







Bahasa
Halaman
Hukum


1
Synthesisofrecombinantantibacterial
proteinsintheChlamydomonasreinhardtii
chloroplast
MaximilianEdwardAlfredBlanshard
UCL
AthesissubmittedforthedegreeofDoctorofPhilosophy
September2017

2
Declaration
I,MaximilianE.A.Blanshard,confirmthattheworkpresentedinthisthesisismy
own.Whereinformationhasbeenderivedfromothersources,Iconfirmthatthis
hasbeenindicatedinthethesis.
…………………………………………………………………

3
Acknowledgments
Firstly,Iwouldliketothankmysupervisor,ProfSaulPurton,fortheopportunityto
studyinhislabforthelastfouryears,andforprovidinghelp,encouragement,and
ideassofreelyandwillingly.Hugethankstomysecondarysupervisor,DrElaineAllan,
whosesupportandkindwords,aswellastimeinhelpingmetocompletemilestones,
are hugely appreciated. Thank you also tomy thesis committee chair,Dr Richard
Hayward, who’s ability to constructively criticise and encourage a methodical
approachwasinvaluable.DrMelindaMayer,mycollaborator,deserveshugethanks
foragreeingtohelpoutwiththeCD27Lworkandrunningsomanyturbidityreduction
assaysforme.
Iwould like to thank allmembersof thePurton Lab, past andpresent. From the
beginningtotheendofmyPhD,DrLauraStoffelshastaughtmehowtobeascientist
andheramazingabilitytoknoweverything(andwhereeverythingis!)neverceases
toamazeme,soIwouldliketothankherforallofthehoursoftimeandfrustration
shehasputintohelpingme,andresistingtheurgetosay“Justlookitup”.Iwould
alsoliketothankDrRosieYoungforteachingmesomuchmolecularbiologyandfor
alloftheencouragementwhenexperimentsfailed.Anenormousthankyoutoour
labmanager,ThushiSivagnanam,withoutwhomItrulybelievenothingwouldever
getdone–yourworkisremarkableandwedonotsayitoftenenough.Thankyouto
DrHenryTaunt,whoestablishedtheendolysinworkandhasplayedan important
part inmyPhDdespitesupposedly leavingthe lab in2013, includingbuyingmore
thanhisfairshareofroundsandgivinginsightintotheworldofhome-brewing.
TomyfriendsandcolleaguesinthePurtonlabwithwhomIhavemoaned,celebrated,
travelled, andgenerallyhad fun– I couldnothavedone itwithout you.Dr Janet
Waterhouse,DrAliceLui,DrPriscillaRajakumar,andDrUmaimaAlHoquanimade
mefeelwelcomefromthedayIarrived.ToJulianadeCostaRamos,XeniaSpencer-
Milne, Fiona Li, SaowalakChankgoandMarco LarreaAlvarez thank you forbeing
greatfriendsandbestofluckwiththerestofyourPhDs.

4
Mostimportantly,Iwouldliketothankmyfamily,inparticularmyMumandDadfor
theirexceptionalgenerosityandunbelievablekindness throughoutmy life,and in
theirpatiencewithallofmylifedecisions.Iamsoluckytohavethecoolestparents
intheworld.Icouldnothavedoneitwithoutyou!
Last,andmostcertainlynotleast,IwouldliketothankSamanthaHunt,whoselove,
companionship and supportmade even themost frustratingmoments in the lab
bearable,andwhohasfilledthelastsevenyearswithadventuresandfun.Thankyou!

5
Abstract
Theriseofantibioticresistanceandthedeclineinantibioticdiscoveryhavebeenwell
publicised. These issues, in combination with a growing global reliance upon
antibiotics for everyday modern life, urgently require the discovery of novel
antibacterialdrugs.
Endolysins are one potential candidate to support, or replace, conventional
antibiotics. Endolysins are lytic enzymes produced by bacteriophage in natura to
enablethereleaseofviralprogenyfrominsidethehostbacterium.Whenapplied
exogenously,endolysinscanlyseGram-positivebacteria,andthuscouldbeusedas
anovelantibacterialforthisgroupofpathogens.
Biologicallyactiveendolysinshavebeensuccessfullyexpressedasrecombinants in
thechloroplastofthegreenalga,Chlamydomonasreinhardtii.C.reinhardtii,andin
particular the chloroplast, has several features as a cell factorywhichmake it an
attractivealternativetothetraditionalrecombinantproteinproductionplatforms.C.
reinhardtii isfreeofendotoxins,canbecultivatedatlowcostinphotobioreactors,
hasGRASstatus,andisgeneticallytractable.
This study initially focuses upon improving the accumulation and activity of one
endolysin,Cpl-1,targetingStreptococcuspneumoniae.RecombinantCpl-1hasbeen
shown previously in the Purton lab to accumulate to moderate levels in the C.
reinhardtii chloroplast.Herewepresent two transgenic linesofC. reinhardtii that
appear to accumulate recombinant Cpl-1 to higher levels – one through the
incorporation of multiple expression cassettes, and one through codon pair
optimization. To improve the activity of Cpl-1 as an enzyme, Cpl-1 binding site
mutagenesis,Cpl-1dimerization,andtheproductionofapotentiallysynergisticholin
proteinwereallattempted.
Finally,anendolysinagainstClostridiumdifficile,CD27L,wassuccessfullyproduced
intheC.reinhardtiichloroplastandshowntobeactiveinvitro.Anotherendolysin,
thistimetargetingPropionibacteriumacnes, failedtoexpress inC.reinhardtii,but
wasexpressedinE.coli,albeitwithoutobviouslyticactivity.

6
Abbreviations
aadA aminoglycoside-3’’-adenyl-transferasegene
Amp ampicillin
AMPS ammoniumpersulphate
BHI brainheartinfusion
BME β-mercaptoethanol
bp basepairs
BSA bovineserumalbumin
CBD cellbindingdomain
CD catalyticdomain
CES controlbyepistasisofsynthesis
CFU colonyformingunit
CPK Corey,Pauling,Koltun
CTB choleratoxinB
DNA deoxyribonucleicacid
dNTP 2’deoxynucleoside5’-triphosphate
EDTA ethylenediaminetetraaceticacid(disodiumsalt)
e.g. exempligratia=forexample
ELISA enzyme-linkedimmunosorbentassay
g gram
GC guanineandcytosine
GFP greenfluorescentprotein
GOI geneofinterest
HA hemagglutinin

7
His histidine
HSM highsaltminimalmedium
IPTG Isopropylβ-D-1-thiogalactopyranoside
kb kilobasepairs
kDa kiloDalton
LB luria-bertanimedium
mg milligram
min minute
ml millilitre
mM milimolar
mRNA messengerribonucleicacid
mt mating-type
NCBI NationalCentreforBiotechnologyInformation
OD opticaldensity
ORF openreadingframe
PCR polymerasechainreaction
PDB proteindatabank
PEG polyethyleneglycol
PG peptidoglycan
PMF protonmotiveforce
PSI photosystemI
PSII photosystemII
psi poundspersquareinch
RCSB ResearchCollaboratoryforStructuralBioinformatics

8
RNA ribonucleicacid
rRNA ribosomalribonucleicacid
SD Shine-Dalgarnoconsensussequence
SDS sodiumdodecylsulphate
SDS-PAGE sodiumdodecylsulphatepolyacrylamidegelelectrophoresis
STGG skimmedmilktryptoneglycerolglucosemedium
Strep streptavidin
TAP trisacetatephosphatemedium
TBS trisbufferedsaline
TBS-T trisbufferedsaline–tween20
TCP totalcellprotein
TEMED N,N,N’,N’-tetramethylethylenediamine
tris tris(hydroxymethyl)aminomethane
TRA turbidityreductionassay
tRNA transferribonucleicacid
TSP totalsolubleprotein
TSY trypticasesoyyeastextractmedium
UTR untranslatedregion
v/v volumeforvolume
w/v weightforvolume
WGS wholegenomesequencing
WHO WorldHealthOrganisation
WT wildtype

9
CHAPTER1INTRODUCTION 15
1.1 Anewgenerationofantibiotics 15
1.1.1 Whyareantibioticssoimportant? 15
1.1.2 Whydoweneednewantibiotics? 19
1.1.3 Whatalternativestoantibioticscurrentlyexist? 24
1.2 Anoverviewofbacteriophageendolysins 28
1.2.1 Thebiologicalroleofbacteriophageendolysins 28
1.2.2 Thetherapeuticpotentialofendolysinsand“exolysis” 32
1.2.3 Otherapplications 38
1.3 Producingrecombinanttherapeuticproteins 39
1.3.1 Commonexpressionplatforms 39
1.3.2 Microalgaeasexpressionplatforms 41
1.3.3 Chlamydomonasreinhardtiiasanexpressionplatform 44
1.4 Targets/applications 51
1.4.1 Streptococcuspneumoniae 51
1.4.2 Clostridiumdifficile 53
1.4.3 Propionibacteriumacnes 54
1.5 Summary,aimsandobjectives 55
CHAPTER2 MATERIALSANDMETHODS 57
2.1 Strains,media,cultureconditionsandcelldensityquantification 57
2.1.1 Chlamydomonasreinhardtii 57
2.1.2 Escherichiacoli 63
2.1.3 Streptococcuspneumoniae 64
2.1.4 Clostridiumdifficile 65
2.1.5 Propionibacteriumacnes 66
2.2 Plasmids 67

10
2.3 Molecularbiology 69
2.3.1 Buffers 69
2.3.2 Genedesignandsynthesis 70
2.3.3 Polymerasechainreaction(PCR) 70
2.3.4 PCRDNAPurification 71
2.3.5 AgarosegelElectrophoresis 71
2.3.6 DNAgelextraction 71
2.3.7 SitedirectedmutagenesisbyPCR 72
2.3.8 DephosphorylationusingAntarcticPhosphatase 73
2.3.9 Blunt-endDNAcloning 73
2.3.10 DNARestrictionendonucleasedigestion 73
2.3.11 DNALigation 73
2.3.12 CompetentE.colitransformation–Heatshock 73
2.3.13 C.reinhardtiitransformation–glassbead 74
2.3.14 E.colicolonyPCR 75
2.3.15 E.coliplasmidisolation–CrudeandKit 75
2.3.16 E.colitestdigest 75
2.3.17 C.reinhardtiigenomicDNAisolation 75
2.3.18 DNASequencing 76
2.4 Proteinanalysis 76
2.4.1 Preparationoftotalproteinextracts 76
2.4.2 Sodiumdodecylsulphatepolyacrylamidegels(SDS-PAGE)� 77
2.4.3 Proteinpreparationfordotblot 78
2.4.4 CoomassieBrilliantBlueRstaining 78
2.4.5 Westernblotanalysis 79
2.4.6 Immuno-detection 79

11
2.4.7 Proteinquantificationassay 80
2.5 C.reinhardtiiproteinpurification 80
2.5.1 Crudeextractpreparation 80
2.5.2 Ammoniumsulphateprecipitation 81
2.5.3 Anti-StrepIIpurificationcolumn 81
2.6 E.coliproteinpreparation 81
2.6.1 IPTGinduction 81
2.6.2 BugBuster™(Novagen) 82
2.6.3 HighPressureHomogenizer 82
2.6.4 Sonication 82
2.6.5 Disulphidebondformationinvitro 83
2.7 Antibacterialactivityassays 83
2.7.1 Colonyformingunitassay 83
2.7.2 Turbidityreductionassay 84
CHAPTER3 IMPROVINGACCUMULATIONLEVELSOFCPL-1INTHECHLOROPLASTOFCHLAMYDOMONASREINHARDTII 86
3.1 Introduction 86
3.1.1 IntroductiontobacteriophagelysinCpl-1 86
3.1.2 Factorstoconsiderinproteinaccumulation 88
3.1.3 EpistasyofsynthesisintheC.reinhardtiichloroplast 89
3.1.4 CodonusageintheC.reinhardtiichloroplastgenome 90
3.2 Aimsandobjectives 90
3.3 Resultsanddiscussion 91
3.3.1 Investigationintomeasuringproteinaccumulationlevels 91
3.3.2 Investigating control by epistasy of synthesis in proteinaccumulation 105
3.3.3 Codonpairoptimizationforimprovementofproteinaccumulationlevels 124

12
3.4 Conclusionandfuturework 140
3.4.1 Multipleexpressioncassettestrategy 140
3.4.2 Identicaltransformanttransgenevariability 142
3.4.3 Codonpairoptimisation 144
CHAPTER4 IMPROVING THE ACTIVITY OF CPL-1 AGAINST STREPTOCOCCUSPNEUMONIAE 146
4.1 Introduction 146
4.1.1 ManipulatingCpl-1bindingactivity 146
4.1.2 Dimerisationasamethodtoimproveproteinstability 147
4.1.3 Theroleofholinsinthebacteriophagelyticcycle 150
4.2 Aimsandobjectives 152
4.3 Resultsanddiscussion 153
4.3.1 ManipulationoftheCpl-1cholinebindingsitestoimproveactivity 153
4.3.2 ImprovingCpl-1stabilitythroughdimerisation 176
4.3.3 ExpressionoftheCpl-1associatedholin,Cph-1 186
4.4 Conclusionandfuturework 193
4.4.1 Cellwallbindingsitemutagenesis 193
4.4.2 Cpl-1dimerisation 196
4.4.3 Holinproduction 197
CHAPTER5 EXTENDING THE CHLAMYDOMONAS REINHARDTII CHLOROPLASTPLATFORMTOOTHERENDOLYSINS 200
5.1 Introduction 200
5.1.1 PreviousendolysinexpressionintheC.reinhardtiichloroplast 200
5.1.2 Theneedformoreendolysins 201
5.2 Aimsandobjectives 202
5.3 Resultsanddiscussion 203
5.3.1 Clostridiumdifficile,ΦCD27andCD27L1-179 203

13
5.3.2 C.difficileendolysinCD27L 203
5.3.3 Propionibacterium acnes, bacteriophage PA6 and its endolysinGp20 218
5.4 Conclusionandfuturework 228
5.4.1 CD27Lendolysin 228
5.4.2 Gp20endolysin 232
5.4.3 Futureendolysins 238
CHAPTER6 FINALDISCUSSIONANDFUTUREPROSPECTS 240
6.1 Summary 240
6.1.1 Summaryofresults 240
6.1.2 Summaryofshort-termfuturework 241
6.2 Discussionandlong-termfutureprospects 243
6.2.1 Alternative strategies to improving expression levels in C.reinhardtiichloroplast 243
6.2.2 Othermicroalgalhostsforscale-up 246
6.3 Concludingremarks 248
6.3.1 Viewsonthefutureofantibiotics 248
6.3.2 Viewsonthefutureofmicroalgae-producedtherapeutics 249
REFERENCES 250
CHAPTER7 APPENDICES 274
7.1 ResultsChapter3Appendices 274
7.1.1 AppendixA 274
7.1.2 AppendixB 275
7.1.3 AppendixC 276
7.1.4 AppendixD 277
7.1.5 AppendixE 277
7.1.6 AppendixF 278

14
7.1.7 AppendixG 280
7.2 ResultsChapter4Appendices 284
7.2.1 AppendixH 284
7.2.2 AppendixI 285
7.3 ResultsChapter5Appendices 286
7.3.1 AppendixJ 286
7.3.2 AppendixK 287
7.3.3 AppendixL 287
7.3.4 AppendixM 288
7.3.5 AppendixN 289
7.3.6 AppendixO:ListofPrimers 289

15
Chapter1 Introduction
1.1 Anewgenerationofantibiotics
1.1.1 Whyareantibioticssoimportant?
1.1.1.1 History
SirAlexanderFleming(1881-1955)isbestknownforhisdiscoveryofbenzylpenicillin
(PenicillinG)in1928,anobservationthatshapedthemodernworldandopenedup
hugevistasofmodernmedicalandsurgical techniques thathavebecomeroutine
across the globe. After the initial discovery of penicillin by Fleming at St Mary’s
Hospital in London, it took a further 16 years of refinement, optimization and
collaborationbetweenindustryandacademiabeforepenicillincouldbeproducedin
sufficientvolumes to treathumans (Zaffirietal.2012). Incontrast to thepopular
storyofFlemingstumblingacrosspenicillinonhisreturnfromholiday,thescalingup
ofproductionwasnomeanfeat.After12yearsofpromotingpenicillinandsearching
forsolutionstothepurificationandstabilityissuespenicillinfaced,Fleminghadall
butgivenuponhisdiscovery(Aminov2010).However,duetothepressuresofthe
secondworldwar,theworkwascontinued,thoughstillitdidnotcomeeasy:asJohn
L.SmithofPfizersaidatthetime,“Themouldisastemperamentalasanoperasinger,
theyieldsarelow,theisolationisdifficult,theextractionismurder,thepurification
invitesdisaster,and theassay isunsatisfactory.” (Werth2014).By1945however,
massproductionanddistributionofpenicillinhadbeenachieved–theworld’sfirst
commerciallyavailableantibiotic.ThechemicalstructureofPenicillinGanditsmode
ofactionasdisplayedinFigure1.1.

16
FigureRemoved
Figure1.1Thesmallmoleculebenzylpenicillin(PenicillinG),knownasaβ-lactamduetothepresenceofaβ-lactamringinitsmolecularstructure(Drugbank2017).Aswithmostβ-lactamantibiotics, benzylpenicillin functions throughdisrupting cellwall biosynthesis inbacteria.The image on the right shows benzylpenicilin inhibiting formation of crosslinks in thebacterial peptidoglycan cell wall. Normally transpeptidases (also called penicilin-bindingproteins) bind to D-alanyl-D-alanine pentapeptide of the peptidoglycan monomers andcatalyse cross-linking via a peptide bridge. However, penicillin resembles the D-alanyl-D-alaninepentapeptideandblockstheactivesiteofthetranspeptidases,preventingmonomerpolymerisation.Thelackofcross-linkingweakensthecellwall,causingosmoticlysis.ImagereproducedfromKaiser(2011).
PenicillinGwasthefirstofmorethan20distinctantibioticclassestobediscovered
fromthe1940stothe1960s,inwhatisnowrecognisedasagoldeneraofantibiotic
discovery (Arias andMurray 2015). These smallmoleculeswere all isolated from
bacteria and fungi for their ability to block key enzymatic steps in bacteria, and
presumablyservetogivemicroorganismsacompetitiveadvantageinenvironmental
niches(Lewis2013).However,therearealsotheoriesthatthesesmallmoleculesact
ascell-cellcommunicationsystemsatlowconcentrations,giventheirvastdiversity
andtheirabilitytomodulatecelltranscripts(Davies2006).Regardlessoftheirnatural
role,theseidentified antibiotics targeted several different aspects of bacterial
machineryessentialforcellmaintenanceandreproduction,anillustrationofwhich
ispresented inFigure1.2.The incredibleefficacyofantibiotics, theirrapidrateof
discoveryandtheirabilitytocurepreviouslydeadlyinfectionsinamatterofdaysled
tothembeinghailedasa“magicbullet”(Senguptaetal.2013).Theywerenotonly
valuableascurestobacterialinfections,butinpreventiontoo,thusopeningupvast
avenuesformedicineandindustrywhichwillbefurtherexploredinthenextsection.

17
FigureRemoved
Figure1.2Thevariousmodesofactionofdifferentantibioticclassestokillbacteria.Itshouldbe noted that many classes share the same targets, for example cell-wall synthesis isinhibitedbyfiveseparateclassesofantibiotics.ReproducedfromLewis(2013).
1.1.1.2 Applications
Themostobviousapplicationforantibioticsisintreatingbacterialinfections.When
penicillinwas firstmass produced,WorldWar IIwas still raging and stockswere
earmarkedforsoldiersonthefrontline,sufferingfromstaphylococcal,streptococcal,
andpneumococcalwoundinfections(MailerandMason2001).Whileinfectionsof
minorcutsandinjuriescanstillwarrantantibioticsonoccasion,betterknowledgeof
hygiene,vaccinations,andafter-carehavereducedthismarket.
Prophylactic antibiotics on the other hand are still of immense importance and
enableawholesuiteofsurgeriesandchemotherapieswhichmanytakeforgranted.

18
Allinvasivesurgeries,fromorgantransplantstodentalwork,areaccompaniedwith
aprophylacticcourseofantibiotics.Immunosuppressingchemotherapiesforcancer
treatmentsrelyonantibioticstostopbacterialinfectionswhilethebody’simmune
systemisoutofaction(O’Neilletal.2015).Inshort,accesstoantibioticsunderpins
vastnumbersofsurgeriesandmedicaltechniquesusedinmodernmedicine.
Oneofthemostcontroversialusesofantibioticsisasgrowthpromotersinlivestock.
This stems from the discovery that sub-therapeutic levels of antibiotic cause
increasedgrowth.Themechanismforthisremainsunclear,butthemostlikelytheory
seems to be that growth is more efficient without gut bacteria competing for
nutrients(Gaskinsetal.2002).Notonlydolowlevelsofantibioticsimprovegrowth
rates, but also conception rates and feed conversion ratios, altogether having a
pronouncedimpactuponglobalmeatproduction(Haoetal.2014).
Veterinarymedicine is another large consumerof antibiotics, enabling farmers to
rearanimalsincloserproximitytooneanothersuchasinbarnswherepreviouslya
build-upofpotentiallyinfectiousbacteriawouldpreventhighanimaldensitiesand
continuousfarming.Economically,thishasbeenadvantageous–increasingfarming
efficienciesandloweringfoodprices.Thesameistrueoffishfarming:thegrowthof
aquaculture to industrial scales has been almostwholly reliant uponprophylactic
antibioticuse,enablingthehighdensityfarmingoffishtomeetthegrowingglobal
demand(Cabello2006).
The remarkable reliance that the modern world has built upon antibiotic
consumption leads us to worry about what would happen if they ceased to be
effective.Wehavementionedbrieflyjustafewofthehugeindustriesthatuse,and
over-use,antibiotics.Aswewillseeinthefollowingsection,antibioticsareafinite
resourceduetotheemergenceofresistantbacteriaand if these industriesareto
continuetogrow,thennewantibioticsmustbesought.

19
1.1.2 Whydoweneednewantibiotics?
1.1.2.1 Resistance
TheWorldHealthOrganisation(WHO)definesantibioticresistanceasachangewhich
occurstobacteriawhenexposedtoantibiotics,resultingintheantibioticbecoming
ineffective and the persistence of the infection (WHO 2016). This definition is
valuableatatherapeuticlevel,butthissectionwillgiveanoverviewofhowantibiotic
resistancefunctionsamolecularlevel.
ThefivemechanismsofantibioticactionillustratedinFigure1.2representtheentire
variability within our antibiotic arsenal. Given how important antibiotics are to
modernsociety,thisisanalarminglysmallnumberofbacterialtargetstorelyupon,
giventhatthereareknowntobeapproximately200conservedessentialbacterial
proteins(Lewis2013).Resistancetoeachofthesetargetshasarisenquicklyaseach
newdrughasbeenusedonalargescale,withresistancetomorerecentantibiotics
suchasLevofloxacin(asecondgenerationfluoroquinolone)beingreportedin1996,
the same year as it was introduced (Ventola 2015). It is therefore important to
understand how resistance is acquired both at the molecular level, so that new
therapiesanddrugcombinationscanbeproduced,andatthepopulationlevel,so
thatpreventativemeasuresandprocedurescanbeimplemented.

20
FigureRemoved
Figure1.3Thesixmajorantibioticresistancemechanisms.Resistancemechanismsarenotnecessarilyspecific toacertainclassofantibiotics,withmanyantibioticsbeingsubjecttomultipleresistancemechanisms.Sometherapiestargettheresistancemechanisminordertorestoreantibioticefficacy,forexampletheapplicationofβ-lactamaseinhibitorsalongsideβ-lactamantibiotics.ReproducedfromLewis(2013).
Resistancearisesduetothehighselectivepressurethatantibioticsputuponbacterial
populations.Eachmodeofresistancetoeachofthefivesitesofantibacterialaction
isillustratedinFigure1.3.Forexample,penicillinisaβ-lactam,aclassofantibiotics
that inhibit cell wall synthesis in bacteria. β-lactams bind to penicillin-binding
proteins(PBPs),whichareresponsibleforbacterialcellwallcross-linking,andinhibit
theiraction.Theresultinginabilityofthebacteriatocorrectlyformacellwallleads
toautolysis(Fernandesetal.2013).Severalmethodsofresistancehavedeveloped
towards β-lactams. Firstly, target modification: modification of PBPs to exhibit a
muchloweraffinitytopenicillin.Secondistheproductionofβ-lactamases,enzymes
whichdegradeormodifyβ-lactams,andthirdlyarechangestopermeabilityinthe

21
cellwalltoβ-lactams,whichcaneitherblockentryoractivelyremovetheβ-lactams
via efflux proteins (Fernandes et al. 2013). In this single example three different
resistancemechanismsexistforasingleantibiotic,highlightingtheremarkableability
ofbacteriatoevolveunderantibioticinducedselectivepressureandunderliningthe
realpotentialforsomeantibioticstobecomeobsolete.
Itisreasonabletoaskhowantibioticresistanceevolvesandspreadssorapidly.Two
categories for resistance can be formed: endogenous resistance and exogenous
resistance. Endogenous resistance refers to resistance arising within the bacteria
itself i.e. bymutationand selection. Exogenous resistance involves thepassingof
resistance genes between different bacterial species via horizontal gene transfer
(Silver2011),eachofwhichisexploredbelow.
Endogenousresistanceisoftengeneticallyrecessiveasittendstoresultinalossof
function, for example reduced permeability to antibiotics, which have other
(negative)consequencesfornormalcelloperation.Ifantibioticselectioniscontinued
foralongtime,thensecondarymutationsmayeventuallyevolvetocompensatefor
lossoffunction.However,generallythesemutationswilleventuallybelostafterthe
removalofselectivepressure.
Exogenously acquired resistance on the other hand generally involves a gain of
functionandisthereforemorestable,forexampletheabilitytosynthesizeantibiotic-
degradingenzymes.Apartfromaslightmetabolicburden,thesegainsoffunctionare
not generally detrimental to the bacterium. Exogenous resistance is therefore of
mostconcernclinically,asresistancepersistsafterantibiotictreatmenthasfinished
(SilverandBostian1993).
Themanyusesofantibioticsdescribedin1.1.1.2provideperfectopportunitiesfor
resistance to be selected. For example, the lack of distinction between “human
antibiotics” and “veterinary antibiotics” has dangerous implications for emerging
resistanceviahorizontalgenetransfer.Furthermore,themannerinwhichantibiotics
aregenerallydeliveredtolivestock,suchasintheirfoodorwater,canleadtosub-
inhibitory concentrations of antibiotic which provides an ideal environment for
resistanceselection(Meeketal.2015).

22
FigureRemoved
1.1.2.2 Antibioticdevelopment
Duringthegoldeneraofantibioticdiscovery,arangeofbroad-spectrumantibiotics
werequickly identified,patentedandscaled toproduction,but today thesesame
chemicalsstillformthebasisofourantibioticarsenal.Fromthemid-1960sonwards,
onlysixnew,approved,antibioticclasseshavebeendiscovered,andnonesincethe
discoveryofdaptomycinin1986.
Figure1.4The“GoldenEra”ofantibioticdiscoveryproducedthebasisforalloftheantibioticsuponwhichwestill rely today.Modificationsandsyntheticvariantsof theseclasseshaveextendedthelifeofmanyofthesedrugs,butalackoftrulynovelantibioticspersists.Veryrecentlynewbacterialculturingtechniqueshaveraisedhopesofnewantibioticclassesbeingdiscoveredandapproved(e.g.teixobactin),andarediscussedlaterinthissection(Lingetal.2015).ImagereproducedfromSilver(2011).
Thisalarmingdrop-off inantibioticdiscovery isduetoseveral factors. Intheearly
daysofantibioticresearch,discoverywaslargelythroughempiricalscreening–this
amounted to testing of various microorganism extracts for bacterial growth
inhibition, requiring little understanding of their action. This method worked

23
extremelywellinthe1940s,50sand60s,butsoonthese“lowhangingfruits”hadall
been discovered. These methods soon led to replication of discovery; common
antibiotics were being repeatedly discovered, thus reducing the efficiency of the
discoverysystem.Themethoddeployedshiftedtowardstarget-directedscreensand
eventually big pharma companies reallocated resources to more lucrative
therapeutics(Livermore2011;Silver2011).Economically,theantibioticmarkethas
been somewhat a victim of its own success: withmany competing drugs on the
market, the price pharmaceutical companies can charge per prescription of
antibioticsismuchlowerthanthatofmanyotherdrugs(Bax1997).
However, the news is not all bad. Although no (or few) new classes have been
discovered since the 1980s, there have been improvements within the existing
classes.Whilesuchmodificationsdoincreasetheefficacyoftheseantibioticsfora
limited period of time, the fundamental resistance mechanisms still exist and
resistanceoftenquicklyre-emerges(Payneetal.2007).Othertacticshaveinvolved
blockingtheresistancemechanisms,suchasthroughuseofβ-lactamaseinhibitors
(WalshandWencewicz2014),anddeliveringtheseincombinationwithexistingβ-
lactam antibiotics. But none of these techniques broaden the limited number of
targetsthatourantibioticarsenalcurrentlypossessesandhavebeenincrementalin
nature.
New strategies to specifically target virulence factors, thus applying less selective
pressureandwithresistancebeingatthecostofvirulence,presentneatalternative
targetsforantibiotics,butremainalongwayfromtheclinic(Clatworthyetal.2007).
Combinationsofantibioticsarenowthenorm,andcanachievesynergisticeffects.In
somecases,resistancetooneantibioticmayincreasesusceptibilitytoanother–for
example colistin acts as a highly synergistic combination with several other
antibiotics,byimprovingthepermeabilityofthebacterialoutermembraneinGram
negativebacteria(Tängdénetal.2014).
Oneof thegreatest restraints to the“GoldenEra”ofantibioticdiscoverywas the
limitednumberofmicrobesthatcouldbegrowninthelaboratory,andthentested
for antimicrobial properties. In fact, it is estimated that 99% of bacteria are

24
“uncultured”andthusuntapped.Thedevelopmentofnewtechniquesforculturing
soilmicrobesinsituenablestheproductsofthesemicroorganismstobestudiedfor
the first time. This technology has already produced a new antibiotic candidate
teixobactinwhichbindslipidIIandIII,therespectiveprecursorsofpeptidoglycanand
cellwallteichoicacid,andthusinhibitscellwallsynthesis(Lingetal.2015).
Itisclearthat,despiterecentinnovations,thedearthofrecentantibioticdiscovery
combinedwiththeriseofantibioticresistanceisaworryingstateofaffairsforour
modernwayoflife.Bettercontrolofcurrentantibioticusagemayslowtherateof
resistanceoccurrence,butnewantibioticsaredesperatelyneededtocompeteinour
evolutionaryarmsraceagainstbacteria.
1.1.3 Whatalternativestoantibioticscurrentlyexist?
Thearmsraceagainstbacteriawillneverend,andevenwhennewantibioticswith
newtargetsarediscovered,resistancewilleventuallyarise.Itisthereforevitalthata
robustpipelineforantibioticproductionisinplace,andgiventhelackindiscoveryof
conventionalantibioticsoverthepast50years, itmakessensetolooktoplugthe
gaps with alternative antimicrobials. With the advancement of knowledge and
technologyinthesenovelantibioticfields,itislikelythateventuallyone,ormany,of
these“alternativeantibiotics”willbecomethenew“conventionalantibiotics”.
1.1.3.1 Non-lethaltreatments
Whilenotstrictly“antibiotics”,non-bactericidaltherapiesdeserveabriefmentionin
this literature review as they provide an alternative treatment to traditional
antibiotics.Theyalsoopenupadifferentwayofthinkingaboutbacterialinfections,
namelythatdamagetohostorganismswhensufferingfrombacterialinfectioncan
becausedasmuchbyhostresponseasbymicrobialfactors(PirofskiandCasadevall
2008). Efforts to prevent the host response, through tactics such as anti-
inflammatorymonoclonalantibodiescanpotentiallyminimisehostdamagewhilethe
body’simmunesystemfightstheinfection.
Othermethodsinvolvehinderingtheactivityofthebacteriathroughcompetitionfor
resources, either by the introduction of other bacteria (probiotics) or the

25
sequestration of host nutrients (Spellberg 2014). Furthermore, bacterial toxin
inhibitorsandbiofilminhibitorswhichdonotdirectlyharmthebacteria,butreduce
theirpathogenicityofferapromisingalternativepath.Amajoradvantageofthese,
alongwithhost-responsemodulation, is that theydonotapply the same levelof
selective pressure on the bacteria in question to evolve resistance as traditional
bactericidalmeasures(Chengetal.2014).
1.1.3.2 Predatorybacteria
Somebacteria,suchasBdellovibrio,PeredibacterandVampirococcus,preyonother
bacteria and therefore may offer a mechanism for active control of bacterial
infections.Thesebacteriagoonestep further thancommensal ‘probiotics’,which
generally compete for nutrients and resources thus reducing the scope for
opportunistic pathogenic bacterial colonisation, by entering host bacteria and
causing cell lysis (Dwidar et al. 2012). Predatory bacteria, widely known as
Bdellovibrio-and-like-organisms (BALOs), have demonstrated killing of pathogenic
bacteriainvitro(VanEsscheetal.2011),andrecentinvivostudiesshowareduction
inbacterialburdenofKlebsiellapneumoniaeinratlungs(Shatzkesetal.2016).
AninterestingstrengthofBALOsistheirabilitytopreyuponGram-negativebacteria,
whichareamajorsourceofglobaldisease.Theouter-membraneofGram-negative
bacteria presents a barrierwhich severalmajor antibiotics (e.g. vancomycin) and
antibiotic-alternatives(e.g.endolysins)struggletopenetrate(Miller2016).Another
advantageisthatpreyresistanceisextremelyrare,withonlyonecasesofarreported
(Varon1979),suggestingaderivedtherapycouldbeeffectiveforasubstantialperiod
oftime.
However, safety concerns around inoculating humans with bacteria must be
addressed. A potentially toxic response to the lipopolysaccharides in the Gram-
negative outer-membrane of BALOs is yet to be solved. Furthermore, predatory
bacteriaareunabletoeradicatealltheirprey,evenwhenappliedinveryhighratios
(SockettandLambert2004),andarealsostrictlyaerobicthuslimitingtheirpotential
for internalhumanapplication.Theymayhowever findaplace in therapieswhen

26
usedtogetherwith,forexample,β-lactamstowhichtheyaretolerant(Dwidaretal.
2012).
1.1.3.3 Antimicrobialpeptidesandbacteriocins
Antimicrobialpeptides(AMPs)aresmallpeptides(10-50aminoacids)thatformpart
oftheinnateimmunesystem,andarefoundinallclassesoflife.AMPshavebeen
shown to act independently as antimicrobials, but also to have wide ranging
immunomodulatory functions in mammals, including altering gene expression,
promotingwound healing andmodulating dendritic cell response (Bowdish et al.
2005;Fernebro2011).Furthermore,AMPscandisplayactivityagainstvirusesand
fungitoo,thusearningtheuseoftheword“antimicrobial”,ratherthan itssubset
“antibiotic”.AMPsalsohaveactivityagainstbiofilms,whichcanharbourbacteriain
chronicdiseases andare thereforean interesting routeof research. Themodeof
actionofAMPs is stillunclear,although it isbelievedthat theycausecell lysisvia
permeationofthecytoplasmicmembrane(ChungandKhanum2017).
However,thehightoxicityofAMPstomammaliancellsandtheirpoorstabilityhave
ledtopoorresultsinclinicaltrials.Thishasguidedresearchintothemodificationof
AMPs, to produce Structurally Nanoengineered Antimicrobial Peptide Polymers
(SNAPPs),whicharemorestableanddisplaygreaterlevelsofantimicrobialactivity
(Lametal.2016).Whileasyetunprovenclinically,SNAPPshavebeenshowntobe
particularlyactiveagainstGram-negativebacteriaandshowpromisingsynergywith
conventionalantibiotics.
Antimicrobialpeptides fromprokaryotesareknownas “bacteriocins”and tend to
haveanarroweractivity spectrum.Bacteriocinshavebeenused for some time in
foodpreservatives–forexamplenisinisusedtotreatcheese,meatandvegetables
to improve shelf life (Yang et al. 2014).While effective as inhibitors ofmicrobial
colonisation, the instability and potency of bacteriocins has to date limited their
potentialastherapeutics.

27
FigureRemoved
1.1.3.4 Bacteriophage
Bacteriophage,or“phage”,arethesubsetofvirusesthaninfectbacteria.Theyare
believedtobethemostnumerousbiologicalentitiesontheplanet,outnumbering
bacteria10-to-1anddisplayinghugevariability(Oliveiraetal.2013).Phage-therapy,
whichinvolvesusingphagetocombatbacterialinfections,hasbeeninvestigatedas
apotentialantibioticsincetheearly1900sbutinterestinthefielddeclinedacross
much of the globe after the discovery of penicillin. However, phage therapy
continuedtobepracticed in theUSSRdueto their limitedaccess toconventional
antibiotics,andeventodaytheEliava Institute inGeorgiastill treatspatientswith
phagetherapy(Fernebro2011).
Phage therapymakesuseof the lytic cycleof livebacteriophage to lyse infecting
bacteria.Thelyticcycle,illustratedinFigure1.5,involvesspecificattachmentofthe
bacteriophagetothehostbacterialcellwall,injectionofviralDNAintothehostcell,
andrecruitmentofthehostproteinsynthesismachinerytoproducebacteriophage
progeny.Aftersufficientbacteriophageprogenysynthesis,hostcelllysisisinitiated
bythecombinedeffectsofholinandendolysinproteins,thuskillingthehostcelland
releasingthephageprogenyintotheenvironment(ReindelandFiore2017).
Figure 1.5 The bacteriophage lytic cycle. Phage specifically bind to their target bacterialspecies and inject viral DNA. The host cell protein synthesis machinery produces phageprogeny,thenholinstoformaporeinthecellmembraneandfinallyendolysinstohydrolysethepeptidoglycanlayer.ReproducedfromFeineretal.(2015).

28
Sincetheriseofantibioticresistanceandthedecline innewantibioticdiscoveries
however,interestinphagetherapyhasbeenrenewed.In2006phagetreatmentof
cookedmeatswasapprovedbytheFDAtopreventpost-processingcontamination
with Listeriamonocytogenes (Fernebro 2011).However, no FDA approval has yet
beenmadeforhumantherapies,althougharecent“emergencyinvestigationalnew
drugapplication”was approved for the treatmentof apatient in SanDiego,USA
suffering from a multi-drug resistance bacterial infection, which resulted in the
successfultreatmentofthatpatient(LaFeeandBuschman2017).
Thehighspecificityofbacteriophageresultsinanarrow-spectrumofactivity.Thisis
excellentnews for theprotectionof commensal flora in thegut forexample,but
relieson rapidandeffectivediagnosis techniqueswhicharenot readily available.
Furthermore,occurrenceofresistancetophageishigh,andpoordataexistonthe
efficacy of treatment and their safety profile (Fernebro 2011). However, if these
obstacles can be overcome, then the sheer number, diversity and availability of
bacteriophagemakethemaveryattractivesourceofnovelantibiotics.
1.1.3.5 Bacteriophageendolysins
Manyofthehurdlesfacedbybacteriophagetherapycanbeovercomebytheuseof
their endolysins. These are the lytic enzymes which degrade the host bacterial
peptidoglycancellwallduringcelllysis.Theuseoftheseproteinsasnovelantibiotics
holds great promise and is the focus of this thesis. Endolysins are introduced in
greaterdetailinsection1.2.
1.2 Anoverviewofbacteriophageendolysins
1.2.1 Thebiologicalroleofbacteriophageendolysins
1.2.1.1 Background
Peptidoglycanhydrolase is a genericnamegiven to anenzyme that catalyses the
hydrolysisof thebacterialpeptidoglycancellwall.Peptidoglycanhydrolasescome
frommanydifferent sources,withdifferent functions:autolysinsareproducedby
bacteriatoremodeltheircellwallorcauseapoptosis;exolysinsareaweaponused

29
by bacteria to attack competing bacteria of another species; lysozymes can be
produced by eukaryotes as part of their innate immune response; tail-associated
lysins enable bacteriophage to inject their DNA into a new host bacterial cell;
endolysinsarepeptidoglycanhydrolases,alsoofviralorigin,thatlysethebacterial
hostcellwallfromwithinattheendofthelyticcycle.
Upon injectionofbacteriophageDNAintothehostbacterium,bacteriophageviral
progenyaresynthesizedandassembledbythehostmachinery.Twootherproteins
arealsoproduced,whichactinsynchronytooptimizethetimingofhostbacterium
lysis.Oneistheholin,whicholigomerisesintheinnermembranetoformapore.The
otherisanendolysinwhichisabletoaccessthehostpeptidoglycancellwall,viathe
poreformedbytheholins,degradingthecellwallandcausingcelllysis.
1.2.1.2 Structure
Endolysinsaregenerallyencodedbyasinglegeneandtwodistinctcategoriesexist:
those fromGram-positive infectingbacteriophage,and those fromGram-negative
infectingbacteriophage.EndolysinstargetingGram-positivebacteriahaveamodular
structure and arehighly species specific. Theirmodular structure consists of aN-
terminalcatalyticdomainandaC-terminalcellwallbindingdomain(CBD),generally
connectedbyaflexibleacidiclinker(Figure1.6).Themajorityofendolysinspecificity
is conferredby thecellwallbindingdomain,although in somecases thecatalytic
domain has been shown to bestow specificity via recognition of the enzymatic
substrate(Mayeretal.2011).RemovaloftheCBDinsomeinstancescanimprovethe
activityandspectrumofanendolysin,but inothercasestheCBDappearstobea
requirementforactivity,perhapsbyachievingthecorrectorientationofthecatalytic
domaintothesubstrate.
Figure1.6ThecanonicalmodularstructureofaGram-positiveendolysin.Thecellwallbindingdomain (CBD) is responsible for conferring specificity to the enzyme andmodification orremovaloftheCBDhassometimesresultedinimprovedactivityofthetruncatedendolysin(ChengandFischetti2007).
CellwallbindingdomainCatalyticDomain Acidic
linkerN C

30
Gram-negative endolysins on the other hand tend to be smaller with a globular
structure,andlackthehighbindingaffinityandspecificityforthecellwall.Thereason
for this is believed to be that, while it may be advantageous for Gram-positive
targetingendolysinstobindtightlytothepeptidoglycancellwallinordertoprevent
them from lysing surrounding bacteria whichmight be future hosts for the viral
progeny, the surrounding Gram-negative bacteria are protected by their outer
membranesonosuchrestraintisrequired(Loessneretal.2002).
Thesedescriptionsareofcoursesimplifications,andtherearemanyexceptionsthat
provetherule.ForexampleseveralstaphylococcalendolysinshavetwoN-terminal
catalytic domains, the streptococcal λSA2 endolysin has a central CBD with two
flankingcatalyticdomainsateachterminal,andPlyC,anotherstreptococcallysinand
oneofthemostactiveendolysinstestedtodate,isencodedbytwoseparategenes
formingamultimericlysin(Schmelcheretal.2012).
Afewthree-dimensionalcrystalstructuresofendolysinshavebeensolvedtodate,
butformationofcrystalshasproveddifficultpartiallyduetotheflexibleacidiclinker
betweenthedomains.However,T4lysozyme(SagermannandMatthews2002),T7
amidase(Chengetal.1994),PSAendolysin(Korndörferetal.2006),Cpl-1(Hermoso
etal.2003),CTP1L(Dunneetal.2016)andpartsofCD27L(Dunneetal.2014)have
allbeenelucidated.ThecrystalstructureofCpl-1willbediscussedingreaterdetail
in4.3.1.
1.2.1.3 Mechanismofaction
Endolysinsdisplayhugevariationintheirmechanismofaction.Whiletheyallattack
thepeptidoglycan,therehavebeenendolysinsidentifiedwhichtargetalmostevery
bondwithinthepeptidoglycannetwork.Thesecanbebroadlysplitintoglycosidases
whichhydrolysetheaminosugarlinkages,andamidases/peptidaseswhichattackthe
cross-linkingpeptidestemsandinterpeptidebridges(Loessner2005).

31
Figure1.7ThepeptidoglycanfromStreptococcalcellwall(A3α).Hexagonsrepresentsugarsandovalsareaminoacids.Redarrowsindicatesitesofactionforglycosidases(A:N-acetyl-β-D-muramidase;B: N-acetyl-β-D-glucosaminidase). Green arrows indicate endopeptidasetargets (C: D-glutamyl-L-lysine endopeptidase; D: D-alanoyl-L-alanine endopeptidase).ModifiedfromSchmelcheretal.(2012).
CBD domains are present in Gram-positive targeting endolysins, and recruit the
catalyticdomaintothepeptidoglycancellwall.TheCBDislargelyresponsibleforthe
specificityobservedinendolysinsandisabletorecognizeandbind(non-covalently)
arangeof ligands inthecellwall.These ligandscaneitherbeadirectpartof the
peptidoglycan,ortheycanbeothercomponentsofthecellwall.Anexampleofthe
firstistheLysMdomain,oneofthemostcommonconservedbindingmodules,which
binds the GlcNAc residues in the peptidoglycan. Meanwhile, the SH3b domain
presentintheCpl-1endolysinbindscholinemoietiesincell-wallassociatedteichoic
acidsofStreptococcuspneumoniae.Thespecificityconferredtoendolysinsbytheir
CBDcanrangefromanentirebacterialgenus,downtoserovarorstrainandoften
thebindingisexceptionallytight–inthepico-tonano-molarrange(Loessneretal.
2002;Briersetal.2009).
GlcNAc GlcNAcMurNAc MurNAc
D-Ala
GlcNAc MurNAc MurNAc
L-Ala L-Ala D-Ala
L-Lys
D-Glu
L-Ala
D-Glu
L-Lys
GlcNAc
NH2
H2N
L-Ala
A B
C D

32
FigureRemoved
1.2.2 Thetherapeuticpotentialofendolysinsand“exolysis”
1.2.2.1 Theoryandhistory
Gram-positive bacteria, which have no outer membrane, are also susceptible to
endolysins applied exogenously as the endolysin can directly access the
peptidoglycan cell wall. The osmotic pressure inside the bacterial cell (20-50
atmospheres) then causes cell lysis. This sensitivity enables endolysins to be
exploitedasnovelantibacterialagents,witharangeofadvantagesoverconventional
antibiotics.
Gram-negativebacteriaontheotherhandhaveaprotectiveoutermembrane(Figure
1.8) which prevents the endolysins from accessing the peptidoglycan layer when
appliedexogenously.However,recentadvancementshaveopenedupthefieldfor
endolysin therapy against Gram-negative bacterial infections. The addition of a
polycationic peptide, able to cross the lipopolysaccharide outer-membrane, to a
globular Gram-negative endolysin has yielded exciting results, and the chimeric
proteinshavebeennamedArtilysins®(Briersetal.2014).However,Gram-positive
targetingendolysinsarethefocusofthisthesis.
Figure 1.8 The cellwall structural differences betweenGram-positive andGram-negativebacteria. The noticeable differences are the thicker peptidoglycan cell wall in the Gram-postivebacteria,andthepresenceofanoutermembraneinGram-negativebacteria.Inbothinstances the cytoplasm is at thebottomof thediagram.Reproduced from (Brownetal.2015).

33
The first in vivo demonstration of endolysin activity was performed by Vincent
Fischetti’s group at Rockefeller University in 2001. The group used an endolysin
prophylacticallytopreventaStreptococcalinfectioninthemousepharynx,showing
thatafterasingletreatmentnostreptococcalbacteriaremainedintheoralpharynx
whileleavingtheindigenousoralfloraunaffected(Nelsonetal.2001).Sincethenthe
fieldhasgrownsignificantly,withonecommerciallyavailableendolysintherapeutic
andseveralmoreinthepipelineaswillbediscussedinthefollowingsection.
1.2.2.2 Currenttherapeuticuses
TheDutchcompanyMicreosproducedthefirstendolysin-basedtherapytoreceive
FDAapproval.Theproduct,namedStaphefektandmarketedunderthebrandname
GladSkin, has been approved for topical treatment of Staphylococcus aureus on
unbroken skin, treating diseases such as acne, rosacea, eczema and furunculosis
(Micreos).
AnothernotablecompanyinthetherapeuticendolysinfieldisContraFect.ContraFect
arealsodevelopinganendolysintherapyagainstStaphylococcusaureusnamedCF-
301,whichisinphaseIIclinicaltrialsandhasbeengrantedFastTrackDesignationby
theFDAhavingpreviouslybeenputonclinicalholdduetoconcernsovertrialdesign
(Parmley 2014).Notably, ContraFect are focusinguponendolysin purity,with the
intentionoftreatingbacteraemiasystemically.ContraFectalsohaveaGram-negative
endolysinatdiscoverystage(ContraFect).
Totheauthor’sknowledge,thesearetheonlytwoendolysin-basedtherapeuticsin
clinical trials. However, there are other companies such as Lysando, who have
licencedArtilysins®,withnewproductsinsight.
BothMicreosandContraFectuseE.coliastheirendolysinproductionplatform,and
ContraFect inparticularhavefocussedonengineeringtheirendolysintobehighly
expressed, soluble and readily purifiedwhen produced in E. coli (Parmley 2014).
While this appears to have been a suitable approach for producing endolysins
targetingGram-positivebacteria,itisunclearwhetherthesecompanieswillseekto
use an alternative expression platform for producing endolysins targeting Gram-

34
negativebacteria.SomeGram-negativetargetingendolysinshavebeenshowntobe
highly toxic toE. coli, suchasSPN9CC (Limetal. 2014), and thereforepotentially
renderE.colianinefficientproductionplatform.
1.2.2.3 Advantagesofendolysinsoverconventionalantibiotics
Endolysins hold great promise as novel antibiotics and they have numerous
advantagesoverconventionalantibiotics.
Firstly,endolysinskillbacteriaatasignificantlyfasterratethanthatofconventional
antibiotics.Indeed,purifiedlysinsinnanogramquantitieshavebeenshowntoreduce
107Streptococcuspyogenesbymorethan6logsinonlyamatterofseconds(Fischetti
2008).InvivomodelshaveshownthattheendolysinClySachievesa3logreduction
ofbacteriain30minutes,whiledaptomycintakes6hoursandvancomycin>24hours
to achieve similar levels of killing (Pastagia et al. 2013).Generally, endolysins act
much faster thanconventionalantibiotics,which forblood infections inparticular
suchassepsisandbacteraemia,couldbelifesaving.
The specificity of endolysins presents an enormous advantage over conventional
antibioticsiftheinfectingbacteriahasbeendiagnosed.Theadvantagesaretwo-fold:
firstly, a specificantibioticwillnotdisrupt thebody’s indigenous “good”bacteria,
whilekillingthepathogen.Thisminimisestheopportunityforsecondaryinfections
such as Clostridium difficile infection and yeast infections which are common
complications after a course of broad-spectrum antibiotics (U.S. Department of
HealthandHumanServices2013).Secondly,asthevastmajorityofbacteriainthe
bodyorenvironmentareunaffectedbytheendolysin,noselectivepressureisapplied
to them. This is in contrast to broad-spectrum antibiotics, which put a selective
pressureforresistanceuponallbacterialspeciesintheenvironment,thusincreasing
thechancesofresistanceemerging.Highspecificityisalsoalimitationofendolysins
however,aswillbediscussedinsection1.2.2.4.
Oneofthegreatestproblemsthatconventionalantibioticsfaceistheirlimitedability
tokillpersistercellsinbiofilms.Biofilmscanforminvariouslocations–fromthegut
lining, to water treatment plant equipment. Bacterial cells in biofilms are in a

35
dormant state, designed to withstand harsh environments and therefore do not
undergoreplication.Duetothemechanismofactionofconventionalantibiotics,such
asdisruptingcellwallformation,thesebiofilm-dwellingbacteriaareunaffectedby
them.Aftertreatmentwithantibiotics,theunaffectedbiofilmcellsarethenableto
recolonisetheenvironment.Endolysinsontheotherhandactivelylysebacterialcell
walls, regardless of the state of the cell and have been shown to be extremely
effectiveagainstbiofilms(Gutiérrezetal.2014).
Oneofthegreatestandmostoftenquotedadvantagesofendolysinsisthat,sofar,
bacterial resistance to them has been unreported, despite efforts to produce it
(Loeffler et al. 2001). It is believed that several factors contribute to endolysin-
resistancebeingarareevent.Firstly,thenaturalevolutionofendolysinshasledto
theadoptionofhighlyconserved targets in thepeptidoglycancellwall,whichare
essentialforbacterialviability(Fischetti2008).Secondly,theexogenousapplication
andactionofendolysinsavoidsmanyoftheresistancemechanismsobservedagainst
conventional antibiotics (efflux pumps and deactivating enzymes). Finally, with
regardstoreallifeapplications,thespecificityofendolysinsmeansthatnoselective
pressureisbeingappliedtonon-targetbacteria.
Itisinevitablethatoneday,ifendolysinsbecomewidelyused,thensomeresistance
will eventually be observed. In the case of conventional antibiotics, as we have
discussed, discoveringnewantibiotics requires serendipity andahugeamountof
time,andhasrecentlyprovenverydifficult.Incontrast,giventhediversityandsize
ofthebacteriophagepopulation,thereisahugestoreofendolysinstotapwhenthe
needarises.
Waterpollutionbyconventionalantibioticsisagrowingconcern.Whenantibiotics
arenotfullymetabolisedbythebody,thehighstabilityofsomeantibioticscanlead
them to remain active throughout the sewerage system and, eventually,
watercourses too. These low levels of antibiotics in the environment create ideal
conditions forresistanceemergence,and indeedantibiotic resistantbacteriahave
beendetectedinBritishwatercoursesforsometime(HughesandMeynell1974).The
proteinaceousendolysinsontheotherhandarerelativelyunstable,withahalf-life

36
ofabout20minutesinthebody,soareunlikelytosurviveintootherenvironments,
andgiventheirspecificityandlackofresistance,areaconsiderablylowerrisk.
Finally,thoughnotatrueadvantageoverconventionalantibiotics, istheabilityof
endolysins to act synergistically with antibiotics to reduce antibiotic usage and
improvetherapeuticefficacy.Forexample,theendolysinClyShasbeenshowntoact
synergisticallywithoxacillintokillMRSA invivo.Asoxacillinisacellwallsynthesis
inhibitor,andcausescelllysisbytheresultingupregulationofautolysins,itisbelieved
thatClySfurtherexacerbatesthedegradationofthecellwall,thuspromotingeven
morebacterial autolysin activity (Daniel et al. 2010). Such synergistic effectsmay
enable discontinued antibiotics to be reinstatedwhen usedwith endolysins, and
reduceoverallantibioticintakeorcourselength.
1.2.2.4 Limitations
Thebacteriaposing thegreatest threat tomankindhavebeengiven theacronym
ESKAPE by the Infections Diseases Society of America, standing for Enterococcus
faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumanni,
Pseudomonasaeruginosa,andEnterobacterspecies(Rice2008).Ofthese,thefinal
four are all Gram-negative. Conventional antibiotics currently in clinical trials are
skewedtowardsGram-positiveactivity.Onelimitationofendolysinsistheirinability
tocauseGram-negativelysisduetotheirinabilitytopenetratetheoutermembrane.
However,asmentionedearlier,Artilysins®havemadesomeprogresstowardsbeing
able to tackleGram-negative infections, and for topical applications EDTA can be
usedtopermeablisetheGram-negativeoutermembrane(BriersandLavigne2015).
Therearetwofactorstobeconsideredintermsofimmuneresponsetoapplication
ofendolysins.Thefirst istotheendolysin itself:endolysinsareaproteinandthus
maytriggeranimmuneresponseincludingantibodygeneration,potentiallylimiting
futureuseofthatendolysin.Anattempttoreducethepostulatedimmuneresponse
toCpl-1wasmadebyPEGylationoftheprotein,awell-knownmethodofminimising
immune response and increasing half-life, but this completely inhibited the lytic
activityoftheendolysin(Reschetal.2011b).However,ithasbeenobservedinvivo
thatalthoughantibodiesare indeedraisedagainsttheCpl-1endolysin inmice,on

37
reapplicationnoadverseeffectswereobservedandactivitywasonlyslightlyslowed
(Jadoetal.2003).Itispostulatedthattheendolysinactivityismuchfasterthanthe
host’s antibody mediated secondary immune response, and is thus largely
unaffected.
Thesecondimmuneresponseofconcernistothelargevolumeofbacterialcelldebris
rapidly released by endolysin activity. Two studies have been undertaken to
investigate this issue, with varying results: the first study found that the Cpl-1
endolysinproducedanaccentuatedcytokineresponsewhencomparedtothesame
treatmentusingvancomycin(Entenzaetal.2005);thelatterstudyobservednomajor
difference incytokineproductionbetweenCpl-1andamoxicillin(Witzenrathetal.
2009).Thisdiscrepancyisthoughttobeanissueofendolysindosing:whenahigh
dose is given, then the bacterial cellwall is fragmented thus producing a greater
response,comparedtoalowdosewhichmerelypuncturesthebacterialcellswith
minimalfragmentation(Fischetti2008).
Bioavailability of endolysins is a concern given their large size compared to
conventional antibiotics. In particular, intracellular bacterial infections such as
chlamydiaandtuberculosisareparticularlyhardtoaccess,andpresentachallenge
evenforconventionalantibiotics.However,arecentdemonstrationthatPlyCisable
to enter human throat cells and kill internalized S. pyogenes demonstrates the
diverseabilitiesoftheendolysins(Shenetal.2016).Inthisinstanceitappearsthata
specificcomponentofthethroatcellmembrane,aphosphatidylserine,grantsaccess
tothePlyCendolysin–howeverit isalsonotedinthestudythatotherendolysins
couldnotbeexpectedtofunctioninthisway.Thisfindingdoeshoweverraisethe
prospectthatevenintracellularbacteriamayeventuallybetreatedwithendolysins.
Theonlyendolysincurrentlyonthemarket,Staphefekt,isdesignedfortopicaluse
and isdeliveredeitherasacreamoragel (Micreos). It isunclearhowContraFect
intendtodeliver theirendolysindrug,howevertheirstudiesanddiscussionshave
mostlyfocussedaroundintranasalandaerosoldeliverymethods(Doehnetal.2013;
Loeffleretal.2003).Oraldeliveryofendolysinswillbeharder,astheproteinaceous
natureofendolysinsmeansthattheyaresusceptibletodigestionanddenaturingin

38
thestomach,andwouldneedtobeencapsulatedtoensurereleaseintotheintestine
forexample.
The final limitationofendolysins,whichwascontrastinglypresentedearlierasan
advantage, is theirspecificity.Thenarrow-spectrumoftargetbacteriakilledbyan
endolysinpresentsachallengeforcliniciansinthattheinfectionmustbeaccurately
diagnosedbeforeanendolysintreatmentcanbeprescribed.Thisdiagnosisisnotyet
easilypossible,butthepotentialtocreateendolysin“cocktails”,whichtargetallthe
mostcommonbacteriaassociatedwithacertaindisease,mayworkaroundthis.
To conclude, although various challenges havebeen laid in thepathof endolysin
research,nonehasyetpresentedaseriousflawtothetechnologyasatherapeutic,
andtechniquesareemergingtosolvethemajorityofissues.
1.2.3 Otherapplications
The remarkable ability of endolysins to identify and strongly bind highly specific
bacterialtargetshasproducedinterestinfieldsotherthandirecttherapeutics.
Forexample,workbytheLoessnergroupatETH,Zurichhasusedendolysinstocreate
rapid diagnostic tools for food safety. Their technology uses electrochemical
impedance spectroscopy (EIS) to rapidlydetectListeria cells binding toendolysin-
derivedCBDsimmobilizedonagoldscreenprintedelectrode.Thisenablesextremely
rapid, accurate, and sensitive detection of contaminating Listeria cells in food
products(Tolbaetal.2012).
Nelson’s group at theUniversity ofMaryland have designed two novel endolysin
basedplatforms,designedtoaidthebody’snaturalimmunesystem.Thefirstisthe
generation of Infection Site Targeted Antitoxin antibodies (ISTAbs), which are
endolysinCBDsfusedtospecificantitoxinneutralizingmonoclonalantibodies.The
CBDtargetstheantibodytothepathogencellwall,whereitsequestersthereleased
toxins, thus reducing the toxin load and furthermore, functionally opsonises the
bacterium for opsonophagocytic killing by the host immune system. The second
approach,namedInfectionSiteTargeteduniversalBridgingAntigen(ISTuBA),fuses

39
CBDs to antigens for which most people have existing immunity (e.g. childhood
vaccinetargets).Thisanchorstheantigentothepathogencellwall,anddirectsthe
existingimmunitytowardsthenewpathogen(Nelson2016).
Otherapplicationsincludeendolysinbaseddisinfectants,whichhavebeenshownto
be 1,000xmore active than virkon-S against Streptococcus equi over 30minutes
(Hoopesetal.2009).Foodbiopreservationandbiofilmeliminationhavealsobeen
showntobesuccessfulapplicationsofendolysins(Ajueboretal.2016).
1.3 Producingrecombinanttherapeuticproteins
Thesuccessfuladministrationoftherapeutic insulintoaseverelydiabetic14-year-
oldboy in1922markedthefirstuseofaproteinasapharmaceutical (Joshietal.
2007). Since then, the ability to produce proteins recombinantly in a number of
biological platforms has led to lower costs of production and the rise of protein
therapies.In1977Somatostatin,averysmallinhibitorpeptide,wasthefirsthuman
recombinantproteintobeproducedinEscherichiacoli,byGenentechInc(Brooker
2010). Other platforms have since been developed to meet the various protein
requirements, such as producing larger proteins, glycosylation patterns, and
disulphide bonds. Here we provide a very brief overview of some of the most
commonexpressionplatformsusedtoday.
1.3.1 Commonexpressionplatforms
E.coli,asalreadymentioned,wasthefirstrecombinantproteinexpressionplatform
andremainswidelyused.Duetomanyyearsofresearchanddevelopmentresulting
in an excellent understanding of E. coli genetics, genetic engineering of E. coli is
cheap,easyandextremelyfast.Highyieldsofrecombinantproteinscanbereadily
achievedandmanygenetictools,suchasinducibleexpressionsystemsandprotein
secretion,enableitsprecisemanipulation.However,thesystemisnotwellsuitedto
producinglargeproteins(over50kDa).Furthermore,theproteinscanforminactive
inclusion bodies, E. coli does not naturally glycosylate proteins, and endotoxin
contaminationrequiresexpensivedown-streampurificationprocedures(Guptaand
Shukla2015).

40
ChineseHamsterOvary(CHO)cellsareprobablythemostcommonlyusedexpression
platform for current therapeutic proteins, in particular for the production of
antibodies (Dumont et al. 2015). As amammalian system, CHO cells are able to
produce large proteins (over 100 kDa) and glycosylate and fold them correctly -
although they do produce some post-translational modifications that are not
expressed in humans, and subsequently against which humans have antibodies
(Dumontetal.2015).Onthewhole,CHOcellsareusedtoproducelargerandmore
complexbiologicsthanothersystems.Theirdrawbacks includepotentialDNAand
viralcontamination,veryhighcostsofproductionandloweryieldsthansomeother
systems(DemainandVaishnav2011).
Yeast,asasinglecellulareukaryote,sharessomeoftheadvantagesofamicrobial
platform, such as cheap growthmedia and high yields, as well as advantages of
mammalians cells, such as protein glycosylation and disulphide bond formation.
Yeast therefore presents a very attractive platform for recombinant protein
expression,althougha few issuesprevent it frombeingusing forallproteins.For
example,yeast’shigh-mannose typeN-glycosylationpatterncan lead toa shorter
protein half-life and reduced therapeutic efficacy, and its lack of necessary
chaperonescanoftenresultinpoorproteinfolding(Nielsen2013).
Plant-basedplatformshavelaggedbehindothersoverthepastdecades,despitethe
numerousadvantagestheyholdasarecombinantproteinproductionplatform.Plant
cellsareabletofoldcomplexproteinsandperformpost-translationalmodifications
including glycosylation. Either whole plants or plant cell cultures can be used as
expression platforms. Both are devoid of the endotoxins and potential viral
contamination of other platforms, but otherwise have different advantages and
pitfalls.Wholeplants,suchastobacco(Nicotianatabacum)arehighlyscalable,and
costeffectiveatscale.Transientgeneexpressioninleaftissueofwholeplantscanbe
achievedveryrapidlyandhasbeenusedtosuccessfullyproduceZMapp, theonly
successfuldrugproducedtotreatEbolaviral infectioninhumans,aswellasbeing
regularlyusedforproducinginfluenzavaccines(Yaoetal.2015).However,several
issues have limitedwhole plant use to date, including the relatively long time to
produce stable transgenic plants desired for long-term production, variable

41
expression levels,and issuesofbiocontainmentat largescale (ParkandWi2016).
Plantcellculturesoffersolutionstomanyoftheseissues,suchprecisemonitoring
andcontrolofgrowthconditions, consistent cell yieldsandbiocontainmentwhen
growninbioreactors.Severalplantcelllinesareusedroutinely,suchasthosederived
fromN. tabacum, aswell as those fromedible crops including rice (Oriza sativa),
carrot(Daucuscarota)andtomato(Lycopersiconesculentum)(XuandZhang2014),
and in 2012 taliglucerase alfa, the first plant-made pharmaceutical approved for
humanuse,wasmadeinacarrotcelllinetotreatGaucher’sdisease(Grabowskiet
al.2014).
Like plant cell cultures, microalgae offer many similar advantages over other
platforms and higher plants, but have additional advantages too.Microalgae are
photosynthetic (unlikecarrotcell cultures forexample),enabling thepotential for
using cheaper, less complex media that is less susceptible to contamination.
Microalgaealsogenerallygrowfasterthanplantcellculturesandaremorerobustin
termsofresistingstressesfrompHchangesandshearstress(SunandLinden1999).
Finally,thegenerationofnewtransplastomiclinesisfasterinmicroalgae(2-3weeks)
thanplantcellcultures(4-5weeks)(Govea-Alonsoetal.2017).
1.3.2 Microalgaeasexpressionplatforms
1.3.2.1 Whataremicroalgae?
The term microalgae can generally refer to any unicellular organism capable of
oxygenic photosynthesis, both eukaryotic (e.g. green algae) and prokaryotic (e.g.
cyanobacteria)(Walkeretal.2005).Microalgaearethereforeanextremelydiverse
groupoforganismsandcanbefoundinbothfreshandsaltwaterenvironments,as
wellasterrestrially.Figure1.9illustratesthelevelofdiversityobservedwithinthe
microalgae.

42
FigureRemoved
Figure1.9Aselectionofimagesdisplayingthediversityofmicroalgae.(A)Meridioncirculare(B) Merismopedia elegans (C) Acutodesmus acuminatus (D) Microcystis novacekii (E)Arthrospira jenneri (F) Dissodinium pseudolunula (G) Zygnema circumcarinatum (H)Actinocyclus normanii (I)Pediastrumduplex (J)Klebsormidiumdissectum (K)Diploneis (L)Chlamydomonas reinhardtii. A-F from http://nordicmicroalgae.org/. G-K fromhttp://www.algaterra.org/.Lfromhttp://protist.i.hosei.ac.jp/.Imagesnottoscale.
MicroalgaesuchasArthrospira(Spirulina)andAphanizomenonhavebeenusedasa
sourceoffoodforthousandsofyears(Spolaoreetal.2006),butonlyinthelast50
yearshavemicroalgaebeensubjecttocommercialcultivation.Thefirstcompanyto
produce Chlorella sorokiniana on a commercial scale was Taiwan Chlorella
ManufacturingLtd,whichwasfoundedin1964andstillproducesChlorellatoday,to
besoldasdietarysupplement(Spolaoreetal.2006).WhileSpirulinaandChlorella
remainpopularhealth-foodproducts,theabilityofmicroalgaetoactas“sunlight-
driven cell factories” (Chisti 2008) for producing high value products has spurred
researchinthefieldtowardsharnessingthefullpotentialofthesediverseorganisms.

43
1.3.2.2 Thehistoryofmicroalgaeasanexpressionplatform
Microalgae,andinparticularthegreenalgaChlamydomonasreinhardtii,havebeen
used as a model organism for many years for the investigation of the genetic
mechanismsbehindcellularprocesses(Harris2009).Microalgaegainedprominence
as a recombinant protein production platform with their potential to produce
biofuelswithout affecting food production - as a photosynthetic organism itwas
postulatedthatalgaecouldbegrowninareasunsuitableforcropproduction,thus
producing biomass from cheap substrates on unused land. However, despite the
successful production of biodiesel in microalgae, the economics of this strategy
currentlyfailtocompetewiththatoffossil-fuels(Scrantonetal.2015).
Despitethecurrentshortcomingsofmicroalgaefortheproductionofbiodiesel,the
platformstillholdspromiseforhigh-valueproteinsandhasseveraladvantagesover
higher-plants. Microalgae have the ability, like higher plants, to synthesize and
correctlyfoldhumanantibodies,andmanyareGenerallyRegardedAsSafe(GRAS).
Butinadditiontotheseattributes,microalgaecanbegrowninclosedcontainment
photobioreactors,removingtheriskofgeneflowintotheenvironmentviapollen;
genetically engineered strains can be produced in amatter ofweeks rather than
sometimesyearsforhigher-plants;andscaleupfrombenchtoproductionscaleis
straightforward (Franklin and Mayfield 2004). Microalgae therefore present an
extremelyattractiveplatformforrecombinantproteinexpression.
Severalspeciesofmicroalgaehavetodatebeengeneticallytransformed,including
C. reinhardtii,Haematococcus pluvialis, Chlorella and diatoms (Spicer and Purton
2016). H. pluvialis is a natural producer of astaxanthin, a carotenoid with high
economic and health-related value, and it has been successfully genetically
engineered to produce higher levels of astaxanthin (Steinbrenner and Sandmann
2006).ThegreenalgaChlorellavulgarisisasourceofseveralproductsbelievedtobe
beneficial as health supplements, including high-quality proteins, fatty acids, and
essentialaminoacids,andamethodofagrobacterium-mediatedtransformationhas
beenestablished(Chaetal.2012).

44
However,whilevariousspeciesofmicroalgaehavebeengeneticallymodified, the
specieswith themost advanced genetic toolkit isC. reinhardtii.C. reinhardtii has
been shown to express multiple recombinant proteins using a variety of
transformation methods, and in all three genomes (nuclear, mitochondrial and
chloroplast)(Scaifeetal.2015).ThepotentialofC.reinhardtiiwillbeexploredfurther
inthefollowingsection.
1.3.3 Chlamydomonasreinhardtiiasanexpressionplatform
1.3.3.1 AnintroductiontoChlamydomonasreinhardtii
Chlamydomonas reinhardtii is a unicellular green alga found widely in soil and
freshwaterandbelongstotheChlorophytadivisionofgreenalgae.Figure1.10shows
aschematicofaC.reinhardtiicellaswellasascanningelectronmicroscopyimage,
highlightingthemainmorphologicalfeaturesofthecell.Itisapproximately10μmin
diameter and has two flagella enabling phototaxis, and a large chloroplast filling
approximately 70% of the cell volume (Harris 2009). C. reinhardtii can grow
phototrophicallyor,inthepresenceofacetate,mixotrophicallyorheterotrophically.
Allthreegenomeshavebeensequenced(Vahrenholzetal.1993;Mauletal.2002;
Merchantetal.2010),andasimple(andcontrollable)sexualcycleexists inwhich
nucleargenesareinheritedaccordingtoMendelianrules,chloroplastgenesinherited
uniparentallyfromthemating-type(mt)plusparent,andmitochondrialgenesfrom
themtminusparent.Furthermore,thehaploidnatureofthenucleargenomeand
the fact thatmany nuclear genes are single copy (rather thanmembers of gene
families) makes the recovery and classical genetic analysis of mutants very
straightforward.ThishasledtoC.reinhardtiibeingreferedtoasthe“photosynthetic
yeast” (Rochaix1995).Given theseattributesand the fact that it is relatively fast
growing(5-8hourdoublingtime,dependingonnutrient,CO2andlightavailability
(Hosleretal.1989)), ithasbeenanattractivemodel for studyingphotosynthesis,
circadianrhythms,matingandotherprocessesformanyyears(Harris2009).

45
FigureRemoved
Figure1.10(A)TheChlamydomonasreinhardtiicellrepresentedasaschematicdiagramwithlabelledphysiological features (Shi2013). (B)AScanningElectronMicroscopy imageofC.reinhardtiiclearlyshowingthemainmorphologiesofthecellincludingthetwoflagella(Smithetal.1996).
1.3.3.2 GeneticengineeringofChlamydomonasreinhardtii
C.reinhardtiiisthemostgeneticallywellstudiedofthemicroalgae,andmethodsto
transform the nuclear, chloroplast and mitochondrial genomes have all been

46
developed.Thesexualcyclecanbepreciselycontrolled,enablinggeneticcrossesto
becarriedoutandintroducedtraitstobecombined(Grossmanetal.2003).
The nuclear genome can be transformed using a number of methods including
electroporationandparticlebombardment to randomly incorporateDNA into the
genome(JinkersonandJonikas2015).Antibioticmarkersandrescuingofnutritional
auxotrophshavebeensuccessfulmethodsfornucleartransformationselection,but
positionaleffectsleadtoaspectrumofgeneexpressionlevels(Rochaix1995).The
nucleargenomeisveryGCrich(64%),andcodonoptimizationhasbeenshownto
playan important role in geneexpression levels, improvingGFPexpression levels
approximately 5-fold (Fuhrmann et al. 1999). However, gene silencing and low
protein yields have been problematic for C. reinhardtii nuclear transformants,
althoughrecentwork,suchasthatbyBarahimipouretal.(2016)demonstratinghigh
yields of HIV antigen P24, show that these issues are being gradually overcome.
Furthermore, Lauersen et al. (2015) have created the pOptimized vector system,
which allows the rapid swapping of reporter genes, selectable markers and
regulatory elements to speed up the optimisation of nuclear protein expression,
whileScrantonetal.(2016)havedemonstratedthesuccessfulidentificationanduse
ofsyntheticnuclearpromoterstoimproveheterologousnucleargeneexpression.
The chloroplast on the other hand offers a more attractive target for genetic
modification. Very simple transformation processes exist, for example transient
pores can be formed by vortexing cell wall-lessmutants with glass beads, which
enablesDNAentrytothechloroplast(Kindleetal.1991;Maliga2014).Homologous
recombination then allows precise targeting of expression cassettes into the
plastome, and generally results in greater protein accumulation than nuclear
transformations(Franklinetal.2002).Furthermore,bytransformingphotosynthesis-
deficient mutants, antibiotic-free selection is possible by using the rescue of
photosynthesis as a selection mechanism (Wannathong et al. 2016). The precise
method of transformation is described in more detail in 2.3.13. Gene silencing,
thoughttobeadefencemechanismagainstviralDNAinsertionintothegenome,has
notbeenobservedintheC.reinhardtiichloroplast.However,thechloroplastdoes
notglycosylateproteins.

47
FigureRemoved
AsummaryofthestrengthsandlimitationsoftheC.reinhardtiichloroplastcompared
tothenucleuscanbeseeninFigure1.11.
Figure 1.11 The nucleus and chloroplast genomes represent the major locations fortransgene insertion and recombinant protein synthesis in C. reinhardtii. While themitochondrial genome has been transformed it is not commonly used for recombinantproteinproduction.ReproducedfromRasala&Mayfield(2015).
1.3.3.3 ThesuitabilityoftheC.reinhardtiichloroplastforendolysinsynthesis
Overonebillionyearsagoanendosymbioticevent,resultingintheencapsulationof
aphotosyntheticprokaryoteinsideanonphotosyntheticeukaryotichost,isbelieved
tohavebeenthegenesisofthechloroplastorganelle(Purton2007).Sincethenthe
prokaryoticgenomehasshedmuchofitsunnecessarygeneticmaterial,resultingin
a simplified circular plastome. The C. reinhardtii chloroplast, sequenced in 2002
(Mauletal.2002),contains99genesandhasalowGCcontent(34%),incontrastto
thenucleargenome.
Theprokaryoticbeginningsofthechloroplastresultinanenvironmentwellsuitedfor
the production of endolysins, which are naturally produced in bacteriophage-
infectedprokaryotes.Forexample,thechloroplastappearstocontainproteasesthat

48
aredirecthomologuesofprokaryoticproteases(Adametal.2006).Thisisexpected
toleadtoarespectablehalf-lifeoftheendolysinsinsidethechloroplast,astheyare
adapted to be relatively resistant to prokaryotic proteases. Furthermore, the
chloroplastisnon-glycosylating,whichmayoftenbeseenasadisadvantage,butfor
theproductionofendolysins,andotherprokaryoticproteins,isbeneficial(Almaraz-
Delgadoetal.2014).TheexpressionofendolysinstohighlevelsinE.colihasbeen
reported to have a detrimental effect upon bacterial cell growth, presumably
becauseoflow-levelactivityagainstitspeptidoglycancellwalldespitethespecificity
ofendolysinsandlackofholin.However,plantandgreenalgalchloroplastshavelost
thecellwallduringevolution,andextremelyhighlevelsofendolysinaccumulation
(~70%TSP)havebeenreported in thechloroplastofNicotiana (Oeyetal.2009).
Therefore, the prokaryotic environment, lack of glycosylation, and lack of
peptidoglycanstructuresmakesthechloroplastawellsuitedplatformforendolysin
production(Oeyetal.2009).TheC.reinhardtiichloroplasthassuccessfullyproduced
therapeuticproteins inthepast, includingendolysins,asummaryofwhichcanbe
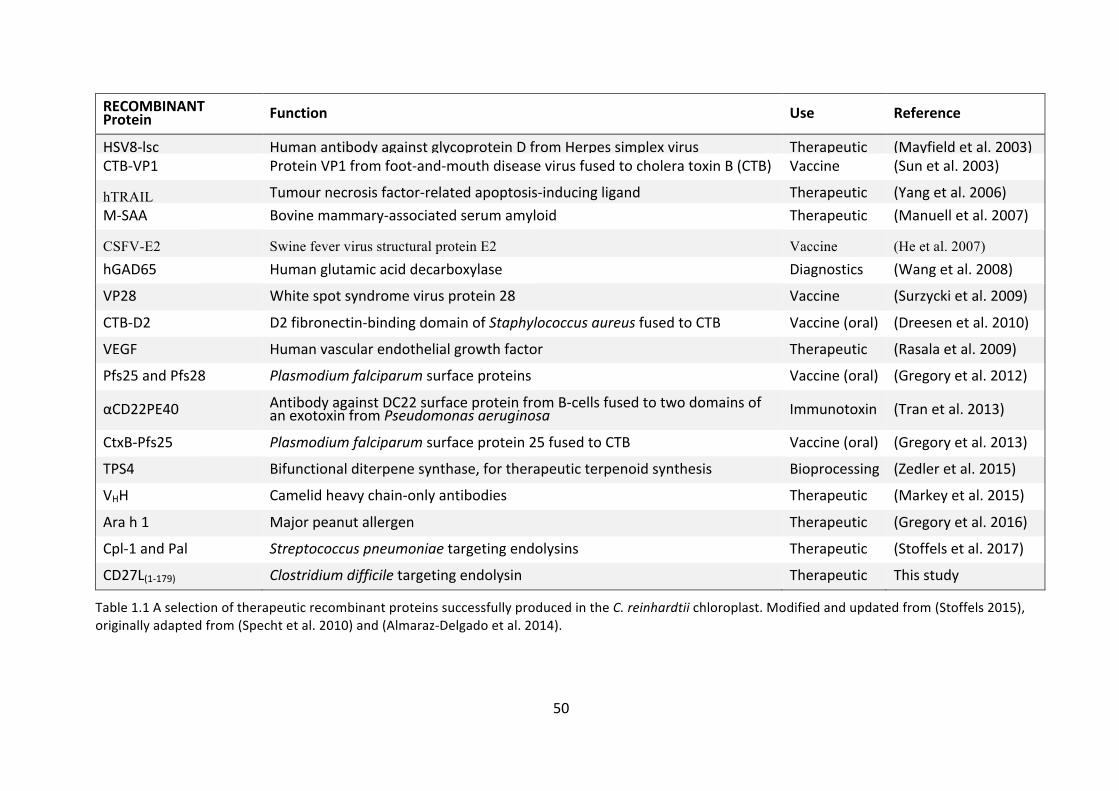
seeninTable1.1.
C.reinhardtiihasbeendesignatedGRASstatusandthereisspeculationinthefield
thatthismayleadtoreduceddownstreamprocessingcosts,oreventheabilitytouse
crudeextractsofrecombinantproteintherapeutically(Almaraz-Delgadoetal.2014).
In fact, the twocompaniesclosest to therapeuticendolysinproductionhaveboth
been hampered by issues of product purity and solubility with their bacterial
production systems.ContraFect, attempting toproduceanendolysin for systemic
use,havesofarfailedtoreceiveFDAapproval,whileMicreoshaveinsteadoptedto
produce an endolysin for topical use, with less regulation around product purity
(Parmley2014).
To date, themajor limitation tomicroalgae as a recombinant protein production
platformhasbeentheirlowproductionyields.CHOcellsarecapableofproducing4-
6 g/L of recombinant protein (Wurm 2004), yeasts 9-12 g/L, E. coli 15-17 g/L,
transgenicanimals(e.g.goatmilk)upto23g/L,andfungalexpressionsystemshave
been reported to achieve 35 g/L (Demain and Vaishnav 2011). These are clearly
optimized yields, and differ depending on the protein in question. However, the

49
highest level of recombinant protein production inC. reinhardtii is 5% TSP under
laboratoryconditions(Manuelletal.2007),whichwouldequatetoapproximately8
mg/Lculturevolumewhenscaledtopilotscale (Gimpeletal.2015).As it is,pilot
scale production of recombinant bovine milk amyloid A (MAA) in C. reinhardtii
achieved 3.28 mg/L (Gimpel et al. 2015). It is clear from these data that the C.
reinhardtii platformhas a longway to gobefore being able to competewith the
platformsnamedabove.However,rateofdevelopmentcanbefast:between1986
and2004therecombinantproteinyieldofCHOcellsrosefrom50mg/Lto4.7g/L
(Wurm2004).Thisvast improvementwasachievedthroughavarietyof improved
techniques such as better selection strategies for high-productivity clones and
identificationandsubsequenttargetingtotransgenestotranscriptional“hotspots”
(Kimetal.2012).Furthermore,differentplatformshavedifferentproteinrecovery
rates,forexample,duetoinclusionbodiesinE.coli,productionratesmaybehigh,
but recovery rates and renaturation rates can be extremely low, leading to the
possibility that another platform with a lower headline production rate is more
suitable(Dataretal.1993).
WhiletheGRASstatusofmanymicroalgaegiveshopethatsubsequentpurification
ofproteinproductswillbecheaperandlessnecessaryincomparisonto,forexample,
E.coli,thereareotherlimitationstotheiruseasaproteinproductionplatform,such
asthepresenceofchlorophyll.Chlorophyllisaproblematicpigmentpresentinalgal
extractsthatrequiresfurtherpurification,althoughthiscanbeperformedrelatively
cheaplybyuseofcolumnchromatographywithactivatedcarbon(Santillan-Jimenez
etal.2016).
50
RECOMBINANTProtein Function Use Reference
HSV8-lsc HumanantibodyagainstglycoproteinDfromHerpessimplexvirus Therapeutic (Mayfieldetal.2003)CTB-VP1 ProteinVP1fromfoot-and-mouthdiseasevirusfusedtocholeratoxinB(CTB) Vaccine (Sunetal.2003)
hTRAIL Tumournecrosisfactor-relatedapoptosis-inducingligand Therapeutic (Yangetal.2006)M-SAA Bovinemammary-associatedserumamyloid Therapeutic (Manuelletal.2007)
CSFV-E2 Swine fever virus structural protein E2 Vaccine (He et al. 2007)
hGAD65 Humanglutamicaciddecarboxylase Diagnostics (Wangetal.2008)
VP28 Whitespotsyndromevirusprotein28 Vaccine (Surzyckietal.2009)
CTB-D2 D2fibronectin-bindingdomainofStaphylococcusaureusfusedtoCTB Vaccine(oral) (Dreesenetal.2010)
VEGF Humanvascularendothelialgrowthfactor Therapeutic (Rasalaetal.2009)
Pfs25andPfs28 Plasmodiumfalciparumsurfaceproteins Vaccine(oral) (Gregoryetal.2012)
αCD22PE40 AntibodyagainstDC22surfaceproteinfromB-cellsfusedtotwodomainsofanexotoxinfromPseudomonasaeruginosa Immunotoxin (Tranetal.2013)
CtxB-Pfs25 Plasmodiumfalciparumsurfaceprotein25fusedtoCTB Vaccine(oral) (Gregoryetal.2013)
TPS4 Bifunctionalditerpenesynthase,fortherapeuticterpenoidsynthesis Bioprocessing (Zedleretal.2015)
VHH Camelidheavychain-onlyantibodies Therapeutic (Markeyetal.2015)
Arah1 Majorpeanutallergen Therapeutic (Gregoryetal.2016)
Cpl-1andPal Streptococcuspneumoniaetargetingendolysins Therapeutic (Stoffelsetal.2017)
CD27L(1-179) Clostridiumdifficiletargetingendolysin Therapeutic Thisstudy
Table1.1AselectionoftherapeuticrecombinantproteinssuccessfullyproducedintheC.reinhardtiichloroplast.Modifiedandupdatedfrom(Stoffels2015),originallyadaptedfrom(Spechtetal.2010)and(Almaraz-Delgadoetal.2014).

51
FigureRemoved
1.4 Targets/applications
1.4.1 Streptococcuspneumoniae
Although S. pneumoniae is a common commensal bacterium found in the
nasopharynx,and isusuallybenign,colonisationand infectioncancauseahostof
diseases, including pneumonia, meningitis, febrile bacteraemia and otitis media
(Bogaert et al. 2004). Pneumococcal infection accounts for the death of
approximately1.2millionpeopleeveryyear,mostofthemchildrenundertheageof
five(ObaroandAdegbola2002).
Figure 1.12 Scanning electron microscope image of an encapsulated strain (R6) of S.pneumoniae,initsstreptococcalformwith2%choline(left)anddiplococcalformwithoutcholine(right).Scalebarsare2μm.Imagereproducedfrom(Paiketal.1999).
S.pneumoniaeisamesophilic,non-motile,Gram-positivebacteriumandwasnamed
after its role in pneumonia. It usually takes a diplococcal form but can form
streptococciinliquidmedium.Dependinguponthestrain,apolysaccharidecapsule
sometimesencasesthecell,belowwhichisthepeptidoglycancellwall.Presenceof
this capsule can increase virility of the strain and interferes with leukocyte
phagocytosis (Hyamset al. 2010).However, it hasbeen shown thatencapsulated

52
FigureRemoved
strainsarenomoreorlesssusceptibletolysisbyendolysinsthanun-encapsulated
strains(Loeffleretal.2001).
The peptidoglycan cell wall is approximately six layers thick and teichoic acid is
attached to approximately every third N-acetylmuramic acid. Specifically to S.
pneumoniae, both the teichoic acid and lipoteichoic acid (attached to the cell
membrane via a lipid moiety) are decorated with phosphoryl-choline residues.
Choline isanessential requirementforpneumococcalactivity,beingthe ligandby
which it attaches to human cells via the platelet-activating-factor receptor and
choline-bindingprotein,CbpA(DworkinandFalkow2006;Gehreetal.2008).Choline
isalsotheligandbywhichtheS.pneumoniaeautolysin,LytA,andtheendolysin,Cpl-
1,bindtothecellwall.
Figure1.13Aschematicofagenericpeptidoglycancellwall.SomestrainsofS.pneumoniaealso have an outer capsule, and both teichoic and lipoteichoic acids are decorated withcholine.ReproducedfromMadigan&Martinko(2006).
While highly effective vaccines have been produced to protect humans from S.
pneumoniaeinfections,theirsuccesshasbeenlimitedduetopoorcoverage,andthe
threat of non-vaccine serotypes requires an active solution (Bogaert et al. 2004).
Studiesagreethatpreventionofnasopharyngealcolonizationisthemostsuccessful
methodof averting infection,however the riseof antibiotic resistant strainsofS.

53
pneumoniae,inparticularpenicillin-resistantS.pneumoniae(PRSP),chloramphenicol
resistance,macrolide resistance, andmultiresistant strainspresentamajor global
healththreat(Appelbaum1992;Adam2002;Bogaertetal.2004;Staneketal.2011).
Thepresenceofavirulence-factortarget(choline)inS.pneumoniae,andtheriseof
multiresistant strains makes it an excellent candidate for endolysin treatment.
Furthermore,fornon-systemicinfections,forexampletopicalapplicationsorinhalers
to reducenasopharyngealcolonization, regulatory requirementsare less stringent
thusbeinganattractivetargetforendolysinproof-of-concept.
1.4.2 Clostridiumdifficile
Clostridium difficile, which inhabits the gastrointestinal tract, was first isolated in
1935(HallandO’Toole1935)andhashaditswholegenomesequenced(Sebaihiaet
al. 2006). It is a Gram-positive anaerobe, has the ability to form spores, and has
shownitselftobeparticularlyadeptatresistingantibiotics.C.difficileinfections(CDI)
generallyoccurnosocomially,andareacommonproblemafterantibiotictreatment,
causingantibiotic-associateddiarrhoeaandpseudomembranouscolitis.
11%oftheC.difficilegenomehasbeenshowntoconsistofmobilegeneticelements,
including conjugative transposons and phage, conferring high mobility to the
genome.ThishighlymobilegenomehasenabledC.difficiletosurviveinthehighly
variablegastrointestinaltractenvironment,andhasbenefittedfromanichewhere
opportunitiestoexchangeDNAwithotherorganismsarefrequent.Itisbelievedthat
manyoftheantibioticresistancegenespresentinC.difficileareencodedbythese
mobilegeneticelements(Sebaihiaetal.2006).Resistanceandmultiresistancehas
been reported to: metronidazole; vancomycin; clindamycin; erythromycin;
tetracycline;moxifloxacin;rifampicin;andfusidicacid,viaanumberofmechanisms
includingribosomalmodification,antibioticeffluxanddruginactivation(Huangetal.
2009). Furthermore, theabilityofC.difficile to formsporeswhen it comesunder
environmentalstresscauseshugehealthcareissues.Theresultantsporesarelong-
livedandresistanttomanydisinfectantsandantibiotics,makingthemasourceof
infectioninmanyhospitalenvironments(Gerdingetal.2008).

54
Spore-formation, alongside huge potential for antibiotic resistance, frequently
resultsinaresidualpopulationofC.difficilesurvivinginthehumangutafteracourse
ofantibiotics.Thispopulationisthenabletorapidlycolonisethegutduetolackof
competition fromnatural gut flora,and the result isCDI. Further treatmentusing
antibiotics isthenrequiredtotreattherecurrent infectionwhichcanincreasethe
rateoffuturerecurrence.Faecaltransplanttherapy,amethodofrepopulatingthe
gutwiththenaturalmicrobiotatocompetewithC.difficileandpreventCDI,aswell
asprobioticshavebeenusedrelativelysuccessfullytocombatthisissue(Allenetal.
2014).EndolysintherapyasatreatmentforCDIisparticularlyattractiveasitwould
notdisturbthenaturalgutfloraagainstwhichC.difficileisusuallyunabletocompete,
whileconventionalantibioticsoftenexacerbatethesituation.
1.4.3 Propionibacteriumacnes
PropionibacteriumacnesisaGram-positivebacilliformandformsasignificantpartof
thehumancommensalmicrobiome.Itcanbefoundontheskin,particularlyinfatty
acidrichareassuchasthefaceandscalp,aswellasoninternalmembranessuchas
thelargeintestineandurinarytract(DessiniotiandKatsambas2017).P.acnesisbest
knownforitsroleinthecommonskinafflictionacnevulgaris,butisalsoassociated
with a wide range of inflammatory diseases and implant-associated infections
(Portilloetal.2013).Acnevulgarisisoneofthemostcommonglobalskindiseases,
affecting45millionpeopleintheUSAandaccountingforapproximately20%ofall
dermatologist appointments (Bhatia et al. 2004). Currently treatments include
topical and systemic antibiotics, as well as chemical treatments such as benzyl
peroxide. However, growing antibiotic resistance and the sometimes severe side
effectsofchemicaltherapiescallforanewsolution.
Duetotheprevalenceandunsightlynatureofacnevulgaris,P.acneshasbeenwidely
treated with antibiotics for many years, and cross-resistance to clindamycin and
erythromycinwas first reported in1979.Sincethen, resistancetooxytetracycline,
doxycycline, andminocycline have also been reportedworld-wide (Dessinioti and
Katsambas2017).While rarely life-threatening in the formofacnevulgaris,other
less-commonP.acnesinfectionsbenefitfromresistancecommonlyacquiredwhen

55
treatingacne,forexample,implant-associatedinfectionsareaseriousthreat(Portillo
etal.2013).ItisthereforehighlydesirabletoreducetheselectivepressureuponP.
acnesduringthetreatmentofacne.
Asatopicalinfectionwithahighprevalenceofantibioticresistanceandlargeafflicted
population,acnevulgaris,andmorepreciselyP.acnes,makesasuitabletargetfor
endolysintherapy.Furthermore,thetargetedkillingofP.acnesintheaffectedregion
ispreferabletoeliminationofthebacteriumacrossthebody,asP.acneshasbeen
demonstratedtobeanonspecificimmunestimulant,somaybebeneficialatsome
level,althoughpreciselyhowthismechanismworksisnotyetunderstood(Bhatiaet
al.2004).
1.5 Summary,aimsandobjectives
The importanceofantibiotics tomodernsocietycannotbeoverstated.Theyhave
wideranginguses,fromenablinginvasivesurgeries,tolargescalefoodproduction.
Forthepast70yearswehavebecomeincreasinglydependentuponthem,butthe
efficacyoftheolddrugsisdwindlingandnewdrugsarebecominghardertofind.
A number of potential alternative antibiotics exists, each with strengths and
weaknesses.Fromreviewing the literature, ithasbecomeevident thatour future
antibiotic use will require a different strategy to that which we have become
accustomed.Wewillnotbeabletorelyonahandfulofbroad-spectrumantibiotics,
butarelikelytorequireavarietyofantimicrobialproductstobestsuittheproblem
inhand.
Oneof thepossible toolsagainstbacteria thatwewillbeable to callupon is the
bacteriophageendolysins.Thequestionremainsofhowtoproducetheminacost-
effectiveandsafemanner.RecombinantexpressionintheC.reinhardtiichloroplast
provides a potential solution to this problem: featuring GRAS status; lack of
glycosylation; quick and precise genetic transformation; and ability to scale to
productionrapidly.However,lowrecombinantproteinyieldshavesofarheldback
thisplatform.Threepathogens,S.pneumoniae,C.difficile,andP.acneshavebeen
selectedassuitabletargetsforendolysintherapy.

56
Thisthesissetsouttoaddresstheissuesdiscussedabove,withtheaimsofadvancing
boththeuseoftheC.reinhardtiichloroplastasarecombinantproteinplatform,and
thepotentialforendolysintherapy,usingthefollowingobjectives:
1. To improve the level of recombinant endolysin accumulation in the C.
reinhardtiichloroplast.
2. To improve the activity of endolysins produced in the C. reinhardtii
chloroplastagainstS.pneumoniae.
3. Todemonstratethesynthesisandactivityoftwoendolysinsneverpreviously
expressedintheC.reinhardtiichloroplast.

57
Chapter2 MaterialsandMethods
2.1 Strains,media,cultureconditionsandcelldensityquantification
2.1.1 Chlamydomonasreinhardtii
2.1.1.1 Strains
StrainsofC.reinhardtiiinthisthesisarenamedinasimilarmannertopreviouswork
(Taunt2013;Stoffels2015),andtheconventionisillustratedinFigure2.1.
Figure2.1AnamingconventionmodifiedfromTaunt(2013)andinlinewithpreviousworkby Stoffels (2015). This convention is used throughout this thesis, and any minormodificationsarenotedinthefollowingsections.
STRAIN DESCRIPTION REFERENCE
TN72 A recipient strain based upon the cell wall-less strain cw15 (mt+). Partial deletion of psbH gene via aadA cassette insertion. PSII-deficient mutant. Spectinomycin resistant.
Ninlayarn (2012)
TN72_SR_Control TN72 transformed with pSRSapI expression vector, containing no GOI, psbH rescued.
Young & Purton (2014)
TN72_SR_cpl1_c-HA
Thecellularbackground
Anabbreviationoftheexpressionvectorused.pSRSapI (SR),pASapI (A),
pWUCA2(WUCA)
Theexpressedgene
Modificationsandmutationsmadetothe
expressedgene
Epitopetag

58
TN72_ SR_cpl1-HA TN72 transformed with pSRSapI containing HA-tagged cpl1, psbH rescued.
Taunt (2013)
TN72_ SR_cpl1_CPO1-HA TN72 transformed with pSRSapI containing HA-tagged cpl1 with a single codon change, psbH rescued.
This study
TN72_ SR_cpl1_CPO2-HA TN72 transformed with pSRSapI containing HA-tagged cpl1 with a single codon change, psbH rescued.
This study
TN72_ SR_cpl1_W202A-HA TN72 transformed with pSRSapI containing HA-tagged cpl1 with tryptophan-202 mutated to an alanine, psbH rescued.
This study
TN72_ SR_cpl1_W209A-HA TN72 transformed with pSRSapI containing HA-tagged cpl1 with tryptophan-209 mutated to an alanine, psbH rescued.
This study
TN72_ SR_cpl1_W230A-HA TN72 transformed with pSRSapI containing HA-tagged cpl1 with tryptophan-230 mutated to an alanine, psbH rescued.
This study
TN72_ SR_cpl1_W202A_W209A-HA
TN72 transformed with pSRSapI containing HA-tagged cpl1 with tryptophan-202 and -209 mutated to alanine residues, psbH rescued.
This study
TN72_ SR_cpl1_W223A_W230A-HA
TN72 transformed with pSRSapI containing HA-tagged cpl1 with tryptophan-223 and -230 mutated to alanine residues, psbH rescued.
This study
TN72_ SR_cpl1_W202A_W209A_-W223A_W230A-HA
TN72 transformed with pSRSapI containing HA-tagged cpl1 with tryptophan-202, -209, -223 and -230 mutated to alanine residues, psbH rescued.
This study
TN72_ SR_cpl1_dimer-HA TN72 transformed with pSRSapI containing HA-tagged cpl1 with mutations to putatively enable dimerization, psbH rescued.
This study
59
TN72_ A_cpl1_c-HA TN72 transformed with pASapI containing HA-tagged cpl1 encoded by a different DNA nucleotide sequence (cpl1_c), psbH rescued.
This study
TN72_SR_cpl1-HA_A_cpl1_c-HA
TN72 transformed with a plasmid containing both the HA-tagged cpl1 under the psaA exon 1 promoter, and the HA-tagged cpl1_c under the atpA promoter, psbH rescued
This study
TN72_ SR_cpl1-StrepII TN72 transformed with pSRSapI containing StrepII-tagged cpl1, psbH rescued
This study
TN72_SR_cpl1-StrepII_A_cpl1-HA
TN72 transformed with a plasmid containing both the StrepII-tagged cpl1 under the psaA exon 1 promoter, and the HA-tagged cpl1-c under the atpA promoter, psbH rescued
This study
TN72_SR_cd27l-HA TN72 transformed with pSRSapI containing HA-tagged cd27l, psbH rescued
This study
TN72_ SR_cd27l-StrepII TN72 transformed with pSRSapI containing StrepII-tagged cd27l, psbH rescued
This study
TN72_ WUCA2_Control TN72 transformed with pWUCA2 expression vector, containing no GOI, psbH rescued.
Young & Purton (2016)
TN72_ WUCA2_cph1-HA TN72 transformed with pWUCA2 containing HA-tagged cph1, psbH rescued
This study
TN72_A_gp20-HA TN72 transformed with pASapI containing HA-tagged gp20, psbH rescued
Taunt (2013)
TN72_ WUCA2_gp20N-HA TN72 transformed with pWUCA2 containing N-terminal HA-tagged gp20, psbH rescued
This study

60
2.1.1.2 Media
2.1.1.2.1 Tris-acetatephosphate(TAP)medium
NH4Cl 0.4 g/L
MgSO4•7H2O 0.1 g/L CaCl2•2H2O 0.05 g/L
K2HPO4 0.108 g/L KH2PO4 0.056 g/L
Tris 2.42 g/L Trace element stock solution 1 ml/L
Glacial acetic acid - to pH 7.0 Bacto agar (Optional) 2 g/L
2.1.1.2.2 Highsaltmedium(HSM)
NH4Cl 0.4 g/L
MgSO4 • 7H2O 0.1 g/L CaCl2 • 2H2O 0.05 g/L
K2HPO4 0.717 g/L KH2PO4 0.363 g/L
Trace element stock solution 1 ml/L HCl - to pH 6.9
Bacto agar (optional) 2 g/L
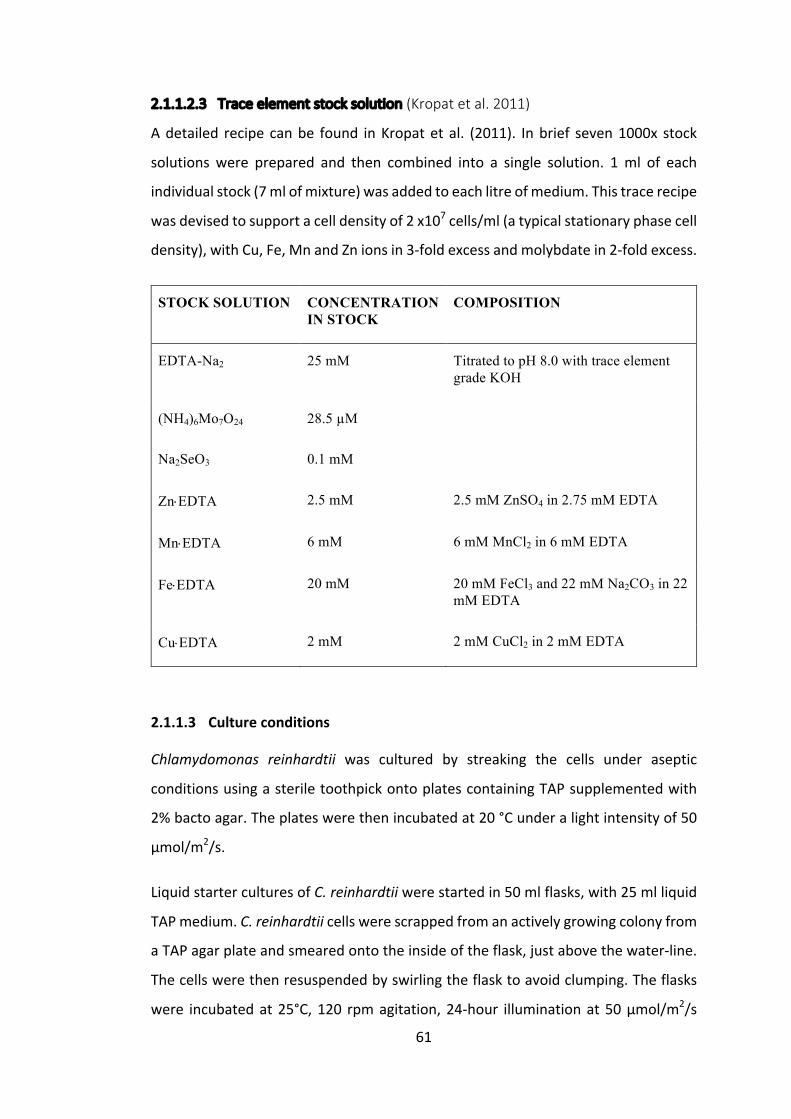
61
2.1.1.2.3 Traceelementstocksolution(Kropatetal.2011)
Adetailed recipe canbe found inKropatetal. (2011). Inbrief seven1000x stock
solutions were prepared and then combined into a single solution. 1ml of each
individualstock(7mlofmixture)wasaddedtoeachlitreofmedium.Thistracerecipe
wasdevisedtosupportacelldensityof2x107cells/ml(atypicalstationaryphasecell
density),withCu,Fe,MnandZnionsin3-foldexcessandmolybdatein2-foldexcess.
STOCK SOLUTION CONCENTRATION IN STOCK
COMPOSITION
EDTA-Na2 25 mM Titrated to pH 8.0 with trace element grade KOH
(NH4)6Mo7O24 28.5 µM
Na2SeO3 0.1 mM
Zn×EDTA 2.5 mM 2.5 mM ZnSO4 in 2.75 mM EDTA
Mn×EDTA 6 mM 6 mM MnCl2 in 6 mM EDTA
Fe×EDTA 20 mM 20 mM FeCl3 and 22 mM Na2CO3 in 22 mM EDTA
Cu×EDTA 2 mM 2 mM CuCl2 in 2 mM EDTA
2.1.1.3 Cultureconditions
Chlamydomonas reinhardtii was cultured by streaking the cells under aseptic
conditionsusingasteriletoothpickontoplatescontainingTAPsupplementedwith
2%bactoagar.Theplateswerethenincubatedat20°Cunderalightintensityof50
μmol/m2/s.
LiquidstarterculturesofC.reinhardtiiwerestartedin50mlflasks,with25mlliquid
TAPmedium.C.reinhardtiicellswerescrappedfromanactivelygrowingcolonyfrom
aTAPagarplateandsmearedontotheinsideoftheflask,justabovethewater-line.
Thecellswerethenresuspendedbyswirlingtheflasktoavoidclumping.Theflasks
were incubatedat25°C,120 rpmagitation,24-hour illuminationat50μmol/m2/s

62
lightintensity,tomid-logphase.A100-folddilutionwasthenperformedforlarger
sub-cultures.
2.1.1.4 Celldensityquantification
Cellcountingwasperformedusingahaemocytometer.1mlsamplewasextracted
fromaflaskand10μltinctureofiodine(19.7mMiodinein95%(v/v)ethanol)was
addedtoimmobilisethecells.Thenumberofcellsontwogridswasthencountedby
eyeat400xmagnification,themeantaken,andmultipliedby10,000toprovidean
estimationofthenumberofcells/mlofculturevolume.
Alternatively,theopticaldensityoftheculturewasmeasuredusingaUnicamUV/Vis
Spectrometerat750nm,withapath-lengthof1cm.
2.1.1.5 Storage
InoculatedTAPplateswerestoredat20°C,underdimlightconditions(5μmol/m2/s)
forupto5weeksbeforebeingre-streakedonfreshplates.
CryopreservationofC.reinhardtiistrainswasbaseduponaprotocolfromCrutchfield
etal.(1999).C.reinhardtiistrainswerere-streakedonTAPagarplatesevery3days
foraweektoensureactivegrowthstates.Thecellswerethenresuspendedin10ml
TAP-Sorbitol (1%),with the sorbitol acting as an osmoprotector for the cell-wall
deficientstrains,toaconcentrationofapproximately107cells/ml.Thesuspension
wasgentlyshakenat120rpmtoensurecelldissociationfor30minutes,andthen
dilutedto6x106cells/ml.Thecellsuspensionwasthengentlymixedina1:1ratio
withTAPcontaining10%methanol,underlowlightconditions.2mlaliquotsofthe
finalcellmixturewerethencooledatanapproximaterateof-1°C/minto-80°Cfor
90minutes,beforebeingstoredpermanentlyinaDewarcontainingliquidnitrogen.

63
2.1.2 Escherichiacoli
2.1.2.1 Strains
STRAIN DESCRIPTION REFERENCE
DH5α fhuA2 Δ(argF-lacZ)U169 phoA glnV44 Φ80 Δ(lacZ)M15 gyrA96 recA1 relA1 endA1 thi-1 hsdR17
Hanahan (1983)
DH5α_ProEX_cpl1_dimer-HA-StrepII
Carrying pProEX HTb containing HA-tagged cpl1 with mutations to induce dimerization
This study
DH5α_ProEX_gp20-HA Carrying pProEX HTb containing gp20 tagged on the C-terminus with an HA tag
This study
2.1.2.2 Media
2.1.2.2.1 Lysogenicbroth(LB)(Bertani1951)
Bacto-tryptone 10 g/L Yeast Extract 5 g/L
Sodium chloride 10 g/L Bacto agar (optional) 15 g/L
Where necessary, ampicillin was added to a final concentration of 100 μg/ml.
Ampicillinstockswerecreatedat100mg/mlinH2Oandstoredinaliquotsat-20°C.
2.1.2.3 Cultureconditions
E.coliwasculturedeitheronsolidLBagarplatesat37°C,orinliquidLBat37°Cwith
constantshaking.

64
2.1.2.4 Storage
25%(v/v)glycerolstockswerepreparedforlongtermstorageofE.colibymixing
500μlofstationaryphasebacterialculturewith500μl50%(v/v)sterileglycerol.The
aliquotswerethensnapfrozeninliquidnitrogenandstoredat–80�.
2.1.3 Streptococcuspneumoniae
2.1.3.1 Strains
STRAIN DESCRIPTION REFERENCE
D39 Serotype 2, Mouse-passaged clinical isolate
McDaniel et al. (1987)
AL2 D39 LytA ̅ . Defined autolysin-deficient lytA mutant of D39; Emr
Berry et al. (1989)
2.1.3.2 Media
2.1.3.2.1 TrypticaseSoyyeast(TSY)extractmedium
Trypticase soy broth 30 g/L Yeast Extract 3 g/L
2.1.3.2.2 Columbiabloodagarplates(SIGMA)
Agar 15 g/L Special nutrient substrate 23 g/L
Sodium chloride 5 g/L Starch 1 g/L
2.1.3.3 Cultureconditions
S.pneumoniaewereculturedinliquidcultureinTSYmediumat35�inananaerobic
CO2richenvironment,usingananaerobic jar fromOxoidandOxoidAGCCO2Gen
Compactgaspacks.S.pneumoniaeculturesweregrownfor16hinTrypticaseSoy
yeastextractmedium,sub-culturedthenextmorningandgrowntomid-logphase.
InoculatedColumbiabloodagarplatesweresealedandincubatedat35°Cfor18h.

65
2.1.3.4 Celldensityquantification
Opticaldensitywasmeasuredin96-wellplatesusingaFLUOstarOmegaplatereader
(BMGLabtech)at595nm.
Serialdilutionfollowedbyacolonyformingunitassaywerealsousedtoestimatecell
density.
2.1.3.5 Storage
Aliquotsofbacterialcultureweresnapfrozeninliquidnitrogenandstoredat–80°C.
2.1.4 Clostridiumdifficile
2.1.4.1 Strains
STRAIN DESCRIPTION REFERENCE
NCTC 11204 Isolated from human neonatal meconium
(Hafiz and Oakley 1976)
2.1.4.2 Media
2.1.4.2.1 RobertsonsCookedMeatMedium(SGL,Corby)
Heart muscle 454.0 g/L
Peptone 10.0 g/L
`Lab-Lemco’ powder 10.0 g/L
Sodium chloride 5.0 g/L
Glucose 2.0 g/L
pH 7.2 ± 0.2 @ 25°C

66
2.1.4.2.2 LiquidBrainHeartInfusion+complements(Mayeretal.2008)
Brain infusion solids 12.5 g/L Beef heart infusion solids 5.0 g/L
Proteose peptone 10.0 g/L Glucose 2.0 g/L
Sodium chloride 5.0 g/L Disodium phosphate 2.5 g/L
pH 7.4 ± 0.2 @ 25°C Complements:
Vitamin K 50 mg/L Hemin 5 mg/L
Resaurin 1 mg/L L-cysteine 0.5 g/L
2.1.4.3 Cultureconditions
The C. difficile NCTC 11204 spore stock was stored in Robertson’s cooked meat
medium(SGL,Corby).Thestockwasthengrownin25mlofliquidBrainHeartInfusion
(BHI)+complements(Mayeretal.2008).After16hofgrowthat37°C,200-500μlofC.difficileculturewasusedtoinoculatefreshbrothandgrowntoanOD595nmof1-1.4
(latelogphase).
2.1.5 Propionibacteriumacnes
2.1.5.1 Strains
STRAIN DESCRIPTION REFERENCE
ATCC 6919 Type strain. Strain Designations: NCTC 737 [VPI 0389]
From Laura Stoffels
2.1.5.2 Media
2.1.5.2.1 LiquidBrainHeartInfusion
See2.1.4.2.2,withoutcomplements.

67
2.1.5.2.2 Columbiabloodagarplates(SIGMA)
See2.1.3.2.2.
2.1.5.3 Cultureconditions
P.acneswasgrownat37°Cfor4-5daysinliquidheartbraininfusionoronblood
agarplatesinananaerobicCO2richenvironment,usingananaerobicjarfromOxoid
andOxoidAGCCO2GenCompactgaspacks.
2.1.5.4 Storage
Aliquotsofbacterialsuspensionweremixedinequalvolumeswith50%(v/v)sterile
glycerolandsnapfrozeninliquidnitrogenbeforebeingstoredat–80°C.
2.2 Plasmids
PLASMID DESCRIPTION REFERENCE
pSRSapI Based on the pUC8 backbone, containing the endogenous psaA promoter and 5’ UTR, with the rbcL terminator and 3’ UTR. Includes flanking regions around the expression cassette for homologous recombination with the chloroplast genome, and a functional psbH gene
Young and Purton (2014)
pSRSapI_Control pSRSapI expression vector, containing no gene of interest
Young & Purton (2015)
pSRSapI _cpl1-HA pSRSapI containing HA-tagged cpl1 Taunt (2013)
pSRSapI _cpl1_CPO1-HA pSRSapI containing HA-tagged cpl1 with a single codon change
This study
pSRSapI _cpl1_CPO2-HA pSRSapI containing HA-tagged cpl1 with a single codon change
This study
pSRSapI _cpl1_W202A-HA pSRSapI containing HA-tagged cpl1 with tryptophan-202 mutated to an alanine
This study

68
pSRSapI _cpl1_W209A-HA pSRSapI containing HA-tagged cpl1 with tryptophan-209 mutated to an alanine
This study
pSRSapI _cpl1_W230A-HA pSRSapI containing HA-tagged cpl1 with tryptophan-230 mutated to an alanine
This study
pSRSapI _cpl1_W202A_W209A-HA
pSRSapI containing HA-tagged cpl1 with tryptophan-202 and -209 mutated to alanines
This study
pSRSapI _cpl1_W223A_W230A-HA
pSRSapI containing HA-tagged cpl1 with tryptophan-223 and -230 mutated to alanines
This study
pSRSapI _cpl1_W202A_W209A_-W223A_W230A-HA
pSRSapI containing HA-tagged cpl1 with tryptophan-202, -209, -223 and -230 mutated to alanines
This study
pSRSapI _cpl1_dimer-HA pSRSapI containing HA-tagged cpl1 with mutations to induce dimerization
This study
pASapI Based on the pUC8 backbone, containing the endogenous atpA promoter and 5’ UTR, with the rbcL terminator and 3’ UTR. Includes flanking regions around the expression cassette for homologous recombination with the chloroplast genome, and a functional psbH gene
(Wannathong et al. 2016)
pASapI_cpl1_c-HA pASapI containing HA-tagged cpl1 with a different DNA nucleotide sequence (cpl1_c
This study
pSRSapI _cpl1-HA_A_cpl1_c-HA
pSRSapI containing both the HA-tagged cpl1 under the psaA exon 1 promoter, and the HA-tagged cpl1-c under the atpA promoter
This study
pSRSapI _cpl1-StrepII pSRSapI containing StrepII-tagged cpl1
This study
pSRSapI _cpl1-StrepII_A_cpl1-HA
pSRSapI containing both the StrepII-tagged cpl1 under the psaA exon 1 promoter, and the HA-tagged cpl1-c under the atpA promoter
This study

69
pSRSapI _cd27l-HA pSRSapI containing HA-tagged cd27l This study
pSRSapI _cd27l-StrepII pSRSapI containing StrepII-tagged cd27l
This study
pWUCA2 Based upon pSRSapI, with the introduction of a sythetic trnWUCA gene
Young & Purton (2015)
pWUCA2_Control pWUCA2 expression vector, containing no GOI
Young & Purton (2015)
pWUCA2_cph1-HA pWUCA2 containing HA-tagged cph1 This study
pWUCAC2_gp20N-HA pWUCA2 containing N-terminal HA-tagged gp20
This study
pProEX HTb An expression vector confering Ampicilin resistance and under trc promoter
Gift from J. Santini, Birkbeck College
pProEX_cpl1_dimer-HA-StrepII pProEX HTb containing cpl1 with mutations to induce dimerization. Tagged on the C-terminus with an HA-tag, followed by a StrepII tag
This study
pProEX_gp20-HA pProEX HTb containing gp20 tagged on the C-terminus with an HA tag
This study
2.3 Molecularbiology
2.3.1 Buffers
2.3.1.1 Sodiumphosphate(Na-Pi)buffer
NaH2PO4 59.99 g/L
Adjust pH to 7.4 with NaOH
2.3.1.2 Tris-HCl
Tris 121.14 g/L Adjust pH to 8.0 (unless otherwise stated) with HCl

70
2.3.2 Genedesignandsynthesis
GenesequenceswereacquiredfromGenbank(www.ncbi.nlm.nih.gov/genbank)and
verifiedwithBlastn. The sequence for an epitope tagwas addedbefore the stop
codon,unlessotherwiseindicated.
EPITOPE TAG AMINO ACID SEQUENCE
NUCLEOTIDE SEQUENCE
HA tag YPYDVPDYA
5' TAC CCA TAC GAT GTT CCA GAT TAC GCT 3'
StrepII tag WSHPQFEK 5’ TGG TCT CAC CCA CAA TTC GAA AAA 3’
His tag HHHHHH 5’ CAT CAC CAT CAC CAT CAC 3’
ASapIandaSphIrestrictionsitewere includedatthe5’and3’endsofthegene,
respectively,forsubsequentcloningunlessotherwiseindicated.Thesequenceswere
thencodonoptimisedusingCodonUsageOptimizerBeta0.92(Kong2013)software
developedspecificallyforC.reinhardtiichloroplastgenesinthePurtonlabbyKhai
Kong.GeneswerethensynthesisedbyIntegratedDNATechnologies.
2.3.3 Polymerasechainreaction(PCR)
DNAamplification,whencarriedout invitro,wasperformedusingthePolymerase
ChainReaction(PCR).Thereactionwasgenerallycarriedoutin50μlvolumes,and
usingtheFermentasPhusionpolymerase.Mix:
5x HF Buffer 10 µl
Template DNA 50-100 ng Primers 10 pmol each
20mM mixed dNTPs 1 µl Phusion polymerase 0.5 µl
ddH2O 35.5 µl

71
PolymeraseChainReactioncyclingparameterswerevariedslightlydependingupon
theprecisenatureofthereactionbeingperformed:theannealingtemperaturewas
calculateddependinguponthemeltingtemperature(Tm)oftheprimersbeingused,
and the extension time was altered depending upon the length of the expected
amplicon, according to the Fermentas Phusion polymerase instructions. As an
example,foramplifyingtheentireCpl-1geneusingaTechneTCF3000Xthermocycler
thefollowingconditionswereused:
98°C 2 min
98°C 10 s
X 30 cycles 60°C 30 s
72°C 30 s
72°C 5 min
10°C Hold
2.3.4 PCRDNAPurification
Following PCR, if the product was not to be loaded onto an agarose gel, it was
“cleanedup”bytheQiagenPCRclean-upkit,asperthemanufacturer’sinstruction.
Theclean-upremovedunwantedprimers,bufferandpolymerasefromthereaction
mixture. It was also used following restriction digests to remove small (<50 bp)
fragmentsofDNA.
2.3.5 AgarosegelElectrophoresis
1%agarosegelswereused for gel electrophoresis. 10μgofDNA sampleand1x
loadingdyewererunat10voltspercentimetreuntiltheladderiswellseparated.
EitherThermoScientificGeneruler1kbladderorGenerulerMixladderwereusedto
confirmDNAlength,forexpectedlargeorsmallbandsrespectively.
2.3.6 DNAgelextraction
Followingagarosegelelectrophoresis,DNAbandsofinterestwereexcisedfromthe
gelusingasterilescalpelandplacedintoamicrocentrifugetubeofpredetermined

72
mass.TheexcisedsliceofagarosewastheweighedandextractedusingtheQIAquick
GelExtractionKitfromQiagen,followingthemanufacturer’sinstructions.
2.3.7 SitedirectedmutagenesisbyPCR
PrimersweresynthesisedbyEurofinsandusedinconjunctionwithPolymeraseChain
ReactiontochangearequiredDNAnucleotide.
Figure2.2describestheprocess.
Figure2.2Processforsitedirectedmutagenesis.TemplateDNAisinblackandprimersareinblue.The‘X’indicatesapointmutation.Primersa+bandc+dwereinitiallyuseinseparatereactions.Thenthetworeactionswerecombinedand10cyclesofannealingwasperformed.Finally,afurther25cyclesofextensionwithprimersaanddwereperformed.
a b
c d
a
d
TwoseparatePCRreactions(a+b,c+d)
Anneal
PCRextensionandamplification

73
2.3.8 DephosphorylationusingAntarcticPhosphatase
InordertopreventtheligationoflinearizedplasmidDNAafterrestrictiondigestion
withouttherequiredDNAinsertion,the5’phosphatewasremoved.For1pmolof
DNAends,5unitsofAntarcticphosphatasewereaddedin1XAntarcticphosphatase
reactionbuffer.Afterincubationat37°Cfor30minutes,thereactionwasstopped
byheatinactivationat80°Cfor2minutes.
2.3.9 Blunt-endDNAcloning
Blunt-endDNAcloningintothepositiveselectioncloningvectorpJET1.2/bluntwas
carried out using a Thermo Scientific CloneJET PCR Cloning Kit according to the
manufacturer’sinstructions.
2.3.10 DNARestrictionendonucleasedigestion
LinearizationofthepSRSapIexpressionvector,andexcisionoftheGeneofInterest
(GoI) frompJetwas carriedout as shown in the tablebelow, followedby1hour
incubationat37°C.
Tango Buffer 4 µl
LguI (SapI) 1.2 µl PaeI (Sph1) 1 µl
DNA 2 µl
ddH2O 31.8 µl
2.3.11 DNALigation
DNAinsertandvectorconcentrationswereadjustedsuchthatthereisa3:1molar
ratioandapproximately20ngofvector.Then2μlof10xT4DNAligasebuffer,1μl
ofT4ligase(NewEnglandBiolabs)andddH2Oupto20μltotalvolumewasadded.
Thismixturewasthenincubatedatroomtemperaturefor1hour.
2.3.12 CompetentE.colitransformation–Heatshock
Competent E. coli (DH5α) stocks, stored at -80°C, were thawed on ice. For
transformationusingligationmixes,thewhole10μlligationreactionmixwasadded

74
to 100 μl of E. coli stock in a 1.5 ml microcentrifuge tube. For intact plasmid
transformations,approximately10ngofDNAwassufficient.Anadditionaltubeof
competent E. coli was also included as a negative control. The tubes were then
incubatedonicefor25minutes,followedbyaheatshockfor70sina42°Cwater
bath,followedbyanadditional2minonice.0.5mlofliquidLBwasaddedtoeach
tubebeforeincubationat37°Cfor1hourwithshaking.100μlfromeachtubewas
thenspreadontoLBamp100orLBkan100plates(dependingonantibioticresistance
geneintheplasmid)andincubatedat37°Covernight.
2.3.13 C.reinhardtiitransformation–glassbead
TN72isaphotosynthesis-deficient,cellwall-deficient,C.reinhardtiirecipientstrain
thatcanbegrownheterotrophicallyonmediacontainingacarbonsourcee.g.TAP
medium.Itisalsophotosensitivesoculturesgrowbetterwhenkeptinthedark.TN72
colonieswereregularlyrestreakedonfreshTAPagarplatesoverthreeweeksuntil
growinghealthilybeforestartinga400mlliquidTAPculture.
ThevectorpSRSapI (YoungandPurton2014)containsanexpressioncassette into
which a geneof interest can be inserted between SapI and SphI restriction sites.
pSRSapIcanthenrescuephotosyntheticactivity inTN72aftertransformationas it
carries thephotosystem II genepsbH,which isnon-functional inTN72.Successful
transformantscanthenbeselectedforonminimalmediaunderbrightlight,bytheir
abilitytogrowphototrophically.
Theprotocol followed forglassbeadtransformationwasbaseduponKindleetal.
(1991).TheTN72cellswereharvestedbycentrifugationat3,800xgfor15minutes,
oncetheculturedensityreached1-2x106cells/ml.Thepelletwasthenresuspended
inliquidHSMtogiveafinalconcentrationof2x108cells/ml.300μlofresuspended
cellsweretransferredtoanautoclavedglasstesttubecontainingapproximately300
mgof0.4mmdiameterpre-washedglassbeads.10μgofDNAwereaddedpertube
to11tubes,andonewasusedasanegativecontrol-withnoDNAadded.Thetubes
werevortexedfor15s,andthen3.5mlofwarmsoftHSMagarwereadded,quickly
swirledandpouredontoHSMplates.After20minutestheplateswereinvertedand

75
sealedwithParafilm,makingtwosmallpiercingsinthefilmwithasterilepipettetip.
Theplateswerethenplacedunderabrightlightfor21days.Allcoloniespresentafter
thistimeweretwicerestreakedontofreshHSMplates.
2.3.14 E.colicolonyPCR
E. coli colonies were picked using a sterile pipette tip and mixed well in a tube
containingTaqPolymerasePCRmixcontainingrelevantprimers.PCRwascarriedout
asdescribedpreviously,butwithanextrapreliminary2minutesofcelllysisat98°C.
Confirmedtransformantswerepickedagainfromtheplateandculturedforfurther
work.
2.3.15 E.coliplasmidisolation–CrudeandKit
Forplasmidpreparationsoflessthan12μgaQiagenMiniprepkitwasusedasper
the manufacturer’s instruction, while for larger preparations (<200 μg) a Qiagen
Midiprepkitwasused.
2.3.16 E.colitestdigest
Tango Buffer 1 µl
LguI (SapI) 0.2 µl PaeI (SphI) 0.2 µl
DNA sample 2 µl ddH2O 6.6 µl
A digestion was performed on plasmid mini/midi preparations as above and
incubatedat37°C for1hour.Gelelectrophoresiswasusedtoensurethecorrect
insertionhadbeenexcisedbydigestion.
2.3.17 C.reinhardtiigenomicDNAisolation
Thismethodallowscheckingforhomoplasmicity,thatis,toensurethatall~80copies
ofthechloroplastplastomehavebeensuccessfullytransformed.C.reinhardtiicells
werepickedusingasterilepipettetipandresuspendedin20μlofH2O.Then20μlof

76
100%ethanoland200μlof5%CHELEXwereaddedandmixed.Afterincubationfor
10minutesina98°Cheatblockandthencentrifugationat13,000gfor10minutes,
the supernatant was used in a PCR using the following primers (unless stated
otherwise):
Flank1: GTCATTGCGAAAATACTGGTGC
rbcL.Fn: CGGATGTAACTCAATCGGTAG
RY-psaR: CATGGATTTCTCCTTATAATAAC (ForpSRSapItransformants)
atpA.R: ACGTCCACAGGCGTCGT (ForpASapItransformants)
Successfulrecombination:primersFlank1andRY-psaRgavea1,135bpproduct,and
Flank1andatpa.Rgavea1,204bpproduct.Parentalsequence(e.g.TN72):primers
Flank1andrbcL.Fngavean850bpproduct.
2.3.18 DNASequencing
SourceBiosciences carriedoutDNA sequencinguponprovidingDNA samplesand
primersaccordingtotheirinstructions.
2.4 Proteinanalysis
2.4.1 Preparationoftotalproteinextracts
TheOD740ofC.reinhardtiiculturewasmeasured.Theculturewasthencentrifuged
(3,795g,20minutes)andthepelletresuspendedinavolumecorrespondingtothe
obtainedOD.Forexample,a10mlC.reinhardtiiculture,withanODof0.569,would
beresuspendedin0.569mlofbuffer(20mMNaPibuffer+Proteaseinhibitorunless
otherwisestated).Then10μlof4xproteinloadingdye(200mMTris-HClpH6.8,8%
(w/v)SDS,40%(v/v)glycerol,4%(v/v)β-mercaptoethanol,50mMEDTA,0.08%
(w/v) bromophenol blue) was added to 30 μl of the sample to a final 1x
concentration.Themixturewas thenboiled for3minat98°Candcentrifugedat11,000gfor2min.Theresultingsupernatantwasusedasthepreparedsample.

77
2.4.2 Sodiumdodecylsulphatepolyacrylamidegels(SDS-PAGE)�
TheBio-Radmini-PROTEANTetraSystemwasusedtocastSDS-PAGEgels.Thegels
consisted of a 15 % polyacrylamide resolving layer, topped with a 3.75 %
polyacrylamidestackinglayer,asdetailedbelow.
RESOLVING GEL (FOR 2 GELS)
Acrylamide/bisacrylamide (Sigma)�40% stock, acrylamide:bisacrylamide = 37:1
3.75 ml
Resolving buffer (8x stock)�0.25M Tris, 1.92M glycine, 1% SDS, pH8.3
1.25 ml
10 % SDS 0.1 ml ddH2O 4 ml
10 % ammonium persulphate 0.375 ml
Tetramethylethlenediamine 3.75 μl
STACKING GEL (FOR 2 GELS)
Acrylamide/bisacrylamide (Sigma)�40 % stock, acrylamide:bisacrylamide = 37:1
0.47 ml
Stacking buffer (4x stock) 0.5M Tris-HCl (pH 6.8)
1.25 ml
10 % SDS 0.05 ml
ddH2O 3 ml 10 % ammonium persulphate 0.25 ml
Tetramethylethlenediamine 3.75 μl
RESERVOIR BUFFER (10X STOCK)
Tris 0.25 M Glycine 1.92 M SDS 1 %
HCl To pH 8.3

78
Thepreparedsample(20μlfor10-wellgelor12μlfor15-wellgel)and5μlofproteinladder(ColorPrestainedProteinStandard,BroadRange(11-245kDa),(NewEngland
Biolabs))wereloadedontothegelandruninreservoirbufferfor120minutesat120
V.
2.4.3 Proteinpreparationfordotblot
TheC. reinhardtii culturewas resuspended as for the SDS-PAGEpreparation. The
freeze-thawmethodwasthenusedtomechanicallybreakthecells(3xfreezingin
liquidnitrogen,thawingat35°C).Thenthesampleswerecentrifuged(16,200g,2
min) and 2 μl of supernatant was pipetted directly onto the Hybond-ECLnitrocellulosemembranes(GEHealthcare).Themembranewasallowedtodryfor10
minandthenimmuno-detectionwasperformedasbelow.
2.4.4 CoomassieBrilliantBlueRstaining
TheSDSgelwas incubated inCoomassieBrilliantBlueR solution for1hour,with
gentle agitation, followed bywashing in destaining solution overnight. Gelswere
photographedusingtheLiCorOdyssey®CLximagingsystem.
CoomassieBlueSolution:
Coomassie Brilllant Blue R250 2.5 g/L
Acetic acid 100 ml/L Methanol 450 ml/L
ddH2O 450 ml/L
DestainingSolution:
Acetic acid 100 ml/L
Methanol 450 ml/L
ddH2O 450 ml/L

79
2.4.5 Westernblotanalysis
Semi-dry electrophoresiswas used to transfer separatedproteins fromSDS-PAGE
ontoHybond-ECLnitrocellulosemembranes(GEHealthcare).Initiallythegel,3MM
WhatmanpaperandnitrocelluloseweresoakedinTowbinbuffer(25mMTris,192
mMglycineand20%(v/v)methanol)atroomtemperaturefor10minutes.Atransfer
stackwasbuilt, consisting (inorder fromtop tobottom)of4 sheetsofWhatman
paper,SDS-PAGEgel,nitrocellulosemembrane,4moresheetsofWhatmanpaper.
ThiswasthenplacedintoaBio-RadTrans-BlotSDsemi-dryelectrophoretictransfer
system,andallairbubbleswererolledoutandexcessbufferremoved.Transferwas
carriedoutaccordingtothemanufacturer’sinstructions,ataconstantvoltageof20
V(FisonsFEC570powerpack)for1hour.
2.4.6 Immuno-detection
2.4.6.1.1 Antibodies
ANTIBODY SOURCE DILUTION
Primary antibodies
Anti-HA (polyclonal, rabbit)
Sigma Aldrich, #H6908 1:2,000
Anti-rbcL (polyconal, rabbit)
Gift from J. Gray,
University of
Cambridge
1:20,000
Anti-StrepII (polyclonal, rabbit)
Abcam, #ab76949 1:2,000
Secondary antibodies
Goat anti-Rabbit IgG (H+L) Secondary Antibody, DyLight 488
ThermoFisher Scientific, #35552
1:20,000

80
2.4.6.1.2 Protocol
Aftertransfer,thenitrocellulosemembranewasblockedin3%(w/v)skimmedmilk
powderinTBS-T(20mMTrisbase,pHadjustedto7.4with5MHCl,137mMNaCl,
0.1%(v/v)Tween-20)for1houratroomtemperature,orovernightat4°C.Thiswas
followedbyprimaryantibodytreatmentin1.5%skimmedmilkpowderinTBS-Tfor
1houratroomtemperaturewhileshakingat60rpm.Themembranewasthenrinsed
twiceinTBS-T,followedbya10minwashinTBS-Tat60rpm.Thesecondaryantibody
treatment was then applied 1.5 % milk solution, but diluted according to the
manufacturer’s instructions.Detectionwasperformedusinga LiCorOdyssey®CLx
SystemandanalysedusingOdyssey®ImageStudioLiteVersion5.5.
2.4.7 Proteinquantificationassay
Inordertodeterminethetotalsolubleproteinconcentrationofsamples,aBradford
assay was performed in a 96 well plate. This was performed according to the
manufacturer’s instructions (SigmaAldrich).Bovineserumalbumin (BSA)dilutions
were used to generate a standard curve. Then various dilutions of the unknown
samplewerepreparedtofindonewithaconcentrationintherangeof0.1-1.4mg/ml.
250μlofBradfordreagentwasaddedtoeachwellcontainingtheBSAorunknownsampleandtheplatewas incubatedatroomtemperaturefor10minutes.Aplate
readerwas used tomeasure absorbance at 595 nm. The net absorbance vs. the
proteinconcentrationofeachstandardwereplotted,andthenbycomparingthenet
A595valuesofthesampleagainstthestandardcurveitispossibletodeterminethe
concentrationofproteinintheunknownsample.
2.5 C.reinhardtiiproteinpurification
2.5.1 Crudeextractpreparation
C. reinhardtii cell cultures were grown to an OD740nm of 2 – 3 and harvested by
centrifugationat5,000gfor20minutes.Thepelletedcellswerethenresuspended
in20mMNaPi-buffer(NaH2PO4,thepHwasadjustedto6.9withNaOH),orBinding
buffer(20mMNaH2PO4,280mMNaCl,6mMpotassiumchloride,pH7.4)ifbeing

81
usedforlatercolumnpurification,to50xconcentration.RochecOmplete,EDTA-free
proteaseinhibitorwasalsoadded.Thecellswereeitherbrokenthroughfree-thawing
(3cyclesoffreezinginliquidnitrogenandthawingina37°Cwaterbath),orviaglass
bead abrasion (1 g of 0.4 mm diameter glass beads was added per 10 ml of
concentratedcells,followedbyvigorousshakingat4°Cfor10minutes).Subsequently
the broken cell suspension was centrifuged at 21,000 g for 10 minutes and the
supernatantcollected.Thesupernatantisreferredtoascrudeproteinextract.
To further remove cell debris and insoluble proteins from the supernatant,
ultracentrifugationat100,000gfor1hourwasperformed,producingasupernatant
referredtoasultracentrifugedcrudeproteinextract.
2.5.2 Ammoniumsulphateprecipitation
Ultracentrifuged crude protein extracts were stirred on ice and powdered
ammonium sulphate was added until the desired percentage of saturation was
reached.After30minutesofstirringthesolutionwascentrifugedfor30minutesat
3,000g.Thepelletwascollectedforfurtheranalysisandtheprocesswasrepeated
tothesupernatanttocollectallthedesiredproteinfractions.
2.5.3 Anti-StrepIIpurificationcolumn
Ultracentrifugedcrudeproteinextracts,inbindingbuffer,werepassedthrougha1ml
StrepTrapHPHiTrap™column(GEHealthcare)at1mlperminuteusingaperistaltic
pump and eluted using 2.5 mM desthiobiotin in binding buffer, as per the
manufacturer’sinstructions.
2.6 E.coliproteinpreparation
Three different methods of protein preparation were used for E. coli-produced
recombinantproteins:
2.6.1 IPTGinduction
ForE.colitransformedwiththepProEXHTbplasmid,transgeneexpressionmustbe
inducedusingIsopropylβ-D-1-thiogalactopyranoside(IPTG).Anovernightcultureof

82
E.coliwasdiluted1:100andgrownin2mlLB+amp100for4hoursat37°C.After4
hours,1mlwassnapfrozeninliquidnitrogentoprovidethe“uninduced”control.1
mlofLB+amp100+1mMIPTGwasthenaddedtothecultureandincubatedat20°C
overnight, to provide an induced culture which was then analysed by western
blottingorusedforfurtherinvestigation.
2.6.2 BugBuster™(Novagen)
BugBusterProteinExtractionReagentwasusedasaccordingtothemanufacturer’s
instructions.Inbrief,E.colicellswereharvestedbycentrifugation(16,000gfor10
minutes)andresuspendedinBugBusterreagent(2.5mlofBugBusterforevery50ml
of bacterial culture). The suspension was incubated for 10 minutes at room
temperaturewithgentleshaking.Centrifugationat16,000gfor20minutesat4°C
removedtheinsolublefractionandthesupernatantwasusedforSDS-PAGEanalysis
orpurification.
2.6.3 HighPressureHomogenizer
E.coliwereharvestedbycentrifugation(16,000gfor10minutes)andresuspended
in1/5thculturevolumeofNaPibuffer.Apre-equilibratedhighpressurehomogenizer
(StanstedFluidPower)wasusedtolysethecellsat14psiandwashedwithbuffer
betweenruns.Theresultingcellextractwascentrifuged(16,000gfor30minutesat
4°C)toremovecelldebrisandtheresultingsupernatantwasfiltersterilised(0.22
μm)beforebeingusedforcolonyformingunitassaysorSDS-PAGEanalysis.
2.6.4 Sonication
Overnight cultures of E. coli were harvested by centrifugation (16,000 g for 10
minutes).Theresultingpelletwasresuspendedin1/5thculturevolumeof10mMTris-
HClbuffer(pH8.0)andplacedinapolystyrenemicrocentrifugetube.Sonicationwas
performedusingaQ700cup-hornSonicator(QSonica).Thebathwaspre-cooledto4
°C and sonication performed at 100% amplitude for a total of fourminutes (30
secondson,30secondsoff).Thelysedcellswerecentrifuged(16,000gfor30minutes

83
at4°C)andthepelletandsupernatantanalysedbySDS-PAGEorusedfor further
analysis.
2.6.5 Disulphidebondformationinvitro
Whiledisulphidebondformationinvitrowasunsuccessfulinthisstudy,thefollowing
method was used, adapted from Hyvonen (2002). Sonication was used to lyse
inducedE.colicellsresuspended in10mMTris-HClbuffer (pH8.0)asdescribed in
2.6.4,andtheresultingsupernatantwasusedforthisanalysis.Tocreateanoxidising
environment conducive to disulphide bond formation, glutathionewas used as a
redoxreagent,bothinitsoxidisedandreducedforms(GSSGandGSHrespectively).
Solutionsof10mMGSSGandGSHweremadefreshfrompowderedstocks(SIGMA).
25μlofE.colicellextractwascombinedwithGSSGandGSHinordertoachievea
rangeofratiosbetweenthetwo.Ingeneral,thefinalconcentrationofGSHwas2-5
mM,with10-foldlessoftheoxidisedform.Themixtureswereincubatedovernight
at20°CbeforebeingseparatedbySDS-PAGEandanalysedbywesternblotting.No
β-mercaptoethanolwasincludedintheloadingdyeandthemixturewasnotboiled
beforerunningonthegel.
2.7 Antibacterialactivityassays
2.7.1 Colonyformingunitassay
2.7.1.1 Streptococcuspneumoniae
S.pneumoniaeculturesweregrownasdescribedabove.100μlofculturewasmixed
with endolysin samples and incubated at 37 °C for 1 hour. The mixture was
subsequentlydilutedinten10-folddilutionsteps.20μlofdilutionnumbers6-10were
spotted, in triplicate, onto Columbia blood agar plates and incubated at 37 °C
overnight.Thenumberofcoloniesineachspotwerecountedbyeyeandthenumber
ofcolonyformingunitspermlwascalculated.
2.7.1.2 Propionibacteriumacnes
CulturesofP.acnesweregrownanaerobicallyasdescribedabove.Thecultureswere
centrifugedat5,000gfor20minutesandresuspendedin20mMNaPibuffer.50μl

84
of bacterial suspension and 50 μl of filter sterilized cell lysate or buffer were
incubatedat37°Cfor0,30,60,90,120or150minutes.10μlsamplesweretaken
after the incubation time from each combination and spotted in triplicate onto
Columbia blood agar plates. After anaerobic incubation at 37 °C for one week a
comparisonwasmadesubjectivelybetweengrowthasindividualcolonygrowthwas
notvisible.
2.7.2 Turbidityreductionassay
2.7.2.1 Streptococcuspneumoniae
S.pneumoniaecultureswereharvestedbycentrifugation(15min,5,500g)andthe
cellswereresuspendedin20mMNaPi-buffertoanOD600nmof~0.8.
The turbidity reductionassayswereperformed in96-wellmicrotitreplateswitha
finalassayvolumeof300μl.AnELx808microplatereader(BIO-TEKINSTRUMENTS
INC.)measuredtheOD595nmeveryminutefor200minutes,including2sofshaking
beforeeachmeasurement,at37°C.
Unless otherwise stated, 20μl of antimicrobial sample,with varying total soluble
proteinquantitieswereaddedto280μlofsuspensionofthetargetbacterium.Asa
positive lysis control for S. pneumoniae turbidity reduction assays, 20 μl of
deoxycholate was added. As a negative control, samples were prepared from
TN72_SR_Control.
2.7.2.2 Escherichiacoli
E.coliculturesweregrowninLBtostationaryphaseandpelletedbycentrifugation
(3,800g,5min). The followingprotocol todisrupt E. colimembranes isbasedon
Nakimbugweetal.(2006):
ChloroformcontainingTris-HClbufferwaspreparedbyadding30%(v/v)chloroform
toTri-HCl(50mM,pH7.0)andshakingfor30min,200rpm,at25°C.Thetopphase
ofthismixturewasthenusedtoresuspendtheE.colipelletinitsoriginalvolume.
Thiswasthenshakenfor45minat25°Candthencentrifugedagain(8000g,20min,
4°C).ThepelletwasthenwashedwithTris-HClandresuspendedinTris-HClbufferto

85
anOD600nmbetween0.7-0.8.100Uofeggwhite lysozymewasusedperwell asa
positivelysiscontrol.
TheassaywasthenperformedasdescribedforS.pneumoniaeabove.
2.7.2.3 Clostridiumdifficile
C.difficilecellswereharvestedbycentrifugation(2minat5,000g)andresuspended
inavolumeof20mMNaPibufferequivalenttotheirOD(e.g.1.2mlifanOD595nmof
1.2wasmeasured).10-50μgofC.reinhardtiiextractin30μlfinalvolumewasadded
to270μlofpreparedC.difficilecellsinbuffer.Positivecontrolswere100U/wellof
mutanolysin and 30 μl of CD27LNiNTA-purified protein (unknown concentration)
fromtheMayerlab.ALabsystemsBioscreenC(ThermoFisherScientific)measured
theODevery2minutesfor18h,at37°C,inaThermoFisher100-wellhoneycomb
plate.

86
Chapter3 ImprovingaccumulationlevelsofCpl-1inthechloroplast
ofChlamydomonasreinhardtii
3.1 Introduction
3.1.1 IntroductiontobacteriophagelysinCpl-1
ThebacteriophageCp1was first isolated in1981, from throat samplesofhealthy
children(Rondaetal.1981).Cp1infectsthebacteriumStreptococcuspneumoniae,
whichcausesarangeofgloballysignificantdiseases.S.pneumoniae isacausative
agentofpneumonia,meningitisandfebrilebacteraemia,causingatotalannualdeath
toll of 1.6 million people, mainly young children and the elderly in developing
countries(WHO2007).
In1987the39kDaproteinofCp1wascharacterisedasamuramidaseandnamed
Cpl-1 (Garcíaetal.1987).Thesamestudynoted thatcholinewas required in the
pneumococcalcellwallforCpl-1tobeactive,andin2003thecrystalstructureofCpl-
1wasdetermined,showingtwoactivecholinebindingsites(Hermosoetal.2003).
Cpl-1,displayedinFigure3.1,isa339-residuepolypeptidechainconsistingofanN-
terminalcatalyticdomainandaC-terminalcellwallbindingdomain(CBD),joinedby
a10residueacidlinker.
Cpl-1belongstotheglycosylhydrolase(GH)25family,basedupontheaminoacid
sequencesimilarityof itscatalyticdomain,and isbuiltofmoduleshomologousto
LytC,amajorS.pneumoniaeautolysin(Monterrosoetal.2008).Thecatalyticdomain
displaysacharacteristicpeptidoglycanbindingsiteandadeepholeofhighlynegative
electrostatic potential located at the C-terminal end of the catalytic barrel that
constitutestheactivesite.
TheCBDisknowntobindthecholinemoietyintheteichoicandlipoteichoicacidof
theS.pneumoniaecellwall(seeFigure1.13),withtwoidentifiedaccessiblecholine
bindingdomainsontheprotein.Theadditionoffreecholine,orthesubstitutionof
cholineforethanolamineintheS.pneumoniaecellwall,inhibitsCpl-1activity.The
CBDiscomposedoftwowell-definedregions,namedCIandCII,containingatotalof

87
sixhomologousrepeatsofabout20aminoacids.CIconsistsoffourrepeatsinaleft-
handed superhelical arrangement and includes both active choline binding sites,
whileCIIhastworepeatsarrangedasanantiparallelsix-strandedβ-sheet.
Figure3.1Cpl-1crystalstructure.N-terminalcatalyticdomain(green)islinkedviaaflexibleacidiclinker(orange)totheCIdomain(blue)andtheCIIdomain(pink).Theboundcholinemoleculesaredisplayedinsitu,inball-and-stickrepresentation.ImageproducedusingJmol(PDB1oba).
ProductionofCpl-1isattractiveduetoitsantimicrobialpotential.VincentFischetti
wasthefirsttoproposeusingCpl-1asanovelantibioticandhisgroupshowedits
efficacyinbacteremicmice(Jadoetal.2003).However,aspreviouslydiscussedin
1.1.2,theantibioticmarketisoneofnotoriouslylowmargins,andproducingbiologics
iscurrentlyanexpensiveundertaking.Thisisdueinnosmallparttotheexpensive
purificationprocess involvedinremovingantigensandendotoxinsassociatedwith
traditionalproteinproductionplatforms,suchasE.coli.GiventheGRASstatusofC.

88
reinhardtii,we speculate that purifying recombinant Cpl-1 from theC. reinhardtii
chloroplastmaynotberequiredtobeasrigorous,andthereforebecheaper.
3.1.2 Factorstoconsiderinproteinaccumulation
The relatively low levels of recombinant protein accumulation achieved in theC.
reinhardtiichloroplast,whencomparedtoexistingplatformssuchasE.coli,offeran
area for improvement. This chapter discusses two techniques investigated to
improveaccumulationoftheCpl-1proteinintheC.reinhardtiichloroplast.
Whenproteinaccumulationisconsidered,onemustbreaktheissuedownintotwo
key factors, protein synthesis and protein degradation, and investigate how the
balanceofthetwoisleadingtotheobservedaccumulation.
Figure3.2Proteinturnoveriscomposedofproteinsynthesis(transcriptionandtranslation)andproteindegradation.
TheliteraturehasdescribedthatanincreasedquantityofmRNA,i.e.byincreasing
levels of transcription, does not appear to improve the final levels of protein
accumulation(Coragliottietal.2011).GiventhepolyploidnatureoftheC.reinhardtii
chloroplast and the strength of promoters from chloroplast genes encoding
photosyntheticcomponents,onecanimaginethattherateofsynthesisoftransgene
transcriptsislikelytobehigh(Hosleretal.1989;Blowersetal.1990).Thesamestudy
pointstotranslation,andinparticularribosomebindingandtranslationelongation,
as likely limiting factors.WorkbyTaunt (2013) indicates thatCpl-1degradation is
minimalintheC.reinhardtiichloroplast.
Thischapter,therefore,investigatestwopotentialmolecularmethodsforimproving
proteinaccumulation.Firstly,constructscontainingtwosimilargenesunderdifferent
Transcription Translation Degradation
DNA mRNA Activeprotein Aminoacids

89
promoterswereinsertedintothechloroplastinordertopotentiallyovercomethe
process of Control by Epistasy of Synthesis (Choquet et al. 1998). Secondly,
continuing upon previous work by Taunt (2013), codon-pair optimization is
investigatedinordertoimprovetranslation.
ExperimentsandcalculationsbyTaunt(2013)estimatedthatamid-logcultureofC.
reinhardtiicanaccumulateCpl-1toapproximately8mgL-1,or17mgg-1dryweight.
A comparable endolysin, Pal, produced by Stoffels (2015) achieved slightly lower
levelsofaccumulation:5mgL-1or13mgg-1dryweight.Theseofferabenchmark
uponwhichweshouldaimtoimprove.However,inthisthesisdrycellweightwas
not considered, as generallywewere interested in relative improvements, rather
thanactualyieldsofprotein.However,movingforwardaclearunderstandingofdry
cell weights achieved and their reliable quantification will be important in
understandingthecommercialviabilityoftheplatform.
3.1.3 EpistasyofsynthesisintheC.reinhardtiichloroplast
IthasbeenreportedthatintheC.reinhardtiichloroplastthe5’UTRofpetA,which
encodestheapoproteinofcytochromef,mediatesitsautoregulationoftranslation
viainteractionwiththeC-terminaldomainoftheunassembledprotein(Choquetet
al.1998).ThisprocessiscalledControlbyEpistasyofSynthesis(CES)andeffectively
meansthatproteinshavetheabilitytocontroltherateoftranslationoftheirown
mRNA.Therefore,whenusinganendogenous5’UTRfortransgeneexpression,the
nativeassociatedproteinlevelsmaycontrolthelevelofthetransgeneproduct.
We hypothesize in this chapter that the CES process is limiting the level of
recombinant protein synthesis, due to the presence of associated endogenous
proteins.Byintroducingtwocopiesofcpl1andexpressingthemunderthecontrolof
different5’promoter/UTRsintheC.reinhardtiichloroplastweaimtoincreaseoverall
levelsofCpl-1accumulation.

90
3.1.4 CodonusageintheC.reinhardtiichloroplastgenome
Redundancyinthegeneticcoderesultsinmultiplecodonscodingforthesameamino
acid.Itiswellestablishedthatthereisadifferenceinpreferenceamongstorganisms
forwhichparticularcodoncodesforwhichaminoacid,andthisisknownascodon
bias.Byoptimizingnucleotidesequencessuchthatpreferredcodonsareused,itis
possibletoimprovegeneexpressioncomparedtoun-optimizedversions(Franklinet
al.2002).
Protein coding sequence analysis inEscherichia coli byGutman&Hatfield (1989)
revealedthatthereisalsoastrongbiasincodonpairs,inthattheydonotappearto
berandomlydistributed.This isbelievedtobeduetothevaryingcompatibilityof
adjacentaminoacyl-tRNAacceptorsattheAandP-sitesofaribosome.This,inturn,
ledtothesuggestionthatcodonpairscanbeoptimizedinasimilarwaytocodons,
andthusfurtherimprovegeneexpressionlevels.Indeed,Irwinetal.(1995)produced
evidencetosuggestthattheinfluencesofcodonpairchoicearemoreimportantthan
thechoiceofindividualcodonsinE.coli,exemplifiedbythefactthatACCandCUG
are frequently used codons, but ACC:CUG and CUG:ACC are translated at very
differentrates.
Previous work by Taunt (2013) recognised a similar, non-random distribution of
codonpairs intheC.reinhardtiichloroplastplastome.Onepair inparticularstood
outasappearingextremelynon-random,thephenylalanine:argininepair,UUU:CGU.
Under a random codon distribution, one would expect to observe this pair
approximately 28 times in the chloroplast plastome, but in fact it never occurs.
Interestingly, the UUU:CGU pair is present in the well expressed cpl1 gene. This
projectinvestigatestheimpactofthiscodonpairuponCpl-1expression.
3.2 Aimsandobjectives
1. Toidentifyandoptimizeamethodforcomparingproteinaccumulationlevels
betweendifferenttransgenicC.reinhardtiilines.
2. ToproducetransgeniclinesofC.reinhardtiiinwhichtwocopiesofcpl1are
introducedunderthecontrolofdifferent5’UTRsequences.

91
3. Toshowimprovedlevelsofproteinaccumulationinthenewtransgeniclines
comparedwithexistinglines.
4. Toproduce two transgenic linesofC. reinhardtiiwithdifferent codonpair
optimizationstrategies.
5. To compare protein accumulation levels of the two codon pair optimized
versions.
3.3 Resultsanddiscussion
3.3.1 Investigationintomeasuringproteinaccumulationlevels
The first hurdle to overcome when attempting to improve protein accumulation
levels in the C. reinhardtii chloroplast was to identify and optimize an accurate
methodforcomparingtheproteinaccumulationlevelsbetweentwocelllines.Given
thealmostimpossibletaskofstartingtwoormorecultureswithanidenticalnumber
ofcells,inthesamegrowthphase,andgrowingthemunderidenticalphysiological
conditions,itwasapparentthatasuitablenormalisationtechniquewasrequired.Of
particularconcernistheabilitytoestimatetheerrorintroducedateachstageofthe
proteinpreparationandquantificationprocess.
3.3.1.1 Quantifyingcellspermillilitreofculture
When comparing two ormore algal cultures, onemust be sure to inoculate the
culturewiththesamenumberofcells.Thisimmediatelyintroducesanopportunity
forerror,whichhereweaimtoquantify.Thereareessentiallytwopractisedmethods
to estimate the cell density of an algal culture – either direct counting using a
haemocytometerandmicroscope,orbyusingopticaldensity(OD)asaproxyforcell
number.Bothofthesemethodsareinvestigatedandthissectionexaminestheerror
associatedwitheachmethod.
Thefollowingsetofexperimentswerecarriedoutasfollows:threeflaskscontaining
400ml TAPmediumwere inoculatedwith equal starter volumes from a singleC.
reinhardtii starter culture. These were incubated and measured in the Algem
photobioreactorfor96hours.Afterthis,thethreeflasks(labelled:a,bandc)were

92
used to provide samples to test all of the subsequent techniques. Therefore, the
followingsetofexperimentswereperformedusingpreciselythesamealgalcultures,
enablingthemtobecompared.
Twoerrorswillbemeasuredwitheachtechnique.Firstly,theerrorbetweenidentical
samples–e.g.measuringthesamecuvettethreetimes.Thisgivesanindicationof
themachine/techniqueerror.Secondly,theerrorbetweenflasks,whichwillprovide
an understanding of the pipetting error, a combination of measuring out of the
mediumandinaddingthestarterculture.

93
3.3.1.1.1 AlgemPhotobioreactor
TheAlgemPhotobioreactorautomaticallymeasuresOpticalDensityat740nm,while
maintaining near identical physiological conditions in three separate flasks. By
startingthreeC.reinhardtiicultures,andcomparingtheirOD740over96hours,itis
possibletoseetheerrorintroduced.
Figure3.3ThreeflaskscontainingthesamevolumeofTAPmediumandthesamevolumeofstarterculture(400mlTAPmediumwith2mlofstarterculture)weregrownintheAlgemPhotobioreactorfor96hours.TheaverageOD740ofthethreeflaskswascalculatedandtheerrorbarsindicatethestandarddeviationbetweentheflasks.
InordertoestimatetheerrorintroducedbytheAlgemphotobioreactor,thestandard
deviation between every threemeasurements in flask Awas calculated. As each
measurementwastakenat30-minuteintervals,thenforthepurposesofestimation,
threeconsecutivemeasurementswillbeconsideredtobefromthesametimepoint.
Theaveragepercentageerrorcalculatedinthismannerwas1.99%.
0
0.5
1
1.5
2
2.5
3
0 20 40 60 80 100
OpticalDensity(740nm
)
Time(h)

94
Meanwhile, theaverageerrorbetweenthethreeseparatecultures,shownbythe
errorbarsinFigure3.3,was4.17%.
3.3.1.1.2 Opticaldensityerror
Threebiological replicatesofC.reinhardtiicultures,a,bandc,weresampledand
measuredthreetimeseachusingaUnicamUV/VisSpectrometer(ThermoElectron
Corporation,USA)at750nm(OD740):
Three different samples were then taken from the same flasks and their OD740
measuredinthesamemanner:
These results indicate that a Unicam UV/Vis Spectrometer (Thermo Electron
Corporation,USA)hasanaverageerrormarginof0.35%whenmeasuringtheexact
samesamplethreetimes.Takingdifferentsamplesfromthesameflask,atthesame
time-point,raisestheerrorto0.74%–potentiallyduetothedifferentsettlingofcells
and non-uniform distribution of cells through the culture. This is lower than the
Algemphotobioreactor,butherethetimepointsareidentical,whiletoaveragethe
Algemtheywere90minutesapart.It isexpectedthatthetrueerroroftheAlgem
photobioreactorissimilartothis,asitusesasimilartechnology.
Theaverageerrormeasuredbetweentheculturesis4.15%,virtuallyidenticaltothat
observedintheAlgemphotobioreactor.
Count Calculations1 2 3 St.Dev. %error
Culturea 252 322 267 36.86 4.38%b 223 281 220 34.39 4.75%c 248 275 220 27.50 3.70%
Av. 32.91 4.28%
OD750 Calculations1 2 3 St.dev. %error
Culturea 0.541 0.532 0.540 0.0040 0.25%b 0.479 0.499 0.480 0.0092 0.63%c 0.499 0.503 0.505 0.0025 0.17%
Av. 0.0052 0.35%
OD750 Calculations1 2 3 St.dev. %error
Culturea 0.532 0.569 0.538 0.0162 0.99%b 0.479 0.499 0.480 0.0092 0.63%c 0.499 0.503 0.505 0.0024 0.17%
Av. 0.0093 0.60%
Count Calculations1 2 3 St.Dev. %error
Culturea 252 322 267 36.86 4.38%b 223 281 220 34.39 4.75%c 248 275 220 27.50 3.70%
Av. 32.91 4.28%
OD750 Calculations1 2 3 St.dev. %error
Culturea 0.541 0.532 0.540 0.0040 0.25%b 0.479 0.499 0.480 0.0092 0.63%c 0.499 0.503 0.505 0.0025 0.17%
Av. 0.0052 0.35%
OD750 Calculations1 2 3 St.dev. %error
Culturea 0.532 0.569 0.538 0.0162 0.99%b 0.509 0.499 0.522 0.0094 0.62%c 0.503 0.521 0.499 0.0095 0.63%
Av. 0.0117 0.74%

95
3.3.1.1.3 Haemocytometererror
AhaemocytometercanbeusedtocountthenumberofC.reinhardtiicellsinacertain
volumeandthenthatnumbercanbeusedtoestimatetheoverallconcentrationof
cellsintheculture,asexplainedin2.1.1.4.
Inorderto investigatetheerror introducedwhencountingC.reinhardtiicells,the
samesamplesasaboveweredilutedinaratioof1:4withsterileTAPmedia,andthen
iodinetincturewasaddedtoimmobilizethecells.
Tocalculatethetotalcells/mlofculture,thecountednumberwasmultipliedbythe
dilutionfactorandthenby10,000asaccordingtothemanufacturer’sinstructions–
thisdoesnotaffectthepercentageerror.
Theaveragestandarddeviationwithinthethreecountsofthesamesamplesis1.65
x106cells/ml,representing~4.3%error.
Meanwhile,theaverageerrorcalculatedbetweentheculturesis8.16%,almosttwice
thatoftheAlgemphotobioreactorandUnicamSpectrometer.
3.3.1.1.4 Celldensityquantificationconclusions
Theseinvestigationsindicatethatthehaemocytometerintroducesthelargestdegree
oferrortocelldensityestimations,andtheUnicamUV/VisSpectrometerintroduces
theleast.Thisismostlikelytobeduetotheslightlyobjectivenatureofcountingcells
onagrid,todecidewhetherthecellisonoroffthegrid,andalsothehumanerror
whencountinglargenumbers.Itcouldalsobeduetotheunevendispersalofcells
acrossthehaemocytometer,orasettlingofcellsintheflaskbeforethesamplewas
Count Calculations1 2 3 St.Dev. %error
Culturea 252 322 267 36.86 4.38%b 223 281 220 34.39 4.75%c 248 275 220 27.50 3.70%
Av. 32.91 4.28%
OD750 Calculations1 2 3 St.dev. %error
Culturea 0.541 0.532 0.540 0.0040 0.25%b 0.479 0.499 0.480 0.0092 0.63%c 0.499 0.503 0.505 0.0025 0.17%
Av. 0.0052 0.35%
OD750 Calculations1 2 3 St.dev. %error
Culturea 0.532 0.569 0.538 0.0162 0.99%b 0.479 0.499 0.480 0.0092 0.63%c 0.499 0.503 0.505 0.0024 0.17%
Av. 0.0093 0.60%

96
taken.However,theAlgemphotobioreactorhasaleveloferrorataround2%,but
asmentionedearlier,this iscertainlyanoverestimationduetothefactthatthree
timepointsacross90minuteswereused,andwithaC.reinhardtiidoublingrateof
approximately5hours, this isasignificantperiodof time.Additionally, theAlgem
photobioreactorisabletoautomaticallymeasureuptofourculturessimultaneously
acrossanextendedperiodoftime.
Therefore,theUnicamUV/VisSpectrometerwillbeusedforcelldensityestimations
andcomparisonsfor infrequentandone-offmeasurements.Otherwise,theAlgem
photobioreactorwillbeusedwherepossible.
Themanualcountingmethodofthehaemocytometerstillhasavalueinofferinga
realnumberforcelldensity,butforthepurposesofcomparingtwoormorecultures,
anopticaldensityreadingissuperior.
3.3.1.2 Westernblotquantification
3.3.1.2.1 Anti-HAprimaryantibodyfordetectionandquantification
Toinvestigatetheerrorintroducedwhenquantifyingwesternblotsignals,thethree
separateculturesofTN72_SR_cpl1-HA,grownintheAlgemPhotobioreactor,were
preparedforwesternblotting.Equalvolumesfromeachculturewereloadedinthree
technical repeats onto an SDS gel and probed with anti-HA primary antibodies,
followedbyGoatanti-RabbitIgG(H+L)SecondaryAntibodyIRDye®800CW(referred
to henceforth as IRDye®). The resulting signal was quantified using the LiCor
Odyssey®CLxSystem.

97
Figure3.4ThewesternblotwasquantifiedusingOdyssey® ImageStudioLitev5.Thebarchart shows the raw data and percentage errors. The table shows the signal from eachreplicatewithineachcultureandthecorrespondingpercentageerror.
InFigure3.4averyhighpercentageerrorwasobservedbetweentechnicalreplicates,
despiteanequalvolumeofsamplebeingloadedontotheSDSgel.Itispossiblethat
differentquantitiesofproteinwereincludedineachreplicate,duetopipettingand
loadingerror.
3.3.1.2.2 Anti-RbcLprimaryantibodyasloadingcontrol
Inordertotesttheassertionthatpipettingandloadingerrorcausedthehighlevelof
error in Figure 3.4, the endogenous protein RbcL (which is the large subunit of
Ribulose-1,5-bisphosphatecarboxylase/oxygenase(RuBisCo),theproductoftherbcL
gene), was probed with an anti-RbcL primary antibody on the same blot and
CultureA CultureB CultureC
Replicate1 22800 59300 348002 50700 51900 274003 39400 35800 25500 Average
%Error 37.3% 24.5% 16.8% 26.2%
CultureA CultureB CultureC
4632 Cpl-1
0
10000
20000
30000
40000
50000
60000
70000
80000
CultureA CultureB CultureC
Signal(IRFluorescence)
(kDa)

98
quantifiedinthesamemanner.Ifloadingerrorisindeedtoblame,thenonewould
expecttoseecorrelatingvariationindetectionofthiscontrol.
Figure3.5ThewesternblotwasquantifiedusingOdyssey® ImageStudioLitev5.Thebarchartshowstherawdata.Thetableshowsthesignalfromeachreplicatewithineachcultureandthecorrespondingpercentageerror.
There appears to be little correlation between the RbcL andHA detection levels,
althoughsimilarlevelsoferrorareobservedbetweenreplicates.Thiscouldbedue
toanumberoffactors.Possiblytheblottingefficiencyvariedacrossthemembrane,
so proteins were unevenly blotted from the SDS gel onto the nitrocellulose
membrane.Itcouldalsobeduetounevenantibodydecorationoftheblot,suggesting
a longerblotting time isnecessary. Finally, thequantification techniqueusing the
CultureA CultureB CultureCHA RbcL HA RbcL HA RbcL
Rep.1 22800 20600 59300 22500 34800 156002 50700 19700 51900 24000 27400 49303 39400 23000 35800 18000 25500 17100 HAAv. RbcL Av.
%Error 37.3% 8.1% 24.5% 14.5% 16.8% 52.9% 26.2% 25.2%
CultureA CultureB CultureC
4632
58 RbcL
Cpl-1
0
10000
20000
30000
40000
50000
60000
70000
HA RbcL HA RbcL HA RbcL
CultureA CultureB CultureC
Signal(IRFluorescence)
(kDa)

99
ImageStudioLitesoftwarerequiresthedrawingofadiscreetboxaroundtheband,
whichrequiressomejudgementandintroduceserror.
3.3.1.2.3 Anti-PsbOantibodyasloadingcontrol
Tofurtherconfirmtheresultabove,thesameblotwasadditionalprobedwithaPsbO
primaryantibody,whichisexpectedtobindtoanotherphotosynthesisprotein:the
33 kDa subunit of the oxygen evolving system of photosystem II. As a highly
conservedprotein,theaimwastouseitsdetectionasaloadingcontrol.However,as
seeninFigure3.6,no33kDaproteincouldbedetected.
Figure3.6Furtherprobingwithananti-PsbOantibodyfailedtoprovideausefulbandfornormalization.
Failure todetectPsbO left thequestionofwhy suchhigh variationwasobserved
betweenwesternblot technical repeatsunanswered,buttheRbcLresultsuggests
thatusingasingleproteinasaloadingcontrolisunreliable.
3.3.1.2.4 TotalProteinStainasloadingcontrol
REVERT™TotalProteinStainisspecificallydesignedforwesternblotnormalization
and can be easily quantified using the LiCor Odyssey® CLx system, in a separate
channeltotheproteinofinterest.
Toinvestigatetheusabilityandaccuracyofthissystem,atestwasrunusingasample
ofTN72_SR_cpl1-HAinvariousdilutions,asshowninFigure3.7.
SampleA
46PsbO32
SampleB SampleC
(kDa)
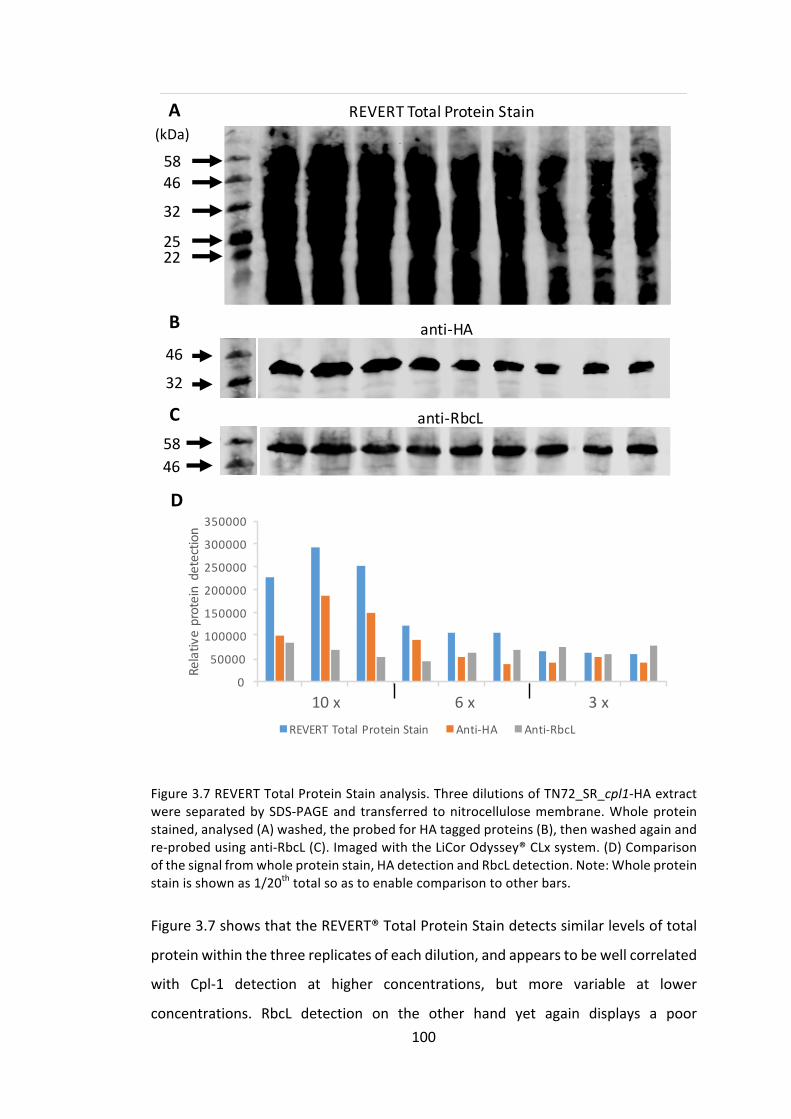
100
Figure3.7REVERTTotalProteinStainanalysis.ThreedilutionsofTN72_SR_cpl1-HAextractwereseparatedbySDS-PAGEand transferred tonitrocellulosemembrane.Wholeproteinstained,analysed(A)washed,theprobedforHAtaggedproteins(B),thenwashedagainandre-probedusinganti-RbcL(C).ImagedwiththeLiCorOdyssey®CLxsystem.(D)Comparisonofthesignalfromwholeproteinstain,HAdetectionandRbcLdetection.Note:Wholeproteinstainisshownas1/20thtotalsoastoenablecomparisontootherbars.
Figure3.7showsthattheREVERT®TotalProteinStaindetectssimilarlevelsoftotal
proteinwithinthethreereplicatesofeachdilution,andappearstobewellcorrelated
with Cpl-1 detection at higher concentrations, but more variable at lower
concentrations. RbcL detection on the other hand yet again displays a poor
5846
32
2522
32
A
C
B46
D
5846
0
50000
100000
150000
200000
250000
300000
350000
100% 100% 100% 66% 66% 66% 33% 33% 33%
Relativeproteindetection
REVERTTotalProteinStain Anti-HA Anti-RbcL
anti-HA
anti-RbcL
REVERTTotalProteinStain
10x 6 x 3x
(kDa)

101
correlation to anti-HA tagged protein detection. This is most likely due to the
interaction of the RbcL protein and anti-RbcL antibody – for example, the RbcL
proteinmayrequiremoreboilingtofullydenatureandenabletheanti-RbcLantibody
tobind,andwearethereforeseeingerraticbinding.
Overall,thisanalysissuggeststhatREVERT®TotalProteinStainisagoodmethodof
normalisingwesternblotsignalsprovidedthesignalisrelativelystrong.Therefore,a
tentimes(10x)concentrationofC.reinhardtiicultures,atagrowthstagewithan
OD740ofapproximately0.750usingtheUnicamUV/VisSpectrometer,willbeused
forproteinquantificationpurposesunlessotherwisespecified.
3.3.1.2.5 Usingaproteinstandardforquantification
Aproteinstandardenablesobservedproteinaccumulationlevelstobequantifiedby
comparingthemtoastandardofknownconcentration.Furthermore,by including
the same standard in all western blot analyses, one can normalize between
membranes,ifforexampletheyhavebeenblottedforslightlydifferentamountsof
time. A standard encoding two ormore epitopes will also enable comparison of
differentlytaggedproteins.
PreviousattemptsatquantifyingaccumulationofHA-taggedrecombinantproteinin
C.reinhardtiiinthePurtonlabhaveusedaMultiTagprotein(AbCam)thatincludes
anHAepitope,oracommerciallyavailableHA-taggedprotein (CalciumRegulated
HeatStableProtein1(CARHSP1))asproteinstandardsagainstwhichtheHA-tagged
proteins inC. reinhardtii cell extracts have been compared. However, the results
obtained in this manner have been unreliable due to several factors including
degradation of the protein standards, the unknown efficiency of blotting to
nitrocellulosemembrane and unknown accessibility of the HA tag to the anti-HA
antibodies.Furthermore,estimated levelsofaproteinobtained inthisway,when
expressedas‘percentageoftotalsolubleproteinincellextracts’werenotsupported
byCoomassieBrilliantBluestainedSDS-PAGEanalysisoftheextracts(Stoffels2015)
andappearedtooverestimatethequantityofaccumulatedproteininC.reinhardtii
extracts.

102
3.3.1.2.6 ProducingabespokeCpl-1proteinstandard
Inordertominimisetheproblemsstatedabovewithcommerciallyavailableprotein
standards,abespokeCpl-1proteinstandardwasproducedinE.coli.
A slightlymodified version of Cpl-1was used (cpl1_dimer), for compatibilitywith
otherprojects,see4.3.2,andbothHAandStrepIIepitopetagswereincludedonthe
C-terminus.ThegenewascodonoptimizedforE.coliandsynthesizedbyIntegrated
DNATechnologies.NcoIandNotIrestrictionsiteswereusedtoclonethegene in-
frameintotheIPTGinducibleexpressionvectorpProEX-HTb.Theresultingplasmid
was sequenced to confirm the correct insertion and used to transform DH5α
competentE.colicells.
TheresultingE.colistrainwasnamedDH5α_ProEX_cpl1_dimer-HA-StrepII,and its
recombinant protein product, purified using StrepTactin affinity to the StrepII
epitopetagandquantifiedusingaBradfordassay,would,therefore,produceamore
reliableproteinstandardforquantifyingCpl-1accumulationinC.reinhardtiithanthe
Multi-tagorCARHSP1standards.
IPTGinductionwasperformed(2.6.1)andcpl1_dimer-HA-StrepIIaccumulationwas
confirmedbySDS-PAGEandCoomassieBrilliantBlueRstaining(Figure3.8).
Figure3.8WholecellextractswereseparatedbySDS-PAGEandtotalproteinstainingwasperformedusingaCoomassieBrilliantBlueRstain.Note:despitethenameCpl1_dimer,thisisastandard39kDaCpl-1monomer(thename“dimer”refersto itsabilitytodimerize innon-denaturingconditions,asdiscussedin4.3.2).
The IPTG-induced DH5α_ProEX_cpl1_dimer-HA-StrepII cells were lysed using
BugBuster™ (2.6.2). The StrepII epitope tag on the Cpl1_dimer-HA-StrepII protein
x10
32
46
x5 x2 x1 x0.25
x10
x5 x2 x1
IPTGInducedxculturevolume
Uninducedxculturevolume
Cpl-1_dimer(39kDa)
(kDa)

103
was then used for protein enrichment via a commercially available StrepTactin
column(ThermoFisherScientific).ThepurificationfractionsareshowninFigure3.9.
Figure 3.9 The various fractions involved using the StrepTactin purification columnwereseparatedbySDS-PAGEandstainedwithCoomassieBrilliantBlueRstain.
CoomassieBrilliantBluestaininginFigure3.9indicatesthatotherproteinsasidefrom
Cpl-1arestillpresentintheelutedfractions,sotheCpl1-enrichedelutionwasthen
further purified and concentrated by ammonium sulphate precipitation – the
fractionsofwhichareshowninFigure3.10.
Figure3.10CoomassieBrilliantBlueRstainedSDS-PAGE.The20–40%ammoniumsulphatefractionwaschosenasthefractionwiththemostCpl-1comparedtoothervisibleproteins.
CellLysate
32
46
Flow
through
Wash1
Wash2
Elution1
Elution2
Elution3
Cpl1(39kDa)
55
Cpl-1(39kDa)
40-60%
20-40%
0-20%
46
32
23
Ammoniumsulphatepercentagesaturation
(kDa)
(kDa)
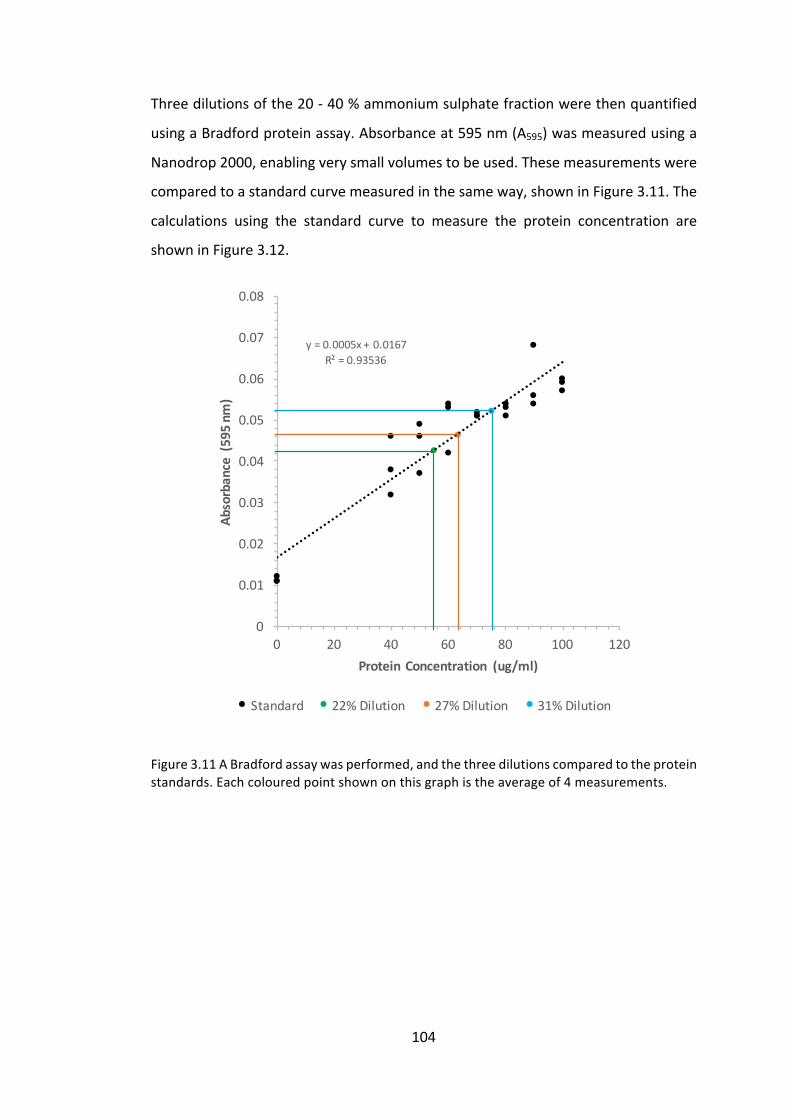
104
Threedilutionsofthe20-40%ammoniumsulphatefractionwerethenquantified
usingaBradfordproteinassay.Absorbanceat595nm(A595)wasmeasuredusinga
Nanodrop2000,enablingverysmallvolumestobeused.Thesemeasurementswere
comparedtoastandardcurvemeasuredinthesameway,showninFigure3.11.The
calculations using the standard curve to measure the protein concentration are
showninFigure3.12.
Figure3.11ABradfordassaywasperformed,andthethreedilutionscomparedtotheproteinstandards.Eachcolouredpointshownonthisgraphistheaverageof4measurements.
y=0.0005x+0.0167R²=0.93536
0
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
0 20 40 60 80 100 120
Absorbance(595n
m)
ProteinConcentration (ug/ml)
Standard 22%Dilution 27%Dilution 31%Dilution

105
Figure3.12BradfordAssaycalculations.TheequationwasderivedfromthelineofbestfitfromthegraphinFigure3.11.
ItwasinferredfromFigure3.12thatthestocksolutionofpurifiedcpl1_dimer-HA-
StrepIIhadaconcentrationof230±30μg/ml.
Thesynthesisofthisproteinstandardwillenablecomparisonofdifferentwestern
blot analyses aswell as compare theaccumulation levels of proteins taggedwith
differentepitopes,inthiscaseHAandStrepII.
3.3.2 Investigatingcontrolbyepistasyofsynthesisinproteinaccumulation
ThelowyieldsofrecombinantproteinaccumulationintheC.reinhardtiichloroplast
areamajorfactorimpedingitasaproductionplatform.Hereweattempttoimprove
proteinaccumulationlevelsofCpl-1byexpressingthecpl1geneunderthecontrolof
twodifferentpromoter/5’UTRs.
3.3.2.1 ExistingC.reinhardtiilines
ThissectionusestwoC.reinhardtiilinescreatedbyTaunt(2013)ascontrols.Oneis
anegativecontrolthatwillbeusedthroughoutthisthesis,namedTN72_SR_Control,
asshowninFigure3.13thisistherecipientstrainofC.reinhardtiitransformedwith
anemptypSRSapIplasmid.ThesecondC.reinhardtiilineisTN72_SR_cpl1-HAwhich
willbeusedasthe“benchmark”forthischapter-i.e.alinedisplayingamoderate
levelofCpl-1accumulationuponwhichweaimtoimprove.
A595AverageA595
St.Dev.A595
(y=0.0005x+0.0167) StockConc.Average
StockConc.St.Dev.
AverageProtein
StDev.Protein
Dilution1(22.3 %)
0.0500.0380.0430.039 0.0425 0.128 51.6 6.61 231.9 29.7
Dilution2(26.7 %)
0.0510.0530.0370.045 0.0465 0.155 59.6 9.21 223.2 34.5
Dilution3(31.2%)
0.0570.0530.0460.053 0.0523 0.088 71.1 6.22 228.3 20.0
Average: 227.8 28.1

106
3.3.2.1.1 C.reinhardtiilineTN72_SR_Control
Figure 3.13 TN72 transformedwith an empty pSRSapI plasmid. Unlike the original TN72recipient,thecontroliswildtypeforphotosynthesis,likethecpl1transformants,asthepsbHdeletionhasbeenrescued.
3.3.2.1.2 C.reinhardtiilineTN72_SR_cpl1-HA
Figure3.14TN72 transformedwith thepSRSapIexpressionplasmidcontainingHA-taggedcpl1.
3.3.2.1.3 Westernblotconfirmation
ToshowthatCpl-1accumulationoccursinTN72_SR_cpl1-HAandthatnoproteinof
a similar size is detected in TN72_SR_Control, whole cell extracts of each were
preparedandseparatedbySDS-PAGE.Ablotofthegelwasthenprobedwithananti-
HA primary antibody and an anti-RbcL primary antibody, followed by IRDye®
secondaryantibodies.TheLiCorOdyssey®CLxSystemwasthenusedfordetection.
The55kDaRbcLbandactsasanapproximateloadingcontrolinthisinstance,toshow
thatcellextractwaspresentinthenegativecontrollane.The39kDaCpl-1protein
canonlybeseenintheTN72_SR_cpl1-HAtransformedline.
trnE2 psbH
5’psaA 1 3’rbcL
TN72_SR_Controltransformant MluI
trnE2 psbH
5’psaA 1 3’rbcL
TN72_SR_cpl1-HAtransformant MluI
cpl1-HA

107
Figure3.15Westernblotanalysisofwholecellextracts.AColorPrestainedProteinStandard,BroadRange(NewEnglandBiolabs,Inc.)wasusedtoestimateproteinsize.
3.3.2.2 TheproductionofC.reinhardtiilineswithimprovedCpl-1accumulation
WehavehypothesisedthatCESlimitsthelevelofrecombinantproteinaccumulation,
duetotheuseofendogenous5’UTRsforrecombinantproteinexpressionintheC.
reinhardtiichloroplast.Byintroducingthegeneofinterestintothechloroplastinside
two different expression cassettes, we speculate that the level of protein
accumulationpercellcanbeimproved.Inthiscase,wewillusetheendogenouspsaA
exon15’UTRandtheatpA5’UTR,whichhavebothbeenshowntoachievemoderate
levelsofrecombinantproteinaccumulationintheC.reinhardtiichloroplast(Taunt
2013).
However,giventhatwewishtotransformC.reinhardtiiusingtheconvenientTN72
system for targeted insertion into the chloroplast, we have only one plastome
insertionsite;namely,theneutralsitebetweenpsbHandtrnE2.Thetwoexpression
cassettes,containingthesamegeneofinterest,will,therefore,needtobeadjacent
to one another in the transformation plasmid, and be integrated by a single
transformationeventintotheplastomebyhomologousrecombination.
Thegeneof interest in this instance iscpl1which is~1kb in length.Homologous
recombination between DNA direct repeats in the C. reinhardtii chloroplast is
efficient–indeed,itisthispropensitytorecombinewhichweexploitforthetargeted
insertionoftransgeneswhenperformingC.reinhardtiichloroplasttransformations.
TN72_SR_
Control
4632
58 RbcLCpl-1
TN72_SR_
cpl1-HA
(kDa)

108
However, two identical copies of cpl1 in close proximity to one another are
considered tobeat veryhigh riskofundergoing recombinationbetween the two
copies,resultinginaplastomerearrangementandlossofonecopyoftheGeneof
Interest(GOI)(Figure3.16).Muddetal.(2014)suggestthatdirectrepeatsof~600bp
ormoreresultinveryhighfrequenciesofhomologousrecombination.Additionally,
sucharecombinationeventwouldstillproducerecombinantprotein,somaygoun-
noticedifitoccursinatransformantlinesubsequenttomolecularanalysisconfirming
thetwo-GOIinsertion.
Figure3.16Twoexpressioncassettesinparallelorientationmighthomologouslyrecombineto leaveaviableexpressioncassette in the chloroplastplastome, thus still produceCpl-1protein.
However, as illustrated in Figure 3.17, by arranging the expression cassettes in
antiparalleldirections,recombinationeventswillmerelyresultinaswitchingofthe
promoterregionsandnolossofDNAwilloccur.
5’psaA 1 cpl1-HA 3’rbcL5’atpA 3’rbcLcpl1-HA
cpl1-HA 3’rbcL5’atpA
cpl1-HA

109
Figure3.17Whenarranged in antiparallel directions, the recombinationeventwouldnotresultinanylossofDNAfromthetransformedplastome.
Therefore,theantiparallelconfigurationwasusedwhentransformingC.reinhardtii
withtwoexpressioncassettesinasingleplasmid,inparticularwhenthetwogenes
are highly similar. Furthermore, Coragliotti et al. (2011) report no detectable
differenceinexpressionasaresultoftransgeneorientation.Infact,theC.reinhardtii
chloroplastcontainstwolargeinvertedrepeatregionswhichallowsimilar“flip-flop”
recombination of the entire plastome, and it is believed that the two possible
orientationsexistinapproximatelyequalproportion,withnoapparentadverseeffect
(Harris2009).
3.3.2.2.1 C.reinhardtiilineTN72_A_cpl1_c-HA
However,toreducethechanceofDNArecombinationeventsbetweenthedirectly
repeated cpl1 genes, one gene was differently codon optimized to reduce the
sequence similarity. Thedifferent versionofcpl1wasnamedcpl1_c,with the “c”
standingforcodonchanged.
3.3.2.2.1.1 Cpl-1_cgenedesign
The original genetic sequence for cpl1 as used by Taunt (2013), and present in
TN72_SR_cpl1-HA,wasuploadedtoCodonUsageOptimizerBeta0.92(Kong2013)
software and codonsweremanually edited to ones of similar “scores” for theC.
reinhardtii chloroplast. In total 148nucleotideswere changedoutof 1050 (14%)
withoutaffectingtheoverallscoreforcodonusage.SapIandSphIrestrictionsites
wereincludedforcloning.
5’psaA 1 cpl1-HA 3’rbcL5’atpA3’rbcL cpl1-HA
cpl1-HA 3’rbcL5’psaA 1 5’atpA3’rbcL cpl1-HA

110
3.3.2.2.1.2 ExpressioncassetteproductionandE.colitransformation
Thefull-lengthcpl1_cgenewassynthesizedby IntegratedDNATechnologies.SapI
andSphIrestrictionenzymeswereusedtoclonethegeneintothepASapIexpression
vectorin-frame,producingtheplasmidpASapI_cpl1_c-HA.DH5αcompetentE.coli
cells were transformed with the resulting plasmid and a plasmid midi-prep was
performedtoproducesuitablequantitiesofplasmidforglassbeadtransformation.
3.3.2.2.1.3 C.reinhardtiitransformation
ThepASapI_cpl1_c-HAplasmidwasusedtotransformrecipientC.reinhardtiistrain
TN72via theglassbeadmethodasdescribed in2.3.13.Figure3.18 illustrates the
resultingC.reinhardtiicellline,TN72_A_cpl1_c-HA
Figure3.18TN72recipientstrainsuccessfullytransformedtocarrycpl1_cundertheatpA5’UTRinthechloroplast,andwithpsbHfunctionalityrestored.
Theplastomesoftheresultingtransformantswereconfirmedtobehomoplasmicas
describedin2.3.17.Inthisinstance,thethreeprimersusedwereFlank1,atpa.Rand
psbH.R. Figure 3.19 shows the resulting PCR products separated by gel
electrophoresis.TN72wasusedasanegativecontrol.Asafinalstep,thesequence
integrity of the cassettewas confirmed by DNA sequencing of the PCR-amplified
plastomeregion,asin2.3.18.
trnE2 psbH
3’rbcL
TN72_A_cpl1_c-HAtransformant MluI
5’atpA cpl1_c-HA

111
Figure3.19PCRscreeningforsuccessfultransformationofTN72andreachingahomoplasmicstate.(A)Schematicshowingtheproductexpectedwitheachprimercombination.(B)Gelelectrophoresis of thePCRproducts confirms that the transformant is homoplasmic as itlacksthe350bpbandoftherecipientstrain.
3.3.2.2.1.4 Westernblotquantification
AwholecellextractofTN72_A_cpl1_c-HAwaspreparedandseparatedbySDS-PAGE.
Afterblottingtonitrocellulosemembraneasdescribedin2.4.5,themembranewas

112
probedwithanti-HAandanti-RbcLprimaryantibodies,followedbyIRDye®secondary
antibodiesandfluorescencewasobservedusingtheLiCorOdyssey®CLxsystem.
Figure3.20WesternblotanalysisshowsthatsimilarlevelsofCpl-1proteinaccumulationaredetectedinthetwoC.reinhardtiilines.RbcLisanapproximateloadingcontroltoshowthatsimilar quantities of cell lysate were loaded onto the gel and a negative control,TN72_SR_Controlisalsoincluded.
TheresultsshowsimilarlevelsofCpl-1proteinaccumulation,andtheRbcLloading
controlconfirmsthatsimilarquantitiesofcelllysatewereused.
TN72_SR_
cpl1-HA
TN72_A
_cpl1_c-HA
Cpl-1(39kDa)
TN72_SR_
Control
RbcL58
48
32
0
5000
10000
15000
20000
25000
TN72_A_cpl1_c-HA TN72_SR_Control TN72_SR_cpl1-HA
Signal(IRFluoresence)
Anti-HA Anti-rubisco
TN72_SR_cpl1-HA TN72_A_cpl1_c-HATN72_SR_Control
Anti-HA Anti-RbcL

113
3.3.2.2.2 C.reinhardtiilineTN72_SR_cpl1-HA_A_cpl1_c-HA
Toseeifhigherlevelsofproteinaccumulationcouldindeedbeachievedwithtwo
differentcpl-1expressioncassettesinsertedintotheC.reinhardtiichloroplast,the
SR_cpl1-HAandtheA_cpl1_c-HAcassetteswerecombinedintoasingleplasmidand
usedtotransformC.reinhardtiirecipientstrainTN72.
3.3.2.2.2.1 Expressionplasmidproduction
The finalassemblyofpSRSapI_cpl1-HA_A_cpl1_c-HAwasachievedasdescribed in
Figure3.21.TheexistingMluIsiteinpSRSapI_cpl1-HAwasusedtoopenthevector,
andtheinsertwascreatedbysite-directedmutagenesisPCRto introduceaBssHII
restrictionsiteat the3’endof theA_cpl1-c-HAcassette.AsMluIandBssHII form
compatible restriction sites, the cpl1-c containing expression cassettewas ligated
intotheopenvector,producingoneconservedMluIsite.Theresultingplasmidwas
called pSRSapI_cpl1-HA_A_cpl1_c-HA. It is believed that the alternative
arrangement,i.e.openingthepASapI_cpl1-c-HAplasmidandinsertingtheSR_cpl1-
HA cassette,would have also been possible, butwhen bothwere attempted the
cloningsucceededwiththeformerfirstandthereforethisroutewascontinued.
The primers used for the PCR reaction are indicated below. The BssHII site is
underlined,andthemutatednucleotideisshowninlowercase.
Cpl1.F:CAGGCAATTTGCTTACACCTTT
Cpl1_BssHII.R:TTAAGATAAgCGCGCTACATCC
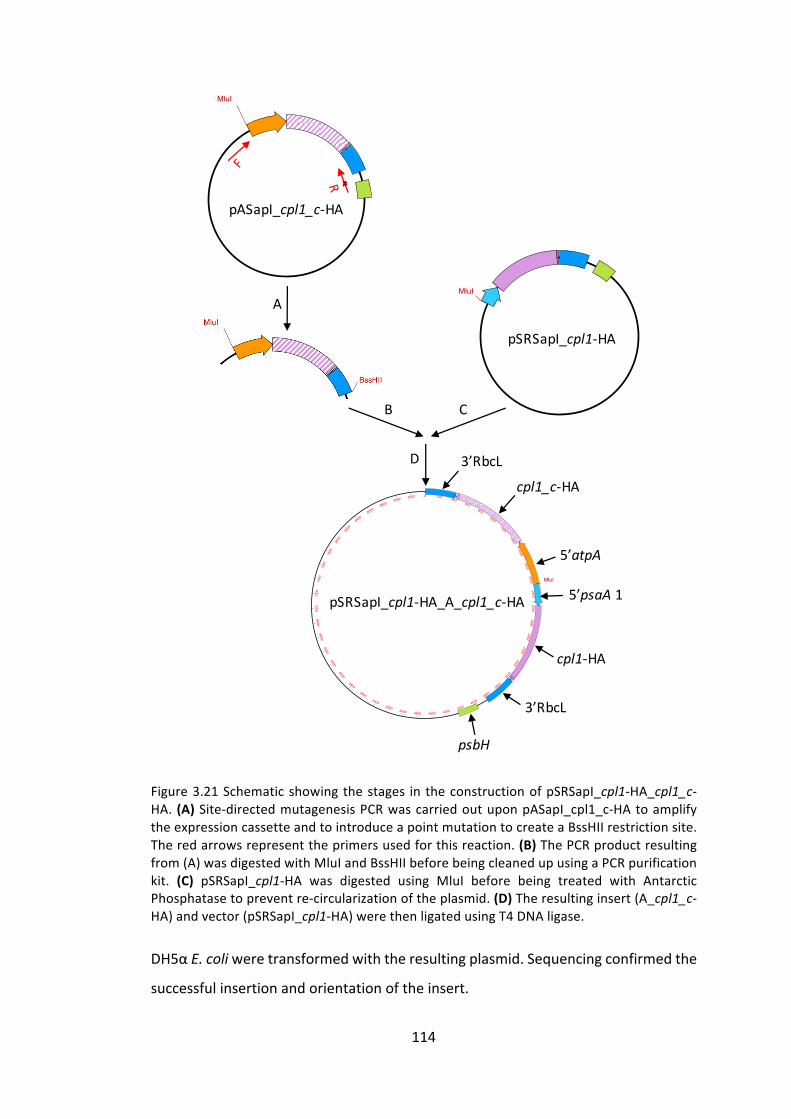
114
Figure3.21Schematicshowingthestages in theconstructionofpSRSapI_cpl1-HA_cpl1_c-HA. (A)Site-directedmutagenesisPCRwascarriedoutuponpASapI_cpl1_c-HAtoamplifytheexpressioncassetteandtointroduceapointmutationtocreateaBssHIIrestrictionsite.Theredarrowsrepresenttheprimersusedforthisreaction.(B)ThePCRproductresultingfrom(A)wasdigestedwithMluIandBssHIIbeforebeingcleanedupusingaPCRpurificationkit. (C) pSRSapI_cpl1-HA was digested using MluI before being treated with AntarcticPhosphatasetopreventre-circularizationoftheplasmid.(D)Theresultinginsert(A_cpl1_c-HA)andvector(pSRSapI_cpl1-HA)werethenligatedusingT4DNAligase.
DH5αE.coliweretransformedwiththeresultingplasmid.Sequencingconfirmedthe
successfulinsertionandorientationoftheinsert.
MluI (7035)
pASapI_cpl1_c-HA
A
B C
D
pSRSapI_cpl1-HA
MluI (7035)
0 200 400600
8001000
1200
14001600
18002000
2200
2400
2600
2800
3000
3200
3400
3600
3800
4000
4200440046004800
50005200
5400
5600
5800
6000
6200
6400
66006800
7000
7200
7400
7600
7800
8000
8200
8400
8600
88009000
9200
MluI (2065)
pSRSapI_cpl1-HA_A_cpl1_c-HAMluI (7035)
MluI (5979)
3’RbcLcpl1_c-HA
5’atpA
5’psaA 1
cpl1-HA
3’RbcL
psbH

115
3.3.2.2.2.2 C.reinhardtiitransformation
ThepSRSapI_cpl1-HA_A_cpl1_c-HAplasmidwasusedtotransformtherecipientC.
reinhardtii strainTN72via theglassbeadmethodasdescribed2.3.13.Figure3.22
illustratestheresultingC.reinhardtiicellline,TN72_SR_cpl1-HA_A_cpl1_c-HA.
Figure 3.22 TN72 recipient strain transformed with the pSRSapI_cpl1-HA_A_cpl1_c-HAplasmid,thusrescuingpsbHactivity.Carryingcpl1-HAunderthepsaA5’UTR,andcpl1_c-HAundertheatpA5’UTRinthechloroplast.
The resulting transformants were confirmed to be homoplasmic as described in
2.3.17andrepresentedinFigure3.23.Finally,correctinsertionintotheplastomewas
confirmed by PCR amplification from transformant genomic DNA and DNA
sequencing,asin2.3.18.
trnE2 psbH
TN72_SR_cpl1-HA_A_cpl1_c-HAtransformant MluI
5’psaA 1 cpl1-HA 3’rbcL5’atpAcpl1_c-HA3’rbcL

116
Figure 3.23 (A) Schematic describing the expected band when testing homoplasmictransformants.Thecombinationwhichproduces thenegativebandcanbeseen inFigure3.19. It istheabsenceofthenegative,350bp,bandthat indicateshomoplasmicity i.e.nocopies of the untransformed plastid remain (B) PCR products separated by gelelectrophoresisshowthecorrectexpectedlengthsandthusshowthatthenewtransformantisnowhomoplasmic.
3.3.2.2.2.3 Westernblotquantification
InordertocomparetherelativeCpl-1accumulationlevelsbetweenTN72_SR_cpl1-
HAandTN72_SR_cpl1-HA_A_cpl1_c-HA,andinvestigatewhethertwocassetteswith
the samegenedoes indeed improveoverall proteinaccumulation levels in theC.
reinhardtiichloroplast,westernblotanalysiswasperformed.Wholecellextractsof
TN72_SR_cpl1-HA and TN72_SR_cpl1-HA_A_cpl1_c-HA were prepared, and five
technical replicates of each were separated by SDS-PAGE. After blotting to
3Kb
TN72
TN72_SR_
cpl1-HA
_A_cpl1_c-HA
1Kb
0.5Kb
psbH
TN72_A_cpl1_c-HAtransformant
MluIatpA.R psbH.R
2500bp
A
B
5’psaA 1 cpl1-HA 3’rbcL5’atpAcpl1_c-HA3’rbcL
atpA.R
psbH.R

117
nitrocellulose membrane as described in 2.4.5, the membrane was stained with
REVERT™WholeProteinStainwhichconfirmedsimilarquantitiesofcellpreparation
were loaded onto the gel (Appendix A) andwas used to normalize small loading
differences. After removal of the REVERT stain and blocking, themembranewas
probedwiththeanti-HAprimaryantibody,followedbyIRDye®secondaryantibody
andfluorescencewasobservedusingtheLiCorOdyssey®CLxsystem.Thewestern
blotimageandanalysisareshowninFigure3.24.
Figure3.24WesternblotanalysisenabledthequantificationofCpl-1accumulationinthetwoC. reinhardtii lines. Cpl-1 detection was normalised using the signal from REVERT TotalProteinStain.
Atwosamplet-testwascarriedouttocomparethelevelsofproteinaccumulationin
TN72_SR_cpl1-HA and TN72_SR_cpl1-HA_A_cpl1_c-HA, in order to identify any
statisticalsignificance.Thisisacomparisonofthemeanproteinaccumulationlevels,
0
0.1
0.2
0.3
0.4
0.5
0.6
WT1 WT2 WT3 WT4 WT5 2Cpl1R1
2Cpl1R2
2Cpl1R3
2Cpl1R4
2Cpl1R5
Relativedetectedfluoresence
***
TN72_SR_cpl1-HA TN72_SR_cpl1-HA_A_cpl1_c-HA
TN72_SR_cpl1-HATN72_SR_cpl1-HA_
A_cpl1_c-HA
Cpl-1(39kDa)

118
anditisatwo-tailedtest.Thenullhypotheses(H0)isthatthemeansarethesame.
Thealternativehypothesis(H1)isthatthemeansaredifferent.
H0:μ1=μ2
H1:μ1≠μ2
The full results can be seen in Figure 3.25, but in brief, there was a significant
difference in accumulation for TN72_SR_cpl1-HA (M = 0.247, Var = 0.001) and
TN72_SR_cpl1-HA_A_cpl1_c-HA(M=0.451,Var=0.007);t(8)=-5.14,p<0.001.
Figure3.25StatisticalreportgeneratedusingMicrosoftExceltocomparethelevelsofproteinaccumulationinthetwolinesofC.reinhardtii.
This result supports our original hypothesis - that protein accumulation can be
improvedintheC.reinhardtiichloroplastbyintroducingseveralcopiesoftheGOIas
differentexpressioncassettes.ItsuggeststhatCESislimitingtheleveloftranslation
foreachendogenous5’UTRandthatbyhavingthesamegeneunderthecontrolof
twodifferent5’UTRs,thiscanbeimproved.
t-Test:Two-SampleAssumingEqualVariancesTN72_A_cpl1-HA
TN72_SR_cpl1-HA_A_cpl1_c-HA
Mean 0.2472 0.4512Variance 0.0006 0.0073Observations 5 5PooledVariance 0.0039HypothesizedMeanDifference 0
df 8tStat -5.1423P(T<=t)one-tail 0.0004tCriticalone-tail 1.8595P(T<=t)two-tail 0.0009
tCriticaltwo-tail 2.3060

119
3.3.2.2.3 C.reinhardtiilineTN72_SR_cpl1-StrepII
Inthepreviousexperiment,itisnotpossibletodeterminehowtheproteinsynthesis
loadisbeingsharedbythetwoexpressioncassettes,astheproteinsarebothtagged
withanHAepitopeandarethereforeindistinguishableonawesternblot.
WethereforeproducedasimilarC.reinhardtiicelllinebutwithonecpl1encodinga
StrepIIepitopetaginsteadofanHAtag.ThisenablestheCpl-1proteinfromthetwo
different promoters/5’UTRs to be distinguished on a western blot and the
contributionofthetwocassettescompared.
ThefirststageinthisprocesswastoproducetheC.reinhardtiicelllineTN72_SR_cpl1-
StrepII,tobeusedasacontrolbutalsotoensurethatthistagcanbedetectedon
westernblotsandthattheproteinaccumulatestodetectablelevels.
3.3.2.2.3.1 Expressioncassetteproduction
Thefull-lengthcpl1-StrepIIgenewassynthesizedby IntegratedDNATechnologies.
SapI and SphI restriction enzymeswereused to clone the gene in-frame into the
pSRSapI expression vector, producing the plasmid pSRSapI_cpl1-StrepII. DH5α
competentE.colicellsweretransformedwiththeresultingplasmid.
3.3.2.2.3.2 C.reinhardtiitransformation
ThepSRSapI_cpl1-StrepIIplasmidwasusedtotransformtherecipientC.reinhardtii
strainTN72viatheglassbeadmethodasdescribedin2.3.13.Figure3.26illustrates
theresultingC.reinhardtiicellline,TN72_SR_cpl1-StrepII.
Figure3.26TN72 recipient strain successfully transformed to carrycpl1-StrepII under thepsaA5’UTRinthechloroplast,andwithpsbHfunctionalityrestored.
trnE2 psbH
5’psaA 1 3’rbcL
TN72_SR_cpl1-StrepIItransformant MluI
cpl1-StrepII

120
The resulting transformants were confirmed for homoplasmicity as described in
2.3.17 (Appendix B). Finally, the sequence integrity of the inserted cassette was
confirmedbyDNAsequencingasin2.3.18.
3.3.2.2.3.3 Westernblotdetection
WholecellpreparationsofTN72_SR_cpl1-HA(asacontrol)andTN72_SR_cpl1-StrepII
were prepared and separated, in duplicate, by SDS-PAGE. After blotting to
nitrocellulosemembrane and blocking as described in 2.4.5, onemembranewas
probedwithananti-HAprimaryantibody,whilethesecondmembranewasprobed
withanti-StrepIIprimaryantibody.BothmembraneswerethenincubatedbyIRDye®
secondary antibody and fluorescencewasobservedusing the LiCorOdyssey®CLx
system.TheresultingimagesareshowninFigure3.27.
Figure 3.27 Two duplicatemembranes were probedwith anti-HA or anti-StrepII primaryantibodiesandCpl-1accumulationwasdetectedintheexpectedcelllinesi.e.Cpl-1-HAwasdetectedwiththeanti-HAantibodyandnotwiththeanti-StrepIIantibodyandviceversa.Thenon-specificbandseen in the lower imageconfirmsthatasimilarquantityofextractwasloadedontothegelforbothstrains.
3.3.2.2.4 C.reinhardtiilineTN72_SR_cpl1-StrepII_A_cpl1_c-HA
3.3.2.2.4.1 Transformationplasmidproduction
The plasmid was produced in the same manner as in 3.3.2.2.2.1, but using
pSRSapI_cpl1-StrepIIinsteadofpSRSapI_cpl1-HA.Theresultingplasmidwasnamed
pSRSapI_SR_cpl1-StrepII_A_cpl1_c-HA.
SR_Cpl1-HA
SR_Cpl1-StrepII
Cpl1(39kDa)
Anti-HA1ºantibody
Non-specific
Cpl1(39kDa)
Anti-strepII 1ºantibody

121
3.3.2.2.4.2 C.reinhardtiitransformation
The pSRSapI_SR_cpl1-StrepII_A_cpl1_c-HA plasmid was used to transform the
recipientC.reinhardtiistrainTN72viatheglassbeadmethodasdescribedin2.3.13.
Figure3.28illustratestheresultingC.reinhardtiicellline,TN72_SR_cpl1-StrepII.
Figure3.28TN72 recipient strain successfully transformed to carrycpl1-StrepII under thepsaA 5’ UTR, and cpl1_c-HA under the atpA 5’ UTR in the chloroplast, and with psbHfunctionalityrestored.
Correct insertion of the two GOIs into the plastome and homoplasmy of the
transformantlineswereconfirmedasdescribedabove.
3.3.2.2.4.3 Westernblotquantification
Inordertocompareaccumulationofproteinstaggedwithdifferentepitopetags,and
thereforedetectedviadifferentantibodies,aproteinstandardtaggedwithbothHA
and StrepII epitope tags was used to normalize the signal. This standard was
producedspecificallyforthispurpose,asdescribedin3.3.1.2.6.
C.reinhardtiiwholecellextractsfromthevariouscelllinesconstructedinthischapter
wereseparatedbySDS-PAGEinduplicate.Theproteinswereblottedtonitrocellulose
membranes as described in 2.4.5 and REVERT™ Total Protein stain was used to
normalise loading error (Appendix C). One membrane was probed with anti-HA
primaryantibody,whilethesecondmembranewasprobedwithanti-StrepIIprimary
antibody.Bothmembraneswere then incubatedwith IRDye® secondaryantibody
andfluorescencewasobservedusingtheLiCorOdyssey®CLxsystem.Theresulting
imagesareshowninFigure3.29.
trnE2 psbH
TN72_SR_cpl1-StrepII_A_cpl1_c-HAtransformant MluI
5’psaA 1 3’rbcL5’atpAcpl1_c-HA3’rbcL cpl1-StrepII

122
Theproteinstandard,whichistaggedwithbothHAandStrepIIepitopetagswasthen
usedtonormalizethetwomembranes,aswastheREVERT™totalproteinstain.The
relativeproteinaccumulationlevelsofeachC.reinhardtiistrainwerecomparedin
Figure3.29(B).TN72_SR_cpl1-HA,whichhasbeenusedthroughoutthischapterasa
benchmarkforCpl-1proteinaccumulationlevel,uponwhichtoimprove,wassetto
100%andotherlevelsarerepresentedaspercentagesofthis.

123
Figure3.29(A)WesternblotanalysisofC.reinhardtiiwholecellextractsprobedwithanti-HAoranti-StrepIIprimaryantibodies.IRdetectionwasperformedbytheLiCorOdyssey®CLx.(B)ProteinaccumulationlevelswerequantifiedusingtheOdyssey®ImageStudiov5.0andthesignalwasnormalisedusingtheproteinstandardandREVERT™totalproteinstaining.TN72_SR_cpl1-HAisthebenchmarkstraininthisinvestigationandwasthereforesetto100%accumulation.Other strains are represented relative to this. Protein accumulation of theStrepIItaggedCpl-1appearstobeconsiderablylowerthanthatoftheHAtaggedprotein.
As observed in Figure 3.24, the level of Cpl-1 protein accumulation in the
TN72_SR_cpl1-HA_A_cpl1_c-HAisapproximatelyequaltothesumofTN72_SR_cpl1-
HAandTN72_A_cpl1_c-HA.However,itisinterestingtonotethataccumulationof
StrepII-taggedCpl-1isconsiderablylowerthanitsHA-taggedequivalent.Thiscould
SR_cpl1-HA
SR_cpl1-StrepII
Cpl-1
Anti-HA1ºantibody
Non-specific
Cpl-1
Anti-strepII1ºantibody
A_cpl1_c-HA
SR_cpl1-HA
_A_
cpl1_c-HA
SR_cpl1-StrepII_
A_cpl1_c-HA
SR_Contro
l
Standard(10ug/ml)
0%
50%
100%
150%
200%
250%
300%
SR_cpl1-HA
SR_cpl1-StrepII
A_cpl1_c-HA
SR_cpl1-HA
_A_
cpl1_c-HA
SR_cpl1-StrepII_
A_cpl1_c-HA
SR_Contro
l
Anti-HA Anti-StrepII
A
B

124
beduetoeithertheStrepIItagdestabilizingtheprotein,oritcouldpossiblybean
artefactofthenormalizationmethod.
Theadditionofanypeptidetoaproteinhasthepotentialtodestabiliseitbyaffecting
theabilityorrateoffoldingoftheproteinintoastableform,andthismaybethe
casewiththeStrepIItaggedCpl-1.
Alternatively,itispossiblethattheStrepIItagontheproteinstandardismoreeasily
accessiblethanthatonCpl-1,orconverselythattheHAtagislessaccessibleonthe
standardthanonCpl-1.Thisscenariowould leadtoanunderestimationofStrepII
taggedproteins,oranoverestimationofHAtaggedproteins,respectively.Itseems
plausible that a little of both scenariosmay be occurring, given that the protein
standardistaggedwithanHAtagattheC-terminalend,followedbytheStrepIItag
beyondthat.Anti-HAantibodiescouldthereforebeslightlyhinderedbythepresence
of the StrepII tag, while anti-StrepII antibodiesmay find the epitope to bemore
accessibleduetoitsslightlyincreasedprotuberancefromtheCpl-1protein.
To investigatewhich of these theories is correct, two separate protein standards
couldbecreatedforeachtaggedCpl-1.IfmuchlowerlevelsofStrepIItaggedCpl-1
arestillobserved,thenitcouldbeconcludedthattheStrepIItagisdestabilizingCpl-
1,butifaccumulationlevelsappeartobesimilarthenitwasmerelyanartefactof
thenormalizationprocess.
Ineithercase,itappearsthattheinsertionofmultipleexpressioncassettesintothe
C.reinhardtiichloroplastresultsinasimpleadditionoftheproteinlevelsobserved
whencassettesareinsertedsingly.
3.3.3 Codonpairoptimizationforimprovementofproteinaccumulationlevels
The second aspect of this chapter looks at using codon pair optimization as a
techniqueforimprovingrecombinantproteinaccumulationlevelsintheC.reinhardtii
chloroplast.Codonoptimizationisawell-knownapproachtoimproverecombinant
protein yields, and has been shown to be particularly important for transgene
expression in theC. reinhardtii chloroplast – Franklin et al. (2002b) increasedgfp

125
expression by 80-fold in a codon optimized version compared to a unoptimized
version. The codon frequency chart for theC. reinhardtii chloroplast is shown in
AppendixD.Taunt(2013) investigatedtherolethatcodonpairsplayandwhether
these too couldbeoptimized to improveexpression. The study identified several
codonpairswhichhadhighpredictedoccurrencebutwererarelyorneverobserved
in the C. reinhardtii chloroplast plastome. These pairs were terms Unexpectedly
Unseen Zero Scoring Codon Pairs (uuZSCP). Two such uuZSCP stood out as being
particularlysignificant,aPhenylalanine:Argininepair,andanAsparticAcid:Leucine
pair, shown in Figure 3.30. The Phe:Arg pair was chosen for investigation in this
projectasithappenedtobeincludedintheinitialcodonoptimizationforcpl1,andis
thereforepresentinTN72_SR_cpl1-HA.
Figure3.30TwouuZSCPareparticularlyrareintheC.reinhardtiichloroplast,despitetheirconstitutivecodonsbeingrelativelycommon.Therelativefrequencyreferstothefrequencywithwhich this codonwouldbechosen to represent thisaminoacid in theC. reinhardtiichloroplast.ThePredictedOccurrenceishowoften,giventhefrequencyofaminoacidpairs,onewouldexpecttoseethiscodonpairinthetotal2579codonpairsintheC.reinhardtiichloroplast.
FirstCodon SecondCodon Codon PairAminoAcidPair
Code RelativeFreq. Code Relative
Freq.
CombinedRelativeFreq.
PredictedOccurrence
ActualOccurrence
Phe:Arg UUU 0.67 CGU 0.73 0.49 28.29 0Asp:Leu GAU 0.76 CUU 0.14 0.1 9.1 0

126
Figure3.31TheaminoacidsequenceofCpl-1.ThePhe:Argpairishighlightedinyellow,whilethepurpleboxindicatestheHAtag.
AccordingtoMauletal(2002),theC.reinhardtiichloroplastplastomecontains30
identifiedtRNAgenes,providingasmallyetsufficientsetforproteinsynthesis.After
examination of the data and checking the chloroplast genome (GenBank:
BK000554.2) for tRNA genes using tRNAscan-SE (http://lowelab.ucsc.edu/cgi-
bin/tRNAscan-SE),acoupleofinaccuraciesinthetRNAdatawerenoticed.Firstly,the
trnEgeneiscountedthreetimes,wheninfactthereappeartobeonlytwocopies
identified on the plastome. There are therefore only 29 tRNA genes in the C.
reinhardtiichloroplast.Secondly,thetrnR2islabelledwiththeincorrectanti-codon
–itshouldreadtrnR2(UCU),nottrnR2(AGA)–likelyjustaconfusionbetweencodon
andanticodon.Thirdly,whilenotamistakeperse,thenomenclatureisunclearfor
tRNAgenesand seems tobe inconsistent. Forexample, thereare trnR and trnR2
genes,whichidentifytRNAswithdifferentanticodons,butnotrnR1exists.Atableof
allthetRNAgenesintheC.reinhardtiichloroplasthasbeencompiledinFigure3.33.
TheC. reinhardtii chloroplast chromosomemapbyMauletal. (2002) is shown in
MVKKNDLFVDVSSHNGYDITGILEQMGTTNTIIKISEST
TYLNPCLSAQVEQSNPIGFYHFARFGGDVAEAEREAQFF
LDNVPMQVKYLVLDYEDDPSGDAQANTNACLRFMQMIAD
AGYKPIYYSYKPFTHDNVDYQQILAQFPNSLWIAGYGLN
DGTANFEYFPSMDGIRWWQYSSNPFDKNIVLLDDEEDDK
PKTAGTWKQDSKGWWFRRNNGSFPYNKWEKIGGVWYYFD
SKGYCLTSEWLKDNEKWYYLKDNGAMATGWVLVGSEWYY
MDDSGAMVTGWVKYKNNWYYMTNERGNMVSNEFIKSGKG
WYFMNTNGELADNPSFTKEPDGLITVAYPYDVPDYA

127
Figure3.34.ItshowsthelocationofallthetRNAgenesontheplastomeandexplains
thatthetwocopiesoftrnAandtrnIareduetotheirlocationontheinvertedrepeat
sequences.
InordertotestwhetherthepresenceoftheuuZSCPPhe:Argcodonpairisaffecting
thelevelofproteinaccumulationinTN72_SR_cpl1-HA,twocpl1genemutantswere
createdbysite-directedmutagenesis.Thefirst,CPOCv1(CodonPairOptimizedCpl-1
version1),wasamutationofthephenylalaninecodonfromUUUtoUUC–thereis
onlyonetrnFgene,sothiscodonwouldbeservedbythesametRNA“wobbling”as
cpl1.Thesecondmutant,CPOCv2, includesamutationoftheargininecodonfrom
CGUtoAGG–thiswasdesignedtobeservedbythetRNAencodedbytrnR2,rather
thanthatencodedbytrnR1whichservescpl1.
N.B.AGAisaslightlymorefrequentlyseenArgininecodonthanAGG,asshownin
Figure7.4,andwouldnotrelyontRNAwobble.However,UUU:AGAisalsoacodon
pairthatisneverobservedinthechloroplast,soforthisreason,UUU:AGGwhichis
observed,waschosen.
Phe Arg CPAI Presentin…UUU CGU 0 cpl1
UUU CGC 0 UUU CGA 0 UUU CGG 0 UUU AGA 0 UUU AGG 0.0625 cpov2
UUC CGU 1 cpov1
UUC CGC 0 UUC CGA 0.0625 UUC CGG 0 UUC AGA 0.0625 UUC AGG 0
Figure3.32AllpossiblecodoncombinationsforaPhe:Argpair.Thecodonpairadaptationindex(CPAI)istheratioofcodonpairfrequencytothefrequencyofthemostobservedcodonpair.Thedataabovearefroma“hand-pick”databaseintheCodonUsageOptimizerBeta
0.92 (Kong 2013) software, whereby the codon pair usage in highly expressed genes iscollected.

128
Giventhetheoryforcodonpairbiaslaidoutin3.1.4,itwashypothesisedthatCPOCv1
wouldbeunaffectedby thecodonchange,whileCPOCv2maydisplayadifferent,
possiblyhigher,levelofCpl-1proteinaccumulation.
Figure3.33A listoftRNAgenes identifiedbyMauletal. (2002).TheyellowrowsindicateduplicatetRNAgenes,i.e.theyhavethesameanticodon.GreenrowsindicatetRNAgeneswithdifferentanti-codons,butcodingforthesameaminoacid.
No. Name Anticodon Codon AminoAcid
1trnA UGC
GCA AlatrnA UGC
2 trnC GCA UGC Cys3 trnD GUC GAC Asp
4trnE UUC
GAA GlutrnE UUC
5 trnF GAA UUC Phe6 trnG1 UCC GGA Gly7 trnG2 GCC GGC Gly8 trnH GUG CAC His
9trnI GAU
AUC IletrnI GAU
10 trnK UUU AAA Lys11 trnL1 UAG CUA Leu12 trnL2 UAA UUA Leu13 trnM1 CAU AUG fMet14 trnM2 CAU AUG Met15 trnM3 LAU AUA Ile16 trnN GUU AAC Asn17 trnP UGG CCA Pro18 trnQ UUG CAA Gln19 trnR1 ACG CGU Arg20 trnR2 UCU AGA Arg21 trnS1 UGA UCA Ser22 trnS2 GCU AGC Ser23 trnT UGU ACA Thr24 trnV UAC GUA Val25 trnW CCA UGG Trp26 trnY GUA UAC Tyr
Phenylalanine
Arginine

129
Several naming changes have been made in Figure 3.33 to the original names
publishedinMauletal.(2002).Thenewnamesfallinlinewiththeannotationsmade
to the chloroplast genome available at NCBI
(https://www.ncbi.nlm.nih.gov/nuccore/BK000554).Thenamesmeanthatidentical,
duplicategenes (trnA, trnIand trnE)have thesameanticodon,whilegeneswhich
encode similarly charged tRNAs,butwithdifferinganticodons, arenumberede.g.
trnL1andtrnL2.TheonlychangemadefromtheNCBInamingistonamebothtrnE
genesthesame,astheyareidenticalduplicates.Itshouldalsobenotedthatsome
discrepancyexistsbetween thedefinedgene lengthonNCBIand the tRNAgenes
detectedusing the tRNAscan software,most likelydue to incorrect annotationof
NCBI.

130
FigureRemoved
Figure 3.34 TheC. reinhardtii chloroplast chromosome. tRNA genes are identified on theouterringinblackandlabelledwithasingleletter.e.g.V,whichindicatestrnV.TwotRNAgenes (trnA and trnI) are located in the Inverted Repeat (IR) regions of the plastome,indicatedinthickblack,thusexplainingtheirduplication.ThesecondcopyoftrnL(2o’clock)shouldbeblacktoindicatetRNA,asshouldtrnIandtrnAontheright-handIR.Reproducedfrom Maul et al. (2002). Note: As an aside, the neutral site used for chloroplasttransformationthroughoutthisthesisislocatedbetweentrnEandpsbH,5o’clock.

131
3.3.3.1 CodonPairOptimizedCpl-1version1and2(CPOCv1andCPOCv2)
CPOCv1wasdesignedsuchthatadifferentcodoncodedforphenylalanine,butthat
codonwouldstillbeservedwiththesametRNAaswasusedintheoriginal,through
the phenomenon known as tRNA wobble (Alkatib et al. 2012). CPOCv1 would
thereforeactasacontrol for this investigation,as the theorybeing tested is that
tRNAsinteractintheAandthePsiteoftheribosomeinamannerthatcaninfluence
translationefficiency.Here, the same tRNAswill beused in thismutant as in the
originalversionandthusweexpecttoseenochangeintranslationefficiency.
CPOCv2wasdesignedsuchthatadifferentcodoncodedforarginine,andthatthat
codonwouldbeservedwithadifferenttRNAtotheoriginal,astherearetwotrnR
genes in the C. reinhardtii chloroplast with different anti-codons. CPOCv2 was
producedinanattempttoinfluenceaccumulationlevelsofCpl-1,asdifferenttRNAs
willbeinteractingintheAandthePsiteoftheribosomecomparedtothoseinthe
originalversion,andthereforetranslationmayprogressatafasterrate.
3.3.3.1.1 Codonchangebysite-directedmutagenesis
TocreateCPOCv1,primersweredesignedtomutateU→Cinthethirdpositionof
thephenylalaninecodonthroughPCR,andforCPOv2tomutateC→Ainthefirst
positionandT→Ginthethirdpositionoftheargininecodon.Thestepstakento
performthismutagenesisareshowninFigure3.35andFigure3.36respectively.The
primersusedareindicatedbelow.Thelowercaselettersshowthesitesformutation.
cpl1.F: GCTCTTCAATGGTTAAAAAAAATGATTTATTC
cpocv1_SDM.F: AGGTGGTGGTTcCGTCGTAA
cpocv1_SDM.R: TTACGACGgAACCACCAACCT
cpocv2_SDM.F: GTTGGTGGTTTaGgCGTAATAATG
cpocv2_SDM.R: CATTATTACGcCtAAACCACCAAC
cpl1.R: GCATGCTTATTAAGCATAATCTGGAAC

132
Figure3.35Site-directedmutagenesiswasperformedasabovetomodifythephenylalaninecodontomakeCPOCv1.PrimersareshowninblueandPCRconditionsaredescribedin2.3.7.
Figure3.36Site-directedmutagenesiswasperformedasabovetomodifytheargininecodontomakeCPOCv2.PrimersareshowninblueandPCRconditionsaredescribedin2.3.7.
3.3.3.1.2 Expressioncassetteproduction
The expression cassette was assembled as with previous examples. The gene,
includingtheHAtagattheCterminus,wasclonedintothepSRSapIvectorinframe
usingSapIandSphIrestrictionsites.TheinsertionwasconfirmedbyDNAsequencing
TTT CGTAAA GCA
AAG GCA
TTC CGT
TTC CGTAAG GCA
TTC CGTAAG GCA
1st PCR
2nd PCR
Finalproduct
Cpl-1
CPOCv1
cpl1.F
cpl1.R
cpocv1_SMD.F
cpocv1_SMD.R
TTT CGTAAA GCA
AAA TCC
TTT AGG
TTT AGGAAA TCC
TTT AGGAAA TCC
1st PCR
2nd PCR
Finalproduct
Cpl-1
CPOCv2
cpl1.F
cpl1.R
cpocv2_SMD.F
cpocv2_SMD.R

133
of the plasmid as described in 2.3.18. The resulting plasmids were called
pSRapI_CPOCv1-HAandpSRapI_CPOCv2-HA.
3.3.3.1.3 C.reinhardtiitransformation
Two TN72 transformations were performed, using pSRSapI_CPOCv1-HA and
pSRSapI_CPOCv2-HA plasmids. Transformed C. reinhardtii lines were checked for
homoplasmicitybyPCRandfinallyconfirmedbysequencing.
3.3.3.2 Western blot quantification of TN72_SR_CPOCv1-HA and
TN72_SR_CPOCv2-HA
Itishypothesisedthatthecodonchangesmadeabovewillresultindifferentlevels
ofproteinaccumulationbetween the two transformant lines.To test this, fourC.
reinhardtiicultures,TN72_SR_cpl1-HA,TN72_SR_CPOCv1-HA,TN72_SR_CPOCv2-HA
and TN72_SR_Control, were grown in an Algem Photobioreactor simultaneously,
withthesamestartingcelldensityandwiththesamephysiologicalconditions.OD740
wasmeasuredautomaticallyevery30minutesandsamplesweretakenatintervals
foranalysisbywesternblotting.1mlsampleswerecollectedfromtheculturevolume
andimmediatelysnap-frozeninliquidnitrogenandstoredat-80°C.Theculturewas
grownfor145hoursintotal.Attheendofthistimethefrozensampleswereanalysed
togetherbywesternblotting.Whole-cellextractswereseparatedbySDS-PAGE,with
nocompensationmadeforcelldensity.Afterblottingtonitrocellulosemembrane
andblocking,themembraneswereprobedwithanti-HAprimaryantibody,followed
by IRDye® secondary antibody and fluorescence was observed using the LiCor
Odyssey®CLxsystem.
Figure3.37showsthewesternblotanalysis–ariseinproteinaccumulationovertime
ascelldensityincreasesistobeexpected.

134
Figure3.37WesternblotanalysisoffourC.reinhardtiistrainsatfourtimepoints.Theknownstandardwasusedtonormaliseanydifferencebetweenblottingonseparatemembranes.
The quantified and normalised data is displayed in Figure 3.38, alongside optical
density data collected by theAlgemphotobioreactor. Thewhole experimentwas
repeatedasecondtime,andthefinalresultscanbeseeninFigure3.39,withwestern
blotanalysisinAppendixE.
3.3.3.2.1 Statisticalanalysisofwesternblotdata
A one-way ANOVA test was carried out on the recombinant protein yields of
TN72_SR_cpl1-HA, TN72_SR_CPOCv1-HA and TN72_SR_CPOCv2-HA in order to
identify any statistical difference between the levels of protein accumulation at
differenttimepoints,detailedinAppendixF.Inthefirstrunoftheexperiment,itwas
foundthattheproteinaccumulationat45hoursand58hourswassignificantlyhigher
inTN72_SR_CPOCv2-HAthanintheothercelllines,butthattherewasnosignificant
difference beyond this point.On the other hand, the second performance of the
experimentwasrunforlongerandlikewise,suggeststhatproteinaccumulationof
TN72_SR_CPOCv2issignificantlyhigherthe45and56-hourtimepoints.However,
CPOCv1 and CPOCv2 then continue to rise, while the protein accumulation in
TN72_SR_cpl1-HArisesatamuchlowerrate,beingsignificantlylowerthanthecodon
pair optimised versions at the 112, 139 and 194-hour time points. The statistical
methodandresultsaredetailedinAppendixG.
TN72_SR_cpl1-HA
45h
Standard
(10ug/ml)
TN72_SR_CPOCv1-HA
TN72_SR_Control
TN72_SR_CPOCv2-HA
58h
77h
119h

135
Figure3.38Algemgrowthdata(linechart,lefty-axis)andwesternblotquantification(barchart,righty-axis).TheAlgemphotobioreactorrecordedopticaldensityat740nmevery90seconds.AtOD740=~2.5,thephotobioreactorisunabletorecordanyhigherODduetothelongpath-lengthacrossa400mlculture.However,asindicatedbytheproteinaccumulationlevels,theculturesarereachingtheirstationaryphasearoundthispointanyway.TherelativedetectedfluorescenceforHA-taggedproteinaccumulationistakenfromFigure3.37,andnormalisedusingtheproteinstandardandtheOD740.
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0
0.5
1
1.5
2
2.5
3
0 12 24 36 48 60 72 84 96 108 120 132 144
Relativ
edetectedflu
oresence
OpticalDensity(740nm
)
Time(hours)SR_cpl1-HAProtein SR_CPOCv1-HAProtein SR_CPOCv2-HAProtein SR_ControlProteinSR_Cpl1-HA SR_CPOCv1-HA SR_CPOCv2-HA SR_Control
****
****

136
Figure3.39Algemgrowthdata(linechart,lefty-axis)andwesternblotquantification(barchart,righty-axis).ThestartingODwaslowerinthisexperiment,sotheculturetakeslongertoreachthelogarithmicgrowthstage.TheoriginalwesternblotanalysisisshowninAppendixE,inthisinstancethe25kDabandoftheproteinladderwasusedtonormalisethemembranes,thisenabledmoresamplestobefittedontoasinglegelandthusmeasureaccumulationoveralongertimeperiod.
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
0
0.5
1
1.5
2
2.5
3
0 12 24 36 48 60 72 84 96 108 120 132 144 156 168 180 192
Relativ
edetectedflu
oresence
OpticalDensity(740nm
)
Time(hours)
SR_cpl1-HAProtein SR_CPOCv1-HAProtein SR_CPOCv2-HAProtein SR_ControlProteinSR_Cpl1-HA SR_CPOCv1-HA SR_CPOCv2-HA SR_Control
** **
* *
*

137
Figure3.38indicatesthatCPOCv1isgrowingslightlyslowerthanthetwocontrols,
andCPOCv2isgrowingslightlyfaster.This ismost likelyduetoaslightlydifferent
numberofcellsusedtoinoculatethecultures,despiteeffortstoensuretheywere
the same. However, to compensate for thiswhen comparing protein yields from
westernblotanalysis,thedetectedsignalwasdividedbytheOD,thusnormalising
thedataforcelldensity.Eachofthefourmembranesusedforwesternblotanalysis
alsoshowedslightlydifferentsignalsfortheproteinstandard,probablyduetonon-
uniformtransferfromthegeltothemembrane,slightlydifferentantibodyincubation
times and slightly different dilutions of milk and antibody. Assuming that these
variables are the same within membranes, but can vary from membrane to
membrane, the datawere normalised by division of the average level of protein
standarddetected.ThesameistrueforFigure3.39,butthistimeusingthe25kDa
bandoftheproteinladdertonormaliseforinter-membranedifferences,astherewas
noroomonthemembranetoincludetheproteinstandardtoo.
Whilethetwographscannotbecompareddirectly,duetothelongerlagtimeinthe
latter and different inter-membrane normalisation techniques, they do appear to
displaytwotrends.Firstly,thatCPOCv2seemstoaccumulatefasterthantheother
Cpl-1variantsthroughoutthe logarithmicgrowthstage,andsecondlythattheun-
optimisedversionofCpl-1consistentlyaccumulatestoalowerlevelthaneitherof
theCPOversions.
This is an interesting observation, as according to the theory of codon pair
optimisation,CPOCv1accumulationshouldbenodifferenttotheunchangedCpl-1,
as the tRNAs serving the mRNA are identical. However, particularly in the later
growth stages in Figure 3.39, there is a consistently and significantly higher
accumulationofCPOCv1andCPOCv2.
Given the evidence from section 3.3.1, in which datawas collected on the error
introducedatvariouspointsofmeasuringproteinaccumulation,thesedifferences
seem too large to be artefacts of variability in western blotting and cell density
measuring techniques. Therefore, alternative explanations for this resultmust be
sought.

138
The first thing to consider is the fact that in CPOv2, another codon pair is being
unintentionallyaltered.Theaminoacidimmediatelyfollowingthecodonoptimized
ArginineisanotherArginine,ascanbeseeninFigure3.31.ThisArg:Argcodonpairis
beingunintentionallyalteredwhentheprecedingPhe:Argcodonpairisoptimizedin
CPOv2.Thispresentsanothervariablethathasnotbeenaccountedfor,andasshown
in Figure 3.40, the new pair is never observed in the “hand-picked” database,
consistingofhighlyexpressedproteinsintheC.reinhardtiichloroplast,generatedby
Taunt(2013).Thispresentsanissue,asalthoughoneuuZSCP(UUU:CGU)hasbeen
replaced,anotherunseenpairisintroduced(AGG:CGU).Itis,therefore,possiblethat
thebeneficialeffectsof the formerarenegatedby thedetrimentaleffectsof the
latter. There is no similar effectmade by CPOv1, as all Phenylalanine codons are
servedbyasingletRNA.
Arg Arg CAI Presentin…CGU CGU 1 cpl1&cpov1CGC CGU 0 CGA CGU 0 CGG CGU 0 AGG CGU 0 AGA CGU 0 cpov2
Figure3.40Datatakenfromthe“hand-pick”databaseofCodonUsageOptimizerBeta0.92(Kong 2013). This is a database of codon usage in highly expressed proteins, a commonmethodofselectingthebestcodonusage.
Anotherpossibilityisthatwhilegeneexpressionappearstobeprimarilycontrolled
attheleveloftranslationintheC.reinhardtiichloroplast,thetranscriptionallevelstill
mayplayapartandassuchthelevelsofmRNAproducedforCPOCv1andCPOCv2
shouldbeinvestigated.Ifthesearefoundtoremainsimilarthenitcanbediscounted,
butshouldbeinvestigatednevertheless.
Additionally, the unexpectedly unseen nature of UUU:CGU could be just a
coincidence.Althoughthisseemedunlikely,giventheconvincingstatisticalanalysis
performedby Taunt (2013), the assertion that this pair is purposefully avoided is
slightlyunderminedbythefrequentpresenceofUUC:CGU.UUC:CGUaccountsfor33
ofthe59observedinstancesofPhe:ArgintheC.reinhardtiichloroplast,butgiven

139
thefactthatUUUandUUCareservedbythesametRNA,thedifferentcodonshould
confer no difference in translational efficiency or codon selection. It could be
thereforethatsomeneutralmechanismhasresultedinthisbias,notselection,for
example, preferentialmutationbias duringDNA replicationor repair (Lucks et al.
2008).Anotheralternativeisthatthesinglebasechangeinthecpl1transgenehas
created an unexpected local area of secondary structure within the mRNA, thus
altering the translationalefficiency (Gasparetal.2013).Thiskindof interaction is
very difficult to predict, as the mRNA folding events may involve two distant
sequences,andchangingonebasemightstrengthen/weakentheseinteractions.
Thefinalandpossiblymostlikelyexplanationisonewhichislittleunderstood.Thisis
thefrequentlyobservedandreportedphenomenonofvariabletransgeneexpression
between“identical”C. reinhardtiichloroplast transformants (MayfieldandSchultz
2004; Surzycki et al. 2009; Michelet et al. 2011). The differences can be quite
remarkable: Surzycki et al. (2009) reported that recombinant protein expression
levelsofVP28,aWhiteSpotSyndromevirusprotein,variedbetweentransformants
from0.88%to20.9%TCP.Whenonlyalownumberoftransformantsaregenerated,
as is frequently thecasewiththeglassbeadmethodregularlyused in thePurton
group, a significant difference in transgene expression could be incorrectly
apportioned to a factor which really has little effect. The reason put forward by
Surzycki et al. (2009) for this observation is transformation-associated genotypic
modifications,termedthe“transformosome”.Thepaperpostulatesthatduringthe
process of genetic transformation, fragments of the transformation plasmid are
unknowingly inserted into theplastomeor nuclear genome, thus alteringnuclear
genes that regulate chloroplast gene expression, protease expression, or sites of
regulatory gene action in the chloroplast. Evidence is presented that the level of
steadystatetransgenemRNAisunaffected,givingfurtherweighttothebeliefthat
generegulationoccursprimarilyatthetranslationlevelinC.reinhardtii.
C. reinhardtii is extremely good at resourcemanagement, and as such is able to
prioritise theproductionofkeyproteinsoverothers, forexamplewhenCopper is
limitingthecellwillpreferentiallyproducecytochromeCoxidaseoverplastocyanin
foruseintheelectrontransferchainofPS1(MerchantandBogorad1986).However,

140
asCpl-1isarecombinantprotein,itisnotinvolvedinthishierarchyandistherefore
likelytobethefirstproteintobesacrificedinthecaseofametaboliclimitation.As
the value for protein accumulation is relative (i.e. it has been normalised for the
opticaldensityoftheculture),Figures3.38and3.39bothshowincreasinglevelsof
protein accumulation as the culture progresses into stationary phase and then it
plateaus. Therefore, metabolic limitation does not appear to be an issue in this
instance.
Toconclude,itappearsthatadifferenceinCpl-1accumulationhasbeenobserved,
butitisunclearastowhetherthisdifferenceisduetothecodonpairoptimisation,
astheresultsdonotsupportthetheorythatcodonpairscanbeoptimisedduetothe
differentinteractionsoftRNAsattheAandP-sitesoftheribosome,andtheobserved
differencesaresmallenoughtobeaccountedforbyotherfactors.
3.4 Conclusionandfuturework
This chapter has investigated two putative methods of improving recombinant
protein expression in the C. reinhardtii chloroplast: the introduction of multiple
expression cassettes; and the application of codon pair optimisation. The former
appearstobeamethodtorobustlyimproveproteinaccumulation,whilethelatter
method remains unclear as the complex and incremental nature of the change
appearstobemaskedbyotherfactorsinplay.Interestinglytheconcernaddressed
atthestartofthechapter,namelyhowtoaccuratelycompareproteinaccumulation
between transformant lines, re-emerged as an issue towards the end of the
investigation.
3.4.1 Multipleexpressioncassettestrategy
TheoverallconclusionfromthisstudywasthatC.reinhardtiilinestransformedwith
multiple expression cassettes produce a recombinant protein yield approximately
equal to the sum of the yields observed in lines transformedwith each cassette
individually.ThisfindingcouldpotentiallybeusedtodoubleexistingyieldsintheC.
reinhardtiichloroplast.However,somefurtherworkintowhatthelimitationsofthis
methodarewillenableustobetterunderstanditspotential.

141
Thefirstandmostobvioustestistoinvestigatewhetherthisresultcanberepeated
withothertransgenes.Thiswouldbearelativelystraightforwardtesttoperform,but
itwouldbeinterestingtotestbothanotherhighlyexpressedgene,aswellasagene
whichiscurrentlyonlyweaklyexpressedintheC.reinhardtiichloroplast.Thecpl1
geneiswellexpressed,andthereforeitispossiblethatCESmechanismsarelimiting
its accumulation under a single 5’ UTR, thusmaking it a good candidate for this
technique. On the other hand, proteins displaying poor accumulation might be
limitedbyproteaseactivityforexample,andwillthereforeremainatlowlevelsof
accumulationdespitedifferentmRNAtranscripts.
Thesecondquestionthatonewouldliketoaskistowhatpointcanthistheorybe
extended.Willthreeorfourcassettestripleorquadrupletheproteinaccumulation,
for instance? Going beyond two copies of the gene raises the issue discussed in
section3.3.2.2–whentwoduplicategenesareinparalleldirectionstoeachother,
thereisapotentialforDNAexcisiontooccurbetweentherepeats.Avoidingthisby
increasingthedistancebetweenthecopieswouldmostlikelyrequireanotherneutral
site to be used and targeted for transformation, requiring the use of a second
selectablemarker, enabling sequential transformations. Two options exist in this
case, either the addition of a positive selectable marker, such as aadA and
maintaining the algae on spectinomycin containing media, or the addition of a
negativeselectablemarker,suchascodA,totherecipientlinewhichisthenreplaced
inthesecondtransformationevent(YoungandPurton2014).Thelatterwouldresult
in the formation of a marker-less mutant which is desirable for further
transformationsbutrequiresmoretimetomaketheconstructs.Asaninitialtestof
how far themultiple-cassette theory can be taken, it is suggested that using an
antibiotic resistancemarker ismorestraightforward.Furthermore, thepracticeof
usingcodonmanipulationtominimisethepotentialforDNAexcision,asperformed
in3.3.2.2.1.1,mayenablemorecassettestobeinsertedintothesamesite.However,
given that codon optimisation has been shown to be very influential in protein
accumulation theC. reinhardtii chloroplast (Surzycki et al. 2009), this canonlybe
takensofarwithoutaconsiderablereductionintranslation.

142
Asdiscussedin3.3.2.2.4.3,aninvestigationintowhytheStrepIIepitopeappearsto
beaffectingproteinaccumulationsoprofoundlyshouldbeperformed.Thesimplest
explanation is that normalising to the protein standard is skewing the results by
havingadifferentepitopeaccessibilitythanthatofthesinglytaggedproteins.This
wasanissueencounteredpreviouslyinthePurtongroup(Taunt2013;Stoffels2015),
wherebythecommercialmulti-taggedproteinstandardsuggestedlevelsofprotein
accumulationthatcouldnotbeconfirmedbySDS-PAGEandCoomassieBluestaining.
Furthermore,asimilarresultin5.3.2.4.3suggeststhatthisisthecase,whereanother
endolysin,CD27L,alsoappears toaccumulate toamuch lower levelwhenStrepII
tagged.However,itmaybethattheendolysinisextremelysensitivetoC-terminal
epitopetagsintermsofstability,inwhichcaseaninvestigationintotheaccumulation
usingdifferentepitopetagscouldbecarriedoutandoneshouldalsoconsidertagging
theN-terminusinstead.
Finally,acalculationofrecombinantproteinyieldasapercentageofTotalSoluble
Protein(TSP)orTotalCellProtein(TCP)wouldbepossibleoncetheissuesurrounding
theproteinstandardhasbeenresolved.Thiswouldallowcomparisontootheryields
achieved in the literature. Alternatively, an ELISA assay could be performed but
antibodies would have to be raised against Cpl-1, which is expensive. A robust
methodofestimatingproteinyieldusinganepitopetagandwesternblotanalysis
wouldbecheaperandmoreflexible,enablingquantificationofotherproteinsinthe
future.
3.4.2 Identicaltransformanttransgenevariability
Theissueoflargevariabilityintransgeneexpressionbetweensupposedlyidentical
transformants, also known as “clonal variation”, has the potential to mask or
exaggerate investigations into othermethods of improving protein accumulation.
Untilthisissuehasbeenaddressed,“fine-tuning”techniqueswillremaindifficultto
assayandareunlikelytobeaseffectiveassimplyscreeningmanytransformantsand
selecting the best, as many protein production companies working with Pichia
pastoriscurrentlydo(Awetal.2017).

143
The most useful tool to aid investigation into this phenomenon would be the
development of a sensitive and robust reporter gene for C. reinhardtii. Reporter
genes such asGFP have beenused for this purpose inmany other organisms, as
fluorescence is graded and easily measured in vivo, enabling rapid quantitative
screening of many transformants. However, while some studies have produced
reasonablelevelsofGFPintheC.reinhardtiichloroplast(Franklinetal.2002),dueto
thehighlevelsofauto-fluorescenceinC.reinhardtii,GFPisunsuitableasitmustbe
producedathighlevelstobedetectable(MayfieldandSchultz2004).Luciferasehas
beendevelopedasaviablealternative,withfluorescenceshowntobeproportional
toproteinaccumulation,andnointerferencebycellauto-fluorescence(Mayfieldand
Schultz 2004). Surzycki et al. (2009) demonstrated that co-introduced selectable
markergenesareexpressedinamannerproportionaltotheGOI.Therefore,theco-
introductionofluciferasewithaGOIshouldenableitsfluorescencetobeusedasa
reliable proxy for recombinant protein accumulation. This in turnwill enable the
rapidscreeningofmanydifferenttransformants,andtheproductionofabell-curve
fortransgeneexpression.Oncethismethodcanbeusedtocreateareproduciblebell-
curveofluciferasedetection,thenstatisticalanalysiscanreliablydeducehowsmaller
changesareinfluencingproteinaccumulationlevels.
Figure 3.41 A comparison of means. By producing two bell curves with the luciferaseexpressionlevelmeasuredfrommanytransformants, itwillbepossibletoidentifysubtlerimprovementsinproteinaccumulation.Here!isthedifferencebetweentwotransformant
Num
bero
ftransform
ants
Luciferaseexpression
.

144
lines,withthebluelinerepresentingallthetransformantsofonetreatment,andtheredlineallthetransformantsofanother.
Development of the above will enable more precise testing of other strategies
including codon and codon pair optimization. The above also depends upon the
developmentofamoreefficientsystemoftransformingC.reinhardtiichloroplast,as
many transformantsneed tobe rapidlyproduced for thismethod tobeeffective.
Current work in the Purton lab by Saowalak Changko aims to improve current
transformationmethods.Anotherfutureadvantageofsuchasystemisthatitwould
allowtheidentificationofoutliers,cellswhichproduceveryhighorverylowlevelsof
recombinantprotein.Withthecostofwholegenomesequencing(WGS)fallingall
thetime,theseoutlierscouldbesubjectedtoWGSinanattempttounderstandwhat
factorsareinfluencingproteinexpressionsoheavily,andthenattempttospecifically
introducethosechanges.WGShasalreadybeenusedtosuccessfullyidentifypoint
mutationsinC.reinhardtii(Dutcheretal.2012).Thiswillimproveourunderstanding
ofthesubtletiesofcurrenttransformationmethodsandthecomplexroleofnuclear
factorsinchloroplastgeneexpression.
3.4.3 Codonpairoptimisation
As discussed in the previous section, attempting to manipulate and detect
differencesincodonpairoptimisationstrategiesmaybeprematureatthisstageof
developingtheC.reinhardtiichloroplastasaproteinproductionplatform.However,
giventheimportanceofcodonoptimizationoftenreportedintheliterature(Franklin
etal.2002;Surzyckietal.2009;Coragliottietal.2011),andtheclearexistenceofa
codonpairbiasintheC.reinhardtiichloroplast(Taunt2013),itispossiblethatcodon
pairoptimisationwillbecomeanimportanttoolinthefuture.
Codonandcodonpairoptimisationarecurrentlyjusta‘bestguess’athowtoexpress
arecombinantproteintoahighlevel.Itappearsthatcodonoptimisationbythehost
initsownendogenousgenesenablescontrolled,delicateadjustmentsofexpression.
Thediscoveryofcodonpairoptimisationfurthermuddiesthewatersaroundderiving
which codons to use in a transgene. It seems intuitive that just using the most
commoncodonswith the largestpoolof available tRNAswill result in the fastest

145
translationandthereforethegreatestlevelofproteinproduction.However,thereis
evidencetosuggestthatmoresubtlemechanismsareinplay,suchastheuseofrare
codonstoslowdowntranslationinsectionsoftheproteinthatrequirecomplexco-
translationalfoldingforexample(ShermanandQian2013).
Whenone is tryingtoexpressproteins forcommercialuses,generally thehighest
possiblelevelofproteinaccumulationisdesired,somethingthatthehostcellrarely
attemptstodoinnatura.Ontheotherhand,bacteriophage,whichinfecthostcells
and whose genomes follow similar codon usage patterns to their host, often do
attempttoproduceasmuchproteinaspossible,tomakeasmanyphageprogenies
aspossible(Lucksetal.2008).Inthecaseofendolysins,astheyaredesignedtobe
highlyexpressedinnatura,itcanbeassumedthattheyarealreadycodedinanear-
optimalmannerforthehostbacterium.Thisraisesthepossibilityofgeneratingan
inter-species codon map, by which one can map a rare codon in Streptococcus
pneumoniaetoararecodoninC.reinhardtiiforexample,thusconservingthepattern
of“fast”and“slow”codonsthathasbeenoptimisedovermillennia.
Overall, this study demonstrates that there is huge potential to improve protein
accumulation yields in the C. reinhardtii chloroplast, and the technique of using
multipleexpressioncassetteinsertionsappearstobearobustmethodofimproving
proteinyields.However,othermethodsofimprovingproteinaccumulationsuchas
codon pair optimisation firstly require improvements in understanding of clonal
variation and transformosome effects before they can be properly assayed and
implemented.

146
Chapter4 ImprovingtheactivityofCpl-1againstStreptococcus
pneumoniae
4.1 Introduction
ThischapteraimstoimprovetheactivityoftheStreptococcuspneumoniaetargeting
endolysin, Cpl-1, in its ability to lyseS. pneumoniae cells. Several strategieswere
testedwiththisaiminmind,namely:manipulatingthecellwallbindingsiteofCpl-1;
attemptingtoformpre-dimerisedCpl-1endolysinsintheC.reinhardtiichloroplast;
and investigating holin production in theC. reinhardtii chloroplast.While Cpl-1 is
alreadyahighlyactiveendolysin,withprovenefficacy invivousingmouseandrat
models(Loeffleretal.2003;Entenzaetal.2005;McCullersetal.2007;Grandgirard
etal.2008),anincreaseinactivitywouldenableareductionindose,thusminimising
some of the potential limitations discussed in 1.2.2.4 involving immunogenic
reactions.
4.1.1 ManipulatingCpl-1bindingactivity
Thefunctionofanendolysin,whenproducedbyabacteriophage,istolysethehost
bacterium, thus allowing the release of phage progeny into the environment in
search of another bacterial host. The endolysin, therefore, should lyse only the
current host cell, and not neighbouring bacterial cells, which could be potential
futurehosts for theprogeny.Onecharacteristicofendolysins is that theytendto
bindstrongly,ofteninthepico-tonanomolarrange(Loessneretal.2002),totheir
substrate - it has been postulated that this is a tethering mechanism to reduce
unintentionaldamagetonearbybacteria(Chapot-Chartier2014).
Thisfeature,whilebeneficialtobacteriophage,oftenreducestheactivitylevelofthe
endolysin as an enzyme as it restricts its lytic activity to a localised area of the
peptidoglycan cell wall (Nelson et al. 2001). For some endolysins, such as Cpl-1,
removal of the C-terminal cell wall binding domain (CBD) results in an almost
completelossoffunction(Sanzetal.1992),butforotherssuchasCD27L(aclostridial
endolysin)removingtheCBDcanimprovelyticactivityandbroadenthespectrumof

147
bacteria against which it is active (in this case, to include two more species, in
additiontothesixspeciessusceptibletothefull-lengthCD27Lprotein)(Mayeretal.
2011).
Whileitisclearfromtheliterature(Sanzetal.1992;Garcíaetal.1999;Monterroso
etal.2008)thatacompleteremovaloftheCpl-1CBDresultsinareductioninactivity,
we propose that if binding could be weakened, activity may be improved. With
weakerbinding,theendolysinmaybeabletoreleaseafterlysisandrepeatitslytic
actionselsewhere,thusmakingtheendolysinmoresuitedtoatherapeuticrole.This
project involved mutating the cell wall binding site of Cpl-1 via site-directed
mutagenesis,andcomparingtheactivityof theresultingproteinswiththatof the
original.
Itisworthnotingthatinthiscaseofmanipulatingthecellwallbindingsite,weare
usingaslightlydifferentdefinitionof“activity”.TheSIunitforenzymeactivityisthe
katal,1katal=1mols-1,i.e.thenumberofmolesofsubstrateconvertedpersecond.
However,inthisinstance,wearemoreinterestedinthenumberofmolesconverted
perendolysinmolecule,whichiswhatistermed“activity”inthischapter.
From a protein engineering perspective, Gram-positive-targeting endolysins are
particularly attractive due to their modular structure, and the decoupling of the
bindingandcatalyticdomains.Inmanyglobularenzymes,thebindingandcatalytic
sitesarethesame,suchthatchangestobindingsitesoftenreducetheabilityofthe
enzymetocatalysethereaction.Whiletheendolysincatalyticdomainmuststillbind
its substrate, the majority of binding specificity and affinity is conferred by the
bindingdomain(Mayeretal.2011).
4.1.2 Dimerisationasamethodtoimproveproteinstability
Cpl-1exhibitsextensivesimilaritieswithbothmajorpneumococcalautolysins,LytA
andLytC.Inthepresenceofcholine,LytAdimerises–becomingstableandactive,
whileLytCisactiveasamonomerbutisstabilizedinthepresenceofcholine.Cpl-1is
believedtodimerisethroughitsC-terminal(cellwallbinding)domainwhencholine
ispresent.Theadditionalactivityofthedimersislikelytobeduetothedoublingof

148
FigureRemoved
activesitesspatiallyorientatednexttothepneumococcalcellwall(Monterrosoetal.
2008).
Figure4.1ThestableandactiveLytAdimer.Cellbindingdomainsareshowninorange,thecatalyticdomaininblueandthe10cholineligandsshowninCPKcolourscheme.Reschetal.(2011)postulatethatCpl-1coulddimerise inasimilarmannergiventhehomologyof thefinal13aminoacidsattheC-terminus.ImagereproducedfromMaestro&Sanz(2016).
Cpl-1 is known tobe cleared from thebloodplasmaofmice in approximately20
minutes(Witzenrathetal.2009),presentingapossibleissueforsystemicuse.Resch
etal.(2011)hypothesisedthata“pre-dimerised”Cpl-1homodimerwouldbeamore
stable and active therapeutic than thenativemonomer. LytAdimerisationoccurs
throughthe13aminoacidsattheC-terminus(Fernández-Torneroetal.2002),and
10ofthese13aminoacidswerefoundtobeidenticaltotheC-terminusofCpl-1.By
themutationofeachofthe13aminoacidsinturn,Reschetal.(2011)produceda
seriesofmutatedCpl-1proteins,inanattempttofindonewhichformsadisulphide
bridgebetweentwomonomerstoproduceahomodimer.Thesuccessfulmutated
Cpl-1dimerwasshowntohavea10-foldreductioninplasmaclearanceandatwo-
foldincreaseinactivity,comparedtothemonomericversion(Reschetal.2011a).
Cpl-1naturallydimerisesonbindingcholine,thecrystalstructureofwhichhasbeen
reportedbyBueyetal.(2007)andisdisplayedinFigure4.2.Thisdimerisationoccurs
notattheC-terminus,asthepre-dimerisingmutantcreatedbyReschetal.(2011)

149
FigureRemoved
does,butbetweenthefinalcholinebindingrepeatsattheN-terminusofthecellwall
binding domain – i.e. the choline binding repeat closest to the acidic linker and
catalyticdomain.
Figure4.2TheC-terminusoftheCBDiscircledinredineachmonomer.Theboxshowstheside-chainof thearomatic residue involved indimerisation.Reproduced fromBueyet al.(2007).
Figure4.2doesnotshowtheacidiclinkerjoiningthetwodomainsofeachmonomer
as this could not be accuratelymodelled. This observedmode of dimerisation is
similartothatofthedimer-dimerinteractionsofLytAtoformatetramer(Bueyetal.
2007).
ThequaternarystructureofLytAandLytCiscrucialtotheirhydrolyticactivity,and
given the homology each displays to Cpl-1, it is reasonable to believe that the
quaternarystructureofCpl-1isalsoimportant.However,giventhestabilityofCpl-1

150
bothasamonomerandwithoutthepresenceofcholine,itmaynotbeasimportant
aswiththepneumococcalautolysins.
4.1.3 Theroleofholinsinthebacteriophagelyticcycle
Inthecanonicaltwo-genemodelofbacteriophage-mediatedcelllysis,theholinplays
anessentialroleinenablingtheendolysintoaccessthepeptidoglycancellwall,by
creatingahole in thecytoplasmicmembraneof thebacterium(Young1992).The
timingof this event is critically important to the success of thebacteriophageby
controlling the length of the cycle, and thus the holin gene is under enormous
selectivepressure(Wangetal.2000).Itisperhapsduetothisselectivepressurethat
holins are such a diverse functional group, comprising seven superfamilies
encompassing52recognisedfamilieswithnoclearconservedsequencemotif(Reddy
and Saier 2013). However, all holins have certain characteristics including the
possessionofatleastonetransmembraneα-helicalsequenceandahighlycharged
hydrophilicC-terminaldomain(Guetal.2014).
The release of endolysins beyond the cytoplasmic membrane is a carefully
orchestratedevent,andonewhichisnotwhollyunderstood.Severaltheoriesexist
onhowthistimingiscontrolled,butthemostwell-establishedtheoryisthatthereis
alateexpressionofholins,whichaccumulateinthecytoplasmicmembrane.When
theconcentrationoftheseholinsreachesacriticalthreshold,araft isformedand
thenaconformationalchangecausestheformationofahole(Whiteetal.2011).This
sequenceofeventsisillustratedinFigure4.3.

151
FigureRemoved
Figure 4.3 An illustration of the holin triggering mechanism, and the consequentialdepolarisationofthemembrane.pmf=protonmotiveforce.
Afurthermethodofholincontrolexistsinsomebacteriophagesystems,involvingan
antiholin.Anantiholinisalmostidenticaltotheholin,butwithoneortwoadditional
N-terminalaminoacids,oneofwhichisapositivelychargedresidue(usuallylysine)
andistranslatedfromadualstartmotifencoding:Met-Lys-(x)-Met-….Theantiholin
thenbindstotheholinresultinginheterodimersunabletoformtheraftdisplayedin
Figure4.3.However, thisbinding isPMF (protonmotive force)-dependent:as the
antiholin isproducedataslightly lowerrate thantheholin,aholewilleventually
form,resultinginaloweringofthePMFandtheunbindingoftheanti-holinanda
positive-feedbackresultinginthecollapseofthePMFandthe‘allornothing’release
ofendolysinsbeyondthecell(Pangetal.2013;Lellaetal.2016).
Thegenecph1,encodingaholin,islocatedimmediatelyupstreamofcpl1intheCp-
1 bacteriophage genome. The two genes are transcribed from two tandem
promotersupstreamofcph1andareexpressedfromlatephagetranscripts(Martín
etal.1998).Thecph1genedoesnotdisplaytheclassicaldual-startmotif,althoughit
doesencodeanothermethionineresidueatpositionsix,butno lysineresidue lies
betweenthem.Cph-1belongstotheHolinIVsuperfamilyandhasthreepredicted
transmembraneα-helicalsegments(ReddyandSaier2013).
Recent literature has suggested that holins can be applied exogenously to
Streptococcussuis(Shietal.2012)andStaphylococcusaureus(Songetal.2016)in

152
conjunctionwithendolysinstoproduceanenhancedantibacterialeffect,improving
thekillingefficacyandalsobroadeningthespectrumofsusceptiblepathogens(Song
etal.2016).ThestudiesbyShietal.(2012)demonstratethatthetransmembrane
domainsoftheholinareessentialforitsantimicrobialactivity,anditisassumedthat
theformationoflargeholesinthemembranecausescelldeath.However,itisnot
understoodhowtheholingainsaccesstothecellmembranegiventhepresenceof
the peptidoglycan layer, nor how capsules and slime-layers affect holin access.
Furtherinvestigationofholinstructureanditspotentialasanovelantibacterialhas
been hindered by the difficulties involved in successfully expressing holins in
prokaryoticsystems,giventhecytotoxicityofholins(Garrettetal.1990).Therefore
weaimtoproduceholinsintheC.reinhardtiichloroplasttoenablefurtherresearch.
4.2 Aimsandobjectives
1. To identify and optimize a method for comparing activity levels between
differentmutantCpl-1endolysinsagainstS.pneumoniae.
2. ToproduceseventransgenicC.reinhardtiilinesexpressingcpl-1geneswith
differentmutationsintheircholinebindingsites(CBS).
3. ToconfirmaccumulationoftheCBSmutantCpl-1proteinsintheC.reinhardtii
chloroplastbywesternblotting.
4. To compare the activity of the CBS mutant Cpl-1 proteins against S.
pneumoniae.
5. To produce a transgenicC. reinhardtii line expressing a variant cpl-1 gene
encodingadimerisingmutant.
6. ToconfirmaccumulationofaCpl-1dimerintheC.reinhardtiichloroplastby
westernblotting.
7. To produce the holin Cph-1 in the C. reinhardtii chloroplast and confirm
accumulationbywesternblotting.

153
4.3 Resultsanddiscussion
4.3.1 ManipulationoftheCpl-1cholinebindingsitestoimproveactivity
The two choline binding sites on the C-terminus of the Cpl-1 protein recruit the
endolysintothecellwallbybindingtotheintegral(lipo)teichoicacids.Herethekey
residuesinvolvedinCpl-1cholinebindingareidentifiedandmutatedbysitedirected
mutagenesis.TheresultingCpl-1mutantsareassayedforlyticactivity.
4.3.1.1.1 Identificationofpotentiallyimportantresiduesincholinebinding
ThebindingaffinityoftheCpl-1cellbindingdomaintocholineisrelativelyweakfor
anendolysin,Kd≈3.6mM (Monterrosoetal. 2008),however the requirementof
bacteriophagestotethertheirendolysinstoonlythehost’scellwallsuggeststhatit
is unlikely to completely dissociate, and Cpl-1 possibly transits across the
glycopeptidenetworktoreachnewcleavablebonds(Jervisetal.1997;Monterroso
etal.2008).Inordertotestwhetherthereisanoptimalbindingaffinitythatimproves
activity,andliesbetweenthetwoextremesofcompleteCBDremovalandthenative
tetheringCBD,agradedweakeningoftheCBDwastested.
ThecrystalstructureofCpl-1bindingcholine,showninFigure3.1,wasanalysedusing
Jmol1.Interactionsoflessthan5angstromsfromthecholinemoietywereconsidered
to be of importance for hydrogen bonding during endolysin binding and two
tryptophanresiduesandonetyrosinewereidentifiedineachbindingsitetofitthese
criteria. This confirmed the original analysis by Hermoso et al. (2003)which also
identifiedthesethreeresiduesasimportant,formingahydrophobiccavityintowhich
thecholinemethylgroupsits.Hydrogenbondsformbetweenthethreeresiduesand
thecholineandtheseinteractionsareshowninFigure4.4.Forthepurposesofthis
investigation, the two tryptophan residues in each binding sitewere selected for
mutation.
1 Jmol is an open-source Java viewer for chemical structures in 3D.http://www.jmol.org/

154
ThecellwallbindingdomainofCpl-1hassixcholinebindingrepeats(p1-6,seeFigure
4.5),at the interfaceofwhichaputativecholinebindingsiteexists.However, the
crystalstructureofCpl-1suggeststhatonlythefirsttwocholinebindingsitesarein
factactive,namely:p1/p2andp2/p3(Hermosoetal.2003).Atthep1/p2site,there
aretwokeyinteractions:thenitrogenatomsonTrp202andTrp209interactingwith
thecholinemoiety.Atthesecondcholinebindingsite,thep2/p3site,itisTrp223and
Trp230thatinteractwiththecholine.WhenthefactthatanO-HbondandN-Hbond
are approximately one Angstrom in length is accounted for, the hydrogen bonds
formed in these interactions cover approximately 3-4 Angstroms. According to
Jeffrey (1997), hydrogen bonds over this distance range from “moderate,mostly
electrostatic”to“weak,electrostatic”.
Figure 4.4 The two active choline binding sites in the CBD of Cpl-1. Distances are inAngstroms.Itshouldbenotedthatthedistancesarebetweenthenitrogen(blue)andoxygen(red)atoms,soarenotadirectrepresentationofahydrogenbondlengthasthehydrogenmust be accounted for too in each case. However, these remain “moderate/weak andelectrostatic”hydrogenbonds.
Given that tryptophan is a polar amino acid, and it is this polaritywhich creates
hydrogenbonds,itwasdecidedtomutatethesetotheaminoacidalanine,whichis
non-polar.Thismethodofsystematicallysubstitutingresiduesforalanineisknown
as ‘alaninescanning’andisawidelyusedmutagenesisapproach(Kristensenetal.
p2/p3bindingsitep1/p2bindingsite

155
FigureRemoved
1997).Thedisruptionofthesehydrogenbondswasdesignedtoweakentheaffinity
ofCpl-1tocholine,andpotentiallyimprovethelyticactivityoftheendolysin.
4.3.1.2 ProductionofC.reinhardtiilinesexpressingcpl1CBSmutants
4.3.1.2.1 Site-directedmutagenesisofcpl1
Ofthefouridentifiedtryptophanresidues,shownaspartofthewholeCpl-1amino
acidsequenceinFigure4.6,noneofferedamoreorlessobvioustargetformutation
and therefore sevenC. reinhardtii cell lines expressing cpl1with either individual
tryptophanmutationsorcombinationsoftryptophanmutationsweregenerated.
Figure 4.5 The six putative choline binding repeats of Cpl-1. Asterisks indicate strictlyconservedresidues,whileacolonindicatesconservativesubstitutions.Redcircleshighlightthemutatedresidues.ReproducedandannotatedfromMonterrosoetal.(2008).

156
Figure4.6identifiesthefourmutatedtryptophanresiduesinthecpl1gene.Theseven
genemutantswhichwereintendedtobecreatedaredisplayedinFigure4.7,with
each tryptophan codon (TGG) beingmutated by site directedmutagenesis to an
alaninecodon(GCT).
Figure4.6Thefourtryptophanresiduesidentifiedtobeofimportanceincholinebindingarehighlightedinyellowinthecontextoftheentireproteinsequence.W202andW209formpartofthep1/p2cholinebindingsite,andW223andW230formpartofthep2/p3site.TheHAtagusedforwesternblotdetectionishighlightedinpurple.
MVKKNDLFVDVSSHNGYDITGILEQMGTTNTIIKISEST
TYLNPCLSAQVEQSNPIGFYHFARFGGDVAEAEREAQFF
LDNVPMQVKYLVLDYEDDPSGDAQANTNACLRFMQMIAD
AGYKPIYYSYKPFTHDNVDYQQILAQFPNSLWIAGYGLN
DGTANFEYFPSMDGIRWWQYSSNPFDKNIVLLDDEEDDK
PKTAGTWKQDSKGWWFRRNNGSFPYNKWEKIGGVWYYFD
SKGYCLTSEWLKDNEKWYYLKDNGAMATGWVLVGSEWYY
MDDSGAMVTGWVKYKNNWYYMTNERGNMVSNEFIKSGKG
WYFMNTNGELADNPSFTKEPDGLITVAYPYDVPDYA
202 209 223 230

157
Figure4.7Thegenecpl1wassubjectedtositedirectedmutagenesistocreatesevenuniquemutants. The naming of each construct indicates themutation location and nature, e.g.cpl1_W202A-HAisthecpl1genewithtryptophan-202mutatedtoanalanineresidue,andtaggedwithanHAtag.Multiplemutationsareseparatedbyanunderscore.
A pair of primers was designed for eachmutation, as displayed below, with the
mutationsiteinlowercase,andstartandstopcodonsunderlined.cpl1.Fandcpl1.R
aretheprimersatthestartandendofthegene.
cpl1.F: GCTCTTCAATGGTTAAAAAAAATGATTTATTC
cpl1.R: GCATGCTTATTAAGCATAATCTGGAAC
W202A_SDM.F: GTACAgctAAACAAGATTCAAAAG
W202A_SDM.R: CTTTTGAATCTTGTTTagcTGTAC
W209A_SDM.F: CAAAAGGTgctTGGTTTC
W209A_SDM.R: GAAACCAagcACCTTTTG
W223A_SDM.F: CCATATAATAAAgctGAAAAAATTG
Trp202 Trp209 Trp223 Trp230
TGG TGG TGG TGG
GCT TGG TGG TGG
TGG GCT TGG TGG
TGG TGG GCT TGG
TGG TGG TGG GCT
GCT GCT TGG TGG
TGG TGG GCT GCT
GCT GCT GCT GCT
cpl1_W202A-HA
cpl1_W209A-HA
cpl1_W223A-HA
cpl1_W230A-HA
cpl1_W202A_W209A-HA
cpl1_W223A_W230A-HA
cpl1_W202A_W209A_-W223A_W230A-HA
cpl1-HA

158
W223A_SDM.R: CAATTTTTTCagcTTTATTATATGG
W230A_SDM.F: GTGGTGTTgctTATTATTTCG
W230A_SDM.R: CGAAATAATAagcAACACCAC
Thefirstfourmutantgenes,whichhaveonlyoneW→Achange,wereproducedusing
site directed mutagenesis (SDM) as shown in Figure 4.8. These mutated DNA
sequenceswerethenusedastemplateDNAinsubsequentroundsofSMDPCRto
producethefinalthreegenes,whichhavemorethanonemutatedcodon.
Figure 4.8 Site directedmutagenesiswas performed as above tomutate the tryptophanencodingcodon,TGG,tothealanineencodingone,GCT.PrimersareshowninblueandPCRconditionsaredescribedin2.3.7.
Of thesevenmutants, sixweresuccessfully created.The transformationofE. coli
withcpl1_W223A-HAinanintermediatevector,pJet,wasrepeatedlyunsuccessful.It
isnotclearwhythisparticularmutantwassoproblematic,anditisdiscussedfurther
in4.4.1,butduetotimeconstraintsitwasnotpossibletoinvestigatethisfurther.
TGGACC
GCT
CGA
GCTCGA
GCTCGA
1st PCR
2nd PCR
Finalproduct
Cpl-1

159
4.3.1.2.2 Expressioncassetteproduction
TheresultingPCRproductsweredigestedwithSapIandSphIand ligated in-frame
intothepSRSapIvector.TheinsertionwasconfirmedbyDNAsequencing.
4.3.1.2.3 C.reinhardtiitransformation
TheC. reinhardtii recipient lineTN72was transformedby theglassbeadmethod,
using the pSRSapI_cpl1 plasmids (2.3.13). The resulting transformed lines are
representedinFigure4.9.PhotosystemIIfunctionalityisrestoredbypsbHandthe
resulting transformants were capable of growing phototrophically on minimal
medium.Eachofthesixtransformedlineswasshowntoreachahomoplasmicstate
by PCR as described in 2.3.17. As a final step, the integrity of the cassette was
confirmedbyDNAsequencingofthePCR-amplifiedplastomeregion,asin2.3.18.
Figure 4.9 The resulting C. reinhardtii lines express the mutated cpl1 gene under theendogenous psaA exon 1 5’UTR/promoter. The successfully transformed colonies areselectedbytheirabilitytogrowphototrophicallyonminimalmedium.
4.3.1.2.4 Westernblotconfirmation
WholecellextractfromeachofthesixC.reinhardtiicelllineswasseparatedbySDS-
PAGE and blotted to nitrocellulose membrane for western blot analysis. The
membrane was probed with anti-HA primary antibodies followed by IRDye®
secondary antibody to enabledetectionusing the LiCorOdyssey®CLx system.An
imageofthewesternblotanalysisisshowninFigure4.10andrelativequantification
ofCpl-1accumulationisshowninFigure4.11.TheerrorbarsinFigure4.11represent
thestandarddeviationinsignaldetectionwhenthisblotwasrepeated.
trnE2 psbH
5’psaA 1 3’rbcL
TN72_SR_cpl1_WxxxA-HAtransformant
MluI
cpl1_WxxxA-HA

160
Figure4.10WesternblotanalysisofeachofthesixC.reinhardtii linesexpressingmutatedcpl1. Visible levels of accumulation are seen in all cases. TN72_SR_cpl1-HA andTN72_SR_Controlareincludedaspositiveandnegativecontrols,respectively.
Figure4.11Quantificationoftwowesternblotanalyses.Ingeneral,thefirstfourcell linesproduce levels of protein accumulation comparable to the unmodified version, whileTN72_SR_cpl1_W223A_W230A-HA and TN72_SR_cpl1_W202A_W209A_W223A_W230A-HAaccumulatetolowerlevels,approximately30%ofthatobservedfortheothers.
TN72
_SR_
cpl1_W
202A
-HA
TN72
_SR_
cpl1_W
209A
-HA
TN72
_SR_
cpl1_W
230A
-HA
TN72
_SR_
cpl1_W
202A
_W20
9A-HA
TN72
_SR_cpl1_W
223A
_W23
0A-HA
TN72
_SR_cpl1_W
202A
_W20
9A_-22
3A_W
230A
-HA
TN72
_SR_cpl1-HA
TN72
_SR_Co
ntrol
Cpl-1(39kDa)
0
0.2
0.4
0.6
0.8
1
1.2
TN72
_SR_cpl1_W20
2A-HA
TN72
_SR_cpl1_W20
9A-HA
TN72
_SR_cpl1_W23
0A-HA
TN72
_SR_cpl1_W20
2A_
W20
9A-HA
TN72
_SR_cpl1_W22
3A_
W23
0A-HA
TN72
_SR_cpl1_W20
2A_
W20
9A_-W22
3A_W
230A
-HA
TN72
_SR_cpl1-HA
TN72
_SR_Co
ntrol
RelativeFluorescence

161
As discussed at length in 3.3.2.2.4.3, the reason for the difference in protein
accumulation levels isdisputable.However, for thepurposesof this investigation,
onlytherelativeaccumulationlevelsarenecessarytoensurethatequalquantitiesof
proteinwereusedinfutureassays.
4.3.1.3 ActivityofCpl-1CBSmutantsagainstS.pneumoniae
ThesixC.reinhardtiilinesdescribedabovewerecreatedwiththeintentionofaltering
Cpl-1 activity. To measure this activity several approaches were taken, namely:
turbidity reduction assays and colony forming unit assays, using both crude cell
extracts, and extracts enriched and concentrated by ammonium sulphate
precipitationorepitopetagenrichment.
4.3.1.3.1 Activityofcrudeextracts–turbidityreductionassay
OneoftheproposedadvantagesofusingC.reinhardtiiasarecombinantendolysin
productionplatformisthat,duetoitsGRASstatus,simplecrudeextractscouldbe
used therapeutically, particularly for topical applications, thus removing the
expensiverequirementtopurifythefinalprotein.
Itwas therefore investigatedwhethercrudeextractsof thesixCpl-1CBSmutants
described in this section display different levels of lytic activity towards S.
pneumoniae,whencomparedtotheactivityoftheun-mutatedCpl-1protein.
Crudeextractswerepreparedasdescribedin2.4.1.Inbrief,thesixC.reinhardtiicell
lines were grown simultaneously and their OD740 measured after 72 hours. The
cultureswerecentrifugedandthepelletresuspendedin20mMNaPibuffertogive
suspensionsofequivalentcelldensitymeasuredatOD740.Thecellswere lysedby
freeze-thaw and ultracentrifuged to remove the cell debris. The supernatantwas
stored at 4 °C and thiswas considered the “crude extract”. Fresh S. pneumoniae
cultures were prepared as described in 2.7.1.1 and resuspended in 20mMNaPi
buffertoanOD595=~0.8.
Ina96-wellplate,S.pneumoniaecellsandC.reinhardtiicrudeextractwerecombined
andthechangeinOD595measuredwithaplatereaderoveraperiodof150minutes
at37°C.200μlofresuspendedS.pneumoniaecellswereincubatedwith~50μlof

162
crudeextract.TheprecisevolumewasadjustedaccordingtodatashowninFigure
4.11,toensureanequalquantityofCpl-1proteinwasincludedineachwell.Crude
TN72_SR_Controlwasusedasanegativecontrol.

163
Figure4.12TurbidityreductionassayofS.pneumoniaewhenchallengedwithcrudeC.reinhardtiiextractscontainingdifferentCpl-1CBSmutants.ChangeinOD595isshownratherthanmeasuredOD595duetothedifferentvolumesofcrudeextractineachwell.Eachcombinationwasperformedintriplicateandtheaverageisshown.
-0.3
-0.25
-0.2
-0.15
-0.1
-0.05
00 15 30 45 60 75 90 105 120 135
ChangeinOD(595nm)
Time(minutes)
TN72_SR_cpl1_W202A-HA TN72_SR_cpl1_W209A-HATN72_SR_cpl1_W230A-HA TN72_SR_cpl1_W202A_W209A-HATN72_SR_cpl1_W223A_W230A-HA TN72_SR_cpl1_W202A_W209A_W223A_W230A-HATN72_SR_Control TN72_SR_cpl1-HANaPiBuffer(20mM)

164
From Figure 4.12, it appears that the un-modified TN72_SR_cpl1-HA is themost
active endolysin, while all the mutated endolysins showed activity intermediate
between this and the negative control, TN72_SR_Control. The high level of lysis
observedbythebufferaloneissurprising.However,ithasbeennotedinprevious
endolysinactivityassaysusingcrudeextractsthatan increase inosmoticpressure
duetothepresenceoflargequantitiesofproteininthesuspensioncanleadtoan
inhibitionofcelllysis(Mayeretal.2010)-thisisexemplifiedherebythehighlevelof
autolysisobservedwhenNaPibuffer(20mM)isadded,comparedtothatseeninthe
negativecontrol.
FirstinspectionofthedatashowninFigure4.12suggeststhatalteringtheCBDofCpl-
1hasadetrimentaleffectuponenzymaticactivity.Whilethisresult isnotentirely
unexpected, given the issue of osmotic pressure affecting bacterial cell lysis,
combinedwiththedifferentlevelsofproteinaccumulationobservedinFigure4.11,
itwasdecidedthatturbidityreductionassayswithcrudeextractsareinsufficiently
precisetoenableacomparisonofthevariousCpl-1CBSmutantactivities.
4.3.1.3.2 EnrichmentofCBSmutantCpl-1byammoniumsulphateprecipitation
Inordertoaddresstheaboveconcerns,ammoniumsulphateprecipitationwasused
to partially purify and concentrate the Cpl-1 CBSmutants, providing an enriched
crude extract. The enriched extracts were separated by SDS-PAGE, blotted to
nitrocellulose membrane and probed with antibodies for western analysis. The
resultinganalysisandrelativequantificationaredisplayedinFigure4.13.

165
Figure4.13ThewesternblotanalysisofsixCpl-1CBSmutantsconcentratedandenrichedbyammoniumsulphateprecipitation,alongsidepositiveandnegativecontrols(TN72_SR_cpl1-HAandTN72_SR_Control,respectively).ImageStudioLite5.0softwareandLiCorOdyssey®CLxsystemenabledthequantificationofeachband,shownbeloweachlaneasabarchart.
4.3.1.3.3 Activityofextractsenrichedbyammoniumsulphateprecipitation-Colony
formingunitassay
The turbidity reduction assay shown in Figure 4.12 required fresh S. pneumoniae
culturestobegrownonthemorningofeachassay,whichoftenleadstodifficulties
inthereproducibilityoftheassay.Ontheotherhand,colonyformingunitassays,as
described in2.7.1.1,wereperformedusingsnap-frozenaliquotsofS.pneumoniae
suspensions,thusenablingamuchmorereproducibleandrobustassay.Thenumber
of colony forming units was counted by eye and when an uncountable lawn of
indistinguishablecolonieswaspresent,thiswascountedas“100”.Serialdilutionsof
TN72
_SR_
cpl1_W
202A
-HA
TN72
_SR_
cpl1_W
209A
-HA
TN72
_SR_
cpl1_W
230A
-HA
TN72
_SR_
cpl1_W
202A
_W20
9A-HA
TN72
_SR_cpl1_W
223A
_W23
0A-HA
TN72
_SR_cpl1_W
202A
_W20
9A_-22
3A_W
230A
-HA
TN72
_SR_cpl1-HA
TN72
_SR_Co
ntrol
0
20000
40000
60000
80000
100000
120000
140000
TN72
_SR_cpl1_W20
2A-HA
TN72
_SR_cpl1_W20
9A-HA
TN72
_SR_cpl1_W23
0A-HA
TN72
_SR_cpl1_W20
2A_
W20
9A-HA
TN72
_SR_cpl1_W22
3A_
W23
0A-HA
TN72
_SR_cpl1_W20
2A_
W20
9A_W
223A
_W23
0A-HA
TN72
_SR_cpl1-HA
TN72
_SR_Co
ntrol
Relativefluorescence

166
bacterial suspension containing endolysin were performed after one hour of
incubationat37°C,andthree10μldropsofeachdilutionwerespottedontoblood
agarplates.Themeancountacrossthethreespotswascalculated.Thedatafrom
Figure 4.13 enabled similar quantities of HA-tagged protein to be added to the
bacterialsuspension.

167
Figure4.14ColonyformingunitassayofS.pneumoniaecellstreatedwithCpl-1CBSmutantammoniumsulphate-enrichedcrudeextracts.
0
10
20
30
40
50
60
70
80
90
100
10^2 10^3 10^4 10^5 10^6 10^7
Colonyfo
rmingunits
DilutionFactor
TN72_SR_cpl1_W202A-HA
TN72_SR_cpl1_W209A-HA
TN72_SR_cpl1_W230A-HA
TN72_SR_cpl1_W202A_W209A-HA
TN72_SR_cpl1_W223A_W230A-HA
TN72_SR_cpl1_W202A_W209A_W223A_W230A-HATN72_SR_Control
TN72_SR_cpl1-HA
20mMNaPiBuffer

168
ThecolonyformingunitassayinFigure4.14confirmsthatTN72_SR_cpl1-HAismore
activethananyoftheCpl-1CBSmutants,howevertherestillexiststheproblemof
varyingosmoticpressureduetothevaryingconcentrationsofproteinpresent.While
equal quantitiesof endolysinhavebeenachieved, this hasprobably resulted in a
different levelof totalprotein,becauseammoniumsulphateprecipitationenables
the enrichment of all proteins within a range of solubility, which undoubtedly
includesmanyotherproteins.InordertoincludesimilarquantitiesoftheHA-tagged
Cpl-1CBSmutant protein, differing volumesof the ammonium sulphate enriched
extractwereused, thusaltering theosmoticpressureonthebacterialcells in the
suspension.Inthiscasetoo,itisworthnotingthatthe20mMNaPibufferiscausing
lysis to a greater extent than all of the extracts except for the unmodified
TN72_SR_cpl1-HA.
4.3.1.3.4 Proteinenrichmentviaepitopetags–HAtags
AllCpl-1CBSmutantsweretaggedwithanHAepitopetagfordetectioninwestern
blotanalysis,butitisalsopossibletousethisepitopetopurifytheprotein.Eachof
thesixCpl-1CBSmutantswaspurifiedusingananti-HAresin,andthefractionswere
spottedontoanitrocellulosemembraneforadotblot,asdescribedin2.4.3.After
probing with anti-HA antibodies and IR secondary antibody, the membrane was
analysedusingtheLiCorOdyssey®CLxsystem,showninFigure4.15

169
Figure4.15DotblotanalysisofHA-resinpurificationfractions.TheC.reinhardtiilineswereabbreviatedtojustthemutationforclaritye.g.‘TN72_SR_cpl1_W202A-HA’isjustlabelled‘W202A’. Mixed results were achieved, with W202A, W202A_W209A,W202A_W209A_W223A_W230A,andtheun-mutatedCpl-1beingtheonlystrainswheretheelutionhadvisiblequantitiesofHA-taggedprotein.
ItisnoteworthythatTN72_SR_Controlgivesastrongsignalforthecrudeextractand
flow-through,which suggests that there are background proteins fluorescing and
disguising the HA-tagged proteins in these fractions. However, these background
proteins are not present in the elution fractions. To confirm that the fluorescing
proteinisindeedtheendolysin,themostpromisingexamplesfromthedotblotwere
separatedbySDS-PAGE.Westernblotanalysisallowsrelativequantificationofthe
yieldachievedwiththispurificationmethod,showninFigure4.16.
W202A
W209A
W230A
W202A_W209A
W223A_W230A
W202A_W209A_W223A_W230A
TN72_SR_cpl1-HA
TN72_SR_Control
Crud
eExtract
Flow
through
Wash1
Wash2
Elution1
Elution2
Elution3
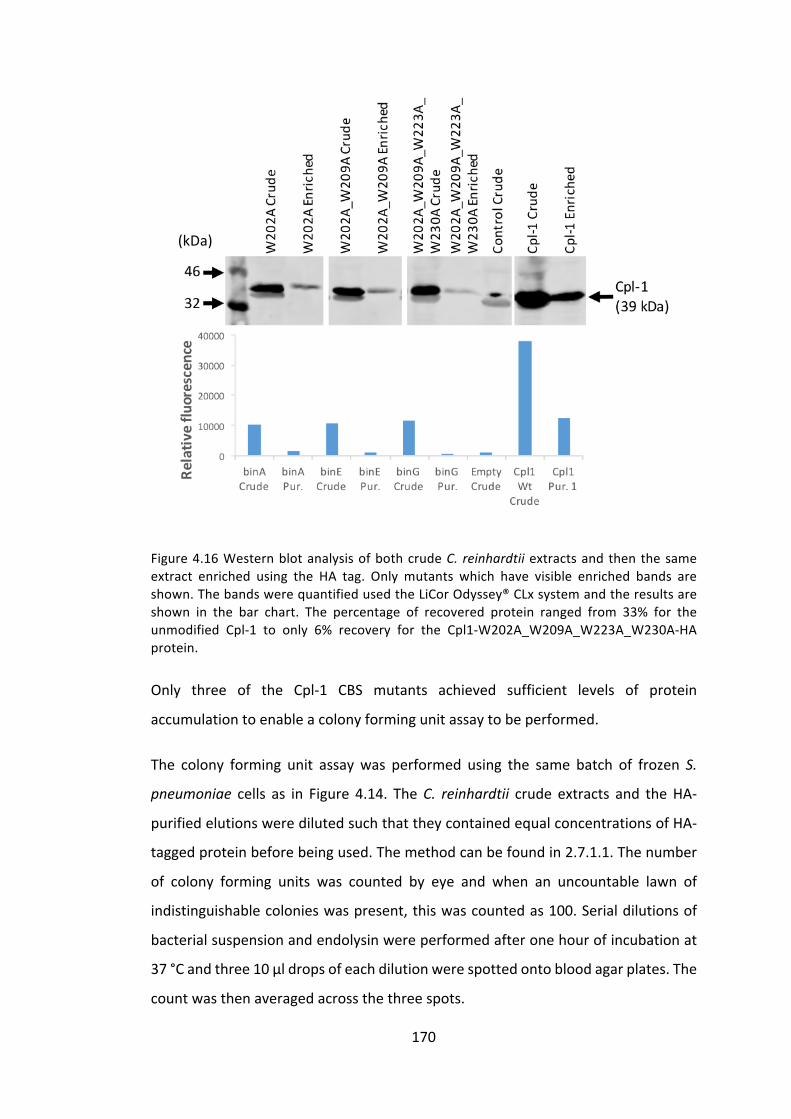
170
Figure4.16WesternblotanalysisofbothcrudeC. reinhardtiiextractsandthenthesameextract enriched using the HA tag. Only mutants which have visible enriched bands areshown.ThebandswerequantifiedusedtheLiCorOdyssey®CLxsystemandtheresultsareshown in the bar chart. The percentage of recovered protein ranged from 33% for theunmodified Cpl-1 to only 6% recovery for the Cpl1-W202A_W209A_W223A_W230A-HAprotein.
Only three of the Cpl-1 CBS mutants achieved sufficient levels of protein
accumulationtoenableacolonyformingunitassaytobeperformed.
The colony forming unit assaywas performed using the same batch of frozen S.
pneumoniae cells as in Figure 4.14. TheC. reinhardtii crude extracts and theHA-
purifiedelutionsweredilutedsuchthattheycontainedequalconcentrationsofHA-
taggedproteinbeforebeingused.Themethodcanbefoundin2.7.1.1.Thenumber
of colony forming units was counted by eye and when an uncountable lawn of
indistinguishablecolonieswaspresent,thiswascountedas100.Serialdilutionsof
bacterialsuspensionandendolysinwereperformedafteronehourofincubationat
37°Candthree10μldropsofeachdilutionwerespottedontobloodagarplates.The
countwasthenaveragedacrossthethreespots.
(kDa)
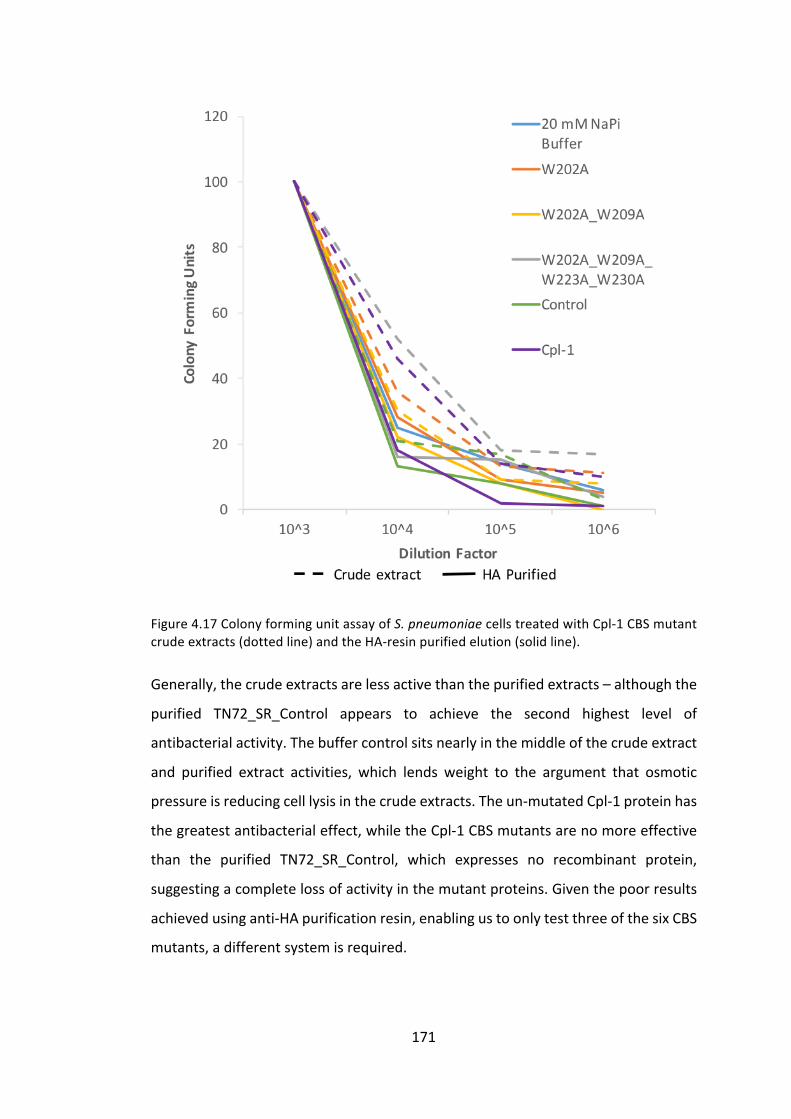
171
Figure4.17ColonyformingunitassayofS.pneumoniaecellstreatedwithCpl-1CBSmutantcrudeextracts(dottedline)andtheHA-resinpurifiedelution(solidline).
Generally,thecrudeextractsarelessactivethanthepurifiedextracts–althoughthe
purified TN72_SR_Control appears to achieve the second highest level of
antibacterialactivity.Thebuffercontrolsitsnearlyinthemiddleofthecrudeextract
and purified extract activities, which lends weight to the argument that osmotic
pressureisreducingcelllysisinthecrudeextracts.Theun-mutatedCpl-1proteinhas
thegreatestantibacterialeffect,whiletheCpl-1CBSmutantsarenomoreeffective
than the purified TN72_SR_Control, which expresses no recombinant protein,
suggestingacompletelossofactivityinthemutantproteins.Giventhepoorresults
achievedusinganti-HApurificationresin,enablingustoonlytestthreeofthesixCBS
mutants,adifferentsystemisrequired.

172
4.3.1.3.5 Proteinenrichmentviaepitopetags–StrepIItags
TheHAepitopehasbeenwidelyused in thePurton group, aswell as other algal
research groups. The original expression of cpl1 in C. reinhardtii encoded an HA
epitope tag on its C-terminus (Taunt 2013), and this was continued into this
investigation.However,whileconvenientforwesternblotdetection,usingtheHA-
tag for protein purification is expensive compared to some other epitopes, as it
involvesaffinitypurificationusingananti-HAmonoclonalantibody.TheStrepIItag
waschosenasanalternativeas ithasbeenused inthealgal fieldtopurifynative
chloroplast proteins from C. reinhardtii (Derrien et al. 2012; Derrien 2013).
Furthermore, as the system is based on the affinity of biotin and streptavidin,
purificationcolumnsarerelativelycheap.3.3.2.2.3describestheproductionoftheC.
reinhardtii line TN72_SR_cpl1-StrepII, which is used again here to investigate the
StrepII-tagforproteinpurificationandenrichment.Polyhistidinetags(His-tags)are
themostcommonepitopetagusedforthispurposebutaStrepIItagwaschosenin
thisinstanceduetoitsprevioussuccessintheC.reinhardtiichloroplast,aswellas
thefactthatitcanbereadilyusedfordetectioninwesternblotting,whileantibodies
toHistagsaremoreproblematic.
4.3.1.3.6 EnrichmentofCpl1-StrepIIusingHiTrapStrepIIColumn
C.reinhardtiicellextractswerepreparedasdescribedin2.5.1,andultracentrifuged
at100,000gfor1hourtoremovecelldebris.Thisultracentrifuged(UC)extractwas
then enriched for StrepII-taggedproteins using the commercially availableHiTrap
StrepIIColumn(GELifesciences)andelutedwithdesthiobiotin,asdescribedin2.5.3.

173
Figure4.18SamplesfromextractpreparationandtheHiTrapStrepIIcolumnwerecollectedandseparatedbySDS-PAGE.WesternblotanalysisandrelativequantificationusingtheLiCorOdyssey® CLx system enabled the StrepII-tagged protein recovery to be estimated as apercentageoftheultracentrifuged(UC)extractpassedthroughthecolumn.
Approximately one-third of the total protein remains in the supernatant on
ultracentrifugation,butvirtually100%ofthiswasrecoveredusingtheHiTrapStrepII
column. Further optimisation of ultracentrifugation speeds and durations could
increase the proportion remaining in the supernatant further, but for this
investigationmaximisingtotalproteinyieldisnottheaim.
Asanegativecontrol,HiTrapStrepIIpurificationwasalsoperformedwithanextract
ofTN72_SR_Control.Thiscontrols foranyC.reinhardtiicellcomponentsthatmay
havenon-specificallyboundtothecolumnandbepresentintheelutedfraction.

174
4.3.1.3.7 ActivityofpurifiedCpl1-StrepII
AS.pneumoniaecolonyformingunitassaywasperformedasdescribedpreviously
(4.3.1.3.4),usingtheelutedfraction,describedhereasan“enrichedextract”,from
bothTN72_SR_cpl1-StrepIIandTN72_SR_Control.Bacterialsuspensionandextract
were combined in a 1:1 ratio and serial dilutions of bacterial suspension and
endolysinwereperformedafteronehourofincubationat37°C,whereuponthree
10μldropsofeachdilutionwerespottedontobloodagarplates.
Figure 4.19 Colony forming unit assay of S. pneumoniae cells treated with Cpl1-StrepIIenrichedextracts.“Control”referstoapurifiedextractfromTN72_SR_Control.Thenumberof colony forming units was counted by eye and when an uncountable lawn ofindistinguishable colonies was present, this was counted as 100. The count was thenaveragedacrossthethreespots.Threedilutionsoftheenrichedextractweretested(100%,75 % and 25 %), and the percentage indicates the initial concentration, not the finalconcentration(i.e.priortomixingwiththebacterialsuspension).

175
The colony forming unit assay in Figure 4.19 indicates a measurable difference
betweenthethreeCpl-1dilutions,andtheyareclearlymoreactivethantheextract
fromTN72_SR_Controlatthetwohigherconcentrations.Thesedatasuggestthatthis
method of enrichment removes both the mild antibacterial activity of the C.
reinhardtiicrudeextractandtheosmoticpressureeffectthatseemedtobemasking
resultsinprevioustrials.
Unfortunately,timelimitationspreventedtheapplicationofthismorerobustassay
totheinvestigationofbindingsitemutation.ByreproducingeachoftheC.reinhardtii
linesexpressingCBSmutants,thistimeencodingaStrepIIepitopeinsteadofanHA
epitope, itwouldbepossibletomoreaccuratelycomparetherelativeactivitiesof
the mutants. However, given the problems encountered above and the longer
transformationtimeofC.reinhardtiilines,inthisparticularcaseinvestigationscould
bemoreeasilycarriedoutusingrecombinantproteinsproducedinE.coli.E.coliisan
excellentplatformforrapidlyproducingrecombinantproteinstoahighlevelandin
this case would allow easy comparison of different mutants. The best of these
mutants,ifindeedcellwallbindingsitemutagenesiscanimproveendolysinactivity,
couldthenbeexpressedinC.reinhardtiiforthefinaltherapeuticdrugproduction.In
addition, proteins toxic to E. coli, such as the endolysin SPN9CC, which can be
expressedinC.reinhardtii(YoungandPurton2015)couldbeinvestigatedusingthis
method.

176
4.3.2 ImprovingCpl-1stabilitythroughdimerisation
TheC. reinhardtii chloroplast is known to produce and regulate the formation of
disulphidebonds(Kim1997),andresearchsuggeststhatyieldsofproteinscontaining
disulphidebondscanbeimprovedintheC.reinhardtiichloroplastbytheadditionof
selenocystaminetotheculturemedium(Ferreira-Camargoetal.2015).Furthermore,
Resch et al. (2011) showed that Cpl-1 can be modified to “pre-dimerise”, thus
decreasingplasmaclearanceandimprovingitsclinicalpotential.
Cpl-1 is stable as amonomer, but Cpl-1 dimerisation occurs naturally on choline
binding (Buey et al. 2007). We expect that the addition of a disulphide bridge
between two Cpl-1 monomers will reduce the rate of degradation in storage by
limitingbothproteaseaccessandthedenaturingoftheprotein,aswellasachieving
thedecreasedplasmaclearancereportedbyReschetal.(2011).Theaimisalsoto
show that the C. reinhardtii chloroplast is a suitable platform for producing
disulphide-bondedendolysinsastherapeutics.
4.3.2.1.1 StabilityofCpl-1undervaryingconditions
AninitialinvestigationintothestabilityofCpl-1instoragewasconducted.Anextract
fromtheC.reinhardtiilineTN72_SR_cpl1-HAwaspreparedasdescribedin2.5.1.The
cellswere either resuspended and lysed in 20mMNaPi buffer or 20mMNaPi +
ProteaseInhibitorbuffer,thenbothwereultracentrifugedforonehourat100,000g.
Eachsupernatantwasthensplitintotwotubesandstoredat25°Cor4°Cforseven
days.Thefourtubesweresampledat24-hourintervalsandthesamplesweresnap-
frozeninliquidnitrogenandstoredat-80°C.Attheendoftheseven-dayperiod,all
samples were thawed and separated by SDS-PAGE before being blotted to
nitrocellulosemembraneandanalysedbywesternblotting.Relativequantification
usingtheLiCorOdyssey®CLxsystemproducedthedatadisplayedinFigure4.20.
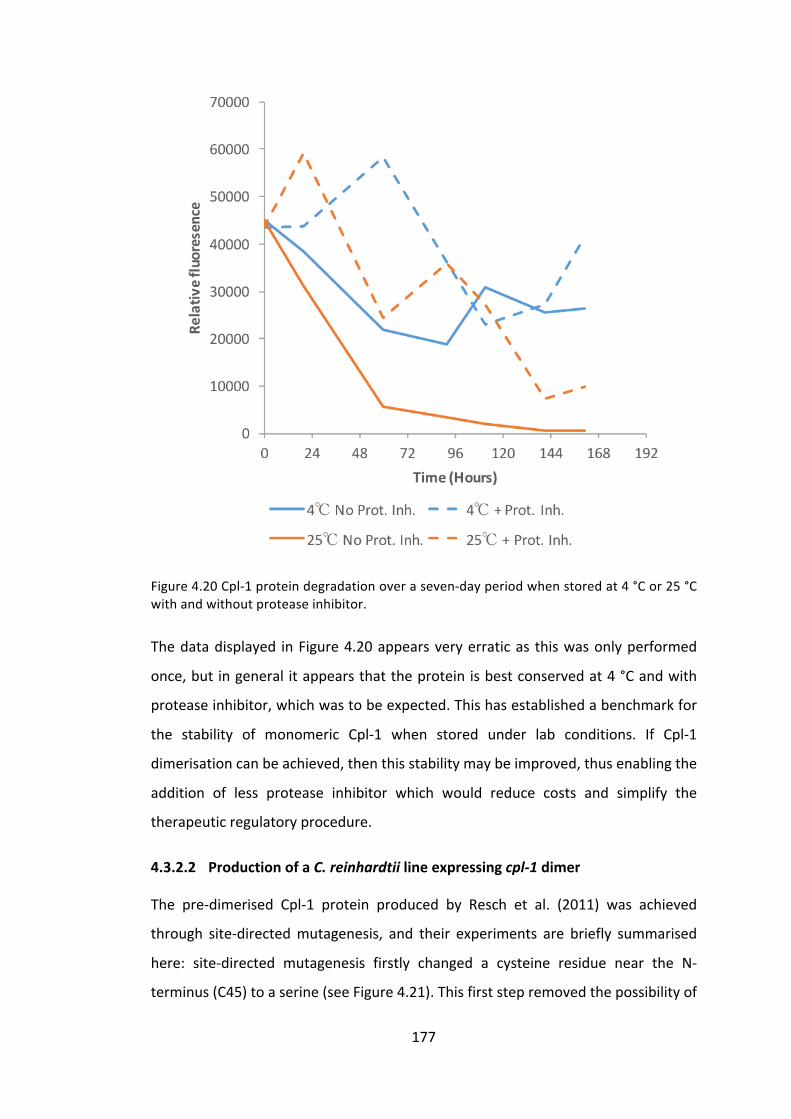
177
Figure4.20Cpl-1proteindegradationoveraseven-dayperiodwhenstoredat4°Cor25°Cwithandwithoutproteaseinhibitor.
Thedatadisplayed inFigure4.20appearsveryerraticas thiswasonlyperformed
once,butingeneral itappearsthattheproteinisbestconservedat4°Candwith
proteaseinhibitor,whichwastobeexpected.Thishasestablishedabenchmarkfor
the stability of monomeric Cpl-1 when stored under lab conditions. If Cpl-1
dimerisationcanbeachieved,thenthisstabilitymaybeimproved,thusenablingthe
addition of less protease inhibitor which would reduce costs and simplify the
therapeuticregulatoryprocedure.
4.3.2.2 ProductionofaC.reinhardtiilineexpressingcpl-1dimer
The pre-dimerised Cpl-1 protein produced by Resch et al. (2011) was achieved
through site-directedmutagenesis, and their experiments are briefly summarised
here: site-directed mutagenesis firstly changed a cysteine residue near the N-
terminus(C45)toaserine(seeFigure4.21).Thisfirststepremovedthepossibilityof

178
interactions with this solvent-accessible cysteine. Then an aspartic acid residue
(D324), inaposition13aminoacidsawayfromtheC-terminus,wasmutatedtoa
cysteine. The 13 final amino acids are highly conserved and, in the case of the
homologouslysinLytA,responsibleforLytAdimerisation,sowerenotmodified.
4.3.2.2.1 Site-directedmutagenesisofcpl1
Sitedirectedmutagenesiswasperformedasdescribedin2.3.7.Theprimersusedin
thisinstancearebelow,andmutationsitesareinlowercase:
cpl1.F: GCTCTTCAATGGTTAAAAAAAATGATTTATTC
cpl1_C45S.F ACATATTTAAATCCATcaTTATCAGCTC
cpl1_C45S.R GAGCTGATAAtgATGGATTTAAATATGT
cpl1_D324C.F GTGAATTAGCAtgtAATCCTTCATTT
cpl1_D324C.R AAATGAAGGATTacaTGCTAATTCAC
cpl1.R: GCATGCTTATTAAGCATAATCTGGAAC

179
Figure4.21Thetwoaminoacidsselectedformutationarehighlighted inyellow.C45wasmutated to a serine to prevent unwanted interactions, as this cysteine is believed to besolvent-accessible. D324 was mutated to a cysteine to form a disulphide bridge in ahomodimer.TheHAtagishighlightedinpurple.
4.3.2.2.2 Expressioncassetteproduction
ThefinalPCRproduct,namedcpl1_dimer-HA,wasinsertedintothepSRSapIvector
usingSapIandSphIaspreviouslydescribed(4.3.1.2.2).CompetentDH5αE.coliwere
then transformedwith the resultant plasmid and DNA sequencing confirmed the
correctinsertionandthattheintendedmutationshadbeensuccessfullyintroduced.
4.3.2.2.3 C.reinhardtiitransformation
The TN72 recipient line was transformed with the pSRSapI_cpl1_dimer-HA
transformation plasmid. Transformed C. reinhardtii lines, named
TN72_SR_cpl1_dimer-HAwerecheckedforhomoplasmicitybyPCR(AppendixI)and
finallyconfirmedbysequencing.

180
4.3.2.2.4 Westernblotconfirmation
Whole cell extracts of TN72_SR_cpl1-HA and TN72_SR_Control (as controls) and
TN72_SR_cpl1_dimer-HAwereprepared,bothwithandwithoutβ-mercaptoethanol
(BME), and separated by SDS-PAGE. The BME reduces disulphide bonds so was
includedtotestifthedimerisedCpl-1couldbedetectedwhenBMEwasabsentbut
not when present – this would confirm disulphide presence. After blotting to
nitrocellulose membrane and blocking as described in 2.4.5, the membrane was
probed with anti-HA primary antibody, then incubated with IRDye® secondary
antibody. Fluorescence was observed using the LiCor Odyssey® CLx system. The
resultinganalysisisshowninFigure4.22.
Figure4.22Westernblot analysisof TN72_SR_cpl1_dimer-HA. The loadingmixtureeithercontainedβ-mercaptoethanolordidnot,indicatedby+/-BME.ThepredictedbandlocationforCpl-1_dimerisindicatedbutnobandswereobserved.
No spontaneously forming Cpl-1 dimerswere observed in thewhole cell extract.
However, monomeric Cpl-1 still accumulates to similar levels as seen in
TN72_SR_cpl1-HA.Thepresenceofthemonomersuggeststhatthereisnoissuewith
synthesis,butforsomereasondimerisationisnotoccurring,perhapsduetosteric
issuesinvolvingtheC-terminalHAtag,orsimplythat,attheconcentrationsobserved,
theequilibriumisshiftedtowardsmonomersanddimersarenotformed.
(kDa)
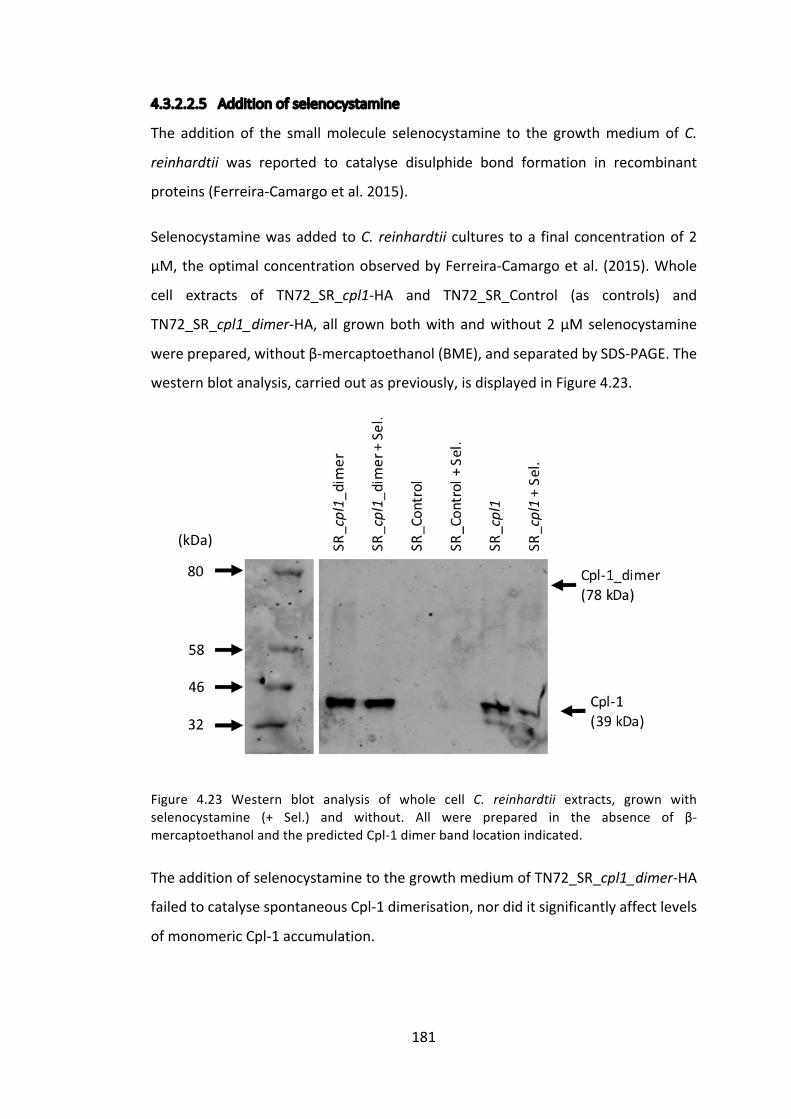
181
4.3.2.2.5 Additionofselenocystamine
The addition of the smallmolecule selenocystamine to the growthmediumofC.
reinhardtii was reported to catalyse disulphide bond formation in recombinant
proteins(Ferreira-Camargoetal.2015).
SelenocystaminewasaddedtoC. reinhardtii cultures toa finalconcentrationof2
μM,theoptimalconcentrationobservedbyFerreira-Camargoetal. (2015).Whole
cell extracts of TN72_SR_cpl1-HA and TN72_SR_Control (as controls) and
TN72_SR_cpl1_dimer-HA, all grownbothwithandwithout2μMselenocystamine
wereprepared,withoutβ-mercaptoethanol(BME),andseparatedbySDS-PAGE.The
westernblotanalysis,carriedoutaspreviously,isdisplayedinFigure4.23.
Figure 4.23 Western blot analysis of whole cell C. reinhardtii extracts, grown withselenocystamine (+ Sel.) and without. All were prepared in the absence of β-mercaptoethanolandthepredictedCpl-1dimerbandlocationindicated.
TheadditionofselenocystaminetothegrowthmediumofTN72_SR_cpl1_dimer-HA
failedtocatalysespontaneousCpl-1dimerisation,nordiditsignificantlyaffectlevels
ofmonomericCpl-1accumulation.
(kDa)
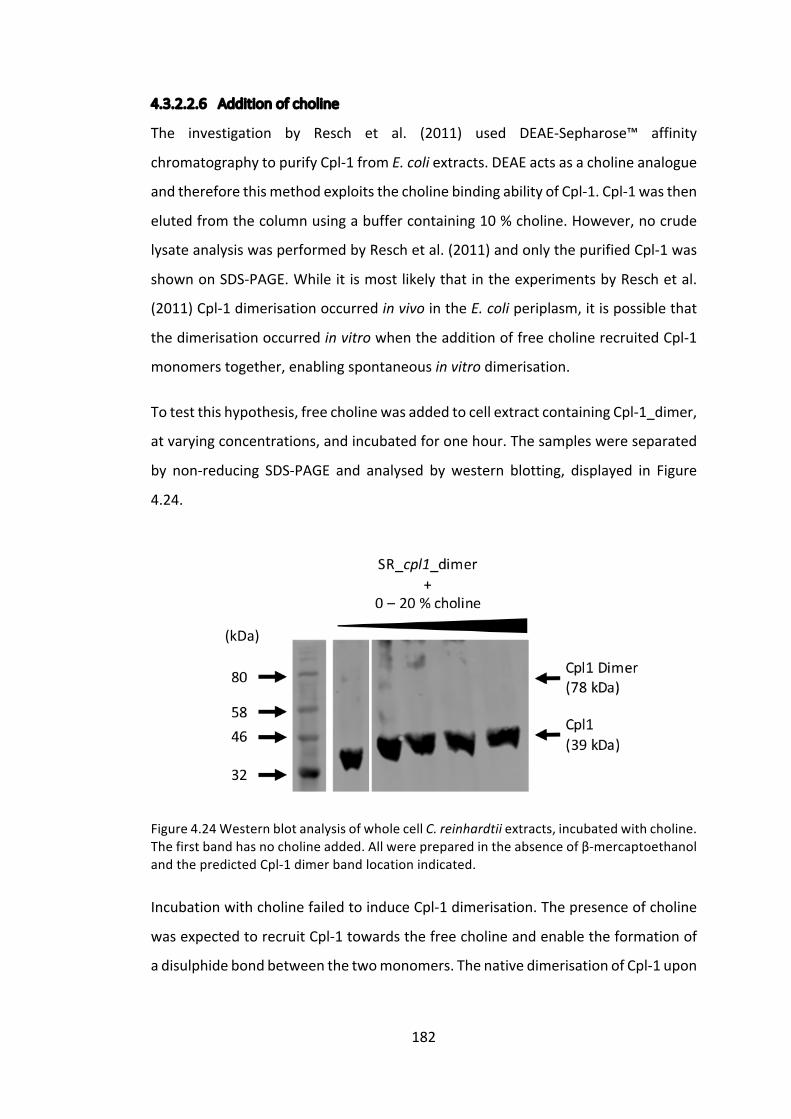
182
4.3.2.2.6 Additionofcholine
The investigation by Resch et al. (2011) used DEAE-Sepharose™ affinity
chromatographytopurifyCpl-1fromE.coliextracts.DEAEactsasacholineanalogue
andthereforethismethodexploitsthecholinebindingabilityofCpl-1.Cpl-1wasthen
elutedfromthecolumnusingabuffercontaining10%choline.However,nocrude
lysateanalysiswasperformedbyReschetal.(2011)andonlythepurifiedCpl-1was
shownonSDS-PAGE.WhileitismostlikelythatintheexperimentsbyReschetal.
(2011)Cpl-1dimerisationoccurredinvivointheE.coliperiplasm,itispossiblethat
thedimerisationoccurredinvitrowhentheadditionoffreecholinerecruitedCpl-1
monomerstogether,enablingspontaneousinvitrodimerisation.
Totestthishypothesis,freecholinewasaddedtocellextractcontainingCpl-1_dimer,
atvaryingconcentrations,andincubatedforonehour.Thesampleswereseparated
by non-reducing SDS-PAGE and analysed bywestern blotting, displayed in Figure
4.24.
Figure4.24WesternblotanalysisofwholecellC.reinhardtiiextracts,incubatedwithcholine.Thefirstbandhasnocholineadded.Allwerepreparedintheabsenceofβ-mercaptoethanolandthepredictedCpl-1dimerbandlocationindicated.
IncubationwithcholinefailedtoinduceCpl-1dimerisation.Thepresenceofcholine
wasexpectedtorecruitCpl-1towardsthefreecholineandenabletheformationof
adisulphidebondbetweenthetwomonomers.ThenativedimerisationofCpl-1upon
(kDa)

183
theadditionofcholinereportedbyMonterrosoetal.(2005)wasnotexpectedtobe
observed,giventhedenaturingconditionsofSDS-PAGE.
4.3.2.2.7 ProductionofCpl-1_dimerinE.coliandenrichment
As shown in Figure 3.8, the E. coli line DH5α_pProEX_cpl1_dimer-HA-StrepII was
shown to express Cpl-1 to a high level, and the application ofHiTrap StrepTactin
purification columns enabled the production of an enriched extract. This was
performedtoreproducetheworkofReschetal.(2011),aswellastoactasaprotein
standardasdescribedin3.3.1.2.6.WhenseparatedbySDS-PAGEundernon-reducing
conditions,nodimerisationcouldbeobserved,howeverthiswastobeexpectedas
disulphidebondformationrequiresanoxidisingenvironment inordertooxidisea
pairof cysteines. InGram-negativeprokaryotes, suchasE. coli, thisoccurs in the
periplasmic space, where enzymes catalyse the reaction. Recombinant proteins
thereforerequireanN-terminalsignalpeptidetotargetthemtotheperiplasmfor
disulphidebond formation,whichwasnot includedonourcpl1_dimer transgene.
Resch et al. (2011) expressed cpl1 using a plasmid based upon pIN-IIIA, which
attachesanoutermembraneproteinA(OmpA)signalpeptidetotheN-terminusof
clonedproteinstopromotesecretionoftherecombinantproteinintotheperiplasm.
Giventhattheplasmidusedhere,pProEX_HTb,doesnotattachanN-terminalsignal
peptide,itwasnecessarytoperformdimerisationinvitro.
4.3.2.2.8 DimerisationofE.coli-producedCpl-1usingoxidisedglutathioneinvitro
Disulphidebondformationcanoccurspontaneouslyiftheenvironmentisoxidising,
andcan thereforebe formed invitro (Berkmen2012).Glutathione isacommonly
used reagent in its reduced (GSH) and oxidised (GSSH) form to produce a redox
environmentbeneficialtodisulphidebondformation.Theprotocolisdescribedfully
in2.6.5,adaptedfromHidakaetal.(1998)andHyvonen(2002).
A crude cell lysateofE. coli cells induced toexpresscpl1_dimerwasobtainedby
sonication,andultracentrifugationremovedtheinsolublefraction.AnexistingCpl-
1_StrepIIenrichedextractfromC.reinhardtiiwasusedasacontrol(from4.3.1.3.6).
The soluble fractionand thecontrolwere then incubatedovernightat20 °Cwith
varyingGSH:GSSGratios.SDS-PAGEwasusedtoseparatethefractions,withnoβ-
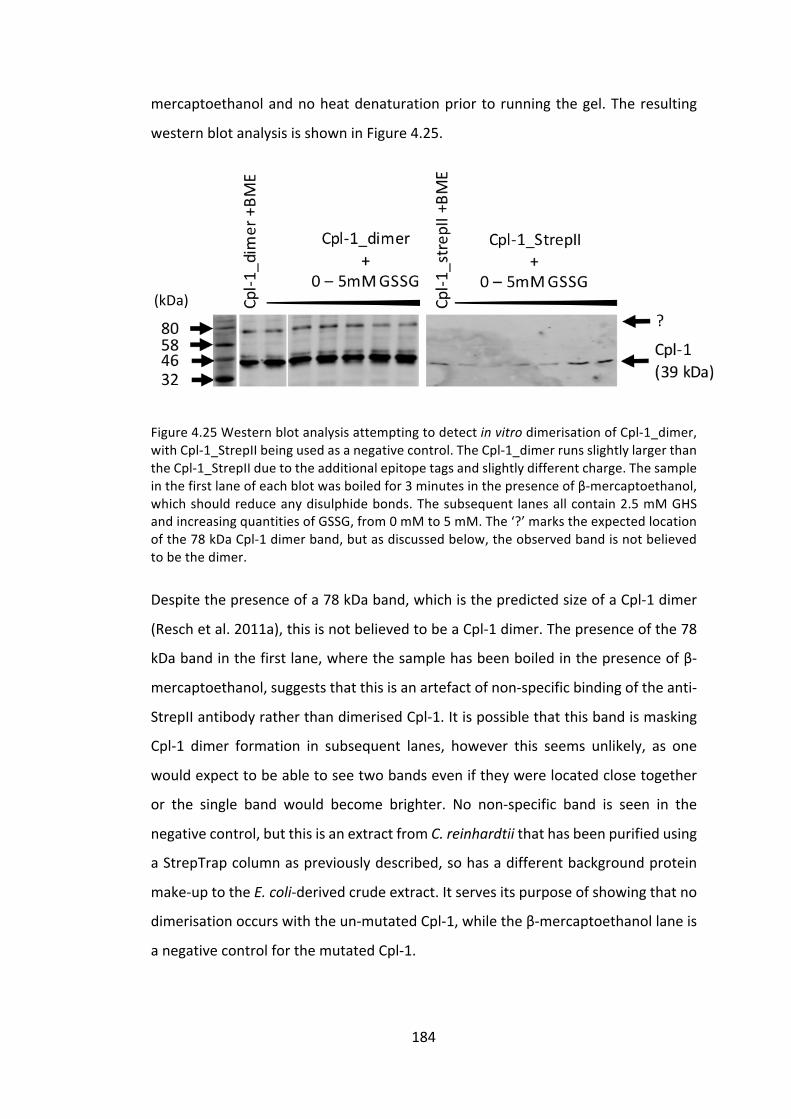
184
mercaptoethanolandnoheatdenaturationprior torunningthegel.Theresulting
westernblotanalysisisshowninFigure4.25.
Figure4.25WesternblotanalysisattemptingtodetectinvitrodimerisationofCpl-1_dimer,withCpl-1_StrepIIbeingusedasanegativecontrol.TheCpl-1_dimerrunsslightlylargerthantheCpl-1_StrepIIduetotheadditionalepitopetagsandslightlydifferentcharge.Thesampleinthefirstlaneofeachblotwasboiledfor3minutesinthepresenceofβ-mercaptoethanol,whichshouldreduceanydisulphidebonds.Thesubsequent lanesallcontain2.5mMGHSandincreasingquantitiesofGSSG,from0mMto5mM.The‘?’markstheexpectedlocationofthe78kDaCpl-1dimerband,butasdiscussedbelow,theobservedbandisnotbelievedtobethedimer.
Despitethepresenceofa78kDaband,whichisthepredictedsizeofaCpl-1dimer
(Reschetal.2011a),thisisnotbelievedtobeaCpl-1dimer.Thepresenceofthe78
kDabandinthefirstlane,wherethesamplehasbeenboiledinthepresenceofβ-
mercaptoethanol,suggeststhatthisisanartefactofnon-specificbindingoftheanti-
StrepIIantibodyratherthandimerisedCpl-1.Itispossiblethatthisbandismasking
Cpl-1 dimer formation in subsequent lanes, however this seems unlikely, as one
wouldexpecttobeabletoseetwobandseveniftheywerelocatedclosetogether
or the single band would become brighter. No non-specific band is seen in the
negativecontrol,butthisisanextractfromC.reinhardtiithathasbeenpurifiedusing
aStrepTrapcolumnaspreviouslydescribed,sohasadifferentbackgroundprotein
make-uptotheE.coli-derivedcrudeextract.Itservesitspurposeofshowingthatno
dimerisationoccurswiththeun-mutatedCpl-1,whiletheβ-mercaptoethanollaneis
anegativecontrolforthemutatedCpl-1.
(kDa)

185
ThereareacoupleoffactorsthatcouldbepreventingdimerisationofCpl-1_dimer,
eveninvitro.ThefirstandmostlikelyissueisthepresenceofC-terminalepitopetags
causingsterichindrance.TheHAtagintheC.reinhardtiiversionoftheproteinand
theHAandStrepIItagsintheE.coliversionarebothontheC-terminusoftheprotein.
Inhindsight,locatingtheseontheN-terminusmayhavebeenabetterapproachas
theCpl-1proteinhasbeenengineeredtoformadisulphidebridgeclosetotheC-
terminus.Reschetal.(2011)notethatthepositioningofthenewcysteineresidue
neartheC-terminusisofcrucialimportancetodimerisation.Givenallofthis,itisnot
surprisingthattheinclusionofan8-9aminoacidepitopetagattheC-terminuswould
interferewithdimerisation.
ThesameCpl-1_dimerproteinwasexpressedasarecombinant inE.coliafterthe
failure to achieve dimerisation inC. reinhardtii to testwhether this failurewas a
propertyoftheproteinperseoraresultoftheexpressionplatform.Inessence,if
dimerisationofCpl-1_dimercouldbeachievedinE.colibutnotinC.reinhardtiithen
wewouldconcludethattheC.reinhardtiichloroplastisunsuitableforpre-dimerised
Cpl-1production.However, ifdimerisationcouldnotbeachieved inE.colieither,
thentheconclusionwouldbethattheproteindesignwasinsomewaypreventing
dimerisation.
In this study, the lackof targeting to theE.coliperiplasm likelyprevented invivo
dimerisationfromoccurring.FuturestudiesshouldusevectorsuchaspIN-III,which
includesanN-terminalOmpAtargetpeptideandmayrectifythis,orconsiderusing
anengineeredstrainofcompetentE.colisuchasRosetta-gami™(Novagen)which
displaysenhanceddisulphidebondformationinthecytoplasm.Thefailureofinvitro
dimerisation could be due to a lack of condition-optimisation of the dimerisation
protocol,asonlyarelativelynarrowrangeofconditionsweretested.Forexample,
differentreagentscouldbeused,suchascysteineandcystine,andindifferentratios
tocreatetheredoxenvironment,aswellasdifferenttemperaturesandincubation
times.OptimisationofthisprotocolforCpl-1_dimermayhaveenableddimerisation.

186
4.3.3 ExpressionoftheCpl-1associatedholin,Cph-1
4.3.3.1 ProductionofC.reinhardtiilinesexpressingcph-1
TheC.reinhardtiiderivedpsaA5’UTR/promoterisfunctionalinE.coli,asareseveral
chloroplastpromotersprobablyduetothechloroplast’sprokaryoticorigins(Young
and Purton 2015). Given the ability of holins to form holes in the cytoplasmic
membraneofE.coli,andthusinhibitcellgrowth(Catalãoetal.2011),theuseofthe
pSRSapIplasmidtoclonetheseinE.coliisunlikelytobepossible.
AstrategydevelopedbyYoung&Purton (2015)makesuseofa ‘spare’opal stop
codon,UGA,thatisunusedintheC.reinhardtiichloroplast.Thecodonisreassigned
tocodeforatryptophanbytheco-introductionofamodifiedtryptophantRNAgene
(trnW)intotheC.reinhardtiichloroplast,alongwiththegeneofinterest.Thegene
of interest is modified to contain several UGA codons in the place of UGG
(Tryptophan)–thesearereadas“Stop”codonsbytheE.colitranslationalmachinery,
but as tryptophan codons in the chloroplast. A plasmid, named pWUCA2 was
developedtoperformthiscloningstrategy.Theplasmid isverysimilar topSRSapI
(2.2)butwiththeadditionofatrnWUCAgeneupstreamofthegeneofinterest(GOI)
expressioncassette,illustratedinFigure4.26.
Figure4.26ThepWUCA2 transformationvector isbaseduponpSRSapIandhas thesamecloning sites for the GOI and the same flanking regions for homologous recombination.However, an additional synthetic gene, trnWUCA, has been inserted upstream of the GOIexpressioncassette.
This strategy enables the cloning inE. coli of genes under functional/constitutive
promoters,whichwouldotherwiseproduceproteinsthataretoxicordetrimentalto

187
E. coli growth. In this case, the E. coli translational machinery will stop short of
producingthewholelengthproteinduetothepresenceofstopcodonsinthemiddle
oftheGOI.
ProducingholinsintheC.reinhardtiichloroplastmayavoidtheproblemofE.colilysis
thatoccurswhenholinsareexpressedasrecombinantproteinsinbacterialplatforms
(Smithetal.1998;Shietal.2012;Songetal.2016).Thisproblemhasbeenlargely
overcome by the use of induction systems, but allows only 10 – 15 minutes of
accumulationbeforethemetabolicactivityfails(Smithetal.1998).Furthermore,the
natureoftheproteinmeansthatitmustbepurifiedfromthebacterialmembraneto
which it localises, which further reduces yields and increases costs. While C.
reinhardtiiachievesonlylowlevelsofrecombinantproteinexpressioncomparedto
E.coliformanyproteins,giventhesetwoissuesitmaybethatitcancompeteasan
expressionplatformwhenproducingholins.
4.3.3.1.1 Genedesignandcodonoptimization
CodonUsageOptimizerBeta0.92(Kong2013)wasusedtooptimizethecph1gene
forC.reinhardtiichloroplastexpression.Twotryptophancodonsearly inthegene
werechangedfromUGGtoUGA.SapIandSphI restrictionsiteswere includedfor
cloning into the pWUCA expression vector. The full-length cph1-HA gene was
synthesizedbyIntegratedDNAtechnologies(AppendixH).
4.3.3.1.2 Expressioncassetteproduction
SapIandSphIrestrictionenzymeswereusedtodigestandligatethecph1geneinto
pWUCA2 in-frame,producing theplasmidpWUCA2_cph1-HA.DH5αcompetentE.
colicellsweretransformedwiththeresultingplasmidandsequencingconfirmedthe
correctplasmidassembly.
4.3.3.1.3 C.reinhardtiitransformation
ThepWUCA2_cph1-HAplasmidwasused to transform the recipientC. reinhardtii
strainTN72viatheglassbeadmethodasdescribedin2.3.13.Figure4.27illustrates
theresultingC.reinhardtiicellline,pWUCA2_cph1-HA.

188
Figure4.27TN72recipientstrainsuccessfullytransformedtocarrycph1-HAunderthepsaAexon1promoter/5’UTRinthechloroplast,andwithpsbHfunctionalityrestored.AmodifiedtrnWgenehasbeenco-introducedtocorrectlycodefortheintroducedUGAcodonsinthecph1gene.TheapproximatepositionsoftheUGAcodonsareindicatedinred.
The resulting transformants were confirmed for homoplasmicity as described in
2.3.17.Inthisinstance,duetotheadditionofthetrnWUCAgene,theexpectedband
sizeforhomoplasmicitywillbe291bplarger,givingatotalexpectedsizeof1421bp
(Primers:Flank1&psa.R (AppendixO:ListofPrimers)).Gelelectrophoresisof the
resultingPCRproductfromthreetransformantsisshowninFigure4.28.
Figure4.28TheresultingPCRproductsareapproximately1.4kbinsizeasexpected.Thelackofan850bpproductconfirmsthatallthreeofthesetransformantsarehomoplasmic.
Finally,theintegrityoftheinsertedcassettewasconfirmedbyDNAsequencingasin
2.3.18.
4.3.3.1.4 Westernblotconfirmation
WholecellextractsofTN72_WUCA2_cph1-HAandTN72_SR_Control(asanegative
control)werepreparedandseparatedbySDS-PAGE.Afterblottingtonitrocellulose
membraneandblockingasdescribedin2.4.5,themembranewasprobedwithanti-
HAprimaryantibody,thenincubatedwithIRDye®secondaryantibody.Fluorescence
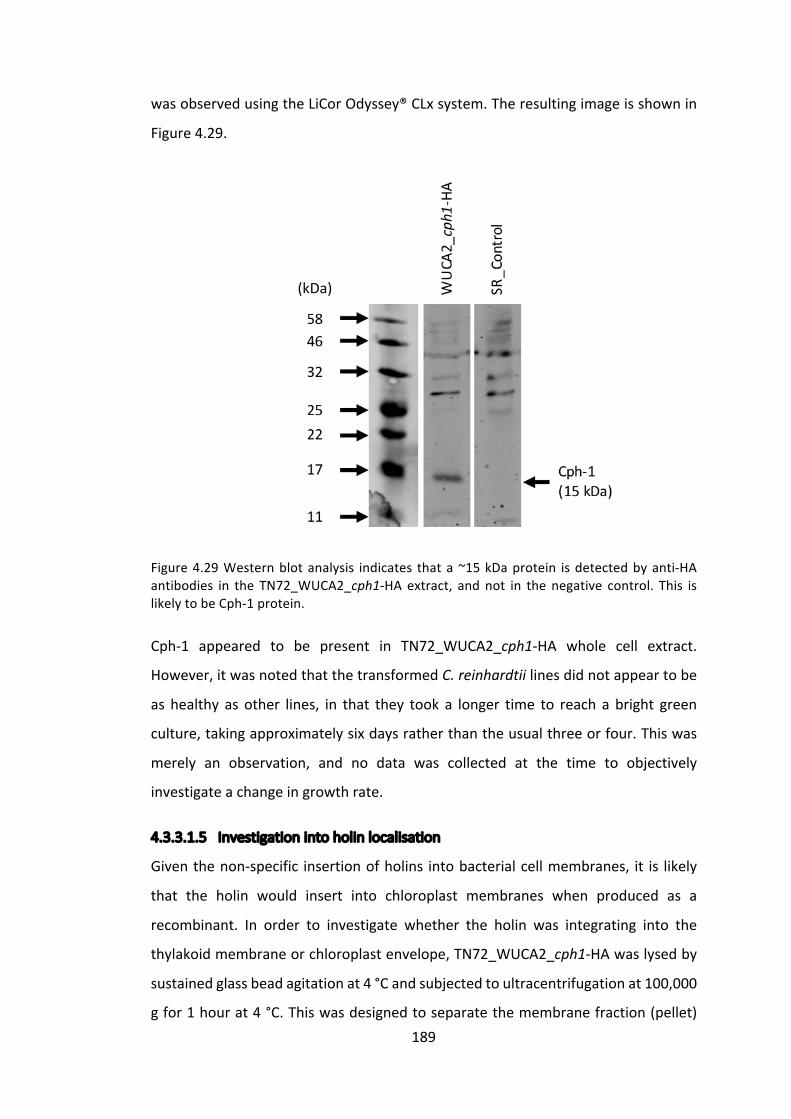
189
wasobservedusingtheLiCorOdyssey®CLxsystem.Theresultingimageisshownin
Figure4.29.
Figure4.29Westernblotanalysis indicates thata~15kDaprotein isdetectedbyanti-HAantibodies in the TN72_WUCA2_cph1-HAextract, andnot in thenegative control. This islikelytobeCph-1protein.
Cph-1 appeared to be present in TN72_WUCA2_cph1-HA whole cell extract.
However,itwasnotedthatthetransformedC.reinhardtiilinesdidnotappeartobe
as healthy as other lines, in that they took a longer time to reach a bright green
culture,takingapproximatelysixdaysratherthantheusualthreeorfour.Thiswas
merely an observation, and no data was collected at the time to objectively
investigateachangeingrowthrate.
4.3.3.1.5 Investigationintoholinlocalisation
Giventhenon-specific insertionofholins intobacterialcellmembranes, it is likely
that the holin would insert into chloroplast membranes when produced as a
recombinant. In order to investigate whether the holin was integrating into the
thylakoidmembraneorchloroplastenvelope,TN72_WUCA2_cph1-HAwaslysedby
sustainedglassbeadagitationat4°Candsubjectedtoultracentrifugationat100,000
gfor1hourat4°C.Thiswasdesignedtoseparatethemembranefraction(pellet)
(kDa)

190
and cytoplasmic fraction (supernatant). However, when the two fractions were
separated by SDS-PAGE and analysed by western blotting as before, no 15 kDa
proteincouldbedetected.
InanattempttodetectCph-1accumulationagain,veryhighconcentrationsofcell
extractwereseparatedonSDS-PAGEandanalysedbywesternblotting,aswellas
usingammoniumsulphateprecipitationtoconcentratelargevolumesofcellextract,
butnoCph-1wasdetected.Again,onlyobservationally,itwasnotedthatatthetime
ofthesefurtherexperiments,theTN72_WUCA2_cph1-HAcelllineseemedtogrow
atanormal rateand ithadrecovered from itsearlierunhealthyappearance.This
observationandthelackofdetectableCph-1accumulationsuggestedthatthegene
hadbeensilenced insomeway,either throughpointmutationsor recombination
eventsbetweensimilarsequences.
4.3.3.1.6 Investigatingcph1silencing
Figure 4.28 demonstrates that the three TN72_WUCA2_cph1-HA lines were
homoplasmic,meaningthatallofthe~70chloroplastgenomecopiescontainedthe
desiredexpressioncassette.Thiswasachievedbyseveralroundsofselectionupon
minimal medium, thus selecting for photosynthetically-competent cells and
promoting the presence of the transformed plastome copies. However, once
homoplasmicityhadbeenachievedandconfirmed,cellsweregrownmixotrophically
upon acetate and under light which promotes faster growth. Had the cell been
heteroplasmic at this stage and the GOI was sufficiently toxic, it is possible that
selection could be driven the otherway, back to the photosynthetically deficient
recipientline.However,theselectionforphotosynthesisissostrongthatthisseems
quiteunlikelyandthelossofpsbHfunctionalityalsoconfersphotosensitivity,further
increasingthepressureontheplastometogainpsbHfunction.However,giventhat
thelineswerehomoplasmic,thiscannotbethecase.ThesensitivityofPCRmeans
thatevenifoneplastomecopywerewithouttheexpressioncassette,thisislikelyto
bevisibleinFigure4.28.
Thereareseveralsitesintheinsertedexpressioncassettethatcouldbesubjectto
recombination events within the chloroplast as they are of endogenous origin,

191
namelythepsaAexon1promoterand5’UTR,rbcL3’UTRandtrnW.However,the
nativerbcLgeneandthepsaAexon1arerespectively46and47kbpawayfromthe
insertionsite,andthetrnWgeneis61kbpaway,whichwouldbeexpectedtoreduce
recombinationrates,althoughthisrelationshiphasnotbeenconfirmed(Muddetal.
2014). Furthermore,eachof these repeated sequences is relatively small: trnW is
73bp;psaA exon1promoter and5’UTR is 294bp; and rbcL 3’UTR is 407bp. For
context,Muddetal.(2014)suggestthatrepeatsof650bpseparatedby5kbpresults
inefficientrecombination,butnostrictrulehasbeensuggested.Anotheralternative
isthatpointmutationsineitherthemodifiedtrnWgeneorcph1genehaveresulted
infailedtranslationorinstabilityoftheprotein.
Totesttheabovetheories,thecassetteregionwasamplifiedbyPCRfromgenomic
DNAandsequenced(regionsbandcinFigure4.30),asperformedaftertheinitial
confirmation of homoplasmicity. Sequencing confirmed the presence of both the
holin and trnWUCA genes, with nomutations. The psbH.R primer binds inside the
downstreamflankingregionofthetransformationplasmid,and123abindsupstream
of the trnWUCA gene,which therefore suggests that thewhole cassette is present.
However,theFlank1primerliesoutsidethetransformationplasmid,upstreamofthe
cassette,andnoproductcouldbeseenwhenthisprimerwasused.

192
Figure4.30Reactionsa-cproducedtheexpectedamplifiedPCRproduct,whiledandefailedtoproduceanyproduct.Thesuccessfulamplificationofa-cshowsthatthecassetteispresentsomewhereinthegenome.ThesuccessofamplificationinFigure4.28(thesameprimersaseabove)suggeststhatthecassettewasoriginallyinthecorrectlocus.However,thefailureofd&eheresuggests that thecassette isno longerpresent in the intendedneutral sitebetween trnE2 and psbH in the chloroplast. The two lanes show two differentTN72_WUCA_cph1-HAtransformants.PrimersequencescanbefoundinAppendixO:ListofPrimers.
It ispossiblethataverysmallnumberofplastomecopiesremainwiththecorrect
cassetteinsertion,butthatthemajorityofcopieshaverecombinedinamannerso
astosilencegeneexpression.However,asmentionedpreviously,thesensitivityof
PCRwouldnormallyenableevenasinglecopytobedetected.Muddetal. (2014)
does note that recombination has been observedwith relatively small repeats in
Chlamydomonas, for example, 216 bp direct repeats in the chlLgene are able to

193
recombine. The most likely explanation is therefore that some combination of
recombinationeventshasledtotherelocationoftheexpressioncassettetoasilent
siteintheplastome,whichwasthenhighlyselectedfor,giventheapparenttoxicity
ofCph-1.
4.4 Conclusionandfuturework
This chapter has examined three strategies to improve Cpl-1 activity against S.
pneumoniae,thefirsttwoofwhichdealwithdirectproteinengineeringandthethird
attemptstoexploitsynergywithanotherprotein,aholin.
4.4.1 Cellwallbindingsitemutagenesis
SixbindingsitemutatedCpl-1proteinswereexpressedinC.reinhardtiitodetectable,
albeitvarying, levels.However,difficultiesarosewhenattemptingtocomparethe
activityoftheseproteinsaslyticenzymesagainstS.pneumoniae.Firstly,thepotential
forimprovementsaroundthemutationsthemselveswillbediscussed,followedbya
discussionofhowtheactivityassaysmightbeadaptedandsuggestionsofpossible
alternativeassays.
It is noted in 4.3.1.1.1 and illustrated in Figure 4.4 that there are three residues
believed to be of key importance in each choline binding site, two tryptophan
residuesandatyrosineresidue.Hermosoetal.(2003)alsosuggestthatalysineis
involved in capping the site and stabilising the phosphate group of phosphoryl-
choline.Thisinvestigationonlyconsideredthetwotryptophanresiduesofeachsite,
simply due to time constraints and the fact that they appear to be closer to the
choline residue. It appears that mutating the tryptophan residues did indeed
negativelyaffect cholinebinding,but that this resulted in reduced lyticactivity. It
would,therefore,beinterestingtoconsiderthetyrosineandlysineineachbinding
siteas theymayenablea subtlermutation tobeproduced.Alanine scanning isa
commonapproachtomutatingresiduesofinterest,asiteliminatestheroleofthe
polarsidechain,however,inthisinstance,thismayresultintoodrasticachangein
polarity.Therefore,amoreconservativemutationmightbeconsidered,forexample,
the mutation of tryptophan to tyrosine. The binding site is clearly sensitive to

194
mutationsandhighlyconserved,andthealaninechangesappeartohavebeentoo
severeandresultedinreducedactivity.
Furthermore,theCpl1_W223A-HAconstructwasneversuccessfullysynthesised,for
reasons unknown. It is interesting to note that the Cpl1_W223A_W230A-HA and
Cpl1_W202A_W209A_W223A_W230A-HA,whichbothcontaintheW223Amutation
inadditiontoothers,produceconsistentlydetectablelevelsofproteinaccumulation.
Thisissomewhatunexpected,asW223isnotastrictlyconservedresidueincholine
bindingrepeats(Figure4.5)whileW209andW230are,thoughnoproblemswere
observedwhentheseweremutated.TheissuesaroseafterPCRwhenattemptingto
insert the gene into pJet, a vector from a commercially available PCR cloning kit
(ThermoFischer),enablingbluntendcloningandtransformationofE.coli.NoE.coli
colonies were observed in any of the attempts to produce cpl1_W223A-HA,
suggestingthismutatedgeneiseitherhighlytoxictoE.coliorforsomereasonunable
toligateintothepJetvector.Infutureeitheradifferentintermediatevector,suchas
pGemTEasycouldbeusedortheintermediatestepcouldbeskippedandattemptto
clonedirectlyintopSRSapIusingadigestedPCRproduct.
An interesting and related point of discussion iswhy the inactive choline binding
repeats(p4-p6)remainsohighlyconserved.Astheserepeatsappeartobesterically
unable to bind choline, it would be reasonable to assume that there was little
evolutionarypressure for themtoremainunaltered.The fact that theyremainso
highlyconservedindicatesthattheyalsohavesomeotherimportantfunction.This
maysuggestthatthethree-dimensionalshapeoftheproteinishighlydependenton
theinteractionoftheserepeatsandthatmutationstothemhaveeffectsmorewide-
rangingthatmerelyalteringcholinebinding.
Giventhatinitialattemptstodisruptthecholinebindingsitesappearedtoresultina
reductionofactivity,andthatthep4-p6repeatsarehighlyconserveddespitetheir
purported inactivity, an alternative protein engineering focus could be taken. For
example,theflexibleacidiclinkerthatjoinsthecellwallbindingdomainandcatalytic
domainwouldbeaninterestingsiteforimprovement.Ifitisindeedthecasethatthe
binding domain remains bound to choline in the cell wall, then the number of

195
FigureRemoved
peptidoglycancleavagesthatcanbeperformedbythecatalyticdomainislimitedby
thelengthandnatureofthislinker.WhenproducingArtilysins®,Briersetal.(2014)
investigatedtheeffectofincreasinglinkerlengthbetweenthepeptideandendolysin.
Itwasfoundthatantibacterialactivityincreasedinastepwisefashionasthelinker
was extended, then began to plateau at around 18 amino acids in length (Figure
4.31).
Figure4.31L0,L1,L2,L3,andL4correspondto linkeraminoacidsequencesofAGAS (4),AGAGAS (6),AGAGAGAGAS (10),AGAGAGAGAGAGAS (14), andAGAGAGAGAGAGAGAGAS(18).ReproducedfromBriersetal.(2014).
Theacidic linker forCpl-1 comprises residues189-199,withaminoacid sequence
DDEEDDKPKTA.WhilenotdirectlyanalogoustothelinkerinvestigatedbyBriersetal.
(2014), itwouldbe interesting toattempt toextend theCpl-1 linker to see ifany
improvement in activity can be achieved. However, it is unlikely that much
improvementtoactivitycanbegleanedbyextendingtheacidiclinker,asthiswould
haveprobablybeenhighlyselectedforthroughoutevolution.Thisisincontrastto
choline binding which, as discussed previously, appears to act as a tethering
mechanismtopreventaccidentallysisofpotentialfuturehostbacterialcells.

196
DEAEcelluloseactsasacholineanalogueandhasthereforebeenexploitedforion
exchangechromatographytopurifyCpl-1,usingaprotocolfirstdescribedbyLoeffler
etal.(2001).Cpl-1bindstoDEAEandcanthenbeelutedusingafreecholine.This
method was not used in this investigation, as choline binding was the object of
investigation. However, the method could be adapted into an assay for choline
bindingability.Forexample,amutatedCpl-1proteindisplayingweakenedcholine
bindingwouldbeelutedfromtheDEAEcolumnmoreeasily thanstronglybinding
Cpl-1.Thisassaycouldbeusedtoconfirmthatanychangeinactivitywasindeeddue
tocholinebindingalteration,andnotduetosomeotherunknownfactororthree-
dimensionalshapechange.
Theproteinengineeringstepstakeninthissectionwerehighlyspeculativeandwould
have benefitted from testing a larger number of mutants more quickly. For this
reason,thechoiceofC.reinhardtiiasaplatformlimitedtheseinitialinvestigations,
as transformations take more time, recombinant protein yields are lower, and
background antimicrobial effects disrupted activity assays compared to other
platformssuchasE.coli.ThisworkwouldhavebeenbettersuitedtoE.coli,which
enablesrapidprototypingandtestingofrecombinantproteins.C.reinhardtiishould
thenbeusedfurtherdowntheproductionline,oncetheproteinsequencehasbeen
optimised.Atthatpoint,themanybenefitsofusingC.reinhardtiiasaplatformfor
theproductionoftherapeuticrecombinantproteinscanstillbeharnessed.
4.4.2 Cpl-1dimerisation
Cpl-1dimerisationwasattemptedfortworeasons.Firstly,areportbyReschetal.
(2011) demonstrated that pre-dimerised Cpl-1 had a 10-fold reduction in plasma
clearanceandatwo-foldincreaseinactivity.Secondly,theC.reinhardtiichloroplast
isknowntocontrolredoxreactionsandiscapableofcatalysingdisulphidebondsin
recombinant proteins (Tran et al. 2009), which can be further increased by the
additionofselenocystaminetothegrowthmedium(Ferreira-Camargoetal.2015).
However,ourattempttoproduceaCpl-1dimerintheC.reinhardtiichloroplastwas
unsuccessful,aswasinvitroformationofdisulphidebondsusinganE.coliproduced
Cpl-1.

197
Themostlikelyreasonforthisfailureisthepresenceofepitopetagsontheprotein
C-terminus,aspreviouslydiscussed.Thefirststeptowardstestingthistheoryisto
redesign the cpl1_dimer gene such that it encodes an N-terminal tag, with no
additionalaminoacidsontheC-terminus.Thegeneshouldalsobetransformedinto
E.coliusingaplasmidsuchaspIN-IIIwhichwilltargettheproteintotheperiplasm
fordisulphidebondcatalysis,orintoanengineeredstrainofcompetentE.colisuch
asRosetta-gami™.IfthissuccessfullyproducesaCpl-1dimer,whichitshouldasitis
asimplereplicationoftheworkbyReschetal.(2011),thenthesamegeneshouldbe
transformedintotheC.reinhardtiichloroplast.Atthispoint,itwillbeclearthatifno
dimercanbeseen in theC. reinhardtii transformants, then it isan issuewith the
platform,ratherthantheprotein.
IfCpl-1dimerscanaccumulateintheC.reinhardtiichloroplast,theninvestigations
intotheuseofselenocystaminecanbeuntaken.Itmaybethatthisstrategycanbe
usedtoincreaseaccumulationlevelsofCpl-1intotal,particularlyifthedimerismore
stable than themonomer. An investigation can also be carried out intowhether
dimerised Cpl-1 is less susceptible to degradation whilst in storage, as it would
potentiallybelessaccessibletoproteasesandlesspronetodenaturation.Thiswould
beanimportantpropertyforthefuturecommercialisationanddistributionofCpl-1
asatherapeutic.
4.4.3 Holinproduction
Holinsenableendolysinstocrossthecellmembraneduringthebacteriophagelytic
cycle. The treatment of some bacteriawith both endolysins and holins has been
showntohaveasynergisticeffectuponcelllysis(Shietal.2012;Songetal.2016).
However,theproductionofholinsinbacterialsystemsisproblematicastheyinsert
intocellmembranesanddepolarisethecell.Herewehavereportedthesuccessful
accumulation of the holin Cph-1 in the C. reinhardtii chloroplast but found that
accumulationquicklyceased.
The‘silencing’ofCph-1isworthyofinvestigation,aschloroplasttransformationsare
not usually susceptible to gene silencing, although it is common in nuclear
transformations.Themost likelyexplanation is that in facthomoplasmicityof the

198
plastomewasneverachievedandthatsomenon-specificPCRamplificationoccured,
whichledtoFigure4.28appearingtoconfirmhomoplasmicity.SequencingofthePCR
productcouldhaveconfirmedthisandaTN72negativecontrolshouldhavebeen
includedtodemonstratethatheteroplasmicitywouldhavebeendetected.However,
asdiscussedpreviously,even if thiswas thecase it seemsunlikely that thestrain
would revert to the parental sequence, as loss of psbH function results in
photosensitivity,sogrowthunderbrightlightwouldalsoencouragehomoplasmicity.
Furthermore,aninvestigationbyPCRofthegenomicDNAisdisplayedinFigure4.30,
andclearlydemonstratesthatthecassetteispresentsomewhereintheC.reinhardtii
genome. However, the same investigation failed to confirm, or refute, that the
cassettewas located in the targeted location,namelybetweenthe trnEandpsbH
genesinthechloroplast.AprimerlocatedbeyondthepsbHgeneisrequiredsothat
the entire region can be amplified to confirm that the cassette is not present or
otherwise-thiswouldbeverystraightforward.
We know that over the millennia since the first photosynthetic prokaryote was
engulfedintoaeukaryotetobecomeachloroplast,thevastmajorityofthegenetic
information fromthechloroplastgenomehasbeeneliminatedormigrated to the
nucleus,leavingonlygenesessentialforphotosynthesisinthechloroplast(Howeand
Purton2007).Itispossiblethatthehighlyundesirablecph1-containingcassettehas
been translocated to the nucleus under a similar mechanism, and once there,
silencingmechanismsareprolific.ThepotentialmechanismforsuchaDNArelocation
is not understood, but such a mechanism is known to exist (Cullis et al. 2009).
Furthermore, such transfers have been shown to occur at high frequencies in
tobacco:morethan1in5millioncellswereabletosuccessfullytransferamarker
fromthechloroplasttothenucleus(Stegemannetal.2003).Suchaneventwould
explainthepresenceofthecassette,yetlackofexpression.Amethodsuchasinverse
PCRcouldthenbeusedtodiscoverthelocationofthecassette.
Regardless of the mechanism used by the C. reinhardtii cells to prevent Cph-1
accumulation, it clearly is not tolerated by the cell. It is likely that the holinwas
inserting into the thylakoid membranes, given their similarity to bacterial cell
membranes.AsshowninFigure4.3,alargenumberofholinmonomersarerequired

199
toformaraftandthenoligomeriseintoaholebeforeanydepolarisationwilloccur.
Itmaybethattheholinwasonlyweaklyexpressedandthereforenotlethaltothe
cell,butcausedenoughselectivepressurethatitwashighlyundesirable.Thiswould
explainwhytransformantswereobtainedbutquicklylosttheirCph-1expression.
ItislikelythatanyrepeatedattempttoexpressCph-1intheC.reinhardtiichloroplast
will result in the activation of a mechanism to prevent its expression. However,
holins,unlikeendolysins,arenotspeciesspecificinnatureandaregenerallyableto
insertintothemembranesofawiderangeofbacterialspecies(Savvaetal.2008).It
wouldbeinterestingtoexpressaholinandanti-holinpair,suchasS105andS107
fromthelambdabacteriophage,aswellastheholinalone,toseewhetherexpressing
thepairpreventssilencingoftheholingene.Thiswouldenableacontinuationofthe
investigationintowhethertheadditionofaholinalongsideCpl-1canimproveitslytic
activity.

200
Chapter5 ExtendingtheChlamydomonasreinhardtiichloroplast
platformtootherendolysins
5.1 Introduction
5.1.1 PreviousendolysinexpressionintheC.reinhardtiichloroplast
DrHenryTaunt,workinginthePurtonLab,wasthefirsttosuccessfullyexpressan
activeendolysinintheC.reinhardtiichloroplast(Taunt2013).Theworkshowedthat
Cpl-1 accumulated to a relatively high level, approximately 1% of total soluble
protein, in the chloroplast and that it was active against S. pneumoniae in vitro.
However,DrTauntwasunabletoshowexpressionoftwootherendolysins,Gp20and
Lys16thattargetPropionibacteriumacnesandStaphylococcusaureus,respectively.
TheworkbyTaunt (2013)alsoattemptedto identifyelementsofendolysingenes
thatresultedinsuccessfulorunsuccessfulexpressionintheC.reinhardtiichloroplast.
However,while several fusion proteinswere created to test this theory, no clear
conclusions could be drawn. Additionally, work by Dr Laura Stoffels, also of the
Purtongroup,achievedexpressionandactivityoftheS.pneumoniaeendolysinPal,
but failed to show activity of theStaphylococcus aureus targeting endolysinφ11,
despitedetectingexpression (Stoffels2015).AnotherendolysinSPN9CC, targeting
Salmonellatyphimurium,wasexpressedbyDrRosieYoungasaproofofconceptthat
recombinantproteinshighlytoxictoE.colicanbesynthesisedintheC.reinhardtii
chloroplast using a synthetic tRNA system, but the activity of SPN9CC was not
investigated(YoungandPurton2015).AsaGram-negativetargetingendolysin,itis
unlikelytobeactiveagainstthetargetbacteriumwithoutmodificationowingtothe
presenceoftheoutermembrane.
Current work in the Purton Group by Juliana de Costa Ramos (unpublished) is
focussed upon producing endolysins targeting Gram-negative bacteria. Two such
endolysins (LysAB2 and LoGT-023) have been successfully expressed in the C.
reinhardtii chloroplast, and although the ‘target bacteria’ for both endolysins is
Acinetobacterbaumannii,Gram-negativeendolysins tend tobemuch less specific
than those targeting Gram-positive bacteria and therefore have a much broader

201
spectrumoflyticactivity.Forexample,theendolysinLysAB2hasbeenreportedto
effectively lyse Staphylococcus aureus, Bacillus subtilis, Streptococcus sanguis,
Escherichia coli, Citrobacter freundii and Salmonella enterica, all in addition to
Acinetobacter baumannii, which the bacteriophage producing LysAB2 specifically
infects(Laietal.2011).Table5.1listsallknownattemptsatexpressingendolysinsin
theC.reinhardtiichloroplasttodate.
Endolysin Targetpathogen SuccessfullyexpressedinC.reinhardtiichloroplast
Activeagainsttarget
pathogen
Reference
Cpl-1 Streptococcuspneumoniae
Yes Yes (Taunt2013)
Gp20 Propionibacteriumacnes
No N/A (Taunt2013)
Lys16 Staphylococcusaureus
No N/A (Taunt2013)
Pal Streptococcuspneumoniae
Yes Yes (Stoffels2015)
φ11 Staphylococcusaureus
Yes No (Stoffels2015)
SPN9CC Salmonellatyphimurium
Yes Nottested (YoungandPurton2015)
LoGT-023(Artilysin®)
Acinetobacterbaumannii
Yes Yes JulianadeCostaRamos(unpublished)
LysAB2 Acinetobacterbaumannii
Yes Yes JulianadeCostaRamos(unpublished)
CD27L1-179 Clostridiumdifficile Yes Yes Thisstudy
Table5.1ThefulllistofendolysinexpressionattemptsbythePurtonGroup.SPN9CC,LoGT-023 and LysAB2 target Gram-negative bacteria,while the others all target Gram-positivebacteria.
5.1.2 Theneedformoreendolysins
Asdiscussedin1.2.2.4,Gram-positiveendolysinshaveaverynarrowspectrumoflytic
activity. This property is highly advantageous in some respects, yet highly
problematicinothers.Theadvantageofanarrowspectrumantibioticisthatonlythe

202
target pathogenic bacteria are killed, while the commensal bacteria remain
unharmed.Thispropertylimitsthedevelopmentofresistanceandtheopportunity
forpathogenicre-colonisation.Thedisadvantageisthatapreciseidentificationofthe
pathogen causing thediseasemust bemadebefore the correct endolysin canbe
prescribedbyaclinician,andadistinctendolysintherapeuticmustbeproducedfor
everycommonbacterialinfection.
Unlike conventional antibiotics, where a single new drug discovery (such as
teixobactin 1.1.2.2) has the potential to treat hundreds of different bacterial
infections, endolysin therapy requires thedevelopment of a large arsenal of very
specificproteinpharmaceuticals.AsshowninTable5.1,theabilitytoproduceactive
endolysinsinC.reinhardtiiisfarfromroutine,withonlya~56%successratesofar.It
isimportant,therefore,tocontinuetoattempttoexpressnewendolysinsaswellas
totrytounderstandthefactorsaffectingexpression.Thischapterdemonstratesthe
successful expression of CD27L in the C. reinhardtii chloroplast, as well as
documentinganattempttoachieveexpressionofGp20,whichhasfailedtoexpress
inpreviousefforts.
5.2 Aimsandobjectives
1. Toproduceatransgenic lineofC.reinhardtii inwhichtheendolysinCD27L
accumulates.
2. TodemonstratetheactivityofCD27LonC.difficileinvitro.
3. ToproduceatransgeniclineofC.reinhardtiiinwhichGp20accumulates.
4. TodemonstratetheactivityofGp20onP.acnesinvitro.

203
5.3 Resultsanddiscussion
5.3.1 Clostridiumdifficile,ΦCD27andCD27L1-179
ClostridiumdifficileisaGram-positivebacteriumofparticularimportanceduetoits
prevalenceasacommonhospital-acquiredinfection.C.difficile infection(CDI) isa
toxin-mediatedinfection,andisthemajorcauseofpseudomembranouscolitis(Best
et al. 2012). The ability of C. difficile to form spores, which canwithstand harsh
conditionsandantibiotics,meansthat it isoftenthefirstbacteriumtore-colonise
thehumangutaftertreatmentwithbroad-spectrumantibiotics.Whileresistanceto
antibiotics has been reported in C. difficile, vancomycin remains the first line of
treatmentanduntil2014resistancetovancomycinwasrarelyobserved(Hargreaves
andClokie2014).However,recentlytherehavebeenreportsofreducedvancomycin
susceptibility(Spigaglia2016),emphasizingtheneedfornewtreatments.
5.3.2 C.difficileendolysinCD27L
The bacteriophage ΦCD27 infects C. difficile, and its endolysin, CD27L, has been
identifiedanddemonstratedtocauseC.difficile lysis invitrowhenexpressedasa
recombinantproteininE.coli(Mayeretal.2008).CD27Lhasasimilarconformation
tomanyendolysins–aC-terminalcellbindingdomain,andanN-terminalcatalytic
domain. Removal of the C-terminal domain, to create CD27L1-179 was found to
improveenzymeactivity andmarginally extend the lytic rangeof theendolysin –
enablingittolysemorebacterialspecies(Mayeretal.2008).
The C. reinhardtii chloroplast has been successfully used to express two active
endolysinstodate,Cpl-1andPal,bothofwhicharespecifictoS.pneumoniae.To
developtheC.reinhardtiichloroplastasaplatformtoproduceendolysins,andgiven
thespecificnatureofendolysins, it is importanttoshowproductionofendolysins
that are active against a range of bacteria. To this end, the truncated endolysin
CD27L1-179,whichhasapartiallyresolvedcrystalstructure(PDBcode4CU5and3QAY)
andhasbeenshowntobeactiveagainstasmallrangeofbacteriawhenproducedin
E.coli,waschosenasanovelcandidateforC.reinhardtiichloroplastexpression.

204
5.3.2.1 ProductionofaC.reinhardtiilineexpressingCD27L
From hence forth, CD27L will refer to the truncated version of the endolysin,
previouslyreferredtoasCD27L1-179.
5.3.2.1.1 Genedesignandcodonoptimization
TheCD27LproteinisaccessiblebyPDBcodes3QAY(catalyticdomain)and4CU5(Cell
bindingdomain).The179-aminoacidsequence(catalyticdomain)wasobtainedfrom
RCSBPDB(ahistidineaminoacidwasremovedfromtheN-terminus,whichremained
fromtheHis-tag)andthegenesequencewascodonoptimizedfortheC.reinhardtii
chloroplast as described in 2.3.2. AnHA tagwas added to the C-terminus of the
proteincodingsequence forwesternblotdetection,andSapIandSphI restriction
siteswere included for cloningpurposes.The full-lengthgenewas synthesizedby
IntegratedDNATechnologiesasagBlock®GeneFragment(AppendixJ).
5.3.2.1.2 Expressioncassetteproduction
GiventhatCD27LhaspreviouslybeenexpressedinE.coliwithoutdetrimentaleffects
toE. coli growth, therewerenoconcernsabout its toxicityduringcloning, so the
pSRSapI transformation vector could be safely used instead of the variant vector
pWUCA2thatallowsUGAstopcodonstobeintroducedintothecodingregion(Young
andPurton2015).ThesynthesizedDNAwasdigestedwithSapIandSphIrestriction
enzymesandligatedintothepSRSapIvectorandtheinsertionwasconfirmedbyDNA
sequencing.
5.3.2.1.3 C.reinhardtiitransformation
The recipient line TN72 was transformed by the glass bead method using the
pSRSapI_cd27l-HAplasmid.Theresultingtransformedline isrepresented inFigure
5.1.PhotosystemIIfunctionalityisrestoredbypsbHandtheresultingtransformants
werecapableofgrowingphototrophicallyonminimalmedia.Transformantswere
showntoreachahomoplasmicstatebyPCR(AppendixK)asdescribedin2.3.17.

205
Figure5.1TN72recipientstrainsuccessfullytransformedtocarrycd27l-HAunderthepsaAexon1promoter/5’UTRinthechloroplast,andwithpsbHfunctionalityrestored.
5.3.2.1.4 Westernblotconfirmation
WholecellpreparationsofthreeTN72_SR_cd27l-HAtransformants(T1-3),aswellas
TN72_SR_cpl1-HA and TN72_SR_Control (as positive and negative controls,
respectively) were prepared and separated by SDS-PAGE. After blotting to a
nitrocellulose membrane and blocking as described in 2.4.5, the membrane was
probedwith anti-HAprimary antibody, then incubatedwith an IRDye® secondary
antibody. Fluorescence was observed using the LiCor Odyssey® CLx system. The
resultingimageisshowninFigure5.2.
Figure 5.2 Western blot analysis shows that a ~21 kDa protein is detected by anti-HAantibodiesinthethreeTN72_SR_cd27l-HAextracts,andnotinthenegativecontrol.ThisislikelytobeCD27Lprotein.
(kDa)

206
CD27L protein accumulates to easily detectable levels in all three transformant
clones,andisoftheexpectedsize.
5.3.2.2 ActivityofCD27Lcrudeextractagainst‘stripped’E.coli
SinceitwasnotfeasibletosetupC.difficilecultivationfacilitiesinourlab,anassay
using‘stripped’E.coliwasusedsinceCD27Lhadbeenshowntobeactiveagainstthis
bacterium(Mayeretal.2011).TheE.colihadtobestrippedoftheirouter-membrane
toenableaccesstothecellwallbytheendolysinasdescribedin2.7.2.2(Nakimbugwe
etal.2006).
Crude extracts of TN72_SR_cd27l-HA and TN72_SR_Control were prepared as
describedin2.5.1,andaBradfordassaywasperformedtomeasurethetotalprotein
ineachsample.5μgofeachcrudeextract,aswellas5μgofBSAtoactasanosmotic
pressurecontrol,wereaddedtoa96-wellplate,containing200μlofstrippedE.coli
ineachwell.ThechangeinOD595wasmeasuredbyaFLUOstarOmegaplatereader
(BMGLabtech)overaperiodof200minutesat37°C.TheresultingchangeinOD595
isdisplayedinFigure5.3.
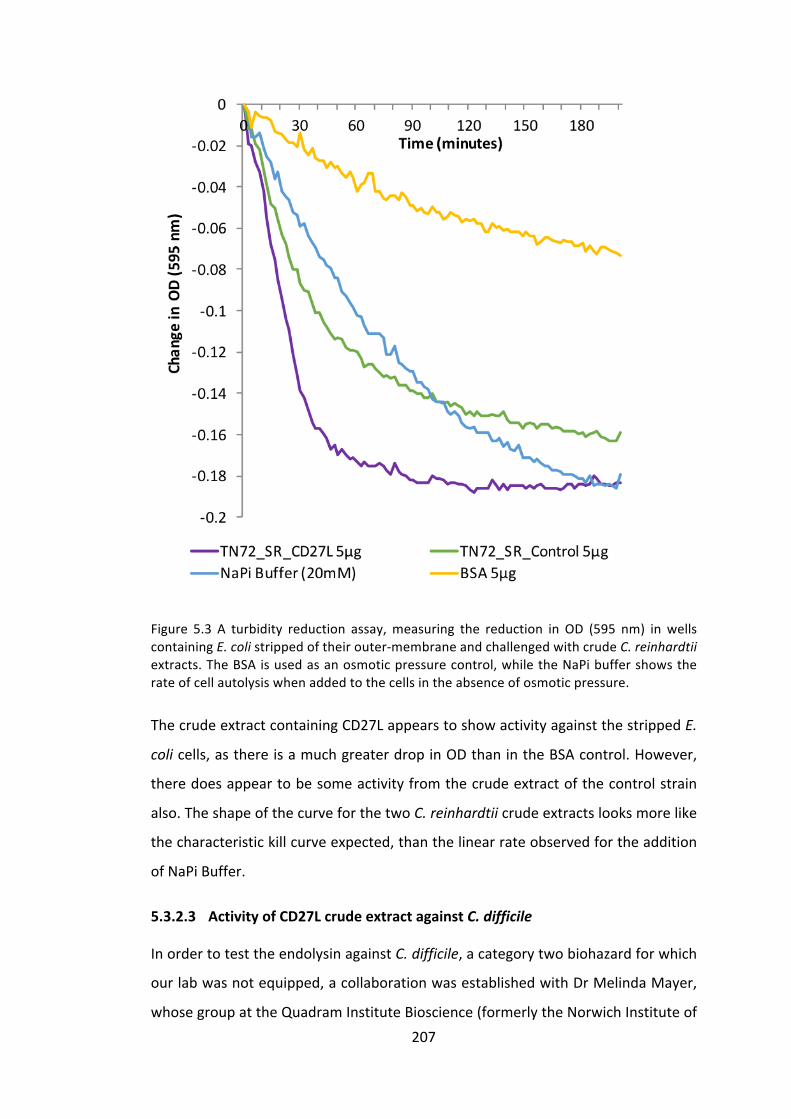
207
Figure 5.3 A turbidity reduction assay,measuring the reduction in OD (595 nm) inwellscontainingE.colistrippedoftheirouter-membraneandchallengedwithcrudeC.reinhardtiiextracts.TheBSAisusedasanosmoticpressurecontrol,whiletheNaPibuffershowstherateofcellautolysiswhenaddedtothecellsintheabsenceofosmoticpressure.
ThecrudeextractcontainingCD27LappearstoshowactivityagainstthestrippedE.
colicells,asthereisamuchgreaterdropinODthanintheBSAcontrol.However,
theredoesappeartobesomeactivityfromthecrudeextractofthecontrolstrain
also.TheshapeofthecurveforthetwoC.reinhardtiicrudeextractslooksmorelike
thecharacteristickillcurveexpected,thanthelinearrateobservedfortheaddition
ofNaPiBuffer.
5.3.2.3 ActivityofCD27LcrudeextractagainstC.difficile
InordertotesttheendolysinagainstC.difficile,acategorytwobiohazardforwhich
ourlabwasnotequipped,acollaborationwasestablishedwithDrMelindaMayer,
whosegroupattheQuadramInstituteBioscience(formerlytheNorwichInstituteof

208
FoodResearch)werethefirsttoidentifyCD27L(Mayeretal.2008).Thisenabledthe
testing of the CD27L-containing crudeC. reinhardtii extract directly onC. difficile
usingtheMayergroup’sestablishedprotocol.
A turbidity reduction assay was performed as described in 2.7.2.3. Two
concentrationsof crudeextract, inNaPibuffer,wereused: 10μgand50μg. The
purifiedCD27LwasprovidedbyDrMayerandusedasapositivecontrol.Itwasan
old(2008)NiNTA-purifiedCD27LsampleproducedinE.coli,sotheconcentrationwas
deemed irrelevant due to its age as much of it was thought to have degraded,
however,itstillservesasausefulpositivecontrol.
Figure5.4Clostridiumdifficileturbidityreductionassay.ThepurifiedCD27LwasprovidedbyDrMayerasapositivecontrolandTN72_SR_Controlasanegativecontrol.PerformedbyDrMelindaMayeratQuadramInstituteBioscience,Norwich,UK.

209
Figure5.4showsthattheTN72_SR_Control,whichwasusedasanegativecontrol,
appearstohaveaninhibitoryeffectuponcelllysiswhencomparedtotheNaPibuffer
control.Thisisbelievedtobeduetotheeffectofahigherosmoticpressureonthe
C.difficilecellsduetothepresenceofotherproteinsinthecrudeextract,andhas
beenreportedpreviously(Mayeretal.2010).
5.3.2.4 EnrichedCD27Lcrudeextract
TheosmoticpressureeffectsseenintheC.difficileturbidityreductionassay(Figure
5.4),andtheapparentactivityofthecontrolextractobservedinFigure5.3against
strippedE.colicells,hinderedourabilitytosatisfactorilymeasuretheactivityofthe
C. reinhardtiiproducedendolysin.Consequently, itwasdecidedtochangetheHA
epitopetagontheC-terminusoftheCD27LproteintoaStrepIItagtobetterenable
enrichmentor purification. Thiswasperformed to enable abetter comparisonof
endolysinactivitieswithouttheeffectsoftheC.reinhardtiicrudeextract.
5.3.2.4.1 ExpressionofCD27L-StrepIIinC.reinhardtii
PCRwasusedtochangetheepitopetagonthecd27lgene.Thereverseprimerwas
designedtohaveanoverhangwiththeStrepIItagsequenceattached,thusremoving
theHAtagandaddingtheStrepIItag.TheStrepIItagisshowninlowercase,andthe
startandstopcodonsareunderlinedontheprimersbelow:
cd27l.F: TGGTATTGCTCTTCAATGAAAATTTGTATTACT
cd27l_StrepII.R: ACTGACGCATGCTTATTAtttttcgaattgtgggtgagacca GTTAATGTTTTTGTTTAAAACACCTTCAAC
ThePCRproductwasdigestedwithSphIandSapIrestrictionenzymesandligatedinto
pSRSapIasdescribedin2.3.10and2.3.11.Sequencingconfirmedtheadditionofthe
correctencodingsequencefortheStrepIItag.
The recipient line TN72 was transformed by the glass bead method using the
pSRSapI_cd27l-StrepII plasmid. The resulting transformed line is represented in
Figure 5.5. Photosystem II functionality was restored by psbH and the resulting
transformants were capable of growing phototrophically on minimal media.

210
Transformantswere shown to reachahomoplasmic state (Appendix L)byPCRas
describedin2.3.17.
Figure5.5 TN72 recipient strain successfully transformed to carrycd27l-StrepII under thepsaAexon1promoter/5’UTRinthechloroplast,andwithpsbHfunctionalityrestored.
5.3.2.4.2 ConfirmationbyWesternblotting
WholecellpreparationsofTN72_SR_cd27l-StrepII,aswellasTN72_SR_cd27l-HA,and
TN72_SR_Control(aspositiveandnegativecontrolsrespectively)werepreparedand
separatedbySDS-PAGE.Afterblottingtonitrocellulosemembraneandblockingas
described in2.4.5, themembranewasprobedwith anti-StrepII primary antibody,
thenincubatedwithIRDye®secondaryantibody.Fluorescencewasobservedusing
the LiCorOdyssey® CLx system. Themembranewas then re-probedwith anti-HA
primary antibody and IRDye® secondary antibody. The two images are shown in
Figure5.6.
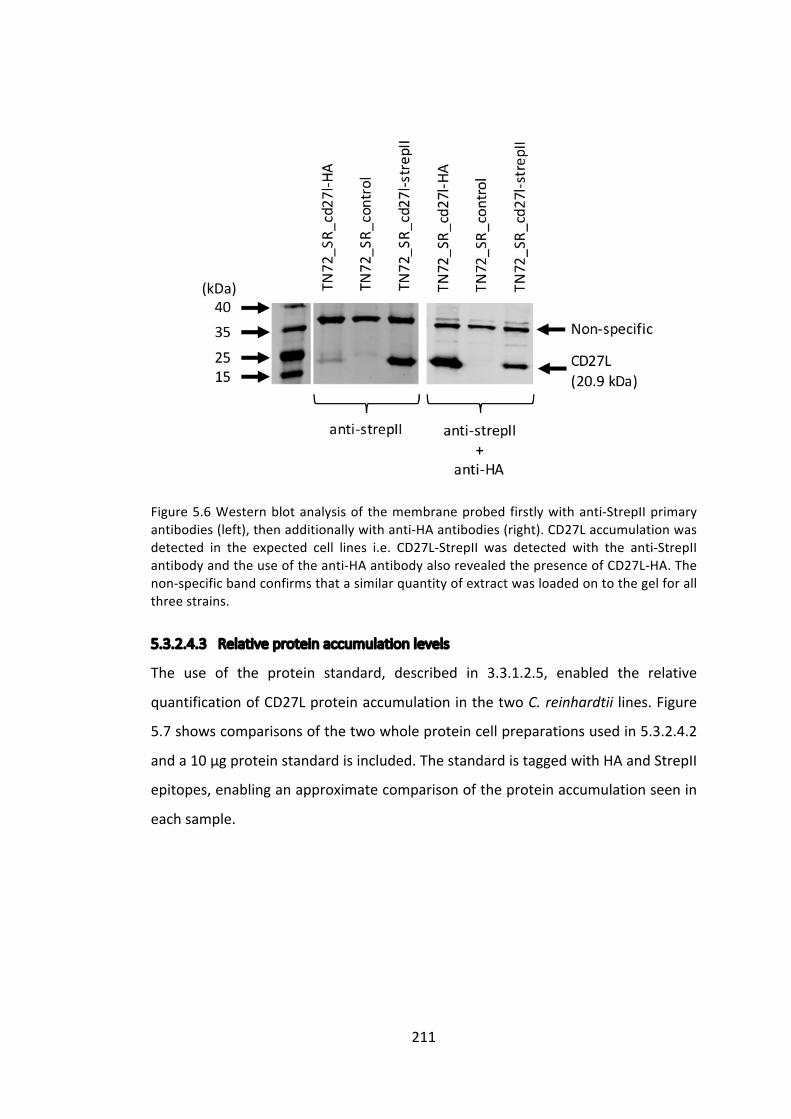
211
Figure5.6Westernblotanalysisof themembraneprobedfirstlywithanti-StrepIIprimaryantibodies(left),thenadditionallywithanti-HAantibodies(right).CD27Laccumulationwasdetected in the expected cell lines i.e. CD27L-StrepII was detected with the anti-StrepIIantibodyandtheuseoftheanti-HAantibodyalsorevealedthepresenceofCD27L-HA.Thenon-specificbandconfirmsthatasimilarquantityofextractwasloadedontothegelforallthreestrains.
5.3.2.4.3 Relativeproteinaccumulationlevels
The use of the protein standard, described in 3.3.1.2.5, enabled the relative
quantificationofCD27LproteinaccumulationinthetwoC.reinhardtii lines.Figure
5.7showscomparisonsofthetwowholeproteincellpreparationsusedin5.3.2.4.2
anda10μgproteinstandardisincluded.ThestandardistaggedwithHAandStrepII
epitopes,enablinganapproximatecomparisonoftheproteinaccumulationseenin
eachsample.
(kDa)

212
Figure5.7Westernblotanalysisofwholecellextracts,firstnormalisedbycultureOD740andcompared to a 10 μg protein standard. Quantification was performed using the LiCorOdyssey®CLxsystem,afterwhichthestandardwasusedtonormalisethetwoblotsandthesignalfromthe21kDabandscomparedinabarchart.
The protein accumulation level of the HA-tagged protein appears to be
approximately six times greater than the StrepII-tagged protein. However, as
discussed in 3.3.2.2.4.3, the difference observed is possibly due to the different
accessibility of the epitope tags on the Cpl_1-HA-StrepII protein standard. This is
supportedbythatfactthatthispatternalsoappearsbetweentheCpl1-HAandCpl1-
StrepIIaccumulationlevelsanditseemsunlikelythattheStrepIItagishavingsucha
consistentlydetrimentaleffectuponaccumulationlevels.
(kDa)

213
5.3.2.4.4 EnrichmentofCD27L-StrepIIusingHiTrapStrepIIColumn
TheinclusionofaStrepIItagonCD27Lenablesenrichmentoftheproteinandwill
thusallowaclearerinvestigationintoitsantibacterialproperties.C.reinhardtiicell
extractswerepreparedasdescribedin2.5.1,andultracentrifugedat100,000gfor1
hourtoremovecelldebris.Theresultingultracentrifuged(UC)supernatantwasthen
enrichedforStrepII-taggedproteinsusingthecommerciallyavailableHiTrapStrepII
Column, as described in 2.5.3.Western blot analysis of the resulting fractions is
showninFigure5.8.
Figure5.8Westernblotanalysisusinganti-StrepIIprimaryantibodyandIRDye®secondaryantibody.FractionsfromsamplepreparationandtheHiTrapStrepIIcolumnwerecollectedandseparatedbySDS-PAGE.Theelutedfractionhasnonon-specificband,and ‘Elution2’wasusedforfurtherexperiments.
ItisclearfromFigure5.8thatenrichmenthasbeensuccessful,andthattheeluted
fractionscontainhighquantitiesofCD27Landlowquantitiesofthenon-specifically
recognisedprotein.ItisalsonoticeablethatalargeproportionoftheCD27Lprotein
islostintheultracentrifugedpellet,butforthepurposesofthisinvestigation,thisis
not important. In the future,optimisationof thisprotocolmay result inagreater
yield.
5.3.2.4.5 ActivityofenrichedCD27LcrudeextractagainststrippedE.coli
TheenrichedCD27Lextractwaspreparedasabove(5.3.2.4.4)andanenrichedCpl-1
extractwasusedasanegativecontrol.ABradfordassaywasperformedtomeasure
thetotalproteinineachsample.
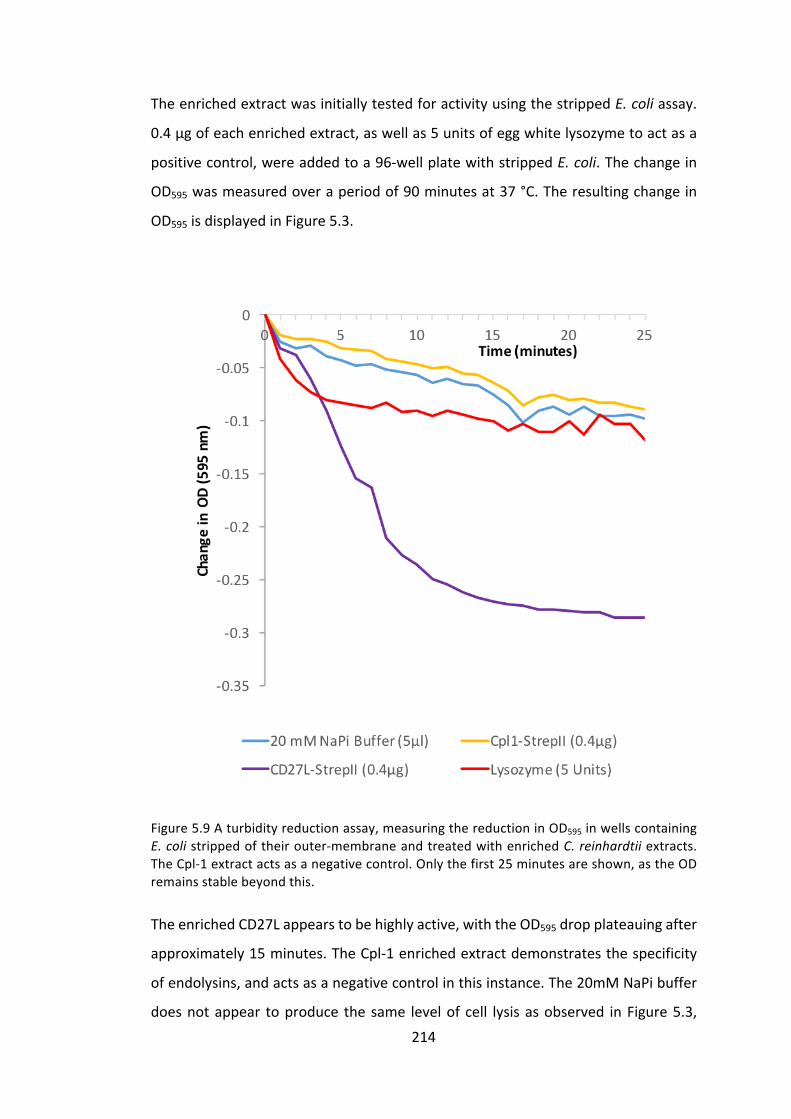
214
TheenrichedextractwasinitiallytestedforactivityusingthestrippedE.coliassay.
0.4μgofeachenrichedextract,aswellas5unitsofeggwhitelysozymetoactasa
positivecontrol,wereaddedtoa96-wellplatewithstrippedE.coli.Thechangein
OD595wasmeasuredoveraperiodof90minutesat37°C.Theresultingchangein
OD595isdisplayedinFigure5.3.
Figure5.9Aturbidityreductionassay,measuringthereductioninOD595inwellscontainingE.colistrippedoftheirouter-membraneandtreatedwithenrichedC.reinhardtiiextracts.TheCpl-1extractactsasanegativecontrol.Onlythefirst25minutesareshown,astheODremainsstablebeyondthis.
TheenrichedCD27Lappearstobehighlyactive,withtheOD595dropplateauingafter
approximately15minutes.TheCpl-1enrichedextractdemonstratesthespecificity
ofendolysins,andactsasanegativecontrolinthisinstance.The20mMNaPibuffer
doesnotappear toproduce thesame levelof cell lysisasobserved inFigure5.3,

215
probablybecausesmallersamplevolumeswereusedinthisinstance–around5μl
asopposedto~30μl.ThereislittledifferenceobservedbetweentheCpl-1andNaPi
buffer, suggesting that this enrichment has removedmany of the other proteins
contributing to the osmotic pressure effect described previously. The lysozyme,
whichwasincludedtoactasapositivecontrol,doescausesomelevelofcelllysisbut
plateauswithanOD595dropofapproximately0.1.
5.3.2.5 ActivityofenrichedCD27LcrudeextractagainstC.difficile
GiventhesuccessofenrichedCD27LagainstE.colishowninFigure5.9,similarfresh
samples were sent to Norwich for in vitro testing upon C. difficile. The turbidity
reductionassaywasperformedasdescribedin2.7.2.3.

216
Figure5.10Aturbidityreductionassay,measuringthereductioninOD600inwellscontainingC.difficilechallengedwithenrichedC.reinhardtiiextracts.TheCpl-1extractactsasanegativecontrol.Thebufferused istheStrep-Tactin®elutionbufferprovidedbytheHiTrapStrepIIcolumnkit,containingdesthiobiotin(abiotinanalogue).0.3μgofeachenrichedextractwereused. TheCD27L (E. coli) refers to apurifiedCD27L recombinantproteinproducedbyDrMayer in E. coli, to act here as a positive control. This turbidity reduction assay wasperformedbyDrMelindaMayeratQuadramInstituteBioscience,Norwich,UK.
The data show convincing lysis of C. difficile cells in vitro, despite the fact that
autolysis appears to be relatively high, as is characteristic of C. difficile when
incubatedforextendedperiodsoftimeinoxygenatedconditions,asC.difficileisan
obligateanaerobe(Mayeretal.2011).Thesampleswere,unfortunately,muchmore
dilutethanDrMayercommonlyusesforturbidityreductionassays,andinthefuture
atechniquesuchasammoniumsulphateprecipitationmayenablemoreendolysin
(~10μgratherthan0.3µg)tobeaddedtoeachwell.

217
TheconclusionsofalloftheseCD27Lexperimentsisthatanotherbiologicallyactive
andstableendolysincanbeproducedintheC.reinhardtiichloroplast.Theaddition
of a StrepII tag to the recombinant protein enables the fast and relatively cheap
enrichmentofcrudeextracts,whichinturnhasallowedustoobservemoreclearly
theactivityoftheendolysin.

218
5.3.3 Propionibacteriumacnes,bacteriophagePA6anditsendolysinGp20
Propionibacteriumacnesisaprevalent,Gram-positive,commensalbacteriumknown
bestforitsroleinthecommonskinconditionacnevulgaris.Acnevulgarisisbelieved
toaffectover80%ofadolescentsandassuch,antibioticstotreatithavebeenover-
usedandresistancehasbecomecommonplace(Leydenetal.1983;BojarandHolland
2004;Tayloretal.2014).Somebroad-spectrumantimicrobialagents,suchasbenzoyl
peroxide, while still effective, have side effects that make their prolonged use
unadvisable(Fultonetal.1974;Bojaretal.1995).
Thetreatmentofacnevulgarisoffersalargemarketandunmetneed,whichcould
possiblybeservedbyendolysintreatment.Asacnevulgarisisatopicalaffliction,it
alsopresentsaplausibletargetfortreatmentwithaC.reinhardtiicrudeextract.
The bacteriophage PA6 is known to infect P. acnes and in 2007 its genomewas
sequenced(Farraretal.2007).ThesamestudyshowedthatPA6wasabletoinfect
32differentP.acnesisolatesincludingantibioticresistantstrains.PA6belongstothe
Siphoviridae, the same virus family as Dp-1 (from which the endolysin Pal was
isolated)andΦ11(fromwhichaStaphylococcalendolysinwasisolated).Theresulting
genomeanalysis identifiedageneencodingaputativeamidase,namedgp20.The
identificationofgp20asencodinganendolysinisfurthersupportedbythefactthat
thegeneimmediatelydownstreamofit,gp21,appearstoencodeaholin:aclassic
endolysin-holingenearrangement.
Todate,therearenoreportsintheliteraturedescribingthesuccessfulexpressionof
Gp20asarecombinantprotein,orademonstrationofpeptidoglycancleavageofP.
acnesorcelllysis.However,aUSpatent(US_2012_0195872_A1)wasfiledin2010by
Lysando(anaforementionedendolysincompany)describingthefusionofGp20toan
18aminoacidpeptidesequence,whichwasshowntobeactiveagainsttwoP.acnes
strains, presumablyworking in a similarmanner to theArtilysins®, alsodiscussed
earlier(1.2.2.1).

219
5.3.3.1 PreviousattemptstoexpressGp20intheC.reinhardtiichloroplast
SeveralpreviousattemptstoexpressGp20intheC.reinhardtiichloroplasthavebeen
madeinthePurtonlab.Thesehaveincludedinsertionintothechloroplastindifferent
expressioncassettes,withdifferentpromoter/5’UTRs,andevenaschimericgenes,
butdetectionhasnotbeenachieved(Taunt2013).
5.3.3.2 NewattempttoproduceaC.reinhardtiilineexpressinggp20
Onevariablethathasnotbeenalteredhowever is theC-terminalHAtag.Epitope
tags,suchastheHAtag,areanextremelyusefultoolfordetectingproteinsforwhich
noantibodyisavailable,suchasnewproteins.Traditionallyepitopetagsareadded
toaproteinterminusinthehopethattheywillnotinterferewithproteinfunction.
However, as seen in this thesis and elsewhere in the literature (e.g. Brault et al.
(1999)), epitope choice and position can alter protein accumulation levels and
function.Aterminaltagremainedasensiblechoice,giventhatthestructureofthe
Gp20proteinisunknown,soasimpleswitchfromtheC-terminustotheN-terminus
oftheproteinwasproposed.
5.3.3.2.1 Genedesignandcodonoptimizationofgp20N
The success of the pWUCA2 expression vector (which allows stop codons to be
insertedintoagene-of-interest,therebycircumventingtoxicityissueswhencloning
inE.coli(YoungandPurton2015))inachievingsimilarlevelsofproteinaccumulation
intheC.reinhardtiichloroplast,andtheunknowntoxicityoftheGp20endolysininE.
coliledustousethepWUCA2plasmidasaprecaution.CodonUsageOptimizerBeta
0.92(Kong2013)wasusedtooptimizethegp20geneforC.reinhardtiichloroplast
expression. Three tryptophan codons spaced throughout the genewere changed
fromUGGtoUGA.TheHAtagcodingsequencewasincludedattheNterminus.SapI
andSphIrestrictionsiteswereincludedforcloningintothepWUCA2vector.Thefull-
lengthgp20N-HA(the‘N’indicatesthattheHAtagisplacedontheNterminus)gene
wassynthesizedbyIntegratedDNATechnologies(AppendixM).
5.3.3.2.2 Expressioncassetteproduction
SapI and SphI restriction enzymes were used to digest and ligate the gene into
pWUCA2inframe,producingtheplasmidpWUCA2_gp20N-HA.DH5αcompetentE.

220
colicellsweretransformedwiththeresultingplasmidandsequencingconfirmedthe
correctinsertion.
5.3.3.2.3 C.reinhardtiitransformation
ThepWUCA2_gp20N-HAplasmidwasusedtotransformrecipientC.reinhardtiistrain
TN72 via the glass bead method as described 2.3.13. Figure 5.11 illustrates the
resultingC.reinhardtiicellline,pWUCA2_gp20N-HA.
Figure5.11TN72recipientstrainsuccessfullytransformedtocarrygp20N-HAunderthepsaAexon1promoter/5’UTRinthechloroplast,andwithpsbHfunctionalityrestored.AmodifiedtrnWgenehasbeenco-introducedtotranslatetheintroducedUGAcodons(indicatedasredbars)inthegp20Ngeneastryptophans.
TheresultingtransformantswereconfirmedforhomoplasmicitybyPCRasdescribed
in 2.3.17 (Appendix N). Finally, the correct insertion was confirmed by DNA
sequencingasin2.3.18.
5.3.3.2.4 Westernblottingunabletoconfirmaccumulation
To test for the accumulation ofGp20 inC. reinhardtii,whole cell preparations of
TN72_WUCA2_gp20N-HA,TN72_A_gp20-HA(Taunt2013),andTN72_SR_Control(as
twonegativecontrols)werepreparedandseparatedbySDS-PAGE.Afterblottingto
nitrocellulose membrane and blocking as described in 2.4.5, the membrane was
probed with anti-HA primary antibody, then incubated with IRDye® secondary
antibody. Fluorescence was observed using the LiCor Odyssey® CLx system. The
resultingimageisshowninFigure5.12.
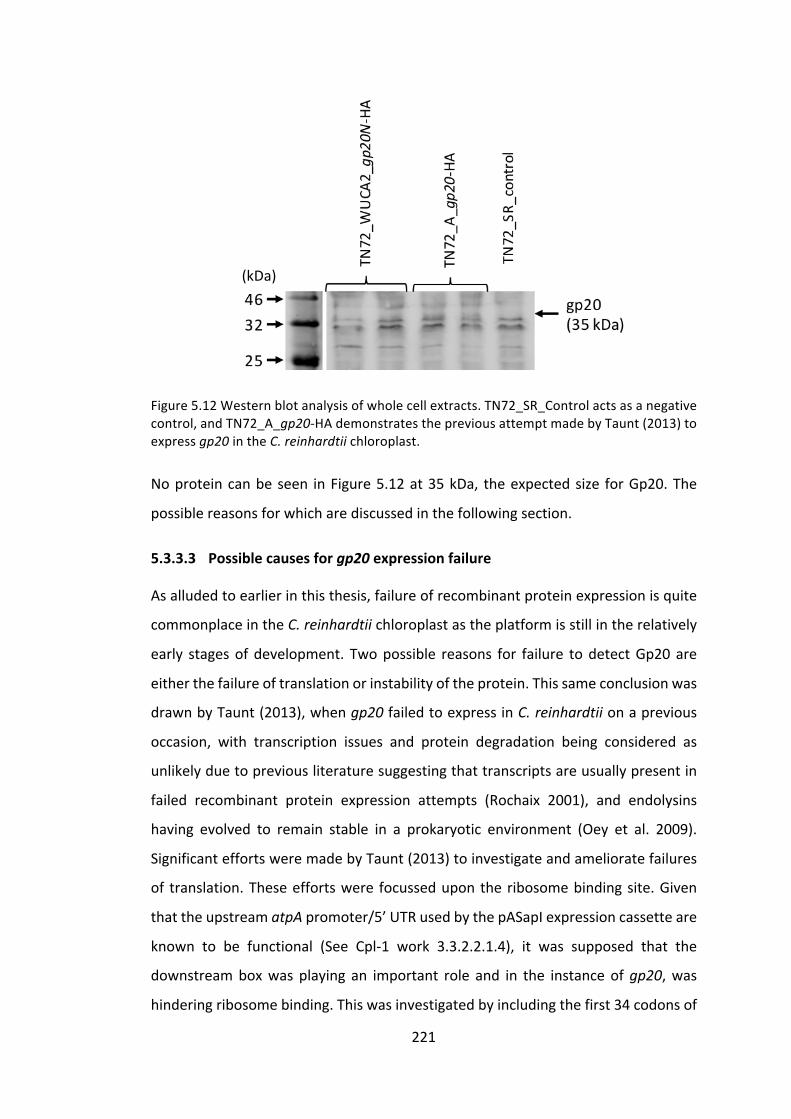
221
Figure5.12Westernblotanalysisofwholecellextracts.TN72_SR_Controlactsasanegativecontrol,andTN72_A_gp20-HAdemonstratesthepreviousattemptmadebyTaunt(2013)toexpressgp20intheC.reinhardtiichloroplast.
Noproteincanbeseen inFigure5.12at35kDa, theexpectedsize forGp20.The
possiblereasonsforwhicharediscussedinthefollowingsection.
5.3.3.3 Possiblecausesforgp20expressionfailure
Asalludedtoearlierinthisthesis,failureofrecombinantproteinexpressionisquite
commonplaceintheC.reinhardtiichloroplastastheplatformisstillintherelatively
early stagesofdevelopment. Twopossible reasons for failure todetectGp20are
eitherthefailureoftranslationorinstabilityoftheprotein.Thissameconclusionwas
drawnbyTaunt(2013),whengp20failedtoexpressinC.reinhardtiionaprevious
occasion, with transcription issues and protein degradation being considered as
unlikelyduetopreviousliteraturesuggestingthattranscriptsareusuallypresentin
failed recombinant protein expression attempts (Rochaix 2001), and endolysins
having evolved to remain stable in a prokaryotic environment (Oey et al. 2009).
SignificanteffortsweremadebyTaunt(2013)toinvestigateandamelioratefailures
of translation.Theseeffortswere focussedupontheribosomebindingsite.Given
thattheupstreamatpApromoter/5’UTRusedbythepASapIexpressioncassetteare
known to be functional (See Cpl-1 work 3.3.2.2.1.4), it was supposed that the
downstreamboxwas playing an important role and in the instanceofgp20,was
hinderingribosomebinding.Thiswasinvestigatedbyincludingthefirst34codonsof
(kDa)

222
theendogenousatpAgenefollowedbytheGOI,tocreateafusion(separatedbya
stromalprocessingpeptidase).TheplasmidusedtoperformthiswasnamedpASap2
andwascreatedbyDrChloeEconomou(Taunt2013).However, thisstrategyalso
failedtoproduceanyvisiblelevelofproteinaccumulation.
5.3.3.4 ProductionofanE.colilineexpressingGp20
The failure to detect Gp20 accumulation in the C. reinhardtii chloroplast after
attempting several strategies led us to investigatewhether Gp20was stable and
activewhenexpressedasarecombinantproteininE.coli.Apreviousinvestigation
byTaunt(2013)failedtoobserveexpressionofGp20inE.colilinestransformedwith
pASapI containing the gp20 gene, believing a lack of optimisation of the atpA
promoter/5’UTRforE.coliexpressiontoberesponsible(Wannathongetal.2016).
Weinvestigatethisfurtherhere,asifproteinaccumulationcanbeachievedinE.coli,
then it suggests that the protein is likely to be stable in C. reinhardtii and that
translation is likely to be the key issue in the failure to detect the recombinant
protein.
5.3.3.4.1 Expressioncassetteproduction
Thegp20N-HAgeneusedpreviously couldnotbe reused for expression inE. coli
owingtothepresenceofstopcodonsinthegene,asitwasoriginallydesignedfor
use with the pWUCA2 plasmid. A gp20 gene with a C-terminal HA tag, codon
optimized for Synechocystis expression in another project and synthesized by
Integrated DNA technologies, was used as a template. Primerswere designed to
introduceaNcoIrestrictionsiteupstreamoftheATG.ABamHIsitealreadyexisted
below the stop codons so did not needmodifying. The introducedmutations are
showninlowercase,whilethestartandstopcodonsareunderlined.
gp20_NcoI.F: CGTATACccATGGTGCGTTA
gp20.R: GATTGCGGATCCTTATTAGGC
ThePCRproductwasdigestedandligatedintothehighexpression,IPTGinducible,
plasmidpProEXHTb(Figure5.13)andtransformedintoE.colistrainDH5a.
223
FigureRemoved
Figure 5.13 Structural plasmid map of pProEX HTb and details of the multiple cloning site. Reproduced fromhttp://www.biofeng.com/zaiti/dachang/pProEXHTb.html.

224
5.3.3.4.2 Coomassiestainconfirmationofexpression
SynthesisoftherecombinantproteinwasinducedusingIPTGasdescribedin2.6.1.
E. coli lysateswere then createdusingBugBuster™asdescribed in2.6.2 forboth
inducedanduninducedculturesandpreparedforproteinanalysis.Thepreparations
wereseparatedbySDS-PAGEandstainedwithCoomassieBrilliantBlueasdescribed
in2.4.4.ThestainedgelwasanalysedusingtheLiCorOdyssey®CLxsystemandthe
resultingimageisdisplayedinFigure5.14.
Figure5.14WholecellpreparationsofinducedanduninducedE.colitransformedwith
pProEX_gp20-HAwereseparatedbySDS-PAGEandtotalproteinstainingwasperformed
usingaCoomassiestain.
HighaccumulationisobservedintheinducedE.coliattheexpectedsizeforGp20,
whichis~35kDa.Thereisnoproteinseenintheuninducedsampleswhichgivesus
confidencethattheobservedproteinisindeedGp20.
5.3.3.5 ActivityofGp20
AcrudelysateoftheinducedE.coliexpressinggp20wasproducedusingaStansted
‘Pressure cell’ homogeniser (2.6.3), and centrifuged at 5,000 g for 20minutes to
remove cell debris. The lysate was then filter sterilized using a 0.22 μm filter to
remove any remaining viable E. coli cells. The same procedure was applied to a
culture of untransformedDH5α cells as a negative control. UntransformedDH5α
wereusedinsteadofuninducedDH5α_ProEX_gp20-HAtoavoidissuesaroundleaky
expressionofgp20intheuninducedcontrol.
25
32
46
Induced Uninduced
gp20(35kDa)
x10
x5 x1 x10
x5 x1

225
5.3.3.5.1 Crudelysateforactivityassay
Thepellet, lysateand filter sterilized lysatewerepreparedandseparatedbySDS-
PAGE.Afterblottingtonitrocellulosemembraneandblockingasdescribedin2.4.5,
the membrane was probed with anti-HA primary antibody, then incubated with
IRDye®secondaryantibody.FluorescencewasobservedusingtheLiCorOdyssey®CLx
system.TheresultingimageisshowninFigure5.15.
Figure5.15WesternblotanalysisshowsHA-taggedGp20inallthreelanesoftheinduced
DH5α_ProEX_gp20-HAlysate,andnotinthenegativecontrollanes.
AlthoughitappearsthatalargeproportionofGp20proteinislostinthepellet,there
isstilladetectablequantityinthefiltersterilisedlysate.Thisisanencouragingresult,
asitsuggeststhatrecombinantGp20canexistinastableform.Thisfindingreduces
thelikelihoodthatGp20instabilityisthemajorcauseofthelackofdetectableprotein
the C. reinhardtii chloroplast, and requires us to look once again at translation
elongationasthemostlikelyissuepreventingaccumulation.
5.3.3.5.2 ColonyformingunitassayagainstP.acnes
InordertotestwhethertherecombinantGp20isactive,acolonyformingunitassay
againstP.acneswasperformedusingthefiltersterilizedlysatesasshowninFigure
5.15.
(kDa)

226
CulturesofP.acnesweregrownanaerobicallyasdescribedin2.1.5.3.Thecultures
werecentrifugedat5,000gfor20minutesandresuspendedin20mMNaPibuffer.
50 μl of bacterial suspension and 50 μl of filter sterilized lysate or buffer were
incubatedat37°Cfor0,30,60,90,120or150minutes.Three10μlsampleswere
taken after the stated incubation time from each combination and spotted onto
blood agar plates. After anaerobic incubation at 37 °C for oneweek, growthwas
visibleandtheplateswerephotographed,showninFigure5.16.
Figure5.16P.acnesgrowthonbloodagarplatesafterincubationwithcrudeE.colilysateswithandwithoutGp20,orjustwith20mMNaPibuffer.AsisoftenthecasewithP.acnes,individualcoloniestendnottobevisibleandasmalllawnofcellsappearsinstead(Stoffels
2015).
Thepresenceofalawn,ratherthandistinctcoloniesofP.acnesmakesquantification
of the antimicrobial effect of Gp20 difficult to measure. However, visually there
appeared to be no difference in growth between any of the spots regardless of
substrateandincubationtime.Infact,thebufferaloneappearedtoreducethelawn

227
themost,perhapsduetotheosmoticpressureeffectsdescribedpreviouslyinthis
thesis. Unfortunately, this experiment lacks a positive control, without whichwe
cannotbesurethatGp20isnotworkingas,forexample,thevisiblebacteriamaybe
somethingotherthanP.acnesorthedosagemightnotbehighenough.
ThisresulttentativelysuggeststhatGp20,whenproducedasarecombinantprotein
inE.coli,doesnotdisplaystrongantibacterialpropertiestowardsP.acnes,however,
as mentioned, the assay requires improvements for this conclusion to be more
convincing.SinceTaunt(2013)initiallyinvestigatedgp20expressioninC.reinhardtii,
13 other P. acnes targeting endolysins have been identified from other
bacteriophages(Marinellietal.2012).Theseputativeendolysinsdisplayremarkable
sequence similarity, as do the whole bacteriophage genomes themselves. This is
unusualforbacteriophageandunexpected,asusuallyahighlevelofgeneticdiversity
exists amongst bacteriophage (Pride et al. 2012). The differences that do occur
betweenendolysinsequencesaremostlyintheflexiblelinkerregionseparatingthe
twodomains.ThissignificantlevelofsimilarityledMarinellietal.(2012)tosuggest
that any of the 14 endolysins chosen for recombinant production and endolysin
therapywouldbesuccessful.

228
5.4 Conclusionandfuturework
This chapter has attempted to express two endolysins in the C. reinhardtii
chloroplast. The first, CD27L, has been shown in the literature tobe activewhen
producedinE.coliandtestedinvitroagainstC.difficile(Mayeretal.2008).Herewe
haveshownthatCD27LcanbeexpressedinC.reinhardtiitoarelativelyhighlevel
and is active in vitro against both strippedE. coli andC. difficile. In contrast, the
secondendolysin,Gp20,hasnoliteraturedemonstratingitsrecombinantsynthesis
oractivityinvitroorotherwise,andpreviousattemptstoexpressgp20inE.coliand
C.reinhardtiihavefailed,forunknownreasons(Taunt2013).Afterfailingtoachieve
expressioninC.reinhardtiiwithadifferentepitopetagposition,thefocusshiftedto
achievingexpressioninE.coliandassayingforactivityagainstP.acnes.Expressionin
E.coliwassuccessfulbutactivitywasnotsatisfactorilyassayed,aswillbediscussed
furtherbelow.
5.4.1 CD27Lendolysin
TheinvestigationintoexpressingCD27LinC.reinhardtiihasbeenrelativelysuccessful
todate.Expressionhasbeenachievedand theuseof theStrepII taghasenabled
enrichmentofCD27Lfromcrudeextracts,allowingmorepreciseactivityassaystobe
performed.Activityhasbeenconfirmed invitroagainstbothC.difficile, itsnatural
target,andE.colicellswhenstrippedoftheirouter-membrane,whichactsasauseful
assaywhenC.difficilefacilitiesareunavailable.
AswithCpl-1inChapter3,attemptstoquantifyCD27Laccumulationhaveproduced
varied results. The StrepII-tagged CD27L protein appears to accumulate to
significantlylowerlevelsthantheHA-taggedversion–apatternalsoseenwithCpl-
1.The finding thatStrepII-taggedproteinsconsistentlyappear toaccumulate toa
lowerlevelthantheirHA-taggedcounterpartshasledustoquestionthevalidityof
thedouble-taggedproteinstandardproducedin3.3.1.2.6,ashasbeendiscussedin
more detail in 3.3.2.2.4.3. For both this CD27L work and the Cpl-1 work, it is
important toaddress this issueand finda suitablemethodofquantifyingprotein

229
accumulation,suchasraisingantibodiestoCD27Litselfandusingthepurifiedprotein
asastandard.
WhiletheresultsshowninFigure5.10appearcredible,demonstratingtheactivityof
C.reinhardtii-producedCD27LagainstC.difficilerequiresafewminormodifications
totheassaytomakeitmoreconvincing.Thefirstistoperformtheassaywithhigher
concentrationsofendolysin.DrMayerusuallyincludes10μgofendolysinperwell,
whilewewere only able to use 0.3 μg perwell due to the diluted nature of the
extract. This can be easily remedied by performing an ammonium sulphate
precipitationupon theextract to concentrate theprotein. It is expected that this
improvementwillwidenthegapbetweenthepositiveandnegativecontrols,thus
makingtheassaymoreconvincing.Secondly,adistinctionmustbemadebetween
celllysisandcellviability.Whileitisreasonabletoassumethatthetwoarerelated,
aminorreductioninturbiditydoesnotnecessarilymeanthatcellviabilityisequally
unaffected.Forexample,anendolysinmaycleavethepeptidoglycaninonlyafew
locations,enoughtolysethecell,butleavingthewallotherwisemostlyintact.This
willregisterasalowlevelofturbidityreduction,buthasreducedcellviabilitytothe
samelevelasanendolysinthatbreaksthewallintomanypieces.Inordertoconfirm
theefficacyofanendolysin,itisthereforeimportanttoincludeadirectmeasurement
ofcellviability,suchasaCFUassay.Byplatingthecontentsofthewellsfromthe
turbidity reduction assay onto nutrient agar it would be possible to quantify the
remainingnumberofviablebacteria.
Arecentlydiscoveredpropertyofsomeendolysins,suchasCD27LandCTP1L(Dunne
etal.2016)andLys170(Proençaetal.2015),istheirencodingofasecond,internal,
translation start site. This second translation start site results in the additional
synthesisoftheC-terminaldomaini.e.afreecellwallbindingdomain.ThisfreeCBD
formsamultimericendolysinwiththefull-lengthendolysintoenableittobecome
fullyactive.ItwasnotedbyDunneetal.(2016)thattherecombinantendolysinCTP1L
produced froma codon-optimised genedisplayed significantly lower activity than
thatproducedfromthewild-typenucleotidesequence.Thecodonoptimisedversion
produced much lower levels of free CBD, and the addition of recombinantly
synthesisedfreeCBDrescuedactivitylevels.

230
Thecd27lgeneexpressedhereinC.reinhardtiiisthetruncatedversionofthefull-
lengthgene,asdiscussed in5.3.2.1,andassuchdoesnotcontain theCBDor the
putativeinternalribosomebindingsiteidentifiedbyDunneetal.(2016).Dunneetal.
(2016) noted that the free CBD of CD27L was not necessary for activity, but
postulated that it may play a role when the endolysin is under physiological
conditionswhichhaveyettobeinvestigated.
Itwouldbeinterestingtoexpressthefull-lengthCD27Lendolysin inC.reinhardtii,
usingacodonoptimisedversionbutensuringthattheShineDalgarno(SD)andRBS
oftheputative internal translationsiteremainunchanged.Thiswouldanswerthe
questionofwhethertheC.reinhardtiichloroplastmachineryisabletorecognisethis
internaltranslationsite.Itseemsunlikelythattheinternalsitewillberecognisedby
the chloroplast machinery, as although the chloroplast system does share many
featureswithitsprokaryoticancestors,translationinitiationislesswellunderstood
in the C. reinhardtii chloroplast. For example, although chloroplast 16S rRNA
molecules display a highly conserved anti-SD sequence near their 3’ end, the
complementary SD sequences on chloroplast mRNA UTRs are hypervariable, if
presentatall(Rochaixetal.1998).AC-terminalStrepIItagonthefull-lengthCD27L
proteinwillenableenrichmentanddetectionofboththefull-lengthproteinandany
productsynthesisedfromtheinternaltranslationsite.Iftheinternaltranslationsite
fails to produce a freeCBD, then itwould alsobeof interest to express theCBD
independently,underanalternativepromoter/5’UTR.
TheinvestigationbyDunneetal.(2016)concludedthatthefreeCBDoligomerised
withthefull-lengthCD27L,butforthisendolysinitdidnotappeartogreatlyaffect
activity. Therefore, it may be more relevant to look at CTP1L, an endolysin that
targetsClostridiumtyrobutyricum,andwasshowntorequirethefreeCBDforactivity.
Thepotential forCTP1Las a future candidate forC. reinhardtii expressionwill be
discussedlaterinthischapter.
Usually,atthisstageofanendolysinproject,mostfurtherworkwouldinvolvethe
useofananimalmodelforinvivotesting.However,inthecaseofC.difficilemodels,
themosthelpfulnext stage isprobablymoreadvanced in vitro testing,using, for

231
example,atriple-stagechemostatmodelofthehumangut.Thereasonforthisisthat
the most well-used animal model for CDI, the Syrian Hamster, is not fully
standardised and presents several important and problematic differences in
pathophysiologytohumaninfection(Bestetal.2012).Thetriple-stagehumangut
modelwasdesignedbyMacfarlaneetal.(1998)andhassincebeenimprovedand
updated,butessentiallyconsistsofthreeconnected,pHcontrolled,anaerobically-
maintainedvessels,withgrowthmediafedintothetopataspecificflowrate.The
model emulates the characteristics (spatial, temporal, nutritional and
physicochemical)oftheproximaltothedistalbowel (Bestetal.2012).Thetriple-
stagechemostathumangutmodelhasbeenusedextensivelyfortestingC.difficile
treatmentefficacy(Freemanetal.2007;Bainesetal.2008)andwillenablevarious
questions around the CD27L endolysin to be asked and answered, such as: how
quicklydoestheendolysindegrade/denatureinthehumangutenvironment?How
muchendolysinisrequiredtoachieveareductioninC.difficilebacterialloadandhow
oftenmustdosesbegiven?Whatdeliverymechanismislikelytoenablethedelivery
of CD27L to the key C. difficile infection sites? How does CD27L compare to
vancomycintreatment?CanCD27Lactsynergisticallywithvancomycintreatment?
However,majorissueswithevenanadvancedinvitromodelstillexist,suchasthe
absenceofimmunologicalinfluence.Toaddressthis,eventuallysomeformofinvivo
modelwill doubtlessbe required,but theuseof advanced in vitro testing should
reducetherelianceupontheseand informmorepreciselywhichtestsneedtobe
carriedout.
An interesting final considerationofC. reinhardtii-producedCD27L is thedelivery
mechanism. To date, C. reinhardtii-produced therapeutics have been considered
mostviablefortopicalapplicationsduetothepotentialcostsavingsmadebyusinga
crude extract, on the assumption that a crude extract would not achieve FDA
approval for systemic use. However, C. difficile has recently emerged as an
increasinglyprevalentpathogeninfoodanimals(Songer2004;Keeletal.2007),such
aspigs,calvesandostrichesaswellaspetssuchascats,dogsandhorses(Gouldand
Limbago2010).Theseofferapotentialmarketthatmaybelesstightlyregulatedand
enableC.reinhardtiiproducedtherapeuticstoshowaproof-of-concept.WorkbyDr

232
PriscillaRajakumarinthePurtonLabinvestigatedtheadministrationoforalvaccines
produced in C. reinhardtii to chickens and fish, and discussed the use of the C.
reinhardtiicellasadeliverycapsule(Rajakumar2016).Whilethisconcepthasnot
been practically tested, Rajakumar (2016) suggests that the cell wall and the
chloroplast of C. reinhardtii protect the therapeutic recombinant proteins from
degradationthroughtheupperlevelsofthedigestivesystem,enablingthemtoreach
the gut. This will, of course, depend upon which animal is being treated, as for
examplethechickensstudiedbyRajakumar(2016)haveaveryshortintestinaltract,
whilethesamestrategymaynotworkincattle.
5.4.2 Gp20endolysin
TheGp20endolysinhasbeenproblematic since recombinantexpressionwas first
attempted by Taunt (2013). As an endolysin targeting a topical and prevalent
bacterialspecies,itwouldbeanidealcandidateforproductioninC.reinhardtiiasa
treatmentforacnevulgaris.However,previousattemptstoachievegp20expression
intheC.reinhardtiichloroplast,aswellasnewattemptsdescribedinthischapter,
haveallfailedtoproducedetectablelevelsofexpression.TotestwhethertheGp20
protein is stable and active outside the P. acnes intracellular environment, the
endolysinwasexpressed inE.coli. InE.coliGp20accumulated tohigh levels,but
colonyformingunitassaysusingacrudelysateagainstP.acnesfailedtoshowany
activity.GivenhighlevelsofinterestinP.acnes,andtheidentificationofatleast14
relevant putative endolysins, it does seem remarkable that no reports of a
recombinantly produced endolysin targeting P. acnes exist, other than the
aforementionedpatentapplicationbyLysando.Herewewilldiscussinmoredetail
thepossiblereasonsforissuesfacedinexpressionandactivity,andpotentialfuture
linesofinvestigation.
Afterfailedinitialattemptstoexpressgp20 intheC.reinhardtiichloroplast,Taunt
(2013) investigated whether translation initiation was the issue by creating a
cpl1:gp20 fusion, but again expression failed, while a positive control cpl1:pal
succeeded.Thissuggestedthattheribosomewasstallingatsomepointwithinthe
gp20gene,anissuemostcommonlyassociatedwithcodonusage.However,further

233
codon optimisation failed to solve the problem. Taunt (2013)was also unable to
detectgp20expressioninE.coli,leadingtoasuggestionthatrapidproteinturnover
maybetoblame.However,stronggp20expressioninE.coliwasdescribedearlierin
this chapter,making this a less likely suspect, althoughadmittedly theE. coli and
chloroplastenvironmentsarenotidentical.
Asmentionedearlier,theremarkablesimilaritybetweenthe14P.acnes targeting
endolysinsdescribedsofarmeansthattestingtheexpressionofadifferentendolysin
isunlikelytomakeanydifference.ThepotentialdemandforsolutionstoP.acnes
infection from both the cosmetic and pharmaceutical industries means that this
endolysinremainsworthyofinvestigation.
ThealignmentperformedbyMarinellietal. (2012) (Figure5.17)providesauseful
indicationofthetwoendolysindomains,astheareaofleasthomologyislikelytobe
the glycine rich linker domain (circa position 200). This is further supported by
databasecomparisonspredictingthattheN-terminaldomainisassociatedwithN-
acetylmuramoyl-1-alanineamidaseactivity(Farraretal.2007).Giventhatendolysin
modulesgenerallyremainstablewhenseparated(Mayeretal.2008;Dunneetal.
2016),itwouldbeinterestingtoattempttoexpressthetwoseparatedomainsinthe
C. reinhardtii chloroplast. This would give an insight into where the ribosome is
stalling,andcouldpotentiallybesubjectedtofurthercodonoptimisationtominimise
this.Furthermore,asdemonstratedwithCD27L,occasionallytheendolysincatalytic
domainaloneissufficientforcelllysiswhichcouldbetestedhereifexpressioncan
beachieved.

234
FigureRemoved
Figure5.17Proteinsequencealignmentof14putativeendolysinsfromP.acnesbacteriophages.Thealignmentshowsthatthesequenceishighlyconserved,withthemajorityofdifferenceslocatedintheglycine-richlinkerataroundposition200.TheC-terminuscellwallbindingmoduleislesshighlyconservedthanthecatalyticN-terminusmodule.Gp20fromthePA6bacteriophageisthe4thfromthebottom,labelledPA6_rev_gp20,asitisarevisedannotationofthePA6genome.ReproducedfromMarinellietal.(2012).

235
ThenextissuetoresolveistheapparentlackofactivityoftheE.coli-producedGp20
proteinagainstP.acnes.ThefirstquestiontoaddressinthiscaseistheCFUassay
itself,whichwasnotfullysatisfyingandwasnotrepeatedduetotimeconstraints.
The investigation requiresa suitablepositivecontrol, theobviouschoicebeingan
antibiotic such as erythromycin, the first line of treatment in a clinical setting
(Zaenglein et al. 2016). However, it would be preferable to have amore specific
antibacterial,whichwillpartiallyconfirmthatthebacteriaseenareindeedP.acnes
andnotacontaminatingspecies.Thepeptideadrenomedullinissecretedbyhuman
epithelial cells and has been demonstrated to have an antimicrobial effect upon
severalmicroorganisms foundontheskinandrespiratory tract,but isparticularly
effectiveagainstP.acnes,M.luteus,P.gingivalisandH.pylori(Allakeretal.1999).It
thereforerepresentsanarrower-spectrumantimicrobial,whenusedatanoptimal
concentration, than erythromycin and may provide a useful positive control. To
furtherconfirmthatthebacteriuminquestionisP.acnes,Gramstain(+),catalase(+)
andindole(+)tests,followedbysequencingofthe16SrRNAgeneforspecies-level
identificationcanbeperformed(Butler-Wuetal.2011).
Assumingthattheaboveassayisimprovedtoanacceptablestandardbutactivityis
stillnotobserved,severalissuescouldbeatplay.ThefirstisthatP.acnesisknown
tosecretemanyprotectiveenzymeswhichallowittosurviveintheharshhumanskin
environment, includinghydrolases andproteases (Holland et al. 2010), aswell as
propionic and acetic acid which inhibit the growth of competing species (Wilson
2005).Itisplausiblethatsomecombinationofthesefactorsiscausingtheendolysin
todenatureorbedegradedbeforeitcanbindandlysethecellwall.Astheendolysins
have evolved to be active from within the bacterial cell, it is possible that such
extrinsicbarriersareblockingitsaction.Anotherpossibilityisthattheholin,whichis
normallyexpressedalongsidetheendolysin, isrequiredforcell lysis,althoughthis
seemsunlikelyandhasnotbeenreportedbefore,totheauthor’sknowledge.
Asseenearlier in thischapter,whilemostendolysinsconsistofa simplemodular
monomer,otherssuchasPlyCaremultimeric,andstillotherssuchasCTP1Lrequire
oligomerisationwiththeirowntruncatedC-terminaldomain.Itispossiblethatthe
lackof activity observedwithGp20 is due to the requirement tooligomerise like

236
CTP1L for full activity. Figure 5.17 shows a conserved methionine residue at
approximatelyposition193whichcouldbeaninternalstartcodon.Alsoatposition
210inFigure5.17thealternativestartcodon,GTG,codesforavaline.Thisresidueis
conserved as either a methionine or a valine in the other putative endolysins,
howeverananalysisofthenucleotidesequenceisrequiredtoidentifywhetherthis
isapotentialinternalstartcodon.ThislocationissimilartothatobservedinCTP1L
andCD27Landwouldsimilarly result in thesynthesisofa freeCBD.However,no
ShineDalgarnoconsensuscanbeseenupstreamofeitherthemethionine-encoding
ATGorvaline-encodingGTGwhenthenucleotidesequenceofgp20isexamined,and
analysis using the NCBI Open Reading Frame Finder
(https://www.ncbi.nlm.nih.gov/orffinder/) software did not identify either as a
potentialORF(Figure5.18).

237
Figure5.18Resultsfromhttps://www.ncbi.nlm.nih.gov/orffinder/withtheputativeendolysinsequencegeneID5247034.OnlyORF1isinframeandinthe
samedirectionasthefull-lengthendolysin.ORF1is654nucleotidesinlength,producinga217aminoacidprotein.

238
TheinternalribosomebindingsiteandORFidentifiedinFigure5.18wouldresultin
thesynthesisofa freeCBD, likeCTP1L,but includesamuch largerportionof the
catalyticdomainthanwouldbeexpected.Nevertheless, itwouldbe interestingto
attempttoexpressgp20 inE.coliusingthewild-typenucleotidesequence,which
mayidentifywhetheraninternalribosomebindingsiteisactive.Suchafindingmay
resultintheimprovementofactivityagainstP.acnes.
5.4.3 Futureendolysins
Asdiscussedearlierinthischapter,theproblemssurroundingGp20cannotbesolved
bymerelyattemptingtoexpressanalternativeP.acnestargetingendolysin,asthe
levelofhomologybetweentheproteinsissohighthatitisextremelyunlikelythat
this will make any difference. In this case the strategy needs to be to better
understand why the endolysin is failing to express and attempting to solve any
problemswithactivity.
ThereareseveralotherendolysinswhichwouldbeinterestingtoexpressintheC.
reinhardtii chloroplast in the future. CTP1L is an interesting endolysin due to its
requirement tooligomerisewith its freeCBD,but is also a commercially relevant
endolysin, as it targets Clostridium tyrobutyricum. Clostridium tyrobutyricum
contaminationcanleadtoextensivecostsinthecheeseproductionindustrythrough
‘late-blowing’, a phenomenon affecting high-pH cheeses such as Edammer and
Gouda,andresultinginexcessbubblesandanabnormalflavour.Anapplicationof
CTP1L istherefore infoodsafetyandproduction,andgiventhatC.reinhardtiihas
GRASstatus,thismaywellbeaverywellsuitedmarketforC.reinhardtii-produced
endolysins.
WorkbyJadoetal.(2003)demonstratedthattwoS.pneumoniaeendolysins,Paland
Cpl-1,areabletoactsynergistically.Therearenumerousadvantagestoexploiting
drug synergies therapeutically, such as lower dosages, greater efficacy, and
minimising the opportunity for resistance to occur. It would be interesting to
investigate whether a similar synergy can be observed with endolysins targeting
otherbacteria,suchasC.difficile.Wangetal. (2015) identifiedanotherC.difficile
endolysin,namedPlyCD,whichwasdemonstratedtobeactiveagainstC.difficilein

239
vitroand,similarlytoCD27L,moreactiveinitstruncatedform,PlyCD1-174.Successful
expressionofPlyCDintheC.reinhardtiichloroplastwouldallowtestingofbothPlyCD
as a stand-alone endolysin, as well as enabling an investigation into whether a
synergisticeffectcanbeobservedbetweenPlyCDandCD27L.
As a final note on selecting future endolysins, one method might be termed a
‘disease-orientated’ approach. This approach looks at the bacterial species
associatedwithcertainidentifiablediseasesanddesigning‘endolysincocktails’tokill
allofthosespecies.Forexample,theskindiseaseimpetigoistypicallytreatedwitha
broad-spectrumantibioticcream(GeorgeandRubin2003),despitethefactthatitis
knowntobecausedbyoneofonlytwobacterialspecies:Staphylococcusaureusor
Streptococcuspyogenes. It is thereforeconceivablethatacocktailof twoormore
endolysins targetingS. aureus andS. pyogeneswouldbe a viable treatment. This
particular example, as a topical application, would again be well suited to C.
reinhardtii production. Two potential endolysins, in this case, could be LysH5
(targetingS.aureus)(Gutiérrezetal.2014)andPlyC(targetingS.pyogenes)(Shenet
al.2013).
Overall,thischapterhasdemonstratedthatwhiletheC.reinhardtiichloroplastcan
beaverywellsuitedplatformforendolysinproduction,thereremainseveralissues
and unexplored areas which leave recombinant endolysin expression far from
routine.However,non-expressingendolysingenessuchasgp20offeranopportunity
tounderstandmoreabout factorsaffectingchloroplastexpression, suchascodon
optimisationandtranslationelongation.Thesuggestedfutureworklaidoutinthis
section may help us to progress the C. reinhardtii chloroplast platform towards
achievingmorereliableproteinproduction.

240
Chapter6 Finaldiscussionandfutureprospects
6.1 Summary
6.1.1 Summaryofresults
Theresearchdescribedinthisthesissoughttoaddresstwooverarchingissues.The
first is the improvement of the genetic engineering technology underpinning the
developmentof theC. reinhardtiichloroplastasanovelplatformforrecombinant
proteinproduction.Thesecondistheadvancementofendolysinsasanalternative
toconventionalantibiotics.
Chapter3investigatestheissueofrelativelylowlevelsoftransgeneexpressionseen
inC.reinhardtiianddemonstratestechniquesavailabletoimprovethis.Asdemand
grows for protein therapeutics, such as monoclonal antibodies, hormones (e.g.
humaninsulin),andvaccines,sodoestherequirementforsuitableplatformsfortheir
recombinantproduction.Asdiscussedin1.3.1,therearevariousplatformscurrently
used for therapeutic protein production, each with different advantages and
limitations.ThegreenalgaC.reinhardtiioffersanovelalternativecellfactory,with
importantadvantagesover severalmorewidelyusedplatforms.However, several
limitationsofC.reinhardtii,includinglowlevelsofexpression,needtobeaddressed
beforeitcanrealisticallycompetewiththeestablishedplatforms.Initially,methods
used to compare protein accumulation levels between different transgenic C.
reinhardtii lines were investigated. The chapter highlights the limitations of
comparingproteinaccumulationlevelswhilstclonalvariationappearstobehaving
such a large effect, as well as proposing and testing improved methods of
comparison.Assuch,attemptstoinvestigatetheeffectsofcodonpairoptimisation
remaininconclusive.However,thechapterdoesdemonstratethattheintroduction
oftwoexpressioncassettesintotheC.reinhardtiichloroplastplastomeresultsina
robusttwo-foldincreaseinproteinaccumulationcomparedwithasingleexpression
cassette.

241
Thegrowingthreatofantibioticresistancehasencouragedresearchintodeveloping
alternativestoconventionalantibiotics.Endolysinsareofincreasinginterestandthe
first endolysin based therapies arenowavailable (1.3.3.3). Chapter 4 investigates
whether the activity of these endolysins can be improved against their target
bacterium, S. pneumoniae, either by direct mutation of the protein or through
synergisticeffectswithholins.Sitedirectedmutagenesiswasusedtodisruptthecell
wallbindingdomainofCpl-1andexpressionwasachievedforallsixmutants inC.
reinhardtii.However,suchmodificationsappearedonlytodecreasetheactivityof
theendolysin,demonstratingthatthefunctionalityofthetwoactivecholinebinding
sitesisessentialfortheactivityoftheendolysininthisinstance.DimerisationofCpl-
1viaadisulphidebridgewasalsoinvestigated,butcouldnotbeachievedintheC.
reinhardtii chloroplast. Several reasons for the failure of Cpl-1 to dimerise were
suggested, along with possible solutions, mostly revolving around the steric
hindrance of the C-terminal epitope tag. Finally, the holin protein Cph-1 was
expressedintheC.reinhardtiichloroplast.However,expressionquicklyceasedand
thepossiblereasonsforthiswerediscussed.
Chapter 5 describes attempts at increasing the suite of endolysins that can be
producedintheC.reinhardtiichloroplast.TheC.difficiletargetingendolysin,CD27L,
wassuccessfullyexpressedintheC.reinhardtiichloroplastanditsactivityagainstC.
difficile was demonstrated in vitro. Meanwhile, the P. acnes targeting endolysin,
Gp20, failed to express in transformed C. reinhardtii lines. However, it was
successfullyexpressedinE.coliunderaninduciblepromoter,butactivitycouldnot
beobservedagainstP.acnes.
6.1.2 Summaryofshort-termfuturework
Futureworkhasbeenroughlydividedintotwocategoriesinthisthesis:‘short-term’
futureworkand‘long-term’futurework.Whilebothofthesetermsarerelative,the
goalofthissystemistodistinguishbetweenworkthatwillenabletheprogressionor
conclusionofcurrentprojects(short-term),andworkthatlooksfurthertothefuture
andwouldserveasastand-aloneprojectinitsownright(long-term).Relevantshort-

242
termfutureworkhasbeensuggestedattheendofeachresultschapter,muchof
whichisveryspecific.However,themostinterestingareathateachresultschapter
highlightedforfurtherinvestigationissummarisedbrieflybelow.Potentialpathsfor
long-termfutureprojectsarediscussedin6.2below.
TheissueofclonalvariationhighlightedinChapter3limitsourabilitytoaccurately
investigate strategies for improving recombinant protein accumulation in the C.
reinhardtiichloroplast,butalsooffersanopportunitytounderstandwhatleadstoa
veryhighor very lowexpressing ‘clone’. Inorder toenable the investigation into
subtlermethodsofimprovingrecombinantproteinaccumulation,in3.4.2wesuggest
thattheuseofareportergene,suchasluciferase,andtheuseofhigh-throughput
techniques will enable ‘comparison-of-means’ analysis. Advancements in whole
genome sequencing, and importantly its falling cost, may eventually enable a
methodicalwholegenomeanalysisofhigh-expressingclonestoidentifymutationsor
insertionsthatareresultinginhighlevelsofproteinaccumulation.Thesemutations
might be affecting nuclear genes for chloroplast regulatory factors, proteases, or
chaperones. These changes can thenbe purposefully introduced alongside future
transgenes,thusimprovingC.reinhardtiiasaproteinproductionplatform.
TheendolysinCpl-1wasthemainfocusof investigation inChapter4,andassuch
much of the proposed future work was specific to this endolysin. However, the
unexpected andunexplained result thatCph-1 expressionwas achieved and then
ceased isworthyof further investigation.Silencingmechanisms,whilecommon in
nuclear transformations, have not previously been described in the chloroplast
(Danielletal.2005).Itisthereforeimportanttounderstandhowtheterminationof
expression has occurred, the findings of which may help prevent this from
reoccurring,butalsocouldbecomeausefulmolecularswitch.
Chapter5demonstratedthesuccessfulexpressionandactivityofasyntheticgene
encodingCD27L,andalsodocumentedtheunsuccessfulattemptstoexpressgp20in
C. reinhardtii. However, it is thegp20 result that remains interesting in terms of
gainingagreaterunderstandingofC. reinhardtiiexpressionand itssuitabilityasa
platformforproducingendolysins.Despiterepeatedattemptsandfailurestoachieve

243
gp20expressionintheC.reinhardtiichloroplast,thereisstillsomewaytogobefore
we can deduce the precise reason for this and attempt to ameliorate it. Codon
optimisation, translation initiation, and epitope tag location have all been
investigatedanddonotappear tobecausing failureofexpression.Attempting to
expressthetwodomainsoftheendolysinseparatelymayshedsomelightontothe
problem, and aid us in a broader understanding of expression failure in the C.
reinhardtiichloroplast.
6.2 Discussionandlong-termfutureprospects
6.2.1 AlternativestrategiestoimprovingexpressionlevelsinC.reinhardtii
chloroplast
6.2.1.1 Syntheticpromotersand5’UTRs
Syntheticpromotersand5’UTRsarebecomingwidelyusedinsyntheticbiologyand
in recombinant protein production, due to their frequent ability to providemore
consistent and higher final protein yields than endogenous regulatory elements
(Gilman and Love 2016). As discussedpreviously in this thesis (3.1.2), chloroplast
gene expression appears to bemostly controlled at the level of translation in C.
reinhardtii,ratherthantranscription,andassuchtheimprovementof5’UTRsisof
greater interestthanthatofpromotersatthisstage.Todate,themostconsistent
levelsofgeneexpressionintheC.reinhardtiichloroplasthavebeenachievedusing
endogenouspromoter/5’UTRsequencesfromhighlyexpressedproteins,andinsome
caseshigherlevelshavebeenachievedthroughcombinationswithnuclearmutants
(Micheletetal.2011)andalteredCESmechanisms(Manuelletal.2007)whichwill
bediscussedisthenextsection(6.2.1.2).
ResearchbySpecht&Mayfield(2013)useda luciferasereportergene(Figure6.1)
andhigh-throughputanalysistocompileanunbiaseddatasetofmutatedpsaAand
psbD5’UTRs,fromwhichthefirstsuccessfulsyntheticC.reinhardtiichloroplast5’UTR
was designed. The resulting synthetic 5’UTR improved protein accumulation by
approximately66%,comparedtoendogenous5’UTRs.

244
FigureRemoved
Figure6.1C.reinhardtiitransformedwithexpressioncassettescontainingmutated5’UTRscontrolling luciferase expression in the chloroplast. This false colour image illustrates thedifferencesinluciferaseexpressionunderdifferent5’UTRs.TechniquessuchasthisenabletheprocessingandexpressionquantificationofhundredsofC.reinhardtiicoloniesinaverysmall amount of time. The positive control (wild type UTR) and negative control (no-luciferase)areindicatedonthebottomleftoftheplate.ReproducedfromSpecht&Mayfield(2013).
Ashigh-throughputtechniquesbecomemoreaccessibleanddefinedinC.reinhardtii,
thepotentialforproducinglargeunbiaseddatasetsimproves.Theavailabilityofsuch
datasetsmayenabletheidentificationofconsensusmotifsinboththepromoterand
5’UTRs of endogenous genes and pave the way towards producing synthetic
promotersand5’UTRsthatdramaticallyimproveproteinyieldsaswellasproviding
consistentlevelsofgeneexpression.TheworkbySpecht&Mayfield(2013)isthefirst
step in achieving this, but their use of particle bombardment to transform C.
reinhardtiirecipientlinesremainssubjecttotheissueofclonalvariation,andassuch
veryprecisedeductionsabout5’UTRconsensussequencesaredifficulttodrawand
anevenlargerdatasetislikelytoberequired.Recentadvancementsinprecisiongene
editingmayprovidethesolutiontothisissue,aswillbediscussedinthefollowing
section.
6.2.1.2 ThepotentialofCRISPR/Cas9geneediting
Asanalternativetomodifyingthe5’UTRwhichisregulatedbyvariouselementsin
the C. reinhardtii chloroplast (a cis-acting element), one could also modify the

245
regulatory elements themselves (trans-acting factors). Such an experiment was
performedbyMicheletetal.(2011),wherechloroplasttransgeneswereshowntobe
upregulated when expressed in a cellular background that included a nuclear
mutation.Themutationinthisinstancewaswithinatrans-splicingfactorthatwas
importedtothechloroplastfromthenucleus,thuspreventingtrans-splicinginthe
chloroplast.However,thismutationalsoledtoalossofphotoautotrophymeaning
thattheC.reinhardtiicellsmustbegrowninthedarkandrequireacetateasacarbon
source.ThediscoveryofCRISPR/Cas9geneediting,andtherecentapplicationofthis
technologytotheC.reinhardtiinucleus(Baeketal.2016;Shinetal.2016),willresult
inexperimentssuchasthatdescribedbyMicheletetal.(2011)becomingfareasier
to perform since targeted knock-outs of nuclear geneswouldbepossible. Rather
thanknockingouttrans-actingelementssuchassplicingfactorswhich,asdescribed,
result in other undesirable phenotypes, CRISPR/Cas9 could be used to knock-out
other factors that indirectly control protein accumulation, such as chloroplast-
targetedproteases.Theeasewithwhichsuchmutationscouldbeintroducedshould
quicklyenableresearcherstomanipulatethechloroplastenvironmentintoonethat
is more amenable to recombinant protein accumulation without having large
detrimentaleffectsoncellphysiology.
6.2.1.3 Expressionofnovelchaperones
As discussed earlier, the primary suspect for causing low rates of successful
recombinantproteinexpressionintheC.reinhardtiichloroplasthasbeentranslation
initiationandelongation.However,poorproteinstabilityorhighproteolysiscould
alsobeplayingarole inthis.Misfoldedproteinsandthosethathavenotreached
theirnativestatearetargetsofproteases.Whileknockingoutproteasesmaybepart
of the solution to improving protein accumulation, the quality of protein being
produced is equally important, and disruption of proteases may have other
detrimentaleffectsuponthecell(RamundoandRochaix2014).
WhilerecombinantproteinyieldsinE.colihavebeensteadilyimprovedoverthepast
four decades, through a process of largely trial and error, we must attempt to
reproduce these advances inC. reinhardtiiby analysing those improvements and

246
specificallyintroducing.ManyofthesuccessfulimprovementsinE.coliweredueto
theco-expressionofchaperones(Makinoetal.2011),andthereforethisisworthyof
investigation. Many chaperones are known to be expressed in the C. reinhardtii
nucleus and targeted to the chloroplast to aid protein folding (Schroda 2004).
However,somenovelchaperonesmayhavespecificabilitiestoaidinthefoldingof
certain recombinant proteins, and studies in E. coli have found that co-
overproductionofcertainchaperonesintheE.colicytoplasmhaveaidedinprotein
solubility and yields (deMarco et al. 2007). Itwould, therefore, be of interest to
identifyrelevantchaperones,suchasHeatShockProtein70(Willmundetal.2008),
andattempttoexpressthemalongsideendolysins intheC.reinhardtiichloroplast
withthegoalofimprovingproteinaccumulationandexpressionsuccessrates.
6.2.2 Othermicroalgalhostsforscale-up
C.reinhardtiihasbeenanimportantmodelorganismforthestudyofphotosynthesis,
chloroplastbiogenesis,andflagellarfunctionfordecades(Rochaix1995).Giventhe
years of research behind it and its well-defined genetics, C. reinhardtii was the
obvious initial candidate for proving the concept ofmicroalgae as a recombinant
protein production platform. However, when it comes to scaling up microalgae
production for commercial exploitation, other speciesmaybemore suitable, and
researchperformedonC.reinhardtiishouldbereadilytransferabletothesespecies.
Forexample,somespeciessuchasthoseofSpirulina,Chlorella,Haematococcus,and
Dunaliellahaveallbeengrownoncommercialscalesforsometime(Figure6.2)and
arewellsuitedtocultivation(Rosenbergetal.2008).

247
FigureRemoved
Figure 6.2 Spirulina growing in open ponds (raceways) for commercial production atCyanotech’sfacilitiesinHawaii2.
Theadvantagetousingoneoftheabovemicroalgaeisthatthetechnologyalready
exists fortheir largescaleproduction,aswellasdataongrowthoptimisationand
commercialviability.WhileyieldsoftherapeuticproteinsfromC.reinhardtiiarevery
low, hovering around 5-8mg L-1culture volume (1.3.3.3), the potential for cheap
cultivationandhighvolumesgoessomewaytowardsnegatingthisissue.Incontrast
CHO cells, as an example, must be grown in highly controlled environments in
extremelyexpensivemedia,itisunderstandabletheiryieldperlitremustbeveryhigh
tomakethiseconomicallyviable.Itisthereforeplausibletoexpectthatmovingtoa
newalgalplatformcouldbeeconomicallyfeasiblewithoutrequiringvastincreasesin
proteinyield.
Othermicroalgaeofinterestincludeextremophiles,becausetheircultureconditions
generallyminimisecontamination,whichisabigissuewhengrowingmicroalgaeat
the commercial scales and open raceways shown in Figure 6.2. For example,
Cyanidioschyzonmerolaeisamicroalgathatinhabitshot(40–56°C)andacidic(pH
0.2-4)environments,anditschloroplasthasrecentlybeentransformedforthefirst
time(Zienkiewiczetal.2017).Attemptingtoexpressrecombinantproteins,suchas
Cpl-1,inC.merolaeandsimilarorganismswillbeanimportantstepindetermining
2https://www.cyanotech.com/company/facility.html

248
thebest option for commercial scale-up.However, useof high temperatures and
extremophiles will require investigation into the thermostability of the proteins
lookingtobeproduced,andwillnotbesuitableforallproducts.Furthermore,the
cost of maintaining cultures at such temperatures may negate or minimize the
savingsmadebyadoptingsuchasystem.
6.3 Concludingremarks
6.3.1 Viewsonthefutureofantibiotics
As with many emerging technologies, the adoption of endolysins as antibiotics
dependsheavilyuponthestatusoftheexistingtechnology:conventionalantibiotics.
Therecentdiscoveryofanewantibiotic,teixobactin,andthenovelmannerinwhich
it was discovered (see 1.1.2.2) may yet extend the ruling era of small molecule
antibiotics.Whilethediscoveryofnewantibioticsisclearlyagoodthing,itisvitally
importantthatcomplacencydoesnot limittheresearch intoendolysinsandother
antibiotic alternatives. Conventional antibiotics have clear disadvantages when
compared toendolysins–namely thehugeselectivepressure theyexertuponall
bacteria to form resistance, thus making us increasingly dependent upon new
discoveries. It is of great importance, therefore, that research continues into the
developmentofmoreadvancedantibioticssothatwecanspreadourrelianceacross
several strategies rather than continuing to depend upon the discovery of new
conventionalantibiotics.Withapproximately1031bacteriophageinexistenceonthe
planet,thereisavast,andconstantlyevolving,supplyofpotentialantibioticsinthe
formofphage-therapyandendolysins,offeringamuchmorestableandsustainable
sourceoffuturetherapies.
For full adoption of endolysins and replacement of conventional antibiotics, both
much improved point-of-care diagnostics, and much more research into tackling
Gram-negativebacteriaare required.Bothof theseare stumblingblocks thatwill
take time and resources to overcome, but the final result will be a far superior
solution to tackling bacterial infections. Furthermore, if endolysins, or any other
technology, do become the first-line treatment for bacterial infections, research

249
shouldconstantlystrivetofindalternatives,asthe‘arms-race’withbacteriaisnever-
ending,andresistancetoanynewtechnologieswilleventuallyemerge.
6.3.2 Viewsonthefutureofmicroalgae-producedtherapeutics
Hypothetical conversationswith drug regulatory bodies and thought-experiments
around the advantages of algae-based therapeutics are clearly important initial
stages of developing a new recombinant protein platform, and currently more
immediate issues such as low recombinant protein yields are preventing
advancementoftheplatform.However,toperformacostcomparisonofmicroalgae
and other platforms, and to establish the viability of the platform, it is vitally
important to understand the extent to which drug regulatory bodies will look
favourablyuponproteinsproducedinGRASorganismssuchasC.reinhardtii.Ifitis
the case that crude or enriched algal extracts can be approved, then thiswill be
enormouslyencouragingforthefutureoftheplatform.However, ifthis isnotthe
case, thentheuseofestablishedplatformswillalmostalwaysbemore financially
favourable. In this situation, the future of microalgae as recombinant protein
production platforms must shift towards alternative markets, such as cosmetics,
veterinarymedicine, and food safety, and look to other proteins that cannot be
expressedinothersystemsorhavemarkedsynergieswithalgalcrudeextracts.Other
proteinproductionplatformswithGRASstatus,suchasspinach(Schulzetal.2015),
andpossiblyeventhebacterialsystemLactococcuslactis(Cano-Garridoetal.2014),
arelikelytoreachthepointofcommercialviabilitysoonerthanmicroalgaeandthis
willoffervaluableinsightsintohowtheseproteinsarereceivedandregulatedbythe
relevantregulatorybodies.

250
References
Adam, D. 2002. Global antibiotic resistance in Streptococcus pneumoniae. TheJournalofantimicrobialchemotherapy50:1–5.
Adam,Z.,A.Rudella&K.J.vanWijk.2006.RecentadvancesinthestudyofClp,FtsHandotherproteases located inchloroplasts.CurrentOpinion inPlantBiology9:234–240.
Ahmad,V.,M.S.Khan,Q.M.S. Jamal,M.A.Alzohairy,M.A.AlKaraawi&M.U.Siddiqui.2017.Antimicrobialpotentialofbacteriocins: in therapy,agricultureandfoodpreservation.InternationalJournalofAntimicrobialAgents49:1–11.
Ajuebor, J., O. McAuliffe, J. O’Mahony, R. P. Ross, C. Hill & A. Coffey. 2016.Bacteriophageendolysinsandtheirapplications.ScienceProgress99:183–199.
Alkatib,S.,L.B.Scharff,M.Rogalski,T.T.Fleischmann,A.Matthes,S.Seeger,M.A.Schöttler, S. Ruf & R. Bock. 2012. The Contributions of Wobbling andSuperwobblingtotheReadingoftheGeneticCode.PLoSGenetics8:11.
Allaker,R.P.,C.Zihni&S.Kapas.1999.Aninvestigationintotheantimicrobialeffectsof adrenomedullin on members of the skin, oral, respiratory tract and gutmicroflora.FEMSImmunologyandMedicalMicrobiology23:289–293.
Allen,H.K.,J.Trachsel,T.Looft&T.A.Casey.2014.Findingalternativestoantibiotics.AnnalsoftheNewYorkAcademyofSciences1323:91–100.
Almaraz-Delgado,A.L.,J.Flores-Uribe,V.H.Pérez-España,E.Salgado-Manjarrez&J.A.Badillo-Corona.2014.ProductionoftherapeuticproteinsinthechloroplastofChlamydomonasreinhardtii.AMBExpress4:57.
Aminov, R. I. 2010. A brief history of the antibiotic era: Lessons learned andchallengesforthefuture.FrontiersinMicrobiology1:1–7.
Appelbaum,P.C.1992.AntimicrobialResistanceinStreptococcuspneumoniae:AnOverview.ClinicalInfectiousDiseases15:77–83.
Arias, C. A. & B. E. Murray. 2015. clinical implications of basic research A NewAntibioticandtheEvolutionofResistance:1168–1170.
Aw,R.,G.R.Barton&D.J.Leak.2017.Insightsintotheprevalenceandunderlyingcauses of clonal variation through transcriptomic analysis in Pichia pastoris.AppliedMicrobiologyandBiotechnology101:5045–5058.
Baek,K.,D.H.Kim,J.Jeong,S.J.Sim,A.Melis,J.-S.Kim,E.Jin&S.Bae.2016.DNA-free two-gene knockout in Chlamydomonas reinhardtii via CRISPR-Cas9ribonucleoproteins.ScientificReports6:30620.
Baines,S.D.,R.O’Connor,K.Saxton,J.Freeman&M.H.Wilcox.2008.Comparison

251
of oritavancin versus vancomycin as treatments for clindamycin-inducedClostridiumdifficilePCRribotype027infectioninahumangutmodel.JournalofAntimicrobialChemotherapy62:1078–1085.
Barahimipour, R., J. Neupert & R. Bock. 2016. Efficient expression of nucleartransgenesinthegreenalgaChlamydomonas:synthesisofanHIVantigenanddevelopmentofanewselectablemarker.PlantMolecularBiology90:403–418.
Bax, R. 1997. P. Antibiotic resistance: a view from the pharmaceutical industry.ClinicalInfectiousDiseases24:5151–5153.
Berkmen, M. 2012. Production of disulfide-bonded proteins in Escherichia coli.ProteinExpressionandPurification82:240–251.
Berry,A.M.,R.A.Lock,D.Hansman&J.C.Paton.1989.ContributionofautolysintovirulenceofStreptococcuspneumoniae.InfectionandImmunity57:2324–2330.
Bertani,G.1951.Studiesonlysogenesis.I.ThemodeofphageliberationbylysogenicEscherichiacoli.Journalofbacteriology62:293–300.
Best, E. L., J. Freeman&M.H.Wilcox.2012.Models for the studyofClostridiumdifficileinfection.GutMicrobes3:145–167.
Bhatia,A., J.-F.Maisonneuve&D.H.Persing.2004.PropionibacteriumAcnesandChronic Diseases. The infectious etiology of chronic diseases: defining therelationship, enhancing the research, and mitigating the effects: workshopsummary.
Blowers,A.D.,G.S.Ellmore,U.Klein&L.Bogorad.1990.TranscriptionalanalysisofendogenousandforeigngenesinchloroplasttransformantsofChlamydomonas.PlantCell2:1059–1070.
Bogaert, D., R. De Groot & P.W.M. Hermans. 2004. Streptococcus pneumoniaecolonisation: The key to pneumococcal disease. Lancet Infectious Diseases4:144–154.
Bojar,R.A.,W.J.Cunliffe&K.T.Holland.1995.Theshort-termtreatmentofacnevulgariswithbenzoylperoxide:effectsonthesurfaceandfollicularcutaneousmicroflora.BritishJournalofDermatology132:204–208.
Bojar, R. A. & K. T. Holland. 2004. Acne and propionibacterium acnes. Clinics inDermatology22:375–379.
Bowdish,D.M.E.,D.J.Davidson&R.E.W.Hancock.2005.Are-evaluationoftheroleofhostdefencepeptides inmammalian immunity.Currentprotein&peptidescience6:35–51.

252
Brault,V.,U.Sauder,M.C.Reedy,U.Aebi&C.aSchoenenberger.1999.DifferentialepitopetaggingofactinintransformedDrosophilaproducesdistincteffectsonmyofibrilassemblyandfunctionoftheindirectflightmuscle.Molecularbiologyofthecell10:135–49.
Briers,Y.&R.Lavigne.2015.Breakingbarriers:expansionoftheuseofendolysinsasnovel antibacterials against Gram-negative bacteria. Future microbiology10:377–90.
Briers,Y.,M.Schmelcher,M.J.Loessner,J.Hendrix,Y.Engelborghs,G.Volckaert&R. Lavigne. 2009. The high-affinity peptidoglycan binding domain ofPseudomonas phage endolysin KZ144. Biochemical and Biophysical ResearchCommunications383:187–191.
Briers,Y.,M.Walmagh,V.VanPuyenbroeck,A.Cornelissen,W.Cenens,A.Aertsen,H. Oliveira, J. Azeredo, G. Verween, J. P. Pirnay, S.Miller, G. Volckaert & R.Lavigne. 2014. Engineered endolysin-based “Artilysins” to combatmultidrug-resistantgram-negativepathogens.AmericanSocietyforMicrobiology5.
Brooker,R.J.2010.GeneticPrincipals.TheMcGraw-HillCompanies,S.r.l.-PublishingGroupItalia.
Brown, L., J.M.Wolf, R. Prados-Rosales&A.Casadevall. 2015. Through thewall:extracellularvesiclesinGram-positivebacteria,mycobacteriaandfungi.Naturereviews.Microbiology13:620–30.
Buey,R.M.,B.Monterroso,M.Menéndez,G.Diakun,P.Chacón,J.A.Hermoso&J.F. Díaz. 2007. Insights into Molecular Plasticity of Choline Binding Proteins(Pneumococcal Surface Proteins) by SAXS. Journal of Molecular Biology365:411–424.
Butler-Wu, S.M., E.M.Burns, P. S. Pottinger, A. S.Magaret, J. L. Rakeman, F. A.Matsen & B. T. Cookson. 2011. Optimization of periprosthetic culture fordiagnosis of Propionibacterium acnes prosthetic joint infection. Journal ofClinicalMicrobiology49:2490–2495.
Cabello,F.C.2006.Heavyuseofprophylacticantibioticsinaquaculture:Agrowingproblemforhumanandanimalhealthandfortheenvironment.EnvironmentalMicrobiology8:1137–1144.
Cano-Garrido,O.,F.L.Rueda,L.Sànchez-García,L.Ruiz-Ávila,R.Bosser,A.Villaverde& E. García-Fruitós. 2014. Expanding the recombinant protein quality inLactococcuslactis.MicrobialCellFactories13:167.
Catalão,M.J.,F.Gil,J.Moniz-Pereira&M.Pimentel.2011.FunctionalanalysisoftheHolin-Like proteins of mycobacteriophage Ms6. Journal of Bacteriology193:2793–2803.
Cha,T.S.,W.Yee&A.Aziz.2012.AssessmentoffactorsaffectingAgrobacterium-

253
mediatedgenetictransformationoftheunicellulargreenalga,Chlorellavulgaris.WorldJournalofMicrobiologyandBiotechnology28:1771–1779.
Chapot-Chartier,M.-P.P.2014.Interactionsofthecell-wallglycopolymersoflacticacidbacteriawiththeirbacteriophages.FrontiersinMicrobiology5:1–10.
Cheng, G., H. Hao, S. Xie, X.Wang,M. Dai, L. Huang& Z. Yuan. 2014. Antibioticalternatives:Thesubstitutionofantibiotics inanimalhusbandry?Frontiers inMicrobiology5:1–15.
Cheng, Q. & V. A. Fischetti. 2007. Mutagenesis of a bacteriophage lytic enzymePlyGBS significantly increases its antibacterial activity against group Bstreptococci.AppliedMicrobiologyandBiotechnology74:1284–1291.
Cheng, X., X. Zhang, J. W. Pflugrath & F. W. Studier. 1994. The structure ofbacteriophage T7 lysozyme, a zinc amidase and an inhibitor of T7 RNApolymerase. Proceedings of theNational Academy of Sciences of theUnitedStatesofAmerica91:4034–4038.
Chisti,Y.2008.Biodieselfrommicroalgaebeatsbioethanol.TrendsinBiotechnology26:126–131.
Choquet,Y.,D.B.Stern,K.Wostrikoff,R.Kuras,J.Girard-Bascou&F.A.Wollman.1998.Translationofcytochromefisautoregulatedthroughthe5ʹuntranslatedregion of petA mRNA in Chlamydomonas chloroplasts. Proceedings of theNationalAcademyofSciences95:4380–4385.
Chung, P. Y. & R. Khanum. 2017. Antimicrobial peptides as potential anti-biofilmagents against multi-drug resistant bacteria. Journal of Microbiology,ImmunologyandInfection50:405-410.
Clatworthy,A.E.,E.Pierson&D.T.Hung.2007.Targetingvirulence:anewparadigmforantimicrobialtherapy.Naturechemicalbiology3:541–548.
ContraFect. no date. Product Pipeline. ContraFect Corporation (CFRX).<http://www.contrafect.com/pipeline/overview>(15July2017).
Coragliotti,A.T.,M.V.Beligni,S.E.Franklin&S.P.Mayfield.2011.Molecularfactorsaffecting the accumulation of recombinant proteins in the chlamydomonasreinhardtiichloroplast.MolecularBiotechnology48:60–75.
Crutchfield, A., K. Diller & J. Brand. 1999. Cryopreservation of Chlamydomonasreinhardtii(Chlorophyta).EuropeanJournalofPhycology34:43–52.
Cullis,C.A.,B.J.Vorster,C.VanDerVyver&K.J.Kunert.2009.Transferofgeneticmaterial between the chloroplast and nucleus: How is it related to stress inplants?AnnalsofBotany103:625–633.

254
Daniel, A., C. Euler, M. Collin, P. Chahales, K. J. Gorelick & V. a Fischetti. 2010.Synergismbetweenanovelchimericlysinandoxacillinprotectsagainstinfectionby methicillin-resistant Staphylococcus aureus. Antimicrobial agents andchemotherapy54:1603–12.
Daniell,H.,S.Kumar&N.Dufourmantel.2005.Breakthroughinchloroplastgeneticengineeringofagronomicallyimportantcrops.Trendsinbiotechnology23:238–45.
Datar,R.V,T.Cartwright&C.G.Rosen.1993.Processeconomicsofanimalcellandbacterialfermentations:acasestudyanalysisoftissueplasminogenactivator.Bio/technology(NaturePublishingCompany)11:349–357.
Davies, J. 2006. Are antibiotics naturally antibiotics? Journal of IndustrialMicrobiologyandBiotechnology33:496–499.
Demain,A.L.&P.Vaishnav.2011.ProductionofRecombinantProteinsbyMicrobesandHigherOrganisms.ComprehensiveBiotechnology, SecondEdition3:333–345.
Derrien,B.2013.StrepTagIIpurificationofC.reinhardtiichloroplastcomplex.Bio-Protocol3:2–6.
Derrien,B.,W.Majeran,G.Effantin,J.Ebenezer,G.Friso,K.J.vanWijk,A.C.Steven,M. R. Maurizi & O. Vallon. 2012. The purification of the Chlamydomonasreinhardtii chloroplast ClpP complex: Additional subunits and structuralfeatures.PlantMolecularBiology80:189–202.
Dessinioti, C. & A. Katsambas. 2017. Propionibacterium acnes and antimicrobialresistanceinacne.ClinicsinDermatology35:163–167.
Doehn,J.M.,K.Fischer,K.Reppe,B.Gutbier,T.Tschernig,A.C.Hocke,V.A.Fischetti,J. Löffler, N. Suttorp, S. Hippenstiel &M.Witzenrath. 2013. Delivery of theendolysinCpl-1byinhalationrescuesmicewithfatalpneumococcalpneumonia.TheJournalofantimicrobialchemotherapy68:2111–7.
Dreesen,I.A.J.,G.C.E.Hamri&M.Fussenegger.2010.Heat-stableoralalga-basedvaccine protects mice from Staphylococcus aureus infection. Journal ofBiotechnology145:273–280.
Drugbank. "Benzylpenicillin." <https://www.drugbank.ca/drugs/DB01053>(accessed31July2017).
Dumont, J.,D.Euwart,B.Mei,S.Estes&R.Kshirsagar.2015.Humancell linesforbiopharmaceutical manufacturing: history, status, and future perspectives.CriticalreviewsinBiotechnology1:1–13.

255
Dunne,M.,S. Leicht,B.Krichel,H.D.T.Mertens,A.Thompson, J.Krijgsveld,D. I.Svergun,N.Gómez-Torres,S.Garde,C.Uetrecht,A.Narbad,M.J.Mayer&R.Meijers.2016.CrystalstructureoftheCTP1Lendolysinrevealshowitsactivityisregulatedbyasecondarytranslationproduct.JournalofBiologicalChemistry291:4882–4893.
Dunne,M.,H.D.T.Mertens,V.Garefalaki,C.M.Jeffries,A.Thompson,E.A.Lemke,D.I.Svergun,M.J.Mayer,A.Narbad&R.Meijers.2014.TheCD27LandCTP1LEndolysins Targeting Clostridia Contain a Built-in Trigger and Release Factor.PLoSPathogens10(7).
Dutcher,S.K.,L.Li,H.Lin,L.Meyer,T.H.Giddings,a.L.Kwan,B.L.Lewis&J.Berman.2012.Whole-GenomeSequencing to IdentifyMutants andPolymorphisms inChlamydomonasreinhardtii.Genes|Genomes|Genetics2:15–22.
Dwidar,M.,A.K.Monnappa&R.J.Mitchell.2012.ThedualprobioticandantibioticnatureofBdellovibriobacteriovorus.BMBReports45:71–78.
Dworkin, M. & S. Falkow. 2006. The prokaryotes: a handbook on the biology ofbacteria.Chapter10.1TheFamilyChloroflexceae.NewYork:Springer-Verlag.
Entenza, J.M., J.M.Loeffler,D.Grandgirard,V.A.Fischetti&P.Moreillon.2005.Therapeutic effects of bacteriophage Cpl-1 lysin against Streptococcuspneumoniae endocarditis in rats. Antimicrobial Agents and Chemotherapy49:4789–4792.
Van Essche,M.,M. Quirynen, I. Sliepen, G. Loozen, N. Boon, J. Van Eldere&W.Teughels.2011.Killingofanaerobicpathogensbypredatorybacteria.MolecularOralMicrobiology26:52–61.
Farrar,M.D.,K.M.Howson,R.aBojar,D.West,J.C.Towler,J.Parry,K.Pelton&K.T.Holland.2007.GenomesequenceandanalysisofaPropionibacteriumacnesbacteriophage.Journalofbacteriology189:4161–7.
Feiner,R.,T.Argov,L.Rabinovich,N.Sigal,I.Borovok&A.A.Herskovits.2015.Anewperspectiveon lysogeny:prophagesasactiveregulatoryswitchesofbacteria.NatureReviewsMicrobiology13:641–650.
Fernandes,R.,P.Amador&C.Prudêncio.2013.β-Lactams:chemicalstructure,modeofactionandmechanismsofresistance.ReviewsinMedicalMicrobiology24:7–17.
Fernández-Tornero,C.,E.García,R.López,G.Giménez-Gallego&A.Romero.2002.Two new crystal forms of the choline-binding domain of the majorpneumococcalautolysin: Insights intothedynamicsoftheactivehomodimer.JournalofMolecularBiology321:163–173.

256
Fernebro, J. 2011. Fighting bacterial infections-future treatment options. Drugresistanceupdates :reviewsandcommentariesinantimicrobialandanticancerchemotherapy14:125–39.
Ferreira-Camargo, L. S., M. Tran, J. Beld, M. D. Burkart & S. P. Mayfield. 2015.Selenocystamine improvesproteinaccumulation inchloroplastsofeukaryoticgreenalgae.AMBExpress5:126.
Fischetti,V.a.2008.Bacteriophagelysinsaseffectiveantibacterials.Currentopinioninmicrobiology11:393–400.
Franklin,S.E.&S.P.Mayfield.2004.ProspectsformolecularfarminginthegreenalgaChlamydomonasreinhardtii.CurrentOpinioninPlantBiology7:159–165.
Franklin,S.,B.Ngo,E.Efuet&S.P.Mayfield.2002.DevelopmentofaGFPreportergeneforChlamydomonasreinhardtiichloroplast.ThePlantjournal :forcellandmolecularbiology30:733–44.
Freeman,J.,S.D.Baines,K.Saxton&M.H.Wilcox.2007.EffectofmetronidazoleongrowthandtoxinproductionbyepidemicClostridiumdifficilePCRribotypes001and027inahumangutmodel.JournalofAntimicrobialChemotherapy60:83–91.
Fuhrmann,M.,W.Oertel&P.Hegemann.1999.Asyntheticgenecodingforthegreenfluorescentprotein(GFP)isaversatilereporterinChlamydomonasreinhardtii.ThePlantjournal :forcellandmolecularbiology19:353–361.
Fulton,J.E.,A.Farzad-Bakshandeh&S.Bradley.1974.StudiesontheMechanismofActionofTopicalBenzoylPeroxideandVitaminAAcidinAcneVulgaris.JournalofCutaneousPathology1:191–200.
García, J. L., E. García, a Arrarás, P. García, C. Ronda& R. López. 1987. Cloning,purification, and biochemical characterization of the pneumococcalbacteriophageCp-1lysin.Journalofvirology61:2573–80.
García, P.,M.P.González, E.García, J. L.García&R. López. 1999. Themolecularcharacterizationof the first autolytic lysozymeof Streptococcus pneumoniaerevealsevolutionarymobiledomains.MolecularMicrobiology33:128–138.
Garrett,J.,C.Bruno&R.Young.1990.LysisproteinSofphagelambdafunctionsinSaccharomycescerevisiae.JournalofBacteriology172:7275–7277.
Gaskins, H. R., C. T. Collier & D. B. Anderson. 2002. Antibiotics As GrowthPromotants:ModeofAction.AnimalBiotechnology13:29–42.
Gaspar,P.,G.Moura,M.A.S.Santos&J.L.Oliveira.2013.MRNAsecondarystructureoptimization using a correlated stem-loopprediction.Nucleic Acids Research41:3–7.

257
Gehre,F.,S.L.Leib,D.Grandgirard,J.Kummer,A.Bhlmann,F.Simon,R.Gumann,A.S. Kharat, M. G. Tuber & A. Tomasz. 2008. Essential role of choline forpneumococcal virulence in an experimental model of meningitis. Journal ofInternalMedicine264:143–154.
George,A.&G.Rubin.2003.Asystematicreviewandmeta-analysisoftreatmentsforimpetigo.BritishJournalofGeneralPractice53:480–487.
Gerding,D.N.,C.A.Muto&R.C.Owens.2008.MeasurestocontrolandpreventClostridiumdifficileinfection.ClinicalInfectiousDiseases46:S43-49.
Gilman, J. & J. Love. 2016. Synthetic promoter design for newmicrobial chassis.BiochemicalSocietyTransactions44:731–737.
Gimpel, J. A., J. S. Hyun, N. G. Schoepp & S. P. Mayfield. 2015. Production ofrecombinant proteins inmicroalgae at pilot greenhouse scale. BiotechnologyandBioengineering112:339–345.
Gould,L.H.&B.Limbago.2010.ClostridiumdifficileinFoodandDomesticAnimals:ANewFoodbornePathogen?ClinicalInfectiousDiseases51:577–582.
Govea-Alonso,D.O.,M.A.Tello-Olea,J.Beltrán-López,E.Monreal-Escalante,J.A.Salazar-Gonzalez, B. Bañuelos-Hernández & S. Rosales-Mendoza. 2017.AssessmentofCarrotCallusasBiofactoriesofanAtherosclerosisOralVaccinePrototype.MolecularBiotechnology59:482–489.
Grandgirard,D.,J.M.Loeffler,V.A.Fischetti&S.L.Leib.2008.PhageLyticEnzymeCpl-1 for Antibacterial Therapy in Experimental Pneumococcal Meningitis197:1519–1522.
Grabowski,G.A.,M.Golembo&Y. Shaaltiel. 2014.Taliglucerasealfa:Anenzymereplacementtherapyusingplantcellexpressiontechnology.MolecularGeneticsandMetabolism1:1-8.
Gregory,J.A.,F.Li,L.M.Tomosada,C.J.Cox,A.B.Topol,J.M.Vinetz&S.Mayfield.2012.Algae-producedpfs25elicitsantibodiesthatinhibitmalariatransmission.PLoSONE7(5).
Gregory,J.A.,A.Shepley-McTaggart,M.Umpierrez,B.K.Hurlburt,S.J.Maleki,H.A.Sampson, S. P. Mayfield & M. C. Berin. 2016. Immunotherapy using algal-produced Ara h 1 core domain suppresses peanut allergy in mice. PlantBiotechnologyJournal14:1541–1550.
Gregory,J.A.,A.B.Topol,D.Z.Doerner&S.Mayfield.2013.Alga-producedcholeratoxin-Pfs25 fusion proteins as oral vaccines. Applied and EnvironmentalMicrobiology79:3917–3925.

258
Grossman,A.R.,E.E.Harris,C.Hauser,P.A.Lefebvre,D.Martinez,D.Rokhsar, J.Shrager, C. D. Sil, D. Stern, O. Vallon & Z. Zhang. 2003. ChlamydomonasreinhardtiiattheCrossroadsofGenomics.Society2:1137–1150.
Gu,J.,Y.Feng,X.Feng,C.Sun,L.Lei,W.Ding,F.Niu,L.Jiao,M.Yang,Y.Li,X.Liu,J.Song,Z.Cui,D.Han,C.Du,Y.Yang,S.Ouyang,Z.J.Liu&W.Han.2014.StructuralandBiochemicalCharacterizationRevealsLysGH15asanUnprecedented“EF-Hand-Like”Calcium-BindingPhageLysin.PLoSPathogens10.
Gupta,S.K.&P.Shukla.2015.Advancedtechnologies for improvedexpressionofrecombinantproteinsinbacteria:perspectivesandapplications.CriticalReviewsinBiotechnology0:1–10.
Gutiérrez,D.,P.Ruas-Madiedo,B.Martínez,A.Rodríguez&P.García.2014.EffectiveRemoval of Staphylococcal Biofilms by the Endolysin LysH5. PloS ONE9:e107307.
Gutman, G. A. & G. W. Hatfield. 1989. Nonrandom utilization of codon pairs inEscherichiacoli.ProceedingsoftheNationalAcademyofSciencesoftheUnitedStatesofAmerica86:3699–703.
Hafiz, S. & C. L. Oakley. 1976. Clostridium difficile: isolation and characteristics.Journalofmedicalmicrobiology9:129–36.
Hall,I.&E.O’Toole.1935.Intestinalflorainnew-borninfants:Withadescriptionofanewpathogenicanaerobe,Bacillusdifficilis.AmericanJournalofDiseasesofChildren49:390–402.
Hanahan, D. 1983. Studies on transformation of Escherichia coli with plasmids.JournalofMolecularBiology166:557–580.
Hao,H.,G.Cheng,Z.Iqbal,X.Ai,H.I.Hussain,L.Huang,M.Dai,Y.Wang,Z.Liu&Z.Yuan.2014.Benefitsandrisksofantimicrobialuseinfood-producinganimals.FrontiersinMicrobiology5:1–11.
Hargreaves,K.R.&M.R.J.Clokie.2014.Clostridiumdifficilephages:Stilldifficult?FrontiersinMicrobiology5:1–14.
Harris, E. H. 2009. The Chlamydomonas sourcebook. Volume 1. 2nd edition. SanDiego,CA:AcademicPress.
He,D.M.,K.X.Qian,G.F.Shen,Z.F.Zhang,Y.N.LI,Z.L.Su&H.B.Shao.2007.Recombinationandexpressionof classical swine fever virus (CSFV) structuralprotein E2 gene in Chlamydomonas reinhardtii chroloplasts. Colloids andSurfacesB:Biointerfaces55:26–30.

259
Hermoso, J. A., B. Monterroso, A. Albert, B. Galán, O. Ahrazem, P. García, M.Martínez-Ripoll,J.L.García&M.Menéndez.2003.StructuralbasisforselectiverecognitionofpneumococcalcellwallbymodularendolysinfromphageCp-1.Structure11:1239–1249.
Hidaka,Y.,M.Ohno,B.Hemmasi,O.Hill,W.G.Forssmann&Y.Shimonishi.1998.Invitro disulfide-coupled folding of guanylyl cyclase-activating peptide and itsprecursorprotein.Biochemistry37:8498–8507.
Holland,C.,T.N.Mak,U.Zimny-Arndt,M.Schmid,T.F.Meyer,P.R.Jungblut&H.Brüggemann. 2010. Proteomic identification of secreted proteins ofPropionibacteriumacnes.BMCmicrobiology10:230.
Hoopes,J.T.,C.J.Stark,H.A.Kim,D.J.Sussman,D.M.Donovan&D.C.Nelson.2009.Use of a bacteriophage lysin, PlyC, as an enzyme disinfectant againstStreptococcusequi.AppliedandEnvironmentalMicrobiology75:1388–1394.
Hosler,J.P.,E.A.Wurtz,E.H.Harris,N.W.Gillham&J.E.Boynton.1989.Relationshipbetween gene dosage and gene expression in the chloroplast ofChlamydomonasreinhardtii.PlantPhysiology91:648–655.
Howe,C.J.&S.Purton.2007.TheLittleGenomeofApicomplexanPlastids:itsraisond’etreandaPossibleExplanationforthe“DelayedDeath”Phenomenon.Protist158:121–133.
Huang, H., A.Weintraub, H. Fang& C. E. Nord. 2009. Antimicrobial resistance inClostridiumdifficile.InternationalJournalofAntimicrobialAgents34:516–522.
Hughes, C. & G. G. Meynell. 1974. High frequency of antibiotic-resistantenterobacteriaintheRiverStour,Kent.Lancet(London,England)2:451–3.
Hyams,C.,E.Camberlein,J.M.Cohen,K.Bax&J.S.Brown.2010.TheStreptococcuspneumoniaecapsuleinhibitscomplementactivityandneutrophilphagocytosisbymultiplemechanisms.InfectionandImmunity78:704–715.
Hyvonen, M. 2002. Refolding of disulfide rich proteins.<http://camelot.bioc.cam.ac.uk/~marko/methods/refold.pdf>.
Irwin,B.,J.D.Heck&G.W.Hatfield.1995.CodonPairUtilizationBiasesInfluenceTranslationalElongationStepTimes.Biochemistry270:22801–22806.
Jado,I.,R.López,E.García,A.Fenoll,J.Casal&P.García.2003.Phagelyticenzymesas therapy for antibiotic-resistant Streptococcus pneumoniae infection in amurinesepsismodel.TheJournalofantimicrobialchemotherapy52:967–73.
Jeffrey, G. A. 1997. An Introduction to Hydrogen Bonding. Topics in PhysicalChemistry.NewYork:OxfordUniveristyPress.

260
Jervis,E.J.,C.A.Haynes,G.Kilburn,E.J.Jervis,C.A.Haynes&D.G.Kilburn.1997.ENZYMOLOGY : Surface Diffusion of Cellulases and Their Isolated BindingDomainsonCelluloseSurfaceDiffusionofCellulasesandTheirIsolatedBindingDomainsonCellulose.TheJournalofBiologicalChemistry272:24016–24023.
Jinkerson,R.E.&M.C.Jonikas.2015.MoleculartechniquestointerrogateandedittheChlamydomonasnucleargenome.PlantJournal82:393–412.
Joshi,S.R.,R.M.Parikh&A.K.Das.2007.Insulin--history,biochemistry,physiologyand pharmacology. The Journal of the Association of Physicians of India 55Suppl:19–25.
Kaiser, G. E. 2011. BIOL 230 Lecture Guide - Mode of Action of Penicillins.<http://faculty.ccbcmd.edu/courses/bio141/lecguide/unit2/control/penresillus.html>(31July2017).
Keel,K.,J.S.Brazier,K.W.Post,S.Weese&J.G.Songer.2007.PrevalenceofPCRribotypes among Clostridium difficile isolates from pigs, calves, and otherspecies.Journalofclinicalmicrobiology45:1963–4.
Kim,J.1997.ProteinDisulfideIsomeraseasaRegulatorofChloroplastTranslationalActivation.Science278:1954–1957.
Kim,J.Y.,Y.G.G.Kim&G.M.Lee.2012.CHOcellsinbiotechnologyforproductionof recombinant proteins: current state and further potential. AppliedMicrobiologyandBiotechnology93:917–930.
Kindle,K.L.,K.L.Richards&D.B.Stern.1991.Engineeringthechloroplastgenome:techniquesandcapabilitiesforchloroplasttransformation inChlamydomonasreinhardtii. Proceedings of the National Academy of Sciences of the UnitedStatesofAmerica88:1721–1725.
Kong, K. 2013. Codon Usage Optimizer (CUO). GitHub. <https://github.com/khai-/CUO>.
Korndörfer,I.P.,J.Danzer,M.Schmelcher,M.Zimmer,A.Skerra&M.J.Loessner.2006.ThecrystalstructureofthebacteriophagePSAendolysinrevealsauniquefold responsible for specific recognition of Listeria cell walls. Journal ofMolecularBiology364:678–89.
Kristensen,C.,T.Kjeldsen,F.C.Wiberg,L.Schäffer,M.Hach,S.Havelund,J.Bass,D.F.Steiner&A.S.Andersen.1997.Alaninescanningmutagenesisofinsulin.TheJournalofbiologicalchemistry272:12978–83.
Kropat, J.,A.Hong-Hermesdorf,D.Casero,P.Ent,M.Castruita,M.Pellegrini,S.S.Merchant&D.Malasarn.2011.ArevisedmineralnutrientsupplementincreasesbiomassandgrowthrateinChlamydomonasreinhardtii.ThePlantjournalforcellandmolecularbiology66:770–80.

261
LaFee,S.&H.Buschman.2017.NovelPhageTherapySavesPatientwithMultidrug-Resistant Bacterial Infection. UC San Diego Health.<https://health.ucsd.edu/news/releases/Pages/2017-04-25-novel-phage-therapy-saves-patient-with-multidrug-resistant-bacterial-infection.aspx> (14July2017).
Lai,M. J., N. T. Lin, A.Hu, P. C. Soo, L. K. Chen, L. H. Chen& K. C. Chang. 2011.Antibacterial activity of Acinetobacter baumannii phage PhiAB2 endolysin(LysAB2) against both Gram-positive and Gram-negative bacteria. AppliedMicrobiologyandBiotechnology90:529–539.
Lam,S.J.,N.M.O’Brien-Simpson,N.Pantarat,A.Sulistio,E.H.H.Wong,Y.-Y.Chen,J. C. Lenzo, J. A. Holden, A. Blencowe, E. C. Reynolds & G. G. Qiao. 2016.Combating multidrug-resistant Gram-negative bacteria with structurallynanoengineeredantimicrobialpeptidepolymers.NatureMicrobiology1:16162.
Lauersen, K. J., O. Kruse& J. H.Mussgnug. 2015. Targeted expression of nucleartransgenes in Chlamydomonas reinhardtii with a versatile, modular vectortoolkit.AppliedMicrobiologyandBiotechnology99:3491–3503.
Lella,M.,S.Kamilla,V.Jain&R.Mahalakshmi.2016.MolecularMechanismofHolinTransmembrane Domain i in Pore Formation and Bacterial Cell Death. ACSChemicalBiology11:910–920.
Lewis, K. 2013. Platforms for antibiotic discovery.NatureReviewsDrugDiscovery12:371–387.
Leyden,J.J.,K.J.McGinley,S.Cavalieri,G.F.Webster,O.H.Mills&A.M.Kligman.1983.Propionibacteriumacnesresistancetoantibioticsinacnepatients.JournaloftheAmericanAcademyofDermatology8:41–45.
Lim,J.-A.,H.Shin,S.Heu&S.Ryu.2014.ExogenouslyticactivityofSPN9CCendolysinagainst gram-negative bacteria. Journal of microbiology and biotechnology24:803–11.
Ling,L.L.,T.Schneider,A.J.Peoples,A.L.Spoering,I.Engels,B.P.Conlon,A.Mueller,D.E.Hughes,S.Epstein,M.Jones,L.Lazarides,V.aSteadman,D.R.Cohen,C.R.Felix,K.A.Fetterman,W.P.Millett,A.G.Nitti,A.M.Zullo,C.Chenetal.2015.Anewantibiotickillspathogenswithoutdetectableresistance.Nature517:455–459.
Livermore,D.M.2011.Discoveryresearch:Thescientificchallengeof findingnewantibiotics.JournalofAntimicrobialChemotherapy66:1941–1944.
Loeffler,J.M.,S.Djurkovic&V.A.Fischetti.2003.PhageLyticEnzymeCpl-1asaNovelAntimicrobialforPneumococcalBacteremia.InfectionandImmunity71:6199–6204.

262
Loeffler, J. M., D. Nelson & V. a Fischetti. 2001. Rapid killing of Streptococcuspneumoniaewithabacteriophagecellwallhydrolase.Science294:2170–2172.
Loessner, M. J. 2005. Bacteriophage endolysins--current state of research andapplications.Currentopinioninmicrobiology8:480–7.
Loessner,M.J.,K.Kramer,F.Ebel&S.Scherer.2002.C-terminaldomainsofListeriamonocytogenes bacteriophage murein hydrolases determine specificrecognition and high-affinity binding to bacterial cell wall carbohydrates.Molecularmicrobiology44:335–49.
Lucks,J.B.,D.R.Nelson,G.R.Kudla&J.B.Plotkin.2008.Genomelandscapesandbacteriophagecodonusage.PLoSComputationalBiology4.
Macfarlane,G.T.,S.Macfarlane&G.R.Gibson.1998.ValidationofaThree-StageCompoundContinuousCultureSystemforInvestigatingtheEffectofRetentionTimeontheEcologyandMetabolismofBacteriaintheHumanColon.MicrobialEcology35:180–187.
Madigan,M.T.&J.M.Martinko.2006.BrockBiologyofMicroorganisms,11thedn.InternationalMicrobiology.UpperSaddleRiver,NJ;London:PearsonPrenticeHall.
Maestro, B. & J. M. Sanz. 2016. Choline Binding Proteins from Streptococcuspneumoniae: A Dual Role as Enzybiotics and Targets for the Design of NewAntimicrobials.Antibiotics5.
Mailer,J.S.&B.Mason.2001.Penicillin:Medicine’sWartimeWonderDrugandItsProduction at Peoria, Illinois. Illinois Periodicals Online.<http://www.lib.niu.edu/2001/iht810139.html>(13July2017).
Makino, T., G. Skretas & G. Georgiou. 2011. Strain engineering for improvedexpressionofrecombinantproteinsinbacteria.MicrobialCellFactories.10:32-42.
Maliga, P. 2014. Chloroplast Biotechnology :Methods and Protocols. Totowa, NJ:HumanaPress.
Manuell, A. L., M. V. Beligni, J. H. Elder, D. T. Siefker, M. Tran, A. Weber, T. L.McDonald&S.P.Mayfield.2007.RobustexpressionofabioactivemammalianproteininChlamydomonaschloroplast.PlantBiotechnologyJournal5:402–412.
deMarco,A.,E.Deuerling,A.Mogk,T.Tomoyasu&B.Bukau.2007.Chaperone-basedproceduretoincreaseyieldsofsolublerecombinantproteinsproducedinE.coli.BMCBiotechnology7:32.
Marinelli, L. J., S. Fitz-gibbon & C. Hayes. 2012. Propionibacterium acnesBacteriophagesDisplayLimitedGenetic3:1–13.

263
Markey,P.M.,S.E.Racine,C.N.Markey,C.J.Hopwood,P.K.Keel,S.A.Burt,M.C.Neale,C. L. Sisk,M. Steven,K. L. Klump,E. Lansing&E. Lansing. 2015.Algalchloroplast produced camelid VH H antitoxins are capable of neutralizingbotulinumneurotoxin.PlantBiotechnologyJournal6:300–308.
Martín, aC.,R.López&P.García.1998.Functionalanalysisofthetwo-gene lysissystemofthepneumococcalphageCp-1inhomologousandheterologoushostcells.Journalofbacteriology180:210–7.
Maul,J.E.,J.W.Lilly,L.Cui,C.W.DePamphilis,W.Miller,E.H.Harris&D.B.Stern.2002.TheChlamydomonasreinhardtiiplastidchromosome:islandsofgenesinaseaofrepeats.ThePlantcell14:2659–2679.
Mayer,M.J.,V.Garefalaki,R.Spoerl,A.Narbad&R.Meijers.2011.Structure-basedmodification of a Clostridium difficile-targeting endolysin affects activity andhostrange.JournalofBacteriology193:5477–5486.
Mayer, M. J., A. Narbad & M. J. Gasson. 2008. Molecular characterization of aClostridiumdifficilebacteriophageanditsclonedbiologicallyactiveendolysin.JournalofBacteriology190:6734–6740.
Mayer,M. J., J. Payne,M. J. Gasson & A. Narbad. 2010. Genomic sequence andcharacterization of the virulent bacteriophage CTP1 from Clostridiumtyrobutyricum and heterologous expression of its endolysin. Applied andEnvironmentalMicrobiology76:5415–5422.
Mayfield,S.P.,S.E.Franklin&R.A.Lerner.2003.Expressionandassemblyofafullyactiveantibodyinalgae.ProceedingsoftheNationalAcademyofSciencesoftheUnitedStatesofAmerica.100:438–442.
Mayfield,S.P.&J.Schultz.2004.Developmentofaluciferasereportergene,luxCt,forChlamydomonasreinhardtiichloroplast.PlantJournal37:449–458.
McCullers,J.A.,Å.Karlström,A.R.Iverson,J.M.Loeffler&V.A.Fischetti.2007.Novelstrategy to prevent otitis media caused by colonizing Streptococcuspneumoniae.PLoSPathogens3:1–3.
McDaniel,L.S.,J.Yother,M.Vijayakumar,L.McGarry,W.R.Guild&D.E.Briles.1987.Use of insertional inactivation to facilitate studies of biological properties ofpneumococcalsurfaceproteinA(PspA).TheJournalofexperimentalmedicine165:381–394.
Meek,R.W.,H.Vyas&L.J.V.Piddock.2015.NonmedicalUsesofAntibiotics:TimetoRestrictTheirUse?PLoSBiology13:1–11.
Merchant, S.& L. Bogorad. 1986. Rapid degradation of apoplastocyanin in Cu(II)-deficient cells of Chlamydomonas reinhardtii. Journal of Biological Chemistry261:15850–15853.

264
Merchant,S.S.,S.E.Prochnik,O.Vallon,E.H.Harris,J.Karpowicz,G.B.Witman,A.Terry,A.Salamov,L.K.Fritz-laylin,L.Maréchal-Drouard,W.F.Marshall,L.Qu,D.R.Nelson,A.Sanderfoot,M.H.Spalding,V.V.Kapitonov,Q.Ren,P.Cardol,H.Ceruttietal.2010.TheChlamydomonasGenomeReveals theEvolutionofKeyAnimalandPlantFunctions.Science318:245–250.
Michelet, L., L. Lefebvre-Legendre, S. E. Burr, J. D. Rochaix & M. Goldschmidt-Clermont.2011.EnhancedchloroplasttransgeneexpressioninanuclearmutantofChlamydomonas.PlantBiotechnologyJournal9:565–574.
Micreos. Staphefekt Product Fact Sheets.<http://www.micreos.com/content/human-health.aspx>(15July2017).
Miller,S.I.2016.Antibioticresistanceandregulationofthegram-negativebacterialoutermembranebarrierbyhostinnateimmunemolecules.mBio7:e01541-16.
Monterroso,B.A.,J.L.Sáiz,P.García,J.L.García&M.Menéndez.2008.Insightsintothestructure-functionrelationshipsofpneumococcalcellwalllysozymes,LytCandCpl-1.JournalofBiologicalChemistry283:28618–28628.
Monterroso,B.,C.López-Zumel,J.L.García,J.L.Sáiz,P.García,N.E.Campillo&M.Menéndez. 2005. Unravelling the structure of the pneumococcal autolyticlysozyme.BiochemicalJournal391:41–49.
Mudd,E.A.,P.Madesis,E.M.Avila&A.Day.2014.Excisionofplastidmarkergenesusing directly repeated DNA sequences. Methods in Molecular Biology1132:107–123.
Nakimbugwe, D., B. Masschalck, D. Deckers, L. Callewaert, A. Aertsen & C. W.Michiels. 2006. Cell wall substrate specificity of six different lysozymes andlysozyme inhibitory activity of bacterial extracts. FEMS Microbiology Letters259:41–46.
Nelson,D.2016.ImmunotherapeuticsDerivedfromBacteriophageEndolysins.TheLysinMeeting.NewYork,USA.
Nelson,D., L. Loomis&V. a Fischetti. 2001. Prevention and elimination of upperrespiratory colonization of mice by group A streptococci by using abacteriophagelyticenzyme.ProceedingsoftheNationalAcademyofSciencesoftheUnitedStatesofAmerica98:4107–12.
Nielsen,J.2013.Productionofbiopharmaceuticalproteinsbyyeast.Bioengineered4:207–211.
O’Neill, J.,C.By,J. I.M.O.Neill&J.O’Neill.2015.SecuringNewDrugsforFutureGenerations: The Pipeline of Antibiotics. The Review on AntimicrobialResistance:42.

265
Obaro,S.&R.Adegbola.2002.Thepneumococcus:Carriage,diseaseandconjugatevaccines.JournalofMedicalMicrobiology51:98–104.
Oey,M.,M.Lohse,B.Kreikemeyer&R.Bock.2009.Exhaustionof thechloroplastprotein synthesis capacity by massive expression of a highly stable proteinantibiotic.PlantJournal57:436–445.
Oliveira,H.,L.D.R.Melo,S.B.Santos,F.L.Nobrega,E.C.Ferreira,N.Cerca,J.Azeredo& L. D. Kluskens. 2013. Molecular Aspects and Comparative Genomics ofBacteriophageEndolysins.JournalofVirology87:4558–4570.
Paik, J., I. Kern, R. Lurz & R. Hakenbeck. 1999. Mutational analysis of theStreptococcus pneumoniae bimodular class A penicillin-binding proteins.JournalofBacteriology181:3852–3856.
Pang,T.,T.C.Fleming,K.Pogliano&R.Young.2013.Visualizationofpinholinlesionsinvivo.ProceedingsoftheNationalAcademyofSciencesoftheUnitedStatesofAmerica110:E2054-63.
Park, K. Y. & S. J. Wi. 2016. Potential of plants to produce recombinant proteinproducts.JournalofPlantBiology59:559–568.
Parmley,S.2014.Lysininwait.Science-BusinesseXchange7.
Pastagia,M., R. Schuch, V. a Fischetti & D. B. Huang. 2013. Lysins: the arrival ofpathogen-directedanti-infectives.Journalofmedicalmicrobiology62:1506–16.
Payne,D.J.,M.N.Gwynn,D.J.Holmes&D.L.Pompliano.2007.Drugsforbadbugs:confronting the challenges of antibacterial discovery. Nature Reviews. DrugDiscovery6:29–40.
Pirofski,L.A.&A.Casadevall.2008.Thedamage-responseframeworkofmicrobialpathogenesisandinfectiousdiseases.AdvancesinExperimentalMedicineandBiology635:135–146.
Portillo,M.E.,S.Corvec,O.Borens&A.Trampuz.2013.Propionibacteriumacnes:Anunderestimated pathogen in implant-associated infections.BioMed ResearchInternational.
Pride,D.T.,J.Salzman,M.Haynes,F.Rohwer,C.Davis-Long,R.A.White,P.Loomer,G. C. Armitage & D. A. Relman. 2012. Evidence of a robust residentbacteriophage population revealed through analysis of the human salivaryvirome.TheISMEJournal6:915–926.
Proença,D.,C.Velours,C.Leandro,M.Garcia,M.Pimentel&C.São-José.2015.Atwo-component, multimeric endolysin encoded by a single gene. MolecularMicrobiology95:739–753.
Purton, S. 2007. Tools and techniques for chloroplast transformation ofChlamydomonas.Advancesinexperimentalmedicineandbiology616:34–45.

266
Rajakumar,P.D.2016.ThechloroplastofChlamydomonasreinhardtiiasaplatformfor recombinant vaccine production. PhD thesis. London: University CollegeLondon.
Ramundo,S.& J.D.Rochaix.2014.Chloroplastunfoldedprotein response,anewplastidstresssignalingpathway?PlantSignaling&Behavior9:e972874.
Rasala,B.A.&S.P.Mayfield.2015.Photosyntheticbiomanufacturingingreenalgae;productionofrecombinantproteinsforindustrial,nutritional,andmedicaluses.Photosynthesisresearch123:227–239.
Rasala,B.A.,M.Muto,P.A.Lee,M.Jager,R.M.F.Cardoso,C.A.Behnke,P.Kirk,C.A. Hokanson, R. Crea, M. Mendez & S. P. Mayfield. 2009. Production oftherapeuticproteinsinalgae,analysisofexpressionofsevenhumanproteinsinthe chloroplast of Chlamydomonas reinhardtii. Plant biotechnology journal8:719–33.
Reddy,B.L.&M.H.Saier.2013.Topologicalandphylogeneticanalysesofbacterialholinfamiliesandsuperfamilies.BiochimicaetBiophysicaActa-Biomembranes1828:2654–2671.
Reindel,R.&C.R.Fiore.2017.PhageTherapy:ConsiderationsandChallenges forDevelopment.ClinicalInfectiousDiseases64:1589–1590.
Resch,G.,P.Moreillon&V.A.Fischetti.2011a.Astablephage lysin (Cpl-1)dimerwith increased antipneumococcal activity and decreased plasma clearance.InternationalJournalofAntimicrobialAgents38:516–21.
Resch,G.,P.Moreillon&V.A.Fischetti.2011b.PEGylatingabacteriophageendolysininhibitsitsbactericidalactivity.AMBExpress1:29.
Rice, L. B. 2008. Federal Funding for the Study of Antimicrobial Resistance inNosocomial Pathogens: No ESKAPE. The Journal of Infectious Diseases197:1079–1081.
Rochaix,J.-D.1995.ChlamydomonasreinhardtiiasthePhotosyntheticYeast.Annualreviewofgenetics29:209–230.
Rochaix, J.-D. 2001. Assembly, function, and dynamics of the photosyntheticmachineryinChlamydomonasreinhardtii.Plantphysiology127:1394–8.
Rochaix, J.-D., M. Goldschmidt-Clermont & S. Merchant. 1998. Advances inPhotosynthesis and Respiration: The molecular biology of chloroplasts andmitochondriainChlamydomonas.Netherlands:Springer.
Ronda, C., R. Lopez & E. Garcia. 1981. Isolation and characterization of a newbacteriophage,Cp-1, infectingStreptococcuspneumoniae.JournalofVirology40:551–559.

267
Rosenberg,J.N.,G.A.Oyler,L.Wilkinson&M.J.Betenbaugh.2008.Agreenlightforengineeredalgae: redirectingmetabolism to fuel abiotechnology revolution.CurrentOpinioninBiotechnology19:430–436.
Sagermann, M. & B. W. Matthews. 2002. Crystal structures of a T4-lysozymeduplication-extension mutant demonstrate that the highly conserved beta-sheetregionhaslowintrinsicfoldingpropensity.Journalofmolecularbiology316:931–940.
Santillan-Jimenez,E.,R.Pace,S.Marques,T.Morgan,C.McKelphin,J.Mobley&M.Crocker.2016.Extraction,characterization,purificationandcatalyticupgradingofalgaelipidstofuel-likehydrocarbons.Fuel180:668–678.
Sanz,J.M.,E.Díaz&J.L.García.1992.StudiesonthestructureandfunctionoftheN-terminal domain of the pneumococcal murein hydrolases. MolecularMicrobiology6:921–931.
Savva,C.G.,J.S.Dewey,J.Deaton,R.L.White,D.K.Struck,A.Holzenburg&R.Young.2008. The holin of bacteriophage lambda forms rings with large diameter.Molecularmicrobiology69:784–793.
Scaife,M.A.,G.T.D.T.Nguyen,J.Rico,D.Lambert,K.E.Helliwell&A.G.Smith.2015.Establishing Chlamydomonas reinhardtii as an industrial biotechnology host.PlantJournal82:532–546.
Schmelcher,M.,D.M.Donovan&M.J.Loessner.2012.Bacteriophageendolysinsasnovelantimicrobials.FutureMicrobiology7:1147–1171.
Schroda,M.2004.TheChlamydomonasgenomerevealsitssecrets:Chaperonegenesandthepotentialrolesoftheirgeneproductsinthechloroplast.PhotosynthesisResearch82:221–240.
Schulz,S.,A.Stephan,S.Hahn,L.Bortesi,F.Jarczowski,U.Bettmann,A.-K.Paschke,D.Tusé,C.H.Stahl,A.Giritch&Y.Gleba.2015.Broadandefficientcontrolofmajor foodborne pathogenic strains of Escherichia coli bymixtures of plant-produced colicins. Proceedings of the National Academy of Sciences of theUnitedStatesofAmerica112:e5454-60.
Scranton,M.A.,J.T.Ostrand,F.J.Fields&S.P.Mayfield.2015.Chlamydomonasasamodelforbiofuelsandbio-productsproduction.PlantJournal82:523–531.
Scranton,M.A.,J.T.Ostrand,D.R.Georgianna,S.M.Lofgren,D.Li,R.C.Ellis,D.N.Carruthers,A.Dräger,D.L.Masica&S.P.Mayfield.2016.Syntheticpromoterscapable of driving robust nuclear gene expression in the green algaChlamydomonasreinhardtii.AlgalResearch15:135–142.

268
Sebaihia,M.,B.W.Wren,P.Mullany,N.F.Fairweather,N.Minton,R.Stabler,N.R.Thomson,A.P.Roberts,A.M.Cerdeño-Tárraga,H.Wang,M.T.G.Holden,A.Wright,C.Churcher,M.aQuail,S.Baker,N.Bason,K.Brooks,T.Chillingworth,A. Cronin et al. 2006. The multidrug-resistant human pathogen Clostridiumdifficilehasahighlymobile,mosaicgenome.Naturegenetics38:779–786.
Sengupta,S.,M.K.Chattopadhyay&H.P.Grossart.2013.Themultifacetedrolesofantibioticsandantibioticresistanceinnature.FrontiersinMicrobiology4:1–13.
Shatzkes,K.,E.Singleton,C.Tang,M.Zuena,S.Shukla,S.Gupta,S.Dharani,O.Onyile,J.Rinaggio,N.D.Connell&D.E.Kadouri.2016.PredatorybacteriaattenuateKlebsiellapneumoniaeburdeninratlungs.AmericanSocietyforMicrobiology7:1–9.
Shen, Y., M. Barros, T. Vennemann, D. T. Gallagher, Y. Yin, S. B. Linden, R. D.Heselpoth,D.J.Spencer,D.M.Donovan,J.Moult,V.A.Fischetti,F.Heinrich,M.Lische & D. C. Nelson. 2016. A bacteriophage endolysin that eliminatesintracellularstreptococci.eLife5:1–26.
Shen, Y., T. Köller, B. Kreikemeyer & D. C. Nelson. 2013. Rapid degradation ofStreptococcuspyogenesbiofilmsbyPlyC,abacteriophage-encodedendolysin.JournalofAntimicrobialChemotherapy68:1818–1824.
Sherman,M.Y.&S.B.Qian.2013.Lessismore:Improvingproteostasisbytranslationslowdown.TrendsinBiochemicalSciences38:585–591.
Shi, N. 2013. Cross section of a Chlamydomonas reinhardtii algae cell, a 3Drepresentation.<https://commons.wikimedia.org/wiki/File:Cross_section_of_a_Chlamydomonas_reinhardtii_algae_cell,_a_3D_representation.jpg>(16July2017).
Shi, Y.,N. Li, Y. Yan,H.Wang, Y. Li, C. Lu& J. Sun. 2012.CombinedAntibacterialactivity of phage lytic proteins holin and lysin from streptococcus suisbacteriophageSMP.CurrentMicrobiology65:28–34.
Shin,S.-E.,J.-M.Lim,H.G.Koh,E.K.Kim,N.K.Kang,S.Jeon,S.Kwon,W.-S.Shin,B.Lee,K.Hwangbo,J.Kim,S.H.Ye,J.-Y.Yun,H.Seo,H.-M.Oh,K.-J.Kim,J.-S.Kim,W.-J.Jeong,Y.K.Changetal.2016.CRISPR/Cas9-inducedknockoutandknock-inmutationsinChlamydomonasreinhardtii.ScientificReports6:27810.
Silver,L.L.2011.Challengesofantibacterialdiscovery.ClinicalMicrobiologyReviews24:71–109.
Silver,L.L.&K.A.Bostian.1993.DiscoveryandDevelopmentofNewAntibiotics:theProblem of Antibiotic Resistance. Antimicrobial Agents and Chemotherapy37:377–383.
Smith,D.L.,D.K.Struck,J.M.Scholtz&R.Young.1998.Purificationandbiochemicalcharacterizationofthelambdaholin.Journalofbacteriology180:2531–40.

269
Smith,E.,L.Howard&E.Dymek.1996.Scanningelectronmicroscopeimage,showingan example of green algae (Chlorophyta).<http://remf.dartmouth.edu/images/algaeSEM/source/4.html>(16July2017).
Sockett, R. E. & C. Lambert. 2004. Opinion: Bdellovibrio as therapeutic agents: apredatoryrenaissance?NatureReviewsMicrobiology2:669–675.
Song,J.,F.Xia,H.Jiang,X.Li,L.Hu,P.Gong,L.Lei,X.Feng,C.Sun,J.Gu&W.Han.2016. Identification and characterization of HolGH15: The Holin ofStaphylococcus Aureus bacteriophage GH15. Journal of General Virology97:1272–1281.
Songer, J. G. 2004. The emergence of Clostridium difficile as a pathogen of foodanimals.AnimalHealthResearchReviews5:321–326.
Specht,E.A.&S.P.Mayfield.2013.Syntheticoligonucleotidelibrariesrevealnovelregulatory elements in chlamydomonas chloroplast mRNAs. ACS SyntheticBiology2:34–46.
Specht, E., S. Miyake-Stoner & S. Mayfield. 2010. Micro-algae come of age as aplatformforrecombinantproteinproduction.BiotechnologyLetters32:1373–1383.
Spellberg,B.2014.Thefutureofantibiotics.CriticalCare18:228.
Spicer,A.&S.Purton.2016.GeneticEngineeringofMicroalgae.CurrentStatusandFutureProspects.Pp.139–163inMicroalgalProductionforBiomassandHigh-ValueProducts(S.P.Slocombe&J.R.Benemann,eds.).CRCPress.
Spigaglia,P.2016.RecentadvancesintheunderstandingofantibioticresistanceinClostridiumdifficileinfection.TherapeuticAdvancesinInfectiousDisease3:23–42.
Spolaore, P., C. Joannis-Cassan, E. Duran & A. Isambert. 2006. Commercialapplicationsofmicroalgae.JournalofBioscienceandBioengineering101:87–96.
Stanek,R.J.,M.B.Maher,N.B.Norton&M.A.Mufson.2011.Emergenceofauniquepenicillin-resistantStreptococcuspneumoniaeserogroup35strain. JournalofClinicalMicrobiology49:400–404.
Stegemann,S.,S.Hartmann,S.Ruf&R.Bock.2003.High-frequencygenetransferfrom the chloroplast genome to the nucleus. Proceedings of the NationalAcademyofSciencesoftheUnitedStatesofAmerica100:8828–8833.
Steinbrenner, J. & G. Sandmann. 2006. Transformation of the green algaHaematococcus pluvialis with a phytoene desaturase for acceleratedastaxanthin biosynthesis. Applied and Environmental Microbiology 72:7477–7484.

270
Stoffels,L.2015.Microalgaeasanovelproductionplatformforantibacterialproteins.PhDthesis.London:UniversityCollegeLondon.
Stoffels, L., H. N. Taunt, B. Charalambous & S. Purton. 2017. Synthesis ofbacteriophage lytic proteins against Streptococcus pneumoniae in thechloroplast of Chlamydomonas reinhardtii. Plant Biotechnology Journal15:1130–1140.
Sun,M.,K.Qian,N.Su,H.Chang,J.Liu&G.Chen.2003.Foot-and-mouthdiseasevirus VP1 protein fused with cholera toxin B subunit expressed inChlamydomonasreinhardtiichloroplast.BiotechnologyLetters25:1087–1092.
Sun,X.&J.C.Linden.1999.Shearstresseffectsonplantcellsuspensionculturesinarotating wall vessel bioreactor. Journal of Industrial Microbiology &Biotechnology22:44–47.
Surzycki,R.,K.Greenham,K.Kitayama,F.Dibal,R.Wagner,J.D.Rochaix,T.Ajam&S. Surzycki. 2009. Factors effecting expression of vaccines in microalgae.Biologicals37:133–138.
Tängdén,T.,R.A.Hickman,P.Forsberg,P.Lagerbäck,C.G.Giske&O.Cars.2014.Combinationantibiotictherapyformultidrug-resistantGram-negativebacteria.Antimicrobialagentsandchemotherapy9734:1–5.
Taunt, H. N. 2013. The Synthesis of Novel Antibacterial Proteins in theChlamydomonasreinhardtiiChloroplast.PhDthesis.London:UniversityCollegeLondon.
Taylor, E. J. M., Y. Yu, J. Champer & J. Kim. 2014. Resveratrol DemonstratesAntimicrobial Effects Against Propionibacterium acnes In Vitro. DermatologyandTherapy4:249–257.
Tolba,M.,M.U.Ahmed,C.Tlili,F.Eichenseher,M.J.Loessner,M.Zourob,L.Yang,R.Bashir, V. Ramaswamy, V. M. Cresence, J. S. Rejitha, M. U. Lekshmi, K. S.Dharsana, S. P. Prasad, H.M. Vijila, P.Mook, S. J. O’Brien, I. A. Gillespie, C.Genigeorgis et al. 2012. A bacteriophage endolysin-based electrochemicalimpedance biosensor for the rapid detection of Listeria cells. The Analyst137:5749.
Tran,M.,C.Van,D.J.Barrera,P.L.Pettersson,C.D.Peinado,J.Bui&S.P.Mayfield.2013. Production of unique immunotoxin cancer therapeutics in algalchloroplasts. Proceedingsof theNationalAcademyof Sciencesof theUnitedStatesofAmerica110:E15-22.
Tran,M.,B.Zhou,P.L.Pettersson,M.J.Gonzalez&S.P.Mayfield.2009.Synthesisandassemblyofafull-lengthhumanmonoclonalantibodyinalgalchloroplasts.BiotechnologyandBioengineering104:663–673.

271
US.DepartmentofHealthandHumanServices2013.Antibioticresistancethreatsinthe United States, 2013. Centres for Disease Control and Prevention.<http://www.cdc.gov/drugresistance/threat-report-2013/index.html>.
Vahrenholz,C.,G.Riemen,E.Pratje,B.Dujon&G.Michaelis.1993.MitochondrialDNAofChlamydomonasreinhardtii:thestructureoftheendsofthelinear15.8-kbgenomesuggestsmechanismsforDNAreplication.CurrentGenetics24:241–247.
Varon, M. 1979. Selection of predation-resistant bacteria in continuous culture.Nature277:386–388.
Ventola,C.L.2015.Theantibioticresistancecrisis:part1:causesandthreats.P&T :Apeer-reviewedjournalforformularymanagement40:277–83.
Walker,T. L.,S.Purton,D.K.Becker&C.Collet.2005.Microalgaeasbioreactors.Plantcellreports24:629–41.
Walsh, C. T.& T. A.Wencewicz. 2014. Prospects for new antibiotics: amolecule-centeredperspective.TheJournalofAntibiotics67:7–22.
Wang, I.-N. N., D. L. Smith & R. Young. 2000. Holins: the protein clocks ofbacteriophageinfections.Annualreviewofmicrobiology54:799–825.
Wang,Q.,C.W.Euler,A.Delaune&V.A.Fischetti.2015.UsinganovellysintohelpcontrolClostridiumdifficileinfections.AntimicrobialAgentsandChemotherapy59:AAC.01357-15.
Wang,X.,M.Brandsma,R.Tremblay,D.Maxwell,A.M.Jevnikar,N.Huner&S.Ma.2008. A novel expression platform for the production of diabetes-associatedautoantigenhumanglutamicaciddecarboxylase(hGAD65).BMCBiotechnology8:87.
Wannathong,T.,J.C.Waterhouse,R.E.B.Young,C.K.Economou&S.Purton.2016.New tools for chloroplast genetic engineering allow the synthesis of humangrowth hormone in the green alga Chlamydomonas reinhardtii. AppliedMicrobiologyandBiotechnology100:5467–5477.
Werth,B.(2014).Thebillion-dollarmolecule.NewYork:Simon&Schuster,p.122.
White,R.,S.Chiba,T.Pang,J.S.Dewey,C.G.Savva,A.Holzenburg,K.Pogliano&R.Young.2011.Holintriggeringinrealtime.ProceedingsoftheNationalAcademyofSciencesoftheUnitedStatesofAmerica108:798–803.
WHO.2007.Pneumococcal conjugatevaccine for childhood immunization–WHOpositionpaper.Weeklyepidemiologicalrecord82:93–104.
WHO. 2016. Antimicrobial resistance Fact sheet. World Health Organization.<http://www.who.int/mediacentre/factsheets/fs194/en/>(14July2017).

272
Willmund,F.,K.V.Dorn,M.Schulz-Raffelt&M.Schroda.2008.TheChloroplastDnaJHomolog CDJ1 of Chlamydomonas reinhardtii Is Part of a MultichaperoneComplexContainingHSP70B, CGE1, andHSP90C. Plant Physiology 148:2070–2082.
Wilson,M.2005.Microbialinhabitantsofhumans :theirecologyandroleinhealthanddisease.Cambridge,UK:CambridgeUniversityPress.
Witzenrath,M.,B.Schmeck,J.M.Doehn,T.Tschernig,J.Zahlten,J.M.Loeffler,M.Zemlin,H.Muller,B.Gutbier,H.Schutte,S.Hippenstiel,V.aFischetti,N.Suttorp&S.Rosseau.2009.SystemicuseoftheendolysinCpl-1rescuesmicewithfatalpneumococcalpneumonia.CriticalCareMedicine37:642–649.
Wurm, F.M. 2004. Production of recombinant protein therapeutics in cultivatedmammaliancells.NatureBiotechnology22:1393–1398.
Xu,J.&N.Zhang.2014.Onthewaytocommercializingplantcellcultureplatformforbiopharmaceuticals:presentstatusandprospect.PharmaceuticalBioprocessing2:499–518.
Yang, S.-C., C.-H. Lin, C. T. Sung & J.-Y. Fang. 2014. Antibacterial activities ofbacteriocins: application in foods and pharmaceuticals. Frontiers inMicrobiology5:241.
Yang,Z.,Y.Li,F.Chen,D.Li,Z.Zhang,Y.Liu,D.Zheng,Y.Wang&G.Shen.2006.ExpressionofhumansolubleTRAILinChlamydomonasreinhardtiichloroplast.ChineseScienceBulletin51:1703–1709.
Yao, J., Y. Weng, A. Dickey & K. Y. Wang. 2015. Plants as factories for humanpharmaceuticals: Applications and challenges. International Journal ofMolecularSciences16:28549–28565.
Young, R. 1992. Bacteriophage lysis: mechanism and regulation. Microbiologicalreviews56:430–81.
Young,R.E.B.&S.Purton.2014.Cytosinedeaminaseasanegativeselectablemarkerforthemicroalgalchloroplast:Astrategyfortheisolationofnuclearmutationsthataffectchloroplastgeneexpression.PlantJournal80:915–925.
Young, R. E. B. & S. Purton. 2015. Codon reassignment to facilitate geneticengineering and biocontainment in the chloroplast of Chlamydomonasreinhardtii.PlantBiotechnologyJournal:1–10.
Young, R. E. B. & S. Purton. 2016. Codon reassignment to facilitate geneticengineering and biocontainment in the chloroplast of Chlamydomonasreinhardtii.PlantBiotechnologyJournal14:1251–1260.

273
Zaenglein,A.L.,A.L.Pathy,B.J.Schlosser,A.Alikhan,H.E.Baldwin,D.S.Berson,W.P.Bowe,E.M.Graber,J.C.Harper,S.Kang,J.E.Keri,J.J.Leyden,R.V.Reynolds,N.B.Silverberg,L.F.SteinGold,M.M.Tollefson,J.S.Weiss,N.C.Dolan,A.A.Sagan et al. 2016. Guidelines of care for the management of acne vulgaris.JournaloftheAmericanAcademyofDermatology74:945–973e33.
Zaffiri, L., J. Gardner & L. H. Toledo-Pereyra. 2012. History of antibiotics. Fromsalvarsantocephalosporins.JournalofInvestigativeSurgery25:67–77.
Zedler,J.A.Z.Z.,D.Gangl,B.B.Hamberger,S.Purton&C.Robinson.2015.Stableexpression of a bifunctional diterpene synthase in the chloroplast ofChlamydomonasreinhardtii.JournalofAppliedPhycology27:2271–2277.
Zienkiewicz, M., T. Krupnik, A. Droak, A. Golke & E. Romanowska. 2017.TransformationoftheCyanidioschyzonmerolaechloroplastgenome:prospectsfor understanding chloroplast function in extreme environments. PlantMolecularBiology93:171–183.

274
Chapter7 Appendices
7.1 ResultsChapter3Appendices
7.1.1 AppendixA
Figure 7.1 Total protein staining of five technical repeats of TN72_SR_cpl1-HA andTN72_SR_cpl1-HA_A_cpl1_c-HA. These data were used for normalisation of the anti-HAsignaltoaccountforloadingerror.
(kDa)
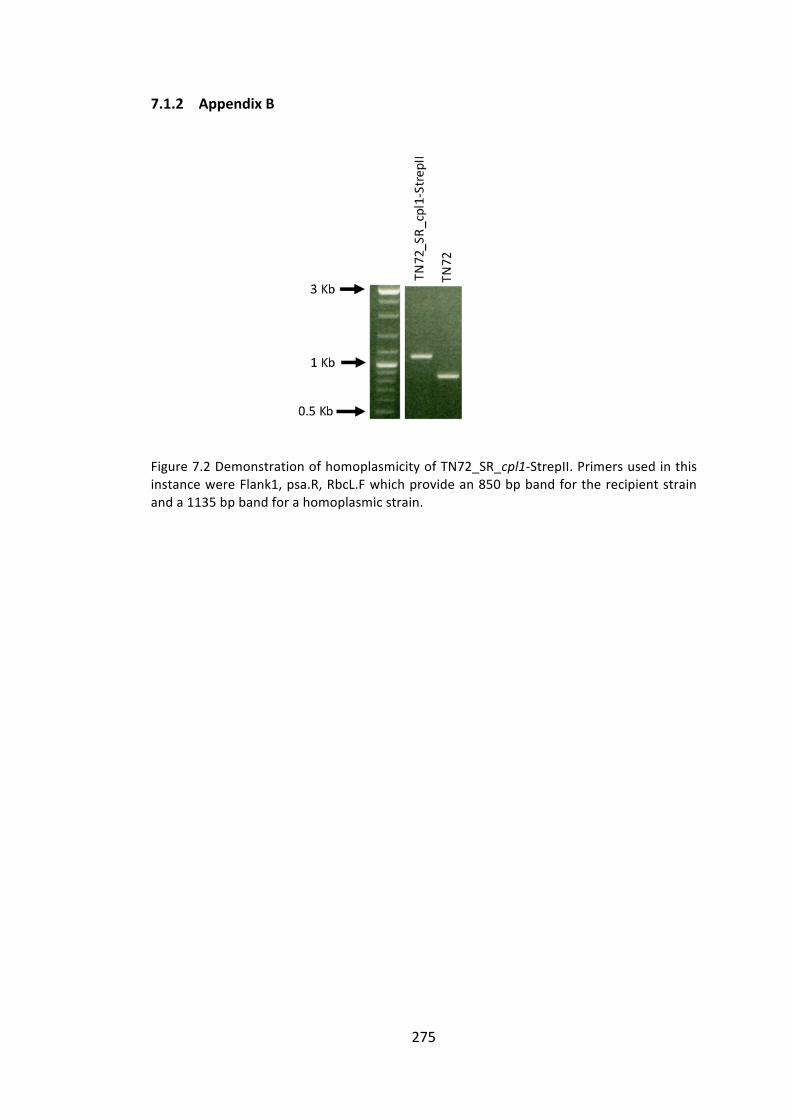
275
7.1.2 AppendixB
Figure7.2DemonstrationofhomoplasmicityofTN72_SR_cpl1-StrepII.PrimersusedinthisinstancewereFlank1,psa.R,RbcL.Fwhichprovidean850bpbandfortherecipientstrainanda1135bpbandforahomoplasmicstrain.

276
7.1.3 AppendixC
Figure7.3TotalproteinstainingusedtonormaliseforloadingerrorswhencomparingtotalproteinaccumulationbetweentheaboveC.reinhardtiilines.
(kDa)
277
FigureRemoved
7.1.4 AppendixD
Figure7.4CodonfrequencychartgeneratedforC.reinhardtiichloroplasttransgenecodonoptimization,fromCodonUsageOptimizerBeta0.92(Kong2013).
7.1.5 AppendixE
Figure 7.5 Western blot analysis demonstrating the varying levels of HA-tagged proteinaccumulationovera194hourcultureperiod.
0 unidentified codons.Nucleotide frequency: A=29 U=45 G=15 C=10 X=0 GC Content: 25.25% 1st letter GC: 36.36% 2nd letter GC: 27.27% 3rd letter GC: 12.12% Summary table:Sequence length: 21279 Number of protein genes: 32 Total codons: 7093
0 unidentified codons.Nucleotide frequency: A=5880 U=7318 G=4106 C=3975 X=0 GC Content: 37.98% 1st letter GC: 50.53% 2nd letter GC: 41.41% 3rd letter GC: 21.99% Weight table from reference set:
CodonUsageOptimizerBeta 0.6
Amino acid | Codon | Number | Frequency per thousandPhe UUU 1 30.3 Ser UCU 0 0.0 Tyr UAU 1 30.3 Cys UGU 0 0.0
UUC 0 0.0 UCC 0 0.0 UAC 1 30.3 UGC 0 0.0Leu UUA 6 181.8 UCA 0 0.0 STOP UAA 1 30.3 STOP UGA 0 0.0
UUG 0 0.0 UCG 0 0.0 UAG 0 0.0 Trp UGG 0 0.0Leu CUU 3 90.9 Pro CCU 0 0.0 His CAU 0 0.0 Arg CGU 0 0.0
CUC 0 0.0 CCC 0 0.0 CAC 0 0.0 CGC 0 0.0CUA 0 0.0 CCA 0 0.0 Gln CAA 0 0.0 CGA 0 0.0CUG 0 0.0 CCG 0 0.0 CAG 0 0.0 CGG 0 0.0
ile AUU 2 60.6 Thr ACU 1 30.3 Asn AAU 0 0.0 Ser AGU 1 30.3AUC 2 60.6 ACC 0 0.0 AAC 0 0.0 AGC 0 0.0AUA 0 0.0 ACA 2 60.6 Lys AAA 2 60.6 Arg AGA 0 0.0
Met AUG 1 30.3 ACG 0 0.0 AAG 0 0.0 AGG 0 0.0Val GUU 2 60.6 Ala GCU 1 30.3 Asp GAU 0 0.0 Gly GGU 4 121.2
GUC 0 0.0 GCC 0 0.0 GAC 0 0.0 GGC 0 0.0GUA 2 60.6 GCA 0 0.0 Glu GAA 0 0.0 GGA 0 0.0GUG 0 0.0 GCG 0 0.0 GAG 0 0.0 GGG 0 0.0
Amino acid | Codon | Number | Frequency per thousandPhe UUU 171 24.1 Ser UCU 147 20.7 Tyr UAU 87 12.3 Cys UGU 61 8.6
UUC 285 40.2 UCC 0 0.0 UAC 152 21.4 UGC 3 0.4Leu UUA 544 76.7 UCA 144 20.3 STOP UAA 30 4.2 STOP UGA 0 0.0
UUG 5 0.7 UCG 9 1.3 UAG 2 0.3 Trp UGG 170 24.0Leu CUU 152 21.4 Pro CCU 119 16.8 His CAU 23 3.2 Arg CGU 245 34.5
CUC 0 0.0 CCC 2 0.3 CAC 148 20.9 CGC 10 1.4CUA 53 7.5 CCA 200 28.2 Gln CAA 218 30.7 CGA 2 0.3CUG 9 1.3 CCG 13 1.8 CAG 10 1.4 CGG 0 0.0
ile AUU 343 48.4 Thr ACU 210 29.6 Asn AAU 83 11.7 Ser AGU 80 11.3AUC 102 14.4 ACC 3 0.4 AAC 198 27.9 AGC 30 4.2AUA 4 0.6 ACA 193 27.2 Lys AAA 240 33.8 Arg AGA 8 1.1
Met AUG 182 25.7 ACG 12 1.7 AAG 9 1.3 AGG 2 0.3Val GUU 258 36.4 Ala GCU 433 61.0 Asp GAU 135 19.0 Gly GGU 586 82.6
GUC 0 0.0 GCC 9 1.3 GAC 115 16.2 GGC 24 3.4GUA 261 36.8 GCA 173 24.4 Glu GAA 311 43.8 GGA 19 2.7GUG 6 0.8 GCG 23 3.2 GAG 20 2.8 GGG 7 1.0
TN72_SR_cpl1-HA
45h
TN72_SR_CPOCv1-HA
TN72_SR_Control
TN72_SR_CPOCv2-HA
53.5h
66.5h
97h
122h
139h
194h
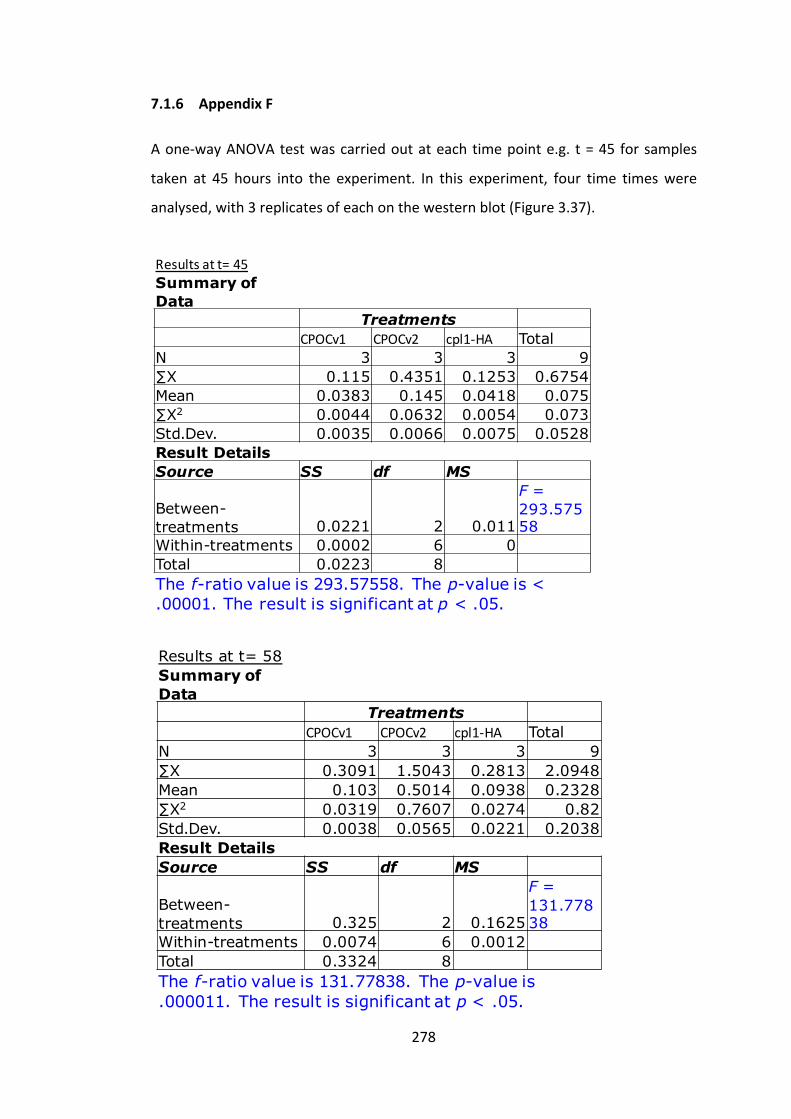
278
7.1.6 AppendixF
Aone-wayANOVAtestwascarriedoutateachtimepointe.g. t=45 forsamples
taken at 45 hours into the experiment. In this experiment, four time timeswere
analysed,with3replicatesofeachonthewesternblot(Figure3.37).
Resultsatt=45Summary of Data
TreatmentsCPOCv1 CPOCv2 cpl1-HA Total
N 3 3 3 9∑X 0.115 0.4351 0.1253 0.6754Mean 0.0383 0.145 0.0418 0.075∑X2 0.0044 0.0632 0.0054 0.073Std.Dev. 0.0035 0.0066 0.0075 0.0528Result DetailsSource SS df MS
Between-treatments 0.0221 2 0.011
F = 293.57558
Within-treatments 0.0002 6 0Total 0.0223 8The f-ratio value is 293.57558. The p-value is < .00001. The result is significant at p < .05.
Results at t= 58Summary of Data
TreatmentsCPOCv1 CPOCv2 cpl1-HA Total
N 3 3 3 9∑X 0.3091 1.5043 0.2813 2.0948Mean 0.103 0.5014 0.0938 0.2328∑X2 0.0319 0.7607 0.0274 0.82Std.Dev. 0.0038 0.0565 0.0221 0.2038Result DetailsSource SS df MS
Between-treatments 0.325 2 0.1625
F = 131.77838
Within-treatments 0.0074 6 0.0012Total 0.3324 8The f-ratio value is 131.77838. The p-value is .000011. The result is significant at p < .05.
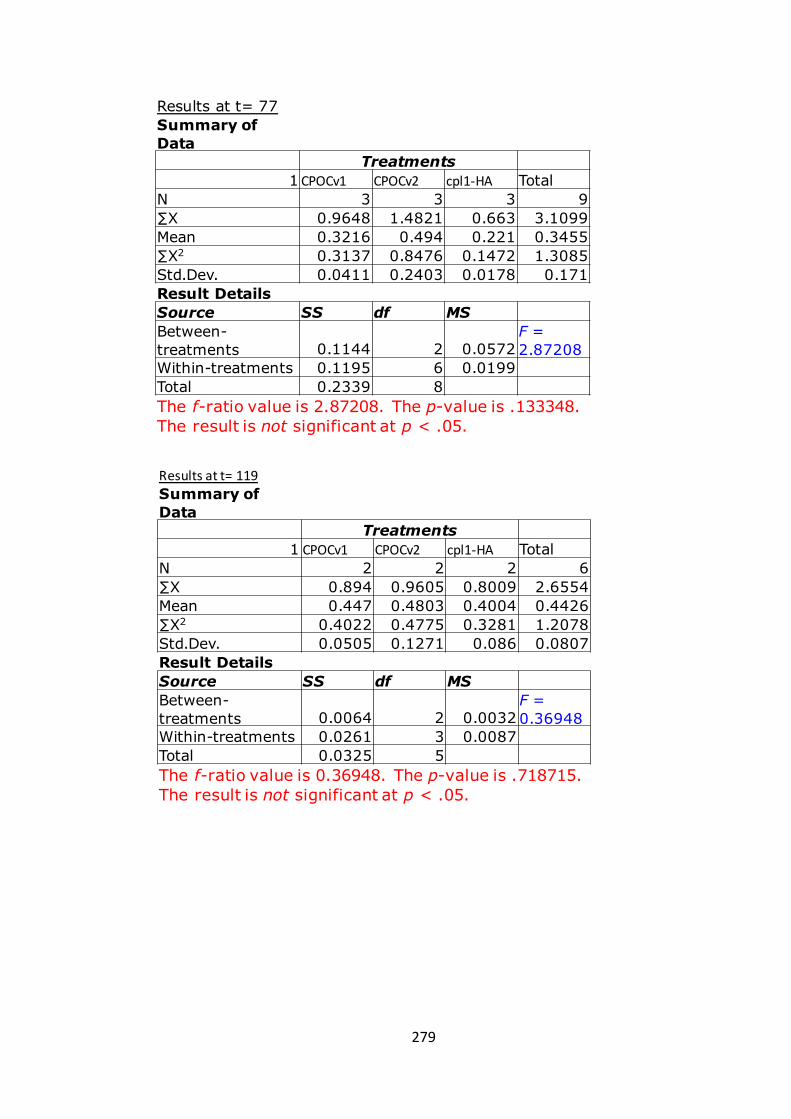
279
Results at t= 77Summary of Data
Treatments1 CPOCv1 CPOCv2 cpl1-HA Total
N 3 3 3 9∑X 0.9648 1.4821 0.663 3.1099Mean 0.3216 0.494 0.221 0.3455∑X2 0.3137 0.8476 0.1472 1.3085Std.Dev. 0.0411 0.2403 0.0178 0.171Result DetailsSource SS df MSBetween-treatments 0.1144 2 0.0572
F = 2.87208
Within-treatments 0.1195 6 0.0199Total 0.2339 8The f-ratio value is 2.87208. The p-value is .133348. The result is not significant at p < .05.
Resultsatt=119Summary of Data
Treatments1 CPOCv1 CPOCv2 cpl1-HA Total
N 2 2 2 6∑X 0.894 0.9605 0.8009 2.6554Mean 0.447 0.4803 0.4004 0.4426∑X2 0.4022 0.4775 0.3281 1.2078Std.Dev. 0.0505 0.1271 0.086 0.0807Result DetailsSource SS df MSBetween-treatments 0.0064 2 0.0032
F = 0.36948
Within-treatments 0.0261 3 0.0087Total 0.0325 5The f-ratio value is 0.36948. The p-value is .718715. The result is not significant at p < .05.
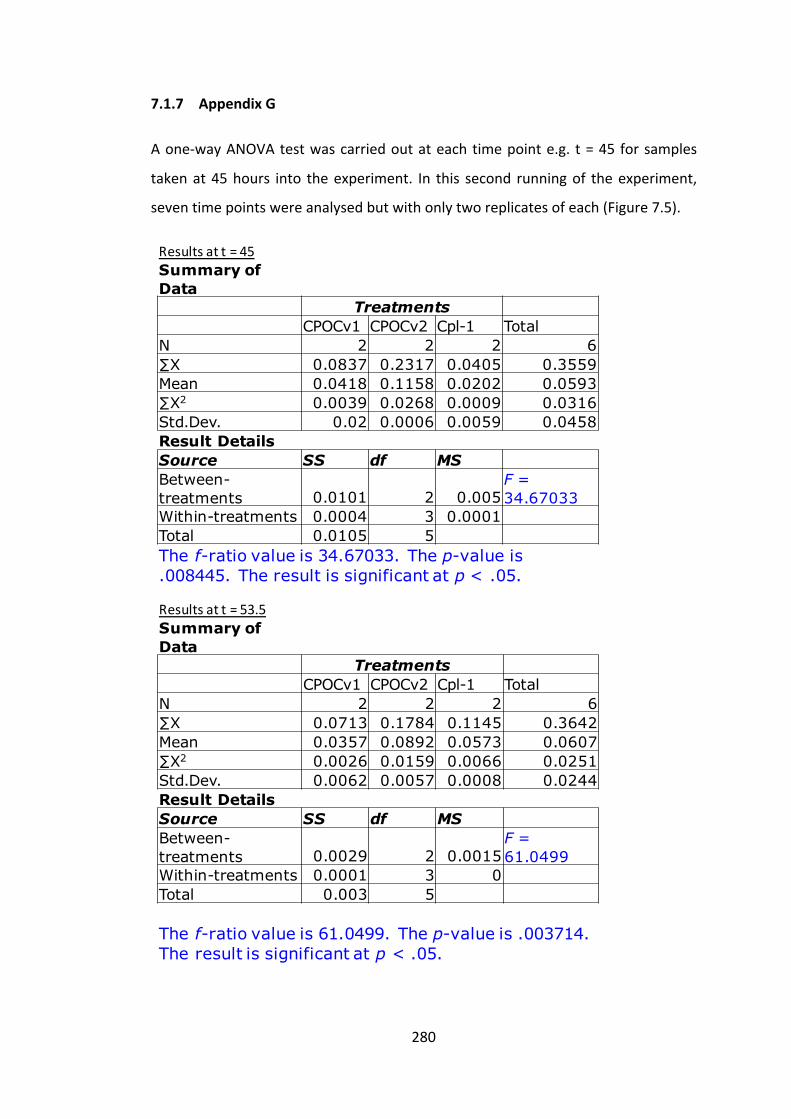
280
7.1.7 AppendixG
Aone-wayANOVAtestwascarriedoutateachtimepointe.g. t=45 forsamples
takenat45hours into theexperiment. In this second runningof theexperiment,
seventimepointswereanalysedbutwithonlytworeplicatesofeach(Figure7.5).
Resultsatt=45Summary of Data
TreatmentsCPOCv1 CPOCv2 Cpl-1 Total
N 2 2 2 6∑X 0.0837 0.2317 0.0405 0.3559Mean 0.0418 0.1158 0.0202 0.0593∑X2 0.0039 0.0268 0.0009 0.0316Std.Dev. 0.02 0.0006 0.0059 0.0458Result DetailsSource SS df MSBetween-treatments 0.0101 2 0.005
F = 34.67033
Within-treatments 0.0004 3 0.0001Total 0.0105 5The f-ratio value is 34.67033. The p-value is .008445. The result is significant at p < .05.
Resultsatt=53.5Summary of Data
TreatmentsCPOCv1 CPOCv2 Cpl-1 Total
N 2 2 2 6∑X 0.0713 0.1784 0.1145 0.3642Mean 0.0357 0.0892 0.0573 0.0607∑X2 0.0026 0.0159 0.0066 0.0251Std.Dev. 0.0062 0.0057 0.0008 0.0244Result DetailsSource SS df MSBetween-treatments 0.0029 2 0.0015
F = 61.0499
Within-treatments 0.0001 3 0Total 0.003 5
The f-ratio value is 61.0499. The p-value is .003714. The result is significant at p < .05.

281
Resultsatt=66.5Summary of Data
TreatmentsCPOCv1 CPOCv2 Cpl-1 Total
N 2 2 2 6∑X 0.1001 0.2606 0.1458 0.5065Mean 0.05 0.1303 0.0729 0.0844∑X2 0.005 0.0356 0.011 0.0516Std.Dev. 0.0003 0.0405 0.0193 0.0421Result DetailsSource SS df MSBetween-treatments 0.0068 2 0.0034
F = 5.09397
Within-treatments 0.002 3 0.0007Total 0.0089 5
The f-ratio value is 5.09397. The p-value is .108497. The result is not significant at p < .05.
Resultsatt=97Summary of Data
TreatmentsCPOCv1 CPOCv2 Cpl-1 Total
N 2 2 2 6∑X 0.1803 0.2498 0.1553 0.5854Mean 0.0901 0.1249 0.0776 0.0976∑X2 0.0168 0.0313 0.0121 0.0603Std.Dev. 0.0235 0.0102 0.0095 0.0251Result DetailsSource SS df MSBetween-treatments 0.0024 2 0.0012
F = 4.82318
Within-treatments 0.0007 3 0.0002Total 0.0031 5
The f-ratio value is 4.82318. The p-value is .11554. The result is not significant at p < .05.

282
Resultsatt=122Summary of Data
TreatmentsCPOCv1 CPOCv2 Cpl-1 Total
N 2 2 2 6∑X 0.9233 0.8594 0.3558 2.1385Mean 0.4617 0.4297 0.1779 0.3564∑X2 0.4288 0.3714 0.0634 0.8636Std.Dev. 0.0505 0.0458 0.0095 0.1424Result DetailsSource SS df MSBetween-treatments 0.0966 2 0.0483
F = 30.59972
Within-treatments 0.0047 3 0.0016Total 0.1014 5
The f-ratio value is 30.59972. The p-value is .010101. The result is significant at p < .05.
Resultsatt=139Summary of Data
TreatmentsCPOCv1 CPOCv2 Cpl-1 Total
N 2 2 2 6∑X 1.1899 1.1952 0.3528 2.7379Mean 0.595 0.5976 0.1764 0.4563∑X2 0.7086 0.7301 0.0634 1.5021Std.Dev. 0.0245 0.126 0.0342 0.2248Result DetailsSource SS df MSBetween-treatments 0.2351 2 0.1175
F = 20.00184
Within-treatments 0.0176 3 0.0059Total 0.2527 5
The f-ratio value is 20.00184. The p-value is .018426. The result is significant at p < .05.

283
Resultsatt=194Summary of Data
TreatmentsCPOCv1 CPOCv2 Cpl-1 Total
N 2 2 2 6∑X 1.4863 1.4019 0.6651 3.5533Mean 0.7431 0.7009 0.3326 0.5922∑X2 1.1065 0.9986 0.222 2.327Std.Dev. 0.0445 0.1261 0.0284 0.211Result DetailsSource SS df MSBetween-treatments 0.204 2 0.102
F = 16.38062
Within-treatments 0.0187 3 0.0062Total 0.2227 5
The f-ratio value is 16.38062. The p-value is .024298. The result is significant at p < .05.

284
7.2 ResultsChapter4Appendices
7.2.1 AppendixH
Figure7.6cph1sequenceasorderedfromIntegratedDNATechnologies.Thestartcodonishighlightedingreen.TheintroducedTGAcodonsfortryptophanarehighlightedinorange.Thestopcodonsarehighlighted inred.TheHAtag ishighlighted inpurple.SapIandSphIrestrictionsitesareindicatedbybluearrows.
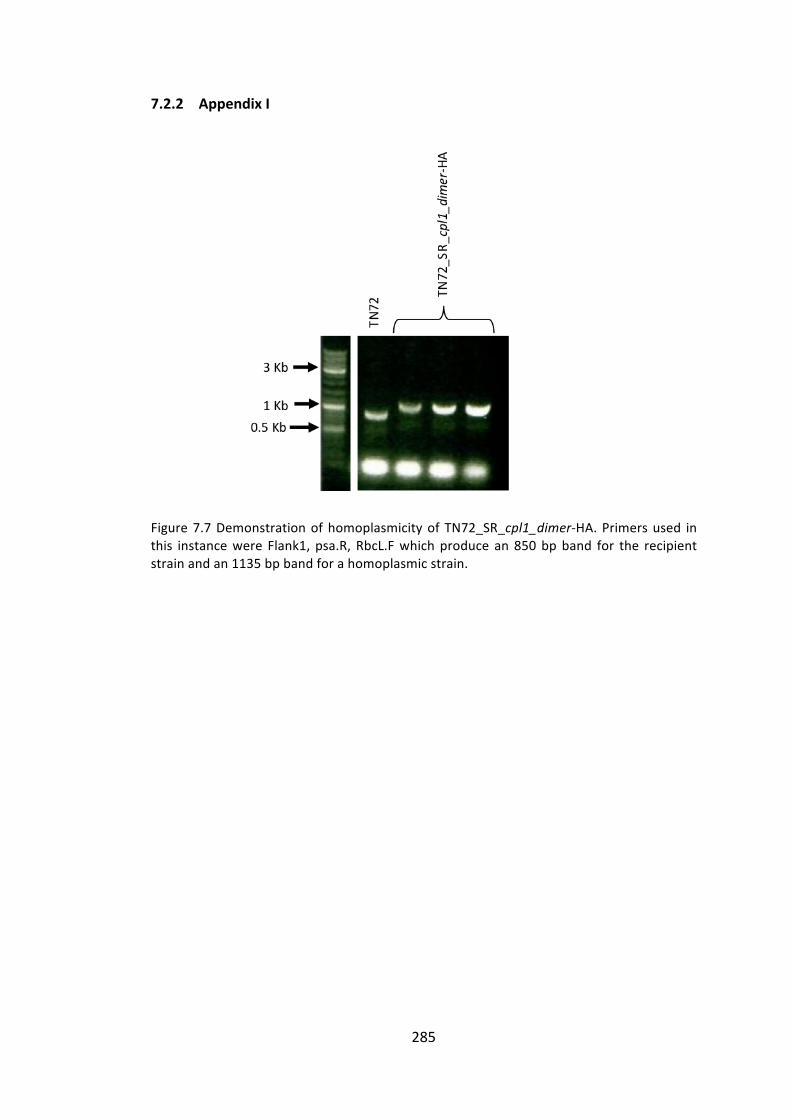
285
7.2.2 AppendixI
Figure7.7DemonstrationofhomoplasmicityofTN72_SR_cpl1_dimer-HA.Primersused inthis instancewere Flank1, psa.R, RbcL.Fwhichproduce an850bpband for the recipientstrainandan1135bpbandforahomoplasmicstrain.

286
7.3 ResultsChapter5Appendices
7.3.1 AppendixJ
Figure7.8cd27lsequenceasorderedfromIntegratedDNATechnologies.Thestartcodonishighlighted ingreen.The stopcodonsarehighlighted in red.TheHA tag ishighlighted inpurple.SapIandSphIrestrictionsitesareindicatedbybluearrows.

287
7.3.2 AppendixK
Figure 7.9 Demonstration of homoplasmicity of TN72_SR_cd27l-HA. Primers used in thisinstancewereFlank1,psa.R,RbcL.Fwhichproducean850bpbandfortherecipientstrainandan1135bpbandforahomoplasmicstrain.Sixtransformantsweretested.
7.3.3 AppendixL
Figure7.10DemonstrationofhomoplasmicityofTN72_SR_cd27l-StrepII.PrimersusedinthisinstancewereFlank1,psa.R,RbcL.Fwhichproducean850bpbandfortherecipientstrainandan1135bpbandforahomoplasmicstrain.Twotransformantsweretested.
3Kb
1Kb0.5Kb
TN72
TN72_SR_
cd27l-H
A
3Kb
1Kb0.5Kb
TN72 TN
72_SR_
cd27l-S
trepII

288
7.3.4 AppendixM
Figure7.11gp20NsequenceasorderedfromIntegratedDNATechnologies.Thestartcodonishighlightedingreen.TheintroducedTGAcodonsfortryptophanarehighlightedinorange.Thestopcodonsarehighlighted inred.TheHAtag ishighlighted inpurple.SapIandSphIrestrictionsitesareindicatedbybluearrows.
TG GTA TGC TCT TCA ATG GCA TAC CCA TAT GAT
GTA CCT GAC TAT GCT GTT CGT TAT ATT CCA GCT
GCT CAT CAC TCA GCA GGT TCT AAC AAT CCT GTT
AAT CGT GTA GTA ATT CAT GCA ACA TGT CCA GAT
GTA GGA TTT CCT AGT GCT AGC CGT AAA GGT CGT
GCT GTT TCA ACT GCT AAT TAC TTC GCA TCA CCA
TCT AGT GGT GGT AGC GCT CAC TAT GTG TGT GAC
ATT GGA GAA ACA GTA CAA TGT TTA TCT GAA AGT
ACT ATT GGA TGG CAC GCA CCA CCA AAC CCT CAT
TCA TTA GGT ATT GAA ATT TGT GCT GAT GGT GGT
TCT CAT GCT AGC TTT CGT GTG CCT GGT CAC GCA
TAT ACA CGT GAA CAA TGA TTA GAT CCA CAA GTT
TGG CCA GCT GTT GAA CGT GCT GCA GTA TTA TGT
CGT CGT TTA TGT GAC AAA TAC AAT GTA CCT AAA
CGT AAA TTA TCA GCT GCT GAC TTA AAA GCA GGT
CGT CGT GGT GTT TGT GGT CAT GTT GAT GTA ACT
GAC GCT TGA CAC CAA AGC GAT CAT GAT GAT CCA
GGA CCA TGG TTT CCT TGG GAC AAA TTC ATG GCT
GTT GTA AAC GGT GGT AGC GGT GAC AGC GGA GAA
TTA ACA GTT GCA GAT GTA AAA GCT TTA CAC GAT
CAA ATT AAA CAA TTA TCA GCA CAA TTA ACT GGT
AGC GTG AAC AAA TTA CAT CAC GAT GTT GGT GTT
GTA CAA GTG CAA AAC GGT GAC TTA GGA AAA CGT
GTA GAC GCT TTA AGC TGG GTG AAA AAT CCA GTG
ACT GGA AAA TTA TGA CGT ACA AAA GAT GCT TTA
TGG TCT GTT TGG TAT TAT GTA TTA GAA TGT CGT
AGC CGT TTA GAT CGT TTA GAA AGC GCT GTA AAT
GAC TTA AAA AAA TAA TAA GCA TGC GTC AGT ttt
t
SapI
SphI

289
7.3.5 AppendixN
Figure7.12DemonstrationofhomoplasmicityofTN72_SR_gp20N-HA.PrimersusedinthisinstancewereFlank1,psa.R,RbcL.Fwhichproducean850bpbandfortherecipientstrainandan1135bpbandforahomoplasmicstrain.Twotransformantsweretested.
7.3.6 AppendixO:ListofPrimers
Primersinorderofappearanceinthisthesis.
NAME SEQUENCE NOTES
Flank1 GTCATTGCGAAAATACTGGTGC
Primers for general sequencing, PCR and
homoplasmicity testing
RbcL.F CGGATGTAACTCAATCGGTAG
Rbcl.R CAAACTTCACATGCAGCAGC
Psa.R CATGGATTTCTCCTTATAATAAC
atpA.R ACGTCCACAGGCGTCGT
123a GGAGGGAAGTAGGCAGTAGC
psbH.R GCAACAGGAACTTCTAAAGC
psaSEQ AACTATTTGTCTAATTTAATAACC
3Kb
1Kb
0.5Kb
TN72
TN72_SR_
gp20N-HA
290
cpl1.F GCTCTTCAATGGTTAAAAAAAATGATTTATTC
Primers for the site-directed mutageneis of the cell wall binding domain
cpl1.R GCATGCTTATTAAGCATAATCTGGAAC
W202A_SDM.F GTACAGCTAAACAAGATTCAAAAG
W202A_SDM.R CTTTTGAATCTTGTTTAGCTGTAC
W209A_SDM.F CAAAAGGTGCTTGGTTTC
W209A_SDM.R GAAACCAAGCACCTTTTG
W223A_SDM.F CCATATAATAAAGCTGAAAAAATTG
W223A_SDM.R CAATTTTTTCAGCTTTATTATATGG
W230A_SDM.F GTGGTGTTGCTTATTATTTCG
W230A_SDM.R CGAAATAATAAGCAACACCAC
Cpl1_BssHII.F CAGGCAATTTGCTTACACCTTT Primers to introduce a BssHII restriction site into Cpl-1 Cpl1_BssHII.R TTAAGATAAGCGCGCTACATCC
cpl1_C45S.F ACATATTTAAATCCATCATTATCAGCTC
Primers to remove and introduce cysteine residues to create Cpl1_dimer
cpl1_C45S.R GAGCTGATAATGATGGATTTAAATATGT
cpl1_D324C.F GTGAATTAGCATGTAATCCTTCATTT
cpl1_D324C.R AAATGAAGGATTACATGCTAATTCAC
cd27l.F TGGTATTGCTCTTCAATGAAAATTTGTATTACT Primers for the replacement of the HA tag with a StrepII tag on cd27l
cd27l_StrepII.R ACTGACGCATGCTTATTATTTTTCGAATTGTGGGTGAGACCAGTTAATGTTTTTGTTTAAAACACCTTCAAC
gp20_NcoI.F CGTATACCCATGGTGCGTTA Primers to introduce an NcoI restriction site to gp20 gp20.R GATTGCGGATCCTTATTAGGC
Top Related
Copyright © 2022 FDOKUMEN

