







Bahasa
Halaman
Hukum


Generation of Affinity Reagents to Challenging
Targets Through Phage Display
BY
JENNIFER ELISE MCGINNIS B.S., University of Illinois at Urbana-Champaign, 2014
Thesis
Submitted as partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biological Sciences
in the Graduate College of the University of Illinois at Chicago, 2019
Chicago, IL
Defense Committee:
Brian Kay, Advisor David Stone, Chair Teresa Orenic Yury Polikanov Andrei Karginov, Pharmacology

For my mother, Marla, who has been my mentor when I have needed guidance, my
cheerleader when I have needed encouragement, and my best friend when I have needed
companionship.
ii

ACKNOWLEDGEMENTS
I would first like to thank my advisor, Dr. Brian Kay. Over the past five years, Brian
has consistently made my education his priority. He frustratingly never gave me
immediate answers to problems in the lab (even when he already knew the answer), and
instead sat with me, and talked at length until I had expanded my mind and thought of
new, creative solutions. Earning a Ph.D. is not just about becoming extremely
knowledgeable in a very specific research area. It is also about becoming a scientist: a
person capable of independent and creative thinking and using this to develop solutions
and solve problems. Because of Brian’s expertise and mentorship, I am now a budding
scientist. I only hope one day I can be as creative, intelligent, and ambitious as he is. A
single paragraph certainly does not describe how impactful Brian has been to me. I am
so honored to have been invited into his lab, so humbled to have learned so much from
him, and so thankful to have gained a lifelong friend and mentor.
I would like to thank my committee members for the guidance they have provided
throughout my studies at UIC. Dr. Stone’s thoughtful questions and high expectations
have challenged me to think critically about my experiments. Dr. Orenic has always been
there to encourage me and celebrate my successes. It has made getting through
graduate school struggles so much more manageable. Dr. Polikanov’s and Dr. Karginov’s
expertise in protein structures and protein engineering have been instrumental in guiding
the direction of my thesis and helping me interpret my experimental results.
I would not have survived graduate school without all the members of the Kay lab,
both past and present. They have provided forever treasured guidance, friendship, and
iii

support. Specifically, I would like to acknowledge Dr. Kevin Gorman and Dr. Leon
Venegas who took time out of their busy days to train me and always answer my
questions. Sehar Khosla began this journey with me. She was there to celebrate my
successes and encourage me after my failures. Christina Miller has brought so much joy
into the lab this past year. Between all our hard work, our inside jokes and funny moments
have lessened my stress while preparing to graduate. I do not doubt that we will be friends
for life.
Finally, I want to thank my close friends and family. Stephanie Czarnik, Christina
Norman, and Grace Aldrich are always there when I need support, love, or just a good
laugh. My stepfather and stepmother have spoiled and treated me as if I was one of their
own. Most importantly, my mother, father, and sister have made sure I am surrounded by
love, happiness, and encouragement every single day of my life. From piano recitals and
gymnastics meets, to presentations and graduations, they have been there through it all,
both big and small, cheering me on. Thank you!
iv

CONTRIBUTION OF AUTHORS
Chapter 1 is a literature review of the affinity reagent generation and protein engineering
fields. Portions of section 1.5 Phage Display were previously published in:
Gorman, K., McGinnis, J. & Kay, B. (2018) Generating FN3-Based Affinity
Reagents Through Phage Display, Current Protocols in Chemical Biology. 10,
e39.
Dr. Kevin Gorman provided figures 3 and 4 and contributed to writing the protocol section
of the manuscript. The portion of the work duplicated in this thesis can be found in the
commentary section of the manuscript and was written by me and edited by my research
mentor, Dr. Brian Kay.
Chapter 2 represents the published manuscript:
McGinnis, J. E. & Kay, B. K. (2018) Generation of recombinant affinity reagents
against a two-phosphosite epitope of ATF2, New biotechnology. 45, 45-50.
for which I is was the primary author and main researcher. Dr. Kay contributed to
experimental design and editing of the manuscript.
Chapter 3 represents the published manuscript:
McGinnis, J. E., Venegas, L. A., Lopez, H. & Kay, B. K. (2018) A Recombinant Affinity
Reagent Specific for a Phosphoepitope of Akt1, Int J Mol Sci. 19, 3305.
for which I was the primary author and main researcher. Dr. Venegas contributed figures
2, 3, and 5 and wrote the methods section of the manuscript. Dr. Kay contributed to
experimental design and editing of the manuscript.
Chapter 4 represents my unpublished experimental results that contribute to improving
v

diagnostic and biochemical sandwich assays. With the addition of a few experiments, I
presume this work will soon be published as a co-authored manuscript.
Chapter 5 summarizes the work presented in this dissertation, potential future
experiments, and the overall impact of this work.
Dr. Brian Kay has been instrumental in editing each chapter and providing financial
(through federal grants) and intellectual support for this dissertation.
vi

TABLE OF CONTENTS
CHAPTER PAGE
1. INTRODUCTION .............................................................................. 1
1.2 Applications of Antibodies .......................................................... 1
1.3 Targets of Affinity Reagents ...................................................... 3
1.4 Methods for Generating Affinity Reagents ................................. 5
1.5 Phage Display ........................................................................... 9
1.6 Displayed Proteins ................................................................... 13
1.7 Thesis Goals ............................................................................ 19
1.8 Literature Cited ........................................................................ 22
2. GENERATION OF RECOMBINANT AFFINITY REAGENTS AGAINST A TWO-PHOSPHOSITE EPITOPE OF ATF2 ................ 35
2.1 Abstract ................................................................................... 35
2.2 Introduction .............................................................................. 36
2.3 Materials and Methods ............................................................ 38
2.3.1 Reagents ........................................................................ 38
2.3.2 Cloning and Bacterial Expression ................................... 39
2.3.3 Enzyme-Linked Immunosorbent Assay (ELISA) ............. 41
2.3.4 Affinity Selection of the Primary Library .......................... 41
2.3.5 Secondary Library Construction and Affinity Selection ... 43
2.3.6 Western Blot ................................................................... 43
2.4 Results and discussion ............................................................ 44
2.4.1 The FHA Domain Scaffold Can Recognize a Dual-Phosphorylated Epitope ................................................. 44
2.4.2 Characterization of the Interaction Between an FHA
Variant Selective for the Dual-Phosphorylated ATF2
Peptide ........................................................................... 44
2.4.3 Generation of FHA Affinity Reagents to Specifically
Recognize the Mono-Phosphorylated Forms of the
ATF2 Peptide ................................................................. 50
2.4.4 Using an FHA Reagent to Detect Phospho-ATF2
in a Western Blot ............................................................ 53
2.5 Conclusions ............................................................................. 55
2.6 Literature Cited ........................................................................ 56
vii

TABLE OF CONTENTS (continued)
CHAPTER PAGE
3. A RECOMBINANT AFFINITY REAGENT SPECIFIC FOR A PHOSPHOEPITOPE OF AKT1 ...................................................... 60
3.1 Abstract ................................................................................... 60
3.2 Introduction .............................................................................. 60
3.3 Materials and Methods ............................................................ 62
3.3.1 Peptides ......................................................................... 62
3.3.2 Cloning and Bacterial Expression of Proteins................. 63
3.3.3 Affinity Selections ........................................................... 63
3.3.4 Enzyme-Linked Immunosorbent Assay (ELISA) ............. 65
3.3.5 Surface Plasmon Resonance ......................................... 65
3.4 Results and Discussion ........................................................... 65
3.4.1 Directed Evolution of the FHA1 Domain Yielded ... Variants that Recognize an Akt1 Phosphopeptide ....................... 65
3.4.2 An Isolated FHA Clone binds the Akt1 Peptide with Unique Specificity ....................................................................... 70
3.5 Conclusions ............................................................................. 74
3.6 Literature Cited ........................................................................ 75
4. STREAMLING THE VALIDATION PROCESS FOR BINDING REAGENTS ISOLATED BY MEGASTAR ...................................... 79
4.1 Abstract ................................................................................... 79
4.2 Introduction .............................................................................. 80
4.3 Materials and Methods ............................................................ 84
4.3.1 Cloning and Overexpression of FN3 Fusion Proteins ..... 84
4.3.2 Isolation of binding pairs by MegaSTAR ........................ 85
4.3.3 Sandwich ELISA with FN3 fusion binding pairs .............. 85
4.3.4 Monobody labeling via sortase reaction ......................... 86
4.3.5 Sandwich ELISA with monobodies labeled ....... via sortase reaction .......................................................................... 86
4.4 Results and Discussion ........................................................... 87
4.4.1 MegaSTAR is robust and reproducible ........................... 87
4.4.2 Improving binding pairs by assay development .............. 87
4.4.3 Easy Use of Pairs for Heterogenous Assays .................. 93
4.5 Conclusions ............................................................................. 97
4.5 Literature Cited ........................................................................ 98
viii

TABLE OF CONTENTS (continued)
CHAPTER PAGE
5. CONCLUSIONS ........................................................................... 102
5.1 Thesis Summary .................................................................... 102
5.2 Future Directions ................................................................... 104
5.2.1 Incorporate Multiple Scaffolds and Linker ....... Lengths into MegaSTAR ................................................................... 104
5.2.2 Generating Reagents to Cell-Free Expressed ........ Targets ..................................................................................... 106
5.2.3 Use of Sortase-Mediated Ligation to Format ......... Pairs for Homogeneous Assays ...... Error! Bookmark not defined.
5.3 Overall Impact ....................................................................... 110
5.7 Literature cited ....................................................................... 112
Chapter 6 APPENDIX ................................................................................... 114
Chapter 7 VITA ............................................................................................. 117
ix

LIST OF TABLES
TABLE PAGE
I. IN VITRO DISPLAY TECHNOLOGIES ........................................................... 8
II. ALTERNATIVE BINDING SCAFFOLD PROTEINS FOR ...... PHAGE DISPLAY ......................................................................................................................15
III. LIST OF PRIMERS AND THEIR SEQUENCES USED IN THIS STUDY .......40
IV. ATF2 PHOSPHOPEPTIDES ........................................................................45
V. OUTPUT SEQUENCES OF CLONES ISOLATED FROM SELECTIONS ......52
VI. AFFINITY MEASUREMENTS ........................................................................69
VII. SANDWICH ELISA METRICs WITH MONOBODY FUSION .................. PAIRS ......................................................................................................................92
x

LIST OF FIGURES
FIGURE PAGE
1.1. The M13 phage particle ............................................................................. 9
1.2. The M13 phage life cycle ......................................................................... 11
1.3. Generation of affinity reagents through phage display. ............................ 12
1.4. Antibody and antibody variant structures. ................................................ 14
1.5. FHA1 interacts with phosphothreonine containing peptides through its β loops. .................................................................................. 18
1.6. PyMOL representations of the FN3 monobody ........................................ 19
2.1. FHA variants can recognize the dual-phosphorylated ATF2 ............ epitope ................................................................................................................. 46
2.2. The FHA library contains variants that recognize a pT-X-p(S/T) motifs ................................................................................... 46
2.3. Alanine-scanning of β4-β5 and β10-β11 loop residues from FHAαATF2 F6 ................................................................................. 48
2.4. Determining FHA variant F6′s affinity for its target ................................... 50
2.5. Generation of reagents that specifically recognized the mono-phosphorylated targets ............................................................................ 51
2.6. Detection of full-length ATF2 in a western blot ........................................ 54
3.1. Primary structure of the Akt1 protein ....................................................... 66
3.2. Affinity selection process and ELISA of 12 output clones ........................ 66
3.3. Amino acid sequence analysis of two loops randomized in the phage-displayed scaffold ............................................................... 68
3.4. Comparing the relative affinity of four clones ........................................... 69
3.5. Binding of an FHA variant to a set of related peptides ............................. 70
xi

LIST OF FIGURES (continued)
FIGURE PAGE
3.6. Identification of important residues on the peptide by alanine scanning. ................................................................................................................. 72
3.7. Binding of an FHA variant to corresponding peptides from Akt2 and Akt3 ................................................................................................................. 73
4.1. Sandwich ELISA ...................................................................................... 81
4.2. Megaprimer shuffling for tandem affinity reagents ................................... 83
4.3. Loop sequences of tandem clones isolated against COPS5 ................... 88
4.4. Monobody fusion proteins for improved sandwich assays ....................... 90
4.5. Sandwich ELISA with NanoLuc readout .................................................. 91
4.6. Sandwich ELISA with alkaline phosphatase (AP_ readout ...................... 92
4.7. Site-specific monobody labeling via sortase-mediated ligation ............... 94
4.8. Sortase-mediated ligation efficiency ........................................................ 95
4.9. Sandwich ELISA with monobody binding pairs labeled via ............ sortase-mediated ligation ..................................................................................... 96
4.10. Workflow for identifying and characterizing binding reagents through MegaSTAR .............................................................................................. 97
5.1. An array of tandem vectors for improved binding pairs .......................... 105
5.2. Target immobilization via HaloTag technology ...................................... 106
5.3. Homogeneous assays utilizing sortase-mediated ligation...................... 108
5.4. Generating recombinant enzyme labels for sortase-mediated ligation .. 109
xii

LIST OF ABBREVIATIONS
A Alanine
Ab Antibody
ABTS 2,2-azinobis(3-ethylbenzthiazoline-6–sulfonic acid)
Akt1 Protein kinase B
ATF2 Activating Transcription Factor 2
CB Carbenicillin
CDR Complementarity-determining region
COPS5 Cop9-signalosome subunit 5
DARPin Designed ankyrin repeat protein
DNA deoxyribonucleic acid
dsDNA double-stranded DNA
E. coli Escherichia coli
ELISA Enzyme linked immunosorbent assay
epPCR error-prone Polymerase Chain Reaction
ERK Extracellular-regulated kinase
Fab Fragment antigen-binding
FHA1 Forkhead-associated I domain
FN3 Fibronectin type III domain
G Glycine
HRP Horseradish peroxidase
IgG Immunoglobulin G
xiii

KN Kanamycin
M13KO7 Helper phage
mAb Monoclonal antibody
MegaSTAR Megaprimer Shuffling for Tandem Affinity Reagents
MEK Mitogen-activated protein kinase-kinase
Tm Melting temperature
p38 p38 mitogen-activated protein kinases
pAb Polyclonal antibody
PBS Phosphate buffered saline
PBST Phosphate buffered saline with 0.1% Tween
PCR Polymerase Chain Reaction
PDB Protein Data Bank
PEG Polyethylene glycol
PNK Polynucleotide kinase
pS Phosphoserine
pT Phosphothreonine
PTM Post-translational modification
pY Phosphotyrosine
Raf Raf protein kinase
RalGDS Ral guanine nucleotide dissociation stimulator
S Serine
scFv Single-chain variable fragment
SDS-PAGE Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
xiv

SPR Surface Plasmon Resonance
ssDNA single-stranded DNA
SUMO Small ubiquitin-like modifier
T Threonine
TM13KO7 Trypsin cleavable helper phage
WT Wild-type
Y Tyrosine
xv

Summary
Antibodies are essential tools in both the research and medical communities, which
can be utilized in a variety of biochemical experiments, diagnostic assays, and therapies.
However, they sometimes are cross-reactive in specificity, have a modest affinity for their
target, and are time consuming and expensive to produce. In recent years, alternative
methods for generating recombinant affinity reagents have overcome many of the
limitations of polyclonal and monoclonal antibodies. During my Ph.D. research, I applied
the phage display technology to generate high-quality affinity reagents to a variety of
phosphopeptides and folded proteins and demonstrated their utility in biochemical
assays. Furthermore, I have designed and successfully implemented several
experimental strategies to reduce the cost needed to generate and validate these
reagents.
Antibodies have been extremely useful probes to monitor post-translational
modifications, such as protein phosphorylation in cells. Protein phosphorylation provides
a method of activating or inhibiting proteins, altering their cellular location, modulating
their stability, or facilitating protein-protein interactions. Unfortunately, many of the
antibodies generated against peptides carrying phosphoserine or phosphothreonine are
cross-reactive, which limits their usefulness.
To overcome the limitations of phosphospecific antibodies, I screened a library of
phage that displayed variants of the engineered Forkhead Associated (FHA) domain to
generate recombinant affinity reagents that bind selectively to several different
phosphopeptides of human proteins. First, I generated FHA reagents to phosphopeptides
xvi

SUMMARY (continued)
of Activating Transcription Factor 2 (ATF2), a transcription factor involved proliferation,
apoptosis, and DNA repair. Because ATF2 contains two neighboring phosphothreonines,
my goal was to isolate reagents that distinguish between its various phosphostates. I was
successful in developing through directed evolution one affinity reagent that specifically
recognized the ATF2 peptide when di-phosphorylated and another that only recognized
one of the two mono-phosphorylated states. On the contrary, a commercial antibody to
the di-phosphopeptide was cross reactive between all three phosphopeptides, illustrating
that phage display can be a better source for generating high quality, phosphospecific
reagents. Additionally, I showed that an engineered FHA domain could detect native,
phosphorylated ATF2 protein in a western blot.
I also generated FHA affinity reagents to a phosphopeptide from Akt1, a protein that
is observed to be phosphorylated in many cancer patients. Not only were these reagents
uniquely specific for the peptide - capable of distinguishing between Akt1 and its highly
conserved isoforms - but they also had high affinity for the Akt1 phosphopeptide,
illustrating their potential success in a variety of different assays.
In the last portion of my Ph.D. research, I focused on developing affinity reagents that
work as binding pairs in sandwich assays. The sandwich assay requires two separate
affinity reagents to bind an analyte simultaneously, which substantially lowers false
positive results. Using a phage library displaying engineered fibronectin type III (FN3)
monobodies, I isolated 6 different pairs with a new technique in the laboratory,
Megaprimer Shuffling for Tandem Reagents (MegaSTAR). I streamlined the validation of
xvii

SUMMARY (continued)
pairs isolated by MegaSTAR by eliminating the need to purify large quantities of each
binding reagent separately, which reduces time and cost, and allows testing of more pairs
compared to past methods. Additionally, I labeled binding reagents in vitro with various
tags through site-specific, enzymatic ligation, making these reagents suitable for
heterogenous and homogenous sandwich assays.
While assays with pairs of antibodies are very common in basic research and
diagnostics, they are difficult to develop, and they depend on a steady supply of each
antibody. The phage display technology provides an alternative method to generate high
quality binding reagents quickly and inexpensively. My work advances this technology in
generating binding reagents against challenging targets and improving their utility in
assays.
xviii

1
Chapter 1 1. INTRODUCTION
Portions of this chapter have been published in Current Protocols in Chemical Biology
[1].
This work was done in collaboration with Dr. Kevin Gorman and Dr. Brian Kay. Dr. Kevin
Gorman provided figures and helped write the protocol section of the article. Dr. Kay
helped with editing.
1.1 Antibodies and Antigens
Antibodies are plasma proteins that are naturally produced by the immune system in
response to foreign agents that have entered the body. They circulate throughout the
blood and bind to these foreign agents, termed “antigens”, and render them inactive
through neutralization, aggregation, or internalization by cells. Antibodies protect the body
from harmful pathogens, viruses, and infections [2, 3]. The immune system has the ability
to generate billions of antibodies that vary in amino acid sequence at their antigen-binding
sites, which allows them to recognize a multitude of antigens [4].
1.2 Applications of Antibodies
Probes that recognize biological targets are invaluable tools for biomedical and basic
laboratory research and have assisted in analysis of the structure and function of the cell
[5]. The most commonly used probes of proteins are antibodies due to their natural
binding properties. Antibodies are used in a variety of biochemical assays such as
western blots [6], enzyme linked immunosorbent assays (ELISA) [7], and pull-downs [8]
to detect or quantify particular proteins in complex mixtures, such as blood or cell lysates.
Additionally, scientists have employed antibodies in immunohistochemistry [9] and
immunofluorescence [10] experiments to detect particular proteins in fixed cells or tissue
samples. An antibody’s ability to selectively recognize and bind these proteins provides

2
information regarding the protein’s location within a cell or tissue and insight into the
protein’s possible function.
Antibodies also serve a crucial role in diagnostics. While doctors rely on a patient’s
symptoms and medical history to assess their illness, without confirmatory tests, the
patient could be misdiagnosed. Many diseases are caused by similar environmental and
genetic factors and/or produce similar symptoms, but require different treatments [11].
Antibodies help in such a situation because they can monitor biomarkers in a patient’s
blood, serum, urine, stool, or tissue and indicate a pathological biological process is taking
place in the body as a result of a specific disease [12]. Biomarkers can range from the
presence, absence, or change in levels of specific antigens in the body, a change in a
normal enzymatic activity, neoepitopes generated by genetic mutations, post-
translational modifications, or a change in a protein’s conformation [13, 14]. There are
currently many tests where biomarkers are detected with antibodies. These include tests
for pregnancy [15, 16], cancer [17-19], myocardial infarction [20], and bacterial and viral
infections [21, 22]. Antibodies have been instrumental in detecting and monitoring the
progression of a disease, as well as monitoring a patient’s response to treatment [11].
In recent years, antibodies have also served as therapeutics to treat various diseases,
such as inflammatory, respiratory, and cardiovascular diseases, cancer, and viral and
bacterial infections [23]. Humira (adalimumab), an anti-tumor necrosis factor alpha
(TNFα) antibody, is one of the most popular pharmaceuticals, with global sales of $18
billion in 2017. Humira has been shown to treat a variety of autoimmune diseases,
including rheumatoid arthritis [24], plaque psoriasis [25], and Crohn’s disease [26].
Antibody therapies have become a leading product in the biopharmaceutical industry,

3
with > 30 antibody therapies in the market, contributing to > $100 billion in global sales in
2017. With hundreds more in development and clinical trials, it is likely that global sales
of therapeutic antibodies will grow substantially in the future [27, 28].
Antibody therapeutics rely on several different mechanisms to combat diseases and
infections. The natural functions of antibody-mediated immune responses have been
exploited by many of these reagents. One such way is through their neutralization
capabilities; in this case, antibodies bind a hormone or receptor, blocking a cell signaling
pathway that promotes proliferation, growth, stress responses, etc. [23, 29-31]. Other
reagents utilize an antibody-dependent cell-mediated cytotoxic (ADCC) response in
which the antibody binds the antigen on the cell surface and also recruits immune effector
cells to lyse and destroy the target cell [32, 33]. Of course, researchers have not limited
themselves to these natural methods of action and have further engineered antibodies to
drive certain responses. For example, reagents conjugated to a toxin or other drug guide
their selective and efficient degradation of the antigen without harming the rest of the
body [34]. Other antibodies have been genetically engineered to bind two targets
simultaneously, such as antigens on tumor and immune cells, to drive the immune
response by forcing the antigen and immune cell into proximity [35]. Many of these
approaches have been clinically effective and will likely be exploited more in the future.
1.3 Targets of Affinity Reagents
Affinity reagents that detect and track protein post-translational modifications (PTMs)
are extremely useful in research. PTMs such as phosphorylation, methylation,
acetylation, glycosylation, ubiquitination, lipidation, and nitrosylation influence almost all
aspects of cell biology [36-41]. For example, affinity reagents that monitor protein

4
phosphorylation are of great interest because phosphorylation plays a crucial role in
regulating, activating, or inhibiting a protein, altering a protein’s cellular location, signaling
a protein for degradation, or creating or destroying specific binding interactions [39, 40].
Over 100,000 phosphosites have been mapped on > 13,000 human proteins [42, 43],
illustrating their significance in cell function, growth, and survival.
Most proteins have multiple phosphorylation sites, and the effects of these
modifications cannot be fully understood by examining only individual phosphorylation
sites for a protein [44]. Many signaling pathways depend on multi-site phosphorylation
events to maintain proper temporal regulation of cells as well as regulate a complex
system of signaling cascades. Thus, affinity reagents that selectively recognize
phosphoproteins in their various phospho-states are needed to study the triggers, timing,
and downstream effects of these events.
Reagents with this level of specificity are challenging to find, especially for multiple
phosphorylation sites in proximity to one another in a protein. Cells contain a “priming”
protein kinase that phosphorylates one residue in a protein, which leads to
phosphorylation of a nearby residue by a second protein kinase. A classic example
highlighting this mechanism involves phosphorylation of target proteins by Glycogen
Synthase Kinase 3 (GSK-3). A priming kinase phosphorylates one residue of a GSK-3
substrate, whereupon GSK-3 binds the priming phosphate group (through a positively
charged pocket adjacent to the kinase’s active site) and then phosphorylates a serine or
threonine four residues N-terminal of the priming phosphate [45]. Depending on the
priming site, GSK-3 has different affinities for its targets, leading to differences in the
efficiency and timing of how quickly a target is phosphorylated [46]. While mass

5
spectrometry is a useful tool in determining the timing of each of these phosphorylation
events, it does not provide information about the location of these events in cells and
tissue. Alternatively, affinity reagents to particular PTM epitopes provide both spatial and
temporal information regarding the phosphorylation of particular residues in proteins.
There is a considerable need for affinity reagents that recognize specific
conformational epitopes as well. Changes in a protein’s conformation can result from a
variety of factors along with genetic changes including temperature, pH, post-translational
modifications, binding of a ligand (i.e., allostery), solvent polarity, and ion concentration
[47-49]. Thus, reagents that recognize a conformational epitope are useful for monitoring
the effects of these factors on a protein’s structure or determining if a protein is denatured
or misfolded. Additionally, many diseases, such as neurodegenerative disorders and
cancers, are caused by protein misfolding, thereby rendering these proteins toxic [50, 51].
For example, in Huntington’s disease, the huntingtin protein is misfolded and interacts
with other correctly folded copies of itself to catalyze their transition to the toxic
configuration [52]. Other neurodegenerative disorders including Alzheimer’s disease,
dementia, and amyotrophic lateral sclerosis (ALS) progress through similar paths. Affinity
reagents that can distinguish between these native and misfolded proteins have been
instrumental tools in studying samples from deceased patients [53]. Additionally, affinity
reagents that recognize epitopes only found in misfolded proteins have potential
therapeutic value for sequestering the misfolded, but not the native, forms of proteins.
1.4 Methods for Generating Affinity Reagents
Traditionally, antibody generation requires immunizing an animal with an antigen,
which induces the animal’s immune system to produce immunoglobulin subtype G (IgG)

6
antibodies specific for that antigen. After waiting several weeks for the response to occur,
the animal’s serum can be harvested, and the desired antibodies purified by affinity
chromatography (via the immobilized antigen) [54-56]. This pool of antibodies, described
as polyclonal, contains a population of IgGs that can be used in various biochemical
assays. While polyclonal antibodies have been critical tools for research, they have their
limitations. Polyclonal antibodies are not renewable and are only regenerated by
repeating the immunization process. Not only is this time-consuming and expensive, but,
but the quality, specificity, and affinity of polyclonal antibodies vary from batch to batch,
sometimes leading to experimental irreproducibility [57].
Rather than harvesting the animal’s serum, scientists discovered they could isolate
antibody-producing B-cells from the spleen and fuse them to immortal, myeloma cells to
create “hybridomas” [58]. The creation of these hybrid cells eliminates some of the major
issues with polyclonal antibodies. First, because these cells are immortal, they secrete
antibodies as they grow and divide in culture flasks, eliminating the constant need for
animal immunization. Additionally, as each B-cell only secretes a single antibody, the
antibodies produced by the hybridoma will also only have a single amino acid sequence,
also known as a monoclonal antibody.
Monoclonal antibodies still have their limitations, though. Ideally, they are perpetually
secreted from hybridomas and do not vary from batch to batch as they have a single
identity. Unfortunately, this is not usually the case. After many passages, genetic drift and
mutations can occur in the monoclonal antibody or cells stop secreting the antibody [59].
Furthermore, a recent study analyzed over 185 hybridoma cell lines and discovered that
over 30% secreted two or more antibodies. While it is unclear why this happens - perhaps

7
due to multiple B-cells fusing to generate heterokaryons or innate characteristics of the
myeloma cells – a mixture of monoclonal antibodies leads to off-target binding and
decreases the binding signals for the desired target [60].
To circumvent the problems that arise from monoclonal antibody generation, in vitro
methods for antibody generation has been developed. In vitro display methods have
taken advantage of the polymerase chain reaction (PCR) to amplify and clone millions of
antibody genes (See section 1.5 for types of recombinant antibody libraries) and “display”
them recombinantly. [61-64]. These recombinant antibody libraries can then be screened
against targets of interest without animal immunization [65, 66]. Unlike, the traditional
methods of antibody generation, recombinant antibodies provide complete control over
screening conditions including antigen conformation, buffer environment, and the addition
of competitors to drive binding towards a specific epitope. Additionally, because these
libraries are recombinantly expressed, alternative, non-antibody proteins, or scaffolds,
with unique characteristics can be used in place of antibody libraries (See section 1.5 for
types of scaffolds and their advantages) [67, 68]. This increases the probability of isolating
reagents with high specificity and affinity for antigens.
Most significantly, display technologies, notably phage display [69, 70], yeast display
[71-73], mRNA display [74], and ribosome display [75] link each antibody or binding
protein variant to its corresponding DNA or RNA sequence through several genetic fusion
methods (Table 1). By directly attaching each variant’s genotype to its phenotype, the
primary structure of selected binders can be quickly identified by DNA sequencing.
Because of this advantage, the reagents are renewable and inexpensive to produce by
recombinant expression in bacteria or Chinese Hamster Ovary (CHO) cells. Furthermore,

8
the display reagents can be engineered by directed evolution to increase their specificity
and affinity [76-78]
.Table I. IN VITRO DISPLAY TECHNOLOGIES
TABLE I. IN VITRO DISPLAY TECHNOLOGIES
Name Display Methodology Recovery Average library
size
mRNA display
Recombinant protein with puromycin-mRNA fusion incorporated into C-terminal end
mRNA from selected binders is converted to cDNA and expressed as soluble protein for characterization
1013
Phage display
Recombinant protein genetically fused to bacteriophage coat protein that displays on surface of phage. Phage houses genome containing recombinant protein gene
Recovered virions infect E. coli cells and utilize their quick growth rate to propagate more bacteriophage
1010
Ribosome Display
Recombinant protein attached to ribosome-mRNA complex due to addition of a genetic spacer that prevents release factors from binding and triggering disassembly during translation
mRNA from selected binders is converted to cDNA and expressed as soluble protein for characterization
1013
Yeast Display
Recombinant protein genetically fused to yeast surface protein, and thus, displayed on the surface of yeast cells that house genome containing recombinant protein gene
Cell sorting 107

9
1.5 Phage Display
Since its inception in 1985 [65], researchers have spent a great amount of effort to
improve and expand upon the phage display technique. In fact, in 2018 George Smith
won the Nobel Prize in Chemistry for his invention of phage display. It is now a mature
tool that is used to generate binding partners (i.e., peptides, antibody fragments, scaffold
proteins, protein fragments) for a wide variety of targets. Bacteriophage M13 is the most
popular vehicle for display because its structure is well understood [79]. The virion houses
a circular, single-stranded DNA that is covered with five coat proteins: 2700 copies of
pVIII and five copies each of pIII, pVI, pVII, and pIX (Figure 1.1) [80, 81].
Figure 1.1. The M13 phage particle. Cartoon of an M13 phage particle with its capsid
proteins as well as its single stranded genome. Copy numbers are listed per virion.

10
The life cycle of the M13 bacteriophage begins with infection by protein III (pIII) of a
virion binding the F pilus of a male E. coli cell [80]. This interaction leads to a pore forming
on the bacterial cell surface that allows the virion’s DNA to enter the cell, where it is
converted into double-stranded DNA, and synthesis of the M13 proteins begins [82]. Coat
proteins, are co-translationally inserted into the periplasm, while new, single stranded
phage DNA is synthesized and coated by pV, a single-stranded DNA-binding protein [80,
82]. The protein-coated, single-stranded DNA then interacts with export machinery,
including other capsid proteins, to assemble into virions that are secreted from the cell
(Figure 1.2) [83-85].
In phage display, the coding region for a peptide, an antibody fragment, or scaffold
protein is fused to the coding region of one of the five capsid proteins by molecular
cloning, and when the chimeric gene is transcribed and translated, it is assembled into
the virion and displayed on the viral particle surface. Each of the five coat proteins has
been successfully used for display, with pIII and pVIII protein fusions being the most
commonly used [65, 86-91]. Each coat protein has its distinct advantages and
disadvantages. In a virion, there are 2700 copies of pVIII. This is useful for displaying
peptides that have weak affinity for their targets because they will be present at a high
copy number on each virion which can create an avidity effect (increased apparent
affinity). However, only small peptides can be displayed on all 2700 copies of pVIII without
steric hindrance. Conversely, in a virion there are five copies of pIII, and it tolerates display
of large peptides and proteins. This also helps in the discrimination of tight binders
because the avidity effect is greatly reduced [70].

11
Figure 1.2. The M13 phage life cycle. The M13 virion interacts with the F-pilus on the
bacterial cell membrane, allowing the virion to insert its single-stranded DNA (ssDNA)
into the bacterial cell. The M13 genome is converted to dsDNA by host cell machinery
and transcription/translation of phage proteins is initiated. Additionally, single stranded
copies of the M13 genome are synthesized through rolling circle amplification. Once a
sufficient amount of the capsid protein, pV, has been synthesized, it coats the M13
ssDNA, preventing its conversion to dsDNA. The pV-DNA complex is recognized by
phage proteins, which have already accumulated in the inner cell membrane, and it is
exported to the periplasm. Here, pV is removed and capsid proteins assemble around the
DNA genome to secrete a mature virion from the bacterial cell. Not to scale.

12
For affinity selection experiments, a phage library is first mixed with a target of interest
(Figure 1.3). Phage variants that do not bind or bind weakly are washed away, while
variants that are bound tightly are eluted and amplified for subsequent rounds of
selection. After two or three rounds of affinity selection (each increasing in stringency with
additional washes, less target, and/or negative selection steps), individual clones are
tested for binding to the target in an ELISA. Because the gene coding for the binding
element protein is encoded in the phage genome, the identity of “positive hits” is deduced
by DNA sequencing.
Figure 1.3. Generation of affinity reagents through phage display. A phage library is
incubated with immobilized target. Non-binding phage are washed away, the remaining
phage are eluted, amplified, and subjected to further rounds of selection. Individual clones
are examined for binding to the target by ELISA. It typically takes 2-3 weeks to go from
target to isolation of binding phage clones.

13
Over the years, the phage display system has been modified. In the first version of
phage display, the fusion protein or peptide was displayed from every copy of the coat
protein to which it was fused [65]. To circumvent the avidity effect caused by this type of
multivalent display, phagemid systems were developed. Phagemids are plasmids
containing the capsid fusion sequence, bacteria and phage origins of replication, and the
phage packaging signal. However, they lack coding for all other phage proteins needed
for assembly and release. Upon infection of bacterial cells with helper phage, which
supplies in trans the other viral proteins, replication and virion assembly begins [92]. The
“3+3 display system" allows for a small number of copies of the chimeric coat protein to
be displayed along with the wild-type capsid protein [93].
The phagemid display technique has been applied to a variety of directed evolution
experiments. Not only have they been utilized for affinity reagent generation, but also to
improve protein stability [94], generate inhibitors [95], provide insight into potential protein
binding interactions [96], and map transcription factor–DNA interactions [97]. As the
technology continues to develop, the opportunities involving phage display will as well.
1.6 Displayed Proteins
Antibodies are large molecules (150 kDa) that consist of two heavy and two light chains
connected by disulfide bonds. Each polypeptide chain contains constant (CH and CL)
regions, needed for cell surface receptor binding, and variable (VH and VL) regions,
responsible for binding antigens [98]. Displaying a molecule of this size on the phage
surface is challenging, and so researchers have eliminated the antibody constant regions
while retaining its variable regions, which specify antigen binding (Figure 1.4) [99]. Two
types of antibody fragments are commonly displayed on virions. The 50 kDa antigen-

14
binding fragment (Fab) only contains the heavy and light chain variable regions and a
portion of the heavy and light chain constant regions. Single-domain variable fragments
(scFv) are even smaller (i.e., 25 kDa) and consist of only the light and heavy chain
variable regions, connected by a fifteen amino acid linker [98].
Figure 1.4. Antibody and antibody fragment structures. Cartoon of a whole antibody
(IgG) (Left), Fab (Middle), and scFv (Right). Variable regions (V) are shown in green and
constant regions (C) are shown in blue. Heavy chains (H) are represented as dark green
and dark blue, and light chains (L) are shown as light green and light blue. Each number
represents a single domain. Single black lines represent amino acid linkers and red lines
indicate interdomain disulfide bonds.

15
In 1989, it was discovered that a scFv could be displayed on the surface of virions
[100, 101]. This proof-of-concept demonstration led to the generation of phage displaying
libraries of naïve scFvs to discover scFvs that bound to many different targets through
affinity selection [102]. Since then, phage libraries have incorporated a number a different
antibody formats including divalent scFvs and Fabs as display proteins [66]. Scientists
have made great strides in therapeutics, diagnostics, and research assays because of
these advances. Nevertheless, these antibody formats have several disadvantages. They
are difficult to express in bacteria due to inter- and intra-domain disulfide bonds, thus,
requiring labor intensive and expensive expression in mammalian CHO cells. Additionally,
these molecules have low thermal stability (Tm = 50-70°C), are prone to aggregation, and
are often cross-reactive [103-105].
Recombinant affinity reagents generated using alternative binding proteins, or
scaffolds, have eliminated many of the disadvantages associated with antibodies.
Typically, scaffolds are natural proteins that have inherent binding properties. Desirable
scaffolds are also small, easy to express in bacteria, thermally stable, highly soluble, and
lacking disulfide bonds [66, 103]. Their binding properties are exploited to generate a
library of variants in which amino acids that participate in the natural binding interaction
but do not participate in maintaining the molecules structural integrity are randomized.
There are numbers of different scaffold proteins that have successfully displayed as
libraries on the phage surface and produced binders to targets of interest (Table II) in
order to bind a variety of different targets with femtomolar to micromolar affinities [1, 94,
106-111].
II. ALTERN ATIVE BINDIN G AFFOLD PROTEIN S FOR PH AGE D ISPLAY

16
This work will focus specifically on generating FHA and FN3 scaffold binding reagents.
The Forkhead Associated (FHA) 1 domain, a naturally occurring phosphothreonine
binding domain, has been used as a scaffold in phage display to select for variants that
recognize phosphothreonine peptide targets. The domain is made of two β-sheets which
fold into a twisted β-sandwich. The imidazole side chain of the conserved Histidine 88
residue interacts with Serine 85, Isoleucine 104, and Glycine 108 to create a pocket to
accommodate the phosphothreonine residue. This pocket allows for discrimination of
phosphothreonine from phosphoserine because it precisely fits the γ-methyl group from
the threonine residue, and Serine 85 and Arginine 70 provide additional contact with the
phosphate group. Furthermore, the FHA domain interacts with other residues on the
phosphopeptide target through two loop regions that connect the β-strands together, the
TABLE II. ALTERNATIVE BINDING SCAFFOLD PROTEINS FOR PHAGE DISPLAY
Scaffold Origin Structure Size (kDa)
Tm (°C) Types of targets
Affibody Z domain of protein A
three alpha helices
6 >90 Proteins, peptides, post-translational modifications
Designed ankyrin repeat protein (DARPin)
Mammalian ankryin proteins
3-7 repeat motifs
14-18 >90 Proteins, peptides, post-translational modifications
Forkhead-associated domain (FHA)
FHA domain from Saccharomyces cerevisiae
β-sandwich 25 70-80 Phosphothreonine peptides
Fibronectin type-III (FN3/ monobody)
Fibronectin domain from
many animal proteins involved in ligand binding
β-sandwich; resembles immunoglobulin domains
10 >90 Conformational epitopes

17
β4-β5 and β10-β11 loops, making them good candidates for randomization (Figure 1.5)
[112-114]. A phage display library was created by Dr. Kritika Pershad in the Kay Lab by
randomizing amino acids 82-84 and 133-139, present in the β4-β5 and β10-β11 loops,
respectively, of the FHA1 domain of the yeast Rad9 protein. The library has successfully
generated FHA variants that recognize an array of mono- and dual-phosphothreonine
peptide targets at an 82% success rate. Additionally, each reagent selectively recognizes
targets containing phosphothreonine and cannot recognize the targets when that
particular residue is changed to phosphoserine, phosphotyrosine, or is unphosphorylated
[115-117]. Thus, the FHA1 library proves to be a valuable tool to generate reagents that
selectively recognize a phosphothreonine post-translational modification.
The human fibronectin 10th type III (FN3) domain has a folding pattern similar to that
of immunoglobulin variable domains (Figure 1.6) [109, 118]. However, the FN3 has
several significant biochemical advantages over antibodies: it lacks disulfide bonds, can
be easily overexpressed in E. coli, is thermally stable (Tm = 88°C), and retains binding
when absorbed onto microtiter plate wells, unlike 95% of monoclonal antibodies, which
lose functionality when adsorbed onto plastic [119, 120]. Additionally, protein engineering
experiments have shown that it is possible to randomize residues within three loops (BC,
DE, FG) on one side of the 10 kDa domain without impacting stability or folding [118,
121]. The BC, DE, and FG loops mimic the complementarity determining regions (CDR)
of the variable domains of the light and heavy chains of antibodies [109, 122]. FN3
variants, also known as “monobodies,” have been generated via phage and yeast display
to a wide variety of targets, such as Abl [123], β-catenin [124], EphA2 [110], estrogen
receptor [125, 126], Fyn [95], integrin [127], Pak1 [128], VEGF-R [129], and several other

18
human cell-signaling proteins [128]. In every case, the FN3 appears to recognize a
conformational, rather than a linear, epitope making it an ideal scaffold for current needs
in research and diagnostics.
Figure 1.5. FHA1 interacts with phosphothreonine containing peptides through its
β loops. FHA1 domain (grey) bound to the Rad9 phosphothreonine peptide (green) (PDB
1G6G). Residues randomized in the β4-5 and β10-11 loops of the FHA phage library are
shown in purple. (Left) Cartoon of the FHA1 domain interacting with the Rad9
phosphopeptide. (Right) Zoomed in surface view representation of FHA1 β loops bound
to the Rad9 phosphopeptide. The phosphate group of the phosphothreonine (pT) residue
in the peptide interacts with Ser85 and Arg70 of the FHA1 domain through electrostatic
interactions. The FHA1 domain also contains a hydrophobic pocket that accommodates
the γ-methyl group of the pT residue in the phosphopeptide. Residues C-terminal to the
pT residue of the peptide, especially +3 (The third amino acid C-terminal to the pT),
strongly interact with residues in the β4-5 and β10-11 FHA1 loops.

19
Figure 1.6. PyMOL representations of the FN3 monobody. FG, BC, and DE loops are
shown in yellow, blue, and red, respectively (PDB: 1ttg). All other residues are shown in
grey. (Left) Schematic showing β-sheets and loops. (Middle) Surface representation.
(Right) Surface representation, viewed from the top.
1.7 Thesis Goals
The overarching goal of this thesis was to develop recombinant affinity reagents to
challenging targets through phage display. I used phage-display libraries of two different
scaffolds to generate high-quality affinity reagents to phosphothreonine peptides and
conformational epitopes and reformatted some of them to work in sensitive biochemical
assays.
In Chapter 2, I successfully screened a phage library displaying variants of the
Forkhead Associated (FHA) domain and produced affinity reagents that bind
phosphopeptides corresponding to the human protein, Activation Transcription Factor 2

20
(ATF2). This segment of ATF2 contains two phosphosites and it exists in various
phosphorylated (i.e., monophosphorylated, diphosphorylated) states in the cell. Unlike a
commercial antibody, the FHA affinity reagents generated in the chapter are capable of
distinguishing between these phosphostates. I use alanine scanning to identify the
molecular recognition elements of an FHA variant that binds only to the fully
phosphorylated ATF2 peptide, and I further improve the selectivity of other FHA reagents
through mutagenesis to distinguish between ATF2’s partially phosphorylated states.
Finally, I demonstrate that an engineered FHA domain can recognize native,
phosphorylated ATF2 protein in a cell lysate analyzed by western blotting.
In Chapter 3, I generate FHA-based affinity reagents to a phosphorylated peptide of
human AKT1. These are the first FHA domains engineered to recognize a target
containing glycine at the position three residues C-terminal to the phosphothreonine
(pT+3). While the pT+3 position has previously been shown to be extremely important for
FHA binding, it was surprising to find that glycine, a small and flexible residue, could
contribute to binding at this position. Analysis by surface plasmon resonance (SPR) and
alanine scanning reveal an unusually high affinity and specificity for the phosphopeptide,
respectively. These results demonstrate the potential of developing high quality FHA-
based affinity reagents to a wide array of phosphopeptides.
In Chapter 4, I streamline the validation process of FN3 binding pairs in sandwich
ELISAs. These pairs were previously isolated through Megaprimer Shuffling for Tandem
Reagents (MegaSTAR), a technique to quickly and inexpensively generate tandem
binding reagents with potential use as sandwich assay binding pairs. My new protocols
eliminate the need to purify the affinity reagents from overexpressing bacterial cells and

21
instead use crude cell lysates directly in ELISAs. This new strategy permits testing more
pairs to discover the ones that yield the best assays. Additionally, I label FN3 proteins in
vitro with various tags through sortase-mediated ligation. Because of the simple labeling
method, researchers will be able to use these binding reagents in a variety of different
assays.
In the final chapter, I summarize my work and discuss future experiments to improve
MegaSTAR by incorporating multiple scaffolds and linkers into the selection process as
well as generate targets of selection by cell free protein synthesis. I then discuss using
sortase-mediated ligation to format binding pairs for highly sensitive homogeneous
assays. Finally, I examine the impact of my work on basic research and diagnostics.

22
1.8 Literature Cited
1. Gorman, K., McGinnis, J. & Kay, B. (2018) Generating FN3-Based Affinity Reagents Through Phage Display, Current Protocols in Chemical Biology. 10, e39.
2. Forthal, D. N. (2014) Functions of Antibodies, Microbiology spectrum. 2, 1-17.
3. Wootla, B., Denic, A. & Rodriguez, M. (2014) Polyclonal and Monoclonal Antibodies in Clinic in Human Monoclonal Antibodies: Methods and Protocols (Steinitz, M., ed) pp. 79-110, Humana Press, Totowa, NJ.
4. Litman, G. W., Rast, J. P., Shamblott, M. J., Haire, R. N., Hulst, M., Roess, W., Litman, R. T., Hinds-Frey, K. R., Zilch, A. & Amemiya, C. T. (1993) Phylogenetic diversification of immunoglobulin genes and the antibody repertoire, Molecular Biology and Evolution. 10, 60-72.
5. Kennedy, P. J., Oliveira, C., Granja, P. L. & Sarmento, B. (2018) Monoclonal antibodies: technologies for early discovery and engineering, Critical Reviews in Biotechnology. 38, 394-408.
6. Kurien, B. T. & Scofield, R. H. (2006) Western blotting, Methods. 38, 283-293.
7. Reen, D. J. (1994) Enzyme-Linked Immunosorbent Assay (ELISA) in Basic Protein and Peptide Protocols (Walker, J. M., ed) pp. 461-466, Humana Press, Totowa, NJ.
8. Dong, Q. & Li, F. (2018) Antibody Pull-Down Experiments in Fission Yeast, Methods Mol Biol. 1721, 117-123.
9. Scanziani, E. (1998) Immunohistochemical Staining of Fixed Tissues in Mycoplasma Protocols (Miles, R. & Nicholas, R., eds) pp. 133-140, Humana Press, Totowa, NJ.
10. Odell, I. D. & Cook, D. (2013) Immunofluorescence Techniques, Journal of Investigative Dermatology. 133, 1-4.
11. Leow, C. H., Fischer, K., Leow, C. Y., Cheng, Q., Chuah, C. & McCarthy, J. (2017) Single Domain Antibodies as New Biomarker Detectors, Diagnostics (Basel). 7, 52.

23
12. Hulka, B. S. & Wilcosky, T. (1988) Biological Markers in Epidemiologic Research, Archives of Environmental Health: An International Journal. 43, 83-89.
13. Aronson, J. K. & Ferner, R. E. (2017) Biomarkers—A General Review, Current Protocols in Pharmacology. 76, 9.23.1-9.23.17.
14. Zhao, X., Modur, V., Carayannopoulos, L. N. & Laterza, O. F. (2015) Biomarkers in Pharmaceutical Research, Clinical Chemistry. 61, 1343-1353.
15. Cole, L. A. (2010) Biological functions of hCG and hCG-related molecules, Reprod Biol Endocrinol. 8, 102-102.
16. Gnoth, C. & Johnson, S. (2014) Strips of Hope: Accuracy of Home Pregnancy Tests and New Developments, Geburtshilfe Frauenheilkd. 74, 661-669.
17. Kierny, M. R., Cunningham, T. D. & Kay, B. K. (2012) Detection of biomarkers using recombinant antibodies coupled to nanostructured platforms, Nano Rev. 3, 10.3402/nano.v3i0.17240.
18. Baird, C. L., Fischer, C. J., Pefaur, N. B., Miller, K. D., Kagan, J., Srivastava, S. & Rodland, K. D. (2010) Developing recombinant antibodies for biomarker detection, Cancer Biomarkers. 6, 271-279.
19. Zhu, J., Zou, N., Mao, H., Wang, P., Zhu, D., Ji, H., Cong, H., Sun, C., Wang, H., Zhang, F., Qian, J., Jin, Q. & Zhao, J. (2013) Evaluation of a modified lateral flow immunoassay for detection of high-sensitivity cardiac troponin I andmyoglobin, Biosensors and Bioelectronics. 42, 522-525.
20. Agnibh, M., Uzair, A., Martin, B., Ibrahim, A. & Michael, B. (2017) Clinically Relevant Biomarkers in Acute Heart Failure: An Update, Current Pharmaceutical Biotechnology. 18, 482-490.
21. Ji, M., Cho, B., Cho, Y. S., Park, S.-Y., Cho, S.-N., Jeon, B.-Y. & Yoon, B.-S. (2014) Development of a quantitative sandwich enzyme-linked immunosorbent assay for detecting the MPT64 antigen of Mycobacterium tuberculosis, Yonsei Med J. 55, 746-752.

24
22. Arai, H., Petchclai, B., Khupulsup, K., Kurimura, T. & Takeda, K. (1999) Evaluation of a rapid immunochromatographic test for detection of antibodies to human immunodeficiency virus, J Clin Microbiol. 37, 367-370.
23. Suzuki, M., Kato, C. & Kato, A. (2015) Therapeutic antibodies: their mechanisms of action and the pathological findings they induce in toxicity studies, J Toxicol Pathol. 28, 133-139.
24. Goodman, S. M. (2015) Rheumatoid arthritis: Perioperative management of biologics and DMARDs, Seminars in Arthritis and Rheumatism. 44, 627-632.
25. Burness, C. B. & McKeage, K. (2015) Adalimumab: A Review in Chronic Plaque Psoriasis, Drugs. 75, 2119-2130.
26. Faubion, W. A., Dubinsky, M., Ruemmele, F. M., Escher, J., Rosh, J., Hyams, J. S., Eichner, S., Li, Y., Reilly, N., Thakkar, R. B., Robinson, A. M. & Lazar, A. (2017) Long-term Efficacy and Safety of Adalimumab in Pediatric Patients with Crohn's Disease, Inflamm Bowel Dis. 23, 453-460.
27. Ecker, D. M., Jones, S. D. & Levine, H. L. (2015) The therapeutic monoclonal antibody market, MAbs. 7, 9-14.
28. Lai, Y., Wang, R., Chen, X., Tang, D., Hu, Y., Cai, J., Zhang, Q. & Hu, H. (2017) Emerging trends and new developments in monoclonal antibodies: A scientometric analysis (1980-2016), Hum Vaccin Immunother. 13, 1-10.
29. Cavallo, F., Calogero, R. A. & Forni, G. (2007) Are oncoantigens suitable targets for anti-tumour therapy?, Nature Reviews Cancer. 7, 707.
30. Ferguson, K. M. (2004) Active and inactive conformations of the epidermal growth factor receptor, Biochemical Society Transactions. 32, 742-745.
31. Albanell, J., Codony, J., Rovira, A., Mellado, B. & Gascón, P. (2003) Mechanism of Action of Anti-HER2 Monoclonal Antibodies: Scientific Update on Trastuzumab and 2C4 in New Trends in Cancer for the 21st Century: Proceedings of the International Symposium on Cancer: New Trends in Cancer for the 21st Century, held November 10–13, 2002, in Valencia, Spain (Llombart-Bosch, A. & Felipo, V., eds) pp. 253-268, Springer US, Boston, MA.

25
32. Cartron, G., Dacheux, L., Salles, G., Solal-Celigny, P., Bardos, P., Colombat, P. & Watier, H. (2002) Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcγRIIIa gene, Blood. 99, 754-758.
33. Zafir-Lavie, I., Michaeli, Y. & Reiter, Y. (2007) Novel antibodies as anticancer agents, Oncogene. 26, 3714.
34. de Graaf, M., Boven, E., Oosterhoff, D., van der Meulen-Muileman, I. H., Huls, G. A., Gerritsen, W. R., Haisma, H. J. & Pinedo, H. M. (2002) A fully human anti-Ep-CAM scFv-beta-glucuronidase fusion protein for selective chemotherapy with a glucuronide prodrug, British journal of cancer. 86, 811-818.
35. Sedykh, S. E., Prinz, V. V., Buneva, V. N. & Nevinsky, G. A. (2018) Bispecific antibodies: design, therapy, perspectives, Drug Des Devel Ther. 12, 195-208.
36. Blanc, R. S. & Richard, S. (2017) Arginine Methylation: The Coming of Age, Molecular cell. 65, 8-24.
37. Casey, P. J. & Seabra, M. C. (1996) Protein Prenyltransferases, Journal of Biological Chemistry. 271, 5289-5292.
38. Dalziel, M., Crispin, M., Scanlan, C. N., Zitzmann, N. & Dwek, R. A. (2014) Emerging Principles for the Therapeutic Exploitation of Glycosylation, Science. 343, 1235681.
39. Humphrey, S. J., James, D. E. & Mann, M. (2015) Protein Phosphorylation: A Major Switch Mechanism for Metabolic Regulation, Trends in Endocrinology & Metabolism. 26, 676-687.
40. L N Johnson, a. & Barford, D. (1993) The Effects of Phosphorylation on the Structure and Function of Proteins, Annual Review of Biophysics and Biomolecular Structure. 22, 199-232.
41. Zhao, S., Xu, W., Jiang, W., Yu, W., Lin, Y., Zhang, T., Yao, J., Zhou, L., Zeng, Y., Li, H., Li, Y., Shi, J., An, W., Hancock, S. M., He, F., Qin, L., Chin, J., Yang, P., Chen, X., Lei, Q., Xiong, Y. & Guan, K.-L. (2010) Regulation of cellular metabolism by protein lysine acetylation, Science (New York, NY). 327, 1000-1004.

26
42. Goel, R., Harsha, H. C., Pandey, A. & Prasad, T. S. K. (2012) Human Protein Reference Database and Human Proteinpedia as resources for phosphoproteome analysis, Mol Biosyst. 8, 453-463.
43. Khoury, G. A., Baliban, R. C. & Floudas, C. A. (2011) Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database, Scientific reports. 1, 90.
44. Suwanmajo, T. & Krishnan, J. (2015) Mixed mechanisms of multi-site phosphorylation, Journal of The Royal Society Interface. 12, 20141405.
45. Aoki, M., Yokota, T., Sugiura, I., Sasaki, C., Hasegawa, T., Okumura, C., Ishiguro, K., Kohno, T., Sugio, S. & Matsuzaki, T. (2004) Structural insight into nucleotide recognition in tau-protein kinase I/glycogen synthase kinase 3[beta], Acta Crystallographica Section D. 60, 439-446.
46. Jope, R. S., Yuskaitis, C. J. & Beurel, E. (2007) Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics, Neurochem Res. 32, 577-595.
47. van den Berg, B., Wain, R., Dobson, C. M. & Ellis, R. J. (2000) Macromolecular crowding perturbs protein refolding kinetics: implications for folding inside the cell, The EMBO journal. 19, 3870-3875.
48. Dobson, C. M. (2003) Protein folding and misfolding, Nature. 426, 884-890.
49. Hartl, F. U. (1996) Molecular chaperones in cellular protein folding, Nature. 381, 571-580.
50. Chaudhuri, T. K. & Paul, S. (2006) Protein-misfolding diseases and chaperone-based therapeutic approaches, The FEBS Journal. 273, 1331-1349.
51. Nin, D. S., Li, F., Visvanathan, S. & Khan, M. (2015) Misfolded N-CoR is Linked to the Ectopic Reactivation of CD34/Flt3-Based Stem-Cell Phenotype in Promyelocytic and Monocytic Acute Myeloid Leukemia, Frontiers in Oncology. 5.
52. Arrasate, M. & Finkbeiner, S. (2012) Protein aggregates in Huntington's disease, Exp Neurol. 238, 1-11.

27
53. Gregoire, S., Irwin, J. & Kwon, I. (2012) Techniques for Monitoring Protein Misfolding and Aggregation in Vitro and in Living Cells, Korean J Chem Eng. 29, 693-702.
54. Hanly, W. C., Artwohl, J. E. & Bennett, B. T. (1995) Review of Polyclonal Antibody Production Procedures in Mammals and Poultry, ILAR Journal. 37, 93-118.
55. Dunbar, B. S. & Schwoebel, E. D. (1990) Preparation of polyclonal antibodies in Methods in Enzymology (Deutscher, M. P., ed) pp. 663-670, Academic Press.
56. Liu, S., Li, S., Zhang, Y., Wang, Y., Zhu, Y., Wang, B. & Chen, Z.-N. (2017) Purification of a polyclonal antibody against CD147 for ELISA using antigen‑immunoaffinity chromatography, Mol Med Rep. 15, 4035-4040.
57. Nelson, P. N., Reynolds, G. M., Waldron, E. E., Ward, E., Giannopoulos, K. & Murray, P. G. (2000) Monoclonal antibodies, Mol Pathol. 53, 111-117.
58. KÖHler, G. & Milstein, C. (1975) Continuous cultures of fused cells secreting antibody of predefined specificity, Nature. 256, 495-497.
59. Corrêa, A. L., Senna, J. P. M. & de Sousa, Á. P. B. (2016) Effects of passage number on growth and productivity of hybridoma secreting MRSA anti-PBP2a monoclonal antibodies, Cytotechnology. 68, 419-427.
60. Bradbury, A. R. M., Trinklein, N. D., Thie, H., Wilkinson, I. C., Tandon, A. K., Anderson, S., Bladen, C. L., Jones, B., Aldred, S. F., Bestagno, M., Burrone, O., Maynard, J., Ferrara, F., Trimmer, J. S., Görnemann, J., Glanville, J., Wolf, P., Frenzel, A., Wong, J., Koh, X. Y., Eng, H.-Y., Lane, D., Lefranc, M.-P., Clark, M. & Dübel, S. (2018) When monoclonal antibodies are not monospecific: Hybridomas frequently express additional functional variable regions, MAbs. 10, 539-546.
61. Skerra, A. & Pluckthun, A. (1988) Assembly of a functional immunoglobulin Fv fragment in Escherichia coli, Science. 240, 1038-1041.
62. Larrick, J. W., Danielsson, L., Brenner, C. A., Abrahamson, M., Fry, K. E. & Borrebaeck, C. A. K. (1989) Rapid cloning of rearranged immunoglobulin genes from human hybridoma cells using mixed primers and the polymerase chain reaction, Biochemical and biophysical research communications. 160, 1250-1256.

28
63. Orlandi, R., Güssow, D. H., Jones, P. T. & Winter, G. (1989) Cloning immunoglobulin variable domains for expression by the polymerase chain reaction, Proceedings of the National Academy of Sciences of the United States of America. 86, 3833-3837.
64. Sastry, L., Alting-Mees, M., Huse, W. D., Short, J. M., Sorge, J. A., Hay, B. N., Janda, K. D., Benkovic, S. J. & Lerner, R. A. (1989) Cloning of the immunological repertoire in Escherichia coli for generation of monoclonal catalytic antibodies: construction of a heavy chain variable region-specific cDNA library, Proceedings of the National Academy of Sciences of the United States of America. 86, 5728-5732.
65. Smith, G. (1985) Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface, Science. 228, 1315-1317.
66. Bradbury, A. R. M., Sidhu, S., Dübel, S. & McCafferty, J. (2011) Beyond natural antibodies: the power of in vitro display technologies, Nature biotechnology. 29, 245-254.
67. Binz, H. K., Amstutz, P. & Plückthun, A. (2005) Engineering novel binding proteins from nonimmunoglobulin domains, Nature Biotechnology. 23, 1257-1268.
68. Binz, H. K. & Plückthun, A. (2005) Engineered proteins as specific binding reagents, Current Opinion in Biotechnology. 16, 459-469.
69. Paschke, M. (2006) Phage display systems and their applications, Applied Microbiology and Biotechnology. 70, 2-11.
70. Kehoe, J. W. & Kay, B. K. (2005) Filamentous Phage Display in the New Millennium, Chemical Reviews. 105, 4056-4072.
71. Boder, E. T. & Wittrup, K. D. (1997) Yeast surface display for screening combinatorial polypeptide libraries, Nature Biotechnology. 15, 553-557.
72. Feldhaus, M. J., Siegel, R. W., Opresko, L. K., Coleman, J. R., Feldhaus, J. M. W., Yeung, Y. A., Cochran, J. R., Heinzelman, P., Colby, D., Swers, J., Graff, C., Wiley, H. S. & Wittrup, K. D. (2003) Flow-cytometric isolation of human antibodies from a nonimmune Saccharomyces cerevisiae surface display library, Nature Biotechnology. 21, 163-170.

29
73. Boder, E. T., Midelfort, K. S. & Wittrup, K. D. (2000) Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity, Proceedings of the National Academy of Sciences of the United States of America. 97, 10701-10705.
74. Seelig, B. (2011) mRNA display for the selection and evolution of enzymes from in vitro-translated protein libraries, Nature Protocols. 6, 540-552.
75. Zahnd, C., Amstutz, P. & Plückthun, A. (2007) Ribosome display: selecting and evolving proteins in vitro that specifically bind to a target, Nature Methods. 4, 269.
76. Razai, A., Garcia-Rodriguez, C., Lou, J., Geren, I. N., Forsyth, C. M., Robles, Y., Tsai, R., Smith, T. J., Smith, L. A., Siegel, R. W., Feldhaus, M. & Marks, J. D. (2005) Molecular Evolution of Antibody Affinity for Sensitive Detection of Botulinum Neurotoxin Type A, Journal of molecular biology. 351, 158-169.
77. Hanes, J., Schaffitzel, C., Knappik, A. & Plückthun, A. (2000) Picomolar affinity antibodies from a fully synthetic naive library selected and evolved by ribosome display, Nature Biotechnology. 18, 1287-1292.
78. Schier, R., McCall, A., Adams, G. P., Marshall, K. W., Merritt, H., Yim, M., Crawford, R. S., Weiner, L. M., Marks, C. & Marks, J. D. (1996) Isolation of Picomolar Affinity Anti-c-erbB-2 Single-chain Fv by Molecular Evolution of the Complementarity Determining Regions in the Center of the Antibody Binding Site, Journal of molecular biology. 263, 551-567.
79. Marvin, D. A. (1998) Filamentous phage structure, infection and assembly, Current Opinion in Structural Biology. 8, 150-158.
80. Barbas, C. F. (2001) Phage display : a laboratory manual.
81. van Wezenbeek, P. M. G. F., Hulsebos, T. J. M. & Schoenmakers, J. G. G. (1980) Nucleotide sequence of the filamentous bacteriophage M13 DNA genome: comparison with phage fd, Gene. 11, 129-148.
82. Nakamura, M., Tsumoto, K., Kumagai, I. & Ishimura, K. (2003) A morphologic study of filamentous phage infection of Escherichia coli using biotinylated phages, FEBS letters. 536, 167-172.

30
83. Feng, J. N., Russel, M. & Model, P. (1997) A permeabilized cell system that assembles filamentous bacteriophage, Proceedings of the National Academy of Sciences of the United States of America. 94, 4068-4073.
84. Marciano, D. K., Russel, M. & Simon, S. M. (1999) An Aqueous Channel for Filamentous Phage Export, Science. 284, 1516-1519.
85. Rakonjac, J. & Model, P. (1998) Roles of pIII in filamentous phage assembly11Edited by M. Gottesman, Journal of molecular biology. 282, 25-41.
86. Fuh, G. & Sidhu, S. S. (2000) Efficient phage display of polypeptides fused to the carboxy-terminus of the M13 gene-3 minor coat protein, FEBS letters. 480, 231-234.
87. Gao, C., Mao, S., Kaufmann, G., Wirsching, P., Lerner, R. A. & Janda, K. D. (2002) A method for the generation of combinatorial antibody libraries using pIX phage display, Proceedings of the National Academy of Sciences of the United States of America. 99, 12612-12616.
88. Gao, C., Mao, S., Lo, C.-H. L., Wirsching, P., Lerner, R. A. & Janda, K. D. (1999) Making artificial antibodies: A format for phage display of combinatorial heterodimeric arrays, Proceedings of the National Academy of Sciences. 96, 6025-6030.
89. Hayhurst, A. & Harris, W. J. (1999) Escherichia coliSkp Chaperone Coexpression Improves Solubility and Phage Display of Single-Chain Antibody Fragments, Protein Expression and Purification. 15, 336-343.
90. Hufton, S. E., Moerkerk, P. T., Meulemans, E. V., de Bruı̈ne, A., Arends, J.-W. & Hoogenboom, H. R. (1999) Phage display of cDNA repertoires: the pVI display system and its applications for the selection of immunogenic ligands, Journal of Immunological Methods. 231, 39-51.
91. Weiss, G. A. & Sidhu, S. S. (2000) Design and evolution of artificial M13 coat proteins11Edited by G. Von Heijne, Journal of molecular biology. 300, 213-219.
92. Zhong, G., Smith, G. P., Berry, J. & Brunham, R. C. (1994) Conformational mimicry of a chlamydial neutralization epitope on filamentous phage, Journal of Biological Chemistry. 269, 24183-24188.

31
93. Lund, P. E., Hunt, R. C., Gottesman, M. M. & Kimchi-Sarfaty, C. (2010) Pseudovirions as vehicles for the delivery of siRNA, Pharm Res. 27, 400-420.
94. Pershad, K., Wypisniak, K. & Kay, B. K. (2012) Directed evolution of the forkhead-associated domain to generate anti-phosphospecific reagents by phage display, Journal of molecular biology. 424, 88-103.
95. Huang, R., Fang, P. & Kay, B. K. (2012) Isolation of monobodies that bind specifically to the SH3 domain of the Fyn tyrosine protein kinase, New biotechnology. 29, 526-533.
96. Halperin, I., Wolfson, H. & Nussinov, R. (2003) SiteLight: binding-site prediction using phage display libraries, Protein Sci. 12, 1344-1359.
97. Freckleton, G., Lippman, S. I., Broach, J. R. & Tavazoie, S. (2009) Microarray profiling of phage-display selections for rapid mapping of transcription factor-DNA interactions, PLoS Genet. 5, e1000449-e1000449.
98. Nelson, A. L. (2010) Antibody fragments: hope and hype, MAbs. 2, 77-83.
99. Hammers, C. M. & Stanley, J. R. (2014) Antibody phage display: technique and applications, J Invest Dermatol. 134, 1-5.
100. Skerra, A. & Plückthun, A. (1988) Assembly of a Functional Immunoglobulin Fragment in Escherichia coli, Science. 240, 1038-1041.
101. Better, M., Chang, C. P., Robinson, R. R. & Horwitz, A. H. (1988) Escherichia coli Secretion of an Active Chimeric Antibody Fragment, Science. 240, 1041-1043.
102. McCafferty, J., Griffiths, A. D., Winter, G. & Chiswell, D. J. (1990) Phage antibodies: filamentous phage displaying antibody variable domains, Nature. 348, 552-554.
103. Yu, X., Yang, Y.-P., Dikici, E., Deo, S. K. & Daunert, S. (2017) Beyond Antibodies as Binding Partners: The Role of Antibody Mimetics in Bioanalysis, Annu Rev Anal Chem (Palo Alto Calif). 10, 293-320.

32
104. Bradbury, A. & Pluckthun, A. (2015) Reproducibility: Standardize antibodies used in research., Nature. 518, 27-29.
105. Voskuil, J. (2014) Commercial antibodies and their validation, F1000Res. 3, 232-232.
106. Kummer, L., Parizek, P., Rube, P., Millgramm, B., Prinz, A., Mittl, P. R., Kaufholz, M., Zimmermann, B., Herberg, F. W. & Pluckthun, A. (2012) Structural and functional analysis of phosphorylation-specific binders of the kinase ERK from designed ankyrin repeat protein libraries, Proceedings of the National Academy of Sciences of the United States of America. 109, E2248-57.
107. McGinnis, J. E., Venegas, L. A., Lopez, H. & Kay, B. K. (2018) A Recombinant Affinity Reagent Specific for a Phosphoepitope of Akt1, Int J Mol Sci. 19, 3305.
108. McGinnis, J. & Kay, B. K. (2017) Generation of recombinant affinity reagents against a two-phosphosite epitope of ATF2, New biotechnology.
109. Koide, A., Wojcik, J., Gilbreth, R. N., Hoey, R. J. & Koide, S. (2012) Teaching an old scaffold new tricks: monobodies constructed using alternative surfaces of the FN3 scaffold, Journal of molecular biology. 415, 393-405.
110. Park, S.-H., Park, S., Kim, D.-Y., Pyo, A., Kimura, R. H., Sathirachinda, A., Choy, H. E., Min, J.-J., Gambhir, S. S. & Hong, Y. (2015) Isolation and Characterization of a Monobody with a Fibronectin Domain III Scaffold That Specifically Binds EphA2, PLoS One. 10, e0132976-e0132976.
111. Friedman, M., Nordberg, E., Höidén-Guthenberg, I., Brismar, H., Adams, G. P., Nilsson, F. Y., Carlsson, J. & Ståhl, S. (2007) Phage display selection of Affibody molecules with specific binding to the extracellular domain of the epidermal growth factor receptor, Protein Engineering, Design and Selection. 20, 189-199.
112. Huang, Y.-M. M. & Chang, C.-E. A. (2011) Mechanism of PhosphoThreonine/Serine Recognition and Specificity for Modular Domains from All-atom Molecular Dynamics, BMC biophysics. 4, 12-12.
113. Huang, Y.-M. M. & Chang, C.-E. A. (2014) Achieving peptide binding specificity and promiscuity by loops: case of the forkhead-associated domain, PLoS One. 9, e98291-e98291.

33
114. Durocher, D., Taylor, I. A., Sarbassova, D., Haire, L. F., Westcott, S. L., Jackson, S. P., Smerdon, S. J. & Yaffe, M. B. (2000) The molecular basis of FHA domain:phosphopeptide binding specificity and implications for phospho-dependent signaling mechanisms, Molecular cell. 6, 1169-82.
115. Pershad, K., Wypisniak, K. & Kay, B. K. (2012) Directed evolution of the forkhead-associated domain to generate anti-phosphospecific reagents by phage display, Journal of molecular biology. 424, 88-103.
116. Venegas, L. A., Kall, S. L., Bankole, O., Lavie, A. & Kay, B. (2018) Generating a recombinant phosphothreonine-binding domain for a phosphopeptide of the human transcription factor, c-Myc, New biotechnology.
117. Venegas, L. A., Pershad, K., Bankole, O., Shah, N. & Kay, B. K. (2016) A comparison of phosphospecific affinity reagents reveals the utility of recombinant Forkhead-associated domains in recognizing phosphothreonine-containing peptides, New biotechnology. 33, 537-43.
118. Koide, A., Bailey, C. W., Huang, X. & Koide, S. (1998) The fibronectin type III domain as a scaffold for novel binding proteins11Edited by J. Wells, Journal of molecular biology. 284, 1141-1151.
119. Butler, J. E., Ni, L., Brown, W. R., Joshi, K. S., Chang, J., Rosenberg, B. & Voss, E. W. (1993) The immunochemistry of sandwich elisas—VI. Greater than 90% of monoclonal and 75% of polyclonal anti-fluorescyl capture antibodies (CAbs) are denatured by passive adsorption, Molecular Immunology. 30, 1165-1175.
120. Jacobs, S. A., Diem, M. D., Luo, J., Teplyakov, A., Obmolova, G., Malia, T., Gilliland, G. L. & O'Neil, K. T. (2012) Design of novel FN3 domains with high stability by a consensus sequence approach, Protein Engineering, Design and Selection. 25, 107-117.
121. Batori, V., Koide, A. & Koide, S. (2002) Exploring the potential of the monobody scaffold: effects of loop elongation on the stability of a fibronectin type III domain, Protein Engineering, Design and Selection. 15, 1015-1020.
122. De Genst, E., Silence, K., Decanniere, K., Conrath, K., Loris, R., Kinne, J., Muyldermans, S. & Wyns, L. (2006) Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies, Proceedings of the National Academy of Sciences of the United States of America. 103, 4586-4591.

34
123. Wojcik, J., Hantschel, O., Grebien, F., Kaupe, I., Bennett, K. L., Barkinge, J., Jones, R. B., Koide, A., Superti-Furga, G. & Koide, S. (2010) A potent and highly specific FN3 monobody inhibitor of the Abl SH2 domain, Nat Struct Mol Biol. 17, 519-527.
124. Yeh, J. T. H., Binari, R., Gocha, T., Dasgupta, R. & Perrimon, N. (2013) PAPTi: a peptide aptamer interference toolkit for perturbation of protein-protein interaction networks, Scientific reports. 3, 1156-1156.
125. Koide, A., Abbatiello, S., Rothgery, L. & Koide, S. (2002) Probing protein conformational changes in living cells by using designer binding proteins: application to the estrogen receptor, Proceedings of the National Academy of Sciences of the United States of America. 99, 1253-1258.
126. Huang, J., Koide, A., Nettle, K. W., Greene, G. L. & Koide, S. (2006) Conformation-specific affinity purification of proteins using engineered binding proteins: Application to the estrogen receptor, Protein Expression and Purification. 47, 348-354.
127. Richards, J., Miller, M., Abend, J., Koide, A., Koide, S. & Dewhurst, S. (2003) Engineered Fibronectin Type III Domain with a RGDWXE Sequence Binds with Enhanced Affinity and Specificity to Human αvβ3 Integrin, Journal of molecular biology. 326, 1475-1488.
128. Huang, R., Gorman, K. T., Vinci, C. R., Dobrovetsky, E., Gräslund, S. & Kay, B. K. (2015) Streamlining the Pipeline for Generation of Recombinant Affinity Reagents by Integrating the Affinity Maturation Step, Int J Mol Sci. 16, 23587-23603.
129. Fellouse, F. A., Esaki, K., Birtalan, S., Raptis, D., Cancasci, V. J., Koide, A., Jhurani, P., Vasser, M., Wiesmann, C., Kossiakoff, A. A., Koide, S. & Sidhu, S. S. (2007) High-throughput Generation of Synthetic Antibodies from Highly Functional Minimalist Phage-displayed Libraries, Journal of molecular biology. 373, 924-940.

35
Chapter 2 2. GENERATION OF RECOMBINANT AFFINITY REAGENTS AGAINST A TWO-
PHOSPHOSITE EPITOPE OF ATF2
A portion of this work was previously published in New Biotechnology [1]
This work was done in collaboration with Dr. Brian Kay. Dr. Kay helped with experimental
design and editing.
2.1 Abstract
Activating Transcription Factor 2 (ATF2) plays an important role in mammalian cell
proliferation, apoptosis and DNA repair. Its activation is dependent on the sequential
phosphorylation of residue threonine 71 (T71) followed by threonine 69 (T69) in its
transactivation domain. While these modifications can be directed by a variety of kinases,
the time to reach full phosphorylation is dependent on which signaling pathway has been
activated, which is thought to be important for proper temporal regulation. To explore this
phenomenon further, there have been ongoing efforts to generate affinity reagents for
monitoring phosphorylation events in cellular assays. While phospho-specific antibodies
have been valuable tools for monitoring cell signaling events, those raised against a
peptide containing two or more adjacent phosphosites tend to cross-react with that
peptide’s various phospho-states, rendering such reagents unusable for studying
sequential phosphorylation. As an alternative, we have employed the N-terminal
Forkhead-associated 1 (FHA1) domain of yeast Rad53p as a scaffold to generate
recombinant affinity reagents via phage display and were successful in generating a set
of reagents that can distinguish between the dual-phosphorylated epitope, 63-
IVADQpTPpTPTRFLK-77, and the mono-phosphorylated epitope, 63-
IVADQpTPTPTRFLK-77, in the human ATF2 transactivation domain.

36
2.2 Introduction
Protein phosphorylation is an important post-translational modification that allows cell
signaling pathways to activate or inhibit proteins, alter protein cellular locations, change
a protein’s stability or create new protein binding interactions [2]. Over the years, mass
spectrometry has mapped >100,000 phosphosites on >13,000 human proteins [3],
illustrating the prevalence of protein phosphorylation. However, in recent years it has
become apparent that phosphorylation is not always used as a simple switch to alter
protein function. Due to extensive regulatory networks, cells utilize multisite protein
phosphorylation to coordinate signal timing and strength, which is imperative for proper
pathway crosstalk and gene expression [4]. Sequential phosphorylation, in which multiple
residues on a protein are modified over time, provides a mechanism to delay or develop
a graded response to maintain temporal regulation [5-7].
One well-studied example illustrating the potential importance of sequential
phosphorylation is the human protein, Activating Transcription Factor 2 (ATF2). In
unstimulated conditions, ATF2 is ubiquitously expressed and maintained in an inactive
state due to intramolecular interactions between its N-terminal and C-terminal domains
[8]. Upon signaling, ATF2 is phosphorylated on threonine 71 (T71) and threonine 69 (T69)
of the protein, which disrupts an intramolecular interaction between the N- and C-termini,
allowing co-activating transcription factors, such as c-Jun, to bind the ATF2 B-Zip domain
and create a transcriptionally active heterodimer [9]. Multiple signaling pathways are
responsible for these ATF2 phosphorylation events [10], most notably stress and growth
activated pathways. When the MAPK pathway is stimulated upon stress signals, T71 is
phosphorylated by c-Jun N-terminal kinase (JNK), and, shortly thereafter, T69 is

37
phosphorylated by p38 mitogen activated protein kinase. This near simultaneous, two-
step mechanism activates ATF2 as an “early response” transcription factor [11].
Conversely, signaling that is initiated by growth factors involves a longer time to fully
phosphorylate this ATF2 epitope. This delay is caused by a first requirement for
phosphorylation of T71 through the Raf–MEK–ERK pathway. Once this modification
occurs, it takes several minutes for T69 to become phosphorylated in the Ral–RalGDS–
Src-p38 pathway [12]. While it is apparent that these two-step phosphorylation
mechanisms are utilized to alter the timing of full ATF2 activation depending on which
pathway is activated, it is unclear how the timing differences between pathways affect the
cell’s response. Monitoring these phosphorylation events in the cell would be highly useful
in exploring this further.
While mass spectrometry is a powerful method for monitoring protein phosphorylation,
antibodies that recognize phospho-epitopes have also been instrumental in tracking
individual phosphorylation events in cells [13, 14]. Unfortunately, antibodies that
recognize two or more adjacent phosphosites tend to cross-react with the singly
phosphorylated states of that epitope, and vice versa [15], limiting their usefulness in
studying dual-phosphorylation events. Consequently, there has been growing interest in
utilizing engineered protein scaffolds as alternatives to antibodies, because their target
specificity can be altered through protein engineering techniques [16]. Several protein
domains have been employed as scaffolds to engineer phospho-specific recombinant
affinity reagents, among them the Src Homology 2 domain (SH2) [17], the 10th fibronectin
type III domain (10FnIII) [18], antigen binding fragments (Fab) [19], single chain variable
fragments (scFv) [20], designed ankyrin repeat proteins (DARPins) [21] and the

38
Forkhead-associated 1 (FHA1) domain [22]. As we have previously succeeded in
generating FHA domains to a variety of pT-containing peptides, we investigated in this
study whether we could identify FHA domain variants that discriminate between mono-
and dual-phosphorylated ATF2. Given that a functionally and structurally similar FHA
domain, Dun1-FHA, requires a dual-phosphorylated target for recognition [23], we
hypothesized that we could evolve the FHA1 scaffold to behave in a similar manner. Our
FHA phage-display library, which displayed FHA domains randomized at residues 82–84
in the β4-β5 loop and residues 133–139 in the β10-β11 loop [24] was screened by affinity
selection to isolate binders to ATF2 phospho-peptides. Through phage-display, we
isolated FHA domain variants that recognize the ATF2 dual-phosphosite epitope,
containing pT at only position 69, or at both positions 69 and 71, with greater selectivity
than a commercially available antibody.
2.3 Materials and Methods
2.3.1 Reagents
The peptides were synthesized at the University of Illinois at Chicago's Research
Resource Center with >90% purity. Each peptide contained a biotin molecule at their N-
terminus and an amine group at their C-terminus. The peptide targets used for affinity
selection and reagent characterization included IVADQpTPpTPTRFLKY (pT69-pT71),
IVADQTPTPTRFLKY (T69-T71), IVADQpTPTPTRFLKY (pT69-T71), and
IVADQTPpTPTRFLKY (T69-pT71). Peptide targets used for the phosphoserine
substitution study included IVADQpSPpTPTRFLKY (pS69-pT71),
IVADQpTPpSPTRFLKY (pT69-pS71), and IVADQpSPpSPTRFLKY (pS69-pS71).

39
The commercial antibody used to compare against the FHA variants was a mouse
monoclonal antibody (mAb) generated against the dual-phosphorylated form of an ATF2
peptide (Millipore Sigma, catalog# 05-891). A goat anti-mouse IgG, conjugated to
horseradish peroxidase (HRP; ThermoFisher Scientific, catalog# 62–6520), was used to
detect the mAb retained in washed microtiter plate wells. The anti-Flag IgG, clone M2,
conjugated to HRP (Sigma-Aldrich, catalog# A8592), was used to detect the FHA variants
retained in microtiter plate wells. An anti-ATF2 IgG, conjugated to Alexa Fluor 790 (Santa-
Cruz, catalog #sc-8398) was used to detect both unphosphorylated and phosphorylated
ATF2 on a membrane blot and an anti-FLAG IgG, conjugated to DyLight™ 800 (Rockland
Immunochemicals, catalog #600-445-383) was used to detect the FHA clone F6 bound
to the membrane blot.
2.3.2 Cloning and Bacterial Expression
The phagemid DNA isolated from affinity selection against the ATF2 dual-
phosphorylated peptide was digested with NcoI and NotI restriction endonucleases and
the FHA coding region was subcloned into the pKP600 phagemid vector [22]. This
construct included a C-terminally fused gene III coding sequence, flanked by AscI
restriction sites, an N-terminally-fused DsbA leader sequence, and a FLAG epitope tag.
Upon digestion with the AscI restriction endonuclease, the gene III coding region dropped
out, and when the plasmid was ligated back together, an in-frame His6-tag at the C-
terminal (pKP600ΔIII) was generated. All vectors were sequenced using the DsbA-Fw
oligonucleotide primer. All primers and oligonucleotides were ordered from Integrated
DNA Technologies, and their sequences can be found in Table III.

40
To generate each alanine-scanning mutant, the Kunkel mutagenesis protocol [25] was
followed using the F6 clone coding region in the pKP600 vector as the template and one
of ten oligonucleotides. Each oligonucleotide converted one of the ten amino acids in the
β4-5 and β10-11 loops to alanine.
TABLE III. LIST OF PRIMERS AND THEIR SEQUENCES USED IN THIS STUDY
Primer Sequence
DsbA-Fw 5'- CGC TGG CTG GTT TAG TTT TAG CGT -3'
P82A 5'- GAC AAA TAC GCA ATG TTA CCT AAG TG -3'
Y83A 5'- GTG TTT ATT AGA CAA CGC AGG AAT GTT ACC -3'
L84A 5'- CCC AGG AGG ATT TGA AAG TGT TTA TTA GAC GCA TAA GGA ATG -3'
A133G 5'- GAT CAT TAG GCT GCC CTA CCG TAA TTT CG -3'
Q134A 5'- TGA TCA TTA GGC GCC GCT ACC GTA ATT TCG -3'
P135A 5'- GAT CAT TCG CCT GCG CTA CCG TAA TTT CGT C -3'
N136A 5'- AAA TCC GAT GAT CCG CAG GCT GCG CTA C -3'
D137A 5' GCT TAA AAT CCG ATG CGC ATT AGG CTG CG -3'
H138A 5'- GCG CAG CCT AAT GAT GCC CGG ATT TTA AGC -3'
R139A 5'- GAC TAA GCT TAA AAT CGC ATG ATC ATT AGG CTG C -3'
epβ4-β5-FW 5'- GCG CCA TGG CGA TGG AAA ATA TTA -3'
epβ4-β5-RV 5'- CCA TTT GTT GAG ATG TCG TTG -3'
epβ10-β11-FW 5'- GGC TCA ACG GTC AAA AAG TAG -3'
epβ10-β11-RV 5'- TGC GGC CGC GGT ATT TTT AAG -3'
Protein expression and purification of the FHA variants was performed as described
previously [26]. Protein purity was assessed by sodium dodecyl sulfate-polyacrylamide
gel electrophoresis (SDS-PAGE) and the protein concentration was determined with a
NanoDrop spectrophotometer.

41
2.3.3 Enzyme-Linked Immunosorbent Assay (ELISA)
Biotinylated peptides (100 μL, 5 μg/μL) were immobilized on Nunc Maxisorp™ flat-
bottom 96 well plates coated with NeutrAvidin (100 μL, 5 μg/μL) and blocked with 2%
skim milk in Phosphate Buffered Saline (PBS; 137 mM NaCl, 3 mM KCl, 8 mM Na2HPO4,
1.5 mM KH2PO4). After washing the wells with PBS with Tween 20 (PBST; 137 mM NaCl,
3 mM KCl, 8 mM Na2HPO4, 1.5 mM KH2PO4, 0.1% Tween 20), purified FHA variants in
PBST (at a range of concentrations from 0.01 μM to 1 μM) or the anti-phospho-ATF2
(Thr69/71) antibody (Millipore Sigma) in PBST (1:1000 dilution) were added and
incubated for 1 h (h). The plates were washed three times with PBST and incubated with
the anti-FLAG M2-HRP antibody or goat-anti-mouse IgG (HRP) to monitor binding of the
FHA variants and anti-ATF2 antibody, respectively. Wells were washed three times with
PBST, and 100 μL of ABTS in 50 mM sodium citrate (pH 4), 0.03% hydrogen peroxide
was added to each well. The absorbance of the color change was measured at 405 nm
on a POLARstar OPTIMA microtiter plate reader (BMG Labtech)
2.3.4 Affinity Selection of the Primary Library All selection steps were performed at room temperature. One hundred μL of
Dynabeads® MyOne™ Streptavidin T1 magnetic beads (Invitrogen) were washed three
times with PBS and incubated with 1 μg of the biotinylated phosphopeptide for 30 min.
The beads were washed once with PBS to remove unbound target and incubated with
blocking buffer (2% skim milk in PBS. with 1 μM free biotin) for 1 h. The phage library
[22], with 2 × 109 unique members, was diluted in an equal volume of 4% skim milk in
PBS and incubated with the blocked beads for 1 h. The beads were washed three times
with PBST followed by two washes with PBS to remove weak-binding or non-binding

42
phage particles. Virions were eluted from the beads by incubating with 40 μg of TPCK-
treated trypsin (Sigma-Aldrich), diluted in 400 μL of 50 mM Tris-HCl (pH 8) and 1 mM
CaCl2, and used to infect 800 μL of TG1 cells (at mid-logarithmic growth phase) for 1 h at
37 °C. The cells were plated on one 15 cm 2 × YT/Carbenicillin (CB) agar plate and
incubated overnight at 30 °C. The next day, cells were scraped with 5 mL of 2xYT/CB
medium and approximately 109 cells were inoculated into 60 mL of 2xYT/CB medium.
Once the cells had grown to mid-logarithmic phase, 30 mL was infected with trypsin
cleavable helper phage (TM13KO7) [22]. Infected cells were centrifuged and
resuspended in 30 mL of 2xYT/CB/Kanamycin (Kan) medium and amplified for 24 h at
25°C/200 rpm, and precipitated with a polyethylene glycol (PEG)/NaCl mixture to
concentrate the virions 30-fold.
The second and third rounds of selection were completed in a similar manner,
however, 0.75 μg and 0.5 μg of immobilized target peptide was incubated with the beads,
respectively. Additionally, the number of washes before phage elution were increased,
and in the third round of selection, the phage/immobilized target mixture was incubated
with free dual-phosphorylated and mono-phosphorylated peptides at a 10-fold increase
as compared to the immobilized peptide concentration. After third round phage had
infected TG1 cells at mid-logarithmic phase, serial dilutions were made and plated on
2xYT/CB containing petri plates to produce single colonies. Ninety-six clones were
propagated as phage and used as probes in a phage ELISA as previously described [22].
Positive clones were sequenced and further characterized for specificity by protein
ELISA.

43
2.3.5 Secondary Library Construction and Affinity Selection
Phagemid DNA was recovered from the third round of selection. The pooled clones
were used as a template and epβ4-β5-Fw, epβ4-β5-RV, epβ10-β11-Fw, epβ10-β11-RV
as primers for PCR. The error-prone PCR method and construction of the secondary FHA
libraries were completed as described [27]. The amplified secondary libraries were
screened through three more rounds of selection following the primary selection scheme.
Additionally, a 100-fold and 1000-fold increase of non-biotinylated mono-phosphorylated
and dual-phosphorylated peptide (as compared to the immobilized peptide target
concentration) was incubated with the phage/target mixtures for the 2nd and 3rd rounds
of secondary library selection, respectively. Individual clones were propagated and tested
for binding in an ELISA.
2.3.6 Western Blot
25 μg of Tumor necrosis factor alpha (TNFα) stimulated and 25 μg of unstimulated
HeLa cell lysates (Santa Cruz) were run on an 8-16% gradient SDS-PAGE gel and the
resolved proteins were transferred to a polyvinylidene fluoride (PVDF) membrane. After
the transfer, the blot was blocked in 5% skim milk for 1 hr at room temperature with
rocking. The blot was then probed with the anti-ATF2 IgG conjugated to Alexa Fluor 790
(1:200 in PBST) or the FHA clone F6 (1μM in PBST) for 1 hr at room temperature with
rocking. The blot probed with the FHA was washed three times with PBST and then
incubated with anti-FLAG IgG, conjugated to DyLight™ 800 (1:2000 in PBST) for 1 hr at
room temperature. Each blot was washed three times with PBST and then imaged with
the Odyssey Fc imaging system (LI-COR).

44
2.4 Results and discussion
2.4.1 The FHA Domain Scaffold Can Recognize a Dual-Phosphorylated Epitope Discovering the importance of multiple phosphorylation states in single protein
epitopes has created a need for affinity reagents to monitor such states in cells.
Considering the inability of some antibodies to distinguish between the various
phosphorylation states, we utilized our FHA domain library to search for specific binders.
To determine if the phage library, displaying 2×109 FHA domain variants, contained
clones capable of recognizing a dual-phosphorylated epitope, we performed three rounds
of affinity selection with a biotinylated synthetic phosphopeptide whose sequence was
identical to a portion of the human ATF2 sequence, including phosphorylated threonine
69 (pT69) and phosphorylated threonine 71 (pT71) (Table IV). Three FHA variants were
isolated from affinity selection with the dual-phosphorylated peptide, which is consistent
with FHA reagent generation against other mono-phosphorylated targets [22]. Although
two of the clones and a commercial monoclonal antibody, generated against the same
peptide sequence, showed cross-reactive binding to the two mono-phosphorylated forms
of the peptide, clone F6 uniquely recognized the dual-phosphorylated peptide with little
or no binding to the other peptides (Figure 2.1). These results were consistent at varying
FHA concentrations (data not shown).
2.4.2 Characterization of the Interaction Between an FHA Variant Selective for the
Dual-Phosphorylated ATF2 Peptide
To examine the interaction between clone F6 and the target ATF2 peptide further, we
utilized ATF2 dual-phosphopeptide targets that contained pS substitutions at positions 69
(pS69-pT71), 71 (pT69-pS71), or both 69 and 71 (pS69-pS71) to determine recognition

45
TABLE IV. ATF2 PHOSPHOPEPTIDES
Nomenclature and respective sequences of the ATF2 peptide targets used in the study.
Sequences were taken from residues 64-77 of human ATF2 sequence. Note an additional
Tyrosine added to the C-terminus to determine concentration by absorbance. A “p”
preceding a “T” indicates phosphothreonine.
patterns for these residues (Figure 2.2). This FHA variant had a strong preference for the
pT69-pT71 and pT69-pS71 peptides, with almost a complete loss in signal when pT69
was replaced by pS. Because all known FHA domains and the FHA1 variants, which we
have previously generated, differentiate between pS and pT residues [7, 28], it is likely
that clone F6 recognizes pT69 of the ATF2 peptide in the same manner. This is unlike
clone F6′s recognition for pT71, which requires the presence of a phosphate group but
appears to not discriminate between phospho-threonine or phospho-serine. This is
surprising given that the Dun1-FHA domain requires the second phosphorylated residue
to also be pT for binding [23]; thus, it appears that FHA domains are capable of
recognizing a broad range of dual-phosphorylated epitopes through more than one
mechanism.

46
Figure 2.1. FHA variants can recognize the dual-phosphorylated ATF2 epitope.
Biotinylated peptides were captured via NeutrAvidin-coated microtiter plates and probed
with FHA variants and an anti-phospho-ATF2 (T69/71) monoclonal mouse antibody
(mAb). Complexes were detected by the M2-HRP and goat α-rabbit-HRP antibodies,
respectively. Error bars represent standard deviation of triplicate measurements.
Figure 2.2. The FHA library contains variants that recognize a pT-X-p(S/T) motifs.
ATF2 peptides, pT69-pT71, T69-T71, and peptides in which one or both threonine
residues were replaced with pS (pT69-pS71, pS69-pT71, pS69-pS71), were used as
targets in an ELISA. Clone F6 was used as a probe. Complexes were detected with the
M2-HRP antibody. The binding signal of clone F6 for the pT69-pT71 peptide was

47
normalized to 100% and the percentage binding of all other circumstances was calculated
accordingly. Error bars represent standard deviation of triplicate measurements.
To further unpack clone F6′s interaction with the peptide, we created a series of
mutations in clone F6, in which the β4-β5 and β10-β11 loop residues were systematically
replaced with alanine, in order to determine which residues in the loops contribute to
binding the dual-phosphorylated peptide [29]. The binding of each alanine-scanned
variant was assessed in an enzyme linked immunosorbent assay (ELISA; Figure 2.3).
Most residues when mutated to alanine appeared to have little effect on binding, however,
mutating tyrosine at position 83 to alanine (Y83A) led to a complete loss of binding of
clone F6 to the dual-phosphorylated ATF2 peptide. Previous studies have determined
that the FHA1 domain and other FHA variants are highly dependent on two characteristics
of the target for recognition: the presence of pT and a specific residue C-terminally
adjacent to pT. Position 83 on the FHA1 domain is important for its interaction with the
pT + 3 residue (the third residue C-terminal to pT) on the Rad9 target [24]. Our result is
consistent with this finding, and suggests that Y83 is important for maintaining the FHA-
target complex through interaction with a C-terminally adjacent residue, such as pT71, as
well.
Interestingly, mutating L84, N136, or H138 to alanine in clone F6 led to an increase in
binding to the pT69-T71 mono-phosphorylated peptide. Considering that asparagine
contains an amide group that often plays a role in hydrogen bonding and histidine is
positively charged at physiological pH, we speculate that N136 and H138 maintain a
selective dual-phosphosite recognition of the target through hydrogen bonding and
electrostatic interactions with the negatively charged phosphate group of pT71. When
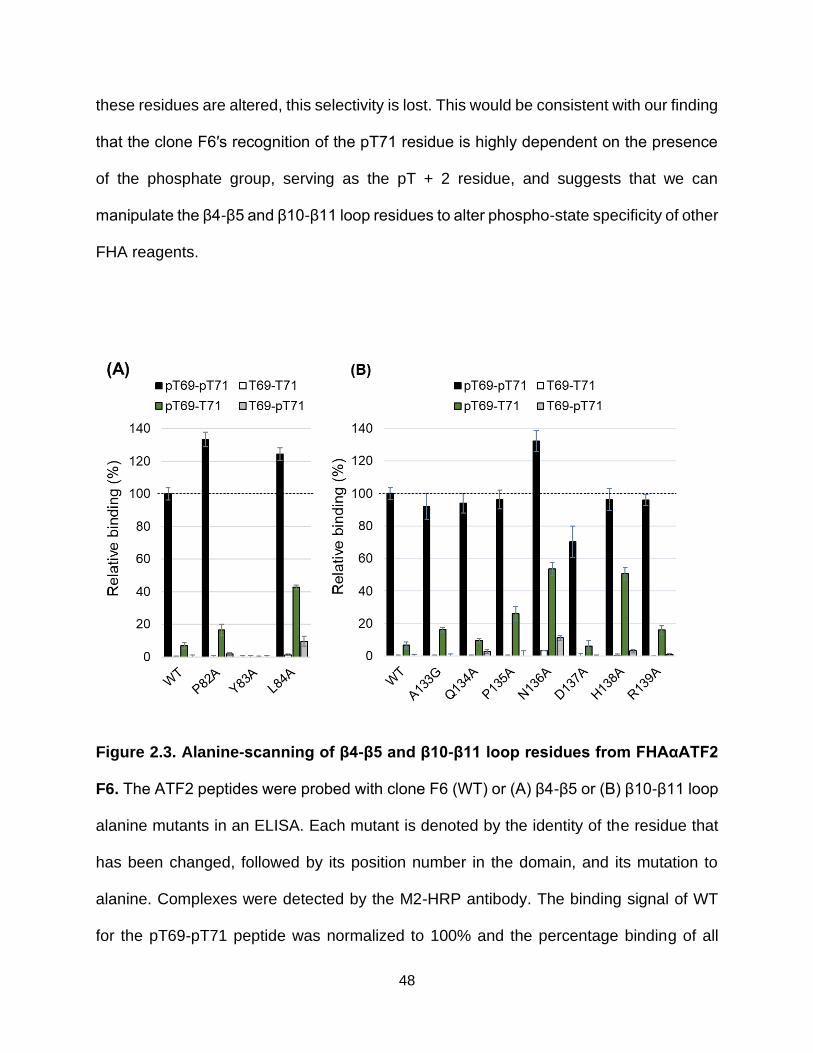
48
these residues are altered, this selectivity is lost. This would be consistent with our finding
that the clone F6′s recognition of the pT71 residue is highly dependent on the presence
of the phosphate group, serving as the pT + 2 residue, and suggests that we can
manipulate the β4-β5 and β10-β11 loop residues to alter phospho-state specificity of other
FHA reagents.
Figure 2.3. Alanine-scanning of β4-β5 and β10-β11 loop residues from FHAαATF2
F6. The ATF2 peptides were probed with clone F6 (WT) or (A) β4-β5 or (B) β10-β11 loop
alanine mutants in an ELISA. Each mutant is denoted by the identity of the residue that
has been changed, followed by its position number in the domain, and its mutation to
alanine. Complexes were detected by the M2-HRP antibody. The binding signal of WT
for the pT69-pT71 peptide was normalized to 100% and the percentage binding of all
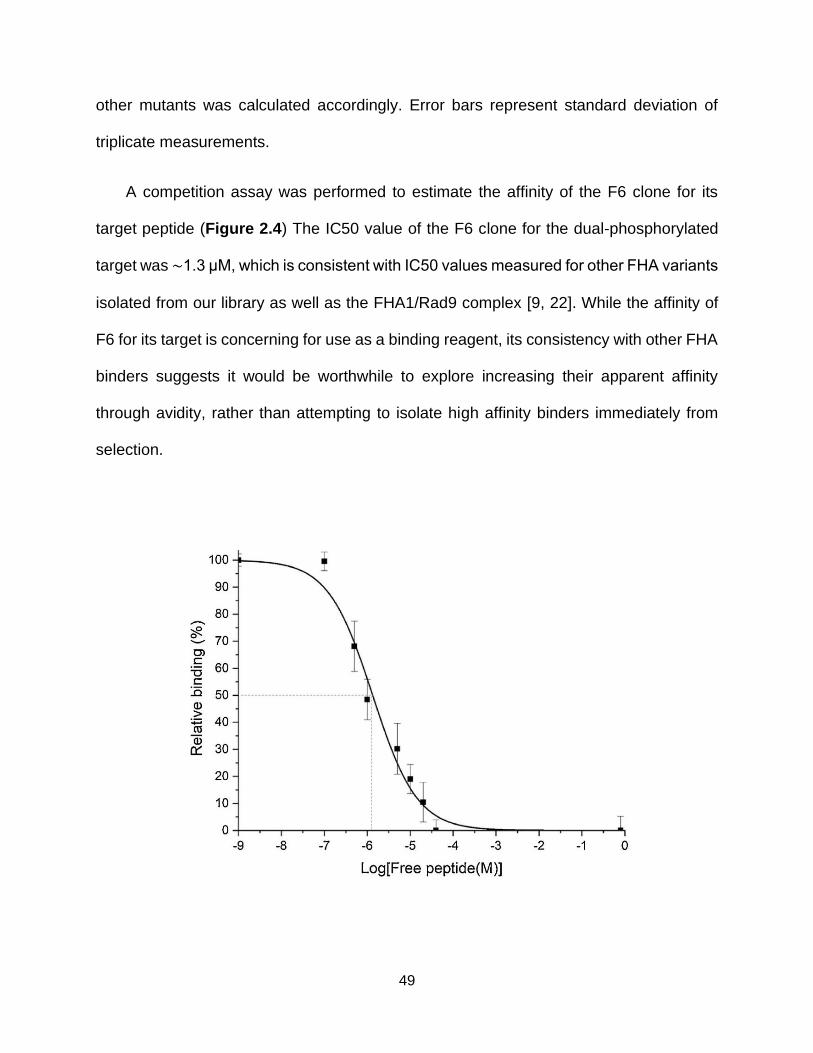
49
other mutants was calculated accordingly. Error bars represent standard deviation of
triplicate measurements.
A competition assay was performed to estimate the affinity of the F6 clone for its
target peptide (Figure 2.4) The IC50 value of the F6 clone for the dual-phosphorylated
target was ∼1.3 μM, which is consistent with IC50 values measured for other FHA variants
isolated from our library as well as the FHA1/Rad9 complex [9, 22]. While the affinity of
F6 for its target is concerning for use as a binding reagent, its consistency with other FHA
binders suggests it would be worthwhile to explore increasing their apparent affinity
through avidity, rather than attempting to isolate high affinity binders immediately from
selection.

50
Figure 2.4. Determining FHA variant F6′s affinity for its target. Competition binding
of clone F6 to immobilized dual-phosphorylated ATF2 peptide in the presence of free
dual-phosphorylated peptide. The dashed lines indicate an IC50 of 1.3 μM for clone F6
to the dual-phosphorylated peptide.
2.4.3 Generation of FHA Affinity Reagents to Specifically Recognize the Mono-
Phosphorylated Forms of the ATF2 Peptide
Given that alanine scanning of clone F6 revealed residues in the β4-β5 and β10-β11
loops play an important role in phospho-state specific recognition of the ATF2 peptide,
we hypothesized that we could screen the FHA library for complementary clones that
specifically recognized the two mono-phosphorylated forms of the ATF2 peptides. To our
surprise, no binders specific for either of the mono-phosphorylated peptides were
isolated, which prompted us to modify our affinity selection protocol. Instead, the library
was affinity selected over three rounds (with each peptide), secondary libraries were
generated through error-prone polymerase chain reaction (PCR), and the secondary
libraries, containing ∼2×107 variants, were affinity selected for three additional rounds
(Figure 2.5A). During the second and third rounds of affinity selection, beads were
incubated with a 100-fold and 1000-fold excess of the other phosphorylated ATF2
peptides, respectively [27]. By this method, two binders were isolated against the mono-
phosphorylated peptide, pT69-T71, of which one variant, G7, appeared to be specific
(Figure 2.5B). Aside from position 82 in the β4-β5 loop, which previously did not appear
to be a contributor to phospho-state specificity, the sequences of G7 and the other
isolated clone, C1, were identical with one notable difference: G7 carries a R164C
mutation (Table V). While we observed no change in binding pattern of clone G7 in the

51
presence of the reducing reagent, dithiothreitol (DTT), upon reverting residue 164 back
to arginine, selectivity for pT69-T71 peptide was lost and the binding pattern mimicked
that of the C1 clone (not shown). The importance of this residue is surprising given that
position 164 is located on the opposing surface of the β4-β5 and β10-β11 loops, however,
it is possible that this change slightly alters FHA folding and consequently its binding
surface.
Figure 2.5. Generation of reagents that specifically recognized the mono-
phosphorylated targets. (A) Workflow used to isolate FHA reagents against pT69-T71
peptide. (B) Reagents isolated after round 6 against pT69-T71 were characterized in an
ELISA in which immobilized ATF2 peptides were used as targets. The FHA variants were
used as probes. Complexes were detected by the M2-HRP antibody. The binding signal
of the variants to their cognate target peptide were normalized to 100% and the

52
percentage binding to all other peptides was calculated accordingly. Error bars represent
standard deviation of triplicate measurements.
TABLE V. OUTPUT SEQUENCES OF CLONES ISOLATED FROM SELECTIONS
β4-β5 and β10-β11 loops are the variable regions. Framework mutations were generated
by error-prone PCR.
Our new scheme (Figure. 2.5A) was not successful in yielding unique binders for the
second mono-phosphorylated ATF2 peptide, T69-pT71. While two FHA domain variants
were isolated and demonstrated capable of binding this peptide, they also bound the dual-
phosphorylated peptide to some degree (data not shown). The crystal structure of the
FHA1 domain in complex with the phosphorylated Rad9 peptide shows that the residues
N-terminally adjacent to the pT point away from the domain and have little interaction with
it [24]. We conclude that the FHA1 scaffold cannot be used to generate variants that can
discriminate between peptides with variations that are N-terminally adjacent to the pT

53
residue and that other alternatives must be explored. First, expanding the β4-β5 loop to
increase the domain’s proximity to the N-terminally adjacent residues or using another
naturally occurring FHA domain as a binding scaffold deserves examination. Secondly,
“affinity clamping”, in which two different affinity reagents are linked together in a
polypeptide, and can bind both sides of a target peptide, is another experimental option
[30]. Another scaffold, such as the fibronectin type III (FN3) monobody, could be linked
to the FHA variant and serve to bind the exposed N-terminal residues. Thirdly, it may also
be worthwhile to explore alternative scaffolds for generating recombinant affinity reagents
to proteins with and without various post-translational modifications. For example,
DARPins have been generated that can distinguish between non-phosphorylated and
dual-phosphorylated ERK [21].
2.4.4 Using an FHA Reagent to Detect Phospho-ATF2 in a Western Blot
Our FHA reagents were screened against phosphopeptides to ensure they were
targeting the desired phosphoresidues on ATF2. However, to have value in the scientific
community, we needed to demonstate that FHA variants are capable of recognizing full-
length phosphorylated cognate proteins. We performed a western blot and probed for
phosphorylated ATF2 (expected to resolve as a 60 kDa species in an SDS-PAGE gel) in
tumor necrosis factor alpha (TNFα) stimulated and unstimulated HeLa cell lysates. ATF2
remains unphosphorylated in unstimulated HeLa cells, and is rapidly di-phosphorylated
in stimulated cells [31].
A commercial antibody that recognizes both unphosphorylated and phosphorylated
ATF2 was used as a positive control; it detected the full-length protein in both stimulated
and unstimulated cell lysates as expected (Figure 2.6A). However, a commercial

54
antibody, which was generated against a phospho-ATF2 peptide, failed to bind ATF2 in
either stimulated or unstimulated cell lysates (data not shown). Conversely, when the blot
was probed with the FHA variant, clone F6, a reactive species was detected at ATF2’s
expected size in only the lysate prepared from TNFα stimulated cells. This was the only
band detected, illustrating that clone F6 is highly specific for full-length, dully-
phosphorylated ATF2. This further indicates that FHA recombinant affinity reagents are
not only comparable but can yield more accurate results than some commercial
antibodies.
Figure 2.6. Detection of full-length ATF2 in a western blot. TNFα stimulated and
unstimulated HeLa cell lysates were probed with (A) a commercial anti-ATF2 IgG
antibody conjugated to Alexa Fluor 790 or (B) the FHA clone F6, followed by an anti-
FLAG IgG antibody conjugated to DyLight 800. Blots were imaged with the Odyssey Fc
imaging system (LI-COR).

55
2.5 Conclusions
We demonstrate here that the FHA domain can distinguish between two of three
phospho-states of a peptide segment of the human transcription factor, ATF2 and can
recognize full-length phospho-ATF2 in a western blot. Due to our ability to manipulate the
FHA sequence to increase specificity, this domain is an appealing alternative to traditional
antibodies that lack such specificity and often cannot be further engineered because there
is limited knowledge about their sequence [32]. We anticipate that the FHA scaffold may
also prove useful for generating phospho-state specific reagents against other targets.
Proteins such as Extracellular signal-regulated kinase1/2 (ERK1/2) [33] and Checkpoint
kinase 2 (CHK2) [34] make intriguing candidate targets because they are also activated
by nearby sequential phosphorylation events containing phosphothreonine. Thus,
engineered FHA domains have the potential to complement commercial antibodies in
monitoring mono- and dual-phosphorylation of nearby threonines in eukaryotic proteins.
The field of recombinant affinity reagent generation continues to grow at an increasing
rate with unique and exciting ideas to overcome existing specificity and affinity limitations
of antibodies. Our FHA reagents have potential value as phospho-state specific probes.
By combining the promising possibilities that are developing in the field and our success
thus far, we hope to utilize a set of recombinant affinity reagents in cellular assays to
detect phosphorylated ATF2 protein and provide a tool for cell biologists to study
sequential phosphorylation.

56
2.6 Literature Cited
1. McGinnis, J. E. & Kay, B. K. (2018) Generation of recombinant affinity reagents against a two-phosphosite epitope of ATF2, New biotechnology. 45, 45-50.
2. Humphrey, S. J., James, D. E. & Mann, M. (2015) Protein Phosphorylation: A Major Switch Mechanism for Metabolic Regulation, Trends in Endocrinology & Metabolism. 26, 676-687.
3. Goel, R., Harsha, H. C., Pandey, A. & Prasad, T. S. K. (2012) Human Protein Reference Database and Human Proteinpedia as resources for phosphoproteome analysis, Mol Biosyst. 8, 453-463.
4. Nishi, H., Demir, E. & Panchenko, A. R. (2015) Crosstalk between Signaling Pathways Provided by Single and Multiple Protein Phosphorylation Sites, Journal of molecular biology. 427, 511-520.
5. Ferreon, J. C., Lee, C. W., Arai, M., Martinez-Yamout, M. A., Dyson, H. J. & Wright, P. E. (2009) Cooperative regulation of p53 by modulation of ternary complex formation with CBP/p300 and HDM2, Proceedings of the National Academy of Sciences. 106, 6591-6596.
6. Patwardhan, P. & Miller, W. T. (2007) Processive phosphorylation: Mechanism and biological importance, Cellular signalling. 19, 2218-2226.
7. Kholodenko, B. N., Hancock, J. F. & Kolch, W. (2010) Signalling ballet in space and time, Nature Reviews Molecular Cell Biology. 11, 414.
8. Liu, H., Deng, X., Shyu, Y. J., Li, J. J., Taparowsky, E. J. & Hu, C.-D. (2006) Mutual regulation of c-Jun and ATF2 by transcriptional activation and subcellular localization, The EMBO journal. 25, 1058-1069.
9. Durocher, D., Henckel, J., Fersht, A. R. & Jackson, S. P. (1999) The FHA Domain Is a Modular Phosphopeptide Recognition Motif, Molecular cell. 4, 387-394.
10. Bhoumik, A. & Ronai, Z. e. (2008) ATF2: A transcription factor that elicits oncogenic or tumor suppressor activities, Cell cycle. 7, 2341-2345.

57
11. Zhang, J.-Y., Jiang, H., Gao, W., Wu, J., Peng, K., Shi, Y.-F. & Zhang, X.-J. (2008) The JNK/AP1/ATF2 pathway is involved in H2O2-induced acetylcholinesterase expression during apoptosis, Cellular and Molecular Life Sciences. 65, 1435-1445.
12. Ouwens, D. M., de Ruiter, N. D., van der Zon, G. C. M., Carter, A. P., Schouten, J., van der Burgt, C., Kooistra, K., Bos, J. L., Maassen, J. A. & van Dam, H. (2002) Growth factors can activate ATF2 via a two-step mechanism: phosphorylation of Thr71 through the Ras–MEK–ERK pathway and of Thr69 through RalGDS–Src–p38, The EMBO journal. 21, 3782-3793.
13. Nagata, K.-i., Izawa, I. & Inagaki, M. (2001) A decade of site- and phosphorylation state-specific antibodies: recent advances in studies of spatiotemporal protein phosphorylation, Genes to Cells. 6, 653-664.
14. Jczernik, A., Girault, J.-A., Nairn, A. C., Chen, J., Snyder, G., Kebabian, J. & Greengard, P. (1991) [23] Production of phosphorylation state-specific antibodies in Methods in Enzymology pp. 264-283, Academic Press.
15. Mandell, J. W. (2003) Phosphorylation State-Specific Antibodies: Applications in Investigative and Diagnostic Pathology, The American Journal of Pathology. 163, 1687-1698.
16. Bradbury, A. R. M., Sidhu, S., Dübel, S. & McCafferty, J. (2011) Beyond natural antibodies: the power of in vitro display technologies, Nature biotechnology. 29, 245-254.
17. Kaneko, T., Huang, H., Cao, X., Li, X., Li, C., Voss, C., Sidhu, S. S. & Li, S. S. (2012) Superbinder SH2 domains act as antagonists of cell signaling, Science signaling. 5, ra68.
18. Olson, C. A., Liao, H.-I., Sun, R. & Roberts, R. W. (2008) mRNA Display Selection of a High-Affinity, Modification-Specific Phospho-IκBα-Binding Fibronectin, ACS chemical biology. 3, 480-485.
19. Shih, H. H., Tu, C., Cao, W., Klein, A., Ramsey, R., Fennell, B. J., Lambert, M., Ni Shuilleabhain, D., Autin, B., Kouranova, E., Laxmanan, S., Braithwaite, S., Wu, L., Ait-Zahra, M., Milici, A. J., Dumin, J. A., LaVallie, E. R., Arai, M., Corcoran, C., Paulsen, J. E., Gill, D., Cunningham, O., Bard, J., Mosyak, L. & Finlay, W. J. (2012) An ultra-specific avian antibody to phosphorylated tau protein reveals a unique mechanism for phosphoepitope recognition, The Journal of biological chemistry. 287, 44425-34.

58
20. Vielemeyer, O., Yuan, H., Moutel, S., Saint-Fort, R., Tang, D., Nizak, C., Goud, B., Wang, Y. & Perez, F. (2009) Direct Selection of Monoclonal Phosphospecific Antibodies without Prior Phosphoamino Acid Mapping, Journal of Biological Chemistry. 284, 20791-20795.
21. Kummer, L., Parizek, P., Rube, P., Millgramm, B., Prinz, A., Mittl, P. R., Kaufholz, M., Zimmermann, B., Herberg, F. W. & Pluckthun, A. (2012) Structural and functional analysis of phosphorylation-specific binders of the kinase ERK from designed ankyrin repeat protein libraries, Proceedings of the National Academy of Sciences of the United States of America. 109, E2248-57.
22. Pershad, K., Wypisniak, K. & Kay, B. K. (2012) Directed evolution of the forkhead-associated domain to generate anti-phosphospecific reagents by phage display, Journal of molecular biology. 424, 88-103.
23. Lee, H., Yuan, C., Hammet, A., Mahajan, A., Chen, E. S., Wu, M. R., Su, M. I., Heierhorst, J. & Tsai, M. D. (2008) Diphosphothreonine-specific interaction between an SQ/TQ cluster and an FHA domain in the Rad53-Dun1 kinase cascade, Molecular cell. 30, 767-78.
24. Durocher, D., Taylor, I. A., Sarbassova, D., Haire, L. F., Westcott, S. L., Jackson, S. P., Smerdon, S. J. & Yaffe, M. B. (2000) The Molecular Basis of FHA Domain:Phosphopeptide Binding Specificity and Implications for Phospho-Dependent Signaling Mechanisms, Molecular cell. 6, 1169-1182.
25. Kunkel, T. A. (1985) Rapid and efficient site-specific mutagenesis without phenotypic selection, Proceedings of the National Academy of Sciences. 82, 488-492.
26. Kierny, M. R., Cunningham, T. D., Bouhenni, R. A., Edward, D. P. & Kay, B. K. (2015) Generating Recombinant Antibodies against Putative Biomarkers of Retinal Injury, PLoS One. 10, e0124492-e0124492.
27. Huang, R., Gorman, K. T., Vinci, C. R., Dobrovetsky, E., Gräslund, S. & Kay, B. K. (2015) Streamlining the Pipeline for Generation of Recombinant Affinity Reagents by Integrating the Affinity Maturation Step, Int J Mol Sci. 16, 23587-23603.
28. Venegas, L. A., Pershad, K., Bankole, O., Shah, N. & Kay, B. K. (2016) A comparison of phosphospecific affinity reagents reveals the utility of recombinant Forkhead-associated domains in recognizing phosphothreonine-containing peptides, New biotechnology. 33, 537-43.

59
29. Morrison, K. L. & Weiss, G. A. (2001) Combinatorial alanine-scanning, Current Opinion in Chemical Biology. 5, 302-307.
30. Huang, J., Makabe, K., Biancalana, M., Koide, A. & Koide, S. (2009) Structural Basis for Exquisite Specificity of Affinity Clamps, Synthetic Binding Proteins Generated through Directed Domain-interface Evolution, Journal of molecular biology. 392, 1221-1231.
31. Winston, B. W., Lange-Carter, C. A., Gardner, A. M., Johnson, G. L. & Riches, D. W. (1995) Tumor necrosis factor alpha rapidly activates the mitogen-activated protein kinase (MAPK) cascade in a MAPK kinase kinase-dependent, c-Raf-1-independent fashion in mouse macrophages, Proceedings of the National Academy of Sciences. 92, 1614-1618.
32. Venegas, L. (2017) Reagents for detecting phosphosites within proteins, Journal of Bioinformatics, Genomics and Proteomics.
33. Ahmed, S., Grant, K. G., Edwards, L. E., Rahman, A., Cirit, M., Goshe, M. B. & Haugh, J. M. (2014) Data-driven modeling reconciles kinetics of ERK phosphorylation, localization, and activity states, Mol Syst Biol. 10, 718-718.
34. Guo, X., Ward, M. D., Tiedebohl, J. B., Oden, Y. M., Nyalwidhe, J. O. & Semmes, O. J. (2010) Interdependent Phosphorylation within the Kinase Domain T-loop Regulates CHK2 Activity, Journal of Biological Chemistry. 285, 33348-33357.

60
Chapter 3 3. A RECOMBINANT AFFINITY REAGENT SPECIFIC FOR A
PHOSPHOEPITOPE OF AKT1
This research has previously been published in International journal of molecular
sciences [1].
This work was done in collaboration with Dr. Leon Venegas and Dr. Brian Kay. Dr.
Venegas helped to generate binding reagents to Akt1 and write the methods section and
Dr. Kay helped with experimental design and editing.
3.1 Abstract
The serine/threonine-protein kinase, Akt1, plays an important part in mammalian cell
growth, proliferation, migration and angiogenesis, and becomes activated through
phosphorylation. To monitor phosphorylation of threonine 308 in Akt1, we developed a
recombinant phosphothreonine-binding domain that is highly selective for the Akt1
phosphopeptide. A phage-display library of variants of the Forkhead-associated 1 (FHA1)
domain of yeast Rad53p was screened by affinity selection to the phosphopeptide, 301-
KDGATMKpTFCGTPEY-315, and yielded 12 binding clones. The strongest binders have
equilibrium dissociation constants of 160-180 nanomolar and are phosphothreonine-
specific in binding. The specificity of one FHA variant was compared to commercially
available polyclonal antibodies (pAbs) generated against the same phosphopeptide. The
clone was either as equal to or better than three pAbs, in detecting the Akt1 pT308
phosphopeptide in ELISAs.
3.2 Introduction
The serine/threonine-protein kinase, Akt1, is responsible for regulating a range of
biochemical pathways involved in cell proliferation and survival. It contains an N-terminal

61
pleckstrin homology (PH) domain, a serine/threonine kinase catalytic domain, and a C-
terminal regulatory domain [2, 3]. Activation of Akt1 is dependent on recruitment of the
protein, through its PH domain [4], to the inner side of the plasma membrane, which
causes a conformational change [5], allowing PDK1 to phosphorylate threonine 308
(T308) in Akt1’s catalytic domain and mTORC2 to phosphorylate serine 473 (S473) in
Akt1’s regulatory domain [6, 7]. Once these two residues are phosphorylated, Akt1 is fully
active and phosphorylates a range of intracellular proteins involved in cell survival,
growth, proliferation, cell migration and angiogenesis [8].
Given the critical roles that Akt1 serves in the cell, biologists are extremely interested
in understanding its involvement in cancer. Mass spectrometry and phospho-specific
antibodies have been essential tools in pursuing this question by tracking Akt1’s
phosphorylation state and levels in cells and tissues [9, 10]. Such methods have shown
a strong link between the hyperactivation of Akt1 through increased phosphorylation
levels in breast, prostate [11], ovarian [12], and pancreatic cancer [13]. Additionally,
studying phosphorylation of specific residues within a protein can provide valuable
information as some diseases are marked by the excessive phosphorylation of only one
or a few of these residues. For example, the phosphorylation of T308, but not of S473,
has been characterized as a marker of lung cancer [14]. Thus, antibodies that recognize
specific phosphorylated residues as part of their epitopes serve as valuable diagnostic
tools to distinguish between diseases caused by Akt1 deregulation.
Unfortunately, mass spectrometry is not well suited to monitoring protein
phosphorylation at the cytological level, and antibodies are often poorly validated,
unsequenced, and not amenable to protein engineering [15]. To circumvent these

62
limitations, current efforts have been focused on generating engineered protein scaffolds
that recognize phosphoepitopes, such as the 10th fibronectin type III domain (10FnIII)
[16], designed ankyrin repeat proteins (DARPins) [17], the Src Homology 2 domain (SH2)
[18], single chain variable fragments (scFv) [19], antigen binding fragments (Fab) [20],
and the Forkhead-associated (FHA) domain [21]. Unlike other scaffolds and most
antibodies, FHA domains are selective for phosphothreonine (pT)-containing targets due
to a pocket on the domain that interacts with phosphate and γ-methyl group of
phosphothreonine (pT) [22, 23]. Because of this unique characteristic, a phage library
displaying FHA1 variants randomized at residues 82-84 in the β4-β5 loop and residues
133-139 in the β10-β11 loop has been employed to generate affinity reagents to a variety
of targets [21, 22, 24]. Here, we describe the isolation and characterization of Akt1
phosphothreonine 308 (pT308)-binding reagents. We show that these reagents are pT-
dependent, bind with high affinity, and recognize the phosphopeptide with comparable or
better specificity than commercially made antibodies.
3.3 Materials and Methods
3.3.1 Peptides
Peptides were synthesized at University of Illinois at Chicago’s Research Resource
Center with >90% purity. All peptides are biotinylated at their N-terminus and amidated at
their C-termini. A phosphopeptide corresponding to human Akt1 was
KDGATMKpTFCGTPEY (Akt1-pT308) used as the target in phage display affinity
selection. In addition, a number of related peptides were synthesized:
KDGATMKTFCGTPEY (T308), KDGATMKpSFCGTPEY (pT308pS),
KDGATMKpYFCGTPEY (pT308pY), KDGATMKDFCGTPEY (pT308D), and

63
KDGATMKEFCGTPEY (pT308E), KDGAAMKpTACGTPEY (T305A),
KDGATAKpTFCGTPEY (M306A), KDGATMApTFCGTPEY (K307A),
KDGATMKpTACGTPEY (F309A), KDGATMKpTFAGTPEY (C310A),
KDGATMKpTFCATPEY (G311A), KDGATMKpTFCGAPEY (T312A), and
KDGAAAAApTAAAAAPEY (Ala).
Polyclonal antibodies (pAbs) to the Akt1-pT308 peptide were purchased from Abcam,
Cell Signaling Technology, Millipore, and ThermoFisher Scientific. The secondary
reagent for Akt1-pTBD detection was the anti-Flag epitope mAb, M2, conjugated to
horseradish peroxidase (HRP), was purchased from Sigma–Aldrich. The secondary
reagent for the pAbs was a goat anti-rabbit immunoglobulin G (IgG), conjugated to HRP
(Abcam).
3.3.2 Cloning and Bacterial Expression of Proteins
The phagemid DNA isolated from affinity selection against the Akt1-pT308 peptide
were subcloned into the pKP600ΔIII vector [25], and expressed and purified as described
elsewhere [23]. Protein purity was assessed by sodium dodecyl sulfate-polyacrylamide
gel electrophoresis (SDS-PAGE), and protein concentrations were measured with a
NanoDrop A280 spectrophotometer.
3.3.3 Affinity Selections
To isolate FHA variants, the FHA1G2 library [21] was screened against the Akt1-
pT308 peptide through three rounds of affinity selection using a modified version of a
previously described protocol [21]. Affinity selection was performed at room temperature
with the KingFisher™ mL device Purification System (ThermoFisher Scientific). The
biotinylated peptide (3 ng/μL, 400 μL) was immobilized with Dynabeads™ M-270

64
Streptavidin (ThermoFisher Scientific) and blocked with 2% skim milk in phosphate
buffered saline (PBS; 137 mM NaCl, 3 mM KCl, 8 mM Na2HPO4, 1.5 mM KH2PO4). A
ten-fold excess of the phage library containing 2×109 members was incubated with the
blocked target for 1 hour. Weak or non-binding phage variants were removed by washing
the mixture three times with PBS plus 0.1% Tween 20 (PBST) followed by three times
with PBS. Virions, which contain a trypsin-cleavable site in the helper M13 viral coat
protein III [26], were eluted from the beads with 40 µg of tosyl phenylalanyl chloromethyl
ketone-treated trypsin (Sigma-Aldrich), diluted in 400 µL of 50 mM Tris-HCl (pH 8) and 1
mM CaCl2, and used to infect 800 μL of Escherichia coli TG1 cells (at mid-logarithmic
growth phase) for 1 h at 37°C. The cells were plated onto one 15 cm petri plates
containing 2xYT medium (per liter 16 g tryptone, 10 g yeast extract, 5 g NaCl), 1.4% agar,
and 0.5 µg/µL carbenicillin, incubated overnight at 37ºC, scraped the next day, and phage
amplified. Phage particles were precipitated with 24% polyethylene glycol 8000, 3 M NaCl
and the pellet was resuspended in 0.6 mL of PBS. and virions concentrated 30-fold. The
second and third rounds of affinity selection were performed in a similar manner; however,
the Akt1-pT308 peptide concentration for rounds two and three were reduced to 750 nM
and 500 nM in 400 μL of PBS, respectively. Additionally, in the third round of affinity
selection, a 10-fold excess of non-biotinylated Akt1-pT308 peptide was added during the
wash steps. After the third round of affinity selection, 96 individual clones were
propagated as phage, and used in an ELISA to identify clones that bind to the peptide
target. The DNA inserts of positive, binding clones were sequenced.

65
3.3.4 Enzyme-Linked Immunosorbent Assay (ELISA)
Biotinylated peptides diluted in PBS were incubated overnight in 0.5 mM DTT at 4°C.
ELISAs were performed as previously described [22, 24] using the peptide targets
incubated with dithiothreitol (DTT) at a concentration of 2.5 µM in 100 μL and FHA
variants at concentrations varying from 0.01 to 10 μM in PBST. The absorbance was read
at 405 nm.
3.3.5 Surface Plasmon Resonance
The affinity of FHA variants E12 and H11 to the pT308 and T308 biotinylated peptides
was measured as previously described [27].
3.4 Results and Discussion
3.4.1 Directed Evolution of the FHA1 Domain Yielded Variants that Recognize an
Akt1 Phosphopeptide
To generate recombinant affinity reagents that recognize Akt1-pT308, a peptide with
a phosphothreonine (pT) residue at position 308 in a 13-mer peptide (positions 302 to
314) was synthesized with a C-terminal biotin (Figure 3.1). A phage-display FHA library,
containing 2 × 109 members [21], was affinity selected with the Akt1-pT308 peptide. After
three rounds of affinity selection, 96 recovered clones were analyzed by ELISA and 12
were confirmed to bind to the Akt1-pT308 peptide (Figure 3.2). None of the 12 binding
clones demonstrated any binding to the non-phosphorylated form of the 302–314 Akt1
peptide.

66
Figure 3.1. Primary structure of the Akt1 protein. The kinase consists of an N-terminal
pleckstrin homology (PH) domain (blue), a serine/threonine kinase catalytic domain
(yellow), and a C-terminal regulatory domain (pink). The amino acid sequence from
residues 301 to 314, including pT308, was used in peptide form for affinity selection
experiments.
Figure 3.2. Affinity selection process and ELISA of 12 output clones. (A) An M13
bacteriophage library, displaying variants of a thermostable Forkhead-associated 1
(FHA1) H domain, was screened through three rounds (R1–R3) of affinity selection.

67
During R3, excess phosphopeptide was added for off-rate competition, and recovered
clones were examined for binding (B) Biotinylated Akt1-pT308 peptide was captured on
a neutravidin-coated plate and probed with FHA variants that were detected by an M2-
HRP conjugated antibody. Error bars represent standard deviation of triplicate
measurements.
DNA sequencing of 12 selected FHA domains revealed that all were unique. A
comparison of their coding sequences demonstrated that many of them shared amino
acid sequences in the two regions that were randomized in the FHA domain scaffold
(Figure 3.3A). The consensus motif in the β4-β5 and β10-β11 loops determined by a
WebLogo plot [28, 29] is (S/A)Y(Y/R) and (S/T)(P/A)x(R/I)(P/E)(S/D)(H/A), respectively
(Figure 3.3B). We infer from this finding that the semiconserved positions contribute to
binding of the Akt1-pT308 peptide. Correlated with this observation, clones E12 and
binding of the Akt1-pT308 peptide. Correlated with this observation, clones E12 and H11
closely resemble the consensus sequence, and are the strongest binders, whereas
clones E1 and B3 are more diverged and are the weakest binders. Among the residues
shared most often among the binders is tyrosine at position 83, consistent with previous
conclusions that position 83 is extremely important for FHA domain interactions with their
phosphopeptide targets [24, 27, 30].
Two of the affinity selected FHA domains bind the Akt1 phosphopeptide with high
affinity. To estimate the binding strength of the affinity selected FHA domain clones for
the pT308 peptide, we performed competition binding assays. Half maximal inhibitory
concentration (IC50) values for clones B3, E1, E12 and H11 were 100 μM, 1.95 μM, 45
nM and 90 nM, respectively. (Figure 3.4A). These relative values correlated well with

68
ELISA results: clone B3 was the weakest, clones E12 and H11 were the strongest
binders, and clone E1 was an intermediate binder (Figure 3.4B). We then performed
surface plasmon resonance (SPR) for two clones, E12 and H11, and determined
equilibrium dissociation constants (KD) of 162 ± 12 nM and 178 ± 8 nM, respectively
(Table VI). These clones are among the tightest binders that we have isolated [19].
Figure 3.3. Amino acid sequence analysis of two loops randomized in the phage-
displayed scaffold. (A) Primary sequences of the wild-type (WT) form of the scaffold
and 12 variants that bind the Akt1 phosphopeptide. Three and seven residues in the β4-
β5 or β10-β11 loops, respectively, were randomized with NNK codons, where N is an
equimolar mixture of A, C, G and T and K is an equimolar mixture of G and T. Residues
that differ from the wild-type sequences are show in red. (B) WebLogo plots of the
frequency of particular residues at each position (82–84 or 133–139). The height of a
residue refers to probability of the residue at the given position. Hydrophobic, polar, and
charged residues are shown in black, green, and blue, respectively.

69
Figure 3.4. Comparing the relative affinity of four clones. (A) Competition binding of
clone B3, E1, H11 and E12 to immobilized phosphorylated Akt1 peptide in the presence
of free phosphorylated peptide. Error bars represent standard deviation of triplicate
measurements. (B) IC50 values for each clone to the Akt1 phosphorylated peptide.
TABLE VI. AFFINITY MEASUREMENTS
Affinity measurements of FHA clone E12 and H11 to the phosphorylated (Akt1-pT308)
and unphosphorylated (Akt1-T308) peptides determined by SPR.
FHA Variant
E12 H11
Peptide Target Ka (M-1s-1) Kd (s-1) KD (nM) Ka (M-1s-1) Kd (s-1) KD (nM)
Akt1-pT308 4.830*104 7.821*10-3 162±12 4.157*104 7.401*10-3 178±8
Akt1-T308 2.302 2.144*10-3 9.31*105 2.165 1.01083*10-2 5.002*106

70
3.4.2 An Isolated FHA Clone binds the Akt1 Peptide with Unique Specificity
Due to its unexpected high affinity for its phosphopeptide ligand, we chose to evaluate
clone E12 further. The specificity of this FHA variant was examined by testing for binding
to peptides that substituted phosphothreonine (pT) in the phosphopeptide with
phosphoserine (pS) or phosphotyrosine (pY). As seen in Figure 3.5, E12 only binds the
peptide when position 308 is pT. On the other hand, three commercially made polyclonal
antibodies, which were generated against the same or a similar Akt1 phosphopeptide,
varied in their specificity. While one antibody (pAb 1) demonstrated a similar level of
selectivity as clone E12, another (pAb 2) lacked specificity, and a third did not bind any
of the peptides (data not shown).
Figure 3.5. Binding of an FHA variant to a set of related peptides. A set of biotinylated
Akt1 phosphopeptides were synthesized with threonine (Akt-T308), pS (Akt-pS308) or pY
(Akt-pY308) substituted at the pT position and immobilized by NeutrAvidin. Binding of the
E12 variant and two commercially produced polyclonal antibodies were determined by an
ELISA. Error bars represent standard deviation of triplicate measurements.

71
To learn about what residues in the peptide ligand contributed to binding to clone E12,
we tested a set of alanine scanned phosphopeptides. Seven biotinylated peptides, with
each position replaced one at a time by alanine, were captured in neutravidin-coated
microtiter plate wells and probed separately with a pAb and clone E12 (Figure 3.6). The
pAb bound most alanine-scanned peptides as well as or better than the wild-type
sequence, except for alanine replacement at positions −3 and +2 in the phosphopeptide
sequence. By convention the phosphorylated amino acid is defined as 0 and resides N-
terminal and C-terminal to are numbered − and +, respectively. The pAb did not bind,
however, to the non-phosphorylated peptide or to a peptide with alanine flanking the
central pT position. Conversely, the E12 clone demonstrated very little or no binding to
all of the test peptides. Additionally, it had a reduced ability to bind corresponding peptides
from isoforms Akt2 or Akt3 whose sequences differ by only one or two amino acids in
positions not tested by the alanine scan (Figure 3.7). This was surprising since previous
observations show that FHA domains recognize only a few nearby residues in addition to
the central pT. In this case, though, it appears that many of the residues that flank the
central pT in the Akt1 phosphopeptide contribute directly or indirectly to binding.
Preliminary molecular dynamic studies of the peptide and the solved structure of
activated Akt [31] suggest hairpin loops form on both sides of the pT residue, which could
explain why so many adjacent residues are important directly or indirectly for the binding
interaction. While more biophysical experiments are needed to dissect further details
regarding the binding interaction, the apparent contribution of multiple flanking residues
to the interaction correlates with the unusually tight binding observed for the E12 variant
and suggests further mutagenesis could increase its specificity for the Akt1 isoform.

72
Figure 3.6. Identification of important residues on the peptide by alanine scanning.
Alanine (A) was substituted one at a time at the −3, −2, −1, +1, +2, +3 and +4 positions
of the Akt1 peptide. Binding of E12 and a polyclonal antibody (pAb) to the wild type Akt1
peptide target was set to 100%, and the phosphopeptide variants were normalized
against it in an ELISA. Error bars represent standard deviation of triplicate measurements.

73
Figure 3.7. Binding of an FHA variant to corresponding peptides from Akt2 and
Akt3. A set of biotinylated phosphopeptides containing the Akt1 sequence, the Akt2
corresponding sequence (SDGATMKpTFCGTPE) and the Akt3 corresponding sequence
(TDAATMKpTFCGTPE) were immobilized by neutravidin. Binding of the E12 variant and
a commercially produced polyclonal antibody (pAb) were determined by an ELISA. Error
bars represent standard deviation of triplicate measurements.
Published results demonstrate that the three residues C-terminally adjacent to the pT
residue, most notably the +3 residue, contribute significantly to FHA domain interactions
with peptides [21, 22, 24, 27]. While many of the 20 amino acids occur among
phosphopeptide ligands for FHA domains, the Akt1 pT308 FHA variant is the first one
observed to have glycine at the +3 position. Glycine’s small side group and
conformational flexibility together may explain why it is not commonly positioned at
protein-protein interfaces [32]. Thus, it seems likely that these FHA variants are
compensating by interacting more strongly with the remaining peptide residues, and
therefore, have a higher specificity and affinity for Akt1 peptide target than other reported
FHA binding domains. Given this, clone E12 may serve as an attractive reagent for
probing full length phosphorylated Akt1 in western blots, fixed cells, or tissue sections.
Such experiments will provide further insight into clone E12’s potential as a diagnostic
reagent.

74
3.5 Conclusions
Herein, we demonstrate the directed evolution of the FHA domain to bind a
phosphorylated peptide that corresponds to a segment of the phosphorylated,
oncoprotein, Akt1. The work represented in this paper bolsters the utility of the
recombinant FHA domain to serve as an alternative to traditional antibodies for detecting
phosphothreonine in peptide sequences. Among the 12 variants isolated from a phage-
display FHA domain library, we discovered two that bind the Akt1 phosphopeptide,
KDGATMKpTFCGTPEY, with 160–180 nM affinity. This is the strongest interaction
between a peptide target and an FHA variant isolated from our library to date.
While these FHA variants have yet to be employed as binding reagents against full
length protein targets, this FHA probe has the potential to detect phosphorylated Akt1
protein in vivo and/or in vitro due to this high affinity. Thus, we have achieved an early
milestone in our goal to replace anti-phosphothreonine antibodies with engineered
recombinant affinity reagents. Further work to optimize assay detection methods will
prove this FHA variant’s potential as a detection and diagnostic reagent.

75
3.6 Literature Cited
1. McGinnis, J. E., Venegas, L. A., Lopez, H. & Kay, B. K. (2018) A Recombinant Affinity Reagent Specific for a Phosphoepitope of Akt1, Int J Mol Sci. 19, 3305.
2. Liu, P., Wang, Z. & Wei, W. (2014) Phosphorylation of Akt at the C-terminal tail triggers Akt activation, Cell cycle. 13, 2162-4.
3. Manning, B. D. & Toker, A. (2017) AKT/PKB Signaling: Navigating the Network, Cell. 169, 381-405.
4. Stephens, L., Anderson, K., Stokoe, D., Erdjument-Bromage, H., Painter, G. F., Holmes, A. B., Gaffney, P. R., Reese, C. B., McCormick, F., Tempst, P., Coadwell, J. & Hawkins, P. T. (1998) Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B, Science. 279, 710-4.
5. Huang, B. X. & Kim, H. Y. (2006) Interdomain conformational changes in Akt activation revealed by chemical cross-linking and tandem mass spectrometry, Molecular & cellular proteomics : MCP. 5, 1045-53.
6. Alessi, D. R., Andjelkovic, M., Caudwell, B., Cron, P., Morrice, N., Cohen, P. & Hemmings, B. A. (1996) Mechanism of activation of protein kinase B by insulin and IGF-1, The EMBO journal. 15, 6541-51.
7. Sarbassov, D. D., Guertin, D. A., Ali, S. M. & Sabatini, D. M. (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex, Science. 307, 1098-101.
8. Manning, B. D. & Cantley, L. C. (2007) AKT/PKB signaling: navigating downstream, Cell. 129, 1261-74.
9. Kuo, Y. C., Huang, K. Y., Yang, C. H., Yang, Y. S., Lee, W. Y. & Chiang, C. W. (2008) Regulation of phosphorylation of Thr-308 of Akt, cell proliferation, and survival by the B55alpha regulatory subunit targeting of the protein phosphatase 2A holoenzyme to Akt, The Journal of biological chemistry. 283, 1882-92.
10. Wang, F., Zhang, W., Guo, L., Bao, W., Jin, N., Liu, R., Liu, P., Wang, Y., Guo, Q. & Chen, B. (2014) Gambogic acid suppresses hypoxia-induced hypoxia-inducible factor-1alpha/vascular endothelial growth factor expression via inhibiting phosphatidylinositol 3-kinase/Akt/mammalian target protein of rapamycin pathway in multiple myeloma cells, Cancer science. 105, 1063-70.

76
11. Nakatani, K., Thompson, D. A., Barthel, A., Sakaue, H., Liu, W., Weigel, R. J. & Roth, R. A. (1999) Up-regulation of Akt3 in estrogen receptor-deficient breast cancers and androgen-independent prostate cancer lines, The Journal of biological chemistry. 274, 21528-32.
12. Yuan, Z. Q., Sun, M., Feldman, R. I., Wang, G., Ma, X., Jiang, C., Coppola, D., Nicosia, S. V. & Cheng, J. Q. (2000) Frequent activation of AKT2 and induction of apoptosis by inhibition of phosphoinositide-3-OH kinase/Akt pathway in human ovarian cancer, Oncogene. 19, 2324-30.
13. Altomare, D. A., Tanno, S., De Rienzo, A., Klein-Szanto, A. J., Tanno, S., Skele, K. L., Hoffman, J. P. & Testa, J. R. (2002) Frequent activation of AKT2 kinase in human pancreatic carcinomas, Journal of Cellular Biochemistry. 87, 470-476.
14. Vincent, E. E., Elder, D. J., Thomas, E. C., Phillips, L., Morgan, C., Pawade, J., Sohail, M., May, M. T., Hetzel, M. R. & Tavare, J. M. (2011) Akt phosphorylation on Thr308 but not on Ser473 correlates with Akt protein kinase activity in human non-small cell lung cancer, British journal of cancer. 104, 1755-61.
15. Bradbury, A. & Pluckthun, A. (2015) Reproducibility: Standardize antibodies used in research., Nature. 518, 27-29.
16. Olson, C. A., Liao, H. I., Sun, R. & Roberts, R. W. (2008) mRNA display selection of a high-affinity, modification-specific phospho-IkappaBalpha-binding fibronectin, ACS chemical biology. 3, 480-5.
17. Kummer, L., Parizek, P., Rube, P., Millgramm, B., Prinz, A., Mittl, P. R., Kaufholz, M., Zimmermann, B., Herberg, F. W. & Pluckthun, A. (2012) Structural and functional analysis of phosphorylation-specific binders of the kinase ERK from designed ankyrin repeat protein libraries, Proceedings of the National Academy of Sciences of the United States of America. 109, E2248-57.
18. Kaneko, T., Huang, H., Cao, X., Li, X., Li, C., Voss, C., Sidhu, S. S. & Li, S. S. (2012) Superbinder SH2 domains act as antagonists of cell signaling, Science signaling. 5, ra68.
19. Vielemeyer, O., Yuan, H., Moutel, S., Saint-Fort, R., Tang, D., Nizak, C., Goud, B., Wang, Y. & Perez, F. (2009) Direct selection of monoclonal phosphospecific antibodies without prior phosphoamino acid mapping, The Journal of biological chemistry. 284, 20791-5.

77
20. Shih, H. H., Tu, C., Cao, W., Klein, A., Ramsey, R., Fennell, B. J., Lambert, M., Ni Shuilleabhain, D., Autin, B., Kouranova, E., Laxmanan, S., Braithwaite, S., Wu, L., Ait-Zahra, M., Milici, A. J., Dumin, J. A., LaVallie, E. R., Arai, M., Corcoran, C., Paulsen, J. E., Gill, D., Cunningham, O., Bard, J., Mosyak, L. & Finlay, W. J. (2012) An ultra-specific avian antibody to phosphorylated tau protein reveals a unique mechanism for phosphoepitope recognition, The Journal of biological chemistry. 287, 44425-34.
21. Pershad, K., Wypisniak, K. & Kay, B. K. (2012) Directed evolution of the forkhead-associated domain to generate anti-phosphospecific reagents by phage display, Journal of molecular biology. 424, 88-103.
22. Venegas, L. A., Pershad, K., Bankole, O., Shah, N. & Kay, B. K. (2016) A comparison of phosphospecific affinity reagents reveals the utility of recombinant Forkhead-associated domains in recognizing phosphothreonine-containing peptides, New biotechnology. 33, 537-43.
23. Huang, Y. M. & Chang, C. E. (2011) Mechanism of PhosphoThreonine/Serine Recognition and Specificity for Modular Domains from All-atom Molecular Dynamics, BMC biophysics. 4, 12.
24. McGinnis, J. & Kay, B. K. (2017) Generation of recombinant affinity reagents against a two-phosphosite epitope of ATF2, New biotechnology.
25. Pershad, K., Sullivan, M. A. & Kay, B. K. (2011) Drop-out phagemid vector for switching from phage displayed affinity reagents to expression formats, Analytical biochemistry. 412, 210-6.
26. Goletz, S., Christensen, P. A., Kristensen, P., Blohm, D., Tomlinson, I., Winter, G. & Karsten, U. (2002) Selection of large diversities of antiidiotypic antibody fragments by phage display, Journal of molecular biology. 315, 1087-97.
27. Venegas, L. A., Kall, S. L., Bankole, O., Lavie, A. & Kay, B. (2018) Generating a recombinant phosphothreonine-binding domain for a phosphopeptide of the human transcription factor, c-Myc, New biotechnology.
28. Crooks, G. E., Hon, G., Chandonia, J.-M. & Brenner, S. E. (2004) WebLogo: A Sequence Logo Generator, Genome Research. 14, 1188-1190.
29. Schneider, T. D. & Stephens, R. M. (1990) Sequence logos: a new way to display consensus sequences, Nucleic acids research. 18, 6097-6100.

78
30. Durocher, D., Taylor, I. A., Sarbassova, D., Haire, L. F., Westcott, S. L., Jackson, S. P., Smerdon, S. J. & Yaffe, M. B. (2000) The Molecular Basis of FHA Domain:Phosphopeptide Binding Specificity and Implications for Phospho-Dependent Signaling Mechanisms, Molecular cell. 6, 1169-1182.
31. Yang, J., Cron, P., Good, V. M., Thompson, V., Hemmings, B. A. & Barford, D. (2002) Crystal structure of an activated Akt/Protein Kinase B ternary complex with GSK3-peptide and AMP-PNP, Nature Structural Biology. 9, 940.
32. Peri, C., Morra, G. & Colombo, G. (2016) Surface energetics and protein-protein interactions: analysis and mechanistic implications, Scientific reports. 6, 24035.

79
Chapter 4 4. STREAMLING THE VALIDATION PROCESS FOR BINDING REAGENTS
ISOLATED BY MEGASTAR
4.1 Abstract
The “sandwich” assay, in which the antigen is sandwiched between two binding
reagents, is considered the “gold standard” for diagnostics and other biochemical tests.
However, pairs are difficult to find. A technique invented in the lab, Megaprimer Shuffling
for Tandem Affinity Reagents (MegaSTAR), minimizes the cost and time required to find
non-overlapping binding pairs by isolating simultaneous binders during the phage
display affinity selection process. In MegaSTAR, two binding reagents are displayed in
tandem on the surface of an M13 bacteriophage particle, and clones in which both
reagents simultaneously contribute to binding, causing increased apparent affinity
(avidity), are selected under stringent conditions. To demonstrate that a tandem pair can
work in a sandwich assay format, one subclones each coding region, overexpresses
each in Escherichia coli, purifies them, and tests them pair-wise in an assay. To
streamline this validation process, we shortened the time it takes to test reagents in a
sandwich assay format by eliminating the need to purify the binding pairs, and at the
same time, maintain performance metrics of a 6-log dynamic range and a 10 picomolar
limit of detection. Additionally, we in vitro labeled FN3 proteins with various tags through
sortase-mediated ligation, which allows binders to quickly be reformatted for a variety of
different sandwich assays.

80
4.2 Introduction
The concentrations of proteins, hormones, and signaling molecules vary in cells and
organisms in response to environmental stimuli. However, their concentrations can also
change drastically due to a condition or disease, making such molecules excellent
biomarkers [1-3]. Evaluating this change in concentration of a biomarker in a patient
sample is a useful and quick diagnostic method. There are many such assays on the
market, including those that test for pregnancy [4, 5], cancer [6, 7], myocardial infarction
[8], and bacterial and viral infections [9, 10] that rely on a specific molecule’s deviation
from a normal concentration for diagnosis. For example, the cardiac troponin I and T
proteins exist at very low quantities in the blood of healthy patients. However, both are
released into the blood when there is heart damage (i.e., myocardial infarction) in
patients. The levels of troponin I or T directly in the blood correlate with the amount of
damage to the heart, making them excellent biomarkers for monitoring heart disease in
patients [11].
Currently, the most common diagnostic assay format is the enzyme-linked
immunosorbent assay (ELISA) [12]. This is a microtiter plate-based assay in which the
antigen is immobilized on the bottom of wells and detected by an antibody conjugated
to an enzyme. The antigen’s concentration is quantified by the conjugated enzyme’s
conversion of a substrate to a measurable product [13]. More specifically, the sandwich
ELISA, in which the antigen is “ sandwiched" between two antibodies, is considered the
gold standard in diagnostics because it requires two separate antibody binding events
to occur in order to produce a signal (Figure 4.1) [14, 15]. Requiring that two different
antibodies bind to an analyte increases the assay’s specificity and minimizes false

81
positive results. The sandwich assay is a valuable tool in basic and clinical research in
which it can be used to monitor changes in a protein’s levels upon addition of various
hormones or drugs to cells, tissues, or patients [16, 17].
Figure 4.1. Sandwich ELISA. The capture antibody (Ab, blue) is adsorbed to the
microtiter plate’s surface. The well is then incubated with a complex mixture containing
the analyte (yellow), washed, and incubated with the detection antibody (black). The
amount of analyte retained in the well is quantified by the addition of a substrate that is
converted to a measurable product by the enzyme. For this assay to work, the capture
and detection antibodies must be able to bind simultaneously to the analyte (i.e., at non-
overlapping epitopes). Analytes can be proteins or small molecules.

82
Often, non-overlapping binding pairs for a sandwich assay are identified by generating
antibodies and then many are tested in pair-wise combinations. For example, to build an
assay to a particular analyte, one would buy ten antibodies (> $3000) to that protein, and
test in 45 different pairwise combinations. If lucky, one working pair may be discovered.
This trial and error strategy is time consuming, expensive, and while the best single
binding reagents are isolated in this process, these are not necessarily the best
reagents. To improve this process, our lab has invented Megaprimer Shuffling for
Tandem Reagents (MegaSTAR) [18]. This new technology involves four steps. First,
target binders are isolated by phage display. Second, binder coding regions are
amplified by PCR. These “megaprimers” are annealed to a new, single-stranded,
uricilated DNA “tandem display” phagemid vector that contains two places for the binding
variant genes to anneal, which will be done at random, with a linker between the two
binding spots. Third, the megaprimers are extended, ligated, and the heteroduplex DNA
is transformed into E. coli cells, where the uricilated parental DNA strand is degraded by
cellular enzymes [19]. Fourth, the new, tandem display library is amplified and screened
against the analyte under stringent conditions. Pairs binding simultaneously to the
analyte are selected due to their bivalent binding, which increases their apparent affinity,
or avidity. Recovered clones are confirmed for binding by ELISA and are DNA
sequenced [18].
The MegaSTAR process eliminates much of the time and cost required to generate
pairs that can bind simultaneously to an analyte. However, there remains a downstream
bottleneck - confirming and identifying the best pairs for sandwich assays - which can

83
take a considerable amount of effort. In this Chapter, I describe my improvements to the
validation steps downstream of MegaSTAR.
Figure 4.2. Megaprimer shuffling for tandem affinity reagents. ① The primary
phage library is screened against an analyte and ② the coding regions of the selection
output are PCR amplified and used as “megaprimers" to anneal to a single-stranded,
uracilated, tandem vector. ③ The primers are extended, ligated, and the DNA is
transformed into E. coli, where the parental strand is preferentially degraded. ④ The
new tandem library is amplified and screened for binding to the analyte under stringent
conditions. Virions displaying pairs that can bind simultaneously to the analyte are affinity
selected by avidity.

84
4.3 Materials and Methods
4.3.1 Cloning and Overexpression of FN3 Fusion Proteins
To generate the FN3-AviTag fusion construct, the coding region of one of the FN3
variants from the tandem clone was PCR-amplified and subcloned into a modified
version of the pET-14b vector (Novagen) that contains the AviTag sequence in frame.
The vector also includes a hexahistidine tag for purification [20] and a small ubiquitin-
like modifier (SUMO) tag for improved expression [21]. This was repeated with the other
FN3 variant from the tandem dimer, but instead, it was subcloned into a modified pET-
14b vector to generate a C-terminal NanoLuc fusion [22]. This FN3 was also subcloned
into pKP300ΔIII [23] which contains a DsbA signal sequence to direct the C-terminal
alkaline phosphatase fusion protein to the E. coli periplasm [24].
For expression of the various FN3 fusion proteins, the FN3-Avitag fusion construct
was transformed in AVB101 E. coli cells (Avidity, LLC), which promote in vivo
biotinylation during protein expression [25], and the FN3-Nanoluc fusion construct was
transformed into BL21-CodonPlus cells. Transformed cells were inoculated in 5 mL 2xYT
medium (16 g tryptone, 10 g yeast extract, 5 g NaCl per liter) cultures, supplemented
with carbenicillin (50 µg/mL), and Overnight Express Autoinduction System 1 (Novagen).
Cultures were grown for 24 hr at 30°C, 300 revolutions per min (rpm). The FN3-Alkaline
fusion construct was transformed in TG1 cells (genotype: F' [traD36 proAB+ lacIq
lacZΔM15] supE thi-1 Δ(mcrB- hsdSM)5(rK-mK-) Δ(lac-proAB) and expressed for 24 hr
in 5 mL of 2xYT supplemented with carbenicillin (50 µg/mL) at 30°C, 300 rpm.
After 24 hours of overexpression, cells were spun down at 10,000 xg for 10 minutes.
Pellets were resuspended in 1 mL of BugBuster Protein Extraction Reagent (Millipore)

85
and incubated at room temperature for 15 min, while rocking. Lysates were spun down
again and the supernatant was used immediately in a sandwich ELISA.
4.3.2 Isolation of binding pairs by MegaSTAR
Soluble COP9 Signalosome Subunit 5 (COPS5) target protein was a gift from Dr.
Susanne Gräslund’s group from the Structural Genomics Consortium in Toronto,
Canada. The FN3 phage library was subjected to two rounds of affinity selection against
the COPS5 target, as previously described [26]. The output pool of clones was used to
generate a secondary, tandem dimer phage-display library. The COPS5 target was then
screened against the secondary, tandem library through two more rounds of affinity
selection. Generation of the secondary library and selection of tandem dimers was
performed as previously described [18].
Ninety-six output clones were screened in an ELISA as previously described [18].
DNA was prepared from the ten clones that yielded the strongest ELISA signal with the
Wizard DNA Miniprep kit (Promega) and sequenced using the oligonucleotide primer,
5’-CGCTGGCTGGTTTAGTTTTAGCGT-3’. Sequences were then compared to those
previously generated [18].
4.3.3 Sandwich ELISA with FN3 fusion binding pairs
Lysates of bacteria expressing FN3-AviTag fusion proteins were added to a Nunc
Maxisorp™ flat-bottom 96 well plate, which was coated with NeutrAvidin (100 μL, 5
μg/μL) and blocked with 2% skim milk in phosphate buffered saline (PBS; 137 mM NaCl,
3 mM KCl, 8 mM Na2HPO4, 1.5 mM KH2PO4). After washing the wells with PBS+0.1%
Tween 20 (PBST), bacterial lysates spiked with recombinant COPS5 protein were added
to the plate for a 1 hr incubation. The plates were washed three times with PBST and

86
incubated with lysates overexpressing FN3-NanoLuc or FN3-alkaline phosphatase
fusions for 1 hr. Wells were again washed three times with PBST. To quantify the amount
of NanoLuc present, Nano-Glo® Luciferase substrate (Promega) was added to the plate
and luminescence was measured on the POLARstar OPTIMA microtiter plate reader
(BMG Labtech). Alkaline phosphatase was quantified by addition of p-Nitrophenyl
Phosphate substrate and the absorbance of the wells was measured at 405 nm with the
POLARstar OPTIMA (BMG Labtech) plate reader.
4.3.4 Monobody labeling via sortase reaction
The Sortase-Tag, LPxTG, was added C-terminal of FN3 coding regions by PCR and
the amplified DNA products were subcloned into the pET-14b vector. These fusion
proteins were then expressed and purified as previously described [27].
The FN3-Sortase-Tag fusion proteins (3.75 mM) were incubated with 1 unit of Sortase
A5 enzyme (Active Motif), and 125 μM of (Glycine)5-Biotin label or 25 μM of (Glycine)5-
HRP label (Active Motif) for 1 hr at 30°C, 1000 rpm. After incubation, the reactions were
applied to Amicon® Ultra Centrifugal Filters (Millipore) with 10 and 50 kDa thresholds,
respectively. Unconjugated (Glycine)5-Biotin label and (Glycine)5-HRP label (Active
Motif) were removed from the reaction product with three PBS washes. Purity of the
monobodies was analyzed by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE).
4.3.5 Sandwich ELISA with monobodies labeled via sortase reaction
Purified, FN3-(Glycine)5-Biotin (100 μL, 10 μg/μL) was added to a Nunc Maxisorp™
flat-bottom 96 well plate coated with NeutrAvidin (100 μL, 5 μg/μL) and blocked with 2%
skim milk in PBS. After washing the wells with PBST, bacterial lysates spiked with

87
recombinant COPS5 protein were added to the plate for a 1 hr incubation. The plates
were washed three times with PBST and incubated with 0.1 μM of purified, FN3-
(Glycine)5-HRP for 1 h. Wells were again washed three times with PBST, and 100 μL of
2’,2’-Azino-Bis 3-Ethylbenzothiazoline-6-Sulfonic Acid (ABTS) in 50 mM sodium citrate
(pH 4), 0.03% hydrogen peroxide was added to each well. The absorbance of the color
change was measured at 405 nm on a POLARstar OPTIMA microtiter plate reader.
4.4 Results and Discussion
4.4.1 MegaSTAR is robust and reproducible
Binding pairs to the human proteins Cop9 signalosome subunit 5 (COPS5), p21-
associated kinase 1 (PAK1), and Rho GTPase-activating protein 32 (RICS) have
successfully been generated through MegaSTAR [18]. To determine that this technique
is robust and reproducible, we repeated the entire selection and MegaSTAR process
with the human target protein, COPS5, a subunit of an important regulator in multiple
signaling pathways. Both FN3 motifs and frequency of the tandem clones were highly
conserved with the previous results (Figure 4.3). Because of this reproducibility, we are
likely isolating the strongest tandem binders from the library.
4.4.2 Improving binding pairs by assay development
MegaSTAR minimizes the time and cost involved in generating binding pairs that
work in sandwich assays. However, these binding pairs require improvements to work
in diagnostic tests that target analytes existing in complex mixtures and low, endogenous
concentrations. We, therefore, wanted to improve the sandwich binding reagents’
sensitivity and specificity for the analyte.

88
Figure 4.3. Loop sequences of tandem clones isolated against COPS5. Comparison
of FN3 clones isolated previously (Left) [18] to repeated results (Right). Each motif
determined by the randomized BC and FG loops are represented by a different color.
The frequency with which each tandem clone appears and number of total clones
analyzed are listed. FN3a and FN3b do not distinguish between orientation for each pair.
Conserved positions are noted by their single-letter amino acid abbreviations while
positions that varied are noted with an “X.”
To improve the sensitivity and specificity of binding pairs, we wanted to reformat
many tandem clones as two separate binding reagents and test each pair in the
sandwich assay set up. The current validation process after MegaSTAR and secondary
selection requires testing clones for analyte binding as tandem dimers in an indirect
ELISA. Only the few clones producing strong positive signals are sequenced. The clones
whose sequences appear at high frequency are subcloned, expressed, and purified as
separate, soluble proteins and finally tested in the sandwich ELISA setup. This

89
problematic strategy assumes high affinity and high frequency tandem dimers produce
the best separate binding pairs for a sensitive sandwich assay with a large dynamic
range. It would be highly desirable to test many more clones by sandwich ELISA.
However, the bottleneck for testing hundreds of pairs is due to the extensive time and
cost required to sequence many tandem dimers, reformat as separate binders, and
purify each monobody.
To lessen this bottleneck, we devised a strategy to eliminate the need to purify each
monobody, and instead, use them directly in crude cell lysates for assays. After isolating
tandem dimers, we cloned the FN3 coding regions into separate capture and detection
fusion vectors (Figure 4.4). The capture FN3 fusion vector contains an AviTag, allowing
for protein biotinylation in vivo, when expressed in E. coli cells that also contain an
isopropyl-β-D-thiogalactopyranoside (IPTG) inducible plasmid for BirA biotin ligase
expression [25]. Biotinylating the capture FN3 monobody as it is expressed in vivo
makes its capture by NeutrAvidin possible while in cell lysates. The detection vectors
genetically fuse NanoLuc or alkaline phosphatase enzymes, which express well in E.
coli cells, directly to the monobody. These enzymes convert substrates to measurable
products to make the monobody fusions directly detectable [22, 28]. Often in ELISAs,
the primary detection antibody or binding reagent is not conjugated to a read-out enzyme
and requires a secondary antibody for detection. Genetically fusing the enzyme directly
to the primary detection binder eliminates the need for a secondary reagent, which
reduces assay time and the number of wash steps. These improvements facilitate better
binding between the analyte and the detection monobody such that they can also be
used directly from cell lysates.

90
Figure 4.4. Monobody fusion proteins for improved sandwich assays. (A) Flow
chart outlining the generation and characterization of binding pairs isolated by
MegaSTAR. (B) Cartoons of the sub-elements in the capture and detection monobody
fusion proteins. Not to scale.
Once the FN3 coding regions were transferred to the capture and detection fusion
vectors, expressed in small volume cultures, and lysed, they were used immediately in
a sandwich ELISA (Figures 4.5A, 4.6A). The sandwich ELISAs measured the amount
of COPS5 analyte spiked over a range of concentrations into a bacterial cell lysate
(Figures 4.5B, 4.6B). Both sandwich assays successfully determined the analyte
concentration over approximately a 6-log dynamic range, which is comparable, if not
better, to other sandwich ELISAs [29-31].

91
Figure 4.5. Sandwich ELISA with NanoLuc readout. (A) Set up of the sandwich ELISA
with detection by NanoLuc. Plates are coated with NeutrAvidin, which then bind
biotinylated, capture FN3 from bacterial cell lysates. The immobilized complex is then
incubated with the analyte, COPS5, whose presence is detected by the FN3 fused to
NanoLuc and the addition of substrate. As the NanoLuc enzyme converts the furimazine
to furimamide, light is released. (B) Detection of the COPS5 analyte spiked over a range
of concentrations into a bacterial cell lysate. “Cognate pair” indicates both FN3 binding
reagents used in the assay originated from a tandem clone isolated against COPS5,
while “unrelated capture reagent” indicates use of a capture FN3 to an unrelated target
(i.e., negative control). Error bars represent standard deviation of triplicate
measurements. Data analyzed with OriginLab software.

92
Figure 4.6. Sandwich ELISA with alkaline phosphatase (AP) readout. (A) Set up of
the sandwich ELISA with AP detection. Plates are coated with NeutrAvidin, which then
bind biotinylated, capture FN3 from bacterial cell lysates. The complex is then incubated
with the analyte, COPS5, whose presence is detected by the FN3-AP fusion binder and
addition of substrate. The AP enzyme converts the p-Nitrophenyl Phosphate substrate
to p-nitrophenol, a chromogenic product. (B) Sandwich ELISA detecting COPS5 over a
range of concentrations with FN3 fusion reagents. “Cognate pair” indicates both FN3
binders originated from a tandem clone isolated against COPS5, while “unrelated
capture reagent” indicates use of a capture FN3 to an unrelated target. Error bars denote
standard deviation of triplicate measurements. Data analyzed with OriginLab software.
Additionally, both assays were able to detect the analyte at concentrations as low as
5-20 pM (Table VII), which is physiologically relevant. Many proteins that circulate
throughout the blood exist at similar, picomolar to nanomolar concentrations such as C-
reactive protein (CRP), β2 microglobulin, and Cardiac troponin I [32]. Therefore, this
assay format has the potential to be applied to a variety of different plasma proteins.

93
TABLE VII. SANDWICH ELISA METRICS WITH MONOBODY FUSION PAIRS
Metrics AP Readout NanoLuc Readout
Limit of detection 20 pM 5 pM
Dynamic range 6 logs 6 logs
R2 0.974 0.982
4.4.3 Easy Use of Pairs for Heterogenous Assays To reduce assay development time once binding pairs are confirmed, we wanted to
implement a quick and easy system to conjugate the binding pairs to a variety of labels
for sandwich assays. Rather than cloning the FN3 coding regions into multiple different
expression vectors with different genetic fusions, and expressing and purifying each
monobody fusion separately, we decided to conjugate the tags enzymatically to purified
monobodies via sortase-mediated ligation. The Sortase A enzyme, naturally found in
gram positive bacteria for cell wall biosynthesis, catalyzes the conjugation of proteins
carrying an LPxTG motif, where x is any amino acid, by cleaving between threonine and
glycine and then joining threonine to a poly-glycine peptidoglycan through a trans-
peptide ligation reaction [33]. However, this reaction can be applied to site-specific
protein labeling in vitro through the joining of a protein with a pentapeptide tag, LPxTG,
to a protein or peptide carrying poly-glycine at its N-terminus [34].
To test the sortase-catalyzed protein ligation method, we first evaluated its efficiency
in tagging monobodies for a sandwich ELISA. We genetically fused each FN3 from the
tandem MegaSTAR clone to a LPxTG peptide, and ligated the tagged proteins to an N-
terminated-(glycine)5 biotin-conjugated peptide, to create a capture reagent, and an N-
terminated-(glycine)5 horseradish peroxidase (HRP) enzyme, to create a detection
reagent, via the sortase-catalyzed ligation reaction (Figure 4.7). Almost all of the

94
detection FN3 was converted to an HRP-fusion protein and almost all unlabeled FN3
was efficiently removed by centrifugal filtration (Figure 4.8A). The removal of
unconjugated FN3 detection reagent is extremely important to eliminate its competitive
binding for the analyte. Additionally, excess (G)5-biotin carrying peptide was efficiently
removed after tagging (Figure 4.8B), which is important for eliminating its competitive
binding to NeutrAvidin for immobilization.
Figure 4.7. Site-specific monobody labeling via sortase-mediated ligation. (A)
Tandem vector open reading frame. FN3a and FN3b denote the FN3 N- and C-terminal
to the linker in MegaSTAR constructs, respectively. (B) The Sortase A enzyme from
Gram-positive bacteria catalyze the trans-peptization of a poly-Glycine (G)5 tag to the
LPxTG sortase recognition sequence. FN3a is converted into the capture FN3 by
conjugating it to a biotin-labeled peptide while FN3b is converted into the detection FN3
by conjugating it to HRP. (C) FN3 ligation is reversed. Cartoon is not to scale.

95
Figure 4.8. Sortase-mediated ligation efficiency. (A). SDS-PAGE gel showing
unlabeled FN3-LPxTG (~27 kDa) conversion to FN3-HRP (~67 kDa) fusion via sortase-
mediated ligation. Unconjugated HRP is approximately 40 kDa. (B) FN3-LPxTG
incubated with (G)5-Biotin peptide in the presence or absence of sortase enzyme and
passively fused to a microtiter plate. Biotin detected by HRP-conjugated Streptavidin.
The labeled monobody pairs were used in sandwich ELISAs where the FN3-(G)5-
Biotin peptide conjugate was captured on NeutrAvidin coated plates to then capture
COPS5. The FN3b-(G)5-HRP conjugate was then added to the complex to detect
COPS5 at the second epitope (Figure 4.9AC). Both FN3 orientations were tested to
determine if there was any difference in assay performance (Figure 4.9BD) The two
ELISA formats varied slightly in dynamic range and limit of detection (Figure 4.9E),
suggesting that both orientations should be tested to identify the best format for a robust
assay.
Overall, the sandwich assays relying on monobody labeled binding pairs via sortase-
mediated ligation were very comparable to the sandwich ELISAs with genetically fused
monobody pairs. This opens the door to quickly testing monobody pairs with many other
labels, such as IR dyes, oligonucleotides, NanoLuc, and alkaline phosphotase to further
improve assay metrics or provide researchers with their desired measuring system.

96
Figure 4.9. Sandwich ELISA with binding pairs labeled by sortase-mediated
ligation. (A and C) Set up of the sandwich ELISA with FN3 reagents labeled by sortase-
mediated ligation. Plates are coated with NeutrAvidin, which complexes with biotinylated,
capture FN3. The plate is washed and then incubated with the analyte, COPS5, which
has been spiked at various amounts in bacteria cell lysates. The analyte is detected with
a second FN3, conjugated to HRP, and addition of the ABTS substrate. (B and D)
Sandwich ELISA detecting COPS5 over a range of concentrations with capture reagent
[FN3-(G)5-Biotin] and the detection reagent [FN3-(G)5)-HRP]. Error bars denote standard
deviation of triplicate measurements. (E) Performance metrics for set up 1 and 2.

97
4.5 Conclusions
MegaSTAR is a useful and innovative technique to quickly and inexpensively
generate tandem binding reagents with potential use as sandwich assay binding pairs.
By separating and genetically fusing each binding reagent to a tag for capture and
detection, we have further streamlined this process (Figure 4.10) through improving the
sandwich assay format to validate the binding pairs. This new format promotes testing
many possible pairs simultaneously to achieve an assay with a broad dynamic range
and low limit of detection in complex mixtures. Furthermore, we have introduced site-
specific, sortase-mediated enzymatic labeling to easily prepare high performance
binding pairs for a variety of heterogeneous assays. Overall, the process reduces the
time required to isolate and validate pairs, improves performance of pairs, and offers a
user-friendly assay system.
Figure 4.10. Workflow for identifying and characterizing binding reagents through
MegaSTAR. An estimated timeline is shown for a specific number of targets and their
corresponding pairs.

98
4.5 Literature Cited
1. Zini, R., Riant, P., Barré, J. & Tillement, J.-P. (1990) Disease-Induced Variations in Plasma Protein Levels Implications for Drug Dosage Regimens (Part I), Clinical Pharmacokinetics. 19, 147-159.
2. Rifai, N., Gillette, M. A. & Carr, S. A. (2006) Protein biomarker discovery and validation: the long and uncertain path to clinical utility, Nature Biotechnology. 24, 971-983.
3. Fu, Y., Wang, N., Yang, A., Law, H. K.-w., Li, L. & Yan, F. (2017) Highly Sensitive Detection of Protein Biomarkers with Organic Electrochemical Transistors, Advanced Materials. 29, 1703787.
4. Cole, L. A. (2010) Biological functions of hCG and hCG-related molecules, Reprod Biol Endocrinol. 8, 102-102.
5. Gnoth, C. & Johnson, S. (2014) Strips of Hope: Accuracy of Home Pregnancy Tests and New Developments, Geburtshilfe Frauenheilkd. 74, 661-669.
6. Kierny, M. R., Cunningham, T. D. & Kay, B. K. (2012) Detection of biomarkers using recombinant antibodies coupled to nanostructured platforms, Nano Rev. 3, 10.3402/nano.v3i0.17240.
7. Baird, C. L., Fischer, C. J., Pefaur, N. B., Miller, K. D., Kagan, J., Srivastava, S. & Rodland, K. D. (2010) Developing recombinant antibodies for biomarker detection, Cancer Biomarkers. 6, 271-279.
8. Agnibh, M., Uzair, A., Martin, B., Ibrahim, A. & Michael, B. (2017) Clinically Relevant Biomarkers in Acute Heart Failure: An Update, Current Pharmaceutical Biotechnology. 18, 482-490.
9. Ji, M., Cho, B., Cho, Y. S., Park, S.-Y., Cho, S.-N., Jeon, B.-Y. & Yoon, B.-S. (2014) Development of a quantitative sandwich enzyme-linked immunosorbent assay for detecting the MPT64 antigen of Mycobacterium tuberculosis, Yonsei Med J. 55, 746-752.
10. Arai, H., Petchclai, B., Khupulsup, K., Kurimura, T. & Takeda, K. (1999) Evaluation of a rapid immunochromatographic test for detection of antibodies to human immunodeficiency virus, J Clin Microbiol. 37, 367-370.

99
11. Zhu, J., Zou, N., Mao, H., Wang, P., Zhu, D., Ji, H., Cong, H., Sun, C., Wang, H., Zhang, F., Qian, J., Jin, Q. & Zhao, J. (2013) Evaluation of a modified lateral flow immunoassay for detection of high-sensitivity cardiac troponin I andmyoglobin, Biosensors and Bioelectronics. 42, 522-525.
12. Lequin, R. M. (2005) Enzyme Immunoassay (EIA)/Enzyme-Linked Immunosorbent Assay (ELISA), Clinical Chemistry. 51, 2415-2418.
13. Reen, D. J. (1994) Enzyme-Linked Immunosorbent Assay (ELISA) in Basic Protein and Peptide Protocols (Walker, J. M., ed) pp. 461-466, Humana Press, Totowa, NJ.
14. Kragstrup, T. W., Vorup-Jensen, T., Deleuran, B. & Hvid, M. (2013) A simple set of validation steps identifies and removes false results in a sandwich enzyme-linked immunosorbent assay caused by anti-animal IgG antibodies in plasma from arthritis patients, Springerplus. 2, 263-263.
15. Kohl, T. O. & Ascoli, C. A. (2017) Immunometric Double-Antibody Sandwich Enzyme-Linked Immunosorbent Assay, Cold Spring Harbor Protocols. 2017, pdb.prot093724.
16. Ayoub, M. A., Trebaux, J., Vallaghe, J., Charrier-Savournin, F., Al-Hosaini, K., Gonzalez Moya, A., Pin, J.-P., Pfleger, K. D. G. & Trinquet, E. (2014) Homogeneous time-resolved fluorescence-based assay to monitor extracellular signal-regulated kinase signaling in a high-throughput format, Front Endocrinol (Lausanne). 5, 94-94.
17. Li, C., Tao, Y., Yang, Y., Feng, C., Xiang, Y. & Li, G. (2017) Dynamic sandwich-type electrochemical assay for protein quantification and protein–protein interaction, Analyst. 142, 4399-4404.
18. Gorman, K. T., Roby, L. C., Giuffre, A., Huang, R. & Kay, B. K. (2017) Tandem phage-display for the identification of non-overlapping binding pairs of recombinant affinity reagents, Nucleic acids research. 45, e158.
19. Kunkel, T. A. (1985) Rapid and efficient site-specific mutagenesis without phenotypic selection, Proceedings of the National Academy of Sciences of the United States of America. 82, 488-492.
20. Porath, J. (1992) Immobilized metal ion affinity chromatography, Protein Expression and Purification. 3, 263-281.

100
21. Panavas, T., Sanders, C. & Butt, T. R. (2009) SUMO Fusion Technology for Enhanced Protein Production in Prokaryotic and Eukaryotic Expression Systems in SUMO Protocols (Ulrich, H. D., ed) pp. 303-317, Humana Press, Totowa, NJ.
22. England, C. G., Ehlerding, E. B. & Cai, W. (2016) NanoLuc: A Small Luciferase Is Brightening Up the Field of Bioluminescence, Bioconjug Chem. 27, 1175-1187.
23. Pershad, K., Sullivan, M. A. & Kay, B. K. (2011) Drop-out phagemid vector for switching from phage displayed affinity reagents to expression formats, Analytical biochemistry. 412, 210-6.
24. Schierle, C. F., Berkmen, M., Huber, D., Kumamoto, C., Boyd, D. & Beckwith, J. (2003) The DsbA signal sequence directs efficient, cotranslational export of passenger proteins to the Escherichia coli periplasm via the signal recognition particle pathway, J Bacteriol. 185, 5706-5713.
25. Ashraf, S. S., Benson, R. E., Payne, E. S., Halbleib, C. M. & Grøn, H. (2004) A novel multi-affinity tag system to produce high levels of soluble and biotinylated proteins in Escherichia coli, Protein Expression and Purification. 33, 238-245.
26. Huang, R., Gorman, K. T., Vinci, C. R., Dobrovetsky, E., Gräslund, S. & Kay, B. K. (2015) Streamlining the Pipeline for Generation of Recombinant Affinity Reagents by Integrating the Affinity Maturation Step, Int J Mol Sci. 16, 23587-23603.
27. Gorman, K., McGinnis, J. & Kay, B. (2018) Generating FN3-Based Affinity Reagents Through Phage Display, Current Protocols in Chemical Biology. 10, e39.
28. Neumann, H. & Van Vreedendaal, M. (1967) An improved alkaline phosphatase determination with p-nitrophenyl phosphate, Clinica Chimica Acta. 17, 183-187.
29. Ebai, T., Souza de Oliveira, F. M., Löf, L., Wik, L., Schweiger, C., Larsson, A., Keilholtz, U., Haybaeck, J., Landegren, U. & Kamali-Moghaddam, M. (2017) Analytically Sensitive Protein Detection in Microtiter Plates by Proximity Ligation with Rolling Circle Amplification, Clinical Chemistry. 63, 1497-1505.
30. Vashist, S. K., Schneider, E. M. & Luong, J. H. T. (2015) Rapid sandwich ELISA-based in vitro diagnostic procedure for the highly-sensitive detection of human fetuin A, Biosensors and Bioelectronics. 67, 73-78.

101
31. Tang, X., Liang, Q., Liu, L., Sheng, X., Xing, J. & Zhan, W. (2018) An optimized double-antibody sandwich ELISA for quantitative detection of WSSV in artificially infected crayfish, Journal of Virological Methods. 251, 133-138.
32. Hortin, G. L. & Sviridov, D. (2010) The dynamic range problem in the analysis of the plasma proteome, Journal of Proteomics. 73, 629-636.
33. Mazmanian, S. K., Liu, G., Ton-That, H. & Schneewind, O. (1999) Staphylococcus aureus Sortase, an Enzyme that Anchors Surface Proteins to the Cell Wall, Science. 285, 760-763.
34. Mao, H., Hart, S. A., Schink, A. & Pollok, B. A. (2004) Sortase-Mediated Protein Ligation: A New Method for Protein Engineering, Journal of the American Chemical Society. 126, 2670-2671.

102
Chapter 5 5. CONCLUSIONS
5.1 Thesis Summary
The central goals of this thesis were to develop recombinant affinity reagents to
challenging targets through phage display and demonstrate their utility in a biochemical
assay. These goals were achieved through two projects. The first was to generate affinity
reagents that had high selectivity and affinity for phosphothreonine peptide targets using
a phage library developed by our lab’s former member, Dr. Kritika Pershad. The second
was to format and utilize pairs of fibronectin type III (FN3) monobodies, isolated through
a new technique, Megaprimer Shuffling for Tandem Reagents (MegaSTAR), in sandwich
assays.
In Chapter 2, I isolated FHA affinity reagents that had unique specificities for the
Activating Transcription Factor 2 (ATF2) phosphopeptide targets. ATF2 is a challenging
target because it contains two neighboring phosphothreonine residues and can exist as
an unphosphorylated, mono-phosphorylated, or di-phosphorylated protein in cells.
Because ATF2 has multiple phosphorylation states that are biologically relevant, I was
motivated to generate Forkhead Associated (FHA) domain variants that would distinguish
between the fully and partially phosphorylated states of ATF2 peptides. I successfully
isolated affinity reagents that selectively recognized the di-phosphorylated peptide and
one of the two mono-phosphorylated forms of the peptide. Additionally, I showed that an
FHA binding reagent could detect full-length, di-phosphorylated ATF2 in a western blot.
In another aspect of this work, reported in Chapter 3, I generated FHA affinity reagents
to a mono-phosphothreonine peptide of human Akt1, an oncogenic protein that is often
phosphorylated in cancer patients. These affinity reagents are noteworthy because of

103
their low nanomolar affinity for the Akt1 phosphopeptide. They also selectively recognize
the Akt1 phosphopeptide, with little to no binding to sequences in the highly conserved
isoforms, Akt2 and Akt3.
Given the desirable characteristics the binding reagents discussed in Chapters 2 and
3 provide, we found it important to investigate their molecular recognition properties
through alanine scanning and DNA sequencing. As more FHA affinity reagents are
generated to phosphopeptides in the future, the compilation of what works and what does
not as peptide ligands will help elucidate the recognition range of the engineered FHA1
domain.
Many diagnostic assays detect molecules or proteins in blood or urine samples.
Because analytes typically remain folded in such mixtures, I focused in Chapter 4 on FN3
affinity reagents that recognize conformational epitopes of protein targets. Specifically,
this chapter concentrated on developing FN3 affinity reagents that work as pairs in
sandwich assays. The sandwich assay format is ideal because it requires two separate
binding events to occur to produce a signal, which significantly limits false positive results.
FN3 pairs were isolated through MegaSTAR, a technique developed by a previous
member of our lab, Dr. Kevin Gorman, that modifies the phage-display process to
generate tandem binding reagents with potential use in sandwich assays [1]. I streamlined
the validation process for these binding pairs. In this new process, FN3 binders are
expressed at small scale and used directly in crude cell lysates, eliminating the need to
express and purify large quantities of the binding reagents. By greatly reducing the time,
cost, and labor of validating pairs, more pairs can be tested.

104
Often, high quality binding reagents are not used to their full capacity because they
must be reformatted to work in each assay. To remove this bottleneck, I genetically fused
FN3 binders to the pentapeptide motif, LPxTG, allowing researchers to quickly conjugate
labels of their choice to the binding reagents through enzymatic, sortase-mediated
ligation. This simple labeling method makes these binding reagents accessible for a
variety of assays.
5.2 Future Directions
5.2.1 Incorporate Multiple Scaffolds and Linker Lengths into MegaSTAR
MegaSTAR has shown promising results in generating binding pairs of monobodies
that can bind targets simultaneously. In its original version, pairs that were generated
recognized conformational epitopes. To expand on early efforts with MegaSTAR, we
propose to generate a suite of tandem vectors that incorporate other scaffolds and binding
reagents, such as the FHA domain, a designed ankyrin repeat protein (DARPin), and an
scFv (Figure 5.1). These new vectors would be constructed through standard cloning
methods.
First, we would perform primary selection on the target with each of the individual
libraries. Because of the variety of binding capabilities associated with each scaffold, we
can pursue a range of difficult targets, such as post translational modifications and
membrane receptor proteins. The DNA from the output clones would be PCR amplified
and annealed to the appropriate tandem vectors. After synthesizing the heteroduplex
DNA and transforming into bacterial cells, the clones would be pooled together to create
a large secondary library with tandem scaffolds. This pool would be amplified for a
combined selection, in which the best possible pairs would be isolated.

105
Figure 5.1. An array of tandem vectors for improved binding pairs. Schematic
showing (A) the tandem vector coding region and corresponding phage particle and (B)
possible tandem scaffold combinations that could be used in MegaSTAR. Not to scale.
To improve the success rate for generating binding pairs to difficult targets, we
propose to also incorporate an array of linkers between the tandem binders. Currently,
the tandem vector contains a 25 amino acid linker with the repeated GGGGS motif. If the
epitopes that two binding reagents recognize are far apart, this short linker may hinder
their ability to bind the target simultaneously. We would generate two additional tandem
vectors that incorporate 50 amino acid and 75 amino acid linkers to eliminate this issue.
Additionally, we anticipate difficulties with expressing clones that contain a long, highly
repetitive GGGGS linker due to the inherent instability of repetitive sequences in bacteria
cells. The clones may incur mutations caused by recombination or replication slippage
[2]. We propose to test alternative linkers such as the 95 amino acid disordered region of
Calcineurin [3].

106
5.2.2 Generating Reagents to Cell-Free Expressed Targets
Many eukaryotic proteins require chaperones for proper folding in the cells. Such
protein chaperons are absent in bacteria, which necessitates overexpression of human
proteins in mammalian cell lines. This is laborious, expensive, and requires technical
competency. As an alternative, we propose to use cell-free protein synthesis to generate
these target proteins in vitro and use them directly in phage-display affinity selection
experiments. HaloTagged fusion proteins would be generated through coupled in vitro
transcription-translation in HeLa cell extracts [4]. HaloTag is a modified dehalogenase
that forms a covalent bond with suicide substrates. We would capture the HaloTagged
targets directly from the protein synthesis reaction by covalently conjugating them to resin
or magnetic beads coated with suicide substrate for HaloTag (Figure 5.2) [5]. Because
no purification step is required, the production of correctly folded target proteins can be
miniaturized. Additionally, the targets would be prepared fresh before each selection,
eliminating harmful freeze-thaw cycles.
Figure 5.2. Target immobilization via HaloTag technology. Cartoon showing nucleophilic
displacement of the resin’s terminal chloride by an aspartic acid residue on HaloTag leading to a
covalent, irreversible alkyl-enzyme intermediate. Not to scale.

107
‘
We have shown that the FN3 binding reagents can be enzymatically labeled by site-
specific sortase-mediated ligation and utilized in sandwich ELISAs. Additionally, these
binding pairs can be applied to homogeneous assays to further improve sensitivity and
robustness while greatly decreasing assay time, as no washing steps are required. I
describe two popular homogenous assays below.
The proximity ligation assay (PLA) is unmatched in sensitivity and is capable of
detecting zeptomole levels of an analyte [6]. Two binding reagents are conjugated to DNA
oligonucleotides, and when they are brought into proximity to each other (due to binding
same analyte or two analytes interacting with each other) and the required substrates and
enzymes are added, the DNA oligonucleotides will anneal, be extended by DNA
polymerase, and permit rolling circle DNA amplification. Fluorescent, complementary
oligonucleotides are annealed to the DNA once it is amplified, resulting in a robust
fluorescent signal.
DNA can be randomly conjugated to binding reagents quite easily [7], But it is
imperative that oligonucleotides conjugated to the FN3 binders do not interfere with the
FN3’s binding mechanism. Because sortase-mediated ligation promotes site-specific
labeling, we propose incorporating this labeling technique to tag FN3 binding reagents
with peptide-DNA oligonucleotide conjugates and perform proximity ligation assays to
detect analytes that exist at extremely low concentrations in human samples (Figure
5.3A).
Another popular homogenous assay format is the bioluminescence resonance energy
transfer (BRET) assay (Figure 5.3B) [8]. The assay requires that one of the FN3 binding

108
Figure 5.3. Homogeneous assays utilizing sortase-mediated ligation. Overview of
homogeneous assays. FN3 binders are labeled by sortase-mediated ligation with (A)
oligonucleotides and used in a proximity ligation assay or (B) NanoLuc and the 618 ligand
(via HaloTag) and used in a BRET assay. Not to scale.

109
reagents be labeled with the NanoLuc enzyme (the energy donor) and the other be
labeled with a ligand that carries 618 nanometer wavelength emission Alexa dye (618
Ligand; the energy acceptor). The 618 ligand is formatted to covalently bond to the
HaloTag enzyme [9]. We would express and purify NanoLuc and HaloTag with N-terminal
SUMO fusions followed by a poly-glycine tag. Cleavage by site-specific SUMO protease
will yield NanoLuc and HaloTag proteins with N-terminal poly-glycine tags (Figure 5.4).
These glycine-terminated enzymes can then be ligated to FN3 monobodies by sortase-
mediated ligation. The FN3-HaloTag binder would further be altered to also be
enzymatically labeled with the 618 ligand through HaloTag enzymatic conjugation (as
discussed in section 5.2.2). Like PLA, the two binders are brought into proximity by
binding the same analyte. Upon adding the NanoLuc substrate, NanoLuc emits a signal
at 460 nM which excites the Alexa dye conjugated to the ligand, and in turn, emits a signal
at 618 nM that can be measured with a luminometer [8].
Figure 5.4. Generating recombinant enzyme labels for sortase-mediated ligation.
Cartoon illustrating the removal of a SUMO tag by site-specific cleavage to produce
recombinant NanoLuc and HaloTag enzymes with N-terminal poly-glycine tags. Not to
scale.

110
5.3 Overall Impact
While biomarker discovery has exploded in recent years, it has not translated to the
production of many sensitive and specific diagnostic and biochemical tests. This is in part
due to the technical challenge of producing robust assays with low false positive and low
false negative results [10]. Antibody binding reagents needed for these assays are difficult
to obtain as animal immunizations, antibody collection and purification, and validation
require extensive time and labor and often still result in non-specific, off-target, and/or low
affinity binders. In this work we demonstrate that by deploying phage display and
MegaSTAR to generate recombinant affinity reagents, we can reduce the time needed to
select binding pairs and improve their specificity and affinity through directed evolution,
resulting in robust, sensitive sandwich assays.
Recombinant affinity reagent generation has demonstrated success in helping
quantify the amount of an analyte present in a complex mixture. However, this is only the
beginning of what these improvements can provide. By incorporating many scaffolds into
the selection and MegaSTAR process, they will be helpful in developing a variety of other
diagnostic and biochemical tests. Not only is it useful to determine the absence or
presence of a molecule, but detecting biomarkers such as protein mutants, changes in a
protein’s folding through allosteric interactions, or post-translational modifications would
be useful in quickly diagnosing many diseases. For example, the RAS GTPase is part of
a signaling cascade and is activated by binding GTP, which ultimately leads to increased
expression of genes involved in cell growth and proliferation. Upon hydrolysis of GTP to
GDP, RAS is inactivated and the signaling pathway is turned off. However, RAS mutants
that are constitutively active are commonly observed in cancer patients [11, 12]. Through

111
use of many scaffold libraries, it is possible to distinguish between GTP and GDP bound
RAS by detecting conformational changes, and between wild-type and mutant RAS by
detecting sequence changes. These reagents could then be used in a multiplex sandwich
assay that diagnoses a patient with RAS driven cancer.
As genomic and biomarker testing becomes more reliable and routine, diagnostics will
radically reshape the healthcare landscape. Cancer, heart disease, and other disorders
will be detected in their early stages which will both reduce cost of treatment and save
more lives. Additionally, diagnostics provide personalized healthcare. Biomarkers and
gene mutations offer information about how a patient will respond to a therapeutic,
allowing doctors to tailor treatments to each patient. My work has the potential to improve
preventative and personalized health care, and ultimately enhance the lives of many.

112
5.7 Literature cited
1. Gorman, K. T., Roby, L. C., Giuffre, A., Huang, R. & Kay, B. K. (2017) Tandem phage-display for the identification of non-overlapping binding pairs of recombinant affinity reagents, Nucleic acids research. 45, e158.
2. Bzymek, M. & Lovett, S. T. (2001) Instability of repetitive DNA sequences: The role of replication in multiple mechanisms, Proceedings of the National Academy of Sciences. 98, 8319-8325.
3. Creamer, T. P. (2013) Transient disorder: Calcineurin as an example, Intrinsically Disord Proteins. 1, e26412-e26412.
4. Gagoski, D., Polinkovsky, M. E., Mureev, S., Kunert, A., Johnston, W., Gambin, Y. & Alexandrov, K. (2016) Performance benchmarking of four cell-free protein expression systems, Biotechnology and Bioengineering. 113, 292-300.
5. Ohana, R. F., Hurst, R., Vidugiriene, J., Slater, M. R., Wood, K. V. & Urh, M. (2011) HaloTag-based purification of functional human kinases from mammalian cells, Protein Expression and Purification. 76, 154-164.
6. Fredriksson, S., Gullberg, M., Jarvius, J., Olsson, C., Pietras, K., Gústafsdóttir, S. M., Östman, A. & Landegren, U. (2002) Protein detection using proximity-dependent DNA ligation assays, Nature Biotechnology. 20, 473-477.
7. Gullberg, M., Gústafsdóttir, S. M., Schallmeiner, E., Jarvius, J., Bjarnegård, M., Betsholtz, C., Landegren, U. & Fredriksson, S. (2004) Cytokine detection by antibody-based proximity ligation, Proceedings of the National Academy of Sciences of the United States of America. 101, 8420-8424.
8. Machleidt, T., Woodroofe, C. C., Schwinn, M. K., Méndez, J., Robers, M. B., Zimmerman, K., Otto, P., Daniels, D. L., Kirkland, T. A. & Wood, K. V. (2015) NanoBRET—A Novel BRET Platform for the Analysis of Protein–Protein Interactions, ACS chemical biology. 10, 1797-1804.
9. Benink, H. A. & Urh, M. (2015) HaloTag Technology for Specific and Covalent Labeling of Fusion Proteins in Site-Specific Protein Labeling: Methods and Protocols (Gautier, A. & Hinner, M. J., eds) pp. 119-128, Springer New York, New York, NY.

113
10. Frangogiannis, N. G. (2012) Biomarkers: hopes and challenges in the path from discovery to clinical practice, Transl Res. 159, 197-204.
11. Zhou, B., Der, C. J. & Cox, A. D. (2016) The role of wild type RAS isoforms in cancer, Semin Cell Dev Biol. 58, 60-69.
12. Zeitouni, D., Pylayeva-Gupta, Y., Der, C. J. & Bryant, K. L. (2016) KRAS Mutant Pancreatic Cancer: No Lone Path to an Effective Treatment, Cancers (Basel). 8, 45.

114
Chapter 6 APPENDIX
GLOBAL JOURNAL PERMISSIONS TO REUSE PREVIOUSLY PUBLISHED
MATERIALS
Permission for reuse of article, Generating FN3-based Affinity Reagents through Phage
Display, originally published by Current Protocols in Chemical Biology (Wiley publication)
Copyright policy
The Article, including all figures, illustrations, and tabular and other supplementary material, shall be considered a work made for hire for Current Protocols, and we shall own the copyright and all of the rights comprised in the copyright. If the Article or any such material does not qualify as a work made for hire, then you hereby transfer to us during the full term of the copyright and all extensions thereof the full and exclusive rights comprised in the copyright and all other rights in and to the Article, and in any such material, in all media, worldwide.

115
Permission for reuse of article, Generation of Recombinant Affinity Reagents against a
Two-phosphosite epitope of ATF2, originally published by New Biotechnology

116
The article, A Recombinant Affinity Reagent Specific for a Phosphoepitope of Akt1,
originally published in International journal of molecular sciences is an open access article
distributed under the Creative Commons Attribution License which permits unrestricted
use, distribution, and reproduction in any medium, provided the original work is properly
cited (CC BY 4.0).

117
Chapter 7 VITA
NAME: Jennifer Elise McGinnis
EDUCATION: Ph.D., Biological Sciences, University of Illinois at Chicago, Chicago,
IL, 2019
B.S., Molecular and Cellular Biology, University of Illinois at Urbana-
Champaign, Urbana, IL, 2014
TEACHING Department of Biological Sciences, University of Illinois at Chicago,
Chicago, Illinois: Genetics Laboratory, Teaching Assistant, 2014-
2017
RESEARCH Department of Biological Sciences, University of Illinois at Chicago,
Chicago IL: Laboratory of Dr. Brian Kay, Graduate Research
Assistant, 2014-2019
Downstream Process Development, Bristol-Myers Squibb, Devens,
Massachusetts: Co-Op, 2018.
Department of Molecular and Cellular Biology, University of Illinois at
Urbana-Champaign, Urbana, IL: Laboratory of Dr. Isaac Cann,
Undergraduate Research Assistant, 2012-2014.
POSTERS Jennifer E. McGinnis, Kevin T. Gorman, and Brian K. Kay. (2019)
Direct Recovery of Pairs by Phage Display. Affinity 2019. Stockholm,
Sweden.
Jennifer E. McGinnis and Brian K. Kay. (2016) Generation of an
Affinity Reagent that Recognizes a Dually Phosphorylated Epitope in
a Human Transcription Factor. Gordon Research Conference:
Chemistry and Biology of Peptides. Ventura, CA.
PUBLICATIONS McGinnis, J. E., Venegas, L. A., Lopez, H. & Kay, B. K. (2018) A
Recombinant Affinity Reagent Specific for a Phosphoepitope of Akt1,
Int J Mol Sci. 19, 3305.
Gorman, K. T., McGinnis, J. E. & Kay, B. K. (2018) Generating FN3-Based Affinity Reagents Through Phage Display, Current Protocols in Chemical Biology. 10, e39.
Kokoszka, M. E., Kall, S. L., Khosla, S., McGinnis, J. E., Lavie, A. &
Kay, B. K. (2018) Identification of two distinct peptide-binding

118
pockets in the SH3 domain of human mixed-lineage kinase 3, The
Journal of biological chemistry. 293, 13553-13565.
McGinnis, J. E. & Kay, B. K. (2017) Generation of recombinant
affinity reagents against a two-phosphosite epitope of ATF2, New
biotechnology.
AWARDS Department of Biological Sciences, University of Illinois at Chicago,
Chicago, Chicago, IL: Excellence in Teaching, 2017.
Affinity 2019 Conference, Stockholm, Sweden: Affinity Poster Award
(2nd place), 2019
Top Related
Copyright © 2022 FDOKUMEN

