
A Scalable Parallel Approach for Peptide Identification from Large-Scale Mass Spectrometry Data
8
A Scalable Parallel Approach for Peptide Identification from Large-scale Mass Spectrometry Data Gaurav Kulkarni, Ananth Kalyanaraman School of Electrical Engineering and Computer Science Washington State University Pullman, WA, USA Email: [email protected], [email protected] William R. Cannon, Douglas Baxter Pacific Northwest National Laboratory Richland, WA, USA Email: [email protected], [email protected] Abstract—Identifying peptides, which are short polymeric chains of amino acid residues in a protein sequence, is of fundamental importance in systems biology research. The most popular approach to identify peptides is through database search. In this approach, an experimental spectrum (“query”) generated from fragments of a target peptide using mass spectrometry is computationally compared with a database of already known protein sequences. The goal is to detect database peptides that are most likely to have generated the target peptide. The exponential growth rates and overwhelming sizes of biomolecular databases make this an ideal application to benefit from parallel computing. However, the present generation of software tools is not expected to scale to the magnitudes and complexities of data that will be generated in the next few years. This is because they are all either serial algorithms or parallel strategies that have been designed over inherently serial methods, thereby requiring high space- and time-requirements. In this paper, we present an efficient parallel approach for peptide identification through database search. Three key factors distinguish our approach from that of existing solutions: i) (space) Given p processors and a database with N residues, we provide the first space-optimal algorithm (O( N p )) under distributed memory machine model; ii) (time) Our algorithm uses a combination of parallel techniques such as one-sided communication and masking of communication with computation to ensure that the overhead introduced due to parallelism is minimal; and iii) (quality) The run-time savings achieved using parallel processing has allowed us to incorporate highly accurate statistical models that have previously been demonstrated to ensure high quality prediction albeit on smaller scale data. We present the design and evaluation of two different algorithms to implement our approach. Experimental results using 2.65 million microbial proteins show linear scaling up to 128 processors of a Linux commodity cluster, with parallel efficiency at ∼50%. We expect that this new approach will be critical to meet the data-intensive and qualitative demands stemming from this important application domain. Keywords-parallel peptide identification; mass spectrometry; I. I NTRODUCTION A fundamental problem in systems biology research is to identify the set of proteins, or more generally peptides, expressed in a specific organism or a community of or- ganisms (metagenomic communities) under certain envi- ronmental conditions. As proteins constitute the molecular basis for cellular functions, addressing this problem helps in the understanding of the cellular dynamics of organisms under various environmental conditions. Mass spectrometry (“MS”) is a powerful and now a standard technique to identify peptides. In this technique, multiple copies of an unknown (target) peptide are experimentally fragmented and the distribution of fragments (called intensity peaks) over a range of mass-to-charge ratio values (m/z) is recorded. The resulting plot of peak intensities (y-axis) to m/z values (x-axis) is called an experimental spectrum for the target peptide. The subsequent computational task is to deduce the peptide sequence from its experimental spectrum. This can be achieved by comparing the experimental spectrum against model spectra generated from a database of known peptides. Databases include conventional protein sequences (e.g., UniProt/Swiss-Prot [3]) and/or unconventional peptide se- quences derived from putative open reading frames (ORFs) of genomic/metagenomic DNA (e.g., GenBank [1]). Col- lectively, these collections are growing at exponential rates (e.g., see Figure 1a), and as a result, the number of spectrum- to-peptide comparisons to be dealt with during database search is starting to overwhelm current software ability. Figure 1b shows the magnitudes of peptides that need to be evaluated as “candidates” for a match against each input spectrum. If a sample involves any unsequenced genome(s) corresponding to the target peptides, which is typically the case in metagenomic projects, the number of candidates for evaluation increases by orders of magnitudes. Additional requirements to take into consideration post-translation mod- ifications (PTMs) further exacerbate the situation. A. Related Work The task of generating peptides for evaluation against an experimental spectrum can be done in two distinct and complementary ways. The first approach is called database searching and refers to deriving protein sequences from genomic DNA sequences [11], and then using empirical rules to determine which peptides should be present in the proteins. The advantage to this approach is that the database provides an independent evidence of the peptide. The caveats 2009 International Conference on Parallel Processing Workshops 1530-2016/09 $26.00 © 2009 IEEE DOI 10.1109/ICPPW.2009.41 423
Transcript of A Scalable Parallel Approach for Peptide Identification from Large-Scale Mass Spectrometry Data

A Scalable Parallel Approach for Peptide Identification from Large-scale MassSpectrometry Data
Gaurav Kulkarni, Ananth Kalyanaraman
School of Electrical Engineering and Computer ScienceWashington State University
Pullman, WA, USAEmail: [email protected], [email protected]
William R. Cannon, Douglas Baxter
Pacific Northwest National LaboratoryRichland, WA, USA
Email: [email protected], [email protected]
Abstract—Identifying peptides, which are short polymericchains of amino acid residues in a protein sequence, is offundamental importance in systems biology research. The mostpopular approach to identify peptides is through databasesearch. In this approach, an experimental spectrum (“query”)generated from fragments of a target peptide using massspectrometry is computationally compared with a databaseof already known protein sequences. The goal is to detectdatabase peptides that are most likely to have generated thetarget peptide. The exponential growth rates and overwhelmingsizes of biomolecular databases make this an ideal applicationto benefit from parallel computing. However, the presentgeneration of software tools is not expected to scale to themagnitudes and complexities of data that will be generatedin the next few years. This is because they are all eitherserial algorithms or parallel strategies that have been designedover inherently serial methods, thereby requiring high space-and time-requirements. In this paper, we present an efficientparallel approach for peptide identification through databasesearch. Three key factors distinguish our approach from that ofexisting solutions: i) (space) Given p processors and a databasewith N residues, we provide the first space-optimal algorithm(O(N
p)) under distributed memory machine model; ii) (time)
Our algorithm uses a combination of parallel techniques suchas one-sided communication and masking of communicationwith computation to ensure that the overhead introduced dueto parallelism is minimal; and iii) (quality) The run-time savingsachieved using parallel processing has allowed us to incorporatehighly accurate statistical models that have previously beendemonstrated to ensure high quality prediction albeit onsmaller scale data. We present the design and evaluation of twodifferent algorithms to implement our approach. Experimentalresults using 2.65 million microbial proteins show linear scalingup to 128 processors of a Linux commodity cluster, withparallel efficiency at ∼50%. We expect that this new approachwill be critical to meet the data-intensive and qualitativedemands stemming from this important application domain.
Keywords-parallel peptide identification; mass spectrometry;
I. INTRODUCTION
A fundamental problem in systems biology research is
to identify the set of proteins, or more generally peptides,
expressed in a specific organism or a community of or-
ganisms (metagenomic communities) under certain envi-
ronmental conditions. As proteins constitute the molecular
basis for cellular functions, addressing this problem helps
in the understanding of the cellular dynamics of organisms
under various environmental conditions. Mass spectrometry
(“MS”) is a powerful and now a standard technique to
identify peptides. In this technique, multiple copies of an
unknown (target) peptide are experimentally fragmented and
the distribution of fragments (called intensity peaks) over
a range of mass-to-charge ratio values (m/z) is recorded.
The resulting plot of peak intensities (y-axis) to m/z values
(x-axis) is called an experimental spectrum for the target
peptide. The subsequent computational task is to deduce thepeptide sequence from its experimental spectrum. This can
be achieved by comparing the experimental spectrum against
model spectra generated from a database of known peptides.
Databases include conventional protein sequences (e.g.,
UniProt/Swiss-Prot [3]) and/or unconventional peptide se-
quences derived from putative open reading frames (ORFs)
of genomic/metagenomic DNA (e.g., GenBank [1]). Col-
lectively, these collections are growing at exponential rates
(e.g., see Figure 1a), and as a result, the number of spectrum-
to-peptide comparisons to be dealt with during database
search is starting to overwhelm current software ability.
Figure 1b shows the magnitudes of peptides that need to
be evaluated as “candidates” for a match against each input
spectrum. If a sample involves any unsequenced genome(s)
corresponding to the target peptides, which is typically the
case in metagenomic projects, the number of candidates for
evaluation increases by orders of magnitudes. Additional
requirements to take into consideration post-translation mod-
ifications (PTMs) further exacerbate the situation.
A. Related Work
The task of generating peptides for evaluation against
an experimental spectrum can be done in two distinct and
complementary ways. The first approach is called databasesearching and refers to deriving protein sequences from
genomic DNA sequences [11], and then using empirical
rules to determine which peptides should be present in the
proteins. The advantage to this approach is that the database
provides an independent evidence of the peptide. The caveats
2009 International Conference on Parallel Processing Workshops
1530-2016/09 $26.00 © 2009 IEEE
DOI 10.1109/ICPPW.2009.41
423
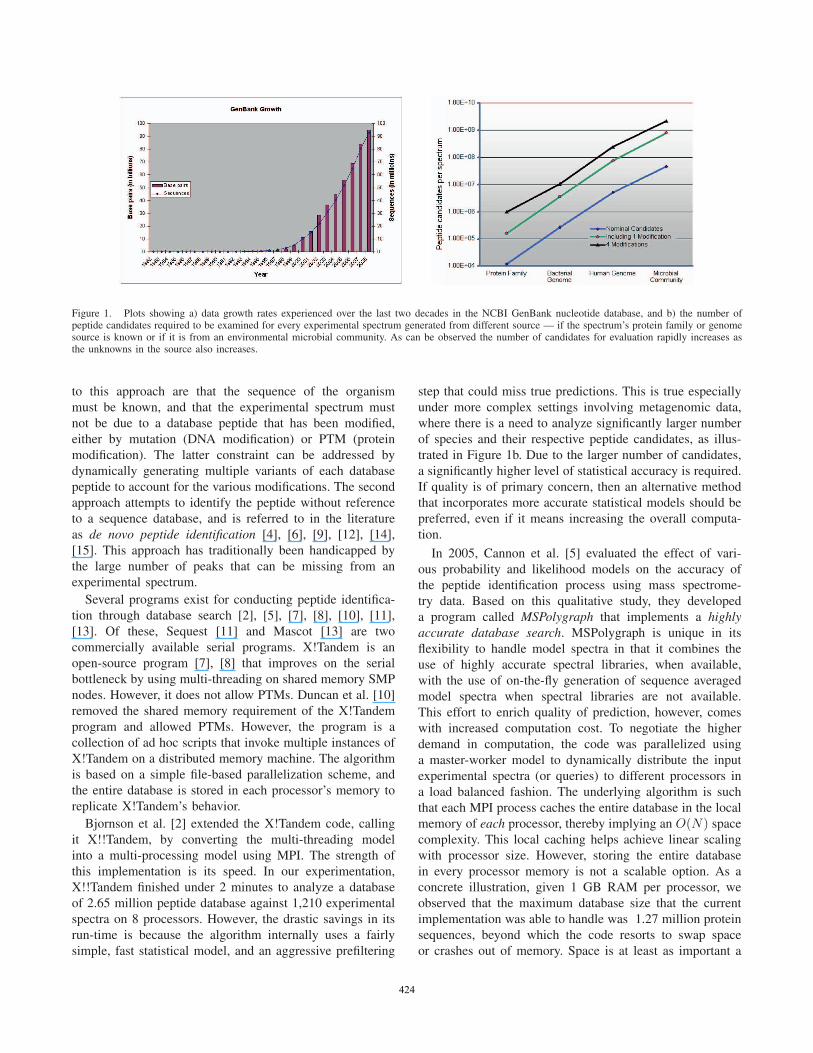
Figure 1. Plots showing a) data growth rates experienced over the last two decades in the NCBI GenBank nucleotide database, and b) the number ofpeptide candidates required to be examined for every experimental spectrum generated from different source — if the spectrum’s protein family or genomesource is known or if it is from an environmental microbial community. As can be observed the number of candidates for evaluation rapidly increases asthe unknowns in the source also increases.
to this approach are that the sequence of the organism
must be known, and that the experimental spectrum must
not be due to a database peptide that has been modified,
either by mutation (DNA modification) or PTM (protein
modification). The latter constraint can be addressed by
dynamically generating multiple variants of each database
peptide to account for the various modifications. The second
approach attempts to identify the peptide without reference
to a sequence database, and is referred to in the literature
as de novo peptide identification [4], [6], [9], [12], [14],
[15]. This approach has traditionally been handicapped by
the large number of peaks that can be missing from an
experimental spectrum.
Several programs exist for conducting peptide identifica-
tion through database search [2], [5], [7], [8], [10], [11],
[13]. Of these, Sequest [11] and Mascot [13] are two
commercially available serial programs. X!Tandem is an
open-source program [7], [8] that improves on the serial
bottleneck by using multi-threading on shared memory SMP
nodes. However, it does not allow PTMs. Duncan et al. [10]
removed the shared memory requirement of the X!Tandem
program and allowed PTMs. However, the program is a
collection of ad hoc scripts that invoke multiple instances of
X!Tandem on a distributed memory machine. The algorithm
is based on a simple file-based parallelization scheme, and
the entire database is stored in each processor’s memory to
replicate X!Tandem’s behavior.
Bjornson et al. [2] extended the X!Tandem code, calling
it X!!Tandem, by converting the multi-threading model
into a multi-processing model using MPI. The strength of
this implementation is its speed. In our experimentation,
X!!Tandem finished under 2 minutes to analyze a database
of 2.65 million peptide database against 1,210 experimental
spectra on 8 processors. However, the drastic savings in its
run-time is because the algorithm internally uses a fairly
simple, fast statistical model, and an aggressive prefiltering
step that could miss true predictions. This is true especially
under more complex settings involving metagenomic data,
where there is a need to analyze significantly larger number
of species and their respective peptide candidates, as illus-
trated in Figure 1b. Due to the larger number of candidates,
a significantly higher level of statistical accuracy is required.
If quality is of primary concern, then an alternative method
that incorporates more accurate statistical models should be
preferred, even if it means increasing the overall computa-
tion.
In 2005, Cannon et al. [5] evaluated the effect of vari-
ous probability and likelihood models on the accuracy of
the peptide identification process using mass spectrome-
try data. Based on this qualitative study, they developed
a program called MSPolygraph that implements a highlyaccurate database search. MSPolygraph is unique in its
flexibility to handle model spectra in that it combines the
use of highly accurate spectral libraries, when available,
with the use of on-the-fly generation of sequence averaged
model spectra when spectral libraries are not available.
This effort to enrich quality of prediction, however, comes
with increased computation cost. To negotiate the higher
demand in computation, the code was parallelized using
a master-worker model to dynamically distribute the input
experimental spectra (or queries) to different processors in
a load balanced fashion. The underlying algorithm is such
that each MPI process caches the entire database in the local
memory of each processor, thereby implying an O(N) space
complexity. This local caching helps achieve linear scaling
with processor size. However, storing the entire database
in every processor memory is not a scalable option. As a
concrete illustration, given 1 GB RAM per processor, we
observed that the maximum database size that the current
implementation was able to handle was 1.27 million protein
sequences, beyond which the code resorts to swap space
or crashes out of memory. Space is at least as important a
424

consideration as run-time efficiency, given that the analysis
of metagenomic data sets that contain peptides from many
organisms is becoming increasingly common.
B. Our Contribution
In this paper, we present a new parallel approach for large-
scale peptide identification. The contributions are:
• (Space) Our approach is space-optimal — i.e., if mand N denote the sizes of the queries and the database,
respectively, then its space complexity is O(m+Np ).
• (Time) The algorithm uses a combination of paral-
lel techniques such as one-sided communication and
communication-computation masking to ensure that the
overhead introduced due to data distribution is minimal.
We implemented (in C/MPI) and evaluated two algo-
rithmic variants of our approach.
• (Results) Experimental results demonstrate strong linear
scaling of our new method up to 128 processors on
a commodity Linux cluster, in which each processor
has access to 1 GB RAM. Under this setting, our
implementation allows us to scale up the database size
by ∼420K sequences for every new processor added to
the system.
The above improvements have made it now feasible to
incorporate the highly accurate statistical models and on-the-
fly model spectra generation capabilities of MSPolygraph for
significantly large data sets.
II. METHODS
A. Definitions and Problem Statement
Let q denote an unknown peptide sequence, which is
fragmented using MS. A fragment of a peptide typically
captures the mass-spectrum of either a prefix or a suffix
sequence of q. MS generates an experimental spectrum of
q, which is a plot of the relative abundance of its fragments
against a range of m/z ratio values. It also reports the m/z
of the whole parent peptide q, denoted by m(q). Given
q, a suffix or prefix of another (known) peptide sequence
is said to be a candidate for q if the suffix’s/prefix’s m/z
is m(q) ± δ. (δ is a tolerance constant.) An experimental
spectrum for q is said to match with a candidate peptide
if it can be shown that the candidate is most likely to
generate a model spectrum similar to that of the experimental
spectrum. This is achieved in MSPolygraph by generating
two different spectra [5] — one a model spectrum for the
candidate and the other being a spectrum generated for a
random peptide — and then comparing both against the
experimental spectrum. The result is a likelihood ratio score,
and if the score is above a user-specified cutoff then the
corresponding matching peptide is reported as a “hit”.
Let Q = {q1, q2, . . . qm} denote a set of m input ex-
perimental spectra generated from m unknown peptides,
and let D = {d1, d2, . . . dn} denote a database of nalready known peptide sequences. For convenience, let
N =∑n
i=1 length(di). We use p to denote the number of
processors, and label them P0, P1, . . . Pp−1. Also, the terms
“experimental spectrum” and “query” are used interchange-
ably.
The Peptide Identification Problem: Given input sets Qand D, the peptide identification problem is to identify a
list of at most τ top database hits for every input spectrum
q ∈ Q.
In practice, τ is assigned a value between 10 and 1,000.
The algorithmic steps in MSPolygraph can be summarized
as follows:
S1) Instantiate one master processor and p − 1 worker
processors. The master processor loads Q into its local
memory, while all workers load the entire database Din their respective local memory.
S2) The master processor starts by distributing small, fixed
size batches of experimental spectra (or queries) to
individual worker processors.
S3) Each worker processor works on the assigned batch of
queries, processing one query at a time, reporting at
most τ hits per query to an output file, and informing
the master processor upon completion.
S4) Steps S3 and S4 are repeated iteratively until all the
queries at the master processor have been processed.
The above parallelization scheme has two main advan-
tages: i) the processing of every query is strictly localized
within each worker and generates practically no communica-
tion during processing; and ii) the master processor plays the
role of a load distributor and since the queries are allocated
to worker processors in small batches based on demand,
the workload is balanced. While these factors yield linear
scaling of the algorithm with processor size, the input data
size cannot be scaled with processor size because it is O(N).
B. Our Parallel Approach for Peptide Identification
We designed a new approach to parallelize the peptide
identification process that partitions the database evenly
among the p processors, such that each processor stores
a distinct O(Np ) fraction of the database. Distributing the
database, however, introduces challenges related to commu-
nication and overhead. If a database sequence can generate
a valid candidate for a given query, then it has to be made
available to the processor handling that query. In the worst
case, a query may need the entire database and such a
worst-case is not far from practical expectations either (as
corroborated in our experiments). If a query on processor
Pi requires a database sequence that is resident in a remote
processor Pj’s memory, then there are two design options:
i) (Database transport) Communicate the database se-
quence from Pj to Pi so that the query can be locally
processed; or
ii) (Query transport) Communicate the query from Pi to
Pj for remote query processing.
425

The query transport model can help, especially since m is
expected to be much smaller than n. However, the challenge
with such a scheme is that a query can get processed in
multiple processor locations, and the results have to be sent
to one root processor for merging. Whereas, the database
transport model does not generate such serialization issues,
and allows each processor to take full responsibility of
any given query. We chose the database transport model to
develop our approach. The trick here is to efficiently mask
the communication costs introduced by the transport of the
database.
Here, we propose two different algorithms that implement
the database transport model.
Algorithm A:The pseudocode for this algorithm is shown in Figure 2.
Briefly, the loading step loads the database sequence file in
parallel such that processor Pi receives roughly the ith Np
byte chunk of the file. Care is taken to ensure sequences at
the boundaries are fully read. This step ensures a balanced
partitioning of the database sequences across processors.
The query file is read similarly, such that each Pi receives
roughly mp queries. In the next step, the queries are
processed over p iterations. At any step s, processor Pi
compares all its queries against Dj , where j = (i + s)%p.
Before the queries are processed, a non-blocking request
to receive the database portion for the next iteration is
issued. This is achieved using the MPI Get() one-sided
communication primitive, so that the communication is
achieved without disturbing the remote processor. Also, at
every iterative step, Pi keeps a separate running list of the
τ topmost hits for every query in Qi. Upon termination,
this list is output.
Analysis: For memory management, each Pi keeps three
O(Np ) buffers: i) Di stores the local portion of the database;
ii) Drecv is the communication buffer. It is over-written by
the non-blocking MPI Get() primitive at every iteration;
and iii) Dcomp is the buffer against which local queries are
compared at any given iteration. This is also over-written
at every iteration. Therefore, the space complexity of this
algorithm is O(N+mp ).
As for computation complexity, steps A1 and A3 col-
lectively take O(N+mp + m
p × τ) for input loading and
output reporting. For step A2, let r be the average number
of candidates evaluated against each query. Let ρ be the
constant time it takes to compare each query against each
candidate. Then, the total computation complexity for query
comparison at every processor is O(mp ×r×ρ). In addition,
the cost of maintaining a running list of the top τ hits for
every query is O(mp ×r×τ). The amortized cost of fetching
database sequences in step A2 over p different stages is
O(n). Therefore, the overall computation complexity is:
O(N+mp + m
p × r × (ρ + τ) + n).
Figure 2. Pseudocode for Algorithm A. The main idea is to distribute thedatabase and queries across processors and then to transport the databasefragments as and when needed to ensure O(m+N
p) space per processor.
The non-blocking request in step A2 is for masking communication withcomputation.
As for communication complexity, let λ be the network
latency and μ be the time to transfer one byte over the
network. Then the total communication complexity is
O(λ × p + μ × N). However, due to masking, the net
observed communication cost is expected to be significantlyless, as shown by our results in Section III.
Algorithm B:The pseudocode for this algorithm is shown in Figure 3.
The main idea of this algorithm is as follows: Prior to
query processing, the database is sorted in parallel based on
the sequences’ m/z values (step B2). The output is a non-
decreasingly sorted array, generated such that each processor
receives O(Np ) characters from the sorted database. During
subsequent query processing (B3), the sorted order could
help identify only that subset of processors which have
sequences with candidates to offer the local batch of queries,
implying that it is sufficient to restrict the communication
to the identified subset. More specifically: Given a query q,
its candidates can originate only from any database peptide
d such that m(d) ≥ m(q). Therefore, if processor Pi
precomputes m(q)min = min|Qi|j=1(m(qj)), then candidates
for any query in Qi can originate from only database
sequences with m/z values m(d) ≥ m(q)min. Let Pi′ be
the lowest ranked processor which has database sequences
in the sorted order satisfying this property. Then the sender
426

Figure 3. Pseudocode for Algorithm B. The idea is similar to AlgorithmA’s, except that a database sorting step is added as a preprocessingstep. This way, each processor needs to query only a range of otherprocessors for database transport as determined by its local set of queries.The non-blocking request in step B3 is for masking communication withcomputation.
group for Pi is the set {Pi′ . . . Pp−1}.We were able to implement this sorting step using par-
allel counting sort because the m/z values are bounded in
practice within the range [1, ..., 300000]. Our parallel sorting
algorithm is as follows:
S1) Each processor computes the parent m/z value of each
sequence in Di. The processors then compute the
global maximum of the m/z values (m/zmax) using the
MPI Allreduce primitive.
S2) Each processor creates a local “count” array of size
m/zmax in which it records the frequency occurrence
of each m/z value in Di. Subsequently, using the
MPI Allreduce primitive on the local count arrays, the
processors compute a global count array, which they
use as a reference to redistribute the sequences in Di.
Sequences with the same m/z are sent to the same
processor, and the sum of the lengths of the sequences
resulting in each processor is O(Np ). This data exchange
is implemented using the MPI Alltoallv primitive. Let
Dsi denote the output of sorting in processor Pi.
Based on the partitioning pivots used in sorting, all pro-
cessors store an array of p tuples of the form (begini, endi)
to keep track of m/z boundary values at every processor.
This tuple information can be used to determine the value
of i′.The query processing step is almost identical to that
of Algorithm A, with a minor addition: To accelerate
identifying the range of queries that require any database
sequence, we maintain the local query set Qi also sorted
by their m/z values and then use binary search.
Analysis: Along with the buffer to store Qi, this algorithm
requires at most three of the following four database buffers,
each of size O(Np ), at any given execution point: Di, Ds
i ,
Dcomp, and Drecv . This, with the p tuples stored at every
processor for indexing yields a net space complexity of
O(N+mp + p). — i.e., it is O(N+m
p ) if p2 < N + m;
otherwise, it is O(p).Relative to Algorithm A, the only addition to computation
complexity in this algorithm is the computation performed
during sorting in step B2. As we use integer sorting, this
additive factor is only O(np ) which is dominated by O(N
p ).In addition, as only those database sequences with at least
one candidate to offer for a given query are compared against
that query, that cost is O(mp × r), which in the worst case
equals O(n). Therefore, the overall computation complexity
is: O(N+mp + m
p × r × (ρ + τ)).The communication complexity within the parallel
counting sort routine is dominated by the cost of
the MPI Alltoallv primitive. As each processor sends
and receives approximately Np characters, this cost is
O(λ × p + μNp ). During query processing, each processor
performs at most p − 1 MPI Get function calls, and
each such call fetches O(Np ) characters. Overall, the
worst-case communication complexity of Algorithm B is
O(λ×p+μ×N). In practice, it could be less depending on
the distribution of the queries and the database sequences
that they need.
Implementation: The code is written in C/MPI, and can be
obtained by contacting one of the authors.
III. EXPERIMENTAL RESULTS & DISCUSSION
All experiments were conducted on a 24-node, 192-
processor Linux commodity cluster. Each node contains
8 2.33 GHz Xeon CPUs and a shared 8 GB RAM. The
network interconnect is a gigabit ethernet, and all nodes
share an NFS mounted file system. To mimic a modest
setting under most commodity clusters, we set the RAM
usage limit to 1 GB RAM per MPI process.
Input Data: We tested our implementations on two
collections of data (see Table I): i) (Human) 88,333 human
protein sequences; and ii) (Microbial) ∼2.65 million
microbial protein sequences, both downloaded from NCBI
GenBank [1]. The human database was used for validating
427

Human Microbial#Protein Sequences 88,333 2,655,064
Total seq. length (in residues) 26,647,093 834,866,454Avg. seq. length (in residues) 301.66 314.44
Table IINPUT DATABASE STATISTICS.
our results against MSPolygraph’s results as published
in [5]. The microbial database was used for large-scale
performance studies. To conduct scalability tests on this
data set, we extracted arbitrary subsets of sizes 1K, 2K,
4K, . . . up to 2.65 million. A collection of 1,210 human
experimental spectra was used as queries in all experiments.
Validation: Upon validation, we found that both
implementations A & B successfully reproduceMSPolygraph’s output on the human protein collection.
This validates the correctness of the programs because
internally we use the same scoring functions and statistical
modeling as MSPolygraph.
Performance analysis of Algorithm A: We evaluated
the performance of Algorithm A using the 1,210 human
experimental spectra as queries and for varying subset sizes
of the 2.65 million protein sequence database. Table II
shows the parallel run-time of Algorithm A over the entire
range of input sizes and processor sizes tested. Although
the asymptotic run-time is data-dependent (depends on the
number of candidates evaluated for all queries), the prac-
tical expectation is that the run-time scales linearly with
the database size. This expectation is consistent with our
observations within each column of Table II. The run-time
growth as a function of processor size can be explained as
follows: Our parallel run-time can be broken down into two
parts: computation time and “residual communication” time.
Residual communication time is defined as the time spent by
the code waiting for the next batch of data, and is equal to
the total communication time minus its portion masked by
computation. In practice, it is only the residual communi-
cation that matters for the total time. We observed that the
mean±std. deviation of the ratio of residual communicationto computation time to be 0.36 ±0.11, for all processor sizes
greater than 2 on all input sizes. In other words, the overhead
due to communication is approximately 25% of the total
time even for larger processor sizes.
The parallel speedup and efficiency are shown in Fig-
ures 4a and 4b, respectively, for data sizes 16K or more1.
Because any run of our Algorithm A at p = 1 is equiva-
lent to the uni-worker processor run of MSPolygraph, the
speedup values in Figure 4a represent real speedup. As can
1For input sizes < 16K, the algorithm scales only until 8 processors.For p > 8, the efficiency starts to deteriorate expectedly as the input sizebecomes too small for the processor size.
p 8 16 32 64 128#Candidates 41,429 76,057 159,220 271,294 522,331per sec.
Table IIINUMBER OF CANDIDATES EVALUATED PER SECOND AS A FUNCTION OF
PROCESSOR SIZE (FOR THE 2.65 MILLION MICROBIAL DATABASE).
be observed, the speedup approximately doubles whenever
processor size is also doubled, with the only exception being
when p is increased from 2 to 4. This near-linear scaling
behavior overall is captured in the parallel efficiency plot.
As Figure 4b shows, the efficiency is near perfect at p = 2,
but reduces to ∼ 50% at p = 4. Thereafter, it is maintained
at ∼ 50% until p = 64, and gradually dips to 41.51% at
p = 128. The reason for the one-time loss in efficiency from
p = 2 to p = 4 is as follows: At p = 2, Algorithm A requires
that each processor communicate with the other processor
and the masking of communication by computation in the
first iteration ensures that the residual communication is
practically neglible. However, at p = 4 each processor is
required to communicate with three processors (3X-fold
increase) and due to the high latency costs involved, the
contribution of the residual communication to the total run-
time increases from almost 0% to nearly 35%. Except for
this anomaly, the efficiency for all processor sizes between 4
and 128 is maintained at ∼50% — implying strong scaling.
To assess the positive effect of masking, we implemented
a second version of the algorithm that does not mask
communication with computation. Results showed that the
masking technique reduces the total run-time by a factor
of 72.75% ± 0.02%. For example, if the parallel run-time
without masking for a given input is 100s, then the same
analysis with masking will take only 27.25s.
Finally, we also measured the candidate evaluation rate
of our algorithm. This result is shown in Table III. From
an application point of view, this is likely to be the most
interesting performance measure as it directly conveys the
effect of parallel processing on peptide identification. As
shown, our implementation achieves linear scaling of the
number of candidates processed every second with processor
size.
Performance analysis of Algorithm B: We analyzed Algo-
rithm B’s performance and observed that it was consistently
outperformed by Algorithm A in speedup and efficiency.
Table IV shows a concrete example of this behavior. Upon
investigation, we found that the decline in speedup for
Algorithm B was because the overhead due to its sorting step
was becoming dominant as processor size was increased.
This is also shown in Table IV. In addition, the input
queries were such that each processor had to communi-
cate and fetch database segments from a majority of the
other p − 1 processors, thereby defeating the purpose of
sorting. However, note that the set of 1,210 experimental
428
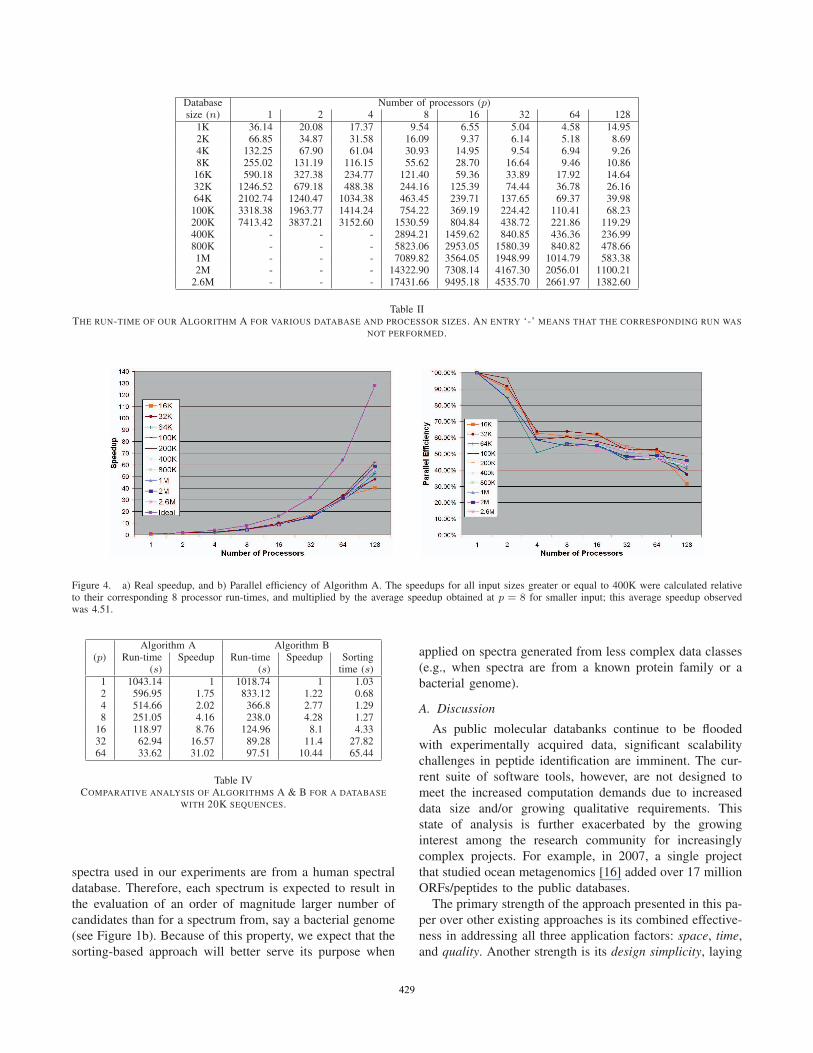
Database Number of processors (p)size (n) 1 2 4 8 16 32 64 128
1K 36.14 20.08 17.37 9.54 6.55 5.04 4.58 14.952K 66.85 34.87 31.58 16.09 9.37 6.14 5.18 8.694K 132.25 67.90 61.04 30.93 14.95 9.54 6.94 9.268K 255.02 131.19 116.15 55.62 28.70 16.64 9.46 10.86
16K 590.18 327.38 234.77 121.40 59.36 33.89 17.92 14.6432K 1246.52 679.18 488.38 244.16 125.39 74.44 36.78 26.1664K 2102.74 1240.47 1034.38 463.45 239.71 137.65 69.37 39.98100K 3318.38 1963.77 1414.24 754.22 369.19 224.42 110.41 68.23200K 7413.42 3837.21 3152.60 1530.59 804.84 438.72 221.86 119.29400K - - - 2894.21 1459.62 840.85 436.36 236.99800K - - - 5823.06 2953.05 1580.39 840.82 478.661M - - - 7089.82 3564.05 1948.99 1014.79 583.382M - - - 14322.90 7308.14 4167.30 2056.01 1100.21
2.6M - - - 17431.66 9495.18 4535.70 2661.97 1382.60
Table IITHE RUN-TIME OF OUR ALGORITHM A FOR VARIOUS DATABASE AND PROCESSOR SIZES. AN ENTRY ‘-’ MEANS THAT THE CORRESPONDING RUN WAS
NOT PERFORMED.
Figure 4. a) Real speedup, and b) Parallel efficiency of Algorithm A. The speedups for all input sizes greater or equal to 400K were calculated relativeto their corresponding 8 processor run-times, and multiplied by the average speedup obtained at p = 8 for smaller input; this average speedup observedwas 4.51.
Algorithm A Algorithm B(p) Run-time Speedup Run-time Speedup Sorting
(s) (s) time (s)1 1043.14 1 1018.74 1 1.032 596.95 1.75 833.12 1.22 0.684 514.66 2.02 366.8 2.77 1.298 251.05 4.16 238.0 4.28 1.27
16 118.97 8.76 124.96 8.1 4.3332 62.94 16.57 89.28 11.4 27.8264 33.62 31.02 97.51 10.44 65.44
Table IVCOMPARATIVE ANALYSIS OF ALGORITHMS A & B FOR A DATABASE
WITH 20K SEQUENCES.
spectra used in our experiments are from a human spectral
database. Therefore, each spectrum is expected to result in
the evaluation of an order of magnitude larger number of
candidates than for a spectrum from, say a bacterial genome
(see Figure 1b). Because of this property, we expect that the
sorting-based approach will better serve its purpose when
applied on spectra generated from less complex data classes
(e.g., when spectra are from a known protein family or a
bacterial genome).
A. Discussion
As public molecular databanks continue to be flooded
with experimentally acquired data, significant scalability
challenges in peptide identification are imminent. The cur-
rent suite of software tools, however, are not designed to
meet the increased computation demands due to increased
data size and/or growing qualitative requirements. This
state of analysis is further exacerbated by the growing
interest among the research community for increasingly
complex projects. For example, in 2007, a single project
that studied ocean metagenomics [16] added over 17 million
ORFs/peptides to the public databases.
The primary strength of the approach presented in this pa-
per over other existing approaches is its combined effective-
ness in addressing all three application factors: space, time,
and quality. Another strength is its design simplicity, laying
429

the foundation for further improvements and extensions. The
space-optimality result will allow application scientists to
scale to very large input sizes than was possible before,
by exploiting the vast, aggregate memory easily available
from large-scale distributed memory supercomputers. For
example, we were able to store and analyze 2.65 million
sequences using as little as 8 processors.
It is to be noted, however, the application of our approach
will make sense only for inputs that do not fit in local mem-
ory. For small inputs that fit within a processor’s memory, the
older version of MSPolygraph is more appropriate because
it will output the same result with no added communication
delays. For medium range inputs, however, it could be worth
exploring an extension of our approach in which processors
can divide themselves into smaller sub-groups, where the
database is partitioned within each sub-group and the query
set is partitioned across sub-groups.
A dominant fraction of the query processing time is spent
on generating candidates on-the-fly. Each query, in practice,
may require generation of hundreds of thousands to even
millions of candidates (as shown in Figure 1b). From this
perspective, it may be worth exploring an alternative strategy
in which candidates, and not the database sequences, are
stored in-memory and are communicated on demand to
worker processors. This strategy could drastically reduce
the overall computation time. While current approaches are
not designed to store such large magnitudes of candidates
in memory, our algorithm, because of its space-optimality,
makes the investigation of this alternative approach feasible.
Furthermore, the sorting version of our approach (Algorithm
B) could prove more useful under this setting.
IV. CONCLUSIONS
In this paper, we presented the design and development of
a new parallel algorithm for conducting large-scale peptide
identification using mass spectrometry data. The approach
proposed here is better equipped than any other contempo-
rary software tool for meeting the scalability demands of the
peptide identification application. The highlights of our new
algorithm are its space-optimality, the ability to maintain
run-time efficiency through a combination of known parallel
techniques, and its incorporation of accurate statistical mod-
els for improved accuracy. Using the approach developed
here, we plan to conduct a full-scale application and inves-
tigation on the largest available metagenomic collections.
Such a large-scale application would not only advance
scientific pursuit, but also layout a strong foundation for the
proteomics community to benefit from parallel processing.
REFERENCES
[1] D.A. Benson, I. Karsch-Mizrachi, et al. GenBank. NucleicAcids Research, 35:D21–D25, 2007.
[2] R.D. Bjornson, N.J. Carriero, et al. X!!Tandem, an improvedmethod for running X!Tandem in parallel on collections ofcommodity clusters. Journal of Proteome Research, 7:293–299, 2008.
[3] B. Boeckmann, A. Bairoch, et al. The SWISS-PROT proteinknowledgebase and its supplement TrEMBL in 2003. NucleicAcids Research, 31:365–370, 2003.
[4] W.R. Cannon and K.H. Jarman. Improved peptide sequencingusing isotope information inherent in tandem mass spectra.Rapid Communications in Mass Spectrometry, 17:1793–1801,2003.
[5] W.R. Cannon, K.H. Jarman, et al. Comparison of probabilityand likelihood models for peptide identification from tandemmass spectrometry data. Journal of Proteome Research,4:1687–1698, 2005.
[6] T. Chen, M.Y. Kao, et al. A dynamic programming approachto de novo peptide sequencing via tandem mass spectrometry.Journal of Computational Biology, 8(3):325–337, 2001.
[7] R. Craig and R.C. Beavis. A method for reducing the timerequired to match protein sequences with tandem mass spectra.Rapid Communications in Mass Spectrometry, 17(20):2310–2316, 2003.
[8] R. Craig and R.C. Beavis. TANDEM: matching proteins withtandem mass spectra. Bioinformatics, 20(9):1466–1467, 2004.
[9] V. Dancik, T.A. Addona, et al. De novo peptide sequencing viatandem mass spectrometry. Journal of Computational Biology,6(3/4):327–342, 1999.
[10] D.T. Duncan, R. Craig, and A.J. Link. Parallel tandem: aprogram for parallel processing of tandem mass spectra usingPVM or MPI and X!Tandem. Journal of Proteome Research,4(5):1842–1847, 2005.
[11] A.L. McCormack J.K. Eng and J.R. Yates. An approach tocorrelate tandem mass spectral data of peptides with aminoacid sequences in a protein database. Journal of The AmericanSociety for Mass Spectrometry, 5(11):976–989, 1994.
[12] C. Liu, Y. Song, et al. Fast de novo peptide sequencingand spectral alignment via tree decomposition. In PacificSymposium on Biocomputing, volume 11, pages 255–66, 2006.
[13] D.N. Perkins, D.J. Pappin, et al. Probability based proteinidentification by searching sequence databases using massspectrometry data. Electrophoresis, 20(18):3551–3567, 1999.
[14] P.A. Pevzner, Z. Mulyukov, et al. Efficiency of databasesearch for identification of mutated and modified proteins viamass spectrometry. Genome Research, 11(2):290–299, 2001.
[15] J.A. Taylor and R.S. Johnson. Sequence database searchesvia de novo peptide sequencing by tandem mass spectrometry.Rapid Communications in Mass Spectrometry, 11:1067–1075,1997.
[16] S. Yooseph, G. Sutton, et al. The Sorcerer II Global OceanSampling Expedition: Expanding the Universe of Protein Fam-ilies. PLoS Biology, 5:e16, 2007.
430



















