







Bahasa
Halaman
Hukum


1
The role of activated MEK-ERK pathway in quercetin-induced growth inhibition and apoptosis in A549 lung cancer cells.
Nguyen, T.T.T., Tran, E., Nguyen, T.H., Do, P.T., Huynh, T.H., and Huynh*, H.
Laboratory of Molecular Endocrinology, Division of Cellular and Molecular Research, National Cancer Centre of Singapore, Singapore 169610
*Correspondence to:Hung HuynhLaboratory of Molecular EndocrinologyDivision of Cellular and Molecular ResearchNational Cancer Centre of SingaporeSingapore 169610Phone: +65 6436 8347Fax: +65 6226 5694E-mail: [email protected]
Running Title: Quercetin induces apoptosis in A549 cells.Key words: Activated ERK, growth inhibition, apoptosis, quercetin
; a .# Oxford University Press ll rights reserved2003arcinogenesisC
Carcinogenesis Advance Access published December 19, 2003 by guest on D
ecember 23, 2013
http://carcin.oxfordjournals.org/D
ownloaded from

2
Abstract
Dietary phytochemicals have been shown to be protective against various types of cancers.
However, the precise underlying protective mechanisms are poorly understood. In the present study,
we report that treatment of A549 cells with quercetin resulted in a dose-dependent reduction in cell
viability and DNA synthesis with the rate of apoptosis equivalent to 1.2 ± 0.8, 6.3 ± 0.9, 16.5 ± 1.5,
36.4 ± 2.6, and 42.5 ± 5.8 % on treatment with 0.1% DMSO, 14.5, 29.0, 43.5 and 58.0 µM
quercetin, respectively. Concomitantly, quercetin treatments led to a 1.1-, 1.1-, 2.5- and 3.5-fold
increase in Bax. Similar elevations were also observed in Bad, which increased 1.1-, 2.1-, 2.2- and
2.3-fold respectively as compared to control. While Bcl-2 was decreased by 30%, Bcl-xL was
elevated in a dose-dependent fashion. Quercetin also induced the cleavage of caspase-3, caspase-7
and PARP (poly ADP-ribose polymerase). While Akt-1 and phosphorylated Akt-1 were inhibited,
the extracellular signal-regulated kinase (ERK) was phosphorylated following quercetin treatment
in a dose-dependent fashion. Phosphorylation of ERK and c-Jun occurred at 3 h and was sustained
over 14 h. Phosphorylation of MEK1/2 was increased in concordance with ERK activation.
Quercetin-induced phosphorylation of JNK and cleavage of caspase-3 occurred 6 h after quercetin
exposure and before cleavage of caspase-7 and PARP was detected. Inhibition of MEK1/2 but not
PI-3 kinase, p38 kinase or JNK abolished quercetin-induced phosphorylation of c-Jun, cleavage of
caspase-3 and -7, cleavage of PARP, and apoptosis. Inhibition of caspase activation completely
blocked quercetin-induced apoptosis. Expression of constitutively activated MEK1 in A549 cells
led to activation of caspase-3 and apoptosis. The results suggest that in addition to inactivation of
Akt-1 and alteration in the expression of the Bcl-2 family of proteins, activation of MEK-ERK is
required for quercetin-induced apoptosis in A549 lung carcinoma cells.
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

3
Introduction
Lung cancer is one of the most common cancers in many countries and accounts for 28% of all
cancer death (Reviewed in (1,2)). Approximately 75-85% of lung cancer is non-small cell lung
cancer and the rest is small cell lung cancer (3). The average five-year survival rate for localized
lung cancer was 48% compared to 14.5 % overall and 2.5 % for a metastatic lung cancer. Five-year
survival rate after surgical resection was approximately 60% for stage I lung cancer patients (4).
Unfortunately, only 15% of people are diagnosed at an early, localized stage because most lung
cancer begins to grow silently without any symptoms until the cancer is in an advanced stage (5).
Thus, there is an urgent need for novel diagnosis, prevention and/or treatment of lung cancer.
Epidemiological studies have shown that the consumption of vegetables, fruits and tea is associated
with a low risk of cancer (6). Many natural dietary phytochemicals found in fruits, vegetables,
spices and tea have been shown to be protective against cancer in various animal models (7,8). The
most common flavonoid glycones found in the diet are quercetin, rutin and robinin (9). Although
the mechanisms by which quercetin exerts its anti-proliferative and apoptotic activities remain to be
elucidated, there is evidence suggesting that the action of flavonoids is probably mediated by either
interaction with the type II estrogen binding sites (10) or aryl hydrocarbon receptor (11). Quercetin
has been shown to inhibit the enzymes involving in proliferation and signal transduction pathway
including protein kinase C (12), tyrosine kinase (13), cdc25 phosphatase (14), PI-3 kinase (15),
DNA topoisomerase II (16), proline-directed protein kinase fatty acid in human prostate carcinoma
cells (17), Na+K+ATPase (18) and JNK (19). Quercetin has a wide range of biological activities
including inhibition of mutant p53 expression (20), and androgen receptor expression and function
in LnCap cells (21). It has anti-proliferative activity in vitro against several cancer cells of human
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

4
origin (22-26). It blocks the cell cycle at the G1/S transition in colon and gastric cancer as well as in
leukaemic cells (27,28) but causes a G2/M block in breast and laryngeal cancer cell lines or in non-
oncogenic fibroblasts (20,29,30). Quercetin potentiates the cytotoxic action of 1-ß-D-
arabinofuranosylcytosine (31). Quercetin also inhibits cell invasion and induces apoptosis through a
pathway involving heat shock proteins (32). These activities of quercetin make it a promising
candidate for treatment and prevention of various cancers.
One of the most frequent targets downstream of receptor and non-receptor tyrosine kinases and the
ras family of GTP-binding proteins is the extracellular related kinase (ERK) signal transduction
pathway (33). Constitutive MEK1 activation contributes to cell survival (33), migration (34),
transformation of fibroblasts and epithelial cells (35). Studies with small molecule inhibitors of
MEK activity (36) demonstrate a role for MEK in mediating expression of proteinases implicated in
invasion and metastasis (37), and disruption of normal epithelial morphology (38). Treatment of
cells with various growth factors or chemotherapeutic agents produces activation of MEK1/2 and
its downstream target, ERK, resulting in proliferation and differentiation (Reviewed in (33)). ERK
activation may exert either anti-apoptotic (Reviewed in (33)) or pro-apoptotic (39) influence
depending upon the cellular context.
Regulation of apoptosis is a complex process and involves a number of cellular genes, including
Bcl-2 (40), and Bcl-2 related family members such as Bcl-xL, Bcl-xs, Bad, and Bax (41). The Bcl-2
gene product protects cells against apoptosis in a variety of experimental systems. Suppression of
Bcl-2 has been shown to promote apoptosis in response to a number of stimuli, including anti-
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

5
cancer drugs (40). Bcl-2 and Bcl-xL exert their anti-apoptotic effect, at least in part by binding to
Bax and related pro-apoptotic proteins. They also prevent Bax and pro-apoptotic proteins from
inducing the release of cytochrome c and activation of the caspase-9. Recent work into apoptosis
has demonstrated the importance of PI-3 kinase and its downstream substrate protein kinase B
(Akt) (42). Akt exerts an anti-apoptosis effect against various stimuli (42) and confers resistance to
taxol (43). A direct link between the PI-3 kinase and apoptosis-regulating proteins was established
through Akt phosphorylation of Bad (44,45).
We demonstrate herein that quercetin inhibited the A549 cell growth and induced apoptosis.
Quercetin-induced apoptosis was associated with inactivation of Akt-1 protein, altered expression
of Bcl-2 family of proteins and phosphorylation of ERK. Our studies, employing transfection study
and pharmacological inhibitors for MEK-ERK and caspase revealed that MEK-ERK activation was
required to trigger caspase-3 activation and that MEK-ERK blockage modified the cytotoxicity of
quercetin, indicating that MEK-ERK activation in A549 cells linked to cell death.
Materials and Methods
Reagents: U0126, PD98059, LY294002, rabbit anti-phospho MEK1/2 (Ser217/221), rabbit anti-
cleaved caspase-7 (20 kDa), rabbit anti-caspase-3, rabbit anti-phospho Akt (Ser473), mouse anti-
phospho p44/42 ERK (Thr202/Tyr204), rabbit anti-Akt, mouse anti-ERK and rabbit anti-cleaved
PARP antibodies were purchased from New England Biolabs, Beverly, MA. Caspase inhibitor Z-
VAD-FMK was from Promega, Madison, MI. A c-Jun N-terminal kinase (JNK) specific inhibitor
SP600125, a p38 kinase inhibitor SB203580 and G418 were supplied by Calbiochem, La Jolla, CA.
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

6
Mouse anti-Bax, mouse anti-phospho-c-Jun (Ser63), mouse anti-phospho-JNK (Thr183/Tyr185),
mouse anti-α-tubulin, rabbit anti-Bcl-2, rabbit anti-Bcl-xL, rabbit anti-Bad antibodies were obtained
from Santa Cruz Inc., Santa Cruz, CA. Horseradish peroxidase-conjugated donkey anti-mouse or
anti-rabbit secondary antibodies were purchased from Pierce, Rockford, Illinois. Chemiluminescent
detection system was supplied from Amersham, Pharmacia Biotech, Arlington Heights, IL. Tissue
culture petri-dishes, 6-well plates, 96-well plates, 100 mm tissue culture petri-dish, and 8-chamber
slides were purchased from Nunc Inc. Naperville, IL. Quercetin and 3-(4,5-dimethylthiazol-2-yl)-
2,5-diphenyltetrazolium bromide (MTT) was purchased from Sigma, Saint Louis, MO. Cell
Proliferation ELISA Kit (BrdU, colorimetric assay) and In Situ Cell Death Detection Kit
(Fluorescein) were supplied from Roche Diagnostics Corporation, Indianapolis, IN. RPMI 1640
medium, fetal bovine serum (FBS), Lipofectamine reagent and penicillin-streptomycin were from
Gibco-BRL, Grand Island, NY.
U0126, PD98059, SP600125, SB203580, LY294002 and caspase inhibitor Z-VAD-FMK
compounds were dissolved in dimethylsulfoxide (DMSO) with final concentration never exceeding
0.1%. Quercetin was dissolved in DMSO at a concentration of 100 mg/ml. They were stored frozen
under light-protected conditions at -20 0C.
Cell culture and treatment: Human A549 lung epithelial cells were obtained from American Type
Culture Collection and cultured in RPMI 1640 medium supplemented with 10% FBS, 1% penicillin
and streptomycin (growth medium) at 37 0C in a 5% CO2 incubator. It has been documented that
A549 cells are pRB-positive (46) and express wild-type p53 (47). To study the effects of quercetin
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

7
on cell morphology, A549 cells were seeded onto 6-well plates at a density of 5 x 104 per well in
the growth medium. Confluent cells were washed with serum-free RPMI 1640 (SRF) medium and
then incubated further with SRF medium for 6 h. Cells were then treated with 14.5, 29.0 and 58.0
µM of quercetin in SRF medium for 14 h. Photographs were taken using the inverse microscope
(Nikon TMS, Tokyo, Japan).
Cell transfection: To examine whether constitutively activated MEK1 had any impact on the
survival of lung cancer cells, A549 cells were transfected with 10 µg of the pß-MEKDD or
pcDNA-3 control plasmid DNA and 28 µl of Lipofectamine reagent following the manufacturer’s
recommendation. The pß-MEKDD was the constitutively activated MEK1 cDNA, kindly provided
by Professor Phillip Leder (48). Forty-eight hours post-transfection, cells were subcultured at a ratio
of 1:10 and replaced with selective growth medium containing 800 µg/ml G418. Three weeks post-
transfection, pooled clones were expanded. To study the effects activated MEK1 on quercetin-
induced apoptosis, constitutively activated MEK1-transfected or pcDNA-transfected cells were
seeded at a density of 1 x 106 cells per 100 mm petri-dish and allowed to grow in growth medium
for 24 h. Cells were treated with indicated concentrations of quercetin in serum free RPMI for 14 h
and then harvested for analysis.
Detection of apoptosis: A549 cells were plated onto 8-chamber slides at a density of 5 x 103 cells
per well and allowed to grow in the growth medium for 24 h. Cells were then washed once with
SRF medium and treated with indicated concentrations of quercetin in fresh SRF medium for 14 h.
Cells were fixed with phosphate buffer saline (PBS) containing 4% formaldehyde for 1 h at room
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

8
temperature, washed with PBS and stored at -80 0C until analysis. Apoptosis was detected by the
terminal deoxynucleotidyl transferase-mediated dUTP nick-end labelling (TUNEL) assay using the
In Situ Cell Death Detection Kit (Roche) as described by the manufacturer. Slides were visualized
under fluorescent microscope (Olympus BX60) equipped with an FITC filter. Labelling indices
were obtained by counting cell number of labelled cells among at least 500 cells per region and
expressed as a percentage values.
Cell viability and proliferation: To study the effects of quercetin on cell proliferation and
viability, A549 cells were plated at 1x104 cells per well in 96-well plates and allowed to grow in the
growth medium for 24 h. Cells were then washed once with SRF medium and allowed to grow in
SRF medium for another 24 h. Cells then treated with indicated concentrations of quercetin in fresh
SRF medium for 24 or 48 h. Cell proliferation and cell viability were determined at 24 and 48 h
after treatment using the Cell Proliferation ELISA Kit and the MTT assay respectively as described
(49). Experiments were repeated at least 3 times, and the data were expressed as the mean ± SD.
Western blot analysis: To examine the effects of quercetin on Bax, Bad, Bcl-2, Bcl-xL, cleaved
caspase-3 and -7, cleaved PARP, PI-3 kinase, Akt, and phosphorylation of ERK, c-Jun, JNK, and
Akt. A549 cells were plated at a density of 5 x 106 cells per 100 mm petri-dish in the growth media.
After 24 h, the cell monolayer was washed and then treated with indicated concentrations of
quercetin in SRF medium as described above. Following the treatment, cells were harvested at
indicated times, lysed in a lysis buffer and Western blot analysis was performed using total cell
lysate as described (50). For each lane 100 µg of protein were loaded. Blots were incubated with the
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

9
indicated antibodies and 1:7500 horseradish peroxidase-conjugated donkey anti-mouse or anti-
rabbit secondary antibody. All the primary antibodies were used at the final concentration of 1
µg/ml. Blots were then visualized with a chemiluminescent detection system as described by the
manufacturer.
Statistical analysis: For quantitation analysis, the sum of the density of bands corresponding to
protein blotting with the antibody under study was calculated, and normalized to the amount of α-
tubulin. After normalisation with α-tubulin, changes in the expression of the protein under study in
treated samples were expressed relative to the basal levels of this protein in untreated sample.
Differences in cell proliferation, apoptosis and the levels of proteins under study were analyzed by
the Kruskal-Wallis test.
Results
For the time-course and dose-response experiments, human A549 lung cancer cells were treated
with 14.5, 29.0, 43.5 and 58.0 µM of quercetin for 24 and 48 h. Cell viability and cell growth were
the assessed by the MTT assay and BrdU incorporation, respectively. Control cells were treated
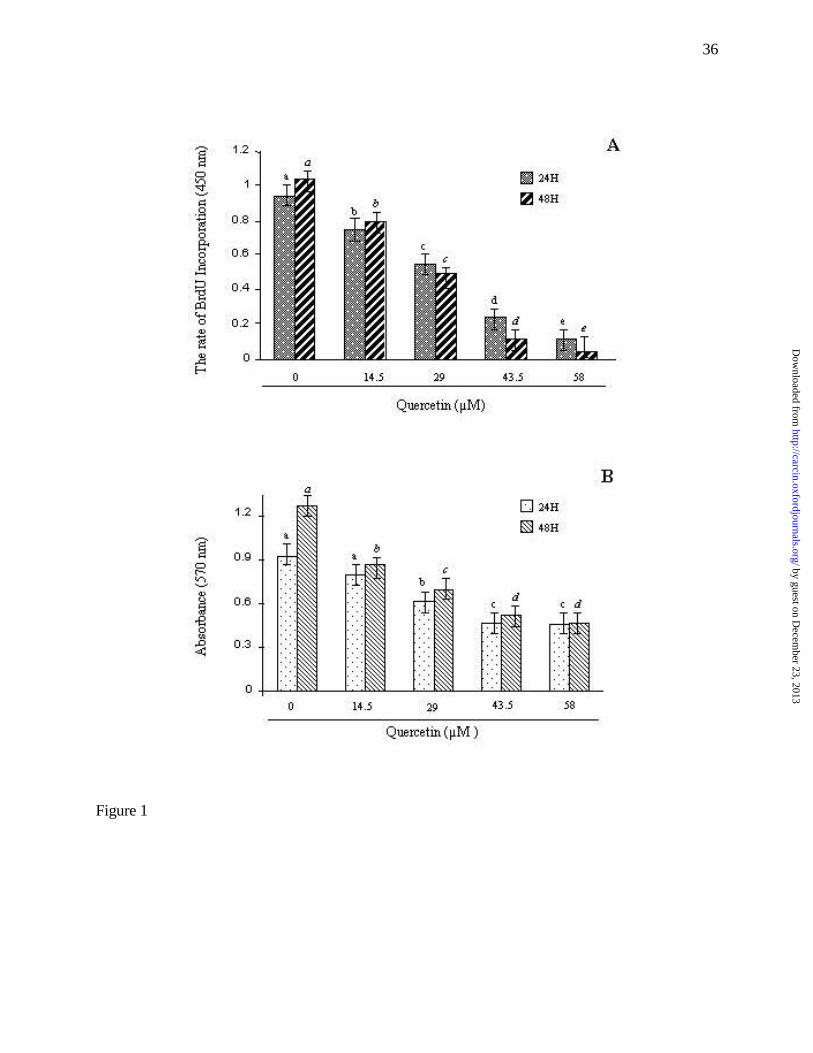
with 0.1% DMSO alone. Figure 1 shows that quercetin caused a dose-dependent reduction in DNA
synthesis (Fig. 1A) and cell viability (Fig. 1B). Significant inhibition in BrdU incorporation was
observed as early as 24 hours post-treatment (p<0.01). Fifty percent reduction in cell viability was
seen at a dose of 29.0 µM after 48 h incubation (Fig. 1B). To determine if quercetin reduced cell
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

10
number by inducing apoptosis, TUNEL assay was performed. Figure 2A shows that DNA
fragmentation was detected in quercetin-treated cells. In cells treated with 0.1% DMSO, 14.5, 29.0,
43.5 and 58.0 µM of quercetin for 24 h, approximately 1.2 ± 0.8, 6.3 ± 0.9, 16.5 ± 1.5, 36.4 ± 2.6,
and 42.5 ± 5.8% of apoptotic cells were observed, respectively (Fig. 2B). The percentage of
apoptotic cells in the treatment groups compared to the control group were statistically significant at
p<0.01 as determined by the Kruskal-Wallis test.
Because apoptosis in mammalian cells has been shown to be regulated by Bax, Bcl-xL, Bad, Bcl-2
(41), we determined whether quercetin-induced apoptosis in A549 cells was also associated with
the modulation of these proteins. To test this possibility, cell lysate from A549 cells treated with
different concentrations of quercetin was examined by Western blot analysis. As shown in figure 3,
quercetin induced a significant elevation in the expression of pro-apoptotic Bax and Bad. Treatment
of A549 cells with 14.5, 29.0, 43.5 and 58.0 µM of quercetin for 14 h led to 1.1-, 1.1-, 2.5- and 3.5-
fold increase in Bax. Similar elevations were observed in Bad levels, which increased 1.1-, 2.1-,
2.2- and 2.3-fold respectively in the quercetin treatment as compared to controls. While Bcl-2
expression was decreased by approximate 30%, the anti-apoptotic Bcl-xL expression was increased
by 2.0-, 2.3-, 2.4-, and 2.5-fold on treatment with 14.5, 29.0, 43.5 and 58.0 µM of quercetin,
respectively (Fig. 3D). Because phosphorylation of Bad at Serine 112 and 136 created consensus
sites for interaction with 14-3-3 protein; phosphorylated Bad then bound to 14-3-3 instead of Bcl-2
or Bcl-xL, resulting in the liberation of the anti-apoptotic proteins and the consequent promotion of
cell survival (51), we determined the phosphorylation status of Bad following quercetin treatment.
Using anti-phospho-specific Bad (Ser112) and Bad (Ser136) antibodies, we observed that Bad was
not phosphorylated at these positions (data not shown). The results indicate that quercetin-induced
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

11
apoptosis in A549 cells is associated with the accumulation on Bax, Bad, Bcl-xL and the decrease in
Bcl-2 proteins.
It has been demonstrated that the proteolytic cleavage of PARP, which synthesizes poly(ADP-ribo)
from ß-nicotinamide adenine dinucleotide (NDA) in response to DNA strand breaks, is an
biochemical event during apoptosis (52). Since PARP cleavage is a hallmark of caspase activation,
we determined whether the apoptosis machinery was activated by quercetin treatment, using an
anti-specific-cleaved PARP antibody that detects only cleaved products of PARP. As shown in
Figure 4E, the 89 kDa cleaved PARP fragment was detected in quercetin-treated samples. Since
caspase-3 and -7 have a central role in PARP cleavage (52), we determined the activation of these
two caspases by Western blot analysis using antibodies capable of detecting activated (cleaved)
caspase-3 and -7. Figure 4C shows that the cleaved forms of caspase-3 (19 and 17 kDa fragments)
were readily detectable at the dose as low as 14.5 µM of quercetin and reached high levels at the
dose of 29.0 µM. Cleaved caspase-7 fragments (19 and 20 kDa) were also observed in concordance
with caspase-3 activation (Fig. 4D). The data indicate that quercetin induced the activation of
caspase -3 and -7.
PI-3 kinase pathway is regulated by a variety of growth factors (43,44). Recent work on apoptosis
signalling has demonstrated the importance of PI-3 kinase and its downstream substrate, Akt
(42,53). In addition, a direct link between PI-3 kinase and apoptosis-regulating protein Bcl-2 family
of proteins has been established through Akt phosphorylation of Bad (44,45). We determined the
effects of quercetin on the levels of p85 subunit of PI-3 kinase, Akt-1 and phosphorylated Akt-1 in
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

12
A549 cells. Figure 5B shows that the expression of p85 subunit of PI-3 kinase was not altered
following quercetin treatment. However, treatment of A549 cells with 14.5, 29.0, 43.5 and 58.0 µM
of quercetin for 14 h resulted in a 30, 44, 48 and 58% decrease in total Akt protein (Fig. 5D). The
basal phosphorylation of Akt-1 was sharply reduced and barely detectable in cells treated with 29.0
µM of quercetin (Fig. 5C). The results indicate that quercetin was more effective in inhibiting the
phosphorylation of Akt-1 than Akt-1 expression. Subsequent blotting with anti-α-tubulin antibody
showed relatively equal amounts of total protein loaded per lane.
It has been demonstrated that the ERK signalling pathway is activated in response to certain cellular
stresses (Reviewed in (54)). To investigate whether quercetin induced growth arrest and apoptosis
in A549 cell was associated with the activation of ERK, cell lysate from quercetin-treated cells at
different times were subjected to Western blot analysis using an anti-phospho-ERK antibody to
detect phosphorylated (and therefore activated) ERK. The same blots were subsequently stripped
and reblotted with an antibody that recognized total ERK to verify equal amounts of the protein in
various samples. As shown in figure 6, treatment of A549 cell with 14.5, 29.0, 43.5 and 58.0 µM of
quercetin, all of which induced apoptosis, led to a dose-dependent activation of ERK (Fig. 6D). The
phosphorylation of MEK1/2 was increased in a dose-dependent manner following quercetin
treatment over the same time frame as seen for ERK (Fig. 6B). Because c-Jun is a target for ERK
activity, the levels of c-Jun phosphorylation were determined. Figure 6F shows that c-Jun was
phosphorylated following quercetin treatment and the pattern of c-Jun phosphorylation was similar
to ERK activation suggesting that phosphorylation of ERK by MEK1/2 increased ERK activity.
Since JNK activation is thought to be a determining factor in cell cycle arrest and apoptosis (55),
we set out to determine whether quercetin-induced apoptosis in A549 lung cancer cells was also
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

13
associated with the activation of JNK/SAPK pathway. The activation of JNK was determined by
examining its degree of phosphorylation by Western blot analysis with anti-phospho-JNK1/2
antibody. Figure 6G shows that quercetin significantly enhanced the phosphorylation of a 54 kDa
JNK2 without significant increase in the basal phosphorylation of 46 kDa JNK1. Blocking of JNK
activity with JNK inhibitor SP600125 did not prevent quercetin-induced apoptosis (Fig. 9)
suggesting that JNK is not a mediator of quercetin-induced apoptosis.
To study the time-course of MEK1/2, ERK, c-Jun, caspase-3, caspase-7 and JNK activation, cells
were treated with 0.1% DMSO or 58.0 µM of quercetin and then harvested at different times for
Western blot analysis. As shown in Figure 7D, activation of ERK was detected as early as 3 hours,
reached maximum levels 6 h after quercetin treatment and sustained over the 14 h period (Fig. 7D).
The result was corroborated by demonstrating that ERK kinase activity was increased by 4-fold 6 h
after quercetin treatment as determined by the levels of c-Jun phosphorylation (Fig. 7F). MEK1/2
was also phosphorylated by quercetin treatment over the same time frame as seen for ERK (Fig.
7B). Phosphorylation of the 54 kDa JNK2 was detectable 6 h after quercetin exposure (Fig. 7G).
Cleaved forms of caspase-3 and -7 were clearly detected 9 h after quercetin treatment and became
more pronounced at 14 h (Fig. 7H and 7I). Significant PARP (89 kDa) cleavage was detected 14 h
after quercetin treatment (Fig. 7K).
It has been demonstrated that ERK activation may exert pro-apoptotic influence depending upon
the cellular context (39,55). To determine whether quercetin-induced apoptosis is mediated by the
activation of ERK, two approaches were used. First strategy was to treat the cells with
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

14
pharmacologic blockers of MEK1/2 to inhibit quercetin-induced ERK activation and their
downstream effects. Second approach was to express the constitutively activated MEK1 in these
cells. Quercetin, in combination with MEK1/2 inhibitors U0126 and PD98059, or a PI-3 kinase
inhibitor LY294002, or p38 kinase inhibitor SB203580, or JNK inhibitor SP600125 or caspase
inhibitor Z-VAD-FMK were also used to treat human A549 lung carcinoma cells. The apoptosis
was measured by TUNEL assay. The levels of cleaved caspase-3, cleaved caspase-7, and cleaved
PARP were also investigated. TUNEL assay showed that quercetin and combined quercetin-
LY294002 caused apoptosis in A549 cells (Fig. 8D & 8F). Co-treatment of A549 cells with
quercetin and U0126 or PD98059 completely blocked quercetin-induced apoptosis as determined
by TUNEL assay (Fig. 8E). Western blot analysis revealed that quercetin alone significantly
increased phosphorylation of ERK, c-Jun, cleaved PARP, cleaved caspase-3 and cleaved caspase-7
(Fig 9). Like quercetin, both LY294002 and U0126 were capable of inducing 54 kDa JNK2
phosphorylation. Unlike U0126 and PD98059, LY294002 alone caused a mild increase in cleaved
PARP, cleaved caspase-7, cleaved caspase-3 (Fig. 9) and apoptosis (Fig. 8C). Co-treatment of cells
with quercetin and U0126 or PD98059 prevented quercetin-induced phosphorylation of ERK,
phosphorylation of c-Jun, cleavage of caspase-3 and-7 and cleavage of PARP (Fig. 9). Quercetin-
induced 54 kDa JNK2 phosphorylation was also attenuated by U0126-quercetin or PD98059-
quercetin treatment (Fig. 9E). Blocking PI-3 kinase, p38 kinase and JNK activity neither potentiated
nor prevented quercetin-induced apoptosis, cleavage of caspase-3 and -7 and cleavage of PARP
(Fig. 9). Inhibition of caspase activation by Z-VAD-FMK abolished quercetin-induced cleavage of
caspase-3 and PARP and apoptosis (Fig. 9). The results suggest that activation of MEK-ERK plays
an important role in quercetin-induced apoptosis and ERK acts upstream of caspase-3 and -7 to
exert its apoptotic influence in the quercetin-treated A549 cells.
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

15
Since blocking of MEK activity by chemical inhibitors prevented quercetin-induced apoptosis in
A549 cells, we wished to determine if constitutive activation of MEK1 in A549 cells would make
the cells more sensitive to quercetin-induced apoptosis. A549 cells were transfected with pcDNA-3
or constitutively activated MEK1 (48). After 3 weeks of selection in G418, pooled mock-
transfected and constitutively activated MEK1-transfected cells were treated with 0.1% DMSO or
58 µM of quercetin for 14 h and harvested for analysis. Because cleaved PARP by activated
caspase-3 and -7 has been proposed as one of the events in the execution phase of apoptosis, the
levels of cleaved caspase-3, -7 and PARP were used as markers for apoptosis. As shown in figure
10, cleaved caspase-3, -7 and PARP were detected in activated MEK1-transfected cells grown in
serum free medium. In contrast, these apoptotic markers were not detected in mock-transfected
cells in the absence of quercetin. Furthermore, activated MEK1-transfected cells were more
susceptible to quercetin-induced apoptosis than mock-transfected cells as determined by the
increased levels of apoptotic markers and TUNEL assay (Fig. 10G). The results further support the
important role of activated MEK-ERK in quercetin-induced apoptosis in A549 cells.
Discussion
The relationship between diet and cancers has been implicated in several epidemiological studies
(6). The cancer incidence is significantly lower in people whom diet consists largely of fruits and
vegetables than people whom diet consists mainly of animal products (6). The results from several
studies indicate that vegetables and fruits contain components that have anti-proliferative and anti-
neoplastic properties (56,57). In the present study, we have shown that quercetin inhibits human
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

16
A549 lung cancer cell proliferation and induces apoptosis. TUNEL assay clearly shows DNA
fragmentation. In addition to the inhibition of Akt-1 phosphorylation, alteration in the expression of
Bcl-2 family of proteins, sustained activation of ERK is required for quercetin-induced apoptosis in
A549 cells. Quercetin treatment results in dose- and time-dependent activation of ERK. The
elevated ERK activity contributed to apoptosis by quercetin is supported by the observations that
activation of ERK by expression of activated MEK1 induces apoptosis while inhibition of ERK by
MEK inhibitors blocks quercetin-induced cell death. Quercetin-induced apoptosis is associated with
phosphorylation of c-Jun, PARP cleavage, caspase -3 and -7 cleavage, all of which can be reduced
by treatment of A549 cells with the MEK1/2 inhibitor. Our findings suggest that in addition to the
inhibition of Akt activation and alteration in the ratio between the anti-apoptotic and pro-apoptotic
proteins, ERK activation plays an important role in mediating quercetin-induced apoptosis in A549
cells and ERK functions upstream of the caspase activation to initiate the apoptotic signal.
After ingestion, the major circulating metabolites in rat blood are quercetin glycosides, quercetin
glucoronides and quercetin sulfates. It remains to be determined whether quercetin is metabolised
to more or less active metabolites. Our preliminary data indicate that the concentration of quercetin
and its metabolites was 25.1, 43.3 and 54.3 µM after 10 days of treatment with 50, 100 and 150 mg
of quercetin per kg body weight respectively (our unpublished data). The results suggest that the
concentrations of quercetin employed in the present study can be achieved in vivo.
Two major distinct apoptotic pathways have been described for mammalian cells. One involves
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

17
caspase-8, which is recruited by the adapter molecule Fas/APO-1 associated death domain protein
to death receptors upon extracellular ligand binding (58). We do not observe any change in either
Fas or FasL expression in quercetin-treated A549 cells (data not shown). Administration of
quercetin results in a significant increase in the expression of Bax and Bad. Furthermore, the levels
of Bcl-xL are elevated while the expression of Bcl-2 protein is reduced by quercetin. Overall, there
is a shift in the ratio between the anti-apoptotic and pro-apoptotic proteins following quercetin
treatment. Although the contribution of Bcl-2 family of proteins to quercetin-induced apoptosis is
not examined in the present study, it is possible that the reduction in Bcl-2 by quercetin allows less
Bcl-2-Bax complex. Furthermore, inactivation of Akt prevents Akt-1 from phosphorylating Bad on
the serine 112 and serine 136. As a result, Bad becomes bound to Bcl-2, and its pro-apoptotic
activity is effectively increased from the death-regulation equation. Elevation of non-
phosphorylated Bad by quercetin further helps to sequester more Bcl-2 and Bcl-xL. The net effect is
more free Bax which then translocates to the mitochondrial membrane and induces the opening of
the mitochondrial permeability transition pore, a critical event in the loss of cell viability (59).
Anti-apoptotic effects of PI-3K are due to its activation of serine/threonine protein kinase Akt. This
kinase has been shown to block apoptosis via several mechanisms (60). In the present report, we
observe that quercetin inhibits Akt and Akt phosphorylation. Because Akt is a downstream target of
PI-3 kinase, the observed inhibition of Akt phosphorylation suggests that quercetin also inhibits PI-
3 kinase. This argument is supported by previous studies showing that quercetin is an inhibitor of
PI-3 kinase and serine/threonine protein kinases (12,13,15,19). By suppressing the activation of
Akt-1, quercetin can promote apoptosis via several pathways. Inactivation of Akt would prevent
Akt-1 from phosphorylating Bad on the serine 136. As a result, Bad becomes bound to Bcl-2, and
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

18
its pro-apoptotic activity is effectively increased in the death-regulation equation. Although the
effects of quercetin on the gene encoding A1/Bf11 protein is not examined in the present study, it is
possible that inhibition of Akt-1 phosphorylation by quercetin might impair the ability of Akt to
transactivate the gene encoding A1/Bf11 protein which, under certain experimental conditions, is
essential for the release of cytochrome c and/or AIF from the mitochondria (61).
Previous studies indicate that JNK/SAPK activation is associated with apoptosis (55,62). In our
own study using A549 lung cancer epithelial cells, we also find that 54 kDa JNK2 is activated in
response to quercetin, LY294002 and U0126 treatments. Our observation is different from the
previous study (19) showing that JNK activation by angiotensin II in cultured rat aortic smooth
muscle cells is inhibited by quercetin. Therefore, the effects quercetin on JNK may differ
significantly depending on the cell context. In U0126- and LY294002-treated cells, phosphorylation
of 54 kDa JNK2 takes place in the absence and in the presence of mild apoptosis respectively.
Furthermore, inhibition of JNK activity by SP600125 did not prevent quercetin-induced apoptosis.
Therefore, it is unlikely that activation of JNK plays an important role in quercetin-induced
apoptosis of A549 cells.
In the present study, we have provided the evidence that activation of MEK-ERK plays a dominant
role in quercetin-induced activation of caspase-3 and -7 which is necessary for cleavage of PARP
and apoptosis in A549 lung cancer cells. Quercetin treatment results in high and sustained
activation of ERK in A549 cells. One important difference between the quercetin and IGF-I
induced ERK activation is the time and duration of activity (data not shown). In the case of IGF-I,
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

19
ERK activation is rapid, occurring within minutes of treatment, and transient. With quercetin,
significant activation occurs at 3 h, but the activity remains highly elevated throughout the
experiment (up to 14 h). Utilizing U0126 or PD98059 to modulate ERK activity, we find that
inhibition of MEK-ERK activation abolishes quercetin-induced apoptosis. Our results are similar to
other studies which demonstrate that abrogation of ERK pathway by taxol delayed or failed to
prevent taxol-induced apoptosis (63,64). However, it is not a universal feature of mammalian cells
as activation of MEK-ERK pathway has been shown to contribute to cell proliferation and survival
(33), migration (34) and transformation (35). Furthermore, inhibition of stress-induced signalling
via the MEK-ERK pathway can potentiate the toxic effects of chemotherapic drugs and irradiation
(65). Therefore, the ability of MEK-ERK pathway to regulate proliferation versus survival appears
to depend on cell types and the amplitude and duration of ERK activation. A short activation of
MEK-ERK cascade by growth factors such as IGF-I is associated with proliferation while
prolonged activation of ERK activity inhibits DNA synthesis.
The questions remaining are: how does quercetin induce apoptosis of A549 cells and what are the
mechanisms responsible for transmitting the signal to the cell nucleus. It has been proposed that the
anti-proliferative and apoptotic effects of quercetin are also mediated via non-estrogen receptor
regulated mechanisms (20,29,30). Although the precise mechanisms of the anti-proliferation and
apoptosis of quercetin are unknown, there is evidence suggesting that the action of flavonoids is
probably mediated by interaction with the aryl hydrocarbon receptor (11) or type II estrogen binding
sites (10) which have been shown to occupy by a flavonoid-like molecule with growth inhibitory
properties (66). In addition to activation of MEK-ERK as we report in this study, quercetin is
shown to induce apoptosis through a pathway involving heat shock proteins (32). Other effects of
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

20
quercetin may also contribute to its anti-proliferative activity seen in certain experimental systems
including inhibition of mutant p53 expression in breast cancer cells (20) and blockage of the cell
cycle at the G1/S or G2/M transition (27,28). Quercetin also inhibits various enzymes involved in
proliferation and apoptosis including protein kinase C (12), tyrosine kinase (13), cdc25 phosphatase
(14), PI-3 kinase (12,15), and DNA topoisomerase II (16). This suggests that quercetin exerts
multiple effects on cellular growth and apoptosis. The target proteins observed under one
experimental condition or cell type may differ from one another depending on cell context.
Our data support a role for ERK activation in quercetin-induced growth inhibition and apoptosis in
lung cancer cells. Since quercetin can act in synergy with cisplatin to inhibit lung cancer xenograft
growth (67) and potentiate the cytotoxic action of 1-ß-D-arabinofuranosylcytosine (31), it raises the
possibility that quercetin can be used either as a single agent or in combination with other
chemotherapeutic agents such as cisplatin or 1-ß-D-arabinofuranosylcytosine in treatment and/or
prevention of lung cancer.
Acknowledgements
We thank Professor Phillip Leder (Department of Genetics, Harvard Medical School) for the
plasmid pß-MEKDD plasmid. This work was supported by grants from National Medical Research
Council of Singapore (NMRC/0541/2001), A*STAR-BMRC (LS/00/019) and A*STAR-BMRC
(LS/00/017) to Huynh Hung.
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

21
Reference
1. Bilello,K.S., Murin,S., and Matthay,R.A. (2002) Epidemiology, etiology, and prevention of
lung cancer. Clin. Chest Med., 23, 1-25.
2. Alavanja,M.C. (2002) Biologic damage resulting from exposure to tobacco smoke and from
radon: implication for preventive interventions. Oncogene, 21, 7365-7375.
3. Midthun,D.E. and Jett,J.R. (1997) Chemotherapy for advanced lung cancer. When to expect
a response. Postgrad. Med, 101, 187-2, 194.
4. Feng,G., Xu,X., Youssef,E.M., and Lotan,R. (2001) Diminished expression of S100A2, a
putative tumor suppressor, at early stage of human lung carcinogenesis. Cancer Res., 61,
7999-8004.
5. Gargiullo P, Wingo PA, Coates RJ, and Thompson TD. Recent trends in mortality rates for
four major cancers, by sex and race/ethnicity - United States. MMWR 51, 49-53. 2002.
6. Block,G., Patterson,B., and Subar,A. (1992) Fruit, vegetables, and cancer prevention: a
review of the epidemiological evidence. Nutr. Cancer, 18, 1-29.
7. Zhou,J.R., Mukherjee,P., Gugger,E.T., Tanaka,T., Blackburn,G.L., and Clinton,S.K. (1998)
Inhibition of murine bladder tumorigenesis by soy isoflavones via alterations in the cell
cycle, apoptosis, and angiogenesis. Cancer Res., 58, 5231-5238.
8. Shao,Z.M., Wu,J., Shen,Z.Z., and Barsky,S.H. (1998) Genistein exerts multiple suppressive
effects on human breast carcinoma cells. Cancer Res., 58, 4851-4857.
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

22
9. Anton,R. (1988) Flavonoids and Traditional Medicine. Alan R.Liss, Inc., New York.
10. Ranelletti,F.O., Ricci,R., Larocca,L.M., Maggiano,N., Capelli,A., Scambia,G., Benedetti-
Panici,P., Mancuso,S., Rumi,C., and Piantelli,M. (1992) Growth-inhibitory effect of
quercetin and presence of type-II estrogen- binding sites in human colon-cancer cell lines
and primary colorectal tumors. Int. J. Cancer, 50, 486-492.
11. Ashida,H., Fukuda,I., Yamashita,T., and Kanazawa,K. (2000) Flavones and flavonols at
dietary levels inhibit a transformation of aryl hydrocarbon receptor induced by dioxin. FEBS
Lett., 476, 213-217.
12. Agullo,G., Gamet-Payrastre,L., Manenti,S., Viala,C., Remesy,C., Chap,H., and Payrastre,B.
(1997) Relationship between flavonoid structure and inhibition of phosphatidylinositol 3-
kinase: a comparison with tyrosine kinase and protein kinase C inhibition. Biochem.
Pharmacol., 53, 1649-1657.
13. Hagiwara,M., Inoue,S., Tanaka,T., Nunoki,K., Ito,M., and Hidaka,H. (1988) Differential
effects of flavonoids as inhibitors of tyrosine protein kinases and serine/threonine protein
kinases. Biochem. Pharmacol., 37, 2987-2992.
14. Aligiannis,N., Mitaku,S., Mitrocotsa,D., and Leclerc,S. (2001) Flavonoids as cycline-
dependent kinase inhibitors: inhibition of cdc 25 phosphatase activity by flavonoids
belonging to the quercetin and kaempferol series. Planta Med., 67, 468-470.
15. Gamet-Payrastre,L., Manenti,S., Gratacap,M.P., Tulliez,J., Chap,H., and Payrastre,B.
(1999) Flavonoids and the inhibition of PKC and PI 3-kinase. Gen. Pharmacol., 32, 279-
286.
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

23
16. Constantinou,A., Mehta,R., Runyan,C., Rao,K., Vaughan,A., and Moon,R. (1995)
Flavonoids as DNA topoisomerase antagonists and poisons: structure- activity relationships.
J. Nat. Prod., 58, 217-225.
17. Lee,S.C., Kuan,C.Y., Yang,C.C., and Yang,S.D. (1998) Bioflavonoids commonly and
potently induce tyrosine dephosphorylation/inactivation of oncogenic proline-directed
protein kinase FA in human prostate carcinoma cells. Anticancer Res., 18, 1117-1121.
18. Suolinna,E.M., Lang,D.R., and Racker,E. (1974) Quercetin, an artificial regulator of the
high aerobic glycolysis of tumor cells. J.Natl.Cancer Inst., 53, 1515-1519.
19. Yoshizumi,M., Tsuchiya,K., Kirima,K., Kyaw,M., Suzaki,Y., and Tamaki,T. (2001)
Quercetin inhibits Shc- and phosphatidylinositol 3-kinase-mediated c- Jun N-terminal
kinase activation by angiotensin II in cultured rat aortic smooth muscle cells.
Mol.Pharmacol., 60, 656-665.
20. Avila,M.A., Velasco,J.A., Cansado,J., and Notario,V. (1994) Quercetin mediates the down-
regulation of mutant p53 in the human breast cancer cell line MDA-MB468. Cancer Res.,
54, 2424-2428.
21. Xing,N., Chen,Y., Mitchell,S.H., and Young,C.Y. (2001) Quercetin inhibits the expression
and function of the androgen receptor in LNCaP prostate cancer cells. Carcinogenesis, 22,
409-414.
22. Scambia,G., Ranelletti,F.O., Benedetti,P.P., Piantelli,M., Rumi,C., Battaglia,F.,
Larocca,L.M., Capelli,A., and Mancuso,S. (1990) Type-II estrogen binding sites in a
lymphoblastoid cell line and growth- inhibitory effect of estrogen, anti-estrogen and
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

24
bioflavonoids. Int. J. Cancer, 46, 1112-1116.
23. Markaverich,B.M., Roberts,R.R., Alejandro,M.A., Johnson,G.A., Middleditch,B.S., and
Clark,J.H. (1988) Bioflavonoid interaction with rat uterine type II binding sites and cell
growth inhibition. J. Steroid Biochem., 30, 71-78.
24. Larocca,L.M., Piantelli,M., Leone,G., Sica,S., Teofili,L., Panici,P.B., Scambia,G.,
Mancuso,S., Capelli,A., and Ranelletti,F.O. (1990) Type II oestrogen binding sites in acute
lymphoid and myeloid leukaemias: growth inhibitory effect of oestrogen and flavonoids. Br.
J. Haematol., 75, 489-495.
25. Hosokawa,N., Hosokawa,Y., Sakai,T., Yoshida,M., Marui,N., Nishino,H., Kawai,K., and
Aoike,A. (1990) Inhibitory effect of quercetin on the synthesis of a possibly cell- cycle-
related 17-kDa protein, in human colon cancer cells. Int. J. Cancer, 45, 1119-1124.
26. Kuo,M.L., Lin,J.K., Huang,T.S., and Yang,N.C. (1994) Reversion of the transformed
phenotypes of v-H-ras NIH3T3 cells by flavonoids through attenuating the content of
phosphotyrosine. Cancer Lett., 87, 91-97.
27. Ranelletti,F.O., Ricci,R., Larocca,L.M., Maggiano,N., Capelli,A., Scambia,G., Benedetti-
Panici,P., Mancuso,S., Rumi,C., and Piantelli,M. (1992) Growth-inhibitory effect of
quercetin and presence of type-II estrogen- binding sites in human colon-cancer cell lines
and primary colorectal tumors. Int. J. Cancer, 50, 486-492.
28. Yoshida,M., Yamamoto,M., and Nikaido,T. (1992) Quercetin arrests human leukemic T-
cells in late G1 phase of the cell cycle. Cancer Res., 52, 6676-6681.
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

25
29. Plaumann,B., Fritsche,M., Rimpler,H., Brandner,G., and Hess,R.D. (1996) Flavonoids
activate wild-type p53. Oncogene, 13, 1605-1614.
30. Ferrandina,G., Almadori,G., Maggiano,N., Lanza,P., Ferlini,C., Cattani,P., Piantelli,M.,
Scambia,G., and Ranelletti,F.O. (1998) Growth-inhibitory effect of tamoxifen and quercetin
and presence of type II estrogen binding sites in human laryngeal cancer cell lines and
primary laryngeal tumors. Int. J. Cancer, 77, 747-754.
31. Teofili,L., Pierelli,L., Iovino,M.S., Leone,G., Scambia,G., De Vincenzo,R., Benedetti-
Panici,P., Menichella,G., Macri,E., and Piantelli,M. (1992) The combination of quercetin
and cytosine arabinoside synergistically inhibits leukemic cell growth. Leuk. Res., 16, 497-
503.
32. Wei,Y.Q., Zhao,X., Kariya,Y., Fukata,H., Teshigawara,K., and Uchida,A. (1994) Induction
of apoptosis by quercetin: involvement of heat shock protein. Cancer Res., 54, 4952-4957.
33. Ballif,B.A. and Blenis,J. (2001) Molecular mechanisms mediating mammalian mitogen-
activated protein kinase (MAPK) kinase (MEK)-MAPK cell survival signals. Cell Growth
Differ., 12, 397-408.
34. Krueger,J.S., Keshamouni,V.G., Atanaskova,N., and Reddy,K.B. (2001) Temporal and
quantitative regulation of mitogen-activated protein kinase (MAPK) modulates cell motility
and invasion. Oncogene, 20, 4209-4218.
35. Montesano,R., Soriano,J.V., Hosseini,G., Pepper,M.S., and Schramek,H. (1999)
Constitutively active mitogen-activated protein kinase kinase MEK1 disrupts
morphogenesis and induces an invasive phenotype in Madin-Darby canine kidney epithelial
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

26
cells. Cell Growth Differ., 10, 317-332.
36. Dudley,D.T., Pang,L., Decker,S.J., Bridges,A.J., and Saltiel,A.R. (1995) A synthetic
inhibitor of the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. U.S.A, 92,
7686-7689.
37. Liu,E., Thant,A.A., Kikkawa,F., Kurata,H., Tanaka,S., Nawa,A., Mizutani,S., Matsuda,S.,
Hanafusa,H., and Hamaguchi,M. (2000) The Ras-mitogen-activated protein kinase pathway
is critical for the activation of matrix metalloproteinase secretion and the invasiveness in v-
crk-transformed 3Y1. Cancer Res., 60, 2361-2364.
38. Chen,Y., Lu,Q., Schneeberger,E.E., and Goodenough,D.A. (2000) Restoration of tight
junction structure and barrier function by down- regulation of the mitogen-activated protein
kinase pathway in ras- transformed Madin-Darby canine kidney cells. Mol. Biol. Cell, 11,
849-862.
39. Moos,P.J. and Fitzpatrick,F.A. (1998) Taxanes propagate apoptosis via two cell populations
with distinctive cytological and molecular traits. Cell Growth Differ., 9, 687-697.
40. Fisher,T.C., Milner,A.E., Gregory,C.D., Jackman,A.L., Aherne,G.W., Hartley,J.A., Dive,C.,
and Hickman,J.A. (1993) bcl-2 modulation of apoptosis induced by anticancer drugs:
resistance to thymidylate stress is independent of classical resistance pathways. Cancer
Res., 53, 3321-3326.
41. Boise,L.H., Gonzalez-Garcia,M., Postema,C.E., Ding,L., Lindsten,T., Turka,L.A., Mao,X.,
Nunez,G., and Thompson,C.B. (1993) bcl-x, a bcl-2-related gene that functions as a
dominant regulator of apoptotic cell death. Cell, 74, 597-608.
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

27
42. Franke,T.F., Yang,S.I., Chan,T.O., Datta,K., Kazlauskas,A., Morrison,D.K., Kaplan,D.R.,
and Tsichlis,P.N. (1995) The protein kinase encoded by the Akt proto-oncogene is a target
of the PDGF-activated phosphatidylinositol 3-kinase. Cell, 81, 727-736.
43. Page,C., Lin,H.J., Jin,Y., Castle,V.P., Nunez,G., Huang,M., and Lin,J. (2000)
Overexpression of Akt/AKT can modulate chemotherapy-induced apoptosis. Anticancer
Res., 20, 407-416.
44. Datta,S.R., Dudek,H., Tao,X., Masters,S., Fu,H., Gotoh,Y., and Greenberg,M.E. (1997) Akt
phosphorylation of BAD couples survival signals to the cell- intrinsic death machinery.
Cell, 91, 231-41.
45. Zha,J., Harada,H., Yang,E., Jockel,J., and Korsmeyer,S.J. (1996) Serine phosphorylation of
death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L)
(see comments). Cell, 87, 619-628.
46. Niculescu,A.B., III, Chen,X., Smeets,M., Hengst,L., Prives,C., and Reed,S.I. (1998) Effects
of p21(Cip1/Waf1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical
determinant in blocking DNA replication and in preventing endoreduplication. Mol. Cell
Biol., 18, 629-643.
47. Rom,W.N. and Tchou-Wong,K.M. (2003) Molecular and genetic aspects of lung cancer.
Methods Mol. Med., 75, 3-26.
48. Pinkas,J. and Leder,P. (2002) MEK1 signaling mediates transformation and metastasis of
EpH4 mammary epithelial cells independent of an epithelial to mesenchymal transition.
Cancer Res, 62, 4781-4790.
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

28
49. Lim,I.J., Phan,T.T., Song,C., Tan,W.T.L., and Longaker,M.T. (2001) Investigation of the
influence of keloid-derived keartinocytes on fibroblast growth and proliferation in vitro.
Plast. Reconstr. Surg., 107.
50. Huynh,H., Chow,P.K., Ooi,L.L., and Soo,K.C. (2002) A possible role for insulin-like
growth factor-binding protein-3 autocrine/paracrine loops in controlling hepatocellular
carcinoma cell proliferation. Cell Growth Differ., 13, 115-122.
51. Downward,J. (1999) How BAD phosphorylation is good for survival (news). Nat. Cell
Biol., 1, E33-E35.
52. Germain,M., Affar,E.B., D'Amours,D., Dixit,V.M., Salvesen,G.S., and Poirier,G.G. (1999)
Cleavage of automodified poly(ADP-ribose) polymerase during apoptosis. Evidence for
involvement of caspase-7. J. Biol. Chem., 274, 28379-28384.
53. Kulik,G., Klippel,A., and Weber,M.J. (1997) Antiapoptotic signalling by the insulin-like
growth factor I receptor, phosphatidylinositol 3-kinase, and Akt. Mol. Cell Biol., 17, 1595-
1606.
54. Walter kolch, Ashwin Kotwaliwale, Keith Vass, and Petra Janosch. The role of Raf kinases
in malignant transformation. Expert Reviews in molecular medicine , 1-18. 25-4-2002. UK,
Cambridge University Press.
55. MacKeigan,J.P., Collins,T.S., and Ting,J.P. (2000) MEK inhibition enhances paclitaxel-
induced tumor apoptosis. J. Biol. Chem., 275, 38953-38956.
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

29
56. Leighton T, GInther C, Fluss L, Harter WK, Cansado J, and Notario V (1992) Phenolic
compounds in food and their effects on health II. American Chemical Society, Washington.
57. Messina,M.J., Persky,V., Setchell,K.D., and Barnes,S. (1994) Soy intake and cancer risk: a
review of the in vitro and in vivo data. Nutr. Cancer, 21, 113-131.
58. Muzio,M., Stockwell,B.R., Stennicke,H.R., Salvesen,G.S., and Dixit,V.M. (1998) An
induced proximity model for caspase-8 activation. J. Biol. Chem., 273, 2926-2930.
59. Chao,D.T. and Korsmeyer,S.J. (1998) BCL-2 family: regulators of cell death. Annu. Rev.
Immunol., 16, 395-419.
60. Khwaja,A. (1999) Akt is more than just a Bad kinase (news; comment). Nature, 401, 33-34.
61. Wang,C.Y., Guttridge,D.C., Mayo,M.W., and Baldwin,A.S., Jr. (1999) NF-kappaB induces
expression of the Bcl-2 homologue A1/Bfl-1 to preferentially suppress chemotherapy-
induced apoptosis. Mol. Cell Biol., 19, 5923-5929.
62. Yujiri,T., Sather,S., Fanger,G.R., and Johnson,G.L. (1998) Role of MEKK1 in cell survival
and activation of JNK and ERK pathways defined by targeted gene disruption. Science, 282,
1911-1914.
63. Lieu,C.H., Liu,C.C., Yu,T.H., Chen,K.D., Chang,Y.N., and Lai,Y.K. (1998) Role of
mitogen-activated protein kinase in taxol-induced apoptosis in human leukemic U937 cells.
Cell Growth Differ., 9, 767-776.
64. Kalechman,Y., Longo,D.L., Catane,R., Shani,A., Albeck,M., and Sredni,B. (2000)
Synergistic anti-tumoral effect of paclitaxel (Taxol)+AS101 in a murine model of B16
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

30
melanoma: association with ras-dependent signal- transduction pathways. Int. J. Cancer, 86,
281-288.
65. Yano,T., Pinski,J., Groot,K., and et al. (1992) Stimulation by bombesin and inhibition by
bombesin/GRP antagonist RC-3905 of growth of human breast cancer cell lines
Cancer Res., 52, 4545-4547.
66. Markaverich,B.M., Roberts,R.R., Alejandro,M.A., and Clark,J.H. (1984) An endogenous
inhibitor of (3H)estradiol binding to nuclear type II estrogen binding sites in normal and
malignant tissues. Cancer Res., 44, 1515-1519.
67. Hofmann,J., Doppler,W., Jakob,A., Maly,K., Posch,L., Uberall,F., and Grunicke,H.H.
(1988) Enhancement of the antiproliferative effect of cis- diamminedichloroplatinum(II)
and nitrogen mustard by inhibitors of protein kinase C. Int. J. Cancer, 42, 382-388.
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

31
Figure Legends
Figure 1. Effects of quercetin on the viability and proliferation of A549 cells. A549 lung cancer
cells were grown and treated with serum free RPMI 1640 (SRF) medium containing either 0.1%
DMSO or indicated doses of quercetin for 24 h and 48 h as described under Materials and Methods.
Cell proliferation (A) and cell viability (B) were determined by Bromo-uridine incorporation and
MTT assay respectively as described under Materials and Methods. Experiments were performed in
quadruplicate, with the results reflecting the mean and standard deviation of the quadruplicate of
each group. Bars with different letters are significantly different from one another at p<0.01 as
determined by the Kruskal-Wallis test. The experiments were repeated 3 times with similar results.
Figure 2. Induction of apoptosis by quercetin in A549 cells. A549 cells were grown and treated
with escalating doses of quercetin in SRF medium for 14 h. Apoptotic cells were determined by
TUNEL assay as described in Materials and Methods. (A) Apoptotic cells were visualized under a
fluorescent microscope. The rate of apoptosis expressed as percentage of total cells counted is
shown in (B). Bars with different letters are significantly different from one another at p<0.01 as
determined by the Kruskal-Wallis test. Experiments were repeated 3 times with similar results.
Figure 3. Effects of quercetin on the levels of Bcl-2, Bax, Bad and Bcl-xL in A549 cells. A549
cells were cultured as described under Materials and Methods. Cells were treated with 0.1% DMSO
or indicated concentrations of quercetin in SRF medium for 14 h. Cells were harvested and lysed
for Western blot analysis as described under Materials and Methods. Blots were incubated with
mouse anti-α-tubulin (A), mouse anti-Bax (B), rabbit anti-Bad (C), rabbit anti-Bcl-xL (D) and
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

32
rabbit anti-Bcl-2 (E) antibodies. Changes in the levels of Bax, Bad, Bcl-2 and Bcl-xL proteins after
being normalized to the levels of α-tubulin are shown below each blot. Experiments were repeated
3 times with similar results.
Figure 4. Effects of quercetin on cleavage of caspase-3 and -7 and PARP in A549 cells. A549
cells were cultured as described under Materials and Methods. Cells were treated with SRF medium
containing 0.1% DMSO or indicated concentrations of quercetin for 14 h. Cells were harvested and
lysed for Western blot analysis as described under Materials and Methods. Blots were incubated
with mouse anti-α-tubulin (A), rabbit anti-cleaved caspase-3 (B), rabbit anti-cleaved caspase-7 (20
kDa) (C) and rabbit anti-cleaved PARP (D) antibodies. Changes in the levels of cleaved caspase-3,
cleaved caspase-7 and cleaved PARP after being normalized to the levels of α-tubulin are shown
below each blot. Experiments were repeated 3 times with similar results.
Figure 5. Effects of quercetin on the basal levels of p85 subunit of PI-3 kinase, Akt-1 and
phosphorylated Akt (Ser473) in A549 cells. A549 cells were cultured as described in Materials
and Methods. Cells were treated with SRF medium containing 0.1% DMSO or indicated
concentrations of quercetin for 14 h. Cells were harvested and lysed for Western blot analysis as
described under Materials and Methods. Blots were incubated with mouse anti-α-tubulin (A), rabbit
anti- p85 subunit of PI-3 kinase (B), rabbit anti-phospho Akt-1 (Ser473) (C), and rabbit anti-Akt-1
(D) antibodies. Changes in the levels of the Akt-1 and phospho-Akt-1 after being normalized to the
levels of α-tubulin are shown below each blot. Experiments were repeated 3 times with similar
results.
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

33
Figure 6. Effects of quercetin on the levels of MEK1, ERK, and phosphorylated MEK1/2
(Ser217/221), phosphorylated ERK (Thr202/Tyr204), phosphorylated JNK (Thr183/Tyr185),
and phosphorylated c-Jun (Ser63) in A549 cells. A549 cells were cultured as described under
Materials and Methods. Cells were treated with SRF medium containing 0.1% DMSO or indicated
concentrations of quercetin for 14 h. Cells were harvested and lysed for Western blot analysis as
described under Materials and Methods. Blots were incubated with mouse anti-α-tubulin (A), rabbit
anti-phospho MEK1/2 (Ser217/221) (B), rabbit anti-MEK1 (C), mouse anti-phospho p44/42 ERK
(Thr202/Tyr204) (D), rabbit anti-ERK (E), and mouse anti-phospho c-Jun (Ser63) (F) and mouse
anti-phospho JNK (Thr183/Tyr185) (G). Changes in the levels of phosphorylated ERK,
phosphorylated MEK1/2, phosphorylated JNK and phosphorylated c-Jun after being normalized to
the levels of a-tubulin are shown below each blot. Experiments were repeated 3 times with similar
results.
Figure 7. Time-dependent phosphorylation of MEK1/2, ERK, JNK and c-Jun, and cleavage
of caspase-3 and -7 and PARP in A549 cells. A549 cells were cultured as described under
Materials and Methods. Cells were treated with SRF medium containing 0.1% DMSO or 58.0 µM
of quercetin for 3, 6, 9 and 14 h. Cells were harvested at indicated time and lysed for Western blot
analysis as described under Materials and Methods. Blots were incubated with mouse anti-α-
tubulin (A), rabbit anti-phospho MEK 1/2 (Ser217/221) (B), rabbit anti-MEK1 (C), mouse anti-
phospho p44/42 ERK (Thr202/Tyr204) (D), rabbit anti-ERK (E), mouse anti-phospho c-Jun
(Ser63) (F), mouse anti-phospho JNK (Thr183/Tyr185) (G), rabbit anti-cleaved caspase-3 (H),
rabbit anti-cleaved caspase-7 (I) and rabbit anti-cleaved PARP (K) antibodies. Experiments were
repeated 3 times with similar results.
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

34
Figure 8. Effects of MEK1/2 inhibitor U0126 and PI-3 kinase inhibitor LY294002 on
quercetin-induced apoptosis in A549 cells. A549 lung cancer cells were grown and treated with
SRF medium containing 0.1% DMSO (A), 10.0 µM of U0126 (B), 10.0 µM of LY294002 (C),
58.0 µM of quercetin (D), 58.0 µM of quercetin plus 10.0 µM of U0126 (E), and 58.0 µM of
quercetin plus 10.0 µM of LY294002 (F) for 14 h. Cells were subjected to TUNEL assay as
described under Materials and Methods. Apoptotic cells were visualized under a fluorescent
microscope. Original magnification, X 200.
Figure 9. Effects of MEK1/2 inhibitor U0126 or PD98059, p38 kinase inhibitor SB203580,
JNK inhibitor SP600125, caspase inhibitor Z-VAD-FMK and PI-3 kinase inhibitor LY294002
on quercetin-induced phosphorylation of ERK, c-Jun, JNK, and cleavage of caspase-3, -7,
and PARP in A549 cells. A549 lung cancer cells were grown and treated with SRF medium
containing 0.1% DMSO and indicated concentration of drugs for 14 h. Cells were harvested and
lysed for Western blot analysis as described under Materials and Methods. Blots were incubated
with mouse anti-α-tubulin (A), mouse anti-phospho p44/42 ERK (Thr202/Tyr204) (B), rabbit anti-
ERK (C), mouse phospho c-Jun (Ser63) (D), mouse anti-phospho JNK (Thr183/Tyr185) (E), rabbit
anti-cleaved caspase-3 (F), rabbit anti-cleaved caspase-7 (G) and rabbit anti-cleaved PARP (H)
antibodies. Experiments were repeated 3 times with similar results.
Figure 10. Effects of constitutively activated MEK1 on quercetin-induced ERK, phospho-
ERK (Thr202/Tyr204), cleavage caspase-3, caspase-7 and PARP and apoptosis in mock-
transfected and activated MEK1-transfected A549 cells. Mock-transfected and constitutively
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

35
activated MEK1-transfected A549 cells were cultured as described under Materials and Methods.
Cells were incubated with PSF medium containing 0.1% DMSO or indicated concentrations of
quercetin for 14 h. Total cell lysate was prepared for Western blot analysis as described under
Materials and Methods. Blots were incubated with mouse anti-∝-tubulin (A), mouse anti-phospho-
ERK (Thr202/Tyr204) (B), mouse anti-ERK (C), rabbit anti-cleaved caspase-3 (D), rabbit anti-
cleaved caspase-7 (E) and rabbit anti-cleaved PARP (F) antibodies. All the antibodies were used at
a final concentration of 1 µg per ml. Apoptotic cells were determined by TUNEL assay as described
in Materials and Methods. The rate of apoptosis expressed as percentage of total cells counted is
shown in (G). Bars with different letters are significantly different from one another at p<0.01 as
determined by the Kruskal-Wallis test. Experiments were repeated 3 times with similar results.
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from
36
Figure 1
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

37
Figure 2
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

38
Figure 3
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

39
Figure 4
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

40
Figure 5
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

41
Figure 6
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

42
Figure 7
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

43
Figure 8
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

44
Figure 9
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

45
Figure 10
by guest on Decem
ber 23, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from
Top Related
Copyright © 2022 FDOKUMEN

