
Virus-Host and CRISPR Dynamics in Archaea-Dominated Hypersaline Lake Tyrrell, Victoria, Australia
14
Hindawi Publishing Corporation Archaea Volume , Article ID , pages http://dx.doi.org/.// Research Article Virus-Host and CRISPR Dynamics in Archaea-Dominated Hypersaline Lake Tyrrell, Victoria, Australia Joanne B. Emerson, 1,2 Karen Andrade, 3 Brian C. Thomas, 1 Anders Norman, 1,4 Eric E. Allen, 5,6 Karla B. Heidelberg, 7 and Jillian F. Banfield 1,3 Department of Earth and Planetary Science, University of California, Berkeley, McCone Hall, Berkeley, CA - , USA Cooperative Institute for Research in Environmental Sciences, University of Colorado, Boulder, CO, USA Department of Environmental Science, Policy, and Management, University of California, Berkeley, Mulford Hall, Berkeley, CA , USA Department of Biology, University of Copenhagen, Copenhagen, Denmark Marine Biology Research Division, Scripps Institution of Oceanography, La Jolla, CA, USA Division of Biological Sciences, University of California, San Diego, La Jolla, CA -, USA Department of Biological Sciences, University of Southern California, Los Angeles, CA , USA Correspondence should be addressed to Joanne B. Emerson; [email protected] Received December ; Revised May ; Accepted May Academic Editor: Yoshizumi Ishino Copyright © Joanne B. Emerson et al. is is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. e study of natural archaeal assemblages requires community context, namely, a concurrent assessment of the dynamics of archaeal, bacterial, and viral populations. Here, we use lter size-resolved metagenomic analyses to report the dynamics of archaeal and bacterial OTUs and viral populations across samples collected over dierent timescales – from Australian hypersaline Lake Tyrrell (LT). All samples were dominated by Archaea (–%). Archaeal, bacterial, and viral populations were found to be dynamic on timescales of months to years, and dierent viral assemblages were present in planktonic, relative to host-associated (active and provirus) size fractions. Analyses of clustered regularly interspaced short palindromic repeat (CRISPR) regions indicate that both rare and abundant viruses were targeted, primarily by lower abundance hosts. Although very few spacers had hits to the NCBI nr database or to the LT viral populations, % had hits to unassembled LT viral concentrate reads. is suggests local adaptation to LT-specic viruses and/or undersampling of haloviral assemblages in public databases, along with successful CRISPR-mediated maintenance of viral populations at abundances low enough to preclude genomic assembly. is is the rst metagenomic report evaluating widespread archaeal dynamics at the population level on short timescales in a hypersaline system. 1. Introduction As the most abundant and ubiquitous biological entities, viruses inuence host mortality and community structure, food web dynamics, and geochemical cycles [, ]. In order to better characterize the potential inuence that viruses have on archaeal evolution and ecology, it is important to under- stand the coupled dynamics of viruses and their archaeal hosts in natural systems. Although previous studies have demonstrated dynamics in virus-host populations, most of these studies have focused on bacterial hosts, oen restricted to targeted groups of virus-host pairs, and little is known about archaeal virus-host dynamics in natural systems. Community-scale virus-host analyses have oen been based on low-resolution measurements of the whole com- munity, relying on techniques such as denaturing gradient gel electrophoresis (DGGE), pulsed-eld gel electrophoresis (PFGE), and microscopic counts (e.g., [–]). One excep- tion is a study that examined viral and microbial dynam- ics through single read-based metagenomic analyses in four aquatic environments, including an archaea-dominated
Transcript of Virus-Host and CRISPR Dynamics in Archaea-Dominated Hypersaline Lake Tyrrell, Victoria, Australia

Hindawi Publishing CorporationArchaeaVolume !"#$, Article ID $%"&%#, #! pageshttp://dx.doi.org/#".##''/!"#$/$%"&%#
Research ArticleVirus-Host and CRISPR Dynamics in Archaea-DominatedHypersaline Lake Tyrrell, Victoria, Australia
Joanne B. Emerson,1,2 Karen Andrade,3 Brian C. Thomas,1 Anders Norman,1,4
Eric E. Allen,5,6 Karla B. Heidelberg,7 and Jillian F. Banfield1,3
! Department of Earth and Planetary Science, University of California, Berkeley, "#$McCone Hall, Berkeley, CA %&$'#-&$($, USA'Cooperative Institute for Research in Environmental Sciences, University of Colorado, Boulder, CO, USA"Department of Environmental Science, Policy, and Management, University of California, Berkeley, )&Mulford Hall, Berkeley,CA %&$'#, USA
&Department of Biology, University of Copenhagen, Copenhagen, Denmark)Marine Biology Research Division, Scripps Institution of Oceanography, La Jolla, CA, USA(Division of Biological Sciences, University of California, San Diego, La Jolla, CA %'#%"-#'#', USA$Department of Biological Sciences, University of Southern California, Los Angeles, CA %##*%, USA
Correspondence should be addressed to Joanne B. Emerson; [email protected]
Received % December !"#!; Revised #%May !"#$; Accepted !%May !"#$
Academic Editor: Yoshizumi Ishino
Copyright © !"#$ Joanne B. Emerson et al. (is is an open access article distributed under the Creative Commons AttributionLicense, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properlycited.
(e study of natural archaeal assemblages requires community context, namely, a concurrent assessment of the dynamics ofarchaeal, bacterial, and viral populations. Here, we use )lter size-resolved metagenomic analyses to report the dynamics of #"#archaeal and bacterial OTUs and #*" viral populations across #% samples collected over di+erent timescales !""%–!"#" fromAustralian hypersaline Lake Tyrrell (LT). All samples were dominated by Archaea (%'–,'%). Archaeal, bacterial, and viralpopulations were found to be dynamic on timescales of months to years, and di+erent viral assemblages were present in planktonic,relative to host-associated (active and provirus) size fractions. Analyses of clustered regularly interspaced short palindromic repeat(CRISPR) regions indicate that both rare and abundant viruses were targeted, primarily by lower abundance hosts. Although veryfew spacers had hits to the NCBI nr database or to the #*" LT viral populations, !#% had hits to unassembled LT viral concentratereads.(is suggests local adaptation to LT-speci)c viruses and/or undersampling of haloviral assemblages in public databases, alongwith successful CRISPR-mediated maintenance of viral populations at abundances low enough to preclude genomic assembly.(isis the )rstmetagenomic report evaluating widespread archaeal dynamics at the population level on short timescales in a hypersalinesystem.
1. Introduction
As the most abundant and ubiquitous biological entities,viruses in-uence host mortality and community structure,food web dynamics, and geochemical cycles [#, !]. In orderto better characterize the potential in-uence that viruses haveon archaeal evolution and ecology, it is important to under-stand the coupled dynamics of viruses and their archaealhosts in natural systems. Although previous studies havedemonstrated dynamics in virus-host populations, most ofthese studies have focused on bacterial hosts, o.en restricted
to targeted groups of virus-host pairs, and little is knownabout archaeal virus-host dynamics in natural systems.
Community-scale virus-host analyses have o.en beenbased on low-resolution measurements of the whole com-munity, relying on techniques such as denaturing gradientgel electrophoresis (DGGE), pulsed-)eld gel electrophoresis(PFGE), and microscopic counts (e.g., [$–']). One excep-tion is a study that examined viral and microbial dynam-ics through single read-based metagenomic analyses infour aquatic environments, including an archaea-dominated

! Archaea
hypersaline crystallizer pond [/]. In that work, it was pro-posed that microorganisms and viruses persisted over time atthe level of individual taxa (species) but were highly dynamicat the genotype (strain) level. However, in a reanalysisof some of those data by our group using metagenomicassembly, we concluded that viruses were actually dynamicat the population (taxon) level in that system [%]. (is resultsuggests that further analyses are necessary to determinewhether archaeal populations tend to be dynamic or stablein hypersaline systems on short timescales.
Of the relatively few metagenomic analyses of virus-hostdynamics that have been reported, several have consideredthe clustered regularly interspaced short palindromic repeat(CRISPR) system, which provides an opportunity to studyhosts’ responses to viral predation and to link viruses to hosts[&–#!]. (e CRISPR system is a genomic region in nearlyall archaea and some bacteria, and CRISPRs (at least in allsystems that have been biochemically characterized to date)have been shown to confer adaptive immunity to virusesand/or other mobile genetic elements through nucleotidesequence identity between the host CRISPR system andinvading nucleic acids [#$, #*]. (e hallmarks of CRISPRregions are spacers, generally derived from foreign nucleicacids, including plasmid, viral DNA, and short palindromicrepeat sequences between each spacer (reviewed in [#', #/]).Di+erent strains of the same species can have highly divergentCRISPR regions (e.g., [#%]). A highly genomically resolvedstudy of virus-host dynamics in an archaea-dominated acid-mine drainage system demonstrated that only the mostrecently acquiredCRISPR spacersmatched coexisting virusesand showed that viruses rapidly recombined to evadeCRISPRtargeting, indicating that community stability was achievedby the rapid coevolution of host resistance to viruses andviral resistance to the host CRISPR system [,]. ArchaealCRISPR dynamics have also been investigated in Sulfolobusislandicus populations [#&, #,], indicating clear biogeographyof viral populations and adaptation of CRISPR sequences tolocal viral populations. Whether similar dynamics occur inarchaea-dominated hypersaline systems is not well under-stood.
Previously, our group tracked the dynamics of $' viralpopulations in eight viral concentrates (representing the$" kDa–".#!m size fraction) collected during three sum-mers from archaea-dominated hypersaline Lake Tyrrell (LT),Victoria, Australia [%]. We demonstrated that viruses inthe LT system were generally stable on the timescale ofdays and dynamic over years. In this study, we sought toexpand our analyses to include LT viruses frommetagenomiclibraries generated from ".#, ".&, and $."!m )lters, potentiallyincluding proviruses, viruses larger than ".# !m, activelyinfecting viruses, and viruses otherwise retained on the)lters.(is allowed us to increase the temporal and spatial scope ofour study to #% samples, including four winter samples fromwhich viral concentrate DNA was not sequenced. To givecontext to the current and previous viral analyses from LTand to test the prevailing theory that microbial taxa are stableat the species level in archaea-dominated hypersaline systems(presented in [/]), in this studywe also characterized archaealand bacterial dynamics through #/S rRNA gene analyses, and
we used CRISPR analyses to assist in the interpretation of theresults.
2. Materials and Methods
'.!. Sample Collection and Preparation. Sample collection,DNA extraction, and sequencing methods have beendescribed previously [%, !", !#]. Brie-y, #" L surface watersamples were collected from LT and sequentially )lteredthrough !", $.", ".&, and ".#!m )lters. Post-".#!m )ltrateswere concentrated through tangential -ow )ltration andretained for viral DNA extraction. Viral concentrates and".#, ".&, and $." !m )lters were retained for each sample,and sequencing was undertaken for di+erent size ranges,depending on the sample (Table #). Sample names includethe month (J for January, A for August), year, site (A or B,"$""m apart), and time point if the sample was part of adays-scale time series (e.g., t#, t!, etc.). Where necessary, thesize fraction is also indicated in the sample name ($.", ".&,or ".# for )lter size in !m, or VC for viral concentrate). SitesA and B are isolated pools in the summer (January samples)but continuous with the lake in the winter (August samples).GPS coordinates for sites A and B are $'∘ #," ",./"" S, #*!∘ *%"',.%"" E and $'∘ #," #&.%#"" S, #*!∘ *&" *.!$"" E, respectively.'.'. Recovery of !&# Viral Contigs >!# kb and Detection ofViruses in Each Sample. In addition to the $' LT viral andvirus-like (meaning virus or plasmid) populations that wedescribed previously [%], we incorporated all contigs >#" kbfrom a new IDBA UD [!!] assembly of the six Illumina-sequenced viral concentrate samples (default parameters).We also sought to include as many assembled viral sequencesas possible from libraries from larger size-fraction )lters.To do that, we )rst attempted to assemble reads from allIllumina-sequenced )lter samples and reads from at leastone ".& !m )lter per sample (regardless of sequencing type),using IDBA UD with default parameters [!!] for Illumina-sequenced samples and gsAssembler [!$] with default param-eters for *'* sequenced samples. (e assemblies were gen-erally fragmentary, and, as such, no viral contigs larger than#" kb were identi)ed from most assemblies. However, werecovered viral contigs >#" kb from metagenomic assembliesof all ".#!m )lters from January !"#", identi)ed throughannotation that could be con)dently assigned to viruses orproviruses (e.g., contigs that included viral capsid proteins,tail proteins, and/or terminases). Annotation parameterswere the same as those described previously [%]. We usedBLASTn to identify duplicate viral sequences across assem-blies and samples, and we removed any contigs that shared>! kb at >,'%nt identity (all contigs that were removedactually shared #,,%nt identity because no contigs shared>! kb at &&–,&%nt identity).(e remaining #*" viruses shareup to ! kb at up to &%%nt identity, with some smaller sharedregions at higher identity.
To determine whether or not a given virus was present ina given sample, we used the #*" viral contig sequences >#" kbas references for fragment recruitment (i.e., recruitment of

Archaea $
0
10
20
30
40
50
VC onlyVC (plus 0.1)
0.8 and/or 3 only0.1 only
Viru
ses d
etec
ted
J200
7At1
J200
7At2
J201
0Bt2
J201
0Bt3
J201
0A(a)
0
10
20
30
40
50
VC onlyVC (plus 0.1)
0.8 and/or 3 only
Viru
ses d
etec
ted
J200
7At1
J200
7At2
J200
9B
J201
0Bt2
J201
0Bt3
J201
0Bt4
J201
0A
(b)
0
0.5
1
1.5
2
J200
7At1
J200
7At2
J200
9B
J201
0Bt2
J201
0Bt3
J201
0Bt4
J201
0A
Det
ecte
d vi
rus r
atio
(%!l
ter/V
C%)
(c)
F01234 #
Illumina sequencing reads or equivalent #"" bp read frag-ments from other sequencing technologies, as described in[%]), using the Burrows-Wheeler Aligner (bwa) with defaultparameters [!*]. We required at least #x read coverage acrossat least '"%of a given reference contig sequence for detection.For hierarchical clustering (Pearson correlation, average link-age clustering), the number of reads that mapped to a givenviral contig sequence in a given samplewas normalized by thelength of the viral sequence and the number of reads in thesample, as described previously [%].
'.". Generation of !(S rRNA Gene Data and Calculations ofHost Relative Abundance. To generate a reference databaseof #/S rRNA gene sequences, we used the EMIRGE algo-rithm [!'] to reconstruct near full-length #/S rRNA genesfrom Illumina metagenomic data. All ".# and ".&!m )lters,from which DNA was Illumina sequenced and from whichviral concentrate DNA was also sequenced from the samesample, underwent EMIRGE analysis in order to generate areference #/S rRNA gene database for the LT system. (efollowing )lter sample metagenomes were EMIRGE ana-lyzed: !""%At# (".&!m), !"#"Bt$ (".& !m), !"#"Bt# (".# !m),
!"#"Bt! (".# !m), !"#"Bt$ (".# !m), and !"#"A (".# !m). Weclustered all EMIRGE-generated #/S rRNA gene sequencesat ,%% nt identity, using UCLUST [!/], resulting in adatabase of #"# #/S rRNA gene sequences. Taxonomy wasassigned to theseOTUs, using the SILVA Incremental Aligner(SINA) [!%] on the SILVA website [!&, !,]. Using bwa withdefault parameters [!*], wemappedmetagenomic reads (splitinto #"" bp lengths to limit biases associated with di+erentsequencing technologies, as described above and in [%]) fromall )lter samples to the reference database of #"# OTUs,in order to generate relative abundance estimates for eachOTU across samples. For a given OTU to be detected in agiven library, we required ##x coverage across #%"% of thelength of the EMIRGE-generated #/S rRNA gene sequence.In order to account for di+erences in sequencing throughput,we used relative abundances; that is, we estimated the percentabundance of each OTU in each sample as the number ofreads that mapped to that OTU divided by the total numberof reads that mapped to any OTU in that sample times#"". (ose relative abundances were used for hierarchicalclustering (Pearson correlation, average linkage clustering) #

* Archaea
0 10000 500000
J2010A
VCJ2007A
t2VC
A2007A
t20.8
J2007A
t20.1
A2008A
t20.8
J2009B
t10.8
J2009B
t20.8
J2009B
t30.8
A2008A
t13
J2009A
t10.1
J2009A
t20.1
J2009A
t10.8
J2009A
t20.8
J2010A
0.1
J2007A
t10.1
J2007A
t20.8
J2007A
t10.8
J2010A
3A2
007A
t10.1
A2007A
t10.8
A2007A
t13
A2007A
t23
J2010B
t30.8
J2010B
t23
J2010B
t43
J2007A
t1VC
J2009B
VCJ2010B
t20.1
J2010B
t3.5
0.1
J2010B
t10.1
J2010B
t30.1
J2010B
t3VC
J2010B
t1VC
J2010B
t2VC
J2010B
t4VC
(a)
0 2 20
A2007A
t13
IIIA2
007A
t20.1
Sang
A2007A
t10.1454
A2007A
t10.8454
A2007A
t20.8
Sang
A2007A
t23
IIIJ2007A
t10.1
Sang
J2007A
t20.1
Sang
J2010A
0.1
IIIJ2010B
t20.1
IIIJ2010B
t3.5
0.1
IIIJ2010B
t10.1
IIIJ2010B
t30.1
IIIJ2007A
t20.8
Sang
A2008A
t20.8454
J2009B
t10.8454
IIIJ2009B
t20.8454
J2009B
t30.8454
J2009A
t20.1454
J2009A
t10.1454
J2009A
t10.8454
J2009A
t20.8454
J2007A
t10.8
Sang
IIIA2
008A
t13
IIIJ2010A
3III
J2010B
t30.8
IIIJ2010B
t23
IIIJ2010B
t43
III
(b)
F01234 !
and appear in Table S# (see Supplementary Material availableonline at http://dx.doi.org/#".##''/!"#$/$%"&%#).
(ese sequences (#*" viral contigs >#" kb and #"# #/SrRNA gene sequences) have been submitted to NCBI underBioProject accession no. PRJNA&#&'#.
!'.&. Correlation with CRISPR Spacers. We used Crass [$"] toidentify clustered regularly interspaced short palindromicrepeat (CRISPR) and spacer sequences in each sample. Forthis analysis, we considered all sequenced )lter sizes fora given water sample together (i.e., ".#, ".&, and $." !m)lters). Using BLASTn with an $-value cuto+ of 1% & 10, weassessed the number of CRISPR spacers from each samplethat matched (#) any of the #*" viral contig sequences >#" kb,(!) reads from viral concentrates collected from the samesample (applicable only to the eight samples from whichviral concentrates were sequenced), and ($) reads from viralconcentrates collected from other samples.
3. Results and Discussion$
* ".!. Relative Abundances of Viral Populations across SizeFractions. Contigs >#" kb from #"' new viruses were recon-structed, increasing the number of genomically characterized
viruses from Lake Tyrrell (LT), Victoria, Australia, from $'to #*". (e #*" contigs, including seven previously reportedcomplete genomes, range in size from #","'" to ,$,!&$ bp.Weanalyzed the relative abundances of these #*" viral genotypesin metagenomic libraries from viral concentrates and )ltersize fractions (".#–".&, ".&–$.", and $."–!"!m) across timeand between locations in LT. As few as *! and as many as ##/viruses were detected in a given sample (any size fraction),and more viruses were detected in samples from which viralconcentrates were sequenced. (ough we acknowledge thata comparison of #/S rRNA gene microbial OTUs to >#" kbviral contig OTUs is an imperfect proxy for comparing thediversity of these groups, we )nd approximately #" viralpopulations in the viral concentrate size fraction per hostOTU (any )lter size fraction) in most samples, suggest-ing that planktonic virus diversity and host diversity scaleapproximately with abundance, which has previously beenestablished to be approximately #" : # in most environments(e.g., [$#]).
In an attempt to distinguish among free (planktonic)viruses physically trapped on )lters and host-associatedviruses (i.e., active viruses and/or proviruses), we assumedthat the ".& and $." !m )lter pore sizes would be too largeto retain signi)cant numbers of viral particles and should
Archaea '
0 10 20 30 40 50 60 70 80 90 100
A2007At1
J2009At1
J2009At2
J2010A
J2010Bt1
J2010Bt2
J2010Bt3.5
J2010Bt3
0.1 !
m !
lter s
ampl
e
(a)
0 10 20 30 40 50 60 70 80 90 100
A2007At1A2008At2J2007At1J2009At1J2009At2J2009Bt1J2009Bt2J2009Bt3J2010Bt3
0.8 !
m !
lter s
ampl
e
(b)
0 10 20 30 40 50 60 70 80 90 100
A2007At2
A2008At1
J2010A
J2010Bt2
J2010Bt4
(%)
Halorubrum 1Halorubrum 2Halorubrum 3Halorubrum 4Halorubrum 5Halorubrum 6Haloquadratum 1Halorubrum 7Haloquadratum 2Haloquadratum 1walsbyi
Haloquadratum 2walsbyiHaloquadratum 3Haloquadratum 4Haloquadratum 5Haloquadratum 3walsbyiHaloquadratum 6Candidatus NanosalinaUnclassi!ed HalobacteriaceaeHalorhabdus 1Halorhabdus 2
Halorhabdus 3Halorhabdus 4HalorientalisHalogranumHalobellusCandidatus NanosalinarumSalinibacter 1Salinibacter 2Unclassi!ed Sphingobacteriales
3 !m
!lte
r sam
ple
(c)
F01234 $
predominantly include host-associated viruses. We assumedthat the ".# !m )lters could retain both host-associated andplanktonic viruses and that the viral concentrates ($" kDa–".# !m size fraction) should generally exclude host cells andbe dominated by planktonic viruses. (erefore, we consid-ered viral detection in libraries from three )lter size groupsseparately: (#) viral concentrates, (!) ".# !m )lters, and ($)".& and/or $." !m )lters. For )ve samples from which at least
one library from each of those groups was sequenced, thenumber of viruses detected only in the viral concentrateswas always higher than the number detected only on )lters(Figure #(a)).(at trend is robust to the addition of twomoresamples, from which libraries from viral concentrates andat least one ".& and/or $."!m )lter sample were sequenced(Figure #(b)). Although we detected more unique viruses inthe viral concentrates, the ratio of total viruses detected on ".&

/ Archaea
0
250
500
750
1000
1250
1500
1750
2000
J2007A timeseries
J2010B timeseries
All VC timeseries
TotalSingle sample or seriesTwo samples or series
"ree samples or seriesFour samples or series
Spac
er h
its to
VCs
(a)
0
2
4
6
8
10
12
14
16
18
Hits to same sampleMin. hits to other samplesMax. hits to other samples
Spac
er h
its to
VCs
(%)
J200
7At1
J200
7At2
J200
9B
J201
0Bt1
J201
0Bt2
J201
0Bt3
J201
0Bt4
J201
0A
(b)
0
2
4
6
8
10
12
14
16
18
20
Min. hits to other samplesMax. hits to other samples
Spac
er h
its to
VCs
(%)
A200
7At1
A200
7At2
A200
8At1
A200
8At2
J200
9At1
J200
9At2
J201
0Bt3
.5
(c)
F01234 *
or $." !m )lters, relative to total viruses detected in the viralconcentrates, tended to be approximately equal (Figure #(c)),meaning that the richness of viruses in viral concentratestended to be similar to viral richness in the host-associatedsize fractions.
We used hierarchical clustering (Figure !(a)) to deter-mine whether patterns in the relative abundances of the#*" viruses (i.e., the #*" viral contigs >#" kb) could beobserved, according to season, )lter size, and/or sample site.No universal patterns were observed, but there was some
clustering according to)lter type and/or for samples collectedfrom the same site during the same week. Speci)cally, allfour viral concentrate samples from January !"#", site Bclustered together, and they clustered more closely with allof the ".# !m )lters from the same time series than with otherviral concentrate samples. Two additional viral concentratesamples collected during the same season but at di+erent sitesand years (J!""%At# and J!"",B) complete a larger clusterfor that group, indicating similarity among planktonic viralfractions, relative to viruses on larger )lter sizes. Interestingly,

Archaea %
T5674 #: Sequencing and sample information, all )lter Sizes and viral concentrates.
Sample Date Time ' (∘C) pH TDS Filter Sequencing Library Reads(wt%) size technology type(s)
J!""%At# Jan. !$, !""% 888 !! %.! $#".# Sanger &–#" kb /'"'//".& Sanger, Illumina fosmid, &–#" kb, and #"" cycles SR !!#,!$,&VC Illumina #"" cycles PE !*$/$$"
J!""%At! Jan. !', !""% 888 !& %.# $#".# Sanger &–#" kb ,"'#*!".& Sanger fosmid, &–#" kb &$!&&"VC Illumina #"" cycles PE %$$"",,
A!""%At# Aug. %, !""% 888 !* nm !'".# *'* SR '''&,&!".& *'* SR '$$$'$!$ Illumina #"" cycles SR #!,%&#"
A!""%At! Aug. ,, !""% 888 !$ nm !'".# Sanger &–#" kb #!/",%//".& Sanger &–#" kb %*/$,*$ Illumina #"" cycles SR $,*,*!%
A!""&At# Aug. ##, !""& 888 #! %.! !' $ Illumina #"" cycles SR #'%&/"'/A!""&At! Aug. #!, !""& 888 ## nm !' ".& *'* SR and PE #"$',!&"
J!"",At# Jan. $, !"", 888 !" % !& ".# *'* SR and PE /$"$!&$".& *'* SR ',!"!%/
J!"",At! Jan. %, !"", 888 !% /., !% ".# *'* SR ''#,,*/".& *'* SR /#&#'**
J!"",Bt# Jan. ', !"", %:!# #& /., !* ".& *'*, Illumina SR, #"" cycles SR /&***$/J!"",Bt! Jan. ', !"", 888 $" %.# !/ ".& *'* SR %$%!#',J!"",Bt$ Jan. ', !"", 888 $/ % !% ".& *'* SR %'*/*!&J!"",B∗ Jan. ', !"", VC Illumina #"" cycles PE #,'/%*/&
J!"#"Bt# Jan. %, !"#" %:*' !" %.! $! ".# Illumina #"" cycles PE #$!#$!**VC *'* SR !$%$"!#
J!"#"Bt! Jan. %, !"#" 888 $! %.$ $/".# Illumina #"" cycles PE !%$""/$*$ Illumina #"" cycles PE !$$%'$#'VC Illumina #"" cycles PE $$#!%&%
J!"#"Bt$ Jan. &, !"#" &:"" !# %.! $*".# Illumina #"" cycles PE $&!&%,/&".& Illumina #"" cycles PE #$&"&',,VC *'* SR !!*$,#/
J!"#"Bt$.'∗∗ Jan. ,, !"#" 888 *' %.# !% ".# Illumina #"" cycles PE !#%*%/,!
J!"#"Bt*∗∗ Jan. #", !"#" 888 $$ %.! $! $ Illumina #"" cycles PE #'*/'//*VC Illumina #"" cycles PE ,/#"!$$
J!"#"A Jan. #", !"#" 888 $% %.# $'".# Illumina #"" cycles PE '!'!"$!&$ Illumina #"" cycles PE /&'*$'&VC Illumina #"" cycles PE ,!/&$&*
VC: viral concentrate.Nm: not measured.PE: paired-end sequencing.SR: single-read sequencing.∗VC from J!"",B is a pool of DNA from three viral concentrates collected throughout a single day.∗∗Tomaintain consistent sample naming with previous publications, we are retaining sample names that were based on consecutive viral concentrate samples.Sample $.' was actually collected fourth in the series and sample * was ).h.
the remaining two viral concentrate samples (the only samplecollected in January !"#" from site A and another samplefrom site A collected in January !""%) clustered separatelyfrom each other and from the rest of the dataset. Overall, thissuggests that viral concentrates represent a di+erent part ofthe viral community than is sampled by other size fractions,particularly fractions >".&!m. Interestingly, this is despite
the similarity in richness described above (i.e., the numberof viral OTUs remains relatively similar across samples, butthe composition di+ers in viral concentrates, relative to hostsize fractions). '
Although the most obvious trend in the viral assemblagehierarchical clustering analysis was the separation of viralconcentrates from other )lter sizes described above, some

& Archaea
clustering was observed for ".#, ".&, and $."!m )lter sizefractions. Speci)cally, all four )lter samples collected fromJanuary !"", site A cluster together, as do all of the ".& and$." !m )lters from January !"#" site B, suggesting stability ofthe active viral and/or proviral assemblages over four daysin both cases. Although the ".# and ".& !m )lter samplescollected in August !""% from site A time # cluster together,they are separate from )lters of the same size collected twodays later, suggesting a shi. in the active viral assemblageover days or possibly the induction of proviruses on thattimescale. Together, these data suggest that, although someturnover in active viruses and/or proviruses was observed,most active viruses and/or proviruses were stable within thearchaea-dominated LT system over days.
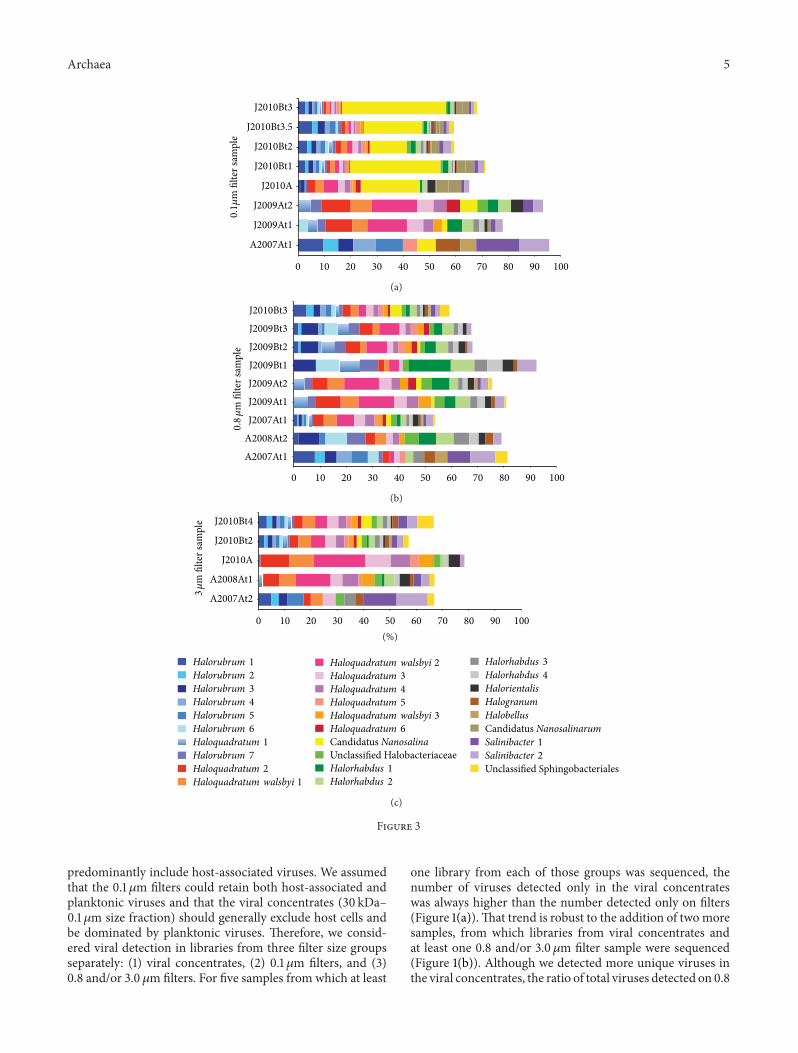
".'. Archaeal and Bacterial OTUs. We also characterized therelative abundances of potential host OTUs across time andbetween sites in the LT system. Of #"# total archaeal andbacterial #/S rRNA gene OTUs at ,%% nt identity, !, weredetected at '% abundance or higher on any )lter in anysample. For easier visualization, Figure $ shows only those !,OTUs, and it includes only samples from which at least )veof those OTUs were detected.
In the )lter size-resolved plots (Figures $(a)–$(c)), it isclear that even the most abundant archaeal groups changessigni)cantly over time and space, in terms of both pres-ence/absence and relative abundance. For example, the rela-tive proportions ofHalorubrum-like andHaloquadratum-likeOTUs tend to di+er signi)cantly across samples, from almostexclusively Halorubrum-like organisms (e.g., in the August!""% time series) to almost exclusively Haloquadratum-likeorganisms (e.g., in the January !"",, site A, time seriesand in January !"#", site A). Some changes in the relativeabundances of these groups can even be observed overhours on ".&!m )lters from the January !"",, site B, timeseries (Figure $(b)). A signi)cant change in temperature wasmeasured between the)rst and second samples of the January!"",, site B, time series (Table #), and we hypothesize thatthis shi. in community structure may mark a response tothe temperature change. With the exception of the January!"#", site B, time series, the diversity and relative abundanceof Haloquadratum-like and Halorubrum-like organisms aregenerally anticorrelated, suggesting that these archaea maycompete for a similar niche in the LT system. (e obser-vation of a relatively low diversity and abundance of Halo-quadratum-like organisms in some samples (i.e., A!""%At#-t!, A!""&At!, and J!"",Bt#) suggests that Haloquadratumspecies are more dynamic in hypersaline systems than hasbeen previously appreciated [$!].
In addition to trends within organism types, speci)cOTUs also exhibited interesting dynamics. For example,OTUHaloquadratum walsbyi !, which belongs to a species gener-ally considered to be among the most abundant organisms inhypersaline lakes [$!], is at relatively low abundance or notdetected on ".& and $." !m )lters from August !""%, site A,and January !"#", site B, though it is at high abundance inthat size fraction at site A in January !"", and January !"#".Interestingly, that OTU also appears dynamic on a days scale
in August !""& at site A, appearing at high abundance on the$." !m )lter at time # but not detected on the ".& !m )lterat time !. (ree OTUs, including two related to Salinibacter,weremore abundant in theAugust !""%, site A, time series (all)lter sizes) than in any other sample.(e Nanohaloarchaeon,Candidatus Nanosalina [!#], was detected at reasonably highabundance on all ".#!m )lters from January !"#" sites A andB but was less abundant at other sites and times (Figure $(a)).Consistent with the small cell size for that organism, itand the other abundant Nanohaloarchaeal OTU, CandidatusNanosalinarum,were found at low abundance or not detectedon )lters larger than ".#!m.
Overall, analyses of the !, most abundant archaealand bacterial OTUs indicate dynamics at the populationlevel across time and space, particularly on months-to-yearstimescales, with some dynamics indicated over hours todays. (ese results indicate dynamics in the most abundantarchaeal and bacterial populations at the taxon level, incontrast to a previous study, which predicted that the mostabundant microbial taxa were stable over timescales ofweeks to one month in a hypersaline crystallizer pond nearSan Diego (SD), CA, USA [/]. (at study was based ontaxonomic a9liations predicted from "#"" bp metagenomicreads. It is possible that di+erences in sampling timescales,geochemistry, and/or community composition could resultin true di+erences in microbial dynamics between thesetwo systems. Also, LT microbial eukaryotic communities aredominated by a predatory Colpodella sp., and it is unclearwhat e+ect top-down grazing may have on shi.s in bacterialand archaeal community structure [!"]. However, the SDstudy also suggested stability of viral populations in thatsystem, and we previously demonstrated through a reanalysisof the SD data that the most abundant viral populationsfrom that study were in fact dynamic [&], so it is possiblethat di+erent sequencing and analytical methods would haverevealed dynamics in archaeal and bacterial populations atthe SD site as well. Unfortunately, a reanalysis of the existingSD data using the methods in this study is not possible,due to the incompatibility of *'* sequencing reads withthe EMIRGE algorithm, which is designed to reconstructnear-complete #/S rRNA genes from paired-end Illuminametagenomic data.
Based on the relative abundances of the #"# archaeal andbacterial OTUs (including lower abundance LT organisms),we used hierarchical clustering to determine whether simi-larities in overall archaeal and bacterial community structurewould group LT samples according to season, sample type,or )lter size (Figure !(b)). In general, archaeal and bacterialcommunities sampled from the same location over daysclustered more closely than did viral assemblages from thesame samples (see above and Figure !(a)), indicating greaterstability of host populations than viral populations over days.(e only samples for which no within-time series (i.e., daysscale) clustering was observed for host communities weresamples from August !""&, site A. For that time series,DNA from di+erent )lter sizes was sequenced from eachsample, and di+erent sequencing technologies were used (*'*and Illumina), so it is possible that similarities between thesamples exist but were masked by di+erent methodologies.

Archaea ,
However, interestingly, all samples and )lter sizes from theAugust !""%, site A, time series clustered together, includingDNA from ".#, ".&, and $." !m size fractions sequenced by allthree technologies (Sanger, *'*, and Illumina).(is indicatesa distinct community at site A in August !""%, which wasstable over days and robust to methodological di+erences inlibrary construction and sequencing.
In some cases, the archaeal and bacterial communitieson the ".#!m )lters clustered together and separately fromother )lters from the same time point, re-ecting enrichmentfor Nanohaloarchaea. Speci)cally, both ".#!m )lters fromJanuary !""%, site A, cluster together, and they belong toa larger cluster that includes all ".#!m )lters from January!"#", site B. (e prevalence of Nanohaloarchaea in thesesamples but not in others is evident in Figure $(a), which isbased on the !, most abundant OTUs described above. Inall other cases, the ".# !m )lters clustered with larger )ltersfrom the same time series. For the January !"",, site B, single-day time series, from which only DNA from ".&!m )lters(and a viral concentrate) was sequenced, the morning sampleclusters separately from the a.ernoon and evening samples,which cluster together, indicating a shi. in communitystructure over a single day.(is is consistent with the shi. inrelative abundance of Halorubrum-like and Haloquadratum-like organisms from that time series (Figure $) and may berelated to a temperature shi., as suggested above.
Interestingly, a seasonal trend was not indicated in eitherthe analysis of the !, most abundant OTUs or in the #"#OTU hierarchical clustering analysis (seasonal trends inviral populations would be di9cult to infer, as no viralconcentrates were sequenced from winter samples). Samplesfrom site A in August !""% have quite di+erent archaeal andbacterial community structures from samples at the same sitein August !""&, and January samples from site A are alsofairly distinct year to year. Although some samples from siteB, collected in January !"", and January !"#", might appearto be the exception (Figure $(b)), there was as much of a shi.in community structure over hours in January !"", at site Bas there was across samples collected over years.
As with any metagenomic study, we cannot say forcertain to what extent abundance in metagenomic librariesre-ects true abundances. However, we have taken care toavoid biases that are o.en associated with sequencing-basedcommunity analyses. Speci)cally, by focusing on #/S rRNAgene sequences from metagenomic data, we have avoidedPCRampli)cation biases in our estimates of archaeal and bac-terial dynamics. Similarly, we were able to generate enoughviral concentrate metagenomic DNA for sequencing withoutmultiple-displacement ampli)cation (MDA),which is knownto generate signi)cant biases, especially in viralmetagenomes(discussed in [$$, $*]).
".". CRISPR Analyses. Using the Crass algorithm [$"], weidenti)ed '*, unique repeats and &,",' unique spacersequences from clustered regularly interspaced short palin-dromic repeat (CRISPR) regions in the LT dataset. Apartfrom a single sample from January !"",, site A, the onlylibraries with spacers that matched any of the #*" complete
and near-complete viral genomes were those from samplesfrom which viral concentrates were also sequenced (Table !).(is may indicate that di+erent planktonic viral assemblagesexisted in LT at time points from which viral concentrateswere not sequenced, which would be consistent with theviral population dynamics observed across the sequencedviral concentrates [%]. Notably, no spacers from LT Augustmetagenomesmatched any of the #*" viruses, consistent witha possible seasonal shi. in viral community structure (noviral concentrates were sequenced from August samples), ashas been observed in marine systems (e.g., [$']). However, aconcomitant seasonal shi. in host community structure wasnot supported, so it is also possible that the August samplescould harbor di+erent planktonic viral assemblages each year.
(e only spacer matches to any of the seven previouslydescribed complete LT viral and virus-like genomes [%] wereto LTV! (%/,%#/ bp) and LTVLE$ (%#, $*# bp) in librariesfrom three samples from the January !"#", site B, time series(Table !). Of the seven viral concentrate genomes, those twoachieved the highest abundance by approximately one orderof magnitude [%], and they were at their most abundancein the time series from which spacers targeting them weredetected. (is indicates that LT CRISPRs can actively targetabundant, coexisting viruses, consistent with previous obser-vations of CRISPR sampling of coexisting viruses in an acid-mine drainage system [,, ##]. However, it should be noted thatthe vast majority of spacers do not match any of the #*" near-complete viral genomes, which we assume to be among themost abundant viruses because they assembled signi)cantly.(is suggests that most CRISPRs target rare viruses. An alter-native explanationmight be a preponderance of inactive, ves-tige CRISPR regions, but if that were the case, then we wouldexpect to see the same CRISPR spacers duplicated acrosssamples (i.e., CRISPR regions would be clonally inheritedover multiple generations, as opposed to actively integratingnew spacers on subgenerational timescales). Given that only$'$ spacer sequences were duplicated across samples, relativeto &,",' unique spacer sequences in the dataset, we infereither that most CRISPR regions were highly dynamic and/orthat we detected di+erent CRISPR regions over time, due toshi.s in host community structure. Regardless, there is a highdiversity of spacers in the LT system, consistent with the highdiversity of viruses.
Of the CRISPR regions that had matches to the #*"LT viral contigs, only )ve had repeats with matches toassemblies from ".#, ".&, and/or $." !m )lter sequences (amatch would be indicative of the host organism), and nonematched any of the #! complete and near-complete bacterialand archaeal genomes assembled from the LT January !""%site A time series [$/]. (ree matches to the January !"#"site B assemblies (Andrade et al., unpublished data) wereto contigs that could only be identi)ed at the order level ofHalobacteriales, based on best BLAST hits throughout thecontigs, and one repeat matched a contig predicted to belongto a Natronomonas-like organism. (e most interestingCRISPR repeat sequence match was to a contig from a likelyNanohaloarchaeon, based on the taxonomy of best BLASThits throughout the contig. A spacer associated with thatrepeat matched LTVLE$, a virus or plasmid described in

#" Archaea
T5674 !: CRISPR analyses by sample.
Sample Unique Unique Hits to Viral contig Predictedrepeats spacers #*" viruses match(es) host
J!""%At# /% /&# # sca+old #/J!""%At! &! /$$ # Contig,,,""*A!""%At# $" #*# "A!""%At! /* ''* "A!""&At# !" !%' "A!""&At! !$ #!# "J!"",At# *# #/$ # sca+old ##%J!"",At! *" #%$ "J!"",B !, ##* "J!"#"Bt# !! '"# "
J!"#"Bt! %, #%,& *
LTV!LTV!
LTVLE$Contig,,,""*
J!"#"Bt$ '& ##%# &
LTVLE$Contig##"""', Natronomonas-likeContig,,&,%'sca+old ''sca+old !,LTVLE$LTVLE$ NanohaloarchaeaLTVLE$ Nanohaloarchaea
J!"#"Bt$.' /" ,$" *
LTV!LTVLE$ Nanohaloarchaea
Contig,,,""*Contig,,&,%'
J!"#"Bt* *$ &'$ # Contig,,,""*J!"#"A !" $*" "
[%], strongly suggesting that LTVLE$ is a virus or plasmidof the Nanohaloarchaea. Interestingly, while LTVLE$ wasonly at high abundance in viral concentrates from theJanuary !"#" site B four-day time series, from which theonly spacers targeting it were identi)ed, the most abundantNanohaloarchaea, CandidatusNanosalina [!#], was detectedin most LT samples and is as abundant at site A as it is atsite B (isolated pools $""m apart) in !"#" (Figure $(a)).(issuggests that the Nanohaloarchaea at site B may have beenadapted locally to the presence of abundant LTVLE$ at site Bin January !"#"./
BLAST searches of all &,",' LT CRISPR spacers and '*,repeat sequences revealed essentially no hits to the NCBI nrand environmental databases, and no hits were detected forany of the repeat sequences.(ree spacers had #""%matchesHomo sapiens, indicating that they are likely either to beerroneous spacers or representative of mobile genetic ele-ments that are not present in public databases. Overall, thesedata demonstrate that the LT CRISPR spacers target mobilegenetic elements that have not been previously identi)ed,suggesting that LT archaea may be highly adapted to localviral predation, though we cannot rule out the possibility of
public database bias (i.e., perhaps LT CRISPRs could targetviruses from other locations that are not represented inpublic databases). In support of local adaptation, despite therelatively small number of spacer matches to the #*" LT viralcontigs >#" kb, #,%!, spacers (!#%) had matches to unassem-bled LT viral concentrate reads. (is may indicate selectionfor CRISPR spacers that target locally adapted, potentiallycoexisting viruses, consistent with previously observed localCRISPR adaptation to coexisting viruses in an acid-minedrainage system and in geographically distinct sludge biore-actors [&, ,]. Because these matches are to unassembledreads, we infer that most targeted LT viruses are at relativelylow abundance, at least at the times and locations sampledby this study (with the exception of LTV! and LTVLE$,as described above). (is may indicate successful CRISPR-mediated maintenance of viral populations at abundanceslow enough to preclude genomic assembly.
In terms of the timescales on which CRISPR spacersare retained and match coexisting, low-abundance virusesin the LT system, we found several lines of evidence thatsupport the stability of many CRISPR spacers and low-abundance viruses over all timescales sampled by this study

Archaea ##
(up to three years). LT spacers tended to match reads frommultiple viral concentrate metagenomes collected over thecourse of days almost as o.en as (January !""%, site A) ormore o.en than (January !"#", site B) they matched readsfrom a single viral concentrate metagenome from the sametime series (Figure *(a)). (is suggests that many spacersand their targeted, low-abundance viruses are stable overdays. On the timescale of years, spacers and their targetedcoexisting viruses were found most o.en across more thanone of the four sites and times sampled by viral concentrates(January !""%, site A; January !"",, site B; January !"#",site A; January !"#", site B). Similarly, in all eight samplesfrom which viral concentrates were sequenced, nearly asmany (and o.en more) spacers had hits to viral concentratereads from other samples as matched viral concentrate readsfrom the same sample (Figure *(b)). A similar number ofmatches to the eight viral concentrates were observed forsamples from which viral concentrates were not sequenced(Figure *(c)). Overall, these results demonstrate that CRISPRregions generally retain spacer targets against low-abundanceviruses on timescales of #–$ years. We also infer that low-abundance viruses are likely more stable in the LT systemthan their highly dynamic, higher abundance counterparts.
4. Conclusions
Both virus (#*" contigs >#" kb) and host (#"# #/S rRNA geneOTUs) assemblage structures were highly dynamic over timeand space in the archaea-dominated LT system, particularlyover months to years. However, archaeal and bacterial pop-ulations were generally more stable than viral populations,and lower abundance viruses were inferred to be more stablethan abundant viruses. Filter size-resolved analyses of viralpopulations revealed di+erent viral assemblages in the plank-tonic and host-associated (i.e., active and provirus) fractions,suggesting that a higher diversity of viruses may have thepotential to infect than are actively infecting at any given time.Consistent with that hypothesis, CRISPR analyses revealedpersistent targeting of lower abundance viral populations,along with a high diversity of spacer sequences. However,interestingly, the most abundant viruses ("!% of the viralcommunity, relative to (".#% for most viruses, [%]) werealso targeted by CRISPRs, indicating that archaeal hostsstrike a balance between protection against persistent, low-abundance viruses and those viruses potentially abundantenough to a+ect catastrophic changes in host communitystructure.
Acknowledgments
Funding for this work was provided by the National ScienceFoundation Award "/!/'!/ and DE-FG"!-"%ER/*'"' fromthe Department of Energy. (anks to Cheetham Salt Works(Victoria, Australia) for permission to collect samples; JohnMoreau, Jochen Brocks, and Mike Dyall-Smith for assistancein the )eld; Matt Lewis and the J. Craig Venter Institute(JCVI) for library construction and sequencing; ShannonWilliamson and Doug Fadrosh (JCVI) for training Joanne
B. Emerson in virus-related laboratory techniques; ConnorSkennerton and Gene Tyson for early access to and assis-tance with the Crass program; Christine Sun for helpfuldiscussions; and two anonymous reviewers for constructivecomments that improved the paper.
References
[#] F. Rohwer, D. Prangishvili, and D. Lindell, “Roles of viruses inthe environment,”EnvironmentalMicrobiology, vol. ##, no. ##, pp.!%%#–!%%*, !"",.
[!] C. A. Suttle, “Marine viruses—major players in the globalecosystem,”Nature Reviews Microbiology, vol. ', no. #", pp. &"#–&#!, !""%.
[$] K. Porter, B. E. Russ, and M. L. Dyall-Smith, “Virus-hostinteractions in salt lakes,” Current Opinion in Microbiology, vol.#", no. *, pp. *#&–*!*, !""%.
[*] R.-A. Sandaa and A. Larsen, “Seasonal variations in virus-host populations in Norwegian coastal waters: focusing on thecyanophage community infecting marine Synechococcus spp,”Applied andEnvironmentalMicrobiology, vol. %!, no. %, pp. */#"–*/#&, !""/.
['] S. M. Short and C. A. Suttle, “Temporal dynamics of naturalcommunities of marine algal viruses and eukaryotes,” AquaticMicrobial Ecology, vol. $!, no. !, pp. #"%–##,, !""$.
[/] B. Rodriguez-Brito, L. Li, L. Wegley et al., “Viral and microbialcommunity dynamics in four aquatic environments,” ISMEJournal, vol. *, no. /, pp. %$,–%'#, !"#".
[%] J. B. Emerson, B. C. (omas, K. Andrade, E. E. Allen, K. B.Heidelberg, and J. F. Ban)eld, “Dynamic viral populations inhypersaline systems as revealed by metagenomic assembly,”Applied and Environmental Microbiology, vol. %&, pp. /$",–/$!", !"#!.
[&] V. Kunin, S. He, F. Warnecke et al., “A bacterial metapopulationadapts locally to phage predation despite global dispersal,”Genome Research, vol. #&, no. !, pp. !,$–!,%, !""&.
[,] A. F. Andersson and J. F. Ban)eld, “Virus population dynamicsand acquired virus resistance in natural microbial communi-ties,” Science, vol. $!", no. '&%,, pp. #"*%–#"'", !""&.
[#"] R. E. Anderson, W. J. Brazelton, and J. A. Baross, “UsingCRISPRs as ametagenomic tool to identify microbial hostsof a di+use -ow hydrothermal vent viral assemblage,” FEMSMicrobiology Ecology, vol. %%, no. #, pp. #!"–#$$, !"##.
[##] G. W. Tyson and J. F. Ban)eld, “Rapidly evolving CRISPRsimplicated in acquired resistance ofmicroorganisms to viruses,”Environmental Microbiology, vol. #", no. #, pp. !""–!"%, !""&.
[#!] J. F. Heidelberg, W. C. Nelson, T. Schoenfeld, and D. Bhaya,“Germ warfare in a microbial mat community: CRISPRs pro-vide insights into the co-evolution of host and viral genomes,”PLoS ONE, vol. *, no. #, Article ID e*#/,, !"",.
[#$] R. Barrangou, C. Fremaux, H. Deveau et al., “CRISPR providesacquired resistance against viruses in prokaryotes,” Science, vol.$#', no. '&#,, pp. #%",–#%#!, !""%.
[#*] F. J. M. Mojica, C. Dıez-Villasenor, J. Garcıa-Martınez, and E.Soria, “Intervening sequences of regularly spaced prokaryoticrepeats derive from foreign genetic elements,” Journal of Molec-ular Evolution, vol. /", no. !, pp. #%*–#&!, !""'.
[#'] R. Sorek, V. Kunin, and P. Hugenholtz, “CRISPR—a widespreadsystem that provides acquired resistance against phages inbacteria and archaea,” Nature Reviews Microbiology, vol. /, no.$, pp. #&#–#&/, !""&.

#! Archaea
[#/] B.Wiedenhe., S. H. Sternberg, and J. A. Doudna, “RNA-guidedgenetic silencing systems in bacteria and archaea,” Nature, vol.*&!, no. %$&', pp. $$#–$$&, !"#!.
[#%] R. T. DeBoy, E. F. Mongodin, J. B. Emerson, and K. E. Nelson,“Chromosome evolution in the (ermotogales: large-scaleinversions and strain diversi)cation of CRISPR sequences,”Journal of Bacteriology, vol. #&&, no. %, pp. !$/*–!$%*, !""/.
[#&] N. L. Held, A. Herrera, H. C. Quiroz, and R. J. Whitaker,“CRISPR associated diversity within a population of Sulfolobusislandicus,” PLoS ONE, vol. ', no. ,, Article ID e#!,&&, !"#".
[#,] N. L. Held and R. J. Whitaker, “Viral biogeography revealedby signatures in Sulfolobus islandicus genomes,” EnvironmentalMicrobiology, vol. ##, no. !, pp. *'%–*//, !"",.
[!"] K. B. Heidelberg, W. C. Nelson, J. B. Holm, N. Eisenkolb, K.Andrade, and J. B. Emerson, “Characterization of eukaryoticmicrobial diversity in hypersaline Lake Tyrrell, Australia,”Frontiers in Microbiology, !"#$.
[!#] P. Narasingarao, S. Podell, J. A. Ugalde et al., “De novometagenomic assembly reveals abundant novel major lineage ofArchaea in hypersaline microbial communities,” ISME Journal,vol. /, no. #, pp. &#–,$, !"#!.
[!!] Y. Peng, H. C. M. Leung, S. M. Yiu, and F. Y. L. Chin, “IDBA-UD: a de novo assembler for single-cell and metagenomicsequencing data with highly uneven depth,” Bioinformatics, vol.!&, no. ##, pp. #*!"–#*!&, !"#!.
[!$] M. Margulies, M. Egholm, W. E. Altman et al., “Genomesequencing in microfabricated high-density picolitre reactors,”Nature, vol. *$%, pp. $%/–$&", !""'.
[!*] H. Li and R. Durbin, “Fast and accurate short read alignmentwith Burrows-Wheeler transform,” Bioinformatics, vol. !', no.#*, pp. #%'*–#%/", !"",.
[!'] C. S. Miller, B. J. Baker, B. C. (omas, S. W. Singer, and J.F. Ban)eld, “EMIRGE: reconstruction of full-length ribosomalgenes from microbial community short read sequencing data,”Genome Biology, vol. #!, no. ', article R**, !"##.
[!/] R. C. Edgar, “Search and clustering orders of magnitude fasterthan BLAST,” Bioinformatics, vol. !/, no. #,, pp. !*/"–!*/#,!"#".
[!%] E. Pruesse, J. Peplies, and F. O. Glockner, “SINA: accurate high-throughput multiple sequence alignment of ribosomal RNAgenes,” Bioinformatics, vol. !&, pp. #&!$–#&!,, !"#!.
[!&] E. Pruesse, C. Quast, K. Knittel et al., “SILVA: a comprehensiveonline resource for quality checked and aligned ribosomal RNAsequence data compatible with ARB,” Nucleic Acids Research,vol. $', no. !#, pp. %#&&–%#,/, !""%.
[!,] C. Quast, E. Pruesse, P. Yilmaz et al., “(e SILVA ribosomalRNA gene database project: improved data processing andweb-based tools,” Nucleic Acids Research, vol. *#, pp. D',"–D',/,!"#$.
[$"] C. T. Skennerton, M. Imelfort, and G. W. Tyson, “Crass:identi)cation and reconstruction of CRISPR fromunassembledmetagenomic data,” Nucleic Acids Research, vol. *#, no. #", p.e#"', !"#$.
[$#] R. Danovaro, C. Corinaldesi, A. Dell’Anno et al., “Marineviruses and global climate change,” FEMSMicrobiology Reviews,vol. $', no. /, pp. ,,$–#"$*, !"##.
[$!] M. L. Dyall-Smith, F. Pfei+er, K. Klee et al., “Haloquadratumwalsbyi: limited diversity in a global pond,” PLoS ONE, vol. /,no. /, Article ID e!",/&, !"##.
[$$] M. B. Duhaime and M. B. Sullivan, “Ocean viruses: rigorouslyevaluating the metagenomic sample-to-sequence pipeline,”Virology, vol. *$*, pp. #&#–#&/, !"#!.
[$*] M. B. Duhaime, L. Deng, B. T. Poulos, and M. B. Sullivan,“Towards quantitative metagenomics of wild viruses and otherultra-low concentration DNA samples: a rigorous assessmentand optimization of the linker ampli)cation method,” Environ-mental Microbiology, vol. #*, pp. !'!/–!'$%, !"#!.
[$'] R. J. Parsons, M. Breitbart, M. W. Lomas, and C. A. Carlson,“Ocean time-series reveals recurring seasonal patterns of viri-oplankton dynamics in the northwestern Sargasso Sea,” ISMEJournal, vol. /, no. !, pp. !%$–!&*, !"#!.
[$/] S. Podell, J. A. Ugalde, P. Narasingarao, J. F. Ban)eld, K. B.Heidelberg, and E. E. Allen, “Assembly-driven communitygenomics of a hypersalinemicrobial ecosystem,”PLoSONE, vol.&, Article ID e/#/,!, !"#$.

Archaea #$
Composition Comments
1. Please provide a short description (in paragraph style)of the Supplementary Material. This description along withthe Supplementary Material will be available online.
2. We removed the part after the highlighted one for thesake of clarity. Please check.
3. We redrew some parts in Figure 2. Please check.
4. We reduced the font size of some labels in Figure 2.Please check.
5. Please check the reformat of Tables 1 and 2.6. We made the highlighted change(s) for the sake ofconsistency. Please check.

Author(s) Name(s)
�� �� �� �� ����� �� ������ �� �������� ���� ��� ���� ���� �� �� �� � ��� ���� �������� �� ����� �� ��� �� �� �� ������� ���������
Author 1���� ����� ���� !"#��� ���� $����
Author 2���� ����� %��#��� ���� &����
Author 3���� ����� !��� '"#��� ���� (�����
Author 4���� ����� &���#��� ���� �����
Author 5���� ����� $�� $"#��� ���� &���
Author 6���� ����� %��� !"#��� ���� )�����
Author 7���� ����� ������ *"#��� ���� !������
�� �� ���� �� �� ����� �� ��� ����� �� ���� �� +,'�- �+ � ,���� ��� '���������-�" +,'�- ���� �� ���� �� ��� ��������� ���� �� �������� �������������� �� ������� ����� �� ������ ���.� �������� �� ���������� �����"
(� ���� �� +,'�-/ ��� �� �� �� &������ 0 ��� �� ���� �11���"�������"���1� ���1��� �� 2������ � (��3��� 4���� ��� ��� ��� ��� ����� �� ����3 �� �� +,'�- ���3 �� ���� �� �� ��" (��� ���3 ���� ��3 ��� �� �� +,'�- ����� �� ��� ���� � ��� �� ��� ��������� �� ������" +�� ��� ��� ��� ��/ ��� �� +,'�- ���� � ���� �� �� 2������ �(��3��� 4���� �������������"




















