







Bahasa
Halaman
Hukum


ORIGINAL ARTICLE
Death of Hypothalamic Astrocytes in Poorly Controlled Diabetic Rats isAssociated with Nuclear Translocation of Apoptosis Inducing FactorC. Garcıa-Caceres,*�� A. Lechuga-Sancho,* J. Argente,*�� L. M. Frago*��1 and J. A. Chowen*�1
*Hospital Infantil Universitario Nino Jesus, Servicio de Endocrinologıa, Madrid, Spain.
�Universidad Autonoma de Madrid, Departamento de Pediatrıa, Madrid, Spain.
�CIBER Fisiopatologıa Obesidad y Nutricion (CIBEROBN), Instituto de Salud Carlos III, Madrid, Spain.
Many of the secondary complications arising from poorly controlled
diabetes mellitus (DM) involve increased cell death (1, 2). Indeed,
the hormonal imbalances that occur in poorly controlled diabetic
subjects (3) may be due, at least in part, to increased cell death in
the anterior pituitary (4). In addition, the increase in cell death at
the level of the hypothalamus (5, 6) may also be involved. Neuronal
death is reported to occur in the hypothalamus after 3 months (6),
whereas astrocytes are affected as early as 4 weeks after the onset
of diabetes (5). Not only is there increased death of these glial cells,
but also their proliferation is reduced, resulting in a significant
reduction in the number of astrocytes in the arcuate nucleus. As
hypothalamic glial cells modulate neuroendocrine functions (7),
these changes, as well as the reduction in the projections per astro-
cyte (5), could participate in the hormonal changes observed in
poorly controlled diabetes.
Astrocytes are involved in the maintenance of the neuronal
homeostatic environment (8), provide metabolic and trophic support
to neurones (9), regulate neurogenesis and participate in neuropro-
tection (10). Synaptic number and activity are also modulated by
glial cells (7, 11–13) and, in the hypothalamus, this is associated
with physiological changes in endocrine function (7, 12, 13). Thus,
a reduction in hypothalamic astrocytes and their projections could
modulate endocrine function, as well as render neurones more sus-
ceptible to adverse situations. Furthermore, as astrocytes modulate
Journal ofNeuroendocrinology
Correspondence to:
Julie A. Chowen, Servicio de
Endocrinologıa, Hospital Infantil
Universitario Nino Jesus, Avenida
Menendez Pelayo 65, 28009 Madrid,
Spain (e-mail: jachowen@
telefonica.net).1These authors contributed equally to
this study.
Astrocytes in the hypothalamus of poorly controlled diabetic rats are reduced in number, due to
increased apoptosis and decreased proliferation, and undergo morphological changes, including
a decrease in projections. These changes are associated with modifications in synaptic proteins
and most likely affect neuroendocrine signalling and function. The present study aimed to deter-
mine the intracellular mechanisms underlying this increase in hypothalamic cell death. Adult
male Wistar rats were injected with streptozotocin (70 mg ⁄ kg, i.p) and controls received vehicle.
Rats were killed at 1, 4, 6 and 8 weeks after diabetes onset (glycaemia > 300 mg ⁄ dl). Cell death,
as detected by enzyme-linked immunosorbent assay, increased at 4 weeks of diabetes. Immuno-
histochemistry and terminal dUTP nick-end labelling (TUNEL) assays indicated that these cells
corresponded to glial fibrillary acidic protein (GFAP) positive cells. No significant change in frag-
mentation of caspases 2, 3, 6, 7, 8, 9, or 12 was observed with western blot analysis. However,
enzymatic assays indicated that caspase 3 activity increased significantly after 1 week of diabe-
tes and decreased below control levels thereafter. In the hypothalamus, cell bodies lining the
third ventricle, fibres radiating from the third ventricle and GFAP positive cells expressed frag-
mented caspase 3, with this labelling increasing at 1 week of diabetes. However, because no
nuclear labelling was observed and this increase in activity did not correlate temporally with the
increased cell death, this caspase may not be involved in astrocyte death. By contrast, nuclear
translocation of apoptosis inducing factor (AIF) increased significantly in astrocytes in parallel
with the increase in death and AIF was found in TUNEL positive cells. Thus, nuclear translocation
of AIF could underlie the increased death, whereas fragmentation of caspase 3 could be associ-
ated with the morphological changes found in hypothalamic astrocytes of diabetic rats.
Key words: astrocytes, caspase, apoptosis.
doi: 10.1111/j.1365-2826.2008.01795.x
Journal of Neuroendocrinology 20, 1348–1360
ª 2008 The Authors. Journal Compilation ª 2008 Blackwell Publishing Ltd
Journal of NeuroendocrinologyFrom Molecular to Translational Neurobiology

glucose metabolism and glucose sensing in the central nervous sys-
tem (9), their affectation in diabetes could contribute to further
deregulation of glucose homeostasis. Hence, understanding the
mechanisms underlying glial apoptosis in metabolic conditions such
as poorly controlled diabetes mellitus could afford valuable infor-
mation regarding the role of these cells in hypothalamic function.
In diabetic animals, cell death by classical apoptotic pathways
involving caspases occurs in numerous tissues (1, 2, 5, 14). In the
intrinsic cell death pathway, initiator caspases such as caspase 9
are activated and these in turn activate effector caspases, including
caspases 3, 6 and 7. Activation of these effector caspases then trig-
gers processes that ultimately result in cell death. This pathway is
triggered by external signals or internal changes, with the mito-
chondria being a very important component of this cascade (15).
The balance between pro- and anti-apoptotic members of the Bcl-2
protein family has a crucial role in determining the integrity of the
mitochondria and its release of apoptogenic factors, and hence, cell
death (15).
The extrinsic cell death pathway is activated by ligand binding to
extra-cellular death receptors that belong to the tumour necrosis
factor (TNF) receptor superfamily (15, 16). This pathway, which
entails cleavage of procaspase-8 that can subsequently either acti-
vate executioner caspases or act directly to induce apoptosis (17),
is activated in the anterior pituitary of diabetic rats in a cell specific
manner (18, 19).
These pathways, however, are not independent because there is
extensive cross-talk between them, with many of the same intracel-
lular proteins being involved in both processes. Furthermore, apop-
totic cell death can also occur by caspase independent mechanisms
with specific mitochondrial factors also playing a fundamental role
(20, 21). For example, apoptosis-inducing factor (AIF) is normally
located in the mitochondria but, when confronted with a fatal
insult, can be translocated to the nucleus where it induces chroma-
tin condensation and DNA fragmentation (21). The present study
aimed to determine the intracellular mechanisms involved in the
increased death of hypothalamic astrocytes in response to poorly
controlled diabetes.
Materials and methods
Materials
All chemicals and reagents were purchased from Sigma or Merck (Barcelona,
Spain) unless otherwise indicated.
Animals
Young adult male Wistar rats (weighing approximately 250 g) were housed
under a 12 : 12 h light ⁄ dark cycle and given free access to rat chow and tap
water. Rats were injected with streptozotocin (70 mg ⁄ kg, i.p.) or vehicle (0.1 M
citrate buffer, pH 4.5). Blood glucose levels were measured (Glucocard Memory
2; Menarini Diagnostic, Florence, Italy) at baseline and then daily to determine
the onset of diabetes (glycaemia > 300 mg ⁄ dl) and then when killed. Rats
were killed by asphyxiation with CO2 and decapitation 1, 4, 6 or 8 weeks later.
All rats were killed between 10.00 h and 12.00 h. The brains were removed
and rapidly frozen on dry ice and stored at )70 �C until processed.
The following groups of rats were established: diabetic for 1 week
(DB1W; n = 6), diabetic for 4 weeks (DB4W; n = 6), diabetic for 6 weeks
(DB6W; n = 6) and diabetic for 8 weeks (DB8W; n = 6). Control rats were
killed at each time point but, as no differences in any of the studied vari-
ables were found (data not shown), control data were analysed as one
group for statistical analysis (n = 8). Three independent studies were per-
formed and hypothalamic cell death was verified in each study.
For all studies, rats were treated according to the European Community
laws for animal care and the study was approved by the appropriate institu-
tional ethical committee.
Protein extraction
For western blotting and enzyme-linked immunosorbent assay (ELISA), hypo-
thalami were isolated on ice using the following boundaries: an anterior cut
was made at the level of the optic chiasm, a posterior coronal section ante-
rior to the mammilary bodies, two sagittal cuts parallel to the lateral ventri-
cles, and a dorsal horizontal cut at the level of the anterior commissure.
Each hypothalamus was then divided into two symmetric halves. One hemi-
hypothalamus was employed for cell death detection ELISA assays and the
other half for western blotting. Tissue for ELISA was homogenised in lysis
buffer provided by the manufacturer of the commercial kit (Roche Diagnos-
tics, Mannheim, Germany). Tissue for western blotting was processed as pre-
viously described (5). Total protein concentration was determined by the
method of Bradford (Protein Assay; Bio-Rad Laboratories, Hercules, CA,
USA).
Insulin ELISA
This assay was performed according to the manufacturer’s instructions
(Linco Research, Inc., St Charles, MO, USA). Briefly, microtitre plates
coated with pre-titred monocloncal mouse anti-rat insulin antibody were
washed three times with wash buffer [50 mM Tris buffered saline (TBS)
containing Tween-20]. Ten microlitres of prediluted standards, quality con-
trol samples and serum samples were added to wells in duplicate. Detec-
tion antibody (biotinylated anti-insulin, 80 ll) was added and the plate
sealed and incubated at room temperature for 2 h while shaking. The
wells were then washed, 100 ll of enzyme solution (streptavidin-horse-
radish peroxidase conjugate) added and incubated (30 min). After wash-
ing, 100 ll of substrate solution (3,3¢,5,5¢-tetramethylbenzidine) was
added and the colour allowed to develop (approximately 15 min). Stop
solution (0.3 M HCl) was added and the plates were read at 450
and 590 nm on an automatic plate reader (Infinite M200; Tecan, Grodig,
Austria). The intra- and inter-assay coefficients of variation were 1.9 and
7.6, respectively. The limit of sensitivity of this assay is 0.2 ng ⁄ ml. For
those diabetic samples below the limit of detection, 0.2 ng ⁄ ml was used
for statistical analysis.
Cell death detection ELISA
This photometric enzyme immunoassay for the quantitative in vitro determi-
nation of cytoplasmic histone-associated DNA fragments (mono- and oligo-
nucleosomes) after induced cell death was carried out according to the
manufacturer’s instructions (Cell Death Detection ELISA; Roche Diagnostics)
and as previously described (5). Each sample was measured in duplicate in
each assay. Background measurements were made and this value was sub-
tracted from the mean value of each sample. This assay has a detection
limit of approximately 50 dead cells ⁄ well and results were normalised to
protein levels in each sample and are reported as relative levels of cell death
compared to controls. The inter- and intra-assay coefficients of variation
were 8.5% and 4.3%, respectively.
AIF induced death of astrocytes 1349
ª 2008 The Authors. Journal Compilation ª 2008 Blackwell Publishing Ltd, Journal of Neuroendocrinology, 20, 1348–1360

Caspase activity
Caspase 9, 8 and 3 ⁄ 7 activities were measured by using Caspase-Glo Assays
(Promega, Madison, WI, USA). These assays provide a luminogenic substrate
that, in the presence of the specific caspase in question, is cleaved to
release aminoluciferin, a substrate for luciferase. The luminescent signal pro-
duced is proportional to the amount of caspase activity present. Briefly,
10 lg of sample protein were added in a total volume of 25 ll of distilled
water in duplicate to a white-walled 96-well assay plate. Caspase-Glo
reagent (25 ll) was then added to each sample. The plate was then covered
and mixed gently at 300–500 r.p.m. for 30 s and incubated at room temper-
ature for 20–180 min. Luminescence was measured by using an Infinite
M200 automatic plate reader (Tecan).
Western blotting
Depending on the specific protein to be detected, either 20, 40 or 60 lg of
protein were resolved on a 12% SDS-polyacrylamide gel under denaturing
conditions. The proteins were then electro-transferred to poly(vinylidene di-
fluoride) membranes (Bio-Rad). Membranes were blocked in TBS (20 mM)
containing 5% nonfat dried milk or 5% bovine serum albumin (BSA) and
0.1% Tween 20 for 2 h. Primary antibodies, used at a concentration of
1 : 1000 unless otherwise stated, were: Anti-caspase 8, anti-caspase 7, anti-
caspase 2, anti-p53 and anti-Bax (1 : 5000), all purchased from Neomarkers
(Fremont, CA, USA). Anti-caspase 9, anti-caspase 3, anti-phosphorylated (p)-
Akt and anti-p-glycogen synthase kinase (GSK)3b (1 : 500) were from Cell
Signaling Technology (Beverly, MA, USA). Anti-caspase 12 was from Sigma
(St Louis, MO, USA), anti-caspase 6 from Medical & Biological Laboratories
(Woburn, MA, USA), anti-c-Jun and anti-pMAPK from Upstate Cell Signaling
Solutions (Lake Placid, NY, USA), anti-Bcl2alpha from Thermo Scientific (Fre-
mont, CA, USA), anti-PARP, anti-calpain-I and anti-AIF from Chemicon Inter-
national (Temecula, CA, USA). Anti-pJNK and anti-p-p38 (1 : 750) were from
Promega, anti-JNK1 and anti-p38 (1 : 500) from Santa Cruz Biotechnology
(Santa Cruz, CA, USA), anti-X-linked apoptosis inhibiting protein (XIAP) was
from BD Transduction Laboratories (Franklin Lakes, NJ, USA), anti-heat shock
protein (HSP)70 and HSP25 from Stressgen (Ann Arbor, MI, USA) and anti-
Bcl-2phosphoTyr129 was purchased from Labvision (Fremont, CA, USA). All
primary antibodies were incubated overnight at 4 �C under agitation. The
membranes were washed and incubated with the corresponding secondary
antibody conjugated with peroxidase (Pierce, Rockford, IL, USA). Bound per-
oxidase activity was visualised by chemiluminiscence (Perkin Elmer Life Sci-
ence, Boston, MA, USA) and quantified by densitometry using the BIO-1D
system (Vilber Lournat, Marne la Vallee, France). All results were first norma-
lised to actin levels in each lane (anti-actin 1 : 1000, Santa Cruz Biotechnol-
ogy) and then to control values on each blot. All experiments were
performed a minimum of two times.
Tissue sectioning
The brains were allowed to equilibrate in the cryostat chamber ()17 �C),
trimmed and embedded in OCT (Tissue-Tek, Elkhart, IN, USA). Coronal sec-
tions were cut at 20 lm throughout the entire arcuate nucleus and thaw-
mounted onto positively-charged slides. The slides were then stored at
)70 �C until immunohistochemistry was performed.
Double immunohistochemistry
After fixation in 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4),
sections were washed in phosphate buffer and then equilibrated in TBS
(0.1 M, pH 7.4) or phosphate-buffered saline (PBS; 0.1 M, pH 7.4) with 0.1%
Triton X-100 and 0.1% BSA for 20 min. The same buffer was used in all
subsequent washes. Sections were blocked for 90 min at room temperature
in TBS or PBS with 1% Triton X-100 and 3% BSA and then incubated for
48 h at 4 �C in a humid chamber with the primary antibodies. Antibodies
included anti-cleaved-caspase 3 antiserum (1 : 250; Cell Signaling Technol-
ogy), anti-pJNK (1 : 500), anti-Bax (1 : 500), anti-AIF (1 : 1000), anti-Bcl-
2alpha (1 : 500), anti-pBcl-2 (1 : 500) or anti-calpainI (1 : 250) combined
with either rabbit anti-GFAP (Neomarkers) or mouse anti-GFAP (Sigma) at a
dilution of 1 : 1000 or mouse anti-NeuN (Chemicon) at a dilution of
1 : 250. Sections were washed and incubated for 2 h under dark conditions
with the appropriate secondary antibodies that included Alexa Fluor 633
goat anti-mouse IgG (1 : 2000), Alexa Fluor 488 goat anti-mouse IgG
(1 : 1000), Alexa Fluor 633 goat anti-rabbit IgG (1 : 1000) and Alexa Fluor
488 goat anti-rabbit IgG (1 : 1000) from Molecular Probes (Leiden, Nether-
lands), and biotin conjugated goat anti-mouse IgG (1 : 1000) and biotin
conjugated goat anti-rabbit IgG (1 : 1000) from Pierce. The slides were
washed and incubated in the dark during 90 min with Alexa Fluor 633 con-
jugated streptavidin or Alexa Fluor 488 conjugated streptavidin (Molecular
Probes) at a dilution of 1 : 2000.
Preliminary assays were performed to determine the concentration of
antibodies to be used and specificity. For every experiment, sections for all
groups were incubated in parallel. In each assay, control slides consisted of
the omission of primary antibodies and verification that immunofluores-
cence was absent. Immunofluorescence was visualised by using a confocal
microscope (Leica model DMIRB; Leica, Wetzlar, Germany).
Terminal dUTP nick-end labelling (TUNEL) plusimmunohistochemistry
Cell death detection assays by TUNEL were performed following the manu-
facturer’s instructions (Promega). Briefly, sections were fixed in 4% parafor-
maldehyde in 0.1 M phosphate buffer (pH 7.4) for 20 min, washed three
times in buffer and incubated twice with PBS containing 0.3% BSA for
5 min. Sections were incubated in permeabilisation solution (PBS containing
1% Triton X-100) for 60 min and the labelling was performed with the ter-
minal deoxynucleotidyl transferase enzyme in a buffer containing fluores-
cein-12-dUTP for 60 min at 37 �C. Sections were washed, blocked with PBS
containing 3% BSA and 1% Triton X-100 for 60 min and incubated over-
night in a humid chamber at 4 �C with primary antibodies for mouse anti-
GFAP (1 : 500) or NeuN (1 : 250) in PBS containing 3% BSA and 1% Triton
X-100. After washing, the slides were incubated with Alexa Fluor goat anti-
mouse 633 in blocking buffer at a dilution of 1 : 1000. Finally, the slides
were again washed three times before mounting in glycerol. The results
were visualised by confocal microscopy.
Negative controls for TUNEL staining and immunohistochemistry were
performed by omitting terminal deoxynucleotidyl-transferase from the label-
ling mixture and the primary antibody, respectively, and resulted in no spe-
cific labelling.
Statistical analysis
All experiments were performed a minimum of two times. When a sample was
analysed more than once in separate assays (western blots) or repeated mea-
sures in the same assay (ELISA), the mean value was used for statistical analy-
sis; hence, ‘n’ represents the number of animals used in each group and no
pooling of samples was performed (n = 6 for diabetic groups and n = 8 for
controls unless otherwise stated). Testing for normality was performed by the
Lilliefor’s test. Bartlett’s test was used to determine that the groups have equal
variances. A one-way ANOVA followed by a Scheffe F-test was performed to
determine difference between experimental groups. P < 0.05 was considered
statistically significant. All results are reported as the mean � SEM. All wes-
tern blot results are reported as the percent of control value.
1350 C. Garcıa-Caceres et al.
ª 2008 The Authors. Journal Compilation ª 2008 Blackwell Publishing Ltd, Journal of Neuroendocrinology, 20, 1348–1360

Results
Blood glucose levels
Glucose levels remained significantly elevated in diabetic rats com-
pared to controls throughout the study (ANOVA: P < 0.0001) and no
significant differences were found between the diabetic groups. At
the time when the rats were killed, mean morning blood glucose
levels were: Control = 81.3 � 2.1 mg ⁄ dl; DB1W = 495.9 � 29.4
mg ⁄ dl; DB4W = 515.3 � 39.0 mg ⁄ dl; DB6W = 521.1 � 32.8 mg ⁄ dl;
DB8W = 549 � 23.6 mg ⁄ dl (n = 18 for each diabetic group, n = 24
for controls).
Insulin levels
In diabetic rats, insulin levels remained significantly reduced com-
pared to controls throughout the study (ANOVA: P < 0.01) and no sig-
nificant differences were found between the diabetic groups. At the
time when the rats were killed, mean morning blood insulin levels
were: Control = 1.89 � 0.23 ng ⁄ ml; DB1W = 0.43 � 0.04 ng ⁄ ml;
DB4W = 0.41 � 0.01 ng ⁄ ml; DB6W = 0.40 � 0.02 ng ⁄ ml; DB8W =
0.45 � 0.05 ng ⁄ ml (n = 18 for each diabetic group, n = 24 for
controls).
Cell death
There was a significant increase in cell death in the hypothalamus
of diabetic rats starting at 4 weeks of diabetes evolution [Con-
trol = 100 � 11 arbitrary units (AU); DB1W = 99 � 9 AU;
DB4W = 152 � 19 AU; DB6W = 160 � 8 AU; DB8W = 165 � 12
AU; ANOVA: P < 0.01 (n = 12 for each diabetic group, n = 16 for
controls)] as detected by ELISA, thereby confirming our previously
reported results (5).
At all stages of diabetes, the majority of TUNEL positive hypotha-
lamic cells were found in the arcuate nucleus. In control animals,
very few or no TUNEL positive cells were found. As demonstrated in
Fig. 1, TUNEL labelling found in the hypothalamus of DB4W rats
was associated with GFAP immunostained cells (Fig. 1A–C, solid
arrow). In the hypothalamus of DB8W rats, GFAP immunostaining
was also found in the majority of TUNEL labelled cells (Fig. 1D–F,
solid arrows). In addition, some TUNEL positive cells were not GFAP
positive (open arrow); however, these cells were few in number and
were not demonstrated to be NeuN positive (data not shown), sug-
gesting that they were not neurones.
Caspase activation
Although caspase 9 activation tended to increase at 1 week of dia-
betes and decrease at 4 weeks, these changes were not statistically
significant either by the Caspase-Glo assay (Fig. 2A) or western blot
analysis (data not shown).
Caspase 3 ⁄ 7 activation was significantly increased at 1 week of
diabetes and decreased thereafter as detected by the Caspase-Glo
assay (ANOVA: P < 0.05; Fig. 2B). In an attempt to differentiate
between activation of caspase 3 and 7, western blot analysis was
performed. There was no significant change in the levels of frag-
mented caspase 7 (Control = 100 � 10; DB1W = 113 � 21;
DB4W = 96 � 17; DB6W = 98 � 7; DB8W = 87 � 26). Western
blot analysis indicated that the fragmented form of caspase 3
increased after 1 week of diabetes, although this change was not
significant (Control = 100 � 8; DB1W = 141 � 21; DB4W =
106 � 9; DB6W = 117 � 17; DB8W = 116 � 17). These results
suggest that the more sensitive Caspase-Glo assay was most likely
detecting activation of caspase 3.
Immunohistochemistry for cleaved caspase 3 also suggested an
increase in the activation of this caspase in the hypothalamus after
1 week of diabetes. In control rats, immunoreactivity for cleaved cas-
pase 3 was low in the hypothalamus, with infrequent colocalisation
of GFAP and cleaved caspase 3 being found (Fig. 3A–C). Immuno-
reactivity for both GFAP and cleaved caspase 3 increased in the
TUNEL
TUNEL
GFAP
GFAP
Merge
Merge
DB4W
DB8W
(A) (B) (C)
(D) (E) (F)
DB4W
DB8W
DB4W
DB8W
Fig. 1. Terminal dUTP nick-end labelling (TUNEL) plus immunohistochemistry for glial fibrillary acidic protein (GFAP) in the hypothalamus of a rat diabetic for
4 weeks (DB4W; A–C) or for 8 weeks (DB8W; D–F). Cells that are positive for both TUNEL and GFAP are indicated by solid arrows. TUNEL positive cells that were
not GFAP positive were infrequently observed in DB8W rats (open arrow). Scale bar = 50 lm.
AIF induced death of astrocytes 1351
ª 2008 The Authors. Journal Compilation ª 2008 Blackwell Publishing Ltd, Journal of Neuroendocrinology, 20, 1348–1360

hypothalamus of DB1W rats, with colocalisation of immunoreactivity
for these two proteins also increasing (Fig. 3D–F). In diabetic rats,
cleaved caspase 3 labelling remained cytoplasmic. Projections origi-
nating at the base of the third ventricle were also immunopositive for
cleaved caspase 3. These projections were sparse in control rats and
did not colocalise with GFAP immunostaining (Fig. 3G–I, open arrow).
Cleaved caspase 3 immunolabelling of these fibres increased in DB1W
rats, but they remained GFAP negative (Fig. 3J–L, open arrows). Cells
lining the third ventricle were more highly immunoreactive for
cleaved caspase 3 in diabetic rats (Fig. 3P–R, asterisks) compared to
control rats (Fig. 3M–O, asterisks), with these cells being GFAP negative
in both experimental groups. Immunoreactive cleaved caspase 3 cells
were also negative for NeuN (data not shown).
Activation of caspase 8 was not detected in any of the experi-
mental groups by the Caspase Glo assay, nor was caspase 8 frag-
mentation detected by western blot analysis (data not shown).
There was no significant change in the activation levels of cas-
pase 6 (Control = 100 � 12; DB1W = 84 � 21; DB4W = 93 � 15;
DB6W = 88 � 11; DB8W = 91 � 29), caspase 12 (Control =
100 � 12; DB1W = 96 � 6; DB4W = 66 � 7; DB6W = 116 � 9;
DB8W = 67 � 15) or caspase 2 (Control = 100 � 8; DB1W =
138 � 23; DB4W = 110 � 12; DB6W = 95 � 20; DB8W = 130
� 29) as determined by western blot analysis.
Bcl family
Relative levels of the pro-apoptotic factor Bax increased signifi-
cantly at 1 week of diabetes compared to control rats and then
decreased (ANOVA: P < 0.0001; Fig. 4A). In the control group, Bax
immunostaining was not found to colocalise with GFAP. In DB1W
rats, Bax immunostaining increased, with the majority of Bax posi-
tive cells being GFAP negative, although an occasional cell immuno-
positive for both GFAP and Bax was found (data not shown). After
4 weeks of diabetes, when increased cell death could be detected,
relative Bax levels in the hypothalamus were not significantly
different from control rats (Fig. 4A) and differences in the cellular
localisation as detected by immunohistochemistry were not seen
(data not shown).
Bcl-2a protein levels increased at 4 weeks of diabetes, becoming
significant at 6 weeks and remaining so at 8 weeks (ANOVA:
P < 0.05; Fig. 4B). This anti-apoptotic protein was found in both
neurones and glia in all experimental groups (data not shown).
The anti-apoptotic properties of Bcl-2 can be blocked by phos-
phorylation (22); however, pBcl-2 levels did not change in response
to diabetes (Fig. 4C). Immunohistochemistry indicated that the
phosphorylated form of this protein was found predominantly in
GFAP positive cells in the hypothalamus of all experimental groups
(data not shown).
HSP and inhibitors of apoptosis
HSP can prevent both neuronal and glial cell death (23) and there
was an increase in HSP70 as early as 1 week after diabetes onset,
which gradually declined to control levels by 8 weeks of diabetes
(ANOVA P < 0.01; Fig. 4D). In both control and diabetic rats, HSP70
was localised mainly with NeuN positive cells, whereas colocalisa-
tion with GFAP positive cells was infrequent. Although HSP25 levels
tended to increase at 4 weeks of diabetes, this change was not sig-
nificant (Control = 100 � 12; DB1W = 97 � 20; DB4W = 172 �18; DB6W = 114 � 30; DB8W = 99 � 12).
There was no difference between any of the experimental groups
in the levels of XIAP (Control = 100 � 3; DB1W = 103 � 19;
DB4W = 116 � 12; DB6W = 103 � 2; DB8W = 125 � 13).
Phosphokinases
No significant change in the levels of pAkt (Control = 100 � 8;
DB1W = 119 � 8; DB4W = 94 � 5; DB6W = 112 � 22; DB8W =
119 � 15), pERK1 ⁄ 2 (Control = 100 � 11; DB1W = 89 � 18;
DB4W = 98 �11; DB6W = 117 � 8; DB8W = 109 � 7) or p-p38
levels (Control = 100 � 3; DB1W = 96 � 8; DB4W = 81 � 4;
DB6W = 88 � 7; DB8W = 106 � 14) were found in the hypothala-
mus at any stage of diabetes.
Cas
pase
3/7
act
ivat
ion
(% c
ontr
ol)
Ct DB1W DB4W DB6W DB8W0
100
200
300
400
(A)
(B)*
* *
Cas
pase
9 a
ctiv
atio
n (%
con
trol
)
0
50
100
150
200
Ct DB1W DB4W DB6W DB8W
NS
Fig. 2. (A) Caspase 9 activity in the hypothalamus of control and diabetic
rats as determined by a caspase activity assay. (B) Caspase 3 ⁄ 7 activity in
the hypothalamus of control and diabetic rats. There was a significant
increase at 1 week of diabetes, whereas, at 4 and 8 weeks of diabetes,
caspase 3 ⁄ 7 activity was significantly below control levels (ANOVA: P < 0.05 =
significant compared to control groups). NS, not significant; Ct, control;
DB1W, diabetic for 1 week; DB4W, diabetic for 4 weeks; DB6W, diabetic for
6 weeks; DB8W, diabetic for 8 weeks.
1352 C. Garcıa-Caceres et al.
ª 2008 The Authors. Journal Compilation ª 2008 Blackwell Publishing Ltd, Journal of Neuroendocrinology, 20, 1348–1360

Cl- Casp 3 GFAP Merge
Cl- Casp 3 GFAP Merge
Cl- Casp 3 GFAP Merge
Cl- Casp 3 GFAP Merge
(A)
(D)
(G)
(J)
3V 3V 3V
3V 3V 3V
Control Control Control
Control Control Control
Control Control Control
DB1W DB1W DB1W
DB1W DB1W DB1W
DB1W DB1W DB1W
3V 3V 3V
3V 3V 3V
*
**
*
*
*
**
*
*
*
**
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
Cl- Casp 3 GFAP Merge
Cl. Casp 3 GFAP Merge
(M)
(P)
(B)
(E)
(H)
(K)
(N)
(Q)
(C)
(F)
(I)
(L)
(O)
(R)
Fig. 3. Immunolabelling for cleaved caspase 3 (Cl-Casp3; green) was found to colocalise with glial fibrillary acidic protein (GFAP; red) in the arcuate nucleus
of both control groups (A–C) and rats that were diabetic for 1 week (DB1W; D–F) as indicated by the solid arrows. Cl-Casp3 labelled fibres (open arrows) were
seen radiating from the third ventricle (3V) in both control (G–I) and DB1W (J–L) rats. The labelling of these fibres clearly increased in DB1W rats. Cells lining
the 3V (asterisks) were also found to express Cl-Casp3 in both control (M–O) and DB1W (P–R) rats, with the intensity of this labelling increasing in diabetic rats.
AIF induced death of astrocytes 1353
ª 2008 The Authors. Journal Compilation ª 2008 Blackwell Publishing Ltd, Journal of Neuroendocrinology, 20, 1348–1360
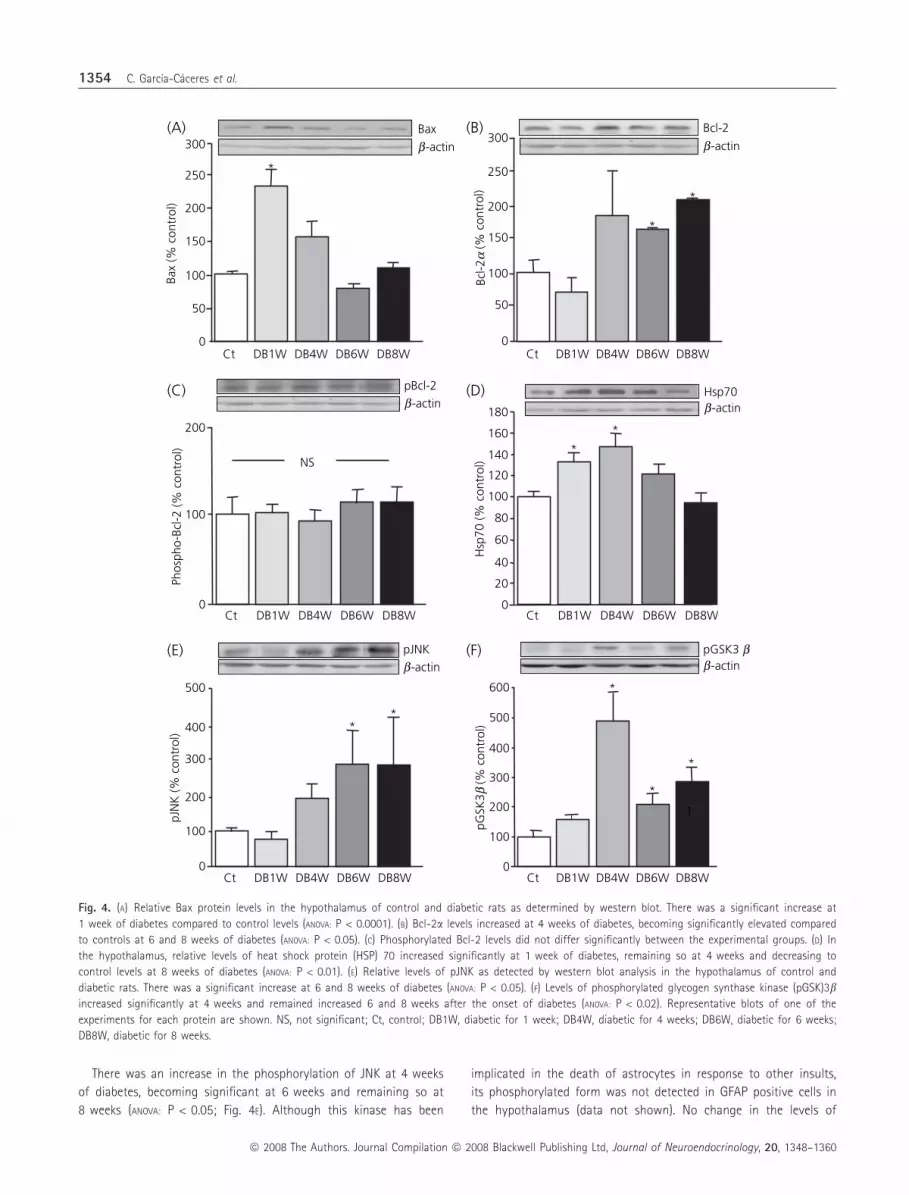
There was an increase in the phosphorylation of JNK at 4 weeks
of diabetes, becoming significant at 6 weeks and remaining so at
8 weeks (ANOVA: P < 0.05; Fig. 4E). Although this kinase has been
implicated in the death of astrocytes in response to other insults,
its phosphorylated form was not detected in GFAP positive cells in
the hypothalamus (data not shown). No change in the levels of
Ct DB1W DB4W DB6W DB8W Ct DB1W DB4W DB6W DB8W
Ct DB1W DB4W DB6W DB8W Ct DB1W DB4W DB6W DB8W
Ct DB1W DB4W DB6W DB8W Ct DB1W DB4W DB6W DB8W
50
100
150
200
250
0
300(A) (B)
(C) (D)
(E) (F)
*
Bax
b-actin300
Bcl-2
a (%
con
trol
)
0
100
200
*
*
Bcl-2
b-actin
pBcl-2 Hsp70
Hsp
70 (%
con
trol
)
0
20
40
60
80
100
120
140
160
180
*
*
b-actinb-actin
0
Phos
pho-
Bcl-2
(% c
ontr
ol)
Bax
(% c
ontr
ol)
100
200
NS
pJNK
pJN
K (%
con
trol
)
b-actin
100
200
300
400
0
500
**
0
1
100
200
300
400
500
600 *
*
*
pGSK
3b (%
con
trol
)
b-actinpGSK3 b
50
150
250
Fig. 4. (A) Relative Bax protein levels in the hypothalamus of control and diabetic rats as determined by western blot. There was a significant increase at
1 week of diabetes compared to control levels (ANOVA: P < 0.0001). (B) Bcl-2a levels increased at 4 weeks of diabetes, becoming significantly elevated compared
to controls at 6 and 8 weeks of diabetes (ANOVA: P < 0.05). (C) Phosphorylated Bcl-2 levels did not differ significantly between the experimental groups. (D) In
the hypothalamus, relative levels of heat shock protein (HSP) 70 increased significantly at 1 week of diabetes, remaining so at 4 weeks and decreasing to
control levels at 8 weeks of diabetes (ANOVA: P < 0.01). (E) Relative levels of pJNK as detected by western blot analysis in the hypothalamus of control and
diabetic rats. There was a significant increase at 6 and 8 weeks of diabetes (ANOVA: P < 0.05). (F) Levels of phosphorylated glycogen synthase kinase (pGSK)3bincreased significantly at 4 weeks and remained increased 6 and 8 weeks after the onset of diabetes (ANOVA: P < 0.02). Representative blots of one of the
experiments for each protein are shown. NS, not significant; Ct, control; DB1W, diabetic for 1 week; DB4W, diabetic for 4 weeks; DB6W, diabetic for 6 weeks;
DB8W, diabetic for 8 weeks.
1354 C. Garcıa-Caceres et al.
ª 2008 The Authors. Journal Compilation ª 2008 Blackwell Publishing Ltd, Journal of Neuroendocrinology, 20, 1348–1360
p-cJun, a downstream target of JNK, were found (Control =
100 � 11; DB1W = 77 � 12; DB4W = 84 � 3; DB6W = 82 � 10;
DB8W = 122 � 13).
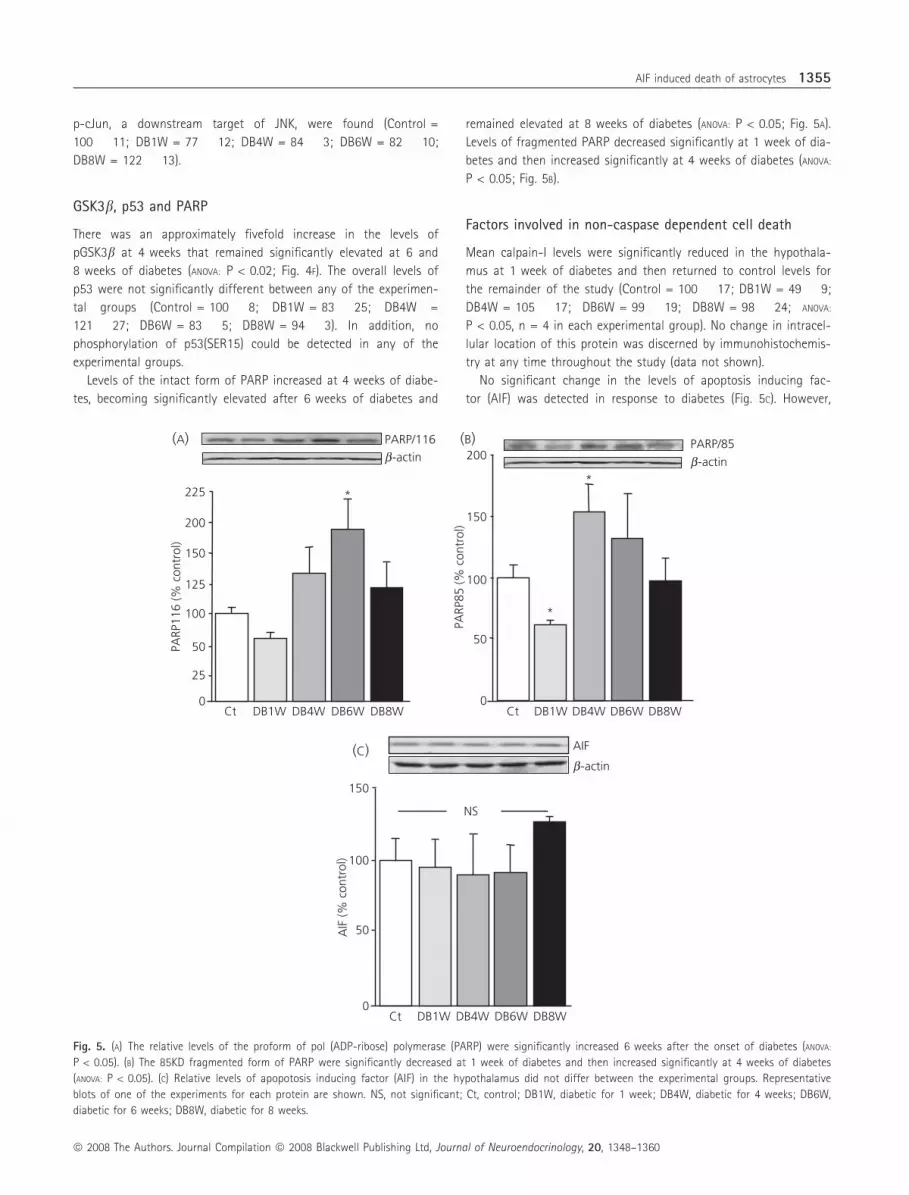
GSK3b, p53 and PARP
There was an approximately fivefold increase in the levels of
pGSK3b at 4 weeks that remained significantly elevated at 6 and
8 weeks of diabetes (ANOVA: P < 0.02; Fig. 4F). The overall levels of
p53 were not significantly different between any of the experimen-
tal groups (Control = 100 � 8; DB1W = 83 � 25; DB4W =
121 � 27; DB6W = 83 � 5; DB8W = 94 � 3). In addition, no
phosphorylation of p53(SER15) could be detected in any of the
experimental groups.
Levels of the intact form of PARP increased at 4 weeks of diabe-
tes, becoming significantly elevated after 6 weeks of diabetes and
remained elevated at 8 weeks of diabetes (ANOVA: P < 0.05; Fig. 5A).
Levels of fragmented PARP decreased significantly at 1 week of dia-
betes and then increased significantly at 4 weeks of diabetes (ANOVA:
P < 0.05; Fig. 5B).
Factors involved in non-caspase dependent cell death
Mean calpain-I levels were significantly reduced in the hypothala-
mus at 1 week of diabetes and then returned to control levels for
the remainder of the study (Control = 100 � 17; DB1W = 49 � 9;
DB4W = 105 � 17; DB6W = 99 � 19; DB8W = 98 � 24; ANOVA:
P < 0.05, n = 4 in each experimental group). No change in intracel-
lular location of this protein was discerned by immunohistochemis-
try at any time throughout the study (data not shown).
No significant change in the levels of apoptosis inducing fac-
tor (AIF) was detected in response to diabetes (Fig. 5C). However,
AIF
(% c
ontr
ol)
PARP/116b-actin
0
50
100
150
200PARP/85
b-actin
*
*
PARP
116
(% c
ontr
ol)
0
50
25
125
150
100
225
(A) (B)
(C)
200
*
PARP
85 (%
con
trol
)
Ct DB1W DB4W DB6W
0
50
100
150
AIF
b-actin
NS
Ct DB1W DB4W DB6W DB8W
DB8W Ct DB1W DB4W DB6W DB8W
Fig. 5. (A) The relative levels of the proform of pol (ADP-ribose) polymerase (PARP) were significantly increased 6 weeks after the onset of diabetes (ANOVA:
P < 0.05). (B) The 85KD fragmented form of PARP were significantly decreased at 1 week of diabetes and then increased significantly at 4 weeks of diabetes
(ANOVA: P < 0.05). (C) Relative levels of apopotosis inducing factor (AIF) in the hypothalamus did not differ between the experimental groups. Representative
blots of one of the experiments for each protein are shown. NS, not significant; Ct, control; DB1W, diabetic for 1 week; DB4W, diabetic for 4 weeks; DB6W,
diabetic for 6 weeks; DB8W, diabetic for 8 weeks.
AIF induced death of astrocytes 1355
ª 2008 The Authors. Journal Compilation ª 2008 Blackwell Publishing Ltd, Journal of Neuroendocrinology, 20, 1348–1360

the intracellular localisation of this protein differed between
experimental groups. In the hypothalamus of control rats, AIF
was not found to be translocated to the nucleus of GFAP posi-
tive cells (Fig. 6C,G). In rats that were diabetic for only 1 week,
AIF was not found in the nucleus of GFAP positive cells
(Fig. 6H). After 4, 6 and 8 weeks of diabetes, AIF was found in
the nucleus of numerous GFAP positive cells, as well as non-
nuclear parts in others (Fig. 6 I, J, F, K, respectively). Cells with AIF
translocated to the nucleus were mainly localised to the arcuate
nucleus.
AIF immunolabelling was also found associated with TUNEL posi-
tive cells; although not all TUNEL positive cells were AIF positive
(Fig. 6L–M).
Discussion
In diabetes mellitus, apoptotic cell death occurs in numerous tissues
(1, 2, 4–6, 14, 18, 19) with increased glucose levels (24, 25),
decreased insulin or insulin-like growth factor signalling (26) or an
increase in cytokines such as TNFa (27) being the triggering factor.
Astrocytes in the arcuate nucleus are activated by changes in glu-
cose concentrations (28) and are involved in glucose sensing at the
brain level (9). Furthermore, glial cells are reported to control the
supply of glucose and its metabolites to neurones (9), indicating
that they are the first line of defence against changes in glucose
concentrations. Indeed, when glucose availability is reduced, glyco-
gen stored in astrocytes serves as a fuel source for neurones (29).
Control
AIF
DB
DB8W
AIF
GFAP
GFAP AIF GFAP
AIF GFAP
AIF GFAP DB8WControl DB6WDB4W
(A) (B) (C)
(D) (E) (F)
(G) (K)
DB1W AIF GFAP AIF GFAP AIF GFAP AIF GFAP
TUNELDB6W AIFDB6W AIF TUNELDB6W
(H) (I) (J)
(L) (M) (N)
Fig. 6. Immunoreactivity for AIF (green) was not found in the nucleus of glial fibrillary acidic protein (GFAP; red) positive cells in the arcuate nucleus of con-
trol rats (A–C, G). In the arcuate nucleus of rats after 8 weeks of diabetes, AIF immunolabelling was observed to be non-nuclear in some GFAP positive cells
and nuclear in others (D–F, K). In DB1W rats, AIF was found to be mainly non-nuclear in GFAP positive cells (H). In DB4W (I) and DB6W (J) rats, AIF was non-
nuclear in some GFAP positive cells and translocated to the nucleus of others. Solid arrows indicate GFAP positive cells with non-nuclear localisation of AIF.
Open arrows indicate GFAP positive cells with AIF localised to the nucleus. AIF (red) colocalization in terminal dUTP nick-end labelled (TUNEL, green) positive
cells. DB1W, diabetic for 1 week; DB4W, diabetic for 4 weeks; DB6W, diabetic for 6 weeks; DB8W, diabetic for 8 weeks. Scale bar = 30 lm.
1356 C. Garcıa-Caceres et al.
ª 2008 The Authors. Journal Compilation ª 2008 Blackwell Publishing Ltd, Journal of Neuroendocrinology, 20, 1348–1360

Thus, it is possible that prolonged exposure to elevated glucose lev-
els underlies the increased death of hypothalamic astrocytes.
Indeed, high glucose levels have been shown to directly induce
death of different glial cell types (30, 31). However, other circulat-
ing factors, such as increased TNFa (18), decreased insulin (19) or
insulin growth factor (IGF)-I (32), or decreased central IGF-I as well
as its receptor (L. M. Frago, J. A. Chowen; unpublished observation),
could also be involved.
Activation of caspases 9 and 3 (14, 25, 33), as well as caspase 6
(14, 34) and 8 (18, 33, 34) have been implicated in diabetes induced
cell death. However, these classical apoptotic pathways cannot
explain the death of astrocytes observed in the present study
because no activation of caspases 8, 2, 12 or 6 was detected.
Changes in caspase 9 activity temporally paralleled caspase 3 ⁄ 7activity, but did not reach statistical significance. Although the
enzymatic method employed to detect caspase activity did not dis-
tinguish between caspases 3 and 7, western blot analysis suggested
that caspase 3 was activated, as no fragmentation of caspase 7
was detected, whereas changes in fragmentation of caspase 3
expressed the same temporal pattern as caspase activity. This was
further supported by our immunohistochemical data.
Caspase 3 activity increased 1 week after diabetes onset, when
no increase in cell death was observed, and then decreased below
control levels at 4 weeks of diabetes, when cell death increased. The
lack of temporal coordination and also the absence of nuclear local-
isation of cleaved caspase 3 suggest that this caspase is most likely
not inducing the death of astrocytes. In control rats, cleaved cas-
pase 3 was found in the cytoplasm of some GFAP positive cells in
the arcuate nucleus, in GFAP negative cellular projections originat-
ing at the base of the third ventricle, as well as in GFAP negative
cell bodies lining the third ventricle. After 1 week of diabetes,
immunoreactivity of cleaved caspase 3 increased in all of these cel-
lular locations. Caspase 3 is implicated in functions other than
induction of cell death, including cleavage of substrates involved in
cell proliferation and migration (35, 36), or cellular plasticity
through cleavage of cytoskeletal components, including actin,
vimentin and GFAP (37–39). Indeed, cleaved caspase 3 is expressed
in astrocytes in the normal adult brain where it is not associated
with cell death (40, 41). In the normal adult cerebellum, caspase 3 is
suggested to participate in the differentiation or proliferation of
Bergman glia (42), although, in diabetic rats, the proliferation of
hypothalamic astrocytes is reduced (5). Caspase 3 mediated cleavage
of GFAP has also been associated with turnover of GFAP in reactive
astrocytes (38, 39). In our experimental paradigm, not only do GFAP
protein levels decline in the hypothalamus, but also the mean num-
ber of GFAP positive projections per astrocyte in the arcuate nucleus
also decreases significantly (5). Whether caspase 3 activation is
involved in this cytoskeletal reorganisation in hypothalamic astro-
cytes remains to be demonstrated. However, because cytoplasmic
caspase 3 can also act an as initiator caspase, activating down-
stream apoptosis inducing factors that are then translocated to the
nucleus, participation of this increase in caspase 3 activation at
1 week of diabetes in cell death processes cannot be ruled out.
Proteins of the Bcl-2 family were also modified in response to
diabetes. The anti-apoptotic protein Bcl-2a increased at 4 weeks
and remained significantly increased at 6 and 8 weeks of diabetes,
precisely when cell death increased, and was found in both neuro-
nes and astrocytes. However, phosphorylation of Bcl-2, which can
inhibit its anti-apoptotic properties (22), was found mainly in astro-
cytes. Hence, although overall levels of phosphorylated Bcl-2 did
not change in response to diabetes, its inactivation in some astro-
cytes could render them more susceptible to further assaults and
possible death. Indeed, streptozotocin induced diabetes results in
astrocytes being more susceptible to death in response to ischaemia
(43).
Overall, levels of the pro-apoptotic protein Bax increased signifi-
cantly at 1 week of diabetes and were found mainly in neurones.
As neuronal death was not observed at this time, translocation of
this protein to the mitochondrial membrane and its apoptotic
actions must have been impeded. One possibility is inhibition of the
intrinsic cell death pathway by HSP (44). Indeed, changes in the
levels of HSP70 paralleled those of Bax, increasing significantly at
1 week of diabetes and declining gradually thereafter. Furthermore,
HSP70 was found mainly in neurones, with very few GFAP positive
cells expressing this protein in control or diabetic rats. By contrast,
XIAP, which can inhibit both the intrinsic and extrinsic cell death
pathways, was not modified.
The ubiquitous serine ⁄ threonine kinase GSK3b is constitutively
active in resting cells, with its actions being tightly controlled
through inactivation by phosphorylation. When active, GSK3b can
phosphorylate members of the Bcl-2 family and other mitochon-
drial proteins to promote cell death (45). Phosphorylation of GSK3bincreased significantly in the hypothalamus at 4 weeks of diabetes,
in accordance with changes in other mitochondrial proteins moving
towards an anti-apoptotic balance. Taken together, the data
obtained in the present study indicate that the intrinsic cell death
pathway is not involved in the increased cell death in the hypothal-
amus and that, indeed, anti-apoptotic processes are activated that
could explain the lack of neuronal death at this time.
Activation of distinct MAP kinases involved in cell survival or
induction of apoptosis were analysed to further understand the
intracellular changes occurring in this experimental paradigm. No
change in the activation of Akt or ERKs was observed. These pro-
teins are generally involved in cell survival and proliferative pro-
cesses in both neurones and glia (46), although ERKs have also
been associated with induction of glial death (47, 48). Similarly, no
modification was found in the activation of p38, a kinase involved
in both neuronal (49) and glial (50) cell death. JNK has been associ-
ated with both induction and protection (46, 48, 50) of astrocytic
cell death, as well as glial induced neurone cell death (51). A signif-
icant increase in phosphorylation of JNK occurred at 4 weeks of
diabetes, coincident with the increase in cell death, but was found
almost exclusively in non-GFAP positive cells. Indeed, phosphoryla-
tion of JNK occurs in neurones in response to a decrease in insulin
(52) and participates in neuronal protection in diabetes and in
response to glucose ⁄ oxidative stress (53). In addition, JNK has
recently been shown to modulate mitochondrial bioenergetics
through phosphorylation of pyruvate dehydrogenase (54). Thus, the
observed activation of JNK could be involved in changes in neuro-
nal metabolism and protection from the diabetic state.
AIF induced death of astrocytes 1357
ª 2008 The Authors. Journal Compilation ª 2008 Blackwell Publishing Ltd, Journal of Neuroendocrinology, 20, 1348–1360

Nuclear translocation of AIF is involved in caspase-independent
cell death in response to a variety of signals, including oxidative
stress, glutamate toxicity, and ischaemia (21). Loss of mitochon-
drial membrane integrity results in the release of this factor,
which can then be translocated to the nucleus where it induces
DNA fragmentation and chromatin condensation (21). Some stud-
ies suggest that this mechanism may be especially involved in cell
death in the adult brain (55). Nuclear translocation of AIF has
also been shown to induce caspase-independent cell death in reti-
nal neurones in response to high glucose levels (56), suggesting
that this mechanism may be involved in diabetes induced cell
death. Although we found no overall increase in AIF levels,
increased nuclear localisation of this factor was observed at the
same experimental time points where increased astrocytic death
was found. Furthermore, AIF was found to colocalise in most,
although not all, TUNEL positive cells, indicating that it is most
likely one of the mechanisms involved in diabetes induced cell
death in the hypothalamus.
Elucidation of the molecular events involved in the release of
AIF from the mitochondria remains an active area of investigation,
with many aspects of this process yet to be determined. Activa-
tion of p53 and its downstream proteins such as Bax (21) have
been implicated in AIF translocation from the mitochondria; how-
ever, we found no activation of p53 and Bax was not found to
be increased when AIF translocation was seen. Calpain I has
recently been shown to be involved in AIF translocation in ischae-
mia induced neuronal cell death (57), but in rat hypothalamus
calpain I was actually decreased after 1 week of diabetes, return-
ing to control levels thereafter. Furthermore, no changes in its
subcellular distribution were detected. Under basal conditions,
PARP1 is involved in the detection of DNA damage and repair
(58). However, in conditions of severe cellular damage, increased
PARP activation can result in cellular energy depletion and thus
lead to increased cell death, and one mechanism by which PARP
induces cell death is through liberation of AIF (21). In the hypo-
thalamus of diabetic rats, an increase in overall PARP1 levels, as
well as increased fragmentation of PARP, was coincident with the
increase in cell death. Furthermore, when PARP levels were
increased, translocation of AIF to the nucleus was found in hypo-
thalamic astrocytes. Together, these observations suggest that
PARP induced translocation of AIF may be involved in the
increased death of astrocytes in the hypothalamus of poorly con-
trolled diabetic rats. Indeed, primary astrocyte cultures have been
used to demonstrate that AIF-mediated cell death in astrocytes
involves PARP1 activation and requires NAD+ depletion, although
how PARP1 activation is associated with AIF release remains to
be determined (59).
The results reported in the present study demonstrate that
numerous intracellular signalling mechanisms are modulated in a
time specific manner in the hypothalamus in response to poorly
controlled diabetes and that many of these changes are cell type
specific (Table 1). The original aim of the study was to determine
the intracellular mechanism underlying the increased death of as-
trocytes. We report that this process most likely involves nuclear
translocation of AIF, although other mechanisms may also be
involved. In addition, we demonstrate that intracellular mechanisms
involved with cell protection are also activated and that many of
these processes are neurone specific, which is congruent with the
delay in neuronal death.
Acknowledgements
This work was funded by grants from Fondo de Investigacion Sanitaria
(PI040817, PI051268 and PI070182), CIBER Fisiopatologıa de Obesidad y
Nutricion (CIBEROBN) Instituto de Salud Carlos III and Fundacion de Endo-
crinologıa y Nutricion. C.G.-C. is supported by a predoctoral fellowship from
the Ministerio de Educacion y Ciencia, A.M.L.-S. was supported by a post-
doctoral fellowship from Fondo de Investigacion Sanitaria. J.A.C. is supported
by the biomedical investigation programme of the Consejerıa de Sanidad y
Consumo de la Comunidad de Madrid. The authors would like to thank San-
dra Canelles and Francisca Dıaz for the excellent technical support.
Received 30 May 2008,
revised 11 August 2008,
accepted 26 August 2008
Table 1. Summary of the Major Changes in Pro- and Anti-Apoptotic Proteins Observed in the Hypothalamus of Diabetic Rats Compared to Control Rats.
DB1 DB4 DB6 DB8 Cellular localisation
Cell death M › › › Astrocytes, mainly arcuate nucleus
Caspase 3 ⁄ 7 activation (pro-apoptotic) › fl M fl Astrocytes (cytoplasmic), projections from and cells lining third ventricle
Bcl-2a (anti-apoptotic) M › › › Neurones and astrocytes
pBcl-2a (pro-apoptotic) M M M M Astrocytes
Bax (pro-apoptotic) › M M M Mainly neurones, few astrocytes
Hsp70 (anti-apoptotic) › › M M Mainly neurones, few astrocytes
pJNK (can be either) M M › › Mainly neurones, few astrocytes
pGSKb (anti-apoptotic) M › › › ND
Calpain 1 (pro-apoptotic) fl M M M Cytoplamic localisation
PARP116 (pro-apoptotic) M M › M ND
PARP85 (pro-apoptotic) fl › M M ND
AIF nuclear translocation (pro-apoptotic) M › › › Astrocytes
DB1W, diabetic for 1 week; DB4W, diabetic for 4 weeks; DB6W, diabetic for 6 weeks; DB8W, diabetic for 8 weeks; ND, not done.
1358 C. Garcıa-Caceres et al.
ª 2008 The Authors. Journal Compilation ª 2008 Blackwell Publishing Ltd, Journal of Neuroendocrinology, 20, 1348–1360

References
1 Nishikawa T, Araki E. Impact of mitochondrial ROS production in the
pathogenesis of diabetes mellitus and its complications. Antioxid Redox
Signal 2007; 9: 343–353.
2 Allen DA, Yaqoob MM, Harwood SM. Mechanisms of high glucose-
induced apoptosis and its relationship to diabetic complications. J Nutr
Biochem 2005; 16: 705–713.
3 Steger RW, Rabe MM. The effect of diabetes mellitus on endocrine and
reproductive function. Proc Soc Exp Biol Med 1997; 214: 1–11.
4 Arroba AI, Frago LM, Paneda C, Argente J, Chowen JA. The number of
lactotrophs is reduced in the anterior pituitary of streptozotocin-induced
diabetic rats. Diabetologıa 2003; 465: 634–638.
5 Lechuga-Sancho AM, Arroba AI, Frago LM, Garcıa-Caceres C, Delgado
Rubin de Celix A, Argente J, Chowen JA. Reduction in the number of as-
trocytes and their projections is associated with increased synaptic pro-
tein density in the hypothalamus of poorly controlled diabetic rats.
Endocrinology 2006; 147: 5314–5324.
6 Klein JP, Hains BC, Craner MJ, Black JA, Waxman SG. Apoptosis of vaso-
pressinergic hypothalamic neurons in chronic diabetes mellitus. Neurobi-
ol Dis 2004; 15: 221–228.
7 Garcia-Segura LM, McCarthy MM. Minireview: role of glia in neuroendo-
crine function. Endocrinology 2004; 145: 1082–1086.
8 Walz W. Role of astrocytes in the clearance of excess extracellular
potassium. Neurochem Int 2000; 36: 291–300.
9 Pellerin L, Bouzier-Sore AK, Aubert A, Serres S, Merle M, Costalat R,
Magistretti PJ. Activity-dependent regulation of energy metabolism by
astrocytes: an update. Glia 2007; 55: 1251–1262.
10 Azcoitia I, Garcia-Ovejero D, Chowen JA, Garcia-Segura LM. Astroglia
play a key role in the neuroprotective actions of estrogen. Prog Brain
Res 2001; 132: 469–478.
11 Ullian EM, Sapperstein SK, Christopherson KS, Barres BA. Control of syn-
apse number by glia. Science 2001; 291: 657–660.
12 Theodosis DT, Trailin A, Poulain DA. Remodeling of astrocytes, a prere-
quisite for synapse turnover in the adult brain? Insights from the oxyto-
cin system of the hypothalamus. Am J Physiol Regul Integr Comp
Physiol 2006; 290: R1175–R1182.
13 Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, Friedman
JM, Horvath TL. Rapid rewiring of arcuate nucleus feeding circuits by
leptin. Science 2004; 304: 110–115.
14 Lechuga-Sancho AM, Arroba AI, Frago LM, Paneda C, Garcıa-Caceres C,
Delgado Rubın de Celix A, Argente J, Chowen JA. Activation of the
intrinsic cell death pathway, increased apoptosis and modulation of as-
trocytes in the cerebellum of diabetic rats. Neurobiol Dis 2006; 23: 290–
299.
15 Green DR, Droemer G. The pathophysiology of mitochondrial cell death.
Science 2004; 305: 626–629.
16 Wajant H. Death receptors. Essays Biochem 2003; 39: 53–71.
17 Krammer PE. CD95’s deadly mission in the immune system. Nature
2000; 407: 789–795.
18 Arroba AI, Frago LM, Argente J, Chowen JA. Activation of caspase 8 in
the pituitaries of streptozotocin-induced diabetic rats: implication in
increased apoptosis of lactotrophs. Endocrinology 2005; 146: 4417–
4424.
19 Arroba AI, Lechuga-Sancho AM, Frago LM, Argente J, Chowen JA. Cell-
specific expression of X-linked inhibitor of apoptosis in the anterior pitu-
itary of streptozotocin-induced diabetic rats. J Endocrinol 2007; 192:
215–227.
20 Hetz CA, Torres V, Quest AF. Beyond apoptosis: nonapoptotic cell death
in physiology and disease. Biochem Cell Biol 2005; 83: 579–588.
21 Krantic S, Mechawar N, Reix S, Quirion R. Apoptosis-inducing factor: a
matter of neuron life and death. Prog Neurobiol 2007; 81: 179–196.
22 Srivastava RK, Srivastava AR, Korsmeyer SJ, Nesterova M, Cho-Chung YS,
Longo DL. Involvement of microtubules in the regulation of Bcl2 phos-
phorylation and apoptosis through cyclic AMP-dependent protein kinase.
Mol Cell Biol 1998; 18: 3509–3517.
23 Yenari MA, Liu J, Zheng Z, Vexler ZS, Lee JE, Giffard RG. Antiapoptotic
and anti-inflammatory mechanisms of heat-shock protein protection.
Ann NY Acad Sci 2005; 1053: 74–83.
24 Romero G, Liu WH, Asnaghi V, Kern TS, Lorenzi M. Activation of
nuclear factor-kappaB induced by diabetes and high glucose regulates
a proapoptotic program in retinal pericytes. Diabetes 2002; 51: 2241–
2248.
25 Anitha M, Gondha C, Sutliff R, Parsadanian A, Mwangi S, Sitaraman SV,
Srinivasan S. GDNF rescues hyperglycemia-induced diabetic enteric neu-
ropathy through activation of the PI3K ⁄ Akt pathway. J Clin Invest 2006;
116: 344–356.
26 Ishii DN. Implication of insulin-like growth factors in the pathogenesis
of diabetic neuropathy. Brain Res Brain Res Rev 1995; 20: 47–67.
27 Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science
2002; 296: 1634–1635.
28 Guillod-Maximin E, Lorsignol A, Alquier T, Penicaud L. Acute intracarotid
glucose injection towards the brain induces specific c-fos activation in
hypothalamic nuclei: involvement of astrocytes in cerebral glucose-sens-
ing in rats. J Neuroendocrinol 2004; 16: 464–471.
29 Brown AM, Ransom BR. Astrocyte glycogen and brain energy metabo-
lism. Glia 2007; 55: 1263–1271.
30 Xi X, Gao L, Atala DA, Smith DG, Codispoti MC, Gong B, Kern TS, Zhang
JZ. Chronically elevated glucose-induced apoptosis is mediated by inacti-
vation of Akt in cultured Muller cells. Biochem Biophys Res Commun
2005; 326: 548–553.
31 Delaney CL, Russell JW, Cheng HL, Feldman EL. Insulin-like growth
factor-I and over-expression of Bcl-xL prevent glucose-mediated
apoptosis in Schwann cells. J Neuropathol Exp Neurol 2001; 60:
147–160.
32 Busiguina S, Chowen JA, Argente J, Torres-Aleman I. Specific alterations
of the insulin-like growth factor I system in the cerebellum of diabetic
rats. Endocrinology 1996; 137: 4980–4987.
33 Al-Mashat HA, Kandru S, Liu R, Behl Y, Desta T, Graves DT. Diabetes
enhances mRNA levels of proapoptotic genes and caspase activity,
which contribute to impaired healing. Diabetes 2006; 55: 487–495.
34 Bojunga J, Nowak D, Mitrou PS, Hoelzer D, Zeuzem S, Chow KU. Antioxi-
dative treatment prevents activation of death-receptor- and mitochon-
drion-dependent apoptosis in the hearts of diabetic rats. Diabetologia
2004; 47: 2072–2080.
35 Yan XX, Najbauer J, Woo CC, Dashtipour K, Ribak CE, Leon M. Expression
of active caspase-3 in mitotic and postmitotic cells of the rat forebrain.
J Comp Neurol 2001; 433: 4–22.
36 Kuranaga E, Miura M. Nonapoptotic functions of caspases: caspases as
regulatory molecules for immunity and cell-fate determination. Trends
Cell Biol 2007; 17: 135–144.
37 Morishima N. Changes in nuclear morphology during apoptosis correlate
with vimentin cleavage by different caspases located either upstream or
downstream of Bcl-2 action. Genes Cells 1999; 4: 401–414.
38 Mouser PE, Head E, Ha KH, Rohn TT. Caspase-mediated cleavage of glial
fibrillary acidic protein within degenerating astrocytes of the Alzheimer’s
disease brain. Am J Pathol 2006; 168: 936–946.
39 Acarin L, Villapol S, Faiz M, Rohn TT, Castellano B, Gonzalez B. Caspase-
3 activation in astrocytes following postnatal excitotoxic damage corre-
lates with cytoskeletal remodeling but not with cell death or prolifera-
tion. Glia 2007; 55: 954–965.
40 Noyan-Ashraf MH, Brandizzi F, Juurlink BH. Constitutive nuclear localiza-
tion of activated caspase 3 in subpopulations of the astroglial family of
cells. Glia 2005; 49: 588–593.
AIF induced death of astrocytes 1359
ª 2008 The Authors. Journal Compilation ª 2008 Blackwell Publishing Ltd, Journal of Neuroendocrinology, 20, 1348–1360

41 Delgado-Rubın del Celix A, Chowen JA, Argente J, Frago LM. Growth
hormone releasing peptide-6 acts as a survival factor in glutamate-
induced excitotoxicity. J Neurochem 2006; 99: 839–849.
42 Oomman S, Strahlendorf H, Finckbone V, Strahlendorf J. Non-lethal
active caspase-3 expression in Bergmann glia of postnatal rat cerebel-
lum. Brain Res Dev Brain Res 2005; 160: 130–145.
43 Muranyi M, Ding C, He Q, Lin Y, Li PA. Streptozotocin-induced diabetes
causes astrocyte death after ischemia and reperfusion injury. Diabetes
2006; 55: 349–355.
44 Stankiewicz AR, Lachapelle G, Foo CP, Radicioni SM, Mosser DD. Hsp70
inhibits heat-induced apoptosis upstream of mitochondria by preventing
Bax translocation. J Biol Chem 2005; 280: 38729–38739.
45 Beurel E, Jope RS. The paradoxical pro- and anti-apoptotic actions of
GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog
Neurobiol 2006; 79: 173–189.
46 Ciccarelli R, D¢Alimonte I, Ballerini P, D¢Auro M, Nargi E, Buccella S,
Di Iori P, Bruno V, Nicoletti F, Caciagli F. Molecular signalling mediat-
ing the protective effect of A1 adenosine and mGlu3 metabotropic
glutamate receptor activation against apoptosis by oxygen ⁄ glucose
deprivation in cultured astrocytes. Mol Pharmacol 2007; 71: 1369–
1380.
47 Shinozaki Y, Koizumi S, Ohno Y, Nagao T, Inoue K. Extracellular ATP
counteracts the ERK1 ⁄ 2-mediated death-promoting signaling cascades
in astrocytes. Glia 2006; 54: 606–618.
48 Kawasaki T, Kitao T, Nakagawa K, Fujisaki H, Takegawa Y, Koda K, Ago Y,
Baba A, Matsuda T. Nitric oxide-induced apoptosis in cultured rat astro-
cytes: protection by edaravone, a radical scavenger. Glia 2007; 55:
1325–1333.
49 Redman PT, He K, Hartnett KA, Jefferson BS, Hu L, Rosenberg PA, Levitan
ES, Aizenman E. Apoptotic surge of potassium currents is mediated by
p38 phosphorylation of Kv2.1. Proc Natl Acad Sci USA 2007; 104:
3568–3573.
50 Wang Y, Luo W, Stricker R, Reiser G. Protease-activated receptor-1 pro-
tects rat astrocytes from apoptotic cell death via JNK-mediated release
of the chemokine GRO ⁄ CINC-1. J Neurochem 2006; 98: 1046–1060.
51 Xie Z, Smith CJ, Van Eldik LJ. Activated glia induce neuron death via
MAP kinase signaling pathways involving JNK and p38. Glia 2004; 45:
170–179.
52 Schechter R, Beju D, Miller KE. The effect of insulin deficiency on tau
and neurofilament in the insulin knockout mouse. Biochem Biophys Res
Commun 2005; 334: 979–986.
53 Price SA, Onusom L, Purves-Tyson TD, Fernyhough P, Tomlinson DR. Acti-
vation of JNK in sensory neurons protects against sensory neuron cell
death in diabetes and on exposure to glucose ⁄ oxidative stress in vitro.
Ann NY Acad Sci 2003; 1010: 95–99.
54 Zhou Q, Lam PY, Han D, Cadenas E. c-Jun N-terminal kinase regulates
mitochondrial bioenergetics by modulating pyruvate dehydrogenase
activity in primary cortical neurons. J Neurochem 2008; 104: 325–335.
55 Cao G, Clark RS, Pei W, Tin W, Zhang F, Sun FY, Grahm SH, Chen J.
Translocation of apoptosis-inducing factor in vulnerable neurons after
transient cerebral ishcemia and neuronal cultures after oxygen-glucose
deprivation. J Cereb Blood Flow Metab 2003; 23: 1137–1150.
56 Santiago AR, Cristovao AJ, Santos PF, Carvalho CM, Ambrosio AF. High
glucose induces caspase-independent cell death in retinal neural cells.
Neurobiol Dis 2007; 25: 464–472.
57 Cao G, Xing J, Xiao X, Liou AKF, Gao Y, Yin X-M, Clark RSB, Graham SH,
Chen J. Critical role of calpain I in mitochondrial release of apoptosis-
inducing factor in ischemic neuronal injury. J Neurosci 2007; 27: 9278–
9293.
58 Kauppinen TM. Multiple roles for poly(ADP-ribose)polymerase-1 in neu-
rological disease. Neurochem Int 2007; 50: 954–958.
59 Alano CC, Ying W, Swanson RA. Poly(ADP-ribose) polymerase-1-mediated
cell death in astrocytes requires NAD+ depletion and mitochondrial per-
meability transition. J Biol Chem 2004; 279: 18895–18902.
1360 C. Garcıa-Caceres et al.
ª 2008 The Authors. Journal Compilation ª 2008 Blackwell Publishing Ltd, Journal of Neuroendocrinology, 20, 1348–1360
Top Related
Copyright © 2022 FDOKUMEN

