
34248.pdf - Corpus UL
350
© Alice Anaïs Varlet, 2018 Analyse fonctionnelle du remodelage des structures à base d'actine qui dirigent la division cellulaire par le complexe chaperon HSPB8-BAG3 Thèse Alice Anaïs Varlet Doctorat en biologie cellulaire et moléculaire Philosophiæ doctor (Ph. D.) Québec, Canada
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of 34248.pdf - Corpus UL

© Alice Anaïs Varlet, 2018
Analyse fonctionnelle du remodelage des structures à base d'actine qui dirigent la division cellulaire par le
complexe chaperon HSPB8-BAG3
Thèse
Alice Anaïs Varlet
Doctorat en biologie cellulaire et moléculaire
Philosophiæ doctor (Ph. D.)
Québec, Canada

Analyse fonctionnelle du remodelage des structures à base d’actine qui dirigent la division cellulaire par le
complexe chaperon HSPB8-BAG3
Thèse
Alice-Anaïs Varlet
Sous la direction de :
Josée N. Lavoie, directrice de recherche

i
Résumé
Les changements dans la forme des cellules sont essentiels à de nombreux processus qui
déterminent le destin de la cellule, notamment la division cellulaire. Ils sont orchestrés par
le remodelage de structures mécanosensibles à base d’actine contrôlant la tension
cellulaire. Les évidences récentes laissent croire que le contrôle de qualité des protéines
pourrait contribuer à la régulation spatiotemporelle du remodelage des structures d’actine
par des mécanismes de séquestration, de recyclage et de dégradation protéiques. Les
chaperons moléculaires de la famille des HSPB apparaissent comme des modulateurs des
structures à base d’actine en conditions physiologiques. Durant un stress protéotoxique,
ces protéines séquestrent des composantes cellulaires endommagées afin de prévenir leur
agrégation. Bien que leur implication dans différentes pathologies soit démontrée, le mode
d’action des HSPB demeure encore peu compris. Les travaux effectués dans le cadre de
cette thèse adressent l’hypothèse centrale que le complexe chaperon formé d’HSPB8 et de
son co-chaperon BAG3 régule le remodelage des structures à base d’actine qui dirigent la
division cellulaire.
Nos travaux ont établi que lors de la mitose, BAG3, d’une manière dépendante de son
association avec HSPB8, facilite l’arrondissement mitotique requis pour le positionnement
du fuseau et la ségrégation adéquate des chromosomes. Nous montrons que la réduction
des niveaux d’HSPB8 ou de BAG3 interfère également avec le désassemblage de l’anneau
contractile d’actomyosine et l’abscission des cellules filles lors de la cytokinèse. Cet effet
est corrélé avec une augmentation de la prévalence de cellules multi-nucléées, laquelle est
restaurée à des niveaux contrôles par des drogues qui normalisent la dynamique de l’actine
dans les cellules déplétées en HSPB8. Ceci établit un lien de cause à effet entre la
dérégulation de la dynamique de l’actine et les défauts de division cellulaire observés dans
ces cellules. De plus, l’inhibition du désassemblage de l’anneau d’actomyosine est corrigée
par la rapamycine, une drogue qui active la dégradation par autophagie. Inversement, ce
phénotype est reproduit par les cellules contrôles traitées avec des drogues qui réduisent le
flux autophagique. Ces observations suggèrent que la régulation de la morphodynamique
mitotique par HSPB8-BAG3 impliquerait sa fonction dans l’autophagie sélective. En outre,

ii
pendant la mitose et la cytokinèse, HSPB8-BAG3 limiterait la polymérisation de l’actine
branchée. Notamment, HSPB8-BAG3 semble faciliter l’arrondissement mitotique et la
dynamique d’un réseau sous-cortical d’actine qui dépend d’Arp2/3, en modulant la
déacétylase HDAC6 et son substrat la cortactine, tous deux intervenants dans les processus
d’autophagie sélective.
L’ensemble de ces travaux contribue à une meilleure compréhension des mécanismes par
lesquels le complexe chaperon HSPB8-BAG3 facilite les transitions dans la forme des
cellules qui contrôlent des fonctions essentielles, notamment la division cellulaire. Ils
identifient de nouvelles cibles cellulaires du complexe chaperon impliquées dans le
remodelage des structures à base d’actine, lesquelles pourraient contribuer au
développement de pathologies associées avec une dérégulation du complexe, notamment à
la progression tumorale.

iii
Abstract
Changes in cell shape are essential to key cellular processes that determine the cell fate,
including cell division. They are orchestrated by the remodeling of mechanosensitive actin-
based structures guiding cellular tension. Recent data suggest that protein quality control
may contribute to the spatiotemporal remodeling of actin-based structures through protein
sequestration, recycling and degradation mechanisms. Incidentally, the molecular
chaperones of the HSPB family appear as modulator of actin-based structures under
physiological processes. These proteins are known to sequester damaged cellular
constituents to prevent their toxic aggregation during proteotoxic stress. Whilst their
implications in human pathologies have been clearly established, their mode of action is
still poorly understood. The work presented in this thesis addresses the central hypothesis
that the chaperone complex formed by HSPB8 and its co-chaperone BAG3 would facilitate
the remodeling of actin-based structures, which is deemed instrumental for proper mitotic
progression.
We have shown that during mitosis, BAG3, in a manner requiring its association with
HSPB8, facilitates mitotic rounding, spindle positioning and accurate chromosomes
segregation. We further showed that depletion of HSPB8 or BAG3 silencing also interferes
with disassembly of the contractile actomyosin ring during cytokinesis, thereby impairing
daughter cells abscission. Such an effect is correlated with accumulation of multinucleated
cells, a defect which is corrected by drugs that normalize actin dynamics in HSPB8-
depleted cells. These results established a cause-effect relationship between deregulation
of actin dynamics and the cell division defects induced by HSPB8 silencing. Further, we
found that such a phenotype can be rescued by rapamycin, a drug that stimulates
autophagy, while it is recapitulated in control cells by inhibitors of autophagic degradation.
These results, and others presented in this thesis, suggest that the regulation of cell shape
remodelling by HSPB8-BAG3 may involve their function in the selective targeting of
proteins for autophagic degradation. Finally, evidence was obtained that during mitosis,
like during cytokinesis, HSPB8-BAG3 would act by limiting branched actin
polymerization. We found that HSPB8-BAG3 can modulate mitotic rounding and the

iv
dynamics of an Arp2/3-dependent subcortical actin pool that contributes to spindle
positionning, by restricting the activity of HDAC6 deacetylase maybe on its target
cortactin, both having a role in selective autophagy processes.
Together, this work contributed to a better understanding of the mechanisms by which the
chaperone complex HSPB8-BAG3 would facilitate transitions in cell shape during cell
division and uncovered novel targets of HSPB8-BAG3 that may be implicated in the
development of human disorders associated with deregulation of the chaperon complex,
notably cancer.

v
Table des matières
Résumé ................................................................................................................................ i
Abstract ............................................................................................................................. iii
Table des matières ............................................................................................................. v
Liste des tableaux .............................................................................................................. x
Liste des figures ................................................................................................................ xi
Liste des abréviations .................................................................................................... xiii
Dédicace .......................................................................................................................... xvi
Remerciements .............................................................................................................. xvii
Avant-propos ................................................................................................................... xx
1. Chapitre 1 : Introduction ......................................................................................... 1
1.1. Rôle du cytosquelette d’actine dans la réponse cellulaire aux forces mécaniques . 1 1.2. Les chaperons de la famille HSPB et la régulation de la dynamique et de
l’intégrité des structures d’actine ............................................................................................. 3 1.2.1. Le contrôle de qualité des protéines et les chaperons moléculaires ...................... 3 1.2.2. Modèle prévalent des HSPB : l’oligomérisation pour prévenir de l’agrégation
protéique 6 1.2.3. Les HSPB dans le contrôle de la dynamique de l’actine et les pathologies
associées 7 1.3. Le complexe chaperon HSPB8-BAG3 dans la régulation de structures
contractiles d’actine et de l’autophagie sélective .................................................................. 13 1.3.1. HSPB8 : un membre particulier de la famille des HSPB avec une spécificité de
substrats? 13 1.3.2. Structure et fonctions du co-chaperon BAG3 ...................................................... 15 1.3.3. Les fonctions assurées par BAG3 en tant que protéine d’échafaudage ............... 17
1.3.3.1. Le co-chaperon BAG3 module l’apoptose à travers la régulation de la stabilité de
facteurs anti-apoptotiques ................................................................................................................ 18 1.3.3.2. BAG3 régule l’adhésion, la motilité cellulaire et la transition
épithéliomésenchymateuse .............................................................................................................. 19

vi
1.3.3.3. Lien fonctionnel entre l’activité signalétique de BAG3 et son rôle en tant que co-
chaperon d’HSP70 ........................................................................................................................... 21 1.3.4. Fonctions assurées par BAG3 lors d’un stress protéotoxique ............................. 22
1.3.4.1. L’Autophagie sélective et ses déterminants p62/SQSTM1 et HDAC6 ....................... 23 1.3.4.2. BAG3 cible les protéines poly-ubiquitinées ou dénaturées à la dégradation par
autophagie sélective ......................................................................................................................... 27 1.3.5. Régulation de structures contractiles d’actine par le complexe chaperon HSPB8-
BAG3 31 1.3.5.1. BAG3 assure l’intégrité des myofibrilles musculaires grâce à sa fonction de co-
chaperon d’HSP70 ........................................................................................................................... 31 1.3.5.2. Régulation de l’intégrité du cytosquelette d’actine dépendant de l’autophagie sélective
induite par HSPB8-BAG3 dans la cellule musculaire ..................................................................... 32 1.4. Contrôle de la division cellulaire par le remodelage des structures à base d’actine
35 1.4.1. Les régulateurs de la dynamique de l’actine impliqués dans la division cellulaire
37 1.4.1.1. Le nucléateur de l’actine banchée : le complexe Arp2/3 ............................................. 37 1.4.1.2. Les formines et la formation de faisceaux d’actine ...................................................... 39 1.4.1.3. La myosine II et la contractilité cellulaire .................................................................... 40 1.4.1.4. Les Rho GTPases ......................................................................................................... 43
1.4.2. L’arrondisssement mitotique et son implication dans l’orientation du fuseau
mitotique et la ségrégation des chromosomes ....................................................................... 45 1.4.2.1. Démantèlement des adhésions focales et mise en place des fibres de rétraction ......... 48 1.4.2.2. Fonctions essentielles de RhoA et de la myosine II dans la mise en place et le maintien
de la tension corticale à l’orgine de l’arrondissement mitotique ..................................................... 49 1.4.2.3. Contrôle du positionnement du fuseau mitotique par le nuage sous-cortical d’actine et
l’implication du complexe Arp2/3 dans ce processus ..................................................................... 52 1.4.2.4. Promotion de la rigidité corticale et du positionnement du fuseau mitotique par les
protéines ERM ................................................................................................................................. 54 1.4.3. Les mécanismes de contrôle de l’anneau contractile en cytokinèse .................... 56
1.4.3.1. Spécification et formation de l’AC : un rôle crucial pour RhoA et mDia2 ................. 57 1.4.3.2. La contraction de l’anneau contractile ......................................................................... 60 1.4.3.3. Désassembler l’AC pour permettre l’abscission .......................................................... 61
1.4.4. Rôle émergeant de la dégradation ciblée de protéines comme mécanisme de
contrôle spatiotemporel du remodelage de l’actine pendant la division cellulaire ............... 65

vii
1.4.5. Un rôle pour le complexe chaperon HSPB8-BAG3 dans la dynamique de
l’actine, l’orientation du fuseau mitotique et la ségrégation des chromosomes pendant la
mitose 68 1.5. Objectifs de travail ..................................................................................................... 72
2. Chapitre 2: Fine-tuning of actin dynamics by the HSPB8-BAG3 chaperone
complex facilitates cytokinesis and contributes to its impact on cell division ........... 74
2.1. Avant-propos .............................................................................................................. 75 2.2. Résumé ........................................................................................................................ 77 2.3. Abstract ....................................................................................................................... 78 2.4. Introduction ................................................................................................................ 79 2.5. Material and methods ................................................................................................ 81
2.5.1. Cell culture, synchronization, drug treatments and siRNA experiments ............. 81 2.5.2. Transfection, infection, adenovirus and vectors .................................................. 82 2.5.3. Antibodies and chemicals .................................................................................... 82 2.5.4. Immunofluorescence and live-cell imaging ......................................................... 83 2.5.5. Western blotting ................................................................................................... 84 2.5.6. Statistical analyses ............................................................................................... 85
2.6. Results .......................................................................................................................... 85 2.6.1. Silencing of HSPB8 or BAG3 perturbs abscission of daughter cells .................. 85 2.6.2. Knockdown of HSPB8 causes abnormal F-actin accumulation at the ICB ......... 88 2.6.3. Down modulation of actin dynamics can normalize F-actin accumulation at the
ICB and correct cytokinetic defects caused by depletion of HSPB8 .................................... 92 2.6.4. Inhibition of Arp2/3 complex can normalize F-actin at the ICB in HSPB8-
depleted cells ......................................................................................................................... 95 2.6.5. Drugs that affect lysosomal function recapitulate the HSPB8-dependent
phenotype, whereas induction of autophagy signaling can restore the F-actin defects caused
by depletion of HSPB8 .......................................................................................................... 98 2.7. Discussion .................................................................................................................. 101 2.8. References ................................................................................................................. 105
3. Chapitre 3 : Le complexe chaperon HSPB8-BAG3 facilite le remodelage des
structures mitotiques à base d’actine en modulant l’activité d’HDAC6 ................. 113

viii
3.1. La déplétion de l’une ou l’autre des protéines du complexe mitotique HSPB8-
BAG3-p62/SQSTM1 interfère avec la dynamique du nuage sous-cortical d’actine qui
dépend du complexe Arp2/3 ................................................................................................. 114 3.2. De faibles doses d’un inhibiteur d’HDAC6 peut normaliser la dynamique du
nuage sous-cortical d'actine et l'arrondissement mitotique dans les cellules déplétées en
BAG3, HSPB8 ou p62/SQSTM1 .......................................................................................... 121 3.3. L’activité d’HDAC6 et son association à la cortactine sont régulées de façon
mitose-spécifique, d’une manière qui pourrait impliquer l’intervention du complexe
chaperon HSPB8-BAG3-p62/SQSTM1 ............................................................................... 126 3.4. Modification du phénotype BAG3-dépendant par l’expression de mutants
cortactine acétyl-mimétique et non-acétylable ................................................................... 131 3.5. Figures supplémentaires .......................................................................................... 137 3.6. Matériel et méthode .................................................................................................. 138
3.6.1. Culture cellulaire, synchronisation, traitements et transfection de siARN ........ 138 3.6.2. Transfection, infection, vecteurs et adénovirus ................................................. 139 3.6.3. Anticorps et produits chimiques ........................................................................ 141 3.6.4. Immunoprécipitation et immunobuvardage de type « Western » ...................... 142 3.6.5. Immunofluorescences, PLA (« proximity ligation assay »), imagerie en temps
réel, microscopie et statistiques ........................................................................................... 142 3.6.6. Analyses statistiques .......................................................................................... 144
4. Chapitre 4 : Lien fonctionnel entre l’activité autophagique du complexe
chaperon HSPB8-BAG3 et la régulation de structures à base d’actine pendant la
division cellulaire .......................................................................................................... 145
4.1. La déplétion d’ATG7 récapitule les défauts de remodelage de l’actine observés
suivant la déplétion de BAG3, HSPB8 ou p62/SQSTM1 lors de la division cellulaire ... 146 4.2. La déplétion d’HSPB8 interfère avec le flux autophagique dans les cellules en
cytokinèse ............................................................................................................................... 155 4.2.1. La déplétion d’HSPB8 cause une augmentation de l’intensité et du volume des
vésicules LC3 ...................................................................................................................... 155 4.2.2. La déplétion d’HSPB8 est associée avec une accumulation de protéines poly-
ubiquitinées sur K63 dans les cellules en cytokinèse .......................................................... 160 4.3. L’inhibition d’HDAC6 normalise l’intensité et le volume des autophagosomes
ainsi que l’accumulation de F-actine au PI dans les cellules déplétées en HSPB8 .......... 164

ix
4.4. Matériel et méthode .................................................................................................. 170 4.4.1. Culture cellulaire, synchronisation, traitements et transfection de siARN ........ 170 4.4.2. Anticorps et drogues .......................................................................................... 171 4.4.3. Immunobuvardage de type « Western » ............................................................ 171 4.4.4. Immunofluorescence, microscopie et analyses statistiques ............................... 172
5. Chapitre 5 : Discussion ......................................................................................... 174
5.1. HSPB8-BAG3 agit en limitant la polymérisation de l’actine et/ou en stimulant son
renouvellement pendant la division cellulaire .................................................................... 174 5.2. Le complexe HSPB8-BAG3 facilite le remodelage des structures mitotiques à base
d’actine à travers la modulation d’HDAC6 ........................................................................ 179 5.3. Implication potentielle de l’autophagie sélective dépendante d’HSPB8-BAG3
dans la progression de la division cellulaire ........................................................................ 182
6. Conclusion ............................................................................................................. 186
7. Bibliographie ......................................................................................................... 187
8. Annexes .................................................................................................................. 259
8.1. Annexe 1 .................................................................................................................... 259 8.2. Annexe 2 .................................................................................................................... 291 8.3. Annexe 3 .................................................................................................................... 313

x
Liste des tableaux Chapitre 1 Tableau 1.1 : Tableau récapitulatif des fonctions de certaines HSPB associées à la progression tumorale et au développement de pathologies présentant une désorganisation du cytosquelette d’actine.

xi
Liste des figures Chapitre 1 Figure 1.1 : Schéma représentatif de l’axe de signalisation p38MAPK/MAPKAPK2 à l’origine de la phosphorylation d’HSP27. Figure 1.2 : Modèle décrivant comment HSPB1 faciliterait la motilité à base d'actine. Figure 1.3 : Schéma représentatif des différents domaines qui constituent les protéines BAG humaines. Figure 1.4 : Schéma de la structure modulaire du co-chaperon BAG3, mettant en évidence des interactions avec des partenaires impliqués dans la signalisation intracellulaire, le remodelage du cytosquelette d’actine, et l’autophagie sélective. Figure 1.5 : Les étapes de formation et de dégradation de l’autophagosome. Figure 1.6 : Schéma représentant le mécanisme par lequel le co-chaperon BAG3 aiderait au ciblage de protéines endommagées ou poly-ubiquitinées à la machinerie autophagique. Figure 1.7 : Schéma du mécanisme moléculaire de la CASA. Figure 1.8 : Schématisation des différentes étapes de la division cellulaire. Figure 1.9 : Schéma du mécanisme par lequel le complexe Arp2/3 induit la polymérisation d’un nouveau filament d’actine le long d’un filament préexistant. Figure 1.10 : Schéma structurel d’un dimère de myosine II. Figure 1.11 : Schéma du mouvement assuré par la myosine le long du filament d’actine. Figure 1.12 : Représentation schématique des fonctions assurées par les microtubules et leurs protéines motrices dans le positionnement du fuseau mitotique et la ségrégation des chromosomes. Figure 1.13 : Schéma représentatif de la voie de signalisation impliquée dans la formation du cortex mitotique et le maintien de sa rigidité. Figure 1.14 : Schéma modèle du mouvement du nuage sous-cortical d’actine durant la mitose. Figure 1.15 : Modèle selon lequel la Myo10 assure la communication des fibres de rétraction, du cortex et du nuage sous-cortical d’actine avec le centrosome. Figure 1.16 : Représentation schématique de la voie de signalisation dépendante du complexe « centralspindlin » et responsable de la spécification et de la formation de l’AC. Figure 1.17 : Modèle traditionnel de la contraction de l’AC. Figure 1.18 : Mécanismes moléculaires de désassemblage de l’AC dépendant des GTPases Rab11 et Rab35. Figure 1.19 : Modèle selon lequel SCFFbxw5 régule les niveaux d'Eps8 pour contrôler la dynamique de l'actine pour une progression mitotique normale. Figure 1.20 : Réprésentation schématique du ciblage de la forme active de RhoA (RhoA-GTP) par p62/SQSTM1 à l’autophagie sélective au sillon de clivage. Chapitre 2 Figure 2.1: Depletion of HSPB8 or BAG3 perturbs daughter cells spreading and ICB disappearance during cytokinesis. Figure 2.2: Silencing of HSPB8 interferes with cytokinetic abscission and markedly decreases the mitotic levels of BAG3.

xii
Figure 2.3: Silencing of HSPB8 causes abnormal F-actin accumulation at the ICB. Figure 2.4: Down modulation of actin dynamics can rescue phenotypes caused by depletion of HSPB8. Figure 2.5: Co-distribution of abnormal F-actin and Arp2/3, and restoration of F-actin organization at the ICB in HSPB8-depleted cells upon inhibition of Arp2/3 complex. Figure 2.6: Inhibition of lysosomal function mimics, whereas autophagic signaling can correct, the cytokinetic defect in F-actin caused by depletion of HSPB8. Chapitre 3 Figure 3.1 : La déplétion de BAG3, HSPB8 ou p62/SQSTM1 perturbe le nuage sous-cortical d’actine qui dépend d’Arp2/3. Figure 3.2 : L’activité d’HDAC6 régule la dynamique du nuage sous-cortical d’actine : la réduction de l’activité de HDAC6 par de faibles doses de tubacine peut normaliser les défauts mitotiques associés à la déplétion de BAG3, HSPB8 ou p62/SQSTM1. Figure 3.3 : L’activité d’HDAC6 et son association à la cortactine sont régulées de façon différentielle en mitose ; un rôle pour le complexe chaperon HSPB8-BAG3. Figure 3.4 : Les mutants cortactine-9KQ et 9KR restaure et mime respectivement les défauts d’organisation du cytosquelette d’actine associés à la déplétion de BAG3 ou d’HSPB8. Figure S3.1 : Mesure de l’efficacité de la déplétion de BAG3, HSPB8 ou p62/SQSTM1 par immunobuvardage de type « Western ». Figure S3.2 : L'inhibition d’Arp2/3 normalise les défauts d'arrondissement des cellules mitotiques dans les cellules HeLa déplétées en BAG3. Chapitre 4 Figure 4.1: La déplétion d’ATG7 se traduit par un réarrangement du cytosquelette d’actine en interphase. Figure 4.2: Effet de la déplétion d’ATG7 sur la progression mitotique des cellules HeLa. Figure 4.3 : La déplétion d’ATG7 conduit à l’accumulation de cellules partiellement arrondies en métaphase. Figure 4.4: La déplétion d’ATG7 augmente la proportion de cellules présentant une accumulation anormale de F-actine au PI. Figure 4.5: La déplétion d’HSPB8 interfère avec le flux autophagique en cytokinèse. Figure 4.6: Les cellules déplétées en HSPB8 présentent une accumulation d’agrégats K63. Figure 4.7 : L’inhibition de l’activité d’HDAC6 restaure un flux autophagique normal et normalise la quantité de F-actine au PI dans les cellules déplétées en HSPB8.

xiii
Liste des abréviations ABP « Actin Binding Proteins » AC Anneau contractile d’actomyosine aPKC « atypical protein kinase C » Arf6 « ADP-ribosylation factor 6 » Arp2/3 « Actin related proteins 2 and 3» ATG « AuTophaGy » BAG « Bcl2-associated athanogen » BAG3 « Bcl2-associated athanogen 3 » Bcl-2 « B-cell lymphoma 2 » Bcl-XL « B-cell lymphoma-extra large » Cas9 « CRISPR associated protein 9 » CASA « Chaperone-Assisted Selective Autophagy » CCN1 « Cyr61- CTGF- NOV protein 1 » CCT « Chaperonin Containing T-complex polypeptide » Cdc42 « cell division cycle 42 » CDK1 « Cyclin-Dependent Kinase 1 » CHIP « Carboxyl terminus of Hsc70-Interacting Protein » CK2 « Casein Kinase 2 » CRIB « Cdc42-Rac interactive binding » CRISPR « Clustered Regularly Interspaced Short Palindromic Repeats » DAAM « Dishevelled-Associated Activators of Morphogenesis » DEPDC1B « DEP domain containing 1B » Dia « Diaphanous » ECT2 « Epithelial Cell Transforming 2 » ERK « Extracellular-Related Kinase » ERM Ezrine-Radixine-Moésine ESCRT-III «Endosomal Sorting Complexes Required for Transport II» FAK « Focal Adhesion Kinase » FH1 « Formin Homology domain 1 » FHOD « Formin-Homology Domain proteins » FIP3/FIP4 « rab11 Family of Interacting Proteins 3/4 » FLN Filamine FMN Formine FRAP « Fluorescence Recovery After Photobleaching » FRL « Formin-Related proteins in Leukocytes » GAP « GTPase Activating Proteins » GDI « Guanine nucleotide Dissociation Inhibitor » GEF « Guanine nucleotide Exchange Factor » HDAC Histone DéAcétylases HDAC6 Histone DéAcétylase 6 HSC70 « Heat Shock Cognate 70 » HSF-1 « Heat Schock Factor 1 » HSP « Heat Shock Protein »

xiv
HSP70 « Heat Shock Protein 70 » HSPB « Heat Shock Protein familly B » HSPB8 « Heat Shock Protein familly B member 8 » Htt43Q « Huntigtin 43 glutamin » INF « INverted-Formin » IQGAP1 « IQ Motif Containing GTPase Activating Protein 1 » Keap1 « Kelch-like ECH-associated protein 1 » LC3 « microtubule-associated protein 1A/1B-Light Chain 3 » LGN Leucine–Glycine–Asparagine LIMK1 « LIM motif-containing protein Kinase 1 » LIR « LC3 Interacting Region » MAPKAPK2 « p38 MAP kinase-activated protein kinase 2 ») Mcl-1 « Myeloid cell leukemia sequence 1 » MEF « Mouse Embryonic Fibroblast » MICAL1 « Molecule Interacting with CasL 1 » MKLP1 « Mitotic Kinesin-Like Protein 1 » MLC2 « Myosin Light Chain 2 » MLCK « Myosin Light Chain Kinase » MMP-2 « Matrix Metalloproteinase-2 » MRCK «Myotonic dystrophy kinase-Related Cdc42-binding Kinase» MTOC « MicroTubules Organization Center » mTORC1 « mammalian Target Of Rapamycin Complex 1 » MyoII Myosine II Myo10 Myosine X NBR1 « Next to BRCA1 gene 1 protein » NDP52 « Nuclear Dot 52 kDa Protein » NF-kB « Nuclear Factor-kappa B » NPF « Nucleation-Promoting Factor » Nrf2 « Nuclear Factor (Erythroid-Derived 2)-Like 2 » NTA « N-Terminal Acidic domain » NuMA « Nuclear and Mitotic Apparatus » OCRL « OculoCerebroRenal syndrome of Lowe » OPTN « OPTiNeurin » p38MAPK « p38 Mitogen Activated Protein Kinase » PAK « p21-activated kinase » PAR6 « partitioning-defective protein 6 » PB1 Phox et Bem 1 PDZGEF2 «PDZ Domain-Containing Guanine Nucleotide Exchange Factor 2» PE PhosphatidylÉthanolamine PI Pont Intercellulaire PI3K « PhosphatIdylinositol-4,5-bisphosphate 3-Kinase » PI3KC3–C1 « class III PI 3-Kinase Complex I » PI(3)P PhosphatIdylinositol-3-Phosphate PI(4,5)P2 PhosphatIdylinositole 4,5 biPhosphate

xv
PIP5K « PhosphatIdylinositol 4-Phosphate 5-Kinase » PKCd « Protein Kinase C delta » PLCg Phospholipase C gamma PTPRF « receptor Protein Tyrosine Phosphatase, Receptor type, F » Rab11/35 « Ras-related protein 11 or 35 » Rac1 « Ras-related C3 botulinum toxin substrate 1 » Rap1 « Ras-proximate-1 » ou « Ras-related protein 1 » RhoA « Ras homolog gene family, member A » ROCK « Rho-associated, Coiled-coil containing protein Kinase » SCAMP2/3 « Secretory Carrier-Associated Membrane Protein 2 and 3 » SCAR « Suppressor of cAMP Receptor » SCFFbxw5 « Skp1–Cul1–F-box F-Box and WD Repeat Domain Containing 5 » SH3 « Src Homology 3 Domain » SLK « Sterile 20-Like Kinase » SOD1 « Super Oxyde Dismutase 1 » SQSTM1 SeQueSTosoMe 1 SSH1 « SlingSHot 1 » STAT3 « Signal Transducer and Activator of Transcription 3 » STX 17 Syntaxine 17 SYNPO2 SYNaptoPOdine-2 TAX1BP1 « TAX1 Binding Protein 1 » TAZ « Tafazzin » TSC1/2 « Tuberous SClerosis 1/2 » UBA « UBiquitin Association » UBL « UBiquitin-Like » ULK1 « Unc-51-Like autophagy activating Kinase 1 » WASP « Wiskott–Aldrich Syndrome Protein » WAVE « WASP-family VErprolin-homologous protein » YAP « Yes-Associated Protein »

xvi
Dédicace
« Il n’y a rien dont la patience ne vienne à bout quand elle est secondée de persévérance »
Tite-Live, Maximes et sentences, Ier siècle ap. J-C

xvii
Remerciements
Je tiens, en premier lieu, à remercier chaudement ma directrice de thèse Josée N. Lavoie.
Je la remercie de m’avoir accueilli dans son laboratoire et de m’avoir permis de m’y
épanouir au fils des années. Je lui suis extrêmement reconnaissante pour la confiance
qu’elle m’a témoignée, tout au long de ces années, qui m’a permis de développer ma
confiance et mon « savoir faire ». Je la remercie pour son écoute attentive tant sur le plan
professionnel que personnel. Je lui suis reconnaissante de ses conseils avisés et de sa
minutie qu’elle emploi au quotidien et qu’elle m’a enseignée durant ces six années de
doctorat. Grâce à sa passion pour la recherche et son habileté à la transmettre, je me sens
aujourd’hui grandie de mon expérience et en confiance pour mon avenir.
Je tiens également à remercier le Dr Jacques Landry pour m’avoir dirigé lors des
premières années de mon doctorat, en co-direction avec Dre Josée N. Lavoie, et pour
m’avoir introduit au monde des petites HSP.
Je souhaite également remercier l’ensemble des membres, passés et présents, du
laboratoire Lavoie. En particulier Herman Lambert et Margit Fuchs, les professionnels de
recherche du laboratoire, qui ont toujours répondu présents lorsque j’avais besoin d’eux ;
qui m’ont donné énormément de conseils pour m’assurer de l’efficacité et de la
reproductibilité de mes expériences ; qui ont tendu l’oreille durant les jours sombres de
mon doctorat et qui ont toujours su apporter des réponses à mes questions. À Émilie Pic,
post-doctorante du laboratoire, avec qui je pouvais m’exprimer dans mon patois lorrain.
Merci pour ses conseils, son écoute et ses blagues cocasses. Merci à Marc-Antoine
Rodrigue, étudiant au doctorat du laboratoire, qui porte toujours au party de Noël des
pulls qui font jaser. Un grand merci à ma collègue et amie, Solenn Guilbert, alias « Soso »,
pour les discussions scientifiques et les soirées passées à décompresser. Je ne remercierai
jamais assez mes deux collègues et amies Carole Luthold, alias « Zozo », et Claire
Dziengelwski, alias « Clairon », qui ont été mes piliers durant mon doctorat. J’ai toujours
pu compter sur leur soutien, leur écoute et leurs conseils. Même à distance elles ont
répondu à mes interrogations, m’ont donné des conseils avisés et m’ont soutenu durant les

xviii
moments difficiles. Merci à elles de m’avoir souvent ouvert leur porte les jours où je
n’avais pas le courage de rentrer chez moi ou lorsque je devais commencer tôt le « lab ».
Merci à Claire de rire à mes « jokes plates » et de m’avoir appris à relativiser faces aux
problèmes du quotidien. Je tiens également à remercier Carole, avec qui je forme un duo
inébranlable. Elle m’a apporté bien plus que du réconfort, grâce à elle j’ai appris à être
confiante, positive et davantage minutieuse. Merci à elle pour nos discussions
interminables concernant nos projets scientifiques et de vies, et pour avoir été ma
collaboratrice dans différents projets qui ont notamment menés à une étude qui sera
présentée dans cette thèse.
Je suis également très reconnaissante de l’ensemble des personnes qui m’ont formé et
supporté à un moment de mon doctorat, notamment Carl Saint-Pierre de la plateforme
d’imagerie. Je remercie aussi le Dr Norman Marceau pour ses conseils avisés à une
période où je ne croyais plus en moi. Je pense aussi à Anne Loranger qui m’a apporté des
conseils lors de mes séances au microscope et qui m’a également permis de partager ma
passion pour la recherche à des jeunes du primaire et du secondaire.
Tout ce travail n’aurait pu voir le jour sans l’aide et le soutien de mes amis et de mes
proches. Je pense tout d’abord à ma famille et particulièrement à mes parents, qui ont su
rendre la distance moins pénible par leurs appels hebdomadaires, leurs visites « icite » et
leurs colis remplis de douceurs. Merci à ma mère qui a toujours eut, à sa façon, les mots
pour me pousser à me surpasser. Merci à ma grand-mère, Lucie, qui a été d’un grand
soutien moral et financier. Merci à mes oncles, tantes, cousins et cousines, pour les petits
mots de réconfort, les visites qu’ils m’ont faites et les repas de famille auxquels ils m’ont
fait participer même à plus de 6000 km de distance. Je remercierai aussi Nicolas, « mon
cha », pour avoir été mon partenaire de vie, pour tout le soutien moral qu’il m’a apporté
durant les dernières années de mon doctorat. Merci à lui de m’avoir offert un toit, du
réconfort quotidien, des bons petits plats, de magnifiques vacances en France et une
famille (avec notre chien Junior) qui m’ont aidé à passer au travers des jours sombres du
doctorat. Merci à mes « P », Sophie et Céline, je leur suis reconnaissante de leur soutien,
de leur écoute et surtout de leur grain de folie qui m’a souvent aidé à décompresser. Merci

xix
à Lucile, alias « Boulette » et Lucie-Marie, alias « Lulu », elles sont à mes côtés depuis le
Lycée et le sont restées durant mon doctorat. Merci à elles pour leur écoute et leurs petits
messages qui m’ont réconforté et m’ont remonté plus d’une fois le moral. Je suis également
très reconnaissante du soutien que mes amis rencontrés à Québec m’ont apporté :
Alexandra, alias « mon Roux », et Gaëlle, dont le grain de folie a été un réconfort bien des
fois ; Emmanuelle, alias « Manu », et Mélany, alias « Poussin », dont l’humour, l’écoute
et les conseils ont été importants ; Clémence et Sébastien, alias « Sebounet », avec qui les
discussions et les soirées autour d’un bon petit plat ont toujours été d’une grande aide.
Merci à ma colocataire et amie Françoise et à son fils Noah, qui m’ont transmis leur
énergie positive durant cette période éprouvante qu’est la rédaction. Je souhaite également
remercier mes amis de l’épicerie J.A Moisan, avec qui j’ai travaillé durant mes 6 derniers
mois de vie à Québec, qui ont été une vraie bouffée d’air frais dans mon quotidien et qui
m’ont aidé à penser à d’autres choses que la thèse et l’incertitude qu’elle implique.
Pour finir, je remercierai les membres de mon jury, à savoir le Dr Jacques Huot, le Dr
Marc-Étienne Huot et le Dr Steve Jean, pour avoir accepté d’évaluer mon travail.
Bonne lecture !

xx
Avant-propos
Au cours de mon doctorat, j’ai participé à la rédaction d’un manuscrit en tant que première
auteure, d’un chapitre de livre en tant que co-première auteure et j’ai contribué à
l’élaboration d’un second manuscrit en tant que co-seconde auteure.
Le premier objectif de mon projet de doctorat, qui fait l’objet du chapitre 2, a été de
déterminer l’importance de la modulation du cytosquelette d’actine par le complexe
chaperon HSPB8-BAG3 lors de la division cellulaire. J’ai montré que le complexe
chaperon facilite le désassemblage d’une structure d’actine appelée anneau comtractile en
cytokinèse, que cette fonction contribue à la séparation adéquate des cellules filles et
qu’une telle fonction pourrait nécessiter la modulation de la machinerie autophagique par
HSPB8-BAG3. Ces travaux ont donné naissance à un article publié dans le journal Cell
stress and Chaperones. (Varlet, A.A., Fuchs, M., Luthold, C., Lambert, H., Landry, J.,
Lavoie, J.N. (2017) Fine-tuning of actin dynamics by the HSPB8-BAG3 chaperone
complex facilitates cytokinesis and contributes to its impact on cell division. Cell stress
and chaperones, doi:10.1007/s12192-017-0780-2). Le format PDF de cet article se trouve
dans l’Annexe 3.
Le second objectif de mon doctorat, qui fait l’objet du chapitre 3, a été de caractériser le
mécanisme moléculaire par lequel le complexe chaperon HSPB8-BAG3 pourrait réguler
la dynamique du cytosquelette d’actine pendant la mitose. Ces travaux ont montré
qu’HSPB8-BAG3, en association avec le récepteur autophagique p62/SQSTM1, pourrait
moduler l’activité de la déacétylase HDAC6 et qu’une telle modulation pourrait impliquer
une cible majeure d’HDAC6 : la cortactine, dont la dynamique d’activation/inhibition
semble être nécessaire à la régulation de structures d’actine en mitose.
Le Dre Lavoie m’a également laissé l’opportunité d’analyser plus en détail l’existence d’un
lien fonctionnel entre la régulation du cytosquelette d’actine par le complexe chaperon
HSPB8-BAG3 et sa fonction autophagique. Ces travaux exploratoires font l’objet du
chapitre 4.

xxi
À mon arrivée au laboratoire, j’ai eu également la chance de participer activement aux
travaux montrant que le complexe chaperon HSPB8-BAG3 module des structures à base
d’actine qui dirigent le positionnement du fuseau mitotique et la progression en mitose des
cellules cancéreuses. L’article issu de ces recherches, où je suis co-deuxième auteure, a été
publié dans le journal PLoS Genetics et est disponible à l’Annexe 1. (Fuchs, M., Luthold,
C., Guilbert, M., Varlet, A.A., Lambert, H., Jetté, A., Elowe, S., Landry, J., Lavoie, J.N.
(2015) A Role for the Chaperone Complex BAG3-HSPB8 in Actin Dynamics, Spindle
Orientation and Proper Chromosome Segregation during Mitosis. PLoS Genet. 23;11(10):
e1005582.)
Le Dre Lavoie m’a aussi donné la possibilité de participer à la rédaction d’un chapitre de
livre qui porte sur l’implication des chaperons moléculaires de la famille de petites
protéines de choc thermique (HSPB) dans la régulation de la dynamique d’actine. Ce
chapitre, disponible dans l’Annexe 2, a été effectué pour le livre The Big Book on Small
Heat Shock Proteins (Tanguay, R.M., Hightower, L.E., Springer, 2015 - 610 p). (Varlet,
A.A., Guilbert, S.M., Fuchs, M., Lambert, H., Landry, J., Lavoie, J.N. (2015) Regulation
of Actin-Based Structure Dynamics by HspB Proteins and Partners. The Big Book on
Small Heat Shock Proteins, Volume 8 of the series Heat Shock Proteins pp 435-456)
L’ensemble de ces travaux n’aurait pu voir le jour sans l’aide des co-auteurs cités ci-dessus.

1
1. Chapitre 1 : Introduction
1.1. Rôle du cytosquelette d’actine dans la réponse cellulaire aux forces mécaniques
Les changements morphologiques de la cellule sont nécessaires au bon déroulement
d’activités cellulaires cruciales telles que la migration et l’adhésion cellulaires, la transition
épithéliomésenchymateuse, la polarisation cellulaire ou encore la division et la
différenciation cellulaires. La morphodynamique cellulaire est déterminée par des
transitions dans l’organisation des réseaux d’actine. Par exemple, lors de la transition
épithéliomésenchymateuse, le réseau cortical d’actine se réorganise en structures rigides
appelées fibres de stress qui promouvoient la migration et l’adhésion cellulaires (Shankar
et Nabi 2015; Haynes et al. 2011; Bhowmick et al. 2001). Le cytosquelette d’actine est au
cœur du contrôle de la mécanique cellulaire en assurant la rigidité cellulaire et la
transmission des forces perçues par la cellule. En effet, à travers la formation de structures
à base d’actine, telles que le cortex ou les fibres de stress, le cytosquelette d’actine assure
la résistance cellulaire face aux tensions mécaniques (Chalut et Paluch 2016). Notamment,
la réorganisation du cytosquelette d’actine en un cortex d’actomyosine rigide permet à la
cellule mitotique de s’arrondir contre son environnement et de maintenir une géométrie
optimale pour la progression mitotique (Lancaster et al. 2013). En outre, des éléments du
cytosquelette d'actine, comprenant les moteurs de myosine et des agents de reticulation
d'actine comme la filamine, se remodèlent et activent des voies de signalisation en réponse
aux contraintes mécaniques qui permettront à la cellule d’y répondre (Kee et al. 2012;
Chowdhury et al. 2010; Ehrlicher et al. 2011; Effler et al. 2006). Par exemple, la contrainte
mécanique augmente l’interaction de la filamine aux intégrines, ce qui conduit à un
renforcement des adhésions focales en raison du maintient de l’activation des intégrines
(Ehrlicher et al. 2011).
Ainsi, la morphodynamique des cellules implique un contrôle spatiotemporel fin de la
dynamique de l’actine. Les composantes majeures ainsi que les régulateurs clés des

2
structures à base d’actine ont été bien caractérisés suite à d’innombrables études.
Cependant, comment toutes ces protéines fonctionnent comme un ensemble intégré et
modulable par la signalisation intracellulaire reste peu compris (Roybal et al. 2016). Peu
d’éléments sont connus quand aux mécanismes qui assurent la ségrégation des différents
complexes multi-protéiques responsables du remodelage spatiotemporel de structures
distinctes à l’intérieur d’une même cellule. De plus, les mécanismes par lesquels la cellule
assure le maintien de l’intégrité de telles structures soumises à des cycles
d’assemblage/désassemblage et des stress mécaniques, demeurent méconnus. Bien qu’on
suppose l’existence de mécanismes de contrôle de qualité protéique régulant l’intégrité de
telles structures, ceux-ci demeurent peu étudiés à ce jour (Baird et al. 2014).
Notre laboratoire s’intéresse aux fonctions des chaperons moléculaires dans le contrôle de
qualité des structures à base d’actine et dans la régulation spatiotemporelle des réseaux
d’actine. Pour étudier ces fonctions, nous avons choisi un modèle d’étude paradigme qui
implique un remodelage drastique et rapide des structures à base d’actine : la division
cellulaire. Ce processus par lequel une cellule mère donne deux cellules filles dépend d’une
régulation spatiotemporelle fine de la dynamique du cytosquelette d’actine par des
mécanismes qui sont peu compris (section 1.4). Cette thèse s’inscrit dans l’optique
d’approfondir notre compréhension des mécanismes qui assurent une régulation
spatiotemporelle fine des transitions dans l’organisation des structures à base d’actine lors
de la division cellulaire. Pour ce faire, nous avons adressé le rôle et le mode d’action d’un
complexe chaperon formé des protéines HSPB8 et BAG3 durant la division cellulaire.
Dans ce chapitre, les notions de base et les concepts émergeants concernant le rôle des
chaperons de la famille HSPB dans le remodelage du cytosquelette d’actine seront
présentés (section 1.2). Nous décrirons ensuite les fonctions connues du chaperon HSPB8
et celles de son co-chaperon BAG3 dans le contrôle de qualité des protéines, la
signalisation et le remodelage de l’actine, et nous discuterons des questions actuellement
soulevées par ces travaux (section 1.3). Finalement, nous présenterons les mécanismes
moléculaires impliqués dans le remodelage des structures à base d’actine qui dirigent la
division cellulaire et les questions non résolues dans ce domaine (section 1.4).

3
1.2. Les chaperons de la famille HSPB et la régulation de la dynamique et de l’intégrité
des structures d’actine
1.2.1. Le contrôle de qualité des protéines et les chaperons moléculaires
La fonction des protéines est gouvernée par leur structure tridimensionnelle assurée par le
repliement adéquat de la chaîne polypeptidique qui les constitue. Un repliement incorrect
ou une dénaturation de la structure tridimensionnelle des protéines affecte leur fonction et
peut conduire à des conséquences catastrophiques au niveau tissulaire. En effet, dans la
cellule, les protéines mal repliées peuvent avoir des activités délétères de « gain de
fonction », en partie en raison de leur tendance accrue à l'agrégation, et mener à des
dysfonctionnements importants des activités cellulaires (Dubnikov, Ben-Gedalya, et
Cohen 2017; Kumar et al. 2016). Bien que les mécanismes précis de la toxicité ne soient
pas bien compris, il est clair que les protéines dénaturées s'engagent dans des interactions
inappropriées avec d'autres composantes cellulaires et peuvent s'accumuler dans des
inclusions potentiellement toxiques de protéines (Lansbury et Lashuel 2006). La
dénaturation protéique apparaît ainsi comme un mécanisme majeur de maladies humaines,
comme le souligne la liste croissante de « maladies conformationnelles », résultant de
l'accumulation cellulaire de protéines mal repliées (Muchowski 2002; Sakahira et al. 2002;
Dubnikov, Ben-Gedalya, et Cohen 2017; Kumar et al. 2016). Parmi celles-ci, on peut citer
une gamme substantielle de pathologies, comme le cancer, la fibrose kystique, ou encore
de nombreux troubles neurodégénératifs tels que la maladie d'Alzheimer, les maladies de
Parkinson et d’Huntington (Morimoto 2011; Manecka et al. 2017; Klaips, Jayaraj, et Hartl
2017; Chiti et Dobson 2017; Hartl 2017; Sontag, Samant, et Frydman 2017). Ces
pathologies mettent donc en évidence l’importance de maintenir l’intégrité du protéome
quelles que soient les conditions environnementales auxquelles la cellule est soumise : il
s’agit du contrôle de l’homéostasie protéique ou du contrôle de qualité des protéines
(Sontag, Samant, et Frydman 2017).
Le maintien de l’intégrité du protéome est assuré par un ensemble de mécanismes de
contrôle de qualité, qui sont sous la gouverne des chaperons moléculaires. Ils contrôlent le

4
repliement protéique, aident à l’assemblage de complexes protéiques et dirigent les
protéines endommagées vers la dégradation protéique. La cellule dispose de deux voies de
dégradation qui appartiennent aussi au système de contrôle de qualité des protéines: la
dégradation par le système ubiquitine-protéasome et par la machinerie autophagique
(section 1.3.4.1). Ainsi, un chaperon moléculaire se définit par ses capacités à intéragir et
à aider au repliement d’une protéine ou à l’assemblage d’un complexe protéique, ou de
structures polymériques comme celles qui contrôlent l’organisation des cytosquelettes,
sans être une partie intégrante de leurs structures finales.
Les chaperons moléculaires de la famille des protéines de choc thermique ou HSP (« Heat
Shock Protein ») sont des régulateurs clés de l’homéostasie protéique. Il s’agit pour la
plupart de protéines de stress induites en réponse à une variété de stimuli externes ou
internes qui endommagent la structure des protéines. Le facteur de transcription du stress
cellulaire HSF-1 (« Heat Shock Factor 1 ») contrôle l’expression des protéines de choc
thermique (DiDomenico, Bugaisky, et Lindquist 1982; Goldenberg et al. 1988). Il existe
deux classes de chaperons moléculaires essentiels : les chaperons à activité dépendante ou
indépendante de l’ATP. Les chaperons moléculaires à domaine ATPase sont ceux qui vont
stimuler le repliement des protéines dénaturées lors de cycles d’hydrolyse de l’ATP
souvent régulés par des co-chaperons, ou les diriger vers les voies de dégradation le cas
échéant. Il s’agit des chaperons HSP40, HSP60, HSP70, HSP90 ou encore HSP100.
Les chaperons moléculaires dont la fonction est indépendante de l’ATP forment la famille
des petites protéines de choc thermique ou HSPB (« Heat Shock Protein familly B »). Ces
chaperons moléculaires se caractérisent par la présence d’un domaine a-cristallin
hautement conservé et flanqué de séquences N- et C-terminales variables et peu conservées
(de Jong, Caspers, et Leunissen 1998; Kriehuber et al. 2010; van Montfort et al. 2001). Le
domaine a-cristallin se caractérise par un empilement de feuillets b dont la structure est
essentielle à la fonction des HSPB (Baranova et al. 2009; K. K. Kim, Kim, et Kim 1998).
Les HSPB ont été trouvées dans toutes les niches évolutives où leurs nombres et leurs poids
moléculaires varient de 12 à 42 kDa selon les espèces (de Jong, Caspers, et Leunissen
1998). Chez l’humain, dix HSPB ont été identifiées (HSPB1 à 10) et leur expression dans

5
l’organisme peut être ubiquitaire, comme c’est le cas pour HSPB1 (aussi connue sous le
nom d’HSP27) et HSPB8, ou spécifique à un tissu comme HSPB9 retrouvée
majoritairement dans les testicules (Morrow et Tanguay 2012).
Fonctionnellement, ces chaperons moléculaires forment la première ligne de défense de la
cellule lors d’un stress protéotoxique où ils préviennent l'agrégation irréversible des
protéines dénaturées indépendamment de l'ATP. De ce fait, les HSPB ont été impliquées
dans la prévention de l’agrégation de protéines impliquées dans différentes pathologies
comme la maladie d’Alzheimer et la maladie de Parkinson (Shammas et al. 2011; King et
al. 2009; Cox et Ecroyd 2017; Dehle et al. 2010; Golenhofen et Bartelt-Kirbach 2016). En
outre, certaines HSPB ont été impliquées dans la séquestration de protéines agrégées. En
effet, les cellules ont développé un certain nombre de mécanismes pour résister à
l’accumulation de protéines endommagées dont la séquestration de celles-ci dans des
inclusions cellulaires localisées à des endroits précis dans la cellule (Wolff, Weissman, et
Dillin 2014). C’est le cas par exemple de l’aggrésome qui est formé lors d’une surchage du
protéasome (Johnston, Ward, et Kopito 1998). Ce mécanisme de compartimentation de
dépôts agrégés permet à la cellule de séquestrer les protéines endommagées, de se protéger
de leur toxicité et facilite leur élimination par dégradation. Ainsi, il a été noté qu’HSP42
agit par son domaine N-terminal pour se co-agréger avec des protéines mal repliées et peut-
être les relier à d'autres facteurs de tri chez la levure S. cerevisiae (Specht et al. 2011).
Par ailleurs, en conditions physiologiques, il apparaît qu’HSPB1, le membre prototype de
cette famille, pourrait constituer un réservoir de monomères d’actine qui serait mobilisé de
façon dépendante de la signalisation intracellulaire, suivant sa phosphorylation (During et
al. 2007; Lavoie, Hickey, et al. 1993; Huot et al. 1995; Lavoie et al. 1995; Lavoie, Hickey,
et al. 1993; Lavoie, Gingras-Breton, et al. 1993; McLaughlin et al. 1996; Huot et al. 1995;
Stokoe et al. 1992; Ahlers et al. 1994; Zhou, Lambert, et Landry 1993). En somme, on
attribue aux membres de cette famille des rôles grandissants dans la signalisation cellulaire.
Cependant, leur mode d’action demeure peu caractérisé. Dans cette section, nous
présenterons le modèle prévalent quant à leur fonction en réponse au stress protéotoxique

6
et nous décrirons les données montrant une fonction encore peu comprise des HSPB dans
la régulation de la dynamique du cytosquelette d’actine.
1.2.2. Modèle prévalent des HSPB : l’oligomérisation pour prévenir de
l’agrégation protéique
En général, les HSPB sont très poly-dispersées, ce qui signifie qu'elles peuvent exister sous
la forme de dimères considérés comme le « bloc de construction » pour des homo- ou
hétéro-oligomères qui comportent jusqu'à 50 sous-unités (Dudich et al. 1995). La
formation des dimères est dépendante du domaine a-cristallin qui les caractérisent et d’un
motif IXI/V (isoleucine-X-isoleucine ou valine) localisé en C-terminal des HSPB essentiel
à l’oligomérisation (Pasta et al. 2004; Zhou et al. 2016). La formation des larges oligomères
est aussi dépendante des extrémités N- et C-terminales des HSPB qui déterminent la taille
des oligomères (Ghosh, Shenoy, et Clark 2006; Salerno et al. 2003; Lindner et al. 2000;
Hilton et al. 2013;Heirbaut et al. 2017; Rutsdottir et al. 2017; Moutaoufik et al. 2017;
Strózecka et al. 2012; Chen et al. 2010; McDonald et al. 2012; Mani et al. 2016).
L’oligomérisation est suggérée être le mode d’action canonique de la famille des HSPB où
les oligomères forment des réservoirs d’intermédiaires de repliement qui seront pris en
charge par les chaperons moléculaires dépendant de l’ATP (Lee et al. 1997; Ehrnsperger
et al. 1997; Żwirowski et al. 2017; Ungelenk et al. 2016; Leroux et al. 1997).
L'oligomérisation est un processus très dynamique et l'échange de sous-unités peut être
accéléré par des contraintes mécaniques, thermiques, ou encore de pH. Dans les cellules,
des modifications post-traductionnelles (en particulier des phosphorylations) sont
supposées stimuler davantage la dynamique d’oligomérisation qui, selon toute
vraisemblance, permettra aux HSPB de répondre de façon appropriée aux stimuli de stress.
En effet, plusieurs membres de la famille HSPB contiennent des sites de phosphorylation
qui conduit généralement à une dissociation des oligomères en dimères, tandis que la
déphosphorylation favorise l’oligomérisation en espèces de haut poids moléculaires
(Arrigo et Gibert 2012; Lanneau et al. 2008; Parcellier et al. 2005; Rogalla et al. 1999;
Sluchanko, Chebotareva, et Gusev 2015; Suss et Reichmann 2015; Haslbeck et Vierling

7
2015). Ainsi, la formation de différentes structures (dimères ou oligomères) pourrait
fournir un mécanisme de régulation de l’activité et/ou de la localisation des HSPB (van
Montfort et al. 2001; Acunzo, Katsogiannou, et Rocchi 2012; Boncoraglio, Minoia, et
Carra 2012, 2012; Bakthisaran et al. 2016; Aquilina et al. 2004; Hayes et al. 2009) .
Cependant, il apparaît que tous les chaperons de la famille HSPB ne peuvent pas
s’oligomériser in vivo. En effet, certains membres de la famille, dont HSPB8, n’arborent
pas de motif IXI/V essentiel à leur oligomérisation (Zhou et al. 2016; Studer et al. 2002;
Quinlan et al. 2013; Carra et al. 2008). De ce fait, ces HSPB sont retrouvées principalement
sous la forme de dimères (Carra et al. 2008). Il semble donc que le mode d’action des
HSPB ne soit pas ubiquitaire à tous les membres de la famille. Cela soulève la question de
savoir quel est le mode d’action des HSPB qui ne forment pas de larges oligomères et si ce
mode d’action est commun à toutes les HSPB.
1.2.3. Les HSPB dans le contrôle de la dynamique de l’actine et les pathologies
associées
En conditions physiologiques, il apparaît que les membres de la famille HSPB pourraient
avoir des fonctions dans la régulation de la dynamique des cytosquelettes, notamment celle
du cytosquelette d’actine. En effet, il a été mis en évidence qu’HSPB1, le membre le plus
étudié et utilisé comme paradigme de la famille, module la dynamique du cytosquelette
d’actine en séquestrant les monomères d’actines dans de larges complexes qui sont
dissociés en réponse à un stimulus de stress ou physiologique (Annexe 1).
Les études pionières de Lavoie et al. ont mis en évidence pour la première fois qu’HSPB1
pourrait favoriser la survie cellulaire en présence de l'inhibiteur de la polymérisation
d'actine : cytochalasine D, de manière dépendante de sa phosphorylation (Lavoie, Gingras-
Breton, et al. 1993). Ces effets protecteurs ont été corrélés avec des changements dans
l'organisation du cytosquelette d’actine lors de la surexpression d’HSPB1, fournissant un
argument pour une activité du chaperon moléculaire sur l'actine (Lavoie et al. 1995). En
outre, Lavoie et al. trouva que la phosphorylation d’HSPB1 agit non seulement pour

8
prévenir les altérations sévères de l'architecture du cytosquelette d'actine en réponse à
différents stress, mais aussi pour moduler le remodelage de l’actine et la dynamique de la
membrane induits par des facteurs de croissance (Lavoie et al. 1995; Lavoie, Hickey, et al.
1993). Ces travaux apportèrent les premières évidences d’un rôle physiologique d’HSPB1
dans la régulation du remodelage de l’actine.
Il fût aussi noté que l'activité inhibitrice de la polymérisation de l’actine par HSPB1 dépend
du degré de sa phosphorylation et de son organisation structurelle (Benndorf et al. 1994).
Effectivement, in vitro, les monomères non phosphorylés inhibent la polymérisation de
l'actine, alors que les monomères phosphorylés et les particules multimériques non
phosphorylées en sont incapables. Ces travaux ne concordaient donc pas avec ceux
obtenues in vivo montrant que l’expression de la forme sauvage d’HSPB1 stimule le
remodelage de l’actine alors qu’un mutant non phosphorylable d’HSPB1 exerce un effet
dominant négatif sur cette fonction du chaperon (Lavoie et al. 1995; Lavoie, Hickey, et al.
1993). Malgré cette incohérence, il fût proposé qu’HSPB1 sous sa forme non phosphorylée
inhibe l'assemblage de l’actine en coiffant l’extrémité à croissance rapide des filaments
d’actine (Benndorf et al. 1994; Mounier et Arrigo 2002).
Ce modèle fût contesté à la suite des travaux de During et al. qui décryptèrent le mécanisme
moléculaire par lequel HSPB1 contrôle la dynamique d’actine in vivo (During et al. 2007).
En effet, à la suite des nombreux travaux qui ont contribué à identifier l’axe de signalisation
p38MAPK (« p38 Mitogen Activated Protein Kinase ») /MAPKAPK2 (« p38 MAP kinase-
activated protein kinase 2 ») comme étant le signal responsable de la phosphorylation
d’HSPB1 (Figure 1.1), l’équipe du Dr Southwick utilisa la toxine létale de l’anthrax comme
modèle d’induction du remodelage de l’actine (During et al. 2005, 2007; McLaughlin et
al. 1996; Rouse et al. 1994; Huot et al. 1995; Stokoe et al. 1992; Ahlers et al. 1994; Zhou,
Lambert, et Landry 1993).

9
Figure 1.1 : Schéma représentatif de l’axe de signalisation p38MAPK/MAPKAPK2 à l’origine de la phosphorylation d’HSPB1.
Les travaux montrèrent notamment que l’inhibition de l’assemblage d’actine par la toxine
est médiée par le blocage de la phosphorylation d’HSPB1 et qu’un tel processus pouvait
être reproduit suite à l’inhibition de la p38MAPK. De façon remarquable, les auteurs
notèrent que la forme non-phosphorylée d’HSPB1 interagit et séquestre les monomères
d’actine in vitro. In vivo, les résultats montrèrent que la déplétion d’HSPB1 conduisait à
une réduction des effets de l’anthrax sur le cytosquelette d’actine. Un tel phénotype fut
restauré par l’induction de l’expression d’HSPB1 WT mais pas d’un mutant non
phosphorylable. Ainsi, ces travaux mirent en évidence le mode d’action d’HSPB1 qui sous
la forme d’oligomères dans la cellule fournirait un réservoir de monomères d’actine
localisé dans la région péri-nucléaire (Figure 1.2) (During et al. 2007). Suite à la
phosphorylation du chaperon moléculaire, par la MAPKAPK 2/3, les oligomères se
dissocieraient en dimères capables de larguer l’actine monomérique aux sites de
polymérisation de la F-actine comme par exemple au front de migration (Figure 1.2). Grâce
à cette étude, la phosphorylation d’HSPB1 a été fermement établie comme un mécanisme

10
clé pour réguler la motilité dans les cellules non musculaires, 20 ans après la découverte
initiale de son activité modulatrice de l’actine.
Figure 1.2: Modèle décrivant comment HSPB1 faciliterait la polymérisation de l’actine et la motilité cellulaire (adapté de Guilbert et al. 2015).
Une telle fonction pourrait être commune aux autres membres de la famille des HSPB
puisque certains d’entre eux arborent, tout comme HSPB1, un rôle protecteur du
cytosquelette d’actine (Mounier et Arrigo 2002; Clarke et Mearow 2013; Miron, Wilchek,
et Geiger 1988; Verschuure et al. 2002; Brophy, Lamb, et Graham 1999). En outre, les
HSPB exercent un rôle modulateur clé lors de processus impliquant un remodelage de
l’actine comme la migration, l’adhésion et la différenciation cellulaires, ou encore la
transition épithéliomésenchymateuse, l’angiogénèse et l’apoptose (Tableau 1.1)

11
(Montagna, Matuschewski, et Buscaglia 2012; Shimizu, Tanaka, et Atomi 2016; Park et
al. 2016; Davidson et Morange 2000; Lahvic et al. 2013; Lee et al. 2008; Piotrowicz,
Hickey, et Levin 1998). Via ces rôles de modulateurs du cytosquelette d’actine, les HSPB
qui sont souvent suractivées dans des lignées cancéreuses, sont associées à la progression
et l’invasion tumorales (Tableau 1.1) (Wei et al. 2011; Matsushima-Nishiwaki et al. 2016;
M. Suzuki et al. 2015; Doshi, Hightower, et Lee 2009; Piccolella et al. 2017; Tessier et al.
2003; Huang et al. 2013; Moyano et al. 2006; Schootbrugge et al. 2013). De plus, des
mutations des HSPB ont largement été associées au développement de pathologies sévères
présentant une désorganisation du cytosquelette d’actine comme des neuropathies et des
myopathies (Tableau 1.1) (Cappola et al. 2010; Stoevring, Frederiksen, et Christiansen
2007; Irobi et al. 2004; Evgrafov et al. 2004; Bova et al. 1999).
Malgré toutes ces évidences, le mode d’action des autres membres HSPB sur la régulation
de la dynamique de l’actine reste élusif mais pourrait être distinct de celui utilisé par
HSPB1. Notamment, dans des modèles in vitro et in vivo de fibrillation auriculaire, une
étude a démontré que certaines HSPB pouvaient prévenir les réarrangements du
cytosquelette d’actine associés à ce processus (Ke et al. 2011). Les auteurs ont observé que
la surexpression d’HSPB1, HSPB6, HSPB7 et HSPB8 protègent du remodelage de l’actine
dans un tel contexte. Les résultats suggèrent qu’HSPB1, HSPB6 et HSPB7 préviennent la
formation de F-actine en empêchant l’insertion de la G-actine dans les filaments, alors
qu’HSPB8 réduirait l’activation de RhoA. En outre, HSPB8 a récemment émergé en tant
que membre particulier de cette famille avec un mode d'action clairement distinctif pour la
régulation des structures cellulaires à base d'actine. Des études qui ont révélé des relations
fonctionnelles entre l’élimination de protéines par autophagie sélective et le contrôle de
l’organisation du cytosquelette d’actine seront décrites dans la section 1.3.5.2.

12
HSPB Fonctions cellulaires Pathologies Références
HSPB1 Remodelage de l’actine, Stabilisation de protéines, Migration cellulaire, Apoptose, Angiogenèse, Transition épithéliomésenchy-mateuse
Maladie de Charcot Marie Tooth et neuropathie ; Cancer du sein, colon, de la prostate, poumon, des os, de la tête et du cou, nasopharyngiale, des ovaires, hépatique
(Ylikallio et al. 2015; Evgrafov et al. 2004; James, Rankin, et Talbot 2008; Capponi et al. 2011; Kang et al. 2008; O’Callaghan-Sunol, Gabai, et Sherman 2007; Andrieu et al. 2010; Vahid et al. 2016; Guo et al. 2009; Wei et al. 2011; Rocchi et al. 2005; Lu et al. 2016; Xu, Chen, et Bergan 2006; Doshi, Hightower, et Lee 2009; Choi et al. 2014; Pavan et al. 2014; Voll et al. 2014)
HSPB5 Remodelage de l’actine, Migration cellulaire, Angiogenèse, Transition épithélio-mésenchymateuse
Dystrophie musculaire et myopathie ; Cancer des ovaires, poumons, de la prostate, lymphome métastaique
(Sitterding et al. 2008; Malin et al. 2015; Schootbrugge et al. 2013; Del Bigio et al. 2011; Forrest et al. 2011)
HSPB6 Remodelage de l’actine, Apoptose
Cardiomyopathie ; Carcinome hépatique
(Nicolaou et al. 2008; Nagasawa et al. 2014)
HSPB8 Remodelage de l’actine, Migration cellulaire
Maladie de Charcot Marie Tooth et neuropathie ; Cancer de l’ovaire
(Capponi et al. 2011; Irobi et al. 2004; Tang et al. 2005; Wilhelmus et al. 2006; Kim et al. 2006; Suzuki et al. 2015)
Tableau 1.1 : Tableau récapitulatif des fonctions de certaines HSPB associées à la progression tumorale et au développement de pathologies présentant une désorganisation du cytosquelette d’actine.

13
1.3. Le complexe chaperon HSPB8-BAG3 dans la régulation de structures contractiles
d’actine et de l’autophagie sélective
1.3.1. HSPB8 : un membre particulier de la famille des HSPB avec une spécificité
de substrats?
HSPB8, dont le poids moléculaire est de 22 kDa, appartient à la famille des petites
protéines de choc thermique. Historiquement, HSPB8 fût identifiée comme la protéine
kinase H11 similaire à la sérine-thréonine kinase ICP10 du virus de l’herpès (Smith et al.
2000). Plus tard, l’activité kinase de H11 fût discutée et des analyses d’homologies
établirent qu’H11 était en réalité un chaperon moléculaire de la famille HSPB (Kappé et
al. 2001; Gober, Depre, et Aurelian 2004; Kim, Seit-Nebi, et Gusev 2004). L’activité
chaperon d’HSPB8 fût ensuite caractérisée in vitro par (Chowdary et al. 2004) et in vivo
par (Carra et al. 2005). HSPB8 est considérée comme ubiquitaire et sa surexpression dans
différents types de cancers a été associée à la progression et la résistance tumorales
(Tableau 1.1) (Yu et al. 2001; Xiao-shan Li et al. 2014; Suzuki et al. 2015; Hamouda et al.
2014; Piccolella et al. 2017). Comme pour les autres membres de sa famille, des mutations
d’HSPB8, les mutations K141E et K141N situées dans le domaine a-cristallin de la
protéine, ont largement été associées au développement de maladies neurologiques et de
myopathies sévères dont la maladie de Charcot-Marie Tooth (Tableau 1.1) (Irobi et al.
2004; Tang et al. 2005; Wilhelmus et al. 2006; Kim et al. 2006; Hu et al. 2007).
Cependant, HSPB8 est un membre particulier de sa famille. En effet, bien qu’il présente
un domaine a-cristallin caractéristique de sa famille, ce chaperon moléculaire ne dispose
pas des motifs IXI/V essentiels à l’oligomérisation (Zhou et al. 2016; Studer et al. 2002;
Quinlan et al. 2013). Les travaux du laboratoire du Dr Landry ont permis d’établir
qu’HSPB8 existe principalement dans les cellules sous la forme d’un complexe stable avec
le co-chaperon BAG3 (« Bcl2-associated athanogen 3 ») (Carra et al. 2008), bien que des
études récentes suggèrent que le complexe est régulé de façon dynamique en réponse au
stress (Guilbert et al. 2018). Par des expériences de co-sédimendation, les auteurs ont noté
que le complexe HSPB8-BAG3 présentait la stoechiométrie de deux molécules d’HSPB8

14
pour une molécule de BAG3. De plus, par des expériences de déplétion à l’aide de siARN,
l’équipe montra que le co-chaperon BAG3 est essentiel à la stabilité d’HSPB8.
Effectivement, la déplétion de BAG3 mène à une réduction importante des niveaux
d’expression du chaperon HSPB8; cependant l’inverse n’est pas observé. Dans la plupart
des conditions, la déplétion d’HSPB8 a peu d’effet sur les niveaux d’expression de BAG3
(à l’exception de la mitose, voir Chapitre 2, Figure 2.2F). Par la suite, Fuchs et al. ont
identifié deux motifs de type IPV dans la région N-terminale du co-chaperon BAG3 avec
lesquels HSPB8 peut intéragir directement (Figure 1.3) (Fuchs et al. 2009). Cette étude
montra que la présence de ces motifs de BAG3 est essentielle pour maintenir l’activité
chaperon d’HSPB8. Ainsi, les résultats du laboratoire du Dr Landry suggèrent que les
fonctions cellulaires d’HSPB8 dépendent de BAG3. À l’inverse, BAG3 possède des
fonctions dépendantes et indépendantes d’HSPB8. Cependant, le rôle exact d’HSPB8 au
sein des fonctions de BAG3 demeure très peu compris et peu d’études s’y sont intéressées.
À titre de complexe, HSPB8-BAG3 est principalement associé au contrôle de qualité des
protéines. Notamment, le complexe est impliqué dans la régulation de la dégradation ciblée
de protéines par un processus appelé macroautophagie (autophagie) (section 1.3.4.1); ces
études seront décrites dans la section 1.3.4 (Carra et al. 2008; Crippa, Sau, et al. 2010;
Crippa, Carra, et al. 2010). Plus récemment, nous avons précisé l’action d’HSPB8 au sein
du complexe pendant un stress protéotoxique. Nous avons montré qu’HSPB8, en
coopération avec BAG3, coordonne la séquestration de protéines nuisibles et la réponse
cellulaire adaptative lors de l’inhibition du protéasome, ce qui faciliterait leur transport à
l’agrésome (Guilbert et al. 2018). Mécanistiquement, nos travaux suggèrent qu’HSPB8
pourrait agir en amont de BAG3, dans la régulation de la fonction de p62/SQSTM1, une
protéine d’échafaudage impliquée dans la formation d’inclusions cellulaires pour la
séquestration de protéines polyubiquitinées (section 1.3.4.1). La déplétion d’HSPB8 inhibe
la phosphorylation de p62/SQSTM1 et la formation de micro-aggrégats de protéines
ubiquitinées, ce qui interfère avec leur ciblage à l’agrésome via un mécanisme qui implique
BAG3. En outre, une étude récente suggère que le complexe chaperon, via l’action
d’HSPB8 en amont de BAG3, pourrait également participer au désassemblage de structures
agrégées nommées granules de stress, de façon indépendante de son activité autophagique

15
(Ganassi et al. 2016). Ces travaux suggèrent qu’HSPB8 et BAG3 peuvent agir
indépendemment de leur association. Dans ce contexte, il est critique de déterminer leur
rôle exact en tant que complexe protéique.
Dans le cadre de cette thèse, ce sont les fonctions physiologiques d’HSPB8 en association
avec BAG3 qui nous ont intéressées, notamment dans le remodelage des structures à base
d’actine. En effet, BAG3 a été impliquée dans de nombreux processus via sa structure
modulaire qui pourrait lui permettre, en principe, de relier le contrôle de qualité des
protéines à la signalisation et aux processus de remodelage de l’actine. De telles fonctions
de BAG3 seront présentées dans les sections suivantes et nous soulèverons les questions
que ces travaux ont mis en avant.
1.3.2. Structure et fonctions du co-chaperon BAG3
BAG3 fait partie d’une famille de protéines nommée BAG (« Bcl2-associated athanogen »)
qui sont connues pour moduler la fonction des systèmes chaperons HSP70 (Antoku et al.
2001; Doong, Vrailas, et Kohn 2002). Chez l’humain, il existe six protéines BAG (BAG1
à BAG6) qui possèdent toutes un domaine BAG situé dans leur région C-terminale, à
l’exception de BAG5 qui en possède quatre (Figure 1.3). Fonctionnellement, le domaine
BAG permet aux membres de cette famille de moduler négativement l’activité d’HSP70.
Une telle fonction des protéines BAG les implique dans le triage des substrats du chaperon
moléculaire. Par exemple, BAG1 a été largement impliquée dans l’induction de la
dégradation des substrats d’HSP70 par le protéasome (Lüders, Demand, et Höhfeld 2000;
Alberti et al. 2002; Demand et al. 2001). La différence entre les protéines BAG réside dans
la diversité de leur extrémité N-terminale qui leur permet de cibler HSP70 à différentes
localisations et machineries cellulaires (Figure 1.3). Notamment, BAG1 et BAG6 arborent
un domaine UBL (« ubiquitin-like ») qui leur permet d’intéragir avec et d’induire la
dégradation des cibles d’HSP70 par le protéasome (Lüders, Demand, et Höhfeld 2000).
BAG3 est unique parmi les membres de sa famille puisqu’elle est la seule à arborer des
motifs IPV, ainsi qu’un domaine riche en motifs PxxP et un domaine WW (Figure 1.3). En
outre, il est à noter que BAG3 est la seule membre de sa famille dont l’expression est

16
induite au cours d’un stress protéotoxique par l’intermédiaire du facteur de transcription
HSF-1, suggérant une fonction importante de ce co-chaperon dans le contrôle de qualité
des protéines au cours d’un stress (Franceschelli et al. 2008; Du et al. 2009; Wang et al.
2008). En tant que co-chaperon d’HSP70, il est reconnu que BAG3 inhibe son activité dans
le ciblage de ses substrats au protéasome et pourrait conduire à leur redirection à la
machinerie autophagique (section 1.3.4.1). L’implication de l’association à HSPB8 dans
une telle fonction de BAG3 a été suggérée par différentes études que nous décrirons dans
la section 1.3.4. De ce fait, BAG3 est perçue comme une plateforme unique qui pourrait
relier deux systèmes chaperons (HSP70 et HSPB8) (Rauch et al. 2016). BAG3 pourrait
coordonner leurs fonctions notamment pendant les processus d’autophagie sélective.
Cependant, les évidences mécanistiques qui mettent en avant une telle fonction de BAG3
sont encore déficientes.
Figure 1.3 : Schéma représentatif des différents domaines qui constituent les protéines BAG humaines.

17
Outre sa fonction dans la dégradation ciblée des protéines, BAG3 est reconnue comme une
protéine d’échafaudage qui intervient dans de nombreux processus de signalisation. Par ses
différents domaines, BAG3 est capable d’intéragir avec des enzymes de signalisation,
notamment PLCg (Phospholipase C gamma), la kinase oncogénique Src et des protéines
associées à l’actine (Figure 1.4) (Doong et al. 2000; Colvin et al. 2014). Ces interactions
vont l’impliquer dans des processus biologiques majeurs, c'est-à-dire l'apoptose, le
développement, l'organisation du cytosquelette d’actine et l'autophagie sélective (Figure
1.4). Une telle fonction de BAG3 permettrait de diriger les réponses adaptées des cellules
aux stimuli stressants. En outre, un nombre grandissant d’évidences laissent croire que ces
fonctions de BAG3 contribuent à soutenir la survie cellulaire, la résistance au traitement
et/ou la motilité et la progression métastatique dans différents types de cancer (Habata et
al. 2015; Liu et al. 2009; Ammirante et al. 2011; Habata et al. 2016; Rosati et al. 2015;
Zhang et al. 2012; Guerriero et al. 2014; Esposito et al. 2017; Festa et al. 2011; Maria
Fiammetta Romano et al. 2003; Shi et al. 2016; Lee et al. 2014; Franco et al. 2012; Yang
et al. 2016; Huayuan Zhu et al. 2014; Boiani et al. 2013).
1.3.3. Les fonctions assurées par BAG3 en tant que protéine d’échafaudage
De part sa structure particulière, le co-chaperon BAG3 a été impliqué dans la modulation
de différentes voies de signalisation intracellulaire (Figure 1.4) (Chen et al. 2013).
Toutefois, on se doit de souligner que peu d’études ont adressé le rôle des chaperons
associés à BAG3 dans de telles fonctions. BAG3 intervient notamment dans la voie de
signalisation dépendante de la kinase ERK (« Extracellular-Related Kinase ») où il module
la phosphorylation et en conséquence l’activation de la kinase (Falco et al. 2012; Yunoki
et al. 2015). Une telle fonction de BAG3 a été impliquée dans la régulation de
l’angiogénèse et de la transition épithéliomésenchymateuse. De plus, une étude suggère
que BAG3 est impliquée dans la modulation de la concentration intracellulaire de calcium
à travers son interaction avec PLCg (Figure 1.4) (Doong et al. 2000). Dans les sections ci-
après, nous nous intéresserons aux fonctions signalétiques de BAG3 associées aux
processus qui requièrent un remodelage de structures à base d’actine. Nous discuterons

18
également d’un potentiel rôle fonctionnel entre l’activité signalétique de BAG3 et son rôle
en tant que co-chaperon d’HSP70 et d’HSPB8.
Figure 1.4 : Schéma de la structure modulaire du co-chaperon BAG3 mettant en évidence des interactions avec des partenaires impliqués dans la signalisation intracellulaire, le remodelage du cytosquelette d’actine et l’autophagie sélective. Les nombres représentent les positions des acides aminés dans la séquence de BAG3; P209L correspond à la mutation liée à une myopathie sévère chez l'homme (adapté de Guilbert et al. 2015).
1.3.3.1. Le co-chaperon BAG3 module l’apoptose à travers la régulation de la
stabilité de facteurs anti-apoptotiques
BAG3 a été associée à la régulation de l’apoptose dans différents types de lignées
cancéreuses telles que des cellules du cancer de l’ovaire, de glioblastome, de carcinome
pulmonaire et de neuroblastome ( Zhang et al. 2012; Romano et al. 2003; Lee et al. 1999;
Jacobs et Marnett 2009; Chiappetta et al. 2007; Boiani et al. 2013). Une telle fonction de
BAG3 a été corrèlée à sa capacité à stabiliser des facteurs anti-apoptotiques dont Bcl-2
(« B-cell lymphoma 2 »), Bcl-XL (« B-cell lymphoma-extra large ») et Mcl-1 (« Myeloid
cell leukemia sequence 1 ») ( Zhang et al. 2012; Lee et al. 1999; Karpel-Massler et al. 2015;

19
Jacobs et Marnett 2009; Habata et al. 2016; Boiani et al. 2013). La stabilisation de ces
facteurs anti-apoptotiques passe par une interaction de BAG3 avec ceux-ci. Il est à noter
que les expériences de co-immunoprécipitation de BAG3 avec ces facteurs anti-
apoptotiques ont révélé la présence d’HSP70 dans les immunocomplexes. En outre,
l’interaction de BAG3 avec Bcl-2 implique le domaine BAG du co-chaperon (Antoku et
al. 2001). De ce fait, il est proposé que la fonction anti-apoptotique de BAG3 requiert son
activité de co-chaperon. Cependant, aucune évidence à l’heure actuelle n’a mis en avant
un lien fonctionnel entre l’activité anti-apoptotique de BAG3 et sa fonction de co-chaperon
d’HSP70 ou d’HSPB8.
1.3.3.2. BAG3 régule l’adhésion, la motilité cellulaire et la transition
épithéliomésenchymateuse
BAG3 a également été impliquée dans la signalisation de l’adhésion et de la motilité
cellulaires. Notamment, la surexpression de BAG3 induit une réduction de la migration
cellulaire, de la quantité d’adhésions focales et en conséquence de l’adhésion cellulaire
(Kassis et al. 2006). Ces résultats suggèrent que BAG3 peut réguler négativement
l'adhérence, l'assemblage d'adhésions focales, la signalisation et la migration via son
domaine PxxP. Une telle fonction de BAG3 impliquerait le contrôle de l’expression de la
protéine CCN1 (« Cyr61- CTGF- NOV protein 1 »), une protéine qui interagit avec et
augmente l’activité des adhésions focales, provoquant ainsi un phénotype adhésif (Chen et
al. 2004; Kassis et al. 2009).
Les travaux d’Iwasaki et al. s’opposent à ces conclusions. En effet, les auteurs notèrent que
la surexpression de BAG3 dans des lignées cancéreuses augmente leur mobilité alors que
sa déplétion la réduit (Iwasaki et al. 2007). En outre, des cellules MEF (« mouse embryonic
fibroblast ») KO pour BAG3 présentent un retard dans la formation de filopodes et des
adhésions focales, et une réduction de la motilité cellulaire. Dans ce contexte, la réduction
de la mobilité cellulaire a été associée à une réduction de l’activation de la GTPase Rac1.
Les auteurs ont observé que cette fonction dépendait du domaine WW de BAG3, puisque
la surexpression d’un mutant de ce domaine réduit la motilité cellulaire (Iwasaki et al.

20
2010). Ces derniers résultats indiquent que par ce domaine BAG3 est capable d’intéragir
et de réguler l’activité de la GEF de Rap1 (« Ras-proximate-1 » ou « Ras-related protein
1 ») : PDZGEF2 (« PDZ Domain-Containing Guanine Nucleotide Exchange Factor 2 »).
De cette manière, BAG3 contrôlerait l’activation de Rap1 et la mise en place des adhésions
focales et de leur signalisation subséquente (Figure 1.4).
Bien que les résultats d’Iwasaki et al. et de Kassis et al. soient contradictoires, il est possible
d’envisager que la surexpression ou la déplétion de BAG3 débalance les complexes
protéiques régulés par le co-chaperon. De cette façon, suivant le contexte du protéome
(cellules cancéreuses, MEF, cellules épithliales immortalisées, etc…), des complexes
favorisant ou inhibant l’adhésion et/ou la motilité cellulaires vont se former
majoritairement donnant lieu à des effets cellulaires contradictoires.
D’autre part, BAG3 a été impliquée dans la progression tumorale à travers sa fonction dans
la régulation de la transition épithéliomésenchymateuse dans des lignées de cancer
thyroïdien, hépatique ou de gliome (Li et al. 2013; Xiao et al. 2014; Lee et al. 2014; Meng
et al. 2014). Notamment, BAG3 régulerait positivement ce processus de façon dépendante
de sa phosphorylation par PKCd (« Protein Kinase C delta ») (Li et al. 2013). Une telle
fonction de BAG3 serait également reliée à sa capacité à moduler l’expression de
marqueurs mésenchymateux comme la N-cadhérine et la vimentine (Xiao et al. 2014; Shi
et al. 2016). De plus, BAG3 stabiliserait le facteur de transcription STAT3 (« Signal
transducer and activator of transcription 3 ») impliqué dans la transition
épithéliomésenchymateuse (Im et al. 2016; Yuan, Zhang, et Niu 2015). Par cette fonction,
BAG3 interviendrait dans la régulation de gènes codant pour des marqueurs
mésenchymateux tels que SNAIL et la métaloprotéinase MMP-2 (« Matrix
Metalloproteinase-2 »).
Dans l’ensemble de ces études, l’activité co-chaperon de BAG3 n’a pas été abordée
directement. Bien que le domaine BAG et les motifs IPV de BAG3 n’aient pas été étudiés
dans sa fonction régulatrice de l’adhésion et de la migration cellulaires ou de la transition
épithéliomésenchymateuse, il est envisageable que son activité co-chaperon soit requise

21
pour de telles fonctions. L’implication de la stabilisation de facteurs tels que STAT3 par
BAG3 suggère que son rôle de co-chaperon pourrait être impliqué. Ces hypothèses restent
à être testées.
1.3.3.3. Lien fonctionnel entre l’activité signalétique de BAG3 et son rôle en tant
que co-chaperon d’HSP70
Mise à part nos travaux récents (Fuchs et al. 2015; Annexe 1; section 1.4.5), une seule
étude, à notre connaissance, s’est intéressée à l’implication fonctionnelle de l’activité de
co-chaperon de BAG3 dans sa fonction signalétique. Cette étude a démontré que le
complexe formé de BAG3 et d’HSP70 est impliqué dans la modulation de plusieurs voies
de signalisation associées à la progression tumorale (Colvin et al. 2014). Notamment, les
auteurs, par des expériences de co-immunoprécipitation et de « pull-down », ont montré
que BAG3 interagit avec la kinase oncogénique Src de façon dépendante d’HSP70. En
effet, un mutant ponctuel de BAG3, le mutant R480A qui n’interagit plus avec HSP70,
n’est plus capable d’intéragir avec le domaine SH3 (« Src Homology 3 ») de Src. De même,
cette interaction entre BAG3 et Src est perdue lorsque le domaine PxxP de BAG3 est délété.
En outre, la déplétion de BAG3 ou d’HSP70 interfère avec la phosphorylation activatrice
de Src. La déplétion d’HSP70 induit une activation de la voie de signalisation Src, alors
que la déplétion de BAG3 n’induit pas d’activation de Src, mais l’action d’HSP70 sur Src
requiert BAG3. Ce résultat suggère donc qu’HSP70 régule négativement la voie de
signalisation Src et que cette fonction du chaperon moléculaire est modulée par son
association à BAG3. Dans cette même étude, les auteurs se sont aussi intéressés à l’impact
du complexe BAG3-HSP70 sur l’activation de la voie de signalisation NF-kB (« Nuclear
Factor-kappa B »). Comme pour la voie de signalisation Src, les auteurs ont montré que la
déplétion d’HSP70 induit une activation de la voie NF-kB qui peut être inhibée suivant la
déplétion de BAG3.
En résumé, ces résultats indiquent que le complexe chaperon formé d’HSP70 et de BAG3
est impliqué dans la régulation d’un réseau complexe de signalisation, qui comprend
notamment Src et NF-kB, dans un contexte de lignées cancéreuses. De façon importante,

22
cette étude a démontré pour la première fois un lien fonctionnel entre l’activité signalétique
de BAG3 et sa fonction de co-chaperon d’HSP70. Cette étude identifia également un petite
molecule inhibitrice de l’interaction de BAG3 à HSP70 dont l'administration in vivo est
suffisante pour supprimer la croissance tumorale chez la souris. Néanmoins, l’ensemble
des études analysant la fonction signalétique de BAG3 suggère, mais n’a jamais démontré,
que l’activité de co-chaperon de BAG3 pouvait être impliquée. De même, ces études n’ont
pas analysé le rôle fonctionnel de l’association de BAG3 à HSPB8 dans ces processus
cellulaires.
Ainsi, les fonctions cellulaires du complexe chaperon HSPB8-BAG3 sont très peu étudiées
dans un contexte physiologique, ou dans les cellules cancéreuses qui surexpriment BAG3
et HSPB8. Les fonctions d’HSPB8-BAG3 ont été caractérisées principalement dans un
contexte de stress protéotoxique où il est impliqué dans le ciblage sélectif de protéines à la
machinerie autophagique. C’est ce que nous allons décrire dans les prochaines sections.
1.3.4. Fonctions assurées par BAG3 lors d’un stress protéotoxique
L’activité de co-chaperon assurée par BAG3 a été étudiée majoritairement dans un contexte
de stress protéotoxique où son expression est induite par HSF-1 (Franceschelli et al. 2008;
Du et al. 2009; Wang et al. 2008). Ainsi, la plupart des études initiales sont basées sur la
surexpression de BAG3, considérant qu’elle reproduit les conditions d’un stress
protéotoxique. Il convient donc de mentionner ici que certains partenaires potentiels de
BAG3 dans ces fonctions pourraient en principe se retrouver en quantités limitantes,
compliquant l’interprétation de ces données.
La fonction de co-chaperon de BAG3 fut étudiée pour la première fois par l’équipe du Dre
Kohn E.C. qui montra que la surexpression de BAG3 prévient la perte de la kinase Akt et
d’autres clients connus d’HSP70, suite au traitement avec la geldanamycine (un inhibiteur
d’HSP90), tandis que la surexpression d’un mutant de délétion du domaine BAG de BAG3
est inactif (Doong et al. 2003). Cette étude suggérait donc que BAG3 intervient comme un
inhibiteur de la dégradation des substrats d’HSP70 par le protéasome en condition de stress

23
cellulaire. Suite à cette étude, il fut noté qu’HSPB8-BAG3 module la dégradation de
protéines par la machinerie autophagique (section 1.3.4.2) (Carra et al. 2008). Ces travaux
ont par la suite été corroborés dans d’autres systèmes comme le vieillissement, l’hypoxie
et la réponse à l’agrésome (Minoia et al. 2014; Rapino, Jung, et Fulda 2014; Gamerdinger
et al. 2011, 2009; Zhang et al. 2016). Cela dit, dans l’ensemble de ces études, le rôle exact
d’HSPB8 demeure imprécis bien qu’il ait été montré que la surexpression d’HSPB8 peut
reproduire le phénotype BAG3 sur la dégradation de substrats par autophagie (Carra et al.
2008; Minoia et al. 2014). En revanche, la déplétion d’HSPB8 n’inhibe pas l’action de
BAG3 surexprimée, suggérant qu’HSPB8 n’est pas requise, ou bien qu’elle pourrait agir
en amont de BAG3, comme le suggère nos plus récents travaux (Guilbert et al. 2018).
Le rôle physiologique d’HSPB8-BAG3 dans l’autophagie sélective a été étudié dans le
contexte du muscle squelettique où les deux protéines sont abondamment exprimées
(section 1.3.5.2). Avant de passer en revue ces études et leur implication, il convient de
présenter les généralités concernant la dégradation par autophagie sélective.
1.3.4.1. L’Autophagie sélective et ses déterminants p62/SQSTM1 et HDAC6
Historiquement, l’autophagie a été décrite comme un processus non sélectif conduisant à
l’engouffrement du contenu cytoplasmique et à sa dégradation lysosomale pour soutenir
les besoins métaboliques de la cellule (Auteri et al. 1983). De nombreux travaux ont ensuite
décrit que le mécanisme d’autophagie n’était pas unique et qu’il pouvait se diviser en trois
types différents : la macroautophagie (nommée ci-après autophagie), la microautophagie
et l’autophagie dépendante des chaperons (Schneider et Cuervo 2014).
L’autophagie implique la formation d’une vésicule à double membrane appelée
autophagosome autour du cargo cytoplasmique qui sera transportée vers le lysosome pour
sa dégradation (Figure 1.5). Il s’agit d’un flux qui se divise en trois étapes dépendantes des
protéines ATG (« AuTophaGy »). Ces étapes consistent en l’initiation de l’autophagosome,
l’allongement membranaire et la maturation de l’autophagosome, et la fusion de
l’autophagosome avec le lysosome (Figure 1.5). Il est à noter que la maturation de

24
l’autophagosome implique l’insertion dans sa double membrane de la protéine LC3
(« microtubule-associated protein 1A/1B-light chain 3 ») sous sa forme lipidée (passage de
LC3 I à LC3 II; Figure 1.5; Tanida et al. 2004, 2002; Ichimura et al. 2000; Kirisako et al.
2000). Ce processus permet de suivre la maturation du phagophore en autophagosome
biochimiquement ou par immunofluorescence.
Figure 1.5 : Les étapes de formation et de dégradation de l’autophagosome (de http://www.invivogen.com/autophagy). L’initiation de l’autophagie dépend de l’activation de deux complexes majeurs : le complexe ULK1 (« Unc-51-Like autophagy activating Kinase 1 ») et le complexe PI3KC3–C1 (« class III PI 3-Kinase Complex I »). Leurs activation et relocalisation permettent la création d’une région membranaire enrichie en PI(3)P (phosphatidylinositol-3-phosphate). Une fois l’initiation engagée, la membrane enrichie en PI(3)P se courbe pour former le précurseur de l’autophagosome appelé phagophore. À la suite de l’étape d’initiation, le phagophore est enrichi des marqueurs autophagiques spécifiques ATG12 et LC3, c’est l’étape de maturation. Au cours de l'enrichissement de ces marqueurs à la membrane, le phagophore s’allonge et englobe la cargaison cytoplasmique. La fermeture de la double membrane du phagophore conduit à la formation de l'autophagosome. Enfin, l'autophagosome fusionne avec le lysosome, formant le compartiment de dégradation connu sous le nom d'autophagolysosome, dans lequel la cargaison engloutie sera digérée par les enzymes lysosomales.
L'autophagie peut être sélective ou non sélective. L'autophagie non sélective est déclenchée
par la privation des cellules en nutriments et elle est essentielle pour le maintien d'un apport
cellulaire de lipides, d'acides aminés, de glucides et de nucléotides en situation de stress.

25
Ce processus permettrait aux cellules cancéreuses de survivre dans les régions hypoxiques
des tumeurs solides (Zhu et al. 2014; Vitale et al. 2015; Feng et al. 2016; Tan et al. 2016;
Marcucci, Ghezzi, et Rumio 2017). L'autophagie sélective, quant à elle, constitue un
mécanisme de contrôle de qualité cellulaire : elle permet l’élimination et le recyclage de
matériaux nocifs ou simplement inutiles à la cellule. Ceux-ci comprennent les agrégats de
protéines, les mitochondries endommagées, les peroxysomes et les ribosomes en excès, le
réticulum endoplasmique et les endosomes, les gouttelettes lipidiques, les agents
pathogènes intracellulaires et les protéines poly-ubiquitinées sur K63 (Zaffagnini et
Martens 2016; Tan et al. 2008; F. Huang et al. 2013; Clough et al. 2016; McKeon et al.
2015). C’est ainsi qu’actuellement différents types d’autophagies sélectives sont retrouvés
et nommés d’après la nature du cargo, incluant la mitophagie (mitochondies), l’agréphagie
(agrégats protéiques), la réticulophagie (réticulum endoplasmique), la pexophagie
(péroxisome) etc. (Kissová et al. 2004; Schuck, Gallagher, et Walter 2014; Zientara-Rytter
et Subramani 2016a; Anding et Baehrecke 2017; Galluzzi et al. 2017). Des défauts dans ce
mécanisme de contrôle de qualité cellulaire sont associés au développement de nombreuses
pathologies dont le cancer, l’infection chronique ou encore la neurodégénérescence
(Mancias et Kimmelman 2016).
Ainsi, l’autophagie sélective implique la reconnaissance de cargos spécifiques. Cela
suppose que la cellule exprime des récepteurs spécifiques qui pourront reconnaître ces
cargos et les cibler à la machinerie autophagique. Ce sont les adaptateurs autophagiques
qui comprennent la protéine NBR1 (« Next to BRCA1 gene 1 protein »), TAX1BP1
(« TAX1 Binding Protein 1 »), NDP52 (« Nuclear Dot 52 kDa Protein »), OPTN
(« Optineurin ») et p62/SQSTM1 (Sequestosome 1) (Rogov et al. 2014).
La protéine p62/SQSTM1 est le premier adapteur identifié et donc le plus étudié. Elle
comprend plusieurs domaines structurels dont le domaine UBA (« Ubiquitin Association »)
qui lui permet de lier des protéines poly-ubiquitinées et le domaine PB1 (Phox et Bem 1)
impliqué dans l’auto-oligomérisation de la protéine (Vadlamudi et al. 1996; Lamark et al.
2003; Isogai et al. 2011). Il a été noté, in vitro, que par son domaine UBA p62/SQSTM1
se lie préférentiellement aux protéines poly-ubiquitinées sur K63, une marque post-

26
traductionnelle qui permet le ciblage à la dégradation par autophagie (Tan et al. 2008;
Huang et al. 2013; Clough et al. 2016; McKeon et al. 2015; Xu et al., 2009). De plus, in
vivo, la phosphorylation sur la sérine 403 de p62/SQSTM1 par CK2 (« Casein Kinase 2 »)
stimule une telle liaison (Wooten et al. 2008). p62/SQSTM1 arbore aussi un domaine LIR
(« LC3 Interacting Region ») qui permet à l’adaptateur d'intéragir directement avec la
protéine LC3 sur la membrane de l’autophagosome (Pankiv et al. 2007; Ichimura et al.
2008). De façon générale, les trois domaines de p62/SQSTM1 sont essentiels pour sa
fonction dans l’autophagie sélective. En effet, p62/SQSTM1, grâce à son domaine UBA,
reconnaît les protéines marquées par l’ubiquitine K63 et déclenche leur agrégation par
auto-oligomérisation à travers son domaine PB1. Puis, p62/SQSTM1 cible les agrégats de
protéines vers l'autophagosome via son domaine LIR (Wurzer et al. 2015; Bjørkøy et al.
2005; Ciuffa et al. 2015; Paine et al. 2005).
Les processus d’autophagie sélective semblent faire intervenir également l’action d’une
déacétylase cytoplasmique : HDAC6 (« Histone deacetylase 6 »). En effet, HDAC6 est
fortement impliquée dans le contrôle de qualité des protéines. Cette fonction a été établie
dans le contexte de la formation de l’agrésome, une structure de protection cellulaire où
sont recrutées les protéines poly-ubiquitinées en réponse à un stress protéotoxique extrême
(Johnston, Ward, et Kopito 1998). Dans ce contexte, la formation d'agrégats de protéines
est stimulée de façon active, via une aggrégation dite fonctionnelle, ce qui les rend plus
enclines à la dégradation par autophagie (agréphagie). Les études ont montré qu’HDAC6
est en mesure de reconnaître les protéines ubiquitinées et dénaturées à travers son domaine
BUZ. HDAC6 charge alors ces protéines sur la dynéine qui se déplace vers le MTOC
(« microtubules organization center ») afin de former l’agrésome (Ouyang et al. 2012; Liu
et al. 2012; Kawaguchi et al. 2003; Olzmann et al. 2007; Gal et al. 2013; Du et al. 2010;
Into et al. 2010). Ceci suggère qu’HDAC6 est impliquée dans des voies de signalisation
régulatrices de l’autophagie sélective. Dans ce sens, il a été suggéré que la modulation de
la stabilité des microtubules par HDAC6 est utilisée par les cellules pour stabiliser les
microtubules et promouvoir le transport des vésicules autophagiques (Iwata et al. 2005).
Par la suite, des travaux ont indiqué qu’HDAC6 facilite la dégradation de substrats
protéiques potentiellement nocifs par la voie autophagique et ont impliqué une telle

27
fonction dans la protection neuronale (Pandey et al. 2007, 2007). Cependant, ces travaux
n’avaient pas encore élucidé le mécanisme moléculaire par lequel HDAC6 module
l’autophagie.
Ce sont (Lee et al. 2010) qui ont suggéré qu’HDAC6 est une composante centrale de
l'autophagie sélective. Ces travaux ont montré qu’HDAC6 favorise l'autophagie en
recrutant une machinerie de remodelage d'actine dépendante de la cortactine qui, à son
tour, stimule l’assemblage d’un réseau de F-actine qui promouvoit la fusion des
autophagosomes avec le lysosome. Ces travaux remarquables ont aussi démontré que
l'assemblage de F-actine dépendant d’HDAC6 et sa cible cortactine est entièrement
dispensable pour l'autophagie non sélective alors qu’il est requis pour l’agréphagie. Il est
intéressant de noter que dans ce contexte, p62/SQSTM1 régulerait négativement l’activité
déacétylase d’HDAC6 suggérant que p62/SQSTM1 joue probablement un double rôle dans
l'autophagie sélective, non seulement dans la formation de l'autophagosome, mais aussi
dans la régulation de la fusion des autophagosomes avec le lysosome dépendante
d’HDAC6 (Yan et al. 2013).
En résumé, l’autophagie sélective dépend d’un ciblage sélectif assuré par des adaptateurs
autophagiques aux autophagosomes. Ce processus repose également sur la régulation de la
dynamique de l’actine par la déacétylase HDAC6 via la modulation de la cortactine.
Cependant, les mécanismes d’activation et les voies de signalisation à l’origine de
l’induction de la dégradation sélective de substrats par autophagie restent encore très peu
connus.
1.3.4.2. BAG3 cible les protéines poly-ubiquitinées ou dénaturées à la dégradation
par autophagie sélective
Ce sont Carra et al. qui notèrent pour la première fois que le co-chaperon BAG3 module la
dégradation de protéines par autophagie. En effet, la surexpression de BAG3 se traduit par
une accélération de la dégradation de la protéine Htt43Q (« Huntigtin 43 glutamin »), une
forme pathogène de la protéine huntingtine qui s’agrège spontanément (Carra et al. 2008).

28
L’inhibition de l’autophagie, à l’aide d’inhibiteurs, diminue considérablement la
dégradation de Htt43Q dans les cellules surexprimant BAG3 ou HSPB8, suggérant que la
dégradation de la protéine mutante est dépendante de la machinerie autophagique. Aussi,
ces travaux ont montré que la réduction de l’expression de BAG3 réduit la lipidation de
LC3 tandis que sa surexpression ou celle de son partenaire HSPB8 induit une augmentation
du niveau de LC3 II. Ces résultats suggéraient alors que le complexe chaperon HSPB8-
BAG3 pourrait réguler la dégradation autophagique de protéines agrégées au cours d’un
stress protéotoxique.
Dans la même période, l’équipe du Dr Behl montra qu’une telle fonction de BAG3 était
induite au cours du vieillissement cellulaire (Gamerdinger et al. 2009). En effet, en criblant
les différences d'expression des co-chaperons de la famille BAG pendant le vieillissement
des cellules humaines, Gamerdinger et al. identifièrent une réduction de l'expression de
BAG1 en faveur de celle de BAG3 durant le vieillissement cellulaire ainsi que dans des
conditions de stress oxydatif ou protéasomal (Gamerdinger et al. 2009). Ce transfert
d’expression des co-chaperons fut associé à un passage fonctionnel de l'activité
protéasomale médiée par BAG1 à l'activité autophagique médiée par BAG3 (Gamerdinger
et al. 2009; Minoia et al. 2014). Ce changement de BAG1 vers BAG3 pour le contrôle de
la dégradation des protéines poly-ubiquitinées serait lié à une compétition des deux co-
chaperons pour l’interaction avec HSP70 (Minoia et al. 2014). De plus, une telle fonction
de BAG3 semble être dépendante de l’adaptateur autophagique p62/SQSTM1 avec lequel
le co-chaperon interagit à travers une région indéterminée actuellement. Cette observation
suggère donc que BAG3 via son association à p62/SQSTM1 est impliquée dans la
modulation de l’autophagie sélective (Figure 1.6).
Plus tard, Gamerdinger et al. utilisèrent la réponse à l’agrésome comme modèle d’étude de
la fonction autophagique de BAG3. Cette étude montra que BAG3 facilite la formation de
l’agrésome en y dirigeant spécifiquement les substrats d’HSP70 via le chargement des
protéines dénaturées sur la protéine motrice dynéine (Gamerdinger et al. 2011). Ces
travaux suggérèrent que le domaine PxxP est important pour l’interaction de BAG3 avec
la dynéine et que le ciblage des protéines dénaturées requérait son domaine BAG.
Cependant, l’interaction de BAG3 avec la dynéine fût par la suite montrée pour être

29
également dépendante de son association avec la protéine adaptatrice 14-3-3 (Figure 1.6)
(Xu et al. 2013). En effet, 14-3-3 participe au ciblage de protéines qui s’agrègent
spontanément de façon dépendante de son interaction à la dynéine et à BAG3 via les sérines
phosphorylées 136 et 173 localisées dans la région N-terminale du co-chaperon. Par
ailleurs, le domaine WW de BAG3 a également été impliqué dans sa fonction autophagique
lors d’une étude dans des cellules de gliome (Figure 1.6) (Merabova et al. 2015).
Figure 1.6 : Schéma représentant le mécanisme par lequel le co-chaperon BAG3 aiderait au ciblage de protéines endommagées, agrégées ou poly-ubiquitinées à la machinerie autophagique.
Le mécanisme moléculaire par lequel BAG3 module l’activation de la dégradation par
autophagie est encore peu connu à l’heure actuelle. Cependant, certaines évidences sont
apparues dans les dernières années. Notamment, des travaux ont montré que le niveau

30
d’expression de BAG3 affecte la lipidation de LC3 suggérant que BAG3 pourrait contrôler
ce mécanisme moléculaire essentiel au maintien d’un flux autophagique (Carra et al. 2008;
Merabova et al. 2015; Liu et al. 2013). Il semble qu’une telle fonction de BAG3 implique
également sa capacité à réguler la traduction de l’ARNm codant pour LC3 sans affecter ni
la transcritpion de son gène, ni la dégradation de la protéine par le protéasome ou par
autophagie (Rodríguez et al. 2016). Par ailleurs, une étude très récente a démontré que
BAG3 coordonne la synthèse protéique et l'autophagie par la régulation spatiale de
mTORC1 (« mammalian Target Of Rapamycin Complex 1 ») qui gère de multiples voies
de biosynthèse et favorise la croissance cellulaire lorsque les nutriments sont en abondance
et dont l’inhibition est essentielle à l’activation de l’autophagie (Dibble et Manning 2013;
Dunlop et Tee 2014; Kathage et al. 2016). Les résultats obtenus dans cette étude seront
détaillés davantage dans la section 1.3.5.2.
En résumé, il semble que les trois domaines d’interaction de BAG3 : BAG, PxxP et WW
soient importants pour la fonction autophagique de BAG3 (Figure 1.6). Les résultats de
Carra et al. suggèrent que l’association de BAG3 à HSPB8, incluant ainsi les motifs IPV
de BAG3, est impliquée dans sa fonction autophagique (Carra et al. 2008). De même que
la nécessité de l’association de BAG3 à p62/SQSTM1 pour assurer la fonction
autophagique du co-chaperon suggère que ce dernier est impliqué dans le ciblage sélectif
de protéines à l’autophagie. Bien que certaines évidences soient apparues dans les dernières
années, le mécanisme moléculaire par lequel BAG3 module l’autophagie est encore à
élucider. La démonstration la plus probante pour un mécanisme a été faite dans le contexte
des cellules musculaires lors de la contraction musculaire. Dans ce contexte, il semble que
BAG3, en association avec HSPB8 et HSP70, soit requise pour le ciblage d’une protéine
de réticulation du cytosquelette d’actine, la filamine (FLN), à la dégradation autophagique
(section 1.3.5.2). C’est ce que nous allons présenter dans les prochaines sections.

31
1.3.5. Régulation de structures contractiles d’actine par le complexe chaperon
HSPB8-BAG3
1.3.5.1. BAG3 assure l’intégrité des myofibrilles musculaires grâce à sa fonction
de co-chaperon d’HSP70
Le co-chaperon BAG3 régulerait la polymérisation du cytosquelette d’actine à travers des
mécanismes moléculaires impliquant sa fonction de co-chaperon d’HSP70. En effet, il a
été démontré, dans le contexte du muscle squelettique, que BAG3 régule la stabilité de la
myofibrille, la structure de contraction du muscle squelettique constituée d’actine et de
myosine notamment (Hishiya, Kitazawa, et Takayama 2010; Schafer, Hug, et Cooper
1995). En absence de BAG3, l’intégrité des myofibrilles soumises à une contraction
mécanique est perturbée. Un tel phenotype a été associé à une promotion par BAG3 de
l’association de CapZb1 à HSC70 (« Heat Shock Cognate 70 ») ce qui assurerait sa stabilité
et sa bonne localisation. La déplétion de BAG3 conduit à une dégradation de CapZ par le
système ubiquitine-protéasome, suggérant que BAG3 pourrait inhiber le ciblage de la
protéine de coiffe au protéasome par HSC70. Ainsi, BAG3 est cruciale pour le maintien de
l'intégrité du cytosquelette d’actine de la cellule musculaire lors d’un stress mécanique en
assurant la stabilité de la sous-unité β1 de CapZ, entrainant ainsi l'association et la
stabilisation de la F-actine par la protéine de coiffe.
Par ailleurs, le domaine BAG de BAG3 a également été impliqué dans la modulation de la
polymérisation de la F-actine. En effet, il a été démontré qu’à travers ce domaine BAG3
interagit avec la protéine CCT (« Chaperonin Containing T-complex polypeptide »), un
chaperon de la G-actine (Fontanella et al. 2010). De cette manière, BAG3 pourrait réguler
la migration cellulaire et influencer le repliement de l’actine monomérique dépendante de
CCT.
Par ces fonctions de régulation de l’intégrité du cytosquelette d’actine, des mutations de
BAG3 ont été associées au développement de myopathies présentant une désorganisation
myofibrillaire (Homma et al. 2006; Lee et al. 2012; Arimura et al. 2011; Selcen et al. 2009).

32
Notamment, la mutation P209L de BAG3 a été corrélée au développement d’une
myopathie myofibrilaire fulgurante chez l’être humain (Selcen et al. 2009; Figure 1.4). Du
fait que cette mutation soit localisée dans le second motif IPV de BAG3, les études
proposent que l’association de BAG3 à HSPB8 est requise pour sa fonction de modulateur
du cytosquelette d’actine (Figure 1.4). Cependant, aucune de ces études n’ont démontré
une dépendance de BAG3 à HSPB8 pour une telle fonction. De plus, l’impact réel de la
mutation P209L sur l’association de BAG3 à HSPB8 n’a pas été démontré. Des travaux
récents du laboratoire suggèrent que la mutation n’abolit pas l’association de BAG3 à
HSPB8, mais qu’elle pourrait la déréguler (Guilbert et al. 2018). En outre, les études
n’avaient pas encore analysé un lien fonctionnel entre l’activité autophagique de BAG3 et
sa fonction de modulateur du cytosquelette d’actine. Récemment, des travaux pionniers ont
adressé un lien potentiel entre HSPB8 et BAG3 et l’activité de BAG3 dans le ciblage de
protéines du cytosquelette d’actine à l’autophagie et seront présentés dans la section ci-
après.
1.3.5.2. Régulation de l’intégrité du cytosquelette d’actine dépendant de
l’autophagie sélective induite par HSPB8-BAG3 dans la cellule musculaire
C’est l’équipe du Dr Höfheld J. qui a établi que BAG3 régule l’intégrité du cytosquelette
d’actine dans les cellules musculaires, de façon dépendante de sa fonction autophagique
lorsque la cellule est soumise à une tension mécanique (Arndt et al. 2010; Ulbricht et al.
2013; Kathage et al. 2016).
En effet, dans ces travaux, les auteurs ont montré que BAG3 contrôle la formation d’un
complexe chaperon multiprotéique constitué d’HSC70, HSPB8, de la E3 ubiquitine ligase
CHIP (« Carboxyl terminus of Hsc70-Interacting Protein ») et de l’adaptateur autophagique
p62/SQSTM1 lors d’un stress mécanique (Arndt et al. 2010; Ulbricht et al. 2013) Figure
1.7). L’équipe démontra dans un premier temps que ce complexe chaperon aide au ciblage
de la protéine dénaturée FLN à la dégradation par autophagie sélective de façon dépendante
de son ubiquitination impliquant CHIP et nomma ce mécanisme moléculaire CASA
(« Chaperone-Assisted Selective Autophagy ») (Arndt et al. 2010). Dans un second temps,

33
l’équipe montra que la CASA est une voie d'autophagie sélective induite par la tension,
essentielle à la mécano-transduction dans les cellules musculaires (Ulbricht et al. 2013).
Dans cette étude, les auteurs ont montré que la formation d'autophagosomes pendant la
CASA dépend d'une interaction de BAG3 avec la SYNPO2 (Synaptopodine-2), une
protéine adaptatrice du cytosquelette d’actine qui se lie à la FLN, via son domaine WW
(Figure 1.7). De plus, ces travaux révèlent que BAG3 utilise également son domaine WW
pour s'engager dans la voie de signalisation YAP (« Yes-associated protein »)/TAZ
(« Tafazzin »). Grâce à cette voie, le co-chaperon contribue à la translocation des facteurs
YAP/TAZ au noyau et stimule ainsi la transcription de la FLN (Figure 1.7). Un tel
mécanisme moléculaire permet donc d’une part, l’élimination de la FLN partiellement
dénaturée par le stress mécanique, et d’autre part, son remplacement via la transcription
dépendante de YAP/TAZ.
Figure 1.7: Schéma du mécanisme moléculaire de la CASA (tiré de Ulbricht et Höhfeld 2013). Le seul substrat démontré de la CASA est la protéine de réticulation de l’actine FLN, qui devient dénaturée et endommagée sous la tension mécanique, ce qui conduit à sa reconnaissance par HSPB8 en association avec BAG3, et à son ubiquitination par le

34
complexe CASA. Au cours de la dégradation de la FLN, la formation d'autophagosomes est déclenchée par la coopération entre BAG3 et SYNPO2 qui facilite le ciblage et la fusion des autophagosomes. BAG3 utilise également son domaine WW pour contacter LATS1/2 qui sont impliquées dans la séquestration cytoplasmique des régulateurs transcriptionnels YAP et TAZ (WWTR1). La liaison de BAG3 à LATS1/2 abroge la séquestration YAP/TAZ et induit la transcription FLN sous tension. Ig, type immunoglobuline; ECM, matrice extracellulaire.
Plus récemment, il a été montré que lors de la contrainte mécanique dans les cellules
musculaires lisses, BAG3 pourrait coordonner la synthèse protéique et la dégradation par
autophagie par la régulation spatiale de mTORC1 (Kathage et al. 2016). En effet, il a été
montré que par son domaine WW le co-chaperon interagit avec la protéine TSC1
(« Tuberous sclerosis 1 ») qui fonctionne comme un inhibiteur de mTORC1 en association
avec TSC2 (« Tuberous sclerosis 2 »). Cette interaction entraînerait un recrutement de
complexes TSC dans des fibres de stress, où les complexes inhibent une sous-population
de vésicules mTOR positives associées au cytosquelette d’actine. Ainsi, l'inhibition locale
de mTORC1 permettrait d’initier l'autophagie sur les sites de dénaturation de la FLN. Dans
le même temps, la séquestration de TSC1/TSC2 médiée par BAG3 soulage l'inhibition de
mTORC1 dans le cytoplasme restant, ce qui stimule la traduction des protéines. Ceci
suggère un modèle selon lequel une fonction de BAG3 dans la séquestration des protéines
permettrait une régulation spatiotemporelle de la signalisation cellulaire et de l’autophagie.
En résumé, ces observations indiquent un rôle central de BAG3 dans l’induction de la
dégradation par autophagie sélective de la FLN afin d’assurer l’organisation du
cytosquelette d’actine au cours d’un stress mécanique. Dans l’ensemble de ces travaux, les
auteurs suggèrent que le chaperon moléculaire HSPB8 est impliqué dans le mécanisme de
la CASA et aiderait à la reconnaissance de la FLN dénaturée. Néanmoins, aucun résultat
ne l’a démontré de façon probante. Ainsi, dans le contexte d’un stress mécanique, il semble
exister un lien fonctionnel entre l’activité signalétique de BAG3 et sa fonction dans
l’autophagie sélective qui permet au co-chaperon de moduler le cytosquelette d’actine. Il
reste à savoir si la CASA pourrait cibler d’autres substrats, comme des protéines
« mécanosensibles », et si son implication peut être élargie aux cellules non-musculaires.

35
Dans ce contexte, nous nous sommes demandés si certaines des activités de BAG3 dans le
remodelage du cytosquelette d’actine pourraient être spécifiées par HSPB8 dans les
cellules cancéreuses, et pourraient faire intervenir son action dans l’autophagie sélective.
Pour répondre à cette question, nous avons choisi comme modèle paradigme : la division
cellulaire. En effet, durant ce processus cellulaire, les structures à base d’actine sont
soumises à des forces mécaniques importantes. Nous avons donc émis l’hypothèse, que
BAG3, en association avec HSPB8, participe au maintien et à l’organisation des structures
à base d’actine qui dirigent la division cellulaire. Ainsi, les prochaines sections auront pour
but d’introduire les mécanismes moléculaires et les régulateurs clés qui assurent la
formation et l’intégrité des structures contractiles d’actine lors de la division cellulaire.
1.4. Contrôle de la division cellulaire par le remodelage des structures à base d’actine
La division cellulaire est le processus par lequel une cellule mère donne deux cellules filles.
Cet événement est gouverné par l’activation de la kinase CDK1 (« cyclin-dependent kinase
1 »), dépendante de son association à la cycline B, qui régule l’engagement de la cellule
dans la division cellulaire, assure la progression mitotique et la ségrégation correcte du
matériel génétique (Lindqvist, Rodríguez-Bravo, et Medema 2009). La mitose se divise en
trois étapes : la prophase, la prométaphase et la métaphase (Figure 1.8). Elle est suivie de
la cytokinèse qui se divise en deux étapes : l’anaphase et la télophase dont l’aboutissement
conduit à l’étape d’abscission c’est-à-dire la séparation physique des cellules filles (Figure
1.8). Deux processus clés qui gouvernent la mitose et la cytokinèse, respectivement,
dépendent d’un remodelage précis de structures à base d’actine : l’arrondissement
mitotique à l’entrée des cellules en mitose, et la formation de l’anneau contractile
d’actomyosine (AC) qui contrôle la cytokinèse. Ces processus, ainsi que les régulateurs
clés identifiés à ce jour, seront décrits dans les sections suivantes.

36
Figure 1.8: Schématisation des différentes étapes de la division cellulaire. La mitose se divise en cinq étapes. En prophase, l’ADN se condense, l’enveloppe nucléaire se rompt et les pôles du futur fuseau mitotique appelés centrosomes se séparent. En prométaphase, les chromosomes sont capturés au niveau des kinétochores par les microtubules. Une fois que ces derniers sont attachés aux deux centrosomes via les microtubules appelés fibres k, ils s’alignent à la plaque métaphasique en métaphase. À ce stade les deux centrosomes sont positionnés de part et d’autre de la cellule de façon symétrique grâce aux microtubules astraux et à la protéine motrice dynéine. Les chromatides sœurs sont séparées et tractées vers les centrosomes en anaphase A. En anaphase B, une séparation physique du matériel génétique des futures cellules filles est assurée par la formation du fuseau central fait de microtubules antiparallèles appelés microtubules intermédiaires enchevêtrés à la zone médiane. Durant cette phase, l’AC se forme et contracte le cortex formant ainsi le sillon de clivage. En télophase, l’anneau se contracte jusqu’à former une structure compacte bordée de microtubules denses formant le PI (pont intercellulaire) séparé en deux par une structure dense formée d’un échafaudage de protéines appelé « midbody ring ».

37
1.4.1. Les régulateurs de la dynamique de l’actine impliqués dans la division
cellulaire
In vitro, la polymérisation des filaments d’actine se déroule selon un processus nommé
« treadmilling » qui consiste en l’allongement et la dissociation du filament simultanés aux
extrémités (+) et (-) du filament respectivement (Lodish et al. 2000). La polymérisation de
l’actine pendant le « treadmilling » est extrêmement lente et ne peut pas expliquer à elle
seule la dynamique de structures à base d’actine qui dirigent les propriétés mécaniques de
la cellule et les changements morphologiques qui y sont associés. Il est donc évident que
la cellule a développé différentes stratégies pour pouvoir modeler le cytosquelette d’actine
face aux contraintes et aux stimuli environnementaux. Pour ce faire, la cellule dispose d’un
grand nombre de protéines particulières appelées ABP (« Actin Binding Proteins »)
(Pollard, Blanchoin, et Mullins 2000). Ces protéines interagissent directement avec l’actine
et peuvent d’une manière ou d’une autre modifier les propriétés dynamiques et/ou
structurales du cytosquelette d’actine. Plus de 60 familles de protéines ABP ont été
identifiées chez les eucaryotes comprenant des protéines de coiffe et des nucléateurs de
l’actine. Dans les sections suivantes nous décrirons des ABP qui sont impliquées dans la
progression de la division cellulaire.
1.4.1.1. Le nucléateur de l’actine banchée : le complexe Arp2/3
Le complexe Arp2/3 constitué de 7 sous-unités, dont les protéines Arp2 et Arp3, fût le
premier nucléateur de l’actine découvert par Machesky et al. 1994. Ce nucléateur permet
de créer un réseau branché d’actine requis dans une multitude de fonctions biologiques.
Notamment, ces réseaux branchés sont requis pour la formation du lamellipode au front de
migration cellulaire, la phagocytose, l’établissement de la polarité cellulaire, ou encore la
division asymétrique (Lai et al. 2008; Sun et al. 2011; Georgiou et al. 2008; Insall et al.
2001; May et al. 2000; Machesky et al. 1997; Suraneni et al. 2012; Hable et Kropf 2005;
Xiong, Mohler, et Soto 2011; Bailly et al. 2001). Le complexe Arp2/3 régule également la
motilité intracellulaire des endosomes, des lysosomes, des vésicules pinocytaires et des

38
mitochondries (Boldogh et al. 2001; Georgiou et al. 2008; Moore et al. 2016; Insall et al.
2001; Zhou, Sumigray, et Lechler 2015; Moreau et al. 1997; Duleh et Welch 2010).
Biochimiquement, il a été montré qu’Arp2/3 se lie sur le côté des filaments d’actine où il
initie la polymérisation d’un nouveau filament avec un angle d’environ 70° (Figure 1.9)
(Bailly et al. 2001; Mullins, Heuser, et Pollard 1998). In vivo, l’activation d’Arp2/3 est
dépendante de facteurs favorisant la nucléation (NPF : « Nucleation-Promoting Factor »)
(Symons et al. 1996; Rohatgi et al. 1999a; Ma, Rohatgi, et Kirschner 1998; Torres et Rosen
2006; Ten Klooster et al. 2006). Les NPF comprennent des membres de la famille des
protéines du syndrome de Wiskott-Aldrich comme les protéines WASP (« Wiskott–
Aldrich syndrome protein »), SCAR (« suppressor of cAMP receptor »), et WAVE
(« WASP-family verprolin-homologous protein »), lesquelles sont régulées par les petites
GTPases de la famille Rho en réponse à la signalisation cellulaire (section 1.4.1.4) (Welch
et Mullins 2002; Goode et Rodal 2001). L'ajout d’un NPF à Arp2/3 induit des
réarrangements structurels majeurs du complexe qui lui permettent de former une pseudo-
actine servant de base pour la nucléation de l’actine (Figure 1.9) (Volkmann et al. 2001;
Rouiller et al. 2008; Goley et al. 2004; Rodal et al. 2005).
Il est à noter que la cortactine, une protéine n’appartenant pas à la famille des NPF, peut
également intéragir et activer le complexe Arp2/3 (Weed et al. 2000; Uruno et al. 2001;
Weaver et al. 2001). En effet, la cortactine est une protéine adaptatrice qui possède un
domaine NTA (« N-terminal acidic domain ») commun aux NPF lui permettant d’intéragir
avec le complexe Arp2/3 (Wu et al. 1991; Weed et al. 2000). De plus, la cortactine lie
l’actine ce qui lui permettrait de contribuer à la stabilisation des filaments branchés formés
suite à l’activation du complexe Arp2/3 (Wu et Parsons 1993; Weaver et al. 2001; Weed
et al. 2000). Il est à noter que la cortactine possède également un domaine SH3 qui lui
permet de s’associer avec une multitude de partenaires l’impliquant de cette manière dans
un grand nombre de processus physiologiques comme la migration cellulaire et la mise en
place des jonctions cellulaires (Helwani et al. 2004; Kowalski et al. 2005; Zhang et al.
2009). Notamment, son association et sa phosphorylation par la kinase Src contribuent à
stimuler son association à Arp2/3 et son activation subséquente (Wu et al. 1991; Tehrani

39
et al. 2007). Un tel mécanisme a été associé à la migration et l’invasion tumorale (Mader
et al. 2011; Wu et al. 2016; Mezi et al. 2012; Oser et al. 2010). La cortactine est également
acétylée et déacétylée par différentes enzymes dont la déacétylase HDAC6 (Zhang et al.
2007). L’acétylation de la cortactine empêche son association à la F-actine et induit sa
localisation nucléaire ce qui réduierait la migration cellulaire (Zhang et al. 2007; Ito et al.
2015).
Figure 1.9: Schéma du mécanisme par lequel le complexe Arp2/3 induit la polymérisation d’un nouveau filament d’actine le long d’un filament préexistant. Le complexe Arp2/3 se lie sur le côté d’un filament d’actine dans un état inactif, la liaison du complexe à un NPF ou à la cortactine conduit à un changement de conformation du complexe lui permettant d’acquérir une structure proche d’un noyau d’actine à partir de laquelle un nouveau filament sera polymérisé le long du filament préexistant avec un angle de 70 °.
1.4.1.2. Les formines et la formation de faisceaux d’actine
Dans les cellules animales, les formines sont connues pour la conduite de l'assemblage de
filaments et de faisceaux d'actine linéaires dans les filopodes, les lamellipodes, ou encore
les fibres de stress (Watanabe et al. 2008; Satoh et Tominaga 2001; Yang et al. 2007;
Schirenbeck et al. 2005; Pellegrin et Mellor 2005; Block et al. 2008). Des études proposent
que la polymérisation des filaments d’actine conduite par les formines est à l’origine de la
production des forces requisent pour la déformation des membranes dans le filopode et le
lamellipode (Schirenbeck et al. 2005). En outre, les réseaux linéaires d’actine produits par
les formines servent d’échafaudage pour les protéines motrices de l’actine : les myosines

40
(section 1.4.1.3). Ces réseaux linéaires d’actine contenant des myosines génèrent ainsi de
fortes contraintes de traction à l’origine des forces motrices et de la contraction cellulaire
(Aratyn-Schaus, Oakes, et Gardel 2011; Case et Waterman 2015).
Au niveau structurel, les formines se caractérisent par la présence de domaines conservés
FH1 (« formin homology domain 1 ») et FH2 dans leur région C-terminale qui ont été
impliqués dans l’interaction des formines avec l’actine et les microtubules (Castrillon et
Wasserman 1994). Ces protéines se répartissent en sept sous-classes différentes basées sur
la divergence de la séquence du domaine FH2: Dia (« Diaphanous »), FRL (« Formin-
Related proteins in Leukocytes »), DAAM (« Dishevelled-Associated Activators of
Morphogenesis »), FHOD (« Formin-Homology Domain proteins »), FMN (Formine),
Delphilin et INF (« Inverted-Formin ») (Higgs et Peterson 2005).
Biochimiquement, les domaines FH2 se lient aux extrémités barbées des filaments
d’actines et agissent comme des coiffes processives sur les filaments en élongation. En
conséquence, les formines préviennent de l’arrêt de l’élongation induit par d’autres
protéines de coiffe (Harris, Li, et Higgs 2004; Romero et al. 2004). Le domaine FH1 des
formines permet d’accélérer la vitesse de polymérisation de la F-actine en aidant à la
livraison rapide des formines à l'extrémité barbée (+) de la F-actine (Paul et al. 2008; Paul
et Pollard 2009). Grâce à cette interaction, les formines peuvent ainsi accélérer de 19 fois
l’ajout de monomères d’actine à l’extrémité barbée coiffée (Kovar et al. 2006; Romero et
al. 2004; Vidali et al. 2009).
1.4.1.3. La myosine II et la contractilité cellulaire
Les myosines sont les protéines motrices de l’actine qui utilisent de l'énergie dérivée de
l’hydrolyse de l'ATP pour générer de la force et des mouvements le long des filaments
d'actine. Chez l’humain, 38 gènes codant pour des myosines séparées en douze classes
(classes I à XII) ont été identifiés (Masters, Kendrick-Jones, et Buss 2017). La myosine II
(MyoII) présente un intérêt particulier pour nos travaux puisqu’elle est fortement impliquée
dans les mécanismes de régulation du cytosquelette d’actine lors de la division cellulaire.

41
De plus, elle gouverne la contractilité des réseaux d’actine et la tension cellulaire dans les
cellules non musculaires. Ainsi, la MyoII est essentielle pour la motilité et l’adhésion
cellulaires, la morphogénèse tissulaire ainsi que l’invasion tumorale et la progression
métastatique (Franke, Montague, et Kiehart 2005; Bertet, Sulak, et Lecuit 2004; Franke,
Montague, et Kiehart 2005; Beningo et al. 2001; Walker et al. 2010; Even-Ram et al. 2007;
Vicente-Manzanares et al. 2009; Betapudi, Licate, et Egelhoff 2006).
Toutes les myosines sont composées d'une ou deux chaînes lourdes qui peuvent dimériser
et de plusieurs chaînes légères qui régulent l’activité ATPase des myosines. La MyoII est
constituée de deux chaînes lourdes associées à deux chaînes légères régulatrices (Figure
1.10). Comme toutes les chaînes lourdes des myosines, celles de la MyoII contiennent trois
domaines structurellement et fonctionnellement différents (Figure 1.10). Le domaine de la
tête globulaire contient des sites de liaison à l’actine et à l’ATP et est responsable de la
génération de forces (Mornet et al. 1979; Cope et al. 1996). À côté du domaine de la tête
se trouve la région du col α-hélicoïdal qui est associée aux chaînes légères régulatrices
(Figure 1.10). Le domaine de la queue contient une hélice a super-enroulée importante
pour la dimérisation de la MyoII. Il est à noter que dans les cellules les dimères de MyoII
se rassemblent dans des filaments bipolaires de taille variable (de 14 à 300 myosines) qui
vont contribuer à l’organisation et à la contraction de structures à base d’actine.
Figure 1.10 : Schéma structurel d’un dimère de myosine II (adapté de Walck-Shannon et Hardin 2014). La myosine peut être divisée en trois parties : (1) la tête qui contient le domaine moteur et le domaine de liaison à l’actine, (2) le col constitué des chaînes légères qui contribuent à

42
la stabilité de la protéine et à la régulation de son activité, (3) et la queue principalement constituée d’hélices a super-enroulées impliquées dans la dimérisation de la myosine.
Le mouvement assuré par la MyoII se fait suite à un cycle d’hydrolyse de l’ATP par les
têtes du dimère qui va contrôler son association à l’actine (Figure 1.11) (Whittaker et al.
1995; Jontes, Wilson-Kubalek, et Milligan 1995; Uyeda, Abramson, et Spudich 1996). Ce
modèle de mouvement de la myosine est appelé « swinging lever-arm » et permet à la
myosine de faire glisser le filament d’actine et ainsi de créer un mouvement de ce dernier
conduisant à la contraction du cytosquelette d’actine (Figure 1.11) (Holmes 1997).
Figure 1.11: Schéma du mouvement assuré par la myosine le long du filament d’actine. Initialement, la protéine motrice est dans l'état de rigueur c’est-à-dire liée à l'actine mais pas au nucléotide ATP. La liaison à ce dernier provoque le détachement du domaine moteur de l'actine et son hydrolyse bloque le domaine moteur qui revient à l'actine. La libération du phosphate inorganique déclenche l'exécution du mouvement du bras levier de la myosine. Finalement l’ADP est relâché.
Il est intéressant de noter qu’en plus d’assurer la motricité de la F-actine, la MyoII serait
aussi impliquée dans le contrôle de son renouvellement par un mécanisme dépendant de sa
concentration (Wilson et al. 2010; Haviv et al. 2008). En effet, un examen attentif de la
dynamique de certains processus cellulaires a révélé une relation intime entre l'activité
ATPase de la MyoII et le taux de renouvellement de l'actine (Pelham et Chang 2002;
Medeiros, Burnette, et Forscher 2006; Guha, Zhou, et Wang 2005; Murthy et Wadsworth
2005; Zumdieck et al. 2007; Miklavc et al. 2015). Notamment, la MyoII joue un rôle dans
le renouvellement des filaments d’actine dans les neurones en croissance et dans le
recyclage de ceux-ci pendant la cytokinèse (voir section 1.4.3.2).

43
L’activité motrice, l’activation et l’état d’assemblage de la MyoII sont déterminés par la
phosphorylation de ses chaînes légères. En effet, chez les mammifères, la phosphorylation
des chaînes légères régulatrices de MyoII stimule son activité enzymatique et déclenche
son assemblage en des filaments d'ordre supérieur (Matsumura 2005). Les résidus
hautement conservés sérine 19 (S19) et thréonine 18 (T18) des chaînes légères constituent
respectivement les sites de phosphorylation primaire et secondaire responsables de la
régulation des myosines. In vitro, il a été montré que la mono-phosphorylation de S19
améliore l'activité ATPase de la myosine, son activité motrice ainsi que l'assemblage des
filaments (Somlyo et Somlyo 2003; Kim et al. 2005; Scholey, Taylor, et Kendrick-Jones
1980; Sellers, Pato, et Adelstein 1981). La di-phosphorylation simultanée de T18 et S19
améliore d’avantage l'activité ATPase et l'assemblage des filaments (Umemoto, Bengur, et
Sellers 1989; Ikebe, Koretz, et Hartshorne 1988). Il est à noter que les kinases
phosphorylant les chaînes légères de la MyoII peuvent être activées par deux voies de
signalisation : la voie de signalisation dépendante de la calmoduline/Ca2+ ou la voie de
signalisation des GTPases RhoA ou Cdc42 (section 1.4.1.4). Ces kinases comprennent les
enzymes MLCK (« Myosin Light Chain Kinases »), la kinase ROCK (« RHO-associated,
Coiled-coil Containing Protein Kinase ») ou encore la MRCK (« Myotonic dystrophy
kinase-Related Cdc42-binding Kinase ») (Ikebe et Hartshorne 1985; Amano et al. 1996;
Leung et al. 1998).
1.4.1.4. Les Rho GTPases
La famille des Rho GTPases est une famille conservée de 20 petites GTPases qui agissent
comme des interrupteurs moléculaires régulant de nombreux processus cellulaires
essentiels, notamment ceux qui dépendent de la dynamique de l'actine. La plupart des Rho
GTPases se situent entre un état actif, reliées au GTP et un état inactif, liées au GDP. Dans
leur conformation active (liée au GTP), les Rho GTPases s’associent avec des membranes
cellulaires et peuvent intéragir avec et activer des protéines effectrices spécifiques, ce qui
entraîne des effets localisés sur le cytosquelette d’actine.

44
Les Rho GTPases sont activées par des facteurs d'échange de nucléotides (GEF : « guanine
nucleotide exchange factor ») et sont inactivées par des protéines stimulant leur activité
GTPase (GAP : « GTPase activating proteins ») (Hodge et Ridley 2016). De plus, les
inhibiteurs de dissociation de nucléotides (GDI : « guanine nucleotide dissociation
inhibitor ») contribuent à l'inactivation des Rho GTPases en les extrayant de la membrane
plasmique, en liant les GTPases inactives et en empêchant leur réactivation et leur
dégradation. Par conséquent, l'emplacement et l'étendue de l'activité des Rho GTPases et
leur signalisation dépendent fortement de la localisation et de l'activité des GEF, GAP et
GDI, ainsi que de la disponibilité de leurs effecteurs.
La famille des Rho GTPases chez les mammifères comprend huit membres dont RhoA
(« Ras homolog gene family, member A »), Cdc42 (« cell division cycle 42 ») et Rac1
(« Ras-related C3 botulinum toxin substrate 1 ») qui sont les plus étudiés (Jansen et al.
2017). Il est à noter que les Rho GTPases présentent une myriade de protéines effectrices
qui vont assurer leur fonction de régulation de la dynamique du cytosquelette d’actine. Par
exemple, RhoA est connue pour activer les myosines via l’activation de la kinase ROCK
ce qui lui permet de médier la contractilité de l'actomyosine par la phosphorylation de la
chaîne légère de la myosine (Amano et al. 1996; Kimura et al. 1996; Kureishi et al. 1997).
RhoA active également la polymérisation de l’actine via l’activation des formines
(Watanabe et al. 1997). La plupart des effecteurs de Cdc42 et Rac1 arborent un domaine
typique CRIB (« Cdc42-Rac interactive binding ») qui permet leur ciblage spécifique par
ces GTPases et leur activation subséquente (Burbelo, Drechsel, et Hall 1995). Ce motif a
été retrouvé dans de nombreuses protéines interagissant avec ces GTPases comme les
kinases régulatrices du cytosquelette d’actine PAK (« p21-activated kinase »), PI3K
(« phosphatidylinositol-4,5-bisphosphate 3-kinase ») et MRCK, ou encore les protéines
d’organisation de ce cytosquelette telles que IQGAP1 (« IQ Motif Containing GTPase
Activating Protein 1 »), les formines, WAVE et WASP (Cotteret et Chernoff 2002;
Bagrodia, Laudano, et Shalloway 1994; Vidal et al. 2002; Bashour et al. 1997; Rohatgi et
al. 1999b; Higgs et Pollard 2000; Eden et al. 2002).

45
Grâce à ces nombreuses protéines effectrices, il est admis que RhoA régule l’assemblage
des fibres de stress, Rac1 contrôle la polymérisation de l’actine à la périphérie de la cellule
pour produire les lamellipodes, alors que Cdc42 stimule la formation des filopodes (Ridley
et al. 1992; Ridley et Hall 1992; Kozma et al. 1995; Nobes et Hall 1995; Puls et al. 1999).
De plus, ces trois GTPases sont impliquées dans la formation des adhésions focales qui
sont intimement liées au cytosquelette d’actine (Nobes et Hall 1995; Hotchin et Hall 1995).
En outre, RhoA et Cdc42 présentent également des fonctions importantes durant la division
cellulaire (description dans les sections 1.4.2.2 et 1.4.3.1).
1.4.2. L’arrondisssement mitotique et son implication dans l’orientation du fuseau
mitotique et la ségrégation des chromosomes
Il a longtemps été pensé que la séparation adéquate du matériel génétique lors de la division
cellulaire reposait uniquement sur le contrôle du cytosquelette de microtubules. En effet,
ce processus implique la capture des chromosomes au niveau des kinétochores par des
kinésines situées sur les fibres k (Figure 1.12) (West, Malmstrom, et McIntosh 2002;
Wargacki et al. 2010; Mayr et al. 2007; Tolić 2017). La capture des chromosomes dépend
également du positionnement adéquat du fuseau mitotique dépendant des microtubules
astraux et de la protéine motrice dynéine (Figure 1.12) (Kiyomitsu et Cheeseman 2012,
2012; Laan et al. 2012; Kotak, Busso, et Gönczy 2012; Lee et al. 2000). Cependant, des
études récentes ont fourni des informations importantes sur la façon dont les
réarrangements du cytosquelette d’actine en mitose sont impliqués dans la fidélité de la
ségrégation chromosomique.

46
Figure 1.12 : Représentation schématique des fonctions assurées par les microtubules et leurs protéines motrices dans le positionnement du fuseau mitotique et la ségrégation des chromosomes. La dynéine grâce à sa capacité de motricité vers l’extrémité (-) des microtubules produit des forces qui tirent les microtubules astraux vers le cortex cellulaire et contribue de cette manière à tirer sur les centrosomes du fuseau mitotique pour le positionner de façon adéquate. Les kinésines localisées sur les fibres k du fuseau mitotique interagissent spécifiquement avec les kinétochores (cercles rouges) des chromosomes leur permettant ainsi la capture des chromosomes et leur transport vers le centrosome. Les flèches indiquent la direction des protéines motrices.
Notamment, l'arrondissement mitotique des cellules, dépendant du cortex d’actomyosine,
contribue à créer une géométrie optimale pour l'assemblage et le positionnement du fuseau
mitotique, facilitant la capture et l’alignement des chromosomes par les microtubules
(Kiyomitsu et Cheeseman 2012; Cadart et al. 2014; Fink et al. 2011; Nestor-Bergmann,
Goddard, et Woolner 2014; Minc, Burgess, et Chang 2011; Bird, Heald, et Weis 2013;
Théry et al. 2005; Magidson et al. 2011; Lancaster et Baum 2014; Magidson et al. 2011;
Cattin et al. 2015; Rosa et al. 2015). Ce processus important est observé tant dans les
cellules en culture qu’à l’intérieur d’un tissu où il a été associé à la morphogénèse des
vilosités intestinales, la régulation de la croissance et de la forme des cavités épithéliales
ainsi que l’invagination épithéliale (Freddo et al. 2016; Kondo et Hayashi 2013; Hoijman
et al. 2015).

47
Aussi, en conditions de culture cellulaire, le positionnement du fuseau mitotique dépend
du désassemblage des adhésions focales et de la mise en place de structures adhésives
d’actine appelées fibres de rétraction, qui permettent de faire un lien entre le substrat (i.e.
la matrice extracellulaire) et le cortex de la cellule. Le positionnement du fuseau mitotique
est aussi contrôlé par une structure hautement dynamique nommée nuage sous-cortical
d’actine, influencée par les fibres de rétraction (Mitsushima et al. 2010; Fink et al. 2011;
Théry et al. 2005).
Ainsi, interférer avec la formation de l’une de ces trois structures à base d’actine altère le
positionnement du fuseau mitotique et affecte la capture, l’alignement et la ségrégation des
chromosomes conduisant à une instabilité génétique. De ce fait, le positionnement du
fuseau mitotique doit être finement régulé et définit le contenu, la position et le devenir des
cellules filles dans les tissus (Siller et Doe 2009). Sa dérégulation a été corrélée avec
différentes pathologies, y compris la microcéphalie et le cancer (Noatynska, Gotta, et
Meraldi 2012). Il est donc important de comprendre les mécanismes moléculaires associés
au positionnement du fuseau mitotique.
Bien que l’arrondissement mitotique, la mise en place des fibres de rétraction et le nuage
sous-cortical d’actine aient été identifiés comme des processus impliqués dans le
positionnement du fuseau mitotique, les mécanismes moléculaires à l’origine de leur
formation et de leur maintien sont encore peu compris. Notamment, les voies de
signalisation spécifiques qui contrôlent la dynamique de l’actine dans de telles structures
sont peu étudiées. Comment est régulée la communication entre le cytosquelette d’actine
et les microtubules reste à être déterminé. Comment la dynamique de l’actine est régulée
dans le temps et dans l’espace au sein de ces structures demeure peu compris. Dans les
sections suivantes nous présenterons les processus moléculaires qui contrôlent
l’arrondissement mitotique et discuterons des questions encore non résolues.

48
1.4.2.1. Démantèlement des adhésions focales et mise en place des fibres de
rétraction
Le désassemblage des adhésions focales est la première étape conduisant à
l’arrondissement mitotique et au positionnement du fuseau. Interférer avec ce processus
altère l’arrondissement mitotique, la morphologie du fuseau mitotique, la ségrégation
chromosomique et la progression mitotique (Lancaster et al. 2013). Un joueur important
dans le démontage mitotique des adhésions focales est la petite GTPase Rap1 (Dao et al.
2009). En effet, l'inactivation de Rap1 par un mécanisme encore inconnu est nécessaire
pour le désassemblage des adhésions focales puisqu’une forme constitutivement active de
Rap1 inhibe un tel désassemblage et force les cellules en culture à maintenir une
morphologie plus plate tout au long de la mitose (Dao et al. 2009; Lancaster et al. 2013).
Il est intéressant de souligner ici que Rap1 a été identifiée comme étant un partenaire de
BAG3, impliquée dans ses effets sur l’adhérence cellulaire (Iwasaki et al. 2010).
De plus, le désassemblage des adhésions focales requiert également la protéine DEPDC1B
(« DEP domain containing 1B ») (Marchesi et al. 2014). En effet, DEPDC1B s'accumule
spécifiquement à la phase G2 du cycle cellulaire et inhibe le recrutement et l'activation de
RhoA par la protéine associée aux adhésions focales PTPRF (« receptor protein tyrosine
phosphatase, receptor type, F »). Par ce mécanisme, DEPDC1B fonctionne comme un
inhibiteur de la voie de signalisation RhoA/ROCK/MLC2 (« Myosin Light Chain 2 »)
pendant la transition G2/M, ce qui permet le démantèlement des adhésions focales et le
détachement des cellules.
Bien que le désassemblage des structures d’adhésion soit un prérequis à l’arrondissement
mitotique, les cellules mitotiques en culture doivent garder un contact avec leur substrat.
Ce contact est dépendant de la mise en place de structures d’actine hautement dynamiques
qui permettraient de faire un pont entre le substrat et le cortex mitotique : les fibres de
rétraction. En effet, par le contrôle de la répartition spatiale de la matrice extracellulaire
sur le substrat et donc du positionnement des fibres de rétractions, l’équipe de Bornens M.
démontra que cette répartition joue un rôle dans l'orientation du fuseau mitotique des

49
cellules HeLa (Théry et al. 2005). Les résultats de l’équipe indiquèrent que la matrice
extracellulaire contrôle l'emplacement de la dynamique de l'actine à la membrane en dictant
la répartition hétérogène de composants corticaux impliqués dans cette dynamique comme
l’ezrine (voir section 1.4.2.4). Les auteurs conclurent que les fibres de rétractions
permettent de lier le substrat au cortex d’actomyosine en mitose et assurent la
« communication » entre ces deux structures.
Ceci fût confirmé par des expériences d'ablation au laser qui montrèrent que la répartition
polarisée des fibres de rétraction pendant la mitose constitue une mémoire du modèle
d'adhérence en interphase et influence l'orientation du fuseau mitotique (Fink et al. 2011).
Le mécanisme moléculaire par lequel les fibres de rétraction contrôlent le positionnement
du fuseau mitotique implique la régulation spatiotemporelle de la dynamique du nuage
sous-cortical d’actine. Ceci sera décrit dans le pargaraphe 1.4.2.3. Il est à noter qu’à l’heure
actuelle, la composition des fibres de rétraction est inconnue. Seules quelques protéines
ont été localisées par immunofluorescence dans de telles structures notamment la F-actine,
l’ezrine et la cortactine (Théry et al. 2005).
Ainsi, de nos jours, bien que leur importance ait été établie, les mécanismes moléculaires
contrôlant le démantèlement des adhésions focales et la mise en place et le contrôle de la
dynamique des fibres de rétraction restent encore à être élucidés.
1.4.2.2. Fonctions essentielles de RhoA et de la myosine II dans la mise en place et
le maintien de la tension corticale à l’orgine de l’arrondissement mitotique
En parallèle avec le démantèlement des structures d’adhérence, les structures d'actine
interphasiques, telles que les fibres de stress, se désassemblent et se réorganisent en un
cortex riche en actomyosine sous la membrane plasmique (Maddox et Burridge 2003). Ce
réseau cortical d'actomyosine contribue à l'augmentation de la rigidité corticale requise
pour l'arrondissement mitotique contre son environnement que ce soit en culture ou dans
un tissu (Sorce et al. 2015). En outre, le cortex mitotique fournit des sites de fixation pour
les microtubules astraux, aidant ainsi à positionner et à séparer les centrosomes du fuseau

50
mitotique (Rosenblatt et al. 2004; Busson et al. 1998; O’Connell et Wang 2000). En
conséquence, le cortex mitotique contribue à la formation d’une géométrie cellulaire
favorable au positionnement du fuseau mitotique et à la ségrégation adéquate des
chromosomes (Cadart et al. 2014; Minc, Burgess, et Chang 2011; Lancaster et al. 2013).
RhoA, à travers la régulation de la MyoII, a un rôle majeur à jouer dans la formation et le
maintien de la rigidité du cortex d'actomyosine en mitose (Figure 1.13). En conséquence,
dès l’entrée des cellules en mitose, l’activité de RhoA doit être inhibée au niveau des
adhésions focales, mais stimulée au niveau du cortex cellulaire (Matthews et al. 2012;
Maddox et Burridge 2003). L’activation de RhoA à l’entrée en mitose dépend d’une part
de l'inactivation de son inhibiteur p190RhoGAP, et d’autre part de l’activation de la GEF
ECT2 (« epithelial cell transforming 2 ») (Tatsumoto et al. 1999; Niiya et al. 2006; Maddox
et Burridge 2003). Une étude réalisée chez la mouche indique que l’activation de la GEF
ECT2 conduit à l’activation de RhoA et de Cdc42 (Figure 1.13) (Rosa et al. 2015). Dans
ce contexte, RhoA est essentielle à l’activation de MyoII, tandis que Cdc42, en
collaboration avec les protéines de polarité PAR6 (« partitioning-defective protein 6 ») et
aPKC (« atypical protein kinase C »), contrôle la localisation et l’activation corticale de
mDia1 (Figure 1.13). Ainsi, RhoA agit sur l'arrondissement mitotique et la rigidité corticale
par l'activation de la kinase ROCK et la phosphorylation subséquente des chaînes légères
de la MyoII. L’activation locale de mDia1 par Cdc42 quant à elle permet d’induire la
polymérisation de l’actine et de former un réseau d’actine favorable au recrutement de
MyoII au cortex (Ramanathan et al. 2015).
Il est à noter que dans cette même étude, les auteurs ont également démontré, par imagerie
en temps réel, qu’il existe un changement d’organisation de l’actine corticale à l’entrée des
cellules en mitose qui devient strictement dépendant de mDia1 et non plus d’Arp2/3. En
conséquence, il semblerait que le complexe Arp2/3 ne soit pas nécessaire à la formation du
cortex mitotique et au maintien de la rigidité corticale en mitose (Bovellan et al. 2014). En
revanche, Arp2/3 a récemment été impliqué dans le positionnement du fuseau mitotique en
contrôlant la dynamique du nuage sous-cortical d’actine (section 1.4.2.3).

51
Figure 1.13 : Schéma représentatif de la voie de signalisation impliquée dans la formation du cortex mitotique et le maintien de sa rigidité.
En résumé, l’activation de la MyoII par RhoA, de concert avec une architecture de l’actine
favorable dépendante de mDia1 et de Cdc42, contribuent à l’arrondissement mitotique et
au maintien d’une tension optimale du cortex mitotique requise pour le positionnement
adéquat du fuseau mitotique. Ainsi, un mécanisme moléculaire à l’origine de la formation
du cortex mitotique a émergé des récentes études. Cependant, les données obtenues chez
la mouche, quant à l’activation et la localisation de mDia1 dépendante de Cdc42, n’ont pas
encore été corrélées chez les mammifères. L’activité de Cdc42 est-elle limitée à l’activation
de mDia1 ou d’autres cibles de la GTPase sont-elles requises pour la formation du cortex
mitotique, est une question non répondue également. De plus, le mécanisme moléculaire à
l’origine de l’inactivation de p190RhoGAP n’a pas encore été élucidé. Une question
critique qui demeure concerne les mécanismes par lesquels la régulation spatiotemporelle

52
de différents types de structures d’actine, notamment via la ségrégation des activités Rho
GTPases, est assurée.
1.4.2.3. Contrôle du positionnement du fuseau mitotique par le nuage sous-cortical
d’actine et l’implication du complexe Arp2/3 dans ce processus
Comme mentionné précédemment, le complexe Arp2/3 ne serait pas essentiel à la
formation du cortex mitotique. En revanche, l’activité d’Arp2/3 est nécessaire pour réguler
la dynamique du nuage sous-cortical d’actine impliqué dans le positionnement du fuseau
mitotique (Mitsushima et al. 2010).
En effet, des travaux ont montré que ce nuage d'actine est hautement dynamique durant la
métaphase. Le renouvellement rapide de l'actine dans les filaments se déroulerait partout
dans le nuage et serait également nécessaire pour sa rotation pendant la mitose (Figure
1.14). La rotation du nuage est en réalité liée à une dynamique de
polymérisation/dépolymérisation locale qui serait dépendante du nucléateur puisque la
déplétion d’Arp3 abolie la formation du nuage sous-cortical. La fonction mitotique de ce
nuage d'actine est de contribuer à l'orientation du fuseau mitotique. Effectivement, des
analyses dynamiques suggèrent que le nuage régule la rotation du fuseau mitotique d'une
manière dépendante des microtubules astraux (Fink et al. 2011). En outre, par des
expériences d'ablation au laser, il a été montré que la répartition des fibres de rétraction
influence la distribution et les mouvements polarisés de ces nuages d'actine.

53
Figure 1.14 : Schéma modèle du mouvement du nuage sous-cortical d’actine durant la mitose. La zone (rose) dans la partie inférieure de la cellule indique la zone dans laquelle la polymérisation de l'actine et la dépolymérisation se produisent activement. Rose, filaments d'actine; Flèches jaunes, la direction du mouvement de rotation. Inspiré de Mitsushima et al. 2010.
La communication du nuage sous-cortical d’actine avec les microtubules astraux passerait
par la myosine non conventionnelle myosine X (Myo10) (Figure 1.15). En effet,
récemment, Kwon et coll ont noté que la liaison directe de Myo10 aux microtubules est
essentielle pour son rôle dans le positionnement des centrosomes (Kwon et al. 2015). Leurs
analyses quantitatives démontrèrent que Myo10 régule la dynamique des microtubules
astraux et est requise pour les interactions des microtubules avec le cortex. Ainsi, ces
résultats suggérèrent que Myo10 couple les forces dépendantes des fibres de rétraction et
les nuages sous-corticaux d'actine aux centrosomes, afin de permettre le positionnement
adéquat du fuseau mitotique (Figure 1.15).

54
Figure 1.15 : Modèle selon lequel la Myo10 assure la communication des fibres de rétraction, du cortex et du nuage sous-cortical d’actine avec le centrosome.
L’ensemble de ces résultats met en évidence un rôle majeur de la dynamique de l’actine
dans le contrôle du positionnement du fuseau mitotique, négligé pendant plusieurs années.
Il semble de plus en plus clair qu’une régulation spatiotemporelle fine des différents
intervenants du remodelage de l’actine est cruciale pour la progression normale de la
mitose. Cependant, les mécanismes moléculaires qui les dirigent demeurent peu compris.
La découverte du nuage sous-cortical d’actine soulève plusieurs questions laissées sans
réponses à ce jour. Par quelle voie de signalisation la cellule mitotique régule-t-elle
l’activation d’Arp2/3 dans le nuage sous-cortical? Comment la dynamique de
polymérisation/dépolymérisation locale est-elle contrôlée? La Myo10 est-elle la seule
protéine couplant les forces dépendantes des fibres de rétraction et les nuages sous-
corticaux d'actine aux centrosomes?
1.4.2.4. Promotion de la rigidité corticale et du positionnement du fuseau mitotique
par les protéines ERM
Comme nous l’avons décrit précédemment, il est connu que la MyoII est responsable du
maintien de la rigidité corticale dans les cellules en mitose grâce à son recrutement

55
dépendant de mDia1 et de RhoA. De récentes études suggérèrent que les protéines ERM
(Ezrine-Radaxine-Moésine) favorisent la rigidité corticale en se liant aux filaments
corticaux d'actine (Kunda et al. 2008; Carreno et al. 2008).
Les protéines ERM sont une famille d'agents de réticulation de l’actine et de la membrane
plasmique qui contrôlent la rigidité et la stabilité du cortex cellulaire (Fehon, McClatchey,
et Bretscher 2010). En mitose, il a été noté que la réduction des niveaux d’expression de
moésine, le seul membre de la famille présent chez la drosophile, conduit à une instabilité
corticale massive et à un bourgeonnement des cellules. Il en résulte des oscillations
exagérées du fuseau mitotique et un mauvais positionnement de ce dernier (Kunda et al.
2008; Carreno et al. 2008). Ceci fût confirmé dans les cellules de mammifères où les
membres de la famille ERM sont cruciaux pour organiser le cortex mitotique (Carreno et
al. 2008; Kunda et al. 2008; Machicoane et al. 2014). En effet, lors de l'entrée en mitose,
les ERM sont activées par la kinase SLK (« sterile 20-like kinase ») et aident alors à
l'interaction entre la membrane plasmique et le réseau d'actomyosine (Machicoane et al.
2014). Ainsi, grâce à leur capacité de réticulation, les protéines ERM assurent la répartition
de la tension corticale, empêchant ainsi la contraction locale du cortex et en conséquence
la déformation cellulaire.
De plus, les protéines ERM ont récemment été étudiées pour leur rôle dans l'orientation du
fuseau mitotique dans les cellules de vertébrés. Lorsque des cellules humaines sont
cultivées sur des matrices micro-fabriquées en forme de L, les protéines ERM sont réparties
asymétriquement, avec un enrichissement dans le domaine cortical face aux fibres de
rétraction (Théry et al. 2005; Machicoane et al. 2014). La réduction des niveaux
d’expression des trois protéines ERM ainsi que l'altération de leur activation par déplétion
de la kinase SLK entraînent une désorientation du fuseau mitotique dans l'espace
(Machicoane et al. 2014). Ce phénotype a été associé à la perte de la localisation corticale
de LGN (leucine–glycine–asparagine) et de NuMA (« nuclear and mitotic apparatus »), qui
ont des fonctions centrales dans l'orientation et le positionnement du fuseau mitotique
(Morin et Bellaïche 2011). Ces observations suggèrent que les protéines ERM activées sont
nécessaires pour le recrutement cortical du complexe LGN ou sa stabilité dans ce contexte.

56
Il est important de noter que, contrairement aux effets observés lors de la déplétion de la
moésine dans la mouche, la réduction des niveaux protéiques des protéines ERM ne génère
pas d'altération évidente de la forme cellulaire et de la morphologie du fuseau mitotique
dans les cellules humaines, argumentant dans le sens d'un rôle spécifique de ces protéines
dans l’orientation du fuseau par le contrôle de la localisation du complexe LGN. Par
ailleurs, des évidences montrent que les protéines ERM peuvent aussi contrôler le
positionnement du fuseau mitotique en stabilisant l'interaction des microtubules astraux
avec le cortex (Kunda et al. 2008; Solinet et al. 2013).
En résumé, un rôle des protéines ERM dans le maintien de la rigidité corticale et le
positionnement du fuseau en mitose émerge depuis les dernières années. Cependant, de
telles fonctions sont encore peu comprises. Le complexe est-il vraiment requis pour le
positionnement du fuseau mitotique ? Est-ce leur capacité de réticulation qui leur permet
de maintenir la tension corticale en mitose ? Comment la localisation des ERM en mitose
est-elle contrôlée ? Sont-elles impliquées dans la « communication » entre les fibres de
rétraction et le nuage sous-cortical d’actine ? Telles sont les questions qui restent à être
élucidées.
1.4.3. Les mécanismes de contrôle de l’anneau contractile en cytokinèse
Une fois que le fuseau mitotique est positionné et les chromosomes capturés correctement,
la cycline B est progressivement dégradée permettant ainsi l’engagement de la cellule dans
la cytokinèse (Wheatley et al. 1997; Sigrist et al. 1995). Cette étape requiert la séparation
du matériel génétique assurée par les microtubules du fuseau mitotique ainsi que la
formation des futures cellules filles qui dépend du contrôle accru de la dynamique du
cytosquelette d’actine spatialement et temporellement. En effet, ce processus se caractérise
par la formation et la contraction de l’AC, au centre de la cellule en division (Figure 1.8).
La division cellulaire facilitée par un anneau contractile d’actomyosine est une
caractéristique presque universelle dans toutes les niches évolutives.

57
Le processus de cytokinèse peut être divisé en quatre étapes distinctes. Tout d'abord, la
cellule doit spécifier un emplacement pour placer l’AC afin d’assurer une séparation
adéquate du contenu de la cellule en deux cellules filles. Deuxièmement, la cellule doit
pouvoir transporter toutes les composantes nécessaires dans cette région et construire un
AC de manière fiable et efficace. Troisièmement, l’AC doit générer un stress contractile
de manière régulée, pour cliver physiquement la cellule mère en deux cellules filles. Enfin,
l'anneau doit être désassemblé pour permettre le recrutement de la machinerie de
l'abscission et en conséquence permettre la séparation physique des cellules filles.
Ainsi, l’échec de l’une de ces quatre étapes conduit immanquablement à l’avortement de
l’abscission, à une forte instabilité génétique et à une augmentation de l’aneuploïdie
(Sagona et Stenmark 2010; Urzúa et al. 2016; McKenzie et D’Avino 2016; Lv et al. 2012).
Les principaux régulateurs et les questions actuelles concernant les 4 étapes de la
cytokinèses seront décrits ci-après.
1.4.3.1. Spécification et formation de l’AC : un rôle crucial pour RhoA et mDia2
Dans les cellules de métazoaires, la sélection du site de division se produit au cours de
l'anaphase et dépend du ciblage de la forme active de RhoA à la membrane équatoriale
(Bement, Benink, et von Dassow 2005). En effet, l’inhibition de RhoA ou l’expression
d’un dominant négatif de la GTPase inhibe la formation du sillon de clivage en cytokinèse
dans de multiples systèmes comme la drosophile, l’embryon de xénoppe et les cellules de
mammifères (Kishi et al. 1993; Prokopenko et al. 1999; Drechsel et al. 1997; Crawford et
al. 1998; Moore et al. 1994; Zhong et al. 2005). Des études d’immuno-localisation ont
montré que la protéine RhoA s'accumule au niveau du sillon de clivage avant même que la
cellule ne commence à se contracter (Takaishi et al. 1995; Madaule et al. 1998; Nishimura
et Yonemura 2006; Yüce, Piekny, et Glotzer 2005; Kamijo et al. 2006). Cette localisation
spatiale et temporelle de la GTPase suggérait alors que la protéine devait spécifier le plan
de division de la cellule.

58
Deux mécanismes moléculaires ont été associés à la spécification de ce plan par le contrôle
du ciblage de RhoA: les microtubules astraux et le complexe « centralspindlin ». En effet,
des études ont montré qu'un sous-ensemble de microtubules astraux stables fournit des
composantes du sillon de clivage au cortex pour spécifier la position de la GTPase
(Canman et al. 2003; Foe et von Dassow 2008; Odell et Foe 2008). À l’inverse, les
microtubules astraux dynamiques permettraient d’exclure des pôles cellulaires la MyoII et
RhoA (Rankin et Wordeman 2010; Zanin et al. 2013). De cette façon, il est proposé que
les microtubules astraux permettent de recruter au sillon de clivage des facteurs clés dans
l’assemblage et la constriction de l’AC, tels que la MyoII et RhoA.
Le complexe « centralspindlin » est également l’un de ces facteurs clés qui contribue
majoritairement à l’activation locale de la GTPase RhoA au sillon de clivage. Il s’agit d’un
hétéro-tétramère formé de la kinésine MKLP1 (« mitotic kinesin-like protein 1 ») et de la
protéine CYK-4 (ou MgcRacGAP). La découverte d'une interaction directe entre la sous-
unité CYK4 et ECT2 a immédiatement conduit à une voie linéaire pour l'induction de la
formation de l’AC: « centralspindlin » → recrutement d’ECT2 → activation de RhoA →
induction de l’AC (Figure 1.16) (Yüce, Piekny, et Glotzer 2005; Nishimura et Yonemura
2006; Kamijo et al. 2006; Dean et al. 2005; Zhao et Fang 2005).
L’activation de RhoA se traduit alors par l’activation de la kinase ROCK et de la formine
mDia2 au sillon de clivage (Figure 1.16) (Castrillon et Wasserman 1994; Watanabe et al.
2008)). En effet, des travaux ont montré que la déplétion de mDia2, qui se localise au sillon
de clivage en anaphase et télophase, conduit à une augmentation de la quantité de cellules
binuclées caractérisant une abscission inefficace et provoque une contraction dans des sites
aberrants (Watanabe et al. 2008). Dans cette même étude, les auteurs ont noté que le
traitement avec la blebbistatine, un inhibiteur de la MyoII, en plus de supprimer cette
contraction anormale, corrige également la localisation de la GTPase RhoA et de la MyoII.
Cette étude révéla également que la quantité de F-actine dans la région équatoriale pendant
l'anaphase/télophase est significativement diminuée suite à la déplétion de mDia2. Ces
résultats démontrèrent ainsi que la formine est essentielle pour la cytokinèse des cellules
de mammifères. Le réseau de F-actine conduit par mDia2 forme un échafaudage pour les

59
protéines de l’AC et permet le maintien de la position de l’AC au centre de la cellule en
division.
Il est à noter que bien d’autres protéines sont impliquées dans la formation et l’organisation
de l’AC. Notamment, l’ancrage de l’AC à la membrane plasmique serait assuré par
l’anilline (D’Avino 2009; Field et al. 2005). Cette protéine d’échafaudage est aussi
importante pour l'organisation et le recrutement de protéines structurales et de signalisation
dans l'AC (Maddox et al. 2005; Hickson et O’Farrell 2008; Piekny et Glotzer 2008;
Maddox et al. 2005; Thomas et Wieschaus 2004).
Figure 1.16: Représentation schématique de la voie de signalisation dépendante du complexe « centralspindlin » et responsable de la spécification et de la formation de l’AC.
En somme, il apparaît que la GTPase RhoA joue un rôle prédominant dans la spécification
et la formation de l’AC. Le contrôle précis de la localisation et de l’activation de RhoA au
sillon de clivage est nécessaire pour le recrutement et l’activation des protéines mDia2 et
MyoII à l’AC. Un échec du positionnement ou de l’activation de RhoA ou de ses effecteurs
au sillon de clivage conduit immanquablement à un avortement de l’abscission. Il est
important de noter que les protéines effectrices de RhoA dans le contexte de l’AC sont les

60
mêmes que celles requises pour l’arrondissement mitotique, à savoir une formine et la
MyoII, suggérant un mécanisme commun aux deux processus. Cependant, aucune étude à
l’heure actuelle n’a démontré un tel mécanisme. De plus, l’implication des GTPases Cdc42
et Rac1 dans le positionnement et la formation de l’AC restent encore mal comprise. En
effet, la surexpression de mutants dominant positif de Cdc42 ou de Rac1 conduisent aussi
à un échec de l’abscission (Dutartre et al. 1996; Muris et al. 2002). Ces observations
suggèrent qu’un contrôle fin de l’activité des GTPases de la famille Rho et de la dynamique
de l’actine qui en découle doit avoir lieu durant le positionnement et la formation de l’AC.
1.4.3.2. La contraction de l’anneau contractile
Une fois que la position de l’AC est spécifiée et que celui-ci est formé, l’AC se contracte
progressivement. Le modèle traditionnel de la contraction de l’AC est que les filaments
bipolaires de MyoII utilisent leur activité motrice pour se déplacer le long de deux
filaments antiparallèles d'actine, causant ainsi un glissement des filaments d’actines
(Figure 1.17). Dans le cadre d'un AC, de nombreux moteurs MyoII glissant sur de multiples
filaments d'actine conduiraient à une constriction de l’AC.
Figure 1.17: Modèle traditionnel de la contraction de l’AC.
L’implication de la MyoII dans la contraction de l’AC a été largement confirmée dans
différentes espèces où son inactivation stoppe la cytokinèse (Mabuchi et Okuno 1977;

61
Straight et al. 2003). Fait important, à mesure que l'anneau se rétrécit, la section
transversale de l'anneau contractile reste constante, ce qui suggère que le désassemblage
de la F-actine doit équilibrer la polymérisation en cours (Schroeder 1972). En effet, l'actine
et la myosine s’échangent rapidement au sein du sillon de clivage avec leur « pool »
cytoplasmique, ce qui implique que leur concentration locale dépend d'un équilibre
dynamique entre polymérisation/recrutement et dépolymérisation/dissociation (Yumura
2001; Murthy et Wadsworth 2005; Chew et al. 2017; Oelz, Rubinstein, et Mogilner 2015;
Oelz et Mogilner 2016; Stachowiak et al. 2014). Le mécanisme précis par lequel la
dépolymérisation de l'actine est contrôlée au sillon de clivage reste controversé. Il semble
cependant que l'activité de séparation des filaments d'actine de la cofiline et l'activité
ATPase de la MyoII soient nécessaires pour le désassemblage de la F-actine dans l’AC
(Mendes Pinto et al. 2012; Gunsalus et al. 1995).
En somme, les composantes de l’AC sont hautement dynamiques. Ceci suggère l’existence
de mécanismes, encore méconnus, de contrôle du renouvellement des composantes de l’AC
qui contribuent à la contraction de cette structure.
1.4.3.3. Désassembler l’AC pour permettre l’abscission
À la suite de la contraction de l’AC, les protéines rémanentes de cette structure à base
d’actine, dont la F-actine, doivent être enlevées pour permettre l’abscission. Ceci permet
vraisemblablement d'amincir le PI reliant les cellules filles. L’élimination des structures
d'actine serait également nécessaire pour permettre le recrutement de la machinerie
impliquée dans la fusion des membranes plasmiques opposées et donc l’abscission.
Notamment, interférer avec le démantèlement de l’AC en cytokinèse inhibe le recrutement
du complexe ESCRT-III (« endosomal sorting complexes required for transport III ») qui
coordonne les événements de remodelage de la membrane et du cytosquelette essentiels à
l'achèvement de la division cellulaire (Eikenes et al. 2015; Stoten et Carlton 2017). Les
mécanismes moléculaires contrôlant le désassemblage de l’AC en fin de cytokinèse sont
encore peu compris, mais des études récentes indiquent un rôle important pour le
métabolisme des phosphatidylinositols et le trafic membranaire.

62
Il a été montré que la modification de l’organisation de la membrane lipidique est requise
pour signaler le désassemblage de l’AC (Emoto et Umeda 2000). En effet, il a été constaté
que le PE (phosphatidyléthanolamine) est exposé à la surface cellulaire uniquement durant
les stades tardifs de la cytokinèse (Emoto et al. 1996; Emoto et Umeda 2000). Sa
perturbation mène à une inhibition du désassemblage de l’actine au sillon de clivage et à
la fusion ultérieure des membranes, suggérant que la redistribution des phospholipides de
la membrane plasmatique est une étape cruciale pour la cytokinèse. Il a été démontré que
l'immobilisation du PE à la surface cellulaire par un peptide de liaison bloque l'inactivation
de RhoA au stade tardif de la cytokinèse (Emoto et al. 2005).
De plus, il a été observé que le PI(4,5)P2 (Phosphatidylinositole 4,5 biphosphate) accumule
au sillon de clivage de façon dépendante de la kinase effectrice de RhoA PIP5K
(« Phosphatidylinositol 4-phosphate 5-kinase ») (Emoto et al. 2005; Field et al. 2005).
Cette accumulation contribue à la stabilisation de l’AC, ainsi que la liaison du cytosquelette
d'actine à la membrane plasmique en vertu de sa liaison aux protéines septines et ERM
(Janetopoulos et Devreotes 2006). Conformément à cela, les niveaux de PI(4,5)P2 au PI
diminuent lorsque le cytosquelette d'actine est démantelé avant que les cellules ne
progressent vers l'abscision (Wong et al. 2005).
Les travaux de (Dambournet et al. 2011) ont permis de découvrir un mécanisme
moléculaire par lequel les niveaux de PI(4,5)P2 sont réduits en cytokinèse tardive. Après
avoir déterminé dans une première étude le recrutement et l’importance de la petite GTPase
Rab35 (« Ras-related protein 35 ») au sillon de clivage lors des étapes tardives de la
cytokinèse, les auteurs ont démontré que la phosphatase OCRL (« oculocerebrorenal
syndrome of Lowe ») est un effecteur de Rab35 (Figure 1.18) (Kouranti et al. 2006). OCRL
est une PI(4,5)P2-phosphatase dont la déplétion est marquée par une inhibition de
l'abscission, une accumulation locale de PI(4,5)P2 et une accumulation anormale de F-
actine au PI (Dambournet et al. 2011).

63
Par ailleurs, très récemment, il a été proposé que l’oxydation de la F-actine est requise pour
le démantèlement de l’AC en cytokinèse tardive et que ceci nécessite également la GTPase
Rab35 (Frémont et al. 2017). Cette étude montra que l'oxydation médiant la
dépolymérisation de l'actine par l'enzyme oxydante MICAL1 (« Molecule Interacting with
CasL 1 ») est nécessaire au recrutement des effecteurs de l’abscission comme le complexe
ESCRT-III. MICAL1 serait recrutée sur le site d'abscission via une interaction directe avec
la GTPase Rab35 (Figure 1.18). Sur le plan mécanistique, les essais in vitro sur des
filaments d'actine uniques démontrèrent que MICAL1 est activée par Rab35. De plus, dans
les conditions expérimentales employées, MICAL1 n'agit pas comme une enzyme de
séparation des filaments d’actine, comme cela avait été montré initialement, mais induit
plutôt la dépolymérisation de la F-actine à partir de ses deux extrémités (Hung, Pak, et
Terman 2011; Grintsevich et al. 2016).
Un rôle pour la GTPase Rab11 a également été proposé pour la dépolymérisation de
l’actine au sillon de clivage en cytokinèse tardive. En effet, il a été démontré que les
endosomes de recyclage, associés à Rab11, sont importants pour la cytokinèse dans les
cellules de mammifères (Pelissier, Chauvin, et Lecuit 2003; Riggs et al. 2003; Wilson et
al. 2005). Une série d'études, a notamment observé que les protéines de liaison à Rab11 :
FIP3 et FIP4 (« Rab11 Family of Interacting Proteins 3 and 4 ») se localisent au PI et
interagissent simultanément avec Rab11 ainsi qu'une autre petite GTPase, Arf6 (« ADP-
ribosylation factor 6 ») (Wilson et al. 2005; Fielding et al. 2005; Hickson et al. 2003).
L'analyse spatiotemporelle des endosomes contenant FIP3 pendant la cytokinèse a
démontré que ces derniers sont une sous-population spécifique d'endosomes de recyclage
qui sont dirigés au PI au cours de la télophase tardive et qui sont nécessaires pour générer
une constriction secondaire du PI permettant l'achèvement complet de l'abscission (Schiel
et al. 2011; Wilson et al. 2005; Schiel et al. 2012; Simon et al. 2008). Il a été montré que
les protéines transportées par les endosomes Rab11-FIP3 : SCAMP2/3 (« Secretory carrier-
associated membrane protein 2 and 3 ») et p50RhoGAP, ont des rôles importants dans la
dépolymérisation de la F-actine corticale au PI pour permettre la constriction secondaire
du pont intercellulaire (Figure 1.18) (Schiel et al. 2012). p50RhoGAP permet
vraisemblablement de réduire la polymérisation de la F-actine en inactivant la petite

64
GTPase RhoA, tandis que le mécanisme moléculaire impliquant SCAMP2/3 reste peu
défini.
Figure 1.18: Mécanismes moléculaires de désassemblage de l’AC dépendant des GTPases Rab11 et Rab35.
Finalement, la protéine de coiffe cofiline qui stimule la dépolymérisation de l’actine est
également impliquée dans le désassemblage de l’AC en cytokinèse. En fait, son activité,
régulée par phosphoryaltion, apparaît contrôlée de façon mitose-spécifique par la kinase
LIMK1 (« LIM motif-containing protein kinase 1 »). La phosphorylation par LIMK1 de la
cofiline est importante en mitose pour le positionnement précis du fuseau mitotique
dépendant de la stabilité de l’actine corticale (Kaji, Muramoto, et Mizuno 2008).
Cependant, il a été établi que sa déphosphorylation et son activation subséquente en
cytokinèse sont requises pour la progression des cellules et leur abscission efficace (Amano
et al. 2002; Kaji et al. 2003). En effet, il a été montré qu’une expression ectopique de la
kinase LIMK1, impliquée dans la réorganisation du cytosquelette d’actine en inactivant la
cofiline, conduit à une accumulation de cellules multinuclées (Normand et King 2010;
Amano et al. 2002). Il a été mis en avant que la déphosphorylation de la cofiline en
cytokinèse dépend de l’activation de la phosphatase SSH1 (« Slingshot 1 ») dont la
déplétion conduit aux mêmes phénotypes que la surexpression d’un mutant

65
phosphomimétique de la cofiline et à l’accumulation aberrante de F-actine au PI (Amano
et al. 2002; Kaji et al. 2003). Ces travaux suggérèrent donc que la cofiline et la phosphatase
SSH1 sont impliquées dans le désassemblage de l’AC en cytokinèse.
Toutes ces observations montrent la complexité par laquelle est contrôlé le désassemblage
de l’AC en cytokinèse et la modulation subséquente de l’abscission. Le désassemblage
d’une telle structure pourrait aussi impliquer d’autres mécanismes à l’origine du contrôle
de la dynamique du cytosquelette d’actine. Notamment, on peut imaginer que le
démantèlement de l’AC implique des voies de dégradation des protéines. Certains travaux
ont mis en évidence l’implication de ces voies de dégradation dans la régulation de la
dynamique d’actine durant la division cellulaire. C’est ce dont nous discuterons dans la
section suivant.
1.4.4. Rôle émergeant de la dégradation ciblée de protéines comme mécanisme de
contrôle spatiotemporel du remodelage de l’actine pendant la division
cellulaire
De façon intéressante, le système ubiquitine-protéasome a été impliqué dans le remodelage
du cytosquelette d’actine au cours de la mitose. En effet, des travaux ont montré que
l’arrondissement cellulaire nécessite la dégradation de la protéine de coiffe Eps8 par le
protéasome de façon dépendante de son ubiquitination par SCFFbxw5 (« Skp1–Cul1–F-box
F-Box And WD Repeat Domain Containing 5 ») (Disanza et al. 2004; Werner et al. 2013).
Les auteurs ont notamment observé qu’interférer avec la dégradation d’Eps8 en phase G2,
par inhibition du protéasome ou déplétion de SCFFbxw5, prolonge sa localisation au cortex
cellulaire, retarde considérablement l'arrondissement mitotique des cellules et prolonge la
durée de la prométaphase. De plus, pendant les stades tardifs de la mitose et de la
cytokinèse, une activité de coiffe de la F-actine d’Eps8 semble être nécessaire pour éviter
les déformations de la membrane et de la forme de la cellule. En effet, l’expression d’un
dominant négatif d’Eps8 pour son activité de coiffe induit une instabilité corticale qui se
traduit par des déformations de la membrane et des déformations de la forme des cellules
(i.e. bourgeonnements membranaires) en anaphase et en télophase.

66
Ainsi, ces résultats identifient la fluctuation des niveaux d’Eps8 comme un mécanisme
important qui contribue aux changements de forme cellulaire lors de l'entrée et de la sortie
de la mitose (Figure 1.19). Ces résultats mettent aussi en évidence, un mécanisme
moléculaire de contrôle de la dynamique d’actine en mitose dépendant du contrôle fin
spatiotemporel de la dégradation d’une protéine de coiffe de l’actine.
Figure 1.19 : Modèle selon lequel SCFFbxw5 régule les niveaux d'Eps8 pour contrôler la dynamique de l'actine pour une progression mitotique normale. Inspiré de Werner et al. 2013.
En outre, il a longtemps été assumé que l’activité autophagique était fortement diminuée
durant la mitose et reprenait en cytokinèse tardive (Eskelinen et al. 2002; Furuya et al.
2010). Cependant, de récents travaux ont montré que le flux autophagique est maintenu
lors de la mitose bien que le nombre d’autophagosomes soit réduit durant cette période (Li
et al. 2016). Cette étude a notamment démontré que le flux autophagique est très actif en
mitose dans différentes lignées cellulaires y compris les cellules HeLa. Les auteurs ont
également noté que de multiples protéines impliquées dans l’autophagie, comme la
protéine LC3, sont induites lors de la mitose. Loukil et al. ont aussi observé que LC3,
p62/SQSTM1 et des marqueurs lysosomaux localisent avec des foyers de cycline A2
pendant la mitose et ont constaté que l'autophagie contribue partiellement à la dégradation
mitotique de la cycline A2 (Loukil et al. 2014). Ces travaux ont ainsi révélé que
l’autophagie reste active au cours de la division cellulaire et supposent que l’autophagie
sélective pourrait être requise dans le contrôle de la progression mitotique.

67
Dans ce sens, en cytokinèse, l’adaptateur autophagique p62/SQSTM1 est impliqué dans le
contrôle de cette étape à travers la régulation de la dynamique d’actine dépendante de RhoA
(Figure 1.20) (Belaid et al. 2013). En effet, par inhibition de la machinerie autophagique
(déplétion de la protéine ATG5 ou ATG7 ou KO de la v-ATPase a3), il a été établi que
l’autophagie est le mécanisme le plus important pour restreindre l’activité de la GTPase
RhoA au PI en cytokinèse (Figure 1.20). L’inhibition du flux autophagique a permis
d’observer l'accumulation de RhoA-GTP dans des autolysosomes, proches du PI de
cellules en cytokinèse. Au niveau moléculaire, les résultats de l’étude démontrèrent que la
GTPase est séquestrée sous sa forme active de façon dépendante de l’adaptateur
autophagique p62/SQSTM1 (Figure 1.20). En conséquence, interférer avec un tel
mécanisme, par inhibition de l’autophagie ou par déplétion de p62/SQSTM1, conduit à une
accumulation au PI de RhoA-GTP et à son expansion en dehors de cette zone (Figure 1.20).
Des cellules ayant subi ces traitements se caractérisent alors par des aneuploïdies
conséquentes à une accumulation nucléaire caractéristique d’un avortement de
l’abscission.
Figure 1.20: Réprésentation schématique du ciblage de la forme active de RhoA (RhoA-GTP) par p62/SQSTM1 à l’autophagie sélective au sillon de clivage.
En somme, seulement deux études ont clairement associé le ciblage de protéines aux voies
de dégradation aux mécanismes de remodelage de l’actine durant la division cellulaire. En

68
mitose, la dégradation de la protéine de coiffe Eps8 a été associée à la progression mitotique
en aidant au remodelage du cytosquelette d’actine requis pour l’arrondissement mitotique.
En cytokinèse, l’autophagie sélective semble être impliquée dans la régulation spatiale de
l’activité de la GTPase RhoA au PI. Ces observations suggèrent donc que les voies de
dégradation protéique et le contrôle de qualité des protéines qui y est associé seraient des
mécanismes utilisés pour le remodelage fin spatiotemporel des structures à base d’actine
qui dirigent la division cellulaire. Ces résultats soulèvent de nombreuses questions. Quelles
sont les voies de signalisation qui régulent ces mécanismes moléculaires durant la division
cellulaire? Ces fonctions sont-elles requises durant d’autres processus cellulaires
dépendant du remodelage de l’actine? Ces mécanismes de remodelage de l’actine font-ils
appel aux chaperons de la famille des HSPB? Est-ce que l’autophagie est contrôlée de
façon cycle cellulaire-dépendante?
1.4.5. Un rôle pour le complexe chaperon HSPB8-BAG3 dans la dynamique de
l’actine, l’orientation du fuseau mitotique et la ségrégation des chromosomes
pendant la mitose
Nous avons vu que les multiples étapes de la division cellulaire sont dépendantes d’une
régulation spatiotemporelle fine du cytosquelette d’actine qui depend en partie du ciblage
de protéines aux voies de dégradation protéique. Or, le co-chaperon BAG3 présente une
implication forte dans le remodelage du cytosqulette d’actine de façon dépendante de son
activité autophagique en condition de stress mécanique. De plus, il est important pour
assurer la stabilité d’HSPB8, un chaperon de la famille des HSPB qui émergent comme
des régulateurs du remodelage et de l’intégrité des structures d’actine en conditions
physiologiques. Ainsi, notre laboratoire a émis l’hypothèse que le complexe chaperon
HSPB8-BAG3 pourrait contribuer au remodelage des structures à base d’actine lors de la
division de cellules à croissance rapide telles que les cellules cancéreuses. À mon arrivée
au laboratoire, des évidences que le co-chaperon BAG3 est impliqué dans la progression
mitotique avaient déjà été obtenues. J’ai pu contribuer à la progression de ces travaux qui
sont présentés dans l’annexe 1 de cette thèse et qui seront résumés dans cette section.

69
En premier lieu, nous avons découvert que le co-chaperon BAG3 est hyperphosphorylé à
l’entrée des cellules en mitose et que ceci s’accompagne d’une relocalisation de la protéine
aux pôles du fuseau mitotique (Annexe 1, Figure 1). Nous avons ensuite investigué si la
déplétion du co-chaperon interfère avec la progression des cellules dans la division
cellulaire (Annexe 1, Figure 2). Nous avons notamment observé que la déplétion de BAG3
s’accompagne d’une élévation des anomalies nucléaires (micronoyaux et multi-nucléation)
suggérant un rôle du co-chaperon dans la modulation de la ségrégation des chromosomes
et de l’abscission. De plus, plus de 60% des cellules déplétées en BAG3 présentent des
défauts en métaphase et ceci se traduit par un retard considérable des cellules en
prométaphase-métaphase (Annexe 1, Figure 2). Par des expériences d’imagerie en temps
réel, nous avons également montré que la déplétion de BAG3 interfère considérablement
avec l’alignement des chromosomes à la plaque métaphasique et le positionnement du
fuseau mitotique qui subit des rotations anormales dans ces conditions (Annexe 1, Figure
3). Ces observations indiquent donc que le co-chaperon BAG3 est requis pour la
progression mitotique des cellules HeLa et qu’il interviendrait dans le contrôle du
positionnement du fuseau mitotique et l’alignement des chromosomes en métaphase.
Pour renforcer notre étude, nous avons réalisé une analyse structure-fonction de BAG3 en
mitose. Nous avons donc exprimé des mutants des domaines d’interaction de BAG3 et
analysé leur effet sur le positionnemment du fuseau mitotique, par déplétion-restoration de
phénotypes (Annexe 1, Figure 3). Nous avons pu voir de façon surprenante que le domaine
BAG n’est pas requis pour la fonction mitotique de BAG3. En effet, comme la protéine
WT, l’expression du mutant du domaine BAG est capable de restaurer le positionnement
du fuseau mitotique dans les cellules déplétées en BAG3 (Annexe 1, Figure 3). À l’inverse,
l’expression des protéines BAG3 mutées pour les motifs IPV ou le domaine PxxP sont
incapables de restaurer ce phénotype dans les cellules déplétées en BAG3 (Annexe 1,
Figure 3). Ceci nous indique donc que l’interaction de BAG3 à HSPB8 à travers ses motifs
IPV et son association à des voies de signalisation à travers son domaine PxxP sont
essentielles, tandis que son interaction avec HSP70 est dispensable pour sa fonction
mitotique. Ceci contraste avec une grande majorité des fonctions précédemment décrites
pour BAG3 qui dépendent d’HSP70.

70
Pour confirmer l’importance d’HSPB8 dans la fonction mitotique de BAG3, nous avons
analysé l’impact de la déplétion du chaperon sur la progression mitotique. Ainsi, nous
avons vu que la déplétion d’HSPB8, tout comme celle de BAG3, interfère avec le
positionnement du fuseau mitotique dans les cellules en métaphase (Annexe 1, Figure 4).
De plus, la déplétion du chaperon s’accompagne d’une délocalisation de BAG3 aux
centrosomes ainsi que d’une réduction de son hyperphosphorylation (Annexe 1, Figure 4).
Ces résultats argumentent dans le sens qu’HSPB8 est requise pour la fonction mitotique de
BAG3 en aidant à sa bonne localisation et à sa phosphorylation adéquate en mitose.
De façon intéressante, par des expériences de co-immunprécipitation, nous avons mis en
avant qu’HSPB8, p62/SQSTM1 et HDAC6 s’associent préférentiellement avec BAG3 en
mitose (Annexe 1, Figure 5). Ces observations nous suggèrent que l’hyperphosphorylation
de BAG3 s’accompagne d’une modulation de ses partenaires en mitose. Ces changements
de partenariat pourraient être requis pour la fonction mitotique de BAG3. Pour appuyer
cette hypothèse, nous avons par la suite évalué l’impact de la déplétion de p62/SQSTM1
sur le positionnement du fuseau mitotique. Ainsi, nous avons montré que comme la
déplétion de BAG3 ou d’HSPB8, celle de p62/SQSTM1 interfère avec le positionnment
correct du fuseau mitotique en métaphase (Annexe 1, Figure 5). Ces résultats tendent donc
à suggérer que les partenaires de BAG3 en mitose, à savoir HSPB8 et p62/SQSTM1, sont
requis pour sa fonction mitotique.
Finalement, comme le co-chaperon BAG3 est impliqué dans la modulation du remodelage
du cytosquelette d’actine, nous avons investigué si une telle fonction de BAG3 était requise
en mitose. Nous avons donc évalué l’effet de la déplétion de BAG3 sur l’arrondissement
mitotique, la dynamique des fibres de rétraction et la rigidité corticale. Ainsi, nos résultats
montrent que la déplétion de BAG3 s’accompagne d’un défaut d’arrondissement
mitotique, de dynamique des fibres de rétraction et de rigidité corticale qui se traduit par
un bourgeonnement excessif du cortex des cellules en métaphase (Annexe 1, Figure 6). De
façon remarquable, la dynamique des fibres de rétraction et la rigidité corticale sont
également altérés suivant la déplétion de p62/SQSTM1 ou d’HSPB8 (Annexe 1, Figure 6).
Ces observations nous suggèrent donc que BAG3 facilite le remodelage des structures à

71
base d’actine en mitose, et que cette fonction pourrait faire intervenir ses partenaires
mitotiques HSPB8 et p62/SQSTM1.
L’arrondissement mitotique, la dynamique des fibres de rétraction et la rigidité corticale
sont requis pour le positionnement du fuseau mitotique ainsi que la capture et la ségrégation
des chromosomes. Nous avons voulu savoir si de tels défauts observés suivant la déplétion
de BAG3 pouvaient être la cause des défauts de positionnement du fuseau mitotique. Pour
ce faire, nous avons restauré la rigidité corticale des cellules déplétées en BAG3 à l’aide
de la concaviline A et analysé son effet sur le positionnement du fuseau mitotique. De
façon intéressante, nous avons vu que le positionnement du fuseau mitotique est largement
restauré dans les cellules déplétes en BAG3 suite à leur traitement avec la concaviline A
(Annexe 1, Figure 7). Ainsi, ce résultat tend à suggérer que les défauts de rigidité corticale
observés suite à la déplétion de BAG3 contribuent aux anomalies de positionnement du
fuseau mitotique et d’alignement des chromosomes en métaphase.
En résumé, ces travaux ont permis pour la première fois d’établir une fonction pour le
complexe chaperon HSPB8-BAG3 dans la régulation de la progression mitotique. Ces
observations suggèrent que cette activité pourrait être reliée à la capacité du complexe à
réguler le remodelage des structures d’actine qui dirigent le positionnment du fuseau
mitotique et l’alignement des chromosomes en métaphase. Une telle fonction d’HSPB8-
BAG3 impliquerait l’association du complexe à l’adaptateur autophagique p62/SQSTM1
et à des protéines encore non identifiées via le domaine PxxP de BAG3. De plus, ces
travaux ont pour la première fois mis en évidence un système (la mitose) dans lequel
l’association de BAG3 au chaperon moléculaire HSP70 est dispensable alors que son
association à HSPB8 est requise. Ces travaux ont donc établi la mitose comme un modèle
paradigme pour étudier les fonctions de BAG3 qui dépendent du chaperon HSPB8 et le
lien fonctionnel avec le remodelage de l’actine, offrant un modèle unique pour disséquer
le mode d’action du complexe chaperon.

72
1.5. Objectifs de travail
De plus en plus d’études suggèrent que les chaperons moléculaires soutiennent la survie
des cellules cancéreuses. Cependant, les mécanismes moléculaires associés à une telle
fonction des chaperons moléculaires sont très peu connus. HSPB8-BAG3 est un complexe
chaperon responsable du ciblage de protéines endommagées à la machinerie autophagique
lors d’un stress protéotoxique. BAG3 a été impliquée dans la progression tumorale via sa
fonction régulatrice du cytosquelette d’actine dans différentes lignées cancéreuses.
Toutefois, l’importance de son association au chaperon HSPB8 n’a jamais été étudiée. Des
travaux du laboratoire auxquels j’ai participé ont montré que BAG3, de façon dépendante
d’HSPB8, régule la progression mitotique et le remodelage de structures d’actine qui
contrôlent le positionnement adéquat du fuseau mitotique et la ségrégation des
chromosomes.
Nous avons émis l’hypothèse que le complexe chaperon HSPB8-BAG3 facilite la division
cellulaire via son activité dans le remodelage des structures à base d’actine. Nous avons
postulé que cette activité fait intervenir, du moins en partie, l’activité de son partenaire
mitotique HDAC6 et le rôle du complexe chaperon dans l’autophagie sélective.
Ainsi, l’objectif principal de mon doctorat était de déterminer l’importance de la
modulation du cytosquelette d’actine par le complexe chaperon HSPB8-BAG3 sur la
division cellulaire et d’explorer les mécanismes moléculaires à l’origine de cette
modulation pouvant impliquer HDAC6.
1) Déterminer la contribution du remodelage de l’actine dans la régulation de la division
cellulaire par le complexe HSPB8-BAG3.
Pour cet objectif, nous avons étudié l’effet de la déplétion, dans des cellules HeLa,
d’HSPB8 et de BAG3 sur la progression de la cytokinèse : une phase de la division
cellulaire où la dynamique d’actine est hautement régulée. Nous avons testé l’effet de
drogues inhibant la dynamique d’actine et l’autophagie sur les phénotypes mis en évidence
dans cette étude. Cette étude est présentée au chapitre 2 et suggère que le complexe

73
chaperon agit en partie, en limitant la polymérisation de l’actine dépendante d’Arp2/3 et/ou
en stimulant son renouvellement et en facilitant la dégradation par autophagie sélective.
2) Étudier l’importance du partenariat entre HSPB8-BAG3 et HDAC6 dans la modulation
de la dynamique d’actine au cours de la mitose
Dans cette étude, nous avons validé l’impact du complexe sur la polymérisation de l’actine
dépendante d’Arp2/3 et mis en évidence un rôle pour la déacétylase HDAC6 et son substrat
cortactine, comme cibles de l’action mitotique du complexe chaperon dans la régulation
du remodelage des structures à base d’actine. Ces travaux sont présentés au chapitre 3 et
suggèrent un rôle important pour le complexe HSPB8-BAG3 dans la régulation spatiale de
l’activité d’HDAC6 pendant la mitose.
3) Explorer l’effet du complexe sur l’activité autophagique au cours de la mitose et le lien
potentiel avec sa fonction mitotique.
Pour ce dernier objectif, nous avons analysé l’influence de l’inhibition du flux
autophagique sur la dynamique du cytosquelette d’actine au cours du cycle cellulaire,
observé l’effet de la déplétion d’HSPB8 sur le flux autophagique, et étudié l’impact de
l’inhibition d’HDAC6 sur le flux autophagique et la dynamique d’actine en cytokinèse
après déplétion d’HSPB8 (Chapitre 4).
L’ensemble de ce travail vise à mieux comprendre les mécanismes par lesquels le complexe
chaperon HSPB8-BAG3 peut soutenir la progression tumorale et identifier de potentielles
nouvelles cibles thérapeutiques.

74
2. Chapitre 2: Fine-tuning of actin dynamics by the HSPB8-BAG3 chaperone
complex facilitates cytokinesis and contributes to its impact on cell division
Alice Anaïs Varlet 1, 2, Margit Fuchs 1, 2, Carole Luthold 1 ,2 , Herman Lambert 1, 2,
Jacques Landry 1, 2, 3 and Josée N. Lavoie 1, 2, 3 *
1 Centre de recherche sur le cancer de l’Université Laval & 2 Oncology, Centre de
recherche du CHU de Québec-Université Laval, Canada, G1R 3S3 ; 3 Département de
biologie moléculaire, biochimie médicale et pathologie Université Laval, Québec, Canada,
G1V OA6
Running Head: HSPB8 Regulates F-actin Dynamics during Cytokinesis
Keywords: cytokinesis, BAG3, HSPB8, actin, autophagy,
* Author for correspondence ([email protected])

75
2.1. Avant-propos
Lorsque j’ai commencé mon doctorat, le complexe chaperon HSPB8-BAG3 avait été
impliqué dans le ciblage des protéines agrégées et poly-ubiquitinées à la machinerie
autophagique. Les travaux de Arndt et al. 2010 suggéraient un rôle pour le complexe
chaperon dans le contrôle de l’intégrité du cytosquelette d’actine dans des cellules
musculaires soumises à un stress mécanique. Au laboratoire, le Dre Fuchs M. avait mis en
évidence l’implication de BAG3 dans la progression mitotique et sa déplétion avait été
associée à une dérégulation du positionnement du fuseau mitotique et une élévation de
l’instabilité génétique. L’importance de son partenariat avec HSPB8 et le mécanisme par
lequel le complexe contrôle la progression mitotique n’avaient pas encore été découverts.
C’est dans ce contexte que nous avons investigué l’impact d’une déplétion d’HSPB8 ou de
BAG3 sur la progression de la division cellulaire et évalué le mécanisme par lequel le
complexe chaperon facilite celle-ci. En collaboration avec le Dre Fuchs M., j’ai réalisé des
expériences d’imagerie en temps réel de cellules HeLa déplétées en BAG3 ou en HSPB8
dans le but d’analyser l’impact de la déplétion de BAG3 ou d’HSPB8 sur l’étape ultime de
la division cellulaire : l’abscission. J’ai ensuite analysé en temps réel la dynamique du
cytosquelette d’actine dans des cellules synchronisées en cytokinèse et déplétées en
HSPB8. Puis, par des protocoles que j’ai développés, j’ai tenté de restaurer ou de mimer
les phénotypes associés à la déplétion d’HSPB8 à l’aide de drogues inhibant la
polymérisation de l’actine ou inhibant/activant l’autophagie. Les résultats obtenus ont
démontré pour la première fois que le complexe chaperon HSPB8-BAG3 aide à la
progression des cellules cancéreuses dans la division cellulaire en limitant la dynamique
de l’actine. Ces travaux suggèrent également que le complexe chaperon pourrait faciliter
la dynamique d’actine au cours de la division cellulaire de façon dépendante de sa fonction
dans l’autophagie sélective.
J’ai participé activement à la conception de cet article et j’ai réalisé l’ensemble des figures
qui y sont présentées, à l’exception de la figure 2.2F réalisée par Mr Lambert H. Les
expériences d’imagerie en temps réel de la progression mitotique de cellules déplétées en

76
BAG3 ont été réalisées par le Dre Fuchs M. Ce projet a donné lieu à un article publié dans
le journal Cell Stress and Chaperones publié en mars 2017.
Le format PDF de ces travaux est disponible en Annexe 3.

77
2.2. Résumé
La petite protéine de choc thermique HSPB8 et son co-chaperon BAG3 ont été associés au
contrôle de l’intégrité du cytosquelette d’actine dans le contexte d’un stress mécanique. Ici,
nous montrons qu’HSPB8-BAG3 est essentiel au démantèlement de l’anneau contractile
lors de la cytokinèse, un processus nécessaire pour permettre l'abscission des cellules filles.
Nous avons noté qu’une diminution considérable de la forme hyperphosphorylée de BAG3,
qui contribue à son rôle crucial en mitose, accompagne la réduction des niveaux endogènes
d’HSPB8 par ARN interférence dans les cellules HeLa. En outre, les cellules déplétées en
HSPB8 sont retardées dans la cytokinèse, restent connectées par un pont intercellulaire (PI)
désorganisé et présentent une incidence accrue d'anomalies nucléaires résultant d’un échec
de l’abscission (à savoir bi- et multi-nucléation). De tels phénotypes ont été associés à une
accumulation anormale de F-actine au PI des cellules en télophase. De façon remarquable,
la drogue séquestrant les monomères d’actine, la latrunculine A, et l'inhibiteur d’Arp2/3,
le CK666, normalisent la F-actine au PI pendant la cytokinèse. La latrunculine A restaure
également l’incidence des anomalies nucléaires dans les cellules appauvries en HSPB8,
impliquant une dynamique d'actine dérégulée comme cause d'échec de l’abscision. De plus,
l’accumulation anormale de F-actine au PI semble être corrigée par la rapamycine, une
drogue activant l'autophagie, alors qu'elle est mimée par des drogues inhibant le flux
autophagique. Ensemble, ces résultats appuient le rôle du complexe chaperon HSPB8-
BAG3 dans le contrôle de qualité de la dynamique de structures à base d'actine qui sont
soumises à de fortes tensions, notamment lors de la cytokinèse. Ils développent une
connexion jusqu'à maintenant très peu appréciée entre l'autophagie sélective et les
changements morphologiques qui guident la division cellulaire.

78
2.3. Abstract
The small heat shock protein HSPB8 and its co-chaperone BAG3 are proposed to regulate
cytoskeletal proteostasis in response to mechanical signaling in muscle cells. Here, we
show that in dividing cells, the HSPB8-BAG3 complex is instrumental to the accurate
disassembly of the actin-based contractile ring during cytokinesis, a process required to
allow abscission of daughter cells. Silencing of HSPB8 markedly decreased the mitotic
hyperphosphorylated forms of BAG3 in HeLa cells, supporting its crucial role in BAG3
mitotic functions. Cells depleted of HSPB8 were delayed in cytokinesis, remained
connected via a disorganized intercellular bridge and exhibited increased incidence of
nuclear abnormalities that result from failed cytokinesis (i.e. bi-, multi-nucleation). Such
phenotypes were associated with abnormal accumulation of F-actin at the intercellular
bridge of daughter cells at telophase. Remarkably, the actin sequestering drug latrunculin
A, like the inhibitor of branched actin polymerization CK666, normalized F-actin during
cytokinesis and restored proper cell division in HSPB8-depleted cells, involving
deregulated actin dynamics as a cause of abscission failure. Moreover, this HSPB8-
dependent phenotype could be corrected by rapamycin, an autophagy-promoting drug,
whereas it was mimicked by drugs impairing lysosomal function. Together, the results
further support a role for the HSPB8-BAG3 chaperone complex in quality control of actin-
based structure dynamics that are put under high tension, notably during cell cytokinesis.
They expand a so-far under-appreciated connection between selective autophagy and
cellular morphodynamics that guide cell division.

79
2.4. Introduction
Fine-tuned protein quality control (PQC) systems are governed by molecular chaperones
of the heat shock protein (HSP) family. Chaperones of the small HSP family (HSPB)
formed an eclectic family of proteins, which differ in their expression profiles and
biochemical properties. Nonetheless, they are proposed to act mainly as ATPase-
independent holdase or unfoldase by virtue of their dynamic quaternary structure, to
segregate damaged proteins and prevent their irreversible aggregation. In addition to their
cytoprotective function under proteotoxic stress conditions, they appear to be instrumental
to the normal functioning of complex proteinous structures of the cytoskeleton during
biological processes relying on acute cellular remodeling. For instance, a phosphorylation-
regulated function of HSPB1 has been reported, which regulates actin polymerization and
cancer cell migration. Furthermore, HSPB8 has been involved in the recognition of
damaged filamin during muscle cell contraction and its segregation for degradation by
selective autophagy (Ulbricht, Arndt, et Höhfeld 2013). The later mechanism is proposed
to maintain muscle fibers integrity by promoting clearance of actin-binding proteins
undergoing partial misfolding in response to mechanical strain. Expectedly, several HSPB
chaperones are up-regulated in cancer cells where they could contribute to the fitness of
actin-based structures that support the tumor phenotype. Thus, by enabling targeted protein
storage, recycling or degradation, HSPB proteins would play important roles in
cytoskeletal PQC, to maintain the integrity of cytoskeletal structures during stress, as well
as to facilitate cellular morphodynamics that guide cell division and differentiation. In line
with this concept, we have recently uncovered a unique role for HSPB8 in mitotic cell
remodelling (Fuchs et al. 2015).
In spite of their known implications in human diseases such as cancer, the mechanisms
underlying the contribution of HSPB proteins to cytoskeletal dynamics/proteostasis is
poorly understood. In that regard, the co-chaperone and scaffold protein BCL2-associated
athanogene 3 (BAG3) is of particular interest. In the context of cancer cells, BAG3 is
viewed as a molecular scaffold owing to its interactions with signaling proteins and several
regulators of actin dynamics, and its implication in key biological processes, including

80
apoptosis, development, cell motility and cell division (Behl 2016; Rosati et al. 2011). Yet,
the significance of defined chaperone partners for BAG3 functions in intracellular
signaling has remained unexplored. BAG3 can associate with several HSPB proteins but
shows a preferred association with, and regulates the stability of HSPB8 (Carra et al. 2008;
Fuchs et al. 2010; Taipale et al. 2014). BAG3 also acts as a co-chaperone for
HSC70/HSP70 chaperone system and modulates its function in proteasomal degradation
and aggrephagy (Chen et al. 2013; Gamerdinger et al. 2009; Gamerdinger et al. 2011;
Minoia et al. 2014). Accumulating evidence suggests that BAG3 serves to bridge important
components of the chaperone network (Rauch et al. 2017). Nonetheless, BAG3 may
function without recruiting HSPB8 in aggrephagy (Minoia et al. 2014). In contrast, its
interaction with HSPB8 may be instrumental in typifying BAG3 functions in cytoskeletal
remodelling. This is supported by our recent findings that BAG3 regulates remodelling of
the mitotic actin-based structures that guide spindle orientation in a manner that requires
its physical binding to HSPB8, but not to HSC70/HSP70 (Fuchs et al. 2015). Because the
same mitotic phenotype can be recapitulated by depletion of HSPB8, BAG3 or the
autophagic receptor p62/SQSTM1, which shows enhanced mitotic association with BAG3,
we have proposed that they act within a so-far unrecognized cytoskeletal PQC mechanism
during mitosis. Evidence suggests that targeted cytoskeletal protein degradation can
provide a mechanism to limit signaling pathways in time and space during mitotic cell
remodelling. Little is known, however, on the mechanism of quality control autophagy that
provides “built in” selectivity for components of cytoskeletal remodelling pathways.
Whether the HSPB8-BAG3 chaperone complex may contribute for such mechanism during
mitosis remains to be demonstrated.
Here, we investigate the impact of HSPB8 in cytoskeletal PQC during cytokinesis, a
process that is orchestrated by highly organized actin-based structures that are put under
high tensile forces. We show that HSPB8 regulates BAG3 levels during mitosis and the
accurate dynamics of the cytokinetic actin ring, via a mechanism that may involve effects
on branched actin nucleation and autophagy signaling. Importantly, strong evidence is
provided that first link HSPB8 function in cytoskeletal PQC to its modulation of mitotic
cell division in cancer cells.

81
2.5. Material and methods
2.5.1. Cell culture, synchronization, drug treatments and siRNA experiments
Parental HeLa cells, HeLa-GFP-H2B cells (gift from Laurence Pelletier) and HeLa-RFP-
H2B cells (gift from Sabine Elowe) (Klebig et al. 2009), were maintained in a-minimal
essential medium supplemented with 10% foetal bovine serum. All cell lines were grown
in a humidified atmosphere with 5% CO2 at 37°C. Cells were synchronized by a double
thymidine block and were released for 8 h to 10h in fresh medium, as described previously
(Fuchs et al. 2015). For knockdown experiments, HeLa cells were transfected with 25 nM
to 50 nM siRNA duplexes in calcium-phosphate transfection buffer (125 mM MgCl2, 140
mM NaCl, 25 mM HEPES, 0.75 mM Na2HPO4) overnight. Analyses were performed 48 h
to 88 h later by western blot, immunofluorescence or live cell imaging. For cell cycle
analyses of the impact of HSPB8-specific siRNAs on BAG3 protein levels, HeLa cells
were synchronized in G2/M or M-phase by a 16 h-treatment with the selective Cdk1
inhibitor (RO3306, 8 µM) or with nocodazole (400 ng/ml), respectively, and mitotic cells
were recovered by a mitotic shake-off, as described before (Fuchs et al. 2015). Cells were
lysed by 3 cycles of freezing/thawing in 4 volumes of lysis buffer (20 mM TRIS-HCl pH
7.6, 150 mM NaCl, 1 mM EDTA, 0,1% IGEPAL, 1 mM NaVO4, 10 mM NaF, 40 mM β-
glycerophosphate, 1X Complete [Roche], 1 mM DTT), centrifuged at 15,000 g for 15 min,
and the supernatants were processed for western blot analyses. The following drugs were
added to cells that have been synchronized in mitosis with a double thymidine block, during
the last hour of second release period before cell fixation as followed: latrunculin A, 20
nM; CK666, 40 µM; rapamycin, 150 nM; E-64D and pepstatin A, 10 µg/ml; bafilomycin
A1, 200 nM. For multinucleation assays, unsynchronized HeLa-GFP-H2B cells were
grown in the presence of 1 nM latrunculin A for 48 h before cell fixation. The siRNA
duplexes were based on human sequences and were purchased from Qiagen (HPP grade
siRNA) or Thermo Fisher Scientific (standard A4 grade). Sequences of the sense strands
are as followed: siBAG3 #1: CGAAGAGTATTTGACCAAA -3’;
siBAG3 #2: 5’-GCAAAGAGGTGGATTCTAA-3’;
siHSPB8 #1: 5’-CAGATAGGCTAGTGGTATT-3’;

82
siHSPB8 #2: 5’-GCAGTGAATGCAAGGGTTATT-3’;
siHSPB8 #3: 5’-CAGAGGAGTTGATGGTGAATT-3’.
si AllStars Negative Control, target sequence: 5’-CAGGGTATCGACGATTACAAA-3’.
2.5.2. Transfection, infection, adenovirus and vectors
HeLa cells were seeded on fibronectin-coated dishes and were synchronized by the double
thymidine block method. During the second release period, cells were transfected with
GFP-Arp3 (human Arp3 in pEGFP-N1 vector, gift from Dr John A. Cooper, ST Louis,
USA) (Welch et al. 1997) using Lipofectamine 2000 at the ratio Lipo:DNA of 2:1 and
following the manufacturer’s instructions (Invitrogen) for a 10 h-period before cell
fixation. To follow actin dynamics in vivo, cells were seeded on fibronectin-coated glass
dishes (MatTek Corp.) and synchronized by the double thymidine block method.
Adenofection was performed during the first thymidine block using Ad-LifeAct-TagGFP2
(60121, IBIDI) at a multiplicity of infection (MOI) of 1 plaque-forming units per cell
(pfu/cell), BacMam 2.0 reagents at 4 pfu/cell in a-MEM-minus and siRNAs in calcium-
phosphate transfection buffer, using the method described in (Fuchs et al. 2016). Sixteen
hours after adenofection, cells were washed 3 times with HEPES (6.7 mM KCl, 150 mM
NaCl, 10 mM HEPES, pH 7.3) and were released in fresh medium for 8 h before adding
back 2mM thymidine.
2.5.3. Antibodies and chemicals
The following antibodies were used for western blot analyses: rabbit anti-BAG3 LP11 was
raised against full length recombinant human BAG3 fused with glutathione S transferase
(LP11) (Fuchs et al. 2015), rabbit anti-HSPB8 against a C-terminal peptide
(NELPQDSQEVTCT) (Carra et al. 2008); anti-Vinculin (V9131) was from Sigma-
Aldrich; anti-Arp3 (ab49671) was from Abcam; anti-Calreticulin (#612136) was from BD
Biosciences; and anti-GAPDH Clone 6C5 (Fitzgerald, #10R-G109a) was from Millipore.
Secondary antibodies (anti-mouse-HRP 115-035-146 and anti-rabbit-HRP 111-055-003)
were from Jackson ImmunoResearch. The following antibodies were used for

83
immunofluorescence: anti-aurora B (ab2254) was from Abcam; Alexa-Fluor® 488
Phalloidin (A12379) and Texas Red®-X Phalloidin (T-7471 were from Molecular
Probes/Thermo Fisher. Goat-anti-rabbit-594 (A11037) was from Molecular Probes.
Hoechst Bisbenzimide H 33342 (B2261), thymidine (T9250), fibronectin (F1141-5 mg),
RO-3306 (SML0569), E-64D (E8640), pepstatin A (P5318), bafilomycin A1 (B1793) and
CK666 (SML0006) were from Sigma-Aldrich; latrunculin A (428021-100UG) was from
Calbiochem.
2.5.4. Immunofluorescence and live-cell imaging
Cells were gently washed once with LUFTIG (0,2 M sucrose, 35 mM Pipes, pH 7,4, 5 mM
EGTA, 5 mM MgSO4) and fixed with 3,7 % formaldehyde in LUFTIG or 4% PFA in PBS
(137 mM NaCl, 2,7 mM KCl, 4.3 mM Na2HPO4, 1.47 mM KH2PO4) for 20 min at room
temperature. DNA was stained with cell permeable Hoechst (150 ng/mL). Specimens were
processed for immunofluorescence as described (Lavoie et al. 2000) and were post-fixed
with 3.7% formaldehyde in PBS for 20 min at room temperature. Phenotypes were
monitored routinely by at least two independent investigators by visual inspection of fixed
specimens, using an AxioObserver Z1 system using a 60x 1.25NA objective and a charged-
coupled device (CCD) camera Axiocam MRm controlled by the Zen software (Carl Zeiss).
Confocal microscopy in live cells and fixed samples was performed with a PerkinElmer
Life Science Ultraview Spinning Disk Confocal microscope, equipped with a 40x 0,75NA
objective, an EMCCD cooled charge-coupled camera at -50°C (Hamamatsu Photonics K.
K) and driven by Volocity software version 6.01. The system was equipped with a
humidified/5% CO2 /thermoregulated chamber. Bi- and Multi-nucleation was scored as the
number of cells presenting 2, or >2 nuclei per cell; single cells were delineated by staining
of the cortex with F-actin. To quantify abnormal F-actin accumulation at the intercellular
bridge, we scored the number of daughter cells connected by a bridge labelled by Aurora
B staining that had completely de-condensed DNA (late telophase) and abnormal F-actin
structures at the bridge.

84
Long-term live-cell imaging was performed using a Nikon TE-2000 inverted microscope
equipped with CO2/thermo-regulated chamber with a 40x 0.6 NA objective. Images were
captured as 16-bit TIFF files with a photometrix Collsnap FX cooled CCD camera (-30°C)
(Rooper Scientific, Tuscon AZ) driven by the Metaview software version 4.5 (Universal
Imaging Corp., Downington, PA). Images were acquired at 90 sec intervals for a 48 h-
period, or at 10 min intervals for a 72h period, starting at 40 h after siRNA transfection.
Phenotypes associated with cytokinesis were estimated by visual inspection of time-
sequences. Spreading of daughter cells and ICB disappearance were evaluated by
monitoring the time spent between closure of the actin and cell spreading, or between AR
closure and disappearance of the dense midbody ring, respectively. Defects in daughter cell
spreading were considered as followed: (1) prolonged cortex blebbing; (2) uncoordinated
spreading of daughter cells; (3) no spreading. To quantify the number of cells that re-
entered mitosis synchronously, we determined that cells are derived from the same mother
cell by visual inspection of time-sequences; cells were considered synchronized if they
entered mitosis at the same time-point, or separated by less than 2 time-points. AR
thickness and width were expressed as the mean intensities from F-actin staining and were
evaluated by delineating AR manually on confocal slices determined as the center of the
cell, using the volocity software version 6.0 (Quorum Technologies); cells were considered
only after AR closure and DNA de-condensation. To quantify GFP-Arp3 recruitment, AR
and daughter cells were delineated manually using F-actin staining as the reference for cell
shape using the same software. The mean fluorescence intensities of GFP-Arp3 at AR and
in the cytoplasm were measured from confocal image stacks acquired at 0,5 µm z-steps
and expressed as a ratio of AR over cytoplasm. The volocity software version 6.0 (Quorum
Technologies) and Image J 1.48v (National Institute of Health) were used for processing
entire images before cropping to emphasize the main point of the image. Processing was
limited to background subtraction and brightness/contrast adjustment.
2.5.5. Western blotting
Cells were lysed in SDS sample buffer (62.5 mM Tris-HCl, pH 6.8, 2.3% SDS, 10%
glycerol, 5% β-mercaptoethanol, 0.05% bromphenol blue, 1 mM phenylmethylsulfonyl

85
fluoride), equal amounts of protein from whole cell lysates were separated by SDS-PAGE
on 10% acrylamide gels and transferred onto nitrocellulose membranes. Immunoblotting
was performed as described (Champagne et al. 2004). Protein concentrations were
determined with DC protein assay reagent and densitometric analyses were performed from
FluorS MAX MultiImager-captured images using the QuantityOne software version 4.5.0
(Bio-Rad Laboratories).
2.5.6. Statistical analyses
For experiments examining (a) the time spent for daughter cells spreading or (b) AR
thickness/width, the mean values from individual cells from at least 3 independent
experiments were analyzed using the Mann Whitney test, which is a non-parametric test
that compares two unpaired groups of ordinal or numerical variables. For experiments
examining the proportions of cells with: (a) cell spreading defects (b) ICB defects, (c)
persistent connections (d) synchronized daughter cell mitosis, (e) nuclear abnormalities
and (f) abnormal F-actin accumulation at the ICB, data from individual experiments were
analyzed using the Fisher's exact test, which is used for small sample sizes with
nominal/categorical variables. Statistical calculations were performed using Prism 6.0
(GraphPad Software) statistical software. P-values <0.05 considered significant (*, p<0.05;
**, p<0.01; ***, p<0.001 ; ****, p<0,0001).
2.6. Results
2.6.1. Silencing of HSPB8 or BAG3 perturbs abscission of daughter cells
We sought to characterize the function of HSPB8-BAG3 chaperone complex in the
extreme cell morphodynamic changes that guide cytokinesis. To that end, unsynchronized
HeLa-GFP-H2B were treated with control small interfering RNA (siRNA), or HSPB8- or
BAG3-specific siRNAs that achieved >75% depletion of the protein in these cells, and
were imaged for a period of 72 h starting at 40 h after transfection of siRNAs (Figure 2.1A).
In agreement with our previous findings, cells depleted of HSPB8 or BAG3 eventually

86
progressed through anaphase, but exhibited higher incidence of defects during cytokinesis
(Fuchs et al. 2015). Such defects were associated with 2 major phenotypes: delayed cell
spreading at telophase, and delayed abscission of daughter cells. HSPB8-depleted cells
showed a ~1.3-fold increase in the mean time required to achieve spreading (35 min;
siHSPB8) relative to control cells (26 min; siCtl) (Figure 2.1B, 2.1C), frequently associated
with prolonged cortex blebbing (Figure 1B, siBAG3 490’; supplemental movies S1-S3).
This phenotype was observed in 24% to 30% of HSPB8- or BAG3-depleted cells,
respectively, as compared to 12% of the control cell population (Figure 2.1C). The data are
consistent with previous work involving BAG3 in modulation of cell adhesion, a function
that may engage its association with HSPB8 at mitotic exit (Iwasaki et al. 2007; Iwasaki et
al. 2010; Kassis et al. 2006).
Most remarkably, depletion of HSPB8 or BAG3 was associated with increased occurrence
of abnormally thick and long intercellular bridges (ICB) connecting nascent daughter cells,
which persisted for longer periods of time (Figure 2.1D, arrowheads; Figure 2.1E;
supplemental movies S4-S6). Indeed, BAG3-depleted cells showed a ~1.7-fold increase in
the mean time required for ICB disappearance relative to control cells (Figure 2.1E).
Moreover, a larger proportion of HSPB8-depleted cells remained connected by an ICB
comprising a typical midbody ring (Figure 2.2A, red arrowhead and inset), and even re-
entered mitosis synchronously (Figure 2.2B, 2.2C). These phenotypes are typical of cells
that fail abscission and were also observed in BAG3-depleted cells (not shown) (Belaid et
al. 2013; Schiel et al. 2012). In further support, a significant proportion of cells transfected
with 2 distinct HSPB8-specific siRNA sequences exhibited bi-nucleation, a defect that
arises upon cytokinetic failure and collapsing back into a single bi-nucleated cell, which
may be due to impairment of abscission (Figure 2.2D, 2.2E). Together, these observations
suggest that HSPB8, in complex with BAG3, facilitates some late steps during cytokinesis
that control the abscission of daughter cells.

87
Figure 2.1: Depletion of HSPB8 or BAG3 perturbs daughter cells spreading and ICB disappearance during cytokinesis. (A) Representative western blot of HeLa-GFP-H2B cells lysed in SDS-sample buffer, showing the efficiency of HSPB8 and BAG3 depletion after transfection with HSPB8- and BAG3-specific siRNAs. Depletion was estimated at > 75% by loading increasing amounts of control extracts (siCtl). Western blot analysis was performed with anti-HSPB8 and anti-BAG3 antibodies; GAPDH: loading control. (B) Unsynchronised HeLa-GFP-H2B cells were depleted of HSPB8 or BAG3 using the indicated siRNAs and were imaged starting 40 h post-transfection for 72 h at 10 min intervals. First row of panels: representative time sequence of a control cell showing normal daughter cells spreading; second and third rows of panels: representative time sequences of HSPB8- or BAG3-depleted cells showing delayed spreading. Scale bar, 10

88
µm. (C) On the left: graph showing the average time of daughter cells spreading for control (siCtl) versus HSPB8 depleted cells (siHSPB8). Data are the means +/- SE from 3 independent experiments; the Mann-Whitney’s test was performed relative to control ****, p < 0.0001. On the right: graph depicting the percentage of cells with abnormal daughter cells spreading in HSPB8- (siB8) or BAG3-depleted cells (siBAG3) compared to control cells (siCtl), means +/- SE from 2 or 3 independent experiments. *, p < 0.05 compares siHSPB8 #3 to siCtl by the Fischer exact test. (D) Representative time sequences of HeLa-GFP-H2B cells transfected with the indicated siRNAs and imaged for 72 h starting at 40 h after transfection. Enlarged views of the boxed regions emphasize the structure of the ICB, which is abnormally long and/or thick in cells depleted of HSPB8 or BAG3, as designated by the white and red arrows, respectively; time sequences are in min; Bars, 10 µm. (E) Graphs depicting the percentages of cells with abnormal ICB (on the left) or the time spent between early telophase and disappearance of the ICB (on the right); the data are the means +/- SE of 2 to 3 independent experiments as indicated. **, p < 0.01 and ****, p < 0.0001 compare siHSPB8 to siCtl by the Fisher exact test.
2.6.2. Knockdown of HSPB8 causes abnormal F-actin accumulation at the ICB
Having shown that silencing of either BAG3 or HSPB8 causes similar cytokinetic defects,
and considering that HSPB8 levels depend on BAG3 (Fig 2.1A) (Carra et al. 2008),
subsequent analyses were performed in HSPB8-depleted cells. Besides, we found that
depletion of HSPB8 reduced BAG3 levels by ~ 50% to 75% in cells arrested at the G2/M
border and at the M-phase, respectively (Figure 2.2F, G2/M and M). In mitotic cells, BAG3
was mainly detected as a supershifted, hyperphophorylated protein band on SDS-PAGE as
previously shown (Figure 2.2F, red arrow) (Fuchs et al. 2015). This was contrasting with
the modest reduction of BAG3 levels seen upon silencing of HSPB8 in asynchronous cells
(~ 30%), as shown before (Figure 2.2F, AS) (Carra et al. 2008). Together with our previous
findings that HSPB8 facilitates BAG3 phosphorylation and localization at mitotic entry
(Fuchs et al. 2015), these results suggest that cell mitosis offers a unique model to uncover
the HSPB8-dependent functions of BAG3.

89
Figure 2.2: Silencing of HSPB8 interferes with cytokinetic abscission and markedly decreases the mitotic levels of BAG3. (A) Unsynchronized HeLa-GFP-H2B cells transfected with various HSPB8-specifique siRNA sequences were imaged for 72 h at 10 min intervals. The red arrowhead designates a persistent ICB connecting sister cells, comprises a dense midbody (emphasized in the enlarged view); bar 10 µm. The graph shows the percentage of cells remaining connected by an ICB; means +/- SE. *, p < 0.05 compares siHSPB8 #3 to siCtl by the Fisher exact test. (B) Time-sequences showing sister cells entering mitosis synchronously (red arrowheads); bar 20 µm; P: prophase; M: metaphase; T: telophase. (C) Graph depicting the percentages of cells undergoing synchronous mitosis; means +/- SE. ***, p < 0.001 compares siHSPB8 #3 to siCtl by the Fisher exact test. (D) Representative western blot of HeLa-GFP-H2B cells that have been lysed in SDS-sample buffer extracts, showing the efficiency of HSPB8 depletion, 88 h after transfection of the indicated siRNA sequences; calreticulin: loading control. (E) Confocal image of HeLa-GFP-H2B cells transfected with HSPB8-specific siRNAs, showing multi-nucleation; F-actin was stained with Texas-Red-Phalloidin; bar 30 µm. The graph shows

90
the incidence of bi- and multi-nucleation; means +/- SE from 3 independent experiments. ****, p < 0.0001 compares siHSPB8 #2 to siCtl, and **, p < 0.01 compares siHSPB8 #3 to siCtl by the Fisher exact test. (F) Representative western blot of HeLa cells extracts isolated from unsynchronized cells (As), or cells that have been synchronized at the G2/M border (G2/M), or M-phase (M), after transfection with HSPB8-specific siRNA or control siRNA (siCtl) for 88 h, showing the levels of BAG3, HSPB8 and GAPDH (loading control). Cells have been lysed by 3 cycles of freezing/thawing in 4 volumes of 0.1% IGEPAL lysis buffer.
Based on our previous work linking BAG3-HSPB8 to the remodelling of actin-based
structures at mitotic entry (Fuchs et al. 2015), we then sought to determine the impact of
HSPB8 on actin dynamics during cytokinesis. In animal cells, cytokinesis begins with the
assembly and contraction of an actomyosin ring that leads to the formation of an
intercellular bridge (von Dassow and Bement 2005). Final cleavage and fusion of this
intercellular bridge requires thinning of the ICB (secondary ingression) that critically relies
on actin depolymerization within the ICB (Schiel et al. 2013; Schiel et al. 2012). We
hypothesized that the chaperone complex might assist in accurate assembly and/or
disassembly of actin-based cytokinetic structures.
To test this idea, we took advantage of LifeAct-GFP — a probe that binds to F-actin (Riedl
et al. 2008), to visualize the dynamics of actin at the actomyosin ring by spinning confocal
microscopy. HeLa-RFP-H2B cells were adenofected with recombinant adenovirus driving
the expression of LifeAct-GFP together with control or HSPB8-specific siRNAs, and then
synchronized by a double thymidine block, using a method developed for knockdown-
rescue experiments (Figure 2.3A) (Fuchs et al. 2016). Live cell imaging revealed that
assembly and constriction of the actomyosin was not significantly perturbed by depletion
of HSPB8 (Figure 2.3C). Nonetheless, in HSPB8-depleted cells, the AR appeared
abnormally enriched in F-actin. This was revealed by an increase of AR thickness at
telophase (Figure 2.3C, supplemental movies S7-S8). Remarkably, higher proportions of
HSPB8-depleted cells (~ 43% +/- 2.5; 43% +/- 4.2; 58% +/-2.5) displayed abnormal F-
actin accumulation at the ICB marked by Aurora B (Murata-Hori et al. 2002) — a mitotic
kinase that relocates to the ICB at telophase, as compared to cells transfected with control
siRNAs (~ 24% +/- 1.3). Such disorganized actin structures persisted after cell spreading
and DNA de-condensation, and were observed using 3 different HSPB8-specific siRNA

91
sequences in depletion experiments. The data suggest that HSPB8-BAG3 chaperone
complex facilitates accurate AR dynamics to enable timely abscission of daughter cells.
Figure 2.3: Silencing of HSPB8 causes abnormal F-actin accumulation at the ICB. (A) Schematic of the protocol used for adenofection of HeLa-RFP-H2B cells with Ad-LifeAct-GFP together with HSPB8-specific siRNAs. (B) Western blot of HeLa-RFP H2B cell extracts prepared in SDS-sample buffer showing the efficiency of HSPB8 depletion 48 h after transfection of the indicated HSPB8-specific siRNAs; depletion was estimated at >

92
75% by loading increasing amounts of control extracts (1/4, ½, 1, siCtl); vinculin: loading control. (C) Live-cell imaging was performed for a 2 h-period at 5 min intervals by spinning disk confocal microscopy. Representative time-sequences show F-actin organization at the actin ring (pointed by white arrowheads) in cells transfected with control siRNAs (siCtl) compared to cells transfected with HSPB8-specfic siRNAs; bar, 10 µm. Quantification of the actin ring thickness over width; means +/- SE from 3 independent experiments. The Mann-Whitney’s test was performed relative to siCtl; ****, p < 0.0001. (D) Confocal images of HeLa-RFP-H2B cells that have been synchronized in mitosis after transfection with HSPB8-specific siRNAs, showing abnormal F-actin accumulation stained by Alexa-488-phalloidin at the ICB (green) marked by immunostaining of Aurora B (red). White arrowheads designate enlarged viewed that emphasize actin organization at the bridge of daughter cells; bar, 10 µm. The graph shows the percentages of cells with abnormal F-actin accumulation at the ICB in late cytokinesis, means +/- SE from 3 independent experiments. ****, p < 0.0001 compares HSPB8-specific siRNAs to siCtl by the Fischer exact test.
2.6.3. Down modulation of actin dynamics can normalize F-actin accumulation at
the ICB and correct cytokinetic defects caused by depletion of HSPB8
Abnormal accumulation of F-actin at the ICB suggested that HSPB8 depletion could
perturb actin dynamics, i.e. polymerization-depolymerization. To obtain evidence for a
causal relationship, we used the actin drug latrunculin A (LatA), which inhibits actin
dynamics by sequestering G-actin, hence limiting the actin pool available for F-actin
assembly (Coue et al. 1987; Morton et al. 2000). Cells were treated with a low
concentration (20 nM) during the last hour of the release period before cell fixation. Under
these conditions, the organization of F-actin in control cells was not noticeably affected.
Significantly, such treatment inhibited F-actin accumulation at the ICB in HSPB8-depleted
cells, bringing it in line with cells transfected with control siRNAs (Figure 2.4B, 4C). The
results suggest that HSPB8-depleted cells have deregulated actin polymerization at the ICB
that can be normalized by limiting the pool of G-actin.
We then took advantage of the hypersensibility of HSPB8-depleted cells to LatA to
examine the cause-effect relationship between deregulated actin dynamics and cytokinetic
failure, by measuring the occurrence of bi- and multi-nucleation. We determined that
incubation of HeLa-GFP-H2B cells in the presence of 1 nM LatA for a 48 h-period of time
modestly affected cell division and increased the incidence of nuclear abnormalities by
~1.8-fold (Figure 2.4D, siCtl LatA). However, this treatment reduced the incidence of bi-

93
and multi-nucleation in HSPB8-depleted cells to a level comparable to the one observed in
cells transfected with control siRNAs (Fg. 2.4D, siB8 LatA). Thus, the data suggest that
perturbation of actin dynamics upon depletion of HSPB8 contributes to its impact on cell
division.

94
Figure 2.4: Down modulation of actin dynamics can rescue phenotypes caused by depletion of HSPB8. (A) Schematic of the protocol used to modulate actin dynamic in HeLa-RFP-H2B cells synchronized in late mitosis that have been transfected with HSPB8-specific or control siRNAs. (B) Representative confocal images of fixed HSPB8-depleted HeLa-RFP-H2B cells, showing normalized F-actin at the ICB (emphasized in enlarged views pointed by white arrowheads), after treatment with latrunculin A (LatA, 20 nM); immunostaining of Autora B (red) was performed to detect ICB and F-actin was stained

95
with Alexa-488-phalloidin (green). Bar, 10 µm. (C) The graph depicts the percentages of cells with abnormal F-actin accumulation at the ICB; means +/- SE from 4 independent experiments. ****, p < 0.0001 compares siHSPB8 #3 treated with the vehicle (DMSO) to siCtl + DMSO by the Fisher exact test; n.s.: non significant compares siHSPB8 #3 + LatA to siCtl + LatA. (D) Unsynchronized HeLa-GFP-H2B that have been transfected with HSPB8-specific or control siRNAs (siB8 vs siCtl) were grown in the presence of 1 nM LatA for 48 h and the percentages of nuclear abnormalities were scored; means +/- SE from 3 independent experiments. ****, p < 0.0001 compares siHSPB8 #2 treated with the vehicle (DMSO) to siCtl + DMSO by the Fisher exact test; n.s.: non significant difference between siHSPB8 #3 + LatA and siCtl + LatA.
2.6.4. Inhibition of Arp2/3 complex can normalize F-actin at the ICB in HSPB8-
depleted cells
Accurate assembly and disassembly of the cortical AR during cytokinesis is proposed to
rely on the combined action of two F-actin nucleators: the formin Diaphanous (Dia) and
the Arp2/3 complex and require dynamic actin turnover at the equatorial AR (Bovellan et
al. 2014; Murthy and Wadsworth 2005). We noted that the BAG3 interactome from HeLa
cells contains proteins that could link BAG3 to Arp2/3 complex, notably ArpC2, a
component of Arp2/3 complex, and ELMO1, which provides a connection with upstream
regulators (Chen et al. 2013). To explore a potential relationship with the Arp2/3 complex,
we first analyzed its subcellular distribution during cytokinesis. Low amounts of a
fluorescent probe, GFP-Arp3, were expressed in HeLa cells for a short period of time after
silencing HSPB8 to avoid perturbing actin dynamics. The subcellular distribution of GFP-
Arp3 was analyzed by confocal microscopy after cell fixation (Figure 2.5A, experimental
scheme; Figure 2.5B). Cells transfected with control siRNAs undergoing cytokinesis
exhibited a rather diffuse localization of Arp3-GFP with no significant enrichment at the
ICB (Figure 2.5C). This was revealed by quantification of the GFP fluorescence ratio of
ICB over cytoplasm (Figure 2.5C, ~0.89). In marked contrast, in cells transfected with
HSPB8-specific siRNAs, GFP-Arp3 accumulated at the ICB and co-distributed with
aberrant F-actin structures, as revealed by a ~1.6-fold increase in the ICB/cytoplasmic GFP
fluorescence ratio (Figure 2.5C, arrowhead).

96
We then sought to acutely perturb Arp2/3 activity as cells entered cytokinesis and monitor
the impact on ICB-associated aberrant F-actin. To that end, HSPB8-depleted cells that have
been synchronized in mitosis-cytokinesis were incubated in the presence of the selective
Arp2/3 inhibitor CK666 (Nolen et al. 2009) or with the vehicle only, during the last hour
of the released period from the thymidine-block. Using this protocol, inhibition of the
Arp2/3 complex did not impact cytokinesis in cells treated with control siRNAs, consistent
with recent findings (Bovellan et al. 2014; Rosa et al. 2015). Nonetheless, such treatment
inhibited the occurrence of abnormal F-actin at the ICB in HSPB8-depleted cells (Figure
2.5D). These data suggest that by limiting Arp2/3-mediated actin polymerization, an
accurate balance between actin assembly and turnover at the ICB can be restored in
HSPB8-depleted cells.

97
Figure 2.5: Co-distribution of abnormal F-actin and 1 Arp2/3, and restoration of F-actin organization at the ICB in HSPB8-depleted cells upon inhibition of Arp2/3 complex. (A) Schematic of the protocol used to visualize Arp2/3 localization in Hela-RFP-H2B cells during cytokinesis. (B) Western blot of HeLa-RFP-H2B cell extracts prepared in SDS-sample buffer showing the levels of transfected GFP-Arp3 and endogenous Arp3; calreticulin levels are shown as loading control. (C) Representative confocal images of HSPB8-depleted HeLa-RFP-H2B cells, showing accumulation of GFP-Arp3 together with F-actin; staining of Aurora B (purple) was performed to localize ICBs that are emphasized

98
in enlarged views designated by white arrowheads; bar, 10 µm. Quantification of the fluorescence intensity ratios of GFP-Arp3 is shown on the right graph, means +/- SE of at least 98 cells from 4 independent experiments. The Mann- Whitney’s test was performed relative to control; ****, p < 0.0001. (D) Confocal images showing synchronized HeLa-RFP-H2B cells depleted of HSPB8 treated with the vehicle only (DMSO) or with a selective Arp2/3 inhibitor (CK666, 40 µM) 1 h before cell fixation. F-actin organization at the ICB (phalloidin staining: green, Aurora B staining: red) is emphasized in the enlarged views designated by white arrowheads; bar, 10 µm. A schematic of the protocol used is shown below. The graph depicts the percentages of cells with abnormal F-actin accumulation at the ICB; means +/- SE from 2 or 4 independent experiments; n.s.: non-significant difference between siHSPB8 #3 + CK666 versus siCtl + CK666 by the Fisher exact test.
2.6.5. Drugs that affect lysosomal function recapitulate the HSPB8-dependent
phenotype, whereas induction of autophagy signaling can restore the F-actin
defects caused by depletion of HSPB8
A specialized role for a multichaperone complex organized by BAG3 has been proposed
in the clearance of damaged proteins undergoing partial misfolding in response to
mechanical strain during skeletal muscle contraction (Arndt et al. 2010; Ulbricht et al.
2013). Given that the actin-based AR is also put under high tensile forces during
cytokinesis, we hypothesized that HSPB8-BAG3 could be mobilized to facilitate the
autophagic clearance of components of actin-based cytokinetic structures.
To explore this idea, we used two distinct experimental approaches. We first asked whether
impairing lysosomal function could interfere with AR dynamics. HeLa cells that have been
synchronized in mitosis by a double thymidine block were incubated in the presence of
drugs that are commonly used to disrupt the autophagic flux; these include bafilomycin A
(BafA), which inhibits V-ATPases and blocks autophagosome-lysosome fusion, or
pepstatin A and E64D, two inhibitors of lysosomal proteases (Klionsky et al. 2016). The
drugs were added during the last hour of the release period before cell fixation, and cells
in late cytokinesis were detected by immunostaining of the ICB using anti-Aurora B
antibody. Analysis of F-actin distribution revealed that a larger proportion of cells treated
with the drugs accumulated aberrant actin structures at the ICB that persist after cell
spreading and DNA de-condensation relative to cells treated with the vehicle only (~1.8-

99
fold increase; Figure 6A). Thus, the data suggest that inhibition of vacuolar acidification
or lysosomal enzymes, like depletion of HSPB8, can recapitulate similar defects in actin
dynamics at the ICB.
As a second approach, we submitted HSPB8-depleted cells that have been synchronized in
late mitosis to a 1 h-incubation period with rapamycin — a drug that inhibits mTORC1 and
stimulates autophagy signaling (Crespo and Hall 2002). Remarkably, such treatment
corrected F-actin accumulation at the ICB and brought the proportions of HSPB8- depleted
cells bearing F-actin defects in line with cells transfected with control siRNAs (Figure 6B).
Collectively, our results suggest that defects in the dynamics of the actin-based AR caused
by depletion of HSPB8 may result, at least in part, from its function in the segregation of
cytoskeletal protein clients to selective autophagy.
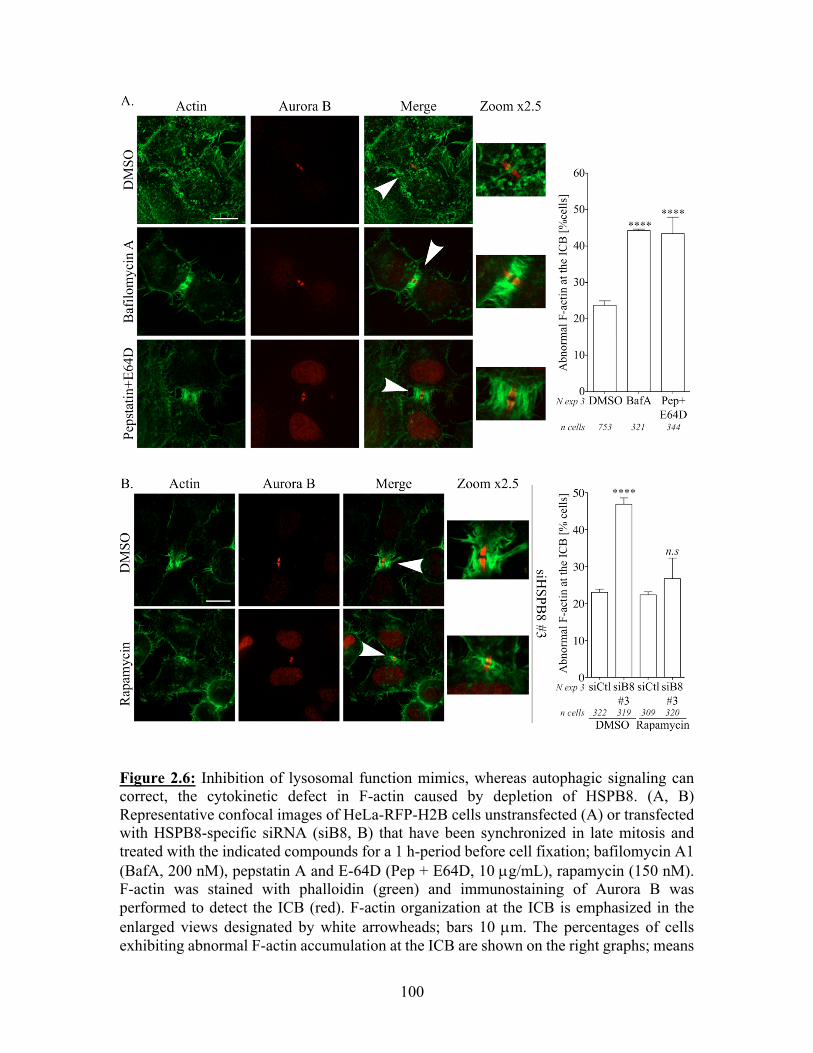
100
Figure 2.6: Inhibition of lysosomal function mimics, whereas autophagic signaling can correct, the cytokinetic defect in F-actin caused by depletion of HSPB8. (A, B) Representative confocal images of HeLa-RFP-H2B cells unstransfected (A) or transfected with HSPB8-specific siRNA (siB8, B) that have been synchronized in late mitosis and treated with the indicated compounds for a 1 h-period before cell fixation; bafilomycin A1 (BafA, 200 nM), pepstatin A and E-64D (Pep + E64D, 10 µg/mL), rapamycin (150 nM). F-actin was stained with phalloidin (green) and immunostaining of Aurora B was performed to detect the ICB (red). F-actin organization at the ICB is emphasized in the enlarged views designated by white arrowheads; bars 10 µm. The percentages of cells exhibiting abnormal F-actin accumulation at the ICB are shown on the right graphs; means

101
+/- SE 1 from 3 independent experiments. ****, p < 0.0001, compare BafA or Pepstatin + E64D to DMSO, or siB8 + DMSO to siCtl by the Fisher exact test; n.s.: non significant difference by comparing siB8 + rapamycin to siCtl + rapamycin.
2.7. Discussion
BAG3, in association with its chaperone partners, has been linked to the selection of
damaged proteins for autophagic degradation during proteotoxic stress and muscle cell
contraction (Minoia et al. 2014; Ulbricht et al. 2013). While BAG3 controls the stability of
HSPB8, it may also function without HSPB8 during stress, questioning the exact role of
HSPB8 (our unpublished data) (Ganassi et al. 2016; Minoia et al. 2014). Recently, we
uncovered an HSPB8-dependent function of BAG3 in a basic and fundamental activity of
cancer cells: mitosis, in the remodelling of actin-based structures that guide spindle
dynamics (Fuchs et al. 2015). In the current study, we present further evidence that in
response to mitotic signaling, HSPB8 and BAG3 act together to modulate actin dynamics
during cytokinesis and facilitate abscission of daughter cells. Because drugs that selectively
perturb actin polymerization can correct the multinucleation phenotype caused by
depletion of HSPB8, which also depletes the mitotic forms of BAG3, we suggest that an
HSPB8-BAG3 complex promotes accurate cell division mainly by limiting actin
polymerization and/or promoting actin turnover. Whether such process may involve
HSPB8-BAG3 function in autophagic sequestration is suggested by some results presented
here, and further discussed below.
Our finding that HSPB8 maintains BAG3 levels mainly during M-phase suggests that it
may serve to stabilize particular conformations of BAG3 induced by mitotic
phosphorylation. This could promote interactions between BAG3 and client proteins that
would connect the chaperone complex to actin-based structures, perhaps, via BAG3 Pro-
rich motifs, which we have identified as crucial determinants of BAG3 mitotic activity in
previous work (Fuchs et al. 2015). BAG3 was reported to interact with several proteins that
could modulate actin assembly or promote actin turnover; for instance CapZ — an actin-
capping protein that prevents filament growth (Cooper and Schafer 2000; Hishiya et al.
2010; Pollard and Cooper 2009), or non muscle myosin II — the molecular motor that

102
generates forces for constriction of the actin-based AR (Hong et al. 2016; Maupin and
Pollard 1986; Schiel et al. 2013). It has been shown that actin at the AR is highly dynamic
and myosin II was involved in the dynamic turnover of actin during cytokinesis (Murthy
and Wadsworth 2005). Given that BAG3 can interact with a plethora of potential client
proteins in HeLa cells (Chen et al. 2013), its mitotic activity likely involves complex
interactions with client proteins that could be regulated in time and space by HSPB8 and
multisite phosphorylation of BAG3.
The results presented here provide important mechanistic clue by showing that silencing
of HSPB8 causes accumulation of branched actin at the ICB. Assembly of branched actin
networks is mediated by the actin nucleator complex Arp2/3 that appeared to accumulate
at the ICB of HSPB8-depleted cells (Goley and Welch 2006). Furthermore, inhibition of
Arp2/3 using CK666 can prevent abnormal F-actin accumulation at the ICB in these cells,
suggesting that HSPB8-BAG3 act by limiting Arp2/3 activity. It has been proposed that a
switch from Arp2/3 to Diaphanous-mediated actin nucleation is required to drive assembly
of a rigid actin cortex in metaphase cells (Rosa et al. 2015). Too much Arp2/3-mediated
actin polymerization may also lead to cytokinesis failure (Chircop 2014). Indeed,
deregulation of the antagonism between the Rho GTPases Rho and Rac, which is believed
to control AR dynamics during cytokinesis, lead to defective cytokinesis and aberrant
distribution of F-actin (D'Avino et al. 2004). We have shown that depletion of HSPB8 or
BAG3 interferes with cell rounding and assembly of a mechanically rigid actin cortex at
mitotic entry, in addition to its perturbing effect during cytokinesis (Fuchs et al. 2015).
Based on the overall data, it can be argued that failure to accurately limit the activity of
Arp2/3 upon depletion of HSPB8 or BAG3 contributes to defects in mitotic cortical
rigidity, spindle positioning and cytokinesis, leading to impaired cell division and
multinucleation.
Our findings also suggest that a phosphorylation-activated function of HSPB8-BAG3
would promote actin turnover rather than enhancing actin polymerization, as it is the case
for HSPB1 (During et al. 2007; Lavoie et al. 1995). This is in line with the proposed role
of HSPB8-BAG3 within a pathway induced by mechanical cues for the clearance of

103
cytoskeletal protein in the muscle sarcomere — the actin contractile unit of myofibrils
(Ulbricht and Hohfeld 2013). Such pathway could be mobilized in dividing cells by mitotic
signaling, via BAG3 phosphorylation, to facilitate accurate turnover of non muscle
contractile structures that are submitted to high tensile forces. It has been hypothesized that
the organization of actin and myosin II in the cytokinetic AR is similar to that seen in
muscle sarcomere; recent evidence supports a sliding filament-based purse string
contraction mechanism for AR constriction (Fenix et al. 2016; Pollard and Wu 2010).
Although it has been established that the AR needs to be disassembled to allow final
abscission of daughter cells, the mechanisms remain poorly understood. A role for selective
autophagy has been proposed, in the localized inactivation of RhoA involving the
autophagic receptor p62/SQSTM1 (Belaid et al. 2014; Mizuno 2013; Piekny et al. 2005;
Theriot 1997). In further support of a role for autophagy, we observed here that treatment
of cells with commonly used inhibitory drugs recapitulated abnormal accumulation of F-
actin at the ICB, similar to the phenotype caused by depletion of HSPB8. Recently, we
have shown that HSPB8, BAG3 and p62/SQSTM1 form a mitotic complex and that
depletion of any of the proteins resulted in the same phenotypes in early mitosis (Fuchs et
al. 2015). Here, we provide further evidence for a connection between the activity of
HSPB8-BAG3 during cytokinesis and autophagy signaling by showing that treatment of
HSPB8-depleted cells with rapamycin could prevent F-actin accumulation. This is in line
with recent work suggesting that BAG3 modulates the spatial activity of mTORC1 — the
target of rapamycin and a key regulator of autophagy signaling, in addition to its direct
interaction with the selective autophagic machinery via p62/SQSTM1 (Gamerdinger et al.
2011; Kathage et al. 2016). Overall, though the exact mechanism whereby HSPB8 and
BAG3 promote disassembly of cytokinetic actin structures remain to be established, we
think that we can reasonably speculate that the chaperone complex could act in the
autophagic sequestration of proteins that regulate assembly of actin networks, a function
that would be modulated by BAG3 mitotic phosphorylation.
In conclusion, the present study further supports a specialized role for HSPB chaperones
as members of a quality control network that would assist in the assembly and disassembly
of macromolecular complexes controlling the dynamic architecture of actin during various

104
mechanical processes, including cell-shape changes that guide mitosis in cancer cells
(Guilbert et al. 2015). Deciphering the mitotic interactome of BAG3 and the molecular
impact of BAG3 multisite phosphorylation is crucial to our understanding of HSPB8-
BAG3 function in actin dynamics and is the object of our current work. Given that HSPB8
and BAG3 are expressed in a wide variety of cancer cell types, their activity in cell division
may sustain tumor expansion (Rosati et al. 2011). Thus, uncovering their mode of action
is of great interest for the development of targeted therapeutic approaches to inhibit the
HSPB8-dependent function of BAG3 and further enhance mitotic stress in cancer cells.

105
2.8. References
Arndt V, Dick N, Tawo R, Dreiseidler M, Wenzel D, Hesse M, Furst DO, Saftig
P, Saint R, Fleischmann BK, Hoch M, Hohfeld J (2010) Chaperone-
assisted selective autophagy is essential for muscle maintenance. Curr
Biol 20: 143-8. doi: 10.1016/j.cub.2009.11.022
Behl C (2016) Breaking BAG: The Co-Chaperone BAG3 in Health and Disease.
Trends Pharmacol Sci 37: 672-88. doi: 10.1016/j.tips.2016.04.007
Belaid A, Cerezo M, Chargui A, Corcelle-Termeau E, Pedeutour F, Giuliano
S, Ilie M, Rubera I, Tauc M, Barale S, Bertolotto C, Brest P, Vouret-
Craviari V, Klionsky DJ, Carle GF, Hofman P, Mograbi B (2013)
Autophagy plays a critical role in the degradation of active RHOA, the
control of cell cytokinesis, and genomic stability. Cancer Res 73: 4311-
22. doi: 10.1158/0008-5472.CAN-12-4142
Belaid A, Ndiaye PD, Cerezo M, Cailleteau L, Brest P, Klionsky DJ, Carle GF,
Hofman P, Mograbi B (2014) Autophagy and SQSTM1 on the RHOA(d)
again: emerging roles of autophagy in the degradation of signaling
proteins. Autophagy 10: 201-8. doi: 10.4161/auto.27198
Bovellan M, Romeo Y, Biro M, Boden A, Chugh P, Yonis A, Vaghela M,
Fritzsche M, Moulding D, Thorogate R, Jegou A, Thrasher AJ, Romet-
Lemonne G, Roux PP, Paluch EK, Charras G (2014) Cellular control of
cortical actin nucleation. Curr Biol 24: 1628-35. doi:
10.1016/j.cub.2014.05.069
Carra S, Seguin SJ, Lambert H, Landry J (2008) HspB8 chaperone activity
toward poly(Q)-containing proteins depends on its association with
Bag3, a stimulator of macroautophagy. J Biol Chem 283: 1437-44. doi:
10.1074/jbc.M706304200
Champagne C, Landry MC, Gingras MC, Lavoie JN (2004) Activation of
adenovirus type 2 early region 4 ORF4 cytoplasmic death function by
direct binding to Src kinase domain. J Biol Chem 279: 25905-15.

106
Chen Y, Yang LN, Cheng L, Tu S, Guo SJ, Le HY, Xiong Q, Mo R, Li CY,
Jeong JS, Jiang L, Blackshaw S, Bi LJ, Zhu H, Tao SC, Ge F (2013)
Bcl2-associated athanogene 3 interactome analysis reveals a new role in
modulating proteasome activity. Mol Cell Proteomics 12: 2804-19. doi:
10.1074/mcp.M112.025882
Chircop M (2014) Rho GTPases as regulators of mitosis and cytokinesis in
mammalian cells. Small GTPases 5. doi: 10.4161/sgtp.29770
Cooper JA, Schafer DA (2000) Control of actin assembly and disassembly at
filament ends. Curr Opin Cell Biol 12: 97-103.
Coue M, Brenner SL, Spector I, Korn ED (1987) Inhibition of actin
polymerization by latrunculin A. FEBS Lett 213: 316-8.
Crespo JL, Hall MN (2002) Elucidating TOR signaling and rapamycin action:
lessons from Saccharomyces cerevisiae. Microbiol Mol Biol Rev 66:
579-91, table of contents.
D'Avino PP, Savoian MS, Glover DM (2004) Mutations in sticky lead to
defective organization of the contractile ring during cytokinesis and are
enhanced by Rho and suppressed by Rac. J Cell Biol 166: 61-71. doi:
10.1083/jcb.200402157
During RL, Gibson BG, Li W, Bishai EA, Sidhu GS, Landry J, Southwick FS
(2007) Anthrax lethal toxin paralyzes actin-based motility by blocking
Hsp27 phosphorylation. Embo J 26: 2240-50. doi:
10.1038/sj.emboj.7601687
Fenix AM, Taneja N, Buttler CA, Lewis J, Van Engelenburg SB, Ohi R,
Burnette DT (2016) Expansion and concatenation of non-muscle myosin
IIA filaments drive cellular contractile system formation during
interphase and mitosis. Mol Biol Cell. doi: 10.1091/mbc.E15-10-0725
Finka A, Mattoo RU, Goloubinoff P (2016) Experimental Milestones in the
Discovery of Molecular Chaperones as Polypeptide Unfolding Enzymes.
Annu Rev Biochem 85: 715-42. doi: 10.1146/annurev-biochem-060815-
014124

107
Fuchs M, Boulanger MC, Lambert H, Landry J, Lavoie JN (2016)
Adenofection: A Method for Studying the Role of Molecular Chaperones
in Cellular Morphodynamics by Depletion-Rescue Experiments. J Vis
Exp. doi: 10.3791/54557
Fuchs M, Luthold C, Guilbert SM, Varlet AA, Lambert H, Jetté A, Elowe S,
Landry J, Lavoie JN (2015) A Role for the Chaperone Complex BAG3-
HSPB8 in Actin Dynamics, Spindle Orientation and Proper Chromosome
Segregation during Mitosis. PLoS Genetics, e1005582. doi: 10.1371 11.
Fuchs M, Poirier DJ, Seguin SJ, Lambert H, Carra S, Charette SJ, Landry J
(2010) Identification of the key structural motifs involved in
HspB8/HspB6-Bag3 interaction. Biochem J 425: 245-55. doi:
10.1042/BJ20090907
Gamerdinger M, Hajieva P, Kaya AM, Wolfrum U, Hartl FU, Behl C (2009)
Protein quality control during aging involves recruitment of the
macroautophagy pathway by BAG3. Embo J 28: 889-901.
Gamerdinger M, Kaya AM, Wolfrum U, Clement AM, Behl C (2011) BAG3
mediates chaperone-based aggresome-targeting and selective autophagy
of misfolded proteins. EMBO Rep 12: 149-56. doi:
10.1038/embor.2010.203
Ganassi M, Mateju D, Bigi I, Mediani L, Poser I, Lee HO, Seguin SJ, Morelli
FF, Vinet J, Leo G, Pansarasa O, Cereda C, Poletti A, Alberti S, Carra
S (2016) A Surveillance Function of the HSPB8-BAG3-HSP70
Chaperone Complex Ensures Stress Granule Integrity and Dynamism.
Mol Cell 63: 796-810. doi: 10.1016/j.molcel.2016.07.021
Goley ED, Welch MD (2006) The ARP2/3 complex: an actin nucleator comes
of age. Nat Rev Mol Cell Biol 7: 713-26. doi: 10.1038/nrm2026
Hishiya A, Kitazawa T, Takayama S (2010) BAG3 and Hsc70 interact with
actin capping protein CapZ to maintain myofibrillar integrity under
mechanical stress. Circ Res 107: 1220-31. doi:
10.1161/CIRCRESAHA.110.225649

108
Hong J, Park JS, Lee H, Jeong J, Hyeon Yun H, Yun Kim H, Ko YG, Lee JH
(2016) Myosin heavy chain is stabilized by BCL-2 interacting cell death
suppressor (BIS) in skeletal muscle. Exp Mol Med 48: e225. doi:
10.1038/emm.2016.2
Iwasaki M, Homma S, Hishiya A, Dolezal SJ, Reed JC, Takayama S (2007)
BAG3 regulates motility and adhesion of epithelial cancer cells. Cancer
Res 67: 10252-9. doi: 10.1158/0008-5472.CAN-07-0618
Iwasaki M, Tanaka R, Hishiya A, Homma S, Reed JC, Takayama S (2010)
BAG3 directly associates with guanine nucleotide exchange factor of
Rap1, PDZGEF2, and regulates cell adhesion. Biochem Biophys Res
Commun 400: 413-8. doi: 10.1016/j.bbrc.2010.08.092
Kassis JN, Guancial EA, Doong H, Virador V, Kohn EC (2006) CAIR-1/BAG-
3 modulates cell adhesion and migration by downregulating activity of
focal adhesion proteins. Exp Cell Res 312: 2962-71. doi:
10.1016/j.yexcr.2006.05.023
Kathage B, Gehlert S, Ulbricht A, Ludecke L, Tapia VE, Orfanos Z, Wenzel
D, Bloch W, Volkmer R, Fleischmann BK, Furst DO, Hohfeld J (2017)
The cochaperone BAG3 coordinates protein synthesis and autophagy
under mechanical strain through spatial regulation of mTORC1. Biochim
Biophys Acta 1864: 62-75. doi: 10.1016/j.bbamcr.2016.10.007
Klebig C, Korinth D, Meraldi P (2009) Bub1 regulates chromosome
segregation in a kinetochore-independent manner. J Cell Biol 185: 841-
58. doi: 10.1083/jcb.200902128
Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo
Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, Adhihetty PJ,
Adler SG, Agam G, Agarwal R, Aghi MK, Agnello M, Agostinis P,
Aguilar PV, Aguirre-Ghiso J, Airoldi EM, Ait-Si-Ali S, Akematsu T,
Akporiaye ET, Al-Rubeai M, Albaiceta GM, Albanese C, Albani D,
Albert ML, Aldudo J, Algul H, Alirezaei M, Alloza I, Almasan A,
Almonte-Beceril M, Alnemri ES, Alonso C, Altan-Bonnet N, Altieri DC,
Alvarez S, Alvarez-Erviti L, Alves S, Amadoro G, Amano A, Amantini

109
C, Ambrosio S, Amelio I, Amer AO, Amessou M, Amon A, An Z, Anania
FA, Andersen SU, Andley UP, Andreadi CK, Andrieu-Abadie N, Anel
A, Ann DK, Anoopkumar-Dukie S, Antonioli M, Aoki H, Apostolova N,
Aquila S, Aquilano K, Araki K, Arama E, Aranda A, Araya J, Arcaro A,
Arias E, Arimoto H, Ariosa AR, Armstrong JL, Arnould T, Arsov I,
Asanuma K, Askanas V, Asselin E, Atarashi R, Atherton SS, Atkin JD,
Attardi LD, Auberger P, Auburger G, Aurelian L, Autelli R, Avagliano
L, Avantaggiati ML, Avrahami L, Awale S, Azad N, Bachetti T, Backer
JM, Bae DH, Bae JS, Bae ON, Bae SH, Baehrecke EH, Baek SH,
Baghdiguian S, Bagniewska-Zadworna A, et al. (2016) Guidelines for
the use and interpretation of assays for monitoring autophagy (3rd
edition). Autophagy 12: 1-222. doi: 10.1080/15548627.2015.1100356
Lavoie JN, Champagne C, Gingras MC, Robert A (2000) Adenovirus E4 open
reading frame 4-induced apoptosis involves dysregulation of src family
kinases. J Cell Biol 150: 1037-56.
Lavoie JN, Lambert H, Hickey E, Weber LA, Landry J (1995) Modulation of
cellular thermoresistance and actin filament stability accompanies
phosphorylation-induced changes in the oligomeric structure of heat
shock protein 27. Mol Cell Biol 15: 505-16.
Maupin P, Pollard TD (1986) Arrangement of actin filaments and myosin-like
filaments in the contractile ring and of actin-like filaments in the mitotic
spindle of dividing HeLa cells. J Ultrastruct Mol Struct Res 94: 92-103.
Minoia M, Boncoraglio A, Vinet J, Morelli FF, Brunsting JF, Poletti A, Krom
S, Reits E, Kampinga HH, Carra S (2014) BAG3 induces the
sequestration of proteasomal clients into cytoplasmic puncta:
implications for a proteasome-to-autophagy switch. Autophagy 10:
1603-21. doi: 10.4161/auto.29409
Mizuno K (2013) Signaling mechanisms and functional roles of cofilin
phosphorylation and dephosphorylation. Cell Signal 25: 457-69. doi:
10.1016/j.cellsig.2012.11.001

110
Morton WM, Ayscough KR, McLaughlin PJ (2000) Latrunculin alters the
actin-monomer subunit interface to prevent polymerization. Nat Cell
Biol 2: 376-8. doi: 10.1038/35014075
Murata-Hori M, Tatsuka M, Wang YL (2002) Probing the dynamics and
functions of aurora B kinase in living cells during mitosis and
cytokinesis. Mol Biol Cell 13: 1099-108. doi: 10.1091/mbc.01-09-0467
Murthy K, Wadsworth P (2005) Myosin-II-dependent localization and
dynamics of F-actin during cytokinesis. Curr Biol 15: 724-31. doi:
10.1016/j.cub.2005.02.055
Nolen BJ, Tomasevic N, Russell A, Pierce DW, Jia Z, McCormick CD,
Hartman J, Sakowicz R, Pollard TD (2009) Characterization of two
classes of small molecule inhibitors of Arp2/3 complex. Nature 460:
1031-4. doi: 10.1038/nature08231
Piekny A, Werner M, Glotzer M (2005) Cytokinesis: welcome to the Rho zone.
Trends Cell Biol 15: 651-8. doi: 10.1016/j.tcb.2005.10.006
Pollard TD, Cooper JA (2009) Actin, a central player in cell shape and
movement. Science 326: 1208-12. doi: 10.1126/science.1175862
Pollard TD, Wu JQ (2010) Understanding cytokinesis: lessons from fission
yeast. Nat Rev Mol Cell Biol 11: 149-55. doi: 10.1038/nrm2834
Rauch JN, Tse E, Freilich R, Mok SA, Makley LN, Southworth DR, Gestwicki
JE (2017) BAG3 Is a Modular, Scaffolding Protein that physically Links
Heat Shock Protein 70 (Hsp70) to the Small Heat Shock Proteins. J Mol
Biol 429: 128-141. doi: 10.1016/j.jmb.2016.11.013
Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke
F, Jenne D, Holak TA, Werb Z, Sixt M, Wedlich-Soldner R (2008)
Lifeact: a versatile marker to visualize F-actin. Nat Methods 5: 605-7.
doi: 10.1038/nmeth.1220
Rosa A, Vlassaks E, Pichaud F, Baum B (2015) Ect2/Pbl acts via Rho and
polarity proteins to direct the assembly of an isotropic actomyosin
cortex upon mitotic entry. Dev Cell 32: 604-16. doi:
10.1016/j.devcel.2015.01.012

111
Rosati A, Graziano V, De Laurenzi V, Pascale M, Turco MC (2011) BAG3: a
multifaceted protein that regulates major cell pathways. Cell Death Dis
2: e141. doi: 10.1038/cddis.2011.24
Schiel JA, Childs C, Prekeris R (2013) Endocytic transport and cytokinesis:
from regulation of the cytoskeleton to midbody inheritance. Trends Cell
Biol 23: 319-27. doi: 10.1016/j.tcb.2013.02.003
Schiel JA, Simon GC, Zaharris C, Weisz J, Castle D, Wu CC, Prekeris R (2012)
FIP3-endosome-dependent formation of the secondary ingression
mediates ESCRT-III recruitment during cytokinesis. Nat Cell Biol 14:
1068-78. doi: 10.1038/ncb2577
Solenn M. Guilbert, Alice-Anaïs Varlet, Margit Fuchs, Herman Lambert, and
JL, Lavoie JN (2015) Regulation of Actin-Based Structure Dynamics by
HspB Proteins and Partners. . In: Hightower ERTaL (ed) The Big Book
of Small Heat Shock Proteins, vol ISBN: 978-3-319-16076-4. Springer
international Publishing AG
Taipale M, Tucker G, Peng J, Krykbaeva I, Lin ZY, Larsen B, Choi H, Berger
B, Gingras AC, Lindquist S (2014) A quantitative chaperone interaction
network reveals the architecture of cellular protein homeostasis
pathways. Cell 158: 434-48. doi: 10.1016/j.cell.2014.05.039
Theriot JA (1997) Accelerating on a treadmill: ADF/cofilin promotes rapid
actin filament turnover in the dynamic cytoskeleton. J Cell Biol 136:
1165-8.
Ulbricht A, Arndt V, Hohfeld J (2013) Chaperone-assisted proteostasis is
essential for mechanotransduction in mammalian cells. Commun Integr
Biol 6: e24925. doi: 10.4161/cib.24925
Ulbricht A, Hohfeld J (2013) Tension-induced autophagy: may the chaperone
be with you. Autophagy 9: 920-2. doi: 10.4161/auto.24213
von Dassow G, Bement WM (2005) A ring-like template for abscission. Dev
Cell 9: 578-80. doi: 10.1016/j.devcel.2005.10.003

112
Welch MD, Iwamatsu A, Mitchison TJ (1997) Actin polymerization is induced
by Arp2/3 protein complex at the surface of Listeria monocytogenes.
Nature 385: 265-9.

113
3. Chapitre 3 : Le complexe chaperon HSPB8-BAG3 facilite le remodelage des
structures mitotiques à base d’actine en modulant l’activité d’HDAC6
Nous avons établi que la mitose représente un modèle paradigme pour étudier les fonctions
de BAG3 qui dépendent d’HSPB8 dans le remodelage des structures à base d’actine (Fuchs
et al. 2015 ; Annexe 1). En effet, au cours de la mitose, nous avons montré que la
localisation, la phosphorylation et la stabilité du co-chaperon BAG3 dépendent de son
chaperon HSPB8 (Fuchs et al., 2015 ; Annexe 1 ; Chapitre 2). Lors de la cytokinèse, le
complexe HSPB8-BAG3 semble agir en partie en limitant la dynamique de l’actine
dépendante d’Arp2/3 (Chapitre 2). Nous avons également démontré qu’en mitose, BAG3
est associée préférentiellement à p62/SQSTM1 et que la déplétion de l’adaptateur
autophagique peut récapituler les mêmes phénotypes sur le remodelage des structures
d’actine, suggérant qu’HSPB8, BAG3 et p62/SQSTM1 agissent ensembles (Gamerdinger
et al. 2009; Fuchs et al. 2015); Annexe 1). En outre, HDAC6, un partenaire de
p62/SQSTM1, est également recrutée par le complexe mitotique (Fuchs et al. 2015 ;
Annexe1, Figure 6D). Il a été rapporté que p62/SQSTM1, en s’associant avec HDAC6,
régule négativement l’activité catalytique de la déacétylase pendant un stress protéotoxic,
et ce faisant, contrôle le processus d’assemblage de F-actine entourant l’agrésome (Yan et
al. 2013).
Sur la base de ces résultats, nous avons émis l'hypothèse qu’un complexe mitotique formé
de BAG3-HSPB8 et p62/SQSTM1 pourrait moduler l’activité mitotique d’HDAC6, ce qui
faciliterait le remodelage des structures à base d’actine pendant la mitose. Ceci implique
que l’activité d’HDAC6 soit modulée par des signaux mitotiques et qu’elle contribue
normalement à réguler l’assemblage/désassemblage des structures mitotiques à base
d’actine. HDAC6 a été impliquée en G2/M où elle est requise pour le démantèlement du
cil primaire préalablement à l’entrée des cellules en mitose (Ran et al. 2015). Notamment,
la surexpression d’HDAC6 stimule le désassemblage du cil primaire dans les cellules
synchronisées en G2/M alors que sa déplétion interfère avec ce processus. Cependant, à
notre connaissance, une étude systématique de son activité à l’entrée des cellules en mitose
n’a pas été réalisée jusqu’à maintenant. Nous avons donc étudié la relation entre

114
l'expression de BAG3, HSPB8 et p62/SQSTM1 et l'activité d’HDAC6 sur le remodelage
des structures mitotiques à base d'actine.
Dans ce chapitre, nous présenterons des résultats suggérant que le complexe chaperon
HSPB8-BAG3-p62/SQSTM1 limite l’activité d’Arp2/3 dans une structure hautement
dynamique d’actine qui dirige le positionnement du fuseau mitotique : le nuage sous-
cortical d’actine. Des résultats indiquant que l’activité d’HDAC6 est requise pour la
formation et la dynamique du nuage sous-cortical d’actine seront ensuite montrés. Nous
montrerons aussi que les défauts de dynamique du nuage sous-cortical d’actine et
d’arrondissement mitotique peuvent être corrigés par l’inhibition d’HDAC6. Nous
présenterons ensuite des résultats qui supportent une interaction fonctionnelle entre le
complexe chaperon, HDAC6 et la cortactine, laquelle pourrait limiter l’activité spatiale de
la cortactine sur la polymérisation de l’actine dépendante d’Arp2/3, en inhibant la
déacétylation de la cortactine.
J’ai réalisé la majorité des expériences qui sont présentées dans ce chapitre, en
collaboration avec Carole Luthold. Ces travaux feront l’objet d’une publication ou nous
serons co-premières auteurs. Spécifiquement, les résultats présentés dans les figures 3.2,
3.4, S3.1 ont été obtenus par cette collaboration. Les données présentées dans la figure 3.1
ont été obtenues par Carole Luthold et celles présentées dans la figure 3.3A et 3.3C, par
Herman Lambert. J’ai réalisé les expériences présentées dans la figure 3.3B.
3.1. La déplétion de l’une ou l’autre des protéines du complexe mitotique HSPB8-
BAG3-p62/SQSTM1 interfère avec la dynamique du nuage sous-cortical d’actine
qui dépend du complexe Arp2/3
Un contrôle serré du positionnement du fuseau mitotique parallèle au substrat dépend de
structures hautement dynamiques d’actine : les fibres de rétraction (Mitchison 1992; Fink
et al. 2011). Celles-ci pemettraient des connections physiques entre la matrice
extracellulaire et le fuseau mitotique, assurant ainsi la transmission des signaux de tension
provenant de la matrice aux pôles du fuseau mitotique. Notamment, il a été montré que les

115
forces émises par les fibres de rétraction influencent la dynamique du nuage sous-cortical
d’actine en mitose (Fink et al. 2011). Le nuage sous-cortical d’actine est une structure
hautement dynamique soumise à un renouvellement rapide par
polymérisation/dépolymérisation résultant en un mouvement circulaire (Chapitre 1, Figure
1.14). La formation et le positionnement du nuage sous-cortical d’actine qui dépendent de
l’activité d’Arp2/3, produirait des forces de traction sur le fuseau mitotique et faciliterait
son positionnement en fonction des forces transmises par les fibres de rétraction
(Mitsushima et al. 2010; Fink et al. 2011). Nous avons montré dans notre première étude
que la déplétion de BAG3, qui organise le complexe mitotique HSPB8-BAG3-
p62/SQSTM1, interfère avec l’organisation des fibres de rétraction et conduit à des
oscillations anormales du fuseau mitotique ce qui perturbe la progression mitotique (Fuchs
et al. 2015; Annexe 1). En outre, les travaux présentés dans le chapitre 2 suggèrent
qu’HSPB8-BAG3 facilite le remodelage de l’actine en partie en limitant l’activité d’Arp2/3
(Chapitre 2, Figure 2.5). Ainsi, nous avons émis l’hypothèse que le complexe chaperon
HSPB8-BAG3-p62/SQSTM1 pourrait réguler la dynamique d’actine associée à la
polarisation du nuage sous-cortical d’actine, en limitant l’activité d’Arp2/3.
Pour tester notre hypothèse, nous avons réalisé des expériences d’imagerie en temps réel
dans des cellules HeLa synchronisées en mitose, déplétées en BAG3 et exprimant la sonde
LifeAct-GFP spécifique de la F-actine. Notez que les niveaux de déplétion protéique
obtenus avec les différents siARN utilisés dans cette étude sont présentés dans la figure
supplémentaire S3.1. En accord avec les travaux précédemment publiés, dans les cellules
contrôles, nous avons observé la présence d’un regroupement de F-actine situé sous le
cortex cellulaire et dont la dynamique au cours du temps suit un mouvement circulaire, i.e.
polarisé (Figure 3.1A, flèches blanches) (Mitsushima et al. 2010). En effet, il a été décrit
que la dynamique du nuage suit une rotation constante qui est due à la dynamique spatiale
de polymérisation/dépolymérisation qui semble déplacer le nuage de façon circulaire. La
structure sous-corticale dynamique de F-actine a été retrouvée dans ~60 % des cellules
contrôles, dont une majorité ~42 % présentait un mouvement polarisé (Figure 3.1B, siCtl,
DMSO). Pour déterminer si cette structure d’actine correspond au nuage sous-cortical
d’actine décrit par Mitsushima et al., nous avons traité les cellules avec le CK666, un

116
inhibiteur d’Arp2/3, 30 min avant l’acquisition en temps réel (Nolen et al. 2009;
Mitsushima et al. 2010). Le traitement des cellules contrôles avec le CK666 a conduit à
une augmentation de 1,5 fois du pourcentage de cellules n’arborant plus de structure sous-
corticale dynamique de F-actine (>60 % ; Figure 3.1B). Ce résultat montre que la formation
de cette structure peut être inhibée rapidement suivant une réduction de l’activité d’Arp2/3,
en accord avec les travaux de Mitsuchima et al. (Mitsushima et al. 2010).
Figure 3.1 : La déplétion de BAG3, HSPB8 ou p62/SQSTM1 perturbe le nuage sous-cortical d’actine qui dépend d’Arp2/3. (A) Séquences temporelles d’images confocales de cellules HeLa-RFP-H2B traitées avec des siARN et exprimant la sonde LifeAct-GFP, montrant un mouvement polarisé (siCtl, panel du haut) comparé à un mouvement désordonné et erratique (siBAG3_3, panel du bas) du nuage sous-cortical d’actine. Les cellules ont été synchronisées en mitose et l’acquisition s’est faite sur 75 min à un intervalle de 90 secondes. Les flèches blanches indiquent le nuage sous-corticale d'actine et les flèches jaunes son mouvement ; barres d'échelle, 10 µm. (B) Le graphique décrit le pourcentage de cellules présentant un mouvement polarisé ou erratique du nuage sous-cortical d’actine ou ne présentant pas de nuage sous-cortical d’actine (absent). Des cellules HeLa-RFP-H2B ont été synchronisées

117
en mitose, infectées avec l’Ad-Life-Act-GFP (1PFU) et le bacculovirus α-tubuline-RFP (4PFU) et transfectées avec un siCtl ou un siBAG3 (siBAG3_3). Trente minutes avant l’acquisition, les cellules ont été traitées avec du CK666 (40 µM) et les images ont été acquises par microscopie confocale de type « spinning disc » pendant une période de 75 minutes à des intervalles de 90 secondes. Les résultats +/- SEM sont représentatifs de 3 expériences indépendantes où 113 à 146 cellules ont été quantifiées. L'analyse statistique a été effectuée par le test de Fisher et compare les conditions indiquées à la condition siCtl DMSO. (C) Reconstitution 3D d’images confocales déconvoluées montrant des cellules HeLa-RFP-H2B en métaphase présentant une augmentation de la densité et une réorganisation de la F-actine en réponse à la déplétion de BAG3 (siBAG3_1) en comparaison aux cellules transfectées avec le siCtl. Des cellules HeLa-RFP-H2B ont été synchronisées en mitose et transfectées avec un siARN dirigé contre BAG3 (siBAG3_1) ou contrôle (siCtl). L’immunofluorescence a été réalisée avec un anti-a-tubuline pour marquer le fuseau mitotique et la phalloïdoine-488 pour marquer la F-actine ; barre d’échelle, 10 µm. (D) Graphique montrant le pourcentage de cellules présentant une mitose défectueuse (oscillation du fuseau mitotique et retard de la progression des cellules en prométa- et métaphase). Le protocole décrit en B a été employé pour l’obtention des résultats +/- SEM représentés provenant de 3 expériences indépendantes où ou au moins 112 cellules ont été quantifiées. L'analyse statistique a été effectuée par le test de Fisher et compare les conditions indiquées à la condition siCtl DMSO. (E) Histogramme représentant le pourcentage de cellules arborant un mouvement erratique du nuage sous-cortical d’actine. Des images de cellules HeLa-RFP-H2B synchronisées en mitose, déplétées en BAG3, HSPB8 ou p62/SQSTM1 et exprimant les sondes LifeAct-GFP et a-tubuline-RFP ont été acquises en temps réel par microscopie confocale de type « spinning disc » durant une période de 75 min à un intervalle de 90 secondes. Les résultats +/-SEM proviennent de 3 expériences indépendantes où au minimum 111 cellules ont été analysées. L'analyse statistique a été effectuée par le test de Fisher et compare les conditions indiquées à la condition siCtl. (F) Graphique montrant le pourcentage de cellules avec une mitose défectueuse (oscillation du fuseau mitotique et retard de la progression des cellules en prométa- et métaphase). Les résultats +/-SEM proviennent de 3 expériences indépendantes où au minimum 93 cellules ont été quantifiées. L'analyse statistique a été effectuée par le test de Fisher et compare les conditions indiquées à la condition siCtl.
Suivant la déplétion de BAG3, nous avons d’abord noté que le nuage sous-cortical d’actine
était observable dans une plus grande proportion de cellules ~90 % (Figure 3.1B). De façon
remarquable, plus de 50 % de ces cellules montraient un nuage d’actine « non-polarisé »
et « erratique » (Figure 3.1B, siBAG3_3, DMSO). En effet, tel que montré dans la figure
3.1A, le mouvement du nuage sous-cortical d’actine ne suivait pas une rotation soutenue
et montrait des changements désordonnés d’orientation, suggérant une dérégulation de la
dynamique de polymérisation/dépolymérisation de l’actine. L’augmentation de la présence
de la structure sous-corticale dynamique de F-actine dans les cellules déplétées en BAG3
pourrait donc refléter une dérégulation de la dynamique de l’actine qui se traduirait par des

118
mouvements anarchiques de celle-ci. A l’appui de cette hypothèse, le traitement avec le
CK666 des cellules déplétées en BAG3, a provoqué une augmentation du pourcentage de
cellules n’arborant aucun nuage sous-cortical d’actine (passage de ~15 % à ~32%),
corroborant ainsi la dépendance à Arp2/3. Cependant et de façon remarquable, nous avons
vu que pour les ~70 % des cellules déplétées en BAG3 et traitées avec le CK666 présentant
toujours un nuage sous-cortical d’actine, seulement ~25 % d’entre elles présentaient un
mouvement erratique du nuage sous-cortical d’actine comparé à plus de 50 % pour les
cellules traitées avec le contrôle DMSO (Figure 3.1B). Ainsi, le traitement avec le CK666
des cellules déplétées en BAG3 conduit à une réduction significative de la proportion de
cellules arborant un mouvement erratique du nuage sous-cortical d’actine se rapprochant
de la condition contrôle. Ces résultats indiquent qu’en réduisant l’activité Arp2/3 à l’aide
de l’inhibiteur CK666, on normalise la dynamique du nuage sous-cortical d’actine qui est
altérée en réponse à la déplétion de BAG3. Nos résultats suggèrent que les mouvements
erratiques du nuage sous-cortical d’actine seraient la conséquence d’une polymérisation
excessive de l’actine branchée induite par Arp2/3. Il semble donc que le co-chaperon
BAG3 pourrait réguler la dynamique d’actine au sein du nuage sous-cortical d’actine en
limitant l’activité d’Arp2/3, en lien avec les résultats présentés dans le chapitre 2 suggérant
que le complexe chaperon HSPB8-BAG3 limite l’activité d’Arp2/3 lors de la cytokinèse.
En outre, une accumulation ectopique de F-actine autour du fuseau mitotique a été
corroborée en marquant les structures d’actine endogènes à l’aide de la phalloïdine. Nous
avons noté que dans les cellules contrôles, la F-actine s’organise en de petits filaments fins
à l’intérieur du cytoplasme entourant le fuseau mitotique (Figure 3.1C). En revanche, dans
les cellules déplétées en BAG3, nous avons observé une élévation importante de l’intensité
du marquage de la phalloïdine montrant une augmentation de la densité de la F-actine dans
ces cellules (Figure 3.1C). De plus, nous avons vu que les filaments d’actine sont davantage
épais et nombreux en comparaison aux cellules contrôles et s’organisent en une structure
ectopique très dense apparaissant comme une cage entourant le fuseau mitotique que nous
appellerons dans les sections suivantes cage de F-actine (Figure 3.1C). Il est à noter ici que
le même genre de structure ectopique a été observé en réponse à la déplétion d’HSPB8
(Figure 3.2B). Ce résultat suggère que la polymérisation de l’actine se fait en excès dans

119
les cellules déplétées en HSPB8 et en BAG3. Il serait intéressant d’analyser si cette
structure ectopique d’actine endogène est la conséquence d’un excès de polymérisation
d’actine branchée. Pour cela, nous pourrions traiter des cellules déplétées en BAG3 ou en
HSPB8 avec du CK666. Une normalisation de la densité de F-actine dans la cage de F-
actine suggèrerait que cette structure est la conséquence d’une activation excessive
d’Arp2/3 comme cela est suggéré pour la dynamique du nuage sous-cortical d’actine.
Puisqu’il a été montré que le nuage sous-cortical d’actine dirige le positionnement du
fuseau mitotique et que la dynamique de celui-ci est dérégulée dans les cellules déplétées
en BAG3, nous avons souhaité savoir si cet effet contribue aux défauts de positionnement
du fuseau. Pour ce faire, nous avons analysé ce processus par imagerie en temps réel dans
des cellules HeLa synchronisées en mitoses déplétées en BAG3 et exprimant l’a-tubuline-
RFP ; 30 min avant l’acquisition en temps réel les cellules ont été traitées avec le CK666
afin de réduire l’activité du complexe Arp2/3. Dans les cellules traitées avec le contrôle
DMSO, nous avons observé que ~54 % des cellules déplétées en BAG3 présentent une
mitose défectueuse, caractérisée par des anomalies de positionnement du fuseau mitotique
et/ou des délais de la progression des cellules en mitose, contre seulement ~29 % des
cellules contrôles (Figure 3.1D), ce qui est consistent avec nos travaux précédents (Fuchs
et al. 2015 ; Annexe 1).
Suite au traitement avec le CK666, nous avons constaté une augmentation de 2 fois de la
proportion de cellules présentant une mitose défectueuse dans les cellules contrôles (Figure
3.1D). Cette observation montre que la disparition du nuage sous-cortical d’actine suite au
traitement avec le CK666 (Figure 3.1B) corrèle avec une augmentation de défauts
mitotiques dont des défauts de positionnement du fuseau. Ceci est en accord avec les
données de la littérature qui suggèrent que le nuage sous-cortical d’actine dirige le
positionnement du fuseau mitotique (Mitsushima et al. 2010). À l’inverse, les cellules
déplétées en BAG3 présentent une réduction de 1,5 fois de la proportion de cellules
présentant une mitose défectueuse en réponse au CK666 (Figure 3.1D). Ce résultat montre
qu’en réduisant l’activité Arp2/3, on peut normaliser les défauts de positionnement du
fuseau mitotique et les retards dans la progression mitotique conséquents à la déplétion de
BAG3. Ce résultat suggère que de tels défauts sont, au moins en partie, la conséquence

120
d’une dérégulation de l’activité d’Arp2/3, liés à la dynamique défectueuse du nuage sous-
cortical d’actine observée dans les cellules déplétées en BAG3.
Nous avons montré que la fonction de BAG3 en mitose dépend de son association avec
HSPB8 et pourrait impliquer l’adaptateur autophagique p62/SQSTM1 (Fuchs et al. 2015 ;
Annexe 1). Notamment, la déplétion de ces deux protéines conduit aux mêmes phénotypes
mitotiques que celle de BAG3. Nous avons donc voulu savoir si la fonction de BAG3 dans
la modulation de la dynamique du nuage sous-cortical d’actine implique ses partenaires
mitotiques p62/SQSTM1 et HSPB8. Pour cela, nous avons effectué des expériences
d’imagerie en temps réel de cellules HeLa synchronisées en mitose, déplétées en BAG3,
en HSPB8 ou en p62/SQSTM1 et exprimant les sondes LifeAct-GFP et a-tubuline-RFP
(Figure supplémentaire S3.1). Comme nous l’avons montré précédemment, la déplétion
d’une de ces trois protéines se traduit par une élévation de défauts mitotiques (i.e.
oscillation du fuseau mitotique et/ou retard dans la progression mitotique) montrant que
BAG3, HSPB8 et p62/SQSTM1 facilitent la progression mitotique (Figure 3.1F ; Fuchs et
al. 2015 ; Annexe 1). De façon remarquable, nous avons noté que la déplétion d’HSPB8
ou de p62/SQSTM conduit à une augmentation de presque 3 fois de la proportion de
cellules arborant un mouvement erratique du nuage sous-cortical d’actine en comparaison
aux cellules contrôles (Figure 3.1E). Ces observations montrent que comme la déplétion
de BAG3, la déplétion d’HSPB8 ou de p62/SQSTM1 interfère avec le mouvement polarisé
du nuage sous-cortical d’actine.
En conclusion, nos résultats sont en accord avec les résultats présentés dans le chapitre 2
qui suggèrent que le complexe chaperon HSPB8-BAG3 facilite le remodelage de l’actine
en limitant l’activité d’Arp2/3 (Chapitre 2, Figure 2.5). En effet, nos travaux montrent que
le co-chaperon BAG3 facilite la polarisation du nuage sous-cortical d’actine la seule
structure mitotique décrite dans la littérature comme étant dépendante du complexe Arp2/3
(Mitsushima et al. 2010). Nous concluons que le complexe chaperon HSPB8-BAG3-
p62/SQSTM1 pourrait faciliter le positionnement du fuseau mitotique à travers la
limitation de l’activité d’Arp2/3 requise pour la polarisation du nuage sous-cortical
d’actine.

121
Afin d’adresser le mécanisme impliqué nous nous sommes tournés vers l’étude de la
contribution potentielle d’HDAC6, un partenaire du complexe chaperon HSPB8-BAG3 en
mitose.
3.2. De faibles doses d’un inhibiteur d’HDAC6 peut normaliser la dynamique du nuage
sous-cortical d'actine et l'arrondissement mitotique dans les cellules déplétées en
BAG3, HSPB8 ou p62/SQSTM1
HDAC6 est une déacétylase unique qui présente des fonctions importantes dans le contrôle
de la migration cellulaire (Valenzuela-Fernández et al. 2008; Zhang et al. 2014; Kanno et
al. 2012; Zuo et al. 2012). Cette fonction implique notamment la régulation de la
dynamique de l'actine à travers la modulation de l'acétylation de la cortactine, une protéine
liant l'actine et activant le complexe Arp2/3 (Zhang et al. 2007; Uruno et al. 2001; Weaver
et al. 2001). De plus, il a été montré dans le contexte de la réponse à l’agrésome que
p62/SQSTM1 agit comme un inhibiteur de l’activité d’HDAC6 en interagissant avec la
déacétylase (Yan et al. 2013). Ainsi, nous avons postulé que le complexe chaperon HSPB8-
BAG3 par son association avec p62/SQSTM1 et HDAC6, pourrait limiter l’activité
d’Arp2/3 en partie, en limitant la déacétylation de la cortactine.
Nos résultats préliminaires ont indiqué que la déplétion d’HDAC6 perturbe
significativement l’entrée des cellules en mitose, rendant irréalisable cette approche
(résultats non montrés). En conséquence, nous avons tiré avantage de la tubacine, une
drogue qui est hautement sélective pour HDAC6, pour inhiber l’activité de la déacétylase
sur de courts temps et évaluer son effet sur la dynamique du nuage sous-cortical d’actine
(Haggarty et al. 2003; Schölz et al. 2015). Lorsque les cellules ont été traitées avec 5 µM
de tubacine, une dose qui inhibe l’activité d’HDAC6, pendant la seconde relâche d’un
double blocage à la thymidine sur une période de 8 h, la formation du nuage sous-cortical
d’actine a été inhibée dans plus de 70 % des cellules contrôles, comme dans les cellules
déplétées en BAG3 (Figure 3.2A ; Haggarty et al. 2003). Ainsi, ce résultat indique que
l’inhibition d’HDAC6 même sur de courts temps inhibe la formation du nuage sous-

122
cortical d’actine. En outre, ce traitement a causé la disparition des structures ectopiques
d’actine marquées par la phalloïdine dans les cellules mitotiques déplétées en HSPB8 ou
en BAG3 (Figure 3.2B). Ces résultats suggèrent fortement qu’HDAC6 est essentielle pour
la formation du nuage sous-cortical d’actine et que son activité est dérégulée suivant la
déplétion du complexe chaperon, causant une accumulation de F-actine.
Figure 3.2 : L’activité d’HDAC6 régule la dynamique du nuage sous-cortical d’actine : la réduction de l’activité de HDAC6 par de faibles doses de tubacine peut normaliser les défauts mitotiques associés à la déplétion de BAG3, HSPB8 ou p62/SQSTM1. (A) Histogramme représentant le pourcentage de cellules arborant un mouvement polarisé ou erratique du nuage sous-cortical d’actine ou ne présentant pas de nuage sous-cortical d’actine (absent) en réponse au traitement avec la tubacine (tub) ou le contrôle DMSO. Des cellules HeLa-RFP-H2B ont été synchronisées en mitose par un double blocage à la thymidine, infectées avec l’Ad-Life-Act-GFP (1PFU) et le bacculovirus α-tubuline-RFP (4PFU) et transfectées avec un siCtl ou un siBAG3 (siBAG3_3). Durant les 8 h de la seconde relâche les cellules ont été traitées avec la tubacine (5 µM) et les images ont été acquises par microscopie confocale de type « spinning disc » pendant une période de 75 minutes à des intervalles de 90 secondes. Les résultats sont représentatifs d’une expérience.

123
(B) Reconstitution 3D d’images confocales déconvoluées montrant des cellules HeLa-RFP-H2B en métaphase déplétées en HSPB8 présentant une normalisation de la densité de la F-actine en réponse au traitement avec la tubacine. L’immunofluorescence a été réalisée avec un anti-a-tubuline pour marquer le fuseau mitotique et la phalloïdoine-488 pour marquer la F-actine ; barre d’échelle, 10 µm. (C) Graphique représentant (gauche) le pourcentage de cellules arborant un mouvement polarisé ou erratique du nuage sous-cortical d’actine ou ne présentant pas de nuage sous-cortical d’actine (absent) et (droite) le pourcentage de cellules avec une mitose défectueuse, en réponse au traitement avec la tubacine (tub) ou le contrôle DMSO. Des cellules HeLa-RFP-H2B ont été synchronisées en mitose par un double blocage à la thymidine, infectées avec l’Ad-Life-Act-GFP (1PFU) et le bacculovirus α-tubuline-RFP (4PFU) et transfectées avec un siCtl ou un siBAG3 (siBAG3_3). 30 minutes avant l’acquisition en temps réel les cellules ont été traitées avec la tubacine (20 nM) et les images ont été acquises par microscopie confocale de type « spinning disc » pendant une période de 75 minutes à des intervalles de 90 secondes. Les résultats sont représentatifs d’une expérience. Les résultats +/- SEM sont représentatifs de 2 ou 3 expériences indépendantes où au moins 69 cellules ont été quantifiées. L'analyse statistique a été effectuée par le test de Fisher et compare les conditions indiquées à la condition siCtl DMSO (D) Images d'épifluorescence déconvoluées montrant une cellule MCF7 mitotique ronde typique (siCtl) par rapport à une cellule mitotique plate défectueuse (siBAG3_1); la F-actine a été marquée par la phalloïdine-488, le fuseau mitotique par l'anti-a-tubuline et l'ADN par le Hoechst; barres d'échelle, 10 µm. (F) Histogramme représentant le pourcentage de cellules HeLa-RFP-H2B et MCF7 avec des défauts d'arrondissement mitotique (cellules partiellement ou non arrondies) après 48 h de déplétion de BAG3 ou d’HSPB8 (gauche) avec les siBAG3_1, siBAG3_2 et siHSPB8_2 ou 24 h de déplétion d’HSPB8 et de p62/SQSTM1 (droite) avec les sip62_1, sip62_2 et siHSPB8_1. Les résultats +/- SEM sont représentatifs de 3 expériences indépendantes où 605 à 846 cellules ont été analysées. L'analyse statistique a été effectuée par le test de Fisher et compare les conditions indiquées à la condition siCtl DMSO.
En outre, nos observations nous ont indiqué que le nuage sous-cortical d’actine est très
sensible à des variations de l’activité d’HDAC6. Nous avons donc effectué des essais de
dose/réponse pour déterminer une dose ayant peu d’impact sur la formation du nuage sous-
cortical d’actine dans les cellules contrôles (résultat non montrés). Ainsi, nous avons établi
qu’un traitement avec 20 nM de tubacine 30 minutes avant l’acquisition d’images en temps
réel permet de préserver le nuage sous-cortical d’actine dans les cellules HeLa exprimant
les sondes LifeAct-GFP et a-tubuline-RFP synchronisées en mitose ; cependant, nous
avons observé une augmentation de la proportion de cellules montrant un mouvement
désordonné du nuage sous-cortical d’actine (Figure 3.2C, erratique).

124
Lorsqu’un même traitement a été appliqué aux cellules déplétées en BAG3, une réduction
de ~2 fois du pourcentage de cellules présentant un mouvement erratique du nuage sous-
cortical d’actine a été mesurée (Figure 3.2C). De plus, ce traitement a réduit la proportion
de cellules présentant des défauts mitotiques, notamment de positionnement du fuseau
mitotique dans les cellules déplététes en BAG3, alors qu’il a augmenté ces défauts
mitotiques dans les cellules contrôles (Figure 3.2C). Ces résultats indiquent qu’une
réduction de l’activité d’HDAC6 a un effet opposé dans les cellules contrôles versus les
cellules déplétées en BAG3, suggérant qu’un contrôle spatial fin de son activité est requis
pour favoriser la dynamique polarisée du nuage sous-cortical d’actine dirigée par la
polymérisation/dépolymérisation de l’actine branchée. Par conséquent, ce résultat suggère
que les mouvements erratiques du nuage sous-cortical d’actine sont en partie la
conséquence d’une dérégulation de l’activité d’HDAC6 suivant la déplétion de BAG3.
Nous avons montré que le complexe chaperon influence l’arrondissement mitotique (Fuchs
et al. 2015 ; Annexe 1, Figure 7A et B). Ce processus implique notamment une transition
dans l’activité des nucléateurs de l’actine, passant d’Arp2/3 à la formine mDia1 dont
l’activité est requise pour la formation du cortex mitotique (Rosa et al. 2015). Ainsi, il est
possible qu’un dérèglement de l’activité mitotique du complexe Arp2/3 interfère avec
l’arrondissement mitotique. Afin de déterminer si l’action du complexe chaperon sur
l’activité HDAC6 et Arp2/3 contribue à son effet sur l’arrondissement mitotique, les
cellules HeLa ou les cellules MCF7 ont été synchronisées en mitose et déplétées en BAG3,
HSPB8 ou p62/SQSTM1, puis traitées avec 5 µM de tubacine durant 8 h. Pour consolider
nos observations, nous avons utilisé deux séquences de siARN différentes pour chacune
des protéines ciblées dans les cellules HeLa ou MCF7 (Figure S3.1). Par
immunofluorescence, nous avons ensuite examiné la proportion de cellules arrondies en
mitose versus la proportion de cellules partiellement ou non arrondies (plate, Figure 3.2D).
Nous avons observé que le traitement avec la tubacine des cellules transfectées avec le
siCtl conduit à un doublement de la proportion de cellules avec des défauts
d’arrondissement mitotique dans les cellules HeLa ; par contre ce même traitement a eu
peu d’effet sur l’arrondissement mitotique des cellules MCF7 (Figure 3.2E). Puisque
l’arrondissement mitotique est dépendant entre autre de modifications dans les structures

125
d’adhérence cellulaire, il est possible que des effets dépendants du type cellulaires
interviennent (Marchesi et al. 2014; Margadant, Opstal, et Boonstra 2007). Par exemple,
une étude a suggéré qu’en contrôlant le niveau d’acétylation de la tubuline, HDAC6
pourrait aider au renouvellement des adhésions focales (Tran et al. 2007). Dans les cellules
HeLa, mais pas dans les cellules MCF7, ce processus pourrait être requis pour
l’arrondissement mitotique et pourrait expliquer l’effet différentiel du traitement avec la
tubacin dans les deux lignées cellulaires.
Par contre, que ce soit dans les cellules HeLa ou dans les cellules MCF7, nous avons
observé une augmentation significative du pourcentage de cellules avec des défauts
d’arrondissement mitotique en réponse à la déplétion de BAG3, d’HSPB8 ou de
p62/SQSTM1 (Figure 3.2E). Ce résultat corrobore nos travaux précédents et indique que
la déplétion de l’une de ces trois protéines interfère avec l’arrondissement mitotique dans
les deux lignées cellulaires (Fuchs et al. 2015 ; Annexe 1). Dans ces cellules, le traitement
avec la tubacine a causé une réduction significative d’au moins 2 fois de la proportion de
cellules présentant des défauts d’arrondissement mitotique (Figure 3.2E). Ceci suggère
qu’une réduction de l’activité d’HDAC6 peut normaliser les défauts d’arrondissement
mitotique observés en réponse à la déplétion des protéines BAG3, HSPB8 et p62/SQSTM1.
En outre, un inhibiteur de l’activité Arp2/3 peut également corriger les défauts
d’arrondissement mitotique dans les cellules HeLa (Figure supplémentaire S3.2).
Collectivement, les résultats suggèrent que le complexe chaperon HSPB8-BAG3-
p62/SQSTM1 pourrait faciliter le remodelage de l’actine en mitose à travers la modulation
de l’activité d’HDAC6. Une telle fonction pourrait aider à la régulation de l’activité
d’Arp2/3 requise pour la formation et la polarisation du nuage sous-cortical d’actine. La
modulation d’HDAC6 par HSPB8-BAG3-p62/SQSTM1 aiderait également aux processus
conduisant l’arrondissement mitotique. Récemment, l'activité d’HDAC6 a été impliquée
dans la progression de la cytokinèse puisque son inhibition ou sa déplétion se caractérise
par une inhibition de la formation de l’anneau contractile (Li et al. 2017). Il semble donc
que l'activité d’HDAC6 soit finement régulée pendant la progression du cycle cellulaire

126
par des mécanismes qui pourraient faire intervenir le complexe chaperon. Les résultats
présentés ci-après suggèrent que son activité sur la cortactine serait impliquée.
3.3. L’activité d’HDAC6 et son association à la cortactine sont régulées de façon
mitose-spécifique, d’une manière qui pourrait impliquer l’intervention du
complexe chaperon HSPB8-BAG3-p62/SQSTM1
Jusque-là, nos résultats suggèrent qu’un débalancement de l’activité d’HDAC6 interfère
avec les processus de remodelage du cytosquelette d’actine en mitose et que le complexe
chaperon HSPB8-BAG3-p62/SQSTM1 pourrait intervenir dans sa régulation. Pour
appuyer ces résultats fonctionnels, nous avons d’abord tenté d’obtenir des évidences
biochimiques supportant la modulation d’HDAC6 dans les cellules en mitose. Pour ce
faire, nous avons traité des cellules asynchrones ou relâchées d’un double blocage à la
thymidine avec 5 µM de tubacine, pendant toute la seconde relâche (8 h), ou pendant les 4
dernières heures avant l’entrée des cellules en mitose. Les niveaux d’acétylation des
protéines totales, ainsi que les niveaux d’acétylation de la cortactine ont été analysés par
immunobuvardage de type « Western », en utilisant des anticorps anti-acétyl à large
spectre, ou spécifiquement dirigés contre la lysine 309 de la cortactine, déacétylée par
HDAC6 (Zhang et al. 2007).
Les résultats indiquent que le niveau total d'acétylation des protéines augmente
proportionnellement au temps de traitement avec la tubacine dans les deux conditions,
démontrant qu’HDAC6 est active dans les cellules asynchrones comme dans les cellules
mitotiques (Figure 3.3A, Ac-K totale). De manière intéressante, nous avons noté
l’apparition de bandes protéiques qui semblent plus fortement acétylées dans les cellules
mitotiques. Ceci suggère que l’activité d’HDAC6 sur ses différents substrats pourrait être
stimulée ou modulée dans les cellules en mitose (Figure 3.3A, M, Ac-K totale).

127

128
Figure 3.3 : L’activité d’HDAC6 et son association à la cortactine sont régulées de façon différentielle en mitose ; un rôle pour le complexe chaperon HSPB8-BAG3. (A) Des cellules HeLa-RFP-H2B asynchrones et synchronisées en mitose ont été traitées pendant 0, 4 ou 8 h avec de la tubacine (5 µM) puis extraites pour analyse par immunobuvardage de type « Western ». Les immunobuvardages de type « Western » ont été réalisés en utilisant un anti-acétylation totale (Ac-K), un anti-acétyl-cortactine (Ac-K309) ; niveaux de cycline B1 : contrôle de synchronisation mitotique; cortactine: contrôle de dépôt. (B) Images confocales de cellules transfectées avec le siCtl asynchrones (AS) et mitotiques (M) et de cellules mitotiques déplétées en BAG3 (siBAG3_1) ou en HSPB8 (siHSPB8_2) montrant des spots d'interaction entre HDAC6 et la cortactine par PLA. Des cellules MCF7 ont été synchronisées en mitose, déplétées en BAG3 ou HSPB8 avec des siARN spécifiques pendant 48 h et transfectées avec les vecteurs GFP-HDAC6 et myc-cortactine durant 24 h. Barre d'échelle, 10 µm. Le graphique montre le nombre de spots de PLA par cellule ; résultats +/- SEM représentatifs de 3 expériences indépendantes où 30 à 101 cellules ont été anaylisées et l’analyse statistique a été effectuée par le test de Mann Whitney. (C) Des cellules HeLa-RFP-H2B ont été synchronisées en mitose par un traitement de 16 h avec le nocodazole et ont été infectées avec des adénovirus recombinants conduisant l'expression de GFP ou de mutants BAG3-GFP (IPV et DPxxP) suivant la déplétion sur 48 h de BAG3 à l’aide d’un siARN spécifique. Les extraits cellulaires ont été préparés à partir des cellules asynchrones (AS) ou synchronisées en mitose et récupérées par une agitation mécanique (M). Pour toutes les conditions, les immunoprécipitations ont été obtenues en utilisant un anticorps anti-GFP. Des analyses en immunobuvardage de type « Western » ont été réalisées avec les anticorps indiqués. Les niveaux de GFP, BAG3, HSPB8, p62/SQSTM1, HDAC6 et cortactine dans les extraits cellulaires totaux sont montrés (Input).
Puisque HDAC6 régule la dynamique de l'actine dépendante du complexe Arp2/3 via la
déacétylation de la cortactine, nous avons également comparé les niveaux d'acétylation de
la cortactine en interphase et en mitose (Zhang et al. 2007; Gao et al. 2007; Kaluza et al.
2011; Kozyreva et al. 2014). En utilisant un anticorps dirigé contre la cortactine acétylée,
nous avons observé une augmentation de l'acétylation de la cortactine en réponse à
l’inhibition d’HDAC6 de façon proportionnelle au temps de traitement avec la tubacine
dans les extraits cellulaires asynchrones et mitotiques (Figure 3.3A, Ac-K309). Ce résultat
confirme que la déacétylase cible la cortactine dans les cellules en interphase et en mitose.
De façon surprenante, dans les cellules mitotiques, le taux d'acétyl-cortactine suivant
l’inhibition d’HDAC6 est substantiellement inférieur à celui des cellules asynchrones,
même après 8 heures de traitement avec la tubacine (Figure 3.3A, M, Ac-K309). Ce résultat
suggère que la déacétylation de la cortactine par HDAC6 est diminuée pendant la mitose ;
ceci est corroboré par un niveau d’acétylation plus important en mitose (Figure 3.3A, M, 0

129
h). Nous avons conclu que l'activité d’HDAC6 sur sa cible la cortactine est limitée à
l’entrée des cellules en mitose, et donc que l’activité de la cortactine pourrait être inhibée.
Pour investiguer l’implication du complexe chaperon HSPB8-BAG3, nous avons cherché
à corroborer la diminution d’activité HDAC6 sur la cortactine pendant la mitose à l’échelle
cellulaire, et ensuite évalué l’impact de la déplétion de BAG3 ou d’HSPB8. Pour ce faire,
nous avons effectué des expériences de PLA (« proximity ligation assay ») qui permettent
de visualiser in situ des interaction protéine-protéine (Söderberg et al. 2008). Nous avons
examiné l’interaction d’HDAC6 avec la cortactine dans des cellules MCF7 asynchrones
ou mitotiques exprimant des formes exogènes de GFP-HDAC6 et myc-cortactine, et
déplétées en HSPB8 ou en BAG3.
Les résultats indiquent qu’en moyenne 74 spots de PLA, représentant une potentielle
interaction entre HDAC6 et la cortactine, ont été dénombrés dans des cellules contrôles
asynchrones tandis que ~30 spots ont été quantifiés dans des cellules mitotiques (Figure
3.3B, siCtl AS versus siCtl M). Cette observation suggère qu’il y a une réduction
significative de l’interaction de la déacétylase HDAC6 avec sa cible la cortactine dans les
cellules mitotiques, corroborant l’augmentation d’acétylation de la cortactine (Figure 3.3A,
M, 0 h). Notamment, dans les cellules mitotiques, nous avons observé qu’une déplétion de
BAG3 ou d’HSPB8 se traduit par une augmentation significative d’au moins 2 fois du
nombre de spots de PLA en comparaison aux cellules contrôles (Figure 3.3B). Ce résultat
suggère que le complexe chaperon pourrait limiter l’association d’HDAC6 avec la
cortactine à l’entrée des cellules en mitose.
Pour analyser davantage la relation fonctionnelle entre le complexe chaperon HSPB8-
BAG3-p62/SQSTM1, HDAC6 et la cortactine durant la mitose, nous avons effectué des
expériences de co-immunoprécipitations de BAG3-GFP dans des cellules déplétées de
BAG3 endogène, afin d’identifier les domaines fonctionnels responsables de l’interaction
BAG3-HDAC6. Les immunoprécipitations ont été réalisées à partir d'extraits de cellules
HeLa asynchrones (AS) ou mitotiques (M), après déplétion de BAG3 et réintroduction de
protéines BAG3-GFP portant des mutations qui abolissent la liaison à HSPB8 (IPV) ou la

130
délétion des motifs PxxP (ΔPxxP) (Figure 3.3C ; Fuchs et al. 2015 ; Annexe 1). Comme
nous l'avons démontré dans notre première étude, HDAC6 et p62/SQSTM1 co-précipitent
préférentiellement avec BAG3-GFP WT dans les cellules mitotiques (Figure 3.3C, M ;
Fuchs et al., 2015 ; Annexe 1). De façon intéressante, nous avons observé que la cortactine
co-précipite également avec BAG3-GFP WT davantage dans les extraits cellulaires
mitotiques que dans les extraits cellulaires asynchrones comme HDAC6 et p62/SQSTM1
(Figure 3.3C, M). Ainsi, ce résultat montre que BAG3 interagit préférentiellement avec
p62/SQSTM1, HDAC6 et sa cible la cortactine pendant la mitose. Cette observation
soutient l'importance d'un complexe formé des quatre protéines dans la progression
mitotique.
Nous avons précédemment démontré que l’activité mitotique de BAG3 dépend des motifs
IPV qui médient sont interaction avec HSPB8 et des motifs PXXP, qui sont présumés
permettre l’association de BAG3 à des protéines de signalisation (Fuchs et al. 2015 ;
Annexe 1 ; Behl 2016). Nous avons donc évalué la contribution de ces domaines au
recrutement d’HDAC6 et de la cortactine. Les résultats indiquent que le mutant BAG3 IPV
semble avoir moins d’affinité pour HDAC6 dans les extraits mitotiques (Figure 3.3C, M).
Une diminution plus marquée de l'interaction BAG3-HDAC6 concernant le mutant du
domaine PxxP a également été observée dans les extraits mitotiques (Figure 3.3C, M). De
manière significative, la même diminution a été détectée pour les interactions des mutants
de BAG3 avec p62/SQSTM1 et la cortactine (Figure 3.3C, M). Ces résultats montrent que
les motifs IPV et le domaine PxxP de BAG3 régulent les interactions avec p62/SQSTM1,
HDAC6 et la cortactine. Puisque le mutant IPV est incapable d'interagir avec HSPB8, ces
résultats ont identifié l'association à HSPB8 et le domaine riche en motifs PxxP, qui sont
les domaines fonctionnels mitotiques de BAG3, comme déterminants pour son interaction
avec p62/SQSTM1, HDAC6 et la cortactine pendant la mitose. Ce résultat renforce
l’hypothèse d’une fonction importante pour le complexe HSPB8-BAG3-p62/SQSTM1-
HDAC6-cortactine dans la progression mitotique.
En conclusion, nos résultats indiquent que l’activité d’HDAC6 est régulée de façon
différentielle en mitose en comparaison à l’interphase. De façon intéressante, l’activité

131
d’HDAC6 sur la cortactine est réduite en mitose à l’inverse d’un grand nombre des cibles
de la déacétylase. Ceci s’accompagne d’une réduction significative de l’association
d’HDAC6 à la cortactine. Nos résultats suggèrent que le complexe chaperon HSPB8-
BAG3 pourrait limiter l’association d’HDAC6 avec la cortactine en mitose par un
mécanisme qui reste à déterminer. Ce mécanisme pourrait impliquer un recrutement de la
déacétylase par BAG3 de façon dépendante de son association à HSPB8 et de son domaine
PxxP qui semble être essentiel à l’association de BAG3 avec HDAC6, cortactine et
p62/SQSTM1. Une protéine intermédiaire, qui interagit avec le domaine PXXP de BAG3,
pourrait jouer un rôle clé pour ponter ces interactions, laquelle reste à identifier.
Il nous faudra confirmer que le complexe chaperon module l’activité d’HDAC6 sur la
cortactine. Pour cela, nous pourrions analyser l’impact de la déplétion de BAG3 ou
d’HSPB8 sur les niveaux d’acétylation de la cortactine, par des expériences
d’immunobuvardage de type « Western », à partir d’extraits cellulaires mitotiques traités
ou non avec la tubacine. Une élévation des niveaux d’acétylation de la cortactine en
réponse à la déplétion de BAG3 ou d’HSPB8 et au traitement avec la tubacine confirmera
l’hypothèse. Cela dit, nos résultats préliminaires indiquent que cette approche pourrait
s’avérer mitigée, due aux effets hétérogènes mesurés sur une population cellulaire
présentant des phénotypes hypomorphes (i.e. déplétion de BAG3 ou de HSPB8). Pour
pallier à ces effets, nous pourrions tirer avantage de la technologie CRISPR (« Clustered
Regularly Interspaced Short Palindromic Repeats »)/Cas9 (« CRISPR associated protein
9 ») pour obtenir des lignées cellulaires KO pour BAG3 ou HSPB8. Alternativement une
approche basée sur la détection des niveaux de cortactine acétylée in situ, par
immunofluorescence est envisageable.
3.4. Modification du phénotype BAG3-dépendant par l’expression de mutants
cortactine acétyl-mimétique et non-acétylable
Nos résultats suggèrent que le complexe chaperon HSPB8-BAG3 pourrait moduler
l'activité d’HDAC6 sur la cortactine pendant la mitose et qu’une telle fonction est

132
nécessaire pour le remodelage de l'actine dans les cellules mitotiques. Nous avons donc
tenté de valider ce modèle par l’utilisation de mutants de la cortactine.
La cortactine peut être acétylée dans des régions répétitives enrichies en lysines situées en
aval de son domaine NTA (Zhang et al. 2007). Il a été montré que son acétylation prévient
sa localisation au front de migration et inhibe la motilité cellulaire, tandis que sa
déacétylation par HDAC6 entraîne une liaison accrue de la cortactine à la F-actine et
stimule ainsi la migration cellulaire (Zhang et al. 2007). Ainsi, pour tester notre hypothèse,
nous avons tout d’abord étudié les effets de l'expression de mutants non-acétylable
(cortactine-9KR) ou acétyl-mimétique (cortactine-9KQ) de la cortactine sur la densité de
la F-actine et la cage de F-actine observée suivant la déplétion de BAG3 ou d’HSPB8
(Figure 3.1C ; Zhang et al. 2007). Pour cela, nous avons transfecté sur 32 h des cellules
HeLa synchronisées en mitose et déplétées en HSPB8 ou en BAG3 avec les mutants de
cortactine (Figure 3.4A). Comme le montre la figure 3.4B, l'expression du mutant
cortactine-9KQ n'induit pas de changements dans l'organisation de la F-actine dans les
cellules mitotiques contrôles. À l’inverse, l'expression du mutant cortactine-9KR conduit
à un enrichissement de F-actine dans une structure en forme de cage qui entoure le fuseau
mitotique dans les cellules mitotiques, mimant le phénotype observé suivant la déplétion
de l’une ou l’autre des protéines du complexe HSPB8-BAG3. De plus, alors que
l'expression du mutant cortactine-9KR n'affecte pas l'organisation de cette structure
d'actine dans les cellules déplétées en BAG3 ou en HSPB8, une réduction significative de
la densité en F-actine a été observée dans les cellules déplétées et exprimant le mutant
cortactine-9KQ (Figure 3.4B). Ce résultat montre que les structures d’actine ectopiques qui
s’accumulent autour du fuseau mitotique suivant la déplétion de BAG3 ou d’HSPB8
peuvent être normalisées par la surexpression du mutant acétyl-mimétique de cortactine
(9KQ).

133

134
Figure 3.4 : Les mutants cortactine-9KQ et 9KR restaure et mime respectivement les défauts d’organisation du cytosquelette d’actine associés à la déplétion de BAG3 ou d’HSPB8. (A) Immunobuvardage de type « Western » d’extraits protéiques de cellules HeLa-RFP-H2B synchronisées en mitose et transfectées durant 32 h avec des vecteurs codant pour des mutants de cortactine (9KQ et 9KR) étiquetés 3X-FLAG et montrant leur niveau d'expression par rapport à l'expression endogène de la cortactine (EV). L'analyse par immunoburadage de type « Western » a été réalisée avec un anti-cortactine; GAPDH: contrôle de dépôt. (B) Reconstitution 3D d'images confocales déconvoluées de cellules traitées selon le protocole décrit en A et montrant une normalisation pour le mutant cortactine-9KR et un mime pour le mutant cortactine-9KQ de la densité et de l’organisation de la F-actine. L'immunofluorescence a été réalisée avec un anti-a-tubuline, la phalloïdine-488 et le Hoechst pour marquer respectivement le fuseau mitotique, la F-actine et l'ADN; barre d'échelle; 10 µm. (C) Les images déconvoluées d'épifluorescence de cellules traitées selon le protocole décrit en A montrent une restauration de l'arrondissement mitotique de cellules déplétées en BAG3 ou en HSPB8 après expression du mutant cortactine-9KQ et un mime du défaut d’arrondissement mitotique par les cellules contrôles (siCtl) après l'expression du mutant cortactine-9KR. L’immunofluorescence a été réalisée avec un anti-FLAG pour marquer la cortactine exogène, un anti-a-tubuline pour marquer les microtubules, la phalloïdine-488 pour marquer la F-actine et le Hoechst pour marquer l'ADN; barres d'échelle, 10 µm. (D) Le graphique représente le pourcentage de cellules arborant des défauts d'arrondissement mitotique après la transfection des mutants cortactine-9KR et 9KQ des cellules HeLa-RFP-H2B synchronisées en mitose et transfectées avec un siARN dirigé contre BAG3 (siBAG3_1) ou HSPB8 (siHSPB8_2) ou un contrôle (siCtl) ; résultats +/- SEM représentatifs de 3 expériences indépendantes où au moins 609 cellules ont été analysées. L’analyse statistique a été effectuée par le test de Fisher et compare les conditions indiquées à la conditiontion siCtl EV.
Nous avons également étudié l’impact des mutants de cortactine sur les défauts
d’arrondissement mitotique induits par la déplétion d’HSPB8 ou de BAG3. Pour cela, nous
avons transfecté des cellules HeLa synchronisées en mitose et déplétées en BAG3 ou en
HSPB8 avec les vecteurs codant pour les mutants de cortactine sur une période de 32 h.
Nous avons constaté que l’expression du mutant cortactine-9KR dans les cellules HeLa
mitotiques induit un doublement du pourcentage de cellules présentant des défauts
d'arrondissement mitotique par rapport aux cellules contrôles transfectées avec le vecteur
vide (EV) (Figure 3.4C et D). Ainsi, l'expression d'un mutant de cortactine non-acétylable
interfère avec l'arrondissement des cellules mitotiques tout comme le fait la déplétion de
BAG3 ou d’HSPB8 (Figures 3.4C et D). À l’inverse, l'expression du mutant cortactine-
9KQ ne semble pas induire de défauts d'arrondissement mitotique dans les cellules
contrôles (Figures 3.4C et D). De façon remarquable, nous avons noté que l'expression de

135
ce mutant corrige les défauts d'arrondissement mitotique dans les cellules déplétées en
BAG3 ou en HSPB8 (Figures 3.4C et D). Ces résultats indiquent qu’un mutant acétyl-
mimétique de cortactine peut restaurer l'arrondissement mitotique des cellules déplétées en
BAG3 ou en HSPB8.
Ensembles, ces résultats nous suggèrent qu’une perturbation de la régulation mitotique de
l’acétylation de la cortactine contribue aux défauts de réarrangement du cytosquelette
d’actine observés suivant la déplétion de BAG3 ou d’HSPB8. Ces observations sont en
accord avec l’hypothèse selon laquelle HSPB8-BAG3 en modulant l’activité d’HDAC6
sur la cortactine facilite le remodelage des structures à base d’actine qui conduisent la
progression mitotique. Nos résultats suggèrent que l’acétylation de la cortactine est requise
pour limiter la dynamique de l’actine dans les cellules mitotiques et pour l’arrondissement
mitotique. Ainsi, nos résultats suggèrent pour la première fois que la dynamique
d’acétylation de la cortactine doit être finement régulée lors de la progression mitotique.
Comme l’acétylation de la cortactine inhibe son activité sur la polymérisation de l’actine
dépendante d’Arp2/3, nos résultats associés aux données de la littérature suggèrent que le
complexe chaperon limiterait l’activité d’Arp2/3 en modulant l’activité d’HDAC6 sur la
cortactine (Zhang et al. 2007; Uruno et al. 2001; Weaver et al. 2001).
Pour confirmer cette hypothèse, nous pourrions analyser l’effet d’un mutant de cortactine
incapable d’activer Arp2/3 sur les défauts mitotiques associés à la déplétion de BAG3,
HSPB8 ou p62/SQSTM1. Une correction de l’arrondissement mitotique et de la dynamique
du nuage sous-cortical d’actine dans de telles conditions permettrait de confirmer notre
hypothèse.
En conclusion, dans la présente étude, nous fournissons des évidences que le complexe
chaperon, en recrutant p62/SQSTM1 à l'entrée en mitose, modulerait l'activité mitotique
d’HDAC6 sur la cortactine. Un modèle dans lequel la fonction de BAG3 dans la
séquestration des protéines permettrait la régulation spatiotemporelle de la signalisation
cellulaire et de l'autophagie a été récemment proposé par (Kathage et al. 2016). En effet,
les auteurs ont décrit que BAG3 par interaction avec TSC1 induit le recrutement de

136
complexes TSC dans des fibres de stress, où les complexes inhibent une sous-population
de vésicules mTORC1-positives associées au cytosquelette d'actine. Ainsi, l'inhibition
locale de mTORC1 déclencherait l'autophagie de la filamine, une protéine de réticulation
de l'actine, aux sites de dénaturation. En même temps, la séquestration de TSC1/TSC2
induite par BAG3 soulage l'inhibition de mTORC1 dans le cytoplasme, ce qui stimule la
traduction des protéines. Un tel processus pourrait être nécessaire pendant la mitose pour
contrôler spécifiquement dans le temps et l'espace l'activité d’HDAC6 sur la cortactine. En
séquestrant des complexes contenant p62/SQSTM1, HDAC6 et la cortactine, BAG3-
HSPB8 serait capable d'inhiber localement l'activité d’HDAC6 sur la cortactine et par
conséquent de limiter la polymérisation branchée de l'actine. Selon ce modèle, grâce à la
séquestration de ses partenaires mitotiques, BAG3-HSPB8 modulerait les transitions de
structures à base d'actine qui favorisent la progression mitotique.

137
3.5. Figures supplémentaires
Figure S3.1 : Mesure de l’efficacité de la déplétion de BAG3, HSPB8 ou p62/SQSTM1 par immunobuvardage de type « Western ». (A) Immunobuvardage de type « Western » représentatif d’extraits protéiques provenant de cellules HeLa-RFP-H2B montrant l'efficacité de la déplétion de BAG3, HSPB8 et p62/SQSTM1 48 h après la transfection de siRNAs spécifiques. Le niveau de déplétion a été estimé à plus de 75% par un dépôt de quantités croissantes d'extraits de contrôle (siCtl). L'analyse par immunobuvardage a été réalisée avec des anticorps anti-BAG3, anti-HSPB8 et anti-p62/SQSTM1 ; vinculine : contrôle de dépôt. (B) Immunobuvardage de type « Western » représentatif d’extraits protéiques provenant de cellules HeLa-RFP-H2B montrant l'efficacité de la déplétion de BAG3 et HSPB8, 48 h ou 24 h après la transfection de siRNAs spécifiques. La déplétion a été estimée à plus de 75% par un dépôt de quantités croissantes d'extraits de contrôle (siCtl). L'analyse par immunobuvardage a été réalisée avec des anticorps anti-BAG3 et anti-HSPB8 ; vinculine : contrôle de dépôt. (C) Immunobuvardage de type « Western » représentatif d’extraits protéiques provenant de cellules HeLa-RFP-H2B montrant l'efficacité de la déplétion de p62/SQSTM1 24 h après la transfection de siRNAs spécifiques. La déplétion a été estimée à plus de 75% par un dépôt de quantités croissantes d'extraits de contrôle (siCtl). L'analyse par immunobuvardage a été réalisée avec un anticorps anti-p62/SQSTM1 ; vinculine : contrôle de dépôt.

138
Figure S3.2 : L'inhibition d’Arp2/3 normalise les défauts d'arrondissement des cellules mitotiques dans les cellules HeLa déplétées en BAG3. L'histogramme montre le pourcentage de cellules mitotiques avec des défauts d’arrondissement (i.e. cellules partiellement ou non arrondies) dans des cellules HeLa-H2B-RFP transfectées avec un siRNA contrôle (siCtl) ou un siARN spécifique de BAG3 (siBAG3_1) pendant 48 h, synchronisées en mitose et traitées pendant 8 h avec le CK666 (40 µM).
3.6. Matériel et méthode
3.6.1. Culture cellulaire, synchronisation, traitements et transfection de siARN
Des cellules HeLa-RFP-H2B (don de Sabine Elowe) et des cellules MCF7 ont été
maintenues dans du milieu essentiel a-minimal (a-MEM) additionné de 10% de sérum
bovin fœtal. Les lignées cellulaires ont été cultivées dans une atmosphère humidifiée avec
5 % de CO2 à 37 °C. Les cellules ont été synchronisées par un double blocage à la
thymidine et ont été relâchées pendant 8 à 10 h dans du milieu frais, comme décrit
précédemment (Fuchs et al., 2015). Pour les expériences de déplétion, les cellules ont été
transfectées sur la nuit avec des siRNA duplex à 50 nM dans du tampon de transfection
calcium-phosphate (MgCl2 125 mM, NaCl 140 mM, HEPES 25 mM, Na2HPO4 0,75 mM)
pour les cellules HeLa et le réactif RNAimax (Invitrogen) en suivant les instructions du
fabricant pour les cellules MCF7. L'efficacité de la déplétion a été analysée 24 à 48 h plus
tard par immunobuvardage de type « Western » ou par immunofluorescence. Les siRNA

139
duplex sont basés sur les séquences humaines et ont été achetés auprès de Qiagen (siRNA
de qualité HPP) ou de Thermo Fisher Scientific (qualité A4 standard). Les séquences des
brins sens sont les suivantes : siBAG3_1 : 5’-CGAAGAGTATTTGACCAAA-3′ ;
siBAG3_2 : 5′-GCAAAGAGGTGGATTCTAA-3′ ; siBAG3_3 : 5’-
GATGTGTGCTTTAGGGAAT-3 ; siHSPB8_1 : 5′-CAGATAGGCTAGTGGTATT-3′ ;
siHSPB8_2 : 5′-GCAGTGAATGCAAGGGTTATT-3′ ; sip62_1 : 5’-
GGAAATGGGTCCACCAGGATT 3 ’; sip62_2 : 5’- AGACCAAGAACTATGACAT -
3’. La séquence cible du siCtl (si Allstars Negative Control) provient de Qiagen et est la
suivante : 5’-CAGGGTATCGACGATTACAAA-3’.
Pour évaluer la restauration de l'arrondissement mitotique et de la cage de F-actine et
l’activité d’HDAC6 par immunoburdage de type « Western », les drogues suivantes ont été
ajoutées aux cellules qui ont été synchronisées en mitose par un double blocage à la
thymidine, pendant 4 à 8 heures lors la seconde période de relâche avant la fixation
cellulaire ou l’extraction protéique : tubacine, 5 µM ou CK666, 40 µM. Pour analyser la
correction de la dynamique du nuage sous-cortical d'actine par imagerie en temps réel, les
drogues ont été ajoutées aux cellules qui ont été synchronisées en mitose par un double
blocage à la thymidine 30 min avant l'imagerie en temps réel comme suit : tubacine, 20
nM; CK666, 40 µM.
3.6.2. Transfection, infection, vecteurs et adénovirus
Les adénovirus recombinants conduisant l’expression de la protéine BAG3 étiquetée GFP
(WT, IPV [I9V/V98G/I208G/V210G] or ΔPXXP (Δ302–418) ; décrit dans Fuchs et al.
2015), pour les expériences de co-immunoprécipitation, ont été produits par Welgen Inc,
Worcester, MA, et ont été amplifiés dans des cellules HEK293VR, comme décrit dans
Fuchs et al. 2015. Tous les adénovirus recombinants ont été confirmés par séquençage des
séquences insérées. Les titres de virus ont été déterminés en utilisant le kit de titrage
AdenoX Rapid (Clontech Laboratories, #631028) en suivant les instructions du fabricant.
Des cellules HeLa-RFP-H2B ont été infectées avec les adénovirus Ad-BAG3-GFP à une
multiplicité d'infection (MOI) de 2 unités formant des plaques par cellule (pfu/cellule) dans

140
de l'a-MEM complet comme décrit dans Fuchs et al. 2016. Deux heures plus tard, un
tampon de transfection calcium-phosphate a été ajouté au milieu. La transfection-infection
simultanée permet une transduction virale efficace à faible MOI. Seize heures après
l'infection, les cellules ont été lavées trois fois avec de l'HEPES (KCl 6,7 mM, NaCl 150
mM, HEPES 10 mM, pH 7,3) et ont été laissées dans du milieu a-MEM frais pendant 8h
avant de rajouter du nocodazole (400 ng/ml) pour une période de 16h.
Pour suivre la dynamique de l'actine et de la tubuline par imagerie en temps réel, des
cellules ont été ensemencées sur des pétris à fond de verre recouvertes de fibronectine
(MatTek Corp.) et synchronisées par la méthode de double blocage à la thymidine.
L'infection a été réalisée dans l'a-MEM-minus au cours du premier blocage à la thymidine
en utilisant l’adénovirus Ad-LifeAct-TagGFP2 (60121, IBIDI) à une MOI de 1 pfu/cellule
et le bacculovirus Bac-RFP-αtubuline (C10614) à 4 pfu/cellule dilué dans le réactif
BacMam 2.0. Le siARN dirigé contre BAG3 a été transfecté durant la même période dans
un tampon de transfection calcium-phosphate, en utilisant la méthode décrite dans Fuchs
et al., 2016. 16 h après l'infection et la transfection simultanées, les cellules ont été lavées
trois fois avec de l'HEPES et ont été relâchées dans du milieu frais pendant 8 h avant de
rajouter 2 mM de thymidine. Pour les expériences de PLA le vecteur d’expression
peGFP.N1-HDAC6 WT provenant d’Addgene (#36188) et le vecteur d’expression myc-
cortactin WT don du Dr Zhan, Department of Experimental Pathology, Jerome H. Holland
Laboratory for the Biomedical Sciences, American Red Cross, Rockville, MD 20855,
USA, ont été utilisés ; les constructions 3XFLAG-cortactine (9KQ or 9KR; les lysines (K)
situées dans des régions de répétition ont été mutées en glutamine (Q) ou en arginine (R)
utilisées pour les expériences de restauration de l’arrondissement mitotique et de la cage
de F-actine sont un don du Dr Seto, Washington University School of Medicine and Health
Sciences, Washington, DC 20037, USA. Pour induire la surexpression de la cortactine et
d’HDAC6 pour les expériences de PLA, des MCF7 ont été synchronisées par un double
blocage à la thymidine et transfectées durant la seconde période de blocage à la thymidine
en utilisant la Lipofectamine 2000 (Invitrogen) en suivant les instructions du fabricant. 16
h après la transfection les cellules ont été lavées avec du PBS (137 mM NaCl, 2.7 mM KCl,
4.3 mM Na2HPO4, 1.47 mM KH2PO4) et relâchées dans du milieu frais durant 8 à 9 h avant

141
d’être fixées. Pour induire la surexpression des mutants 3X-FLAG de cortactine pour les
expériences de restauration de l'arrondissement mitotique et de la structure ectopique
enrichie en F-actine entourant le fuseau mitotique, des cellules HeLa-RFP-H2B
synchronisées en mitose par un double blocage à la thymidine ont été transfectées durant
la première période de relâche en utilisant la Lipofectamine 2000 (Invitrogen) en suivant
les instructions du fabricant. 8 h après la transfection le milieu des cellules a été changé et
2 mM de thymidine y ont été ajoutés.
3.6.3. Anticorps et produits chimiques
Les anticorps suivants ont été utilisés pour les expériences de co-immunoprécipitation et
les analyses par immunobuvardage de type « Western » : l'anticorps anti-BAG3 LP11 de
lapin a été développé contre un BAG3 recombinant humain complet fusionné avec la
glutathion S transférase (LP11) (Fuchs et al., 2015), anti-HSPB8 de lapin contre un peptide
C-terminal (NELPQDSQEVTCT) (Carra et al. 2008) ; anti-p62/SQSTM1 (sc-28359) et
anti-GFP utilisé pour les immunobuvardages de type « Western » (sc-9996) proviennent
de Santa Cruz ; anti-HDAC6 (# 7612) et anti-lysines-acétylées (ac-K-100, #9814)
proviennent de Cell Signaling Technologies ; anti-GFP employé pour les essais de co-
immunoprécipitation (A-11120) provient de Molecular Probes/Thermo Fisher; anti-
Vinculin (V9131) provient de Sigma-Aldrich ; l'anti-cortactine (IF et WB) (ab33333)
provient d'Abcam ; anti-acétyl-cortactine K309 (ABT1378) provient de Millipore. Les
anticorps secondaires (anti-souris-HRP 115-035-146 et anti-lapin-HRP 111-055-003)
proviennent de Jackson ImmunoResearch. Les anticorps suivants ont été utilisés pour
l'immunofluorescence et le PLA : l'anti-GFP (sc-9996) provient de Santa Cruz ; anti-a-
tubulin souris (T5168) provient de Sigma Aldrich et anti-a-tubulin lapin (ab18251)
provient d’Abcam ; la Alexa-Fluor® 488 Phalloidin (A12379) provient de Molecular
Probes/Thermo Fisher. La tubacine (S2239) provient de Selleckchem ; le bisbenzimide H
33342 de Hoechst (B2261), la thymidine (T9250), la fibronectine (F1141-5 mg) et le
CK666 (SML0006) proviennent de Sigma-Aldrich ; le nocodazole (487928) provient de
Calbiochem.

142
3.6.4. Immunoprécipitation et immunobuvardage de type « Western »
Des quantités égales de cellules ont été lavées dans du PBS glacé et ont été lysées par 3
cycles de congélation/décongélation dans 4 volumes de tampon de lyse (20 mM TRIS-HCl
pH 7.6, 150 mM NaCl, 1 mM EDTA, 0,1% IGEPAL, 1 mM NaVO4, 10 mM NaF, 40 mM
β-glycerophosphate, 1X Complete [Roche], 1 mM DTT). Les extraits cellulaires ont été
centrifugés à 15 000 g pendant 15 min, les surnageants ont été transférés dans un tube
propre et incubés avec un anticorps spécifique pendant 60 min à 4 °C, après quoi ils ont
été transférés dans un tube contenant des billes magnétiques recouvertes de protéine A
(Dynabeads protien A, Life Technologies) préalablement équilibrées dans le tampon de
lyse. Après incubation pendant 30 minutes sous agitation douce, les immunocomplexes ont
été recueillis en utilisant un support magnétique et lavés trois fois dans du tampon de lyse.
Des quantités égales de complexes immuns ont été chargées sur SDS-PAGE et analysées
par immunobuvardage de type « Western ».
Pour les immunobuvardage de type « Western », les cellules ont été lysées dans un tampon
SDS (62.5 mM Tris-HCl, pH 6.8, 2.3% SDS, 10% glycérol, 5% β-mercaptoéthanole,
0.05% bleu de bromphénole, 1 mM phénylméthylsulfonylfluoride) et des quantités
équivalentes de protéines provenant des lysats cellulaires totaux ont été séparés par
électrophorèse SDS-PAGE de gels à 8 % d’acrylamide puis transférés sur membranes de
nitrocellulose. L’immunobuvardage a été effectué selon le protocole décrit par Champagne
et al., 2004. La concentration protéique des échantillons a été déterminée à l’aide du kit «
DC protein assay » (Laboratoires Bio-Rad) et l’analyse densitométrique a été effectuée à
partir d’images acquises avec l’appareil FluorS MAX MultiImager contrôlé par le logiciel
QuantityOne version 4.5.0 (Laboratoires Bio-Rad).
3.6.5. Immunofluorescences, PLA (« proximity ligation assay »), imagerie en
temps réel, microscopie et statistiques
Pour les expériences d’immunofluorescence et de PLA, les cellules ont été lavées dans du
PBS avec précaution puis fixées dans un tampon contenant 4% de formaldéhyde dans 0,2%

143
de Triton X-100, 20 mM Pipes, pH 6,8, 1 mM MgCl2, 10 mM EGTA, pH 8,0, pendant 10
min à température pièce. L'ADN a été marqué avec le Hoechst perméable aux cellules et
la F-actine a été marquée avec la phalloïdine. Les spécimens ont ensuite été traités pour
l’immunofluorescence comme décrit dans Lavoie et al., 2000, en utilisant les anticorps
primaires indiqués et les anticorps anti-lapin, anti-chèvre ou anti-chèvre Alexa Fluor. Pour
les essais de PLA, les échantillons ont été incubés avec des anticorps secondaires fournis
par le kit in situ Duolink (Invitrogen, DUO92002-100RXN ; anti-mouse minus #82004;
anti-rabbit plus #82002) durant 1 h à 37 °C dans du PBS 1 % lait. Les spécimens ont été
lavés trois fois pendant 5 minutes dans du tampon A (Tris 0,001 M, NaCl 0,15 M, 0,05%
de Tween 20) et incubés avec la ligase (Invitrogen, # 82027) à 0,025 U/µl dans un tampon
de ligation (# 82009) fourni par le Kit de réactifs de détection rouge (Invitrogen,
DUO92008-100RXN) pendant 30 minutes à 37 °C. Après trois lavages pendant 2 minutes
dans le tampon A, les échantillons ont été incubés avec la polymérase (# 82028) à 0,1 U/µl
dans du tampon rouge d'amplification (# 82011) pendant 90 minutes à 37 ° C. Enfin, les
échantillons ont été lavés trois fois dans du tampon B (Tris 0,2 M, NaCl 0,1 M) pendant
10 min puis lavés deux fois dans le tampon B 0,001X. Le montage des lamelles de verre a
été effectué à l'aide du milieu de montage fourni par le kit.
Les phénotypes ont été observés en routine par au moins deux investigateurs indépendants
par inspection visuelle d'échantillons fixés et ont été quantifiés de manière aveugle au
traitement, en utilisant un système AxioObserver Z1 utilisant un objectif 60x 1,25 NA et
une caméra CCD Axiocam MRm contrôlée par le logiciel Zen (Carl Zeiss). Les images
d'épifluorescence ont été acquises avec le même système de microscope en utilisant un
objectif 40x Plan-Neofluoar 0.6 NA. La microscopie confocale des cellules vivantes et
fixées a été réalisée avec un appareil Spinning Disc Confocal Perkin Elmer UltraVIEW
(40x 0,75 NA ou 60x huile 1,4 NA, respectivement), équipé d'une caméra à couplage de
charge refroidie par EMCCD à -50 ° C (Hamamatsu Photonics KK) et piloté par la version
6.01 du logiciel Volocity. Le système était équipé d'une chambre humidifiée à 5 % CO2 et
thermorégulée. Pour l'imagerie en temps réel, les images ont été acquises à des intervalles
de 90 secondes pendant une période de 75 minutes commençant à 48 heures après la
transfection des siARN. Les versions des logiciels Volocity 6.01 (technologies de Quorum)

144
et Image J 1.48v (institut national de la santé) ont été employées pour le traitement sur des
images entières avant le recadrage pour accentuer le point principal de l'image. Le
traitement s'est limité à la soustraction de l'arrière-plan, à l'ajustement de la luminosité/du
contraste et à la déconvolution. L’outil « 3D opacity » du logiciel Volocity a servi à la
reconstitution 3D des images confocales.
Les défauts d'arrondissement mitotique ont été estimés par inspection visuelle des défauts
de la mitose comme suit : cellules mitotiques partiellement arrondies ou plates et ont été
estimées à partir d'expériences indépendantes. Les phénotypes de nuage sous-cortical
d’actine ont été estimés par inspection visuelle des séquences temporelles comme suit :
mouvement rotatif unidirectionnel = polarisé ; changements dans la direction du
mouvement de rotation pendant la mitose = erratique ; immobile ou absent = absent. Les
phénotypes de défauts mitotiques ont été estimés par inspection visuelle des séquences
temporelles comme suit : oscillation du fuseau mitotique et retard dans la progression en
prométa- et métaphase. Le nombre de spots PLA ont été quantifiés en utilisant l'outil « Find
object » du logiciel Volocity. Un seuil minimal de volume d'objets de 0,1 µm3 a été calibré
et les cellules ont été considérées selon un seuil d'intensité moyenne de GFP-HDAC6
compris entre 50 et 250.
3.6.6. Analyses statistiques
Pour l’analyse de comparaison du nombre de spots de PLA par cellule, les données
provenant d’au moins trois expériences indépendantes ont été analysées en utilisant le test
statistique Mann Whitney. Pour l’analyse statistique de (a) du nuage sous-cortical d’actine,
(b) des mitoses défectueuses, (c) des défauts d’arrondissement mitotique, les données
provenant d’au moins trois expériences indépendantes ont été analysées en utilisant le test
statistique Fischer. Le logiciel statistique Prism 6.0 (GraphPad Software) a été utilisé pour
effectuer les tests statistiques. Les valeurs de p<0,05 ont été considérées comme
significatives (*, p <0,05; **, p <0,01; ***, p <0,001; ****, p <0,0001).

145
4. Chapitre 4 : Lien fonctionnel entre l’activité autophagique du complexe chaperon
HSPB8-BAG3 et la régulation de structures à base d’actine pendant la division
cellulaire
Dans les chapitres 2 et 3 nos résultats suggèrent que le complexe chaperon HSPB8-BAG3
facilite, au cours de la division cellulaire, le remodelage du cytosquelette d’actine d’une
manière qui pourrait faire intervenir son rôle dans l’autophagie sélective, d’une part, et la
modulation de l’activité de la déacétylase HDAC6 notamment sur la cortactine, d’autre
part. En outre, il a été démontré qu’HDAC6 régule la fusion des autophagosomes avec les
lysosomes lors de l’autophagie sélective par un mécanisme qui implique la cortactine et la
polymérisation de l’actine (Lee et al. 2010). Il est donc possible que la régulation
d’HDAC6 par le complexe HSPB8-BAG3 contribue à moduler la dégradation des
protéines par un mécanisme d’autophagie sélective, lequel pourrait jouer un rôle clé dans
la régulation spatiotemporelle du remodelage du cytosquelette d’actine. Nous avons donc
voulu confirmer qu’il existe bien un lien fonctionnel entre la fonction autophagique du
complexe chaperon HSPB8-BAG3 et son activité sur le cytosquelette d’actine, et explorer
si ce lien implique la modulation de l’activité d’HDAC6.
Dans ce chapitre sont présentés des résultats préliminaires suggérant qu’au moins en partie
la fonction autophagique du complexe HSPB8-BAG3 est impliquée dans la régulation de
la dynamique d’actine et qu’une telle activité requiert la modulation de l’activité
déacétylase d’HDAC6.
Dans un premier temps, nous verrons des résultats complémentaires concernant l’influence
de l’inhibition du flux autophagique sur la modulation de la dynamique d’actine au cours
de la division cellulaire. Ensuite, nous analyserons l’impact de la déplétion d’HSPB8 sur
le flux autophagique et sur l’autophagie sélective lors de la cytokinèse. Finalement, nous
étudierons l’effet de l’inhibition d’HDAC6 sur la régulation du flux autophagique et du
désassemblage de l’AC dans des cellules déplétées en HSPB8 lors de la cytokinèse.

146
4.1. La déplétion d’ATG7 récapitule les défauts de remodelage de l’actine observés
suivant la déplétion de BAG3, HSPB8 ou p62/SQSTM1 lors de la division
cellulaire
Il a été montré que l’autophagie est toujours active à l’entrée des cellules en mitose bien
que la formation des autophagosomes soit réduite (Furuya et al. 2010; Li et al. 2016).
Cependant les fonctions assurées par la machinerie autophagique au cours de la division
cellulaire sont très peu connues. Il a été montré que le ciblage par p62/SQSTM1 de la
GTPase RhoA à la dégradation autophagique contribue à l’abscission des cellules filles et
au désassemblage de l’AC (Belaid et al. 2013). De plus, en mitose, l’adaptateur
autophagique participe au ciblage de la cycline A2 à la machinerie autophagique lors de
transition métaphase-anaphase (Loukil et al. 2014). Ainsi, ces études ont révélé certains
mécanismes dans lesquels l’autophagie sélective est requise pour la progression de la
division cellulaire. Or, le complexe chaperon HSPB8-BAG3 a été impliqué dans la
régulation de l’autophagie sélective dans les cellules musculaires et nous avons montré
qu’il est important pour la progression mitotique des cellules cancéreuses. Nous avons donc
émis l’hypothèse que la fonction mitotique du complexe HSPB8-BAG3 impliquerait sa
fonction régulatrice de l’autophagie sélective.
Dans ce sens, dans le chapitre 2, des résultats suggèrent que l’inhibition de l’activité
lysosomale ainsi que l’inhibition de la fusion des autophagosomes avec le lysosome
conduit à une accumulation anormale de F-actine au PI mimant la déplétion d’HSPB8 en
cytokinèse (Chapitre 2, Figure 2.6A). En outre, l’activation de l’autophagie non sélective
par traitement avec la rapamycine peut corriger ce défaut dans les cellules déplétées en
HSPB8 (Chapitre 2, Figure 2.6B). Ceci nous suggère que la modulation de la dynamique
du cytosquelette d’actine par le complexe chaperon HSPB8-BAG3 serait dépendante de sa
fonction dans le ciblage de protéines à la machinerie autophagique. Pour corroborer cette
hypothèse et renforcer nos résultats, nous avons décidé d’inhiber le flux autophagique de
façon plus spécifique en transfectant des cellules HeLa synchronisées par un double
blocage à la thymidine avec des siARN ciblant la protéine ATG7 impliquée dans la
formation des autophagosomes (Suzuki et al. 2001; Kim et al. 1999).

147
Il a été montré que des MEF (« mouse embryonic fibroblasts ») KO pour ATG5 ainsi que
des cellules pulmonaires cancéreuses déplétées d’ATG7 ou de p62/SQSTM1 présentent
une sur-activation de RhoA qui se traduit par une polymérisation excessive de la F-actine
(Belaid et al. 2013). Pour confirmer l’efficacité de nos siARN contre ATG7 nous avons
choisi d’évaluer si la déplétion d’ATG7 conduit aux mêmes phénotypes de réorganisation
du cytosquelette d’actine dans les cellules HeLa. Pour cela, nous avons transfecté nos
cellules sur une période de 24 h ou 48 h avec deux siARN ciblant ATG7 et réalisé un
marquage de la F-actine endogène à l’aide de la phalloïdine. Que ce soit 48 h ou 24 h après
la transfection des siARN, où les niveaux d’expression d’ATG7 sont réduits à moins de 25
% et 50 % respectivement (Figure 4.1A), nos observations ont été les mêmes. Nous avons
vu que les cellules transfectées avec le siCtl présentent des structures corticales et ventrales
fines de F-actine semblables aux cellules non transfectées, alors que les cellules déplétées
en ATG7 arborent des câbles épais et denses de F-actine au cortex et à la face ventrale
(Figure 4.1B). Ce résultat montre donc que la déplétion d’ATG7 s’accompagne d’un
enrichissement de F-actine au cortex et à la face ventrale des cellules comme cela a été
observé dans les MEF KO pour ATG5 (Belaid et al. 2013). Ceci nous permet donc de
confirmer l’efficacité des siARN ciblant ATG7 utilisés après 24 h ou 48 h de déplétion.

148
Figure 4.1: La déplétion d’ATG7 se traduit par un réarrangement du cytosquelette d’actine en interphase (A) Immunobuvardage de type « Western » d’extraits protéiques de cellules HeLa-RFP-H2B qui ont été transfectées avec des siARN spécifiques contre ATG7 (siATG7 #1 et siATG7 #2) pendant 24 h ou 48 h. La déplétion a été estimée à 50 % et plus de 75 % après 24 h et 48 h de transfection respectivement par le dépôt de quantités croissantes d’extraits protéiques contrôles (siCtl). L’analyse par immunobuvardage a été effectuée avec un anticorps anti-ATG7; GAPDH : contrôle de dépôt. (B) Images d’une coupe confocale de la face ventrale de cellules HeLa-RFP-H2B asynchrones déplétées en ATG7 pour une période de 24 h montrant un enrichissement ventral et cortical de F-actine; les flèches indiquent la position où ont été réalisées les coupes pour le zoom x2 présenté dans les encadrés; l’ADN et la F-actine ont été marqués avec le Hoechst et la phalloïdine-488 respectivement; échelle, 30 µm. Par la suite, nous avons testé si la déplétion d’ATG7 provoque un ralentissement de la
progression mitotique comme nous avons pu l’observé dans des cellules déplétées en
BAG3 ou en HSPB8 (Annexe 1, Figure 2E). Nous avons donc quantifié l’index mitotique
des cellules HeLa synchronisées par un double blocage à la thymidine, préalablement

149
transfectées avec des siARN dirigés contre ATG7 pour une période de 48 h. Dans ce
contexte, plus de 70 % des cellules contrôles sont en division cellulaire (mitose et
cytokinèse) tandis que cette proportion est drastiquement réduite suite à la transfection du
siATG7 #1; dans ces conditions, plus de 90 % des cellules sont en interphase (Figure 4.2A).
En revanche, les cellules transfectées avec le siATG7 #2 présentent les mêmes proportions
de cellules en division cellulaire que les cellules contrôles (Figure 4.2A). Nous avons
également constaté que ~20 % des cellules contrôles sont en mitose (prométaphase-
métaphase) après 8 h 30 de relâche tandis que ~30 % des cellules déplétées avec le siATG7
#2 sont dans cette phase (Figure 4.2B). Ces observations montrent que le siATG7 #2
n’interfère pas avec l’entrée et peu avec la progression des cellules en mitose,
contrairement au siATG7 #1. Une telle différence entre les deux siARN ciblant ATG7
pourrait dépendre d’une différence d’efficacité des siARN dans la déplétion d’ATG7 ou
d’un ciblage non spécifique (« off target ») conduisant à des phénotypes différents. Par
ailleurs, après 24 h de déplétion, lorsque 50 % des niveaux protéiques d’ATG7 sont réduits
(Figure 4.1A), aucun ralentissement de la progression des cellules dans les différents stades
de la division cellulaire n’a pu être observé avec les deux différents siARN ciblant ATG7
(Figure 4.2C). Ces résultats indiquent donc que la forte déplétion d’ATG7 (75 % de
déplétion) peut conduire soit à un fort ralentissement des cellules dans le cycle cellulaire,
soit à un léger ralentissement des cellules en mitose. Tandis qu’une plus faible déplétion
d’ATG7 (50 % de déplétion) ne semble pas affecter la progression des cellules mitotiques.
Figure 4.2: Effet de la déplétion d’ATG7 sur la progression mitotique des cellules HeLa. (A) Le graphique représente la proportion de cellules HeLa-RFP-H2B en interphase et en division cellulaire (mitose + cytokinèse) après la déplétion d’ATG7 avec deux siARN

150
différents (siATG7 #1 et siATG7 #2) sur 48 h et après une relâche de 8 h 30 suite à un double blocage à la thymidine; les résultats représentent la moyenne de deux expériences indépendantes +/- SEM où 33 à 573 cellules ont été analysées. (B) Histogramme du pourcentage de cellules HeLa-RFP-H2B dans les différentes phases mitotiques; les cellules ont été transfectées avec le siCtl ou le siATG7 #2 sur une période de 48 h et synchronisées par un double blocage à la thymidine en mitose; les résultats représentent la moyenne de deux expériences indépendantes +/- SEM où 29 à 424 cellules ont été quantifiées. (C) Graphique montrant le pourcentage de cellules HeLa-RFP-H2B synchronisées par un double blocage à la thymidine dans les différentes phases mitotiques suite à une relâche de 8 h 30 et 24 h après la transfection d’un siARN contrôle (siCtl) ou de siARN ciblant ATG7 (siATG7 #1 et siATG7 #2); les résultats représentent la moyenne de trois expériences indépendantes +/- SEM où 38 à 642 cellules ont été analysées. Aucune différence significative n’a été calculée entre la condition siCtl et les conditions siATG7 quelque soit la phase de la division cellulaire analysée par le test statistique Fisher.
Nous avons observé que la déplétion d’HSPB8, comme la déplétion de BAG3 ou de
p62/SQSTM1, interfère avec l’arrondissement mitotique des cellules en métaphase
suggérant que le complexe protéique faciliterait le remodelage du réseau cortical d’actine
et de myosine II (Annexe 1, Figure 7A, Chapitre 3, Figure 3.2E et F). Nous avons donc
testé si la déplétion d’ATG7 pouvait récapituler ce phénotype. Pour cela, nous avons
synchronisé des cellules HeLa en mitose et les avons déplétées pour une période de 24 h
avec les siARN ciblant ATG7. Nous avons observé que ~10 % des cellules contrôles en
métaphase démontrent un défaut d’arrondissement mitotique (Figure 4.3). De façon
intéressante, la déplétion d’ATG7, quelque soit le siARN utilisé, induit un doublement de
la proportion de cellules présentant ce phénotype anormal comme nous avons pu l’observer
suite à la déplétion de BAG3 ou d’HSPB8 (Figure 4.3). Par conséquent, la déplétion
d’ATG7 semble interférer avec l’arrondissement des cellules en métaphase suggérant que
la dégradation autophagique est importante pour le remodelage du cytosquelette d’actine
qui dirige l’arrondissement mitotique.

151
Figure 4.3 : La déplétion d’ATG7 conduit à l’accumulation de cellules partiellement arrondies en métaphase Images confocales de HeLa-RFP-H2B synchronisées en métaphase 24 h après la transfection d’un siARN contrôle (siCtl) ou ciblant ATG7 (siATG7 #2) présentant une géométrie mitotique normale, ou anormale, respectivement ; un anticorps anti-a-tubuline a été utilisé pour marquer le fuseau mitotique, l’ADN et la F-actine ont été marqués avec le Hoechst et la phalloïdine-488 respectivement ; échelle, 10 µm. Le graphique représente le pourcentage de cellules partiellement rondes en métaphase suite à la transfection sur 24 h des siCtl, siATG7 #1 ou siATG7 #2 ; moyenne de deux expériences indépendantes +/-SEM où au moins 189 cellules ont été analysées. Finalement, nous avons testé si la déplétion d’ATG7 récapitule les phénotypes observés
suivant la déplétion d’HSPB8 en cytokinèse. En effet, dans le chapitre 2, nos résultats
montrent que la déplétion d’HSPB8 se caractérise par une accumulation anormale de F-
actine au PI qui provoque une augmentation significative des défauts de cytokinèse (i.e. bi-
et multi-nucléation; Chapitre 2, Figure 2.2E et 2.3D). Pour tester notre hypothèse, nous
avons synchronisé des cellules HeLa en cytokinèse et les avons déplétées sur 24 h en
ATG7. Nos résultats montrent que ~23 % des cellules transfectées avec le siCtl présentent
une accumulation anormale de F-actine au PI, comparativement à 40 à 45 % pour les
cellules déplétées en ATG7 (Figure 4.4). Ce résultat indique que la déplétion d’ATG7
s’accompagne d’une accumulation anormale de F-actine au PI et suggère que le flux
autophagique est impliqué dans le désassemblage de l’AC en cytokinèse.

152
Figure 4.4: La déplétion d’ATG7 augmente la proportion de cellules présentant une accumulation anormale de F-actine au PI Images confocales de cellules HeLa-RFP-H2B synchronisées en cytokinèse et transfectées avec des siARN spécifiques à ATG7 montrant une accumulation anormale de F-actine au PI (flèches) dont le zoom x4 est dans les boîtes juxtaposées. Le PI a été marqué à l’aide d’un anticorps anti-Aurora B et la F-actine avec la phalloïdine-488 ; échelle, 10 µm. L’histogramme présente le pourcentage de cellules arborant une accumulation anormale de F-actine au PI en cytokinèse. Les données représentent la moyenne de quatre expériences indépendantes +/- SEM où au moins 521 cellules ont été analysées. Le test statistique Fisher a été employé et compare les deux siATG7 avec le siCtl.
En résumé, nos résultats indiquent que la déplétion d’ATG7, en mitose ou en cytokinèse,
récapitule les défauts de remodelage du cytosquelette d’actine observés suivant la déplétion
de BAG3, d’HSPB8 ou de p62/SQSTM1. Ceci suggère un lien potentiel entre le
remodelage mitotique de l’actine et le processus d’autophagie sélective modulé par le
complexe HSPB8-BAG3, en association avec l’adaptateur autophagique p62/SQSTM1.
Nous avons vu que la déplétion d’ATG7 provoque un ralentissement plus ou moins
important des cellules en mitose ou dans le cycle cellulaire suivant les niveaux de déplétion
de la protéine et les siARN utilisés. Ceci est en accord avec la littérature où il a été montré

153
que la déplétion d’ATG7 dans des cellules MCF7 conduit à un ralentissement des cellules
en mitose (Loukil et al. 2014). Il est à noter que seul le siATG7 #1 provoque un important
ralentissement des cellules dans le cycle cellulaire après 48 h de déplétion. Ceci pourrait
indiquer une variation de la pénétrance du phénotype associé à la déplétion d’ATG7. Ce
fort ralentissement pourrait également suggérer que ce phénotype est indépendant de la
déplétion d’ATG7 et lié à un « off-target » du siARN. Pour confirmer ou non ce résultat il
nous faudra réaliser d’autres expériences avec de nouvelles séquences de siARN dirigés
contre ATG7. Par ailleurs, en utilisant des siARN ciblant d’autres protéines ATG
impliquées dans des stades plus tardifs du flux autophagique nous pourrons appuyer nos
résultats et confirmer l’implication de l’autophagie dans la progression mitotique.
Nos résultats suggèrent que la déplétion d’ATG7 interfère avec l’arrondissement normal
des cellules en métaphase ainsi que le démantèlement de l’AC en cytokinèse, tout comme
nous avons pu le voir suivant la déplétion de BAG3, HSPB8 ou p62/SQSTM1 (Annexe 1,
Figure 7A; Chapitre 2 Figure 2.3D; Chapitre 3, Figure 3.2E et F). Ce résultat renforce ceux
présentés dans le chapitre 2 et notre hypothèse d’une inhibition du flux autophagique en
réponse à la déplétion de l’une de ces trois protéines.
Par ailleurs, ces observations sont en accord avec celles de Belaid et al. montrant
qu’interférer avec le flux autophagique provoque une accumulation de F-actine au PI
inhibant la progression de la cytokinèse (Belaid et al. 2013). Il est à noter que
l’arrondissement des cellules en métaphase ainsi que l’assemblage et la contraction de l’AC
sont deux mécanismes impliquant la régulation fine spatiotemporelle de la GTPase RhoA
(Maddox et Burridge 2003; Matthews et al. 2012; Rosa et al. 2015; Cheffings, Burroughs,
et Balasubramanian 2016). Or, il a été montré que RhoA sous sa forme active est ciblée à
la dégradation autophagique par l’adaptateur autophagique et partenaire du complexe
chaperon : p62/SQSTM1 (Belaid et al. 2013; Fuchs et al. 2015; Gamerdinger et al. 2011).
Ceci nous suggère que le complexe chaperon HSPB8-BAG3 pourrait faciliter le ciblage de
RhoA-GTP par p62/SQSTM1 à la machinerie autophagique permettant le contrôle fin de
son activation durant la division cellulaire. Pour confirmer cette hypothèse, nous pourrions
réaliser des expériences de déplétion de BAG3 ou d’HSPB8 et analyser l’effet d’une telle

154
déplétion sur la stabilité de RhoA-GTP par « pull-down » à partir d’extraits de cellules
synchronisées en mitose ou en cytokinèse. Par ailleurs, le domaine WW de BAG3 a été
impliqué dans sa fonction de régulation de l’autophagie sélective (Merabova et al. 2015;
Arndt et al. 2010; Ulbricht, Arndt, et Höhfeld 2013). Nous pourrions étudier l’impact de
l’expression d’un mutant de délétion du domaine WW de BAG3 sur la stabilité de RhoA-
GTP par « pull-down » également. Nous pourrions aussi analyser l’effet de l’expression
d’un tel mutant sur les défauts de remodelage de l’actine observés au cours de la division
cellulaire suite à la déplétion de BAG3. De plus, il serait intéressant de vérifier que la
déplétion d’ATG7 ou d’autres protéines ATG s’accompagne d’une accumulation de RhoA-
GTP au PI comme cela a été montré dans la littérature (Belaid et al. 2013). Nous pourrions
également examiner si la déplétion d’HSPB8 ou de BAG3 se caractérise aussi par
l’accumulation de RhoA-GTP au PI à l’aide d’un anticorps spécifique (Selva et Egea 2011;
Winitthana, Lawanprasert, et Chanvorachote 2014). Un tel résultat permettrait de corréler
l’implication de l’autophagie sélective dans le phénotype d’accumulation de F-actine au PI
dans les cellules déplétées en HSPB8 et appuierait l’hypothèse d’un lien fonctionnel entre
la fonction autophagique du complexe HSPB8-BAG3 et son activité sur le cytosquelette
d’actine.
Finalement, l’arrondissement des cellules en métaphase requiert l’activation du complexe
ERM qui assure la distribution correcte de la tension corticale (Roubinet et al. 2011; Kunda
et al. 2008). Or, une étude de l’interactome de BAG3 a montré que l’ezrine est un partenaire
du co-chaperon en condition basale (Chen et al. 2013). Il est donc possible que le complexe
chaperon HSPB8-BAG3 module l’activité du complexe ERM en mitose via le ciblage de
l’ezrine à la machinerie autophagique ce qui assurerait le renouvellement de la protéine.
Pour confirmer cette hypothèse, nous pourrions réaliser des expériences de déplétion de
BAG3 ou d’HSPB8 et analyser l’effet d’une telle déplétion sur la stabilité de l’ezrine par
immunobuvardage de type « Western ». Par ailleurs, nous pourrions effectuer des
expériences de FRAP « Fluorescence Recovery After Photobleaching » en utilisant la
protéine ezrine étiquetée GFP dans des cellules déplétées en BAG3 ou en HSPB8 et
synchronisées en mitose. Une augmentation du temps de recouvrement de la fluorescence

155
au lieu de photoblanchiment suggèrera que les protéines du complexe chaperon sont
impliqués dans le renouvellement de l’ezrine.
4.2. La déplétion d’HSPB8 interfère avec le flux autophagique dans les cellules en
cytokinèse
4.2.1. La déplétion d’HSPB8 cause une augmentation de l’intensité et du volume
des vésicules LC3
Nos résultats suggèrent que les phénotypes de réorganisation anormale du cytosquelette
d’actine observés suite à la déplétion d’une des protéines du complexe chaperon HSPB8-
BAG3 sont liés un défaut dans le ciblage de protéines à la dégradation autophagique. Afin
de valider cette hypothèse, nous avons analysé l’effet de la déplétion d’HSPB8 sur le
marqueur typique de l’autophagie : LC3. En effet, celui-ci s’insère dans la membrane des
autophagosomes et sa dégradation est le reflet de l’efficacité du flux autophagique. Ainsi
la comparaison du nombre de vésicules LC3 par immunofluorescence avant et après
l’inhibition du flux autophagique à l’aide de drogues permet de déterminer l’état
d’activation du flux autophagique (Klionsky et al. 2016).
Pour ce faire, nous avons réalisé des immunofluorescences de LC3 dans des cellules HeLa
synchronisées en cytokinèse et déplétées en HSPB8. Nos quantifications ont montré qu’en
condition basale la déplétion d’HSPB8 conduit à une réduction significative du nombre de
vésicules LC3 en comparaison à la condition contrôle suggérant que la déplétion d’HSPB8
pourrait influencer la biogenèse des autophagosomes et/ou leur dégradation (Figure 4.5A).
Afin de déterminer l’impact sur le flux autophagique en cytokinèse, nous avons traité les
cellules HeLa synchronisées en cytokinèse et déplétées en HSPB8 durant 1 h avec des
inhibiteurs de l’activité lysosomale (Pep+E64d) ou un inhibiteur de la fusion des
autophagosomes avec le lysosome (BafA). Pour les cellules contrôles, quelque soit la
drogue utilisée nous avons observé une augmentation d’au minimum 1,5 fois du nombre
d’autophagosomes par cellule permettant d’appuyer la présence d’un flux autophagique
dans ces conditions (Figure 4.5A). Une augmentation de ~2,3 fois du nombre

156
d’autophagosomes est également observable dans les cellules déplétées en HSPB8 suite au
traitement avec les différents inhibiteurs, ce qui suggère un flux autophagique robuste dans
ces cellules (Figure 4.5A). Nous avons aussi quantifié le nombre d’autophagosomes suite
à l’activation de l’autophagie par un traitement d’une heure à la rapamycine (Figure 4.5A).
Dans les cellules contrôles, une réduction du nombre d’autophagosomes est observable en
réponse à la rapamycine, ce qui est consistent avec une augmentation de flux autophagique
(~2,3 fois; Figure 4.5A). Le traitement avec la rapamycine induit une réduction de ~1,5
fois du nombre d’autophagosomes dans les cellules transfectées avec le siHSPB8 #3
suggérant que l’activation du flux autophagique a été efficace dans ces conditions, bien
que réduite (Figure 4.5A). Ainsi, la quantification du nombre d’autophagosomes révèle que
le flux autophagique est actif dans les cellules en cytokinèse comme cela a été démontré
dans des études précédentes (Belaid et al. 2013; Li et al. 2016). En outre, nos observations
suggèrent qu’un flux autophagique est maintenu dans les cellules déplétées en HSPB8.
Ceci contraste avec les données de la littérature indiquant que le chaperon activerait le flux
autophagique (Carra et al. 2008; Hamouda et al. 2014; Li et al. 2017; Crippa, Sau, et al.
2010). Néanmoins, la réduction du nombre d’autophagosomes en condition basale ou suite
aux traitements dans les cellules déplétées en HSPB8 en comparaison aux cellules
contrôles (Figure 4.5A) suggère que des étapes du flux autophagique (i.e. initiation ou
maturation) pourraient être affectées suivant la déplétion d’HSPB8 (Klionsky et al. 2016).
Dans ce sens, nous avons observé que l’intensité moyenne et le volume moyen des
autophagosomes sont significativement plus élevés dans les cellules déplétées en HSPB8
que dans les cellules transfectées avec le siCtl en condition basale (Figure 4.5B et C). Ceci
suggère que la déplétion en HSPB8 conduit à un enrichissement de la protéine LC3 dans
les vésicules autophagiques et à une augmentation de leur volume. Suite aux traitements
avec des inhibiteurs du flux autophagique, nous avons observé une augmentation d’au
minimum 2 fois de l’intensité et de ~1,2 fois du volume des autophagosomes dans les
celules contrôles (Figure 4.5B et C). Ces observations suggèrent donc qu’inhiber le flux
autophagique dans les cellules contrôles induit une augmentation l’intensité et du volume
des vésicules autophagiques ce qui est en accord avec la littérature (Tumbarello et al. 2012;
Klionsky et al. 2016). Il est intéressant de noter que l’intensité moyenne des

157
autophagosomes suite à l’inhibition du flux autophagique dans les cellules contrôles se
rapproche de celle observée en condition basale dans les cellules déplétées en HSPB8.
Cette observation suggère que l’augmentation de l’intensité des autophagosomes observée
suite à la déplétion d’HSPB8 pourrait être liée à une réduction ou une inefficacité du flux
autophagique. Dans les cellules déplétées en HSPB8, l’inhibition du flux autophagique
conduit à une augmenation plus mesurée de l’intensité moyenne des vésciules LC3 (BafA :
~1,2 fois; Pep+E64d : ~1,3 fois) suggérant que le flux autophagique n’est pas inhibé mais
pourrait être moins efficace suivant la déplétion d’HSPB8 (Figure 4.5B). En revanche, nous
avons observé que le traitement à la BafA conduit à une augmentation de ~1,5 fois du
volume des vésicules LC3 tandis que le traitement à la Pepstatine et E64d affecte peu leur
volume (Figure 4.5C). Ceci indique qu’inhiber la fusion des autophagosomes avec le
lysosome accentu le phénotype provoqué par la déplétion d’HSPB8 sur le volume des
vésicules LC3 alors que l’inhibition de l’activité des enzymes lysosomales non. Cette
observation suggère qu’HSPB8 pourrait agir sur la même voie que la BafA et donc pourrait
faciliter l’étape de fusion des autophagosomes avec le lysosome comme cela a été proposé
(Li et al. 2017; Kwok et al. 2011). Alternativement, la déplétion d’HSPB8 pourrait agir sur
la maturation des autophagolysosomes. En effet, une augmentation du volume pourrait
traduire une perturbation de la maturation des autophagolysosomes comme cela a été
observé suivant la déplétion de Rab7 dans les cellules de mammifères (Kuchitsu et al.
2018).
En conclusion, nos résultats préliminaires suggèrent que la déplétion d’HSPB8 interfère
avec l’activité de la machinerie autophagique dans les cellules en cytokinèse, mais ne nous
permettent pas de déterminer à quelle étape du flux autophagique elle interviendrait. Les
résultats présentés suggèrent que le chaperon pourrait faciliter la biogénèse des
autophagosomes et/ou la maturation des autophagosomes et/ou des autophagolysosomes
(Klionsky et al. 2016; Kuchitsu et al. 2018). Il est intéressant de noter que le complexe
Arp2/3 est proposé pour être central dans les processus de formation, de maturation et de
fusion des autophagosomes avec le lysosome (Monastyrska et al. 2008; Kast et Dominguez
2015; Mi et al. 2015; Höhfeld 2016; Zientara-Rytter et Subramani 2016b; Coutts et La
Thangue 2016; Lee et al. 2010). En effet, la polymérisation d’actine branchée induite par

158
Arp2/3 serait nécessaire pour le trafic intracellulaire de membranes contenant ATG9
(phagophores), pour la courbure membranaire lors de l’étape d’initiation, le contrôle du
volume des autophagosomes, la motilité des autophagosomes et leur fusion avec le
lysosome. Les résultats présentés aux chapitres 2 et 3 suggèrent que le complexe chaperon
HSPB8-BAG3 limiterait l’activité d’Arp2/3 en mitose et en cytokinèse. La perturbation de
l’organisation des vésicules LC3 observées suivant la déplétion d’HSPB8 pourrait donc
être une conséquence de la dérégulation de l’activité d’Arp2/3. Par ce mécanisme le
complexe pourrait faciliter la dégradation autophagique de protéines endommagées lors de
l’assemblage/désassemblage des structures du cytosquelette d’actine pendant la division
cellulaire, comme cela a été observé dans le muscle squelettique (Arndt et al. 2010;
Ulbricht et al. 2013).
Il nous faudra vérifier nos résultats en répétant nos expériences. En outre, afin de
déterminer si une ou plusieurs étapes du flux autophagique sont affectées par la dépltion
d’HSPB8, il sera essentiel d’effectuer des immunofluorescences de marqueurs de
phagophores comme ATG9, d’autophagosomes immatures comme ATG16 et
d’autophagolysosomes matures comme LAMP-1 (Klionsky et al. 2016; Kuninori Suzuki
et al. 2007; Kuchitsu et al. 2018). Ceci est essentiel afin de déterminer la nature des
vésicules autophagiques présentant une augmentation de volume et d’intensité LC3, i.e.
autophagosomes (positifs pour ATG16) versus autophagolysosomes (positifs pour LAMP-
1). De plus, nous pourrions tirer avantage de l’outil LC3-clover-rubby pour déterminer si
le flux autophagique est perturbé dans les cellules déplétées en HSPB8. En effet, cet outil
permet in situ de suivre le flux autophagique en suivant la localisation de LC3. Le capteur
en tandem clover (vert) et rubby (rouge) permet de déterminer si LC3 est localisée dans un
autophagosome ou dans un autolysosome en raison de l’instabilité de clover à un pH faible
tel que le pH du lysosome (Klionsky et al. 2016). LC3-clover-rubby pourrait nous
permettre de déterminer de façon si la fusion des autophagosomes avec le lysosome peut
être réduite en réponse à la déplétion d’HSPB8 comme cela a été proposé dans la littérature
(Kwok et al. 2011; Li et al. 2017).

159
Figure 4.5: La déplétion d’HSPB8 interfère avec le flux autophagique en cytokinèse. (A) Des cellules HeLa-RFP-H2B ont été synchronisées en cytokinèse et déplétées sur 48 h en HSPB8. Les images confocales montrent la distribution des autophagosmes après 1 h de traitement avec 10 µg/ml de pepstatine et 10 µg/ml de E64d, ou 200 nM de BafA, ou 150 nM de rapamycine, ou le contrôle DMSO. Les microtubules ont été marqués avec un anticorps anti-a-tubuline et les autophagosomes avec un anticorps anti-LC3 ; échelle, 10 µm. Le graphique représente le nombre d’autophagosomes par cellule quantifié pour

160
chacune des conditions indiquées ; représentatif d’une à deux expériences +/- SEM où au moins 21 cellules ont été analysées. L’analyse statistique compare la condition siCtl à la condition siHSPB8 pour le traitement indiqué par le test Mann Whitney. (B) Représentation graphique de l’intensité moyenne des autophagosomes par cellule suite à la transfection de cellules HeLa-RFP-H2B synchronisées en cytokinèse du siCtl sur 48 h et traitées durant 1 h avec 10 µg/ml de pepstatine et 10 µg/ml de E64d ou 200 nM de BafA ; représentatif d’une à deux expériences +/- SEM où au moins 21 cellules ont été analysées. L’analyse statistique compare la condition siCtl à la condition siHSPB8 pour le traitement indiqué par le test Mann Whitney. (C) Représentation graphique de la distribution du volume moyen et du nombre d’autophagosomes par cellule suite à la transfection sur 48 h de cellules HeLa-RFP-H2B synchronisées en cytokinèse des siCtl ou siHSPB8 #3 ; représentatif d’une à deux expériences +/- SEM où au moins 21 cellules ont été analysées. L’analyse statistique compare la condition siCtl à la condition siHSPB8 pour le traitement indiqué par le test Mann Whitney.
4.2.2. La déplétion d’HSPB8 est associée avec une accumulation de protéines
poly-ubiquitinées sur K63 dans les cellules en cytokinèse
Il est connu que l’autophagie sélective implique généralement l’ubiquitination des substrats
sur K63 (Tan et al. 2008; Huang et al. 2013; Clough et al. 2016; McKeon et al. 2015; Xu
et al., 2009). p62/SQSTM1 qui forme un complexe avec HSPB8 et BAG3 dans les cellules
en mitose (Annexe 1, Figure 6D; Chapitre 3, Figure 3.3C), se lie préférentiellement aux
substrats portant cette modification (Wooten et al. 2008). Nous avons donc analysé l’effet
de la déplétion d’HSPB8 sur les protéines poly-ubiquitinées sur K63.
Pour cela, nous avons réalisé des immunofluorescences de l’ubiquitine K63 dans des
cellules HeLa synchronisées en cytokinèse et déplétées en HSPB8. Nos résultats montrent
qu’en condition basale, dans les cellules contrôles, les protéines poly-ubiquitinées sur K63
forment de petits agrégats (ci-après identifiés sous le nom d’agrégats K63) compris entre
0,2 et 0,4 µm3 dispersés dans la cellule et dont le nombre moyen est de 100 par cellule
(Figure 4.6A et C). Après inhibition de la fusion des autophagosomes avec le lysosome par
un traitement avec la BafA, une augmentation du volume (entre 0,3 et 0,5 µm3) et du
nombre (~200 par cellule) de ces agrégats est observable (Figure 4.6A et C). De façon
remarquable, la déplétion d’HSPB8 en condition basale s’accompagne d’un doublement
du nombre d’agrégats K63 par cellule en comparaison aux cellules contrôles montrant une
accumulation de protéines poly-ubiquitinées sur K63 dans ces conditions (Figure 4.6B et

161
C). De plus, il n’est pas rare d’observer de gros agrégats K63 qui font augmenter
légèrement le volume moyen de ceux-ci (~0,3 µm3; Figure 4.6C et D). Ceci suggère qu’en
l’absence d’HSPB8, les protéines ubiquitinées sur K63 s’accumulent et seraient prises en
charge moins efficacement. Suite au traitement avec la BafA, nous avons observé que le
volume moyen des agrégats K63 augmente de façon moins drastique dans les cellules
déplétées en HSPB8 que dans les cellules transfectées avec le siCtl (1,09 fois contre 1,2
fois; Figure 4.6D), corroborant ainsi l’effet observé précédemment sur les vésicules LC3.
Suite au traitement avec la BafA, nous avons observé que le volume moyen des agrégats
K63 augmente de façon moins drastique dans les cellules déplétées en HSPB8 que dans les
cellules transfectées avec le siCtl (1,09 fois contre 1,2 fois; Figure 4.6D). Il semble donc
que le traitement avec la BafA ait un effet plus restreint dans les cellules déplétées en
HSPB8 en comparaison aux cellules contrôles. Il est intéressant de noter que les résultats
obtenus suite à l’inhibition de l’autophagie dans les cellules contrôles sont proches de ceux
obtenus en condition basale dans les cellules déplétées en HSPB8 en termes de volume et
de nombre d’agrégats K63 (Figure 4.6A et B). Ces observations suggèrent que la déplétion
d’HSPB8 est mimée par un traitement avec la BafA et suggèrent donc qu’une telle
déplétion inhibe la fusion des autophagosomes avec le lysosome ou la maturation des
autophagolysosomes. De façon intéressante, nous avons aussi noté que le nombre
d’agrégats K63 est réduit en réponse au traitement avec la BafA dans les cellules déplétées
en HSPB8 à la différence des cellules contrôles et nos quantifications indiquent qu’il s’agit
du nombre de petits agrégats K63 dont le volume est inférieur à 0,3 µm3 qui est réduit
(Figure 4.6E).
Nos résultats suggèrent donc que l’inhibition du flux autophagique induit une accumulation
d’agrégats K63 en cytokinèse. Ce résultat est concordant avec ce qui a été observé dans la
littérature où l’inhibition du flux autophagique conduit à une agrégation des protéines poly-
ubiquitinées et suggère que l’autophagie sélective est active dans ces conditions (Hara et
al. 2006; Riley et al. 2010; Komatsu et al. 2006; Lee et al. 2011). L’ensemble de nos
résultats suggèrent qu’en condition basale, dans les cellules déplétées en HSPB8,
l’autophagie sélective est moins efficace ce qui va dans le sens de ce qui a été proposé par
les travaux de l’équipe du Dr Höhfeld dans le muscle squelettique (Ulbricht, Arndt, et

162
Höhfeld 2013; Arndt et al. 2010). En outre, il est connu que les chaperons de la famille
HSPB sont importants pour contrôler l’agrégation protéique au cours d’un stress
protéotoxique (Haslbeck et Vierling 2015). L’inactivation de cette fonction se caractérise
par la formation de gros agrégats protéiques poly-ubiquitinés toxiques pour la cellule.
Ainsi, la disparition des petits agrégats K63 au profit d’agrégats plus volumineux suivant
la déplétion d’HSPB8 est en accord avec la fonction chaperon de la protéine (Carra et al.
2005). Aussi, nos résultats tendent à indiquer qu’en réponse à la déplétion d’HSPB8 en
cytokinèse il y a une réduction de l’efficacité de l’autophagie sélective.
Il nous faudra répéter ces expériences afin d’appuyer nos résultats. Par ailleurs,
l’autophagie sélective implique des adaptateurs autophagiques comme p62/SQSTM1 qui
est un partenaire important du complexe chaperon HSPB8-BAG3 dans les cellules en
mitose (Fuchs et al. 2015). Il est connu que l’oligomérisation de p62/SQSTM1 est
essentielle à l’agrégation des protéines ubiquitinées et à leur ciblage à la machinerie
autophagique (Matsumoto et al. 2011; Wurzer et al. 2015). Ainsi interférer avec
l’oligomérisation de l’adaptateur autophagique réduirait l’agrégation et l’élimination des
protéines ubiquitinées sur K63 par la machinerie autophagique, tout comme nous l’avons
observé suite à la déplétion en HSPB8 en cytokinèse. Afin de comprendre l’impact
d’HSPB8, il sera donc important d’analyser l’effet de sa déplétion sur la distribution et
l’organisation de p62/SQSTM1 en cytokinèse. Aussi, la FLN est la seule cible connue du
complexe chaperon HSPB8-BAG3 au cours de l’autophagie sélective (Ulbricht, Arndt, et
Höhfeld 2013; Arndt et al. 2010). Il serait intéressant de suivre la dégradation autophagique
de la FLN, grâce à une construction FLN-clover-ruby, en réponse à la déplétion d’HSPB8
en cytokinèse (Klionsky et al. 2016).

163

164
Figure 4.6: Les cellules déplétées en HSPB8 présentent une accumulation d’agrégats K63. (A) Représentation graphique de la distribution des agrégats K63 en fonction de leur volume moyen et de leur nombre par cellule suite à la transfection de cellules HeLa-RFP-H2B synchronisées en cytokinèse avec le siCtl et traitées avec la BafA (200 nM) ou avec le contrôle DMSO ; les résultats sont représentatifs d’une expérience où 33 cellules pour la condition DMSO et 39 cellules pour la condition BafA ont été analysées. (B) Représentation graphique de la distribution des agrégats K63 en fonction de leur volume moyen et de leur nombre par cellule suite à la transfection de cellules HeLa-RFP-H2B synchronisées en cytokinèse avec le siCtl ou le siHSPB8 #3 ; les résultats sont représentatifs d’une expérience où 33 et 23 cellules ont été analysées pour les conditions siCtl et siHSPB8 #3 respectivement. (C) Des cellules HeLa-RFP-H2B ont été synchronisées en cytokinèse et traitées durant la dernière heure de relâche avec 200 nM de BafA suite à la déplétion sur 48 h d’HSPB8 puis une immunofluorescence de l’ubiquitine K63 a été effectuée. Les images confocales représentent le marquage typique des protéines poly-ubiquitinées sur K63 dans des cellules contrôles ou déplétées en HSPB8 traitées avec la BafA ou avec le contrôle DMSO. Les flèches jaunes indiquent les petits agrégats K63 et les flèches rouges ceux de plus gros volume. Les microtubules et les protéines poly-ubiquitinées sur K63 ont été marquées avec des anticorps anti-a-tubuline et anti-ubiquitine K63 respectivement ; échelle, 10 µm. (D) Graphique montrant le volume moyen par cellule des agrégats K63 après 1 h de traitement avec la BafA ou le contrôle DMSO de cellules HeLa-RFP-H2B synchronisées en cytokinèse 48 h après la déplétion d’HSPB8 avec un siARN spécifique ; médiane représentative d’une expérience. (E) Graphique représentant le nombre d’agrégats K63 dont le volume est inférieur à 0,3 µm3 par cellule après 1 h de traitement avec la BafA ou le contrôle DMSO de cellules HeLa-RFP-H2B synchronisées en cytokinèse 48 h après la déplétion d’HSPB8 avec un siARN spécifique ; médiane représentative d’une expérience.
4.3. L’inhibition d’HDAC6 normalise l’intensité et le volume des autophagosomes
ainsi que l’accumulation de F-actine au PI dans les cellules déplétées en HSPB8
Dans le chapitre 3, nous avons montré que le complexe HSPB8-BAG3 avec p62/SQSTM1
facilite le remodelage du cytosquelette d’actine en partie en limitant l’activité de la
déacétylase HDAC6. Or, HDAC6 a été impliquée dans trois fonctions importantes: (i) elle
régule la migration cellulaire en contrôlant la dynamique des cytosquelettes d’actine et de
microtubules à travers la déacétylation de la cortactine et de l’a-tubuline, respectivement;
(ii) elle régule l’autophagie sélective en contrôlant le transport des protéines poly-
ubiquitinées le long des microtubules à travers le ciblage de celles-ci à la protéine motrice
dynéine et, (iii) en contrôlant la fusion des autophagosomes avec le lysosome via la
régulation de la dynamique d’actine à l’autophagosome dépendante de la cortactine (Lee
et al. 2010; Kawaguchi et al. 2003; Zhang et al. 2007; Tran et al. 2007; Zuo et al. 2012;

165
Kaluza et al. 2011). Aussi, l’activité du nucléateur Arp2/3, dont l’activité serait limitée par
le complexe chaperon lors de la division cellulaire de façon dépendante d’HDAC6-
cortactine (chapitres 2 et 3), serait centrale pour la régulation de multiples étapes du flux
autophagique dont le trafic intracellulaire de membranes contenant ATG9 (phagophores),
la courbure membranaire lors de l’étape d’initiation, le contrôle du volume des
autophagosomes, la motilité des autophagosomes et leur fusion avec le lysosome (Zientara-
Rytter et Subramani 2016b; Mi et al. 2015; Kast et al. 2015; Kast et Dominguez 2015;
Aguilera, Berón, et Colombo 2012; Monastyrska et al. 2008; Höhfeld 2016). En outre, une
étude suggère qu’Arp2/3 pourrait participer à la neutralisation du lysosome en régulant le
recyclage de la V-ATPase, suggérant que le nucléateur pourrait également moduler la
maturation des autophagolysosomes (Carnell et al. 2011). Ainsi, nous avons émis
l’hypothèse que l’effet du complexe chaperon HSPB8-BAG3 sur le flux autophagique des
cellules en cytokinèse pourrait être dû en partie à la régulation de l’activité d’HDAC6.
Afin de répondre à notre hypothèse, nous avons traité des cellules HeLa synchronisées en
cytokinèse et déplétées en HSPB8 avec la tubacine (40 µM), un inhibiteur spécifique
d’HDAC6, durant 1 h et avons réalisé des immunofluroscences de LC3 (Figure 4.7A).
Comme nous l’avons observé précédemment, l’intensité moyenne des autophagosomes
dans les cellules déplétées en HSPB8 est plus élevée que dans les cellules contrôles en
condition basale (Figure 4.7A, DMSO). Dans les conditions expérimentales employées, le
traitement avec la tubacine provoque une augmentation de ~1,5 fois de l’intensité moyenne
des autophagosomes, suggérant ainsi que la drogue induit une réduction du flux
autophagique dans les cellules contrôles (Figure 4.7A, tubacine). De plus, dans la condition
contrôle (DMSO) le volume des autophagosomes est compris entre 0,5 et 1 µm3, alors
qu’après le traitement avec la tubacine il est compris entre 0,6 et 1,4 µm3. L’inhibition
d’HDAC6 se caractérise donc par l’augmentation du volume des vésicules LC3 dans les
cellules contrôles comme le fait un traitement avec la BafA (Figure 4.7B). Ce résultat est
concordant avec l’implication d’HDAC6 dans la fusion des autophagosomes avec le
lysosome qui a été démontrée par Lee et al. 2010. À l’inverse, le traitement avec la tubacine
conduit à une réduction de ~1,6 fois de l’intensité moyenne des autophagosomes dans les
cellules déplétées en HSPB8 (passage de 5400 à 3550; Figure 4.7A). Aussi, la proportion

166
des autophagosomes dont le volume est compris entre 0,5 et 1 µm3 (volumes de la condition
siCtl DMSO) est augmentée dans les cellules déplétées en HSPB8, tout comme le nombre
d’autophagosomes par cellule en réponse à la tubacine (Figure 4.7C). Ces résultats
indiquent que l’inhibition d’HDAC6 conduit à une réduction de l’intensité moyenne des
autophagosomes dans les cellules déplétées en HSPB8 comme le fait un traitement des
cellules à la rapamycine (Figure 4.5A). Le traitement avec la tubacine semble également
provoquer une réduction du volume moyen des autophagosomes tout en augmentant leur
nombre dans les cellules déplétées en HSPB8 (Figure 4.7C).
Ainsi, ces résultats nous suggèrent que l’inhibition de l’activité d’HDAC6 améliore le flux
autophagique dans les cellules déplétées en HSPB8, alors qu’elle le réduirait dans les
cellules contrôles. Par ailleurs, nos résultats suggèrent que le traitement avec la tubacine
des cellules contrôles, comme le traitement avec la BafA, pourrait mimer l’augmentation
du volume et de l’intensité des autophagosomes observées dans les cellules déplétées en
HSPB8. Ce résultat suggère qu’une réduction de l’efficacité du flux autophagique dans les
cellules déplétées en HSPB8 pourrait être reliée à une dérégulation d’HDAC6 dans les
cellules en cytokinèse, à l’instar de la dérégulation de HDAC6 pendant la mitose (Chapitre
3). Ces résultats ne sont pas sans rappeler l’effet de la déplétion de p62/SQSTM1 rapporté
par Yan et al. 2013, est associée avec une sur-activation d’HDAC6 et à une accumulation
anormale de F-actine dans les cellules cancéreuses (Belaid et al. 2013). Ceci indique
qu’HSPB8-BAG3, par interaction avec l’adaptateur autophagique, pourrait moduler la
fusion des autophagosomes avec le lysosome dépendante de la déacétylase en régulant son
activité au cours de la division cellulaire.
Il sera important de vérifier ces observations en répétant l’expérience. De plus, pour
confirmer la modulation par HSPB8 de l’étape de fusion dépendante d’HDAC6 et de
l’activation subséquente de la cortactine, nous pourrions réaliser un essai de restauration
du flux autophagique, dans des cellules déplétées en HSPB8, en sur-exprimant un mutant
acétyl-mimétique de cortactine. L’utilisation de l’outil LC3-clover-rubby nous permettra
de déterminer de façon claire si la fusion des autophagosomes peut être restaurée dans ces
conditions. Par ailleurs, bien que nous n’ayons pas observé d’anomalies dans la distribution

167
des agrégats K63 au sein du cytoplasme, nous ne pouvons pas exclure que le blocage partiel
du flux autophagique observé dans les cellules déplétées en HSPB8 implique également
une inhibition de la fonction autophagique d’HDAC6 dans le transport de protéines poly-
ubiquitinées agrégées. Pour répondre à cette question nous pourrions réaliser le même type
d’expérience avec la ciliobrevine D, un inhibiteur de la dynéine, ou un mutant d’HDAC6
abolissant sa liaison à la dynéine (Firestone et al. 2012; Roossien, Miller, et Gallo 2015).
Ceci nous permettrait de déterminer si interférer avec le transport des agrégats de protéines
poly-ubiquitinées dépendant de la dynéine et/ou d’HDAC6 influerait sur le flux
autophagique comme le fait la déplétion d’HSPB8.
Il semble que la capacité à moduler la dynamique du cytosquelette d’actine du complexe
chaperon HSPB8-BAG3 soit liée à son activité autophagique. Comme l’inhibition
d’HDAC6 semble restaurer le flux autophagique dans les cellules déplétées en HSPB8,
nous nous sommes demandés si une telle inhibition pouvait également corriger
l’accumulation anormale de F-actine associée à la déplétion d’HSPB8 en cytokinèse.
Pour ce faire, nous avons effectué nos comptes à partir des mêmes expériences réalisées
pour la restauration de l’arrondissement des cellules en métaphase et dont le protocole est
schématisé dans la figure 4.7 D (Chapitre 3, Figure 3.2E et F). Nous avons choisi ces
conditions afin de ne pas interférer avec la progression des cellules contrôles dans le cycle
cellulaire en utilisant une faible dose de tubacine (5 µM), comme nous l’avons montré dans
le chapitre 3. De plus, l’utilisation de ces conditions permettra de déterminer si la tubacine
peut corriger deux phénotypes différents associés à la désorganisation du cytosquelette
d’actine en réponse à la déplétion d’HSPB8. Ainsi, pour ~16 % et ~17 % des cellules
tranfectées avec le siCtl nous avons observé une accumulation anormale de F-actine au PI
en présence de DMSO et de tubacine respectivement (Figure 4.7D). Ceci indique que la
dose faible de tubacine utilisée n’affecte pas l’étape de désassemblage de l’anneau d’actine
en cytokinèse dans les cellules contrôles. De façon intéressante, une réduction de ~1,4 fois
et de ~1,9 fois de la proportion de cellules présentant ces défauts a été observée suite au
traitement avec la tubacine dans les cellules transfectées avec le siHSPB8 #3 et le siHSPB8
#1 respectivement (Figure 4.7D). Par conséquent, tout comme nous l’avons vu pour la

168
proportion de cellules présentant un défaut d’arrondissement en métaphase, le traitement
avec de faibles doses de tubacine permet de réduire le pourcentage de cellules présentant
une accumulation de F-actine au PI en cytokinèse dans les cellules déplétées en HSPB8.
Ce résultat nous indique que l’inhibition d’HDAC6 corrige les phénotypes associés à une
désorganisation du cytosquelette d’actine qui sont observés suite à la déplétion d’HSPB8
en mitose et en cytokinèse. Ceci suggère donc que la régulation de la dynamique d’actine
en cytokinèse par le complexe chaperon HSPB8-BAG3 implique la modulation de
l’activité d’HDAC6.
L’ensemble de ces résultats tend à suggérer que la modulation de l’activité d’HDAC6 par
le complexe chaperon HSPB8-BAG3 est importante pour ses fonctions de régulation du
flux autophagique et de la dynamique de l’actine. Nos résultats appuient donc un lien
fonctionnel entre la fonction autophagique du complexe HSPB8-BAG3 et son activité sur
le cytosquelette d’actine, et un tel lien semble impliquer la modulation de l’activité
d’HDAC6 par le complexe.
Toutefois, nos résultats ne nous permettent pas d’exclure un effet direct d’HDAC6 sur la
dynamique d’actine au PI indépendamment de sa fonction autophagique. En effet, les
résultats obtenus dans le chapitre 2 suggèrent fortement que le phénotype d’accumulation
de F-actine à l’AC suite à la déplétion d’HSPB8 est la conséquence d’une activation
anormale d’Arp2/3 au PI. Comme HDAC6 active la cortactine qui elle-même active le
complexe Arp2/3, il est envisageable que l’activation anormale d’Arp2/3 au PI qui est
observée dans les cellules déplétées en HSPB8 soit une conséquence de l’activation
anormale d’HDAC6. L’utilisation d’un mutant de liaison d’HDAC6 aux protéines poly-
ubiquitinées qui abolie sa fonction autophagique sans affecter son activité déacétylase
pourrait nous permettre de répondre à cette question (Kawaguchi et al. 2003).

169
Figure 4.7 : L’inhibition de l’activité d’HDAC6 restaure un flux autophagique normal et normalise la quantité de F-actine au PI dans les cellules déplétées en HSPB8. (A) Des cellules HeLa-RFP-H2B ont été synchronisées en cytokinèse et déplétées sur 48 h en HSPB8. Les images confocales montrent la distribution des autophagosmes après 1 h de traitement avec 40 µM de tubacine ou le contrôle DMSO. Les microtubules ont été

170
marqués avec un anticorps anti-a-tubuline et les autophagosomes avec un anticorps anti-LC3, échelle, 10 µm. Schéma représentatif du protocole expérimental employé pour l’expérience. Graphique représentant l’intensité moyenne des autophagosomes par cellule quantifiée pour chacune des conditions de traitement et de siARN transfectés ; médiane représentative d’une expérience où au moins 26 cellules ont été analysées. (B) Représentation graphique de la distribution du volume moyen et du nombre des autophagosomes par cellule suite à la transfection sur 48 h de cellules HeLa-RFP-H2B en cytokinèse avec le siCtl et traitées durant 1 h avec 40 µM de tubacine ou le contrôle DMSO ; représentatif d’une expérience où 30 et 26 cellules ont été analysées pour les conditions DMSO et tubacine respectivement. (C) Représentation graphique de la distribution du volume moyen et du nombre des autophagosomes par cellule suite à la transfection sur 48 h de cellules HeLa-RFP-H2B synchronisées en cytokinèse avec le siHSPB8 #3 et traitées 1 h avec 40 µM de tubacine ou le contrôle DMSO ; représentatif d’une expérience où 30 et 35 cellules ont été analysées dans les conditions DMSO et tubacine respectivement. (D) Des cellules HeLa-RFP-H2B synchronisées en cytokinèse ont été transfectées avec le siHSPB8 #3 sur 48 h et traitées durant la seconde relâche de 8 h avec 5 µM de tubacine. Les images confocales montrent une restauration de l’accumulation de F-actine au PI (indiqué par les flèches et zoomé x2 dans les boîtes juxtaposées) présentée par les cellules déplétées en HSPB8 suite à un traitement avec la tubacine. Le PI et l’actine ont été marqués respectivement à l’aide d’un anticorps anti-Aurora B et de la phalloïdine-488 ; échelle, 10 µm. Schéma du protocole expérimental employé pour cette expérience. L’histogramme représente le pourcentage de cellules avec une accumulation anormale de F-actine au PI suite à la déplétion ou non d’HSPB8 et au traitement des cellules sur 8 h avec 5 µM de tubacine ou le contrôle DMSO ; représentatif d’une expérience où au moins 110 cellules ont été analysées.
4.4. Matériel et méthode
4.4.1. Culture cellulaire, synchronisation, traitements et transfection de siARN
Des cellules HeLa-RFP-H2B, ont été maintenues dans un milieu a-MEM (« a-minimal
essential medium ») supplémenté avec 10 % de sérum de veau fœtal et dans une atmosphère
humide à 5 % CO2 et à 37°C. Les cellules ont été synchronisées par la technique de double
blocage à la thymidine et relâchées durant 8 h ou 8 h30 dans du milieu frais selon le
protocole employé dans Fuchs et al. 2015. Pour les expériences de déplétion, 50 nM de
siARN ont été transfectés en utilisant la méthode de transfection au calcium phosphate
(125 mM CaCl2, 140 mM NaCl, 25 mM HEPES, 0.75 mM Na2HPO4). L’efficacité des
siARN a été analysée 24 h ou 48 h plus tard par immunobuvardage de type « Western » ou
par immunofluorescence. Pour la mesure du flux autophagique les drogues suivantes ont

171
été ajoutées durant la dernière heure de relâche avant la fixation des cellules : pepstatine A
(10 µg/ml) et E64d (10 µg/ml), bafilomycine A1 (200 nM), rapamycine (150 nM) et
tubacine (40 µM). Pour la restauration de l’accumulation de F-actine au PI, la tubacine (5
µM) a été ajouté durant les 8 h de la seconde relâche avant la fixation des cellules. Les
séquences de siARN employés sont basées sur les séquences humaines des protéinés
ciblées et ont été obtenues par Thermo Fisher Scientific. Les séquences des brins sens sont
les suivant : siATG7 #1 : 5’-GGTCAAAGGACGGAAGATAATT -3’; siATG7 #2 : 5’-
CCAAGGACATTAAGGGTTATT -3’ ; siHSPB8 #1 : 5’-
CAGATAGGCTAGTGGTATT-3’ ; siHSPB8 #3 : 5’-
CAGAGGAGTTGATGGTGAATT-3’. La séquence cible du siCtl (si Allstars Negative
Control) provient de Qiagen et est la suivante : 5’-CAGGGTATCGACGATTACAAA-3’.
4.4.2. Anticorps et drogues
Les anticorps suivant ont été utilisés : l’anti-ATG7 (#2631) et l’anti-LC3 (#3868)
proviennent de Cell Signaling Technology ; l’anti-ubiquitine K63 (#05-1308) et l’anti-
GAPDH Clone 6C5 (Fitzgerald, #10R-G109a) sont de Millipore ; l’anti-Aurora B
(ab2254) provient d’Abcam ; l’anti-a-tubuline (T5168) est de Sigma-Aldricht ; la
phalloïdine Alexa-Fluor® 488 (A12379) est de Molecular Probes/Thermo Fisher. Le
Hoechst bisbenzimide H 33342 (B2261), la thymidine (T9250), la pepstatine A (P5318),
le E64D (E8640) et la bafilomycine A1 (B1793) proviennent de Sigma-Aldrich ; la
rapamycine (553210) est de Calbiochem ; et la tubacine (S2239) provient de Selleckchem.
4.4.3. Immunobuvardage de type « Western »
Les cellules ont été lysées dans un tampon SDS (62.5 mM Tris-HCl, pH 6.8, 2.3 % SDS,
10 % glycérol, 5% β-mercaptoéthanole, 0.05 % bleu de bromphénole, 1 mM
phénylméthylsulfonylfluoride) et des quantités équivalentes de protéines provenant des
lysats cellulaires totaux ont été séparés par électrophorèse SDS-PAGE de gels à 8 %
d’acrylamide puis transférés sur membranes de nitrocellulose. L’immunobuvardage a été
effectué selon le protocole décrit par Champagne et al., 2004. La concentration protéique

172
des échantillons a été déterminée à l’aide du kit « DC protein assay » (Laboratoires Bio-
Rad) et l’analyse densitométrique a été effectuée à partir d’images acquises avec l’appareil
FluorS MAX MultiImager contrôlé par le logiciel QuantityOne version 4.5.0 (Laboratoires
Bio-Rad).
4.4.4. Immunofluorescence, microscopie et analyses statistiques
Les cellules après avoir été doucement laver au PBS (137 mM NaCl, 2,7 mM KCl, 4.3 mM
Na2HPO4, 1.47 mM KH2PO4) ont été fixées soit avec de la PFA 4 % (paraformaldéhyde)
dans du PBS durant 20 min à température pièce, soit dans du méthanol 100 % à -20°C.
L’ADN a été marqué à l’aide de l’agent perméable Hoechst (150 ng/ml). Les échantillons
ont été traités pour l’immunofluorescence selon le protocole décrit par Lavoie et al., 2000
et post-fixés durant 20 min à température pièce dans du PBS 3,7 % formaldéhyde. Les
phénotypes ont été analysés en routine par inspection visuelle et quantifiés de manière
aveugle aux conditions de traitement, en utilisant le microscope AxioObserver Z1 équipé
d’un objectif 60x/1.25NA et d’une caméra à dispositif couplé-chargé (CCD) Axiocam
MRm contrôlé par le logiciel Zen (Carl Zeiss). Les images en microscopie confocale ont
été effectuées avec le microscope PerkinElmer Life Science Ultraview Spinning Disk
Confocal équipé d’un objectif 60x/1.25NA, une caméra à dispositif couplé-chargé
EMCCD à -50 °C et contrôlé par le logiciel Volocity 6.01 (Quorum Technologies). Pour
quantifier les cellules en métaphase partiellement arrondie, nous nous sommes assurés que
les cellules prises en compte étaient en métaphase par inspection du fuseau mitotique
marqué par l’a-tubuline et de l’ADN marqué par le Hoechst et avons déterminé
l’arrondissement cellulaire à l’aide du marquage de la F-actine à la phalloïdine. Pour
quantifier l'accumulation anormale de F-actine au niveau du PI des cellules filles, nous
avons quantifié les cellules connectées par un PI coloré par Aurora B présentant un ADN
décondensé (télophase tardive) et des structures anormales de F-actine au PI marquées par
la phalloïdine. Pour quantifier l’intensité, le volume et le nombre d’agrégats de protéines
K63 ou des autophagosomes colorés par LC3, nous avons utilisé l’outil « find object » du
logiciel Volocity. Pour trouver les objets le seuil d’intensité a été calibré à partir de la
condition la plus intense pour chaque expérience et un seuil de volume minimal des objets

173
de 0,3 µm3 pour les autophagosomes et de 0,1 µm3 pour les agrégats K63 ont été calibrés.
Les logiciels Volocity et ImageJ 1.48v (National Institute of Health) ont été utilisés pour
le traitement des images avant de les recadrer pour souligner le point principal de l'image.
Le traitement des images a été limité à la soustraction du bruit de fond et au réglage des
luminosité et contraste.
Pour l’analyse statistique de (a) la distribution dans les phases mitotiques des cellules
déplétées en ATG7, (b) du pourcentage de cellules avec une accumulation anormale de F-
actine au PI après déplétion d’ATG7, les données provenant d’au moins trois expériences
indépendantes ont été analysées en utilisant le test statistique Fischer et le logiciel
statistique Prism 6.0 (GraphPad Software). Les valeurs de p<0,05 ont été considérées
comme significatives (*, p <0,05; **, p <0,01; ***, p <0,001; ****, p <0,0001).

174
5. Chapitre 5 : Discussion
L’objectif principal de mon doctorat était de déterminer si l’impact du complexe chaperon
HSPB8-BAG3 sur le remodelage des structures à base d’actine contribue à son effet sur la
progression mitotique des cellules cancéreuses d’une part, et d’investiguer les mécanismes
moléculaires impliqués, d’autre part. Les résultats obtenus suggèrent que le complexe
chaperon agit, en partie, en limitant la polymérisation de l’actine dépendante d’Arp2/3
et/ou en stimulant son renouvellement. En outre, les évidences laissent croire que l’action
du complexe pourrait relever de son activité modulatrice de la déacétylase HDAC6 et de
son action dans les processus de dégradation des protéines par autophagie sélective. Nous
discuterons de ces trois principaux résultats dans les sections suivantes.
5.1. HSPB8-BAG3 agit en limitant la polymérisation de l’actine et/ou en stimulant son
renouvellement pendant la division cellulaire
Nous avons vu que la déplétion d’HSPB8, de BAG3 ou de son partenaire p62/SQSTM1,
peut récapituler les mêmes phénotypes mitotiques associés avec une dérégulation du
remodelage des structures à base d’actine qui dirigent la division cellulaire (Fuchs et al.
2015; Champion, Linder, et Kutay 2017; Annexe 1). De plus, nos résultats indiquent que
la réduction des niveaux d’expression de BAG3 ou d’HSPB8 conduit à une augmentation
de la prévalence des cellules multi-nucléées, démontrant ainsi l’impact du complexe
chaperon dans la progression de la division cellulaire (Hayashi et Karlseder 2013; S
Pedersen et al. 2016; Chapitre 2). Nous avons observé qu’en limitant la polymérisation de
l’actine, notamment par de faibles doses de latrunculine A, il est possible de corriger ces
défauts de division cellulaire dans les cellules déplétées en HSPB8, lequel est requis pour
l’activité de BAG3 et le recrutement de p62/SQSTM1 lors de la mitose (Fuchs et al. 2015;
Annexe 1; Chapitre 2). Ces observations suggèrent qu’un complexe tertiaire formé
d’HSPB8-BAG3-p62/SQSTM1, dont la formation est régulée par les signaux mitotiques,
pourrait faciliter la progression mitotique en contrôlant localement la dynamique d’actine.
Cette fonction chaperon pourrait permettre un désassemble/assemblage limité dans le
temps et l’espace des structures à base d’actine qui dirigent la division cellulaire. L’action

175
du complexe chaperon pourrait également favoriser le maintien de l’intégrité de telles
structures qui sont soumises à des forces mécaniques importantes en facilitant leur
renouvellement; c’est le cas notamment du cortex d’actomyosine qui supporte
l’arrondissement mitotique, des fibres de rétraction qui transmettent les forces mécaniques
au cortex pour favoriser l’orientation du fuseau mitotique, ainsi que de l’anneau contractile
d’actomyosine qui conduit la séparation des cellules filles lors de la cytokinèse. Bien que
le ou les mécanismes précis restent à déterminer, il nous est permis de spéculer sur des
pistes potentielles à explorer sur la base des résultats présentés dans cette thèse et de la
nouvelle littérature, qui seront discutés plus bas.
Sur la base de nos résultats, on peut prédire que des composantes et/ou régulateurs du
complexe Arp2/3 pourraient constituer de nouveaux « clients mitotiques » du complexe
chaperon. En effet, nous avons mis en évidence que l’inhibition du complexe Arp2/3 peut
restaurer les défauts associés à l’accumulation de structures d’actine ectopiques suivant la
déplétion de BAG3, HSPB8 ou p62/SQSTM1, incluant les défauts de cytokinèse,
l’arrondissement mitotique, et la dynamique du nuage sous-cortical d’actine qui influence
l’orientation du fuseau mitotique (Mitsushima et al. 2010; Chapitre 2; Chapitre 3). Ces
résultats suggèrent qu’HSPB8-BAG3 en association avec p62/SQSTM1 limiterait
l’activité spatiale du complexe Arp2/3 à l’entrée des cellules en mitose. Il a été montré
qu’un transfert de l’activité d’Arp2/3 en faveur de la formine mDia1 est requis pour
l’assemblage du cortex en mitose essentiel à l’arrondissement mitotique (Rosa et al. 2015).
D’autre part, il a été suggéré qu’Arp2/3 est essentiel à la régulation de la dynamique
d’actine au sein du nuage sous-cortical d’actine qui dirige le positionnement du fuseau
mitotique (Mitsushima et al. 2010; Théry et al. 2005), un résultat corroboré par nos travaux
(Chapitre 3). Ainsi, il est envisageable que dans ce contexte le complexe formé d’HSPB8,
BAG3 et p62/SQSTM1 faciliterait l’inhibition d’Arp2/3 en séquestrant des protéines clés
au cours de la division cellulaire. En conséquence, en ségrégant localement l’activité du
complexe Arp2/3, HSPB8-BAG3-p62/SQSTM1 faciliterait à la fois le transfert d’activité
des nucléateurs à l’entrée des cellules en mitose et la dynamique du nuage sous-cortical
d’actine en mitose. Une telle activité pourrait intervenir en cytokinèse pour limiter la
polymérisation d’actine branchée à l’anneau contractile d’actomyosine; à l’appui de ce

176
modèle, nous avons observé que la déplétion d’HSPB8 conduit à une accumulation
anormale d’Arp2/3 au pont intercellulaire lors de la cytokinèse tardive (Chapitre 2).
ArpC2 est un candidat intéressant comme cible potentielle de l’action inhibitrice d’HSPB8-
BAG3-p62/SQSTM1. ArpC2 est une sous-unité du complexe Arp2/3 essentielle à la
formation du complexe (Machesky et al. 1994; Winter, Choe, et Li 1999; Rotty et al. 2015).
Il a été démontré par spectrométrie de masse que la sous-unité ArpC2 du complexe Arp2/3
est un partenaire de BAG3 en conditions physiologiques (Chen et al. 2013). D’autre part,
la GTPase Rac1 pourrait également être ciblée par l’action inhibitrice du complexe
chaperon. Il a été démontré que la GTPase Rac1 est inactivée à l’entrée des cellules en
mitose et que la surexpression d’un mutant dominant positif de Rac1 interfère avec la
progression de la cytokinèse et conduit ainsi à un avortement de l’abscission (Bastos et al.
2012; Muris et al. 2002; Oceguera-Yanez et al. 2005). Rac1 serait régulée par le co-
chaperon BAG3 lors de la migration et de l’adhésion cellulaires dans le contexte de cellules
cancéreuses (Iwasaki et al. 2007; Hüfner et al. 2002; Chen et al. 2017). La séquestration de
Rac1 ou de ses régulateurs à l’entrée des cellules en mitose aurait pour conséquence de
favoriser l’échange d’activité des nucléateurs (Arp2/3 vers mDia1). Il serait donc
intéressant d’analyser la présence de foyers ArpC2 et/ou Rac1 dans les cellules mitotiques
et d’évaluer l’impact du complexe chaperon HSPB8-BAG3-p62/SQSTM1 sur la
distribution de tels foyers.
D’autre part, l’action régulatrice du complexe HSPB8-BAG3-p62/SQSTM1 pourrait
s’exercer au niveau de la GTPase RhoA. En effet, de récents travaux suggèrent que la
régulation spatiotemporelle de la GTPase RhoA lors de la division cellulaire pourrait
dépendre en partie de la formation de corpuscules contenant la GTPase ainsi que la cycline
A2 qui serait nécessaire à ce processus (Loukil et al. 2016). De tels foyers seraient en partie
dégradés par autophagie sélective via le recrutement de p62/SQSTM1 et LC3 ce qui
contribuerait à la transition métaphase-anaphase (Loukil et al. 2014). D’autre part, en
cytokinèse, il est suggéré que le ciblage sélectif de la forme active de RhoA par
p62/SQSTM1 à la dégradation par autophagie est essentiel pour limiter l’activité de la
GTPase au sillon de clivage (Belaid et al. 2013). Il est donc envisageable que le complexe

177
tertiaire HSPB8-BAG3-p62/SQSTM1 module des corpuscules contenant la GTPase RhoA,
qui ont été reliés à sa régulation fine. RhoA est une GTPase clé qui contrôle la mise en
place du cortex en mitose et la formation et la contraction de l’anneau contractile en
cytokinèse, dont l’activation excessive en mitose et cytokinèse est mimée par la déplétion
de BAG3, HSPB8 ou p62/SQSTM1 (Belaid et al. 2013; Marchesi et al. 2014; Maddox et
Burridge 2003; Liu et Weiner 2016; Chapitres 2 et 3). En outre, les travaux de Ke et al.
suggèrent qu’HSPB8 module l’activité de RhoA dans un contexte de stress cellulaire
provoqué par la fibrillation auriculaire (Ke et al. 2011). Il serait donc important d’analyser
une possible interaction fonctionnelle entre le complexe tertiaire mitotique HSPB8-BAG3-
p62/SQSTM1 et la GTPase RhoA.
En outre, la régulation spatiale de la dynamique de l’actine pourrait faire intervenir un
mécanisme impliquant l’activité du complexe tertiaire HSPB8-BAG3-p62/SQSTM1 sur la
séquestration des protéines. Des travaux récents obtenus au laboratoire ont montré
qu’HSPB8, en coopération avec BAG3, coordonne la séquestration de protéines nuisibles
lors de l’inhibition du protéasome, ce qui faciliterait leur transport à l’agrésome (Guilbert
et al. 2018). De plus, la séquestration locale du complexe TSC par BAG3 lors de la
contraction musculaire a été reliée à la régulation spatiotemporelle de l’activité de
mTORC1 (Kathage et al. 2016). En outre, l’activité protectrice de l’adaptateur
autophagique p62/SQSTM1 lors d’un stress protéotoxique se caractérise par son action sur
l’agrégation fonctionnelle des protéines nuisibles (Bjørkøy et al. 2005; Wooten et al. 2008;
Wurzer et al. 2015). Les protéines en voie de dénaturation sont ainsi séquestrées
rapidement dans des structures réversibles appelées corpuscules (« p62 bodies »), lesquels
sont organisés par l’oligomérisation de p62/SQSTM1. La formation de ces corpuscules par
p62/SQSTM1 a été associée à sa fonction signalétique où la séquestration de protéines clés
permet de reprogrammer et compartimenter des voies de signalisation dans la cellule
(Rantanen et al. 2013; Houslay et Christian 2010). Par exemple, la séquestration de la
phosphatase C1-Ten par p62/SQSTM1 permettrait son inactivation spécifique requise pour
la différenciation musculaire (Koh et al. 2014). p62/SQSTM1 active également la réponse
anti-oxydante dépendante de Nrf2 (« Nuclear Factor (Erythroid-Derived 2)-Like 2 ») en

178
séquestrant l’ubiquitine ligase Keap1 (« Kelch-like ECH-associated protein 1 ») (Komatsu
et al. 2010).
Ainsi, il est raisonnable d’envisager que dès l’entrée des cellules en mitose, la formation
de corpuscules orchestrée par l’oligomérisation de p62/SQSTM1 et régulée par le
complexe chaperon HSPB8-BAG3, pourrait créer une plateforme de séquestration
protéique. Ceci pourrait permettre la ségrégation de complexes de signalisation pour une
action locale, ou la séquestration de protéines endommagées par des cycles répétitifs
d’assemblage/désassemblage ou dont l’action doit être inhibée. Nos récents travaux
suggèrent qu’HSPB8 et BAG3 coopèrent pour promouvoir la phosphorylation de
p62/SQSTM1 sur des résidus spécifiques qui sont prédits comme essentiels pour son
oligomérisation et qui pourraient réguler des voies de signalisation intracellulaires
contrôlant l'état métabolique et oxydatif de la cellule (Guilbert et al. 2018). Nous avons
corrélé l’effet promoteur d’HSPB8-BAG3 sur la phosphorylation de p62/SQSTM1 à son
effet dans la séquestration de protéines poly-ubiquitinées, ainsi qu’à son effet régulateur
dans la séquestration de Keap1. Ainsi, à l’instar de son rôle dans le contrôle local de
l’activité de mTORC1 dans les cellules musculaires lisses, HSPB8-BAG3 semble contrôler
l’activation de Nrf2 et la réponse au stress oxydant dans les premières heures du stress
protéotoxique (Kathage et al. 2016; Guilbert et al. 2018). On peut donc penser qu’un
mécanisme similaire, impliquant la régulation de la phosphorylation de p62/SQSTM1 et
de son oligomérisation en mitose, pourrait intervenir. Dans ce sens, il est pertinent de noter
ici que p62/SQSTM1 est phosphorylée par Cdk1 à l’entrée des cellules en mitose et que
cette phosphorylation modulerait la progression mitotique (Linares et al. 2011). De façon
intéressante, nos résultats préliminaires suggèrent que la déplétion d’HSPB8 ou de BAG3
interfère avec la phosphorylation mitotique de p62/SQSTM1. De plus, l’existence de
corpuscules dynamiques dans les cellules en mitose, associés à la séquestration de protéines
mitotiques et au contrôle de l’activité de RhoA, a été mis en évidence récemment par Loukil
et al. (Loukil et al. 2016, 2014).
Sur la base de nos études récentes et de la littérature actuelle, il est envisageable que la
formation de corpuscules mitotiques par p62/SQSTM1 pourrait être facilitée par l’activité

179
chaperon d’HSPB8 qui aiderait à la reconnaissance des substrats mitotiques de
p62/SQSTM1. La fonction d’adaptateur autophagique de p62/SQSTM1 se caractérise par
la reconnaissance de protéines marquées par l’ubiquitine K63, grâce à son domaine UBA,
puis au déclenchement de leur agrégation par auto-oligomérisation à travers son domaine
PB1. Dans ce sens, nous avons observé l’existence de corpuscules dynamiques caractérisés
par la présence de p62/SQSTM1 dans les cellules mitotiques dont la formation serait
dépendante du complexe chaperon HSPB8-BAG3 puisque la déplétion d’HSPB8, BAG3
ou p62/SQSTM1 conduit à une réduction drastique du nombre de corpuscules
p62/SQSTM1 (Herman Lambert, résultats non publiés). Il sera intéressant d’analyser la
composition de ces structures, par purification d’affinité, puisqu’elles pourraient permettre
d’identifier plusieurs nouveaux clients du complexe chaperon.
Finalement, nos résultats suggèrent que la régulation fine de la dynamique d’atomyosine
ferait intervenir de nouveaux régulateurs mitotiques : soit la déacétylase HDAC6 et son
substrat prototype la cortactine.
5.2. Le complexe HSPB8-BAG3 facilite le remodelage des structures mitotiques à base
d’actine à travers la modulation d’HDAC6
Afin d’identifier des cibles potentielles de l’action du complexe chaperon HSPB8-BAG3-
p62/SQSTM1 dans le remodelage des structures à base d’actine, nous avons concentré nos
efforts à l’étude d’une structure d’actine mitotique suggérée pour dépendre de l’activité
dynamique d’Arp2/3 : le nuage sous-cortical d’actine dans les cellules en prométaphase-
métaphase (Mitsushima et al. 2010). C’est ainsi que nous avons émis l’hypothèse que la
déacétylase HDAC6, interagissant préférentiellement avec BAG3 en mitose et connue pour
réguler la dynamique du cytosquelette d’actine, pourrait être à l’origine de l’activité du
complexe HSPB8-BAG3 sur la dynamique d’actine (Fuchs et al. 2015; Kaluza et al. 2011;
Zhang et al. 2007; Kozyreva et al. 2014; Annexe 1).
Ainsi nous avons obtenu des résultats qui suggèrent que l’activité d’HDAC6 est régulée de
manière différentielle pendant la mitose et qu’un tel processus impliquerait le complexe

180
tertiaire mitotique HSPB8-BAG3-p62/SQSTM1 avec lequel HDAC6 s’associe de façon
dépendante d’HSPB8 (Fuchs et al. 2015; Annexe1; Chapitre 3). Cependant, les
mécanismes précis restent à déterminer. Nos travaux suggèrent que la cortactine pourrait
être une cible d’HDAC6 finement régulée en mitose (Uruno et al. 2001; Weaver et al. 2001;
Chapitre 3). En effet, nos résultats montrent que le « turnover » de l’acétylation de la
cortactine ainsi que son association avec HDAC6 sont réduites dans les cellules mitotiques,
comparativement aux cellules en croissance asynchrone. La déacétylation de la cortactine
est connue pour activer sa fonction activatrice de la polymérisation de l’actine branchée à
travers l’activation d’Arp2/3. Limiter l’activité d’HDAC6 sur la cortactine dans le temps
et l’espace pourrait ainsi contribuer au contrôle de la dynamique d’actine en mitose à
travers la régulation précise de la dynamique d’activation/inhibition d’Arp2/3. Ce
mécanisme moléculaire pourrait être requis pour contrôler la dynamique du nuage sous-
cortical d’actine en mitose et aider au transfert d’Arp2/3 en faveur de mDia1 à l’entrée des
cellules en mitose requis pour l’arrondissement mitotique (Mitsushima et al. 2010; Rosa et
al. 2015). Dans ce sens, nous avons vu que la surexpression d’un mutant non acétylable de
la cortactine interfère avec l’arrondissement mitotique (Chapitre 3).
En outre, la cortactine a été associée au contrôle du démantèlement des adhésions focales
via une dynamique d’association/dissociation avec la tyrosine kinase FAK (« Focal
Adhesion Kinase ») (Tomar et al. 2012). La phosphorylation sur tyrosine de la cortactine
qui stimule le désassemblage des adhésions focales, semble être un mécanisme qui
contribue à stimuler sa déacétylation connue pour être dépendante d’HDAC6 (Zhang et al.
2007; Meiler, Nieto-Pelegrín, et Martinez-Quiles 2012; Tomar et al. 2012). De plus, une
activité d’HDAC6 sur la cortactine a été caractérisée en G2/M où elle serait requise pour
le démantèlement du cil primaire, étape à laquelle se déroule le désassemblage des
adhésions focales requis pour l’arrondissement mitotique (Ran et al. 2015). Il est donc
envisageable qu’une régulation spatiotemporelle fine de la déacétylation de la cortactine
spécifiquement au niveau des adhésions focales par HDAC6 pourrait faciliter le
démantèlement des adhésions focales ce qui l’impliquerait dans le processus
d’arrondissement mitotique. Il serait donc important de préciser le lien fonctionnel entre

181
l’activité d’HDAC6 sur son substrat prototype la cortactine et la régulation des structures
à base d’actine en mitose.
La fonction mitotique d’HDAC6 pourrait également faire intervenir d’autres cibles
connues de la déacétylase qui modulent la dynamique du cytosquelette d’actine.
Notamment, la chaîne lourde de la MyoII, dont l’acétylation stimule la liaison à l’actine,
est une cible démontrée d’HDAC6 (Zhang et al. 2015). La MyoII joue un rôle central dans
la formation et le maintien de la rigidité du cortex requis pour l’arrondissement mitotique
et elle est nécessaire à la contraction de l’anneau contractile en cytokinèse (Mabuchi et
Okuno 1977; Straight et al. 2003; Ramanathan et al. 2015; Rosa et al. 2015). Une telle
fonction de MyoII en cytokinèse implique à la fois son activité motrice et sa capacité à
induire la dépolymérisation de la F-actine (Mendes Pinto et al. 2012). Ainsi en régulant
l’acétylation de la chaîne lourde de la MyoII et donc en réduisant son association à l’actine,
HDAC6 pourrait contrôler spécifiquement l’activité de la MyoII au cours de la division
cellulaire. En mitose, cette fonction aiderait à l’arrondissement mitotique et au maintien de
la rigidité corticale. En cytokinèse, la déacétylation de la chaîne lourde de la MyoII par
HDAC6 serait requise pour coordonner la contraction de l’anneau contractile et la
dépolymérisation simultanée de la F-actine. Il serait donc important de déterminer
l’acétylome d’HDAC6 en mitose afin d’identifier l’ensemble de ses cibles mitotiques et de
préciser leurs fonctions mitotiques.
En somme, la modulation de l’activité d’HDAC6 par le complexe chaperon HSPB8-
BAG3-p62/SQSTM1 facilite le remodelage des structures à base d’actine en mitose et en
cytokinèse. Le mécanisme moléculaire par lequel HDAC6 contrôle la dynamique d’actine
en mitose semble impliquer sa cible la cortactine. Il est évident que d’autres cibles
d’HDAC6 pourraient être également requises pour le remodelage de l’actine en mitose et
celles-ci restent à être identifiées. Outre sa fonction modulatrice de l’activité d’HDAC6,
les résultats obtenus dans le cadre des études montrées dans le chapitre 4 supportent un rôle
pour la fonction autophagique du complexe chaperon HSPB8-BAG3 dans la régulation du
remodelage de l’actine lors de la cytokinèse.

182
5.3. Implication potentielle de l’autophagie sélective dépendante d’HSPB8-BAG3
dans la progression de la division cellulaire
Le complexe chaperon HSPB8-BAG3 a été impliqué dans le contrôle du ciblage de
protéines agrégées, dénaturées ou poly-ubiquitinées à la machinerie autophagique dans le
contexte d’un stress protéotoxique (Carra et al. 2008; Gamerdinger et al. 2011, 2009;
Minoia et al. 2014; Xu et al. 2013). Dans le muscle, cette fonction du complexe chaperon
a été associée au contrôle de l’intégrité de la structure d’actine qui dirige la contraction
cellulaire : la myofibrille (Arndt et al. 2010; Ulbricht et al. 2013). La division cellulaire
implique des structures contractiles, soumises aux tensions mécaniques, qui sont
déstabilisées par la déplétion d’HSPB8 ou de BAG3 (Fuchs et al. 2015, Annexe 1; Chapitre
2). Nous avons supposé que la fonction dans l’autophagie sélective du complexe chaperon
est impliquée dans les processus de remodelage des structures contractiles qui dirigent la
division cellulaire. Nous avons montré que l’inhibition du flux autophagique mime les
phénotypes associés à la déplétion d’HSPB8 ou de BAG3 (i.e. inhibition de
l’arrondissement mitotique et du désassemblage de l’anneau contractile). À l’inverse, nos
résultats indiquent qu’activer le flux autophagique par un traitement avec la rapamycine
restaure le désassemblage de l’anneau contractile dans les cellules déplétées en HSPB8.
L’ensemble de ces résultats suggère que le ciblage sélectif de protéines à la machinerie
autophagique est une fonction du complexe HSPB8-BAG3 requise pour le remodelage du
cytosquelette d’actine au cours de la division cellulaire.
Les travaux de l’équipe du Dr Höfehld ont caractérisé une fonction importante du co-
chaperon BAG3 dans l’assemblage d’un complexe requis pour le ciblage de la filamine à
l’autophagie sélective (Arndt et al. 2010; Ulbricht et al. 2013). Cette fonction est
coordonnée avec une action du co-chaperon dans le renouvellement de la filamine
dénaturée par le stress mécanique, permettant ainsi de préserver l’intégrité de la protéine
au cours de la contraction de la cellule musculaire. En plus de proposer que ce mécanisme
moléculaire intervienne en réponse à tous types de tensions mécaniques, les auteurs ont
aussi suggéré que le chaperon HSPB8 est requis pour la reconnaissance spécifique de la
filamine dénaturée. Le mécanisme moléculaire de la CASA fait intervenir plusieurs

183
protéines clés, dont p62/SQSTM1 et HSC70, recrutées spécifiquement par BAG3. Comme
les fonctions mitotiques de BAG3 ne requièrent pas son association à HSC70, il est
envisageable qu’un mécanisme similaire au processus de CASA, mais distinct car
indépendant d’HSC70, soit impliquer dans le remodelage des structures à base d’actine
lors de la division cellulaire. D’autre part, il est possible que lors de la cytokinèse, HSC70
soit un partenaire important du complexe chaperon. En effet, l’architecture du complexe
pourrait être différente de celle observée en mitose, compte-tenu de l’état de
phosphorylation de BAG3 dont l’hyperphosphorylation est stimulée à l’entrée des cellules
en mitose, alors qu’elle est inhibée au début de la cytokinèse (Fuchs et al. 2015 ; Annexe1).
Reste à identifier les cibles de l’activité autophagique du complexe chaperon lors de la
division cellulaire.
La filamine pourrait être une cible potentielle du complexe HSPB8-BAG3 puisqu’une
fonction en mitose et en cytokinèse a été associée à la protéine de réticulation de l’actine.
En mitose, la filamine serait importante pour réticuler le cytosquelette d’actine de façon
dépendante de sa phosphorylation par CDK1 (Cukier, Li, et Lee 2007). En cytokinèse, une
fonction pour la filamine a été associée au recrutement de protéines requises pour la
machinerie de l’abscission (Mondal et al. 2012). Ainsi, en aidant au maintien de l’état natif
de la filamine, en induisant sa dégradation par autophagie sélective et sa synthèse
simultanée, le complexe HSPB8-BAG3-p62/SQSTM1 permettrait de maintenir l’intégrité
corticale en mitose via son activité de réticulation et le recrutement des protéines de
l’abscission en cytokinèse. Il serait intéressant de tirer avantage d’une construction
filamine-clover-rubby qui nous permettrait de suivre le ciblage de la protéine à la
machinerie autophagique et d’analyser l’influence du complexe chaperon HSPB8-BAG3
sur un tel processus au cours de la division cellulaire.
Telle que mentionnée dans la section précédente, Belaid et al. ont montré qu’en cytokinèse,
p62/SQSTM1 est impliquée dans le ciblage sélectif de la forme active de RhoA à la
machinerie autophagique en contrôlant l’agrégation de la GTPase (Belaid et al. 2013).
Interférer avec un tel processus conduit à une activation excessive de RhoA, à une
expansion de sa localisation et à une accumulation anormale de F-actine, qui seraient

184
responsables d’une instabilité génétique caractéristique d’un avortement de l’abscission.
De plus, HSPB8 modulerait négativement l’activité de RhoA dans un contexte de stress
cellulaire provoqué par la fibrillation auriculaire (Ke et al. 2011). Ainsi en aidant au ciblage
spécifique de la forme active de RhoA à l’autophagie sélective via son association à
p62/SQSTM1, le complexe chaperon HSPB8-BAG3 pourrait faciliter les processus de
remodelage de l’actine requis pour le désassemblage de l’anneau contractile en cytokinèse.
Il serait donc intéressant d’analyser l’implication du complexe HSPB8-BAG3 dans le
ciblage à la machinerie autophagique de RhoA-GTP en cytokinèse.
En outre, nos résultats préliminaires suggèrent que la déplétion d’HSPB8 interfère avec le
flux autophagique. Sur la base de l’ensemble de nos résultats, il est possible de suggérer
un modèle selon lequel la régulation d’HDAC6-cortactine puisse avoir un rôle dans la
modulation du flux autophagique. En effet, Lee et al. a montré que le cytosquelette d’actine
et la déacétylation de la cortactine par HDAC6 sont requis pour l’étape de fusion de
l’autophagosome avec le lysosome uniquement au cours des processus d’autophagie
sélective (Lee et al. 2010). De plus, les réseaux d’actine branchée régulés par Arp2/3, dont
l’activité semble être limitée par HSPB8-BAG3 au cours de la division cellulaire de façon
dépendante d’HDAC6 (chapitres 2 et 3), seraient au cœur de la régultation de multiples
étapes du flux autophagique (Monastyrska et al. 2008; Kast et Dominguez 2015; Kast et
al. 2015; Mi et al. 2015; Höhfeld 2016; Coutts et La Thangue 2016; Zientara-Rytter et
Subramani 2016b). Or, il semble que l’inhibition de l’activité d’HDAC6 améliore le flux
autophagique normal dans les cellules déplétées en HSPB8. Cela suggère que la
modulation de l’activité d’HDAC6 sur la cortactine par le complexe HSPB8-BAG3
pourrait facilter une ou plusieurs étapes du flux autophagique dépendantes de l’activité
d’Arp2/3. Il est intéressant de noter ici que p62/SQSTM1 régulerait négativement l’activité
déacétylase d’HDAC6 suggérant que l’adaptateur autophagique joue un rôle dans la
régulation de la fusion des autophagosomes avec le lysosome dépendante d’HDAC6 lors
du processus d’autophagie sélective (Yan et al. 2013). De récentes études suggèrent aussi
que le chaperon HSPB8 faciliterait la fusion des autophagosomes avec le lysosome à
travers un mécanisme moléculaire indéterminé (Li et al. 2017; Kwok et al. 2011).
Notamment, les travaux de Kwok et al. ont montré que la surexpression d’un mutant

185
d’HSPB8, le mutant K141E retrouvé chez des patients atteints de neuropathie motrice
héréditaire, interfère avec la livraison des autophagosomes au lysosome (Kwok et al. 2011).
Ainsi au cours de la division cellulaire HSPB8, en association avec BAG3 et p62/SQSTM1,
pourrait faciliter le flux autophagique en assistant l’étape de fusion. Un tel mécanisme
moléculaire pourrait impliquer la modulation de l’activité d’HDAC6 sur la cortactine
requise pour la fusion des autophagosomes avec le lysosome au cours du processus
d’autophagie sélective. Il sera intéressant de tester ce modèle par analyse de la distribution
de la construction LC3-clover-rubby en réponse à la déplétion d’HSPB8 ou de BAG3.
En résumé, les résultats présentés dans cette thèse suggèrent un modèle selon lequel le
complexe chaperon pourrait moduler le cytosquelette d’actine en facilitant deux étapes
distinctes du flux autophagique. Premièrement, il pourrait aider à la séquestration et au
ciblage sélectif de protéines mécanosensibles à la machinerie autophagique tel qu’il l’a été
proposé pour le processus de CASA. Deuxièmement, en modulant l’activité de la
déacétylase HDAC6 sur sa cible la cortactine, il pourrait faciliter l’étape de fusion des
autophagosomes avec le lysosome.

186
6. Conclusion
Les travaux présentés dans cette thèse ont mis en avant une fonction encore non appréciée
du complexe chaperon HSPB8-BAG3 dans la modulation de la dynamique des structures
à base d’actine qui dirigent la division cellulaire dans le contexte de cellules cancéreuses.
Tout d’abord, nous avons déterminé que la limitation de la dynamique d’actine par le
complexe HSPB8-BAG3 est un mécanisme par lequel le complexe chaperon aide à la
progression normale de la division cellulaire dans le contexte de cellules cancéreuses.
L’exploration des mécanismes moléculaires par lesquels le complexe HSPB8-BAG3 peut
moduler la dynamique d’actine a révélé deux modèles qui pourraient être interdépendants.
D’une part, nous avons vu que le complexe chaperon facilite la dynamique d’actine au sein
de structures qui dirigent la division cellulaire en modulant l’activité d’HDAC6. D’autre
part, le complexe chaperon HSPB8-BAG3, par le ciblage de protéines à la dégradation
autophagique et/ou en facilitant la fusion des autophagosomes avec le lysosome,
modulerait la transition dans l’organisation des structures d’actine au cours de la division
cellulaire.
L’ensemble de ces travaux appuient l’hypothèse que les chaperons de la famille des HSPB
contrôlent la dynamique de l’actine dans un contexte physiologique et qu’une telle fonction
pourrait favoriser la survie des cellules cancéreuses et l’expansion tumorale. Par ailleurs,
ces travaux ont mis en évidence la déacétylase HDAC6 comme un partenaire important du
complexe chaperon HSPB8-BAG3 dans ses fonctions régulatrices du cytosquelette
d’actine. HDAC6 pourrait donc être une potentielle cible thérapeutique pour des
pathologies impliquant une dérégulation du complexe chaperon. En conséquence, il serait
important de déterminer l’ensemble processus cellulaires dans lesquels HDAC6 et ses
cibles potentielles sont régulées par le complexe chaperon HSPB8-BAG3.

187
7. Bibliographie
• Acunzo, Julie, Maria Katsogiannou, et Palma Rocchi. 2012. « Small Heat Shock
Proteins HSP27 (HspB1), ΑB-Crystallin (HspB5) and HSP22 (HspB8) as Regulators of
Cell Death ». The International Journal of Biochemistry & Cell Biology 44 (10): 1622‑31.
https://doi.org/10.1016/j.biocel.2012.04.002.
• Aguilera, Milton Osmar, Walter Berón, et María Isabel Colombo. 2012. « The Actin
Cytoskeleton Participates in the Early Events of Autophagosome Formation upon
Starvation Induced Autophagy ». Autophagy 8 (11): 1590‑1603.
https://doi.org/10.4161/auto.21459.
• Ahlers, A., C. Belka, M. Gaestel, N. Lamping, C. Sott, F. Herrmann, et M. A. Brach.
1994. « Interleukin-1-Induced Intracellular Signaling Pathways Converge in the Activation
of Mitogen-Activated Protein Kinase and Mitogen-Activated Protein Kinase-Activated
Protein Kinase 2 and the Subsequent Phosphorylation of the 27-Kilodalton Heat Shock
Protein in Monocytic Cells ». Molecular Pharmacology 46 (6): 1077‑83.
• Alberti, Simon, Jens Demand, Claudia Esser, Niels Emmerich, Hansjorg Schild, et
Jorg Hohfeld. 2002. « Ubiquitylation of BAG-1 Suggests a Novel Regulatory Mechanism
during the Sorting of Chaperone Substrates to the Proteasome ». The Journal of Biological
Chemistry 277 (48): 45920‑27. https://doi.org/10.1074/jbc.M204196200.
• Amano, M., M. Ito, K. Kimura, Y. Fukata, K. Chihara, T. Nakano, Y. Matsuura, et
K. Kaibuchi. 1996. « Phosphorylation and Activation of Myosin by Rho-Associated Kinase
(Rho-Kinase) ». The Journal of Biological Chemistry 271 (34): 20246‑49.
• Amano, Toru, Noriko Kaji, Kazumasa Ohashi, et Kensaku Mizuno. 2002.
« Mitosis-Specific Activation of LIM Motif-Containing Protein Kinase and Roles of
Cofilin Phosphorylation and Dephosphorylation in Mitosis ». The Journal of Biological
Chemistry 277 (24): 22093‑102. https://doi.org/10.1074/jbc.M201444200.
• Ammirante, M., V. De Laurenzi, V. Graziano, M. C. Turco, et A. Rosati. 2011.
« BAG3 Is Required for IKKα Nuclear Translocation and Emergence of Castration
Resistant Prostate Cancer ». Cell Death & Disease 2 (mars): e139.
https://doi.org/10.1038/cddis.2011.23.

188
• Anding, Allyson L., et Eric H. Baehrecke. 2017. « Cleaning House: Selective
Autophagy of Organelles ». Developmental Cell 41 (1): 10‑22.
https://doi.org/10.1016/j.devcel.2017.02.016.
• Andrieu, C., D. Taieb, V. Baylot, S. Ettinger, P. Soubeyran, A. De-Thonel, C.
Nelson, et al. 2010. « Heat Shock Protein 27 Confers Resistance to Androgen Ablation and
Chemotherapy in Prostate Cancer Cells through EIF4E ». Oncogene 29 (13): 1883‑96.
https://doi.org/10.1038/onc.2009.479.
• Antoku, K., R. S. Maser, W. J. Scully, S. M. Delach, et D. E. Johnson. 2001.
« Isolation of Bcl-2 Binding Proteins That Exhibit Homology with BAG-1 and Suppressor
of Death Domains Protein ». Biochemical and Biophysical Research Communications 286
(5): 1003‑10. https://doi.org/10.1006/bbrc.2001.5512.
• Aquilina, J. Andrew, Justin L. P. Benesch, Lin Lin Ding, Orna Yaron, Joseph
Horwitz, et Carol V. Robinson. 2004. « Phosphorylation of AlphaB-Crystallin Alters
Chaperone Function through Loss of Dimeric Substructure ». The Journal of Biological
Chemistry 279 (27): 28675‑80. https://doi.org/10.1074/jbc.M403348200.
• Aratyn-Schaus, Yvonne, Patrick W. Oakes, et Margaret L. Gardel. 2011.
« Dynamic and structural signatures of lamellar actomyosin force generation ». Molecular
Biology of the Cell 22 (8): 1330‑39. https://doi.org/10.1091/mbc.E10-11-0891.
• Arimura, Takuro, Taisuke Ishikawa, Shinichi Nunoda, Sachio Kawai, et Akinori
Kimura. 2011. « Dilated Cardiomyopathy-Associated BAG3 Mutations Impair Z-Disc
Assembly and Enhance Sensitivity to Apoptosis in Cardiomyocytes ». Human Mutation 32
(12): 1481‑91. https://doi.org/10.1002/humu.21603.
• Arndt, Verena, Nikolaus Dick, Riga Tawo, Michael Dreiseidler, Daniela Wenzel,
Michael Hesse, Dieter O. Fürst, et al. 2010. « Chaperone-Assisted Selective Autophagy Is
Essential for Muscle Maintenance ». Current Biology: CB 20 (2): 143‑48.
https://doi.org/10.1016/j.cub.2009.11.022.
• Arrigo, A.-P., et B. Gibert. 2012. « HspB1 Dynamic Phospho-Oligomeric Structure
Dependent Interactome as Cancer Therapeutic Target ». Current Molecular Medicine 12
(9): 1151‑63.
• Auteri, J. S., A. Okada, V. Bochaki, et J. F. Dice. 1983. « Regulation of Intracellular

189
Protein Degradation in IMR-90 Human Diploid Fibroblasts ». Journal of Cellular
Physiology 115 (2): 167‑74. https://doi.org/10.1002/jcp.1041150210.
• Bagrodia, S., A. P. Laudano, et D. Shalloway. 1994. « Accessibility of the C-Src
SH2-Domain for Binding Is Increased during Mitosis ». The Journal of Biological
Chemistry 269 (14): 10247‑51.
• Bailly, M., I. Ichetovkin, W. Grant, N. Zebda, L. M. Machesky, J. E. Segall, et J.
Condeelis. 2001. « The F-Actin Side Binding Activity of the Arp2/3 Complex Is Essential
for Actin Nucleation and Lamellipod Extension ». Current Biology: CB 11 (8): 620‑25.
• Baird, Nathan A., Peter M. Douglas, Milos S. Simic, Ana R. Grant, James J.
Moresco, Suzanne C. Wolff, John R. Yates, Gerard Manning, et Andrew Dillin. 2014.
« HSF-1-Mediated Cytoskeletal Integrity Determines Thermotolerance and Life Span ».
Science (New York, N.Y.) 346 (6207): 360‑63. https://doi.org/10.1126/science.1253168.
• Bakthisaran, Raman, Kranthi Kiran Akula, Ramakrishna Tangirala, et Ch Mohan
Rao. 2016. « Phosphorylation of ΑB-Crystallin: Role in Stress, Aging and Patho-
Physiological Conditions ». Biochimica Et Biophysica Acta 1860 (1 Pt B): 167‑82.
https://doi.org/10.1016/j.bbagen.2015.09.017.
• Baranova, E. V., S. Beelen, N. B. Gusev, et S. V. Strelkov. 2009. « The Taming of
Small Heat-Shock Proteins: Crystallization of the Alpha-Crystallin Domain from Human
Hsp27 ». Acta Crystallographica. Section F, Structural Biology and Crystallization
Communications 65 (Pt 12): 1277‑81. https://doi.org/10.1107/S1744309109044571.
• Bashour, Anne-Marie, Aaron T. Fullerton, Matthew J. Hart, et George S. Bloom.
1997. « IQGAP1, a Rac- and Cdc42-binding Protein, Directly Binds and Cross-links
Microfilaments ». The Journal of Cell Biology 137 (7): 1555‑66.
• Bastos, Ricardo Nunes, Xenia Penate, Michelle Bates, Dean Hammond, et Francis
A. Barr. 2012. « CYK4 Inhibits Rac1-Dependent PAK1 and ARHGEF7 Effector Pathways
during Cytokinesis ». The Journal of Cell Biology 198 (5): 865‑80.
https://doi.org/10.1083/jcb.201204107.
• Behl, Christian. 2016. « Breaking BAG: The Co-Chaperone BAG3 in Health and
Disease ». Trends in Pharmacological Sciences 37 (8): 672‑88.
https://doi.org/10.1016/j.tips.2016.04.007.

190
• Belaid, Amine, Michaël Cerezo, Abderrahman Chargui, Elisabeth Corcelle-
Termeau, Florence Pedeutour, Sandy Giuliano, Marius Ilie, et al. 2013. « Autophagy Plays
a Critical Role in the Degradation of Active RHOA, the Control of Cell Cytokinesis, and
Genomic Stability ». Cancer Research 73 (14): 4311‑22. https://doi.org/10.1158/0008-
5472.CAN-12-4142.
• Bement, William M., Hélène A. Benink, et George von Dassow. 2005. « A
Microtubule-Dependent Zone of Active RhoA during Cleavage Plane Specification ». The
Journal of Cell Biology 170 (1): 91‑101. https://doi.org/10.1083/jcb.200501131.
• Beningo, K. A., M. Dembo, I. Kaverina, J. V. Small, et Y. L. Wang. 2001. « Nascent
Focal Adhesions Are Responsible for the Generation of Strong Propulsive Forces in
Migrating Fibroblasts ». The Journal of Cell Biology 153 (4): 881‑88.
• Benndorf, R., K. Hayess, S. Ryazantsev, M. Wieske, J. Behlke, et G. Lutsch. 1994.
« Phosphorylation and Supramolecular Organization of Murine Small Heat Shock Protein
HSP25 Abolish Its Actin Polymerization-Inhibiting Activity ». The Journal of Biological
Chemistry 269 (32): 20780‑84.
• Bertet, Claire, Lawrence Sulak, et Thomas Lecuit. 2004. « Myosin-Dependent
Junction Remodelling Controls Planar Cell Intercalation and Axis Elongation ». Nature
429 (6992): 667‑71. https://doi.org/10.1038/nature02590.
• Betapudi, Venkaiah, Lucila S. Licate, et Thomas T. Egelhoff. 2006. « Distinct Roles
of Nonmuscle Myosin II Isoforms in the Regulation of MDA-MB-231 Breast Cancer Cell
Spreading and Migration ». Cancer Research 66 (9): 4725‑33.
https://doi.org/10.1158/0008-5472.CAN-05-4236.
• Bhowmick, N. A., M. Ghiassi, A. Bakin, M. Aakre, C. A. Lundquist, M. E. Engel,
C. L. Arteaga, et H. L. Moses. 2001. « Transforming Growth Factor-Beta1 Mediates
Epithelial to Mesenchymal Transdifferentiation through a RhoA-Dependent Mechanism ».
Molecular Biology of the Cell 12 (1): 27‑36.
• Bird, Stephen L., Rebecca Heald, et Karsten Weis. 2013. « RanGTP and CLASP1
Cooperate to Position the Mitotic Spindle ». Molecular Biology of the Cell 24 (16):
2506‑14. https://doi.org/10.1091/mbc.E13-03-0150.
• Bjørkøy, Geir, Trond Lamark, Andreas Brech, Heidi Outzen, Maria Perander, Aud

191
Overvatn, Harald Stenmark, et Terje Johansen. 2005. « P62/SQSTM1 Forms Protein
Aggregates Degraded by Autophagy and Has a Protective Effect on Huntingtin-Induced
Cell Death ». The Journal of Cell Biology 171 (4): 603‑14.
https://doi.org/10.1083/jcb.200507002.
• Block, J., T. E. B. Stradal, J. Hänisch, R. Geffers, S. A. Köstler, E. Urban, J. V.
Small, K. Rottner, et J. Faix. 2008. « Filopodia Formation Induced by Active
MDia2/Drf3 ». Journal of Microscopy 231 (3): 506‑17. https://doi.org/10.1111/j.1365-
2818.2008.02063.x.
• Boiani, Mariana, Cristina Daniel, Xueyuan Liu, Michael D. Hogarty, et Lawrence
J. Marnett. 2013. « The Stress Protein BAG3 Stabilizes Mcl-1 Protein and Promotes
Survival of Cancer Cells and Resistance to Antagonist ABT-737 ». The Journal of
Biological Chemistry 288 (10): 6980‑90. https://doi.org/10.1074/jbc.M112.414177.
• Boldogh, I. R., H. C. Yang, W. D. Nowakowski, S. L. Karmon, L. G. Hays, J. R.
Yates, et L. A. Pon. 2001. « Arp2/3 Complex and Actin Dynamics Are Required for Actin-
Based Mitochondrial Motility in Yeast ». Proceedings of the National Academy of Sciences
of the United States of America 98 (6): 3162‑67. https://doi.org/10.1073/pnas.051494698.
• Boncoraglio, Alessandra, Melania Minoia, et Serena Carra. 2012. « The Family of
Mammalian Small Heat Shock Proteins (HSPBs): Implications in Protein Deposit Diseases
and Motor Neuropathies ». The International Journal of Biochemistry & Cell Biology 44
(10): 1657‑69. https://doi.org/10.1016/j.biocel.2012.03.011.
• Bova, M. P., O. Yaron, Q. Huang, L. Ding, D. A. Haley, P. L. Stewart, et J. Horwitz.
1999. « Mutation R120G in AlphaB-Crystallin, Which Is Linked to a Desmin-Related
Myopathy, Results in an Irregular Structure and Defective Chaperone-like Function ».
Proceedings of the National Academy of Sciences of the United States of America 96 (11):
6137‑42.
• Bovellan, Miia, Yves Romeo, Maté Biro, Annett Boden, Priyamvada Chugh,
Amina Yonis, Malti Vaghela, et al. 2014. « Cellular Control of Cortical Actin Nucleation ».
Current Biology: CB 24 (14): 1628‑35. https://doi.org/10.1016/j.cub.2014.05.069.
• Brophy, C. M., S. Lamb, et A. Graham. 1999. « The Small Heat Shock-Related
Protein-20 Is an Actin-Associated Protein ». Journal of Vascular Surgery 29 (2): 326‑33.

192
• Burbelo, P. D., D. Drechsel, et A. Hall. 1995. « A Conserved Binding Motif Defines
Numerous Candidate Target Proteins for Both Cdc42 and Rac GTPases ». The Journal of
Biological Chemistry 270 (49): 29071‑74.
• Busson, Sylvie, Denis Dujardin, Anne Moreau, Jim Dompierre, et Jan R. De Mey.
1998. « Dynein and dynactin are localized to astral microtubules and at cortical sites in
mitotic epithelial cells ». Current Biology 8 (9): 541‑44. https://doi.org/10.1016/S0960-
9822(98)70208-8.
• Cadart, Clotilde, Ewa Zlotek-Zlotkiewicz, Maël Le Berre, Matthieu Piel, et Helen
K. Matthews. 2014. « Exploring the Function of Cell Shape and Size during Mitosis ».
Developmental Cell 29 (2): 159‑69. https://doi.org/10.1016/j.devcel.2014.04.009.
• Canman, Julie C., Lisa A. Cameron, Paul S. Maddox, Aaron Straight, Jennifer S.
Tirnauer, Timothy J. Mitchison, Guowei Fang, Tarun M. Kapoor, et E. D. Salmon. 2003.
« Determining the Position of the Cell Division Plane ». Nature 424 (6952): 1074‑78.
https://doi.org/10.1038/nature01860.
• Cappola, Thomas P., Mingyao Li, Jing He, Bonnie Ky, Joan Gilmore, Liming Qu,
Brendan Keating, et al. 2010. « Common Variants in HSPB7 and FRMD4B Associated
with Advanced Heart Failure ». Circulation. Cardiovascular Genetics 3 (2): 147‑54.
https://doi.org/10.1161/CIRCGENETICS.109.898395.
• Capponi, Simona, Alessandro Geroldi, Paola Fossa, Marina Grandis, Paola Ciotti,
Rossella Gulli, Angelo Schenone, Paola Mandich, et Emilia Bellone. 2011. « HSPB1 and
HSPB8 in Inherited Neuropathies: Study of an Italian Cohort of DHMN and CMT2
Patients ». Journal of the Peripheral Nervous System: JPNS 16 (4): 287‑94.
https://doi.org/10.1111/j.1529-8027.2011.00361.x.
• Carnell, Michael, Tobias Zech, Simon D. Calaminus, Seiji Ura, Monica Hagedorn,
Simon A. Johnston, Robin C. May, Thierry Soldati, Laura M. Machesky, et Robert H.
Insall. 2011. « Actin Polymerization Driven by WASH Causes V-ATPase Retrieval and
Vesicle Neutralization before Exocytosis ». The Journal of Cell Biology 193 (5): 831‑39.
https://doi.org/10.1083/jcb.201009119.
• Carra, Serena, Samuel J. Seguin, Herman Lambert, et Jacques Landry. 2008.
« HspB8 Chaperone Activity toward Poly(Q)-Containing Proteins Depends on Its

193
Association with Bag3, a Stimulator of Macroautophagy ». The Journal of Biological
Chemistry 283 (3): 1437‑44. https://doi.org/10.1074/jbc.M706304200.
• Carra, Serena, Mitchel Sivilotti, Aura T. Chávez Zobel, Herman Lambert, et
Jacques Landry. 2005. « HspB8, a Small Heat Shock Protein Mutated in Human
Neuromuscular Disorders, Has in Vivo Chaperone Activity in Cultured Cells ». Human
Molecular Genetics 14 (12): 1659‑69. https://doi.org/10.1093/hmg/ddi174.
• Carreno, Sébastien, Ilektra Kouranti, Edith Szafer Glusman, Margaret T. Fuller,
Arnaud Echard, et François Payre. 2008. « Moesin and Its Activating Kinase Slik Are
Required for Cortical Stability and Microtubule Organization in Mitotic Cells ». The
Journal of Cell Biology 180 (4): 739‑46. https://doi.org/10.1083/jcb.200709161.
• Case, Lindsay B., et Clare M. Waterman. 2015. « Integration of Actin Dynamics
and Cell Adhesion by a Three-Dimensional, Mechanosensitive Molecular Clutch ». Nature
Cell Biology 17 (8): 955‑63. https://doi.org/10.1038/ncb3191.
• Castrillon, D. H., et S. A. Wasserman. 1994. « Diaphanous Is Required for
Cytokinesis in Drosophila and Shares Domains of Similarity with the Products of the Limb
Deformity Gene ». Development 120 (12): 3367‑77.
• Cattin, Cedric J., Marcel Düggelin, David Martinez-Martin, Christoph Gerber,
Daniel J. Müller, et Martin P. Stewart. 2015. « Mechanical Control of Mitotic Progression
in Single Animal Cells ». Proceedings of the National Academy of Sciences of the United
States of America 112 (36): 11258‑63. https://doi.org/10.1073/pnas.1502029112.
• Chalut, Kevin J., et Ewa K. Paluch. 2016. « The Actin Cortex: A Bridge between
Cell Shape and Function ». Developmental Cell 38 (6): 571‑73.
https://doi.org/10.1016/j.devcel.2016.09.011.
• Champion, Lysie, Monika I. Linder, et Ulrike Kutay. 2017. « Cellular
Reorganization during Mitotic Entry ». Trends in Cell Biology 27 (1): 26‑41.
https://doi.org/10.1016/j.tcb.2016.07.004.
• Cheffings, Thomas H., Nigel J. Burroughs, et Mohan K. Balasubramanian. 2016.
« Actomyosin Ring Formation and Tension Generation in Eukaryotic Cytokinesis ».
Current Biology: CB 26 (15): R719-737. https://doi.org/10.1016/j.cub.2016.06.071.
• Chen, Baoyu, Hui-Ting Chou, Chad A. Brautigam, Wenmin Xing, Sheng Yang,

194
Lisa Henry, Lynda K. Doolittle, Thomas Walz, et Michael K. Rosen. 2017. « Rac1 GTPase
Activates the WAVE Regulatory Complex through Two Distinct Binding Sites ». ELife 6
(septembre). https://doi.org/10.7554/eLife.29795.
• Chen, Jin, Matthias J. Feige, Titus M. Franzmann, Alexander Bepperling, et
Johannes Buchner. 2010. « Regions Outside the Alpha-Crystallin Domain of the Small
Heat Shock Protein Hsp26 Are Required for Its Dimerization ». Journal of Molecular
Biology 398 (1): 122‑31. https://doi.org/10.1016/j.jmb.2010.02.022.
• Chen, Ningyu, Shr-Jeng Leu, Viktor Todorović, Stephen C.-T. Lam, et Lester F.
Lau. 2004. « Identification of a Novel Integrin Αvβ3 Binding Site in CCN1 (CYR61)
Critical for Pro-Angiogenic Activities in Vascular Endothelial Cells ». Journal of
Biological Chemistry 279 (42): 44166‑76. https://doi.org/10.1074/jbc.M406813200.
• Chen, Ying, Li-Na Yang, Li Cheng, Shun Tu, Shu-Juan Guo, Huang-Ying Le, Qian
Xiong, et al. 2013. « Bcl2-Associated Athanogene 3 Interactome Analysis Reveals a New
Role in Modulating Proteasome Activity ». Molecular & Cellular Proteomics: MCP 12
(10): 2804‑19. https://doi.org/10.1074/mcp.M112.025882.
• Chew, Ting Gang, Junqi Huang, Saravanan Palani, Ruth Sommese, Anton Kamnev,
Tomoyuki Hatano, Ying Gu, Snezhana Oliferenko, Sivaraj Sivaramakrishnan, et Mohan
K. Balasubramanian. 2017. « Actin Turnover Maintains Actin Filament Homeostasis
during Cytokinetic Ring Contraction ». J Cell Biol, juin, jcb.201701104.
https://doi.org/10.1083/jcb.201701104.
• Chiappetta, Gennaro, Massimo Ammirante, Anna Basile, Alessandra Rosati,
Michela Festa, Mario Monaco, Emilia Vuttariello, et al. 2007. « The Antiapoptotic Protein
BAG3 Is Expressed in Thyroid Carcinomas and Modulates Apoptosis Mediated by Tumor
Necrosis Factor-Related Apoptosis-Inducing Ligand ». The Journal of Clinical
Endocrinology and Metabolism 92 (3): 1159‑63. https://doi.org/10.1210/jc.2006-1712.
• Chiti, Fabrizio, et Christopher M. Dobson. 2017. « Protein Misfolding, Amyloid
Formation, and Human Disease: A Summary of Progress Over the Last Decade ». Annual
Review of Biochemistry 86 (juin): 27‑68. https://doi.org/10.1146/annurev-biochem-
061516-045115.
• Choi, Seo-hyun, Hae-June Lee, Yeung Bae Jin, Junho Jang, Ga-young Kang,

195
Minyoung Lee, Chun-Ho Kim, et al. 2014. « MMP9 Processing of HSPB1 Regulates
Tumor Progression ». PloS One 9 (1): e85509.
https://doi.org/10.1371/journal.pone.0085509.
• Chowdary, Tirumala Kumar, Bakthisaran Raman, Tangirala Ramakrishna, et
Chintalagiri Mohan Rao. 2004. « Mammalian Hsp22 Is a Heat-Inducible Small Heat-Shock
Protein with Chaperone-like Activity ». The Biochemical Journal 381 (Pt 2): 379‑87.
https://doi.org/10.1042/BJ20031958.
• Chowdhury, Farhan, Sungsoo Na, Dong Li, Yeh-Chuin Poh, Tetsuya S. Tanaka,
Fei Wang, et Ning Wang. 2010. « Cell material property dictates stress-induced spreading
and differentiation in embryonic stem cells ». Nature materials 9 (1): 82‑88.
https://doi.org/10.1038/nmat2563.
• Ciuffa, Rodolfo, Trond Lamark, Abul K. Tarafder, Audrey Guesdon, Sofia Rybina,
Wim J. H. Hagen, Terje Johansen, et Carsten Sachse. 2015. « The Selective Autophagy
Receptor P62 Forms a Flexible Filamentous Helical Scaffold ». Cell Reports 11 (5):
748‑58. https://doi.org/10.1016/j.celrep.2015.03.062.
• Clarke, Joseph P., et Karen M. Mearow. 2013. « Cell Stress Promotes the
Association of Phosphorylated HspB1 with F-Actin ». PloS One 8 (7): e68978.
https://doi.org/10.1371/journal.pone.0068978.
• Clough, Barbara, Joseph D. Wright, Pedro M. Pereira, Elizabeth M. Hirst, Ashleigh
C. Johnston, Ricardo Henriques, et Eva-Maria Frickel. 2016. « K63-Linked Ubiquitination
Targets Toxoplasma Gondii for Endo-Lysosomal Destruction in IFNγ-Stimulated Human
Cells ». PLoS Pathogens 12 (11): e1006027. https://doi.org/10.1371/journal.ppat.1006027.
• Colvin, Teresa A., Vladimir L. Gabai, Jianlin Gong, Stuart K. Calderwood, Hu Li,
Suryaram Gummuluru, Olga N. Matchuk, et al. 2014. « Hsp70-Bag3 Interactions Regulate
Cancer-Related Signaling Networks ». Cancer Research 74 (17): 4731‑40.
https://doi.org/10.1158/0008-5472.CAN-14-0747.
• Cope, M Jamie TV, James Whisstock, Ivan Rayment, et John Kendrick-Jones.
1996. « Conservation within the myosin motor domain: implications for structure and
function ». Structure 4 (8): 969‑87. https://doi.org/10.1016/S0969-2126(96)00103-7.
• Cotteret, Sophie, et Jonathan Chernoff. 2002. « The evolutionary history of

196
effectors downstream of Cdc42 and Rac ». Genome Biology 3 (2): reviews0002.1-
reviews0002.8.
• Coutts, Amanda S., et Nicholas B. La Thangue. 2016. « Regulation of Actin
Nucleation and Autophagosome Formation ». Cellular and Molecular Life Sciences:
CMLS 73 (17): 3249‑63. https://doi.org/10.1007/s00018-016-2224-z.
• Cox, Dezerae, et Heath Ecroyd. 2017. « The Small Heat Shock Proteins ΑB-
Crystallin (HSPB5) and Hsp27 (HSPB1) Inhibit the Intracellular Aggregation of α-
Synuclein ». Cell Stress & Chaperones, mars. https://doi.org/10.1007/s12192-017-0785-x.
• Crawford, J. M., N. Harden, T. Leung, L. Lim, et D. P. Kiehart. 1998.
« Cellularization in Drosophila Melanogaster Is Disrupted by the Inhibition of Rho Activity
and the Activation of Cdc42 Function ». Developmental Biology 204 (1): 151‑64.
https://doi.org/10.1006/dbio.1998.9061.
• Crippa, Valeria, Serena Carra, Paola Rusmini, Daniela Sau, Elena Bolzoni, Caterina
Bendotti, Silvia De Biasi, et Angelo Poletti. 2010. « A Role of Small Heat Shock Protein
B8 (HspB8) in the Autophagic Removal of Misfolded Proteins Responsible for
Neurodegenerative Diseases ». Autophagy 6 (7): 958‑60.
https://doi.org/10.4161/auto.6.7.13042.
• Crippa, Valeria, Daniela Sau, Paola Rusmini, Alessandra Boncoraglio, Elisa
Onesto, Elena Bolzoni, Mariarita Galbiati, et al. 2010. « The Small Heat Shock Protein B8
(HspB8) Promotes Autophagic Removal of Misfolded Proteins Involved in Amyotrophic
Lateral Sclerosis (ALS) ». Human Molecular Genetics 19 (17): 3440‑56.
https://doi.org/10.1093/hmg/ddq257.
• Cukier, I. Howard, Yun Li, et Jonathan M. Lee. 2007. « Cyclin B1/Cdk1 Binds and
Phosphorylates Filamin A and Regulates Its Ability to Cross-Link Actin ». FEBS Letters
581 (8): 1661‑72. https://doi.org/10.1016/j.febslet.2007.03.041.
• Dambournet, Daphné, Mickael Machicoane, Laurent Chesneau, Martin Sachse,
Murielle Rocancourt, Ahmed El Marjou, Etienne Formstecher, Rémi Salomon, Bruno
Goud, et Arnaud Echard. 2011. « Rab35 GTPase and OCRL Phosphatase Remodel Lipids
and F-Actin for Successful Cytokinesis ». Nature Cell Biology 13 (8): 981‑88.
https://doi.org/10.1038/ncb2279.

197
• Dao, Vi Thuy, Aurélien Guy Dupuy, Olivier Gavet, Emmanuelle Caron, et Jean de
Gunzburg. 2009. « Dynamic Changes in Rap1 Activity Are Required for Cell Retraction
and Spreading during Mitosis ». Journal of Cell Science 122 (Pt 16): 2996‑3004.
https://doi.org/10.1242/jcs.041301.
• Davidson, S. M., et M. Morange. 2000. « Hsp25 and the P38 MAPK Pathway Are
Involved in Differentiation of Cardiomyocytes ». Developmental Biology 218 (2): 146‑60.
https://doi.org/10.1006/dbio.1999.9596.
• D’Avino, Pier Paolo. 2009. « How to Scaffold the Contractile Ring for a Safe
Cytokinesis - Lessons from Anillin-Related Proteins ». Journal of Cell Science 122 (Pt 8):
1071‑79. https://doi.org/10.1242/jcs.034785.
• Dean, Sara O., Stephen L. Rogers, Nico Stuurman, Ronald D. Vale, et James A.
Spudich. 2005. « Distinct Pathways Control Recruitment and Maintenance of Myosin II at
the Cleavage Furrow during Cytokinesis ». Proceedings of the National Academy of
Sciences of the United States of America 102 (38): 13473‑78.
https://doi.org/10.1073/pnas.0506810102.
• Dehle, Francis C., Heath Ecroyd, Ian F. Musgrave, et John A. Carver. 2010. « ΑB-
Crystallin Inhibits the Cell Toxicity Associated with Amyloid Fibril Formation by κ-
Casein and the Amyloid-β Peptide ». Cell Stress & Chaperones 15 (6): 1013‑26.
https://doi.org/10.1007/s12192-010-0212-z.
• Del Bigio, Marc R., Albert E. Chudley, Harvey B. Sarnat, Craig Campbell, Sharan
Goobie, Bernard N. Chodirker, et Duygu Selcen. 2011. « Infantile Muscular Dystrophy in
Canadian Aboriginals Is an ΑB-Crystallinopathy ». Annals of Neurology 69 (5): 866‑71.
https://doi.org/10.1002/ana.22331.
• Demand, J., S. Alberti, C. Patterson, et J. Höhfeld. 2001. « Cooperation of a
Ubiquitin Domain Protein and an E3 Ubiquitin Ligase during Chaperone/Proteasome
Coupling ». Current Biology: CB 11 (20): 1569‑77.
• Dibble, Christian C., et Brendan D. Manning. 2013. « Signal Integration by
MTORC1 Coordinates Nutrient Input with Biosynthetic Output ». Nature Cell Biology 15
(6): 555‑64. https://doi.org/10.1038/ncb2763.
• DiDomenico, B. J., G. E. Bugaisky, et S. Lindquist. 1982. « The Heat Shock

198
Response Is Self-Regulated at Both the Transcriptional and Posttranscriptional Levels ».
Cell 31 (3 Pt 2): 593‑603.
• Disanza, Andrea, Marie-France Carlier, Theresia E. B. Stradal, Dominique Didry,
Emanuela Frittoli, Stefano Confalonieri, Assunta Croce, Jurgen Wehland, Pier Paolo Di
Fiore, et Giorgio Scita. 2004. « Eps8 Controls Actin-Based Motility by Capping the Barbed
Ends of Actin Filaments ». Nature Cell Biology 6 (12): 1180‑88.
https://doi.org/10.1038/ncb1199.
• Doong, H., J. Price, Y. S. Kim, C. Gasbarre, J. Probst, L. A. Liotta, J. Blanchette,
K. Rizzo, et E. Kohn. 2000. « CAIR-1/BAG-3 Forms an EGF-Regulated Ternary Complex
with Phospholipase C-Gamma and Hsp70/Hsc70 ». Oncogene 19 (38): 4385‑95.
https://doi.org/10.1038/sj.onc.1203797.
• Doong, Howard, Kathryn Rizzo, Shengyun Fang, Vyta Kulpa, Allan M. Weissman,
et Elise C. Kohn. 2003. « CAIR-1/BAG-3 Abrogates Heat Shock Protein-70 Chaperone
Complex-Mediated Protein Degradation: Accumulation of Poly-Ubiquitinated Hsp90
Client Proteins ». The Journal of Biological Chemistry 278 (31): 28490‑500.
https://doi.org/10.1074/jbc.M209682200.
• Doong, Howard, Alysia Vrailas, et Elise C. Kohn. 2002. « What’s in the “BAG”?-
-A Functional Domain Analysis of the BAG-Family Proteins ». Cancer Letters 188 (1‑2):
25‑32.
• Doshi, Bindi M., Lawrence E. Hightower, et Juliet Lee. 2009. « The Role of Hsp27
and Actin in the Regulation of Movement in Human Cancer Cells Responding to Heat
Shock ». Cell Stress & Chaperones 14 (5): 445‑57. https://doi.org/10.1007/s12192-008-
0098-1.
• Drechsel, D. N., A. A. Hyman, A. Hall, et M. Glotzer. 1997. « A Requirement for
Rho and Cdc42 during Cytokinesis in Xenopus Embryos ». Current Biology: CB 7 (1):
12‑23.
• Du, Guiping, Xiang Liu, Xinping Chen, Mei Song, Yan Yan, Renjie Jiao, et Chih-
Chen Wang. 2010. « Drosophila Histone Deacetylase 6 Protects Dopaminergic Neurons
against {alpha}-Synuclein Toxicity by Promoting Inclusion Formation ». Molecular
Biology of the Cell 21 (13): 2128‑37. https://doi.org/10.1091/mbc.E10-03-0200.

199
• Du, Zhen-Xian, Hai-Yan Zhang, Xin Meng, Yan-Yan Gao, Ren-Long Zou, Bao-
Qin Liu, Yifu Guan, et Hua-Qin Wang. 2009. « Proteasome Inhibitor MG132 Induces
BAG3 Expression through Activation of Heat Shock Factor 1 ». Journal of Cellular
Physiology 218 (3): 631‑37. https://doi.org/10.1002/jcp.21634.
• Dubnikov, Tatyana, Tziona Ben-Gedalya, et Ehud Cohen. 2017. « Protein Quality
Control in Health and Disease ». Cold Spring Harbor Perspectives in Biology 9 (3):
a023523. https://doi.org/10.1101/cshperspect.a023523.
• Dudich, I. V., V. P. Zav’yalov, W. Pfeil, M. Gaestel, G. A. Zav’yalova, A. I.
Denesyuk, et T. Korpela. 1995. « Dimer Structure as a Minimum Cooperative Subunit of
Small Heat-Shock Proteins ». Biochimica Et Biophysica Acta 1253 (2): 163‑68.
• Duleh, Steve N., et Matthew D. Welch. 2010. « WASH and the Arp2/3 Complex
Regulate Endosome Shape and Trafficking ». Cytoskeleton (Hoboken, N.J.) 67 (3):
193‑206. https://doi.org/10.1002/cm.20437.
• Dunlop, E. A., et A. R. Tee. 2014. « mTOR and autophagy: A dynamic relationship
governed by nutrients and energy ». Seminars in Cell & Developmental Biology,
Development of the urogenital system & mTOR Signalling & Tight Junctions in Health
and Disease, 36 (décembre): 121‑29. https://doi.org/10.1016/j.semcdb.2014.08.006.
• During, Russell L., Bruce G. Gibson, Wei Li, Ellen A. Bishai, Gurjit S. Sidhu,
Jacques Landry, et Frederick S. Southwick. 2007. « Anthrax Lethal Toxin Paralyzes Actin-
Based Motility by Blocking Hsp27 Phosphorylation ». The EMBO Journal 26 (9):
2240‑50. https://doi.org/10.1038/sj.emboj.7601687.
• During, Russell L., Wei Li, Binghua Hao, Joyce M. Koenig, David S. Stephens,
Conrad P. Quinn, et Frederick S. Southwick. 2005. « Anthrax Lethal Toxin Paralyzes
Neutrophil Actin-Based Motility ». The Journal of Infectious Diseases 192 (5): 837‑45.
https://doi.org/10.1086/432516.
• Dutartre, H., J. Davoust, J. P. Gorvel, et P. Chavrier. 1996. « Cytokinesis Arrest
and Redistribution of Actin-Cytoskeleton Regulatory Components in Cells Expressing the
Rho GTPase CDC42Hs ». Journal of Cell Science 109 ( Pt 2) (février): 367‑77.
• Eden, Sharon, Rajat Rohatgi, Alexandre V. Podtelejnikov, Matthias Mann, et Marc
W. Kirschner. 2002. « Mechanism of Regulation of WAVE1-Induced Actin Nucleation by

200
Rac1 and Nck ». Nature 418 (6899): 790‑93. https://doi.org/10.1038/nature00859.
• Effler, Janet C., Yee-Seir Kee, Jason M. Berk, Minhchau N. Tran, Pablo A. Iglesias,
et Douglas N. Robinson. 2006. « Mitosis-Specific Mechanosensing and Contractile-Protein
Redistribution Control Cell Shape ». Current Biology: CB 16 (19): 1962‑67.
https://doi.org/10.1016/j.cub.2006.08.027.
• Ehrlicher, A. J., F. Nakamura, J. H. Hartwig, D. A. Weitz, et T. P. Stossel. 2011.
« Mechanical Strain in Actin Networks Regulates FilGAP and Integrin Binding to Filamin
A ». Nature 478 (7368): 260‑63. https://doi.org/10.1038/nature10430.
• Ehrnsperger, M., S. Gräber, M. Gaestel, et J. Buchner. 1997. « Binding of Non-
Native Protein to Hsp25 during Heat Shock Creates a Reservoir of Folding Intermediates
for Reactivation ». The EMBO Journal 16 (2): 221‑29.
https://doi.org/10.1093/emboj/16.2.221.
• Eikenes, Åsmund H., Lene Malerød, Anette Lie Christensen, Chloé B. Steen,
Juliette Mathieu, Ioannis P. Nezis, Knut Liestøl, Jean-René Huynh, Harald Stenmark, et
Kaisa Haglund. 2015. « ALIX and ESCRT-III Coordinately Control Cytokinetic
Abscission during Germline Stem Cell Division in Vivo ». PLoS Genetics 11 (1):
e1004904. https://doi.org/10.1371/journal.pgen.1004904.
• Emoto, K., T. Kobayashi, A. Yamaji, H. Aizawa, I. Yahara, K. Inoue, et M. Umeda.
1996. « Redistribution of Phosphatidylethanolamine at the Cleavage Furrow of Dividing
Cells during Cytokinesis ». Proceedings of the National Academy of Sciences of the United
States of America 93 (23): 12867‑72.
• Emoto, K., et M. Umeda. 2000. « An Essential Role for a Membrane Lipid in
Cytokinesis. Regulation of Contractile Ring Disassembly by Redistribution of
Phosphatidylethanolamine ». The Journal of Cell Biology 149 (6): 1215‑24.
• Emoto, Kazuo, Hironori Inadome, Yasunori Kanaho, Shuh Narumiya, et Masato
Umeda. 2005. « Local Change in Phospholipid Composition at the Cleavage Furrow Is
Essential for Completion of Cytokinesis ». The Journal of Biological Chemistry 280 (45):
37901‑7. https://doi.org/10.1074/jbc.M504282200.
• Eskelinen, Eeva-Liisa, Alan R. Prescott, Jennifer Cooper, Saskia M. Brachmann,
Lijun Wang, Xiuwen Tang, Jonathan M. Backer, et John M. Lucocq. 2002. « Inhibition of

201
Autophagy in Mitotic Animal Cells ». Traffic (Copenhagen, Denmark) 3 (12): 878‑93.
• Esposito, Veronica, Carlo Baldi, Pio Zeppa, Michelina Festa, Luana Guerriero,
Morena d’Avenia, Massimiliano Chetta, et al. 2017. « BAG3 Protein Is Over-Expressed in
Endometrioid Endometrial Adenocarcinomas ». Journal of Cellular Physiology 232 (2):
309‑11. https://doi.org/10.1002/jcp.25489.
• Even-Ram, Sharona, Andrew D. Doyle, Mary Anne Conti, Kazue Matsumoto,
Robert S. Adelstein, et Kenneth M. Yamada. 2007. « Myosin IIA Regulates Cell Motility
and Actomyosin-Microtubule Crosstalk ». Nature Cell Biology 9 (3): 299‑309.
https://doi.org/10.1038/ncb1540.
• Evgrafov, Oleg V., Irena Mersiyanova, Joy Irobi, Ludo Van Den Bosch, Ines
Dierick, Conrad L. Leung, Olga Schagina, et al. 2004. « Mutant Small Heat-Shock Protein
27 Causes Axonal Charcot-Marie-Tooth Disease and Distal Hereditary Motor
Neuropathy ». Nature Genetics 36 (6): 602‑6. https://doi.org/10.1038/ng1354.
• Falco, A., M. Festa, A. Basile, A. Rosati, M. Pascale, F. Florenzano, S. L. Nori, et
al. 2012. « BAG3 Controls Angiogenesis through Regulation of ERK Phosphorylation ».
Oncogene 31 (50): 5153‑61. https://doi.org/10.1038/onc.2012.17.
• Fehon, Richard G., Andrea I. McClatchey, et Anthony Bretscher. 2010.
« Organizing the Cell Cortex: The Role of ERM Proteins ». Nature Reviews. Molecular
Cell Biology 11 (4): 276‑87. https://doi.org/10.1038/nrm2866.
• Feng, Helin, Jin Wang, Wei Chen, Baoen Shan, Yin Guo, Jianfa Xu, Ling Wang,
Peng Guo, et Yingze Zhang. 2016. « Hypoxia-Induced Autophagy as an Additional
Mechanism in Human Osteosarcoma Radioresistance ». Journal of Bone Oncology 5 (2):
67‑73. https://doi.org/10.1016/j.jbo.2016.03.001.
• Festa, Michelina, Luis Del Valle, Kamel Khalili, Renato Franco, Giosuè
Scognamiglio, Vincenzo Graziano, Vincenzo De Laurenzi, Maria Caterina Turco, et
Alessandra Rosati. 2011. « BAG3 Protein Is Overexpressed in Human Glioblastoma and
Is a Potential Target for Therapy ». The American Journal of Pathology 178 (6): 2504‑12.
https://doi.org/10.1016/j.ajpath.2011.02.002.
• Field, Christine M., Margaret Coughlin, Steve Doberstein, Thomas Marty, et
William Sullivan. 2005. « Characterization of Anillin Mutants Reveals Essential Roles in

202
Septin Localization and Plasma Membrane Integrity ». Development 132 (12): 2849‑60.
https://doi.org/10.1242/dev.01843.
• Field, Seth J., Nikki Madson, Monica L. Kerr, Kenneth A. A. Galbraith, Caitlin E.
Kennedy, Mamta Tahiliani, Andrew Wilkins, et Lewis C. Cantley. 2005. « PtdIns(4,5)P2
Functions at the Cleavage Furrow during Cytokinesis ». Current Biology 15 (15): 1407‑12.
https://doi.org/10.1016/j.cub.2005.06.059.
• Fielding, Andrew B., Eric Schonteich, Johanne Matheson, Gayle Wilson, Xinzi Yu,
Gilles R. X. Hickson, Sweta Srivastava, Stephen A. Baldwin, Rytis Prekeris, et Gwyn W.
Gould. 2005. « Rab11-FIP3 and FIP4 Interact with Arf6 and the Exocyst to Control
Membrane Traffic in Cytokinesis ». The EMBO Journal 24 (19): 3389‑99.
https://doi.org/10.1038/sj.emboj.7600803.
• Fink, Jenny, Nicolas Carpi, Timo Betz, Angelique Bétard, Meriem Chebah, Ammar
Azioune, Michel Bornens, et al. 2011. « External Forces Control Mitotic Spindle
Positioning ». Nature Cell Biology 13 (7): 771‑78. https://doi.org/10.1038/ncb2269.
• Firestone, Ari J., Joshua S. Weinger, Maria Maldonado, Kari Barlan, Lance D.
Langston, Michael O’Donnell, Vladimir I. Gelfand, Tarun M. Kapoor, et James K. Chen.
2012. « Small-Molecule Inhibitors of the AAA+ ATPase Motor Cytoplasmic Dynein ».
Nature 484 (7392): 125‑29. https://doi.org/10.1038/nature10936.
• Foe, Victoria E., et George von Dassow. 2008. « Stable and Dynamic Microtubules
Coordinately Shape the Myosin Activation Zone during Cytokinetic Furrow Formation ».
The Journal of Cell Biology 183 (3): 457‑70. https://doi.org/10.1083/jcb.200807128.
• Fontanella, Bianca, Leila Birolo, Giuseppe Infusini, Claudia Cirulli, Liberato
Marzullo, Pietro Pucci, Maria Caterina Turco, et Alessandra Tosco. 2010. « The Co-
Chaperone BAG3 Interacts with the Cytosolic Chaperonin CCT: New Hints for Actin
Folding ». The International Journal of Biochemistry & Cell Biology 42 (5): 641‑50.
https://doi.org/10.1016/j.biocel.2009.12.008.
• Forrest, Katharine M. L., Safa Al-Sarraj, Caroline Sewry, Stefan Buk, S. Veronica
Tan, Matthew Pitt, Andrew Durward, et al. 2011. « Infantile Onset Myofibrillar Myopathy
Due to Recessive CRYAB Mutations ». Neuromuscular Disorders: NMD 21 (1): 37‑40.
https://doi.org/10.1016/j.nmd.2010.11.003.

203
• Franceschelli, Silvia, Alessandra Rosati, Rosa Lerose, Serena De Nicola, Maria
Caterina Turco, et Maria Pascale. 2008. « Bag3 Gene Expression Is Regulated by Heat
Shock Factor 1 ». Journal of Cellular Physiology 215 (3): 575‑77.
https://doi.org/10.1002/jcp.21397.
• Franco, Renato, Giosuè Scognamiglio, Vincenzo Salerno, Adolfo Sebastiani,
Giovanni Cennamo, Paolo A. Ascierto, Gerardo Botti, Maria C. Turco, et Alessandra
Rosati. 2012. « Expression of the Anti-Apoptotic Protein BAG3 in Human Melanomas ».
The Journal of Investigative Dermatology 132 (1): 252‑54.
https://doi.org/10.1038/jid.2011.257.
• Franke, Josef D., Ruth A. Montague, et Daniel P. Kiehart. 2005. « Nonmuscle
Myosin II Generates Forces That Transmit Tension and Drive Contraction in Multiple
Tissues during Dorsal Closure ». Current Biology: CB 15 (24): 2208‑21.
https://doi.org/10.1016/j.cub.2005.11.064.
• Freddo, Andrew M., Suzanne K. Shoffner, Yue Shao, Kenichiro Taniguchi, Ann S.
Grosse, Margaux N. Guysinger, Sha Wang, et al. 2016. « Coordination of Signaling and
Tissue Mechanics during Morphogenesis of Murine Intestinal Villi: A Role for Mitotic
Cell Rounding ». Integrative Biology: Quantitative Biosciences from Nano to Macro 8 (9):
918‑28. https://doi.org/10.1039/c6ib00046k.
• Frémont, Stéphane, Hussein Hammich, Jian Bai, Hugo Wioland, Kerstin Klinkert,
Murielle Rocancourt, Carlos Kikuti, et al. 2017. « Oxidation of F-Actin Controls the
Terminal Steps of Cytokinesis ». Nature Communications 8 (février): 14528.
https://doi.org/10.1038/ncomms14528.
• Fuchs, Margit, Marie-Chloé Boulanger, Herman Lambert, Jacques Landry, et Josée
N. Lavoie. 2016. « Adenofection: A Method for Studying the Role of Molecular
Chaperones in Cellular Morphodynamics by Depletion-Rescue Experiments ». Journal of
Visualized Experiments: JoVE, no 115 (septembre). https://doi.org/10.3791/54557.
• Fuchs, Margit, Carole Luthold, Solenn M. Guilbert, Alice Anaïs Varlet, Herman
Lambert, Alexandra Jetté, Sabine Elowe, Jacques Landry, et Josée N. Lavoie. 2015. « A
Role for the Chaperone Complex BAG3-HSPB8 in Actin Dynamics, Spindle Orientation
and Proper Chromosome Segregation during Mitosis ». PLoS Genetics 11 (10).

204
https://doi.org/10.1371/journal.pgen.1005582.
• Fuchs, Margit, Dominic J. Poirier, Samuel J. Seguin, Herman Lambert, Serena
Carra, Steve J. Charette, et Jacques Landry. 2009. « Identification of the Key Structural
Motifs Involved in HspB8/HspB6-Bag3 Interaction ». The Biochemical Journal 425 (1):
245‑55. https://doi.org/10.1042/BJ20090907.
• Furuya, Tsuyoshi, Minsu Kim, Marta Lipinski, Juying Li, Dohoon Kim, Tao Lu,
Yong Shen, et al. 2010. « Negative Regulation of Vps34 by Cdk Mediated
Phosphorylation ». Molecular Cell 38 (4): 500‑511.
https://doi.org/10.1016/j.molcel.2010.05.009.
• Gal, Jozsef, Jing Chen, Kelly R. Barnett, Liuqing Yang, Erin Brumley, et Haining
Zhu. 2013. « HDAC6 Regulates Mutant SOD1 Aggregation through Two SMIR Motifs
and Tubulin Acetylation ». The Journal of Biological Chemistry 288 (21): 15035‑45.
https://doi.org/10.1074/jbc.M112.431957.
• Galluzzi, Lorenzo, Eric H. Baehrecke, Andrea Ballabio, Patricia Boya, José Manuel
Bravo-San Pedro, Francesco Cecconi, Augustine M. Choi, et al. 2017. « Molecular
Definitions of Autophagy and Related Processes ». The EMBO Journal 36 (13): 1811‑36.
https://doi.org/10.15252/embj.201796697.
• Gamerdinger, Martin, Parvana Hajieva, A. Murat Kaya, Uwe Wolfrum, F. Ulrich
Hartl, et Christian Behl. 2009. « Protein Quality Control during Aging Involves
Recruitment of the Macroautophagy Pathway by BAG3 ». The EMBO Journal 28 (7):
889‑901. https://doi.org/10.1038/emboj.2009.29.
• Gamerdinger, Martin, A. Murat Kaya, Uwe Wolfrum, Albrecht M. Clement, et
Christian Behl. 2011. « BAG3 Mediates Chaperone-Based Aggresome-Targeting and
Selective Autophagy of Misfolded Proteins ». EMBO Reports 12 (2): 149‑56.
https://doi.org/10.1038/embor.2010.203.
• Ganassi, Massimo, Daniel Mateju, Ilaria Bigi, Laura Mediani, Ina Poser, Hyun O.
Lee, Samuel J. Seguin, et al. 2016. « A Surveillance Function of the HSPB8-BAG3-HSP70
Chaperone Complex Ensures Stress Granule Integrity and Dynamism ». Molecular Cell 63
(5): 796‑810. https://doi.org/10.1016/j.molcel.2016.07.021.
• Gao, Ya-sheng, Charlotte C. Hubbert, Jianrong Lu, Yi-Shan Lee, Joo-Yong Lee, et

205
Tso-Pang Yao. 2007. « Histone Deacetylase 6 Regulates Growth Factor-Induced Actin
Remodeling and Endocytosis ». Molecular and Cellular Biology 27 (24): 8637‑47.
https://doi.org/10.1128/MCB.00393-07.
• Georgiou, Marios, Eliana Marinari, Jemima Burden, et Buzz Baum. 2008. « Cdc42,
Par6, and APKC Regulate Arp2/3-Mediated Endocytosis to Control Local Adherens
Junction Stability ». Current Biology: CB 18 (21): 1631‑38.
https://doi.org/10.1016/j.cub.2008.09.029.
• Ghosh, Joy G., Ananth K. Shenoy, et John I. Clark. 2006. « N- and C-Terminal
Motifs in Human AlphaB Crystallin Play an Important Role in the Recognition, Selection,
and Solubilization of Substrates ». Biochemistry 45 (46): 13847‑54.
https://doi.org/10.1021/bi061471m.
• Gober, M. D., C. Depre, et L. Aurelian. 2004. « Correspondence Regarding M.V.
Kim et Al. “Some Properties of Human Small Heat Shock Protein Hsp22 (H11 or
HspB8)” ». Biochemical and Biophysical Research Communications 321 (2): 267‑68.
https://doi.org/10.1016/j.bbrc.2004.06.103.
• Goldenberg, C. J., Y. Luo, M. Fenna, R. Baler, R. Weinmann, et R. Voellmy. 1988.
« Purified Human Factor Activates Heat Shock Promoter in a HeLa Cell-Free Transcription
System ». The Journal of Biological Chemistry 263 (36): 19734‑39.
• Golenhofen, Nikola, et Britta Bartelt-Kirbach. 2016. « The Impact of Small Heat
Shock Proteins (HspBs) in Alzheimer’s and Other Neurological Diseases ». Current
Pharmaceutical Design 22 (26): 4050‑62.
• Goley, Erin D., Stacia E. Rodenbusch, Adam C. Martin, et Matthew D. Welch.
2004. « Critical Conformational Changes in the Arp2/3 Complex Are Induced by
Nucleotide and Nucleation Promoting Factor ». Molecular Cell 16 (2): 269‑79.
https://doi.org/10.1016/j.molcel.2004.09.018.
• Goode, B. L., et A. A. Rodal. 2001. « Modular Complexes That Regulate Actin
Assembly in Budding Yeast ». Current Opinion in Microbiology 4 (6): 703‑12.
• Grintsevich, Elena E., Hunkar Gizem Yesilyurt, Shannon K. Rich, Ruei-Jiun Hung,
Jonathan R. Terman, et Emil Reisler. 2016. « F-Actin Dismantling through a Redox-Driven
Synergy between Mical and Cofilin ». Nature Cell Biology 18 (8): 876‑85.

206
https://doi.org/10.1038/ncb3390.
• Guerriero, L., K. Chong, R. Franco, A. Rosati, F. De Caro, M. Capunzo, M. C.
Turco, et D. S. B. Hoon. 2014. « BAG3 Protein Expression in Melanoma Metastatic Lymph
Nodes Correlates with Patients’ Survival ». Cell Death & Disease 5 (avril): e1173.
https://doi.org/10.1038/cddis.2014.143.
• Guha, Minakshi, Mian Zhou, et Yu-li Wang. 2005. « Cortical Actin Turnover
during Cytokinesis Requires Myosin II ». Current Biology 15 (8): 732‑36.
https://doi.org/10.1016/j.cub.2005.03.042.
• Guilbert, Solenn M., Herman Lambert, Marc-Antoine Rodrigue, Margit Fuchs,
Jacques Landry, et Josée N. Lavoie. 2018. « HSPB8 and BAG3 Cooperate to Promote
Spatial Sequestration of Ubiquitinated Proteins and Coordinate the Cellular Adaptive
Response to Proteasome Insufficiency ». FASEB Journal: Official Publication of the
Federation of American Societies for Experimental Biology, février, fj201700558RR.
https://doi.org/10.1096/fj.201700558RR.
• Guilbert, Solenn M., Alice-Anaïs Varlet, Margit Fuchs, Herman Lambert, Jacques
Landry, et Josée N. Lavoie. 2015. « Regulation of Actin-Based Structure Dynamics by
HspB Proteins and Partners ». Dans The Big Book on Small Heat Shock Proteins, édité par
Robert M. Tanguay et Lawrence E. Hightower, 435‑56. Heat Shock Proteins 8. Springer
International Publishing. https://doi.org/10.1007/978-3-319-16077-1_18.
• Gunsalus, K. C., S. Bonaccorsi, E. Williams, F. Verni, M. Gatti, et M. L. Goldberg.
1995. « Mutations in Twinstar, a Drosophila Gene Encoding a Cofilin/ADF Homologue,
Result in Defects in Centrosome Migration and Cytokinesis ». The Journal of Cell Biology
131 (5): 1243‑59.
• Guo, Kun, Nan Xiao Kang, Yan Li, Lu Sun, Lin Gan, Feng Jie Cui, Mei Dong Gao,
et Kun Yin Liu. 2009. « Regulation of HSP27 on NF-KappaB Pathway Activation May Be
Involved in Metastatic Hepatocellular Carcinoma Cells Apoptosis ». BMC Cancer 9
(mars): 100. https://doi.org/10.1186/1471-2407-9-100.
• Habata, Shutaro, Masahiro Iwasaki, Asuka Sugio, Miwa Suzuki, Masato Tamate,
Seiro Satohisa, Ryoichi Tanaka, et Tsuyoshi Saito. 2015. « BAG3 Increases the
Invasiveness of Uterine Corpus Carcinoma Cells by Suppressing MiR-29b and Enhancing

207
MMP2 Expression ». Oncology Reports 33 (5): 2613‑21.
https://doi.org/10.3892/or.2015.3831.
• ———. 2016. « BAG3-Mediated Mcl-1 Stabilization Contributes to Drug
Resistance via Interaction with USP9X in Ovarian Cancer ». International Journal of
Oncology 49 (1): 402‑10. https://doi.org/10.3892/ijo.2016.3494.
• Hable, Whitney E., et Darryl L. Kropf. 2005. « The Arp2/3 Complex Nucleates
Actin Arrays during Zygote Polarity Establishment and Growth ». Cell Motility and the
Cytoskeleton 61 (1): 9‑20. https://doi.org/10.1002/cm.20059.
• Haggarty, Stephen J., Kathryn M. Koeller, Jason C. Wong, Christina M. Grozinger,
et Stuart L. Schreiber. 2003. « Domain-Selective Small-Molecule Inhibitor of Histone
Deacetylase 6 (HDAC6)-Mediated Tubulin Deacetylation ». Proceedings of the National
Academy of Sciences of the United States of America 100 (8): 4389‑94.
https://doi.org/10.1073/pnas.0430973100.
• Hamouda, Mohamed-Amine, Nathalie Belhacene, Alexandre Puissant, Pascal
Colosetti, Guillaume Robert, Arnaud Jacquel, Bernard Mari, Patrick Auberger, et Frederic
Luciano. 2014. « The Small Heat Shock Protein B8 (HSPB8) Confers Resistance to
Bortezomib by Promoting Autophagic Removal of Misfolded Proteins in Multiple
Myeloma Cells ». Oncotarget 5 (15): 6252‑66. https://doi.org/10.18632/oncotarget.2193.
• Hara, Taichi, Kenji Nakamura, Makoto Matsui, Akitsugu Yamamoto, Yohko
Nakahara, Rika Suzuki-Migishima, Minesuke Yokoyama, et al. 2006. « Suppression of
Basal Autophagy in Neural Cells Causes Neurodegenerative Disease in Mice ». Nature
441 (7095): 885‑89. https://doi.org/10.1038/nature04724.
• Harris, Elizabeth S., Fang Li, et Henry N. Higgs. 2004. « The Mouse Formin, FRLα,
Slows Actin Filament Barbed End Elongation, Competes with Capping Protein,
Accelerates Polymerization from Monomers, and Severs Filaments ». Journal of
Biological Chemistry 279 (19): 20076‑87. https://doi.org/10.1074/jbc.M312718200.
• Hartl, F. Ulrich. 2017. « Protein Misfolding Diseases ». Annual Review of
Biochemistry 86 (juin): 21‑26. https://doi.org/10.1146/annurev-biochem-061516-044518.
• Haslbeck, Martin, et Elizabeth Vierling. 2015. « A First Line of Stress Defense:
Small Heat Shock Proteins and Their Function in Protein Homeostasis ». Journal of

208
Molecular Biology 427 (7): 1537‑48. https://doi.org/10.1016/j.jmb.2015.02.002.
• Haviv, Lior, David Gillo, Frederic Backouche, et Anne Bernheim-Groswasser.
2008. « A Cytoskeletal Demolition Worker: Myosin II Acts as an Actin Depolymerization
Agent ». Journal of Molecular Biology 375 (2): 325‑30.
https://doi.org/10.1016/j.jmb.2007.09.066.
• Hayashi, M. T., et J. Karlseder. 2013. « DNA Damage Associated with Mitosis and
Cytokinesis Failure ». Oncogene 32 (39): 4593‑4601.
https://doi.org/10.1038/onc.2012.615.
• Hayes, David, Vanessa Napoli, Andrew Mazurkie, Walter F. Stafford, et Philip
Graceffa. 2009. « Phosphorylation Dependence of Hsp27 Multimeric Size and Molecular
Chaperone Function ». The Journal of Biological Chemistry 284 (28): 18801‑7.
https://doi.org/10.1074/jbc.M109.011353.
• Haynes, Jennifer, Jyoti Srivastava, Nikki Madson, Torsten Wittmann, et Diane L.
Barber. 2011. « Dynamic actin remodeling during epithelial–mesenchymal transition
depends on increased moesin expression ». Molecular Biology of the Cell 22 (24):
4750‑64. https://doi.org/10.1091/mbc.E11-02-0119.
• Heirbaut, Michelle, Frederik Lermyte, Esther M. Martin, Steven Beelen, Frank
Sobott, Sergei V. Strelkov, et Stephen D. Weeks. 2017. « Specific Sequences in the N-
Terminal Domain of Human Small Heat-Shock Protein HSPB6 Dictate Preferential
Hetero-Oligomerization with the Orthologue HSPB1 ». The Journal of Biological
Chemistry 292 (24): 9944‑57. https://doi.org/10.1074/jbc.M116.773515.
• Helwani, Falak M., Eva M. Kovacs, Andrew D. Paterson, Suzie Verma, Radiya G.
Ali, Alan S. Fanning, Scott A. Weed, et Alpha S. Yap. 2004. « Cortactin Is Necessary for
E-Cadherin-Mediated Contact Formation and Actin Reorganization ». The Journal of Cell
Biology 164 (6): 899‑910. https://doi.org/10.1083/jcb.200309034.
• Hickson, Gilles R. X., Johanne Matheson, Blake Riggs, Valerie H. Maier, Andrew
B. Fielding, Rytis Prekeris, William Sullivan, Francis A. Barr, et Gwyn W. Gould. 2003.
« Arfophilins Are Dual Arf/Rab 11 Binding Proteins That Regulate Recycling Endosome
Distribution and Are Related to Drosophila Nuclear Fallout ». Molecular Biology of the
Cell 14 (7): 2908‑20. https://doi.org/10.1091/mbc.E03-03-0160.

209
• Hickson, Gilles R. X., et Patrick H. O’Farrell. 2008. « Anillin: A Pivotal Organizer
of the Cytokinetic Machinery ». Biochemical Society Transactions 36 (Pt 3): 439‑41.
https://doi.org/10.1042/BST0360439.
• Higgs, H. N., et T. D. Pollard. 2000. « Activation by Cdc42 and PIP(2) of Wiskott-
Aldrich Syndrome Protein (WASp) Stimulates Actin Nucleation by Arp2/3 Complex ».
The Journal of Cell Biology 150 (6): 1311‑20.
• Higgs, Henry N., et Kevin J. Peterson. 2005. « Phylogenetic Analysis of the Formin
Homology 2 Domain ». Molecular Biology of the Cell 16 (1): 1‑13.
https://doi.org/10.1091/mbc.E04-07-0565.
• Hilton, Gillian R., Georg K. A. Hochberg, Arthur Laganowsky, Scott I.
McGinnigle, Andrew J. Baldwin, et Justin L. P. Benesch. 2013. « C-Terminal Interactions
Mediate the Quaternary Dynamics of ΑB-Crystallin ». Philosophical Transactions of the
Royal Society of London. Series B, Biological Sciences 368 (1617): 20110405.
https://doi.org/10.1098/rstb.2011.0405.
• Hishiya, Akinori, Toshio Kitazawa, et Shinichi Takayama. 2010. « BAG3 and
Hsc70 Interact with Actin Capping Protein CapZ to Maintain Myofibrillar Integrity under
Mechanical Stress ». Circulation Research 107 (10): 1220‑31.
https://doi.org/10.1161/CIRCRESAHA.110.225649.
• Hodge, Richard G., et Anne J. Ridley. 2016. « Regulating Rho GTPases and Their
Regulators ». Nature Reviews. Molecular Cell Biology 17 (8): 496‑510.
https://doi.org/10.1038/nrm.2016.67.
• Höhfeld, Jörg. 2016. « Autophagy: Press and Push for Destruction ». Current
Biology: CB 26 (15): R703-705. https://doi.org/10.1016/j.cub.2016.06.017.
• Hoijman, Esteban, Davide Rubbini, Julien Colombelli, et Berta Alsina. 2015.
« Mitotic Cell Rounding and Epithelial Thinning Regulate Lumen Growth and Shape ».
Nature Communications 6 (juin): 7355. https://doi.org/10.1038/ncomms8355.
• Holmes, Kenneth C. 1997. « The swinging lever-arm hypothesis of muscle
contraction ». Current Biology 7 (2): R112‑18. https://doi.org/10.1016/S0960-
9822(06)00051-0.
• Homma, Sachiko, Masahiro Iwasaki, G. Diane Shelton, Eva Engvall, John C. Reed,

210
et Shinichi Takayama. 2006. « BAG3 Deficiency Results in Fulminant Myopathy and
Early Lethality ». The American Journal of Pathology 169 (3): 761‑73.
https://doi.org/10.2353/ajpath.2006.060250.
• Hotchin, N. A., et A. Hall. 1995. « The Assembly of Integrin Adhesion Complexes
Requires Both Extracellular Matrix and Intracellular Rho/Rac GTPases ». The Journal of
Cell Biology 131 (6 Pt 2): 1857‑65.
• Houslay, Miles D., et Frank Christian. 2010. « P62 (SQSTM1) Forms Part of a
Novel, Reversible Aggregate Containing a Specific Conformer of the CAMP Degrading
Phosphodiesterase, PDE4A4 ». Autophagy 6 (8): 1198‑1200.
https://doi.org/10.4161/auto.6.8.13479.
• Hu, Zhiping, Lan Chen, Jie Zhang, Ting Li, Jianguang Tang, Niangui Xu, et Xiang
Wang. 2007. « Structure, Function, Property, and Role in Neurologic Diseases and Other
Diseases of the SHsp22 ». Journal of Neuroscience Research 85 (10): 2071‑79.
https://doi.org/10.1002/jnr.21231.
• Huang, Fangtian, Xuemei Zeng, Woong Kim, Manimalha Balasubramani, Arola
Fortian, Steven P. Gygi, Nathan A. Yates, et Alexander Sorkin. 2013. « Lysine 63-Linked
Polyubiquitination Is Required for EGF Receptor Degradation ». Proceedings of the
National Academy of Sciences of the United States of America 110 (39): 15722‑27.
https://doi.org/10.1073/pnas.1308014110.
• Huang, Xiao-Yong, Ai-Wu Ke, Guo-Ming Shi, Xin Zhang, Chi Zhang, Ying-Hong
Shi, Xiao-Ying Wang, et al. 2013. « ΑB-Crystallin Complexes with 14-3-3ζ to Induce
Epithelial-Mesenchymal Transition and Resistance to Sorafenib in Hepatocellular
Carcinoma ». Hepatology 57 (6): 2235‑47. https://doi.org/10.1002/hep.26255.
• Hüfner, Katharina, Barbara Schell, Martin Aepfelbacher, et Stefan Linder. 2002.
« The Acidic Regions of WASp and N-WASP Can Synergize with CDC42Hs and Rac1 to
Induce Filopodia and Lamellipodia ». FEBS Letters 514 (2‑3): 168‑74.
• Hung, Ruei-Jiun, Chi W. Pak, et Jonathan R. Terman. 2011. « Direct Redox
Regulation of F-Actin Assembly and Disassembly by Mical ». Science (New York, N.Y.)
334 (6063): 1710‑13. https://doi.org/10.1126/science.1211956.
• Huot, J., H. Lambert, J. N. Lavoie, A. Guimond, F. Houle, et J. Landry. 1995.

211
« Characterization of 45-KDa/54-KDa HSP27 Kinase, a Stress-Sensitive Kinase Which
May Activate the Phosphorylation-Dependent Protective Function of Mammalian 27-KDa
Heat-Shock Protein HSP27 ». European Journal of Biochemistry 227 (1‑2): 416‑27.
• Ichimura, Yoshinobu, Takayoshi Kirisako, Toshifumi Takao, Yoshinori Satomi,
Yasutsugu Shimonishi, Naotada Ishihara, Noboru Mizushima, et al. 2000. « A Ubiquitin-
like System Mediates Protein Lipidation ». Nature 408 (6811): 488‑92.
https://doi.org/10.1038/35044114.
• Ichimura, Yoshinobu, Taichi Kumanomidou, Yu-shin Sou, Tsunehiro Mizushima,
Junji Ezaki, Takashi Ueno, Eiki Kominami, Takashi Yamane, Keiji Tanaka, et Masaaki
Komatsu. 2008. « Structural Basis for Sorting Mechanism of P62 in Selective Autophagy ».
The Journal of Biological Chemistry 283 (33): 22847‑57.
https://doi.org/10.1074/jbc.M802182200.
• Ichimura, Yoshinobu, Satoshi Waguri, Yu-Shin Sou, Shun Kageyama, Jun
Hasegawa, Ryosuke Ishimura, Tetsuya Saito, et al. 2013. « Phosphorylation of P62
Activates the Keap1-Nrf2 Pathway during Selective Autophagy ». Molecular Cell 51 (5):
618‑31. https://doi.org/10.1016/j.molcel.2013.08.003.
• Ikebe, M., et D. J. Hartshorne. 1985. « Phosphorylation of Smooth Muscle Myosin
at Two Distinct Sites by Myosin Light Chain Kinase ». The Journal of Biological
Chemistry 260 (18): 10027‑31.
• Ikebe, M., J. Koretz, et D. J. Hartshorne. 1988. « Effects of Phosphorylation of
Light Chain Residues Threonine 18 and Serine 19 on the Properties and Conformation of
Smooth Muscle Myosin ». The Journal of Biological Chemistry 263 (13): 6432‑37.
• Im, Chang-Nim, Hye Hyeon Yun, Byunghoo Song, Dong-Ye Youn, Mei Nu Cui,
Hong Sug Kim, Gyeong Sin Park, et Jeong-Hwa Lee. 2016. « BIS-Mediated STAT3
Stabilization Regulates Glioblastoma Stem Cell-like Phenotypes ». Oncotarget 7 (23):
35056‑70. https://doi.org/10.18632/oncotarget.9039.
• Insall, R., A. Müller-Taubenberger, L. Machesky, J. Köhler, E. Simmeth, S. J.
Atkinson, I. Weber, et G. Gerisch. 2001. « Dynamics of the Dictyostelium Arp2/3 Complex
in Endocytosis, Cytokinesis, and Chemotaxis ». Cell Motility and the Cytoskeleton 50 (3):
115‑28. https://doi.org/10.1002/cm.10005.

212
• Into, Takeshi, Megumi Inomata, Shumpei Niida, Yukitaka Murakami, et Ken-ichiro
Shibata. 2010. « Regulation of MyD88 Aggregation and the MyD88-Dependent Signaling
Pathway by Sequestosome 1 and Histone Deacetylase 6 ». The Journal of Biological
Chemistry 285 (46): 35759‑69. https://doi.org/10.1074/jbc.M110.126904.
• Irobi, Joy, Katrien Van Impe, Pavel Seeman, Albena Jordanova, Ines Dierick,
Nathalie Verpoorten, Andrej Michalik, et al. 2004. « Hot-Spot Residue in Small Heat-
Shock Protein 22 Causes Distal Motor Neuropathy ». Nature Genetics 36 (6): 597‑601.
https://doi.org/10.1038/ng1328.
• Isogai, Shin, Daichi Morimoto, Kyohei Arita, Satoru Unzai, Takeshi Tenno, Jun
Hasegawa, Yu-shin Sou, et al. 2011. « Crystal Structure of the Ubiquitin-Associated
(UBA) Domain of P62 and Its Interaction with Ubiquitin ». The Journal of Biological
Chemistry 286 (36): 31864‑74. https://doi.org/10.1074/jbc.M111.259630.
• Ito, Akihiro, Tadahiro Shimazu, Satoko Maeda, Asad Ali Shah, Tatsuhiko Tsunoda,
Shun-Ichiro Iemura, Toru Natsume, et al. 2015. « The Subcellular Localization and
Activity of Cortactin Is Regulated by Acetylation and Interaction with Keap1 ». Science
Signaling 8 (404): ra120. https://doi.org/10.1126/scisignal.aad0667.
• Iwasaki, Masahiro, Sachiko Homma, Akinori Hishiya, Samuel J. Dolezal, John C.
Reed, et Shinichi Takayama. 2007. « BAG3 Regulates Motility and Adhesion of Epithelial
Cancer Cells ». Cancer Research 67 (21): 10252‑59. https://doi.org/10.1158/0008-
5472.CAN-07-0618.
• Iwasaki, Masahiro, Ryoichi Tanaka, Akinori Hishiya, Sachiko Homma, John C.
Reed, et Shinichi Takayama. 2010. « BAG3 Directly Associates with Guanine Nucleotide
Exchange Factor of Rap1, PDZGEF2, and Regulates Cell Adhesion ». Biochemical and
Biophysical Research Communications 400 (3): 413‑18.
https://doi.org/10.1016/j.bbrc.2010.08.092.
• Iwata, Atsushi, Brigit E. Riley, Jennifer A. Johnston, et Ron R. Kopito. 2005.
« HDAC6 and Microtubules Are Required for Autophagic Degradation of Aggregated
Huntingtin ». Journal of Biological Chemistry 280 (48): 40282‑92.
https://doi.org/10.1074/jbc.M508786200.
• Jacobs, Aaron T., et Lawrence J. Marnett. 2009. « HSF1-Mediated BAG3

213
Expression Attenuates Apoptosis in 4-Hydroxynonenal-Treated Colon Cancer Cells via
Stabilization of Anti-Apoptotic Bcl-2 Proteins ». The Journal of Biological Chemistry 284
(14): 9176‑83. https://doi.org/10.1074/jbc.M808656200.
• James, P. A., J. Rankin, et K. Talbot. 2008. « Asymmetrical Late Onset Motor
Neuropathy Associated with a Novel Mutation in the Small Heat Shock Protein HSPB1
(HSP27) ». Journal of Neurology, Neurosurgery, and Psychiatry 79 (4): 461‑63.
https://doi.org/10.1136/jnnp.2007.125179.
• Janetopoulos, Chris, et Peter Devreotes. 2006. « Phosphoinositide Signaling Plays
a Key Role in Cytokinesis ». J Cell Biol 174 (4): 485‑90.
https://doi.org/10.1083/jcb.200603156.
• Jansen, Sepp, Reinoud Gosens, Thomas Wieland, et Martina Schmidt. 2017.
« Paving the Rho in Cancer Metastasis: Rho GTPases and Beyond ». Pharmacology &
Therapeutics, septembre. https://doi.org/10.1016/j.pharmthera.2017.09.002.
• Johnston, J. A., C. L. Ward, et R. R. Kopito. 1998. « Aggresomes: A Cellular
Response to Misfolded Proteins ». The Journal of Cell Biology 143 (7): 1883‑98.
• Jong, W. W. de, G. J. Caspers, et J. A. Leunissen. 1998. « Genealogy of the Alpha-
Crystallin--Small Heat-Shock Protein Superfamily ». International Journal of Biological
Macromolecules 22 (3‑4): 151‑62.
• Jontes, J. D., E. M. Wilson-Kubalek, et R. A. Milligan. 1995. « A 32 Degree Tail
Swing in Brush Border Myosin I on ADP Release ». Nature 378 (6558): 751‑53.
https://doi.org/10.1038/378751a0.
• Kaji, Noriko, Aya Muramoto, et Kensaku Mizuno. 2008. « LIM Kinase-Mediated
Cofilin Phosphorylation during Mitosis Is Required for Precise Spindle Positioning ». The
Journal of Biological Chemistry 283 (8): 4983‑92.
https://doi.org/10.1074/jbc.M708644200.
• Kaji, Noriko, Kazumasa Ohashi, Mika Shuin, Ryusuke Niwa, Tadashi Uemura, et
Kensaku Mizuno. 2003. « Cell Cycle-Associated Changes in Slingshot Phosphatase
Activity and Roles in Cytokinesis in Animal Cells ». The Journal of Biological Chemistry
278 (35): 33450‑55. https://doi.org/10.1074/jbc.M305802200.
• Kaluza, David, Jens Kroll, Sabine Gesierich, Tso-Pang Yao, Reinier A. Boon,

214
Eduard Hergenreider, Marc Tjwa, et al. 2011. « Class IIb HDAC6 Regulates Endothelial
Cell Migration and Angiogenesis by Deacetylation of Cortactin ». The EMBO Journal 30
(20): 4142‑56. https://doi.org/10.1038/emboj.2011.298.
• Kamijo, Keiju, Naoya Ohara, Mitsuhiro Abe, Takashi Uchimura, Hiroshi Hosoya,
Jae-Seon Lee, et Toru Miki. 2006. « Dissecting the Role of Rho-Mediated Signaling in
Contractile Ring Formation ». Molecular Biology of the Cell 17 (1): 43‑55.
https://doi.org/10.1091/mbc.E05-06-0569.
• Kang, Se Hun, Keon Wook Kang, Kyung-Hee Kim, Bumi Kwon, Seok-Ki Kim,
Ho-Young Lee, Sun-Young Kong, Eun Sook Lee, Sang-Geun Jang, et Byong Chul Yoo.
2008. « Upregulated HSP27 in Human Breast Cancer Cells Reduces Herceptin
Susceptibility by Increasing Her2 Protein Stability ». BMC Cancer 8 (octobre): 286.
https://doi.org/10.1186/1471-2407-8-286.
• Kanno, Kiminori, Shoji Kanno, Hiroyuki Nitta, Noriyuki Uesugi, Tamostu Sugai,
Tomoyuki Masuda, Go Wakabayashi, et Chihaya Maesawa. 2012. « Overexpression of
Histone Deacetylase 6 Contributes to Accelerated Migration and Invasion Activity of
Hepatocellular Carcinoma Cells ». Oncology Reports 28 (3): 867‑73.
https://doi.org/10.3892/or.2012.1898.
• Kappé, G., P. Verschuure, R. L. Philipsen, A. A. Staalduinen, P. Van de Boogaart,
W. C. Boelens, et W. W. De Jong. 2001. « Characterization of Two Novel Human Small
Heat Shock Proteins: Protein Kinase-Related HspB8 and Testis-Specific HspB9 ».
Biochimica Et Biophysica Acta 1520 (1): 1‑6.
• Karpel-Massler, Georg, Chang Shu, Lily Chau, Matei Banu, Marc-Eric Halatsch,
Mike-Andrew Westhoff, Yulian Ramirez, et al. 2015. « Combined Inhibition of Bcl-2/Bcl-
XL and Usp9X/Bag3 Overcomes Apoptotic Resistance in Glioblastoma in Vitro and in
Vivo ». Oncotarget 6 (16): 14507‑21. https://doi.org/10.18632/oncotarget.3993.
• Kassis, Jareer N., Elizabeth A. Guancial, Howard Doong, Victoria Virador, et Elise
C. Kohn. 2006. « CAIR-1/BAG-3 Modulates Cell Adhesion and Migration by
Downregulating Activity of Focal Adhesion Proteins ». Experimental Cell Research 312
(15): 2962‑71. https://doi.org/10.1016/j.yexcr.2006.05.023.
• Kassis, Jareer N., Victoria M. Virador, Elizabeth A. Guancial, Daniel Kimm, Allen

215
S. Ho, Mark Mishra, Eric Y. Chuang, John Cook, David Gius, et Elise C. Kohn. 2009.
« Genomic and Phenotypic Analysis Reveals a Key Role for CCN1 (CYR61) in BAG3-
Modulated Adhesion and Invasion ». The Journal of Pathology 218 (4): 495‑504.
https://doi.org/10.1002/path.2557.
• Kast, David J., et Roberto Dominguez. 2015. « WHAMM Links Actin Assembly
via the Arp2/3 Complex to Autophagy ». Autophagy 11 (9): 1702‑4.
https://doi.org/10.1080/15548627.2015.1073434.
• Kast, David J., Allison L. Zajac, Erika L. F. Holzbaur, E. Michael Ostap, et Roberto
Dominguez. 2015. « WHAMM Directs the Arp2/3 Complex to the ER for Autophagosome
Biogenesis through an Actin Comet Tail Mechanism ». Current Biology: CB 25 (13):
1791‑97. https://doi.org/10.1016/j.cub.2015.05.042.
• Kathage, Barbara, Sebastian Gehlert, Anna Ulbricht, Laura Lüdecke, Victor E.
Tapia, Zacharias Orfanos, Daniela Wenzel, et al. 2016. « The Cochaperone BAG3
Coordinates Protein Synthesis and Autophagy under Mechanical Strain through Spatial
Regulation of MTORC1 ». Biochimica Et Biophysica Acta 1864 (1): 62‑75.
https://doi.org/10.1016/j.bbamcr.2016.10.007.
• Kawaguchi, Yoshiharu, Jeffrey J. Kovacs, Adam McLaurin, Jeffery M. Vance,
Akihiro Ito, et Tso Pang Yao. 2003. « The Deacetylase HDAC6 Regulates Aggresome
Formation and Cell Viability in Response to Misfolded Protein Stress ». Cell 115 (6):
727‑38.
• Ke, Lei, Roelien A. M. Meijering, Femke Hoogstra-Berends, Katarina
Mackovicova, Michel J. Vos, Isabelle C. Van Gelder, Robert H. Henning, Harm H.
Kampinga, et Bianca J. J. M. Brundel. 2011. « HSPB1, HSPB6, HSPB7 and HSPB8 Protect
against RhoA GTPase-Induced Remodeling in Tachypaced Atrial Myocytes ». PloS One
6 (6): e20395. https://doi.org/10.1371/journal.pone.0020395.
• Kee, Yee-Seir, Yixin Ren, Danielle Dorfman, Miho Iijima, Richard Firtel, Pablo A.
Iglesias, et Douglas N. Robinson. 2012. « A Mechanosensory System Governs Myosin II
Accumulation in Dividing Cells ». Molecular Biology of the Cell 23 (8): 1510‑23.
https://doi.org/10.1091/mbc.E11-07-0601.
• Kim, J., V. M. Dalton, K. P. Eggerton, S. V. Scott, et D. J. Klionsky. 1999.

216
« Apg7p/Cvt2p Is Required for the Cytoplasm-to-Vacuole Targeting, Macroautophagy,
and Peroxisome Degradation Pathways ». Molecular Biology of the Cell 10 (5): 1337‑51.
• Kim, K. K., R. Kim, et S. H. Kim. 1998. « Crystal Structure of a Small Heat-Shock
Protein ». Nature 394 (6693): 595‑99. https://doi.org/10.1038/29106.
• Kim, Kye-Young, Mihály Kovács, Sachiyo Kawamoto, James R. Sellers, et Robert
S. Adelstein. 2005. « Disease-Associated Mutations and Alternative Splicing Alter the
Enzymatic and Motile Activity of Nonmuscle Myosins II-B and II-C ». The Journal of
Biological Chemistry 280 (24): 22769‑75. https://doi.org/10.1074/jbc.M503488200.
• Kim, Maria V., Alexei S. Kasakov, Alim S. Seit-Nebi, Steven B. Marston, et
Nikolai B. Gusev. 2006. « Structure and Properties of K141E Mutant of Small Heat Shock
Protein HSP22 (HspB8, H11) That Is Expressed in Human Neuromuscular Disorders ».
Archives of Biochemistry and Biophysics 454 (1): 32‑41.
https://doi.org/10.1016/j.abb.2006.07.014.
• Kim, Maria V., Alim S. Seit-Nebi, et Nikolai B. Gusev. 2004. « The Problem of
Protein Kinase Activity of Small Heat Shock Protein Hsp22 (H11 or HspB8) ».
Biochemical and Biophysical Research Communications 325 (3): 649‑52.
https://doi.org/10.1016/j.bbrc.2004.10.074.
• Kimura, K., M. Ito, M. Amano, K. Chihara, Y. Fukata, M. Nakafuku, B. Yamamori,
et al. 1996. « Regulation of Myosin Phosphatase by Rho and Rho-Associated Kinase (Rho-
Kinase) ». Science (New York, N.Y.) 273 (5272): 245‑48.
• King, Michael, Firoozeh Nafar, Joseph Clarke, et Karen Mearow. 2009. « The
Small Heat Shock Protein Hsp27 Protects Cortical Neurons against the Toxic Effects of
Beta-Amyloid Peptide ». Journal of Neuroscience Research 87 (14): 3161‑75.
https://doi.org/10.1002/jnr.22145.
• Kirisako, T., Y. Ichimura, H. Okada, Y. Kabeya, N. Mizushima, T. Yoshimori, M.
Ohsumi, T. Takao, T. Noda, et Y. Ohsumi. 2000. « The Reversible Modification Regulates
the Membrane-Binding State of Apg8/Aut7 Essential for Autophagy and the Cytoplasm to
Vacuole Targeting Pathway ». The Journal of Cell Biology 151 (2): 263‑76.
• Kishi, K., T. Sasaki, S. Kuroda, T. Itoh, et Y. Takai. 1993. « Regulation of
Cytoplasmic Division of Xenopus Embryo by Rho P21 and Its Inhibitory GDP/GTP

217
Exchange Protein (Rho GDI) ». The Journal of Cell Biology 120 (5): 1187‑95.
• Kissová, Ingrid, Maïka Deffieu, Stéphen Manon, et Nadine Camougrand. 2004.
« Uth1p Is Involved in the Autophagic Degradation of Mitochondria ». The Journal of
Biological Chemistry 279 (37): 39068‑74. https://doi.org/10.1074/jbc.M406960200.
• Kiyomitsu, Tomomi, et Iain M. Cheeseman. 2012. « Chromosome- and Spindle-
Pole-Derived Signals Generate an Intrinsic Code for Spindle Position and Orientation ».
Nature Cell Biology 14 (3): 311‑17. https://doi.org/10.1038/ncb2440.
• Klaips, Courtney L., Gopal Gunanathan Jayaraj, et F. Ulrich Hartl. 2017.
« Pathways of Cellular Proteostasis in Aging and Disease ». The Journal of Cell Biology,
novembre. https://doi.org/10.1083/jcb.201709072.
• Klionsky, Daniel J., Kotb Abdelmohsen, Akihisa Abe, Md Joynal Abedin, Hagai
Abeliovich, Abraham Acevedo Arozena, Hiroaki Adachi, et al. 2016. « Guidelines for the
Use and Interpretation of Assays for Monitoring Autophagy (3rd Edition) ». Autophagy 12
(1): 1‑222. https://doi.org/10.1080/15548627.2015.1100356.
• Koh, Ara, Dohyun Park, Heeyoon Jeong, Jiyoun Lee, Mi Nam Lee, Pann-Ghill Suh,
et Sung Ho Ryu. 2014. « Regulation of C1-Ten Protein Tyrosine Phosphatase by
P62/SQSTM1-Mediated Sequestration and Degradation ». Cellular Signalling 26 (11):
2470‑80. https://doi.org/10.1016/j.cellsig.2014.07.033.
• Komatsu, Masaaki, Hirofumi Kurokawa, Satoshi Waguri, Keiko Taguchi, Akira
Kobayashi, Yoshinobu Ichimura, Yu-Shin Sou, et al. 2010. « The Selective Autophagy
Substrate P62 Activates the Stress Responsive Transcription Factor Nrf2 through
Inactivation of Keap1 ». Nature Cell Biology 12 (3): 213‑23.
https://doi.org/10.1038/ncb2021.
• Komatsu, Masaaki, Satoshi Waguri, Tomoki Chiba, Shigeo Murata, Jun-ichi Iwata,
Isei Tanida, Takashi Ueno, et al. 2006. « Loss of Autophagy in the Central Nervous System
Causes Neurodegeneration in Mice ». Nature 441 (7095): 880‑84.
https://doi.org/10.1038/nature04723.
• Kondo, Takefumi, et Shigeo Hayashi. 2013. « Mitotic Cell Rounding Accelerates
Epithelial Invagination ». Nature 494 (7435): 125‑29.
https://doi.org/10.1038/nature11792.

218
• Kotak, Sachin, Coralie Busso, et Pierre Gönczy. 2012. « Cortical Dynein Is Critical
for Proper Spindle Positioning in Human Cells ». J Cell Biol 199 (1): 97‑110.
https://doi.org/10.1083/jcb.201203166.
• Kouranti, Ilektra, Martin Sachse, Nassim Arouche, Bruno Goud, et Arnaud Echard.
2006. « Rab35 Regulates an Endocytic Recycling Pathway Essential for the Terminal Steps
of Cytokinesis ». Current Biology: CB 16 (17): 1719‑25.
https://doi.org/10.1016/j.cub.2006.07.020.
• Kovar, David R., Elizabeth S. Harris, Rachel Mahaffy, Henry N. Higgs, et Thomas
D. Pollard. 2006. « Control of the Assembly of ATP- and ADP-Actin by Formins and
Profilin ». Cell 124 (2): 423‑35. https://doi.org/10.1016/j.cell.2005.11.038.
• Kowalski, Jennifer R., Coumaran Egile, Susana Gil, Scott B. Snapper, Rong Li, et
Sheila M. Thomas. 2005. « Cortactin Regulates Cell Migration through Activation of N-
WASP ». Journal of Cell Science 118 (Pt 1): 79‑87. https://doi.org/10.1242/jcs.01586.
• Kozma, R., S. Ahmed, A. Best, et L. Lim. 1995. « The Ras-Related Protein
Cdc42Hs and Bradykinin Promote Formation of Peripheral Actin Microspikes and
Filopodia in Swiss 3T3 Fibroblasts ». Molecular and Cellular Biology 15 (4): 1942‑52.
• Kozyreva, Varvara K., Sarah L. McLaughlin, Ryan H. Livengood, Robin A.
Calkins, Laura C. Kelley, Anuradha Rajulapati, Ryan J. Ice, Matthew B. Smolkin, Scott A.
Weed, et Elena N. Pugacheva. 2014. « NEDD9 Regulates Actin Dynamics through
Cortactin Deacetylation in an AURKA/HDAC6-Dependent Manner ». Molecular Cancer
Research: MCR 12 (5): 681‑93. https://doi.org/10.1158/1541-7786.MCR-13-0654.
• Kriehuber, Thomas, Thomas Rattei, Thomas Weinmaier, Alexander Bepperling,
Martin Haslbeck, et Johannes Buchner. 2010. « Independent Evolution of the Core Domain
and Its Flanking Sequences in Small Heat Shock Proteins ». FASEB Journal: Official
Publication of the Federation of American Societies for Experimental Biology 24 (10):
3633‑42. https://doi.org/10.1096/fj.10-156992.
• Kuchitsu, Yoshihiko, Yuta Homma, Naonobu Fujita, et Mitsunori Fukuda. 2018.
« Rab7 Knockout Unveils Regulated Autolysosome Maturation Induced by Glutamine
Starvation ». Journal of Cell Science 131 (7). https://doi.org/10.1242/jcs.215442.
• Kumar, Vijay, Neha Sami, Tara Kashav, Asimul Islam, Faizan Ahmad, et Md

219
Imtaiyaz Hassan. 2016. « Protein Aggregation and Neurodegenerative Diseases: From
Theory to Therapy ». European Journal of Medicinal Chemistry 124 (novembre): 1105‑20.
https://doi.org/10.1016/j.ejmech.2016.07.054.
• Kunda, Patricia, Andrew E. Pelling, Tao Liu, et Buzz Baum. 2008. « Moesin
Controls Cortical Rigidity, Cell Rounding, and Spindle Morphogenesis during Mitosis ».
Current Biology 18 (2): 91‑101. https://doi.org/10.1016/j.cub.2007.12.051.
• Kureishi, Y., S. Kobayashi, M. Amano, K. Kimura, H. Kanaide, T. Nakano, K.
Kaibuchi, et M. Ito. 1997. « Rho-Associated Kinase Directly Induces Smooth Muscle
Contraction through Myosin Light Chain Phosphorylation ». The Journal of Biological
Chemistry 272 (19): 12257‑60.
• Kwok, Alice S., Kanchan Phadwal, Bradley J. Turner, Peter L. Oliver, Annie Raw,
Anna Katharina Simon, Kevin Talbot, et Vishwas R. Agashe. 2011. « HspB8 Mutation
Causing Hereditary Distal Motor Neuropathy Impairs Lysosomal Delivery of
Autophagosomes ». Journal of Neurochemistry 119 (6): 1155‑61.
https://doi.org/10.1111/j.1471-4159.2011.07521.x.
• Kwon, Mijung, Maria Bagonis, Gaudenz Danuser, et David Pellman. 2015. « Direct
Microtubule-Binding by Myosin-10 Orients Centrosomes toward Retraction Fibers and
Subcortical Actin Clouds ». Developmental Cell 34 (3): 323‑37.
https://doi.org/10.1016/j.devcel.2015.06.013.
• Laan, Liedewij, Nenad Pavin, Julien Husson, Guillaume Romet-Lemonne, Martijn
van Duijn, Magdalena Preciado López, Ronald D. Vale, Frank Jülicher, Samara L. Reck-
Peterson, et Marileen Dogterom. 2012. « Cortical Dynein Controls Microtubule Dynamics
to Generate Pulling Forces That Position Microtubule Asters ». Cell 148 (3): 502‑14.
https://doi.org/10.1016/j.cell.2012.01.007.
• Lahvic, Jamie L., Yongchang Ji, Paloma Marin, Jonah P. Zuflacht, Mark W.
Springel, Jonathan E. Wosen, Leigh Davis, Lara D. Hutson, Jeffrey D. Amack, et Martha
J. Marvin. 2013. « Small Heat Shock Proteins Are Necessary for Heart Migration and
Laterality Determination in Zebrafish ». Developmental Biology 384 (2): 166‑80.
https://doi.org/10.1016/j.ydbio.2013.10.009.
• Lai, Frank P. L., Malgorzata Szczodrak, Jennifer Block, Jan Faix, Dennis

220
Breitsprecher, Hans G. Mannherz, Theresia E. B. Stradal, Graham A. Dunn, J. Victor
Small, et Klemens Rottner. 2008. « Arp2/3 Complex Interactions and Actin Network
Turnover in Lamellipodia ». The EMBO Journal 27 (7): 982‑92.
https://doi.org/10.1038/emboj.2008.34.
• Lamark, Trond, Maria Perander, Heidi Outzen, Kurt Kristiansen, Aud Øvervatn,
Espen Michaelsen, Geir Bjørkøy, et Terje Johansen. 2003. « Interaction Codes within the
Family of Mammalian Phox and Bem1p Domain-Containing Proteins ». The Journal of
Biological Chemistry 278 (36): 34568‑81. https://doi.org/10.1074/jbc.M303221200.
• Lancaster, Oscar M., et Buzz Baum. 2014. « Shaping up to Divide: Coordinating
Actin and Microtubule Cytoskeletal Remodelling during Mitosis ». Seminars in Cell &
Developmental Biology 34 (octobre): 109‑15.
https://doi.org/10.1016/j.semcdb.2014.02.015.
• Lancaster, Oscar M., Maël Le Berre, Andrea Dimitracopoulos, Daria Bonazzi, Ewa
Zlotek-Zlotkiewicz, Remigio Picone, Thomas Duke, Matthieu Piel, et Buzz Baum. 2013.
« Mitotic Rounding Alters Cell Geometry to Ensure Efficient Bipolar Spindle Formation ».
Developmental Cell 25 (3): 270‑83. https://doi.org/10.1016/j.devcel.2013.03.014.
• Lanneau, D., M. Brunet, E. Frisan, E. Solary, M. Fontenay, et C. Garrido. 2008.
« Heat Shock Proteins: Essential Proteins for Apoptosis Regulation ». Journal of Cellular
and Molecular Medicine 12 (3): 743‑61. https://doi.org/10.1111/j.1582-
4934.2008.00273.x.
• Lansbury, Peter T., et Hilal A. Lashuel. 2006. « A Century-Old Debate on Protein
Aggregation and Neurodegeneration Enters the Clinic ». Nature 443 (7113): 774‑79.
https://doi.org/10.1038/nature05290.
• Lavoie, J. N., G. Gingras-Breton, R. M. Tanguay, et J. Landry. 1993. « Induction
of Chinese Hamster HSP27 Gene Expression in Mouse Cells Confers Resistance to Heat
Shock. HSP27 Stabilization of the Microfilament Organization ». The Journal of
Biological Chemistry 268 (5): 3420‑29.
• Lavoie, J. N., E. Hickey, L. A. Weber, et J. Landry. 1993. « Modulation of Actin
Microfilament Dynamics and Fluid Phase Pinocytosis by Phosphorylation of Heat Shock
Protein 27 ». The Journal of Biological Chemistry 268 (32): 24210‑14.

221
• Lavoie, J. N., H. Lambert, E. Hickey, L. A. Weber, et J. Landry. 1995. « Modulation
of Cellular Thermoresistance and Actin Filament Stability Accompanies Phosphorylation-
Induced Changes in the Oligomeric Structure of Heat Shock Protein 27 ». Molecular and
Cellular Biology 15 (1): 505‑16.
• Lee, G. J., A. M. Roseman, H. R. Saibil, et E. Vierling. 1997. « A Small Heat Shock
Protein Stably Binds Heat-Denatured Model Substrates and Can Maintain a Substrate in a
Folding-Competent State ». The EMBO Journal 16 (3): 659‑71.
https://doi.org/10.1093/emboj/16.3.659.
• Lee, H. C., S. W. Cherk, S. K. Chan, S. Wong, T. W. Tong, W. S. Ho, A. Y. Chan,
K. C. Lee, et C. M. Mak. 2012. « BAG3-Related Myofibrillar Myopathy in a Chinese
Family ». Clinical Genetics 81 (4): 394‑98. https://doi.org/10.1111/j.1399-
0004.2011.01659.x.
• Lee, J. H., T. Takahashi, N. Yasuhara, J. Inazawa, S. Kamada, et Y. Tsujimoto.
1999. « Bis, a Bcl-2-Binding Protein That Synergizes with Bcl-2 in Preventing Cell
Death ». Oncogene 18 (46): 6183‑90. https://doi.org/10.1038/sj.onc.1203043.
• Lee, Joong-Won, Hee-Jin Kwak, Je-Jung Lee, Yong-Nyun Kim, Jung Weon Lee,
Myung-Jin Park, Seung Eun Jung, Seok-Il Hong, Jeong-Hwa Lee, et Jae-Seon Lee. 2008.
« HSP27 Regulates Cell Adhesion and Invasion via Modulation of Focal Adhesion Kinase
and MMP-2 Expression ». European Journal of Cell Biology 87 (6): 377‑87.
https://doi.org/10.1016/j.ejcb.2008.03.006.
• Lee, Joo-Yong, Hiroshi Koga, Yoshiharu Kawaguchi, Waixing Tang, Esther Wong,
Ya-Sheng Gao, Udai B. Pandey, et al. 2010. « HDAC6 Controls Autophagosome
Maturation Essential for Ubiquitin-Selective Quality-Control Autophagy ». The EMBO
Journal 29 (5): 969‑80. https://doi.org/10.1038/emboj.2009.405.
• Lee, Junho, Kyu-Hwan Yang, Cheol O. Joe, et Seok-Seong Kang. 2011.
« Formation of Distinct Inclusion Bodies by Inhibition of Ubiquitin-Proteasome and
Autophagy-Lysosome Pathways ». Biochemical and Biophysical Research
Communications 404 (2): 672‑77. https://doi.org/10.1016/j.bbrc.2010.12.040.
• Lee, L., J. S. Tirnauer, J. Li, S. C. Schuyler, J. Y. Liu, et D. Pellman. 2000.
« Positioning of the Mitotic Spindle by a Cortical-Microtubule Capture Mechanism ».

222
Science (New York, N.Y.) 287 (5461): 2260‑62.
• Lee, Young Dae, Mei Nu Cui, Hye Hyeon Yoon, Hye Yun Kim, Il-Hoan Oh, et
Jeong-Hwa Lee. 2014. « Down-Modulation of Bis Reduces the Invasive Ability of Glioma
Cells Induced by TPA, through NF-ΚB Mediated Activation of MMP-9 ». BMB Reports
47 (5): 262‑67.
• Leroux, M. R., R. Melki, B. Gordon, G. Batelier, et E. P. Candido. 1997.
« Structure-Function Studies on Small Heat Shock Protein Oligomeric Assembly and
Interaction with Unfolded Polypeptides ». The Journal of Biological Chemistry 272 (39):
24646‑56.
• Leung, Thomas, Xiang-Qun Chen, Ivan Tan, Edward Manser, et Louis Lim. 1998.
« Myotonic Dystrophy Kinase-Related Cdc42-Binding Kinase Acts as a Cdc42 Effector in
Promoting Cytoskeletal Reorganization ». Molecular and Cellular Biology 18 (1): 130‑40.
• Li, N., Z.-X. Du, Z.-H. Zong, B.-Q. Liu, C. Li, Q. Zhang, et H.-Q. Wang. 2013.
« PKCδ-Mediated Phosphorylation of BAG3 at Ser187 Site Induces Epithelial-
Mesenchymal Transition and Enhances Invasiveness in Thyroid Cancer FRO Cells ».
Oncogene 32 (38): 4539‑48. https://doi.org/10.1038/onc.2012.466.
• Li, Xiao-Cheng, Qi-Kuan Hu, Ling Chen, Si-Yang Liu, Shi Su, Hong Tao, Lin-Na
Zhang, Tao Sun, et Lan-Jie He. 2017. « HSPB8 Promotes the Fusion of Autophagosome
and Lysosome during Autophagy in Diabetic Neurons ». International Journal of Medical
Sciences 14 (13): 1335‑41. https://doi.org/10.7150/ijms.20653.
• Li, Xiao-shan, Qing Xu, Xiang-yang Fu, et Wei-sheng Luo. 2014. « Heat Shock
Protein 22 Overexpression Is Associated with the Progression and Prognosis in Gastric
Cancer ». Journal of Cancer Research and Clinical Oncology 140 (8): 1305‑13.
https://doi.org/10.1007/s00432-014-1698-z.
• Li, Xuehui, Yang Mei, Bowen Yan, Eric Vitriol, Suming Huang, Peng Ji, et Yi Qiu.
2017. « Histone Deacetylase 6 Regulates Cytokinesis and Erythrocyte Enucleation through
Deacetylation of Formin Protein MDia2 ». Haematologica 102 (6): 984‑94.
https://doi.org/10.3324/haematol.2016.161513.
• Li, Zhiyuan, Xinmiao Ji, Dongmei Wang, Juanjuan Liu, et Xin Zhang. 2016.
« Autophagic Flux Is Highly Active in Early Mitosis and Differentially Regulated

223
throughout the Cell Cycle ». Oncotarget 7 (26): 39705‑18.
https://doi.org/10.18632/oncotarget.9451.
• Linares, Juan F., Ramars Amanchy, Kenneth Greis, Maria T. Diaz-Meco, et Jorge
Moscat. 2011. « Phosphorylation of P62 by Cdk1 Controls the Timely Transit of Cells
through Mitosis and Tumor Cell Proliferation ». Molecular and Cellular Biology 31 (1):
105‑17. https://doi.org/10.1128/MCB.00620-10.
• Lindner, R. A., J. A. Carver, M. Ehrnsperger, J. Buchner, G. Esposito, J. Behlke,
G. Lutsch, A. Kotlyarov, et M. Gaestel. 2000. « Mouse Hsp25, a Small Shock Protein. The
Role of Its C-Terminal Extension in Oligomerization and Chaperone Action ». European
Journal of Biochemistry 267 (7): 1923‑32.
• Lindqvist, Arne, Verónica Rodríguez-Bravo, et René H. Medema. 2009. « The
Decision to Enter Mitosis: Feedback and Redundancy in the Mitotic Entry Network ». The
Journal of Cell Biology 185 (2): 193‑202. https://doi.org/10.1083/jcb.200812045.
• Liu, Bao-Qin, Zhen-Xian Du, Zhi-Hong Zong, Chao Li, Ning Li, Qiang Zhang, De-
Hui Kong, et Hua-Qin Wang. 2013. « BAG3-Dependent Noncanonical Autophagy Induced
by Proteasome Inhibition in HepG2 Cells ». Autophagy 9 (6): 905‑16.
https://doi.org/10.4161/auto.24292.
• Liu, Peng, Bei Xu, Jianyong Li, et Hua Lu. 2009. « BAG3 Gene Silencing
Sensitizes Leukemic Cells to Bortezomib-Induced Apoptosis ». FEBS Letters 583 (2):
401‑6. https://doi.org/10.1016/j.febslet.2008.12.032.
• Liu, Wei, Lucy X. Fan, Xia Zhou, William E. Sweeney, Ellis D. Avner, et Xiaogang
Li. 2012. « HDAC6 Regulates Epidermal Growth Factor Receptor (EGFR) Endocytic
Trafficking and Degradation in Renal Epithelial Cells ». PloS One 7 (11): e49418.
https://doi.org/10.1371/journal.pone.0049418.
• Liu, Zairan, et Orion D. Weiner. 2016. « Positioning the Cleavage Furrow: All You
Need Is Rho ». J Cell Biol 213 (6): 605‑7. https://doi.org/10.1083/jcb.201606010.
• Lodish, Harvey, Arnold Berk, S. Lawrence Zipursky, Paul Matsudaira, David
Baltimore, et James Darnell. 2000. « The Dynamics of Actin Assembly ».
https://www.ncbi.nlm.nih.gov/books/NBK21594/.
• Loukil, Abdelhalim, Fanny Izard, Mariya Georgieva, Shaereh Mashayekhan, Jean-

224
Marie Blanchard, Andrea Parmeggiani, et Marion Peter. 2016. « Foci of Cyclin A2 Interact
with Actin and RhoA in Mitosis ». Scientific Reports 6 (juin): 27215.
https://doi.org/10.1038/srep27215.
• Loukil, Abdelhalim, Manuela Zonca, Cosette Rebouissou, Véronique Baldin,
Olivier Coux, Martine Biard-Piechaczyk, Jean-Marie Blanchard, et Marion Peter. 2014.
« High-Resolution Live-Cell Imaging Reveals Novel Cyclin A2 Degradation Foci
Involving Autophagy ». Journal of Cell Science 127 (Pt 10): 2145‑50.
https://doi.org/10.1242/jcs.139188.
• Lu, Hao, Chaoyang Sun, Ting Zhou, Bo Zhou, Ensong Guo, Wanying Shan, Meng
Xia, et al. 2016. « HSP27 Knockdown Increases Cytoplasmic P21 and Cisplatin Sensitivity
in Ovarian Carcinoma Cells ». Oncology Research 23 (3): 119‑28.
https://doi.org/10.3727/096504015X14496932933656.
• Lüders, J., J. Demand, et J. Höhfeld. 2000. « The Ubiquitin-Related BAG-1
Provides a Link between the Molecular Chaperones Hsc70/Hsp70 and the Proteasome ».
The Journal of Biological Chemistry 275 (7): 4613‑17.
• Lv, Lei, Tianwei Zhang, Qiyi Yi, Yun Huang, Zheng Wang, Heli Hou, Huan Zhang,
et al. 2012. « Tetraploid Cells from Cytokinesis Failure Induce Aneuploidy and
Spontaneous Transformation of Mouse Ovarian Surface Epithelial Cells ». Cell Cycle
(Georgetown, Tex.) 11 (15): 2864‑75. https://doi.org/10.4161/cc.21196.
• Ma, L., R. Rohatgi, et M. W. Kirschner. 1998. « The Arp2/3 Complex Mediates
Actin Polymerization Induced by the Small GTP-Binding Protein Cdc42 ». Proceedings of
the National Academy of Sciences of the United States of America 95 (26): 15362‑67.
• Mabuchi, I., et M. Okuno. 1977. « The Effect of Myosin Antibody on the Division
of Starfish Blastomeres ». The Journal of Cell Biology 74 (1): 251‑63.
https://doi.org/10.1083/jcb.74.1.251.
• Machesky, L. M., S. J. Atkinson, C. Ampe, J. Vandekerckhove, et T. D. Pollard.
1994. « Purification of a Cortical Complex Containing Two Unconventional Actins from
Acanthamoeba by Affinity Chromatography on Profilin-Agarose ». The Journal of Cell
Biology 127 (1): 107‑15.
• Machesky, L. M., E. Reeves, F. Wientjes, F. J. Mattheyse, A. Grogan, N. F. Totty,

225
A. L. Burlingame, J. J. Hsuan, et A. W. Segal. 1997. « Mammalian Actin-Related Protein
2/3 Complex Localizes to Regions of Lamellipodial Protrusion and Is Composed of
Evolutionarily Conserved Proteins ». The Biochemical Journal 328 ( Pt 1) (novembre):
105‑12.
• Machicoane, Mickael, Cristina A. de Frutos, Jenny Fink, Murielle Rocancourt,
Yannis Lombardi, Sonia Garel, Matthieu Piel, et Arnaud Echard. 2014. « SLK-Dependent
Activation of ERMs Controls LGN-NuMA Localization and Spindle Orientation ». The
Journal of Cell Biology 205 (6): 791‑99. https://doi.org/10.1083/jcb.201401049.
• Madaule, P., M. Eda, N. Watanabe, K. Fujisawa, T. Matsuoka, H. Bito, T. Ishizaki,
et S. Narumiya. 1998. « Role of Citron Kinase as a Target of the Small GTPase Rho in
Cytokinesis ». Nature 394 (6692): 491‑94. https://doi.org/10.1038/28873.
• Maddox, Amy Shaub, et Keith Burridge. 2003. « RhoA Is Required for Cortical
Retraction and Rigidity during Mitotic Cell Rounding ». The Journal of Cell Biology 160
(2): 255‑65. https://doi.org/10.1083/jcb.200207130.
• Maddox, Amy Shaub, Bianca Habermann, Arshad Desai, et Karen Oegema. 2005.
« Distinct Roles for Two C. Elegans Anillins in the Gonad and Early Embryo ».
Development 132 (12): 2837‑48. https://doi.org/10.1242/dev.01828.
• Mader, Christopher C., Matthew Oser, Marco A. O. Magalhaes, Jose Javier Bravo-
Cordero, John Condeelis, Anthony J. Koleske, et Hava Gil-Henn. 2011. « An EGFR-Src-
Arg-Cortactin Pathway Mediates Functional Maturation of Invadopodia and Breast Cancer
Cell Invasion ». Cancer Research 71 (5): 1730‑41. https://doi.org/10.1158/0008-
5472.CAN-10-1432.
• Magidson, Valentin, Christopher B. O’Connell, Jadranka Lončarek, Raja Paul,
Alex Mogilner, et Alexey Khodjakov. 2011. « The Spatial Arrangement of Chromosomes
during Prometaphase Facilitates Spindle Assembly ». Cell 146 (4): 555‑67.
https://doi.org/10.1016/j.cell.2011.07.012.
• Malin, D., E. Strekalova, V. Petrovic, H. Rajanala, B. Sharma, A. Ugolkov, W. J.
Gradishar, et V. L. Cryns. 2015. « ERK-Regulated ΑB-Crystallin Induction by Matrix
Detachment Inhibits Anoikis and Promotes Lung Metastasis in Vivo ». Oncogene 34 (45):
5626‑34. https://doi.org/10.1038/onc.2015.12.

226
• Mancias, Joseph D., et Alec C. Kimmelman. 2016. « Mechanisms of Selective
Autophagy in Normal Physiology and Cancer ». Journal of Molecular Biology 428 (9 Pt
A): 1659‑80. https://doi.org/10.1016/j.jmb.2016.02.027.
• Manecka, Destiny-Love, Benoît Vanderperre, Edward A. Fon, et Thomas M.
Durcan. 2017. « The Neuroprotective Role of Protein Quality Control in Halting the
Development of Alpha-Synuclein Pathology ». Frontiers in Molecular Neuroscience 10:
311. https://doi.org/10.3389/fnmol.2017.00311.
• Mani, Nandini, Spraha Bhandari, Rodolfo Moreno, Liya Hu, B. V. Venkataram
Prasad, et Kaza Suguna. 2016. « Multiple Oligomeric Structures of a Bacterial Small Heat
Shock Protein ». Scientific Reports 6 (avril): 24019. https://doi.org/10.1038/srep24019.
• Marchesi, Stefano, Francesca Montani, Gianluca Deflorian, Rocco D’Antuono,
Alessandro Cuomo, Serena Bologna, Carmela Mazzoccoli, Tiziana Bonaldi, Pier Paolo
Di Fiore, et Francesco Nicassio. 2014. « DEPDC1B Coordinates De-Adhesion Events and
Cell-Cycle Progression at Mitosis ». Developmental Cell 31 (4): 420.
https://doi.org/10.1016/j.devcel.2014.09.009.
• Marcucci, Fabrizio, Pietro Ghezzi, et Cristiano Rumio. 2017. « The Role of
Autophagy in the Cross-Talk between Epithelial-Mesenchymal Transitioned Tumor Cells
and Cancer Stem-like Cells ». Molecular Cancer 16 (1): 3. https://doi.org/10.1186/s12943-
016-0573-8.
• Margadant, Coert, Angelique van Opstal, et Johannes Boonstra. 2007. « Focal
Adhesion Signaling and Actin Stress Fibers Are Dispensable for Progression through the
Ongoing Cell Cycle ». Journal of Cell Science 120 (1): 66‑76.
https://doi.org/10.1242/jcs.03301.
• Masters, Thomas A., John Kendrick-Jones, et Folma Buss. 2017. « Myosins:
Domain Organisation, Motor Properties, Physiological Roles and Cellular Functions ».
Handbook of Experimental Pharmacology 235: 77‑122.
https://doi.org/10.1007/164_2016_29.
• Matsumoto, Gen, Koji Wada, Misako Okuno, Masaru Kurosawa, et Nobuyuki
Nukina. 2011. « Serine 403 Phosphorylation of P62/SQSTM1 Regulates Selective
Autophagic Clearance of Ubiquitinated Proteins ». Molecular Cell 44 (2): 279‑89.

227
https://doi.org/10.1016/j.molcel.2011.07.039.
• Matsumura, Fumio. 2005. « Regulation of Myosin II during Cytokinesis in Higher
Eukaryotes ». Trends in Cell Biology 15 (7): 371‑77.
https://doi.org/10.1016/j.tcb.2005.05.004.
• Matsushima-Nishiwaki, Rie, Hidenori Toyoda, Tomoaki Nagasawa, Eisuke
Yasuda, Naokazu Chiba, Seiji Okuda, Atsuyuki Maeda, Yuji Kaneoka, Takashi Kumada,
et Osamu Kozawa. 2016. « Phosphorylated Heat Shock Protein 20 (HSPB6) Regulates
Transforming Growth Factor-α-Induced Migration and Invasion of Hepatocellular
Carcinoma Cells ». PloS One 11 (4): e0151907.
https://doi.org/10.1371/journal.pone.0151907.
• Matthews, Helen K., Ulysse Delabre, Jennifer L. Rohn, Jochen Guck, Patricia
Kunda, et Buzz Baum. 2012. « Changes in Ect2 Localization Couple Actomyosin-
Dependent Cell Shape Changes to Mitotic Progression ». Developmental Cell 23 (2):
371‑83. https://doi.org/10.1016/j.devcel.2012.06.003.
• May, R. C., E. Caron, A. Hall, et L. M. Machesky. 2000. « Involvement of the
Arp2/3 Complex in Phagocytosis Mediated by FcgammaR or CR3 ». Nature Cell Biology
2 (4): 246‑48. https://doi.org/10.1038/35008673.
• Mayr, Monika I., Stefan Hümmer, Jenny Bormann, Tamara Grüner, Sarah Adio,
Guenther Woehlke, et Thomas U. Mayer. 2007. « The Human Kinesin Kif18A Is a Motile
Microtubule Depolymerase Essential for Chromosome Congression ». Current Biology:
CB 17 (6): 488‑98. https://doi.org/10.1016/j.cub.2007.02.036.
• McDonald, Ezelle T., Marco Bortolus, Hanane A. Koteiche, et Hassane S.
Mchaourab. 2012. « Sequence, Structure, and Dynamic Determinants of Hsp27 (HspB1)
Equilibrium Dissociation Are Encoded by the N-Terminal Domain ». Biochemistry 51 (6):
1257‑68. https://doi.org/10.1021/bi2017624.
• McKenzie, Callum, et Pier Paolo D’Avino. 2016. « Investigating Cytokinesis
Failure as a Strategy in Cancer Therapy ». Oncotarget 7 (52): 87323‑41.
https://doi.org/10.18632/oncotarget.13556.
• McKeon, Jeanne E., Di Sha, Lian Li, et Lih-Shen Chin. 2015. « Parkin-Mediated
K63-Polyubiquitination Targets Ubiquitin C-Terminal Hydrolase L1 for Degradation by

228
the Autophagy-Lysosome System ». Cellular and Molecular Life Sciences: CMLS 72 (9):
1811‑24. https://doi.org/10.1007/s00018-014-1781-2.
• McLaughlin, M. M., S. Kumar, P. C. McDonnell, S. Van Horn, J. C. Lee, G. P.
Livi, et P. R. Young. 1996. « Identification of Mitogen-Activated Protein (MAP) Kinase-
Activated Protein Kinase-3, a Novel Substrate of CSBP P38 MAP Kinase ». The Journal
of Biological Chemistry 271 (14): 8488‑92.
• Medeiros, Nelson A., Dylan T. Burnette, et Paul Forscher. 2006. « Myosin II
Functions in Actin-Bundle Turnover in Neuronal Growth Cones ». Nature Cell Biology 8
(3): 215‑26. https://doi.org/10.1038/ncb1367.
• Meiler, Eugenia, Elvira Nieto-Pelegrín, et Narcisa Martinez-Quiles. 2012.
« Cortactin Tyrosine Phosphorylation Promotes Its Deacetylation and Inhibits Cell
Spreading ». PloS One 7 (3): e33662. https://doi.org/10.1371/journal.pone.0033662.
• Mendes Pinto, Inês, Boris Rubinstein, Andrei Kucharavy, Jay R. Unruh, et Rong
Li. 2012. « Actin Depolymerization Drives Actomyosin Ring Contraction during Budding
Yeast Cytokinesis ». Developmental Cell 22 (6): 1247‑60.
https://doi.org/10.1016/j.devcel.2012.04.015.
• Meng, X., D.-H. Kong, N. Li, Z.-H. Zong, B.-Q. Liu, Z.-X. Du, Y. Guan, L. Cao,
et H.-Q. Wang. 2014. « Knockdown of BAG3 Induces Epithelial-Mesenchymal Transition
in Thyroid Cancer Cells through ZEB1 Activation ». Cell Death & Disease 5 (février):
e1092. https://doi.org/10.1038/cddis.2014.32.
• Merabova, Nana, Ilker Kudret Sariyer, A. Sami Saribas, Tijana Knezevic, Jennifer
Gordon, M. Caterina Turco, Alessandra Rosati, Michael Weaver, Jacques Landry, et
Kamel Khalili. 2015. « WW Domain of BAG3 Is Required for the Induction of Autophagy
in Glioma Cells ». Journal of Cellular Physiology 230 (4): 831‑41.
https://doi.org/10.1002/jcp.24811.
• Mezi, Silvia, Laura Todi, Errico Orsi, Antonio Angeloni, et Patrizia Mancini. 2012.
« Involvement of the Src-Cortactin Pathway in Migration Induced by IGF-1 and EGF in
Human Breast Cancer Cells ». International Journal of Oncology 41 (6): 2128‑38.
https://doi.org/10.3892/ijo.2012.1642.
• Mi, Na, Yang Chen, Shuai Wang, Mengran Chen, Mingkun Zhao, Guang Yang,

229
Meisheng Ma, et al. 2015. « CapZ Regulates Autophagosomal Membrane Shaping by
Promoting Actin Assembly inside the Isolation Membrane ». Nature Cell Biology 17 (9):
1112‑23. https://doi.org/10.1038/ncb3215.
• Miklavc, Pika, Konstantin Ehinger, Ayesha Sultan, Tatiana Felder, Patrick Paul,
Kay-Eberhard Gottschalk, et Manfred Frick. 2015. « Actin Depolymerisation and
Crosslinking Join Forces with Myosin II to Contract Actin Coats on Fused Secretory
Vesicles ». Journal of Cell Science 128 (6): 1193‑1203.
https://doi.org/10.1242/jcs.165571.
• Minc, Nicolas, David Burgess, et Fred Chang. 2011. « Influence of Cell Geometry
on Division-Plane Positioning ». Cell 144 (3): 414‑26.
https://doi.org/10.1016/j.cell.2011.01.016.
• Minoia, Melania, Alessandra Boncoraglio, Jonathan Vinet, Federica F. Morelli,
Jeanette F. Brunsting, Angelo Poletti, Sabine Krom, Eric Reits, Harm H. Kampinga, et
Serena Carra. 2014. « BAG3 Induces the Sequestration of Proteasomal Clients into
Cytoplasmic Puncta: Implications for a Proteasome-to-Autophagy Switch ». Autophagy 10
(9): 1603‑21. https://doi.org/10.4161/auto.29409.
• Miron, T., M. Wilchek, et B. Geiger. 1988. « Characterization of an Inhibitor of
Actin Polymerization in Vinculin-Rich Fraction of Turkey Gizzard Smooth Muscle ».
European Journal of Biochemistry 178 (2): 543‑53.
• Mitchison, T. J. 1992. « Actin Based Motility on Retraction Fibers in Mitotic PtK2
Cells ». Cell Motility and the Cytoskeleton 22 (2): 135‑51.
https://doi.org/10.1002/cm.970220207.
• Mitsushima, Masaru, Kazuhiro Aoki, Miki Ebisuya, Shigeru Matsumura, Takuya
Yamamoto, Michiyuki Matsuda, Fumiko Toyoshima, et Eisuke Nishida. 2010.
« Revolving Movement of a Dynamic Cluster of Actin Filaments during Mitosis ». The
Journal of Cell Biology 191 (3): 453‑62. https://doi.org/10.1083/jcb.201007136.
• Monastyrska, Iryna, Congcong He, Jiefei Geng, Adam D. Hoppe, Zhijian Li, et
Daniel J. Klionsky. 2008. « Arp2 Links Autophagic Machinery with the Actin
Cytoskeleton ». Molecular Biology of the Cell 19 (5): 1962‑75.
https://doi.org/10.1091/mbc.E07-09-0892.

230
• Mondal, Gourish, Matthew Rowley, Lucia Guidugli, Jianmin Wu, Vernon S.
Pankratz, et Fergus J. Couch. 2012. « BRCA2 Localization to the Midbody by Filamin A
Regulates Cep55 Signaling and Completion of Cytokinesis ». Developmental Cell 23 (1):
137‑52. https://doi.org/10.1016/j.devcel.2012.05.008.
• Montagna, Georgina N., Kai Matuschewski, et Carlos A. Buscaglia. 2012. « Small
Heat Shock Proteins in Cellular Adhesion and Migration: Evidence from Plasmodium
Genetics ». Cell Adhesion & Migration 6 (2): 78‑84. https://doi.org/10.4161/cam.20101.
• Montfort, R. L. van, E. Basha, K. L. Friedrich, C. Slingsby, et E. Vierling. 2001.
« Crystal Structure and Assembly of a Eukaryotic Small Heat Shock Protein ». Nature
Structural Biology 8 (12): 1025‑30. https://doi.org/10.1038/nsb722.
• Moore, Andrew S., Yvette C. Wong, Cory L. Simpson, et Erika L. F. Holzbaur.
2016. « Dynamic Actin Cycling through Mitochondrial Subpopulations Locally Regulates
the Fission-Fusion Balance within Mitochondrial Networks ». Nature Communications 7
(septembre): 12886. https://doi.org/10.1038/ncomms12886.
• Moore, G. D., T. Ayabe, P. E. Visconti, R. M. Schultz, et G. S. Kopf. 1994. « Roles
of Heterotrimeric and Monomeric G Proteins in Sperm-Induced Activation of Mouse
Eggs ». Development (Cambridge, England) 120 (11): 3313‑23.
• Moreau, V., J. M. Galan, G. Devilliers, R. Haguenauer-Tsapis, et B. Winsor. 1997.
« The Yeast Actin-Related Protein Arp2p Is Required for the Internalization Step of
Endocytosis ». Molecular Biology of the Cell 8 (7): 1361‑75.
• Morimoto, Richard I. 2011. « The Heat Shock Response: Systems Biology of
Proteotoxic Stress in Aging and Disease ». Cold Spring Harbor Symposia on Quantitative
Biology 76 (janvier): 91‑99. https://doi.org/10.1101/sqb.2012.76.010637.
• Morin, Xavier, et Yohanns Bellaïche. 2011. « Mitotic Spindle Orientation in
Asymmetric and Symmetric Cell Divisions during Animal Development ». Developmental
Cell 21 (1): 102‑19. https://doi.org/10.1016/j.devcel.2011.06.012.
• Mornet, D., P. Pantel, E. Audemard, et R. Kassab. 1979. « The Limited Tryptic
Cleavage of Chymotryptic S-1: An Approach to the Characterization of the Actin Site in
Myosin Heads ». Biochemical and Biophysical Research Communications 89 (3): 925‑32.
• Morrow, Geneviève, et Robert M. Tanguay. 2012. « Small Heat Shock Protein

231
Expression and Functions during Development ». The International Journal of
Biochemistry & Cell Biology 44 (10): 1613‑21.
https://doi.org/10.1016/j.biocel.2012.03.009.
• Mounier, Nicole, et André-Patrick Arrigo. 2002. « Actin Cytoskeleton and Small
Heat Shock Proteins: How Do They Interact? » Cell Stress & Chaperones 7 (2): 167‑76.
• Moutaoufik, Mohamed Taha, Geneviève Morrow, Stéphanie Finet, et Robert M.
Tanguay. 2017. « Effect of N-Terminal Region of Nuclear Drosophila Melanogaster Small
Heat Shock Protein DmHsp27 on Function and Quaternary Structure ». PloS One 12 (5):
e0177821. https://doi.org/10.1371/journal.pone.0177821.
• Moyano, Jose V., Joseph R. Evans, Feng Chen, Meiling Lu, Michael E. Werner,
Fruma Yehiely, Leslie K. Diaz, et al. 2006. « AlphaB-Crystallin Is a Novel Oncoprotein
That Predicts Poor Clinical Outcome in Breast Cancer ». The Journal of Clinical
Investigation 116 (1): 261‑70. https://doi.org/10.1172/JCI25888.
• Muchowski, Paul J. 2002. « Protein Misfolding, Amyloid Formation, and
Neurodegeneration: A Critical Role for Molecular Chaperones? » Neuron 35 (1): 9‑12.
• Mullins, R. D., J. A. Heuser, et T. D. Pollard. 1998. « The Interaction of Arp2/3
Complex with Actin: Nucleation, High Affinity Pointed End Capping, and Formation of
Branching Networks of Filaments ». Proceedings of the National Academy of Sciences of
the United States of America 95 (11): 6181‑86.
• Muris, D. F. R., T. Verschoor, N. Divecha, et R. J. a. M. Michalides. 2002.
« Constitutive Active GTPases Rac and Cdc42 Are Associated with Endoreplication in
PAE Cells ». European Journal of Cancer (Oxford, England: 1990) 38 (13): 1775‑82.
• Murthy, Kausalya, et Patricia Wadsworth. 2005. « Myosin-II-Dependent
Localization and Dynamics of F-Actin during Cytokinesis ». Current Biology: CB 15 (8):
724‑31. https://doi.org/10.1016/j.cub.2005.02.055.
• Nagasawa, Tomoaki, Rie Matsushima-Nishiwaki, Hidenori Toyoda, Junya
Matsuura, Takashi Kumada, et Osamu Kozawa. 2014. « Heat Shock Protein 20 (HSPB6)
Regulates Apoptosis in Human Hepatocellular Carcinoma Cells: Direct Association with
Bax ». Oncology Reports 32 (3): 1291‑95. https://doi.org/10.3892/or.2014.3278.
• Nestor-Bergmann, Alexander, Georgina Goddard, et Sarah Woolner. 2014. « Force

232
and the Spindle: Mechanical Cues in Mitotic Spindle Orientation ». Seminars in Cell &
Developmental Biology 34 (octobre): 133‑39.
https://doi.org/10.1016/j.semcdb.2014.07.008.
• Nicolaou, Persoulla, Ralph Knöll, Kobra Haghighi, Guo-Chang Fan, Gerald W.
Dorn, Gerd Hasenfub, et Evangelia G. Kranias. 2008. « Human Mutation in the Anti-
Apoptotic Heat Shock Protein 20 Abrogates Its Cardioprotective Effects ». The Journal of
Biological Chemistry 283 (48): 33465‑71. https://doi.org/10.1074/jbc.M802307200.
• Niiya, F., T. Tatsumoto, K. S. Lee, et T. Miki. 2006. « Phosphorylation of the
Cytokinesis Regulator ECT2 at G2/M Phase Stimulates Association of the Mitotic Kinase
Plk1 and Accumulation of GTP-Bound RhoA ». Oncogene 25 (6): 827‑37.
https://doi.org/10.1038/sj.onc.1209124.
• Nishimura, Yukako, et Shigenobu Yonemura. 2006. « Centralspindlin Regulates
ECT2 and RhoA Accumulation at the Equatorial Cortex during Cytokinesis ». Journal of
Cell Science 119 (Pt 1): 104‑14. https://doi.org/10.1242/jcs.02737.
• Noatynska, Anna, Monica Gotta, et Patrick Meraldi. 2012. « Mitotic Spindle
(DIS)Orientation and DISease: Cause or Consequence? » J Cell Biol 199 (7): 1025‑35.
https://doi.org/10.1083/jcb.201209015.
• Nobes, Catherine D, et Alan Hall. 1995. « Rho, Rac, and Cdc42 GTPases regulate
the assembly of multimolecular focal complexes associated with actin stress fibers,
lamellipodia, and filopodia ». Cell 81 (1): 53‑62. https://doi.org/10.1016/0092-
8674(95)90370-4.
• Nolen, B. J., N. Tomasevic, A. Russell, D. W. Pierce, Z. Jia, C. D. McCormick, J.
Hartman, R. Sakowicz, et T. D. Pollard. 2009. « Characterization of Two Classes of Small
Molecule Inhibitors of Arp2/3 Complex ». Nature 460 (7258): 1031‑34.
https://doi.org/10.1038/nature08231.
• Normand, Guillaume, et Randall W. King. 2010. « Understanding Cytokinesis
Failure ». Advances in experimental medicine and biology 676: 27‑55.
• O’Callaghan-Sunol, Cornelia, Vladimir L. Gabai, et Michael Y. Sherman. 2007.
« Hsp27 Modulates P53 Signaling and Suppresses Cellular Senescence ». Cancer Research
67 (24): 11779‑88. https://doi.org/10.1158/0008-5472.CAN-07-2441.

233
• Oceguera-Yanez, Fabian, Kazuhiro Kimura, Shingo Yasuda, Chiharu Higashida,
Toshio Kitamura, Yasushi Hiraoka, Tokuko Haraguchi, et Shuh Narumiya. 2005. « Ect2
and MgcRacGAP Regulate the Activation and Function of Cdc42 in Mitosis ». The Journal
of Cell Biology 168 (2): 221‑32. https://doi.org/10.1083/jcb.200408085.
• O’Connell, C. B., et Y. L. Wang. 2000. « Mammalian Spindle Orientation and
Position Respond to Changes in Cell Shape in a Dynein-Dependent Fashion ». Molecular
Biology of the Cell 11 (5): 1765‑74.
• Odell, Garrett M., et Victoria E. Foe. 2008. « An Agent-Based Model Contrasts
Opposite Effects of Dynamic and Stable Microtubules on Cleavage Furrow Positioning ».
The Journal of Cell Biology 183 (3): 471‑83. https://doi.org/10.1083/jcb.200807129.
• Oelz, Dietmar B., Boris Y. Rubinstein, et Alex Mogilner. 2015. « A Combination
of Actin Treadmilling and Cross-Linking Drives Contraction of Random Actomyosin
Arrays ». Biophysical Journal 109 (9): 1818‑29.
https://doi.org/10.1016/j.bpj.2015.09.013.
• Oelz, Dietmar, et Alex Mogilner. 2016. « Actomyosin Contraction, Aggregation
and Traveling Waves in a Treadmilling Actin Array ». Physica D. Nonlinear Phenomena
318‑319 (avril): 70‑83. https://doi.org/10.1016/j.physd.2015.10.005.
• Olzmann, James A., Lian Li, Maksim V. Chudaev, Jue Chen, Francisco A. Perez,
Richard D. Palmiter, et Lih-Shen Chin. 2007. « Parkin-Mediated K63-Linked
Polyubiquitination Targets Misfolded DJ-1 to Aggresomes via Binding to HDAC6 ». The
Journal of Cell Biology 178 (6): 1025‑38. https://doi.org/10.1083/jcb.200611128.
• Oser, Matthew, Christopher C. Mader, Hava Gil-Henn, Marco Magalhaes, Jose
Javier Bravo-Cordero, Anthony J. Koleske, et John Condeelis. 2010. « Specific Tyrosine
Phosphorylation Sites on Cortactin Regulate Nck1-Dependent Actin Polymerization in
Invadopodia ». Journal of Cell Science 123 (Pt 21): 3662‑73.
https://doi.org/10.1242/jcs.068163.
• Ouyang, Hui, Yousuf O. Ali, Mani Ravichandran, Aiping Dong, Wei Qiu, Farrell
MacKenzie, Sirano Dhe-Paganon, Cheryl H. Arrowsmith, et R. Grace Zhai. 2012. « Protein
Aggregates Are Recruited to Aggresome by Histone Deacetylase 6 via Unanchored
Ubiquitin C Termini ». The Journal of Biological Chemistry 287 (4): 2317‑27.

234
https://doi.org/10.1074/jbc.M111.273730.
• Paine, Michael G., J. Ramesh Babu, M. Lamar Seibenhener, et Marie W. Wooten.
2005. « Evidence for P62 Aggregate Formation: Role in Cell Survival ». FEBS Letters 579
(22): 5029‑34. https://doi.org/10.1016/j.febslet.2005.08.010.
• Pandey, Udai Bhan, Yakup Batlevi, Eric H. Baehrecke, et J. Paul Taylor. 2007.
« HDAC6 at the Intersection of Autophagy, the Ubiquitin-Proteasome System and
Neurodegeneration ». Autophagy 3 (6): 643‑45.
• Pankiv, Serhiy, Terje Høyvarde Clausen, Trond Lamark, Andreas Brech, Jack-
Ansgar Bruun, Heidi Outzen, Aud Øvervatn, Geir Bjørkøy, et Terje Johansen. 2007.
« P62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated
Protein Aggregates by Autophagy ». The Journal of Biological Chemistry 282 (33):
24131‑45. https://doi.org/10.1074/jbc.M702824200.
• Parcellier, Arnaud, Elise Schmitt, Mathilde Brunet, Arlette Hammann, Eric Solary,
et Carmen Garrido. 2005. « Small Heat Shock Proteins HSP27 and AlphaB-Crystallin:
Cytoprotective and Oncogenic Functions ». Antioxidants & Redox Signaling 7 (3‑4):
404‑13. https://doi.org/10.1089/ars.2005.7.404.
• Park, Ah-Mee, Kyosuke Kanai, Tatsuki Itoh, Takao Sato, Tatsuya Tsukui, Yutaka
Inagaki, Moises Selman, Kouji Matsushima, et Osamu Yoshie. 2016. « Heat Shock Protein
27 Plays a Pivotal Role in Myofibroblast Differentiation and in the Development of
Bleomycin-Induced Pulmonary Fibrosis ». PloS One 11 (2): e0148998.
https://doi.org/10.1371/journal.pone.0148998.
• Pasta, Saloni Yatin, Bakthisaran Raman, Tangirala Ramakrishna, et Ch Mohan
Rao. 2004. « The IXI/V Motif in the C-Terminal Extension of Alpha-Crystallins:
Alternative Interactions and Oligomeric Assemblies ». Molecular Vision 10 (septembre):
655‑62.
• Paul, Aditya S., Aditya Paul, Thomas D. Pollard, et Thomas Pollard. 2008. « The
Role of the FH1 Domain and Profilin in Formin-Mediated Actin-Filament Elongation and
Nucleation ». Current Biology: CB 18 (1): 9‑19.
https://doi.org/10.1016/j.cub.2007.11.062.
• Paul, Aditya S., et Thomas D. Pollard. 2009. « Energetic Requirements for

235
Processive Elongation of Actin Filaments by FH1FH2-Formins ». The Journal of
Biological Chemistry 284 (18): 12533‑40. https://doi.org/10.1074/jbc.M808587200.
• Pavan, Simona, Daniele Musiani, Erica Torchiaro, Giorgia Migliardi, Marta Gai,
Ferdinando Di Cunto, Jessica Erriquez, Martina Olivero, et Maria Flavia Di Renzo. 2014.
« HSP27 Is Required for Invasion and Metastasis Triggered by Hepatocyte Growth
Factor ». International Journal of Cancer 134 (6): 1289‑99.
https://doi.org/10.1002/ijc.28464.
• Pelham, Robert J., et Fred Chang. 2002. « Actin Dynamics in the Contractile Ring
during Cytokinesis in Fission Yeast ». Nature 419 (6902): 82‑86.
https://doi.org/10.1038/nature00999.
• Pelissier, Anne, Jean-Paul Chauvin, et Thomas Lecuit. 2003. « Trafficking through
Rab11 Endosomes Is Required for Cellularization during Drosophila Embryogenesis ».
Current Biology: CB 13 (21): 1848‑57.
• Pellegrin, Stéphanie, et Harry Mellor. 2005. « The Rho Family GTPase Rif Induces
Filopodia through MDia2 ». Current Biology: CB 15 (2): 129‑33.
https://doi.org/10.1016/j.cub.2005.01.011.
• Piccolella, Margherita, Valeria Crippa, Riccardo Cristofani, Paola Rusmini,
Mariarita Galbiati, Maria Elena Cicardi, Marco Meroni, et al. 2017. « The Small Heat
Shock Protein B8 (HSPB8) Modulates Proliferation and Migration of Breast Cancer
Cells ». Oncotarget, janvier. https://doi.org/10.18632/oncotarget.14422.
• Piekny, Alisa J., et Michael Glotzer. 2008. « Anillin Is a Scaffold Protein That Links
RhoA, Actin, and Myosin during Cytokinesis ». Current Biology: CB 18 (1): 30‑36.
https://doi.org/10.1016/j.cub.2007.11.068.
• Piotrowicz, R. S., E. Hickey, et E. G. Levin. 1998. « Heat Shock Protein 27 KDa
Expression and Phosphorylation Regulates Endothelial Cell Migration ». FASEB Journal:
Official Publication of the Federation of American Societies for Experimental Biology 12
(14): 1481‑90.
• Pollard, T. D., L. Blanchoin, et R. D. Mullins. 2000. « Molecular Mechanisms
Controlling Actin Filament Dynamics in Nonmuscle Cells ». Annual Review of Biophysics
and Biomolecular Structure 29: 545‑76.

236
https://doi.org/10.1146/annurev.biophys.29.1.545.
• Prokopenko, S. N., A. Brumby, L. O’Keefe, L. Prior, Y. He, R. Saint, et H. J.
Bellen. 1999. « A Putative Exchange Factor for Rho1 GTPase Is Required for Initiation of
Cytokinesis in Drosophila ». Genes & Development 13 (17): 2301‑14.
• Puls, A., A. G. Eliopoulos, C. D. Nobes, T. Bridges, L. S. Young, et A. Hall. 1999.
« Activation of the Small GTPase Cdc42 by the Inflammatory Cytokines TNF(Alpha) and
IL-1, and by the Epstein-Barr Virus Transforming Protein LMP1 ». Journal of Cell Science
112 ( Pt 17) (septembre): 2983‑92.
• Quinlan, Roy A., Yan Zhang, Andrew Lansbury, Ian Williamson, Ehmke Pohl, et
Fei Sun. 2013. « Changes in the Quaternary Structure and Function of MjHSP16.5
Attributable to Deletion of the IXI Motif and Introduction of the Substitution, R107G, in
the α-Crystallin Domain ». Philosophical Transactions of the Royal Society of London.
Series B, Biological Sciences 368 (1617): 20120327.
https://doi.org/10.1098/rstb.2012.0327.
• Ramanathan, Subramanian P., Jonne Helenius, Martin P. Stewart, Cedric J. Cattin,
Anthony A. Hyman, et Daniel J. Muller. 2015. « Cdk1-Dependent Mitotic Enrichment of
Cortical Myosin II Promotes Cell Rounding against Confinement ». Nature Cell Biology
17 (2): 148‑59. https://doi.org/10.1038/ncb3098.
• Ran, Jie, Yunfan Yang, Dengwen Li, Min Liu, et Jun Zhou. 2015. « Deacetylation
of α-Tubulin and Cortactin Is Required for HDAC6 to Trigger Ciliary Disassembly ».
Scientific Reports 5 (août): 12917. https://doi.org/10.1038/srep12917.
• Rankin, Kathleen E., et Linda Wordeman. 2010. « Long Astral Microtubules
Uncouple Mitotic Spindles from the Cytokinetic Furrow ». The Journal of Cell Biology
190 (1): 35‑43. https://doi.org/10.1083/jcb.201004017.
• Rantanen, Krista, Juha-Pekka Pursiheimo, Heidi Högel, Petra Miikkulainen, Jari
Sundström, et Panu M. Jaakkola. 2013. « P62/SQSTM1 Regulates Cellular Oxygen
Sensing by Attenuating PHD3 Activity through Aggregate Sequestration and Enhanced
Degradation ». Journal of Cell Science 126 (Pt 5): 1144‑54.
https://doi.org/10.1242/jcs.115667.
• Rapino, F., M. Jung, et S. Fulda. 2014. « BAG3 Induction Is Required to Mitigate

237
Proteotoxicity via Selective Autophagy Following Inhibition of Constitutive Protein
Degradation Pathways ». Oncogene 33 (13): 1713‑24.
https://doi.org/10.1038/onc.2013.110.
• Rauch, Jennifer N., Eric Tse, Rebecca Freilich, Sue-Ann Mok, Leah N. Makley,
Daniel R. Southworth, et Jason E. Gestwicki. 2016. « BAG3 Is a Modular, Scaffolding
Protein That Physically Links Heat Shock Protein 70 (Hsp70) to the Small Heat Shock
Proteins ». Journal of Molecular Biology, novembre.
https://doi.org/10.1016/j.jmb.2016.11.013.
• Ridley, A. J., et A. Hall. 1992. « The Small GTP-Binding Protein Rho Regulates
the Assembly of Focal Adhesions and Actin Stress Fibers in Response to Growth Factors ».
Cell 70 (3): 389‑99.
• Ridley, Anne J., Hugh F. Paterson, Caroline L. Johnston, Dagmar Diekmann, et
Alan Hall. 1992. « The small GTP-binding protein rac regulates growth factor-induced
membrane ruffling ». Cell 70 (3): 401‑10. https://doi.org/10.1016/0092-8674(92)90164-8.
• Riggs, Blake, Wendy Rothwell, Sarah Mische, Gilles R. X. Hickson, Johanne
Matheson, Thomas S. Hays, Gwyn W. Gould, et William Sullivan. 2003. « Actin
Cytoskeleton Remodeling during Early Drosophila Furrow Formation Requires Recycling
Endosomal Components Nuclear-Fallout and Rab11 ». The Journal of Cell Biology 163
(1): 143‑54. https://doi.org/10.1083/jcb.200305115.
• Riley, Brigit E., Stephen E. Kaiser, Thomas A. Shaler, Aylwin C. Y. Ng, Taichi
Hara, Mark S. Hipp, Kasper Lage, et al. 2010. « Ubiquitin Accumulation in Autophagy-
Deficient Mice Is Dependent on the Nrf2-Mediated Stress Response Pathway: A Potential
Role for Protein Aggregation in Autophagic Substrate Selection ». The Journal of Cell
Biology 191 (3): 537‑52. https://doi.org/10.1083/jcb.201005012.
• Rocchi, Palma, Eliana Beraldi, Susan Ettinger, Ladan Fazli, Robert L. Vessella,
Colleen Nelson, et Martin Gleave. 2005. « Increased Hsp27 after Androgen Ablation
Facilitates Androgen-Independent Progression in Prostate Cancer via Signal Transducers
and Activators of Transcription 3-Mediated Suppression of Apoptosis ». Cancer Research
65 (23): 11083‑93. https://doi.org/10.1158/0008-5472.CAN-05-1840.
• Rodal, Avital A., Olga Sokolova, Deborah B. Robins, Karen M. Daugherty, Simon

238
Hippenmeyer, Howard Riezman, Nikolaus Grigorieff, et Bruce L. Goode. 2005.
« Conformational Changes in the Arp2/3 Complex Leading to Actin Nucleation ». Nature
Structural & Molecular Biology 12 (1): 26‑31. https://doi.org/10.1038/nsmb870.
• Rodríguez, Andrea E., Camila López-Crisosto, Daniel Peña-Oyarzún, Daniela
Salas, Valentina Parra, Clara Quiroga, Tobias Morawe, Mario Chiong, Christian Behl, et
Sergio Lavandero. 2016. « BAG3 Regulates Total MAP1LC3B Protein Levels through a
Translational but Not Transcriptional Mechanism ». Autophagy 12 (2): 287‑96.
https://doi.org/10.1080/15548627.2015.1124225.
• Rogalla, T., M. Ehrnsperger, X. Preville, A. Kotlyarov, G. Lutsch, C. Ducasse, C.
Paul, et al. 1999. « Regulation of Hsp27 Oligomerization, Chaperone Function, and
Protective Activity against Oxidative Stress/Tumor Necrosis Factor Alpha by
Phosphorylation ». The Journal of Biological Chemistry 274 (27): 18947‑56.
• Rogov, Vladimir, Volker Dötsch, Terje Johansen, et Vladimir Kirkin. 2014.
« Interactions between Autophagy Receptors and Ubiquitin-like Proteins Form the
Molecular Basis for Selective Autophagy ». Molecular Cell 53 (2): 167‑78.
https://doi.org/10.1016/j.molcel.2013.12.014.
• Rohatgi, R., L. Ma, H. Miki, M. Lopez, T. Kirchhausen, T. Takenawa, et M. W.
Kirschner. 1999a. « The Interaction between N-WASP and the Arp2/3 Complex Links
Cdc42-Dependent Signals to Actin Assembly ». Cell 97 (2): 221‑31.
• ———. 1999b. « The Interaction between N-WASP and the Arp2/3 Complex
Links Cdc42-Dependent Signals to Actin Assembly ». Cell 97 (2): 221‑31.
• Romano, M. F., M. Festa, G. Pagliuca, R. Lerose, R. Bisogni, F. Chiurazzi, G.
Storti, et al. 2003. « BAG3 Protein Controls B-Chronic Lymphocytic Leukaemia Cell
Apoptosis ». Cell Death and Differentiation 10 (3): 383‑85.
https://doi.org/10.1038/sj.cdd.4401167.
• Romano, Maria Fiammetta, Michelina Festa, Antonello Petrella, Alessasndra
Rosati, Maria Pascale, Rita Bisogni, Vincenzo Poggi, et al. 2003. « BAG3 Protein
Regulates Cell Survival in Childhood Acute Lymphoblastic Leukemia Cells ». Cancer
Biology & Therapy 2 (5): 508‑10.
• Romero, Stéphane, Christophe Le Clainche, Dominique Didry, Coumaran Egile,

239
Dominique Pantaloni, et Marie-France Carlier. 2004. « Formin Is a Processive Motor That
Requires Profilin to Accelerate Actin Assembly and Associated ATP Hydrolysis ». Cell
119 (3): 419‑29. https://doi.org/10.1016/j.cell.2004.09.039.
• Roossien, Douglas H., Kyle E. Miller, et Gianluca Gallo. 2015. « Ciliobrevins as
Tools for Studying Dynein Motor Function ». Frontiers in Cellular Neuroscience 9: 252.
https://doi.org/10.3389/fncel.2015.00252.
• Rosa, André, Evi Vlassaks, Franck Pichaud, et Buzz Baum. 2015. « Ect2/Pbl Acts
via Rho and Polarity Proteins to Direct the Assembly of an Isotropic Actomyosin Cortex
upon Mitotic Entry ». Developmental Cell 32 (5): 604‑16.
https://doi.org/10.1016/j.devcel.2015.01.012.
• Rosati, Alessandra, Anna Basile, Raffaella D’Auria, Morena d’Avenia, Margot De
Marco, Antonia Falco, Michelina Festa, et al. 2015. « BAG3 Promotes Pancreatic Ductal
Adenocarcinoma Growth by Activating Stromal Macrophages ». Nature Communications
6 (novembre): 8695. https://doi.org/10.1038/ncomms9695.
• Rosenblatt, Jody, Louise P. Cramer, Buzz Baum, et Karen M. McGee. 2004.
« Myosin II-Dependent Cortical Movement Is Required for Centrosome Separation and
Positioning during Mitotic Spindle Assembly ». Cell 117 (3): 361‑72.
• Rotty, Jeremy D., Congying Wu, Elizabeth M. Haynes, Cristian Suarez, Jonathan
D. Winkelman, Heath E. Johnson, Jason M. Haugh, David R. Kovar, et James E. Bear.
2015. « Profilin-1 Serves as a Gatekeeper for Actin Assembly by Arp2/3-Dependent and -
Independent Pathways ». Developmental Cell 32 (1): 54‑67.
https://doi.org/10.1016/j.devcel.2014.10.026.
• Roubinet, Chantal, Barbara Decelle, Gaëtan Chicanne, Jonas F. Dorn, Bernard
Payrastre, François Payre, et Sébastien Carreno. 2011. « Molecular Networks Linked by
Moesin Drive Remodeling of the Cell Cortex during Mitosis ». The Journal of Cell Biology
195 (1): 99‑112. https://doi.org/10.1083/jcb.201106048.
• Rouiller, Isabelle, Xiao-Ping Xu, Kurt J. Amann, Coumaran Egile, Stephan Nickell,
Daniela Nicastro, Rong Li, Thomas D. Pollard, Niels Volkmann, et Dorit Hanein. 2008.
« The Structural Basis of Actin Filament Branching by the Arp2/3 Complex ». The Journal
of Cell Biology 180 (5): 887‑95. https://doi.org/10.1083/jcb.200709092.

240
• Rouse, J., P. Cohen, S. Trigon, M. Morange, A. Alonso-Llamazares, D. Zamanillo,
T. Hunt, et A. R. Nebreda. 1994. « A Novel Kinase Cascade Triggered by Stress and Heat
Shock That Stimulates MAPKAP Kinase-2 and Phosphorylation of the Small Heat Shock
Proteins ». Cell 78 (6): 1027‑37.
• Roybal, Kole T., Taráz E. Buck, Xiongtao Ruan, Baek Hwan Cho, Danielle J.
Clark, Rachel Ambler, Helen M. Tunbridge, et al. 2016. « Computational spatiotemporal
analysis identifies WAVE2 and Cofilin as joint regulators of costimulation-mediated T cell
actin dynamics ». Science signaling 9 (424): rs3.
https://doi.org/10.1126/scisignal.aad4149.
• Rutsdottir, Gudrun, Johan Härmark, Yoran Weide, Hans Hebert, Morten I.
Rasmussen, Sven Wernersson, Michal Respondek, et al. 2017. « Structural Model of
Dodecameric Heat-Shock Protein Hsp21: Flexible N-Terminal Arms Interact with Client
Proteins While C-Terminal Tails Maintain the Dodecamer and Chaperone Activity ». The
Journal of Biological Chemistry 292 (19): 8103‑21.
https://doi.org/10.1074/jbc.M116.766816.
• S Pedersen, Ronni, Gopal Karemore, Thorkell Gudjonsson, Maj-Britt Rask, Beate
Neumann, Jean-Karim Hériché, Rainer Pepperkok, et al. 2016. « Profiling DNA Damage
Response Following Mitotic Perturbations ». Nature Communications 7 (décembre):
13887. https://doi.org/10.1038/ncomms13887.
• Sagona, Antonia P., et Harald Stenmark. 2010. « Cytokinesis and Cancer ». FEBS
Letters 584 (12): 2652‑61. https://doi.org/10.1016/j.febslet.2010.03.044.
• Sakahira, Hideki, Peter Breuer, Manajit K. Hayer-Hartl, et F. Ulrich Hartl. 2002.
« Molecular Chaperones as Modulators of Polyglutamine Protein Aggregation and
Toxicity ». Proceedings of the National Academy of Sciences of the United States of
America 99 Suppl 4 (décembre): 16412‑18. https://doi.org/10.1073/pnas.182426899.
• Salerno, John C., Cheryl L. Eifert, Kathleen M. Salerno, et Jane F. Koretz. 2003.
« Structural Diversity in the Small Heat Shock Protein Superfamily: Control of
Aggregation by the N-Terminal Region ». Protein Engineering 16 (11): 847‑51.
• Satoh, Sachie, et Tomoko Tominaga. 2001. « MDia-Interacting Protein Acts
Downstream of Rho-MDia and Modifies Src Activation and Stress Fiber Formation ».

241
Journal of Biological Chemistry 276 (42): 39290‑94.
https://doi.org/10.1074/jbc.M107026200.
• Schafer, D. A., C. Hug, et J. A. Cooper. 1995. « Inhibition of CapZ during
Myofibrillogenesis Alters Assembly of Actin Filaments. » The Journal of Cell Biology 128
(1): 61‑70. https://doi.org/10.1083/jcb.128.1.61.
• Schiel, John A., Kristin Park, Mary K. Morphew, Evan Reid, Andreas Hoenger, et
Rytis Prekeris. 2011. « Endocytic Membrane Fusion and Buckling-Induced Microtubule
Severing Mediate Cell Abscission ». Journal of Cell Science 124 (Pt 9): 1411‑24.
https://doi.org/10.1242/jcs.081448.
• Schiel, John A., Glenn C. Simon, Chelsey Zaharris, Julie Weisz, David Castle,
Christine C. Wu, et Rytis Prekeris. 2012. « FIP3-Endosome-Dependent Formation of the
Secondary Ingression Mediates ESCRT-III Recruitment during Cytokinesis ». Nature Cell
Biology 14 (10): 1068‑78. https://doi.org/10.1038/ncb2577.
• Schirenbeck, Antje, Till Bretschneider, Rajesh Arasada, Michael Schleicher, et Jan
Faix. 2005. « The Diaphanous-Related Formin DDia2 Is Required for the Formation and
Maintenance of Filopodia ». Nature Cell Biology 7 (6): 619‑25.
https://doi.org/10.1038/ncb1266.
• Schneider, Jaime L., et Ana Maria Cuervo. 2014. « Autophagy and Human Disease:
Emerging Themes ». Current Opinion in Genetics & Development 26 (juin): 16‑23.
https://doi.org/10.1016/j.gde.2014.04.003.
• Scholey, J. M., K. A. Taylor, et J. Kendrick-Jones. 1980. « Regulation of Non-
Muscle Myosin Assembly by Calmodulin-Dependent Light Chain Kinase ». Nature 287
(5779): 233‑35.
• Schölz, Christian, Brian T. Weinert, Sebastian A. Wagner, Petra Beli, Yasuyuki
Miyake, Jun Qi, Lars J. Jensen, et al. 2015. « Acetylation Site Specificities of Lysine
Deacetylase Inhibitors in Human Cells ». Nature Biotechnology 33 (4): 415‑23.
https://doi.org/10.1038/nbt.3130.
• Schootbrugge, Chantal van de, Johan Bussink, Paul N. Span, Fred CGJ Sweep,
Reidar Grénman, Hanneke Stegeman, Ger JM Pruijn, Johannes HAM Kaanders, et Wilbert
C. Boelens. 2013. « ΑB-Crystallin Stimulates VEGF Secretion and Tumor Cell Migration

242
and Correlates with Enhanced Distant Metastasis in Head and Neck Squamous Cell
Carcinoma ». BMC Cancer 13 (1): 128. https://doi.org/10.1186/1471-2407-13-128.
• Schroeder, Thomas E. 1972. « THE CONTRACTILE RING: II. Determining Its
Brief Existence, Volumetric Changes, and Vital Role in Cleaving Arbacia Eggs ». The
Journal of Cell Biology 53 (2): 419‑34. https://doi.org/10.1083/jcb.53.2.419.
• Schuck, Sebastian, Ciara M. Gallagher, et Peter Walter. 2014. « ER-Phagy
Mediates Selective Degradation of Endoplasmic Reticulum Independently of the Core
Autophagy Machinery ». Journal of Cell Science 127 (Pt 18): 4078‑88.
https://doi.org/10.1242/jcs.154716.
• Selcen, Duygu, Francesco Muntoni, Barbara K. Burton, Elena Pegoraro, Caroline
Sewry, Anna V. Bite, et Andrew G. Engel. 2009. « Mutation in BAG3 Causes Severe
Dominant Childhood Muscular Dystrophy ». Annals of Neurology 65 (1): 83‑89.
https://doi.org/10.1002/ana.21553.
• Sellers, J. R., M. D. Pato, et R. S. Adelstein. 1981. « Reversible Phosphorylation of
Smooth Muscle Myosin, Heavy Meromyosin, and Platelet Myosin ». The Journal of
Biological Chemistry 256 (24): 13137‑42.
• Selva, Javier, et Gustavo Egea. 2011. « Ethanol Increases P190RhoGAP Activity,
Leading to Actin Cytoskeleton Rearrangements ». Journal of Neurochemistry 119 (6):
1306‑16. https://doi.org/10.1111/j.1471-4159.2011.07522.x.
• Shammas, Sarah L., Christopher A. Waudby, Shuyu Wang, Alexander K. Buell,
Tuomas P. J. Knowles, Heath Ecroyd, Mark E. Welland, John A. Carver, Christopher M.
Dobson, et Sarah Meehan. 2011. « Binding of the Molecular Chaperone ΑB-Crystallin to
Aβ Amyloid Fibrils Inhibits Fibril Elongation ». Biophysical Journal 101 (7): 1681‑89.
https://doi.org/10.1016/j.bpj.2011.07.056.
• Shankar, Jay, et Ivan R. Nabi. 2015. « Actin Cytoskeleton Regulation of Epithelial
Mesenchymal Transition in Metastatic Cancer Cells ». PLoS ONE 10 (3).
https://doi.org/10.1371/journal.pone.0119954.
• Shi, Huiyong, Haidong Xu, Zengjun Li, Yanan Zhen, Bin Wang, Shoujun Huo,
Ruixue Xiao, et Zhongfa Xu. 2016. « BAG3 Regulates Cell Proliferation, Migration, and
Invasion in Human Colorectal Cancer ». Tumour Biology: The Journal of the International

243
Society for Oncodevelopmental Biology and Medicine 37 (4): 5591‑97.
https://doi.org/10.1007/s13277-015-4403-1.
• Shimizu, Miho, Mikihito Tanaka, et Yoriko Atomi. 2016. « Small Heat Shock
Protein ΑB-Crystallin Controls Shape and Adhesion of Glioma and Myoblast Cells in the
Absence of Stress ». PloS One 11 (12): e0168136.
https://doi.org/10.1371/journal.pone.0168136.
• Sigrist, S., H. Jacobs, R. Stratmann, et C. F. Lehner. 1995. « Exit from Mitosis Is
Regulated by Drosophila Fizzy and the Sequential Destruction of Cyclins A, B and B3 ».
The EMBO Journal 14 (19): 4827‑38.
• Siller, Karsten H., et Chris Q. Doe. 2009. « Spindle Orientation during Asymmetric
Cell Division ». Nature Cell Biology 11 (4): 365‑74. https://doi.org/10.1038/ncb0409-365.
• Simon, Glenn C., Eric Schonteich, Christine C. Wu, Alisa Piekny, Damian Ekiert,
Xinzi Yu, Gwyn W. Gould, Michael Glotzer, et Rytis Prekeris. 2008. « Sequential Cyk-4
Binding to ECT2 and FIP3 Regulates Cleavage Furrow Ingression and Abscission during
Cytokinesis ». The EMBO Journal 27 (13): 1791‑1803.
https://doi.org/10.1038/emboj.2008.112.
• Sitterding, Stephanie M., William R. Wiseman, Carol L. Schiller, Chunyan Luan,
Feng Chen, Jose V. Moyano, William G. Watkin, Elizabeth L. Wiley, Vincent L. Cryns, et
Leslie K. Diaz. 2008. « AlphaB-Crystallin: A Novel Marker of Invasive Basal-like and
Metaplastic Breast Carcinomas ». Annals of Diagnostic Pathology 12 (1): 33‑40.
https://doi.org/10.1016/j.anndiagpath.2007.02.004.
• Sluchanko, Nikolai N., Natalia A. Chebotareva, et Nikolai B. Gusev. 2015.
« Quaternary Structure of Human Small Heat Shock Protein HSPB6 (Hsp20) in Crowded
Media Modeled by Trimethylamine N-Oxide (TMAO): Effect of Protein
Phosphorylation ». Biochimie 108 (janvier): 68‑75.
https://doi.org/10.1016/j.biochi.2014.11.001.
• Smith, C. C., Y. X. Yu, M. Kulka, et L. Aurelian. 2000. « A Novel Human Gene
Similar to the Protein Kinase (PK) Coding Domain of the Large Subunit of Herpes Simplex
Virus Type 2 Ribonucleotide Reductase (ICP10) Codes for a Serine-Threonine PK and Is
Expressed in Melanoma Cells ». The Journal of Biological Chemistry 275 (33): 25690‑99.

244
https://doi.org/10.1074/jbc.M002140200.
• Söderberg, Ola, Karl-Johan Leuchowius, Mats Gullberg, Malin Jarvius, Irene
Weibrecht, Lars-Gunnar Larsson, et Ulf Landegren. 2008. « Characterizing Proteins and
Their Interactions in Cells and Tissues Using the in Situ Proximity Ligation Assay ».
Methods (San Diego, Calif.) 45 (3): 227‑32. https://doi.org/10.1016/j.ymeth.2008.06.014.
• Solinet, Sara, Kazi Mahmud, Shannon F. Stewman, Khaled Ben El Kadhi, Barbara
Decelle, Lama Talje, Ao Ma, Benjamin H. Kwok, et Sébastien Carreno. 2013. « The Actin-
Binding ERM Protein Moesin Binds to and Stabilizes Microtubules at the Cell Cortex ».
The Journal of Cell Biology 202 (2): 251‑60. https://doi.org/10.1083/jcb.201304052.
• Somlyo, Andrew P., et Avril V. Somlyo. 2003. « Ca2+ Sensitivity of Smooth
Muscle and Nonmuscle Myosin II: Modulated by G Proteins, Kinases, and Myosin
Phosphatase ». Physiological Reviews 83 (4): 1325‑58.
https://doi.org/10.1152/physrev.00023.2003.
• Sontag, Emily Mitchell, Rahul S. Samant, et Judith Frydman. 2017. « Mechanisms
and Functions of Spatial Protein Quality Control ». Annual Review of Biochemistry 86
(juin): 97‑122. https://doi.org/10.1146/annurev-biochem-060815-014616.
• Sorce, Barbara, Carlos Escobedo, Yusuke Toyoda, Martin P. Stewart, Cedric J.
Cattin, Richard Newton, Indranil Banerjee, et al. 2015. « Mitotic Cells Contract
Actomyosin Cortex and Generate Pressure to Round against or Escape Epithelial
Confinement ». Nature Communications 6 (novembre): 8872.
https://doi.org/10.1038/ncomms9872.
• Specht, Sebastian, Stephanie B. M. Miller, Axel Mogk, et Bernd Bukau. 2011.
« Hsp42 Is Required for Sequestration of Protein Aggregates into Deposition Sites in
Saccharomyces Cerevisiae ». The Journal of Cell Biology 195 (4): 617‑29.
https://doi.org/10.1083/jcb.201106037.
• Stachowiak, Matthew R., Caroline Laplante, Harvey F. Chin, Boris Guirao, Erdem
Karatekin, Thomas D. Pollard, et Ben O’Shaughnessy. 2014. « Mechanism of Cytokinetic
Contractile Ring Constriction in Fission Yeast ». Developmental Cell 29 (5): 547‑61.
https://doi.org/10.1016/j.devcel.2014.04.021.
• Stoevring, Birgitte, Jette L. Frederiksen, et Michael Christiansen. 2007. « CRYAB

245
Promoter Polymorphisms: Influence on Multiple Sclerosis Susceptibility and Clinical
Presentation ». Clinica Chimica Acta; International Journal of Clinical Chemistry 375
(1‑2): 57‑62. https://doi.org/10.1016/j.cca.2006.06.017.
• Stokoe, D., K. Engel, D. G. Campbell, P. Cohen, et M. Gaestel. 1992.
« Identification of MAPKAP Kinase 2 as a Major Enzyme Responsible for the
Phosphorylation of the Small Mammalian Heat Shock Proteins ». FEBS Letters 313 (3):
307‑13.
• Stoten, Caroline Louise, et Jeremy Graham Carlton. 2017. « ESCRT-Dependent
Control of Membrane Remodelling during Cell Division ». Seminars in Cell &
Developmental Biology, août. https://doi.org/10.1016/j.semcdb.2017.08.035.
• Straight, Aaron F., Amy Cheung, John Limouze, Irene Chen, Nick J. Westwood,
James R. Sellers, et Timothy J. Mitchison. 2003. « Dissecting Temporal and Spatial
Control of Cytokinesis with a Myosin II Inhibitor ». Science 299 (5613): 1743‑47.
https://doi.org/10.1126/science.1081412.
• Strózecka, Joanna, Elżbieta Chrusciel, Emilia Górna, Aneta Szymanska, Szymon
Ziętkiewicz, et Krzysztof Liberek. 2012. « Importance of N- and C-Terminal Regions of
IbpA, Escherichia Coli Small Heat Shock Protein, for Chaperone Function and
Oligomerization ». The Journal of Biological Chemistry 287 (4): 2843‑53.
https://doi.org/10.1074/jbc.M111.273847.
• Studer, Sonja, Markus Obrist, Nicolas Lentze, et Franz Narberhaus. 2002. « A
Critical Motif for Oligomerization and Chaperone Activity of Bacterial Alpha-Heat Shock
Proteins ». European Journal of Biochemistry 269 (14): 3578‑86.
• Sun, Shao-Chen, Zhen-Bo Wang, Yong-Nan Xu, Seung-Eun Lee, Xiang-Shun Cui,
et Nam-Hyung Kim. 2011. « Arp2/3 Complex Regulates Asymmetric Division and
Cytokinesis in Mouse Oocytes ». PloS One 6 (4): e18392.
https://doi.org/10.1371/journal.pone.0018392.
• Suraneni, Praveen, Boris Rubinstein, Jay R. Unruh, Michael Durnin, Dorit Hanein,
et Rong Li. 2012. « The Arp2/3 Complex Is Required for Lamellipodia Extension and
Directional Fibroblast Cell Migration ». The Journal of Cell Biology 197 (2): 239‑51.
https://doi.org/10.1083/jcb.201112113.

246
• Suss, Ohad, et Dana Reichmann. 2015. « Protein Plasticity Underlines Activation
and Function of ATP-Independent Chaperones ». Frontiers in Molecular Biosciences 2.
https://doi.org/10.3389/fmolb.2015.00043.
• Suzuki, K., T. Kirisako, Y. Kamada, N. Mizushima, T. Noda, et Y. Ohsumi. 2001.
« The Pre-Autophagosomal Structure Organized by Concerted Functions of APG Genes Is
Essential for Autophagosome Formation ». The EMBO Journal 20 (21): 5971‑81.
https://doi.org/10.1093/emboj/20.21.5971.
• Suzuki, Kuninori, Yuka Kubota, Takayuki Sekito, et Yoshinori Ohsumi. 2007.
« Hierarchy of Atg Proteins in Pre-Autophagosomal Structure Organization ». Genes to
Cells: Devoted to Molecular & Cellular Mechanisms 12 (2): 209‑18.
https://doi.org/10.1111/j.1365-2443.2007.01050.x.
• Suzuki, Mariko, Rie Matsushima-Nishiwaki, Gen Kuroyanagi, Noriko Suzuki,
Reika Takamatsu, Tatsuro Furui, Naoki Yoshimi, Osamu Kozawa, et Ken-ichirou
Morishige. 2015. « Regulation by Heat Shock Protein 22 (HSPB8) of Transforming
Growth Factor-α-Induced Ovary Cancer Cell Migration ». Archives of Biochemistry and
Biophysics 571 (avril): 40‑49. https://doi.org/10.1016/j.abb.2015.02.030.
• Symons, M., J. M. Derry, B. Karlak, S. Jiang, V. Lemahieu, F. Mccormick, U.
Francke, et A. Abo. 1996. « Wiskott-Aldrich Syndrome Protein, a Novel Effector for the
GTPase CDC42Hs, Is Implicated in Actin Polymerization ». Cell 84 (5): 723‑34.
• Takaishi, K., T. Sasaki, T. Kameyama, S. Tsukita, S. Tsukita, et Y. Takai. 1995.
« Translocation of Activated Rho from the Cytoplasm to Membrane Ruffling Area, Cell-
Cell Adhesion Sites and Cleavage Furrows ». Oncogene 11 (1): 39‑48.
• Tan, Jeanne M. M., Esther S. P. Wong, Donald S. Kirkpatrick, Olga Pletnikova,
Han Seok Ko, Shiam-Peng Tay, Michelle W. L. Ho, et al. 2008. « Lysine 63-Linked
Ubiquitination Promotes the Formation and Autophagic Clearance of Protein Inclusions
Associated with Neurodegenerative Diseases ». Human Molecular Genetics 17 (3):
431‑39. https://doi.org/10.1093/hmg/ddm320.
• Tan, Qian, Marina Wang, Man Yu, Junyan Zhang, Robert G. Bristow, Richard P.
Hill, et Ian F. Tannock. 2016. « Role of Autophagy as a Survival Mechanism for Hypoxic
Cells in Tumors ». Neoplasia (New York, N.Y.) 18 (6): 347‑55.

247
https://doi.org/10.1016/j.neo.2016.04.003.
• Tang, Bei-sha, Guo-hua Zhao, Wei Luo, Kun Xia, Fang Cai, Qian Pan, Ru-xu
Zhang, et al. 2005. « Small Heat-Shock Protein 22 Mutated in Autosomal Dominant
Charcot-Marie-Tooth Disease Type 2L ». Human Genetics 116 (3): 222‑24.
https://doi.org/10.1007/s00439-004-1218-3.
• Tanida, Isei, Yu-shin Sou, Junji Ezaki, Naoko Minematsu-Ikeguchi, Takashi Ueno,
et Eiki Kominami. 2004. « HsAtg4B/HsApg4B/Autophagin-1 Cleaves the Carboxyl
Termini of Three Human Atg8 Homologues and Delipidates Microtubule-Associated
Protein Light Chain 3- and GABAA Receptor-Associated Protein-Phospholipid
Conjugates ». The Journal of Biological Chemistry 279 (35): 36268‑76.
https://doi.org/10.1074/jbc.M401461200.
• Tanida, Isei, Emiko Tanida-Miyake, Masaaki Komatsu, Takashi Ueno, et Eiki
Kominami. 2002. « Human Apg3p/Aut1p Homologue Is an Authentic E2 Enzyme for
Multiple Substrates, GATE-16, GABARAP, and MAP-LC3, and Facilitates the
Conjugation of HApg12p to HApg5p ». The Journal of Biological Chemistry 277 (16):
13739‑44. https://doi.org/10.1074/jbc.M200385200.
• Tatsumoto, T., X. Xie, R. Blumenthal, I. Okamoto, et T. Miki. 1999. « Human
ECT2 Is an Exchange Factor for Rho GTPases, Phosphorylated in G2/M Phases, and
Involved in Cytokinesis ». The Journal of Cell Biology 147 (5): 921‑28.
• Tehrani, Shandiz, Nenad Tomasevic, Scott Weed, Roman Sakowicz, et John A.
Cooper. 2007. « Src Phosphorylation of Cortactin Enhances Actin Assembly ».
Proceedings of the National Academy of Sciences of the United States of America 104 (29):
11933‑38. https://doi.org/10.1073/pnas.0701077104.
• Ten Klooster, Jean Paul, Eva E. Evers, Lennert Janssen, Laura M. Machesky, Frits
Michiels, Peter Hordijk, et John G. Collard. 2006. « Interaction between Tiam1 and the
Arp2/3 Complex Links Activation of Rac to Actin Polymerization ». The Biochemical
Journal 397 (1): 39‑45. https://doi.org/10.1042/BJ20051957.
• Tessier, Deron J., Padmini Komalavilas, Alyssa Panitch, Lokesh Joshi, et Colleen
M. Brophy. 2003. « The Small Heat Shock Protein (HSP) 20 Is Dynamically Associated
with the Actin Cross-Linking Protein Actinin ». The Journal of Surgical Research 111 (1):

248
152‑57.
• Théry, Manuel, Victor Racine, Anne Pépin, Matthieu Piel, Yong Chen, Jean-
Baptiste Sibarita, et Michel Bornens. 2005. « The Extracellular Matrix Guides the
Orientation of the Cell Division Axis ». Nature Cell Biology 7 (10): 947‑53.
https://doi.org/10.1038/ncb1307.
• Thomas, Jeffrey H., et Eric Wieschaus. 2004. « Src64 and Tec29 Are Required for
Microfilament Contraction during Drosophila Cellularization ». Development 131 (4):
863‑71. https://doi.org/10.1242/dev.00989.
• Tolić, Iva M. 2017. « Mitotic Spindle: Kinetochore Fibers Hold on Tight to
Interpolar Bundles ». European Biophysics Journal, juillet, 1‑13.
https://doi.org/10.1007/s00249-017-1244-4.
• Tomar, Alok, Christine Lawson, Majid Ghassemian, et David D. Schlaepfer. 2012.
« Cortactin as a Target for FAK in the Regulation of Focal Adhesion Dynamics ». PloS
One 7 (8): e44041. https://doi.org/10.1371/journal.pone.0044041.
• Torres, Eduardo, et Michael K. Rosen. 2006. « Protein-Tyrosine Kinase and
GTPase Signals Cooperate to Phosphorylate and Activate Wiskott-Aldrich Syndrome
Protein (WASP)/Neuronal WASP ». The Journal of Biological Chemistry 281 (6):
3513‑20. https://doi.org/10.1074/jbc.M509416200.
• Tran, Andy Dong-Anh, Timothy P. Marmo, Ambar A. Salam, Sally Che, Erik
Finkelstein, Rafi Kabarriti, Harry S. Xenias, et al. 2007. « HDAC6 Deacetylation of
Tubulin Modulates Dynamics of Cellular Adhesions ». Journal of Cell Science 120 (Pt 8):
1469‑79. https://doi.org/10.1242/jcs.03431.
• Tumbarello, David A., Bennett J. Waxse, Susan D. Arden, Nicholas A. Bright, John
Kendrick-Jones, et Folma Buss. 2012. « Autophagy-receptors link myosin VI to
autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with
the lysosome ». Nature cell biology 14 (10): 1024‑35. https://doi.org/10.1038/ncb2589.
• Ulbricht, Anna, Verena Arndt, et Jörg Höhfeld. 2013. « Chaperone-Assisted
Proteostasis Is Essential for Mechanotransduction in Mammalian Cells ». Communicative
& Integrative Biology 6 (4): e24925. https://doi.org/10.4161/cib.24925.
• Ulbricht, Anna, Felix J. Eppler, Victor E. Tapia, Peter F. M. van der Ven, Nico

249
Hampe, Nils Hersch, Padmanabhan Vakeel, et al. 2013. « Cellular Mechanotransduction
Relies on Tension-Induced and Chaperone-Assisted Autophagy ». Current Biology: CB 23
(5): 430‑35. https://doi.org/10.1016/j.cub.2013.01.064.
• Ulbricht, Anna, et Jörg Höhfeld. 2013. « Tension-Induced Autophagy: May the
Chaperone Be with You ». Autophagy 9 (6): 920‑22. https://doi.org/10.4161/auto.24213.
• Umemoto, S., A. R. Bengur, et J. R. Sellers. 1989. « Effect of Multiple
Phosphorylations of Smooth Muscle and Cytoplasmic Myosins on Movement in an in Vitro
Motility Assay ». The Journal of Biological Chemistry 264 (3): 1431‑36.
• Ungelenk, Sophia, Fatemeh Moayed, Chi-Ting Ho, Tomas Grousl, Annette Scharf,
Alireza Mashaghi, Sander Tans, Matthias P. Mayer, Axel Mogk, et Bernd Bukau. 2016.
« Small Heat Shock Proteins Sequester Misfolding Proteins in Near-Native Conformation
for Cellular Protection and Efficient Refolding ». Nature Communications 7 (novembre):
13673. https://doi.org/10.1038/ncomms13673.
• Uruno, T., J. Liu, P. Zhang, null Fan Yx, C. Egile, R. Li, S. C. Mueller, et X. Zhan.
2001. « Activation of Arp2/3 Complex-Mediated Actin Polymerization by Cortactin ».
Nature Cell Biology 3 (3): 259‑66. https://doi.org/10.1038/35060051.
• Urzúa, Ulises, Sandra Ampuero, Katherine F. Roby, Garrison A. Owens, et David
J. Munroe. 2016. « Dysregulation of Mitotic Machinery Genes Precedes Genome
Instability during Spontaneous Pre-Malignant Transformation of Mouse Ovarian Surface
Epithelial Cells ». BMC Genomics 17 (Suppl 8): 728. https://doi.org/10.1186/s12864-016-
3068-5.
• Uyeda, T. Q., P. D. Abramson, et J. A. Spudich. 1996. « The Neck Region of the
Myosin Motor Domain Acts as a Lever Arm to Generate Movement ». Proceedings of the
National Academy of Sciences of the United States of America 93 (9): 4459‑64.
• Vadlamudi, R. K., I. Joung, J. L. Strominger, et J. Shin. 1996. « P62, a
Phosphotyrosine-Independent Ligand of the SH2 Domain of P56lck, Belongs to a New
Class of Ubiquitin-Binding Proteins ». The Journal of Biological Chemistry 271 (34):
20235‑37.
• Vahid, Sepideh, Daksh Thaper, Kate F. Gibson, Jennifer L. Bishop, et Amina
Zoubeidi. 2016. « Molecular Chaperone Hsp27 Regulates the Hippo Tumor Suppressor

250
Pathway in Cancer ». Scientific Reports 6 (août): 31842.
https://doi.org/10.1038/srep31842.
• Valenzuela-Fernández, Agustín, J. Román Cabrero, Juan M. Serrador, et Francisco
Sánchez-Madrid. 2008. « HDAC6: A Key Regulator of Cytoskeleton, Cell Migration and
Cell-Cell Interactions ». Trends in Cell Biology 18 (6): 291‑97.
https://doi.org/10.1016/j.tcb.2008.04.003.
• Verschuure, Pauline, Yvonne Croes, Paul R. L. A. van den IJssel, Roy A. Quinlan,
Wilfried W. de Jong, et Wilbert C. Boelens. 2002. « Translocation of Small Heat Shock
Proteins to the Actin Cytoskeleton upon Proteasomal Inhibition ». Journal of Molecular
and Cellular Cardiology 34 (2): 117‑28. https://doi.org/10.1006/jmcc.2001.1493.
• Vicente-Manzanares, Miguel, Xuefei Ma, Robert S. Adelstein, et Alan Rick
Horwitz. 2009. « Non-Muscle Myosin II Takes Centre Stage in Cell Adhesion and
Migration ». Nature Reviews. Molecular Cell Biology 10 (11): 778‑90.
https://doi.org/10.1038/nrm2786.
• Vidal, Catherine, Blandine Geny, Josiane Melle, Martine Jandrot-Perrus, et
Michaëla Fontenay-Roupie. 2002. « Cdc42/Rac1-Dependent Activation of the P21-
Activated Kinase (PAK) Regulates Human Platelet Lamellipodia Spreading: Implication
of the Cortical-Actin Binding Protein Cortactin ». Blood 100 (13): 4462‑69.
https://doi.org/10.1182/blood.V100.13.4462.
• Vidali, Luis, Peter A. C. van Gisbergen, Christophe Guérin, Paula Franco, Ming Li,
Graham M. Burkart, Robert C. Augustine, Laurent Blanchoin, et Magdalena Bezanilla.
2009. « Rapid Formin-Mediated Actin-Filament Elongation Is Essential for Polarized Plant
Cell Growth ». Proceedings of the National Academy of Sciences of the United States of
America 106 (32): 13341‑46. https://doi.org/10.1073/pnas.0901170106.
• Vitale, Ilio, Gwenola Manic, Vito Dandrea, et Ruggero De Maria. 2015. « Role of
Autophagy in the Maintenance and Function of Cancer Stem Cells ». The International
Journal of Developmental Biology 59 (1‑3): 95‑108.
https://doi.org/10.1387/ijdb.150082iv.
• Volkmann, N., K. J. Amann, S. Stoilova-McPhie, C. Egile, D. C. Winter, L.
Hazelwood, J. E. Heuser, R. Li, T. D. Pollard, et D. Hanein. 2001. « Structure of Arp2/3

251
Complex in Its Activated State and in Actin Filament Branch Junctions ». Science (New
York, N.Y.) 293 (5539): 2456‑59. https://doi.org/10.1126/science.1063025.
• Voll, Eric A., Irene M. Ogden, Janet M. Pavese, XiaoKe Huang, Li Xu, Borko D.
Jovanovic, et Raymond C. Bergan. 2014. « Heat Shock Protein 27 Regulates Human
Prostate Cancer Cell Motility and Metastatic Progression ». Oncotarget 5 (9): 2648‑63.
https://doi.org/10.18632/oncotarget.1917.
• Walck-Shannon, Elise, et Jeff Hardin. 2014. « Cell Intercalation from Top to
Bottom ». Nature Reviews Molecular Cell Biology 15 (1): 34‑48.
https://doi.org/10.1038/nrm3723.
• Walker, Andrea, Hua Su, Mary Anne Conti, Nicole Harb, Robert S. Adelstein, et
Noboru Sato. 2010. « Non-Muscle Myosin II Regulates Survival Threshold of Pluripotent
Stem Cells ». Nature Communications 1 (septembre): 71.
https://doi.org/10.1038/ncomms1074.
• Wang, Hua-Qin, Hai-Mei Liu, Hai-Yan Zhang, Yifu Guan, et Zhen-Xian Du. 2008.
« Transcriptional Upregulation of BAG3 upon Proteasome Inhibition ». Biochemical and
Biophysical Research Communications 365 (2): 381‑85.
https://doi.org/10.1016/j.bbrc.2007.11.001.
• Wargacki, Megan M., Jessica C. Tay, Eric G. Muller, Charles L. Asbury, et Trisha
N. Davis. 2010. « Kip3, the Yeast Kinesin-8, Is Required for Clustering of Kinetochores
at Metaphase ». Cell Cycle (Georgetown, Tex.) 9 (13): 2581‑88.
https://doi.org/10.4161/cc.9.13.12076.
• Watanabe, N., P. Madaule, T. Reid, T. Ishizaki, G. Watanabe, A. Kakizuka, Y.
Saito, K. Nakao, B. M. Jockusch, et S. Narumiya. 1997. « P140mDia, a Mammalian
Homolog of Drosophila Diaphanous, Is a Target Protein for Rho Small GTPase and Is a
Ligand for Profilin ». The EMBO Journal 16 (11): 3044‑56.
https://doi.org/10.1093/emboj/16.11.3044.
• Watanabe, Sadanori, Yoshikazu Ando, Shingo Yasuda, Hiroshi Hosoya, Naoki
Watanabe, Toshimasa Ishizaki, et Shuh Narumiya. 2008. « MDia2 Induces the Actin
Scaffold for the Contractile Ring and Stabilizes Its Position during Cytokinesis in NIH 3T3
Cells ». Molecular Biology of the Cell 19 (5): 2328‑38. https://doi.org/10.1091/mbc.E07-

252
10-1086.
• Weaver, A. M., A. V. Karginov, A. W. Kinley, S. A. Weed, Y. Li, J. T. Parsons, et
J. A. Cooper. 2001. « Cortactin Promotes and Stabilizes Arp2/3-Induced Actin Filament
Network Formation ». Current Biology: CB 11 (5): 370‑74.
• Weed, S. A., A. V. Karginov, D. A. Schafer, A. M. Weaver, A. W. Kinley, J. A.
Cooper, et J. T. Parsons. 2000. « Cortactin Localization to Sites of Actin Assembly in
Lamellipodia Requires Interactions with F-Actin and the Arp2/3 Complex ». The Journal
of Cell Biology 151 (1): 29‑40.
• Wei, Li, Tsung-Ta Liu, Hsiu-Huan Wang, Hui-Mei Hong, Alice L. Yu, Hsiang-Pu
Feng, et Wen-Wei Chang. 2011. « Hsp27 Participates in the Maintenance of Breast Cancer
Stem Cells through Regulation of Epithelial-Mesenchymal Transition and Nuclear Factor-
ΚB ». Breast Cancer Research: BCR 13 (5): R101. https://doi.org/10.1186/bcr3042.
• Welch, Matthew D., et R. Dyche Mullins. 2002. « Cellular Control of Actin
Nucleation ». Annual Review of Cell and Developmental Biology 18: 247‑88.
https://doi.org/10.1146/annurev.cellbio.18.040202.112133.
• Werner, Achim, Andrea Disanza, Nina Reifenberger, Gregor Habeck, Janina
Becker, Matthew Calabrese, Henning Urlaub, et al. 2013. « SCFFbxw5 Mediates Transient
Degradation of Actin Remodeller Eps8 to Allow Proper Mitotic Progression ». Nature Cell
Biology 15 (2): 179‑88. https://doi.org/10.1038/ncb2661.
• West, Robert R., Terra Malmstrom, et J. Richard McIntosh. 2002. « Kinesins
Klp5(+) and Klp6(+) Are Required for Normal Chromosome Movement in Mitosis ».
Journal of Cell Science 115 (Pt 5): 931‑40.
• Wheatley, S. P., E. H. Hinchcliffe, M. Glotzer, A. A. Hyman, G. Sluder, et Y. l
Wang. 1997. « CDK1 Inactivation Regulates Anaphase Spindle Dynamics and Cytokinesis
in Vivo ». The Journal of Cell Biology 138 (2): 385‑93.
• Whittaker, M., E. M. Wilson-Kubalek, J. E. Smith, L. Faust, R. A. Milligan, et H.
L. Sweeney. 1995. « A 35-A Movement of Smooth Muscle Myosin on ADP Release ».
Nature 378 (6558): 748‑51. https://doi.org/10.1038/378748a0.
• Wilhelmus, Micha M. M., Wilbert C. Boelens, Irene Otte-Höller, Bram Kamps,
Benno Kusters, Marion L. C. Maat-Schieman, Robert M. W. de Waal, et Marcel M.

253
Verbeek. 2006. « Small Heat Shock Protein HspB8: Its Distribution in Alzheimer’s Disease
Brains and Its Inhibition of Amyloid-Beta Protein Aggregation and Cerebrovascular
Amyloid-Beta Toxicity ». Acta Neuropathologica 111 (2): 139‑49.
https://doi.org/10.1007/s00401-005-0030-z.
• Wilson, Cyrus A., Mark A. Tsuchida, Greg M. Allen, Erin L. Barnhart, Kathryn T.
Applegate, Patricia T. Yam, Lin Ji, Kinneret Keren, Gaudenz Danuser, et Julie A. Theriot.
2010. « Myosin II contributes to cell-scale actin network treadmilling via network
disassembly ». Nature 465 (7296): 373‑77. https://doi.org/10.1038/nature08994.
• Wilson, Gayle M., Andrew B. Fielding, Glenn C. Simon, Xinzi Yu, Paul D.
Andrews, Rebecca S. Hames, Andrew M. Frey, Andrew A. Peden, Gwyn W. Gould, et
Rytis Prekeris. 2005. « The FIP3-Rab11 Protein Complex Regulates Recycling Endosome
Targeting to the Cleavage Furrow during Late Cytokinesis ». Molecular Biology of the Cell
16 (2): 849‑60. https://doi.org/10.1091/mbc.E04-10-0927.
• Winitthana, Thidarat, Somsong Lawanprasert, et Pithi Chanvorachote. 2014.
« Triclosan Potentiates Epithelial-To-Mesenchymal Transition in Anoikis-Resistant
Human Lung Cancer Cells ». PLOS ONE 9 (10): e110851.
https://doi.org/10.1371/journal.pone.0110851.
• Winter, D. C., E. Y. Choe, et R. Li. 1999. « Genetic Dissection of the Budding
Yeast Arp2/3 Complex: A Comparison of the in Vivo and Structural Roles of Individual
Subunits ». Proceedings of the National Academy of Sciences of the United States of
America 96 (13): 7288‑93.
• Wolff, Suzanne, Jonathan S. Weissman, et Andrew Dillin. 2014. « Differential
Scales of Protein Quality Control ». Cell 157 (1): 52‑64.
https://doi.org/10.1016/j.cell.2014.03.007.
• Wong, Raymond, Irene Hadjiyanni, Ho-Chun Wei, Gordon Polevoy, Rachel
McBride, Kai-Ping Sem, et Julie A. Brill. 2005. « PIP2 Hydrolysis and Calcium Release
Are Required for Cytokinesis in Drosophila Spermatocytes ». Current Biology 15 (15):
1401‑6. https://doi.org/10.1016/j.cub.2005.06.060.
• Wooten, Marie W., Thangiah Geetha, J. Ramesh Babu, M. Lamar Seibenhener,
Junmin Peng, Nancy Cox, Maria-T. Diaz-Meco, et Jorge Moscat. 2008. « Essential Role

254
of Sequestosome 1/P62 in Regulating Accumulation of Lys63-Ubiquitinated Proteins ».
The Journal of Biological Chemistry 283 (11): 6783‑89.
https://doi.org/10.1074/jbc.M709496200.
• Wu, H., et J. T. Parsons. 1993. « Cortactin, an 80/85-Kilodalton Pp60src Substrate,
Is a Filamentous Actin-Binding Protein Enriched in the Cell Cortex ». The Journal of Cell
Biology 120 (6): 1417‑26.
• Wu, H., A. B. Reynolds, S. B. Kanner, R. R. Vines, et J. T. Parsons. 1991.
« Identification and Characterization of a Novel Cytoskeleton-Associated Pp60src
Substrate ». Molecular and Cellular Biology 11 (10): 5113‑24.
• Wu, Huo, Xiaoqian Jing, Xi Cheng, Yonggang He, Lei Hu, Haoxuan Wu, Feng Ye,
et Ren Zhao. 2016. « Asporin Enhances Colorectal Cancer Metastasis through Activating
the EGFR/Src/Cortactin Signaling Pathway ». Oncotarget 7 (45): 73402‑13.
https://doi.org/10.18632/oncotarget.12336.
• Wurzer, Bettina, Gabriele Zaffagnini, Dorotea Fracchiolla, Eleonora Turco,
Christine Abert, Julia Romanov, et Sascha Martens. 2015. « Oligomerization of P62
Allows for Selection of Ubiquitinated Cargo and Isolation Membrane during Selective
Autophagy ». ELife 4 (septembre): e08941. https://doi.org/10.7554/eLife.08941.
• Xiao, Heng, Shaobing Cheng, Rongliang Tong, Zheng Lv, Chaofeng Ding, Chengli
Du, Haiyang Xie, Lin Zhou, Jian Wu, et Shusen Zheng. 2014. « BAG3 Regulates
Epithelial-Mesenchymal Transition and Angiogenesis in Human Hepatocellular
Carcinoma ». Laboratory Investigation; a Journal of Technical Methods and Pathology 94
(3): 252‑61. https://doi.org/10.1038/labinvest.2013.151.
• Xiong, Huajiang, William A. Mohler, et Martha C. Soto. 2011. « The Branched
Actin Nucleator Arp2/3 Promotes Nuclear Migrations and Cell Polarity in the C. Elegans
Zygote ». Developmental Biology 357 (2): 356‑69.
https://doi.org/10.1016/j.ydbio.2011.07.008.
• Xu, L., S. Chen, et R. C. Bergan. 2006. « MAPKAPK2 and HSP27 Are
Downstream Effectors of P38 MAP Kinase-Mediated Matrix Metalloproteinase Type 2
Activation and Cell Invasion in Human Prostate Cancer ». Oncogene 25 (21): 2987‑98.
https://doi.org/10.1038/sj.onc.1209337.

255
• Xu, Zhe, Kourtney Graham, Molly Foote, Fengshan Liang, Raed Rizkallah, Myra
Hurt, Yanchang Wang, Yuying Wu, et Yi Zhou. 2013. « 14-3-3 Protein Targets Misfolded
Chaperone-Associated Proteins to Aggresomes ». Journal of Cell Science 126 (Pt 18):
4173‑86. https://doi.org/10.1242/jcs.126102.
• Yan, Jin, Michael Lamar Seibenhener, Luis Calderilla-Barbosa, Maria-Theresa
Diaz-Meco, Jorge Moscat, Jianxiong Jiang, Marie W. Wooten, et Michael C. Wooten.
2013. « SQSTM1/P62 Interacts with HDAC6 and Regulates Deacetylase Activity ». PloS
One 8 (9): e76016. https://doi.org/10.1371/journal.pone.0076016.
• Yang, Changsong, Lubov Czech, Silke Gerboth, Shin-ichiro Kojima, Giorgio Scita,
et Tatyana Svitkina. 2007. « Novel Roles of Formin mDia2 in Lamellipodia and Filopodia
Formation in Motile Cells ». PLOS Biology 5 (11): e317.
https://doi.org/10.1371/journal.pbio.0050317.
• Yang, Dong, Ji Zhou, Hao Wang, Yutao Wang, Ge Yang, et Yundong Zhang. 2016.
« High Expression of BAG3 Predicts a Poor Prognosis in Human Medulloblastoma ».
Tumour Biology: The Journal of the International Society for Oncodevelopmental Biology
and Medicine 37 (10): 13215‑24. https://doi.org/10.1007/s13277-016-5197-5.
• Ylikallio, Emil, Svetlana Konovalova, Yogesh Dhungana, Taru Hilander, Nella
Junna, Juhani V. Partanen, Jussi P. Toppila, Mari Auranen, et Henna Tyynismaa. 2015.
« Truncated HSPB1 Causes Axonal Neuropathy and Impairs Tolerance to Unfolded
Protein Stress ». BBA Clinical 3 (juin): 233‑42.
https://doi.org/10.1016/j.bbacli.2015.03.002.
• Yu, Y. X., A. Heller, T. Liehr, C. C. Smith, et L. Aurelian. 2001. « Expression
Analysis and Chromosome Location of a Novel Gene (H11) Associated with the Growth
of Human Melanoma Cells ». International Journal of Oncology 18 (5): 905‑11.
• Yuan, Jie, Fei Zhang, et Ruifang Niu. 2015. « Multiple Regulation Pathways and
Pivotal Biological Functions of STAT3 in Cancer ». Scientific Reports 5 (décembre):
17663. https://doi.org/10.1038/srep17663.
• Yüce, Ozlem, Alisa Piekny, et Michael Glotzer. 2005. « An ECT2-Centralspindlin
Complex Regulates the Localization and Function of RhoA ». The Journal of Cell Biology
170 (4): 571‑82. https://doi.org/10.1083/jcb.200501097.

256
• Yumura, Shigehiko. 2001. « Myosin II Dynamics and Cortical Flow during
Contractile Ring Formation in Dictyostelium Cells ». The Journal of Cell Biology 154 (1):
137‑46. https://doi.org/10.1083/jcb.200011013.
• Yunoki, Tatsuya, Yoshiaki Tabuchi, Atsushi Hayashi, et Takashi Kondo. 2015.
« BAG3 Protects against Hyperthermic Stress by Modulating NF-ΚB and ERK Activities
in Human Retinoblastoma Cells ». Graefe’s Archive for Clinical and Experimental
Ophthalmology = Albrecht Von Graefes Archiv Fur Klinische Und Experimentelle
Ophthalmologie 253 (3): 399‑407. https://doi.org/10.1007/s00417-014-2874-1.
• Zaffagnini, Gabriele, et Sascha Martens. 2016. « Mechanisms of Selective
Autophagy ». Journal of Molecular Biology 428 (9 Pt A): 1714‑24.
https://doi.org/10.1016/j.jmb.2016.02.004.
• Zanin, Esther, Arshad Desai, Ina Poser, Yusuke Toyoda, Cordula Andree, Claudia
Moebius, Marc Bickle, Barbara Conradt, Alisa Piekny, et Karen Oegema. 2013. « A
Conserved RhoGAP Limits M-phase Contractility and Coordinates with Microtubule
Asters to Restrict Active RhoA to the Cell Equator During Cytokinesis ». Developmental
cell 26 (5): 496‑510. https://doi.org/10.1016/j.devcel.2013.08.005.
• Zhang, Jiankai, Zhangyou He, Wenjian Xiao, Qingqing Na, Tianxiu Wu, Kaixin
Su, et Xiaojun Cui. 2016. « Overexpression of BAG3 Attenuates Hypoxia-Induced
Cardiomyocyte Apoptosis by Inducing Autophagy ». Cellular Physiology and
Biochemistry: International Journal of Experimental Cellular Physiology, Biochemistry,
and Pharmacology 39 (2): 491‑500. https://doi.org/10.1159/000445641.
• Zhang, Linlin, Ningning Liu, Songbo Xie, Xianfei He, Jun Zhou, Min Liu, et
Dengwen Li. 2014. « HDAC6 Regulates Neuroblastoma Cell Migration and May Play a
Role in the Invasion Process ». Cancer Biology & Therapy 15 (11): 1561‑70.
https://doi.org/10.4161/15384047.2014.956632.
• Zhang, Linlin, Shanshan Liu, Ningning Liu, Yong Zhang, Min Liu, Dengwen Li,
Edward Seto, Tso-Pang Yao, Wenqing Shui, et Jun Zhou. 2015. « Proteomic Identification
and Functional Characterization of MYH9, Hsc70, and DNAJA1 as Novel Substrates of
HDAC6 Deacetylase Activity ». Protein & Cell 6 (1): 42‑54.
https://doi.org/10.1007/s13238-014-0102-8.

257
• Zhang, Xiaohong, Zhigang Yuan, Yingtao Zhang, Sarah Yong, Alexis Salas-
Burgos, John Koomen, Nancy Olashaw, et al. 2007. « HDAC6 Modulates Cell Motility by
Altering the Acetylation Level of Cortactin ». Molecular Cell 27 (2): 197‑213.
https://doi.org/10.1016/j.molcel.2007.05.033.
• Zhang, Xiaoying, Ulka Shrikhande, Bethany M. Alicie, Qing Zhou, et Robert L.
Geahlen. 2009. « Role of the Protein Tyrosine Kinase Syk in Regulating Cell-Cell
Adhesion and Motility in Breast Cancer Cells ». Molecular Cancer Research: MCR 7 (5):
634‑44. https://doi.org/10.1158/1541-7786.MCR-08-0371.
• Zhang, Yong, Jian-Hua Wang, Qiang Lu, et Yun-Jie Wang. 2012. « Bag3 Promotes
Resistance to Apoptosis through Bcl-2 Family Members in Non-Small Cell Lung Cancer ».
Oncology Reports 27 (1): 109‑13. https://doi.org/10.3892/or.2011.1486.
• Zhao, Wei-meng, et Guowei Fang. 2005. « MgcRacGAP Controls the Assembly of
the Contractile Ring and the Initiation of Cytokinesis ». Proceedings of the National
Academy of Sciences of the United States of America 102 (37): 13158‑63.
https://doi.org/10.1073/pnas.0504145102.
• Zhong, Zhi-Sheng, Li-Jun Huo, Cheng-Guang Liang, Da-Yuan Chen, et Qing-Yuan
Sun. 2005. « Small GTPase RhoA Is Required for Ooplasmic Segregation and Spindle
Rotation, but Not for Spindle Organization and Chromosome Separation during Mouse
Oocyte Maturation, Fertilization, and Early Cleavage ». Molecular Reproduction and
Development 71 (2): 256‑61. https://doi.org/10.1002/mrd.20253.
• Zhou, Kang, Kaelyn D. Sumigray, et Terry Lechler. 2015. « The Arp2/3 Complex
Has Essential Roles in Vesicle Trafficking and Transcytosis in the Mammalian Small
Intestine ». Molecular Biology of the Cell 26 (11): 1995‑2004.
https://doi.org/10.1091/mbc.E14-10-1481.
• Zhou, M., H. Lambert, et J. Landry. 1993. « Transient Activation of a Distinct
Serine Protein Kinase Is Responsible for 27-KDa Heat Shock Protein Phosphorylation in
Mitogen-Stimulated and Heat-Shocked Cells ». The Journal of Biological Chemistry 268
(1): 35‑43.
• Zhou, Qiuhu, Xiaodong Shi, Kaiming Zhang, Chao Shi, Lixin Huang, et Zhenzhan
Chang. 2016. « The Function of Ile-X-Ile Motif in the Oligomerization and Chaperone-

258
Like Activity of Small Heat Shock Protein AgsA at Room Temperature ». The Protein
Journal 35 (6): 401‑6. https://doi.org/10.1007/s10930-016-9681-y.
• Zhu, Haitao, Dongqing Wang, Lirong Zhang, Xiaodong Xie, Yingying Wu,
Yanfang Liu, Genbao Shao, et Zhaoliang Su. 2014. « Upregulation of Autophagy by
Hypoxia-Inducible Factor-1α Promotes EMT and Metastatic Ability of CD133+ Pancreatic
Cancer Stem-like Cells during Intermittent Hypoxia ». Oncology Reports 32 (3): 935‑42.
https://doi.org/10.3892/or.2014.3298.
• Zhu, Huayuan, Wei Wu, Yuan Fu, Wenyi Shen, Kourong Miao, Min Hong, Wei
Xu, Ken H. Young, Peng Liu, et Jianyong Li. 2014. « Overexpressed BAG3 Is a Potential
Therapeutic Target in Chronic Lymphocytic Leukemia ». Annals of Hematology 93 (3):
425‑35. https://doi.org/10.1007/s00277-013-1883-1.
• Zientara-Rytter, Katarzyna, et Suresh Subramani. 2016a. « Autophagic
Degradation of Peroxisomes in Mammals ». Biochemical Society Transactions 44 (2):
431‑40. https://doi.org/10.1042/BST20150268.
• ———. 2016b. « Role of Actin in Shaping Autophagosomes ». Autophagy 12 (12):
2512‑15. https://doi.org/10.1080/15548627.2016.1236877.
• Zumdieck, Alexander, Karsten Kruse, Henrik Bringmann, Anthony A. Hyman, et
Frank Jülicher. 2007. « Stress Generation and Filament Turnover during Actin Ring
Constriction ». PloS One 2 (8): e696. https://doi.org/10.1371/journal.pone.0000696.
• Zuo, Qinqin, Wenjing Wu, Xu Li, Le Zhao, et Wei Chen. 2012. « HDAC6 and
SIRT2 Promote Bladder Cancer Cell Migration and Invasion by Targeting Cortactin ».
Oncology Reports 27 (3): 819‑24. https://doi.org/10.3892/or.2011.1553.
• Żwirowski, Szymon, Agnieszka Kłosowska, Igor Obuchowski, Nadinath B.
Nillegoda, Artur Piróg, Szymon Ziętkiewicz, Bernd Bukau, Axel Mogk, et Krzysztof
Liberek. 2017. « Hsp70 Displaces Small Heat Shock Proteins from Aggregates to Initiate
Protein Refolding ». The EMBO Journal 36 (6): 783‑96.
https://doi.org/10.15252/embj.201593378.
•

259
8. Annexes
8.1. Annexe 1

260

261

262

263

264

265

266

267

268

269

270

271

272

273

274

275

276

277

278

279

280

281

282

283

284

285

286

287

288

289

290

291
8.2. Annexe 2

292

293

294

295

296

297

298

299

300

301

302

303

304

305

306

307

308

309

310

311

312

313
8.3. Annexe 3
SMALL HEAT SHOCK PROTEINS
Fine-tuning of actin dynamics by the HSPB8-BAG3 chaperonecomplex facilitates cytokinesis and contributes to its impacton cell division
Alice Anaïs Varlet1,2 & Margit Fuchs1,2 & Carole Luthold1,2 &
Herman Lambert1,2 & Jacques Landry1,2,3 & Josée N. Lavoie1,2,3
Received: 16 January 2017 /Revised: 13 February 2017 /Accepted: 14 February 2017 /Published online: 8 March 2017# Cell Stress Society International 2017
Abstract The small heat shock protein HSPB8 and its co-chaperone BAG3 are proposed to regulate cytoskeletalproteostasis in response to mechanical signaling in musclecells. Here, we show that in dividing cells, the HSPB8-BAG3 complex is instrumental to the accurate disassemblyof the actin-based contractile ring during cytokinesis, a pro-cess required to allow abscission of daughter cells. Silencingof HSPB8 markedly decreased the mitotic levels of BAG3 inHeLa cells, supporting its crucial role in BAG3 mitotic func-tions. Cells depleted of HSPB8 were delayed in cytokinesis,remained connected via a disorganized intercellular bridge,and exhibited increased incidence of nuclear abnormalitiesthat result from failed cytokinesis (i.e., bi- and multi-nucle-ation). Such phenotypes were associated with abnormal accu-mulation of F-actin at the intercellular bridge of daughter cellsat telophase. Remarkably, the actin sequestering druglatrunculin A, like the inhibitor of branched actin polymeriza-tion CK666, normalized F-actin during cytokinesis and re-stored proper cell division in HSPB8-depleted cells, implicat-ing deregulated actin dynamics as a cause of abscission fail-ure. Moreover, this HSPB8-dependent phenotype could becorrected by rapamycin, an autophagy-promoting drug,
whereas it was mimicked by drugs impairing lysosomal func-tion. Together, the results further support a role for theHSPB8-BAG3 chaperone complex in quality control ofactin-based structure dynamics that are put under high tension,notably during cell cytokinesis. They expand a so-far under-appreciated connection between selective autophagy and cel-lular morphodynamics that guide cell division.
Keywords Cytokinesis . BAG3 .HSPB8 .Actin . Autophagy
Introduction
Fine-tuned protein quality control (PQC) systems are governedby molecular chaperones of the heat shock protein (HSP) fam-ily. Chaperones of the small HSP family (HSPB) form aneclectic family of proteins, which differ in their expressionprofiles and biochemical properties (Bakthisaran et al. 2015).Nonetheless, they are proposed to act mainly as ATPase-independent holdase or unfoldase by virtue of their dynamicquaternary structure, to segregate damaged proteins and pre-vent their irreversible aggregation (Finka et al. 2016; Haslbeckand Vierling 2015). In addition to their cytoprotective functionunder proteotoxic stress conditions, they appear to be instru-mental to the normal functioning of complex proteinous struc-tures of the cytoskeleton during biological processes relying onacute cellular remodeling (Guilbert et al. 2015). For instance, aphosphorylation-regulated function of HSPB1 has been report-ed, which may regulate actin polymerization and cancer cellmigration (During et al. 2007; Kostenko and Moens 2009;Lavoie et al. 1993). Furthermore, HSPB8 has been involvedin the recognition of damaged filamin, an actin-binding pro-tein, during muscle cell contraction and its segregation fordegradation by selective autophagy (Ulbricht et al. 2013).The later mechanism is proposed to maintain muscle fiber
Electronic supplementary material The online version of this article(doi:10.1007/s12192-017-0780-2) contains supplementary material,which is available to authorized users.
* Josée N. [email protected]
1 Centre de recherche sur le cancer de l Université Laval,Québec, Canada
2 Oncology, Centre de recherche du CHU deQuébec-Université Laval,Québec G1R 3S3, Canada
3 Département de Biologie Moléculaire, Biochimie Médicale etPathologie Université Laval, Québec G1V OA6, Canada
Cell Stress and Chaperones (2017) 22:553–567DOI 10.1007/s12192-017-0780-2

314
integrity by promoting clearance of actin-binding proteins un-dergoing partial misfolding in response to mechanical strain.Expectedly, several HSPB chaperones are up-regulated in can-cer cells where they could contribute to the fitness of actin-based structures that support the tumor phenotype. Thus, byenabling targeted protein storage, recycling, or degradation,HSPB proteins would play important roles in cytoskeletalPQC, to maintain the integrity of cytoskeletal structures duringstress, as well as to facilitate cellular morphodynamics thatguide cell division and differentiation. In line with this concept,we have recently uncovered a novel role for HSPB8 in mitoticcell remodeling (Fuchs et al. 2015).
In spite of their known implications in human diseases,such as cancer, the mechanisms underlying the contributionof HSPB proteins to cytoskeletal dynamics/proteostasis arepoorly understood. In that regard, the co-chaperone and scaf-fold protein BCL2-associated athanogene 3 (BAG3) is ofparticular interest. BAG3 can associate with several HSPBproteins but show a preferred association with and regulatethe stability of HSPB8 (Carra et al. 2008; Fuchs et al. 2010;Taipale et al. 2014). In the context of cancer cells, BAG3 isviewed as a molecular scaffold owing to its interactions withsignaling proteins and several regulators of actin dynamicsand its implication in key biological processes, including ap-optosis, development, cell motility, and cell division (Behl2016; Rosati et al. 2011). Yet, the significance of distinctchaperone partners for BAG3 functions in intracellular signal-ing has remained unexplored. BAG3 also acts as a co-chaperone for HSC70/HSP70 chaperone system and modu-lates its function in proteasomal degradation, aggrephagy, andsignaling (Chen et al. 2013; Colvin et al. 2014; Gamerdingeret al. 2009; Gamerdinger et al. 2011; Minoia et al. 2014).Accumulating evidence suggests that BAG3 serves to bridgeimportant components of the chaperone network (Rauch et al.2017). Nonetheless, BAG3 may function without recruitingHSPB8 in aggrephagy (Minoia et al. 2014). In contrast, itsinteraction with HSPB8 seems to be instrumental in typifyingBAG3 functions in cytoskeletal remodeling. This is supportedby our recent findings that BAG3 regulates remodeling of themitotic actin-based structures that guide spindle orientation ina manner that requires binding to HSPB8, but not to HSC70/HSP70 (Fuchs et al. 2015). Because the same mitotic pheno-type can be recapitulated by depletion of HSPB8, BAG3, orthe autophagic receptor p62/SQSTM1, which shows en-hancedmitotic association with BAG3, we have proposed thatthey act within a so-far unrecognized cytoskeletal PQC mech-anism during mitosis. Evidence suggests that targeted cyto-skeletal protein degradation can provide a mechanism to limitsignaling pathways in time and space during mitotic cell re-modeling (Belaid et al. 2013; Werner et al. 2013). Little isknown, however, on the mechanism of quality control autoph-agy that provides Bbuilt in^ selectivity for components of cy-toskeletal remodeling pathways. Whether the HSPB8-BAG3
chaperone complex may contribute for such mechanism dur-ing mitosis remains to be demonstrated.
Here, we investigate the impact of HSPB8 in cytoskeletalPQC during cytokinesis, a process that is orchestrated byhighly organized actin-based structures that are put under hightensile forces. We show that HSPB8 regulates BAG3 levelsduring mitosis and the accurate dynamics of the cytokineticactin ring, via a mechanism that may involve effects onbranched actin nucleation and autophagy signaling.Importantly, strong evidence is provided that first linkHSPB8 function in cytoskeletal PQC to its modulation ofmitotic cell division in cancer cells.
Material and methods
Cell culture, synchronization, drug treatments, and siRNAexperiments
Parental HeLa cells, HeLa-GFP-H2B cells (gift fromLaurence Pelletier), and HeLa-RFP-H2B cells (gift fromSabine Elowe) (Klebig et al. 2009) were maintained in α-minimal essential medium supplemented with 10% fetal bo-vine serum. All cell lines were grown in a humidified atmo-sphere with 5% CO2 at 37 °C. Cells were synchronized by adouble thymidine block and were released for 8 to 10 h infresh medium, as described previously (Fuchs et al. 2015).For knockdown experiments, HeLa cells were transfectedwith 25 to 50 nM siRNA duplexes in calcium-phosphatetransfection buffer (125 mM MgCl2, 140 mM NaCl, 25 mMHEPES, 0.75 mM Na2HPO4) overnight. Analyses were per-formed 48 to 88 h later byWestern blot, immunofluorescence,or live cell imaging. For cell cycle analyses of the impact ofHSPB8-specific siRNAs on BAG3 protein levels, HeLa cellswere synchronized in G2/M or M-phase by a 16-h treatmentwith the selective CDK1 inhibitor (RO3306, 8 μM) or withnocodazole (400 ng/ml), respectively, and mitotic cells wererecovered by a mitotic shake-off, as described before (Fuchset al. 2015). Cells were lysed by 3 cycles of freezing/thawingin 4 volumes of lysis buffer (20 mM TRIS-HCl pH 7.6,150 mM NaCl, 1 mM EDTA, 0.1% IGEPAL, 1 mMNaVO4, 10 mM NaF, 40 mM β-glycerophosphate, 1XComplete [Roche], 1 mM DTT), centrifuged at 15,000g for15 min, and the supernatants were processed for Western blotanalyses. The following drugs were added to cells that havebeen synchronized in mitosis with a double thymidine block,during the last hour of the second release period before cellfixation as followed: latrunculin A, 20 nM; CK666, 40 μM;rapamycin, 150 nM; E-64D and pepstatin A, 10 μg/ml;bafilomycin A1, 200 nM. For multi-nucleation assays, unsyn-chronized HeLa-GFP-H2B cells were grown in the presenceof 1 nM latrunculin A for 48 h before cell fixation. The siRNAduplexes were based on human sequences and were purchased
554 A. A. Varlet et al.

315
from Qiagen (HPP grade siRNA) or Thermo Fisher Scientific(standard A4 grade). Sequences of the sense strands are asfollows: siBAG3 #1: CGAAGAGTATTTGACCAAA-3′;
siBAG3 #2: 5′-GCAAAGAGGTGGATTCTAA-3′;siHSPB8 #1: 5′-CAGATAGGCTAGTGGTATT-3′;siHSPB8 #2: 5′-GCAGTGAATGCAAGGGTTATT-3′;siHSPB8 #3: 5′-CAGAGGAGTTGATGGTGAATT-3′.si AllStars Negative Control, target sequence: 5′-CAGG
GTATCGACGATTACAAA-3′.
Transfection, infection, adenovirus, and vectors
HeLa cells were seeded on fibronectin-coated dishes and weresynchronized by the double thymidine block method. Duringthe second release period, cells were transfected with GFP-Arp3 (human Arp3 in pEGFP-N1 vector, gift fromDr John A.Cooper, ST Louis, USA) (Welch et al. 1997) usingLipofectamine 2000 at the ratio Lipo/DNA of 2:1 and follow-ing the manufacturer’s instructions (Invitrogen) for a 10-hperiod before cell fixation. To follow the dynamics of actin,cells were seeded on fibronectin-coated glass dishes (MatTekCorp.) and synchronized by the double thymidine block meth-od. Adenofection was performed during the first thymidineblock using Ad-LifeAct-TagGFP2 (60121, IBIDI) at a multi-plicity of infection (MOI) of 1 plaque-forming units per cell(pfu/cell), BacMam 2.0 reagents at 4 pfu/cell in α-MEM-mi-nus, and siRNAs in calcium-phosphate transfection buffer,using the method described in Fuchs et al. (2016). Sixteenhours after adenofection, cells were washed 3 times withHEPES (6.7 mM KCl, 150 mM NaCl, 10 mM HEPES,pH 7.3) and were released in fresh medium for 8 h beforeadding back 2 mM thymidine.
Antibodies and chemicals
The following antibodies were used forWestern blot analyses:rabbit anti-BAG3 LP11 was raised against full length recom-binant human BAG3 fused with glutathione S transferase(LP11) (Fuchs et al. 2015), rabbit anti-HSPB8 against a C-terminal peptide (NELPQDSQEVTCT) (Carra et al. 2008);anti-Vinculin (V9131) was from Sigma-Aldrich; anti-Arp3(ab49671) was from Abcam; anti-Calreticulin (#612136)was from BD Biosciences; and anti-GAPDH Clone 6C5(Fitzgerald, #10R-G109a) was from Millipore. Secondary an-tibodies (anti-mouse-HRP 115-035-146 and anti-rabbit-HRP111-055-003) were from Jackson ImmunoResearch. The fol-lowing antibodies were used for immunofluorescence: anti-Aurora B (ab2254) was from Abcam; Alexa-Fluor® 488Phalloidin (A12379) and Texas Red®-X Phalloidin (T-7471)were fromMolecular Probes/Thermo Fisher. Goat-anti-rabbit-594 (A11037) was from Molecular Probes. HoechstBisbenzimide H 33342 (B2261), thymidine (T9250), fibro-nectin (F1141-5 mg), RO-3306 (SML0569), E-64D
(E8640), pepstatin A (P5318), bafilomycin A1 (B1793), andCK666 (SML0006) were from Sigma-Aldrich; latrunculin A(428021-100UG) was from Calbiochem.
Immunofluorescence and live-cell imaging
Cells were gently washed once with LUFTIG (0.2 M sucrose,35 mMPipes, pH 7.4, 5 mMEGTA, 5 mMMgSO4) and fixedwith 3.7% formaldehyde in LUFTIG or 4% PFA in PBS(137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.47 mMKH2PO4) for 20 min at room temperature. DNAwas stainedwith cell permeable Hoechst (150 ng/ml). Specimens wereprocessed for immunofluorescence as described (Lavoieet al. 2000) and were post-fixed with 3.7% formaldehyde inPBS for 20 min at room temperature. Phenotypes were mon-itored routinely by at least two independent investigators byvisual inspection of fixed specimens and were scored in amanner blind to treatment, using an AxioObserver Z1 systemusing a 60 × 1.25NA objective and a charged-coupled device(CCD) camera AxiocamMRm controlled by the Zen software(Carl Zeiss). Confocal microscopy in live cells and fixed sam-ples was performed with a PerkinElmer Life ScienceUltraview Spinning Disk Confocal microscope, equippedwith a 40 × 0.75NA objective, an EMCCD cooled charge-coupled camera at −50 °C (Hamamatsu Photonics K.K), anddriven by the Volocity software version 6.01. The system wasequipped with a humidified/5% CO2/thermoregulated cham-ber. Bi- and multi-nucleation was scored as the number ofcells presenting 2 or >2 nuclei per cell; single cells were de-lineated by staining of the cortex with F-actin. To quantifyabnormal F-actin accumulation at the intercellular bridge ofdaughter cells, we scored the number of daughter cells con-nected by a bridge labeled by Aurora B staining that hadcompletely de-condensed DNA (late telophase) and abnormalF-actin structures at the bridge.
Long-term live-cell imaging was performed using a NikonTE-2000 inverted microscope equipped with CO2/thermo-regulated chamber with a 40 × 0.6 NA objective. Images werecaptured as 16-bit TIFF files with a photometrix Collsnap FXcooled CCD camera (−30 °C) (Rooper Scientific, Tuscon AZ)driven by the Metaview software version 4.5 (UniversalImaging Corp., Downington, PA). Images were acquired at90 s intervals for a 48-h period or at 10 min intervals for a72-h period, starting at 40 h after siRNAs transfection.Phenotypes associated with cytokinesis were estimated by vi-sual inspection of time sequences. Spreading of daughter cellsand disappearance of the intercellular bridge were evaluatedby monitoring the time spent between closure of the actin ringand cell spreading and/or disappearance of the dense midbodyring, respectively. Defects in daughter cell spreading wereconsidered as follows: (1) prolonged cortex blebbing, (2) un-coordinated spreading of daughter cells, and (3) no spreading.To quantify the number of cells that re-entered mitosis
HSPB8 regulates F-actin dynamics during cytokinesis 555

316
synchronously, we determined that cells are derived from thesame mother cell by visual inspection of time sequences; cellswere considered synchronized if they entered mitosis at thesame time point or separated by less than 2 time points. Actinring (AR) thickness and width were expressed as the meanintensities from F-actin staining and were evaluated by delin-eating AR manually on confocal slices determined as the cen-ter of the cell, using the Volocity software version 6.0(Quorum Technologies); cells were considered only afterAR closure and DNA de-condensation. To quantify GFP-Arp3 recruitment, AR and daughter cells were delineatedmanually using F-actin staining as the reference for cell shapeusing the same software. The mean fluorescence intensities ofGFP-Arp3 at AR and in the cytoplasm were measured fromconfocal image stacks acquired at 0.5 μm z-steps andexpressed as a ratio of AR over cytoplasm. The Volocity soft-ware version 6.0 (Quorum Technologies) and ImageJ 1.48v(National Institute of Health) were used for processing entireimages before cropping to emphasize the main point of theimage. Processing was limited to background subtraction andbrightness/contrast adjustment.
Western blotting
Cells were lysed in SDS sample buffer (62.5 mM Tris-HCl,pH 6.8, 2.3% SDS, 10% glycerol, 5% β-mercaptoethanol,0.05% bromphenol blue, 1 mM phenylmethylsulfonyl fluo-ride); equal amounts of protein from whole cell lysates wereseparated by SDS-PAGE on 10% acrylamide gels and trans-ferred onto nitrocellulose membranes. Immunoblotting wasperformed as described (Champagne et al. 2004). Protein con-centrations were determined with DC protein assay reagent,and densitometric analyses were performed from FluorSMAX MultiImager-captured images using the QuantityOnesoftware version 4.5.0 (Bio-Rad Laboratories).
Statistical analyses
For experiments examining (a) the time spent for daughter cellsspreading or (b) AR thickness/width, the mean values from in-dividual cells from at least three independent experiments wereanalyzed using the Mann-Whitney test, which is a non-parametric test that compares two unpaired groups of ordinalor numerical variables. For experiments examining the propor-tions of cells with (a) cell spreading defects, (b) ICB defects, (c)persistent connections, (d) synchronized daughter cell mitosis,(e) nuclear abnormalities, and (f) abnormal F-actin accumulationat the ICB, data from individual experiments were analyzedusing the Fisher’s exact test, which is used for small sample sizeswith nominal/categorical variables. Statistical calculations wereperformed using Prism 6.0 (GraphPad Software) statistical soft-ware. p values <0.05 considered significant (*p < 0.05;**p < 0.01; ***p < 0.001; ****p < 0.0001).
Results
Silencing of HSPB8 or BAG3 perturbs abscissionof daughter cells
We sought to characterize the function of HSPB8-BAG3chaperone complex in the extreme cell morphodynamicchanges that guide cytokinesis. To that end, unsynchro-nized HeLa-GFP-H2B were treated with control small in-terfering RNA (siRNA), or HSPB8- or BAG3-specificsiRNAs that achieved >75% depletion of the protein inthese cells, and were imaged for a period of 72 h startingat 40 h after transfection of siRNAs (Fig. 1a). In agree-ment with our previous findings, cells depleted of HSPB8or BAG3 eventually progressed through anaphase, butexhibited higher incidence of defects during cytokinesis(Fuchs et al. 2015). Such defects were associated with twomajor phenotypes: delayed cell spreading at telophase anddelayed abscission of daughter cells. HSPB8-depletedcells showed a ∼1.3-fold increase in the mean time re-quired to achieve spreading (35 min; siHSPB8) relativeto control cells (26 min; siCtl) (Fig. 1b, c), frequentlyassociated with prolonged cortex blebbing (Fig. 1b,siBAG3 490′; supplemental movies S1–S3). This pheno-type was observed in 24 to 30% of HSPB8- or BAG3-depleted cells, respectively, as compared to 12% of thecontrol cell population (Fig. 1c). The data are consistentwith previous work involving BAG3 in modulation of celladhesion, a function that may engage its association withHSPB8 at mitotic exit (Iwasaki et al. 2007; Iwasaki et al.2010; Kassis et al. 2006).
Most remarkably, depletion of HSPB8 or BAG3 wasassociated with increased occurrence of abnormally thickand long intercellular bridges (ICB) connecting nascentdaughter cells, which persisted for longer periods of time(Fig. 1d, arrowheads; Fig. 1e; supplemental movies S4–S6).Indeed, BAG3-depleted cells showed a ∼1.7-fold increasein the mean time required for ICB disappearance relative tocontrol cells (Fig. 1e). Moreover, a larger proportion ofHSPB8-depleted cells remained connected by an ICB com-prising a typical midbody ring (Fig. 2a, red arrowhead andinset) and even re-entered mitosis synchronously (Fig. 2b,c). These phenotypes are typical of cells that fail abscissionand were also observed in BAG3-depleted cells (not shown)(Belaid et al. 2013; Schiel et al. 2012). In further support, asignificant proportion of cells transfected with two distinctHSPB8-specific siRNA sequences exhibited bi-nucleation,a defect that arises upon cytokinetic failure and collapsingback into a single bi-nucleated cell, which may be due toimpairment of abscission (Fig. 2d, e). Together, these ob-servations suggest that HSPB8, in complex with BAG3,facilitates some late steps during cytokinesis that controlthe abscission of daughter cells.
556 A. A. Varlet et al.

317
Fig. 1 Depletion of HSPB8 or BAG3 perturbs daughter cells spreadingand ICB disappearance during cytokinesis. a Representative Western blotof HeLa-GFP-H2B cells lysed in SDS-sample buffer, showing theefficiency of HSPB8 and BAG3 depletion after transfection withHSPB8- and BAG3-specific siRNAs. Depletion was estimated at >75%by loading increasing amounts of control extracts (siCtl). Western blotanalysis was performed with anti-HSPB8 and anti-BAG3 antibodies;GAPDH: loading control. b Unsynchronized HeLa-GFP-H2B cellswere depleted of HSPB8 or BAG3 using the indicated siRNAs andwere imaged starting 40 h post-transfection for 72 h at 10-min intervals.First row of panels: representative time sequence of a control cellshowing normal daughter cells spreading; second and third rows ofpanels: representative time sequences of HSPB8- or BAG3-depletedcells showing delayed spreading. Scale bar, 10 μm. c On the left: graphshowing the average time of daughter cells spreading for control (siCtl)versus HSPB8-depleted cells (siHSPB8). Data are the means ± SE fromthree independent experiments; the Mann-Whitney’s test was performed
relative to control ****p < 0.0001. On the right: graph depicting thepercentage of cells with abnormal daughter cells spreading in HSPB8-(siB8) or BAG3-depleted cells (siBAG3) compared to control cells(siCtl), means ± SE from two or three independent experiments.*p < 0.05 compares siHSPB8 #3 to siCtl by the Fischer exact test. dRepresentative time-sequences of HeLa-GFP-H2B cells transfectedwith the indicated siRNAs and imaged for 72 h starting at 40 h aftertransfection. Enlarged views of the boxed regions emphasize thestructure of the ICB, which is abnormally long and/or thick in cellsdepleted of HSPB8 or BAG3, as designated by the white and redarrows, respectively; time sequences are in min; bars, 10 μm. e Graphsdepicting the percentages of cells with abnormal ICB (on the left) or thetime spent between early telophase and disappearance of the ICB (on theright); the data are the means ± SE of two to three independentexperiments as indicated. **p < 0.01 and ****p < 0.0001 comparesiHSPB8 to siCtl by the Fisher exact test
HSPB8 regulates F-actin dynamics during cytokinesis 557

318
Knockdown of HSPB8 causes abnormal F-actinaccumulation at the intercellular bridge of daughter cells
Having shown that silencing of either BAG3 or HSPB8 causessimilar cytokinetic defects, and considering that HSPB8 levelsdepend on BAG3 (Fig. 1a) (Carra et al. 2008), subsequentanalyses were performed in HSPB8-depleted cells. Besides,we found that depletion of HSPB8 reduced BAG3 levels by∼50 to 75% in soluble fractions recovered from cells arrested atthe G2/M border and at the M-phase, respectively, withoutaffecting BAG3 levels seen in the insoluble fractions (Fig. 2f,G2/M andM). In mitotic cells, BAG3was mainly detected as a
supershifted, hyperphophorylated protein band on SDS-PAGEas previously shown (Fig. 2f, red arrow) (Fuchs et al. 2015).This was contrasting with themodest reduction of BAG3 levelsseen upon silencing of HSPB8 in asynchronous cells (~30%),as shown before (Fig. 2f, AS) (Carra et al. 2008). Together withour previous findings that HSPB8 facilitates BAG3 phosphor-ylation and localization at mitotic entry (Fuchs et al. 2015),these results suggest that cell mitosis offers a unique model touncover the HSPB8-dependent functions of BAG3.
Based on our previous work linking BAG3-HSPB8 to theremodeling of actin-based structures at mitotic entry (Fuchset al. 2015), we then sought to determine the impact of
Fig. 2 Silencing of HSPB8 interferes with cytokinetic abscission andmarkedly decreases the mitotic levels of BAG3. a UnsynchronizedHeLa-GFP-H2B cells transfected with various HSPB8-specific siRNAsequences were imaged for 72 h at 10-min intervals. The redarrowhead designates a persistent ICB connecting sister cellsthat comprises a dense midbody (emphasized in the enlarged view); bar10 μm. The graph shows the percentage of cells remaining connectedby an ICB; means ± SE. *p < 0.05 compares siHSPB8 #3 to siCtl by theFisher exact test. b Time sequences showing sister cells entering mitosissynchronously (red arrowheads); bar 20 μm; P prophase,M metaphase,T telophase. c Graph depicting the percentages of cells undergoingsynchronous mitosis; means ± SE. ***p < 0.001 compares siHSPB8 #3to siCtl by the Fisher exact test. d Representative Western blot of HeLa-GFP-H2B cells that have been lysed in SDS-sample buffer extracts,
showing the efficiency of HSPB8 depletion, 88 h after transfection ofthe indicated siRNA sequences; calreticulin: loading control. bConfocal image of HeLa-GFP-H2B cells transfected with HSPB8-specific siRNAs, showing multi-nucleation; F-actin was stained withTexas-Red-Phalloidin; bar 30 μm. The graph shows the incidence ofbi- and multi-nucleation; means ± SE from three independentexperiments. ****p < 0.0001 compares siHSPB8 #2 to siCtl, and**p < 0.01 compares siHSPB8 #3 to siCtl by the Fisher exact test. fRepresentative Western blot of HeLa cell extracts isolated fromunsynchronized cells (As), or cells that have been synchronized at theG2/M border (G2/M), or M-phase (M), after transfection with HSPB8-specific siRNA or control siRNA (siCtl) for 88 h, showing the levels ofBAG3, HSPB8, and GAPDH (loading control). Cells have been lysed by3 cycles of freezing/thawing in 4 volumes of 0.1% IGEPAL lysis buffer
558 A. A. Varlet et al.

319
HSPB8 on actin dynamics during cytokinesis. In animalcells, cytokinesis begins with the assembly and contractionof an actomyosin ring that leads to the formation of an in-tercellular bridge (von Dassow and Bement 2005). Finalcleavage and fusion of this intercellular bridge require thin-ning of the ICB (secondary ingression) that critically relieson actin depolymerization within the ICB (Schiel et al.2013; Schiel et al. 2012). We hypothesized that the chaper-one complex might assist in accurate assembly and/or dis-assembly of actin-based cytokinetic structures.
To test this idea, we took advantage of LifeAct-GFP—aprobe that binds to F-actin (Riedl et al. 2008), to visualizethe dynamics of actin at the actomyosin ring by spinningconfocal microscopy. HeLa-RFP-H2B cells wereadenofected with recombinant adenovirus driving the ex-pression of LifeAct-GFP together with control or HSPB8-specific siRNAs, and then synchronized by a double thy-midine block using a protocol developed for knockdown-rescue experiments (Fig. 3a) (Fuchs et al. 2016). Live-cellimaging revealed that assembly and constriction of the ac-tomyosin ring were not significantly perturbed by depletionof HSPB8 (Fig. 3c). Nonetheless, in HSPB8-depleted cells,the AR appeared abnormally enriched in F-actin. This wasrevealed by an increase of AR thickness at telophase(Fig. 3c, supplemental movies S7–S8). Remarkably, higherproportions of HSPB8-depleted cells (∼43 ± 2.5%;43 ± 4.2%; 58 ± 2.5%) displayed abnormal F-actin accu-mulation at the ICB marked by Aurora B (Murata-Horiet al. 2002)—a mitotic kinase that relocates to the ICB attelophase, as compared to cells transfected with controlsiRNAs (∼24 ± 1.3%). Such disorganized actin structurespersisted after cell spreading and DNA de-condensationand were observed using three different HSPB8-specificsiRNA sequences in depletion experiments (Fig. 3b, c).The data suggest that HSPB8-BAG3 chaperone complexfacilitates accurate AR dynamics to enable timely abscis-sion of daughter cells.
Down-modulation of actin dynamics can normalizeF-actin accumulation at the intercellular bridgeand correct cytokinetic defects caused by depletionof HSPB8
Abnormal accumulation of F-actin at the ICB suggestedthat HSPB8 depletion could perturb actin dynamics, i.e.,polymerization-depolymerization. To obtain evidence fora causal relationship, we used the actin drug latrunculin A(LatA), which inhibits actin dynamics by sequestering G-actin, hence limiting the actin pool available for F-actinassembly (Coue et al. 1987; Morton et al. 2000). Cells weretreated with a low concentration (20 nM) during the lasthour of the release period before cell fixation (Fig. 4a).Under these conditions, the organization of F-actin in
control cells was not noticeably affected. Significantly,such treatment inhibited F-actin accumulation at the ICBin HSPB8-depleted cells, bringing it in line with cellstransfected with control siRNAs (Fig. 4b, c). The resultssuggest that HSPB8-depleted cells have deregulated actinpolymerization at the ICB that can be normalized by limit-ing the pool of G-actin.
We then took advantage of the hypersensibility ofHSPB8-depleted cells to LatA to examine the cause-effectrelationship between deregulated actin dynamics and cyto-kinetic failure, by measuring the occurrence of bi- andmulti-nucleation. We determined that incubation of HeLa-GFP-H2B cells in the presence of 1 nM LatA for a 48-hperiod of time modestly affected cell division and in-creased the incidence of nuclear abnormalities by ∼1.8-fold(Fig. 4d, siCtl LatA). However, this treatment reduced theincidence of bi- and multi-nucleation in HSPB8-depletedcells to a level comparable to the one observed in cellstransfected with control siRNAs (Fg. 4d, siB8 LatA).Thus, the data suggest that perturbation of actin dynamicsupon depletion of HSPB8 contributes to its impact on celldivision.
Inhibition of Arp2/3 complex can normalize F-actinat the ICB in HSPB8-depleted cells
Accurate assembly and disassembly of the cortical AR duringcytokinesis are proposed to rely on the combined action of twoF-actin nucleators, the formin diaphanous (Dia) and theArp2/3 complex, and require dynamic actin turnover at theequatorial AR (Bovellan et al. 2014; Murthy and Wadsworth2005). We noted that the BAG3 interactome from HeLa cellscontains proteins that could link BAG3 to Arp2/3 complex,notably ArpC2, a component of Arp2/3 complex, andELMO1, which provides a connection with upstream regula-tors (Chen et al. 2013). To explore a potential relationship withthe Arp2/3 complex, we first analyzed its subcellular distribu-tion during cytokinesis. Small amounts of a fluorescent probe,GFP-Arp3, were expressed in HeLa cells for a short period oftime after silencing HSPB8 to avoid perturbing actin dynam-ics. The subcellular distribution of GFP-Arp3was analyzed byconfocal microscopy after cell fixation (Fig. 5a, experimentalscheme; Fig. 5b). Cells transfected with control siRNAs un-dergoing cytokinesis exhibited a rather diffuse localization ofArp3-GFPwith no significant enrichment at the ICB (Fig. 5c).This was revealed by quantification of the GFP fluorescenceintensity ratio of ICB over cytoplasm (Fig. 5, ∼0.89). Inmarked contrast, in cells transfected with HSPB8-specificsiRNAs, GFP-Arp3 accumulated at the ICB and co-distributed with aberrant F-actin structures, as revealed by a∼1.6-fold increase in the ICB/cytoplasmic GFP fluorescenceintensity ratio (Fig. 5c, arrowhead).
HSPB8 regulates F-actin dynamics during cytokinesis 559

320
We then sought to acutely perturb Arp2/3 activity ascells entered cytokinesis and monitor the impact on ICB-associated aberrant F-actin. To that end, HSPB8-depletedcells that have been synchronized in late mitosis were in-cubated in the presence of the selective Arp2/3 inhibitorCK666 (Nolen et al. 2009) or with the vehicle only, duringthe last hour of the release period from the thymidine-block. Using this protocol, inhibition of the Arp2/3
complex did not impact cytokinesis in cells treated withcontrol siRNAs, consistent with recent findings (Bovellanet al. 2014; Rosa et al. 2015). Nonetheless, such treatmentinhibited the occurrence of abnormal F-actin at the ICB inHSPB8-depleted cells (Fig. 5d). These data suggest that bylimiting Arp2/3-mediated actin polymerization, an accuratebalance between actin assembly and turnover at the ICBcan be restored in HSPB8-depleted cells.
Fig. 3 Silencing of HSPB8 causes abnormal F-actin accumulation at theICB. a Schematic of the protocol used for adenofection of HeLa-RFP-H2B cells with Ad-LifeAct-GFP together with HSPB8-specific siRNAs.b Western blot of HeLa-RFP-H2B cell extracts prepared in SDS-samplebuffer showing the efficiency of HSPB8 depletion 48 h after transfectionof the indicated HSPB8-specific siRNAs; depletion was estimated at>75% by loading increasing amounts of control extracts (1/4, ½, 1,siCtl); vinculin: loading control. c Live-cell imaging was performed fora 2-h period at 5-min intervals by spinning disk confocal microscopy.Representative time sequences show F-actin organization at the actinring (pointed by white arrowheads) in cells transfected with controlsiRNAs (siCtl) compared to cells transfected with HSPB8-specficsiRNAs; bar, 10 μm. Quantification of the actin ring thickness over
width; means ± SE from three independent experiments. The Mann-Whitney’s test was performed relative to siCtl; ****p < 0.0001. dConfocal images of HeLa-RFP-H2B cells that have been synchronizedin mitosis after transfection with HSPB8-specific siRNAs, showingabnormal F-actin accumulation stained by Alexa-488-phalloidin at theICB (green) marked by immunostaining of Aurora B (red). Whitearrowheads designate enlarged viewed that emphasize actinorganization at the bridge of daughter cells; bar, 10 μm. The graphshows the percentages of cells with abnormal F-actin accumulation atthe ICB in late cytokinesis, means ± SE from three independentexperiments. ****p < 0.0001 compares HSPB8-specific siRNAs tosiCtl by the Fischer exact test
560 A. A. Varlet et al.

321
Drugs that affect lysosomal function recapitulatethe HSPB8-dependent phenotype, whereas inductionof autophagy signaling can restore the F-actin defectscaused by depletion of HSPB8
A specialized role for a multichaperone complex organizedby BAG3 has been proposed in the clearance of damaged
proteins undergoing partial misfolding in response to me-chanical strain during skeletal muscle contraction (Arndtet al. 2010; Ulbricht et al. 2013). Given that the actin-based AR is also put under high tensile forces during cyto-kinesis, we hypothesized that HSPB8-BAG3 could be mo-bilized to facilitate the autophagic clearance of componentsof actin-based cytokinetic structures.
Fig. 4 Down-modulation of actin dynamics can rescue phenotypes causedby depletion of HSPB8. a Schematic of the protocol used to modulate actindynamic in HeLa-RFP-H2B cells synchronized in late mitosis that havebeen transfected with HSPB8-specific or control siRNAs. bRepresentativeconfocal images of fixed HSPB8-depleted HeLa-RFP-H2B cells, showingnormalized F-actin at the ICB (emphasized in enlarged views pointed bywhite arrowheads), after treatment with latrunculin A (LatA, 20 nM);immunostaining of Autora B (red) was performed to detect ICB and F-actin was stained with Alexa-488-phalloidin (green). Bar, 10 μm. c Thegraph depicts the percentages of cells with abnormal F-actin accumulation
at the ICB; means ± SE from four independent experiments.****p < 0.0001 compares siHSPB8 #3 treated with the vehicle (DMSO)to siCtl ± DMSO by the Fisher exact test; n.s.: non-significant comparessiHSPB8 #3 + LatA to siCtl ± LatA. d Unsynchronized HeLa-GFP-H2Bthat have been transfectedwithHSPB8-specific or control siRNAs (siB8 vssiCtl) were grown in the presence of 1 nM LatA for 48 h and thepercentages of nuclear abnormalities were scored; means ± SE from threeindependent experiments. ****p < 0.0001 compares siHSPB8 #2 treatedwith the vehicle (DMSO) to siCtl ± DMSO by the Fisher exact test; n.s.:non-significant difference between siHSPB8 #3 + LatA and siCtl ± LatA
HSPB8 regulates F-actin dynamics during cytokinesis 561

322
To explore this idea, we used two distinct experimentalapproaches. We first asked whether impairing lysosomal func-tion could interfere with AR dynamics. HeLa cells that havebeen synchronized in mitosis by a double thymidine blockwere incubated in the presence of drugs that are commonlyused to disrupt the autophagic flux; these include bafilomycin
A (BafA), which inhibits V-ATPases and blocksautophagosome-lysosome fusion, or pepstatin A and E64D,two inhibitors of lysosomal proteases (Klionsky et al. 2016).The drugs were added during the last hour of the release pe-riod before cell fixation, and cells in late cytokinesis weredetected by immunostaining of the ICB using anti-Aurora B
Fig. 5 Co-distribution of abnormal F-actin and Arp2/3, and restorationof F-actin organization at the ICB in HSPB8-depleted cells uponinhibition of Arp2/3 complex. a Schematic of the protocol used tovisualize Arp2/3 localization in Hela-RFP-H2B cells duringcytokinesis. b Western blot of HeLa-RFP-H2B cell extracts prepared inSDS-sample buffer showing the levels of transfected GFP-Arp3 andendogenous Arp3; calreticulin levels are shown as loading control. cRepresentative confocal images of HSPB8-depleted HeLa-RFP-H2Bcells, showing accumulation of GFP-Arp3 together with F-actin;staining of Aurora B (purple) was performed to localize ICBs that areemphasized in enlarged views designated by white arrowheads; bar,10 μm. Quantification of the fluorescence intensity ratios of GFP-Arp3is shown on the right graph, means ± SE of at least 98 cells from four
independent experiments. The Mann-Whitney’s test was performedrelative to control; ****p < 0.0001. d Confocal images showingsynchronized HeLa-RFP-H2B cells depleted of HSPB8 treated with thevehicle only (DMSO) or with a selective Arp 2/3 inhibitor (CK666,40 μM) 1 h before cell fixation. F-actin organization at the ICB(phalloidin staining: green, Aurora B staining: red) is emphasized in theenlarged views designated by white arrowheads; bar, 10 μm. Aschematic of the protocol used is shown below. The graph depicts thepercentages of cells with abnormal F-actin accumulation at the ICB;means ± SE from two or four independent experiments; n.s.: non-significant difference between siHSPB8 #3 + CK666 versussiCtl ± CK666 by the Fisher exact test
562 A. A. Varlet et al.

323
antibody. Analysis of F-actin distribution revealed that a largerproportion of cells treated with the drugs accumulated aber-rant actin structures at the ICB that persist after cell spreadingand DNA de-condensation relative to cells treated with thevehicle only (∼1.8-fold increase; Fig. 6a). Thus, the data sug-gest that inhibition of vacuolar acidification or lysosomal en-zymes, like depletion of HSPB8, can recapitulate similar de-fects in actin dynamics at the ICB.
As a second approach, we submitted HSPB8-depletedcells that have been synchronized in late mitosis to a 1-hincubation period with rapamycin—a drug that inhibitsmTORC1 and stimulates autophagy signaling (Crespo andHall 2002). Remarkably, such treatment corrected F-actinaccumulation at the ICB and brought the proportions ofHSPB8-depleted cells bearing F-actin defects in line withcel ls transfected with control s iRNAs (Fig. 6b) .Collectively, our results suggest that defects in the dynam-ics of the actin-based AR caused by depletion of HSPB8may result, at least in part, from its function in the segrega-tion of cytoskeletal protein clients to selective autophagy.
Discussion
BAG3, in association with its chaperone partners, has beenlinked to the selection of damaged proteins for autophagic deg-radation during proteotoxic stress and muscle cell contraction(Minoia et al. 2014; Ulbricht et al. 2013).While BAG3 controlsthe stability of HSPB8, it may also function without HSPB8during stress, questioning the exact role of HSPB8 in the re-ported functions of BAG3 (our unpublished data) (Ganassiet al. 2016; Minoia et al. 2014). Recently, we uncovered anHSPB8-dependent function of BAG3 in a basic and fundamen-tal activity of cancer cells: mitosis, in the remodeling of actin-based structures that guide spindle dynamics (Fuchs et al.2015). In the current study, we present further evidence thatin response to mitotic signaling, HSPB8 and BAG3 act togeth-er to modulate actin dynamics during cytokinesis and facilitateabscission of daughter cells. Depletion of HSPB8 or BAG3caused levels of cytokinetic defects, notably multi-nucleation,that compare favorably with those reported in the literatureupon silencing of p62/SQSTM1 or key effectors of cytokinesis,using similar approaches based on analyses of hypomorphicphenotypes (5 to 25% of multi-nucleated cells on average)(Belaid et al. 2013; Ma and Chircop 2012; Schiel et al. 2012).This suggests that the HSPB8-BAG3 complex plays a signifi-cant role in the context of HeLa cells. Because drugs that se-lectively perturb actin polymerization can correct the multi-nucleation phenotype caused by depletion of HSPB8, whichalso depletes the mitotic forms of BAG3, we suggest that anHSPB8-BAG3 complex promotes accurate cell division main-ly by limiting actin polymerization and/or promoting actin turn-over. Whether such process may involve HSPB8-BAG3
function in autophagic sequestration is suggested by some re-sults presented here and is further discussed below.
Our finding that HSPB8 maintains BAG3 levels mainlyduring M-phase suggests that it may serve to stabilize partic-ular conformations of BAG3 induced by mitotic phosphory-lation. This could promote interactions between BAG3 andclient proteins that would connect the chaperone complex toactin-based structures, perhaps, via BAG3 Pro-rich motifs,which we have identified as crucial determinants of BAG3mitotic activity in previous work (Fuchs et al. 2015). BAG3was reported to interact with several proteins that could mod-ulate actin assembly or promote actin turnover, for instanceCapZ—an actin-capping protein that prevents filamentgrowth (Cooper and Schafer 2000; Hishiya et al. 2010;Pollard and Cooper 2009), or non muscle myosin II—themolecular motor that generates forces for constriction of theactin-based AR (Hong et al. 2016; Maupin and Pollard 1986;Schiel et al. 2013). It has been shown that actin at the AR ishighly dynamic and myosin II was involved in the dynamicturnover of actin during cytokinesis (Murthy and Wadsworth2005). Given that BAG3 can interact with a plethora of po-tential client proteins in HeLa cells (Chen et al. 2013), itsmitotic activity likely involves complex interactions with cli-ent proteins that could be regulated in time and space byHSPB8 and multisite phosphorylation of BAG3.
The results presented here provide important mechanisticclue by showing that silencing of HSPB8 causes accumulationof branched actin at the ICB. Assembly of branched actinnetworks is mediated by the actin nucleator complex Arp2/3that appeared to accumulate at the ICB of HSPB8-depletedcells (Goley and Welch 2006). Furthermore, inhibition ofArp2/3 using CK666 can prevent abnormal F-actin accumu-lation at the ICB in these cells, suggesting that HSPB8-BAG3act by limiting Arp2/3 activity. It has been proposed that aswitch from Arp2/3 to diaphanous-mediated actin nucleationis required to drive assembly of a rigid actin cortex in meta-phase cells (Rosa et al. 2015). Too much Arp2/3-mediatedactin polymerization may also lead to cytokinesis failure(Chircop 2014). Indeed, deregulation of the antagonism be-tween the Rho GTPases Rho and Rac, which is believed tocontrol AR dynamics during cytokinesis, leads to defectivecytokinesis and aberrant distribution of F-actin (D Avinoet al. 2004). We have shown that depletion of HSPB8 orBAG3 interferes with cell rounding and assembly of a me-chanically rigid actin cortex at mitotic entry, in addition to itsperturbing effect during cytokinesis (Fuchs et al. 2015). Basedon the overall data, it can be argued that failure to accuratelylimit the activity of Arp2/3 upon depletion of HSPB8 orBAG3 contributes to defects in mitotic cortical rigidity, spin-dle positioning, and cytokinesis, leading to impaired cell divi-sion and multi-nucleation.
Our findings also suggest that a phosphorylation-activatedfunction of HSPB8-BAG3 would promote actin turnover
HSPB8 regulates F-actin dynamics during cytokinesis 563
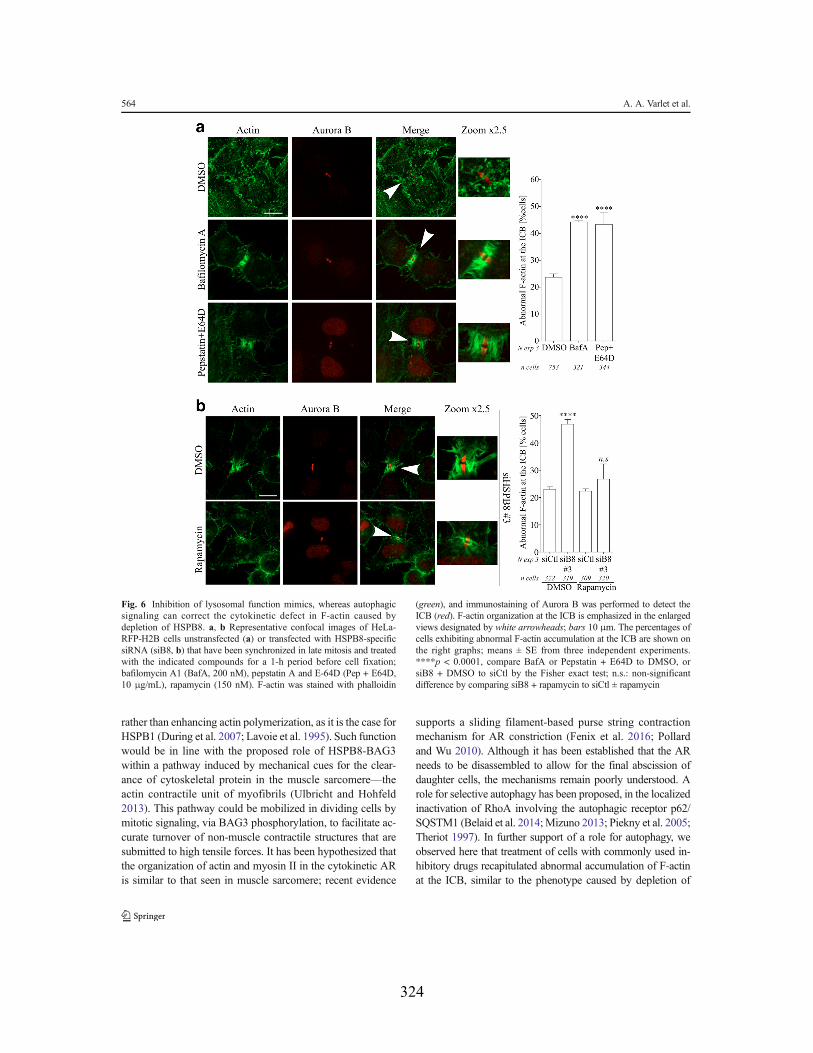
324
rather than enhancing actin polymerization, as it is the case forHSPB1 (During et al. 2007; Lavoie et al. 1995). Such functionwould be in line with the proposed role of HSPB8-BAG3within a pathway induced by mechanical cues for the clear-ance of cytoskeletal protein in the muscle sarcomere—theactin contractile unit of myofibrils (Ulbricht and Hohfeld2013). This pathway could be mobilized in dividing cells bymitotic signaling, via BAG3 phosphorylation, to facilitate ac-curate turnover of non-muscle contractile structures that aresubmitted to high tensile forces. It has been hypothesized thatthe organization of actin and myosin II in the cytokinetic ARis similar to that seen in muscle sarcomere; recent evidence
supports a sliding filament-based purse string contractionmechanism for AR constriction (Fenix et al. 2016; Pollardand Wu 2010). Although it has been established that the ARneeds to be disassembled to allow for the final abscission ofdaughter cells, the mechanisms remain poorly understood. Arole for selective autophagy has been proposed, in the localizedinactivation of RhoA involving the autophagic receptor p62/SQSTM1 (Belaid et al. 2014;Mizuno 2013; Piekny et al. 2005;Theriot 1997). In further support of a role for autophagy, weobserved here that treatment of cells with commonly used in-hibitory drugs recapitulated abnormal accumulation of F-actinat the ICB, similar to the phenotype caused by depletion of
Fig. 6 Inhibition of lysosomal function mimics, whereas autophagicsignaling can correct the cytokinetic defect in F-actin caused bydepletion of HSPB8. a, b Representative confocal images of HeLa-RFP-H2B cells unstransfected (a) or transfected with HSPB8-specificsiRNA (siB8, b) that have been synchronized in late mitosis and treatedwith the indicated compounds for a 1-h period before cell fixation;bafilomycin A1 (BafA, 200 nM), pepstatin A and E-64D (Pep + E64D,10 μg/mL), rapamycin (150 nM). F-actin was stained with phalloidin
(green), and immunostaining of Aurora B was performed to detect theICB (red). F-actin organization at the ICB is emphasized in the enlargedviews designated by white arrowheads; bars 10 μm. The percentages ofcells exhibiting abnormal F-actin accumulation at the ICB are shown onthe right graphs; means ± SE from three independent experiments.****p < 0.0001, compare BafA or Pepstatin + E64D to DMSO, orsiB8 + DMSO to siCtl by the Fisher exact test; n.s.: non-significantdifference by comparing siB8 + rapamycin to siCtl ± rapamycin
564 A. A. Varlet et al.

325
HSPB8. Recently, we have shown that HSPB8, BAG3, andp62/SQSTM1 form a mitotic complex and that depletion ofany of the proteins resulted in the same phenotypes in earlymitosis (Fuchs et al. 2015). Here, we provide further evidencefor a connection between the activity of HSPB8-BAG3 duringcytokinesis and autophagy signaling by showing that treatmentof HSPB8-depleted cells with rapamycin could prevent F-actinaccumulation. This is in line with recent work suggesting thatBAG3 modulates the spatial activity of mTORC1—the targetof rapamycin and a key regulator of autophagy signaling, inaddition to its direct interaction with the selective autophagicmachinery via p62/SQSTM1 (Gamerdinger et al. 2011;Kathage et al. 2017). Overall, though the exact mechanismwhereby HSPB8 and BAG3 promote disassembly of cytoki-netic actin structures remains to be established, we think thatwe can reasonably speculate that the chaperone complex couldact in the autophagic sequestration of proteins that regulateassembly of actin networks, a function that would be modulat-ed by BAG3 mitotic phosphorylation.
In conclusion, the present study further supports a special-ized role for HSPB chaperones as members of a quality controlnetwork that would assist in the assembly and disassembly ofmacromolecular complexes controlling the dynamic architec-ture of actin during various mechanical processes, includingcell-shape changes that guide mitosis in cancer cells (Guilbertet al. 2015). Deciphering the mitotic interactome of BAG3 andthe molecular impact of BAG3 multisite phosphorylation iscrucial to our understanding of HSPB8-BAG3 function in actindynamics and is the object of our current work. Given thatHSPB8 and BAG3 are expressed in a wide variety of cancercell types, their activity in cell division may sustain tumor ex-pansion (Rosati et al. 2011). Thus, uncovering their mode ofaction is of great interest for the development of targeted ther-apeutic approaches to inhibit the HSPB8-dependent function ofBAG3 and further enhance mitotic stress in cancer cells.
Acknowledgements We are grateful to Sabine Elowe (CentreHospitalier de l’Université Laval, Quebec, Canada), John A. Cooper(Dept of Biochemistry and Molecular Biophysics WashingtonUniversity, St Louis, USA), and Laurence Pelletier (the Lunenfeld-Tanenbaum Research Institute, Toronto, Canada) for providing criticalreagents. We thank Carl St-Pierre and Anne Loranger (Centre derecherche sur le cancer de l’Université Laval, HDQ) for their assistancein microscopic analyses. This work was supported by the CanadianInstitutes of Health Research Grant No 7088.
References
Arndt V, Dick N, Tawo R, Dreiseidler M, Wenzel D, Hesse M, Furst DO,Saftig P, Saint R, Fleischmann BK, Hoch M, Hohfeld J (2010)Chaperone-assisted selective autophagy is essential for musclemaintenance. Curr Biol 20:143–148. doi:10.1016/j.cub.2009.11.022
Bakthisaran R, Tangirala R, Rao ChM (2015) Small heat shock proteins:role in cellular functions and pathology. Biochim Biophys Acta1854:291–319. doi:10.1016/j.bbapap.2014.12.019
Behl C (2016) Breaking BAG: the Co-chaperone BAG3 in health anddisease. Trends Pharmacol Sci 37:672–688. doi:10.1016/j.tips.2016.04.007
Belaid A, Cerezo M, Chargui A, Corcelle-Termeau E, Pedeutour F,Giuliano S, Ilie M, Rubera I, Tauc M, Barale S, Bertolotto C,Brest P, Vouret-Craviari V, Klionsky DJ, Carle GF, Hofman P,Mograbi B (2013) Autophagy plays a critical role in the degrada-tion of active RHOA, the control of cell cytokinesis, and genomicstability. Cancer Res 73:4311–4322. doi:10.1158/0008-5472.CAN-12-4142
Belaid A, Ndiaye PD, Cerezo M, Cailleteau L, Brest P, Klionsky DJ,Carle GF, Hofman P, Mograbi B (2014) Autophagy and SQSTM1on the RHOA(d) again: emerging roles of autophagy in the degra-dation of signaling proteins. Autophagy 10:201–208. doi:10.4161/auto.27198
Bovellan M, Romeo Y, Biro M, Boden A, Chugh P, Yonis A, Vaghela M,Fritzsche M, Moulding D, Thorogate R, Jegou A, Thrasher AJ,Romet-Lemonne G, Roux PP, Paluch EK, Charras G (2014)Cellular control of cortical actin nucleation. Curr Biol 24:1628–1635. doi:10.1016/j.cub.2014.05.069
Carra S, Seguin SJ, Lambert H, Landry J (2008) HspB8 chaperone activ-ity toward poly(Q)-containing proteins depends on its associationwith Bag3, a stimulator of macroautophagy. J Biol Chem 283:1437–1444. doi:10.1074/jbc.M706304200
Champagne C, LandryMC, GingrasMC, Lavoie JN (2004) Activationof adenovirus type 2 early region 4 ORF4 cytoplasmic deathfunction by direct binding to src kinase domain. J Biol Chem279:25905–25915
Chen Y, Yang LN, Cheng L, Tu S, Guo SJ, Le HY, Xiong Q, Mo R, LiCY, Jeong JS, Jiang L, Blackshaw S, Bi LJ, Zhu H, Tao SC, Ge F(2013) Bcl2-associated athanogene 3 interactome analysis reveals anew role in modulating proteasome activity. Mol Cell Proteomics12:2804–2819. doi:10.1074/mcp.M112.025882
Chircop M (2014) Rho GTPases as regulators of mitosis and cytokinesisin mammalian cells. Small GTPases 5. doi:10.4161/sgtp.29770
Colvin TA, Gabai VL, Gong J, Calderwood SK, Li H, Gummuluru S,Matchuk ON, Smirnova SG, Orlova NV, Zamulaeva IA, Garcia-Marcos M, Li X, Young Z, Rauch JN, Gestwicki JE, Takayama S,Sherman MY (2014) Hsp70-Bag3 interactions regulate cancer-related signaling networks. Cancer Res. doi:10.1158/0008-5472.CAN-14-0747
Cooper JA, Schafer DA (2000) Control of actin assembly and disassem-bly at filament ends. Curr Opin Cell Biol 12:97–103
Coue M, Brenner SL, Spector I, Korn ED (1987) Inhibition of actinpolymerization by latrunculin a. FEBS Lett 213:316–318
Crespo JL, Hall MN (2002) Elucidating TOR signaling and rapamycinaction: lessons from Saccharomyces cerevisiae. Microbiol Mol BiolRev 66: 579–91, table of contents.
D'Avino PP, Savoian MS, Glover DM (2004) Mutations in sticky lead todefective organization of the contractile ring during cytokinesis andare enhanced by rho and suppressed by Rac. J Cell Biol 166:61–71.doi:10.1083/jcb.200402157
During RL, Gibson BG, Li W, Bishai EA, Sidhu GS, Landry J,Southwick FS (2007) Anthrax lethal toxin paralyzes actin-basedmotility by blocking Hsp27 phosphorylation. EMBO J 26:2240–2250. doi:10.1038/sj.emboj.7601687
Fenix AM, Taneja N, Buttler CA, Lewis J, Van Engelenburg SB, Ohi R,Burnette DT (2016) Expansion and concatenation of non-musclemyosin IIA filaments drive cellular contractile system formationduring interphase and mitosis. Mol Biol Cell. doi:10.1091/mbc.E15-10-0725
Finka A, Mattoo RU, Goloubinoff P (2016) Experimental milestones inthe discovery of molecular chaperones as polypeptide unfolding
HSPB8 regulates F-actin dynamics during cytokinesis 565

326
enzymes. Annu Rev Biochem 85:715–742. doi:10.1146/annurev-biochem-060815-014124
Fuchs M, Boulanger MC, Lambert H, Landry J, Lavoie JN (2016)Adenofection: a method for studying the role of molecular chaper-ones in cellular morphodynamics by depletion-rescue experiments. JVis Exp. doi:10.3791/54557
FuchsM, Luthold C, Guilbert SM, Varlet AA, Lambert H, Jetté A, EloweS, Landry J, Lavoie JN (2015) A Role for the Chaperone ComplexBAG3-HSPB8 in Actin Dynamics, Spindle Orientation and ProperChromosome Segregation during Mitosis. PLoS Genetics,e1005582. doi: 10.1371 11.
Fuchs M, Poirier DJ, Seguin SJ, Lambert H, Carra S, Charette SJ, LandryJ (2010) Identification of the key structural motifs involved inHspB8/HspB6-Bag3 interaction. Biochem J 425:245–255. doi:10.1042/BJ20090907
Gamerdinger M, Hajieva P, Kaya AM, Wolfrum U, Hartl FU, Behl C(2009) Protein quality control during aging involves recruitment ofthe macroautophagy pathway by BAG3. EMBO J 28:889–901
Gamerdinger M, Kaya AM, Wolfrum U, Clement AM, Behl C (2011)BAG3mediates chaperone-based aggresome-targeting and selectiveautophagy of misfolded proteins. EMBO Rep 12:149–156. doi:10.1038/embor.2010.203
Ganassi M, Mateju D, Bigi I, Mediani L, Poser I, Lee HO, Seguin SJ,Morelli FF, Vinet J, LeoG, Pansarasa O, Cereda C, Poletti A, AlbertiS, Carra S (2016) A surveillance function of the HSPB8-BAG3-HSP70 chaperone complex ensures stress granule integrity and dy-namism. Mol Cell 63:796–810. doi:10.1016/j.molcel.2016.07.021
Goley ED,WelchMD (2006) The ARP2/3 complex: an actin nucleatorcomes of age. Nat Rev Mol Cell Biol 7:713–726. doi:10.1038/nrm2026
Guilbert SM, Varlet A-A, Fuchs M, Lambert H, Landry Ja, Lavoie JN(2015) Regulation of Actin-Based Structure Dynamics by HspBProteins and Partners.. In: Hightower ERTaL (ed) The Big Bookof Small Heat Shock Proteins, vol ISBN: 978–3–319-16076-4.Springer international Publishing AG
Haslbeck M, Vierling E (2015) A first line of stress defense: small heatshock proteins and their function in protein homeostasis. J Mol Biol427:1537–1548. doi:10.1016/j.jmb.2015.02.002
Hishiya A, Kitazawa T, Takayama S (2010) BAG3 and Hsc70 interactwith actin capping protein CapZ to maintain myofibrillar integrityunder mechanical stress. Circ Res 107:1220–1231. doi:10.1161/CIRCRESAHA.110.225649
Hong J, Park JS, Lee H, Jeong J, Hyeon Yun H, Yun KimH, Ko YG, LeeJH (2016) Myosin heavy chain is stabilized by BCL-2 interactingcell death suppressor (BIS) in skeletal muscle. Exp Mol Med 48:e225. doi:10.1038/emm.2016.2
Iwasaki M, Homma S, Hishiya A, Dolezal SJ, Reed JC, Takayama S(2007) BAG3 regulates motility and adhesion of epithelial cancercells. Cancer Res 67:10252–10259. doi:10.1158/0008-5472.CAN-07-0618
Iwasaki M, Tanaka R, Hishiya A, Homma S, Reed JC, Takayama S(2010) BAG3 directly associates with guanine nucleotide ex-change factor of Rap1, PDZGEF2, and regulates cell adhesion.Biochem Biophys Res Commun 400:413–418. doi:10.1016/j.bbrc.2010.08.092
Kassis JN, Guancial EA, Doong H, Virador V, Kohn EC (2006) CAIR-1/BAG-3 modulates cell adhesion and migration by downregulatingactivity of focal adhesion proteins. Exp Cell Res 312:2962–2971.doi:10.1016/j.yexcr.2006.05.023
Kathage B, Gehlert S, Ulbricht A, Ludecke L, Tapia VE, Orfanos Z,Wenzel D, Bloch W, Volkmer R, Fleischmann BK, Furst DO,Hohfeld J (2017) The cochaperone BAG3 coordinates protein syn-thesis and autophagy under mechanical strain through spatial regu-lation of mTORC1. Biochim Biophys Acta 1864:62–75. doi:10.1016/j.bbamcr.2016.10.007
Klebig C, Korinth D, Meraldi P (2009) Bub1 regulates chromosomesegregation in a kinetochore-independent manner. J Cell Biol 185:841–858. doi:10.1083/jcb.200902128
Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H,Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K,Adhihetty PJ, Adler SG, Agam G, Agarwal R, Aghi MK, AgnelloM, Agostinis P, Aguilar PV, Aguirre-Ghiso J, Airoldi EM, Ait-Si-AliS, Akematsu T, Akporiaye ET, Al-Rubeai M, Albaiceta GM,Albanese C, Albani D, Albert ML, Aldudo J, Algul H, AlirezaeiM, Alloza I, Almasan A, Almonte-Beceril M, Alnemri ES, AlonsoC, Altan-Bonnet N, Altieri DC, Alvarez S, Alvarez-Erviti L, AlvesS, Amadoro G, Amano A, Amantini C, Ambrosio S, Amelio I,Amer AO, Amessou M, Amon A, An Z, Anania FA, AndersenSU, Andley UP, Andreadi CK, Andrieu-Abadie N, Anel A, AnnDK, Anoopkumar-Dukie S, Antonioli M, Aoki H, Apostolova N,Aquila S, Aquilano K, Araki K, Arama E, Aranda A, Araya J,Arcaro A, Arias E, Arimoto H, Ariosa AR, Armstrong JL,Arnould T, Arsov I, Asanuma K, Askanas V, Asselin E, AtarashiR, Atherton SS, Atkin JD, Attardi LD, Auberger P, Auburger G,Aurelian L, Autelli R, Avagliano L, Avantaggiati ML, AvrahamiL, Awale S, Azad N, Bachetti T, Backer JM, Bae DH, Bae JS, BaeON, Bae SH, Baehrecke EH, Baek SH, Baghdiguian S,Bagniewska-Zadworna A et al (2016) Guidelines for the use andinterpretation of assays for monitoring autophagy (3rd edition).Autophagy 12:1–222. doi:10.1080/15548627.2015.1100356
Kostenko S, Moens U (2009) Heat shock protein 27 phosphorylation:kinases, phosphatases, functions and pathology. Cell Mol Life Sci66:3289–3307. doi:10.1007/s00018-009-0086-3
Lavoie JN, Champagne C, Gingras MC, Robert A (2000) Adenovirus E4open reading frame 4-induced apoptosis involves dysregulation ofsrc family kinases. J Cell Biol 150:1037–1056
Lavoie JN, Hickey E, Weber LA, Landry J (1993) Modulation of actinmicrofilament dynamics and fluid phase pinocytosis by phosphory-lation of heat shock protein 27. J Biol Chem 268:24210–24214
Lavoie JN, Lambert H, Hickey E, Weber LA, Landry J (1995)Modulation of cellular thermoresistance and actin filament stabilityaccompanies phosphorylation-induced changes in the oligomericstructure of heat shock protein 27. Mol Cell Biol 15:505–516
Ma MP, Chircop M (2012) SNX9, SNX18 and SNX33 are required forprogression through and completion of mitosis. J Cell Sci 125:4372–4382. doi:10.1242/jcs.105981
Maupin P, Pollard TD (1986) Arrangement of actin filaments andmyosin-like filaments in the contractile ring and of actin-like fila-ments in themitotic spindle of dividingHeLa cells. J Ultrastruct MolStruct Res 94:92–103
Minoia M, Boncoraglio A, Vinet J, Morelli FF, Brunsting JF, Poletti A,Krom S, Reits E, Kampinga HH, Carra S (2014) BAG3 induces thesequestration of proteasomal clients into cytoplasmic puncta: impli-cations for a proteasome-to-autophagy switch. Autophagy 10:1603–1621. doi:10.4161/auto.29409
Mizuno K (2013) Signaling mechanisms and functional roles of cofilinphosphorylation and dephosphorylation. Cell Signal 25:457–469.doi:10.1016/j.cellsig.2012.11.001
Morton WM, Ayscough KR, McLaughlin PJ (2000) Latrunculin altersthe actin-monomer subunit interface to prevent polymerization. NatCell Biol 2:376–378. doi:10.1038/35014075
Murata-Hori M, Tatsuka M, Wang YL (2002) Probing the dynamics andfunctions of aurora B kinase in living cells during mitosis and cyto-kinesis. Mol Biol Cell 13:1099–1108. doi:10.1091/mbc.01-09-0467
Murthy K, Wadsworth P (2005) Myosin-II-dependent localization anddynamics of F-actin during cytokinesis. Curr Biol 15:724–731.doi:10.1016/j.cub.2005.02.055
Nolen BJ, Tomasevic N, Russell A, Pierce DW, Jia Z, McCormick CD,Hartman J, Sakowicz R, Pollard TD (2009) Characterization of twoclasses of small molecule inhibitors of Arp2/3 complex. Nature 460:1031–1034. doi:10.1038/nature08231
566 A. A. Varlet et al.

327
Piekny A, Werner M, Glotzer M (2005) Cytokinesis: welcome to the rhozone. Trends Cell Biol 15:651–658. doi:10.1016/j.tcb.2005.10.006
Pollard TD, Cooper JA (2009) Actin, a central player in cell shape andmovement. Science 326:1208–1212. doi:10.1126/science.1175862
Pollard TD, Wu JQ (2010) Understanding cytokinesis: lessons from fis-sion yeast. Nat Rev Mol Cell Biol 11:149–155. doi:10.1038/nrm2834
Rauch JN, Tse E, Freilich R, Mok SA, Makley LN, Southworth DR,Gestwicki JE (2017) BAG3 is a modular, scaffolding protein thatphysically links heat shock protein 70 (Hsp70) to the small heatshock proteins. J Mol Biol 429:128–141. doi:10.1016/j.jmb.2016.11.013
Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M,Bradke F, Jenne D, Holak TA, Werb Z, Sixt M, Wedlich-Soldner R(2008) Lifeact: a versatile marker to visualize F-actin. Nat Methods5:605–607. doi:10.1038/nmeth.1220
Rosa A, Vlassaks E, Pichaud F, Baum B (2015) Ect2/Pbl acts via rho andpolarity proteins to direct the assembly of an isotropic actomyosincortex upon mitotic entry. Dev Cell 32:604–616. doi:10.1016/j.devcel.2015.01.012
Rosati A, Graziano V, De Laurenzi V, Pascale M, Turco MC (2011)BAG3: a multifaceted protein that regulates major cell pathways.Cell Death Dis 2:e141. doi:10.1038/cddis.2011.24
Schiel JA, Childs C, Prekeris R (2013) Endocytic transport andcytokinesis: from regulation of the cytoskeleton to midbodyinheritance. Trends Cell Biol 23:319–327. doi:10.1016/j.tcb.2013.02.003
Schiel JA, Simon GC, Zaharris C, Weisz J, Castle D, Wu CC, Prekeris R(2012) FIP3-endosome-dependent formation of the secondary in-gression mediates ESCRT-III recruitment during cytokinesis. NatCell Biol 14:1068–1078. doi:10.1038/ncb2577
Taipale M, Tucker G, Peng J, Krykbaeva I, Lin ZY, Larsen B, Choi H,Berger B, Gingras AC, Lindquist S (2014) A quantitative chap-erone interaction network reveals the architecture of cellular pro-tein homeostasis pathways. Cell 158:434–448. doi:10.1016/j.cell.2014.05.039
Theriot JA (1997) Accelerating on a treadmill: ADF/cofilin promotesrapid actin filament turnover in the dynamic cytoskeleton. J CellBiol 136:1165–1168
Ulbricht A, Arndt V, Hohfeld J (2013) Chaperone-assisted proteostasis isessential for mechanotransduction in mammalian cells. CommunIntegr Biol 6:e24925. doi:10.4161/cib.24925
Ulbricht A, Hohfeld J (2013) Tension-induced autophagy: may the chap-erone be with you. Autophagy 9:920–922. doi:10.4161/auto.24213
von Dassow G, Bement WM (2005) A ring-like template for abscission.Dev Cell 9:578–580. doi:10.1016/j.devcel.2005.10.003
Welch MD, Iwamatsu A, Mitchison TJ (1997) Actin polymerization isinduced by Arp2/3 protein complex at the surface of Listeriamonocytogenes. Nature 385:265–269
Werner A, Disanza A, Reifenberger N, HabeckG, Becker J, CalabreseM,Urlaub H, Lorenz H, Schulman B, Scita G, Melchior F (2013)SCFFbxw5 mediates transient degradation of actin remodellerEps8 to allow proper mitotic progression. Nat Cell Biol 15:179–188. doi:10.1038/ncb2661
HSPB8 regulates F-actin dynamics during cytokinesis 567




















