







Bahasa
Halaman
Hukum


1 23
Interdisciplinary Sciences:Computational Life SciencesComputational Life Sciences ISSN 1913-2751Volume 7Number 2 Interdiscip Sci Comput Life Sci (2015)7:157-167DOI 10.1007/s12539-015-0018-x
In Silico Approach to Support that p-Nitrophenol Monooxygenase fromArthrobacter sp. Strain JS443 Catalyzes theInitial Two Sequential Monooxygenations
Monika Kallubai, UmamaheswariAmineni, Megharaj Mallavarapu &Venkateswarlu Kadiyala

1 23
Your article is protected by copyright
and all rights are held exclusively by
International Association of Scientists in
the Interdisciplinary Areas and Springer-
Verlag Berlin Heidelberg. This e-offprint is
for personal use only and shall not be self-
archived in electronic repositories. If you wish
to self-archive your article, please use the
accepted manuscript version for posting on
your own website. You may further deposit
the accepted manuscript version in any
repository, provided it is only made publicly
available 12 months after official publication
or later and provided acknowledgement is
given to the original source of publication
and a link is inserted to the published article
on Springer's website. The link must be
accompanied by the following text: "The final
publication is available at link.springer.com”.

ORIGINAL RESEARCH ARTICLE
In Silico Approach to Support that p-Nitrophenol Monooxygenasefrom Arthrobacter sp. Strain JS443 Catalyzes the Initial TwoSequential Monooxygenations
Monika Kallubai1 • Umamaheswari Amineni2 • Megharaj Mallavarapu3 •
Venkateswarlu Kadiyala1
Received: 19 November 2013 / Revised: 25 June 2014 / Accepted: 29 October 2014 / Published online: 14 August 2015
� International Association of Scientists in the Interdisciplinary Areas and Springer-Verlag Berlin Heidelberg 2015
Abstract p-Nitrophenol (PNP), used primarily for man-
ufacturing pesticides and dyes, has been recognized as a
priority environmental pollutant. It is therefore important
to reduce the input of this toxicant into the environment
and to establish approaches for its removal from the con-
taminated sites. PNP monooxygenase, a novel enzyme
from Gram-positive bacteria like Arthrobacter sp. and
Bacillus sp., that comprises two components, a flavoprotein
reductase and an oxygenase, catalyzes the initial two
sequential monooxygenations to convert PNP to trihy-
droxybenzene. Accurate and reliable prediction of this
enzyme–substrate interactions and binding affinity are of
vital importance in understanding these catalytic mecha-
nisms of the two sequential reactions. As crystal structure
of the enzyme has not yet been published, we built a
homology model for PNP monooxygenase using crystal-
lized chlorophenol 4-monooxygenase from Burkholderia
cepacia AC1100 (3HWC) as the template. The model was
assessed for its reliability using PROCHECK, ERRAT and
ProSA. Molecular docking of the physiological substrates,
PNP and 4-nitrocatechol (4-NC), was carried out using
Glide v5.7 implemented in Maestro v9.2, and the binding
energies were calculated to substantiate the prediction.
Docking complexes formed by molecular level interactions
of PNP monooxygenase-PNP/4-NC without or with the
cofactors, FAD and NADH, showed good correlation with
the established experimental evidence that the two-com-
ponent PNP monooxygenase catalyzes both the hydroxy-
lation of PNP and the oxidative release of nitrite from
4-NC in B. sphaericus JS905. Furthermore, molecular
dynamics simulations performed for docking complexes
using Desmond v3.0 showed stable nature of the interac-
tions as well.
Keywords PNP monooxygenase � Arthrobacter sp. strainJS443 � Bacillus sphaericus JS905 � Homology modeling �Molecular docking � Molecular dynamics simulations
1 Introduction
With the rapid thrust in industrial and agricultural activity,
the recent times have witnessed vast quantities of soil and
groundwater resources becoming contaminated with haz-
ardous chemicals. p-Nitrophenol (PNP) is probably the
most important contaminant among the mononitrophenols
since 27 % of its use in industry is for pesticide manu-
facture and 13 % is for synthesis of dye components [1].
PNP is also found as a major metabolite of microbial
degradation of organophosphorous insecticides, parathion
and methyl parathion [2] and is thus an important envi-
ronmental pollutant [3, 4] posing even odor problems to
water bodies [5, 6]. PNP is, therefore, one of the eleven
phenolic compounds listed as priority pollutants by the US
Environmental Protection Agency [7].
Electronic supplementary material The online version of thisarticle (doi:10.1007/s12539-015-0018-x) contains supplementarymaterial, which is available to authorized users.
& Venkateswarlu Kadiyala
1 Department of Microbiology, Sri Krishnadevaraya
University, Anantapur 515055, India
2 Department of Bioinformatics, Sri Venkateswara Institute for
Medical Sciences, Tirupati 517507, India
3 Centre for Environmental Risk Assessment and Remediation,
and CRC for Contamination Assessment and Remediation of
the Environment, University of South Australia, Adelaide,
SA 5095, Australia
123
Interdiscip Sci Comput Life Sci (2015) 7:157–167
DOI 10.1007/s12539-015-0018-x
Author's personal copy

Two alternative pathways for aerobic bacterial degra-
dation that convert PNP to maleylacetate have been elu-
cidated [8]. The first pathway that results in the formation
of hydroquinone from PNP, probably via 1,4-benzo-
quinone, with concomitant release of nitrite [9], is more
common in Gram-negative isolates. In the second catabolic
pathway, Bacillus sphaericus JS905 [10] hydroxylates PNP
to produce 4-nitrocatechol (4-NC), and subsequent oxida-
tive removal of the nitro group from 4-NC as nitrite yields
1,2,4-trihydroxybenzene (THB) as shown in Fig. 1. PNP
monooxygenase, a novel enzyme from B. sphaericus JS905
that belongs to a two-component flavin-diffusible
monooxygenase family, consisting of two components—a
flavoprotein reductase and an oxygenase, catalyzes the first
two sequential reactions in the degradation of PNP to THB
via 4-NC. The enzyme uses loosely bound FAD as the
redox chromophore and is NADH dependent. Genetic
evidence that the oxygenase component of the enzyme
hydroxylates PNP as well as 4-NC following two sequen-
tial monooxygenations has been provided for Arthrobacter
sp. strain JS443 [11] and Pseudomonas sp. 1-7 [12]. The
nitro substituent from 4-NC is directly removed from the
aromatic ring in the form of nitrite.
Understanding the microbial metabolism of nitroaro-
matic compounds will assist in management options to
minimize their persistence in the environment, and one
needs to have specific information on various components
involved in catalysis that explains the overall function of
the system. Our present study concerning analysis of
functional sites in PNP monooxygenase relies on the bio-
chemical information provided for B. sphaericus JS905 and
the genetic information reported for Arthrobacter sp. strain
JS443, since the proposed pathways for PNP degradation in
both the bacterial strains are the same involving two
sequential monooxygenation reactions. Experimental evi-
dence also implicates four conserved residues (Arg100,
Gln158, Arg161 and Thr193) for the substrate binding with
oxygenase component of the enzyme [13]. The objective of
the present study was primarily to verify whether the same
amino acid residues at the catalytic site of the enzyme are
involved in the two sequential monooxygenations. Since
molecular docking study using 3D structure of an enzyme
is one way to predict interaction between the substrate and
the active site, we also report the 3D model of PNP
monooxygenase and prediction of interaction sites for the
substrates, PNP and 4-NC, through molecular modeling,
docking and molecular dynamics (MD) simulations.
2 Methodology
2.1 Homology Modeling of PNP Monooxygenase
The protein sequence of PNP monooxygenase from
Arthrobacter sp. strain JS443 (accession number ABL7
5143.1)was retrieved from theNCBI protein database (http://
www.ncbi.nlm.nih.gov/protein). BLASTp [14, 15] search for
PNP monooxygenase was performed against the protein
databank [16] to find a structural template for homology
modeling. Chlorophenol 4-monooxygenase of Burkholderia
cepacia AC1100 (3HWC) was selected as the structural
template and aligned with PNP monooxygenase using
Fig. 1 Proposed pathway for
degradation of PNP by Bacillus
sphaericus JS905
158 Interdiscip Sci Comput Life Sci (2015) 7:157–167
123
Author's personal copy

ClustalX 1.83 [17]. Modeller9v7 [18, 19] that performs
comparative modeling based on satisfaction of spatial
restraints was used to build 3D structure of PNP monooxy-
genase. Twenty 3D structures of PNP monooxygenase were
generated, and the structure with the lowest discrete opti-
mized protein energy (DOPE) score was chosen for further
analysis [20]. The stereochemical excellence of the selected
homology model was confirmed by PROCHECK analysis
[21]. The structure of each residue in the model was ensured
for their authenticity by WHATCHECK [22], ERRAT [23]
and ProSA [24].
2.2 Molecular Docking
As PNP monooxygenase catalyzes the two sequential
monooxygenations in both B. sphaericus JS905 [10] and
Arthrobacter sp. strain JS443 [11], we performed docking
studies for PNP monooxygenase from strain JS443 with the
established physiological substrates (PNP, 4-NC) and the
cofactors (FAD and NADH) for the enzyme in order to
gain insights into the most probable binding interactions.
Each atom of the protein and substrate or cofactor needs
to be fixed before molecular docking for any potential
aberrations, if any, and for accurate prediction of interac-
tions with better binding affinity. The 3D model of PNP
monooxygenase was preprocessed with the protein prepa-
ration workflow in the Maestro v9.2 [25]. Hydrogens were
added to all the atoms of PNP monooxygenase for subse-
quent minimization using OPLS 2005 force field by con-
verging heavy atoms with maximum root-mean-square
deviation (RMSD) of 0.30 A. Minimization was performed
restraining the heavy atoms with the hydrogen torsion
parameters turned off, to allow free rotation of the hydro-
gen atoms. The structures of PNP, 4-NC, FAD and NADH
(Fig. 2) were imported to Maestro v9.2. LigPrep module
along with Epik was used to prepare multiple 3D confor-
mations of the four ligand molecules in a pH range of
7 ± 2.
Molecular docking was carried out using Glide v5.7
applying extra precision (XP) method [26, 27] that gener-
ates 10000 poses for each substrate during docking and
reports the best pose based on the energy term Emodel.
Residues of PNP monooxygenase specific for substrates
and cofactors were selected to generate a grid of
20� 20� 20 A using Glide. The best poses of each ligand
were further ranked based on XP Gscore. Lower XP
Gscore for a ligand indicates better binding affinity toward
the protein. The cutoff XP Gscore parameter for XP
docking was set to 0.0 kcal/mol, a constraint set to discard
ligands with positive XP Gscore from the final docking
output. Binding affinity of 4-NC and PNP with PNP
monooxygenase was estimated using Prime/MM-GBSA
analysis.
2.3 Molecular Dynamics (MD) Simulations
MD simulations were performed for docking complexes
(PNP monooxygenase?PNP?FAD?NADH and PNP
monooxygenase?4-NC?FAD?NADH) to evaluate the
stability and conformational changes and to gain insights
into the natural dynamics on different timescales in solu-
tion. Simulations were carried out using Desmond v3.0 [28,
29], implemented in Maestro v9.2 graphical user interface.
The system was embedded with simple point charge (SPC)
water model and neutralized by replacing solvent mole-
cules with Naþ ions. The final system was simulated
through a multistep protocol devised in Maestro v9.2. In
brief, the full system was minimized with maximum 2000
interactions of a hybrid of the steepest descent and the
limited-memory Broyden–Fletcher–Goldfarb–Shanno
(LBFGS) algorithms, with a convergence threshold of
50.0 kcal/mol/A2 followed by a similar unrestrained min-
imization with a convergence threshold of 5.0 kcal/mol/A2.
The minimized system was relaxed with four subsequent
short-span simulations: (a) 12-ps simulation in NVT
ensemble (temperature 10 K) restraining nonhydrogen
solute atoms, (b) 12-ps simulation in the NPT ensemble
(temperature 10 K) restraining nonhydrogen solute atoms,
(c) 24-ps simulation in the NPT ensemble restrained with
solute nonhydrogen atoms (temperature 300 K) and
(d) 24-ps simulation in the NPT ensemble (temperature
300 K) with no restraints. The temperatures and pressures in
these initial simulations were controlled using Berendsen
thermostats and barostats, respectively. These initial mini-
mization and simulations were performed to relax the model
before implementing a longer simulation time. The relaxed
system was simulated for a simulation time of 5 ns with a
time step of 2 fs, NPT ensemble using a Nose–Hoover
thermostat at 300 K and Martyna–Tobias–Klein barostat at
1.01325 bar pressure. The simulated systems were analyzed
for stability of the docking complex. Energy fluctuations
and RMSD of the complexes in each trajectory were ana-
lyzed with respect to simulation time. The root-mean-square
fluctuations (RMSF) of backbone and side chains atoms of
PNP monooxygenase were analyzed for each residue. The
docking complex was analyzed and monitored for consis-
tency in hydrogen bonding interactions.
3 Results and Discussion
3.1 3D Model of PNP Monooxygenase
The overall sequence identity between PNP monooxyge-
nase and the template chlorophenol 4-monooxygenase of
B. cepacia AC1100 was 45 % with query coverage of
Interdiscip Sci Comput Life Sci (2015) 7:157–167 159
123
Author's personal copy
99 %. The sequence alignment for these two sequences
enabled us to identify sporadically spread conserved resi-
dues all over the sequence (Fig. 3). Conserved regions or
positions indicate residues supposedly under stronger
evolutionary constraints and thus might be more important
for the protein to fulfill its function. Moreover, residues
that are specifically conserved in subfamilies point to
sequence changes that occurred during the divergence of a
common ancestor, and they imply functional changes or the
acquisition of modified specificity [30].
Twenty PNP monooxygenase homology models were
constructed based on the template structure. All models
were assigned a predicted Genetic algorithm 341
(GA341) score and DOPE score. GA341 score of � 0.7
suggests that fold assignments in the homology model
are correct. GA341 scores for the 20 predicted models
were 1.0. Therefore, fold assignments in the homology
models were declared accurate. Selection of the best
model from the twenty was done based on DOPE score.
Measure of conformational energy is represented as
DOPE score in homology modeling. Lower DOPE score
represents relatively more stable 3D conformation [31].
Thus, the 20th model with the lowest DOPE score
(�54799.56 kcal/mol) was selected for further structural
validation.
The overall geometric and stereochemical quality of the
final modeled structure of PNP monooxygenase was ana-
lyzed using PROCHECK, which showed that 92.9 %
residues lie in the most favored region and 6.1 % in
additionally allowed regions (Fig. 4a). Evaluation of PNP
monooxygenase model with ProSA-web revealed the Z-
score value as �8.56, and the overall residue energy of
PNP monooxygenase model was largely negative except
for some peaks at the end region (Fig. 4b, c). The residue
energies of active site and allosteric site regions of the
protein were highly negative. The overall quality factor
checked from the ERRAT graph was 85.09, indicating the
acceptable protein environment (Fig. 4d). The structural
superimposition of the model of PNP monooxygenase with
the template structure showed backbone RMSD of 0.66 A.
The low backbone RMSD value represents that the pre-
dicted structure is reliable. The validated model was sub-
mitted to protein model database [32] with ID number
PM0077668 (Fig. 5).
(a)
(b) (c) (d)
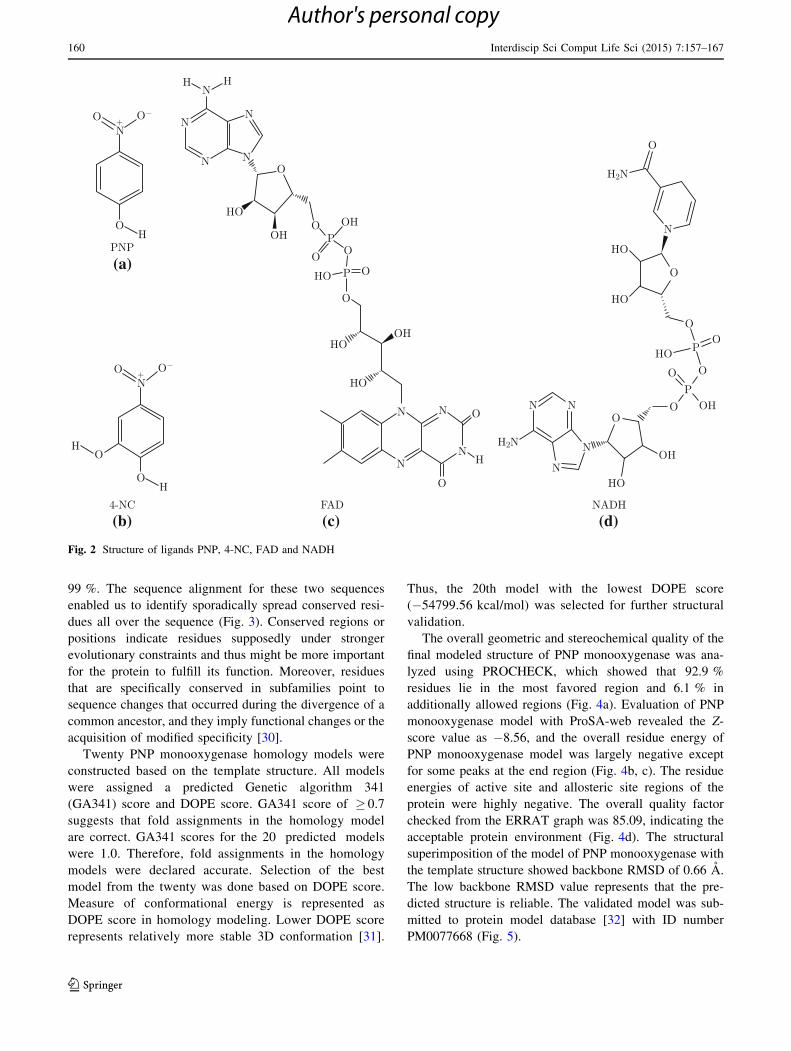
Fig. 2 Structure of ligands PNP, 4-NC, FAD and NADH
160 Interdiscip Sci Comput Life Sci (2015) 7:157–167
123
Author's personal copy

Fig. 3 Alignment of target and template using ClustalX
(a) (b)
(d)
(c)
Fig. 4 Validation of PNP monooxygenase. a PROCHECK evaluation, b energy plot obtained through ProSA-web analysis, c ProSA-web Z-plot
showing the model that corroborates with crystal structures of similar length, d ERRAT server validation
Interdiscip Sci Comput Life Sci (2015) 7:157–167 161
123
Author's personal copy

3.2 Docking of Substrates into the Active Site
of PNP Monooxygenase
Molecular docking helps in determining binding affinity,
binding orientations and various bonded and nonbonded
enzyme–substrates and enzyme–cofactors interactions in
various enzymatic reactions. Docking analysis was imple-
mented for PNP monooxygenase with its two physiological
substrates, PNP and 4-NC, and the cofactors, FAD and
NADH, to corroborate with the experimental findings of
monooxygenation reactions.
Earlier experimental findings showed that active site
residues, Arg100 and Thr193, are important for substrate
(PNP or 4-NC) binding, while allosteric site residues such
as Gln158 and Arg161 are specific for cofactor (FAD or
NADH) interaction [13]. Multiple sequence alignment of
PNP monooxygenase with homologous proteins,
chlorophenol 4-monooxygenase and hydroxyphenylacetate
hydoxylase, revealed that residues of the active site and
allosteric site are conserved.
A 20� 20� 20 A grid was created around centroid of
the active site of PNP monooxygenase for PNP and 4-NC
docking. Both the substrates, PNP (complex 1) and 4-NC
(complex 3), were docked with PNP monooxygenase at the
active site with XP Gscore of �3.434 and �3.828 kcal/-
mol, respectively. The (O) atom of hydroxyl group of PNP
formed hydrogen bond (h-bond) with residue Thr193, and
pi-cation interacted with Arg100 of PNP monooxygenase.
The hydrogen atom of one hydroxyl group of 4-NC showed
one hydrogen bond with Thr193 of PNP monooxygenase
and Arg100 involved in pi-cation interaction with 4-NC
(Table 1; Supplementary Fig. 1a, d). Similar binding ori-
entations were observed in both the substrate–PNP
monooxygenase complexes.
Further, a docking grid was generated centering on
allosteric site of the PNP–PNP monooxygenase complex
(complex 1) and 4-NC–PNP monooxygenase complex
(complex 3). The grids were generated to determine the
probable site of interactions of PNP, 4-NC, FAD and
NADH with PNP monooxygenase. Docking interactions of
PNP (complex 2) (Fig. 6a) and 4-NC (complex 4) (Fig. 6b)
with PNP monooxygenase in the presence of FAD and
NADH remained same as that of complex 1 and complex 2,
respectively (Table 1; Fig. 6a, b; Supplementary Fig. 1a,
d). Binding affinity of NADH and FAD was expressed in
terms of XP Gscore in complex 2 and complex 4 (Table 1).
NADH was observed to have more binding affinity toward
PNP monooxygenase compared to FAD in both the com-
plexes. This result corroborates well with the experimental
evidence that FAD is loosely bound to PNP monooxyge-
nase compared to NADH [10]. The docking interactions of
FAD and NADH in complex 2 revealed that FAD is
interacting with Gln158 and Arg161 through hydrogen
bonds while NADH is also forming hydrogen bond with
Arg161, and good van der Waals interaction with Gln158
of PNP monooxygenase (Fig. 6a; Supplementary Fig. 1a–
d). Similar interactions were also observed in complex 4.
FAD was observed to form intermolecular hydrogen bond
with 4-NC and good van der Waals interaction with
Gln158, while NADH was involved in intermolecular
hydrogen bond formation with Arg161 and van der Waals
interaction with Gln158 (Fig. 6b; Supplementary Fig. 1d–
f). Interaction between FAD and NADH through one
intermolecular hydrogen bond was observed in complex 2
(Fig. 6a), while that of complex 4 did not reveal any
hydrogen bond (Fig. 6b). Prime/MM-GBSA calculations
for PNP showed DG score of �28.79 kcal/mol and that of
4-NC showed DG score of �27.35 kcal/mol.
3.3 MD Simulation Studies of Docked Complexes
The molecular interactions of PNP monooxygenase with
PNP, FAD and NADH, and with 4-NC, FAD and NADH
(Fig. 6a, b) corroborated well with the experimental evi-
dences reported in the literature for the substrate and
cofactor [13]. Therefore, 5-ns molecular dynamics simu-
lations (MD) were performed for complex 2 (Fig. 7a) and
complex 4 (Fig. 7b) to understand the stability of the
interactions during PNP degradation in closer-to-natural
environmental condition. The interactions obtained through
MD simulations are more convincing than docking com-
plexes. Initially, conformational stability of complex 2 and
complex 4 was evaluated. The energy plots of complex 2
and complex 4 showed that the energy of the respective
systems were stable throughout the simulations (Fig. 8a,
b). Analysis of the RMSD plot for protein backbone,
cofactors (FAD, NADH) and the substrates (PNP, 4-NC)
Fig. 5 Three-dimensional structure of PNP monooxygenase
162 Interdiscip Sci Comput Life Sci (2015) 7:157–167
123
Author's personal copy

Table 1 Docking interactions and MD simulations of PNP, 4-NC, FAD, NADH with PNP monooxygenase
S. no. Docked molecules Hydrogen bonds Pi-cation XP
GScore
(kcal/mol)
MD simulation
Hydrogen bonds van der
Waal
interactions
1 PNP monooxygenase ? PNP (docking
in absence of FAD and NADH)
(complex 1)
Thr193, Pro152 Arg100 �3.434 Thr193, Ser151 Arg100
2 NADH (PNP monooxygenase ? PNP ?
FAD ? NADH complex) (complex 2)
Asp156, Val157, Tyr159,
Arg161, Phe448 (2)
– �9.288 Arg161, Val157,
Gln211, Gln215,
Water (19)
Gln158,
Phe148,
Asp156
3 FAD (PNP monooxygenase ? PNP ?
FAD ? NADH complex) (complex 2)
Asp156 (2), Gln158,
Tyr159, Arg161, Thr451
– �8.492 Arg161, Trp207,
Water (19)
Thr451
4 PNP (PNP monooxygenase ? PNP ?
FAD ? NADH complex) (complex 2)
Thr193, Pro152 Arg100 �3.434 Water (2) PNP monooxygenase
(Phe154, Val155, Ile191) through
one water molecule (SPC 4299)
connected to PNP
NO2 group of PNP through the path
SPC12001-SPC9446-SPC4359
connected to FAD. Similarly, NO2
group of PNP through the path
SPC12001-SPC9446-SPC3736 to
NADH
FAD and NADH are connected
through inter molecular hydrogen
bonds
5 PNP monooxygenase ? 4-NC (docking
in absence of FAD and NADH)
(complex 3)
Thr193 Arg100 �4.139 – Arg100
6 NADH (PNP monooxygenase ? 4-NC ?
FAD ? NADH complex) (complex 4)
Asp156 (2), Val157,
Tyr159, Arg161,
Phe448, Leu206
– �10.455 – Gln158,
Phe448,
Leu206
7 FAD (PNP monooxygenase ? 4-NC ?
FAD ? NADH complex) (complex 4)
Asp156, Trp207 – �8.332 Asp156 (2), Arg161,
Trp207, Arg208
Gln158
8 4-NC (PNP monooxygenase ? 4-NC ?
FAD ? NADH complex) (complex 4)
Thr193 Arg100 �4.139 PNP monooxygenase connected to
FAD through hydrogen bond; FAD
and NADH are connected through
hydrogen bonds; NADH is
connected to 4-NC through
hydrogen bond
Fig. 6 Docking interactions. a PNP monooxygenase with PNP, FAD and NADH, b PNP monooxygenase with 4-NC, FAD and NADH
Interdiscip Sci Comput Life Sci (2015) 7:157–167 163
123
Author's personal copy

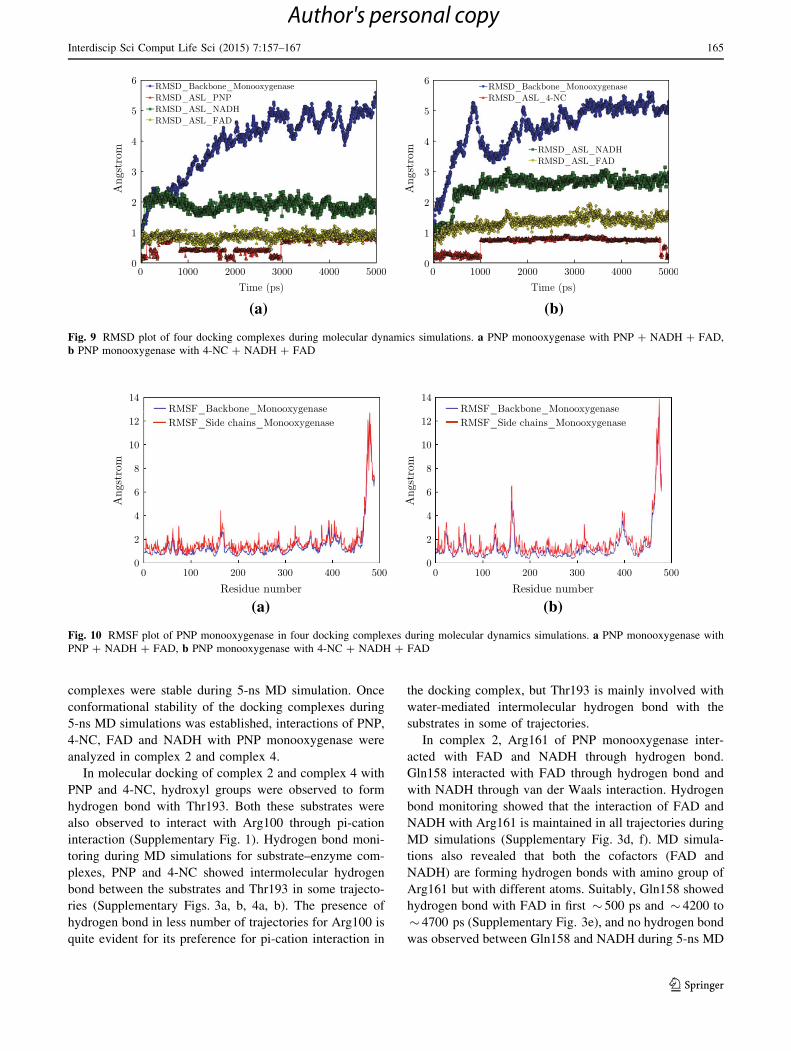
for complex 2 (Fig. 9a) and complex 4 (Fig. 9b) showed
that the RMSD was relatively consistent throughout the
MD simulations period. During 5-ns MD simulations of
both the complexes, 4-NC showed lower structural rear-
rangement with PNP monooxygenase compared to PNP.
Similarly, FAD showed less structural rearrangement when
compared to NADH. Lower RMSD of docking complex
during MD simulations represents higher conformational
stability of the docking interactions. The root-mean-square
fluctuations (RMSF) of a given residue in the MD trajec-
tory were calculated for protein backbone and all side chain
atoms of each residue (Fig. 10a, b). RMSF for most of the
residues of both the complexes were within the limit of
2.5 A. The atomic fluctuations of the active site and
allosteric site residue were consistent, and backbone resi-
dues were also consistent with small conformational peak
regions. Analysis of energy plots, RMSD plots and RMSF
plots for both the complexes revealed that the two docking
Fig. 7 MD simulations of docking complexes. a PNP monooxygenase with PNP, FAD and NADH, b PNP monooxygenase with 4-NC, FAD and
NADH
(a)
(b)
Fig. 8 Energy plot for docking
complexes during molecular
dynamics simulations. a PNP
monooxygenase with
PNP ? NADH ? FAD, b PNP
monooxygenase with
4-NC ? NADH ? FAD
164 Interdiscip Sci Comput Life Sci (2015) 7:157–167
123
Author's personal copy
complexes were stable during 5-ns MD simulation. Once
conformational stability of the docking complexes during
5-ns MD simulations was established, interactions of PNP,
4-NC, FAD and NADH with PNP monooxygenase were
analyzed in complex 2 and complex 4.
In molecular docking of complex 2 and complex 4 with
PNP and 4-NC, hydroxyl groups were observed to form
hydrogen bond with Thr193. Both these substrates were
also observed to interact with Arg100 through pi-cation
interaction (Supplementary Fig. 1). Hydrogen bond moni-
toring during MD simulations for substrate–enzyme com-
plexes, PNP and 4-NC showed intermolecular hydrogen
bond between the substrates and Thr193 in some trajecto-
ries (Supplementary Figs. 3a, b, 4a, b). The presence of
hydrogen bond in less number of trajectories for Arg100 is
quite evident for its preference for pi-cation interaction in
the docking complex, but Thr193 is mainly involved with
water-mediated intermolecular hydrogen bond with the
substrates in some of trajectories.
In complex 2, Arg161 of PNP monooxygenase inter-
acted with FAD and NADH through hydrogen bond.
Gln158 interacted with FAD through hydrogen bond and
with NADH through van der Waals interaction. Hydrogen
bond monitoring showed that the interaction of FAD and
NADH with Arg161 is maintained in all trajectories during
MD simulations (Supplementary Fig. 3d, f). MD simula-
tions also revealed that both the cofactors (FAD and
NADH) are forming hydrogen bonds with amino group of
Arg161 but with different atoms. Suitably, Gln158 showed
hydrogen bond with FAD in first � 500 ps and � 4200 to
� 4700 ps (Supplementary Fig. 3e), and no hydrogen bond
was observed between Gln158 and NADH during 5-ns MD
(a) (b)
Fig. 10 RMSF plot of PNP monooxygenase in four docking complexes during molecular dynamics simulations. a PNP monooxygenase with
PNP ? NADH ? FAD, b PNP monooxygenase with 4-NC ? NADH ? FAD
(a) (b)
Fig. 9 RMSD plot of four docking complexes during molecular dynamics simulations. a PNP monooxygenase with PNP ? NADH ? FAD,
b PNP monooxygenase with 4-NC ? NADH ? FAD
Interdiscip Sci Comput Life Sci (2015) 7:157–167 165
123
Author's personal copy
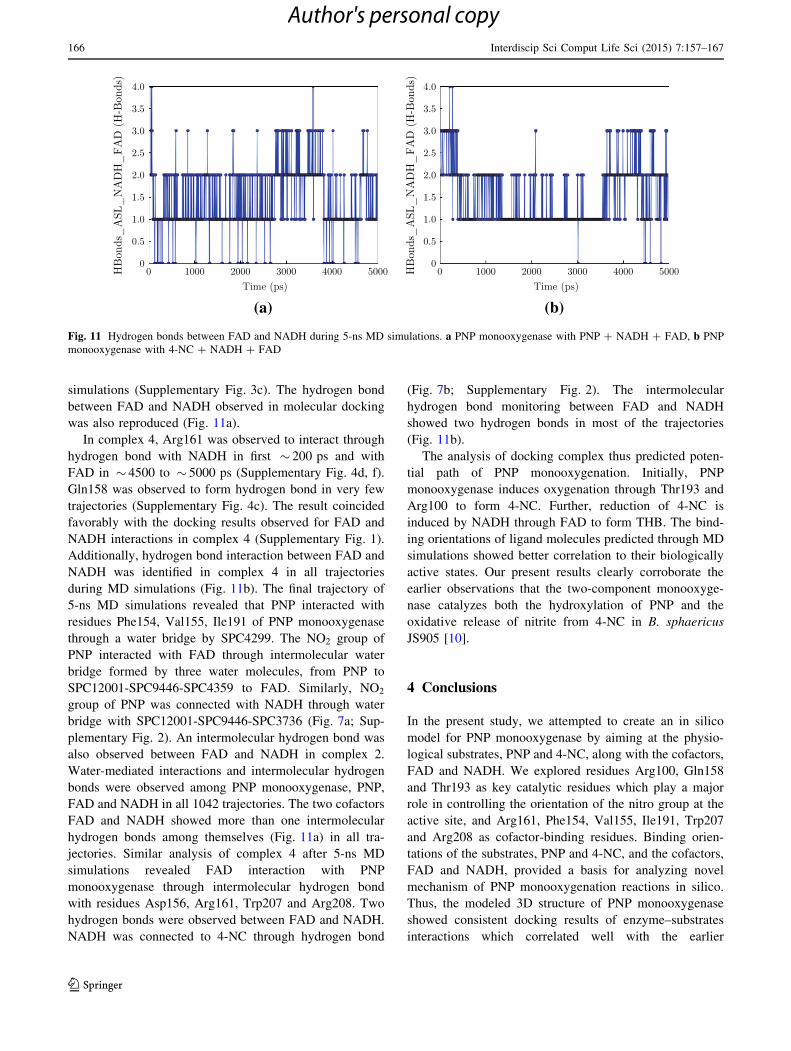
simulations (Supplementary Fig. 3c). The hydrogen bond
between FAD and NADH observed in molecular docking
was also reproduced (Fig. 11a).
In complex 4, Arg161 was observed to interact through
hydrogen bond with NADH in first � 200 ps and with
FAD in � 4500 to � 5000 ps (Supplementary Fig. 4d, f).
Gln158 was observed to form hydrogen bond in very few
trajectories (Supplementary Fig. 4c). The result coincided
favorably with the docking results observed for FAD and
NADH interactions in complex 4 (Supplementary Fig. 1).
Additionally, hydrogen bond interaction between FAD and
NADH was identified in complex 4 in all trajectories
during MD simulations (Fig. 11b). The final trajectory of
5-ns MD simulations revealed that PNP interacted with
residues Phe154, Val155, Ile191 of PNP monooxygenase
through a water bridge by SPC4299. The NO2 group of
PNP interacted with FAD through intermolecular water
bridge formed by three water molecules, from PNP to
SPC12001-SPC9446-SPC4359 to FAD. Similarly, NO2
group of PNP was connected with NADH through water
bridge with SPC12001-SPC9446-SPC3736 (Fig. 7a; Sup-
plementary Fig. 2). An intermolecular hydrogen bond was
also observed between FAD and NADH in complex 2.
Water-mediated interactions and intermolecular hydrogen
bonds were observed among PNP monooxygenase, PNP,
FAD and NADH in all 1042 trajectories. The two cofactors
FAD and NADH showed more than one intermolecular
hydrogen bonds among themselves (Fig. 11a) in all tra-
jectories. Similar analysis of complex 4 after 5-ns MD
simulations revealed FAD interaction with PNP
monooxygenase through intermolecular hydrogen bond
with residues Asp156, Arg161, Trp207 and Arg208. Two
hydrogen bonds were observed between FAD and NADH.
NADH was connected to 4-NC through hydrogen bond
(Fig. 7b; Supplementary Fig. 2). The intermolecular
hydrogen bond monitoring between FAD and NADH
showed two hydrogen bonds in most of the trajectories
(Fig. 11b).
The analysis of docking complex thus predicted poten-
tial path of PNP monooxygenation. Initially, PNP
monooxygenase induces oxygenation through Thr193 and
Arg100 to form 4-NC. Further, reduction of 4-NC is
induced by NADH through FAD to form THB. The bind-
ing orientations of ligand molecules predicted through MD
simulations showed better correlation to their biologically
active states. Our present results clearly corroborate the
earlier observations that the two-component monooxyge-
nase catalyzes both the hydroxylation of PNP and the
oxidative release of nitrite from 4-NC in B. sphaericus
JS905 [10].
4 Conclusions
In the present study, we attempted to create an in silico
model for PNP monooxygenase by aiming at the physio-
logical substrates, PNP and 4-NC, along with the cofactors,
FAD and NADH. We explored residues Arg100, Gln158
and Thr193 as key catalytic residues which play a major
role in controlling the orientation of the nitro group at the
active site, and Arg161, Phe154, Val155, Ile191, Trp207
and Arg208 as cofactor-binding residues. Binding orien-
tations of the substrates, PNP and 4-NC, and the cofactors,
FAD and NADH, provided a basis for analyzing novel
mechanism of PNP monooxygenation reactions in silico.
Thus, the modeled 3D structure of PNP monooxygenase
showed consistent docking results of enzyme–substrates
interactions which correlated well with the earlier
(a) (b)
Fig. 11 Hydrogen bonds between FAD and NADH during 5-ns MD simulations. a PNP monooxygenase with PNP ? NADH ? FAD, b PNP
monooxygenase with 4-NC ? NADH ? FAD
166 Interdiscip Sci Comput Life Sci (2015) 7:157–167
123
Author's personal copy

experimental results that the enzyme catalyzes two
sequential monooxygenation reactions to yield THB with
the concomitant release of nitro group as nitrite.
Acknowledgments KM thanks the University Grants Commission,
New Delhi, India, for the financial assistance.
References
1. Markle RA, Fentiman AF, Steadman JR, Mayer RA (1980)
Potentially toxic and hazardous substances in the industrial
organic chemicals and organic dyes in pigment industries.
National Technical Information Service, Springfield, VA.
EPA600/2-80056
2. Barik S, Sethunathan N (1978) Metabolism of p-nitrophenol in
flooded soils. J Environ Qual 7:349–352
3. Ramakrishnan B, Megharaj M, Venkateswarlu K, Naidu R,
Sethunathan N (2010) The impacts of environmental pollutants
on microalgae and cyanobacteria. Crit Rev Environ Sci Technol
40:699–821
4. Ramakrishnan B, Megharaj M, Venkateswarlu K, Sethunathan N,
Naidu R (2011) Mixtures of environmental pollutants: effects on
microorganisms and their activities. Rev Environ Contam Toxi-
col 211:63–120
5. Bruhn C, Lenke H, Hooper DJ (1987) Nitrosubstituted aromatic
compounds as nitrogen source for bacteria. Appl Environ
Microbiol 7:208–210
6. Jones SH, Alexander M (1986) Kinetics of mineralization of
phenol in lake water. Appl Environ Microbiol 51:891–897
7. Keith LH (1980) EPA priority pollutants: where they come from,
where they are going? AIChE Symp Ser 77:209–249
8. Spain JC (1995) Biodegradation of nitroaromatic compounds.
Annu Rev Microbiol 49:523–555
9. Spain JC, Gibson DT (1991) Pathway for biodegradation for p-
nitrophenol inMoraxella sp. Appl Environ Microbiol 57:812–819
10. Kadiyala V, Spain JC (1998) A two-component monooxygenase
catalyzes both the hydroxylation of p-nitrophenol and the
oxidative release of nitrite from 4-nitrocatechol in Bacillus
sphaericus JS905. Appl Environ Microbiol 64:2479–2484
11. Perry LL, Zylstra GJ (2007) Cloning of a gene cluster involved in
the catabolism of p-nitrophenol by Arthrobacter sp. strain JS443
and characterization of the p-nitrophenol monooxygenase.
J Bacteriol 189:7563–7572
12. Zhang S, Sun W, Xu L, Zheng X, Chu X, Tian J, Wu N, Fan Y
(2012) Identification of the para-nitrophenol catabolic pathway,
and characterization of three enzymes involved in the hydro-
quinone pathway, in Pseudomonas sp. 1–7. BMC Microbiol
12:27
13. Liu PP, Zhang JJ, Zhou NY (2010) Characterization of mutage-
nesis of a two-component monooxygenase involved in para-ni-
trophenol degradation by an Arthrobacter strain. Int Biodeterior
Biodegrad 64:293–299
14. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller
W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new
generation of protein database search algorithms. Nucleic Acids
Res 50:3389–3402
15. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990)
Basic local alignment search tool. J Mol Biol 215:403–410
16. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN,
Weissig H, Shindyalov IN, Bourne PE (2000) The protein data
bank. Nucl Acids Res 28:235–242
17. Chenna R, Sugawana H, Koike T, Lopez R, Gibson TJ, Higgins
DG, Thompson JD (2003) Multiple sequence alignment with the
clustal series of programs. Nucl Acid Res 31:3497–3500
18. Sali AL, Overington JP (1994) Derivation of rules for compara-
tive protein modeling from a database of protein structure
alignments. Protein Sci 3:1582–1596
19. Sali AL, Potterton F, van Vlijmen H, Karplus M (1995) Evalu-
ation of comparative protein modeling by MODELLER. Proteins
23:318–326
20. Umamaheswari A, Pradhan D, Hemanthkumar M (2010) Virtual
screening for potential inhibitors of homology modeled Lep-
tospira interrogans MurD ligase. J Chem Biol 3:175–187
21. Laskowski RA, Rullmann JA, MacArthur MW, Kaptein R,
Thrornton JM (1996) AQUA and PROCHECK-NMR: programs
for checking the quality of protein structures solved by NMR.
J Biomol NMR 8:477–486
22. Hooft R, Vriend WWG, Sander C, Abola EE (1996) Errors in
protein structure. Nature 38:272
23. Colovos C, Yeates TO (1993) Verification of protein crystal
structures: patterns of non-bonded atomic interactions. Protein
Sci 2:1511–1519
24. Tomii K, Hirokawa T, Motono C (2005) Protein structure pre-
diction using a variety of profile libraries and 3D verification.
Proteins 61:114–121
25. Maestro v9.0 (2009) Schrodinger, LLC, Portland, OR
26. Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ
(2004) Glide: a new approach for rapid, accurate docking and
scoring. 1. Method and assessment of docking accuracy. J Med
Chem 47:1739–1749
27. Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR
(2006) Extra precision glide: docking and scoring incorporating a
model of hydrophobic enclosure for protein-ligand complexes.
J Med Chem 49:6177–6196
28. Priyadarshini V, Pradhan D, Munikumar M, Umamaheswari A,
Rajasekhar D Srinivasa, Rao PVLN (2011) Docking and
molecular dynamic simulations of Legionella pneumophila MurB
reductase for potential inhibitor design. Biochem Anal Biochem.doi:10.4172/2161-1009.1000101
29. Sandeep S, Priyadarshini V, Pradhan D, Munikumar M,
Umamaheswari A (2012) Docking and molecular dynamics
simulations studies of human protein kinase catalytic subunit
alpha with antagonist. J Clin Sci Res 1:15–23
30. Lopez-Romero P, Gomez MJ, Gomez-Puertas P, Valencia A
(2004) Prediction of functional sites in proteins by evolutionary
methods. In: Kamp RM, Calvette JJ, Choli-Papadopoulou T (eds)
Principles and practice: methods in proteome and protein analy-
sis. Springer, Berlin, pp 319-340
31. Umamaheswari A, Muni Kumar M, Pradhan D, Hemanth Kumar
M (2011) Docking studies towards exploring antiviral com-
pounds against envelope protein of yellow fever virus. Interdiscip
Sci Comput Life Sci 3:64–77
32. Castrignano T, De Meo PD, Cozzetto D, Talamo IG, Tramontano
A (2006) The PMDB protein model database. Nucl Acids Res
34:306–309
Interdiscip Sci Comput Life Sci (2015) 7:157–167 167
123
Author's personal copy
Top Related
Copyright © 2022 FDOKUMEN

