







Bahasa
Halaman
Hukum


ARTICLE
Received 28 Jan 2013 | Accepted 22 Jul 2013 | Published 22 Aug 2013
Wall teichoic acid structure governs horizontalgene transfer between major bacterial pathogensVolker Winstel1,2, Chunguang Liang3, Patricia Sanchez-Carballo4, Matthias Steglich5, Marta Munar3,w,
Barbara M. Broker6, Jose R. Penades7, Ulrich Nubel5, Otto Holst4, Thomas Dandekar3, Andreas Peschel1,2
& Guoqing Xia1,2
Mobile genetic elements (MGEs) encoding virulence and resistance genes are widespread in
bacterial pathogens, but it has remained unclear how they occasionally jump to new host
species. Staphylococcus aureus clones exchange MGEs such as S. aureus pathogenicity islands
(SaPIs) with high frequency via helper phages. Here we report that the S. aureus ST395
lineage is refractory to horizontal gene transfer (HGT) with typical S. aureus but exchanges
SaPIs with other species and genera including Staphylococcus epidermidis and Listeria mono-
cytogenes. ST395 produces an unusual wall teichoic acid (WTA) resembling that of its HGT
partner species. Notably, distantly related bacterial species and genera undergo efficient HGT
with typical S. aureus upon ectopic expression of S. aureus WTA. Combined with genomic
analyses, these results indicate that a ‘glycocode’ of WTA structures and WTA-binding
helper phages permits HGT even across long phylogenetic distances thereby shaping the
evolution of Gram-positive pathogens.
DOI: 10.1038/ncomms3345 OPEN
1 Cellular and Molecular Microbiology Division, Interfaculty Institute of Microbiology and Infection Medicine, University of Tubingen, Elfriede-Aulhorn-Stra�e6, 72076 Tubingen, Germany. 2 German Center for Infection Research (DZIF), partner site Tubingen, 72076 Tubingen, Germany. 3 Bioinformatik, Biozentrum,University of Wurzburg, Am Hubland, 97074 Wurzburg, Germany. 4 Division of Structural Biochemistry, Research Center Borstel, Leibniz-Center for Medicineand Biosciences, Parkallee 4a/c, 23845 Borstel, Germany. 5 Robert Koch Institute 38855 Wernigerode, Germany. 6 Institute of Immunology and TransfusionMedicine, University Medicine Greifswald, F.-Sauerbruchstra�e, 17475 Greifswald, Germany. 7 Instituto de Biomedicina de Valencia (IBV-CSIC), 46010Valencia, Spain. w Present address: Universite Libre de Bruxelles, Genomics and Structural Bioinformatics, Campus du Solbosch, Batiment U, Porte D,Niveau 3, CP165/61, Avenue F.D. Roosevelt 50, 1050 Bruxelles, Belgium. Correspondence and requests for materials should be addressed to A.P.(email: [email protected]).
NATURE COMMUNICATIONS | 4:2345 | DOI: 10.1038/ncomms3345 | www.nature.com/naturecommunications 1
& 2013 Macmillan Publishers Limited. All rights reserved.
Major parts of bacterial genomes consist of geneticmaterial originating from other organisms. Many ofthese elements can be mobilized and exchanged by
horizontal gene transfer (HGT) thereby shaping bacterial genomeplasticity and permitting rapid adaptation to changing environ-mental challenges1. HGT of mobile genetic elements (MGEs)usually occurs at high frequency only among closely relatedbacterial clones because the transfer mechanisms, phage-mediated transduction or plasmid conjugation, rely on specificrecognition of cognate recipient strains1,2. However, HGT alsooccurs between members of different species or even genera albeitwith lower frequency. Such rare events are responsible for theimport of new genes into the species’ genetic pool along with theemergence of new phenotypic properties; they are particularly
important for evolution of new bacterial pathogen lineages withnew virulence and antibiotic resistance traits.
The major human pathogen Staphylococcus aureus represents aparadigm for studying the roles of ‘short-distance’ HGT betweenstrains of the same species and ‘long-distance’ HGT with otherspecies or genera. MGEs and non-mobile genomic islandsconstitute ca. 22% of the S. aureus genomes and govern thevirulence and colonization capacities, host-specificity and anti-biotic resistance of the various clonal complexes3,4. Methicillin-resistant S. aureus carrying staphylococcal cassette chromosomeswith mecA genes represent the most frequent cause of severecommunity- or healthcare-associated infections in manydeveloping and developed countries5,6. While conjugation anduptake of naked DNA by natural transformation seem to occur
10–4
10–3
10–2
10–1
10–5
10–4
10–3
10–2
S. aureus strains(sequence type)
NRS229 w.t. (ST1)
a
b
N315 w.t. (ST5)
USA300 w.t. (ST8)
NRS184 w.t. (ST22)
A860325 w.t. (ST25)
NRS71 w.t. (ST30)
PS187 w.t. (ST395)
PS187 ΔsauUSI ΔhsdR (ST395)
PS187 ΔsauUSI (ST395)
TRU/PFU TRU/PFU
SaPIbov1 (Φ11) SaPI1 (Φ80α)
NRS111 w.t. (ST395)
BKI066 w.t. (ST395)
BKI084 w.t. (ST395)
T132-1 w.t. (ST395)
T166-1 w.t. (ST395)
None obtained None obtained
None obtained None obtained
None obtained None obtained
None obtained None obtained
None obtained None obtained
None obtained None obtained
None obtained None obtained
None obtained None obtained
S. aureus strain pT181 (transformation)
CFU per μg DNA
PS187 w.t.
PS187 ΔsauUSI
PS187 ΔsauUSI ΔhsdR
**
1 × 105 2 × 105
*
0
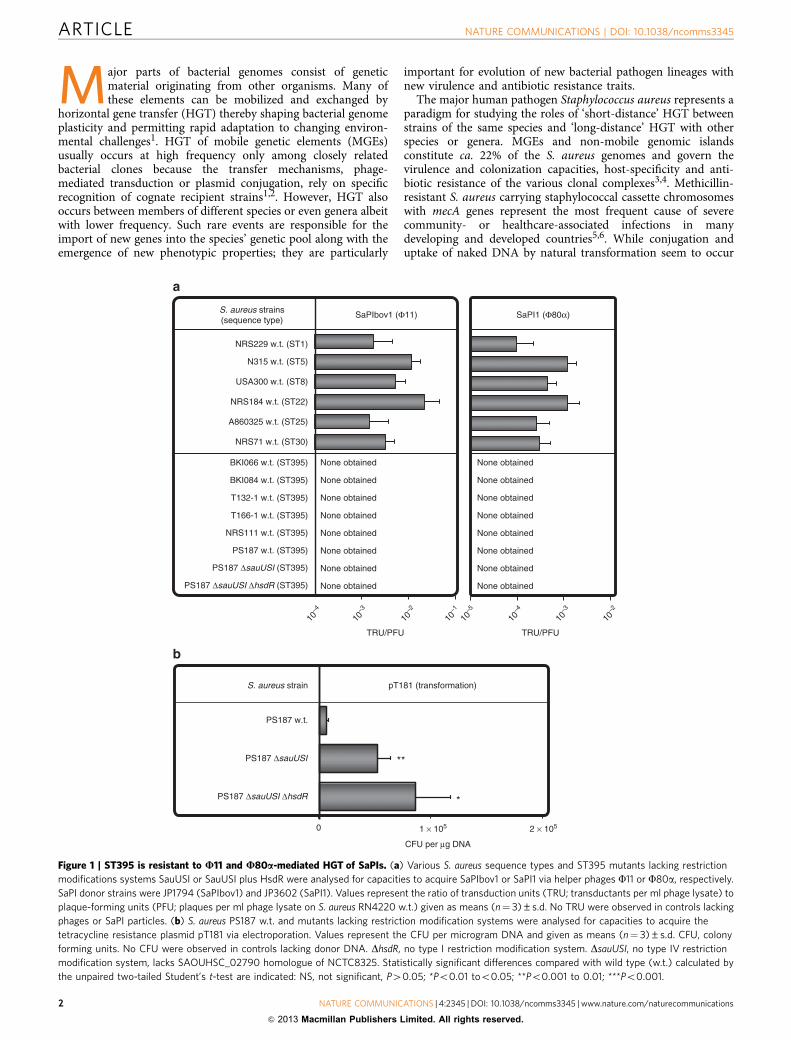
Figure 1 | ST395 is resistant to U11 and U80a-mediated HGT of SaPIs. (a) Various S. aureus sequence types and ST395 mutants lacking restriction
modifications systems SauUSI or SauUSI plus HsdR were analysed for capacities to acquire SaPIbov1 or SaPI1 via helper phages F11 or F80a, respectively.
SaPI donor strains were JP1794 (SaPIbov1) and JP3602 (SaPI1). Values represent the ratio of transduction units (TRU; transductants per ml phage lysate) to
plaque-forming units (PFU; plaques per ml phage lysate on S. aureus RN4220 w.t.) given as means (n¼ 3)±s.d. No TRU were observed in controls lacking
phages or SaPI particles. (b) S. aureus PS187 w.t. and mutants lacking restriction modification systems were analysed for capacities to acquire the
tetracycline resistance plasmid pT181 via electroporation. Values represent the CFU per microgram DNA and given as means (n¼ 3)±s.d. CFU, colony
forming units. No CFU were observed in controls lacking donor DNA. DhsdR, no type I restriction modification system. DsauUSI, no type IV restriction
modification system, lacks SAOUHSC_02790 homologue of NCTC8325. Statistically significant differences compared with wild type (w.t.) calculated by
the unpaired two-tailed Student’s t-test are indicated: NS, not significant, P40.05; *Po0.01 too0.05; **Po0.001 to 0.01; ***Po0.001.
ARTICLE NATURE COMMUNICATIONS | DOI: 10.1038/ncomms3345
2 NATURE COMMUNICATIONS | 4:2345 | DOI: 10.1038/ncomms3345 | www.nature.com/naturecommunications
& 2013 Macmillan Publishers Limited. All rights reserved.

rarely4,7, staphylococcal HGT of MGEs is generally believed todepend largely on transducing helper phages4. Certain temperatephages of serogroup B such as F11 or F80a have been shown tobe capable of transducing DNA between S. aureus clones and toemploy the N-acetyl-D-glucosamine (GlcNAc) residues on wallteichoic acid (WTA), a surface-exposed glycopolymer, asreceptor4,8. WTA is produced by most Gram-positive bacteriaand usually has species- or strain-specific structure9. S. aureusproduces a WTA polymer composed of ca. 40 ribitol-phosphate(RboP) repeating units modified with a- and/or b-linked GlcNAcand D-alanine9,10 while the various coagulase-negative staphylococcalspecies (CoNS) produce WTA with glycerophosphate (GroP) orhexose-containing, complex repeating units modified with differenttypes of sugars11.
S. aureus pathogenicity islands (SaPIs) are exchanged amongS. aureus lineages with high frequency by SaPI particles consistingof SaPI genomes and structural proteins from helper phages12,13.While such ‘short-distance’ HGT events occur with highfrequency, antibiotic resistance-mediating MGEs have beenacquired only occasionally from other bacterial species. Of note,b-lactam antibiotic resistance genes from CoNS have frequentlybeen imported into S. aureus14 whereas enterococcal vancomycinresistance genes have emerged in staphylococci only in a fewexceptional cases15 suggesting that there are mechanismsinvolved that favour or disfavour specific ‘long-distance’ HGTevents. Restriction modification systems16–18 and rarely occurringclustered, regularly interspaced, short palindromic repeat(CRISPR) sequences19 have been shown to interfere with HGTefficiency in staphylococci but the major determinants permittingHGT with other bacterial species and genera have remainedunknown.
We demonstrate here that the variable structure of glycosylatedWTA constitutes a ‘glycocode’ that is sensed by transducingphages thereby defining the routes of HGT. Similar WTAstructures enable DNA exchange via helper bacteriophages even
across the boundaries of species or genera, whereas S. aureusclones producing altered WTA become separated from thespecies’ genetic pool and may initiate new routes of HGT withother bacterial species and genera that share related WTA. Thus,related WTA structures are sufficient to initiate HGT even acrosslong phylogenic distances.
ResultsST395 cannot undergo HGT with other S. aureus lineages. Thevarious S. aureus clonal complexes differ largely in their epidemicpotential and number of MGEs4. We compared several S. aureuslineages for capability to acquire SaPI1 or SaPIbov1 originatingfrom sequence types ST8 and ST151, respectively13. Derivativesof these SaPIs with antibiotic resistance gene markers20 weretransferred from S. aureus ST8 to a variety of potential recipientstrains using helper phages F11 (for SaPIbov1) or F80a (forSaPI1). The majority of the sequence types acquired SaPIs albeitwith varying efficiency, probably as a consequence of differentrestriction modification systems16–18 (Fig. 1a). In contrast, severalindependent clones of the ST395 lineage from various parts of theworld including isolates from the lung or blood stream infectionsand nasal swabs (Supplementary Table S1)21–23 were completelyresistant to HGT of SaPIs (Fig. 1a). Restriction modificationsystems were obviously not responsible for HGT resistance ofST395 because consecutive inactivation of the genes for type I(DhsdR) and type IV (DsauUSI) restriction systems in the ST395strain PS187 did not enable transfer of SaPIs (Fig. 1a), whereas itconsiderably increased the rates of plasmid electroporation(Fig. 1b).
U187 mediates HGT between ST395 and other bacterialspecies. While strain PS187 was resistant to infection by F80a orF11 (Supplementary Fig. S1a), it has previously been shown to besusceptible to phage F187 (refs 21,24). Interestingly, most ST395
Species Strain SaPIbov1 (Φ187)
None obtained
L. monocytogenes ATCC19118 w.t.
S. capitis
PS187 w.t.
PS187 ΔtagO
NRS111w.t.
PS187 ΔO c-tagO
RN4220 w.t.
TM300 w.t.
ATCC27840 w.t.
TÜ3298 w.t.
1457 w.t.
VRE392 w.t.
V583 w.t.
ATCC25401 w.t.
S. aureus
S. carnosus
S. epidermidis
E. faecium
E. faecalis
L. grayi None obtained
None obtained
None obtained
None obtained
None obtained
None obtained
None obtained
None obtained
None obtained
10–8
10–7
10–6
10–5
10–4
10–7
10–6
10–5
10–4
10–3
TRU/PFU TRU/PFU
ND
ND
10–8
ND
SaPI187β (Φ187)
Figure 2 | U187 mediates HGT of SaPIs among ST395 and to other species and genera. S. aureus ST395 strains with or without WTA or other
Gram-positive bacteria were analysed for capacities to acquire SaPIbov1 or SaPI187b via helper phage F187. SaPI donor strains were VW1 (SaPIbov1) and
VW7 (SaPI187b). Values represent the ratio of transduction units (TRU; transductants per ml phage lysate) to plaque-forming units (PFU; plaques
per ml phage lysate on S. aureus PS187 w.t.) given as means (n¼ 3)±s.d. No TRU were observed in controls lacking phages or SaPI particles. DtagO, no
WTA. c-tagO, DtagO complemented with tagO. ND, not determined due to marker resistance or spontaneous occurring clones.
NATURE COMMUNICATIONS | DOI: 10.1038/ncomms3345 ARTICLE
NATURE COMMUNICATIONS | 4:2345 | DOI: 10.1038/ncomms3345 | www.nature.com/naturecommunications 3
& 2013 Macmillan Publishers Limited. All rights reserved.
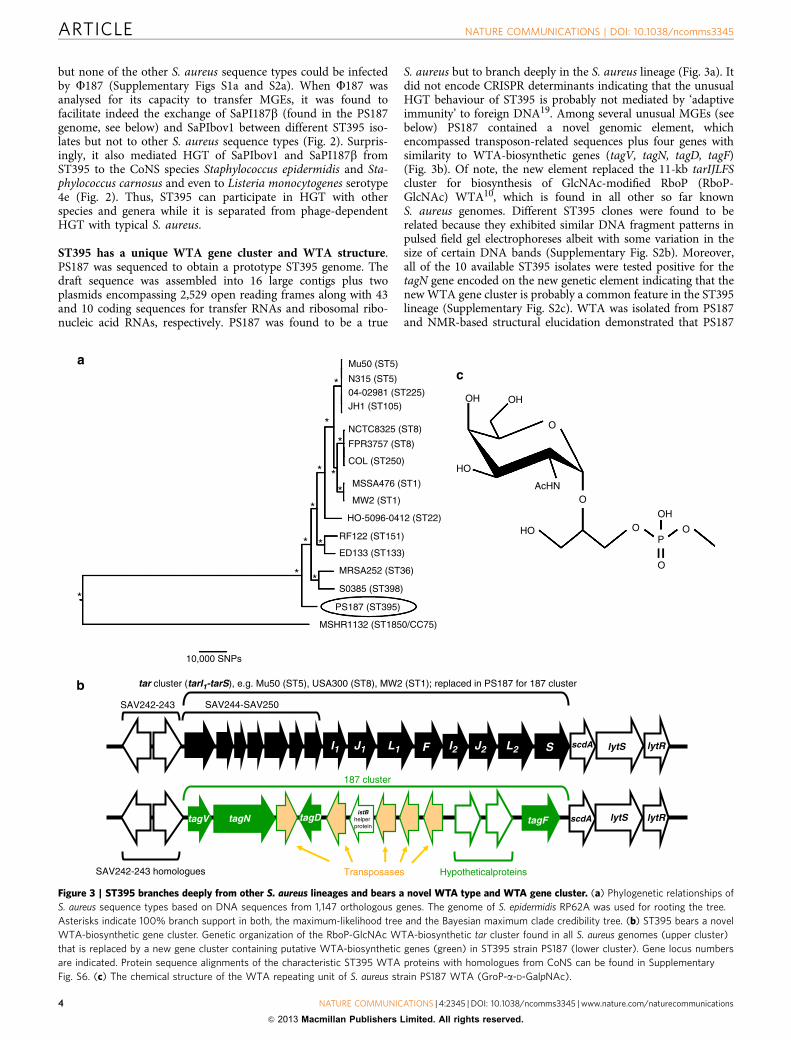
but none of the other S. aureus sequence types could be infectedby F187 (Supplementary Figs S1a and S2a). When F187 wasanalysed for its capacity to transfer MGEs, it was found tofacilitate indeed the exchange of SaPI187b (found in the PS187genome, see below) and SaPIbov1 between different ST395 iso-lates but not to other S. aureus sequence types (Fig. 2). Surpris-ingly, it also mediated HGT of SaPIbov1 and SaPI187b fromST395 to the CoNS species Staphylococcus epidermidis and Sta-phylococcus carnosus and even to Listeria monocytogenes serotype4e (Fig. 2). Thus, ST395 can participate in HGT with otherspecies and genera while it is separated from phage-dependentHGT with typical S. aureus.
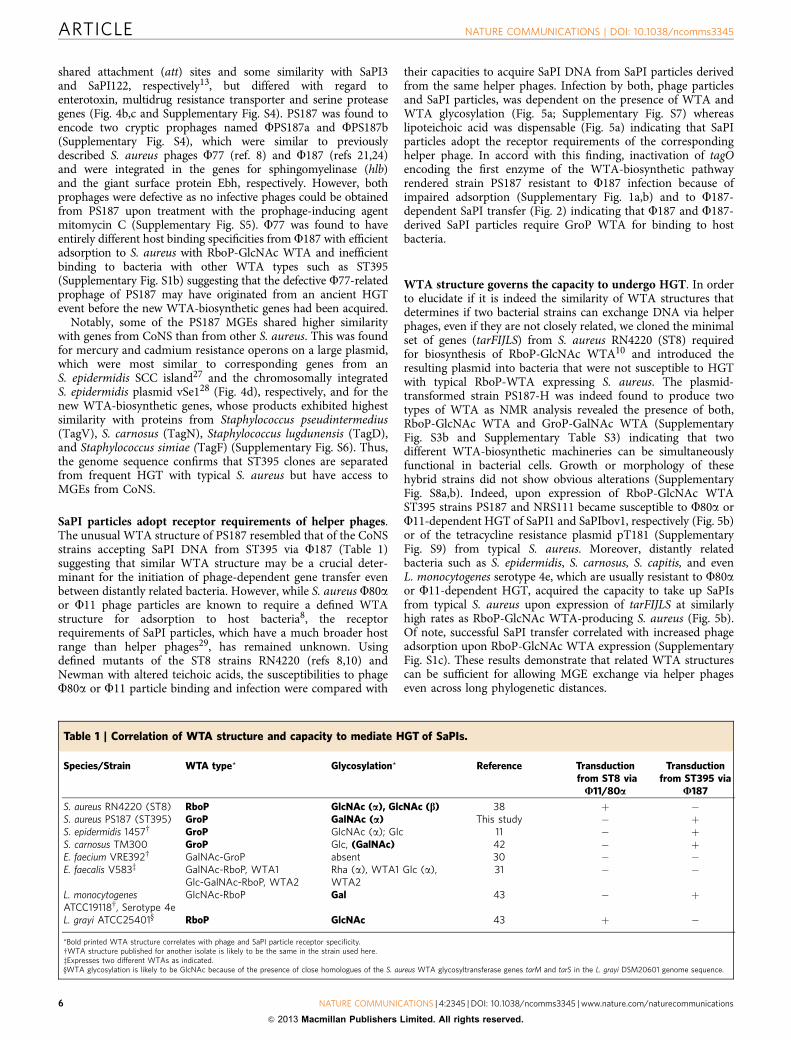
ST395 has a unique WTA gene cluster and WTA structure.PS187 was sequenced to obtain a prototype ST395 genome. Thedraft sequence was assembled into 16 large contigs plus twoplasmids encompassing 2,529 open reading frames along with 43and 10 coding sequences for transfer RNAs and ribosomal ribo-nucleic acid RNAs, respectively. PS187 was found to be a true
S. aureus but to branch deeply in the S. aureus lineage (Fig. 3a). Itdid not encode CRISPR determinants indicating that the unusualHGT behaviour of ST395 is probably not mediated by ‘adaptiveimmunity’ to foreign DNA19. Among several unusual MGEs (seebelow) PS187 contained a novel genomic element, whichencompassed transposon-related sequences plus four genes withsimilarity to WTA-biosynthetic genes (tagV, tagN, tagD, tagF)(Fig. 3b). Of note, the new element replaced the 11-kb tarIJLFScluster for biosynthesis of GlcNAc-modified RboP (RboP-GlcNAc) WTA10, which is found in all other so far knownS. aureus genomes. Different ST395 clones were found to berelated because they exhibited similar DNA fragment patterns inpulsed field gel electrophoreses albeit with some variation in thesize of certain DNA bands (Supplementary Fig. S2b). Moreover,all of the 10 available ST395 isolates were tested positive for thetagN gene encoded on the new genetic element indicating that thenew WTA gene cluster is probably a common feature in the ST395lineage (Supplementary Fig. S2c). WTA was isolated from PS187and NMR-based structural elucidation demonstrated that PS187
I2 J2L1 lytSscdASL2FJ1I1 lytR
tar cluster (tarI1-tarS), e.g. Mu50 (ST5), USA300 (ST8), MW2 (ST1); replaced in PS187 for 187 cluster
scdA lytS lytRtagFistB
helper protein
tagDtagV
187 cluster
tagN
SAV244-SAV250
SAV242-243 homologues
SAV242-243
Transposases Hypotheticalproteins
Mu50 (ST5)
*
*
*
*
*
**
* *
*
*
*
N315 (ST5)
04-02981 (ST225)
JH1 (ST105)
NCTC8325 (ST8)
FPR3757 (ST8)
COL (ST250)
MSSA476 (ST1)
MW2 (ST1)
HO-5096-0412 (ST22)
RF122 (ST151)
ED133 (ST133)
MRSA252 (ST36)
S0385 (ST398)
PS187 (ST395)
MSHR1132 (ST1850/CC75)
10,000 SNPs
ac
b
OH OH
O
O
O
O
OP
HO
HO
OH
AcHN
Figure 3 | ST395 branches deeply from other S. aureus lineages and bears a novel WTA type and WTA gene cluster. (a) Phylogenetic relationships of
S. aureus sequence types based on DNA sequences from 1,147 orthologous genes. The genome of S. epidermidis RP62A was used for rooting the tree.
Asterisks indicate 100% branch support in both, the maximum-likelihood tree and the Bayesian maximum clade credibility tree. (b) ST395 bears a novel
WTA-biosynthetic gene cluster. Genetic organization of the RboP-GlcNAc WTA-biosynthetic tar cluster found in all S. aureus genomes (upper cluster)
that is replaced by a new gene cluster containing putative WTA-biosynthetic genes (green) in ST395 strain PS187 (lower cluster). Gene locus numbers
are indicated. Protein sequence alignments of the characteristic ST395 WTA proteins with homologues from CoNS can be found in Supplementary
Fig. S6. (c) The chemical structure of the WTA repeating unit of S. aureus strain PS187 WTA (GroP-a-D-GalpNAc).
ARTICLE NATURE COMMUNICATIONS | DOI: 10.1038/ncomms3345
4 NATURE COMMUNICATIONS | 4:2345 | DOI: 10.1038/ncomms3345 | www.nature.com/naturecommunications
& 2013 Macmillan Publishers Limited. All rights reserved.

produces a unique WTA type with an N-acetyl-D-galactosamine(GalNAc) modified GroP backbone (GroP-GalNAc) (Fig. 3c;Supplementary Fig. S3a and Supplementary Table S2), which wasin agreement with an earlier analysis of PS187-related strains11.
Some unusual MGEs from ST395 may be originating fromCoNS. Because of its inability to exchange DNA with typicalS. aureus, we assumed that ST395 may be genetically isolated.ST395 clones have been occasionally described as human com-mensals and invasive pathogens22,23 and a recent study hasfound that ST395 accounts for 5 and 2% of S. aureus nasal andblood culture isolates, respectively, in Northeastern Germany23.However, the actual numbers may be higher because ST395isolates are usually methicillin susceptible and such clones are
hardly collected and typed. Interestingly, early reports from the1960s have pointed to a canine reservoir of PS187-related S.aureus25. In accord with this notion, we found the sequences ofsome of the host range-determining proteins of PS187 (IsdB andVwBP)26 to differ from typical human strains whereas thepresence of the strictly human-specific chp, scn and sak genes26
suggests that PS187 is at least in part adapted to the human host.Most MGEs of PS187 were distinct in synteny and composition
from those found in other S. aureus genomes and some were evenmore related to MGEs found in CoNS. The genomic islands vSAaand vSAb found in all previously sequenced S. aureus genomeswere also present in PS187 but lacked typical enterotoxin orlantibiotic gene clusters, respectively (Fig. 4a and SupplementaryFig. S4). Two novel PS187 SaPIs named SaPI187a and SaPI187b
SaPI187 α
SaPI187β
SaPI3
SaPI122
vSAα
vSAβ
pV187(PS187)
vSE1(RP62A)
SCC island(ATCC12228)
SERP2223
SERP2222
SERP2221
SERP2220
SERP2224
SERP2225
SE0087
SE0085
SE0084
SE0083
SE0082
SE0081
SE0086
Organomercurial lyaseMercuric ion reductase
Mercuric transporter
Cadmium resistance protein
Cadmium efflux protein
ArsR transcriptional regulator
Metallothionein2-related protein
Putative lipoprotein
Virulence-associatedprotein VapE
a
b
c
d
Transcriptionalregulator, Cro/CIfamily
Enterotoxin
Putative primase
Multidrug resistanceproteine
Exotoxins
Common to allvSAα/β types
Putative β-lactamase
Leukocidine
Hyaluronate lyaseprecursor
Type I restrictionmodification system
Hypothetical protein
Putative DNA helicase
Serine protease
tRNA cluster
Transposase
Ear protein
Integrase
Putative terminase
ArsR regulator &metallothionein 2-related protein
Mercury R cluster
Cadmium R cluster
Replication-associatedprotein
vapE
att site: TTATTTAGCAGGAATAA
sew ear
att site: TTATTTAGCAGGAATAA
vapE earsek seq seb
guaA hsdM hsdS
lukD lukEhsdS
att site: GTTTTACATCATTCCTGGCAT att site: GTTTTACATCATTCCTGGCAT
hsdM
Figure 4 | Characteristic MGEs of S. aureus PS187. (a) Genetic organization of the two genomic islands found in PS187. The organization of PS187 vSAa is
similar to that of other S. aureus genomes. The vSAb of PS187 lacks typical enterotoxin or lantibiotic clusters found in all other vSAb types sequenced
so far. (b) Comparison of SaPI187a from PS187 and SaPI3 from typical S. aureus genomes. SaPI187a shares the att site with SaPI1, SaPI3 and SaPI5 and was
found to be similarly organized as SaPI3 from S. aureus Col. Note the presence of a novel enterotoxin in SaPI187a designated as enterotoxin SeW.
(c) Comparison of SaPI187b from PS187 and SaPI122 from S. aureus RF122. Although SaPI187b shares the att site with SaPI122 the SaPI122-characteristic
gene encoding a multidrug resistance protein is absent in SaPI187b. Instead SaPI187b encodes a new serine protease. (d) Comparative map of mercury and
cadmium resistance operons on a large plasmid from PS187 and corresponding genes from the S. epidermidis ATCC12228 SCC composite island (right)
and the S. epidermidis RP62A integrated plasmid vSE1 (left). Dotted lines indicate sequence identities above 492% (DNA level) and 93% (protein level).
Gene locus numbers are listed and ORFs are coloured according to functional categories as indicated.
NATURE COMMUNICATIONS | DOI: 10.1038/ncomms3345 ARTICLE
NATURE COMMUNICATIONS | 4:2345 | DOI: 10.1038/ncomms3345 | www.nature.com/naturecommunications 5
& 2013 Macmillan Publishers Limited. All rights reserved.
shared attachment (att) sites and some similarity with SaPI3and SaPI122, respectively13, but differed with regard toenterotoxin, multidrug resistance transporter and serine proteasegenes (Fig. 4b,c and Supplementary Fig. S4). PS187 was found toencode two cryptic prophages named FPS187a and FPS187b(Supplementary Fig. S4), which were similar to previouslydescribed S. aureus phages F77 (ref. 8) and F187 (refs 21,24)and were integrated in the genes for sphingomyelinase (hlb)and the giant surface protein Ebh, respectively. However, bothprophages were defective as no infective phages could be obtainedfrom PS187 upon treatment with the prophage-inducing agentmitomycin C (Supplementary Fig. S5). F77 was found to haveentirely different host binding specificities from F187 with efficientadsorption to S. aureus with RboP-GlcNAc WTA and inefficientbinding to bacteria with other WTA types such as ST395(Supplementary Fig. S1b) suggesting that the defective F77-relatedprophage of PS187 may have originated from an ancient HGTevent before the new WTA-biosynthetic genes had been acquired.
Notably, some of the PS187 MGEs shared higher similaritywith genes from CoNS than from other S. aureus. This was foundfor mercury and cadmium resistance operons on a large plasmid,which were most similar to corresponding genes from anS. epidermidis SCC island27 and the chromosomally integratedS. epidermidis plasmid vSe128 (Fig. 4d), respectively, and for thenew WTA-biosynthetic genes, whose products exhibited highestsimilarity with proteins from Staphylococcus pseudintermedius(TagV), S. carnosus (TagN), Staphylococcus lugdunensis (TagD),and Staphylococcus simiae (TagF) (Supplementary Fig. S6). Thus,the genome sequence confirms that ST395 clones are separatedfrom frequent HGT with typical S. aureus but have access toMGEs from CoNS.
SaPI particles adopt receptor requirements of helper phages.The unusual WTA structure of PS187 resembled that of the CoNSstrains accepting SaPI DNA from ST395 via F187 (Table 1)suggesting that similar WTA structure may be a crucial deter-minant for the initiation of phage-dependent gene transfer evenbetween distantly related bacteria. However, while S. aureus F80aor F11 phage particles are known to require a defined WTAstructure for adsorption to host bacteria8, the receptorrequirements of SaPI particles, which have a much broader hostrange than helper phages29, has remained unknown. Usingdefined mutants of the ST8 strains RN4220 (refs 8,10) andNewman with altered teichoic acids, the susceptibilities to phageF80a or F11 particle binding and infection were compared with
their capacities to acquire SaPI DNA from SaPI particles derivedfrom the same helper phages. Infection by both, phage particlesand SaPI particles, was dependent on the presence of WTA andWTA glycosylation (Fig. 5a; Supplementary Fig. S7) whereaslipoteichoic acid was dispensable (Fig. 5a) indicating that SaPIparticles adopt the receptor requirements of the correspondinghelper phage. In accord with this finding, inactivation of tagOencoding the first enzyme of the WTA-biosynthetic pathwayrendered strain PS187 resistant to F187 infection because ofimpaired adsorption (Supplementary Fig. 1a,b) and to F187-dependent SaPI transfer (Fig. 2) indicating that F187 and F187-derived SaPI particles require GroP WTA for binding to hostbacteria.
WTA structure governs the capacity to undergo HGT. In orderto elucidate if it is indeed the similarity of WTA structures thatdetermines if two bacterial strains can exchange DNA via helperphages, even if they are not closely related, we cloned the minimalset of genes (tarFIJLS) from S. aureus RN4220 (ST8) requiredfor biosynthesis of RboP-GlcNAc WTA10 and introduced theresulting plasmid into bacteria that were not susceptible to HGTwith typical RboP-WTA expressing S. aureus. The plasmid-transformed strain PS187-H was indeed found to produce twotypes of WTA as NMR analysis revealed the presence of both,RboP-GlcNAc WTA and GroP-GalNAc WTA (SupplementaryFig. S3b and Supplementary Table S3) indicating that twodifferent WTA-biosynthetic machineries can be simultaneouslyfunctional in bacterial cells. Growth or morphology of thesehybrid strains did not show obvious alterations (SupplementaryFig. S8a,b). Indeed, upon expression of RboP-GlcNAc WTAST395 strains PS187 and NRS111 became susceptible to F80a orF11-dependent HGT of SaPI1 and SaPIbov1, respectively (Fig. 5b)or of the tetracycline resistance plasmid pT181 (SupplementaryFig. S9) from typical S. aureus. Moreover, distantly relatedbacteria such as S. epidermidis, S. carnosus, S. capitis, and evenL. monocytogenes serotype 4e, which are usually resistant to F80aor F11-dependent HGT, acquired the capacity to take up SaPIsfrom typical S. aureus upon expression of tarFIJLS at similarlyhigh rates as RboP-GlcNAc WTA-producing S. aureus (Fig. 5b).Of note, successful SaPI transfer correlated with increased phageadsorption upon RboP-GlcNAc WTA expression (SupplementaryFig. S1c). These results demonstrate that related WTA structurescan be sufficient for allowing MGE exchange via helper phageseven across long phylogenetic distances.
Table 1 | Correlation of WTA structure and capacity to mediate HGT of SaPIs.
Species/Strain WTA type* Glycosylation* Reference Transductionfrom ST8 via
U11/80a
Transductionfrom ST395 via
U187
S. aureus RN4220 (ST8) RboP GlcNAc (a), GlcNAc (b) 38 þ �S. aureus PS187 (ST395) GroP GalNAc (a) This study � þS. epidermidis 1457w GroP GlcNAc (a); Glc 11 � þS. carnosus TM300 GroP Glc, (GalNAc) 42 � þE. faecium VRE392w GalNAc-GroP absent 30 � �E. faecalis V583z GalNAc-RboP, WTA1
Glc-GalNAc-RboP, WTA2Rha (a), WTA1 Glc (a),WTA2
31 � �
L. monocytogenesATCC19118w, Serotype 4e
GlcNAc-RboP Gal 43 � þ
L. grayi ATCC25401y RboP GlcNAc 43 þ �
*Bold printed WTA structure correlates with phage and SaPI particle receptor specificity.wWTA structure published for another isolate is likely to be the same in the strain used here.zExpresses two different WTAs as indicated.yWTA glycosylation is likely to be GlcNAc because of the presence of close homologues of the S. aureus WTA glycosyltransferase genes tarM and tarS in the L. grayi DSM20601 genome sequence.
ARTICLE NATURE COMMUNICATIONS | DOI: 10.1038/ncomms3345
6 NATURE COMMUNICATIONS | 4:2345 | DOI: 10.1038/ncomms3345 | www.nature.com/naturecommunications
& 2013 Macmillan Publishers Limited. All rights reserved.

In accord with this finding, a systematic analysis with severalGram-positive bacteria revealed a strong correlation betweenWTA structure and the capacity to exchange resistance andvirulence genes with either typical S. aureus via F11 or F80a (forexample, Listeria grayi) or with ST395 via F187 (for example,S. epidermidis and other CoNS) (Table 1). The susceptibility ofL. monocytogenes serotype 4e to F187-mediated HGT wasreflected by efficient binding of F187 (Supplementary Fig. S1b).It may be due to the decoration of WTA with galactose, whichmay facilitate binding of F187-derived SaPI particles in a similarway as GalNAc. Of note, vancomycin-resistant Enterococcusfaecium or Enterococcus faecalis could not undergo HGT withany S. aureus probably because of the very complex enterococcalWTA structures30,31.
DiscussionHGT between S. aureus and other bacterial species and generacontributes substantially to the evolution of new epidemic clones,but it has remained unclear if it occurs accidentally or followscertain rules. While restriction modification and CRISPR systemshave been shown to limit HGT efficiency16–19, no data on thecriteria that need to be fulfilled by the HGT partners to initiatephage-dependent MGE exchange have been available. Our studieswith naturally occurring and engineered bacterial strains withatypical WTA demonstrate that related WTA structures of MGEdonor and recipient are sufficient to permit HGT even across longphylogenetic distances. While helper phage particles are knownto have quite narrow host ranges for parasitic reproduction29,we found that their receptor specificities govern the capacity to
Species Strain SaPIbov1 (Φ11)
TRU/PFU TRU/PFU
PS187 w.t. None obtained
L. monocytogenes ATCC19118 w.t.
PS187-H
NRS111-H
NRS111 w.t.
TM300-H
TM300 w.t.
ATCC27840-H
ATCC27840 w.t.
1457-H
1457 w.t.
ATCC19118-H
L. grayi ATCC25401 w.t.
S. epidermidis
S. capitis
S. carnosus
S. aureus
None obtained
None obtained
None obtained
None obtained
None btained
10–4
10–3
10–2
10–1
10–4
10–3
10–2
None obtained
None obtained
None obtained
None obtained
None obtained
None obtained
None obtained
SaPI1 (Φ80α)
S. aureus strain SaPIbov1 (Φ11) SaPI1 (Φ80α)
TRU/PFUTRU/PFU
RN4220 w.t.
a
b
RN4220 ΔltaS (4S5)
RN4220 ΔtarM ΔtarS
RN4220 ΔMS c-tarM
RN4220 ΔtagO
RN4220 ΔO c-tagO
RN4220 ΔMS c-tarS
None obtained None obtained
10–7 10–6 10–5 10–4 10–3 10–210–5 10–4 10–3 10–2 10–1
Figure 5 | WTA structure determines the capacity of SaPIs to traverse even long phylogenetic distances. S. aureus RN4220 (ST8) strains with, with
altered or without WTA (a) or S. aureus PS187 (ST395) or other Gram-positive bacterial species expressing genes for biosynthesis of RboP-GlcNAc
WTA (b) were analysed for capacities to acquire SaPIbov1 or SaPI1 via helper phages F11 or F80a, respectively. SaPI donor strains were JP1794 (SaPIbov1)
and JP3602 (SaPI1). Values represent the ratio of transduction units (TRU; transductants per ml phage lysate) to plaque-forming units (PFU; plaques
per ml phage lysate on S. aureus RN4220 w.t.) given as means (n¼ 3)±s.d. No TRU were observed in strains expressing WTA other than RboP-GlcNAc.
DtagO, no WTA; DltaS (4S5), no lipoteichoic acid; c-tagO, DtagO complemented with tagO; DtarM DtarS, no WTA glycosylation; c-tarM and c-tarS,
DtarM DtarS complemented either with tarM or tarS. WTA hybrid strains expressing additional RboP-GlcNAc WTA are indicated with H.
NATURE COMMUNICATIONS | DOI: 10.1038/ncomms3345 ARTICLE
NATURE COMMUNICATIONS | 4:2345 | DOI: 10.1038/ncomms3345 | www.nature.com/naturecommunications 7
& 2013 Macmillan Publishers Limited. All rights reserved.

transmit MGEs to a broad range of recipient strains expressingcognate surface ligands. On the other hand, changes in helperphage receptor structures can enable binding of new types ofhelper phages and redirect the routes of HGT, which mayfacilitate the development of new clonal lineages and species.
The structurally diverse WTA molecules represent a species orlineage-specific signature at the surface of Gram-positive bacteria.WTA has several important roles for bacterial physiology such asthe control of autolytic and peptidoglycan-biosynthetic enzymes9,but all these functions could be achieved without extensivevariation of its structure. Our data indicate that WTA structureconstitutes a species- or lineage-defining ‘glycocode’ governingthe bacterial access to a common genetic pool via WTA-specifichelper phages. A thorough investigation of WTA structures andcognate phage receptor specificities will enable to assess howlikely the transfer of new antibiotic resistance genes across theboundaries of species and genera will be in the future. Antibioticstress is known to activate prophages thereby probably contri-buting to phage-mediated HGT32. In contrast, recently developedcompounds blocking the biosynthesis of WTA33 may help toreduce the frequency of phage-dependent HGT for example, inchronic polymicrobial infections.
MethodsBacterial strains and growth media. The various bacterial strains listed inSupplementary Table S1 were grown in BM broth (1% tryptone, 0.5% yeast extract,0.5% NaCl, 0.1% K2HPO4, 0.1% glucose) or Luria Bertani Broth supplemented withappropriate antibiotics at a concentration of 10 mg ml� 1 (chloramphenicol),3 mg ml� 1 (tetracycline) or 100 mg ml� 1 (ampicillin). In order to assess the growthand microscopic properties of mutant strains, phenotypic characterization of WTAhybrid strains overnight cultures were diluted to OD578 0.1 in BM and grown at37 �C on a shaker. Samples for microscopy were analysed after 3-h growth using aLeica DMRE microscope and Leica HCX 100� objective. Bacterial growth wasmonitored for 8 h.
Molecular genetic methods. For the construction of marker-less DtagO, DsauUSIand DsauUSI DhsdR mutants in S. aureus PS187, the pKOR1 shuttle vector wasused according to standard procedures34. For knockout plasmid construction,primers listed in Supplementary Table S4 were used. To label SaPI187b with anantibiotic resistance marker, the ermB gene was integrated into SaPI187b using thepKOR1 system. Knock-in primers are listed in Supplementary Table S4.
For amplification of the genomic region coding for tarF, tarI2, tarJ2, tarL2 andtarS from genomic DNA (RN4220 w.t.) primers, Tar-up and Tar-dn were used.The PCR product was cloned in the E. coli/S. aureus shuttle vector pRB47435 at theBamHI/EcoRI restriction sites resulting in plasmid pRB474-tarFI2J2L2S. Theplasmid was isolated from E. coli TOP10 or E. coli DC10B and used to transformtarget strains resulting in hybrid WTA-producing strains S. aureus PS187-H andNRS111-H, S. carnosus TM300-H, S. capitis ATCC27840-H and S. epidermidis1457-H. L. monocytogenes ATCC19118 was transformed by electroporation36
resulting in strain L. monocytogenes ATCC19118-H.For plasmid transformation, 50 ml competent cells were mixed with 500 ng
pT181 plasmid DNA, incubated at room temperature for 30 min, transferred into a1-mm gap electroporation cuvette and pulsed at 1,000 volts. Immediately afterelectroporation, 950ml BM medium was added followed by 70-min incubation at37 �C. Cells were finally plated onto selective media and transformants werecounted to calculate electroporation efficiency.
Pulsed field gel electrophoresis typing was performed according to Goerkeet al.37 tagN genotyping of available ST395 isolates was performed using primersN-up and N-dn (Supplementary Table S4).
Experiments with phages and SaPI particles. For rapid determination of phagesusceptibility, the soft agar spot assay was performed according to Xia et al.38 Aphage panel encompassing broad host range phage FK (ref. 38), serogroup L phageF187 (ref. 39), two serogroup B phages F11 (ref. 39) and F80a40 and serogroup Fphage F77 (ref. 8) (Supplementary Table S5) was used. The various phages werepropagated on S. aureus RN4220 (FK, F11, F80a, F77) or PS187 (F187). Mito-mycin C induction experiments confirmed that these two propagation strainsreleased no phage particles from endogenous prophages thereby ensuring that noother than the propagated phages were present in the obtained lysates. In order todetermine phage adsorption rates, the multiplicity of infection was set to 0.005 (forF187) or 0.1 (for F80a and F77). Adsorption was calculated by determining thePFU of the unbound phage in the supernatant and subtracting it from the totalnumber of input PFU. Adsorption efficiency was indicated relative to the
adsorption on the parental wild-type strain (RN4220 or PS187), which was set to100%. Prophages or SaPI particles were induced with 1 mg ml� 1 mitomycin C.
For phage-mediated SaPI and plasmid transfer, recipient strains were grownovernight and used for HGT experiments. Approximately 8.0� 107 bacteria weremixed with 100ml of SaPI lysates produced from S. aureus strains JP1794 or JP3602bearing the resistance marker-labelled SaPIbov1 (B1.0� 106 PFU ml� 1) or SaPI1(B3.0� 107 PFU ml� 1), respectively (Supplementary Table S1), incubated for15 min at 37 �C, diluted, and plated on BM agar supplemented with appropriateantibiotics.
For F187-mediated SaPIbov1 transfer, the PS187-H strain producing RboP andGroP WTA was transduced by F11 with SaPIbov1. The resulting strain VW1 wasinfected with F187 and 100ml of the resulting lysate (B1.3� 1010 PFU ml� 1) wasused to infect recipient strains as mentioned above. Note that for F187-mediatedSaPIbov1 transduction of Listeria strains, the used lysate contained onlyB5.0� 105 PFU ml� 1. For F187-mediated SaPI187b transfer, SaPI187b waslabelled with the ermB resistance marker in S. aureus PS187 wild type resulting instrain VW7, which was subsequently infected with F187 and 100 ml of the obtainedlysate (B3.0� 107 PFU ml� 1) was used to infect recipient strains as mentionedabove. To exclude that transductants resulted from spontaneous uptake of nickedDNA, SaPI-containing lysates were treated with 20 U DNase I for 1 h at 37 �C andused for SaPI transfer experiments. For plasmid transduction, pT181-bearingRN4220 was infected with F80a and the lysate was used to infect recipient strains.Approximately 8.0� 107 bacteria were resuspended in phage buffer (100 mMMgSO4, 100 mM CaCl2, 1 M Tris–HCl, pH 7.8, 0.59% NaCl, 0.1% gelatine) andmixed with 100 ml lysate (B1.1� 108 PFU ml� 1), incubated for 10 min at 37 �C,mixed with soft agar and poured onto BM agar plates containing 12.5 mg ml� 1
tetracycline. Transductants were counted after overnight incubation (SaPIs) orafter 48 h at 37 �C (pT181 and Listeria assays) and transduction efficiency wascalculated. SaPI transfer was confirmed by molecular typing of resistance marker.
WTA extraction and purification. WTA was isolated as described previously withminor modifications41. Briefly, overnight cultures were washed and disrupted in acell disrupter (Euler). Cell lysates were incubated at 37 �C overnight in the presenceof DNase and RNase. SDS was added to a final concentration of 2% followed byultrasonication for 15 min. Cell walls were washed several times to remove SDS. Torelease WTA from cell walls samples were treated with 5% trichloroacetic acid for4 h at 65 �C. Peptidoglycan debris was separated via centrifugation (10 min,14,000g). Determination of inorganic phosphate as described previously41 was usedfor WTA quantification. These crude WTA extracts were further purified asdescribed previously with minor modifications38. Briefly, the pH of the crudeextract was adjusted to 5.5 with NaOH and dialyzed against water with a Spectra/Por3 dialysis membrane (MWCO of 3.5 kDa; VWR International GmbH,Darmstadt). Samples were concentrated in a SpeedVac concentrator at 45 �C to5 ml and applied to DEAE-Sephadex A25 matrix according to Xia et al.38 Forelution from DEAE-Sephadex A25 matrix 20 mM Tris–HCl pH 7.2 0.35 M NaClwas used. The eluate was dialyzed again against water and concentrated to 1 ml.The purified WTA samples were stored at 20 �C for further analysis. All generalanalytical chemistry and NMR spectroscopy methods for WTA characterizationare described in detail in the supplementary section.
Genome sequencing and analyses. S. aureus PS187 was sequenced initially byRoche 454 pyrosequencing (Roche GS-FLX system), then resequenced by usingIllumina technology (Illumina HiSeq2000) at a 150-fold coverage. Details aredescribed in supplementary section.
References1. Thomas, C. M. & Nielsen, K. M. Mechanisms of, and barriers to, horizontal
gene transfer between bacteria. Nat. Rev. Microbiol. 3, 711–721 (2005).2. Frost, L. S., Leplae, R., Summers, A. O. & Toussaint, A. Mobile genetic
elements: the agents of open source evolution. Nat. Rev. Microbiol. 3, 722–732(2005).
3. Malachowa, N. & DeLeo, F. R. Mobile genetic elements of Staphylococcusaureus. Cell. Mol. Life Sci. 67, 3057–3071 (2010).
4. Lindsay, J. A. Genomic variation and evolution of Staphylococcus aureus. Int. J.Med. Microbiol. 300, 98–103 (2010).
5. Chambers, H. F. & Deleo, F. R. Waves of resistance: Staphylococcus aureus inthe antibiotic era. Nat. Rev. Microbiol. 7, 629–641 (2009).
6. Mediavilla, J. R., Chen, L., Mathema, B. & Kreiswirth, B. N. Globalepidemiology of community-associated methicillin-resistant Staphylococcusaureus (CA-MRSA). Curr. Opin. Microbiol. 15, 588–595 (2012).
7. Morikawa, K. et al. Expression of a cryptic secondary sigma factor gene unveilsnatural competence for DNA transformation in Staphylococcus aureus. PLoSPathog. 8, e1003003 (2012).
8. Xia, G. et al. Wall teichoic acid-dependent adsorption of staphylococcalsiphovirus and myovirus. J. Bacteriol. 193, 4006–4009 (2011).
9. Weidenmaier, C. & Peschel, A. Teichoic acids and related cell-wallglycopolymers in Gram-positive physiology and host interactions. Nat. Rev.Microbiol. 6, 276–287 (2008).
ARTICLE NATURE COMMUNICATIONS | DOI: 10.1038/ncomms3345
8 NATURE COMMUNICATIONS | 4:2345 | DOI: 10.1038/ncomms3345 | www.nature.com/naturecommunications
& 2013 Macmillan Publishers Limited. All rights reserved.

10. Brown, S. et al. Methicillin resistance in Staphylococcus aureus requires glycosylatedwall teichoic acids. Proc. Natl. Acad. Sci. USA 109, 18909–18914 (2012).
11. Endl, J., Seidl, P. H., Fiedler, F. & Schleifer, K. H. Determination of cell wallteichoic acid structure of staphylococci by rapid chemical and serologicalscreening methods. Arch. Microbiol. 137, 272–280 (1984).
12. Tallent, S. M., Langston, T. B., Moran, R. G. & Christie, G. E. Transducingparticles of Staphylococcus aureus pathogenicity island SaPI1 are comprised ofhelper phage-encoded proteins. J. Bacteriol. 189, 7520–7524 (2007).
13. Novick, R. P., Christie, G. E. & Penades, J. R. The phage-related chromosomalislands of Gram-positive bacteria. Nat. Rev. Microbiol. 8, 541–551 (2010).
14. Otto, M. Coagulase-negative staphylococci as reservoirs of genes facilitatingMRSA infection: staphylococcal commensal species such as Staphylococcusepidermidis are being recognized as important sources of genes promotingMRSA colonization and virulence. Bioessays 35, 4–11 (2012).
15. Perichon, B. & Courvalin, P. VanA-type vancomycin-resistant Staphylococcusaureus. Antimicrob. Agents. Chemother. 53, 4580–4587 (2009).
16. Corvaglia, A. R. et al. A type III-like restriction endonuclease functions as amajor barrier to horizontal gene transfer in clinical Staphylococcus aureusstrains. Proc. Natl. Acad. Sci. USA 107, 11954–11958 (2010).
17. Waldron, D. E. & Lindsay, J. A. Sau1: a novel lineage-specific type I restriction-modification system that blocks horizontal gene transfer into Staphylococcusaureus and between S. aureus isolates of different lineages. J. Bacteriol. 188,5578–5585 (2006).
18. Monk, I. R., Shah, I. M., Xu, M., Tan, M. W. & Foster, T. J. Transforming theuntransformable: application of direct transformation to manipulate geneticallyStaphylococcus aureus and Staphylococcus epidermidis. mBio 3 pii e00277-11 (2012).
19. Marraffini, L. A. & Sontheimer, E. J. CRISPR interference limits horizontal genetransfer in staphylococci by targeting DNA. Science 322, 1843–1845 (2008).
20. Tormo, M. A. et al. Staphylococcus aureus pathogenicity island DNA ispackaged in particles composed of phage proteins. J. Bacteriol. 190, 2434–2440(2008).
21. Asheshov, E. A. & Jevons, M. P. The effect of heat on the ability of a host strain tosupport the growth of a Staphylococcus phage. J. Gen. Microbiol. 31, 97–107 (1963).
22. Francois, P. et al. Evaluation of three molecular assays for rapid identification ofmethicillin-resistant Staphylococcus aureus. J. Clin. Microbiol. 45, 2011–2013(2007).
23. Holtfreter, S. et al. Clonal distribution of superantigen genes in clinicalStaphylococcus aureus isolates. J. Clin. Microbiol. 45, 2669–2680 (2007).
24. Rosenblum, E. D. & Tyrone, S. Serology, Density, and Morphology ofStaphylococcal Phages. J. Bacteriol. 88, 1737–1742 (1964).
25. Meyer, W. A proposal for subdividing the species Staphylococcus aureus. Int.J. Syst. Evol. Microbiol. 17, 387–389 (1967).
26. McCarthy, A. J. et al. The distribution of mobile genetic elements (MGEs) inMRSA CC398 is associated with both host and country. Genome Biol. Evol. 3,1164–1174 (2011).
27. Mongkolrattanothai, K., Boyle, S., Murphy, T. V. & Daum, R. S. Novel non-mecA-containing staphylococcal chromosomal cassette composite islandcontaining pbp4 and tagF genes in a commensal staphylococcal species: apossible reservoir for antibiotic resistance islands in Staphylococcus aureus.Antimicrob. Agents. Chemother. 48, 1823–1836 (2004).
28. Gill, S. R. et al. Insights on evolution of virulence and resistance from thecomplete genome analysis of an early methicillin-resistant Staphylococcusaureus strain and a biofilm-producing methicillin-resistant Staphylococcusepidermidis strain. J. Bacteriol. 187, 2426–2438 (2005).
29. Chen, J. & Novick, R. P. Phage-mediated intergeneric transfer of toxin genes.Science 323, 139–141 (2009).
30. Bychowska, A. et al. Chemical structure of wall teichoic acid isolated fromEnterococcus faecium strain U0317. Carbohydr. Res. 346, 2816–2819 (2011).
31. Geiss-Liebisch, S. et al. Secondary cell wall polymers of Enterococcus faecalis arecritical for resistance to complement activation via mannose-binding lectin.J. Biol. Chem. 287, 37769–37777 (2012).
32. Ubeda, C. et al. Antibiotic-induced SOS response promotes horizontaldissemination of pathogenicity island-encoded virulence factors instaphylococci. Mol. Microbiol. 56, 836–844 (2005).
33. Suzuki, T., Swoboda, J. G., Campbell, J., Walker, S. & Gilmore, M. S. In vitroantimicrobial activity of wall teichoic acid biosynthesis inhibitors againstStaphylococcus aureus isolates. Antimicrob. Agents. Chemother. 55, 767–774(2011).
34. Bae, T. & Schneewind, O. Allelic replacement in Staphylococcus aureus withinducible counter-selection. Plasmid 55, 58–63 (2006).
35. Bruckner, R. A series of shuttle vectors for Bacillus subtilis and Escherichia coli.Gene 122, 187–192 (1992).
36. Promadej, N., Fiedler, F., Cossart, P., Dramsi, S. & Kathariou, S. Cell wallteichoic acid glycosylation in Listeria monocytogenes serotype 4b requires gtcA,a novel, serogroup-specific gene. J. Bacteriol. 181, 418–425 (1999).
37. Goerke, C. et al. Diversity of prophages in dominant Staphylococcus aureusclonal lineages. J. Bacteriol. 191, 3462–3468 (2009).
38. Xia, G. et al. Glycosylation of wall teichoic acid in Staphylococcus aureus byTarM. J. Biol. Chem. 285, 13405–13415 (2010).
39. Pantucek, R. et al. Identification of bacteriophage types and their carriage inStaphylococcus aureus. Arch. Virol. 149, 1689–1703 (2004).
40. Christie, G. E. et al. The complete genomes of Staphylococcus aureusbacteriophages 80 and 80alpha--implications for the specificity of SaPImobilization. Virology 407, 381–390 (2010).
41. Weidenmaier, C. et al. Role of teichoic acids in Staphylococcus aureus nasalcolonization, a major risk factor in nosocomial infections. Nat. Med. 10,243–245 (2004).
42. Schleifer, K. H. &. Fischer, U. Description of a new species of the genusStaphylococcus: Staphylococcus carnosus. Int. J. Syst. Bacteriol. 32, 153–156(1982).
43. Fiedler, F. Biochemistry of the cell surface of Listeria strains: a locating generalview. Infection 16(Suppl 2): S92–S97 (1988).
AcknowledgementsWe thank Jacques Schrenzel, Gabriele Bierbaum, Christiane Wolz, Angelika Grundling,Matthias Marschal and Ian Monk for providing bacterial strains and phages and PeterBauer and Claudia Bauer for help with genome sequencing. This work was supported bygrants TRR34 to A.P., B.M.B., and T.D. and SFB766 to G.X. and A.P. from the GermanResearch Council, grants from the German Center for Infectious Disease Research toA.P. and G.X., and SkinStaph and Menage grants to A.P. from the German Ministry ofEducation and Research.
Author contributionsV.W., C.L., P. S.C., G.X., M.S. and M.M. performed the experiments and analysed thedata; B.M.B. and J.R.P. provided essential materials; V.W., J.R.P., U.N., O.H., T.D., A.P.and G.X. conceived the study; V.W., A.P. and G.X. wrote the manuscript.
Additional informationAccession codes: The Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession number ARPA00000000 (BioProjectPRJNA197438). The version described in this paper is the first version, ARPA01000000.
Supplementary Information accompanies this paper at http://www.nature.com/naturecommunications
Competing financial interests: The authors declare no competing financial interests.
Reprints and permission information is available online at http://npg.nature.com/reprintsandpermissions/
How to cite this article: Winstel, V. et al. Wall teichoic acid structure governs horizontalgene transfer between major bacterial pathogens. Nat. Commun. 4:2345 doi: 10.1038/ncomms3345 (2013).
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of
this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
NATURE COMMUNICATIONS | DOI: 10.1038/ncomms3345 ARTICLE
NATURE COMMUNICATIONS | 4:2345 | DOI: 10.1038/ncomms3345 | www.nature.com/naturecommunications 9
& 2013 Macmillan Publishers Limited. All rights reserved.
1
Supplementary Figures
Supplementary Figure S1: WTA-dependent S. aureus phage adsorption and infection. (a) Soft agar phage susceptibility assay for various wild-type or engineered S. aureus or CoNS strains. No S. aureus helper phage Φ11 or Φ80α infection was observed in strains expressing WTA other than RboP-GlcNAc. (b) Phage Φ187 and Φ77 adsorption rates to various Gram-positive bacteria. (c) Determination of phage Φ80α adsorption rates of different Gram-positive bacteria and corresponding WTA hybrid strains. Phage adsorption correlates with WTA structure. Values are given as means (n=3) ± SD. Based on the unpaired two-tailed Student’s t-test, statistically significant differences vs. the phage Φ187 propagation strains PS187 w.t. or vs. the phage Φ80α and Φ77 propagation strain RN4220 w.t. are indicated: ns, not significant with P > 0.05; *, P < 0.01 to < 0.05; **, P < 0.001 to 0.01; ***, P < 0.001. c-tagO, ∆tagO complemented with tagO. WTA hybrid strains are indicated with H. ∆hsdR, no type I restriction modification system. ∆sauUSI, no type IV restriction modification system (lacks SAOUHSC_02790 homolog of NCTC8325).
2
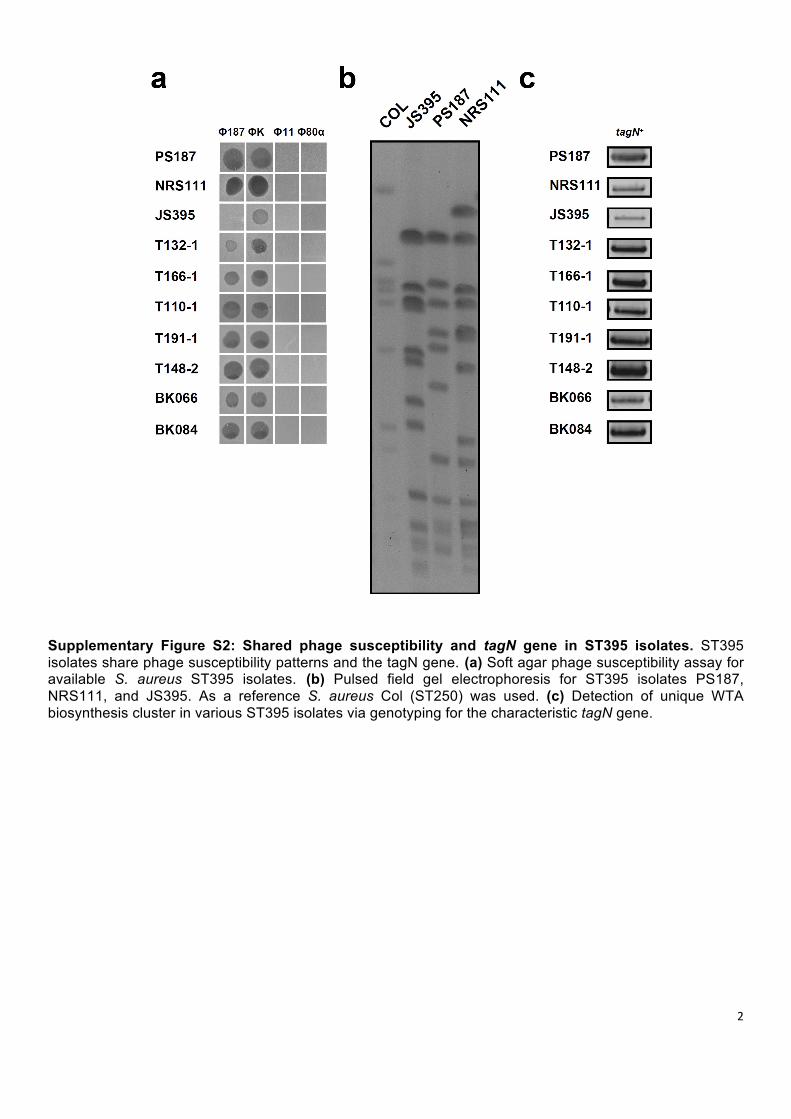
Supplementary Figure S2: Shared phage susceptibility and tagN gene in ST395 isolates. ST395 isolates share phage susceptibility patterns and the tagN gene. (a) Soft agar phage susceptibility assay for available S. aureus ST395 isolates. (b) Pulsed field gel electrophoresis for ST395 isolates PS187, NRS111, and JS395. As a reference S. aureus Col (ST250) was used. (c) Detection of unique WTA biosynthesis cluster in various ST395 isolates via genotyping for the characteristic tagN gene.

3
Supplementary Figure S3: NMR analysis of WTA from ST395 strains PS187 and PS187-H. (a) Heteronuclear 13C,1H single quantum correlation (HSQC) spectrum from purified S. aureus PS187 (ST395) WTA repeating unit reveals the GroP- α-GalpNAc structure. Arabic numerals label the proton/carbon atoms of the residues. Spectra were recorded at 700 MHz and 300 K relative to acetone (δH 2.225; δC 31.50). 1H and 13C chemical shifts are indicated in Supplementary Table S2. (b) HSQC spectrum from purified S. aureus PS187-H WTA repeating unit reveals the presence of both WTAs, GroP- α-GalpNAc and RboP-β-GlcpNAc. Arabic numerals label the proton/carbon atoms of the residues. Spectra were recorded at 700 MHz and 333 K relative to acetone (δH 2.225; δC 31.50). 1H and 13C chemical shifts are indicated in Supplementary Table S3.
4
WTA cluster
WTA cluster
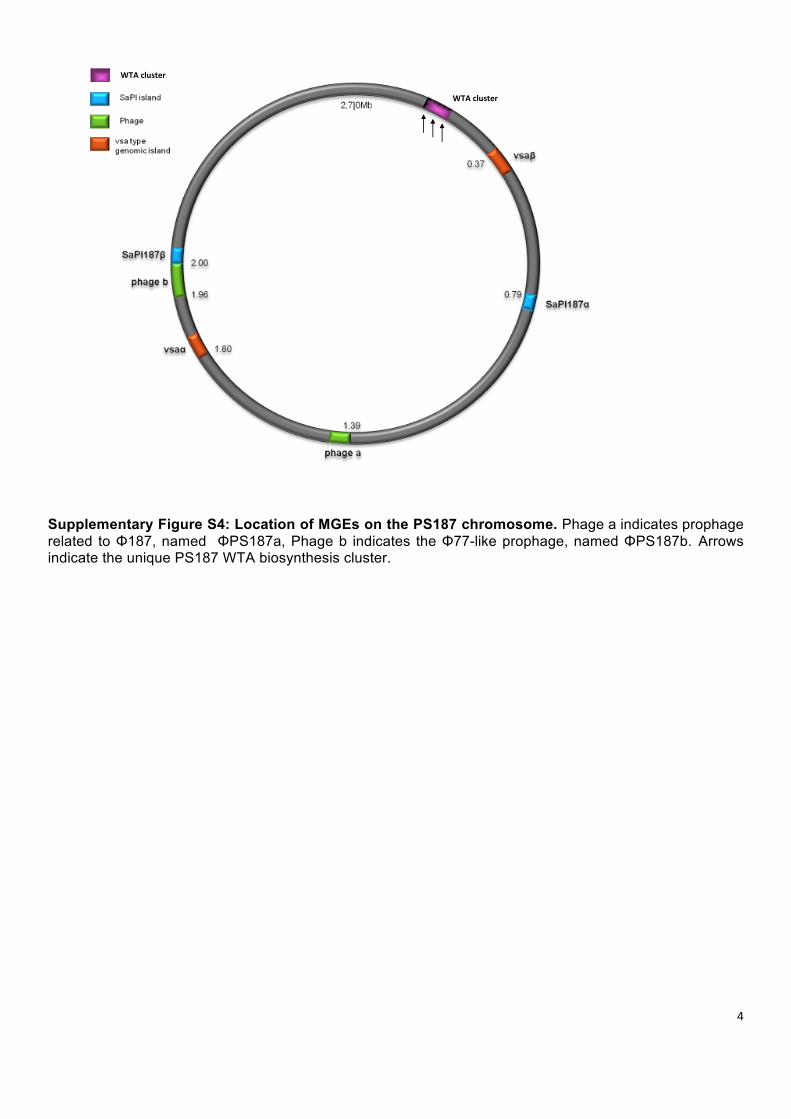
Supplementary Figure S4: Location of MGEs on the PS187 chromosome. Phage a indicates prophage related to Φ187, named ΦPS187a, Phage b indicates the Φ77-like prophage, named ΦPS187b. Arrows indicate the unique PS187 WTA biosynthesis cluster.

5
Supplementary Figure S5: Both prophages of PS187 are non-inducible. Both prophages found in the PS187 genome are cryptic and not inducible. Mitomycin C inducibility of PS187 prophages. Sterile filtered supernatants of mitomycin C-induced PS187 cultures were spotted on bacterial lawns and analyzed for plaque formation. One representative experiment is shown.

6

7
Supplementary Figure S6: ST395-specific WTA enzymes are related to proteins encoded by CoNS. Protein sequence alignment of (a) TagV with its closest homologue YP_006014466.1 from S. pseudintermedius ED99 (60.13% identity); (b) TagN with YP_002635271.1 from S. carnosus subsp. carnosus TM300 (60.96% identity); (c) TagF with ZP_09145406.1 from S. simiae CCM 7213 (67.77% identity); and (d) TagD with YP_003472398.1 from S. lugdunensis HKU09-01 (71.21% identity). For homology searches blastp was used and alignments were performed using ClustalW. Sequence identity (*), strong similarity (:), weak similarity (.) and difference ( ) are indicated.
8
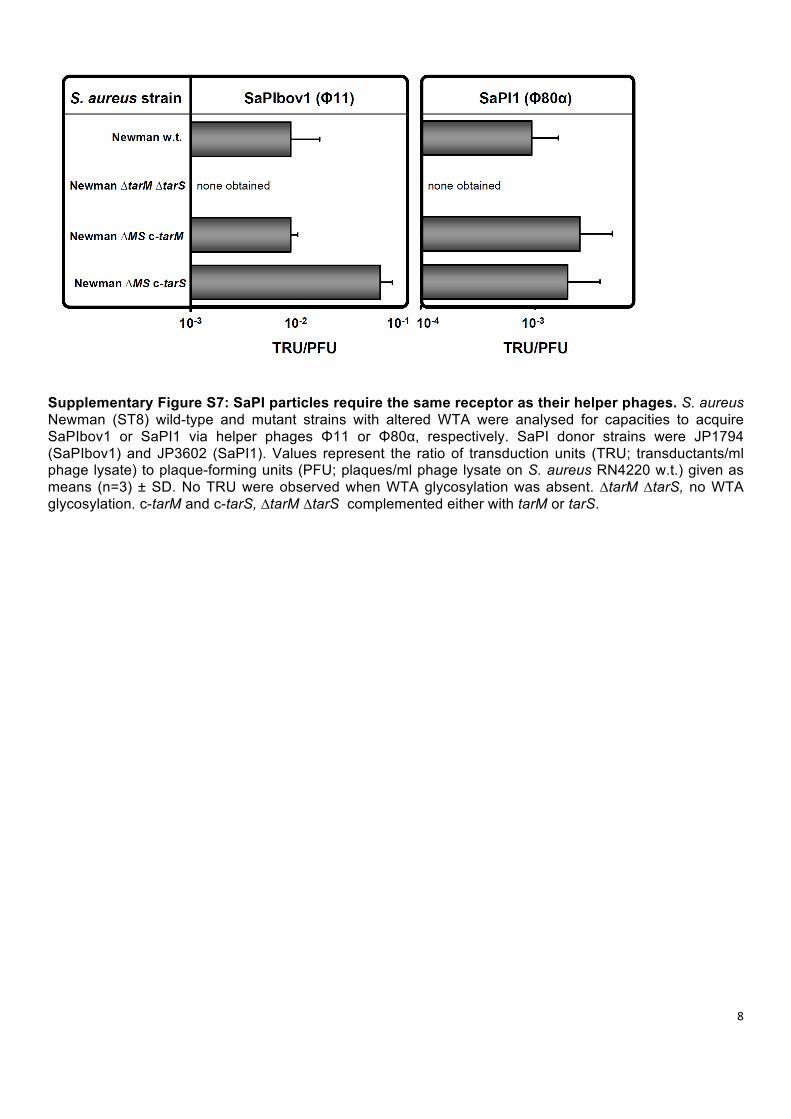
Supplementary Figure S7: SaPI particles require the same receptor as their helper phages. S. aureus Newman (ST8) wild-type and mutant strains with altered WTA were analysed for capacities to acquire SaPIbov1 or SaPI1 via helper phages Φ11 or Φ80α, respectively. SaPI donor strains were JP1794 (SaPIbov1) and JP3602 (SaPI1). Values represent the ratio of transduction units (TRU; transductants/ml phage lysate) to plaque-forming units (PFU; plaques/ml phage lysate on S. aureus RN4220 w.t.) given as means (n=3) ± SD. No TRU were observed when WTA glycosylation was absent. ∆tarM ∆tarS, no WTA glycosylation. c-tarM and c-tarS, ∆tarM ∆tarS complemented either with tarM or tarS.
9
a
b
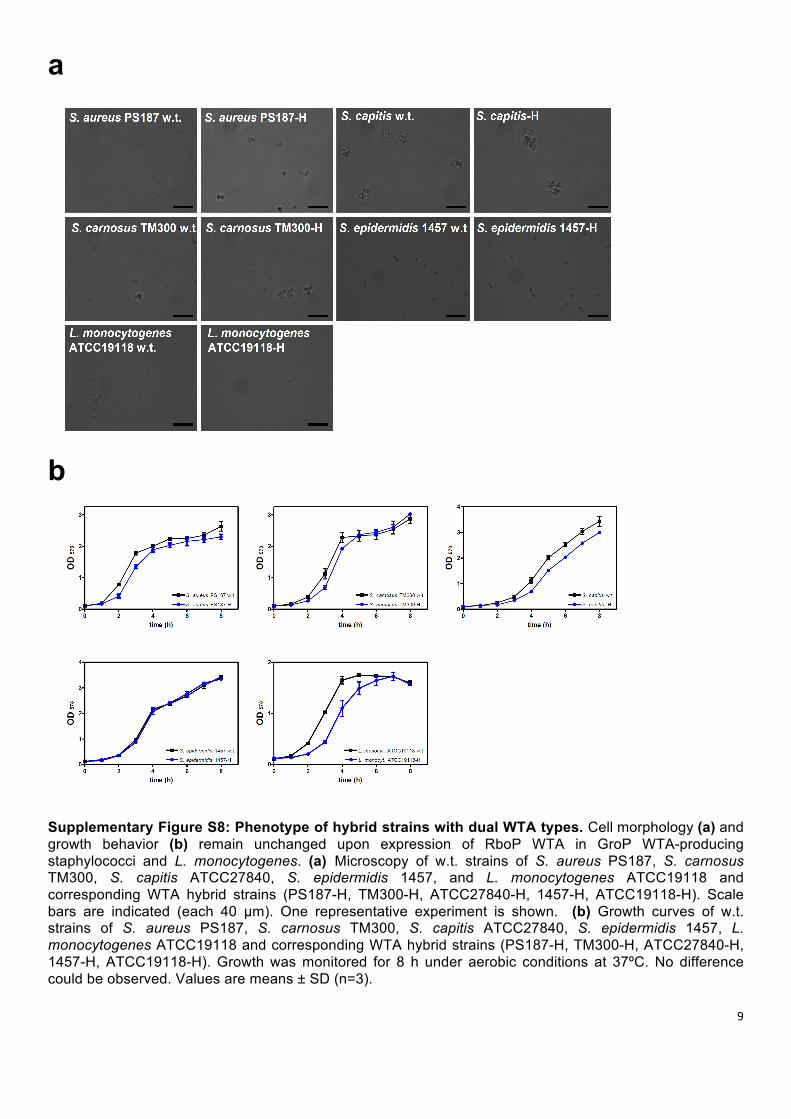
Supplementary Figure S8: Phenotype of hybrid strains with dual WTA types. Cell morphology (a) and growth behavior (b) remain unchanged upon expression of RboP WTA in GroP WTA-producing staphylococci and L. monocytogenes. (a) Microscopy of w.t. strains of S. aureus PS187, S. carnosus TM300, S. capitis ATCC27840, S. epidermidis 1457, and L. monocytogenes ATCC19118 and corresponding WTA hybrid strains (PS187-H, TM300-H, ATCC27840-H, 1457-H, ATCC19118-H). Scale bars are indicated (each 40 µm). One representative experiment is shown. (b) Growth curves of w.t. strains of S. aureus PS187, S. carnosus TM300, S. capitis ATCC27840, S. epidermidis 1457, L. monocytogenes ATCC19118 and corresponding WTA hybrid strains (PS187-H, TM300-H, ATCC27840-H, 1457-H, ATCC19118-H). Growth was monitored for 8 h under aerobic conditions at 37ºC. No difference could be observed. Values are means ± SD (n=3).
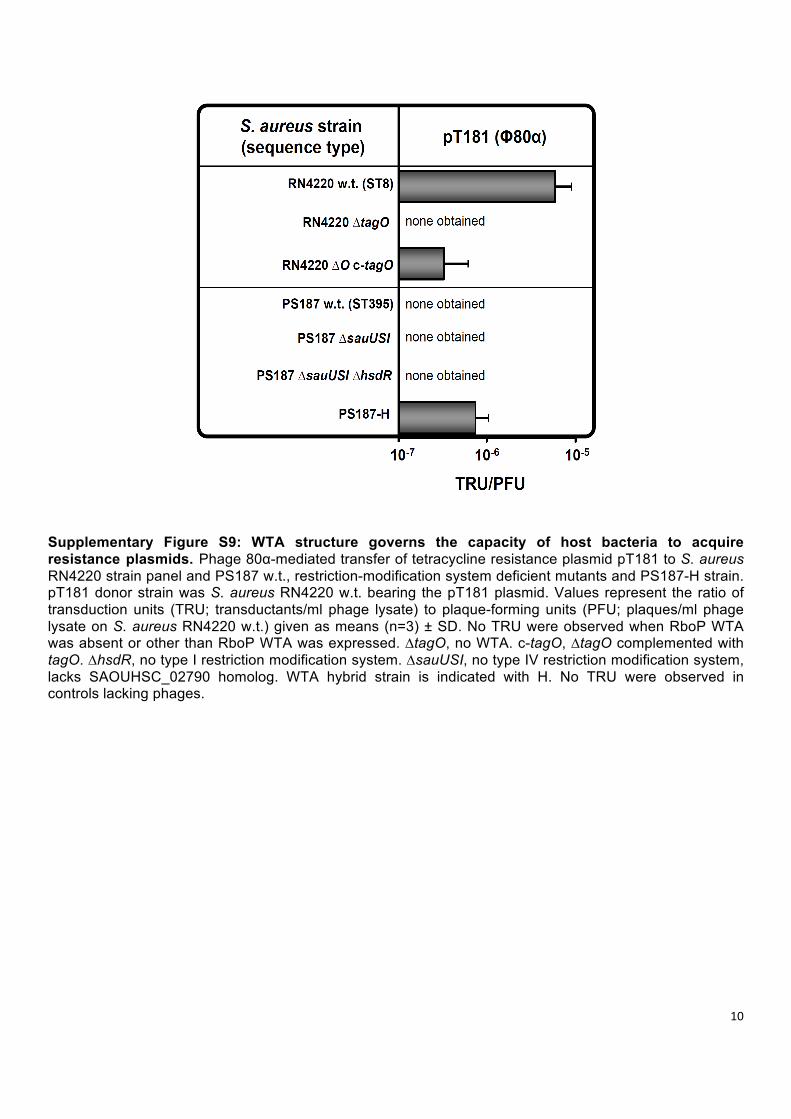
10
Supplementary Figure S9: WTA structure governs the capacity of host bacteria to acquire resistance plasmids. Phage 80α-mediated transfer of tetracycline resistance plasmid pT181 to S. aureus RN4220 strain panel and PS187 w.t., restriction-modification system deficient mutants and PS187-H strain. pT181 donor strain was S. aureus RN4220 w.t. bearing the pT181 plasmid. Values represent the ratio of transduction units (TRU; transductants/ml phage lysate) to plaque-forming units (PFU; plaques/ml phage lysate on S. aureus RN4220 w.t.) given as means (n=3) ± SD. No TRU were observed when RboP WTA was absent or other than RboP WTA was expressed. ∆tagO, no WTA. c-tagO, ∆tagO complemented with tagO. ∆hsdR, no type I restriction modification system. ∆sauUSI, no type IV restriction modification system, lacks SAOUHSC_02790 homolog. WTA hybrid strain is indicated with H. No TRU were observed in controls lacking phages.
11
Supplementary Tables
Supplementary Table S1: Strains used in this study Bacterial strain
Description
Reference
E. coli TOP10 One Shot® TOP10 chemically competent E.coli
Invitrogen
E. coli OmniMax One Shot® OmniMAX™ 2 T1 Phage-Resistant Cells (C8540-03)
Invitrogen
E. coli DC10B E. coli DH10B Δdcm 18 S. aureus RN4220 Wild type, deficient in restriction,
capsule, and prophage
44
S. aureus RN4220 ∆tagO RN4220 ∆tagO 8 S. aureus RN4220 ∆tagO pRB474-tagO
∆tagO complemented with tagO 8
S. aureus RN4220 ∆tarM ∆tarS
RN4220 ∆tarM ∆tarS
This study
S. aureus RN4220 ∆tarM ∆tarS pRB474-tarM
RN4220 ∆tarM ∆tarS complemented with tarM
This study
S. aureus RN4220 ∆tarM ∆tarS pRB474-tarS
RN4220 ∆tarM ∆tarS complemented with tarS
This study
S. carnosus TM300 Wild type 42 S. carnosus TM300-H pRB474-tarFI2J2L2S
S. carnosus TM300 hybrid expressing tarFI2J2L2S
This study
S. aureus PS187 Wild type, clinical ST395 isolate from elderly patient with post-influenzal pneumonia
ATCC collection, ID 15564, 21
S. aureus PS187 ∆tagO PS187 ∆tagO This study S. aureus PS187 ∆tagO pRB474-tagO
∆tagO complemented with tagO This study
S. aureus PS187-H pRB474-tarFI2J2L2S
PS187 hybrid expressing tarFI2J2L2S This study
S. aureus PS187-H VW1 pRB474-tarFI2J2L2S
PS187 hybrid expressing tarFI2J2L2S; transductant received after transduction with SaPIbovI particle
This study
S. aureus PS187∆sauUSI PS187 deficient in type IV restriction modification system
This study
S. aureus PS187∆sauUSI ∆hsdR PS187 deficient in type IV and type I restriction modification system
This study
S. aureus JP1794 RN451(SaPIbov1 tst::tetM) 20 S. aureus JP3602 RN10359(SaPI1 tst::tetM) 20 S. aureus Newman Wild type ATCC collection, ID 13420 S. aureus Newman ∆tarM ∆tarS
Newman ∆tarM ∆tarS
This study
S. aureus Newman ∆tarM ∆tarS pRB474-tarM
Newman ∆tarM ∆tarS complemented with tarM
This study
S. aureus Newman ∆tarM ∆tarS pRB474-tarS
Newman ∆tarM ∆tarS complemented with tarS
This study
S. aureus JS395 Wild type, clinical ST395 isolate, renamend as JS395
22
S. aureus NRS111 wildtype NARSA strain collection S. aureus NRS111-H pRB474-tarFI2J2L2S
NRS111 hybrid expressing tarFI2J2L2S This study
S. capitis ATCC27840 Wild type, human skin isolate ATCC collection, ID27840 S. capitits ATCC27840-H pRB474-tarFI2J2L2S
S. capitis ATCC27840 hybrid expressing tarFI2J2L2S
This study
S. aureus COL Wild type, HA-MRSA 45 S. aureus 4S5 RN4220 ∆spa ∆ltaS 8 S. epidermidis TÜ3298 Wild type Strain collection Peschel laboratory S. epidermidis 1457 Wild type 46 S. epidermidis 1457-H 1457 hybrid expressing tarFI2J2L2S This study S. aureus RN4220 Wild type, transformed with pT181 This study
12
pT181 plasmid E. faecalis V583 Wild type, vancomycin resistant Strain collection diagnostics Medical
Microbiology and Hygiene Department, University of Tuebingen
E. faecium VRE392 Wild type, vancomycin resistant Strain collection diagnostics Medical Microbiology and Hygiene Department, University of Tuebingen
S. aureus NRS71 Wild type, clonal complex 30 NARSA strain collection S. aureus NRS184 Wild type, clonal complex 22 NARSA strain collection S. aureus A860325 Wild type, clonal complex 25, NRS187 NARSA strain collection S. aureus NRS229 Wild type, clonal complex 1 NARSA strain collection S. aureus N315 Wild type, clonal complex 5 NARSA strain collection S. aureus USA300 lac w.t. Wild type, clonal complex 8 47 L. monocytogenes ATCC19118 Wild type, serotype 4e ATCC collection, ID19118 L. monocytogenes ATCC19118 pRB474-tarFI2J2L2S
ATCC19118 hybrid expressing tarFI2J2L2S
This study
L. grayi ATCC25401 Wild type ATCC collection, ID25401 S. aureus PS187 SaPI187beta::ermB VW7
SaPI187β ermB labeled PS187 w.t. strain
This study
S. aureus T132-1 Wild type, nasal isolate, ST395 23 S. aureus T148-2 Wild type, nasal isolate, ST395 23 S. aureus T191-1 Wild type, nasal isolate, ST395 23 S. aureus T110-1 Wild type, nasal isolate, ST395 23 S. aureus T166-1 Wild type, nasal isolate, ST395 23 S. aureus BK084 Wild type, blood culture isolate, ST395 23 S. aureus BK066 Wild type, blood culture isolate, ST395 23 Supplementary Table S2: 1H and 13C chemical shifts of WTA from S. aureus PS187 w.t. 1a 1b 2 3a 3b 4 5 6a 6b Gro 1a 1H 3.92 3.85 4.01 3.92 3.85
13C 66.03 69.46 66.03 Gro 2a 1H 3.96 3.93 3.85 3.96 3.93
13C 64.41 78.05 66.03 GalN 1H 5.04 4.17 3.93 4.00 4.22 3.73 3.73
13C 97.76 49.73 67.50 68.08 68.83 61.50 NAc: 2.04/22.45 Terminal Gro CH2OH: 3.68/59.23 a Gro1 indicates unglycosylated Gro polymer, Gro2 indicates GalNAc modified Gro polymer.
Supplementary Table S3: 1H and 13C chemical shifts of WTAs from S. aureus PS187-H. 1st WTAa 1a 1b 2 3a 3b 4 5 6a 6b Gro 1 1H 4.01 3.97 3.88 4.01 3.97
13C 65.69 79.26 65.69 GalN 1H 5.07 4.21 3.95 4.04 4.23 3.76 3.76
13C 98.81 51.16 68.85 69.39 71.15 62.36 NAc: 2.08/23.41 Terminal Gro CH2OH: 3.73/60.8 2nd WTAa 1a 1b 2 3 4 5a 5b 6a 6b Rbo 1H 4.08 4.08 3.89 3.91 4.14 4.14 4.14
13C 67.92 72.20 71.60 80.60 66.21 GlcN 1H 4.75 3.74 3.56 3.46 3.45 3.90 3.92
13C 102.39 56.80 75.10 71.20 77.00 61.98 NAc: 2.08/23.41 a Minor signals indicate the presence of unglycosylated Gro or Rbo polymer

13
Supplementary Table S4: Primers used in this study
Primer Sequence Application tagO-F1-up 5’ GGGGACAAGTTTGTACAAAAAAGCAGGC TGACAACTGAGAACTCTTCGCCACC 3’ tagO deletion in PS187
tagO-F1-dn 5’ ′GTCATCTAGACTTTATAGA ATG ATA CAT CTA TTT TCT ACG TAT ATC TTA TAG 3’ tagO deletion in PS187 tagO-F2-up 5′ GTCATCTAGAGCATAG CTG TAT GGG ATA ATT TGT ATT ATA TGG 3’ tagO deletion in PS187 tagO-F2-dn 5 ′GGGGACCAC TTTGTACAAGAAAGCTGG GTCGATGT TCA TAATGC GTG TGT CGT AG 3’ tagO deletion in PS187 N-up 5’ ATGAAA AAATTTCGAAAAGTAATTAAGAATC 3’ tagN typing N-dn 5’ TCA CAA TAA ATT ATA AAATTC AGA CAT AGC 3’ tagN typing Tar-up 5’ atc ggatcc ATCAAAAGCAATCACCTTCCAACTGAATTG 3’ tar cluster cloning
Tar-dn 5’ gaga GAATTC ATTGAGACGCTGTAATAATTCGTTCAAATCTA 3’ tar cluster cloning Type4-F1up 5’ GGGGACAAGT TTG TAC AAA AAA GCA GGC T ACCTTCTCGCTGTTCAATGTCAGATT 3’ sauUSI deletion in PS187 Type4-F1dn 5’ GTCA tctaga AAC ACC CTT ATT GCT TAA ATC TAA TAG CTG A 3’
sauUSI deletion in PS187
Type4-F2up 5’ GTCA tctaga AGAAGCCAAGAAATGCCAATATTTATCACT 3’
sauUSI deletion in PS187 Type4-F2dn 5’ GGGGACCACTTTGTACAAGAA AGCTGG GT ATCTCAGAATCAACAATCCAAGTGCCT 3’ sauUSI deletion in PS187 Type1-F1up 5’ GGGGACAAGTTTGTACAAAAAAGCAGGCTATCTAAATGAACTAATACGA AGGTGCTAAA 3’ hsdR deletion in PS187 Type1-F1dn 5’ GTCAAAGCTTCACTCTCGAAAATACTTTTCCCATCAATCAT 3’ hsdR deletion in PS187 Type1-F2up 5’ GTCA AAGCTTAGGCTCATGTGTTTGGCAATCATCACTT 3’
hsdR deletion in PS187
Type1-F2dn 5’ GGGGACCACTTTGTACAAGAAAGCTGGGTAGAAGATATTATCAAACATCCGCAATCAGA 3’
hsdR deletion in PS187 187bF1-up 5’ GGGG ACA AGTTTG TAC AAA AAA GCA GGC T ATGTCTTCTTCATCCGCATAGGTTAAA 3’ SaPI187β ermB labeling
187bF1-dn 5’ GTCA AAGCTTTATTTATTGCGCTCGCTTTTAATAGCCT 3’ SaPI187β ermB labeling 187bF2-up 5’ GTCA tctagaACCTTTAATTCCTAAGAAATAAGTAAGGAC 3’ SaPI187β ermB labeling 187bF2-dn 5’ GGGGACCACTTT GTA CAA GAA AGCTGGGT TTGTTGAAGTTTCTTGTTAAGTCTCTAACT 3’ SaPI187β ermB labeling Supplementary Table S5: Phages used in this study
Phage Description Reference ΦK Myoviridae, Serogroup D Gift from G. Bierbaum Φ187 Siphoviridae, Serogroup L Gift from G. Bierbaum Φ11 Siphoviridae, Serogroup B Gift from G. Bierbaum Φ80α Siphoviridae, Serogroup B Gift from C. Wolz Φ77 Siphoviridae, Serogroup F Gift from C. Wolz
14
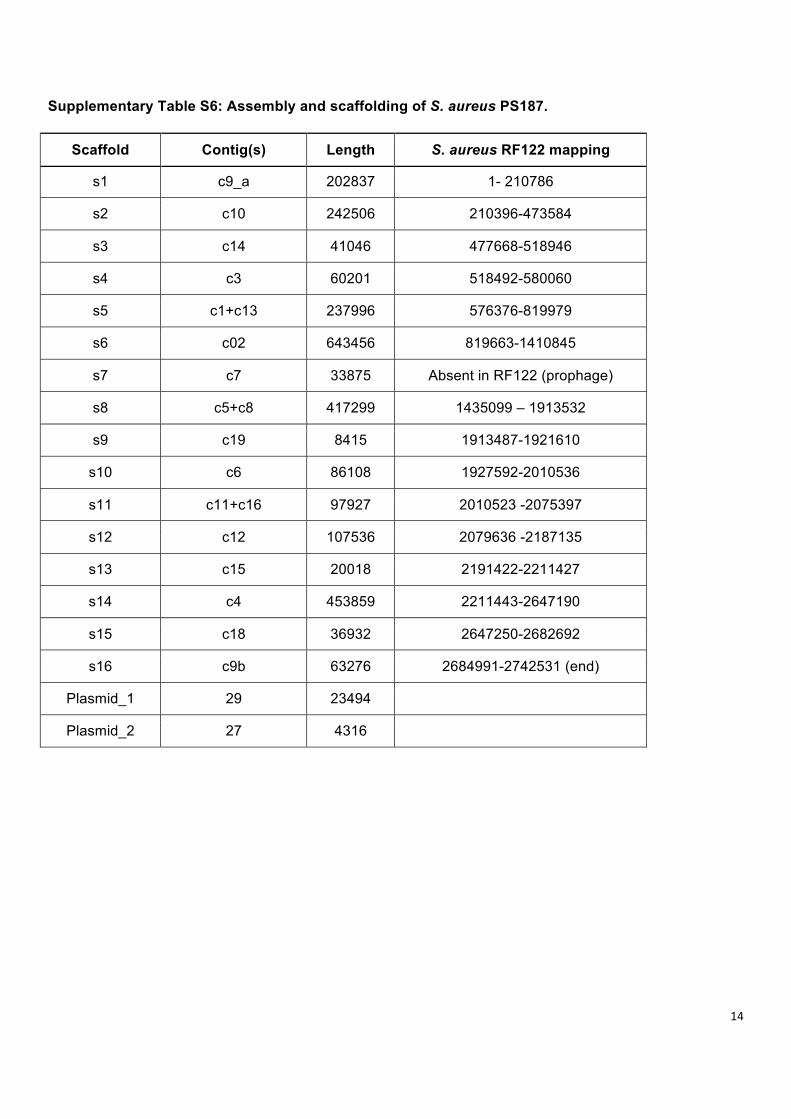
Supplementary Table S6: Assembly and scaffolding of S. aureus PS187.
Scaffold Contig(s) Length S. aureus RF122 mapping
s1 c9_a 202837 1- 210786
s2 c10 242506 210396-473584
s3 c14 41046 477668-518946
s4 c3 60201 518492-580060
s5 c1+c13 237996 576376-819979
s6 c02 643456 819663-1410845
s7 c7 33875 Absent in RF122 (prophage)
s8 c5+c8 417299 1435099 – 1913532
s9 c19 8415 1913487-1921610
s10 c6 86108 1927592-2010536
s11 c11+c16 97927 2010523 -2075397
s12 c12 107536 2079636 -2187135
s13 c15 20018 2191422-2211427
s14 c4 453859 2211443-2647190
s15 c18 36932 2647250-2682692
s16 c9b 63276 2684991-2742531 (end)
Plasmid_1 29 23494
Plasmid_2 27 4316
15
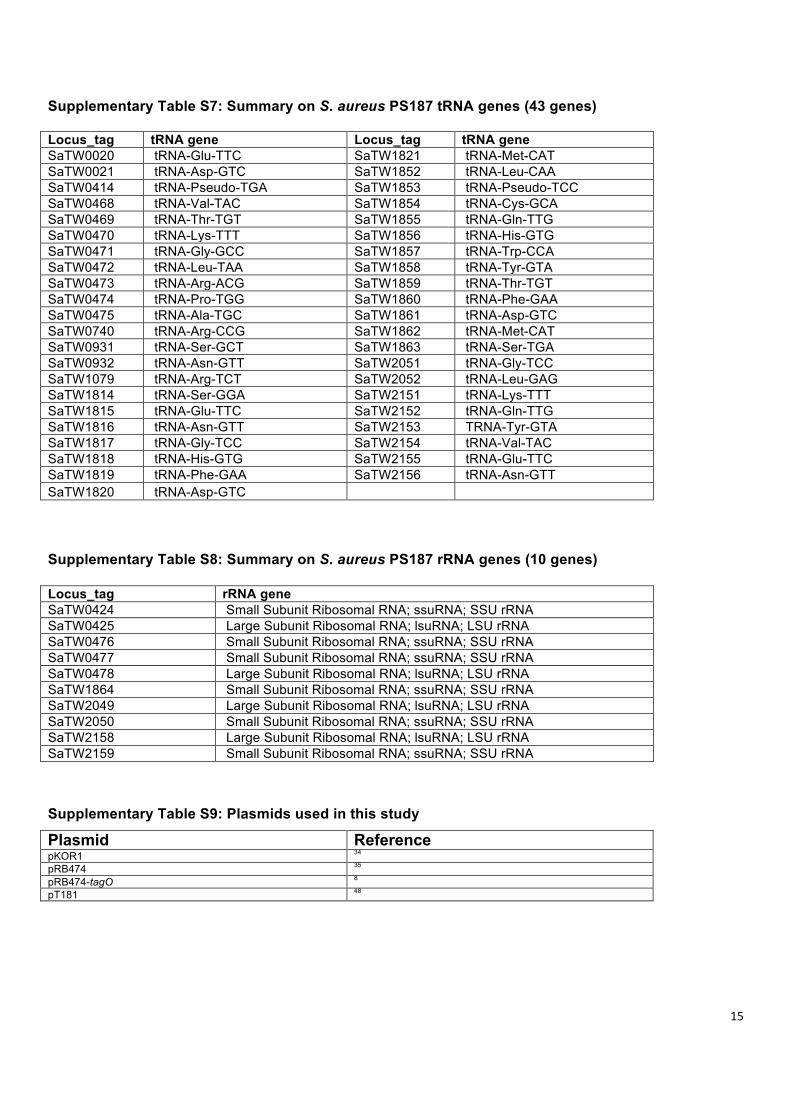
Supplementary Table S7: Summary on S. aureus PS187 tRNA genes (43 genes) Locus_tag tRNA gene Locus_tag tRNA gene SaTW0020 tRNA-Glu-TTC SaTW1821 tRNA-Met-CAT SaTW0021 tRNA-Asp-GTC SaTW1852 tRNA-Leu-CAA SaTW0414 tRNA-Pseudo-TGA SaTW1853 tRNA-Pseudo-TCC SaTW0468 tRNA-Val-TAC SaTW1854 tRNA-Cys-GCA SaTW0469 tRNA-Thr-TGT SaTW1855 tRNA-Gln-TTG SaTW0470 tRNA-Lys-TTT SaTW1856 tRNA-His-GTG SaTW0471 tRNA-Gly-GCC SaTW1857 tRNA-Trp-CCA SaTW0472 tRNA-Leu-TAA SaTW1858 tRNA-Tyr-GTA SaTW0473 tRNA-Arg-ACG SaTW1859 tRNA-Thr-TGT SaTW0474 tRNA-Pro-TGG SaTW1860 tRNA-Phe-GAA SaTW0475 tRNA-Ala-TGC SaTW1861 tRNA-Asp-GTC SaTW0740 tRNA-Arg-CCG SaTW1862 tRNA-Met-CAT SaTW0931 tRNA-Ser-GCT SaTW1863 tRNA-Ser-TGA SaTW0932 tRNA-Asn-GTT SaTW2051 tRNA-Gly-TCC SaTW1079 tRNA-Arg-TCT SaTW2052 tRNA-Leu-GAG SaTW1814 tRNA-Ser-GGA SaTW2151 tRNA-Lys-TTT SaTW1815 tRNA-Glu-TTC SaTW2152 tRNA-Gln-TTG SaTW1816 tRNA-Asn-GTT SaTW2153 TRNA-Tyr-GTA SaTW1817 tRNA-Gly-TCC SaTW2154 tRNA-Val-TAC SaTW1818 tRNA-His-GTG SaTW2155 tRNA-Glu-TTC SaTW1819 tRNA-Phe-GAA SaTW2156 tRNA-Asn-GTT SaTW1820 tRNA-Asp-GTC Supplementary Table S8: Summary on S. aureus PS187 rRNA genes (10 genes) Locus_tag rRNA gene SaTW0424 Small Subunit Ribosomal RNA; ssuRNA; SSU rRNA SaTW0425 Large Subunit Ribosomal RNA; lsuRNA; LSU rRNA SaTW0476 Small Subunit Ribosomal RNA; ssuRNA; SSU rRNA SaTW0477 Small Subunit Ribosomal RNA; ssuRNA; SSU rRNA SaTW0478 Large Subunit Ribosomal RNA; lsuRNA; LSU rRNA SaTW1864 Small Subunit Ribosomal RNA; ssuRNA; SSU rRNA SaTW2049 Large Subunit Ribosomal RNA; lsuRNA; LSU rRNA SaTW2050 Small Subunit Ribosomal RNA; ssuRNA; SSU rRNA SaTW2158 Large Subunit Ribosomal RNA; lsuRNA; LSU rRNA SaTW2159 Small Subunit Ribosomal RNA; ssuRNA; SSU rRNA
Supplementary Table S9: Plasmids used in this study
Plasmid Reference pKOR1 34 pRB474 35 pRB474-tagO 8 pT181 48

16
Supplementary Methods
General and analytical chemistry methods. The amino sugars composition of isolated WTA
components was determined as their amino alditols obtained after hydrolysis of the samples with 3.6 M HCl
at 100°C for 20 h, followed by acetylation using acetic anhydride and pyridine (1:1, v/v) at 85°C for 10 min -
prior to extraction of fatty acids with 10% (v/v), ether/hexane, carbonyl reduction with NaBH4 at 25°C in the
dark for 14 h, and further acetylation as described above. Detection followed by gas-liquid chromatography
(GLC). Further analysis of the samples was made by fractionation of the phosphodiester bonds using 48%
HF at 4°C for 14 h, acetylation of the resulting fragments as described above, and methylation using CH2N2
at 25°C for 5 min following detection by GLC and GLC-mass spectrometry (MS). The systems used were
an Agilent Technologies 7890A GC instrument equipped with a HP-5MS capillary column (30 m x 0.25 mm,
film thickness 0.25 µm) and applying a temperature gradient of 150 (kept for 3 min) - 260°C at 3°C/min for
GLC, or a Agilent Technologies 5975 using Inert XL Mass Selective Detector instrument equipped with the
same capillary column and applying a temperature gradient of 70°C (kept for 1.5 min) – 150°C at 60°C/min
– 150°C (kept for 3 min) – 320°C at 5°C/min for GLC-MS.
NMR spectroscopy. NMR experiments were carried in D2O at 300 K (333K for WTA from PS187-H
strain). All one-dimensional (1H and 13C) and two-dimensional homo- (COSY, TOCSY and ROESY) and
heteronuclear (HSQC-DEPT, HMBC and 1H,31P-HMQC-TOCSY) experiments were recorded with a Bruker
DRX Avance III 700 MHz spectrometer (operating frequencies of 700.75 MHz for 1H NMR, 176.2 MHz for
13C NMR, and 283.7 for 31P) using standard Bruker software. COSY, TOCSY and ROESY were recorded
using data sets (t1 by t2) of 4,096 by 512 points, and 12, 24, 4 and 4 scans were acquired for each t1 value
in the case of COSY for WTA samples from PS187 w.t. (sample V01) and PS187-H (sample V07)
respectively, while 16, 32, 8 and 8 scans were acquired in the case of TOCSY and ROESY for V01 and
V07 respectively. The TOCSY experiment was carried out in the phase-sensitive mode with mixing times of
100 ms for all samples. The 1H,13C correlations were measured in the 1H-detected mode via HSQC-DEPT
with proton decoupling in the 13C domain acquired using data sets of 4,096 by 512 points and 40, 56, 4 and
10 scans for each t1 value and for V01, and V07 respectively. The HMBC spectra were acquired using data
sets of 4,096 by 512 points, and 40, 56, 40 and 48 scans for each t1 value and for V01 and V07

17
respectively. The 1H,31P-HMQC-TOCSY experiment was recorded using a data set of 2,048 by 512 points
(16, 16, 2 and 4 scans for each t1 value and for V01 and V07 respectively using a mixing time of 100 ms for
all samples. Chemical shifts were reported relative to acetone (δH 2.225; δC 31.50).
Genome sequencing, annotation, and phylogenetic analyses. Sequence reads from Illumina
(raw reads were in average 51 bp in length) were analysed with FASTX toolkit (Hannon Lab, CSH), the
sequencing adapters were removed and low-quality reads were filtered out. These processed reads were
then de novo assembled with Velvet software49. During assembly different kmer values were approached
and best was determined by highest N50 value. Afterwards we splitted the harvested contigs into sequence
reads of 5,000 bp and each consecutive two sequences shared 2,000 bp as overlapping region. This
Illumina sequence library was then in silico considered as Sanger reads and assembled together with 454
reads by Mira software50. Alignment for scaffolding was performed with MUMmer51. The mapping result was
painted and studied using Circos52, Assembly led to a library of 516 contigs (N50 = 232, 175 bp), based on
these 16 scaffolds were selected and constructed for the genome and two putative plasmids. The resulting
assembly sequences were deposited in NCBI with accession number ARPA01000000 under the BioProject
PRJNA197438. Genome annotation was carried out using Maker 253 and the functional annotation was
accomplished by combining the results of Interproscan54 and RAST55. In total 2.714.790 base pairs were
harvested, which is 93.6% of average S. aureus genome size. The annotation effort reveals 2,529 ORFs
including 43 tRNA (41 regular tRNA, 2 Pseudo tRNA) gene and 10 rRNA genes (Supplementary Tables S6,
S7, S8 and Supplementary Fig. S4).
For phylogenetic analyses DNA sequence alignment was generated from staphylococcal genome
sequences by employing a series of shell scripts as follows. Protein-coding DNA sequences were retrieved
from the genome sequence from isolate N315 (sequence accession number BA000018). At this step, we
excluded genes annotated in the N315 genome as parts of mobile genetic elements including SCCmec,
prophage, pathogenicity islands, transposons, and insertion sequences. Then, by running locally installed
BLASTN56, orthologous genes in each of 15 other genomes (S. aureus isolate Mu50 -- sequence accession
number BA000017; 04-02981 -- CP001844; JH1 -- CP000736; NCTC8325 -- CP000253; FPR3757 --
CP000255; COL -- CP000046; MSSA476 -- BX571857; MW2 -- BA000033; HO-5096-0412 -- HE681097;
RF122 -- AJ938182; ED133 -- CP001996; MRSA252 -- BX571857; S0385 -- AM990992; MSHR1132 --

18
FR821777; S. epidermidis isolate RP62A -- CP000029) were identified on the basis of sequence similarity.
Genes, which either were shorter than 90% of the queried genes from N315, or which yielded more than
one hit with ≥90% length in any single genome (i. e. duplicate genes), or which were not found in each of
the investigated genomes, were excluded from further analyses. In contrast, sequences from orthologous
genes (n=1,147) present in each of the genomes were retrieved and concatenated resulting in contiguous
sequences of 1,095,318 to 1,101,819 base pairs from each of 16 genomes. By using SMALT 0.6.2
(available at http://www.sanger.ac.uk/resources/software/ smalt/), Illumina sequencing reads from isolate
PS187 (ST395) were mapped against concatenated ortholog sequences from three alternative references
(i. e., N315, COL, and S0385) to avoid any bias that otherwise potentially could have resulted from the
choice of reference. This procedure resulted in the discovery of high-quality polymorphisms, which were
then introduced to the respective reference sequences to construct three alternative sequences for PS187.
A total of 19 concatenated ortholog sequences were aligned by using MAFFT v6.814b57 implemented as a
plugin in Geneious 5.5.7 (www.biomatters.com) and subjected to phylogenetic analyses. We constructed a
maximum likelihood tree by using PhyML 3.0.158 as implemented in the Seaview software package59, with
the HKY85 nucleotide substitution model and the approximated likelihood ratio test for estimating branch
support. In addition, we used the software package BEAST v1.6.160 to calculate a Bayesian maximum
clade credibility tree, with the HKY85 nucleotide substitution model, a strict molecular clock, and otherwise
default parameters in BEAUTi v1.6.1. An MCMC chain length of 107 iterations resulted in effective sample
size values greater than 250. Posterior clade probabilities were used to judge branch support.

19
Supplementary references
44 Kreiswirth, B. N. et al. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305, 709-‐712 (1983).
45 Shafer, W. M. & Iandolo, J. J. Genetics of staphylococcal enterotoxin B in methicillin-‐resistant isolates of Staphylococcus aureus. Infection and immunity 25, 902-‐911 (1979).
46 Mack, D., Siemssen, N. & Laufs, R. Parallel induction by glucose of adherence and a polysaccharide antigen specific for plastic-‐adherent Staphylococcus epidermidis: evidence for functional relation to intercellular adhesion. Infection and immunity 60, 2048-‐2057 (1992).
47 Diep, B. A. et al. Complete genome sequence of USA300, an epidemic clone of community-‐acquired meticillin-‐resistant Staphylococcus aureus. Lancet 367, 731-‐739, doi:10.1016/S0140-‐6736(06)68231-‐7 (2006).
48 Khan, S. A. & Novick, R. P. Complete nucleotide sequence of pT181, a tetracycline-‐resistance plasmid from Staphylococcus aureus. Plasmid 10, 251-‐259 (1983).
49 Zerbino, D. R. & Birney, E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome research 18, 821-‐829, doi:10.1101/gr.074492.107 (2008).
50 Chevreux B., W. T., Suhai S. Genome Sequence Assembly Using Trace Signals and Additional Sequence Information. Proceedings of the German Conference on Bioinformatics (GCB) 99, 45-‐56 (1999).
51 Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome biology 5, R12, doi:10.1186/gb-‐2004-‐5-‐2-‐r12 (2004).
52 Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome research 19, 1639-‐1645, doi:10.1101/gr.092759.109 (2009).
53 Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-‐database management tool for second-‐generation genome projects. BMC bioinformatics 12, 491, doi:10.1186/1471-‐2105-‐12-‐491 (2011).
54 Zdobnov, E. M. & Apweiler, R. InterProScan-‐-‐an integration platform for the signature-‐recognition methods in InterPro. Bioinformatics 17, 847-‐848 (2001).
55 Aziz, R. K. et al. The RAST Server: rapid annotations using subsystems technology. BMC genomics 9, 75, doi:10.1186/1471-‐2164-‐9-‐75 (2008).
56 Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. Journal of molecular biology 215, 403-‐410, doi:10.1016/S0022-‐2836(05)80360-‐2 (1990).
57 Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic acids research 30, 3059-‐3066 (2002).
58 Guindon, S. et al. New algorithms and methods to estimate maximum-‐likelihood phylogenies: assessing the performance of PhyML 3.0. Systematic biology 59, 307-‐321, doi:10.1093/sysbio/syq010 (2010).
59 Gouy, M., Guindon, S. & Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Molecular biology and evolution 27, 221-‐224, doi:10.1093/molbev/msp259 (2010).
60 Drummond, A. J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC evolutionary biology 7, 214, doi:10.1186/1471-‐2148-‐7-‐214 (2007).
Top Related
Copyright © 2022 FDOKUMEN

