







Bahasa
Halaman
Hukum


Journal of the Neurological Sciences 308 (2011) 35–40
Contents lists available at ScienceDirect
Journal of the Neurological Sciences
j ourna l homepage: www.e lsev ie r.com/ locate / jns
The involvement of Na+, K+-ATPase activity and free radical generation in thesusceptibility to pentylenetetrazol-induced seizures after experimental traumaticbrain injury
Luiz Fernando Almeida Silva c, Maurício Scopel Hoffmann a, Leonardo Magno Rambo b,Leandro Rodrigo Ribeiro b, Frederico Diniz Lima c, Ana Flavia Furian a, Mauro Schneider Oliveira a,Michele Rechia Fighera d, Luiz Fernando Freire Royes a,c,⁎a Centro de Ciências da Saúde, Laboratório de Neurotoxicidade e Psicofarmacologia, Departamento de Fisiologia, Universidade Federal de Santa Maria, 97105-900 Santa Maria, RS, Brazilb Programa de Pós-graduação em Ciências Biológicas: Bioquímica, Universidade Federal de Santa Maria, 90035-003 Porto Alegre, RS, Brazilc Centro de Educação Física e Desportos, Departamento de Métodos e Técnicas Desportivas, Universidade Federal de Santa Maria, 97105-900 Santa Maria, RS, Brazild Instituto do Cérebro (InsCer), Pontifícia Universidade Católica do Rio Grande do Sul— PUC, Porto Alegre,90619-900 Brazil
⁎ Corresponding author at: Departamento deMétodosde Educação Física e Desportos, Universidade Federal deMaria, RS, Brazil. Fax: +55 55 3220 8031.
E-mail address: [email protected] (L.F.F. Ro
0022-510X/$ – see front matter. Crown Copyright © 20doi:10.1016/j.jns.2011.06.030
a b s t r a c t
a r t i c l e i n f oArticle history:Received 13 December 2010Received in revised form 10 June 2011Accepted 16 June 2011Available online 6 July 2011
Keywords:Na+,K+-ATPaseFluid percussion brain injuryOxidative damageSeizuresProtein carbonylationTBARS
Although the importance of brain trauma as risk factor for the development of epilepsy is well established, themechanisms of epileptogenesis are not well understood. In the present study, we revealed that the injection of asubthreshold dose of PTZ (30 mg/Kg, i.p.) after 5 weeks of injury induced by Fluid Percussion Brain Injury (FPI)decreased latency for first clonic seizures, increased the time of spent generalized tonic–clonic seizures andelectrocorticographic (EEG)waveamplitude. In addition, statistical analysis revealed thatN-acetylcysteine (NAC)(100 mg/kg) supplementation during 5 weeks after neuronal injury protected against behavioral andelectrographical seizure activity elicited by subthreshold dose of PTZ. The supplementation of this antioxidantcompound also protected against theNa+,K+-ATPase activity inhibition and concomitant increase in the levels ofoxidative stress markers (protein carbonylation and thiobarbituric acid-reactive substances-TBARS) in site andperi-contusional cortical tissue. In summary, the current experiments clearly showed that FPImodel induces earlyposttraumatic seizures and suggest that an alteration in the lipid/protein oxidation,membrane fluidity, and Na+,K+-ATPase activitymay be correlatedwith neuronal excitability, a significant component of the secondary injurycascade that accompanies TBI.
e Técnicas Desportivas, CentroSanta Maria, 97105-900 Santa
yes).
11 Published by Elsevier B.V. All rights res
Crown Copyright © 2011 Published by Elsevier B.V. All rights reserved.
1. Introduction
Traumatic brain injury (TBI) is a devastating disease thatcommonly causes disability and strongly affects the quality of life ofpatients [1,2]. TBI also exacerbates seizure severity in individuals withpreexisting epilepsy [28], being one example of the process ofepileptogenesis [3]. In this context, it has been demonstrated thatearly lesion in Central Nervous System (CNS) alter the transportdynamic of the blood–brain barrier (BBB) and deteriorate the balanceof inhibitory and excitatory neurotransmitter system [4]. Thisneuronal dysfunction predisposes to subsequent development ofspontaneous recurrent seizures in the presence of prior subtle brainmalformation [5]. Nevertheless, there is great need for the identifi-cation of biomarkers that provide quantitative measures of the
process of post-traumatic epileptogenesis and predict which TBIpatients are likely to develop epilepsy.
In this context, oxidative stress, an imbalance between oxidantsand antioxidants, contributes to the pathogenesis of TBI [6,1]. Recentstudies have demonstrated that the oxidative damage mediated byreactive oxygen species (ROS) and nitrogen species (RNS) is asignificant component of the secondary injury cascade that accom-panies TBI [7–11]. Although several evidences supporting the ideathat any cellular constituent may be a target for free radical damage[12], the inhibition of some selected targets such as Na+,-K+-ATPasemay play an important role in the hyperexcitability induced by TBI[13]. Na+,K+-ATPase is a membrane bound enzyme known to play apivotal role in cellular ionic gradient maintenance and is particularlysensitive to reactive species [14,15]. Several studies have suggestedthat ROS inhibit the activity of Na+, K+-ATPase by oxidation of SHgroups and alteration of the membrane fluidity [16–19]. Recently, ithas demonstrated that experimental Fluid Percussion Injury (FPI)induces impairment in animal performance in a spatial learning,increase of the oxidative stress markers, and decrease in Na+,K+-ATPase activity suggesting that cognitive impairment following TBI
erved.

36 L.F.A. Silva et al. / Journal of the Neurological Sciences 308 (2011) 35–40
may result, at least in part, from an increase of oxidative stressmarkers that occurs concomitantly with a decrease in Na+,K+-ATPaseactivity [20].
It is well known that the overproduction of free radicals may leadto the development of epileptic focus (neurons) by the disruption ofthe antioxidant activity [21,22] and by the oxidative inactivation ofmembrane Na+,K+-ATPase. Thesemechanismswould be a key reasonbehind enhanced neuronal excitability and seizuremanifestation [23],since Na+,K+-ATPase activity is inhibited in brain of epileptic patientspost-mortem[24] and mutation in the Na+,K+-ATPase α subunit genehas been associated with epilepsy in humans [25]. Therefore, thepresent study was performed to investigate the effect of the freeradicals generation and Na+,K+-ATPase activity in the process ofpost-traumatic epileptogenesis.
2. Materials and methods
2.1. Animal and reagents
Adult male Wistar rats (270–300 g) were maintained undercontrolled light and environment (12:12 h light/dark cycle, 24±1 °C, 55% relative humidity), with standard laboratory chow andwater ad libitum. Animal utilization reported in this study has beenconducted in accordance with the policies of the National Institute ofHealth Guide for the Care and Use of Laboratory Animals (NIHPublications No. 80-23) revised in 1996, and with the approval of theEthics Committee for Animal Research of the Federal University ofSanta Maria (23081.018516/2006-57).
2.2. Traumatic brain injury and drug treatment
FPI was carried out as described previously [26,27]. Briefly, animalswere anesthetized with a single i.p. injection of Equithesin (6 ml/kg),a mixture containing sodium pentobarbital (58 mg/kg), chloralhydrate (60 mg/kg), magnesium sulfate (127.2 mg/kg), propyleneglycol (42.8%), and absolute ethanol (11.6%) and placed in a rodentstereotaxic apparatus. A burr hole of 3 mm in diameter was drilled onthe right convexity, 2 mm posterior to the bregma and 3 mm lateral tothe midline, taking care to keep the dura mater intact. A plastic injurycannula was placed over the craniotomy with dental cement. Whenthe dental cement hardened, the cannula was filled with Chloram-phenicol, closed with a proper plastic cap, and the animal wasremoved from the stereotaxic device and returned to its homecage.After 24 h, the animals were anesthetized with Halothane, and hadthe injury cannula attached to the fluid percussion device and placedin a heatpad maintained at 37±0.2 °C. TBI was produced by a fluid-percussion device developed in our laboratory. A brief (10–15 ms)transient pressure fluid pulse (4.05±0.17 atm) impact was appliedagainst the exposed dura. Pressure pulses were measured extracra-nially by a transducer (Fluid Control Automação Hidráulica, BeloHorizonte, MG, Brazil) and recorded on a storage oscilloscope (GouldLtd., Essex, UK). Sham-operated animals underwent an identicalprocedure, with the exception of FPI.
In order to evaluate the combination between electrographicseizures, oxidative and neurochemical alterations in cerebral cortexafter TBI, immediately after FPI procedure, a subset of animals weresupplemented with NAC (100 mg/kg) or its vehicle (distillated H2O)by intragastric gavage during 5 weeks after FPI procedure.
2.3. Seizure evaluation
After 4 weeks of FPI procedure, all animals were deeply anesthe-tized and two screw electrodes were placed bilaterally over theparietal cortex, along with a ground lead positioned over the nasalsinus as described by [22]. The experiments for electrocorticographic(EEG) recordings were performed 5 days after surgery as described by
[23]. Briefly, the rat was connected to the lead socket in a swivel insidea Faraday's cage, and the EEG was recorded using a digitalencephalographer (Neuromap EQSA260, Neurotec LTDA, Brazil).EEG signals were amplified, filtered (0.1 to 70.0 Hz, bandpass),digitalized (sampling rate 256 Hz) and stored in a PC for off-lineanalysis. Routinely, a 10 min baseline recording was obtained toestablish an adequate control period. After baseline recording, thesham or TBI animals received an injection of saline (0.9% NaCl,1 ml/kg,i.p) and/or subconvulsant dose of PTZ (30 mg/Kg, i.p.) [41]. Theanimals were observed for the appearance of generalized tonic–clonicconvulsive episodes for 20 min according to Ferraro et al. [28], whodescribe generalized convulsive episodes as generalized whole-bodyclonus involving all four limbs and tail, rearing, wild running andjumping, followed by sudden loss of upright posture and autonomicsigns, such as hypersalivation and defecation. PTZ-induced general-ized convulsions typically lasted between 30 and 60 s, and werefollowed by a quiescent period. During the 20-min observation period,the latency for generalized tonic–clonic convulsions was measured.EEG recordings were visually analyzed for seizure activity, whichwere defined by the occurrence of the following alterations in therecording leads: isolated sharp waves (≥1.5×baseline); multiplesharp waves (≥2×baseline) in brief spindle episodes (≥1 s≥5 s);multiple sharpwaves (≥2×baseline) in long spindle episodes (≥5 s);spikes (≥2×baseline) plus slow waves; multispikes (≥2×baseline,≥3 spikes/complex) plus slowwaves; major seizure (repetitive spikesplus slow waves obliterating background rhythm, ≥5 s). For quanti-tative analysis of EEG amplitude, we averaged EEG amplitude over the20 min of observation.
2.4. Tissue processing for neurochemical analyses
Immediately after the EEG and behavioral evaluation, the animalswere killed by decapitation and their brain was exposed by removingthe parietal bone. The brains were quickly removed and a coronalsection (7 mm) of the injured hemisphere corresponding to theimpact site of injury was dissected (see Fig. 1). The cortical tissuessurrounding the injured core were homogenized in cold 10 mM Tris–HCl buffer (pH 7.4) and used for determination of carbonyl content,TBARS and Na+,K+-ATPase activity.
2.5. Measurement of thiobarbituric acid-reactive substances (TBARS)content and protein carbonyl
For the TBARS assay, the cortical tissues surrounding the injuredcore were homogenized in ultra-pure water and mixed with the TBAreagent (15% of trichloroacetic acid, 0.375% of thiobarbituric acid and2.5% v/v of HCl). After 30 min of incubation, samples were centrifuged(3000 ×g, 15 min) and then TBARS levels were measured at 532 nmaccording to Ohkawa et al.[29]. For the protein carbonyl assay, thecortical tissues surrounding the injured core were homogenized incold 10 mM Tris–HCl buffer (pH 7.4) and carbonyl protein contentwas determined by Yan et al.[30] adapted for brain tissue by Lima etal.[13]. Briefly, homogenates were diluted to 750–800 μg/ml ofprotein in each sample, and 1 ml aliquots were mixed with 0.2 ml of2,4 dinitrophenylhydrazine (10 mM DNPH)or 0.2 ml HCl (2 M). Afterincubation at room temperature for 1 h in a dark ambient, 0.6 ml ofdenaturing buffer (150 mM sodium phosphate buffer, pH 6.8 contain-ing 3% SDS),1.8 ml of heptanes(99.5%) and 1.8 ml of ethanol (99.8%)were added sequentially, andmixedwith vortex agitation for 40 s andcentrifuged for 15 min. Next, the protein isolated from the interfacewas washed two times with 1 ml of ethyl acetate/ethanol 1:1 (v/v)and suspended in 1 ml of denaturing buffer. Each DNPH sample wasread at 370 nm in a Hitaschi U-2001 spectrophotometer against thecorresponding HCL sample (blank), and total carbonylation calculatedusing a molar extinction coefficient of 22,000 M−1 cm−1.
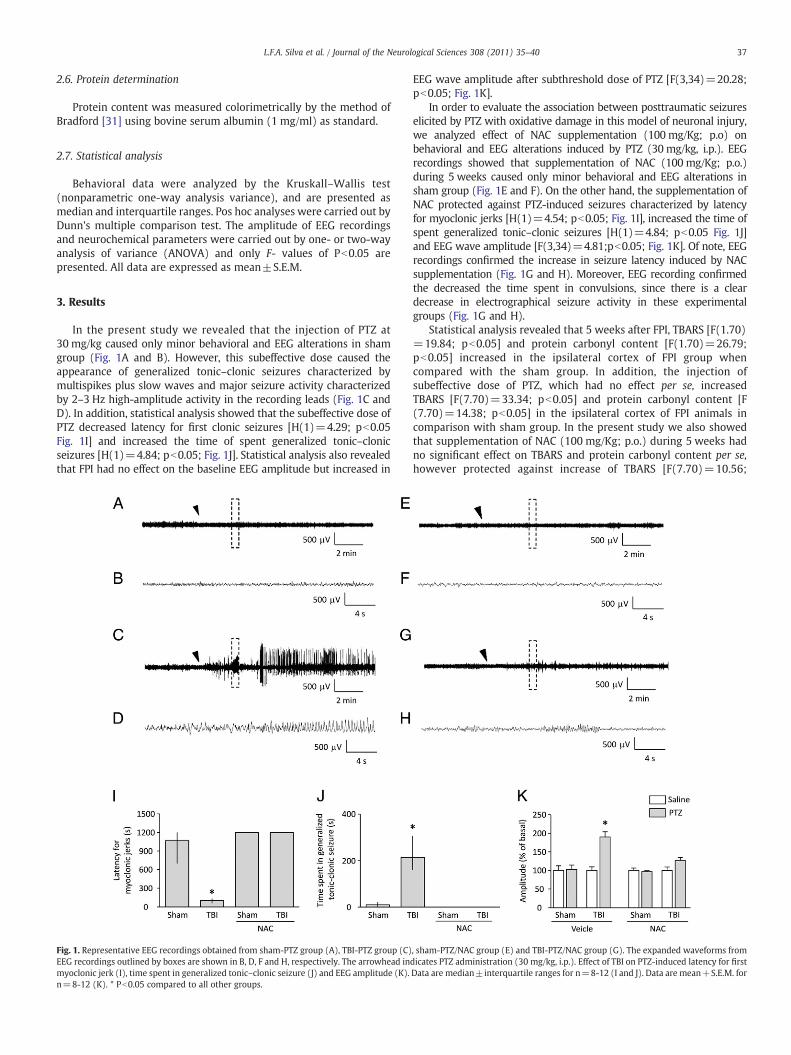
37L.F.A. Silva et al. / Journal of the Neurological Sciences 308 (2011) 35–40
2.6. Protein determination
Protein content was measured colorimetrically by the method ofBradford [31] using bovine serum albumin (1 mg/ml) as standard.
2.7. Statistical analysis
Behavioral data were analyzed by the Kruskall–Wallis test(nonparametric one-way analysis variance), and are presented asmedian and interquartile ranges. Pos hoc analyses were carried out byDunn's multiple comparison test. The amplitude of EEG recordingsand neurochemical parameters were carried out by one- or two-wayanalysis of variance (ANOVA) and only F- values of Pb0.05 arepresented. All data are expressed as mean±S.E.M.
3. Results
In the present study we revealed that the injection of PTZ at30 mg/kg caused only minor behavioral and EEG alterations in shamgroup (Fig. 1A and B). However, this subeffective dose caused theappearance of generalized tonic–clonic seizures characterized bymultispikes plus slow waves and major seizure activity characterizedby 2–3 Hz high-amplitude activity in the recording leads (Fig. 1C andD). In addition, statistical analysis showed that the subeffective dose ofPTZ decreased latency for first clonic seizures [H(1)=4.29; pb0.05Fig. 1I] and increased the time of spent generalized tonic–clonicseizures [H(1)=4.84; pb0.05; Fig. 1J]. Statistical analysis also revealedthat FPI had no effect on the baseline EEG amplitude but increased in
Fig. 1. Representative EEG recordings obtained from sham-PTZ group (A), TBI-PTZ group (C)EEG recordings outlined by boxes are shown in B, D, F and H, respectively. The arrowhead inmyoclonic jerk (I), time spent in generalized tonic–clonic seizure (J) and EEG amplitude (K).n=8-12 (K). * Pb0.05 compared to all other groups.
EEG wave amplitude after subthreshold dose of PTZ [F(3,34)=20.28;pb0.05; Fig. 1K].
In order to evaluate the association between posttraumatic seizureselicited by PTZ with oxidative damage in this model of neuronal injury,we analyzed effect of NAC supplementation (100 mg/Kg; p.o) onbehavioral and EEG alterations induced by PTZ (30 mg/kg, i.p.). EEGrecordings showed that supplementation of NAC (100 mg/Kg; p.o.)during 5 weeks caused only minor behavioral and EEG alterations insham group (Fig. 1E and F). On the other hand, the supplementation ofNAC protected against PTZ-induced seizures characterized by latencyfor myoclonic jerks [H(1)=4.54; pb0.05; Fig. 1I], increased the time ofspent generalized tonic–clonic seizures [H(1)=4.84; pb0.05 Fig. 1J]and EEG wave amplitude [F(3,34)=4.81;pb0.05; Fig. 1K]. Of note, EEGrecordings confirmed the increase in seizure latency induced by NACsupplementation (Fig. 1G and H). Moreover, EEG recording confirmedthe decreased the time spent in convulsions, since there is a cleardecrease in electrographical seizure activity in these experimentalgroups (Fig. 1G and H).
Statistical analysis revealed that 5 weeks after FPI, TBARS [F(1.70)=19.84; pb0.05] and protein carbonyl content [F(1.70)=26.79;pb0.05] increased in the ipsilateral cortex of FPI group whencompared with the sham group. In addition, the injection ofsubeffective dose of PTZ, which had no effect per se, increasedTBARS [F(7.70)=33.34; pb0.05] and protein carbonyl content [F(7.70)=14.38; pb0.05] in the ipsilateral cortex of FPI animals incomparison with sham group. In the present study we also showedthat supplementation of NAC (100 mg/Kg; p.o.) during 5 weeks hadno significant effect on TBARS and protein carbonyl content per se,however protected against increase of TBARS [F(7.70)=10.56;
, sham-PTZ/NAC group (E) and TBI-PTZ/NAC group (G). The expanded waveforms fromdicates PTZ administration (30 mg/kg, i.p.). Effect of TBI on PTZ-induced latency for firstData are median±interquartile ranges for n=8-12 (I and J). Data are mean+S.E.M. for

38 L.F.A. Silva et al. / Journal of the Neurological Sciences 308 (2011) 35–40
pb0.05] and protein carbonyl content increase [F(7.70)=15.64;pb0.05] of FPI animals injected with PTZ (Table 1).
Considering that Na+,K+-ATPase enzyme plays a pivotal role incellular ionic gradient maintenance and is particularly sensitive toreactive species [14,15], we decided to evaluate the involvement ofthis enzyme in the progression and manifestation of seizure elicitedby PTZ in this post-traumatic model of epilepsy. Statistical analysisrevealed a decrease in Na+,K+-ATPase activity in the ipsilateralcerebral cortex of FPI animals [F(7.70)=37.90; pb0.05] whencompared with sham animals. Post hoc analysis also revealed thatthe injection of subeffective dose of PTZ decreased Na+,K+-ATPaseactivity in sham animals when compared with the control group(sham-NaCl group), and that the decrease in the activity of this en-zyme was larger in FPI animals. Statistical analysis also revealed thatNAC supplementation (100 mg/Kg; p.o) protected against Na+,K+-ATPase inhibition in the ipsilateral cerebral cortex of FPI [F(7.70)=18.72; pb0.05] and FPI-PTZ group [F(7.70)=5.63; pb0.05].
4. Discussion
The sequelae of TBI, including posttraumatic epilepsy, represent asocietal problemwhere the importance of brain trauma as a risk factorfor the development of epilepsy is well established [32,33]. Never-theless, clinical trials aiming the prevention of epilepsy following TBIhave failed [34] due to the multiple pathophysiologic epileptogenicprocesses that are likely activated simultaneously or sequential bybrain trauma [35]. Another limiting factor is the heterogeneity ofpatient populations. For instance, it is difficult to match both theseverity and locations of TBI between placebo and treatment groups.Thus, significant resources are required to develop a better under-standing of the pathophysiology mechanism as targets for potentialprophylactic therapies. In line of this view, most of the dataconcerning the progression of damage have been collected duringthe first 1–2 months post-injury, which also corresponds to the timeperiod when most of the neuronal alterations, implying increasedexcitability of injured tissue, have been performed [36,37].
In the present study we revealed that a simple FPI episode in ratparietal cortex decreases Na+,K+-ATPase activity with a concomitantincrease in the levels of oxidative stressmarkers (protein carbonylationand TBARS), 5 weeks after the injury. Our results also revealed that theinjection of subeffective dose of PTZ (30 mg/Kg, i.p.) caused earlyposttraumatic seizures suggesting that an alteration in the lipid/proteinoxidation, membrane fluidity, and Na+,K+-ATPase activity may becorrelatedwith posttraumatic epilepsy development. In fact, the resultspresented in this report showed that N-acetylcysteine (NAC) supple-mentation, a powerful antioxidant both in the peripheral tissues andcentral nervous system (CNS) [38,39], was effective in preventing ofbehavioral and electrographic seizures induced by PTZ in this post-traumatic model of epilepsy. The supplementation of this compoundalso protected against the increase in the levels of oxidative stressmarkers (protein carbonylation and TBARS) and Na+,K+-ATPase
Table 1Effect of FPI on Na+,K+-ATPase activity, protein carbonyl content and TBARS content.
Na+,K+-ATPase activity(nmol Pi/mg protein/min)
Protein ca(nmol/mg
Treatment Sham TBI Sham
NaCl 93.90±4.56 51.44±2.35a 5.13±0.6PTZ 68.06±5.38a 39.73±4.06a,c 6.59±0.4NAC 104.0±3.62 103.45±4.4b 5.48±0.5NAC/PTZ 106.7±4.49c 99.09±4.06d 7.00±0.4
Data are mean±SEM for n=8–12 in each group.a pb0.05 compared with NaCl-Sham group.b pb0.05 compared with NaCl-TBI group.c pb0.05 compared with PTZ-Sham group.d pb0.05 compared with PTZ-TBI group.
activity inhibition. These experimental findings suggest that a dysregu-lation of both inhibitory and excitatory neurotransmission followingtraumatic injury is responsive to antioxidant treatment. Furthermore, itis plausible to propose that these alterations play an important role inneuronal cell loss following TBI, and thus contribute to the pathophys-iology of cerebral damage following brain injury [40].
These results are in full agreement with the view that free radicalscontribute to neural injury following cerebral ischemia [41] and TBI[20]. In addition, oxidative stress in CNS is involved in thedegeneration of neurons in several rodent models of experimentalepilepsy and seizure, such as the amygdale kindling model [42], thekainic acid model [43], the PTZ kindling model [44], and the acutePTZ-induced seizure [45]. In line of this view, a substantial body ofevidence has suggested that a cascade of biological events, includingreactive species generation, underlies the development and propaga-tion of epilepsy [46–48,45]. In addition, the total activity of superoxidedismutase (SOD) and the content of the antioxidant α-tocopherolhave been found to be reduced in rat brain homogenates after acutePTZ-induced convulsion [49]. Previous studies from our group havedemonstrated that the injection of PTZ-induced convulsive activity isaccompanied by ROS generation and inhibition of local Na+,K+-ATPase activity [50,51,13,22,52]reinforcing the assumption that theinhibition of some selected targets for free radicals increases cellularexcitability [53–55].
Nevertheless, since there is no information available describingwhether posttraumatic seizures are an epiphenomenon of braininjury or whether they exacerbate damage for a given injury severity,there is still some debate about specific mechanisms that initiate orsustain epileptogenesis. While the weight-drop model of TBI wasfound to show a persistent susceptibility to PTZ-evoked behavioralseizures for at least 15 weeks with no spontaneously seizures [56],spontaneous chronic seizures in rats originated from the neocortex atthe side of injury have been observed [26]. Such seizures have beenalso found to be progressively worsened and spread over timecorrelating with hyperexcitability in the injury cortex [27]. Inaddition, increasing neuronal excitability by negative modulation ofan inhibitory neurotransmitter system (GABA) after TBI has showedto present a positive effect on the recovery of cognitive function [57].Although the determining factor for such a discrepancy is unknown,these apparent contradictions may be explained by differentexperimental conditions (species, tissue analyzed and type of modelTBI), however, there are caveats to consider when interpreting theexperimental data available [37].
On the other hand, the results presented in this report suggest thata significant increase of protein carbonylation and TBARS that occursconcomitantly with a decrease in Na+,K+-ATPase activity mayinfluence in the membrane electrophysiology by altering the actionpotential production, decreasing, this way, the threshold to seizuresinduced by sub effective dose of PTZ (30 mg/Kg; i.p). In concordancewith this view, the results presented in this report demonstrated thatNAC supplementation protected against electroencephalographic
rbonyl contentprotein)
TBARS content(μmol MDA/mg protein)
TBI Sham TBI
2 8.50±0.34a 0.140±0.75 0.336±0.09a
0 12.09±0.95a,b 0.240±0.10a 0.530±0.20a,b,c
5 6.36±0.65b 0.158±0.02 0,161±0.03b
2 6.75±0.56d 0.145±0.04 0,149±0.07d

39L.F.A. Silva et al. / Journal of the Neurological Sciences 308 (2011) 35–40
seizures and oxidative damage in this model of TBI. Furthermore, theobservation available from in vitro slice studies support the idea thatcortical and hippocampal excitability are increased in a variety of TBImodels [58–60]. In addition, a considerable body of evidence hassuggested that oxidative damage, mediated by reactive oxygen andnitrogen species, including the superoxide ion, hydrogen peroxide,hydroxyl radical, nitric oxide, and peroxynitrite, is well recognized asa significant component of the secondary injury cascade thataccompanies TBI [7]. In fact, while free-radical-induced damage hasbeen proposed to be involved in traumatic cell injury and cell death,free radical scavengers, such as catalase and glutathione peroxidase(GSH-Px) are associated with partial amelioration of traumatic injury[61–63].
Another point to be addressed is that the neurotoxicity effectexerted by subeffective dose of PTZ (30 mg/kg, i.p) in FPI animals aswell as the effective protection exerted by NAC supplementationsuggests that convulsive activity and neurochemical parameters areclosely linked events in this model of TBI. In addition, it is plausible topropose that important changes in the brain after FPI may affect thesusceptibility to seizure or events followed by epileptiform activityinduced by PTZ. With respect to the effects of subconvulsant dose ofPTZ (30 mg/kg,i.p) in cerebral cortex after FPI , data from our studysuggest that changes in this structure could be related to the specificfunction of PTZ as selective blocker of the chloride ionophore complex[64,65] or to cellular thiol homeostasis [45]. Furthermore, the PTZ-induced excitability proposed in this report is in accordance withpreviously report data showing that single convulsive dose of PTZresults in significant changes in many parameters such as GABAA
receptor density and function [64,65], whole brain hydroxyl radicals[49], free fatty acids, and glutathione peroxidase activity in specificbrain areas [48]. However, this explanation remains speculative innature, and further studies are necessary to determine thismechanism.
In conclusion, the present study reports that a simple FPI episodein rat parietal cortex decreases Na+,K+-ATPase activity with aconcomitant increase in the levels of oxidative stressmarkers (proteincarbonylation and TBARS), 5 weeks after the injury. In addition, theearly posttraumatic seizures caused by injection of subeffective doseof PTZ (30 mg/Kg, i.p.) and the significant protection elicited byantioxidant compound (NAC) suggest that failure of some selectedtargets, such as Na+,K+-ATPase elicited by ROS generation, mayincrease cellular excitability and facilitate the appearance and/orpropagation of convulsions after TBI.
References
[1] Langlois JA, Kegler SR, Butler JA, Gotsch KE, Johnson RL, et al. Traumatic brain injury-related hospital discharges. Results from a 14-state surveillance system, 1997.MMWR Surveill Summ 2003;52:1–20.
[2] Ashman TA, Gordon WA, Cantor JB, Hibard MR. Neurobehavioural consequences oftraumatic brain injury. Mt Sinai J Med 2006;73:999–1005.
[3] Christensen J, Pedersen MG, Pedersen CB, Sidenius P, Olsen J, Vestergaard M. Long-term risk of epilepsy after traumatic brain injury in children and young adults: apopulation-based cohort study. Lancet 2009;373:1105–10.
[4] Scantlebury MH, Gibbs SA, Foadjo B, Lema P, Psarropoulou C, Carmant L. Febrileseizures in the predisposed brain: a new model of temporal lobe epilepsy. AnnNeurol Jul. 2005;58(1):41–9.
[5] Love R. Two hit hypothesis for temporal lobe epilepsy. Lancet Neurol Aug. 2005;4(8):458.
[6] Tyurin VA, Tyurina YY, Borisenko GG, Sokolova TV, Ritov VB, Quim PJ, et al.Oxidative stress following traumatic brain injury in rats: quantitation ofbiomarkers and detection of free radical intermediates. J Neurochem 2000;75:2178–89.
[7] Potts MB, Koh SE, Whetstone WD, Walker BA, Yoneyama T, Claus CP, et al.Traumatic injury to the immature brain: inflammation, oxidative injury, and iron-mediated damage as potential therapeutic targets. Neuro Rx 2006;3:143–53.
[8] Lau A, Arundine M, Sun HS, Jones M, Michael Tymianski. Inhibition of caspase-mediated apoptosisby peroxynitrite in traumatic brain injury. J Neurosci 2006;26:11540–50.
[9] Singh IN, Sullivan PG, Deng Y, Mbye LH, Hall ED. Time course of post-traumaticmitochondrial oxidative damage and dysfunction in a mouse model of focal
traumatic brain injury: implications for neuroprotective therapy. J Cereb BloodFlow Metab 2006;26:1407–18.
[10] Shao C, Roberts KN, Markesbery WR, Scheff SW, Lovell MA. Oxidative stress inhead trauma in aging. Free Radic Biol Med 2006;41:77–85.
[11] Davis LM, Pauly JR, Readnower RD, Rho JM, Sullivan PG. Fasting is neuroprotectivefollowing traumatic brain injury. J Neurosci Res 2008;86:1812–22.
[12] Dawson VL, Dawson TM. Nitric oxide in neural degeneration. Proc Soc Exp BiolMed 1996;211:33–40.
[13] Lima FD, Oliveira MS, Furian AF, SouzaMA, Rambo LM, Ribeiro LR, et al. Adaptationto oxidative challenge induced by chronic physical exercise prevents Na+, K+−ATPase activity inhibition after traumatic brain injury. Brain Res 2009;1279:147–55.
[14] Morel P, Tallineau C, PontcharraudR, PiriouA,Huguet F. Effects of 4-hydroxynonenal,a lipid peroxidation product, on dopamine transport and Na+/K+ ATPase in ratstriatal synaptosomes. Neurochem Int 1998;33:531–40.
[15] Petrushanko I, BogdanovN, BulyginaE,Grenacher B, Leinsoo T, BoldirevA, et al. Na-K-ATPase in rat cerebelar granule cells is redox sensitive. Am J Physiol Regul IntegrComp Physiol 2006;209:R916–25.
[16] Boldyrev AA, Bulygina ER, Kramarenko GG, Vanin AF. Effect of nitroso compounds onNa/K-ATPase. Biochim Biophys Acta 1997;20:243–51.
[17] Muriel P, Sandoval G. Nitric oxide and peroxynitrite anion modulate liver plasmamembrane fluidity and Na(+), K(+)-ATPase activity. Nitric Oxide 2000;4:333–42.
[18] Muriel P, Castaneda G, OrtegaM, Noel F. Insights into themechanisms of erythrocyteNa+/K+−ATPase inhibition by nitric oxide and peroxynitrite anion. J Appl Toxicol2003;23:275–8.
[19] BarrivieraML, Fontes Carlos Frederico L, AídaHassón-Voloch, Louro Sonia RenauxW.Influence of nitric oxide donors on the intrinsic fluorescence of Na+,K+-ATPase andeffects on the membrane lipids. Nitric Oxide 2005;13:10–20.
[20] Lima FD, Souza MA, Furian AF, Rambo LM, Ribeiro LR, Martignoni FV, et al. Na+, K+−ATPase activity impairment after experimental traumatic brain injury: relationshipto spatial learning deficits and oxidative stress. Behav Brain Res 2008;193:306–10.
[21] Samuelson C, Kumlien E, Elfving A, LindhomD, Ronne-Engstron E. The effects of PBN(phenyl-butil-nitrone) onGLT-1 levels and on extracellular levels of amino acids andenergy metabolites in a model of iron-induced posttraumatic epilepsy. Epilepsy Res2003;56:165–73.
[22] Rambo LM, Ribeiro LR, Oliveira MS, Furian AF, Lima FD, Souza MA, et al. Additiveanticonvulsants effect of creatine supplementation and physical exercise againstpentylenetetrazol-induce seizures. Neurochem Int 2009;55:333–40.
[23] Souza MA, Oliveira MS, Furian AF, Rambo LM, Ribeiro LR, Lima FD, et al. Swimmingtraining prevents pentylenetetrazol-induced inhibition Na+, K+−ATPase, seizuresand oxidative stress. Epilepsia 2009;50:811–23.
[24] Grisar T, Guillaume D, Delgado-Escueta AV. Contribution of Na+, K(+)-ATPase tofocal epilepsy: a brief review. Epilepsy Res 1992;12:141–9.
[25] Jurkat-Rott K, Freilinger T, Dreier JP, Herzog J, Gobel H, Petzold GC, et al. Variability offamilial hemiplegic migraine with novel A1A2 Na+/K+−ATPase variants. Neurol-ogy 2004;62:1857–61.
[26] D'Ambrosio R,Maris DO, GradyMS,WinnHR, Janigro D. Impaired K(+) homeostasisand altered electrophysiological properties of post-traumatic hippocampal glia. JNeurosci 1999;19:8152–62.
[27] D'Ambrosio R, Fairbanks JP, Fender JS, Born DE, Doyle DL,Miller JW. Post-traumaticepilepsy following fluid percussion injury in the rat. Brain 2004;127:304–14.
[28] Ferraro TN, Golden GT, Smith GG, St Jean P, Schork NJ, Mulholland N, et al.Mapping loci for Pentilenetetrazol-induce seizures susceptibility in mice. JNeurosci 1999;19:6733–9.
[29] Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues bythiobarbituric acid reaction. Anal Biochem 1979;95:351–8.
[30] Yan LJ, Traber MG, Packer L. Spectrophotometric method for determination ofcarbonyls in oxidatively modified apolipoprotein B of human low-densitylipoproteins. Anal Biochem 1995;228:349–51.
[31] Bradford MM. A rapid and sensitive method for the quantitation of microgramquantities of protein utilizing the principle of protein-dye binding. Anal Biochem1976;72:248–54.
[32] Salazar AM, Jabbari B, Vance SC, Grafman J, Amin D, Dillon JD. Epilepsy afterpenetrating head injury. I. Clinical correlates: a report of Vietnam Head InjuryStudy. Neurology 1985;35:1406–14.
[33] Lowestein DH. Epilepsy after head injury: an overview. Epilepsia 2009;50(Suppl2):4–9.
[34] Temkin MR. Preventing and treating posttraumatic seizures: the humanexperience. Epilepsia 2009;50(Suppl 2):10–3.
[35] Garga N, Lowenstei DH. Posttraumatic epilepsy: a major problem in desperateneed of major advances. Epilepsy Curr 2006;6:1–5.
[36] Thompson HJ, Lifshitz J, Marklund N, Grady MS, Graham DI, Hovda DA, et al.Lateral fluid percussion brain injury: a 15-year review and evaluation. JNeurotrauma 2005;22:42–75.
[37] Pitkanen A, Immonen RJ, Grohl OH, Kharatishvily I. From treaumatic brains injuryto posttraumatic epilepsy: what animal models tell us about the process andtreatments options. Epilepsia 2009;50(Suppl 2):21–9.
[38] Uma Devi P, Pillai KK, Voroha D. Modulation of pentilenotetrazole-inducedseizures and oxidative stress parameters by sodium valproate in the absence andpresence of N-acetylcysteine. Fundam Clin Pharmacol 2006;20:247–53.
[39] Chen G, Shi J, Hu Z, Hang C. Inhibitory effect on cerebral inflammatory responsefollowing TBI in rats: a potential neuroprotective mechanism of N-acetylcysteine.Mediators Inflamm 2008;71:1–8.
[40] Yi JH, Hoover R, McIntosh TK, Hazell AS. Early, transient increase in complexin Iand complexin II in the cerebral cortex following traumatic brain injury isattenuated by N-acetylcysteine. J Neurotrauma Jan. 2006;23(1):86–96.

40 L.F.A. Silva et al. / Journal of the Neurological Sciences 308 (2011) 35–40
[41] Willmore LJ, Ueda Y. Posttraumatic epilepsy: hemorrhage, free radicals and themolecular regulation of glutamate. Neurochem Res 2009;34:688–97.
[42] Frantseva MV, Peres Velazquez JL, Tsoraklides G, Mendonça A, Adamchik Y, MillsRL, et al. Oxidative stress is involved in seizures-induced neurodegeneration in thekindling model of epilepsy. Neuroscience 2000;97:431–5.
[43] Gluck MR, Jayatilleke E, Shaw S, Rowan AJ, Haroutunian V. CNS oxidative stressassociated with the kainic acid rodent model of experimental epilepsy. EpilepsyRes 2000;39:63–71.
[44] Gupta YK, Veerendra Kumar MH, Srivastava AK. Effect of Centella asiatica onpentylenetetrazole-induced kindling, cognition and oxidative stress in rats.Pharmacol Biochem Behav 2003;74:579–85.
[45] Patsoukis N, Zervoudakis G, Georgiou CD, Angelatou F, Matsokis NA, PanagopoulosN. Thiol redox state and lipid and protein oxidation in the mouse striatum afterpentylenetetrazol-indi=uced epileptic seizure. Epylepsia 2005;46:1205–11.
[46] Armstead WM, Mirro R, Zuckerman SR, Shibata M, Lefler CW. Transforming growfactor-beta attenuates ischemia-induced alterations in cerebrovascular responses.Am J Physiol 1993;264:H381–5.
[47] Bruce AJ, Baundry M. Oxygen free radicals in rat limbic structures after kainate-induced seizures. Free Radic Biol Med 1995;18:992–1002.
[48] Erakovic V, Zulpan G, Varljen J, Sominic A. Pentilenetetrazol-induced seizures andkindling: changes in free fatty acids, superoxide dismutase and glutathioneperoxidase activity. Neurochem Int 2003;42:173–8.
[49] Rauca C, Zerbe R, Jantze H. Formation of free hydroxyl radicals after pentilenete-trazol-induced seizure and kindling. Brain Res 1999;847:347–51.
[50] Oliveira MS, Furian AF, Royes LF, Fighera MR, de Carvalho Myskiw J, Fiorenza NG.Ascorbate modulates pentylenetetrazol-induced convulsions biphasically. Neuro-science 2004;128:721–8.
[51] Ribeiro MC, de Avila DS, Schneider DY, Hermes FS, Furian AF, Schneider MS, et al.Alpha-Tocopherol protects against pentylenetetrazol— and methylmalonate-induced convulsions. Epilepsy Res 2005;66:185–94.
[52] Fighera MR, Royes LF, Furian AF, Oliveira MS, Fiorenza NG, Frussa-Filho R, et al.GM1 ganglioside prevents seizures, Na+K+−ATPase activity inhibition andoxidative stress induced by glutaric acid and pentilenetetrazole. Neurobiol Dis2006;22:611–23.
[53] Jamme I, Petit E, Divoux D, Gerbi A, Maixent JM, Nouvelot A. Modulation of mousecerebral Na+,K(+)-ATPase activity by oxygen free radicals. Neuroreport 1995;7:333–7 1995.
[54] Danbolt NC. Glutamate uptake. Prog Neurobiol 2001;65:1–105.[55] Prigol M, Wilhelm EA, Schneider CC, Rocha JB, Nogueira CW, Zeni G. Involvement
of oxidative stress in seizures induced by diphenyl diselenide in rats pups. BrainRes 2007;1147:226–32.
[56] Golarai G, Greenwood AC, Feeney DM, Connor JA. Physiological and structuralevidence for hippocampal involvement in persistent seizures susceptibility aftertraumatic brain injury. J Neurosci 2001;21:8523–37.
[57] Hamm RJ, Pike BR, Temple MD, O'Dell DM, Lyeth BG. The effect of postinjurykindled seizures on cognitive performance of traumatically brain-injuried rats.Exp Neurol 1995;136:143–8.
[58] Coulter DA, Rafiq A, Shumate M, Gong QZ, DeLorenzo RJ, Lyeth BG. Brain injury-induced enhanced limbic epileptogenesis: anatomical and physiological parallel-sto an animal model of temporal lobe epilepsy. Epilepsy Res 1996;26:81–91.
[59] Santakhumar V, Ratzliff AD, Jeng J, Toht Z, Soltesz I. Long-term hyperescitabilityin the hippocampus after experimental head trauma. Ann Neurol 2001;50:708–17.
[60] Tran LD, Lifshitz J, Witgen BM, Schwartzbach E, Cohen AS, Grady MS. Responde ofthe contralateral hippocampus to lateral flud percussion brain injury. JNeurotrauma 2006;23:1330–42.
[61] Ikeda Y, Brelsford KL, Ikeda K, Long DM. Oxygen-free radicals in traumatic brainoedema. Neurol Res 1989;11:213–6.
[62] Marmarou CR, Walker SA, Davis CL, Povlishock JT. Quantitative analysis of therelationship between intra- axonal neurofilament compaction and impairedaxonal transport following diffuse traumatic brain injury. J Neurotrauma 2005;22:1066–80.
[63] Soustiel JF, Palzur E, Nevo O, Thaler I, Vlodavsky E. Neuroprotective anti-apoptosiseffect of estrogens in traumatic brain injury. J Neurotrauma 2005;22:345–52.
[64] Psarropoulou C, Matsokis, Angelatou F, Kostopoulos G. Pentylenetetrazol-inducedseizures decrease gamma aminobutyric acid-mediatedrecurrent inhibition andenhance adenosine-mediated depression. Epilepsia 1994;35:12–9.
[65] Walsh LA, Li M, Zhao TJ, Chiu TH, Rosenberg HC. Acute pentylenetetrazol injectionreduces rat GABAA receptor mRNA levels and GABA stimulation of benzodiaze-pinebinding with No effect on benzodiazepine binding site density. J PharmacolExp Ther 1999;289:1626–33.
Top Related
Copyright © 2022 FDOKUMEN

