
Disclosures-on-Risk-Based-Capital-Adequacy-Basel-III-for-the ...
Upload
khangminh22Category
view
6download
0
Cholestatic liver diseases of Infancy – a liver
biopsy pattern-based approach
Basel Seminars in Pathology Paediatric Pathology
and Genetics
Pierre Russo, MD
Director, Division of Anatomic Pathology
The Children’s Hospital of Philadelphia
Professor, Department of Pathology and Laboratory Medicine
Perelman School of Medicine at the University of Pennsylvania

Objectives
• To define neonatal cholestasis and neonatal hepatitis
• To outline the evaluation of neonatal cholestasis
• To review the potentially fatal / treatable etiologies of
neonatal cholestasis where the correlation between
clinical features and histopathology is critical
• To illustrate various histologic patterns in liver biopsies
from cholestatic infants and their differential diagnosis

Pediatric Liver Disease
• Children are not little adults
• However, many adult liver diseases are also seen in
pediatric patients
– Hepatitis B
– Hepatitis C
– Autoimmune hepatitis
• Unique to pediatric hepatology
– Neonatal cholestasis
– Emphasis on metabolic and genetic disorders

Definitions
• Cholestasis – impairment of bile formation or reduction of bile
flow from the liver.
– Bile acids
– Bilirubin
– Cholesterol
– Organic anions, drugs and toxins
• Hepatitis - inflammation of the liver
• Jaundice – discoloration of skin, sclera or mucous
membranes resulting from elevated unconjugated or
conjugated bilirubin
• Conjugated hyperbilirubinemia
> 2.0 mg/dl or >15% of total bilirubin

15% Of Neonates Develop Jaundice
Physiologic vs pathologic jaundice
• Physiologic jaundice – essentially means unconjugated hyperbilirubinemia - common and nearly never dangerous
• Neonatal cholestasis is always pathologic, and essentially means conjugated hyperbilirubinemia (with a few exceptions)
• Neonatal cholestasis - an emergency with potential for fatal outcome and chronic disease. It is never normal.
• Any infant with jaundice after 2 weeks of age should have a fractionated bilirubin to assess for conjugated hyperbilirubinemia

Differential Diagnosis Of Neonatal Cholestasis
Neonatal Hepatitis
• Idiopathic NH
• Viral NH
– CMV
– Herpes
– Rubella
– Reovirus
– Adenovirus
– Enteroviruses
– Parvovirus B19
– Paramyxovirus
– Hepatitis B
– HIV
• Bacterial and parasitic
– bacterial sepsis
– UTI
– Syphilis
– Listeriosis
– Toxoplasmosis
– Tuberculosis
– Malaria
Bile duct obstruction
• Cholangiopathies
– Biliary atresia
– Choledochal cysts
– Nonsyndromic Paucity
– Alagille syndrome
– Sclerosing Cholangitis
– Spontaneous duct perforation
– Caroli disease
– Congenital hepatic fibrosis
– Bile duct stenosis
• Other
– Inspissated bile/mucus
– Cholelithiasis
– Tumors
– Masses
Cholestatic syndromes
• PFIC
– Type 1 Byler P-type ATPase
– Type 2 Canalicular Bile Acid Tx
– Type 3 MDR3 deficiency
• Aagenaes cholestasis lymphedema
• N. Am. Indian Cholestasis
• Nielsen Greenland Eskimo cholestasis
• Benign Recurrent Intrahepatic cholestasis
• Dubin Johnson MRP2 cMOAT deficiency
• Rotor syndrome
Metabolic disorders
• a1-antitrypsin deficiency
• Cystic fibrosis
• Neonatal iron storage disease
• Endocrinopathies
– Hypopituitarism
– Hypothyroidism
• Amino acid disorders
– Tyrosinemia
– Hypermethionemia
– Mevalonate kinase deficiency
• Lipid disorders
– Niemann-Pick A, B
– Niemann-Pick C
– Gaucher
– Wolman
– Cholesterol ester storage ds
• Urea cycle disorders
– Arginase deficiency
• Carbohydrate disorders
– Galactosemia
– Fructosemia
– Glycogen storage IV
• Mitochondrial disorders
– Oxidative phosphorylation
• Peroxisomal disorders
– Zellweger
– Infantile Refsum
– Other enzymopathies
• Bile acid synthetic disorders
– 3b-hydroxysteroid dehydrogenase/i
– D4-3-oxosteroid 5b-reductase
– Oxosterol 7a-hydroxylase
Toxic
• Drugs
• Parenteral alimentation
• Aluminum
Miscellaneous associations
• Shock/hypoperfusion
• Histiocytosis X
• Neonatal lupus erythematosus
• Indian childhood cirrhosis
• Autosomal trisomies 17, 18, 21
• Graft v host disease
• Erythrophagocytic lymphohistiocytosis
• ECMO
• Veno-occlusive disease
• Donahue leprechaunism
• Arthrogryposis cholestasis
• Erythroblastosis fetalis

Classification of Neonatal Cholestasis
Extrahepatic Intrahepatic
Biliary atresia
Choledochal cyst
Bile duct stenosis
Stones
Tumors
Neonatal hepatitis
Alagille syndrome
Galactosemia
CMV
TPN
Α1- antitrypsin deficiency
There is overlap in clinical presentation, laboratory abnormalities, and histology.

Guiding Principles for Evaluation of the
cholestatic infant
• There are over 100 etiologies
• Many complex, rare metabolic diseases which have new and
effective therapies
• Design initial evaluation according to:
– What is dangerous and treatable?
– What is common?
– What patterns provide clues for specific etiologies?

Etiologies Of Neonatal Conjugated Hyperbilirubinemia
n= %age
Biliary atresia 377 34.7%
Idiopathic neon. hepatitis 331 30.5%
a1-antitrypsin deficiency 189 17.4%
Other hepatitis 94 8.7%
Alagille syndrome 61 5.6%
Choledochal cyst 34 3.1%
Mieli-Vergani G, et al. Lancet 1989. King’s College
Hospital

Important Critical Etiologies:
Early Treatment Improves Outcome
• Biliary atresia Kasai portoenterostomy
• Extrahepatic obstruction Surgery
• Galactosemia Lactose restriction
• Tyrosinemia NTBC
• Hypopituitarism Cortisol, thyroxine
• Bile acid synthetic defects Cholic acid
• Sepsis and urinary tract infection Antibiotics
• Syphilis Antibiotics
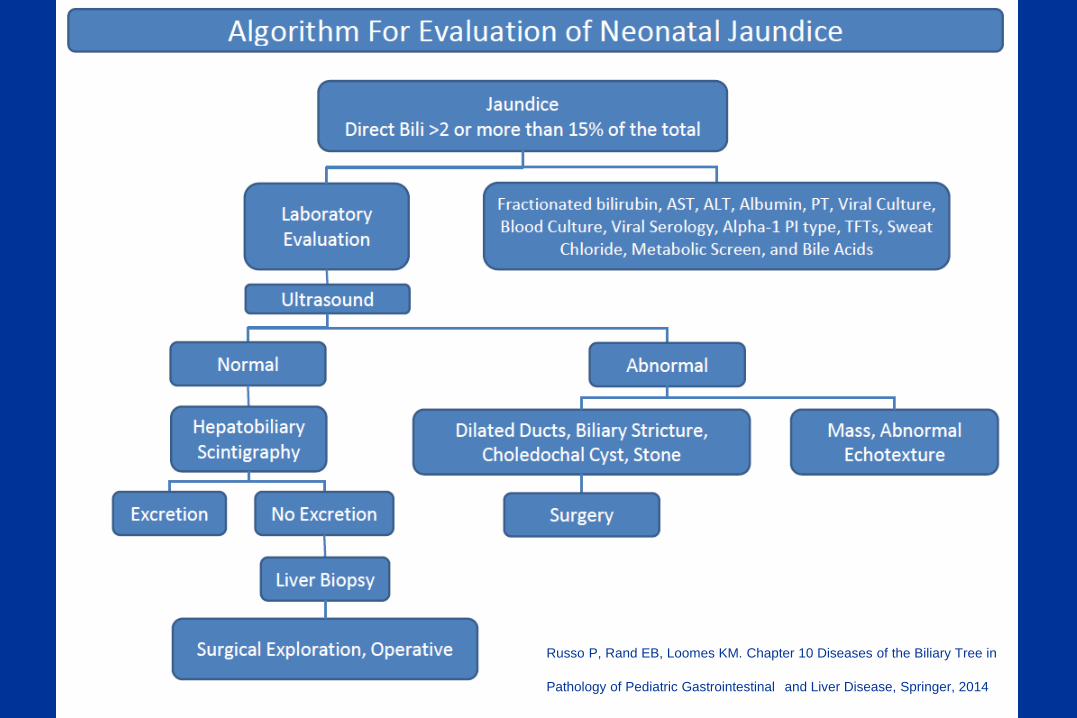
Russo P, Rand EB, Loomes KM. Chapter 10 Diseases of the Biliary Tree in
Pathology of Pediatric Gastrointestinal and Liver Disease, Springer, 2014

Percutaneous Liver Biopsy
A useful, commonly essential, and many times mandatory step in the evaluation of neonatal and infantile liver disease prior to OR cholangiogram.
Collaboration – communication
Amount of tissue needed
Special studies or assays
PCR or cultures
Special stains
Electron microscopy

The Clues to Diagnosis
Jaundice Physiologic --- Pathologic
Bilirubin Unconjugated --- Conjugated
Site Intrahepatic --- Extrahepatic
Type Metabolic --- Anatomic
Histology Hepatitis,Paucity--- Proliferation
GGT Low --- High
Systemic Abnormalities --- Isolated
Synthesis Normal --- Abnormal

The liver biopsy – major histologic patterns in
neonatal cholestatic disorders
• Obstructive pattern
• Giant cell Hepatitis
• Intrahepatic bile duct paucity
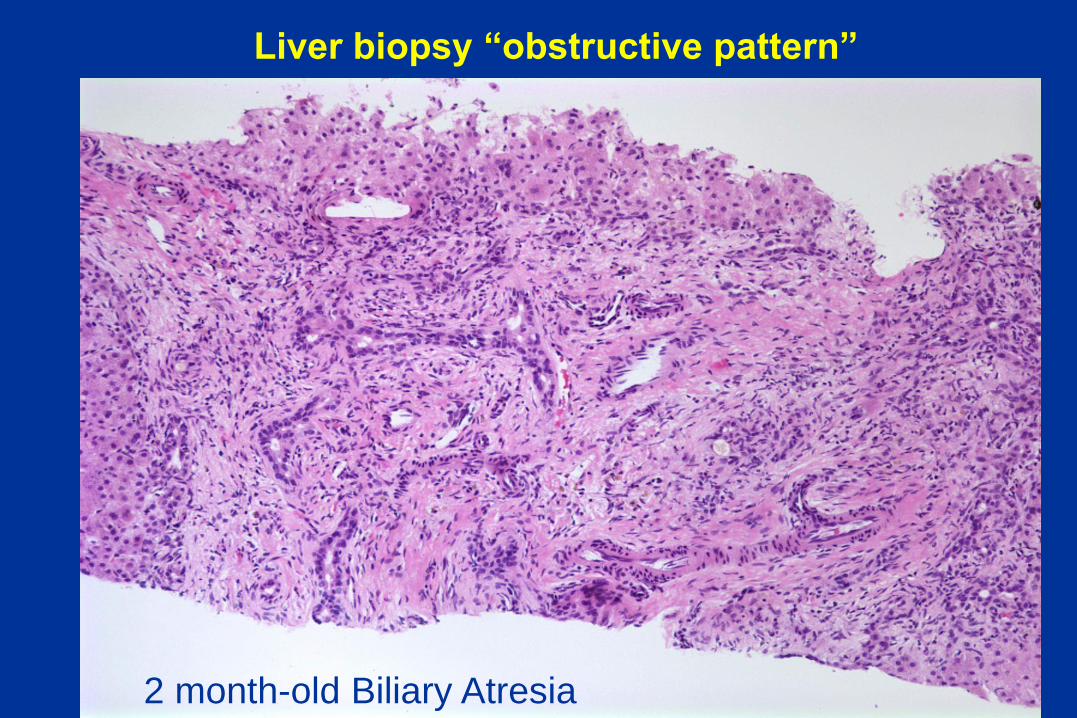
Liver biopsy “obstructive pattern”
2 month-old Biliary Atresia

Biliary Atresia
• Most severe chronic liver
disease of infancy
• Fibro-inflammatory
obliteration of the biliary
tree
• Etiology unknown (viral,
autoimmune, toxic)
• 1/8,000 live births in US
(400 cases/yr); higher in
Asia
• Untreated, death from
cirrhosis < 1 year of age
• Most common cause of
pediatric liver
transplantation worldwide

Diagnosis Of Biliary Atresia
Typical scenario
Well appearing child
Days to weeks old
Acholic stools, dark urine
Mild icterus
Hepatosplenomegaly
Conjugated bilirubin
Mildly elevated ALT
Elevated GGT
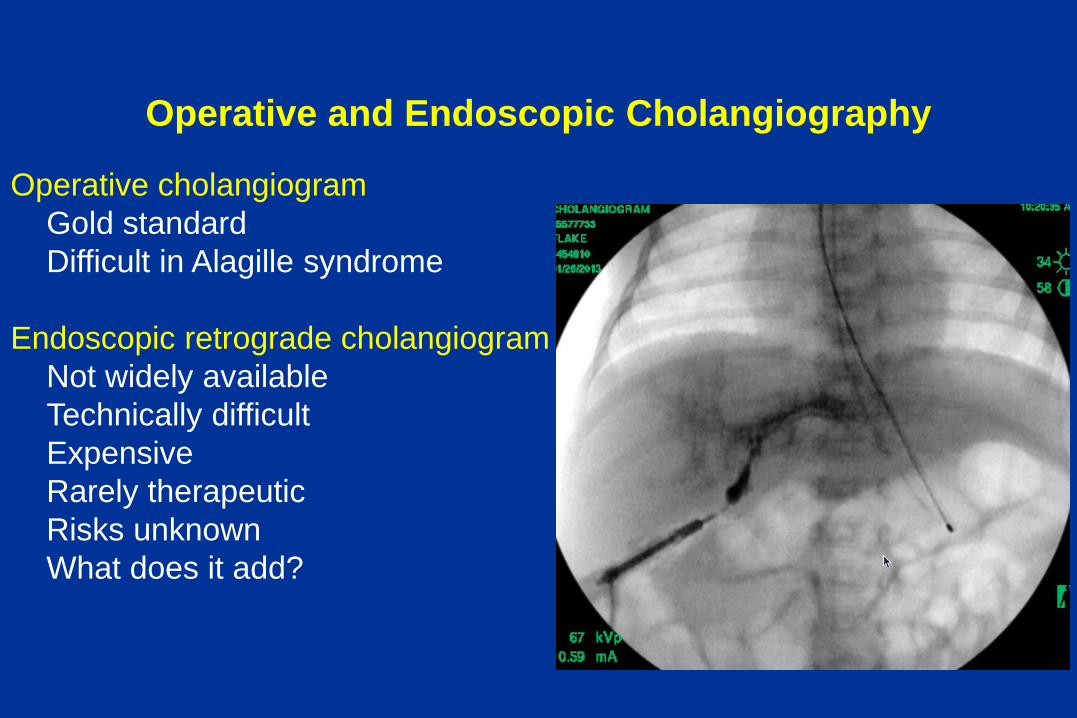
Operative and Endoscopic Cholangiography
Operative cholangiogram
Gold standard
Difficult in Alagille syndrome
Endoscopic retrograde cholangiogram
Not widely available
Technically difficult
Expensive
Rarely therapeutic
Risks unknown
What does it add?
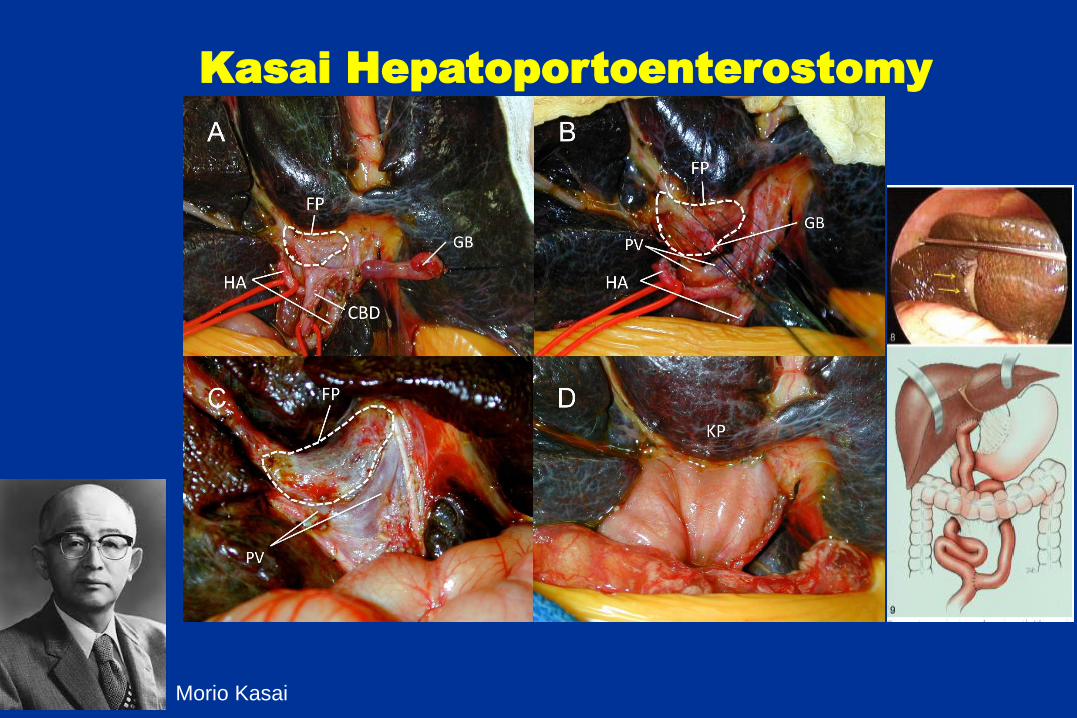
Kasai Hepatoportoenterostomy
Morio Kasai

Biliary Atresia – the remnant
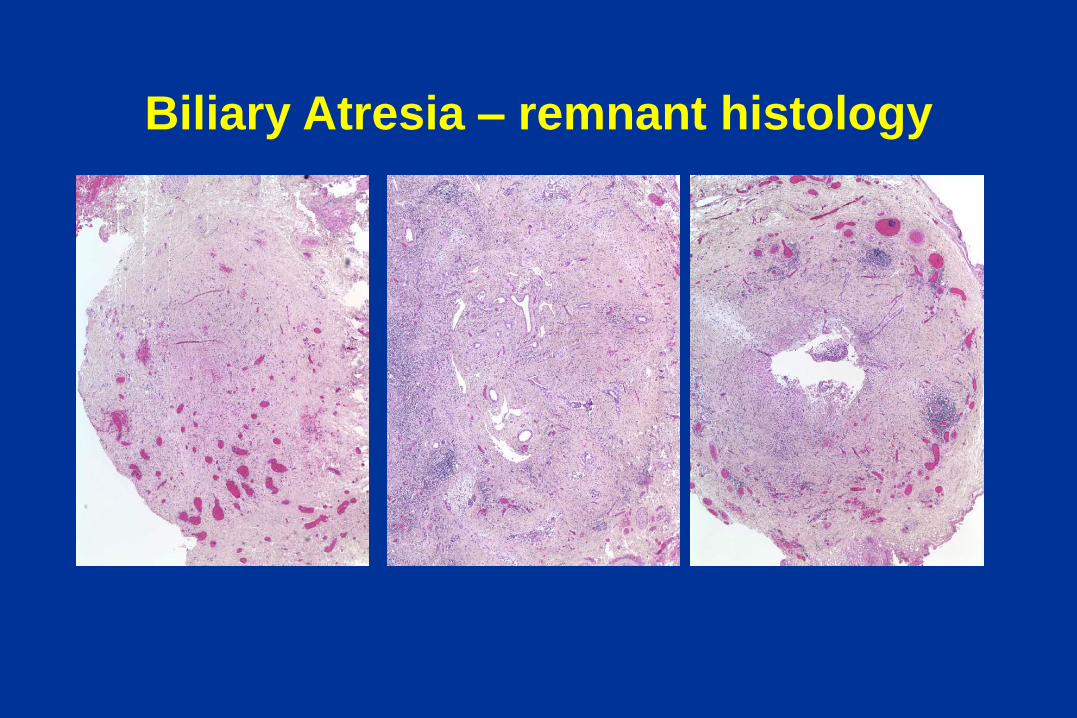
Biliary Atresia – remnant histology

Hepatic ducts
Normal, 40 weeks BA, 2 months
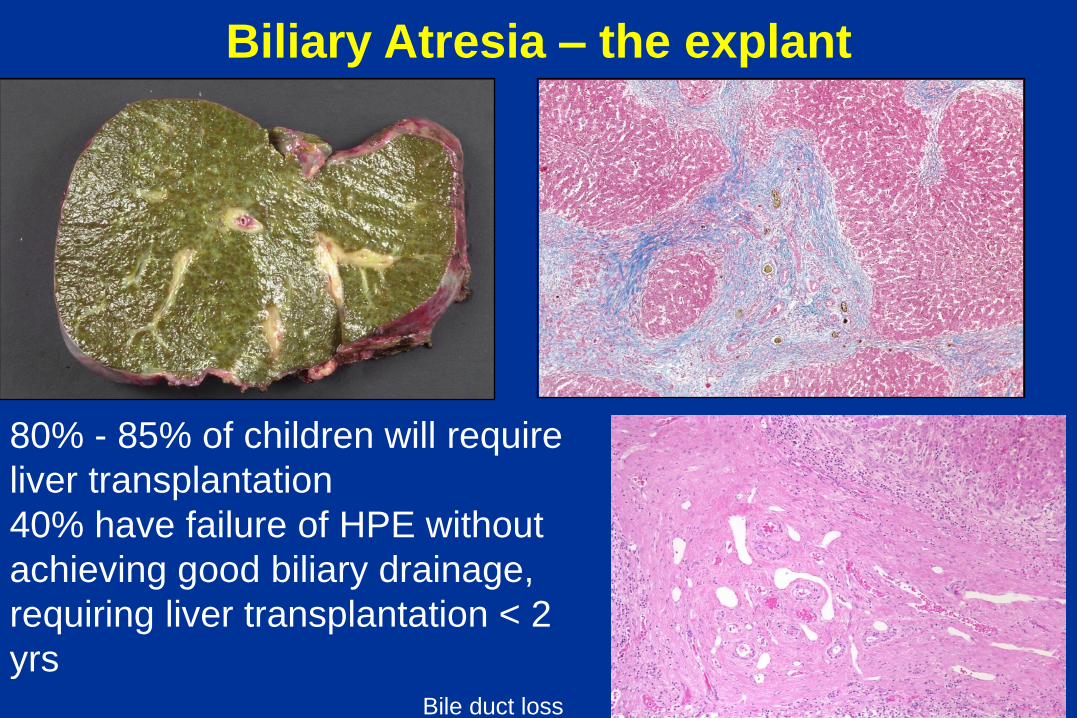
Biliary Atresia – the explant
80% - 85% of children will require
liver transplantation
40% have failure of HPE without
achieving good biliary drainage,
requiring liver transplantation < 2
yrs Bile duct loss

Differential Diagnosis of Obstructive Cholestasis
on Liver Biopsy
Alpha-1 antitrypsin deficiency low or absent serum alpha-1 antitrypsin level,
PiZZ phenotype
Choledochal cyst
Ultrasonographic and cholangiographic
findings
Alagille syndrome (early) Associated malformations, mutation in JAG1
or Notch2 gene
Cystic fibrosis History of meconium ileus, failure to thrive,
positive sweat test
TPN hepatopathy
History of prematurity and TPN use
Neonatal sclerosing cholangitis Ichthyosis, scarring alopecia, dysmorphism,
Claudin-1 mutations
Progressive familial cholestasis
type 3, ABCB4 disease
Usually older infants at presentation; absence
of immunohistochemical staining for MDR3
along canaliculi
Galactosemia Extensive fatty change
Tumor, stone or other mechanical
obstruction

a1 ANTITRYPSIN DEFICIENCY
Emphysema Neonatal hepatitis Cirrhosis
Disease due to mutations of protease inhibitor (Pi) gene (chr 14)

a1-Antitrypsin Deficiency
7 week-old with a1-AT deficiency

α1 Antitrypsin Deficiency
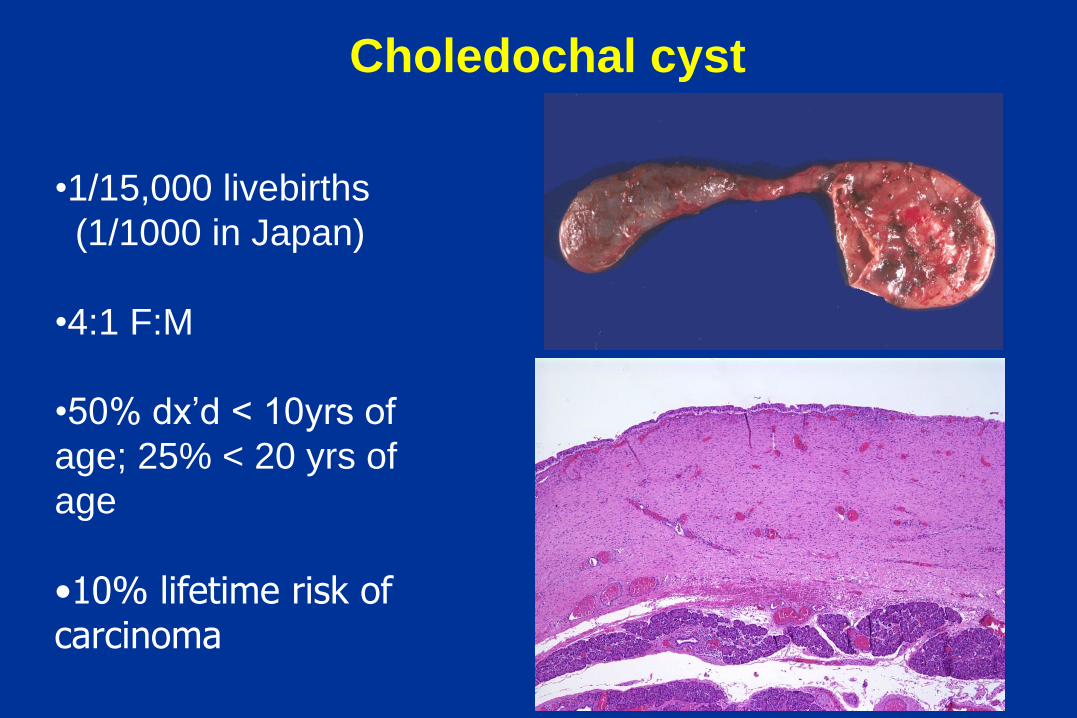
Choledochal cyst
•1/15,000 livebirths
(1/1000 in Japan)
•4:1 F:M
•50% dx’d < 10yrs of
age; 25% < 20 yrs of
age
•10% lifetime risk of carcinoma

Obstructive cholestasis - Parenteral
Nutrition
3 months

Parenteral Nutrition
• 2 month-old ex 32 week premie
with jejunal atresia s/p resection
and reanastomosis, on TPN
majority of his life
• Now off TPN but still cholestatic,
please evaluate for BA
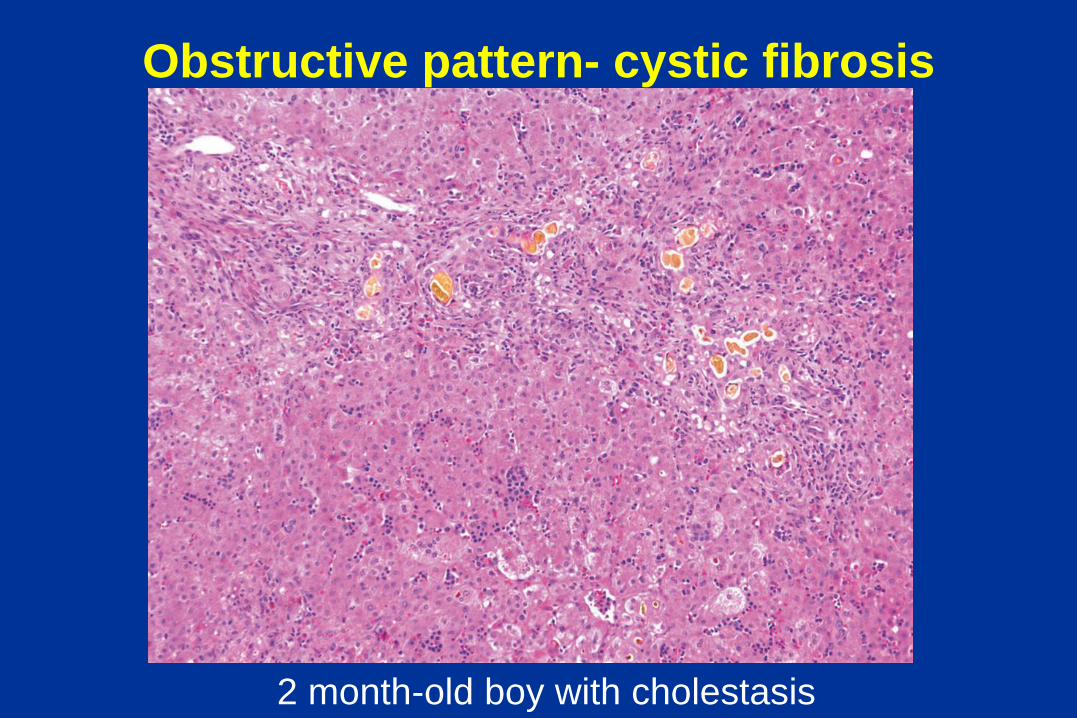
Obstructive pattern- cystic fibrosis
2 month-old boy with cholestasis

Obstructive pattern - Galactosemia
• Autosomal Recessive
• FTT, vomiting, diarrhea
• E. coli septicemia
• Hemolytic anemia
• Progression to cirrhosis in
6 months if untreated
• Treatment is dietary
removal of lactose
• Neonatal screening
available
Obstructive pattern with fat in liver
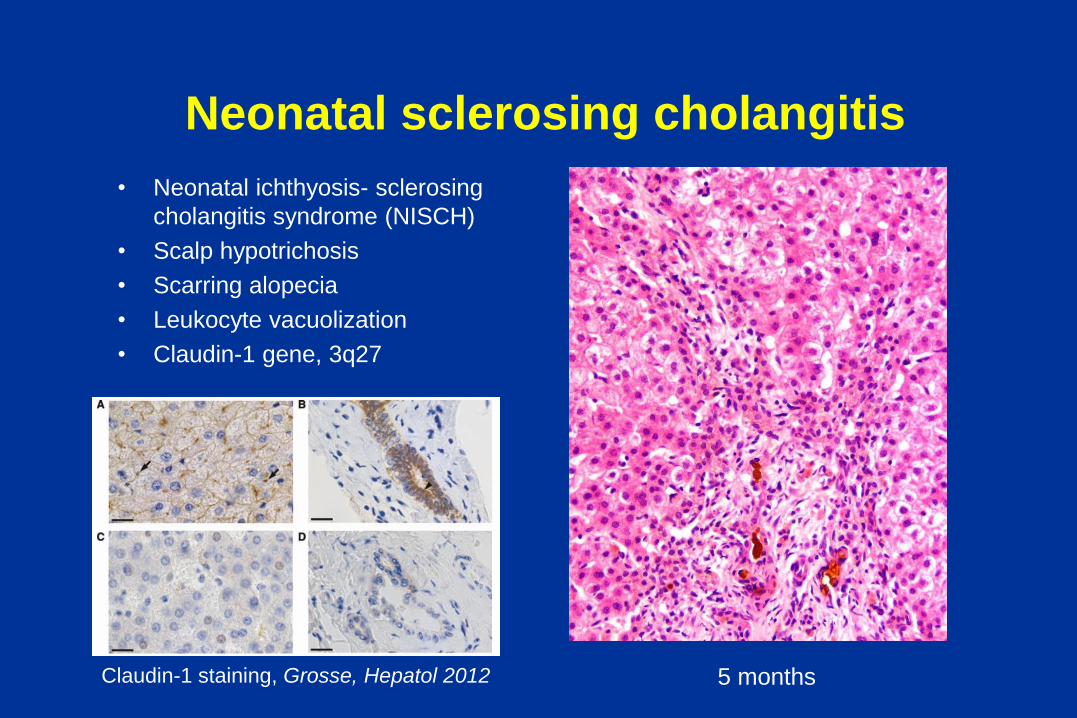
Neonatal sclerosing cholangitis
• Neonatal ichthyosis- sclerosing
cholangitis syndrome (NISCH)
• Scalp hypotrichosis
• Scarring alopecia
• Leukocyte vacuolization
• Claudin-1 gene, 3q27
5 months Claudin-1 staining, Grosse, Hepatol 2012

Obstructive pattern - Langerhans cell
histiocytosis
8 month-old girl with cholestasis
and skin rash
CD1a
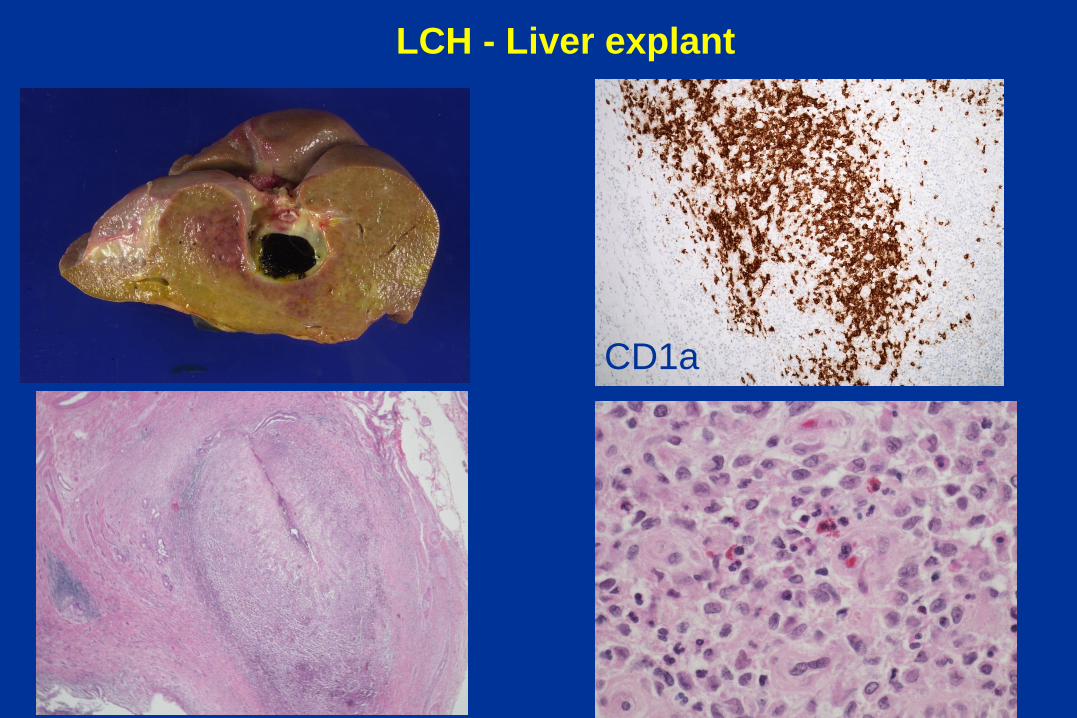
LCH - Liver explant
CD1a
The liver biopsy – major histologic patterns in
neonatal cholestatic disorders
• Obstructive pattern
• Giant cell Hepatitis
• Intrahepatic bile duct paucity

Neonatal Giant Cell Hepatitis

Disorders with a Neonatal Giant Cell Hepatitis
phenotype
• Idiopathic
• Infections (TORCH)
• Progressive familial cholestasis
• Bile acid synthetic disorders
• Metabolic disease
• Hypopituitarism
• Biliary Atresia

Torbenson, AJSP 2010

PC
ABCB4
MDR3
OA
ABCC2
ABCB11
BSEP
BA
ATP8B1
PS?
Cholangiocyte
CFTR
TJ 2
Cl -
BA
SLC10A2
Bile
flow
PL
ATP8B1
BCS1L
CIRHA1
PS?
BAAT
UGT1A1
Familial Cholestatic Disorders
Hepatocyte

Progressive Familial Intrahepatic
Choestasis type 2 (PFIC 2, ABCB 11)
2 months, Giant cell hepatitis and
low serum GGT cholestasis
BSEP control
BSEP BSEP

J Peds 2007, 150:556

Bile Acid Synthesis Defects
• Nine different defects in primary synthetic pathway identified
• Defects presenting with neonatal hepatitis with low GGT
cholestasis and abnormal bile acids in blood, bile or urine
• Severe disease result from enzyme defects acting on sterol
nucleus and present early in life
– 3β-hydroxy δ5-C27 steroid dehydrogenase
– Δ 4-3-oxosteroid 5β-reductase
• Disorders of bile acid conjugation present later in life with
failure to thrive and fata malabsorption
• Mass spectrometry analysis of urine bile acids
• Treatment with bile acids to restore physiologic bile acid
function and to decrease synthesis of toxic abnormal bile
acids

Δ 4-3-oxosteroid 5β-reductase Giant cell hepatitis
Fibrosis
Distorsion of canaliculi
on EM

3β-hydroxy δ5-C27 steroid
dehydrogenase Pre-treatment Post-treatment

A case of Neonatal Giant cell hepatitis
Clinical Vignette
• This full-term 2 month-old male was referred to The Children’s Hospital of Philadelphia for jaundice and scleral icterus
• At 4 days of life, he was noted to have a total bili 12.8 (direct bili 1.8) He had no further labs until his referral to CHOP
• He underwent a work up that revealed hepatosplenomegaly, cholestasis, and elevated transaminases.
• Abdominal ultrasound showed nonspecific hepatosplenomegaly, contracted gallbladder, and trace perihepatic ascites and doppler revealed normal flow.
• DISIDA excreted.
• Work up included normal urinalysis, negative blood cultures and negative testing for CMV, HSV, adenovirus, enterovirus, and parechovirus.
• He underwent a liver biopsy

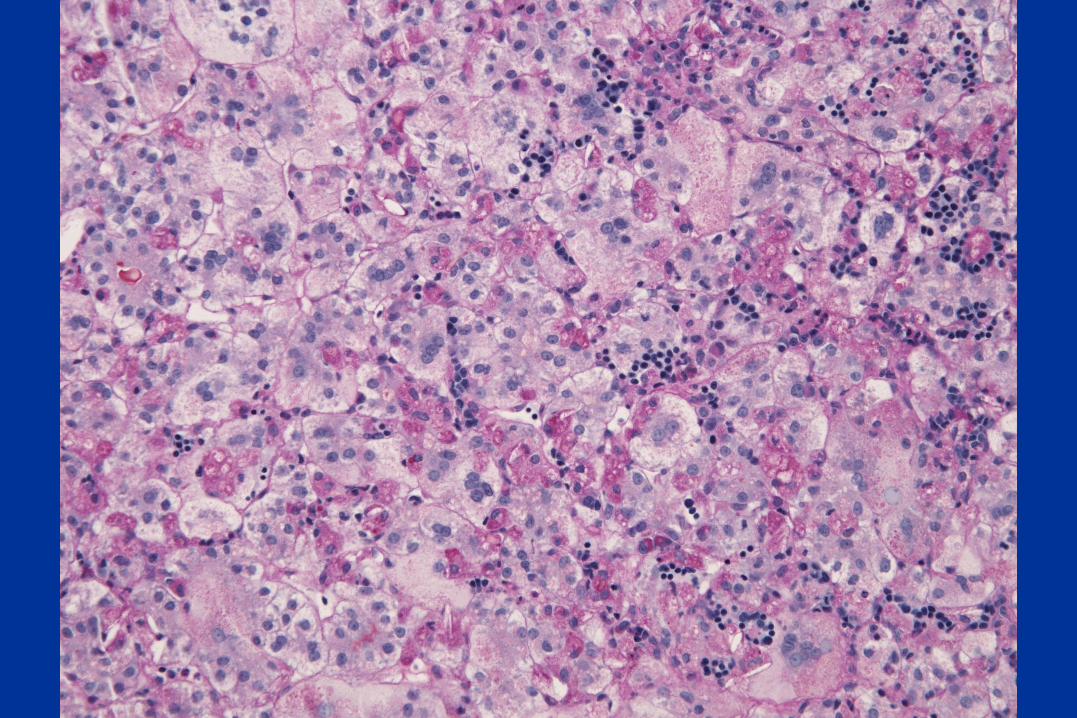
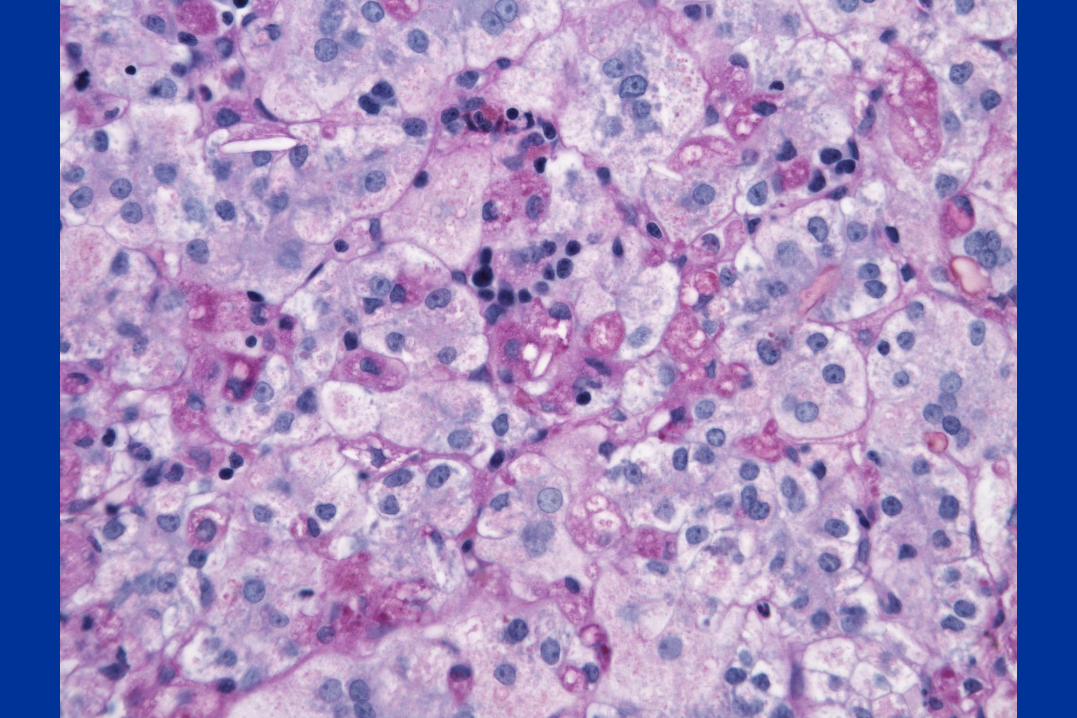

Diagnosis:
Niemann-Pick type C
mutation in NPC 1
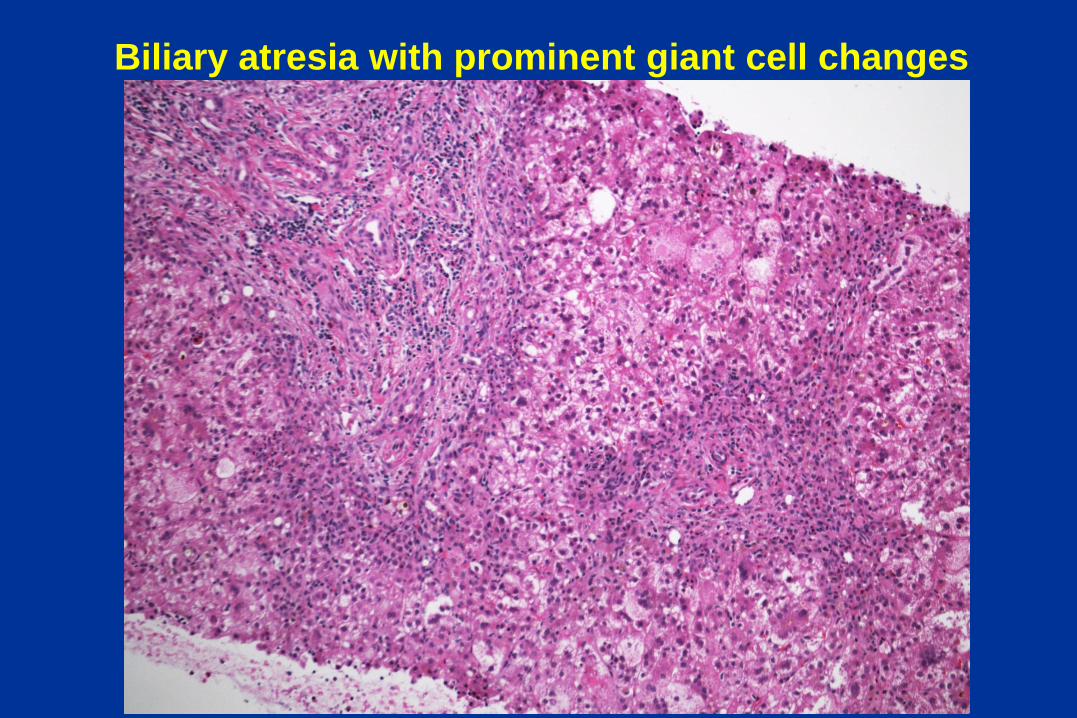
Biliary atresia with prominent giant cell changes
The liver biopsy – major histologic patterns in
neonatal cholestatic disorders
• Obstructive pattern
• Giant cell Hepatitis
• Bile duct paucity

Bile duct paucity

Definition of Bile Duct Paucity
• Duct / portal tract ratio in full-term infants, children and adults is 0.9 to 1.8
• Ratio is lower in premature infants (physiological)
• May be transient in some cases
• Paucity = duct / portal tract ratio < 0.5 (in children)
• Must have adequate # of portal tracts (10)
• Comparison with hepatic artery may be more reliable
– Ratio of BD/HA = 1 with comparable external diameters

Over-reporting of Bile Duct Paucity
• Non-pathologist authors
• Self-perpetuating references
• Sampling or observational error
– Non-representative biopsy
– Sampling of peripheral portal tracts in premature infants and infants < 1month of age (bile ducts develop up to first month of life)

Conditions Associated with Bile Duct Paucity
Well documented association
Alagille syndrome
Alpha-1 antitrypsin deficiency
Peroxisomal disorders (Zellweger)
Congenital cytomegalovirus infection
Biliary Atresia (late)
Secondary feature
Graft versus Host disease
Chronic hepatic allograft rejection
Primary biliary cirrhosis
Primary (or 2ary) sclerosing cholangitis
Sarcoidosis
Hodgkin disease (post-treatment)
Drug-associated (antibiotics)
Occasionally documented associations
Cystic fibrosis
HNF-1β mutations (renal cysts, diabetes)
PFIC
Familial hemophagocytic lymphohistiocytosis
Arthrogryposis-renal dysfunction-cholestasis
Niemann-Pick type C
Mitochondrial DNA depletion syndrome
Trisomy 18
Trisomy 21
Prune Belly syndrome
Williams syndrome
Congenital syphilis
Congenital rubella

•Autosomal Dominant
•Mutation in JAG1
(chr. 20p12) •Multi-system
•Liver, heart,
•Skeleton, eye,
•Face, kidney,
•vasculature
Alagille Syndrome (ALGS)
PA/PPS 67%
PVS 3%
TOF 16%
ASD 4%
PDA 5%
VSD +ASD 1%

Bile duct paucity – Alpha 1 antitrypsin deficiency
2 month-old with cholestasis

Bile duct paucity – Alpha 1 antitrypsin deficiency
Liver explant at 8 months of age

Summary points
• Hepatic histology is an essential component of the
diagnostic algorithm of the cholestatic infant
• The pathologist must be familiar with the differential
diagnosis of various histologic patterns in the liver biopsy
and communicate effectively with the clinician
• Integration of clinical and laboratory findings with histologic
features and close collaboration between hepatologist and
pathologist are essential for accurate and timely diagnosis

You find only what you look for, you seek only what you know
Copyright © 2022 FDOKUMEN




















