
New Machine Learning Methods Demonstrate the Existence of a Human Stylome
Upload
independentCategory
view
2download
0
International Edition in English
I5N-NMR Spectroscopy- New Methods and
By Wolfgang von Philipsborn" and Raffaello Miiller
Applications**
The nitrogen nucleus is the third most important probe (after ' H and I3C) for structural investigations of organic and bioorganic molecules by NMR spectroscopy. For a long time, however, the insufficient sensitivity and low natural abundance of the I5N isotope ham- pered detection of the I5N nucleus, and the quadrupolar I4N nucleus proved unsuitable for the study of larger molecules with several nonequivalent nitrogen atoms. The advent of new techniques, such as pulse sequences and polarization transfer, in conjunction with the use of high-field magnets and large-sample probe heads largely solved the detection problem. As a result, the last few years have seen a dramatic development of "N-NMR spectroscopy as a versatile method for studying molecular structure, both in isotropic (liquid) and aniso- tropic (solid) phases. The scope of chemical applications extends from inorganic, organo- metallic, and organic chemistry to biochemistry and molecular biology, and includes the study of reactive intermediates, biopolymers, enzyme-inhibitor complexes, and nitrogen metabolism. Two-dimensional NMR techniques offer additional possibilities for detailed studies of biological systems.
Volume 25 - Number 5 May 1986
Pages 383-486
1. introduction
Nitrogen belongs to the few elements that form the skel- eton of organic and bioorganic molecules, and functional groups containing nitrogen atoms are of great importance in organic and biochemical reactions. Therefore, nitrogen NMR spectroscopy provides a sensitive method for the study of chemical structure and bonding, reaction mecha- nisms, biosynthesis, nitrogen fixation, and metal coordina- tion, as well as for the study of active sites in biochemical systems.
['I Prof. Dr. W. v a n Philipsborn, Dip1.-Chem. R. Miiller Organisch-chemisches Institut der Universitat Winterthurerstrasse 190, CH-8057 Zurich (Switzerland)
[**I "N-NMR Spectroscopy, Part 16.-Part 15: [I].
New Analytical Methods (28)
In fact, Proctor and Yurzl carried out nitrogen NMR studies in the very early days of nuclear magnetic reson- ance spectroscopy and thereby discovered the chemical shift phenomenon in NH4N0,. For a long time, the quad- rupolar I4N nucleus was preferred by NMR spectrosco- pists because of its high natural abundance (99.63%). The introduction of F T NMR spectroscopy, however, stimu- lated interest in the much less abundant spin-% "N iso- tope. Although the NMR receptivity of the I5N nucleus (0.37% abundance) is 280 times smaller than that of I4N (Table l), there are at least two factors that compensate for this deficiency. First, the line broadening of I4N signals (up to several kHz) is absent in "N resonances, and, sec- ond, the large nuclear Overhauser effect (NOE) (qo= -4.93) enhances the "N signal intensities of proton- bearing I5N atoms when proton noise decoupling is ap- plied. Furthermore, it is sometimes possible to shorten the
Angew Chem. Inr. Ed. Engl. 25 (1986) 383-413 0 VCH Verlagsgesellschajl mbH. 0-6940 Weinheim. 1986 0S70-0833/86/05~15-0383 .$ 02.50/0 383

Table I . Magnetic properties of the "N, '"N, "C, and 'H isotopes
Nucleus v/MHz Nat. abun- Spin Gyromagnetic Relative sensi- [a] dance/% ratio tivity
v:IO'radT. Is-' [b] [c]
' H 400.0 99.98 1/2 26.7510 62.91 5676 100.6 1.11 I12 6.7263 I 1 f y.
"N 28.9 99.63 I 1.9324 0.064 5.69 "N 40.5 0.37 I/2 -2.7107 0.066 0.02
[a] At a field of 9.4 tesla. [bl For the same number of nuclei at constant field B( , . [c] For natural isotopic abundance and constant field Bo.
long spin-lattice relaxation times (TI) of tertiary N atoms by the use of high magnetic field strengths (superconduct- ing magnets) or, alternatively, to circumvent the problem by the use of polarization-transfer from fast-relaxing, sen- sitive nuclei, such as ' H or "F.
I4N-NMR spectroscopy has been employed to study relatively small and inorganic mole- c u l e ~ ~ ~ , ~ ~ and to study molecular motion;[61 the quadrupole moment is an especially important nuclear property for the investigation of dynamic processes. For larger molecules with several nonequivalent N atoms, including biopoly- mers, only the "N isotope yields satisfactory and interpret- able NMR spectra. The results obtained up to 1980 are summarized in three monograph^."^ In many cases, "N- NMR spectra can also be recorded in the proton-coupled mode, thereby yielding both chemical shift and spin-cou- pling information (scalar coupling in solution and dipolar coupling in the solid). Finally, the use of large sample vol- umes in wide-bore high-field magnets has helped consider- ably to overcome the hitherto bad reputation of "N-NMR spectroscopy as being insensitive, time-consuming, and un- suitable for analytical applications in structural chemistry and biochemistry. I t is the purpose of this review article to discuss the experimental progress and to illustrate the broad spectrum of present and future applications of "N- NMR spectroscopy in chemistry.
2. Experimental Techniques
2.1. Standard Pulse Experiments
Conventional FT NMR experiments with a pulse-modu- lated high-frequency field at the I5N resonance are only feasible if the spin-lattice relaxation times (TI) are reason- ably short; this is true for N atoms with directly bonded protons, e.g., in NH2 and NH groups. In such cases (nu- clear-nuclear) dipole-dipole relaxation (DD) is the domi- nant relaxation mechanism, and T , values of less than 20 s are usually observed.['"' Figure 1 shows the spectrum of the dication 1 of 6,7-dimethyltetrahydropterin. Each N atom is bonded to at least one H atom. Moreover, the N atoms have similar relaxation times, which results in comparable signal intensities. However, even tertiary N atoms may be detected with sufficient sensitivity if measurements are conducted in strong magnetic fields (9-1 1 tesla) generated by superconducting magnets. In this case, a chemical shift anisotropy dependent relaxation process (CSA), which in- creases with the square of the field strength, may contri- bute significantly to the spin-lattice relaxation rate (Sec-
3
I . , . . I -250 -300 - 3 5 0
- 6
Flg. 1. Proton-noise-decoupled and phase-adjusted "N-NMR spectrum of the dication 1 of cis-6,7-dimethyl-5,6,7,8-tetrahydropterin (10.1 MHz; CF1COOH, 0.7 M ) [ 9 ] .
tion 3.3). Finally, otherwise long TI values may be reduced by electron-nuclear dipole-dipole interaction when para- magnetic species, so-called relaxation reagents, are added. A convenient and widely used compound is chromium(rri) acetylacetonate, Cr(acac),,[H1 which enhances "N relaxa- tion rates without significant line shifts (< 1 ppm).
'H broadband-decoupling enhances the signal-to-noise ratio not only as a result of multiplet collapse but even more effectively by the large NOE factor (v0= -4.93) for the "N, ' H system. From Equation (a) it is apparent that the full benefit of this (negative) signal enhancement is only obtained if TI is dominated by dipole-dipole interac- tions, which is the case for NH2 and NH groups, and that it leads to signal inversion (Fig. 2) ( M , =z-magnetization; M: = z-magnetization at thermal equilibrium, T:"' = time constant of dipole-dipole relaxation).
?o=O.5 .( - 9.86) = - 4.93
As shown in Figure 2a, inefficient dipolar relaxation for tertiary N atoms may result in loss of the signal. Such cases, frequently encountered in N-heterocycles, can be handled by deliberate suppression of the NOE; this is achieved by switching the decoupler power output on dur- ing acquisition and off during the pulse delay time (inverse gated decoupling). Since spin decoupling becomes effec- tive almost instantaneously when the BZ field is switched on whereas the NOE is built up more slowly with the time constant TYD, 'H-decoupled I5N-NMR spectra may be re- corded with a minimized Overhauser effect (Fig. 2b). This technique allows tertiary and proton-bearing N atoms to
384 Angew. Chem. Int. Ed. Engl. 25 (1986) 383-413

2
3
100 Hz H I
I 1 I I
-150 -200 -250 -300 -5
Fig. 2 "N-NMR spectra of 2.4-diaminopyrimidine 2 (10.1 MHz; (CD+SO): a) ' H-noise-decoupled: b) "inverse-gated"-decoupled (decoupler output "on" during acquisition, "ofr' during acquisition delay time): c ) no 'H decoupling.
be simultaneously observed. Thus, the relative intensities of 'H-decoupled I5N resonances are dependent on the spin-lattice relaxation times TI and the nuclear Over- hauser effects (v I lqol). Quenching of undesired, small Overhauser effects may also be achieved by the use of re- laxation reagents.
The sensitivity of "N-NMR detection may be further enhanced by the use of large-diameter sample tubes (15- 25 mm) and the application of strong magnetic fields (Bo). The excellent performance, in terms of field homogeneity and stability, of modern wide-bore superconducting mag- nets permits the routine measurement of I5N-NMR spectra with natural isotope abundance with the techniques de- scribed above. A typical example is shown by the spectrum of the t4.4. Ilpropellane derivative 3 (Fig. 3).["l
2.2, One-Dimensional Pulse Sequence Experiments (INEPT, D E W )
The rapid development of pulse techniques and their implementation on modern spectrometers provides still
NC--n
3 II I
. . I I I I I I
-2L70 -2L9 0 -251 0 -6
Fig. 3. Proton-coupled DEPT "N-NMR spectrum of the I : 2 adduct 3 of tricyclo[4.4. 1.0'.hlundeca-2,4,7,9-tetraene- I I-carbonitrile with N-methyl- I .2,4- triazoledione) (40.6 MHz: (CD&SO). The cyano doublet (&= - 113.3) is not shown. The assignment of the two weak and two strong signals to the NCHl and NCHCH groups, respectively, follows from the multiplicities and from the values of N,H coupling constants.
more effective and versatile methods for sensitivity im- provement in "N-NMR spectroscopy. In such experi- ments, application of a sequence of nonselective and si- multaneous pulses on a scalar-coupled heteronuclear spin system leads to spin polarization transfer from the sensi- tive ' H nuclei (large abundance, large y ) to the insensitive
N nuclei (small abundance, small y) and a maximum en- hancement factor of IyH/yNI =9.86 for the intensities of the 15N resonances. Furthermore, since the rate at which the entire pulse sequence can be repeated is governed by the short relaxation times of the ' H nuclei, additional time is saved. A typical polarization transfer experiment, the INEPT pulse sequence (Insensitive Nuclei Enhanced by Polarization Transfer) is shown in Scheme l.l"l For a dis-
15
Scheme I . S , 'H; I , 15N; @preparation, @ evolution, @ refocusing, 0 ' H - broadband decoupling, @ detection. See also [12].
cussion of the mechanism of polarization transfer the reader is referred to the review by Benn and Giinrher.L'21 After the polarization transfer (90" pulse) and following the "N detection pulse, antiphase signals can be detected or, by refocusing and decoupling during acquisition, the spin system can be "processed" further. Thus, proton-cou- pled, selectively decoupled, or fully decoupled "N-NMR spectra can be obtained, thereby increasing the informa- tion content of the spectra for structural studies.
Anyen, Chem. Int. Ed. Engl. 25 (1986) 383-413 385

The quality of such spectra, in terms of sensitivity and resolution, in comparison with standard pulse spectra, is exemplified in Figure 4. This example also demonstrates that the multiplicity and phases of the signals in INEPT
I
H
CH3 $$ 5
H
10 Hz
0 ..... H CH3\ / \
CH,-C - C / \ /"-"
CH3 /c=c\"
Fig. 5. INEPT "N-NMR spectra of 5-methylisoxazole 5 (20.3 MHz; (CD,)2SO): a) proton-coupled; b) refocused and selectively 'H-decoupled (H-C4); c) same as b) but irradiated at the CH3 resonance 1151.
Fig. 4. l5N-NMR spectra of the conformer shown of (Z)-l-amino-4,4-di- methyl-I-penten-3-one 4 (20.3 MHz; C6D6, 3.7 M): a) standard-pulse NMR spectrum (measuring time 10 h); b) INEPT l5N-NMR spectrum (measuring time 1.9 h) 1131. 'J(N,H)=92.7, 'J(N,H)= 1.7, 'J(N,H)=4.0 Hz.
spectra differ from those in regular spectra. For in- stance, the relative intensities for NH doublets are + 1 and - I , for NH2 triplets + 1 , 0, and - The phases can be adjusted by application of the above-mentioned refocusing procedure. In addition, selective ' H-decoupling may lead to considerably sharper lines and, hence, to still better sen- sitivity. The "N-NMR spectra of 5-methylisoxazole 5 (Fig. 5 ) illustrate the accurate determination of 'J(N,H) and 3J( N,H) through selective 'H-decoupling. Coupling of the I5N nuclei to methyl protons often obscures the desired spin-coupling information, as shown in the case of 2-ace- tylpyridine 12 (Section 3.2.2), and increases the measuring time required.
Polarization transfer to an insensitive nucleus requires scalar-coupling to a sensitive nucleus. In addition to the ' H nucleus, '9Ff171 and 3 ' P have been used; in particular, 3 'P was used successfully to transfer spin polarization to lSN,[lhil] S 7 F e , Io3Rh, and '83W.0hh1 The substantial spin coupling between I9F and "N is especially advantageous in polarization transfer experiments. Figure 6 shows the re-
386
a)
1 . / I
' I I !
!I I I
sult of such an INEPT measurement for perfluoropyridine 6.[17] The large 'J(N,F) coupling (-51.7 Hz) determines the pulse delay s=1/2J(N,F) in the INEPT pulse se- quence (cf. Scheme 1). Here again, the central lines of the triplet are absent, so that the large splitting corresponds to 2 x J(N,F).
The optimum setup of an INEPT experiment is less straightforward if there are no dominant heteronuclear
10 HZ H
Fig. 6. INEPT "N-NMR spectrum of perfluoropyridine 6 (20.3 MHz: (CD,)>CO) with polarization transfer from the IYF nuclei (188.2 MHz). The r value for the INEPT sequence was calculated using 12J(N,F)I=S2 Hz [171.
Angew. Chem. In[ . Ed. Engl. 25 (1986) 383-413

coupling constants. If the "N nucleus is coupled with sev- eral protons, which, in turn, are mutually coupled (spin systems of the type IS'S'. . . S"), the optimal s values can be determined by using analytical expressions."'] This is shown for the nitroethene 7 in Figure-7. It should be noted
E
20
16,
12-
8-
1 -20 , - 0 LO 80 120 160 200 2LO 280
z Imsl
Fig. 7. Right: INEPT "N-NMR spectrum of the NO2 group of (E) -N ,N- dimethyl-2-nitrovinylamine 7 ; left: signal enhancements (E) as a function of r. (-) calculated and (A) experimental values [18]. J(N,H-2)=2.2,
MHz). J (N,H-I )= 1.8, J(H-I,H-2)= 10.6 Hz; v(H-l)-v(H-2)= --290 Hz (20.3
that zopl (190 ms) does not correspond to either 1/2J(N,H') ( t=278 ms) or 1/2J(N,H2) (227 ms), because the homonuclear coupling constant 3J(H',H2) is much larger (10.6 Hz) than the two heteronuclear values (1.8 and 2.2 Hz, respectively). Nevertheless, such spin systems are also amenable to a n effective polarization transfer, which considerably extends the application of the INEPT tech- nique beyond the detection of simple NH and NH, gr0ups['~1 (see also Section 4).
DEPT n12, "I.
S: _n T n T n T
!
Scheme 2 S, I,@, @, 0. @ see legend to Scheme 1. See also [I21
The pulse sequence DEFT (Distortionless Enhancement by Polarization Transfer) was developed"91 as an alterna- tive polarization transfer technique in order to avoid the intensity and phase distortions inherent in the INEPT ex- periment. The DEPT sequence yields spectra (Scheme 2) with positive signals only, which thus resemble standard pulse spectra. In this technique, the sensitivity enhance- ment is described as a function of the delay time I and the pulse angle 8. Only for simple spin systems of the type IS,,
however, are pure phase spectra obtained; spin systems IS'S'. . . S" with hetero- and homonuclear coupling are subject to phase distortions."" Further, when higher-order spin systems are involved, the intensities are also distorted. Decoupled DEPT spectra always show positive phase sig- nals. DEPT "N-NMR spectra of thiazole 8 are shown in Figure 8. Selective decoupling allows the assignment of the small coupling constant (2.2 Hz) to the vicinal N,H inter- action (3J(N,H)).
H
10 Hz
c
H
L 8 Fig. 8. DEPT "N-NMR spectra of thiazole 8 (20.3 MHz; CDC13): a) proton- coupled, b) selective irradiation of the H-5 resonance [15].
In terms of sensitivity enhancement, the INEPT pulse sequence has proven superior to the DEPT sequence;['9h1 for the five-spin system of 2-methoxypyridine the enhance- ment is larger by a factor of 1.8.["] The simple INEPT ex- periment (without refocusing) appears to be less prone to perturbation by additional homo- and heteronuclear cou- plings.
The advantage of the polarization transfer experiments discussed above is the nonselective nature of the 'H-irra- diation. Thus, polarization can be transferred to several nonequivalent N atoms, which allows the structural inves- tigation of complex molecules (cf. Section 4).
An alternative way of achieving sensitivity enhancement is based on a selective population transfer (SPT), in which a single ' H transition is irradiated, resulting in equaliza- tion or inversion of the populations followed by transfer to connected levels.[201 The "N sensitivity enhancement at- tained is the same as in the nonselective INEPT or DEPT experiments; however, the transition-selective nature of the experiment permits the determination of the relative signs of N,H coupling constants.["] Similarly, for non-pro- ton-bearing N atoms a modified INEPT sequence, which achieves a selective polarization transfer, was proposed. It largely suppresses homonuclear multiquantum coherence and allows neglect of N,H long-range coupling to other than the selectively irradiated proton.[221 For this purpose,
Angew. Chem. Inr. Ed. Enyl. 25 (1986) 383-413 387

soft proton pulses (typical pulse duration 5 ms for a 90" flip angle) were used in the detection of the proline "N resonance of a hexapeptide. However, the enhancement is only large if the proton excitation is highly selective, which constitutes a serious limitation for complex molecules, even in high magnetic fields.
The largest sensitivity improvements are achieved when the "N resonance is detected indirectly via the proton re- sonance; theoretically, the sensitivity can be increased by a factor of ( ~ ~ / y ~ ) ~ , i.e., by about 961. Such results have ac- tually been obtained in multiple quantum coherence ex- periments on large peptides by the use of 1 D and 2D data representation^[^^.^^] (see also Section 2.3).
A critical evaluation of the advantages and disadvan- tages of the numerous one-dimensional pulse sequence ex- periments has recently been presented by Turner.'"'
2.3. Heteronuclear Correlated 2D NMR Spectroscopy
During the last decade, 2 D NMR experiments have de- veloped to a very high degree of versatility and sophistica- tion and many of them can be routinely performed on commercial spectrometers. Nevertheless, the application to "N-NMR spectroscopy has been slow and, only recently, owing to improved general NMR instrumentation and the development of multiple quantum coherence experiments, has significant progress been achieved. Of the classical 2D experiments, only heteronuclear 'H,I5N shift correlation has found application in the assignment of 15N resonances via IJ(N,H) and long-range J(N,H). For a review of the classical 2D NMR methods the reader is again referred to the article by Benn and Giinther.1'21
The correlation of ' H and I5N chemical shifts based on multiple quantum coherence has also led to a dramatic im- provement in sensitivity, since determination of the "N frequencies is effected by detection of the scalar-coupled protons.[261 A third significant application of 2 D NMR is found in the analysis of solid-state "N-NMR spectra with dipolar
'H,"N shift correlated (COSY) 2D NMR spectroscopy can be applied to oligopeptides and leads to an assignment of "N resonance lines based on known NH and C,H pro- ton f r e q u e n c i e ~ . ' ~ ~ ~ ~ ~ ~ Measurements of this kind, when car- ried out on nonenriched substrates, are rather time-con- suming compared with 'H,'H and I3C,'H 2D NMR ex- periments. The complete assignment of all "N-NMR sig- nals for the undecapeptide cyclosporin A has been achieved by using the COLOC pulse sequenceL3o1 (Fig. 9). The proton shift assignments were obtained from addi- tional COSY experiments, e.g., 2D homonuclear NH,H(a) and 2D heteronuclear C(a),H(a) correlation.
A 2D NMR experiment specifically designed for the de- tection of proton-coupled "N resonances utilizes the idea of a double transfer of polarization based on an INEPT pulse sequence and allows the determination of I5N chem- ical shifts with the sensitivity of the proton resonance. The original pulse sequence used by Bodenhausen and Rub- en["] was subsequently modified and simplified by Red- fieldL3'] (HAHNDOR pulse sequence) and Bax et al.1241 with the result that this 2D NMR multiple quantum
388
6 ' H l I
I I
-9P
4 P
-10111 .Ll9
- 1 - 6 3
;o L
8 1
- 2
N-CH,
L l i " l ~ ' l ' ' ~ t '
-250 -260 6I5N
Fig. 9. COLOC 'H,"N-heteronuclear shift correlated NMR spectrum of cy- closporin A (50.7 MHz; CDCI,, measuring time 36 h). The spectrum shows correlation peaks for "N atoms with directly bonded protons (NH) and gem- inal protons (N-C,-H and N-CH,), thereby allowing a complete assignment of the eleven N atoms in one experiment [28cI Cyclosporin A IS an undeca- peptide with the following amino acid sequence (MeBmt = N-methylbutenyl- methylthreonine):
c y / o [ MeBmt- Abu-Sar- MeLeu-Val-MeLeu- Ala-D- Ala-MeLeu-MeLeu-MeVal]. 1 2 3 4 5 6 7 8 9 1 0 1 1
method has found wide practical use. The advantages of this experiment are a dramatic sensitivity gain by a factor of ( ~ ~ / y ~ ) ~ =961 and the detection of only those 'H reson- ances that are scalar-coupled to an "N nucleus. This per- mits detection of relevant NH groups in large biomole- ~ ~ l e s . ~ ~ ~ " . ~ ~ ~ The spectrum of "N-labeled E. coli tRNA?"' (Fig. 10) demonstrates the use of this method to identify the six nucleotide units biosynthetically derived from urid- ine and, at the same time, the improved resolution obtain- able in the 2D data representation. It was estimated that "N-NMR shifts for "N, 'H groups in nonenriched sam- ples can be obtained in 2-4 h with a 100 mM sample of the biopolymer.
Recent developments in high-resolution 15N-NMR spec- troscopy of solids (see Section 4.6) permit the measure- ment of spectra of biomolecules which contain precise iso- tropic chemical shift and "N,'H dipolar coupling infor- mation. Since such spectra may be very complex, 2D NMR spectroscopy and graphical representation of the spectra with 6(N) as the F2 and D(N,H) as the F, coordinate is very useful. In one of the first applications, DiVerdi and O ~ e Z l a ~ ~ ~ l reported N-H bond lengths in DNA based on the magnitude of dipolar "N,'H couplings (Fig. 11) . "D- resolved" I5N,'H spectra of oriented virus solutions, ob- tained by spontaneous alignment of these macromolecules in the external magnetic field Bo, have also been re- corded.[3s1
Angew. Chem. Int. Ed. Engl. 25 (1986) 383-413

L S('5N)
-220- f -210 1 -200
15 10 &('HI
Fig. 10. Contour plot of "N,'H heteronuclear chemical shilt correlation NMR spectrum, derived from a multiple quantum coherence experiment, of "N-labeled tRNAy" from E. coli (36.5 MHz; v(H)=360.1 MHz; 65% en- riched at N-3 of uridine and uridine-derived bases, 5 mg sample dissolved in 250 pL of aqueous buffer solution, measuring time 6 h). Projections of the 'H- and "N-NMR spectra are shown opposite the respective chemical shift axes. For further experimental details and assignments of chemical shifts see [33a]. An improved method for the indirect detection of I5N resonances was recently describedl33bI.
J-resolved 2 D "N-NMR spectra are another powerful source for structure elucidation via long-range I5N,'H spin couplings. Such experiments have been performed in com- bination with a n INEPT polarization-transfer pulse se- quence applied during the preparation period of a selec- tive 2D J pulse sequence.[361 In this way, long-range "N,'H spin coupling constants have been determined, with excel- lent sensitivity and resolution, for aromatic and aliphatic
n
b)
0
-150 -200 - 250 -300 - 350 - 6
Fig. I I . CPMAS (cross polarization and magic angle spinning) '%NMR spectra of DNA (15.24 MHz): a) isotropic chemical shifts of B-DNA; b) low- humidity DNA; c) G(N)/D(N,H) 2 D "N-NMR spectrum of low-humidity DNA. For assignments of chemical shifts see 1341.
compounds, and the method has been suggested as a means to obtain precise values for 3J(N,H) in peptides (see Section 3.2.2).
In conclusion, modern FT NMR spectroscopic tech- niques combined with high magnetic field strengths and large sample volumes have largely overcome the sensitivity problem and now permit the routine performance of "N- N M R experiments at natural isotope abundance. Nev- ertheless, depending on the type of I5N resonance to be investigated, the most suitable detection technique has to be found and the experimental conditions carefully matched. This set-up procedure often requires more time than for conventional nuclei such as ' H and I3C. One ma- jor obstacle, the long T , relaxation times of tertiary N atoms and the small NOE, has been successfully removed by polarization transfer from a sensitive, fast-relaxing sca- lar-coupled nucleus, usually a proton. Even higher sensi- tivity can be achieved by indirect detection of the "N nu- clei via the 'H resonance, and corresponding 1D and 2D multiple quantum coherence experiments are particularly suited for large biomolecules. The "N resonance in nonla- beled low-molecular-weight compounds ( M r < 500) can now be directly detected in a few hours with 0.1-0.2 M so- lutions. A variety of applications to organic and inorganic chemistry as well as to biochemistry will be discussed in Section 4.
3. Structural Dependence of Spectral Parameters
High-resolution I5N-NMR spectra provide information on nuclear shielding constants (chemical shift), spin-lat- tice relaxation times (TI), spin-spin relaxation times ( T2), and scalar heteronuclear spin coupling (e.g., "J(N,H)). This constitutes a considerable improvement over I4N- NMR spectroscopy, which mainly yields chemical shift in- formation. Only in a few favorable cases can heteronuclear coupling constants be obtained and line-shape information be used to characterize exchange processes and molecular motion. In this section, the spectral parameters of "N- N M R will be discussed in terms of their structural signifi- cance and potential application in chemistry.
3.1. Chemical Shifts
The shielding range of the "N nucleus in diamagnetic molecules extends over 1000 ppm, corresponding to about 40 kHz in a field of 9.4 tesla (v0=40.5 MHz). Taking into account a line width of 1-2 Hz and the smaller abundance of nitrogen atoms in organic and bioorganic molecules, this leads to an even better dispersion of the resonance lines than in I3C-NMR spectra. This excellent dispersion is not attenuated by the fact that the resonances of the major- ity of N atoms in organic structures lie within a 500-ppm range. An example is provided in Figure 12 by the spec- trum of a mixture of 1- and 2-methyltetrazole, 9 and 10, respectively; well-separated lines for the eight nonequiva- lent N atoms are observed over a range of 170 ppm, but their assignment requires additional experiments (cf. Sec- tion 3.4).
Angew. Chem. lnt . Ed. Engl. 25 (1986) 383-413 389

lo3 lo4, 10'
10.6 -2.4 -11.7 -50.3 -52.1 -75.0 -104.9 -154.2 I, I I , I I I I , 1
I I 0 - 6 -150
Fig. 12. "N-NMR spectrum of a mixture of I- and 2-methyltetrazole, 9 and 10, respectively (40.6 MHz; 200 mg in 2.5 m L of CDCI,, 75 mg of Cr(acach added; measuring time 42 rnin).
3.1.1. Standardization - Reference Compounds
In this review, all chemical shifts (6 values) will be given relative to that of nitromethane (CH3"N02) as pure liquid in a capillary (external standard), without susceptibility correction and with negative signs for low-frequency and positive signs for high-frequency shifts. Literature data ori- ginally measured relative to other reference compounds, such as NH4"N03, HI5No3, or "NH3, are adjusted to the C H 3 N 0 2 We have used nitromethane throughout our own work; its use allows direct comparison with the two extensive data collections compiled by Martin et al.""l and by Witanowski et al.'7b1 The choice of C H 3 N 0 2 has the consequence that most 6 values are negative. This fact, al- though quite common for other nuclei (e.g., metal nuclei), has induced some authors to adopt other references (e.g., NHs(liq.) o r NH?) at the low-frequency end of the nitro- gen-shift scale, either as a primary reference (external standard) or as a secondary artificial reference. However, an arbitrary value for the CH3I5NO2 shift relative to NH3 (liq.) (e.g., 380.2 ppm) should be used since the NH3 reson- ance is strongly temperature dependent.[371 Moreover, as a consequence of concentration, solvent, and temperature effects, especially for different sample alignments in elec- tromagnets and superconducting magnets, and usually ne- glected susceptibility the general reproduci- bility of I5N chemical shifts is not better than f 1 ppm. This value may be a lower limit in cases where relaxation reagents (>0.05 M ) are used to measure the "N reson- ance.
3.1.2. Main Trends in "N-NMR Shi@
The "N-NMR shifts of N atoms with different oxida- tion states, coordination numbers, and hybridizations (sp3, sp2, sp) are summarized in Figure 13. As expected from the similarity of nitrogen and carbon in electronic structure and bonding, the I5N-NMR shifts parallel the I3C-NMR shifts in many respects. Thus, the tricoordinated N atoms (amines) appear a t the low-frequency end of the scale; te- tracoordinated N atoms (ammonium salts and N-oxides) absorb in the same range. The introduction of electronega- tive substituents leads to deshielding; the three principal hybridization states of nitrogen give rise to deshielding in the sequence sp3 < sp < sp2. The ranges for the resonance of carbon-substituted tricoordinated (6= - 400 to - 200)
and dicoordinated N atoms (6= - 100 to + 100) are well separated. The carbonyl analogues - NO and - NOz exhi- bit resonance positions at the high-frequency end of the scale for organic nitrogen compounds. The characteristic position of functionalized N atoms is largely maintained when they act as ligands in metal complexes, i.e., the coor- dination shifts are fairly smallf4] (see Section 4.4). An im- portant feature of the N atom, the lone-pair electrons, have a pronounced influence on the shielding of "N nuclei if low-lying n+n* transitions are available to affect the (A@- I term in the paramagnetic shielding expression. For example, protonation of the azine N atoms in pyridines and pyrimidines leads to low-frequency shifts of 100-120 ppm,[391 such effects being suitable for the study of proton- ation and hydrogen bonding in organic and bioorganic molecules (see Section 4.1). When the lone pair is involved in n,n-delocalization, e.g., in enamines and enamino ke- tones, high-frequency shifts are observed which can be correlated with the extent of conjugation in such sys- tem~.'~'] Figure 14 illustrates the transition between am- ines, enamines, enamino ketones (vinylogous amides), and amides as reflected in the "N chemical shifts. An applica- tion of these principles to a conformational study of enam- ino ketones will be discussed in Section 4.3.
A less phenomenological and more detailed discussion of I5N chemical shifts has to be based on the paramagnetic shielding term, d', which dominates the relative line posi- tions of the heavier nuclei. The observed "N chemical shifts have also been extensively discussed on the basis of quantum chemical c a l ~ u l a t i o n s . ~ ~ ~ ~ ~ ' ] In this context, it is important to note that the shielding values for the I4N and "N isotopes may be readily compared with each other as well as with calculated data since the ratio of the reson- ance frequencies of a series of isotopomeric molecules was found to be constant within i 8 x All available data indicate that primary isotope effects are of the order of a few tenths of a ppm17'l and may thus be neglected for practical purposes (see also ref. [78b]). In doing so, howev- er, one must realize that the error limits of I4N shielding data can be quite large (several ppm) depending on the line width (up to several kHz), whereas I5N-NMR data are usually accurate to 0.5 ppm.
Both "N and I4N chemical shifts are prone to medium effects, i.e., solvent, concentration, and temperature. De- pending on the nature of the solvent and substrate, these effects may be as high as several tens of ppm. An example
390 Angew. Chem. Int. Ed. Engl. 25 (1986) 383-413

Structural element
Y-
,Y - N
Unsaturated heterocycles
“=N -
I- - ,Y-CI
,Y-F
( :Y),C = N ’ - ,Y-co- 7
:r-cs- - (-CO),Y - I
:N - Y: - :N- N = C:
:N-N=N’
- -
,Y -NO,
Y-NO i -4 in azoies
‘Y/ m azines
*N’ in azoles
,W=C=W’ -
m--Y=u--M M--YEN .-_--- . + ,C-NEY ‘C-N=N + Fe-CEY paramagnetic
- Y O f
- Y O -S-YO -ON,-;NYO c_
M - ~ ~ O m-n=o
1100 ?Oh0 900 800 70.3 600 sbo 400 300 200 +lo0 d -100 -200 -300 -400
-s Fig. 13. 15N chemical shift ranges in organic and inorganic compounds relative to CH3N02; positive 6 values correspond to ‘’N resonances shifted to higher frequencies [38]. Ar=aryl.
is given in Figure 15. The substituent-induced shifts in p-substituted N, N-dimethylbenzaldehyde hydrazones 11 correlate well with Hammett substituent constants, and the slope is solvent dependent (which indicates the origin of the effects).[421 The dicoordinated N atoms of’ azines and azoles, frequently encountered in heterocyclic systems, are
@ - 0 .
(3 - I @
- 250 -300 - 350
4
8 t /
4 1 w 11
X: NCH,), OCH, CN NO? t
-0.4 n
-6 Fig. 15. Correlation between AS(N)=Sz-6: and the substituent constants 8=@Io,+pRuR)/@I +pK) for the N-l resonance of p-substituted benzalde- hyde hydrazones 11; measured in CeH,2 (O), (CH,),SO (m), and CH,OH ( A ) 1421.
Fig. 14. “N chemical shift ranges for @ rert.-arnines, @ enamines, Q en- amin6 ketones, and @ am?des.
Angew. Chem. Im. Ed. Engl. 25 (1986) 383-413 39 1

typical examples where the solvent-induced "N shift cor- relates well with the hydrogen bonding properties of the solvent1431 (Table 2). The limiting case, full protonation on the N atom, causes a low-frequency shift of approximately 120 ppm (see ref. [39] and references cited therein). An ex- cellent overview of solvent effects on "N chemical shifts is given in ref. [7a], and some applications will be discussed in Section 4.1.
Table 2. Solvent effects A6(N) for pyridine (in ppmj 1931.
of coupling energies of different nuclei (isotopes or ele- ment~)'~'] [Eq. (c)].
4 ?12
h Y X Y X , K(X,X') = ~ ' J(X,X') [ N . A - ' . ~ I - - ~ ]
This definition eliminates the influence of the gyromag- netic ratio (magnitude and sign). Therefore, K reflects the coupling pathway and the mechanism of the scalar spin- spin interaction. In practice, coupling energies are always reported as J or IJI values.
Gas CeHii CCI, ChHa Neat DMSO phase liquid [a]
~~ ~~ ~
0 -1.5 -4.3 -4.9 -6.3 -6.9
CH2C12 CHCI, CH,OH Hz0 TFE [bl
-9. I -12.5 -24.9 -28.1 -39.9
[a](CH,jiSO. [b] CFICHiOH.
Several authors have investigated deuterium isotope ef- fects on "N chemical shifts, although the data base is still rather small. In an early study, Litchman et al.1441 measured low-frequency shifts of 0.68, 1.29, and 1.96 ppm for mono-, di-, and trideuterated ammonia, respectively, and a value of -0.6 ppm per deuterium atom was observed in [I3C- 2,3-'5N,-guanidino]arginine.1451 Deuterium isotope effects over several bonds have been detected in enamino ketones, but they usually d o not exceed 0.1 ppm.'461 If the deutera- tion affects a fast tautomeric equilibrium, as in 2-hydroxy- azo compounds, much larger shifts (several ppm) can be 0 b ~ e r v e d . l ~ ~ ~ Isotope effects may thus be useful for the de- tection and quantitative study of tautomeric systems.
Because of the manifold influences on "N chemical shifts, the assignment of resonance lines for structure elu- cidations may become a difficult problem. A variety of as- signment techniques, however, are available, which will be discussed collectively in Section 3.4. The possibilities have been considerably expanded by the inclusion of N,H cou- pling constants, which are obtainable from INEPT, DEPT, or 2D NMR heteronuclear correlation experiments. In the course of this development our knowledge of N,H cou- plings over several bonds ("J(N,H), n=2, 3, 4) has pro- gressed to a point where an application to structural anal- ysis becomes possible.
3.2. Spin-Spin Coupling Constants
Contrary to the nearly identical chemical shifts of the two nitrogen isotopes, their coupling constants to a n X nu- cleus depend on the gyromagnetic ratios y(I4N)= 1.9324 and y("N)= -2.7107. From Equation (b), it is apparent that the "N,X coupling constants are 40% larger than the I4N,X coupling constants and have an opposite sign.
J("N,X) = y( I5N)/y( I4N). J ( I4N,X) = - 1 .40,J(l4N,X) (b)
It may be recalled that a "reduced coupling constant" K(X,X') has been defined in order to allow a comparison
3.2. I . One-Bond "N,X Coupling {'J(N,X))
As for C-H bonds in I3C-NMR spectroscopy, the largest coupling constants in "N-NMR spectroscopy are ob- served for N-H bonds; moreover, they depend linearly on the s-character of the hybrid orbital involved in the N-H bond (see Scheme 3). There are several empirical equations that describe this dependence.
'J(N,H)= -2 .94 . (%~) IHzl 'J(N,H)= -2 .33.(%~)+ 6 [Hz] 'J(N,H)= - 1 .69 . (%~) - 17 [Hz]
'J(N,H) found -76.9 1491 -96.0 (491 -76.5 -96.8 [SO] - 136 [491 [Hz] calcd -73.5 -98.0 -73.5 -98.0 - 147
YO S 25 33 25 33 50
Scheme 3
Equation (d) is preferably used for charged species and re- produces the 'J(N,H) values in Scheme 3 within "3.5 Hz with the exception of the value for protonated benzonitrile (- 11 Hz). These correlations can be used to estimate the hybridization of the N atom. For example, compared with the 'J(N,H) value for ammonia ( 6 2 a 2 Hz) , [~"~ the small values for the 1,2-disubstituted triaziridines 24 and 25 (5 1.7 and 58.1 Hz, respectively, see Section 4.2)lS4' indicate a pyramidal geometry at N-3.
'J(N,H) coupling constants in aminopyridines and ami- nopyrimidines are dependent on the position of the NH2 group, a manifestation of the extent of conjugation and, as a consequence, loss of pyramidal geometry at the N atom (Scheme 4). This result can be used for the assignment of amino groups in azines and is consistent with the conclu- sions drawn from chemical The barriers to inter- nal rotation about the N-C bond1"] are 22.1 and 31.3 kJ/mol for the 3- and 4-amino groups, respectively, in 3,4-diaminopyridine, which clearly supports the weaker conjugation and more pyramidal geometry of the 3-amino group. i t should be added that N,H coupling in anilines is also solvent dependent. 'J(N,H) increases with increasing tendency of the solvent to form hydrogen bonds to the NHZ groups; for example, AJFD"? is 4-5 H z . " ~ ~
392 Angew. Chem. Int. Ed. Engl. 2.5 (1986) 383-413

NH,
-82 5 -87.6 -80.5 a""' N H2
-77.4 -84.0 -81.8 -78.0
Scheme 4. The coupling constants 'J(N,H) [Hz] are given below the formulas 1391.
The 'J(N,H) coupling shows no significant H I D isotope effect since the same value ('J(N,D)=9.45*0.2 Hz) was measured for the three species NH2D, NHDZ, and ND3. This value can be used to calculate 'J(N,H) in ammonia, 'J(N,H) = yH/yD.J(N,D)=6.494.J(N,D)= 61.4 Hz, in good agreement with direct measurements.1441
One-bond coupling of "N to other nuclei (e.g., I3C, "N, 29Si, 31P) is expected to decrease with decreasing gyromag- netic ratio of the X nucleus since, for the lighter elements, the Fermi contact mechanism dominates this coupling.'571 In fact, 'J(N,P) shows the largest values (50-95 Hz) in am- i n o p h ~ s p h a n e s . [ ~ ~ I The one-bond coupling between I3C and "N nuclei is much smaller (for alkyl amines, absolute values of 2-5 Hz) but increases with increasing s-character in both the N- and C-orbitals and reaches values of - 17.5 Hz in a ~ e t o n i t r i l e ' ~ ~ ~ and -20.2 Hz in diazome- thane.[6"' No general relationship, however, seems to hold between 'J(N,C) and s-character, although the equation ' J ( N , C ) = 0 . 0 1 2 5 ~ ( % ~ ) ~ . ( % ~ ) , [ ~ ~ ~ was proposed. It is evi- dent from the extensive data collections on 'J(N,C)[7".bJ that factors such as the electronegativity of the substituents on nitrogen, the n-bond order, and the orientation of the lone-pair electrons contribute substantially to the coupling constant. For example, the lone-pair electrons make a po- Sitive contribution to 'J(N,C) in pyridine, which results in a small coupling constant for the free base and a large, ne- gative value for the pyridinium ion and the N-oxidel6'I (Scheme 5). Furthermore, mechanisms other than the Fermi contact type can make considerable positive or ne- gative contributions to coupling between I5N and "C nu- cIei.[621
0 0 0 Y Y I H
*O 62 -11 85 -15.23
Scheme 5. The coupling constants 'J(N,C) [Hz] are given below the formulas b l l .
3.2.2. Long-Range "N,X Coupling (".I(N,X), n - 2, 3)
In this section we will restrict ourselves to the discussion of "N,'H and j5N,I3C coupling constants and their appli-
cation in structure elucidation. Values for these interac- tions are usually < 20 Hz and thus in the same range as corresponding 'H, 'H or 13C,'H coupling constants. Whereas J( "N,'H) can be determined for compounds con- taining a natural abundance of "N, the measurement of "N,I3C coupling constants requires I3C- or "N-labeled components since both nuclei have a very low natural iso- tope abundance (1.1% and 0.37%, respectively). I5N,'H coupling constants are, therefore, useful for analytical pur- poses (e.g., spectral assignments, configurational and con- formational studies), while 15N,"C coupling constants may be expected to aid mechanistic and biosynthetic in- vestigations. With the advent of modern pulse sequences (cf. Section 2.2), a large number of I5N,X coupling con- stants have become readily available, and major trends in their structural dependence have been worked out. Scheme 6 lists some typical examples for 'J(N,H) and 'J(N,H) in organic structures. Geminal N,H coupling is rather large in H-C=N- systems (10-20 Hz); a significant contribution to 2J(N,H) originates from nonbonding electron transfer into the antibonding o* orbital of the C-H bond.1641 Pro- tonation or quaternization of the dicoordinated sp2-hy- bridized N atom leads to a drastic reduction of the value of *J(N,H), as seen in pyridinium and pyrimidinium com- pounds (Scheme 6).L65b1
k -10 76 - 3.01 +O L7 (-) 1L 5
+ 2 7
R- H C O B CH,NH, 7 - O H H %H,
-4 5 -13 9 <2 - 1 0
-1.5 - L 0 1.L (1,3) L.8 (ti,) 1631 6.0 11,L) 1.0 ( H e ] 1.0 12,L)
Scheme 6. The coupling constants "J(N,H), n = 2 , 3 [Hz], are given below and, if necessary, above the formulas [7a, b].
When the INEPT and DEPT pulse sequences were first used to measure proton-coupled "N-spectra, it was recog- nized that ( 2 x 'J(N,H))-' is the optimal T value for a ~ i n e s [ ~ ~ ] and a~oles.[ '~] The following example illustrates the use of 'J(N,H-6)= 11.1 Hz in 2-acetylpyridine 12 to obtain a fully proton-coupled refocused INEPT spectrum. The very complex spectrum (Fig. 16a) can be simplified by applying selective 'H-decoupling during the acquisition period. These experiments (Fig. 16b) permit the determina- tion and assignment of all long-range N,H coupling con- stants ; corresponding studies can help to elucidate the
Angew. Chem. Inr. Ed. Engl. 25 (1986) 383-413 393

substitution pattern in more complex nitrogen heterocycles (see Section 4).
Fig. 16. INEPT "N-NMR spectra of 2-acetylpyridine I2 (20.3 MHz; 90% v/v in (CD&CO): a) proton-coupled, 1 1 000 pulses; b) selective irradiation at the CH, resonance, IS00 pulses [6Sa].
A detailed investigation of N,H coupling constants in azoles1661 has revealed a subtle dependence of 'J(N,H) and 3J(N,H) on the coupling pathway, i.e., connectivity, geom- etry, and bond order (Scheme 7). Such information, even if only semiquantitative, can be useful for structural assign- ments and for the estimation of coupling constants in or- der to optimize delay times for polarization transfer ex- periments and for heteronuclear 2D NMR.
Long-range N,H coupling constants in aliphatic systems are usually small ( < 4 Hz) and, since the spin systems are often complex, the precise values cannot be readily deter- mined. For example, the potential importance of 3J(N,H) for the determination of the torsional angles ty or x in pep- tide systems has raised considerable interest (Scheme 8).1671 Although a torsional angle dependence of the type
3J(N-CO-CH) = ACOS' p+ B cos p + C
was demonstrated both theoretically[681 and experimental- ly,1691 the very narrow range of 'J(N,H) is a serious limita- tion. The fact that small values may have either sign consti- tutes a further uncertainty in stereochemical applications.
Scheme 8.
The relative signs of N,H coupling constants may be de- termined by standard double resonance t e c h n i q u e ~ ~ ~ ~ 1 or spectral analysis and simulation. As expected, the Fermi contact dominated coupling constants 'J(N,H) are always negative, whereas 'J(N,H) and 'J(N,H) may show either sign; in general, N,H coupling constants > 10 Hz are nega- tive.I7'] The relative signs of all N,H coupling constants in pyridine and pyrimidine have been determined by SPT ex- perirnents['"l and spectral simulation,1721 respectively. The consistent results are collected in Table 3.
Table 3. Magnitudes and relative signs of coupling constants "J(N,H) [Hz] in pyridine [21c] and pyrimidine 1721.
\ 2 ~ ( ~ , ~ ) , N-CH3 1.5-1.9
H-2 H-3 H-4 H-S H-6
\ \ / / N-C,
H
\ / N=C
H
7.2- 12.0
10.5- 15.5
/I I1
N 3J(N*,H) *N,, /C-H < 2
[Hz l I I ll
*N\ ,,C--H 1 .o-2.2
7 / ' I
* N C-H 3.0-7.4 / \ 4
C I
Scheme 7.
Pyridine -10.93 -1.48 +0.27 -1.48 -10.93 Pyrimidine - 14.42 - +0.54 -1.07 -10.76
Long-range "N,I3C coupling constants are generally smaller than 10 Hz. The data can be best obtained from the I3C-NMR spectra of I5N-labeled compounds. 'J(N,C), like 'J(N,H), is known to depend upon the orientation of the lone-pair electrons on nitrogen relative to the geminal carbon atom, significantly larger values being observed for synperiplanar than for antiperiplanar arrangements[731 (Scheme 9). The geometrical dependence of the N-C-C coupling is not impaired by the presence of other hetero- atoms, as shown by the spectra of (Q- and (2)-azoxyben- zene. This fact permitted the use of 'J(N,C) in the struc- tural proof of two stereoisomeric azimines, 13 and 14, formed by addition of phthalimidonitrene to (Q- and (2)- azobenzene, respectively (Scheme Since N-2 and N- 3 have similar chemical shifts in 13 and 14, their assign-
394 Angew. Chem. Int. Ed. Engl. 25 (1986) 383-413

Quantum-chemical calculations have predicted that most 'J(N,N) values should have a negative sign["] owing Q 6 %& II %qQ II to the dominant contribution of lone-pair electrons in "s- hybridized" orbitals. A pronounced dependence of the magnitude and sign of this coupling on the lone-pair dihe-
(-1 11 0 [7'1 (+) 1 8 [7L1 (-) 9 L [751 (+) 2.5 [751 dral angle was also A detailed interpretation Of N,N coupling data should be based upon experimental determinations of the sign,"71 but such measurements are still lacking in most cases. Some structural applications of N,N coupling constants will be discussed in Section 4.
/ y. .N HO/N. '* 'OH C6H5 * 'C,H,
Scheme Y. The coupling constants 'J(N,C) (Hz] are given below the formu- las
ments are based upon the very different values of 'J(N,C), in analogy to the azoxybenzenes and on the basis of a the- oretical treatment of 'J(N,C).[6Zr The (@- and (2)-configu- rations clearly follow from the 2J(N,C) values obtained from the 13C-NMR spectra of the [2,3-N,]-labeled sub- strates. 3.3. 15N Relaxation Times
13 14
13 14 'J(N-2,C-2) 13.6 12.3 [Hz] 'J(N-3,C-3) 3.0 2.0 2J(N-3,C-2) 6.9 2.0 'J(N-2,C-3) 1.5 <O.S
Scheme 10
The use of N,C coupling constants in biosynthetic stud- ies will be discussed in Sections 4.1 and 4.5.
3.2.3. I s N, " N Coupling Constants
For sensitivity reasons, I5N,"N coupling can only be observed in the spectra of I5N-labeled compounds. Nev- ertheless, a considerable amount of data has accumulated and is summarized in several review^.^^^.^". bl The coupling constants are small (<25 Hz) since two nuclei with very small gyromagnetic ratios are involved. 'J(N:N) does not seem to show correlations with either s-character in the bonding orbitals or N-N bond length (Scheme 11).
7.8 -
The two principal time constants of nuclear magnetic re- laxation, TI and T2, have very different significance for ''N- and I5N-NMR spectroscopy. For the quadrupolar nu- cleus 14N, it is the transverse relaxation time TI that in- fluences the ease of observation of the NMR signal. T2 de- termines the line width A V , , ~ of the signal, which may vary from a few Hz for a highly symmetrical electronic environ- ment to several kHz for unsymmetrical environments and large molecules. Thus, sensitivity is lost as a result offust relaxation, which results in line broadening (Tz =
1/nAv=65 ms to 65 ps for A v = 5 Hz to 5 kHz). On the other hand, a very different situation arises for
the spin-1/2 isotope I5N, which lacks an effective internal relaxation mechanism. As a nucleus with a small gyromag- netic ratio, 15N depends on the spatial proximity of a sen- sitive nucleus for fast dipolar spin-lattice relaxation rates, ( T P D ) - ' , or, alternatively, on high magnetic fields (Bo) to increase the contribution of the chemical shift anisotropy term to the rate of relaxation, ( TYS") - I. The rate of spin- lattice relaxation ( T I ) - ' determines the pulse repetition rate in single-pulse experiments. Hence, it becomes evident that NH2 and NH groups with efficient 'H,I5N dipolar re- laxation can be more readily detected than tertiary N atoms, which lack the dipolar interaction with a directly bonded proton, resulting in rather long TI values and the loss of a nuclear Overhauser effect (Section 2.1). With the availability of high-field magnets (B , = 9.4 or 1 1.7 tesla), the measurement of tertiary N atoms is less time-consum- ing. The CSA term, which contributes to l/Tl for all larger molecules, increases proportionally to B; [Eq.(g)]f"Ol and thus causes a significant decrease in TI (Table 4).
C6H5 n 8 8 'NH-NH, [C&CH,)5Zr-N~N],N, NEN--CHCO,C,H, Table 4. Field dependence of "N spin-lattice relaxation times T,/a.
Bo= 1.4 4.2 4.7 9.4 T 6 7 6.2 "78al 5.6
NaNO, [a] 19.0 17.5 K C N [b] 21.2 14.5
pyrimidine [c] 170 I14
fa] 99% lSN, 1.1 M in D20, 2 5 T [b]. 95% "N, 7.9 M in DzO, 30°C (from Table 2.3 in [7a]). [cl 99% "N2, 1.6 M in (CD,),CO, 25 "C.
N E N
2.5 I78bI 2-chloro-
Scheme I I . The coupling constants ' J (N,N) [HI] are given below and, if nec- essary, a b w e the formulas [7a, bl. The value for Nz was calculated from the experimental value for '"N''N [78b] by means of Equation (b).
Angew Chem. In!. Ed. Engl. 25 (1986) 383-413 395

It should be mentioned that chemical shift anisotropy, A D = o , , - D ~ , is up to 4 times larger for I5N than for I3C nuclei and, consequently, the Bi-dependent CSA relaxa- tion mechanism becomes more effective for I5N nuclei in high-field spectrometers.
At lower field strengths very fast relaxation can be ob- tained by the addition of paramagnetic relaxation rea- gents, such as Cr(acac),, Ni(acac),, or Gd(dpm), (dpm = dipivalomethanato)[811 or, for aqueous solutions, [Gd(2 :2 : cryptate.L821 An increase in the spin-lattice relaxation rate ( T : ' ) by a factor of 10-100 is frequently observed,["] and measuring times are, therefore, dramati- cally reduced. The most recent and efficient technique to overcome the low sensitivity due to long TI relaxation times is the polarization transfer from an abundant, sensi- tive, fast-relaxing nucleus (e.g., 'H, I9F) to "N in the INEPT or DEPT pulse sequence experiments (Section 2.2). The repetition rate of the pulse sequence is governed by the (fast) proton relaxation and not by the (slow) "N re- laxation." '. "1
An intricate feature of spin-lattice relaxation is its de- pendence on molecular motion, specifically the reorienta- tion times or correlation times zc. TI decreases linearly with increasing correlation time z, for small fast tumbling molecules in the so-called extreme narrowing condition ( T : ( O ~ + W , , ) ~ < l), approaches a minimum for T,= lop9- 10-'s, and it increases again for longer correlation times (Fig. 17). This effect applies to large biomolecules, such as proteins and nucleic acids, and becomes more pronounced at high fields.
5, I T
2 1102 2 -lo2
-2 lo-* i Fig. 17. Dipole-dipole relaxation (T;"') as a function of the correlation time 7, (for isotropic molecular reorientation) for different field strengths Bo [7al.
In conclusion it may be emphasized that a consideration of spin-lattice relaxation is a prerequisite for the optimiza- tion of "N-NMR experiments. Since TI values are strongly dependent on experimental conditions (field strength, concentration, solvent, temperature) and very sensitive to the structural environment, data compilations are only of limited value. TI values of some typical mole- cules are reported in ref. [7a, c, 41.
3.4. Assignment Techniques
The assignment of I5N resonances may become a con- siderable problem for compounds with several N atoms, in particular when repeating structural units are involved, such as in polypeptides or polynucleotides. But even in small molecules the identification of the "N-NMR signals may become a challenging task. The first difficulty arises when the true signals have to be differentiated from spu- rious electronic signals in spectra with low signal-to-noise ratio. As a result of variable TI relaxation times and (nega- tive) nuclear Overhauser enhancements, "N-NMR intensi- ties can be very different, and weak signals are easily lost in the noise. Application of several of the detection meth- ods outlined in Section 2 is often necessary to ensure that all resonances are indeed detected. In particular, experi- ments with and without NOE should be performed and, in the case of polarization transfer experiments with pulse se- quences, the delay time z should be varied.
The second problem concerns the assignment of lines to specific N atoms. Based on the discussion of the spectral parameters in Section 3, the following criteria can be ap- plied :
- chemical shifts, including substituent effects - protonation effects, N-H exchange rates - lanthanide-induced shifts (LIS) - nuclear Overhauser effects - N,H and N,X coupling constants - N,H and N,X correlation experiments (1 D and 2D) - spin-lattice relaxation times - isotopic labeling
The most powerful assignment aid is the analysis of the proton-coupled "N-NMR spectra. It is this area of "N- NMR spectroscopy where rapid progress has been made in the past few years, owing to the development of modern 1 D and 2D pulse sequence experiments. Paramagnetic shift reagents (lanthanide complexes) have also proven useful in several cases[83.39.841 to assign "N resonances. In oligopeptides, the different exchange rates of free and hy- drogen-bonded NH groups are a useful criterion for their assignment. Moreover, additive empirical shift increments are available, e.g., for a z ~ l e s ~ ~ ~ ' and azines.lg6I Finally, ex- tensive molecular orbital calculations have led to the pre- diction of a wide range of "N chemical shifts, which may be used for assignment purpose^.[^'.'^.^'^
4. Chemical Applications
The increased sensitivity of "N-NMR spectroscopy to- gether with the possibility of obtaining 'H-coupled "N- NMR spectra has greatly enlarged the scope of applica- tions in structural chemistry during the past few years. In this section, we will discuss selected examples from the fields of organic chemistry, biochemistry, and organome- tallic and coordination chemistry. They were chosen to il- lustrate the principles discussed in the previous Sections, the wide spectrum of applications, and the general poten- tial of "N-NMR spectroscopy as a probe into molecular structure and reactions.
396 Angew. Chem. Int. Ed. Engl. 25 (1986) 383-413

4.1. Structure and Bonding of Organic Molecules
"N-NMR spectroscopy is particularly suited for studies of isomerism in nitrogen-containing heterocyclic systems. For example, isomeric acetylpyrazoles of the types 15 and 16, which serve as precursors for biologically active sub- stances,'"] are formed by the reaction of monosubstituted hydrazines with 3-(ethoxymethylene)-2,4-pentanedione, the product ratio depending on the nature of R (Scheme 12). Since the tricoordinated and dicoordinated nitrogen
CH&O COCH, R-NHNH, \,-/ L, t
II CHOC2H5
I R
15 16 Scheme 12
atoms, N- 1 and N-2, respectively, are readily distinguisha- ble by their chemical shifts (Fig. 18), the geminal and vici- nal N,H coupling constants obtained from an INEPT ex- periment allow an unambiguous assignment of the iso- mers.["I As expected from the results discussed in Sec- tion 3.2.2, the pyrazoles 16 show large 'J(N-2,H) and 'J(N-I,H) values (e.g., 16a, R=COZC2H,, 13.7 and 9.0 Hz, respectively), whereas the corresponding values for 15a are 3.4 and 1.9 Hz, respectively. Very similar data were found for the N-benzyl derivatives.
C02Et
b -6
0 -159 2 - 7 6 0
+d Fig. 18. Refocused and 'H-decoupled INEPT "N-NMR spectrum of ethyl 4-acetyl-5-methyl- I -pyrazolecarboxylate 1627 (20.3 MHz; (CD&SO): without decoupling (insets) 1151.
4- and 5-Nitroimidazoles present another case of regio- isomerism, and their differentiation is of particular interest since N-substituted 5-nitro isomers are potent antiproto- zoics.[88bl A reliable assignment was possible on the basis of the chemical shifts of the NOZ group (4-N02, 6= - 18 -+ 1 ; 5-NOZ, 6= - 25 Ifr 1) and I5N,'H coupling constants of N-1 and N-3.'15]
Intricate tautomeric equilibria are common in heterocy- clic systems and their elucidation can often be achieved by "N-NMR spectroscopy. For example, the position of the fast tautomeric equilibrium of 1 -hydroxybenzotriazole 17a and its derivatives was determined as a function of solvent on the basis of the chemical shifts of N-3.la9] The range of chemical shifts for N-3 ( - 58 to - 100 ppm) clearly shows that the N-hydroxy form 17a dominates the equilibrium, the contribution of the N-oxide 17b ranging from 6% (DMSO) to 26% (CH,OH) (Scheme 13). 2-Hydroxyazines
OH 0-
Scheme 13
are known to exist in the tautomeric amide form. However, a n unusual iminol-amide equilibrium was recently ob- served for the hydroxyaza[ IO]annulene 18 (Scheme 14). The averaged ''N chemical shift of the tautomeric system (6= -204.3, (CD&SO, 37°C) was compared with data of the N-methyl (6= - 240.6) and 0-methyl (6= - 115.9) der- ivatives, the results indicating an 18a/18b ratio of about 7 :3.f901 The remarkable stability of the iminol form 18b was also evident from the "C-NMR spectrum.
18a 18 b
Scheme 14.
Similar studies have been carried out on a series of sub- s ti tuted azoIes[' 92. 7h1 with special emphasis on imid- a ~ o l e [ ~ ~ l and nucleotides, which are important for the in- vestigation of biological systems.[941
Scheme 15
Hull, Kiinstlinger. and Breitrnaie~-'~~' have reported on the tetrazolo[ 1,5-a]pyrimidine 19 /2-azidopyrimidine 20 valence isomerism, which lends itself to an investigation by I5N-NMR spectroscopy (Scheme 15). This exchange, stud- ied previously by 'H-NMR is so slow that it is possible to assign all nine signals of the nonequivalent N atoms of the n-butyl derivative (as a neat liquid) (Fig. 19); the equilibrium lies on the tetrazole side (85%). The 'J(N,H) values were crucial for the correct assignment. The equilibrium was shown to be strongly solvent depend- ent, the tetrazolo form 19 being favored in polar solvents, while the azido form 20 predominates in chloroform.
Angew. Chem. Int. Ed. Engl. 25 (1986) 383-413 397

12 Hz
N-2.N-3 I N-1 azide
1 I I r 1 1 I 1 T I I I r ----- T - 7 - 1 -100 - 260 -50 - 6 0
Fig. 19. "N-NMR spectrum of the equilibrium mixture of 6-butyltetrazolo[l,5-~~pyrimidine 19 and 2-azido-5-butylpyrimidine 20, R = n-CIHy (40.6 MHz: neat liquid, IS-mm sample tube). The numbering of the N atoms does not correspond to the systematic nomenclature (see Scheme IS) 1951.
Substituent effects on the "N resonances were studied in pyridine~,'~'] pyrimidines,[971 and pyrazine~,'~'] and linear correlations with 13C substituent effects could be estab- lished.
Extensive studies of the 15N-NMR spectra of the biolo- gically important pteridines['I have led to a definite struc- ture assignment of the so-called "activated formaldehyde" (Blakley cofactor), which is formed from (6S)-5,6,7,8-tetra- hydro-L-folic acid and formaldehyde. This compound serves as a cofactor in the enzymatic conversion of glycine into ~ e r i n e . ~ ~ ~ ] Both the 5-hydroxymethyl- and the cyclic 5,IO-methylene structure 21 have been 'Ool
The natural abundance "N-NMR spectrum of the (6RS) compound is shown in Figure 20. The assignments of the seven N atoms are based on the "N-NMR data of tetrahy- drofolic acid derivatives,"' but the signals of the two dia- stereoisomers (at 10.1 MHz) are not resolved.
The 20.3-MHz "N-NMR spectrum of the product la- beled with "N in the 5- and 10-positions (Fig. 21) reveals clearly resolved doublets (A6=0.24) for the (6R) and (6s) diastereoisomers. Since the small 1SN-5,15N-10 coupling of 2 Hz does not disprove the 5-hydroxymethyl structure, the 1 1-position was labeled by reaction with ['3C]formalde- hyde. In the 'H-decoupled I3C-NMR spectrum the methy- lene carbon appears as a double doublet with J( '5N,13C)= 10.8 and 4.7 Hz. Since the signal of "C-11 in the compound with natural "N isotope abundance is not split, the double doublet structure in the triply labeled product is due to 'J(N,C) coupling only, thus clearly prov- ing the cyclic methylene structure 21 for the activated for-
maldehyde['O'l (cf. the other 13C, "N coupling constants in Scheme 16). As expected from the linear correlation be- tween 'J(N,C) and the s-character of the bonding orbitals (see Section 3.2.1), the aniline-type N-10 atom shows the largest 'J(N,C) values, whereas N-5 shows smaller values, indicating a pyramidal (sp3-type) geometry at this nitrogen
N-1 N-8.N-10
1 'j3 amide NHZ-2 i ,i5
l ~ " ' " " ' ' ~ " ' ~ ~ l
-200 -250 -300 -350 - 6
Fig. 20. ISN-NMR spectrum of (6RS)-S.lO-methyIenetetrahydrofolic acid 21 (10.1 MHz; 0 . 8 ~ in H 2 0 / D Z 0 92:s at pH 7, 20-mm tube) [lola].
398 Angew. Chem. Int. Ed. Engl. 25 (1986) 383-413

N -10 N - 5
Fig. 21. "N-NMR spectrum of (5,10-'5N2)-tl (20.3 MHz; 0.05 M in D,O at pD 7 , 10-mm tube); for the assignments of the splittings in the resonances of N-5 and N-10 see text and [IOIb].
atom. For comparison, the 'J(N,C) value for CH3NH2 is 4.5 Hz."O2I
H
Scheme 16 For numerical values see text.
The lone pair of electrons on the nitrogen atom is re- sponsible for the dependence of "N chemical shifts on n,n-delocalization and hydrogen bonding. A further, di- rectly related effect-most of the time annoying but occa- sionally useful-is the solvent dependence of "N shield- ing data (cf. Section 3.1.2). In the following discussion we will give specific examples for the successful use of "N- NMR spectroscopy in studies of electronic structure and hydrogen bonding.
The n,n-interactions in enamines and related com- pounds are largely responsible for the specific physical and chemical properties of enamines as synthons in or- ganic synthesis.['031 In our laboratory, extensive studies have been carried out on "N chemical shifts in amines, enamines, enamino ketones, and other enamines with ex- tended c o n j u g a t i ~ n . [ ~ ~ . ~ ~ . ~ ~ . The chemical shifts of "N nuclei in this class of compounds are found between those of tertiary amines (more shielded) and amides (less shielded) (Fig. 14). In order to separate the n,n-delocaliza- tion from electronic and steric substituent effects, it proved advantageous to define a differential chemical shift A6( "N) =6(amine) - G(enamine). As( "N) correlates well with the free energy of activation AG' for restricted rota- tion about the N-C, bond as determined from variable- temperature 13C-DNMR measurements[40' (Scheme 17, Fig. 22).
Scheme 17.
n 16 -
14 -
12 - * U (3 a
10 -
-30 -40 - 50 - 60 - A&N)
Fig. 22. Correlation of the free energy of activation AG,' for rotation about the N-C(a) bond with the differential chemical shift A6("N)=6(am- ine) -6(enamine) 1401.
OYU\ ,H (a) ,c=c ("-I> L-
Extrapolation of the data in Figure 22 gives a A G + value of 15-20 kJ mol-' for rotation about the N-C, bond in a simple enamine; this is in good agreement with theor- etical predictions for vinylamine (25.5 kJ mol- I)rlo51 and with experimental data on conjugative interactions in other simple enamines.['061 In a similar study, Martin et al.[1071 correlated activation
energies E, with "N chemical shifts in amides and thioam- ides and also advanced a semiempirical explanation for the validity of such correlations. Since enamino ketones may be considered as vinylogous amides, our results lend further support to this approach.
n,n-Delocalization was studied in a similar manner in substituted anilinostyrenes 22 ; the A6(N) values show ex- cellent correlation with the Hammett constants for a se- ries of donor and acceptor substituents (Fig. 23).[401 In this case, the I3C chemical shift of Cr3 also exhibits a similar correlation with A6(N), since this nucleus is in a similar electronic environment and no steric interactions with the substituents occur.
An excellent survey of correlations of 6(15N) with E , and AG' for hindered rotation about N-X bonds and with cr values is given in ref. [7a] and covers the literature up to 1980. More recently, Taft et a1.[1081 analyzed I5N-NMR shifts of amides in solution in terms of three solvent pa-
Angew Chem. Int. Ed. Engl. 25 (1986) 383-413 399

22 0.8 c \
- 0.2 "-1 -35 - A6(N)
Fig. 23. Correlation of A6("N)=6&6Z with Hammett u values for substi- tuted anilinostyrenes 22; ( I ) R=rn-NOI, (2) m-CI, (3) p-CI, (4) m-OCH,, (5) H, (6) p-CH,, and (7) p-OCH, [40].
rameters, n* (polarity/polarizability), a (hydrogen-bond donor acidity), and #? (hydrogen-bond acceptor basicity) using multiparameter linear relationships with 6( I5N). Fur- ther linear free energy correlations involving Hammett (T
constants have been reported for &(NO) values in substi- tuted N-methyl-N-nitrosoanilines,'" for the three N atoms in p-substituted I-phenyl-3,3-pentamethylenetri- azenes,'"'' and in a detailed study of I5N- and "0-NMR shifts versus nI and nR constants as well as calculated elec- tron densities in nitrobenzene derivatives.["']
As expected from the foregoing discussion "N chemical shifts have been used successfully to monitor hydrogen- bond formation and protonation. Particularly large, low- frequency shifts are observed in systems containing the C = N group; pyridine has already been discussed in Sec- tion 3.1.2. Detailed studies on imines and oximes by Rob- erts et a1.[1121 showed that hydrogen-bond effects may be- come as large as 28 ppm (in going from CHCI3 to CF3CH20H as solvent), whereas full protonation (CF3COOH) results in shifts of 110 to 150 ppm (to lower frequencies). I5N chemical shifts were measured in N-reti- nylidenebutylamine and related Schiff bases as models for rhodopsin and bacteriorhodopsin in solvents of increasing hydrogen-bond donor acidity.[' 1 3 ] The protonation effects on the "N-NMR shifts increase with increasing length of the conjugated polyene, and it was concluded that I5N- NMR spectroscopy is a most suitable method for the de- termination of the degree of protonation of the Schiff base linkage. In contrast, small high-frequency shifts ( < 25 ppm) are observed upon protonation (and hydrogen bonding) of aliphatic amines. In cases where hydrogen- bond shifts are too small to permit a reliable interpreta- tion, 'IN spin-lattice relaxation times can be used to mon- itor restricted motion, as recently shown by Yu and Levy for 2,4-pentanediamine.[' 14]
The study of hydrogen bonds between NH and CO groups in peptides showed that 'IN chemical shifts are a sensitive indicator of intramolecular NH . . . O=C interac- tions and thus of the secondary structure of peptides. Gramicidin S, a cyclic decapeptide containing the se- quence Phe-Leu-Om-Val-Pro twice, is a particularly well-
studied case investigated by several research groups." ''I
The four hydrogen bonds between valine and leucine are indeed reflected in the ''N chemical shifts of the two am- ino acids when the decapeptide is measured in solvents of increasing proton acidity; this is due to intermolecular hy- drogen-bond formation between the solvent and the ex- posed carbonyl group of the respective peptide bond.[''61 The four intramolecular hydrogen bonds are also demon- strated by reduced exchange rates for the protons in- volved, which is reflected in a distinct I5N,'H coupling even in basic medium. A comparison of the 15N-NMR spectra of linear oligopeptides with that of gramicidin S led to the same conclusion.~'~7' Intermolecular hydrogen bonding in nucleosides, the predominant mechanism of self-association and base-pairing, is another domain in which "N-NMR spectroscopy has been successfully ap- plied to structure e l ~ c i d a t i o n . " ' ~ ~ An extension to the study of N-H bond lengths in DNA by solid-state 'IN-NMR spectroscopy has also been reported (cf. Section 4.6 and Fig. 11) .
4.2. Reaction Mechanisms and Reactive intermediates
In mechanistic studies 'IN-NMR spectroscopy has proven to be a powerful tool for the structure elucidation of unstable intermediates. Very recently, we reinvestigated the classic reaction of benzenediazonium chloride with li- thium azide, which results in the elimination of nitrogen and the formation of phenyl azide."] This reaction, first de- scribed by Hanlzsch,[' l9 ] was investigated mechanistically in 1956-1958 by Huisgen and Ugi et al.['zol They discovered that the reaction proceeds by two separate mechanisms. For one of them, it was proposed that phenylpentazole 23a is a n intermediate, since it accounts for the equal distribu- tion of the "N-labeled, terminal N atom of the diazonium salt in the decomposition products phenyl azide and ni- trogen (Scheme 18). Although a series of crystalline p-sub-
23
a , R = H; b , R = N(CHJ)2
Scheme 18
stituted phenylpentazoles was subsequently isolated1'*" at low temperature (- 30°C) and despite the determination of the structure of p-dimethylaminophenylpentazole 23b by X-ray c ry~ta l lography,"~~~ a direct proof for the exis- tence of the pentazole ring in solution was still lacking.
400 Angew. Chem. lnt . Ed. Engl. 25 (1986) 383-413

The natural abundance 40.56-MHz I5N-NMR spectrum at -35°C of 23b is shown in Figure 24. Although even at this low temperature the pentazole had partially decom- posed to the azide and NZ, its four resonance signals could be identified. The assignment was achieved by means of the "N-NMR spectrum of the [2(5)-15N]pentazole 23b* (Fig. 25a). The "N-NMR spectrum measured during de- composition of 23b* (Fig. 25b) proves the exclusive forma- tion of I5N=I4N and azide labeled in the N-2 position. The structural integrity and very slow decomposition of the pentazole ring at - 20°C is thus confirmed for the p-dime- thylaminophenyl derivative 23b.'"
31
3' I
I I N2111 2: l
l . . - - ' . . . . l . . - 0.0 -100.0 -200.0 -300.0
- 6 Fig. 24. "N-NMR spectrum of p-dirnethylarninopheny1pent:izole 23b (40.6 MHz; -35°C: 0 . 0 4 ~ in CDCI, with 0 . 0 7 ~ Cr(acac),). The dashed numbers apply to the azide
ref.
1 a> 2
ref.
L---L 1 1 1 1 I
0.0 -100.0 0.0 -100.0
- 6 Fig. 25. I5N-NMR spectra of the [2(5)-1SN]pentazole 23b (40.56 MHz; -20°C; 0 . 0 1 3 ~ in CDCI, with 0 . 0 6 ~ Cr(acac)3); a) directly after prepara- tion of the sample; b) during decomposition to the N-2-labeled azide and "N=I4N. ref=standard CH,N02.
A considerably less reactive nitrogen homocycle, triazir- idine, has been a target of organic synthesis for many years. Dreiding et al. achieved the photochemical cycliza- tion of azimines to substituted t r iazir idine~.[ '~~l Subse- quently, the stereochemistry of substituted triaziridines was investigated both experimentally and theoretically['241 and a series of compounds was characterized by "N-NMR
The chemical shifts of 1,2-dialkyl-substi- tuted triaziridines lie in the range of substituted hydrazines (6 = - 240 to - 290) and are typical for pyramidal, tricoor- dinated N atoms. The N,H coupling constants, however, are unusually small (cf. Section 3.2.1 and Scheme 19). The
significant increase in IIJ(N,H)I in going from the 1,2- trans-diisopropyl derivative 24 to the cis-triaziridine 25 is consistent with a second hyperconjugative lone-pair effect from the inverted N-2 atom. The N,H coupling constant may thus provide a structural criterion for the still un- known all-cis-triaziridine, for which in the absence of any lone-pair effect a 'J(N,H) value of approximately 45 Hz would be expected. These effects are in line with similar observations in N-C-H systems where a syn-orientation of the lone pair and the C-H bond leads to an increase of 'J(C,H), e.g., in l - m e t h y l a ~ i r i d i n e ~ ' ~ ~ ' ~ and in pep-
24
1 (CH,),CH
51.7'05
0
58.1 20.5
Scheme 19. The coupling constants 'J(N,H) [Hz] are given below the formu- las (in CDC13).
The existence of short-lived free radical intermediates in chemical reactions can be demonstrated by "N-CIDNP (chemially induced dynamic nuclear polarization) effects in the stable products if radical pairs are involved.['2h1 This technique provided the first direct experimental proof for the formation of aromatic radical cations in the nitration of N,N-dimethylaniline with H ''NO3 in 88% aqueous H2S04[1271 (Scheme 20). The radical cations are believed to originate in a reaction catalyzed by HN02.11281 As shown in Figure 26, polarization is only present in the p-nitro prod- uct (time-dependent negative signals), whereas the m-nitro product gives a n absorption signal that steadily increases with time, since this product is not formed in the catalyzed reaction.
B I
A
A
Fig. 26. "N-NMR spectra (25°C; H " N 0 , (0.53 M) in aqueous H2S04 (88%)) as a function of time for the nitration of dimethylaniline (0 .55~) : a) after 3 min; b) after 12min; c) after 55 min; A=p-02'5N-CnH4-N(CH~)~, B = m-02'5N-C6H4-N(CH3)2 [ 127al.
Angew. Chem. Int. Ed. Engl. 25 (19861 383-413 40 1

The same authors have reported C I D N P effects in the "N-NMR spectra recorded during the acid-catalyzed rear- rangement of N-nitroanilines to p-nitroanilines; their re- sults support the intermediate formation of [ A r y I N g "NO:] radical pairs.['291 Because of the negative magnetic moment of the "N nucleus, a negative sign has to be inserted into Kaptein's formulas for the calculation of the sign of net polarization.['301
AryiH + NO," \
Scheme 20.
I5N-CIDNP spectra have also been recorded during nu- cleophilic substitution of arenediazonium ions and indi- cate that the process is at least partially h o m o l y t i ~ . ~ ' ~ ' ~ The "N-NMR spectrum of '5N-labeled 4-chlorobenzenedia- zoniurn tetrafluoroborate subjected to decomposition in weakly alkaline aqueous solution (Fig. 27) clearly exhibits enhanced absorption and emission signals for diazonium
27 26
IN2
200 0 - 100 -200 1 1 I I
- 8
jor limitations of the significance of CIDNP experiments for quantitative mechanistic studies.
Several other applications of "N-NMR spectroscopy to reaction mechanisms have been published, e.g., detailed investigations of the Fischer indole the rear- rangement of 1,3-oxazine-2,4-diones into pyrimidines and p y r a ~ o l e s , " ~ ~ ~ and the mechanism of formation of N-ni- trosothiourea."341
4.3. Stereochemistry and Dynamic Processes
Variable temperature 'H- and I3C-NMR studies have proven very useful in investigations of intra- and intermo- lecular dynamic phenomena. Measurements of "N- DNMR parameters are still hampered by the low sensitiv- ity of the "N nucleus. However, it can be anticipated that the experimental improvements discussed in Section 2 will surmount this difficulty. Furthermore, lowering of the tem- perature results in a more effective spin-lattice relaxation, at least for small molecules and in magnetic fields of mod- erate strength.
The high sensitivity of the "N chemical shift to delocal- ization of the lone-pair electrons makes it a useful configu- rational and conformational probe for conjugated rnole- cules. Thus, different chemical shifts were observed for the (Q and (2) isomers of enamino ketones, esters, and am- ides; moreover, different conformers could be detected in the slow exchange limit.['3.
2 8 a
IZ, s-c1sj
28b 2 8 c
I €, 5- CIS) I€ , s-trans1
Fig. 27. ' W N M R spectrum of [I,2-'5N2]-p-chlorobenzenediazonium tetra- fluoroborate (9.1 MHz; 7 2 T , H20, pH 9.7) [131]. The signal for N-2 of 26 is not observed. 'J(N,N) in 26 is < 1 Hz (not resolved), whereas a value of 14.7 k 0.5 Hz is obtained for 27.
ion 26, the trans-diazotate ion 27, and molecular nitrogen. In a detailed interpretation, the authors conclude that the two radical pairs
[AryIN$'oONzAryl] and fAryIo00N2Aryl]
are formed in a very complex reaction sequence. However, other radical pairs cannot be excluded and, most impor- tantly, the experiments "do not allow determination of the extent to which the homolytic pathway contributes to the total reaction." The last statement describes one of the ma-
-292 9 - 2 9 6 5 - 2 8 2 6
- 6
Fig. 28. INEPT "N-NMR spectrum of 4-methyl-l-(methylarnino)-l-penten- 3 - 0 1 ~ 28 (20.3 MHz; -65°C: [Da]dioxane) [104].
For example, the low-temperature spectrum of the en- amino ketone 28 (Fig. 28) exhibits signals that can be as- signed to s-cis and s-trans conformers on account of re- stricted rotation around the C(2)-C(3) Whereas the high-frequency signal (6 = - 282.6, hydrogen bonded
402 Angew. Chem. In?. Ed. Engl. 25 (1986) 383-413

(Z.s-cis) isomer 28a) remains narrow over the entire tem- perature range from +24 to -6O"C, the two other lines exhibit dynamic behavior and, at -6O"C, show the equili- brium 28b+28c in the slow-exchange limit. The weak, low-frequency signal (6= - 296.5) increases in intensity with decreasing size of the C-alkyl group and is therefore assigned to the nonplanar (E.s-trans) conformer 28c. The higher shielding of the N atom in 28c (- 2.6 to - 4.7 ppm relative to the N atom in the (E,s-crs) conformer 28b) is in agreement with expectations based on the paramagnetic shielding term, up= -(A@- (r-3)2,CQ, since inhibition of conjugation in nonplanar 28c is expected to result in a larger A E value (see also Fig. 14).
It should be noted that the dependence of the chemical shift differences O R solvent and temperature requires care- ful consideration before small chemical shift effects can be interpreted.
Nomura and Take~chi~'~~~ observed slow double inver- sion at two N atoms in bicyclic hydrazines by low-temper- ature I5N-NMR spectroscopy. At -45"C, the chemical shift difference of the two diastereotopic N atoms in 29 is 1.08 ppm (10.9 Hz at 10.1 MHz) and AGc" (Tc==2695 1 K) was determined to be 58.4 kJ mol-I, a value that agrees well with the data obtained from 'H- and "C-NMR meas- urements (55.2 kJ mol-I) (Scheme 21). Further conforma- tional studies on ~ i p e r i d i n e ~ ' ~ ~ ] and te t raa~adecal ins[ '~ '~ as well as on restricted C-N bond rotation in the guanidino moiety of ~ - a r g i n i n e [ ' ~ ~ ~ have also been reported.
Scheme 2 1
Although stereochemical applications of "N-DNMR studies are still scarce, the above-mentioned examples show that the considerable sensitivity of the I5N nucleus to subtle stereochemical effects constitutes a promising basis for further investigations, particularly in biochemistry.
In this context, the "N-NMR spectroscopic investiga- tions on chiral substances reported by Roberts et al."391 are worth mentioning. The diastereotopic splittings in the 15N- NMR spectrum of racemic S-benzyI-5,6,7,8-tetrahydroqui- noline in the presence of optically active carboxylic acids were found to be 3-12 Hz at 18.25 MHz and were strongly medium dependent, larger differential shifts being asso- ciated with less polar solvents and lower temperatures.
small (a few tens of ppm) for the majority of nitrogen li- g a n d ~ , [ ~ ] a wide range of shifts has been observed for N2 and NO groups. In addition, the known I5N chemical shifts for organic compounds are extended toward higher resonance frequencies (Fig. 13). Unsaturated ligands with direct metal-nitrogen bonds are observed between 6 = 0 and 900; additional deshielding effects are observed in pa- ramagnetic molecules. Moreover, the coupling constants for "N nuclei directly bonded to metals may vary between a few and several hundred Hz. From a study of these pa- rameters, information about the mode of coordination and the geometry of the complexed ligand can be obtained. The literature of this field up to 1980 is covered in a review by M u s ~ n . [ ~ ~ l
The fundamental biochemical problem of nitrogen fixa- tion and reduction has stimulated the study of dinitrogen complexes of various transition metals, and "N-NMR spectroscopy has proven a powerful tool in their structural investigation.[1401 Dinitrogen was found to coordinate either in a linear dihupto (M-N-N-M) or in a monohapto (M-N-N) fashion. The structure of the first complex con- taining both types of dinitrogen ligands, 30, was estab- lished by X-ray diffraction (Scheme 22). The
Scheme 22. [{[~s-C5(CH,)s]2ZrN212N2] 30.
"N-NMR spectrum of the fully labeled compound exhi- bits a singlet a t 6= 179 for the 1,2-q2-bonded N2 (p-N2) and doublets at 6=80 and 6= 1 I ( 'J(N,N)=6.2 Hz) for the q '-bonded N2. The latter signals are temperature-depend- ent indicating dissociative exchange between bound Nz and free, dissolved N2.1781 The "N chemical shifts of 1,2- q2-N2 complexes extend from 6=300 to 50, whereas the ql-bound N2 is more shielded (6=200 to - 100) with the N, atom resonating at lower The I5N,M coupling constants provide another important criterion for the differentiation of the two binding modes of N2, as shown in the [runs-chloro(dinitrogen)bis(triisopropylphos- phane)rhodium complex 31 ('J(Rh,N( 1)) = 28 Hz, 2J(Rh,N(2))=4 H z ) " ~ ~ ] (Scheme 23).
4.4. Coordination Chemistry
Although the number of significant structural applica- tions of "N-NMR spectroscopy is much smaller in orga- nometallic and inorganic chemistry then in organic chem- istry, there are a few nitrogen-containing ligands that have been studied extensively with respect to metal binding, e.g., NO, CN and, most important, N2. Whereas the differ- ences in chemical shift due to coordination are relatively Scheme 23.
Q N
31
Angew. Chem. I n f . Ed. Engl. 25 (1986) 383-413 403

Reduction of coordinated dinitrogen leads to the diazen- ido complexes M-N=N-R, which can exist in a singly bent or doubly bent configuration (Scheme 24). The trans- oid form is characterized by a strongly deshielded N, nu- cleus (&= 300), which allows a differentiation from the singly bent isomer[1431 (cf. Fig. 13). Further reduction prod- ucts, e.g., hydrazido, nitrido, and imido complexes, have also been characterized by "N-NMR data.1140-'431
Scheme 24. Left: singly bent; right: doubly bent.
The stereochemistry of nitroso complexes was also suc- cessfully studied by means of I5N chemical shifts.11441 As in the case of the M-N=N-R system, bending leads to strong deshielding effects on the "N(0) signal, and availa- ble data allow a correlation between M-N-0 bond angle and "N chemical shift. Linear nitrosyl complexes exhibit shielding values of 6= 0 f 80 and strongly bent complexes (120") 6= 350-800; intermediate cases are also known. Since the strong deshielding parallels a bathochromic shift of the UV/VIS absorption maximum, the effect can be as- cribed to a lower n+n* excitation energy, which results in a n increase in the paramagnetic shielding term. For nitroso complexes of cobalt(m) this is also reflected in decreased shielding of the 59C0 nucleus.114s1
"N chemical shifts of nitroso ligands were used in the structure determination of the Roussin cluster anions, 32 and 3311461 (Scheme 25). The presence of three resonances in the spectrum of "N-enriched (99%) 32 (Fig. 29), two of which are mutually coupled, suggests a threefold symmetry axis and the presence of Fe(NO), and Fe(N0) subunits in the cluster molecule. The "N chemical shifts of the nitroso ligands in 32 and 33 are typical for linear Fe-N=O groups (Fe(NO),,(NO),,: 6=30-80; Fe(NO),,: 6=7-20).
Spin coupling of "N nuclei to complexed metal nuclei depends on the gyromagnetic ratio of the metal and is very sensitive to the bonding orbitals involved. An interesting
Scheme 25
404
1 I 1 80 6 0 40 20 0
- s Fig. 29. "N-NMR spectrum (36.5 MHz) of the NO groups in [(CtiHs)3PNP(CtiHs)3]~[Fe,S3('sN0),]B, a salt of the anion 32; the equatorial and axial NO groups show a 'J(N,N) coupling of 4.3 Hz [146].
example is provided by the lead(I1) nitrate complex of 1,4,8,1 I-tetraazacyclotetradecane, 34, which forms a dis- torted octahedral structure (Scheme 26) with strikingly dif- ferent 207Pb,'sN coupling constants (207.5 and 19.8 Hz) for the two "N resonances at 6= -318.8 and 6 = -325.5, re- spectively, at the low-frequency end of the "N-NMR shift scale. Since the lengths of the equatorial and axial Pb-N bonds (2.45 f 0.02 and 2.50f 0.08 A, respectively) are not appreciably different, the large difference in 'J(Pb,N) is ascribed to differing contributions of the Fermi contact term as a consequence of the axially distorted octahedral geometry. Hence, the assignment of 207.5 Hz to equatorial N-donor ligands (normal octahedral bonds) and 19.8 Hz to axial N-donor ligands (distorted octahedral bonds) was s~gges ted .~ '~ ' ] Such differences in 'J(M, 15N) values were also reported for related Cd", Hg", and Pb" com- p I e ~ e s . [ ' ~ ~ J
34
0
Scheme 26
In favorable circumstances, i.e., very short electron spin relaxation times, "N resonances may even be observed in paramagnetic metal complexes, e.g., "N=CQ ligands in low-spin iron(ir1) cyanide complexes of porphyrin deriva- tives and hemoproteins. A further extension of the "N chemical shift range to higher frequencies is found for the NCe resonance, 6=450 to l100.['491 In low-spin iron(II1)
Angew. Chern. Int. Ed. Engl. 25 (1986) 383-413

tetraphenylporphin complexes extremely short TI and T2 relaxation times (0.2-3.0 ms) were reported for the pyrrole N atoms.lt5"]
4.5. Biochemical Applications
The potential of "N-NMR spectroscopy for the study of biologically relevant molecules and molecular systems has been the major driving force for the rapid development of experimental techniques and for a steady improvement of "N-NMR sensitivity. The last few years, in particular, have seen a significant increase in biological "N-NMR studies. A recent review by Blomberg and Riiterjans"'ll covers detection techniques, spectral parameters, and typ- ical applications in the major biological fields. Further, Kanamori and Roberts"521 reported specific applications with particular emphasis on enzyme systems, nucleotides and nucleic acids, and intact cell systems. Intermolecular interactions in biomolecules were reviewed by Kyogo- k ~ , " ' ~ ' and Coxon reported on "N-NMR studies of amino sugars.["41 Although not primarily biological in nature, a comprehensive account of the I5N-NMR spectroscopic characterization of oligo- and polypeptides by Krichel- ~ f o r f " ~ ~ should be mentioned in this context. Very few studies of nonbiological polymers have so far been re- ported.[""]
We will focus our attention on a few examples from the recent literature in order to illustrate the possibilities and trends of "N-NMR spectroscopy in the study of biological systems. Studies of biosynthesis and metabolism are one field in which isotopic labeling combined with I5N-NMR measurements allows detailed studies, which afford new insights into biochemical mechanisms. Hori and Shimi- Z U [ ' ' ~ ~ have investigated the structures of two related neu- rotoxins using biosynthetically enriched material. They succeeded in demonstrating that neosaxitoxin 35 is the N'-hydroxy derivative of saxitoxin (Scheme 27). The "N-
35
Scheme 27.
36
NMR spectrum (Fig. 30) of an "N-enriched (99%) toxin allows the identification and assignment of the seven N atoms, based on chemical shifts and N,H coupling con- stants, and the elimination of the alternative structure 36. The correct assignment of the I5N-NMR signals of com- plex molecules is an important prerequisite for biosyn- thetic studies. "N,13C double labeling allows l5N,I3C cou- pling constants to be measured, thus providing evidence for biosynthetic routes; for example, the biosynthetic for- mation of the N-C(5) bond in isopenicillin N (which is formed by cyclization of &(a-L-aminoadipoyl)-
L-[3-13C]cysteinyl-D-[ ''Nlvaline) is demonstrated by the one- bond "N, I3C coupling constant (4.4 HZ).'~'~]
a) N e
N(C-14)
3' -280 -290 -300 -3;O '- I - 6
b)
&I-280 -290 -300 -3;O -6
Fig. 30. I5N-NMR spectra of 99% "N-enriched neosaxitoxin 35 (36.5 MHz; 1.8 mM in H 2 0 / D 2 0 95 : 5 ) . a) 'H-decoupled; b) 'H-coupled [157].
The in vivo study of amino acid biosynthesis in fungal and bacterial cells has proven a particularly successful do- main for "N-NMR spectroscopy. In 1975 we observed that fast motion of intracellular small molecules permits the measurement of narrow-line spectra in living cells of Ustilago ~phaerogena . [ ' '~~ Subsequently, the research groups of Roberts[1601 and Lapidot[i6'1 investigated the bio- synthesis of amino acids in Neurospora crassa and Brevi- bacterium lactoferrnentum. The latter organism was used to monitor both nitrogen assimilation from "NH4CI and am- ino acid production throughout the growth cycle.'"'] Un- der optimal growth conditions, i.e., high aeration and low biotin concentration, the glutamic-acid-producing bacteria convert about 50% of the ammonium salt into ~ - [ ' ~ N ] g l u - tamic acid. Spectra were recorded of intact cells and cell media a t different stages of growth and production (Fig. 31). All "N-NMR signals can be clearly assigned to amino acids and metabofites, although a quantitative evaluation is hampered by different nuclear Overhauser effects and spin-lattice relaxation times. However, a semiquantitative comparison of the respective peaks in spectra recorded at various times in the growth cycle is possible. Two phases were distinguished during the fermentation process. At a very early stage (Fig. 3 1 b) glutamic acid is accumulated in the cells, but practically no product is released into the growth medium. Later, the cell walls become permeable
Angew. Chem. Inr. Ed. Engl. 25 (1986) 383-413 405

and amino acids are excreted (Fig. 31a). The alanine/glu- tamic acid intracellular ratio depends on oxygen and bio- tin concentration; low oxygen supply leads to a release of alanine instead of glutamic acid. This sensitive and conve- nient "N-NMR technique has thus proven a powerful probe for monitoring both intra- and infercellular amino acid concentrations in fermentation processes, and it may become an important analytical tool in biotechnology. Furthermore, the metabolic pathways of the intracellular amino acid pool can be followed and elucidated. A num- ber of other studies on the metabolism of important bio- logical precursor molecules have appeared recently.['621 The mobility and environment of intracellular glutamine, alanine, and arginine were studied using "N spin-lattice relaxation times T I , and nuclear Overhauser effects."631
we,,..-*--
I I
1 0 0 -
t N I 1 5 0 -
5
N . - a
N-AcGLN OLU
l t i U l " " I ' " 1 " ' ' l ' ~
- 200 - 250 -300 -350 - 5
GLU 1 L Y S
l ' ' " l " ' ' l ' ~ ' ' ~ ' ' -200 -250 -300 -350
- 6
Fig. 3 I . Proton-decoupled ISN-NMR spectra of 96.7% I5N-enriched B. laefo- fermenturn cells grown under conditions of high oxygen supply in normal biotin concentration (30.4 MHz). a) Spectrum of cell-free culture medium obtained after 31 h of fermentation; a') shows a spectrum from the same sample with gated 'H-decoupling; b) spectrum of growth-phase cells (ob- tained after 10 h of fermentation); c) spectrum of early stationary phase cells (obtained after 16 h of fermentation) (1611.
An important parameter characterizing the intracellular medium, the pH, can be determined from I5N-NMR meas- urements. For example, the two ring atoms of histidine show substantial chemical shift changes between p H 4 and 8; this was used successfully to estimate the p H in the cy- toplasmic and vacuolar environment of intact mycelia of Neurospora c ~ a s s a . ~ ' ~ ~ ~ In addition, pH-dependent line widths for the "N-NMR signals of the terminal NH2 groups of arginine and the a-NH, group of alanine were used. Measurements on arginine and histidine yielded a p H value of 6.1 f 0 . 4 for the vacuoles; the cytoplasmic p H determined by measurements on proline and alanine, on the other hand, was 7.2, in good agreement with other measurements.
L I 1 I I I
5.5 .6.0 6.5 7.0 7.5
PH - Fig. 32. Plots of the line width at half-height, Av,,?, of the ' IN-NMR signal versus pH for alanine in nitrogen-free medium: ( 0 ) 0.01 M, (0) 0.04 M, and (0) 0.005 M proline [164]. a=alanine, p=proline.
The p H dependence of the I5N-NMR line widths for proline and alanine is due to base-catalyzed proton ex- change between the NH2 group and H 2 0 (see Fig. 32). "N-NMR experiments appear to offer definite advantages over other techniques, such as "P-NMR spectroscopy, since amino acids are regiospecifically distributed in sub- cellular units and their structure-dependent NMR parame- ters are more sensitive to pH changes in the weakly acidic region.
As a last example, we will discuss some "N-NMR stud- ies on the binding of drugs to biomolecules, in particular, to proteins and nucleic acids. The antitumor activity of certain platinum(II)-amine complexes has stimulated the investigation of model systems for the presumed DNA
Binding of I5N-enriched c i ~ - [ P t ( N H ~ ) ~ C l ~ l to a nucleoside results in changes in the chemical shifts and ('5N,'95Pt) coupling constants in the "N-NMR spectrum. Interpretation of these spectra is facilitated by the large low-frequency shifts of the resonances for the azine ni- trogen atoms (-N=, N-3) upon coordination (Table 5). Pt complexes containing one or two nucleoside ligands are formed. As expected, cytidine is complexed at N-3, whereas for guanosine, which has several possible sites of coordination, a mixture of mono-complexes is formed. Ex- change of both chloride ligands affords a complex for which the structures in Scheme 28 were proposed in order
406 Angew. Chem. Int. Ed. Engl. 25 (1986) 383-413

Table 5. 15N chemical shifts 6 of free nucleosides and of platinum-nucleoside complexes [I651 [a].
N- I N-3 N-7 N-9 NH2
cytidine 5'-phosphate -227.9 - 179.6 -287.6 ~ is - [Pt(NH,)~(Cyd)~]~ -228.0 -255.4 -276.8 guanosine 5'-phosphate -233.5 -215.7 -145.6 -211.6 -307.9 CIS-[ Pt(NH3)z(G~o)Z]2 -232.5 -215.8 -237.9 -207.1 -304.7
[a] Recalculated relative to CH3NO2
to explain the pH dependence of the resonance of the NH3 ligand trans to the oxygen ligand. The low pK, value of 4.9 for the acidic group suggests the incorporation of a water molecule in the complex rather than deprotonation of the amide function. The most complex coordination behavior was observed for adenosine and involves all four basic N atoms (N-1, N-3, N-7, and 6'-NH,). A few relevant studies on 15N-195Pt interactions have also been reported['661 (see also references cited in ref. 116.51).
J
Scheme 28.
J
The binding of small molecules to large biomolecules does not necessarily lead to broad resonance lines, as might be expected from the long correlation times of these macromolecules. An example of the application of I5N- NMR spectroscopy to enzyme systems was provided by Becker and Roberts in their detailed investigation of a ter- nary inhibitor complex, liver alcohol dehydrogenase- NAD+-pyra~ole."~'l The I5N-NMR spectrum obtained at 18.25 MHz (4.2 tesla) (Fig. 33) demonstrates the nonequi- valence of N-1 and N-2 in the bound [1,2-"N2]pyrazole. The chemical shift of N-1 (S= - 146.5) is relatively close to the corresponding resonance of the adduct between pyra- zole and N-benzylnicotinamide (S= - 140.7), whereas that of N-2 in the enzyme complex (6= - 122.2) is shifted by 41.1 ppm to lower frequencies relative to the model com- pound. These results were interpreted in terms of covalent bonding between N-1 of pyrazole and C-4 of the nicotin- amide group and coordination of N-2 by the Zn atom at the active site of the enzyme, in agreement with a struc-
tural model proposed by 7'heoreN and Yonetani and with X-ray crystallographic studies (see ref. [ 1671).
a)
b)
Fig. 33. l5N-NMR spectra of the dehydrogenase-NADO-pyrazole complex (18.25 MHz, prepared from 99% "NZ-labeled pyrazole; 0.1 M sodium phos- phate buffer; pH 7.0): a) before dialysis; b) after dialysis 11671. The signals at 6= - 122.2, - 136.4, and - 146.5 are assigned to N-2 of bound pyrazole, free pyrazole, and N-l of bound pyrazole, respectively.
The formation of an enzyme-inhibitor complex is also responsible for the pharmacological activity of the dihy- drofolate reductase (DHFR) inhibitors. Typical inhibitors are trimethoprim (2,4-diamino-5-(3,4,5-trimethoxyben- zyl)pyrimidine, TMP) and methotrexate (MTX), the latter of which has a modified fo lk acid structure. The binding mode of T M P and MTX to DHFR has been the subject of extensive investigations by several interdisciplinary re- search groups.['681 The ternary complex between Lactoba- cillus casei DHFR, trimethoprim, and NADP+ exists in so- lution as a mixture of approximately equal amounts of two slowly interconverting conformational states, which have been characterized by multinuclear NMR experiments. The "N-NMR spectrum11681 of a triply I5N-labeled T M P in its ternary complex with DHFR and NADP' is shown in Figure 34. Comparison of this spectrum with the spectra of the binary DHFR-TMP complex and with the free proton- ated trimethoprim confirms the presence of two species in aqueous solution. The chemical shifts of the N-1 and 2-NH21SN resonances in the inhibitor complex are typical for protonated trimethoprim, which had been studied ear- lier by our group.[391 In another study, binding of human carbonic anhydrase B with benzenesulfonamide and cya-
Anyew Chem In<. Ed. Engi. 25 (1986) 383-413 407
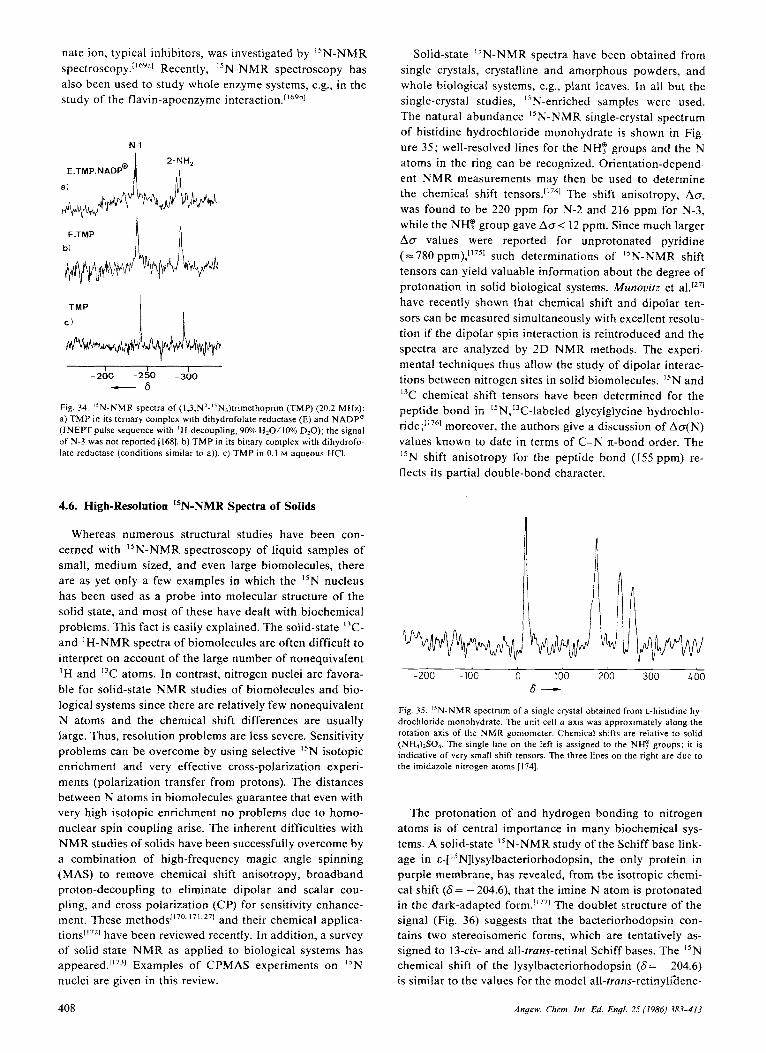
nate ion, typical inhibitors, was investigated by "N-NMR s p e c t r ~ s c o p y . ~ ' " ~ ~ ' ~ Recently, "N-NMR spectroscopy has also been used to study whole enzyme systems, e.g., in the study of the flavin-apoenzyme interaction."69h1
N-l
E.TMP.NADP@
a )
I I -200 -250 -3;o
- 6
Fig. 34. ' IN-NMR spectra of (1,3,N'-'5N,)trimethoprim (TMP) (20.2 MHz): a) T M P in its ternary complex with dihydrofolate reductase (E) and NADPQ (INEPT pulse sequence with 'H-decoupling, 90% H20/10% D20) ; the signal of N-3 was not reported [1681. b) T M P in its binary complex with dihydrofo- late reductase (conditions similar to a)). c) T M P in 0.1 M aqueous HCI.
4.6. High-Resolution "N-NMR Spectra of Solids
Whereas numerous structural studies have been con- cerned with "N-NMR spectroscopy of liquid samples of small, medium sized, and even large biomolecules, there are as yet only a few examples in which the I5N nucleus has been used as a probe into molecular structure of the solid state, and most of these have dealt with biochemical problems. This fact is easily explained. The solid-state I3C- and 'H-NMR spectra of biomolecules are often difficult to interpret on account of the large number of nonequivalent ' H and I3C atoms. In contrast, nitrogen nuclei are favora- ble for solid-state NMR studies of biomolecules and bio- logical systems since there are relatively few nonequivalent N atoms and the chemical shift differences are usually large. Thus, resolution problems are less severe. Sensitivity problems can be overcome by using selective I5N isotopic enrichment and very effective cross-polarization experi- ments (polarization transfer from protons). The distances between N atoms in biomolecules guarantee that even with very high isotopic enrichment no problems due to homo- nuclear spin coupling arise. The inherent difficulties with NMR studies of solids have been successfully overcome by a combination of high-frequency magic angle spinning (MAS) to remove chemical shift anisotropy, broadband proton-decoupling to eliminate dipolar and scalar cou- pling, and cross polarization (CP) for sensitivity enhance- ment. These method^['^^.'^'^^^^ and their chemical applica- ti on^["^] have been reviewed recently. In addition, a survey of solid-state NMR as applied to biological systems has appeared."731 Examples of CPMAS experiments on "N nuclei are given in this review.
Solid-state I5N-NMR spectra have been obtained from single crystals, crystalline and amorphous powders, and whole biological systems, e.g., plant leaves. In all but the single-crystal studies, "N-enriched samples were used. The natural abundance "N-NMR single-crystal spectrum of histidine hydrochloride monohydrate is shown in Fig- ure 35; well-resolved lines for the NH? groups and the N atoms in the ring can be recognized. Orientation-depend- ent NMR measurements may then be used to determine the chemical shift The shift anisotropy, Ao, was found to be 220 ppm for N-2 and 216 ppm for N-3, while the NH? group gave ACT< 12 ppm. Since much larger AD values were reported for unprotonated pyridine (= 780 ppm),1'751 such determinations of "N-NMR shift tensors can yield valuable information about the degree of protonation in solid biological systems. Munouitz et al.1271 have recently shown that chemical shift and dipolar ten- sors can be measured simultaneously with excellent resolu- tion if the dipolar spin interaction is reintroduced and the spectra are analyzed by 2D NMR methods. The experi- mental techniques thus allow the study of dipolar interac- tions between nitrogen sites in solid biomolecules. "N and I3C chemical shift tensors have been determined for the peptide bond in 15N,'3C-labeled glycylglycine hydrochlo- ride;"761 moreover, the authors give a discussion of Ao(N) values known to date in terms of C-N n-bond order. The "N shift anisotropy for the peptide bond (155 ppm) re- flects its partial double-bond character.
-200 -100 0 100 200 300 LOO 6 -
Fig. 35. ' IN-NMR spectrum of a single crystal obtained from L-histidine hy- drochloride monohydrate. The unit cell (I axis was approximately along the rotation axis of the NMR goniometer. Chemical shifts are relative to solid (NH4)2S04. The single line on the left is assigned to the NHF groups; it is indicative of very small shift tensors. The three lines o n the right are due to the imidazole nitrogen atoms 11741.
The protonation of and hydrogen bonding to nitrogen atoms is of central importance in many biochemical sys- tems. A solid-state "N-NMR study of the Schiff base link- age in E-["N]Iysylbacteriorhodopsin, the only protein in purple membrane, has revealed, from the isotropic chemi- cal shift (6= - 204.6), that the imine N atom is protonated in the dark-adapted form.['771 The doublet structure of the signal (Fig. 36) suggests that the bacteriorhodopsin con- tains two stereoisomeric forms, which are tentatively as- signed to 13-cis- and all-trans-retinal Schiff bases. The "N chemical shift of the lysylbacteriorhodopsin (6= - 204.6) is similar to the values for the model all-trans-retinyliaene-
408 Angew. Chem. Int. Ed. Engl. 25 (1986) 383-413

I ---- +250 - 50 -350
- A
Fig. 36. Proton-decoupled MAS "N-NMR spectra of a) retinylidenebu- tyl['SN]amine ("N-RB); b) "N-RBH"CI' ; c) s-['5N]lysylbacleriorhodopsin (29.8 MHz). For clarity the center bands in a) and b) are marked with aster- isks. The inset in c) is an expansion of the Schiff base signal showing the doublet structure ( A s = 5.7) tentatively assigned to the 13-cis and the all-trans stereoisomers. The two remaining lines in c) arise from the peptide backbone N atoms (S= -259.3) and the six lysine N e groups (6= -344.5). Chemical shifts originally referred to external, aqueous 5.6 M NH,CI 11771 were recal- culated relative to C H , N 0 2 ; conversion value -352.9 ppm [;a].
n z -180
c a
I
butyl['5N]ammoniumchloride (S= - 181.2), whereas the "N atom of the model base is strongly deshielded (6= -37.6). The chemical shift of the protonated solid is thus very similar t o the one reported for the hydrochloride in solution (S= - 147)Li'31 (see also section 4.1). Here, too, protonation of nitrogen results in a drastic narrowing of the chemical shift tensor, as shown in the 'H-decoupled "N-NMR powder spectrum (Fig. 37), from Acr=600 ppm for the Schiff base to 270 ppm for the hydrochloride.
- \ \ \ \
b)
-160
A
-
_1__1_/ L -50 - 350
+*5O L h
Fig. 37. Proton-decoupled l5N-NMR powder spectra of all-truns-retinylidene b~ty l [ '~N]amine; a) unprotonated; b) hydrochloride. In a ) the powder pat- tern is e 6 0 0 ppm ( I S kHz) wide with an isotropic shift of 6== -38. Protona- tion narrows the spectrum to a 270 ppm breadth and moves the chemical shift 142 ppm downfield. The chemical shifts were recalculated relative to CH,N02 as standard (see legend to Fig. 36) [177].
Solid-state NMR studies are particularly attractive since they permit a comparison of enzyme structures in aniso- tropic and isotropic phase (i.e., in solid and in solution, respectively) using the same physical method, in contrast to the usual comparisons between X-ray diffraction and
NMR data. For example, the structure of a-lytic protease was studied by the CPMAS technique; initially, the N' atom of HisS7 was "N-labeled, and, in a subsequent ex- periment, both the N" and N' atoms were isN-labeled.[i781 The behavior of the I5N chemical shifts as a function of the pH (Fig. 38) of the solution used to prepare the lyo-
-140 1
PH - Fig. 38. The pH-dependence of the "N chemical shifts of the imidazole ring N atoms of histidine-57 in a-lytic protease in solutions (--) [I791 and in lyo- philized powders (-) [178].
philized samples was found to closely parallel that ob- served in earlier solution s t u d i e ~ , " ~ ~ " ~ except for the slower proton exchange at His57 in the solid state. The local envi- ronment at the His57 close to the active site of the enzyme is shown in Scheme 29. The unusual tautomeric form of
Scheme 29.
histidine with a n N"-H bond was originally p r o p o ~ e d [ " ~ " ~ and later confirmed193c1 on the basis of "N-NMR measure- ments on the enzyme in solution. The specific triad struc- ture involving His" with the closely spaced aspartate lo*
ion on the n-side and the O H group of SerI9' on the T side is responsible for the stabilization of this tautomer. The good agreement between the new solid-state data and the solution results represents convincing structural evidence. A similar study employing I5N-CPMAS and X-ray diffrac- tion data was recently carried out on a carboxypeptidase A complex with g l y c y l t y r o ~ i n e . [ ' ~ ~ ~ ~
The reduced proton exchange rates in the solid state al- low the identification of protonated N atoms in biomole- cules and "N-DNMR studies of proton transfer. A recent investigation by Limbach et a1.['80"1 on tautomerism in I5N- enriched meso-tetra-p-tolylporphins 37 has shown that the process can be conveniently studied as a two-site exchange problem in the temperature range from 210 to 295 K. The CPMAS I5N-NMR spectra are shown in Figure 39; a com- plete line-shape analysis gave first-order rate constants
409 Angrw. Chem. In(. Ed. Engl. 25 (1986) 383-413

300 A 257 nE, 210
2086 f - v t H z 0
37
Fig. 39. CPMAS "N-DNMR spectra of meso-tetra-p-tolylporphin 37 (Ar=p- tolyl; 6.1 MHz; 95% "N-enriched 37); experimental and calculated spectra are superimposed. The small sharp low-frequency line is attributed to an im- purity [ISO].
( k H H ) . Whereas the dynamic behavior of the p-tolyl- and phenylporphins was found to be the same in solution, they show very different solid-state spectra as a function of temperature. The complex cases of the arylporphins were also discussed in conjunction with the results from X-ray diffraction studies.[180b1
I5N-NMR spectroscopy of solids has also been applied to nucleic acids, e.g., to the study of N-H bond lengths in B-DNA[341 (see also Section 2.3, Fig. 11) and to the study of the DNA backbone structures of fd and Pfl bacterio- phages.['*'] The latter investigations were supported by 2 D NMR analysis of the dipolar splittings in the spectra of oriented virus molecules.1351 From the same research group,
Fig. 40. Shoot of a soybean plant (Glycine soja) [I841
410
several solid-state I5N-NMR studies on proteins were also
We will close this review by quoting a paper that demon- strates the almost unlimited potential of I5N-NMR spec- troscopy in biological studies.['R2h1 Schaefer et al.['83"1 re- corded the double cross-polarization MAS 15N-NMR spectra of soybean leaves in order to determine the protein turnover number. This plant was also used to investigate the metabolism of allantoin in the early growth state.['83h1 Figure 40 shows a soybean shoot as a symbol of the ever- growing applications of NMR spectroscopy to chemistry, biochemistry, and biology. New branches are expected to grow and will call for imaginative investigators.
18 1. l*2;11
5. Outlook
During the past few years I5N-NMR spectroscopy has developed as a versatile experimental probe into the struc- ture and reactivity of organic, bioorganic, and inorganic molecules. The majority of studies have been performed without isotopic enrichment of the substrates. Since the sensitivity of the "N-NMR technique is still improving, its application is expected to increase still further in the fu- ture. Modern I D and 2D NMR experiments will undoubt- edly make possible the structural investigations of larger molecules and more complex biomolecules in the liquid and solid states. For example, the heteronuclear "D-re- solved" 2D NMR experiments recently described offer a new dimension for the quantitative study of hydrogen bonding in solids. Although many "N-NMR investiga- tions of active sites in biological systems still require iso- topic enrichment, the first successful attempts to study drug binding, i.e., the mechanism of drug action, have shown a considerable pharmacological potential. The fun- damental problem of biological nitrogen fixation and me- tabolism may be investigated in the future by an extended use of "N-NMR techniques, and other applications may be envisaged. However, the realization of many chemical and biochemical ideas will finally depend on the availabil- ity of versatile multinuclear NMR instrumentation and flexible computing facilities. It is this area where the most significant advances can be expected for the near future.
The senior author (W. v. P.) wishes to express his gratitude and indebtedness to his co-workers who have contributed to the results from the Zurich laboratory thar are described in this review: Dr. W. Schwotzer, Dr. W. Stadeli, Dr. R . Hol- lenstein, Dr. P. Bigler, Dr. L. Kozerski, Dr. G. Cerioni, Dr. M . Tato, B. C . Chen, C . Adams, and R . Muller. Theirscien- trfic enthusiasm, experimental skill, and endurance have had, and continue to have, a major impact on the research and spirit of our group. Generous financial support from the Government of the Kanton Zurich and the Swiss National Science Foundation is gratefully acknowledged.
Received: July 23, 1985; supplemented: October 23, 1985 [A 576 IE]
German version. Angew Chem. 98 (1986) 38 I
Angew. Chem. Inr. Ed. Engl. 25 (1986) 383-413

[I] R. Miiller, J. D. Wallis, W. von Philipsborn, Angew. Chem. 97 (1985)
121 W. G. Proctor, F. C. Yu, Phys. Reu. 77 (1950) 717; 81 (1951) 20. 131 M. Witsnowski, G. A. Webb (Eds.): Nitrogen NMR. Plenum Press,
[4] a) J. Mason, Chem. Reu. 81 (1981) 205; b) Chem. Br. 19 (1983) 654. [5] N. Logan in [3], chapter 6, p. 319, “Applications of “N NMR Data in
the Study of Inorganic Molecules”. 161 J. M. Lehn, J. P. Kintzinger in 131, chapter. 3, p. 79, “Nitrogen-14 Nu-
clear Quadrupole Effects”. 171 a) G. J. Martin, M. L. Martin, J.-P. Gouesnard, “”N-NMR Spectrosco-
py” in P. Diehl, E. Fluck, R. Kosfeld (Eds.): NMR Basic Principles and Progress, Yo/. 18. Springer, Berlin 1981 ; b) M. Witanowski, L. Stefan- iak, G. A. Webb, Annu. Rep. NMR Spectrosc. I I B (1981); c) G. C. Levy, R. L. Lichter: Nitrogen-I5 Nuclear Magnetic Resonance Spectroscopy. Wiley. New York 1979.
[8] L. F. Farnell, E. W. Randall, A. 1 . White, 1. Chem. Soc. Chem. Com- mun. 1972. 1159; 0. A. Gansow, A. R. Burke, G. N. La Mar, ibid. 1972, 456; A. J. DiGioia, R. L. Lichter, J. Magn. Reson. 27 (1977) 431.
191 W. Schwotzer, J. H. Bieri, M. Viscontini, W. von Philipsborn, Helu. Chim. Acra 61 (1978) 2108.
[lo] P. Ashkenazi, M. Kaftory, D. Arad, Y. Apeloig, D. Ginsburg, Helu. Chim. Acta 64 (1981) 579.
[ I I] G. A. Morris, R. Freeman, J. Am. Chem. Soc. I01 (1979) 760. [I21 R. Benn, H. Gunther, Angew. Chem. 95 (1983) 381; Angew. Chem. Int.
1131 L. Kozerski, K. Kamienska-Trela, L. Kania, W. von Philipsborn, Helu.
[ 141 G. A. Morris, J. Am. Chem. SOC. I02 (1980) 428. 1151 B. C. Chen, W. von Philipsborn, K. Nagarajan, Helu. Chim. Acra 66
(1983) 1537. 1161 a) S. J B. Price, M. J. DiMartino, D. T. Hill, R. Kuroda, M. A. Mazid,
P. J. Sddler, Inorg. Chem. 24 (1985) 3425; b) C. Brevard, R. Schimpf, J. Magn Reson. 47 (1982) 528.
515; Angew. Chem. lnt. Ed. Engl. 24 (1985) 513.
London 1973.
Ed. Engl. 22 (1983) 350.
Chim. Acra 66 (1983) 2113.
[I71 A. Costa, M. Tato, R. S. Matthews, Magn. Reson. Chent.. in press. 1181 K. V. Schenker, W. von Philipsborn, J . Magn. Reson. 61 (1985) 294. 1191 a) D. M. Doddrell, D. T. Pegg, M. R. Bendall, J . Mugn. Reson. 48
(1982) 323; b) D. T. Pegg, D. M. Doddrell, M. R. Bendall, J. Chem. Phy.5. 77 (1982) 2745; c) D. T. Pegg, M. R. Bendall, J . Magn. Reson. 53 (1983) 229.
[201 K. G. R. Pachler, P. L. Wessels, J . Magn. Reson. 28 (1977) 53; H. J. Jakobsen, H. Bildsoe, ibid. 26 (1977) 183.
1211 a) H. J. Jakobsen, W. S. Brey, J . Am. Chem. SOC. I01 (1979) 774; b) J . Chem Soc. Chem. Commun. 1979. 478; c) H. J. Jakob:.en, P.-I. Yang, W. S. Brey, Org. Magn. Reson. 17 (1981) 290; d) 0. W. Sorensen, S. Scheibye, S.-0. Lawesson, H. J. Jakobsen, ibid. 16 (1981) 322.
1221 A. Bax, Ch.-H. Niu, D. Live, J. Am. Chem. SOC. 106 (1’984) 1150. 1231 D. H. Live, D. G. Davis, W. C. Agosta, D. Cowburn, J . .4m. Chem. SOC.
[24] A. Hax, R. H. Griffey, B. L. Hawkins, J Am. Chem. Soc. 105 (1983)
[25] C . J. Turner, Prog. Nucl. Magn. Reson. Specrrosc. 16 (1984) 31 1 [26] A. Hax. R. H. Griffey, B. L. Hawkins, J. Magn. Reson. 55 (1983) 301. [27] M. G . Munovitz, H. H. Huang, C. M. Dobson, R. G. Griffin, J. Magn.
Rrcon 57 (1984) 56. 1281 a) H. Kessler, W. Hehlein, R. Schuck, J. Am. Chem. SOC. 104 (1982)
4534: b) H. Kessler, H. R. Loosli, H. Oschkinat, Helu. Chim. Acta 68 (1985) 661; c) H. Kessler, H. Oschkinat, H. R. LOOS~I, ,,LD NMR Spec- troscopy”, in R. M. Carlson, W. R. Croasmun (Eds.): Methods in Ster- eochemical Analysu. VCH Verlagsgesellschaft, Weinheim, 1986.
I06 ( 1984) 6 104.
7188.
[29] G. A. Gray, Org. Magn. Reson. 21 (1983) I I I. [30] H. Kessler, C . Griesinger, J. Zarbock, H. R. Loosli, J. Magn. Reson. 57
1311 G. Bodenhausen, D. J. Ruben, Chem. Phys. Lett. 69 (1980) 185. 1521 A. G. Redfield, Chem. Phys. Lett. 96 (1983) 537. [33] a) R. H. Griffey, C. D. Poulter, A. Bax, B. L. Hawkins, Z. Yamaizumi,
S. Nishimura. Proc. Null. Acad. Sci. USA 80 (1983) 5895; R. H. Griffey, D. Davis, Y. Yamaizumi, S. Nishimura, A. Bax, B. Hawkins, C. D. Poulter, J. Biol. Chem. 260 (1985) 9734; b) L. Miiller, R. A. Schiksnis, S. J. Opella, J . Magn. Reson. 66 (1986) 379.
(1984) 33 I .
1341 J. A. DiVerdi, S. J. Opella, J. Am. Chem. SOC. 104 (1982) 1761. 1351 T. A. Cross, P. Tsang, S. J. Opella, Biochemistry 22 (1983) 721. [36] D. G Davis, W. C. Agosta, D. Cowburn, J. Am. Chem. Soc. /OS (1983)
6189.
1371 P. R. Srinivasan, R. L. Lichter, J. Magn. Reson. 28 (1977) 227. 1381 IUPAC Recommendations for nuclei other than ‘H, cf. Pure Appl.
[39] W. Stadeli, A. Wick, I. KompiS, W. von Philipsborn, Helu. Chim. Acta
1401 W. Schwotzer, W. von Philipsborn, Helu. Chim. Acta 60 (1977) 1501. [41] K. A. K. Ebraheem, G . A. Webb, Prog. Nucl. Magn. Reson. Spectrosc.
I 1 (1977) 149; M. Bremond, G. J. Martin, G. A. Webb, D. J. Reynolds, Org. Magn. Reson. 22 (1984) 640; K. A. K. Ebraheem, G. A. Webb, M. Witanowski, ibid. I 1 (1978) 27; K. A. K. Ebraheem, G. A. Webb, ibid. 9 (1977) 248.
1421 W. Stadeli, Dissertation. Universitat Zurich 198 1. [43] See [7a], p. 60. 1441 W. M. Litchman, M. Alei, A. E. Florin, J . Chem. Phys. 50 (1969)
1451 R. E. London, T. E. Walker, T. W. Whaley, N. A. Matwiyoff, Org.
[46] L. Kozerski, W. von Philipsborn, Helu. Chim. Acta 65 (1982) 2077. [47] A. LyTka, P. E. Hansen, Org. Magn. Reson. 22 (1984) 569. [48] J. A. Pople, D. P. Santry, Mol. Phys. 8 (1964) I. [49] See [3], p. 269. [ S O ] G. A. Olah, A. W. White, J. Am. Chem. Sac. 90 (1968) 6087. [Sl] A. J . R. Bourn, E. W. Randall, Mol. Phys. 8 (1964) 567. 1521 G. Binsch, J. B. Lambert, B. W. Roberts, J. D. Roberts, J. Am. Chem.
1531 R. Wasylishen, T. Schaefer, Can. J . Chem. 49 (1971) 3627. [54] H. Hilpert, R. Hollenstein, Helu. Chim. Acta. 69 (1986) 136. [55] J. Dorie, B. Mechin, G. Martin, Org. Magn. Reson. 12 (1979) 229. [56] L. Paolillo, E. D. Becker, J. Magn. Reson. 2 (1970) 168. 1571 See [7b], p. 12. [58] See [7a]. p. 310f. 1591 W. McFarlane, Mol. Phys. I0 (1965) 603. I601 W. Runge, J. Firl, Z . Naturforsch. B 31 (1976) 1515. [61] T. Bundgaard, H. J. Jakobsen, E. J. Rahkamaa, J. Magn. Reson. 19
(1975) 345. 1621 J. M. Schulman, T. Venanzi, J . Am. Chem. SOC. 98 (1976) 4701,6739; T .
Khin, G. A. Webb, Org. Magn. Reson. 10 (1977) 175; J. M. Schulman, J. Magn. Reson. 28 (1977) 137; T . Axenrod, C. M. Watnick, M. J. Wied- er, S. Duangthai, G. A. Webb, H. J. C. Yeh, S. Bulusu, M. M. King, Org. Magn. Reson. 20 (1982) I I .
1631 M. J. 0. Anteunis, F. A. M. Borremans, J. Gelan, A. P. Marchand, R. W. Allen, J. Am. Chem. SOC. 100 (1978) 4050; the ‘J(14N,’H) values were recalculated by use of Equation (b).
1641 R. Wasylishen in T. Axenrod, G. A. Webb (Eds.): Nuclear Magnefic Resonance Spectroscopy of Nuclei Other Than Protons. Wiley, New York 1974, chapter 8.
1651 a) W. Stadeli, P. Bigler, W. von Philipsborn, Org. Magn. Reson. 16 (1981) 170; b) M. Schumacher, H. Gunther, Chem. Ber. 116 (1983) 200 I .
1661 R. Miiller, Diplomarbeit. Universitat Zurich 1983; R. Miiller, W. von Philipsborn, unpublished.
[67] V. F. Bystrov, Prog. Nucl. Magn. Reson. Specfrosc. I0 (1976) 41. 1681 M. Barfield, H. L. Gearhart, Mof. Phys. 2 7 ( 1974) 899; V. N. Solkan, V.
F. Bystrov, lru. Akad. Nauk SSSR, Ser. Khim. 1974. 102. [69] V. F. Bystrov, Yu. D. Gavrilov, V. N. Solkan, J. Magn. Reson. 19 (1975)
123; J. A. Sogn, W. A. Gibbons, E. W. Randall, Biochemistry 12 (1973) 2100; A. J. Fischman, H. R. Wyssbrod, W. C. Agosta, D. Cowburn, J. Am. Chem. SOC. 100 (1978) 54; A. J. Fischman, D. H. Live, H. R. Wyss- brad, W. C. Agosta, D. Cowburn, bid. I02 (1980) 2533; A. de Marco. M. Llinas, K. Wiithrich, Biopolymers 17 (1978) 2727.
[70] W. von Philipsborn, Angew. Chem. 83 (1971) 470; Angew. Chem. lnr.
Ed Engi. 10 (1971) 472. [71] T. Axenrod in [3], chapter 5 , p. 261. 1721 C. M. Adams, W. von Philipsborn, Magn. Reson. Chem. 23 (1985)
[73] S. Nagata, T. Yamabe, K. Htrao, K. Fukui, J. Phys. Chem. 79 (1975)
[74] R. L. Lichter, D. E. Dorman, R. Wasylishen, J. Am. Chem. Soc. 96
[75] W. Schwotzer, C. Leuenberger, L. Hoesch, A. S. Dreiding, W. von Phil-
Chem. 45 (1976) 219.
63 (1980) 504, and references cited therein.
1897.
Magn. Reson. 9 (1977) 598.
SOC. 86 (1964) 5564.
130.
I863
(1974) 930.
ipsborn, Org. Magn. Reson. 9 (1977) 382.
[76] H. Schultheiss, E. Fluck, Z . Naturforsch. B 32 (1977) 257. [77] a) T. Berkhoudt, H. J. Jakobsen, J . Magn. Reson. 50 (1984) 323; b) Y.
Kuroda, Y. Fujiwara, K. Matsushita, ibid. 62 (1985) 218; c) M. Gruner,
Angew. Chem. fnt. Ed. Engl. 25 (1986) 383-413 41 1

D. Pfeifer, H. G. 0. Becker, R. Radeglia, J . Epperlein, J. Prakt. Chem. 327 (1985) 63.
1781 a) J. M. Manriquez, D. R. McAlister, E. Rosenberg, A. M. Shiller, K. L. Williamson, S. I. Chan, J. E. Bercaw, J. Am. Chem. SOC. I00 (1978) 3078; b) J. 0. Friedrich, R. E. Wasylishen, J. Chem. Phys. 83 (1985) 3707.
1791 a) T. Khin, G. A. Webb, J . Magn. Reson. 33 (1979) 159; b) J. M. Schul- man, J. Ruggio, T. J. Venanzi, J. Am. Chem. SOC. 99 (1977) 2045.
[SO] J. R. Lyerla, Jr., in G. C. Levy (Ed.): Topics in Carbon-13 NMR Spec- troscopy. Vol. 1. Wiley, New York 1974, p. 79.
[Sl] G. C. Levy, C. E. Holloway, R. C. Rosanske, J. M. Hewitt, C. H. Brad- ley, Org. Magn. Reson. 8 (1976) 643; G. C. Levy, T. Pehk, P. R. Srini- vasan, ibid. 14 (1980) 129; see also [7a], p. 7.
[82] 0. A. Gansow, K. M. Triplett, T. T. Peterson, R. E. Botto, J. D. Ro- berts, Org. Magn. Reson. 13 (1980) 77.
1831 H. R. Kricheldorf, W. E. Hull, Makromol. Chem. 181 (1980) 507. [84] T. A. Scahill, S. L. Smith, Org. Magn. Reson. 21 (1983) 621, 662. [SS] See [7b], p- 77. 1861 See [7b], p. 84. 1871 L. Stefaniak, J. D. Roberts, M. Witanowski, G. A. Webb, Org. Magn.
Reson. 22 (1984) 201, 215; L. Stefaniak, J. D. Roberts, M. Witanowski, B. T. Hamdi, G. A. Webb, ibid. 22 (1984) 209.
[88] a) K . Nagarajan, V. P. Arya, J. Sci. Ind. Res. 41 (1982) 232; b) M. D. Nair, K. Nagarajan in E. Jucker (Ed.): Progress in Drug Research, Vol. 27. Birkhauser Verlag, Basel 1983, p. 163.
1891 W. Schilf, L. Stefaniak, M. Witanowski, G. A. Webb, Magn. Reson. Chem. 23 (1985) 181.
1901 P. Schmitt, K. Finneiser, H. Giinther, personal communication. [9l] E. Bojarska-Olejnik, L. Stefaniak, M. Witanowski, B. T. Hamdi, G. A.
Webb, M a p . Reson. Chem. 23 (1985) 166. 1921 D. S. Wofford, D. M. Forkey, J. G. Russell, J. Org. Chem. 47 (1982)
5132. 1931 a) W. W. Bachovchin, J. D. Roberts, J. Am. Chem. SOC. 100 (1978)
8041; b) 1.1. Schuster, J. D. Roberts, J. Org. Chem. 44 (1979) 3864; c) J. D. Roberts, Chun Yu, C. Flanagan, R. T. Birdseye, J. Am. Chem. SOC. 104 (1982) 3945.
[94] N. C. Gonnella, H. Nakanishi, J. B. Holtwick, D. S. Horowitz, K. Ka- namori. N. J. Leonhard, J . D. Roberts, J. Am. Chem. SOC. I05 (1983) 2050; N. C. Gonnella, J. D. Roberts, ibid. 104 (1982) 3162.
[95] W. E. Hull, M. Kiinstlinger, E. Breitmaier, Angew. Chem. 92 (1980) 957; Angew. Chem. Inf. Ed. Engl. 19 (1980) 924.
1961 See references in [95]. [97] W. Stadeli, W. von Philipsborn, Org. Magn. Reson. IS (1981) 106. 1981 S. Tobias, P. Schmitt, H. Giinther, Chem. Ber. 115 (1982) 2015. 1991 R. L. Blakley, Biochem. J. 58 (1954) 448.
[loo] R. L. Kisliuk, W. Sakami, J. Am. Chem. SOC. 76 (1954) 1456; M. J. Osborn, P. T. Talbert, F. M. Huennekens, ibid. 82 (1960) 4921; R. G. Kallen, W. P. Jencks, J. Biol. Chem. 241 (1966) 5851.
[ lo l l a) R. Kalbermatten, W. Stadeli, J. H. Bieri, M. Viscontini, Helu. Chim. Acfa 64 (1981) 2627: b) R. Kalbermatten, Dissertation. Universitat Zu- rich 1981.
11021 L. Paolillo, E. D. Becker, J. Magn. Reson. 3 (1970) 200. [I031 S. K. Malhotra in A. G. Cook (Ed.): Enamines. M. Dekker, New York
[I041 L. Kozerski, W. von Philipsborn, Org. Magn. Reson. 17(1981) 306. [I051 a) R. A. Eades, D. A. Weil, M. R. Ellenberger, W. E. Farneth, D. A.
Dixon, C. H. Douglas, Jr., J. Am. Chem. SOC. 103 (1981) 5372; b) W. von E. Doering, personal communication.
1969.
{I061 J. Dorie, B. Mechin, G. Martin, Org. Magn. Reson. 12 (1979) 229.
[I071 G. J. Martin, J. P. Gouesnard, J. Dorie, C. Rabiller, M. L. Martin, J.
[IOS] M. J. Kamlet, C. Dickinson, R. W. Taft, J. Chem. SOC. Perkin Trans. 2
[I091 R. Kupper, B. D. Hilton, M. B. Kroger-Koepke, S . R. Koepke, C. J.
Am. Chem. Sac. 99 (1977) 1381.
1981. 353.
Michejda, J. Org. Chem. 49 (1984) 3781. [I101 T. B. Patrick, R. P. Willaredt, J. Org. Chem. 48 (1983) 4415. [ I I l l D. 1. Craik, G. C. Levy, R. T. C. Brownlee, J. Org. Chem. 48 (1983)
(1121 M. Alien, J. D. Roberts, J . Org. Chem. 45 (1980) 130, and references
[ I 131 D. D. Muccio, W. G. Copan, W. W. Abrahamson, G. D. Mateescu, Org.
[ I 141 C. Yu, G. C. Levy, Org. Magn. Reson. 22 (1984) 131.
1601.
cited therein.
Magn. Reson. 22 (1984) 121.
11151 For references see: D. H. Live, D. G. Davis, W. G. Agosta, D. Cow-
[I161 G. E. Hawkes, E. W. Randall, W. E. Hull, 0. Convert, Biopolymers 19
11 I71 H. R. Kricheldorf, Org. Magn. Reson. I S (1981) 162. [I181 C. Dyllick-Brenzinger, G. R. Sullivan, P. P. Pang, J . D. Roberts, Proc.
[ I 191 A. Hantzsch, Chem. Ber. 36 (1903) 2056. 11201 R. Huisgen, 1. Ugi, Chem. Ber. 90 (19.57) 2914; 1. Ugi, R. Huisgen, ibid.
91 (1958) 53 I ; 1. Ugi, R. Huisgen, K. Clusius, M. Vecchi, Angew. Chem. 68 (1956) 753.
burn, J Am. Chem. Soc. 106 (1984) 1939.
(1980) 1815.
Nafl. Acad. Sci. USA 77 (1980) 5580.
11211 1. Ugi, H. Perlinger, L. Behringer, Chem. Ber. 91 (1958) 2324. 11221 J. D. Wallis, J. D. Dunitz, J. Chem. SOC. Chem. Commun. 1983, 910. I1231 L. Hoesch, C. Leuenberger, H. Hilpert, A. S. Dreiding, Helu. Chim.
Acfa 65 (1982) 2682; C. Leuenberger, L. Hoesch, A. S. Dreiding, J. Chem Soc. Chem. Commun. 1980. 1197.
[I241 M. T. Nguyen, J. Kaneti, L. Hoesch, A. S. Dreiding, Helu. Chim. Acfa 67(1984) 1918.
[I251 a) T. Yonezawa, 1. Morishima, J. Mol. Specfrosc. 27 (1968) 210; b) H. Egli, W. von Philipsborn, Helu. Chim. Acta 64 (1981) 976.
[I261 C. Richard, P. Granger in P. Diehl, E. Fluck, R. Kosfeld (Eds.): NMR Basic Principles and Progress, Vol. 8, Springer, Berlin 1974.
11271 a) J. H. Ridd, J. P. B. Sandall, J. Chem. SOC. Chem. Commun. 1981. 402; b) A. H. Clemens, P. Helsby, J. H. Ridd, F. Al-Omran, J. P. B. Sandall, J. Chem. SOC. Perkin Trans. 2 1985. 1217; c) A. H. Clemens, J. H. Ridd, J. P. B. Sandall, ibid. 2 1985. 1227.
I1281 J. C. Giffney, J. H. Ridd, J. Chem. SOC. Perkin Trans. 2 1979. 618. I1291 J. H. Ridd, J. P. B. Sandall, J. Chem. Soc. Chem. Commun. 1982. 261. [I301 N. A. Porter, G. R. Dubay, J. G. Green, J. Am. Chem. SOC. 100 (1978)
[I311 E:L. Dreher, P. Niederer, A. Rieker, W. Schwarz, H. Zollinger, Helu.
11321 A. W. Douglas, J. Am. Chem. Soe. 101 (1979) 5676. 11331 M. Yogo, K. Hirota, S. Senda, J. Chem. Soc. Perkin Trons. 1 1982.
11341 J. W. Lown, S. M. S. Chauhan, J. Org. Chem. 48 (1983) 513. 11351 Y. Nomura, Y. Takeuchi, J Chem. SOC. Chem. Commun. 1979. 295. 11361 G. T. Furst, R. L. Lichter, F. W. Vierhapper, J . Org. Chem. 45 (1980)
I1371 R. L. Willer, D. W. Moore, L. F. Johnson, J. Am. Chem. SOC. I04 (1982)
11381 K. Kanamori. J. D. Roberts, J. Am. Chem. Soc. I05 (1983) 4698. 11391 R. Dyllick-Brenzinger, J. D. Roberts, J. Am. Chem. Soc. I02 (1980)
1166. [I401 a) J. R. Dilworth, S. Donovan-Mtunzi, C. T. Kan, R. L. Richards, J.
Mason, fnorg. Chim. Acfa 53 (1981) L161; b) J. Chatt, M. E. Fakley, R. L. Richards, J. Mason, I . A. Stenhouse, J. Chem. Res. (S) 1979. 322; J. Chem. Res. (MI 1979. 3701.
[I411 R. D. Sanner, J. M. Manriquez, R. E. Marsh, J. E. Bercaw, J. Am. Chem. SOC. 98 (1976) 8351, 3042.
11421 D. L. Thorn, T. H. Tulip, J. A. Ibers, J. Chem. Soc. Dalton Trans. 1979. 2022.
11431 J. R. Dilworth, C. T. Kan, R. L. Richards, J. Mason, I . A. Stenhouse, J . Organornet. Chem. 201 (1980) C24.
11441 a) L. K. Bell, D. M. P. Mingos, D. G. Tew, L. F. Larkworthy, B. San- dell, D. C. Povey, J. Mason, J. Chem. Soc. Chem. Commun. 1983. 125; D. H. Evans, D. M. P. Mingos, J. Mason, A. Richards, J. Organornet. Chem. 249 (1983) 293; b) J. Mason, D. M. P. Mingos, J. Schafer, D. Sherman, E. 0. Stejskal, J . Chem. SOC. Chem. Commun. 1985. 444.
114.51 J. Bultitude, L. F. Larkworthy, J . Mason, D. C. Povey, B. Sandell, Inorg. Chem. 23 (1984) 3629.
11461 A. R. Butler, C. Glidewell, A. R. Hyde, J. McGinnis, Polyhedron 3 (1984) 1165.
[I471 N. W. Alcock, N. Herron, P. Moore, J. Chem. SOC. Dalton Trans. 1979. 1486.
11481 E. H. Curzon, N. Herron, P. Moore, J. Chem. SOC. Dalton Trans. 1980.
[149] I . Morishima, T. Inubushi, J. Am. Chem. SOC. 100 (1978) 3568; FEBS
[I501 E. von Goldammer, Z. Nafurjorsch. C 34 (1979) 1106. IlSl] F. Blomberg, H. Riiterjans in L. J. Berliner, J. Reuben (Eds.): Biological
1152) K. Kanamori, J. D. Roberts, Arc. Chem. Res. 16 (1983) 35. [I531 Y. Kyogoku, Appl. Spectrosc. Reu. 17 (1981) 279.
920.
Chim. Acfa 64 (I98 1) 488.
473.
1521.
395 I .
721.
Letf. 81 (1977) 57.
Magnetrc Resonance. Vol. 5. Plenum Press, New York 1983, p. 21.
41 2 Angew. Chem. Int. Ed. Engl. 25 (1986) 383-413

[I541 B. Coxon, Pure Appl. Chem. 49 (1977) 1151. [I551 H. R. Kricheldorf, Pure Appl. Chem. 54 (1982) 467. 11561 J. R. Ebdon, P. E. Heaton, T. N. Huckerby, W. T. S. ORourke, J. Par-
kin, Po[ymer 25 (1984) 821 : R. A. McKay, J. Schaefer, E. 0. Stejskal, R. Ludicky, C. N. Matthews, Macromolecules 17 (1984) 1124; D. E. Axel- son, S. L. Blake, J. Polym. Sci. Polym. Chem. Ed. 23 (1985) 2507; H. R. Kricheldorf, W. E. Hull, D. Miiller, Macromolecules 18 (1985) 2135.
[I571 A. Hori, Y. Shimizu, J . Chem. Soc. Chem. Commun. 1983, 790. [IS81 R. L. Baxter, C. J. McGregor, G. A. Thomson, A. 1. Scott, J. Chem. SOC.
Perkrn Trans. I 1985. 369. (1591 M. Llinas, K. Wiithrich, W. Schwotzer, W. von Philipsborn, Nature
iLondonj 257 (1975) 817. [I601 T. L. Legerron, K. Kanamori, R. L. Weiss, J. D. Roberts, Proc. Nail.
Acad. Sci. USA 78 (1981) 1495; K. Kanamori, T. L. Legerton, R. L. Weiss, J. D. Roberts, J. Biol. Chem. 257 (1982) 14168.
[ 1611 N. Haran, Z. E. Kahana, A. Lapidot, J. Biol. Chem. 256' (1983) 12929. (1621 NHJNO'! (Nitrosomonas): K. K. Anderson, S. B. Philson, A. B.
Hooper, Proc. Nut/. Acud. Sci. USA 79 (1982) 5871; D-lysine (Neuros- pora crassa): N. Fangmeier, E. Leistner, J. BiO/. Chem. 255 (1980) 10205: nitrate (Neurospora crassa): G. S . Jacob, J. Schaeler, E. 0. Stejs- kal, R. A. McKay, Biochem. Biophys. Res. Commun. 97 (1980) 1176; NH? (Streptomyces oenezuelae): S. Shapiro, L. C. Vining, M. Laycock, A. G. Mclnnes, J. A. Walter, Can. J. Microbiol. 31 (19x5) 629; Nitro- propanoic acid (Penicillium atroveneturn): R. L. Baxter, S. L. Green- wood, J. Chem. SOC. Chem. Commun. 1986, 175.
11631 K. Kanamori, T. L. Legerton, R. L. Weiss, J. D. Robem, Biochemistry
[I641 T. L. Legerton, K. Kanamori, R. L. Weiss, J. D. Roberts, Biochemistry
11651 M. Nee, J. D. Roberts, Biochemzsfry 21 (1982) 4920.
11661 M. Alei, Jr., P. J. Vergamini, W. E. Wageman, J. Am. (:hem. SOC. 101 (1979) 5415: A. Albinati, H. Moriyama, H. Riiegger, P. S Pregosin, A. Togni, Inorg. Chem. 24 (1985) 4430.
21 (1982) 4916.
22 (1983) 899.
[I671 N. N. Becker, J. D. Roberts, Biochemistry 23 (1984) 3336.
[I681 B. Birdsall, A. W. Bevan, C. Pascual, G. C. K. Roberts, J. Feeney, A. Gronenborn, G. M. Clove, Biochemistry 23 (1983) 4733; A. W. Bevan,
G. C. K. Roberts, J. Feeney, L. Kuyper, Eur. Biophys. J. 1 1 (1985) 21 I.
[I691 a) K. Kanamori, J. D. Roberts, Biochemistry 22 (1983) 2658; b) J. Ver- voort, F. Miiller, J. LeGall, A. Bacher, H. Sedlmaier, Eur. J. Biochem. I51 (1985) 49; W.-D. Beinert, H. Riiterjans, F. Miiller, ibid. I52 (1985) 573.
11701 M. Mehring: Principles of High-Resolution NMR in Solids. 2nd ed., Springer, Berlin 1983.
11711 C. S. Yannoni, Acc. Chem. Res. 15 (1982) 201. [I721 J . R. Lyerla, C. S. Yannoni, C. A. Fyfe. Acc. Chem. Res. 15 (1982)
[I731 S. J. Opella, Annu. Rev. Phys. Chem. 33 (1982) 533. 11741 G. Harbison, J. Herzfeld, R. G. Griffin, J. Am. Chem. SOC. 103 (1981)
11751 D. Schweitzer, H. W. Spiess, J. Magn. Reson. 15 (1974) 529. 11761 G. S. Harbison, L. W. Jelinski, R. E. Stark, D. A. Torchia, J. Herzfeld,
R. G. Griffin, J. Mugn. Reson. 60 (1984) 79; R. E. Stark, L. W. Jelinski, D. J. Ruben, D. A. Torchia, R. G. Griffin, ibid. 55 (1983) 266.
11771 G. S. Harbison, J. Herzfeld, R. G. Griffin, Biochemistry22 (1983) 1. [I781 T.-H. Huang, W. W. Bachovchin, R. G. Griffin, Ch. M. Dobson, Bio-
chemistry 23 (1984) 5933. 11791 a) W. W. Bachovchin, J. D. Roberts, J. Am. Chem. SOC. 100 (1978)
8041; b) N. E. Mackenzie, P. E. Fagerness, A. I. Scott. J. Chem. SOC. Chem. Commun. 1985. 635.
[I801 a) H. H. Limbach, J. Hennig, R. Kendrick, C. S. Yannoni, J. Am. Chem. SOC. 106 (1984) 4059; b) R. J. Butcher, G. B. Jameson, C. B. Storm, ibid. (1985) 2978; M. J. Hamor, T. A. Hamor, J. L. Hoard (1964) 1938.
[IS11 T. A. Cross, J. A. DiVerdi, S. J. Opella, J. Am. Chem. SOC. 104 (1982) 1759.
[I821 a) T. A. Cross, S. J. Opella, J. Am. Chem. Soc. 105 (1983) 306; T. A. Cross, M. H. Frey, S. J. Opella, ibid. 105 (1983) 7471 ; T. A. Cross, S. J. Opella, J. Mol. Biol. 182 (1985) 367; b) F. Martin, Physiol. Veg. 23 (1985) 463.
11831 a) J. Schaefer, T. A. Skokut, E. 0. Stejskal, R. A. McKay, J. E. Varner, J . Biol. Chem. 256 (1981) 11574; b) G. T. Coker, 111, J. Schaefer, Plant Physiol. 77 (1985) 129.
208.
4152.
11841 H. G. Baker: Plants and Civilization. Macmillan, London 1970.
Angew. Chem. Int. Ed. Engl. 25 (1986) 383-413 413
Copyright © 2022 FDOKUMEN




















