
2-Hydroxyethylammonium Carboxylates - Highly Biodegradable and Slightly Toxic Ionic Liquids

Journal of Molecular Liquids 194 (2014) 20–29
Contents lists available at ScienceDirect
Journal of Molecular Liquids
j ourna l homepage: www.e lsev ie r .com/ locate /mol l iq
A quantum chemical study on themolecular interaction between pyrroleand ionic liquids
Hanee Farzana Hizaddin, Ramalingam Anantharaj ⁎, Mohd. Ali HashimUniversity of Malaya Centre for Ionic Liquids (UMCiL), Department of Chemical Engineering, Faculty of Engineering, University of Malaya, 50603 Kuala Lumpur, Malaysia
⁎ Corresponding author. Tel.: +60 3 7967 7687.E-mail address: [email protected] (R. Ananthara
0167-7322/$ – see front matter © 2014 Elsevier B.V. All rihttp://dx.doi.org/10.1016/j.molliq.2013.12.041
a b s t r a c t
a r t i c l e i n f oArticle history:Received 26 September 2013Received in revised form 20 December 2013Accepted 22 December 2013Available online 17 January 2014
Keywords:Ionic liquidDenitrificationQuantum chemicalHartree–FockInteraction energyHOMO/LUMO
Requirement to produce Ultra Low Sulphur Diesel is becomingmore stringent with the target of zero emission. Thepresence of nitrogen compound in crude oil is undesirable because it inhibits the efficiency of desulphurisationprocess besides being environmentally hazardous when found in transportation fuels. Extractive denitrificationwith ionic liquids (ILs) as solvents has been found to be applicable for denitrification butmore insights are requiredto explain the interaction between ILs and nitrogen compounds. In this study, the interaction between pyrrole asmodel nitrogen compound with 18 ILs built from 6 cations and 3 anions is investigated by means of quantumchemical calculation. Geometry optimisation was done for individual molecules of pyrrole, cations and anions, aswell as complexes of ILs, pyrrole–cation, pyrrole–anion, pyrrole–IL at Hartree–Fock level and 6-31G* basis set.NBOanalysiswas performed from the optimised geometry for eachmolecule and complex. The interaction betweenpyrrole and ILs is investigated via HOMO and LUMO energy values and gaps, global scalar properties, interactionenergies and partial charges. It was found that [EPY][EtSO4] is the most favourable IL for the removal of pyrrole.
© 2014 Elsevier B.V. All rights reserved.
1. Introduction
With the growing concern towards reducing emission fromtransportation fuels to the environment, the requirement to producelow-sulphur fuels is becoming more stringent. The current limit forsulphur content in diesel fuel is less than 10 ppm for the productionof Ultra Low-Sulphur Diesel (ULSD) with the target of zero emission inthe future. However, the presence of nitrogen compounds even at lowconcentration hinders the efficiency of sulphur removal from dieselfuels. Laredo et al. [1,2] reported that the presence of nitrogen compoundsas low as b30 ppm inhibits the efficiency of hydrodesulphurisationprocess through competitive adsorption, making it more difficult tomeet the low-sulphur requirement. Besides the inhibition effect, thepresence of nitrogen compounds in diesel is also non-desirable, becauseit can cause coke formation and lead to catalyst deactivation. Also, thepresence of nitrogen compoundsmay possibly affect the stability of dieselduring storage. Therefore, in order to achieve high sulphur removal,knowledge on the removal of nitrogen compounds is essential [3].
The common practice in industry for removal of nitrogen ishydrodenitrification (HDN) and for removal of sulphur is hydro-desulphurisation (HDS). These processes are energy intensive and arecostly due to the high operating temperature (300–400 °C) and elevat-ed pressure (20–100 atm of H2). This drives the research community to
j).
ghts reserved.
search for alternative processes for the removal of sulphur and nitrogencompounds. Solvent extraction process such as extractive desul-phurisation and extractive denitrification is seen as a potentially prom-ising candidate to replace HDS and HDN, since it is a much simplerprocess, it can be operated at ambient temperature and pressurewithout much modification on the equipment, thus lowering theoperating cost and energy consumption. The efficiency of extractionprocess however depends upon the performance of the solvent. Solventselection is therefore crucial in order to achieve high nitrogen removalfrom liquid fuels.
Ionic liquids have emerged as a potentially versatile solvent forvarious applications including for liquid extraction process. Propertiesof ionic liquids include high thermal stability, high electrochemicalstability, wide liquid range, and high ionic conductivity [4]. Ionic liquidis also known as “designer solvent” as it can be designed to suit apreferred separation process by predetermining its compositionthrough altering the anion–cation combination and its substituents.The tunability of ionic liquids is also the main reason why it is exploredfor various applications. In recent years, several authors have reportedthe use of ionic liquids as solvents to remove nitrogen compoundsfrom gasoline and diesel. Kedra-Krolik et al. [5,6], Zhang et al. [7] andAlonso et al. [8] reported the use of imidazolium-based ionic liquidswith various anions to remove aromatic sulphur and nitrogencompounds frommodel diesel oils and concluded that these ILs demon-strated good capacity for desulphurisation as well as denitrification.Chloride-based ILs [9,10] and dicyanamide-based ILs [11,12] have alsobeen used for selective extraction of nitrogen compounds; with

21H.F. Hizaddin et al. / Journal of Molecular Liquids 194 (2014) 20–29
competitive selectivity and capacity towards nitrogen compounds.Thus, it is evident that ionic liquids have great potential to be furtherutilised as solvent for denitrification of liquid fuels.
However, although the number of publications reporting on the useof various ionic liquids for denitrification purposes is increasing, it is stillconsidered lowwhen compared to the infinite number of possible ionicliquids which could be synthesised. Furthermore, these works onlyreport the suitable ILs for the purpose of denitrification process basedon phase equilibria data, selectivity and capacity of the systems withILs without giving much insights on the interactions between thespecies involved. In order to design an IL for denitrification, a systematicapproachmust be taken and knowledge on the reactivity and stability ofinteracting system is essential. Ab initio method such as quantumchemical calculation provides an alternative to achieve this objectivewith the advantage of not requiring a huge base of experimental data.Instead, only the structure of the species involved is required as aninput to the calculation.
In recent times, the number of literature related to ab initio calcula-tions describing the interactions between organic solute and ionicliquids for separation processes including desulphurisation and denitri-fication has increased. Zhang et al. [13] investigated the interactions be-tween thiophene and the ionic liquids 1-butyl-3-methylimidazoliumtetrafluoroborate ([BMIM][BF4]) and 1-butyl-3-methylimidazoliumhexafluorophosphate ([BMIM][PF6]) by performing geometry optimisa-tion on five clusters of thiophene–cation, thiophene–anions and thio-phene–ILs. It was discovered that the interaction of thiophene with ILsis mainly via Coulombian attraction and that stronger electron donationto the fluorine atoms and compactness of the ILs influences the
Table 1Structure of the species involved in this work.
Compound Name Structure
Nitrogen compound Pyrrole
Cations 1-Ethyl-3-methylimidazolium
1-Ethylpyridinium
1-Ethyl-1-methylpyrrolidinium
1-Ethyl-1-methylpiperidinium
4-Ethyl-4-methylmorpholinium
1,2,4-Trimethylpyrazolium
Anions Acetate
Methylsulphonate
Ethyl sulphate
interactionwith thiophene. Anantharaj et al. reportedHOMO/LUMOen-ergy values and gapwith global scalar properties to investigate simulta-neous interaction for the systemsof ILwith thiophene andpyridine [14],where geometry optimisation calculations were done for themoleculesthiophene, pyridine, the cations 1-butyl-3-methylpyrrolidinium([BMPYRO]), 1-butyl-3-methylpyridinium ([BMPY]) and benzy-limidazolium ([BeMIM]), and the anions tetrafluoroborate ([BF4])and hexafluorophosphate ([PF6]), as well as the complexes ofthiophene–pyridine–IL, thiophene–IL and pyridine–IL. The samegroup later performed quantum chemical studies consisting of determi-nation of partial charges, interaction energies and sigma profile genera-tion (based on COSMO-RS theory) to study simultaneous interactionsthiophene and pyridine with the ILs [BMPYRO][BF4], [BMPYRO][PF6],[BMPY][BF4], [BMPY][PF6] and [BeMIM][BF4] [15]. The result based oneach approach agreeswellwith each other and confirms the descriptionof the interactions in each system which demonstrates the dominanceof CH–π interaction. More recently, geometry optimisation based onDFT calculations for the interactions between the ILs [BMIM][PF6] and[BMIM][BF4] with pyridine/hexane was performed and it was conclud-ed that the interactions between the studied ILswith pyridine are stron-ger than that of between ILs and hexane [16].
In this work, the interactions of a 5-membered non-basic nitrogencompound, pyrrole, with 18 ionic liquids built from the combinationof 6 cations and 3 anions are investigated based on quantum chemicalcalculations by determining the HOMO/LUMO energy values andenergy gap, scalar properties, interaction energies and atomic chargesof the species and its complexes. The structure of pyrrole, cations andanions is presented in Table 1.
Abbreviation Chemical formula MW
PYR C4H5N 67
[EMIM] C6H11N2 111
[EPY] C7H10N 108
[EMPYRO] C7H16N 114
[EMPIPE] C8H18N 128
[EMMOR] C7H16O 116
[TMPYRA] C6H11N2 111
[Ac] CH3COO 59
[MeSO3] CH3SO3 95
[EtSO4] C2H5SO4 125

Fig. 1. HOMO (a) and LUMO (b) of PYR.
22 H.F. Hizaddin et al. / Journal of Molecular Liquids 194 (2014) 20–29
2. Background theory
2.1. HOMO/LUMO energy values and energy gap
When dealing with interacting molecular orbitals, the two orbitalsthat interact are generally the HOMO of one molecule and the LUMOof another molecule. HOMO and LUMO are also known as frontierorbitals and they are essential in determining the amount of energy re-quired to add or remove electrons in a molecule. HOMO is associatedwith the tendency of a species to donate electron and is characteristicfor nucleophile components while LUMO is associated with the tenden-cy to receive electron and is characteristic for electrophilic components.
Ionisation potential (I) is defined as the ability of a ligand to donateprecisely an electron to an acceptorwhile electron affinity (A) is definedas the ability of a ligand to accept precisely an electron from a donor.HOMO energy is related to the ionisation potential as approximatedby Koopmann's theorem while LUMO energy is used to estimate theelectron affinity in a similar way as shown in Eqs. (1) and (2), where− ϵHOMO and − ϵLUMO are the one electron eigenvalues related to thefrontier orbitals as defined in Kohn–Sham Density Functional Theory.Species with low HOMO energy indicates high ionisation potential(better electron donor) while those with high LUMO energy indicatehigh electron affinity (better electron acceptor). It should be notedthat these equations are only exact for hydrogenic atoms, and serveonly as an approximation for other atoms since the electron movementafter ionisation occurred is not taken into account [17].
I ¼ −ϵHOMO ð1Þ
A ¼ −ϵLUMO ð2Þ
The HOMO–LUMO energy gap can be determined from the differ-ence in HOMO and LUMO energy values, and further, the importantproperties in quantum chemical calculation such as the hardness (η),softness (S), chemical potential (μ), electronegativity (χ) and electro-philicity index (ω) can be determined.
Fig. 2. HOMO (a) and LUM
2.2. Global hardness and softness
The hardness of a molecule (η) is related to the energy gap betweenthe HOMO and LUMO by the following expression:
2η ¼ ϵLUMO−ϵHOMO ¼ I−A: ð3Þ
Molecules with a large HOMO–LUMO gap are hard moleculeswhich mean it is highly stable and not keen to charge transferbecause they resist changes in their electron number and distribu-tion. Soft molecules have a small HOMO–LUMO gap which meansthey only require small amount of energies to get them to the excit-ed states and thus are more polarizable. In terms of reactivity, softmolecules are more reactive than hard molecules. Softness (S) isthe opposite definition to molecule hardness and its representationis the inverse of hardness:
S ¼ 1=η ¼ 2= ϵLUMO−ϵHOMOð Þ ¼ 2= I−Að Þ: ð4Þ
2.3. Electronegativity
Electronegativity (χ) describes the ability of a molecule to attractelectrons to itself and can be determined by the average value of theHOMO and LUMO energy values as defined by Mulliken [18]:
χ ¼ − ϵHOMO−ϵLUMOð Þ=2 ¼ I þ Að Þ=2: ð5Þ
2.4. Chemical potential
Chemical potential (μ) describes the tendency of an electron to es-cape and is defined as thefirst derivative of the total energywith respectto the number of electrons in a molecule. In terms of molecular orbitaltheory, chemical potential is simply the negative of electronegativityvalue.
μ ¼ ϵHOMO−ϵLUMOð Þ=2 ¼ − I þ Að Þ=2 ð6Þ
2.5. Electrophilicity index
The ability of a ligand to accept as many electrons as possible istermed as electrophilicity index (ω). As elaborated by Parr et al. [19],while electron affinity describes the capability of a ligand to acceptonly one electron from the environment, electrophilicity index mea-sures the energy lowering of a ligand due to maximal electron flowbetween donor and acceptor. As suggested by Parr and co-workers,
O (b) of [EPY][EtSO4].

23H.F. Hizaddin et al. / Journal of Molecular Liquids 194 (2014) 20–29
the mathematical expression for electrophilicity index is described asfollows:
ω ¼ μ2=2η ¼ ϵHOMO−ϵLUMO
2
� �2= ϵLUMO−ϵHOMOð Þ: ð7Þ
2.6. Interaction energy
When two molecules interact, a certain part of energy will decreaseand this decreased energy corresponds to the interaction energy (ΔE)between the two molecules [20]. In actuality, it is still a challenge tomeasure the interaction energy between interacting molecules, espe-cially for the interaction between the cation and anion of an IL [21].However, the interaction energy can be quantified and approximatedby the difference between the energy of the system or complex andthe sum of energy of individual molecules. For the systems in thisstudy, it is the difference between the energy of the complex pyrrole–cation–anion and the individual energy of pyrrole, cation and anion:
ΔE ¼ EPYR−CAT−AN− EPYR þ ECAT þ EANð Þ ð8Þ
where EPYR, ECAT and EAN are the total energy of pyrrole, cation and anion,respectively. Negative values of interaction energy indicate favourableinteraction between the molecules in the system as this means thatmore energy is required to break the molecule or complexes.
2.7. Charge distribution
Charge distribution in a system which relates to the molecular andatomic orbitals and atomic centres can be determined by doing a popu-lation analysis. In population analysis methods, each atom is assigned a“partial charge”. The charge distribution is qualitatively differentdepending on different method used. Most common population analy-sis methods are Mulliken population analysis, Natural Bond Orbital(NBO) analysis and Bader's Atoms in Molecules (AIM) analysis. NBOanalysis is a popularmethodwhere the natural atomic orbitals in the ac-tual molecular environment are determined for the population analysis.It is also proven that NBO analysis has been successfully used to investi-gate the partial charge transfer within the parent compound [13,20].From the partial charges, the chemical reactivity can be explained andthe differences in hydrogen bonding, electrostatic interaction andCH–π interaction are considered [15].
Fig. 3. HOMO (a) and LUMO
3. Computational details
Geometry optimisation job was performed for each individualspecies (each heterocyclic nitrogen compound, cation and anion), neationic liquid complex, and complexes of heterocyclic nitrogen compoundwith neat ionic liquids. Initial structures for each job were drawn andthe job was executed to perform geometry optimisation at Hartree–Fock level and 6-31G* basis set. Calculation of geometry optimisationat Hartree–Fock level gives more meaningful and accurate values forthe orbital energy [16], while the 6-31G* basis set accounts forpolarisation effect of the species in the complexes. NBOpopulation anal-ysis is also performed for the complexes at the same level and basis set.All of these computationswere performed using the Turbomole 6.4 soft-ware package [22]. From the calculation results, the total energy of theoptimised geometries, as well as the HOMO and LUMO energy values,is retrieved and used for determination of global hardness (η), globalsoftness (S), electronegativity (χ), and electrophilicity index (ω) asdiscussed below.
4. Results and discussion
4.1. HOMO and LUMO energy values
TheHOMO and LUMOof optimised pyrrole are shown in Fig. 1. Fig. 2shows the HOMO and LUMO of optimised [EPY][EtSO4] where it isshown that theHOMOcomes from the anion and the LUMO from cation.This is consistentwith the result reported by Lu et al. [20] andNishi et al.[23]. The optimised geometry of PYR–[EPY][EtSO4] complex in Fig. 3shows the HOMO from pyrrole and LUMO from the cation, consistentwith the report by Wagle et al. [24] in their study on the interactionbetween ionic liquids and polyaromatic hydrocarbons.
From the optimised geometry, the HOMO and LUMO energy valuesfor each molecule and complex are retrieved and represented in Fig. 4and the values can be referred to in the supplementary information.The optimised geometries of pyrrole, cations, anions, and all complexesare given in the supplementary information which is available online.As discussed in previous section, HOMO and LUMO are the two mostimportant molecular orbitals which are useful in describing the chemi-cal behaviour of species. At Hartree–Fock level, Koopmans' theoremsuggests that the energy of the HOMO is a good approximation to thenegative of ionisation potential (I) and similarly LUMO is a good approx-imation to the negative of electron affinity (A) [17]. Low HOMO energy
(b) of PYR–[EPY][EtSO4].
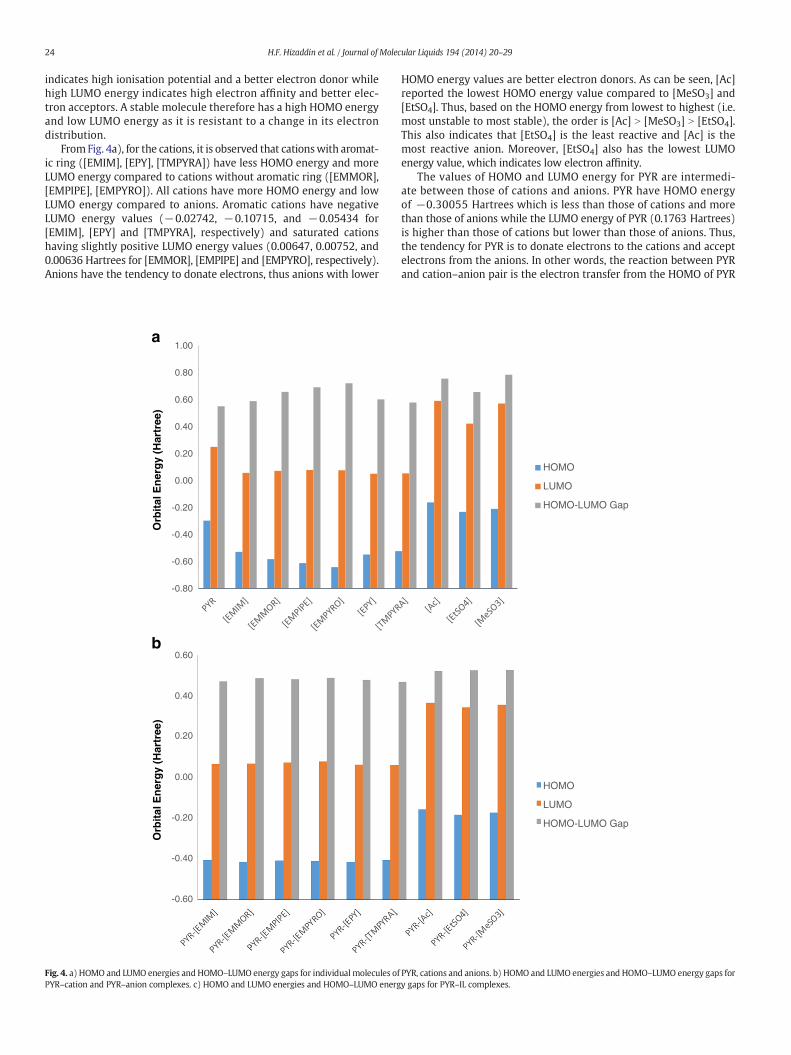
24 H.F. Hizaddin et al. / Journal of Molecular Liquids 194 (2014) 20–29
indicates high ionisation potential and a better electron donor whilehigh LUMO energy indicates high electron affinity and better elec-tron acceptors. A stable molecule therefore has a high HOMO energyand low LUMO energy as it is resistant to a change in its electrondistribution.
FromFig. 4a), for the cations, it is observed that cationswith aromat-ic ring ([EMIM], [EPY], [TMPYRA]) have less HOMO energy and moreLUMO energy compared to cations without aromatic ring ([EMMOR],[EMPIPE], [EMPYRO]). All cations have more HOMO energy and lowLUMO energy compared to anions. Aromatic cations have negativeLUMO energy values (−0.02742, −0.10715, and −0.05434 for[EMIM], [EPY] and [TMPYRA], respectively) and saturated cationshaving slightly positive LUMO energy values (0.00647, 0.00752, and0.00636 Hartrees for [EMMOR], [EMPIPE] and [EMPYRO], respectively).Anions have the tendency to donate electrons, thus anions with lower
-0.80
-0.60
-0.40
-0.20
0.00
0.20
0.40
0.60
0.80
1.00
Orb
ital
En
erg
y (H
artr
ee)
0.60
0.40
0.20
0.00
-0.20
-0.40
-0.60
Orb
ital
En
erg
y (H
artr
ee)
a
b
Fig. 4. a) HOMO and LUMO energies and HOMO–LUMO energy gaps for individual molecules ofPYR–cation and PYR–anion complexes. c) HOMO and LUMO energies and HOMO–LUMO energ
HOMO energy values are better electron donors. As can be seen, [Ac]reported the lowest HOMO energy value compared to [MeSO3] and[EtSO4]. Thus, based on the HOMO energy from lowest to highest (i.e.most unstable to most stable), the order is [Ac] N [MeSO3] N [EtSO4].This also indicates that [EtSO4] is the least reactive and [Ac] is themost reactive anion. Moreover, [EtSO4] also has the lowest LUMOenergy value, which indicates low electron affinity.
The values of HOMO and LUMO energy for PYR are intermedi-ate between those of cations and anions. PYR have HOMO energyof −0.30055 Hartrees which is less than those of cations and morethan those of anions while the LUMO energy of PYR (0.1763 Hartrees)is higher than those of cations but lower than those of anions. Thus,the tendency for PYR is to donate electrons to the cations and acceptelectrons from the anions. In other words, the reaction between PYRand cation–anion pair is the electron transfer from the HOMO of PYR
HOMO
LUMO
HOMO-LUMO Gap
HOMO
LUMO
HOMO-LUMO Gap
PYR, cations and anions. b) HOMO and LUMO energies and HOMO–LUMO energy gaps fory gaps for PYR–IL complexes.

0.60
0.50
0.40
0.30
0.20
0.10
0.00
-0.10
-0.20
-0.30
-0.40
Orb
ital
En
erg
y (H
artr
ee)
HOMO
LUMO
HOMO-LUMO Gap
c
Fig. 4 (continued).
25H.F. Hizaddin et al. / Journal of Molecular Liquids 194 (2014) 20–29
to the LUMO of cation and electron transfer from the HOMO of anion tothe LUMO of PYR.
EHOMO−CATIONSNEHOMO−PYRNEHOMO−ANIONS
−ϵHOMOELUMO−ANIONSNELUMO−PYRNELUMO−CATIONS
Fig. 4b) shows HOMO and LUMO energy values for complexes ofPYR–cations and PYR–anions. In the complexes of PYR–cations, theHOMO and LUMO energy values are brought to a value somewhere inbetween the values of HOMO and LUMO energy for individual mole-cules. The values of HOMO energy for PYR–cations are similar with avery small range of difference from the lowest to the highest values(−0.406 Hartrees to −0.417 Hartrees). On the other hand, the LUMO
Table 2Rank of ILs for interaction with pyrrole from most favourable to least favourable based on diffe
LUMO energy HOMO LUMO gap Gl
(Lowest to highest) (Lowest to highest) (H
Most favourable 1 [EPY][EtSO4] [EPY][EtSO4] [EP2 [EPY][MeSO3] [EPY][Ac] [EP3 [EPY][Ac] [EPY][MeSO3] [EP4 [TMPYRA][Ac] [TMPYRA][EtSO4] [TM5 [TMPYRA][EtSO4] [TMPYRA][Ac] [TM6 [TMPYRA][MeSO3] [EMIM][Ac] [EM7 [EMIM][Ac] [TMPYRA][MeSO3] [TM8 [EMPYRO][EtSO4] [EMMOR][MeSO3] [EM9 [EMIM][EtSO4] [EMIM][EtSO4] [EM10 [EMIM][MeSO3] [EMPYRO][EtSO4] [EM11 [EMPIPE][EtSO4] [EMPIPE][EtSO4] [EM12 [EMMOR][MeSO3] [EMMOR][Ac] [EM13 [EMPYRO][MeSO3] [EMPIPE][MeSO3] [EM14 [EMPIPE][Ac] [EMIM][MeSO3] [EM15 [EMMOR][Ac] [EMPYRO][MeSO3] [EM16 [EMMOR][EtSO4] [EMPIPE][Ac] [EM17 [EMPIPE][MeSO3] [EMPYRO][Ac] [EM
Least favourable 18 [EMPYRO][Ac] [EMMOR][EtSO4] [EM
energy values for PYR–cation complexes show similar trend with theLUMO energy values of the individual cations. Overall, the LUMOenergyvalues of cations become less negative when combined with PYR.
Comparable observation is also reported in the complexesof PYR–anions, where the HOMO energy values are similar inone complex to another with very small range of difference(−0.157 Hartrees to −0.182 Hartrees). Similarly, the LUMO energyvalues of PYR–anion complexes show the same trend with LUMO ener-gy values of individual anions, but when combinedwith PYR, the LUMOenergy values decrease.
Complexes of PYR–ILs reportmoreHOMOenergy than LUMOenergybut with smaller values of HOMO and LUMO energy, as shown inFig. 4c). In complexes involving ILs, the LUMO energies aremore impor-tant since the LUMO sites lead to the overlapping of the incomingHOMO orbitals from the nitrogen compound molecule [14]. It is
rent parameters.
obal softness Electrophilicity Index Interaction energy
ighest to lowest) (Highest to lowest) (Most negative to least negative)
Y][EtSO4] [EPY][MeSO3] [TMPYRA][Ac]Y][Ac] [EPY][Ac] [EMIM][Ac]Y][MeSO3] [EPY][EtSO4] [EPY][EtSO4]PYRA][EtSO4] [EPY][MeSO3] [EMPIPE][EtSO4]PYRA][Ac] [TMPYRA][EtSO4] [TMPYRA][MeSO3]IM][Ac] [TMPYRA][MeSO3] [EMIM][EtSO4]PYRA][MeSO3] [EMM][MeSO3] [TMPYRA][EtSO4]MOR][MeSO3] [EMMOR][EtSO4] [EMMOR][MeSO3]IM][EtSO4] [EMPYRO][MeSO3] [EMPYRO][EtSO4]PYRO][EtSO4] [EMIM][Ac] [EMMOR][Ac]PIPE][EtSO4] [EMPIPE][Ac] [EPY][MeSO3]MOR][Ac] [EMPYRO][Ac] [EPY][Ac]PIPE][MeSO3] [EMPIPE][MeSO3] [EMIM][MeSO3]IM][MeSO3] [EMPYRO][EtSO4] [EMPIPE][Ac]PYRO][MeSO3] [EMIM][EtSO4] [EMPIPE][MeSO3]PIPE][Ac] [EMPIPE][EtSO4] [EMMOR][EtSO4]PYRO][Ac] [EMMOR][Ac] [EMPYRO][MeSO3]MOR][EtSO4] [EMMOR][MeSO3] [EMPYRO][Ac]

Fig. 5. a) Global hardness and softness for individual molecule. b) Global hardness andsoftness for PYR–cation and PYR–anion complexes. c) Global hardness and softness forPYR–IL complexes.
26 H.F. Hizaddin et al. / Journal of Molecular Liquids 194 (2014) 20–29
desirable to have the LUMO energies of the complexes in highernegative values indicative of favourable interaction. The complexPYR–[EPY][EtSO4] reports LUMO energy with the highest nega-tive value, thus it is considered the most favourable IL for separa-tion of pyrrole based on LUMO energy criterion. The ranking ofother ILs for separation of pyrrole based on this criterion isshown in Table 2.
4.2. HOMO–LUMO energy gap
The gap between HOMO and LUMO energy values for a singlemolecule measures the stability of a molecule through the lowest elec-tronic excitation energy (i.e. the lowest energy gap), while the HOMO–LUMO energy gap for pairs of molecules measures the reactivitythrough the thermochemical electronic hopping energy among themolecules involved [25]. The energy difference between the HOMOand LUMO corresponds to the lowest excitation. Thus, a small energygap indicates low stability in themolecules and explains its high reactiv-ity in chemical reactions.
Fig. 4a) shows HOMO–LUMO energy gap for individual molecules ofPYR, cations and anions. The energy gap for aromatic cations ([EMIM],[EPY] and [TMPYRA]) is smaller than those of saturated cations([EMMOR], [EMPIPE], [EMPYRO]) and their values are almost similarto each other (0.5033, 0.4441, 0.4715 Hartrees, respectively). Thus, itcan be deduced that aromatic cations are more reactive than saturatedcations. [EPY] reports the lowest HOMO–LUMOgapwhile [EMPYRO] re-ports the highest gap (0.6531 Hartrees). Among the anions, [Ac] has thesmallest HOMO–LUMO energy gap (0.5201 Hartrees) which indicatesthat it is the most reactive anions followed by [EtSO4] and [MeSO3](0.5417 and 0.5488 Hartrees, respectively). The energy gap for pyrroleis 0.4769 Hartrees.
Complexes of PYR with aromatic cations report smaller HOMO–LUMO gap than complexes with saturated cations, as shown in Fig. 4b).The energy gap for complexes with saturated cations however has littledifference among themselves (0.4397, 0.4302, and 0.4299 Hartreesfor complexes of PYR with [EMMOR], [EMPIPE], and [EMPYRO], respec-tively). The small energy gap for complexes of PYR with aromatic cationsindicates that PYR is more reactive with aromatic cations. On the otherhand, PYR–anions report similar values of HOMO–LUMO energy gaps,which could indicate that PYR is equivalently reactive with all of the an-ions investigated in this work. It is noted that smaller gaps are reportedfor PYR–cation complexes compared to PYR–anion complexes.
From Fig. 4c), it is observed that the HOMO–LUMO energy gaps forcomplexes of PYR–ILs are lower than those of neat ILs. This is consistentwith the results obtained by Wagle et al. [24], where it was reportedthat the values of energy gap for ILs with polyaromatic hydrocarbonsare lower than those of neat ILs. This indicates that potentially, all ILshave favourable interaction with pyrrole due to the lowering of the en-ergy gap. However, the complex which reports the smallest energy gapis considered as the most favourable combination for separation of pyr-role. On this basis, [EPY][EtSO4] is the best IL for pyrrole removal. Therank of other ILs based on this parameter is presented in Table 2.
4.3. Global hardness and global softness
Global hardness and softness are directly related to the HOMO–LUMO energy gap. By definition, hardness is indicative of a molecule'sability to be polarised while softness is indicative of its ability to acceptelectron. Fig. 5a)–c) represents the global hardness against global soft-ness for individual molecules and complexes. In general, the trendshows increasing global softness with decreasing global hardness. It isdesired to have maximum softness as this can lead to an extended mo-lecular interaction through hydrogen bond network or increasing the πdensity in aromatic compounds [14].
From Fig. 5a), it is observed that all molecules i.e. pyrrole, cations,anions, havemore softness than hardness. Aromatic cations have highersoftness than saturated cations, while the softness of anions is interme-diate between saturated and aromatic cations. [EPY] has the highestglobal softness among all the molecules (4.5036 Hartrees) while[EMPYRO] has least softness and the highest hardness. Fig. 5b) showsglobal hardness and global softness of complexes PYR–cations andPYR–anions. The overall trend is showing increased global softnesswhen cations and anions are combined with pyrrole. PYR-aromatic cat-ion complexes show higher softness than PYR-saturated cation

Fig. 6. a) Electronegativity vs. electrophilicity index for individual molecules.b) Electronegativity vs. electrophilicity index for PYR–cation and PYR–anioncomplexes. c) Electronegativity vs. electrophilicity index for PYR–IL complexes.
27H.F. Hizaddin et al. / Journal of Molecular Liquids 194 (2014) 20–29
complexes. This is consistent with earlier results where complexes ofPYR-aromatic cations have smaller HOMO–LUMO gaps than those ofPYR-saturated cations.
Similar trend was observed for PYR–IL complexes in Fig. 5c) wherepyrrole complexes with ILs containing aromatic cations possess higherglobal softness than complexes with ILs containing saturated cations.However, the order of neat ILs and ILs in pyrrole complexes differsbased on global softness. Results for neat ILs can be obtained from the
Supplementary material. For example, the IL with the highest globalsoftness is [EPY][Ac] while in Fig. 5c), the complex with highest globalsoftness is PYR–[EPY][EtSO4]. This difference in softness and hardnessindicates the importance of cation–anion combination for the removalof pyrrole. The most favourable ILs for removal of pyrrole would bethe oneswhose complexes show the largest softness. Based on the glob-al softness from the PYR–IL complexes, [EPY][EtSO4] is considered themost favourable IL for pyrrole removal. The rank of other ILs is shownin Table 2.
4.4. Electronegativity and electrophilicity index
As discussed in Section 2.1, electronegativity describes the ability ofa molecule to attract electrons to itself, while electrophilicity index is ameasure of a molecule's ability to saturate itself with as many electronsas possible. Fig. 6a)–c) shows the plot of electronegativity against elec-trophilicity index for individualmolecules and complexes. From Fig. 6a),cations have high electronegativity and electrophilicity index ascompared to anions. This is expected as it is generally known thatcations accept electrons and anions are electron donors. Higher electro-philicity index is indicative of a better electrophile and fromFig. 6a) we can see that the order of electrophilic cations is[EPY] N [TMPYRA] N [EMPYRO] N [EMIM] N [EMPIPE] N [EMMOR].As for anions, lower electrophilicity index indicates a better nucleophileand electron donor, and the order of nucleophilic anions is[Ac] N [MeSO3] N [EtSO4].
On the other hand, pyrrole has a small value in electronegativity andelectrophilicity index (0.008089 and 0.06212, respectively), which indi-cates its tendency to donate electron rather than receiving. Pyrrole hasa lower electrophilicity index than those of cations and anions. Similartrend is also observed for complexes of PYR–cations and PYR–anionsin Fig. 6b), where the effect of combining pyrrole with cations showsa reduced gap in electrophilicity index for the cations while for anions,combination with pyrrole reduces both the electronegativity andelectrophilicity index. For complexes of PYR–cations, highest globalelectrophilicity index is reported by the complex PYR–[EPY], followedby PYR–[TMPYRA] and PYR–[EMIM]. On the other hand, complexesPYR–[EMMOR], PYR–[EMPIPE] and PYR–[EMPYRO] report close valuesfor both electronegativity and electrophilicity index. For complexes ofPYR–anions, the trend is similar to that of individual anions in termsof nucleophilicity, i.e. PYR–[Ac] N PYR–[MeSO3] N PYR–[EtSO4].
Complexes of ILs and pyrrole show a decrease in electronegativi-ty and electrophilicity index, which indicates stabilisation of PYR–ILcomplexes as compared to IL complexes (which can be referred tofrom the Supplementary material). There is a large difference be-tween the electrophilicity index and electronegativity of [EPY]-based ILs and those of other ILs as there is a big gap shown in thevalues from Fig. 6c). Based on the values of electrophilicity indexwhere higher values indicate better preference of IL for pyrrole re-moval, [EPY][MeSO3] is the most favourable IL for this purpose.However, its value is close to those ILs with [EPY] cations, i.e. [EPY][Ac] and [EPY][EtSO4].
4.5. Interaction energies
Solubility of nitrogen compounds in ionic liquids is dependent uponthe distance between the cation and anion of the ionic liquids. The dis-tance between the ion pair is given by the interaction energy of the ILs.The distance between cation and anion is almost inversely proportionalto the calculated interaction energy due to domination of charge–charge interaction between the ion pair [26]. For example, Zhang et al.[13] evaluated the performance of ILs 1-butyl-3-methylimidazoliumtetrafluoroborate ([BMIM][BF4]) and 1-methyl-3-methylimidazoliumhexafluorophosphate ([BMIM][PF6]) for separation of thiophene, andit was found that [BMIM][PF6] has higher solubility of thiophene than[BMIM][BF4]. It was concluded that the compactness of the IL plays an

-450.00
-400.00
-350.00
-300.00
-250.00
-200.00
-150.00
-100.00
-50.00
0.00
PYR- ILComplex
Neat IL[E
MIM
][Ac]
[EM
IM][E
tSO
4]
[EM
IM][M
eSO
3]
[EM
MO
R][A
c]
[EM
MO
R][E
tSO
4]
[EM
MO
R][M
eSO
3]
[EM
PIP
E][A
c]
[EM
PIP
E][E
tSO
4]
[EM
PIP
E][M
eSO
3]
[EM
PY
RO
][Ac]
[EM
PY
RO
][EtS
O4]
[EM
PY
RO
][MeS
O3]
[EP
Y][A
c]
[EP
Y][E
tSO
4]
[EP
Y][M
eSO
3]
[TM
PY
RA
][Ac]
[TM
PY
RA
][EtS
O4]
[TM
PY
RA
][MeS
O3]
Inte
ract
ion
En
erg
y (k
J/m
ol)
Fig. 7. Interaction energy of the ionic liquids and their complexes with pyrrole.
Table 3Interaction energies for neat ionic liquids and their complexes with pyrrole and partialcharges of nitrogen atom of pyrrole in complexes. Rows in bold indicates a decrease inpartial charge of N-atom in pyrrole.
IL Interaction energy(kJ/mol)
Partial charge ofN-atom in pyrrole
[EMIM][Ac] −389.80 N/A[EMIM][EtSO4] −379.77 N/A[EMIM][MeSO3] −396.64 N/A[EMMOR][Ac] −429.23 N/A[EMMOR][EtSO4] −371.91 N/A[EMMOR][MeSO3] −396.03 N/A[EMPIPE][Ac] −417.04 N/A[EMPIPE][EtSO4] −361.28 N/A[EMPIPE][MeSO3] −379.42 N/A[EMPYRO][Ac] −419.52 N/A[EMPYRO][EtSO4] −372.46 N/A[EMPYRO][MeSO3] −396.49[EPY][Ac] −415.90 N/A[EPY][EtSO4] −359.32 N/A[EPY][MeSO3] −382.74 N/A[TMPYRA][Ac] −393.94 N/A[TMPYRA][EtSO4] −372.15 N/A[TMPYRA][MeSO3] −396.50 N/APYR–[EMIM][Ac] −64.11 −0.6280PYR–[EMIM][EtSO4] −58.46 −0.6153PYR–[EMIM][MeSO3] −13.53 −0.6170PYR–[EMMOR][Ac] −37.80 −0.6242PYR–[EMMOR][EtSO4] −1.78 −0.6126PYR–[EMMOR][MeSO3] −45.29 −0.6279PYR–[EMPIPE][Ac] −12.17 −0.6177PYR–[EMPIPE][EtSO4] −60.38 −0.6210PYR–[EMPIPE][MeSO3] −4.75 −0.6101PYR–[EMPYRO][Ac] −1.40 −0.6161PYR–[EMPYRO][EtSO4] −38.76 −0.6220PYR–[EMPYRO][MeSO3] −1.71 −0.6169PYR–[EPY][Ac] −16.80 −0.6161PYR–[EPY][EtSO4] −61.46 −0.6196PYR–[EPY][MeSO3] −16.93 −0.6133PYR–[TMPYRA][Ac] −77.05 −0.6289PYR–[TMPYRA][EtSO4] −47.82 −0.6251PYR–[TMPYRA][MeSO3] −60.34 −0.6237
28 H.F. Hizaddin et al. / Journal of Molecular Liquids 194 (2014) 20–29
important role, where [BMIM][PF6] is less compact and has smaller in-teraction energy as compared to [BMIM][BF4], and this leads to facilerestructuring of the cation and anion to accommodate thiophenemolecules.
Fig. 7a) shows the calculated interaction energy of ILs sorted accord-ing values. Molecular interaction between the cation and anion in an ILis mostly attributed to electrostatic interaction and hydrogen bond in-teraction via CH (cation)–n (anion) where n is the anion heteroatom(i.e. S, O, or N in this case) [27]. The numerical values can be referredto in Table 3. [EtSO4]-based ILs report the lowest interaction energycompared to [MeSO3]- and [Ac]-based ILs. Stronger interaction energyfor the ILs is reported for [Ac]-based ILs. Considering the cation factor,it is observed that for a specific anion, ILs based on aromatic cationhave smaller interaction energy than those with saturated cations.Aromatic cations have smaller electrostatic strength due to chargedelocalisation in aromatic rings, while saturated cations have largerelectrostatic strength due to localised charge on the heteroatom of thesaturated rings, thus aromatic-based ILs have lower interaction strengthvalues compared to saturated-based ILs [28]. In addition, increase in cat-ion size from five-membered to six-membered ring (for both aromaticand saturated cores) leads to increase in the distance between thecharges and thus decrease in interaction energy [28]. This explainswhy [EPY]-based ILs have smaller interaction energy than [EMIM]-and [TMPYRA]-based ILs among the aromatic ringed cations. Anions in-fluences the interaction strength in the ion pair where ILs with smalland higher symmetrical anion have low anion configurational variationand thus higher connectivity with the cations and results in relativelystronger interactions than ILs with larger and less symmetrical anion[29]. The sizes of anions in this study follow the order from smallest tolargest: [Ac] b [MeSO3] b [EtSO4]. Consequently, ILs made of six-membered aromatic cations combined with large anion are expectedto have smaller interaction energy and may allow facile restructuringof the ion pairs to accommodate incoming molecules.
Therefore, ILs with smaller interaction energy is expected to pro-vide better capacity for solute extraction. Nevertheless, for com-plexes with ILs and the aromatic solute, it is preferable to have

29H.F. Hizaddin et al. / Journal of Molecular Liquids 194 (2014) 20–29
higher interaction energy between the ILs and the solute whichdemonstrate that extra amount of energy is required to disassemblethe complex [15]. Fig. 7 and Table 3 show the interaction energy ofcomplexes PYR–ILs. It can be seen that in general, complexes witharomatic cations have stronger interaction energy than those withsaturated cations. On the basis of choosing an IL with strongest inter-action energy with pyrrole, [TMPYRA][Ac] is considered as the mostfavourable IL.
4.6. Charge distribution
For favourable interaction between pyrrole and ionic liquids, pyrrolemolecule should be able to form cation–π or hydrogen bond by over-coming the cation–anion interaction. To form an efficient CH–π interac-tion, the aromatic π density needs to be increased. A decrease in thepartial charge of the nitrogen atom in the complex versus its charge inindividual pyrrole indicates a better cation–π interaction as thedecreasein partial charge will cause the density of the π system to increase. Thepartial charge of nitrogen atom in individual pyrrole and in pyrrolemol-ecule when in complexes of ILs is obtained from NBO analysis andshown in Table 3. The partial charge of N in PYR is −0.6129. Thus, thepartial charge of N atom of pyrrole in the complex should be less than−0.6129 (in magnitude) to indicate favourable interaction betweenpyrrole and the ILs. Unfortunately, based on the results in Table 3,only two ILs show a decrease in the partial charge of N atom in pyrrole,which are [EMMOR][EtSO4] and [EMPIPE][MeSO3]. Between these twoILs, [EMPIPE][MeSO3] is the more favourable IL due to a larger decreasein the charge of N-atom in pyrrole.
4.7. Combination of parameters
The quantum chemical descriptors studied in this work cannot berelated and compared to one another because of the different definitionand criteria they have at quantum level. Nevertheless, they can providea guidance to select themost suitable IL for removal of pyrrole based onthe criteria of each parameter. For instance, based on HOMO–LUMO en-ergy gap, the ILs can be ranked according to the smallest values whichindicate higher softness and polarizability. Table 2 shows the rankingof all ILs based on each parameter. It is observed that [EPY][EtSO4] dom-inated the overall rank. The result which indicates [EPY]-based IL as themost favourable IL for removal of pyrrole is also supported by experi-mental work by earlier publications [11,30]. Pyridinium-based ILs areproven to have better extraction capacity for aromatic sulphur and ni-trogen compounds than imidazolium-based ILs with the same anion[11,30]. Pyridinium cation possesses a pronounce π system and this fa-cilitate interactionwith aromatic compounds through cation–π andπ–πinteraction [31]. Moreover, the IL [EPY][EtSO4] have among the smallestinteraction energy for the ILs. As discussed above, ILswith small interac-tion energy have lower packing and may allow for incoming moleculesto accommodate between the large distance of cation and anion thusbetter affinity and capacity for extraction.
5. Conclusion
Geometry optimisation at Hartree–Fock level has been used to de-termine thequantum chemical descriptors to investigate the interactionbetween pyrrole and 18 ionic liquids. Parameters such as HOMO/LUMOenergy, HOMO–LUMO energy gap, global hardness and softness, elec-trophilicity index, interaction energy and partial charges are used to de-termine themost favourable IL for interactionwith pyrrole as themodelnon-basic aromatic nitrogen compound. From theHOMO/LUMO energyvalues of PYR–IL complexes, themost favourable IL is [EPY][EtSO4] sinceit has the smallest LUMO energy value. HOMO–LUMO energy gapamong the ILs have small variation in values. However, when combined
with PYR in complexes, there variation is more apparent and [EPY][EtSO4] is found to be the most favourable IL for pyrrole removalbased on its smallest HOMO–LUMO energy gap. Since global softnesshas direct correlation with HOMO–LUMO gap, [EPY][EtSO4] also topsthe rank for the most favourable IL based on its largest global softnessvalue. In terms of electrophilicity which measures the ability of a mole-cule or complex to saturate itself with electrons, [EPY][MeSO3] makesthe most favourable IL for pyrrole removal. Interaction energies be-tween pyrrole and ILs show small variations in values, but from thevalues, [TMPYRA][Ac] shows the most negative interaction energywith pyrrole which indicates the most favourable interaction. As forthe partial charges parameter, only two ILs show favourable interactionwith pyrrolewhich is indicated by thedecrease in the charge of nitrogenatom in pyrrole when in PYR–IL complex, and these are [EMMOR][EtSO4] and [EMPIPE][MeSO3], with [EMPIPE][MeSO3] being the morefavourable IL. Overall, [EPY][EtSO4] is chosen as the most favourableIL for pyrrole removal and this is also supported by published exper-imental work which reports the performance of pyridinium-basedIL.
Acknowledgement
This research was carried with funding from the University ofMalaya under the HIR grant number HIR-MOHE (D000003-16001).
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.molliq.2013.12.041.
References
[1] G.C. Laredo, E.n. Altamirano, J.A. De los Reyes, Appl. Catal. A (2003) 207–214.[2] G.C. Laredo, J.A. De los Reye, J. Luis Cano, J. Jesús Castillo, Appl. Catal. A (2001)
103–112.[3] U.T. Turaga, X. Ma, C. Song, Catal. Today (2003) 265–275.[4] T. Welton, Chem. Rev. (1999) 2071–2084.[5] K. Ke ̧dra-Królik, M. Fabrice, J.-N.l. Jaubert, Ind. Eng. Chem. Res. 50 (2011)
2296–2306.[6] K. Kedra-Krolik, F. Mutelet, J.-C. Moise, J.-N.l. Jaubert, Energy Fuels (2011)
1559–1565.[7] J. Zhang, C. Huang, B. Chen, P. Ren, Z. Lei, Energy Fuels (2007) 1724–1730.[8] L. Alonso, A. Arce, M.a. Francisco, A. Soto, J. Chem. Eng. Data (2010) 3262–3267.[9] L.-L. Xie, A. Favre-Reguillon, S. Pellet-Rostaing, X.-X. Wang, X. Fu, J. Estager, M.
Vrinat, M. Lemaire, Ind. Eng. Chem. Res. (2008) 8801–8807.[10] L.-L. Xie, A. Favre-Reguillon, X.-X. Wang, X. Fu, S. Pellet-Rostaing, G. Toussaint, C.
Geantet, M. Vrinat, M. Lemaire, Green Chem. (2008) 524–531.[11] A.R. Hansmeier, G.W. Meindersma, A.B. de Haan, Green Chem. (2011)
1907–1913.[12] C. Asumana, G. Yu, Y. Guan, S. Yang, S. Zhou, X. Chen, Green Chem. (2011) 3300–3305.[13] J. Zhou, J. Mao, S. Zhang, Fuel Process. Technol. (2008) 1456–1460.[14] R. Anantharaj, T. Banerjee, Fluid Phase Equilib. (2010) 22–31.[15] R. Anantharaj, T. Banerjee, AIChE J (2011) 749–764.[16] R. Lü, Z. Qu, H. Yu, F. Wang, S. Wang, Chem. Phys. Lett. (2012) 13–18.[17] C.-G. Zhan, J.A. Nichols, D.A. Dixon, J. Phys. Chem. A (2003) 4184–4195.[18] R.G. Pearson, Proc. Natl. Acad. Sci. U. S. A. (1986) 8440–8441.[19] R.G. Parr, L.v. Szentpály, S. Liu, J. Am. Chem. Soc. (1999) 1922–1924.[20] R. Lü, Z. Qu, J. Lin, J. Mol. Liq. (2013) 207–214.[21] K. Fumino, R. Ludwig, J. Mol. Liq. (2013), http://dx.doi.org/10.1016/j.
molliq.2013.07.009.[22] TURBOMOLE Users' Manual2012.[23] T. Nishi, T. Iwahashi, H. Yamane, Y. Ouchi, K. Kanai, K. Seki, Chem. Phys. Lett. (2008)
213–217.[24] D. Wagle, G. Kamath, G.A. Baker, J. Phys. Chem. C (2013) 4521–4532.[25] J.-M. Martínez-Magadán, R. Oviedo-Roa, P. García, R. Martínez-Palou, Fuel Process.
Technol. (2012) 24–29.[26] S. Tsuzuki, H. Tokuda, M. Mikami, Phys. Chem. Chem. Phys. (2007) 4780–
4784.[27] N.R. Dhumal, H.J. Kim, J. Kiefer, J. Phys. Chem. A 113 (2009) 10397–10404.[28] A.M. Fernandes, M.A.A. Rocha, M.G. Freire, I.M. Marrucho, J.o.A.P. Coutinho,
L.s.M.N.B.F. Santos, J. Phys. Chem. B (2011) 4033–4041.[29] P.A. Hunt, I.R. Gould, B. Kirchner, Aust. J. Chem. (2007) 9–14.[30] V.R. Ferro, E. Ruiz, J. de Riva, J. Palomar, Sep. Purif. Technol. (2012) 195–204.[31] S. Tsuzuki, M. Mikami, S. Yamada, J. Am. Chem. Soc. (2007) 8656–8662.
Copyright © 2022 FDOKUMEN




















