
A new environmentally benign technology for transforming wood pulp into paper. Engineering...
26
ELSEVIER Journal of Molecular Catalysis A: Chemical 116 (1997) 59-84 JOURNALOF MOLECULAR CATALYSIS A: CHEMICAL A new environmentally benign technology pulp into paper Engineering polyoxometalates as catalysts for transforming wood for multiple processes Ira A. Weinstock a,* , Rajai H. Atalla a , Richard S. Reiner a , Mark A. Moen a , Kenneth E. Hammel a , Carl J. Houtman b , Craig L. Hill * c , Mason K. Harrup c a USDA Forest Sercice, Forest Products Laboratory, One Gifford Pinchot Drive, Madison, WI 53705, USA b Department of Chemical Engineering, University of Wisconsin-Madison, Madison, WI 53706, USA c Department of Chemistry, Emory University, Atlanta, GA 30322, USA Received 4 January 1996; accepted 28 February 1996 Abstract A new environmentally benign technology, based on the use of polyoxometalate (POM) salts and oxygen, is being developed to bleach wood pulps for use in the manufacture of paper. Details of POM chemistry relating to the unit operations of an effluent-free bleaching process are reported. These include anaerobic delignification of wood pulp, aerobic reoxidation of reduced POMs for their cyclic reuse and POM catalyzed and initiated aerobic mineralization (wet oxidation) of lignin fragments removed from pulp during bleaching. The results of bleaching trials using a series of isostructural POM complexes of the Keggin family are reported. Key structural components of the POMs are varied to determine the effects of these changes on POM performance. Homogeneous reactions of lignin-model compounds with select POMs are used to help interpret kinetic data obtained in the heterogeneous reaction of POM solutions with pulp fibers. Finally, new directions in catalyst design that promise to expand the potential of the technology are discussed. Keywords: Polyoxometalates; Oxygen; Wood pulp; Paper; Oxidative bleaching; Oxidative delignification 1. Introduction Some unique characteristics of the polyox- ometalates (POMs) have made possible their use as the cornerstone of a new technology for the delignification of wood and wood pulp fibers. This technology promises both environ- mental and economic advantages over current practice in the industry. The Achilles heal of most processes currently in use is their rather * Corresponding authors. limited selectivity with respect to delignifica- tion. The most desirable condition is that a process remove the lignin, which is the primary encrustant of cell wall matter in higher plants, with little or no damage to the polysaccharides, particularly the cellulose, which is the primary substance of the cell wall and the key con- stituent of the natural fibers from which paper is made. Most systems for delignification become quite aggressive, particularly as the level of lignin declines, so that delignification to desired levels 1381-1169/97/$17.00 Copyright 1997 Elsevier Science B.V. All rights reserved. PII S 1 3 8 1-1 1 6 9 (9 6 ) 0 0 0 7 4 - X
-
Upload
independent -
Category
Documents
-
view
7 -
download
0
Transcript of A new environmentally benign technology for transforming wood pulp into paper. Engineering...

ELSEVIER Journal of Molecular Catalysis A: Chemical 116 (1997) 59-84
JOURNALOFMOLECULARCATALYSISA: CHEMICAL
A new environmentally benign technologypulp into paper
Engineering polyoxometalates as catalysts
for transforming wood
for multiple processes
Ira A. Weinstock a,*, Rajai H. Atalla a, Richard S. Reiner a, Mark A. Moen a,Kenneth E. Hammel a, Carl J. Houtman b, Craig L. Hill * c, Mason K. Harrup c
a USDA Forest Sercice, Forest Products Laboratory, One Gifford Pinchot Drive, Madison, WI 53705, USAb Department of Chemical Engineering, University of Wisconsin-Madison, Madison, WI 53706, USA
c Department of Chemistry, Emory University, Atlanta, GA 30322, USA
Received 4 January 1996; accepted 28 February 1996
Abstract
A new environmentally benign technology, based on the use of polyoxometalate (POM) salts and oxygen, is beingdeveloped to bleach wood pulps for use in the manufacture of paper. Details of POM chemistry relating to the unitoperations of an effluent-free bleaching process are reported. These include anaerobic delignification of wood pulp, aerobicreoxidation of reduced POMs for their cyclic reuse and POM catalyzed and initiated aerobic mineralization (wet oxidation)of lignin fragments removed from pulp during bleaching. The results of bleaching trials using a series of isostructural POMcomplexes of the Keggin family are reported. Key structural components of the POMs are varied to determine the effects ofthese changes on POM performance. Homogeneous reactions of lignin-model compounds with select POMs are used to helpinterpret kinetic data obtained in the heterogeneous reaction of POM solutions with pulp fibers. Finally, new directions incatalyst design that promise to expand the potential of the technology are discussed.
Keywords: Polyoxometalates; Oxygen; Wood pulp; Paper; Oxidative bleaching; Oxidative delignification
1. Introduction
Some unique characteristics of the polyox-ometalates (POMs) have made possible theiruse as the cornerstone of a new technology forthe delignification of wood and wood pulpfibers. This technology promises both environ-mental and economic advantages over currentpractice in the industry. The Achilles heal ofmost processes currently in use is their rather
* Corresponding authors.
limited selectivity with respect to delignifica-tion. The most desirable condition is that aprocess remove the lignin, which is the primaryencrustant of cell wall matter in higher plants,with little or no damage to the polysaccharides,particularly the cellulose, which is the primarysubstance of the cell wall and the key con-stituent of the natural fibers from which paper ismade.
Most systems for delignification become quiteaggressive, particularly as the level of lignindeclines, so that delignification to desired levels
1381-1169/97/$17.00 Copyright 1997 Elsevier Science B.V. All rights reserved.PII S 1 3 8 1-1 1 6 9 (9 6 ) 0 0 0 7 4 - X

60 I.A. Weinstock et al. /Journal of Molecular Catalysis A: Chemical 116 (1997) 59-84
frequently carries the penalty of some degrada-tion in fiber properties. The goal of the programbased on POMs has been to focus on theirapplication under conditions wherein a far higherlevel of selectivity is achieved. A key addedbenefit is that the same POMs can be used in acomplementary process to catalyze the wet oxi-dation of the organic byproducts of the deligni-fication stage.
In this report we provide an account of thedevelopment of the POM delignification tech-nology. We begin with some background infor-mation on wood, pulping and bleaching technol-ogy, and the lines of inquiry that led to identifi-cation of POMs as the promising candidatereagents for this work. We then describe ex-ploratory bleaching and related studies carriedout with particular POMs and ongoing develop-mental work. Finally, we consider new direc-tions in catalyst design that promise to expandthe potential of the technology.
2. Background
2.1. Wood
In the context of utilization technologies,wood is regarded as a material or process feed-stock. In fact, it is a biological tissue that ishighly organized at the cellular and molecularlevels [1]. Its structural components consist pri-
marily of polysaccharides (cellulose and hemi-celluloses) and lignin. Lignin, which constitutesroughly 20–35% by weight of most woods, is ahighly cross-linked hydroxylated and methoxyl-ated phenylpropane polymer formed by oxida-tive coupling of para-hydroxycinnamyl alco-hols biochemically derived from the amino acidphenylalanine (Fig. 1 and Fig. 2) [2,3]. Celldivision and growth are limited to the cambiumlayer at the outer circumference of a tree. Uponmaturation, wood cells die, leaving behind rigidcell walls constructed of lamellar layers of poly-meric polysaccharides. Lignin is found betweencells and within the cell walls, providing bothresistance to biological attack and structural

I.A. Weinstock et al./Journal of Molecular Catalysis A: Chemical 116 (1997) 59-84 61
rigidity. In the manufacture of paper, individualwood cells, or fibers, are separated from oneanother in a pulping process and then bleached.The bleached wood pulps are slurried in waterand formed into thin sheets.
2.2. Pulp
Pulps can be made in two ways, either bymechanical separation of wood fibers (mechani-cal pulps) or by chemical removal of lignin(chemical pulps) [4]. The amount of lignin re-maining in the pulp affects both the physicalproperties, such as strength and flexibility, andthe chemical stability of the finished paper.Mechanical (high yield) pulping results in somefiber damage and a high lignin content. Suchpulps are bleached using oxidative or reductiveagents that remove chromophoric groups, butleave much of the lignin in the fibers, and areused for the manufacture of newsprint and lowerquality paper. The ‘yellowing’ that occurs uponexposure of these products to sunlight arisesfrom photochemical changes in the lignin com-ponent of the paper.
Chemical pulps are used for the manufactureof high strength paper products, such as card-board, and for high quality printing and writingpapers. During chemical pulping, most (but notall) of the lignin is removed. A high proportionof chemical pulps produced in the United Statesand in much of Europe are manufactured fromconifers, such as pine, using the kraft process.Kraft pulping, which entails cooking wood chipsat high temperature (ea. 170°C) in an aqueousliquor consisting of sodium hydroxide and sul-fide, removes roughly 80–90% of the originallignin by nucleophilic cleavage and concomitantfragmentation and solubilization of the polymer.The remaining lignin (residual kraft lignin) isstructurally distinct from native lignin. Whilepossessing structures already present in nativelignin, it is enriched in condensed structures thateither survive the kraft cook (biphenyl struc-tures) or arise from it (diarylmethane structures,Fig. 3) [5]. Approximately 20–40% of the phenyl
rings in residual kraft lignin are hydroxylated,as opposed to roughly 15% in native lignin [6].In addition, some highly conjugated structuresare generated, which, even at low concentra-tions, impart a dark brown color to the kraftpulp. Brown paper bags, familiar in the UnitedStates, and corrugated cardboard boxes familiarworldwide, are usually made directly from un-bleached kraft pulp. However, before this pulpcan be used in the manufacture of high quality,thermally and photochemically stable, whiteprinting and writing papers, it must first bebleached. The purpose of bleaching is to chemi-cally remove residual kraft lignin (delignifica-tion) and chromophores (brightening).
2.3. Oxidative bleaching
Delignification has traditionally been accom-plished using aqueous molecular chlorine at pH1.5–3.0 [7]. The chlorination stage (identifiedby the letter ‘C’) is the first step in a multi-stageprocess denoted CEDED where C refers tomolecular chlorine, E refers to extraction withalkali and D refers to treatment with chlorinedioxide. The D stages are where brighteningoccurs. During the C stage, electrophilic aro-matic substitution of phenyl rings in the ligninpolymer generates chloroaromatic by-productswhich are difficult to degrade effectively in thebiological waste-treatment lagoons located atmill sites [8]. For this reason, the industry inmost developed countries has moved away fromthe use of elemental chlorine and has replaced itwith a combination of extended delignificationduring kraft pulping, alkaline oxygen pretreat-

62 I.A. Weinstock et al./Journal of Molecular Catalysis A: Chemical 116 (1997) 59–84
ment prior to bleaching and chlorine dioxide inplace of elemental chlorine. Chlorine dioxide,though more costly and hazardous than chlo-rine, can also be used for delignification. Al-though it generates far fewer chlorocarbon by-products than does Cl2, some, however, are stillproduced [9].
The most attractive alternatives to chlorineand chlorine dioxide, with respect to both envi-ronmental impact and cost, are oxygen, hydro-gen peroxide and ozone. The limiting issue inindustrial use of these chemicals is often theirintrinsic or operational (process) selectivities forlignin (vs. cellulose); high selectivity is crucialto retaining the strength properties of pulp fibers.Oxygen is the oxidant of first choice based oncost and environmental concerns. It is effectiveat depolymerizing and solubilizing lignin whenapplied under mild pressure to alkaline, aqueouspulp slurries. However, because a variety ofuncontrolled autoxidation reactions occur, theprocess is not sufficiently selective to removemore than half of the residual lignin from kraftpulps [10]. Alkaline hydrogen peroxide, while itreadily and selectively degrades chromophoricgroups such as the many quinonoid compoundspresent in residual kraft lignin, does not delig-nify effectively [8]. More selective delignifica-tion is obtained using organic or inorganicderivatives such as performic or peracetic acids[8] or dioxiranes [10], persulfates [11], or per-oxo-metalates [12], each of which is more ex-pensive than hydrogen peroxide itself. In addi-tion, the use of peroxide compounds requiresthe prior removal or in-situ chelation of metalions that might otherwise lead to rapid loss ofactive oxidant via decomposition reactionswhich often proceed via unselective oxygen rad-ical intermediates that readily degrade pulpfibers [13]. Ozone is very effective at degradingaromatic and other conjugated structures. Al-though the β-D-glucose units in cellulose reactreadily with ozone, the oxidant’s relative rate ofreaction with lignin is substantially greater thanthat with cellulose and, in principle, high selec-tivity is possible [10]. Mass-transfer limitations,
however, imposed by the need to deliver ozoneto the dilute concentrations of lignin presentwithin primarily cellulosic fiber walls, makehigh process selectivity difficult to achieve con-sistently on an industrial scale [14].
Ideally, a means might be found to controlthe action of dioxygen such that it could be usedwith greater selectivity. A good example is thatprovided by the enzymatic processes associatedwith the depolymerization of native lignin bywood-rotting fungi, the only micro-organismscapable of degrading this polymer. A number ofextracellular metalloenzymes are involved. Lac-case, a copper containing enzyme oxidizes phe-nolic moieties in lignin (no mono- or dioxyge-nase activity is reported) and reduces dioxygendirectly to water [15]. In addition, two ligni-nolytic peroxidase systems have been described,lignin peroxidase [16] and manganese peroxi-dase [17]. The hydrogen peroxide needed bythese enzymes is produced from simple aldehy-des and dioxygen by a second copper containingenzyme, glyoxal oxidase [18]. When oxidized,lignin peroxidase, a protoheme containing en-zyme, oxidizes phenolic groups and, more sig-nificantly, generates radical cations from a widevariety of non-hydroxylated phenyl structures.The second protoheme containing peroxidase,manganese peroxidase, oxidizes MnII to MnIII
which is stabilized at or near the enzyme activesite by complexation with α -hydroxy acids.Chelated MnIII ions penetrate the wood-cell walland oxidize phenolic groups within the lignin.These systems demonstrate that delignificationcan be brought about by non-substrate specific,transition-metal catalyzed oxidation reactions.This observation suggests a role for molecularcatalysis in the search for new, environmentallybenign and economically viable bleaching tech-nologies.
In general, however, the behavior and reac-tivity of transition-metal ions in water are diffi-cult to control [19]. Complex equilibria are es-tablished between ionic oxides, hydroxides andhydrates, as well as between accessible oxida-tion states of the metal ions, each of which may

I.A. Weinstock et al./Journal of Molecular Catalysis A: Chemical 116 (1997) 59-84 63
react in unique ways with substrates or oxi-dants. In addition, many transition-metal oxidesand hydroxides have limited solubilities in wa-ter and the metal ions are rapidly lost fromsolution as solid precipitates. With the fungalperoxidases as a model, biomimetic bleachingsystems have been developed using halo-genated, water-soluble metalloporphyrin com-plexes [20]. While elegant and relatively robuststructural mimics of lignin peroxidases, thesesynthetic materials are expensive, require theuse of expensive organic or inorganic peroxidesand are inherently susceptible to oxidativedegradation during bleaching. An alternative ap-proach is to control the behavior and reactivityof redox-active metal ions by incorporating theminto the structures of polyoxometalate anions.
2.4. Polyoxometalates
Early transition metal oxygen anion clusters,or polyoxometalates (POMs for short) are alarge, structurally diverse and rapidly growingclass of inorganic clusters. They are composedof d0 metal cations, and in particular WVI,MO
VI, and VV, in varying combinations, andoxide anions held together with metal oxygenbonds [21–31]. The principal building blocks ofPOMs are MO x, x = 4–6, polyhedra and usu-ally MO6 octahedra that are linked together by
one, two, and occasionally three oxygen atoms.There are two generic classes of POMs, theisopolyanions, that can contain only the d0 metalcations and oxide anions (general formula:MxOJ -), and the heteropolyanions, that containone or more d or p block ‘heteroatom’ cations,X n +, in addition to the metal cations and oxideanions (general formula: Xa M~O~- ). The het-eropolyanions are the focus of the research elab-orated in this paper as they are a larger, moreversatile, and more easily modified class ofPOMs than the isopolyanions [21]. The mostcommon and most thoroughly investigated classof heteropolyanions are the Keggin structures(Fig. 4). While POMs can range in size from 9A (0.9 nm) to over 30 Å (3 nm), the Kegginstructure has a diameter of ca. 1.1 nm, similar tothat of typical phenylpropane units in lignin.The negative charge of a POM can be counter-balanced by hydrophilic cations, such as Li+,N a+, K+, and NH~, that render the complexsoluble in H2O, or by hydrophobic cations, suchas Et4N
+ or Ph4P+, that render the complex
soluble in organic solvents and other hydropho-bic media.
Several attributes make POMs attractive ingeneral and other attributes make them attrac-tive for the bleaching and processing of woodpulp in particular. First, POMs are readily pre-pared, often in a single step in H2O from

64 I.A. Weinstock et al./Journal of Molecular Catalysis A: Chemical 116 (1997) 59-84
inexpensive, minimally toxic, and accessiblecompounds such as WO/’, MoO; -, and PO:-[21 -31]. Second, most of the key physical prop-erties of POMs that control their fundamentalreactivities including redox potentials, acidities,charges, solubilities, etc. can be controlled to amarked degree by choice of precursors andconditions. It is doubtful if another class ofcompounds exists whose properties can be soextensively and so readily altered. Third, and apoint of particular pertinence to the title tech-nology, POMs are simultaneously resistant tooxidation (such do systems as glass, some ce-ramics and other inorganic materials are alreadyin the maximum oxidation state achievable un-der any conventional reaction conditions), yetmany families of POMs, including Kegginderivatives, can be reversibly reduced, often bymany electrons. Finally, one or more of the dometal ions in the parent POM structure can bereplaced by other do metal ions or by d-elec-tron-containing metal ions. Indeed, the profoundcontrol of the chemically significant propertiesof POMs enumerated above vests, in part, inthis rich substitution chemistry. These metal-ionsubstituted POMs can remove electrons fromorganic substrates such as lignin and facilitatetheir transfer to O2. As such, they perform thesame function as do the extracellular enzymesystems of wood-decay fungi. The POMs ad-dressed in the initial research elaborated in thispaper are Keggin derivatives with Pv, SiIV, orB III heteroatoms and with one or more WVI orMO
VI ions substituted with V v (d0) or d-elec-tron-containing (e.g. MnIII) ions.
3. POM bleaching
The POM bleaching processveloped at the US DepartmentForest Service, Forest Products
now being de-of AgricultureLaboratory, in
collaboration with the Emory University, De-partment of Chemistry and the University ofWisconsin–Madison, Department of ChemicalEngineering, uses a highly selective and com-
pletely reusable POM bleaching agent [32-40].In bleaching, fully oxidized POM complexesare reacted with unbleached pulp under anaero-bic conditions. During bleaching, the POMcomplexes are reduced and oxidized lignin frag-ments dissolved by the bleaching liquor. Be-cause the POM bleaching complexes are re-versible, and thus reusable, oxidants, designedto circulate continuously in the mill, the build-upof dissolved organic compounds must beavoided. This is accomplished by deoxidizing(reactivating) the reduced bleaching liquors withdioxygen under conditions that will convert thedissolved lignin fragments to carbon dioxideand water [33,36–40]. Thus, an effluent-free(closed mill) bleaching process, a stated goal ofthe pulp and paper industry, will be achieved[41].
3.1. Closed-mill POM bleaching process
As currently envisioned, an effluent-free(closed) POM bleaching mill will consist offour Unit Operations: A, anaerobic bleaching;B, pulp washing; C, concentration of wash wa-ter and removal of undesired inorganic salts;and D, aerobic, POM catalyzed and/or initi-ated, wet oxidation of dissolved lignin frag-ments with simultaneous regeneration of thePOM to its bleaching-active form (Fig. 5).
In anaerobic bleaching, (unit operation A,Fig. 5) the POMs are dramatically more selec-

I.A. Weinstock et al./Journal of Molecular Catalysis A: Chemical 116 (1997) 59-84 65
tive than molecular oxygen and yet, many havethe attractive property that after use in bleach-ing, the now reduced POMs are thermodynami-cally capable of being deoxidized by oxygen(unit operation D). Bleaching and the regenera-tion of used bleaching liquors are each carriedout in separate steps. In the bleaching step(represented in Eq. (l)) mixtures of water, pulpand a fully oxidized polyoxometalate (POMox)are heated anaerobically. During the reaction,the POM is reduced (POMred) as residual ligninis oxidized. After the reaction, the spent bleach-ing liquor is separated from the pulp and reoxi-dized using oxygen as represented by Eq. (2).By separating aerobic reoxidation of POMred
from anaerobic bleaching, oxygen can be uti-lized as a terminal oxidant while avoiding expo-sure of the pulp to non-specific oxygen radicals.Because the POMs only serve in a mediatingrole for oxygen, water is the only by-product ofthe bleaching chemicals.
pulp + POMox ~ bleached pulp + POMred
(1)
P O Mred + O2 + 4H+ ~ P O Mox + 2H2O (2)
After leaving the bleaching reactor, the pulpis washed (unit operation B, Fig. 5). Becausethe POMs are not adsorbed onto pulp fibers,their removal is controlled by diffusion phe-nomena alone. As a result, the POMs are easilywashed out of the pulp. Industrially, a series ofcountercurrent washers would be used. Washwater would be recycled by evaporation usingheat provided by low grade steam, which isoften abundantly available in pulp mills. Afterconcentration, a small spent liquor stream maybe diverted so that inorganic salts carried inwith the pulp can be removed using cationexchange or crystallization (unit operation C). Itis anticipated that some POM will be removedat this or a separate point and re-refined at a ratedictated by its operational half-life.
The spent liquor is then sent to unit operationD (Fig. 5), the purpose of which is two-fold: tooxidatively degrade dissolved lignin fragments
to volatile organic materials, carbon dioxide andwater (wet oxidation) and to return the POM toits active form (Eq. (2)). Although the POMsact with high selectivity in the anaerobic bleach-ing reaction with pulp, the conditions in the wetoxidation unit will be significantly more aggres-sive, including higher temperatures and the in-troduction of oxygen. Under these conditions,the POMs act both as catalysts and initiators inthe aerobic oxidation and autoxidation of dis-solved organic materials. This is where the highthermal stability and resistance to oxidativedegradation of the POMs are used to their fullestadvantage. The POMs are stable under condi-tions wherein even very robust synthetic metal-loporphyrins are susceptible to oxidative degra-dation. Upon cessation of the wet oxidationreactions, the liquor, now containing activePOMox, is returned to the bleaching reactor.
4. Experimental
4.1. Analytical methods
The kappa number of a wood pulp is astandard pulp and paper industry index of itslignin content. Kappa numbers are determinedby measuring the extent to which a standardizedsolution of permanganate is reduced when vig-orously mixed with a pulp sample at a giventemperature for a given time. Weight-percentlignin in pulp is empirically determined to be(0.15) (kappa number). Kappa numbers of un-bleached kraft pulps vary from approximately20 to 35 (3.00 –5.25% lignin). Microkappa num-bers (obtained from relatively small pulp sam-ples) were determined using Technical Associa-tion of the Pulp and Paper Industry (TAPPI)useful method um-246 [42]. Pulp viscosity, anon-linear function of the degree of polymeriza-tion of the cellulosic component, is an indirectmeasure of the extent to which fibers have beendamaged by degradation of individual cellulosechains. Viscosity measurements were obtained,using a capillary viscometer, from solutions of

66 I.A. Weinstock et al./Jounlal of Molecular Catalysis A: Chemical 116 (1997) 59-84
pulp samples in aqueous solutions of ethylene-diamine and cupric sulfate, a good solvent sys-tem for dissolution of cellulose (TAPPI testmethod T230 om-89) [43]. Viscosities of un-bleached kraft pulps are generally near 30 mPas and those of market-grade bleached pulpsrange from roughly 18–25 mPa s. Routine 31Pand 51V NMR spectra were obtained on a 250MHz Bruker instrument according to publishedmethods and externally referenced to H3PO4
and VOC13, respectively [44]. Reduction ofPOMs was measured by titration with CeIV in0.1 M H2SO4 or by UV–vis spectroscopy.
4.2. Materials
Two unbleached mixed-pine kraft pulps, onewith a kappa number of 33.6 and a viscosity of34.2 mPa s (pulp 1) and one with a kappanumber of 24.1 and a viscosity of 27.8 mPa s(pulp 2) were supplied by Consolidated Papers,Wisconsin Rapids, WI. Bleaching, wet oxida-tion and model studies were performed usingsalts of the α-Keggin anions (all shown here intheir POMox forms): α- [PV 2 M o10O 40]
5- (1)[45], [V10O28]
6- (2) [46], α-[PVW l1 O 40]4- (3)
[47], α-[SiVW 11 O 40 ] 5- (4a) [48], α−[ B V W11O 40]
6- (5) [49], in which V=VV, andα-[SiMnIII(OH 2)Wl1O39]
5- (6) [50]. Compound1 is prepared as an equilibrium mixture of α−N a x H 4 _ x [ P V M o 1 1O 4 0 ] , α −N a x H 5- x [P V 2M o 1 0 O 4 0] and α−Na x H6 - x [PV3 MO9O40] in which the divana-dium species is favored and where the ratiosx/ ( 4 – x ), x/ ( 5 – x ) and x/ ( 6 – x ) are deter-mined by pH [51]. In addition, in the di- andtrivanadium complexes, positional isomers arepresent. Henceforth, this mixture of compounds,in which 1 and its positional isomers dominate,will be referred to as compound 1. The one-electron reduced form (VIV, POMred) of com-pound 4a was prepared by stoichiometric addi-tion of VOSO4 to a solution of α-K8 [SiW11O40],obtained as described in Ref. [48]. The solutionwas then filtered and cooled to collect darkpurple crystalline α-K 6 [SiVW 11O40] 4b. The
one-electron reduced forms of compounds 4a, 5and 6 were oxidized in aqueous solution bysparging with ozone at room temperature. In thepreparation of 4a, a stoichiometric amount ofsulfuric acid was added prior to ozonation (oneequivalent of hydroxide is formed per electron-equivalent of VIV oxidized). Ozonation of 5 and6 was carried out in the presence of pH 5acetate buffer. Solid products were obtained bycrystallization of the acid (1), sodium salt (2) orpotassium salts (4a–6) and were characterizedby FTIR (KBr pellet), 31P and 51 V NMR spec-troscopes. Solutions of 3 were prepared prior touse by direct acidification of stoichiometriccombinations of phosphoric acid, sodium meta-vanadate and sodium tungstate, a method knownto give exclusively 3 [47]. The acid of 3 was theonly phosphorus-containing product observed by31P NMR in these solutions. Compound 6 wascharacterized by FTIR, UV–vis and by titrationto a starch endpoint using potassium iodide andsodium thiosulfate.
4.3. Bleaching reactions
4.3.1. H5[PV2 Mo10O40] (1); MO E and ME se-quences
A series of reactions were performed using 1at several pH values and under both aerobic(Mo) and anaerobic (M) conditions, using thefollowing general method. Pulp 1, obtained as a30% solids mass of hydrated pulp fibers (30%consistency or csc), was combined with an 0.054M solution of 1 to obtain a 3% csc mixture ofpulp fibers in a 0.05 M solution of 1. Thesolution was adjusted to the desired pH byaddition of small amounts of either concentratedH 2S O4 or aq NaHCO3 and the mixture wasplaced in a 2 1 Parr reactor and purged withpurified nitrogen, while stirring, for 30 min. Thereactor was then sealed under nitrogen andheated to 100°C for 4 h. During the reaction,aliquots of the POM solution were removed andthe extent of reduction of the vanadium fromV V to VIV was determined by titration withCeIV to a yellow endpoint. The pulps were then

I.A. Weinstock et al. / Journal of Molecular Catalysis A: Chemical 116 (1997) 59-84 67
washed thoroughly with water in a Büchnerfunnel. Alkaline extraction (E) was performedby heating the pulp at a csc of 3.2% at 85°Cunder nitrogen for 3 h in 0.25 to 1.0 M NaOH.In experiments where oxygen was used (MO
stages), the pulps slurried in POM solutionswere purged with purified O2 gas and thenheated under dynamic O2 pressures of 1.0 atm.After reaction, no reduction of the POM solu-tions was observed. After anaerobic bleachingtrials, moist air was bubbled gently throughreduced POM solutions for 1.5 h at 60°C, andthe pH values of the solutions were then ad-justed to 1.5 with concentrated H2S O4, Thereoxidation was monitored spectrophotometri-cally at 756.9 nm (fully reduced solutions aredeep blue, fully oxidized ones are orange). Afterreoxidation, 31PNMR spectra of the used POMsolutions were obtained. No phosphorus-con-taining decomposition products were observed.Control experiments were carried out underidentical conditions but with no POM added. Insome cases, an acid pretreatment was used toremove metal ions from the pulp prior tobleaching. In such cases, the pulp was soakedfor 15 min in a cold solution of 0.5 wt% H2SO4
at a csc of 8.3% and rinsed thoroughly withdistilled water.
4.3.2. Na6[V10O28 ] (2); ME sequencePulp 1 (O. 10 g odw) was added to a 0.10 M
solution of compound 2 to a final csc of 2.7% ina 15 ml round-bottomed flask. The pH of themixture was adjusted to 2.5 with concentratedH 2SO 4. Air was removed in three freeze-pump-thaw cycles, and the flask was sealedunder purified nitrogen and heated in a 100°Cbath for 4 h. During heating, the solutionchanged from orange to red-brown and precipi-tate of the same color fell out of solution. Themixture of pulp and precipitate was collected ona Büchner funnel and washed with water. Littleif any of the precipitate dissolved. The pulp wassoaked for 3 h at room temperature in 1 MNaOH to dissolve the precipitated vanadates,washed with water, and extracted for 3 h at
85°C in 1.0% aqueous NaOH. Little delignifica-tion was observed [36].
4.3.3. α α-H 4[PVW1lOJO ] (3); MOE sequencePulp 1 (1.0 g odw) was added to a 0.09 M
solution of compound 3 to a final csc of 3.0% ina 100 ml round-bottomed flask. The pH of themixture was adjusted to 1.50 with concentratedH2SO4. The flask was sealed in air (the reducedform of 3, α-H 5[PVW11O40] reacts very slowly,if at all, with dioxygen) and immersed in a100°C oil bath for 4 h (average reaction temper-ature was approximately 95°C). During heating,the solution changed from orange to greenish–brown. The pulp, now somewhat lighter in color,was collected on a Büchner funnel and thepartially reduced polyoxometalate solution (pH1.67) was saved. After reaction, 43.6% of thevanadium was present as VIV. The pulp waswashed three times with water and heated for 3h at 85°C in 0.25 M aqueous NaOH at a csc of3.2% in an open round-bottomed flask. At theend of this time the alkali solution was brown,and the pulp was lighter in color. 31P NMRspectra of used POM solutions were obtainedafter reoxidation by oxone (a potassium monop-ersulfate compound, DuPont). Phosphorus-con-taining products of rearrangement or dispropor-tionation were observed at concentrations ofless than ca. 5.0%. These signals have beententatively assigned t o i s o m e r s o fNa5[PV2W10O40]. No other phosphorus-contain-ing compounds were observed.
4.3.4. α α-K5 [SiVW 11O40] (4a); M1M 2M 3E Se-quence
To model a continuous countercurrent pro-cess and to follow the course of the bleachingreactions, POM treatments of unbleached pulpwere divided into three successive batch reac-tions, M1, M2 and M3. In the first set ofbleaching trials, pulp 1 was used. A mild anaer-obic alkali extraction was performed prior to thePOM bleaching. This treatment had little effectand was dropped in later work. M stages wererun in stirred, glass-lined, 1 or 21 high pressure

68 I.A. Weinstock et al./Journal of Molecular Catalysis A: Chemical 116 (1997) 59-84
Parr reaction vessels. The pulps were slurried at3% csc in bright yellow 0.05 M solutions ofα-K5 [SiVWl1O40] (4a) in 0.2 M pH 7 potassiumphosphate buffer. After purging with nitrogen,the reactor was heated to 125°C (ramp time of40 min) and held at temperature one h for M1,and 2.0 h for M2 and M3. The reactor pressurewas sustained with nitrogen at ca. 50 psig.During bleaching, the pH slowly decreased fromseven to no lower than six. The liquor changedfrom a bright yellow color of fully oxidized 4ato the dark purple of its one-electron reducedform, α-K 6 [SiVWl1O40] (4b). After each batchreaction the pulps were collected in a Büchnerfunnel and washed three times with water. Aparallel control sequence was performed byheating mixtures of pulp, water and phosphatebuffer in three successive batch reactions butwith no POM present. Both sequences werefollowed by anaerobic extraction with 1% NaOHfor 2 h at 85°C with pulp consistencies of1–2%. In addition, two samples of the un-bleached pulp were each partially delignifiedusing elemental chlorine followed by extractionwith alkali (CE). In the first case, 7.4% chlo-rine, 100 · (weight of Cl2/dry weight of pulp),was applied to the unbleached pulp, and 9.0%chlorine was applied to a separate sample of theunbleached pulp. For determination of paper-making properties, a POM delignified pulp wascompared to a pulp delignified using a (C/D)Esequence. Due to an insufficient supply of pulp1, a similar pulp, 2, was used. This pulp wasdelignified in triplicate runs using batch POM(M1M2M3E) sequences as described above andby a (C/D)E sequence (a mixture of C12 andchlorine dioxide in which 30% of the totaloxidative equivalents present are supplied byClO2), which provided a delignified pulp closein kappa number to the POM delignified pulp[7]. A POM-free control was performed once.
4.3.5. Kinetic studies using α-K 5 [SiVW11O40](4a)
Data used for kinetic modeling were obtainedduring a M1M 2M 3 sequence carried out as
reported above, but at 130 rather than 125°C. Inaddition, a single-M-stage experiment was per-formed using untreated pulp 2 according to theprocedure described immediately above, exceptthat the reaction was run at 100°C for 4 h.Progress of reaction was followed by measur-ing, either titrametrically or by UV–vis spec-troscopy (496 nm), concentrations of the one-electron reduced anion α -[ SiVWl1O40]
6- (4b).Concentrations of unreacted 4a, [POMox], (as[ P O Mtotal ] – [4b]) were used in the kineticmodel. In addition, kappa numbers, obtainedafter each batch reaction, were mathematicallytreated as indicating lignin concentrations,[lignin]. These data are presented in Fig. 13 andFig. 14.
4.3.6. NMR evaluation of the stability of[BVW11O40]
6- (5) in carbonate bufferAll NMR spectra were recorded on a General
Electric GN 500 spectrometer. The 51V chemi-cal shifts were referenced to HVO~ - in water,11 B shifts were referenced to NaBH4 in water;referencing was achieved by the substitutionmethod. Chemical shifts downfield from thereference are reported as positive (+ δ). In re-porting NMR data, multiplicity and line widths(in Hz), as applicable, are given in parentheses.The probe temperature was 295 K in all NMRexperiments. The spectral parameters for 51VNMR were acquisition time = 106.5 ms, relax-ation delay = 300 ms, pulse width = 15.0 µs,spectral window = 38462 Hz, digital resolution= 2.35 Hz/point. The spectral parameters for11B NMR were acquisition time = 204.8 ms,relaxation delay = 1.50 s, pulse width = 15.0µs, spectral window = 20000 Hz, digital reso-lution = 1.22 Hz/point.
4.3.7. Effect of carbonate buffer, pH and oxida-tion state of vanadium in 5
Four 200 mg portions of 5 were each dis-solved in 2.0 ml of 0.10 M aqueous carbonatebuffer. Two samples were prepared at pH 7.0and two at pH 8.5. One sample at each pH valuewas reduced with 1.0 eq of hydrazine and all

I.A. Weinstock et al./Journal of Molecular Catalysis A: Chemical 116 (1997) 59-84 69
four were maintained at 25°C for 18 h. Twoadditional samples of 5 (200 mg) were preparedin pure water (2.0 ml) without buffer. One ofthese was adjusted to pH 8.5 using dilute aqNaOH, the other was first reduced with hydra-zine and then adjusted to pH 8.5, and both wererefluxed for ca. 1.5 h. Reduced samples weredeoxidized with a slight excess of Br2 and allsix were analyzed by 51V NMR.
4.3.8. α-K 5 [SiMn III (OH 2)W 11 O 39 ] (6);M1M2M3E sequence
For the M1 stage, pulp 1 was added to asolution of 6 in 0.20 M pH 5 potassium acetatebuffer to give a final csc of 3% and a POMconcentration of 0.05 M. The pH after mixingwas 5.02. The mixture was then placed in aglass lined Parr high pressure reactor and, whilestirred, was purged with purified nitrogen for 40minutes, sealed, and heated to 125°C for 1 h.During this time, the pH of the POM solutiondropped to 4.86. The POM bleaching liquor wasthen recovered by filtration and the pulp washedwith water. The amount of 6 reduced to α -K6[SiMnII(OH2)W11O39] (7) during the bleach-ing reaction (M1 stage) was determined by reac-tion of an aliquot of the bleaching liquor withan excess of potassium iodide and titration to astarch endpoint with sodium thiosulfate. Overthe course of the bleaching reaction, more than98.9% of the salt 6 present had been reduced to7. Upon cooling the bleaching liquor to 0°C forthree days, ca. 50% of orange crystalline 7,characterized by FTIR (KBr pellet), was recov-ered. The UV–vis spectrum of the supernatantwas consistent that of a mixture of 6 and 7.During the M2 stage (1.5 h) the pH droppedfrom 5.14 to 4.95 and 89.2% of the POMpresent was reduced. The M3 stage was run for2 h during which the pH dropped from 5.16 to4.89 and 66.8% of the POM present was re-duced. After the three sequential M stages, analkaline extraction (E) was performed as de-scribed above. A control experiment was per-formed by subjecting pulp to the same proce-dure, but with no POM present. Oxidation of 7
at pH 5 by oxygen, at elevated temperature andO2 pressure, is slow.
4.4. Wet oxidation of dissolved organics (unitoperation D)
Eight bleaching-wet oxidation cycles, eachconsisting of an anaerobic bleaching reactionfollowed by an aerobic wet oxidation reactionof the spent bleaching liquor, were carried outusing a single solution of 1. The bleaching wasdone using kraft pulp 2 at 3% csc. The POMliquor was maintained at a concentration of 0.05M for both the bleaching and wet oxidationreactions through the evaporation or addition ofwater as necessary. A pH of 3 was maintainedby periodic addition of small amounts of dilutesulfuric acid. Bleaching was carried out in astirred high pressure Parr reaction vessel with aglass liner at 100°C for 4 hrs under nitrogen.Kappa reductions of 5 were typical. The darkgreen reduced liquor (typically 14 – 17% of thetotal vanadium present was reduced to V IV) wasthen pressed from the pulp and treated withoxygen (wet oxidation) to degrade the dissolvedorganic compounds removed from the pulp dur-ing bleaching. The wet oxidation reaction wasperformed in a glass lined Parr reactor equippedwith a gas entrainment impeller designed tocirculate head gasses through the solution. Thereaction was carried out at 150°C for 4 h underan oxygen pressure of 100 psig. The red, fully-oxidized liquor was then used for bleaching inthe next bleaching/wet oxidation cycle. Afterevery bleaching and wet oxidation reaction, theamount of carbon dioxide produced and thechemical oxygen demand (mg O2/l, COD) ofthe liquors were measured. Carbon dioxide wasmeasured by acid–base titration of aqueousBa(OH)2 solutions through which reaction ves-sels were vented after completion of bleachingor wet oxidation reactions [52]. Prior to eachventing, a Ba(OH)2 solution, prepared undernitrogen, was placed in a chromatography col-umn. Gases were introduced from the bottomthrough a glass frit using inert atmosphere tech-

70 I.A. Weinstock et al./JournaI of Molecular Catalysis A: Chemical 116 (1997) 59-84
niques to avoid contamination with atmosphericCO2. Small amounts of isopropanol were addedto the Ba(OH)2 solutions to promote foaming,thus increasing gas–liquid contact. For purposesof comparison, carbon dioxide values are re-ported as mg CO2/(1 of POM liquor present inthe reaction). COD determinations were carriedout by exhaustive oxidation of POM liquorsaccomplished by adding c. H2SO4 (a volumeequal to the volume of the aqueous samplebeing tested) and an excess of potassium dichro-mate and heating to reflux (ca. 150°C) for sev-eral hours [53]. The extent of bichromate reduc-tion was then determined by titration with fer-rous ammonium sulfate using a ferroin indica-tor. For bleach liquors containing reduced 1, theconcentrations of VIV ions present prior tobichromate oxidation were accounted for in thecalculation of COD values. The integrity of 1after completion of the 8 bleaching–wet oxida-tion cycles was confirmed by 31P NMR spec-troscopy.
5. Results and discussion
5.1. Physical properties of POM bleaching cat-alysts
POM salts useful in the present context mustbe reversible oxidants, capable of undergoingrepeated cycles of reduction and reoxidationwithout degradation. In addition, their oxidizedforms, POMox, must have reduction potentialssufficiently positive to oxidatively degradelignin, while, at the same time, sufficiently neg-ative such that reoxidation with dioxygen ispossible. Using lignin model compounds, wehave found that POM bleaching occurs via oxi-dation of phenolic moieties in residual ligninand that non-hydroxylated phenyl rings, includ-ing those possessing α-alcohol substituents(benzylic alcohols), react too slowly to con-tribute significantly to delignification. Thus, thePOM reduction potentials necessary for deligni-fication are those needed to oxidize phenolic
structures. In Fig. 6, the reduction potentials ofseveral of the POM salts used in the presentstudy are shown in relation to those for four-electron reduction of dioxygen as a function ofpH. All POM complexes whose reduction po-tentials lie below that of dioxygen, and are atthe same time positive enough to oxidize pheno-lic groups in lignin, are thermodynamically ca-pable of transferring electrons from lignin tooxygen. This is the same task accomplished bythe extracellular ligninolytic metalloenzyme sys-tems of wood-rotting fungi.
Another practical consideration in pulpbleaching is the need to avoid hydrolysis ofpulp fibers that occurs most readily at elevatedtemperatures, such as those encountered in POMbleaching. This hydrolysis is due to the acid-catalyzed cleavage of glycosidic linkages in cel-lulose. At very high pH values, alkaline degra-dation reactions can occur and the optimal pHrange for avoiding either cellulose hydrolysis ordegradation (acid catalyzed or alkaline) is near9–10. This, combined with the knowledge thatPOM bleaching occurs via phenol oxidations,leads to the possibility that optimal rates andeffectiveness might be observed near pH values

I.A. Weinstock et al./Journal of Molecular Catalysis A: Chemical 116 (1997) 59-84 71
of 9–10, which correspond both to the range ofp K a, values of many phenols and to the pHregion in which the least amount of cellulosehydrolysis is expected to occur at elevated tem-peratures.
In general, tungsten(VI, d0)-based POMs areless labile, more stable to alkaline hydrolysisand possess higher reduction potentials thantheir isostructural and isoelectronic molybde-num(VI, d0) analogues. For example, the α-[ P V2M o1 0O 4 0]
5- anion (1) is stable to a pH of3.5 to 4.0, while the isoelectronic tungsten ana-log, α-[PV 2 W10O40]
5- is stable to a pH of near5 [54]. Also, within the Keggin structural class,increases in anion charge density are associatedwith greater resistance to alkaline hydrolysisand with increasingly negative reduction poten-tials. In the series α- [ P V W 11 O 40]
4- (3), α-[SiVWl1O40]
5- (4a) and α-[BVW 11 O 40 ]6- (5),
whose charge increments follow the decrease inpositive charges of their central, tetrahedrallycoordinated heteroatoms, PV, SiIV and BIII, hy-drolytic stabilities increase with increasing neg-ative charge density. The tungstophosphate an-ion, 3, is stable perhaps to a pH of 3, while 4a,and its one-electron reduced form α-[SiVIVW 11O 40]
6- (4b) are stable indefinitely atreflux in water at pH 7. The hydrolytic stabilityof the tungstoborate compound, 5, was deter-mined by potentiometric titration at 80°C of a2.0 mM solution of 5 with dilute aqueous sodiumhydroxide. These preliminary data suggest thatthe anion is stable up to a pH range of approxi-mately 8.5–9.5. At the same time, it has beenobserved that the reduction potentials of fullydeprotonated Keggin anions (i.e., their sodiumsalts) in water decrease linearly with anioncharge [49] (compare for example 1, 4 and 5 inFig. 6).
The standard electrode potential for theV V/ VIV couple in 1.0 M acid is + 1.00 Vversus the normal hydrogen electrode (NHE).The potentials of vanadium-containing POMsuseful in the present context likely lie in therange 0.4–0.8 V. This range might be comparedto the standard reduction potentials of several
other oxidants used to delignify pulp: acid O3
(+2.07 V at pH 0, O3+ 2 H++ 2 e- ~ O2+H 2O), acid Cl2 (+ 1.36 V at pH 0, Cl2 + 2e-
~ 2C1-), acid C1O2 (+ 1.27 V at pH 0; C1O2
+ H++ e- ~ HC1O2), alkaline H2O 2 (+0.87V at pH 14, HO~+HzO+2e- ~ 30 H-) andalkaline O2 (+0.402 V at pH 14; O2 + 2H2O+ 4e- ~ 4OH-) [55,56]. Of these, the bestcomparisons are with acid C1O2 and alkalineO2, both of which delignify to a significantextent via single-electron oxidations of phenolor phenolate lignin substructures [57].
5.2. Bleaching trials
A broad survey of POM bleaching effective-ness was undertaken using vanadium(V)-sub-stituted POM salts possessing PV, SiIV and BIII
heteroatoms. The results are reported here asfollows. First, results obtained using a molyb-dophosphate, α-[PV 2 Mo 10O 40]
5- (1), are re-ported. For comparison, a general control exper-iment was performed using aqueous V v cationsalone, in the form of the decavanadate anion[V10O 28]
6- (2). This is followed by results ob-tained using the series of isostructural tungstencompounds α- [ P V W 1 1O4 0]
4 - (3), α-[S iVW11O 40]
5- (4a) and α-[BVW 11 O 40]6- (5),
possessing PV, SiIV and BIII heteroatoms. Next,the effectiveness of α-[SiMn III (OH2)Wl1O39]
5-
(6), is compared to that of its analog of similarcharge, 4a. Finally, the discussion of bleachingresults closes with a discussion of preliminarydata pertaining to the kinetics and mechanism ofdelignification by 4a.
5.2.1. α-[PV 2 M O IOO 10]5- (1)Early bleaching trials were performed using
POM salt mixture 1, which is stable only at acidpH values. This POM was chosen based onliterature reports that it oxidizes phenols rapidly,that its reduced forms are readily deoxidized bydioxygen in water and that, in organic solvents,it also oxidizes 1-phenylethanol (a non-phenolicbenzyl alcohol) to acetophenone, but not 2-phenylethanol, an aliphatic alcohol, the latter

72 I.A. Weinstock et al./Journal of Molecular Catalysis A: Chemical 116 (1997) 59-84
implying low reactivity with β-D-glucose ringsin cellulose [58]. Using 1 under aerobic condi-tions, extensive delignification was observed(entries 7-10, Table 1). However, pulp qualitywas extremely poor, probably due to the genera-tion of unselective oxygen-centered radicals. Incontrol experiments (entries 8 and 10, Table 1),metal ions present in the pulp and which mightparticipate in autoxidation reactions during aer-obic bleaching (namely manganese salts andiron oxide particles, both present in ca. 50–100ppm quantities in kraft pulp) were largely re-moved by pre-soaking the pulp in cold diluteH2SO4 [59]. Subsequent aerobic POM bleach-ing gave results similar to those obtained usingnon-acid-treated pulps (entries 7 and 9, Table1). This was strong evidence that the observeddrops in viscosity were due to reactions ofoxygen with reduced 1, and not exclusively toradical-chain autoxidation processes initiated bytrace metals. This led to the conclusion that, inorder to maximize the selectivity achievableusing POM oxidants, the bleaching reactionswould have to be run anaerobically.
Under anaerobic conditions, substantial delig-nification occurred but with less fiber degrada-tion than was observed in the aerobic reactions
(entries 1-6, Table 1). Nevertheless, drops inpulp viscosities were substantial, particularly atlower pH values (entries 1 and 2). However,even in organic solvents, where salts of 1 areexpected to be more highly ion-paired and tothus possess higher reduction potentials than inwater, no oxidation of secondary alcohols by 1was observed [58]. Therefore, it appeared likelythat pulp degradation was due primarily toacid-catalyzed cellulose hydrolysis and that op-timization of the bleaching reactions would re-quire moving to higher pH values. At the sametime, because the reduction potential of 1 be-comes more positive at lower pH values, thishypothesis could not be proven until a POM saltstable over a higher pH range was tried.
5.2.2. [V1 0O 2]6- ( 2 )
Meanwhile, a control was performed usingV v ions alone, in the form of [V10O 28]
6- (2),the dominant VV-containing species present atthe pH and concentration used (pH 2.5, 1.0 MV v or 0.1 M 2) [60]. During the reaction a largequantity of red-brown precipitate formed thatwas only dissolved by soaking the pulp forseveral hours in 1 M NaOH. Similarly, a darkgreen insoluble precipitate formed when dilute

I.A. Weinstock et al./Journal of Molecular Catalysis A: Chemical 116 (1997) 59-84 73
sodium metavanadate NaVO3 solutions wereused under aerobic conditions. Notably, how-ever, throughout the series of experiments car-ried out using 1, no vanadium precipitation wasobserved and 31P NMR spectra of spent bleachliquors deoxidized by dioxygen showed no signsof POM degradation, although positional iso-merization was seen. These observations stressthe importance of controlling the speciation oftransition metal ions in water and of providingligand environments that ensure the reversibilityof electron-transfer processes.
5.2.3. α-[PVW11 O40]4 - (3)
Compound 3, which possesses a higher re-duction potential than 1 (0.80 V vs. NHE at pH5 compared to an estimated 0.48 to 0.58 for thesodium salt of the α-[PV 2 Mo 10O40]
5- anion), ismore effective at delignification [49]. Using 3 inan ME sequence at pH 1.5, pulp 1 was deligni-fied to a kappa number of 7.6, while that for theparallel control was 18.9. After the reaction,43.6% of the vanadium present had been re-duced to VIV. However, compound 3 is stableonly at pH values below 2, at which consider-able cellulose degradation occurs at elevatedtemperatures. In addition, its one-electron re-duced form is know to react only very slowlywith dioxygen [61].
5.2.4. α-[SiVW 11 O40]5- (4a)
In an effort to show that losses in pulpquality observed in trials using 1 and 3 weredue to hydrolysis alone, and that POM delignifi-cation could be both effective and highly selec-tive, we sought a POM salt stable at neutral orhigher pH values, which possessed a reductionpotential close to, but below, that of dioxygen.The POM salt chosen was α-K5 [SiVWl1O40](4a), which is stable in water up to a pH ofapproximately 8, beyond which decompositionand precipitation of a light yellow material areobserved. After reflux in pure water at pH 7, nochanges were observed in the position or peak-width at half-height (V1/2 ) of the 51V NMRsignals of 4a or 4b (after oxidation with Br2)
after 55 days (Fig. 7). Assuming an instrumentsensitivity of 99% for the 51V nucleus, thisindicates a minimum half-life for each of 8.6years and demonstrates that the reduction of 4ato 4b is entirely reversible. After 112 days, abroad signal of very low intensity (less than1%) was observed in the solution of 4a (τ 1/2 =21.2 y) and no changes were observed in spec-tra of 4b after 208 days (τ 1/2 = 39.3 y). How-ever, when heated in phosphate buffer at pH 7,an equilibrium is established rapidly between4a, [P2W 5O2~]6- and [SiV x W 12 _ x O 40]
(4 + x)-, x= 2, 3 (both identified by 3] P and 51V NMRand by UV–vis spectroscopy), that is shiftedtowards the diphosphate and polyvanadates byrelative increases in the concentration of phos-phate buffer. During bleaching (0.20 M phos-phate, 0.05 M 4a), the concentration of 4 aremains at 90–95% of its initial value.
The potential of the 4a/4b couple, E, hasbeen measured at 0.64 V vs. NHE at pH 5 [49]and in a later report it was measured to be 0.72V at pH 4.5 [62]. After dissolving 4a in a pH 5acetate buffer solution, we measured a reductionpotential of E = 0.69 V (5.0 mM 4a, 0.2 M

74 I.A. Weinstock et al./Journal of Molecular Catalysis A: Chemical 116 (1997) 59–84
sodium acetate buffer, scan rate = 20 mV/s,E pa = 0.72 V, Epc = 0.66 V, E pa – E pc = 60 mV,E = (E pc + E pa)/2). Remarkably for aqueoussolutions of a transition-metal complex, the re-duction potential of the α -[SiVW l1O40]
5- anionremains nearly constant over a 1 000 000 foldchange in hydrogen ion concentration, from pH2 to 8 (Fig. 6). Thus, at pH 1.8 (a pH at which4a is unlikely to be stable), an irreversiblepotential of 0.68 V (as E pc+ E pa)/2) was mea-sured (5.0 mM 4a, 0.2 M, pH 7 potassiumphosphate buffer to which a small amount ofphosphoric acid was added immediately prior tothe potential sweep experiment, scan rate = 20mV/s, E pa = 0.74 V, E pc = 0.62 V, E pa – E pc
= 120 mV). At pH 8.1, the high end of thestability range of 4a, an irreversible potential of0.64 V was measured (0.1 M sodium acetate,scan rate = 20 mV/s, E pa = 0.70 V, E pc = 0.57V, E pa – E pc = 130 mV). These results suggestthat the p K a of the reaction shown in Eq. (3) isconsiderably less than 1.8 and that over theentire pH range studied, one-electron reductionof 4a to 4b is not accompanied by protonation.
Surprisingly, 4b was not oxidized by dioxy-gen after 208 days at reflux at pH 7 (the con-denser was open to the atmosphere over thecourse of the experiment). In addition, littleoxidation was observed when pH 7 solutions of4b in phosphate buffer were heated to 150°C for4 h under 100 psig of dioxygen. This despiteelectrochemical data indicating that the reaction(Eq. (4)) should be favored by 12.5 kcal/mol(taking E (4a/4b) as 0.68 V and the reductionof O2 to water at pH 7 as 0.815 V) [55].
The rate limiting step in the reaction of 4bcould be the single-electron reduction of O2 tothe superoxide anion radical, O2 ·
—, whichwould have a ∆ G0 at pH 7 of + 23.3 kcal/mol,
the reduction of O2 to O2· — requiring an input
of 0.33 V [56]. If this step were forced to occurvia an outer sphere mechanism, a very lowreaction rate would be expected. This might bethe case if the VIV ion in 4b is coordinativelysaturated by the pentadentate α -[SiW11O39]
8-
ligand, if expansion of the vanadium coordina-tion environment to include a seventh liganddoes not occur and if the vanadyl complexα -[SiV(O)W l1O39]
6- (4b) is not labile at neutralpH. In contrast, aqueous vanadyl ions (V02+),which possess labile aqua ligands and range inoxidation potential from approximately 0.95 to0.75 V over the pH range of 2 to 4, are oxidizedby dioxygen at pH values greater than 2.5 [19].After 1 week at 14°C, conversions of 0.01 Msolutions of V02+, sealed under air and shakenin stoppered bottles at initial pH values of 3.5and 4.9, were 21% and 76%, respectively. Reac-tion rates were much enhanced by heating to70°C [63]. At the same time, the reduced formof 6, α -[ SiMnII(OH2)W11O39]
6-, which at pH 5possesses an oxidation potential very close tothat of 4b, but which contains a labile watermolecule on the sixth coordination site of theMnII ion [64], is also oxidized very slowly atelevated temperature under an O2 pressure ofseveral atmospheres.
Bleaching trials using 4a were carried outanaerobically at pH 7 in phosphate buffer usingpulp 1 (kappa number 33.6 and viscosity of 34.2mPa s). Microkappa numbers and viscosity val-ues for the average of three EM1M 2M 3Ebleaching sequences, a control sequence andtwo different CE bleaching reactions, obtainedusing two C12 loadings are shown in Fig. 8. ThePOM bleaching sequence (Fig. 8, solid line) isas selective as the CE sequence down to akappa number of 10 (7.4% C12) and more selec-tive at lower kappa numbers (9.0% C12). It isnoteworthy that the viscosity of the pulp sub-jected to the control run (no POM present)dropped almost in parallel with that of the POMtrials. This suggests that even at pH 7 somecellulose hydrolysis occurs under the conditionsof the bleaching reaction. (In this regard, it

I.A. Weinstock et al./Journal of Molecular Catalysis A: Chemical 116 (1997) 59-84 75
properties close to those of the C/D delignifiedsample. Papers made from the C/D delignifiedslightly higher tear indices.
should be noted that the pH values of the POMliquors dropped to near 6 during the M stages,while that of the control reaction remained at 7throughout.)
A M1M2M3E sequence was performed on alarger scale to provide pulp for the determina-tion of pulp yield and papermaking properties.As shown in Fig. 9, the kappa-viscosity curvesof the POM trials fall slightly below thoseobtained using the (C/D)E sequence. In indus-try, substitution of chlorine dioxide (D) for ele-mental chlorine (C) is done to improve theselectivity of the delignification step and todecrease the amount of chloro-organic com-pounds produced per ton of pulp. Average yieldof the POM delignified pulps was 89 ± 3%,while that of the POM-free control was 89%and that of the C/D treated pulp was 95%.Significantly, the yield of the POM-free controlreaction is identical to the average yield of theunoptimized POM trials, further evidence thatcellulose hydrolysis is responsible for the dropin viscosity observed in each case. Even so, thePOM delignified pulps possess papermaking
while those made from the POM pulps possessslightly higher tear indices.
These observations lead us to believe that,with optimization of reaction conditions, thepapermaking properties of POM delignifiedpulps could easily match or exceed those ofpulps delignified with elemental chlorine andchlorine dioxide, the industry standard. For ex-ample, after modest attempts at optimization,pulp 2 was delignified by 4a to a microkappaNo. of 4.7 and a viscosity of 23.0 mPa s, afteralkali extraction. In addition, the POM reactionsdescribed here were run at pH 7 (phosphatebuffer). POM complexes stable at pH 9 arecurrently being studied and new ones devel-oped. At pH 9, less cellulose hydrolysis isexpected to occur, while, at the same time, thePOM complexes will likely react more rapidlywith phenolic groups present in residual lignin.Both trends are expected to facilitate furtheroptimization of the POM delignification reac-tions and to allow for selective delignificationdown to very low kappa numbers.
76 I.A. Weinstock et al./Journal of Molecular Catalysis A: Chemical 116 (1997) 59-84
5.2.5. α -[BVW 11O 40]6- (5)
In this regard, a 2.0 mM solution of thetungstoborate complex, α -[BVW llO40]
6- (5), inpure water at 80°C appeared stable up to pHvalues of approximately 9.5 upon addition ofsmall aliquots of a 0.1 M solution of sodiumhydroxide. At the same time, its reduction po-tential is substantially more negative than thatof 4a (Fig. 6) and a bleaching trial was per-formed to determine whether effective delignifi-cation might still occur near the p K a of thephenolic lignin substrate. The bleaching experi-ment (M1) was carried out using conditionsestablished for 4a (0.05 M 5, 125°C, etc.),except at an initial pH of 9.6 using carbonatebuffer. Only modest delignification was ob-served (the kappa number after the M1 stagewas 19.7). However, because the pH of thesolution dropped rapidly to pH 8.5, the effec-tiveness of 5, or more significantly, of POMcomplexes with reduction potentials near that of5, at pH 9–10, has yet to be determined.
The carbonate-buffered bleach liquor was an-alyzed to determine whether the drop in pHreflected degradation of 5. The solution wasdeoxidized by addition of a slight excess of Br2
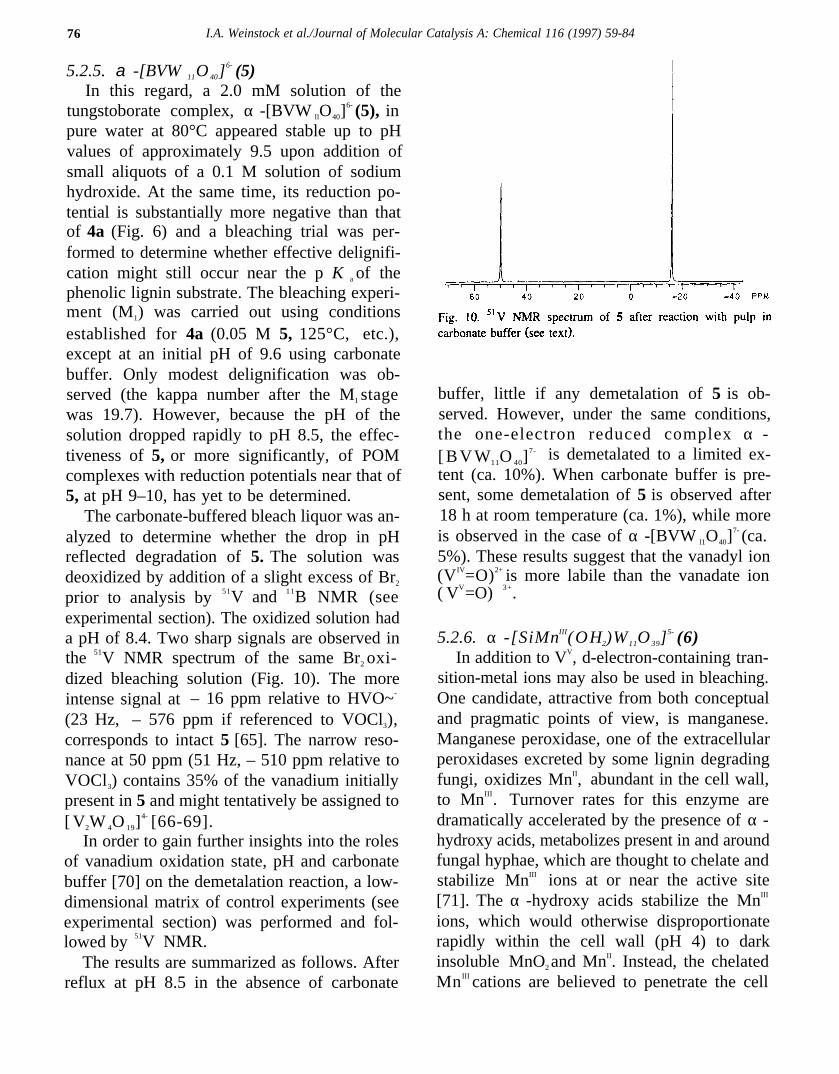
prior to analysis by 51V and 11B NMR (seeexperimental section). The oxidized solution hada pH of 8.4. Two sharp signals are observed inthe 51V NMR spectrum of the same Br2 oxi-dized bleaching solution (Fig. 10). The moreintense signal at – 16 ppm relative to HVO~-
(23 Hz, – 576 ppm if referenced to VOCl3),corresponds to intact 5 [65]. The narrow reso-nance at 50 ppm (51 Hz, – 510 ppm relative toVOCl3) contains 35% of the vanadium initiallypresent in 5 and might tentatively be assigned to[ V2W 4O 19]
4- [66-69].In order to gain further insights into the roles
of vanadium oxidation state, pH and carbonatebuffer [70] on the demetalation reaction, a low-dimensional matrix of control experiments (seeexperimental section) was performed and fol-lowed by 51V NMR.
The results are summarized as follows. Afterreflux at pH 8.5 in the absence of carbonate
buffer, little if any demetalation of 5 is ob-served. However, under the same conditions,the one-electron reduced complex α -[ B V W11O 40]
7- is demetalated to a limited ex-tent (ca. 10%). When carbonate buffer is pre-sent, some demetalation of 5 is observed after18 h at room temperature (ca. 1%), while moreis observed in the case of α -[BVW l1O40]
7- (ca.5%). These results suggest that the vanadyl ion(VIV=O)2+ is more labile than the vanadate ion( VV=O) 3+.
5.2.6. α -[SiMnIII(OH2)W 11O 39]5- (6)
In addition to VV, d-electron-containing tran-sition-metal ions may also be used in bleaching.One candidate, attractive from both conceptualand pragmatic points of view, is manganese.Manganese peroxidase, one of the extracellularperoxidases excreted by some lignin degradingfungi, oxidizes MnII, abundant in the cell wall,to MnIII. Turnover rates for this enzyme aredramatically accelerated by the presence of α -hydroxy acids, metabolizes present in and aroundfungal hyphae, which are thought to chelate andstabilize MnIII ions at or near the active site[71]. The α -hydroxy acids stabilize the MnIII
ions, which would otherwise disproportionaterapidly within the cell wall (pH 4) to darkinsoluble MnO2 and MnII. Instead, the chelatedMn III cations are believed to penetrate the cell

I.A. Weinstock et al./Journal of Molecular Catalysis A: Chemical 116 (1997) 59-84 77
wall where they are capable of oxidizing arange of phenolic lignin substructures.
L i g n i n p e r o x i d a s e s , in t h e i rporphyrinato[FeIV = O] cation radical (com-pound I) and neutral (compound II) forms arecapable of oxidizing many alkyl and methoxylsubstituted benzenes, such as those present inlignin, to cation radicals [72]. On the otherhand, most of the pores within intact fiber walls,defined by loosely oriented 3–4 nm diametercellulose microfibrils containing bulky pendanthemicellulose chains, seem too small for easyaccess by these enzymes. For instance, sphericaldextrans 1.0 nm in diameter can penetrate 84%of the cell-wall volume accessible to water,while those 10 nm in diameter can penetrateonly 14.4% of the water accessible domain [73].(For comparison, Keggin anions are 1.1 nm indiameter and the sizes of POM anions of alltypes range from 0.9–3.0 nm.) Thus, the man-ganese peroxidase system could be used by thefungi to penetrate relatively intact cell walls.
Although bleaching systems have been devel-oped using manganese peroxidases, delignifica-tion rates are too slow for commercial applica-t ion [74]. Bleaching tr ials using α-[SiMnIII( O H2) Wl 1O 39]
5- (6) were carried outto determine whether, when stabilized in aque-ous solution by the α-[SiW 11O39]
8- ligand, MnIII
could be used at elevated temperatures to obtaincommercially relevant delignification rates. An-other impetus for using manganese is its lowcost and high environmental compatibility.
Results of a bleaching trial using 6 at pH 5 inacetate buffer are shown in Fig. 11. The overallkappa number/viscosity relationship is similarto that obtained using 4a under the same condi-tions. This is not surprising considering that: (1)both 4a and 6 are capable of oxidizing phenolicsubstrates, (2) the reduction potential of 6 at PH5 (0.68 V vs. NHE) is close that of 4a at thesame pH (Fig. 6), and (3) model studies stronglysuggest that only phenolic lignin substructuresare substrates for 4a. To our knowledge, nodetailed mechanistic information is yet availableregarding the electron-transfer mechanisms op-
crating in the reduction of 4a or 6 by phenols orphenoxyl radicals in water.
T h e r e d u c e d f o r m o f 6 , α -[SiMnII(OH2)Wl1O39]
6 - (7), is not reoxidizedby oxygen at a rate suitable for commercial use.However, the tungstoborate analog, α -[BMnII(OH2)W11O39]
7-, is reportedly easily ox-idized [50]. This, and other Mn-containingPOMs, such as α-[SiW 11 O39{Mn II(OH 2)} 3]
10-
[75], and new ones under development in ourlaboratories, such as [AlMnIII(OH2)W11O39]
6-,and others prepared using hydrothermal meth-ods (see below), are all promising candidatesfor use in bleaching.
5.3. Mechanism and kinetic analysis
Information concerning the kinetics of POMdelignification of kraft pulps can aid in processoptimization and design of commercial bleach-ing reactors. However, kraft pulp as a substrateis not well-defined, and so, the results of kineticmodeling cannot be used to directly determinereaction mechanisms. Over the last 20 yearsconsiderable progress has been made in definingthe nature and frequency of chemical substruc-tures present in the residual lignin component ofwood fibers subjected to kraft pulping. Al-

78 I.A. Weinstock et al./Journal of Molecular Catalysis A: Chemical 116 (1997) 59-84
though a comprehensive structural model doesnot yet exist, these efforts have provided infor-mation concerning the most abundant structurespresent [5,6]. For example, approximately 15%of the phenyl structures present in native soft-wood lignin are hydroxylated. This figure risesto ca. 20–40% for residual kraft lignin [6]. Thephenolic end-groups arise from the cleavage ofaryl ether linkages during kraft pulping. Basedon model studies, these phenolic groups appearto be the primary substrates for 4a in its reac-tions with kraft pulp.
Additionally, there are uncertainties associ-ated with the macro-molecular structure of woodfibers. Models of woody tissue place the ligninwithin a carbohydrate matrix [1,2]. This matrixis dominated by cellulose microfibrils, 3–4 nmbundles of organized cellulose chains, withlignin polymer forming an extended three-di-mensional network between the microfibrils.From the perspective of bleaching, the nature ofthis matrix is important because it can restrictthe access of oxidant to the lignin substrate, andthus modify the apparent reaction kinetics. Thus,both the complex chemical nature of residuallignin and the heterogeneity and ill-definedphase boundaries of the lignocellulosic matrix,complicate the analysis of kinetic data.
In reactions of 4a with both the non-phenolicand phenolic lignin dimer models I and II (Fig.12), under actual delignification conditions (pH7 phosphate buffer, 3 h at 125°C) no reaction isseen with I, while rapid reaction, leading todimer cleavage, is observed with II [40]. Based
on consideration of the relative reactivities ofthe dominant chemical structures present inresidual kraft lignin, these results strongly sug-gest that only hydroxylated phenyl substructuresare substrates for 4a. However, although thereaction of 4a with lignin model II is very rapid,delignification by this POM requires severalhours of treatment. This observation suggeststhat: (1) residual lignin contains a wide varietyof phenolic structures, some of which react with4a more slowly than others and/or that (2)phenolic moieties are rapidly consumed and thecontinuous generation of new ones is rate limit-ing, and/or that (3) mass transport and otherphenomena associated with the nature and struc-ture of the partially lignified pulp fibers arelimiting. These options are discussed in thecontext of the preliminary kinetic analysis pre-sented below.
Data obtained in bleaching experiments doneon kraft pulp 2 using 4a are shown in Fig. 13and Fig. 14 (see experimental section). Bothfigures indicate that oxidized POM is consumedas the bleaching progresses. The lignin contents,as microkappa numbers, of the pulps before andafter each run were also determined. Data in

I.A. Weinstock et al./Journal of Molecular Catalysis A: Chemical 116 (1997) 59-84 79
Fig. 13 are from a bleaching run done at 100°C.The slow initial reaction rate corresponds to thetime required to heat the reactor to temperature,usually about 45 min. Fig. 14 shows data ob-tained from three successive 125°C batch reac-tions. These data show two discontinuities thatcorrespond to the replacement of the reducedPOM solution with fresh, fully oxidized solu-tion. At each of these points the pulp wasremoved from the cooled reactor, washed withwater, and mixed with fresh POM solution. Thereactor was then reheated to the operating tem-perature. This allowed the bleaching to be ex-tended for more complete removal of ligninfrom the pulp and provided some insight intothe likely behavior of the pulp in a multi-stagebleaching system.
The lines in the figures are model predictionscalculated using kinetic parameters obtainedfrom a simultaneous least squares fit of threedata sets, which include data obtained at twodifferent temperatures. Modeling was done bynumerical integration of the assumed rate ex-pressions using rate constants possessing Arrhe-nius temperature dependencies. Lignin concen-trations in pulp slurries of known consistencies
were represented by kappa numbers. Rate ex-pression parameters were optimized to minimizedeviation between the experimental data and thekinetic model.
The data set was best described by the modelshown in Eq. (5), Eq. (6) and Eq. (7), whichconsists of two independent parallel reactions: afast reaction, first order in both oxidized POMconcentration ([POMox]) and lignin concentra-tion (designated [Lf]), and a slow, zeroth orderreaction in which the rate was unaffected byeither [POMox] or lignin concentration, [Ls].The two reactions can be seen in Fig. 14 wherethe part of the plot showing the progress of theinitial bleaching step has curvature indicative ofa dependence on [POMox] while by the thirdbleaching step the progress is nearly linear withtime, showing no such dependence. After as-suming the functional dependence of the rateexpression on [POMox] and lignin concentra-tion, one can then estimate the activation ener-gies of these two reactions using a least squareserror definition. The activation energies of therate constants that minimize the errors for thewhole data set are 3.7 ± 0.1 and 27.1 ± 0.04kcal/mol for the second order and the zerothorder reactions respectively. The fraction of to-tal lignin that reacts with second order kinetics,Lf, was also adjusted to give the lowest squaredeviation. The best value was 24 ± 2%. Thebalance of the lignin, Ls, was assumed to reactvia the zeroth order kinetics. The uncertaintiesin the fitted parameters were defined as thechange that results in a 10% increase in the sumof the error squares.
80 I.A. Weinstock et al./Journal of Molecular Catalysis A: Chemical 116 (1997) 59-84
modeling are tenuous. The results do, however,provide some useful insights. The activationenergy of the second-order reaction, 3.7kcal/mol, is lower than the values typicallyobserved for oxidation reactions. This suggeststhat the apparent rate of this reaction is likelycontrolled by a mass transport resistance. If thisis the case, then the first-order dependence on[POMox] might result from a linear dependenceof mass transport rate on concentration. Theother reaction, however, exhibits an activationenergy near the value one would expect in achemical-reaction-controlled process. At thesame time, however, the reaction rate unexpect-edly appears to be independent of both [POMox]and [Ls]. One possible explanation of the zerothorder dependence of reaction rate on [POMox]and [Ls] is that there are a fixed number ofphenolic reaction sites responsible for delignifi-cation and that these are saturated with oxidantat all times. Studies designed to better under-stand these and other observations are inprogress. The import of the present analysis isthat full understanding of the reactions of POMswithin pulp fibers must include consideration ofa number of issues arising from the effects ofheterogeneity and surface phenomena.
5.4. Wet oxidation (unit operation D)
During bleaching, lignin and its fragmentsenter the POM bleaching liquor. To achievemill closure, it is necessary to remove them. Inunit operation D, the POM complexes can beused to catalyze and initiate the aerobic degra-dation (wet oxidation) of the dissolved organiccompounds to carbon dioxide and water [76].POM complex 1 was used to demonstrate thefeasibility of wet oxidation because its reducedform reacts rapidly with oxygen. Initial studiesusing 1 in solution with model compounds sug-gested that addition of a ‘sacrificial reductant’might be necessary to maintain a concentrationof energetic oxygen-centered radicals highenough to degrade recalcitrant, partially oxi-dized organic species as the reaction neared
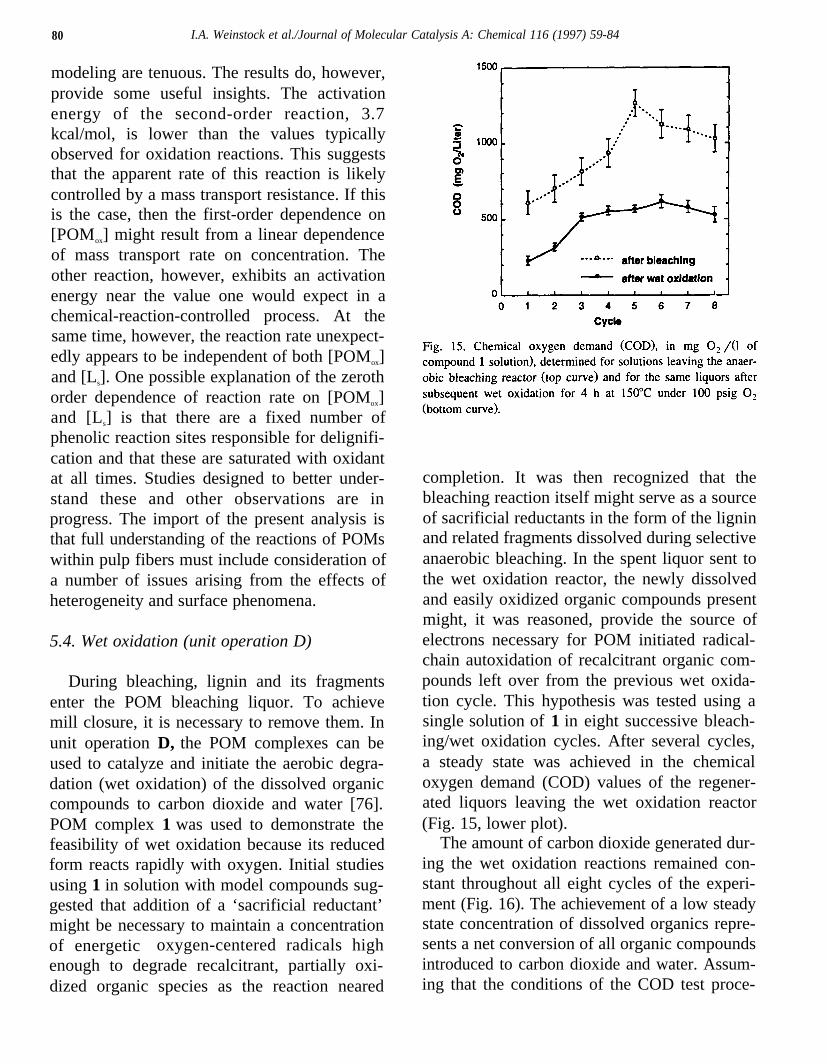
completion. It was then recognized that thebleaching reaction itself might serve as a sourceof sacrificial reductants in the form of the ligninand related fragments dissolved during selectiveanaerobic bleaching. In the spent liquor sent tothe wet oxidation reactor, the newly dissolvedand easily oxidized organic compounds presentmight, it was reasoned, provide the source ofelectrons necessary for POM initiated radical-chain autoxidation of recalcitrant organic com-pounds left over from the previous wet oxida-tion cycle. This hypothesis was tested using asingle solution of 1 in eight successive bleach-ing/wet oxidation cycles. After several cycles,a steady state was achieved in the chemicaloxygen demand (COD) values of the regener-ated liquors leaving the wet oxidation reactor(Fig. 15, lower plot).
The amount of carbon dioxide generated dur-ing the wet oxidation reactions remained con-stant throughout all eight cycles of the experi-ment (Fig. 16). The achievement of a low steadystate concentration of dissolved organics repre-sents a net conversion of all organic compoundsintroduced to carbon dioxide and water. Assum-ing that the conditions of the COD test proce-
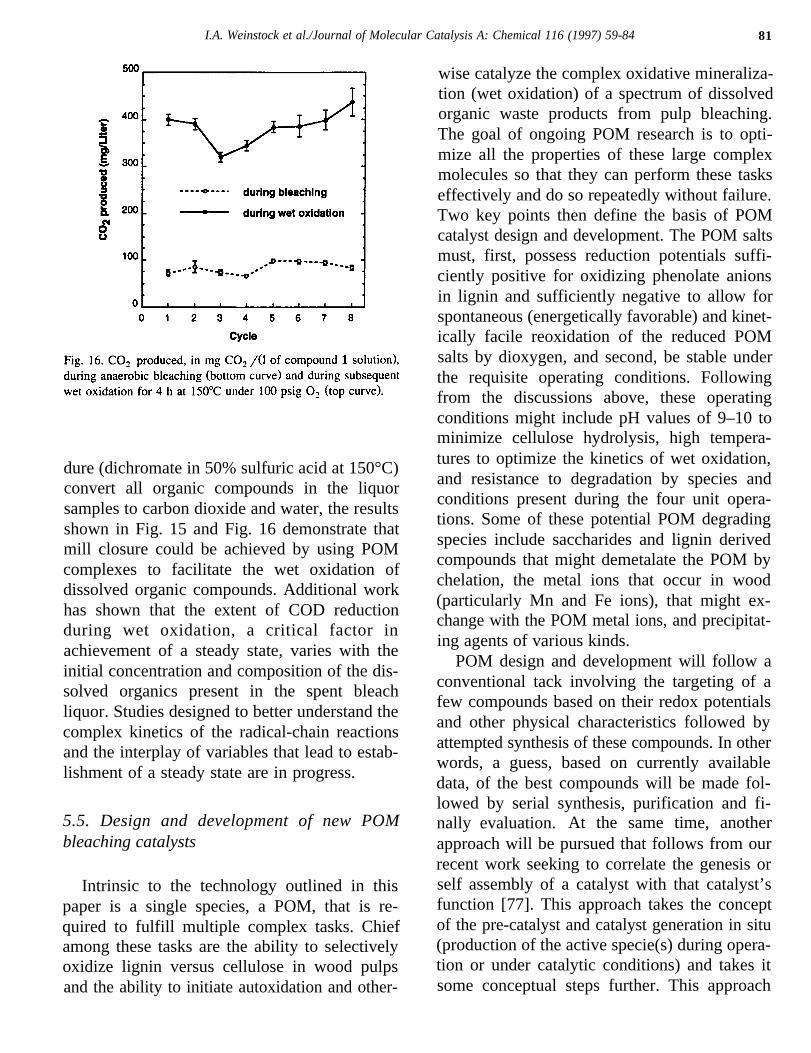
I.A. Weinstock et al./Journal of Molecular Catalysis A: Chemical 116 (1997) 59-84 81
dure (dichromate in 50% sulfuric acid at 150°C)convert all organic compounds in the liquorsamples to carbon dioxide and water, the resultsshown in Fig. 15 and Fig. 16 demonstrate thatmill closure could be achieved by using POMcomplexes to facilitate the wet oxidation ofdissolved organic compounds. Additional workhas shown that the extent of COD reductionduring wet oxidation, a critical factor inachievement of a steady state, varies with theinitial concentration and composition of the dis-solved organics present in the spent bleachliquor. Studies designed to better understand thecomplex kinetics of the radical-chain reactionsand the interplay of variables that lead to estab-lishment of a steady state are in progress.
5.5. Design and development of new POMbleaching catalysts
Intrinsic to the technology outlined in thispaper is a single species, a POM, that is re-quired to fulfill multiple complex tasks. Chiefamong these tasks are the ability to selectivelyoxidize lignin versus cellulose in wood pulpsand the ability to initiate autoxidation and other-
wise catalyze the complex oxidative mineraliza-tion (wet oxidation) of a spectrum of dissolvedorganic waste products from pulp bleaching.The goal of ongoing POM research is to opti-mize all the properties of these large complexmolecules so that they can perform these taskseffectively and do so repeatedly without failure.Two key points then define the basis of POMcatalyst design and development. The POM saltsmust, first, possess reduction potentials suffi-ciently positive for oxidizing phenolate anionsin lignin and sufficiently negative to allow forspontaneous (energetically favorable) and kinet-ically facile reoxidation of the reduced POMsalts by dioxygen, and second, be stable underthe requisite operating conditions. Followingfrom the discussions above, these operatingconditions might include pH values of 9–10 tominimize cellulose hydrolysis, high tempera-tures to optimize the kinetics of wet oxidation,and resistance to degradation by species andconditions present during the four unit opera-tions. Some of these potential POM degradingspecies include saccharides and lignin derivedcompounds that might demetalate the POM bychelation, the metal ions that occur in wood(particularly Mn and Fe ions), that might ex-change with the POM metal ions, and precipitat-ing agents of various kinds.
POM design and development will follow aconventional tack involving the targeting of afew compounds based on their redox potentialsand other physical characteristics followed byattempted synthesis of these compounds. In otherwords, a guess, based on currently availabledata, of the best compounds will be made fol-lowed by serial synthesis, purification and fi-nally evaluation. At the same time, anotherapproach will be pursued that follows from ourrecent work seeking to correlate the genesis orself assembly of a catalyst with that catalyst’sfunction [77]. This approach takes the conceptof the pre-catalyst and catalyst generation in situ(production of the active specie(s) during opera-tion or under catalytic conditions) and takes itsome conceptual steps further. This approach

82 I.A. Weinstock et al./Journal of Molecular Catalysis A: Chemical 116 (1997) 59-84
states that if the system is designed so that thecatalyst forms not only from its components atthe outset of the catalytic process but also dur-ing and at the end of the process, it will have agreater intrinsic stability with respect to thecatalysis itself. Self assembly of a catalyst dur-ing the height of the reaction and at the end ofthe reaction implicates a stability of the catalystwith respect to damaging intermediates as wellas inhibitors or poisons often present at the endof a catalytic process. If such comprehensiveself assembly (spontaneous and kinetically facilecatalyst formation from all catalyst componentsunder all phases of the reaction) is demon-strated, a consequence is that some types ofcatalyst damage, specifically those involvingcatalyst degradation into its original syntheticcomponents, will be effectively ‘sensed’ by thecollective catalyst system and reversed [77]. Inother words such degradation will be followedby catalyst reconstitution, the catalyst will effec-tively ‘heal’ itself.
Two related practical extensions of this ap-proach deserve explication. First, the reactionconditions for the title technology should beused as the conditions of POM synthesis. ThePOMs that form under both the bleaching andwet oxidation conditions may well be moreintrinsically stable (and may have a limitedhealing capability as described above) than thosethat do not. Such syntheses are underway in-cluding the use of hydrothermal conditions simi-lar to those likely to be encountered during wetoxidation (Unit operation D, Fig. 5). Second,there should be more coordination intellectuallyand operationally between the companies thatmake catalysts and the companies that use them.Often one company makes the catalyst and haslittle or no interest in the detailed conditionsunder which the catalyst will be used and asecond company uses the catalyst and pays littleattention to the conditions under which the cata-lyst was made. Coordination of these efforts andfabrication of the catalyst(s) under the condi-tions of operation and processing should lead tobetter catalytic technology.
6. Concluding remarks
This article articulates a new approach, usingcomplex multicomponent molecules, polyox-ometalates (POMs), for the conversion of woodpulp into paper, an enterprise of major andlong-standing import (economic and environ-mental) and current technological challenge. Theapproach is unique by intrinsic design and im-plementation in that the versatile capabilities ofa single POM species are utilized to facilitatemultiple operations that sum to the selecriueconversion of wood pulp to paper and non-toxicproducts (H2O and CO2) using air as the oxi-dant and water as the only solvent. While thisapproach currently promises to be highly ‘green’as well as feasible energetically and otherwise,research will be required in a number of coordi-nated areas including a new approach to catalystsynthesis to bring optimal success to this effort.
Acknowledgements
We thank the USDA Forest Service, the De-partment of Energy, Office of Industrial Tech-nologies and member companies of the USDAForest Service, Forest Products LaboratoryPolyoxometalate Bleaching Consortium for sup-port and Osram Sylvania, and Shieldalloy Met-allurgical Corporation for gifts of tungsten andvanadium, respectively. I.A. Weinstock and K.E.Hammel thank the USDA National ResearchInitiative Competitive Grants Program (93-37103-9270) for support of parts of this work.
References

I.A. Weinstock et al./Journal of Molecular Catalysis A: Chemical 116 (1997) 59-84 83

84 I.A. Weinstock et al./Journal of Molecular Catalysis A: Chemical 116 (1997) 59-84




















