







Bahasa
Halaman
Hukum


ROYAUME DU MAROC UNIVERSITE MOHAMMED V
FACULTE DE MEDECINE ET DE PHARMACIE RABAT
Année 2017 Numéro d’ordre : 22/16 CSVS
THÈSE DE DOCTORAT NATIONAL Centre d’Etude Doctoral Science de la vie et de la Santé
Formation Doctorale : Biologie médicale, Pathologie Humaine et Expérimentale et Environnementale
Intitulé de la thèse
Hajar LEMRISS Présentée et soutenue le 25 juillet 2017
JURY :
Professeur Président Faculté de Médecine et de Pharmacie, Université Mohammed V, Rabat
Professeur Azeddine IBRAHIMI Directeur de thèse Faculté de Médecine et de Pharmacie, Université Mohammed V, Rabat
Professeur Chakib NEJJARI Rapporteur Université Mohammed VI des Science de la Santé, Casablanca
Professeur Anas Kettani Rapporteur Faculté des Sciences Ben M’Sik, Université Hassan II, Casablanca
Professeur Rachid EL JAOUDI Rapporteur Faculté de Médecine et de Pharmacie, Université Mohammed V, Rabat
Analyse génomique de souches multi-résistantes de Staphylococcus capitis isolées dans des services
néonataux de plusieurs pays du monde

Dédicace
Un simple merci ne suffira pas à remercier ma famille.
A mes parents pour leur amour, leur présence et leur soutien sans faille.
Vous avez été des piliers durant ces années d’étude parfois laborieuses, vos encouragements ont
été bénéfiques et m’ont permis d’aller de l’avant.
Vous m’avez toujours poussé à faire ce que j’aimais. Trouvez ici le résultat de vos efforts, avec
toute la reconnaissance et l’amour que je vous porte.
A mon frère pour ta présence pendant les moments difficile, pour ta générosité infinie et ton
soutien. Je te dédie cette thèse et je te souhaite une vie pleine de joie, de bonheur et de succès.
A mes sœurs, qui ont été présentes et compréhensives, leurs appels et leurs mots de soutien m’ont
beaucoup apportés. Que cette thèse soit le témoignage de ma profonde affection.
A mon neveu, Youssouf, mon petit rayon de soleil.
A tout le reste de ma famille, ma grand-mère, ma tante, mon oncle,
mes cousins et mes cousines, pour vous encouragements je vous dédiez cette thèse avec ma
profonde affection.

REMERCIEMENTS
Cette thèse est le fruit de collaborations multiples entre le Laboratoire de
Biotechnologie Médicale (MedBiotech) de la Faculté de Médecine et de Pharmacie de Rabat,
Université Mohammed V, le Centre National de Référence des Staphylocoques (CNRS),
Lyon, France et le Laboratoire de Recherche et d’Analyses Médicales de la Fraternelle de la
Gendarmerie Royale (LRAM) Rabat. De plus, elle est le résultat d’un effort constant qui
n’aurait pu aboutir sans la contribution d’un nombre de personnes. Ainsi se présente
l’occasion de leur exprimer mes remerciements:
A Mon Directeur de thèse le Pr. Azeddine IBRAHIMI, Professeur d’Enseignement
Supérieur et Chef du Laboratoire de Biotechnologie Médicale (Médbiotech) à la Faculté de
Médecine et de Pharmacie de Rabat, Université Mohammed V, pour m’avoir accueilli dans
votre équipe de recherche, pour votre confiance dès le départ, pour votre encadrement, pour
votre disponibilité sans faille malgré un emploi du temps plus que chargé et pour votre
patience. Je tiens également à vous remercier pour la liberté d’action que vous m’avez donnée
à chaque étape, vos précieux conseils, vos connaissances scientifiques et vos encouragements
tout au long des années que j’ai passé au sein de votre laboratoire afin d'aboutir à cette thèse
d'analyse génomique. Veuillez trouver ici toute ma reconnaissance et mon profond respect.
A Monsieur le Dr Saâd EL KEBBAJ, Directeur du Laboratoire de Recherche et
d’Analyses Médicales de la fraternelle de la Gendarmerie Royale (LRAM), pour m’avoir
acceptée en tant que stagiaire à plusieurs reprises dans votre laboratoire afin d'apprendre les
outils de séquençage haut débit et d'analyses bioinformatiques pour aboutir à ce travail. Je
vous remercie pour l'intérêt que vous portez à la recherche scientifique et à l'avancée des
sciences biomédicales au Maroc. Veuillez trouver ici le témoignage de ma reconnaissance,
mes sincères remerciements et ma profonde gratitude.
A mon Co-encadrant de thèse le Pr. Frédéric Laurent, Professeur des Universités
de Microbiologie à la Faculté de Pharmacie de Lyon au sein de l’Université Claude Bernard
Lyon 1 et Co-directeur du Centre National de Référence des Staphylocoques, Lyon, France,
pour m’avoir confié ce sujet qui me paraissait si vaste et complexe au départ et qui s’avère
être si passionnant et enrichissant pour finir, et pour votre aide lors de la rédaction des articles
et pour nous avoir fourni les souches utilisées dans le présent travail. Je vous remercie

également pour votre disponibilité et la qualité de vos relations humaine, votre rigueur
scientifique, votre œil critique et pour tout le temps que vous m’avez consacré lors des
discussions constructives que nous avons échangées tout au long des années de doctorat.
J’espère sincèrement que ce travail sera l’occasion de pouvoir continuer à collaborer avec
votre laboratoire. Veuillez trouver ici l’expression de mes sincères remerciements et ma
profonde reconnaissance.
A ma Co-encadrante de thèse le Dr Sanaâ LEMRISS, Chef du Département de
Biosécurité PCL3 au sein du LRAM. Pour m’avoir si chaleureusement accueillie dans votre
laboratoire, pour m’avoir initié dans ce domaine et pour m’avoir guidée avec compétence. Je
tiens également à vous remercier pour votre patience et soutien tout au long de mon stage de
master et de ces années de thèse. Votre encadrement, vos encouragements et vos conseils
précieux pour l’élaboration de ce travail resteront à jamais marqués dans mon cœur. Merci de
m’avoir orientée vers ce sujet vaste et passionnant que représente le séquençage à haut débit
et l’analyse bioinformatique. Veuillez trouver ici l’expression de ma profonde reconnaissance.
A Mr. Le président de jury ainsi que les membres de jury, malgré vos multiples
occupations vous me faites l’honneur d’évaluer mon travail en acceptant de participer à mon
jury. Je vous exprime ma profonde gratitude.
A Monsieur le Pr. Jamal TAOUFIK, Professeur d’Enseignement Supérieure et
Directeur du Centre d’Etudes Doctorales des Sciences de la Vie et de la Santé à la Faculté de
Médecine et de Pharmacie de Rabat, Université Mohammed V, pour m’avoir inscrit au sein
de cet établissement et pour votre aide administrative. Aussi de me faire l’honneur de présider
le jury de cette soutenance de thèse malgré vos nombreuses occupations. Veuillez trouvez ici
toute ma reconnaissance et mes sincères remerciements.
A Monsieur le Pr. Chakib NEJJARI, Professeur d’enseignement Supérieure et
Président de l’Université Mohammed VI des Sciences de la Santé de Casablanca, pour
m’avoir accordé du temps et d’avoir accepté de participer à ce jury en tant que rapporteur de
cette thèse malgré votre emploi du temps plus que chargé. Veuillez trouver ici le témoignage
de mes respectueuses considérations.
A Monsieur le Pr. Anas Kettani, Professeur d’Enseignement Supérieure à la Faculté
des Sciences Ben M’Sik, Université Hassan II de Casablanca, pour m’avoir accordé du temps
en participant à ce jury et d’avoir la gentillesse de bien vouloir juger ce travail en tant que
rapporteur. Je tiens également à vous remercier pour vos encouragements tout au long de mes

années d’études et de vos précieux conseils. Veuillez trouver ici toute ma reconnaissance et
mon profond respect.
A Monsieur le Pr. Rachid ELJAOUDI, Professeur Agrégé à la Faculté de Médecine
et de Pharmacie de Rabat. Je vous suis reconnaissante d’avoir accepté de participer à ce jury
et de bien vouloir juger ce travail en tant rapporteur. Veuillez trouver ici l’expression de ma
profonde reconnaissance.
A Madame le Dr Patrícia Martins Simões, Ingénieur de recherche en
bioinformatique au laboratoire de bactériologie au Centre International de Recherche en
Infectiologie (CIRI), Hospices Civils de Lyon et fait partie de l’Equipe ”Pathogenèse des
infections à staphylocoques” du Centre National de Référence des Staphylocoques, Lyon,
France, pour avoir accepté de travailler en équipe l’analyse bioinformatique sur les données
de séquençage, pour votre aide, vos suggestions et pour vos judicieux conseils au moment de
la rédaction des articles. Je n’ai pu qu’apprécier votre réactivité au cours de nos discussions,
souvent virtuelles, pour aboutir à ce travail collaboratif, preuve que la science est avant tout
une histoire humaine. Bonne chance pour la suite et j’espère que nos chemins se recroiseront.
Veuillez trouver ici l’expression de ma profonde reconnaissance.
A Monsieur le Dr Jean-Philippe Rasigade, Biologiste Médicale au Centre
International de Recherche en Infectiologie (CIRI), Hospices Civils de Lyon, pour votre aide,
pour votre participation et vos conseils avisés au cours de la correction des articles. Veuillez
trouver ici l’expression de mes sincères remerciements.
A Madame le Dr Marine BUTIN, Pédiatre à Centre Hospitalier Universitaire de
Lyon, France et thésard au laboratoire de bactériologie médicale, Hospices Civils de Lyon.
J’ai beaucoup apprécié la qualité de nos échanges scientifiques et de travailler avec toi la
partie génomique de ce projet, merci pour ta motivation, ton énergie et tes conseils. Veuillez
trouver ici l’expression de mes sincères remerciements.
A Monsieur le Doctorant Yann Dumon, Thésard au laboratoire de bactériologie au
Centre International de Recherche en Infectiologie (CIRI), Hospices Civils de Lyon, qui nous
a rejoint en cours de route, merci pour ta contribution dans l’analyse bioinformatique et ton
aide. Veuillez trouver mes sincères remerciements.

A l’équipe du Centre Nationale de Référence des staphylocoques (CNR), je tiens
à remercier les personnes qui ont participé à ce travail, je vous renouvelle ma gratitude pour
vos efforts et vous souhaitez la réussite que vous méritez.
A Mlle Amal SOURI, Chef adjoint du Département de Biosécurité PCL3 au sein du
LRAM, pour votre aide et vos conseils durant toutes les années de ma thèse. Veuillez trouver
ici l’expression de mes sincères remerciements et l’expression de ma profonde
reconnaissance.
A Madame le Pr. Aicha RIFAI, Professeur Assistant à la Faculté des Sciences
Université Chouaib Doukkali pour avoir pris le temps de relire mon manuscrit. Veuillez
recevoir l’expression de ma profonde reconnaissance.
A l’équipe du Département de Biosécurité PCL3, LRAM, j’ai pu passer mes stages
dans un cadre particulièrement agréable, grâce à l’ensemble des membres de l’équipe de
Département de Biosécurité PCL3. Un immense merci pour leur aide, leur gentillesse, leur
infinie patience…, et leur bonne humeur.
Un grand merci à mes collègues au sein du laboratoire de Biotechnologie Médicale
(MedBiotech) pour leur soutien et leurs encouragements.
Ces remerciements ne seraient pas complets sans une pensée pour mes magnifiques
collègues de la promotion doctorale mes amis de longue date. Merci de m’avoir aidé et
encouragé, et pour m’avoir changé les idées quand j’en avais besoin.
Finalement, au terme de ce travail je tiens à remercier toutes les personnes que j'ai
rencontrées lors de mes études doctorales et qui ont contribué de près ou de loin au bon
déroulement de cette thèse.

LISTE DES PUBLICATIONS
ARTICLES
1. Lemriss, H., Martins Simões P., Lemriss, S., Butin, M., Ibrahimi, A., El Kabbaj, S.,
Rasigade JP., Laurent, F. (2014). Non-contiguous finished genome sequence
of Staphylococcus capitis CR01 (pulsetype NRCS-A). Standards in Genomic
Sciences, 9(3), 1118–1127.
2. Lemriss H., Dumont Y, Lemriss S., Martins-Simoes P., Butin M, Lahlou L., Rasigade JP,
El Kabbaj S., Laurent F., Ibrahimi A. (2015). Genome Sequences of Four Staphylococcus
capitis NRCS-A Isolates from Geographically Distant Neonatal Intensive Care
Units. Genome Announcements, 3(4), e00501–15.
3. Lemriss, H., Lemriss, S., Martins-Simoes, P., Butin, M., Lahlou, L., Rasigade, J.-P., El
Kabbaj, S. (2016). Genome Sequences of Multiresistant Staphylococcus capitis Pulsotype
NRCS-A and Methicillin-Susceptible S. capitis Pulsotype NRCS-C. Genome
Announcements. 4(3):e00541-16.
4. Martins Simões, P., Rasigade, J.-P., Lemriss, H., Butin, M., Ginevra, C., Lemriss, S.,
Goering, R. V., Ibrahimi, A., Picaud, J. C., El Kabbaj, S., Vandenesch, F and Laurent, F.
(2013). Characterization of a Novel Composite Staphylococcal Cassette
Chromosome mec (SCCmec-SCCcad/ars/cop) in the Neonatal Sepsis-Associated
Staphylococcus capitis Pulsotype NRCS-A. Antimicrobial Agents and
Chemotherapy, 57(12), 6354–6357.
5. Martins-Simoes P., Lemriss, H., Dumont, Y., Lemriss, S., Rasigade, J.-P., Assant-
Trouillet, S., Ibrahimi, A., El Kabbaj., Butin, M and Laurent, F. (2016). Single-Molecule
Sequencing (PacBio) of the Staphylococcus capitis NRCS-A Clone Reveals the Basis of
Multidrug Resistance and Adaptation to the Neonatal Intensive Care Unit
Environment. Frontiers in Microbiology, 7, 1991.

6. Butin, M., Rasigade, JP., Martins-Simões, P., Meugnier, H., Lemriss, H., Goering,
RV., Kearns, A., Deighton, MA., Denis, O., Ibrahimi, A., Claris, O., Vandenesch
F., Picaud JC and Laurent, F. (2015) Wide geographical dissemination of the
multiresistant Staphylococcus capitis NRCS-A clone in neonatal intensive-care units.
Clinical Microbiology and Infection, vol. 22, no. 1, pp. 46-52.
COMMUNICATIONS ORALES
1- P. Martins-Simões, Y. Dumont, M. Butin, H. Lemriss, S. Lemriss, A. Ibrahimi, S.El
Kabbaj, F. Vandenesch, J.P. Rasigade, F. Laurent. Caractérisation moléculaire par
séquençage PacBio du clone endémique mondial S. capitis NRCS-A responsable de sepsis
en réanimation néonatale : résistances, virulence et avantage sélectif. RICAI, Paris, 14
Décembre 2015.
2- P. Martins Simoes, H. Lemriss, J.-P. Rasigade, S. Lemriss, F. Vandenesch, S. El Kabbaj,
F. Laurent. Staphylococcus capitis NRCS-A: SCC, SCCMEC and Their Evolutionary
History. 5th Congrees of European Microbiologists, FEMS, Juillet 2013
3- M. Butin, P. Martins Simões, J.P. Rasigade, S. Lemriss, H. Lemriss, S. El Kabbaj, A.
Ibrahimi, S. Tigaud, F. Vandenesch, O. Claris, J.C. Picaud, F. Laurent. Diffusion mondiale
du clone Staphylococcus capitis NRCS-A en réanimation néonatale : caractérisation
moléculaire, analyse génomique et histoire évolutive. RICAI, Paris, 21 Novembre 2013.
4- Martins Simões P, Butin M, Lemriss H, Lemriss S, Deighton MA, Kearns A, Denis O,
Goering R, Ginevra C, Picaud JC, Ibrahimi A, EL Kabbaj S, Vandenesch F, Rasigade J-P,
Laurent F. Worldwide distribution of an endemic multi-resistant NRCS-A Staphylococcus
capitis clone in NICU: molecular typing, whole genome analysis and evolutionary
history.10th International Meeting on Microbial Epidemiological Markers, Paris, 02-05
October 2013.
COMMUNICATIONS AFICHEES

1- Butin M, Martins Simões P, Lemriss H, Lemriss S, Vandenesch F, Claris O, Picaud
JC, Rasigade JP, Laurent F. Staphylococcus capitis in neonatal late-onset sepsis:
unexpected worldwide dissemination of an endemic multi-resistant clone. European
Academy of Pediatric Societies. Barcelone, 17 - 21 October 2014.

RESUME
La prévalence des infections staphylococciques, nosocomiales et communautaires, augmente
régulièrement. Le traitement de ces infections est devenu de plus en plus difficile à cause de
l’émergence de souches multirésistantes. Les Staphylocoques ont la capacité d'acquérir des
déterminants de la résistance aux antimicrobiens rapidement, en particulier après
l'introduction de nouveaux agents antimicrobiens dans la pratique clinique.
Récemment, de nombreuses études ont décrit l’émergence dans les hôpitaux de plusieurs
pays, et particulièrement dans les unités de soins intensifs néonatales (USIN), des isolats de
Staphylococcus capitis résistantes à la méthicilline et ayant une sensibilité réduites à la
vancomycine (VISA). Ces isolats de S. capitis ont causé des septicémies mortelles chez les
nourrissons de faible poids (<1500 g), ce qui a entraîné une vague d'échecs thérapeutiques et
des problèmes socio-économiques.
Une étude de séquençage de haut débit (NGS), d’assemblage, d’analyse bioinformatique des
données NGS, de typage moléculaire et de comparaison des génomes complets de neuf
souches de S. capitis isolées de différents USIN dans le monde à différentes périodes a été mis
en place afin de contribuer à améliorer et à approfondir les connaissances sur le génome
complet de cet espèce peu étudié dans le monde.
Les objectifs étaient d'analyser et de comparer les éléments génétiques communs de ces neuf
souches aux six génomes de l’espèce S. capitis isolés chez des adultes et d'autres souches de
staphylocoques d’une part et de caractériser le clone NRCS-A et de comprendre les origines
de son succès d’autre part.
Les résultats de cette analyse et de comparaison de génomes ont permis d’avoir un premier
génome de référence de la souche S. capitis CR01 et l’identification des transferts horizontaux
des éléments génétiques mobiles (SCCmec-SCCcad / ars / cop et l’élément CRIPR, plasmide
et phage), des gènes de résistance aux antibiotiques, des déterminants de virulences associés à
la pathogénicité, de transposon, des séquences d’insertions et un gène nsr trouvé
exclusivement dans les souches de S. capitis appartenant au clone NRCS-A fonctionnel et
confère une résistance à la nisine.
Les caractéristiques génomiques trouvées mettent l'accent sur la contribution des éléments
génétiques mobiles à l'émergence de la résistance multiple du clone de S. capitis NRCS-A et

sur l'unicité du clone NRCS-A en ce qui concerne la résistance aux antibiotiques, les facteurs
de virulence et les éléments génétiques. Aucun déterminant de virulence connu spécifique à
NRCS-A n'a été détecté, ce qui ne soutient pas le rôle de la virulence en tant que force
motrice de l'émergence du NRCS-A dans les USIN du monde entier.
En conclusion, notre étude a permis de caractériser, d’élucidé et d’enrichir les connaissances
sur les génomes de S.capitis, ainsi que les mécanismes génétique impliqués à l’émergence,
l’évolution, l’adaptation et la dissémination du clone endémique NRCS-A.
Mots-clés : Staphylococcus capitis, NGS, bioinformatique, élément mobile, cassette
SCCmec-SCCcad / ars / cop, résistance aux antibiotiques, virulence, phage, plasmide,
transposon, évolution et dissémination clonale, NRCS-A.

ABSTRACT
The prevalence of staphylococcal nosocomial and community infections increases steadily.
Treatment of these infections has become increasingly difficult due to the emergence of
multidrug resistant strains. Staphylococci have the ability to acquire determinants of
antimicrobial resistance quickly, especially after the introduction of new antimicrobial agents
into clinical practice.
Recently, many studies have described the emergence of methicillin-resistant Staphylococcus
capitis isolates with reduced sensitivity to vancomycin (VISA) in hospitals in several
countries, particularly in the neonatal intensive care units (NICUs). These S. capitis isolates
caused fatal septicemia in low-weight infants (<1500 g), which resulted in a wave of
therapeutic failures and socio-economic problems.
A study using high-throughput sequencing (NGS), assembly, bioinformatics analysis of NGS
data, molecular typing and complete genome comparison of nine S. capitis strains which are
isolated from different NICUs worldwide at different period of time has been set up to
contribute to the improvement and deepening of knowledge about the complete genome of
this little studied species in the world.
The objectives were to analyze and compare the common genetic elements of these nine
strains to six S. capitis genomes isolated from adults and other strains of staphylococci in one
hand, and to characterize the NRCS-A clone and understand the origins of his success on the
other hand.
The results of this analysis and comparison of genomes allowed to have a first reference
genome of the S. capitis CR01 strain and the identification of the horizontal translocations of
the mobile genetic elements (SCCmec-SCCcad / ars / CRIPR, plasmid and phage), antibiotic
resistance genes, pathogenicity-associated virulence determinants, transposon, insert
sequences and an nsr gene found exclusively in S. capitis strains belonging to clone NRCS- A
which is functional and confers resistance to nisin.
The genomic characteristics found in this study emphasize the contribution of mobile genetic
elements to the emergence of multiple resistance of the S. capitis NRCS-A clone and the
uniqueness of the NRCS-A clone concerning antibiotic resistance, virulence factors and
genetic elements.

No known virulence determinant specific to NRCS-A has been detected, which does not
support the role of virulence as a driving force for the emergence of NRCS-A in NICUs
around the world.
In conclusion, our study allowed us to characterize, elucidate and enrich the knowledge on the
genomes of S.capitis, as well as the genetic mechanisms involved in the emergence,
evolution, adaptation and spread of the clone Endemic NRCS-A.
Keywords: NSCS-A, Staphylococcus capitis, NGS, bioinformatics, mobile element, cassette
SCCmec-SCCcad / ars / cop, resistance to antibiotics, virulence, phage, plasmid, transposon,
evolution and clonal dissemination.

H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 1
SOMMAIRE
INTRODUCTION 13
OBJECTIF DELA THESE 17
CHAPITRE I: Revue bibliographique 18
I.1 GENOMIQUE 18
I.1.1 Révolution biologique 18
I.1.2 Séquençage génomique 20
I.1.2.1 Historique 20
I.1.2.2 Constitution de banque d’ADN 22
I.1.2.3 Séquençage 1er
Génération 24
I.1.2.3.1 Méthodes de séquençage 24
I.1.2.3.2 Automatisation de méthode de Sanger 26
I.1.2.3.3 Stratégies de séquençage de génome entier 27
I.1.2.4 Séquençage nouvelle génération (Next Generation Sequencing (NGS)) 30
I.1.2.4.1 Séquençage de 2ème
génération 31
I.1.2.4.2 Séquençage de 3ème
génération 39
I.1.2.4.3 Technologies émergentes 40
I.1.2.5 Application de séquençage à haut débit 41
I.1.2.5.1 Le séquençage de novo 41
I.1.2.5.2 Le reséquençage 41
I.1.2.5.3 La métgénomique 42
I.1.2.6 Génomique comparative et exploitation des génomes 42
I.1.2.6.1 Assemblages des génomes 42
I.1.2.6.2 Annotation des génomes 44
I.1.2.6.3 Apport de la génomique comparative 44
I.2 LES STAPHYLOCOQUES 47
I.2.1 Taxonomie 47
I.2.1.1 Historique 47

H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 2
I.2.1.2 Classification 47
I.2.2 Réservoir et transmission 48
I.2.3 Physiopathologie 48
I.2.4 Infection 49
I.2.5 Caractéristiques intrinsèques 50
I.2.5.1 Génome 50
I.2.5.2 Enveloppe cellulaire 50
I.2.5.3 Capsule 50
I.2.5.4 Facteurs de virulence 51
I.2.5.5 Régulation des facteurs de virulence 52
I.2.6 Antibiothérapie 53
I.2.6.1 Mécanismes d’action des antibiotiques 53
I.2.6.2 Résistance aux antibiotiques 54
I.2.7 Cassettes SCCmec 56
I.2.7.1 Définition 56
I.2.7.2 Structure générale 57
I.2.7.3 Classification et typage 57
I.2.8 Staphylococcus capitis 60
I.2.8.1 Taxonomie 60
I.2.8.2 Habitat 60
I.2.8.3 Pouvoir pathogène 60
I.2.8.4 Résistance aux antibiotiques 60
I.2.8.5 Caractères bactériologiques 61
I.2.8.6 Caractéristiques génomiques 61
CHAPITRE II: Séquence génomique complète de Staphylococcus capitis
CR01 : Génome de référence 63
II.1 Abstract 65
II.2 Introduction 65
II.3 Classification and information 65
II.4 Genome sequencing information 70
II.5 Genome project history 70
II.6 Growth conditions and DNA isolation 70

H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 3
II.7 Genome sequencing and assembly 70
II.8 Genome annotation 71
II.9 Genome properties 71
II.10 Conclusion 73
II.11 Acknowledgments 74
II.12 References 74
CHAPITRE III: Séquençage de huit isolats de Staphylococus capitis isolées
de différentes régions géographiques dans le monde 77
III.1 Genome Sequences of Four Staphylococcus capitis NRCS-A Isolates from
Geographically Distant Neonatal Intensive Care Units 79
III.2 Genome Sequences of multiresistant Staphylococcus capitis pulsotype NRCS-A and
methicillin- 2 susceptible Staphylococcus capitis pulsotype NRCS-C 83
CHAPITRE IV: Génomique comparative de souches de Stapylococcus
capitis 88
IV.1 Characterization of a Novel Composite Staphylococcal Cassette Chromosome mec
(SCCmec-SCCcad/ars/cop) in the Neonatal Sepsis-Associated Staphylococcus capitis
Pulsotype NRCS-A 91
IV.2 Single-Molecule Sequencing (PacBio) of the Staphylococcus capitis NRCS-A Clone
Reveals the Basis of Multidrug Resistance and Adaptation to the Neonatal Intensive
Care Unit Environment 107
IV.2.1 Abstract 107
IV.2.2 Introduction 108
IV.2.3 Methods 109
IV.2.4 Results 113
IV.2.5 Discussion 122
IV.2.6 Author contributions 123

H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 4
IV.2.7 Funding 123
IV.2.8 Acknowledgments 123
IV.2.9 References 124
IV.2.10 Supplementary data 129
CHAPITRE V: Diffusion mondiale du clone Staphylococcus capitis NRCS-A
en réanimation néonatale : caractérisation moléculaire, analyse génomique
et histoire évolutive 136
V.1 Abstract 138
V.2 Introduction 138
V.3 Materials and Methods 139
V.4 Results 142
V.5 Discussion 146
V.6 Conclusion 147
V.7 Transparency declaration 148
V.8 Acknowledgements 148
V.9 References 148
V.10 Supplementary data 150
CHAPITRE VI : DISCUSSION GENERALE ET PERSPECTIVES 154
REFERENCES BIBLIOGRAPHIQUES 166
ANNEXES 181

H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 5
LISTE DES ABREVIATIONS
ADN : Acide désoxyribonucléique
ADNc : ADN complémentaire
Agr : accessory gene regulator
ARN : Acide ribonucléique
ARNm : ARN messager
ARNt : ARN de transfert
AST : Antimicrobial Susceptibility Testing
BAC : Chromosomes Artificiels Bactériens
BBH : Bidirectional best hits
BLAST : Basic Local Alignment Search Tool
BMR : Bactéries Multirésistantes
bp : Base pair (paire de base)
CARD : Comprehensive Antibiotic Resistance Database
ccr : Cassette Chromosome Recombinase
CDS : Coding Sequence (sequence codante)
CHUV : Centre Hospitalier Universitaire Vaudois
CMI : Concentration Minimale Inhibitrice
COG : Cluster of Orthologous Groups (Groupe de Cluster Orthologues)
CoNS : Coagulase-negative staphylococci
CRISPR : Clustered Regularly Interspaced Short Palindromic Repeat
CRT : Cyclic Reversible Termination
dru-typing : Direct repeat units typing
ddNTP : Didésoxyribonucéotides triphosphate
dNTP : Désoxyribonucéotides triphosphate
emPCR : PCR en émulsion
FC : Flow Cell
FRET : Fluorescence Resonance Emission Transfer
GIs : Genomic Islands
GISA : Glycopeptide intermidiate Staphylococcus aureus
GTR : General Time Reversable
IHGSC : International Human Genome Sequencing Consortium
KEGG : Kyoto Encyclopedia of Genes and Genomes

H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 6
LISTE DES ABREVIATIONS (Suite)
LOS : late-onset sepsis
NICU : neonatal intensive care units
MALDI-TOF : Matrix-Assisted Laser Desorption Ionization Time-Of-Flight
MID : Multiplex Identifier
MGE : Mobile Genetic Elements
ML : Maximum Likelihood
MLST : Multi Locus Sequence Typing
MSCRAMM : Microbial Surface Components Recognizing Adhesive Matrix Molecules
NCBI : National center for biotechnology information
NGS : Next-generation sequencing
NTS : Nouvelles Techniques de Séquençages
ORF : Open Reading Frames
ORF : Open Reading Frame
pb : paire de bases
PBP : Penicillin Binding Proteins
PCR : Polymerase Chain Reaction
PFGE : Pulsed-field gel electrophoresis
PGM : Personal Genome Machine
PMS : Phenol Soluble Modulin
PTP : PicoTiterPlate
PVL : Leucocidine de Panton-Valentine
SARM : S. aureus résistants à la méticilline
SARM-C : S. aureus résistants à la méticilline d’origine communautaire
SARM-H : Staphylococcus aureus Résistant à la Méticilline Hospitalier
SASM : S. aureus sensible à la méticilline
SCC : Staphylococcal Chromosomal Cassette
SCCmec : Staphylococcal Chromosomal Cassette mec
SCN : Staphylocoques à Coagulase Négative
SCP : Staphylocoques à Coagulase Positive
SCNRM : SCN résistants à la méticilline
SMS : Single Molecule Sequencing

H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 7
LISTE DES ABREVIATIONS (Suite)
SMRT : Single Molecule Real Time
SNN : Septicémies nosocomiales néonatales
SNP : Single Nucleotide Polymorphism
SOLiD : Sequencing by Oligonucleotide Ligation and Detection
RAST : Rapid annotation using subsystem technology
RDP : Ribosomal Database Project
VFDB : Virulence Factors database
VISA : Vancomycin Intermediate Staphylococcus aureus
WGS : Whole genome sequencing

H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 8
LISTE DES FIGURES
Figure I.1 : Dogme central de la biologie moléculaire.
Figure I.2 : Evolution du nombre de génomes complets disponible
(http://www.genomesonline.org).
Figure I.3 : Quelques étapes importantes démontrant l’évolution des progrès du
séquençage (Blow, 2008).
Figure I.4 : Schéma simplifié de préparation de banques d’ADN génomique et d’ADNc
(http://www.britannica.com/).
Figure I.5 : Schéma récapitulatif de la méthode de séquençage de Maxim et Gilbert
(http://departments.oxy.edu/biology/Stillman/bi221/092200/lecture_notes.htm).
Figure I.6 : Séquençage d’ADN par synthèse enzymatique avec chimie dye primer et dye
terminator (http://www.appliedbiosystems.com/).
Figure I.7 : Schéma de la stratégie du séquençage aléatoire global
(www.bio.davidson.edu/COures/genomics/method/shotgun.html).
Figure I.8 : Schéma simplifiée de la stratégie de séquençage "clone par clone".
Figure I.9 : Chronologie de la commercialisation des séquenceurs à haut-débit
Figure I.10 : Etapes du pyroséquençage (Ahmadianet al., 2006).
Figure I.11 : Préparation de la libraire d’ADN (http://www.454.com/).
Figure I.12 : Préparation des billes où sont fixées les sstDNA pour le séquenceur 454
(http://www.454.com/).
Figure I.13 : Schéma simplifié de Pyroséquençage sur PTP 454 (http://www.454.com/).
Figure I.14 : Amplification en pont de la technologie Illumina ((http://www.illumina.com/).
Figure I.15 : Les principales infections staphylococciques.
Figure I.16 : Facteurs de virulence exprimés à la surface ou excrétés par S. aureus en
fonction de la densité bactérienne (Ferry T et al., 2005).
Figure I.17 : L’histoire d’apparition des antibiotiques et des SARMs (Kos et al.,2012).
Figure I.18 : Représentation des 4 types de cassettes SCCmec (Hiramatsuet al., 2002).
Figure I.19 : Représentation schématique des 8 cassettes SCCmec (Van den Eede et al.,
2009).
Figure II.1 : Reference mass spectrum from Staphylococcus capitis strain (CR01).
Figure II.2 : Transmission electron microscopy of Staphylococcus capitis strain (CR01)
using a JEOL 1400. The scale bar represents 200 nm.

H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 9
LISTE DES FIGURES (Suite)
Figure II.3 : 16S rRNA Phylogenetic tree highlighting the position of Staphylococcus
capitis strain CR01 (indicated by the yellow circle) relative to other type strains
within the genus Staphylococcus.
Figure II.4 : Graphical circular map of the chromosome
Figure IV.1.1 : Comparative structure analysis of the composite staphylococcal chromosome
cassette SCCmec-SCCcad/asr/cop element of Staphylococcus capitis strain
CR01.
Figure IV.1.2 : Phylogenetic analysis of whole genome-sequenced S. capitis strains and
proposed evolutionary history of SCC elements acquisition in S. capitis.
Figure IV.1.3 : Schematic diagram and comparative analysis of the clustered regularly inter-
spaced short palindromic region (CRISPR) region in Staphylococcus capitis
strain CR01 SCCmec element with ST398 LA-MRSA strain 08BA02176.
Figure IV.2.1 : Comparison of the genetic content of the strain CR01 plasmid with the draft
genomes of the other three NRCS-A strains
Figure IV.2.2 : Comparison of the genetic content of the strain CR01 intact prophage present
in the other three draft genomes of NRCS-A strains.
Figure V.1 : Dendrogram and schematic representation of pulsotypes and dru types of
100 Staphylococcus capitis isolates from neonatal intensive-care units (NICUs)
(n = 86) or other settings (n = 14).
Figure V.2 : Single-nucleotide polymorphism (SNP)-based maximum-likelihood tree with
whole genome sequences of neonatal Staphylococcus capitis isolates from four
distinct countries and publicly available S. capitis genomes.
Figure V.S1 : Molecular characterization of S. capitis bloodstream isolates from neonates
(n = 12) and adult patients (n = 2).
Figure V.S2 : Flow diagram of isolate selection.

H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 10
LISTE DES TABLEAUX
Tableau I.1 : Mécanismes d’action des principales familles d’antibiotiques utilisées chez
l’Humain.
Tableau I.2 : Identification biochimique de S. capitis
Tableau I.3 : Lieu, origine et date de séquençage des 3 souches S. capitis.
Tableau I.4 : Caractéristiques générales du génome des trois souches de S. capitis
(http://www.ncbi.nlm.nih.gov/genome/genomes/2054?details=on&
http://www.ncbi.nlm.nih.gov/genome/genomes/155?details=on&project_id=53
751).
Table II.1 : Classification and general features of Staphylococcus capitis strain CR01,
pulsetype-NRCS-A according the MIGS recommendation.
Table II.2 : Project information.
Table II.3 : Nucleotide content and gene count levels of the genome.
Table II.4 : Number of genes associated with general COG functional categories.
Table III.1 : Summary of genome sequencing in the present study.
Table IV.1.S1 : Open reading frames of the novel composite staphylococcal chromosome
cassette (SCC) found in methicillin-resistant Staphylococcus capitis NCRS-A
prototype strain CR01. The first 42 ORFs (orf1 to orf42) were found in the
SCCmec element, while orf43 to orf70 were found in the SCCcad/ars/cop
element.
Table IV.1.S2 : Highest probable matches for the 15 interspersed DNA sequences (spacers)
identified in the clustered regularly inter-spaced short palindromic repeats
(CRISPR) region within the SCCmec element found in S. capitis strain CR01.
Table IV.2.1 : Baterial strains used in nisin susceptibility test.
Table IV.2.2 : General genomic features of S. capitis strain CR01 compared with another
three WGS NRCS-A genomes.
Table IV.2.3 : Comparison of virulence factors in S. capitis NRCS-A, non-NRCS-A, and S.
epidermidis after exclusion of virulence factors present only in S. aureus.
Table IV.2.4 : Genomic profiles of antibiotic resistance for clone NRCS-A strains from four
different countries.
Table IV.2.S1 : Genes found exclusively in the S. capitis NRCS-A clone and not in other S.
capitis public genomes.

H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 11
LISTE DES TABLEAUX (SUITE)
Table IV.2.S2 : Methylated motifs detected for S. capitis NRCS-A strain CR01.
Table V.1 : Antimicrobial susceptibility profile of Staphylococcus capitis bloodstream
isolates from neonates (n = 12) and adults (n = 2).
Table V.S1 : Characteristics of 7 house-keeping genes and related primers 389 used for the
determination of the genetic relationships between 14 S. capitis isolates.
Table V.S2 : Profiles of the MLST-like approach of S. capitis bloodstream isolates from
neonates (n=12) and 409 adults (n=2).

H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 12
LISTES DES ANNEXES
Annexe 1 : Chronologie non exhaustive des événements marquants survenus en
biologie (Rose), et en bioinformatique (Incolore).
Annexe 2 : Espèces et sous-espèces du genre Staphylococcus et hôtes associés.
Annexe 3 : Principales caractéristiques des différentes techniques de séquençage.

INTRODUCTION

INTRODUCTION
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V⎹ 13
Le Staphylocoque est observé pour la première fois par Pasteur en 1879 dans un pus de
furoncle, et ensuite identifié par Ogston en 1881 à travers la description d’observations
cliniques et bactériologiques relatives à son rôle dans le sepsis et la formation d’abcès
(Vandenesch et al., 2007). Actuellement, 50 espèces et sous espèces sont répertoriées dans le
genre Staphylococcus.
La prévalence des infections staphylococciques, nosocomiales et communautaires, augmente
régulièrement. Le traitement de ces infections est devenu de plus en plus difficile à cause de
l’émergence de souches multirésistantes. Les Staphylocoques ont la capacité d'acquérir des
déterminants de la résistance aux antimicrobiens rapidement, en particulier après
l'introduction de nouveaux agents antimicrobiens dans la pratique clinique.
A partir de 1950, dix ans après la découverte de la pénicilline, les Staphylococcus aureus
deviennent un problème thérapeutique majeur par l’acquisition de la résistance plasmidique à
la pénicilline. Peu après l’introduction d’un nouvel antibiotique, la méticilline (découverte en
1959), apparaissent des souches de S. aureus résistantes appelées S. aureus résistants à la
méthicilline (SARM). En fait, les SARM sont résistants non seulement à la méticilline mais
également aux autres antibiotiques de la même classe. Ces souches de SARM sont avant tout
responsables d’infections nosocomiales pouvant évoluer sur un mode épidémique (Mishaan et
al., 2005). En outre, des infections à SARM d’origine communautaire (SARM-C) ont été
également décrites (Dufour et al., 2002, Okuma et al., 2002).
Récemment, de nombreuses études ont décrit l’émergence dans les hôpitaux de plusieurs
pays, et particulièrement dans les unités néonatales de soins intensifs (NICU), des isolats de
Staphylococcus capitis résistantes à la méticilline et ayant une sensibilité réduites à la
vancomycine (Van Der Zwet, 2002 ; Gras-Le Guen et al., 2007). Ces isolats de S. capitis ont
causé des septicémies mortelles chez les nourrissons de faible poids (<1500 g), ce qui a
entraîné une vague d'échecs thérapeutiques et des problèmes socio-économiques (Rasigade et
al, 2012).
Une étude menée dans des NICU françaises a démontré la propagation d'une seule population
clonale de S. capitis (pulsotype NRCS-A) résistante à la méthicilline, associée à une
sensibilité réduite à la vancomycine, antibiotique de première ligne pour le traitement de ces
sepsis néonataux (Rasigade et al, 2012). De plus, ce même clone a été récemment identifié
dans les NICU en Belgique, au Royaume-Uni et en Australie, ce qui suggère une distribution
mondiale (Van Der Zwet et al., 2002 ; Venkatesh et al., 2006 ; D'mello et al., 2008 ;Rasigade

INTRODUCTION
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V⎹ 14
et al., 2012 ; Boghossian et al., 2013 ; Cui et al., 2013). En revanche, dans la bactériémie chez
l'adulte, S. capitis est rarement trouvée et lorsqu'elle est détectée, elle présente une plus
grande diversité en terme de génotypes et de profils de sensibilité aux antimicrobiens que
chez les nouveau-nés (Qin N et al., 2012).
Ce clone présente un profil atypique. En plus d'une résistance à l’ensemble des bêta-
lactamines, il possède une résistance à l’ensemble des aminosides, ainsi qu'une sensibilité
diminuée (hétérorésistance) aux glycopeptides qui constituent le traitement probabiliste de
première intention en cas de sepsis chez le prématuré. Cette combinaison de résistance est très
rarement retrouvée parmi les souches présentes chez l'adulte et reflète la pression de sélection
des antibiotiques utilisés dans les services de néonatologie.
Aujourd’hui, la connaissance scientifique de cette population clonale multirésistantes de S.
capitis (pulsotype NRCS-A), très peu étudiée et disséminée dans les NICU à travers le
monde, devient une urgence primordiale afin de réduire son endémie et limiter sa propagation.
Le séquençage complet du génome (WGS) est devenu un outil essentiel pour comprendre le
pouvoir des bactéries pathogènes. Des avancées majeures ont eu lieu dans le domaine du
séquençage au cours de cette dernière décennie avec le développement de plateformes de
séquençage nouvelle génération, caractérisées par un haut débit et la possibilité d’analyser
simultanément de nombreux échantillons. La génomique comparative des S. aureus est
désormais possible, car plusieurs génomes de S. aureus ont été séquencés et disponibles dans
les bases de données (Holden et al., 2004; Azarian et al., 2015 ; Sabat et al., 2017) . Des
travaux d’analyse génomique ont été également réalisés pour S. epidermidis RP62a et S.
epidermidis ATCC12228 en comparant leurs génomes à ceux des espèces staphylocoques les
plus pathogènes. Ces études ont permis de découvrir de manière approfondie les gènes qui
contribuent probablement à la virulence de S. epidermidis, en particulier la formation de
biofilm et la défense immunitaire (Zhang et al., 2003; Gill et al., 2005). Par la suite, des
projets de génomique comparative similaires ont été effectués pour d'autres espèces de
Staphylocoques à Coagulase Négative (SCN) telles que S. saprophyticus (Kuroda et al.,
2005), S. haemolyticus (Takeuchi et al., 2005 ; Cavanagh et al., 2014) et S. lugdunensis
(Heilbronner et al., 2011).
En revanche, très peu de travaux de recherche ont été entrepris concernant le séquençage du
génome complet de S. capitis. La disponibilité de ce dernier va permettre d’explorer les
caractéristiques génomiques (résistance aux antibiotiques, facteurs de virulence, éléments

INTRODUCTION
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V⎹ 15
génétiques mobiles (MGE) et gènes spécifiques) ayant un rôle dans l'émergence du phénotype
multirésistant du clone NRCS-A dans les différents NICU, et de comprendre l’histoire de son
évolution, son adaptation et sa dissémination dans le monde.
Dans ce contexte et en collaboration avec le Centre Nationale de Référence des
staphylocoques, Lyon, France, notre travail de thèse est un projet de séquençage, d’analyse
bioinformatique et de comparaison génomique de souches multirésistantes de S. capitis
(pulsotype NRCS-A) d'origines géographiques différentes et isolées à différentes périodes.
Le manuscrit est organisé de la façon suivante :
Un premier chapitre présente une revue bibliographique sur la génomique et les
staphylocoques.
Un second chapitre concerne le séquençage, l’assemblage et l’annotation de S. capitis
CR01 (pulsotype NRCS-A) qui représente le premier génome complet de référence de
souches isolées en neonatologie.
Un troisième chapitre traite le séquençage, l’assemblage et l’annotation de huit
souches de S. capitis d'origines géographiques différentes dans le monde et isolées à
différentes périodes.
Le chapitre 4 décrit le reséquençage, par un séquenceur de 3ème génération PacificBio
(SMRT), et l’assemblage du génome de la souche de S. capitis CR01 (pulsotype
NRCS-A), afin d’avoir un génome de référence fermé. De plus, ce chapitre présente
également le travail d’analyse et de comparaison de ce génome de référence par
rapport aux huit autres génomes séquencés de S. capitis, aux six génomes de S. capitis
disponibles sur les bases de données (souches adultes), et aux souches non S. capitis,
en utilisant différents outils et logiciels bioinformatiques, afin de caractériser et de
localiser les éléments génétiques communs (gènes de résistances, facteurs de
virulences, îlots de pathogénicité, îlots génomiques et MGE).
Le chapitre 5 illustre les diverses méthodes moléculaires utilisées (PFGE Sma et SacII,
typage SCCmec, MLST, Dru typing et analyse phylogénétique des génomes complet)
pour prouver la diffusion mondiale et l’unicité du clone épidémique NRCS-A de la
souche S. capitis.

INTRODUCTION
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V⎹ 16
Le chapitre 6 est consacré à une discussion générale des résultats et aux perspectives
ouvertes de ce travail.

OBJECTIF DE LA THESE

OBJECTIF DE LA THESE
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V⎹ 17
L’objectif principal de mon projet de thèse est de contribuer à améliorer et à approfondir les
connaissances du génome complet de l’espèce S. capitis peu étudié dans le monde, et de
caractériser le clone endémique NRCS-A de cette espèce afin de comprendre les origines de
son succès de dissémination et d’adaptation dans les NICU de quatre pays (France, Belgique,
Angleterre et Australie).
La méthodologie du travail s’articule autour de quatre grands axes :
Séquençage, assemblage et annotation d’un premier génome de référence de l’espèce
S. capitis CR01 isolée de l’NICU en France.
Séquençage, assemblage et annotation de huit autres génomes de S. capitis isolées des
NICU de quatre pays du monde (France, Belgique, Angleterre et Australie).
Identification, analyse et comparaison des éléments génétiques communs des neuf
souches séquencées pendant mon travail de thèse aux six génomes de l’espèce S.
capitis isolés chez des adultes et d'autres souches de staphylocoques disponibles sur
NCBI.
Caractérisation du clone endémique NRCS-A de cette espèce, afin de comprendre les
mécanismes génétique de résistances aux antibiotiques impliqués à son émergence,
son évolution, son adaptation et sa dissémination dans le monde.

CHAPITRE I
Revue bibliographique

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 18
I.1 GENOMIQUE
En l’espace de 50 ans seulement, la biologie est passée de l’étude d’un seul gène à l’étude de
génomes, de transcriptomes ou de protéomes d’organismes complets, avec une granularité
d’analyse allant de la simple molécule isolée à l’organisme dans sa globalité. Ceci a été rendu
possible par les avancées technologiques réalisées conjointement en biologie et en
bioinformatique qui ont été motivées par la volonté des scientifiques de soutirer
toujours davantage d’informations de notre génome. L’annexe 1 regroupe les événements
majeurs qui sont intervenus en biologie, et en bioinformatique depuis 1944.
I.1.1 Révolution biologique
La révolution biologique a été initiée par quatre événements majeurs : la découverte de l’ADN
en tant que support de l’information génétique (Avery et al., 1944) et de sa structure en double
hélice (Watson et Crick, 1953), ainsi que la mise en place du dogme central de la biologie
moléculaire et le déchiffrage du code génétique (Matthaei et al., 1962). Ces événements
constituent les fondements de la génomique.
Le dogme central est la modélisation simplifiée du flux de l’information génétique à travers
différentes molécules de la cellule et se résume en trois processus (Figure I.1).
ADN ARNm Protéine
Figure I.1:Dogme central de la biologie moléculaire
Grâce à ces principes, de nouvelles techniques de biologie moléculaire ont pu être
développées, et en synergie avec la montée en puissance et la disponibilité des ordinateurs, une
nouvelle discipline a émergé : la bioinformatique.
L’apparition de deux nouvelles approches de séquençage de l’ADN (Maxam et al., 1977;
Sanger et al., 1977) a permis à la bioinformatique prend réellement son envol. La
production de séquences par ces méthodes est l’occasion de créer les nouvelles banques de
données EMBL (Cochrane et al., 2009) et GenBank (Benson et al., 2009), afin de
répertorier ces séquences nucléiques, et de développer de nouveaux algorithmes
Réplication
Transcription Traduction

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 19
permettant de traiter les données biologiques. Ces derniers ont abouti aux outils majeurs
de la bioinformatique que sont FASTA (Pearson et Lipman, 1988), CLUSTALW (Thompson
et al., 1994), et BLAST (Altschul et al., 1997).
Avec l’apparition des séquenceurs automatiques et de nouveaux outils de biologie
moléculaire, la production des séquences s’accélère et l’on voit apparaître des projets de
séquençage de génomes complets qui aboutissent vers la fin du siècle.
Après dix années d’efforts, l’arrivée des premières séquences préliminaires du génome
Humain (Lander e tal., 2001; Venter et al., 2001) marque la fin de l’ère génomique et
l’entrée dans l’ère post-génomique. Cependant, il a fallu attendre jusqu’en 2004 pour
obtenir de la part de l’IHGSC Consortium une version que l’on peut considérer comme
finalisée (IHGSC, 2004).
Actuellement, grâce aux techniques de séquençage haut-débit, les projets de séquençage se
sont multipliés (environ 60000) de sorte que la communauté scientifique a accès à 1530
génomes complets et publiés (Figure I.2).
Figure I.2 : Evolution du nombre de génomes complets disponible
(http://www.genomesonline.org)

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 20
I.1.2 Séquençage génomique
Le séquençage de l’ADN constitue une méthode dont le but est de déterminer la succession
linéaire des bases A, C, G et T prenant part à la structure de l’ADN. La lecture de cette
séquence permet d’étudier l’information biologique contenue par celle-ci. Étant donné
l’unicité et la spécificité de la structure de l’ADN chez chaque individu, la séquence de
l’ADN permet de nombreuses applications dans le domaine de la médecine, comme, par
exemple, le diagnostic, les études génétiques, l’étude de paternité, la criminologie, la
compréhension de mécanismes physiopathologiques, la synthèse de médicaments, les
enquêtes épidémiologiques. L’objectif de la partie ci-dessous est de décrire l’évolution du
séquençage manuel jusqu’aux séquenceurs haut débit qui sont les plus utilisées à l'heure
actuelle.
I.1.2.1 Historique
En 1965, Holley et ses collaborateurs ont séquencé les deux premiers acides nucléiques de
l’histoire, l’ARNt de l’alanine de la bactérie Escherichia coli, puis celui de la levure
Saccharomyces cerevisiae. C’est grâce à la capacité de purifier des ARNt particuliers et à la
connaissance de RNAses, dont la spécificité était connue, que ces premiers séquençages ont
pu avoir lieu. De plus, il a été possible de déterminer la structure secondaire de l’ARNt,
puisque l’hybridation entre les bases était connue à l’époque. C’est en 1971 que la première
molécule d’ADN a été séquencée. Cette molécule consistait en une séquence de 12
nucléotides, soit la séquence des extrémités cohésives du phage lambda (Wu, 1970). Ces
premières séquences ont été obtenues à l’aide de réactions chimiques spécifiques, comme la
dépurination. Ces méthodes permettaient d’obtenir des séquences longues de 10 à 20
nucléotides.
En 1975, Sanger et Coulson ont introduit la méthode de terminaison des chaînes pour le
séquençage de l’ADN (Figure I.3). En 1977, Maxam et Gilbert ont conçu une méthode
similaire à celle de Sanger, mais ils utilisaient plutôt des nucléotides qui ne permettaient pas
l’élongation des chaînes. La même année, Sanger a introduit la méthode des
didéoxynucléotides, méthode qui permettait de séquencer jusqu’à 100 nucléotides. Cette
technique a permis le séquençage du génome du phage PhiX (Sanger et al., 1977).

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 21
La grande innovation suivante dans l’histoire du séquençage a été l’automatisation des
protocoles et de l’analyse (Première génération) (Hutchison, 2007). Cette avancée importante
a permis de démocratiser le séquençage, jusqu’à permettre le séquençage de génomes
complets (95%), dont le génome humain en février 2001 (Lander et al., 2001, Venter et al.,
2001) et le génome des TriTryp en 2005.
La seconde génération d’outils de séquençage est apparue en 2005 en réponse au prix élevé et
au faible débit du séquençage de première génération. Ici, des dizaines de milliers de
séquences sont traitées ensemble et en parallèle. C’est l’apparition du séquençage haut-débit
(« high throughput sequencing » ou encore le « next-generation sequencing »).
Alors que le projet de séquençage du génome humain a coûté trois milliards de dollars et a
duré 13 ans (achevé en 2006), celui de James Watson (âgé de 79 ans, co-découvreur de la
structure de l’ADN) a coûté un million de dollars et a été réalisé en deux mois. Il a été
effectué sur un séquenceur FLX de Roche (société 454 Life Sciences, Baylor College of
Medicine, Houston, Texas, États-Unis,). Quatre mois après, l’institut Craig Venter publiait le
génome complet de Craig Venter (Levy et al., 2007). Contrairement à celui de James Watson,
celui-ci a été séquencé selon la technique classique de Sanger (Figure 3).
En 2009, un des co-fondateurs de la société Helicos Bionsciences, Stephen R. Quake,
séquence son génome (Pushkarev et al., 2009) avec une profondeur de 28x et une couverture
de génome de 90% pour un coût de 48000 dollars. La même année (2009), quatre autres
génomes humains ont été décrits : ceux d’un homme yoruba du Nigeria (Bentley et al., 2008)
séquencé à une profondeur de 30x, de 2 coréens (Ahn et al., 2009 ; Kim et al ., 2009) à une
profondeur de 28 et 29x et d’un chinois Han (Wang et al., 2008) à une profondeur de 36x. Ces
séquençages individuels constituent une étape majeure vers la médecine personnelle
Les plateformes NGS actuellement disponible dans le marché utilisent des technologies de
séquençage à haut débit de la seconde génération proposées par Roche 454 Life Sciences,
Illumina, Solid et Ion Torrent et la troisième génération (« next-next-generation sequencing »)
proposé par Pacific Biosciences (PacBio RS) (Metzker, 2010 ; McAdam et al., 2014).

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 22
Figure I.3 : Quelques étapes importantes démontrant l’évolution des progrès du séquençage
(Blow, 2008).
I.1.2.2 Constitution de banque d’ADN
Tout projet de séquençage commence par la constitution d’une ou plusieurs banques d’ADN.
Cette banque est une collection de fragments d’ADN à séquencer qui ont été intégrés au
génome de cellules hôtes (généralement des microorganismes) à des fins de stockage et de
réplication. L’intégration est réalisée par l’intermédiaire d’une molécule d’ADN, appelée
vecteur de clonage, à l’intérieur de laquelle a été placé un fragment de l’ADN que l’on veut
séquencer (appelé dans le cas présent ‘insert’).
Il existe deux types de banques d’ADN : les banques d’ADN génomique dont les inserts sont
issus de la fragmentation du matériel génétique initial à séquencer, et les banques d’ADNc
dont les inserts sont des ARNm qui ont été « recopiés » en ADN sous l’effet d’une enzyme de
rétrovirus, la transcriptase inverse. Les banques génomiques sont utilisées dans le cadre de
séquençage de génomes, alors que les banques d’ADNc sont utilisées dans des études
d’expression de gènes.
Mis à part une première étape différente entre la construction d’une banque génomique et
celle d’une banque d’ADNc, les autres étapes sont communes aux deux (Figure I.4).

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 23
Première étape :
Banque génomique : consiste à fractionner l’ADN génomique par digestion partielle
avec une endonucléase, ou une enzyme de restriction, mais les méthodes physiques
sont préférées car elles sont plus reproductibles et les fragmentations sont plus
aléatoires. Ces méthodes physiques peuvent mettre en jeu la sonication, la nébulisation
sous haute pression, ou la force de cisaillement (Levinthal et Davison, 1961).
Banque d’ADNc : aucune fragmentation n’est nécessaire. Au contraire, une attention
toute particulière est apportée pour préserver les molécules d’ARN qui sont plus
fragiles que l’ADN, puisqu’elles ne sont constituées que d’un seul brin. Lors de cette
première étape, l’ARNm est rétro-transcrit en ADN sous l’action de la transcriptase
inverse, une enzyme de rétrovirus.
Les autres étapes :
Elles consistent en une séparation par électrophorèse sur gel d’agarose des fragments
d’ADN ou d’ADNc afin de sélectionner et d’extraire du gel les fragments de taille
désirée, puis de les intégrer au vecteur de clonage choisis. Les cellules hôtes sont
ensuite transformées par l’insertion d’un vecteur à leur matériel génétique. Finalement,
les cellules ayant été transformées sont cultivées et isolées en colonies bien distinctes.
Les cellules ayant intégré un vecteur sont sélectionnées à l’aide d’un des marqueurs du
vecteur, généralement un gène de résistance à un antibiotique présent dans le milieu de
culture, et qui empêche la multiplication des cellules non transformées. Chaque colonie
est ensuite repiquée, conservée et étiquetée par un identifiant unique en vue de son
séquençage.

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 24
Figure I.4 : Schéma simplifié de préparation de banques d’ADN génomique et d’ADNc
(http://www.britannica.com/)
I.1.2.3 Séquençage 1er
génération
I.1.2.3.1 Méthodes de séquençage
Il existe deux méthodes de séquençage dites « classiques » : la méthode de Maxam et
Gilbert, par dégradation chimique sélective et la méthode De Sanger par synthèse
enzymatique. Alors que l’utilisation de la première est restée confidentielle, la deuxième a été
largement développée et constitue aujourd’hui la technique de référence.
Méthode chimique (Maxam et al., 1977)
Cette méthode est basée sur une dégradation chimique de l'ADN et utilise les réactivités
différentes des quatre bases A, T, G et C, pour réaliser des coupures sélectives. En
reconstituant l'ordre des coupures, on peut remonter à la séquence des nucléotidesde l'ADN
correspondant. On peut décomposer ce séquençage chimique en six étapes successives :
Marquage : Les extrémités des deux brins d'ADN à séquencer sont marquées par un
traceur radioactif (32
P). Cette réaction se fait en général au moyen d'ATP radioactif et de
polynucléotide kinase.

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 25
Isolement du fragment d'ADN à séquencer : Celui-ci est séparé au moyen d'une
électrophorèse sur un gel de polyacrylamide. Le fragment d'ADN est découpé du gel et
récupéré par diffusion.
Séparation de brins : Les deux brins de chaque fragment d'ADN sont séparés par
dénaturation thermique, puis purifiés par une nouvelle électrophorèse.
Modifications chimiques spécifiques : Les ADN simple-brin sont soumis à des
réactions chimiques spécifiques des différents types de base. Walter Gilbert a mis au
point plusieurs types de réactions spécifiques, effectuées en parallèle sur une fraction de
chaque brin d'ADN marqué : par exemple, une réaction pour les G (alkylation par le
sulfate de diméthyle), une réaction pour les G et les A (dépurination), une réaction pour
les C, ainsi qu'une réaction pour les C et les T (hydrolyse alcaline).
Coupure : Après ces réactions, l'ADN est clivé au niveau de la modification par réaction
avec une base, la pipéridine.
Analyse : Pour chaque fragment, les produits des différentes réactions sont séparés par
électrophorèse en conditions dénaturantes et analysés pour reconstituer la séquence de
l'ADN. Cette analyse est analogue à celle que l'on effectue pour la méthode de Sanger
(Figure I.5).
Les produits chimiques utilisés dans les milieux réactionnels lors des coupures spécifiques
étant excessivement dangereux pour la santé, cette méthode a été abandonnée au profit de la
méthode par synthèse enzymatique.
Figure I.5 : Schéma récapitulatif de la méthode de séquençage de Maxim et Gilbert (http://departments.oxy.edu/biology/Stillman/bi221/092200/lecture_notes.htm)

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 26
Méthode enzymatique (Sanger et al., 1977).
Cette méthode, encore appelée méthode De Sanger en raison de son inventeur, est basée sur
l’activité de l’ADN polymérase qui permet de polymériser un brin d’ADN complémentaire à
un brin matrice, à partir d’un oligonucléotide, appelé amorce. Cette capacité est utilisée pour
synthétiser un brin complémentaire, mais de façon incomplète, en arrêtant aléatoirement la
réaction de manière à obtenir statistiquement des produits issus de réaction interrompue à
chacune des bases du fragment à séquencer.
Le mix réactionnel est constitué du vecteur de clonage contenant le fragment à cloner, de la
polymérase, des amorces et des dNTP. Pour arrêter aléatoirement la réaction, une faible
concentration de ddNTP est ajoutée. Ces ddNTP ne comportant pas de groupement 3’-OH,
ils agissent comme des terminateurs de la réaction de polymérisation en empêchant
l’accomplissement d’une liaison 5'-3' phosphodiester ultérieure.
A l’instar de la méthode chimique, un marquage des produits de réaction est nécessaire pour
pouvoir détecter ces derniers après leur séparation par électrophorèse sur gel. Pour ceci, il
existe deux chimies : dyeprimer et dyeterminator(Figure I.6).
Figure I.6 : Séquençage d’ADN par synthèse enzymatique avec chimie dye primer et dyeterminator (http://www.appliedbiosystems.com/)
I.1.2.3.2 Automatisation de méthode de Sanger
La lecture manuelle de gels de séquençage est fastidieuse et sujette à de multiples erreurs
d’interprétation, surtout après avoir lu les premiers milliers de bases. C’est pourquoi cette
tâche a été automatisée à l’aide des séquenceurs.

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 27
L’étape de séquençage par synthèse enzymatique est toujours nécessaire. Elle est déléguée à
des robots qui la réalisent dans des microplaques de 96 ou 384 puits, correspondant à autant
de clones séquencés. Ces plaques sont ensuite transférées dans un séquenceur, qui va réaliser
l’électrophorèse tout en enregistrant sur ordinateur les profils d’intensités lumineuses des
fluorochromes. Ces profils sont appelés chromatogrammes.
Il existe deux types de séquenceurs automatiques de technologie Sanger :
Les séquenceurs à gel plat disposent d’un gel enserré entre deux plaques de verre en
haut duquel sont disposés des puits.
Des séquenceurs à capillaires tel que ABI 3730 DNA Analyser de Life technologies.
Cette technologie a permis des progrès en génomique depuis sa commercialisation en
2002. En 2004, en utilisant le séquenceur à capillaire automatique ABI3700, dans le
cadre d'un projet collaboratif, le génome de P. atrosepticum SCRI1043 a été séquencé,
ce qui a permis la caractérisation des facteurs de virulence de cette espèce par
comparaison aux pathogènes de plantes ou d'animaux de la famille des
Enterobacteriaceae (Bell et al., 2004).
I.1.2.3.3 Stratégies de séquençage de génome entier
On distingue deux méthodes de séquençage des génomes entiers :
Séquençage aléatoire globale (ou whole-génome shotgun)
Séquençage par ordonnancement hiérarchique
Dans les deux cas, l'ADN génomique est tout d'abord fragmenté par des méthodes soit
enzymatiques (enzymes de restriction), soit physiques (ultrasons).
Séquençage aléatoire globale « whole-génome shotgun »
Le séquençage du premier génome bactérien, Haemophilus influenzae, a introduit la méthode
du séquençage aléatoire global (Fleischman et al., 1995). Cette dernière avait été proposée en
1982 par Sanger et ses collaborateurs et avait été utilisée, à plus petite échelle, pour le
séquençage du génome du phage lambda. C’est la stratégie retenue aujourd’hui pour tous les
génomes bactériens. On l’associe plus volontiers aux sociétés privées, telles que Celera
Genomics ou Syngenta, parce qu’elle est rapide et économique. Celera a ainsi accompli le
séquençage de plusieurs organismes par une stratégie aléatoire globale. Toutefois, il faut
remarquer que, pour de grands génomes comme celui de l'Homme, ces sociétés se sont

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 28
souvent appuyées sur des données de cartographie établies par les chercheurs académiques.
En outre, pour ces grands génomes, on tend aujourd’hui à utiliser des stratégies mixtes,
mêlant séquençage aléatoire global et "clone par clone".
Le séquençage du génome humain a donné lieu à une lutte médiatique entre Celera Genomics
et le consortium international responsable du « projet Genome humain », qui utilisait quant à
lui une stratégie « clone par clone »
Principe
La technique "shotgun" repose sur un principe simple : découper un génome en un grand
nombre de fragments de petite taille (Figure I.7). Les extrémités d'une partie de ces fragments
sont ensuite séquencées, puis ces séquences sont assemblées sur la base de leurs
chevauchements grâce à des programmes informatiques pour essayer de produire une
séquence complète (Ameziane et al., 2005). Les difficultés d'une telle technique sont à deux
niveaux :
1- avoir assez de fragments pour couvrir le génome dans son entier
2- réussir l'assemblage
Figure I.7: Schéma de la stratégie du séquençage aléatoire global
(www.bio.davidson.edu/COures/genomics/method/shotgun.html)

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 29
Séquençage par ordonnancement hiérarchique
Une stratégie différente dite "clone par clone" ou hiérarchique a été adoptée par divers
consortiums internationaux pour le séquençage du génome humain et d’autres grands
génomes (riz, arabette, ver).
Principe :
L’ADN génomique est découpé en fragments de 50 à 200 kb, puis cloné dans un vecteur
adapté (BAC). Le nombre de clones doit permettre une couverture de 5 à 10 fois la longueur
totale du génome étudié. Le chevauchement et l'ordonnancement des clones sont réalisés soit
par hybridation de sondes spécifiques, soit par analyse des profils de restriction, soit plus
fréquemment par un ordonnancement après séquençage et hybridation des extrémités des
BAC. Après ordonnancement des clones, ils sont fragmentés et séquencés individuellement,
puis assemblés par alignement bioinformatique.
Avec un volume de séquences lues équivalant à 5 ou 6 fois la taille d’un génome comme celui
de l’être humain (profondeur de 5X), il est possible d’assembler clone par clone
une"Ébauche" où l’on obtient pour chaque clone quelques dizaines de contigs. Toutefois, ces
contigs initiaux ne sont en général ni ordonnés, ni orientés. En augmentant la profondeur
jusqu’à 10X, on obtient des contigs plus longs, moins nombreux, et surtout ordonnés et
orientés grâce aux paires de lectures ou à d’autres informations (Figure I.8). Reste alors à
entreprendre une étape fastidieuse de lissage et de finition, pour garantir à tout endroit moins
d’une erreur tous les 10 000 nucléotides, et pour boucher les trous résiduels par un travail
local (www.genoscope.cnr.fr).

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 30
Figure I.8 : Schéma simplifié de la stratégie de séquençage "clone par
clone"(www.genoscope.cnr.fr).
I.1.2.4 Séquençage nouvelle génération (Next Generation Sequencing (NGS))
Les nouvelles technologies de séquençage dites NGS (Next Generation Sequencing), ont
permis de séquencer de l'ADN plus rapidement avec une meilleure qualité tout en réduisant le
coût qui est passé de plus de 1000 dollars pour un million de bases en 2001 à moins de 100
dollars en 2015 (http://www.genome.gov/sequencingcosts/).
Des séquenceurs de paillasse (benchtop sequencers) ont été développés pour permettre l’accès
aux technologies de NGS à la plupart des laboratoires, y compris aux laboratoires de
microbiologie médicale, et non plus aux seuls laboratoires spécialisés (Figure I.9). Ils sont
caractérises par un cout moindre, une petite taille et des performances restreintes mais
suffisantes pour la plupart des applications du NGS en laboratoire de recherche standard
(Loman NJ et al., 2012 ; Junemann S et al., 2013)

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 31
Figure I.9 : Chronologie de la commercialisation des séquenceurs à haut-débit
I.1.2.4.1 Séquençage de 2ème
génération
La technique de séquençage De Sanger a atteint ses limites en termes de rendement. Il n’est
plus possible d’accélérer le séquençage qu’en multipliant le nombre de gels, de capillaires ou de
machines, et la vitesse d’électrophorèse peut difficilement être accélérée sans amoindrir la
qualité de séquençage. C’est pourquoi de nouvelles technologies ont vu le jour pour
aboutir à des séquenceurs temps réel (séquençage base par base) massivement
parallélisés, utilisant des techniques très diverses tel que :
Le pyroséquençage à haut-débit (Technologie 454)
Les techniques de terminaison cyclique réversible (Solexa/Illumina et Helicos)
Le séquençage par ligation (SOliD).
Le séquençage par nano-mesure de PH (PGM)
Malgré leurs différences méthodologiques, les techniques de séquençage à haut débit
actuellement offertes suivent un protocole dont les grandes lignes sont similaires (benthly
2008). Même si les méthodes utilisées à chacune des étapes sont différentes, l’ordre dans
lequel elles sont effectuées est invariable :

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 32
Préparation d’une librairie de séquences marquées par des adaptateurs ;
Amplification des séquences marquées de manière à ce qu’elles soient séparées
spatialement ;
Séquençage par réactions enzymatiques cycliques mesurées en temps réel.
Pyroséquençage (technologie 454)
Le pyroséquençage est la technique qui connaît actuellement le plus grand succès
concurrençant la méthode de Sanger. Cette technique de séquençage d’ADN introduite depuis
1988, par Hyman, a été améliorée par introduction d’une étape PCR. Il s’agit d’un séquençage
par synthèse qui se caractérise par la révélation en temps réel de l’activité de l’ADN
polymérase et qui ajoute un seul nucléotide non fluorescent à la fois.
La première étape consiste à préparer le mélange réactionnel, avec les enzymes clés et les
différents substrats. Ici, les nucléotides ne sont pas ajoutés tous ensemble comme dans une
réaction de séquençage de Sanger mais successivement. Si le nucléotide ajouté dans le milieu
réactionnel correspond à celui attendu par la polymérase, il sera incorporé dans le brin en
cours de synthèse (d’élongation) libérant un pyrophosphate. L’ATP sulfurylase vient alors
transformer ce pyrophosphate (Ludwig et al., 2004) en ATP qui est alors utilisé, couplé à une
Luciférine, par une luciférase avec production d’oxyluciférine et d’un signal lumineux.
L’apyrase dégrade les nucléotides en surplus. Le signal lumineux est capté par un capteur
CCD, puis reproduit sous forme d’un pic sur le Pyrogramme. La hauteur de ce pic est fonction
de l’intensité du signal lumineux, elle-même proportionnelle au nombre de nucléotides
incorporés en même temps. On peut donc déduire la séquence à partir de la taille des pics
obtenus. Par ailleurs, en cas de mélange de nucléotides à une même position (polymorphisme
de séquence), la taille des pics permet d’avoir une quantification de la proportion de brins
porteurs de l’un ou l’autre des nucléotides (Figure I.10).

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 33
Figure I.10 : Etapes du pyroséquençage (Ahmadianet al., 2006).
La technologie de 454 Life Sciences conçue par Rothberg (Margulieset al., 2005) est fondée
sur l’intégration de plusieurs techniques :
- le pyroséquençage,
- les technologies des plaques en fibre optique pico-titré (PTP),
- la PCR en émulsion « emPCR » dans des microréacteurs,
- les technologies informatiques de pointe pour l’acquisition, le traitement et
l’analyse des images.
Préparation de la libraire d’ADN (Figure I.11)
L’ADN génomique est fragmenté par nébulisation (1400-1800 pb), des séquences
adaptateurs sont fixées à chaque extrémité des fragments d’ADN à séquencer. Un des
adaptateurs contient un marqueur biotine en 5’ qui sert à fixer les fragments sur des billes
grâce à un système streptavidine/biotine. Les fragments d’ADN sont dénaturés afin de
libérer les brins non-biotinés, pour obtenir une librairie d’ADN simple brin.

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 34
Figure I.11 : Préparation de la libraire d’ADN (http://www.454.com/)
PCR en émulsion « emPCR » (Figure I.12)
Les sstDNA sont ensuite amplifiées par « emPCR ».Ces molécules sont fixées sur des billes
de manière à fixer une seule molécule de sstDNA par bille. Les billes sont, ensuite,
émulsionnées dans une solution eau/huile dans des proportions favorisant l’inclusion d’un
seul couple bille-sstDNA par gouttelette d’eau, qui va agir comme un microréacteur pour la
réaction de PCR. Après plusieurs cycles de PCR, chaque gouttelette contient une bille où sont
fixés tous les clones amplifiés. Un ensemble de clones issus d’une même matrice est souvent
dénommé «polonie» en référence à l’amplification de toute une colonie de clones par la
polymérase.
Figure I.12 : Préparation des billes où sont fixées les sstDNA pour le séquenceur 454
(http://www.454.com/)
Pyroséquençage (Figure I.13)
Afin de paralléliser massivement les réactions de pyroséquençage, le séquenceur 454 utilise
une PTP constituée de puits. Les billes couvertes de sstDNA sont mélangées au mix de
réaction contenant la polymérase et sont étalées sur la PTP. Les puits de cette dernière sont

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 35
d’une forme telle qu’une seule bille peut être déposée dans chaque puits. Les puits et les billes
de sstDNA sont ensuite recouverts de billes d’enzymes immobilisées contenant la sulfurylase
et la luciférase. Au moment de pyroséquençage, chacune des bases A, C, G, et T est
ajoutée séquentiellement sur la PTP. L’image des intensités de lumière émise par la
réaction intervenue dans chaque puits après l’ajout de chaque base est enregistrée. Avant
l’ajout de la base suivante, les bases non intégrées par la polymérase sont dégradées par une
solution d’apyrase. Ce cycle d’ajout des quatre bases est réitéré 42 fois pour obtenir des
séquences d’une longueur moyenne de 700 pb pour un total de 400 à 600 millions de
bases séquencées (Shendure et Shendureet al., 2008; Metzker, 2010).
Figure I.13 : Schéma simplifié de Pyroséquençage sur PTP 454 (http://www.454.com/)
Analyse des images et détermination de la séquence d’ADN
Connaissant l’ordre dans lequel les 4 nucléotides sont ajoutés automatiquement, l’analyse des
différentes images capturées permet la déduction de la séquence des différents fragments
d’ADN (illustré par Pyrogramme). Des logiciels bioinformatiques sont ensuite utilisés pour
reconstituer la séquence initiale grâce au groupage des contigs. Cette technologie produit de
longues séquences qui facilitent l’assemblage de novo des génomes sans utiliser un génome
de références. En quelque année, la longueur des séquences est passé d’environ 300pb à
700pb et aujourd’hui, la mise à jour de l appareil permet d’atteindre des séquences d’environ
1Kb. Ainsi, une expérimentation sur GS-FLX+génère 1Gb de nucléotide en 23h (Loman et
al., 2012; Jûnemann et al., 2013).. Ces capacités technologiques en font aujourd’hui un outil
indispensable dans les études génomiques et transcriptomiques qui requière un assemblage de
novo. En 2013, la société Roche a annoncé qu'elle prévoyait d'arrêter la commercialisation de
la technologie 454 d'ici mi-2016 (www.genomeweb.com).

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 36
Techniques de terminaison cyclique réversible (Illumina /Solexa)
Initialement développée par Solexa, la plateforme Illumina Genome analyser (Sandiego,
USA), a mis en place une technologie de séquençage sur lames de type puces à ADN, fondée
sur l’intégration de plusieurs techniques : les biopuces à ADN, la nanotechnologie, une
variante de la technique de Sanger appelé terminaison cyclique réversible (CRT), ainsi que
des technologies informatiques de pointe pour l’acquisition, le traitement et l’analyse des
images (Fedurco et al., 2006).
La spécificité de cette technologie repose sur une amplification en pont (bridge PCR) des
fragments à séquencer. Elle a lieu sur une surface de verre appelée flow cell (FC), similaire à
une lame de microscope, divisée en huit lignes (à l'origine, une ligne par échantillon). Les
fragments de la librairie à séquencer possèdent des adaptateurs à leurs extrémités. Ceux-ci
vont leur permettre de se fixer de façon aléatoire sur la FC, par hybridation sur les amorces
qui en couvrent la surface (Figure I.14). Un nouveau brin est alors synthétisé par une
polymérase (a) : il est fixé de façon covalente à la FC. Le brin d'origine est alors éliminé par
dénaturation (b) et l'extrémité libre du brin restant s'hybride à une amorce adjacente pour
former un pont (c). La polymérase synthétise à nouveau le brin complémentaire pour former
un pont d'ADN double brin (d) puis les deux copies sont libérées par dénaturation (e). Le
cycle d'amplification en pont (étapes c à e) recommence pour former à terme un regroupement
d'ADN clonal en une zone appelée cluster (f). Les brins anti-sens (correspondant aux amorces
vertes) sont ensuite clivés (g) : c'est la linéarisation.
L'extrémité 3' libre des fragments d'ADN est bloquée et l'amorce de séquençage s'y hybride
(Figure 14). Le séquençage s'effectue sur des centaines de millions de clusters simultanément,
grâce à une chimie de terminateurs réversibles : des nucléotides bloqués marqués par
fluorescence sont ajoutés, l'un d'entre eux est incorporé, la fluorescence émise est relevée puis
le fluorophore et le bloqueur sont clivés permettant l'ajout d'un nouveau nucléotide. A chaque
cycle d'incorporation, une base peut être déterminée. (Eid et al., 2009).

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 37
Figure I.14 : Amplification en pont de la technologie Illumina ((http://www.illumina.com/)

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 38
Aujourd’hui le système HiSeq 2000 possède un rendement de 6 milliards de séquences pour
un total d’environ 600 Gb en 11 jours (avec une taille moyenne de séquence de 100 pb). Cette
technologie est aujourd’hui très largement employée (séquençage, reséquençage, Chip-Seq,
RNA-Seq).
Séquençage par ligation (SOliD)
La plateforme SOLiD (Sequencing by Oligonucleotide Ligation and Detection) du système
décrit par Shendure et al., 2005, emploie des processus de préparation et amplification
d’échantillons similaires à ceux développés pour la technologie 454. Mais le processus de
séquençage diffère considérablement, dans le fait qu’il repose sur un séquençage par
«ligation», où des oligonucléotides de 8 bases marqués par fluorescence sont liés de manière
séquentielle sur l’amorce de séquençage. Chacun des quatre types de sonde porte un
fluorophore distinct en 3’, représentant la séquence de l’échantillon complémentaire aux
quatrièmes et cinquièmes bases de la sonde oligonucléotidique. Le signal fluorescent généré
par l’hybridation de la sonde est alors détecté et enregistré par un scanner laser et un
processus d’acquisition de données similaires au système Solexa. La sonde liée est alors
clivée entre la 5ème
et 6ème
base, enlevant la moitié fluorescente, et des séries répétées de la
ligation de la sonde (Metzker, 2010).
Grâce à cette méthode, il est possible de lire jusqu'à un milliard et demi de séquences en
parallèle de 75 bases de long (des fragments courts). Le système incorporé de correction
d'erreurs associé à l'utilisation de la ligase rend cette technologie très fiable et utilisable dans
des projets de reséquençage de génome et de transcriptome.
Séquençage par nano-mesure de PH (PGM)
En 2007, Jonathan M. Rothberg fonde la compagnie Ion Torrent, rachetée en 2010 par Life
Technologies, qui commercialisera en décembre 2010 son premier séquenceur, le PGM
(Personal Genome Machine) pour 50000 $. La technologie utilise une méthode de séquençage
par synthèse, similaire au pyroséquençage, basée sur la libération naturelle d’un ion H+ lors
de l’incorporation d’un nucléotide à un fragment d’ADN en cours de production. La séquence
est déterminée par mesure des variations de pH suite à l’incorporation des différents
nucléotides, au sein d’une puce semi-conductrice. Ce système de détection « simple » ne

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 39
nécessitant ni système optique ni marqueur fluorescent, font de la technologie Ion Torrent une
méthode de séquençage rapide et peu couteuse par rapport aux autres plateformes NGS.
La dernière version de ce séquenceur (puce 318) lui permet désormais de lire 1 Gb avec une
grande précision en seulement deux heures et la longueur de lecture atteindra les 400 pb en
2012. Le PGM a ainsi servi à décoder le génome de la souche d'Escherichia coli qui a fait des
ravages en Europe et provoqué un scandale sanitaire en mai 2011 (Mellmann et al., 2011).
I.1.2.4.2 Séquençage de 3ème génération
La troisième génération de séquençage de masse est symbolisée par le séquençage d’une
seule molécule d’ADN « SMS » (Gupta., 2008). Contrairement à la seconde génération,
aucune amplification de l’ADN (ou ARN) n’est nécessaire pour effectuer les mesures. Une
seule molécule est « lue ». Ces innovations diminueraient la durée et simplifieraient
considérablement les étapes de préparation des banques d’ADN. De plus, l’élimination de ces
étapes permettrait aussi de diminuer les sources de biais possibles pouvant être introduits par
les phases de PCR (erreurs de polymérisation ou recombinaisons par PCR). Un autre avantage
de cette technique réside dans le fait que des ADN faiblement concentrés voir même dégradés
pourraient ainsi tout de même devenir exploitables (Orlando., 2011).
Technologie HeliScope
Helicos Biosciences a développé le premier séquenceur d'une molécule unique : le HeliScope
Single Molecule Sequencer. A la différence de la chimie utilisée sur la plateforme Illumina,
ici les nucléotides sont marqués avec le même fluorophore et cette technologie apparaît
parfaitement adapté aux études transcriptomiques quantitatives. Helicos ne vend désormais
plus d'appareil et propose un service de séquençage.
Technologie SMRT (Single Molecule Real-Time Analysis)
La technologie de séquençage PacBio, développée par l'entreprise Pacific Biosciences
(Californie, USA), permet de séquencer une molécule d'ADN en temps réel (SMRT : single
molecule real time) à l'aide d'un instrument, le PacBio RS. Le processus de séquençage se fait
sur un support appelé "SMRT Cell" composé de dizaines de milliers de puits sous forme de
structure détecteur appelé ZMW (zero-mode waveguide). Au fond de chaque puits, d'un
diamètre de 100 nm, est fixée une seule polymérase. Ces structures ZMW permettent

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 40
d'enregistrer en temps réel les dNTP incorporés où chacun des 4 nucléotides est lié à un
flurochrome différent.
Les signaux obtenus sont enregistrés et analysés par des méthodes informatiques de pointe
(Eid et al., 2009 ; Sengenès, 2012).
Pour l'assemblage des lectures issues du séquenceur PacBio, l'entreprise a développé plusieurs
algorithmes d'assemblage adaptés selon la taille des lectures et la couverture du séquençage.
Dans le cas des génomes bactériens, il s'agit de HGAP (Hierarchical Genome Assembly
Process) qui comprend plusieurs étapes dans le processus d'assemblage (Chin et al., 2013).
La vitesse de séquençage de cette technologie est de 10 bases par seconde avec une longueur
moyenne de lecture de plus de 1000 pb (jusqu’à 3000 pb). Malgré son succès, la technologie
PacBio génère aussi des erreurs de séquençage liées à la quantification des homopolymères.
Technologies des nanopores
Plusieurs plateformes permettant le séquençage d’une molécule d’ADN par passage à travers
un nanopore d’une membrane biologique ou synthétique sont actuellement en développement.
Les différents nucléotides A, C, G et T sont successivement détectés lors de leur passage à
travers du pore par des systèmes optiques (marquage des nucléotides) ou électriques
(modification du courant en fonction du degré d’occupation du pore et du temps de transit,
spécifiques à chaque nucléotide).
Les nanopores utilises peuvent être solides (graphène) ou biologiques (α-hémolysine,
Mycobacterium smegmatis porin A)
I.1.2.4.3 Technologies émergentes
Une quatrième vague de séquenceurs va voir le jour, s’arguant des qualités de chacune des
plateformes citées plus haut.
Technologie FRET (Fluorescence Resonance Emission Transfer)
Cette technologie de séquençage est basée sur le transfert d’énergie distance-dépendant entre
deux molécules : un donneur et un accepteur. Le séquençage s’effectue par synthèse à l’aide
d’une ADN polymérase (donneur) et de nucléotides marques à l’aide d’un fluorophore
(accepteurs).
La molécule d’ADN à séquencer est déposée sur une surface en présence d’une ADN
polymérase mobile : les nucléotides marqués sont ensuite ajoutés au milieu. Lorsque la

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 41
polymérase incorpore un nucléotide sur un fragment d’ADN en cours de synthèse, il y a
transfert d’énergie entre le groupement donneur et le groupement accepteur par proximité.
Une fluorescence est émise, et détectée.
Une plateforme de séquençage utilisant cette technologie est actuellement en cours de
développement : Starlight (Life Technologies).
Technologie de séquençage in situ
Une équipe a récemment mis au point une technique permettant le séquençage de courts
fragments d’ARN au sein de tissus, afin d’étudier le profil d’expression génique in situ. La
4ème
génération de séquenceurs permettra peut-être elle-aussi le séquençage de molécules
d’ADN ou d’ARN directement à partir d’une cellule ou d’un tissu (Junemann et al., 2013;
McGinn, 2013).
I.1.2.5 Applications de séquençage à haut débit
Les applications disponibles avec le séquençage à haut débit peuvent globalement se
regrouper en trois grandes catégories.
I.1.2.5.1 Le séquençage de novo
Il s’agit du séquençage d’un génome pour lequel il n’existe pas de séquence référence, donc
détermination d’une séquence inconnue. Pour réussir à obtenir des versions de génome de
bonne qualité, il est souvent nécessaire de combiner plusieurs méthodes. Le pyroséquençage
permet grâce à des lectures longues de construire une première version du squelette du
génome quand les méthodes par terminateurs réversibles vont corriger les erreurs présentes
dans cette première reconstruction pour produire un brouillon du génome de qualité. Dans le
domaine médical ces outils sont utilisés pour la découverte de génomes d’agents pathogènes
inconnus ou de nouveaux virus (Gaynor et al., 2007).
I.1.2.5.2 Le reséquençage
Dans les études de génomes, le terme de reséquençage est utilisé à la place de séquençage.
Cette dénomination, essentiellement utilisée en génétique, désigne le séquençage d’un
segment d’ADN suivi de la comparaison du résultat obtenu avec celui d’une séquence de
référence connue. Le séquençage à haut débit est alors employé pour connaître quelles sont
les variations génomiques de l’échantillon qui est étudié en comparaison avec celui pris

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 42
comme référence. Ces approches sont certainement parmi les plus employées actuellement
dans le domaine médical. Ces outils ont typiquement pour vocation de remplacer les
méthodes traditionnelles d’hybridation génomique comparative (CGH) et de permettre de
préciser les diagnostics soit de façon préventive soit pour caractériser une pathologie déjà
déclarée.
I.1.2.5.3 La métagénomique
Le dernier domaine d’application du séquençage à haut débit, à la frontière entre
reséquençage et séquençage de novo, concerne la métagénomique. Le but est de découvrir
dans un mélange complexe l’ensemble des organismes qui le composent comme par exemple
la flore intestinale. On peut ainsi imaginer qu’en séquençant le sérum de patients, il sera
possible de déterminer quels sont les agents pathogènes qui y sont présents et qui sont à
l’origine de la pathologie observée. Il pourrait aussi être possible d’envisager en toxicologie le
suivi de l’incorporation de vecteurs viraux dans différents tissus d’un organisme en dehors de
ceux ciblés par le vecteur de thérapie génique (Chiu et al., 2008).
I.1.2.6 Génomique comparative et exploitation des génomes
I.1.2.6.1 Assemblages des génomes
L’assemblage de séquence est le processus consistant à aligner, orienter et fusionner les
fragments d’ADN obtenus durant la phase de séquençage. Cette étape est nécessaire parce que
le séquençage d’ADN ne peut pas retourner en sortie le génome complet en une lecture. Au
lieu de cela, on obtient de petites séquences de 20bp à 40000bp (selon la technologie utilisée)
en utilisant souvent des fragments venant de séquençage shotgun. A partir des lectures de la
phase de séquençage, l’assembleur (un programme informatique) va essayer de mettre les
lectures ensemble en se basant sur leur chevauchement. L’assemblage est généralement fait en
2 étapes:
D’abord, les fragments sont assemblés en utilisant le chevauchement des lectures pour
construire de plus grandes séquences complètes appelées contigs. L’information
utilisée est ainsi généralement limitée aux lectures simples (sans l’information de
paire).

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 43
Ensuite vient la phase de scaffolding (échafaudage) : cette étape a pour but d’orienter
et de placer les contigs les uns par rapport aux autres pour obtenir une séquence
unique, bien que possédant des lacunes (soit des trous dans la séquence finale). Ces
lacunes seront corrigées lors de l’étape de finition du génome qui vise à combler les
trous restant dans l’assemblage par des méthodes expérimentales, comme la PCR ou
l’extension d’amorces (Pevzner et al., 2001).
L’assemblage de génomes complets destinés à être comparés nécessite une phase de
vérification. Les critères de qualité d’une séquence génomique complète sont une couverture
suffisante, un petit nombre de contigs et un minimum d’incertitude sur l’ordre de ces derniers.
Les étapes de finition et de validation d’un projet de séquençage sont généralement longues et
difficiles, dépendant à la fois de la méthode de séquençage utilisée, de la stratégie
d’assemblage et de la richesse en éléments répétés des génomes. De plus, la construction des
banques et librairies d’ADN génomique ou l’utilisation des programmes d’assemblage (et leur
paramétrage) peuvent générer des erreurs préjudiciables à l’analyse ultérieure des séquences
tels que des contigs chimériques et des décalages de phase de lecture artéfactuels.
Il existe différentes techniques permettant la validation d’un assemblage au niveau
«macroscopique » (scaffolding).
Une approche plus ancienne, utilisant le clonage, consiste à combiner un séquençage de
plusieurs banques de fragments génomiques de tailles différentes. Il existe des banques
d’inserts de taille moyenne (de 5 à 12 kpb) et des banques d’inserts de plus grande taille (50 à
100 kpb) clonés dans des vecteurs BACs (Bacterial Artificial Chromosome). Le séquençage et
l’assemblage d’inserts de différentes tailles permettent d’organiser les contigs entre eux et de
réduire l’incertitude de l’assemblage de certaines zones.
Une autre approche, par des cartes de restrictions obtenues par digestion de l’ADN
génomique par une enzyme, peut être utilisée pour valider un assemblage de génome. Une
migration sur gel ou une lecture optique des produits obtenus permet d’obtenir un profil de
restriction qui peut être comparé à un profil de restriction in silico du génome à valider.
Les cartes optiques sont des outils développés par la société OpGen Technologies (Madison,
WI) depuis une dizaine d’années. Il s’agit de cartes de restriction ordonnées à haute résolution
d’un génome dont l’obtention est grandement automatisée. Elle permet d’ordonner et
d’orienter des contigs lors d’un assemblage de génome et constitue également une méthode de

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 44
validation indépendante d’un projet de séquençage. Comparée à une carte de restriction in
silico d’une séquence connue, ces cartes optiques peuvent aussi être utilisées en génomique
pour identifier des réarrangements génomiques tels que des insertions, des délétions, des
duplications et des inversions (Shukla et al., 2009). Cette méthode peut être d’un grand intérêt
pour la finition de génomes complets (; Lim et al., 2001; Reslewic et al., 2005 Latreille et al.,
2007).
Dans certains cas, les approches et techniques de validation citées ci-dessus, les techniques
classiques de biologie moléculaire (PCR) et d’analyses bioinformatiques (biais de
composition en GC, assembleurs…) ne suffisent pas à valider l’organisation et la structure
d’un chromosome bactérien. Les incertitudes peuvent alors être levées par la mise en place
d’autres stratégies expérimentales faisant l’objet de projet de recherche.
I.1.2.6.2 Annotation des génomes
Le séquençage d’un génome permet de prédire les fonctions de gènes contenus dans un
organisme en annotant les gènes détectés dans une séquence d’ADN. Ce travail d’annotation
peut être décomposé en deux tâches dont l’une, l’annotation structurale, consiste à identifier et
à localiser les éléments génétiques en présence. La deuxième, l’annotation fonctionnelle,
consiste en la détermination de la fonction de ces éléments, en particulier la fonction des
gènes protéiques qui demeurent centraux dans l’annotation des génomes. Cette dernière est
réalisée à l’aide de pipelines d’annotation à savoir la plateforme Microscope (MaGe) de
Genoscope (Vallenet et al., 2006), la plateforme MIGALE AGMIAL (AGMIAL) (Bryson et
al., 2006), la platforme Prokka (Seemann, 2014) et le serveur Rast (Aziz et al., 2008). Ces
outils d’annotation intègre la détection des gènes à partir de la séquence des bases
nucléotidiques, la comparaison avec des génomes connus, un algorithme d’annotation
automatique permettant entre autres d’effectuer un choix, le plus rationnel possible, de la
fonction d’un gène et une interface visuelle permettant une annotation manuelle. Cependant,
cette annotation manuelle expertisée nécessite un travail manuel de vérification, long et
fastidieux (McAdam et al., 2014).
I.1.2.6.3 Apport de la génomique comparative
L’intérêt pour le séquençage a été stimulé par un besoin de connaissances. En effet, séquencer
le génome d’un micro-organisme permet d’avoir accès à la totalité de son information

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 45
génétique. Cependant, cette connaissance a plus de valeur si elle est mise en perspective et
comparée avec d’autres informations génomiques ou phénotypiques.
Le séquençage et la comparaison de plusieurs génomes au sein d’une espèce ou d’un genre
bactérien permet de définir le génome central (ou « core-genome ») et le génome total (ou «
pan-genome ») de l’espèce ou du genre. Le pan-génome décrit le nombre total de gènes
retrouvés au moins une fois dans une espèce ou un genre. En effet, les bactéries peuvent avoir
de grandes variations de contenu en gènes entre souches proches. L’analyse de ces gènes
permet donc de comprendre l’évolution d’un groupe bactérien, particulièrement pertinent dans
l’étude des métagénomes. Cependant, elle peut aussi être utilisée dans un contexte génomique
plus restreint. Le pan-génome inclut le core-génome contenant les gènes présents chez tous
les organismes en question, le génome accessoire contenant les gènes présents dans deux ou
plus des souches ou espèces, et les gènes uniques, spécifiques d’une seule souche ou espèce.
Le core-génome inclut généralement tous les gènes responsables des aspects principaux de la
physiologie et des caractères phénotypiques majeurs d’une espèce ou d’un genre. En
revanche, les gènes accessoires et uniques contribuent à la diversité des souches ou des
espèces et peuvent coder pour des voies métaboliques supplémentaires et des fonctions qui ne
sont pas essentielles à la croissance mais qui peuvent conférer un avantage adaptatif à
l’organisme, comme par exemple l’adaptation à une niche écologique, la résistance aux
antibiotiques ou encore la colonisation d’un hôte particulier. De tels gènes sont généralement
retrouvés dans les îlots génomiques (Medini et al., 2005 Reinhardt et al., 2009).
L’analyse des gènes accessoires permet donc de comprendre les spécificités de chaque
organisme séquencé. Remarquons ici que ces distinctions ne sont pas strictement biologiques
car elles dépendent en partie des souches ou espèces incluses dans l’analyse ainsi que des
paramètres utilisés pour les comparaisons et le regroupement des gènes.
Les études comparatives in silico effectuées sur les séquences des micro-organismes ont
permis de progresser dans de nombreux domaines concernant notamment la structure et la
diversité des génomes et en particulier l'identification de gènes de virulence et de
déterminants de pathogénicité pour les espèces pathogènes. Ces gènes peuvent êtres multiples
et variés. Ce sont principalement les gènes codant pour des toxines, les systèmes d’export de
macromolécules (ou systèmes de sécrétion) et les gènes codant pour des fonctions liées aux
interactions avec l’hôte tels que des gènes codant pour des fonctions d’adhésion, des gènes de
synthèse des polysaccharides de surface et de molécules d’interaction avec le système

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 46
immunitaire de l’hôte et des systèmes de captation du fer (Sidérophores)] qui sont recherchés
comme déterminants de virulence.
Le contenu en gènes d’un organisme reflète ses possibilités d’interactions avec son
environnement. Par exemple, la présence de gènes codant pour les enzymes d’une voie
catabolique donnée dans un génome indique que le micro-organisme est probablement
capable d’utiliser telles ou telles ressources présentes dans son environnement. Egalement,
comparer les contenus de plusieurs micro-organismes en gènes codant pour des systèmes
d’import et d’export de macromolécules tels que les polysaccharides ou les polyamino-acides
peut renseigner sur la capacité de ces micro-organismes à utiliser les ressources et nutriments
présents dans leurs milieux de vie dans les conditions physico-chimiques nécessaires à leurs
croissance. Le contenu en gènes d’un génome de micro-organisme reflète donc sa niche
écologique (Bhattacharyya et al., 2002; Buchrieser et al., 2003; Mellmann et al., 2011).
La comparaison de données génomiques de plusieurs isolats au sein d’une espèce permet
d’identifier des gènes ubiquitaires dans l’espèce. L’apport de génomes d’autres espèces
proches peut permettre d’identifier alors les gènes spécifiques de cette espèce. Ces gènes
spécifiques et ubiquitaires peuvent par exemple être utilisés comme cibles pour la mise au
point de moyens de détection ou de vaccins.

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 47
I.2 LES STAPHYLOCOQUES
I.2.1 Taxonomie
I.2.1.1 Historique
Les staphylocoques ont été l’objet de nombreuses investigations menées par d’éminents
microbiologistes à l’instar de Koch, Pasteur, Ogston et Rosenbach.
En 1878, Koch souligne le rôle pathogène des bactéries se présentant sous forme de cocci
Gram positif (Spicer, 2003). Ces cocci seront ensuite isolées, puis identifiés d’un pus par
Louis Pasteur en 1880. Ils seront baptisés en 1883 par Ogston sous le nom de
Staphylocoque pour décrire ces grains (kokkos) groupés en amas irréguliers à la
façon d'une grappe de raisin (staphylos) (Ogston, 1882).
En 1884, Rosenbach a obtenu des cultures pures de ces bactéries. Il a scindé le genre
Staphylococcus en deux groupes selon que les colonies étaient blanches ou dorées. La même
année, Gram mettait au point une méthode de coloration des bactéries à partir du violet de
gentiane : les staphylocoques étaient classés parmi les cocci à Gram positif (Gram, 1884).
I.2.1.2 Classification
La classification des staphylocoques a connu de nombreux bouleversements. Lors de la
première édition en 1923 du « Bergey’s Manual® of Determinative Bacteriology », les
staphylocoques étaient classés dans la famille des Streptococcaceae puis lors de la deuxième
édition en 1926, dans la famille des Micrococcaceae. En 1948, cette famille comprenait alors
les genres Micrococcus et Staphylococcus. Ce n’est qu’en 1957 que les staphylocoques et les
microcoques furent séparés sur la base de leur capacité à utiliser le glucose en anaérobiose.
Cependant, ce test amena beaucoup de confusion en attribuant certaines espèces du genre
Staphylococcus au genre Micrococcus (Baird-Parker, 1971 ; Pulverer, 1987).
En 1974, avec les débuts de la biologie moléculaire, Baird-Parker sépare définitivement le
genre Staphylococcus du genre Micrococcus, grâce à la différence du contenu en guanine et
en cytosine de leur ADN: 66-75% pour Micrococcus et 30-39% pour Staphylococcus
(Bascomb et Manafi, 1998). Avec l’évolution des techniques génomiques, le genre
Staphylococcus a été reclassé avec les bactéries à Gram positif dont l’ADN présente un GC%
inférieur à 55. Le GC% et la composition de la paroi sont des marqueurs robustes pour séparer

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 48
le genre Staphylococcus des autres coques à Gram positif et à catalase positive. Ainsi, la
famille des Micrococcaceaea été remaniée et dans la seconde édition du « Bergey’s Manual®
of Systematic Bacteriology », le genre Staphylococcus a été reclassé dans l’ordre des
Bacillales et dans la famille des Staphylococcaceae.
A ce jour, cinquante espèces et sous-espèces ont été identifiées au sein du genre
Staphylococcus (Annexe 2). Les espèces sont généralement classées en deux groupes sur la
base de leur capacité à produire une coagulase libre :
les staphylocoques à coagulase positive (SCP) : généralement considérés comme les
plus pathogènes, représentés par une seule espèce (S. aureus)
les staphylocoques à coagulase négative (SCN) : représentés par de multiples espèces
(S. epidermidis, S. saprophyticus, S. hominis, S. capitis ou S. hæmolyticus)
I.2.2 Réservoir et transmission
Les staphylocoques sont des bactéries ubiquitaires présentes sur la peau, les muqueuses et la
sphère rhino-pharingée chez l’Homme et chez les animaux. Ils ont également été isolés de
l’environnement naturel (sol, eau douce et eau de mer, poussière, air), de l’environnement
domestique de l’Homme (cuisine, réfrigérateur), de l’environnement hospitalier et des ateliers
de préparation alimentaires et à partir de denrées alimentaires (Annexe 2).
Chez l’Homme, la transmission est directe à partir des lésions ouvertes, ou indirecte par voie
aérienne et par les objets souillés. En milieu hospitalier, la transmission est essentiellement
manuportée par le personnel soignant.
I.2.3 Physiopathologie
Le pouvoir pathogène d’une souche est lié à sa capacité de survie et à ses nombreux facteurs
de virulence. La pathogénie des infections à S. aureus est liée à la synthèse d’adhésines, de
nombreuses exoprotéines et de toxines (Vandenesch et al., 2007):
Les adhésines, ont le pouvoir de reconnaitre les protéines de la matrice extracellulaire et
sont ainsi impliquées dans les phénomènes de colonisation des tissus de l’hôte.
Les exoprotéines comportent des facteurs qui interférent avec les défenses naturelles :
o La capsule qui joue un rôle antiphagocytaire.
o La protéase dirigée contre les immunoglobulines.
o Les leucocidines destructrices des polynucléaires.

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 49
o La protéine A qui interfère avec l’opsonisation.
o La coagulase favorisant la formation d’un caillot protecteur autour de la
bactérie.
o L’hyaluronidase et lastaphylokinasequi sont des invasines favorisant la
diffusion de l’infection et donc la dissémination de la bactérie.
Les toxines à activité superantigénique et les exfoliantes à tropisme cutané
La pathogénie des infections à SCN est plus incertaine. Leurs principaux facteurs de virulence
sont constitués d’adhésines dont certaines ont des homologies avec celles de S. aureus et sont
liées à la capacité de production d’exopolysaccharides plus ou moins exubérants entrant dans
la composition de biofilm et inhibant la phagocytose.
I.2.4 Infection
Les infections à staphylocoque se présentent souvent sous la forme d’infections suppuratives
superficielles cutanéomuqueuses pouvant se compliquer par diffusion à distance du foyer
infectieux initial (Figure I.15). Il peut parfois s’agir d’infections non suppuratives via un
phénomène toxinique.
Les infections à S. aureus sont liées aux capacités d’adhésion de l’isolat, à l’expression de
certains facteurs de virulence (notamment certaines toxines) et à la susceptibilité de l’hôte. À
l’inverse, les SCN sont peu virulents. L’expression clinique des infections à SCN est souvent
pauvre et celles-ci reposent essentiellement sur la capacité d’adhésion des SCN à du matériel
comme des cathéters ou des valves cardiaques prothétiques.
La gravité des infections à Staphylocoques dépend également du site infecté (Figure I.15),
notamment lorsqu’il concerne un organe vital, et le spectre des infections à SCN estlimité
alors que presque tous les sites anatomiques peuvent être infectés par S. aureus.
L’impact de la résistance à la méticilline sur la gravité clinique est largement débattu dans la
littérature, surtout pour S. aureus (Combes et al., 2004 ; Zahar et al., 2005). La morbidité des
infections à SARM pourrait être supérieure à celle des infections à SASM, plus en raison de
l’allongement de la durée de séjour attribuable à ces infections, aux difficultés thérapeutiques
rencontrées (Combes et al., 2004), aux pathologies associées et au terrain qu’à une plus
grande virulence (Jordens et al.,1989).

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 50
Figure I.15 : Les principales infections staphylococciques
I.2.5 Caractéristiques intrinsèques
I.2.5.1 Génome
Le génome du staphylocoque est constitué d’un chromosome circulaired’environ2800 paires
de bases, avec des prophages, plasmides et transposons. Les gènes de virulence et de
résistance aux antibiotiques sont retrouvés à la fois sur le chromosome et sur des éléments
extra chromosomiques, permettant leur transfert d’une bactérie à l’autre dans la même espèce
et dans des espèces différentes (Lindsay et al., 2004).
I.2.5.2 Enveloppe cellulaire
L’enveloppe cellulaire est formée du peptidoglycane, des acides teichoïques et
lipoteichoïques. Le peptidoglycane qui représente 50% de l’enveloppe du staphylocoque est
formé de polysaccharides auxquels s’attachent de manière covalente des acides teichoïques.
I.2.5.3 Capsule
Des polysaccharides capsulaires sont trouvés chez 90 % des souches. Onze types capsulaires
sont été décrits, et les types 5 et 8 sont les plus fréquents en pathologie humaine. La majorité
des staphylocoques dorés résistants à la méticilline appartiennent au type 5. La capsule permet

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 51
une meilleure résistance des souches à l'opsonisation et à la phagocytose. Certaines souches
produisent un exo polysaccharide, qui entraîne la formation d'un biofilm, engluant les
bactéries et constituant ainsi une forme de résistance au site de colonisation.
Certaines protéines ou glycoprotéines sont responsables de la spécificité de type. Il existe 14
sérotypesmis en évidence par réaction d'agglutination au moyen d'immunosérums (Renaud et
al., 2007).
I.2.5.4 Facteurs de virulence
Protéines de surface : Il existe une grande homologie dans la capacité de produire des
protéines d’adhésion au sein de toutes les espèces du genre Staphylococcus. Plusieurs de
ces protéines de surface jouent un rôle dans la capacité des staphylocoques à coloniser les
tissus en se fixant aux cellules et à la matrice extracellulaire. Ils agissent comme des
adhésines et portent de manière plus générale le nom de MSCRAMM.
Facteur inhibant la phagocytose : La protéine A inhibe l’opsonophagocytose grâce à sa
capacité de fixation du fragment Fc des immunoglobulines. Elle est actuellement
considérée comme une MSCRAMM car elle permet l’attachement de S. aureus au facteur
de Von Willebrand. Il s’agit d’un peptide présent sur l’endothélium lésé, et la protéine A
peut de ce fait jouer le rôle d’adhésine au début de l’infection intra vasculaire (Renaud et
al., 2007).
Toxines : Les SCP à la différence des SCN, produisent de nombreuses toxines :
Cytotoxines : la protéine alpha est une toxine à action membranaire. Après liaison au
récepteur membranaire, elle forme des pores d’où peuvent s’échapper des cations et
petites molécules
Super antigènes: ces toxines pyrogènes se lient au CMH de type II et causent une
prolifération majeure de lymphocytes T avec production de cytokines.
Entérotoxines: elles sont responsables du choc toxique staphylococcique et de
toxiinfection alimentaire (entérotoxines A, B, C1, C2, C3, D et E),
TSST-1 (toxicshocksyndromtoxin 1) : elle est structurellement proche des
entérotoxines B et C. On la retrouve dans 20 % des souches de S.aureus.
Exfoliatines: toxines épidermolytiques A et B. Elles sont responsables d’érythème et
de clivage de l’épiderme, causant l’épidermolyse bulleuse staphylococcique.

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 52
Leucocidine de Panton-Valentine (PVL) : fait partie d’une famille de toxines
synergohyménotropes. Ce sont des protéines à deux composants non associés mais
agissant en synergie sur les membranes cellulaires. Ces toxines ont des cellules cibles
(polynucléaires, monocytes, macrophages) sur lesquelles elles se fixent et
provoquent la formation de canaux membranaires laissant passer les cations divalents
(Renaud et al., 2007).
Enzymes : Les staphylocoques produisent de nombreuses enzymes comme des protéases,
lipases, hyaluronidases qui lysent les tissus et peuvent faciliter l’extension de l’infection
aux tissus adjacents.
La coagulase est une protéine extracellulaire qui se lie à la prothrombine de l’hôte et forme
un complexe appelé staphylothrombine. La thrombine activée transforme donc le
fibrinogène en fibrine. C’est la base du test de la coagulase en tube. C’est un marqueur
classique de l’identification de S. aureus. Il n’y a cependant pas d’argument formel en
faveur du rôle de la coagulase dans la virulence des souches.
Les lactamases inactivent la pénicilline. Les PBP sont situées dans la membrane
cytoplasmique, leur modification confère une résistance aux pénicillines A et M et aux
céphalosporines (Renaud et al., 2007).
I.2.5.5 Régulation des facteurs de virulence
La synthèse des facteurs de virulence est biphasique. A la phase initiale de la croissance
bactérienne, ce sont surtout les gènes codant pour des adhésines qui sont activés.
Secondairement, les gènes des toxines prennent le relais. Cette activation séquentielle est sous
la dépendance du système de régulation de la virulence appelé agr (accessory gene regulator)
(Bronneret al., 2004; Dufour et al., 2002). Le système agr met en jeu un mécanisme de
déclenchement par un seuil «quorum sensing» : il code pour un peptide qui est sécrété dans
l’espace extracellulaire et son accumulation agit comme un signal sur la densité cellulaire
conduisant à l’activation du système agr (Yarwood et Schlievert, 2003) (Figure I.16). Ce
dernier possède un polymorphisme génétique avec quatre groupes alléliques identifiés
(Jarraud et al., 2000).

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 53
Figure I.16: Facteurs de virulence exprimés à la surface ou excrétés par S. aureus en
fonction de la densité bactérienne (Ferry T et al., 2005).
I.2.6 Antibiothérapie
Un antibiotique (du grec anti : « contre », et bios : « la vie ») est une molécule naturelle ou
synthétique qui détruit ou bloque la croissance des bactéries. Dans le premier cas, on qualifie
l’antibiotique de bactéricide et dans le second cas on dira plutôt que l’antibiotique est
bactériostatique. Plusieurs molécules aujourd'hui sur le marché sont des molécules de
synthèse, dérivées ou non d'antibiotiques naturels.
I.2.6.1 Mécanismes d’action des antibiotiques
Les antibiotiques agissent de manière spécifique sur les bactéries en bloquant une étape
essentielle de leur développement. On peut classer ces molécules selon les mécanismes
qu’elles inhibent. Cinq mécanismes sont majoritairement ciblés, soit l’inhibition de la
synthèse de la paroi bactérienne, de la transcription, de la réplication de l’ADN, de la
traduction et finalement le métabolisme des folates (Procopio et al., 2012) (Tableau I.1).

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 54
Tableau I.1 : Mécanismes d’action des principales familles d’antibiotiques utilisées chez
l’Humain
Mécanismes d’action Familles d’antibiotiques
Inhibition de la synthèse de la paroi
bactérienne
Pénicillines, céphalosporines, carbapénèmes,
daptomycines, monobactames, glycopeptides
Inhibition de la transcription Rifampicine
Inhibition de la réplication de l’ADN Fluoroquinolones
Inhibition de la synthèse protéique Tétracyclines, aminoglycosides, streptogramines,
lincosamides, macrolides, kétolides, oxazolidinones
Inhibition du métabolisme des
folates
Sulfonamides, triméthoprime
I.2.6.2 Résistance aux antibiotiques
La découverte de la pénicilline et ses propriétés bactéricides, par Alexander Fleming en 1929,
a ouvert la voie au traitement des maladies infectieuses par les antibiotiques et a donné de
grands espoirs en changeant le pronostic de ces infections. La pénicilline a été utilisée pour la
première fois en 1941 pour traiter un patient atteint de septicémie à staphylocoque.
En 1953, a été isolée pour la première fois au Canada, une souche de S. aureus résistante à la
pénicilline qui était à l'origine de lésions cutanées, de pneumonies chez les enfants ou de
septicémies. L'arrivée d'un autre antibiotique, la méticilline, dans les années 60, a mis fin à
cette épidémie. Par ailleurs, la résistance à la méticilline de souches de S. aureus, isolées en
pathologie humaine, n'a pas mis longtemps à survenir et a été décrite dès 1961 au Royaume-
Uni (Figure I.17).Cette résistance est due à la production d’une protéine liant les pénicillines,
PLP2a, qui a une affinité faible pour toutes les bêtalactamines. La PLP2a est codée par le
gène mecA qui est inclus dans un élément génétique mobile dénommé SCCmec (Lowy, 1981).
Bien que cette cassette soit un élément génétique mobile, son acquisition in vivo est un
phénomène exceptionnellement observé. Ainsi, S. epidermidis représente la majorité des
SCNRM (Bertrand et al., 2005). Quelques clones SARM hospitaliers ont émergé depuis
l’introductionde la méticilline et sont responsables à eux seuls de la majorité des infections
nosocomiales à S. aureus (Robinson, 2004). Il existe cinq grands groupes de SARM
hospitaliers et actuellement, le clone SARM hospitalier dénommé clone Lyon est majoritaire
en France (Robinson, 2004 ;Ferry et al., 2006). Ce clone, le plus souvent sensible à la
gentamicine, résistant à la kanamycine, à la tobramycine, aux macrolides lincosamides et
streptogramines B et aux fluoroquinolones, était responsable de70 % des bactériémies à
SARM dans les services de réanimation de Lyon en 2003 (Ferry et al., 2006). Dans les
services de réanimation, épicentres de la résistance bactérienne, un taux de SARM supérieur à

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 55
50 % était habituel dans les années quatre-vingts et quatre-vingt-dix. Les efforts concernant le
contrôle des SARM à l’hôpital ont abouti à une diminution constante des taux depuis 1994. Il
atteint 22,8 % des souches isolées en réanimation en 2006 à l’Assistance publique-Hôpitaux
de Paris. Depuis la fin des années quatre-vingt-dix, les SARM circulants et particulièrement
les SARM appartenant au clone Lyon, ont plus fréquemment une résistance de type
hétérogène à la méticilline, et globalement une plus grande sensibilité aux antibiotiques, plus
particulièrement à la gentamicine (Aubry-Damon et al., 1997).
Depuis une dizaine d’années, des clones SARM ont émergé dans la communauté. Ces clones
ont acquis la cassette SCCmec et ont la capacité de produire la PVL qui est un facteur de
virulence associé à des pathologies bien particulières (Lina et al., 1999 ;Dufour et al., 2002).
Plusieurs clones SARM-C ont émergé en même temps sur les cinq continents (Vandenesch et
al., 2003). Un de ces clones, USA300, est responsable d’infections cutanées épidémiques
familiales et hospitalières aux États-Unis (Vandenesch et al., 2003 ; Mishaan et al ., 2005).
Selon des données récentes, ce clone serait devenu majoritaire dans certaines villes des États-
Unis et a même été responsable d’infections nosocomiales (Gonzalez et al., 2006). En Europe,
il existe un clone SARM-C différent du clone USA300 mais dont la prévalence reste faible.
La résistance et la sensibilité intermédiaire de S. aureus aux glycopeptides, respectivement
VRSA et GISA, sont exceptionnelles et concernent les SARM hospitaliers (Figure I.17). Une
première souche de SARM ayant une résistance à la vancomycine a été isolée au Japon en
1996 (Hanaki et al., 1999), puis d’autres souches ont été isolées, notamment aux États-Unis et
en Grande-Bretagne. La première souche de S. hæmolyticus résistante à la vancomycine a été
rapportée en 1987. Des résistances cliniques liées à une augmentation des CMI ont été
décrites pour S. hæmolyticuset S. epidermis dans des péritonites. Elles sont apparues le plus
souvent après une longue exposition préalable à la vancomycine. Le mécanisme de cette
résistance est dû à une augmentation de synthèse de la paroi bactérienne limitant la
pénétration de la vancomycine jusqu’à sa cible (Hanakiet al., 1999). Ces souches VISA ne
semblent pas avoir posé de problème clinique, mais leur apparition doit faire redoubler de
prudence, d’autant qu’elles sont très difficiles à dépister au laboratoire. On note tout de même
une augmentation progressive de la CMI des SARM aux glycopeptides dont l’impact en
clinique est encore incertain (Robert et al., 2006).

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 56
Figure I.17 : L’histoire d’apparition des antibiotiques et des SARMs (Kos et al.,2012).
I.2.7 Cassettes SCCmec
I.2.7.1 Définition
La cassette SCCmec est l'élément génétique mobile véhiculant le gène mecA qui code pour la
résistance à la méticilline chez les staphylocoques (Hiramatsu et al., 2002; Ito et al., 1999).
Cet élément possède plus de 50 orfs et est muni d’un site précis d’intégration : orfX ou
attBscc dans le génome de S. aureus. Des insertions jusqu’à 15kB, des délétions, ou des
duplications ont été observées entraînant l’évolution des séquences attB chez les SASM et par
conséquent leur incapacité à acquérir les cassettes SCCmec dans leur chromosome. Au total,
l’intégration d’une cassette SCCmec représente un coût évolutif pour une souche qui n’a
d’intérêt que si elle est soumise à la pression antibiotique. Sinon, elle poursuit son évolution la
rendant inapte à intégrer de tels éléments génétiques (Noto et al., 2008).
Enfin, il convient de noter que ces cassettes SCCmec ne sont pas exclusivement retrouvées
chez S. aureus, mais sont également rencontrées parmi les SCN. Il existe d’ailleurs une très
grande diversité de cassettes SCCmec parmi les SCN (Hanssen et al., 2007).

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 57
I.2.7.2 Structure générale
La structure des cassettes SCCmec est formée de deux éléments essentiels (Figure I.18) :
Le complexe mec : est composé du gène mecA, des éléments de régulation mecI et mecR1
et d’une séquence d’insertion située à proximité du gène mecA : IS431. L’ensemble forme
le complexe mec de classe A. Des variations ont été détectées, notamment des délétions ou
des insertions partielles dans les gènes de régulation de mecA, donnant naissance aux
autres complexes mec: classe B, classe C, classe D et classe E ( Katayama et al., 2001 ;
IWG-SCC 2009).
Les gènes codant les recombinases : sont responsables de l’intégration et de l’excision de
la cassette ccr situés en 5’ en amont du complexe mec, les gènes ccr sont impliqués dans
l’intégration site spécifique des cassettes SCCmec. Différents types ont été définis : ccr1,
ccr2, ccr3. De part et d’autre de ces éléments, sont situées les régions J: J1, J2 et J3 qui
contiennent de nombreux pseudo gènes d’intérêts inconnus pour la bactérie.
Figure I.18 : Représentation des 4 types de cassettes SCCmec (Hiramatsuet al., 2002).
Le complexe ccr est représenté en orange, le complexe mec en rose. Les zones grises correspondent aux éléments non essentiels des
complexes SCCmec et sont divisées en 3 régions : J1 à J3. Différents gènes codant la résistance à différents antibiotiques sont retrouvés dans les régions J de différentes cassettes SCCmec expliquant les phénotypes multirésistants des souches possédant de tels complexes. Il est
à noter qu’environ 70% de la cassette SCCmec IV est composée de seulement les gènes essentiels. La région J1 est différente des autres
cassettes SCCmec, la région J2 est absente et la région J3 est quasi identique à celle de la cassette SCCmec II.
I.2.7.3 Classification et typage

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 58
L’analyse des différentes collections de souches a permis la mise en évidence de huit types de
cassettes SCCmec selon l’International Working Group on the Classification of
Staphylococcal Cassette Chromosome Elements (IWG-SCC 2009) (Figure I.19).
Figure I.19 : Représentation schématique des 8 cassettes SCCmec (Van den Eede et al.,
2009).
De nombreuses méthodologies de typage des cassettes ont été décrites reposant initialement
sur l’association d’analyses southern et l’utilisation de trois enzymes de restriction, de
nombreuses PCR, et de séquençages. Oliveira et al. ont proposé une stratégie d’identification
rapide des 4 cassettes (I, II, III et IV) par une PCR multiplex unique (Oliveira et al., 2002).
L’analyse de leurs séquences a rapidement fait apparaitre de nombreuses différences :
Cassette SCCmec I : associe gène ccr1 avec un complexe mec de classe B, issu de la
délétion d’une partie de gène mecR1 et de son remplacement par une partie de l’IS1272.
Enfin, la région J1 contient de nombreux pseudo gènes et un gène pls codant une protéine
de surface.

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 59
Cassettes SCCmec de type II et III : sont caractérisées par l’association au complexe
mec de classe A de nombreux gènes d’origines plasmidiques codant la résistance aux
aminoglycosides, aux macrolides, aux tétracyclines, au cadmium, etc. Enfin, ces cassettes
présentent fréquemment une mutation du gène mecI, répresseur du gène mecA.
Cassette SCCmec IV : est caractérisée par une taille plus faible en comparaison aux
précédentes, une recombinase ccr2 et un complexe mec de classe B. Elle possède une
région J de taille réduite et l’absence de gènes de résistance à d’autres antibiotiques
(Hiramatsu et al., 2002).
Cependant, l’analyse de nombreuses collections de souches a rapidement démontré la
complexité structurale des autres cassettes SCCmec mettant en lumière les insuffisances de la
technique de caractérisation développée par Oliveira et al. en 2002. En 2006,
Chongtrakool et al. proposèrent de nouvelles techniques d’identification et une nouvelle
nomenclature d’identification des cassettes SCCmec :
Cassette SCCmec de type V (Ito et al., 2004) : découverte sur un SARM provenant
d’Australie associe un complexe mec de classe C2 avec un nouveau gène de recombinase
présent à l’état unique ccrC. De petite taille et ne présentant pas de résistances associées
aux antibiotiques, cette cassette possède un ensemble de gènes facilitant sa stabilisation
dans le chromosome bactérien.
Cassette SCCmec de type VI (Oliveira et al., 2006) : se caractérise par l’association
d’un complexe mec de type B, de recombinases ccrAB4 et non ccrAB2 et d’une région J1
spécifique.
Cassette SCCmecde type VII (Higuchiet al., 2008): a été individualisée, en 2008, dans
une souche de SARM-C. Elle est de grande taille (41,3kb) par rapport aux autres
cassettes SCCmec, cette nouvelle entité est composée de trois grandes régions distinctes :
la région J1 (site attL, le gène codant la recombinase ccrC8, l’IS431 le transposon Tn554,
et l’ORF), la région J2 (le complexe mec qui est homologue avec la cassette SCCmec V)
et région J3 (issue de recombinaisons multiples entre les différentes cassettes
précédemment décrites).
Cassette SCCmecde type VIII (Zhang et al., 2009) : Elle est en cours de
caractérisation (www.sccmec.org/Pages/SCC_TypesEN.html).

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 60
I.2.8 Staphylococcus capitis
Cette espèce représente la souche cible de notre travail.
I.2.8.1 Taxonomie
Le Staphylococcus capitis est une espèce de SCN. En 1991, elle a été subdivisée en deux
sous-espèces : S. capitis subsp. capitis et S. capitis sbspurealyticus (annexe 2). La
classification scientifique de S. capitis est la suivante :
Uni Bactéries
Embranchement Firmicutes
Classe Bacilles
Ordre Bacillales
Famille Staphylococcaceae
Genre Staphylococcus
Espèce Capitis
Sous-espèce capitissubsp. Capitis
capitissbspurealyticus
I.2.8.2 Habitat
Le S. capitis fait partie de la flore cutanée normal de l’homme, elle a été isolée au voisinage
des glandes sébacées du cuir chevelu, du visage, du cou et des oreilles et aussi chez le
chimpanzé et dans le lait de chèvre (Bannerman et al., 1991).
I.2.8.3 Pouvoir pathogène
Il a été décrit que cette espèce a été la cause de plusieurs infections : endocardite infectieuse
et sur prothèse valvulaire, méningites sur valve de dérivation et septicémie tardive.
I.2.8.4 Résistance aux antibiotiques
Il y a très peu d’information sur le niveau de résistance de cette espèce. Récemment, Il a été
décrit l’émergence de souches résistante à la méticiline et ayant une sensibilité réduite à la
vancomycine dans les hôpitaux de plusieurs pays (Van Der et al., 2002, Gras-Le Guen et al.,
2007, Rasigade et al., 2012). Le rôle de la cassette SCCmec n’a jamais été décrit.

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 61
I.2.8.5 Caractères bactériologiques
Il s’agit de cocci à Gram positif qui ils peuvent être isolés en diplocoques ou en amas. Comme
tous les Staphylocoques, cette espèce n’a pas d’exigence nutritive particulière. Son
métabolisme respiratoire est aérobie-anaérobie facultatif. L’aspect et la couleur des colonies
des deux sous espèces diffèrent :
S. capitis subsp. capitis : Diamètre des colonies de1 à 3 mm, lisse, convexes, à centre
surélevés, luisantes, bords réguliers, opaques, non pigmentées (blanc craie)
S.capitis sbs purealyticus : Diamètre des colonies de 4 à 7 mm, surélevées, circulaire,
bords réguliers à dentelés, luisantes, opaques, surfaces lisse ou rugueuse, pigmentées
en jaune ou jaune-orange, pigmentation tardive et dominante en périphérie de la
colonie pour 73% des souches (Renaud et al., 2007).
Les principaux caractères biochimiques d’identification de S. capitis sont présentés dans le
tableau 2.
Tableau I.2 : Identification biochimique de S. capitis
Caractères Espèces et sous-
espèces
Pig
men
t
Uré
ase
Orn
ith
ine
déc
arb
ox
yla
se
Arg
inin
e ar
yla
my
das
e
Ph
osp
hat
ase
alca
lin
e
Py
rro
lydo
ny
lary
lam
idas
e
Bêt
a-G
luco
sid
ase
Bêt
a-G
alac
tosi
das
e
Th
erm
onu
cléa
se
Co
agu
lase
lib
re
Fac
teu
r ag
glu
tin
ant
Production d’acide enaréabiose)
Alp
ha-
Lac
tose
Mal
tose
Man
nit
ol
D-M
ann
ose
Sac
char
ose
D-T
ura
no
se
D-T
réh
alo
se
S.capitissubsp.
Capitis + d d + d d +
S.capitissbspurealytic
us d ± + ±
+, 90% ou plus de souches positives ; ±, 90% ou plus de souches faiblement positives ; , 90% ou plus de souche négative ;
d, 11 à 89% de souches positives ; (), réaction lente ;
I.2.8.6 Caractéristiques génomiques
En utilisant la stratégie de séquençage aléatoire, il a été décrit en 2012 début de ma thèse que
seulement trois centres ont réalisé le séquençage du génome entier de trois espèces de S.
capitis d’origine clinique (tableau I.3). Les caractéristiques générales du génome de ces trois
souches de S. capitis sont citées dans le tableau I.4. Actuellement le nombre des génomes
séquencés est de 37 génomes (9 génomes font partie de ce travail).
(https://www.ncbi.nlm.nih.gov/genome/genomes/2054?)

CHAPITRE I
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 62
Tableau I.3 : Lieu, origine et date de séquençage des 3 souches S. capitis.
Désignation Centre de Séquençage Spécimen
D’origine
Année de
Séquençage
S. captis SK14 Institut J Craig Venter
(HumanMicrobiome Project)
Peau 03/02/2009
S. capitis
VCU116.
Institut national des maladies
allergiques et infectieuses(NIAID) et
centres de séquençagedu génomepour
les maladies infectieuses(GSCID
inconnu 25/07/2011
S. capitis QN1 Zhejiang University(Creative
Research Groups of the National
Natural Science Foundation of China
and the Technology Group Project for
Infectious Disease Control of Zhejiang
Province)
Peau
22/05/2012
Tableau I.4: Caractéristiques générales du génome des trois souches de S.
capitis.(http://www.ncbi.nlm.nih.gov/genome/genomes/2054?details=on&http://www.ncbi.nl
m.nih.gov/genome/genomes/155?details=on&project_id=53751)
Organisme
Type S. capitis SK14 S. capitis QN1 S. capitis VCU116
Bio project PRJNA55415 PRJNA31013 PRJNA53751
Instrument 454 GS FLX Illumina Hiseq2000 454
Statut Draft Draft Draft
Taille du génome
(Mb)
2.44 2.43 2.45
Contenu GC (%) 32.8 32.8 32.8
Gène 2.406 2.430 2.444
Pseudogènes 111
Protéine 2.230 2.374
ARNr 2 5 5
ARNt 57 53 55
Autre ARN 7 10
Nucléotide 33 31 39
Assemblage ASM17413v1 StacapQN11.0 ASM22152v2
Statut (Scaffolds
or contigs)
32 30 38
CDS (predicted
coding sequences)
2.230 2.368

CHAPITRE II
Séquence génomique complète de
Staphylococcus capitis CR01 :
Génome de référence

CHAPITRE II
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 63
Les staphylocoques, membres de notre écosystème cutané, ensemble avec les streptocoques et
les pneumocoques font partie d’un groupe d’agents pathogènes à Gram positif opportunistes
et envahissants, connus sous le nom de cocci pyogènes, responsables d’une variété
d’infections humaines (Lowy FD, 1998).
Les infections à staphylocoques représentent un pourcentage important des infections graves,
notamment au cours des infections nosocomiales, dont la prévalence ne baisse pas malgré les
mesures de prévention. La pathogénicité des staphylocoques pose peu de problème en ce qui
concerne Staphylococcus aureus (SA), mais elle est plus discutée pour les staphylocoques à
coagulase négative (SCN).
Les espèces de SCN les plus fréquentes sont S. capitis, S. epidermidis, S. haemolyticus et S.
hominis (Kloos et Musselwhite, 1975). Leur distribution sur la peau n’est pas uniforme (Kloos
et al., 1976). Il existe des niches préférentielles qui témoignent d’une adaptation de certaines
espèces aux différentes régions de la peau. Les espèces les plus exigeantes se trouvent dans
les zones où se produit une sudation importante, constituant ainsi un excellent milieu de
culture. S. epidermidis est prédominant dans les narines. S. epidermidis et S. hominis sont
persistants sur la tête, les aisselles, les jambes et les bras. Le front est souvent colonisé par S.
capitis et S. haemolyticus. S. saccharolyticus est souvent isolé de la tête et des bras et S.
auricularis de l’oreille. Au niveau des mains se retrouvent les espèces S. epidermidis, S.
haemolyticus et S. hominis (Kloos et Schleifer, 1986)
Staphylococcus capitis est une espèce en pathologie humaine, et qui le plus souvent n'exprime
que très peu de virulence et de résistances aux antibiotiques. Cependant, il a été identifié par
électrophorèse en champs pulsé « PFGE » un clone, nommé NRCS-A, responsable
d'infections tardives chez les nouveaux nés hospitalisés dans les services de réanimation
néonatale (enfants très grands prématurés). Ce clone a été identifié comme spécifiquement
responsable de septicémie dans ce type de service sur une large échelle de temps et dans de
nombreux pays à travers le monde, alors qu’il n’est jamais retrouvé chez les adultes (Van Der
Zwet et al., 2002 ; Venkatesh et al., 2006 ; Rasigade et al., 2012 ; D'mello et al., 2008 ;
Boghossian et al., 2013 ; Cui et al., 2013).
Au début de mes travaux de thèse, aucun génome complet de Staphylococcus capitis isolée
chez les nouveaux nés hospitalisés dans les services de réanimation néonatale n’a été
disponible dans les bases de données. Seulement trois draft génomes de souches isolées de
chez les adultes ont été déposés dans GenBank (Qin N et al., 2012). Il s’agit de S. capitis

CHAPITRE II
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 64
VCU116 (NCBI Ref. Seq : NZ_AFTX00000000.1), S. capitis SK14 (NCBI Ref. Seq :
NZ_ACFR00000000.1) et S. capitis QN1 (NCBI Ref. Seq : NZ_AJTH00000000.1). Dans ce
contexte, nous avons envisagé de séquencer le génome complet d’une souche multi-résistante
S.capitis CR01 (pulsotype NRCS-A) qui a été isolée en 2009 des services de réanimation
néonatale, hospices civils, Lyon, France. L'objectif était d'avoir un génome de référence
permettant de mieux caractériser cette espèce émergente. Pour ce faire, nous avons utilisé
deux technologies de séquençage à haut débit (mixte) : le pyroséquençage GFLX+ titanuim
(454) et le PacBio (SMART). La première technologie de séquençage fait objet de ce chapitre
et la deuxième est détaillée dans le chapitre IV.
Au moment du lancement de notre projet de recherche, la plateforme de séquençage à haut
débit choisie est le séquenceur 454 de chez Life Sciences, en raison de la longueur des
séquences générées. C’est ce paramètre, important à considérer pour une espèce dont le
génome de référence n’est pas encore disponible, qui nous a conduit à privilégier cette
technique par rapport à d’autres plateformes qui donnent des fragments plus courts à savoir
les deux technologies illumina et solide qui sont plus difficiles à assembler et à annoter
(Metzker, 2010).
Les lectures (« reads ») obtenues par le pyroséquençage GFLX+ titanuim, ont été assembler
en utilisant l’assembleur Newbler 2.7, ce qui a permis d'avoir un premier draft génome de la
taille de 2,504,472 bp. Ce draft génome a été annoté par la plateforme d’annotation MaGe
(Vallenet et al., 2006 ; Vallenet et al., 2013) et a été déposé, par la suite, dans GenBank sous
le numéro d’accession CBUB000000000.1
Le séquençage PacBio détaillé dans le chapitre IV, nous a permis de confirmer le scaffolding
qui a été fait par le pyroséquençage GFLX+ titanuim et aussi d'améliorer la séquence du draft
génome en fermant les gaps au sein des scaffolds. Au final, nous avons pu assembler la
séquence de S.capitis CR01 sous forme d'un seul chromosome circulaire de 2,522,871bp et un
plasmide de 26.140 pb.
Le contenu de ce chapitre est présenté sous forme d’un article publié dans la revue Standards
in Genomic Sciences (Lemriss et al., 2014). La référence de cet article est la suivante : Stand
Genomic Sci. 2014 Jun 15; 9(3): 1118–1127 (doi:10.4056/sigs.5491045). Dans cette
publication, j’ai réalisé tout le travail du laboratoire, principalement l’identification de la
souche S. capitis CR01 par spectrométrie de masse MALDI TOF, l’antibiogramme de cette
souche, l’extraction de l’ADN génomique, le séquençage à haut débit par pyroséquençage

CHAPITRE II
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 65
GFLX+ titanuim, l'assemblage génomique en utilisant les différentes méthodologies
bioinformatiques et outils (logiciels et machines) et l’annotation automatique et manuelle.
II.1 Abstract
Staphylococcus capitis is a coagulase-negative staphylococci (CoNS) commonly found in the
human microflora. Recently, a clonal population of Staphylococcus capitis (denominated
NRCS-A) was found to be a major cause of late-onset sepsis (LOS) in several neonatal
intensive care units in France. Here, we report the complete genome sequence and annotation
of the prototype Staphylococcus capitis NCRS-A strain CR01. The 2,504,472 bp long genome
(1 chromosome and no plasmids) exhibits a G+C content of 32.81 %, and contains 2,468
protein-coding and 59 tRNA genes and 4 rRNA genes.
II.2 Introduction
A frequent cause of low-weight newborns mortality and morbidity in Neonatal Intensive Care
Units (NICUs) are late-onset sepsis (LOS), that are de-fined as sepsis occurring after 3 days
of age. The most frequently encountered pathogens are coagulase-negative staphylococci
(CoNS) and within those Staphylococcus epidermidis has been shown to be the most
prevalent [1,2]. However, a few studies have reported the emergence of Staphylococcus
capitis as a main CoNS- and LOS- causative pathogen in NICU settings [2,4]. A study in
French NICUs [2] has demonstrated the spread of a single clonal population of methicillin-
resistant S. capitis (pulsotype NRCS-A) associated to reduced susceptibility to vancomycin,
the first line of antibiotics used in cases of LOS. Moreover, this clone has also been recently
identified in NICUs in Belgium, United Kingdom and Australia, which suggests a worldwide
distribution. In contrast, in adult bacteremia, S. capitis are rarely found and when detected, it
presents a bigger diversity in terms of genotypes as well as antimicrobial susceptibility
profiles than neonates bacteremia.
In order to elucidate the molecular mechanisms behind the wide spreading of the S. capitis
NRCS-A clone in NICUs throughout the world, we sequenced a prototype strain (CR01).
II.3 Classification and information
A strain belonging to the clonal population of Staphylococcus capitis NCRS-A pulsetype
(Table II.1) was isolated from the blood culture of a preterm infant with LOS, hospitalized in
the NICU of the Northern Hospital Group Center (Hospices Civils de Lyon, Lyon, France)
and suffering of LOS.
Species identification of the bacterial isolates and antimicrobial susceptibility testing (AST)
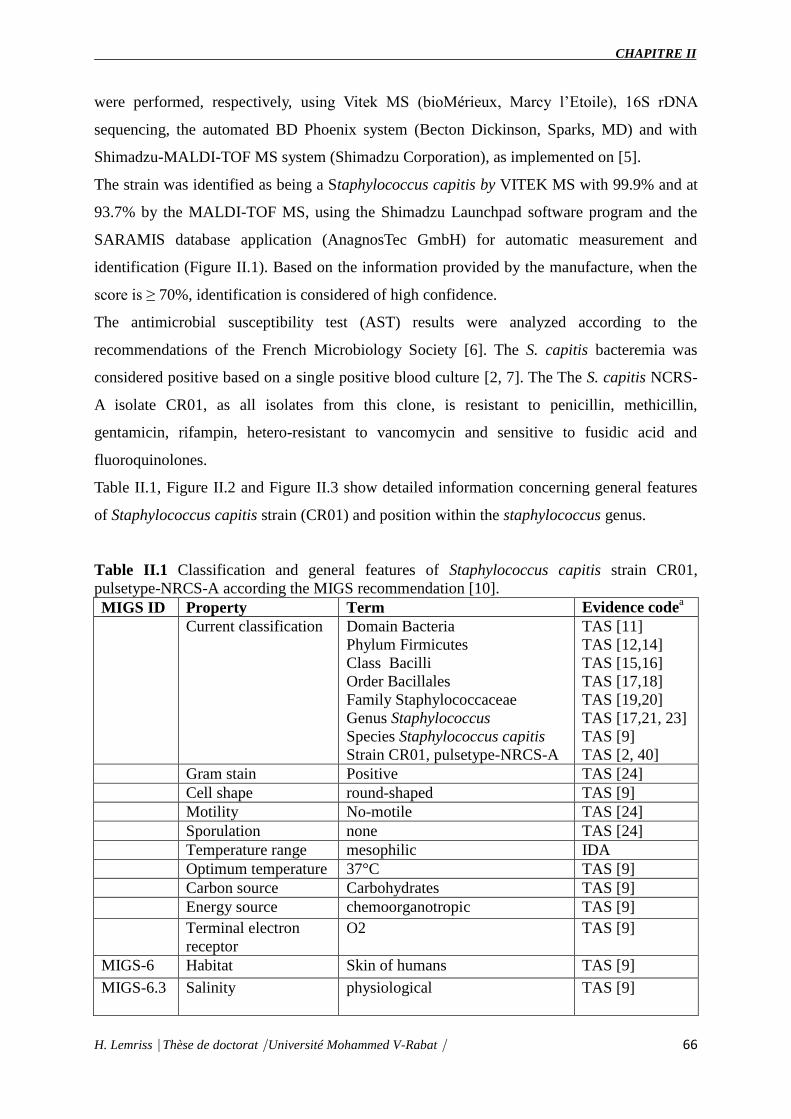
CHAPITRE II
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 66
were performed, respectively, using Vitek MS (bioMérieux, Marcy l’Etoile), 16S rDNA
sequencing, the automated BD Phoenix system (Becton Dickinson, Sparks, MD) and with
Shimadzu-MALDI-TOF MS system (Shimadzu Corporation), as implemented on [5].
The strain was identified as being a Staphylococcus capitis by VITEK MS with 99.9% and at
93.7% by the MALDI-TOF MS, using the Shimadzu Launchpad software program and the
SARAMIS database application (AnagnosTec GmbH) for automatic measurement and
identification (Figure II.1). Based on the information provided by the manufacture, when the
score is ≥ 70%, identification is considered of high confidence.
The antimicrobial susceptibility test (AST) results were analyzed according to the
recommendations of the French Microbiology Society [6]. The S. capitis bacteremia was
considered positive based on a single positive blood culture [2, 7]. The The S. capitis NCRS-
A isolate CR01, as all isolates from this clone, is resistant to penicillin, methicillin,
gentamicin, rifampin, hetero-resistant to vancomycin and sensitive to fusidic acid and
fluoroquinolones.
Table II.1, Figure II.2 and Figure II.3 show detailed information concerning general features
of Staphylococcus capitis strain (CR01) and position within the staphylococcus genus.
Table II.1 Classification and general features of Staphylococcus capitis strain CR01,
pulsetype-NRCS-A according the MIGS recommendation [10].
MIGS ID Property Term Evidence codea
Current classification
Domain Bacteria
Phylum Firmicutes
Class Bacilli
Order Bacillales
Family Staphylococcaceae
Genus Staphylococcus
Species Staphylococcus capitis
Strain CR01, pulsetype-NRCS-A
TAS [11]
TAS [12,14]
TAS [15,16]
TAS [17,18]
TAS [19,20]
TAS [17,21, 23]
TAS [9]
TAS [2, 40]
Gram stain Positive TAS [24]
Cell shape round-shaped TAS [9]
Motility No-motile TAS [24]
Sporulation none TAS [24]
Temperature range mesophilic IDA
Optimum temperature 37°C TAS [9]
Carbon source Carbohydrates TAS [9]
Energy source chemoorganotropic TAS [9]
Terminal electron
receptor
O2 TAS [9]
MIGS-6 Habitat Skin of humans TAS [9]
MIGS-6.3 Salinity
physiological TAS [9]

CHAPITRE II
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 67
MIGS-22 Oxygen
Facultative anaerobes TAS [9]
MIGS-15 Biotic relationship free-living TAS [9]
MIGS-14 Pathogenicity Opportunistic pathogen
(Nosocomial bacteremia in
premature neonates)
TAS[2]
MIGS-4 Geographic location NICU Lyon, France TAS[2]
MIGS-5 Sample collection
time
2007 IDA
MIGS-4.1
MIGS-4.2
Latitude – Longitude 45° 45' 35" N 4° 50' 32" E IDA
MIGS-4.3 Depth Not applicable IDA
MIGS-4.4 Altitude 162 m IDA
a) Evidence codes - IDA: Inferred from Direct Assay; TAS: Traceable Author Statement (i.e.,
a direct re-port exists in the literature); NAS: Non-traceable Author Statement (i.e., not
directly observed for the liv-ing, isolated sample, but based on a generally accepted property
for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology
project [20].
Figure II.1 Reference mass spectrum from Staphylococcus capitis strain (CR01).

CHAPITRE II
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 68
Figure II.2 Transmission electron microscopy of Staphylococcus capitis strain (CR01) using
a JEOL 1400. The scale bar represents 200 nm.

CHAPITRE II
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 69
Figure II.3 16S rRNA Phylogenetic tree highlighting the position of Staphylococcus capitis
strain CR01 (indicated by the yellow circle) relative to other type strains within the genus
Staphylococcus. All 16S rRNA sequences were obtained from the RDP database using as
filtering criteria: sequences with more than 1200 nt and classified as “good” quality
sequences. The tree uses sequences aligned with the MUSCLE software, with the default
parameters as implemented on Seaview version 4 [24], and a tree was inferred based on 1285
sites using the distance model of observed diver-gence, as implemented in the BioNJ
algorithm.
The 16S rRNA sequences were aligned using the MUSCLE software, with the default
parameters as implemented on Seaview version 4 [24], and a tree was inferred based on 1285
sites using the distance model of observed divergence, as imple-mented in the BioNJ
algorithm, and a bootstrap-ping process repeated 500 times.
The final tree was rooted using the 16S rRNA sequence of Macrococcus equipercicus Type
strain that belongs to a closely-related sister genus.

CHAPITRE II
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 70
II.4 Genome sequencing information
The genome sequence of S. capitis strain CR01 was determined by high-throughput
sequencing performed on a Genome Sequencer FLX + system (454 Life Sciences/Roche)
using FLX Titanium reagents according to the manufacturer's protocols and instructions, with
approximately 47 fold coverage of the genome. This platform provides longer read lengths
than other sequencing platforms to obtain raw sequences. De novo assemblies were
performed using the Roche Newbler (v 2.7) software package.
II.5 Genome project history
Table II.2 presents the project information and its association with MIGS version 2.0
compliance[5].
Table II.2 Project information
MIGS ID Property Term
MIGS-31 Finishing quality Non-contiguous finished
MIGS-28 Libraries used 454 pyrosequence rapid library
MIGS-29 Sequencing platforms 454 GS FLX+
MIGS-31.2 Fold coverage 47.0 × pyrosequence
MIGS-30 Assemblers Newbler Assembler 2.7
GenBank CBUB000000000.1
II.6 Growth conditions and DNA isolation
The sample was prepared for sequencing by growing S. capitis CR01, aerobically at 37°C in
Blood Agar for 24-48 hours. Genomic DNA was extracted using the PureLinkTM genomic
DNA kit (InvitrogenTM) according to the manufacturer’s recommended protocol. The
quantity of DNA obtained was determined using a NanoVue TM Plus (HVD Life Sciences),
and 1 μg of DNA was used for sequencing of whole-genome of this strain.
II.7 Genome sequencing and assembly
The isolated DNA of S. capitis CR01 was used to create 454-shotgun libraries following the
GS Rap-id library protocol (Roche 454, Roche). The resulting 454 DNA libraries were
sequenced using a whole-genome shotgun strategy by GS FLX Titanium sequencing kit XL+
[25] (202,108 reads totaling 2.5 Mb, X48 fold coverage of the genome). Genome sequences
were processed by Roche’s sequencing software according to the manufacturer's instructions
(454 Life Science). The resulting shotgun reads were assembled de novo using the Roche
Newbler assembly software 2.7 (454 Life Science) and 26 large contigs (Contig00001 to
Contig00026) were obtained. The N50 was 176239 bp.

CHAPITRE II
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 71
II.8 Genome annotation
An automatic syntactic and functional annotation of the draft genome was performed using
the MicroScope platform pipeline [26,27]. The syntactic analysis combines a set of programs
including AMIGene [28], tRNAscan-SE [29], RNAmmer [30], Rfam scan [31] and Prodigal
software [32] to predict genomic objects that are mainly CDSs and RNA genes. More than 20
bioinformatics methods are then used for functional and relational analyses: homology search
in the generalist databank UniProt [33] and in more specialized databases as COG [34],
InterPro [35], PRIAM profiles for enzymatic classification [36], prediction of protein
localization using TMHMM [37], SignalP [38] and PsortB [39] tools.
II.9 Genome properties
The genome includes one circular chromosome of 2,504,472 bp (32.81% GC content). A total
of 2,565 genes were predicted with 2,453 being protein-coding genes, 59 tRNA-enconding
genes, 4 rRNA-encoding genes (including 2 copies of 5S rRNA, 1 copy of both the large and
the small-subunits, respectively, 23S and 16S rRNA) and 34 other RNA related ORFs. No
plasmid was detected.
Of the 2,453 protein-coding genes, 1,892 genes (76.7%) were assigned to a putative function
with the remaining annotated as hypothetical proteins. The predicted coding density in S.
capitis strain CR01 was 86%.
Table II.3 and Figure II.4 detailed description of the properties and the statistics of
Staphylococcus capitis strain CR01 genome. The distribution of the genes into COGs
functional categories is presented in Table II.4.
Table II.3 Nucleotide content and gene count levels of the genome
Attribute Value % of totala
Genome size (bp) 2.504.472 100.00%
DNA G+C content (bp) 821.717 32.81%
DNA coding region (bp) 2.158.855 6.2%
Number of Scaffolds 26 -
Total genesb 2566 100.00%
RNA genes 97 4.00%
tRNA-enconding genes 59 2.30%
rRNA-encoding genes 4 0.20%
Protein-coding genes (CDS) 2454 96.00%
Genes assigned to COGs 1999 81.00%

CHAPITRE II
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 72
Genes of unknown function 561 23.34%
Genes with transmembrane helicesc 630 25.70%
CRISPR repeats 1 -
a) The total is based on either the size of the genome in base pairs or the total number of
protein coding genes in the annotated genome.
b) Total number of genes includes CDS, RNA genes and pseudogenes.
c) Detection of transmembrane helices was performed using TMHMM v. 2.0 [40].
Table II.4 Number of genes associated with general COG functional categories
Code Value %age 2a Description
J 178 7.21 Translation
K 184 7.46 Transcription
L 169 6.85 Replication, recombination and repair
D 29 1.18 Cell cycle control, mitosis and meiosis
V 85 3.44 Defense mechanisms
T 89 3.61 Signal transduction mechanisms
M 113 4.58 Cell wall/membrane biogenesis
N 23 0.9 Cell motility
W 1 0.04 Extracellular structures
U 33 1.34 Intracellular trafficking and secretion
O 86 3.48 Posttranslational modification, protein
turnover, chaper-ones
C 144 5.83 Energy production and conversion
G 210 8.51 Carbohydrate transport and
metabolism
E 370 14.99 Amino acid transport and metabolism
F 92 3.73 Nucleotide transport and metabolism
H 109 4.42 Coenzyme transport and metabolism
I 92 3.73 Lipid transport and metabolism
P 270 10.94 Inorganic ion transport and metabolism
Q 56 2.27 Secondary metabolites biosynthesis,
transport and catabo-lism
R 425 17.22 General function prediction only
S 203 8.23 Function unknown
- 469 19.00 Not in COGs
a) The total is based on the total number of protein coding genes in the annotated
genome.

CHAPITRE II
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 73
Figure II.4 Graphical circular map of the chromosome. From outside to the center: Genes on
the forward strand (colored by COG categories), genes on the reverse strand colored by COG
categories), RNA genes (tRNAs green, rRNAs blue), GC content, and GC skew.
II.10 Conclusion
Here, we described a new genome sequence of Staphylococcus capitis (strain CR01 belonging
to NRCS-A clone) as a first step toward comparing its content with other sequenced
Staphylococcus capitis genomes as well as CoNS genomes of species associated with late-
onset sepsis. Detailed analyses are in progress to identify virulence fac-tors and mobile
genetic elements (MGE), such as the staphylococcal chromosome cassette (SCCmec) [18],
potentially related to the high specificity of the NRCS-A clone to the NICU environment.

CHAPITRE II
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 74
II.11 Acknowledgments
This work was supported by a grant from the Fondation pour la Recherche Médicale (FRM,
grant ING20111223510) and by the Institut Na-tional de la Recherche Médicale (INSERM)
and the French Ministry of Health.
II.12 References
1. Klingenberg C, Rønnestad A, Anderson AS, Abrahamsen TG, Zorman J, Villaruz A,
Flægstad T, Otto M, Sollid, Ericson J. Persistent strains of coagulase-negative
staphylococci in a neonatal intensive care unit: virulence factors and invasiveness.
Clin Microbiol Infec 2007 Nov;13(11):1100-11.
2. Rasigade J-P, Raulin O, Picaud J-C, Tellini C, Bes M, Grando J, Ben Saïd M, Claris
O, Etienne J, Tigaud S, Laurent F. Methicillin-Resistant Staphylococcus capitis with
Reduced Vancomycin Susceptibility Causes Late-Onset Sepsis in Intensive Care
Neonates. PLoS ONE 2012;7(2):e31548.
3. Ng PC, Chow VC, Lee CH, Ling JM, Wong HL and Chang, RCY. Persistent
Staphylococcus capitis septicemia in a preterm infant. Pediatr Infect Dis J 2006
Jul;25(7):652-4.
4. de Silva GDI, Kantzanou M, Justice A, Massey RC, Wilkinson AR, Day NPJ,
Peacock SJ. The ica Operon and Biofilm Production in Coagulase-Negative
Staphylococci Associated with Carriage and Disease in a Neonatal Intensive Care
Unit. J Clin Microbiol 2002 Feb;40(2):382-8.
5. Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N,
Allen MJ, Angiuoli et al. The minimum information about a genome sequence
(MIGS) specification. Nat Biotechnol 2008; 26:541-547.
6. Woese CR, Kandler O, Wheelis ML. Towards a nat-ural system of organisms:
proposal for the do-mains Archaea, Bacteria, and Eukarya. Proc Natl Acad Sci USA
1990; 87:4576-4579.
7. Gibbons NE, Murray RGE. Proposals Concerning the Higher Taxa of Bacteria. Int J
Syst Bacteriol 1978; 28:1-6. http://dx.doi.org/10.1099/00207713-28-1-1.
8. Murray RGE. The Higher Taxa, or, a Place for Everything...? In: Holt JG (ed),
Bergey's Manual of Systematic Bacteriology, First Edition, Volume 1, The Williams
and Wilkins Co., Baltimore, 1984, p. 31-34.
9. List of new names and new combinations previ- ously effectively, but not validly,
published. List no. 132. Int J Syst Evol Microbiol 2010; 60:469- 472.
http://dx.doi.org/10.1099/ijs.0.022855-0.
10. Ludwig W, Schleifer KH, Whitman WB. Class I. Bacilli class nov. In: De Vos P,
Garrity G, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer KH, Whitman WB
(eds), Bergey's Manual of Systemat- ic Bacteriology, Second Edition, Volume 3,
Springer-Verlag, New York, 2009, p. 19-20.
11. Skerman VBD, McGowan V, Sneath PHA. Ap- proved Lists of Bacterial Names. Int
J Syst Bacteriol 1980; 30:225-420.
12. Prévot AR. In: Hauderoy P, Ehringer G, Guillot G, Magrou. J., Prévot AR, Rosset D,
Urbain A (eds), Dictionnaire des Bactéries Pathogènes, Second Edition, Masson et

CHAPITRE II
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 75
Cie, Paris, 1953, p. 1-692.
13. List Editor. List of new names and new combina- tions previously effectively, but not
validly, pub- lished. List no. 132. Int J Syst Evol Microbiol 2010; 60:469-472.
14. Schleifer KH, Bell JA. Family VIII. Staphylococcaceae fam. nov. In: De Vos P,
Garrity G, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer KH, Whitman WB
(eds), Bergey's Manual of Systematic Bacteriology, Second Edi- tion, Volume 3,
Springer-Verlag, New York, 2009, p. 392.
15. Rosenbach FJ. In: Bergmann JF (ed), Microorganismen bei den Wund-Infections-
Krankheiten des Menschen., Wiesbaden, 1884, p. 1-122.
16. Judicial Commission. Opinion 17. Conservation of the Generic name Staphylococcus
Rosenbach, Designation of Staphylococcus aureus Rosenbach as the Nomenclatural
Type of the Genus Staphy- lococcus Rosenbach, and Designation of the Neotype
culture of Staphylococcus aureus Rosenbach. Int Bull Bacteriol Nomencl Taxon 1958;
8:153-154.
17. Kloos WE, Musselwhite MS. Distribution and Per-sistence of Staphylococcus and
Micrococcus Spe-cies and Other Aerobic Bacteria on Human Skin. Appl Microbiol
1975; 30:381-385.
18. Martins Simões P, Rasigade J-P, Lemriss H, Butin M, Ginevra C, Lemriss S, R. V.
Goering, Ibrahimi A, Picaud JC, Vandenesch F, EL Kabbaj S, Laurent F.
Characterization of a novel staphylococcal chromosome cassette (SCCmec) within a
composite SCC island in neonatal sepsis-associated Staphylococcus capitis pulsotype
NRCS-A. Antimicrob. Agents Chemother 2013; 57(12):6354.
19. Schleifer KH, Bell JA. Family VIII. Staphylococcaceae fam. nov. In: De Vos P,
Garrity G, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer KH, Whitman WB
(eds), Bergey's Manual of Systematic Bacteriology, Second Edi- tion, Volume 3,
Springer-Verlag, New York, 2009, p. 392.
20. Ashburner M, Ball CA, Blake JA, Botstein D, But-ler H, Cherry JM, Davis AP,
Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of
biology. The Gene Ontology Con-sortium. Nat Genet 2000; 25:25-29.
21. Cherkaoui A, Hibbs J, Emonet S, Tangomo M, Girard M, Francois P, Schrenzel J.
Comparison of two matrix-assisted laser desorption ionization-time of flight mass
spectrometry methods with conventional phenotypic identification for routine
identification of bacteria to the species level. J Clin Microbiol 2010; 48:1169-1175.
22. French Society for Microbiology. Recommandations du Comite de l'Antibiogramme
de la Société Française de Microbiologie. 2009. Available at:
wwwsfmasofr/doc/downloadphp?doc=DiU8C&fic =casfm_2009pdf. Accessed 22
February 2009.
23. Hall KK, Lyman JA. Updated review of blood cul-ture contamination. Clin Microbiol
Rev 2006; 19:788-802.
24. Gouy M, Guindon S, Gascuel O. SeaView Ver-sion 4: A Multiplatform Graphical
User Interface for Sequence Alignment and Phylogenetic Tree Building. Mol Biol
Evol 2010; 27:221-224.
25. Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR,
Bult CJ, Tomb JF, Dougherty BA, Merrick JM, et al. Whole-genome random
sequencing and assembly of Haemophilus influenzae Rd. Science 1995 Jul

CHAPITRE II
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 76
28;269(5223):496-512.
26. Vallenet D, Belda E, Calteau A, Cruveiller S, Engelen S, Lajus A, Le Fèvre F, Longin
C, Mornico D, Roche D et al. MicroScope—an integrated microbial resource for the
curation and comparative analysis of genomic and metabolic data. Nucleic Acids Res
2013 Jan;41(Database issue):D636-D647.
27. Vallenet D, Labarre L, Rouy Z, Barbe V, Bocs S, Cruveiller S, Lajus A, Pascal G,
Scarpelli C, Médigue C. MaGe: a microbial genome annotation system supported by
synteny results. Nucleic Acids Res 2006 Jan 10;34(1):53-65.
28. Bocs S, Cruveiller S, Vallenet D, Nuel G, Médigue C. AMIGene: Annotation of
MIcrobial Genes. Nucleic Acids Res 2003; 31:3723-3726.
29. Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer
RNA genes in ge-nomic sequence. Nucleic Acids Res 1997; 25:955-964.
30. Lagesen K, Hallin P, Rødland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer:
consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 2007;
35:3100-3108.
31. Gardner PP, Daub J, Tate JG, Nawrocki EP, Kolbe DL, Lindgreen S, Wilkinson AC,
Finn RD, Grif-fiths-Jones S, Eddy SR, Bateman A. Rfam: updates to the RNA
families database. Nucleic Acids Res 2009; 37:D136-D140.
32. Hyatt D, Chen GL, Locascio PF, Land ML, Lar-imer FW, Hauser LJ. Prodigal:
prokaryotic gene recognition and translation initiation site identifi-cation. BMC
Bioinformatics 2010; 11:119.
33. UnitProt Consortium. The Universal Protein Resource (UniProt) 2009. Nucleic Acids
Res 2009; 37:D169-D174.
34. Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov
DM, Mazumder R, Mekhedov SL, Nikolskaya AN, et al. The COG database: an
updated version includes eukaryotes. BMC Bioinformatics 2003; 4:41.
35. Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Das U,
Daugherty L, Duquenne L, et al. InterPro: the integrative pro-tein signature database.
Nucleic Acids Res 2009; 37:D211-D215.
36. Claudel-Renard C, Chevalet C, Faraut T, Kahn D. Enzyme-specific profiles for
genome annotation: PRIAM. Nucleic Acids Res 2003; 31:6633-6639.
37. Sonnhammer EL, von Heijne G, Krogh A. A hid-den Markov model for predicting
transmembrane helices in protein sequences. Proc Int Conf Intell Syst Mol Biol 1998;
6:175-182.
38. Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal
peptides: SignalP 3.0. J Mol Biol 2004; 340:783-795.
39. Gardy JL, Laird MR, Chen F, Rey S, Walsh CJ, Es-ter M, Brinkman FS. PSORTb
v.2.0: expanded prediction of bacterial protein subcellular locali-zation and insights
gained from comparative pro- teome analysis. Bioinformatics 2005; 21:617-623.
40. Moller S, Croning MDR, Apweiler R. Evaluation of methods for the prediction of
membrane spanning regions. Bioinformatics 2001; 17:646-653.

CHAPITRE III
Séquençage de huit isolats de
Staphylococus capitis isolées de
différentes régions géographiques
dans le monde

CHAPITRE III
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 77
Bien que les techniques de séquençage à haut débit sont restreintes principalement aux études
de recherche, l’étude du génome bactérien d’isolats cliniques devient, actuellement, de plus en
plus réalisé, car le coût de séquençage de l'ADN continue à diminuer, et ce grâce à la
possibilité de séquencer un grand nombre d’échantillon dans un seul run de séquençage
(Koser et al., 2012 ; Salipante et al., 2014 ; Roach et al., 2015). En outre, le séquençage du
génome de ces isolats bactériens a mis en lumière divers aspects d'un certain nombre d'agents
pathogènes, y compris la transmission de la souche globale et intra-hôpital, l'évolution de la
résistance aux antibiotiques, l'échange horizontal des éléments mobiles, la distribution du
matériel génomique à travers une espèce bactérienne et l'identification de nouveaux agents
pathogènes bactériens (Farhat et al., 2013 ; Conlan et al ., 2014; Tong et al., 2015).
Les staphylocoques représentent une proportion importante parmi les bactéries responsables
d’infections graves. Ils sont observés dans de multiples situations cliniques, aussi bien en
pathologie communautaire qu’en pathologie nosocomiale (Zahlane, Haouach et Zouhdi,
2007).
Les staphylocoques à coagulase négative, appelés aussi staphylocoques blancs, représentent
les pathogènes les plus souvent identifiés dans les bactériémies en unités de réanimation
néonatale. L’espèce Staphylococcus epidermidis est la plus prévalente, représentant
classiquement plus de 70 % des staphylocoques blancs isolés dans ce contexte (Klingenberg
et al., 2007). Parmi les autres espèces, Staphylococcus capitis est rarement rapporté dans les
bactériémies et chez l’adulte, il est le plus souvent considéré comme un contaminant en cas
d’isolement dans les hémocultures (Ruhe et al., 2004). Les résultats de l’électrophorèse à
champ pulsé (PFGE) ont permis de mettre en évidence la présence endémique d’un clone
particulier de S. capitis multirésistant aux antibiotiques dans les services de réanimation
néonatale à Lyon, en France et plus généralement dans plusieurs pays d’Europe et en
Australie (Rasigade et al., 2012). La fréquence inhabituellement élevée des bactériémies à S.
capitis, en réanimation néonatale au sein des hospices civils de Lyon et dans plusieurs NICU
dans le monde, a motivé une étude détaillée de ce pathogène en collaboration avec le Centre
Nationale de Référence des staphylocoques (CNRS), Lyon, le Laboratoire de Recherche et
d’analyse Médicale de la Fraternelle de la gendarmerie Royal (LRAM), Rabat et le
Laboratoire de Biotechnologie Médicale (Medbiotech), Rabat.
Dans le chapitre précédent, nous avons décrit une nouvelle séquence génomique de S. capitis
CR01, appartenant au clone NRCS-A, comme une première étape nécessaire afin d’avoir un

CHAPITRE III
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 78
génome de référence. Ce dernier sera une base de comparaison avec d'autres génomes
séquencés de S. capitis, associées à une septicémie tardive, ainsi qu’avec des génomes CoNS
et ce dans le but d'élucider les mécanismes moléculaires de la propagation de ces souches de
S. capitis NRCS-A dans les NICU en France et dans le monde.
Le choix méthodologique de notre étude s’est porté sur l’usage de la technologie de
pyroséquençage du GFLX+ titanium, qui est un outil puissant pour la découverte d’agents
pathogènes. Cette technologie produit des séquences de 400 à 700 pb, facilitant les travaux
d’assemblage des séquences dans une approche de séquençage de novo de génomes complets.
Nous avons séquencé huit souches de S. capitis : CR02, CR03, CR04, CR05, CR06, CR07,
CR08 et CR09. Ces souches ont été isolées de quatre pays (France, Australie, Belgique et
Angleterre) entre les périodes 2002 et 2009. Il s’agit de six isolats cliniques (5 Pulsotypes
NRCS-A et 1 NRCS-C) et de deux souches de laboratoire (souches obtenues après 15
repiquages en présence de la vancomicyne et puis 9 repiquages en son absence « CMI haute
stable »).
La préparation de la librairie est une étape primordiale. En effet, de la qualité de la librairie
dépend la qualité de la PCR en émulsion et par la suite le séquençage. Vu la faisabilité de
séquencé plusieurs génomes dans un seul run de séquençage, nous avons utilisé dans cette
étude des séquences MID (Multiplex Identifier : amorces universelles couplées à un
identifiant) de 6 à 10 pb, spécifique à un échantillon, permettant l’identification d’un génome
lorsque plusieurs isolats sont séquencés et analysés simultanément (Multiplexage).
L’analyse bioinformatique est réalisée à l’aide des logiciels fournis avec le séquenceur
GFLX+ titanium. Les images acquises par la camera au cours du pyroséquencage ont été
converties en signal exploitable pour l’analyse bioinformatique des données par le logiciel GS
Run Processor (Roche Diagnostics). Deux étapes ont été nécessaires : analyse des images
(identification des puits et mesure de la quantité de lumière émise à chaque cycle pour chaque
puits) et analyse des signaux (détermination de la séquence des fragments d’ADN localisés
dans chaque puits à partir des informations précédentes). La qualité du séquençage a été
vérifiée sur le module GS run browser (Roche Diagnostics).
Après démultiplexage des séquences MID (8 fichier), les lectures « read » issues de
séquençage de chaque génome de S. capitis ont été assemblées avec l’assembleur Newbler
version 2.7 par l’approche de novo. Une séquence contiguée a été crée avec une taille et
couverture varie selon chaque génome donnant des draft génomes pour les huit souches. Ces

CHAPITRE III
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 79
derniers ont été annotés par la suite avec la plateforme d’annotation MicroScope (Mage). Les
séquences obtenues des six isolats cliniques ont été déposées dans la base de données
Genbank sous les numéros d’accès : CTEB01000000 pour CR03, CTEM01000000 pour
CR04, CTEO01000000 pour CR05, CTEL01000000 pour CR09, CZWI00000000 pour CR02,
CZWH00000000 pour CR07. En revanche, les données des deux souches de laboratoire
(CR06 et CR08) ne sont pas encore publiées et sont toujours en cours d’étude par des
collègues du CNR, Lyon, France.
Ce chapitre est présenté sous forme de deux articles publié dans Genome Annoucement
(Lemriss et al., 2015, Lemriss et al., 2016 ). Les références de ces articles sont les suivantes :
Genome Anounc 3(4):e00501-15. doi:10.1128/genomeA.00501-15.
Genome Announc 4(3):e00541-16. doi:10.1128/genomeA.00541-16.
Dans ces deux publications (partie III.1 et partie III.2 ci-dessous), j’ai réalisé tout le travail de
paillasse, d’analyse bioinformatique et de rédaction des manuscrits.
III.1 Genome Sequences of Four Staphylococcus capitis NRCS-A Isolates from
Geographically Distant Neonatal Intensive Care Units
Abstract
Staphylococcus capitis pulsotype NRCS-A was previously reported as a frequent cause of
late-onset sepsis in neonatal intensive care units (NICUs) worldwide. Here, we report the
whole-genome shotgun sequences of four S. capitis pulsotype NCRS-A strains, CR03, CR04,
CR05, and CR09, isolated from Belgium, Australia, the United Kingdom, and France,
respectively.
Coagulase-negative staphylococci (CoNS) are the most frequently encountered pathogens in
neonatal intensive care units (NICUs) (1). However, in the NICU setting, recent studies have
indicated that methicillin-resistant Staphylococcus capitis might emerge as a significant
pathogen, causing late-onset sepsis (LOS) in several neonatal intensive care units in France,
the Netherlands, and Australia (2, 4).
A study in French NICUs demonstrated the spread of a single clonal population of
methicillin-resistant S. capitis (pulsotype NRCS-A) associated with reduced susceptibility to
vancomycin, which is the first line of antibiotics used in cases of LOS. Moreover, this clone

CHAPITRE III
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 80
has also been recently identified in NICUs in Belgium, the United Kingdom, and Australia,
suggesting a worldwide distribution (5, 6).
In this report, we present the draft genome sequences of four S. capitis (pulsotype NRCS-A)
strains (CR03, CR04, CR05, and CR09) isolated from blood cultures from four neonates
hospitalized in NICUs in Belgium, Australia, the United Kingdom, and France, respectively.
All S. capitis strains were grown in blood agar at 37°C, and genomic DNA was extracted
using the PureLink genomic DNAkit (Invitrogen), according to the manufacturer’s
recommended protocol. The quantity of DNA was determined using a NanoVue Plus (HVD
Lifesciences), and 1 _g of DNA was used to sequence the whole genome of each strain. The
454-shotgun libraries were prepared from the extracted genomic DNA following GS rapid
library protocol (Roche 454; Roche).
The genome sequence of each S. capitis strain was determined by high-throughput sequencing
performed on a Genome Sequencer FLX_ system (454 Life Sciences/Roche) using FLX
Titanium reagents, according to the manufacturer’s protocols and instructions. De novo
assemblies were performed using the Roche Newbler (version 2.9) software package, and the
sequencing results are summarized in Table III.1.
Table III.1 Summary of genome sequencing in the present study
Strain Country
source
Reads
(MB)
Fold
Covrage
N°of contigs
Genome size (bp)
G+C content
%
Assession no
S.captis CR03 Belgium 141,728 30 31 2,508,352 32 81 CTEB01000000
S.capitis CR04 Australia 132,280 30 38 2,512,289 32.80 CTEM01000000
S.capitis CR05 United Kingdom
139,569 31 39 2,543,917 32.84 CTEO01000000
S.capitis CR09 France 132,205 30 34 2,490,458 32.82 CTEL01000000
An automatic syntactic and functional annotation of the draft genome was performed using
the MicroScope platform pipeline (7, 8). The syntactic analysis combines a set of programs,
including AMIGene (9), tRNAscan-SE (10), RNAmmer (11), Rfam scan (12), and Prodigal
software (13) to predict genomic objects that are mainly coding sequences (CDSs) and RNA
genes. More than 20 bioinformatics methods were used for functional and relational analyses.
The homology search was performed in the generalist databank UniProt (14) and in more
specialized databases, such as COG (15), InterPro (16), PRIAM profiles for enzymatic

CHAPITRE III
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 81
classification (17), prediction of protein localization using TMHMM (18), SignalP (19), and
PSORTb (20) tools.
The chromosome of strain CR03 (ENA accession no. CTEB01000000) contains 2,575 genes,
2,466 coding sequences (CDSs), 4 rRNAs, and 61 tRNAs; the chromosome of strain CR04
(accession no. CTEM01000000) contains 2,566 genes, 2,457 CDSs, 4 rRNAs, and 60 tRNAs;
the chromosome of strain CR05 (accession no. CTEO01000000) contains 2,624 genes, 2,508
CDSs, 4 rRNAs, and 60 tRNAs; and the chromosome of strain CR09 (accession no.
CTEL01000000) contains 2,540 genes, 2,432 CDSs, 4 rRNAs, and 59 tRNAs.
Nucleotide sequence accession numbers. This whole-genome shotgun project has been
deposited at the ENA database under the accession numbers listed in Table 1. The versions
described in this paper are in the first versions, under BioProject designation no. PRJEB8618.
Acknowledgments
This work was supported by a grant from the Fondation pour la Recherche Médicale (FRM,
grant 75 ING20111223510) and by the Institut National de la Recherche Médicale (INSERM)
and the French 76 Ministry of Health. This work was also supported by a grant from the NIH
for H3Africa BioNet.
References
1. Klingenberg C, Rønnestad A, Anderson AS, Abrahamsen TG, Zorman J, Villaruz A,
Flægstad T, Otto M, Sollid, Ericson J. 2007. Persistent strains of coagulase negative
staphylococci in neonatal 80 intensive care unit: virulence factors and invasiveness.
Clin Microbiol Infec. 13(11):1100-11.
2. Rasigade J-P, Raulin O, Picaud J-C, Tellini C, Bes M, Grando J, Ben Saïd M, Claris
O, Etienne J, Tigaud S, Laurent F. 2012. Methicillin-Resistant Staphylococcus capitis
with Reduced Vancomycin Susceptibility Causes Late-Onset Sepsis in Intensive Care
Neonates. PLoS ONE. 7(2):e31548.
3. Wil C. Van Der Zwet, Yvette J. Debets-Ossenkopp, Erik Reinders, Maria Kapi, Paul
H. M. Savelkoul, Ruurd M. Van Elburg, Keiichi Hiramatsu and Christina M. J. E.
Vandenbroucke Grauls. 2002. Nosocomial Spread of a Staphylococcus capitis Strain
with Heteroresistance to Vancomycin in a Neonatal Intensive Care Unit. J Clin
Microbiol. 40(7):2520-2525.
4. D'mello D, J. Daley A, Shihab Rahman M, Qu Y, Garland S , Pearce C and A.
Deighton M. 2008. Vancomycin Heteroresistance in Bloodstream Isolates of
Staphylococcus capitis. J Clin Microbiol. 40(9):3124-3126.

CHAPITRE III
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 82
5. Cui B, Smooker PM, Rouch DA, Daley AJ, Deighton MA. 2013. Differences between
two clinical Staphylococcus capitis subspecies as revealed by biofilm, antibiotic
resistance, and pulsed-field gel electrophoresis profiling. J Clin Microbiol. 51: 9–14.
6. Butin M, Martins Simoes P, Lemriss H, Lemriss S, Vandenesch F, Claris O, Picaud
JC, Rasigade JP and Laurent F. 2014. Staphylococcus capitis in neonatal late-onset
sepsis: unexpected worldwide dissemination of an endemic multi-resistant clone. In:
European Academy of Pediatric Societies - Poster symposium - Session Neonatal
Sepsis - Poster No: 011. Barcelona, Spain.
7. Vallenet D, Belda E, Calteau A, Cruveiller S, Engelen S, Lajus A, Le Fèvre F, Longin
C, Mornico D, Roche D, Rouy Z, Salvignol G, Scarpelli C, Thil Smith A A,Weima,
M, and Médigue C. 2013. MicroScope an integrated microbial resource for the
curation and comparative analysis of genomic and metabolic data. Nucleic Acids Res
41:D636-D647.
8. Vallenet D, Labarre L, Rouy Z, Barbe V, Bocs S, Cruveiller S, Lajus A, Pascal G,
Scarpelli C, Médigue C. 2006. MaGe: amicrobial genome annotation system
supported by synteny results. Nucleic Acids Res 34:53-65.
9. Bocs S, Cruveiller S, Vallenet D, Nuel G, Médigue C. 2003. AMIGene: Annotation of
Microbial Genes. Nucleic Acids Res 31:3723-3726.
10. Lowe TM, Eddy SR. 1997. tRNA scan-SE: a program for improved detection of
transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955-964.
11. Lagesen K, Hallin P, Rødland EA, Staerfeldt HH, Rognes T, Ussery DW. 2007.
RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids
Res 35:3100-3108.
12. Gardner PP, Daub J, Tate JG, Nawrocki EP, Kolbe DL, Lindgreen S, Wilkinson AC,
Finn RD, Grif-fiths-Jones S, Eddy SR, Bateman A. 2009. Rfam: updates to the RNA
families database. Nucleic Acids Res 37:D136-D140.
13. Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal:
prokaryotic gene recognition and translation initiation site identification. BMC
Bioinformatics.11:119.14. UnitProt Consortium. The Universal Protein Re-source
(UniProt) 2009. Nucleic Acids Res 2009; 117 37:D169-D174.
14. UnitProt Consortium. The Universal Protein Re-source (UniProt) 2009. Nucleic Acids
Res 2009; 117 37:D169-D174.
15. Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov
DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV,

CHAPITRE III
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 83
Vasudevan S, Wolf YI, Yin JJ, and Darren AN. 2003. The COG database: an updated
version includes eukaryotes. 121 BMC Bioinformatics. 4:41.
16. Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Das U,
Daugherty L, Duquenne L, Finn RD, Gough J, Haft D, Hulo N, Kahn D, Kelly E,
Laugraud A, Letunic I, Lonsdale D, Lopez R, Madera M, Maslen J, McAnulla C,
McDowall J, Mistry J, Mitchell A, Mulder N, Natale D, Orengo C, Quinn AF,
Selengut JD, Sigrist CJ, Thimma M, Thomas PD, Valentin F, Wilson D, Wu CH and
Yeats C. 2009. InterPro: the integrative protein signature database. Nucleic Acids Res
37:D211-D215.
17. Claudel-Renard C, Chevalet C, Faraut T, Kahn D. 2003. Enzyme-specific profiles for
genome annotation: PRIAM. Nucleic Acids Res 31:6633-6639.
18. Sonnhammer EL, von Heijne G, Krogh A. 1998. A hid-den Markov model for
predicting transmembrane helices in protein sequences. Proc Int ConfIntellSyst Mol
Biol. 6:175-182.
19. Bendtsen JD, Nielsen H, von Heijne G, Brunak S. 2004. Improved prediction of signal
peptides:SignalP 3.0. J Mol Biol. 340:783-795.
20. Gardy JL, Laird MR, Chen F, Rey S, Walsh CJ, Es-ter M, Brinkman FS. 2005.
PSORTb v.2.0: 8 expanded prediction of bacterial protein subcellular localization and
insights gained from comparative 136 proteome analysis. Bioinformatics. 21:617-623.
21. European Nucleotide Archive. http://www.ebi.ac.uk/ena/data/view.
III.2 Genome Sequences of multiresistant Staphylococcus capitis pulsotype NRCS-A and
methicillin- 2 susceptible Staphylococcus capitis pulsotype NRCS-C
Abstract
Here, we report the draft genome sequences of methicillin-susceptible Staphylococcus captis
pulsotype NCRS-C (CR02 strain) and multiresistant Staphylococcus captis pulsotype NCRS-
A (CR07 strain).
Staphylococcus capitis is a Gram-positive coccus belonging to the coagulase-negative
staphylococcus group (CoNS) that is frequently found on the human skin and mucosa (1).
Recently, a few studies have reported the emergence of S. capitis, which was found to be a
major cause of late-onset sepsis (LOS) in several neonatal intensive care units (NICUs) (2, 3).

CHAPITRE III
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 84
A clonal population of methicillin-resistant S. capitis NRCS-A strains has spread into several
geographically distant NICUs (4). These isolates exhibit reduced susceptibility to
vancomycin, which is the most widely used antimicrobial agent in the NICU setting (5, 6).
In order to elucidate the molecular mechanisms behind the wide-spreading of methicillin-
resistant S. capitis and methicillin susceptible S. capitis in NICUs in France, we sequenced
two different pulsotypes (NRCS-A and NRCS-C) of S. capitis strains (CR07 and CR02).
In this report, we present the draft genome sequences of multiresistant S. capitis pulsotype
NRCS-A and methicillin susceptible S. capitis pulsotype NRCS-C isolated from blood
cultures from NICUs in Lyon, France.
These two pulsotype strains were grown in blood agar at 37°C, and genomic DNA was
extracted using the PureLink genomic DNA kit (Invitrogen), according to the manufacturer’s
recommended protocol. The DNA libraries were prepared from 1µg DNA genomic extracted
following GS rapid library protocol (Roche 454; Roche).
The genome sequence of each S. capitis strain was determined by high-throughput sequencing
performed on a Genome Sequencer FLX_system (454 Life Sciences/Roche) using FLX
Titanium reagents, according to the manufacturer’s protocols and instructions. De novo
assemblies were performed using Roche Newbler assembly software (version 2.9).
An automatic syntactic and functional annotation of the draft genome was performed using
the MicroScope platform pipeline (7). The syntactic analysis combines a set of programs
including AMIGene (8), tRNAscan-SE (9), RNAmmer (10), Rfam scan (11), and Prodigal
software (12) to predict genomic objects that are mainly coding sequences (CDSs) and RNA
genes. More than 20 bioinformatics methods were used for functional and relational analyses.
The homology search was performed in the generalist databank UniProt (13) and in more
specialized databases such as COG (14), InterPro (15), PRIAM profiles for enzymatic
classification (16), prediction of protein localization using TMHMM (17), SignalP (18), and
PsortB (19) tools.
The draft genome sequence of methicillin-susceptible S. capitis NRCS-C (CR02 strain) has
33.05% G_C content, is 2,344,750 bp in length which is distributed in 320 contigs (average
coverage, 7.0_) with 2,415 genes, 2,476 CDSs, 42 pseudo genes, 4 rRNAs (5S,16S, 23S), and
59 tRNAs.

CHAPITRE III
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 85
The draft genome sequence of methicillin-resistant S. capitis NRCS-A (CR07 strain) has
32.83% G_C content, is 2,477,101 bp in length which is distributed in 26 contigs (average
coverage, 31x) with 2,411 CDSs, 4 rRNAs (5S,16S, 23S), and 61 tRNAs.
Nucleotide sequence accession numbers. These whole genome sequences were deposited at
GenBank under the accession numbers CZWI00000000 and CZWH00000000 for a
methicillin-susceptible S. capitis NRCS-C strain (CR02) and a multiresistant S. capitis
NRCS-A strain (CR07), respectively. The versions described in this paper are the first
versions.
Acknowledgments
This work was supported by a grant from the Fondation pour la Recherche Médicale (FRM,
grant ING20111223510) and by the Institut National de la Recherche Médicale (INSERM)
and the French Ministry of Health. This work was also supported by a grant from the NIH for
H3Africa BioNet.
References
1. Li X, Lei M, Song Y, Gong K, Li L, Liang H, Jiang X. 2014. Whole genome sequence
and comparative genomic analysis of multidrug-resistant Staphylococcus capitis
subsp. urealyticus strain LNZR-1. Gut Pathogens. 6:45.
2. Klingenberg C, Rønnestad A, Anderson AS, Abrahamsen TG, Zorman J, Villaruz A,
Flægstad T, Otto M, Sollid, Ericson J. 2007. Persistent strains of coagulase negative
staphylococci in neonatal intensive care unit: virulence factors and invasiveness. Clin
Microbiol Infec. 13(11):1100-11.
3. Rasigade J-P, Raulin O, Picaud J-C, Tellini C, Bes M, Grando J, Ben Saïd M, Claris
O, Etienne J, Tigaud S, Laurent F. 2012. Methicillin-Resistant Staphylococcus capitis
with Reduced Vancomycin Susceptibility Causes Late-Onset Sepsis in Intensive Care
Neonates. PLoS ONE. 7(2):e31548.
4. Butin M, Rasigade JP , Martins-Simões P, Meugnier H, Lemriss H, Goering R.V,
Kearns A, Deighton M.A, Denis O, Ibrahimi A, Claris O, Vandenesch F, Picaud JC,
Laurent F. 2016. Wide geographical dissemination of the multiresistant
Staphylococcus capitis NRCS-A clone in neonatal 84 intensive-care units. Clin
Microbiol Infec. 22(1):46-52.
5. . Cui B, Smooker PM, Rouch DA, Daley AJ, Deighton MA. 2013. Differences
between two clinical Staphylococcus capitis subspecies as revealed by biofilm,

CHAPITRE III
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 86
antibiotic resistance, and pulsed-field gel 87 electrophoresis profiling. J Clin
Microbiol. 51: 9–14.
6. Vallenet D, Belda E, Calteau A, Cruveiller S, Engelen S, Lajus A, Le Fèvre F, Longin
C, Mornico D, Roche D, Rouy Z, Salvignol G, Scarpelli C, Thil Smith A A,Weima,
M, and Médiguel C. 2013. MicroScope an integrated microbial resource for the
curation and comparative analysis of 91 genomic and metabolic data. Nucleic Acids
Res 41:D636-D647.
7. Vallenet D, Labarre L, Rouy Z, Barbe V, Bocs S, Cruveiller S, Lajus A, Pascal G,
Scarpelli C, Médigue C. 2006. MaGe: amicrobial genome annotation system
supported by synteny results. Nucleic Acids Res 34:53-65.
8. Bocs S, Cruveiller S, Vallenet D, Nuel G, Médigue C. 2003. AMIGene: Annotation of
Microbial Genes. Nucleic Acids Res 31:3723-3726.
9. Lowe TM, Eddy SR. 1997. tRNA scan-SE: a program for improved detection of
transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955-964.
10. Lagesen K, Hallin P, Rødland EA, Staerfeldt HH, Rognes T, Ussery DW. 2007.
RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids
Res 35:3100-3108.
11. Gardner PP, Daub J, Tate JG, Nawrocki EP, Kolbe DL, Lindgreen S, Wilkinson AC,
Finn RD, Grif-fiths-Jones S, Eddy SR, Bateman A. 2009. Rfam: updates to the RNA
families database. Nucleic Acids Res 37:D136-D140.
12. Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal:
prokaryotic gene recognition and translation initiation site identification. BMC
Bioinformatics.11:119.13.
13. UnitProt Consortium. The Universal Protein Re-source (UniProt) 2009. Nucleic Acids
Res 2009; 37:D169-D174.
14. Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov
DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV,
Vasudevan S, Wolf YI, Yin JJ, and Darren AN. 2003. The COG database: an updated
version includes eukaryotes. BMC Bioinformatics. 4:41
15. Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Das U,
Daugherty L, Duquenne L, Finn RD, Gough J, Haft D, Hulo N, Kahn D, Kelly E,
Laugraud A, Letunic I, Lonsdale D, Lopez R, Madera M, Maslen J, McAnulla C,
McDowall J, Mistry J, Mitchell A, Mulder N, Natale D, Orengo C, Quinn AF,
Selengut JD, Sigrist CJ, Thimma M, Thomas PD, Valentin F, Wilson D, Wu CH and

CHAPITRE III
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 87
Yeats C. 2009. InterPro: the integrative protein signature database. Nucleic Acids Res
37:D211-D215.
16. Claudel-Renard C, Chevalet C, Faraut T, Kahn D. 2003. Enzyme-specific profiles for
genome annotation: PRIAM. Nucleic Acids Res 31:6633-6639.
17. Sonnhammer EL, von Heijne G, Krogh A. 1998. A hid-den Markov model for
predicting transmembrane helices in protein sequences. Proc Int ConfIntellSyst Mol
Biol. 6:175-182.
18. Bendtsen JD, Nielsen H, von Heijne G, Brunak S. 2004. Improved prediction of signal
peptides: SignalP 3.0. J Mol Biol. 340:783-795.
19. Gardy JL, Laird MR, Chen F, Rey S, Walsh CJ, Es-ter M, Brinkman FS. 2005.
PSORTb v.2.0: expanded prediction of bacterial protein subcellular localization and
insights gained from comparative proteome analysis. Bioinformatics. 21:617-623.

87
CHAPITRE IV
Génomique comparative de souches
de Stapylococcus capitis

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 88
Les bactéries pathogènes constituent une menace majeure pour la santé humaine, que ce soit
de manière naturelle ou liée aux activités humaines. De ce fait, elles font l'objet d'études
approfondies afin de comprendre leurs modalités d'évolution, les mécanismes gouvernant
l'acquisition du caractère pathogène, ainsi que la sélection et le transfert de facteurs de
virulence, notamment dans le contexte du développement des résistances aux antibiotiques et
de l'augmentation de la fréquence des infections nosocomiales.
Les staphylocoques à coagulase négative (SCN) sont la cause la plus fréquente de septicémie
tardive dans les unités de soins intensifs néonatals (NICU). L’espèce Staphylococcus
epidermidis est la plus prévalente (> 70 %) dans ce contexte. Cependant, des rapports récents
ont identifié Staphylococcus capitis, un autre SCN, comme une cause émergente de
bactériémie dans les salles de NICU. Les isolats de S. capitis, provenant des nourrissons de
plusieurs NICU dans le monde (France, Royaume-Uni, Belgique et Australie), ont plusieurs
caractéristiques moléculaires (électrophorèse sur gel à champ pulsé) et bactériologiques
frappantes. Ils appartiennent tous au même type, désigné par NRCS-A, ce qui indique leur
relation clonale et leur profil de résistance reflétant leur adaptation aux agents antimicrobiens
spécifiquement utilisés dans les NICU, y compris la résistance aux bêta-lactamines, aux
aminoglycosides et hétérorésistance aux glycopeptides ( Rasigade et al., 2012; de Souza
Rugolo et al., 2014).
Peu de séquences génomiques de S. capitis sont publiquement disponibles à ce jour. Une
seule et unique étude, utilisant le génome d'un isolat de S. capitis recueillie à partir d'une
infection sanguine chez un adulte, a rapporté la comparaison génomique des facteurs de
virulence entre S. capitis et S. epidermidis (Cameron et al., 2015). Compte tenu de
l'importance spécifique de S.capitis NRCS-A dans l'établissement NICU et de sa diffusion
internationale, nous présentons dans ce chapitre la première séquence de génome fermé de la
souche française CR01 de prototype NRCS-A, une comparaison des séquences du génome
entier (WGS) de cette souche avec trois autres souches de prototype NRCS-A recueillies dans
des pays éloignés (Belgique (CR03), Australie (CR04), Royaume-Uni (CR05)) et une
description des caractéristiques génomiques de ce clone NRCS-A.
Le séquençage par la méthode shotgun, l’assemblage et l’annotation (syntaxique et
fonctionnelle) du génome complet de S. capitis CR01, prototype NRCS-A, (souche Française
isolée en 2009) a été réalisé en 2012 comme décrit dans le 2ème chapitre. Ce génome a été le
premier draft génome disponible de cette bactérie. Cette séquence a donc été le point de

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 89
départ de l’étude de ce pathogène par des approches de la génomique comparative, en utilisant
diverses plateformes (publiques et privées), logiciels et outils bioinformatiques.
Les analyses préliminaires du draft génome de cette souche CR01 ont permis la
caractérisation d’une nouvelle cassette SCCmec de type V (numéro d’accession à GenBank
/DDBJ / EMBL : KF049201). Cette dernière est fortement homologue à celle
des S. aureus résistants à la méticilline ST398 (LA-SARM) et portant de façon surprenante un
élément CRISPR (2 CRISPER) d’immunité adaptative et une cassette SCC portant des gènes
de résistance aux métaux lourds (SCCcad/ars/cop). La comparaison avec d’autres génomes
SARM et de S. capitis a permis de proposer un schéma évolutif avec acquisition successive
indépendante des 2 cassettes (SCCmec et SCCcad/ars/cop) de 60,9 kb confirmé par le typage
moléculaire et l’étude phylogénique.
Le séquençage par SMART (Single-Molecule Real-Time) de cette souche française CR01
nous a permis de confirmer le scaffolding qui a été fait par le pyroséquençage 454 et aussi
d'améliorer la séquence du draft génome en fermant les gaps au sein des scaffolds. Au final,
nous avons obtenu une séquence sous forme d'un seul chromosome circulaire de 2,522,871
bp et d’un plasmide de 26.140 bp. Ces séquences ont été corrigées et annotées avec la
plateforme MicroScope (MaGe) (Vallenet et al., 2006. 2013) et le logiciel Mauve (Darling et
al., 2010).
Sur la base des séquençages à haut débit d’un large panel d’isolats cliniques de souches de S.
capitis, provenant d’un échantillonnage à différentes périodes de temps (2000-2009) et à
différentes localisations (France (CR01), Belgique (CR03), Australie (CR04), Royaume-Uni
(CR05)) comme décrit dans le chapitre III, des comparaisons génomiques avec les autres
souches de S. capitis des adultes et d’autres staphylocoques disponibles sur les bases de
données ont été réalisées pour étudier les spécificités génétiques (gènes de virulence, de
résistances, éléments génétiques mobiles, etc.) et l'évolution du clone endémique au cours du
temps.
Le génome fermé de la souche CR01 a été comparé avec trois génomes assemblés des
souches du clone NRCS-A (CR03, CR04 et CR05), six génomes publiques de S.
capitis n’appartenant pas au clone NRCS-A et autres staphylocoques.
Nous avons pu mettre en évidence au sein du clone NRCS-A des gènes de résistance
(mecA, aacA-aphD, ...) associés à des éléments génétiques mobiles ou contenus dans le core-

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 90
genome, expliquant le profil de résistance de ce clone, des facteurs de virulence dans le core-
genome comprenant des Phenol Soluble Modulins, des hémolysines, des protéines de la paroi
fixant le fibrinogène ou la fibronectine, et l'opéron ica. En outre, nous avons noté la présence
spécifique d’un gène de résistance à la nisine (nsr), absent des génomes non-NRCS-A et de la
plupart des génomes de S. aureus et de staphylocoques à coagulase négative (SCN).
Nos résultats soulignent le rôle des éléments génétiques mobiles dans l'émergence du
phénotype multirésistant du clone NRCS-A, lui conférant un profil de résistance parfaitement
adapté à la pression de sélection existant en réanimation néonatale. Par ailleurs le gène de
résistance à la nisine, bactériocine "large-spectre" active sur de nombreuses bactéries à Gram
positif et sécrétée par les bactéries de la flore commensale digestive (Lactococcus,
entérocoques, BGN…), pourrait contribuer à une meilleure fitness au clone NRCS-A et lui
conférer un avantage sélectif au sein du microbiote des prématurés par rapport aux autres
souches de S. capitis et plus largement aux autres souches de SCN ne possédant pas ce gène.
Ce chapitre est présenté sous forme de deux articles publiés dans le journal Antimicrobial
Agents and Chemotherapy (Martins Simões et al., 2013) et celui de Frontiers in Microbiology
(Martins Simões et al., 2016). Les références de ces deux articles sont les suivantes :
Antimicrob. Agents Chemother.December 2013 vol. 57 no. 126354-6357.
doi:10.1128/AAC.01576-13.
Environment. Front. Microbiol. 7:1991. doi: 10.3389/fmicb.2016.01991
Dans ces deux publications (partie IV.1 et partie IV.2 ci-dessous), j’ai réalisé toutes les
analyses de la génomique comparative en double avec l’équipe du CNR de Staphylocoques,
Lyon, France et j’ai contribué à la rédaction des manuscrits.
Le travail de la partie IV.1 a fait également l’objet de deux communications orale ci-dessous
et dont les résumés sont présentés dans la partie publications et communications.
1- P. Martins-Simões, Y. Dumont, M. Butin, H. Lemriss, S. Lemriss, A. Ibrahimi, S.El
Kabbaj, F. Vandenesch, J.P. Rasigade, F. Laurent. Caractérisation moléculaire par
séquençage PacBio du clone endémique mondial S. capitis NRCS-A responsable de sepsis

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 91
en réanimation néonatale : résistances, virulence et avantage sélectif. RICAI, Paris, 14
Décembre 2015.
2- P. Martins Simoes, H. Lemriss, J.-P. Rasigade, S. Lemriss, F. Vandenesch, S. El Kabbaj,
F. Laurent. Staphylococcus capitis NRCS-A: SCC, SCCMEC and Their Evolutionary
History. 5th Congrees of European Microbiologists, FEMS, Juillet 2013.
IV.1 Characterization of a Novel Composite Staphylococcal Cassette Chromosome mec
(SCCmec-SCCcad/ars/cop) in the Neonatal Sepsis-Associated Staphylococcus capitis
Pulsotype NRCS-A
Abstract
Multiresistant Staphylococcus capitis pulsotype NRCS-A has been reported to be a major
pathogen causing nosocomial bacteremia in preterm infants. We report that the NRCS-A
strain CR01 harbors a novel 60.9-kb composite staphylococcal cassette chromosome mec
(SCCmec) element, composed of an SCCmec with strong homologies to Staphylococcus
aureus ST398 SCCmec and of an SCCcad/ars/cop harboring resistance genes for cadmium,
arsenic, and copper. Whole-genome-based comparisons of published S. capitis strains suggest
that strain CR011 acquired the two elements independently.
Coagulase-negative staphylococci (CoNS) are common commensals of the human skin and
mucosa that are now recognized as important opportunistic pathogens that cause nosocomial
bloodstream and indwelling medical device-related infections (1). Recent studies have
reported the emergence of methicillin-resistant, vancomycin-heteroresistant Staphylococcus
capitis as an important cause of late-onset sepsis (LOS) in neonatal intensive care units
(NICUs) in France and other countries (2–6). Surprisingly, all S. capitis strains from intensive
care neonates were reported to belong to a single pulsed-field gel electrophoresis pulsotype,
NRCS-A, and to harbor a type V-related staphylococcal cassette chromosome mec (SCCmec)
element (6). In contrast, S. capitis bloodstream isolates from adult patients belong to several
distinct, non-NRCS-A pulsotypes. To gain insights into the genetic characteristics of this
LOS-specific multiresistant S.capitis clone, we sequenced and characterized the SCCmec
element of one representive starin (CR01) with the NRCS_A plusotype.

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 92
Strain CR01 was isolated from the blood cultures of an infant diagnosed with LOS in 2007 in
the NICU of the Hospices Civils de Lyon, France. The strain was part of a previously
published collection (6), had the pulsotype NRCS-A, and harbored a type V-related
SCCmec element. We performed whole-genome pyrosequencing (454 Life Sciences/Roche)
for this strain and used bioinformatics tools to identify and characterize a novel 60.9-kb
composite SCCmec element (DDBJ/EMBL/GenBank accession no. KF049201). The cassette
was found to be inserted into the characteristic 3′ end of the orfX gene (7, 8). To define the
SCC region, we searched for SCC-specific insertion site sequences (ISSs) (9, 10). Three ISSs
were found, with the furthest downstream from orfX being partially deleted. Because the
predicted SCC region was found in two different contigs, the SCC sequence was closed using
PCR.
SCCmec characterization and similarities to SCCmec elements of livestock-
associated Staphylococcus aureus
A 38.8-kb SCCmec element was identified immediately downstream of orfX. This element
harbored a type V (5C2&5) SCCmec and contained 42 open reading frames (ORFs; orf1 to
orf42 in Table IV.1 in the supplemental material), as predicted using the syntactic and
functional annotation implemented in the MicroScope/MaGe platform pipeline (11, 12).
Using the MicroScope genome browser, the sequence of the CR01 SCCmec element was
aligned with 22 genomes of methicillin-resistant Staphylococcus strains, which are available
in the MaGe database. Two genomes in this database carried SCCmec elements showing a
remarkable similarity to the NRCS-A strain CR01 SCCmec in terms of gene content, gene
order, and nucleotide identity. Both SCCmec elements were carried by livestock-associated
methicillin-resistant S. aureus (LA-MRSA) strains, namely, the S0385 and 08BA02176
strains, which belong to the highly prevalent LA-MRSA sequence type 398 (ST398) (Fig.
IV.1.1; see also Table IV.1). A detailed comparison of gene organization in these three
SCCmec elements revealed that the structures of the joining regions J3 and J2 and
the mec and ccr complexes were identical to those found in the ST398 LA-MRSA strain
S0385, whereas the joining region J1 was identical to that of the ST398 LA-MRSA strain
08BA02176. In both the CR01 and the 08BA02176 SCCmec elements, the J1 region
contained clustered regularly interspaced short palindromic repeat (CRISPR) loci with their
associated cas genes. CRISPR loci are the sole prokaryotic defense mechanism against
foreign DNA conferring an adaptive immune response, inversely to other defense
mechanisms such as restriction-modification (R-M) systems (13). Using the online

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 93
CRISPRFinder software tool (14), we identified two CRISPR elements in the NRCS-A strain
CR01 located immediately before the second ISS (Fig. 1). The first CRISPR element
consisted of 16 direct repeats (DRs) and 15 spacers. The second CRISPR element consisted of
three nonconserved DRs and two spacers. This subtype III-A CRISPR locus (15) (including
the 2 CRISPRs and the cas genes) showed a high similarity to the element found in the ST398
LA-MRSA strain 08BA02176 SCCmec element (see Fig. IV.1.3 table IV.1.S2 in the
supplemental material).
Figure IV.1.1. Comparative structure analysis of the composite staphylococcal
chromosome cassette SCCmec-SCCcad/asr/cop element of Staphylococcus capitis strain
CR01. Sequence of the SCCmec-SCCcad/asr/cop element of strain CR01 was aligned against
whole genomes of ST398 S. aureus strains S0385 and 08BA02176, and of S. capitis strain
SK14. Open reading frames (ORFs) are shown as arrows indicating the transcription direction
and colored according to the SCCmec region to which they belong (J3, mec complex, J2, ccr
complex or J1). Homologous gene clusters in different strains have similar colors. The
chromosomal orfX gene and the transposase IS43 are represented by black and yellow arrows,
respectively. Insertion site sequences (ISSs) are indicated by vertical lines and colored stars as
follows: light yellow star, ccrC recombinase ISS; dark yellow star, ccrAB recombinase ISS
with associated direct repeat (DR) sequences; and red star, vestigial ccrAB ISS and DR

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 94
sequences. The CRISPR associated genes (cas genes) are indicated in light green within the
J3 region, while the ccr complex in the SCCcad/ars/cop element are colored in turquoise and
the resistance genes in dark red.
To further investigate the relationships between the CR01 SCCmec elements and those of
ST398 S. aureus (strains S0385 and 08BA02176), we characterized the SCCmec-associated
direct repeat units (dru) using the scheme described by Goering et al., which has been
described as a discriminant epidemiological tool for differentiating highly uniform epidemic
methicillin-resistant strains (16). Dru analysis showed that the SCCmec cassettes of both the
CR01 strains and the ST398 08BA02176 strain shared the same dru type (5a-2d-4a-0-2d-5b-
3a-2g-4b-4e-3e), namely, dt11c, which further highlighted the close genetic proximity
between these SCCmec elements. Strain S0385 harbored a different dru type, namely, dt10a
(dt10a, 5a-2d-4a-0-2d-5b-3a-2g-3b-4e), but presenting only one mutation in the ninth repeat
and one repeat fewer. The dru type found in strain CR01 was identical to the ones found in
other S. capitis NRCS-A strains (n = 12, data not shown), further corroborating the clonality
within this lineage.
Collectively, these findings suggest that the S. capitis NRCS-A strain CR01 and ST398 LA-
MRSA strains have undergone a recent horizontal transfer of SCCmec elements originating
from a common source.
SCCcad/ars/cop characterization and relationships to other SCC elements
Immediately downstream of the SCCmec domain, we identified a second, 22.2-kb-long SCC
domain (SCCcad/ars/cop) harboring 29 predicted ORFs (orf43 to orf70 in Table IV.1.S1 in
the supplemental material), including a type 8 ccrA1B3 complex with 90% similarity to
the ccr complex recently described in S. aureus strain LGA251 carrying a type IX
SCCmec and the novel mecC allele (17). A comparison of the overall structure of the
SCCcad/ars/cop with other SCC elements revealed only two regions with partial similarities
to previously described SCC elements. The first region encompassed nine ORFs (orf47 to
orf56 in Table IV.1.S1), including the ccrA1B3 gene complex; this region had an organization
similar to the regions found in the SCCpbp4 element of the Staphylococcus epidermidis strain
ATCC 12228 and in the type II SCCmec elements of the S. aureus strains MRSA252 and
N315 (18). The second region encompassed 11 ORFs (orf58 to orf69 in Table IV.1.S1) and
included genes for the detoxification of heavy metals, namely, cadmium (cadDC), arsenic
(arsCBR), and copper (copB). All 11 ORFs were highly homologous to those found in the J1
regions of the SCCmec element of CC398 MRSA S. aureus JCSC6943 (19) and of the

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 95
ΨSCCmec(h1435) domain in Staphylococcus haemolyticus JCSC1435 (18), with nucleotide
identities greater than 97.5%.
Of note, previously published NRCS-A strains have been reported to harbor only a type V-
related SCCmec element (ccrC complex), as determined using the multiplex PCRs described
by Kondo et al. (6, 20). To understand why these PCRs failed to detect the
SCCcad/ars/cop type 8-related ccrAB complex, Kondo's primer sequences were aligned to the
NRCS-A strain CR01 ccrA1 and ccrB3alleles. Several mismatches were observed between
the ccrA primers and their target sequence, which likely prevented amplification of
the ccrAB complex and so explain the misinterpretation of Kondo's scheme.
To assess whether SCC elements are present in other S. capitis strains, we performed a
BLAST search (http://blast.ncbi.nlm.nih.gov) of the composite SCCmec-
SCCcad/ars/cop sequence against publicly available genomes from methicillin-susceptible S.
capitis isolates, namely, those of strains QN1 (GenBank accession no. AJJH00000000) (21),
SK14 (GenBank accession no. ACFR00000000), and VCU116 (GenBank accession
no. AFTX00000000). None of these strains harbored an SCCmec element. Interestingly,
whereas strain QN1 had an intact orfX gene, both the SK14 and the VCU116 strains carried
homologous SCCcad/ars/cop elements inserted into the 3′ end of orfX. These latter elements
were highly similar to the SCCcad/ars/cop found in the NRCS-A strain CR01 (Fig. IV.1.1;
see also Table IV.1.S1 in the supplemental material), except for the first seven ORFs after the
insertion at the end of orfX. Gene organization and nucleotide sequences were identical in 22
remaining ORFs (orf47 to orf49 and orf53 to orf70 in Table IV.1.S1).
Evolutionary history of SCCcad/ars/cop and SCCmec acquisition by S. capitisNRCS-A.
To assess the phylogenetic relationships between the S. capitis strains CR01 (NRCS-A clone),
QN1, SK14, and VCU116, we performed a whole-genome-based single nucleotide
polymorphism (SNP) analysis and constructed a maximum likelihood (ML) SNP tree using
the snpTree online web server (22) and PhyML (23), respectively (see the supplemental
material). Interestingly, all SCCcad/ars/cop-positive strains clustered on a monophyletic
clade, separate from the SCC-negative strain QN1. Furthermore, the sole SCCmec-positive
strain (CR01) was the most divergent compared with strains SK14 and VCU116 (Fig.
IV.1.2a). These results suggest the occurrence of at least two independent SCC acquisition
events within the S. capitis species (Fig. IV.1.2b). The first acquisition of the
SCCcad/ars/copoccurred in the ancestral lineage of strains CR01, SK14, and VCU116, which

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 96
was followed by later acquisition of the SCCmec element in the ancestor lineage of the
NRCS-A clone.
Figure IV.1.2. Phylogenetic analysis of whole genome-sequenced S. capitis strains and
proposed evolutionary history of SCC elements acquisition in S. capitis. (a) SNPs based Maximum likelihood tree of the 4 S. capitis WGS. A total of 27 836 SNPs (indels
excluded) were obtained by alignment against the reference strain S. epidermidis ATCC 12228. Numbers at the branching points represent the aLRT branch support score obtained. Proposed
acquisition events of SCCcad/ars/cop and SCCmec elements are indicated by dark and light arrows,
respectively. (b) Proposed model of sequential acquisition of SCC elements in the S. capitis clade: in a
first event the SCCcad element harboring multiple resistance genes to heavy metals was inserted via site specific recombination at the end of the orfX gene; a second insertion occurred in the
SCCcad/ars/cop positive strain CR01 strain with the insertion of SCCmec at the 3'-end of orfX.
Legend: ancest1 = first ancestor; ancest2 = second ancestor (of CR01,SK14 and VCU116).
Conclusions
The highly clonal population of S. capitis NRCS-A, which is endemic in NICUs in several
countries distant from one another (5), harbors a novel SCCmec-SCCcad/ars/cop composite
island that has likely emerged from two independent acquisition events.
The high degree of structural similarity of the SCCmec type V element and the partial
conservation of genes for heavy metal detoxification strongly suggest the occurrence of
SK14 &
VCU116
Ancest2
CR01
Ancest1
(b)
+
(a)
SCCcad/ars/cop
QN1
+SCCmec
SCCmec
SCCcad/ars/cop
QN1
NRCS-A CR01
VCU116
SK14
0.05
TIME

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 97
genetic shuffling involving S. capitis and CC398 MRSA strains. Whether SCC element
exchanges occurred directly between these strains or indirectly via a common SCC element
donor is presently unclear and warrants further investigation.
Acknowledgements
This work was supported by the Institut National de la Recherche Médicale (INSERM) and
the French Ministry of Health and by a grant from the Fondation pour la Recherche Médicale
(FRM) (grant ING20111223510).
References
1. Otto M. 2009. Staphylococcus epidermidis — the “accidental” pathogen. Nat. Rev.
Microbiol. 7:555–567.
2. Gras-Le Guen C, Fournier S, Andre-Richet B, Caillon J, Chamoux C, Espaze E,
Richet H, Roze JC, Lepelletier D. 2007. Almond oil implicated in a Staphylococcus
capitis outbreak in a neonatal intensive care unit. J. Perinatol. 27:713–717.
3. Ng PC, Chow VC, Lee CH, Ling JM WH, Chang R. 2006. Persistent Staphylococcus
capitis septicemia in a preterm infant. Pediatr Infect Dis J 25:652–654.
4. De Silva GDI, Kantzanou M, Justice A, Massey RC, Wilkinson AR, Day NPJ,
Peacock SJ. 2002. The ica Operon and Biofilm Production in Coagulase-Negative
Staphylococci Associated with Carriage and Disease in a Neonatal Intensive Care
Unit. J Clin Microbiol 40:382–388.
5. Rasigade J-P, Goering R, Kearns A, Hill R, Deighton M, Meugnier H, Bes M, Ben
Saïd M, Claris O, Picaud J-C, Vandenesch F, Etienne J, Laurent F. 2012. Worldwide
epidemic methicillin-resistant Staphylococcus capitis with reduced vancomycin
susceptibility causes late-onset sepsis in intensive care neonates. 15th Isssi Lyon 2012
P 06-075.
6. Rasigade J-P, Raulin O, Picaud J-C, Tellini C, Bes M, Grando J, Ben Saïd M, Claris
O, Etienne J, Tigaud S, Laurent F. 2012. Methicillin-Resistant Staphylococcus capitis
with Reduced Vancomycin Susceptibility Causes Late-Onset Sepsis in Intensive Care
Neonates. Plos One 7:e31548.
7. Ito T, Katayama Y, Asada K, Mori N, Tsutsumimoto K, Tiensasitorn C, Hiramatsu K.
2001. Structural comparison of three types of staphylococcal cassette chromosome
mec integrated in the chromosome in methicillin-resistant Staphylococcus aureus.
Antimicrob. Agents Chemother. 45:1323–1336.

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 98
8. Ma XX, Ito T, Tiensasitorn C, Jamklang M, Chongtrakool P, Boyle-Vavra S, Daum
RS, Hiramatsu K. 2002. Novel Type of Staphylococcal Cassette Chromosome mec
Identified in Community-Acquired Methicillin-Resistant Staphylococcus aureus
Strains. Antimicrob Agents Chemother 46:1147–1152.
9. Ito T, Ma XX, Takeuchi F, Okuma K YH, HiramatsuK. 2004. Novel Type V
Staphylococcal Cassette Chromosome mec Driven by a Novel Cassette Chromosome
Recombinase, ccrC. Antimicrob Agents Chemother 48:2637–2651.
10. Noto MJ, Archer GL. 2006. A subset of Staphylococcus aureus strains harboring
staphylococcal cassette chromosome mec (SCCmec) type IV is deficient in CcrAB-
mediated SCCmec excision. Antimicrob. Agents Chemother. 50:2782–2788.
11. Vallenet D, Labarre L, Rouy Z, Barbe V, Bocs S, Cruveiller S, Lajus A, Pascal G,
Scarpelli C, Médigue C. 2006. MaGe: a microbial genome annotation system
supported by synteny results. Nucleic Acids Res 34:53–65.
12. Vallenet D, Engelen S, Mornico D, Cruveiller S, Fleury L, Lajus A, Rouy Z, Roche D,
Salvignol G, Scarpelli C, Médigue C. 2009. MicroScope: a platform for microbial
genome annotation and comparative genomics. Database J. Biol. Databases Curation
2009:bap021.
13. Sorek R, Lawrence CM, Wiedenheft B. 2013. CRISPR-Mediated Adaptive Immune
Systems in Bacteria and Archaea. Annu. Rev. Biochem. 82:237–266.
14. Makarova KS, Haft DH, Barrangou R, Brouns SJJ, Charpentier E, Horvath P,
Moineau S, Mojica FJM, Wolf YI. 2011. Evolution and classification of the CRISPR-
Cas systems. Nat. Rev. Microbiol. 9:467–477.
15. Grissa I, Vergnaud G, Pourcel C. 2007. CRISPRFinder: a web tool to identify
clustered regularly interspaced short palindromic repeats. Nucleic Acids Res 35:W52–
W57.
16. Goering RV, Morrison D, Al-Doori Z, Edwards GFS, Gemmell CG. 2008. Usefulness
of mec-associated direct repeat unit (dru) typing in the epidemiological analysis of
highly clonal methicillin-resistant Staphylococcus aureus in Scotland. Clin. Microbiol.
Infect. 14:964–969.
17. Shore AC, Deasy EC, Slickers P, Brennan G, O’Connell B, Monecke S, Ehricht R,
Coleman DC. 2011. Detection of staphylococcal cassette chromosome mec type XI
carrying highly divergent mecA, mecI, mecR1, blaZ, and ccr genes in human clinical
isolates of clonal complex 130 methicillin- resistant Staphylococcus aureus.
Antimicrob. Agents Chemother. 55: 3765–3773.

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 99
18. Mongkolrattanothai K, Boyle S, Murphy TV, Daum RS. 2004. Novel non-mecA-
containing staphylococcal chromosomal cassette composite island containing pbp4
and tagF genes in a commensal staphylococcal species: a possible reservoir for
antibiotic resistance islands in Staphylococcus aureus. Antimicrob. Agents
Chemother. 48:1823–1836.
19. Li S, Skov RL, Han X, Larsen AR, Larsen J, S?rum M, Wulf M, Voss A, Hiramatsu
K, Ito T. 2011. Novel Types of Staphylococcal Cassette Chromosome mec Elements
Identified in Clonal Complex 398 Methicillin-Resistant Staphylococcus aureus
Strains. Antimicrob. Agents Chemother. 55:3046–3050.
20. Kondo Y, Ito T, Ma XX, Watanabe S, Kreiswirth BN, Etienne J HK. 2007.
Combination of Multiplex PCRs for Staphylococcal Cassette Chromosome mec Type
Assignment: Rapid Identification System for mec, ccr, and Major Differences in
Junkyard Regions. Antimicrob Agents Chemother 51(1):264–274.
21. Qin N, Ding W, Yao J, Su K, Wu L, Li L. 2012. Genome Sequence of
Staphylococcus capitis QN1, Which Causes Infective Endocarditis. J Bacteriol
194:4469–4470.
22. Leekitcharoenphon P, Kaas RS, Thomsen MCF, Friis C, Rasmussen S, Aarestrup FM.
2012. snpTree - a web-server to identify and construct SNP trees from whole genome
sequence data. Bmc Genomics 13:S6.
23. Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W GO. 2010. New
Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing
the Performance of PhyML 3.0. Syst. Biol. 59(3):307–21.
Supplementary data
Maximum-Likelihood tree building
All four Staphylococcus capitis WGS strains: CR01, QN1, SK14 and VCU116 were aligned
to the reference genome of S. epidermidis ATCC 12228 using the snpTree online web-server
(1). Briefly, the Nucmer and the show-snps applications (as implemented in the MUMmer
v3.23 software package(2)) were used to, respectively, aligned the query genomes to the
reference one and call the SNPs from the resulting alignment. A prunning is then applied by
establishing the minimum distance between SNPs (default value = 10).
The SNPs were then concatenated into a single alignment by ignoring indels.

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 100
The SNPs alignment was then used to build a maximum-likelihood (ML) tree, using the
PhyML v.3 algorithm as implemented in Seaview v.4 (3, 4). The substitution model chosen
was the general time reversable (GTR) and we optimized: (I) the nucleotide equilibrium
frequency, t(ii) he proportion of invariable sites and (iii) the across site rate variation.
The aLRT SH-like test was used as a measure of branch support and the Nearest Neighbor
Interchange (NNI) method was used for improvement of tree topologies.
Table IV.1.S1. Open reading frames of the novel composite staphylococcal chromosome
cassette (SCC) found in methicillin-resistant Staphylococcus capitis NCRS-A prototype strain
CR01. The first 42 ORFs (orf1 to orf42) were found in the SCCmec element, while orf43 to
orf70 were found in the SCCcad/ars/cop element.
ORFa
gene size (bp) Gene Description of productb
protein size (aa)
Homolog (% id)c
GenBank accession no. d Homologs (strains)
ORF X 480 orfX
Ribosomal RNA large subunit methyltransferase
H 159 nap nap nap
SCCmec
orf1 306 conserved protein of
unknown function 101 100 YP_005732838.1 Staphylococcus aureus ST398
(S0385)
orf2 864 conserved protein of
unknown function 287 100 YP_005732839.1 Staphylococcus aureus ST398
(S0385)
orf3 1476 conserved protein of
unknown function 491 100 YP_005732840.1 Staphylococcus aureus ST398
(S0385)
orf4 1101 conserved protein of
unknown function 366 100
YP_005732841.1 Staphylococcus aureus ST398
(S0385)
orf5 372 conserved protein of
unknown function 123 100 YP_005732842.1 Staphylococcus aureus ST398
(S0385)
orf6 1644 conserved protein of
unknown function 547 100 YP_005732843.1 Staphylococcus aureus ST398
(S0385)
orf7 117 conserved protein of
unknown function 38 100 YP_005732844.1 Staphylococcus aureus ST398
(S0385)
orf8 1677 ccrC Cassette chromosome recombinase type C 558 100 YP_005732845.1
Staphylococcus aureus ST398 (S0385)
orf9 339 conserved protein of
unknown function 112 100 (1) Staphylococcus aureus ST398
(S0385)
orf10 312 conserved protein of
unknown function 103 100 YP_005732846.1 Staphylococcus aureus ST398
(S0385)
orf11 507 conserved protein of
168 100 YP_005732847.1 Staphylococcus aureus ST398

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 101
unknown function (S0385)
orf12 675 tnp transposase for IS431 or
IS66families 224 100 YP_005732848.1 Staphylococcus aureus ST398
(S0385)
orf13 168 HMG-CoA synthase 55 100
YP_005732849.1 ,
YP_006708794.1
Staphylococcus aureus ST398 (S0385), Staphylococcus aureus
08BA02176
orf14 744 ugpQ Glycerophosphoryldiester
phosphodiesterase 247 100
YP_0057328450.1,
YP_006708796.1
Staphylococcus aureus ST398 (S0385), Staphylococcus aureus
08BA02176
orf15 429 maoC putative dehydratase with
a MaoC like domain 142 100
YP_005732851.1,
YP_006708797.1
Staphylococcus aureus ST398 (S0385), Staphylococcus aureus
08BA02176
orf16 2007 mecA
Beta-lactam-inducible penicillin-binding protein
2' 668 100
YP_005732852.1,YP_006708798.
1
Staphylococcus aureus ST398 (S0385), Staphylococcus aureus
08BA02176
orf17 144
ΔmecR
1 Truncated methicillin resistance regulator 47 100
YP_005732853.1,
YP_006708799.1
Staphylococcus aureus ST398 (S0385), Staphylococcus aureus
08BA02176
orf18 93 Δtnp Truncated transposase for
IS431 or IS66families 30 100 (2) Staphylococcus aureus ST398
(S0385)
orf19 252 Δtnp Truncated transposase of
IS431 family or IS66 83 100 (3) Staphylococcus aureus ST398
(S0385)
orf20 429
3-demethylubiquinone-9 3-methyltransferase
domain protein 142 100
YP_005732854.1,
YP_006708801.1
Staphylococcus aureus ST398 (S0385), Staphylococcus aureus
08BA02176
orf21 930 Transcriptional regulator
(DeoR family) 309 100
YP_005732855.1,
YP_006708802.1
Staphylococcus aureus ST398 (S0385), Staphylococcus aureus
08BA02176
orf22 1989 conserved protein of
unknown function 662 100
YP_005732856.1,
YP_006708804.1
Staphylococcus aureus ST398 (S0385), Staphylococcus aureus
08BA02176
orf23 1110 conserved protein of
unknown function 369 100
YP_005732857.1,
YP_006708805.1
Staphylococcus aureus ST398 (S0385), Staphylococcus aureus
08BA02176
orf24 369 conserved protein of
unknown function 122 100
YP_005732858.1,YP_006708806.
1
Staphylococcus aureus ST398 (S0385), Staphylococcus aureus
08BA02176
orf25 1617 putative primase 538 100
YP_005732859.1,
YP_006708807.1
Staphylococcus aureus ST398 (S0385), Staphylococcus aureus
08BA02176
orf26 1680 ccrC Cassette chromosome
recombinase 559 100 YP_006708808.1 Staphylococcus aureus
08BA02176
orf27 339 conserved protein of
112 100 YP_006708809.1 Staphylococcus aureus

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 102
unknown function 08BA02176
orf28 240 conserved protein of
unknown function 79 100 YP_006708810.1 Staphylococcus aureus
08BA02176
orf29 504 conserved protein of
unknown function 167 100 YP_006708811.1 Staphylococcus aureus
08BA02176
orf30 222 conserved protein of
unknown function 73 100 YP_006708812.1 Staphylococcus aureus
08BA02176
orf31 132 conserved protein of
unknown function 43 100 (4) Staphylococcus aureus
08BA02176
orf32 276 ΔhsdR
truncated type I restriction modification system
methylase 91 100 YP_006708813.1 Staphylococcus aureus
08BA02176
orf33 117 protein of unknown
function 38 na na na
orf34 906 cas1 CRISPR-associated endonuclease Cas1 301 100 YP_006708814.1
Staphylococcus aureus 08BA02176
orf35 306 cas2 CRISPR-associated
endoribonuclease Cas2 101 100 YP_006708815.1 Staphylococcus aureus
08BA02176
orf36 2286 csm1 CRISPR-associated protein,
Csm1 family 761 100 YP_006708816.1 Staphylococcus aureus
08BA02176
orf37 426 csm2 CRISPR-associated protein,
Csm2 family 141 99.53 YP_006708817.1 Staphylococcus aureus
08BA02176
orf38 645 csm3 CRISPR type III-associated
RAMP protein Csm3 214 100 YP_006708818.1 Staphylococcus aureus
08BA02176
orf39 909 csm4 CRISPR-associated RAMP
protein, Csm4 family 302 100 YP_006708819.1 Staphylococcus aureus
08BA02176
orf40 1023 csm5 CRISPR-associated RAMP
protein, Csm5 family 340 100 YP_006708820.1 Staphylococcus aureus
08BA02176
orf41 1269 csm6 CRISPR-associated protein,
Csm6 family 422 100 YP_006708821.1 Staphylococcus aureus
08BA02176
orf42 735 cas6 CRISPR-associated
endoribonuclease Cas6 244 100 YP_006708822.1 Staphylococcus aureus
08BA02176
SCCcad/ars/cop
orf43 159
conserved protein of unknown function, potential pseudo? 52 100
(5), ZP_14685218.1
Staphylococcus aureus 08BA02176, Staphylococcus
epidermidis NIHLM057
orf44 636
conserved membrane protein of unknown
function 211 83.41 NP_788231.1 Oceanobacillus iheyensis strain
HTE831/DSM 14371
orf45 648 Transcriptional regulator,
215 62.09 ZP_07842269.1 Staphylococcus caprae C87

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 103
TetR family
orf46 660
conserved membrane protein of unknown
function 219 72.25 ZP_07842268.1 Staphylococcus caprae C87
orf47 222 conserved protein of
unknown function 73 89.04 ZP_03613434.1, ZP_12550545.1
Staphylococcus capitis SK14, Staphylococcus capitis VCU116
orf48 510 conserved protein of
unknown function 169 94.67 ZP_03613492.1, ZP_12550495.1
Staphylococcus capitis SK14, Staphylococcus capitis VCU116
orf49 312 conserved protein of
unknown function 103 97.07 ZP_03613450.1, ZP_12550524.1
Staphylococcus capitis SK14, Staphylococcus capitis VCU116
orf50 351 conserved protein of
unknown function 116 ZP_03613375.1, ZP_12550516.1
Staphylococcus capitis SK14, Staphylococcus capitis VCU116
orf51 84 protein of unknown
function 27 100 (6) Staphylococcus aureus COL
orf52 1629 ccrB Cassette chromosome
recombinase B 542 92.25 NC_017341.1 Staphylococcus aureusJKD6008
orf53 1350 ccrA Cassette chromosome
recombinase A 449 93.75 ZP_03613501.1, ZP_12550514.1
Staphylococcus capitis SK14, Staphylococcus capitis VCU116
orf54 258 conserved protein of
unknown function 85 96.47 ZP_03613426.1, ZP_12550528.1
Staphylococcus capitis SK14, Staphylococcus capitis VCU116
orf55 1791 conserved protein of
unknown function 596 98.49 ZP_03613521.1, ZP_12550491.1
Staphylococcus capitis SK14, Staphylococcus capitis VCU116
orf56 150 conserved protein of
unknown function 49 100 ZP_03613420.1, ZP_12550498.1
Staphylococcus capitis SK14, Staphylococcus capitis VCU116
orf57 990 conserved protein of
unknown function 329 (100) NZ_ACFR, NZ_AFTX
Staphylococcus capitis SK14, Staphylococcus capitis VCU116
orf58 165 putative acetyltransferase 54 100 ZP_03613441.1, ZP_12550473.1
Staphylococcus capitis SK14, Staphylococcus capitis VCU116
orf59 348 Transcriptional regulator
(MerR family) 115 100 ZP_03613502.1, ZP_12550548.1
Staphylococcus capitis SK14, Staphylococcus capitis VCU116
orf60 684
conserved protein of unknown function, DJ-
1/PfpI family 227 100 ZP_03613391.1, ZP_12550519.1
Staphylococcus capitis SK14, Staphylococcus capitis VCU116
orf61 666 conserved protein YhfK 221 100 ZP_03613415.1, ZP_12550504.1
Staphylococcus capitis SK14, Staphylococcus capitis VCU116
orf62 1005 yfmJ putative oxidoreductase 334 99.7 ZP_03613481.1, ZP_12550540.1
Staphylococcus capitis SK14, Staphylococcus capitis VCU116
orf63 366 conserved protein of
unknown function 121 100 ZP_03613511.1, ZP_12550508.1
Staphylococcus capitis SK14, Staphylococcus capitis VCU116
orf64 618 cadD Cadmium resistance
205 100 ZP_03613409.1, Staphylococcus capitis SK14,

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 104
transporter family protein ZP_12550544.1 Staphylococcus capitis VCU116
orf65 342 cadC
Cadmium resistance transcriptional regulatory
protein 113 100 ZP_03613548.1, ZP_12550523.1
Staphylococcus capitis SK14, Staphylococcus capitis VCU116
orf66 402 arsC arsenate reductase
(thioredoxin) 133 100 ZP_03613466.1, ZP_12550477.1
Staphylococcus capitis SK14, Staphylococcus capitis VCU116
orf67 1290 arsB arsenical pump membrane
protein 429 100 ZP_03613390.1, ZP_12550530.1
Staphylococcus capitis SK14, Staphylococcus capitis VCU116
orf68 318 arsR arsenical resistance
operon repressor 105 100 ZP_03613475.1, ZP_12550472.1
Staphylococcus capitis SK14, Staphylococcus capitis VCU116
orf69 2061 copB
putative copper-transporting P-type
ATPase B 686 99.71 ZP_03613385.1, ZP_12550486.1
Staphylococcus capitis SK14, Staphylococcus capitis VCU116
orf70 546 ydhK putative lipoprotein 181 93.92 ZP_03613457.1, ZP_12550476.1
Staphylococcus capitis SK14, Staphylococcus capitis VCU116
a ORF: open reading frame, b CRISPR: clustered regularly interspaced short palindromic repeats, c
two adjacent ORFs with 100 % id with distinct regions of the same ORF in strain CR01 are indicated
within parenthesis, d new open reading frames (ORFs) predicted after genomic re-annotation
performed in the MaGe/Miscroscope platform pipeline are indicated within parenthesis.
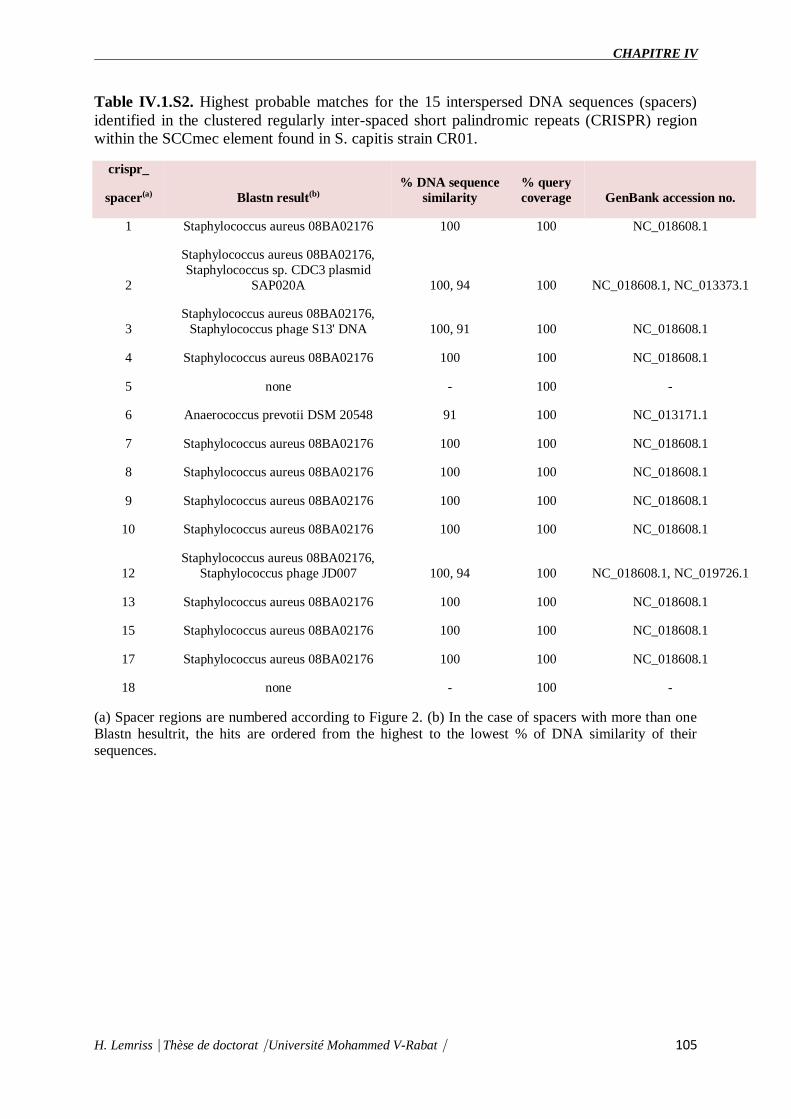
CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 105
Table IV.1.S2. Highest probable matches for the 15 interspersed DNA sequences (spacers)
identified in the clustered regularly inter-spaced short palindromic repeats (CRISPR) region
within the SCCmec element found in S. capitis strain CR01.
crispr_
spacer(a) Blastn result(b)
% DNA sequence
similarity
% query
coverage GenBank accession no.
1 Staphylococcus aureus 08BA02176 100 100 NC_018608.1
2
Staphylococcus aureus 08BA02176,
Staphylococcus sp. CDC3 plasmid
SAP020A 100, 94 100 NC_018608.1, NC_013373.1
3
Staphylococcus aureus 08BA02176,
Staphylococcus phage S13' DNA 100, 91 100 NC_018608.1
4 Staphylococcus aureus 08BA02176 100 100 NC_018608.1
5 none - 100 -
6 Anaerococcus prevotii DSM 20548 91 100 NC_013171.1
7 Staphylococcus aureus 08BA02176 100 100 NC_018608.1
8 Staphylococcus aureus 08BA02176 100 100 NC_018608.1
9 Staphylococcus aureus 08BA02176 100 100 NC_018608.1
10 Staphylococcus aureus 08BA02176 100 100 NC_018608.1
12
Staphylococcus aureus 08BA02176,
Staphylococcus phage JD007 100, 94 100 NC_018608.1, NC_019726.1
13 Staphylococcus aureus 08BA02176 100 100 NC_018608.1
15 Staphylococcus aureus 08BA02176 100 100 NC_018608.1
17 Staphylococcus aureus 08BA02176 100 100 NC_018608.1
18 none - 100 -
(a) Spacer regions are numbered according to Figure 2. (b) In the case of spacers with more than one Blastn hesultrit, the hits are ordered from the highest to the lowest % of DNA similarity of their sequences.

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 106
Figure IV.1.3. Schematic diagram and comparative analysis of the clustered regularly
inter-spaced short palindromic region (CRISPR) region in Staphylococcus capitis strain
CR01 SCCmec element with ST398 LA-MRSA strain 08BA02176. (a) Alignment of S. aureus strain 08BA02176 and S. capitis strain CR01 CRISPR regions
with direct repeats (DR) identified, respectively, by light and dark gray diamonds and spacer
sequences numbered sequentially according to their position in the alignment. The CRISPR
locus found in S. capitis strain CR01 with the CRISPR region (CRISPR1), the CRISPR-
associated (cas) genes – cas1, cas2, cmr1-6, cas6 – and the second CRISPR region
(CRISPR*) is also represented. The cas genes are represented by arrows, indicating the
direction of transcription.
(b) The 36 bp direct repeats found in CRISPR1, represented by light gray diamonds in (a), are
indicated in boldface and the corresponding variable spacer sequences, numbered according
to (a).
References:
1. Leekitcharoenphon P, Kaas RS, Thomsen MCF, Friis C, Rasmussen S, Aarestrup
FM. 2012. snpTree - a web-server to identify and construct SNP trees from whole genome
sequence data. Bmc Genomics 13:S6.
2. Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg
SL. 2004. Versatile and open software for comparing large genomes. Genome Biol.
5:R12.

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 107
3. Gouy M, Guindon S, Gascuel O. 2010. SeaView Version 4: A Multiplatform Graphical
User Interface for Sequence Alignment and Phylogenetic Tree Building. Mol Biol Evol
27:221–224.
4. Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W GO. 2010. New
Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the
Performance of PhyML 3.0 Syst.Biol. 59(3):307-21.
IV.2. Single-Molecule Sequencing (PacBio) of the Staphylococcus capitis NRCS-A Clone
Reveals the Basis of Multidrug Resistance and Adaptation to the Neonatal Intensive
Care Unit Environment
IV.2.1 Abstract
The multi-resistant Staphylococcus capitis clone NRCS-A has recently been described as a
major pathogen causing nosocomial, late-onset sepsis (LOS) in preterm neonates worldwide.
NRCS-A representatives exhibit an atypical antibiotic resistance profile. Here, the complete
closed genome (chromosomal and plasmid sequences) of NRCS-A prototype strain CR01 and
the draft genomes of three other clinical NRCS-A strains from Australia, Belgium and the
United Kingdom are annotated and compared to available non-NRCS-A S. capitis genomes.
Our goal was to delineate the uniqueness of the NRCS-A clone with respect to antibiotic
resistance, virulence factors and mobile genetic elements. We identified 6 antimicrobial
resistance genes, all carried by mobile genetic elements. Previously described virulence genes
present in the NRCS-A genomes are shared with the six non-NRCS-A S. capitis genomes.
Overall, 63 genes are specific to the NRCS-A lineage, including 28 genes located in the
methicillin-resistance cassette SCCmec. Among the 35 remaining genes, 25 are of unknown
function, and 9 correspond to an additional type I restriction modification system (n = 3), a
cytosine methylation operon (n = 2), and a cluster of genes related to the biosynthesis of
teichoic acids (n = 4). Interestingly, a tenth gene corresponds to a resistance determinant for
nisin (nsr gene), a bacteriocin secreted by potential NRCS-A strain niche competitors in the
gut microbiota. The genomic characteristics presented here emphasize the contribution of
mobile genetic elements to the emergence of multidrug resistance in the S. capitis NRCS-A
clone. No NRCS-A-specific known virulence determinant was detected, which does not
support a role for virulence as a driving force of NRCS-A emergence in NICUs worldwide.
However, the presence of a nisin resistance determinant on the NRCS-A chromosome, but not

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 108
in other S. capitis strains and most coagulase-negative representatives, might confer a
competitive advantage to NRCS-A strains during the early steps of gut colonization in
neonates. This suggests that the striking adaptation of NRCS-A to the NICU environment
might be related to its specific antimicrobial resistance and also to a possible enhanced ability
to challenge competing bacteria in its ecological niche..
IV.2.2 Introduction
Coagulase-negative staphylococci (CoNS) are common commensals of the human skin and
mucosa and are also opportunistic pathogens responsible for infections associated with
indwelling medical devices and bloodstream infections (Otto, 2009). Among
CoNSs, Staphylococcus epidermidis is the most prevalent species and is classically involved
in nosocomial bacteremia in patients with co-morbidities including very low-birth weight
preterm infants (Otto, 2009). However, the Staphylococcus capitis has also been incriminated
in sepsis throughout the world in neonatal intensive care units (NICUs) (Van Der Zwet et
al., 2002; Venkatesh et al., 2006; D'mello et al., 2008; Rasigade et al., 2012; Boghossian et
al., 2013; Cui et al., 2013). We recently reported that a group of closely related multidrug-
resistant (MDR) S. capitis strains belonging to the same clone, named NRCS-A, were present
in most French NICUs but absent in the other pediatric or adult ICUs (Rasigade et al., 2012).
In subsequent work, we demonstrated the dissemination of NRCS-A strains in distant NICUs
throughout the world (Butin et al., 2016). This dissemination is of major concern because of
the extensive drug resistance of NRCS-A as well as its ability to become highly endemic in
some NICUs, representing up to 40% of all bacteremia cases (Stoll et al., 2002, 2005;
Rasigade et al., 2012). Due to the presence of the type V-related staphylococcal chromosome
cassette mec (SCCmec), all S. capitis NRCS-A strains are resistant to beta-lactams and exhibit
reduced susceptibility to other antimicrobial agents in common use in NICUs, including
resistance to aminoglycosides and resistance or heteroresistance to vancomycin (Venkatesh et
al., 2006; Rasigade et al., 2012). Overall, the worldwide diffusion of a multidrug-resistant S.
capitis clone that is highly adapted to NICU antibiotic selective pressure raises questions
about the potential genetic support of its epidemiological success.
Few genome sequences of S. capitis are publicly available to date. In addition, only a single
study using the genome of a S. capitis isolate collected from a bloodstream infection in an
adult has reported the genomic comparison of virulence factors between S. capitis and S.
epidermidis (Cameron et al., 2015). Considering the specific importance NRCS-A in the

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 109
NICU setting and its international dissemination, we present here the first closed genome
sequence of the French NRCS-A prototype strain CR01, a comparison of the whole-genome
sequences (WGS) of three other NRCS-A strains collected from distant countries (Australia,
Belgium, the United Kingdom), and a description of the genomic features of this NRCS-A
clone.
IV.2.3 Methods
Genome sequencing and assembly
The four strains of S. capitis, CR01, CR03, CR04, and CR05, used in this study were part of a
previously published collection, with all strains being of the international S. capitis NRCS-A
clone (Butin et al., 2016). The strains were isolated from blood cultures of preterm infants
diagnosed with neonatal sepsis in Neonatal Intensive Care Units (NICUs) from four different
countries: France (isolate CR01), Belgium (isolate CR03), Australia (isolate CR04) and the
United Kingdom (isolate CR05) (for further details see Supplementary Data).
Initially, whole-genome pyrosequencing (454 Life Sciences/Roche) was performed for all 4
isolates, as previously reported (Lemriss et al., 2014, 2015). Single-molecule real-time
(SMRT) sequencing was then performed for strain CR01 to obtain a closed reference genome
for this clone (Supplementary Data).
Sequencing for de novo assembly was performed using PacBio RS II (Menlo Park, CA,
USA). High molecular weight DNA was sheared in a Covaris g-TUBE (Covaris, Woburn,
MA, USA) to obtain 20 kb fragments. After shearing, the DNA size distribution was checked
using a Fragment Analyzer (Advanced Analytical Technologies, Ames, IA, USA), and 5 μg
of the sheared DNA was used to prepare a SMRTbell library with PacBio SMRTbell
Template Prep Kit 1 (Pacific Biosciences, Menlo Park, CA, USA) according to the
manufacturer's recommendations. The resulting library was size selected using a BluePippin
system (Sage Science, Inc. Beverly, MA, USA) for molecules larger than 11 kb. The
recovered library was sequenced using 1 SMRT cell with P6-C4 chemistry and MagBeads
with a PacBio RSII system (Pacific Biosciences, Menlo Park, CA, USA) at 240 min movie
length.
Based on the SMRT cell sequencing, we generated 50,876 post-filter polymerase reads with
an average read length of 15.6 kb and an N50 read length of 21.1 kb. The average coverage
was 265X; for de novo assembly, the PacBio module “RS_HGAP_Assembly.2” in SMRTpipe

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 110
version v2.3.0 was used for continuous long reads (CLR) after polishing and error correction
with Quiver, as described previously (Chin et al., 2013). DNA methylation was determined
using the RS_Modification_and_Motif_Analysis protocol within SMRT Portal v2.30, with a
standardized in silico false positive error of ~1%. Only motifs with a mean modification
quality value (QV) >50 and a mean coverage of >100X were validated as being modified
(https://github.com/PacificBiosciences/Bioinformatics-Training/wiki/Methylome-Analysis-
Technical-Note). Two polished contigs, one corresponding to the chromosome and the other
to the plasmid, were obtained. Circularization of both contigs was achieved by manual
comparison and removal of regions of overlaps.
Computational analysis
Automatic syntactic and functional annotation of the closed, reference genome of strain CR01
[EMBL accession number: LN866849 (chromosome) and LN866850 (plasmid)] was
performed using the MicroScope platform pipeline (Vallenet et al., 2006, 2013). The
annotation of the draft genomes of strains CR03 (EMBL accession number: CVUF01000001-
CVUF01000031), CR04 (EMBL accession number: CTEM01000001 CTEM01000038) and
CR05 (EMBL accession number: CTEO01000001-CTEO01000039) were also performed
using the MicroScope platform pipeline, as published previously (Lemriss et al., 2014).
Visual inspection and manual curation were carried out using the MaGe platform and BLAST
searches against NCBI databases.
Virulence factors were identified using the Virulence Factors database
(VFDB; http://www.mgc.ac.cn/VFs/, Chen et al., 2012) and tblastn (using the ncbi-blast-
2.2.27+ suite, Camacho et al., 2009) searches against in-house databases of known virulence
genes of staphylococci.
Prophage regions were identified using the PHAST server (http://phast.wishartlab.com, Zhou
et al., 2011). The predicted results were confirmed using the MaGe platform.
For comparison, we obtained publicly available assemblies for six additional whole-genome
sequences of S. capitis: strains QN1 (NCBI Ref. Seq.: NZ_AJTH00000000.1), VCU116
(NCBI Ref. Seq.: NZ_AFTX00000000.1), SK14 (NCBI Ref. Seq.: NZ_ACFR00000000.1),
LNZR-1 (NCBI Ref. Seq.: NZ_JGYJ00000000.1), C87 (NCBI Ref. Seq.:
NZ_ACRH00000000.1), and AYP1020 (NCBI Ref. Seq.: NZ_CP007601.1). All six genomes
were aligned against the closed genome of isolate CR01 using Mauve Progressive v.2.3.1
(Darling et al., 2010) with default settings.

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 111
Resistance genes were identified by a combination of ResFinder v.2.1 (Kleinheinz et
al., 2014) and tblastn searches using the complete, curated Antibiotic Resistance Proteins
database (multifasta file) available at the Comprehensive Antibiotic Resistance Database
(CARD) (McArthur et al., 2013). For ResFinder, cutoffs of 80% minimum length and 90%
identity were used to search for resistance genes in CR03, CR04, and CR05 assemblies
previously performed against the reference CR01 using Mauve Progressive to determine their
location and genomic context. For detection of resistance genes using the CARD complete,
curated Antibiotic Resistance Proteins database, tblastn searches against a database composed
of the four genomes of strains CR01, CR03, CR04, and CR05 were performed.
Genes specific to the NRCS-A clone were identified using the MaGe platform “Gene
Phyloprofiler” tool (Vallenet et al., 2013) and the four annotated genomes of NRCS-A and
public S. capitis genomes. Briefly, genomes were compared in terms of gene content using
pre-computed homologies and synteny groups. Pairwise comparisons between predicted
protein sequences of the studied genome and the proteins of another genome allowed
computation of ranked hits and determination of bidirectional best hits (BBH) (for each
protein, the three best hits were retained). Putative orthologous relationships between two
genomes were defined as gene couples satisfying the BBH criterion and an alignment
threshold for homology set as a minimum of 50% sequence identity along 80% of the length
of the smallest protein. Mauve alignments were also used to complement the genomic context,
followed by manual curation of the putative genes exclusively found in only the four NRCS-
A genomes.
Insertion sequences (IS) in the genomes were identified using the ISfinder analysis tool
(Siguier et al., 2006). Both blastn and blastp queries were performed with default settings.
Matches with an e-value smaller than 0.5 were manually curated. To confirm whether a given
IS position was conserved in the other genomes analyzed, the closed genome of isolate CR01
was aligned with the three other NRCS-A genomes and with the six public genomes using
Mauve Progressive v.2.3.1 (Darling et al., 2010) with default settings. ORFs within
immediate proximity of conserved IS loci in the NRCS-A genomes were compared with the S.
capitis reference genome to check for potential alterations in coding sequences, using both
Mauve Progressive and Mage platform. genomic islands (GIs) were predicted using
IslandViewer 3 (Dhillon et al., 2015) and confirmed by alignment with the three NCRS-A
genomes and the six public S. capitis genomes using Mauve Progressive. IS insertions within
GIs were predicted as mentioned above. NRCS-A-specific GIs were searched in the NCBI

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 112
database using megablast (Camacho et al., 2009) query, limiting to the Staphylococcus taxon
(taxid: 1279). Published GIs from reference genomes were searched against Staphylococcus
aureus Mu50 (NCBI Reference Sequence: NC_002758.2), COL (NCBI Reference
Sequence: NC_002951.2), MW2 (NCBI Reference Sequence: NC_003923.1), N315 (NCBI
Reference Sequence: NC_002745.2), FPR3757 (NCBI Reference Sequence: NC_007793.1)
and S0385 (NCBI Reference Sequence: NC_017333.1), and S. epidermidis RP62A (NCBI
Reference Sequence: NC_002976.3) using the Mage platform [15]. Syntonomes of more than
3 ORFs and with identity greater than 33% were considered to be GIs.
Investigation of nsr gene presence in clinical bacterial strains
A panel of 15 strains (Table IV.2.1) comprising clinical strains of S. capitis (NRCS-A and
non-NRCS-A) were used for nisin susceptibility testing. All staphylococcal species were
originally isolated from blood, and species assignment was determined using Matrix-Assisted
Laser Desorption/Ionization Time of Flight Mass Spectrometry (MALDI-TOF Vitek MS,
Biomerieux, France). After isolation, the strains were cultured on Horse Blood Agar
(Biomerieux) and then stored at −80°. Testing for susceptibility to nisin was performed using
the disk diffusion test adapted from EUCAST1. Briefly, nisin solutions were prepared using
2.5% nisin powder (Sigma Aldrich, USA) according to a previously described protocol (Piper
et al., 2009). Sterile paper disks (Thermofisher Diagnostics, France) were soaked with 30 μg
of nisin solution. After an overnight incubation at 36°C on Horse blood Agar (Biomerieux,
France), a 0.5 McF was seeded on Mueller Hinton agar (MHE) plates (Biomerieux, France).
A disk containing 30 μg of nisin was placed in the middle of the plate. The diameter of the
inhibition zones was measured after overnight incubation of the inoculated MHE plates at
36°C. The average diameter of inhibition and SEM were determined from four independent
experiments.
PCRs for detecting the nsr gene were performed using consensus primers: pF–word-5′
GGAGATATGGGACCTATGATTGCA 3’ and pR-word-
5’GCTGTAAkTTCwCCkGAACTkGC 3′. These primers were designed based on alignment
of the nsr gene sequence found in strain CR01 (S. capitis clone NRCS-A) and sequences
retrieved from NCBI after a blastn (ref) search for homologous sequences in staphylococcal
WGS genomes [S. hyicus ATCC 11249 (acc. num. CP008747), Staphylococcus sp. TE8 (acc.
num. NZ_JMGB00000000.1), S. epidermidis MC28 (acc. num. NZ_ATCZ00000000.2), S.
epidermidis VCU128 (acc. num. NZ_AHLI00000000.1)]. PCR amplification was conducted

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 113
in a final volume of 25 μL, with 2.0 μL bacterial DNA (QuickExtract kit, Qiagen), 2.5 μL 10x
Reaction Buffer, 0.75 μL 50 mM MgCl2, 1 μL 10 mM forward primer, 1 μL 100 mM reverse
primer, 4 μL 5 mM dNTPs, 1 μL each primer and 0.125 μL Taq polymerase (Eurobio,
France). The cycling parameters were as follows: (i) initial denaturation for 3 min at 95°C; (ii)
34 cycles, with 1 cycle consisting of 30 s at 95°C, 60 s at 60°C, and 90 s at 72°C; and (iii) a
final extension for 10 min at 72°C.
Table IV.2.1. Bacterial strains used in the nisin susceptibility test.
Species NRCS-A clone Strain Setting Country nsr gene
SC yes CR01 NICU FR +
SC yes CR03 NICU BE +
SC yes CR04 NICU AUS +
SC yes CR05 NICU UK +
SC yes CR07 NICU FR +
SC yes BC69 NICU South K +
SC yes AQ62 NICU NO +
SC yes AW77 NICU NZ +
SC yes BI76 NICU USA +
SC no AB51 ICU AUS −
SC no CR02 ICU FR −
SC no AR22 ICU DK −
SC no AY18 ICU GR −
SC no AZ72 ICU Sing −
SC no BA24 ICU USA − SC, Staphylococcus capitis; FR, France; BE, Belgium; UK, United Kingdom; AUS, Australia; South
K, South Korea; NO, Norway; NZ, New Zealand; USA, United States of America; DK, Denmark; GR,
Greece; Sing, Singapore; NICU, neonatal intensive care unit; ICU, intensive care unit (adults).
IV.2.4 Results
WGSs of four strains belonging to S. capitis clone NRCS-A and originating from NICUs in
four distant countries were obtained to investigate lineage-specific virulomes, resistomes, and
mobilomes as well as the presence of unshared genes using 454 pyrosequencing technology,
as reported previously (Lemriss et al., 2014, 2015). Depending on the isolate, assembly of the
draft genomes produced 26 to 39 contigs. Re-sequencing of strain CR01 using SMRT
technology (PacBio) allowed for the generation of closed complete sequences for the
chromosome and the single plasmid that was subsequently used to produce scaffolds for the
three remaining WGSs.
The length of CR01's chromosome is 2,522,871 bp and exhibits a low G + C content
(33.02%), as is expected for staphylococci. We identified and annotated 2419 protein-coding

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 114
regions, 19 rRNAs, 62 tRNAs, 34 ncRNAs (including RNAIII), and 20 transposases
associated with insertion sequence elements and transposons. Of note, gaps (identified in the
draft genome assemblies of the four NRCS-A isolates (based on 454 pyrosequencing))
frequently occur within the vicinity of transposases, suggesting that some of the 454 short
read assemblies were unable to bridge the gaps associated with repetitive genomic elements.
These data clearly emphasize the suitability of third-generation sequencing technologies, such
as PacBio SMRT, for obtaining fully defined de novo assemblies and, thus, for overcoming
the issue of both local and global repeats (Koren and Phillippy, 2015). The genomic
characteristics of the NRCS-A isolates tested in this study are summarized in Table IV. 2.2.
Tables IV.2.2. General genomic features of S. capitis strain CR01 compared with another
three WGS NRCS-A genomes.
Features S.capitis strain
CR01(Fr)
S.capitis strain CR03
(Be)
S.capitis strain
CR04 (Aus)
S.capitis strain CR05
(UK)
GC% Content 33.02 32.81 32.80 32.84
Nb of phages 1 1 1 3
Nb of plasmids 1 1a 1a 0
Nb and type of Insertion
sequences
(nb on chromosome+ nb on
plasmid)
IS256: 10+0
IS1272: 3+1
IS256: 1+0b
IS1272: 1+0
IS256: 1+0b
IS1272: 1+0
IS256: 1b
IS1272: 0
IS431mec-like: 2+3 IS431mec-like: 2+4 IS431mec-like: 2+0 IS431mec-like: 2 aPutative plasmid, no definitive data about circularization. bInsertion sequence isolated in a small contig, indicating probable multiple insertions in the complete genome.
Virulome
The presence of virulence-associated genes in the closed genome of strain CR01 and
subsequently the draft genomes of strains CR03 (Be), CR04 (Aus), and CR05 (UK) were
inferred by comparison with known virulence factors previously reported for S. aureus, S.
epidermidis, and S. haemolyticus using the VFDB database (Chen et al., 2012). The findings
were augmented with BLAST searches of well-characterized staphylococcal virulence factors
and key regulators (Table IV.2.3). Comparison of the four NRCS-A genomes with the six
published genomes of non-NRCS-A S. capitis, including QN1, VCU116, SK14, LNZR-1,
C87, and AYP1020 strains, showed that none of the known staphylococcal virulence genes
are exclusively carried by the NRCS-A clone.
Interestingly, the virulome of the NRCS-A S. capitis genome was quite similar to that of S.
epidermidis RP62A, including the icaABDCR and capABC biofilm-related operons, Clp
proteases and multiple copies of PSM beta type 1b in tandem (75% aa identity to PSMbeta1b
of strain RP62) and one PSM alpha (59% aa identity) (Otto, 2012). All PSMs were found in

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 115
the same genetic environment as in the S. epidermidis RP62A genome, with conservation of
upstream and downstream genes.
As previously observed for the fully closed S. epidermidis RP62a and S. epidermidis ATCC
12228 genomes (Zhang et al., 2003; Gill et al., 2005), no S. aureus toxins, except beta- and
delta-hemolysins, were identified in any of the 10 S. capitis genomes (four NRCS-A and six
public non-NRCS-A). Similarly, none of the genes associated with secretion systems
(esxA, esxB, esaA, esaB, esaC, essA, essB, and essC) or S. aureus serine proteases (splA, B,
C, D, E, and F) are present in the S. capitis genomes.
Table IV.2.3. Comparison of virulence factors in S. capitis NRCS-A, non-NRCS-A, and S.
epidermidis after exclusion of virulence factors present only in S. aureus.
S. epidermidis S.capitis NRCS-A clone S.capitis non NRCS-A
Function Gene ATCC
12228 RP62A
CR01
(fr) CR03 CR04 CR05 SK14
VCU11
6 C87 QN1 LNZR-1
Adherence atl + + + + + + + + + + +
ebh + + + + + + + + + + +
ebp + + + + + + + + + + +
icaR 0 + + + + + + + + + +
icaA 0 + + + + + + + + + +
icaD 0 + + + + + + + + +
icaB 0 + + + + + + + + + +
icaC 0 + + + + + + + + + +
sdrF + + 0 0 0 0 0 0 0 0 0
sdrG + + 0 0 0 0 0 0 0 0 0
sdrH + + + + + + + + + +
Exozymes sspB + + 0 0 0 0 0 0 0 0 0
sspC 0 + 0 0 0 0 0 0 0 0 0
lip + + + + + + + + + + +
geh + + + + + + + + + + +
sspA + + 0 0 0 0 0 0 0 0 0
nuc + + + + + + + + + + +
Host immune
Invasion capABC + + + + + + + + + + +

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 116
Toxins hlb + + + + + + + + + + +
hld + + + + + + + + + +
hemolysin 0 + 0 0 0 0 + + 0 + 0
hlIII + + + + + + + + + + +
Proteases ClpP + + + + + + + + + + +
ClpB + + + + + + + + + + +
ClpC + + + + + + + + + + +
ClpX + + + + + + + + + + +
Phenol Soluble
Modulins PSM α + + + + + + + + + + +
PSM β1a + + 0 0 0 0 0 0 0 0 0
PSM β1B + + + + + + + + + + +
PSM β2 + + 0 0 0 0 0 0 0 0 0
PSM β3 + + 0 0 0 0 0 0 0 0 0
PSM δ + + + + + + + + + + +
PSM ε + + 0 0 0 0 0 0 0 0 0
PSM-mec + + 0 0 0 0 0 0 0 0 0
Esterases esterase1 + + + + + + + + + + +
esterase2 + + + + + + + + + + +
HTH
transcription
factors
associated with
virulence
SarA + + + + + + + + + + +
SarR + + + + + + + + + + +
SarV + + + + + + + + + + +
SarX + + + + + + + + + + +
SarZ + + + + + + + + + + +
MgrA + + + + + + + + + + +
Rot + + + + + + + + + + +
The presence of virulence genes is indicated by the “+” sign
Resistome

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 117
Several antibiotic resistance-associated genes were identified and correlated with the specific
resistance phenotype previously reported for the NRCS-A clone (Rasigade et al., 2012;
Table IV.2.4). All are located on mobile genetic elements (MGEs). Resistance to
aminoglycosides is related to the bifunctional aminoglycoside-modifying gene aacA-aphD,
which is carried on the transposon Tn4001 (GenBank accession no. AB682805.1; Lyon et
al., 1984). Resistance to methicillin is due to the presence of a composite SCCmec-
SCCcad/ars/cop cassette exclusively present in the NRCS-A clone, as recently published by
our team (Martins Simões et al., 2013). Genomic comparison between the four NRCS-A
strains showed that this mobile element is nearly identical: all ORFs are conserved (100% aa
homology), and the few nucleotide sequence variations occur only in the CRISPR repeat
region, as is expected for these regions. As previously reported (Martins Simões et al., 2013),
no other antibiotic resistance genes were detected for this composite SCC element. Finally,
the plasmidic bla operon, coding for blaZ beta-lactamase and its regulators, is present in all
NRCS-A strains, except strain CR05 (UK).
With regard to other antimicrobial families, for which variability in resistance profiles have
been observed among NRCS-A isolates, phenotypic antimicrobial susceptibility testing
matched the specific resistance gene contents of each NRCS-A strain. Plasmidic msrA
and tetK genes, respectively involved in erythromycin resistance (with a negative D-test) and
tetracycline resistance, were only identified in strain CR04 (Australia). In strain CR05 (UK),
the gene far1, which is responsible for fusidic acid resistance, is carried by a putative phage.
Tables IV.2.4. Genomic profiles of antibiotic resistance for clone NRCS-A strains from four
different countries.
Antibiotic gene Strains Genomic context
CR01
(Fr)
CR03
(Be)
CR04
(Aus)
CR05
(UK)
Penicillin BlaZ + + + − Plasmid
Methicillin mecA + + + + Chromosome
Tetracycline tetK − − + − Plasmid*
Aminoglycosides aac(6′ )-aph(2′′) + + + + Chromosome
Macrolide, Lincosamide and Streptogramin B msr(A) − − + − Plasmid*
Fusidic acid far/fusB − − − + Phage (chromosome)*
All antibiotic resistance genes were found in MGEs. Putative regions in the draft genomes are indicated by an *
Mobile elements
As mentioned previously, the shortest circular sequence obtained by SMRT for strain CR01
corresponds to a plasmid of 26.140 bp (GC content of 29.25%). It harbors 35 ORFs, including
(i) the bla operon coding for penicillinase resistance to penicillins (see above) and (ii) copper
resistance-related genes such as copZ, copA, and csoR (copper transcriptional repressor;

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 118
Figure .1). Genome comparison of strain CR01's plasmid with the three other NRCS-A draft
genomes revealed that the plasmid is not conserved in all NRCS-A strains (Figure IV.2.1). In
strain CR03 (Belgium), one contig presents 31 of the 35 ORFs identified on the CR01
plasmid. Conversely, in strain CR04 (Australia), only 9 of the 35 ORFs (corresponding to
the blaZRI operon and copper resistance operon) were detected in a contig corresponding to a
putative plasmid. Finally, no putative plasmid was detected in strain CR05 (UK).
Figure IV.2.1. Comparison of the genetic content of the strain CR01 plasmid with the
draft genomes of the other three NRCS-A strains. Open reading frames (ORFs) are shown
as arrows indicating the direction of transcription. Homologous gene clusters are indicated by
a light gray shadow connecting ORFs present in distinct plasmid sequences. Light gray
arrows represent unknown proteins, unless specified otherwise. Arrows with dotted lines
represent partial or truncated ORFs. Antibiotic resistance genes are colored with a gray
gradient; genes associated with resistance to heavy metals are colored in dark gray.
Abbreviations: tnp, transposase; copZ, copper insertion chaperone and transporter
component; copA, copper transporter ATPase; csoR, copper-sensing transcriptional
repressor; czcD, potassium/proton-divalent cation antiporter; rep, replication-associated
family protein; repA, replication-associated protein RepA; blaZ, beta-lactamase resistance
gene; blaR, regulatory protein BlaR1; blaI, penicillinase repressor; mntH, divalent metal
cation transporter MntH, tetK, tetracycline resistance gene.

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 119
One intact prophage region was identified in all four NRCS-A genomes. This region shows
100% aa identity to the genomes of strains CR01, CR03, and CR04, but it is degenerate in
CR05 (Figure (Figure2).2). Moreover, this phage exhibits strong homology to a phage present
in strain VCU116 [57.1, 90.2, 76.2% aa homology for regions 1, 2 and 3, respectively
(Figure IV.2.2) and less than 30% homology with phage Staphy_StB20_(NCBI Ref.
Seq. NC_019915), the closest phage found in the NCBI database. Of note, two additional
incomplete prophage regions were also detected in strain CR05.
Figure IV.2.2. Comparison of the genetic content of the strain CR01 intact prophage
present in the other three draft genomes of NRCS-A strains. Prophage prediction was
performed using PHAST software; cR01's prophage is divided into three regions according to
the degree of conservation compared to the other strains. The mean percent of aa identity in
each region is indicated, and a gray gradient is also used to represent the percent similarity.
Nineteen distinct insertion sequence (IS) elements, clustering into three distinct families (type
IS256, n = 4; type IS1272, n = 5; IS431mec, n = 10), were identified in the CR01 genome.
The presence of type IS256 was confirmed in all four NRCS-A strains, with two IS256
elements being associated with the aminoglycosides resistance gene aacA-aphD (′aac(6′)-

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 120
aph(2″) on transposon Tn4001. Type IS1272 was also found in strains CR03 and CR04 but
not in strain CR05, and their genomic locations are not conserved. Two IS431mec cassettes
are present in all NRCS-A genomes, as they are carried within the SCCmec-
SCCcad/ars/cop element that is specific to and conserved among the NRCS-A lineage
(Martins Simões et al., 2013).
Finally, other GIs (n = 5) were predicted for CR01 (Table IV. 2.2) and found in the CR03,
CR04, and CR05 strains. However, only one was found exclusively in the NRCS-A lineage.
This GI (bp position: 150,780–158,140 in the strain CR01 genome) contains six ORFs: one
putative phosphoglycolate phosphatase, one conserved protein of unknown function and three
pseudogenes of phage proteins (bp position: 151,470–152,238). Unexpectedly, two of these
pseudogenes present 40 and 46.5% aa identity to Listeria monocytogenes phage B025 protein
gp27 and the third pseudogene presents 60.6% aa identity to L. monocytogenes phage B025
protein gp28. Both gp27 and gp28 proteins have unknown functions.
Methylome and restriction modification systems
SMRT technology (PacBio) allows for genome-wide detection of modified nucleotides based
on the rate at which DNA polymerase incorporates bases during sequencing (Flusberg et
al., 2010, Nat. Methods; Roberts et al., 2013). Analysis of polymerase kinetic profiles in strain
CR01 identified 1333 methylated positions (Table IV.2.S2) including 94% (n = 1253)
corresponding to adenine methylations (m6A) and 0.006% (n = 8) to cytosine methylations
(m4C). The adenine modifications observed correlate with the presence of two adenine
restriction-modification systems (hsdMSR 1 pos: 476,473–482,360 bp; hsdMSR 2 pos:
686,750–691,866 bp) in the strain CR01 genome, whereas the presence of an mcrBC 5-
methylcytosine restriction system (bp position 487,030–492,276) correlates with the cytosine
modifications detected. Interestingly, the latter is associated in tandem with the specific
additional hdsMSR 1 operon located immediately after the SCCmec-SCCcad/ars/cop element
(see above).
NRCS-A clone specific genes
Comparison of the closed genome of strain CR01 with the draft genomes of the three other
NRCS-A genomes and with the six public non-NRCS-A genomes revealed a unique set of 63
genes present exclusively in the NRCS-A clone genome (Table IV.2.S1). Of these, 28 ORFS
are carried by the composite SCCmec-SCCcad/ars/cop mobile element, which is related to the
acquisition of methicillin resistance (mecA gene). Interestingly, within this cassette, the

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 121
CRISPR element is specific to the NRCS-A lineage (Martins Simões et al., 2013). Of note,
both the clone-specific cassette (28 genes) and its genetic environment (downstream and
upstream regions) are fully conserved in the four NRCS-A strains, which confirmed that the
four isolates belong to the same clonal population, even though they were isolated from
distant countries/continents. Indeed, it is highly unlikely that the same SCCmec element
(100% identity) could be acquired in four independent events in four distant countries.
Among the 35 remaining genes found exclusively in the NRCS-A lineage, 25 are of unknown
function, and 10 correspond to the following: (i) an additional type I restriction modification
system (hsdMSR, n = 3 genes), (ii) a cytosine methylation operon (mcrBC, n = 2), (iii) a
cluster of four genes known to be involved in the biosynthesis of teichoic acids [ispD (2-C-
methyl-D-erythritol 4-phosphate cytidylyltransferase), tarJ (ribitol-5-phosphate
dehydrogenase), tarK (Glycosyl glycerophosphate), and tagF (CDP-glycerol
glycerophosphotransferase), n = 4]. Of note, the tenth gene encodes a 316 aa protein
presenting 41% aa homology with the determinant for resistance to nisin (nsr gene) that is
present in some Lactococcus lactis strains (Froseth and McKay, 1991; Liu et al., 2005) and
which shows protease activity. Contrary to the plasmidic localization of the nsr gene in nisin-
resistant L. lactis strains, the nsr gene in the NRCS-A clone is located on the chromosome
(position: 521,747–522,694 bp) immediately upstream from the potassium (K+)
transporter kdpEDABC operon conserved in all S. capitis isolates and downstream from the
four teichoic acid biosynthesis genes (ispD, tarJ, tarK, and tagF; see above). No IS or
transposon-like region was identified near the nsr gene.
Phenotypic assay of clone NRCS-A nisin resistance
To assess functional expression of the nisin resistance gene (nsr) found exclusively in the
NRCS-A clone, we performed a disk diffusion test with nisin-charged disks using a collection
of S. capitis strains, including nine strains belonging to the NRCS-A clone and six nsr-
negative, non-NRCS-A strains. The S. capitis strains belonging to the NRCS-A clone
presented significantly lower nisin inhibition zones (mean of four independent measurements
± SEM: 8.81 mm ± 1.31) than the strains isolated from adults that do not carry the nsr gene
(16.5 ± 1.5; the Student t-test, p < 0.001 vs. 8.8 mm ± 1.3 in NRCS-A). This confirms that
the nsr gene found exclusively in S. capitis strains belonging to the neonatal NRCS-A clone is
functional and confers resistance to nisin. Nonetheless, it is known (i) that S. aureus (SA) can
modulate its resistance to nisin and other small antibacterial peptides via two-component

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 122
systems (TCSs) GraSR, NsaSR, BraSR in association with ABC transporters (Howden et
al., 2010; Blake et al., 2011; Falord et al., 2011; Kolar et al., 2011; Kawada-Matsuo et
al., 2013) and (ii) that SA strains resistant or heteroresistant to vancomycin (VISA and
hVISA) usually present increased cell wall thickening (Cui et al., 2003). The same
mechanisms have been observed in L. lactis resistant to nisin (Kramer et al., 2006; Shin et
al., 2016). Thus, to assess whether the resistance to nisin observed in the NRCS-A strains is
due exclusively to expression of the nsr gene and not to a cell wall-thickening adaptation,
complementary experiments are required using nsr+/nsr− isogenic isolates. Such analyses are
beyond the scope of the present paper.
IV.2.5 Discussion
The NRCS-A clone is of major interest due to its worldwide high dissemination in NICUs and
its prevalence as an agent of neonatal sepsis in preterm newborns (Butin et al., 2016).
Comparison between the complete genome of the prototype strain belonging to the S.
capitis NRCS-A clone (strain CR01, France) with the draft genomes of clinical S.
capitis isolates belonging or not to the NRCS-A clone revealed the presence of multiple
MGEs mediating the atypical antibiotic resistance profile of this clone. These data confirm (i)
the ability of the NRCS-A clone to adapt to the specific selective pressure of the antibiotics
used in NICUs and (ii) emphasize the ability of WGS to provide accurate predictions of the
resistance phenotypes of staphylococci and to become a promising alternative for culture
methods.
Based on genomic comparisons using previously characterized staphylococcal virulence
genes, the NRCS-A virulome is highly similar to that of other non-NRCS-A S. capitis as well
as S. epidermidis RP62A strains, which suggests that the success of this clone in neonates is
likely not due to increased virulence. Nonetheless, we identified the exclusive presence of a
gene encoding a functional nisin resistance (nsr) gene in the NRCS-A lineage that is seldom
found in staphylococci.
Nisin a 34 aa antimicrobial peptide produced by a group of gram positive
Lactococcus and Streptococcus species (Shin et al., 2016), is active against a wide range of
gram-positive bacteria, including staphylococci. Its mode of action is dual, involving (i)
inhibition of cell wall biosynthesis (transglycosylation step), as it binds to and sequesters lipid
II from its functional location, and (ii) pore formation (Wiedemann et al., 2001; Peschel and
Sahl, 2006; Egan et al., 2016). Nisin has been described as a key player of the gut barrier, and

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 123
it is widely used as food preservative due to its potent bactericidal activity. Moreover, it has
been shown that (i) expression of the nsr gene, identified in Streptococcus agalactiae, in L.
lactis not producing nisin induces resistance to nisin and (ii) the presence of human nisin-
producing lactic acid bacteria in the gut reduces intestinal colonization by vancomycin-
resistant enterococci (Millette et al., 2008).
Taken together, these data and our results strongly suggest that nisin resistance might be
responsible for the potential increase in the ability of the NRCS-A clone to establish itself as
part of the initial microflora of neonates, in which the Lactococcus genus is one of the
dominant bacterial taxa (Park et al., 2005; Morelli, 2008). Expression of the NSR peptidase
might enable NRCS-A isolates to establish themselves in the gut of neonates and also to
colonize it and survive longer than nisin-susceptible bacteria. This may explain the over-
representation of these isolates in sepsis processes, either through direct translocation in blood
(Taft et al., 2015) or via colonization of indwelling devices. Although it remains unknown
how S. capitis NRCS-A strains are able to translocate into the bloodstream of infants, it is
hypothesized that the entry point might be the digestive tract, which is immature in very
preterm infants (Taft et al., 2015). Further studies are needed to fully characterize the
digestive microflora of very low-weight preterm-infants and its potential impact on the
selection of and the fitness advantage to NRCS-A isolates.
IV.2.6 Author contributions
PM: work, study design, data analysis, and manuscript preparation. HL: data analysis and
manuscript preparation. YD: data analysis, work, and manuscript preparation. SL: work and
manuscript preparation. JR, SA, AI, and SE: manuscript preparation. MB and FL: study
design and manuscript preparation.
IV.2.7 Funding
The present work was financed by the French Ministry of Health and the French Institute for
Public Health Surveillance (INVS) - Santé publique France in the framework of the National
Reference Center of Staphylococci and by the grant ING20111223510 from the Fondation
pour la Recherche Medical (FRM).
IV.2.8 Acknowledgments
We would like to thank Michele Bes and Helene Meugnier at the National Reference Center
for Staphylococci in Lyon (France), and our colleagues at the National Reference Center of

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 124
Staphylococci in Belgium (Olivier Denis), in the United Kingdom (Angela Kearns) and in
Australia (Margaret Deighton).
IV.2.9 References
1. Blake K. L., Randall C. P., O'Neill A. J. (2011). In vitro studies indicate a high
resistance potential for the lantibiotic nisin in Staphylococcus aureus and define a
genetic basis for nisin resistance. Antimicrob. Agents Chemother. 55, 2362–2368.
10.1128/AAC.01077-10.
2. Boghossian N. S., Page G. P., Bell E. F., Stoll B. J., Murray J. C., Cotten C. M., et al. .
(2013). Late-onset sepsis in very low birth weight infants from singleton and multiple-
gestation births. J. Pediatr.162, 1120–1124, 1124.e1. 10.1016/j.jpeds.2012.11.089.
3. Butin M., Rasigade J.-P., Simões P. M., Meugnier H., Lemriss H., Goering R. V., et
al. . (2016). Wide geographical dissemination of multiresistant Staphylococcus
capitis NRCS-A clone in neonatal intensive-care units. Clin. Microbiol. Infect. 22, 46–
52. 10.1016/j.cmi.2015.09.008.
4. Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., et al. .
(2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421.
10.1186/1471-2105-10-421.
5. Cameron D. R., Jiang J.-H., Hassan K. A., Elbourne L. D., Tuck K. L., Paulsen I. T.,
et al. . (2015). Insights on virulence from the complete genome of Staphylococcus
capitis. Front. Microbiol. 6:980. 10.3389/fmicb.2015.00980.
6. Chen L., Xiong Z., Sun L., Yang J., Jin Q. (2012). VFDB 2012 update: toward the
genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids
Res. 40, D641–D645. 10.1093/nar/gkr989.
7. Chin C.-S., Alexander D. H., Marks P., Klammer A. A., Drake J., Heiner C., et al. .
(2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT
sequencing data. Nat. Methods 10, 563–569. 10.1038/nmeth.2474.
8. Cui B., Smooker P. M., Rouch D. A., Daley A. J., Deighton M. A. (2013). Differences
between two clinical Staphylococcus capitis subspecies as revealed by biofilm,
antibiotic resistance, and pulsed-field gel electrophoresis profiling. J. Clin.
Microbiol. 51, 9–14. 10.1128/JCM.05124-11.

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 125
9. Cui L., Ma X., Sato K., Okuma K., Tenover F. C., Mamizuka E. M., et al. .
(2003). Cell wall thickening is a common feature of vancomycin resistance in
Staphylococcus aureus. J. Clin. Microbiol.41, 5–14. 10.1128/JCM.41.1.5-14.2003.
10. D'mello D., Daley A. J., Rahman M. S., Qu Y., Garland S., Pearce C., et al. .
(2008). Vancomycin heteroresistance in bloodstream isolates of Staphylococcus
capitis. J. Clin. Microbiol. 46, 3124–3126. 10.1128/JCM.00592-08.
11. Darling A. E., Mau B., Perna N. T. (2010). progressiveMauve: multiple genome
alignment with gene gain, loss and rearrangement. PLoS ONE 5:e11147.
10.1371/journal.pone.0011147.
12. Dhillon B. K., Laird M. R., Shay J. A., Winsor G. L., Lo R., Nizam F., et al. .
(2015). IslandViewer 3: more flexible, interactive genomic island discovery,
visualization and analysis. Nucleic Acids Res. 43, W104–W108. 10.1093/nar/gkv401.
13. Egan K., Field D., Rea M. C., Ross R. P., Hill C., Cotter P. D. (2016). Bacteriocins:
novel solutions to age old spore-related problems? Front. Microbiol. 7:e461.
10.3389/fmicb.2016.00461.
14. Falord M., Mäder U., Hiron A., Débarbouillé M., Msadek T. (2011). Investigation of
the Staphylococcus aureus GraSR regulon reveals novel links to virulence, stress
response and cell wall signal transduction pathways. PLoS ONE 6:e21323.
10.1371/journal.pone.0021323.
15. Flusberg B. A., Webster D. R., Lee J. H., Travers K. J., Olivares E. C., Clark T. A., et
al. . (2010). Direct detection of DNA methylation during single-molecule, real-time
sequencing. Nat. Methods 7, 461–465. 10.1038/nmeth.1459.
16. Froseth B. R., McKay L. L. (1991). Molecular characterization of the nisin resistance
region of Lactococcus lactis subsp. lactis biovar diacetylactis DRC3. Appl. Environ.
Microbiol. 57, 804–811.
17. Gill S. R., Fouts D. E., Archer G. L., Mongodin E. F., Deboy R. T., Ravel J., et al. .
(2005). Insights on evolution of virulence and resistance from the complete genome
analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-
producing methicillin-resistant Staphylococcus epidermidis strain. J. Bacteriol. 187,
2426–2438. 10.1128/JB.187.7.2426-2438.2005.

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 126
18. Howden B. P., Davies J. K., Johnson P. D., Stinear T. P., Grayson M. L.
(2010). Reduced Vancomycin Susceptibility in Staphylococcus aureus, including
Vancomycin-Intermediate and Heterogeneous Vancomycin-Intermediate Strains:
resistance mechanisms, laboratory detection, and clinical implications. Clin.
Microbiol. Rev. 23, 99–139. 10.1128/CMR.00042-09.
19. Kawada-Matsuo M., Yoshida Y., Zendo T., Nagao J., Oogai Y., Nakamura Y., et al. .
(2013). Three distinct two-component systems are involved in resistance to the class I
bacteriocins, Nukacin ISK-1 and nisin A, in Staphylococcus aureus. PLoS
ONE 8:e69455. 10.1371/journal.pone.0069455.
20. Kleinheinz K. A., Joensen K. G., Larsen M. V. (2014). Applying the ResFinder and
VirulenceFinder web-services for easy identification of acquired antibiotic resistance
and E. coli virulence genes in bacteriophage and prophage nucleotide
sequences. Bacteriophage 4:e27943. 10.4161/bact.27943.
21. Kolar S. L., Nagarajan V., Oszmiana A., Rivera F. E., Miller H. K., Davenport J. E., et
al. . (2011). NsaRS is a cell-envelope-stress-sensing two-component system
of Staphylococcus aureus. Microbiol. Read. Engl. 157, 2206–2219.
10.1099/mic.0.049692-0.
22. Koren S., Phillippy A. M. (2015). One chromosome, one contig: complete microbial
genomes from long-read sequencing and assembly. Curr. Opin. Microbiol. 23, 110–
120. 10.1016/j.mib.2014.11.014.
23. Kramer N. E., van Hijum S. A., Knol J., Kok J., Kuipers O. P. (2006). Transcriptome
analysis reveals mechanisms by which Lactococcus lactis acquires nisin
resistance. Antimicrob. Agents Chemother.50, 1753–1761. 10.1128/AAC.50.5.1753-
1761.2006.
24. Lemriss H., Lemriss S., Martins-Simoes P., Butin M., Lahlou L., Rasigade J.-P., et al.
. (2015). Genome sequences of four Staphylococcus capitis NRCS-A isolates from
geographically distant neonatal intensive care units. Genome Announc. 3, e00501–
e00515. 10.1128/genomeA.00501-15.
25. Lemriss H., Martins Simões P., Lemriss S., Butin M., Ibrahimi A., El Kabbaj S., et al.
. (2014). Non-contiguous finished genome sequence of Staphylococcus capitis CR01
(pulsetype NRCS-A). Stand. Genomic Sci. 9, 1118–1127. 10.4056/sigs.5491045.

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 127
26. Liu C.-Q., Su P., Khunajakr N., Deng Y.-M., Sumual S., Kim W. S., et al. .
(2005). Development of food-grade cloning and expression vectors for Lactococcus
lactis. J. Appl. Microbiol. 98, 127–135. 10.1111/j.1365-2672.2004.02441.x.
27. Lyon B. R., May J. W., Skurray R. A. (1984). Tn4001: a gentamicin and kanamycin
resistance transposon in Staphylococcus aureus. Mol. Gen. Genet. 193, 554–556.
28. Martins Simões P., Rasigade J.-P., Lemriss H., Butin M., Ginevra C., Lemriss S., et al.
. (2013). Characterization of a novel composite staphylococcal cassette
chromosome mec (SCCmec-SCCcad/ars/cop) in the neonatal sepsis-
associated Staphylococcus capitis pulsotype NRCS-A. Antimicrob. Agents
Chemother. 57, 6354–6357. 10.1128/AAC.01576-13.
29. McArthur A. G., Waglechner N., Nizam F., Yan A., Azad M. A., Baylay A. J., et al. .
(2013). The comprehensive antibiotic resistance database. Antimicrob. Agents
Chemother. 57, 3348–3357. 10.1128/AAC.00419-13.
30. Millette M., Cornut G., Dupont C., Shareck F., Archambault D., Lacroix M.
(2008). Capacity of human nisin- and pediocin-producing lactic Acid bacteria to
reduce intestinal colonization by vancomycin-resistant enterococci. Appl. Environ.
Microbiol. 74, 1997–2003. 10.1128/AEM.02150-07.
31. Morelli L. (2008). Postnatal development of intestinal microflora as influenced by
infant nutrition. J. Nutr. 138, 1791S–1795S. Available online
at: http://jn.nutrition.org/content/138/9/1791S.full#fn-1.
32. Otto M. (2009). Staphylococcus epidermidis: the “accidental” pathogen. Nat. Rev.
Micro. 7, 555–567. 10.38/nrmicro2182.
33. Otto M. (2012). Molecular basis of Staphylococcus epidermidis infections. Semin.
Immunopathol. 34, 201–214. 10.1007/s00281-011-0296-2.
34. Park H.-K., Shim S.-S., Kim S.-Y., Park J.-H., Park S.-E., Kim H.-J., et al. .
(2005). Molecular analysis of colonized bacteria in a human newborn infant gut. J.
Microbiol. 43, 345–353.
35. Peschel A., Sahl H.-G. (2006). The co-evolution of host cationic antimicrobial
peptides and microbial resistance. Nat. Rev. Microbiol. 4, 529–536.
10.1038/nrmicro1441.

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 128
36. Piper C., Draper L. A., Cotter P. D., Ross R. P., Hill C. (2009). A comparison of the
activities of lacticin 3147 and nisin against drug-resistant Staphylococcus aureus and
Enterococcus species. J. Antimicrob. Chemother. 64, 546–551. 10.1093/jac/dkp221.
37. Rasigade J.-P., Raulin O., Picaud J.-C., Tellini C., Bes M., Grando J., et al. .
(2012). Methicillin-resistant Staphylococcus capitis with reduced Vancomycin
susceptibility causes late-onset sepsis in intensive care neonates. PLoS ONE 7:e31548.
10.1371/journal.pone.0031548.
38. Roberts R. J., Carneiro M. O., Schatz M. C. (2013). The advantages of SMRT
sequencing. Genome Biol. 14:405. 10.1186/gb-2013-14-6-405.
39. Shin J. M., Gwak J. W., Kamarajan P., Fenno J. C., Rickard A. H., Kapila Y. L.
(2016). Biomedical applications of nisin. J. Appl. Microbiol. 120, 1449–1465.
10.1111/jam.13033.
40. Siguier P., Perochon J., Lestrade L., Mahillon J., Chandler M. (2006). ISfinder: the
reference centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32–D36.
10.1093/nar/gkj014.
41. Stoll B. J., Hansen N., Fanaroff A. A., Wright L. L., Carlo W. A., Ehrenkranz R. A., et
al. . (2002). Late-onset sepsis in very low birth weight neonates: the experience of the
NICHD Neonatal Research Network. Pediatrics 110, 285–291.
10.1542/peds.110.2.285.
42. Stoll B. J., Hansen N. I., Higgins R. D., Fanaroff A. A., Duara S., Goldberg R., et al. .
(2005). Very low birth weight preterm infants with early onset neonatal sepsis: the
predominance of gram-negative infections continues in the National Institute of Child
Health and Human Development Neonatal Research Network, 2002-2003. Pediatr.
Infect. Dis. J. 24, 635–639. 10.1097/01.inf.0000168749.82105.64.
43. Taft D. H., Ambalavanan N., Schibler K. R., Yu Z., Newburg D. S., Deshmukh H., et
al. . (2015). Center variation in intestinal Microbiota prior to late-onset sepsis in
preterm infants. PLoS ONE10:e0130604. 10.1371/journal.pone.0130604.
44. Vallenet D., Belda E., Calteau A., Cruveiller S., Engelen S., Lajus A., et al. .
(2013). MicroScope–an integrated microbial resource for the curation and comparative

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 129
analysis of genomic and metabolic data. Nucleic Acids Res. 41, D636–D647.
10.1093/nar/gks1194.
45. Vallenet D., Labarre L., Rouy Z., Barbe V., Bocs S., Cruveiller S., et al. .
(2006). MaGe: a microbial genome annotation system supported by synteny
results. Nucleic Acids Res. 34, 53–65. 10.1093/nar/gkj406.
46. Van Der Zwet W. C., Debets-Ossenkopp Y. J., Reinders E., Kapi M., Savelkoul P. H.,
Elburg R. M., et al. . (2002). Nosocomial spread of a Staphylococcus capitis strain
with heteroresistance to vancomycin in a neonatal intensive care unit. J. Clin.
Microbiol. 40, 2520–2525. 10.1128/JCM.40.7.2520-2525.2002.
47. Venkatesh M. P., Placencia F., Weisman L. E. (2006). Coagulase-negative
staphylococcal infections in the neonate and child: an update. Semin. Pediatr. Infect.
Dis. 17, 120–127. 10.1053/j.spid.2006.06.005.
48. Wiedemann I., Breukink E., van Kraaij C., Kuipers O. P., Bierbaum G., de Kruijff B.,
et al. . (2001). Specific binding of nisin to the peptidoglycan precursor lipid II
combines pore formation and inhibition of cell wall biosynthesis for potent antibiotic
activity. J. Biol. Chem. 276, 1772–1779. 10.1074/jbc.M006770200.
49. Zhang Y.-Q., Ren S.-X., Li H.-L., Wang Y.-X., Fu G., Yang J., et al. .
(2003). Genome-based analysis of virulence genes in a non-biofilm-
forming Staphylococcus epidermidis strain (ATCC 12228). Mol. Microbiol. 49, 1577–
1593. 10.1046/j.1365-2958.2003.03671.x.
50. Zhou Y., Liang Y., Lynch K. H., Dennis J. J., Wishart D. S. (2011). PHAST: a fast
phage search tool. Nucleic Acids Res. 39, W347–W352. 10.1093/nar/gkr485.
IV.2.10 Supplementary data
Bacterial isolates and Growth conditions
All four strain of the S. capitis NRCS-A clone were isolated from blood cultures of preterm
infants with presenting a Late-onset sepsis (LOS) and hospitalized in NICUs from 4 distinct
countries (strain CR01, is from an in infant in an NICU in France, strain CR03 from an in a
NICU in Belgium, strain CR04 from a NICU ion Australia and strain CR05 from an NICU in
the United Kingdom).
Species identification of the bacterial isolates and antimicrobial susceptibility testing (AST)

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 130
were performed, respectively, using Vitek MS (bioMérieux, Marcy l’Etoile), 16S rDNA
sequencing, the automated BD Phoenix system (Becton Dickinson, Sparks, MD) and with
Shimadzu-MALDI-TOF MS system (Shimadzu Corporation), as implemented on [1].
The strain was identified as being a Staphylococcus capitis by VITEK MS with 99.9% and at
93.7% by the MALDI-TOF MS, using the Shimadzu Launchpad software program and the
SARAMIS database application (AnagnosTec GmbH) for automatic measurement and
identification. The antimicrobial susceptibility test (AST) results were analyzed according to
the recommendations of the French Microbiology Society [2]. The S. capitis bacteremia was
considered positive based on a single positive blood culture [3,4]. All 4 isolates were resistant
to penicillin, methicillin, gentamicin, rifampin, hetero-resistant to vancomycin and sensitive
to fusidic acid and fluoroquinolones.
DNA isolation
All samples were prepared for sequencing by growing the S. capitis CR01,CR03, CR04 and
CR05 strains aerobically at 37°C in BloodAgar for 24-48 hours. Genomic DNA was then
extracted using the PureLinkTM genomic DNA kit (InvitrogenTM) according to the
manufacturer’s recommended protocol. DNA quantification was performed using a NanoVue
TM Plus (HVD Life Sciences).
Genome sequencing and assembly
454-shotgun libraries were constructed using 1 µg of DNA, for each sample, and following
the GS Rapid library protocol (Roche 454, Roche) with a mean library size of 1547 bp (strain
CR03), 1874 bp (strain CR04) and 1419 bp (strain CR05). The resulting 454 DNA libraries
were sequenced using a high-throughput whole-genome shotgun strategy performed on a
Genome Sequencer FLX+ system (454 Life Sciences/Roche) using FLX Titanium reagents,
according to the manufacturer’s protocols and instructions. De novo assemblies were
performed using the Roche Newbler (version 2.9) software package, and the sequencing
results are summarized below.
Strain Isolation
country
Read
(Mb)
Fold
Covrage
X
No of
Contig
Genome
Size
G+C
Content%
Accession Number
CR01 France 50,876 265 1 2,522,871 33,02 LN866849 (chromosome)
and LN866850 (plasmid) CR03 Belgium
141,728 30 31 2,508,352 32,81 CTEB1000000 CR04 Australia
132,28 30 38 2,512,289 32,8 CTEM1000000 CR05 United King
139,569 31 39 2,543,917 32,84 CTEO1000000

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 131
References:
1. Gagnaire J, Dauwalder O, Boisset S, Khau D, Freydière A-M, Ader F, et al. (2012) Detection of Staphylococcus aureus Delta-Toxin Production by Whole-Cell MALDI-TOF Mass
Spectrometry. PLoS ONE 7(7): e40660. doi:10.1371/journal.pone.0040660
2. French Society for Microbiology. Recommandations du Comite de l'Antibiogramme de la Societe Francaise de Microbiologie. 2009. Available at:
wwwsfmassofr/doc/downloadphp?doc=DiU8C&fic=casfm_2009pdf. Accessed 22 February
2009.
3. Rasigade J-P, Raulin O, Picaud J-C, Tellini C, Bes M, Grando J, Ben Saïd M, Claris O,
Etienne J, Tigaud S, Laurent F. Methicillin-Resistant Staphylococcus capitis with Reduced
Vancomycin Susceptibility Causes Late-Onset Sepsis in Intensive Care Neonates. PLoS ONE 2012;7(2):e31548.
4. Hall KK, Lyman JA. Updated review of blood culture contamination. Clin Microbiol Rev.
2006 Oct;19(4):788-802.

CHAPITRE IV
132 H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹
Table IV.2.S1 – Genes found exclusively in the S. capitis NRCS-A clone and not in other S. capitis public genomes
CR01 ORFs
annotation Gene Product Genomic context
CR01_v3_0498 hsdM putative type I restriction enzyme HindVIIP M protein
Additional restriction modification system type I, immediately after the composite SCCmec-SCCcad/ars/cop element CR01_v3_0499 hsdS HsdS homologue
CR01_v3_0500 hdsR putative type I restriction enzyme HindVIIP R protein
CR01_v3_0529 nsr Nisin-resistance protein nisin resistance gene
CR01_v3_2157 _ protein of unknown function
Phage region CR01_v3_2158 _ putative lipoprotein
CR01_v3_2159 _ conserved protein of unknown function
CR01_v3_2162 _ protein of unknown function
CR01_v3_0176 _ conserved protein of unknown function
CR01_v3_0520 ispD 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase
possibly a biosynthesic pathway CR01_v3_0521 tarJ putative ribitol-5-phosphate dehydrogenase
CR01_v3_0522 _ conserved protein of unknown function
CR01_v3_0523 _ conserved protein of unknown function
CR01_v3_0429 _ conserved protein of unknown function
SCCmec element of the composite SCCmec-SCCcad/ars/cop element
CR01_v3_0430 _ conserved protein of unknown function
CR01_v3_0431 _ conserved protein of unknown function
CR01_v3_0432 _ conserved protein of unknown function
CR01_v3_0433 _ conserved protein of unknown function
CR01_v3_0434 _ putative primase

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat / 133
CR01_v3_0435 _ Cassette chromosome recombinase C7
CR01_v3_0448 _ Transcriptional regulator, DeoR family
CR01_v3_0449 _ conserved protein of unknown function
CR01_v3_0450 _ conserved protein of unknown function
CR01_v3_0451 _ conserved protein of unknown function
CR01_v3_0452 _ conserved protein of unknown function
CR01_v3_0453 ccrC Cassette chromosome recombinase C
CR01_v3_0458 _ conserved protein of unknown function
CR01_v3_0459 _ truncated hsdR
CR01_v3_0460 cas CRISPR-associated endonuclease Cas1
CR01_v3_0461 cas CRISPR-associated endoribonuclease Cas2
CR01_v3_0462 csm CRISPR-associated Csm1 family protein
CR01_v3_0463 csm CRISPR-associated Csm2 protein
CR01_v3_0464 csm CRISPR type III-associated RAMP protein Csm3
CR01_v3_0465 csm CRISPR-associated RAMP protein
CR01_v3_0466 csm CRISPR-associated RAMP protein, Csm5 family
CR01_v3_0467 csm CRISPR-associated protein, Csm6 family
CR01_v3_0468 cas CRISPR-associated endoribonuclease Cas6
CR01_v3_0469 _ conserved protein of unknown function, potential pseudo?
CR01_v3_0470 _ conserved membrane protein of unknown function
CR01_v3_0471 _ Transcriptional regulator, TetR family
CR01_v3_0472 _ conserved membrane protein of unknown function

CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat / 134
CR01_v3_0310 _ exported protein of unknown function
CR01_v3_0311 _ conserved protein of unknown function
CR01_v3_0312 _ conserved protein of unknown function
CR01_v3_0183 _ protein of unknown function
CR01_v3_0496 _ protein of unknown function
CR01_v3_0497 _ conserved protein of unknown function
CR01_v3_0505 _ conserved protein of unknown function
CR01_v3_0507 _ McrBC 5-methylcytosine restriction system component
CR01_v3_0508 _ protein of unknown function
CR01_v3_0178 _ protein of unknown function
CR01_v3_0179 _ protein of unknown function
CR01_v3_0180 _ protein of unknown function
CR01_v3_0673 _ protein of unknown function
CR01_v3_0674 _ conserved protein of unknown function
CR01_v3_0675 _ conserved protein of unknown function
CR01_v3_0676 _ protein of unknown function
CR01_v3_0677 _ protein of unknown function
CR01_v3_0680 _ conserved protein of unknown function
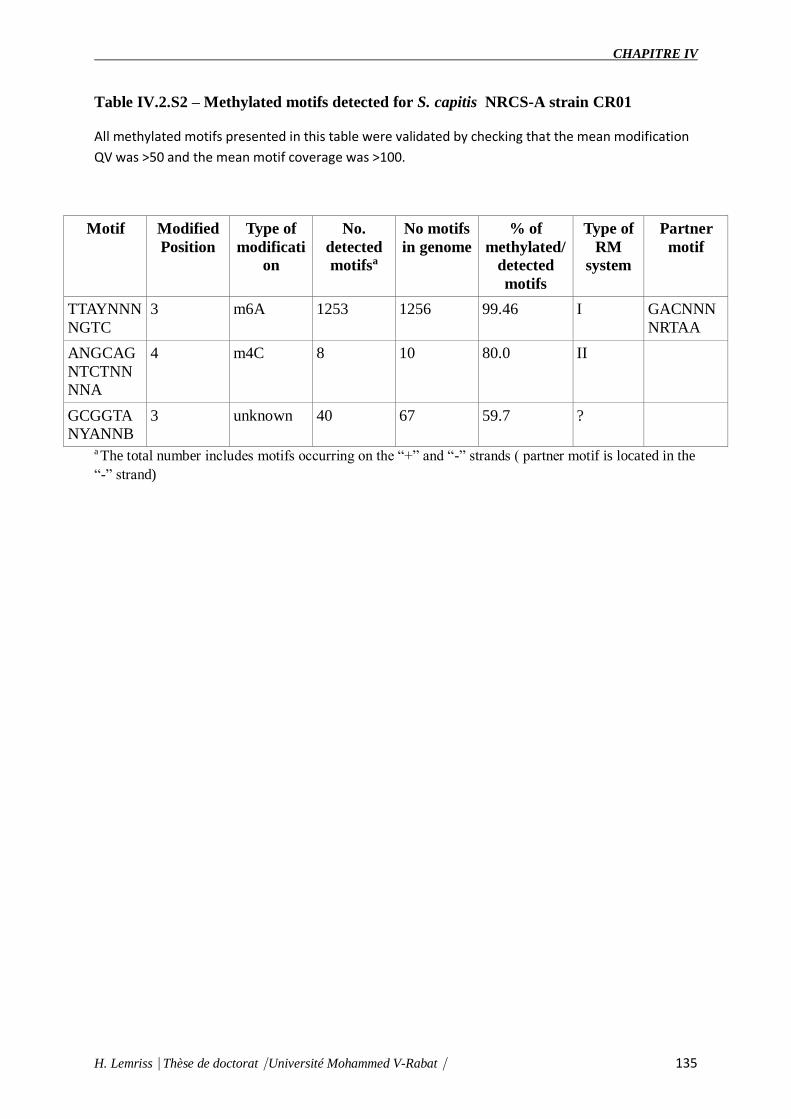
CHAPITRE IV
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 135
Table IV.2.S2 – Methylated motifs detected for S. capitis NRCS-A strain CR01
All methylated motifs presented in this table were validated by checking that the mean modification
QV was >50 and the mean motif coverage was >100.
Motif Modified
Position
Type of
modificati
on
No.
detected
motifsa
No motifs
in genome
% of
methylated/
detected
motifs
Type of
RM
system
Partner
motif
TTAYNNN
NGTC
3 m6A 1253 1256 99.46 I GACNNN
NRTAA
ANGCAG
NTCTNN
NNA
4 m4C 8 10 80.0 II
GCGGTA
NYANNB
3 unknown 40 67 59.7 ?
a The total number includes motifs occurring on the “+” and “-” strands ( partner motif is located in the
“-” strand)

CHAPITRE V
Diffusion mondiale du clone
Staphylococcus capitis NRCS-A en
réanimation néonatale :
caractérisation moléculaire, analyse
génomique et histoire évolutive

CHAPITRE V
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 136
Staphylococcus capitis NRCS-A est un clone endémique impliqué dans les septicémies
nosocomiales néonatales (SNN) à l’échelle mondiale et présente un profil de multirésistance
atypique incluant une sensibilité diminuée à la vancomycine, antibiotique de première ligne
pour le traitement de ces sepsis néonataux. Ce clone multirésistant porte un déterminant de
résistance à la méticilline (staphylococcal cassette chromosome mec, SCCmec) de type V.
Dans cette étude, nous avons cherché à explorer la possibilité de la distribution mondiale du
clone NRCS-A en appliquant diverses méthodes moléculaires (PFGE Sma et SacII, typage
SCCmec, MLST, Dru typing et analyse phylogénétique des génomes complet après WGS)
d’une part, et à savoir la capacité spécifique in vitro de ce clone NRCS-A à acquérir
l’hétérorésistance à la vancomycine sous pression sélective d’autre part.
Quatre vingt six souches NRCS-A isolées de SNN en Angleterre, Australie, Belgique et
France, et quatorze souches de S. capitis non-NRCS-A isolées d’adultes, ont été caractérisées
par électrophorèse en champs pulsés (PFGE) avec les enzymes SmaI et par « direct repeat
unitstyping » (Dru typing).
La grande majorité des souches isolées de SNN ont montré le pulsotype NRCS-A et un dru-
type dt11c (96%) spécifique à ce clone. Ensuite, nous avons sélectionné, un sous-ensemble
d'isolats représentatifs (douze souches isolées de SNN, dont trois par pays, et deux souches
isolées chez des patients adultes), afin de réaliser d’autre typage moléculaire (SacII PFGE,
typage SCCmec et analyse par MLST).
En outre, un arbre phylogénétique a été construit sur la base des génomes complets de cinq
isolats de SNN. Cette analyse phylogénétique de ces cinq isolats des différents pays preuve la
haute parenté génétique dans le clone NRCS-A.
Enfin, l’étude de sensibilité aux antibiotiques montre que toutes les souches NRCS-A ont une
résistance à la méthicilline et aux aminoglycosides et une diminution de sensibilité à la
vancomycine, avec des implications thérapeutiques potentielles pour les nouveau-nés infectés.
Cette étude représente le premier rapport de la dissémination ce clone endémique à grande
échelle géographique.
Des questions restent à propos de l'origine et les moyens de propagation internationale, et les
raisons de la prédilection apparente de ce clone pour les nouveau-nés.

CHAPITRE V
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 137
Le contenu de ce chapitre est présenté sous forme d’un article publié dans la revue « Clinical
Microbiology and Infection » (Butin et al., 2016). La référence de cet article est la suivante :
Clin Microbiol Infect. Jan 2016, 22(1): 46-52 (doi: 10.1016/j.cmi.2015.09.008). Dans cette
publication, j’ai réalisé le travail de séquençage, d’assemblage, d’annotation et d’analyse
phylogénétique des cinq souches sélectionnées pour cette étude : CR01 (CBUB000000000.1),
CR03 (CTEB01000001), CR04 (CTEM01000001), CR05 (CTEO01000001), CR09
(CTEL01000003).
Ce travail a fait objet également des trois communications orales et affichées ci-dessous et
dont les résumés sont présentés dans la partie publications et communications.:
1- Butin M, Martins Simões P, Lemriss H, Lemriss S, Vandenesch F, Claris O, Picaud JC,
Rasigade JP, Laurent F. Staphylococcus capitis in neonatal late-onset sepsis: unexpected
worldwide dissemination of an endemic multi-resistant clone. European Academy of
Pediatric Societies. Barcelone, 17 - 21 October 2014.
2- M. Butin, P. Martins Simões, J.P. Rasigade, S. Lemriss, H. Lemriss, S. El Kabbaj, A.
Ibrahimi, S. Tigaud, F. Vandenesch, O. Claris, J.C. Picaud, F. Laurent. Diffusion mondiale
du clone Staphylococcus capitis NRCS-A en réanimation néonatale : caractérisation
moléculaire, analyse génomique et histoire évolutive. RICAI, Paris, 21 Novembre 2013.
3- Martins Simões P, Butin M, Lemriss H, Lemriss S, Deighton MA, Kearns A, Denis O,
Goering R, Ginevra C, Picaud JC, Ibrahimi A, EL Kabbaj S, Vandenesch F, Rasigade J-P,
Laurent F. Worldwide distribution of an endemic multi-resistant NRCS-A Staphylococcus
capitis clone in NICU: molecular typing, whole genome analysis and evolutionary
history.10th International Meeting on Microbial Epidemiological Markers, Paris, 02-05
October 2013.

CHAPITRE V
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 138
V.1 Abstract
Nosocomial late-onset sepsis represents a frequent cause of morbidity and mortality in
preterm neonates. The Staphylococcus capitis clone NRCS-A has been previously described
as an emerging cause of nosocomial bacteraemia in French neonatal intensive-care units
(NICUs). In this study, we aimed to explore the possible unrecognized dissemination of this
clone on a larger geographical scale. One hundred methicillin-resistant S. capitis strains
isolated from neonates (n = 86) and adult patients (n = 14) between 2000 and 2013 in four
different countries (France, Belgium, the UK, and Australia) were analysed with SmaI pulsed-
field gel electrophoresis (PFGE) and dru typing. The vast majority of NICU strains showed
the NRCS-A pulsotype and the dt11c type (96%). We then randomly selected 14 isolates
(from neonates, n = 12, three per country; from adult patients, n = 2), considered to be a
subset of representative isolates, and performed further molecular typing (SacII PFGE,
SCCmec typing, and multilocus sequence typing-like analysis), confirming the clonality of
the S. capitis strains isolated from neonates, despite their distant geographical origin. Whole
genome single-nucleotide polymorphism-based phylogenetic analysis of five NICU isolates
(from the different countries) attested to high genetic relatedness within the NRCS-A clone.
Finally, all of the NRCS-A strains showed multidrug resistance (e.g. methicillin and
aminoglycoside resistance, and decreased vancomycin susceptibility), with potential
therapeutic implications for infected neonates. In conclusion, this study represents the first
report of clonal dissemination of methicillin-resistant coagulase-negative Stahylococcus
clonale on a large geographical scale. Questions remain regarding the origin and means of
international spread, and the reasons for this clone's apparent predilection for neonates.
V.2 Introduction
Every year, approximately 15 million children are born prematurely worldwide (defined as all
live births before 37 completed weeks). In developed countries, preterm birth is the leading
cause of death in children aged <5 years [1]. In recent decades, technical and medical
advances in neonatal intensive-care units (NICUs) have allowed for an improvement in the
global survival of neonates. Nevertheless, preterm birth-related complications still remain the
major cause of neonatal death [2]. In this context, nosocomial late-onset sepsis (LOS)
(occurring after 3 days of life) represents a frequent cause of morbidity and mortality [3].
Twenty per cent to 30% of very low birthweight preterm babies develop at least one episode
of LOS during their hospitalization in a NICU, resulting in respiratory or neurological

CHAPITRE V
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 139
impairment [4]. Catheter-related bloodstream infections constitute the most frequent cause of
LOS in these vulnerable neonates, owing to multiple indwelling devices and invasive
procedures. Consequently, skin-associated commensal coagulase-negative staphylococci are
the most frequently involved pathogens in LOS, most commonly involving Staphylococcus
epidermidis [3, 5].
However, the predominance of Staphylococcus capitis in several French NICUs has been
reported, whereas this species is rarely reported as a pathogen in adult patients [6]. This
phenomenon is of major concern, for several reasons: first, we have recently shown that all
methicillin-resistant S. capitis strains from NICUs in France belong to a unique clone
(designated NRCS-A), suggesting a high potential for dissemination, whereas
S. capitis isolates from adult patients belong to many distinct clones; second, NRCS-A has
been highly prevalent in our institution (Hospices Civils de Lyon) since at least 2004,
underscoring its ability to persist and become endemic when introduced into NICUs; and
third, NRCS-A strains show decreased susceptibility to all antimicrobial agents that are
commonly used in NICUs [6]. NRCS-A isolates harbour a type V-related staphylococcal
chromosome cassette mec (SCCmec) element conferring resistance to β-lactams [7], and show
a multidrug resistance profile, including resistance or heteroresistance to vancomycin, the
first-line antimicrobial agent in LOS cases [5, 6].
As multiresistant S. capitis LOS cases have been reported in several countries [6, 7, 8, 9], we
sought to explore the possible unrecognized dissemination of the multiresistant NRCS-A
clone on a larger geographical scale. To this end, we collected and characterized
S. capitis bloodstream isolates from NICUs located in geographically diverse countries, by
performing molecular typing and antimicrobial susceptibility testing.
V.3 Materials and Methods
Bacterial isolates
The strain collections of four national reference centres for staphylococci (Australia, Belgium,
France, and the UK) were screened to identify methicillin-resistant S. capitis isolates from
neonatal LOS in NICUs, and from blood cultures of non-neonatal patients (adult or
paediatric). For the period 2000-2013, methicillin-resistant and multiresistant
S. capitis isolates were isolated in NICUs in at least 12 (France), eight (UK), six (Australia)
and four (Belgium) hospitals. Each year during this time period, between two and 70 neonatal

CHAPITRE V
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 140
LOS cases involving S. capitis were identified in each of these hospitals (personal
communication). Each reference laboratory provided a sample of methicillin-
resistant S. capitis strains isolated from its country during the period 2000–2013. In total,
100 S. capitis strains were included in our analysis: 86 from NICUs (France, n = 48;
Belgium, n = 3, Australia, n = 20; and the UK, n = 15) and 14 from other units (paediatric or
adult patients) (France, n = 7; Belgium, n = 3; and Australia, n = 4). These 100 strains were
initially characterized by pulsed-field gel electrophoresis (PFGE) and dru typing.
To confirm our initial results and avoid method-linked bias, we more thoroughly
characterized a subset of isolates belonging to the NRCS-A clone, as it was not feasible to
analyse every isolate in the complete strain collections. Thus, of the 86 NICU isolates, 12
were randomly selected (three NRCS-A isolates per country), and two methicillin-
resistant S. capitis bloodstream isolates from adult patients from France that did not belong to
the NRCS-A clone were selected.
PFGE
PFGE, the reference standard staphylococcal typing and outbreak investigation method, was
performed initially with SmaI restriction digestion, and also with SacII, which has been
recently reported to show increased discriminatory power for S. capitis [6, 9, 10]. Cluster
analysis was performed with BioNumerics software 7.1 (Applied Maths NV, Sint-Martens-
Latem, Belgium). Pattern similarities of >80% (UPGMA, Dice coefficient) were used to
define a pulsotype as previously described [11].
Dru typing
dru typing, a recently developed method based on sequence analysis of a 40-bp variable-
number tandem-repeat region within SCCmec (the mobile genetic element (MGE) harbouring
the mecA gene), was performed as previously described [12].
SCCmec typing
The SCCmec typing was performed as described by Kondo et al. [13].
Multilocus sequence typing

CHAPITRE V
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 141
The genetic relationship between the 14 selected S. capitis isolates was further explored by
multilocus sequence typing (MLST) based on the published S. epidermidis MLST scheme
[14], with appropriately designed consensus primers for amplification of seven housekeeping
genes (Table V.S1). For each allele, sequences were aligned, and single-nucleotide
polymorphisms (SNPs) were detected and compared.
Whole genome sequencing and SNP-based phylogenetic analysis
To explore the genetic conservation inside the NRCS-A clone, five NRCS-A isolates were
selected, including: (a) isolate ST20131582, corresponding to the oldest French isolate
available in our collection (2002) (genome CR09; ENA accession number CTEL01000003)
[15]; and (b) four isolates (one per country) randomly selected from the 12 previously fully
analysed isolates—isolate ST20111775 from France (genome CR01, already published
[16] and considered to be the prototype of the NRCS-A clone; ENA accession number
LN866849), isolate ST20111772 from Belgium (genome CR03; ENA accession number
CTEB01000001) [15], isolate ST20121573 from Australia (genome CR04; ENA accession
number CTEM01000001) [15], and isolate ST20111770 from the UK (genome CR05; ENA
accession number CTEO01000001) [15]. Whole genome sequencing of these five isolates
was performed with pyrosequencing (454; Life Sciences/Roche, Branford, CT, USA).
Moreover, we performed single-molecule real-time sequencing (PacBio, Menlo Park, CA,
USA) for one of these five isolates (ST20111775) to obtain a closed genome (genome CR01).
The mean coverage obtained with the 454 pyrosequencing was 24×, 21×, 20× and 20× for
genomes CR03, CR04, CR05 and CR09, respectively. For the single-molecule real-time
sequencing (genome CR01), a mean coverage of 265× was obtained.
The whole genome sequences (WGSs) of these NRCS-A S. capitis strains were compared
with publicly available S. capitis genomes, which correspond to either skin commensal or
pathogen isolates from adult patients : SK14 (GenBank Reference
Sequence: ACFR00000000.1), VCU116 (GenBank Reference Sequence: AFTX00000000.1),
QN1 (GenBank Reference Sequence: AJTH00000000.1), LNZR-1 (GenBank Reference
Sequence: NZ_JGYJ00000000.1), and C87 (GenBank Reference Sequence :
ACRH00000000.1). To assess the phylogenetic relationships between all S. capitis strains, we
used the REALPHY 1.10 online pipeline [17], with default parameters, to infer phylogenetic
trees from assembled WGS data. We used the closed genome of the S. capitis NRCS-A strain
ST20111775 as the reference genome (genome CR01; ENA accession number LN866849).

CHAPITRE V
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 142
Antimicrobial susceptibility profile
Antimicrobial susceptibility testing of the 14 selected S. capitis isolates (NICU, n = 12; adult
patients, n = 2) was performed with the standard agar diffusion technique as recommended by
the French Society for Microbiology
(www.sfmasso.fr/doc/download.php?doc=DiU8C&fic=casfm_2009.pdf). MICs of
vancomycin, linezolid and daptomycin were determined with the Etest. Strains with a
vancomycin MIC of >2 mg/L were considered to be resistant, and strains with MICs of
≤2 mg/L were tested for vancomycin heteroresistance with the brain–heart infusion agar
method, as previously described [6, 18].
V.4 Results
SmaI PFGE profile
Of the 86 S. capitis isolates from the NICU, all but three (96%) were of the NRCS-A strain
pulsotype (i.e. shared >80% similarity with each other and the NRCS-A pattern)
[6] (Figure. V.1). The three non-NRCS-A strains originated from NICUs in Australia. In
contrast, the 14 non-NICU methicillin-resistant S. capitis isolates showed a diversity of
pulsotypes, only two of which (14%) were NRCS-A: one from a 4-year-old patient
hospitalized in a paediatric intensive-care unit in France, and one from an adult patient in
Australia (Figure. V.1).
Dru typing
With more clonal organisms, PFGE tends to generate related macrorestriction patterns,
causing problems in epidemiological discrimination [12]. Therefore, the 100 S. capitis isolates
were additionally analysed with dru typing for characterization of the MGE harbouring mecA.
Among the 86 NICU isolates, 83 harboured a unique dt11c profile (96%), regardless of their
pulsotype (NRCS-A or not NRCS-A) (Figure. V.1). The three remaining NICU isolates (all
NRCS-A) were dt12r, dt11ba, and dt10h (n = 1 each). Conversely, among the 14 non-NICU
isolates, only two were dt11c (14%), including the NRCS-A paediatric isolate. The
other dru types in non-NICU isolates were diverse: dt10a (n = 1), dt11a (n = 3), dt12af
(n = 1), dt11bd (n = 1), dt3c (n = 2), dt1b (n = 1), and dt9bo (n = 1). Two isolates were non-
typeable.

CHAPITRE V
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 143
Figure V.1. Dendrogram and schematic representation of pulsotypes and dru types of
100 Staphylococcus capitis isolates from neonatal intensive-care units (NICUs) (n = 86)

CHAPITRE V
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 144
or other settings (n = 14). The pulsed-field gel electrophoresis (PFGE) dendrogram was
generated from SmaI PFGE patterns with Bionumerics software 7.1, as previously
described [6] ; [10]. The vertical dotted line represents the 80% limit of similarity. The
pulsotype NRCS-A was defined previously [6]. dru typing was performed as previously
described [12]. *Available genomes.
PFGE with SacII, SCCmec typing, and MLST
To strengthen and complete the data obtained with the set of 100 clinical isolates, we
performed additional analysis on a subset of 14 randomly selected strains: 12 NRCS-A strains
from NICU patients (n = 3 from each country) and two non-NRCS-A strains from adult
patients.
As observed with SmaI, SacII PFGE restriction profiles confirmed the clustering of the 12
NICU isolates into a unique pulsotype (Figure. V.S1). conversely, isolates from adult patients
belonged to two distinct pulsotypes, and shared <80% similarity with NICU isolates.
SCCmec analysis confirmed the genetic closeness of this MGE in the 12 NICU isolates:
indeed, all of these 12 isolates harboured a type V-related SCCmec element, whereas the
isolates from adults harboured a non-typeable SCCmec element (a combination of
a mec complex type C and ccr genes A2/B2/C) (Figure.V.S1). Finally, when an MLST model
encompassing a total of 3020 bp of housekeeping genes was used, the sequence comparison
of the 12 NICU isolates showed that these isolates differed by no more than two SNPs
(Table V.S2), whereas sequences of the two isolates from adult patients showed 32 and 33
SNPs, as compared with the NICU isolates.
Whole genome SNP-based phylogenetic analysis of S. capitis strains
A total of 2 080 323 bp (including coding and intergenic regions) were found to be shared by
all ten genomes (five S. capitis NRCS-A genomes and the five publicly
available S. capitis genomes), and constituted the core genome used for the phylogenetic
reconstruction shown in Figure. V.2. When each genome was compared with the reference
genome of S. capitis NRCS-A CR01, we found a maximum of 162 SNPs and a minimum of
65 SNPs (indels excluded) between the NRCS-A genomes, whereas the non-NRCS-A
genomes harboured at least 7910 SNPs and up to 50 723 SNPs (for LNZR-1 and QN1,
respectively). As seen in Figure. V.2, all five NRCS-A genomes clustered in one clade, which
suggests that they belong to a clonal population.

CHAPITRE V
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 145
Figure V.2. Single-nucleotide polymorphism (SNP)-based maximum-likelihood tree with
whole genome sequences of neonatal Staphylococcus capitis isolates from four distinct
countries and publicly available S. capitis genomes. A total of 2 080 323 homologous sites
from all ten genomes (polymorphic and non-polymorphic sites) were obtained by use of the
REALPHY 1.10 online pipeline with the closed chromosome of S. capitis NRCS-A CR01
(ENA accession number LN866849) as reference. A maximum-likelihood tree was built by
the use of PhyML with default parameters as implemented in REALPHY 1.10. For each
strain, the number of SNPs between each strain and the reference is indicated in grey in
parentheses. Aus, Australia; Be, Belgium; Fr, France.
Antimicrobial susceptibility profiles
Antimicrobial susceptibility profiles of the 14 S. capitis strains are shown in Table V.1. All 12
NICU isolates showed methicillin and aminoglycoside resistance, and either resistance or
heteroresistance to vancomycin, the first-line antimicrobial agent used for LOS. The 12 NICU
isolates remained susceptible to fluoroquinolones.
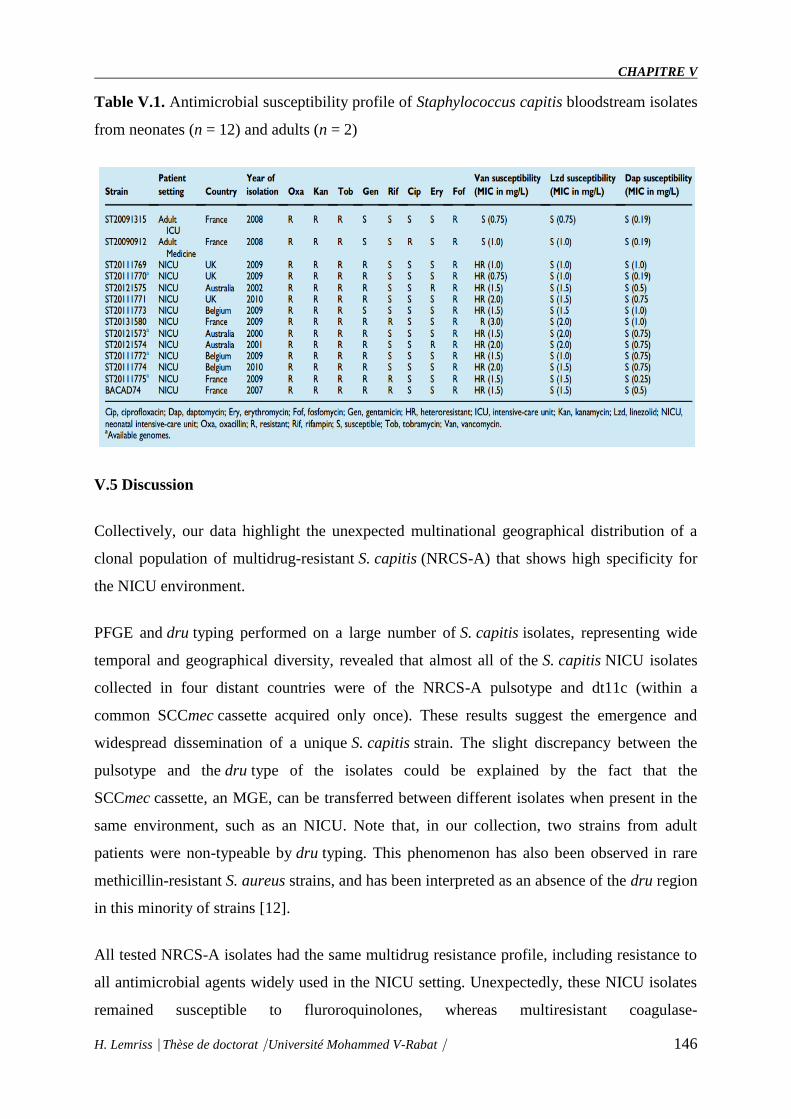
CHAPITRE V
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 146
Table V.1. Antimicrobial susceptibility profile of Staphylococcus capitis bloodstream isolates
from neonates (n = 12) and adults (n = 2)
V.5 Discussion
Collectively, our data highlight the unexpected multinational geographical distribution of a
clonal population of multidrug-resistant S. capitis (NRCS-A) that shows high specificity for
the NICU environment.
PFGE and dru typing performed on a large number of S. capitis isolates, representing wide
temporal and geographical diversity, revealed that almost all of the S. capitis NICU isolates
collected in four distant countries were of the NRCS-A pulsotype and dt11c (within a
common SCCmec cassette acquired only once). These results suggest the emergence and
widespread dissemination of a unique S. capitis strain. The slight discrepancy between the
pulsotype and the dru type of the isolates could be explained by the fact that the
SCCmec cassette, an MGE, can be transferred between different isolates when present in the
same environment, such as an NICU. Note that, in our collection, two strains from adult
patients were non-typeable by dru typing. This phenomenon has also been observed in rare
methicillin-resistant S. aureus strains, and has been interpreted as an absence of the dru region
in this minority of strains [12].
All tested NRCS-A isolates had the same multidrug resistance profile, including resistance to
all antimicrobial agents widely used in the NICU setting. Unexpectedly, these NICU isolates
remained susceptible to fluroroquinolones, whereas multiresistant coagulase-

CHAPITRE V
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 147
negative Staphylococcus strains are typically fluoroquinolone resistant. This atypical
antimicrobial susceptibility profile is similar to that of previously described NRCS-A
strains [6], and probably reflects the specific antimicrobial selective pressure of NICUs,
where β-lactams, vancomycin, and aminoglycosides, but not fluoroquinolones, are widely
used [19]. In our study, all NRCS-A isolates were either resistant or heteroresistant to
vancomycin. Studies of S. capitis isolates from NICUs have reported constant vancomycin
heteroresistance [7] ; [8], and Van der Zwet et al. initially proposed that this could be an
intrinsic feature of S. capitis [8]. However, our recent results obtained by
comparing S. capitis isolates from neonates and adult patients suggest that vancomycin
heteroresistance is more likely to be a specific feature of the NRCS-A clone [7]. This
heteroresistance might have contributed to S. capitis NRCS-A establishment in NICUs.
Alternatively, the frequent use of vancomycin in neonates might have induced/selected this
heteroresistance in the NRCS-A clone. The clinical significance of vancomycin
heteroresistance is controversial, but therapeutic failures of vancomycin in S. capitis-related
LOS have been reported [8]. Thus, the multidrug resistance profile of this emerging clone
raises therapeutic concerns in cases of LOS, and requires substantiation in a clinical study
focusing on S. capitis LOS.
Taken together, these data demonstrate the clonality of S. capitis isolates from neonates,
despite their distant geographical origin, in contrast to the high level of genetic diversity seen
in isolates from non-NICU patients. Thus, this larger dataset, involving an international
collection of strains, mirrors our previous observation of isolates within France [6].
The geographical spread of a clonal population of S. capitis in distant NICUs, although
unexpected, is reminiscent of the extensive diffusion of clonal group B Streptococcus ST-17
causing meningitis and septicaemia in neonates [20]. However, although the global
dissemination of methicillin-resistant Staphylococcus aureus clones has been well described,
to the best of our knowledge this study represents the first report of widespread diffusion of
methicillin-resistant coagulase-negative staphylococci. This observation raises important and
as yet unsolved questions regarding the origin and the dissemination routes involved in the
international spread of NRCS-A S. capitis, as well as the underlying mechanism(s) of
apparent predilection for neonates.
V.6 Conclusion

CHAPITRE V
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 148
In conclusion, we have demonstrated that a methicillin-resistant and multidrug
resistant S. capitis clone is involved in LOS in NICUs from four distant countries. A better
understanding of the epidemiology and pathophysiology of S. capitis NRCS-A infections in
preterm infants is now warranted.
V.7 Transparency declaration
The authors have no conflicts of interest or funding to disclose.
V. 8 Acknowledgements
This work was supported by the French Ministry of Health and the Institut National de la
Santé et de la Recherche Médicale (Inserm). The funders had no role in study design, data
collection, analysis, decision to publish, or preparation of the manuscript.
V.9 References
1. Liu, L., Johnson, H.L., Cousens, S., Perin, J., Scott, S., Lawn, J.E. et al. Global,
regional, and national causes of child mortality: an updated systematic analysis for
2010 with time trends since 2000.Lancet. 2012; 379: 2151–2161.
2. Howson, C.P., Kinney, M.V., McDougall, L., and Lawn, J.E. Born Toon Soon:
preterm birth matters.Reprod Health. 2013; 10: S1.
3. Boghossian, N.S., Page, G.P., Bell, E.F., Stoll, B.J., Murray, J.C., Cotten, C.M. et
al. Late-onset sepsis in very low birth weight infants from singleton and multiple-
gestation births. J Pediatr. 2013; 162: 1120–1124.
4. Cohen-Wolkowiez, M., Moran, C., Benjamin, D.K., Cotten, C.M., Clark, R.H.,
Benjamin, D.K. Jr. et al.Early and late onset sepsis in late preterm infants. Pediatr
Infect Dis J. 2009; 28: 1052–1056.
5. Venkatesh, M.P., Placencia, F., and Weisman, L.E. Coagulase-negative
staphylococcal infections in the neonate and child: an update. Semin Pediatr Infect
Dis. 2006; 17: 120–127.
6. Rasigade, J.-P., Raulin, O., Picaud, J.-C., Tellini, C., Bes, M., Grando, J. et
al. Methicillin-resistantStaphylococcus capitis with reduced vancomycin susceptibility
causes late-onset sepsis in intensive care neonates. PLoS ONE. 2012; 7: e31548.

CHAPITRE V
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 149
7. D’mello, D., Daley, A.J., Rahman, M.S., Qu, Y., Garland, S., Pearce, C. et
al. Vancomycin heteroresistance in bloodstream isolates of Staphylococcus
capitis. J Clin Microbiol. 2008; 46: 3124–3126.
8. Van Der Zwet, W.C., Debets-Ossenkopp, Y.J., Reinders, E., Kapi, M., Savelkoul,
P.H., Van Elburg, R.M. et al. Nosocomial spread of a Staphylococcus capitis strain
with heteroresistance to vancomycin in a neonatal intensive care unit. J Clin
Microbiol. 2002; 40: 2520–2525.
9. Cui, B., Smooker, P.M., Rouch, D.A., Daley, A.J., and Deighton, M.A. Differences
between two clinicalStaphylococcus capitis subspecies as revealed by biofilm,
antibiotic resistance, and pulsed-field gel electrophoresis profiling. J Clin
Microbiol. 2013; 51: 9–14.
10. Goering, R.V. Molecular epidemiology of nosocomial infection: analysis of
chromosomal restriction fragment patterns by pulsed-field gel electrophoresis. Infect
Control Hosp Epidemiol. 1993;14: 595–600.
11. Tenover, F.C., Arbeit, R.D., Goering, R.V., Mickelsen, P.A., Murray, B.E., Persing,
D.H. et al. Interpreting chromosomal DNA restriction patterns produced by pulsed-
field gel electrophoresis: Criteria for bacterial strain typing. J Clin
Microbiol. 1995; 33: 2233–2239.
12. Goering, R.V., Morrison, D., Al-Doori, Z., Edwards, G.F.S., and Gemmell,
C.G. Usefulness of mec-associated direct repeat unit (dru) typing in the
epidemiological analysis of highly clonal methicillin-resistant Staphylococcus
aureus in Scotland. Clin Microbiol Infect. 2008; 14: 964–969.
13. Kondo, Y., Ito, T., Ma, X.X., Watanabe, S., Kreiswirth, B.N., Etienne, J. et
al. Combination of multiplex PCRs for staphylococcal cassette chromosome mec type
assignment: Rapid identification system for mec, ccr, and major differences in
junkyard regions. Antimicrob Agents Chemother. 2007; 51: 264–274.
14. Thomas, J.C., Vargas, M.R., Miragaia, M., Peacock, S.J., Archer, G.L., and Enright,
M.C. Improved multilocus sequence typing scheme for Staphylococcus
epidermidis. J Clin Microbiol. 2007; 45: 616–619.
15. Lemriss, H., Lemriss, S., Martins-Simoes, P., Butin, M., Lahlou, L., Rasigade, J.P. et
al. Genome sequences of four Staphylococcus capitis NRCS-A isolates from
geographically distant neonatal intensive care units. Genome
Announc. 2015; 3: e00501–e00515.

CHAPITRE V
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 150
16. Lemriss, H., Martins Simões, P., Lemriss, S., Butin, M., Ibrahimi, A., El Kabbaj, S. et
al. Non-contiguous finished genome sequence of Staphylococcus capitis CR01
(pulsetype NRCS-A). Stand Genomic Sci. 2014; 9: 1118–1127.
17. Bertels, F., Silander, O.K., Pachkov, M., Rainey, P.B., and van Nimwegen,
E. Automated reconstruction of whole-genome phylogenies from short-sequence
reads. Mol Biol Evol. 2014; 31: 1077–1088.
18. Satola, S.W., Farley, M.M., Anderson, K.F., and Patel, J.B. Comparison of detection
methods for heteroresistant vancomycin-intermediate Staphylococcus aureus, with the
population analysis profile method as the reference method. J Clin
Microbiol. 2011; 49: 177–183.
19. Clark, R.H., Bloom, B.T., Spitzer, A.R., and Gerstmann, D.R. Reported medication
use in the neonatal intensive care unit: data from a large national data
set. Pediatrics. 2006; 117: 1979–1987.
20. Poyart, C., Réglier-Poupet, H., Tazi, A., Billoët, A., Dmytruk, N., Bidet, P. et
al. Invasive group B streptococcal infections in infants, France. Emerg Infect
Dis. 2008; 14: 1647–1649.
V.10 Supplementary data
Table V.S1: Characteristics of 7 house-keeping genes and related primers 389 used for the
determination of the genetic relationships between 14 S. capitis isolates.
To characterize the genetic relationships between the 14 S. capitis isolates from neonatal
intensive care units (NICUs) patients (n=12) and adult patients (n=2), seven housekeeping
genes per isolate, selected on the basis of the published S. epidermidis MLST scheme (10),
were amplified and sequenced. New consensus primers were designed using Primer3 and the
consensus sequence obtained for each gene from the multiple alignments (Muscle algorithm)
of the 4 publicly available S. capitis genomes (GeneBank). Amplification were performed in
a 25 µL volume using Taq polymerase 0.625 U (0.025 U/µL), MgCl 2 1.5mM, dNTP 0.2
mM, buffer 10X, primers 10 pmol (0.4 pmol/µL) for each primer and extracted 398 DNA (2
µL). The amplification program consisted of: an initialization step of heating to 95°C for 3
minutes, 34 amplification cycles (denaturation at 95°C for 30 seconds, annealing at Tm 400 =
58°C for 60 seconds and extension at 72°C for 90 seconds) and a final elongation step at 401
72°C for 10 minutes. For each gene, sequences were aligned against the respective allele in S.
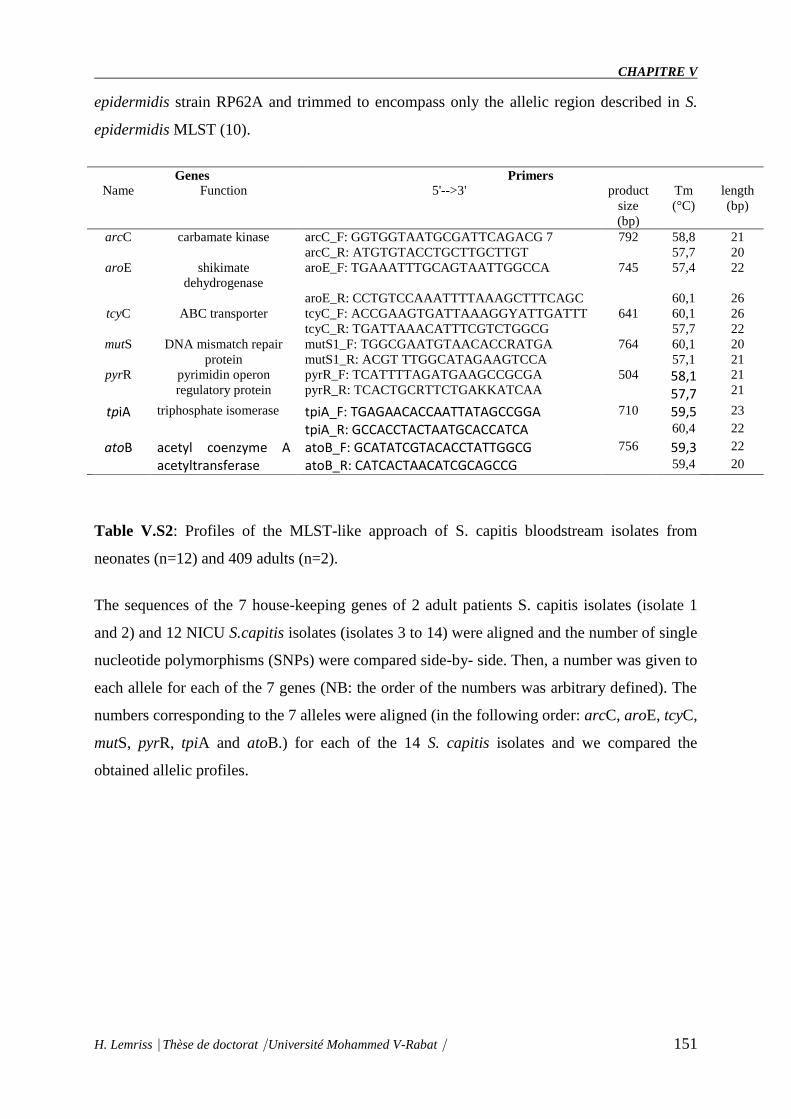
CHAPITRE V
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 151
epidermidis strain RP62A and trimmed to encompass only the allelic region described in S.
epidermidis MLST (10).
Genes Primers
Name Function 5'-->3' product
size
(bp)
Tm
(°C)
length
(bp)
arcC carbamate kinase arcC_F: GGTGGTAATGCGATTCAGACG 7 792 58,8 21
arcC_R: ATGTGTACCTGCTTGCTTGT 57,7 20
aroE shikimate
dehydrogenase
aroE_F: TGAAATTTGCAGTAATTGGCCA 745 57,4 22
aroE_R: CCTGTCCAAATTTTAAAGCTTTCAGC 60,1 26
tcyC ABC transporter tcyC_F: ACCGAAGTGATTAAAGGYATTGATTT 641 60,1 26
tcyC_R: TGATTAAACATTTCGTCTGGCG 57,7 22
mutS DNA mismatch repair
protein
mutS1_F: TGGCGAATGTAACACCRATGA
mutS1_R: ACGT TTGGCATAGAAGTCCA
764 60,1
57,1
20
21
pyrR pyrimidin operon
regulatory protein
pyrR_F: TCATTTTAGATGAAGCCGCGA
pyrR_R: TCACTGCRTTCTGAKKATCAA
504 58,1 57,7
21
21
tpiA triphosphate isomerase tpiA_F: TGAGAACACCAATTATAGCCGGA 710 59,5 23
tpiA_R: GCCACCTACTAATGCACCATCA 60,4 22
atoB acetyl coenzyme A acetyltransferase
atoB_F: GCATATCGTACACCTATTGGCG 756 59,3 22
atoB_R: CATCACTAACATCGCAGCCG 59,4 20
Table V.S2: Profiles of the MLST-like approach of S. capitis bloodstream isolates from
neonates (n=12) and 409 adults (n=2).
The sequences of the 7 house-keeping genes of 2 adult patients S. capitis isolates (isolate 1
and 2) and 12 NICU S.capitis isolates (isolates 3 to 14) were aligned and the number of single
nucleotide polymorphisms (SNPs) were compared side-by- side. Then, a number was given to
each allele for each of the 7 genes (NB: the order of the numbers was arbitrary defined). The
numbers corresponding to the 7 alleles were aligned (in the following order: arcC, aroE, tcyC,
mutS, pyrR, tpiA and atoB.) for each of the 14 S. capitis isolates and we compared the
obtained allelic profiles.
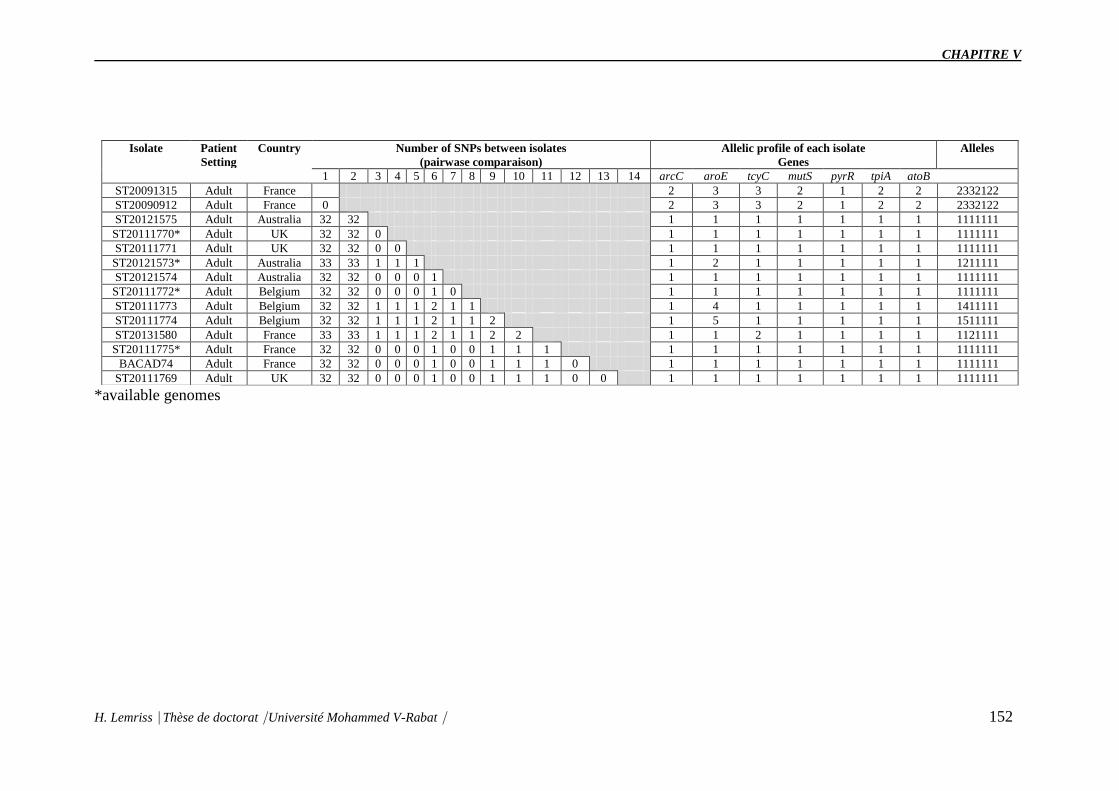
CHAPITRE V
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 152
Isolate Patient
Setting
Country Number of SNPs between isolates
(pairwase comparaison)
Allelic profile of each isolate
Genes
Alleles
1 2 3 4 5 6 7 8 9 10 11 12 13 14 arcC aroE tcyC mutS pyrR tpiA atoB
ST20091315 Adult France 2 3 3 2 1 2 2 2332122
ST20090912 Adult France 0 2 3 3 2 1 2 2 2332122
ST20121575 Adult Australia 32 32 1 1 1 1 1 1 1 1111111
ST20111770* Adult UK 32 32 0 1 1 1 1 1 1 1 1111111
ST20111771 Adult UK 32 32 0 0 1 1 1 1 1 1 1 1111111
ST20121573* Adult Australia 33 33 1 1 1 1 2 1 1 1 1 1 1211111
ST20121574 Adult Australia 32 32 0 0 0 1 1 1 1 1 1 1 1 1111111
ST20111772* Adult Belgium 32 32 0 0 0 1 0 1 1 1 1 1 1 1 1111111
ST20111773 Adult Belgium 32 32 1 1 1 2 1 1 1 4 1 1 1 1 1 1411111
ST20111774 Adult Belgium 32 32 1 1 1 2 1 1 2 1 5 1 1 1 1 1 1511111
ST20131580 Adult France 33 33 1 1 1 2 1 1 2 2 1 1 2 1 1 1 1 1121111
ST20111775* Adult France 32 32 0 0 0 1 0 0 1 1 1 1 1 1 1 1 1 1 1111111
BACAD74 Adult France 32 32 0 0 0 1 0 0 1 1 1 0 1 1 1 1 1 1 1 1111111
ST20111769 Adult UK 32 32 0 0 0 1 0 0 1 1 1 0 0 1 1 1 1 1 1 1 1111111
*available genomes

CHAPITRE V
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 153
Figure V.S1. Molecular characterization of S. capitis bloodstream isolates from neonates
(n = 12) and adult patients (n = 2).
Figure V.S2. Flow diagram of isolate selection.
153

CHAPITRE VI
DISCUSSION GENERALE
ET PERSPECTIVES

CHAPITRE VI
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 154
L'adaptation des bactéries à leur niche écologique constitue un élément majeur permettant
l'évolution de leurs génomes (Altermann, 2012). Cette évolution est un processus complexe
qui se fait par plusieurs mécanismes tels que les mutations, les réarrangements génétiques ou
par les événements de transferts horizontaux de gènes (Juhas et al., 2009). Le transfert
horizontal de gènes joue un rôle important dans l'évolution des bactéries et permet
l'acquisition de nouvelles fonctions ou de nouveaux caractères phénotypiques (Smith et al.,
1992, 1993 ; Koonin et al., 2001). Ce transfert de gène est plus lié à la coexistence des
espèces au sein d'une même niche écologique qu'au rapprochement taxonomique entre les
bactéries (Brochier et al., 2004). L'utilisation répandue d'antibiotiques en association avec une
hygiène hospitalière intense a entraîné l'émergence de bactéries résistantes aux médicaments
qui sont adaptées pour prospérer chez les patients hospitalisés.
Les staphylocoques demeurent immuablement des pathogènes majeurs en pathologie humaine
et animale. Nos connaissances sur leur épidémiologie, leur biologie, leur virulence, et leur
résistance ne cessent de progresser avec l’utilisation d’outils toujours plus précis et
performants.
Les humains sont le principal réservoir des staphylocoques. L'acquisition staphylococcique se
fait directement par contact des mains ou des fluides corporels des individus infectés
(colonisés). La transmission peut également se produire par contact indirect d'objets
colonisés, y compris les stéthoscopes, les vêtements et l'équipement. Les staphylocoques de la
peau du nourrisson peuvent causer une infection, généralement après une rupture de la peau
(lésion) ou l'intégrité des membranes muqueuses avec des cathéters et des tubes (Fonseca et
al., 1994). Il a été suggéré, depuis les années 1960, que les personnes qui travaillent dans la
santé (corps médicaux des hôpitaux) sont une source majeure d'infection staphylococcique,
mais des données récentes ont montré que la colonisation maternelle augmente également le
risque de colonisation staphylococcique chez les nouveau-nés, par transmission verticale et
allaitement (Jimenez-Truque et al., 2012 ; Leshem).
Dans les services de réanimation néonatale, les infections néonatales tardives (survenant après
3 jours de vie) représentent une cause majeure de mortalité et de morbidité (Stoll et al., 2002 ;
Venkatesh et al., 2006). Ces infections nosocomials sont particulièrement fréquentes chez les
prématurés, qui sont à la fois immatures sur le plan immunitaire et exposés à des
thérapeutiques parfois invasives. Plusieurs facteurs de risque ont été identifiées, notamment le
faible âge gestationnel et le faible poids de naissance (Johnson-Robbins et al., 2011). En cas

CHAPITRE VI
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 155
d’infection néonatale précoce, les germes les plus fréquemment identifiés sont Streptococcus
agalactiae (streptocoque du groupe B) et Escherichia coli, et en cas d’infection tardive, plus
de la moitié des bactéries impliquées sont des cocci à Gram positif, en premier lieu desquels
on retrouve les staphylocoques à coagulase négative et dont l’espèce Staphylococcus
epidermidis est la plus prévalente dans ce contexte (Klingenberg et al., 2007). Parmi les autres
espèces, Staphylococcus capitis est rarement rapporté dans les bactériémies et chez l’adulte, il
est le plus souvent considéré comme un contaminant en cas d’isolement dans les
hémocultures (Ruhe et al., 2004). Les souches présentent généralement une faible virulence et
une résistance aux béta-lactamines, justifiant un traitement par la vancomycine en première
intention (en cas de sepsis) (Krediet et al., 2001 ; Huang et al., 2006 ).
Le CNR des staphylocoques a rapporté en 2012 une fréquence particulièrement élevée des
septicémies nosocomiales à Staphylococcus capitis chez les nouveaux nés de petit poids de
naissance au sein des deux services de réanimation néonatale de Lyon, d’autres villes
françaises et dans de nombreux pays à travers le monde sur une large échelle de temps, alors
qu’il n’est jamais retrouvé chez les adultes. La caractérisation moléculaire (électrophorèse en
champs pulsés « PFGE ») et microbiologique (profil phénotypique, biochimique et résistance
aux antibiotiques) de ces souches avait permis de montrer que l’ensemble des souches
appartenait à un même clone NRCS-A (Van Der Zwet et al., 2002 ; Venkatesh et al., 2006 ;
Rasigade et al., 2012 ; D'mello et al., 2012 ; Boghossian et al., 2013 ; Cui et al., 2013). Les
représentants de NRCS-A présentent un profil de résistance atypique aux antibiotiques,
associant une résistance à l’ensemble des béta-lactamines, aux aminosides, ainsi qu’une
sensibilité diminuée (résistance ou hétérorésistance) aux glycopeptides. Cette multi-résistance
peut avoir des impacts en termes de traitement puisque la vancomycine est l’antibiotique de
choix en cas d’infection néonatale à staphylocoque à coagulase négative. Ces résultats ont
motivé l’étude de ce pathogène qui a fait l’objet d’une collaboration avec le Centre Nationale
de Référence des staphylocoques, Lyon, France, le Laboratoire de Recherche et d’analyse
Médicale de la Fraternelle de la Gendarmerie Royal (LRAM), Rabat et le Laboratoire de
Biotechnologie Médicale (Medbiotech), Rabat. Ce travail de doctorat contribue à la
compréhension du profil multi-résistant de cette espèce habituellement sensible et qui a pu
être isolée dans plusieurs pays à travers le monde, spécifiquement en néonatologie, à la
recherche de facteurs de virulence spécifique et à la compréhension des raisons de
dissémination mondiale dans les NICU de ce clone endémique (S. capitis pulsotype NRCS-
A), ainsi que son affinité pour les nouveau-nés (ne sont actuellement pas élucidées). Ce travail

CHAPITRE VI
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 156
permet également d'étudier plus en profondeur son écologie afin de mieux comprendre
l’évolution et les liens phylogénétiques existants entre ces souches d’origines géographiques
variées par l’approche de séquençage haut débit, typage moléculaire et analyse de génomique
comparative en utilisant plusieurs outils bioinformatiques.
Bien qu’il soit principalement restreint aux études de recherche actuellement, le séquençage
du génome bactérien des isolats cliniques devient de plus en plus utilisé car le coût du
séquençage de l'ADN continue de diminuer (Koser et al., 2012 ; McAdam et al., 2014). En
microbiologie, les technologies de séquençage nouvelle génération ont notamment été
appliquées au séquençage de génomes entiers (par exemple, Mycoplasma pneumoniae ou
Pneumocytis jirovecii), (Kenri et al., 2012 ; Cissé et al., 2012) pour la recherche et
l’identification de mutations associées à la résistance aux antirétroviraux, (Dudley et al.,
2012 ; Larrat et al., 2013) à l’étude de la cinétique d’apparition de ces mutations sous
traitement, à l’étude de populations et de variants minoritaires (exemples des virus HCV
(Lauck et al., 2012 ; et de la grippe (Yasugi et al., 2010)) et à l’étude des microbiomes
intestinaux ou pulmonaires (Monira et al., 2011 ; Delhaes et al., 2013). Le potentiel de
l’utilisation de ces technologies en microbiologie clinique est énorme, en particulier en ce qui
concerne le développement de PCR diagnostiques ou de tests sérologiques, la détection de
facteurs de virulence et de gènes de résistance aux antibiotiques et l’épidémiologie de
bactéries pathogènes (Shields et al., 2012; Larrat et al., 2013).
Actuellement, de nombreux génomes bactériens ont été séquencés entièrement ou
partiellement (plus 219 000 projets) sont mentionnés sur le site GOLD
(http://www.genomesonline.org/). Le montage d’un projet de séquençage d’un génome est un
processus long qui demande du temps pour mobiliser des ressources à la fois financières,
techniques, humaines et informatiques. Ce décalage entre d’une part, les innovations et
progrès des NGS et, d’autre part, la réalité de la recherche, constitue également une difficulté
dans le choix de la stratégie de séquençage et d’assemblage d’un génome comme décrit dans
le chapitre I.
La génomique bactérienne a apporté une meilleure compréhension des mécanismes
d’évolution des génomes bactériens. L’obtention d’une culture pure est une étape essentielle
pour relier le phénotype particulier d’une souche bactérienne à son contenu en gènes,
notamment pour étudier spécifiquement sa virulence, sa pathogénicité ou sa résistance. Ainsi,
les comparaisons génomiques ont montré que le transfert horizontal de gènes était chez les
bactéries le principal moyen d’acquérir de nouvelles fonctions. Ces acquisitions peuvent être

CHAPITRE VI
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 157
suivies d’une réduction génomique drastique lors de l’adaptation à l’environnement. L’étude
des génomes bactériens réduits a également permis de comprendre comment les changements
dans la pression de sélection entrainaient l’évolution. Donc la génomique bactérienne est utile
aux microbiologistes, aux infectiologues et aux spécialistes en hygiène hospitalière (Tettelin,
et al., 2005 ; Deschamps et al., 2009).
Les étapes d’un projet de génomique incluent 6 phases principales: le choix de l’espèce et/ou
de l’échantillon à séquencer, l’extraction d’ADN, le séquençage proprement dit, l’assemblage
des séquences obtenues, la fermeture des trous («gap closure » en anglais), l’annotation et
enfin l’exploitation des données. Notons que l’étape de «gap closure» représente plus de 80%
des efforts d’un projet de génomique. Ainsi, dans un but d’application médicale, il est
particulièrement intéressant d’utiliser un génome sans effectuer cette étape, afin de réduire les
coûts et les efforts jusqu’à l’obtention de l’information utile.
Dans le cas de notre étude pour S. capitis, aucun génome complet et de bonne qualité n’était
disponible dans les bases de données publiques. C'est ainsi que le séquençage et l'assemblage
du génome de S. capitis CR01 que nous avons réalisé, constitue une étape importante dans
l'étude de cette espèce émergente (chapitre II). La majorité des projets de séquençage de
S.capitis au début de ma thèse ont utilisé une seule technique de séquençage (NCBI, Chapitre
I). Dans le cas de la souche S.capitis CR01, le séquençage a été réalisé en combinant deux
technologies : Pyroséquençage (GFLX+Titanuim) et PacBio (SMART). Ainsi, nous avons pu
faire un double contrôle de la séquence génomique, obtenir un génome draft et fermer les
scaffolds de la séquence génomique par la suite donnant le premier génome de référence et
éliminer les erreurs de séquençage liées aux homopolymères (Loman et al., 2012). Ces erreurs
qui surviennent au niveau des régions d'homopolymères peuvent biaiser l'analyse et générer
de faux InDels (Chapitre IV). Durant le processus d'assemblage des génomes, plusieurs
critères et indices sont à prendre en considération pour évaluer la qualité de l'assemblage tel
que le N50 des contigs et scaffolds, la couverture, le taux d'erreurs d'assemblage, le nombre de
contigs et scaffolds, et la taille totale de l'assemblage (Ekblom et Wolf, 2014). La séquence
complète de S.capitis CR01 a été assemblée sous forme d'un seul chromosome circulaire de
2,522,871 bp désigné comme le premier génome de référence publié dans les base de données
et un plasmide de 26.140 bp (chapitre II).
Dans une approche similaire nous avons effectué un projet de séquençage à haut débit (un
seul run) pour 8 isolats cliniques de la souche de S.capitis collecté entre 2000 et 2009 des

CHAPITRE VI
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 158
NICU à partir des origines géographique différentes (Chapitre III). L’annotation
automatique et expert des séquences de génome S.capitis CR01, CR02, CR03, CR04, CR05,
CR06, CR07, CR08 et CR09 ont été réalisé sur la platforme MaGe. La souche CR01 et ces
isolats ont été identifiés préalablement par spectrométrie de masse MALDI TOF (chapitre I).
La séquence génomique de S.capitis CR01 a été ensuite utilisée comme référence dans la
recherche des caractéristiques génomiques et les analyses de génomique comparative par
rapport aux 8 souches de S.capitis (CR02, CR03, CR04, CR05, CR06, CR07, CR08 et CR09)
et les autres staphylocoques disponibles dans les bases de données (Chapitre IV, V). Ce
travail est la première étude dans le monde chez S.capitis d’origine néonatale.
Dans ce contexte, les nouvelles techniques de séquençages (NTS) représentent un moyen
indispensable pour comprendre les différents mécanismes de résistance, leur base génétique et
leur dissémination intercontinentale. L’étude des génomes de pathogènes multirésistants a
ainsi permis de mieux décrire les principaux modes de transmission des déterminants de la
résistance par l’échange d’éléments génétiques mobiles, notamment chez des isolats cliniques
de Staphylococcus aureus, d’Enterobacteriaceae, d’Acinetobacter baumannii et
de Pseudomonas aeruginosa (Fournier et al., 2006 ; Diene et al., 2013). Ces éléments
génétiques mobiles, codant parfois pour de multiples gènes de résistance aux antibiotiques et
aux métaux lourds, permettent à ces bactéries multirésistantes (BMR) de persister dans les
milieux hospitaliers et de résister à presque tous les antibiotiques communément prescrits
(Bush et al., 2011).
Les structures hospitalières sont souvent confrontées à des épidémies bactériennes causées par
des pathogènes nosocomiaux tels que les Staphylococcus aureus résistant à la méticilline
(SARM), les BMR composées principalement des entérobactéries (Klebsiella pneumoniae, E.
coli, Enterobacter cloacae, etc.), et des bacilles non fermentatifs tels qu’A. baumannii et P.
aeruginosa (Witney et al., 2014 ; Lee et al., 2014).
Plusieurs études rapportent l’utilisation des NTS comme outils d’investigation pour
comprendre la dissémination de souches à l’échelle d’un hôpital, d’une région, ou même d’un
continent (Rasko et al., 2011; Lavezzo et al., 2013).
Dans une étude publiée en 2012, Vogel et coll. ont rapporté l’usage des NTS pour étudier et
comprendre la persistance et la diffusion d’une souche clonale de S. aureus ST228 au CHUV
sur une période de dix ans (Vogel et al., 2012). Le séquençage et la comparaison génomique

CHAPITRE VI
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 159
de huit souches de S. aureus isolées entre 2001 et 2008 ont mis en évidence des changements
génétiques tels que l’acquisition d’un plasmide, la perte d’un large fragment génomique et des
insertions de transposases. En 2013, Lavezzo et coll. ont étudié une épidémie de N.
meningitidis caractérisée par un taux de mortalité élevé dans le nord-est de l’Italie. Alors que
l’investigation de cette épidémie avec les méthodes conventionnelles telles que le typage
moléculaire (MLST), l’électrophorèse en champ pulsé (PFGE) et les tests sérologiques
concluaient à la diffusion d’un même clone de N. meningitidis ST-11, le séquençage et la
comparaison génomique de certaines souches ont mis en évidence des différences génomiques
et des acquisitions de gènes ayant significativement contribué à la pathogénicité de ces
souches (Lavezzo et al., 2013). Dans une autre étude comparative publiée en 2014, SenGupta
et al., ont typé un panel de 44 SARM collectés sur une période de trois ans à travers l’état de
Washington aux Etats-Unis à l’aide de plusieurs méthodes dont le séquençage du génome
complet. A la différence des techniques de typage moléculaires, le séquençage du génome
complet a établi le caractère unique de chacune des souches, suggérant que les autres
méthodes comportent le risque d’identifier par erreur des épidémies (SenGupta et al., 2014).
Un autre exemple pour les staphylocoques à coagulase négative Staphylococcus
haemolyticus une cause émergente d'infections nosocomiales, affectant principalement les
patients immunodéprimés, une analyse génomique comparative a été effectuée sur des isolats
cliniques de S. haemolyticus pour étudier leur relation génétique et explorer les séquences
génomiques en ce qui concerne les déterminants de la résistance aux antimicrobiens et
l'adaptation hospitalière (Cavanagh et al., 2014).
Les analyses génomiques réalisées pendant ma thèse avec le génome draft de S.capitis CR01
(dans un premier temps), le génome fermé de la même souche, trois génomes des souches
NRCS-A (CR03, CR04, CR05), les six génomes non-NRCS-A S. capitis et les autres
staphylocoques disponible dans les bases de données ont montré une corrélation entre les
caractéristique microbiologique et typage moléculaire (Chapitre IV).
Nous avons identifié six gènes de résistance aux antimicrobiens, tous portés par des éléments
génétiques mobiles. Les gènes de virulence ont montré qu'aucun des gènes connus de la
virulence staphylococcique n'est exclusivement porté par le clone NRCS-A. Fait intéressant,
le virulome du génome NRCS-A S. capitis était assez similaire à celui de S. epidermidis
RP62A, y compris les opérones icaABDCR et capABC liés au biofilm, les protéases Clp et les
copies multiples de Phenol Soluble Modulin (PSM) bêta type 1b en tandem (75% aa Identité
avec PSMbeta1b de la souche RP62) et un PSM alpha (59% aa identité) (Otto, 2012). Tous

CHAPITRE VI
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 160
les PSM ont été trouvés dans le même environnement génétique que dans le génome RP 62A
de S. epidermidis, avec la conservation des gènes en amont et en aval. Les présents dans les
génomes NRCS-A sont partagés avec les six génomes non-NRCS-A S. capitis. Dans
l'ensemble, 63 gènes sont spécifiques à la lignée NRCS-A, y compris 28 gènes situés dans la
cassette à résistance à la méticilline SCCmec. Parmi les 35 gènes restants, 25 sont de fonction
inconnue et 9 correspondent à un système de modification de restriction de type I
supplémentaire (n = 3), un opéron de méthylation de cytosine (n = 2) et un groupe de gènes
liés à la biosynthèse de teichoic Acides (n = 4). Fait intéressant, un dixième gène correspond à
un déterminant de la résistance à la nisine (gène nsr), une bactériocine sécrétée par des
concurrents de niche de souche NRCS-A potentiels dans le microbiote intestinal. Les
caractéristiques génomiques présentées ici soulignent la contribution des éléments génétiques
mobiles à l'émergence d'une résistance multiple aux antibiotiques dans le clone NRCS-A.
Aucun déterminant de virulence connu spécifique de NRCS-A n'a été détecté, ce qui ne
supporte pas un rôle de virulence en tant que force motrice de l'émergence de NRCS-A dans
les NICU dans le monde entier. Cependant, la présence d'un déterminant de la résistance à la
nisine sur le chromosome NRCS-A, mais pas dans d'autres souches de S. capitis et la plupart
des représentants coagulase négatifs, pourrait conférer un avantage concurrentiel aux souches
NRCS-A pendant les premières étapes de la colonisation intestinale chez les nouveau-nés.
Cela suggère que l'adaptation frappante de NRCS-A à l'environnement NICU pourrait être
liée à sa résistance antimicrobienne spécifique et à une capacité possible accrue de défier les
bactéries concurrentes dans sa niche écologique.
Plusieurs gènes associés à la résistance aux antibiotiques ont été identifiés et corrélés avec le
phénotype de résistance spécifique précédemment rapporté pour le clone NRCS-A (Rasigade
et al., 2012). Tous sont situés sur des éléments génétiques mobiles (MGE). La résistance aux
aminoglycosides est liée à l'aminoglycosidéodification bifonctionnelle gène aacA-aphD, qui
est porté sur le transposon Tn4001 (numéro d'accès GenBank AB682805.1, Lyon et al.,
1984). La résistance à la méthicilline est due à la présence d'une cassette composite SCCmec-
SCCcad / ars / cop exclusivement présente dans le clone NRCS-A, publiée par notre équipe
(Chapitre IV). La comparaison génomique entre les quatre souches NRCS-A a montré que
cet élément mobile est presque identique (100% homologie) et quelques variations de
séquence nucléotidique ne se produisent que dans la région répétée CRISPR, comme cela est
prévu pour ces régions et comme indiqué dans le chapitre IV. Enfin, l'opéron bla, codant

CHAPITRE VI
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 161
pour la bêta-lactamase blaZ et ses régulateurs, est présent dans toutes les souches NRCS-A, à
l'exception de la souche CR05 (UK).
Les analyses bioinformatiques ont montré un prophage intacte a été identifiée dans les quatre
génomes NRCS-A. Cette région montre une homologie (100% d'identité) avec les génomes
des souches CR01, CR03 et CR04, mais elle est dégénérée dans CR05. En outre, ce phage
présente une forte homologie à un phage présent dans la souche VCU116 et moins de 30%
d'homologie avec le phage Staphy_StB20_ (NCBI Ref. Seq. NC_019915), le phage le plus
proche trouvé dans la base de données NCBI. Il est à noter que deux régions de prophage
incomplètes supplémentaires ont également été détectées dans la souche CR05.
Dix-neuf éléments de séquence d'insertion distincte (IS), le regroupement en trois familles
distinctes (type IS256, n = 4; type IS1272, n = 5; IS431mec, n = 10) ont été identifiés dans le
génome CR01. La présence du type IS256 a été confirmée dans les quatre souches NRCS-A.
Le type IS1272 était également trouvé dans les souches CR03 et CR04, mais pas dans la
souche CR05, et leurs emplacements génomiques ne sont pas conservés. Deux cassettes
IS431mec sont présentes dans tous les génomes NRCS-A, car ils sont transportés dans
l'élément SCCmec-SCCcad / ars / cop spécifique à et conservé parmi la lignée NRCS-A
(Chapitre IV). Les IS trouvé chez les 3 souches de S. capitis CR03, CRO4 et CR05 sont
moins car la technologie SMART a corrigé les erreurs de pyroséquençage (homoplymère).
En ce qui concerne les autres familles antimicrobiennes, pour lesquelles la variabilité des
profils de résistance a été observée chez les isolats NRCS-A, les tests de sensibilité aux
antimicrobiens phénotypiques correspondent au contenu spécifique du gène de résistance de
chaque souche NRCS-A. Les gènes plasmidic msrA et tetK, impliqués respectivement dans la
résistance à l'érythromycine (avec un test D négatif) et la résistance à la tetracycline, n'ont été
identifiés que dans la souche CR04 (Australie). Dans la souche CR05 (UK), le gène lointain,
qui est responsable de la fusion Résistance à l'acide, est porté par un phage putatif.
Le nisine, un peptide antimicrobien de 34 aa produit par un groupe d'espèces Lactococcus et
Streptococcus Gram-positives (Shin et al., 2016), est actif contre une large gamme de
bactéries gram-positives, y compris les staphylocoques. Son mode d'action est double,
impliquant l'inhibition de la biosynthèse de la paroi cellulaire (étape de transglycosylation),
puisqu'elle se lie et sèche le lipide II à partir de son emplacement fonctionnel et la formation
des pores (Wiedemann et al., 2001; Peschel and Sahl, 2006; Egan et al., 2016). Le Nisin a été
décrit comme un acteur clé de la barrière intestinale, et il est largement utilisé comme

CHAPITRE VI
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 162
conservateur d'aliments en raison de son activité bactéricide puissante. De plus, il a été montré
que l'expression du gène nsr, identifié dans Streptococcus agalactiae, dans L. lactis ne
produisant pas de nisine induit une résistance à la nisine et la présence de bactéries lactiques à
base de nisine dans l'intestin réduit colonisation intestinale par des entérocoques résistant à la
vancomycine (Millette et al., 2008).
L’ensemble de ces données et nos résultats suggèrent fortement que la résistance à la nisine
pourrait être responsable de l'augmentation potentielle de la capacité du clone NRCS-A à
s'établir dans le cadre de la microflore initiale des nouveau-nés, dans laquelle le genre
Lactococcus est l'un des taxons bactériens dominants (Park et al., 2005; Morelli, 2008).
L'expression de la peptidase NSR pourrait permettre aux isolats NRCS-A de s'établir dans
l'intestin des nouveau-nés et aussi de le coloniser et de survivre plus longtemps que les
bactéries sensibles à la nisine. Cela peut expliquer la surreprésentation de ces isolats dans les
processus de séquesse, soit par translocation directe dans le sang (Taft et al., 2015), soit par la
colonisation de dispositifs d'habitation.
Dans cette étude, nous avons cherché à explorer la possibilité de la distribution mondiale du
clone NRCS-A en appliquant diverses méthodes moléculaires (PFGE Sma et SacII, typage
SCCmec, MLST, Dru typing et analyse phylogénétique des génomes complet après WGS)
d’une part, et à savoir la capacité spécifique in vitro de ce clone NRCS-A à acquérir
l’hétérorésistance à la vancomycine sous pression sélective d’autre part.
Dans le cadre de l’étude de sensibilité diminuée du clone NRCS-A aux glycopeptides, des
travaux d’analyse comparative sur les deux souches de S.capitis CR06, et CR08 a permis de
montrer que le clone NRCS-A était capable de s’adapter rapidement sous pression de
sélection par la vancomycine in vitro. Cette acquisition de résistance à la vancomycine était
associée à une résistance croisée avec la teicoplanine et la daptomycine mais pas le linézolide,
et s’accompagnait d’une augmentation de l’épaisseur de la paroi bactérienne des souches
devenues résistantes.
Le traitement de la vancomycine est institué uniquement si une souche de CoNS isolée
d'hémoculture résistante à l'oxacilline et que la détérioration clinique du patient ne peut être
expliquée autrement. L'utilisation fréquente de vancomycine dans l'NICU pourrait avoir
donné à la souche hétérorésistante un avantage sélectif par rapport aux autres souches non-
résistantes (D'Angio et al., 1989 ; Van Der Zwet et al., 2002). L'étude de la vancomycine a été
décrite dans S. aureus, et il a été démontré qu'elle peut être la prédécesseur de

CHAPITRE VI
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 163
l'homorésistance (Hiramatsu et al., 1997). Le mécanisme de résistance à la vancomycine dans
les staphylocoques est complètement différent de celui des entérocoques résistant à la
vancomycine. Les gènes vanA, vanB et vanC n'ont jamais été trouvés dans les
staphylocoques, bien que, expérimentalement, le gène vanA ait été transféré des entérocoques
à S. aureus (Noble et al., 1992). Bien que le mécanisme génétique précis pour la résistance à
la vancomycine dans les staphylocoques attend l'élucidation, on considère que
l'épaississement de la couche de peptidoglycane de la paroi cellulaire est responsable (Hanki
et al., 1998 ; Sieradzki et al., 1997). La vancomycine est capturée dans la paroi cellulaire
épaissie et il est empêché d'atteindre ses cibles sur la membrane cellulaire.
Nous avons effectué un séquençage de génome entier de deux de souches de capitis NRCS-A
CR06 et CR08 inclues dans le modèle d’acquisition de résistance (souche initiale comparée
avec souches devenue résistante sous pression de sélection). La recherche de mutations
ponctuelles dans des gènes connus comme étant associés à la résistance aux glycopeptides
chez S. aureus (Howden et al., 2011), n’a pas permis de mettre en évidence ce type de
mutation chez S. capitis. En revanche, nous avons de façon surprenante mis en évidence une
délétion d’une fraction importante du génome de la bactérie (110 kb, soit une centaine de
gènes concernés), identique dans nos deux souches. L’analyse des génomes entiers de notre
collection internationale de souches cliniques de S. capitis, confrontée aux CMI de
vancomycine de ces mêmes souches, permettra peut-être de mettre en évidence les bases
génétiques du phénotype de résistance/hétérorésistance à la vancomycine chez NRCS-A.
En parallèle, au sein du CNR la technique de dru typing, qui consiste en la caractérisation
d’une zone hypervariable au sein de la cassette SCCmec et qui permet d’étudier la proximité
génétique de souches au sein d’un même clone. Sur 86 souches de S. capitis isolées chez des
nouveau-nés de 4 pays différents, 83 (96%) présentaient le même dru type dt11c. Ce dru type
dt11c n’était en revanche présent que chez 2 des 14 souches isolées de patients hors
réanimation néonatale (1 patient de 4 ans et 1 adulte), et les 12 souches restantes présentaient
une grande variété de dru types. En outre, un arbre phylogénétique a été construit sur la base
des génomes complets de cinq isolats de SNN. Cette analyse phylogénétique de ces cinq
isolats des différents pays preuve la haute parenté génétique dans le clone NRCS-A
La technique de MLST est reconnue comme une puissante méthode pour l’étude génétique
des populations bactériennes. Elle correspond à l’amplification et au séquençage de plusieurs
gènes dits « de ménage », dont la séquence comporte des fragments hautement conservés au

CHAPITRE VI
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 164
sein de l’espèce et des fragments variables. Les séquences obtenues sont comparées avec une
banque d’allèles établie au préalable, afin de déterminer le profil allélique unique ou «
Sequence Type » (ST) de chaque souche. Les avantages de cette technique sont la bonne
reproductibilité et la possibilité de comparer les souches entre les laboratoires (Maiden et al.,
1998 ; Urwin et al., 2003).
Ces différentes techniques ont permis de confirmer la diffusion mondiale d’un clone unique
de S. capitis, spécifiquement en réanimation néonatale. Cette observation constitue la
première description d’une dissémination internationale d’un clone de staphylocoque à
coagulase négative multi-résistant, phénomène jusque là décrit et connu uniquement pour
l’espèce S. aureus.
Ensemble, ces données démontrent de façon incontestable l'endémicité inhabituelle à l'échelle
mondiale du clone NRCS-A résistant aux médicaments multiples dans les NICU. Une fois
endémique dans une NICU, les souches NRCS-A exposent les nouveau-nés infectés à un
risque d'échec thérapeutique car le traitement de la septicémie néonatale impliquant des
staphylocoques à coagulase négative résistant à la méthicilline est habituellement basé sur la
vancomycine et les aminoglycosides auxquels les isolats NRCS-A ne sont pas susceptibles.
Dans de tels cas, les praticiens sont confrontés à des alternatives inconfortables: l'utilisation
continue de la vancomycine malgré le fait que le pathogène a été déclaré sans ambiguïté
comme étant résistant à cette molécule et l'utilisation d'agents antimicrobiens tels que le
linezolid ou la daptomycine qui conservent une efficacité in vitro contre CoNS, L'utilisation
chez les nouveau-nés n'a pas été approuvée par les autorités sanitaires. Pour faire face à ce
défi émergent, des études cliniques sur la tolérance et l'efficacité relatives de la vancomycine,
du linézolide et de la daptomycine chez les nourrissons infectés par l'infection à l'urine atteints
de ce nouveau syndrome de CoNS résistant à la vancomycine ont été urgentes.
Bien qu'il reste inconnu de savoir comment les souches de S. capitis NRCS-A sont capables
de translocation dans le flux sanguin des nourrissons, on suppose que le point d'entrée
pourrait être le tractus digestif, immature chez les nourrissons très prématurés (Taft et al.,
2015) . D'autres études sont nécessaires pour caractériser pleinement la microflore digestive
des prématurés à très faible poids et son impact potentiel sur le choix et l'avantage de forme
physique pour les isolats NRCS-A. Des études sur le niveau de portage de S. capitis NRCS-A
chez le nouveau-né sont en cours notamment au niveau digestif ainsi que des travaux

CHAPITRE VI
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 165
(modèles cellulaires ex vivo) explorant les mécanismes physiopathologiques à l’origine du
déclenchement des sepsis tardifs nosocomiaux.

REFERENCES
BIBLIOGRAPHIQUES

REFERENCES BIBLIOGRAPHIQUES
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 166
Ahn, S.M., Kim, T.H., Lee, S., Kim, D., Ghang, H., Kim, D.S., Kim, B.C., Kim, S.Y.,
Kim, W.Y., Kim, C. et al. (2009) The first Korean genome sequence and analysis: full
genome sequencing for a socio-ethnic group. Genome Res, 19, 1622-1629.
Altermann, E. (2012) Tracing Lifestyle Adaptation in Prokaryotic Genomes. Front.
Microbiol. 3.:48.
Anderson, S. (1981) Shotgun DNA sequencing using cloned DNase I-generated
fragments.Nucleic Acids Res. 9: 3015–3027.
Ahmadian A, Ehn M & Hober S. (2006). Pyrosequencing: history, biochemistry and future.
Clin Chim Acta. 363: 83-94.
Amaro, A., Duarte, E., Amado, A., Ferronha, H., Botelho, A., (2008) Comparison of three
DNA extraction methods for Mycobacterium bovis, Mycobacterium tuberculosis and
Mycobacterium avium subsp. avium. Lett. Appl. Microbiol. 47, 8-11.
Ameziane N, Bogard M, Lamoril J. (2005). Principes de biologie moléculaire en biologie
clinique. Collection Campus Référence Elsevier;
Altschul, S. F., Madden, T.L., Schaffer, A.A., Ahang, Z., Miller, W., et lipman, D.J.
(1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search
programs Nucleic Acids Res 25, 3389-3402.
Artur J. Sabat, Sandra M. Hermelijn, Viktoria Akkerboom, Amadu Juliana, John E.
Degener, Hajo Grundmann & Alexander W. Friedrich. (2017) Complete-genome
sequencing elucidates outbreak dynamics of CA-MRSA USA300 (ST8 spa t008) in an
academic hospital of Paramaribo, Republic of Suriname. Scientific Reports 7,
Article number: 41050
Aubry-Damon H, Legrand P, Brun-Buisson C, Astier A, Soussy CJ, Leclercq R. (1997)
Reemergence of gentamicin-susceptible strains of methicillin-resistant Staphylococcus
aureus. roles of an infection control program and changes in aminoglycoside use. Clin Infect
Dis, 25 (3) : 647-53.
Avery, O. T., Macleod, C. M., et McCarty, M. (1944) Studies on the chemical of inducing
transformation of pneumoccal types. Induction of transformation by a desoxyriboncleic acid
fraction isolated from Pneumococcus type III. 1944. Journal of Experimental Medcine 79,
137-158.
Azarian, T., Cook, R., Johnson, J., Guzman, N., McCarter, Y., Gomez, N., Salemi, M. (2015). Whole-Genome Sequencing for Outbreak Investigations of Methicillin-Resistant
Staphylococcus aureus in the Neonatal Intensive Care Unit: Time for Routine Practice?
Infection Control &Hospital Epidemiology, 36(7), 777-785.
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S,
Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil
LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O,
Vonstein V, Wilke A, Zagnitko O. 2008. The RAST server: Rapid Annotations using
Subsystems Technology. BMC Genomics 9:75.
Baird-Parker, A.C. (1971). The grouping of staphylococci and micrococci. J Clin Pathol.
24, 769-770.
Bannerman T.L., Kloos W.E. (1991) Staphylococcus capitis subsp. ureolyticus subsp. nov.
from human skin, Int. J. Syst. Bacterial. 41: 144-147.

REFERENCES BIBLIOGRAPHIQUES
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 167
Bascomb, S. & Manafi, M. (1998) Use of enzyme tests in characterization and identification
of aerobic and facultative anaerobic Gram-positive cocci. Clin. Microbiol. Rev. 11, 318-340.
Bentley, D.R., Balasubramanian, S., Swerdlow, H.P., Smith, G.P., Milton, J., Brown,
C.G., Hall, K.P., Evers, D.J., Barnes, C.L., Bignell, H.R. et al. (2008) Accurate whole
human genome sequencing using reversible terminator chemistry. Nature, 456, 53-59.
Bentley, D.R. (2006) Whole-genome re-sequencing. Current opinion in genetics &
development, 16, 545-52, 10.1016/j.gde.2006.10.009.
Bertrand X, Costa Y, Pina P. [Surveillance of antimicrobial resistance of bacteria isolated
from bloodstream infections : data of the French National Observatory for Epidemiology of
Bacterial Resistance to Antibiotics (ONERBA), 1998-2003]. Med Mal Infect, 2005, 35 (6) :
329-34.
Berglund EC, Kiialainen A, Syvanen AC. (2011). Next generation sequencing technologies
and applications for human Genetic History and Forensics. Investigative Genetics, 2:23.
Bhattacharyya A, Stilwagen S, Ivanova N, D'Souza M, Bernal A, Lykidis A, Kapatral
V, Anderson I, Larsen N, Los T, Reznik G, Selkov E Jr, Walunas TL, Feil H, Feil
WS, Purcell A, Lassez JL, Hawkins TL, Haselkorn R, Overbeek R, Predki PF, Kyrpides
NC. (2003) Comparison of the genome sequences of Listeria monocytogenes and Listeria
innocua: clues for evolution and pathogenicity,” FEMS Immunol Med Microbiol., vol. 35, no.
3, pp. 207–13.
Bianco C, Arena F, Rossetti B, Tordini G, Migliorini L, Galluzzi P, Cerase A, De Luca
A, Rossolini GM, Montagnani F. (2014) First report of spondylodiscitis due to vancomycin
heteroresistant Staphylococcus capitis in immunocompetent host. J Infect Chemother
20(10):639–642.
Blow. (2008). DNA sequencing: generation next-next. Nat Methods 5:267—74.
Boetzer, M. and Pirovano, W. (2012) Toward almost closed genomes with GapFiller.
Genome Biol. 13: R56.
Boghossian NS, Page GP, Bell EF, et al. (2013) Late-onset sepsis in very low birth weight
infants from singleton and multiple-gestation births. J Pediatr. 162(6):1120–1124. 1124–e1.
Brochier, C., Forterre, P., and Gribaldo, S. (2004) Archaeal phylogeny based on proteins
of the transcription and translation machineries: tackling the Methanopyrus kandleri paradox.
Genome Biol. 5: R17.
Bronner S, Monteil H, Prevost G. (2004) Regulation of virulence determinants in
Staphylococcus aureus: complexity and applications. FEMS Microbiol Rev 28, 183-200.
Bryson K, Loux V, Bossy R, Nicolas P, Chaillou S, van de Guchte M, Penaud S, Maguin
E, Hoebeke M, Bessières P, and Gibrat JF. (2006) AGMIAL: implementing an annotation
strategy for prokaryotegenomes as a distributed system. Nucleic Acids Research. Jan vol. 34,
no. 12, pp. 3533–45.
Bush K, Courvalin P, Dantas G, et al. (2011) Tackling antibiotic resistance. Nat Rev
Microbiol, 9:894-6.
Buchrieser C, Rusniok C, Kunst F, Cossart P, Glaser P, and Consortium L. (2003)
Comparison of the genome sequences of Listeria monocytogenes and Listeria innocua: clues
for evolution and pathogenicity,” FEMS Immunol Med Microbiol., vol. 35, no. 3, pp. 207–13.

REFERENCES BIBLIOGRAPHIQUES
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 168
Cavanagh JP, Hjerde E, Holden MTG, Kahlke T, Klingenberg C, Flaegstad T, Parkhill
J, Bentley SD, Ericson Sollid JU. (2014) Whole-genome sequencing reveals clonal
expansion of multiresistant Staphylococcus heamolyticus in European hospitals. J Antimicrob
Chemother. 69:2920-7.
Chin, C.-S., Alexander, D.H., Marks, P., Klammer, A.A., Drake, J., Heiner, C., et al. (2013) Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing
data. Nat. Methods 10: 563–569.
Chiu, R.W., Chan, K.C., Gao, Y., Lau, V.Y., Zheng, W., Leung, T.Y., Foo, C.H., Xie, B.,
Tsui, N.B., Lun, F.M. et al. (2008) Noninvasive prenatal diagnosis of fetal chromosomal
aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma. Proc Natl
Acad Sci U S A, 105, 20458-20463.
Cissé OH, Pagni M, Hauser PM. (2012) De novo assembly of the Pneumocystis jirovecii
genome from a single bronchoalveolar lavage fluid specimen from a patient. MBio.
4(1):e00428–00412.
Cochrane, G., Akhtar, R., Boniefld, J., Bower, L., Demiralp, F., Faruque, N., Gibson, R.,
Hoad, G., Hubbard, T., Hunter, C., et al. (2009a) Petabyte-scale innovations at the
European Nucleotide Archive. Nucleic Acids Res 37, D19-25.
Combes A, Luyt CE, Fagon JY, et al. (2004).Impact of methicillin resistance on outcome of
Staphylococcus aureus ventilator-associated pneumonia. Am J Respir Crit Care Med, 170 (7)
: 786-92.
Conlan S, Thomas PJ, Deming C, Park M, Lau AF, et al. (2014) Single-molecule
sequencing to track plasmid diversity of hospital-associated carbapenemase-producing
Enterobacteriaceae. Sci Transl Med 6: 254ra126.
Cui B, Smooker PM, Rouch DA, Daley AJ, Deighton MA. (2013) Differences between two
clinical Staphylococcus capitis subspecies as revealed by biofilm, antibiotic resistance, and
pulsed-field gel electrophoresis profiling. J Clin Microbiol. 51(1):9–14.
Darling A. E., Mau B., Perna N. T. (2010). progressiveMauve: multiple genome alignment
with gene gain, loss and rearrangement. PLoS ONE 5:e11147. 10.1371/journal.pone.0011147.
de Souza Rugolo LM, Bentlin MR, Mussi-Pinhata M, de AlmeidaMF, Lopes JM, Marba
ST, et al. (2014) Late-onset sepsis in very low birthweight infants: a Brazilian Neonatal
Research Network Study. JTrop Pediatr. 60:415---21.
D'Angio, C. T., K. L. McGowan, S. Baumgart, J. St. Geme, and M. C. Harris. 1989.
Surface colonization with coagulase-negative staphylococci in premature neonates. J.
Pediatr. 114:1029-1034.
Delhaes L, Botterel F. Que peut-on attendre du microbiote pulmonaire notamment du
mycobiome dans les pathologies pulmonaires chroniques telles que la mucoviscidose ?
Congrès de la Société Française de Mycologie Médicale - Dijon, France; 2013.
Deshpande P, Rodrigues C, Shetty A, Kapadia F, Hedge A, Soman R. (2010) New Delhi
Metallo-β-lactamase (NDM-1) in Enterobacteriaceae: treatment options with carbapenems
compromised. JAssoc Physicans I 58: 14 –149.
Diene SM, Rolain JM. (2013) Investigation of antibiotic resistance in the genomic era of
multidrug-resistant Gram-negative bacilli, especially Enterobacteriaceae, Pseudomonas and
Acinetobacter. Expert Rev Anti Infect Ther. 11:277-96.

REFERENCES BIBLIOGRAPHIQUES
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 169
D’Mello D, Daley AJ, Rahman MS, Qu Y, Garland S, Pearce C, Deighton MA. 2008.
Vancomycin heteroresistance in bloodstream isolates of Staphylococcus capitis. J. Clin.
Microbiol. 46:3124 –3126.
Dufour P, Gillet Y, Bes M, et al. (2002) Community-acquired methicillin-resistant
Staphylococcus aureus infections in France : emergence of a single clone that produces
Panton-Valentine leukocidin. Clin Infect Dis, 35 (7) : 819-24.
Dudley DM, Chin EN, Bimber BN, Sanabani SS, Tarosso LF, Costa PR, et al. (2012)
Low-cost ultra-wide genotyping using Roche/454 pyrosequencing for surveillance of HIV
drug resistance. Plos One. 7(5):e36494.
Egan K., Field D., Rea M. C., Ross R. P., Hill C., Cotter P. D. (2016). Bacteriocins: novel
solutions to age old spore-related problems? Front. Microbiol. 7:e461.
10.3389/fmicb.2016.00461.
Eid, J., Fehr, A., Gray, J., Luong, K., Lyle, J., Otto, G., et al. (2009) Real-Time DNA
Sequencing from Single Polymerase Molecules. Science 323: 133–138.
Ekblom, R. and Wolf, J.B.W. (2014) A field guide to whole-genome sequencing, assembly
and annotation. Evol. Appl. 7: 1026–1042.
Farhat MR, Shapiro BJ, Kieser KJ, Sultana R, Jacobson KR, et al. (2013) Genomic
analysis identifies targets of convergent positive selection in drug-resistant Mycobacterium
tuberculosis. Nat Genet 45: 1183–1189.
Fedurco M, Romieu A, Williams S, Lawrence I & Turcatti G. (2006) BTA, a novel
reagent forDNA attachment on glass and efficient generation of solid-phase amplified DNA
colonies.Nucleic Acids Res. 34: 22.
Ferry T, Perpoint T, Vandenesch F, Etienne J. (2005) Virulence determinants in
Staphylococcus aureus and their involvement in clinical syndromes. Curr Infect Dis Rep, 7
(6) : 420-8.
Ferry T, Bes M, Dauwalder O, et al. (2006) Toxin gene content of the Lyon methicillin-
resistant Staphylococcus aureus clone compared with that of other pandemic clones. J Clin
Microbiol, 44 (7) : 2642-4.
Fleischmann, R.D., Adams, M.D., White, O., Clayton, R.A., Kirkness, E.F., Kerlavage,
A.R., Bult, C.J., Tomb, J.-F., Dougherty, B.A., Merrick, J.M., et al. (1995) Whole-
genome random sequencing and assembly of Haemophilus influenzae Rd. Science, 269, 496-
512.
Fonseca SN, Ehrenkranz RA, Baltimore RS. (1994) Epidemiology of antibiotic use in a
neonatal intensive care unit. Infect Control Hosp Epidemiol. 15(3):156–62. 12.
Fournier PE, Vallenet D, Barbe V, et al. (2006) Comparative genomics of multidrug
resistance in Acinetobacter baumannii. PLoS Genet 2:e7.
Froggatt, J. W., J. L. Johnston, D. W. Galetto, and G. L. Archer. 1989. Antimicrobial
resistance in nosocomial isolates of Staphylococcus haemolyticus. Antimicrob. Agents
Chemother. 33:460-466.
Gram H (1884) Über die isolirte Färbung der Schizomyceten in Schnitt- und
Trockenpräparaten. Fortschritte der Medizin 2.
Gras-Le Guen C, Fournier S, Andre-Richet B, Caillon J, Chamoux C, et al. (2007)
Almond oil implicated in a Staphylococcus capitis outbreak in a neonatal intensive care unit. J
Perinatol 27: 713-717.

REFERENCES BIBLIOGRAPHIQUES
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 170
Greub G. (2013) Genomics of medical importance. Clin Microbiol Infect. 19:781-3.
Gaynor, A.M., Nissen, M.D., Whiley, D.M., Mackay, I.M., Lambert, S.B., Wu, G.,
Brennan, D.C., Storch, G.A., Sloots, T.P. and Wang, D. (2007) Identification of a novel
polyomavirus from patients with acute respiratory tract infections. PLoS Pathog, 3, e64.
Gill,S.R., Fouts,D.E., Archer,G.L., Mongodin,E.F., Deboy, R.T., Ravel, J., et al.
(2005).Insights on evolution of virulence and resistance from the complete genome analysis
of anearly methicillin-resistant Staphylococcus aureus strain and a biofilm-producing
methicillin-resistant Staphylococcus epidermidis strain. J.Bacteriol. 187,2426–2438.
Gonzalez BE, Rueda AM, Shelburne SA, 3rd, Musher DM, Hamill RJ, Hulten KG. (2006). Community-associated strains of methicillin-resistant Staphylococccus aureus as the
cause of healthcare-associated infection. Infect Control Hosp Epidemiol, 27 (10) : 1051-6.
Gupta PK. (2008) Single-molecule DNA sequencing technologies for future genomics
research. Trends Biotechnol. 26: 602-611.
Heilbronner,S., Holden,M.T., vanTonder,A., Geoghegan,J.A., Foster,T.J., Parkhill,J., et
al.(2011).Genome sequence of Staphylococcus lugdunensis N920143 allows identification of
putative colonization and virulence factors. FEMSMicrobiol.Lett. 322,60–67.
Hanaki H, Kuwahara-Arai K, Boyle-Vavra S, Daum RS, Labischinski H, Hiramatsu K.
(1998). Activated cell-wall synthesis is associated with vancomycin resistance in methicillin-
resistant Staphylococcus aureus clinical strains Mu3 and Mu50. J Antimicrob Chemother, , 42
(2) : 199-209.
Hanaki H, Labischinski H, Inaba Y, Kondo N, Murakami H, Hiramatsu K. (1998)
Increase in glutamine-non-amidated muropeptides in the peptidoglycan of vancomycin-
resistant Staphylococcus aureus strain Mu50. J Antimicrob Chemother, 42 (3) : 315-20.
Hanssen AM, Sollid JU. (2007). Multiple staphylococcal cassette chromosomes and allelic
variants of cassette chromosome recombinases in Staphylococcus aureus and coagulase-
negative staphylococci from Norway. Antimicrob Agents Chemother. May;51(5):1671-7.
Harris, T.D., Buzby, P.R., Babcock, H., Beer, E., Bowers, J., Braslavsky, I., et al. (2008)
Single-Molecule DNA Sequencing of a Viral Genome. Science 320: 106–109.
Harris SR, Feil EJ, Holden MT, Quail MA, Nickerson EK, et al. (2010) Evolution of
MRSA during hospital transmission and intercontinental spread. Science 327: 469–474
Higuchi W, Takano T, Teng LJ, Yamamoto T. (2008). Structure and specific detection of
staphylococcal cassette chromosome mec type VII. Biochem Biophys Res Commun. Dec
19;377(3):752-6.
Hiramatsu K, Katayama Y, Yuzawa H, Ito T. (2002) Molecular genetics of methicillin-
resistant Staphylococcus aureus. Int J Med Microbiol. Jul;292(2):67-74.
Hiramatsu, K., N. Aritaka, H. Hanaki, S. Kawasaki, Y. Hosoda, S. Hori, Y. Fukuchi,
and I. Kobayashi. 1997. Dissemination in Japanese hospitals of strains of Staphylococcus
aureus. heterogeneously resistant to vancomycin. Lancet 350:1670-1673.
Holden MT, Feil EJ, Lindsay JA, et al. (2004) Complete genomes of two clinical
Staphylococcus aureus strains: evidence for the rapid evolution of virulence and drug
resistance. Proc Natl Acad Sci USA 101, 9786-9791.
Holden MT, Hsu LY, Kurt K, Weinert LA, Mather AE, Harris SR, Strommenger
B, Layer F, Witte W, de Lencastre H, Skov R, Westh H, Zemlicková H, Coombs
G, Kearns AM, Hill RL, Edgeworth J, Gould I, Gant V, Cooke J, Edwards

REFERENCES BIBLIOGRAPHIQUES
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 171
GF, McAdam PR, Templeton KE, McCann A, Zhou Z, Castillo-Ramírez S, Feil
EJ, Hudson LO, Enright MC, Balloux F, Aanensen DM, Spratt BG, Fitzgerald
JR, Parkhill J, Achtman M, Bentley SD, Nübel U. (2013) A genomic portrait of the
emergence, evolution, and global spread of a methicillin-resistant Staphylococcus aureus
pandemic. Genome Res. Apr;23(4):653-64. doi: 10.1101/gr.147710.112.
Holley, R.W., Apgar, J., Everett, G.A., Madison, J.T., Marquisee, M., Merrill, S.H.,
Penswick, J.R. and Zamir, A. (1965) Structure of a ribonucleic acid. Science, 147, 1462-5.
Holley, R.W., Everett, G.A., Madison, J.T. and Zamir, A. (1965) Nucleotide sequence in
the yeast alanine transfer ribonucleic acid. The Journal of biological chemistry, 240, 2122-8.
Howden BP, McEvoy CR, Allen DL, et al. (2011) Evolution of multidrug resistance during
Staphylococcus aureus infection involves mutation of the essential two component regulator
WalKR. PLOS Pathog.7:e1002359.
Huang Y, Wang YH, Chou YH, Lien RI. (2006) Significance of coagulase-negative
staphylococci isolated from a single blood culture from neonates in intensive care. Ann Trop
Paediatr. 26:311–318.
Huber JA, Mark Welch DB, Morrison HG, Huse SM, Neal PR, Butterfield DA, et al. (2007). Microbial population structures in the deep marine biosphere. Science 318:97-100
Hutchison, C.A. (2007) DNA sequencing: Bench to bedside and beyond. Nucleic acids
research, 35, 6227-37, 10.1093/nar/gkm688.
Hyman ED. (1988). A new method of sequencing DNA. Anal Biochem. 174: 423-436.
IHGSC (2004). Finishing the euchromatic sequence of the human genome. Nature 431, 931-
945.
International Working Group on the Classification of Staphylococcal Cassette
Chromosome Elements (IWG-SCC). 2009. Classification of staphylococcal cassette
chromosome mec (SCCmec): guidelines for reporting novel SCCmec elements. Antimicrob.
Agents Chemother. 53:4961-4967.
Imbeaud S, Graudens E, Boulanger V, Barlet X, Zaborski P, Eveno E, Mueller O,
Schroeder A, Auffray C (2005) Towards standardization of RNA quality assessment using
user-independent classifiers of microcapillary electrophoresis traces. Nucleic Acids Res 30:
e56.
Ito T, Katayama Y, Hiramatsu K. (1999). Cloning and nucleotide sequence determination
of the entire mec DNA of pre-methicillin-resistant Staphylococcus aureus N315. Antimicrob
Agents Chemother. Jun;43(6):1449-58.
Ito T, Ma XX, Takeuchi F, Okuma K, Yuzawa H, Hiramatsu K. (2004). Novel type V
staphylococcal cassette chromosome mec driven by a novel cassette chromosome
recombinase, ccrC. Antimicrob Agents Chemother. Jul;48(7):2637-51.
Jarraud S, Mougel C, Thioulouse J, et al. (2000) Relationships between Staphylococcus
aureus genetic background, virulence factors, agr groups (alleles), and human disease. Infect
Immun 70, 631-641.
Jimenez-Truque N, Tedeschi S, Saye EJ, et al. (2012) Relationship between maternal and
neonatal Staphylococcus aureus colonization. Pediatrics. 129(5):e1252–9. 13.

REFERENCES BIBLIOGRAPHIQUES
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 172
Johnson-Robbins LA, el-Mohandes AE, Simmens SJ, Keiser JF. (1996) Staphylococcus
epidermidis sepsis in the intensive care nursery: a characterization of risk associations in
infants < 1,000 g. Biol Neonate. 69(4):249–256
Jordens JZ, Duckworth GJ, Williams RJ. (1989). Production of « virulence factors » by
« epidemic» methicillin-resistant Staphylococcus aureus in vitro. J Med Microbiol, , 30 (4) :
245-52.
Juhas, M., van der Meer, J.R., Gaillard, M., Harding, R.M., Hood, D.W., and Crook,
D.W. (2009) Genomic islands: tools of bacterial horizontal gene transfer and evolution.
FEMS Microbiol. Rev. 33: 376–393.
Junemann S, Sedlazeck FJ, Prior K, Albersmeier A, John U, Kalinowski J, et al. (2013).
Updating benchtop sequencing performance comparison. Nat. Biotechnol. Apr;31(4):294–6.
Katayama, Y., T. Ito, and K. Hiramatsu. 2001. Genetic organization of the chromosome
region surrounding mecA in clinical staphylococcal strains: role of IS431-
mediated mecI deletion in expression of resistance in mecA-carrying, low-level methicillin-
resistant Staphylococcus haemolyticus. Antimicrob. Agents Chemother. 45:1955-1963.
Kenri T, Horino A, Matsui M, Sasaki Y, Suzuki S, Narita M, et al. (2012) Complete
genome sequence of Mycoplasma pneumoniae type 2a strain 309, isolated in Japan. J.
Bacteriol. Mar;194(5):1253–4.
Kim, J.I., Ju, Y.S., Park, H., Kim, S., Lee, S., Yi, J.H., Mudge, J., Miller, N.A., Hong, D.,
Bell, C.J. et al. (2009) A highly annotated whole-genome sequence of a Korean individual.
Nature, 460, 1011-1015.
Klingenberg C, Ronnestad A, Anderson AS, et al. Persistent strains of coagulase-negative
staphylococci in a neonatal intensive care unit: virulence factors and invasiveness. Clin
Microbiol Infect 2007;13:1100–11.
Kondo Y, Ito T, Ma XX, Watanabe S, Kreiswirth BN, Etienne J, et al. (2007).
Combination of multiplex PCRs for staphylococcal cassette chromosome mec type
assignment: rapid identification system for mec, ccr, and major differences in junkyard
regions. Antimicrob Agents Chemother. Jan;51(1):264-74.
Koonin, E.V., Makarova, K.S., and Aravind, L. (2001) Horizontal gene transfer in
prokaryotes: quantification and classification. Annu. Rev. Microbiol. 55: 709–742.
Kos VN, Desjardins CA, Griggs A, Cerqueira G, Van Tonder A, Holden MT, Godfrey P,
Palmer KL, Bodi K, Mongodin EF, Wortman J, Feldgarden M, Lawley T, Gill SR, Haas
BJ, Birren B, Gilmore MS. (2012 ). "Comparative genomics of vancomycin-resistant
Staphylococcus aureus strains and their positions within the clade most commonly associated
with Methicillin-resistant S. aureus hospital-acquired infection in the United States." mBio.
3(3): Epub 2012 May 22.
Koser CU, Ellington MJ, Cartwright EJ, Gillespie SH, Brown NM, et al. (2012) Routine
use of microbial whole genome sequencing in diagnostic and public health microbiology.
PLoS Pathog 8: e1002824.
Krediet TG, Jones ME, Janssen K, et al. (2011) Prevalence of molecular types and mecA
gene carriage of coagulase-negative Staphylococci in a neonatal intensive care unit: relation
to nosocomial septicemia. J Clin Microbiol, 39:3376–8.
Kuroda,M., Yamashita,A., Hirakawa,H., Kumano,M., Morikawa,K., Higashide, M., et
al. (2005). Whole genome sequence of Staphylococcus saprophyticus reveals the

REFERENCES BIBLIOGRAPHIQUES
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 173
pathogenesis of uncomplicated durinary tract infection. Proc.Natl. Acad.Sci.U.S.A.
102,13272–13277.
Lander, E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody, M.C., Baldwin, J., Devon,
K., Dewar, K., Doyle, M., FitzHugh, W., et al. (2001) Initial sequencing and analysis of the
human genome. Nature, 409, 860-921, 10.1038/35057062.
Larrat S. Did Ultra-Deep pyrosequencing of the NS3 region can help to predict sustained
virological response to pegylated interferon-ribavirine-anti-protease triple therapy in
previously treated chronic HCV genotype 1 infection? 64th annual meeting of the American
Association for the Study of Liver Diseases - Washington DC, USA; 2013.
Latreille P, Norton S, Goldman BS, Henkhaus J, Miller N, Barbazuk B, Bode HB,
Darby C, Du Z, Forst S, Gaudriault S, Goodner B, Goodrich-Blair H, and SlaterS.
(2007) Optical mapping as a routinetool for bacterial genome sequence finishing,” BMC
genomics, vol. 8, p. 321.
Lauck M, Alvarado-Mora MV, Becker EA, Bhattacharya D, Striker R, Hughes AL, et
al. (2012) Analysis of hepatitis C virus intrahost diversity across the coding region by
ultradeep pyrosequencing. J. Virol. Apr;86(7):3952–60.
Lee H, Kim ES, Choi C, et al. (2014) Outbreak among healthy newborns due to a new
variant of USA300-related meticillin-resistant Staphylococcus aureus. J Hosp Infect 87:145-
51.
Lavezzo E, Toppo S, Franchin E, et al. (2013) Genomic comparative analysis and gene
function prediction in infectious diseases: Application to the investigation of a meningitis
outbreak. BMC Infect Dis. 13:554.
Leshem E, Maayan-Metzger A, Rahav G, et al. (2012) Transmission of Staphylococcus
aureus from mothers to newborns. Pediatr Infect Dis J 31(4):360–3.
Levinthal C, Davison PF. (1961). Degradation of deoxyribonucleic acid under
hydrodynamic shearing forces. J. Mol.Bio 3, 674-683.
Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, et al. (2007). The diploid
genome sequence of an individual human. PLoS Biol 5:e254.
Lim A, Dimalanta ET, Potamousis KD, Yen G, Apodoca J, Tao C, Lin J, Qi R, Skiadas
J, Ramanathan A, Perna NT, Iii GP, Burland V, Mau B, Hackett J, Blattner FR,
Anantharaman TS, Mishra B, and Schwartz DC. (2001) Shotgun Optical Maps of the
Whole Escherichia coli O157!: H7Genome. Genome Research, pp. 1584–1593.
Lina G, Piemont Y, Godail-Gamot F, et al. (1999). Involvement of Panton-Valentine
leukocidin-producing Staphylococcus aureus in primary skin infections and pneumonia. Clin
Infect Dis, 29 (5) : 1128-32.
Lindsay J.A and Holden M.T. (2004) Staphylococcus aureus: superbug, super genome.
Trends Microbiol. 12: 378-385.
Loman NJ, Misra RV, Dallman TJ, Constantinidou C, Gharbia SE, Wain J, et al. (2012).Performance comparison of benchtop high-throughput sequencing platforms. Nat.
Biotechnol. May;30(5):434–9.
Lowy FD. (1998) Staphylococcus aureus infections. N Engl J Med, 339 (8) : 520- 32.
Ludwig W, Strunk O, Westram R, et al. (2004). ARB: a software environment for sequence
data. Nucleic Acids Res. 32: 1363-1371.

REFERENCES BIBLIOGRAPHIQUES
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 174
Margulies, M., Egholm, M., Altman, W.E., Attiya, S., Bader, J.S., Bemben, L.A., Berka,
J., Braverman, M.S., Chen, Y.-ju, Chen, Z., et al. (2005) Genome sequencing in
microfabricated high-density picolitre reactors. Nature, 437, 376-81, 10.1038/nature03959.
Maxam, A.M. and Gilbert, W. (1977) A new method for sequencing DNA. Proceedings of
the National Academy of Sciences of the United States of America, 74, 560-4.
McAdam P, Richardson EJ, Fiztgerald JR. (2014) High throughput sequencing for the
study of bacterial pathogen biology. Curr Opin Microbiol. 19:106-13
McGinn S, Gut IG. (2013). DNA sequencing - spanning the generations. New Biotechnol.
May 25;30(4):366–72.
Medini D, Donati C, Tettelin H, Masignani V, and Rappuoli R. (2005) The microbial pan-
genome. Current opinion in genetics & development. Dec vol. 15, no. 6, pp. 589–94.
Maiden MC, Bygraves JA, Feil E, et al. (March 1998). "Multilocus sequence typing: A
portable approach to the identification of clones within populations of pathogenic
microorganisms". Proc. Natl. Acad. Sci. U.S.A. 95 (6): 3140–5
Mellmann, A., Harmsen, D., Cummings, C.A., Zentz, E.B., Leopold, S.R., Rico, A.,
Prior, K., Szczepanowski, R., Ji, Y., Zhang, W. et al. (2011) Prospective genomic
characterization of the German enterohemorrhagic Escherichia coli 0104:H4 outbreak by
rapid next generation sequencing technology. PLoS One, 6, e22751.
Metzker, M.L. (2010) Sequencing technologies - the next generation. Nat Rev Genet, 11, 31-
46.
Miller, J.R., Koren, S., and Sutton, G. (2010) Assembly algorithms for next-generation
sequencing data. Genomics 95: 315–327.
Millette M., Cornut G., Dupont C., Shareck F., Archambault D., Lacroix M.
(2008). Capacity of human nisin- and pediocin-producing lactic Acid bacteria to reduce
intestinal colonization by vancomycin-resistant enterococci. Appl. Environ. Microbiol. 74,
1997–2003. 10.1128/AEM.02150-07.
Mishaan AM, Mason EO, Jr, Martinez-Aguilar G, et al. (2005) Emergence of a
predominant clone of community-acquired Staphylococcus aureus among children in
Houston, Texas. Pediatr Infect Dis J, 24 (3) : 201-6.
Monira S, Nakamura S, Gotoh K, Izutsu K, Watanabe H, Alam NH, et al. (2011) Gut
microbiota of healthy and malnourished children in bangladesh. Front. Microbiol. 2:228.
Mullis, K. (1994) The Polymeras chain reaction. (Boston,birkhaïsur). Pearson, W. R., et
Lipman, D.J. (1998). Improved tools for biological sequence comparaison. Proc. Nati. Acad.
Sci. U.S.A 85, 2444-2448.
Nan Qin, Wenchao Dingb, Jian Yao, Kunkai Su
a, Lingjiao Wu and Lanjuan Li. (2012).
Genome Sequence of Staphylococcus capitis QN1, Which Causes Infective Endocarditis.
10.1128/JB.00827-12 J. Bacteriol. August vol. 194 no. 16 4469-4470.
Noble, W. C., Z. Virani, and R. G. A. Cree. 1992. Co-transfer of vancomycin and other
resistance genes from Enterococcus faecalis NCTC 12201 to Staphylococcus aureus. FEMS
Microbiol. Lett. 93:195-198.

REFERENCES BIBLIOGRAPHIQUES
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 175
Noto MJ, Kreiswirth BN, Monk AB, Archer GL. (2008) Gene acquisition at the insertion
site for SCCmec, the genomic island conferring methicillin resistance in Staphylococcus
aureus. J Bacteriol. Feb;190(4):1276-83.
Ogston A (1882) Microccocus poisoning. J Anat 17, 24-58.
Okuma K, Iwakawa K, Turnidge JD, et al. (2002) Dissemination of New Methicillin-
Resistant Staphylococcus aureus Clones in the Community. Journal of Clinical Microbiology.
40(11):4289-4294.
Oliveira DC, de Lencastre H. (2002) Multiplex PCR strategy for rapid identification of
structural types and variants of the mec element in methicillin-resistant Staphylococcus
aureus. Antimicrob Agents Chemother. Jul;46(7):2155-61.
Oliveira DC, Milheirico C, de Lencastre H. (2006). Redefining a structural variant of
staphylococcal cassette chromosome mec, SCCmec type VI. Antimicrob Agents Chemother.
Oct;50(10):3457-9.
Orlando, L. et al. (2011). True single-molecule DNA sequencing of a pleistocene horse
bone. Genome Res. 21, 1705–1719.
Park H.-K., Shim S.-S., Kim S.-Y., Park J.-H., Park S.-E., Kim H.-J., et al. (2005). Molecular analysis of colonized bacteria in a human newborn infant gut. J.
Microbiol. 43, 345–353.
Peschel A., Sahl H.-G. (2006). The co-evolution of host cationic antimicrobial peptides and
microbial resistance. Nat. Rev. Microbiol. 4, 529–536. 10.1038/nrmicro1441.
Pevzner, P. A. and Tang, H. and Waterman, M. S. (2001) An Eulerian path approach to
DNA fragment assembly. Proc Natl Acad Sci U S A doi:10.1073/pnas.171285098.
Procopio, R. E., I. R. Silva, M. K. Martins, J. L. Azevedo, and J. M. Araujo. (2012)
Antibiotics produced by Streptomyces. The Brazilian journal of infectious diseases : an
official publication of the Brazilian Society of Infectious Diseases 16:466-471.
Pulverer, G., Peters, G. & Schumacher-Perdreau, F. (1987). Coagulase-negative
staphylococci. Zentralbl Bakteriol Mikrobiol Hyg [A]. 264, 1-28.
Pushkarev, D., Neff, N.F. and Quake, S.R. (2009) Single-molecule sequencing of an
individual human genome. Nat Biotechnol, 27, 847- 852.
Qin N, Ding W, Yao J, Su K, Wu L, Li L. (2012) Genome sequence of Staphylococcus
capitis QN1, which causes infective endocarditis. J Bacteriol.194:4469–4470. doi:
10.1128/JB.00827-12
Rasigade JP, Raulin O, Picaud JC, Tellini C, Bes M, Grando J, Ben Saïd M, Claris O,
Etienne J, Tigaud S, Laurent F. (2012). Methicillin-Resistant Staphylococcus capitis with
Reduced Vancomycin Susceptibility Causes Late-Onset Sepsis in Intensive Care Neonates.
PLoS One. 7(2): e31548.
Rasko DA, Webster DR, Sahl JW, et al. (2011) Origins of the E. coli strain causing an
outbreak of hemolytic-uremic syndrome in Germany. N Engl J Med, 365:709-17.
Reinhardt J, Baltrus D, Nishimura M, Jeck W, Jones C, and Dangl J. (2009) De novo
assembly using lowcoverage short read sequence data from the rice pathogen Pseudomonas
syringae pv. oryzae,” Genome Research. Feb vol. 19, no. 2, pp. 294–305.
Renaud F, Hansen Willy, Freney Jean. (2007). Manuel de bactériologie clinique. Ed
Elsevier, 2ème
édition,

REFERENCES BIBLIOGRAPHIQUES
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 176
Reslewic S, Zhou S, Place M, Zhang Y, Briska A, Goldstein S, Churas C, Runnheim
R, Forrest D, Lim A, Lapidus A, Han CS, Roberts GP, Schwartz DC. (2005) Whole-
Genome Shotgun OpticalMapping of Rhodospirillum rubrum,” Applied and Environmental
Microbiology, vol. 71, no. 9, pp. 5511–5522.
Rhee M, Burns MA. (2006). Nanopore sequencing technology: research trends and
applications. Trends Biotechnol;24: 580—6.
Roach DJ, Burton JN, Lee C, Stackhouse B, Butler-Wu SM, Cookson BT, et al. (2015) A
Year of Infection in the Intensive Care Unit: Prospective Whole Genome Sequencing of
Bacterial Clinical Isolates Reveals Cryptic Transmissions and Novel Microbiota. PLoS Genet
11(7): e1005413.
Robert J, Bismuth R, Jarlier V. (2002). Decreased susceptibility to glycopeptides in
methicillin resistant Staphylococcus aureus : a 20 year study in a large French teaching
hospital, 1983-J Antimicrob Chemother, 2006, 57 (3) : 506-10.
Robinson DA, Enright MC. (2004). Multilocus sequence typing and the evolution of
methicillin-resistant Staphylococcus aureus. Clin Microbiol Infect, 10 (2) : 92-7.
Rosenbach FJ (1884) Microorganismen bei den Wund-Infections-Krankheiten des Menschen,
Wiesbaden.
Ruhe J, Menon A, Mushatt D, et al. Non-epidermidis coagulasenegative staphylococcal
bacteremia: clinical predictors of true bacteremia. Eur J Clin Microbiol Infect Dis
2004;23:495–8.
Ryan D, Rahimi M, Lund J, Mehta R, Parviz BA. (2007). Toward nanoscale genome
sequencing. Trends Biotechnol 25:385—9.
Sabat, A. J., Hermelijn, S. M., Akkerboom, V., Juliana, A., Degener, J. E., Grundmann,
H., & Friedrich, A. W. (2017). Complete-genome sequencing elucidates outbreak dynamics
of CA-MRSA USA300 (ST8-spa t008) in an academic hospital of Paramaribo, Republic of
Suriname. Scientific Reports, 7, 41050.
Salipante SJ, Roach DJ, Kitzman JO, Snyder MW, Stackhouse B, et al. (2014) Large-
scale genomic sequencing of extraintestinal pathogenic Escherichia coli strains. Genome Res
25: 119–128.
Sanger, F. and Coulson, A.R. (1975) A rapid method for determining sequences in DNA by
primed synthesis with DNA polymerase. Journal of molecular biology, 94, 441-8.
Sanger, F., Nicklen, S. and Coulson, A.R. (1977) DNA sequencing with chain-terminating
inhibitors. Proceedings of the National Academy of Sciences of the United States of America,
74, 5463-7.
Sanger, F., Coulson, A.R., Hong, G.F., Hill, D.F. and Petersen, G.B. (1982) Nucleotide
sequence of bacteriophage lambda DNA. Journal of molecular biology, 162, 729-73.
Schwalbe, R. S., W. J. Ritz, P. R. Verma, E. A. Barranco, and P. H. Gilligan. 1990.
Selection for vancomycin resistance in clinical isolates of Staphylococcus haemolyticus. J.
Infect. Dis. 161:45-51.
Seemann T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068 –
2069.

REFERENCES BIBLIOGRAPHIQUES
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 177
Sengenès, J. (2012) Développement de méthodes de séquençage de seconde génération
pourl’analyse des profils de méthylation de l’ADN. Thèse de doctorat, Université Pierre et
Marie Curie,216p.
SenGupta DJ, Cummings LA, Hoogestraat DR, Butler-Wu SM, Shendure J, Cookson
BT, et al. Whole-genome sequencing for high-resolution investigation of methicillinresistant
Staphylococcus aureus epidemiology and genome plasticity. J Clin Microbiol.
Aug;52(8):2787–96.
Shendure, J. and Ji, H. (2008) Next-generation DNA sequencing. Nature Biotechnology, 26,
1135-45, 10.1038/nbt1486.
Shendure J. (2008). The beginning of the end for microarrays? Nat Methods. 5: 585-587.
Shields RK, Nguyen MH, Press EG, Kwa AL, Cheng S, Du C, et al. (2012) The presence
of an FKS mutation rather than MIC is an independent risk factor for failure of echinocandin
therapy among patients with invasive candidiasis due to Candida glabrata. Antimicrob.
Agents Chemother. Sep;56(9):4862–9.
Shin J. M., Gwak J. W., Kamarajan P., Fenno J. C., Rickard A. H., Kapila Y. L.
(2016). Biomedical applications of nisin. J. Appl. Microbiol. 120, 1449–1465.
10.1111/jam.13033.
Shukla SK, Kislow J, Briska A, Henkhaus J, Dykes C (2009) Optical mapping reveals a
large geneticinversion between two methicillin-resistant Staphylococcus aureus strains,”
Journal of Bacteriology,vol. 191, no. 18, pp. 5717–23.
Spicer W.J. (2003). Pratique clinique en bactériologie mycologie et parasitologie
Flammarion Médecine-Sciences, Paris. 28-29.
Sieradzki, K., and A. Tomasz. 1997. Inhibition of cell wall turnover and autolysis by
vancomycin in a highly vancomycin-resistant mutant of Staphylococcus aureus. J.
Bacteriol. 179:2557-2566.
Stoll BJ, Hansen N, Fanaroff AA, et al. (2002) late-onset sepsis in very low birth weight
neonates: the experience of the NICHD Neonatal Research Network. Pediatrics. 110:285–291.
Taft D. H., Ambalavanan N., Schibler K. R., Yu Z., Newburg D. S., Deshmukh H., et al. .
(2015). Center variation in intestinal Microbiota prior to late-onset sepsis in preterm
infants. PLoS ONE10:e0130604. 10.1371/journal.pone.0130604
Tettelin, H., V. Masignani, M.J. Cieslewicz, C. Donati, D. Medini, N.L. Ward, S.V.
Angiuoli, J. Crabtree, A.L. Jones, A.S. Durkin, R.T. Deboy, T.M. Davidsen, M. Mora,
M. Scarselli, I. Margarit y Ros, J.D. Peterson, C.R. Hauser, J.P. Sundaram, W.C.
Nelson, R. Madupu, L.M. Brinkac, R.J. Dodson, M.J. Rosovitz, S.A. Sullivan, S.C.
Daugherty, D.H. Haft, J. Selengut, M.L. Gwinn, L. Zhou, N. Zafar, H. Khouri, D.
Radune, G. Dimitrov, K. Watkins, K.J. O'Connor, S. Smith, T.R. Utterback, O. White,
C.E. Rubens, G. Grandi, L.C. Madoff, D.L. Kasper, J.L. Telford, M.R. Wessels, R.
Rappuoli, and C.M. Fraser. (2005) Genome analysis of multiple pathogenic isolates of
Streptococcus agalactiae: implications for the microbial "pangenome". Proc Natl Acad Sci U
S A, 102(39): p. 13950-5
Taj Azarian, Robert L. Cook, Judith A. Johnson, Nilmarie Guzman, Yvette S.
McCarter, Noel Gomez, Mobeen H. Rathore, J. Glenn Morris, Jr. and Marco Salemi.
(2015) Whole-Genome Sequencing for Outbreak Investigations of Methicillin Resistant

REFERENCES BIBLIOGRAPHIQUES
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 178
Staphylococcus aureus in the Neonatal Intensive Care Unit: Time for Routine Practice?.
Infection Control & Hospital Epidemiology / FirstView Article / May 2015, pp 1 – 9.
Takeuchi,F., Watanabe,S., Baba,T., Yuzawa,H., Ito,T., Morimoto,Y., et al. (2005).
Whole-genome sequencing of Staphylococcus haemolyticus uncovers the extreme plasticity of
its genome and the evolution of human-colonizing staphylococcal species. J.Bacteriol.
187,7292–7308.
Tevell S,
Hellmark B,
Nilsdotter-Augustinsson A,
and Söderquist B. (2017)
Staphylococcus capitis isolated from prosthetic joint infections. Eur J Clin Microbiol Infect
Dis. 36(1): 115–122.
Thompson, J. D., Higgins, D. G., et Gibson, T.J., (1994). CLUSTL W: improving the
sensitivity of progressive multiple sequence alignment through sequence weighting position-
specific gap penalities and weight matrix choice. Nucleic Acids Res 22, 4673-4680.
Tong SY, Holden MT, Nickerson EK, Cooper BS, Koser CU, et al. (2015) Genome
sequencing defines phylogeny and spread of methicillin-resistant Staphylococcus aureus in a
high transmission setting. Genome Res 25: 111–118.
Urwin R, Maiden MC; Maiden (October 2003). "Multi-locus sequence typing: a tool for
global epidemiology". Trends Microbiol. 11 (10): 479–87.
Vallenet D, Labarre L, Rouy Z, Barbe V, Bocs S, Cruveiller S, Lajus A, Pascal G,
Scarpelli C, Médigue C. 2006. MaGe: a microbial genome annotation system supported by
synteny results. Nucleic Acids Res 34:53– 65.
Vallenet D, Belda E, Calteau A, Cruveiller S, Engelen S, Lajus A, Le Fèvre F, Longin C,
Mornico D, Roche D et al. MicroScope—an integrated microbial resource for the curation
and comparative analysis of genomic and metabolic data. Nucleic Acids Res 2013
Jan;41(Database issue):D636-D647.
Vandenesch F, Naimi T, Enright MC, et al. (2003). Community-acquired
methicillinresistant Staphylococcus aureus carrying Panton-Valentine leukocidin genes.
worldwide emergence. Emerg Infect Dis, 9 (8) : 978-84.
Vandenesch F., Bes M., Etienne J. (2007). Staphylocoques. EMC (Elsevier Masson SAS,
Paris, Encydopédie Médico-Biologique, 90-05-0255,
Van Der Zwet WC, Debets-Ossenkopp YJ, Reinders E, et al (Juillet 2002). "propagation
nosocomiale d'une souche de Staphylococcus capitis hétérorésistance à la vancomycine dans
une unité néonatale de soins intensifs" . J. Clin. . Microbiol 40 (7):
Van den Eede A, Martens A, Lipinska U, Struelens M, Deplano A, et al. (2009) High
occurrence of methicillin-resistant Staphylococcus aureus ST398 in equine nasal samples. Vet
Microbiol 133: 138-144.
Venkatesh MP, Placencia F, Weisman LE. (2006) Coagulase-negative staphylococcal
infections in the neonate and child: an update. Semin Pediatr Infect Dis, 17:120–7.
Vincent JL, Bihari DJ, Suter PM, Bruining HA, White J, Nicolas-Chanoin MH, et al. (1995). The prevalence of nosocomial infection in intensive care units in Europe. Results of
the European Prevalence of Infection in Intensive Care (EPIC) Study. JAMA 274 : 639-44.
Wang, J., Wang, W., Li, R., Li, Y., Tian, G., Goodman, L., Fan, W., Zhang, J., Li, J.,
Guo, Y. et al. (2008) The diploid genome sequence of an Asian individual. Nature, 456, 60-
65.

REFERENCES BIBLIOGRAPHIQUES
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 179
Vogel V, Falquet L, Calderon-Copete SP, Basset P, Blanc DS. (2012) Short term evolution
of a highly transmissible methicillin-resistant Staphylococcus aureus clone (ST228) in a
tertiary care hospital. PLoS One. 7: e38969.
Waterston, R.H., Lindblad-Toh, K., Birney, E., Rogers, J., Abril, J.F., Agarwal, P.,
Agarwala, R., Ainscough, R., Alexandersson, M., An, P., et al. (2002) Initial sequencing
and comparative analysis of the mouse genome. Nature, 420, 520-62, 10.1038/nature01262.
Watson, J. D., et Crick, F. H. (1953). Molecular structure of nucleic acids, a structure for
deoxyribose nucleic acid. Nature 171, 737-738.
Wiedemann I., Breukink E., van Kraaij C., Kuipers O. P., Bierbaum G., de Kruijff B., et
al. (2001). Specific binding of nisin to the peptidoglycan precursor lipid II combines pore
formation and inhibition of cell wall biosynthesis for potent antibiotic activity. J. Biol.
Chem. 276, 1772–1779.
Willner D, Daly J, Whiley D, Grimwood K, Wainwright CE, Hugenholtz P. (2012).
Comparison of DNA Extraction Methods for Microbial Community Profiling with an
Application to Pediatric Bronchoalveolar Lavage Samples. PLoS ONE 7(4):
e34605. doi:10.1371/journal.pone.0034605.
Witney AA, Gould KA, Pope CF, et al. (2014) Genome sequencing and characterization of
an extensively drugresistant sequence type 111 serotype O12 hospital outbreak strain of
Pseudomonas aeruginosa. Clin Microbiol Infect. Oct;20(10):O609-18.
Wu, R. (1970). Nucleotide sequence analysis of DNA. I. Partial sequence of the cohesive
ends of bacteriophage lambda and 186 DNA. Journal of molecular biology, 51, 501-21.
Wu, R. and Taylor, E. (1971). Nucleotide sequence analysis of DNA. II. Complete
nucleotide sequence of the cohesive ends of bacteriophage lambda DNA. Journal of
molecular biology, 57, 491-511.
Yarwood JM, Schlievert PM. (2003). Quorum sensing in Staphylococcus infections. J Clin
Invest 112, 1620-1625.
Yasugi M, Nakamura S, Daidoji T, Kawashita N, Ramadhany R, Yang C-S, et al. (2012)
Frequency of D222G and Q223R hemagglutinin mutants of pandemic (H1N1) 2009 influenza
virus in Japan between 2009 and 2010. Plos One. 7(2):e30946.
Zahar JR, Clec’h C, Tafflet M, et al. (2005). Is methicillin resistance associated with
aworse prognosis in Staphylococcus aureus ventilator-associated pneumonia ? Clin Infect Dis,
41 (9) : 1224-31.
Zahlane, K. H. (2007). Staphylocoque: état actuel de l’épidémiologie et de l’antibiorésistance
au CHU de Rabat. Maroc Médical, tome 29 n°4 , 279-285.
Zhang, K., J. A. McClure, S. Elsayed, and J. M. Conly. 2009. Novel staphylococcal
cassette chromosome mec type, tentatively designated type VIII, harboring class A mec and
type 4 ccr gene complexes in a Canadian epidemic strain of methicillin-
resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 53:531-540.
Zhang,Y.Q., Ren,S.X., Li,H.L., Wang,Y.X., Fu,G., Yang,J., et al. (2003). Genome-based
analysis of virulence genes in anon-biofilm- forming Staphylococcus epidermidis strain
(ATCC12228). Mol.Microbiol. 49, 1577–1593.

REFERENCES BIBLIOGRAPHIQUES
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 180
Webgraphie
http://www.genomesonline.org
http://departments.oxy.edu/biology/Stillman/bi221/092200/lecture_notes.htm
http://www.appliedbiosystems.com/
www.genoscope.cnr.fr
http://www.454.com
www.sccmec.org/Pages/SCC_TypesEN.html
http://www.bio.davidson.edu/COurses/genomics/method/shotgun.html.
http://www.ncbi.nlm.nih.gov/genome/genomes/2054?details=on&)
http://bioinformatics.ws/index.php/History_of_Biology,
http://bioinformatics.ws/index.php/History_of_Bioinformatics,
http://en.wikipedia.org/wiki/Timeline_of_computing,
http://www.thocp.net/timeline/timeline.htm,
http://seqanswers.com/et http://www.genomesonline.org
http://www.genome.gov/sequencingcosts/

ANNEXES

ANNEXES
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 181
ANNEXE 1
Chronologie non exhaustive des événements marquants survenus en biologie (Rose),
et en bioinformatique (Incolore).
http://bioinformatics.ws/index.php/History_of_Biology,
http://bioinformatics.ws/index.php/History_of_Bioinformatics,
http://en.wikipedia.org/wiki/Timeline_of_computing,
http://www.thocp.net/timeline/timeline.htm,
http://seqanswers.com/ et http://www.genomesonline.org
ANNEE AUTEUR EVENEMENT
1944 Avery Démonstration de l’ADN en tant que support de l’information
génétique (Avery et al., 1994)
1953 Sanger, Thompson Détermination de la séquence des chaines A et B de l’insuline
(Sanger et Tompson, 1953a, 1953b)
Watson, Crick Modèle de la structure en double hélice de l’ADN (Watson et
al., 1953)
1958 Crick Enonciation du dogme central de la biologie moléculaire (Crick,
1958)
1962 Matthaei Déchiffrage du code génétique (Matthaei et al., 1962)
1965 Dayhoff Premier atlas de la séquence et de la structure des protéines
(Dayhoff et national Biomedical Research Foundation., 1965)
1967 Fitch Construction d’arbre phylogénétique (Fitch et Margoliash,
1967)
1970 Smith, Wilcox Première enzyme de restriction spécifique isolée (Smith et
Wilocox, 1970)
1970 Needleman, Wusnch Algorithme d’alignement global optima entre deux séquences
(Needleman, Wusnch, 1970)
1973 Annonce de la protéine Data Bank (PDB), (Protein Data Bank,
1973)
1974 Chou, Fasman Algorithme de prédiction des structures secondaires des
protéines (Chou et Fasman, 1974)
1977 Sanger Méthode de séquençage par synthèse enzymatique de l’ADN
(Sanger, Nicklen, et al., 1977)
Sanger, Air Premier génome complet séquencé : phage X174
Maxam, Gilbert Méthode de séquençage chimique de l’ADN
Staden Suite d’analyse de séquences d’ADN Staden
1980 EMBL Première banque de séquences nucléiques
Anderson Séquence du génome mitochondrial humain
Smith, Waterman Algorithme d’alignement local optimal entre deux séquences
1982 GenBank Banque américaine de séquences nucléique
1983 Mullis Invention de la réaction en chaine de la polymérase (PCR)
1985 Lipman FASTA, programme de recherche de séquences par similarité
Gouy ACNUC, programme d’interrogation des banques de séquences
Bairoch SWISS-PORT, Banque de séquences protéiques
DDBJ Banque Japonaise de séquences protéiques
1987 Applied Biosystems Premier séquenceur automatique (ABI 370)
Burke Création du vecteur de clonage Yeast Artificial Chromosome
(YAC)
Kulesh Apparition de la technologie des puces d’ADN

ANNEXES
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 182
1988 Lancement du projet international de séquençage du génome
humaine (National Research Council (U.S), 1998)
Higgins, Sharp CLUSTAL, programme d’alignement multiple
1989 O’Connor Création du vecteur de clonage Bacterial Artificial Chromosome
(BAC)
1990 Altschul BLAST, programme de recherche de séquences par similarité
1991 Adams Création et utilisation à grande échelle du séquençage partiel
d’ADNc (EST)
Roberts GRAIL, programme de localisation de gène
1993 Etzold, Argos SRS, programme d’interrogation des banques de séquences
1994 Thompson CLUSTALW
1995 Fleischmann Premier organisme vivant séquencé : Haemophilus influenzae
(1.8 Mb)
Fraser Plus petit organisme vivant séquencé : Mycoplasma gentalium
(580 Kb)
1996 Walsh, Barrell Premier organisme eucaryote séquencé : Saccharomyces
cerevisiae (12.1 Mb)
Affymetrix Commercialisation de la première puce d’ADN
1997 Blattner Génome complet de Escherchia coli (4.7 Mb)
Altschul Gapped BLAST et PSI-BLAST
Burge, Karlin GenScan, prediction de la structure complete des gènes dans
l’AND génomique humain
1998 Premier organisme pluricellulaire séquencé : Caenorhabditis
elegans (Mp)
2000 Dennis, Surridge Génome d’Arabidopsis thaliana (100Mb)
Adams Génome de drosophila melanogaster (180 Mb
2001 Lander Publication préliminaire du génome humaine par le Human
Genome Project (2.9 Gb)
Venter Publication préliminaire du génome humain par Celera
Genomics (2.9 Gb)
Ensembl Navigateur de génome Ensembl (Hubbared et al., 2009)
NCBI Navigateur de génome du NCBI
DEBUT DE L’ERE POST-GENOMIQUE
2002 Waterson Séquence préliminaire du génome de la souris (2.5 Gb)
UCSC Navigateur de génome de l’UCSC (Kuhn et al., 2009)
2004 IHGSC Finalisation du génome humaine (IHGSC, 2004) (3.4 Gb)
ENCODE PC ENCODES, projet d’identification de tous les élements
fonctionnels du génome humain (ENCODE Project
Consortium, 2004)
2005 Roche, 454 Séquenceur automatique haut-débit de 2ème
génération par
pyroséquençage : GS20
2007 Illumina, Solexa Séquenceur automatique haut- débit de 2ème
génération par
synthèse microfluidique : Genome Analyzer
Applied Biosystems Séquenceur haut-débit de 2ème
génération par ligation : système
SOLID
2008 Helicos Séquenceur haut-débit de 2ème
génération par synthèse sans
pré-amplification
2009 PRES DE 1000 PROJETS DE SEQUENÇAGE DE GENOME EUCARYOTES EN
COURS…

ANNEXES
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 183
ANNEXE 2
Espèces et sous-espèces du genre Staphylococcus et hôtes associés (Vandenesch et al.,
2007)
Espèce Coagulase Hôte ou source S. arletti - Caprin, volaille
S. aureus subsp. anaerobius S. aureus subsp.aureus
+ Ovin Homme, animaux, environnement
S. auricularis - Homme
S. capitis subsp. capitis
S. capitis subsp. ureolyticus -
- Homme
Homme, primates S. caprae - Homme, caprins
S. carnosus subsp. carnosus
S. carnosus subsp. utilis
- Produits carnés
Aliments S. chromogenes - Animaux, lait S. cohnii subsp. cohnii
S. cohnii subsp. urealyticum
- Homme
Homme, animaux S. condimenti - Sauce au soja
S. delphini + Dauphins S. epidermidis - Homme, animaux, environnement S. equorum subsp. equorum S. equorum subsp. linens
- Chevaux, bétail Surface fromage affiné
S. felis - chats
S. fleurettii - Fromage lait de chèvre S. gallinarum - Volailles, oiseaux S. haemolyticus - Homme, animaux domestique, environnement S. hominis subsp. hominis S. hominis subsp. novobiosepticus
- Homme Homme
S. hyicus +/- Animaux, aliments
S. intermedius + Mammifère, oiseaux, rarement Homme S. kloosii - Animaux sauvages S. lentus - Animaux, rarement Homme S. lugdunensis - Homme S. lutrae + Loutre S. muscae - Mouches, porcs S. nepalensis - chèvre S. pasteuri - Homme, animaux, aliments
S. pettenkoferi - Homme S. piscifermentans - Poisson fermenté S. pseudintermedius (Devriese et al., 2005) + Animaux S. saccharolyticus - Homme S. saprophyticus subsp. bovis
S. saprophyticus subsp. saprophyticus -
- Animaux
Homme, animaux S. schleiferi subsp. coagulans S. schleiferi subsp. schleiferi
+ -
Chiens Homme
S. sciuri subsp. carnaticus
S. sciuri subsp. lentus
S. sciuri subsp. rodentium
S. sciuri subsp. sciuri
-
-
-
-
Produits carnés
Animaux
Rongeurs, animaux
Homme, animaux S. simiae (Pantucek et al., 2005) - Singe
S. simulans - Homme, mammifère
S. succinus subsp. casei - Surface de fromage affiné S. succinus subsp. succinus - ambre S. vitulinus (anciennement S. pulvereri) - Animaux, aliments

ANNEXES
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 184
S. warneri - Homme, primates S. xylosus - Homme, animaux, environnement

ANNEXES
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 185
ANNEXE 3
Principales caractéristiques des différentes techniques de séquençage
Technologie Génération Méthode
d’amplification
Méthode se
séquençage
Détection Particularité
Sanger 1 PCR Synthèse
(terminaison dideoxy)
Radioactivité ou
Fluorescence Technique de référence
Faible débit – Cout élève
Maxam-Gilbert 1 / Dégradation Radioactivité Utilisation confidentielle
454 – Roche
2
PCR en émulsion
Synthèse
(pyroséquencage)
Chimiluminescence
Séquences ≪ longues ≫ : 400-1000 pb
Durée de séquençage : 10-24h
Homopolymères : taux d’erreur élève
Cout relativement élève par rapport aux autres
technologies de 2ème génération
Principal type d’erreur : insertions et délétions
Solexa - Illumina
2
bridge-PCR
Synthèse
(terminaison réversible)
Fluorescence Séquences ≪ courtes ≫ : 100 à 150 pb
Durée de séquençage : 1 - 10j
Faible cout – Faible taux d’erreur
Principal type d’erreur : substitutions
HeliScope –
Helicos
2
/
Synthèse
Fluorescence Séquençage a partir d’une seule molécule
d’ADN : taux d’erreur important. Non
commercialise
SOLiD
Life Technologies PCR en émulsion Ligation (octamères) Fluorescence Séquences ≪ courtes ≫ : 35 à 50 pb
Codage a 2 bases : taux d’erreur limite
Principal type d’erreur : substitutions
Ion Torrent
Life Technologies 2 PCR en émulsion Synthèse Mesure du pH
(détection H+) Fiable cout (1er système < 100 000 $)
Principal type d’erreur : insertions et délétions SMRT
Pacific Biosciences
3
/
Synthèse
Fluorescence
Séquençage en temps réel a partir d’une seule
molécule d’ADN : taux d’erreur important
Séquences longues : 3000 pb
Starlight
Life Technologies 4 /
Synthèse
Fluorescence
Séquençage a partir d’une seule molécule
d’ADN
Possibilité de renouveler l’ADN polymérase
pour des séquences longues. En développement
Nanopores 3 / Variable Nanopores solides ou
biologiques
Séquences longues
Nombreux développements en cours
Sequencage in situ 4 / Synthèse Fluorescence En développement

Publications et
Communications

Standards in Genomic Sciences (2014) 9:1118-1127 DOI:10.4056/sigs.5491045
The Genomic Standards Consortium
Non-contiguous finished genome sequence of Staphylococcus capitis CR01 (pulsetype NRCS-A)
H. Lemriss1,2, Martins Simões P2,3, S. Lemriss1, M. Butin3, A. Ibrahimi4, S. El Kabbaj1, JP Rasigade2,3, F. Laurent2,3,5
1Department of Biosecurity PCL3, Laboratory of Research and Medical Analysis of the Fra-ternal of Gendarmerie Royale, Rabat, Morocco.
2International Centre for Research in Infectious diseases, INSERM U1111, University of Lyon, Lyon, France.
3Department of Clinical Microbiology, Northern Hospital Group, Hospices Civils de Lyon, Lyon, France.
4Medical Biotechnology lab. (MedBiotech), Medical and Pharmacy School, University Mo-hammed V Souissi, Rabat, Morocco.
5National Reference Center for Staphylococci, Hospices Civils de Lyon, Lyon, France.
Correspondence:
Keywords:
Staphylococcus capitis (NCRS-A), draft-genome, methicillin resistance, late-onset sepsis
Staphylococcus capitis is a coagulase-negative staphylococci (CoNS) commonly found in the human microflora. Recently, a clonal population of Staphylococcus capitis (denominated NRCS-A) was found to be a major cause of late-onset sepsis (LOS) in several neonatal inten-sive care units in France. Here, we report the complete genome sequence and annotation of the prototype Staphylococcus capitis NCRS-A strain CR01. The 2,504,472 bp long genome (1 chromosome and no plasmids) exhibits a G+C content of 32.81%, and contains 2,468 pro-tein-coding and 59 tRNA genes and 4 rRNA genes.
Abbreviations: EMBL- European Molecular Biology Laboratory, NCBI- National Center for Biotechnology Information (Bethesda, MD, USA), RDP- Ribosomal Database Project (East Lansing, MI, USA)
IntroductionA frequent cause of low-weight newborns mortali-ty and morbidity in Neonatal Intensive Care Units (NICUs) are late-onset sepsis (LOS), that are de-fined as sepsis occurring after 3 days of age. The most frequently encountered pathogens are coag-ulase-negative staphylococci (CoNS) and within those Staphylococcus epidermidis has been shown to be the most prevalent [1,2]. However, a few studies have reported the emergence of Staphylo-coccus capitis as a main CoNS- and LOS- causative pathogen in NICU settings [2-4]. A study in French NICUs [2] has demonstrated the spread of a single clonal population of methicillin-resistant S. capitis (pulsotype NRCS-A) associated to reduced suscep-tibility to vancomycin, the first line of antibiotics used in cases of LOS. Moreover, this clone has also been recently identified in NICUs in Belgium, United Kingdom and Australia, which suggests a worldwide distribution. In contrast, in adult bac-
teremia, S. capitis are rarely found and when de-tected, it presents a bigger diversity in terms of genotypes as well as antimicrobial susceptibility profiles than neonates bacteremia. In order to elucidate the molecular mechanisms behind the wide spreading of the S. capitis NRCS-A clone in NICUs throughout the world, we se-quenced a prototype strain (CR01).
Classification and information A strain belonging to the clonal population of Staphylococcus capitis NCRS-A pulsetype (Table 1) was isolated from the blood culture of a preterm infant with LOS, hospitalized in the NICU of the Northern Hospital Group Center (Hospices Civils de Lyon, Lyon, France) and suffering of LOS. Species identification of the bacterial isolates and antimicrobial susceptibility testing (AST) were performed, respectively, using Vitek MS

Lemriss et al.
http://standardsingenomics.org 1119
(bioMérieux, Marcy l’Etoile), 16S rDNA sequenc-ing, the automated BD Phoenix system (Becton Dickinson, Sparks, MD) and with Shimadzu-MALDI-TOF MS system (Shimadzu Corporation), as implemented on [21]. The strain was identified as being aStaphylococcus capitis by VITEK MS with 99.9% and at 93.7% by the MALDI-TOF MS, using the Shimadzu Launchpad software program and the SARAMIS database application (AnagnosTec GmbH) for au-tomatic measurement and identification (Figure 1). Based on the information provided by the manufacture, when the score is ≥70%, identifica-tion is considered of high confidence.
The antimicrobial susceptibility test (AST) results were analyzed according to the recommendations of the French Microbiology Society [22]. The S. capitis bacteremia was considered positive based on a single positive blood culture [2,23]. The S. capitis NCRS-A isolate CR01, as all isolates from this clone, is resistant to penicillin, methicillin, gentamicin, rifampicin, hetero-resistant to vancomycin and sensitive to fusidic acid and fluoroquinolones. Table 1, Figure 2 and Figure 3 show detailed in-formation concerning general features of Staphy-lococcus capitis strain (CR01) and position within the genus Staphylococcus.
Table 1. Classification and general features of Staphylococcus capitis strain CR01, pulsetype-NRCS-A ac-cording the MIGS recommendation [5].
MIGS ID Property Term Evidence codea Current classification
Domain Bacteria Phylum Firmicutes Class Bacilli Order Bacillales Family Staphylococcaceae Genus Staphylococcus Species Staphylococcus capitis Strain CR01, pulsetype-NRCS-A
TAS [6] TAS [7,8] TAS [9,10] TAS [11,12] TAS [13,14] TAS [11,15,16] TAS [17] TAS [2,18]
Gram stain Positive TAS [19] Cell shape Coccoid TAS [17] Motility Non-motile TAS [19] Sporulation Non-sporuating TAS [19] Temperature range Mesophilic IDA Optimum temperature 37°C TAS [17] Carbon source Carbohydrates, (glucose, sacharose,
fructose, manitol, mannose) TAS [17]
Energy source Chemoorganotropic TAS [17] Terminal electron recep-
tor O2 TAS [17]
MIGS-6 Habitat Skin of humans TAS [17] MIGS-6.3 Salinity Physiological TAS [17] MIGS-22 Oxygen Facultative anaerobes TAS [17] MIGS-15 Biotic relationship Free-living TAS [17] MIGS-14 Pathogenicity Opportunistic pathogen (Nosocom-
ial bacteremia in premature neo-nates)
TAS [2]
MIGS-4 Geographic location NICU Lyon, France TAS [2] MIGS-5 Sample collection time 2007 IDA MIGS-4.1 MIGS-4.2
Latitude – Longitude 45° 45' 35" N 4° 50' 32" E IDA
MIGS-4.3 Depth Not applicable IDA MIGS-4.4 Altitude 162 m IDA
a) Evidence codes - IDA: Inferred from Direct Assay; TAS: Traceable Author Statement (i.e., a direct re-port exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the liv-ing, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [20].

Staphylococcus capitis
1120 Standards in Genomic Sciences
Figure 1. Reference mass spectrum from Staphylococcus capitis strain (CR01).
Figure 2. Transmission electron microscopy of Staphylococcus capitis strain (CR01) using a JEOL 1400. The scale bar represents 200 nm.

Lemriss et al.
http://standardsingenomics.org 1121
Figure 3. 16S rRNA Phylogenetic tree highlighting the position of Staphylococcus capitis strain CR01 (indicated by the yellow circle) relative to other type strains within the genus Staphylococcus. All 16S rRNA sequences were obtained from the RDP database using as filtering criteria: sequences with more than 1200 nt and classified as “good” quality sequences. The tree uses sequences aligned with the MUSCLE software, with the default parameters as implemented on Seaview version 4 [24], and a tree was inferred based on 1285 sites using the distance model of observed diver-gence, as implemented in the BioNJ algorithm.
The 16S rRNA sequences were aligned using the MUSCLE software, with the default parameters as implemented on Seaview version 4 [24], and a tree was inferred based on 1285 sites using the distance model of observed divergence, as imple-
mented in the BioNJ algorithm, and a bootstrap-ping process repeated 500 times. The final tree was rooted using the 16S rRNA se-quence of Macrococcus equipercicus Type strain that belongs to a closely-related sister genus.

Staphylococcus capitis
1122 Standards in Genomic Sciences
Genome sequencing information The genome sequence of S. capitis strain CR01 was determined by high-throughput sequencing per-formed on a Genome Sequencer FLX + system (454 Life Sciences/Roche) using FLX Titanium re-agents according to the manufacturer's protocols and instructions, with approximately 47-fold cov-erage of the genome. This platform provides long-er read lengths than other sequencing platforms to obtain raw sequences. De novo assemblies were performed using the Roche Newbler (v 2.7) soft-ware package.
Genome project history Table 2 presents the project information and its association with MIGS version 2.0 compliance [5].
Growth conditions and DNA isolation The sample was prepared for sequencing by grow-ing S. capitis CR01, aerobically at 37°C in Blood Agar for 24-48 hours. Genomic DNA was extracted using the PureLinkTM genomic DNA kit (InvitrogenTM) according to the manufacturer’s recommended protocol. The quantity of DNA ob-tained was determined using a NanoVue TM Plus (HVD Life Sciences), and 1 µg of DNA was used for sequencing of whole-genome of this strain.
Genome sequencing and assembly The isolated DNA of S. capitis CR01, was used to create 454-shotgun libraries following the GS Rap-id library protocol (Roche 454, Roche). The result-ing 454 DNA libraries were sequenced using a whole-genome shotgun strategy by GS FLX Titani-um sequencing kit XL+ [25] (202,108 reads total-ing 2.5 Mb, X48 fold coverage of the genome). Ge-nome sequences were processed by Roche’s se-quencing software according to the manufactur-er's instructions (454 Life Science). The resulting shotgun reads were assembled de novo using the Roche Newbler assembly software 2.7 (454 Life
Science) and 26 large contigs (Contig00001 to Contig00026) were obtained. The N50 was 176239 bp.
Genome annotation An automatic syntactic and functional annotation of the draft genome was performed using the Mi-croScope platform pipeline [26,27]. The syntactic analysis combines a set of programs including AMIGene [28], tRNAscan-SE [29], RNAmmer [30], Rfam scan [31] and Prodigal software [32] to pre-dict genomic objects that are mainly CDSs and RNA genes. More than 20 bioinformatics methods are then used for functional and relational anal-yses: homology search in the generalist databank UniProt [33] and in more specialized databases as COG [34], InterPro [35], PRIAM profiles for enzy-matic classification [36], prediction of protein lo-calization using TMHMM [37], SignalP [38] and PsortB [39] tools.
Genome properties The genome includes one circular chromosome of 2,504,472 bp (32.81% GC content). A total of 2,565 genes were predicted with 2,453 being pro-tein-coding genes, 59 tRNA-enconding genes, 4 rRNA-encoding genes (including 2 copies of 5S rRNA, 1 copy of both the large and the small-subunits, respectively, 23S and 16S rRNA) and 34 other RNA related ORFs. No plasmid was detected. Of the 2,453 protein-coding genes, 1,892 genes (76.7%) were assigned to a putative function with the remaining annotated as hypothetical proteins. The predicted coding density in S. capitis strain CR01 was 86%. Table 3 and Figure 4 detailed description of the properties and the statistics of Staphylococcus capitis strain CR01 genome. The distribution of the genes into COGs functional categories is pre-sented in Table 4.
Table 2. Project information MIGS ID Property Term MIGS-31 Finishing quality Non-contiguous finished
MIGS-28 Libraries used 454 pyrosequence rapid li-brary
MIGS-29 Sequencing platforms 454 GS FLX+
MIGS-31.2 Fold coverage 47.0 × pyrosequence
MIGS-30 Assemblers Newbler Assembler 2.7
GenBank CBUB000000000.1
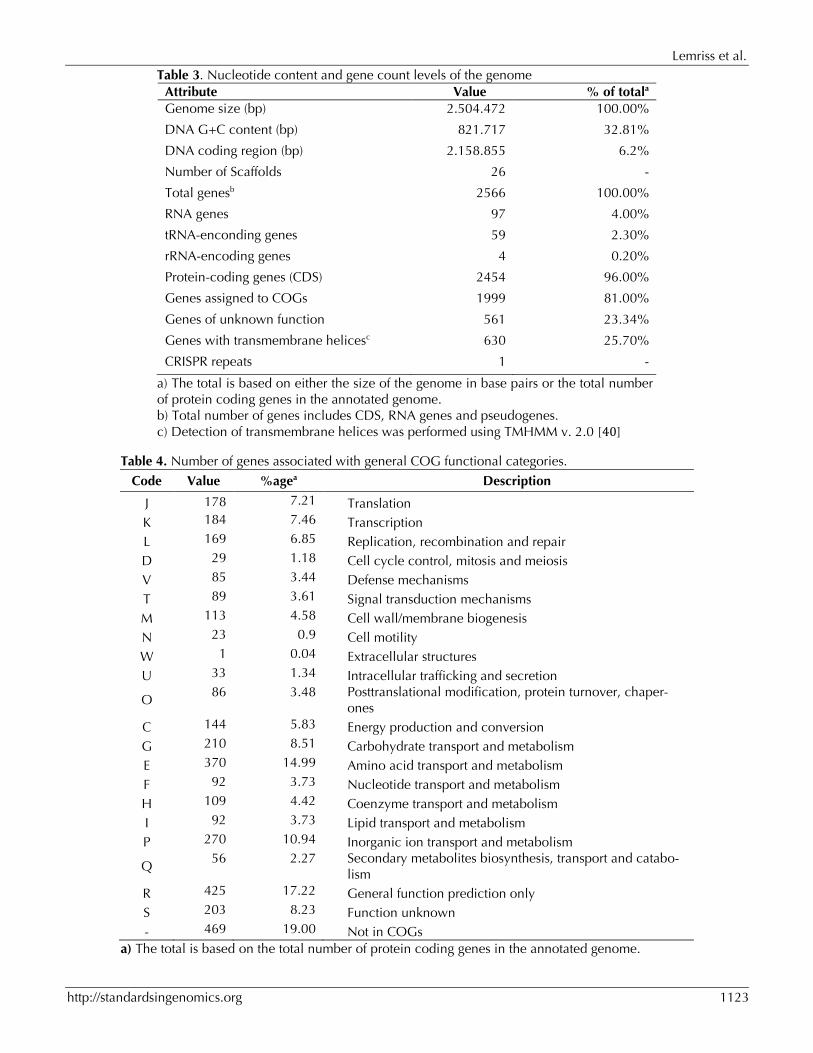
Lemriss et al.
http://standardsingenomics.org 1123
Table 3. Nucleotide content and gene count levels of the genome Attribute Value % of totala Genome size (bp) 2.504.472 100.00%
DNA G+C content (bp) 821.717 32.81%
DNA coding region (bp) 2.158.855 6.2%
Number of Scaffolds 26 -
Total genesb 2566 100.00%
RNA genes 97 4.00%
tRNA-enconding genes 59 2.30%
rRNA-encoding genes 4 0.20%
Protein-coding genes (CDS) 2454 96.00%
Genes assigned to COGs 1999 81.00%
Genes of unknown function 561 23.34%
Genes with transmembrane helicesc 630 25.70%
CRISPR repeats 1 -
a) The total is based on either the size of the genome in base pairs or the total number of protein coding genes in the annotated genome. b) Total number of genes includes CDS, RNA genes and pseudogenes. c) Detection of transmembrane helices was performed using TMHMM v. 2.0 [40]
Table 4. Number of genes associated with general COG functional categories.
Code Value %agea Description
J 178 7.21 Translation K 184 7.46 Transcription L 169 6.85 Replication, recombination and repair D 29 1.18 Cell cycle control, mitosis and meiosis V 85 3.44 Defense mechanisms T 89 3.61 Signal transduction mechanisms M 113 4.58 Cell wall/membrane biogenesis N 23 0.9 Cell motility W 1 0.04 Extracellular structures U 33 1.34 Intracellular trafficking and secretion
O 86 3.48 Posttranslational modification, protein turnover, chaper-
ones C 144 5.83 Energy production and conversion G 210 8.51 Carbohydrate transport and metabolism E 370 14.99 Amino acid transport and metabolism F 92 3.73 Nucleotide transport and metabolism H 109 4.42 Coenzyme transport and metabolism I 92 3.73 Lipid transport and metabolism P 270 10.94 Inorganic ion transport and metabolism
Q 56 2.27 Secondary metabolites biosynthesis, transport and catabo-
lism R 425 17.22 General function prediction only S 203 8.23 Function unknown - 469 19.00 Not in COGs
a) The total is based on the total number of protein coding genes in the annotated genome.

Staphylococcus capitis
1124 Standards in Genomic Sciences
Figure 4. Graphical circular map of the chromosome. From outside to the center: Genes on the forward strand (colored by COG categories), genes on the reverse strand colored by COG categories), RNA genes (tRNAs green, rRNAs blue), GC content, and GC skew
ConclusionHere, we described a new genome sequence of Staphylococcus capitis (strain CR01 belonging to NRCS-A clone) as a first step toward comparing its content with other sequenced Staphylococcus capitis genomes as well as CoNS genomes of spe-cies associated with late-onset sepsis. Detailed analyses are in progress to identify virulence fac-tors and mobile genetic elements (MBE), such as the staphylococcal chromosome cassette (SCCmec) [18], potentially related to the high
specificity of the NRCS-A clone to the NICU envi-ronment.
Acknowledgements This work was supported by a grant from the Fondation pour la Recherche Médicale (FRM, grant ING20111223510) and by the Institut Na-tional de la Recherche Médicale (INSERM) and the French Ministry of Health.

Lemriss et al.
http://standardsingenomics.org 1125
References1. Klingenberg C, Rønnestad A, Anderson AS, Abra-
hamsen TG, Zorman J, Villaruz A, Flægstad T, Ot-to M. Sollid, Ericson J. Persistent strains of coagu-lase-negative staphylococci in a neonatal inten-sive care unit: virulence factors and invasiveness. Clin Microbiol Infect 2007; 13:1100-1111. Pub-Med http://dx.doi.org/10.1111/j.1469-0691.2007.01818.x
2. Rasigade JP, Raulin O, Picaud JC, Tellini C, Bes M, Grando J, Ben Saïd M, Claris O, Etienne J, Tigaud S, Laurent F. Methicillin-ResistantStaphylococcus capitis with Reduced Vancomycin Susceptibility Causes Late-Onset Sepsis in Intensive Care Neonates. PLoS ONE 2012; 7:e31548. PubMed http://dx.doi.org/10.1371/journal.pone.0031548
3. Ng PC, Chow VC, Lee CH, Ling JM, Wong HL, Chang RCY. Persistent Staphylococcus capitis septicemia in a preterm infant. Pediatr Infect Dis J 2006; 25:652-654. PubMed http://dx.doi.org/ 10.1097/01.inf.0000225785.32137.d3
4. de Silva GDI, Kantzanou M, Justice A, Massey RC, Wilkinson AR, Day NPJ, Peacock SJ. The ica Operon and Biofilm Production in Coagulase-Negative Staphylococci Associated with Carriage and Disease in a Neonatal Intensive Care Unit. J Clin Microbiol 2002; 40:382-388. PubMed http://dx.doi.org/10.1128/JCM.40.02.382-388.2002
5. Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541-547. PubMed http://dx.doi.org/10.1038/nbt1360
6. Woese CR, Kandler O, Wheelis ML. Towards a nat-ural system of organisms: proposal for the do-mains Archaea, Bacteria, and Eukarya. Proc Natl Acad Sci USA 1990; 87:4576-4579. PubMed http://dx.doi.org/10.1073/pnas.87.12.4576
7. Gibbons NE, Murray RGE. Proposals Concerning the Higher Taxa of Bacteria. Int J Syst Bacteriol 1978; 28:1-6; http://dx.doi.org/10.1099/00207713-28-1-1.
8. Murray RGE. The Higher Taxa, or, a Place for Everything...? In: Holt JG (ed), Bergey's Manual of Systematic Bacteriology, First Edition, Volume 1, The Williams and Wilkins Co., Baltimore, 1984, p. 31-34.
9. List of new names and new combinations previ- ously effectively, but not validly, published. List
no. 132. Int J Syst Evol Microbiol 2010; 60:469-472. http://dx.doi.org/10.1099/ijs.0.022855-0
10. Ludwig W, Schleifer KH, Whitman WB. Class I. Bacilli class nov. In: De Vos P, Garrity G, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer KH, Whitman WB (eds), Bergey's Manual of Systemat- ic Bacteriology, Second Edition, Volume 3, Springer-Verlag, New York, 2009, p. 19-20.
11. Skerman VBD, McGowan V, Sneath PHA. Ap- proved Lists of Bacterial Names. Int J Syst Bacteriol 1980; 30:225-420. http://dx.doi.org/10.1099/00207713-30-1-225
12. Prévot AR. In: Hauderoy P, Ehringer G, Guillot G, Magrou. J., Prévot AR, Rosset D, Urbain A (eds), Dictionnaire des Bactéries Pathogènes, Second Edition, Masson et Cie, Paris, 1953, p. 1-692.
13. List Editor. List of new names and new combina- tions previously effectively, but not validly, pub- lished. List no. 132. Int J Syst Evol Microbiol 2010; 60:469-472. http://dx.doi.org/10.1099/ijs.0.022855-0
14. Schleifer KH, Bell JA. Family VIII.Staphylococcaceae fam. nov. In: De Vos P, Garrity G, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer KH, Whitman WB (eds), Bergey's Manual of Systematic Bacteriology, Second Edi- tion, Volume 3, Springer-Verlag, New York, 2009, p. 392.
15. Rosenbach FJ. In: Bergmann JF (ed), Microorganismen bei den Wund-Infections- Krankheiten des Menschen., Wiesbaden, 1884, p. 1-122.
16. Judicial Commission. Opinion 17. Conservation of the Generic name Staphylococcus Rosenbach, Designation of Staphylococcus aureus Rosenbach as the Nomenclatural Type of the Genus Staphy- lococcus Rosenbach, and Designation of the Neotype culture of Staphylococcus aureus Rosenbach. Int Bull Bacteriol Nomencl Taxon 1958; 8:153-154.
17. Kloos WE, Musselwhite MS. Distribution and Per-sistence of Staphylococcus and Micrococcus Spe-cies and Other Aerobic Bacteria on Human Skin. Appl Microbiol 1975; 30:381-385. PubMed
18. Martins Simões P, Rasigade JP, Lemriss H, Butin M, Ginevra C, Lemriss S, Goering RV, Ibrahimi A, Picaud JC, Vandenesch FEL, et al. Characteriza-tion of a novel staphylococcal chromosome cas-sette (SCCmec) within a composite SCC island in neonatal sepsis-associated Staphylococcus capitis pulsotype NRCS-A. Antimicrob Agents

Staphylococcus capitis
1126 Standards in Genomic Sciences
Chemother 2013; 57:6354. PubMed http://dx.doi.org/10.1128/AAC.01576-13
19. Schleifer KH, Bell JA. Family VIII.Staphylococcaceae fam. nov. In: De Vos P, Garrity G, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer KH, Whitman WB (eds), Bergey's Manual of Systematic Bacteriology, Second Edi- tion, Volume 3, Springer-Verlag, New York, 2009, p. 392.
20. Ashburner M, Ball CA, Blake JA, Botstein D, But-ler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Con-sortium. Nat Genet 2000; 25:25-29. PubMed http://dx.doi.org/10.1038/75556
21. Cherkaoui A, Hibbs J, Emonet S, Tangomo M, Girard M, Francois P, Schrenzel J. Comparison of two matrix-assisted laser desorption ionization-time of flight mass spectrometry methods with conventional phenotypic identification for routine identification of bacteria to the species level. J Clin Microbiol 2010; 48:1169-1175. PubMed http://dx.doi.org/10.1128/JCM.01881-09
22. French Society for Microbiology. Recommandations du Comite de l'Antibiogramme de la Société Française de Microbiologie. 2009. Available at: wwwsfmasofr/doc/downloadphp?doc=DiU8C&fic =casfm_2009pdf. Accessed 22 February 2009.
23. Hall KK, Lyman JA. Updated review of blood cul-ture contamination. Clin Microbiol Rev 2006; 19:788-802. PubMed http://dx.doi.org/10.1128/CMR.00062-05
24. Gouy M, Guindon S, Gascuel O. SeaView Ver-sion 4: A Multiplatform Graphical User Interface for Sequence Alignment and Phylogenetic Tree Building. Mol Biol Evol 2010; 27:221-224. Pub-Med http://dx.doi.org/10.1093/molbev/msp259
25. Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM, et al. Whole-genome random sequencing and assembly ofHaemophilus influenzae Rd. Science 1995; 269:496-512. Pub-Med http://dx.doi.org/10.1126/science.7542800
26. Vallenet D, Belda E, Calteau A, Cruveiller S, Engelen S, Lajus A, Le Fèvre F, Longin C, Mornico D, Roche D, et al. MicroScope—an integrated microbial resource for the curation and compara-tive analysis of genomic and metabolic data. Nu-cleic Acids Res 2013; 41:D636-D647. PubMed http://dx.doi.org/10.1093/nar/gks1194
27. Vallenet D, Labarre L, Rouy Z, Barbe V, Bocs S, Cruveiller S, Lajus A, Pascal G, Scarpelli C, Médigue C. MaGe: a microbial genome annota-tion system supported by synteny results. Nucleic Acids Res 2006; 34:53-65. PubMed http://dx.doi.org/10.1093/nar/gkj406
28. Bocs S, Cruveiller S, Vallenet D, Nuel G, Médigue C. AMIGene: Annotation of MIcrobial Genes. Nucleic Acids Res 2003; 31:3723-3726. PubMed http://dx.doi.org/10.1093/nar/gkg590
29. Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in ge-nomic sequence. Nucleic Acids Res 1997; 25:955-964. PubMed http://dx.doi.org/10.1093/nar/25.5.0955
30. Lagesen K, Hallin P, Rødland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucle-ic Acids Res 2007; 35:3100-3108. PubMed http://dx.doi.org/10.1093/nar/gkm160
31. Gardner PP, Daub J, Tate JG, Nawrocki EP, Kolbe DL, Lindgreen S, Wilkinson AC, Finn RD, Grif-fiths-Jones S, Eddy SR, Bateman A. Rfam: updates to the RNA families database. Nucleic Acids Res 2009; 37:D136-D140. PubMed http://dx.doi.org/10.1093/nar/gkn766
32. Hyatt D, Chen GL, Locascio PF, Land ML, Lar-imer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identifi-cation. BMC Bioinformatics 2010; 11:119. Pub-Med http://dx.doi.org/10.1186/1471-2105-11-119
33. UnitProt Consortium. The Universal Protein Re-source (UniProt) 2009. Nucleic Acids Res 2009; 37:D169-D174. PubMed http://dx.doi.org/10.1093/nar/gkn664
34. Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, et al. The COG database: an updated version includes eukary-otes. BMC Bioinformatics 2003; 4:41. PubMed http://dx.doi.org/10.1186/1471-2105-4-41
35. Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Das U, Daugherty L, Duquenne L, et al. InterPro: the integrative pro-tein signature database. Nucleic Acids Res 2009; 37:D211-D215. PubMed http://dx.doi.org/10.1093/nar/gkn785
36. Claudel-Renard C, Chevalet C, Faraut T, Kahn D. Enzyme-specific profiles for genome annotation: PRIAM. Nucleic Acids Res 2003; 31:6633-6639. PubMed http://dx.doi.org/10.1093/nar/gkg847

Lemriss et al.
http://standardsingenomics.org 1127
37. Sonnhammer EL, von Heijne G, Krogh A. A hid-den Markov model for predicting transmembrane helices in protein sequences. Proc Int Conf Intell Syst Mol Biol 1998; 6:175-182. PubMed
38. Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 2004; 340:783-795. PubMed http://dx.doi.org/10.1016/j.jmb.2004.05.028
39. Gardy JL, Laird MR, Chen F, Rey S, Walsh CJ, Es-ter M, Brinkman FS. PSORTb v.2.0: expanded prediction of bacterial protein subcellular locali-zation and insights gained from comparative pro-
teome analysis. Bioinformatics 2005; 21:617-623. PubMed http://dx.doi.org/10.1093/bioinformatics/bti057
40. Moller S, Croning MDR, Apweiler R. Evaluation of methods for the prediction of membrane span-ning regions. Bioinformatics 2001; 17:646-653. PubMed http://dx.doi.org/10.1093/bioinformatics/17.7.646

Genome Sequences of Four Staphylococcus capitis NRCS-A Isolatesfrom Geographically Distant Neonatal Intensive Care Units
H. Lemriss,a S. Lemriss,b P. Martins-Simoes,c,d,e M. Butin,c L. Lahlou,a J.-P. Rasigade,c,d A. Kearns,f O. Denis,g M. Deighton,h
A. Ibrahimi,a F. Laurent,c,d,e S. El Kabbajb
Biotechnology Laboratory (Medbiotech), Medical and Pharmacy School, University Mohammed V de Rabat, Rabat, Moroccoa; Department of Biosecurity PCL3, Laboratoryof Research and Medical Analysis of the Fraternal of Gendarmerie Royale, Rabat, Moroccob; International Centre for Research in Infectious diseases, INSERM, University ofLyon, Lyon, Francec; Department of Clinical Microbiology, Northern Hospital Group, Hospices Civils de Lyon, Lyon, Franced; National Reference Center for Staphylococci,Hospices Civils de Lyon, Lyon, Francee; Public Health England, Colindale, London, United Kingdomf; Erasme Hospital, Université Libre de Bruxelles, Brussels, Belgiumg;RMIT University, Bundoora, Victoria, Australiah
Staphylococcus capitis pulsotype NRCS-A was previously reported as a frequent cause of late-onset sepsis in neonatal intensivecare units (NICUs) worldwide. Here, we report the whole-genome shotgun sequences of four S. capitis pulsotype NCRS-Astrains, CR03, CR04, CR05, and CR09, isolated from Belgium, Australia, the United Kingdom, and France, respectively.
Received 15 April 2015 Accepted 16 June 2015 Published 6 August 2015
Citation Lemriss H, Lemriss S, Martins-Simoes P, Butin M, Lahlou L, Rasigade J-P, Kearns A, Denis O, Deighton M, Ibrahimi A, Laurent F, El Kabbaj S. 2015. Genome sequences offour Staphylococcus capitis NRCS-A isolates from geographically distant neonatal intensive care units. Genome Anounc 3(4):e00501-15. doi:10.1128/genomeA.00501-15.
Copyright © 2015 Lemriss et al. This is an open-access article distributed under the terms of the Creative Commons Attribution 3.0 Unported license.
Address correspondence to S. Lemriss, [email protected].
Coagulase-negative staphylococci (CoNS) are the most fre-quently encountered pathogens in neonatal intensive care
units (NICUs) (1). However, in the NICU setting, recent studieshave indicated that methicillin-resistant Staphylococcus capitismight emerge as a significant pathogen, causing late-onset sepsis(LOS) in several neonatal intensive care units in France, the Neth-erlands, and Australia (2–4).
A study in French NICUs demonstrated the spread of a singleclonal population of methicillin-resistant S. capitis (pulsotypeNRCS-A) associated with reduced susceptibility to vancomycin,which is the first line of antibiotics used in cases of LOS. Moreover,this clone has also been recently identified in NICUs in Belgium,the United Kingdom, and Australia, suggesting a worldwide dis-tribution (5, 6).
In this report, we present the draft genome sequences of four S.capitis (pulsotype NRCS-A) strains (CR03, CR04, CR05, andCR09) isolated from blood cultures from four neonates hospital-ized in NICUs in Belgium, Australia, the United Kingdom, andFrance, respectively.
All S. capitis strains were grown in blood agar at 37°C, andgenomic DNA was extracted using the PureLink genomic DNA kit(Invitrogen), according to the manufacturer’s recommended pro-tocol. The quantity of DNA was determined using a NanoVue Plus(HVD Lifesciences), and 1 �g of DNA was used to sequence the
whole genome of each strain. The 454-shotgun libraries were pre-pared from the extracted genomic DNA following GS rapid libraryprotocol (Roche 454; Roche).
The genome sequence of each S. capitis strain was determinedby high-throughput sequencing performed on a Genome Se-quencer FLX� system (454 Life Sciences/Roche) using FLX Tita-nium reagents, according to the manufacturer’s protocols and in-structions. De novo assemblies were performed using the RocheNewbler (version 2.9) software package, and the sequencing re-sults are summarized in Table 1.
An automatic syntactic and functional annotation of the draftgenome was performed using the MicroScope platform pipeline(7, 8). The syntactic analysis combines a set of programs, includ-ing AMIGene (9), tRNAscan-SE (10), RNAmmer (11), Rfam scan(12), and Prodigal software (13) to predict genomic objects thatare mainly coding sequences (CDSs) and RNA genes. More than20 bioinformatics methods were used for functional and relationalanalyses. The homology search was performed in the generalistdatabank UniProt (14) and in more specialized databases, such asCOG (15), InterPro (16), PRIAM profiles for enzymatic classifi-cation (17), prediction of protein localization using TMHMM(18), SignalP (19), and PSORTb (20) tools.
The chromosome of strain CR03 (ENA accession no.CTEB01000000) contains 2,575 genes, 2,466 coding sequences
TABLE 1 Summary of genome sequencing results in the present study
S. capitisstrain Country source
Reads(Mb)
Foldcoverage (�)
No. ofcontigs
Genomesize (bp)
G�Ccontent (%) Accession no.
CR03 Belgium 141,728 30 31 2,508,352 32.81 CTEB01000000CR04 Australia 132,280 30 38 2,512,289 32.80 CTEM01000000CR05 United Kingdom 139,569 31 39 2,543,917 32.84 CTEO01000000CR09 France 132,205 30 34 2,490,458 32.82 CTEL01000000
crossmark
Genome AnnouncementsJuly/August 2015 Volume 3 Issue 4 e00501-15 genomea.asm.org 1
on Novem
ber 15, 2015 by guesthttp://genom
ea.asm.org/
Dow
nloaded from

(CDSs), 4 rRNAs, and 61 tRNAs; the chromosome of strain CR04(accession no. CTEM01000000) contains 2,566 genes, 2,457CDSs, 4 rRNAs, and 60 tRNAs; the chromosome of strain CR05(accession no. CTEO01000000) contains 2,624 genes, 2,508 CDSs,4 rRNAs, and 60 tRNAs; and the chromosome of strain CR09(accession no. CTEL01000000) contains 2,540 genes, 2,432 CDSs,4 rRNAs, and 59 tRNAs.
Nucleotide sequence accession numbers. This whole-genomeshotgun project has been deposited at the ENA database under theaccession numbers listed in Table 1. The versions described in thispaper are in the first versions, under BioProject designation no.PRJEB8618.
ACKNOWLEDGMENTS
This work was supported by a grant from the Fondation pour la RechercheMédicale (FRM) (grant ING20111223510) and by the Institut National dela Recherche Médicale (INSERM) and the French Ministry of Health. Thiswork was also supported by a grant from the NIH for H3Africa BioNet.
REFERENCES1. Klingenberg C, Rønnestad A, Anderson AS, Abrahamsen TG, Zorman
J, Villaruz A, Flægstad T, Otto M, Sollid JE, Ericson J. 2007. Persistentstrains of coagulase-negative staphylococci in a neonatal intensive careunit: virulence factors and invasiveness. Clin Microbiol Infect 13:1100 –1111. http://dx.doi.org/10.1111/j.1469-0691.2007.01818.x.
2. Rasigade J-P, Raulin O, Picaud J-C, Tellini C, Bes M, Grando J, BenSaïd M, Claris O, Etienne J, Tigaud S, Laurent F. 2012. Methicillin-resistant Staphylococcus capitis with reduced vancomycin susceptibilitycauses late-onset sepsis in intensive care neonates. PLoS One 7:e31548.http://dx.doi.org/10.1371/journal.pone.0031548.
3. Van Der Zwet WC, Debets-Ossenkopp YJ, Reinders E, Kapi M,Savelkoul PH, Van Elburg RM, Hiramatsu K, Vandenbroucke-GraulsCM. 2002. Nosocomial spread of a Staphylococcus capitis strain with het-eroresistance to vancomycin in a neonatal intensive care unit. J Clin Mi-crobiol 40:2520 –2525. http://dx.doi.org/10.1128/JCM.40.7.2520-2525.2002.
4. D’mello D, Daley AJ, Rahman MS, Qu Y, Garland S, Pearce C,Deighton MA. 2008. Vancomycin heteroresistance in bloodstream iso-lates of Staphylococcus capitis. J Clin Microbiol 46:3124 –3126. http://dx.doi.org/10.1128/JCM.00592-08.
5. Cui B, Smooker PM, Rouch DA, Daley AJ, Deighton MA. 2013. Differ-ences between two clinical Staphylococcus capitis subspecies as revealed bybiofilm, antibiotic resistance, and pulsed-field gel electrophoresis profil-ing. J Clin Microbiol 51:9 –14. http://dx.doi.org/10.1128/JCM.05124-11.
6. Butin M, Martins Simoes P, Lemriss H, Lemriss S, Vandenesch F, ClarisO, Picaud JC, Rasigade JP, Laurent F. 2014. Staphylococcus capitis inneonatal late-onset sepsis: unexpected worldwide dissemination of an en-demic multi-resistant clone, poster 011. 5th Cong Eur Acad Pediatr Soc,17 to 21 October 2014, Barcelona, Spain.
7. Vallenet D, Belda E, Calteau A, Cruveiller S, Engelen S, Lajus A, LeFèvre F, Longin C, Mornico D, Roche D, Rouy Z, Salvignol G, ScarpelliC, Thil Smith AA, Weiman M, Médigue C. 2013. MicroScope—an
integrated microbial resource for the curation and comparative analysis ofgenomic and metabolic data. Nucleic Acids Res 41:D636 –D647. http://dx.doi.org/10.1093/nar/gks1194.
8. Vallenet D, Labarre L, Rouy Z, Barbe V, Bocs S, Cruveiller S, Lajus A,Pascal G, Scarpelli C, Médigue C. 2006. MaGe: a microbial genomeannotation system supported by synteny results. Nucleic Acids Res 34:53– 65. http://dx.doi.org/10.1093/nar/gkj406.
9. Bocs S, Cruveiller S, Vallenet D, Nuel G, Médigue C. 2003. AMIGene:annotation of microbial genes. Nucleic Acids Res 31:3723–3726. http://dx.doi.org/10.1093/nar/gkg590.
10. Lowe TM, Eddy SR. 1997. tRNA scan-SE: a program for improved detec-tion of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964. http://dx.doi.org/10.1093/nar/25.5.0955.
11. Lagesen K, Hallin P, Rødland EA, Staerfeldt HH, Rognes T, UsseryDW. 2007. RNAmmer: consistent and rapid annotation of ribosomalRNA genes. Nucleic Acids Res 35:3100 –3108. http://dx.doi.org/10.1093/nar/gkm160.
12. Gardner PP, Daub J, Tate JG, Nawrocki EP, Kolbe DL, Lindgreen S,Wilkinson AC, Finn RD, Griffiths-Jones S, Eddy SR, Bateman A. 2009.Rfam: updates to the RNA families database. Nucleic Acids Res 37:D136 –D140. http://dx.doi.org/10.1093/nar/gkn766.
13. Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ.2010. Prodigal: prokaryotic gene recognition and translation initiation siteidentification. BMC Bioinformatics 11:119. http://dx.doi.org/10.1186/1471-2105-11-119.
14. UnitProt Consortium. 2009. The Universal Protein Resource (UniProt)2009. Nucleic Acids Res 37:D169 –D174. http://dx.doi.org/10.1093/nar/gkn664.
15. Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, KooninEV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS,Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA.2003. The COG database: an updated version includes eukaryotes. BMCBioinformatics 4:41. http://dx.doi.org/10.1186/1471-2105-4-41.
16. Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D,Bork P, Das U, Daugherty L, Duquenne L, Finn RD, Gough J, Haft D,Hulo N, Kahn D, Kelly E, Laugraud A, Letunic I, Lonsdale D, Lopez R,Madera M, Maslen J, McAnulla C, McDowall J, Mistry J, Mitchell A,Mulder N, Natale D, Orengo C, Quinn AF, Selengut JD, Sigrist CJ,Thimma M, Thomas PD, Valentin F, Wilson D, Wu CH, Yeats C. 2009.InterPro: the integrative protein signature database. Nucleic Acids Res37:D211–D215. http://dx.doi.org/10.1093/nar/gkn785.
17. Claudel-Renard C, Chevalet C, Faraut T, Kahn D. 2003. Enzyme-specificprofiles for genome annotation: PRIAM. Nucleic Acids Res 31:6633– 6639. http://dx.doi.org/10.1093/nar/gkg847.
18. Sonnhammer EL, von Heijne G, Krogh A. 1998. A hidden Markov modelfor predicting transmembrane helices in protein sequences. Proc Int ConfIntell Syst Mol Biol 6:175–182.
19. Bendtsen JD, Nielsen H, von Heijne G, Brunak S. 2004. Improvedprediction of signal peptides: SignalP 3.0. J Mol Biol 340:783–795. http://dx.doi.org/10.1016/j.jmb.2004.05.028.
20. Gardy JL, Laird MR, Chen F, Rey S, Walsh CJ, Ester M, Brinkman FS.2005. PSORTb v.2.0: expanded prediction of bacterial protein subcellularlocalization and insights gained from comparative proteome analysis.Bioinformatics 21:617– 623. http://dx.doi.org/10.1093/bioinformatics/bti057.
Lemriss et al.
Genome Announcements2 genomea.asm.org July/August 2015 Volume 3 Issue 4 e00501-15
on Novem
ber 15, 2015 by guesthttp://genom
ea.asm.org/
Dow
nloaded from

Genome Sequences of Multiresistant Staphylococcus capitis PulsotypeNRCS-A and Methicillin-Susceptible S. capitis Pulsotype NRCS-C
H. Lemriss,d Y. Dumont,c S. Lemriss,a P. Martins-Simoes,b,c,e M. Butin,c L. Lahlou,d J. P. Rasigade,b,c S. El Kabbaj,a F. Laurent,b,c,e
A. Ibrahimid
Department of Biosecurity PCL3, Research and Medical Analysis Laboratory of Gendarmerie Royale, Rabat, Moroccoa; International Centre for Research in InfectiousDiseases, INSERM U1111, University of Lyon, Lyon, Franceb; Department of Clinical Microbiology, Northern Hospital Group, Hospices Civils de Lyon, Lyon, Francec;Biotechnology Laboratory (Medbiotech), Medical and Pharmacy School, University Mohammed V de Rabat, Rabat, Moroccod; National Reference Center forStaphylococci, Hospices Civils de Lyon, Lyon, Francee
Here, we report the draft genome sequences of methicillin-susceptible Staphylococcus captis pulsotype NCRS-C (CR02 strain)and multiresistant Staphylococcus captis pulsotype NCRS-A (CR07 strain).
Received 28 April 2016 Accepted 29 April 2016 Published 9 June 2016
Citation Lemriss H, Dumont Y, Lemriss S, Martins-Simoes P, Butin M, Lahlou L, Rasigade JP, El Kabbaj S, Laurent F, Ibrahimi A. 2016. Genome sequences of multiresistantStaphylococcus capitis pulsotype NRCS-A and methicillin-susceptible S. capitis pulsotype NRCS-C. Genome Announc 4(3):e00541-16. doi:10.1128/genomeA.00541-16.
Copyright © 2016 Lemriss et al. This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International license.
Address correspondence to S. Lemriss, [email protected].
Staphylococcus capitis is a Gram-positive coccus belonging tothe coagulase-negative staphylococcus group (CoNS) that is
frequently found on the human skin and mucosa (1). Recently, afew studies have reported the emergence of S. capitis, which wasfound to be a major cause of late-onset sepsis (LOS) in severalneonatal intensive care units (NICUs) (2, 3). A clonal populationof methicillin-resistant S. capitis NRCS-A strains has spread intoseveral geographically distant NICUs (4). These isolates exhibitreduced susceptibility to vancomycin, which is the most widelyused antimicrobial agent in the NICU setting (5, 6).
In order to elucidate the molecular mechanisms behind thewide-spreading of methicillin-resistant S. capitis and methicillin-susceptible S. capitis in NICUs in France, we sequenced two dif-ferent pulsotypes (NRCS-A and NRCS-C) of S. capitis strains(CR07 and CR02).
In this report, we present the draft genome sequences ofmultiresistant S. capitis pulsotype NRCS-A and methicillin-susceptible S. capitis pulsotype NRCS-C isolated from bloodcultures from NICUs in Lyon, France.
These two pulsotype strains were grown in blood agar at 37°C,and genomic DNA was extracted using the PureLink genomicDNA kit (Invitrogen), according to the manufacturer’s recom-mended protocol. The DNA libraries were prepared from 1 �gDNA genomic extracted following GS rapid library protocol(Roche 454; Roche).
The genome sequence of each S. capitis strain was determinedby high-throughput sequencing performed on a Genome Se-quencer FLX � system (454 Life Sciences/Roche) using FLX Tita-nium reagents, according to the manufacturer’s protocols and in-structions. De novo assemblies were performed using RocheNewbler assembly software (version 2.9).
An automatic syntactic and functional annotation of the draftgenome was performed using the MicroScope platform pipeline(7). The syntactic analysis combines a set of programs includingAMIGene (8), tRNAscan-SE (9), RNAmmer (10), Rfam scan (11),and Prodigal software (12) to predict genomic objects that are
mainly coding sequences (CDSs) and RNA genes. More than 20bioinformatics methods were used for functional and relationalanalyses. The homology search was performed in the generalistdatabank UniProt (13) and in more specialized databases such asCOG (14), InterPro (15), PRIAM profiles for enzymatic classifi-cation (16), prediction of protein localization using TMHMM(17), SignalP (18), and PsortB (19) tools.
The draft genome sequence of methicillin-susceptible S. capitisNRCS-C (CR02 strain) has 33.05% G�C content, is 2,344,750 bpin length which is distributed in 320 contigs (average coverage,7.0�) with 2,415 genes, 2,476 CDSs, 42 pseudo genes, 4 rRNAs(5S,16S, 23S), and 59 tRNAs.
The draft genome sequence of methicillin-resistant S. capitisNRCS-A (CR07 strain) has 32.83% G�C content, is 2,477,101 bpin length which is distributed in 26 contigs (average coverage,31�) with 2,411 CDSs, 4 rRNAs (5S,16S, 23S), and 61 tRNAs.
Nucleotide sequence accession numbers. These whole-genome sequences were deposited at GenBank under the acces-sion numbers CZWI00000000 and CZWH00000000 for amethicillin-susceptible S. capitis NRCS-C strain (CR02) and amultiresistant S. capitis NRCS-A strain (CR07), respectively. Theversions described in this paper are the first versions.
ACKNOWLEDGMENTS
This work was supported by a grant from the Fondation pour la RechercheMédicale (FRM, grant ING20111223510) and by the Institut National dela Recherche Médicale (INSERM) and the French Ministry of Health. Thiswork was also supported by a grant from the NIH for H3Africa BioNet.
REFERENCES1. Li X, Lei M, Song Y, Gong K, Li L, Liang H, Jiang X. 2014. Whole
genome sequence and comparative genomic analysis of multidrug-resistant Staphylococcus capitis subsp. urealyticus strain LNZR-1. GutPathog 6:45. http://dx.doi.org/10.1186/s13099-014-0045-x.
2. Klingenberg C, Rønnestad A, Anderson AS, Abrahamsen TG, ZormanJ, Villaruz A, Flaegstad T, Otto M, Sollid JE, Ericson J. 2007. Persistentstrains of coagulase negative staphylococci in neonatal intensive care unit:
crossmark
Genome AnnouncementsMay/June 2016 Volume 4 Issue 3 e00541-16 genomea.asm.org 1

virulence factors and invasiveness. Clin Microbiol Infect 13:1100 –1111.http://dx.doi.org/10.1111/j.1469-0691.2007.01818.x.
3. Rasigade J-P, Raulin O, Picaud J-C, Tellini C, Bes M, Grando J, BenSaïd M, Claris O, Etienne J, Tigaud S, Laurent F. 2012. Methicillin-resistant Staphylococcus capitis with reduced vancomycin susceptibilitycauses late-onset sepsis in intensive care neonates. PLoS One 7:e31548.http://dx.doi.org/10.1371/journal.pone.0031548.
4. Butin M, Rasigade JP, Martins-Simões P, Meugnier H, Lemriss H,Goering RV, Kearns A, Deighton MA, Denis O, Ibrahimi A, Claris O,Vandenesch F, Picaud JC, Laurent F. 2016. Wide geographical dissemi-nation of the multiresistant Staphylococcus capitis NRCS-A clone in neo-natal intensive-care units. Clin Microbiol Infect 22:46 –52. http://dx.doi.org/10.1016/j.cmi.2015.09.008.
5. Cui B, Smooker PM, Rouch DA, Daley AJ, Deighton MA. 2013. Differ-ences between two clinical Staphylococcus capitis subspecies as revealed bybiofilm, antibiotic resistance, and pulsed-field gel electrophoresis profil-ing. J Clin Microbiol 51:9 –14. http://dx.doi.org/10.1128/JCM.05124-11.
6. Vallenet D, Belda E, Calteau A, Cruveiller S, Engelen S, Lajus A, LeFèvre F, Longin C, Mornico D, Roche D, Rouy Z, Salvignol G, ScarpelliC, Thil Smith AA, Weiman M, Médigue C. 2013. MicroScope—anintegrated microbial resource for the curation and comparative analysis ofgenomic and metabolic data. Nucleic Acids Res 41:D636 –D647. http://dx.doi.org/10.1093/nar/gks1194.
7. Vallenet D, Labarre L, Rouy Z, Barbe V, Bocs S, Cruveiller S, Lajus A,Pascal G, Scarpelli C, Médigue C. 2006. MaGe: amicrobial genome an-notation system supported by synteny results. Nucleic Acids Res 34:53– 65. http://dx.doi.org/10.1093/nar/gkj406.
8. Bocs S, Cruveiller S, Vallenet D, Nuel G, Médigue C. 2003. AMIGene:annotation of microbial genes. Nucleic Acids Res 31:3723–3726. http://dx.doi.org/10.1093/nar/gkg590.
9. Lowe TM, Eddy SR. 1997. tRNA scan-SE: a program for improved detec-tion of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964. http://dx.doi.org/10.1093/nar/25.5.0955.
10. Lagesen K, Hallin P, Rødland EA, Staerfeldt H-H, Rognes T, UsseryDW. 2007. RNAmmer: consistent and rapid annotation of ribosomalRNA genes. Nucleic Acids Res 35:3100 –3108. http://dx.doi.org/10.1093/nar/gkm160.
11. Gardner PP, Daub J, Tate JG, Nawrocki EP, Kolbe DL, Lindgreen S,
Wilkinson AC, Finn RD, Griffiths-Jones S, Eddy SR, Bateman A. 2009.Rfam: updates to the RNA families database. Nucleic Acids Res 37:D136 –D140. http://dx.doi.org/10.1093/nar/gkn766.
12. Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ.2010. Prodigal: prokaryotic gene recognition and translation initiation siteidentification. BMC Bioinformatics 11:119. http://dx.doi.org/10.1186/1471-2105-11-119.
13. UnitProt Consortium. 2009. The universal protein Re-source (UniProt)2009. Nucleic Acids Res 37:D169 –D174. http://dx.doi.org/10.1093/nar/gkn664.
14. Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, KooninEV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS,Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA.2003. The COG database: an updated version includes eukaryotes. BMCBioinformatics 4:41. http://dx.doi.org/10.1186/1471-2105-4-41.
15. Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D,Bork P, Das U, Daugherty L, Duquenne L, Finn RD, Gough J, Haft D,Hulo N, Kahn D, Kelly E, Laugraud A, Letunic I, Lonsdale D, Lopez R,Madera M, Maslen J, McAnulla C, McDowall J, Mistry J, Mitchell A,Mulder N, Natale D, Orengo C, Quinn AF, Selengut JD, Sigrist CJ,Thimma M, Thomas PD, Valentin F, Wilson D, Wu CH, Yeats C. 2009.InterPro: the integrative protein signature database. Nucleic Acids Res37:D211–D215. http://dx.doi.org/10.1093/nar/gkn785.
16. Claudel-Renard C, Chevalet C, Faraut T, Kahn D. 2003. Enzyme-specificprofiles for genome annotation: PRIAM. Nucleic Acids Res 31:6633– 6639. http://dx.doi.org/10.1093/nar/gkg847.
17. Sonnhammer EL, von Heijne G, Krogh A. 1998. A hidden Markov modelfor predicting transmembrane helices in protein sequences. Proc Int ConfIntell Syst Mol Biol 6:175–182.
18. Bendtsen JD, Nielsen H, von Heijne G, Brunak S. 2004. Improvedprediction of signal peptides: SignalP 3.0. J Mol Biol 340:783–795. http://dx.doi.org/10.1016/j.jmb.2004.05.028.
19. Gardy JL, Laird MR, Chen F, Rey S, Walsh CJ, Ester M, Brinkman FS.2005. PSORTb v.2.0: expanded prediction of bacterial protein subcellularlocalization and insights gained from comparative proteome analysis.Bioinformatics 21:617– 623. http://dx.doi.org/10.1093/bioinformatics/bti057.
Lemriss et al.
Genome Announcements2 genomea.asm.org May/June 2016 Volume 4 Issue 3 e00541-16

Published Ahead of Print 23 September 2013. 10.1128/AAC.01576-13.
2013, 57(12):6354. DOI:Antimicrob. Agents Chemother. S. El Kabbaj, F. Vandenesch and F. Laurent
Picaud,Ginevra, S. Lemriss, R. V. Goering, A. Ibrahimi, J. C. P. Martins Simões, J.-P. Rasigade, H. Lemriss, M. Butin, C. Staphylococcus capitis Pulsotype NRCS-ANeonatal Sepsis-Associated
) in thecop/ars/cad-SCCmec (SCCmecStaphylococcal Cassette Chromosome Characterization of a Novel Composite
http://aac.asm.org/content/57/12/6354Updated information and services can be found at:
These include:
SUPPLEMENTAL MATERIAL Supplemental material
REFERENCEShttp://aac.asm.org/content/57/12/6354#ref-list-1at:
This article cites 22 articles, 14 of which can be accessed free
CONTENT ALERTS more»articles cite this article),
Receive: RSS Feeds, eTOCs, free email alerts (when new
http://journals.asm.org/site/misc/reprints.xhtmlInformation about commercial reprint orders: http://journals.asm.org/site/subscriptions/To subscribe to to another ASM Journal go to:
on Novem
ber 14, 2013 by guesthttp://aac.asm
.org/D
ownloaded from
on N
ovember 14, 2013 by guest
http://aac.asm.org/
Dow
nloaded from

Characterization of a Novel Composite Staphylococcal CassetteChromosome mec (SCCmec-SCCcad/ars/cop) in the NeonatalSepsis-Associated Staphylococcus capitis Pulsotype NRCS-A
P. Martins Simões,a,b,c J.-P. Rasigade,a,b,c H. Lemriss,d,e M. Butin,a,c C. Ginevra,a S. Lemriss,d R. V. Goering,g A. Ibrahimi,e J. C. Picaud,f
S. El Kabbaj,d F. Vandenesch,a,b F. Laurenta,b,c
International Centre for Research in Infectious diseases, INSERM U1111, University of Lyon, Lyon, Francea; Department of Clinical Microbiology, Northern Hospital Group,Hospices Civils de Lyon, Lyon, Franceb; National Reference Center for Staphylococci, Hospices Civils de Lyon, Lyon, Francec; Department of Biosecurity PCL3, Laboratory ofResearch and Medical Analysis of the Fraternal of Gendarmerie Royale, Rabat, Moroccod; Medical and Pharmacy School, University Mohammed V Souissi, Rabat,Moroccoe; Department of Neonatology, Northern Hospital Group, Hospices Civils de Lyon, Lyon, Francef; Department of Medical Microbiology and Immunology,Creighton University School of Medicine, Omaha, Nebraska, USAg
Multiresistant Staphylococcus capitis pulsotype NRCS-A has been reported to be a major pathogen causing nosocomial bactere-mia in preterm infants. We report that the NRCS-A strain CR01 harbors a novel 60.9-kb composite staphylococcal cassette chro-mosome mec (SCCmec) element, composed of an SCCmec with strong homologies to Staphylococcus aureus ST398 SCCmec andof an SCCcad/ars/cop harboring resistance genes for cadmium, arsenic, and copper. Whole-genome-based comparisons of pub-lished S. capitis strains suggest that strain CR01 acquired the two elements independently.
Coagulase-negative staphylococci (CoNS) are common com-mensals of the human skin and mucosa that are now recog-
nized as important opportunistic pathogens that cause nosoco-mial bloodstream and indwelling medical device-related infections(1). Recent studies have reported the emergence of methicillin-resistant, vancomycin-heteroresistant Staphylococcus capitis as animportant cause of late-onset sepsis (LOS) in neonatal intensivecare units (NICUs) in France and other countries (2–6). Surpris-ingly, all S. capitis strains from intensive care neonates were re-ported to belong to a single pulsed-field gel electrophoresis pulso-type, NRCS-A, and to harbor a type V-related staphylococcalcassette chromosome mec (SCCmec) element (6). In contrast, S.capitis bloodstream isolates from adult patients belong to severaldistinct, non-NRCS-A pulsotypes. To gain insights into the ge-netic characteristics of this LOS-specific multiresistant S. capitisclone, we sequenced and characterized the SCCmec element ofone representative strain (CR01) with the NRCS-A pulsotype.
Strain CR01 was isolated from the blood cultures of an infantdiagnosed with LOS in 2007 in the NICU of the Hospices Civils deLyon, France. The strain was part of a previously published col-lection (6), had the pulsotype NRCS-A, and harbored a type V-re-lated SCCmec element. We performed whole-genome pyrose-quencing (454 Life Sciences/Roche) for this strain and usedbioinformatics tools to identify and characterize a novel 60.9-kbcomposite SCCmec element (DDBJ/EMBL/GenBank accessionno. KF049201). The cassette was found to be inserted into thecharacteristic 3= end of the orfX gene (7, 8). To define the SCCregion, we searched for SCC-specific insertion site sequences(ISSs) (9, 10). Three ISSs were found, with the furthest down-stream from orfX being partially deleted. Because the predictedSCC region was found in two different contigs, the SCC sequencewas closed using PCR.
SCCmec characterization and similarities to SCCmec ele-ments of livestock-associated Staphylococcus aureus. A 38.8-kbSCCmec element was identified immediately downstream of orfX.This element harbored a type V (5C2&5) SCCmec and contained42 open reading frames (ORFs; orf1 to orf42 in Table S1 in the
supplemental material), as predicted using the syntactic and func-tional annotation implemented in the MicroScope/MaGe plat-form pipeline (11, 12). Using the MicroScope genome browser,the sequence of the CR01 SCCmec element was aligned with 22genomes of methicillin-resistant Staphylococcus strains, which areavailable in the MaGe database. Two genomes in this databasecarried SCCmec elements showing a remarkable similarity to theNRCS-A strain CR01 SCCmec in terms of gene content, gene or-der, and nucleotide identity. Both SCCmec elements were carriedby livestock-associated methicillin-resistant S. aureus (LA-MRSA)strains, namely, the S0385 and 08BA02176 strains, which belongto the highly prevalent LA-MRSA sequence type 398 (ST398) (Fig.1; see also Table S1). A detailed comparison of gene organizationin these three SCCmec elements revealed that the structures of thejoining regions J3 and J2 and the mec and ccr complexes wereidentical to those found in the ST398 LA-MRSA strain S0385,whereas the joining region J1 was identical to that of the ST398LA-MRSA strain 08BA02176. In both the CR01 and the08BA02176 SCCmec elements, the J1 region contained clusteredregularly interspaced short palindromic repeat (CRISPR) lociwith their associated cas genes. CRISPR loci are the sole prokary-otic defense mechanism against foreign DNA conferring an adap-tive immune response, inversely to other defense mechanismssuch as restriction-modification (R-M) systems (13). Using theonline CRISPRFinder software tool (14), we identified twoCRISPR elements in the NRCS-A strain CR01 located immedi-
Received 24 July 2013 Returned for modification 21 August 2013Accepted 25 August 2013
Published ahead of print 23 September 2013
Address correspondence to P. Martins Simões, [email protected].
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01576-13.
Copyright © 2013, American Society for Microbiology. All Rights Reserved.
doi:10.1128/AAC.01576-13
6354 aac.asm.org Antimicrobial Agents and Chemotherapy p. 6354 – 6357 December 2013 Volume 57 Number 12
on Novem
ber 14, 2013 by guesthttp://aac.asm
.org/D
ownloaded from

ately before the second ISS (Fig. 1). The first CRISPR elementconsisted of 16 direct repeats (DRs) and 15 spacers. The secondCRISPR element consisted of three nonconserved DRs and twospacers. This subtype III-A CRISPR locus (15) (including the 2CRISPRs and the cas genes) showed a high similarity to the ele-ment found in the ST398 LA-MRSA strain 08BA02176 SCCmecelement (see Fig. S1 in the supplemental material).
To further investigate the relationships between the CR01SCCmec elements and those of ST398 S. aureus (strains S0385 and08BA02176), we characterized the SCCmec-associated direct re-peat units (dru) using the scheme described by Goering et al.,which has been described as a discriminant epidemiological toolfor differentiating highly uniform epidemic methicillin-resistantstrains (16). dru analysis showed that the SCCmec cassettes of boththe CR01 strains and the ST398 08BA02176 strain shared the samedru type (5a-2d-4a-0-2d-5b-3a-2g-4b-4e-3e), namely, dt11c,which further highlighted the close genetic proximity betweenthese SCCmec elements. Strain S0385 harbored a different drutype, namely, dt10a (dt10a, 5a-2d-4a-0-2d-5b-3a-2g-3b-4e), but
presenting only one mutation in the ninth repeat and one repeatfewer. The dru type found in strain CR01 was identical to the onesfound in other S. capitis NRCS-A strains (n � 12, data not shown),further corroborating the clonality within this lineage.
Collectively, these findings suggest that the S. capitis NRCS-Astrain CR01 and ST398 LA-MRSA strains have undergone a recenthorizontal transfer of SCCmec elements originating from a com-mon source.
SCCcad/ars/cop characterization and relationships to otherSCC elements. Immediately downstream of the SCCmec domain,we identified a second, 22.2-kb-long SCC domain (SCCcad/ars/cop) harboring 29 predicted ORFs (orf43 to orf70 in Table S1 inthe supplemental material), including a type 8 ccrA1B3 complexwith 90% similarity to the ccr complex recently described in S.aureus strain LGA251 carrying a type IX SCCmec and the novelmecC allele (17). A comparison of the overall structure of theSCCcad/ars/cop with other SCC elements revealed only two re-gions with partial similarities to previously described SCC ele-ments. The first region encompassed nine ORFs (orf47 to orf56 in
FIG 1 Comparative structure analysis of the composite staphylococcal cassette chromosome mec SCCmec-SCCcad/asr/cop element of Staphylococcus capitisstrain CR01. The sequence of the SCCmec-SCCcad/asr/cop element of strain CR01 was aligned against whole genomes of ST398 S. aureus strains S0385 and08BA02176 and of S. capitis strain SK14. Open reading frames (ORFs) are shown as arrows indicating the transcription direction and colored according to theSCCmec region to which they belong (J3, mec complex, J2, ccr complex, or J1). Homologous gene clusters in different strains have similar colors. Thechromosomal orfX gene and the transposase IS431 are represented by black and yellow arrows, respectively. Insertion site sequences (ISSs) are indicated byvertical lines and colored stars as follows: light yellow star, ccrC recombinase ISS; dark yellow star, ccrAB recombinase ISS with associated direct repeat (DR)sequences; red star, vestigial ccrAB ISS and DR sequences. The CRISPR-associated genes (cas genes) are indicated in light green within the J3 region, while the ccrcomplex in the SCCcad/ars/cop element is colored in turquoise and the resistance genes in dark red.
Novel Composite SCCmec Element in S. capitis NRCS-A
December 2013 Volume 57 Number 12 aac.asm.org 6355
on Novem
ber 14, 2013 by guesthttp://aac.asm
.org/D
ownloaded from

Table S1), including the ccrA1B3 gene complex; this region had anorganization similar to the regions found in the SCCpbp4 elementof the Staphylococcus epidermidis strain ATCC 12228 and in thetype II SCCmec elements of the S. aureus strains MRSA252 andN315 (18). The second region encompassed 11 ORFs (orf58 toorf69 in Table S1) and included genes for the detoxification ofheavy metals, namely, cadmium (cadDC), arsenic (arsCBR), andcopper (copB). All 11 ORFs were highly homologous to thosefound in the J1 regions of the SCCmec element of CC398 MRSA S.aureus JCSC6943 (19) and of the �SCCmec(h1435) domain inStaphylococcus haemolyticus JCSC1435 (18), with nucleotide iden-tities greater than 97.5%.
Of note, previously published NRCS-A strains have been re-ported to harbor only a type V-related SCCmec element (ccrCcomplex), as determined using the multiplex PCRs described byKondo et al. (6, 20). To understand why these PCRs failed todetect the SCCcad/ars/cop type 8-related ccrAB complex, Kondo’sprimer sequences were aligned to the NRCS-A strain CR01 ccrA1and ccrB3 alleles. Several mismatches were observed between theccrA primers and their target sequence, which likely preventedamplification of the ccrAB complex and so explain the misinter-pretation of Kondo’s scheme.
To assess whether SCC elements are present in other S. ca-pitis strains, we performed a BLAST search (http://blast.ncbi.nlm.nih.gov) of the composite SCCmec-SCCcad/ars/cop se-quence against publicly available genomes from methicillin-susceptible S. capitis isolates, namely, those of strains QN1(GenBank accession no. AJJH00000000) (21), SK14 (GenBank
accession no. ACFR00000000), and VCU116 (GenBank acces-sion no. AFTX00000000). None of these strains harbored anSCCmec element. Interestingly, whereas strain QN1 had an in-tact orfX gene, both the SK14 and the VCU116 strains carriedhomologous SCCcad/ars/cop elements inserted into the 3= endof orfX. These latter elements were highly similar to the SCCcad/ars/cop found in the NRCS-A strain CR01 (Fig. 1; see also Table S1 inthe supplemental material), except for the first seven ORFs afterthe insertion at the end of orfX. Gene organization and nucleotidesequences were identical in 22 remaining ORFs (orf47 to orf49and orf53 to orf70 in Table S1).
Evolutionary history of SCCcad/ars/cop and SCCmec acqui-sition by S. capitis NRCS-A. To assess the phylogenetic relation-ships between the S. capitis strains CR01 (NRCS-A clone), QN1,SK14, and VCU116, we performed a whole-genome-based singlenucleotide polymorphism (SNP) analysis and constructed a max-imum likelihood (ML) SNP tree using the snpTree online webserver (22) and PhyML (23), respectively (see the supplementalmaterial). Interestingly, all SCCcad/ars/cop-positive strains clus-tered on a monophyletic clade, separate from the SCC-negativestrain QN1. Furthermore, the sole SCCmec-positive strain (CR01)was the most divergent compared with strains SK14 and VCU116(Fig. 2a). These results suggest the occurrence of at least two in-dependent SCC acquisition events within the S. capitis species(Fig. 2b). The first acquisition of the SCCcad/ars/cop occurred inthe ancestral lineage of strains CR01, SK14, and VCU116, whichwas followed by later acquisition of the SCCmec element in theancestor lineage of the NRCS-A clone.
FIG 2 Phylogenetic analysis of whole-genome-sequenced S. capitis strains and proposed evolutionary history of SCC element acquisition in S. capitis. (a)SNP-based maximum likelihood tree of the 4 S. capitis whole-genome sequences. A total of 27,836 SNPs (indels excluded) were obtained by alignment againstthe reference strain S. epidermidis ATCC 12228. Numbers at the branching points represent the aLRT branch support score obtained. Proposed acquisition eventsof SCCcad/ars/cop and SCCmec elements are indicated by dark and light gray arrows, respectively. (b) Proposed model of sequential acquisition of SCC elementsin the S. capitis clade: in a first event, the SCCcad/ars/cop element harboring multiple resistance genes to heavy metals was inserted via site-specific recombinationat the end of the orfX gene; a second insertion occurred in the SCCcad/ars/cop-positive strain CR01 with the insertion of SCCmec at the 3= end of orfX.Abbreviations: Ancest1, first ancestor; Ancest2, second ancestor (of CR01, SK14, and VCU116).
Martins Simões et al.
6356 aac.asm.org Antimicrobial Agents and Chemotherapy
on Novem
ber 14, 2013 by guesthttp://aac.asm
.org/D
ownloaded from

Conclusions. The highly clonal population of S. capitisNRCS-A, which is endemic in NICUs in several countries distantfrom one another (5), harbors a novel SCCmec-SCCcad/ars/copcomposite island that has likely emerged from two independentacquisition events.
The high degree of structural similarity of the SCCmec type Velement and the partial conservation of genes for heavy metaldetoxification strongly suggest the occurrence of genetic shufflinginvolving S. capitis and CC398 MRSA strains. Whether SCC ele-ment exchanges occurred directly between these strains or indi-rectly via a common SCC element donor is presently unclear andwarrants further investigation.
ACKNOWLEDGMENTS
This work was supported by the Institut National de la Recherche Médi-cale (INSERM) and the French Ministry of Health and by a grant from theFondation pour la Recherche Médicale (FRM) (grant ING20111223510).
REFERENCES1. Otto M. 2009. Staphylococcus epidermidis—the “accidental” pathogen.
Nat. Rev. Microbiol. 7:555–567.2. Gras-Le Guen C, Fournier S, Andre-Richet B, Caillon J, Chamoux C,
Espaze E, Richet H, Roze JC, Lepelletier D. 2007. Almond oil implicatedin a Staphylococcus capitis outbreak in a neonatal intensive care unit. J.Perinatol. 27:713–717.
3. Ng PC, Chow VC, Lee CH, Ling JM WH, Chang R. 2006. PersistentStaphylococcus capitis septicemia in a preterm infant. Pediatr. Infect. Dis. J.25:652– 654.
4. De Silva GDI, Kantzanou M, Justice A, Massey RC, Wilkinson AR, DayNPJ, Peacock SJ. 2002. The ica operon and biofilm production in coag-ulase-negative staphylococci associated with carriage and disease in a neo-natal intensive care unit. J. Clin. Microbiol. 40:382–388.
5. Rasigade J-P, Goering R, Kearns A, Hill R, Deighton M, Meugnier H,Bes M, Ben Saïd M, Claris O, Picaud J-C, Vandenesch F, Etienne J,Laurent F. 2012. Worldwide epidemic methicillin-resistant Staphylococ-cus capitis with reduced vancomycin susceptibility causes late-onset sepsisin intensive care neonates, p 06-075. 15th Int. Symp. Staphylococci Staph-ylococcal Infect., Lyon, France, 26 to 30 August 2012.
6. Rasigade J-P, Raulin O, Picaud J-C, Tellini C, Bes M, Grando J, BenSaïd M, Claris O, Etienne J, Tigaud S, Laurent F. 2012. Methicillin-resistant Staphylococcus capitis with reduced vancomycin susceptibilitycauses late-onset sepsis in intensive care neonates. PLoS One 7:e31548.doi:10.1371/journal.pone.0031548.
7. Ito T, Katayama Y, Asada K, Mori N, Tsutsumimoto K, Tiensasitorn C,Hiramatsu K. 2001. Structural comparison of three types of staphylococ-cal cassette chromosome mec integrated in the chromosome in methicil-lin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 45:1323–1336.
8. Ma XX, Ito T, Tiensasitorn C, Jamklang M, Chongtrakool P, Boyle-Vavra S, Daum RS, Hiramatsu K. 2002. Novel type of staphylococcalcassette chromosome mec identified in community-acquired methicillin-resistant Staphylococcus aureus strains. Antimicrob. Agents Chemother.46:1147–1152.
9. Ito T, Ma XX, Takeuchi F, Okuma KYH, Hiramatsu K. 2004. Novel typeV staphylococcal cassette chromosome mec driven by a novel cassette
chromosome recombinase, ccrC. Antimicrob. Agents Chemother. 48:2637–2651.
10. Noto MJ, Archer GL. 2006. A subset of Staphylococcus aureus strainsharboring staphylococcal cassette chromosome mec (SCCmec) type IV isdeficient in CcrAB-mediated SCCmec excision. Antimicrob. Agents Che-mother. 50:2782–2788.
11. Vallenet D, Labarre L, Rouy Z, Barbe V, Bocs S, Cruveiller S, Lajus A,Pascal G, Scarpelli C, Médigue C. 2006. MaGe: a microbial genome anno-tation system supported by synteny results. Nucleic Acids Res. 34:53–65.
12. Vallenet D, Engelen S, Mornico D, Cruveiller S, Fleury L, Lajus A, RouyZ, Roche D, Salvignol G, Scarpelli C, Médigue C. 2009. MicroScope: aplatform for microbial genome annotation and comparative genomics.Database 2009:bap021. doi:10.1093/database/bap021.
13. Sorek R, Lawrence CM, Wiedenheft B. 2013. CRISPR-mediated adaptiveimmune systems in Bacteria and Archaea. Annu. Rev. Biochem. 82:237–266.
14. Makarova KS, Haft DH, Barrangou R, Brouns SJJ, Charpentier E,Horvath P, Moineau S, Mojica FJM, Wolf YI. 2011. Evolution andclassification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 9:467– 477.
15. Grissa I, Vergnaud G, Pourcel C. 2007. CRISPRFinder: a web tool toidentify clustered regularly interspaced short palindromic repeats. NucleicAcids Res. 35:W52–W57.
16. Goering RV, Morrison D, Al-Doori Z, Edwards GFS, Gemmell CG.2008. Usefulness of mec-associated direct repeat unit (dru) typing in theepidemiological analysis of highly clonal methicillin-resistant Staphylo-coccus aureus in Scotland. Clin. Microbiol. Infect. 14:964 –969.
17. Shore AC, Deasy EC, Slickers P, Brennan G, O’Connell B, Monecke S,Ehricht R, Coleman DC. 2011. Detection of staphylococcal cassette chro-mosome mec type XI carrying highly divergent mecA, mecI, mecR1, blaZ,and ccr genes in human clinical isolates of clonal complex 130 methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 55:3765–3773.
18. Mongkolrattanothai K, Boyle S, Murphy TV, Daum RS. 2004. Novelnon-mecA-containing staphylococcal chromosomal cassette compositeisland containing pbp4 and tagF genes in a commensal staphylococcalspecies: a possible reservoir for antibiotic resistance islands in Staphylo-coccus aureus. Antimicrob. Agents Chemother. 48:1823–1836.
19. Li S, Skov RL, Han X, Larsen AR, Larsen J, Sørum M, Wulf M, Voss A,Hiramatsu K, Ito T. 2011. Novel types of staphylococcal cassette chro-mosome mec elements identified in clonal complex 398 methicillin-resistant Staphylococcus aureus strains. Antimicrob. Agents Chemother.55:3046 –3050.
20. Kondo Y, Ito T, Ma XX, Watanabe S, Kreiswirth BN, Etienne J,Hiramatsu K. 2007. Combination of multiplex PCRs for staphylococcalcassette chromosome mec type assignment: rapid identification system formec, ccr, and major differences in junkyard regions. Antimicrob. AgentsChemother. 51(1):264 –274.
21. Qin N, Ding W, Yao J, Su K, Wu L, Li L. 2012. Genome sequence ofStaphylococcus capitis QN1, which causes infective endocarditis. J. Bacte-riol. 194:4469 – 4470.
22. Leekitcharoenphon P, Kaas RS, Thomsen MCF, Friis C, Rasmussen S,Aarestrup FM. 2012. snpTree—a web-server to identify and constructSNP trees from whole genome sequence data. BMC Genomics 13(Suppl7):S6. doi:10.1186/1471-2164-13-S7-S6.
23. Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, GascuelO. 2010. New algorithms and methods to estimate maximum-likelihoodphylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59:307–321.
Novel Composite SCCmec Element in S. capitis NRCS-A
December 2013 Volume 57 Number 12 aac.asm.org 6357
on Novem
ber 14, 2013 by guesthttp://aac.asm
.org/D
ownloaded from

ORIGINAL RESEARCHpublished: 15 December 2016
doi: 10.3389/fmicb.2016.01991
Frontiers in Microbiology | www.frontiersin.org 1 December 2016 | Volume 7 | Article 1991
Edited by:
Benoit Doublet,
National Institute for Agricultural
Research (INRA), France
Reviewed by:
Olin Silander,
Massey University, New Zealand
Ravi Ranjan,
University of Illinois at Chicago, USA
*Correspondence:
Patrícia Martins Simões
Specialty section:
This article was submitted to
Evolutionary and Genomic
Microbiology,
a section of the journal
Frontiers in Microbiology
Received: 07 September 2016
Accepted: 28 November 2016
Published: 15 December 2016
Citation:
Martins Simões P, Lemriss H,
Dumont Y, Lemriss S, Rasigade J-P,
Assant-Trouillet S, Ibrahimi A,
El Kabbaj S, Butin M and Laurent F
(2016) Single-Molecule Sequencing
(PacBio) of the Staphylococcus capitis
NRCS-A Clone Reveals the Basis of
Multidrug Resistance and Adaptation
to the Neonatal Intensive Care Unit
Environment. Front. Microbiol. 7:1991.
doi: 10.3389/fmicb.2016.01991
Single-Molecule Sequencing(PacBio) of the Staphylococcuscapitis NRCS-A Clone Reveals theBasis of Multidrug Resistance andAdaptation to the Neonatal IntensiveCare Unit EnvironmentPatrícia Martins Simões 1, 2, 3*, Hajar Lemriss 4, Yann Dumont 5, Sanâa Lemriss 6,
Jean-Philippe Rasigade 2, 3, Sophie Assant-Trouillet 1, 2, Azeddine Ibrahimi 4,
Saâd El Kabbaj 6, Marine Butin 1, 7 and Frédéric Laurent 1, 2, 3
1Department of Clinical Microbiology, Northern Hospital Group, Hospices Civils de Lyon, Lyon, France, 2 International Centre
for Research in Infectious Diseases, Institut National de la Santé et de la Recherche Médicale U1111, University of Lyon,
Lyon, France, 3National Reference Center for Staphylococci, Hospices Civils de Lyon, Lyon, France, 4Biotechnology
Laboratory (Medbiotech), Medical and Pharmacy School, University Mohammed V de Rabat, Rabat, Morocco, 5Department
of Clinical Microbiology, Eastern Hospital Group, Hospices Civils de Lyon, Lyon, France, 6Department of Biosecurity PCL3,
Laboratory of Research and Medical Analysis of the Fraternal of Gendarmerie Royale, Rabat, Morocco, 7Neonatal Intensive
Care Unit, Eastern Hospital Group, Hospices Civils de Lyon, Lyon, France
The multi-resistant Staphylococcus capitis clone NRCS-A has recently been described
as a major pathogen causing nosocomial, late-onset sepsis (LOS) in preterm neonates
worldwide. NRCS-A representatives exhibit an atypical antibiotic resistance profile.
Here, the complete closed genome (chromosomal and plasmid sequences) of NRCS-A
prototype strain CR01 and the draft genomes of three other clinical NRCS-A strains from
Australia, Belgium and the United Kingdom are annotated and compared to available
non-NRCS-A S. capitis genomes. Our goal was to delineate the uniqueness of the
NRCS-A clone with respect to antibiotic resistance, virulence factors and mobile genetic
elements. We identified 6 antimicrobial resistance genes, all carried by mobile genetic
elements. Previously described virulence genes present in the NRCS-A genomes are
shared with the six non-NRCS-A S. capitis genomes. Overall, 63 genes are specific to
the NRCS-A lineage, including 28 genes located in the methicillin-resistance cassette
SCCmec. Among the 35 remaining genes, 25 are of unknown function, and 9 correspond
to an additional type I restriction modification system (n = 3), a cytosine methylation
operon (n = 2), and a cluster of genes related to the biosynthesis of teichoic acids
(n = 4). Interestingly, a tenth gene corresponds to a resistance determinant for nisin
(nsr gene), a bacteriocin secreted by potential NRCS-A strain niche competitors in the
gut microbiota. The genomic characteristics presented here emphasize the contribution
of mobile genetic elements to the emergence of multidrug resistance in the S. capitis
NRCS-A clone. No NRCS-A-specific known virulence determinant was detected, which
does not support a role for virulence as a driving force of NRCS-A emergence in
NICUs worldwide. However, the presence of a nisin resistance determinant on the

Martins Simões et al. Genomic Characterization of the NRCS-A Clone
NRCS-A chromosome, but not in other S. capitis strains and most coagulase-negative
representatives, might confer a competitive advantage to NRCS-A strains during the
early steps of gut colonization in neonates. This suggests that the striking adaptation of
NRCS-A to the NICU environment might be related to its specific antimicrobial resistance
and also to a possible enhanced ability to challenge competing bacteria in its ecological
niche.
Keywords: bacteremia, multiple drug resistance, late-onset sepsis, SMRT, nisin, comparative genomics,
Staphylococcus capitis
INTRODUCTION
Coagulase-negative staphylococci (CoNS) are commoncommensals of the human skin and mucosa and are alsoopportunistic pathogens responsible for infections associatedwith indwelling medical devices and bloodstream infections(Otto, 2009). Among CoNSs, Staphylococcus epidermidis is themost prevalent species and is classically involved in nosocomialbacteremia in patients with co-morbidities including verylow-birth weight preterm infants (Otto, 2009). However, theStaphylococcus capitis has also been incriminated in sepsisthroughout the world in neonatal intensive care units (NICUs)(Van Der Zwet et al., 2002; Venkatesh et al., 2006; D’melloet al., 2008; Rasigade et al., 2012; Boghossian et al., 2013; Cuiet al., 2013). We recently reported that a group of closelyrelated multidrug-resistant (MDR) S. capitis strains belongingto the same clone, named NRCS-A, were present in mostFrench NICUs but absent in the other pediatric or adult ICUs(Rasigade et al., 2012). In subsequent work, we demonstrated thedissemination of NRCS-A strains in distant NICUs throughoutthe world (Butin et al., 2016). This dissemination is of majorconcern because of the extensive drug resistance of NRCS-Aas well as its ability to become highly endemic in some NICUs,representing up to 40% of all bacteremia cases (Stoll et al., 2002,2005; Rasigade et al., 2012). Due to the presence of the typeV-related staphylococcal chromosome cassette mec (SCCmec),all S. capitis NRCS-A strains are resistant to beta-lactams andexhibit reduced susceptibility to other antimicrobial agents incommon use in NICUs, including resistance to aminoglycosidesand resistance or heteroresistance to vancomycin (Venkateshet al., 2006; Rasigade et al., 2012). Overall, the worldwidediffusion of a multidrug-resistant S. capitis clone that is highlyadapted to NICU antibiotic selective pressure raises questionsabout the potential genetic support of its epidemiological success.
Few genome sequences of S. capitis are publicly availableto date. In addition, only a single study using the genome ofa S. capitis isolate collected from a bloodstream infection inan adult has reported the genomic comparison of virulencefactors between S. capitis and S. epidermidis (Cameron et al.,2015). Considering the specific importance NRCS-A in theNICU setting and its international dissemination, we presenthere the first closed genome sequence of the French NRCS-A prototype strain CR01, a comparison of the whole-genomesequences (WGS) of three other NRCS-A strains collected fromdistant countries (Australia, Belgium, the United Kingdom), anda description of the genomic features of this NRCS-A clone.
METHODS
Genome Sequencing and AssemblyThe four strains of S. capitis, CR01, CR03, CR04, and CR05, usedin this study were part of a previously published collection, withall strains being of the international S. capitis NRCS-A clone(Butin et al., 2016). The strains were isolated from blood culturesof preterm infants diagnosed with neonatal sepsis in NeonatalIntensive Care Units (NICUs) from four different countries:France (isolate CR01), Belgium (isolate CR03), Australia (isolateCR04) and the United Kingdom (isolate CR05) (for furtherdetails see Supplementary Data).
Initially, whole-genome pyrosequencing (454 LifeSciences/Roche) was performed for all 4 isolates, as previouslyreported (Lemriss et al., 2014, 2015). Single-molecule real-time(SMRT) sequencing was then performed for strain CR01 toobtain a closed reference genome for this clone (SupplementaryData).
Sequencing for de novo assembly was performed using PacBioRS II (Menlo Park, CA, USA). High molecular weight DNA wassheared in a Covaris g-TUBE (Covaris, Woburn, MA, USA) toobtain 20 kb fragments. After shearing, the DNA size distributionwas checked using a Fragment Analyzer (Advanced AnalyticalTechnologies, Ames, IA, USA), and 5µg of the sheared DNAwas used to prepare a SMRTbell library with PacBio SMRTbellTemplate Prep Kit 1 (Pacific Biosciences, Menlo Park, CA, USA)according to the manufacturer’s recommendations. The resultinglibrary was size selected using a BluePippin system (Sage Science,Inc. Beverly, MA, USA) for molecules larger than 11 kb. Therecovered library was sequenced using 1 SMRT cell with P6-C4 chemistry and MagBeads with a PacBio RSII system (PacificBiosciences, Menlo Park, CA, USA) at 240 min movie length.
Based on the SMRT cell sequencing, we generated 50,876 post-filter polymerase reads with an average read length of 15.6 kb andanN50 read length of 21.1 kb. The average coverage was 265X; forde novo assembly, the PacBio module “RS_HGAP_Assembly.2”in SMRTpipe version v2.3.0 was used for continuous long reads(CLR) after polishing and error correction with Quiver, asdescribed previously (Chin et al., 2013). DNA methylation wasdetermined using the RS_Modification_and_Motif_Analysisprotocol within SMRT Portal v2.30, with a standardizedin silico false positive error of ∼1%. Only motifs with amean modification quality value (QV) >50 and a meancoverage of >100X were validated as being modified (https://github.com/PacificBiosciences/Bioinformatics-Training/wiki/Methylome-Analysis-Technical-Note). Two polished contigs,
Frontiers in Microbiology | www.frontiersin.org 2 December 2016 | Volume 7 | Article 1991

Martins Simões et al. Genomic Characterization of the NRCS-A Clone
one corresponding to the chromosome and the other to theplasmid, were obtained. Circularization of both contigs wasachieved by manual comparison and removal of regions ofoverlaps.
Computational AnalysisAutomatic syntactic and functional annotation of theclosed, reference genome of strain CR01 [EMBL accessionnumber: LN866849 (chromosome) and LN866850 (plasmid)]was performed using the MicroScope platform pipeline(Vallenet et al., 2006, 2013). The annotations of the draftgenomes of strains CR03 (EMBL accession number:CVUF01000001-CVUF01000031), CR04 (EMBL accessionnumber: CTEM01000001-CTEM01000038) and CR05 (EMBLaccession number: CTEO01000001-CTEO01000039) were alsoperformed using the MicroScope platform pipeline, as publishedpreviously (Lemriss et al., 2014). Visual inspection and manualcuration were carried out using the MaGe platform and BLASTsearches against NCBI databases.
Virulence factors were identified using the Virulence Factorsdatabase (VFDB; http://www.mgc.ac.cn/VFs/, Chen et al., 2012)and tblastn (using the ncbi-blast-2.2.27+ suite, Camacho et al.,2009) searches against in-house databases of known virulencegenes of staphylococci.
Prophage regions were identified using the PHAST server(http://phast.wishartlab.com, Zhou et al., 2011). The predictedresults were confirmed using the MaGe platform.
For comparison, we obtained publicly available assembliesfor six additional whole-genome sequences of S. capitis:strains QN1 (NCBI Ref. Seq.: NZ_AJTH00000000.1),VCU116 (NCBI Ref. Seq.: NZ_AFTX00000000.1), SK14(NCBI Ref. Seq.: NZ_ACFR00000000.1), LNZR-1 (NCBIRef. Seq.: NZ_JGYJ00000000.1), C87 (NCBI Ref. Seq.:NZ_ACRH00000000.1), and AYP1020 (NCBI Ref. Seq.:NZ_CP007601.1). All six genomes were aligned against theclosed genome of isolate CR01 using Mauve Progressive v.2.3.1(Darling et al., 2010) with default settings.
Resistance genes were identified by a combination ofResFinder v.2.1 (Kleinheinz et al., 2014) and tblastn searchesusing the complete, curated Antibiotic Resistance Proteinsdatabase (multifasta file) available at the ComprehensiveAntibiotic Resistance Database (CARD) (McArthur et al., 2013).For ResFinder, cutoffs of 80% minimum length and 90% identitywere used to search for resistance genes in CR03, CR04, andCR05 assemblies previously performed against the referenceCR01 using Mauve Progressive to determine their locationand genomic context. For detection of resistance genes usingthe CARD complete, curated Antibiotic Resistance Proteinsdatabase, tblastn searches against a database composed of thefour genomes of strains CR01, CR03, CR04, and CR05 wereperformed.
Genes specific to the NRCS-A clone were identified using theMaGe platform “Gene Phyloprofiler” tool (Vallenet et al., 2013)and the four annotated genomes of NRCS-A and public S. capitisgenomes. Briefly, genomes were compared in terms of genecontent using pre-computed homologies and synteny groups.Pairwise comparisons between predicted protein sequences of
the studied genome and the proteins of another genomeallowed computation of ranked hits and determination ofbidirectional best hits (BBH) (for each protein, the three besthits were retained). Putative orthologous relationships betweentwo genomes were defined as gene couples satisfying the BBHcriterion and an alignment threshold for homology set as aminimum of 50% sequence identity along 80% of the lengthof the smallest protein. Mauve alignments were also used tocomplement the genomic context, followed by manual curationof the putative genes exclusively found in only the four NRCS-Agenomes.
Insertion sequences (IS) in the genomes were identified usingthe ISfinder analysis tool (Siguier et al., 2006). Both blastn andblastp queries were performed with default settings. Matcheswith an e-value smaller than 0.5 were manually curated. Toconfirm whether a given IS position was conserved in theother genomes analyzed, the closed genome of isolate CR01 wasaligned with the three other NRCS-A genomes and with the sixpublic genomes using Mauve Progressive v.2.3.1 (Darling et al.,2010) with default settings. ORFs within immediate proximityof conserved IS loci in the NRCS-A genomes were comparedwith the S. capitis reference genome to check for potentialalterations in coding sequences, using both Mauve Progressiveand Mage platform. genomic islands (GIs) were predictedusing IslandViewer 3 (Dhillon et al., 2015) and confirmedby alignment with the three NCRS-A genomes and the sixpublic S. capitis genomes using Mauve Progressive. IS insertionswithin GIs were predicted as mentioned above. NRCS-A-specificGIs were searched in the NCBI database using megablast(Camacho et al., 2009) query, limiting to the Staphylococcustaxon (taxid: 1279). Published GIs from reference genomeswere searched against Staphylococcus aureus Mu50 (NCBIReference Sequence: NC_002758.2), COL (NCBI ReferenceSequence: NC_002951.2), MW2 (NCBI Reference Sequence:NC_003923.1), N315 (NCBI Reference Sequence: NC_002745.2),FPR3757 (NCBI Reference Sequence: NC_007793.1) and S0385(NCBI Reference Sequence: NC_017333.1), and S. epidermidisRP62A (NCBI Reference Sequence: NC_002976.3) using theMage platform [15]. Syntonomes of more than 3 ORFs and withidentity greater than 33% were considered to be GIs.
Investigation of nsr Gene Presence inClinical Bacterial StrainsA panel of 15 strains (Table 1) comprising clinical strains ofS. capitis (NRCS-A and non-NRCS-A) were used for nisinsusceptibility testing. All staphylococcal species were originallyisolated from blood, and species assignment was determinedusing Matrix-Assisted Laser Desorption/Ionization Time ofFlight Mass Spectrometry (MALDI-TOF Vitek MS, Biomerieux,France). After isolation, the strains were cultured on HorseBlood Agar (Biomerieux) and then stored at −80◦. Testing forsusceptibility to nisin was performed using the disk diffusiontest adapted from EUCAST1. Briefly, nisin solutions were
1European Committee on antimicrobial Susceptibility Testing, 2015, http://www.
eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Disk_test_documents/
Manual_v_5.0_EUCAST_Disk_Test.pdf
Frontiers in Microbiology | www.frontiersin.org 3 December 2016 | Volume 7 | Article 1991

Martins Simões et al. Genomic Characterization of the NRCS-A Clone
TABLE 1 | Bacterial strains used in the nisin susceptibility test.
Species NRCS-A clone Strain Setting Country nsr gene
SC yes CR01 NICU FR +
SC yes CR03 NICU BE +
SC yes CR04 NICU AUS +
SC yes CR05 NICU UK +
SC yes CR07 NICU FR +
SC yes BC69 NICU South K +
SC yes AQ62 NICU NO +
SC yes AW77 NICU NZ +
SC yes BI76 NICU USA +
SC no AB51 ICU AUS −
SC no CR02 ICU FR −
SC no AR22 ICU DK −
SC no AY18 ICU GR −
SC no AZ72 ICU Sing −
SC no BA24 ICU USA −
SC, Staphylococcus capitis; FR, France; BE, Belgium; UK, United Kingdom; AUS,
Australia; South K, South Korea; NO, Norway; NZ, New Zealand; USA, United States
of America; DK, Denmark; GR, Greece; Sing, Singapore; NICU, neonatal intensive care
unit; ICU, intensive care unit (adults).
prepared using 2.5% nisin powder (Sigma Aldrich, USA)according to a previously described protocol (Piper et al.,2009). Sterile paper disks (Thermofisher Diagnostics, France)were soaked with 30µg of nisin solution. After an overnightincubation at 36◦C on Horse blood Agar (Biomerieux, France),a 0.5 McF was seeded on Mueller Hinton agar (MHE) plates(Biomerieux, France). A disk containing 30µg of nisin wasplaced in the middle of the plate. The diameter of theinhibition zones was measured after overnight incubation ofthe inoculated MHE plates at 36◦C. The average diameter ofinhibition and SEM were determined from four independentexperiments.
PCRs for detecting the nsr gene were performed usingconsensus primers: pF—5′GGAGATATGGGACCTATGATTGCA 3′ and pR—5′GCTGTAAkTTCwCCkGAACTkGC 3′. These primers were designed based on alignment ofthe nsr gene sequence found in strain CR01 (S. capitis cloneNRCS-A) and sequences retrieved from NCBI after a blastn(ref) search for homologous sequences in staphylococcalWGS genomes [S. hyicus ATCC 11249 (acc. num. CP008747),Staphylococcus sp. TE8 (acc. num. NZ_JMGB00000000.1),S. epidermidis MC28 (acc. num. NZ_ATCZ00000000.2),S. epidermidis VCU128 (acc. num. NZ_AHLI00000000.1)].PCR amplification was conducted in a final volume of 25µL,with 2.0µL bacterial DNA (QuickExtract kit, Qiagen), 2.5µL10x Reaction Buffer, 0.75µL 50mM MgCl2, 1 µL 10mMforward primer, 1µL 100mM reverse primer, 4µL 5mMdNTPs, 1 µL each primer and 0.125µL Taq polymerase(Eurobio, France). The cycling parameters were as follows:(i) initial denaturation for 3 min at 95◦C; (ii) 34 cycles,with 1 cycle consisting of 30 s at 95◦C, 60 s at 60◦C,and 90 s at 72◦C; and (iii) a final extension for 10 min at72◦C.
RESULTS
WGSs of four strains belonging to S. capitis clone NRCS-Aand originating from NICUs in four distant countries wereobtained to investigate lineage-specific virulomes, resistomes,and mobilomes as well as the presence of unshared genes using454 pyrosequencing technology, as reported previously (Lemrisset al., 2014, 2015). Depending on the isolate, assembly of thedraft genomes produced 26 to 39 contigs. Re-sequencing ofstrain CR01 using SMRT technology (PacBio) allowed for thegeneration of closed complete sequences for the chromosomeand the single plasmid that was subsequently used to producescaffolds for the three remaining WGSs.
The length of CR01’s chromosome is 2,522,871 bp and exhibitsa low G + C content (33.02%), as is expected for staphylococci.We identified and annotated 2419 protein-coding regions, 19rRNAs, 62 tRNAs, 34 ncRNAs (including RNAIII), and 20transposases associated with insertion sequence elements andtransposons. Of note, gaps (identified in the draft genomeassemblies of the four NRCS-A isolates (based on 454 pyro-sequencing)) frequently occur within the vicinity of transposases,suggesting that some of the 454 short read assemblies wereunable to bridge the gaps associated with repetitive genomicelements. These data clearly emphasize the suitability of third-generation sequencing technologies, such as PacBio SMRT,for obtaining fully defined de novo assemblies and, thus,for overcoming the issue of both local and global repeats(Koren and Phillippy, 2015). The genomic characteristics ofthe NRCS-A isolates tested in this study are summarized inTable 2.
VirulomeThe presence of virulence-associated genes in the closed genomeof strain CR01 and subsequently the draft genomes of strainsCR03 (Be), CR04 (Aus), and CR05 (UK) were inferred bycomparison with known virulence factors previously reportedfor S. aureus, S. epidermidis, and S. haemolyticus using the VFDBdatabase (Chen et al., 2012). The findings were augmented withBLAST searches of well-characterized staphylococcal virulencefactors and key regulators (Table 3). Comparison of the fourNRCS-A genomes with the six published genomes of non-NRCS-A S. capitis, including QN1, VCU116, SK14, LNZR-1, C87, andAYP1020 strains, showed that none of the known staphylococcalvirulence genes are exclusively carried by the NRCS-Aclone.
Interestingly, the virulome of the NRCS-A S. capitis genomewas quite similar to that of S. epidermidis RP62A, including theicaABDCR and capABC biofilm-related operons, Clp proteasesand multiple copies of PSM beta type 1b in tandem (75% aaidentity to PSMbeta1b of strain RP62) and one PSM alpha (59%aa identity) (Otto, 2012). All PSMs were found in the samegenetic environment as in the S. epidermidisRP62A genome, withconservation of upstream and downstream genes.
As previously observed for the fully closed S. epidermidisRP62a and S. epidermidis ATCC 12228 genomes (Zhang et al.,2003; Gill et al., 2005), no S. aureus toxins, except beta- and delta-hemolysins, were identified in any of the 10 S. capitis genomes
Frontiers in Microbiology | www.frontiersin.org 4 December 2016 | Volume 7 | Article 1991

Martins Simões et al. Genomic Characterization of the NRCS-A Clone
TABLE 2 | General genomic features of S. capitis strain CR01 compared with another three WGS NRCS-A genomes.
Features S. capitis strain CR01 (Fr) S. capitis strain CR03 (Be) S. capitis strain CR04 (Aus) S. capitis strain CR05 (UK)
GC% content 33.02 32.81 32.80 32.84
Nb of phages 1 1 1 3
Nb of plasmids 1 1a 1a 0
Nb and type of insertion
sequence
(nb on chromosome + nb on
plasmid)
IS256: 10 + 0 IS256: 1b+ 0 IS256: 1b+ 0 IS256: 1b
IS1272: 3 + 1 IS1272: 1 + 0 IS1272: 1 + 0 IS1272: 0
IS431mec-like: 2 + 3 IS431mec-like: 2 + 4 IS431mec-like: 2 + 0 IS431mec-like: 2
aPutative plasmid, no definitive data about circularization.b Insertion sequence isolated in a small contig, indicating probable multiple insertions in the complete genome.
(four NRCS-A and six public non-NRCS-A). Similarly, none ofthe genes associated with secretion systems (esxA, esxB, esaA,esaB, esaC, essA, essB, and essC) or S. aureus serine proteases(splA, B, C, D, E, and F) are present in the S. capitis genomes.
ResistomeSeveral antibiotic resistance-associated genes were identified andcorrelated with the specific resistance phenotype previouslyreported for the NRCS-A clone (Rasigade et al., 2012; Table 4).All are located on mobile genetic elements (MGEs). Resistanceto aminoglycosides is related to the bifunctional aminoglycoside-modifying gene aacA-aphD, which is carried on the transposonTn4001 (GenBank accession no. AB682805.1; Lyon et al.,1984). Resistance to methicillin is due to the presence of acomposite SCCmec-SCCcad/ars/cop cassette exclusively presentin the NRCS-A clone, as recently published by our team (MartinsSimões et al., 2013). Genomic comparison between the fourNRCS-A strains showed that this mobile element is nearlyidentical: all ORFs are conserved (100% aa homology), and thefew nucleotide sequence variations occur only in the CRISPRrepeat region, as is expected for these regions. As previouslyreported (Martins Simões et al., 2013), no other antibioticresistance genes were detected for this composite SCC element.Finally, the plasmidic bla operon, coding for blaZ beta-lactamaseand its regulators, is present in all NRCS-A strains, except strainCR05 (UK).
With regard to other antimicrobial families, for whichvariability in resistance profiles have been observed amongNRCS-A isolates, phenotypic antimicrobial susceptibility testingmatched the specific resistance gene contents of each NRCS-Astrain. Plasmidic msrA and tetK genes, respectively involved inerythromycin resistance (with a negative D-test) and tetracyclineresistance, were only identified in strain CR04 (Australia). Instrain CR05 (UK), the gene far1, which is responsible for fusidicacid resistance, is carried by a putative phage.
Mobile ElementsAsmentioned previously, the shortest circular sequence obtainedby SMRT for strain CR01 corresponds to a plasmid of 26.140bp (GC content of 29.25%). It harbors 35 ORFs, including (i)the bla operon coding for penicillinase resistance to penicillins(see above) and (ii) copper resistance-related genes such as copZ,copA, and csoR (copper transcriptional repressor; Figure 1).
Genome comparison of strain CR01’s plasmid with the threeother NRCS-A draft genomes revealed that the plasmid is notconserved in all NRCS-A strains (Figure 1). In strain CR03(Belgium), one contig presents 31 of the 35 ORFs identified onthe CR01 plasmid. Conversely, in strain CR04 (Australia), only 9of the 35 ORFs (corresponding to the blaZRI operon and copperresistance operon) were detected in a contig corresponding toa putative plasmid. Finally, no putative plasmid was detected instrain CR05 (UK).
One intact prophage region was identified in all four NRCS-A genomes. This region shows 100% aa identity to the genomesof strains CR01, CR03, and CR04, but it is degenerate inCR05 (Figure 2). Moreover, this phage exhibits strong homologyto a phage present in strain VCU116 [57.1, 90.2, 76.2% aahomology for regions 1, 2 and 3, respectively (Figure 2)] andless than 30% homology with phage Staphy_StB20_(NCBI Ref.Seq. NC_019915), the closest phage found in the NCBI database.Of note, two additional incomplete prophage regions were alsodetected in strain CR05.
Nineteen distinct insertion sequence (IS) elements, clusteringinto three distinct families (type IS256, n = 4; type IS1272, n= 5; IS431mec, n = 10), were identified in the CR01 genome.The presence of type IS256 was confirmed in all four NRCS-A strains, with two IS256 elements being associated with theaminoglycosides resistance gene aacA-aphD (′aac(6′)-aph(2′′) ontransposon Tn4001. Type IS1272 was also found in strains CR03and CR04 but not in strain CR05, and their genomic locations arenot conserved. Two IS431mec cassettes are present in all NRCS-Agenomes, as they are carried within the SCCmec-SCCcad/ars/copelement that is specific to and conserved among the NRCS-Alineage (Martins Simões et al., 2013).
Finally, other GIs (n = 5) were predicted for CR01 (Table 2)and found in the CR03, CR04, and CR05 strains. However, onlyone was found exclusively in the NRCS-A lineage. This GI (bpposition: 150,780–158,140 in the strain CR01 genome) containssix ORFs: one putative phosphoglycolate phosphatase, oneconserved protein of unknown function and three pseudogenesof phage proteins (bp position: 151,470–152,238). Unexpectedly,two of these pseudogenes present 40 and 46.5% aa identity toListeria monocytogenes phage B025 protein gp27 and the thirdpseudogene presents 60.6% aa identity to L. monocytogenes phageB025 protein gp28. Both gp27 and gp28 proteins have unknownfunctions.
Frontiers in Microbiology | www.frontiersin.org 5 December 2016 | Volume 7 | Article 1991

Martins Simões et al. Genomic Characterization of the NRCS-A Clone
TABLE 3 | Comparison of virulence factors in S. capitis NRCS-A, non-NRCS-A, and S. epidermidis after exclusion of virulence factors present only in S.
aureus.
Function Gene S. epidermidis S. capitis NRCS-A clone S. capitis non-NRCS-A
ATCC 12228 RP62A CR01 (Fr) CR03 (Be) CR04 (Aus) CR05 (UK) SK14 VCU116 C87 QN1 LNZR-1
Adherence atl + + + + + + + + + + +
ebh + + + + + + + + + + +
ebp + + + + + + + + + + +
icaR 0 + + + + + + + + + +
icaA 0 + + + + + + + + + +
icaD 0 + + + + + + + + + +
icaB 0 + + + + + + + + + +
icaC 0 + + + + + + + + + +
sdrF + + 0 0 0 0 0 0 0 0 0
sdrG + + 0 0 0 0 0 0 0 0 0
sdrH + + + + + + + + + + +
Exozymes sspB + + 0 0 0 0 0 0 0 0 0
sspC 0 + 0 0 0 0 0 0 0 0 0
lip + + + + + + + + + + +
geh + + + + + + + + + + +
sspA + + 0 0 0 0 0 0 0 0 0
nuc + + + + + + + + + + +
Host immune invasion capABC + + + + + + + + + + +
Toxins hlb + + + + + + + + + + +
hld + + + + + + + + + + +
hemolysin 0 + + + + + + + + + +
hlIII + + + + + + + + + + +
Proteases ClpP + + + + + + + + + + +
ClpB + + + + + + + + + + +
ClpC + + + + + + + + + + +
ClpX + + + + + + + + + + +
Phenol-soluble modulins psm α + + + + + + + + + + +
psm β1a + + 0 0 0 0 0 0 0 0 0
psm β1B + + + + + + + + + + +
psm β2 + + + + + + + + + + +
psm β3 + + 0 0 0 0 0 0 0 0 0
psm δ + + + + + + + + + + +
psmε + + + + + + + + + + +
psm-mec + + 0 0 0 0 0 0 0 0 0
Esterases esterase1 + + + + + + + + + + +
esterase2 + + + + + + + + + + +
HTH transcription factors SarA + + + + + + + + + + +
SarR + + + + + + + + + + +
SarV + + + + + + + + + + +
SarX + + + + + + + + + + +
SarZ + + + + + + + + + + +
MgrA + + + + + + + + + + +
Rot + + + + + + + + + + +
The presence of virulence genes is indicated by the “+” sign.
Frontiers in Microbiology | www.frontiersin.org 6 December 2016 | Volume 7 | Article 1991

Martins Simões et al. Genomic Characterization of the NRCS-A Clone
TABLE 4 | Genomic profiles of antibiotic resistance for clone NRCS-A strains from four different countries.
Antibiotic Gene Strains Genomic context
CR01 (FR) CR03 (Be) CR04 (Aus) CR05 (UK)
Penicillin blaZ + + + − Plasmid
Methicillin mecA + + + + Chromosome
Tetracycline tetK − − + − Plasmid*
Aminoglycosides aac(6′)-aph(2′′) + + + + Chromosome
Macrolide, Lincosamide and Streptogramin B msr(A) − − + − Plasmid*
Fusidic acid far/fusB − − − + Phage (chromosome)*
All antibiotic resistance genes were found in MGEs. Putative regions in the draft genomes are indicated by an *.
FIGURE 1 | Comparison of the genetic content of the strain CR01 plasmid with the draft genomes of the other three NRCS-A strains. Open reading
frames (ORFs) are shown as arrows indicating the direction of transcription. Homologous gene clusters are indicated by a light gray shadow connecting ORFs present
in distinct plasmid sequences. Light gray arrows represent unknown proteins, unless specified otherwise. Arrows with dotted lines represent partial or truncated
ORFs. Antibiotic resistance genes are colored with a gray gradient; genes associated with resistance to heavy metals are colored in dark gray. Abbreviations: tnp,
transposase; copZ, copper insertion chaperone and transporter component; copA, copper transporter ATPase; csoR, copper-sensing transcriptional repressor; czcD,
potassium/proton-divalent cation antiporter; rep, replication-associated family protein; repA, replication-associated protein RepA; blaZ, beta-lactamase resistance
gene; blaR, regulatory protein BlaR1; blaI, penicillinase repressor; mntH, divalent metal cation transporter MntH, tetK, tetracycline resistance gene.
Methylome and Restriction ModificationSystemsSMRT technology (PacBio) allows for genome-wide detectionof modified nucleotides based on the rate at which DNApolymerase incorporates bases during sequencing (Flusberget al., 2010, Nat. Methods; Roberts et al., 2013). Analysis
of polymerase kinetic profiles in strain CR01 identified 1333methylated positions (Table S2) including 94% (n = 1253)
corresponding to adenine methylations (m6A) and 0.006% (n =
8) to cytosine methylations (m4C). The adenine modifications
observed correlate with the presence of two adenine restriction-
modification systems (hsdMSR 1 pos: 476,473–482,360 bp;
Frontiers in Microbiology | www.frontiersin.org 7 December 2016 | Volume 7 | Article 1991

Martins Simões et al. Genomic Characterization of the NRCS-A Clone
FIGURE 2 | Comparison of the genetic content of the strain CR01 intact prophage present in the other three draft genomes of NRCS-A strains.
Prophage prediction was performed using PHAST software; cR01’s prophage is divided into three regions according to the degree of conservation compared to the
other strains. The mean percent of aa identity in each region is indicated, and a gray gradient is also used to represent the percent similarity.
hsdMSR 2 pos: 686,750–691,866 bp) in the strain CR01 genome,whereas the presence of an mcrBC 5-methylcytosine restrictionsystem (bp position 487,030–492,276) correlates with thecytosine modifications detected. Interestingly, the latter isassociated in tandem with the specific additional hdsMSR 1operon located immediately after the SCCmec-SCCcad/ars/copelement (see above).
NRCS-A Clone Specific GenesComparison of the closed genome of strain CR01 with thedraft genomes of the three other NRCS-A genomes and withthe six public non-NRCS-A genomes revealed a unique setof 63 genes present exclusively in the NRCS-A clone genome(Table S1). Of these, 28 ORFS are carried by the compositeSCCmec-SCCcad/ars/copmobile element, which is related to theacquisition of methicillin resistance (mecA gene). Interestingly,within this cassette, the CRISPR element is specific to the NRCS-A lineage (Martins Simões et al., 2013). Of note, both theclone-specific cassette (28 genes) and its genetic environment(downstream and upstream regions) are fully conserved in thefour NRCS-A strains, which confirmed that the four isolatesbelong to the same clonal population, even though they wereisolated from distant countries/continents. Indeed, it is highlyunlikely that the same SCCmec element (100% identity) could beacquired in four independent events in four distant countries.
Among the 35 remaining genes found exclusively in theNRCS-A lineage, 25 are of unknown function, and 10 correspondto the following: (i) an additional type I restriction modificationsystem (hsdMSR, n= 3 genes), (ii) a cytosine methylation operon(mcrBC, n = 2), (iii) a cluster of four genes known to be
involved in the biosynthesis of teichoic acids [ispD (2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase), tarJ (ribitol-5-phosphate dehydrogenase), tarK (Glycosyl glycerophosphate),and tagF (CDP-glycerol glycerophosphotransferase), n = 4].Of note, the tenth gene encodes a 316 aa protein presenting41% aa homology with the determinant for resistance to nisin(nsr gene) that is present in some Lactococcus lactis strains(Froseth and McKay, 1991; Liu et al., 2005) and which showsprotease activity. Contrary to the plasmidic localization of thensr gene in nisin-resistant L. lactis strains, the nsr gene in theNRCS-A clone is located on the chromosome (position: 521,747–522,694 bp) immediately upstream from the potassium (K+)transporter kdpEDABC operon conserved in all S. capitis isolatesand downstream from the four teichoic acid biosynthesis genes(ispD, tarJ, tarK, and tagF; see above). No IS or transposon-likeregion was identified near the nsr gene.
Phenotypic Assay of Clone NRCS-A NisinResistanceTo assess functional expression of the nisin resistance gene(nsr) found exclusively in the NRCS-A clone, we performed adisk diffusion test with nisin-charged disks using a collection ofS. capitis strains, including nine strains belonging to the NRCS-Aclone and six nsr-negative, non-NRCS-A strains. The S. capitisstrains belonging to the NRCS-A clone presented significantlylower nisin inhibition zones (mean of four independentmeasurements± SEM: 8.81mm± 1.31) than the strains isolatedfrom adults that do not carry the nsr gene (16.5 ± 1.5; theStudent t-test, p < 0.001 vs. 8.8mm ± 1.3 in NRCS-A). Thisconfirms that the nsr gene found exclusively in S. capitis strains
Frontiers in Microbiology | www.frontiersin.org 8 December 2016 | Volume 7 | Article 1991

Martins Simões et al. Genomic Characterization of the NRCS-A Clone
belonging to the neonatal NRCS-A clone is functional andconfers resistance to nisin. Nonetheless, it is known (i) thatS. aureus (SA) can modulate its resistance to nisin and othersmall antibacterial peptides via two-component systems (TCSs)GraSR, NsaSR, BraSR in association with ABC transporters(Howden et al., 2010; Blake et al., 2011; Falord et al., 2011;Kolar et al., 2011; Kawada-Matsuo et al., 2013) and (ii) thatSA strains resistant or heteroresistant to vancomycin (VISAand hVISA) usually present increased cell wall thickening (Cuiet al., 2003). The same mechanisms have been observed inL. lactis resistant to nisin (Kramer et al., 2006; Shin et al.,2016). Thus, to assess whether the resistance to nisin observedin the NRCS-A strains is due exclusively to expression ofthe nsr gene and not to a cell wall-thickening adaptation,complementary experiments are required using nsr+/nsr−isogenic isolates. Such analyses are beyond the scope of thepresent paper.
DISCUSSION
The NRCS-A clone is of major interest due to its worldwidehigh dissemination in NICUs and its prevalence as an agentof neonatal sepsis in preterm newborns (Butin et al., 2016).Comparison between the complete genome of the prototypestrain belonging to the S. capitis NRCS-A clone (strain CR01,France) with the draft genomes of clinical S. capitis isolatesbelonging or not to the NRCS-A clone revealed the presenceof multiple MGEs mediating the atypical antibiotic resistanceprofile of this clone. These data confirm (i) the ability of theNRCS-A clone to adapt to the specific selective pressure of theantibiotics used in NICUs and (ii) emphasize the ability of WGSto provide accurate predictions of the resistance phenotypes ofstaphylococci and to become a promising alternative for culturemethods.
Based on genomic comparisons using previouslycharacterized staphylococcal virulence genes, the NRCS-Avirulome is highly similar to that of other non-NRCS-A S. capitisas well as S. epidermidis RP62A strains, which suggests that thesuccess of this clone in neonates is likely not due to increasedvirulence. Nonetheless, we identified the exclusive presence ofa gene encoding a functional nisin resistance (nsr) gene in theNRCS-A lineage that is seldom found in staphylococci.
Nisin, a 34 aa antimicrobial peptide produced by a group ofgram-positive Lactococcus and Streptococcus species (Shin et al.,2016), is active against a wide range of gram-positive bacteria,including staphylococci. Its mode of action is dual, involving (i)inhibition of cell wall biosynthesis (transglycosylation step), asit binds to and sequesters lipid II from its functional location,and (ii) pore formation (Wiedemann et al., 2001; Peschel andSahl, 2006; Egan et al., 2016). Nisin has been described as akey player of the gut barrier, and it is widely used as foodpreservative due to its potent bactericidal activity. Moreover, ithas been shown that (i) expression of the nsr gene, identifiedin Streptococcus agalactiae, in L. lactis not producing nisininduces resistance to nisin and (ii) the presence of human
nisin-producing lactic acid bacteria in the gut reduces intestinalcolonization by vancomycin-resistant enterococci (Millette et al.,2008).
Taken together, these data and our results strongly suggestthat nisin resistance might be responsible for the potentialincrease in the ability of the NRCS-A clone to establishitself as part of the initial microflora of neonates, in whichthe Lactococcus genus is one of the dominant bacterial taxa(Park et al., 2005; Morelli, 2008). Expression of the NSRpeptidase might enable NRCS-A isolates to establish themselvesin the gut of neonates and also to colonize it and survivelonger than nisin-susceptible bacteria. This may explain theover-representation of these isolates in sepsis processes, eitherthrough direct translocation in blood (Taft et al., 2015) orvia colonization of indwelling devices. Although it remainsunknown how S. capitis NRCS-A strains are able to translocateinto the bloodstream of infants, it is hypothesized that theentry point might be the digestive tract, which is immaturein very preterm infants (Taft et al., 2015). Further studiesare needed to fully characterize the digestive microflora ofvery low-weight preterm-infants and its potential impacton the selection of and the fitness advantage to NRCS-Aisolates.
AUTHOR CONTRIBUTIONS
PM: work, study design, data analysis, and manuscriptpreparation. HL: data analysis and manuscript preparation.YD: data analysis, work, and manuscript preparation. SL: workand manuscript preparation. JR, SA, AI, and SE: manuscriptpreparation. MB and FL: study design and manuscriptpreparation.
FUNDING
The present work was financed by the French Ministry ofHealth and the French Institute for Public Health Surveillance(INVS) - Santé publique France in the framework of theNational Reference Center of Staphylococci and by the grantING20111223510 from the Fondation pour la RechercheMedical(FRM).
ACKNOWLEDGMENTS
We would like to thank Michele Bes and Helene Meugnierat the National Reference Center for Staphylococci in Lyon(France), and our colleagues at the National Reference Center ofStaphylococci in Belgium (Olivier Denis), in the United Kingdom(Angela Kearns) and in Australia (Margaret Deighton).
SUPPLEMENTARY MATERIAL
The Supplementary Material for this article can be foundonline at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.01991/full#supplementary-material
Frontiers in Microbiology | www.frontiersin.org 9 December 2016 | Volume 7 | Article 1991

Martins Simões et al. Genomic Characterization of the NRCS-A Clone
REFERENCES
Blake, K. L., Randall, C. P., and O’Neill, A. J. (2011). In vitro studies indicate a
high resistance potential for the lantibiotic nisin in Staphylococcus aureus and
define a genetic basis for nisin resistance. Antimicrob. Agents Chemother. 55,
2362–2368. doi: 10.1128/AAC.01077-10
Boghossian, N. S., Page, G. P., Bell, E. F., Stoll, B. J., Murray, J. C., Cotten,
C. M., et al. (2013). Late-onset sepsis in very low birth weight infants from
singleton and multiple-gestation births. J. Pediatr. 162, 1120–1124, 1124.e1.
doi: 10.1016/j.jpeds.2012.11.089
Butin, M., Rasigade, J.-P., Simões, P. M., Meugnier, H., Lemriss, H., Goering, R. V.,
et al. (2016). Wide geographical dissemination of multiresistant Staphylococcus
capitis NRCS-A clone in neonatal intensive-care units. Clin. Microbiol. Infect.
22, 46–52. doi: 10.1016/j.cmi.2015.09.008
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al.
(2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421.
doi: 10.1186/1471-2105-10-421
Cameron, D. R., Jiang, J.-H., Hassan, K. A., Elbourne, L. D., Tuck, K. L., Paulsen,
I. T., et al. (2015). Insights on virulence from the complete genome of
Staphylococcus capitis. Front. Microbiol. 6:980. doi: 10.3389/fmicb.2015.00980
Chen, L., Xiong, Z., Sun, L., Yang, J., and Jin, Q. (2012). VFDB 2012 update: toward
the genetic diversity and molecular evolution of bacterial virulence factors.
Nucleic Acids Res. 40, D641–D645. doi: 10.1093/nar/gkr989
Chin, C.-S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C.,
et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read
SMRT sequencing data. Nat. Methods 10, 563–569. doi: 10.1038/nmeth.2474
Cui, B., Smooker, P. M., Rouch, D. A., Daley, A. J., and Deighton, M. A. (2013).
Differences between two clinical Staphylococcus capitis subspecies as revealed
by biofilm, antibiotic resistance, and pulsed-field gel electrophoresis profiling.
J. Clin. Microbiol. 51, 9–14. doi: 10.1128/JCM.05124-11
Cui, L., Ma, X., Sato, K., Okuma, K., Tenover, F. C., Mamizuka, E. M.,
et al. (2003). Cell wall thickening is a common feature of vancomycin
resistance in Staphylococcus aureus. J. Clin. Microbiol. 41, 5–14. doi: 10.1128/
JCM.41.1.5-14.2003
D’mello, D., Daley, A. J., Rahman,M. S., Qu, Y., Garland, S., Pearce, C., et al. (2008).
Vancomycin heteroresistance in bloodstream isolates of Staphylococcus capitis.
J. Clin. Microbiol. 46, 3124–3126. doi: 10.1128/JCM.00592-08
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple
genome alignment with gene gain, loss and rearrangement. PLoS ONE
5:e11147. doi: 10.1371/journal.pone.0011147
Dhillon, B. K., Laird, M. R., Shay, J. A., Winsor, G. L., Lo, R., Nizam,
F., et al. (2015). IslandViewer 3: more flexible, interactive genomic island
discovery, visualization and analysis. Nucleic Acids Res. 43, W104–W108.
doi: 10.1093/nar/gkv401
Egan, K., Field, D., Rea, M. C., Ross, R. P., Hill, C., and Cotter, P. D.
(2016). Bacteriocins: novel solutions to age old spore-related problems? Front.
Microbiol. 7:e461. doi: 10.3389/fmicb.2016.00461
Falord, M., Mäder, U., Hiron, A., Débarbouillé, M., and Msadek, T. (2011).
Investigation of the Staphylococcus aureus GraSR regulon reveals novel links
to virulence, stress response and cell wall signal transduction pathways. PLoS
ONE 6:e21323. doi: 10.1371/journal.pone.0021323
Flusberg, B. A., Webster, D. R., Lee, J. H., Travers, K. J., Olivares, E. C., Clark, T.
A., et al. (2010). Direct detection of DNA methylation during single-molecule,
real-time sequencing. Nat. Methods 7, 461–465. doi: 10.1038/nmeth.1459
Froseth, B. R., and McKay, L. L. (1991). Molecular characterization of the nisin
resistance region of Lactococcus lactis subsp. lactis biovar diacetylactis DRC3.
Appl. Environ. Microbiol. 57, 804–811.
Gill, S. R., Fouts, D. E., Archer, G. L., Mongodin, E. F., Deboy, R. T., Ravel, J., et al.
(2005). Insights on evolution of virulence and resistance from the complete
genome analysis of an early methicillin-resistant Staphylococcus aureus
strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis
strain. J. Bacteriol. 187, 2426–2438. doi: 10.1128/JB.187.7.2426-2438.2005
Howden, B. P., Davies, J. K., Johnson, P. D., Stinear, T. P., and Grayson, M.
L. (2010). Reduced Vancomycin Susceptibility in Staphylococcus aureus,
including Vancomycin-Intermediate and Heterogeneous Vancomycin-
Intermediate Strains: resistance mechanisms, laboratory detection, and
clinical implications. Clin. Microbiol. Rev. 23, 99–139. doi: 10.1128/CMR.
00042-09
Kawada-Matsuo, M., Yoshida, Y., Zendo, T., Nagao, J., Oogai, Y., Nakamura, Y.,
et al. (2013). Three distinct two-component systems are involved in resistance
to the class I bacteriocins, Nukacin ISK-1 and nisin A, in Staphylococcus
aureus. PLoS ONE 8:e69455. doi: 10.1371/journal.pone.0069455
Kleinheinz, K. A., Joensen, K. G., and Larsen,M. V. (2014). Applying the ResFinder
and VirulenceFinder web-services for easy identification of acquired antibiotic
resistance and E. coli virulence genes in bacteriophage and prophage nucleotide
sequences. Bacteriophage 4:e27943. doi: 10.4161/bact.27943
Kolar, S. L., Nagarajan, V., Oszmiana, A., Rivera, F. E., Miller, H. K., Davenport,
J. E., et al. (2011). NsaRS is a cell-envelope-stress-sensing two-component
system of Staphylococcus aureus. Microbiol. Read. Engl. 157, 2206–2219.
doi: 10.1099/mic.0.049692-0
Koren, S., and Phillippy, A. M. (2015). One chromosome, one contig: complete
microbial genomes from long-read sequencing and assembly. Curr. Opin.
Microbiol. 23, 110–120. doi: 10.1016/j.mib.2014.11.014
Kramer, N. E., van Hijum, S. A., Knol, J., Kok, J., and Kuipers, O. P.
(2006). Transcriptome analysis reveals mechanisms by which Lactococcus
lactis acquires nisin resistance. Antimicrob. Agents Chemother. 50, 1753–1761.
doi: 10.1128/AAC.50.5.1753-1761.2006
Lemriss, H., Lemriss, S., Martins-Simoes, P., Butin, M., Lahlou, L., Rasigade, J.-P.,
et al. (2015). Genome sequences of four Staphylococcus capitisNRCS-A isolates
from geographically distant neonatal intensive care units. Genome Announc. 3,
e00501–e00515. doi: 10.1128/genomeA.00501-15
Lemriss, H., Martins Simões, P., Lemriss, S., Butin, M., Ibrahimi, A., El
Kabbaj, S., et al. (2014). Non-contiguous finished genome sequence of
Staphylococcus capitis CR01 (pulsetype NRCS-A). Stand. Genomic Sci. 9,
1118–1127. doi: 10.4056/sigs.5491045
Liu, C.-Q., Su, P., Khunajakr, N., Deng, Y.-M., Sumual, S., Kim, W. S., et al. (2005).
Development of food-grade cloning and expression vectors for Lactococcus
lactis. J. Appl. Microbiol. 98, 127–135. doi: 10.1111/j.1365-2672.2004.02441.x
Lyon, B. R., May, J. W., and Skurray, R. A. (1984). Tn4001: a gentamicin and
kanamycin resistance transposon in Staphylococcus aureus. Mol. Gen. Genet.
193, 554–556.
Martins Simões, P., Rasigade, J.-P., Lemriss, H., Butin, M., Ginevra, C., Lemriss,
S., et al. (2013). Characterization of a novel composite staphylococcal cassette
chromosome mec (SCCmec-SCCcad/ars/cop) in the neonatal sepsis-associated
Staphylococcus capitis pulsotype NRCS-A. Antimicrob. Agents Chemother. 57,
6354–6357. doi: 10.1128/AAC.01576-13
McArthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad, M. A., Baylay, A. J.,
et al. (2013). The comprehensive antibiotic resistance database. Antimicrob.
Agents Chemother. 57, 3348–3357. doi: 10.1128/AAC.00419-13
Millette, M., Cornut, G., Dupont, C., Shareck, F., Archambault, D., and Lacroix, M.
(2008). Capacity of human nisin- and pediocin-producing lactic Acid bacteria
to reduce intestinal colonization by vancomycin-resistant enterococci. Appl.
Environ. Microbiol. 74, 1997–2003. doi: 10.1128/AEM.02150-07
Morelli, L. (2008). Postnatal development of intestinal microflora as influenced
by infant nutrition. J. Nutr. 138, 1791S–1795S. Available online at: http://jn.
nutrition.org/content/138/9/1791S.full#fn-1
Otto, M. (2009). Staphylococcus epidermidis: the “accidental” pathogen. Nat. Rev.
Micro. 7, 555–567. doi: 1038/nrmicro2182
Otto, M. (2012). Molecular basis of Staphylococcus epidermidis infections. Semin.
Immunopathol. 34, 201–214. doi: 10.1007/s00281-011-0296-2
Park, H.-K., Shim, S.-S., Kim, S.-Y., Park, J.-H., Park, S.-E., Kim, H.-J., et al.
(2005). Molecular analysis of colonized bacteria in a human newborn infant
gut. J. Microbiol. 43, 345–353.
Peschel, A., and Sahl, H.-G. (2006). The co-evolution of host cationic
antimicrobial peptides and microbial resistance. Nat. Rev. Microbiol. 4,
529–536. doi: 10.1038/nrmicro1441
Piper, C., Draper, L. A., Cotter, P. D., Ross, R. P., and Hill, C. (2009). A comparison
of the activities of lacticin 3147 and nisin against drug-resistant Staphylococcus
aureus and Enterococcus species. J. Antimicrob. Chemother. 64, 546–551.
doi: 10.1093/jac/dkp221
Rasigade, J.-P., Raulin, O., Picaud, J.-C., Tellini, C., Bes, M., Grando, J., et al.
(2012). Methicillin-resistant Staphylococcus capitis with reduced Vancomycin
susceptibility causes late-onset sepsis in intensive care neonates. PLoS ONE
7:e31548. doi: 10.1371/journal.pone.0031548
Roberts, R. J., Carneiro, M. O., and Schatz, M. C. (2013). The advantages of SMRT
sequencing. Genome Biol. 14:405. doi: 10.1186/gb-2013-14-6-405
Frontiers in Microbiology | www.frontiersin.org 10 December 2016 | Volume 7 | Article 1991

Martins Simões et al. Genomic Characterization of the NRCS-A Clone
Shin, J. M., Gwak, J. W., Kamarajan, P., Fenno, J. C., Rickard, A. H., and Kapila, Y.
L. (2016). Biomedical applications of nisin. J. Appl. Microbiol. 120, 1449–1465.
doi: 10.1111/jam.13033
Siguier, P., Perochon, J., Lestrade, L., Mahillon, J., and Chandler, M. (2006).
ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids
Res. 34, D32–D36. doi: 10.1093/nar/gkj014
Stoll, B. J., Hansen, N., Fanaroff, A. A., Wright, L. L., Carlo, W. A., Ehrenkranz,
R. A., et al. (2002). Late-onset sepsis in very low birth weight neonates: the
experience of the NICHDNeonatal Research Network. Pediatrics 110, 285–291.
doi: 10.1542/peds.110.2.285
Stoll, B. J., Hansen, N. I., Higgins, R. D., Fanaroff, A. A., Duara, S.,
Goldberg, R., et al. (2005). Very low birth weight preterm infants with
early onset neonatal sepsis: the predominance of gram-negative infections
continues in the National Institute of Child Health and Human Development
Neonatal Research Network, 2002-2003. Pediatr. Infect. Dis. J. 24, 635–639.
doi: 10.1097/01.inf.0000168749.82105.64
Taft, D. H., Ambalavanan, N., Schibler, K. R., Yu, Z., Newburg, D. S.,
Deshmukh, H., et al. (2015). Center variation in intestinal Microbiota
prior to late-onset sepsis in preterm infants. PLoS ONE 10:e0130604.
doi: 10.1371/journal.pone.0130604
Vallenet, D., Belda, E., Calteau, A., Cruveiller, S., Engelen, S., Lajus, A., et al. (2013).
MicroScope–an integratedmicrobial resource for the curation and comparative
analysis of genomic and metabolic data. Nucleic Acids Res. 41, D636–D647.
doi: 10.1093/nar/gks1194
Vallenet, D., Labarre, L., Rouy, Z., Barbe, V., Bocs, S., Cruveiller, S., et al. (2006).
MaGe: a microbial genome annotation system supported by synteny results.
Nucleic Acids Res. 34, 53–65. doi: 10.1093/nar/gkj406
Van Der Zwet, W. C., Debets-Ossenkopp, Y. J., Reinders, E., Kapi, M., Savelkoul,
P. H., Elburg, R. M., et al. (2002). Nosocomial spread of a Staphylococcus capitis
strain with heteroresistance to vancomycin in a neonatal intensive care unit. J.
Clin. Microbiol. 40, 2520–2525. doi: 10.1128/JCM.40.7.2520-2525.2002
Venkatesh, M. P., Placencia, F., and Weisman, L. E. (2006). Coagulase-negative
staphylococcal infections in the neonate and child: an update. Semin. Pediatr.
Infect. Dis. 17, 120–127. doi: 10.1053/j.spid.2006.06.005
Wiedemann, I., Breukink, E., van Kraaij, C., Kuipers, O. P., Bierbaum, G.,
de Kruijff, B., et al. (2001). Specific binding of nisin to the peptidoglycan
precursor lipid II combines pore formation and inhibition of cell wall
biosynthesis for potent antibiotic activity. J. Biol. Chem. 276, 1772–1779.
doi: 10.1074/jbc.M006770200
Zhang, Y.-Q., Ren, S.-X., Li, H.-L., Wang, Y.-X., Fu, G., Yang, J., et al.
(2003). Genome-based analysis of virulence genes in a non-biofilm-
forming Staphylococcus epidermidis strain (ATCC 12228). Mol. Microbiol. 49,
1577–1593. doi: 10.1046/j.1365-2958.2003.03671.x
Zhou, Y., Liang, Y., Lynch, K. H., Dennis, J. J., and Wishart, D. S. (2011).
PHAST: a fast phage search tool. Nucleic Acids Res. 39, W347–W352.
doi: 10.1093/nar/gkr485
Conflict of Interest Statement: The authors declare that the research was
conducted in the absence of any commercial or financial relationships that could
be construed as a potential conflict of interest.
Copyright © 2016 Martins Simões, Lemriss, Dumont, Lemriss, Rasigade,
Assant-Trouillet, Ibrahimi, El Kabbaj, Butin and Laurent. This is an open-access
article distributed under the terms of the Creative Commons Attribution License (CC
BY). The use, distribution or reproduction in other forums is permitted, provided the
original author(s) or licensor are credited and that the original publication in this
journal is cited, in accordance with accepted academic practice. No use, distribution
or reproduction is permitted which does not comply with these terms.
Frontiers in Microbiology | www.frontiersin.org 11 December 2016 | Volume 7 | Article 1991

ORIGINAL ARTICLE BACTERIOLOGY
Wide geographical dissemination of the multiresistant Staphylococcuscapitis NRCS-A clone in neonatal intensive-care units
M. Butin1,2, J.-P. Rasigade1,2,3,4, P. Martins-Simões1,2,3, H. Meugnier2,3, H. Lemriss5, R. V. Goering6, A. Kearns7, M. A. Deighton8,
O. Denis9, A. Ibrahimi5, O. Claris4,10, F. Vandenesch2,3,4, J.-C. Picaud4,11 and F. Laurent1,2,3,4
1) Department of Clinical Microbiology, Northern Hospital Group, Hospices Civils de Lyon, 2) International Centre for Research in Infectious Diseases (CIRI),
INSERM U1111, CNRS UMR5308, Université Lyon 1, ENS de Lyon, 3) National Reference Centre for Staphylococci, Hospices Civils de Lyon, Lyon, 4) Claude
Bernard University Lyon 1, Villeurbanne, France, 5) Laboratory of Biotechnology, Faculty of Medicine and Pharmacy of Rabat, Mohamed V University of Rabat,
Rabat, Morocco, 6) Creighton University, Omaha, NE, USA, 7) Public Health England, Staphylococcus Reference Service, Colindale, London, UK, 8) RMIT
University, Bundoora, Victoria, Australia, 9) Erasme Hospital, Université Libre de Bruxelles, Laboratoire de Référence MRSA, Staphylocoques, Department of
Microbiology, Brussels, Belgium, 10) Neonatal Intensive Care Unit, Eastern Hospital Group, Hospices Civils de Lyon, Bron and 11) Neonatal Intensive Care Unit,
Northern Hospital Group, Hospices Civils de Lyon, Lyon, France
Abstract
Nosocomial late-onset sepsis represents a frequent cause of morbidity and mortality in preterm neonates. The Staphylococcus capitis clone
NRCS-A has been previously described as an emerging cause of nosocomial bacteraemia in French neonatal intensive-care units (NICUs).
In this study, we aimed to explore the possible unrecognized dissemination of this clone on a larger geographical scale. One hundred
methicillin-resistant S. capitis strains isolated from neonates (n = 86) and adult patients (n = 14) between 2000 and 2013 in four different
countries (France, Belgium, the UK, and Australia) were analysed with SmaI pulsed-field gel electrophoresis (PFGE) and dru typing. The
vast majority of NICU strains showed the NRCS-A pulsotype and the dt11c type (96%). We then randomly selected 14 isolates (from
neonates, n = 12, three per country; from adult patients, n = 2), considered to be a subset of representative isolates, and performed
further molecular typing (SacII PFGE, SCCmec typing, and multilocus sequence typing-like analysis), confirming the clonality of the
S. capitis strains isolated from neonates, despite their distant geographical origin. Whole genome single-nucleotide polymorphism-based
phylogenetic analysis of five NICU isolates (from the different countries) attested to high genetic relatedness within the NRCS-A clone.
Finally, all of the NRCS-A strains showed multidrug resistance (e.g. methicillin and aminoglycoside resistance, and decreased vancomycin
susceptibility), with potential therapeutic implications for infected neonates. In conclusion, this study represents the first report of clonal
dissemination of methicillin-resistant coagulase-negative Staphylococcus clone on a large geographical scale. Questions remain regarding the
origin and means of international spread, and the reasons for this clone’s apparent predilection for neonates.
Clinical Microbiology and Infection © 2015 European Society of Clinical Microbiology and Infectious Diseases. Published by Elsevier Ltd. All
rights reserved.
Keywords: Antibiotic resistance, clone, coagulase-negative Staphylococcus, neonatology, vancomycin
Original Submission: 20 March 2015; Revised Submission: 3 September 2015; Accepted: 8 September 2015
Editor: P.T. Tassios
Article published online: 25 September 2015
Corresponding author: M. Butin, Department of ClinicalMicrobiology, CIRI, INSERM U1111, Hôpital de la Croix Rousse, 103grande rue de la Croix Rousse, 69004 Lyon, FranceE-mail: [email protected]
Introduction
Every year, approximately 15 million children are born pre-
maturely worldwide (defined as all live births before 37completed weeks). In developed countries, preterm birth is the
leading cause of death in children aged <5 years [1]. In recent
Clin Microbiol Infect 2016; 22: 46–52Clinical Microbiology and Infection © 2015 European Society of Clinical Microbiology and Infectious Diseases. Published by Elsevier Ltd. All rights reservedhttp://dx.doi.org/10.1016/j.cmi.2015.09.008

decades, technical and medical advances in neonatal intensive-
care units (NICUs) have allowed for an improvement in theglobal survival of neonates. Nevertheless, preterm birth-related
complications still remain the major cause of neonatal death [2].In this context, nosocomial late-onset sepsis (LOS) (occurring
after 3 days of life) represents a frequent cause of morbidity andmortality [3]. Twenty per cent to 30% of very low birthweightpreterm babies develop at least one episode of LOS during
their hospitalization in a NICU, resulting in respiratory orneurological impairment [4]. Catheter-related bloodstream in-
fections constitute the most frequent cause of LOS in thesevulnerable neonates, owing to multiple indwelling devices and
invasive procedures. Consequently, skin-associated commensalcoagulase-negative staphylococci are the most frequently
involved pathogens in LOS, most commonly involving Staphy-lococcus epidermidis [3,5].
However, the predominance of Staphylococcus capitis in
several French NICUs has been reported, whereas this speciesis rarely reported as a pathogen in adult patients [6]. This
phenomenon is of major concern, for several reasons: first, wehave recently shown that all methicillin-resistant S. capitis
strains from NICUs in France belong to a unique clone(designated NRCS-A), suggesting a high potential for dissemi-
nation, whereas S. capitis isolates from adult patients belong tomany distinct clones; second, NRCS-A has been highly preva-
lent in our institution (Hospices Civils de Lyon) since at least2004, underscoring its ability to persist and become endemicwhen introduced into NICUs; and third, NRCS-A strains show
decreased susceptibility to all antimicrobial agents that arecommonly used in NICUs [6]. NRCS-A isolates harbour a type
V-related staphylococcal chromosome cassette mec (SCCmec)element conferring resistance to β-lactams [7], and show a
multidrug resistance profile, including resistance or hetero-resistance to vancomycin, the first-line antimicrobial agent in
LOS cases [5,6].As multiresistant S. capitis LOS cases have been reported in
several countries [6–9], we sought to explore the possible
unrecognized dissemination of the multiresistant NRCS-Aclone on a larger geographical scale. To this end, we
collected and characterized S. capitis bloodstream isolates fromNICUs located in geographically diverse countries, by per-
forming molecular typing and antimicrobial susceptibilitytesting.
Materials and methods
Bacterial isolatesThe strain collections of four national reference centres forstaphylococci (Australia, Belgium, France, and the UK) were
screened to identify methicillin-resistant S. capitis isolates from
neonatal LOS in NICUs, and from blood cultures of non-neonatal patients (adult or paediatric). For the period
2000–2013, methicillin-resistant and multiresistant S. capitisisolates were isolated in NICUs in at least 12 (France), eight
(UK), six (Australia) and four (Belgium) hospitals. Each yearduring this time period, between two and 70 neonatal LOScases involving S. capitis were identified in each of these hos-
pitals (personal communication). Each reference laboratoryprovided a sample of methicillin-resistant S. capitis strains iso-
lated from its country during the period 2000–2013. In total,100 S. capitis strains were included in our analysis: 86 from
NICUs (France, n = 48; Belgium, n = 3, Australia, n = 20; andthe UK, n = 15) and 14 from other units (paediatric or adult
patients) (France, n = 7; Belgium, n = 3; and Australia, n = 4).These 100 strains were initially characterized by pulsed-field gelelectrophoresis (PFGE) and dru typing.
To confirm our initial results and avoid method-linked bias,we more thoroughly characterized a subset of isolates
belonging to the NRCS-A clone, as it was not feasible to analyseevery isolate in the complete strain collections. Thus, of the 86
NICU isolates, 12 were randomly selected (three NRCS-Aisolates per country), and two methicillin-resistant S. capitis
bloodstream isolates from adult patients from France that didnot belong to the NRCS-A clone were selected.
PFGEPFGE, the reference standard staphylococcal typing and
outbreak investigation method, was performed initially withSmaI restriction digestion, and also with SacII, which has been
recently reported to show increased discriminatory power forS. capitis [6,9,10]. Cluster analysis was performed with Bio-
Numerics software 7.1 (Applied Maths NV, Sint-Martens-Latem, Belgium). Pattern similarities of >80% (UPGMA, Dice
coefficient) were used to define a pulsotype as previouslydescribed [11].
dru typingdru typing, a recently developed method based on sequence
analysis of a 40-bp variable-number tandem-repeat regionwithin SCCmec (the mobile genetic element (MGE) harbouring
the mecA gene), was performed as previously described [12].
SCCmec typingThe SCCmec typing was performed as described by Kondo
et al. [13].
Multilocus sequence typingThe genetic relationship between the 14 selected S. capitisisolates was further explored by multilocus sequence typing
CMI Butin et al. Multiresistant S. capitis clone in NICUs 47
Clinical Microbiology and Infection © 2015 European Society of Clinical Microbiology and Infectious Diseases. Published by Elsevier Ltd. All rights reserved, CMI, 22, 46–52

100
8060
noitalosi foraeYyrtnuoCgnittes tneitaPetalosIepytosluPsnrettap EGFPegatnecrepytiralimiS dru type
NRCS-A BACAD80 NICU France 2008 dt11cNRCS-A BACAE01 NICU France 2008 dt11cNRCS-A BACAD81 NICU France 2009 dt11cNRCS-A ST20111689 NICU France 2011 dt11cNRCS-A ST20111690 NICU France 2011 dt11cNRCS-A ST20111691 NICU France 2011 dt11cNRCS-A ST20111693 NICU France 2011 dt11cNRCS-A ST20111694 NICU France 2011 dt11cNRCS-A ST20111771 NICU UK 2010 dt11cNRCS-A ST20111772 * NICU Belgium 2009 dt11cNRCS-A ST20111773 NICU Belgium 2009 dt11cNRCS-A ST20111774 NICU Belgium 2010 dt11cNRCS-A ST20111732 NICU France 2011 dt11cNRCS-A ST20111734 NICU France 2011 dt11cNRCS-A BACAE06 NICU France 2008 dt11cNRCS-A BACAE03 NICU France 2008 dt11cNRCS-A BACAE12 NICU France 2008 dt11cNRCS-A BACAD75 NICU France 2005 dt11cNRCS-A BACAD78 NICU France 2006 dt11cNRCS-A BACAD73 NICU France 2007 dt11cNRCS-A BACAD74 NICU France 2007 dt11cNRCS-A BACAD76 NICU France 2009 dt11cNRCS-A ST20131580 NICU France 2009 dt11cNRCS-A BACAD71 NICU France 2009 dt11cNRCS-A ST20111775 * NICU France 2009 dt11cNRCS-A ST20141717 NICU France 2008 dt11cNRCS-A ST20141733 NICU France 2009 dt11baNRCS-A ST20141734 NICU France 2008 dt11cNRCS-A ST20141735 NICU France 2009 dt11cNRCS-A ST20141736 NICU France 2009 dt11cNRCS-A ST20141730 NICU France 2007 dt10hNRCS-A ST20141752 NICU France 2007 dt11cNRCS-A ST20141732 NICU France 2008 dt11cNRCS-A ST20141731 NICU France 2008 dt11cNRCS-A ST20090910 France 2009 dt11cNRCS-A ST20111733 NICU France 2011 dt11cNRCS-A ST20141720 NICU UK 2009 dt11cNRCS-A ST20141741 NICU France 2008 dt11cNRCS-A ST20141739 NICU France 2008 dt11cNRCS-A ST20141716 NICU Australia 2000 dt11cNRCS-A ST20141742 NICU France 2008 dt11cNRCS-A ST20141743 NICU France 2008 dt11cNRCS-A ST20141744 NICU France 2008 dt11cNRCS-A ST20141745 NICU France 2008 dt11cNRCS-A ST20141740 NICU France 2008 dt11cNRCS-A ST20090909 NICU France 2008 dt11cNRCS-A ST20090908 NICU France 2008 dt11cNRCS-A BACAD72 NICU France 2009 dt11cNRCS-A ST20111769 NICU UK 2009 dt11cNRCS-A ST20111770 * NICU UK 2009 dt11cNRCS-A ST20141751 NICU France 2006 dt12rNRCS-A BACAE26 NICU France 2008 dt11cNRCS-A BACAC01 NICU UK 2010 dt11cNRCS-A BACAE25 NICU France 2008 dt11cNRCS-A ST20141738 NICU France 2009 dt11cNRCS-A ST20141737 NICU France 2009 dt11cNRCS-A ST20121276 NICU Australia 2012 dt11cNRCS-A BACAC09 NICU UK 2010 dt11cNRCS-A BACAC15 NICU UK 2010 dt11cNRCS-A ST20141727 NICU UK 2010 dt11cNRCS-A BACAB80 NICU UK 2010 dt11cNRCS-A ST20141726 NICU UK 2010 dt11cNRCS-A ST20141722 NICU UK 2009 dt11cNRCS-A ST20141729 NICU UK 2011 dt11cNRCS-A ST20141728 NICU UK 2010 dt11cNRCS-A ST20121261 NICU Australia 2012 dt11cNRCS-A ST20121264 NICU Australia 2012 dt11cNRCS-A ST20121265 NICU Australia 2012 dt11cNRCS-A ST20121268 NICU Australia 2012 dt11cNRCS-A ST20121269 NICU Australia 2012 dt11cNRCS-A ST20121270 NICU Australia 2012 dt11cNRCS-A ST20121273 NICU Australia 2012 dt11cNRCS-A ST20121274 NICU Australia 2012 dt11cNRCS-A ST20121275 NICU Australia 2012 dt11cNRCS-A ST20131582 * NICU France 2002 dt11cNRCS-A ST20121263 NICU Australia 2012 dt11cNRCS-A ST20121272 NICU Australia 2012 dt11cNRCS-A ST20141721 NICU UK 2009 dt11cNRCS-A ST20121262 Adult se ng Australia 2012 dt10aNRCS-A ST20121575 NICU Australia 2002 dt11cNRCS-A ST20111692 NICU France 2011 dt11cNRCS-A ST20121573 * NICU Australia 2000 dt11cNRCS-A ST20121574 NICU Australia 2001 dt11cNRCS-A ST20121576 NICU Australia 2004 dt11cNRCS-A ST20141719 NICU Australia 2001 dt11c
Non-NRCS-A ST20121267 Adult se ng Australia 2012 dt11aNon-NRCS-A ST20121271 Adult se ng Australia 2012 dt11cNon-NRCS-A ST20091315 Adult se ng France 2008 dt11bdNon-NRCS-A ST20111767 NICU Australia 2003 dt11cNon-NRCS-A ST20141724 NICU UK 2010 dt11cNon-NRCS-A ST20111766 NICU Australia 2011 dt11cNon-NRCS-A ST20121260 Adult se ng Australia 2012 dt12afNon-NRCS-A BACAE37 Adult se ng France 2008 dt3cNon-NRCS-A BACAY43 Adult se ng Belgium 2013 dt11aNon-NRCS-A ST20091319 Adult se ng France 2007 dt11aNon-NRCS-A BACAY44 Adult se ng Belgium 2013 dt1bNon-NRCS-A BACAY42 Adult se ng Belgium 2013 dt9boNon-NRCS-A ST20091316 Adult se ng France 2009Non-NRCS-A ST20090912 Adult se ng France 2008 dt3cNon-NRCS-A ST20090917 Adult se ng France 2009
Paediatric ICU
Non-typeable
Non-typeable
48 Clinical Microbiology and Infection, Volume 22 Number 1, January 2016 CMI
Clinical Microbiology and Infection © 2015 European Society of Clinical Microbiology and Infectious Diseases. Published by Elsevier Ltd. All rights reserved, CMI, 22, 46–52

(MLST) based on the published S. epidermidis MLST scheme
[14], with appropriately designed consensus primers foramplification of seven housekeeping genes (Table S1). For each
allele, sequences were aligned, and single-nucleotide poly-morphisms (SNPs) were detected and compared.
Whole genome sequencing and SNP-basedphylogenetic analysisToexplore the genetic conservation inside theNRCS-Aclone,five
NRCS-A isolateswere selected, including: (a) isolate ST20131582,corresponding to the oldest French isolate available in our
collection (2002) (genome CR09; ENA accession numberCTEL01000003) [15]; and (b) four isolates (one per country)
randomly selected from the 12 previously fully analysed iso-lates— isolate ST20111775 from France (genome CR01, alreadypublished [16] and considered to be the prototype of theNRCS-A
clone; ENA accession number LN866849), isolate ST20111772from Belgium (genome CR03; ENA accession number
CTEB01000001) [15], isolate ST20121573 from Australia(genome CR04; ENA accession number CTEM01000001) [15],
and isolate ST20111770 from the UK (genome CR05; ENAaccession number CTEO01000001) [15]. Whole genome
sequencing of these five isolates was performed with pyrose-quencing (454; Life Sciences/Roche, Branford, CT, USA). More-over, we performed single-molecule real-time sequencing
(PacBio, Menlo Park, CA, USA) for one of these five isolates(ST20111775) to obtain a closed genome (genome CR01). The
mean coverage obtained with the 454 pyrosequencing was 24×,21×, 20× and 20× for genomes CR03, CR04, CR05 and CR09,
respectively. For the single-molecule real-time sequencing(genome CR01), a mean coverage of 265× was obtained.
The whole genome sequences (WGSs) of these NRCS-AS. capitis strains were compared with publicly available
S. capitis genomes, which correspond to either skin commensalor pathogen isolates from adult patients : SK14 (GenBankReference Sequence: ACFR00000000.1), VCU116 (GenBank
Reference Sequence: AFTX00000000.1), QN1 (GenBankReference Sequence: AJTH00000000.1), LNZR-1 (GenBank
Reference Sequence: NZ_JGYJ00000000.1), and C87 (GenBankReference Sequence: ACRH00000000.1). To assess the phylo-
genetic relationships between all S. capitis strains, we used theREALPHY 1.10 online pipeline [17], with default parameters, to
infer phylogenetic trees from assembled WGS data. We used
the closed genome of the S. capitis NRCS-A strain ST20111775
as the reference genome (genome CR01; ENA accessionnumber LN866849).
Antimicrobial susceptibility profileAntimicrobial susceptibility testing of the 14 selected S. capitisisolates (NICU, n = 12; adult patients, n = 2) was performed
with the standard agar diffusion technique as recommended bythe French Society for Microbiology (www.sfmasso.fr/doc/
download.php?doc=DiU8C&fic=casfm_2009.pdf). MICs ofvancomycin, linezolid and daptomycin were determined with
the Etest. Strains with a vancomycin MIC of >2 mg/L wereconsidered to be resistant, and strains with MICs of �2 mg/L
were tested for vancomycin heteroresistance with thebrain–heart infusion agar method, as previously described[6,18].
Results
SmaI PFGE profileOf the 86 S. capitis isolates from the NICU, all but three (96%)were of the NRCS-A strain pulsotype (i.e. shared >80% simi-
larity with each other and the NRCS-A pattern) [6] (Fig. 1). Thethree non-NRCS-A strains originated from NICUs in Australia.
In contrast, the 14 non-NICU methicillin-resistant S. capitisisolates showed a diversity of pulsotypes, only two of which
(14%) were NRCS-A: one from a 4-year-old patient hospital-ized in a paediatric intensive-care unit in France, and one from
an adult patient in Australia (Fig. 1).
dru typeWith more clonal organisms, PFGE tends to generate related
macrorestriction patterns, causing problems in epidemiologicaldiscrimination [12]. Therefore, the 100 S. capitis isolates were
additionally analysed with dru typing for characterization of theMGE harbouring mecA. Among the 86 NICU isolates, 83 har-
boured a unique dt11c profile (96%), regardless of their pul-sotype (NRCS-A or not NRCS-A) (Fig. 1). The three remaining
NICU isolates (all NRCS-A) were dt12r, dt11ba, and dt10h(n = 1 each). Conversely, among the 14 non-NICU isolates,only two were dt11c (14%), including the NRCS-A paediatric
isolate. The other dru types in non-NICU isolates were diverse:
FIG. 1. Dendrogram and schematic representation of pulsotypes and dru types of 100 Staphylococcus capitis isolates from neonatal intensive-care units
(NICUs) (n = 86) or other settings (n = 14). The pulsed-field gel electrophoresis (PFGE) dendrogram was generated from SmaI PFGE patterns with
Bionumerics software 7.1, as previously described [6,10]. The vertical dotted line represents the 80% limit of similarity. The pulsotype NRCS-A was
defined previously [6]. dru typing was performed as previously described [12]. *Available genomes.
CMI Butin et al. Multiresistant S. capitis clone in NICUs 49
Clinical Microbiology and Infection © 2015 European Society of Clinical Microbiology and Infectious Diseases. Published by Elsevier Ltd. All rights reserved, CMI, 22, 46–52

dt10a (n = 1), dt11a (n = 3), dt12af (n = 1), dt11bd (n = 1), dt3c
(n = 2), dt1b (n = 1), and dt9bo (n = 1). Two isolates were non-typeable.
PFGE with SacII, SCCmec typing, and MLSTTo strengthen and complete the data obtained with the set of100 clinical isolates, we performed additional analysis on a
subset of 14 randomly selected strains: 12 NRCS-A strains fromNICU patients (n = 3 from each country) and two non-NRCS-A
strains from adult patients.As observed with SmaI, SacII PFGE restriction profiles
confirmed the clustering of the 12 NICU isolates into a uniquepulsotype (Fig. S1). Conversely, isolates from adult patients
belonged to two distinct pulsotypes, and shared <80% similaritywith NICU isolates. SCCmec analysis confirmed the geneticcloseness of this MGE in the 12 NICU isolates: indeed, all of
these 12 isolates harboured a type V-related SCCmec element,whereas the isolates from adults harboured a non-typeable
SCCmec element (a combination of a mec complex type Cand ccr genes A2/B2/C) (Fig. S1). Finally, when an MLST model
encompassing a total of 3020 bp of housekeeping genes wasused, the sequence comparison of the 12 NICU isolates
showed that these isolates differed by no more than two SNPs(Table S2), whereas sequences of the two isolates from adultpatients showed 32 and 33 SNPs, as compared with the NICU
isolates.
Whole genome SNP-based phylogenetic analysis ofS. capitis strainsA total of 2 080 323 bp (including coding and intergenic re-
gions) were found to be shared by all ten genomes (five S. capitisNRCS-A genomes and the five publicly available S. capitis ge-
nomes), and constituted the core genome used for the phylo-genetic reconstruction shown in Fig. 2. When each genome wascompared with the reference genome of S. capitis NRCS-A
CR01, we found a maximum of 162 SNPs and a minimum of65 SNPs (indels excluded) between the NRCS-A genomes,
whereas the non-NRCS-A genomes harboured at least 7910SNPs and up to 50 723 SNPs (for LNZR-1 and QN1, respec-
tively). As seen in Fig. 2, all five NRCS-A genomes clustered inone clade, which suggests that they belong to a clonal
population.
Antimicrobial susceptibility profilesAntimicrobial susceptibility profiles of the 14 S. capitis strains
are shown in Table 1. All 12 NICU isolates showed methicillinand aminoglycoside resistance, and either resistance or heter-
oresistance to vancomycin, the first-line antimicrobial agentused for LOS. The 12 NICU isolates remained susceptible to
fluoroquinolones.
Discussion
Collectively, our data highlight the unexpected multinational
geographical distribution of a clonal population of multidrug-resistant S. capitis (NRCS-A) that shows high specificity forthe NICU environment.
PFGE and dru typing performed on a large number of S. capitisisolates, representing wide temporal and geographical diversity,
revealed that almost all of the S. capitis NICU isolates collectedin four distant countries were of the NRCS-A pulsotype and
dt11c (within a common SCCmec cassette acquired only once).These results suggest the emergence and widespread dissemi-
nation of a unique S. capitis strain. The slight discrepancy be-tween the pulsotype and the dru type of the isolates could be
explained by the fact that the SCCmec cassette, an MGE, can betransferred between different isolates when present in the sameenvironment, such as an NICU. Note that, in our collection,
two strains from adult patients were non-typeable by dru typing.This phenomenon has also been observed in rare methicillin-
resistant S. aureus strains, and has been interpreted as anabsence of the dru region in this minority of strains [12].
Genetic relatedness within the NRCS-A clone was alsoillustrated by the low mutation level seen in the MLST-like
analysis (at most, two SNPs over 3020 bp), and confirmed bythe few SNPs found in the comparison of core genomes of fiveS. capitis NRCS-A strains despite their geographical and tem-
poral diversity. This high conservation of the NRCS-A genomesince at least 2000 (the year of isolation of the Australian strain,
the oldest strain included in the WGS comparison) suggestshigh adaptation of this clone towards the NICU environment,
resulting in few genetic changes inside the clonal population ofNICU isolates. This interesting observation requires further
investigation focusing on the mutation rate, the molecularclock, and the fitness of clinical NRCS-A isolates.
All tested NRCS-A isolates had the same multidrug resis-tance profile, including resistance to all antimicrobial agentswidely used in the NICU setting. Unexpectedly, these NICU
isolates remained susceptible to fluroroquinolones, whereasmultiresistant coagulase-negative Staphylococcus strains are
typically fluoroquinolone resistant. This atypical antimicrobialsusceptibility profile is similar to that of previously described
NRCS-A strains [6], and probably reflects the specific antimi-crobial selective pressure of NICUs, where β-lactams, vanco-
mycin, and aminoglycosides, but not fluoroquinolones, arewidely used [19]. In our study, all NRCS-A isolates were eitherresistant or heteroresistant to vancomycin. Studies of S. capitis
isolates from NICUs have reported constant vancomycin het-eroresistance [7,8], and Van der Zwet et al. initially proposed
that this could be an intrinsic feature of S. capitis [8]. However,
50 Clinical Microbiology and Infection, Volume 22 Number 1, January 2016 CMI
Clinical Microbiology and Infection © 2015 European Society of Clinical Microbiology and Infectious Diseases. Published by Elsevier Ltd. All rights reserved, CMI, 22, 46–52

our recent results obtained by comparing S. capitis isolates fromneonates and adult patients suggest that vancomycin hetero-
resistance is more likely to be a specific feature of the NRCS-Aclone [7]. This heteroresistance might have contributed to
S. capitis NRCS-A establishment in NICUs. Alternatively, thefrequent use of vancomycin in neonates might have induced/
selected this heteroresistance in the NRCS-A clone. The clinicalsignificance of vancomycin heteroresistance is controversial,but therapeutic failures of vancomycin in S. capitis-related LOS
have been reported [8]. Thus, the multidrug resistance profileof this emerging clone raises therapeutic concerns in cases of
LOS, and requires substantiation in a clinical study focusing onS. capitis LOS.
Taken together, these data demonstrate the clonality ofS. capitis isolates from neonates, despite their distant
geographical origin, in contrast to the high level of genetic di-versity seen in isolates from non-NICU patients. Thus, this
larger dataset, involving an international collection of strains,mirrors our previous observation of isolates within France [6].
The geographical spread of a clonal population of S. capitis indistant NICUs, although unexpected, is reminiscent of theextensive diffusion of clonal group B Streptococcus ST-17 causing
meningitis and septicaemia in neonates [20]. However, althoughthe global dissemination of methicillin-resistant Staphylococcus
aureus clones has been well described, to the best of ourknowledge this study represents the first report of widespread
FIG. 2. Single-nucleotide polymorphism (SNP)-
based maximum-likelihood tree with whole
genome sequences of neonatal Staphylococcus
capitis isolates from four distinct countries and
publicly available S. capitis genomes. A total of
2 080 323 homologous sites from all ten ge-
nomes (polymorphic and non-polymorphic
sites) were obtained by use of the REALPHY
1.10 online pipeline with the closed chromo-
some of S. capitis NRCS-A CR01 (ENA acces-
sion number LN866849) as reference. A
maximum-likelihood tree was built by the use
of PhyML with default parameters as imple-
mented in REALPHY 1.10. For each strain, the
number of SNPs between each strain and the
reference is indicated in grey in parentheses.
Aus, Australia; Be, Belgium; Fr, France.
TABLE 1. Antimicrobial susceptibility profile of Staphylococcus capitis bloodstream isolates from neonates (n [ 12) and adults
(n [ 2)
StrainPatientsetting Country
Year ofisolation Oxa Kan Tob Gen Rif Cip Ery Fof
Van susceptibility(MIC in mg/L)
Lzd susceptibility(MIC in mg/L)
Dap susceptibility(MIC in mg/L)
ST20091315 AdultICU
France 2008 R R R S S S S R S (0.75) S (0.75) S (0.19)
ST20090912 AdultMedicine
France 2008 R R R S S R S R S (1.0) S (1.0) S (0.19)
ST20111769 NICU UK 2009 R R R R S S S R HR (1.0) S (1.0) S (1.0)ST20111770a NICU UK 2009 R R R R S S S R HR (0.75) S (1.0) S (0.19)ST20121575 NICU Australia 2002 R R R R S S R R HR (1.5) S (1.5) S (0.5)ST20111771 NICU UK 2010 R R R R S S S R HR (2.0) S (1.5) S (0.75ST20111773 NICU Belgium 2009 R R R S S S S R HR (1.5) S (1.5 S (1.0)ST20131580 NICU France 2009 R R R R R S S R R (3.0) S (2.0) S (1.0)ST20121573a NICU Australia 2000 R R R R S S S R HR (1.5) S (2.0) S (0.75)ST20121574 NICU Australia 2001 R R R R S S R R HR (2.0) S (2.0) S (0.75)ST20111772a NICU Belgium 2009 R R R R S S S R HR (1.5) S (1.0) S (0.75)ST20111774 NICU Belgium 2010 R R R R S S S R HR (2.0) S (1.5) S (0.75)ST20111775a NICU France 2009 R R R R R S S R HR (1.5) S (1.5) S (0.25)BACAD74 NICU France 2007 R R R R R S S R HR (1.5) S (1.5) S (0.5)
Cip, ciprofloxacin; Dap, daptomycin; Ery, erythromycin; Fof, fosfomycin; Gen, gentamicin; HR, heteroresistant; ICU, intensive-care unit; Kan, kanamycin; Lzd, linezolid; NICU,neonatal intensive-care unit; Oxa, oxacillin; R, resistant; Rif, rifampin; S, susceptible; Tob, tobramycin; Van, vancomycin.aAvailable genomes.
CMI Butin et al. Multiresistant S. capitis clone in NICUs 51
Clinical Microbiology and Infection © 2015 European Society of Clinical Microbiology and Infectious Diseases. Published by Elsevier Ltd. All rights reserved, CMI, 22, 46–52

diffusion of methicillin-resistant coagulase-negative staphylococci.
This observation raises important and as yet unsolved questionsregarding the origin and the dissemination routes involved in the
international spread of NRCS-A S. capitis, as well as the under-lying mechanism(s) of apparent predilection for neonates.
Conclusion
In conclusion, we have demonstrated that a methicillin-resistantand multidrug-resistant S. capitis clone is involved in LOS in
NICUs from four distant countries. A better understanding ofthe epidemiology and pathophysiology of S. capitis NRCS-Ainfections in preterm infants is now warranted.
Transparency declaration
The authors have no conflicts of interest or funding to disclose.
Acknowledgements
This work was supported by the French Ministry of Health and
the Institut National de la Santé et de la Recherche Médicale(Inserm). The funders had no role in study design, datacollection, analysis, decision to publish, or preparation of the
manuscript.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://
dx.doi.org/10.1016/j.cmi.2015.09.008.
References
[1] Liu L, Johnson HL, Cousens S, Perin J, Scott S, Lawn JE, et al. Global,regional, and national causes of child mortality: an updated systematicanalysis for 2010 with time trends since 2000. Lancet 2012;379:2151–61.
[2] Howson CP, Kinney MV, McDougall L, Lawn JE. Born Toon Soon:preterm birth matters. Reprod Health 2013;10(Suppl. 1):S1.
[3] Boghossian NS, Page GP, Bell EF, Stoll BJ, Murray JC, Cotten CM, et al.Late-onset sepsis in very low birth weight infants from singleton andmultiple-gestation births. J Pediatr 2013;162:1120–4.
[4] Cohen-Wolkowiez M, Moran C, Benjamin DK, Cotten CM, Clark RH,Benjamin Jr DK, et al. Early and late onset sepsis in late preterm infants.Pediatr Infect Dis J 2009;28:1052–6.
[5] Venkatesh MP, Placencia F, Weisman LE. Coagulase-negative staphy-lococcal infections in the neonate and child: an update. Semin PediatrInfect Dis 2006;17:120–7.
[6] Rasigade J-P, Raulin O, Picaud J-C, Tellini C, Bes M, Grando J, et al.Methicillin-resistant Staphylococcus capitis with reduced vancomycinsusceptibility causes late-onset sepsis in intensive care neonates. PLoSONE 2012;7:e31548.
[7] D’mello D, Daley AJ, Rahman MS, Qu Y, Garland S, Pearce C, et al.Vancomycin heteroresistance in bloodstream isolates of Staphylococcuscapitis. J Clin Microbiol 2008;46:3124–6.
[8] Van Der Zwet WC, Debets-Ossenkopp YJ, Reinders E, Kapi M,Savelkoul PH, Van Elburg RM, et al. Nosocomial spread of a Staphylo-coccus capitis strain with heteroresistance to vancomycin in a neonatalintensive care unit. J Clin Microbiol 2002;40:2520–5.
[9] Cui B, Smooker PM, Rouch DA, Daley AJ, Deighton MA. Differencesbetween two clinical Staphylococcus capitis subspecies as revealed bybiofilm, antibiotic resistance, and pulsed-field gel electrophoresisprofiling. J Clin Microbiol 2013;51:9–14.
[10] Goering RV. Molecular epidemiology of nosocomial infection: analysisof chromosomal restriction fragment patterns by pulsed-field gelelectrophoresis. Infect Control Hosp Epidemiol 1993;14:595–600.
[11] Tenover FC, Arbeit RD, Goering RV, Mickelsen PA, Murray BE,Persing DH, et al. Interpreting chromosomal DNA restriction patternsproduced by pulsed-field gel electrophoresis: Criteria for bacterialstrain typing. J Clin Microbiol 1995;33:2233–9.
[12] Goering RV, Morrison D, Al-Doori Z, Edwards GFS,Gemmell CG. Usefulness of mec-associated direct repeat unit (dru)typing in the epidemiological analysis of highly clonal methicillin-resistant Staphylococcus aureus in Scotland. Clin Microbiol Infect2008;14:964–9.
[13] Kondo Y, Ito T, Ma XX, Watanabe S, Kreiswirth BN, Etienne J, et al.Combination of multiplex PCRs for staphylococcal cassette chromo-some mec type assignment: Rapid identification system for mec, ccr, andmajor differences in junkyard regions. Antimicrob Agents Chemother2007;51:264–74.
[14] Thomas JC, Vargas MR, Miragaia M, Peacock SJ, Archer GL,Enright MC. Improved multilocus sequence typing scheme for Staph-ylococcus epidermidis. J Clin Microbiol 2007;45:616–9.
[15] Lemriss H, Lemriss S, Martins-Simoes P, Butin M, Lahlou L, Rasigade JP,et al. Genome sequences of four Staphylococcus capitis NRCS-A isolatesfrom geographically distant neonatal intensive care units. GenomeAnnounc 2015;3:e00501–15.
[16] Lemriss H, Martins Simões P, Lemriss S, Butin M, Ibrahimi A,El Kabbaj S, et al. Non-contiguous finished genome sequence ofStaphylococcus capitis CR01 (pulsetype NRCS-A). Stand Genomic Sci2014;9:1118–27.
[17] Bertels F, Silander OK, Pachkov M, Rainey PB, van Nimwegen E.Automated reconstruction of whole-genome phylogenies from short-sequence reads. Mol Biol Evol 2014;31:1077–88.
[18] Satola SW, Farley MM, Anderson KF, Patel JB. Comparison of detec-tion methods for heteroresistant vancomycin-intermediate Staphylo-coccus aureus, with the population analysis profile method as thereference method. J Clin Microbiol 2011;49:177–83.
[19] Clark RH, Bloom BT, Spitzer AR, Gerstmann DR. Reported medica-tion use in the neonatal intensive care unit: data from a large nationaldata set. Pediatrics 2006;117:1979–87.
[20] Poyart C, Réglier-Poupet H, Tazi A, Billoët A, Dmytruk N, Bidet P,et al. Invasive group B streptococcal infections in infants, France.Emerg Infect Dis 2008;14:1647–9.
52 Clinical Microbiology and Infection, Volume 22 Number 1, January 2016 CMI
Clinical Microbiology and Infection © 2015 European Society of Clinical Microbiology and Infectious Diseases. Published by Elsevier Ltd. All rights reserved, CMI, 22, 46–52

Caractérisation moléculaire par séquençage PacBio du clone endémique mondial S.
capitis NRCS-A responsable de sepsis en réanimation néonatale : résistances, virulence
et avantage sélectif
P. Martins-Simões3-6-1
, Y. Dumont3-6
, M. Butin5-3-6
, H. Lemriss8, S. Lemriss
7, A. Ibrahimi
7, S.
El Kabbaj7, F. Vandenesch
2-6-1, J.P. Rasigade
6-1-4, F. Laurent
3-6-1
1Centre National de Référence des Staphylocoques
2Laboratoire de Bactériologie,
Groupement Hospitalier Est3Laboratoire de Bactériologie, Groupement Hospitalier
Nord 4Laboratoire de Bactériologie, Groupement Hospitalier Sud
5Service de Réanimation
Néonatale, Groupement Hospitalier Est, Hospices Civils de Lyon6Centre International de
Recherche en Infectiologie, Unité Inserm U1111, Université de Lyon, Lyon,
France7Département de Biosécurité PCL3, Laboratoire de Recherche et d’Analyses
Médicales de la Gendarmerie Royale (LRAM) 8Laboratoire de Biotechnologie Médicale,
École de Médecine et de Pharmacie, Université Mohammed V - Souissi, Rabat, Morocco
Le clone de Staphylococcus capitis NRCS-A a été décrit comme un pathogène nosocomial
majeur responsable de sepsis chez des nouveau-nés prématurés dans de nombreux pays du
monde. Les représentants de ce clone présentent un profil de multirésistance atypique incluant
une sensibilité diminuée à la vancomycine, antibiotique de première ligne pour le traitement
de ces sepsis néonataux. Objectif : explorer les caractéristiques génomiques de ce clone,
(résistance aux antibiotiques, facteurs de virulence et gènes spécifiques).
Le séquençage par SMRT (Single-Molecule Real-Time) de la souche prototype du clone
NRCS-A (CR01) a permis d’obtenir 2 séquences fermées, un chromosome et un plasmide.
Ces séquences ont été corrigées, annotées, puis comparées à i) 3 génomes assemblés de
souches du clone NRCS-A isolées en Australie, Belgique et Royaume-Uni, et séquencées par
pyroséquençage 454, ii) 6 génomes publiques de S. capitis n’appartenant pas au clone NRCS-
A.
Nous avons pu mettre en évidence au sein du clone NRCS-A : i) des gènes de résistance
(mecA, aacA-aphD, ...) associés à des éléments génétiques mobiles ou contenus dans le core-
genome, expliquant le profil de résistance de ce clone, ii) des facteurs de virulence dans le
core-genome comprenant des Phenol Soluble Modulins, des hémolysines, des protéines de la
paroi fixant le fibrinogène ou la fibronectine, et l'opéron ica, iii) la présence spécifique d’un
gène de résistance à la nisine (nsr), absent des génomes non-NRCS-A et de la plupart des
génomes de S. aureus et de staphylocoques à coagulase négative (SCN).
Nos résultats soulignent le rôle des éléments génétiques mobiles dans l'émergence du
phénotype multirésistant du clone NRCS-A, lui conférant un profil de résistance parfaitement
adapté à la pression de sélection existant en réanimation néonatale. Par ailleurs le gène de
résistance à la nisine, bactériocine "large-spectre" active sur de nombreuses bactéries à Gram
positif et sécrétée par les bactéries de la flore commensale digestive (Lactococcus,
entérocoques, BGN…), pourrait contribuer à un meilleur fitness au clone NRCS-A et lui
conférer un avantage sélectif au sein du microbiote des prématurés par rapport aux autres
souches de S. capitis et plus largement aux autres souches de SCN ne possédant pas ce gène.

Abstract Preview - Step 3/4- print version -
Topic: 34 Mobile genetic elements
Title: STAPHYLOCOCCUS CAPITIS NRCS-A: SCC, SCCMEC AND THEIR EVOLUTIONARYHISTORY
Author(s): P. Martins Simoes1,2, H. Lemriss3, J.-P. Rasigade1,2, S. Lemriss3, F. Vandenesch2, S. El Kabbaj3,
F. Laurent1,2
Institute(s): 1Laboratoire de Bactériologie - Hôpital de la Croix-Rousse, Hospices Civils de Lyon, 2CentreInternational de Recherche en Infectiologie (CIRI), INSERM U1111 - Centre National de Référence
des Staphylocoques, Lyon, France, 3Département de Biosécurité PCL3, Laboratoire de Rechercheet d'Analyses Médicales de la Fraternelle de la Gendarmerie Royale, Rabat, Morocco
Text: Background. We have recently shown that Staphylococcus capitis pulsotype NRCS-A is widelyspread worldwide as a cause of late-onset sepsis in intensive care neonates. NRCS-A strainsexhibit methicillin resistance, encoded by mecA gene harbored by a mobile genetic element, thestaphylococcal chromosome cassette mec (SCCmec).Objectives. To gain insights into the evolutionary history of NRCS-A strains, we characterized theSCCmec element of the prototype NRCS-A strain CR01.Methods. Whole-genome sequencing of strain CR01 was performed using 454 pyrosequencing.Available genome sequences of S. capitis and S. aureus strains were collected from the GenBankdatabase and aligned using software Mauve and the MaGe platform. Phylogeny of strain CR01 andof S. capitis strains QN1, SK14 and VCU116 was built using a 10-gene set derived from thestandard multi-locus sequence type approach.Results. Two contiguous SCC elements were found in strain CR01, one mecA-harboring 37kbSCCmec element and one 22,1kb pseudo-SCC element without mecA gene. The SCCmec elementcontained a CRISPR element and was closely related to type V SCCmec of livestock-associated S.aureus ST398, while the pseudo-SCC element was partially identical to those found in methicillin-susceptible S. capitis strains SK14 and VCU116. Phylogenetic analysis suggested the pseudo-SCCelement was first acquired by strains CR01, SK14 and VCU116, and the SCCmec element waslater acquired exclusively by strain CR01.Conclusions. We report the first description of SCC elements harbored by the clinically relevantNRCS-A/CR01 strain. This description could be used in future phylogeographical studies comparingNRCS-A strains.
Author Keywords: Staphylococcus capitis, NICU, SCCmec, CRISPR, evolutionary history
Conference: FEMS2013 - 5th Congress of European Microbiologists · Abstract: A-527-0034-03168 · Status: Draft
FEMS2013 - 5th Congress of European Microbiologists -... http://www.abstractserver.com/fems2013/absmgm/print...
1 of 1 02/18/2013 02:11 PM

Diffusion mondiale du clone Staphylococcus capitis NRCS-A en réanimation néonatale : caractérisation moléculaire, analyse génomique et histoire évolutive. M. Butin1-2, P. Martins Simões1-2, J.P. Rasigade1-2-3, S. Lemriss4, H. Lemriss4, S. El Kabbaj4, A. Ibrahimi4, S. Tigaud1, F. Vandenesch1-2-3, O. Claris1-3, J.C. Picaud1-3, F. Laurent1-2-3 1Hospices Civils de Lyon 2CIRI (Centre International de Recherche en Infectiologie), Inserm U1111, Lyon3Université Claude Bernard Lyon 1, Villeurbanne, France 4Laboratoire de Recherche et analyse clinique, Police Royale, Rabat, Maroc
Introduction : Staphylococcus capitis NRCS-A est un clone endémique impliqué dans les septicémies nosocomiales néonatales (SNN) à l’échelle mondiale. Ce clone multirésistant porte un déterminant de résistance à la méticilline (staphylococcal cassette chromosome mec, SCCmec) de type V. Nous avons exploré la phylogénie de ce clone chez plusieurs souches d’origines géographiques variées, caractérisé son élément SCCmec et précisé son histoire évolutive. Matériels et Méthodes : Douze souches NRCS-A isolées de SNN en Angleterre, Australie, Belgique et France, et 2 souches de S. capitis non-NRCS-A isolées d’adultes, ont été caractérisées par électrophorèse en champs pulsés (ECP) avec les enzymes SmaI et SacII. Un arbre phylogénétique a été construit sur la base des séquences de 7 gènes de ménage. Enfin, l’élément SCCmec de la souche prototype NRCS-A CR01 a été séquencé, caractérisé et comparé à ceux d’autres staphylocoques. Résultats : Les 12 souches NRCS-A appartenaient en ECP SmaI et SacIIà un unique pulsotype (>80% de similarité), différent de ceux des souches d’adultes. Sur 3020 pb séquencées sur 7 gènes de ménage, les souches NRCS-A présentaient au maximum 2 SNPs entre elles, contre 32 ou 33 par-rapport aux souches non-NRCS-A, et étaient regroupées sur un même clade. Le séquençage de l’élément SCCmec de la souche NRCS-A CR01 a révélé une structure composite originale comprenant i) une cassette SCCmec fortement homologue à celle des S. aureus résistants à la méticilline ST398, et portant de façon surprenante un élément CRISPR d’immunité adaptative ; et ii) une cassette SCC portant des gènes de résistance aux métaux lourds (SCCcad/ars/cop). La comparaison avec d’autres génomes de S. capitis a permis de proposer un schéma évolutif avec acquisition successive indépendante des 2 cassettes. Conclusion : La combinaison de plusieurs techniques de typage moléculaire a permis d’affirmer la diffusion internationale de S. capitis NRCS-A, présentant un caractère hautement clonal et portant un élément composite original SCCmec-SCCcad/ars/cop. L’analyse détaillée du génome de la souche prototype CR01 est en cours afin de mettre en évidence des facteurs de virulence pouvant expliquer la pathogénie particulière de ce clone et sa diffusion mondiale atypique.

Worldwide distribution of an endemic multi-resistant NRCS-A
Staphylococcus capitis clone in NICU: molecular typing, whole
genome analysis and evolutionary history
Martins Simões P #(a,b), Butin M (a,b), Lemriss H(c,d), Lemriss S(c),
Deighton MA(e), Kearns A(f), Denis O(g), Goering R (h), Ginevra C (a),
Picaud JC (i), Ibrahimi A (d), Vandenesch F (a), Rasigade J-P(a,b), Laurent
F(a,b)
(a) Centre International de Recherche en Infectiologie (CIRI) - INSERM
U1111, Lyon, France
(b) Centre National de Référence des Staphylocoques, Hôpital de la Croix-
Rousse, Hospices Civils de Lyon, Lyon, France
(c) Département de Biosécurité PCL3, Laboratoire de Recherche et
d'Analyses Médicales de la Fraternelle de la Gendarmerie Royale, Rabat,
Morocco
(d) Equipe de Recherche en Biotechnologie Médicale (MedBiotech), UPR de
#
Corresponding author: [email protected]

Pharmacologie & Toxicologie, Faculté de Médecine & Pharmacie, Université
Mohammed V Souissi, Rabat, Morocco.
(e) School of Applied Sciences, RMIT University, Bundoora, Victoria,
Australia
(f) Staphylococcus Reference Unit, Microbiology Services Colindale, Health
Protection Agency, London, UK
(g) Reference MRSA-Staphylococci Laboratory, Microbiology unit, Erasme
Hospital, Université Libre de Bruxelles, Brussels, Belgium
(h) Department of Medical Microbiology and Immunology, Creighton
University, Omaha, Nebraska, USA
(i) Department of Neonatology, Northern Hospital Group, Hospices Civils de
Lyon, Lyon, France

Objective: Staphylococcus capitis pulsotype NRCS-A is an emerging cause
of nosocomial bacteremia in French neonatal intensive care units (NICUs)
and showed to be multi-resistant (methicillin, aminoglycosides, vancomycin).
In order to investigate the worldwide distribution of NRCS-A clone, we
applied various molecular methods to study S. capitis isolates from several
countries.
Methods: Twelve NICU S. capitis isolates from neonates septicemia
(Australia, Belgium, France, United Kingdom, n=3 each) and 2 S. capitis
isolates from adult patients were analyzed using PFGE (SmaI and SacII),
SCCmec typing and dru-typing. A MLST-like analysis, based on 7 house-
keeping genes, was also performed. After whole-genome sequencing of one
of our clinical NRCS-A strains, we performed a phylogenetic analysis with
other publicly available S. capitis genomes to assess the evolutionary history
of SCC elements acquisition in S. capitis.
Results:All neonatal S. capitis (i) shared >80% similarity when characterized
by SmaI and SacII PFGE and were similar to NRCS-A pulsotype, (ii)
harbored a type V-related SCCmec element, (iii) exhibited the same dru-type
(dt11c), and (iv) formed a monophyletic group using the 7 house keeping

genes. The molecular profiles of both adult isolates differed from those of
NICU strains. Unexpectedly, a CRISPR region was found within the SCCmec
cassette and was detected by PCR, targeting the direct-repeats, in all NICUs
isolates and one adult patient isolate. Finally, sequencing of the SCCmec
element of one of the NICU isolates revealed the presence of a composite
element including a SCCmec cassette and a second SCC carrying genes
related to detoxification of heavy metals (SCCcad/ars). Phylogenetic analysis
of all the 4 available whole-genome sequenced S. capitis strains suggests that
the 2 SCC elements were acquired independently by the NRCS-A clone.
Conclusion: Our analysis demonstrates an unexpected worldwide
distribution of the S. capitis NRCS-A clonal population involved in neonatal
bacteremia. These results suggest that this multi-resistant clone is highly
successful in the specific environment of NICUs. This raises the questions of
the molecular mechanisms behind this success and the way of spreading of
this clonal population. Both questions are currently being addressed by
combining whole genome sequencing, phenotypic features and cellular
models.

Conclusions EOS in term infants is associated with significantlylower maternal and neonatal 25-OHD levels. We also found thatlevels of 25-OHD in neonates were positively correlated withthose in mothers. These data suggest that adequate vitamin Dsupplementation during pregnancy may be helpful to preventEOS in term neonates.
PS-222 STAPHYLOCOCCUS CAPITIS IN NEONATAL LATE-ONSETSEPSIS: UNEXPECTED WORLDWIDE DISSEMINATION OFAN ENDEMIC MULTI-RESISTANT CLONE
1M Butin, 2P Martins Simoes, 3H Lemriss, 3S Lemriss, 2F Vandenesch, 4O Claris, 4JC Picaud,1JP Rasigade, 1F Laurent. 1Clinical Microbiology, Hospices Civils de Lyon, Lyon, France;2U1111 - CIRI, Inserm, Lyon, France; 3Laboratoire de Recherche Et d’Analyses Médicales,Fraternelle de La Gendarmerie Royale, Rabbat, Morocco; 4NICU, Hospices Civils de Lyon,Lyon, France
10.1136/archdischild-2014-307384.521
Background Multi-resistant Staphylococcus capitis NRCS-A isinvolved in late-onset sepsis (LOS) in French NICUs.Aims To investigate the geographical distribution of NRCS-A,and to precise its susceptibility profile.Methods Twelve S. capitis isolates from distant NICUs (Aus-tralia, Belgium, France, United Kingdom, n = 3 each) and 2 S.capitis isolates from adult patients were analysed using PFGE,SCCmec typing, dru-typing, a MLST-like analysis, and antimicro-bial susceptibility testing. To explore impact of vancomycinselective pressure, after 15 daily subcultures with vancomycin,we determined vancomycin, daptomycin and linezolid minimalinhibitory concentrations (MICs).Results All NICU S. capitis (i) shared >80% similarity of PFGEprofile and were similar to NRCS-A profile, (ii) harboured atype V-related SCCmec element, (iii) exhibited the same dru-type, (iv) formed a monophyletic group, (v) harboured a sameantimicrobial susceptibility profile including aminoglycosides andmethicillin resistance, and vancomycin heteroresistance. Thesemolecular and antimicrobial susceptibility profiles differed fromthose of adult isolates. An increase of vancomycin and daptomy-cin (but not linezolid) MICs was observed, significantly faster(p < 0.05) for NRCS-A isolates than other tested strains.Conclusion Our analysis demonstrates an unexpected worldwidedistribution of S. capitis NRCS-A, specifically in NICUs.Recently, we collected complementary NICU isolates belongingto NRCS-A from Norway, Denmark, the Netherlands, USA, Bra-zil, New Zealand and Canada, confirming the worrisome dissem-ination of NRCS-A. Its multi-resistant profile and its ability torapidly adapt to vancomycin selective pressure, constitute aselective advantage to NRCS-A in NICUs, and raise the issue ofpotential therapeutic failure and the need for alternative antimi-crobial regimens.
Neonatology Clinical
PS-223 IMPROVING STAFF COMPETENCE AND CONFIDENCEFOR NEONATAL RESUSCIATION, A QUALITYIMPROVEMENT INITIATIVE
S Hackett, M Casssidy, S Gormally. Neonatal Intensive Care Unit, Our Lady of LourdesHosptial Drogheda Co. Louth, Drogheda, Ireland
10.1136/archdischild-2014-307384.522
Aims To introduce a Quality Improvement Initiative aimed atinstilling confidence and competence with Neonatal Resuscita-tion in Labour Ward (LW) and Neonatal Unit (NICU) staff.Methods A Neonatal Resuscitation Initiative Team (NRIT) con-sisted of LW and NICU midwives, an Advanced Nurse Practi-tioner (ANP) and a Paediatric Consultant. LW, NICU andpaediatric staff were invited to attend a presentation to outlinethe aims and methodology of the initiative. Emphasis was placedon teamwork with effective leadership and communication. Thiswas followed by a demonstration of 2 low fidelity simulatedneonatal resuscitations performed by the NRIT.
Midwifery and NICU staff were then invited to partake in aSimulated Team Response (STR) to 2 simulated resuscitations.
A random selection of participants completed a closed ques-tionnaire before and after the NRIT demonstration and beforeand after their STR. Levels of confidence in all aspects of neona-tal resuscitation, including teamwork and leadership skills wereevaluated using a Likert Scale.Results One hundred and thirteen staff participated in the qual-ity initiative from January 2013 to August 2013. Seventy fourstaff participated in the NRIT demonstrations and 39 in theSTR. Forty three questionnaires were completed following theNRIT and 13 following the STR. Using a Wilcoxon test the pre-post gain in confidence score was statistically significant (p =0.0020) for doctors and midwives.Conclusion Simulation training is a valuable means of maintain-ing skills and enhancing confidence for neonatal resuscitation.This study quantifies the positive impact of a Quality Improve-ment Initiative aimed at improving performance at neonatalresuscitation.
PS-224 VIDEO EDUCATION TO IMPROVE BAG MASKVENTILATION DURING SIMULATED NEWBORNRESUSCITATION
1P Deindl, 2J Schwindt, 2A Berger, 3G Schmoelzer. 1Department of Neonatology andPediatric Intensive Care Medicine, University Medical Center Hamburg-Eppendorf,Hamburg, Germany; 2Department of Pediatrics and Adolescent Medicine Division ofNeonatology Pediatric Intensive Care and Neuropediatrics, Medical University of Vienna,Vienna, Austria; 3Department of Pediatrics and Adolescent Medicine Division ofNeonatology Pediatric Intensive Care and Neuropediatrics, Royal Alexandra Hospital,Edmonton, Canada
10.1136/archdischild-2014-307384.523
Aim To evaluate if video based education could improve qualityof positive pressure ventilation (PPV) performed by novicehealth care providers during neonatal resuscitation.Methods Twenty-eight 4th year medical students were randomlypaired and instructed to give PPV to a modified manikin as sin-gle-person resuscitators, then as two-person paired resuscitatorsusing either an anatomical shaped neonatal face mask with anair cushion rim (IS) or a Laerdal round face mask (LM). Afterwatching a video-tutorial they randomly repeated each maskventilation performance. Airway pressure, gas flow, tidal volume,and mask leak were recorded. PPV performance quality was ana-lysed using video recording.Results Mask leak was lower during one-person ventilationwhen using IS (56 ± 16%) compared to LM (71 ± 19%). LMmask leak during one-person ventilation was significantly lowerwhen using the two point top hold in contrast to the ok rimhold (before training: 63 ± 22% vs. 72 ± 18%, after training:57 ± 17% vs. 77 ± 12%, respectively). Watching a video-tuto-rial improved correct head position (score: 1.4 ± 0.7 vs. 3.8 ±
Poster symposium
Arch Dis Child 2014;99(Suppl 2):A1–A620 A193
group.bmj.com on June 28, 2017 - Published by http://adc.bmj.com/Downloaded from

CloneDissemination Of An Endemic Multi-resistantLate-onset Sepsis: Unexpected Worldwide
Staphylococcus Capitis In Neonatal PS-222
Claris, JC Picaud, JP Rasigade and F LaurentM Butin, P Martins Simoes, H Lemriss, S Lemriss, F Vandenesch, O
doi: 10.1136/archdischild-2014-307384.5212014 99: A193 Arch Dis Child
http://adc.bmj.com/content/99/Suppl_2/A193.1Updated information and services can be found at:
These include:
serviceEmail alerting
box at the top right corner of the online article. Receive free email alerts when new articles cite this article. Sign up in the
CollectionsTopic Articles on similar topics can be found in the following collections
(965)Drugs: infectious diseases (229)Neonatal intensive care
(388)Neonatal and paediatric intensive care
Notes
http://group.bmj.com/group/rights-licensing/permissionsTo request permissions go to:
http://journals.bmj.com/cgi/reprintformTo order reprints go to:
http://group.bmj.com/subscribe/To subscribe to BMJ go to:
group.bmj.com on June 28, 2017 - Published by http://adc.bmj.com/Downloaded from
Copyright © 2022 FDOKUMEN

