
summary of journal open ski policy for small island developing starts
Upload
independentCategory
view
1download
0
THE JOURNAL OF GENE MEDICINE R E S E A R C H A R T I C L EJ Gene Med 2009; 11: 207–219.Published online 2 February 2009 in Wiley InterScience (www.interscience.wiley.com) DOI: 10.1002/jgm.1303
Adenoviral delivery of dominant-negativetransforming growth factor β type II receptorup-regulates transcriptional repressor SKI-likeoncogene, decreases matrix metalloproteinase 2 inhepatic stellate cell and prevents liver fibrosis inrats
Ana Marquez-Aguirre1
Ana Sandoval-Rodriguez1
Jaime Gonzalez-Cuevas1
Miriam Bueno- Topete1
Jose Navarro-Partida1
Immaculada Arellano-Olivera1
Silvia Lucano-Landeros1
Juan Armendariz-Borunda1,2*
1Institute for Molecular Biology inMedicine and Gene Therapy,University of Guadalajara,Department of Molecular Biology andGenomics, Guadalajara, Jalisco,Mexico2OPD Hospital Civil de Guadalajara,Guadalajara, Jalisco, Mexico
*Correspondence to:Juan Armendariz-Borunda,Department of Molecular Biologyand Genomics, CUCS, University ofGuadalajara, PO Box 2-123,Guadalajara, Jalisco 44281, Mexico.E-mail: [email protected]
Received: 2 July 2008Revised: 1 December 2008Accepted: 30 December 2008
AbstractBackground Dominant-negative transforming growth factor β type IIreceptor (TβRII�cyt) is a protein that blocks transforming growth factor(TGF-β) signaling. Because the consequences of blocking TGF-β have not beencompletely elucidated in liver fibrosis, we analysed the effects of adenoviraldelivery of TβRII�cyt on profibrogenic genes and matrix metalloproteinase(MMP) proteins, as well as on TGF-β signal repressor SKI-like oncogene(SnoN), in cultured hepatic stellate cells (HSCs) and in a rat model of liverfibrosis.
Methods To induce liver fibrosis, rats were treated with thioacetamide for7 weeks and administrated once with Ad-TβRII�cyt or Ad-βgal through theiliac vein. Fibrosis was measured by morphometric analysis. We evaluatedSnoN by western blot, immunocytochemistry and immunohistochemistry;MMP activity was determined by zymography and profibrogenic geneexpression by the real-time reverse transcriptase-polymerase chain reactionin cultured HSCs and liver tissue.
Results Profibrogenic gene expression of collagen α1 (I), TGF-β1, platelet-derived growth factor-B, plasminogen activator inhibitor (PAI)-1, tissueinhibitor of matrix metalloproteinase-1 and MMP-2 was down-regulated;whereas MMP-3 was over-expressed in response to Ad-TβRII�cyt in HSCs.Moreover, zymography assays corroborated MMP-2 and MMP-3 changes inactivity. Surprisingly, anti-TGF-β molecular intervention increased nuclearSnoN in HSCs. In vivo, Ad-TβRII�cyt reduced liver fibrosis, increased nuclearSnoN in sinusoidal cells, and also produced significant suppression incollagen α1 (I), TGF-β1, PAI-1, MMP-2 and over-expression in MMP-3 inthioacetamide-intoxicated animals.
Conclusions The results obtained in the present study suggest thatthe molecular mechanism for the blocking effects of Ad-TβRII�cyt inTGF-β signaling acts via up-regulation of the transcriptional repressorSnoN, which antagonizes TGF-β signaling (TGF-β/Smad-pathway inhibitor).Consequently, profibrogenic genes are down-regulated. Copyright 2009John Wiley & Sons, Ltd.
Keywords hepatic stellate cells; liver fibrosis; MMP-2; SnoN; TGF-β signaling;thioacetamide
Copyright 2009 John Wiley & Sons, Ltd.

208 A. L. Marquez-Aguirre et al.
Introduction
In chronic thioacetamide (TAA) intoxication, featuresof human liver cirrhosis have developed after 7 weeksof TAA administration, such as substantial fibrosis,prominent regenerative nodules, portal hypertension andhyperdynamic circulation. Because TAA-induced livercirrhosis in rats has been shown to resemble the humandisease, it has been proposed that it serves as a suitableanimal model for studying the pathogenisis of humanliver fibrosis and cirrhosis [1,2].
The profibrogenic cytokine transforming growth factor(TGF)-β and the resident hepatic stellate cells constitutethe principal mediators for liver fibrosis and the primordialtherapeutic targets [3]. It is well established that TGF-βis the dominant stimuli for hepatic stellate cells (HSCs) toproduce extracellular matrix (ECM) wound-componentsin liver fibrosis [4]. The TGF-β signaling pathway isregulated mainly through Smads [5]. Type II receptor(TβRII) plays a major role in signal transduction dueto its serine/threonine kinase activity [6], which in turnphosphorylates type I receptor, thus promoting activationof Smads. To date, it is well known that Smad2 andSmad3 oligomerize with Smad4 and translocate intothe nucleus to induce the switch-on of profibrogenicgenes that are responsive to TGF-β, such as plasminogenactivator inhibitor (PAI)-1, collagen α1 (I), platelet-derived growth factor (PDGF)-B, tissue inhibitor of matrixmetalloproteinase (TIMP)-1, metalloproteinase (MMP)-2and TGF-β itself [7,8].
Down-regulation of TGF-β1 signaling is carried out inpart by a feedback mechanism involving the inductionof expression of the inhibitory protein Smad7, which,through receptor binding, can prevent receptor-activatedSmad activation [5]. Alternatively, Smad binding todifferent transcription factors may also modulate TGF-β signaling. When co-repressors TGFB-induced factorhomeobox 1, v-ski sarcoma viral oncogene homolog (Ski)and SnoN (SKI-like oncogene) bind to Smad proteins,histone deacetylases are recruited to repress transcription[9,10]. SnoN has been described as a nuclear co-repressorfor Smad4 to maintain TGF-β-responsive gene expressionat a basal level [11]. To enssure a TGF-β signalingpathway that allows transcriptional activation of TGF-β-responsive genes, Smad3-dependent degradation ofSnoN occurs [10,12]. Over-expression of SnoN couldeventually turn off transcriptional activation of TGF-β-responsive genes, effectively limiting the extent of theTGF-β response.
Although there are various studies demonstrating theability of dominant-negative transforming growth factor β
type II receptor (TβRII�cyt) to prevent the developmentof fibrotic processes [13,14], they fail with respect tocompleting the final mechanistic scheme. Thus, in thepresent study, we evaluated the effect of Ad-TβRII�cyt onthe TGF-β signaling blockade and its consequences withrespect to the inhibition of profibrogenic gene expressionand the TGF-β nuclear transcriptional repressor SnoN.
Here, advantage was taken of the fact that adenoviralvectors have been used to effectively deliver genes tohepatic tissue when administered systemically [15].
As expected, we found that the dominant-negativeTGF-β receptor type II down-regulates TGF-β-responsiveprofibrogenic genes. According to our findings, themolecular mechanism for Ad-TβRII�cyt comprises in partthe nuclear up-regulation of the transcriptional repressorSnoN, which antagonizeas TGF-β signaling.
Materials and methods
Construction of Ad-TβRII�cyt andtitration of viral particles
To construct the recombinant adenovirus containingTβRII�cyt [16], we used Kit E from Microbix Biosys-tems, Inc. (Toronto, Ontario, Canada). In brief, plas-mid pCDNA3-TβRII�cyt, which contains the humantruncated receptor cDNA, was subcloned into theSalI/HindIII site of the pDC516, in which TβRII�cytis driven by the cytomegalovirus promoter. Subcloningwas confirmed by the polymerase chain reaction(PCR) with primers flanking pDC516 polylinker (for-ward, 5′-AAAGGGCAGCCAAAACGTAACACC-3′; reverse,5′-CCATTATAAGCTGCAATAAACAAG-3′) (Figure 1A).
Cotransfection of pBHGfrt�E1,3FLP and pDC516-TβRII�cyt plasmids was carried out according to themanufacturer’s instructions [17] on HEK-293 cells, whichconstitutively express Ad-Early Region 1 (E1) [18], result-ing in Ad-TβRII�cyt generation. frt-Site specific recombi-nation between pDC516 and pBHGfrt�E1,3FLP was real-ized by FLP recombinase coded in pBHGfrt�E1,3FLP thatacts in situ [19] generating the pBHGfrt�E1,3-TβRII�cyt(Figure 1B). Ad-TβRII�cyt was grown in 293 cells andpurified by CsCl banding, and transgene insertion in viri-ons was confirmed by PCR using specific primer aligning inHA tag (Figure 1C). Adenovirus serotype (Ad5) was con-firmed with another set of oligonulceotides for Ad5 ITRregion (forward 5′-TCCCACCGTATGATGTTCCTGATT-3′;reverse, 5′-TTCTTTCTCCATACAGCCACACAG-3′) (datanot shown).
Viral particles (vp) were quantified by absorbanceat 260 nm, where one unit is equivalent to 1.1 × 1012
adenoviral vector particles [20]. Viral titer was estimatedin infections units according to the Normalized AdjustedStandard method [21]. Ad-βgal, which contains theEscherichia coli β-galactosidase gene, was also producedas described, and used as the control.
Cell culture
The human HSC-180 line was derived initially from acirrhotic liver and is valuable and useful for addressingissues of HSC biology and function. This line was shownto express α-smooth muscle actin, MMP-2, MMP-3, MMP-9 and several molecules involved in the plasminogen
Copyright 2009 John Wiley & Sons, Ltd. J Gene Med 2009; 11: 207–219.DOI: 10.1002/jgm

Ad-TβRII�cyt prevents liver fibrosis and up-regulates SnoN 209
Figure 1. Construction of recombinant adenovirus Ad-TβRII�cyt. (A) Subcloning of the TβRII�cyt sequence into pDC516 wasconfirmed by PCR with primers flanking pDC516 polylinker. The PCR product in 0.8% agarose gel of 227 bp matches the emptypDC516 and the 997-bp band represents pDC516-TβRII�cyt. (B) Strategy used to generate Ad-TβRII�cyt. HEK-293 cells wereco-transfected with pDC516-TβRII�cyt and pBHGfrt�E1,3FLP bearing a truncated packaging signal flanked by frt sites. Site specificrecombination between these plasmids is catalysed by FLP recombinase and replaces the ��, FLP cassette, �E1 and frt sequencein the pBHGfrt�E1,3FLP, generating an Ad genome including a complete � signal TβRII�cyt cassette. (C) TβRII�cyt sequencepackage into the recombinant Ad5 virus was confirmed by PCR. The 229-bp band corresponds to TβRII�cyt
activator pathway. Furthermore, the cells were foundto have capacity to synthesize the mRNAs for TGF-β1,TIMP-1 and collagen type I [22,23].
Adenoviral transduction in HSCs
Activated HSCs (HSC-180) were used to test TGF-βinhibition [23,24]. HSCs were plated at a density of6.5 × 105 cells per well on 35-mm dishes, in 1.5 ml ofDulbecco’s modified Eagle’s medium (Gibco, Rockville,MD, USA). Atmosphere used was 5% CO2, 95% humidityand 37 ◦C. Media was supplemented with 10% fetalbovine serum (until 1 day before adenovirus infection),100 IU/ml penicillin, 100 µg/ml streptomycin, 2 mM L-glutamine and 1% essential amino acids. Three groups(each n = 3) were set up: (i) control HSC withouttransduction; (ii) HSC with Ad-βgal transduction; and(iii) HSC with Ad-TβRII�cyt transduction. On the fifthday, at 70% confluency, cells were transfected with2 × 108 µi/ml of Ad-TβRII�cyt or Ad-βgal, incubatedfor two additional days, and then cellular lysate andsupernatants were collected.
Ad-TβRII�cyt administration inTAA-intoxicated rats
An in vivo model was intended to assess fibrosisprevention via TβRII�cyt. TAA-induced cirrhosis was
achieved using a dose of 200 mg/kg administratedintraperitoneally three times a week for 7 weeks, asdescribed previously [25,26]. TAA-intoxicated rat groupswere designed to assess the possibility that fibrosisprogression could be halted. Thus, transgene deliverywas carried out in the middle of the TAA intoxicationregimen at 3.5 weeks. Male Wistar rats (150–180 g) wererandomized into six experimental groups (each n = 5):(i) control non-TAA without Ad transduction; (ii) controlnon-TAA with Ad-βgal transduction; (iii) control non-TAAwith Ad-TβRII�cyt transduction; (iv) TAA-intoxicatedrats without Ad transduction; (v) TAA-intoxicated ratswith Ad-βgal transduction; and (vi) TAA-intoxicated ratswith Ad-TβRII�cyt transduction. All animals receivedhumane care and were weighed every week to adjust TAAdosage. At 3.5 weeks, animals were administrated withAd-TβRII�cyt or Ad-βgal (5 × 1011 vp/kg) via the iliacvein, and rat sacrifice was performed at the seventh week.Blood samples were taken immediately before sacrificeand representative liver sections were excised and eitherfixed with 4% buffered paraformaldehyde for histologicalexamination or frozen immediately for RNA and proteinextraction.
β-galactosidase assay
To monitor the degree of transduction for both liverand hepatic stellate cells, β-galactosidase activity was
Copyright 2009 John Wiley & Sons, Ltd. J Gene Med 2009; 11: 207–219.DOI: 10.1002/jgm

210 A. L. Marquez-Aguirre et al.
determined 72 h after Ad-βgal injection, according toprevious reports [15]. In brief, hepatic stellate cellsand liver slices transduced with Ad-βgal were washedthree times with phosphate-buffered saline (PBS) (pH7.5) and fixed for 5 min with 4% paraformaldehyde.After fixation, were washed twice with PBS (pH 8.5)and X-gal staining was performed. To avoid endogenousgalactosidase activity, all reactions were carried out at pH8.5 and incubated in the darkness for a maximum of 2 h.X-gal staining solution contained 1 mg/ml X-gal, 5 mM
K3Fe(CN)6, 5 mM K4Fe(CN)6, 2 mM MgCl2, 0.2% NP-40and 0.1% sodium desoxicholate. Exposure of tissues wasperformed in the dark at 4 ◦C for 16–18 h and was thenobserved by light microscopy.
Western blot analysis for transgenedetection and SnoN expression
To identify the transgene, TβRII�cyt, proteins wereextracted from cultured HSCs and liver from TAA-intoxicated rats was transduced with Ad-TβRII�cyt usinglysis buffer (50 mM Tris-HCl, pH 8.0, 150 nM NaCl, 0.02%NaN3). After centrifugation at 13 000 r.p.m 22000g for5 min at 4 ◦C, supernatant was collected and quantifiedby the Bradford assay. Proteins were stored at −70 ◦Cuntil western blot for TβRII�cyt detection, which has anHA tag. Briefly, 30 µg of total protein was separatedby 10% sodium dodecyl sulphate-polyacrylamide gelelectrophoresis (SDS-PAGE) under reducing conditionsand transferred to poly(vinylidene difluoride) (PVDF)membranes (Bio-Rad Laboratories, Foster City, CA, USA).Blocking was carried out using 2% nonfat dry milkfor 2 h; primary antibody dilution was 1 : 200. Humantruncated receptor was detected in HSCs using anti-HAantibody (Santa Cruz Biotechnology, Santa Cruz, CA,USA) or anti-human TβRII (R&D Systems, Minneapolis,MN, USA) in rats. Antibody binding was revealedwith a secondary antibody diluted 1 : 5000 using a BMChemiluminiscence kit (Roche Diagnostics, Indianapolis,IN, USA). In these samples, the mRNA transgene wasdetected by conventional PCR, as well.
Then, SnoN protein expression was detected by westernblot in nuclear proteins obtained from HSC culturesand liver tissue according to Salazar-Montes et al. [27].Briefly, the samples were homogenized on ice andsupernatant with cytoplasmic proteins was eliminated.Isolated nuclei were subject to a second homogenationto recover nuclear proteins. Western blot was madeusing 10 µg of nuclear proteins in 10% SDS-PAGE underreducing conditions and transferred to PVDF membranes(Bio-Rad Laboratories). Blocking was made using 3%nonfat dry milk for 2 h; primary antibodies againstSnoN and Histone 1 (Santa Cruz Biotechnology) diluted1 : 1000 and 1 : 400, respectively, in PBS and secondaryantibody at 1 : 12 000. Bound antibodies were detectedby the BM Chemiluminiscence kit (Roche Diagnostics).Densitometric analysis was realized with Kodak 1D 3.5Image analyser (Kodak, Rochester, NY, USA).
Quantitative real-time reversetranscriptase (RT)-PCR
RNA was isolated from HSCs and the liver from differentgroups of rats with Trizol reagent (Invitrogen, Carlsbad,CA, USA). Retrotranscription using 2 µg of total RNA wasachieved using M-MLV reverse transcriptase (Invitrogen).Then, 2 µl of cDNAs were subjected to real-time PCRusing a Rotor Gene Termocycler under the conditions:2 min at 50 ◦C, 10 min at 95 ◦C, and 45 cycles of 15 sat 95 ◦C and 1 min at 60 ◦C. Specific probes designed toalign in collagen α1 (I), TGF-β1, PDGF-B, PAI-1, TIMP-1,MMP-2, MMP-3, and SnoN human and rat RNAs wereacquired either from Applied Biosystems (Hammonton,NJ, USA) or from Invitrogen (Table 1). Gene amplificationwas normalized against GAPDH expression. Relativequantification by the 2−��CT method was realized bycomparing to control groups as an internal calibrator[28,29].
Zymography assays for MMP-2 andMMP-3
Zymography assays were performed according toGonzalez-Cuevas et al. [23]. Briefly, they were made withgelatin or casein as substrate to evaluate the functionalactivity of gelatinase A (MMP-2) and stromelysin-1 (MMP-3), respectively. First, 30 µg (for MMP-2) and 50 µg (forMMP-3) of total protein from culture supernatants wasloaded onto a 10% SDS-PAGE containing 1 mg/ml ofgelatin (Sigma-Aldrich, St Louis, MO, USA) for gelati-nase detection or 2 mg/ml of casein (Sigma Aldrich)for stromelysin-1 analysis. Following electrophoresis, gelswere washed overnight in 2.5% Triton X-100 at 4 ◦C,and then incubated for 48 h at 37 ◦C in developing buffer(50 mM Tris-HCl, pH 7.4, 10 mM CaCl2, 5 mM ZnCl2) forgelatinases or for 72 h at 37 ◦C in stromelysin-1 devel-oping buffer (0.1 M glycine and 0.02% NaN3). Gelswere subsequently stained with 0.5% Coomasie Blue-G(Bio-Rad Laboratories) and destained until the desiredcontrast was achieved with a solution containing 10%acetic acid and 20% methanol. The intensity of clearedzones resulting from enzymatic activity was quantified bydensitometry using a Kodak 1D 3.5 Image analyser (datano show).
Immunocytochemistry in HSC culture,histological examination andimmunohistochemistry of liver sections
For immunocytochemistry, HSCs cultures were washedthree times with PBS (pH 7.5) and fixed for 10 min with4% paraformaldehyde. After fixation, were washed twicewith PBS, the endogenous activity of peroxidase wasquenched with 1% hydrogen peroxide diluted in deion-ized H2O, and endogenous biotin was quenched with0.001% avidin. Liver sections were incubated overnight
Copyright 2009 John Wiley & Sons, Ltd. J Gene Med 2009; 11: 207–219.DOI: 10.1002/jgm

Ad-TβRII�cyt prevents liver fibrosis and up-regulates SnoN 211
Table 1. Sequence primer sets in real-time PCR assays
SequenceTargetgene Dye Primer name Forward Reverse
GAPDH FAM Rat NM 017008.3 Propietary (Applied Biosystems)PAI-1 FAM Rat M24067.1 5′-CGTCTTCCTCCACAGCCATTC-3′ 5′-CGGAGGATCGGTCTAAAACCATCTC(FAM)G-3′TIMP-1 JOE Rat NM 053819.1 5′-CGTCGCTAAGATGCTCAAAGGA- 5′-GGTGTAGGCGAACCGGAAAC-3′
TTCGA(JOE)G-3′MMP-2 FAM Rat NM 031054.1 5′-GGCACCACCGAGGACTATGAC-3′ 5′-CGAGACACAGTGGACATAGCGGTCT(FAM)G-3′MMP-3 JOE Rat NM 133523.1 5′-ATTGGCACAAAGGTGGATGCT-3′ 5′-CGGATAAACTCCAACTGTGAAGATC(JOE)G-3′PDGF FAM Rat Z14117.1 5′-CGGATCTGAGTGACCACTCCATC- 5′-TTCGTCTACGGAGTCTCTGTGC-3′
(FAM)G-3′MMP-3 FAM Cell culture NM 002422.3 5′-GGTTCCGCCTGTCTCAAGATG-3′ 5′-CGGAAGTTCTGGAGGGACAGGTTC(FAM)G-3′PDGF FAM Cell culture NM 002608.1 5′-CAAACTCGGGTGACCATTCG-3′ 5′-CGGAATGCGTGTGCTTGAATTTC(FAM)G-3′GAPDH FAM Rat Rn99999916 s1 Propietary (Applied Biosystems)TGF-β1 FAM Rat Rn 00572010 m1 Propietary (Applied Biosystems)COL1 FAM Rat Rn 00670303 g1 Propietary (Applied Biosystems)MMP2 FAM Cell culture Hs 00234422 m1 Propietary (Applied Biosystems)COL1 FAM Cell culture Hs 00166657 m1 Propietary (Applied Biosystems)TGF-β1 FAM Cell culture Hs 00171257 m1 Propietary (Applied Biosystems)TIMP-1 FAM Cell culture Hs 99999139 m1 Propietary (Applied Biosystems)GAPDH FAM Cell culture 4333764T Propietary (Applied Biosystems)PAI-1 FAM Cell culture NM 000602 Propietary (Applied Biosystems)SnoN FAM Cell culture 596258-A11 Propietary (Applied Biosystems)
at 4 ◦C with primary antibody against SnoN (Santa CruzBiotechnology) diluted 1 : 50 in PBS. Bound antibod-ies were detected with peroxidase-labeled rabbit poly-clonal antibodies against mouse immunoglobulins anddiaminobenzidine and counterstained with hematoxylin(Staining System: sc-2018; Santa Cruz Biotechnology).
For the histological study, livers were immediatelyremoved and fixed by immersion in 4% paraformaldehydediluted in PBS, dehydrated in graded ethylic alcohol,and embedded in paraffin. Sections, 5 µm thick, werestained with Masson’s trichrome and analysed bylight microscopy as described previously [30] using acomputer-assisted morphometric analyser (Image-ProPlus5.0; Media Cybernetics, Inc., Bethesda, MD, USA) byanalysing ten random fields per slide and calculatingthe ratio of connective tissue to the whole liver area.For immunohistochemistry, liver specimens from eachanimal section were mounted in silane-covered slides anddeparaffinized, and the analysis continued as previouslydescribed for the immunocytochemistry.
Biochemical assays and hydroxyprolinecontent
Blood was drawn from animals at specified times andserum transaminases ALT, AST, alkaline phosphatase,bilirubins, GT and albumin were determined in an auto-mated Sincron-7 analyser at Hospital Civil de Guadalajara(Guadalajara, Jalisco, Mexico). The hydroxyproline con-tent of liver samples was measured in 150 mg of tissuesubjected to acid hydrolysis, as described previously [31].
Statistical analysis
Normally distributed data were analysed using one-wayanalysis of variance. Then, Holm–Sidak or Dunnett’s
post-hoc test were used for determination of statisticalsignificance (p < 0.05). Data are shown as the mean± SD. For real-time PCR experiments, results areshown as the 2−��CT value (mean ± SD), where thestandard deviation was calculated as: s = √
s(GAPDH)2 +
s(target gene)2, according to user bulletin #2 from Applied
Biosystems.
Results
Transduction efficiency and adenovirusdelivery of TβRII�cyt in cultured HSCsand TAA-intoxicated rats
Transduction efficiency was determined by means of X-gal staining after delivery of the Ad-βgal adenovector.In non-TAA intoxicated rats, the liver was transducedwith greater efficiency (80%) than in TAA intoxicatedrats (40%) using adenoviral vectors (Ad-βgal). In vitro,HSC culture showed 100% transduction (Figures 2B, 2Cand 2E). Generated adenovector Ad-TβRII�cyt codes fora truncated type II TGF-β receptor, which specificallyinhibits TGF-β signaling as a dominant-negative receptor[24]. HSCs transduced with Ad-TβRII�cyt expressedtransgene mRNA as shown in Figure 2F. Furthermore,protein detection using an antibody against the HAepitope present in TβRII�cyt or against the extracellulardomain of human type II TGF-β receptor, demonstrated atruncated receptor of approximately 45 kDa, as shownin Figures 2G and 2H for cultured cells and animals,respectively.
Copyright 2009 John Wiley & Sons, Ltd. J Gene Med 2009; 11: 207–219.DOI: 10.1002/jgm
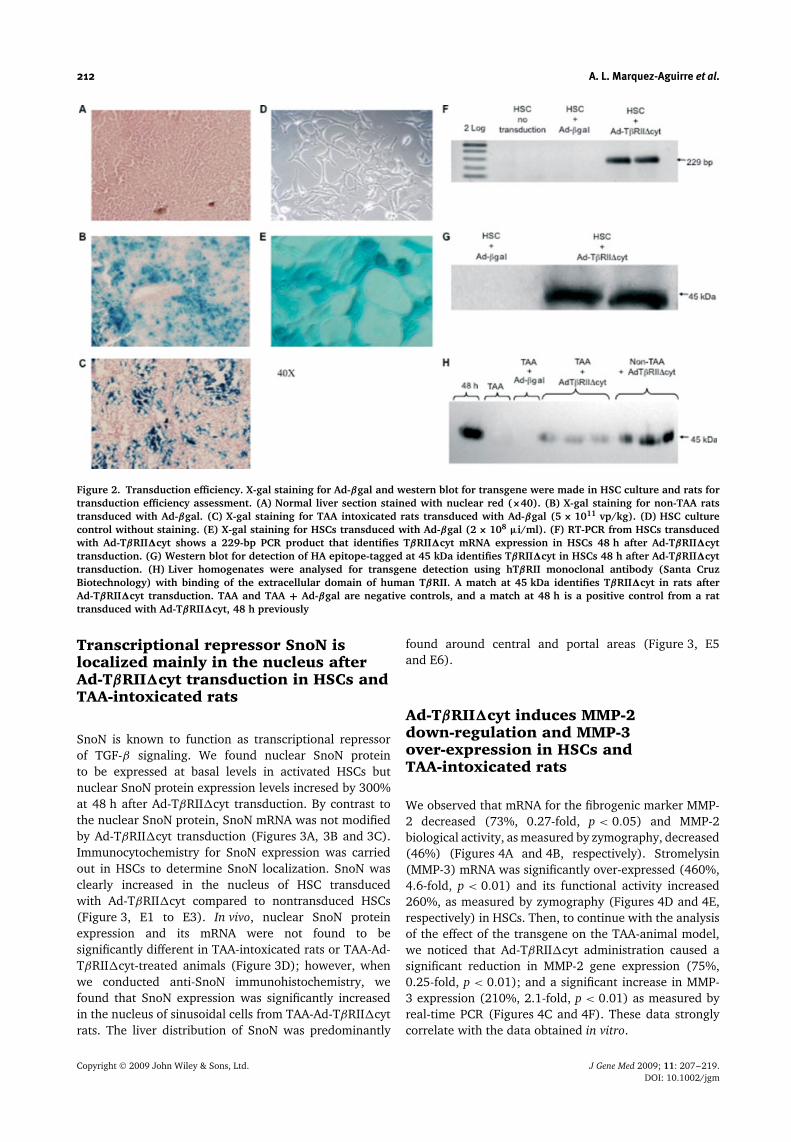
212 A. L. Marquez-Aguirre et al.
Figure 2. Transduction efficiency. X-gal staining for Ad-βgal and western blot for transgene were made in HSC culture and rats fortransduction efficiency assessment. (A) Normal liver section stained with nuclear red (×40). (B) X-gal staining for non-TAA ratstransduced with Ad-βgal. (C) X-gal staining for TAA intoxicated rats transduced with Ad-βgal (5 × 1011 vp/kg). (D) HSC culturecontrol without staining. (E) X-gal staining for HSCs transduced with Ad-βgal (2 × 108 µi/ml). (F) RT-PCR from HSCs transducedwith Ad-TβRII�cyt shows a 229-bp PCR product that identifies TβRII�cyt mRNA expression in HSCs 48 h after Ad-TβRII�cyttransduction. (G) Western blot for detection of HA epitope-tagged at 45 kDa identifies TβRII�cyt in HSCs 48 h after Ad-TβRII�cyttransduction. (H) Liver homogenates were analysed for transgene detection using hTβRII monoclonal antibody (Santa CruzBiotechnology) with binding of the extracellular domain of human TβRII. A match at 45 kDa identifies TβRII�cyt in rats afterAd-TβRII�cyt transduction. TAA and TAA + Ad-βgal are negative controls, and a match at 48 h is a positive control from a rattransduced with Ad-TβRII�cyt, 48 h previously
Transcriptional repressor SnoN islocalized mainly in the nucleus afterAd-TβRII�cyt transduction in HSCs andTAA-intoxicated rats
SnoN is known to function as transcriptional repressorof TGF-β signaling. We found nuclear SnoN proteinto be expressed at basal levels in activated HSCs butnuclear SnoN protein expression levels incresed by 300%at 48 h after Ad-TβRII�cyt transduction. By contrast tothe nuclear SnoN protein, SnoN mRNA was not modifiedby Ad-TβRII�cyt transduction (Figures 3A, 3B and 3C).Immunocytochemistry for SnoN expression was carriedout in HSCs to determine SnoN localization. SnoN wasclearly increased in the nucleus of HSC transducedwith Ad-TβRII�cyt compared to nontransduced HSCs(Figure 3, E1 to E3). In vivo, nuclear SnoN proteinexpression and its mRNA were not found to besignificantly different in TAA-intoxicated rats or TAA-Ad-TβRII�cyt-treated animals (Figure 3D); however, whenwe conducted anti-SnoN immunohistochemistry, wefound that SnoN expression was significantly increasedin the nucleus of sinusoidal cells from TAA-Ad-TβRII�cytrats. The liver distribution of SnoN was predominantly
found around central and portal areas (Figure 3, E5and E6).
Ad-TβRII�cyt induces MMP-2down-regulation and MMP-3over-expression in HSCs andTAA-intoxicated rats
We observed that mRNA for the fibrogenic marker MMP-2 decreased (73%, 0.27-fold, p < 0.05) and MMP-2biological activity, as measured by zymography, decreased(46%) (Figures 4A and 4B, respectively). Stromelysin(MMP-3) mRNA was significantly over-expressed (460%,4.6-fold, p < 0.01) and its functional activity increased260%, as measured by zymography (Figures 4D and 4E,respectively) in HSCs. Then, to continue with the analysisof the effect of the transgene on the TAA-animal model,we noticed that Ad-TβRII�cyt administration caused asignificant reduction in MMP-2 gene expression (75%,0.25-fold, p < 0.01); and a significant increase in MMP-3 expression (210%, 2.1-fold, p < 0.01) as measured byreal-time PCR (Figures 4C and 4F). These data stronglycorrelate with the data obtained in vitro.
Copyright 2009 John Wiley & Sons, Ltd. J Gene Med 2009; 11: 207–219.DOI: 10.1002/jgm
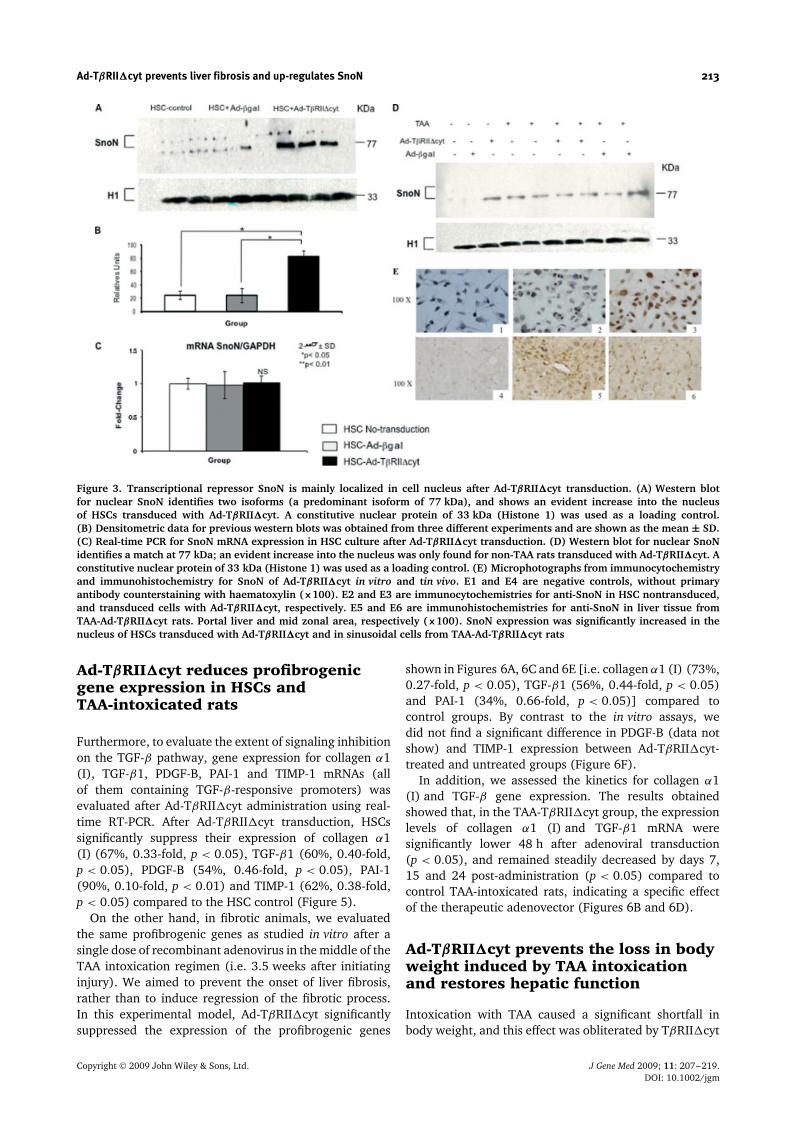
Ad-TβRII�cyt prevents liver fibrosis and up-regulates SnoN 213
Figure 3. Transcriptional repressor SnoN is mainly localized in cell nucleus after Ad-TβRII�cyt transduction. (A) Western blotfor nuclear SnoN identifies two isoforms (a predominant isoform of 77 kDa), and shows an evident increase into the nucleusof HSCs transduced with Ad-TβRII�cyt. A constitutive nuclear protein of 33 kDa (Histone 1) was used as a loading control.(B) Densitometric data for previous western blots was obtained from three different experiments and are shown as the mean ± SD.(C) Real-time PCR for SnoN mRNA expression in HSC culture after Ad-TβRII�cyt transduction. (D) Western blot for nuclear SnoNidentifies a match at 77 kDa; an evident increase into the nucleus was only found for non-TAA rats transduced with Ad-TβRII�cyt. Aconstitutive nuclear protein of 33 kDa (Histone 1) was used as a loading control. (E) Microphotographs from immunocytochemistryand immunohistochemistry for SnoN of Ad-TβRII�cyt in vitro and tin vivo. E1 and E4 are negative controls, without primaryantibody counterstaining with haematoxylin (×100). E2 and E3 are immunocytochemistries for anti-SnoN in HSC nontransduced,and transduced cells with Ad-TβRII�cyt, respectively. E5 and E6 are immunohistochemistries for anti-SnoN in liver tissue fromTAA-Ad-TβRII�cyt rats. Portal liver and mid zonal area, respectively (×100). SnoN expression was significantly increased in thenucleus of HSCs transduced with Ad-TβRII�cyt and in sinusoidal cells from TAA-Ad-TβRII�cyt rats
Ad-TβRII�cyt reduces profibrogenicgene expression in HSCs andTAA-intoxicated rats
Furthermore, to evaluate the extent of signaling inhibitionon the TGF-β pathway, gene expression for collagen α1(I), TGF-β1, PDGF-B, PAI-1 and TIMP-1 mRNAs (allof them containing TGF-β-responsive promoters) wasevaluated after Ad-TβRII�cyt administration using real-time RT-PCR. After Ad-TβRII�cyt transduction, HSCssignificantly suppress their expression of collagen α1(I) (67%, 0.33-fold, p < 0.05), TGF-β1 (60%, 0.40-fold,p < 0.05), PDGF-B (54%, 0.46-fold, p < 0.05), PAI-1(90%, 0.10-fold, p < 0.01) and TIMP-1 (62%, 0.38-fold,p < 0.05) compared to the HSC control (Figure 5).
On the other hand, in fibrotic animals, we evaluatedthe same profibrogenic genes as studied in vitro after asingle dose of recombinant adenovirus in the middle of theTAA intoxication regimen (i.e. 3.5 weeks after initiatinginjury). We aimed to prevent the onset of liver fibrosis,rather than to induce regression of the fibrotic process.In this experimental model, Ad-TβRII�cyt significantlysuppressed the expression of the profibrogenic genes
shown in Figures 6A, 6C and 6E [i.e. collagen α1 (I) (73%,0.27-fold, p < 0.05), TGF-β1 (56%, 0.44-fold, p < 0.05)and PAI-1 (34%, 0.66-fold, p < 0.05)] compared tocontrol groups. By contrast to the in vitro assays, wedid not find a significant difference in PDGF-B (data notshow) and TIMP-1 expression between Ad-TβRII�cyt-treated and untreated groups (Figure 6F).
In addition, we assessed the kinetics for collagen α1(I) and TGF-β gene expression. The results obtainedshowed that, in the TAA-TβRII�cyt group, the expressionlevels of collagen α1 (I) and TGF-β1 mRNA weresignificantly lower 48 h after adenoviral transduction(p < 0.05), and remained steadily decreased by days 7,15 and 24 post-administration (p < 0.05) compared tocontrol TAA-intoxicated rats, indicating a specific effectof the therapeutic adenovector (Figures 6B and 6D).
Ad-TβRII�cyt prevents the loss in bodyweight induced by TAA intoxicationand restores hepatic function
Intoxication with TAA caused a significant shortfall inbody weight, and this effect was obliterated by TβRII�cyt
Copyright 2009 John Wiley & Sons, Ltd. J Gene Med 2009; 11: 207–219.DOI: 10.1002/jgm
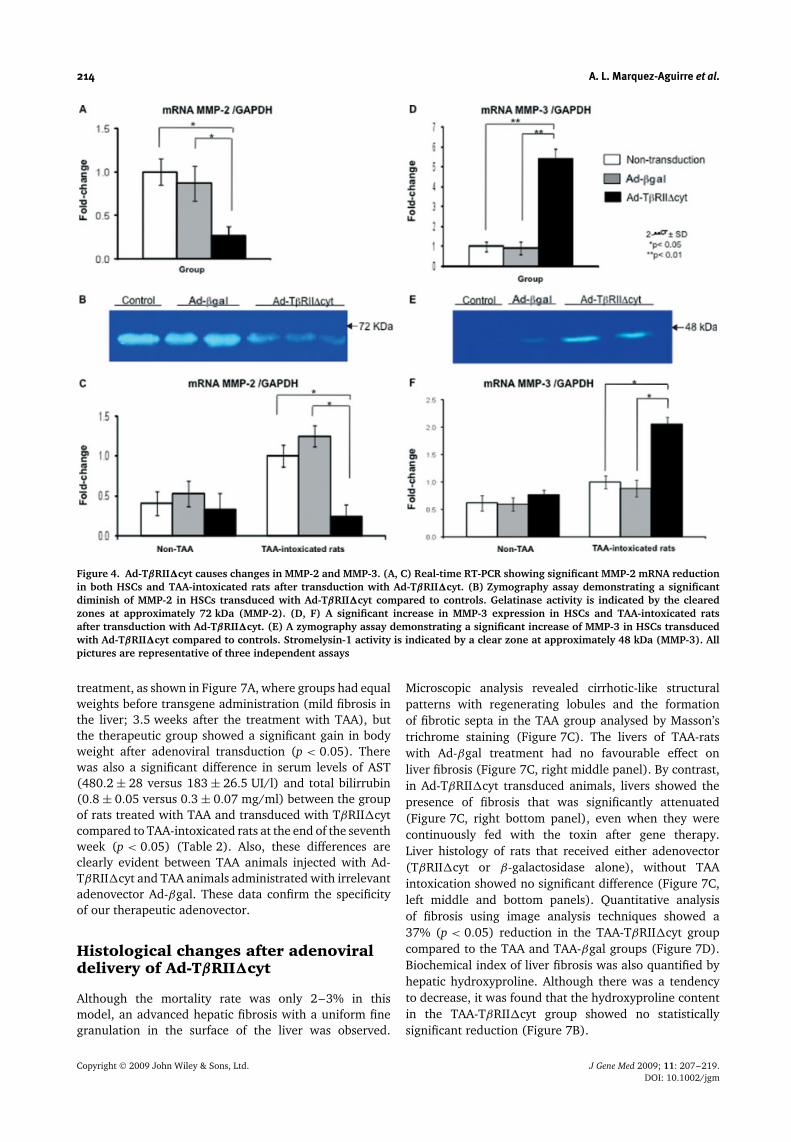
214 A. L. Marquez-Aguirre et al.
Figure 4. Ad-TβRII�cyt causes changes in MMP-2 and MMP-3. (A, C) Real-time RT-PCR showing significant MMP-2 mRNA reductionin both HSCs and TAA-intoxicated rats after transduction with Ad-TβRII�cyt. (B) Zymography assay demonstrating a significantdiminish of MMP-2 in HSCs transduced with Ad-TβRII�cyt compared to controls. Gelatinase activity is indicated by the clearedzones at approximately 72 kDa (MMP-2). (D, F) A significant increase in MMP-3 expression in HSCs and TAA-intoxicated ratsafter transduction with Ad-TβRII�cyt. (E) A zymography assay demonstrating a significant increase of MMP-3 in HSCs transducedwith Ad-TβRII�cyt compared to controls. Stromelysin-1 activity is indicated by a clear zone at approximately 48 kDa (MMP-3). Allpictures are representative of three independent assays
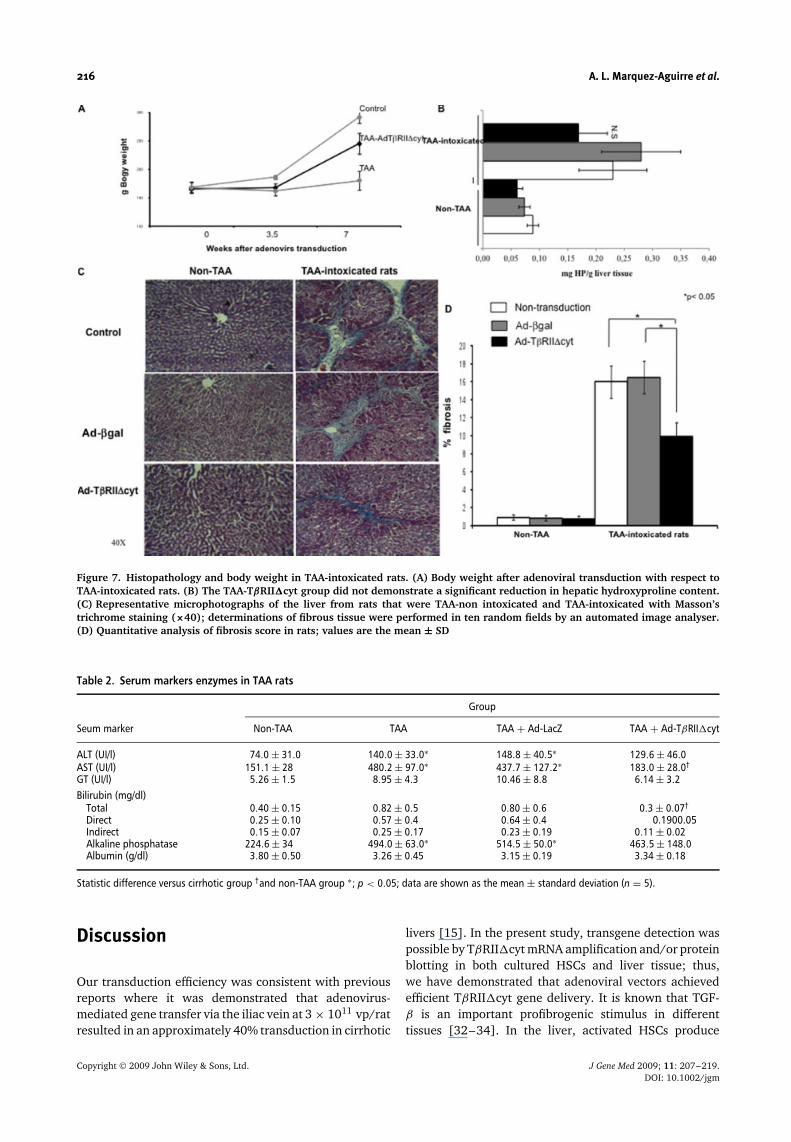
treatment, as shown in Figure 7A, where groups had equalweights before transgene administration (mild fibrosis inthe liver; 3.5 weeks after the treatment with TAA), butthe therapeutic group showed a significant gain in bodyweight after adenoviral transduction (p < 0.05). Therewas also a significant difference in serum levels of AST(480.2 ± 28 versus 183 ± 26.5 UI/l) and total bilirrubin(0.8 ± 0.05 versus 0.3 ± 0.07 mg/ml) between the groupof rats treated with TAA and transduced with TβRII�cytcompared to TAA-intoxicated rats at the end of the seventhweek (p < 0.05) (Table 2). Also, these differences areclearly evident between TAA animals injected with Ad-TβRII�cyt and TAA animals administrated with irrelevantadenovector Ad-βgal. These data confirm the specificityof our therapeutic adenovector.
Histological changes after adenoviraldelivery of Ad-TβRII�cyt
Although the mortality rate was only 2–3% in thismodel, an advanced hepatic fibrosis with a uniform finegranulation in the surface of the liver was observed.
Microscopic analysis revealed cirrhotic-like structuralpatterns with regenerating lobules and the formationof fibrotic septa in the TAA group analysed by Masson’strichrome staining (Figure 7C). The livers of TAA-ratswith Ad-βgal treatment had no favourable effect onliver fibrosis (Figure 7C, right middle panel). By contrast,in Ad-TβRII�cyt transduced animals, livers showed thepresence of fibrosis that was significantly attenuated(Figure 7C, right bottom panel), even when they werecontinuously fed with the toxin after gene therapy.Liver histology of rats that received either adenovector(TβRII�cyt or β-galactosidase alone), without TAAintoxication showed no significant difference (Figure 7C,left middle and bottom panels). Quantitative analysisof fibrosis using image analysis techniques showed a37% (p < 0.05) reduction in the TAA-TβRII�cyt groupcompared to the TAA and TAA-βgal groups (Figure 7D).Biochemical index of liver fibrosis was also quantified byhepatic hydroxyproline. Although there was a tendencyto decrease, it was found that the hydroxyproline contentin the TAA-TβRII�cyt group showed no statisticallysignificant reduction (Figure 7B).
Copyright 2009 John Wiley & Sons, Ltd. J Gene Med 2009; 11: 207–219.DOI: 10.1002/jgm
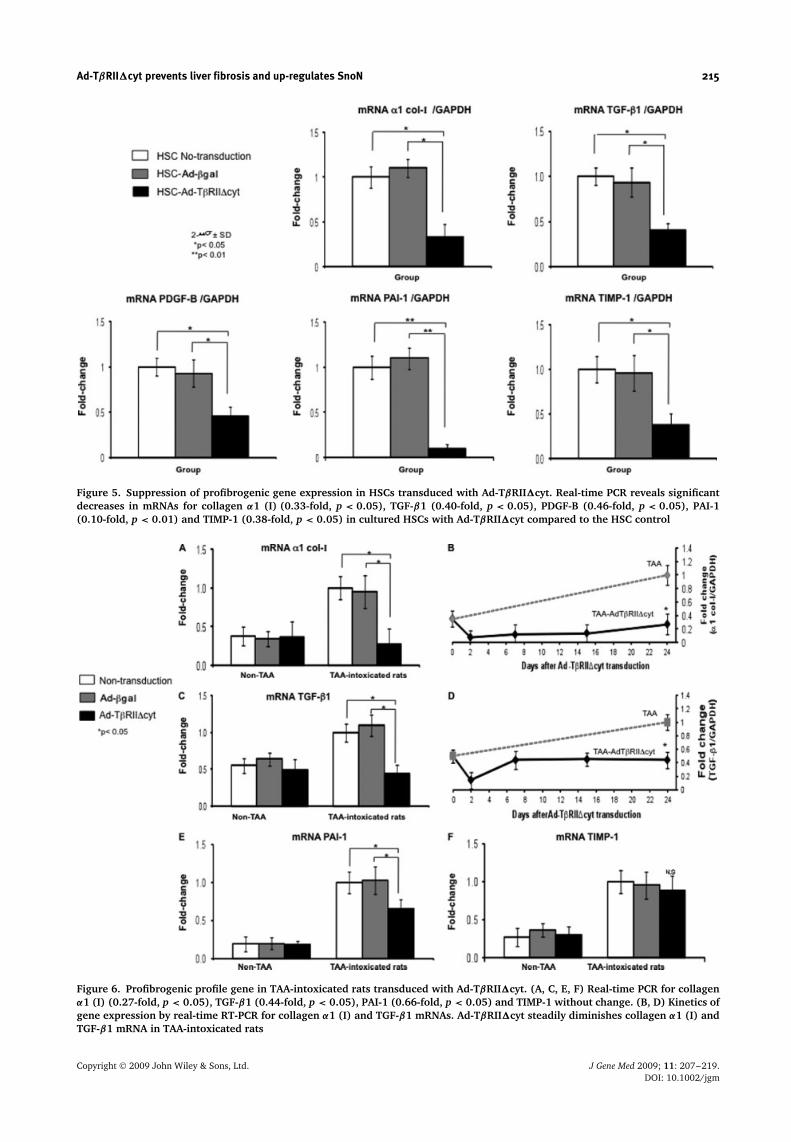
Ad-TβRII�cyt prevents liver fibrosis and up-regulates SnoN 215
Figure 5. Suppression of profibrogenic gene expression in HSCs transduced with Ad-TβRII�cyt. Real-time PCR reveals significantdecreases in mRNAs for collagen α1 (I) (0.33-fold, p < 0.05), TGF-β1 (0.40-fold, p < 0.05), PDGF-B (0.46-fold, p < 0.05), PAI-1(0.10-fold, p < 0.01) and TIMP-1 (0.38-fold, p < 0.05) in cultured HSCs with Ad-TβRII�cyt compared to the HSC control
Figure 6. Profibrogenic profile gene in TAA-intoxicated rats transduced with Ad-TβRII�cyt. (A, C, E, F) Real-time PCR for collagenα1 (I) (0.27-fold, p < 0.05), TGF-β1 (0.44-fold, p < 0.05), PAI-1 (0.66-fold, p < 0.05) and TIMP-1 without change. (B, D) Kinetics ofgene expression by real-time RT-PCR for collagen α1 (I) and TGF-β1 mRNAs. Ad-TβRII�cyt steadily diminishes collagen α1 (I) andTGF-β1 mRNA in TAA-intoxicated rats
Copyright 2009 John Wiley & Sons, Ltd. J Gene Med 2009; 11: 207–219.DOI: 10.1002/jgm
216 A. L. Marquez-Aguirre et al.
Figure 7. Histopathology and body weight in TAA-intoxicated rats. (A) Body weight after adenoviral transduction with respect toTAA-intoxicated rats. (B) The TAA-TβRII�cyt group did not demonstrate a significant reduction in hepatic hydroxyproline content.(C) Representative microphotographs of the liver from rats that were TAA-non intoxicated and TAA-intoxicated with Masson’strichrome staining (×40); determinations of fibrous tissue were performed in ten random fields by an automated image analyser.(D) Quantitative analysis of fibrosis score in rats; values are the mean ± SD
Table 2. Serum markers enzymes in TAA rats
Group
Seum marker Non-TAA TAA TAA + Ad-LacZ TAA + Ad-TβRII�cyt
ALT (UI/l) 74.0 ± 31.0 140.0 ± 33.0∗ 148.8 ± 40.5∗ 129.6 ± 46.0AST (UI/l) 151.1 ± 28 480.2 ± 97.0∗ 437.7 ± 127.2∗ 183.0 ± 28.0†
GT (UI/l) 5.26 ± 1.5 8.95 ± 4.3 10.46 ± 8.8 6.14 ± 3.2Bilirubin (mg/dl)
Total 0.40 ± 0.15 0.82 ± 0.5 0.80 ± 0.6 0.3 ± 0.07†
Direct 0.25 ± 0.10 0.57 ± 0.4 0.64 ± 0.4 0.1900.05Indirect 0.15 ± 0.07 0.25 ± 0.17 0.23 ± 0.19 0.11 ± 0.02Alkaline phosphatase 224.6 ± 34 494.0 ± 63.0∗ 514.5 ± 50.0∗ 463.5 ± 148.0Albumin (g/dl) 3.80 ± 0.50 3.26 ± 0.45 3.15 ± 0.19 3.34 ± 0.18
Statistic difference versus cirrhotic group †and non-TAA group ∗; p < 0.05; data are shown as the mean ± standard deviation (n = 5).
Discussion
Our transduction efficiency was consistent with previousreports where it was demonstrated that adenovirus-mediated gene transfer via the iliac vein at 3 × 1011 vp/ratresulted in an approximately 40% transduction in cirrhotic
livers [15]. In the present study, transgene detection waspossible by TβRII�cyt mRNA amplification and/or proteinblotting in both cultured HSCs and liver tissue; thus,we have demonstrated that adenoviral vectors achievedefficient TβRII�cyt gene delivery. It is known that TGF-β is an important profibrogenic stimulus in differenttissues [32–34]. In the liver, activated HSCs produce
Copyright 2009 John Wiley & Sons, Ltd. J Gene Med 2009; 11: 207–219.DOI: 10.1002/jgm

Ad-TβRII�cyt prevents liver fibrosis and up-regulates SnoN 217
high quantities of TGF-β and respond to it by increasingthe expression of collagen and other profibrogenic genes,such as PDGF, PAI-1 and TIMP-1 [7,35,36]. To avoidthe profibrogenic effect of TGF-β, different anti-TGF-βstrategies have been developed [37,38]. Different animalstudies have shown that adenoviral delivery of thedominant negative TGF-β type II receptor in the fibroticliver impairs TGF-β profibrogenic signals [13,14,39].
On the basis of the efficiency for gene deliverydemonstrated for Ad-TβRII�cyt, we tested the abilityfor this adenovector to block TGF-β signaling. In thepresent study, we showed that TβRII�cyt was ableto block transcription of TGF-β-responsive profibrogenicgenes, and even to improve liver function. The variety ofprofibrogenic transcripts that were suppressed includedcollagen α1 (I), TGF-β, PDGF-B, PAI-1 and TIMP-1, demonstrating an effective TGF-β blockade. Theevaluation of hepatic function via AST and total billirubinindicates significant improved liver functionality in ourAd-TβRII�cyt-transduced rats; whereas, in a previousstudy by Qi et al. [13] using the same transgene, serumlevels of hyaluronic acid and transaminases remained low,whereas untreated rats died of liver dysfunction. Thesedata draw a parallel with other studies of adenovirusexpressing the truncated type II TGF-β receptor indimethylnitrosamine model of liver fibrosis, which alsoobserved attenuated histological fibrosis compared tocontrols [13,14,39].
In different models for cirrhosis, our group hasbeen successful in regressing liver fibrosis, mostly usingadenovector vehicles [15,30,40]. The present study isno exception. The histological data for the fibrosis scoreindicate 37% less fibrosis in Ad-TβRII�cyt transducedanimals (p > 0.05) in comparison with fibrotic animals.Although we observed a significant reduction in fibrosisevaluated by morphometric analysis, hydroxiprolinequantification presented a tendency to diminish, withoutany statistically significant difference. This can beexplained on the basis that, even when collagen isdegraded, fragments could still remain in the liver, andthey will be measured as hydroxyproline [41].
Matrix metalloproteinases are a family of proteasescapable of degrading the hepatic extracellular matrix,thereby playing a central role in tissue remodeling andrepair after injury. HSCs exposed to Ad-TβRII�cyt showeda reduced activity of MMP-2, which has been describedas a fibrotic marker [4]. MMP-2 is suggested to play arole in amplifying liver injury [42], and it is speculatedthat it is required for mitogenic and pro-invasive effectsof reactive oxygen species in HSCs [43]. Both MMP-2mRNA and enzymatic activity are decreased, apparentlyproviding positive evidence of the therapeutic effects ofTβRII�cyt, because continuous MMP-2 over-expressionin HSCs, with its proliferative and migrative sequels, mayfoster the progression of fibrosis.
A principal feature of hepatic fibrosis is the misbalancebetween MMPs and TIMPs. It is known that MMP-3 couldactivate pro-MMPs (e.g. MMP-1,-3 and 13). In the presentstudy, MMP-3 increases 260% of its catabolic action. Thus,
it may be possible that over-expression of MMP-3 couldbe responsible in part for the observed diminished fibrosisdue to its own catalytic action in MEC proteins andactivation of other MMPs. Indeed, the increase in MMP-3activity links with the observed TIMP-1 mRNA reduction,which is a protein that binds to and inhibits activatedcollagenases [36].
The pattern of behaviour of MMPs was preservedin vitro as well as in vivo assays, where MMP-2 had a75% decrement and MMP-3 had an approximately 200%increment. Also, transcriptional TGF-β activation of PAI-1and PDGF-B was found to be almost entirely suppressed,along with an abrogation of collagen type I and TGF-βmRNAs.
In the present study, we quantified SnoN, a tran-scriptional repressor of the Ski family. From currentknowledge, SnoN appears to be involved mainly in theregulation of the Smad/TGF-β pathway [44]. Ski and itsrelated protein, SnoN, are transcriptional co-repressorsthat physically interact with Smads in a TGF-β-dependentmanner and acts to repress TGF-β signaling through therecruitment of histone deacetylases [9,45].
It has been reported that overexpression of SnoNrepresses transcriptional activation by Smad3. Activationof TGF-β signaling leads to rapid degradation of SnoN,and this degradation is likely mediated by cellularproteasomes. These results demonstrate the existenceof a cascade of the TGF-β signaling pathway, which,upon TGF-β stimulation, leads to the destruction ofprotooncoproteins that antagonize the activation of theTGF-β signaling [45].
The data obtained in the present study showthat adenoviral delivery of TβRII�cyt results in anincrease in nuclear SnoN protein in cultured HSCsand in sinusoidal cells of cirrhotic rats, in the absenceof aumented SnoN transcription. It is important tonote that SnoN nuclear protein in vivo evaluated bywestern blot was not sharply increased. This couldbe due to a heterogeneous cell population fromthe liver homogenates. Nonetheless, we confirmednuclear SnoN up-regulation in nonparenchymal cellsby immunohistochemistry and in HSC culture byinmmunocytochemistry (Figure 3E). It is not surprisingthat SnoN expression was found also in nonparenchymalcells because they have shown to be transduced byadenoviral vectors as well [46].
The molecular mechanism by which TβRII�cyt up-regulates SnoN should be eventually clarified, thoughwe might speculate that is not by transcriptional reg-ulation (SnoN mRNA was unchanged by Ad-TβRII�cytboth in vitro and in vivo). Our findings support the notionthat TβRII�cyt is a dominant negative that blocks TGF-β signaling. As a consequence of signal blocking, it ispossible that SnoN molecules cannot be targeted fordestruction by proteasomes and persists in the nucleus.Using this same line of reasoning, Smad3 phosphoryla-tion, and the obliteration of further translocation, couldprevent Smad3-dependent degradation of SnoN, and in,consequence, lead to increased nuclear SnoN, that is
Copyright 2009 John Wiley & Sons, Ltd. J Gene Med 2009; 11: 207–219.DOI: 10.1002/jgm

218 A. L. Marquez-Aguirre et al.
now available to antagonize TGF-β signaling. In otherwords, SnoN ubiquitinization is reduced and, therefore,the stability of nuclear SnoN protein will be incremented,allowing a high efficiency with respect to its anti-TGF-βfunction.
In conclusion, Ad-TβRII�cyt was able to efficientlylimit TGF-β-responsive profibrogenic gene expressionboth in vivo and in vitro. This effect was accomplishedby improving liver function and fibrosis reduction in TAA-induced liver fibrosis. Additionally, our results suggestthat the molecular mechanism for the blocking effectsof Ad-TβRII�cyt in TGF-β signaling acts via nuclear up-regulation of the transcriptional repressor SnoN, whichantagonizes TGF-β signaling (TGF-β/Smad pathwayinhibitor).
Acknowledgements
This work was supported in part by a grant from CONACyT no.25474-2007 awarded to J.A.B. The authors are indebted to DrPedro Diaz for his tenacious and helpful technical assistance
References
1. Mori T, Okanoue T, Sawa Y, et al., Defenestration of thesinusoidal endothelial cell in a rat model of cirrhosis. Hepatology1993; 17(5): 891–7.
2. Muller A, Machnik F, Zimmermann T, et al., Thioacetamide-induced cirrhosis-like liver lesions in rats – usefulness andreliability of this animal model. Exp Pathol 1988; 34(4):229–36.
3. Gressner AM and Weiskirchen R, Modern pathogenetic conceptsof liver fibrosis suggest stellate cells and TGF-beta as majorplayers and therapeutic targets. J Cell Mol Med 2006; 10(1):76–99.
4. Friedman SL, Liver fibrosis - from bench to bedside. J Hepatol2003; 38(Suppl 1): S38–53.
5. Wrana JL and Attisano L, The Smad pathway. Cytokine GrowthFactor Rev 2000; 11(1–2): 5–13.
6. de Caestecker M, The transforming growth factor-betasuperfamily of receptors. Cytokine Growth Factor Rev 2004;15(1): 1–11.
7. Bissell DM, Roulot D and George J, Transforming growth factorbeta and the liver. Hepatology 2001; 34(5): 859–67.
8. Greene RM, Nugent P, Mukhopadhyay P, et al., Intracellulardynamics of Smad-mediated TGFbeta signaling. J Cell Physiol2003; 197(2): 261–71.
9. Luo K, Ski and SnoN: negative regulators of TGF-beta signaling.Curr Opin Genet Dev 2004; 14(1): 65–70.
10. Massague J and Chen YG, Controlling TGF-beta signaling. GenesDev 2000; 14(6): 627–44.
11. Stroschein SL, Wang W, Zhou S, et al., Negative feedbackregulation of TGF-beta signaling by the SnoN oncoprotein.Science 1999; 286(5440): 771–4.
12. Liu X, Sun Y, Weinberg RA, et al., Ski/Sno and TGF-betasignaling. Cytokine Growth Factor Rev 2001; 12(1): 1–8.
13. Qi Z, Atsuchi N, Ooshima A, et al., Blockade of type betatransforming growth factor signaling prevents liver fibrosis anddysfunction in the rat. Proc Natl Acad Sci U S A 1999; 96(5):2345–9.
14. Nakamura T, Sakata R, Ueno T, et al., Inhibition of transforminggrowth factor beta prevents progression of liver fibrosisand enhances hepatocyte regeneration in dimethylnitrosamine-treated rats. Hepatology 2000; 32(2): 247–55.
15. Garcia-Banuelos J, Siller-Lopez F, Miranda A, et al., Cirrhoticrat livers with extensive fibrosis can be safely transduced withclinical-grade adenoviral vectors. Evidence of cirrhosis reversion.Gene Ther 2002; 9(2): 127–34.
16. Wieser R, Attisano L, Wrana JL and Massague J, Signalingactivity of transforming growth factor beta type II receptorslacking specific domains in the cytoplasmic region. Mol Cell Biol1993; 13(12): 7239–47.
17. Hitt M Graham FL, Construction and propagation of humanadenovirus vectors. Cell Biology: a laboratory Handbook 1998;1: 500–512.
18. Graham FL, Smiley J, Russell WC and Nairn R, Characteristics ofa human cell line transformed by DNA from human adenovirustype 5. J Gen Virol 1977; 36(1): 59–74.
19. Ng P, Beauchamp C, Evelegh C, Parks R and Graham FL,Development of a FLP/frt system for generating helper-dependent adenoviral vectors. Mol Ther 2001; 3(5 Pt 1):809–15.
20. Maizel JV, Jr., White DO and Scharff MD, The polypeptides ofadenovirus. II. Soluble proteins, cores, top components and thestructure of the virion. Virology 1968; 36(1): 126–36.
21. Nyberg-Hoffman C, Shabram P, Li W, Giroux D and Aguilar-Cordova E, Sensitivity and reproducibility in adenoviralinfectious titer determination. Nat Med 1997; 3(7): 808–11.
22. Herrmann J, Gressner AM and Weiskirchen R, Immortal hepaticstellate cell lines: useful tools to study hepatic stellate cellbiology and function? J Cell Mol Med 2007; 11(4): 704–22.
23. Gonzalez-Cuevas J, Bueno-Topete M and Armendariz-BorundaJ, Urokinase plasminogen activator stimulates function ofactive forms of stromelysin and gelatinases (MMP-2 and MMP-9) in cirrhotic tissue. J Gastroenterol Hepatol 2006; 21(10):1544–54.
24. Hernandez-Canaveral I, Gonzalez J, Lopez-Casillas F andArmendariz-Borunda J, Amplified expression of dominant-negative transforming growth factor-beta type II receptorinhibits collagen type I production via reduced Smad-3 activity.J Gastroenterol Hepatol 2004; 19(4): 380–7.
25. Dashti H, Jeppsson B, Hagerstrand I, et al., Thioacetamide- andcarbon tetrachloride-induced liver cirrhosis. Eur Surg Res 1989;21(2): 83–91.
26. Li X, Benjamin IS and Alexander B, Reproducible production ofthioacetamide-induced macronodular cirrhosis in the rat withno mortality. J Hepatol 2002; 36(4): 488–93.
27. Salazar-Montes A, Ruiz-Corro L, Sandoval-Rodriguez A, Lopez-Reyes A and Armendariz-Borunda J, Increased DNA bindingactivity of NF-kappaB, STAT-3, SMAD3 and AP-1 inacutely damaged liver. World J Gastroenterol 2006; 12(37):5995–6001.
28. Livak KJ and Schmittgen TD, Analysis of relative gene expressiondata using real-time quantitative PCR and the 2(−Delta DeltaC(T)) Method. Methods 2001; 25(4): 402–8.
29. Yuan JS, Reed A, Chen F and Stewart, CN, Jr., Statisticalanalysis of real-time PCR data. BMC Bioinformatics 2006; 7:85.
30. Siller-Lopez F, Sandoval A, Salgado S, et al., Treatmentwith human metalloproteinase-8 gene delivery amelioratesexperimental rat liver cirrhosis. Gastroenterology 2004; 126(4):1122–33; discussion 949.
31. Rojkind M and Gonzalez E, An improved method fordetermining specific radioactivities of proline-14C andhydroxyproline-14C in collagen and in noncollagenous proteins.Anal Biochem 1974; 57(1): 1–7.
32. Friedman SL, Mechanisms of hepatic fibrogenesis. Gastroenterol-ogy 2008; 134(6): 1655–69.
33. Bonniaud P, Margetts PJ, Ask K, et al., TGF-beta and Smad3signaling link inflammation to chronic fibrogenesis. J Immunol2005; 175(8): 5390–5.
34. Lakos G, Takagawa S, Chen SJ, et al., Targeted disruption ofTGF-beta/Smad3 signaling modulates skin fibrosis in a mousemodel of scleroderma. Am J Pathol 2004; 165(1): 203–17.
35. Zhu YW, Lin Y, Zhang X, Wang YJ, Xie WF, [Transforminggrowth factor beta 1 stimulates the expression of plasminogenactivator inhibitor 1 in hepatic stellate cells.]. Zhonghua GanZang Bing Za Zhi 2008; 16(5): 387–8.
36. Hemmann S, Graf J, Roderfeld M, Roeb E, Expression of MMPsand TIMPs in liver fibrosis - a systematic review with specialemphasis on anti-fibrotic strategies. J Hepatol 2007; 46(5):955–75.
37. Liu X, Hu H and Yin JQ, Therapeutic strategies against TGF-beta signaling pathway in hepatic fibrosis. Liver Int 2006; 26(1):8–22.
Copyright 2009 John Wiley & Sons, Ltd. J Gene Med 2009; 11: 207–219.DOI: 10.1002/jgm

Ad-TβRII�cyt prevents liver fibrosis and up-regulates SnoN 219
38. Breitkopf K, Haas S, Wiercinska E, et al., Anti-TGF-betastrategies for the treatment of chronic liver disease. AlcoholClin Exp Res 2005; 29(11 Suppl): 121S–131S.
39. Ozawa S, Uchiyama K, Nakamori M, et al., Combination genetherapy of HGF and truncated type II TGF-beta receptor for ratliver cirrhosis after partial hepatectomy. Surgery 2006; 139(4):563–73.
40. Salgado S, Garcia J, Vera J, et al., Liver cirrhosis is reverted byurokinase-type plasminogen activator gene therapy. Mol Ther2000; 2(6): 545–51.
41. Iimuro Y, Nishio T, Morimoto T, et al., Delivery of matrixmetalloproteinase-1 attenuates established liver fibrosis in therat. Gastroenterology 2003; 124(2): 445–58.
42. Bansal MB, Kovalovich K, Gupta R, et al., Interleukin-6 protectshepatocytes from CCI4-mediated necrosis and apoptosis in miceby reducing MMP-2 expression. J Hepatol 2005; 42(4): 548–56.
43. Galli A, Svegliati-Baroni G, Ceni E, et al., Oxidative stressstimulates proliferation and invasiveness of hepatic stellate cellsvia a MMP2-mediated mechanism. Hepatology 2005; 41(5):1074–84.
44. Macias-Silva M, Li W, Leu JI, Crissey MA and Taub R, Up-regulated transcriptional repressors SnoN and Ski bind Smadproteins to antagonize transforming growth factor-beta signalsduring liver regeneration. J Biol Chem 2002; 277(32):28483–90.
45. Sun Y, Liu X, Ng-Eaton E, Lodish HF and Weinberg RA, SnoNand Ski protooncoproteins are rapidly degraded in response totransforming growth factor beta signaling. Proc Natl Acad Sci US A 1999; 96(22): 12442–7.
46. Yu Q, Que LG and Rockey DC, Adenovirus-mediated genetransfer to nonparenchymal cells in normal and injured liver.Am J Physiol Gastrointest Liver Physiol 2002; 282(3): G565–72.
Copyright 2009 John Wiley & Sons, Ltd. J Gene Med 2009; 11: 207–219.DOI: 10.1002/jgm
Copyright © 2022 FDOKUMEN




















