







Bahasa
Halaman
Hukum


ATOMIC-MIGRATION-CONTROLLED PROCESSES IN INTERMETALLICS
Rafał Kozubski1, a, Andrzej Biborski1, Mirosław Kozłowski1, Christine Goyhenex2, Veronique Pierron-Bohnes2,b, Mebarek Alouani2,
Marcus Rennhofer3 and Savko Malinov4 1Interdisciplinary Centre for Materials Modelling, M. Smoluchowski Institute of Physics,
Jagellonian University, Reymonta 4, 30-059 Krakow, Poland 2IPCMS, 23, rue du Loess, BP 43, F-67034 Strasbourg, France
3Faculty of Physics, University Vienna, Strudlhofg. 4, A-1090 Vienna, Austria,
4School of Mechanical and Aerospace Engineering, Ashby Building, Queen's University Belfast,
Stranmillis Road, Belfast BT9 5AH, UK
Keywords: intermetallics, superstructure stability, free surface, vacancy thermodynamics, triple defects, ordering kinetics.
Abstract. Chemical ordering kinetics in L10- and B2-ordered AB binary intermetallics was
simulated by means of Monte Carlo (MC) technique implemented with vacancy mechanism of
atomic migration. While vacancy concentration is usually much lower than the antisite defect
concentration in L10-ordered systems, triple defects are generated in particular B2–ordered systems.
The latter definitely affects the chemical ordering process and requires that full thermal vacancy
thermodynamics is involved in B2-ordering simulations. The study on L10-ordered binaries was
dedicated to FePt thin layers considered as a material for ultra-high-density magnetic storage media.
Metastability of the L10 c-variant with monoatomic planes parallel to the layer surface and off-plane
easy magnetization was revealed. Thermal vacancy formation in B2-ordered binaries was modelled
by implementing a mean-field Hamiltonian with a specific formalism of phase equilibria in a lattice-
gas composed of atoms and vacancies. It was demonstrated that for particular pair-interaction
energetics, equilibrium concentrations of vacancies and antisites result mutually proportional in
well-defined temperature ranges. The MC simulations of B2-ordering kinetics involved the
modelled equilibrium vacancy concentration and reproduced the experimentally observed low rate
of the process.
Introduction
Intermetallic compounds are the basis for many promising engineering and functional materials
showing high strength at high temperatures or favourable magnetic properties resulting from the
chemical (atomic) long-range order (LRO) observed in a wide temperature range (see e.g. [1,2] and
numerous references therein). Intelligent design of the related advanced technologies requires basic
investigations focused especially on the mechanism and kinetics of the superstructure formation in
those systems. Physically, the atomic ordering is a specific structural transformation consisting of
generation/elimination of antisite defects and thus, in the case of intermetallics, involving atomic
jumps to the nearest-neighbouring (nn) vacancies. Statistics of these jumps, as well as the
concentration of mediating vacancies depend on the system geometry including the type of
superstructure and the density of other structural defects (e.g. free surfaces limiting the sample).
This short paper focuses on the Monte Carlo (MC) simulation modelling of two experimental
results obtained for FePd, FePt and NiAl representing the L10 and B2 superstructures, respectively:
(i) L10 c-variant metastability and its spontaneous re-orientation in FePd and FePt layers [3,4]; (ii)
unexpectedly sluggish LRO kinetics in B2-ordered NiAl showing a very high vacancy
concentration [5].
Defect and Diffusion Forum Vol. 277 (2008) pp 113-118online at http://www.scientific.net© (2008) Trans Tech Publications, SwitzerlandOnline available since 2008/Apr/07
All rights reserved. No part of contents of this paper may be reproduced or transmitted in any form or by any means without the written permission of thepublisher: Trans Tech Publications Ltd, Switzerland, www.ttp.net. (ID: 213.134.175.55-11/04/08,15:41:49)

Atomic ordering processes in L10 AB layered binaries (FePt)
Monte Carlo Simulations The principles of the applied method have been described in details in
previous papers [6,7]. Standard Glauber dynamics was implemented with vacancy mechanism of
atomic jumps in the simulated c-variant L10 ordered1 thin-layer AB supercell (Fig.1) built of 64000
unit cells and containing a single vacancy. Thin layering was simulated by a removal of periodic
boundary conditions in [001] crystallographic direction – i.e. by limiting the simulated sample with
two parallel (001) free surfaces [7]. The AB intermetallic was modelled by means of an Ising
Hamiltonian with nearest-neighbour (nn) and next-nearest neighbour (nnn) pair interaction energies
evaluated for FePt on the basis of ab-initio calculations combined with a specific Cluster Expansion
(CE) procedure [8]. Interactions with and between vacancies were assumed equal to zero. Simulated
were so-called “order-order” processes: LRO relaxations following sudden temperature changes
from Ti to Tf; both temperatures being lower than the “order-disorder” transition point TC [6].
Results. Free surfaces definitely affected the simulated “order-order” processes in L10 AB binaries.
While single-time-scale homogeneous creation/elimination of antisites was observed in bulk2,
spontaneous (001)→(100) and (001)→(010) re-orientation of the monoatomic A- and B-planes (i.e.
transformation from the c-variant L10 to a mixture of a- and b-variants) predominated the process in
the simulated layer (Fig.1).
free (001) surfaces monoatomic planes
c-variant L10 a(b)-variant L10
Fig.1. Scheme of the transformation of initially c-variant L10 ordered AB (FePt) layer (a) into a
mixture of a- and b-variant L10 domains.
It was observed that the a-and b-variant L10 domains nucleated heterogeneously and selectively
on the Fe surface and then grew inwards the material as discontinuous precipitates [7]. The
phenomenon, recently observed experimentally in FePd and FePt thin layers [3,4], is of great
importance for the magnetic data storage technology as only the c- variant of L10 superstructure
yields off-plane easy magnetisation of the FePt and FePd layers making them candidates for
application.
The phenomenon was quantitatively elucidated within the nn pair-interaction energy
approximation:
• Ideal complete transformation of a homogeneous c-variant L10 ordered AB layer into a
homogeneous a(b)-variant L10 ordered layer (Fig.1) brings about energy gain ∆E per unit cell
proportional to the system ordering energy:
( ) 022
<−−⋅=∆BBAAABVVV
nE (1)
where Vik denotes the nn pair-interaction energies between “i” and “k” atoms and n is the layer
thickness measured in the number of unit-cells.
1 i.e. with (001)-oriented monoatomic A- and B-planes
2 Previously reported weak contribution of a parallel fast relaxation process has been definitely identified as an artefact
of the simulation procedure.
114 Diffusion and Diffusional Phase Transformations in Alloys

The above result means metastability of the c-variant L10 superstructure in any layered AB
intermetallic.
• In reality, the total energy balance of the re-orientation process is composed of the above gain
and the positive L10-variant antiphase boundary energies. The c-variant – a(b)-variant ones
disappear once the re-orientation is completed – i.e. the reaction front percolates the layer. For
example, the configuration energy change ∆E per unit cell related to the transformation shown
in Fig. 2 and calculated within the nn pair interaction energy approximation results as:
( )BBAAVV
nE −⋅=∆
2 (2)
where A-atoms initially build the free surface, at which the process nucleates.
(a) (b)
Fig. 2. Scheme of an initial stage of L10 c-variant → a(b)-variant transformation in a FePt
intermetallic layer.
Eq.2 means that the re-orientation process will nucleate preferentially on the A- monoatomic
surface provided VAA < VBB. As in the applied model VFeFe < VPtPt the result explains the
experimental and simulation observations.
Atomic ordering in B2 AB triple defect binaries
Extensive studies of the “order-order” kinetics in L12 Ni3Al [9] and B2 NiAl [5] revealed much
slower rate of the process in NiAl despite vacancy concentration being by 107 times higher in the
latter system [10]. The proposed explanation of the effect addressed triple defects [11] generated in
NiAl (Fig.3) and capturing vacancies whose concentration may be twice as high as the antisite
concentration [5].
Fig.3. Scheme of triple defect formation: ○ A-atoms, ● B-atoms, vacancies
Atomistic simulation of LRO kinetics in triple defect systems must, therefore, account for high
vacancy concentration, which is LRO degree dependent (i.e. temperature dependent).
Model of thermal vacancies in B2 AB binaries. The applied model was based on the concept of
phase equilibria in a bcc lattice gas composed of A- and B-atoms and vacancies [12-14]. The gas
was modelled within the Bragg-Williams (BW) approximation with diverse sets of nn pair
triple defect
Defect and Diffusion Forum Vol. 277 115

interaction energies (including non-zero atom-vacancy interactions) yielding B2 atomic long-range
ordering within a wide temperature range. Effectively, the following nn pair-interaction energy
parameters were used:
AVBB
VV
BVAV
BBAAas
BBAAAB
VV
V
VV
VVE
VVVW
,
,0
,0
,2
=
>−=
−=
−−=
(3)
Analysed was the BW free energy of the gas as a function of vacancy concentration CV at a fixed
ratio of atomic concentrations CA/CB (the concentrations calculated with respect to the number of
lattice sites in the gas). Except for B2 ordering the lattice gas decomposed into vacancy-poor and
vacancy-rich phases with temperature-dependent vacancy concentrations. The concentration of the
vacancy-poor phase was interpreted as the equilibrium vacancy concentration CV in the system.
Consequently, the BW model calculations resulted in temperature dependences of atomic and
vacancy concentrations on particular sublattices of the B2 superstructure of the lattice gas showing
the correlation between the degree of LRO (i.e. antisite concentration) and vacancy concentration, as
well as vacancy redistribution over sublattices. The calculations were performed both for
stoichiometric (CA/CB = 1) and non-stoichiometric binaries. It should be stressed that no a priori
assumptions of e.g. low concentration of particular defects was made (in contrary to many related
publications – see e.g. [15,16]).
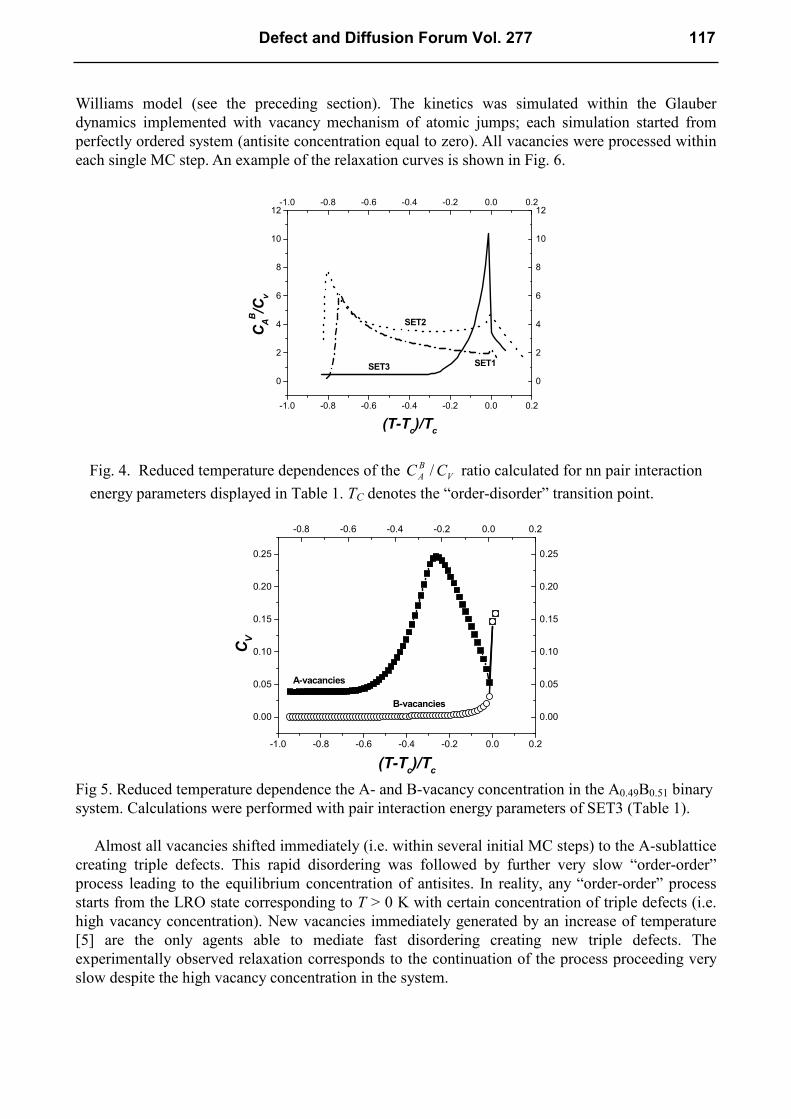
The most interesting result from these calculations was the temperature dependence of a ratio of
A-antisite and vacancy concentrations ( )TCCV
B
A/ calculated for diverse values of nn pair-
interaction energy parameters (Eq. 3). The curves shown in Fig. 4 correspond to three pair-
interaction energy sets given in Table 1.
W [eV] asE [eV]
BBV [eV]
AVV [eV]
SET1 - 0.08 - 0.03 - 0.05 0.0
SET2 - 0.08 - 0.07 - 0.05 0.0
SET3 - 0.08 - 0.07 - 0.05 0.051
Table 1. Values of nn pair-interaction energy parameters used in the calculation of the curves
shown in Figs. 4 and 5.
The plateau in the ( )TCCV
B
A/ curves calculated with SET2 and SET3 parameters indicate the
proportionality between the A-antisite and vacancy concentrations within finite temperature ranges.
In the case of the SET3 curve corresponding to the highest preference for A-vacancy and A-antisite
formation (with respect to B-vacancies and B-antisites) the plateau level equals exactly 0.5, which
corresponds to the ideal case of the generation of triple defects. The plateau, though not at the level
of 0.5, appears, however, also in the case of lower preference for A-vacancy formation (SET2).
The analogous calculations performed for non-stoichiometric binary systems with SET3
parameters resulted in ( )TCCV
B
A/ plateau only in the case of B-rich binaries (CA/CB < 1). The effect
was accompanied by a non-zero A-vacancy concentration remaining at low temperatures, which was
interpreted as the presence of structural vacancies (Fig. 5).
MC simulation of “order-order” kinetics. A B2 ordered AB supercell with periodic boundary
conditions was built of 64000 unit cells. Before an “order-order” kinetics was simulated at
temperature T NV(T) vacancies were introduced by randomly removing the same number of atoms.
NV equaled the equilibrium number of vacancies at temperature T following from the Bragg-
116 Diffusion and Diffusional Phase Transformations in Alloys
Williams model (see the preceding section). The kinetics was simulated within the Glauber
dynamics implemented with vacancy mechanism of atomic jumps; each simulation started from
perfectly ordered system (antisite concentration equal to zero). All vacancies were processed within
each single MC step. An example of the relaxation curves is shown in Fig. 6.
-1.0 -0.8 -0.6 -0.4 -0.2 0.0 0.2
0
2
4
6
8
10
12
SET3
SET2
SET1
CA
B/C
v
(T-Tc)/T
c
0
2
4
6
8
10
12-1.0 -0.8 -0.6 -0.4 -0.2 0.0 0.2
Fig. 4. Reduced temperature dependences of the V
B
ACC / ratio calculated for nn pair interaction
energy parameters displayed in Table 1. TC denotes the “order-disorder” transition point.
-1.0 -0.8 -0.6 -0.4 -0.2 0.0 0.2
0.00
0.05
0.10
0.15
0.20
0.25
B-vacancies
A-vacancies
CV
(T-Tc)/T
c
-0.8 -0.6 -0.4 -0.2 0.0 0.2
0.00
0.05
0.10
0.15
0.20
0.25
Fig 5. Reduced temperature dependence the A- and B-vacancy concentration in the A0.49B0.51 binary
system. Calculations were performed with pair interaction energy parameters of SET3 (Table 1).
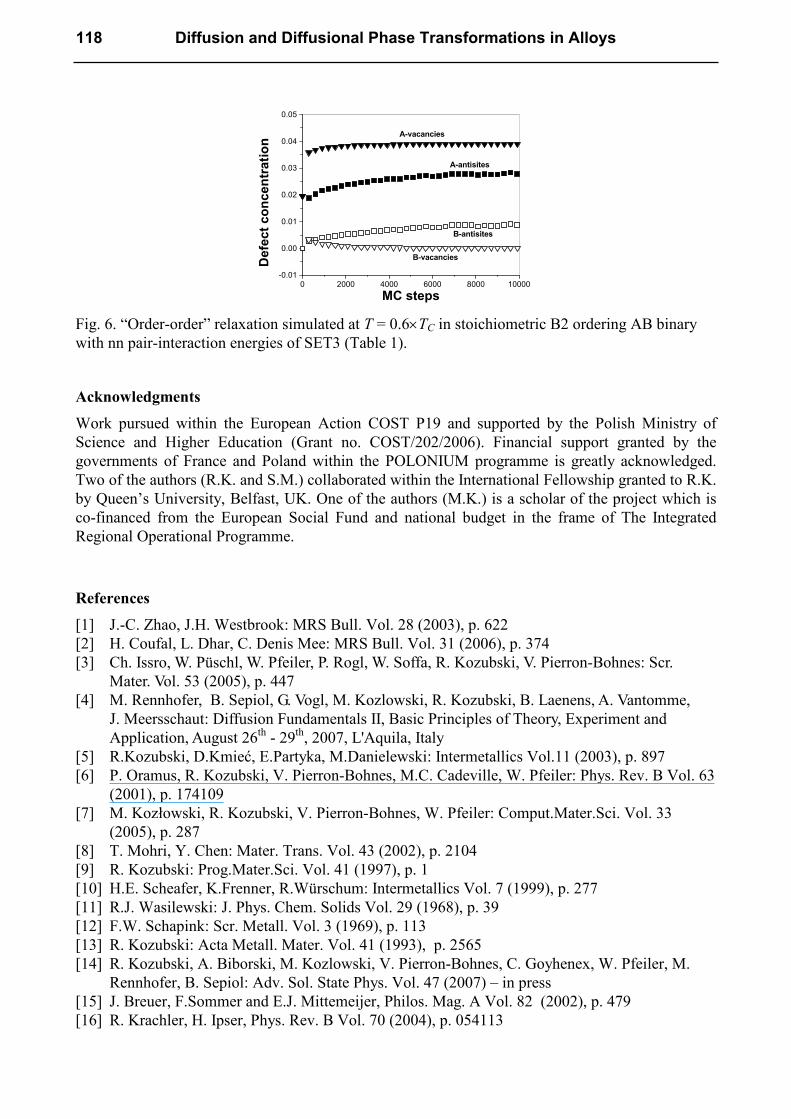
Almost all vacancies shifted immediately (i.e. within several initial MC steps) to the A-sublattice
creating triple defects. This rapid disordering was followed by further very slow “order-order”
process leading to the equilibrium concentration of antisites. In reality, any “order-order” process
starts from the LRO state corresponding to T > 0 K with certain concentration of triple defects (i.e.
high vacancy concentration). New vacancies immediately generated by an increase of temperature
[5] are the only agents able to mediate fast disordering creating new triple defects. The
experimentally observed relaxation corresponds to the continuation of the process proceeding very
slow despite the high vacancy concentration in the system.
Defect and Diffusion Forum Vol. 277 117
0 2000 4000 6000 8000 10000-0.01
0.00
0.01
0.02
0.03
0.04
0.05
B-vacancies
B-antisites
A-antisites
A-vacancies
Defect concentration
MC steps
Fig. 6. “Order-order” relaxation simulated at T = 0.6×TC in stoichiometric B2 ordering AB binary
with nn pair-interaction energies of SET3 (Table 1).
Acknowledgments
Work pursued within the European Action COST P19 and supported by the Polish Ministry of
Science and Higher Education (Grant no. COST/202/2006). Financial support granted by the
governments of France and Poland within the POLONIUM programme is greatly acknowledged.
Two of the authors (R.K. and S.M.) collaborated within the International Fellowship granted to R.K.
by Queen’s University, Belfast, UK. One of the authors (M.K.) is a scholar of the project which is
co-financed from the European Social Fund and national budget in the frame of The Integrated
Regional Operational Programme.
References
[1] J.-C. Zhao, J.H. Westbrook: MRS Bull. Vol. 28 (2003), p. 622
[2] H. Coufal, L. Dhar, C. Denis Mee: MRS Bull. Vol. 31 (2006), p. 374
[3] Ch. Issro, W. Püschl, W. Pfeiler, P. Rogl, W. Soffa, R. Kozubski, V. Pierron-Bohnes: Scr.
Mater. Vol. 53 (2005), p. 447
[4] M. Rennhofer, B. Sepiol, G. Vogl, M. Kozlowski, R. Kozubski, B. Laenens, A. Vantomme,
J. Meersschaut: Diffusion Fundamentals II, Basic Principles of Theory, Experiment and
Application, August 26th - 29
th, 2007, L'Aquila, Italy
[5] R.Kozubski, D.Kmieć, E.Partyka, M.Danielewski: Intermetallics Vol.11 (2003), p. 897
[6] P. Oramus, R. Kozubski, V. Pierron-Bohnes, M.C. Cadeville, W. Pfeiler: Phys. Rev. B Vol. 63
(2001), p. 174109
[7] M. Kozłowski, R. Kozubski, V. Pierron-Bohnes, W. Pfeiler: Comput.Mater.Sci. Vol. 33
(2005), p. 287
[8] T. Mohri, Y. Chen: Mater. Trans. Vol. 43 (2002), p. 2104
[9] R. Kozubski: Prog.Mater.Sci. Vol. 41 (1997), p. 1
[10] H.E. Scheafer, K.Frenner, R.Würschum: Intermetallics Vol. 7 (1999), p. 277
[11] R.J. Wasilewski: J. Phys. Chem. Solids Vol. 29 (1968), p. 39
[12] F.W. Schapink: Scr. Metall. Vol. 3 (1969), p. 113
[13] R. Kozubski: Acta Metall. Mater. Vol. 41 (1993), p. 2565
[14] R. Kozubski, A. Biborski, M. Kozlowski, V. Pierron-Bohnes, C. Goyhenex, W. Pfeiler, M.
Rennhofer, B. Sepiol: Adv. Sol. State Phys. Vol. 47 (2007) – in press
[15] J. Breuer, F.Sommer and E.J. Mittemeijer, Philos. Mag. A Vol. 82 (2002), p. 479
[16] R. Krachler, H. Ipser, Phys. Rev. B Vol. 70 (2004), p. 054113
118 Diffusion and Diffusional Phase Transformations in Alloys
Top Related
Copyright © 2022 FDOKUMEN

