
Minh Quan HO Optimisation de transfert de données pour les ...

1
Vers différents types de règles pour les données d’expression de gènes Application à des données de tumeurs mammaires Marie Agier *,** — Jean-Marc Petit ** — Valérie Chabaud * — Christian
Pradeyrol * — Yves-Jean Bignon *** — Véronique Vidal *
* DIAGNOGENE 83, avenue Charles de Gaulle, 15000 Aurillac
** LIMOS, UMR 6158 CNRS Univ. Clermont-Ferrand II, 63177 Aubière
*** Laboratoire d’Oncologie Moléculaire, UMR 484 INSERM Centre Jean Perrin, 63011 Clermont-Ferrand
[email protected] ; [email protected]
RÉSUMÉ. Les puces à ADN permettent de mesurer le niveau d’expression de milliers de gènes d’un tissu donné dans une seule expérience. De part la nature de ces données (beaucoup de gènes et peu d’expériences), de nouvelles techniques d’analyse de données de biopuces doivent être développées. Dans cet article, nous proposons une technique d’interprétation des données basée sur la notion de règles. L’originalité de notre approche réside dans la définition de trois sémantiques différentes pour les règles répondant à des objectifs biologiques différents. Nous avons intégré ces propositions dans un logiciel libre existant nommé MeV de l’environnement TIGR dédié à l’interprétation des données de biopuces. Grâce à une interface graphique conviviale, les biologistes ont la possibilité d’utiliser nos propositions sur leurs données d’expression de gènes. Nous présentons les premiers résultats obtenus avec une sémantique particulière sur des données d’expression du cancer du sein.
ABSTRACT. Microarrays permit to measure the expression level of thousand genes simultaneously. Because of the nature of these data (many genes and few experiments), new microarray data analysis techniques must be developed. In this article, we propose a microarray data interpretation technique based on the notion of rules. The originality of our approach lies in the definition of three different semantic for rules answering each a particular biological question. We integrated these propositions into an open-source software named MeV of the environment TIGR devoted to the interpretation of microarray data. Thanks to a friendly graphic interface, the biologists have the possibility of using our propositions on their gene expression data. We present the first results obtained with a specific semantics on gene expression data of breast cancer.
MOTS-CLÉS : Puces à ADN, data mining, règles, implications, systèmes de fermeture.
KEYWORDS: Microarrays, data mining, rules, implications, closure systems.

2
1. Introduction
Ces dernières années, la technologie des puces à ADN ou biopuces, a suscité un vif intérêt de la part de la communauté scientifique et industrielle. Cette nouvelle
technologie promet en effet des perspectives sans précédent sur la compréhension
des mécanismes des êtres vivants, puisqu’elle permet de mesurer simultanément
l’activité de milliers de gènes.
Malgré la diversité des puces à ADN, leur coût encore élevé, les difficultés de
leur utilisation, la difficulté de conception des expérimentations et de leurs
interprétations, les chercheurs en biologie les considèrent comme une indispensable
méthode d’investigation (Soularue et al, 2002). La principale caractéristique des
données générées par les puces à ADN, appelées données d’expression de gènes, est qu’elles comportent peu d’expérimentations au regard du nombre de gènes testés
simultanément. De plus, les volumes de données à gérer peuvent être considérables
si l’on considère les autres sources d’information existantes en génomique, données
avec lesquelles il est important de coopérer. De nombreux challenges sont donc
posés aux informaticiens pour fournir des outils adéquats aux biologistes (Piatetsky
et al, 2003). On peut citer l’identification de gènes co-exprimés, la découverte de
groupes de gènes ayant des profils d’expression similaires, l’identification de gènes
dont les profils diffèrent suivant les caractéristiques biologiques ou l’étude de
l’activité des gènes sous différentes conditions de stress. Plus récemment, la
découverte de réseaux de régulation des gènes et la cartographie des données
d’expression selon la localisation chromosomique se sont ajoutées à cette liste.
Dans ce contexte, nous présentons une méthode de fouille de données spécifique
aux données d’expression et aisément utilisable et interprétable par les biologistes et
les médecins. Cette méthode prend ses racines dans la notion d’implication ou règle. Une implication est une expression de la forme X → Y, où X et Y sont des groupes
de gènes, la sémantique étant le sens que l’on souhaite donner à cette implication.
Par exemple, les règles d’association exactes (confiance à 100%) en fouille de
données ou les dépendances fonctionnelles en base de données sont deux types
d’implications. L’idée principale de notre approche est de proposer plusieurs
sémantiques pour les règles afin que les biologistes puissent choisir celle qui
correspond le mieux à leurs objectifs. Cette technique d’analyse apporte de
l’information complémentaire par rapport aux techniques classiques utilisées
jusqu’à présent pour l’analyse de données de biopuces, qui sont essentiellement des
méthodes de classification (Eisen et al, 1998, Kohane et al, 2003).
Exemple 1 Pour illustrer les différentes notions et montrer un exemple de données d’expression de gènes, le tableau 1 décrit les niveaux d’expression de huit
gènes (g1, g2, g3, g4, g5, g6, g7 et g8) pour six expériences (e1, e2, e3, e4, e5 et e6).
Cet exemple sera réutilisé tout au long de l’article.
Dans ce papier, trois principales sémantiques sont proposées pour répondre aux
besoins des biologistes : ils peuvent s’intéresser aux niveaux d’expression des

3
gènes, aux variations des niveaux d’expression des gènes ou à l’évolution des niveaux d’expression des gènes dans le cas de séries d’expériences temporelles.
r g1 g2 g3 g4 g5 g6 g7 g8
e1 1.9 0.4 1.4 -1.5 0.3 1.8 0.8 -1.4
e2 1.7 1.5 1.2 -0.3 1.4 1.6 0.7 0.0
e3 1.8 -0.7 1.3 0.8 -0.1 1.7 0.9 0.6
e4 -1.8 0.4 1.7 1.8 0.6 -0.4 1.0 1.5
e5 -1.7 -1.4 0.9 0.5 -1.8 -0.2 1.2 0.3
e6 0.0 1.9 -1.9 1.7 1.7 -0.5 1.1 1.3
Tableau 1. Exemple de données d’expression de gènes
D’un point de vue algorithmique, une caractéristique importante de notre
proposition est d’utiliser une approche identique quelle que soit la sémantique
choisie. Une fois la sémantique définie, il suffit de vérifier que celle-ci est « bien-
formée », c’est-à-dire qu’elle permet de définir un opérateur de fermeture. Une fois
les accès aux données effectués pour générer une famille génératrice du système de
fermeture sous-jacent, la génération des règles se fait indépendamment de la
sémantique, en se basant sur le calcul des transversaux minimaux (Mannila et al,
1994, Demetrovics et al, 1995). L’intérêt de l’approche est également de pouvoir
ajouter d’autres sémantiques sans difficulté majeure. A long terme, l’objectif
biologique de cette technique est de mieux comprendre le fonctionnement des gènes
pour ensuite retrouver des réseaux de régulation de gènes. La conjecture est de dire qu’un ensemble de règles permet de représenter un réseau de régulation.
Cette nouvelle approche a été implémentée dans une interface homme-machine
conviviale afin de la rendre utilisable par tout biologiste. Nous avons choisi de
l’intégrer comme un module dans un logiciel libre d’analyse de biopuces : TIGR
MeV (Saeed et al, 2003). Cet outil a l’avantage de pouvoir étudier des puces venant
de plusieurs plate-formes (Affymetrix, Stanford...) et de permettre l’utilisation de
plusieurs techniques éprouvées (classification hiérarchique, K-mean, Anova, ACP,
t-tests...). Notons aussi que les règles générées sont stockées dans une base de
données MySQL afin de permettre le requêtage par les biologistes. A terme, nous
intégrerons les informations disponibles sur les banques de données de gènes (e.g.
Swissprot) afin de faciliter l’analyse et de croiser les paramètres fonctionnels
connus des gènes avec les règles produites.
Une première application a été réalisée sur un jeu de données publiques, consti-
tué de biopuces analysant des ARN de tumeurs mammaires (Van’t Veer et al, 2002).
Les premiers résultats obtenus sont très encourageants puisque nous retrouvons des
règles connues des biologistes, qui ont été validées par des techniques plus
anciennes.
Etat de l’art
Les travaux réalisés dans ce domaine concernent essentiellement la génération
des règles d’association en fouille de données et la découverte des dépendances

4
fonctionnelles en bases de données (Mannila et al, 1994, Demetrovics et al, 1995,
Ganter et al, 1999, Caspard et al, 2003).
La génération de règles d’association est une méthode très populaire qui a attiré
un grand nombre de chercheurs ces dix dernières années (cf. (Han et al, 2000) pour
une vue générale). De façon assez surprenante, seulement peu d’équipes ont
appliqué ces techniques aux données d’expression de gènes (Becquet et al, 2002,
Berrar et al, 2002, Icev et al, 2003). Une phase de pré-traitement longue et
fastidieuse est nécessaire pour transformer les données d’expression en relation
binaire (cette étape est dans notre outil complètement transparent à l’utilisateur). De
plus, la génération des motifs fréquents étant un pré-requis à la génération des
règles, ces méthodes ont peu de chances de fonctionner sur les données
d’expression (le nombre de motifs fréquents peut être exponentiel en nombre de
gènes). Pour prendre en compte le petit nombre de lignes, nous avons choisi de ne
pas considérer de support minimum ce qui démarque notre approche des travaux
cités ci-dessus.
La découverte des dépendances fonctionnelles est moins populaire et n’a pas
reçu jusqu’à présent beaucoup d’attention de la part de la communauté de la fouille
de données. Des algorithmes performants existent pour inférer les dépendances
fonctionnelles, nous pouvons par exemple citer Tane (Huhtalav et al, 1998), Fdep
(Flach et al, 1999), Dep-Miner (Lopes et al, 2002), FastFD (Wyss et al, 2001) et
Fun (Novelli et al, 2001). Il est important de noter que sans aucun pré-traitement,
l’inférence des DF serait possible sur des données d’expression de gènes.
Cependant, la connaissance obtenue serait inutile puisque la plupart des valeurs
réelles de la relation diffèrent l’une de l’autre. Autrement dit, pratiquement chaque
gène est une clé, c’est-à-dire une connaissance sans intérêt pour les biologistes.
Néanmoins, nous pensons que l’inférence des DF peut permettre une analyse
intéressante des données de biopuces, lorsque le bruit est pris en compte dans la
définition de satisfaction des DF (Aussem et al, 2002, Bosc et al, 1998). Les
dépendances fonctionnelles approximatives sont étudiées dans (Kivinen et al, 1995)
où plusieurs mesures d’erreurs sont proposées sans toutefois inclure de relation de
proximité entre les valeurs.
2. Cadre de travail
Dans le domaine des puces à ADN, nous sommes confrontés à une « contrainte »
importante : d’abord le nombre d’expériences est petit (pas plus de 100 expériences
en général) tandis que le nombre de gènes est très important (plusieurs milliers).
Cette contrainte est peu souvent rencontrée en fouille de données ou en bases de
données : le nombre de lignes est généralement très important alors que le nombre
d’attributs est très petit. Pour la génération de règles entre gènes (qui sont des
attributs), cette contrainte implique une saturation rapide des temps de calculs.
Les points clés de notre approche peuvent se résumer de la façon suivante :

5
1) Définir une sémantique en collaboration avec les experts du domaine.
2) Vérifier que la sémantique est bien-formée, i.e. qu’elle définit un opérateur de fermeture sur l’ensemble des gènes.
3) Définir une base pour le système de fermeture à partir des données.
4) Caractériser la couverture canonique des règles à partir d’un sous-ensemble minimal et unique de la base du système de fermeture. Ainsi, il est aussi possible,
sans surcoût, de déterminer une couverture pour les implications approximatives
connue sous le nom de couverture de Gottlob et Libkin (Gottlob et al, 1990).
Nous ne développerons pas la méthode utilisée pour la génération des règles, le
lecteur intéressé peut se référer à (Mannila et al, 1994, Demetrovics et al, 1995,
Lopes et al, 2002, Aussem et al, 2002, Agier et al, 2003).
3. Différentes sémantiques pour les règles
L’intérêt de notre projet est de proposer aux biologistes plusieurs sémantiques
correspondant à des objectifs biologiques différents. Naturellement, une grande
partie de la difficulté réside dans l’adéquation entre la sémantique et l’intérêt
biologique auquel elle peut répondre. Nous apportons un début de réponse en
section 5 où une collaboration a été menée.
Pour l’ensemble des sémantiques, une règle se note de façon identique :
Notation 1 Soient ε1 et ε2 deux seuils, r un ensemble d’expériences, X, Y deux
ensembles de gènes et s une sémantique. La règle X → Y satisfaite dans r pour la
sémantique s se note :
r |=s, ε1, ε2 X → Y.
Les trois sémantiques que nous proposons sont détaillées ci-dessous.
3.1. Sémantique 1 : niveaux d’expression similaires entre gènes
La première sémantique consiste à étudier les niveaux d’expression des gènes. Ici, chaque expérience est considérée une fois. Nous appellerons cette sémantique s1 dans la suite. Notons que la définition de cette sémantique est proche de la
définition des règles d’association mais est appliquée ici à des données
quantitatives.
Définition 1 Soient ε1 et ε2 deux seuils, r un ensemble d’expériences et X, Y
deux ensembles de gènes. Une règle X → Y définie avec la sémantique s1, est
satisfaite dans r si et seulement si :
∀ e ∈ r, si ∀ g ∈ X, ε1 ≤ e[g] ≤ ε2 alors ∀ g ∈ Y, ε1 ≤ e[g] ≤ ε2.
Exemple 2 Reprenons l’exemple de données d’expression décrites dans le tableau 1. Supposons que les biologistes souhaitent étudier les relations entre les gènes sur-
exprimés. Pour cela, les seuils doivent être définis par exemple comme suit : ε1 = 1.0
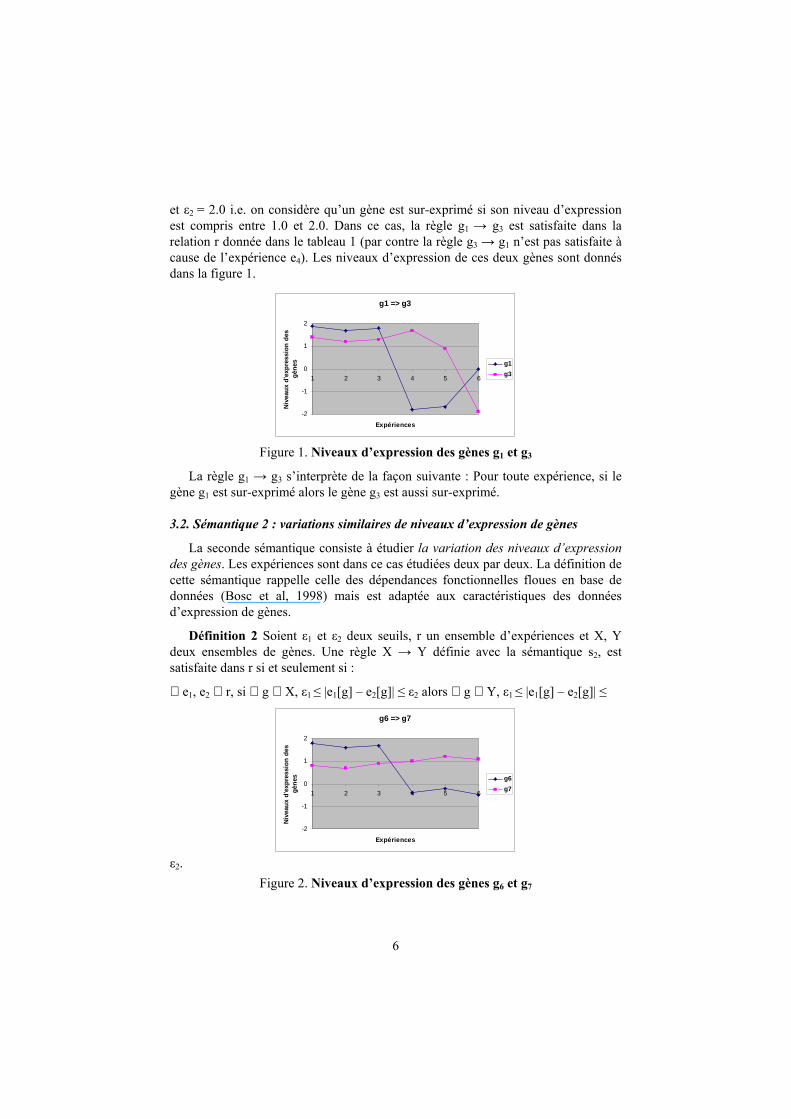
6
et ε2 = 2.0 i.e. on considère qu’un gène est sur-exprimé si son niveau d’expression
est compris entre 1.0 et 2.0. Dans ce cas, la règle g1 → g3 est satisfaite dans la
relation r donnée dans le tableau 1 (par contre la règle g3 → g1 n’est pas satisfaite à
cause de l’expérience e4). Les niveaux d’expression de ces deux gènes sont donnés
dans la figure 1.
Figure 1. Niveaux d’expression des gènes g1 et g3
La règle g1 → g3 s’interprète de la façon suivante : Pour toute expérience, si le
gène g1 est sur-exprimé alors le gène g3 est aussi sur-exprimé.
3.2. Sémantique 2 : variations similaires de niveaux d’expression de gènes
La seconde sémantique consiste à étudier la variation des niveaux d’expression des gènes. Les expériences sont dans ce cas étudiées deux par deux. La définition de cette sémantique rappelle celle des dépendances fonctionnelles floues en base de
données (Bosc et al, 1998) mais est adaptée aux caractéristiques des données
d’expression de gènes.
Définition 2 Soient ε1 et ε2 deux seuils, r un ensemble d’expériences et X, Y
deux ensembles de gènes. Une règle X → Y définie avec la sémantique s2, est
satisfaite dans r si et seulement si :
∀ e1, e2 ∈ r, si ∀ g ∈ X, ε1 ≤ |e1[g] – e2[g]| ≤ ε2 alors ∀ g ∈ Y, ε1 ≤ |e1[g] – e2[g]| ≤
ε2.
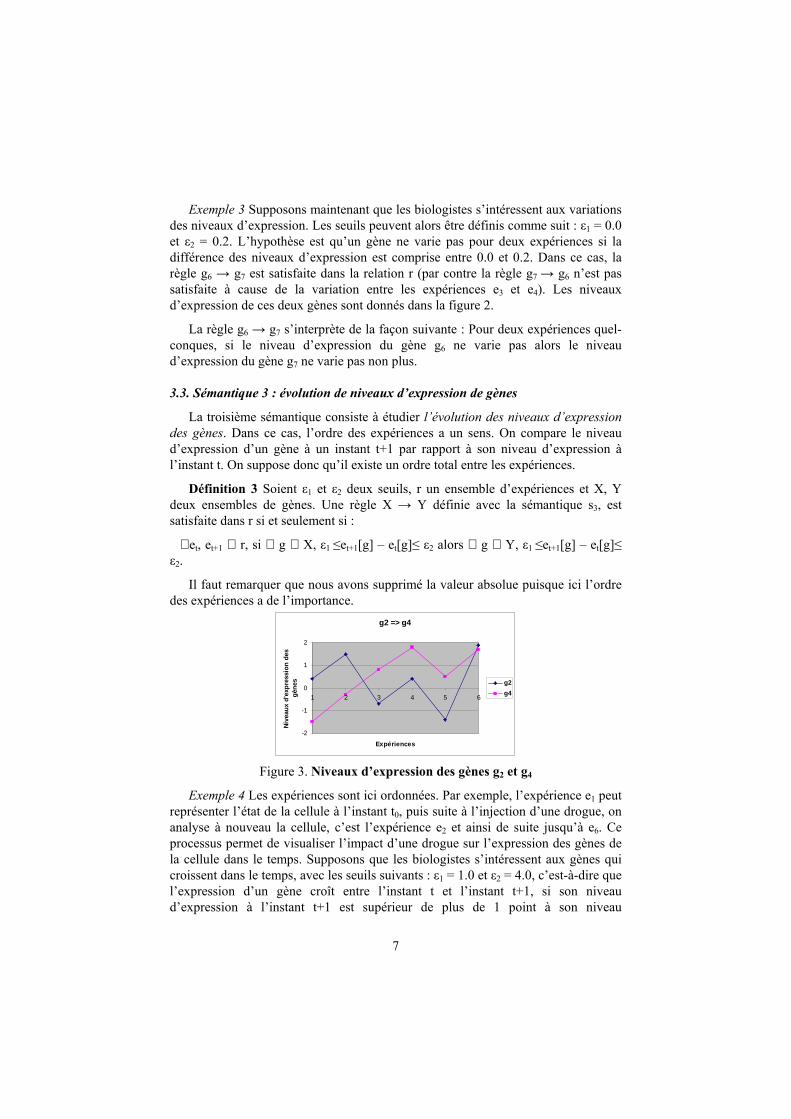
Figure 2. Niveaux d’expression des gènes g6 et g7
g1 => g3
-2
-1
0
1
2
1 2 3 4 5 6
Expériences
Niv
eaux
d'e
xpre
ssio
n de
s gè
nes g1
g3
g6 => g7
-2
-1
0
1
2
1 2 3 4 5 6
Expériences
Niv
eaux
d'e
xpre
ssio
n de
s gè
nes g6
g7
7
Exemple 3 Supposons maintenant que les biologistes s’intéressent aux variations des niveaux d’expression. Les seuils peuvent alors être définis comme suit : ε1 = 0.0
et ε2 = 0.2. L’hypothèse est qu’un gène ne varie pas pour deux expériences si la
différence des niveaux d’expression est comprise entre 0.0 et 0.2. Dans ce cas, la
règle g6 → g7 est satisfaite dans la relation r (par contre la règle g7 → g6 n’est pas
satisfaite à cause de la variation entre les expériences e3 et e4). Les niveaux
d’expression de ces deux gènes sont donnés dans la figure 2.
La règle g6 → g7 s’interprète de la façon suivante : Pour deux expériences quel-
conques, si le niveau d’expression du gène g6 ne varie pas alors le niveau
d’expression du gène g7 ne varie pas non plus.
3.3. Sémantique 3 : évolution de niveaux d’expression de gènes
La troisième sémantique consiste à étudier l’évolution des niveaux d’expression des gènes. Dans ce cas, l’ordre des expériences a un sens. On compare le niveau d’expression d’un gène à un instant t+1 par rapport à son niveau d’expression à
l’instant t. On suppose donc qu’il existe un ordre total entre les expériences.
Définition 3 Soient ε1 et ε2 deux seuils, r un ensemble d’expériences et X, Y
deux ensembles de gènes. Une règle X → Y définie avec la sémantique s3, est
satisfaite dans r si et seulement si :
∀et, et+1 ∈ r, si ∀ g ∈ X, ε1 ≤et+1[g] – et[g]≤ ε2 alors ∀ g ∈ Y, ε1 ≤et+1[g] – et[g]≤ ε2.
Il faut remarquer que nous avons supprimé la valeur absolue puisque ici l’ordre
des expériences a de l’importance.
Figure 3. Niveaux d’expression des gènes g2 et g4
Exemple 4 Les expériences sont ici ordonnées. Par exemple, l’expérience e1 peut représenter l’état de la cellule à l’instant t0, puis suite à l’injection d’une drogue, on
analyse à nouveau la cellule, c’est l’expérience e2 et ainsi de suite jusqu’à e6. Ce
processus permet de visualiser l’impact d’une drogue sur l’expression des gènes de
la cellule dans le temps. Supposons que les biologistes s’intéressent aux gènes qui
croissent dans le temps, avec les seuils suivants : ε1 = 1.0 et ε2 = 4.0, c’est-à-dire que
l’expression d’un gène croît entre l’instant t et l’instant t+1, si son niveau
d’expression à l’instant t+1 est supérieur de plus de 1 point à son niveau
g2 => g4
-2
-1
0
1
2
1 2 3 4 5 6
Expériences
Niv
eaux
d'e
xpre
ssio
n de
s gè
nes g2
g4

8
d’expression à l’instant t. Dans ce cas, la règle g2 → g4 est satisfaite dans la relation
r (contrairement à la règle g4 → g2 contredite par l’évolution de l’expérience e2 à
l’expérience e3). Les niveaux d’expression de ces deux gènes sont donnés dans la
figure 3. La règle g2 → g4 s’interprète de la façon suivante : Entre deux instants
quelconques t et t+1, si le niveau d’expression du gène g2 croît alors le niveau
d’expression du gène g4 croît.
Variantes
Ces sémantiques sont définies avec deux seuils afin de prendre en compte un
aspect du comportement des gènes, par exemple la sur-expression (ou la sous-
expression) pour la sémantique s1. Notons que toutes ces définitions peuvent
s’étendre facilement à plus de seuils. Par exemple, pour la sémantique s1, on peut
choisir 4 seuils ε1, ε2, ε3 et ε4 tels que ε1 ≤ ε2 ≤ ε3 ≤ ε4 et considérer qu’un gène g est
sous-exprimé pour l’expérience e si ε1 ≤ e[g] ≤ ε2 et sur-exprimé pour l’expérience
e’ si ε3 ≤ e’[g] ≤ ε4. Dans ce cas, il est possible de générer des règles du type
(g1sur−exp, g2sous−exp) → g3sous−exp. Ces variantes ont été implémentées dans
l’outil proposé.
3.4. Comparaison avec la classification
Pour comparer notre approche avec une méthode classique d’analyse de données
de biopuces, nous avons réalisé une classification hiérarchique donnée dans la
figure 4 sur notre jeu de données exemple.
Figure 4. Classification hiérarchique sur les données de l’exemple
Les règles obtenues avec les différentes sémantiques lient des gènes qui ne sont
pas forcément regroupés dans les mêmes clusters. On peut notamment remarquer
que les gènes g2 et g4 qui sont reliés par la troisième sémantique, n’ont pas des
profils d’expression similaires. De même, les gènes g6 et g7 qui sont reliés par la
seconde sémantique, ne sont pas dans le même cluster.
Cette approche apporte de la connaissance supplémentaire sur les données et
permet d’aller plus loin dans les relations qui existent entre les gènes. Ceci se
retrouve également dans les données de tumeurs mammaires décrites dans la section
5.

9
4. Implémentation
Le programme de génération de règles est implémenté en C++ et utilise la STL.
Dans sa version actuelle, il utilise une liste d’arbres pour représenter les ensembles
d’ensembles de gènes et l’algorithme de Demetrovics et Thi pour le calcul des
transversaux minimaux (Demetrovics et al, 1995). Pour permettre l’utilisation de cet
outil aux biologistes, nous avons choisi de l’intégrer à un logiciel libre open-source
d’analyse de données de biopuces : TIGR MultiExperimentViewer (Saeed et al,
2003).
Cet outil fait partie d’une suite d’applications, appelée TM4, développée par The
Institute for Genomic Research (TIGR). Ces outils consacrés aux données de
biopuces proposent différentes fonctions : le stockage des données, l’analyse des
images, la normalisation, l’interprétation des résultats... MeV est l’application
consacrée à l’analyse proprement dite des données, il essaie de recenser la plupart
des méthodes pouvant s’appliquer aux données d’expression de gènes. De plus,
MeV prend en entrée plusieurs formats de fichiers provenant de différents logiciels
d’analyse d’images de biopuces. La convivialité de cet outil et le nombre déjà
important de logiciels consacrés aux données de biopuces, nous ont conduits vers ce
choix qui simplifie en plus les manipulations des biologistes.
4.1. Conception de l’interface graphique
Pour l’interface, nous avons choisi de limiter les options proposées aux
utilisateurs pour rendre plus facile l’utilisation du logiciel. L’interface est présentée
dans la figure 5.
Figure 5. Interface du logiciel
Nous proposons d’abord aux utilisateurs de réaliser une sélection sur les gènes.
Ils peuvent indiquer le nombre de gènes qu’ils souhaitent étudier, cette étape permet
outre la limitation des temps de calcul, de se limiter aux règles les plus intéressantes.
L’élagage proposé se fait en fonction de la variance des gènes, nous gardons ceux

10
dont la variance est la plus importante. D’autres méthodes d’élagage sont
actuellement étudiées.
Ensuite, l’utilisateur choisit la sémantique qui le concerne parmi celles
proposées et l’outil lui propose en fonction de ce choix un ensemble de seuils
pertinents. Par défaut, l’outil propose 6 seuils mais l’utilisateur peut sélectionner
que deux ou quatre seuils. Ils sont calculés en fonction soit de la distribution des
niveaux d’expression des gènes pour la première sémantique, soit de la distribution des variations des niveaux d’expression des gènes pour la seconde, soit de la distribution des évolutions des niveaux d’expression des gènes pour la dernière. Par exemple pour la première sémantique, les seuils sont définis de la façon sui-
vante :
Soit max = max(niveaux d’expression) et min = min(niveaux d’expression), alors nous pouvons partager l’intervalle [min, max] en trois parties distinctes (nous
supposons ici que les valeurs extrêmes ont été supprimées pendant la phase de
normalisation des données) :
– pour les gènes sous-exprimés : [min ; min + (max − min)/4]
– pour les gènes « neutres » : [min + (max − min)/4 ; min + (max − min) * (3/4)]
– pour les gènes sur-exprimés : [min + (max − min) * (3/4) ; max]
Pour les autres sémantiques, seule la définition de max et min diffère (par
exemple pour la seconde sémantique, max = max(variations des niveaux d’expression) ).
De plus, pour limiter le nombre de règles, il est possible de ne s’intéresser qu’à
un seul gène ou de donner une taille maximale pour les parties gauches.
4.2. Post-traitement des règles
Le logiciel a été testé sur plusieurs jeux de données et nous nous sommes
naturellement intéressés au post-traitement des règles. Sans être exhaustif, nous
avons pour l’instant choisi de calculer quatre mesures de qualité pour permettre de
réaliser un premier tri des différentes règles (cf figure 6). Notons qu’il existe bien
sûr d’autres indices de qualité (Gras et al, 2001, Jen et al, 2002, Blanchard et al,
2004, Vaillant et al, 2004).
L’ensemble des indices pour une règle X → Y définie avec la sémantique s1 et deux seuils ε1 et ε2, se calculent à partir de quatre paramètres initiaux :
– n : nombre d’expériences dans la relation.
– nX : nombre d’expériences e telles que ∀ g ∈ X, ε1 ≤ e[g] ≤ ε2 (nous dirons par la suite telles que X est satisfait).
– nY : nombre d’expériences e telles que ∀ g ∈ Y, ε1 ≤ e[g] ≤ ε2. – nX,Y : nombre d’expériences e telles que ∀ g ∈ X, ε1 ≤ e[g] ≤ ε2 et ∀ g ∈ Y,
ε1 ≤ e[g] ≤ ε2.
Notons que le calcul de ces quatre paramètres ne nécessite pas de nouvel accès
aux données et s’intègre facilement dans le processus de génération des règles.

11
Dans le cas des sémantiques s2 et s3, les définitions diffèrent puisque nous
comparons les expériences deux à deux. Il ne faut plus compter le nombre
d’expériences mais le nombre de couples d’expériences satisfaisant ou non les conditions correspondant à chaque sémantique.
A partir de ces paramètres, nous pouvons calculer pour la règle X → Y, les
quatre indices suivants :
Support : Le support de la règle X → Y correspond à la probabilité que X et Y
soient satisfaits simultanément.
Support (X → Y) = nX,Y / n.
Figure 6. Interface du logiciel
Confiance : La confiance de la règle X → Y correspond à la probabilité que Y
soit satisfait sachant que X est satisfait.
Confiance (X → Y) = nX,Y / nX. Lorsque cet indice vaut 1, on dit que la règle est exacte, sinon elle est dite approximative.
Intérêt : Pour la règle X → Y , cet indice mesure la dépendance entre X et Y. Il
correspond au quotient entre le nombre observé d’expériences satisfaisant X et Y et
le nombre d’expériences satisfaisant X et Y que l’on obtiendrait si les deux
variables étaient indépendantes.
Intérêt (X → Y) = nX,Y / ( (nX nY) / n ). Un intérêt de 1 signifie que les deux variables sont totalement indépendantes.

12
Dépendance : Cet indice mesure également la dépendance entre X et Y. Il
correspond à la valeur absolue de la différence entre la probabilité que Y soit
satisfait sachant que X est satisfait et la probabilité que Y soit satisfait pour X
satisfait ou non.
Dépendance(X → Y) = | (nX,Y / nX ) - (nY / n) |. Une valeur de dépendance nulle signifie que les deux variables sont totalement
indépendantes.
L’utilisateur ne regarde alors que les règles qu’ils jugent intéressantes aux vues
de ces différents indices.
4.3. Stockage des règles
Hormis le calcul de ces indices de qualité, nous proposons de faciliter davantage
l’interprétation des résultats aux biologistes en stockant les règles obtenues dans une
base de données. Ceci permet en effectuant de simples requêtes, de comparer des
règles obtenues avec différentes sémantiques, avec différents seuils, à partir de plu-
sieurs jeux de données... Cela permet aussi de croiser ces implications avec des
informations disponibles sur le web. Par exemple, un biologiste travaillant sur une
fonction particulière de gènes (par exemple une fonction GO de Gene Ontology
(Gene Ontology Consortium, 2004)) peut sélectionner l’ensemble des règles
concernant les gènes impliqués dans cette fonction. Ce travail est en cours, pour
l’instant seules les règles sont stockées dans la base de données.
5. Application à des données de cancer du sein
Une application a été réalisée sur des données publiques de tumeurs mammaires
(Van’t Veer et al, 2002). Le cancer du sein est aujourd’hui très étudié puisqu’il
constitue en France et dans les pays occidentaux, l’affection tumorale la plus
fréquente de la femme. Il reste néanmoins un cancer très imprévisible et hétérogène
d’un malade à l’autre. Afin d’adapter les traitements au pronostic de la tumeur, des
indicateurs biologiques sensibles sont nécessaires et l’analyse du transcriptome des
tumeurs grâce à la nouvelle technologie des biopuces est une piste explorée par de
nombreuses équipes.
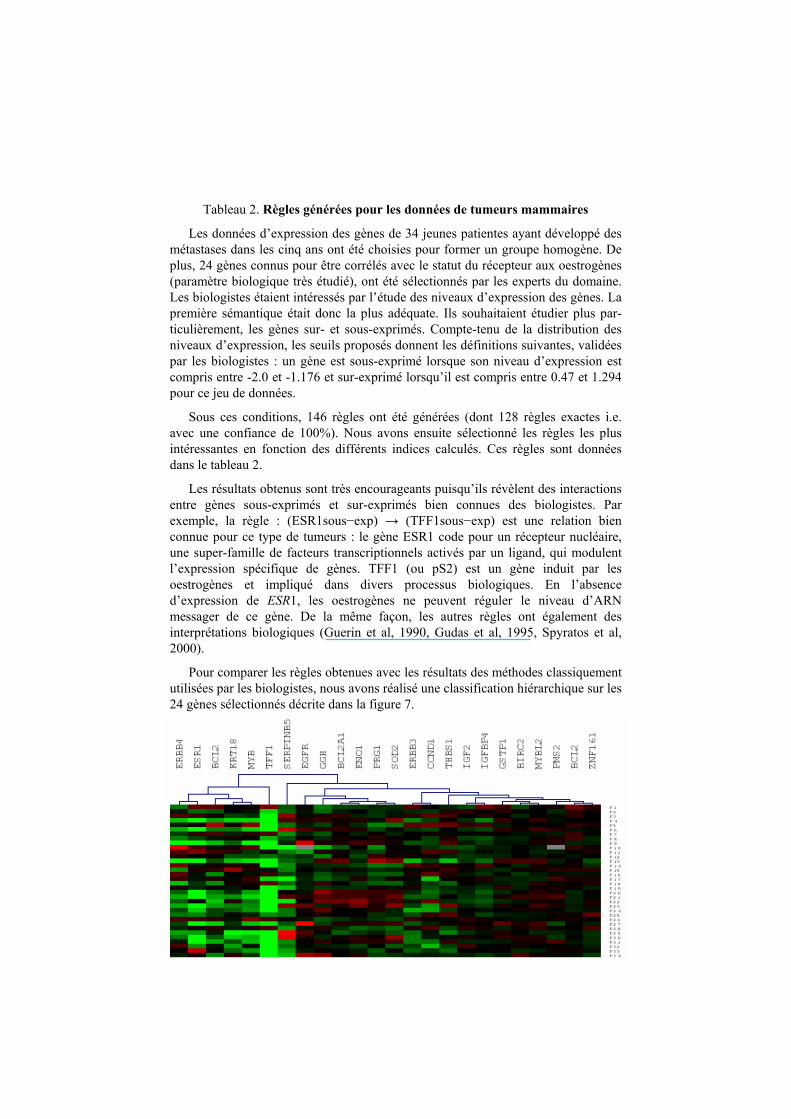
Règle Support Confiance Dépendance Intérêt ESR1- → TFF1- 0.41 1.00 0.26 1.36
MYB- → TFF1- 0.38 1.00 0.26 1.36
ERBB4-, TFF1- → MYB- 0.21 1.00 0.62 2.62
ERBB4-, ESR1- → MYB- 0.21 1.00 0.62 2.62
KRT18- → ESR1- 0.21 1.00 0.59 2.43
ERBB4-, TFF1- → ESR1- 0.21 1.00 0.59 2.43
ERBB4-, MYB- → ESR1- 0.21 1.00 0.59 2.43
BCL2-, ESR1- → MYB- 0.18 1.00 0.62 2.62
BCL2-, MYB- → ESR1- 0.18 1.00 0.59 2.43
ERBB4-, KRT18- → MYB- 0.15 1.00 0.62 2.62
SERPINB5+, ESR1- → MYB- 0.12 1.00 0.62 2.62
SERPINB5+, MYB- → ESR1- 0.12 1.00 0.59 2.43
13
Tableau 2. Règles générées pour les données de tumeurs mammaires
Les données d’expression des gènes de 34 jeunes patientes ayant développé des
métastases dans les cinq ans ont été choisies pour former un groupe homogène. De
plus, 24 gènes connus pour être corrélés avec le statut du récepteur aux oestrogènes
(paramètre biologique très étudié), ont été sélectionnés par les experts du domaine.
Les biologistes étaient intéressés par l’étude des niveaux d’expression des gènes. La
première sémantique était donc la plus adéquate. Ils souhaitaient étudier plus par-
ticulièrement, les gènes sur- et sous-exprimés. Compte-tenu de la distribution des
niveaux d’expression, les seuils proposés donnent les définitions suivantes, validées
par les biologistes : un gène est sous-exprimé lorsque son niveau d’expression est
compris entre -2.0 et -1.176 et sur-exprimé lorsqu’il est compris entre 0.47 et 1.294
pour ce jeu de données.
Sous ces conditions, 146 règles ont été générées (dont 128 règles exactes i.e.
avec une confiance de 100%). Nous avons ensuite sélectionné les règles les plus
intéressantes en fonction des différents indices calculés. Ces règles sont données
dans le tableau 2.
Les résultats obtenus sont très encourageants puisqu’ils révèlent des interactions
entre gènes sous-exprimés et sur-exprimés bien connues des biologistes. Par
exemple, la règle : (ESR1sous−exp) → (TFF1sous−exp) est une relation bien
connue pour ce type de tumeurs : le gène ESR1 code pour un récepteur nucléaire,
une super-famille de facteurs transcriptionnels activés par un ligand, qui modulent
l’expression spécifique de gènes. TFF1 (ou pS2) est un gène induit par les
oestrogènes et impliqué dans divers processus biologiques. En l’absence
d’expression de ESR1, les oestrogènes ne peuvent réguler le niveau d’ARN messager de ce gène. De la même façon, les autres règles ont également des
interprétations biologiques (Guerin et al, 1990, Gudas et al, 1995, Spyratos et al,
2000).
Pour comparer les règles obtenues avec les résultats des méthodes classiquement
utilisées par les biologistes, nous avons réalisé une classification hiérarchique sur les
24 gènes sélectionnés décrite dans la figure 7.

14
Figure 7. Classification hiérarchique sur l’application
Nous voyons que les gènes ESR1 et TFF1 n’ont pas des profils d’expression très
similaires. On peut aussi remarquer que le gène SERPINB5 n’appartient pas au
cluster regroupant les autres gènes auxquels il est lié.
6. Conclusion et Perspectives
Dans le but de se diriger vers des réseaux de régulation à partir de données d’expression de gènes, nous avons intégré une technique dans une plate-forme
existante pour définir différents types de règles entre gènes. Nous avons montré que
de telles implications apportaient de la connaissance supplémentaire par rapport aux
techniques utilisées couramment (Eisen et al, 1998, Kohane et al, 2003).
L’approche mise en place pour générer les règles est, à notre connaissance, la
première contribution traitant de façon identique différentes sémantiques. De plus,
les utilisateurs n’ont pas besoin d’indiquer un seuil de support minimum pour
générer les implications, comme cela est le cas pour les règles d’association.
Une des fonctions clés de cette approche est d’éviter l’encombrante phase de
pré-traitement avant le processus d’extraction des connaissances, en proposant des
seuils pertinents aux utilisateurs et en aidant autant que possible l’utilisateur dans
ces choix grâce à une interface graphique adéquate.
Le prototype décrit dans ce papier sera bientôt disponible pour les biologistes et
médecins. Actuellement, nous souhaitons effectuer des tests supplémentaires sur de
nouvelles données d’expression de tumeurs mammaires pour vérifier si les règles
produites et validées sur le premier jeu se retrouvent aussi sur des données
provenant de laboratoires différents. Malheureusement, il est difficile d’obtenir
toutes les informations nécessaires sur les données pour vérifier notamment que les
tumeurs analysées aient bien des caractéristiques biologiques similaires.
Les principales perspectives sur lesquelles nous nous penchons sont les suivantes :
– Ajouter aux données d’expression des gènes, des données caractérisant les ex-
périences. Par exemple, dans le cadre de patientes atteintes de cancer du sein, il
serait intéressant d’ajouter les données cliniques et anatomo-pathologiques. On
retombe alors dans des techniques de classification supervisée.
– Explorer les relations entre nos travaux et ceux traitant des motifs émergeants à partir de données d’expression de gènes (Li et al, 2003). Nous pourrions ainsi
caractériser des règles qui ont une confiance très élevée pour un certain type
d’expériences et une confiance très faible pour un autre type d’expériences (par
exemple, des patientes atteintes d’une tumeur de type 1 et des patientes atteintes
d’une tumeur de type 2).

15
Bibliographie
Agier M., Petit J.-M., "Génération de différents types de règles à partir de données
d’expression de gènes", Rapport de recherche, LIMOS, 2003.
Aussem A., Petit J.-M., "ε-functional dependency inference : application to DNA microarray expression data", Bases de données avancées (BDA’02), Evry, France, Octobre 2002.
Becquet C., Blachon S., Jeudy B., Boulicaut J.-F., Gandrillon O., "Strong-association-rule
mining for large-scale gene-expression data analysis : a case study on human SAGE
data", Genome Biology, vol. 3, n°12, 2002.
Berrar D.P., Granzow M., Dubitzky W., A Practical Approach to Microarray Data Analysis, Kluwer Academic Publishers, 2002.
Blanchard J., Guillet F., Gras R., Briand H., "Mesurer la qualité des règles et de leurs
contraposées avec le taux informationnel TIC", EGC’04, vol. 1, 2004, p. 287-98.
Bosc P., Dubois D., Prade H., "Fuzzy Functional Dependencies - An Overview and a critical
discussion", Journal of the American Society for Information Science, vol. 49, 1998, p. 217–235.
Caspard N., Monjardet B., "The Lattices of Closure Systems, Closure Operators, and
Implicational Systems on a Finite Set : A Survey", Discrete Applied Mathematics, vol. 127, n°2, 2003, p. 241–269.
Demetrovics J., Thi V., "Some Remarks On Generating Armstrong And Inferring Functional
Dependencies Relation", Acta Cybernetica, vol. 12, n°2, 1995, p. 167-180.
Eisen M., Spellman P., Brown P., Botstein D., "Gene expression profiling predicts clinical
outcome of breast cancer", Proc Natl Acad Sci, vol. 95, n°25, 1998, p. 14863-14868.
Flach P. A., Savnik I., "Database Dependency Discovery : A Machine Learning Approach",
AI Communications, vol. 12, n°3, 1999, p. 139-160.
Ganter B., Wille R., Formal Concept Analysis, Springer-Verlag, 1999.
Gene Ontology Consortium, "The Gene Ontology (GO) database and informatics resource",
Nucleic Acids Research, vol. 32, 2004, p. 258-61.
Gottlob G., Libkin L., "Investigations on Armstrong Relations, Dependency Inference, and
Excluded Functional Dependencies", Acta Cybernetica, vol. 9, n°4, 1990, p. 385–402.
Gras R., Kuntz P., Couturier R., Guillet F., "Une version entropique de l’intensité
d’implication pour les corpus volumineux", EGC’01, vol. 1, 2001, p. 69-80, Hermes.
Gudas J., Klein R., Oka M., Cowan K., "Posttranscriptional regulation of the c-myb proto-
oncogene in estrogen receptor-positive breast cancer cells", Clin Cancer Res, vol. 1, n°2, 1995, p. 235-43.
Guerin M., Sheng Z.-M., Andrieu N., Riou G., "Strong association between c-myb and
oestrogen-receptor expression in human breast cancer", Oncogene, vol. 5, n°1, 1990, p. 131–135.
Han J., Kamber M., Data Mining: Concepts and Techniques, Morgan Kaufmann Publishers, first edition, 2000.

16
Huhtalay., Kärkkäinenj., Porkkap., Toivonenh., "Efficient Discovery of Functional and
Approximate Dependencies Using Partitions", Proc. of the 14thIEEE ICDE, 1998, p. 392–
401.
Icev A., Ruiz C., Ryder E.F., "Distance-enhanced association rules for gene expression",
BIOKDD’03, in conjunction with ACM SIGKDD, Washington, DC, USA, 2003.
Jen T.-Y., Laurent D., Spyratos N., Tanaka Y., "Règles d’association significatives", 18e Journées Bases de Données Avancées (BDA’02), Evry, France, Oct. 2002.
Kohane I.S., Kho A.T., Butte A.J., Microarrays for an integrative genomics, MIT Press, 2003.
Kivinen J., Mannila H., "Approximate inference of functional dependencies from relations",
TCS, vol. 149, n°1, 1995, p. 129–149.
Li J., Liu H., Downing J., Yeoh A., Wong L., "Simple rules underlying gene expression
profiles of more than six subtypes of acute lymphoblastic leukemia (ALL) patients",
Bioinformatics, vol. 19, 2003, p. 71–78.
Lopes S., Petit J.-M., Lakhal L., "Functional and Approximate Dependencies Mining :
Databases and FCA Point of View", Special issue of JETAI, vol. 14, no
2, 2002, p. 93-114.
Mannila H., Räihä K.-J., The Design of Relational Databases, Addison-Wesley, second édition, 1994.
Novelli N., Cicchetti R., "FUN : An Efficient Algorithm for Mining Functional and
Embedded Dependencies", Proc. of the ICDT, UK, vol. 1973, 2001, p. 189-203.
Piatetsky-Shapirov., Tamayop., "Microarray Data Mining : Facing the Challenges", SIGKDD Explorations, Special Issue on Microarray Data Mining, 2003.
Saeed, Sharov, White, Li, Liang, Bhagabati, Braisted, Klapa, Currier, Thiagarajan, Sturn,
Snuffin, Rezantsev, Popov, Ryltsov, Kostukovich, Borisovsky, Liu, Vinsavich, Trush,
Quackenbush, "TM4 : a free, open-source system for microarray data management and
analysis", Biotechniques, vol. 34, n°2, 2003, p. 374-78.
Soularue P., Gidrol X., "Puces à ADN", Techniques de l’Ingénieur, vol. 6, 2002, p. 1-10.
Spyratos F., Andrieu C., Vidaud D., Briffod M., Vidaud, Lidereau, Bieche, "CCND1 mRNA
overexpression is highly related to estrogen receptor positivity but not to proliferative
markers in primary breast cancer", Int J Biol Markers, vol. 15, n°3, 2000, p. 210-214.
Vaillant B., Lenca P., Lallich S., "Etude expérimentale de mesures de qualité de règles
d’association", EGC’04, vol. 2, 2004, p. 341-52, Cépaduès Editions.
Van’t Veer L., Dai H., Vijver M., He Y., Hart A., Mao, Peterse, Kooy, Marton, Witteveen,
Schreiber, Kerkhoven, Roberts, Linsley, Bernards, Friend, "Gene expression profiling
predicts clinical outcome of breast cancer", Nature, vol. 415, n°6871, 2002, p. 530-536.
Wyss C., Giannella C., Robertson E., "FastFDs : A Heuristic-Driven, Depth-First Algorithm
for Mining Functional Dependencies from Relation Instances", Kambayashi, Proc. of the 3rd DAWAK , Germany, September 5-7, vol. 2114, Springer-Verlag, 2001, p. 101-110.
Copyright © 2022 FDOKUMEN




















