
Vitamin E blocks early events induced by 1-methyl-4-phenylpyridinium (MPP+) in cerebellar granule...
11
Vitamin E blocks early events induced by 1-methyl-4-phenylpyridinium (MPP + ) in cerebellar granule cells Rosa A. Gonza ´lez-Polo,* Germa ´n Soler,* Alberto Alvarez, Isabel Fabregatà and Jose ´ M. Fuentes§ *Departamento de Bioquı ´mica y Biologı ´a Molecular y Gene ´tica, Facultad de Veterinaria, Universidad de Extremadura, Ca ´ceres, Spain Centro de Citometrı ´a de Flujo y Microscopı ´a Confocal. Universidad Complutense de Madrid, Madrid, Spain àDepartamento de Bioquı ´mica y Biologı ´a Molecular, Facultad de Farmacia (Centro Mixto CSIC/UCM), Universidad Complutense de Madrid, Madrid, Spain §Departamento de Bioquı ´mica y Biologı ´a Molecular y Gene ´tica, EU Enfermerı ´a y TO, Universidad de Extremadura, Ca ´ceres, Spain Abstract Exposure of cerebellar granule cells (CGCs) to 1-methyl-4- phenylpyridinium (MPP + ) results in apoptotic cell death, which is markedly attenuated by co-treatment of CGCs with the radical scavenger vitamin E. Analysis of free radical produc- tion and mitochondrial transmembrane potential (DY m ), using specific fluorescent probes, showed that MPP + mediates early radical oxygen species (ROS) production without a loss of DY m . Exposure to MPP + also produces an early increase in Bad dephosphorylation and translocation of Bax to the mito- chondria. These events are accompanied by cytochrome c release from mitochondria to cytosol, which is followed by caspase 3 activation. Exposure of the neurons to vitamin E maintains Bad phosphorylation and attenuates Bax translo- cation, inhibiting cytochrome c release and caspase activa- tion. MPP + -mediated cytochrome c release is also prevented by allopurinol, suggesting the participation of xanthine oxidase in the process. Our results indicate that free radicals play an active role in the MPP + -induced early events that culminate with cell death. Keywords: apoptosis, cerebellar granule cells, cytochrome c, 1-methyl-4-phenylpyridinium (MPP + ), vitamin E. J. Neurochem. (2003) 84, 305–315. 1-Methyl-4-phenylpyridinium (MPP + ) is a well-known neu- rotoxin (Langston et al. 1983; Blum et al. 1993), which causes apoptotic cell death at concentrations ranging from 40 to 300 lM (Du et al. 1997). MPP + incorporation into the cells is produced essentially through the dopamine (Chiba et al. 1985; Gonza ´lez-Polo et al. 2001) or the cation amino acid (Gonza ´lez-Polo et al. 2001) transporters. The principal target of MPP + is the mitochondria, where it inhibits Complex I in the mitochondrial respiratory chain, with the consequent cessation of oxidative phosphorylation (Tipton and Singer 1993). Several studies have shown the involvement of radical oxygen species (ROS) in MPP + -induced neurotoxicity (Castagnoli et al. 1985; Rossetti et al. 1988; Kitamura et al. 1998). The importance of MPP + -mediated ROS production is demonstrated in vivo by the fact that transgenic mice that overexpress copper/zinc superoxide dismutase are signifi- cantly more resistant to MPP + -induced toxicity than wild- type mice (Przedborski et al. 1992). In vitro MPP + -induced cell death is abolished by co-treatment with radical scav- engers, such as ascorbic acid (Akaneya et al. 1995), a-tocopherol (Odunze et al. 1990) and others (Akaneya et al. 1995) in several cell types. Free radical production in MPP + -exposure has been classically associated with inhibi- tion of electron transport in mitochondria (Nicklas et al. 1985; Mizuno et al. 1989). However, in other models of programmed cell death cytosolic free radicals have been shown to play active roles in the generation of apoptosis, NADPH-oxidase (Tammariello et al. 2000) or xanthine oxidase (Atlante et al. 2001) being involved in its Received June 4, 2002; revised manuscript received October 2, 2002; accepted October 8, 2002. Address correspondence and reprint requests to Jose ´ M. Fuentes, Departamento de Bioquı ´mica y Biologı ´a Molecular, EU Enfermerı ´a y TO Universidad de Extremadura, Avenida de la Universidad s/n 10071, Ca ´ceres, Spain. E-mail: [email protected] Abbreviations used: CGC, cerebellar granule cells; CMXRos, chlo- romethyl-X-rosamine; DFCH, 2¢-7¢-dichlorodihydrofluorescein; DMEM, Dulbecco’s modified Eagle’s medium; HE, hydroethidine; MCB, mon- ochlorobimane; MPP + , 1-methyl-4-phenylpyridinium; MTT, 3-(4,5- dimehylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; PBS, phosphate- buffered saline; PI, propidium iodide; PVDF, polyvinylidene difluoride; ROS, radical oxygen species; SDS, sodium dodecyl sulphate; DY m : mitochondrial transmembrane potential. Journal of Neurochemistry , 2003, 84, 305–315 Ó 2003 International Society for Neurochemistry, Journal of Neurochemistry , 84, 305–315 305
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Vitamin E blocks early events induced by 1-methyl-4-phenylpyridinium (MPP+) in cerebellar granule...

Vitamin E blocks early events induced by
1-methyl-4-phenylpyridinium (MPP+) in cerebellar granule cells
Rosa A. Gonzalez-Polo,* German Soler,* Alberto Alvarez,� Isabel Fabregat� and Jose M. Fuentes§
*Departamento de Bioquımica y Biologıa Molecular y Genetica, Facultad de Veterinaria, Universidad de Extremadura, Caceres, Spain
�Centro de Citometrıa de Flujo y Microscopıa Confocal. Universidad Complutense de Madrid, Madrid, Spain
�Departamento de Bioquımica y Biologıa Molecular, Facultad de Farmacia (Centro Mixto CSIC/UCM), Universidad Complutense de
Madrid, Madrid, Spain
§Departamento de Bioquımica y Biologıa Molecular y Genetica, EU Enfermerıa y TO, Universidad de Extremadura, Caceres, Spain
Abstract
Exposure of cerebellar granule cells (CGCs) to 1-methyl-4-
phenylpyridinium (MPP+) results in apoptotic cell death, which
is markedly attenuated by co-treatment of CGCs with the
radical scavenger vitamin E. Analysis of free radical produc-
tion and mitochondrial transmembrane potential (DYm), using
specific fluorescent probes, showed that MPP+ mediates early
radical oxygen species (ROS) production without a loss of
DYm. Exposure to MPP+ also produces an early increase in
Bad dephosphorylation and translocation of Bax to the mito-
chondria. These events are accompanied by cytochrome c
release from mitochondria to cytosol, which is followed by
caspase 3 activation. Exposure of the neurons to vitamin E
maintains Bad phosphorylation and attenuates Bax translo-
cation, inhibiting cytochrome c release and caspase activa-
tion. MPP+-mediated cytochrome c release is also prevented
by allopurinol, suggesting the participation of xanthine oxidase
in the process. Our results indicate that free radicals play an
active role in the MPP+-induced early events that culminate
with cell death.
Keywords: apoptosis, cerebellar granule cells, cytochrome c,
1-methyl-4-phenylpyridinium (MPP+), vitamin E.
J. Neurochem. (2003) 84, 305–315.
1-Methyl-4-phenylpyridinium (MPP+) is a well-known neu-
rotoxin (Langston et al. 1983;Blum et al. 1993),which causes
apoptotic cell death at concentrations ranging from 40 to
300 lM (Du et al. 1997). MPP+ incorporation into the cells isproduced essentially through the dopamine (Chiba et al. 1985;
Gonzalez-Polo et al. 2001) or the cation amino acid
(Gonzalez-Polo et al. 2001) transporters. The principal target
ofMPP+ is themitochondria, where it inhibits Complex I in the
mitochondrial respiratory chain, with the consequent cessation
of oxidative phosphorylation (Tipton and Singer 1993).
Several studies have shown the involvement of radical
oxygen species (ROS) in MPP+-induced neurotoxicity
(Castagnoli et al. 1985; Rossetti et al. 1988; Kitamura et al.
1998). The importance of MPP+-mediated ROS production is
demonstrated in vivo by the fact that transgenic mice that
overexpress copper/zinc superoxide dismutase are signifi-
cantly more resistant to MPP+-induced toxicity than wild-
type mice (Przedborski et al. 1992). In vitro MPP+-induced
cell death is abolished by co-treatment with radical scav-
engers, such as ascorbic acid (Akaneya et al. 1995),
a-tocopherol (Odunze et al. 1990) and others (Akaneya
et al. 1995) in several cell types. Free radical production in
MPP+-exposure has been classically associated with inhibi-
tion of electron transport in mitochondria (Nicklas et al.
1985; Mizuno et al. 1989). However, in other models of
programmed cell death cytosolic free radicals have been
shown to play active roles in the generation of apoptosis,
NADPH-oxidase (Tammariello et al. 2000) or xanthine
oxidase (Atlante et al. 2001) being involved in its
Received June 4, 2002; revised manuscript received October 2, 2002;
accepted October 8, 2002.
Address correspondence and reprint requests to Jose M. Fuentes,
Departamento de Bioquımica y Biologıa Molecular, EU Enfermerıa y
TO Universidad de Extremadura, Avenida de la Universidad s/n 10071,
Caceres, Spain. E-mail: [email protected]
Abbreviations used: CGC, cerebellar granule cells; CMXRos, chlo-
romethyl-X-rosamine; DFCH, 2¢-7¢-dichlorodihydrofluorescein; DMEM,Dulbecco’s modified Eagle’s medium; HE, hydroethidine; MCB, mon-
ochlorobimane; MPP+, 1-methyl-4-phenylpyridinium; MTT, 3-(4,5-
dimehylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; PBS, phosphate-
buffered saline; PI, propidium iodide; PVDF, polyvinylidene difluoride;
ROS, radical oxygen species; SDS, sodium dodecyl sulphate; DYm:mitochondrial transmembrane potential.
Journal of Neurochemistry, 2003, 84, 305–315
� 2003 International Society for Neurochemistry, Journal of Neurochemistry, 84, 305–315 305

production. Interestingly, MPP+ has been described as a
substrate for xanthine oxidase and it may lead to ROS
formation (Klaidman et al. 1993). In any case, free radical
production has normally been measured over a relatively
long period (about 1 h after beginning MPP+ or other toxic
treatment). Recently, a new and exciting line of evidence has
emerged reporting, in glutamate-exposed and potassium-
deprived cerebellar granule cells (CGCs), early (before
30 min apoptotic injury) free radicals production, being
these free radicals responsible for cytochrome c release
(Atlante et al. 2000; Valencia and Moran 2001), as a cell
defense system against oxidative stress (Atlante et al. 1999,
2000). However, taking into account that cytochrome c
release has been related to apoptosis in several apoptotic
injuries, including MPP+-treatment (Du et al. 1997; Dodel
et al. 1998; Leist et al. 1998), this release could also activate
the apoptotic machinery, such as caspase activation, DNA
fragmentation and cell death.
The release of cytochrome c is associated with the
translocation to the mitochondria of certain bcl-2-family
proteins, such as Bax or Bad (Desagher and Martinou 2000).
Bax may play a central role in mediating mitochondria-
dependent apoptosis in neurons (Putcha et al. 1999).
Following a death signal, Bax can be translocated from
cytosol to mitochondria, which is rapidly followed by
cytochrome c release. The Bax effect can be prevented by
the presence of Bcl-2 or Bcl-xL proteins in the mitochondria
(Gross et al. 1999). Bad is another member of the pro-
apoptotic Bcl-2 proteins, which is dephosphorylated during
the apoptosis process. Phosphorylated Bad is normally
sequestered in the cytosol through its binding to 14-3-3
protein. Dephosphorylation of Bad by phosphatases of the
PP1 or PP2 families promotes its translocation to mitochon-
dria, where it binds to Bcl-xL, inhibiting its death-repressor
activity (Zha et al. 1996; Desagher and Martinou 2000).
In this paper we describe how vitamin E, a biological anti-
oxidant that is involved in neuroprotection from several
insults, blocks all the early events observed in CGCs after
MPP+-exposure. We propose for the first time that free
radicals, produced through the xanthine oxidase system, play
an active role in the MPP+-induced early events that
culminate with cell death.
Materials and methods
Materials
MPP+ was obtained from Research Biochemicals Inc. (Natick, MA,
USA). Cytosine arabinoside, Dulbecco’s modified Eagle’s medium
(DMEM), fetal calf serum and all the medium and supplements,
3-(4,5-dimehylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT),
poly D-lysine, a-tocopherol (vitamin E), xanthine and xanthine
oxidase, allopurinol and protease inhibitors were obtained from
Sigma (St Louis, MO, USA). Okadaic acid was supplied by Tocris
(London, UK). The fluorescent probes hydroethidine (HE), 2¢-7¢-dichlorodihydrofluorescein diacetate (DFCH-DA) and chloromethyl-
X-rosamine (CMXRos) were from Molecular Probes (Eugene, OR,
USA). Monochlorobimane was supplied by Calbiochem (San
Diego, CA, USA) and caspase 3 substrate (Ac-DEVD-AMC) was
obtained from Pharmingen (San Diego, CA, USA). Anti-Bax
(SC7480) and anti-cytochrome c (SC 7119), anti-actin (SC7210)
and secondary mouse (SC 2005) antibodies were all from Santa
Cruz Biotechnologies (Santa Cruz, CA, USA). Anti-Ser136-phos-
pho-Bad, anti-Ser473-phospho-Akt and cleaved caspase 3 antibod-
ies were obtained from Cell Signalling Technology (Beverly, MA,
USA). The rabbit secondary antibody and electrophoresis reagents
were from Bio-Rad Laboratories (Hercules, CA, USA). ECL Plus
and all the western blot reagents were purchased from Amersham
Biosciences (Buckinghamshire, UK). Other chemicals were of
analytical grade.
Cerebellar granule cell culture
Primary cultures of CGCs were obtained from 7- to 8-day-old
Wistar rats of either sex as described before (Gonzalez-Polo et al.
2001). The animals were housed in a temperature-controlled room
maintained at 12 h light/dark cycles. The standard laboratory animal
food and tap water were available ad libitum for the mothers. The
experimental protocols of this study were approved by the Research
Committee of the University of Extremadura (in accordance with the
National Institutes of Health guidelines) and were designed to
minimize the pain or discomfort of the animals. Briefly, cerebella
dissected free of meninges were chopped into small pieces and
digested with trypsin (2.5 mg/mL, 10 min at 37�C) in a Krebs–Ringer buffer solution, pH 7.4, containing bovine serum albumin
(3 mg/mL). After addition of soybean trypsin inhibitor (0.5 mg/mL)
and DNAse (0.1 mg/mL), the tissue was disrupted by 10 passages
through a fine tip plastic transfer pipette. The resulting cell
suspension was filtered through a 100-lm nylon cloth, centrifuged
and resuspended in DMEM supplemented with 10% fetal calf
serum, 25 mM KCl, 2 mM glutamine, penicillin (50 units/mL) and
streptomycin (50 lg/mL). Then cells were seeded at a density of5 · 105 cells/mL in poly L-lysine-pretreated 24-well or six-well
plates in a humidified atmosphere of 5% CO2 at 37�C. Cytosinearabinoside (10 lM) was added 24 h after plating to arrest thegrowth of non-neuronal cells, mostly astrocytes and microglia.
All the experiments were carried out at 7 days in culture in a
DMEM without fetal calf serum supplemented with 25 mM KCl,
2 mM glutamine, 50 U/mL penicillin, 50 lg/mL streptomycin,
0.1 mg/mL sodium pyruvate, 20 nM progesterone and 5 lg/mLinsulin.
Drug treatment protocol
Cells were exposed to MPP+ (50 lM) and cell viability was
measured 24 h after treatment using the MTT assay (Mosmann
1983). Absorbance at 500 nm in control cultures was used as 100%
viability, typically 0.25 ± 0.005.
DNA fragmentation
After removal of the culture medium, cells grown in six-well plates
were detached with 0.5 mL of 5 mM Tris, 20 mM EDTA pH 7.4 and
transferred to assay tubes. Then, 25 lL Triton X-100 (10% v/v) was
added, and the cells were incubatedwith gentle shaking for 1 h at 4�C.
306 R. A. Gonzalez-Polo et al.
� 2003 International Society for Neurochemistry, Journal of Neurochemistry, 84, 305–315

Nuclei were removed by centrifugation at 15 000 g for 15 min.
The supernatants containing cytosolic DNA were treated with 0.5%
sodium dodecyl sulphate (SDS) and 0.1 mg/mL proteinase K for 3 h
at room temperature. After two phenol–chloroform extractions, the
water-soluble fractions were incubated for 1 h at 37�C with RNAse(15 lg/mL). After two further extractions with phenol–chloroform,DNA was precipitated by adding 0.1 volumes of 2.5 M sodium
acetate, pH 5.2, and three volumes of ice-cold ethanol. DNA pellets
were washed with 70% ethanol, air-dried, and dissolved in 10 mM
Tris, pH 8.0, containing 1 mM EDTA, prior to electrophoresis in
1.5% agarose gels.
Propidium iodide staining
For confocal microscopy visualization of chromatin condensation
and nuclei degradation propidium iodide (PI) was used. After
incubation in the absence or in the presence of 50 lM MPP+, cellswere washed with phosphate-buffered saline (PBS) and incubated
with 0.005% PI in PBS during 10 min. Fluorescence images were
obtained using a MRC-1024 laser confocal microscopy (Bio-Rad,
Hemel Hempstead, UK).
ROS measurement
The oxidation-sensitive fluorescent probes HE and DFCH-DA were
used to analyse the net intracellular generation of ROS by flow
cytometry (Rothe and Valet 1990). CGCs were incubated in the
presence or in the absence of 50 lM MPP+ at different times, cellswere collected in the presence of cold PBS (without calcium and
magnesium salts) with EDTA 1 mM, pH 7.4. Then cells were
centrifuged and diluted in 20 lM HE or 5 lM DFCH-DA in PBS andincubated for 30 min at 37�C. Cells were run in a FACScan flowcytometer (Becton & Dickinson, San Jose, CA, USA) acquiring
10 000 cells per sample.
Analysis of mitochondrial transmembrane potential
The fluorescent probe CMXRos was used to analyse the mito-
chondrial transmembrane potential (DYm) by flow cytometry. CGCswere incubated in the presence or in the absence of 50 lM MPP+.Cells were collected in the presence of cold PBS (without calcium
and magnesium salts) with EDTA 1 mM, pH 7.4. Then cells were
centrifuged and diluted in 0.1 lM CMXRos in PBS for 30 min at
37�C. Cells were run in a FACScan flow cytometer (BD, San Jose,CA) acquiring 10 000 cells per sample.
GSH determination
The glutathion-dependent fluorescent probe monochlorobimane
(MCB) was used to analyse the net intracellular content of GSH
by flow cytometry (Kamencic et al. 2000). CGCs were incubated in
the presence or in the absence of 50 lM MPP+. Cells were collectedin the presence of cold PBS (without calcium and magnesium salts),
with EDTA 1 mM, pH 7.4. Then cells were centrifuged and diluted
in 1 lM MCB in PBS and incubated for 30 min at 37�C. UV laserfrom a LSR flow cytometer (BD) was used to excite MCB, blue
fluorescence was recovered through a 440/40 BP filter and 10 000
cells were acquired per sample.
Caspase 3 activity
Cells were scraped off in PBS, collected by centrifugation and lysed
at 4�C in 5 mM Tris–HCl, pH 8.0, 20 mM EDTA, 0.5% Triton
X-100. Lysates were clarified by centrifugation at 13 000 g for
10 min. The reaction mixture contained 25 lM cellular lysates,
325 lM assay buffer (20 mM HEPES pH 7.5, 10% glycerol, 2 mM
dithiothreitol), and 20 lM caspase 3 substrate (Ac-DEVD-AMC).
After 2 h incubation in the dark, enzymatic activity was measured in
a Luminescence Spectrophotometer (Perkin Elmer LS-50) (kexci-tation, 380 nm; kemission, 440 nm). Caspase activity was repre-sented as percentage over untreated cells.
Western blot analysis
To detect cytochrome c and Bax proteins in mitochondria, attached
cells were scraped off in a buffer containing 25 mM Tris–HCl
pH 6.8, 250 mM sucrose, 1 mM EDTA, 0.05% digitonine, 1 mM
dithiothreitol, 0.1 mM phenylmethylsulfonyl fluoride, 1 lg/mLleupeptine, 1 lg/mL pepstatin and 1 lg/mL aprotinin according tothe method previously described (Pique et al. 2000). Samples were
centrifuged at 13 000 g for 3 min at 4�C, pellets containing themitochondrial fraction were denatured in a lysis buffer and
processed as described below. For phospho-Bad, phospho-Akt,
cleaved caspase 3 and total Bax proteins in total cell lysates, cells
were washed with cold PBS at 4�C and lysed in a lysis buffer
containing 50 mM Tris–HCl pH 6.8, glycerol 10%, 2% SDS, 10 mM
dithiothreitol and 0.005% blue brominphenol. In both, mitochon-
drial and total lysates proteins (30–50 lg/condition) were resolvedin 12% SDS gel electrophoresis and transferred to polyvinylidene
difluoride (PVDF) membranes according to the conventional
methods partially modified by Fuentes et al. (2000). Briefly,
proteins were transferred (250 mA for 60 min) to PVDF membranes
using a Mini Trans-Blot Cell apparatus (Bio-Rad). The procedure
for immunodetection, including transfer, blocking of the membrane
(30 min at 37�C) with TTBS (10 mM Tris–HCl pH 7.5, 150 mM
NaCl and 0.2% Tween-20) containing 10% non-fat dried milk and
incubation (60 min at room temperature) with the primary antibody
(diluted 1 : 1000 in TTBS + 5% non-fat dried milk). After washing
(twice for 5 min each time with TTBS), membranes were incubated
(60 min at room temperature) with peroxidase-conjugated secon-
dary antibodies (1 : 5000 in TTBS with 5% non-fat dried milk).
After washing (twice for 5 min and once for 10 min), detection of
bound antibodies was visualized by chemiluminiscence using the
ECL-plus reagent.
Other methods
Protein concentration was measured according Bradford (1976) for
mitochondrial lysates and Lowry et al. (1951) for total cell lysates
using bovine serum albumin as standard in both. All data were
representative of at least three independent neuronal preparations
(with comparable results) each one in triplicate. Statistical analysis
were performed using the Student’s t-test (*p < 0.05).
Results
As previously reported (Du et al. 1997; Gonzalez-Polo et al.
2001) CGCs are sensitive to the toxic properties of MPP+.
Treatment with MPP+ caused a time- and concentration-
dependent increase in cell death and DNA fragmentation. In
our work, CGCs treated with 50 lM MPP+ for 24 h exhibitedmarked cell death (50% approximately, Fig. 1a) and DNA
Vitamin E blocks early cytochrome c release 307
� 2003 International Society for Neurochemistry, Journal of Neurochemistry, 84, 305–315
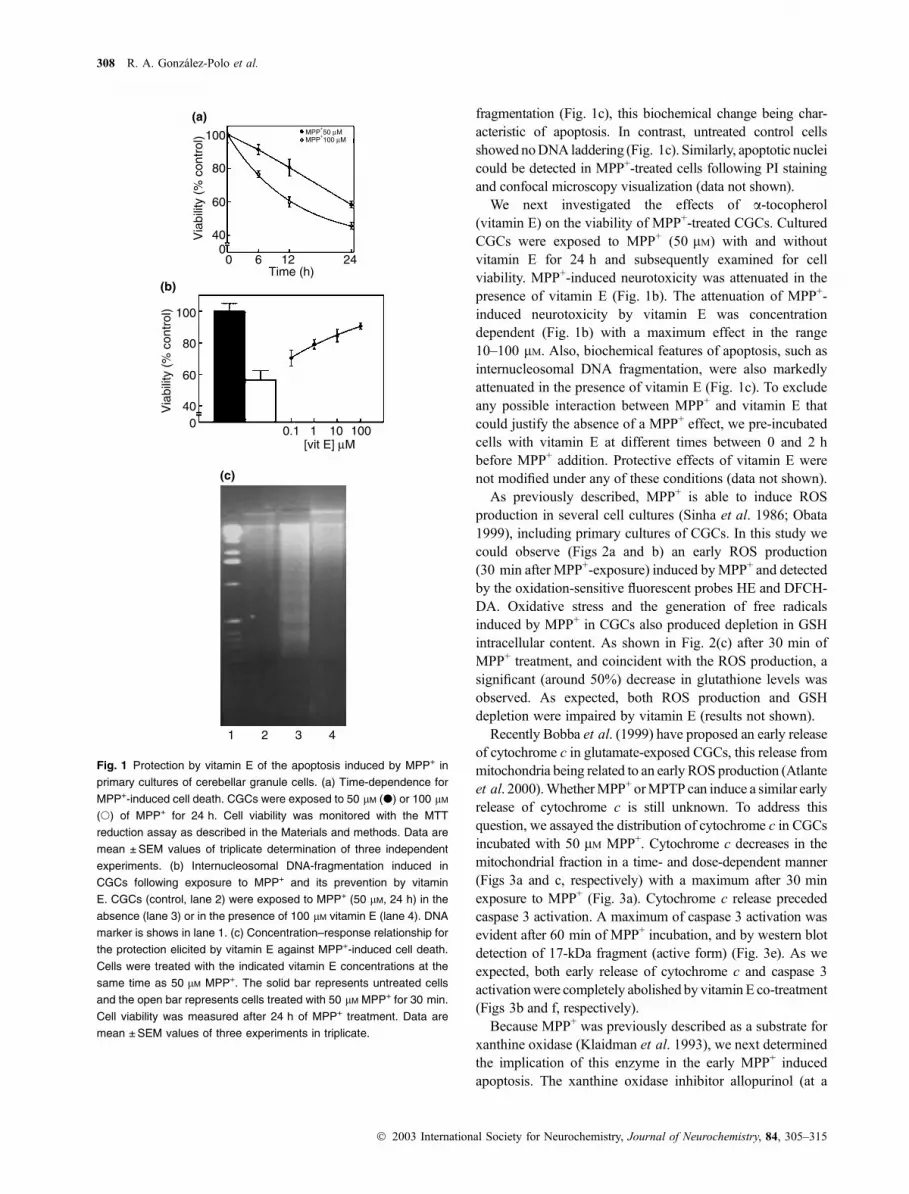
fragmentation (Fig. 1c), this biochemical change being char-
acteristic of apoptosis. In contrast, untreated control cells
showed noDNA laddering (Fig. 1c). Similarly, apoptotic nuclei
could be detected in MPP+-treated cells following PI staining
and confocal microscopy visualization (data not shown).
We next investigated the effects of a-tocopherol(vitamin E) on the viability of MPP+-treated CGCs. Cultured
CGCs were exposed to MPP+ (50 lM) with and withoutvitamin E for 24 h and subsequently examined for cell
viability. MPP+-induced neurotoxicity was attenuated in the
presence of vitamin E (Fig. 1b). The attenuation of MPP+-
induced neurotoxicity by vitamin E was concentration
dependent (Fig. 1b) with a maximum effect in the range
10–100 lM. Also, biochemical features of apoptosis, such asinternucleosomal DNA fragmentation, were also markedly
attenuated in the presence of vitamin E (Fig. 1c). To exclude
any possible interaction between MPP+ and vitamin E that
could justify the absence of a MPP+ effect, we pre-incubated
cells with vitamin E at different times between 0 and 2 h
before MPP+ addition. Protective effects of vitamin E were
not modified under any of these conditions (data not shown).
As previously described, MPP+ is able to induce ROS
production in several cell cultures (Sinha et al. 1986; Obata
1999), including primary cultures of CGCs. In this study we
could observe (Figs 2a and b) an early ROS production
(30 min after MPP+-exposure) induced byMPP+ and detected
by the oxidation-sensitive fluorescent probes HE and DFCH-
DA. Oxidative stress and the generation of free radicals
induced by MPP+ in CGCs also produced depletion in GSH
intracellular content. As shown in Fig. 2(c) after 30 min of
MPP+ treatment, and coincident with the ROS production, a
significant (around 50%) decrease in glutathione levels was
observed. As expected, both ROS production and GSH
depletion were impaired by vitamin E (results not shown).
Recently Bobba et al. (1999) have proposed an early release
of cytochrome c in glutamate-exposed CGCs, this release from
mitochondria being related to an early ROS production (Atlante
et al. 2000).WhetherMPP+ orMPTP can induce a similar early
release of cytochrome c is still unknown. To address this
question, we assayed the distribution of cytochrome c in CGCs
incubated with 50 lM MPP+. Cytochrome c decreases in themitochondrial fraction in a time- and dose-dependent manner
(Figs 3a and c, respectively) with a maximum after 30 min
exposure to MPP+ (Fig. 3a). Cytochrome c release preceded
caspase 3 activation. A maximum of caspase 3 activation was
evident after 60 min of MPP+ incubation, and by western blot
detection of 17-kDa fragment (active form) (Fig. 3e). As we
expected, both early release of cytochrome c and caspase 3
activationwere completely abolished by vitamin E co-treatment
(Figs 3b and f, respectively).
Because MPP+ was previously described as a substrate for
xanthine oxidase (Klaidman et al. 1993), we next determined
the implication of this enzyme in the early MPP+ induced
apoptosis. The xanthine oxidase inhibitor allopurinol (at a
100
80
60
400
100
80
60
400
0.1 1 10 100
0 6 12 24Time (h)
Via
bilit
y (%
con
trol
)
Via
bilit
y (%
con
trol
)
(a)
(b)
1 2 3 4
(c)
MPP+50 µM
MPP+100 µM
[vit E] µM
Fig. 1 Protection by vitamin E of the apoptosis induced by MPP+ in
primary cultures of cerebellar granule cells. (a) Time-dependence for
MPP+-induced cell death. CGCs were exposed to 50 lM (d) or 100 lM
(s) of MPP+ for 24 h. Cell viability was monitored with the MTT
reduction assay as described in the Materials and methods. Data are
mean ± SEM values of triplicate determination of three independent
experiments. (b) Internucleosomal DNA-fragmentation induced in
CGCs following exposure to MPP+ and its prevention by vitamin
E. CGCs (control, lane 2) were exposed to MPP+ (50 lM, 24 h) in the
absence (lane 3) or in the presence of 100 lM vitamin E (lane 4). DNA
marker is shows in lane 1. (c) Concentration–response relationship for
the protection elicited by vitamin E against MPP+-induced cell death.
Cells were treated with the indicated vitamin E concentrations at the
same time as 50 lM MPP+. The solid bar represents untreated cells
and the open bar represents cells treated with 50 lM MPP+ for 30 min.
Cell viability was measured after 24 h of MPP+ treatment. Data are
mean ± SEM values of three experiments in triplicate.
308 R. A. Gonzalez-Polo et al.
� 2003 International Society for Neurochemistry, Journal of Neurochemistry, 84, 305–315

concentration of 100 lM) was assessed by its ability to inhibitapoptotic events in MPP+. This concentration of allopurinol
impaired the mitochondrial cytochrome c release observed at
30 min with 50 lM MPP+ (Fig. 4a). Interestingly, artificial
ROS generation with the xanthine plus xanthine oxidase
system also stimulated early (30 min) cytochrome c release in
CGCs, as detected by western blot analysis (Fig. 4b). These
results are in agreement with those shown in Fig. 4(a) after
MPP+-exposure. In both assays, allopurinol largely prevented
cytochrome c release, indicating the participation of xanthine
oxidase in the early MPP+-induced cytochrome c release.
Cytochrome c release can be produced through the
mitochondrial permeability transition pore, which is fre-
quently coincident with loss of DYm (Petit et al. 1998;
Kroemer and Reed 2000). As MPP+-induced mitochondrial
permeability transition and loss of DYm have been describedin several cell models (Cassarino et al. 1999; Chalmers-
Redman et al. 1999), we decided to analyse whether MPP+
induced early changes in DYm in CGCs, by using the
fluorescent probe CMXRos in a flow cytometry assay. As
shown in Fig. 5, MPP+ had no effect on DYm at 30 min ofMPP+-exposure and it only induced loss of DYm at 6 h (datanot shown). This result demonstrates that MPP+-induced
early cytochrome c release precedes loss of DYm and that novariation in mitochondrial permeability transition is neces-
sary for MPP+-induced early cytochrome c release.
To identify the possible factors responsible for cytochrome
c release in CGCs exposed to MPP+, we prepared a
mitochondria-purified fraction and total cell lysates from
cells incubated with and without 50 lM MPP+. As Bad andBax are Bcl2-related proteins involved in the regulation of
cytochrome c release from mitochondria in several models of
apoptosis (von Harsdorf et al. 1999; Desagher and Martinou
2000), we determined their translocation to the mitochondria
in MPP+-induced cell death. Untreated cells showed unde-
tectable levels of Bax protein in mitochondrial fraction.
However, this protein translocated from cytosol to the
mitochondria (with a maximum over 30 min, data not
shown) after MPP+ exposure. The exact mechanism by
which MPP+ contributes to Bax translocation to mitochon-
dria is as yet unknown. Since this early translocation of Bax
was parallel with cytochrome c release, we next decided to
assay whether Bax translocation was inhibited by vitamin E.
Figure 6(a) shows that co-incubation with 100 lM vitamin Epartially blocked early MPP+-induced Bax translocation.
Total levels of Bax protein in the cell were not altered at the
same times (data not shown). Figure 6(b and c) shows the
changes in the patterns of Bad phosphorylation in MPP+-
exposed CGCs. A strong signal for phosphorylated Bad
protein was detected in lysates from control cells. In contrast,
phosphorylated Bad protein was barely detectable after
30 min of incubation with 50 lM MPP+. Exposure of the
neurons to 100 lM vitamin E (Fig. 6b) or 100 lM allopur-inol (Fig. 6a) maintained Bad phosphorylation even in the
(c)
(b)
(a)
*
*
*
Fig. 2 MPP+ effect on intracellular ROS content and GSH levels in
primary cultures of cerebellar granule cells. (a) and (b) CGCs in the
absence or presence of MPP+ (30 min) were incubated for a further
30 min with 20 lM or 5 lM, respectively, of the oxidation-sensitive
fluorescent probes hydroethidine (DHE) or 2¢-7¢-dichlorodihydrofluo-
rescein (DFCH), measuring the fluorescence intensity by flow cytom-
etry, as described in the Materials and methods. In both, the solid bar
shows untreated cells and the open bar shows the cells treated with
50 lM MPP+ for 30 min. Data are expressed as a percentage of DHE
or DFCH fluorescence with respect to control value and are
mean ± SEM values of three independent experiments. *Significantly
different (p < 0.05) from untreated cells. (c) Effect of MPP+ on GSH
levels in primary cultures of cerebellar granule cells. Cells were treated
(solid bar) or untreated (open bar) with 50 lM MPP+. After 30 min cells
were incubated with 1 lM of monochlorobimane (MCB), measuring the
fluorescence intensity by flow cytometry as described in the Materials
and methods. Data are expressed as a percentage of DHE or MCB
fluorescence with respect to control value and are mean ± SEM values
of three independent experiments. *Significantly different (p < 0.05)
from untreated cells.
Vitamin E blocks early cytochrome c release 309
� 2003 International Society for Neurochemistry, Journal of Neurochemistry, 84, 305–315

presence of MPP+. Total levels of Bad protein in the cell
were not altered at the same times (data not shown). Bad
protein is probably dephosphorylated by protein phospha-
tases as calcineurin, PP1a and others (Desagher and
Martinou, 2000). To evaluate whether participation of protein
phosphatases in the early events implicated MPP+-induced
apoptosis, we assayed the broad-range protein phosphatase
inhibitor okadaic acid. Figure 7(a and b) shows that 1 lMokadaic acid inhibited both Bad dephosphorylation and
cytochrome c release induced by 50 lM MPP+. These data
suggest a protein phosphatase participation in early apoptosis
induction in CGCs by MPP+. Interestingly phospho-Akt
levels (enzyme that phosphorylates Bad) are not modified by
MPP+ exposure (Fig. 7c). Because phospho-Akt is the active
form (Mora et al. 1999) and the medium conditions (high
potassium and insulin) favour this status, the decrease in
phospho-Bad protein shows that an early protein phosphatase
activation is implicated in its dephosphorylation, without
involvement of the PI3-K/Akt pathway.
Discussion
Apoptosis is thought to play an important role in the neuronal
loss in many neurological disorders, including Parkinson’s
disease. Cells undergoing apoptotic death exhibit several
morphological characteristics, such as chromatin condensa-
tion, nuclear fragmentation or apoptotic cell body formation.
Numerous studies suggest that MPP+ is able to induce
apoptosis in vitro in several cell types (Hartley et al. 1994;
Itano and Nomura 1995; Chalmers-Redman et al. 1999;
Yoshinaga et al. 2000) including CGCs (Dipasquale et al.
1991; Gonzalez-Polo et al. 1901; Du et al. 1997), some of
Fig. 3 Vitamin E inhibition of cytochrome c release and caspase 3
activation in primary cultures of cerebellar granule cells. CGC cultures
were exposed to: (a) 50 lM MPP+ at the times indicated, (b) 50 lM
MPP+ or 50 lM MPP+ plus 100 lM vitamin E for 30 min or (c)
increasing concentrations of MPP+ (5–200 lM) for 30 min. In both,
mitochondrial proteins (30–50 lg protein/lane) were size-fractionated
by SDS–PAGE, transferred onto PVDF and probed with cytochrome c
antibody as described in the Materials and methods. Actin content was
analysed as control Blots are representative of at least three inde-
pendent experiments. (d) Caspase 3 activity. CGCs cultures were
exposed to 50 lM MPP+ plus 100 lM vitamin E for 60 min after cells
were lysed and caspase 3 was assayed as described in the Materials
and methods. Data are expressed as a percentage of caspase 3
activity with respect to control value and are mean ± SEM values of
three independent experiments. *Significantly different (p < 0.05) from
untreated cells. (e) Proteolytic activation of caspase 3 and (f) its
prevention by 100 lM vitamin E. Cells were size-fractionated by SDS–
PAGE, transferred onto PVDF and probed with cleaved (active)
caspase 3 (17 kDa) antibody as described in the Materials and
methods. Actin content was analysed as control. Blots are represen-
tative of at least three independent experiments.
310 R. A. Gonzalez-Polo et al.
� 2003 International Society for Neurochemistry, Journal of Neurochemistry, 84, 305–315

the MPP+ effects being mediated by oxidative stress (Rossetti
et al. 1988; Kitamura et al. 1998). However, detection of
early apoptotic events involved in MPP+-cell death has not
been described yet. In this work we show that CGCs exposed
to MPP+ induce an early (in the 30 min after MPP+-
exposure) production of ROS, detected by the oxidation-
sensitive fluorescent probes HE and DFCH-DA. This
production is accompanied by GSH loss and appears to be
responsible for the apoptotic cell death, with the character-
istic cytochrome c release, caspase 3 activation, DNA
fragmentation and morphological changes, such as con-
densed chromatin. Previous works established a relationship
between MPP+-induced ROS production and inhibition of
mitochondrial complex I, but free radical production in these
models always occurred at later times (Mizuno et al. 1987;
Salinas et al. 2001). More recent works describe earlier ROS
generation in CGCs exposed to glutamate (Atlante et al.
1999) or potassium deprivation (Valencia and Moran 2001),
which appear to be directly involved in cytochrome c release
from mitochondria. Experiments reported in Fig. 1 demon-
strate that MPP+ can produce apoptotic cell death in CGCs
and Figs 2 and 3 also reveal an early cytochrome c release
MPP+ 50 µM
Allopurinol 100 µM
actin
actin
Cyt c mitochondria
Cyt c mitochondria
X/XOD
Allopurinol 100 µM
– + +
– – +
– + +
– – +
(a)
(b)
Fig. 4 Allopurinol blocks cytochrome c release from mitochondria in
both, MPP+-exposed and xanthine/xanthine oxidase system. Cells
were incubated for (a) 30 min with 50 lM MPP+ in the absence or the
presence of 100 lM of allopurinol or (b) 30 min with 10 mU/mL xanthine
oxidase plus 10 lM xanthine. In both mitochondrial proteins (30–50 lg
protein/lane) were size-fractionated by SDS–PAGE, transferred onto
PVDF and probed with cytochrome c antibody as described in the
Materials and methods. Actin content was analysed as control. Blot is
representative of at least three independent experiments.
100
Control
CM
xRos
Flu
ores
cenc
e(%
con
trol
)
MPP+
80
60
40
20
0
Fig. 5 MPP+ does not induce early changes in the mitochondrial
transmembrane potential (DYm) in primary cultures of cerebellar
granule cells. CGCs were incubated for 30 min in the absence (solid
bar) or presence (open bar) of 50 lM MPP+. After 30 min of incubation
with 0.1 lM CMXRos, the intracellular fluorescence intensity was
measured in a FACScan flow cytometer as described in the Materials
and methods. Data are expressed as a percentage of CMXRos
fluorescence with respect to control value and are mean ± SEM values
of three independent experiments.
Bax
MPP+ 50 µMvit E 100 µM
actin
– + +– – +
P-Bad
MPP+ 50 µMvit E 100 µM
actin
– + +– – +
P-Bad
MPP+ 50 µM
Allopurinol 100 µM
actin
– + +– – +
(a)
(b)
(c)
Fig. 6 Vitamin E partially prevents MPP+-induced mitochondrial Bax
translocation and vitamin E and allopurinol prevents Bad dephos-
phorylation in primary cultures of cerebellar granule cells. Cells were
incubated with 50 lM MPP+ in the presence or in the absence of
100 lM vitamin E for 30 min. (a) Mitochondrial proteins were pro-
cessed as described for cytochrome c release and analysed by
electrophoresis and western blot as described in the Materials and
methods using Bax antibody. (b) and (c) Cell lysates (30–50 lg protein
lane) obtained as described in the Materials and methods were sub-
jected to electrophoresis and western blot with an anti-Ser136 phos-
pho-Bad antibody. Actin content was analysed as control. Blots are
representative of at least three independent experiments.
Vitamin E blocks early cytochrome c release 311
� 2003 International Society for Neurochemistry, Journal of Neurochemistry, 84, 305–315

(30 min after MPP+-exposure) and caspase 3 activation
(60 min after MPP+-exposure) with an early oxidative stress
(free radical detection and intracellular GSH depletion).
Vitamin E co-incubation blocks both cell death and cyto-
chrome c release/caspase 3 activation, establishing a strong
relationship between early ROS generation and cytochrome c
translocation. These data have not been previously reported
in any cellular model of MPP+-induced neurotoxicity. The
knowledge that mitochondria are a direct target for MPP+
focused the attention on the cell changes that occurred
simultaneously with this event, all the earlier process being
ignored. However, the implication of mitochondrial free
radical production in MPP+-induced apoptotic cell death is
widely reported (Nicklas et al. 1987; Mizuno et al. 1995).
The fast cytosolic ROS production reported here is possibly a
result of MPP+ interaction with the xanthine oxidase enzyme
(Klaidman et al. 1993), which might lead to the formation of
ROS. Experiments reported in Fig. 4 demonstrate that
cytochrome c release is driven by ROS produced in MPP+-
exposure or by exogenous production via the xanthine/
xanthine oxidase system. This conclusion was supported by
the partial prevention of cytochrome c release with the
specific xanthine oxidase inhibitor allopurinol, which would
block ROS formation by impairing xanthine oxidase activity
in both cases. The MPP+ concentration-dependence and
timing in cytochrome c release shown in Fig. 3b could
suggest that this early translocation is a function of ROS
production in cytosol, probably through the xanthine oxidase
system. Blockage by vitamin E and allopurinol of cyto-
chrome c release demonstrates this fact. The cytosolic MPP+-
induced ROS production by xanthine oxidase would occur
earlier than that produced by mitochondrial impairment and
would be compatible with the events observed in the first
30 min after MPP+ exposure: loss of intracellular GSH and
cytochrome c release from mitochondria, without change in
mitochondrial transmembrane potential. Recent studies have
shown that early translocation of cytochrome c, previous to
mitochondrial collapse, is directly implicated in apoptosis
induction (Granville et al. 2001; Herrera et al. 2001; Li et al.
2001; Suen et al. 2001). In good agreement with these data,
our results clearly show that early cytochrome c release
drives cells to apoptotic cell death, because blockade of this
release by vitamin E increases cell survival and impairs
apoptotic events.
It is important to note that cytochrome c release is closely
connected with the Bcl-2 family proteins. Antiapoptotic
members Bcl-2 and Bcl-XL are, for the most part, mito-
chondrial proteins (Hsu et al. 1997) while other related
proteins, such as Bad or Bax, are cytosolic but translocate to
the mitochondria in response to different apoptotic stimuli
(Desagher et al. 1999; Downward 1999). The mechanisms
that promote Bax activation remain unclear, but glutathione
depletion, induced by an increase in free radical concentra-
tion in the cells, could be involved as has been recently
reported (Jungas et al. 2002). In CGCs exposed to MPP+ we
observed an increase in mitochondrial Bax levels, which is
parallel to free radical production and cytochrome c release.
In our conditions Bax translocation is blocked by vitamin E,
linking the Bax translocation to the free radical generation.
This result disagrees with that recently observed by Hart-
mann et al. (2001). In our study Bax is detected in
mitochondrial extract 30 min after incubation with 50 lMMPP+ while in Hartmann’s work Bax localization was
measured at 12 h with a significantly lower MPP+ concen-
tration (1–3 lM). The different time of exposure and MPP+
concentration can explain this disagreement. On the other
hand Bax-induced cytochrome c release is often associated
with changes in DYm (Lotharius et al. 1999; Antonson andMartinou 2000). Loss in DYm is also observed in severalMPP+-induced apoptosis systems (Lambert and Bond 1989;
Cassarino et al. 1999; Chalmers-Redman et al. 1999).
P-Bad
MPP+ 50 µM
Okadaic acid 1 µM
actin
– + +– – +
Cyt c mitochondria
MPP+ 50 µM
Okadaic acid 1 µM
actin
– + +– – +
P-Akt 60kDa
MPP+ 50 µM
actin
– +
(a)
(b)
(c)
Fig. 7 Okadaic acid blocks both MPP+-induced Bad dephosphoryla-
tion and cytochrome c release in primary cultures of cerebellar granule
cells. CGCs were exposed to 50 lM MPP+ for 30 min with or without
preincubation with 1 lM okadaic acid. (a) Cell lysates (30–50 lg pro-
tein/lane) were separated on polyacrylamide gels and western blot
analysis (as described in the Materials and methods) was performed
with an anti-Ser136 phospho-Bad antibody to detect the phosphory-
lation state of Bad. (b) Cytochrome c was detected in mitochondrial
extracts (30–50 lg protein/lane) by western blot as described in Fig. 3
and the Materials and methods using cytochrome c antibody.
(c) Phosphorylation state of Akt. Cells were also incubated for 30 min
in the presence or absence of 50 lM MPP+. After this time cell lysates
were extracted and Akt activity was analysed by electrophoresis
(30–50 lg protein/lane) and western blot using antibodies (specific for
Ser473) against the active form (P-Akt). Actin content was analysed as
control Blots are representative of at least three independent experi-
ments.
312 R. A. Gonzalez-Polo et al.
� 2003 International Society for Neurochemistry, Journal of Neurochemistry, 84, 305–315

However, we observed no changes in DYm coincident withBax translocation and cytochrome release. Our results are in
agreement with other recent studies, which implicated early
events in the development of apoptotic cell death (Annis
et al. 2001; Suen et al. 2001). Bax could form a channel in
mitochondria, allowing cytochrome c release, without mito-
chondrial damage as has been reported (Antonsson et al.
1997).
Another pro-apoptotic Bcl-2-related protein is Bad. In the
absence of apoptotic stimuli Bad is phosphorylated and
sequestered in the cytosol by binding to 14-3-3 protein (Zha
et al. 1996). During apoptosis Bad is dephosphorylated and
promotes cell death by binding to bcl-XL (Zha et al. 1996).
Bad is phosphorylated preferably, but not only, by Akt
(Dudek et al. 1997) and dephosphorylated by PP1 and PP2
phosphatases (Desagher and Martinou, 2000). In our work,
Bad is strongly dephosphorylated at the same time as Bax
translocation to the mitochondria. Bad can bind Bcl-XL and
this fact would contribute to the inhibition of the death-
repressor activity of Bcl-XL, facilitating the Bax-induced
cytochrome c release. Pre-incubation of CGCs with okadaic
acid, a broad-range protein phosphatase inhibitor partially
inhibits both early Bad dephosphorylation and cytochrome c
release. This result clearly indicates that phosphatases are
activated in the 30 min after MPP+ exposure and that this
activation would be involved in cytochrome c release.
Because Bad dephosphorylation is also inhibited by vitamin
E and allopurinol, we could hypothesize that phosphatase
activities are switched on by the ROS produced in the first
minutes after MPP+ exposure. Vitamin E also partially
inhibits Bax translocation to mitochondria, indicating that the
impairment of early MPP+-induced free radical production
eliminates all the apoptotic events initiated by MPP+.
In conclusion, our results (summarized in Fig. 8) demon-
strate that MPP+ produces early Bax translocation, Bad
dephosphorylation, cytochrome c release and caspase acti-
vation, concluding with cell death. All events are blocked by
vitamin E, suggesting that the early events implicated in
MPP+-induced apoptosis in CGCs are related to oxidative
stress probably as a result of activation of the xanthine
oxidase system. These results together point towards the
existence of some early events (30 min) that play an essential
Fig. 8 Summary of the proposed mechanism for the early apoptotic
events induced by MPP+ in primary cultures of cerebellar granule cells.
MPP+ production of ROS could be mediated by xanthine oxidase. The
oxidative stress would contribute to Bad dephosphorylation (by protein
phosphatase activation) and Bax translocation to the mitochondria,
which would mediate cytochrome c release (without loss of DYm),
caspase 3 activation and cell death. Vitamin E is able to block all the
observed events, including cell death.
Vitamin E blocks early cytochrome c release 313
� 2003 International Society for Neurochemistry, Journal of Neurochemistry, 84, 305–315

role in MPP+-induced apoptosis. Further work will be
necessary to understand completely the molecular mechan-
ism by which oxidative stress provokes Bax translocation
and protein phosphatase activation and all the events that
conclude with apoptotic cell death.
Acknowledgements
This work was supported, in part, by grants 2PR01A079, to GS
(from Junta de Extremadura, Spain), 01/51 to JMF (from Junta de
Extremadura, Spain), FIS010797 to IF (from Ministerio de Sanidad
y Consumo, Spain) and CAM-08.1/0078/2000 to IF (from Comu-
nidad de Madrid, Spain). RAGP was supported by a Spanish
Ministerio de Educacion, Cultura y Deporte predoctoral fellowship.
The authors also thanks to J. C. Alonso and J. M. Moran for their
inestimable helpful technical assistance.
References
Akaneya Y., Takahashi M. and Hatanaka H. (1995) Involvement of free
radicals in MPP+ neurotoxicity against rat dopaminergic neurons
in culture. Neurosci. Lett. 193, 53–56.
Annis M. G., Zamzami N., Zhu W., Penn L. Z., Kroemer G. and Leber
B. and Andrews D. W. (2001) Endoplasmic reticulum localized
Bcl-2 prevents apoptosis when redistribution of cytochrome c is a
late event. Oncogene 20, 1939–1952.
Antonsson B. and Martinou J. C. (2000) The Bcl-2 protein family. Exp.
Cell. Res. 256, 50–57.
Antonsson B., Conti F., Ciavatta A., Montessuit S., Lewis S., Martinou
I., Bernasconi L., Bernard A., Mermod J. J., Mazzei G., Maundrell
K., Gambale F. and Sadoul R. and. Martinou J. C. (1997) Inhibition
of Bax channel-forming activity by Bcl-2. Science 277, 370–372.
Atlante A., Calissano P., Bobba A., Azzariti A., Marra E. and Passarella
S. (2000) Cytochrome c is released from mitochondria in a reactive
oxygen species (ROS)-dependent fashion and can operate as a
ROS scavenger and as a respiratory substrate in cerebellar neurons
undergoing excitotoxic death. J. Biol. Chem. 275, 37159–37166.
Atlante A., Calissano P., Bobba A., Giannattasio S., Marra E. and
Passarella S. (2001) Glutamate neurotoxicity, oxidative stress and
mitochondria. FEBS Lett. 497(1), 1–5.
Atlante A., Gagliavoli S., Marra E., Calissano P. and Passavella S.
(1999) Glutamate neurotoxicity in rat cerebellar granule cells
involve cytochrome c release from mitochondria and mitochondrial
shuttle impairment. J. Neurochem. 73(1), 237–246.
Blum D., Torch T., Lambeng N., Nissou M. F., Benabid A. L., Sadoul R.
and Verna. J. R. (2001) Molecular pathways involved in the neu-
rotoxicity of 6-OHDA, dopamine and MPTP, contribution to the
apoptotic theory in Parkinson’s Disease. Prog. Neurobiol. 65, 135–
172.
Bobba A., Atlante A., Giannattasio S., Sgaramella G., Calissano P. and
Marra E. (1999) Early release and subsequent caspase-mediated
degradation of cytochrome c in apoptotic cerebellar granule cells.
FEBS Lett. 457, 126–130.
Bradford M. (1976) A rapid and sensitive method for the quantitation of
microgram quantities of protein utilizing the principle of protein-
dye binding. Anal. Biochem. 72, 248–254.
Cassarino D. S., Parks J. K., Parker W. D. Jr and Bennett J. P. Jr (1999)
The parkinsonian neurotoxin MPP + opens the mitochondrial
permeability transition pore and releases cytochrome c in isolated
mitochondria via an oxidative mechanism. Biochim. Biophys. Acta
1453, 49–62.
Castagnoli N. Jr, Chiba K. and Trevor K. (1985) Potential bioactivation
pathways for the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetra-
hydropyridine (MPTP). Life Sci. 36, 225–230.
Chalmers-Redman R. M., Fraser A. D., Carlile W., Pong A. and Tatton
W. G. (1999) Glucose protection from MPP+-induced apoptosis
depends on mitochondrial membrane potential and ATP synthase.
Biochem. Biophys. Res. Commun. 257, 440–447.
Chiba K., Trevor A. and Castaganoli N. Jr (1985) Active uptake of
MPP+, a metabolite of MPTP by brain synaptosomes. Biochem.
Biophys. Res. Commun. 128, 1228–1232.
Desagher S. and Martinou J. C. (2000) Mitochondria as the central
control point of apoptosis. Trends Cell Biol. 10, 369–377.
Desagher S., Osen-Sand A., Nichols A., Eskes R., Montessuit S.,
Lauper S., Maundrell K., Antonsson B. and Martinou J. C. (1999)
Bid-induced conformational change of Bax is responsible for
mitochondrial cytochrome c release during apoptosis. J. Cell Biol.
144, 891–901.
Dipasquale B., Marini A. M. and Youle R. J. (1991) Apoptosis and DNA
degradation induced by 1-methyl-4-phenylpyridinium in neurons.
Biochem. Biophys. Res. Commun. 181, 1442–1448.
Dodel R. C., Du Y., Bales K. R., Ling Z. D., Carvey P. M. and Paul S. M.
(1998) Peptide inhibitors of caspase-3-like proteases attenuate
1-methyl-4-phenylpyridinum-induced toxicity of cultured fetal rat
mesencephalic dopamine neurons. Neuroscience 86, 701–707.
Downward J. (1999) How BAD phosphorylation is good for survival.
Nat. Cell Biol. 1, E33–E35.
Du Y., Dodel R. C., Bales K. R., Jemmerson R., Hamilton-Bird E. and
Paul S. M. (1997) Involvement of a caspase-3-like cysteine
protease in 1-methyl-4-phenylpyridinium-mediated apoptosis
of cultured cerebellar granule neurons. J. Neurochem. 69, 1382–
1388.
Dudek H., Datta S. R., Franke T. F., Birnbaum M. J., Yao R., Cooper
G. M., Segal R. A., Kaplan D. R. and Greenberg M. E. (1997)
Regulation of neuronal survival by the serine-threonine protein
kinase Akt. Science 275, 661–665.
Fuentes J. M., Lompre A. M., Moller J. V., Falson P., le Maire M. (2000)
Clean Western blots of membrane proteins after yeast heterologous
expression following a shortened version of the method of Perini
et al. Anal. Biochem. 285, 276–278.
Gonzalez-Polo R. A., Mora A., Clemente N., Sabio G., Centeno F., Soler
G. and Fuentes J. M. (2001) Mechanisms of MPP+ incorporation
into cerebellar granule cells. Brain Res. Bull. 56, 119–123.
Granville D. J., Cassidy B. A., Ruehlmann D. O., Choy J. C., Brenner
C., Kroemer G., van Breemen C., Margaron P., Hunt D. W. and
McManus B. M. (2001) Mitochondrial release of apoptosis-indu-
cing factor and cytochrome c during smooth muscle cell apoptosis.
Am. J. Pathol. 159, 305–311.
Gross A., McDonnell J. M. and Korsmeyer S. J. (1999) BCL-2 family
members and the mitochondria in apoptosis. Genes Dev. 13, 1899–
1911.
Hartley A., Stone J. M., Heron C., Cooper J. M. and Schapira A. H.
(1994) Complex I inhibitors induce dose-dependent apoptosis in
PC12 cells: relevance to Parkinson’s disease. J. Neurochem. 63,
1987–1990.
Hartmann A., Michel P. P., Troadec J. C., Mouatt-Prigent A., Faucheux
B. A., Ruberg M., Agid Y., Hirsch E. C. (2001) Is Bax a mito-
chondrial mediator in apoptotic death of dopaminergic neurons in
Parkinson’s disease? J. Neurochem. 76, 1785–1793.
Herrera B., Fernandez M., Alvarez A. M., Roncero C., Benito M., Gil J.
and Fabregat I. (2001) Activation of caspases occurs downstream
from radical oxygen species production, Bcl-xL down-regulation,
and early cytochrome C release in apoptosis induced by trans-
forming growth factor beta in rat fetal hepatocytes. Hepatology 34,
548–556.
314 R. A. Gonzalez-Polo et al.
� 2003 International Society for Neurochemistry, Journal of Neurochemistry, 84, 305–315

Hsu Y. T., Wolter K. G. and Youle R. J. (1997) Cytosol-to-membrane
redistribution of Bax and Bcl-X (L) during apoptosis. Proc. Natl
Acad. Sci. USA 94, 3668–3675.
Itano Y. and Nomura Y. (1995) 1-methyl-4-phenyl-pyridinium ion
(MPP+) causes DNA fragmentation and increases the Bcl-2
expression in human neuroblastoma, SH-SY5Y cells, through
different mechanisms. Brain Res. 704, 240–245.
Jungas T., Motta I., Duffieux F., Fanen P., Stoven V. and Ojcius D. M.
(2002) Glutathione levels and Bax activation during apoptosis due
to oxidative stress in cells expressing wildtype and mutant CFTR.
J. Biol. Chem. 277(31), 27912–27918.
Kamemcic H., Lyon A., Paterson P. G. and Juurlink B. H. (2000)
Monoclorobimane fluorometric method to measure tissue gluta-
thione. Anal. Biochem. 286, 35–37.
Kitamura Y., Kosaka T., Kakimura J. L., Matsuoka Y., Kolmo Y., Nomura
Y. and Taniguchi T. (1998) Protective effects of the antiparkinsonian
drugs talipexole and pramipexole against 1-methyl-4-phenylpyridi-
nium-induced apoptotic cell death in human neuroblastoma
SH-Sy5Y cells. Mol. Pharmacol. 54, 1046–1054.
Klaidman L. K., Adams J. D., Leung A. C., Sam Jim S. and Cadenas E.
(1993) Redox cycling of MPP+: Evidence for a new mechanism
involving hydride transfer with xanthine oxidase, aldehyde dehy-
drogenase, and lipoamide dehydrogenase. Free Radic. Biodiv.
Med. 15, 169–179.
Kroemer G. and Reed J. C. (2000) Mitochondrial control of cell death.
Nat. Med. 6, 513–519.
Lambert C. E. and Bondy S. C. (1989) Effects of MPTP, MPP+ and
paraquat on mitochondrial potential and oxidative stress. Life Sci.
44, 1277–1284.
Langston J. W., Ballard P., Tetrud J. W. and Irwin I. (1983) Chronic
Parkinsonism in humans due to a product of meperidine-analog
synthesis. Science 219, 979–980.
Leist M., Volbracht C., Fava E. and Nicotera P. (1998) 1-Methyl-4-
phenylpyridinium induces autocrine excitotoxicity, protease acti-
vation, and neuronal apoptosis. Mol. Pharmacol. 54, 789–801.
Li P., He Q. P., Ouyang Y. B., Liu C. L., Hu B. R. and Siesjo B. K.
(2001) Early release of cytochrome C and activation of caspase-3
in hyperglycemic rats subjected to transient forebrain ischemia.
Brain Res. 896, 69–76.
Lotharius J., Dugan L. L. and O’Malley K. L. (1999) Distinct mech-
anisms underlie neurotoxin-mediated cell death in cultured dop-
aminergic neurons. J. Neurosci. 19, 1284–1293.
Lowry O. H., Rosebrough N. J., Farr A. L. and Randall R. J. (1951)
Protein measurement with the Folin phenol reagent. J. Biol. Chem.
193, 265–271.
Mizuno Y., Ohta S., Tanaka M., Takamiya S., Suzuki K., Sato T., Oya
H., Ozawa T. and Kagawa K. (1989) Deficiencies in complex I
subunits of the respiratory chain in Parkinson’s Disease. Biochem.
Biophys. Res. Commun. 163, 1450–1455.
Mizuno Y., Saitoh T. and Sone N. (1987b) Inhibition of mitochondrial
NADH-ubiquinone oxidoreductase activity by 1-methyl-4-phe-
nylpyridinium ion. Biochem. Biophys. Res. Commun. 143, 294–
299.
Mora A., Gonzalez-Polo R. A., Fuentes J. M., Soler G. and Centeno F.
(1999) Different mechanisms of protection against apoptosis by
valproate and Li+. Eur J. Biochem. 266, 886–891.
Mosmann T. (1983) Rapid colorimetric assay for cellular growth and
survival: application to proliferation and cytotoxicity assays.
J. Immunol. Meth. 65, 55–63.
Nicklas W. J., Vyas I. and Heikkila R. E. (1985) Inhibition of NADH-
linked oxidation in brain mitochondria by 1-methyl-4-phenyl-
1,2,5,6-tetrahydropyridine. Life Sci. 36, 2503–2508.
Nicklas W. J., Youngster S. K., Kindt M. V. and Heikkila R. E. (1987)
MPTP, MPP+ and mitochondrial function. Life Sci. 40(8), 721–
729.
Obata T. (1999) Reserpine prevents hydroxyl radical formation by MPP+
in rat striatum. Brain Res. 828, 68–73.
Odunze I. N., Klaidman L. K. and Adams J. D. Jr (1990) MPTP toxicity
in the mouse brain and vitamin E. Neurosci. Lett. 108, 346–349.
Putcha G. V., Deshmukh M. and Johnson E. M. Jr. (1999) BAX trans-
location is a critical event in neuronal apoptosis: regulation by
neuroprotectants, BCL-2, and caspases. J. Neurosci. 19(17), 7476–
7485.
Petit P. X., Goubern M., Diolez P., Susin S. A., Zamzami N. and
Kroemer G. (1998) Disruption of the outer mitochondrial
membrane as a result of large amplitude swelling: the impact of
irreversible permeability transition. FEBS Lett. 426, 111–116.
Pique M., Barragan M., Dalmau M., Bellosillo B., Pons G. and Gil J.
(2000) Aspirin induces apoptosis through mitochondrial cyto-
chrome c release. FEBS Lett. 480, 193–196.
Przedborski S., Kostic V., Jackson-Lewis V., Naini A. B., Simonetti S.,
Fahn S., Carlson E., Epstein C. J. and Cadet J. (1992) Transgenic
mice with increased Cu-Zn-superoxide dismutase activity are
resistant to N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced
neurotoxicity. J. Neurosci. 12, 1658–1667.
Rossetti Z., Sotgiu A., Sharp D. E., Hadgiconstantinou M. and Neff
N. H. (1988) 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine (MPTP)
and free radicals in vitro. Biochem. Pharmacol. 37, 4573–4574.
Rothe G. and Valet G. (1990) Flow cytometric analysis of respiratory
burst activity in phagocytes with hydroethidine and 2¢,7¢-dichlo-rofluorescin. J. Leukoc. Biol. 47, 440–448.
Salinas M., Martın D., Alvarez A. and Cuadrado A. (2001) Akt1/PKBaprotects PC12 cells against the Parkinsonism-inducing neurotoxin
1-methyl-4-phenylpyridinium and reduces the levels of oxygen-
free radicals. Mol. Cell. Neurosci. 17, 67–77.
Sinha B. K., Singh Y. and Krishna G. (1986) Formation of su-
peroxide and hydroxyl radicals from 1-methyl-4-phenylpyridi-
nium ion (MPP+): reductive activation by NADPH cytochrome
P-450 reductase. Biochem. Biophys. Res. Commun. 135, 583–
588.
Suen Y. K., Fung K. P., Lee C. Y. and Kong S. K. (2001) Gliotoxin
induces apoptosis in cultured macrophages via production of
reactive oxygen species and cytochrome c release without mito-
chondrial depolarization. Free Radic. Res. 35, 1–10.
Tammariello S. P., Quinn M. T. and Estus S. (2000) NADPH oxidase
contributes directly to oxidative stress and apoptosis in nerve growth
factor-deprived sympathetic neurons. J. Neurosci. 20, 123–132.
Tipton K. F. and Singer T. P. (1993) Advances in our understanding of
the mechanisms of the neurotoxicity of MPTP and related com-
pounds. J. Neurochem. 61, 1191–1206.
Valencia A. and Moran J. (2001) Role of oxidative stress in the apoptotic
cell death of cultured cerebellar granule neurons. J. Neurosci. Res.
64, 284–297.
von Harsdorf R., Li P. F. and Dietz R. (1999) Signaling pathways in
reactive oxygen species-induced cardiomyocyte apoptosis. Circu-
lation 99, 2934–2941.
Yoshinaga N., Murayama T. and Nomura Y. (2000) Apoptosis induction
by a dopaminergic neurotoxin, 1-methyl-4-phenylpyridinium ion
(MPP+), and inhibition by epidermal growth factor in GH3 cells.
Biochem. Pharmacol. 60, 111–120.
Zha J., Harada H., Yang E., Jockel J. and Korsmeyer S. J. (1996) Serine
phosphorylation of death agonist BAD in response to survival
factor results in binding to 14-3-3 not BCL-X (L). Cell 87, 619–
628.
Vitamin E blocks early cytochrome c release 315
� 2003 International Society for Neurochemistry, Journal of Neurochemistry, 84, 305–315



















