2,3,7,8-Tetrachlorodibenzo-p-Dioxin Counteracts the p53 Response to a Genotoxicant by Upregulating...
12
TOXICOLOGICAL SCIENCES 115(2), 501–512 (2010) doi:10.1093/toxsci/kfq082 Advance Access publication March 18, 2010 2,3,7,8-Tetrachlorodibenzo-p-Dioxin Counteracts the p53 Response to a Genotoxicant by Upregulating Expression of the Metastasis Marker AGR2 in the Hepatocarcinoma Cell Line HepG2 Ariane Ambolet-Camoit,* , † Linh Chi Bui,* , † Ste ´phane Pierre,* , † Aline Chevallier,* , † Alexandre Marchand,* , † ,1 Xavier Coumoul,* , † Miche `le Garlatti,* , † Karine Andreau,§ , { Robert Barouki,* , † , ‡ and Martine Aggerbeck* , † ,2 *INSERM UMR-S 747, 75006 Paris, France; †Universite ´ Paris Descartes, Centre Universitaire des Saints-Pe `res, 75006 Paris, France; ‡APHP, Ho ˆ pital Necker Enfants Malades, Service de Biochimie Me ´tabolique, 75015 Paris, France; §CNRS EAC 4413, Unite ´ de Biologie Fonctionnelle et Adaptative (BFA), 75013 Paris, France; and {Universite ´ Paris Diderot-Paris 7, 75013 Paris, France 1 Present address: INSERM UMR-S 956, 75013 Paris, France. 2 To whom correspondence should be addressed at INSERM UMR-S 747, Toxicologie, Pharmacologie et Signalisation Cellulaire, Centre Universitaire des Saints-Pe `res, Universite ´ Paris Descartes, 45 rue des Saints-Pe `res, 75006 Paris, France. Fax: þ33 1-42-86-38-68. E-mail: [email protected]. Received December 7, 2009; accepted March 5, 2010 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) is an environmen- tal pollutant that binds the aryl hydrocarbon receptor (AhR), a transcription factor that triggers various biological responses. In this study, we show that TCDD treatment counteracts the p53 activation (phosphorylation and acetylation) elicited by a genotoxic compound, etoposide, in the human hepatocarcinoma cell line HepG2 and we delineated the mechanisms of this interaction. Using small interfering RNA knockdown experi- ments, we found that the newly described metastasis marker, anterior gradient-2 (AGR2), is involved in this effect. Both AGR2 messenger RNA (mRNA) and protein levels were increased (sixfold and fourfold, respectively) by TCDD treatment, and this effect was mediated by the AhR receptor. The half-life of AGR2 mRNA was unchanged by TCDD treatment. Analysis of the promoter of the AGR2 gene revealed three putative xenobiotic- responsive elements (XREs) in the proximal 3.5-kb promoter. Transient transfection of HepG2 cells by the Gaussia luciferase reporter gene driven by various deleted and mutated fragments of the promoter indicated that only the most proximal XRE was active. Binding of the AhR to the endogenous AGR2 promoter was also triggered by TCDD treatment. These results suggest that AhR ligands such as TCDD might contribute to tumor pro- gression by inhibiting p53 regulation (phosphorylation and acetylation) triggered by genotoxicants via the increased expres- sion of the metastasis marker AGR2. Key Words: AGR2; TCDD; AhR; etoposide; p53. Delineating the effects of combinations of toxicants (cocktail effects) is one of the most challenging issues in current toxicology. Since humans and multiple ecosystems are exposed to hundreds of pollutants, risk assessment should take into consideration the possible interactions among these chemicals. This is clearly impractical for all the possible combinations. However, one feasible approach would be to define the major signaling pathways activated by toxic compounds and to assess the interactions among those pathways using representative activators. In the present study, we focused on the interaction between a genotoxic compound, etoposide, and a cancer- promoting nongenotoxic pollutant, 2,3,7,8-tetrachlorodibenzo- p-dioxin (TCDD), and we found that anterior gradient-2 (AGR2) inhibits the posttranslational modifications of the p53 protein elicited by etoposide. TCDD is a ubiquitous chemical in the environment, which accumulates in animal and human tissues. TCDD has been classified as a group 1 human carcinogen by the International Agency for Research on Cancer and as likely to be carcinogenic to humans by the Environmental Protection Agency in the United States because it is suspected to play a role in cancer development (Kaiser, 2000). Most TCDD effects are mediated by the aryl hydrocarbon receptor (AhR) (Gu et al., 2000). Upon binding to its ligands, cytoplasmic AhR, which is bound to several chaperone proteins (Petrulis and Perdew, 2002; Ramadoss and Perdew, 2005), undergoes a conformational change that leads to its dissociation from the chaperone proteins and its translocation into the nucleus where it dimerizes with its partner Arnt (AhR receptor nuclear translocator; Swanson, 2002). The heterodimer binds to xenobiotic-responsive elements (XREs) in the promoters of target genes resulting in the upregulation of these genes. TCDD is well known for upregulating the genes encoding the enzymes involved in the metabolism of xenobiotics, such as cytochrome P450 (CYP) 1A1 (Xu et al., 2005). However, a growing body of literature indicates that the AhR controls the expression of a wide variety of genes that are unrelated to the metabolism of xenobiotics (reviewed in Kung et al., 2009). Ó The Author 2010. Published by Oxford University Press on behalf of the Society of Toxicology. All rights reserved. For permissions, please email: [email protected] by guest on February 9, 2016 http://toxsci.oxfordjournals.org/ Downloaded from
-
Upload
cnrs-bellevue -
Category
Documents
-
view
3 -
download
0
Transcript of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin Counteracts the p53 Response to a Genotoxicant by Upregulating...

TOXICOLOGICAL SCIENCES 115(2), 501–512 (2010)
doi:10.1093/toxsci/kfq082
Advance Access publication March 18, 2010
2,3,7,8-Tetrachlorodibenzo-p-Dioxin Counteracts the p53 Response toa Genotoxicant by Upregulating Expression of the Metastasis Marker
AGR2 in the Hepatocarcinoma Cell Line HepG2
Ariane Ambolet-Camoit,*,† Linh Chi Bui,*,† Stephane Pierre,*,† Aline Chevallier,*,† Alexandre Marchand,*,†,1
Xavier Coumoul,*,† Michele Garlatti,*,† Karine Andreau,§,{ Robert Barouki,*,†,‡ and Martine Aggerbeck*,†,2
*INSERM UMR-S 747, 75006 Paris, France; †Universite Paris Descartes, Centre Universitaire des Saints-Peres, 75006 Paris, France; ‡APHP, Hopital Necker
Enfants Malades, Service de Biochimie Metabolique, 75015 Paris, France; §CNRS EAC 4413, Unite de Biologie Fonctionnelle et Adaptative (BFA), 75013
Paris, France; and {Universite Paris Diderot-Paris 7, 75013 Paris, France
1 Present address: INSERM UMR-S 956, 75013 Paris, France.2 To whom correspondence should be addressed at INSERM UMR-S 747, Toxicologie, Pharmacologie et Signalisation Cellulaire, Centre Universitaire des
Saints-Peres, Universite Paris Descartes, 45 rue des Saints-Peres, 75006 Paris, France. Fax: þ33 1-42-86-38-68. E-mail: [email protected].
Received December 7, 2009; accepted March 5, 2010
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) is an environmen-
tal pollutant that binds the aryl hydrocarbon receptor (AhR),
a transcription factor that triggers various biological responses. In
this study, we show that TCDD treatment counteracts the
p53 activation (phosphorylation and acetylation) elicited by
a genotoxic compound, etoposide, in the human hepatocarcinoma
cell line HepG2 and we delineated the mechanisms of this
interaction. Using small interfering RNA knockdown experi-
ments, we found that the newly described metastasis marker,
anterior gradient-2 (AGR2), is involved in this effect. Both AGR2
messenger RNA (mRNA) and protein levels were increased
(sixfold and fourfold, respectively) by TCDD treatment, and this
effect was mediated by the AhR receptor. The half-life of AGR2
mRNA was unchanged by TCDD treatment. Analysis of the
promoter of the AGR2 gene revealed three putative xenobiotic-
responsive elements (XREs) in the proximal 3.5-kb promoter.
Transient transfection of HepG2 cells by the Gaussia luciferase
reporter gene driven by various deleted and mutated fragments of
the promoter indicated that only the most proximal XRE was
active. Binding of the AhR to the endogenous AGR2 promoter
was also triggered by TCDD treatment. These results suggest that
AhR ligands such as TCDD might contribute to tumor pro-
gression by inhibiting p53 regulation (phosphorylation and
acetylation) triggered by genotoxicants via the increased expres-
sion of the metastasis marker AGR2.
Key Words: AGR2; TCDD; AhR; etoposide; p53.
Delineating the effects of combinations of toxicants (cocktail
effects) is one of the most challenging issues in current
toxicology. Since humans and multiple ecosystems are exposed
to hundreds of pollutants, risk assessment should take into
consideration the possible interactions among these chemicals.
This is clearly impractical for all the possible combinations.
However, one feasible approach would be to define the major
signaling pathways activated by toxic compounds and to assess
the interactions among those pathways using representative
activators. In the present study, we focused on the interaction
between a genotoxic compound, etoposide, and a cancer-
promoting nongenotoxic pollutant, 2,3,7,8-tetrachlorodibenzo-
p-dioxin (TCDD), and we found that anterior gradient-2
(AGR2) inhibits the posttranslational modifications of the
p53 protein elicited by etoposide.
TCDD is a ubiquitous chemical in the environment, which
accumulates in animal and human tissues. TCDD has been
classified as a group 1 human carcinogen by the International
Agency for Research on Cancer and as likely to be
carcinogenic to humans by the Environmental Protection
Agency in the United States because it is suspected to play
a role in cancer development (Kaiser, 2000). Most TCDD
effects are mediated by the aryl hydrocarbon receptor (AhR)
(Gu et al., 2000). Upon binding to its ligands, cytoplasmic
AhR, which is bound to several chaperone proteins (Petrulis
and Perdew, 2002; Ramadoss and Perdew, 2005), undergoes
a conformational change that leads to its dissociation from the
chaperone proteins and its translocation into the nucleus
where it dimerizes with its partner Arnt (AhR receptor
nuclear translocator; Swanson, 2002). The heterodimer binds
to xenobiotic-responsive elements (XREs) in the promoters of
target genes resulting in the upregulation of these genes.
TCDD is well known for upregulating the genes encoding the
enzymes involved in the metabolism of xenobiotics, such as
cytochrome P450 (CYP) 1A1 (Xu et al., 2005). However,
a growing body of literature indicates that the AhR controls
the expression of a wide variety of genes that are unrelated to
the metabolism of xenobiotics (reviewed in Kung et al.,2009).
� The Author 2010. Published by Oxford University Press on behalf of the Society of Toxicology. All rights reserved.For permissions, please email: [email protected]
by guest on February 9, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

Etoposide is an antineoplastic compound that inhibits
topoisomerase II activity, thus leading to single-strand DNA
breaks and p53 activation. Previous studies have shown that
TCDD attenuates the p53 response to DNA-damaging agents
in HepG2 cells (Paajarvi et al., 2005). In human keratinocytes,
TCDD was shown to attenuate senescence and repress p53
expression leading to immortalization of the cells (Ray and
Swanson, 2004). In AhR�/� fibroblasts transfected with the
AhR under TET-OFF control, AhR activation inhibits E2F1-
induced apoptosis (Marlowe et al., 2008). Although cross talk
between the apoptosis/cell cycle pathways and the AhR
signaling pathway has been described (see Puga et al., 2009
for review), this appears to be a species- or cell type–dependent
phenomena (Marlowe and Puga, 2005) and the molecular
mechanisms are only partially understood.
We hypothesized that the interaction between the TCDD/
AhR pathway and the p53 pathway could be mediated through
the product of an AhR target gene. Recent high-throughput
analyses of TCDD-regulated gene expression in HepG2 cells
have revealed novel targets for this pollutant, which may
account for some features of its toxicity (Frueh et al., 2001;
Kim et al., 2006; Puga et al., 2000). In particular, the
regulation of several genes involved in cell motility may
account for the toxicity of TCDD observed during develop-
ment and for its effects on genes promoting cancer progression
(Diry et al., 2006). For the purposes of this study, we focused
on the AGR2 gene because previous studies showed that
overexpression of AGR2 attenuates p53 serine phosphorylation
(Pohler et al., 2004). Our microarray analyses have revealed
a 10-fold increase of AGR2 gene expression in HepG2 cells
following 30 h of treatment with 25nM TCDD (Marchand and
Garlatti, unpublished results).
Human AGR2, also designated as hAG2 and as GOB-4 in
the mouse (Komiya et al., 1999), are orthologues of the
secreted Xenopus laevis protein (XAG-2) (Aberger et al.,1998). In this species, the protein is involved in the
differentiation of the mucus-secreting cement gland and the
pattern of anterior neural tissues. In humans, AGR2 expression
is increased in various cancers (Innes et al., 2006; Zhang et al.,2005) and, recently, the protein was suggested to be a potential
oncogene in lung cancer (Fritzsche et al., 2007; Zhu et al.,2007). In addition, overexpression of AGR2 silences p53
activity in ultraviolet (UV)-exposed lung cancer cells (Pohler
et al., 2004). In the present study, we found that TCDD
treatment increased AGR2 protein and messenger RNA
(mRNA) levels and elicited an AhR-mediated activation of
the AGR2 gene promoter. We also provide evidence that
TCDD attenuates p53 phosphorylation and acetylation as well
as caspases-3/7 activation by etoposide and, using small
interfering RNA (siRNA) knockdown experiments, we show
that these effects of TCDD are mediated by the upregulation of
AGR2. Thus, AGR2 appears to be a novel participant involved
in the interaction between the TCDD/AhR pathway and the
etoposide/p53 pathway in HepG2 cells.
MATERIALS AND METHODS
Cell culture and chemicals. The human hepatocarcinoma cell line HepG2,
obtained from the ATCC (# HB-8065), was cultured as described previously
(Bui et al., 2009). TCDD was purchased from Promochem (Molsheim, France).
Benzo(a)pyrene (B(a)P) and etoposide were from Sigma-Aldrich (France).
dimethylsulfoxide (DMSO) 0.15% (Merck) was used as a control. Oligonu-
cleotides were purchased from Operon or Eurogentec (France).
RNA preparation. For most experiments, 0.4 million cells were seeded
into six-well plates and treated, or not, 2 days later with the compounds
indicated in the figures. RNA was prepared using the RNeasy mini kit from
Qiagen (France) as described (Bui et al., 2009).
Quantitative reverse transcriptase-PCR. Reverse transcription was per-
formed using the High Capacity cDNA Reverse Transcription Kit (Applied
Biosystems, France) as described (Bui et al., 2009). Complementary DNAs
(cDNAs) were aliquoted and stored at �80�C. Quantitative PCR was
performed with 40 ng of cDNA as described previously (Bui et al., 2009).
The AGR2 and reference gene RPL13A, a ribosomal gene mRNA, amounts
were measured. The primers were: AGR2 forward 5#-GCCATCAGGA-
GAAAGGTG-3# and reverse 5#-GCCAAAAAGGACACAAAGG-3# and
RPL13A forward 5#-CCTGGAGGAGAAGAGGAAAGAGA-3# and reverse
5#-GAGGACCTCTGTGTATTTGTCAA-3#.
Cellular protein extracts. Proteins were prepared from cells seeded under
the same conditions as for the mRNA experiments. The cells were washed
twice with PBS and scraped into 200 ll of M-Per buffer (Pierce, France)
containing 0.15M NaCl and protease and phosphatase inhibitors (Sigma). The
cellular homogenate was centrifuged for 15 min at 13,400 g at 4�C. The
protein concentration was measured by the bicinchoninic acid method (Pierce)
using bovine serum albumin (fraction V) as a standard. The protein extract
was stored at �80�C.
Western blots. Samples (20–40 lg of extract) and 8 lg of protein ladder
(Fermentas, France) were electrophoresed in a 15 or 12% polyacrylamide, 0.1%
SDS gel. Proteins were transferred onto nitrocellulose Hybond C extra
membranes (GE Healthcare, France). Blots were incubated for 120 min in 0.2%
I-Block solution (Applied Biosystems) and 0.1% Tween-20 in 13 PBS at room
temperature. Primary antibodies (anti-AGR2, Abnova, 1/1000; anti-phospho
p53 [Ser15], Cell Signaling, 1/1000; anti-acetylated p53 [Lys382], Abcam,
1/1000; anti-p53, Cell Signaling, 1/1000 or anti-actin, Abcam, 1/20,000; anti-
AhR [ab2770], Abcam, 1/1000, France) were incubated overnight at 4�C and
rinsed five times (5 min) with 0.1% Tween-20 in 13 PBS at room temperature.
Signals were visualized, and quantification was carried out as previously
described using actin to normalize the signals (Bui et al., 2009).
mRNA stability. HepG2 cells were treated with either the vehicle (0.15%
DMSO) or 25nM TCDD for 30 h. 5,6-Dichloro-1b-ribofuranosyl benzimid-
azole (DRB; Sigma-Aldrich) (10lM) was then added to the cells, and mRNA
was prepared at various times after this addition.
Plasmids and transient transfections. Fragments of the AGR2 gene
promoter were obtained by PCR and subcloned into the TOPO TA cloning
vector (Invitrogen, France). Plasmids were prepared (QIAprep Spin Miniprep
Kit; Qiagen), and after DNA digestion, the promoter fragment was cloned into
the pGLuc vector (Biolabs, Ozyme, France). Mutagenesis was performed using
the Quick Change XL site-directed mutagenesis kit from Stratagene (France).
The PCR products were purified (Mini-Elute PCR purification kit; Qiagen) and
cloned in the pGLuc vector as described above. Mutations were verified by
DNA sequencing (GATC, France).
HepG2 cells (0.4 million cells per well in six-well plates) were transiently
transfected in duplicate or triplicate by the lipofectamine-2000 reagent, using
1 lg DNA per well, according to the manufacturer’s instructions (Invitrogen).
Sixteen hours later, fresh medium containing the compounds was added to the
cells for 30 h. The Gaussia luciferase activity was assayed with the Biolabs kit
according to the manufacturer’s instructions in a Berthold luminometer.
502 AMBOLET-CAMOIT ET AL.
by guest on February 9, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

Chromatin immunoprecipitation assay. HepG2 cells were seeded into
150 mm–diameter dishes 48 h before being processed for chromatin
immunoprecipitation (ChIP; ~20 3 106 cells per dish). Treatments with TCDD
(25nM) were performed for 15 and 45 min in Dulbecco’s Modified Eagle
Medium without phenol red and supplemented with 3% charcoal-treated calf
serum. The cells were rinsed with 37�C PBS, and nuclear proteins were cross-
linked to DNA for 10 min at 37�C by adding formaldehyde to a final concentration
of 1%. Cross-linking was stopped by adding glycine to a final concentration of
0.125M for 5 min on ice. The cells were collected in ice-cold PBS supplemented
with complete protease inhibitor tablets (Cat. 1873580; Roche, Mannheim,
Germany) and pelleted. Pellets were lysed in 500 ll of lysis buffer (1% SDS;
10mM EDTA; 50mM Tris-HCl, pH 8.1; and protease inhibitors) for 10 min on
ice. The solution was sonicated 10 times for 30 s each at the high position with
a Dismembrator 300 (Fisher). The soluble chromatin was then centrifuged at
13,400 g for 15 min at 4�C and then diluted 1:10 in ChIP dilution buffer (0.01%
SDS; 1.1% Triton X-100; 1.2mM EDTA; 16.7mM Tris-HCl, pH 8.1; 167mM
NaCl; and protease inhibitors). An aliquot of soluble chromatin was set aside as
the input fraction. Diluted chromatin was precleared with protein A/G Plus
agarose (Cat Sc-2003; Santa Cruz) at 4�C for 2 h with rotation. Protein A/G
agarose beads were pelleted, the supernatant was transferred to a new tube, and
incubated with 5 lg of anti-AhR antibody (Cat SA-210; BioMol) or 5 lg of
normal rabbit IgG (negative control, Cat Sc-2027; Santa Cruz) for immunopre-
cipitation overnight at 4�C with rotation. After immunoprecipitation, fresh
protein A/G agarose beads were added and incubated for 1 h at 4�C with rotation.
Agarose beads were pelleted and washed sequentially for 5 min with the
following buffers: once with buffer I (0.1% SDS; 1% Triton X-100; 2mM EDTA;
200mM Tris-HCl, pH 8.1; 150mM NaCl; and protease inhibitors), twice with
buffer II (0.1% SDS; 1% Triton X-100; 2mM EDTA; 200mM Tris-HCl, pH 8.1;
500mM NaCl; and protease inhibitors), and twice with buffer III (0.25M LiCl; 1%
nonidet-P40; 1% deoxycholate; 1mM EDTA; and 10mM Tris-HCl, pH 8.1).
Protein-DNA complexes were eluted by adding 400ll of elution buffer (1% SDS,
0.1M NaHCO3, and protease inhibitors) with incubation for 20 min at room
temperature with rotation (input samples were included). The cross-linking was
reversed by the addition of NaCl to a final concentration of 0.2M and overnight
incubation at 65�C. The DNA were purified using a PCR purification kit (Cat
28106; Qiagen) and eluted in 25ll. ChIP DNA were subjected to quantitative PCR
using the specific primers covering the region between�807 and�651 bp of the
human AGR2 promoter region (forward primer: 5#-AAATGGTCTTGAGGG-
GAAAAA-3# and reverse primer 5#-GCAACTTCCCTCCACAGAGT-3#). PCR
cycles were proceeded as follows: Taq activation (15 min), denaturation (15 s,
95�C), and annealing and extension (1 min, 60�C). The data were presented
according to the classical comparative Ct method except that we choose to present
the values obtained with the mock antibody instead of using them as a reference
(compared to classical mRNA expression studies). In each condition (Input, AhR
ChIP, and Mock ChIP), an untreated sample was used as a control. The amount of
the AGR2 target gene in TCDD-treated cells, relative to the control sample, was
measured by the equation 2�DCt, where DCt ¼ (Ct TCDD sample � Ct control
sample). The equation thus represents the expression of the target gene in the
TCDD-treated sample, relative to its expression in the control sample.
Nuclear extract preparation and electrophoretic mobility shift assay.
HepG2 cells (3 million/10-cm dish) were treated for 90 min with 25nM TCDD
or 0.15% DMSO. Cells were washed, scraped on ice into PBS, 1mM EDTA,
and centrifuged for 5 min at 1500 3 g at 4�C. The pellet was resuspended in
250 ll of 10mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (Hepes)
(pH 7.9), 50mM NaCl, 0.5M sucrose, 0.1mM EDTA, 0.5% Triton, 1mM DTT,
and 1/100 phosphatase and protease inhibitors, incubated on ice for 5 min,
centrifuged for 10 min at 150 3 g at 4�C. The nuclear pellet was resuspended
in 500 ll of 10mM Hepes (pH 7.9), 10mM KCl, 0.1mM EDTA, 0.1mM
EGTA, 0.5% Triton, 1mM DTT, and phosphatase and protease inhibitors and
centrifuged for 5 min at 150 3 g at 4�C. The pellet was resuspended in 500 ll
of 10mM Hepes (pH 7.9), 500mM NaCl, 0.1mM EDTA, 0.1mM EGTA, 0.5%
Triton, 1mM DTT, and phosphatase and protease inhibitors and mixed for
15 min at 4�C. The solution was centrifuged for 15 min at 14,000 3 g at 4�Cand the supernatant containing nuclear proteins was stored at �80�C.
The electrophoretic mobility shift assay was performed as described
(Gouedard et al., 2004). Briefly, synthetic double-stranded DNA probe
(forward strand: 5#-GGCTTCCTGCTTGCGTGGCAGGAAAGT-3#) was la-
beled with (a-32P) dCTP (PerkinElmer). The nuclear extract (15 lg) was
incubated for 20 min with a 50-fold excess of nonlabeled probe or 3 lg anti-
AhR antibody (Ab63636; Abcam) at room temperature, and the labeled probe
(corresponding to 100,000 cpm) was added for an additional 20-min
incubation. DNA-protein complexes were separated under nondenaturing
conditions on a 6% polyacrylamide gel, detected, and quantified.
AhR and AGR2 gene silencing. HepG2 cells (0.4 million cells per well in
six-well plates) were transfected with 5nM of either control siRNA or two
different siAhR (Qiagen) using HiPerfect transfection reagent according to the
manufacturer’s protocol (Qiagen) as described (Bui et al., 2009). After 12 h, the
medium was replaced by fresh medium, and 16 h later, the cells were treated, or
not, with 25nM TCDD for an additional 30 h. Control siRNA was a custom
GFP siRNA (ref 1027020), siAhR1 was the fluorescent Hs_AHR_5_HP
validated siRNA (ref SI02780148) and siAhR2 was the Hs_AHR_6_HP
validated siRNA (ref SI03043971).
For siAGR2 experiments, HepG2 cells were transfected using the ‘‘Fast-
Forward Protocol’’ (Qiagen). HepG2 cells were seeded and transfected
simultaneously with 5nM of either control siRNA (ref SR-CL000-005;
Eurogentec) or siAGR2 (ref SI00293447; Qiagen), using HiPerfect transfection
reagent. After 16 h, the cells were treated with either 0.15% DMSO or 25nM
TCDD. Twenty-four hours later, the cells were treated, or not, with either 15 or
75lM etoposide and DMSO or TCDD for an additional 24 h.
Caspases-3/7 activities assay. HepG2 cells (5000 cells per well in 96-well
plates) were seeded and transfected, or not, with control siRNA or siAGR2 in
triplicate. Cells were treated as described above in ‘‘AhR and AGR2 gene
silencing.’’ After a 48-h treatment, 100 ll of fresh medium and 100 ll of
Caspase-Glo 3/7 reagent (Promega) were added to each well. After 90-min
incubation at room temperature, the luminescence was measured in a plate-
reading luminometer (ENVision 2101 Multi-label Reader; PerkinElmer).
XTT assay. HepG2 cells (10,000 cells per well in 96-well plates) were
seeded and treated, or not, for 24 h by 25nM TCDD prior to the treatment for an
additional 24 or 48 h by various concentrations of etoposide or DMSO. Cell
viability was measured by the XTT test following the manufacturer’s
instructions (In Cytotox XTT, Xenometrix; Biogenic, France).
Statistical analysis. The data (mean ± SEM) were obtained from at least
three different experiments. A Kruskal-Wallis test (nonparametric comparison
of k independent series) was followed by an ANOVA test (parametric
comparison of k independent series, Fisher’s exact test). A value of p < 0.05 or
less was accepted as being statistically significant.
RESULTS
Interaction between TCDD and Etoposide Treatments
Treatment of HepG2 cells with etoposide led to the activation
of p53 as demonstrated by an increased phosphorylation of serine
15 (Fig. 1A) and by an increased acetylation of lysine
382 (Fig. 1B). Under these experimental conditions, the total
amount of p53 was increased by etoposide treatment as already
described by Paajarvi et al. (2005) (Fig. 1A). p53 activation may
initiate either apoptosis or cell cycle arrest and, ultimately,
senescence, depending on the severity of the damage (Kruse and
Gu, 2009). We, therefore, investigated the regulation of the
effector caspases-3/7, following the same treatment. As shown in
Figure 1C, etoposide treatment increased the caspases-3/7
activities. When combined with etoposide, TCDD prevented,
TCDD-REGULATED AGR2 INHIBITS ETOPOSIDE EFFECT 503
by guest on February 9, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

partially, the increases in p53 phosphorylation and acetylation
and caspases-3/7 activities by the genotoxic agent, whereas
TCDD alone had no effect (Fig. 1). These experiments show that
the distinct signaling pathways activated by these two toxicants
interact at the level of the p53 protein hub. We next evaluated the
effect of a combination of etoposide and TCDD on the viability/
proliferation of the cells as compared to each compound alone,
by measuring the activity of the mitochondrial succinate
dehydrogenase system. As shown in Figure 1D, TCDD does
not modify the enzyme activity as compared to the vehicle
DMSO. Treatment of the cells for 24 and 48 h with 30 and 75lM
etoposide led to a statistically significant decrease of the enzyme
activity. Pretreatment of the cells for 24 h with TCDD prior to
etoposide treatment partially prevented this decrease (Fig. 1D).
In order to determine the mechanism of this cross talk, we
analyzed the gene targets of TCDD as revealed by our
microarray data and we found that the AGR2 gene, which has
previously been shown to counteract p53 activation (Pohler
et al., 2004), was one of the putative targets of TCDD in
HepG2 cells (Marchand et al., 2005; Marchand and Garlatti,
unpublished data). We, therefore, investigated the regulation of
the AGR2 gene by TCDD in order to establish its role in the
etoposide-TCDD interaction.
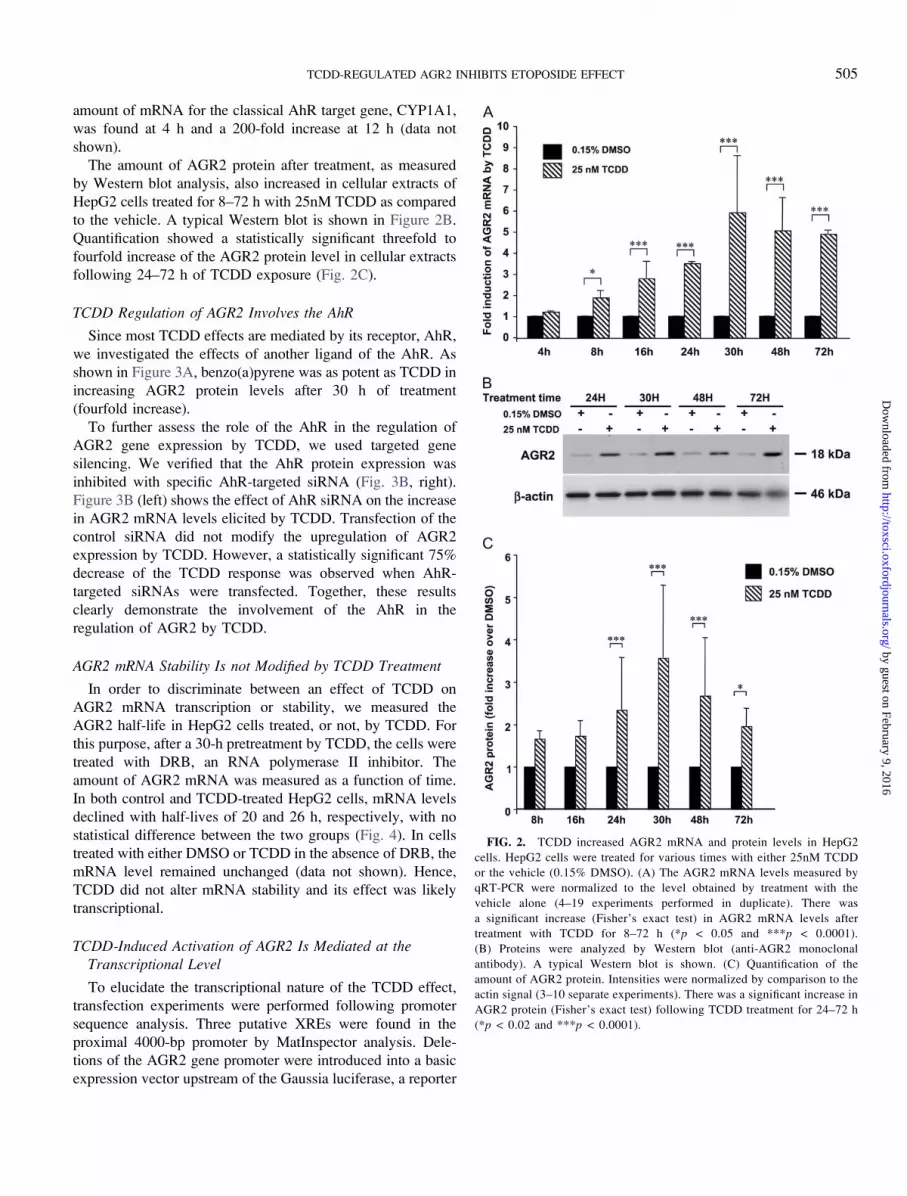
TCDD Increases AGR2 mRNA and Protein Expression inHepG2 Cells
HepG2 cells were treated with vehicle alone (0.15% DMSO)
or with 25nM TCDD within the concentration range typically
used for experiments with human cells (5–100nM). RNA levels
were monitored by quantitative real-time PCR (qRT-PCR). As
shown in Figure 2A, a sixfold increase in RNA levels was
found after 30 h of TCDD treatment. The increase in the
amount of mRNA first became significant at 8 h and remained
stable between 30 and 72 h (Fig. 2A). A 60-fold increase of the
FIG. 1. Interaction between etoposide and TCDD on p53 phosphorylation and acetylation and caspase-3/7 activities in HepG2 cells. (A) HepG2 cells were
pretreated with either 0.15% DMSO or 25nM TCCD for 24 h and incubated, or not, with 15lM etoposide for an additional 24 h. Western blot analysis was
performed on protein extracts, and p53 was identified with antibodies directed against total p53 or phospho p53 (Ser15). Intensities were normalized by comparison
to the actin signal (three separate experiments). There was a statistically significant difference (Fisher’s exact test) in p53 phosphorylation when the cells were treated
with etoposide plus TCDD as compared to etoposide alone (*p < 0.05). (B) HepG2 cells were pretreated with either 0.15% DMSO or 25nM TCCD for 24 h and
incubated, or not, with 75lM etoposide for an additional 24 h. Western blot analysis was performed on protein extracts and p53 was identified with an antibody
directed against acetylated-p53 (Lys382). Intensities were normalized by comparison to the actin signal (four separate experiments). There was a statistically
significant difference (Fisher’s exact test) in p53 acetylation when the cells were treated with etoposide plus TCDD as compared to etoposide alone (**p < 0.001 and
*p < 0.05). (C) HepG2 cells were pretreated with either 0.15% DMSO or 25nM TCDD for 24 h and incubated, or not, with 75lM etoposide for an additional 24 h.
There was a statistically significant increase (Fisher’s exact test) in caspases-3/7 activities following etoposide treatment as compared to DMSO as well as
a significant decrease following treatment with etoposide plus TCDD as compared to etoposide alone (*p < 0.05, **p < 0.001, and ***p < 0.0001). (D) HepG2 cells
were pretreated or not for 24 h with 25nM TCDD and treated for either 24 or 48 h with 15, 30, or 75lM etoposide. Mitochondrial succinate dehydrogenase activity
was measured (three separate experiments in triplicate). There was a significant decrease in enzyme activity following treatment with 30 and 75lM etoposide
(****p < 0.0001, ***p < 0.001, and **p < 0.01). Pretreatment with TCDD partially prevented the decrease in enzyme activity by etoposide (*p < 0.05).
504 AMBOLET-CAMOIT ET AL.
by guest on February 9, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from
amount of mRNA for the classical AhR target gene, CYP1A1,
was found at 4 h and a 200-fold increase at 12 h (data not
shown).
The amount of AGR2 protein after treatment, as measured
by Western blot analysis, also increased in cellular extracts of
HepG2 cells treated for 8–72 h with 25nM TCDD as compared
to the vehicle. A typical Western blot is shown in Figure 2B.
Quantification showed a statistically significant threefold to
fourfold increase of the AGR2 protein level in cellular extracts
following 24–72 h of TCDD exposure (Fig. 2C).
TCDD Regulation of AGR2 Involves the AhR
Since most TCDD effects are mediated by its receptor, AhR,
we investigated the effects of another ligand of the AhR. As
shown in Figure 3A, benzo(a)pyrene was as potent as TCDD in
increasing AGR2 protein levels after 30 h of treatment
(fourfold increase).
To further assess the role of the AhR in the regulation of
AGR2 gene expression by TCDD, we used targeted gene
silencing. We verified that the AhR protein expression was
inhibited with specific AhR-targeted siRNA (Fig. 3B, right).
Figure 3B (left) shows the effect of AhR siRNA on the increase
in AGR2 mRNA levels elicited by TCDD. Transfection of the
control siRNA did not modify the upregulation of AGR2
expression by TCDD. However, a statistically significant 75%
decrease of the TCDD response was observed when AhR-
targeted siRNAs were transfected. Together, these results
clearly demonstrate the involvement of the AhR in the
regulation of AGR2 by TCDD.
AGR2 mRNA Stability Is not Modified by TCDD Treatment
In order to discriminate between an effect of TCDD on
AGR2 mRNA transcription or stability, we measured the
AGR2 half-life in HepG2 cells treated, or not, by TCDD. For
this purpose, after a 30-h pretreatment by TCDD, the cells were
treated with DRB, an RNA polymerase II inhibitor. The
amount of AGR2 mRNA was measured as a function of time.
In both control and TCDD-treated HepG2 cells, mRNA levels
declined with half-lives of 20 and 26 h, respectively, with no
statistical difference between the two groups (Fig. 4). In cells
treated with either DMSO or TCDD in the absence of DRB, the
mRNA level remained unchanged (data not shown). Hence,
TCDD did not alter mRNA stability and its effect was likely
transcriptional.
TCDD-Induced Activation of AGR2 Is Mediated at theTranscriptional Level
To elucidate the transcriptional nature of the TCDD effect,
transfection experiments were performed following promoter
sequence analysis. Three putative XREs were found in the
proximal 4000-bp promoter by MatInspector analysis. Dele-
tions of the AGR2 gene promoter were introduced into a basic
expression vector upstream of the Gaussia luciferase, a reporter
FIG. 2. TCDD increased AGR2 mRNA and protein levels in HepG2
cells. HepG2 cells were treated for various times with either 25nM TCDD
or the vehicle (0.15% DMSO). (A) The AGR2 mRNA levels measured by
qRT-PCR were normalized to the level obtained by treatment with the
vehicle alone (4–19 experiments performed in duplicate). There was
a significant increase (Fisher’s exact test) in AGR2 mRNA levels after
treatment with TCDD for 8–72 h (*p < 0.05 and ***p < 0.0001).
(B) Proteins were analyzed by Western blot (anti-AGR2 monoclonal
antibody). A typical Western blot is shown. (C) Quantification of the
amount of AGR2 protein. Intensities were normalized by comparison to the
actin signal (3–10 separate experiments). There was a significant increase in
AGR2 protein (Fisher’s exact test) following TCDD treatment for 24–72 h
(*p < 0.02 and ***p < 0.0001).
TCDD-REGULATED AGR2 INHIBITS ETOPOSIDE EFFECT 505
by guest on February 9, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

enzyme secreted into the medium of the cell culture. These
constructs were transiently transfected into HepG2 cells to
analyze the putative functional XRE(s) involved in the
regulation of the AGR2 gene by AhR agonists. The cells were
treated with TCDD or the vehicle, and the luciferase activity
released into the culture medium was measured. As expected,
TCDD had no effect on the empty plasmid and the basal
activity was low (0.3 arbitrary units). In contrast, all the AGR2
constructs tested displayed a higher basal activity (2–4 arbitrary
units) and a twofold to fivefold increase following treatment
with 25nM TCDD was observed for the constructs tested
(Fig. 5A).
In order to determine which XREs were responsible for the
induction by TCDD of the luciferase activity, XREs 1 and
2 were mutated. As shown in Figure 5A, the mutation of the
proximal XRE (XRE1) completely abolished the effect of
TCDD on the regulation of the AGR2 promoter, whereas the
mutation in XRE2 did not modify the reporter activity.
Deletion of XRE3 did not modify the regulation of the reporter
gene by dioxin. ChIP experiments demonstrated that treatment
for 45 min with TCDD stimulated AhR recruitment to the
region of the endogenous AGR2 gene promoter that encom-
passes XRE1 (Fig. 5B, left). Moreover, electrophoretic
mobility shift assay experiments revealed a specific band when
a probe encompassing XRE1 was incubated with nuclear
extracts prepared from TCDD-treated HepG2 cells. This band
was displaced by competition with a 50-fold excess of cold
probe and was partially decreased in intensity when an anti-
AhR antibody was used (Fig. 5B, right).
A dose-response curve was performed to determine the
responsiveness of the longest AGR2 construct (pGLuc
AGR2 �3401/þ8) to TCDD. A significant increase of the
Gaussia luciferase activity was obtained at a concentration as
low as 1nM TCDD and up to 50nM TCDD (Fig. 5C). Another
AhR ligand, benzo(a)pyrene (5lM), was as effective as TCDD
(fourfold increase) in increasing the reporter activity of this
construct (Fig. 5C).
TCDD Attenuation of Etoposide Action Is Mediated by AGR2
As shown in Figure 1A, etoposide treatment induced
phosphorylation of Ser15 and acetylation of Lys382 in p53.
Pretreatment with TCDD partially prevented these modifica-
tions of p53 by etoposide. In order to determine whether AGR2
was involved in the TCDD effect, HepG2 cells were
transfected with either control siRNA or AGR2 siRNA prior
to the treatments. AGR2 siRNA transfection significantly
decreased AGR2 mRNA levels of both untreated or TCDD-
treated cells (75–80% decrease, data not shown). AGR2 siRNA
transfection decreased markedly the amount of AGR2 protein
FIG. 4. Half-life of AGR2 mRNA in HepG2 cells. HepG2 cells were
pretreated for 30 h with either 25nM TCDD or the vehicle (0.15% DMSO).
Then, the cells were treated, or not, with 10lM DRB and mRNA was isolated
as a function of time. The AGR2 mRNA level was assessed by qRT-PCR. The
statistical analysis (Fisher’s exact test) showed no statistically significant
difference in the half-life of AGR2 mRNA in cells treated, or not, by TCDD in
the presence of DRB (six experiments performed in duplicate).
FIG. 3. AGR2 regulation by TCDD involves the AhR. (A) HepG2 cells
were treated for 30 h with either 25nM TCDD, 5lM benzo(a)pyrene (BaP), or
the vehicle (0.15% DMSO). Proteins were analyzed by Western blot (anti-
AGR2 monoclonal antibody). (A) A typical Western blot is shown. Intensities
were normalized by comparison to the actin signal (three separate experiments).
There was a significant increase (Fisher’s exact test) in AGR2 protein following
treatment with TCDD and B(a)P (**p < 0.001). (B) Left, siRNAs (control or
AhR) were transiently transfected into HepG2 cells using the HiPerfect reagent.
After 16 h, the cells were treated, or not, with 25nM TCDD for 30 h. The
AGR2 mRNA level was measured by qRT-PCR. The mean fold increase ±
SEM as compared to the control for four independent experiments performed
in duplicate is shown. Statistical analysis (Fisher’s exact test) showed that
the TCDD-induced increase in AGR2 mRNA was significantly decreased with
the two siAhR RNAs as compared to the control siRNA (*p < 0.05,
***p < 0.0001). Right, Western blot of the amount of AhR protein in HepG2
cells transfected with either control siRNA or AhR siRNA and treated or not
with 25nM TCDD is shown. Actin is shown as a control.
506 AMBOLET-CAMOIT ET AL.
by guest on February 9, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

FIG. 5. TCDD increases AGR2 gene promoter activity. (A) AGR2 gene promoter constructs (wild type or mutated in the XRE) in front of the Gaussia luciferase
were transiently transfected into HepG2 cells, which were treated, or not, with 25nM TCDD for 30 h. The reporter activity was expressed in arbitrary units. There
was a statistically significant (Fisher’s exact test) difference in reporter activity between treated and untreated cells (**p < 0.001, ***p < 0.0001, 3–19 experiments
for each construct). (B) ChIP experiment was carried out on the AGR2 promoter XRE1 region. HepG2 cells were treated, or not, with 25nM TCDD for 45 min.
TCDD-REGULATED AGR2 INHIBITS ETOPOSIDE EFFECT 507
by guest on February 9, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

as compared to control siRNA in cells treated with etoposide
alone or combined with TCDD (Fig. 6A). In AGR2-silenced
cells, the inhibitory effect of TCDD on p53 phosphorylation
(serine 15) and acetylation (lysine 382) was abolished, whereas
control siRNA treated cells were still responsive to TCDD
(Figs. 6A and 6B). This indicates that the attenuation of p53
activation by TCDD was dependent, at least in part, on AGR2
expression. We also investigated the role of AGR2 expression
on the effector caspases-3/7 activities. As shown in Figure 6C,
when the cells were transfected by the control siRNA,
pretreatment by TCDD led to a statistically significant 50%
decrease of the effect of etoposide on caspases-3/7 activities.
The negative effect of TCDD on etoposide activation of
caspases-3/7 was considerably reduced in the presence of
AGR2 siRNA.
DISCUSSION
In this paper, we report a cross talk mechanism between
etoposide, a genotoxicant, which is a potent activator of the
p53 pathway, and TCDD, a tumor promoter, which activates
the AhR pathway in the hepatocyte-derived cell line HepG2. In
particular, pretreatment of HepG2 cells by TCDD partially
counteracts the activation of p53 induced by etoposide.
A protective effect of TCDD on the decrease in activity of
mitochondrial succinate dehydrogenase elicited by etoposide
was observed, suggesting a prosurvival/proliferative role for
TCDD in HepG2 cells. The finding that the human AGR2
silences p53 response to DNA damage (Pohler et al., 2004) and
our transcriptomic results showing that TCDD regulates AGR2
mRNA levels in the same cellular model (Marchand and
Garlatti, personal data) led us to hypothesize that this new
TCDD target could be partially responsible for some of the
effects of TCDD on p53 regulation, as proposed in Figure 7.
Therefore, we characterized the positive regulation, by TCDD,
of the gene encoding the human AGR2 and showed that TCDD
attenuates etoposide-mediated p53 phosphorylation and acet-
ylation through AGR2 gene regulation.
Induction of AGR2 by TCDD could have several con-
sequences. One hypothesis is that induction of AGR2 by
TCDD, which is a recognized carcinogen or other AhR ligands
could promote tumor formation. Indeed, recently, AGR2 was
shown to be overexpressed in several human cancers—prostate,
pancreas, lung, and breast cancer (Fritzsche et al., 2006, 2007;
Innes et al., 2006; Missiaglia et al., 2004; Ramachandran et al.,2008; Sitek et al., 2009; Thompson and Weigel, 1998; Wang
et al., 2008; Zhang et al., 2005, 2006). AGR2 also has been
proposed as a novel putative metastasis marker in breast,
prostate, and colorectal cancer (Fletcher et al., 2003; Liu et al.,2005; Smirnov et al., 2005; Thompson and Weigel, 1998), and
it is associated with poor survival of prostate cancer patients
(Zhang et al., 2007b). Stable transfection of lung carcinoma
H1299 cells by a vector expressing AGR2 was shown to
enhance colony formation, an assay that measures the ability of
a gene to influence cell survival. In another study, the
esophageal adenocarcinoma SEG-1 cell line displayed 82%
fewer colonies in soft agar and smaller tumor xenografts
when the AGR2 gene was silenced by short hairpin RNA as
compared to wild-type cells (Wang et al., 2008). Furthermore,
AGR2-expressing NIH3T3 cells showed enhanced foci forma-
tion in soft agar and were able to grow as tumors in nude mice
(Wang et al., 2008). Altogether, both in vivo and in vitrostudies suggest that the expression of this new TCDD target
correlates with a cancerous phenotype.
The data described here indicate that etoposide and TCDD
elicit opposite effects on p53 regulation. The induction of AGR2
by TCDD suggests that the AGR2 gene product may account for
the cross talk between these toxicants, which belong to two
distinct classes. Indeed, the increase in caspases-3/7 activities
and p53 phosphorylation and acetylation by etoposide is partially
blunted by TCDD treatment, and this effect appears to be
correlated with AGR2 induction. Our preliminary experiments
also indicate that constitutive overexpression of AGR2 in HepG2
cells inhibits both phosphorylation (Ser15) and acetylation
(Lys382) of p53 in cells treated with etoposide (Ambolet-Camoit
and Aggerbeck, unpublished results). Thus, the combination of
two toxicants with distinct mechanisms of action could produce
additional harmful effects that result from decreased adaptive
pathways. It will now be of interest to study the effect of other
pollutant combinations on p53 activity and AGR2 induction.
TCDD exposure leads to the activation of several target
genes involved in cell plasticity (Bui et al., 2009; Diry et al.,2006). Other results have shown that the AhR is involved
in cell-cell or cell-substratum adhesion; thus, this receptor
might play a role in cell migration (Barouki et al., 2007;
Carvajal-Gonzalez et al., 2009) and could have a major impact
on cell adhesion and matrix metabolism (Kung et al., 2009).
Gene-specific qPCR analysis with primers spanning the AhR-binding region in the AGR2 gene promoter was performed on genomic DNA immunoprecipitated with
either anti-AhR or mock IgG antibody and an equal amount of input DNA as described in the ‘‘Materials and Methods’’ section. The nontreated sample was used
as control for each condition (***p < 0.0001, three experiments) (left). The electrophoretic mobility shift assay shows the binding of nuclear extracts prepared
from TCDD-treated HepG2 cells (25nM, 90 min) to a probe encompassing the XRE1 (lane 2), after competition with an anti-AhR antibody (lane 3) and after
competition with a 503 nonlabeled probe (lane 4). Lane 1 corresponds to the free probe (right). (C) The longest wild-type construct (�3401/þ8 AGR2) transiently
transfected into HepG2 cells was treated with 0.15% DMSO or increasing TCDD concentrations for 30 h or with 5lM B(a)P. The reporter activity is expressed
in arbitrary units. There was a statistically significant (Fisher’s exact test) difference in reporter activity between treated and untreated cells at concentrations above
1nM TCDD (**p < 0.001, ***p < 0.0001, three to five experiments).
508 AMBOLET-CAMOIT ET AL.
by guest on February 9, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from
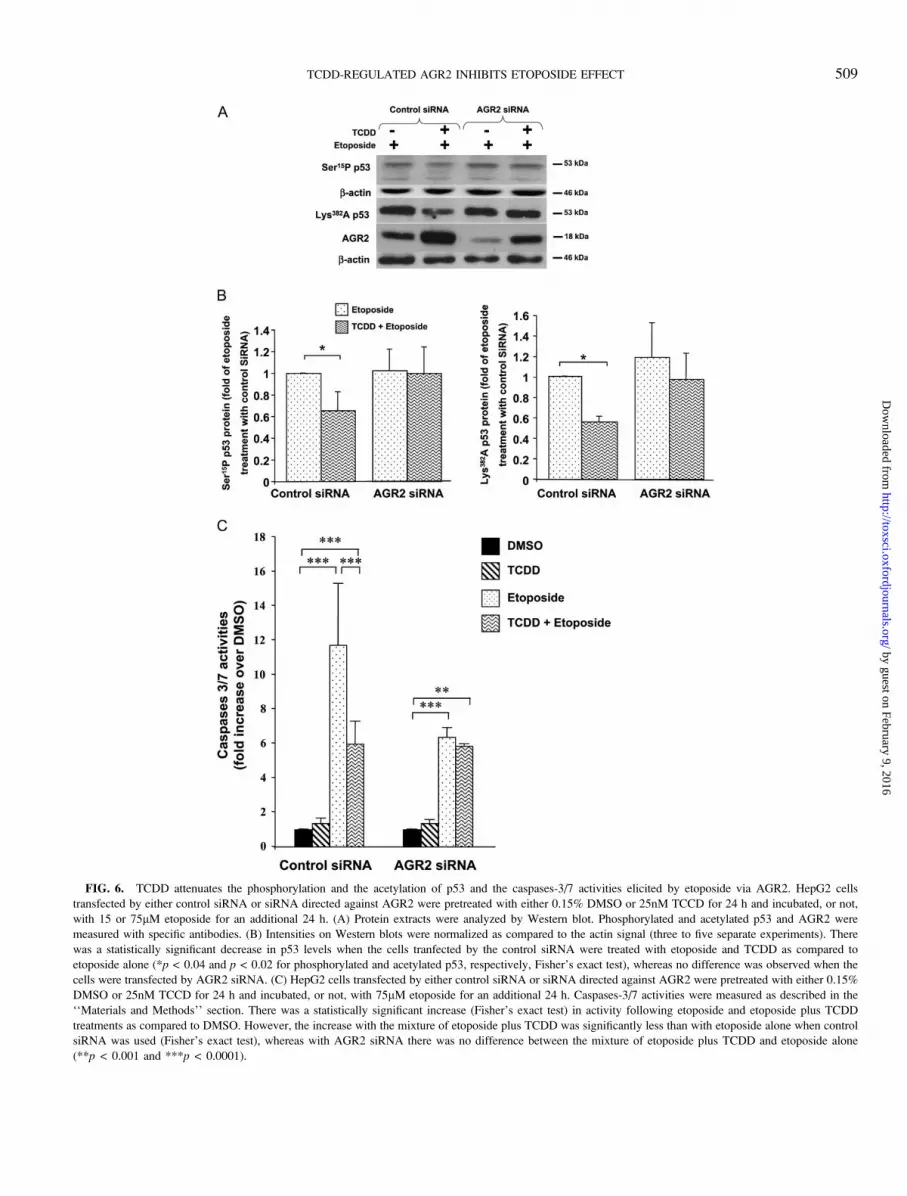
FIG. 6. TCDD attenuates the phosphorylation and the acetylation of p53 and the caspases-3/7 activities elicited by etoposide via AGR2. HepG2 cells
transfected by either control siRNA or siRNA directed against AGR2 were pretreated with either 0.15% DMSO or 25nM TCCD for 24 h and incubated, or not,
with 15 or 75lM etoposide for an additional 24 h. (A) Protein extracts were analyzed by Western blot. Phosphorylated and acetylated p53 and AGR2 were
measured with specific antibodies. (B) Intensities on Western blots were normalized as compared to the actin signal (three to five separate experiments). There
was a statistically significant decrease in p53 levels when the cells tranfected by the control siRNA were treated with etoposide and TCDD as compared to
etoposide alone (*p < 0.04 and p < 0.02 for phosphorylated and acetylated p53, respectively, Fisher’s exact test), whereas no difference was observed when the
cells were transfected by AGR2 siRNA. (C) HepG2 cells transfected by either control siRNA or siRNA directed against AGR2 were pretreated with either 0.15%
DMSO or 25nM TCCD for 24 h and incubated, or not, with 75lM etoposide for an additional 24 h. Caspases-3/7 activities were measured as described in the
‘‘Materials and Methods’’ section. There was a statistically significant increase (Fisher’s exact test) in activity following etoposide and etoposide plus TCDD
treatments as compared to DMSO. However, the increase with the mixture of etoposide plus TCDD was significantly less than with etoposide alone when control
siRNA was used (Fisher’s exact test), whereas with AGR2 siRNA there was no difference between the mixture of etoposide plus TCDD and etoposide alone
(**p < 0.001 and ***p < 0.0001).
TCDD-REGULATED AGR2 INHIBITS ETOPOSIDE EFFECT 509
by guest on February 9, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

These new roles of the AhR suggest that it could have key
functions in human cancer progression. Recent experiments
have pointed out that AhR activation inhibits apoptosis
triggered by UV light (Chopra et al., 2009; Park and
Matsumura, 2006) or by the E2F1 regulator of the cell cycle
(Marlowe et al., 2008) or by diethylnitrosamine (Paajarvi et al.,2005). Culture-induced senescence in keratinocytes is also
suppressed by TCDD or by cigarette smoke (Ray and
Swanson, 2009; Zhang et al., 2007a). Our finding that AGR2
is a novel target of the AhR that downregulates p53 and
caspases-3/7 activities elicited by a genotoxicant is another
argument for the possible involvement of AhR in tumor
progression. This hypothesis is strengthened by the fact that
AGR2 expression is increased in several human cancers and is
correlated with poor survival in breast cancer patients
(Barraclough et al., 2009).
It is noteworthy that AGR2 has been shown to be increased
by different stress conditions, such as serum or oxygen
depletion (Zweitzig et al., 2007) or allergen exposure in
asthma (Di Valentin et al., 2009). In addition, AGR2
expression is increased in the esophageal Barrett’s epithelium,
a tissue that is subjected to various stresses such as acid-bile
reflux, heatshock, and oxidative damage (Pohler et al., 2004).
Our study shows that AGR2 is also induced during xenobiotic
stress. Thus, AGR2 appears to be a target frequently activated
under stressful cellular conditions, and it is possible that the
combination of several stresses leading to AGR2 induction
may result in additional toxic effects. Although little is known
about AGR2 function in mammals, a protein disulfide
isomerase (PDI) function is suspected, based on sequence
homology (Persson et al., 2005), and AGR2 could have
a unique role in intestinal mucus production by forming
disulfide bonds with mucin 2 (Park et al., 2009). The fact that
AGR2 is probably a member of the PDI chaperone family
correlates with the fact that its expression is increased by
a variety of stress conditions, which require chaperone activity
in the endoplasmic reticulum. However, other still unknown
functions of AGR2 may also be important, in addition to the
fine-tuning of p53 activity. Since AGR2 expression is
correlated with cancer aggressiveness, it might constitute
a worthwhile therapeutic target in the future if more extensive
knowledge of its activity and of its regulation by hormones and
chemicals is obtained.
FUNDING
AFSSET (Agence Francxaise de Securite Sanitaire de
l’Environnement et du Travail [ES-2005-003]); ANR (Agence
Nationale de la Recherche [06SETS26, Oncopop]); ARC
(Association pour la Recherche sur le Cancer [3927]);
INSERM (Institut National de la Sante et de la Recherche
Medicale); Universite Paris Descartes; Ministere de l’Enseigne-
ment Superieur et de la Recherche (Bourse doctorale, Ariane
Ambolet-Camoit, Aline Chevallier); Fondation pour la
recherche Medicale (bourse post-doctorale, Lin-Chi Bui);
Ligue contre le Cancer (bourse post-doctorale, Lin-Chi Bui);
Region Ile de France (Bourse doctorale, Stephane Pierre).
ACKNOWLEDGMENTS
We thank Dr L. P. Aggerbeck for critical reading of the
manuscript.
REFERENCES
Aberger, F., Weidinger, G., Grunz, H., and Richter, K. (1998). Anterior
specification of embryonic ectoderm: the role of the Xenopus cement gland-
specific gene XAG-2. Mech. Dev. 72, 115–130.
Barouki, R., Coumoul, X., and Fernandez-Salguero, P. M. (2007). The aryl
hydrocarbon receptor, more than a xenobiotic-interacting protein. FEBS Lett.
581, 3608–3615.
Barraclough, D. L., Platt-Higgins, A., de Silva Rudland, S., Barraclough, R.,
Winstanley, J., West, C. R., and Rudland, P. S. (2009). The metastasis-
associated anterior gradient 2 protein is correlated with poor survival of
breast cancer patients. Am. J. Pathol. 175, 1848–1857.
Bui, L. C., Tomkiewicz, C., Chevallier, A., Pierre, S., Bats, A. S., Mota, S.,
Raingeaud, J., Pierre, J., Diry, M., Transy, C., et al. (2009). Nedd9/Hef1/
Cas-L mediates the effects of environmental pollutants on cell migration and
plasticity. Oncogene 28, 3642–3651.
Carvajal-Gonzalez, J. M., Mulero-Navarro, S., Roman, A. C., Sauzeau, V.,
Merino, J. M., Bustelo, X. R., and Fernandez-Salguero, P. M. (2009). The
dioxin receptor regulates the constitutive expression of the vav3 proto-
oncogene and modulates cell shape and adhesion. Mol. Biol. Cell 20,
1715–1727.
Chopra, M., Dharmarajan, A. M., Meiss, G., and Schrenk, D. (2009). Inhibition
of UV-C light-induced apoptosis in liver cells by 2,3,7,8-tetrachlorodibenzo-
p-dioxin. Toxicol. Sci. 111, 49–63.
Di Valentin, E., Crahay, C., Garbacki, N., Hennuy, B., Gueders, M., Noel, A.,
Foidart, J. M., Grooten, J., Colige, A., Piette, J., et al. (2009). New asthma
biomarkers: lessons from murine models of acute and chronic asthma.
Am. J. Physiol. Lung Cell Mol. Physiol. 296, L185–L97.
Diry, M., Tomkiewicz, C., Koehle, C., Coumoul, X., Bock, K. W., Barouki, R.,
and Transy, C. (2006). Activation of the dioxin/aryl hydrocarbon receptor
FIG. 7. Schematic model for the role of AGR2 expression in the inhibition
by TCDD of etoposide elicited p53 activation.
510 AMBOLET-CAMOIT ET AL.
by guest on February 9, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

(AhR) modulates cell plasticity through a JNK-dependent mechanism.
Oncogene 25, 5570–5574.
Fletcher, G. C., Patel, S., Tyson, K., Adam, P. J., Schenker, M., Loader, J. A.,
Daviet, L., Legrain, P., Parekh, R., Harris, A. L., et al. (2003). hAG-2 and
hAG-3, human homologues of genes involved in differentiation, are
associated with oestrogen receptor-positive breast tumours and interact with
metastasis gene C4.4a and dystroglycan. Br. J. Cancer 88, 579–585.
Fritzsche, F. R., Dahl, E., Dankof, A., Burkhardt, M., Pahl, S., Petersen, I.,
Dietel, M., and Kristiansen, G. (2007). Expression of AGR2 in non small cell
lung cancer. Histol. Histopathol. 22, 703–708.
Fritzsche, F. R., Dahl, E., Pahl, S., Burkhardt, M., Luo, J., Mayordomo, E.,
Gansukh, T., Dankof, A., Knuechel, R., Denkert, C., et al. (2006).
Prognostic relevance of AGR2 expression in breast cancer. Clin. Cancer
Res. 12, 1728–1734.
Frueh, F. W., Hayashibara, K. C., Brown, P. O., and Whitlock, J. P., Jr (2001).
Use of cDNA microarrays to analyze dioxin-induced changes in human liver
gene expression. Toxicol. Lett. 122, 189–203.
Gouedard, C., Barouki, R., and Morel, Y. (2004). Dietary polyphenols increase
paraoxonase 1 gene expression by an aryl hydrocarbon receptor-dependent
mechanism. Mol. Cell Biol. 24, 5209–5222.
Gu, Y. Z., Hogenesch, J. B., and Bradfield, C. A. (2000). The PAS superfamily:
sensors of environmental and developmental signals. Annu. Rev. Pharmacol.
Toxicol. 40, 519–561.
Innes, H. E., Liu, D., Barraclough, R., Davies, M. P., O’Neill, P. A.,
Platt-Higgins, A., de Silva Rudland, S., Sibson, D. R., and Rudland, P. S.
(2006). Significance of the metastasis-inducing protein AGR2 for outcome
in hormonally treated breast cancer patients. Br. J. Cancer. 94, 1057–1065.
Kaiser, J. (2000). Toxicology. Just how bad is dioxin? Science 288,
1941–1944.
Kim, W. K., In, Y. J., Kim, J. H., Cho, H. J., Kim, J. H., Kang, S., Lee, C. Y.,
and Lee, S. C. (2006). Quantitative relationship of dioxin-responsive gene
expression to dioxin response element in Hep3B and HepG2 human
hepatocarcinoma cell lines. Toxicol. Lett. 165, 174–181.
Komiya, T., Tanigawa, Y., and Hirohashi, S. (1999). Cloning of the gene gob-4,
which is expressed in intestinal goblet cells in mice. Biochim. Biophys. Acta.
1444, 434–438.
Kruse, J. P., and Gu, W. (2009). Modes of p53 regulation. Cell 137, 609–622.
Kung, T., Murphy, K. A., and White, L. A. (2009). The aryl hydrocarbon
receptor (AhR) pathway as a regulatory pathway for cell adhesion and matrix
metabolism. Biochem. Pharmacol. 77, 536–546.
Liu, D., Rudland, P. S., Sibson, D. R., Platt-Higgins, A., and Barraclough, R.
(2005). Human homologue of cement gland protein, a novel metastasis
inducer associated with breast carcinomas. Cancer Res. 65, 3796–3805.
Marchand, A., Tomkiewicz, C., Marchandeau, J. P., Boitier, E., Barouki, R.,
and Garlatti, M. (2005). 2,3,7,8-Tetrachlorodibenzo-p-dioxin induces
insulin-like growth factor binding protein-1 gene expression and counteracts
the negative effect of insulin. Mol. Pharmacol. 67, 444–452.
Marlowe, J. L., Fan, Y., Chang, X., Peng, L., Knudsen, E. S., Xia, Y., and
Puga, A. (2008). The aryl hydrocarbon receptor binds to E2F1 and inhibits
E2F1-induced apoptosis. Mol. Biol. Cell 19, 3263–3271.
Marlowe, J. L., and Puga, A. (2005). Aryl hydrocarbon receptor, cell cycle
regulation, toxicity, and tumorigenesis. J. Cell. Biochem. 96, 1174–1184.
Missiaglia, E., Blaveri, E., Terris, B., Wang, Y. H., Costello, E.,
Neoptolemos, J. P., Crnogorac-Jurcevic, T., and Lemoine, N. R. (2004).
Analysis of gene expression in cancer cell lines identifies candidate
markers for pancreatic tumorigenesis and metastasis. Int. J. Cancer 112,
100–112.
Paajarvi, G., Viluksela, M., Pohjanvirta, R., Stenius, U., and Hogberg, J.
(2005). TCDD activates Mdm2 and attenuates the p53 response to DNA
damaging agents. Carcinogenesis 26, 201–208.
Park, S., and Matsumura, F. (2006). Characterization of anti-apoptotic action of
TCDD as a defensive cellular stress response reaction against the cell
damaging action of ultra-violet irradiation in an immortalized normal human
mammary epithelial cell line, MCF10A. Toxicology 217, 139–146.
Park, S. W., Zhen, G., Verhaeghe, C., Nakagami, Y., Nguyenvu, L. T.,
Barczak, A. J., Killeen, N., and Erle, D. J. (2009). The protein disulfide
isomerase AGR2 is essential for production of intestinal mucus. Proc. Natl.
Acad. Sci. U.S.A. 106, 6950–6955.
Persson, S., Rosenquist, M., Knoblach, B., Khosravi-Far, R., Sommarin, M.,
and Michalak, M. (2005). Diversity of the protein disulfide isomerase family:
identification of breast tumor induced Hag2 and Hag3 as novel members of
the protein family. Mol. Phylogenet. Evol. 36, 734–740.
Petrulis, J. R., and Perdew, G. H. (2002). The role of chaperone proteins in the
aryl hydrocarbon receptor core complex. Chem. Biol. Interact. 141, 25–40.
Pohler, E., Craig, A. L., Cotton, J., Lawrie, L., Dillon, J. F., Ross, P., Kernohan, N.,
and Hupp, T. R. (2004). The Barrett’s antigen anterior gradient-2 silences the p53
transcriptional response to DNA damage. Mol. Cell. Proteomics. 3, 534–547.
Puga, A., Ma, C., and Marlowe, J. L. (2009). The aryl hydrocarbon receptor
cross-talks with multiple signal transduction pathways. Biochem. Pharmacol.
77, 713–722.
Puga, A., Maier, A., and Medvedovic, M. (2000). The transcriptional signature
of dioxin in human hepatoma HepG2 cells. Biochem. Pharmacol. 60,
1129–1142.
Ramachandran, V., Arumugam, T., Wang, H., and Logsdon, C. D. (2008). Anterior
gradient 2 is expressed and secreted during the development of pancreatic cancer
and promotes cancer cell survival. Cancer Res. 68, 7811–7818.
Ramadoss, P., and Perdew, G. H. (2005). The transactivation domain of the Ah
receptor is a key determinant of cellular localization and ligand-independent
nucleocytoplasmic shuttling properties. Biochemistry 44, 11148–11159.
Ray, S. S., and Swanson, H. I. (2004). Dioxin-induced immortalization of
normal human keratinocytes and silencing of p53 and p16INK4a. J. Biol.
Chem. 279, 27187–27193.
Ray, S., and Swanson, H. I. (2009). Activation of the aryl hydrocarbon receptor
by TCDD inhibits senescence: a tumor promoting event? Biochem.
Pharmacol. 77, 681–688.
Sitek, B., Sipos, B., Alkatout, I., Poschmann, G., Stephan, C., Schulenborg, T.,
Marcus, K., Luttges, J., Dittert, D. D., Baretton, G., et al. (2009). Analysis of
the pancreatic tumor progression by a quantitative proteomic approach and
immunhistochemical validation. J. Proteome. Res. 8, 1647–1656.
Smirnov, D. A., Zweitzig, D. R., Foulk, B. W., Miller, M. C., Doyle, G. V.,
Pienta, K. J., Meropol, N. J., Weiner, L. M., Cohen, S. J., Moreno, J. G.,
et al. (2005). Global gene expression profiling of circulating tumor cells.
Cancer Res. 65, 4993–4997.
Swanson, H. I. (2002). DNA binding and protein interactions of the AHR/
ARNT heterodimer that facilitate gene activation. Chem. Biol. Interact. 141,
63–76.
Thompson, D. A., and Weigel, R. J. (1998). hAG-2, the human homologue of the
Xenopus laevis cement gland gene XAG-2, is coexpressed with estrogen receptor
in breast cancer cell lines. Biochem. Biophys. Res. Commun. 251, 111–116.
Wang, Z., Hao, Y., and Lowe, A. W. (2008). The adenocarcinoma-associated
antigen, AGR2, promotes tumor growth, cell migration, and cellular
transformation. Cancer Res. 68, 492–497.
Xu, C., Li, C. Y., and Kong, A. N. (2005). Induction of phase I, II and III drug
metabolism/transport by xenobiotics. Arch. Pharm. Res. 28, 249–268.
Zhang, J. S., Gong, A., Cheville, J. C., Smith, D. I., and Young, C. Y. (2005).
AGR2, an androgen-inducible secretory protein overexpressed in prostate
cancer. Genes Chromosomes Cancer. 43, 249–259.
Zhang, L., Wu, R., Dingle, R. W., Gairola, C. G., Valentino, J., and
Swanson, H. I. (2007a). Cigarette smoke condensate and dioxin suppress
culture shock induced senescence in normal human oral keratinocytes. Oral
Oncol. 43, 693–700.
TCDD-REGULATED AGR2 INHIBITS ETOPOSIDE EFFECT 511
by guest on February 9, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

Zhang, Y., Forootan, S. S., Liu, D., Barraclough, R., Foster, C. S.,
Rudland, P. S., and Ke, Y. (2007b). Increased expression of anterior
gradient-2 is significantly associated with poor survival of prostate cancer
patients. Prostate Cancer Prostatic Dis. 10, 293–300.
Zheng, W., Rosenstiel, P., Huse, K., Sina, C., Valentonyte, R., Mah, N.,
Zeitlmann, L., Grosse, J., Ruf, N., Nurnberg, P., et al. (2006). Evaluation of
AGR2 and AGR3 as candidate genes for inflammatory bowel disease. Genes
Immun. 7, 11–18.
Zhu, H., Lam, D. C., Han, K. C., Tin, V. P., Suen, W. S., Wang, E.,
Lam, W. K., Cai, W. W., Chung, L. P., and Wong, M. P. (2007). High
resolution analysis of genomic aberrations by metaphase and array
comparative genomic hybridization identifies candidate tumour genes in
lung cancer cell lines. Cancer Lett. 245, 303–314.
Zweitzig, D. R., Smirnov, D. A., Connelly, M. C., Terstappen, L. W.,
O’Hara, S. M., and Moran, E. (2007). Physiological stress induces the metastasis
marker AGR2 in breast cancer cells. Mol. Cell. Biochem. 306, 255–260.
512 AMBOLET-CAMOIT ET AL.
by guest on February 9, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from