
Mixing experiment in a stirred tank using blue maize flour suspensions

Macromol. Theory Simul. 5 , 477-497 (1996) 411
A theoretical investigation of the production of branched copolymers in continuous stirred tank reactors
Apostolis Baltsas, Dimitris S. Achilias, Costas Kiparissides *
Chemical Engineering Department and Chemical Process Engineering Research Institute, Aristotle University of Thessaloniki, P. 0. Box 472, 540 06 Thessaloniki, Greece
(Received: August 29, 1995; revised manuscript of November 9, 1995)
SUMMARY: A comprehensive mathematical model for free-radical copolymerization reactions has
been developed for a homogeneous continuous stirred tank reactor. The present model is based on a fairly general copolymerization scheme accounting for the formation of linear and branched copolymer chains. Both chain transfer to polymer and terminal double bond reactions are considered in order to predict the long chain branching frequency. Changes in molecular weight, composition and degree of branching occurring during the copolymeriza- tion reaction are modelled using the method of moments. To break-down the dependence of the moment equations on higher order moments two different closure methods are considered. The predictive capabilities of the model are examined in relation to the solution copolymerization of methyl methacrylate with vinyl acetate. It is shown that both chain transfer to polymer and terminal double bond reactions significantly contribute to the broadening of the molecular weight and degree of branching distributions. Furthermore, the terminal double bond reaction affects significantly the copolymer number-average mole- cular weight and the concentration of terminal double bonds.
Introduction
The synthesis of linear and branched copolymers by free-radical reactions is of significant economic importance to the polymer industry, for the polymerization of two or more comonomers can lead to the production of polymers with desired physical, chemical and mechanical end-use properties. Free-radical random copolymers are typically obtained as a mixture of macromolecules with different composition, chain length, degree of branching and chain sequence characteristics. These molecular and structural features are directly related to the end-use properties of the final product. Controlling the polymer chain microstructure requires a thorough understanding of the polymerization kinetics and a quantitative description of the effects of the reactor operating conditions (e. g., temperature, initiator concentration) and physical transport processes (e.g., mixing, mass and heat transfer) on the polymerization rate and molecular properties. Thus, there is an obvious need for the development of comprehensive mathematical models capable of predicting the compositional and structural molecular properties of polymers produced in batch and continuous reactors.
The present work deals with the modelling of branched polymerization reactions. The presence of long chain branches in the copolymer chains results in the broadening of the molecular weight distribution (MWD) and significantly affects the rheological
0 1996, Huthig & Wepf Verlag, Zug CCC 1022-1344/96/$05.00

478 A. Baltsas, D. S. Achilias, C. Kiparissides
properties (e. g. melt index) of the polymer melts. Therefore, control of the long chain branching frequency is of prime importance in the synthesis of high quality copolymers.
Several mathematical models have been published dealing with free-radical copoly- merization kinetics -30). These kinetic models consider the synthesis of linear copoly- merS6~9,1i .16,19,20,23,27,29) and branched copolymers formed in the presence of chain transfer to polymer reactions ' 3 2, 24, 26). Recently, T ~ b i t a ~ I - ~ ~ ) investigated the homo- polymerization kinetics of long chain branching formation via chain transfer to polymer reactions.
The dynamic behaviour of continuous stirred tank polymerization reactors has been the subject of several investigations by Ray and his coworker^^^-^^) and Schork et al. 40)
In the present study, the kinetics of long chain branching formation by both chain transfer to polymer and terminal double bond reactions is investigated. A dynamic model is developed to describe the copolymerization of methyl methacrylate (MMA) with vinyl acetate (VAc) in an isothermal continuous stirred tank reactor (CSTR). The method of moments is invoked to recast the infinite set of macromolecular species balance equations into a system of ordinary differential equations describing the changes in molecular weight, copolymer composition and degree of branching in the copolymerization reactor. A systematic analysis is carried out to determine the effect of chain transfer to polymer and terminal double bond reactions on the broadening of molecular weight and degree of branching distributions. It should be noted that this is a first attempt to model molecular weight developments in a CSTR copolymerization reactor, including both chain transfer to polymer and terminal double bond reactions. Previously published work on the same subject has exclusively dealt with the homo- polymerization kinetics4' -53).
In what follows, generalized expressions are derived to describe the net rate of appearance of disappearance of individual molecular species for a copolymerization reacting system. General design equations, describing the molecular weight and compositional changes in a continuous stirred tank reactor, are derived and simulation results are presented for the solution copolymerization of MMA with VAc. Finally, the effect of transfer to polymer and terminal double bond rate constants on the molecular weight developments is analysed.
Copolymerization rate functions
Besides the conventional chain addition reactions (e. g., initiation, propagation, termination, chain transfer to monomer and to chain transfer agent), the proposed copolymerization mechanism includes chain transfer to copolymer and terminal double bond reactions that lead to the production of long chain branches. Overall, the following elementary chemical reactions are considered 2,
Initiation:
I & 21'

A theoretical investigation of the production of branched copolymers . . . 419
Propagation:
R: + 2 - j, m + j- I , j k% . R:,rn,i + Mj
Chain transfer to solvent:
R:, m. I + S 'lrS . S' + P , r n
k,*, S' + MJ ' R;-J,J-I,I
Chain transfer to monomer:
R;-j,j-I?j + P n , m k', . ,
R:,rn,i + Mj
Transfer to chain transfer agent:
R:, m,i + T
T ' + M j
"" . T' + P n , m
4, * + R;- j, j- I I j
Inhibition.
P n , m kz# . R:.rn, i + Z
Termination by combination: k,:
R:,rn,i + REq,j . P n + r , m + q
Termination by disproportionation:
Rz,m,i + R E , j k", t P , , + P c q
Chain transfer to poIymer:
R t , j + P n , m "P" . R;,m,i + P c q
Terminal double bond polymerization:
Rz,m,i + P r , q k d ~ g t R:+crn+q,j
(3)
(9)
In the above kinetic scheme, the symbols I, S, T, Z and Mj denote the initiator, solvent, chain transfer agent, inhibitor and monomer molecules, respectively. Radicals formed by the fragmentation of the initiator and by chain transfer reactions to solvent and chain transfer agent are denoted by the symbols I*, S' and T', respectively. The symbols R:, m, and P , rn are used to identify the respective "live" macroradicals of type i and the "dead" copolymer chains, containing n M I and m M, monomer units.

480 A. Baltsas, D. S. Achilias, C. Kiparissides
It should be noted that the ultimate monomer unit in a copolymer radical can be either of type 1 (i = 1) or type 2 (i = 2). The present copolymerization mechanism comprises seven initiation reactions, four propagation reactions, four chain transfer to monomer reactions, two chain transfer to solvent and two chain transfer to agent reactions, two inhibition reactions, three termination by combination and three termination by disproportionation reactions, four chain transfer to polymer and four terminal double bond reactions, that is a total of 35 elementary reactions.
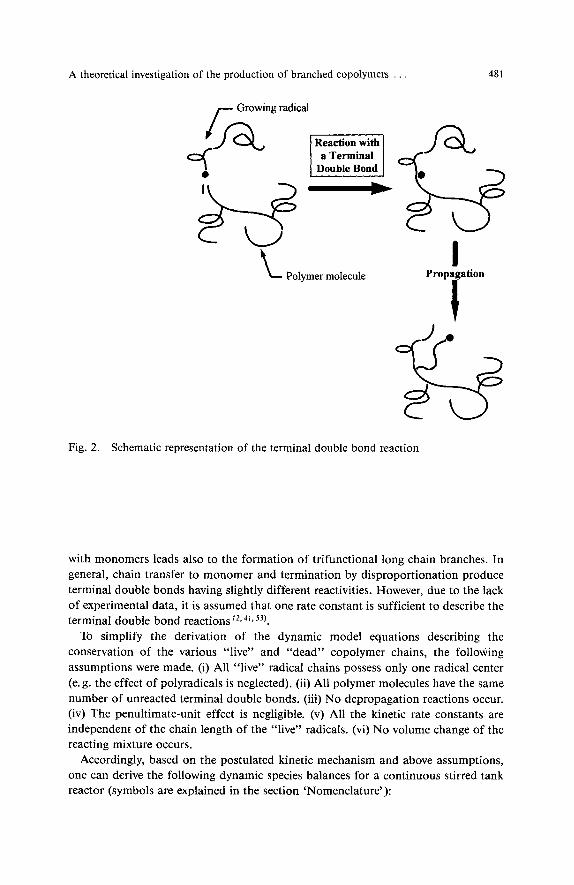
A schematic representation of the reactions leading to the formation of long chain branches is shown in Figs. 1 and 2. In a transfer to polymer reaction, a “live” polymer chain abstracts a hydrogen atom from the backbone of a “dead” copolymer which results in the formation of a radical at a random site of the polymer chain39). Propagation of these internal side radicals with monomers leads to the formation of trifunctional branch points. It must be noticed that the transfer to polymer reaction rate will be proportional to the total number of structural units in the copolymer chains. Terminal double bonds are produced by transfer to monomer and termination by disproportionation reactions. Polymer molecules containing terminal double bonds may further react with macroradicals to form “live” polymer chains with active radical sites located at the copolymer backbone (Fig. 2). Propagation of these side radicals
Growmg radical
t
Fig. 1. Schematic representation of the chain transfer to polymer reaction
A theoretical investigation of the production of branched copolymers 481
r Growing radical
0
a Terminal
t Polymer molecule I
Propagation
Fig. 2. Schematic representation of the terminal double bond reaction
with monomers leads also to the formation of trifunctional long chain branches. In general, chain transfer to monomer and termination by disproportionation produce terminal double bonds having slightly different reactivities. However, due to the lack of experimental data, it is assumed that one rate constant is sufficient to describe the terminal double bond reactions f 2 9 4 1 , J3).
To simplify the derivation of the dynamic model equations describing the conservation of the various “live” and “dead” copolymer chains, the following assumptions were made. (i) All “live” radical chains possess only one radical center (e. g. the effect of polyradicals is neglected). (ii) All polymer molecules have the same number of unreacted terminal double bonds. (iii) No depropagation reactions occur. (iv) The penultimate-unit effect is negligible. (v) All the kinetic rate constants are independent of the chain length of the “live” radicals. (vi) No volume change of the reacting mixture occurs.
Accordingly, based on the postulated kinetic mechanism and above assumptions, one can derive the following dynamic species balances for a continuous stirred tank reactor (symbols are explained in the section ‘Nomenclature’):

482 A. Baltsas, D. S. Achilias, C. Kiparissides
Radicals of type i of chain length one:
where
For modelling purposes, it is not efficient to solve the infinite system of differential equations (14) -(16) describing the conservation of macromolecular species in the reactor. In an attempt to reduce the high dimensionality of the numerical problem, several mathematical techniques have been developed I s 2 ) . In what follows, the method of moments is used to recast the “infinite” set of macromolecular balance equations into a low-order system, which can easily be solved.
Molecular property balances
The method of moments is based on the statistical representation of the average molecular properties (e. g., Mn, a,, etc.) through the use of the leading moments of the molecular property distributions. In the free-radical copolymerization of methyl methacrylate with vinyl acetate, due to the high reactivity ratio of MMA3’), the con- centration of “live” copolymer chains ending in a MMA unit will be very high. It is

A theoretical investigation of the production of branched copolymers . . . 483
further assumed that copolymer macroradicals ending in MMA monomer are terminating exclusively by disproportionation, while the corresponding macroradicals with a VAc terminal unit are terminating by combination. As a result, most “dead” copolymer chains will have one terminal double bond. thus, we can assume that only one chain length distribution will be sufficient to characterize the population of “dead” copolymer chains. Apparently, the last assumption considerably simplified the mathematical model developments. Accordingly, one can define the moments of the total number molecular weight distributions (TNMWDs) of “live” and “dead” copolymer as1,2,8,12):
m m
= c c (nMw, + rnMw2)k R i , , , i i = 1, 2 k = 0, 1, 2 (1 9) n= I m= 1
n= I m= 1
The corresponding rate functions for the moments of the total number molecular weight distributions of “live” and “dead” copolymer chains can be obtained by multiplying each term of Eqs. (15)-(16) by (nMwl + mMw2)k and summing the resulting expressions over the total variation of m and n.

484 A. Baltsas, D. S. Achilias, C . Kiparissides

A theoretical investigation of the production of branched copolymers . . . 485
It should be pointed out that when transfer to polymer reactions are included in the kinetic mechanism, the n-order polymer moment equation will depend on the (n + 1)- order moment. This is due to the fact that the transfer to polymer rate function depends on the total number of monomer units in the copolymer chains. To break-down the dependence of the moment equations on higher order moments several closure methods have been proposed. In the present investigation, two different closure methods were examined, namely the method of Hulburt and K a t ~ ~ ~ ) and the use of the so-called “bulk” moments. The method proposed by Hulburt and K a t ~ ~ ~ ) assumes that the MWD can be represented by truncated (after the first term) series of Laguerre polynomials, using a gamma distribution weighting function, chosen so that the coefficients of the second and third terms are zero. By assuming that the first three terms of the Laguerre polynomials are sufficient for representing the MWD, Hulburt and Katz derived the following approximation for the third moment, p3 :
P2(2P2PO - P3 =
POP 1
According to the second closure method12), a “bulk moment” p j + A: + A,” is defined which includes the contribution of the “live” copolymer chains. Note that the term (pi + A; + A,”) is approximately equal to pi due to the very low contribution of A: and I ,” . Subsequently, the second order “live” radical moment equations (25) and (26) are added to the second order “dead” copolymer moment equation (29). The resulting combined second order moment of the “dead” copolymer MWD is given by
which does not include moments of higher order (e. g., p3) . The two moment closure methods were applied to the present copolymerization system and the results of this comparative study are presented in the ‘Results and Discussion’ section.
The number- and weight-average molecular weights can be expressed in terms of the moments of the TNMWDs of “live” and “dead” copolymer chains as follows:
Number-average molecular weight:
01, + 4 + A:> - PI M“ = - o l o + 4 ,+ Po
- -
Weight-average molecular weight:

486 A. Baltsas, D. S. Achilias, C. Kiparissides
The polydispersity index (D), which is a measure of the breadth of the MWD, is defined as the ratio of the weight- to the number-average molecular weight (D =
The monomer concentration in the copolymer chains, w , , can be calculated by the M,/I@,).
following equation:
dW, (34)
Qf - - - WP1, + kfll) At, + Wp2, + k f 2 ) A l l M , + - (V,f - w , ) dt V
Subscript f denotes reactor feeding conditions. Accordingly, the mass, F,,, and mole fractions, F,, of monomer M, in the
copolymer will be given by:
(35)
Based on the general kinetic scheme considered in this study, Eqs. (1 ) - (1 3), one can identify two additional structural characteristics of the copolymer chains related to the number of long-chain branches (LCB) and the number of terminal double bonds (TDB). To calculate the variation of LCB and TDB per copolymer molecule (B, and T, , respectively) in a continuous stirred tank copolymerization reactor, the following differential equations are considered:
W l
wI + w2 F, =
w , Mw, WIMWI + W2Mw2
Fw, =
Accordingly; the branching density ( E d ) will be given by the ratio of the number- average degree of branching (B, ) over the number-average chain length, X , :
(38) Bn Xn
Bd = -
Detailed material and energy balances
In addition to the above molecular property balances, Eqs. (21)- (38), the following dynamic molar and energy balances must be written for a continuous stirred tank copolymerization reactor:
Initiator molar balance:
dI Q dt V _ - - -kiJ + L(Zf - I )

A theoretical investigation of the production of branched copolymers . . 487
Primary initiator radicals' molar balance:
- 2kiZ - ki,M,Z' - ki2M21' + (%) (ZF - Z') dz' dt
- -
Monomers' molar balance(s):
dM. c (kpU + kfU)Ab+ dt I = 1
j = 1 , 2 (42)
Solvent molar balance:
dS Qr - = -(kfls A; + kfZs Ai)S + - (Sf - S ) dt V
(43)
Solvent radicals' molar balance:
Q dS' dt V - - - (kfls A h + kfzs A6)S - (kis,Ml + kis2M2)S' + (sf - S ' ) (44)
Chain transfer agent molar balance:
Transfer agent radicals' molar balance:
d T' Qr -- - (kf,, Ah + kf2, A;)T - (ki,,MI + kit2M2)T' + - (TF - T') (46)
dt V
Inhibitor molar balance:
dZ Q - = -(kZl A; + kZ2 Ai)Z + (Zf - Z ) dt V
Energy balance for the reaction mixture:
where
(47)
Simulation results and discussion
The predictive capabilities of the present detailed model were examined in relation to the solution copolymerization of methyl methacrylate (MMA) with vinyl acetate (VAc). The polymerization was considered to take place in a well-mixed jacketed continuous vessel, operating isothermally at 80 "C and having a mean residence time of appoximately 6 h.

488 A. Baltsas, D. S. Achilias, C. Kiparissides
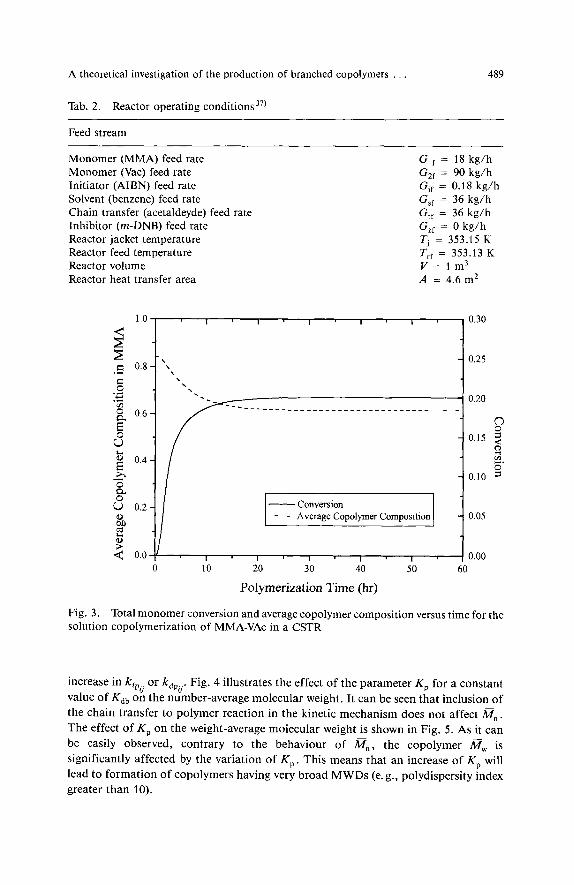
The reaction mixture consisted of MMA, VAc, benzene as a solvent, acetaldehyde (a chain transfer agent), and rn-dinitrobenzene (rn-DNB, a reaction inhibitor). The values of the kinetic rate constants as well as the reactor operating conditions are reported in Tabs. 1 and 2, r e ~ p e c t i v e l y ~ ~ ~ ~ ' ) . It should be noticed that due to the presence of solvent and the low overall monomer conversion (e. g. about 20%) obtained under the present operating conditions, gel and glass effects were considered to be negligible. The time evolution of the overall monomer conversion and average copolymer composition in MMA are plotted in Fig. 3. As it can be observed, conversion and average copolymer composition reach their steady-state values of ca. 20% and 0.6, respectively, in approximately four mean residence times (e. g., 24 h).
The effect of transfer to polymer and terminal double bond reactions on the number- and weight-average molecular weights is illustrated in Figs. 4-7. The nominal values of K , = kfp21 /kPzl = kfPz2/kp2, (i. e., the ratio of the transfer to polymer over the propagation rate constant) and K,, = kdPZ, /kPpl = kdP22/kP22 (i. e., the ratio of the terminal double bond over the propagation rate constant) were set equal to 2.36 x
and 0.66, respectively, which are typical values reported for the homopolymeri- zation of vinyl acetate4'). It should be noted that in all simulations the propagation rate constants remained constant. Thus, an increase in K , or K,, implies an analogous
Tab. 1. Kinetic and thermodynamic parameter^^^'^')
= 4.5 x 1014 exp(-1.25 x IO~/(RT)) s - l = 3.207 x 10' exp(-2.42 x 104/(RT)) m3/(kmol. s) = 1.233 x lo5 exp(-2.42 x 104/(RT)) m3/(kmol. s) = 2.103 x 10' exp(- 1.80 x 104/(RT)) m3/(kmol. s) = 6.308 x lo6 exp(-2.42 x 104/(RT)) m3/(kmol. s) = 32.08 exp(-2.42 x 104/(RT)) m3/(kmol. s) = 1.234 exp(-2.42 x 104/(RT)) m3/(kmol. s) = 5.257 x lo4 exp(-1.80 x 104/(RT)) m3/(kmol.s) = 1577 exp(-1.80 x 104/(RT)) m3/(kmol-s) = 86.6 exp(-2.42 x 104/(RT)) m3/(kmol. s) = 1514 exp(-1.80 x 104/(RT)) m3/(kmol.s) = 2085 exp(-2.42 x 104/(RT)) m3/(kmol-s) = 4.163 x lo5 exp(-2.42 x 104/(RT)) m3/(kmol * s) = 2.2 m3/(kmol. s) = 1.13 x lo5 m3/(kmol. s) = 0 m3/(kmol . s), ktdZZ = 0 m3/(kmol. s) = 1.61 x 109exp(-4.0 x 103/(RT)) m3/(krnol.s) = 4.209 x 10" exp(-2.69 x 104/(RT)) m3/(kmol. s) = 0 m3/(kmol. s), kdPl2 = 0 m3/(kmol * s) = 0.66 kPZ1, kdPZ2 = 0.66 kP22 = 0 m3/(kmol. s), kfP12 = 0 m3/(kmol. s) = 2.36 x = 2.36 x = 54 x lo3 kJ/kmol, -AH,, = 86 x lo3 kJ/kmol
kPZl m3/(kmol. s) kP22 m3/(kmol. s)
I.
= 54 X lo3 kJ/kmol, -AHz2 = 86 x lo3 kJ/kmol = 879 kg/m3, C, = 2.01 kJ/(kg. K), U = 0.06 kJ/(m2. s . K) P
A theoretical investigation of the production of branched copolymers . . . 489
Tab. 2. Reactor operating conditions 37)
Feed stream
Monomer (MMA) feed rate Monomer (Vac) feed rate Initiator (AIBN) feed rate Solvent (benzene) feed rate Chain transfer (acetaldeyde) feed rate Inhibitor (m-DNB) feed rate Reactor jacket temperature Reactor feed temperature Reactor volume Reactor heat transfer area
G , , = 18 kg/h G,, = 90 kg/h Gif = 0.18 kg/h G,, = 36 kg/h Gtf = 36 kg/h G,, = 0 kg/h Tj = 353.15 K T,, = 353.13 K v = i m 3 A = 4.6 m2
0.30
0.25
0.20
s g.
0.15 s 2
0.10 =
0.05
0.00 0 10 20 30 40 50 60
Polymerization Time (hr)
Total monomer conversion and average copolymer composition versus time for the Fig. 3. solution copolymerization of MMA-VAc in a CSTR
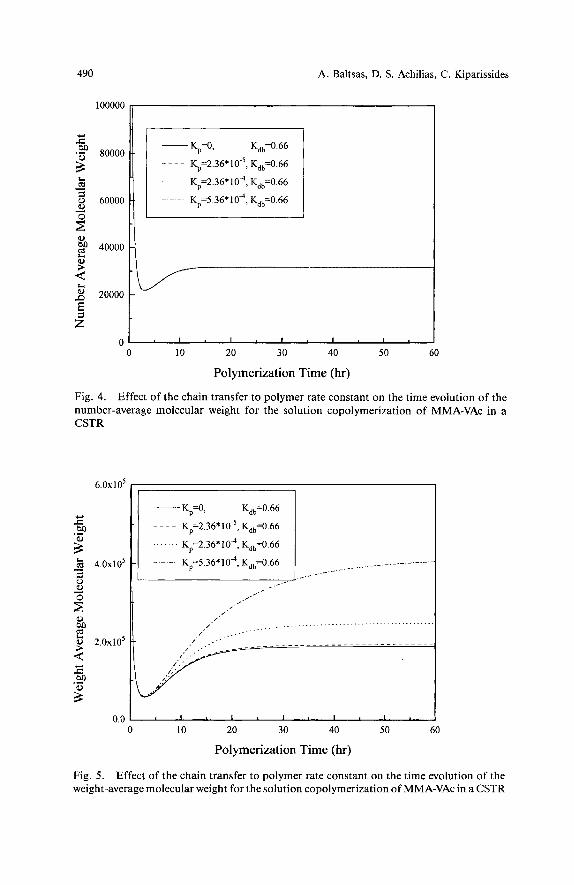
increase in k,,, or kdp... Fig. 4 illustrates the effect of the parameter K , for a constant value of K,, on the ndmber-average molecular weight. It can be seen that inclusion of the chain transfer to polymer reaction in the kinetic mechanism does not affect M, . The effect of K p on the weight-average molecular weight is shown in Fig. 5 . As it can be easily observed, contrary to the behaviour of M,, the copolymer aw is significantly affected by the variation of K , . This means that an increase of K , will lead to formation of copolymers having very broad MWDs (e. g., polydispersity index greater than 10).
490 A. Baltsas, D. S. Achilias, C. Kiparissides
-
-
100000
2 2 80000
3 5
5
< jj 20000
- 60000
e, 4
0
& 0
3 40000
z 0 1 I I I 1 . 1 ,
0 10 20 30 40 50 60
Polymerization Time (hr)
Fig. 4. Effect of the chain transfer to polymer rate constant on the time evolution of the . .
number-average molecular weight for the solution copolymerization of MMA-VAc in a CSTR
n n l 1 1 1 . 1 , I I
0 10 20 30 40 50 60
Polymerization Time (hr)
Fig. 5 . Effect of the chain transfer to polymer rate constant on the time evolution of the weight-average molecular weight for the solution copolymerization of MMA-VAc in a CSTR
A theoretical investigation of the production of branched copolymers . . . 491
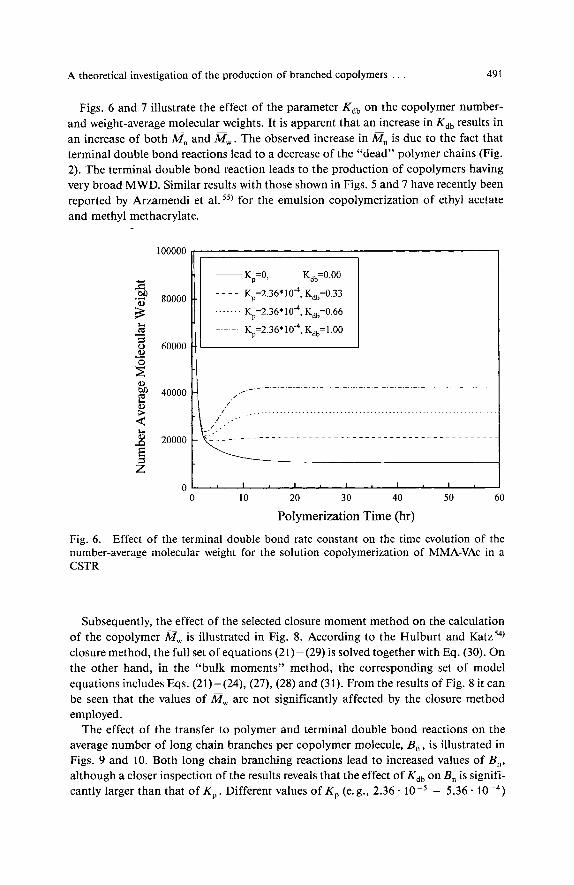
Figs. 6 and 7 illustrate the effect of the parameter Kdb on the copolymer number- and weight-average molecular weights. It is apparent that an increase in K,, results in an increase of both a,, and a,. The observed increase in M, is due to the fact that terminal double bond reactions lead to a decrease of the “dead” polymer chains (Fig. 2). The terminal double bond reaction leads to the production of copolymers having very broad MWD. Similar results with those shown in Figs. 5 and 7 have recently been reported by Arzamendi et al. 5 5 ) for the emulsion copolymerization of ethyl acetate and methyl methacrylate.
100000
@ 80000 s
5 k 2 j 20000
!3
9 c
5 60000 0 - 0 W3 40000
z 0 1 I I I I I I
0 10 20 30 40 50 60
Polymerization Time (hr)
Fig. 6. Effect of the terminal double bond rate constant on the time evolution of the number-average molecular weight for the solution copolymerization of MMA-VAc in a CSTR
Subsequently, the effect of the selected closure moment method on the calculation of the copolymer a, is illustrated in Fig. 8. According to the Hulburt and K a t ~ ~ ~ ) closure method, the full set of equations (21)-(29) is solved together with Eq. (30). On the other hand, in the “bulk moments” method, the corresponding set of model equations includes Eqs. (21)-(24), (27), (28) and (31). From the results of Fig. 8 it can be seen that the values of a, are not significantly affected by the closure method employed.
The effect of the transfer to polymer and terminal double bond reactions on the average number of long chain branches per copolymer molecule, B, , is illustrated in Figs. 9 and 10. Both long chain branching reactions lead to increased values of B,, although a closer inspection of the results reveals that the effect of Kdb on B, is signifi- cantly larger than that of K , . Different values of K , (e. g., 2.36 * - 5.36 -
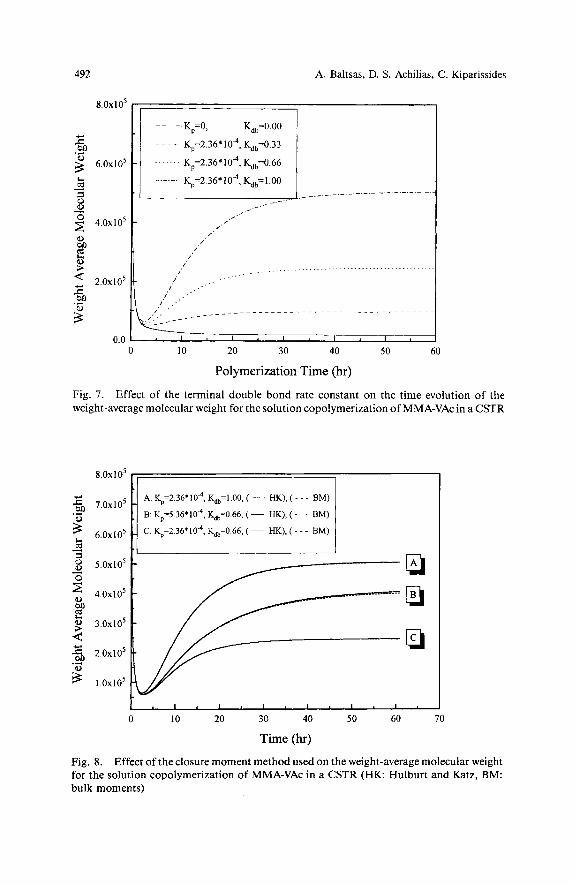
492
8 . 0 ~ 1 0 ~
s 5 6 . 0 ~ 1 0 ~
3 2 - 6) - 5 4 . 0 ~ 1 0 ~ a2 M E
s * 2.0~105
M .-
0.0
A. Baltsas, D. S. Achilias, C. Kiparissides
Kp=2.36* lo4, K,,=0.33
Kdb=0.66 . . . . . . . K,=2.36*
0 10 20 30 40 50 60
Polymerization Time (hr)
Fig. 7. Effect of the terminal double bond rate constant on the time evolution of the weight-average molecular weight for the solution copolymerization of MMA-VAc in a CSTR
8 . 0 ~ 1 0 ~
6 7 . 0 ~ 1 0 ~ ' 6 . 0 ~ 1 0 ~
3 5 . 0 ~ 1 0 ~
4 . 0 ~ 1 0 ~
3 . 0 ~ 1 0 ~
Y
.-
8 - - bo
4 & 2 . 0 ~ 1 0 ~ .3
A: KP-2.36*1O4, K,=1.00, ( - HK), ( - - - BM) B. Kp=5 36*104, Kdb=0.66, (- HK), ( - - - BM) C: KP=2.36*1O4, Kdb=O 66, (- HK), ( - - - BM)
0 10 20 30 40 50 60 70
Time (hr) Fig. 8. Effect of the closure moment method used on the weight-average molecular weight for the solution copolymerization of MMA-VAc in a CSTR (HK: Hulburt and Katz, BM: bulk moments)
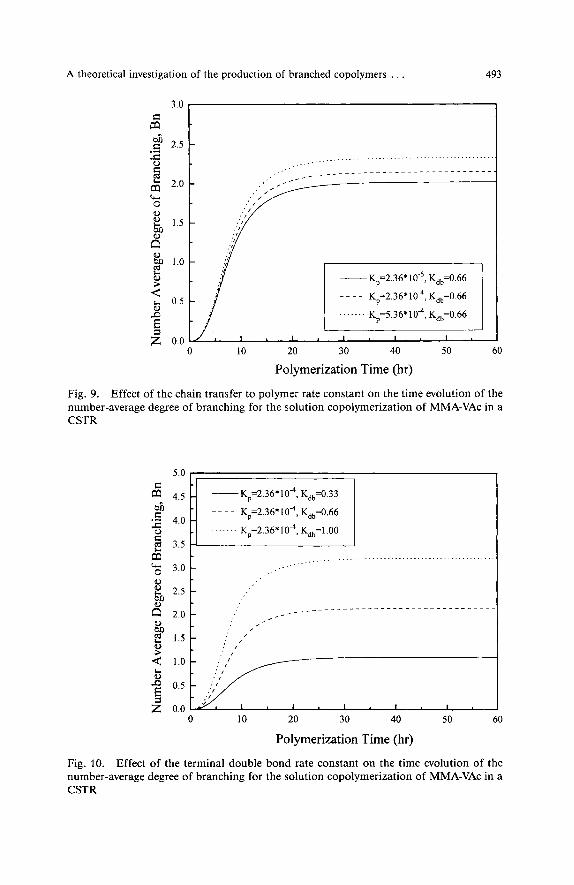
A theoretical investigation of the production of branched copolymers . . . 493
8 $ 2.5 .3
3 -
............................................ ......
5.0 $z a 4.5
-5 4.0 6
5 a 5 3.5
"0 3.0
Polymerization Time (hr)
Fig. 10. Effect of the terminal double bond rate constant on the time evolution of the number-average degree of branching for the solution copolymerization of MMA-VAc in a CSTR
- - Kp=2.36*104, K,,=0.33
_ _ _ _ KP=2.36*1O4, K,,=0.66 - ....... ~ ~ = 2 . 3 6 * 1 0 - ~ , K,,=~.oo
- ...............................................
.... -
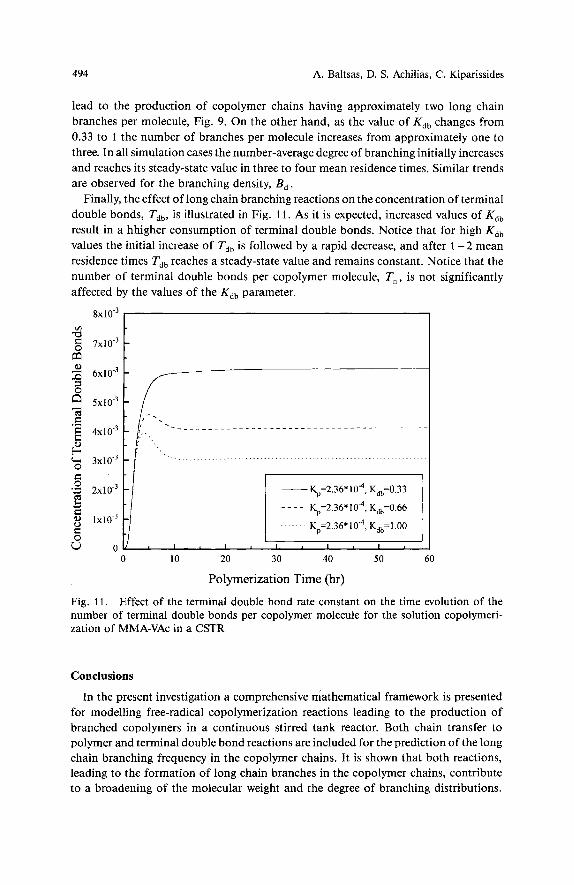
494 A. Baltsas, D. S. Achilias, C. Kiparissides
lead to the production of copolymer chains having approximately two long chain branches per molecule, Fig. 9. On the other hand, as the value of Kdb changes from 0.33 to 1 the number of branches per molecule increases from approximately one to three. In all simulation cases the number-average degree of branching initially increases and reaches its steady-state value in three to four mean residence times. Similar trends are observed for the branching density, B, .
Finally, the effect of long chain branching reactions on the concentration of terminal double bonds, Tdb, is illustrated in Fig. 11. As it is expected, increased values of K,, result in a hhigher consumption of terminal double bonds. Notice that for high K,, values the initial increase of Tdb is followed by a rapid decrease, and after 1 - 2 mean residence times Tdb reaches a steady-state value and remains constant. Notice that the number of terminal double bonds per copolymer molecule, T,, is not significantly affected by the values of the K,, parameter.
F v) a 8 7 ~ 1 0 . ~ m
_ _ _ _ _ _ _ _ _ _ ~ _ _ _ ~ _ _ _ _ _ _ _ - - - - - - - - - - - ~ ~ ~ - ~ ~ - .
.......,. .. ................ . . . .. .. .............. ... . .
2x10-3 - K,=2.36*104, K,,=0.33
Kp=2.36* lo4, K,,=0.66
. . . . . . . Kp=2.36* lo4, K,,=l .OO 1x10-3 - 0 I I 1 . 1 , I
0 10 20 30 40 50 60
Polymerization Time (hr)
Fig. 11. Effect of the terminal double bond rate constant on the time evolution of the number of terminal double bonds per copolymer molecule for the solution copolymeri- zation of MMA-VAc in a CSTR
Conclusions
In the present investigation a comprehensive mathematical framework is presented for modelling free-radical copolymerization reactions leading to the production of branched copolymers in a continuous stirred tank reactor. Both chain transfer to polymer and terminal double bond reactions are included for the prediction of the long chain branching frequency in the copolymer chains. It is shown that both reactions, leading to the formation of long chain branches in the copolymer chains, contribute to a broadening of the molecular weight and the degree of branching distributions.

A theoretical investigation of the production of branched copolymers . . . 495
Furthermore, the terminal double bond reaction significantly affects the number- average molecular weight and the concentration of the terminal double bonds. Finally, it was found that the number-average degree of branching and branching density increase as the values of K p and Kdb increase.
Nomenclature
Reactor heat transfer area in m3 Branching density Number-average number of long chain branches per copolymer molecule Heat capacity in kJ/(kg. K) Polydispersity index Mole fraction of monomer i in the copolymer Weight fraction of monomer i in the copolymer Mass flow rate of component i in kg/h Specific enthalpy of the ij propagation reaction in kJ/kmol Initiator concentration in kmol/m3 Kinetic rate constant ratio kdPIJ/kpl, Kinetic rate constants ratio kfpl / k Termination by combination rate constant of a type-i radical with a type-j radical in m3/(kmol. s) Termination by disproportionation rate constant of type-i radical with a type-j radical in m3/(kmol sec) Terminal double bond rate constant of a type-i radical with a type-j polymer in rn3/(kmol. s) Initiator decomposition rate constant in s Initiation rate constant of monomer i in m3/(kmol. s) Initiation rate constant of monomer i with radical S in m3/(kmol . s) Initiation rate constant of monomer i with radical T in m3/(kmol. s) Propagation rate constant of radical i adding to monomer j in m3/(kmol. s) Chain transfer rate constant of radical type i to monomer of typej in m3/(kmol 3 s) Chain transfer rate constant of radical type i to solvent molecule S in m3/(kmol . s) Chain transfer rate constant of radical type i to agent molecule T in m3/(kmol. s) Chain transfer rate constant of radical type i to polymer of type j in m3/(kmol. s) Inhibition rate constant of radical i with inhibitor Z in m3/(kmol. s) Concentration of monomer M in kmol/m3 Concentration of monomer M2 in kmol/m3
'IJ
MWD Molecular weight distribution ~
MWi Molecular weight of monomer i in kg/kmol Mn Number-average molecular weight in kg/kmol M, Weight-average molecular weight in kg/kmol P, , , , Concentration of dead copolymer chains with n units of monomer M, and rn units
of monomer M, in kmol/m3 Qf Feed volumetric flow rate in m3/s R i , m Concentration of live copolymer radicals with n units of monomer M I and m units
of monomer M, ending in i-type monomer in kmol/m3 S Solvent concentration in kmol/m3 T Chain transfer agent concentration in kmol/m3 Tdb Concentration of terminal double bonds in kmol/m3 Tj Reactor's jacket temperature in K

A. Baltsas, D. S. Achilias, C. Kiparissides
Number-average of terminal double bonds per copolymer molecule Reactor temperature in K Temperature of the feed stream in K Time in h Overall heat transfer coefficient in kJ/(m. s * K) Reactor volume in m3 Number-average chain length Inhibitor concentration in kmol/m3 k-th moment of molecular weight distribution of live copolymer radicals ending in monomer M I k-th moment of molecular weight distribution of live copolymer radicals ending in monomer M, k-th moment of dead copolymer chains Feed density in kg/m3 Concentration of monomer i in the copolymer in kmol/m3
Subscript f denotes reactor feeding conditions
’) D. S. Achilias, C. Kiparissides, J. Macromol. Sci., Rev. Macromol. Chem. Phys. C32,183
’) D. S. Achilias, C. Kiparissides, Polymer 35, 1714 (1994) 3, R. Simha, H. Branson, J. Chem. Phys. 12, 253 (1944) 4, H. M. Melville, B. Noble, W. F. Watson, J. Polym. Sci. 2, 229 (1947) 5 , C. H. Bamforb, W. G. Barb, A. D. Jenkins, D. F. Onyon, “Kinetics of Vinyl
6 , W. H . Ray, Macromolecules 4, 162 (1971) ’) W. H. Ray, T. L. Douglas, E. W. Godsalve, Macromolecules 4, 166 (1971) 8, W. H. Ray, J. Macromol. Sci., Rev. Macromol. Chem. C8, 1 (1972) 9, D. K. Sharma, D. S. Soane, Macromolecules 21, 700 (1988)
”1 A. E. Hamielec, J. F. MacGregor, A. Penlidis, Makromol. Chem., Macromol. Symp. l O / l l , 521 (1987)
‘I) D. N. Butala, W. R. Liang, K. Y. Choi, J. Appl. Polym. Sci. 44, 1759 (1992) D. J. Arriola, Ph. D. Thesis, University of Wisconsin, Madison, WI 1989
1 3 ) W. H. Ray, Can. J. Chem. Eng. 69, 626(1991) 14) W. H. Ray, in “PolymerReaction Engineering’: K. H. Reichert, W. Geiseler, Eds., VCH,
Weinheim 1989, p. 105 15) B. Tilger, G. Luft, in “Polymer Reaction Engineering”, K. H. Reichert, W. Geiseler,
Eds., VCH, Weinheim 1989, p. 84 j 6 ) G. Storti, S. Canegallo, P. Canu, M. Morbidelli, in 4th Intern. Workshop on Polymer
Reaction Engineering, K. H. Reichert, H. U. Moritz, Eds., DECHEMA, Vol. 127, p. 379, VCH, Weinheim 1992
(1 992)
Polymerization by Radical Mechanisms”, Butterworths, London 1958
1 7 ) H. Tobita, A. E. Hamielec, Polymer 32, 2641 (1991) 1 8 ) H. Tobita, Macromolecules 26, 836 (1993) 19) U. Engelmann, G. Schmidt-Naake, Macromol. Theory Simul. 3, 219 (1994) ’O) U. Engelmann, G. Schmidt-Naake, Makromol. Chem., Theory Simul. 2, 275 (1 993)
”1 R. A. Hutchinson, J. P. Congalidis, J. R. Richards, Engineering Foundation Conference
23) T. Y. Xie, A. E. Hamielec, Makromol. Chem., Theory Simul. 2, 421 (1993) 24) T. Y. Xie, A. E. Hamielec, Makromol. Chem., Theory Simul. 2, 455 (1993)
R. A. Hutchinson, Polym. React. Eng. 1, 521 (1993)
on Polymer Reaction Engineering, Saint Augustine, FL 1994

A theoretical investigation of the production of branched copolymers . . . 497
25) T. Y. Xie, A. E. Hamielec, Makromol. Chem., Theory Simul. 2, 777 (1993) 26) G. Verros, M. Papadakis, C. Kiparissides, Polym. React. Eng. 1, 427 (1993) 27) I. A. Maxwell, G. T. Russell, Makromol. Chem., Theory Simul. 2, 95 (1993) 28) 0. Okay, Macromol. Theory Simul. 3, 417 (1994) 29) L. Christov, G. Georgiev, Macromol. Theory Simul. 4, 177 (1995) 30) M. Dube, A. Penlidis, Polymer 36, 587 (1995) 31) H. Tobita, Polym. React. Eng. 1, 357 (1993) 32) H. Tobita, Polym. React. Eng. 1, 379 (1993) 33) H. Tobita, J. Polym. Sci., Part B: Polym. Phys. 31, 1363 (1993) 34) A. D. Schmidt, W. H. Ray, Chem. Eng. Sci. 36, 1401 (1981) 35) J. W. Hamer, T. A. Akramov, W. H. Ray, Chem. Eng. Sci. 36, 1897 (1981) 3 6 ) F. Teymour, W. H. Ray, Chem. Eng. Sci. 47, 4133 (1992) 37) J. P. Congalidis, J. R. Richards, W. H. Ray, AZChE J. 35, 891 (1989) 38) F. Teymour, W. H. Ray, Chaos, Solitons & Fractals 1, 295 (1991) 39) F. Teymour, J. D. Campbell, in 4th Intern. Workshop on Polymer Reaction Engineering,
K. H. Reichert, H. U. Moritz, Eds. DECHEMA, Vol. 127, p. 149, VCH, Weinheim 1992 40) A. K. Adebekun, K. M. Kwalik, F. J. Schork, Chem. Eng. Sci. 44, 2269 (1989) 41) K. Nagasubramanian, W. W. Graessley, Chem. Eng. Sci. 25, 1549 (1970) 42) K. Nagasubramanian, W. W. Graessley, Chem. Eng, Sci. 25, 1559 (1970) 43) A. Chatterjee, W. S. Park, W. W. Graessly, Chem. Eng. Sci. 32, 167 (1977)
G. VerStrate, C. Cozewith, W. W. Graessley, J. Appl. Polym. Sci. 25, 59 (1980) 45) J. C. Hyun, W. W. Graessley, S. G. Bankoff, Chem. Eng. Sci. 31, 945 (1976) 46) W. W. Graessley, H. Mittelhauser, R. Maramba, Makromol. Chem. 86, 129 (1965) 47) 0. Saito, K. Nagasubramanian, W. W. Graessley, J. Polym. Sci., Polym. Phys. Ed. 7, 1937
48) W. W. Graessley, R. D. Hartung, W. C. Uy, J. Polym. Sci., Polym. Phys. Ed. 7, 1919
49) C. Wolf, W. Burchard, Makromol. Chem. 177, 2519 (1976) jO) R. A. Jackson, P. A. Small, K. S. Whiteley, J. Polym. Sci., Polym. Chem. Ed. 11, 1781
j') J. W. Hamer, Ph. D. Thesis, University of Wisconsin-Madison 1983 52) T. W. Taylor, K. H. Reichert, J. Appl. Polym. Sci. 30, 227 (1985) 53) K. Konstantinidis, D. S. Achilias, C. Kiparissides, Polymer 33, 5019 (1992) 54) H. M. Hulburt, S. Katz, Chem. Eng. Sci. 19, 555 (1964) 55) G. Arzamendi, J. Forcada, J. Asua, Macromolecules 27, 6068 (1994)
(1 969)
(1 969)
(1973)
Copyright © 2022 FDOKUMEN




















