MOG-induced EAE in mice treated with/without anti-4-1BB mAba
Treatment
IncidenceMean MaximalClinical Score
(mean � SEM)Mean Day of Onset
(mean � SEM)Severe(3–4)
Mild(0.5–2)
None(0)
Control IgG 10/12 2/12 0/12 3.1 � 0.3 12.7 � 0.6(83.3%) (16.7%) (0%)
Anti-4-1BB 0/12 7/12 5/12 0.4 � 0.1 13.3 � 0.9(0%) (58.3%) (41.7%)
a Mice were immunized with 100 �g of MOG35–55 peptide emulsified in CFA as described in Fig. 1. The differences inclinical score between control and anti-4-1BB mAb-treated mice are statistically significant as determined by Student’s t test( p � 0.001).
1460 REGULATION OF AUTOIMMUNE DISEASES BY ANTI-4-1BB Ab
To determine whether anti-4-1BB mAb treatment impairs invivo MOG peptide-specific responses, active EAE was induced inC57BL/6 mice, as shown in Fig. 1. Mice were then restimulatedin the footpad with MOG35–55 peptide 6 wk after the first immu-nization, and 24 h later footpad thickness was measured. Naivemice were used as negative controls. As shown in Fig. 3B, controlIgG-treated mice, in contrast to naive and 2A-treated mice, re-sponded to the MOG peptide. Therefore, 2A treatment inhibiteddelayed-type hypersensitivity (DTH) responses to MOG peptide,suggesting that anti-4-1BB mAb treatment inhibits the function ofAg-specific T cells.
To further address this possibility, we tested the ability of DLNcells to proliferate in response to MOG peptide stimulation invitro. DLN cells were isolated from mice 10 days after s.c. immu-nization with MOG35–55 peptide and i.p. injection of either controlrat IgG or 2A. The cells were restimulated with various doses ofMOG35–55 peptide in vitro. As shown in Fig. 4A, DLN cells fromcontrol rat IgG-treated mice proliferated in a dose-dependent man-ner to MOG35–55 peptide stimulation, whereas DLN cells from2A-treated mice responded poorly. This result further supports ourin vivo results that anti-4-1BB mAb treatment inhibits the functionof autoreactive MOG-specific T cells.
Th1-type responses are believed to be responsible for EAEpathogenesis, whereas Th2 responses appear to be protective (27,28). To test whether the inhibition of autoreactive T cell function
is due to deviation of Th1 to Th2 cytokine production by anti-4-1BB treatment, we detected cytokine production by DLN cells.IFN-� is an important Th1 cytokine, and the administration ofagonistic anti-4-1BB Ab was reported to enhance the production ofIFN-�. Therefore, we determined the potential effects of in vivoanti-4-1BB mAb treatment on MOG-specific production of IFN-�.C57BL/6 mice were immunized s.c. with MOG35–55 peptide emul-sified in CFA and were treated i.p. with 2A or rat IgG. Ten dayslater, DLN cells were isolated and cultured in medium alone ormedium supplemented with MOG35–55 peptide (25 �g/ml) for 2days. The supernatants were collected, and IFN-� production wasdetermined by ELISA. When cultured with MOG35–55 peptide,DLN cells from mice treated with control rat IgG produced highlevels of IFN-� (Fig. 4B). In contrast, there was a significant de-crease in IFN-� production by DLN cells from mice treated with2A. The levels of IFN-� production by DLN cells cultured withmedium alone were very low (data not shown). To determinewhether this differential response was due to in vitro artifacts, totalRNA was extracted from DLN cells isolated 10 days after immu-nization for quantification of IFN-� mRNA using real-time quan-titative RT-PCR. As shown in Fig. 4C, expression of IFN-�mRNA by DLN cells was significantly reduced in 2A-treated mice.These data demonstrate that 4-1BB engagement results in the re-duced production of the Th1 cytokine, IFN-�. To study whether2A treatment results in increased production of Th2 cytokines,IL-4 and IL-10 production by DLN cells was also measured byELISA. We found that similar low levels of IL-4 was produced byDLN cells from mice treated with either control rat IgG or 2A, andIL-10 production was low and variable within the two groups (datanot shown). These results suggest that agonistic anti-4-1BB doesnot preferentially induce Th2 cytokines, but instead inhibits a Th1response to protect the mice from the development of EAEpathogenesis.
Anti-4-1BB mAb treatment does not inhibit the initial expansionof CD4� T cells, but promotes AICD
The inhibition of Ag-specific CD4� T cell responses by anti-4-1BB treatment could be due to induction of either anergy or de-letion. To discriminate between these two possibilities, we moni-tored in vivo Ag-specific responses using TCR-transgenic T cells.T cells from DO11.10 TCR-transgenic mice (32), which expresstransgenes encoding a TCR specific for chicken OVA peptide
FIGURE 2. Anti-4-1BB treatment does not prevent adoptively trans-ferred EAE. C57BL/6 mice were immunized s.c. with 100 �g of MOG35–55
emulsified in CFA. Ten days postimmunization, splenocytes and DLN cellswere pooled, cultured with MOG35–55 for 4 days, and then transferred i.v.into sublethally irradiated C57BL/6 mice. Recipients were treated i.p. with200 �g of anti-4-1BB or control IgG on the day of transfer (four mice ineach group). Data are representative of two experiments and represent themean clinical score plotted against time.
FIGURE 3. Prevention of the generation of autoreactive T cells by anti-4-1BB treatment in vivo. A, Anti-4-1BB-treated lymphocytes do not transfer EAEto RAG-1�/� mice. C57BL/6 mice were immunized s.c. with 100 �g of MOG35–55 emulsified in CFA twice at a 1-wk interval and were treated with 150�g of purified anti-4-1BB or control rat IgG i.p. on the day of first immunization. One week after the second immunization, DLN cells and splenocyteswere isolated, pooled, and transferred i.v. into RAG-1�/� mice (four mice per group). Data are representative of two experiments and represent the meanclinical score plotted against time. B, Reduced DTH in mice treated with anti-4-1BB. Active EAE was induced in C57BL/6 mice as in Fig. 1 and was theninjected in the footpad with 15 �g of MOG in 15 �l of PBS 6 wk after the first immunization. Twenty-four hours later, thickness of the footpad wasmeasured. Three mice were in each group.
1461The Journal of Immunology
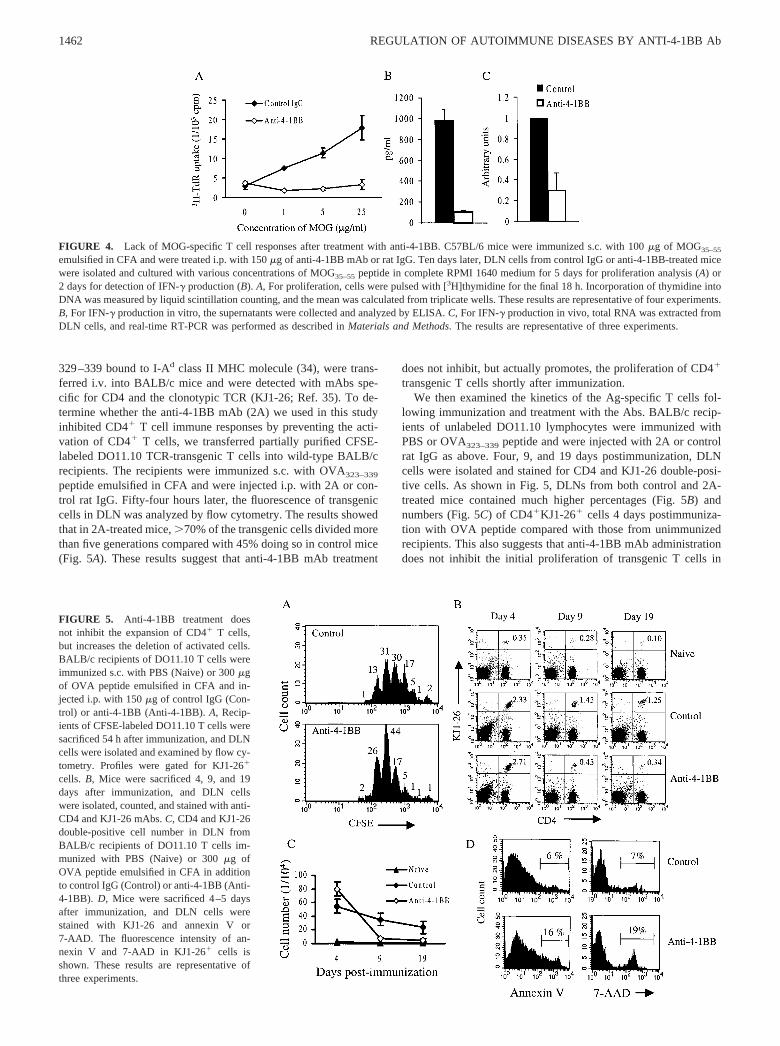
329–339 bound to I-Ad class II MHC molecule (34), were trans-ferred i.v. into BALB/c mice and were detected with mAbs spe-cific for CD4 and the clonotypic TCR (KJ1-26; Ref. 35). To de-termine whether the anti-4-1BB mAb (2A) we used in this studyinhibited CD4� T cell immune responses by preventing the acti-vation of CD4� T cells, we transferred partially purified CFSE-labeled DO11.10 TCR-transgenic T cells into wild-type BALB/crecipients. The recipients were immunized s.c. with OVA323–339
peptide emulsified in CFA and were injected i.p. with 2A or con-trol rat IgG. Fifty-four hours later, the fluorescence of transgeniccells in DLN was analyzed by flow cytometry. The results showedthat in 2A-treated mice, �70% of the transgenic cells divided morethan five generations compared with 45% doing so in control mice(Fig. 5A). These results suggest that anti-4-1BB mAb treatment
does not inhibit, but actually promotes, the proliferation of CD4�
transgenic T cells shortly after immunization.We then examined the kinetics of the Ag-specific T cells fol-
lowing immunization and treatment with the Abs. BALB/c recip-ients of unlabeled DO11.10 lymphocytes were immunized withPBS or OVA323–339 peptide and were injected with 2A or controlrat IgG as above. Four, 9, and 19 days postimmunization, DLNcells were isolated and stained for CD4 and KJ1-26 double-posi-tive cells. As shown in Fig. 5, DLNs from both control and 2A-treated mice contained much higher percentages (Fig. 5B) andnumbers (Fig. 5C) of CD4�KJ1-26� cells 4 days postimmuniza-tion with OVA peptide compared with those from unimmunizedrecipients. This also suggests that anti-4-1BB mAb administrationdoes not inhibit the initial proliferation of transgenic T cells in
FIGURE 4. Lack of MOG-specific T cell responses after treatment with anti-4-1BB. C57BL/6 mice were immunized s.c. with 100 �g of MOG35–55
emulsified in CFA and were treated i.p. with 150 �g of anti-4-1BB mAb or rat IgG. Ten days later, DLN cells from control IgG or anti-4-1BB-treated micewere isolated and cultured with various concentrations of MOG35–55 peptide in complete RPMI 1640 medium for 5 days for proliferation analysis (A) or2 days for detection of IFN-� production (B). A, For proliferation, cells were pulsed with [3H]thymidine for the final 18 h. Incorporation of thymidine intoDNA was measured by liquid scintillation counting, and the mean was calculated from triplicate wells. These results are representative of four experiments.B, For IFN-� production in vitro, the supernatants were collected and analyzed by ELISA. C, For IFN-� production in vivo, total RNA was extracted fromDLN cells, and real-time RT-PCR was performed as described in Materials and Methods. The results are representative of three experiments.
FIGURE 5. Anti-4-1BB treatment doesnot inhibit the expansion of CD4� T cells,but increases the deletion of activated cells.BALB/c recipients of DO11.10 T cells wereimmunized s.c. with PBS (Naive) or 300 �gof OVA peptide emulsified in CFA and in-jected i.p. with 150 �g of control IgG (Con-trol) or anti-4-1BB (Anti-4-1BB). A, Recip-ients of CFSE-labeled DO11.10 T cells weresacrificed 54 h after immunization, and DLNcells were isolated and examined by flow cy-tometry. Profiles were gated for KJ1-26�
cells. B, Mice were sacrificed 4, 9, and 19days after immunization, and DLN cellswere isolated, counted, and stained with anti-CD4 and KJ1-26 mAbs. C, CD4 and KJ1-26double-positive cell number in DLN fromBALB/c recipients of DO11.10 T cells im-munized with PBS (Naive) or 300 �g ofOVA peptide emulsified in CFA in additionto control IgG (Control) or anti-4-1BB (Anti-4-1BB). D, Mice were sacrificed 4–5 daysafter immunization, and DLN cells werestained with KJ1-26 and annexin V or7-AAD. The fluorescence intensity of an-nexin V and 7-AAD in KJ1-26� cells isshown. These results are representative ofthree experiments.
1462 REGULATION OF AUTOIMMUNE DISEASES BY ANTI-4-1BB Ab
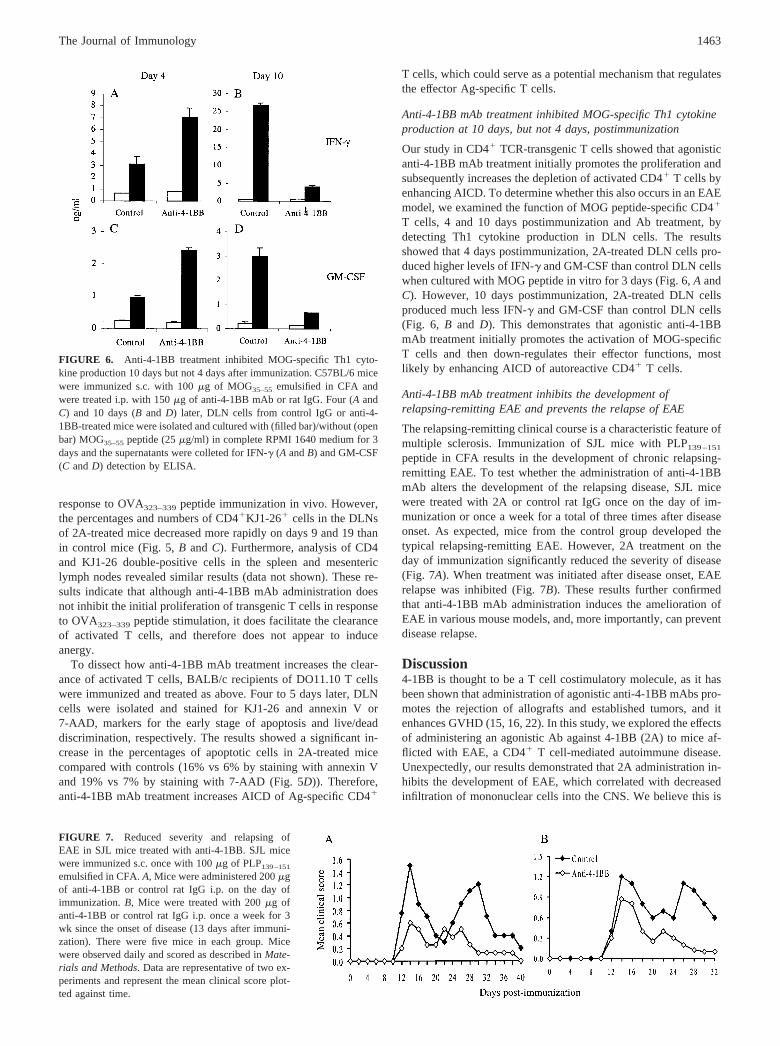
response to OVA323–339 peptide immunization in vivo. However,the percentages and numbers of CD4�KJ1-26� cells in the DLNsof 2A-treated mice decreased more rapidly on days 9 and 19 thanin control mice (Fig. 5, B and C). Furthermore, analysis of CD4and KJ1-26 double-positive cells in the spleen and mesentericlymph nodes revealed similar results (data not shown). These re-sults indicate that although anti-4-1BB mAb administration doesnot inhibit the initial proliferation of transgenic T cells in responseto OVA323–339 peptide stimulation, it does facilitate the clearanceof activated T cells, and therefore does not appear to induceanergy.
To dissect how anti-4-1BB mAb treatment increases the clear-ance of activated T cells, BALB/c recipients of DO11.10 T cellswere immunized and treated as above. Four to 5 days later, DLNcells were isolated and stained for KJ1-26 and annexin V or7-AAD, markers for the early stage of apoptosis and live/deaddiscrimination, respectively. The results showed a significant in-crease in the percentages of apoptotic cells in 2A-treated micecompared with controls (16% vs 6% by staining with annexin Vand 19% vs 7% by staining with 7-AAD (Fig. 5D)). Therefore,anti-4-1BB mAb treatment increases AICD of Ag-specific CD4�
T cells, which could serve as a potential mechanism that regulatesthe effector Ag-specific T cells.
Anti-4-1BB mAb treatment inhibited MOG-specific Th1 cytokineproduction at 10 days, but not 4 days, postimmunization
Our study in CD4� TCR-transgenic T cells showed that agonisticanti-4-1BB mAb treatment initially promotes the proliferation andsubsequently increases the depletion of activated CD4� T cells byenhancing AICD. To determine whether this also occurs in an EAEmodel, we examined the function of MOG peptide-specific CD4�
T cells, 4 and 10 days postimmunization and Ab treatment, bydetecting Th1 cytokine production in DLN cells. The resultsshowed that 4 days postimmunization, 2A-treated DLN cells pro-duced higher levels of IFN-� and GM-CSF than control DLN cellswhen cultured with MOG peptide in vitro for 3 days (Fig. 6, A andC). However, 10 days postimmunization, 2A-treated DLN cellsproduced much less IFN-� and GM-CSF than control DLN cells(Fig. 6, B and D). This demonstrates that agonistic anti-4-1BBmAb treatment initially promotes the activation of MOG-specificT cells and then down-regulates their effector functions, mostlikely by enhancing AICD of autoreactive CD4� T cells.
Anti-4-1BB mAb treatment inhibits the development ofrelapsing-remitting EAE and prevents the relapse of EAE
The relapsing-remitting clinical course is a characteristic feature ofmultiple sclerosis. Immunization of SJL mice with PLP139–151
peptide in CFA results in the development of chronic relapsing-remitting EAE. To test whether the administration of anti-4-1BBmAb alters the development of the relapsing disease, SJL micewere treated with 2A or control rat IgG once on the day of im-munization or once a week for a total of three times after diseaseonset. As expected, mice from the control group developed thetypical relapsing-remitting EAE. However, 2A treatment on theday of immunization significantly reduced the severity of disease(Fig. 7A). When treatment was initiated after disease onset, EAErelapse was inhibited (Fig. 7B). These results further confirmedthat anti-4-1BB mAb administration induces the amelioration ofEAE in various mouse models, and, more importantly, can preventdisease relapse.
Discussion4-1BB is thought to be a T cell costimulatory molecule, as it hasbeen shown that administration of agonistic anti-4-1BB mAbs pro-motes the rejection of allografts and established tumors, and itenhances GVHD (15, 16, 22). In this study, we explored the effectsof administering an agonistic Ab against 4-1BB (2A) to mice af-flicted with EAE, a CD4� T cell-mediated autoimmune disease.Unexpectedly, our results demonstrated that 2A administration in-hibits the development of EAE, which correlated with decreasedinfiltration of mononuclear cells into the CNS. We believe this is
FIGURE 6. Anti-4-1BB treatment inhibited MOG-specific Th1 cyto-kine production 10 days but not 4 days after immunization. C57BL/6 micewere immunized s.c. with 100 �g of MOG35–55 emulsified in CFA andwere treated i.p. with 150 �g of anti-4-1BB mAb or rat IgG. Four (A andC) and 10 days (B and D) later, DLN cells from control IgG or anti-4-1BB-treated mice were isolated and cultured with (filled bar)/without (openbar) MOG35–55 peptide (25 �g/ml) in complete RPMI 1640 medium for 3days and the supernatants were colleted for IFN-� (A and B) and GM-CSF(C and D) detection by ELISA.
FIGURE 7. Reduced severity and relapsing ofEAE in SJL mice treated with anti-4-1BB. SJL micewere immunized s.c. once with 100 �g of PLP139–151
emulsified in CFA. A, Mice were administered 200 �gof anti-4-1BB or control rat IgG i.p. on the day ofimmunization. B, Mice were treated with 200 �g ofanti-4-1BB or control rat IgG i.p. once a week for 3wk since the onset of disease (13 days after immuni-zation). There were five mice in each group. Micewere observed daily and scored as described in Mate-rials and Methods. Data are representative of two ex-periments and represent the mean clinical score plot-ted against time.
1463The Journal of Immunology

due to the dual role of the agonistic anti-4-1BB mAb, which ini-tially promotes T cell activation, and subsequently augments theclearance of CD4� T cells. Therefore, the administration of ago-nistic anti-4-1BB mAb may provide a new immunotherapeutic ap-proach to treating CD4� T cell-mediated autoreactive diseases.
Anti-4-1BB mAb administration may promote T cell clearanceby complement- or FcR-mediated depletion of the activated T cellsthat express 4-1BB, by blockade of 4-1BB-4-1BBL interactions,and by deliverance of an agonistic signal through the 4-1BB re-ceptor. A high dose of anti-4-1BB Ab (2A) Fab (500 �g/injection)enhanced tumor immunity, suggesting that the Fc fragment thatinitiates complement activation or that FcR-mediated depletion isnot essential for T cell costimulation (L. Chen, unpublished data).In addition, we observed that the same anti-4-1BB Ab prolongedCD8� T cell activation while increasing CD4� T cell apoptosis,which is inconsistent with a nonspecific cytolysis mechanism. Fur-thermore, we could detect 4-1BB-expressing cells 4 days postim-munization in the DLNs of mice injected with MOG peptide andtreated with 2A (data not shown). Therefore, we believe that 2Adoes not cause complement- or Fc-mediated depletion of T cellsexpressing 4-1BB.
2A could also inhibit CD4� T cell responses by blocking thesignals between 4-1BBL and 4-1BB expressed on activated Tcells. We found that EAE mice treated with anti-4-1BBL mAbcould not inhibit MOG-specific T cell responses (data not shown).This strongly suggests that just blocking 4-1BB-4-1BBL interac-tions may not be sufficient to prevent the activation of MOG-spe-cific T cells, and, therefore, 2A must be functioning through someother mechanism.
Our data support the third possibility that 2A delivers an ago-nistic signal through 4-1BB, as demonstrated by increased in vivoproliferation of DO11.10 TCR-transgenic T cells (Fig. 5A) andaugmented in vivo cytokine production by MOG-specific T cellsshortly after immunization (Fig. 6, A and C). Although the ago-nistic signal through 4-1BB was able to initially activate CD4� Tcells, we unexpectedly observed that it subsequently increasedtheir clearance (Fig. 5, B and C). Therefore, we have demonstratedthat 4-1BB can costimulate CD4� T cells and then promote theirAICD. Our results differ from previously published data that dem-onstrated that 4-1BB signaling provides a survival signal for acti-vated lymphocytes (18, 23, 36). One possible explanation is thatvarious T cell subsets could differentially respond to 4-1BB-me-diated costimulation in vivo. It has been shown that the adminis-tration of agonistic anti-4-1BB mAb induces CD8-mediated rejec-tion of established tumors and enhances allograft rejection andGVHD (15, 16). It appears that anti-4-1BB-mediated CD8� T cellresponses last longer than CD4� T cells do (15). Takahashi et al.(18) demonstrated that 4-1BB ligation is much more effective inpreventing the death of superantigen-activated CD8� T cells thanCD4� T cells in vivo. Our study clearly shows that treatment withagonistic anti-4-1BB mAb promotes the deletion of activatedCD4� T cells. Taken together, these results suggest that 4-1BBengagement may preferentially provide survival signals for CD8�
T cells in vivo.In accordance with the majority of studies, we found that 4-1BB
ligation by an agonistic mAb promotes the activation of CD4� Tcells by increasing their ability to proliferate and produce cyto-kines. By using the TCR-transgenic DO11.10 mice, Cannon andcolleagues (23) demonstrate that 4-1BBL promotes CD4� T cellsurvival in vitro. However, in this study we used DO11.10 miceand observed the dual roles of 4-1BB on CD4� T cell-mediatedimmune responses in vivo. 4-1BB initially provided T cells withcostimulatory signals, and then it unexpectedly decreased their
longevity via AICD. This discrepancy can be attributed to the com-plex nature of an in vivo immune response.
Our result is in contrast to the studies by Blazar and colleagues(22), which showed that CD4� T cell-mediated alloresponses wereenhanced by agonistic anti-4-1BB mAb. This could be due to dif-ferences among various immune responses. During an allogenic Tcell response in which MHC molecules are mismatched across theMHC, a high proportion of T cell repertoires are involved, and theresponse could be more acute than the Ag-specific autoimmunedisease model we used. Therefore, 4-1BB may have several func-tions that depend on the activation status of the cell, subset of cellinvolved, and the type of immune response initiated. Further stud-ies are needed to elucidate mechanisms by which anti-4-1BB stim-ulation promotes the AICD of CD4� T cells in vivo.
In summary, we were able to inhibit autoreactive T cell re-sponses, and therefore prevent EAE pathogenesis, by treating micewith an agonistic anti-4-1BB mAb. The down-regulation of auto-reactive T cell responses could be due to regulation of the Th1/Th2balance, activation of various subsets of regulatory cells, inductionof T cell unresponsiveness (anergy), or deletion of activated cellsvia AICD. In this study, we have demonstrated that 4-1BB doesnot appear to affect the Th1/Th2 balance or anergize CD4� T cells.Instead, we have clearly shown that the administration of agonisticanti-4-1BB mAb initially increases T cell activation and then pro-motes the clearance of these activated CD4� T cells, which resultsin the attenuation of their effector functions. In support of this dualrole of a costimulatory molecule, a recently published studyshowed that the use of agonistic anti-CD40 Ab initially increasedthe number of tumor-specific CD8� T cells, and then it acceleratedtheir deletion in a tumor model system (37). In addition, anothergroup has reported that agonistic mAbs against CD40 can success-fully control chronic autoimmune inflammatory processes (38).These studies and ours suggest that opposing effects can be elicitedby strong T cell stimulation in vivo, and the use of agonistic Absagainst costimulatory molecules may help us to reveal the complexfunctions of these molecules. The agonistic Ab against 4-1BB wasable to inhibit two different EAE models, including the murinerelapsing model, which is clinically relevant, suggesting that it ispossible to preferentially delete activated autoreactive T cells. Theuse of agonistic Abs against costimulatory molecules may providea novel strategy for treating T cell-mediated autoimmune diseases.
AcknowledgmentsWe thank Zhong Guo for her help in real-time PCR. We thank Lisa Hoffman,James Lo, and Robert Chin for their comments on the manuscript.
References1. Lenschow, D. J., T. L. Walunas, and J. A. Bluestone. 1996. CD28/B7 system of
T cell costimulation. Annu. Rev. Immunol. 14:233.2. Shahinian, A., K. Pfeffer, K. P. Lee, T. M. Kundig, K. Kishihara, A. Wakeham,
K. Kawai, P. S. Ohashi, C. B. Thompson, and T. W. Mak. 1993. Differential Tcell costimulatory requirements in CD28-deficient mice. Science 261:609.
3. Green, J. M., P. J. Noel, A. I. Sperling, T. L. Walunas, G. S. Gray,J. A. Bluestone, and C. B. Thompson. 1994. Absence of B7-dependent responsesin CD28-deficient mice. Immunity 1:501.
4. DeBenedette, M. A., A. Shahinian, T. W. Mak, and T. H. Watts. 1997. Costimu-lation of CD28 T lymphocytes by 4-1BB ligand. J. Immunol. 158:551.
5. Pollok, K. E., Y. J. Kim, Z. Zhou, J. Hurtado, K. K. Kim, R. T. Pickard, andB. S. Kwon. 1993. Inducible T cell antigen 4-1BB: analysis of expression andfunction. J. Immunol. 150:771.
6. Melero, I., J. V. Johnston, W. W. Shufford, R. S. Mittler, and L. Chen. 1998.NK1.1 cells express 4-1BB (CDw137) costimulatory molecule and are requiredfor tumor immunity elicited by anti-4-1BB monoclonal antibodies. Cell. Immu-nol. 190:167.
7. Goodwin, R. G., W. S. Din, T. Davis-Smith, D. M. Anderson, S. D. Gimpel,T. A. Sato, C. R. Maliszewski, C. I. Brannan, N. G. Copeland, N. A. Jenkins, etal. 1993. Molecular cloning of a ligand for the inducible T cell gene 4-1BB: amember of an emerging family of cytokines with homology to tumor necrosisfactor. Eur. J. Immunol. 23:2631.
1464 REGULATION OF AUTOIMMUNE DISEASES BY ANTI-4-1BB Ab

8. Pollok, K. E., Y. J. Kim, J. Hurtado, Z. Zhou, K. K. Kim, and B. S. Kwon. 1994.4-1BB T-cell antigen binds to mature B cells and macrophages, and costimulatesanti-�-primed splenic B cells. Eur. J. Immunol. 24:367.
9. Alderson, M. R., C. A. Smith, T. W. Tough, T. Davis-Smith, R. J. Armitage,B. Falk, E. Roux, E. Baker, G. R. Sutherland, and W. S. Din. 1994. Molecular andbiological characterization of human 4-1BB and its ligand. Eur. J. Immunol.24:2219.
10. DeBenedette, M. A., N. R. Chu, K. E. Pollok, J. Hurtado, W. F. Wade,B. S. Kwon, and T. H. Watts. 1995. Role of 4-1BB ligand in costimulation of Tlymphocyte growth and its upregulation on M12 B lymphomas by cAMP. J. Exp.Med. 181:985.
11. Salih, H. R., S. G. Kosowski, V. F. Haluska, G. C. Starling, D. T. Loo, F. Lee,A. A. Aruffo, P. A. Trail, and P. A. Kiener. 2000. Constitutive expression offunctional 4-1BB (CD137) ligand on carcinoma cells. J. Immunol. 165:2903.
12. Chu, N. R., M. A. DeBenedette, B. J. Stiernholm, B. H. Barber, and T. H. Watts.1997. Role of IL-12 and 4-1BB ligand in cytokine production by CD28� andCD28� T cells. J. Immunol. 158:3081.
13. Saoulli, K., S. Y. Lee, J. L. Cannons, W. C. Yeh, A. Santana, M. D. Goldstein,N. Bangia, M. A. DeBenedette, T. W. Mak, Y. Choi, and T. H. Watts. 1998.CD28-independent, TRAF2-dependent costimulation of resting T cells by 4-1BBligand. J. Exp. Med. 187:1849.
14. Vinay, D. S., and B. S. Kwon. 1998. Role of 4-1BB in immune responses. Semin.Immunol. 10:481.
15. Shuford, W. W., K. Klussman, D. D. Tritchler, D. T. Loo, J. Chalupny,A. W. Siadak, T. J. Brown, J. Emswiler, H. Raecho, C. P. Larsen, et al. 1997.4-1BB costimulatory signals preferentially induce CD8� T cell proliferation andlead to the amplification in vivo of cytotoxic T cell responses. J. Exp. Med.186:47.
16. Melero, I., W. W. Shuford, S. A. Newby, A. Aruffo, J. A. Ledbetter,K. E. Hellstrom, R. S. Mittler, and L. Chen. 1997. Monoclonal antibodies againstthe 4-1BB T-cell activation molecule eradicate established tumors. Nat. Med.3:682.
17. Melero, I., N. Bach, K. E. Hellstrom, A. Aruffo, R. S. Mittler, and L. Chen. 1998.Amplification of tumor immunity by gene transfer of the co-stimulatory 4-1BBligand: synergy with the CD28 co-stimulatory pathway. Eur. J. Immunol. 28:1116.
18. Takahashi, C., R. S. Mittler, and A. T. Vella. 1999. Cutting edge: 4-1BB is a bonafide CD8 T cell survival signal. J. Immunol. 162:5037.
19. DeBenedette, M. A., T. Wen, M. F. Bachmann, P. S. Ohashi, B. H. Barber,K. L. Stocking, J. J. Peschon, and T. H. Watts. 1999. Analysis of 4-1BB ligand(4-1BBL)-deficient mice and of mice lacking both 4-1BBL and CD28 reveals arole for 4-1BBL in skin allograft rejection and in the cytotoxic T cell response toinfluenza virus. J. Immunol. 163:4833.
20. Tan, J. T., J. K. Whitmire, R. Ahmed, T. C. Pearson, and C. P. Larsen. 1999.4-1BB ligand, a member of the TNF family, is important for the generation ofantiviral CD8 T cell responses. J. Immunol. 163:4859.
21. Gramaglia, I., D. Cooper, K. T. Miner, B. S. Kwon, and M. Croft. 2000. Co-stimulation of antigen-specific CD4 T cells by 4-1BB ligand. Eur. J. Immunol.30:392.
22. Blazar, B. R., B. S. Kwon, A. Panoskaltsis-Mortari, K. B. Kwak, J. J. Peschon,and P. A. Taylor. 2001. Ligation of 4-1BB (CDw137) regulates graft-versus-host
disease, graft-versus-leukemia, and graft rejection in allogeneic bone marrowtransplant recipients. J. Immunol. 166:3174.
23. Cannons, J. L., P. Lau, B. Ghumman, M. A. DeBenedette, H. Yagita,K. Okumura, and T. H. Watts. 2001. 4-1BB ligand induces cell division, sustainssurvival, and enhances effector function of CD4 and CD8 T cells with similarefficacy. J. Immunol. 167:1313.
24. Mittler, R. S., T. S. Bailey, K. Klussman, M. D. Trailsmith, and M. K. Hoffmann.1999. Anti-4-1BB monoclonal antibodies abrogate T cell-dependent humoral im-mune responses in vivo through the induction of helper T cell anergy. J. Exp.Med. 190:1535.
25. Gonatas, N. K., M. I. Greene, and B. H. Waksman. 1986. Genetic and molecularaspects of demyelination. Immunol. Today 7:121.
26. Wekerle, H. 1991. Immunopathogenesis of multiple sclerosis. Acta Neurol.(Napoli) 13:197.
27. Liblau, R. S., S. M. Singer, and H. O. McDevitt. 1995. Th1 and Th2 CD4� T cellsin the pathogenesis of organ-specific autoimmune diseases. Immunol. Today 16:34.
28. Adorini, L., and F. Sinigaglia. 1997. Pathogenesis and immunotherapy of auto-immune diseases. Immunol. Today 18:209.
29. Rodriguez, M., R. P. Roos, D. McGavern, L. Zoecklein, K. Pavelko, H. Sang, andX. Lin. 2000. The CD4-mediated immune response is critical in determining theoutcome of infection using Theiler’s viruses with VP1 capsid protein point mu-tations. Virology 275:9.
30. Heid, C. A., J. Stevens, K. J. Livak, and P. M. Williams. 1996. Real-time quan-titative PCR. Genome Res. 6:986.
31. Overbergh, L., D. Valckx, M. Waer, and C. Mathieu. 1999. Quantification ofmurine cytokine mRNAs using real-time quantitative reverse transcriptase PCR.Cytokine 11:305.
32. Murphy, K. M., A. B. Heimberger, and D. Y. Loh. 1990. Induction by antigen ofintrathymic apoptosis of CD4�CD8�TCRlow thymocytes in vivo. Science 250:1720.
33. Kearney, E. R., K. A. Pape, D. Y. Loh, and M. K. Jenkins. 1994. Visualizationof peptide-specific T cell immunity and peripheral tolerance induction in vivo.Immunity 1:327.
34. Shimonkevitz, R., S. Colon, J. W. Kappler, P. Marrack, and H. M. Grey. 1984.Antigen recognition by H-2-restricted T cells. II. A tryptic ovalbumin peptide thatsubstitutes for processed antigen. J. Immunol. 133:2067.
35. Haskins, K., R. Kubo, J. White, M. Pigeon, J. Kappler, and P. Marrack. 1983. Themajor histocompatibility complex-restricted antigen receptor on T cells. I. Isola-tion with a monoclonal antibody. J. Exp. Med. 157:1149.
36. Hurtado, J. C., Y. J. Kim, and B. S. Kwon. 1997. Signals through 4-1BB arecostimulatory to previously activated splenic T cells and inhibit activation-in-duced cell death. J. Immunol. 158:2600.
37. Kedl, R. M., M. Jordan, T. Potter, J. Kappler, P. Marrack, and S. Dow. 2001.CD40 stimulation accelerates deletion of tumor-specific CD8� T cells in theabsence of tumor-antigen vaccination. Proc. Natl. Acad. Sci. USA 98:10811.
38. Mauri, C., L. T. Mars, and M. Londei. 2000. Therapeutic activity of agonisticmonoclonal antibodies against CD40 in a chronic autoimmune inflammatory pro-cess. Nat. Med. 6:673.
1465The Journal of Immunology


![Agonistic depictions of communication: Vaikeneminen [silence] vs. puhuminen [speaking] in classroom settings for adult education in Finland.](https://static.fdokumen.com/doc/165x107/631ef79a7509c0131f095b3e/agonistic-depictions-of-communication-vaikeneminen-silence-vs-puhuminen-speaking.jpg)

















