






Bahasa
Halaman
Hukum


UCP1 and Defense against Oxidative Stress4-HYDROXY-2-NONENAL EFFECTS ON BROWN FAT MITOCHONDRIA ARE UNCOUPLINGPROTEIN 1-INDEPENDENT *
Received for publication, February 13, 2006 Published, JBC Papers in Press, March 16, 2006, DOI 10.1074/jbc.M601387200
Irina G. Shabalina, Natasa Petrovic, Tatiana V. Kramarova, Joris Hoeks1, Barbara Cannon, and Jan Nedergaard2
From the Wenner-Gren Institute, The Arrhenius Laboratories F3, Stockholm University, SE-106 91 Stockholm, Sweden
Uncoupling proteins have been ascribed a role in defense againstoxidative stress, particularly by being activated by products of oxi-dative stress such as 4-hydroxy-2-nonenal (HNE). We have investi-gated here the ability of HNE to activate UCP1. Using brown fatmitochondria from UCP1�/� and UCP1�/� mice to allow for iden-tification ofUCP1-dependent effects, we found thatHNE could nei-ther (re)activate purine nucleotide-inhibited UCP1, nor induceadditional activation of innately active UCP1. The aldehyde non-enal had a (re)activating effect only if converted to the correspond-ing fatty acid by aldehyde dehydrogenase; the presence of a carboxylgroupwasthusanabsoluterequirementfor(re)activation.TheUCP1-dependent proton leakwas not increased byHNEbutHNE changedbasal proton leak characteristics in a UCP1-independent manner.In agreement with the in vitro results, we found, as compared withUCP1�/� mice, no increase in HNE/protein adducts in brown fatmitochondria isolated fromUCP1�/� mice, irrespective of whetherthey were adapted to thermoneutral temperature (30 °C) or to thecold (4 °C). The absence of oxidative damage in UCP1�/� mito-chondria was not due to enhanced activity of antioxidant enzymes.Thus, HNE did not affect UCP1 activity, and UCP1 would appearnot to be physiologically involved in defense against oxidativestress. Additionally, it was concluded that at least in brown adiposetissue, conditions of high mitochondrial membrane potential, highoxygen tension, and high substrate supply do not necessarily lead toincreased oxidative damage.
A generally accepted view of the molecular mechanism of UCP1function and regulation has not been achieved (reviewed in Refs. 1–4).Roles for purine nucleotides as inhibitors and for fatty acids as (re)acti-vators are generally accepted, but the issue has been raised as towhetheralternative physiological activators exist and/or whether certain cofac-tors are necessary for UCP1 activity. These discussions may also berelevant concerning the phylogenetically related mitochondrial mem-brane proteins UCP2 and UCP3 (for review, see Refs. 5–8).Particularly, to explain the lack of constitutive uncoupling activity of
uncoupling proteins reconstituted in liposomes, or the lack of basaluncoupling (inhibitable by GDP) in skeletal muscle mitochondriaexpressing UCP3 (9), it has been suggested that non-fatty acid cofactorsare required for activation, ubiquinone (10) and superoxide (11) beingthose originally suggested. Although the necessity for additional activa-
tors has been questioned in reconstituted systems (12), in isolatedmito-chondria (13) and in animals (14), several other cofactor candidateshave been discussed. A series of compounds that are formed down-stream of superoxide interaction with the mitochondrial membranehave been proposed to be the true activators of UCPs (or to be directlyinvolved in the uncoupling mechanism). These include carbon-cen-tered radicals (15), hydroperoxy fatty acids (16, 17), and lipid peroxida-tion products, e.g. 4-hydroxy-2-nonenal (HNE)3 (3, 6, 24). The questionof a cofactor necessity for UCP function has thus been linked to theearlier suggestion that UCP activity can control the formation of reac-tive oxygen species (ROS) (18). A much discussed (8) scheme has beenproposed where conditions leading to increased oxygen stress lead tosuperoxide production, which results in the formation of compoundssuch asHNE that could activateUCPs. This should lower themitochon-drial membrane potential and, consequently, diminish the risk of oxi-dative damage (3, 6).Due to the broad implications of this hypothesis for pathological
processes, we have here studied the proposed interaction betweenHNE and the originally identified uncoupling protein UCP1. Thechoice of UCP1 was made because it is uncontroversial that UCP1functions as an uncoupling protein; the proton leak associated withUCP1 is thus easily observable. We have used the availability ofUCP1-ablated mice (19) to enable us to dissociate the UCP1-depend-ent effects of HNE fromUCP1-independent effects, whichmay occur inthe mitochondria.We conclude that the reported HNE effects on brown fat mitochon-
dria are UCP1-independent. In vivo studies confirm that no protectiveeffect of UCP1 can be identified physiologically. Our results may havesignificance for the understanding of the regulation not only of UCP1but also of the other uncoupling proteins andmay also contribute to theidentification of physiological conditions associated with the increasedrisk of oxidative damage.
EXPERIMENTAL PROCEDURES
Animals—UCP1-ablatedmice (progeny of those described in Ref. 19)were backcrossed to C57Bl/6 mice for 10 generations and after inter-crossing were maintained as UCP1�/� and UCP1�/� strains. The micewere fed ad libitum (R70 Standard Diet, Lactamin), had free access towater, and were kept on a 12:12-h light:dark cycle, routinely at normal(24 °C) animal house temperature. Adult (8–12-week-old) male micewere routinely used for the experiments.For experiments onwarm-acclimated animals, UCP1�/� andUCP1�/�
adult male mice were divided into age- (7–8-week-old) and body weight(23–24 g)-matched groups, one per cage, and acclimated at 30 °C (i.e.* This work was supported by the Swedish Research Council and the EU program
“Dlarfid” (Dietary Lipid as Risk Factor in Development). The costs of publication of thisarticle were defrayed in part by the payment of page charges. This article must there-fore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734solely to indicate this fact.
1 Visiting student from the Nutrition and Toxicology Research Institute Maastricht, Maas-tricht University, The Netherlands.
2 To whom correspondence should be addressed. Tel.: 46-8-164128; Fax: 46-8-156756;E-mail: [email protected].
3 The abbreviations used are: HNE, 4-hydroxy-2-nonenal; UCP, uncoupling protein;ROS, reactive oxygen species; BSA, bovine serum albumin; DHE, dihydroethidium;SOD, superoxide dismutase; MDA, malonyl dialdehyde; Tes, 2-{[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]amino}ethanesulfonic acid; FCCP, carbonyl cyanide p-tri-fluoromethoxyphenylhydrazone.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 281, NO. 20, pp. 13882–13893, May 19, 2006© 2006 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A.
13882 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 20 • MAY 19, 2006
at Stockholm
Universitetsbibliotek on D
ecember 7, 2006
ww
w.jbc.org
Dow
nloaded from

thermoneutral temperature for both wild-type and UCP1-ablatedmice)4 for at least 1month before the start of the experiment. For exper-iments on cold-acclimated animals, UCP1�/� and UCP1�/� adult malemice were similarly either acclimated (one per cage) at 24 °C or succes-sively acclimated to cold by first placing them at 18 °C for 4 weeks withthe following 4 weeks at 4 °C (the intermediate 18 °C step was requiredto allow for survival of the UCP1�/� animals at 4 °C (20)). The experi-ments were approved by the Animal Ethics Committee of the NorthStockholm region.
Mitochondrial Preparation—Brown fat mitochondria were preparedprincipally as described (21) with some modifications (22). Mitochon-drial protein concentration was measured using the fluorescaminemethod (23), and the suspensions were diluted to stock concentrationsof 25 mg of mitochondrial protein/ml of 125 mM sucrose with 0.2% (1%in experiments with warm-acclimated mice) fatty acid-free bovineserum albumin (BSA).
Oxygen Consumption—Isolated mitochondria, at final concentra-tions of 0.5 or 0.3 mg (indicated in figure legends) of mitochondrialprotein/ml, were added to 1.1 ml of a continuously stirred incubationmedium consisting of 125 mM sucrose, 20 mM K�-Tes (pH 7.2), 2 mM
MgCl2, 1 mM EDTA, 4 mM potassium Pi, and 1.3 �g of oligomycin/ml.The substrates were 5 mM pyruvate plus 3 mM malate or 5 mM glycerol3-phosphate in the presence of 2 �g of rotenone/ml. UCP1�/� andUCP1�/� brown fatmitochondria exhibit identical oxidative capacities,estimated as maximal rates of FCCP-stimulated respiration (22). Thefinal concentration of fatty acid-free BSAwas adjusted to 0.1% (w/v) foroxygen consumptionmeasurements performed inmitochondria respir-ing on pyruvate plus malate. This was increased to 1% (w/v) in experi-ments where glycerol 3-phosphate was used as substrate (as indicated)(i.e. similarly to Ref. 24). Oxygen consumption rates were monitoredwith a Clark-type oxygen electrode (Yellow Springs Instrument Co.) ina sealed chamber at 37 °C, as described (22). Data for GDP concentra-tion-response curves were analyzed with the general fit option of theKaleidaGraph application for Macintosh for adherence to simpleMichaelis-Menten kinetics,V(x)�Vmax � �Vmax � (x/(Km � x)), wherex is the concentration of GDP.
Mitochondrial Membrane Potential—Measurements were per-formed in brown fat mitochondria with the dye safranin O (25). Mito-chondria were incubated with 2 �g/ml oligomycin, 5 �M safranin, 0.1%(w/v) fatty acid-free BSA, and 5 mM glycerol 3-phosphate in the pres-ence of 2 �g of rotenone/ml. The changes in absorbance of safranin Owere followed at 37 °C in an Aminco DW-2 dual-wavelength spectro-photometer at 511–533 nm with a 3-nm slit. Signals were recordedevery 0.5 s via a PowerLab/ADInstrument. The data were stored andanalyzed using theChart version 5.1.1 program.Calibration curvesweremade for each mitochondrial preparation in K�-free medium and wereobtained from traces in which the extramitochondrial K�, [K�]out, wasaltered by addition of KCl in a 0.1–20mM final concentration range. Thechange in absorbance then caused by the addition of 3 �M valinomycinwas plotted against [K�]out. The intramitochondrial K�, [K�]in, wasestimated by extrapolation of the line to the zero uptake point, asdescribed in Ref. 26. The absorbance readings were used to calculate themembrane potential (mV) by the Nernst equation according to: ��m �61 mV�log ([K�]in/[K�]out). To determine the basal proton leak, mito-chondrial membrane potential and oxygen consumption measure-ments were performed in parallel using the samemedia and conditions,but in the presence of increasing amounts of antimycin A, as indicated.
Western Blotting—ForHNE-adduct detection, aliquots of freshly iso-lated mitochondrial suspension were stored under nitrogen at �80 °Cafter supplementation with protease inhibitormixture (CompleteMini,Roche). Protein concentrations of the thawed mitochondrial sampleswere requantified using the Lowry method. Equal amounts of mito-chondrial protein were loaded on SDS-polyacrylamide gel. After elec-trophoresis, proteins were transferred by electroblotting to a polyvinyli-dene difluoride membrane. HNE protein adducts were detected withpolyclonal antibodies fromAlpha Diagnostics (HNE12-S, dilution 1:1000).After incubation with horseradish peroxidase-conjugated secondaryantibodies, the membrane was incubated with detection reagent (ECL,Amersham Biosciences) and the chemiluminescence signal was de-tected with a CCD camera (Fuji). Quantifications were performed withthe Image Gauge 3 software.For UCP1 determination, the membrane used for detection of the
HNE adductswas stripped and blottedwithUCP1 polyclonal antibodies(prepared in rabbit from the C-terminal decapeptide of mouse UCP1),dilution 1:3000. For cytochrome oxidase subunit 1 determination, themembrane used for detection of HNE adducts and UCP1 was blotted,after stripping, with cytochrome oxidase subunit 1-monoclonal anti-bodies (Molecular Probes), diluted 1:2000.
Superoxide Measurement—Net superoxide release rates were assesseddirectly in isolated brown fatmitochondria by fluorescencemeasurementswith thedyedihydroethidium(DHE) (MolecularProbes), theconversionofwhich to ethidium is superoxide-induced (14, 27). The fluorescence emit-ted by the ethidium formed was followed on a spectrophotometer (SigmaZES II) at 37 °Cusing anexcitationwavelengthof 495nmandcollecting theemission via a cutoff filter at 580 nm. The data were acquired, stored, andanalyzed using the Chart 4.1.1 program (PowerLab/ADInstrument).Chemical and biological validations of thismethodwere reported in Ref.14: generation of superoxide by the xanthine plus xanthine oxidase sys-tem or by mitochondria in the presence of DHE resulted in a significantincrease in ethidium-emitted fluorescence, which was blocked by addi-tion of recombinant SOD (14). When estimated by this method, super-oxide generation was diminished in brown fat mitochondria fromhSOD2� mice (i.e. mitochondrial superoxide dismutase-overexpress-ing mice) as compared with wild-type mice (14). The assay thusdetected mitochondrial superoxide release.
Aconitase and Citrate Synthase Activities—Aconitase activity wasmeasured spectrophotometrically as NADPH formation, monitored at340 nm using a coupled assay (28). The frozen mitochondrial samples(the same as those used for Western blotting) were rapidly thawedimmediately prior to assay, and 2 �l of 10 times diluted sample wereadded to 500�l of assay buffer (50mMTris-HCl, pH 7.4, 0.6mMMnCl2,5 mM sodium citrate, 0.2 mM NADP�, 0.1% (v/v) Triton X-100, and 0.4units/ml isocitrate dehydrogenase (Sigma)) pre-equilibrated to 30 °C.Each sample was assayed in duplicate; readings were taken at 15-s inter-vals over 7 min, and the resulting linear slopes were averaged to give ameasurement of aconitase activity for that sample.Superoxide inactivates the Krebs cycle enzyme aconitase, whereas
citrate synthase, another Krebs cycle enzyme, is insensitive to superox-ide; therefore, the aconitase/citrate synthase ratio is a convenient meas-ure of oxidative damage in mitochondria (28). Citrate synthase activitywas determined as in Ref. 29. To validate the assay and to induce exten-sive oxidative damage, brown fat mitochondria were also exposed to asuperoxide-generating system (370 �M xanthine plus 23 �g/ml xan-thine oxidase) for 15 min at 30 °C. Confirmation that this system pro-vides a high level of superoxide was obtained in Ref. 14.
Lipid Peroxidation—Interscapular brown adipose tissue was rapidlydissected fromUCP1�/� andUCP1�/�mice and immediately frozen in4 H. Feldmann, B. Cannon, and J. Nedergaard, unpublished observations.
UCP1 and Defense against Oxidative Stress
MAY 19, 2006 • VOLUME 281 • NUMBER 20 JOURNAL OF BIOLOGICAL CHEMISTRY 13883
at Stockholm
Universitetsbibliotek on D
ecember 7, 2006
ww
w.jbc.org
Dow
nloaded from
liquid nitrogen. The wet weight of brown adipose tissue was measuredand the left lobe of tissue was used for the lipid peroxidation assay andthe right lobe for the determination of antioxidant enzyme activities.The amount of peroxidative reactants was estimated as the forma-tion of thiobarbituric acid-reactive substances (mainly the lipid peroxi-dation product, malonyl dialdehyde (MDA) (30)). Brown adipose tissuehomogenates were prepared in 50 mM Tris-HCl buffer (pH 7.4) in aratio of 1:50 (w/v). Basal MDA levels were measured in freshly homog-enized samples. Inducible peroxidative reactions were stimulated byaddition of 312.5 �M ascorbic acid and 6.25 �M FeSO4 (final concentra-tions) and incubation at 37 °C for 1 h. Spontaneous peroxidative reac-tions were analyzed in samples incubated in parallel without addition ofascorbic acid and FeSO4.
Antioxidant Enzyme Assays—The right lobe of interscapular brownadipose tissue was homogenized in buffer containing 250 mM sucrose,50 mM Tris-HCl, and 1 mM EDTA (pH 7.2), in a 1:10 ratio (w/v) andsonicated three times at 100 watts for 20 s with 10-s pauses in a Bronsonmodel B-12 sonicator. The samples were then centrifuged at 20,000� gin an Eppendorf centrifuge for 60 min. Supernatants were used fordetermination of manganese-containingmitochondrial superoxide dis-mutase 2 (Mn-SOD), CuZn-containing cytosolic superoxide dismutase1 (CuZn-SOD), and catalase.SOD activity was measured by the adrenaline method (31). 100 �l
of acidified adrenaline solution was added to 3 ml of alkaline carbon-ate buffer (50 mM Na2CO3, 0.1 mM EDTA, pH 10.2), and absorbanceof adrenochrome at 480 nm was monitored for 4 min. The decrease
in the rate of change of the absorbance caused by the samples wasfollowed. One unit of SOD was defined as the amount of enzymereducing the rate of autoxidation of adrenaline by 50%. Mn-SOD-specific activity was obtained by inhibiting CuZn-SOD for 20 min atroom temperature with 4 mM KCN (final concentration) before thedismutase assay. CuZn-SOD activity was calculated by subtractingMn-SOD activity from the total SOD activity. Catalase activity wasmeasured spectrophotometrically by a method (32) based on the rateof hydrogen peroxide degradation by the catalase contained in theexamined samples.
Chemicals—Fatty acid-free bovine serum albumin, fraction V, wasfrom Roche Diagnostics GmbH (Germany). HNE was from CaymanChemical (AnnArbor,MI).Nonanoic acid and fluorescamine (4-phenylspiro-[furan-2(3H),1-phthalan]-3,3�-dione) were from Fluka ChemieGmbh. 2-Nonenoic acid was from CHEMOS Gmbh (Regenstauf).Other chemicals were all from Sigma. GDP was dissolved in 20 mM Tes(pH 7.2) and the pH of the solution readjusted. FCCP was dissolved in95% ethanol and diluted in 50% ethanol; oligomycin and rotenone weredissolved in 95% ethanol. Nonanoic acid, 2-nonenoic acid, 2-nonenal,all-trans-retinal, and HNE were dissolved in 95% ethanol, divided intosmall aliquots, and stored under nitrogen at �80 °C. Ethanol in a finalconcentration of 0.1%did not in itself have any effects on the parametersmeasured.
Statistics—All data are expressed as mean � S.E. Statistical analysisfor the comparison of two groups was performed using Student’s t test.
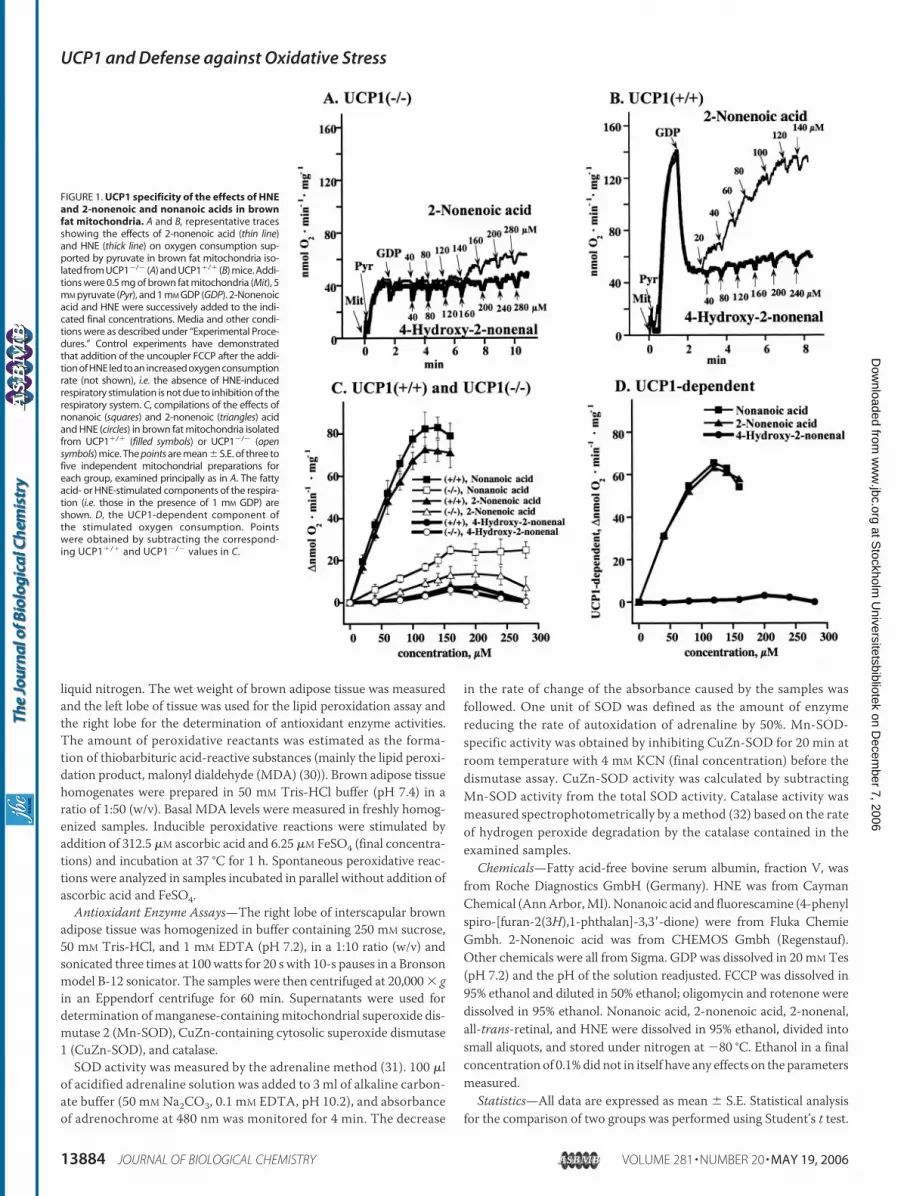
FIGURE 1. UCP1 specificity of the effects of HNEand 2-nonenoic and nonanoic acids in brownfat mitochondria. A and B, representative tracesshowing the effects of 2-nonenoic acid (thin line)and HNE (thick line) on oxygen consumption sup-ported by pyruvate in brown fat mitochondria iso-lated from UCP1�/� (A) and UCP1�/� (B) mice. Addi-tions were 0.5 mg of brown fat mitochondria (Mit), 5mM pyruvate (Pyr), and 1 mM GDP (GDP). 2-Nonenoicacid and HNE were successively added to the indi-cated final concentrations. Media and other condi-tions were as described under “Experimental Proce-dures.” Control experiments have demonstratedthat addition of the uncoupler FCCP after the addi-tion of HNE led to an increased oxygen consumptionrate (not shown), i.e. the absence of HNE-inducedrespiratory stimulation is not due to inhibition of therespiratory system. C, compilations of the effects ofnonanoic (squares) and 2-nonenoic (triangles) acidand HNE (circles) in brown fat mitochondria isolatedfrom UCP1�/� (filled symbols) or UCP1�/� (opensymbols) mice. The points are mean � S.E. of three tofive independent mitochondrial preparations foreach group, examined principally as in A. The fattyacid- or HNE-stimulated components of the respira-tion (i.e. those in the presence of 1 mM GDP) areshown. D, the UCP1-dependent component ofthe stimulated oxygen consumption. Pointswere obtained by subtracting the correspond-ing UCP1�/� and UCP1�/� values in C.
UCP1 and Defense against Oxidative Stress
13884 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 20 • MAY 19, 2006
at Stockholm
Universitetsbibliotek on D
ecember 7, 2006
ww
w.jbc.org
Dow
nloaded from
RESULTS
The Presence of a Carboxyl Group Is an Absolute Requirement forCompounds (Re)activating UCP1—HNE, 2-nonenal, and all-trans-reti-nal have been suggested as general UCP activators (24). We have exam-ined their effects on UCP1, i.e. the uncoupling protein found in brownfat mitochondria and the only uncoupling protein with a verified phys-iological uncoupling ability.We recently demonstrated a role for fatty acids as kinetically compet-
itive (re)activators of UCP1 in GDP-inhibited brown fat mitochondria(22). Our interpretation of those experiments was that fatty acids onlyfunction to overcome the GDP inhibition, in our hands in a kineticallysimple competitivemanner, and that fatty acids do not participate in theuncoupling function of UCP1 as such. We therefore initially examinedthe ability of HNE to reactivate GDP-inhibited UCP1, i.e. to have amechanism of action similar to that of fatty acids. To identify theUCP1-dependent effects, we examined the effect of HNE in brown fat mito-chondria isolated from both UCP1�/� and UCP1�/� mice.
For comparison of activating efficiency, we first determined theeffects of the corresponding fatty acids 2-nonenoic and nonanoic acid.In brown fat mitochondria without UCP1, small uncoupling effects ofthese fatty acids were observed (Fig. 1, A–C, thin lines), indicating aminor UCP1-independent uncoupling mediated by these fatty acids.Both fatty acids were able to (re)stimulate oxygen consumption (pre-inhibited byGDP) inUCP1�/�mitochondria (Fig. 1,B andC, thin lines).
The UCP1-dependent effect was estimated as the difference betweenthe fatty acid effects in the two mitochondrial preparations (UCP1�/�
and UCP1�/�) (Fig. 1D). As seen, the efficiency and apparent affinitywere essentially identical for 2-nonenoic and nonanoic acid. The (re)ac-tivation ability was thus independent of the presence or absence of adouble bond in the fatty acid.The activator candidate, the aldehyde HNE, had practically no effect
in UCP1�/� mitochondria (Fig. 1,A–C, heavy lines). Similarly but nota-bly, no ability to stimulate oxygen consumption was found in UCP1�/�
mitochondria (Fig. 1, B and C, heavy lines). Thus, there was no UCP1-dependent effect of HNE at all (Fig. 1D), i.e. HNE cannot (re)activateGDP-inhibited UCP1. Consequently, in contrast to the correspondingfatty acids, aldehydes were not able to reactive GDP-inhibited UCP1 inbrown fat mitochondria. Because this inhibited state of UCP1 probablymimics the UCP1 state in unstimulated brown fat cells, it is not evidenthow HNE could activate UCP1 in situ.
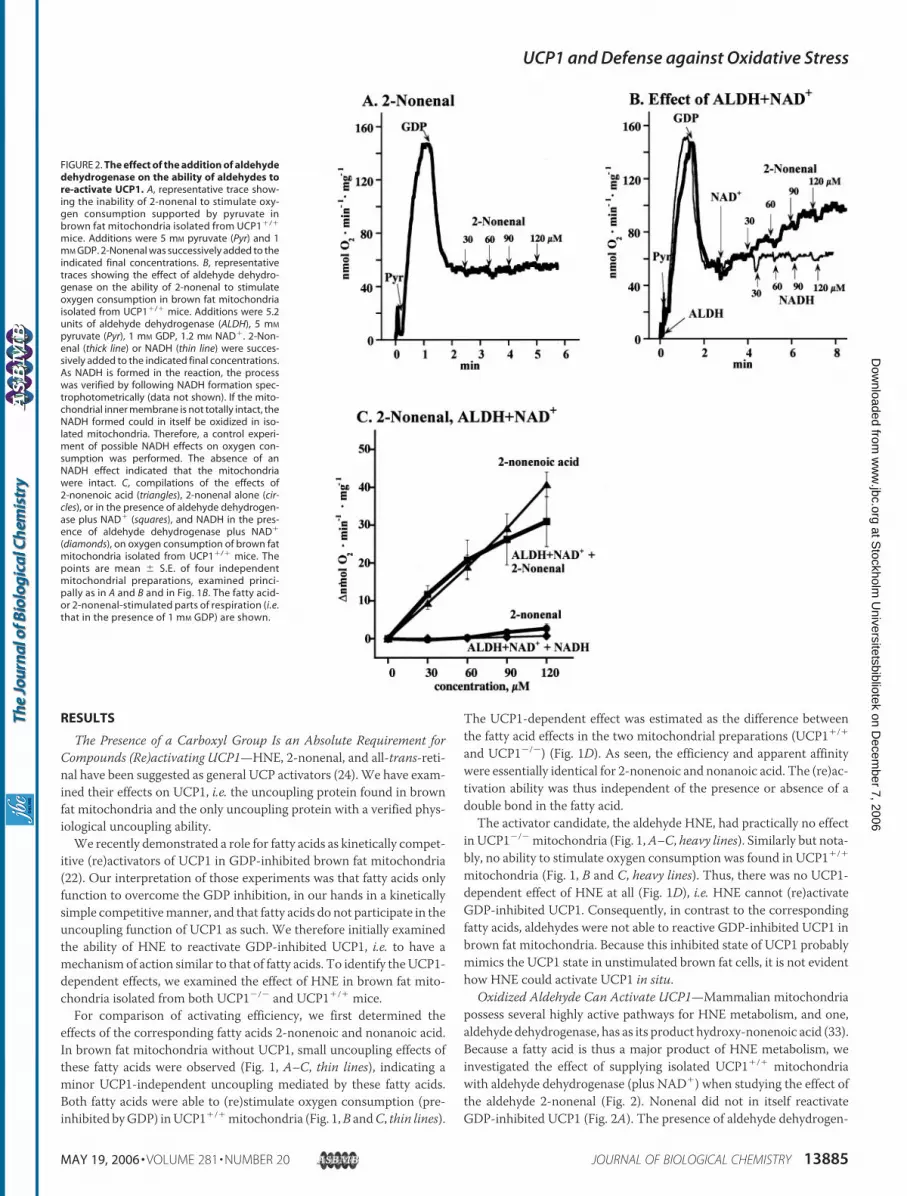
Oxidized Aldehyde Can Activate UCP1—Mammalian mitochondriapossess several highly active pathways for HNE metabolism, and one,aldehyde dehydrogenase, has as its product hydroxy-nonenoic acid (33).Because a fatty acid is thus a major product of HNE metabolism, weinvestigated the effect of supplying isolated UCP1�/� mitochondriawith aldehyde dehydrogenase (plus NAD�) when studying the effect ofthe aldehyde 2-nonenal (Fig. 2). Nonenal did not in itself reactivateGDP-inhibited UCP1 (Fig. 2A). The presence of aldehyde dehydrogen-
FIGURE 2. The effect of the addition of aldehydedehydrogenase on the ability of aldehydes tore-activate UCP1. A, representative trace show-ing the inability of 2-nonenal to stimulate oxy-gen consumption supported by pyruvate inbrown fat mitochondria isolated from UCP1�/�
mice. Additions were 5 mM pyruvate (Pyr) and 1mM GDP. 2-Nonenal was successively added to theindicated final concentrations. B, representativetraces showing the effect of aldehyde dehydro-genase on the ability of 2-nonenal to stimulateoxygen consumption in brown fat mitochondriaisolated from UCP1�/� mice. Additions were 5.2units of aldehyde dehydrogenase (ALDH), 5 mM
pyruvate (Pyr), 1 mM GDP, 1.2 mM NAD�. 2-Non-enal (thick line) or NADH (thin line) were succes-sively added to the indicated final concentrations.As NADH is formed in the reaction, the processwas verified by following NADH formation spec-trophotometrically (data not shown). If the mito-chondrial inner membrane is not totally intact, theNADH formed could in itself be oxidized in iso-lated mitochondria. Therefore, a control experi-ment of possible NADH effects on oxygen con-sumption was performed. The absence of anNADH effect indicated that the mitochondriawere intact. C, compilations of the effects of2-nonenoic acid (triangles), 2-nonenal alone (cir-cles), or in the presence of aldehyde dehydrogen-ase plus NAD� (squares), and NADH in the pres-ence of aldehyde dehydrogenase plus NAD�
(diamonds), on oxygen consumption of brown fatmitochondria isolated from UCP1�/� mice. Thepoints are mean � S.E. of four independentmitochondrial preparations, examined princi-pally as in A and B and in Fig. 1B. The fatty acid-or 2-nonenal-stimulated parts of respiration (i.e.that in the presence of 1 mM GDP) are shown.
UCP1 and Defense against Oxidative Stress
MAY 19, 2006 • VOLUME 281 • NUMBER 20 JOURNAL OF BIOLOGICAL CHEMISTRY 13885
at Stockholm
Universitetsbibliotek on D
ecember 7, 2006
ww
w.jbc.org
Dow
nloaded from
ase plusNAD� dramatically altered the response of themitochondria tothe aldehyde. Addition of 2-nonenal now stimulated UCP1-dependentoxygen consumption to the same extent as did 2-nonenoic acid (Fig. 2,B and C). This demonstrated that the aldehyde has a stimulatory effectonly if converted to a fatty acid.Thus, HNE (Fig. 1), 2-nonenal (Fig. 2), or all-trans-retinal (data not
shown) could not stimulate oxygen consumption in an UCP1-depend-entmanner in brown fatmitochondria. Conversion of the aldehyde intoa fatty acid led to stimulation of oxygen consumption inUCP1�/�mito-chondria. The presence of a carboxyl group is thus an absolute require-ment for compounds (re)activating UCP1.
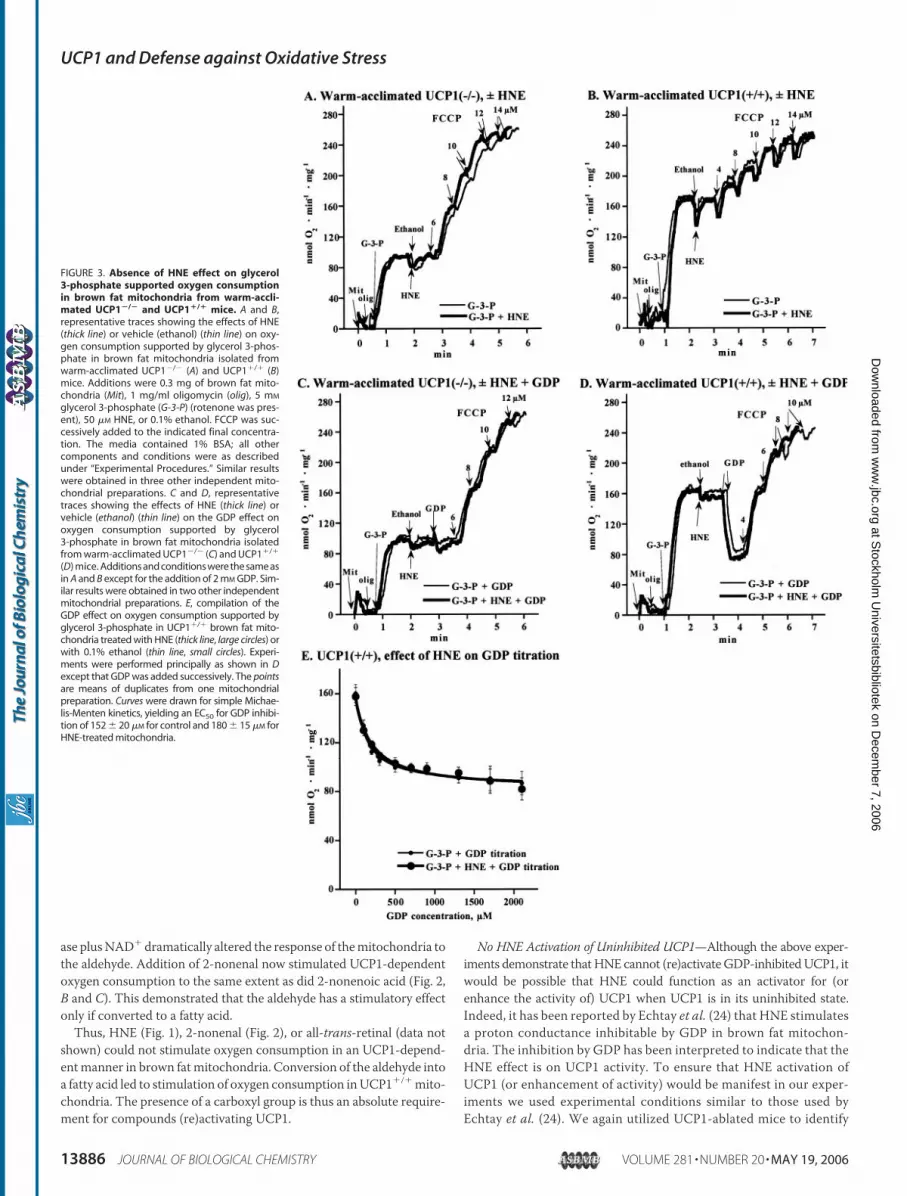
No HNE Activation of Uninhibited UCP1—Although the above exper-iments demonstrate thatHNE cannot (re)activateGDP-inhibitedUCP1, itwould be possible that HNE could function as an activator for (orenhance the activity of) UCP1 when UCP1 is in its uninhibited state.Indeed, it has been reported by Echtay et al. (24) that HNE stimulatesa proton conductance inhibitable by GDP in brown fat mitochon-dria. The inhibition by GDP has been interpreted to indicate that theHNE effect is on UCP1 activity. To ensure that HNE activation ofUCP1 (or enhancement of activity) would be manifest in our exper-iments we used experimental conditions similar to those used byEchtay et al. (24). We again utilized UCP1-ablated mice to identify
FIGURE 3. Absence of HNE effect on glycerol3-phosphate supported oxygen consumptionin brown fat mitochondria from warm-accli-mated UCP1�/� and UCP1�/� mice. A and B,representative traces showing the effects of HNE(thick line) or vehicle (ethanol) (thin line) on oxy-gen consumption supported by glycerol 3-phos-phate in brown fat mitochondria isolated fromwarm-acclimated UCP1�/� (A) and UCP1�/� (B)mice. Additions were 0.3 mg of brown fat mito-chondria (Mit), 1 mg/ml oligomycin (olig), 5 mM
glycerol 3-phosphate (G-3-P) (rotenone was pres-ent), 50 �M HNE, or 0.1% ethanol. FCCP was suc-cessively added to the indicated final concentra-tion. The media contained 1% BSA; all othercomponents and conditions were as describedunder “Experimental Procedures.” Similar resultswere obtained in three other independent mito-chondrial preparations. C and D, representativetraces showing the effects of HNE (thick line) orvehicle (ethanol) (thin line) on the GDP effect onoxygen consumption supported by glycerol3-phosphate in brown fat mitochondria isolatedfrom warm-acclimated UCP1�/� (C) and UCP1�/�
(D) mice. Additions and conditions were the same asin A and B except for the addition of 2 mM GDP. Sim-ilar results were obtained in two other independentmitochondrial preparations. E, compilation of theGDP effect on oxygen consumption supported byglycerol 3-phosphate in UCP1�/� brown fat mito-chondria treated with HNE (thick line, large circles) orwith 0.1% ethanol (thin line, small circles). Experi-ments were performed principally as shown in Dexcept that GDP was added successively. The pointsare means of duplicates from one mitochondrialpreparation. Curves were drawn for simple Michae-lis-Menten kinetics, yielding an EC50 for GDP inhibi-tion of 152 � 20 �M for control and 180 � 15 �M forHNE-treated mitochondria.
UCP1 and Defense against Oxidative Stress
13886 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 20 • MAY 19, 2006
at Stockholm
Universitetsbibliotek on D
ecember 7, 2006
ww
w.jbc.org
Dow
nloaded from
possible UCP1-independent effects of HNE, allowing us to clearlyidentify the contribution of UCP1 to the response to HNE in UCP1-possessing brown fat mitochondria.For experiments examining possible augmentation of UCP1 activity,
it is necessary to investigate brown fat mitochondria, which have a lowinitial degree of uncoupling, i.e. brown fat mitochondria where theinnate respiratory rate is clearly limited by UCP1 activity and not bytotal respiratory chain activity. To be certain of this, we used brown fatmitochondria from warm-acclimated mice (Fig. 3) (also similar toEchtay et al. (24)). In such mitochondria from UCP1�/� mice, glycerol3-phosphate-supported respiration was rather low and could beincreased severalfold by FCCP (Fig. 3A, thin line). In thesemitochondrialacking UCP1, there was, as expected, no effect of HNE (Fig. 3A, heavyline). In UCP1�/� mitochondria, the basal rate of glycerol 3-phosphateoxidation was twice as high as in UCP1�/� mitochondria (Fig. 3, A andB), principally as expected because of the presence and activity of UCP1.As a further stimulation by FCCP could be observed (Fig. 3B), the initialrespiration was limited by the level of activity of UCP1. Thus, if thisactivity was limited by the lack of activator, any activator effect of HNEshould be easilymanifest. However, even here, where UCP1 activity wasrate-limiting for respiration and noGDPwas present, HNEwas withoutstimulatory effect (Fig. 3B). Thus, HNE could not enhance uninhibitedUCP1 activity.We further analyzed whether brown fat mitochondria treated with
HNE became less sensitive to GDP (physiologically this would meanbeing more resistant to the inhibition by the high purine nucleotideconcentration that normally occurs in the cell). There was no GDPeffect and noHNEmodulation inUCP1�/�mitochondria (Fig. 3C). Theexpected GDP inhibitory effect was observed in the UCP1�/� mito-
chondria (Fig. 3D), decreasing the respiratory rate to that observed inUCP1�/� mitochondria. Notably, the degree of inhibition was notchanged by the presence of HNE in UCP1�/� mitochondria (Fig. 3D).The GDP concentration-response curve was also unaffected by HNE (Fig.3E). Thus, HNE was neither able to augment the activity of uninhibitedUCP1 nor to modulate the sensitivity of UCP1 to GDP inhibition.
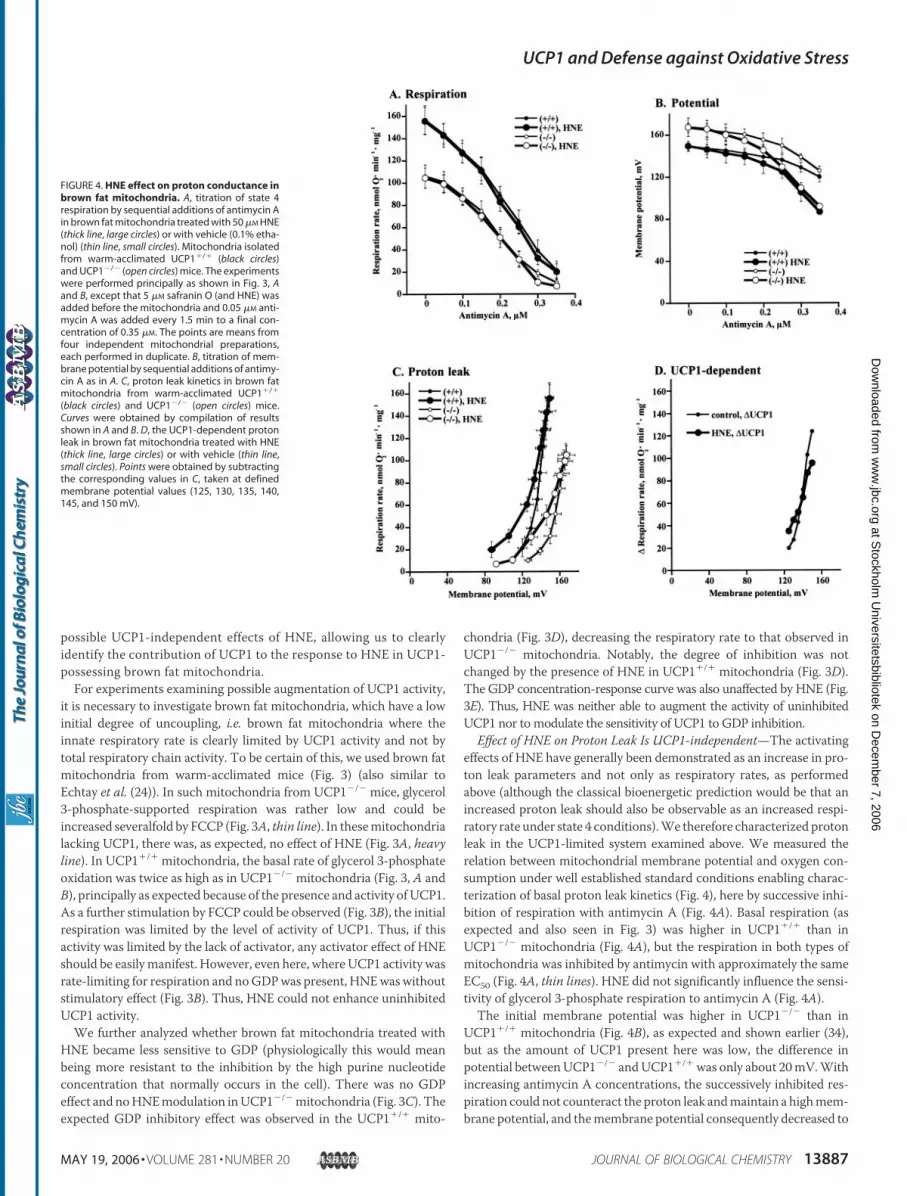
Effect of HNE on Proton Leak Is UCP1-independent—The activatingeffects of HNE have generally been demonstrated as an increase in pro-ton leak parameters and not only as respiratory rates, as performedabove (although the classical bioenergetic prediction would be that anincreased proton leak should also be observable as an increased respi-ratory rate under state 4 conditions).We therefore characterized protonleak in the UCP1-limited system examined above. We measured therelation between mitochondrial membrane potential and oxygen con-sumption under well established standard conditions enabling charac-terization of basal proton leak kinetics (Fig. 4), here by successive inhi-bition of respiration with antimycin A (Fig. 4A). Basal respiration (asexpected and also seen in Fig. 3) was higher in UCP1�/� than inUCP1�/� mitochondria (Fig. 4A), but the respiration in both types ofmitochondria was inhibited by antimycin with approximately the sameEC50 (Fig. 4A, thin lines). HNE did not significantly influence the sensi-tivity of glycerol 3-phosphate respiration to antimycin A (Fig. 4A).The initial membrane potential was higher in UCP1�/� than in
UCP1�/� mitochondria (Fig. 4B), as expected and shown earlier (34),but as the amount of UCP1 present here was low, the difference inpotential betweenUCP1�/� andUCP1�/�was only about 20mV.Withincreasing antimycin A concentrations, the successively inhibited res-piration could not counteract the proton leak andmaintain a highmem-brane potential, and themembrane potential consequently decreased to
FIGURE 4. HNE effect on proton conductance inbrown fat mitochondria. A, titration of state 4respiration by sequential additions of antimycin Ain brown fat mitochondria treated with 50 �M HNE(thick line, large circles) or with vehicle (0.1% etha-nol) (thin line, small circles). Mitochondria isolatedfrom warm-acclimated UCP1�/� (black circles)and UCP1�/� (open circles) mice. The experimentswere performed principally as shown in Fig. 3, Aand B, except that 5 �M safranin O (and HNE) wasadded before the mitochondria and 0.05 �M anti-mycin A was added every 1.5 min to a final con-centration of 0.35 �M. The points are means fromfour independent mitochondrial preparations,each performed in duplicate. B, titration of mem-brane potential by sequential additions of antimy-cin A as in A. C, proton leak kinetics in brown fatmitochondria from warm-acclimated UCP1�/�
(black circles) and UCP1�/� (open circles) mice.Curves were obtained by compilation of resultsshown in A and B. D, the UCP1-dependent protonleak in brown fat mitochondria treated with HNE(thick line, large circles) or with vehicle (thin line,small circles). Points were obtained by subtractingthe corresponding values in C, taken at definedmembrane potential values (125, 130, 135, 140,145, and 150 mV).
UCP1 and Defense against Oxidative Stress
MAY 19, 2006 • VOLUME 281 • NUMBER 20 JOURNAL OF BIOLOGICAL CHEMISTRY 13887
at Stockholm
Universitetsbibliotek on D
ecember 7, 2006
ww
w.jbc.org
Dow
nloaded from
120mV in both types ofmitochondria (Fig. 4B, thin lines). There was noeffect of HNE at low concentrations of antimycin but as the antimycinconcentration was successively increased, a more pronounced decreaseof membrane potential was observed in the HNE-treated mitochondria(Fig. 4B, heavy line). Unexpectedly, this was observed not only inUCP1�/� mitochondria but also in brown fat mitochondria withoutUCP1 (Fig. 4B).These data (Fig. 4, A and B) are plotted to show classical proton leak
kinetics in Fig. 4C. As expected, proton leak was higher in UCP1�/�
mitochondria than in UCP1�/� mitochondria (Fig. 4C, thin lines). Theproton leak thus ascribable to UCP1 can be identified by subtraction(UCP1�/� � UCP1�/�) and is shown in Fig. 4D (thin line). In the non-treated brown fat mitochondria, the UCP1-mediated leak is thus char-acterized by a virtually pseudo-ohmic currentwith a slope of 4.4 nmol ofO2�min�1�mg�1 per mV (pseudo-ohmic because, in a diode-like fash-ion, it is not opened until �120 mV).When plotted as a proton leak, there were also clear effects of the
presence of HNE (Fig. 4C, heavy lines). However, the effect of HNE onleak current characteristics seemed similar in UCP1�/� mitochondriaand inUCP1�/� mitochondria (Fig. 4C). An activating effect of HNE onUCP1 would be expected to increase the proton leak ascribable to
UCP1. However, the proton leak ascribable to UCP1 in HNE-treatedmitochondria was not increased (Fig. 4D); if anything, it was slightlylower (2.5 nmol of O2�min�1�mg�1 per mV) than that observed in theabsence of HNE (4.4). Thus, HNE did not increase the proton leak thatcan be ascribed to UCP1.
No UCP1-dependent Formation of HNE Adducts in Brown AdiposeTissueMitochondria—HNE is amajor product of endogenous lipid per-oxidation (35) and expected to be formed under conditions of increasedoxidative stress. In the established view, superoxide production fromthe respiratory chain is increased whenmembrane potential is high andrespiration is low (i.e. in coupled mitochondria), when there is also ahigh oxygen tension and a high concentration of mitochondrial sitescapable of generating superoxide (36). Under such conditions, superox-ide should be formed, leading to the generation of HNE. HNE is a highlyreactive compound that can react with several functional groups inmitochondrial proteins to form thioesters and Michael adducts (37–39). In contrast, in uncoupled mitochondria, superoxide productionshould be low, HNE should not be formed and thus no HNE/proteinadducts should be generated.The HNE/protein adducts are stable compounds, and post-mortem
analysis of adduct density in the proteins of isolated mitochondria
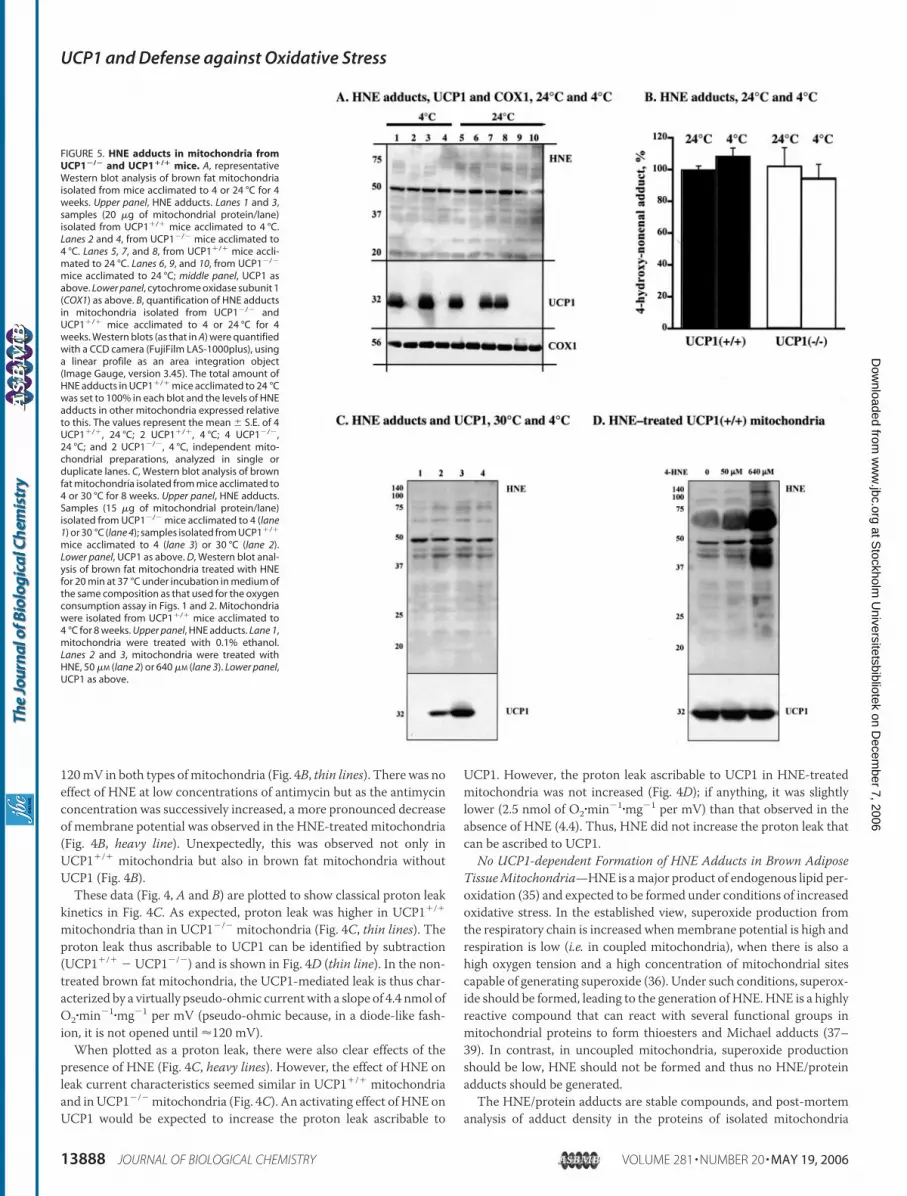
FIGURE 5. HNE adducts in mitochondria fromUCP1�/� and UCP1�/� mice. A, representativeWestern blot analysis of brown fat mitochondriaisolated from mice acclimated to 4 or 24 °C for 4weeks. Upper panel, HNE adducts. Lanes 1 and 3,samples (20 �g of mitochondrial protein/lane)isolated from UCP1�/� mice acclimated to 4 °C.Lanes 2 and 4, from UCP1�/� mice acclimated to4 °C. Lanes 5, 7, and 8, from UCP1�/� mice accli-mated to 24 °C. Lanes 6, 9, and 10, from UCP1�/�
mice acclimated to 24 °C; middle panel, UCP1 asabove. Lower panel, cytochrome oxidase subunit 1(COX1) as above. B, quantification of HNE adductsin mitochondria isolated from UCP1�/� andUCP1�/� mice acclimated to 4 or 24 °C for 4weeks. Western blots (as that in A) were quantifiedwith a CCD camera (FujiFilm LAS-1000plus), usinga linear profile as an area integration object(Image Gauge, version 3.45). The total amount ofHNE adducts in UCP1�/� mice acclimated to 24 °Cwas set to 100% in each blot and the levels of HNEadducts in other mitochondria expressed relativeto this. The values represent the mean � S.E. of 4UCP1�/�, 24 °C; 2 UCP1�/�, 4 °C; 4 UCP1�/�,24 °C; and 2 UCP1�/�, 4 °C, independent mito-chondrial preparations, analyzed in single orduplicate lanes. C, Western blot analysis of brownfat mitochondria isolated from mice acclimated to4 or 30 °C for 8 weeks. Upper panel, HNE adducts.Samples (15 �g of mitochondrial protein/lane)isolated from UCP1�/� mice acclimated to 4 (lane1) or 30 °C (lane 4); samples isolated from UCP1�/�
mice acclimated to 4 (lane 3) or 30 °C (lane 2).Lower panel, UCP1 as above. D, Western blot anal-ysis of brown fat mitochondria treated with HNEfor 20 min at 37 °C under incubation in medium ofthe same composition as that used for the oxygenconsumption assay in Figs. 1 and 2. Mitochondriawere isolated from UCP1�/� mice acclimated to4 °C for 8 weeks. Upper panel, HNE adducts. Lane 1,mitochondria were treated with 0.1% ethanol.Lanes 2 and 3, mitochondria were treated withHNE, 50 �M (lane 2) or 640 �M (lane 3). Lower panel,UCP1 as above.
UCP1 and Defense against Oxidative Stress
13888 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 20 • MAY 19, 2006
at Stockholm
Universitetsbibliotek on D
ecember 7, 2006
ww
w.jbc.org
Dow
nloaded from

should therefore reveal the intensity of the oxidative stress that hadoccurred in situ. We therefore analyzed whether the expected superox-ide overproduction, due to lack of UCP1, resulted in an increased con-tent of HNE adducts in brown fat mitochondria in UCP1-ablated mice.Protein modification by HNE was determined by immunoblotting withan antibody against the sulf-HNE-adduct (37, 38, 40).We examined the presence of HNE adducts in brown fat mitochon-
dria isolated from UCP1�/� and UCP1�/� mice that had been living at24 °C (Fig. 5A, right side lanes). As expected, the antibody detectedseveral bands at different molecular weights. One protein at about 50kDa was dominantly labeled; in the heart it has been indicated that thisprotein is the �-subunit of ATP-synthase (41). However, it was evidentthat there was no obvious difference in the amount of adduct formed inthe mitochondria from UCP1�/� and UCP1�/� mice (Fig. 5A), andquantification of the total labeling of the gel verified this (Fig. 5B).To examine conditions expected to further increase the risk of oxi-
dative stress and lipid peroxidation, we analyzed the effect of cold accli-mation on the formation of HNE/protein adducts. In UCP1-ablatedmice, cold acclimation leads to an increased polyunsaturation of mito-chondrialmembrane fatty acids5 (as it does inwild-typemice (42)). Thisshould increase the risk of lipid peroxidation (43) and thus potentiallylead to increased formation of HNE and HNE/protein adducts. How-ever, cold acclimation did not result in higher levels of HNE/proteinadducts in mitochondria from UCP1-ablated mice (Fig. 5, A and B).
To investigate whether conditions with coupled brown fatmitochon-dria due to the absence of UCP1, and coupled brown fat mitochondriadue to the absence of physiological stimulation, resulted in differentdegrees of oxidative damage, we compared mitochondria from wild-type and UCP1-ablated mice, living either at thermoneutrality (whereUCP1 would not be physiologically stimulated) or at 4 °C. Again, wefound no evidence for alterations in the levels of HNE/protein adducts(Fig. 5C). Thus, taken together, we could not establish an essential roleof UCP1 in the defense against oxidative damage in brown fat mito-chondria, as there was no increased oxidative damage in UCP1�/�
brown fat mitochondria, despite the expected higher generation ofsuperoxide and the concurrent absence of the proposed UCP1-medi-ated elimination of superoxide in these animals (3, 44).
No Formation of HNE/UCP1 Adducts—We compared the Westernblots of HNE adducts with the Western blot of UCP1 (Fig. 5) in anattempt to identify bands of HNE adducts that matched the UCP1molecular mass of 32 kDa, or bands with another molecular mass thatwere present in UCP1�/� but absent in UCP1�/� samples (adductionmay change the apparent molecular mass) (Fig. 5,A andC). None of theHNE adduct bands had an apparent molecular weight of UCP1 proteinor were related to the presence/absence of UCP1 (Fig. 5, A and C).
Treatment of UCP1�/� brown fatmitochondria with increasing con-centrations of HNE in vitro led to formation of HNE adducts withmito-chondrial proteins (as well with the albumin present in the incubationmedium; the band of 69 kDa) in a concentration-dependent manner(Fig. 5D). However, although UCP1 is one of the most abundant pro-teins in brown fat mitochondria, we found no evidence for specificformation of HNE/protein adducts in the 32-kDa region; if anything,there was remarkably little HNE adduct formation at thismolecular size(Fig. 5D). Thus, we found no evidence for a specific, perhaps activating,HNE modification of UCP1.
UCP1�/� and UCP1�/� Brown Fat Mitochondria Exhibit EqualRates of Net Superoxide Release—The absence of increased levels ofHNE adducts in UCP1�/� mitochondria could have two explanations:
superoxide generation by the respiratory chain is not reduced by UCP1activity, even though UCP1 activity leads to low membrane potential(this possibility contrasts with the effect on superoxide production ear-lier proposed (3, 44)), or superoxide generation is normally reduced byactive UCP1, but in the UCP1�/� mice, antioxidant systems are com-pensatorily up-regulated and successfully eliminate excess ROS. Eitherof these possibilitieswould explain the absence of increasedoxidative stressin the UCP1�/� animals.We therefore examined these possibilities.
To analyze the net superoxide release in isolated brown fatmitochon-dria, we used the dye DHE, which is converted to ethidium by superox-ide; the formation of superoxide can therefore be followed as fluores-cence (14, 27). We first examined UCP1�/� brown fat mitochondria inthe absence of GDP, i.e. the mitochondria were uncoupled.When thesemitochondriawere incubatedwith glycerol 3-phosphate in the presenceof rotenone there was a linear increase in fluorescence (Fig. 6A). Anti-mycin A was then added as a positive control to enhance mitochon-drial superoxide generation. This resulted in a pronounced increase influorescence that was inhibited by addition of recombinant superoxidedismutase (Fig. 6A). Thus, although UCP1 is uninhibited in these mito-chondria, resulting in a low membrane potential (Fig. 4) and a highrespiration rate (flux) (Figs. 3 and 4), mitochondrial ROS productionwas not totally abolished.We then examined under identical conditionsbrown fat mitochondria from UCP1-ablated mice, i.e. brown fat mito-chondria with high membrane potential (Fig. 4) and low respiration(Figs. 3 and 4). These conditions have been proposed to lead to highproduction of mitochondrial ROS (3, 44). Surprisingly, we found thatthe coupled, low-fluxUCP1�/� brown fatmitochondria exhibited equalrates of net superoxide release as did the UCP1-containing, uncoupledbrown fat mitochondria (Fig. 6, A and B).Our second approach to estimate superoxide release in brown fat
mitochondria was to measure the activity of the mitochondrial matrixenzyme aconitase. Aconitase activity is readily and specifically impairedby free radicals (28) and therefore widely used as a marker of oxidativedamage ofmitochondria (13, 14, 45). Brown fatmitochondrial aconitasewas, as expected, sensitive to superoxide, as its activity was dramaticallydecreased in the presence of the superoxide-generating systemxanthineplus xanthine oxidase (Fig. 6C). However, we could see no effect of theabsence of UCP1 on aconitase activity (Fig. 6C) and no UCP1-depend-ent difference in the ability of exogenous superoxide to inhibit the activ-ity (Fig. 6C). Thus, all results obtained fromUCP1�/� mitochondria: noextensive formation of HNE/protein adducts (Fig. 5), no enhancedsuperoxide release (Fig. 6B), and no lowered aconitase activity (Fig. 6C),as compared with wild-type mitochondria, are contradictory to a pro-tective role of UCP1 in oxidative stress.
Tolerance toOxidativeAgents andActivities ofAntioxidantEnzymesAreNot Enhanced in Brown Adipose Tissue from UCP1-ablated Mice—Thesecond possible explanation for the absence of increased levels of HNE-adducts in UCP1�/� mitochondria is that the absence of UCP1 inUCP1�/� mitochondria may induce compensatory mechanisms lead-ing to an increased mitochondrial tolerance to oxidative agents and/orenhanced activity of antioxidant systems.Possible recruited mechanisms in protection against lipid peroxida-
tion were analyzed by measurement of the rates of lipid peroxidation,estimated as the formation of the lipid peroxidation product, MDA,under basal, spontaneous, and stimulated conditions of oxidative stressin brown adipose tissue from UCP1�/� and UCP1�/� mice (Fig. 6D).Basal levels of MDA in brown adipose tissue were very low (hardlymeasurable) and were not different in UCP1�/� and UCP1�/� mice.Levels of spontaneous lipid peroxidation were higher compared withbasal but were not different between UCP1�/� and UCP�/� brown5 A. Ocloo, I. G. Shabalina, J. Nedergaard, and M. D. Brand, unpublished observations.
UCP1 and Defense against Oxidative Stress
MAY 19, 2006 • VOLUME 281 • NUMBER 20 JOURNAL OF BIOLOGICAL CHEMISTRY 13889
at Stockholm
Universitetsbibliotek on D
ecember 7, 2006
ww
w.jbc.org
Dow
nloaded from
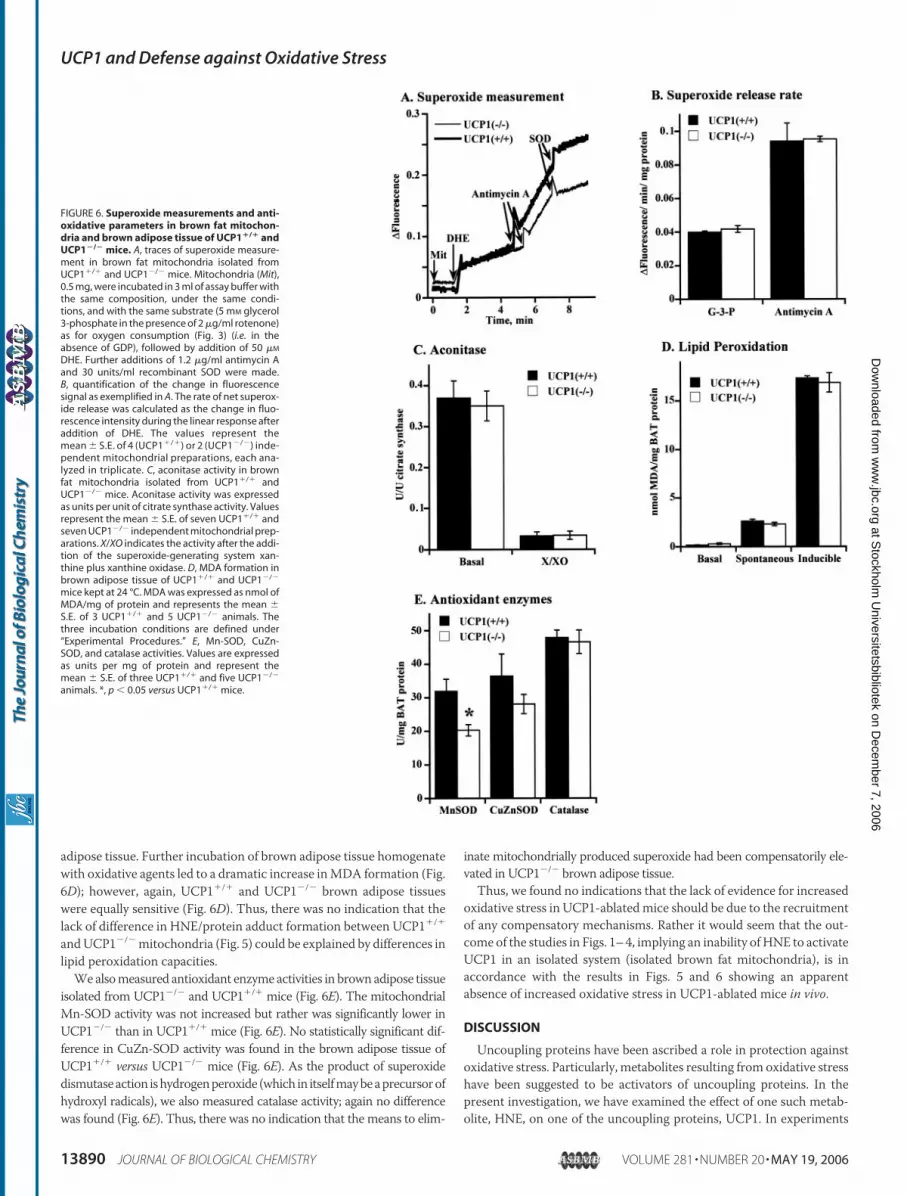
adipose tissue. Further incubation of brown adipose tissue homogenatewith oxidative agents led to a dramatic increase inMDA formation (Fig.6D); however, again, UCP1�/� and UCP1�/� brown adipose tissueswere equally sensitive (Fig. 6D). Thus, there was no indication that thelack of difference in HNE/protein adduct formation between UCP1�/�
andUCP1�/�mitochondria (Fig. 5) could be explained by differences inlipid peroxidation capacities.We alsomeasured antioxidant enzyme activities in brown adipose tissue
isolated from UCP1�/� and UCP1�/� mice (Fig. 6E). The mitochondrialMn-SOD activity was not increased but rather was significantly lower inUCP1�/� than in UCP1�/� mice (Fig. 6E). No statistically significant dif-ference in CuZn-SOD activity was found in the brown adipose tissue ofUCP1�/� versus UCP1�/� mice (Fig. 6E). As the product of superoxidedismutaseaction ishydrogenperoxide (which in itselfmaybeaprecursorofhydroxyl radicals), we also measured catalase activity; again no differencewas found (Fig. 6E). Thus, there was no indication that the means to elim-
inate mitochondrially produced superoxide had been compensatorily ele-vated in UCP1�/� brown adipose tissue.Thus, we found no indications that the lack of evidence for increased
oxidative stress in UCP1-ablatedmice should be due to the recruitmentof any compensatory mechanisms. Rather it would seem that the out-come of the studies in Figs. 1–4, implying an inability ofHNE to activateUCP1 in an isolated system (isolated brown fat mitochondria), is inaccordance with the results in Figs. 5 and 6 showing an apparentabsence of increased oxidative stress in UCP1-ablated mice in vivo.
DISCUSSION
Uncoupling proteins have been ascribed a role in protection againstoxidative stress. Particularly, metabolites resulting from oxidative stresshave been suggested to be activators of uncoupling proteins. In thepresent investigation, we have examined the effect of one such metab-olite, HNE, on one of the uncoupling proteins, UCP1. In experiments
FIGURE 6. Superoxide measurements and anti-oxidative parameters in brown fat mitochon-dria and brown adipose tissue of UCP1�/� andUCP1�/� mice. A, traces of superoxide measure-ment in brown fat mitochondria isolated fromUCP1�/� and UCP1�/� mice. Mitochondria (Mit),0.5 mg, were incubated in 3 ml of assay buffer withthe same composition, under the same condi-tions, and with the same substrate (5 mM glycerol3-phosphate in the presence of 2 �g/ml rotenone)as for oxygen consumption (Fig. 3) (i.e. in theabsence of GDP), followed by addition of 50 �M
DHE. Further additions of 1.2 �g/ml antimycin Aand 30 units/ml recombinant SOD were made.B, quantification of the change in fluorescencesignal as exemplified in A. The rate of net superox-ide release was calculated as the change in fluo-rescence intensity during the linear response afteraddition of DHE. The values represent themean � S.E. of 4 (UCP1�/�) or 2 (UCP1�/�) inde-pendent mitochondrial preparations, each ana-lyzed in triplicate. C, aconitase activity in brownfat mitochondria isolated from UCP1�/� andUCP1�/� mice. Aconitase activity was expressedas units per unit of citrate synthase activity. Valuesrepresent the mean � S.E. of seven UCP1�/� andseven UCP1�/� independent mitochondrial prep-arations. X/XO indicates the activity after the addi-tion of the superoxide-generating system xan-thine plus xanthine oxidase. D, MDA formation inbrown adipose tissue of UCP1�/� and UCP1�/�
mice kept at 24 °C. MDA was expressed as nmol ofMDA/mg of protein and represents the mean �S.E. of 3 UCP1�/� and 5 UCP1�/� animals. Thethree incubation conditions are defined under“Experimental Procedures.” E, Mn-SOD, CuZn-SOD, and catalase activities. Values are expressedas units per mg of protein and represent themean � S.E. of three UCP1�/� and five UCP1�/�
animals. *, p 0.05 versus UCP1�/� mice.
UCP1 and Defense against Oxidative Stress
13890 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 20 • MAY 19, 2006
at Stockholm
Universitetsbibliotek on D
ecember 7, 2006
ww
w.jbc.org
Dow
nloaded from

with isolated brown fatmitochondria, we found thatHNEwas unable to(re)activate UCP1 when it was inhibited by purine nucleotides. HNEwas also unable to augment the activity of uninhibitedUCP1 as it did notincrease the proton leak ascribable to UCP1 (but it did increase theproton leak of the mitochondria in a UCP1-independent way). In theintact animal, we found no indications of increased oxidative stress-related damage due to the absence of UCP1. These results thus questionthe suggested protective role ofUCP1 andmay have implications for thesuggested function of the other uncoupling proteins and for the under-standing of the nature of the physiological conditions associated withoxidative stress in general.
HNE Cannot (Re)activate UCP1—In the cell, uncoupling proteins,including UCP1, are exposed to cytosolic purine nucleotides, mainlyATP and ADP. UCP1 activity is thought to be chronically inhibited bythese nucleotides, and the ability of fatty acids to re-activateUCP1 in thepresence of purine nucleotides is the basis for the concept that fattyacids are the physiological activators of UCP1 (reviewed in Ref. 4). Wefound that irrespective of concentration added, the lipid metaboliteHNEwas unable to overcome inhibition caused by GDP, the nucleotideclassically used experimentally tomimic the situation in the cell (Fig. 1).Thus, the present experiments indicate that HNE cannot overcome thechronic in situ inhibition of UCP1. Physiologically, this means thatUCP1 cannot be part of a self-regulating system where an increasedlevel of HNE would activate UCP1, thus decreasing the membranepotential and through this the generation of ROS, leading to a lower rateof generation of HNE. Basically, HNE lacks the necessary property ofbeing a UCP1 (re)activator.
Oxidized Aldehydes Activate UCP1-independent and UCP1-depend-ent Proton Leak—Mitochondria detoxify HNE through several path-ways: glutathione conjugation (33, 46), formation of 1,4-dihydroxy-trans-2-nonene (33), and oxidation to 4-hydroxy-2-nonenoic acid bytheNAD�-dependent aldehyde dehydrogenases ALDH2 andALDH5A(47, 48). The mitochondrial aldehyde dehydrogenases are highlyexpressed and active in many tissues (liver, heart, kidney, muscle, brain,lung, spleen, and stomach) (48). Thus, in mitochondrial preparationsfrom these tissues, the aldehyde dehydrogenase present could oxidizeHNE into a fatty acid. However, in brown adipose tissue, the expressionof aldehyde dehydrogenase is remarkably low (49).We did not observe signs of spontaneous fatty acid formation from
HNE in brown fat mitochondria (Figs. 1 and 2), in agreement with thelow expression of aldehyde dehydrogenase. The situation in our exper-iments with addition of exogenous aldehyde dehydrogenase (Fig. 2)maythus be said to mimic the situation in many other tissues. In thosetissues, the endogenous aldehyde dehydrogenase may convert a signif-icant amount of the added HNE to 4-hydroxy-2-nonenoic acid; e.g. inisolated intact kidney mitochondria about 80% of HNE is degradedwithin 3 min, and the main product is hydroxynonenoic acid (50). Asseen in Fig. 1A, such fatty acids can uncouplemitochondria in anUCP1-independent way. There is therefore a possibility that apparent alde-hyde-induced increases in proton conductance in mitochondria fromdifferent tissues (24) could in part be because of the effect of formationof hydroxynonenoic acid.
4-Hydroxy-2-nonenal Is Not a Regulatory Cofactor for UCP1—Whendirectly added to brown fat mitochondria, HNE did not augment theactivity of uninhibitedUCP1 (Fig. 3B). In experiments investigating pro-ton leak in the standard manner by successive addition of respiratoryinhibitor, results corresponding to an increase in proton leak caused byHNE were observed, but the effect was essentially the same whetherUCP1was present or not (Fig. 4C). Indeed, when the proton leak ascrib-able to UCP1 was calculated, the result was that UCP1 activity was
nearly halved in the presence of HNE (Fig. 4D). This evidence for non-involvement of UCP1 in response to HNE of brown fat mitochondriaquestions earlier indications that UCP1 is activated by HNE (24).It has also been discussedwhetherHNE could be the activating cofac-
tor explaining the difference between the functional statues of UCP1 inreconstituted systems (where UCP1 is inactive) and in brown fat mito-chondria (where UCP1 is innately active); HNE could be present in thebrown fat mitochondria but absent in the reconstituted systems (10).The inability of added HNE to augment the activity of noninhibitedUCP1 (Figs. 4 and 5) indicates that the amount of HNE present inisolated brown fat mitochondria cannot be rate-limiting for the activityof UCP1. Thus, the present experiments cannot preclude the possibilitythat HNE (or similar compounds) present in brown fat mitochondria isnecessary for the activity of native UCP1, but if so, HNE is clearly pres-ent endogenously in sufficient amounts to allow full UCP1 activity, andHNE can therefore not have a controlling role. The alternative possibil-ity would be that HNE does not have a role as a cofactor for UCP1activity.
The Nature of the UCP1-independent, HNE-induced Proton Leak—Al-though we clearly observed no ability of HNE to activate a proton leakascribable to UCP1 in brown fat mitochondria (Fig. 4D), we observedeffects of HNE addition that were independent of UCP1 but had char-acteristics (Fig. 4C) similar to those earlier demonstrated in brown fatmitochondria (24). The nature of these proton leaksmay be discussed. Itis noteworthy thatHNE can cause direct permeabilization ofmitochon-drial membranes, revealed as high amplitude swelling (51), i.e. HNE-induced mitochondrial swelling represents a so-called permeabilitytransition. There thus seems to be a possibility that such a permeabili-zation of the mitochondrial membrane may also cause the UCP1-inde-pendent increase in proton conductance in mitochondria (24) afterHNE treatment.
Relation to HNE Activation of Other UCPs—We have only examinedthe ability of HNE to activate one of the members of the uncouplingprotein family, UCP1. HNE activation has been suggested as a generalmechanism for uncoupling protein activation (24); it has, e.g. been sug-gested that this was the original mode of action of all members of theuncoupling protein family and that the ability of UCP1 to be thermo-genic was a later evolutionary event (3, 11). The inability shown here ofHNE to activate UCP1 would then be considered a consequence of thisdevelopment, i.e. an evolutionary loss of function. Concerning the otherUCPs, results published indicate that the HNE effect in muscle mito-chondria from UCP3-ablated mice is smaller than in wild-type mice(24), which would support a contribution from UCP3. Direct data con-cerning UCP2 have not been published. The possibility that a UCP1-mediatedHNEeffect in ourwild-typemitochondria has been taken overby an increased amount of UCP3 in the UCP1-ablated mice is unlikely,as UCP3 gene expression is decreased in UCP1-ablated mice (34). Con-cerning UCP2, it would seem that althoughmRNA levels are high in thebrown adipose tissue of UCP1-ablated mice (34), the protein is nottranslated in these mice (52). It is therefore very unlikely that UCP2/UCP3 has taken over as the mediator of HNE effects on proton leak inbrown fat mitochondria of UCP1-ablated mice. Based on our data wecan, thus, at least conclude that activation by HNE is not a generalfeature of all uncoupling proteins.
Covalent Modification of UCP1 Is Not Involved in Regulation of ItsActivity—HNE is a highly reactive compound and several functionalgroups on mitochondrial proteins are modified by interaction withHNE, particularly sulfhydryl groups to form thioester adducts (53), andhistidine and lysine residues of proteins to form stable Michael adducts(37–39). Covalent modification of UCPs by HNE has been proposed as
UCP1 and Defense against Oxidative Stress
MAY 19, 2006 • VOLUME 281 • NUMBER 20 JOURNAL OF BIOLOGICAL CHEMISTRY 13891
at Stockholm
Universitetsbibliotek on D
ecember 7, 2006
ww
w.jbc.org
Dow
nloaded from

a molecular mechanism of activation of UCPs (3, 24). However, theexistence of UCP1 adducts has not so far been demonstrated. UCP1constitutes a large fraction of the mitochondrial membrane proteins,and ifHNE/UCP1 adductswere formed to any significant degree in vivo,as part of the activation process, they should reasonably be identifiable.However, we could not identify any protein/HNE adducts that wouldcorrespond to UCP1 (Fig. 5). This implies that covalent modification ofUCP1 is not directly involved in regulation of UCP1 activity.
Absence of Increased Oxidative Damage in UCP1-ablated MiceImplies That UCP1Does Not Have an Antioxidative Effect in Vivo—Theabove conclusions have been based on in vitro data and were restricted toexamination of HNE effects, and these conditions may not reflect physio-logical conditions.However,wewere also unable to observe any increase inoxidative damage (measured as total amount ofHNEadducts) in brown fatmitochondria from UCP1-ablated mice in different physiological condi-tions (Fig. 5).The absence of effect ofUCP1 ablation could have been dueto a compensatory activation of alternative antioxidant pathways in thebrown adipose tissue of UCP1-ablated mice, but we found no evidencefor this (Fig. 6). In extension of this, it may be concluded that not onlyHNE but also other compounds associatedwith oxidative stress, such assuperoxide (11) and compounds downstream of superoxide (carbon-centered radicals (15), hydroperoxy fatty acids (16, 17), and lipid peroxi-dation product in addition to HNE (24)), which have been suggested toactivate UCP1 in a self-regulatory feed-back process, either do notinduce such a process or do not depend onUCP1 for this process. Thus,physiologically, UCP1 does not seem to be an indispensable antioxidant.
Absence of Increased Oxidative Damage in Cold-acclimated UCP1-ab-lated Mice May Have Implications for Theories for Oxidative Damage—At a general level, the observation that no difference was found in thedegree of oxidative damage between mitochondria from cold-accli-mated UCP1�/� and UCP1�/� mice is surprising considering generallyaccepted ideas about the causes of oxidative damage.Oxidative damage is generally believed to occur under conditions in
which a large pool of electrons is accumulated in the respiratory chain,at highmembrane potential and high oxygen tension, and the damage isthought to be alleviated by a decrease in membrane potential (throughATP-synthase activity or uncoupling), high flux of electrons throughthe system and low oxygen tension (36). The brown adipose tissue ofcold-acclimated UCP1�/� and UCP1�/� mice should constitute asmuch of a contrast between these conditions as can be physiologicallyachieved. Thus, in the brown adipose tissue of UCP1�/� mice in thecold, UCP1 is constantly activated, leading to a lowered membranepotential and a rapid flux of electrons through the respiratory chain, andto a high utilization of oxygen resulting in extremely low oxygen tension(54). In contrast, in the brown adipose tissue of UCP1�/� mice, themitochondrial membrane potential is very high (as even ATP synthesisis very low in these mitochondria (2)), there is ample substrate beingproduced for combustion (as lipolysis in the brown adipocytes proceedsunabrogated (55)) and the oxygen tension is high, simply because oxy-gen is not being utilized. Thus, expectations would be that the tissue inthe UCP1�/� mice would be extremely exposed to oxidative stress, anda high level of oxidative products would be expected to be observable.The result was, however, that there was no difference in the amount ofHNE/protein adducts between UCP1�/� and UCP1�/� mice.
It is clearly not possible to dismiss the basic concepts of the condi-tions causing oxidative damage based on this experiment alone. Theimplication of the experimental result is, nonetheless, that the generallyaccepted view concerning conditions leading to oxidative stress is notsufficient and that further factorsmust be involved in the determinationof the degree of oxidative stress occurring in vivo.
Acknowledgments—We thank Irina Sabanova for establishing and verifyingthe mouse strains and Zdenek Drahota for helpful comments.
REFERENCES1. Ricquier, D., and Bouillaud, F. (2000) Biochem. J. 345, 161–1792. Nedergaard, J., Golozoubova, V., Matthias, A., Asadi, A., Jacobsson, A., and Cannon,
B. (2001) Biochim. Biophys. Acta 1504, 82–1063. Brand, M. D., Affourtit, C., Esteves, T. C., Green, K., Lambert, A. J., Miwa, S., Pakay,
J. L., and Parker, N. (2004) Free Radic. Biol. Med. 37, 755–7674. Cannon, B., and Nedergaard, J. (2004) Physiol. Rev. 84, 277–3595. Nedergaard, J., and Cannon, B. (2003) Exp. Physiol. 88, 65–846. Esteves, T. C., and Brand, M. D. (2005) Biochim. Biophys. Acta 1709, 35–447. Krauss, S., Zhang, C. Y., and Lowell, B. B. (2005)Nat. Rev. Mol. Cell Biol. 6, 248–2618. Nedergaard, J., Ricquier, D., and Kozak, L. P. (2005) EMBO Rep. 6, 917–9219. Cadenas, S., and Brand, M. D. (2000) Biochem. J. 348, 209–21310. Echtay, K. S., Winkler, E., and Klingenberg, M. (2000) Nature 408, 609–61311. Echtay, K. S., Roussel, D., St-Pierre, J., Jekabsons, M. B., Cadenas, S., Stuart, J. A.,
Harper, J. A., Roebuck, S. J., Morrison, A., Pickering, S., Clapham, J. C., and Brand,M. D. (2002) Nature 415, 96–99
12. Jaburek, M., and Garlid, K. D. (2003) J. Biol. Chem. 278, 25825–2583113. Couplan, E., del Mar Gonzalez-Barroso, M., Alves-Guerra, M. C., Ricquier, D.,
Goubern, M., and Bouillaud, F. (2002) J. Biol. Chem. 277, 26268–2627514. Silva, J. P., Shabalina, I. G., Dufour, E., Petrovic, N., Backlund, E. C., Hultenby, K.,
Wibom, R., Nedergaard, J., Cannon, B., and Larsson, N.-G. (2005) EMBO J. 24,4061–4070
15. Murphy, M. P., Echtay, K. S., Blaikie, F. H., Asin-Cayuela, J., Cocheme, H. M., Green,K., Buckingham, J. A., Taylor, E. R., Hurrell, F., Hughes, G., Miwa, S., Cooper, C. E.,Svistunenko, D. A., Smith, R. A., and Brand, M. D. (2003) J. Biol. Chem. 278,48534–48545
16. Goglia, F., and Skulachev, V. P. (2003) FASEB J. 17, 1585–159117. Jaburek, M., Miyamoto, S., Di Mascio, P., Garlid, K. D., and Jezek, P. (2004) J. Biol.
Chem. 279, 53097–5310218. Negre-Salvayre, A., Hirtz, C., Carrera, G., Cazenave, R., Troly, M., Salvayre, R.,
Penicaud, L., and Casteilla, L. (1997) FASEB J. 11, 809–81519. Enerback, S., Jacobsson, A., Simpson, E.M., Guerra, C., Yamashita, H., Harper,M.-E.,
and Kozak, L. P. (1997) Nature 387, 90–9420. Golozoubova, V., Hohtola, E., Matthias, A., Jacobsson, A., Cannon, B., and Neder-
gaard, J. (2001) FASEB J. 15, 2048–205021. Cannon, B., and Nedergaard, J. (2001)Methods Mol. Biol. 155, 295–30322. Shabalina, I. G., Jacobsson, A., Cannon, B., and Nedergaard, J. (2004) J. Biol. Chem.
279, 38236–3824823. Udenfriend, S., Stein, S., Bohlen, P., Dairman, W., Leimgruber, W., and Weigele, M.
(1972) Science 178, 871–87224. Echtay, K. S., Esteves, T. C., Pakay, J. L., Jekabsons,M. B., Lambert, A. J., Portero-Otin,
M., Pamplona, R., Vidal-Puig, A. J., Wang, S., Roebuck, S. J., and Brand, M. D. (2003)EMBO J. 22, 4103–4110
25. Akerman, K. E., and Wikstrom, M. K. (1976) FEBS Lett. 68, 191–19726. Nedergaard, J. (1983) Eur. J. Biochem. 133, 185–19127. Benov, L., Sztejnberg, L., and Fridovich, I. (1998) Free Radic. Biol. Med. 25, 826–83128. Gardner, P. R. (2002)Methods Enzymol. 349, 9–2329. Alp, P. R., Newsholme, E. A., and Zammit, V. A. (1976) Biochem. J. 154, 689–70030. Yagi, K. (1998)Methods Mol. Biol. 108, 101–10631. Misra, H. P., and Fridovich, I. (1972) J. Biol. Chem. 247, 3170–317532. Beutler, E. (1982) inRedCellMetabolism, aManual of BiochemicalMethods (Beutler,
E., ed) pp. 105–106, Grune and Stratton, New York33. Siems, W., and Grune, T. (2003)Mol. Aspects Med. 24, 167–17534. Matthias, A., Jacobsson, A., Cannon, B., and Nedergaard, J. (1999) J. Biol. Chem. 274,
28150–2816035. Esterbauer, H., Schaur, R. J., and Zollner, H. (1991) Free Radic. Biol. Med. 11, 81–12836. Balaban, R. S., Nemoto, S., and Finkel, T. (2005) Cell 120, 483–49537. Cohn, J. A., Tsai, L., Friguet, B., and Szweda, L. I. (1996)Arch. Biochem. Biophys. 328,
158–16438. Chen, J., Schenker, S., Frosto, T. A., and Henderson, G. I. (1998) Biochim. Biophys.
Acta 1380, 336–34439. Reinheckel, T., Noack, H., Lorenz, S., Wiswedel, I., and Augustin, W. (1998) Free
Radic. Res. 29, 297–30540. Waeg, G., Dimsity, G., and Esterbauer, H. (1996) Free Radic. Res. 25, 149–15941. Choksi, K. B., Boylston, W. H., Rabek, J. P., Widger, W. R., and Papaconstantinou, J.
(2004) Biochim. Biophys. Acta 1688, 95–10142. Ricquier, D., Mory, G., Nechad, M., and Hemon, P. (1978) J. Physiol. 74, 695–70243. Barja deq Uiroga, G., Lopez-Torres, M., Perez-Campo, R., Abelenda, M., Paz Nava,
M., and Puerta, M. L. (1991) Biochem. J. 277, 289–29244. Skulachev, V. P. (1998) Biochim. Biophys. Acta 1363, 100–124
UCP1 and Defense against Oxidative Stress
13892 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 20 • MAY 19, 2006
at Stockholm
Universitetsbibliotek on D
ecember 7, 2006
ww
w.jbc.org
Dow
nloaded from

45. Bernal-Mizrachi, C., Gates, A. C.,Weng, S., Imamura, T., Knutsen, R. H., DeSantis, P.,Coleman, T., Townsend, R. R., Muglia, L. J., and Semenkovich, C. F. (2005) Nature435, 502–506
46. Meyer, M. J., Mosely, D. E., Amarnath, V., and Picklo, M. J., Sr. (2004) Chem. Res.Toxicol. 17, 1272–1279
47. Murphy, T. C., Amarnath, V., and Picklo, M. J., Sr. (2003) J. Neurochem. 84,1313–1321
48. Ohta, S., Ohsawa, I., Kamino, K., Ando, F., and Shimokata, H. (2004)Ann. N. Y. Acad.Sci. 1011, 36–44
49. Unami, A., Shinohara, Y., Kajimoto, K., and Baba, Y. (2004) Biochem. Pharmacol. 67,555–564
50. Ullrich, O., Grune, T., Henke,W., Esterbauer, H., and Siems,W. G. (1994) FEBS Lett.352, 84–86
51. Vieira,H. L., Belzacq,A. S., Haouzi, D., Bernassola, F., Cohen, I., Jacotot, E., Ferri, K. F.,El Hamel, C., Bartle, L. M., Melino, G., Brenner, C., Goldmacher, V., and Kroemer, G.(2001) Oncogene 20, 4305–4316
52. Pecqueur, C., Alves-Guerra, M. C., Gelly, C., Levi-Meyrueis, C., Couplan, E., Collins,S., Ricquier, D., Bouillaud, F., and Miroux, B. (2001) J. Biol. Chem. 276, 8705–8712
53. Petersen, D. R., and Doorn, J. A. (2004) Free Radic. Biol. Med. 37, 937–94554. Foster, D. O., and Frydman, M. L. (1978) Can. J. Physiol. Pharmacol. 56, 110–12255. Matthias, A., Ohlson, K. E. B., Fredriksson, J. M., Jacobsson, A., Nedergaard, J., and
Cannon, B. (2000) J. Biol. Chem. 275, 25073–25081
UCP1 and Defense against Oxidative Stress
MAY 19, 2006 • VOLUME 281 • NUMBER 20 JOURNAL OF BIOLOGICAL CHEMISTRY 13893
at Stockholm
Universitetsbibliotek on D
ecember 7, 2006
ww
w.jbc.org
Dow
nloaded from
Top Related

Copyright © 2022 FDOKUMEN

