
validatie van een vloeistofchromatografische methode voor
74
UNIVERSITEIT GENT FACULTEIT FARMACEUTISCHE WETENSCHAPPEN Vakgroep Farmaceutische Analyse Laboratorium voor Analytische Chemie Academiejaar 2010-2011 VALIDATIE VAN EEN VLOEISTOFCHROMATOGRAFISCHE METHODE VOOR DE BEPALING VAN ETHYLPARABEEN EN LITERATUURONDERZOEK – BELANG VAN HET TOLERANTIE-INTERVAL BIJ DE FARMACEUTISCHE METHODEVALIDATIE Arno VERMOTE Eerste Master in de Geneesmiddelenontwikkeling Promotor Dr. K. Van Uytfanghe Commissarissen Prof. Dr. L. Thienpont Prof. Dr. B. De Spiegeleer
-
Upload
khangminh22 -
Category
Documents
-
view
2 -
download
0
Transcript of validatie van een vloeistofchromatografische methode voor

UNIVERSITEIT GENT
FACULTEIT FARMACEUTISCHE WETENSCHAPPEN
Vakgroep Farmaceutische Analyse
Laboratorium voor Analytische Chemie
Academiejaar 2010-2011
VALIDATIE VAN EEN VLOEISTOFCHROMATOGRAFISCHE METHODE VOOR
DE BEPALING VAN ETHYLPARABEEN EN LITERATUURONDERZOEK – BELANG
VAN HET TOLERANTIE-INTERVAL BIJ DE FARMACEUTISCHE
METHODEVALIDATIE
Arno VERMOTE
Eerste Master in de Geneesmiddelenontwikkeling
Promotor
Dr. K. Van Uytfanghe
Commissarissen
Prof. Dr. L. Thienpont
Prof. Dr. B. De Spiegeleer


UNIVERSITEIT GENT
FACULTEIT FARMACEUTISCHE WETENSCHAPPEN
Vakgroep Farmaceutische Analyse
Laboratorium voor Analytische Chemie
Academiejaar 2010-2011
VALIDATIE VAN EEN VLOEISTOFCHROMATOGRAFISCHE METHODE VOOR
DE BEPALING VAN ETHYLPARABEEN EN LITERATUURONDERZOEK – BELANG
VAN HET TOLERANTIE-INTERVAL BIJ DE FARMACEUTISCHE
METHODEVALIDATIE
Arno VERMOTE
Eerste Master in de Geneesmiddelenontwikkeling
Promotor
Dr. K. Van Uytfanghe
Commissarissen
Prof. Dr. L. Thienpont
Prof. Dr. B. De Spiegeleer

AUTEURSRECHT
“De auteur en de promotor geven de toelating deze masterproef voor consultatie beschikbaar te stellen
en delen ervan te kopiëren voor persoonlijk gebruik. Elk ander gebruik valt onder de beperkingen van
het auteursrecht, in het bijzonder met betrekking tot de verplichting uitdrukkelijk de bron te vermelden
bij het aanhalen van de resultaten uit deze masterproef.”
7 juni 2011
Promotor Auteur
Dr. K. Van Uytfanghe Arno Vermote

i
DANKWOORD
Voor u de inhoud van deze thesis leest, had ik graag nog even benadrukt dat deze scriptie niet had
kunnen geschreven worden zonder de hulp van een aantal mensen. Zij verdienen elk individueel een
speciaal woordje van dank.
In de eerste plaats zou ik graag Prof. Dr. L. Thienpont bedanken. Dankzij haar was het voor mij mogelijk
om deze thesis in het Laboratorium voor Analytische Chemie te schrijven. Daarnaast wil ik haar ook nog
bedanken voor de algemene leiding van deze meesterproef.
Te dikwijls behoren professoren tot het mannelijke geslacht, maar laat één ding duidelijk zijn: ik acht Prof.
Dr. L. Thienpont zeer hoog. Heel slimme vrouwen verdienen het om te doceren aan een universiteit om
trots op te zijn.
In je opleiding heb je niet alleen geduldige pedagogen nodig, maar ook kritische geesten die je
tegenspreken. Daarom ben ik Dr. D. Stöckl speciale dank verschuldigd. Uiteraard dank ik hem voor de
intensieve begeleiding en de lessen statistiek, maar daarnaast zeker en vast ook voor het delen van zijn
levenservaring. De werkelijkheid is niet altijd glorieus. De wetenschap reikt postiche een succesverhaal
zonder weerga aan. Geluk, succes, symbiose, maar “wetenschap maakt niet knap”!
Overigens, wat een zuur dankwoord, wat een cultuurpessimisme een pauselijke encycliek waardig. Dr. D.
Stöckl wordt bedankt om ons bij te brengen dat we vandaag onszelf moeten realiseren en onze toekomst
in handen moeten nemen.
Wie ik zeker niet mag vergeten is mijn promotor Dr. K. Van Uytfanghe. Deze sterke dame, die schijnbaar
nooit eigenwijs, knorrig, trots of vermoeid is, heeft mij heel wat praktische zaken bijgebracht. Haar
positieve ingesteldheid heeft een diepe indruk nagelaten. Graag zou ik haar ook bedanken voor het
nalezen van de thesis en de algemene begeleiding.
De doctoraatstudenten Hedwig Stepman en Sofie Van Houcke dank ik voor hun hulp en tips bij het
uitvoeren van de experimenten in het laboratorium. Zij weten de sfeer op het laboratorium ondanks alle
tegenslagen toch op te krikken.
Graag had ik ook nog mijn dank betuigd aan het personeel van het laboratorium voor Analytische
Chemie: Tania, Hilde, Linde en Sara. In het bijzonder zou ik Tania, die meermaals als een freule Francina
Fazant doorheen het labo laveerde, willen bedanken. Een allerfijnste vrouw, een fonkelende fee. Veel
fraaier dan een pauw.
Natuurlijk mag ik ook Manon niet vergeten. Samen met haar heb ik tijdens deze meesterproef een heel
leerrijke en aangename tijd gehad. Zij heeft mede gezorgd voor een aangename werksfeer.
Als laatste bedank ik ook nog mijn moeder en mijn zus. Ik dank hen voor de steun en vele
aanmoedigingen.

ii
INHOUDSOPGAVE
DANKWOORD ..................................................................................................................................... i
INHOUDSOPGAVE .............................................................................................................................. ii
LIJST MET GEBRUIKTE AFKORTINGEN ................................................................................................. iv
DEFINITIES ........................................................................................................................................ vi
1. INLEIDING ................................................................................................................................... 1
1.1. PARABENEN ................................................................................................................................... 1
1.1.1. Structuur en eigenschappen ......................................................................................... 1
1.1.2. Controverse en toxiciteit .............................................................................................. 2
1.2. VALIDATIE ...................................................................................................................................... 4
2. OBJECTIEVEN .............................................................................................................................. 8
3. MATERIALEN EN METHODEN ....................................................................................................... 9
3.1. MATERIALEN .................................................................................................................................. 9
3.1.1. Oplosmiddel en eluens ................................................................................................. 9
3.1.2. Bereiding van de stockoplossing, standaarden en stalen ................................................ 9
3.1.2.1. Lineariteit en kalibratie ............................................................................................... 10
3.1.2.2. Imprecisie .................................................................................................................... 11
3.1.2.3. Detectielimiet .............................................................................................................. 11
3.1.2.4. Juistheid ....................................................................................................................... 11
3.1.2.5. Systeemgeschiktheidstest ........................................................................................... 12
3.1.3. Apparatuur ................................................................................................................ 12
3.1.3.1. Analyse ........................................................................................................................ 12
3.1.3.2. Randapparatuur........................................................................................................... 13
3.2. METHODEN.................................................................................................................................. 13
3.2.1. Systeemfunctiecontrole .............................................................................................. 13
3.2.2. Systeemgeschiktheidscontrole .................................................................................... 13
3.2.3. Analyse ...................................................................................................................... 15
3.2.4. Validatie-experimenten .............................................................................................. 15
3.2.4.1. Lineariteit ..................................................................................................................... 16
3.2.4.2. Kalibratie ...................................................................................................................... 17
3.2.4.3. Imprecisie .................................................................................................................... 18
3.2.4.4. Detectielimiet .............................................................................................................. 20
3.2.4.5. Juistheid ....................................................................................................................... 21
3.2.4.6. Methodevergelijking ................................................................................................... 22
3.2.5. Dataverwerking en statistiek ...................................................................................... 24
3.2.6. Specificaties ............................................................................................................... 25
3.2.7. Literatuuronderzoek ................................................................................................... 25

iii
4. RESULTATEN EN DISCUSSIE ........................................................................................................ 26
4.1. EXPERIMENTEN ........................................................................................................................... 26
4.1.1. Systeemfunctiecontrole .............................................................................................. 26
4.1.2. Systeemgeschiktheidscontrole .................................................................................... 26
4.1.3. Lineariteit .................................................................................................................. 27
4.1.4. Kalibratie ................................................................................................................... 28
4.1.5. Imprecisie .................................................................................................................. 30
4.1.6. Detectielimiet ............................................................................................................ 32
4.1.7. Juistheid .................................................................................................................... 33
4.1.8. Methodevergelijking .................................................................................................. 35
4.1.9. Resultaten: samenvatting ........................................................................................... 39
4.2. LITERATUURONDERZOEK ............................................................................................................ 39
4.2.1. Introductie tot het statistisch concept van het tolerantie-interval ................................ 39
4.2.2. Het tolerantie-interval binnen het (bio)farmaceutische veld ........................................ 42
4.2.3. Belang van het tolerantie-interval bij de farmaceutische methodevalidatie ................. 44
5. CONCLUSIE ............................................................................................................................... 50
6. LITERATUURLIJST ...................................................................................................................... 51
APPENDIX: Detection decisions defined by the standard deviation of the blank – Questions from
“analytical freshmen”

iv
LIJST MET GEBRUIKTE AFKORTINGEN
βETI “β-expectation” tolerantie-interval
βCTI “β-content” tolerantie-interval
ANOVA Variantie-analyse (“Analysis of Variance”)
CI Confidentie-interval (“Confidence Interval”)
CL Betrouwbaarheidslimiet (“Confidence Limit”)
CLSI “Clinical and Laboratory Standards Institute”
CV Variatiecoëfficiënt (“Coefficient of Variation”)
CVwr Binnen-analyse variatiecoëfficiënt (“within run” CV)
CVT Totale variatiecoëfficiënt (“total” CV)
ELISA “Enzyme-Linked Immunosorbent Assay”
EP Evaluatie protocol (“Evaluation Protocol”)
FDA “Food and Drug Administration”
HPLC Hoge druk vloeistofchromatografie (“High Performance Liquid
Chromatography”)
ICH “International Conference on Harmonisation”
IQC Interne kwaliteitscontrole (“Internal Quality Control”)
ISO “International Organization for Standardization”
IUPAC “International Union of Pure and Applied Chemistry”
LCL Onderste betrouwbaarheidslimiet (“Lower Confidence Limit”)
LLoQ Onderste kwantificatielimiet (“Lower Limit of Quantitation”)
LoD Detectielimiet (“Limit of Detection”)
LoQ Kwantificatielimiet (“Limit of Quantitation”)
OLR Gewone lineaire regressie (“Ordinary Linear Regression”)
PI Predictie-interval (“prediction interval”)
psi “Pound-force per square Inch” (1 psi = 6,9 kPa)
p-waarde Probabiliteitswaarde
S/N Signaal tot ruis verhouding (“signal to noise ratio”)
SE Systematische fout (“Systematic Error”)
SFSTP “Société Française des Sciences et Techniques
Pharmaceutiques”

v
SST Systeemgeschiktheidstest (“System Suitability Test”)
sT Totale standaarddeviatie (“total standarddeviation”)
Swr Binnen-analyse standaarddeviatie (“within run
standarddeviation”)
TE Totale fout (“Total Error”)
TI Tolerantie-interval (“tolerance interval”)
TSH Thyroid Stimulerend Hormoon
UCL Bovenste betrouwbaarheidslimiet (“Upper Confidence Limit”)
ULoQ Bovenste kwantificatielimiet (“Upper Limit of Quantitation”)
USP “United States Pharmacopeia”
UV Ultraviolet
VIS Zichtbaar (“Visible”)
WLR Gewogen lineaire regressie (“Weighted Linear Regression”)

vi
DEFINITIES
De definities van de begrippen “measurand”, “analytical specificity”, “bias of measurements” en
“trueness” werden overgenomen uit EN/ISO 17511:
Measurand
Particular quantity subject to measurement.
Analytical specificity
Ability of a measurement procedure to measure solely the measurand.
Bias of measurements
Difference between the expectation of the results of measurement and a true value of the measurand.
Trueness of a measurement
Closeness of the agreement between the average value, obtained from a large series of results, and a
true value.
NOTE 1 Definition adapted from ISO 3534-1:1993, 3.12 that has ‘...test results and an accepted reference
value’, which can be a theoretical (true), assigned, consensus, or procedure-defined value.
NOTE 3 Trueness of measurement cannot be given a numerical value in terms of the measurand, only
ordinal values (e.g. sufficient, insufficient).
NOTE 4 The degree of trueness is usually expressed numerically by the statistical measure bias that is
inversely related to trueness and is the difference between the expectation of the results of
measurement and a true value of the measurand.
De definities van de begrippen “measurement procedure”, “accuracy”, “repeatability” en
“reproducibility” werden overgenomen uit de “Vocabulaire International des Termes Fondamentaux et
Généraux de Métrologie”:
Measurement procedure
Set of operations, described specifically, used in the performance of particular measurements according
to a given method.

vii
Accuracy of a measurement:
Closeness of the agreement between the result of a measurement and a true value of the measurand.
NOTE 1 Accuracy of measurement is related to both trueness of measurement and precision of
measurement.
NOTE 2 Accuracy cannot be given a numerical value in terms of the measurand, only descriptions such as
‘sufficient’ or ‘insufficient’ for a stated purpose.
NOTE 3 An estimator of an inverse measure of accuracy is “deviation”, defined as ‘value minus a
conventional true value’.
NOTE 4 ISO 3534-1, instead of “a true value” in the definition above, uses the concept “the accepted
reference value”, which can be a theoretical (true), assigned, consensus, or procedure-defined value.
Repeatability
Closeness of the agreement between the results of successive measurements of the same measurand
carried out under the same conditions of measurement.
Reproducibility
Closeness of the agreement between results of measurements of the same measurand carried out under
changed conditions of measurement.
De term “reference measurement procedure” werd overgenomen uit ISO 15193.
Reference measurement procedure
Thoroughly investigated measurement procedure shown to yield values having an uncertainty of
measurement commensurate with its intended use, especially in assessing the trueness of other
measurement procedures for the same quantity and in characterizing reference materials.

viii
Voor de term “precision” werd de definitie overgenomen uit ISO - Statistics - Vocabulary and symbols:
Precision of a measurement
The closeness of agreement between independent results of measurements obtained under stipulated
conditions.
Metrologische termen werden in het Nederlands vertaald volgens:
Accuracy Nauwkeurigheid
Trueness Juistheid
Precision Precisie
Repeatability Herhaalbaarheid
Reproducibility Reproduceerbaarheid
Limit of detection Aantoonbaarheidsgrens
Limit of quantification Bepaalbaarheidsgrens
Uncertainty of measurement Meetonzekerheid

1
1. INLEIDING
1.1. PARABENEN
1.1.1. Structuur en eigenschappen
Parabenen zijn alkylesters van parahydroxybenzoëzuur. Ze vormen een groep van chemische
producten, die gebruikt worden als conserveermiddel in cosmetica, voeding en farmaceutische
preparaten. Het gebruik van parabenen en hun zouten als conserveermiddel is te wijten aan hun
bactericide en fungicide eigenschappen. Ze zijn iets meer actief tegen fungi dan tegen bacteriën, maar
hun effectiviteit als bewaarmiddel, in combinatie met hun lage kostprijs en het feit dat ze al jarenlang
gebruikt worden, zorgt ervoor dat deze producten vandaag nog steeds heel vaak aangewend worden in
diverse producten en levensmiddelen (Soni et al., 2005). De apotheker kent deze parabenen dan ook en
gebruikt ze heel vaak in magistrale bereidingen voor de verduurzaming van bepaalde formulaties. In
zowel topische en vaginale preparaten, als in orale oplossingen en suspensies worden vaak parabenen
verwerkt. Ze zijn echter niet geschikt voor oftalmologische aanwending, gezien deze hydroxybenzoëzure
esters irritatie van de ogen kunnen veroorzaken.
Figuur 1.1. toont de algemene structuur van een parabeen, waarbij R een alkylketen voorstelt.
Parabenen bestaan uit een gesubstitueerde aromatische ring. Deze absorbeert elektromagnetische
straling in het UV-gebied. Hierdoor kunnen parabenen gedetecteerd worden met spectrofotometrische
technieken. De esters zijn witgekleurde, geurloze kristallijne poeders (The Merck Index, 13th edition).
Parabenen zijn actief in het pH-gebied 4 tot 8. De activiteit is iets hoger bij een lage pH. Bij een
pH hoger dan 8 wordt de esterbinding gehydrolyseerd en verliezen ze hun werking. De activiteit stijgt
naarmate de ketenlengte van de alkylgroep toeneemt, maar men moet er zich van vergewissen dat de
wateroplosbaarheid afneemt bij stijgende ketenlengte. Parabenen zijn dan ook slecht oplosbaar in
Figuur 1.1.: Algemene structuur van een parabeen

2
water, maar goed oplosbaar in ethanol en propyleenglycol. Deze laatste worden daardoor vaak gebruikt
als co-solvent in waterige magistrale bereidingen om de oplosbaarheid te verhogen. De oplosbaarheid in
water kan daarnaast ook nog opgekrikt worden door zouten te maken van deze geur- en kleurloze
esters. De zoutvorming heeft als nadeel dat de pH van het preparaat kan stijgen, waardoor de
esterbinding kan verbroken worden. Aqua conservans is een oplossing van methylparabeen en
propylparabeen in propyleenglycol of water; de combinatie van deze parabenen met korte alkylketen
zorgt voor een synergistisch effect en is een effectieve methode om de oplosbaarheid te verhogen.
Onder de commercieel beschikbare parabenen worden methyl-, ethyl-, propyl- en butylparabeen
heel vaak gebruikt. Minder vaak gebruikt zijn isopropyl-, isobutyl- en benzylparabeen. Alle parabenen die
in de handel verkrijgbaar zijn, worden synthetisch aangemaakt door esterificatie van
parahydroxybenzoëzuur met het gewenste alcohol. Sommige parabenen, zoals methylparabeen, komen
ook in de natuur voor.
Het werkingsmechanisme van parabenen als conserveermiddel berust op het feit dat ze de
fosfolipidendubbellaag kunnen penetreren. Op die manier zorgen ze voor een fysische verstoring van de
celmembraan met een desorganisatie van de protonengradiënt en verlies van chemi-osmotische kracht
tot gevolg (Denyer, 1995; Soni et al., 2005).
1.1.2. Controverse en toxiciteit
Het laatste decenium zijn parabenen in opspraak gekomen, omwille van hun mogelijke rol bij het
ontstaan van borstkanker. Eerst en vooral moeten we stellen dat epidemiologische, klinische en
experimentele studies bevestigen dat oestrogenen een belangrijke rol spelen bij de ontwikkeling, de
progressie en de behandeling van borstkanker. (Harvey, 2004). Dit roept de vraag op of chemicaliën, die
de menselijke cel kunnen binnentreden en oestrogene activiteit nabootsen een gelijkaardige rol hebben
bij borstkanker. Alle vaakgebruikte parabenen hebben oestrogene activiteit vertoond in zowel in vitro als
in vivo studies (Darbre & Harvey, 2008). Het is dan ook niet verbazend dat de meting van intacte esters
van zowel methyl-, ethyl-, propyl-, butyl-, als isobutylparabeen in humaan borstkankerweefsel de
internationale discussie rond deze bewaarmiddelen heeft gestimuleerd (Darbre et al., 2004).
Bij dermale applicatie kunnen parabenen doorheen de huid penetreren. De mate waarin de
absorptie plaatsvindt, stijgt met de lengte van de alkylketen, met uitzondering van methylparabeen dat
het makkelijkst doorheen de huid diffundeert (El Hussein et al., 2007). Hoewel er in de huid esterases
aanwezig zijn, die de esterbinding hydrolyseren, hebben studies aangetoond dat er bij applicatie toch

3
intacte parabenen in de diepere lagen van de huid voorkomen (Ishiwatari et al., 2007). Dit suggereert
dat de hydrolyse door de huidesterasen niet volledig is. Andere studies waarbij de concentratie van
intacte parabenen in bloed en urine werd gemeten, tonen gelijkaardige resultaten. De hoeveelheid
intacte parabenen die via de urine worden verwijderd, bleek echter laag (Soni et al., 2005).
Naast de absorptie blijkt ook de oestrogene activiteit van parabenen te stijgen met toenemende
ketenlengte (Byford et al., 2002). Meer recente inzichten tonen aan dat het weghalen van de alkylgroep
de oestrogene activiteit vermindert, maar niet volledig doet verdwijnen. Metabole hydrolyse van
parabenen door esterasen in de huid, lever en nier, met omzetting in parahydroxybenzoëzuur doet de
oestrogeniciteit dus niet volledig verdwijnen, in tegenstelling tot wat men eerst dacht (Darbre et al.,
2008).
Parabenen worden extensief gebruikt in de cosmetische, voedings- en farmaceutische industrie.
Vooral methyl- en propylparabeen zijn aanwezig in een grote meerderheid van cosmetische producten,
waaronder deodorants, crèmes en lotions. Moeten we ons, rekening houdend met de hierboven
beschreven controverse rond parabenen, zorgen maken over ons cosmeticagebruik? Voorlopig is er geen
sluitend antwoord te geven op deze vraag. Er zijn een aantal studies die beweren dat er een associatie
zou bestaan tussen het gebruik van parabenen in antitranspiratiemiddelen en het ontstaan van
borstkanker. Volgens Darbre et al. (2008) is er een disproportioneel hoge incidentie van borstkanker bij
vrouwen, waarbij de tumor zich bevindt in het bovenste, buitenste kwadrant van de borst, de regio het
dichtst bij de onderarm (waar de cosmetica wordt aangebracht). Of er echter een causaal verband
bestaat tussen het gebruik van parabenen in onderarmcosmetica en het ontwikkelen van borstkanker in
de regio het dichtst bij de onderarm is maar de vraag. Andere wetenschappers vermelden dat er een
grotere hoeveelheid epitheliaal borstweefsel aanwezig is in deze regio, wat dan de grotere incidentie zou
verklaren. Bovendien is uit in vivo studies gebleken dat ten opzichte van oestrogenen (als controle) de
parabenen met activiteit vele grootte-ordes minder actief zijn dan oestrogenen zelf (Golden et al., 2005).
Er bestaan ook studies die geen causaal verband hebben kunnen aantonen tussen parabeengebruik en
borstkanker. Het wetenschappelijk comité voor consumentenproducten van de Europese Unie volgt
eerder deze laatste resultaten, die de standpunten van Darbre weerleggen (SCCP, 2005).
In ieder geval is het wachten op gecontroleerde wetenschappelijke studies om onderbouwde
conclusies te kunnen stellen. Er is nood aan een gecontroleerde en gedetailleerde evaluatie van het
risico op borstkanker door het gebruik van cosmetica met parabenen. Verder moet er onderzocht
worden in hoeverre er interindividuele variatie bestaat tussen parabeenabsorptie, ontsnappen aan

4
esterase-activiteit en accumulatie in weefsels (Darbre et al., 2008). Bovendien kunnen enkel in vivo data
evidentie verschaffen over hoe hormoonactieve substanties beïnvloed worden door absorptie,
distributie, metabolisatie en excretie in het lichaam. Een gecombineerde in vitro/in vivo benadering is
nodig om een volledig begrip van de activiteit van de bewaarmiddelen in kwestie te bekomen (Golden et
al., 2005). In afwachting van deze studies laat de Europese Unie het gebruik van parabenen in
cosmetische producten toe met een maximale concentratie van elk 0,4% en een totale maximale
concentratie van 0,8% (EU Cosmetics Directive 76/768/EEC). Parabenen worden ook geregistreerd voor
hun gebruik in voedsel.
1.2. VALIDATIE
Als analyticus moet men weten dat vooraleer een methode kan aangewend worden, deze eerst
ontwikkeld moet worden en daarna gevalideerd. Door de grote verscheidenheid aan stalen, kolommen,
eluentia, operationele parameters… lijkt het ontwikkelen van een chromatografische methode erg
complex. Uiteraard is het kiezen van een doel en het consulteren van de literatuur een goede eerste
stap. Dit kan de onderzoeker heel wat onnodig experimenteel werk besparen. Toch kan men ook hier
nog steeds het overzicht over de vele publicaties verliezen. McDowall et al. hebben (naar USP<1058>)
een analytische kwaliteitsdriehoek opgesteld, waarvan een gereduceerde versie te zien is in Tabel 1.1. op
de volgende pagina (McDowall, 2010). Deze bondige versie van de analytische kwaliteitsdriehoek van
McDowall geeft een beknopt, maar duidelijk overzicht over hoe men van methode-ontwikkeling tot
goede analytische kwaliteit komt.
Het onderste deel van de analytische kwaliteitsdriehoek vormt de basis voor alle verdere
stappen en vertelt dat er eerst moet nagegaan worden of het toestel geschikt is voor het beoogde doel.
De analytische instrumentkwalificatie kan verder onderverdeeld worden in vier fasen, ook gekend als de
vier Q’s:
“Design Qualification”: definieert de vereisten (bijvoorbeeld vereisten voor de kwaliteit die nodig is
voor de analyse) van de gebruiker alvorens het toestel wordt aangekocht.
“Installation Qualification”: demonstreert dat de verschillende elementen correct geïnstalleerd
werden.
“Operation Qualification”: toont aan dat het geïnstalleerde systeem aan de specificaties voldoet.
“Performance Qualification”: toont aan dat het systeem dezelfde prestatie zoals gedefinieerd blijft
behouden.

5
Tabel 1.1.: Gereduceerde versie van de analytische kwaliteitsdriehoek volgens McDowall.
Wanneer wordt het
uitgevoerd?
Wat wordt er
gecontroleerd?
Tijdens de analytische metingen of experimenten
Systeemdrift
Kan bias tussen verschillende metingen of experimenten identificeren
Op de dag van de analyse
Voor de metingen
Bevestigt dat het systeem binnen vooraf bepaalde limieten functioneert
Voor applicatie van de methode
Bevestiging van operationele parameters
Monstervoorberei-ding
Bij het initieel opzetten van het instrument
Op regelmatige intervallen daarna
Geschiktheid van het instrument
Na deze eerste stap komt de analytische methodevalidatie, gevolgd door
systeemgeschiktheidscontroles. In deze thesis worden de systeemgeschiktheidscontroles omwille van
educatieve redenen gecombineerd met de methodevalidatie. Als laatste stap komen dan de interne
kwaliteitscontroles. Dit zou men kunnen zien als “methodeonderhoud”; de prestatie van de methode
moet continu op punt gehouden worden. In wat volgt wordt dieper ingegaan op de methodevalidatie. Er
wordt in deze inleiding niet verder ingegaan op de andere stappen van de analytische kwaliteitsdriehoek.
Deze scriptie is het relaas van de validatie van een vloeistofchromatografische methode voor de
bepaling van ethylparabeen. De International Organisation for Standardisation definieert in ISO-9000
validatie als: “The confirmation, through the provision of objective evidence, that requirements for a
specific intended use or application have been fulfilled.” Wanneer men een methode ontwikkelt, moet
deze dus voldoen aan specificaties voor een vooropgesteld doel (meestal is dat dan het oplossen van een
Analytische instrumentkwalificatie
Analytische methodevalidatie
Systeem-
geschiktheids-
controles
Kwaliteits- controles

6
analytische vraagstelling). Bij de start van een validatie maakt men een validatieplan op. Hierin worden
de verschillende uit te voeren stappen beschreven. Tabel 1.2. somt de verschillende stappen van een
validatieplan op wanneer een nieuw ontwikkelde methode wordt gevalideerd.
Tabel 1.2.: Validatieplan gebruikt bij de validatie van een nieuw ontwikkelde analytische methode (Stöckl, 2007)
1. Definieer het gebruik, het doel en het bereik van de methode
2. Definieer de prestatiekenmerken en de aanvaardbaarheidscriteria
3. Ontwikkel een validatieprotocol of werkprocedure voor de validatie
4. Bepaal de materialen bv. standaarden, reagentia en stalen
5. Voer de validatie-experimenten uit
6. Documenteer de validatie-experimenten en de resultaten in een validatierapport
7. Interpreteer de validatiegegevens en maak op statistiek gebaseerde beslissingen
Bij de validatie van een chromatografische methode worden verschillende
prestatiekarakteristieken onderzocht. Tabel 1.3. vat de verschillende prestatiekarakteristieken samen,
die in deze meesterproef aan bod komen. Deze worden verder in deze scriptie onder 3.2.4. ‘Validatie-
experimenten’ uitgebreider besproken.
Tabel 1.3.: Overzicht van de verschillende prestatiekarakteristieken die in deze thesis worden gecontroleerd
Bij het onderzoeken of aan de verschillende prestatiekenmerken wordt voldaan, wordt er
volgens een bepaald protocol gewerkt. Deze protocollen schrijven voor welke stappen er moeten
gevolgd worden om tot een bepaald resultaat te komen. Tijdens deze meesterproef wordt er gewerkt
volgens experimentele protocollen opgesteld door CLSI (“Clinical and Laboratory Standards Institute”). Er
wordt verder ingegaan op deze protocollen onder 3.2.4. ‘Validatie-experimenten’.
De validatiegegevens worden uiteindelijk met behulp van statistische (significantie)testen
geïnterpreteerd. Voor de validatie wordt onder meer gebruik gemaakt van “Method validation with
confidence”, geschreven door Dr. D. Stöckl. Gezien de concentratie van een staal nooit met absolute
zekerheid kan bepaald worden en altijd een schatting blijft, moet men een confidentie-interval (CI)
Prestatiekarakteristiek
Lineariteit
Imprecisie
Detectielimiet
Juistheid (Terugvinding)
Methodevergelijking

7
rapporteren om, met een bepaalde zekerheid, een idee te krijgen van de ligging van de ware waarde. De
titelkeuze voor het werk van Dr. D. Stöckl is dus niet in het minst toevallig. Tijdens een analytisch proces,
met staalname, staalopzuivering en de uiteindelijke analyse, treden fouten op. Voor elke meting bestaat
er dus een onzekerheid op de resultaten. Door het opstellen van confidentie-intervallen kunnen we toch
betrouwbare conclusies trekken uit de bekomen resultaten.
Het interpreteren van een confidentie-interval vraagt een woordje uitleg. We beschouwen eerst
de situatie waarbij een bepaald resultaat vergeleken wordt met de specificatie voor een stabiel proces.
Wanneer bij de meting van een staal de doelwaarde bekend is via een referentiemethode (in dit geval
bijvoorbeeld 10 ppm) en in het CI ligt, dan kunnen we op het 100(1-α)% significantieniveau besluiten dat
het bekomen resultaat niet significant verschilt van de doelwaarde.
Figuur 1.2. geeft een grafische voorstelling van de hierboven beschreven problematiek. Bij
situatie A en D ligt de doelwaarde (10 ppm) niet in het CI en kunnen we op het 100(1-α)%
significantieniveau besluiten dat de meting significant verschilt van de doelwaarde. In situatie B en C is
de doelwaarde wel inbegrepen in het CI. Deze metingen verschillen niet significant van de 10 ppm.
De interpretatie van deze intervallen verschilt echter wanneer er vergeleken wordt ten opzichte van een
limiet, bijvoorbeeld wanneer er nagegaan wordt of de metingen een bepaalde waarde (opnieuw 10 ppm)
al dan niet overschrijden. Opnieuw wordt Figuur 1.2. beschouwd, maar nu wordt deze anders
geïnterpreteerd. Situatie A in Figuur 1.2. overschrijdt de limiet niet op het 100(1-α)% significantieniveau.
Ofschoon bij situatie B het meetresultaat onder de grens ligt, zou het toch kunnen dat de maximale
concentratie wordt overschreden. In dit geval kunnen meerdere metingen het CI smaller maken en op
die manier uitsluitsel bieden. In geval C en D wordt de limiet van 10 ppm overschreden op het 100(1-α)%
significantieniveau.
Figuur 1.2.: Grafische weergave van verschillende situaties bij het interpreteren van confidentie-intervallen

8
2. OBJECTIEVEN
Deze scriptie bestaat uit twee luiken. Het eerste luik is een experimenteel gedeelte waarin de
validatie van een vloeistofchromatografische methode voor de bepaling van ethylparabeen wordt
uitgevoerd. Het tweede deel bestaat uit een literatuuronderzoek.
Voor het experimenteel gedeelte wordt door het Laboratorium voor Analytische Chemie
gedurende de volledige duur van de meesterproef een HPLC systeem ter beschikking gesteld. Bij de
methodevalidatie worden verschillende prestatiekarakteristieken onderzocht. Deze omvatten lineariteit,
imprecisie, detectielimiet, juistheid en uiteindelijk wordt er nog een methodevergelijking uitgevoerd.
Bovendien wordt er bij deze validatie, na een korte periode van kennismaking met de apparatuur,
zelfstandigheid verwacht bij het onderhouden en controleren van het toestel en materiaal. Voor het
plannen, uitvoeren, interpreteren en rapporteren van de analytische validatie-experimenten wordt in
eerste instantie bijstand verleend, maar finaal wordt ook bij deze zaken zelfstandigheid verwacht. Het
eluens en de verschillende analysestalen worden zelf bereid. Systeemfunctie- en
systeemgeschiktheidscontroles worden dagelijks bij aanvang van de metingen uitgevoerd.
In het tweede luik van deze meesterproef, de literatuurstudie, wordt er op zoek gegaan naar
informatie over het tolerantie-interval. Daarnaast wordt nagegaan of dit statistisch interval applicatie
vindt in de farmaceutische methodevalidatie. Uiteraard heeft de literatuurstudie ook als doel het
efficiënt en doelgericht zoeken naar informatie en het kritisch evalueren van de verschillende
informatiebronnen.

9
3. MATERIALEN EN METHODEN
3.1. MATERIALEN
3.1.1. Oplosmiddel en eluens
Voor het aanmaken van de stockoplossing van ethylparabeen en voor de testmix wordt gebruik
gemaakt van een mengsel van 60% methanol en 40% water als oplosmiddel. Er wordt methanol
(gradiënt kwaliteit) van ROMIL-SpSTM (Cambridge, GB) gebruikt. Methanol heeft een relatieve
moleculemassa van 32.04. De zuiverheid van het gebruikte methanol bedraagt ≥99.9%. Het water
(gradiënt kwaliteit) dat wordt gebruikt, wordt eveneens bekomen bij ROMIL-SpSTM. De relatieve
moleculemassa bedraagt 18.02; het residu is kleiner dan 0.0001%; de resistiviteit is groter dan 18
MOhm.m bij 25°C. De zuiverheid van het oplosmiddel wordt getest door injectie in het chromatografisch
systeem. Hierbij ligt de nadruk op het al dan niet voorkomen van interferenties in het retentiebereik van
3 tot 4 minuten.
Het eluens dat dagelijks wordt aangemaakt en gebruikt, heeft dezelfde samenstelling als het
oplosmiddel (60% methanol en 40% water). Methanol en water worden na mengen aan filtratie
onderworpen. Dit heeft als doel eventuele deeltjes uit de vloeistof te verwijderen. Bij het filtreren wordt
gebruik gemaakt van een “Alltech Solvent Filtration” apparaat (Deerfield, IL, VSA). Membraanfilters van
Durapore met poriëngrootte van 0.45 µm zijn van Millipore (Bedford, MA, VSA). Finaal wordt het eluens
nog in een sonificatiebad geplaatst (Branson 1210, Gent, België) om het te ontgassen. Het eluens wordt
elke morgen opnieuw aangemaakt in glazen flessen van 1000 ml met draaidop van Schott Duran (Mainz,
Duitsland).
De stockoplossing wordt aangemaakt in vials van 14 ml van GRACE (Deerfield, IL, VSA) met
draaidop. De standaarden worden aangemaakt in glazen maatkolven van Schott Duran.
3.1.2. Bereiding van de stockoplossing, standaarden en stalen
Alle oplossingen worden gravimetrisch aangemaakt en bewaard bij 4°C. Voor de stockoplossing
wordt ethylparabeen (“ethyl-4-hydroxybenzoate”) gebruikt, aangekocht bij Sigma-Aldrich (Bornem,
België, St Louis MO). Deze substantie heeft een relatieve moleculemassa van 166.2 en een zuiverheid
van ≥99%. Er wordt 6.160 mg ethylparabeen nauwkeurig afgewogen en opgelost in 9.238 g oplosmiddel.
De concentratie van de stockoplossing bedraagt op die manier 666.4 µg/g. Er wordt daarnaast ook een
aantal tussenverdunningen aangemaakt uit de stockoplossing. Tussenverdunning 1 heeft een

10
concentratie van 18.52 µg/g, tussenverdunning 2 een concentratie van 1.693 µg/g en tussenverdunning
3 heeft als concentratie 0.1713 µg/g.
Voor de verschillende validatie-experimenten worden een aantal standaarden en stalen
aangemaakt.
3.1.2.1. Lineariteit en kalibratie
Voor lineariteit en kalibratie worden primair twee standaarden aangemaakt, één met hoge en
één met lage concentratie. De standaard met lage concentratie wordt standaard 1 genoemd, deze met
hoge concentratie wordt standaard 5 genoemd. Standaarden 2, 3 en 4 worden aangemaakt volgens het
alternatieve mengprotocol zoals beschreven staat in het CLSI EP6 protocol (“Clinical and Laboratory
Standards Institute”). Tabel 3.1. geeft dit mengprotocol weer.
Tabel 3.1. : Alternatief mengprotocol beschreven in het CLSI EP6 protocol
1. Laag
2. Laag medium
3. Medium
4. Hoog medium
5. Hoog
Tussenverdunning 2 wordt 62 keer verdund
laag + medium (1:1)
laag + hoog (1:1)
hoog + medium (1:1)
Tussenverdunning 1 wordt 14 keer verdund
Uit oriënterende experimenten is gebleken dat de concentratie die overeenkomt met de
detectielimiet (“Limit of Detection”, LoD) ongeveer gelijk is aan 0.0050 µg/g. Wanneer we deze
concentratie injecteren, wordt een signaal tot ruis verhouding (“signal to noise ratio”, S/N ratio) van
ongeveer 3 bekomen. De concentratie van standaard 1 moet ongeveer gelijk zijn aan een concentratie
die een S/N ratio van 10 oplevert. Om een voldoende groot dynamisch bereik te bekomen, wordt ervoor
gezorgd dat standaard 1 een concentratie heeft van om en bij 0.015 µg/g (wat overeenstemt met 3 keer
LoD) en standaard 5 een concentratie van ongeveer 50 keer de concentratie van standaard 1 (0.75 µg/g).
Standaard 1 wordt ook soms de onderste kwantificatielimiet (“Lower Limit of Quantification”, LLoQ)
genoemd. Analoog wordt standaard 5 de bovenste kwantificatielimiet (“Upper Limit of Quantification”,
ULoQ) genoemd. Uiteindelijk zijn 0.02711 µg/g en 1.348 µg/g de concentraties van respectievelijk
standaard 1 en 5. De concentratie van standaard 5 is hoger dan wat we theoretisch hebben berekend,
maar dat vormt voor de doeleinden van deze scriptie geen probleem. Voor standaarden 2, 3 en 4 wordt
gebruik gemaakt van het mengprotocol, zoals beschreven in Tabel 3.1. Aan de hand van de massa’s die

11
gepipetteerd worden, kan de exacte concentratie van de standaarden berekend worden. Tabel 3.2. zet
de concentraties van de standaarden op een rij.
Tabel 3.2. : Concentraties van standaarden 1 tot en met 5
Standaard Concentratie (µg/g)
1 0.02711
2 0.3566
3 0.6869
4 1.017
5 1.348
3.1.2.2. Imprecisie
Voor de evaluatie van de imprecisie worden twee interne kwaliteitscontrole stalen (“Internal
Quality Control”, IQC) gemaakt. Eén van de stalen heeft een hoge concentratie (High IQC), de andere een
lage concentratie (Low IQC). De concentratie van het High IQC staal bedraagt 0.8853 µg/g, die van het
Low IQC staal 0.4598 µg/g. Beide stalen worden bereid vertrekkende uit de stockoplossing.
3.1.2.3. Detectielimiet
Voor de evaluatie van deze prestatiekarakteristiek wordt gedurende vijftien dagen elke dag een
nieuw staal aangemaakt met een concentratie die een S/N ratio van 3 tot 6 oplevert. Voor het aanmaken
van deze stalen wordt vertrokken uit standaard 1. Zoals in 3.1.2.1. ‘Lineariteit en kalibratie’ al werd
vermeld, was uit oriënterende experimenten gebleken wat de concentratie ongeveer is die een S/N ratio
van 3 oplevert (ongeveer 0.0050 µg/g). Op die manier kunnen we berekenen hoe sterk we standaard 1
moeten verdunnen.
3.1.2.4. Juistheid
Van de zes stalen, die gebruikt worden voor het nagaan van de juistheid, werden er drie
aangemaakt door het Laboratorium voor Analytische Chemie. Daarnaast waren twee van de zes stalen
dezelfde als de stalen voor imprecisie (High IQC en Low IQC). Het laatste staal wordt door onszelf
aangemaakt. De concentratie van de verschillende stalen voor juistheid staat opgelijst in Tabel 3.3.

12
Tabel 3.3. : Concentraties van de zes verschillende stalen voor juistheid
3.1.2.5. Systeemgeschiktheidstest
Voor het nagaan van de geschiktheid van het systeem wordt een testmix gebruikt. Deze bevat 4-
hydroxybenzoëzuur, methylparabeen, ethylparabeen, propylparabeen en butylparabeen. Deze
componenten hebben allemaal een concentratie van ongeveer 2.5 µg/g. Tabel 3.4. vat de naam, de
fabrikant, relatieve moleculemassa en zuiverheid van de gebruikte componenten in de testmix samen.
Tabel 3.4. : Naam, naam van de fabrikant, relatieve moleculemasse en zuiverheid van de componenten in de testmix
Naam Fabrikant Realtieve moleculemassa Zuiverheid
4-hydroxybenzoëzuur Sigma Aldrich 138.1 ≥99%
Methylparabeen Sigma Aldrich 152.2 99%
Ethylparabeen Sigma Aldrich Fluka 166.2 ≥99%
Propylparabeen Sigma Aldrich 180.2 ≥99%
Butylparabeen Sigma Aldrich 194.2 ≥99%
3.1.3. Apparatuur
3.1.3.1. Analyse
Voor de experimenten wordt een HPLC-systeem van SHIMADZU (Kyoto, Japan) gebruikt, met een
Prominence serie LC-20 AT pomp, Prominence DGU 20A5 ontgasser, Prominence serie SPD-20 A UV/VIS
detector en injector van Rheodyne (Rohnert Park, CA, VSA) model 7725i, voorzien van een 5 µl loop.
SHIMADZU LC Solution® wordt als software gebruikt om de data te registreren.
Er wordt gebruik gemaakt van een ODS Hypersil C18 kolom (150 mm x 4.6 mm interne diameter;
partikeldiameter 5 µm; poriëngrootte 120 Å) aangekocht bij Thermo Electron Corp (Waltham, MA, VSA).
Nummer juistheidsstaal Concentratie (µg/g)
1 0.06106
2 0.1088
3 0.3368
4 (= Low IQC) 0.4598
5 0.7570
6 (= High IQC) 0.8853

13
3.1.3.2. Randapparatuur
Bij het afwegen van de materialen, standaarden en stalen wordt gebruik gemaakt van de AT261
DeltaRange® analytische balans van Mettler Toledo (Griefensee, Zwitserland) met nauwkeurigheid tot op
10-5g. Voor het aanmaken van de stalen worden pipetten van Socorex (Eclubens, Zwitserland) gebruikt.
De standaarden en stalen worden geïnjecteerd met een injectienaald van 50µl van Hamilton,
Bonaduz Schweiz.
3.2. METHODEN
3.2.1. Systeemfunctiecontrole
Vooraleer de eigenlijke metingen te starten, wordt elke dag nagegaan of het systeem nog naar
behoren functioneert. Bij deze systeemfunctiecontrole (“system function check”) wordt gecontroleerd of
het systeem nog voldoet aan een aantal vooropgestelde operationele parameters. Tabel 3.5. vat deze
verschillende parameters en hun specificatie samen.
Tabel 3.5. : Parameters en specificaties die worden nagegaan bij de systeemfunctiecontrole
Parameter Specificatie
Detector Maximaal aantal uur gebruik D2 lamp 2000h
Staalenergie bij 220 nm >800mV
Referentie-energie bij 220 nm >800mV
Maximaal toelaatbare ruis <0.006mV
Maximale variatie op stabiele basislijn gedurende 5 minuten <0.01mV
Pomp Totaal geleverd volume
Nauwkeurigheid van het debiet
<180L
± 5%
3.2.2. Systeemgeschiktheidscontrole
Elke dag wordt bij aanvang van de metingen naast het uitvoeren van de systeemfunctiecontrole
ook gecontroleerd of het systeem nog geschikt is voor de beoogde doelstelling. Een aantal
chromatografische parameters wordt dagelijks vergeleken met vooropgestelde limieten, zoals
aangegeven in Tabel 3.6. Hiervoor wordt een testmix gebruikt, zoals beschreven onder 3.1.2.5.
‘Systeemgeschiktheidstest’. Een aantal van de chromatografische parameters uit Tabel 3.6. wordt nog

14
eens opgelijst in Tabel 3.7., samen met de formules zoals beschreven in “United States Pharmacopeia”
(USP).
Tabel 3.6. : Vooropgestelde limieten voor de chromatografische parameters, gebruikt bij de systeemgeschiktheidscontrole voor ethylparabeen
Chromatografische parameter Vooropgestelde limieten voor ethylparabeen
Retentietijd (min) 3.4 ± 0.2
Piekoppervlakte 80000 ± 50%
Piekhoogte 10000 ± 50%
Theoretisch aantal platen minimum 2000
Tailing factor maximum 2
Resolutie minimum 2
Tabel 3.7. : Chromatografische parameters, gebruikt voor de systeemgeschiktheidscontrole met de formules zoals beschreven in USP
Chromatografische parameter Formule zoals beschreven
in USP
Verklaring van de gebruikte symbolen
Theoretisch plaatgetal N = 16 x (tR/W)² N = theoretisch plaatgetal
tR = retentietijd (min)
W = piekbreedte op de basislijn (min)
Asymmetriefactor
(“tailing factor”)
Tf = W0.05/(2 x a0.05) Tf = asymmetriefactor
W0.05 = piekbreedte op 5% van de
piekhoogte (min)
a0.05 = piekbreedte op 5% piekhoogte
van de start van de piek tot aan het
snijpunt van de loodrechte uit de top
van de piek (min)
Resolutie R = 2 x (tR – tRp)/(W+Wp) R = resolutie
tR = retentietijd van de laatst eluerende
piek (min)
tRp = retentietijd van de voorgaande piek
(min)
W = piekbreedte op de basislijn van de
laatst eluerende piek (min)
Wp = piekbreedte op de basislijn van de
vorige piek (min)

15
3.2.3. Analyse
De mobiele fase die gebruikt wordt tijdens deze meesterproef en dagelijks vers bereid wordt,
bestaat zoals reeds eerder vermeld onder 3.1.1. ‘Oplosmiddel en eluens’ uit 60% methanol en 40%
water. Dit eluens wordt over de kolom gepompt met een debiet van 1.1 ml/min. Er wordt gewerkt met
een isocratische methode. Er wordt geïnjecteerd volgens de overvultechniek, waarbij er met de
injectienaald ongeveer 30 µl staal wordt opgezogen, maar bij elke meting slechts 5 µl geïnjecteerd wordt
(loop van 5 µl).
De UV/VIS detector werd ingesteld op 258 nm voor de metingen. Figuur 3.1. toont een
voorbeeldchromatogram van standaard 4, gemeten op 23/03/2011.
Figuur 3.1.: Voorbeeldchromatogram van standaard 4, gemeten op 23/03/2011. De concentratie van standaard 4 bedraagt 1.017 µg/g
3.2.4. Validatie-experimenten
Om de validatie op een correcte manier uit te voeren, moet er volgens een geschikt protocol
gewerkt worden. Voor deze studie wordt geopteerd voor de experimentele protocollen van CLSI. Voor
de prestatiekarakteristiek imprecisie wordt er volgens een gewijzigd CLSI protocol gewerkt. Tabel 3.8.
somt de verschillende prestatiekarakteristieken op die gevalideerd worden met daarbij het CLSI protocol
dat wordt gebruikt.

16
Tabel 3.8. : De verschillende prestatiekarakteristieken die worden gevalideerd met het (gewijzigde) CLSI protocol
Prestatiekarakteristiek Aard stalen en aantal metingen (n) volgens het
(gewijzigde) protocol
Code van het CLSI
protocol
Lineariteit
Vijf gerelateerde kalibratoren, n = 4 (binnen één dag).
Het experiment wordt één keer herhaald ter
bevestiging
EP6
Imprecisie Twee IQC stalen, duplicaat, 15 dagen (n=15) EP5 (gewijzigd
protocol)
Detectielimiet Singlicaat, 15 dagen (n=15) n.v.t. (generisch
protocol)
Juistheid Singlicaat, 5 dagen (n=5) EP15
Methodevergelijking Hiervoor werden de stalen en resultaten gesimuleerd EP9
3.2.4.1. Lineariteit
Voor de evaluatie van de lineariteit, waarbij wordt nagegaan of het verband tussen de
piekoppervlakte en de concentratie van de analyt lineair is, wordt het CLSI EP6 protocol, zoals
aangegeven in Tabel 3.8. gevolgd. Standaarden 1 tot en met 5 worden vier maal per dag gemeten. Bij de
eerste meting worden de standaarden oplopend gemeten, daarna twee maal in willekeurige volgorde en
bij de vierde meting wordt er in aflopende concentratie gemeten. Het experiment wordt één keer
herhaald ter bevestiging.
De gegevens voor lineariteit worden grafisch weergegeven met behulp van een
spreidingsdiagram en een residuendiagram. Deze diagrammen geven een eerste aanwijzing voor het al
dan niet lineair zijn. Uiteraard moeten er nog statistische significantietesten volgen om te kunnen
besluiten of aan deze prestatiekarakteristiek is voldaan (aan de hand van de diagrammen zou men
slechts intuïtief een vermoeden kunnen uitspreken). Met behulp van het residuendiagram kan men de
data evalueren op de mogelijke aanwezigheid van uitschieters. De dataset met potentiële uitschieters
wordt aan een Grubbs test onderworpen (5% significantieniveau). Wanneer uit deze test blijkt dat de
data een uitschieter bevatten, dan wordt de uitschieter voor verdere interpretatie weggelaten uit de
gegevensreeks.
De lineariteitsgegevens worden statistisch geëvalueerd op twee manieren. Met het programma
CBstat5 wordt enerzijds een “lack-of-fit” test uitgevoerd en anderzijds een tweede orde polynomiale
regressieanalyse.

17
Bij de “lack-of-fit” test wordt enerzijds berekend wat de afstand is van het gemiddelde van de
vier metingen van eenzelfde staal tot de regressielijn. Anderzijds wordt ook bepaald wat de variantie is
tussen de verschillende metingen van hetzelfde staal (de binnen-staal variantie). De spreiding
(=variantie) op de vijf gemiddelden van de standaarden wordt vergeleken met de binnen-staal variantie.
Dit gebeurt met behulp van een eenzijdige F-toets op het 5% significantieniveau. Indien de
probabiliteitswaarde (p-waarde) groter is dan 0.05 is de afwijking statistisch niet significant en kunnen
we besluiten dat er een lineair verband is tussen de piekoppervlakte en de concentratie van de
standaarden. Afhankelijk van het homo- of heteroscedastisch verdeeld zijn van de data, wordt gebruik
gemaakt van respectievelijk de “Ordinary Least Squares” regressieanalyse of de “Weighted” lineaire
regressieanalyse.
Gezien het “lack-of-fit” model gevoelig is aan spreiding van de resultaten, wordt er tevens een
tweede orde polynomiale regressieanalyse uitgevoerd. Bij deze analyse wordt er een
tweedegraadsvergelijking (van de vorm ax² + bx + c) opgesteld, gebruikmakend van de lineariteitsdata.
Daarna wordt met behulp van een tweezijdige t-toets (5% significantieniveau) nagegaan of de coëfficiënt
a in de veelterm ax² + bx + c significant verschillend is van nul. Als de p-waarde groter is dan 0.05 dan kan
de nulhypothese (het lineair zijn) weerhouden worden. Mochten de data niet lineair zijn, dan wordt het
verschil tussen de eerste en tweede orde vergelijking gemaakt. Als dat verschil kleiner is dan 5%, dan
wordt dat als verwaarloosbaar beschouwd en kan er alsnog met een lineaire kalibratiecurve gewerkt
worden (verwaarloosbare fout).
3.2.4.2. Kalibratie
Wanneer de analyticus wil kwantificeren, is het zeer belangrijk een geschikt kalibratiemodel te
kiezen. Afhankelijk van de uitkomst van de bepaling van de lineariteit (zie 3.2.4.1. ‘Lineariteit’) zal een
eerste- of tweedegraads kalibratiecurve worden opgesteld.
Uiteraard volstaat het niet om te zeggen dat er voor een eerste- of tweedegraadsvergelijking
wordt gekozen. Er moet bepaald worden welk regressiemodel het best passend is voor de bepaling van
de concentratie uit de piekoppervlakte. Als de methode voldoet aan de voorwaarden voor lineariteit,
worden er vier verschillende regressiemodellen getest. Deze zijn de standaard gewone lineaire
regressie (“Ordinary Linear Regression”, OLR), de OLR geforceerd door nul, de OLR met het punt
(0,0) ingesloten en de gewogen lineaire regressie (“Weighted Linear Regression”, WLR). Voor vijf
verschillende meetdagen wordt via deze vier regressiemodellen de gemiddelde concentratie van het

18
Low IQC staal, High IQC staal en het juistheidsstaal met de laagste concentratie (juistheidsstaal 1)
berekend. Daarna worden van deze gemiddelde concentraties de variatiecoëfficiënt (“coefficient of
variation”, CV) en het procentueel verschil met de gravimetrisch bepaalde concentratie van de
stalen bepaald (zie Formule 3.1.). De regressieanalyse die resulteert in de laagste CV en het kleinste
procentueel verschil wordt als het meest adequaat beschouwd.
Procentueel verschil (%) = (xgemeten – xdoel) / xdoel x 100 (3.1.)
waarbij: xgemeten = gemeten waarde
xdoel = doelwaarde
Daarnaast wordt voor het OLR model ook nog een 95% CI voor het intercept opgesteld om na te gaan of
deze significant verschillend is van nul. Indien dit het geval is, dan kan OLR door nul gebruikt worden.
Deze methode wordt toegepast wanneer het verschil tussen OLR en OLR geforceerd door nul klein is.
OLR geforceerd door nul laat immers eenvoudiger berekeningen toe.
3.2.4.3. Imprecisie
Bij het nagaan of er voldaan is aan deze prestatiekarakteristiek wordt het gewijzigd CLSI EP5
protocol zoals beschreven in Tabel 3.8. gevolgd. Er worden twee stalen, één met lage en één met hoge
concentratie (respectievelijk Low IQC en High IQC) gedurende vijftien dagen in duplicaat gemeten. In het
oorspronkelijke CLSI protocol worden twintig metingen uitgevoerd in plaats van vijftien.
Voor zowel Low IQC als voor High IQC wordt het verschil tussen de duplicaten berekend. Van
deze verschillen wordt een puntendiagram gemaakt, wat ons een idee geeft over de binnen-analyse
spreiding van de resultaten. Met een Grubbs test wordt nagegaan of er uitschieters aanwezig zijn.
Mogelijk aanwezige uitschieters worden voor verdere interpretatie van de resultaten uit de dataset
verwijderd. In dat geval wordt er verder gewerkt met één van beide meetresultaten en niet met het
gemiddelde van die dag.
Ook wordt er een puntendiagram opgesteld voor de daggemiddelden (of althans voor de
gewijzigde dataset, mochten er uitschieters opgedoken zijn volgens de in de vorige alinea beschreven
methode). Dit puntendiagram laat toe de tussen-dag spreiding visueel waar te nemen. Potentiële
uitschieters springen ook makkelijk in het oog in dit puntendiagram. Een Grubbs test volgt. Uitschieters
worden weggelaten uit de dataset en worden niet gebruikt voor de evaluatie van de imprecisie.

19
De berekeningen voor de CV’s kunnen op twee verschillende manieren gebeuren. Men kan de
formules gebruiken die vermeld staan in het CLSI EP5 protocol of men kan gebruik maken van ANOVA
model II. Zowel voor Low IQC, als voor High IQC worden het gemiddelde, de binnen-analyse (“within
run”) standaarddeviatie (swr), de totale standaarddeviatie (sT), de “within run” CV (CVwr) en de totale CV
(CVT) berekend. Deze CV’s worden vergeleken met de doelwaarden voor een stabiel proces. Voor CVwr is
dat 2%, voor CVT 5%. Er wordt nagegaan of het eenzijdig 95% CI van de variantie van de datareeks de
doelwaarde van de variantie voor een stabiel proces insluit. Er wordt met andere woorden getest of de
onderste limiet van het eenzijdig 95% confidentie-interval (“lower confidence limit”, LCL) onder de
doelwaarde gelegen is. Het berekenen van deze LCL van de standaarddeviatie en CV gebeurt volgens
respectievelijk Formule 3.2. en Formule 3.3.
LCL van s = s x √*(df)/Chi²α,df] (3.2.)
waarbij: s = standaarddeviatie
df = “degrees of freedom”, aantal vrijheidsgraden
Chi²α,df = kritische chi²-waarde (berekend in Excel®) met
α = 0.05
LCL van CV = (LCL van s / xgem) * 100 (3.3.)
waarbij: xgem = gemiddelde van de reeks metingen
De imprecisie van de methode kan ook onderzocht worden aan de hand van het vergelijken van
de experimentele Chi²-waarde (Chi²exp) (zie Formule 3.4.) met de kritische Chi²-waarde (Chi²krit) (wordt
met Excel® berekend). Er wordt aan de specificaties voldaan wanneer de experimentele Chi²-waarde
kleiner of gelijk is aan de kritische Chi²-waarde.
Chi²exp = s²exp × df / s²spec (3.4.)
waarbij: Chi²exp = experimentele Chi²-waarde
s²exp = experimentele variantie
s²spec = specificatie voor de variantie
df = “degrees of freedom”, aantal vrijheidsgraden

20
3.2.4.4. Detectielimiet
De bepaling van de detectielimiet gebeurt volgens het generisch protocol zoals beschreven in
Tabel 3.8. Bij dit protocol, wordt gedurende vijftien dagen elke dag een vers staal (met gravimetrisch
bepaalde concentratie) gemeten, dat een S/N ratio van ongeveer 3 tot 6 oplevert. Voor het bepalen van
de ruis wordt op de basislijn een afstand van vijf maal de piekbreedte op halve hoogte beschouwd.
Figuur 3.2. toont hoe de ruis N wordt afgelezen op een (theoretisch) chromatogram. Het signaal S wordt
bepaald als de loodrechte afstand van het maximum van de piek tot het gemiddelde van de ruis (zie
Figuur 3.2.). Met deze gegevens wordt de experimenteel bepaalde S/N ratio berekend.
Van de gekende concentraties van de stalen wordt het gemiddelde berekend. Aan de hand van
Formule 3.5. wordt dan uiteindelijk een genormaliseerde S/N ratio berekend.
Genormaliseerde S/N ratio = (xgem / xgrav) x S/Nexp (3.5.)
waarbij: xgem = gemiddelde concentratie
xgrav = gravimetrisch bepaalde concentratie
S/Nexp = experimenteel bepaalde S/N ratio
Van deze vijftien S/N ratio’s wordt het gemiddelde en de standaarddeviatie berekend. Er wordt
een puntendiagram opgesteld van de genormaliseerde S/N ratio’s. Dit diagram geeft een visueel beeld
van de spreiding en laat toe uitschieters visueel op te merken. Met een Grubbs test wordt vervolgens
Figuur 3.2.: Bepaling van de ruis en het signaal uit een theoretisch chromatogram

21
getest of er uitschieters aanwezig zijn in de dataset. Potentiële uitschieters worden voor verdere
interpretatie van de gegevens weggelaten. Uiteindelijk wordt er ook een tweezijdig 95% CI opgesteld
rond de gemiddelde genormaliseerde S/N ratio (zie Formule 3.6.).
95% CI = [S/Ngem ± tα/2,n-1 s/√n+ (3.6.)
waarbij: S/Ngem = gemiddelde genormaliseerde S/N ratio
tα/2,n-1 = t-waarde met probabiliteit α (= 0.05) en n-1
vrijheidsgraden
s = standaarddeviatie
n = aantal metingen
De detectielimiet wordt in deze thesis als een descriptieve meting beschouwd. We rapporteren
enkel de gemiddelde gravimetrisch bepaalde concentratie van de stalen, samen met het 95% CI rond de
gemiddelde genormaliseerde S/N ratio.
Finaal wordt de gemiddelde absolute hoeveelheid ethylparabeen, die dagelijks geïnjecteerd
wordt, bepaald. Formule 3.7. toont hoe deze absolute hoeveelheid ethylparabeen berekend wordt.
Gemiddelde absolute hoeveelheid ethylparabeen (pg) = xgem x Vinj x ρ x 1000000 pg/µg (3.7.)
waarbij: xgem = gemiddelde concentratie
Vinj = geïnjecteerd volume
ρ = dichtheid van het oplosmiddel
3.2.4.5. Juistheid
Voor de evaluatie van de prestatiekarakteristiek juistheid wordt het CLSI protocol zoals
beschreven in Tabel 3.8. gevolgd: zes stalen van gekende concentratie worden in singlicaat gemeten
gedurende vijf dagen. De verschillen tussen de berekende dagconcentraties voor de verschillende stalen
en de gemiddelde concentratie worden uitgezet in een puntendiagram. Met dit diagram krijgen we een
idee van de spreiding van de resultaten en kunnen we een vermoeden uitspreken over het al dan niet
aanwezig zijn van uitschieters. Potentiële uitschieters worden aan een Grubbs test onderworpen. Met
uitschieters wordt voor verdere interpretatie van de resultaten geen rekening gehouden.

22
Voor de verdere evaluatie van de juistheid wordt gekeken naar het procentueel
verhoudingsdiagram, waarin de terugvinding van de doelwaarde (zie Formule 3.8.) voor alle stalen
uitgedrukt in procent grafisch weergegeven wordt. De juistheid wordt berekend met Formule 3.8., 3.9.
en 3.10.
Terugvinding = 100 x (xgem. gemeten/xdoel) ± 95% CI (3.8.)
waarbij: xgem. gemeten = gemiddelde gemeten waarde
xdoel = doelwaarde
Absoluut 95% CI = [xgem ± tα,n-1 s/√n+ (3.9.)
Relatief 95% CI (%) = 100 x absoluut CI / xgem (3.10.)
waarbij: xgem = gemiddelde concentratie
tα,n-1 = t-waarde met probabiliteit α (= 0.05) en n-1
vrijheidsgraden
s = standaarddeviatie
n = aantal metingen
Bij de evaluatie van de juistheid mogen de limieten van 95% en 105% niet overschreden worden.
Het al dan niet overschrijden van de limieten wordt niet alleen grafisch waargenomen, maar ook
statistisch getest met een eenzijdige t-toets voor één steekproef op het 5% significantieniveau.
3.2.4.6. Methodevergelijking
Bij een methodevergelijking worden bijvoorbeeld de analyseresultaten, bekomen met een
bepaalde methode, vergeleken met die van een referentiemethode, uitgevoerd op dezelfde stalen. Een
tweede mogelijkheid is dat twee methoden van hetzelfde hiërarchisch niveau worden vergeleken. In dit
laatste geval verstrekt geen van beide methoden een ondubbelzinnig correcte meting. De mate van
overeenkomst tussen de twee methodes wordt dus vergeleken. In deze scriptie worden de
analyseresultaten vergeleken met deze bekomen met een referentiemethode. Het CLSI EP9 protocol
schrijft voor dat er voor methodevergelijking minstens veertig stalen in duplicaat moeten gemeten
worden, gespreid over vijf dagen. Gezien het korte tijdsbestek waarin de meesterproef georganiseerd
wordt, wordt deze methodevergelijking echter slechts theoretisch uitgevoerd. Er wordt een dataset
gesimuleerd door het personeel van het Laboratorium voor Analytische Chemie. Het simuleren gebeurt

23
aan de hand van de DataGeneration Excel® file, ter beschikking gesteld door Dr. Stöckl, STT-consulting.
Het concentratiebereik loopt van 0.02 µg/g tot 1.4 µg/g. Binnen dit bereik worden tachtig
analyseresultaten gesimuleerd, met een realistische standaarddeviatie voor de methodes die
gevalideerd worden.
Om na te gaan of er bij deze methodevergelijking aan de vooropgestelde specificaties wordt
voldaan, wordt er enerzijds gebruik gemaakt van de Bland & Altman benadering en anderzijds van
lineaire regressieanalyse. In een Bland & Altman grafiek wordt het procentueel verschil tussen de
gegenereerde data van de routine- en de referentiemethode uitgezet in functie van de meetresultaten
van de referentiemethode. Het gemiddeld procentueel verschil tussen beide methoden wordt berekend
en weergegeven in de Bland & Altman grafiek, samen met het eenzijdig 95% CI ervan. Formule 3.11.
toont hoe dit eenzijdig 95% CI wordt berekend. Daarnaast wordt ook het 1.96s – interval van de
individuele verschillen berekend, samen met het eenzijdig 95% CI voor de 1.96s – limieten. Formule 3.12.
en 3.13. tonen hoe deze waarden worden bekomen. Ten slotte toont de grafiek ook nog de
vooropgestelde limieten voor de systematische fout (5%) (“systematic error”, SE) en de totale fout (15%)
(“total error”, TE). Voor de interpretatie van de Bland & Altman grafiek en bijgevolg de evaluatie van de
resultaten, wordt nagegaan of het gemiddelde procentueel verschil met zijn 95% CI binnen de limieten
voor de systematische fout valt en of de 1.96s – limieten met hun 95% CI binnen de limieten voor de
totale fout vallen.
CIgemiddeld verschil = xgem ± tα,n-1 s/√n (3.11.)
1.96 sind. verschillen – interval = xgem ± 1.96 s (3.12.)
CI1.96 CV ind. verschillen = xgem ± tα,n-1 (3.13.)
waarbij: xgem = gemiddelde verschil tussen beide methoden
tα,n-1 = t-waarde met probabiliteit α (= 0.05) en n-1
vrijheidsgraden
s = standaarddeviatie op verschil tussen de methoden
n = aantal metingen = 80
Bij lage concentraties zou de variatie op de resultaten groter kunnen zijn dan bij hogere
concentraties. Bovendien zouden bij lage concentraties de procentuele limieten te streng kunnen zijn.

24
Daarom is het misschien beter om de limieten concentratieafhankelijk te maken en wordt er naast een
klassieke Bland & Altman grafiek, die gebruik maakt van het procentueel verschil, ook een gelijkaardig
diagram opgesteld, waarbij het absolute verschil tussen de twee methoden wordt uitgezet ten opzichte
van de referentiemethode. In deze grafiek worden er absolute TE limieten gesteld voor stalen met lage
concentratie: tot een concentratie van 0.25 µg/g (een punt dat educatief gekozen wordt) worden de
absolute limieten gezet op ± 0.0375 µg/g (= 0.15 x 0.25 µg/g). Vanaf een concentratie van 0.25 µg/g
worden opnieuw procentuele limieten voor TE gebruikt (15%).
Bij de lineaire regressieanalyse worden de analyseresultaten bekomen met de routinemethode
uitgezet in functie van deze bekomen met de referentiemethode. Op die manier wordt een
regressievergelijking bekomen, waaruit de richtingscoëfficiënt en het snijpunt met de y-as kunnen
afgelezen worden. Wanneer de richtingscoëfficiënt verschillend is van 1, spreekt men van een
proportionele fout. Wanneer het intercept verschillend is van 0, spreekt men van een constante fout.
Indien één van beide gevallen zich voordoet (of allebei), dan wordt nagegaan of de afwijking binnen de
vooropgestelde specificaties voor de systematische en totale fout ligt. Zowel voor de laagste als voor de
hoogste concentratie bij de referentiemethode wordt de overeenkomstige y-waarde voorspeld aan de
hand van de regressievergelijking. Voor deze voorspelde y-waarden wordt het 95% CI opgesteld. Daarna
wordt het procentueel verschil tussen de bekomen confidentielimieten en de minimum en maximum x-
waarde berekend. Dit percentage moet dan binnen de vooropgestelde specificaties voor de
systematische fout vallen. Ook het 95% predictie-interval rond de voorspelde y-waarden wordt
berekend. Ook hier wordt het procentueel verschil tussen de bekomen predicitie-limieten en de
minimum en maximum x-waarde berekend. Dit percentage moet gelegen zijn binnen de specificaties
voor de totale fout. De calculatie van deze laatste twee intervallen gebeurt aan de hand van zeer
complexe statistische formules, die gehaald worden uit het boek Method validation With confidence van
Dr. Stöckl, STT-consulting. Tabellen en gespecialiseerde software worden gebruikt.
3.2.5. Dataverwerking en statistiek
Voor het optekenen van de kalibratiecurven, het overzichtelijk houden van de gegevensreeksen
en het berekenen van de concentratie die bij een bepaalde piekoppervlakte hoort, wordt gebruik
gemaakt van Microsoft Office Excel® 2007 (Microsoft Corporation, Verenigde Staten).
De statistische evaluatie van de resultaten gebeurt met behulp van de MethVal file opgesteld
door Dr. Stöckl, STT consulting (Horebeke, België). Daarnaast werd ook gebruik gemaakt van twee

25
boeken, die door Dr. Stöckl ter beschikking werden gesteld. Laboratory Statistics & Graphics with Excel®
(Stöckl, 2007a) en Method Validation with Confidence (Stöckl, 2007b) zijn de titels van de boeken die
werden gebruikt bij de statistische interpretatie van de gegevens.
De “lack-of-fit” test en de tweedegraads polynomiale regressieanalyse worden uitgevoerd in
CBstat5 (2005, Kristian Linnet, Charlottenlund, Denemarken).
3.2.6. Specificaties
De verschillende specificaties die horen bij de verscheidene prestatiekarakteristieken die in deze
methodevalidatie worden geëvalueerd worden samengevat in Tabel 3.9.
Tabel 3.9. : Overzicht van de specificaties horende bij de onderzochte prestatiekarakteristieken
3.2.7. Literatuuronderzoek
Voor het literatuuronderzoek wordt op zoek gegaan naar informatie over het tolerantie-interval en de
rol van dit soort interval in het farmaceutisch onderzoek. Daarnaast wordt onderzocht of dit soort
statistisch interval applicaties vindt in de farmaceutische methodevalidatie. Er wordt ook een link gelegd
naar de totale fout (“total error”). In Tabel 3.10. wordt een overzicht gegeven van de verschillende
zoekmachines die werden geraadpleegd bij het zoeken naar deze informatie.
Tabel 3.10. : De verschillende zoekmachines die werden gebruikt bij het literatuuronderzoek
Algemene zoekmachines Google
Wetenschappelijke zoekmachines Pubmed
Web of Science
Prestatiekarakteristiek Specificatie
Lineariteit 5%a
Imprecisie: binnen-analyse CV 2%b
Imprecisie: totale CV 5%b
Juistheid 5%a
Methodevergelijking: systematische fout 5%a
totale fout 15%a a: limiet
b: doelwaarde voor een stabiel proces

26
4. RESULTATEN EN DISCUSSIE
4.1. EXPERIMENTEN
4.1.1. Systeemfunctiecontrole
Bij het uitvoeren van de experimenten varieerde de druk tussen 93 en 98 bar. De ruis fluctueerde
rond een gemiddelde waarde van 0.003 mV, maar was nooit hoger dan 0.006 mV. De maximale variatie
op een stabiele basislijn bedroeg nooit meer dan 0.01 mV. Daarnaast werd ook telkens aan alle andere
specificaties voor de verschillende parameters zoals beschreven in Tabel 3.5. voldaan. Op basis van deze
gegevens vormen we het besluit dat het toestel goed functioneerde gedurende het volledige
experimentengedeelte van de meesterproef.
4.1.2. Systeemgeschiktheidscontrole
Voor de systeemgeschiktheidscontrole wordt elke dag bij aanvang van de metingen de testmix
geïnjecteerd. Figuur 4.1. toont een voorbeeldchromatogram van de testmix, geïnjecteerd op
29/03/2011.
Tabel 4.1. geeft een overzicht van het gemiddelde ± standaarddeviatie van de onderzochte
chromatografische parameters voor ethylparabeen, samen met de vooropgestelde limieten. Voor alle
Figuur 4.1.: Voorbeeldchromatogram van de testmix, geïnjecteerd op 29/03/2011. De concentratie van de componenten bedraagt ongeveer 2.5 µg/g

27
chromatografische parameters voldoet het gemiddelde ± standaarddeviatie aan de vooropgestelde
limieten. We kunnen besluiten dat het systeem geschikt is voor de beoogde doelstelling.
Tabel 4.1.: Overzicht van het gemiddelde met standaarddeviatie en de vooropgestelde limieten voor de onderzochte chromatografische parameters
Parameter Gemiddelde ± SD voor
ethylparabeen
Vooropgestelde limiet voor
ethylparabeen
Retentietijd (min) 3.3 ± 0.05 3.4 ± 0.2
Piekoppervlakte 78103 ± 3029 80000 ± 50%
Piekhoogte 10250 ± 472 10000 ± 50%
Theoretisch aantal platen 4227 ± 143 minimum 2000
Tailing factor 1.45 ± 0.03 maximum 2
Resolutie 4.20 ± 0.11 minimum 2
4.1.3. Lineariteit
Bij de evaluatie van deze prestatiekarakteristiek worden vijf stalen in quadruplicaat gemeten.
Zoals eerder beschreven, wordt het experiment één maal herhaald ter bevestiging. Figuur 4.2.A en 4.2.B
geven als voorbeeld respectievelijk het spreidingsdiagram (inclusief regressievergelijking en R²) en het
residuendiagram weer van de lineariteitsdata van 04/04/2011.
Figuur 4.2.: (A) Spreidingsdiagram van de lineariteitsdata op 04/04/2011, (B) Residuendiagram van de lineariteitsdata op 04/04/2011
A B

28
Er wordt nagegaan of er uitschieters aanwezig zijn in het residuendiagram. Op het eerste zicht
lijkt er een uitschieter aanwezig te zijn bij standaard 5, maar na het uitvoeren van een Grubbs test blijkt
dat niet zo te zijn. Voor de andere meetdag worden er uitschieters vermoed bij standaarden 3, 4 en 5,
maar bij de Grubbs test bleek de p-waarde nooit kleiner te zijn dan 0.05. Er zijn dus geen uitschieters
aanwezig.
Ook al toont het spreidingsdiagram met bijhorende regressielijn een goede R², toch kunnen we
op basis van Figuur 4.2.A geen besluit trekken in verband met de lineariteit van de gegevens. Zoals
eerder onder 3.2.4.1. ‘Lineariteit’ werd besproken, worden hier een “lack-of-fit” test en een tweede orde
polynomiale regressie-analyse doorgevoerd. De p-waarde voor de “lack-of-fit” test bedraagt 0.9802.
Voor de andere meetdag is de p-waarde 0.9706. Op basis van deze “lack-of-fit” test kunnen we besluiten
dat de nulhypothese voor lineariteit kan weerhouden worden. Dit omwille van het feit dat de eenzijdige
F-toets een p-waarde oplevert die groter is dan 0.05. De afwijking van lineariteit is niet significant. Bij de
polynomiale regressie-analyse wordt een p-waarde van 0.1185 bekomen. Voor de andere meetdag is de
p-waarde 0.4004. Vermits de p-waarden groter zijn dan 0.05 kan de nulhypothese van lineariteit ook hier
weerhouden worden. De coëfficiënt bij x² is niet significant verschillend van nul.
4.1.4. Kalibratie
Er wordt nagegaan welk kalibratiemodel het meest geschikt is om de concentratie te bepalen
vertrekkende van de piekoppervlakte. Er worden vier verschillende kalibratiemodellen getest: OLR, OLR
geforceerd door nul, OLR met het punt (0,0) ingesloten en WLR. Met elk model wordt gedurende vijf
meetdagen de gemiddelde concentratie berekend van het Low IQC staal, het High IQC staal en het
juistheidsstaal met de laagste concentratie (juistheidsstaal 1). Verder worden ook de CV en het
procentueel verschil met de gravimetrisch bepaalde doelwaarde berekend (zie Tabel 4.2. op de volgende
pagina voor een overzicht). Op basis van de IQC stalen, zien we (wanneer we kijken naar de CV’s en de
procentuele afwijking) geen wezenlijk verschil tussen de vier verschillende methoden. Wanneer we
kijken naar de CV’s van het juistheidsstaal met de laagste concentratie, dan ligt deze voor OLR
geforceerd door nul lager dan bij de andere modellen. De CV van het juistheidsstaal bij WLR komt het
dichtst in de buurt van de CV van het juistheidsstaal voor OLR geforceerd door nul. De absolute waarde
van de procentuele afwijking van de doelwaarde voor deze twee modellen verschilt niet wezenlijk, dus
zijn we geneigd om voor het model OLR geforceerd door nul te kiezen. De precisie weegt meer door dan
de juistheid. Daarnaast wordt nog een 95% CI opgesteld voor het intercept met de y-as dat bekomen
wordt met OLR. De waarde 0 ligt telkens in dat interval, waardoor we met 95% zekerheid kunnen

29
besluiten dat het intercept niet significant verschilt van nul. OLR geforceerd door nul lijkt aldus het meest
adequate regressiemodel. Het is het meest robuuste model voor de berekening van de concentratie uit
de piekoppervlakte. Figuur 4.3. op de volgende pagina toont een voorbeeld van een kalibratiecurve
(inclusief regressievergelijking en R²), opgesteld met het regressiemodel OLR geforceerd door nul. Deze
kalibratiecurve werd opgesteld met de kalibratiegegevens van 08/04/2011.
Tabel 4.2.: Overzicht van de CV (%) en het procentueel verschil met de gravimetrisch bepaalde doelwaarde (%) voor de verschillende stalen bij elk regressiemodel
Regressiemodel CV (%) Procentuele afwijking van de
doelwaarde (%)
Low IQC OLR 2.48 -1.87
OLR door nul 2.48 -1.23
OLR met (0,0) 2.47 -1.62
WLR 2.47 -1.90
High IQC OLR 2.28 0.417
OLR door nul 2.27 0.487
OLR met (0,0) 2.28 0.445
WLR 2.20 0.411
Juistheidsstaal met
laagste concentratie OLR 10.3 -2.01
OLR door nul 4.95 3.34
OLR met (0,0) 7.11 0.0833
WLR 5.79 -3.19

30
4.1.5. Imprecisie
Voor de evaluatie van de imprecisie worden twee IQC stalen gedurende vijftien dagen in
duplicaat gemeten.
Figuur 4.4.A toont het puntendiagram van de verschillen tussen de twee metingen van elke dag
voor het Low IQC staal. Figuur 4.4.B is het puntendiagram van de daggemiddelden voor het Low IQC
staal. In Figuur 4.4.C en Figuur 4.4.D worden deze puntendiagrammen voor het High IQC staal getoond.
Uit de Grubbs test bleek dat er bij geen enkel diagram uitschieters aanwezig zijn. Het gemiddelde van de
daggemiddelden voor het Low IQC staal bedraagt 0.4516 µg/g. Voor het High IQC staal is het gemiddelde
van de daggemiddelden gelijk aan 0.8883 µg/g.
Naast het gemiddelde worden de binnen-analyse (“within run”) standaarddeviatie (swr), de totale
standaarddeviatie (sT), de “within run” CV (CVwr) en de totale CV (CVT) berekend. De resultaten van deze
berekeningen worden weergegeven in Tabel 4.3. op de volgende pagina.
Figuur 4.3.: Voorbeeld van een kalibratiecurve opgesteld met OLR geforceerd door nul van 08/04/2011
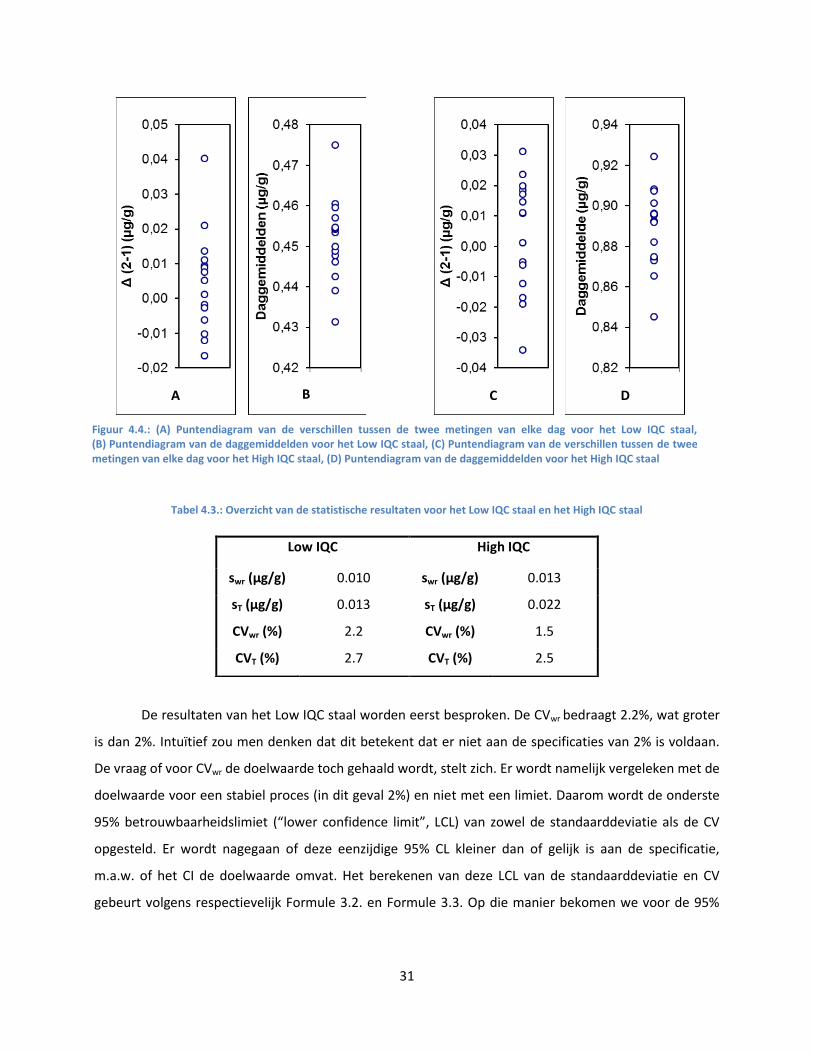
31
Tabel 4.3.: Overzicht van de statistische resultaten voor het Low IQC staal en het High IQC staal
De resultaten van het Low IQC staal worden eerst besproken. De CVwr bedraagt 2.2%, wat groter
is dan 2%. Intuïtief zou men denken dat dit betekent dat er niet aan de specificaties van 2% is voldaan.
De vraag of voor CVwr de doelwaarde toch gehaald wordt, stelt zich. Er wordt namelijk vergeleken met de
doelwaarde voor een stabiel proces (in dit geval 2%) en niet met een limiet. Daarom wordt de onderste
95% betrouwbaarheidslimiet (“lower confidence limit”, LCL) van zowel de standaarddeviatie als de CV
opgesteld. Er wordt nagegaan of deze eenzijdige 95% CL kleiner dan of gelijk is aan de specificatie,
m.a.w. of het CI de doelwaarde omvat. Het berekenen van deze LCL van de standaarddeviatie en CV
gebeurt volgens respectievelijk Formule 3.2. en Formule 3.3. Op die manier bekomen we voor de 95%
Low IQC High IQC
swr (µg/g) 0.010 swr (µg/g) 0.013
sT (µg/g) 0.013 sT (µg/g) 0.022
CVwr (%) 2.2 CVwr (%) 1.5
CVT (%) 2.7 CVT (%) 2.5
Figuur 4.4.: (A) Puntendiagram van de verschillen tussen de twee metingen van elke dag voor het Low IQC staal, (B) Puntendiagram van de daggemiddelden voor het Low IQC staal, (C) Puntendiagram van de verschillen tussen de twee metingen van elke dag voor het High IQC staal, (D) Puntendiagram van de daggemiddelden voor het High IQC staal
A B C D

32
LCL van CVwr de waarde 1.7%. Deze onderste betrouwbaarheidslimiet voor CVwr voldoet aan de
specificatie van ≤2%, ook al is de experimentele CVwr (2.2%) groter dan de limiet (2%).
De imprecisie kan daarnaast ook nog op een andere manier geëvalueerd worden, waarbij de
experimentele Chi²-waarde (Chi²exp) (zie Formule 3.4.) vergeleken wordt met de kritische Chi²-waarde
(Chi²krit). We stellen de nulhypothese op dat CVexp kleiner is dan of gelijk aan CVspec. Wanneer dat zo is,
kunnen we de nulhypothese weerhouden en kunnen we besluiten dat de methode voldoet aan de
vooropgestelde specificaties.
De met Excel® berekende Chi²krit bedraagt 25. De met formule 3.4. bepaalde Chi²exp voor de
“within-run” precisie bedraagt 19. Gezien 19 kleiner is dan 25 kunnen we de nulhypothese weerhouden
en ook via deze weg besluiten dan de methode voldoet aan de vooropgestelde specificaties voor het Low
IQC staal.
CVT van het Low IQC staal bedraagt 2.7% en voldoet aan de specificatie van 5%.
Bij het High IQC staal voldoen zowel CVwr (1.5%) als CVT (2.5%) aan de vooropgestelde
specificaties, respectievelijk 2% en 5%. De methode voldoet aan de vooropgestelde specificaties voor het
High IQC staal.
4.1.6. Detectielimiet
Voor de bepaling van de detectielimiet wordt gedurende vijftien dagen elke dag een nieuw staal
aangemaakt, waarvan de concentratie gravimetrisch wordt bepaald en dat een S/N ratio van 3 tot 6
oplevert. Het gemiddelde van de concentraties van deze stalen bedraagt 0.004196 µg/g. Elke dag wordt
van het geïnjecteerde staal de S/N ratio bepaald. Deze S/N ratio’s worden vermenigvuldigd met de
verhouding van de gemiddelde concentratie op de gravimetrisch bepaalde concentratie van die dag. Van
deze genormaliseerde S/N ratio’s wordt vervolgens het gemiddelde en de standaarddeviatie berekend.
Het gemiddelde bedraagt 4.76. De standaarddeviatie is 0.77. Figuur 4.5. toont het puntendiagram van de
genormaliseerde S/N ratio’s van de vijftien meetdagen. Uit de Grubbs test bleek dat er geen uitschieters
aanwezig zijn in deze dataset.

33
Aan de hand van Formule 3.6. wordt het 95% CI van de gemiddelde S/N ratio berekend. Zo
bekomen we: 95% CI = [4.76 ± 0.426].
Daarnaast wordt bepaald wat de gemiddelde absolute hoeveelheid ethylparabeen is, die
dagelijks geïnjecteerd wordt. Er wordt 5 µl geïnjecteerd en de dichtheid van het oplosmiddel bedraagt
0.9042 g/ml. De gemiddelde absolute hoeveelheid ethylparabeen die wordt geïnjecteerd, bedraagt 18.97
pg. Dit is dus de absolute hoeveelheid ethylparabeen op de kolom die overeenstemt met de LoD.
4.1.7. Juistheid
Voor de evaluatie van de juistheid worden zes stalen gedurende vijf dagen in singlicaat gemeten.
Het puntendiagram van de verschillen tussen de gemeten dagconcentraties van de zes verschillende
juistheidsstalen (zie Figuur 4.6. op de volgende pagina) en hun gemiddelde concentratie geeft een idee
van de spreiding en laat ons toe een vermoeden uit te spreken over het al dan niet aanwezig zijn van
uitschieters. Uit de Grubbs test blijkt bij geen enkel staal een uitschieter aanwezig te zijn.
Figuur 4.5.: Puntendiagram van de genormaliseerde S/N ratio's van de 15 meetdagen

34
De relatieve terugvinding (%) met betrouwbaarheidsinterval van elk staal wordt grafisch
weergegeven in Figuur 4.7. Deze figuur toont een samenvatting van alle juistheidsstalen in een
procentueel verhoudingsdiagram. Uit dit diagram blijkt dat het gemiddelde van elk staal binnen de
limieten van 95% en 105% terugvinding valt. Bij vijf stalen is het zo dat het betrouwbaarheidsinterval van
het gemiddelde de limiet van 5% afwijking van de ware concentratie niet overschrijdt. Enkel de bovenste
betrouwbaarheidslimiet van juistheidsstaal 1 overlapt met de specificatie van 5%. Meer metingen
zouden het betrouwbaarheidsinterval kunnen versmallen en op die manier uitsluitsel kunnen bieden.
Figuur 4.7.: Procentueel verhoudingsdiagram: relatieve terugvinding (%) met betrouwbaarheidsinterval van elk
juistheidsstaal
Figuur 4.6.: Puntendiagram van de verschillen tussen de gemeten concentraties en de gemiddelde concentratie van de zes verschillende juistheidsstalen
1
2
3 4
5 6

35
Het al dan niet overschrijden van de 5% limiet wordt niet enkel grafisch waargenomen. Er wordt
daarnaast ook voor elk staal statistisch getest met een eenzijdige t-toets voor één steekproef of de
gemiddelde relatieve terugvinding significant verschilt van de 95% en 105% limiet (5%
significantieniveau). De p-waarde is hierbij telkens kleiner dan 0.05, behalve voor juistheidstaal 1. Bij dit
staal wordt de 105% limiet overschreden. We kunnen besluiten dat de methode voldoet aan de
vooropgestelde specificaties, op voorwaarde dat de herhaling van de metingen van juistheidsstaal 1
binnen de specificaties valt.
4.1.8. Methodevergelijking
Voor de methodevergelijking worden door het personeel van het Laboratorium voor Analytische
Chemie tachtig analyseresultaten gesimuleerd binnen een concentratiebereik van 0.02 µg/g tot 1.4 µg/g
met een realistische standaarddeviatie. We veronderstellen dat beide methoden dezelfde CV hebben.
Op die manier bekomen we een totale CV die gelijk is aan CV x √2. Voor de doelstellingen van deze
meesterproef is het toegelaten de CV te vervangen door een realistische standaarddeviatie. Voor deze
laatste wordt de waarde 0.022 gekozen, wat gelijk is aan de totale standaarddeviatie van het High IQC
staal.
De mate van overeenkomst tussen de twee methoden wordt niet bestudeerd aan de hand van
de correlatiecoëfficiënt. Deze is immers misleidend. Een hoge correlatiecoëfficiënt betekent niet
onmiddellijk dat de twee methoden vergelijkbaar zijn (Bland & Altman, 1986).
Figuur 4.8. op de volgende pagina toont een Bland & Altman grafiek, waaruit blijkt dat het
gemiddeld procentueel verschil met zijn 95% CI binnen de vooropgestelde limieten van de systematische
fout valt. Het 1.96s – interval van de individuele verschillen ligt met het 95% CI ervan binnen de grenzen
van de totale fout specificaties. Aan de hand van de Bland & Altman benadering zou kunnen besloten
worden dat de methodevergelijking voldoet aan de vooropgestelde specificaties.

36
Het procentueel verschil tussen de routine- en de referentiemethode levert voor lage
concentraties grotere waarden op dan voor hogere concentraties (zie Figuur 4.8.). Voor de individuele
datapunten lijken de procentuele limieten bij deze lage concentraties streng. Daarom wordt er naast een
procentuele Bland & Altman grafiek ook nog een absolute Bland & Altman grafiek opgesteld. Deze is te
zien in Figuur 4.9. op de volgende pagina. In deze grafiek worden er absolute TE limieten gesteld voor
lage concentraties, zoals beschreven onder 3.2.4.6. ‘Methodevergelijking’. Vanaf een concentratie van
0.25 µg/g (een punt dat educatief gekozen wordt) worden opnieuw procentuele limieten gebruikt. We
zien in Figuur 4.9. dat alle individuele datapunten binnen de concentratieafhankelijke limieten liggen.
Bovendien kunnen we aan de hand van deze figuur opmerken dat de CV van stijgend (bij de lage
concentraties) naar constant (bij de hogere concentraties) evolueert, wat moeilijker te zien is in Figuur
4.8.
Figuur 4.8.: Bland & Altman grafiek

37
Figuur 4.9.: Bland & Altman grafiek waarbij in de y-as niet het procentuele verschil tussen de routine- en referentiemethode
uitgezet wordt, maar het absolute verschil tussen beide methoden
Tabel 4.4. geeft de resultaten weer die bekomen worden voor de lineaire regressie. Uit de tabel
blijkt dat de richtingscoëfficiënt significant verschillend is van 1 en het intercept niet significant
verschillend is van 0 op het 5% significantieniveau.
Tabel 4.4.: Resultaten van de lineaire regressieanalyse
Lineaire regressievergelijkinga y = 0.9745x + 0.0034
Richtingscoëfficiënt ± 95% CI 0.9745 ± 0.02269
Intercept ± 95% CI 0.0034 ± 0.01736
a: x stelt de referentiemethode voor, y stelt de routinemethode voor
Mogelijks is er een concentratieafhankelijkheid in de data. Daarom kijken we naar twee extreme
punten op de regressielijn, namelijk de laagste en de hoogste meetwaarde van de referentiemethode.
Dit wordt uitgevoerd om na te gaan of de prestatie van de routinemethode (in termen van
overeenkomst met de referentiemethode) verschillend is bij lage en hoge concentratie. Voor deze
extreme punten wordt het resultaat voor de routinemethode voorspeld aan de hand van de
regressievergelijking. Vervolgens wordt van deze waarden het 95% CI berekend. Daarna wordt het
procentueel verschil tussen de bekomen confidentielimieten en de minimum en maximum x-waarde
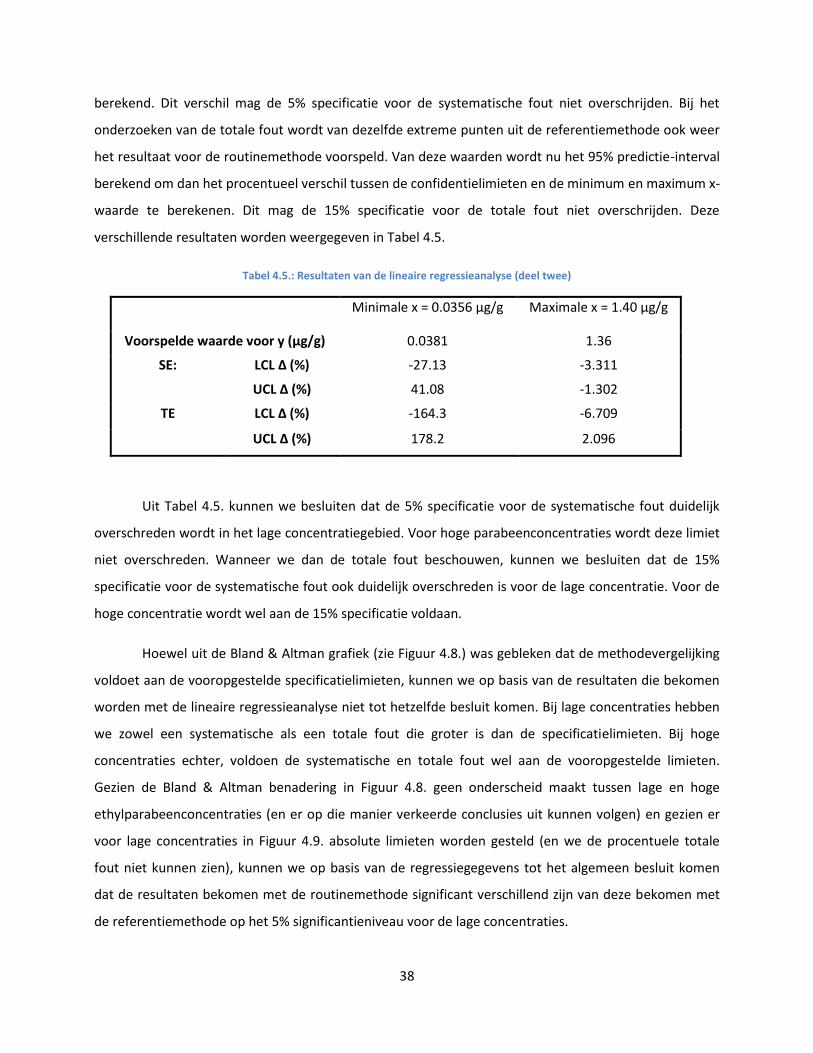
38
berekend. Dit verschil mag de 5% specificatie voor de systematische fout niet overschrijden. Bij het
onderzoeken van de totale fout wordt van dezelfde extreme punten uit de referentiemethode ook weer
het resultaat voor de routinemethode voorspeld. Van deze waarden wordt nu het 95% predictie-interval
berekend om dan het procentueel verschil tussen de confidentielimieten en de minimum en maximum x-
waarde te berekenen. Dit mag de 15% specificatie voor de totale fout niet overschrijden. Deze
verschillende resultaten worden weergegeven in Tabel 4.5.
Tabel 4.5.: Resultaten van de lineaire regressieanalyse (deel twee)
Minimale x = 0.0356 µg/g Maximale x = 1.40 µg/g
Voorspelde waarde voor y (µg/g) 0.0381 1.36
SE: LCL Δ (%) -27.13 -3.311
UCL Δ (%) 41.08 -1.302
TE LCL Δ (%) -164.3 -6.709
UCL Δ (%) 178.2 2.096
Uit Tabel 4.5. kunnen we besluiten dat de 5% specificatie voor de systematische fout duidelijk
overschreden wordt in het lage concentratiegebied. Voor hoge parabeenconcentraties wordt deze limiet
niet overschreden. Wanneer we dan de totale fout beschouwen, kunnen we besluiten dat de 15%
specificatie voor de systematische fout ook duidelijk overschreden is voor de lage concentratie. Voor de
hoge concentratie wordt wel aan de 15% specificatie voldaan.
Hoewel uit de Bland & Altman grafiek (zie Figuur 4.8.) was gebleken dat de methodevergelijking
voldoet aan de vooropgestelde specificatielimieten, kunnen we op basis van de resultaten die bekomen
worden met de lineaire regressieanalyse niet tot hetzelfde besluit komen. Bij lage concentraties hebben
we zowel een systematische als een totale fout die groter is dan de specificatielimieten. Bij hoge
concentraties echter, voldoen de systematische en totale fout wel aan de vooropgestelde limieten.
Gezien de Bland & Altman benadering in Figuur 4.8. geen onderscheid maakt tussen lage en hoge
ethylparabeenconcentraties (en er op die manier verkeerde conclusies uit kunnen volgen) en gezien er
voor lage concentraties in Figuur 4.9. absolute limieten worden gesteld (en we de procentuele totale
fout niet kunnen zien), kunnen we op basis van de regressiegegevens tot het algemeen besluit komen
dat de resultaten bekomen met de routinemethode significant verschillend zijn van deze bekomen met
de referentiemethode op het 5% significantieniveau voor de lage concentraties.

39
4.1.9. Resultaten: samenvatting
In Tabel 4.6. wordt een overzicht gegeven van de verschillende prestatiekarakteristieken die onderzocht
werden, samen met de vooropgestelde specificaties. Er wordt ook vermeld of er al dan niet aan de
specificaties wordt voldaan.
Tabel 4.6.: Overzicht van de verschillende prestatiekarakteristieken die onderzocht werden, samen met de vooropgestelde specificaties en vermelding van het feit of er al dan niet aan die specificaties wordt voldaan
4.2. LITERATUURONDERZOEK
4.2.1. Introductie tot het statistisch concept van het tolerantie-interval
Om financiële, logistieke, organisatorische en andere redenen is het in wetenschappelijke studies
zelden mogelijk de volledige populatie te onderzoeken. Men beperkt zich dan ook vaak tot een kleinere
groep binnen de populatie (de steekproef). Stel dat X de onderzochte variabele is en x1, x2,… xN N
verschillende onafhankelijke observaties zijn van X, dan kunnen er twee situaties worden onderscheiden:
(1) er is niets gekend over de distributie van X, behalve dan misschien dat de observaties continu zijn of
(2) de distributie van X is wel gekend en enkel de numerieke waarden van een eindig aantal parameters
betrokken bij de distributie van X zijn onbekend. Geval (1) wordt een non-parametrisch model genoemd,
terwijl we in geval (2) spreken van het parametrisch model. Bij dit literatuuronderzoek beperken we ons
tot het parametrisch model. Meer bepaald gaan we er hier van uit dat de metingen kunnen beschreven
worden aan de hand van een Gaussiaans model.
Prestatiekarakteristiek Specificatie Wordt er aan de specificatie voldaan?
Lineariteit 5%a Jac
Imprecisie: binnen-analyse CV 2%b Ja, zowel voor Low IQC als voor High IQC
Imprecisie: totale CV 5%b Ja, zowel voor Low IQC als voor High IQC
Juistheid 5%a Ja, mits herhaalde metingen voor
juistheidsstaal 1 binnen de specificatie vallen.
Methodevergelijking: systematische fout 5%a Neend
totale fout 15%a Neend
a: limiet
b: doelwaarde voor een stabiel proces
c: uit de “lack-of-fit” test en de polynomiale regressieanalyse bleek al dat er een lineair verband
bestaat tussen de piekoppervlakte en de concentratie
d: niet bij lage concentraties, wel bij hogere concentraties

40
Op grond van de resultaten die bekomen worden bij een representatieve steekproef, wil men
uitspraken doen over de volledige populatie. Vaak is het zo dat een onbekende parameter van de
populatie (bv. het gemiddelde of de standaarddeviatie) geschat wordt aan de hand van een uit de
steekproef berekende grootheid. Bij dit schatten willen we van die onbekende parameter in de populatie
een indruk krijgen door middel van een puntschatting en/of een confidentie-interval (CI). Anders dan een
puntschatting levert een CI een volledig interval van betrouwbare schattingen van de parameter. Een
waarschijnlijkheidsniveau van 95% is courant. Wat vaak foutief wordt gesteld, is dat er bij een 95% CI
voor een populatieparameter 95% kans is dat die parameter in het interval ligt. Een juistere benadering
van de betekenis van het 95% CI is dat bij herhaling van de procedure mag verwacht worden dat 95% van
de berekende intervallen de populatieparameter zullen bevatten (Vansteelandt, 2009).
Het CI geeft daarnaast ook informatie over de spreiding van de gegenereerde data. De onder- en
bovengrens van het CI worden de confidentielimieten genoemd (“confidence limit”, CL). De breedte van
het CI hangt af van de variabiliteit op de metingen (met als kwantitatieve maat de standaarddeviatie) en
het aantal metingen (algemeen de grootte van de steekproef). Hoe smaller het interval, hoe
nauwkeuriger de schatting van de populatieparameter.
Naast het CI, kan ook gebruik gemaakt worden van een predictie-interval (PI) of een tolerantie-
interval (TI) om schattingen te maken van de populatie. Een PI is een interval dat vertelt waarbinnen
toekomstige observaties, met een zekere probabiliteit, zullen vallen. Wanneer een 95% PI van een
datareeks wordt berekend en daarna één extra meting wordt uitgevoerd, dan wordt verwacht dat bij
herhaling van deze procedure die extra meting in 95% van de gevallen binnen het PI valt. In plaats van de
ligging van enkel het eerstvolgende resultaat te voorspellen, kan ook de ligging van alle volgende n
resultaten of een fractie van alle volgende n resultaten geschat worden. (Technometrics, 1990).
Wat als we nu een voorspelling willen maken van alle 100, 1000 of meer toekomstige resultaten?
Uiteraard zouden de overeenkomstige predictie-intervallen voor die 100 of 1000 observaties kunnen
berekend worden, maar met het toenemen van het aantal toekomstige observaties neemt ook de
breedte van de overeenkomstige predictie-intervallen toe. Gelukkig is er een ander type interval dat
gebruikt kan worden bij dit grote aantal observaties. Zo’n interval wordt een tolerantie-interval (TI)
genoemd (Wald & Wolfowitz, 1946; Hahn & Meeker, 1991).
In plaats van de ligging van een populatieparameter of van een toekomstige meetwaarde te
schatten, kan ook de ligging van een zekere fractie van de populatie geschat worden. Een TI is een

41
statistisch interval waarbinnen met een zekere betrouwbaarheid een vaste proportie van de populatie
valt. De onderste en de bovenste grens van het TI worden de tolerantielimieten (“tolerance limit”, TL)
genoemd. Het TI kan bijvoorbeeld een antwoord geven op de vraag: “Wat als we γ% zeker willen zijn dat
het interval β% van de waarden omvat?”. Om een TI te kunnen berekenen, moeten er dus twee
verschillende percentages gekozen worden (γ en β). Eén percentage (γ) bepaalt het
betrouwbaarheidsniveau, het andere (β) vertelt welke fractie van de waarden het interval insluit. Het
gebeurt dat onderzoekers beweren dat een CI een zekere proportie van de populatie dekt, maar dat is
niet het geval. In situaties waarbij een zekere onderste en bovenste grens vereist zijn om een specifieke
proportie van een populatie te omsluiten, kunnen TI’s aangewezen zijn. Tolerantie-intervallen worden
bijvoorbeeld gebruikt om na te gaan of waarden met een zekere probabiliteit binnen bepaalde
specificatielimieten vallen (NIST/SEMATECH eHandbook of statistical methods).
Alternatief kan men het tolerantie-interval definiëren als een intervalschatting van een bepaald
percentiel. Een percentiel is de waarde binnen een reeks metingen onder dewelke een zeker percentage
van de metingen valt. Een percentiel is met andere woorden de waarde onder dewelke exact p% van de
waarden ligt. Waar een CI een schatting geeft van een populatieparameter, zoals het gemiddelde of de
variantie, is de parameter die geschat wordt bij het TI een percentiel (Mee, 1990; Chakraborti et al.,
2007).
Wanneer noch het populatiegemiddelde µ, noch de populatiestandaarddeviatie ς gekend zijn,
moet het TI rekening houden met de variabiliteit op de schatting van zowel het gemiddelde als de
standaarddeviatie. Dit leidt tot een interval dat minstens een proportie van de distributie omvat met een
zekere probabiliteit. Wanneer het aantal metingen toeneemt, dan zal het TI het interval, dat exact de
proportie p van de normale distributie omvat, benaderen (Odeh & Owen, 1980).
In de wetenschap is het toevoegen van ±3ς aan een bepaalde schatting een gangbare praktijk
om bijvoorbeeld specificatielimieten op te stellen. Voor Normaal verdeelde data is het namelijk zo dat
een ±3ς interval rond het gemiddelde 99.73% van de populatie in zich sluit. Dit is echter slechts correct
wanneer de ware populatieparameter gekend is. Gezien dit heel zelden het geval is, moeten het
gemiddelde en de variantie geschat worden. Waar ligt het verschil met het TI dan? Een tolerantie-
interval wordt ook geconstrueerd als het gemiddelde ± een aantal keer de standaarddeviatie, maar het
houdt rekening met de onzekerheid op de schatting en het aantal metingen via de functie
g(betrouwbaarheid, proportie, aantal metingen) in Formule 4.1.

42
TI = xgem ± g(1-α/2, β, n) . s (4.1.)
waarbij: xgem = gemiddelde van x
g(1-α/2, β, n) = g(betrouwbaarheid (α), proportie (β), aantal metingen (n))
s = standaarddeviatie
Het equivalente 95% TI van een gemiddelde ±3ς interval wordt op die manier gegeven door de
volgende betrekking: xgem ± g(0.975, 0.9973, n) . s. Dit TI zal smaller zijn dan het overeenkomstige PI voor 100 of
1000 metingen, maar toch iets breder dan het gemiddelde ±3ς interval omwille van het feit dat het
rekening houdt met de onzekerheid op de schattingen (Hahn & Meeker, 1991).
4.2.2. Het tolerantie-interval binnen het (bio)farmaceutische veld
Het TI vindt een aantal applicaties in het (bio)farmaceutisch onderzoek. Deze omvatten onder
meer het bepalen van aanvaardbaarheidscriteria in farmaceutische productieprocessen, evaluatie van
dosisuniformiteit en bepaling van de houdbaarheid van geneesmiddelen. In farmaceutische
productieprocessen is de selectie van een adequate statistische benadering heel belangrijk bij het
bepalen van geschikte aanvaardbaarheidscriteria. Deze laatste vormen een set van numerieke limieten
die, wanneer ze overschreden worden, een significante afwijking van normale werkomstandigheden of
productkwaliteit inhouden. Wang et al. (2007) stellen dat bij het bepalen van aanvaardbaarheidscriteria
voor biotechnologische producten het gebruik van het TI adequaat is voor zowel kleine als grote datasets
en bespreken deze twee situaties (kleine en grote dataset) afzonderlijk. Er wordt gesteld dat het gebruik
van gemiddelde ± 3s geen rekening houdt met het feit dat het gemiddelde en de standaarddeviatie
slechts schattingen zijn van de ware waarden. Wanneer deze methode gebruikt wordt bij een relatief
kleine dataset, dan kunnen de ware populatieparameters zowel over- als onderschat worden. Tolerantie-
intervallen worden op een gelijkaardige manier geconstrueerd als het gemiddelde ± g.s. Echter, in plaats
van 3s te gebruiken, wordt gebruik gemaakt van de factor g, die rekening houdt met het aantal
datapunten dat gegenereerd wordt om het gemiddelde en de standaarddeviatie te schatten. Deze factor
g wordt kleiner naarmate de steekproefgrootte toeneemt.
Wanneer het aantal datapunten groter wordt, dan wordt het interval van het gemiddelde ± 3s
vergelijkbaar met het TI. Hoewel deze intervallen vergelijkbaar zijn, is enkel het TI geassocieerd met een
betrouwbaarheidsniveau. Bij beide besproken gevallen (kleine en grote dataset) is het TI de aangewezen
methode om aanvaardbaarheidscriteria te bepalen. Een klein punt van kritiek is dat bij het gebruik van
het TI het belangrijk is om het geschikte betrouwbaarheidsniveau te kiezen (Wang et al., 2007).

43
Het gebruik van het TI is daarnaast ook goed ingeburgerd bij de evaluatie van dosisuniformiteit.
Testen op dosisuniformiteit helpt om te verzekeren dat het gehalte van een therapeutisch product
binnen gespecifieerde aanvaardbaarheidslimieten blijft. Algemeen kan hierbij het TI gebruikt worden om
na te gaan of het gemiddelde ± g.s met een zekere betrouwbaarheid binnen de voorgeschreven
acceptatielimieten valt. Bij dosisuniformiteitstesten is het doel na te gaan of de distributie van de
gemeten dosissen voldoende dicht ligt bij de waarde die op het productlabel wordt vermeld en daarom
worden deze aanvaardbaarheidslimieten berekend als percentages van de hoeveelheid die op het
productlabel staan (Hauck & Shaikh, 2004). Verschillende waarden voor deze limieten werden reeds
voorgesteld. Tot op vandaag is er echter nog steeds geen algemene consensus over de limieten die
gebruikt worden. Het gebruik van het parametrisch TI staat veel minder ter discussie en onder meer
dankzij recente regulatorische initiatieven vindt het TI algemene ingang bij verschillende auteurs en
instanties (Novick et al., 2009).
Farmaceutische bedrijven worden door de autoriteiten verplicht om de houdbaarheidsdatum
aan te geven op de verpakking van elk geneesmiddel. Stabiliteitsstudies van farmaceutische producten
worden uitgevoerd om de houdbaarheid van het product in kwestie te schatten. Deze “shelf life” zorgt
ervoor dat de patiënt zich geen zorgen hoeft te maken over de identiteit, sterkte, kwaliteit en zuiverheid
van het farmaceutisch product gedurende de houdbaarheidsperiode. Voor de bepaling van deze
houdbaarheid raadt de International Conference on Harmonisation (ICH) aan de houdbaarheid te
bepalen op basis van een benadering met een CI met betrouwbaarheidsniveau 100(1-α)%. Komka et al.
(2004) stellen deze benadering in vraag. Voor de stabiliteitsstudie wordt een willekeurige hoeveelheid
tabletten beschouwd. Samen met verschillende andere auteurs (Shao & Chow, 1994; Kiermeier et al.,
2004) komen zij tot de conclusie dat niet de ligging van het gemiddelde van de inhoud van de
verschillende tabletten moet beschouwd worden met zijn CI. In plaats daarvan wil men weten waar de
inhoud van de meerderheid (of algemeen een bepaalde proportie) van de individuele tabletten ligt. Het
geschikte statistische interval is hier opnieuw een TI, dat een specifieke proportie van de populatie met
een hoge betrouwbaarheid insluit (Komka et al., 2010).
Ongetwijfeld zullen er naast de besproken applicaties nog andere velden zijn binnen het
(bio)farmaceutisch onderzoek waar het TI gebruikt wordt, maar deze vallen buiten het bestek van dit
literatuuronderzoek. In verband met methodevalidatie is het TI eerder in een wetenschappelijke fase en
nog niet in een applicatiefase. Hieronder wordt dit meer uitgebreid uit de doeken gedaan.

44
4.2.3. Belang van het tolerantie-interval bij de farmaceutische methodevalidatie
Vooraleer een analytische methode kan gebruikt worden, moet deze uiteraard eerst ontwikkeld
worden en daarna gevalideerd. Het objectief van een kwantitatieve analytische methode is om elke
onbekende hoeveelheid of concentratie van een staal te kwantificeren. Men gaat bij de
methodevalidatie na of de methode geschikt is voor het uiteindelijke doel. Bij de validatie van een
analytische methode wil men er zeker van zijn dat elke toekomstige meting in routine analyse dicht
genoeg zal liggen bij de (onbekende) ware waarde voor de hoeveelheid analyt. M.a.w.: het verschil
tussen de onbekende ware waarde µT en het resultaat dat bekomen wordt met de methode moet
voldoende klein zijn, bijvoorbeeld kleiner dan een vooraf gedefinieerde acceptatielimiet λ (zie Formule
4.2.). Deze acceptatielimiet λ verschilt afhankelijk van de opdrachtgever of het objectief van de
analytische methode. Alternatief krijgt de analyticus de vereiste kwaliteitsspecificaties automatisch uit
hoofde van de discipline waarin het onderzoek kadert (bijvoorbeeld de farmaceutische discipline).
-λ < X - µT < λ |X - µT| < λ (4.2.)
waarbij: λ = vooraf gedefinieerde aanvaardbaarheidslimiet
X = de numerieke waarde van de meting van een staal
µT = de ware waarde
Voor elke te meten onbekende bestaat er theoretisch een ware waarde. Of de analytische
methode in staat is deze te achterhalen, hangt af van de analytische meetfouten en hun omvang. Elke
meting is onderhevig aan meetfouten en blijft daardoor slechts een benadering van de theoretische
ware waarde. Er wordt een onderscheid gemaakt tussen toevallige en systematische meetfouten.
Toevallige meetfouten bepalen de precisie van de methode, systematische fouten zijn bepalend voor de
juistheid (“trueness”) van de methode. De totale fout van één meetresultaat is de resultante van de
systematische en toevallige foutcomponenten.
Verschillende documenten over analytische en bioanalytische methodevalidatie werden reeds
gepubliceerd door regulatoire instanties, zoals de ICH en de Amerikaanse Food and Drug Administration
(FDA). Deze documenten vertellen dat elke analytische methode moet voldoen aan specifieke
aanvaardbaarheidscriteria om als gevalideerd beschouwd te kunnen worden, maar zijn veel minder
duidelijk over het proces en de regels om een beslissing te nemen. Er is met andere woorden geen
consensus over hoe men (op grond waarvan, aan de hand van welke protocollen of statistiek) een

45
beslissing moet nemen voor het al dan niet weerhouden van een analytische methode (Rozet et al.,
2007).
Algemeen kan men wel stellen dat een analytische methode als gevalideerd kan beschouwd
worden wanneer voor elk te kwantificeren staal het “zeer waarschijnlijk” is dat het bekomen resultaat
binnen de acceptatielimieten valt (zie Formule 4.3.). Merk op dat dit een diffuse benadering is. (Rozet et
al., 2007; González & Herrador, 2007)
π = P(|X - µT|) < λ) ≥ πmin (4.3.)
waarbij: π = de kans dat de absolute waarde van het verschil tussen
X en µT kleiner is dan λ
πmin = het kwaliteitsniveau
Wanneer een groot aantal stalen wordt geanalyseerd, dan kan π gezien worden als de proportie
van de resultaten die binnen de acceptatielimieten valt. Dit concept is zeer belangrijk en meteen ook het
centrale idee van Rozet et al. (2007): bij de methodevalidatie gaat het om predictie van de
waarschijnlijkheid (aan de hand van geschikte statistische methoden) dat elke toekomstige meting
accuraat genoeg zal zijn. Er is geen exacte oplossing om π uit Formule 4.3. te schatten, maar er zijn
verschillende auteurs (Rozet et al., 2007; Hoffman et al, 2007; González & Herrador, 2007) die een
oplossing voor dit probleem aanreiken. Het nemen van een beslissing of een analytische methode al dan
niet als gevalideerd kan beschouwd worden, zou volgens deze auteurs moeten gebaseerd zijn op het “β-
expectation” TI (βETI). Wanneer dit βETI volledig omsloten wordt door de acceptatielimieten *-λ, λ+, dan
is de verwachte proportie van de metingen die binnen deze acceptatielimieten ligt ongeveer gelijk aan β.
Om deze reden worden βETI’s ook predictie-intervallen genoemd. Ze geven de locatie weer waar β% van
de toekomstige resultaten verwacht worden (Rozet et al., 2007).
Twee types van tolerantie-intervallen hebben aanzienlijke aandacht gekregen in de literatuur. Er
wordt een onderscheid gemaakt tussen het “β-expectation” TI (βETI) en het “β-content” TI (βCTI).
Wanneer een βETI wordt opgesteld, dan wordt er verwacht dat ongeveer een proportie β van de
populatie binnen dat interval zal liggen. Een βCTI daarentegen wordt zo geconstrueerd dat het minstens
een proportie β van de populatie zal bevatten met probabiliteit γ (Mee, 1990).

46
Het gebruik van het βETI bij de farmaceutische methodevalidatie werd voor het eerst
voorgesteld door de Société Française des Sciences et Techniques Pharmaceutiques (SFSTP) in 2004.
Zoals eerder vermeld zijn er verschillende officiële documenten die de criteria voor een
methodevalidatie beschrijven, maar deze stellen geen experimenteel protocol voor en limiteren zichzelf
meestal tot algemene concepten. Het SFSTP heeft daarom een aantal richtlijnen ontwikkeld om
(industriële) wetenschappers te helpen bij het toepassen van de regulatoire aanbevelingen. Deze
richtlijnen zijn gebaseerd op het gebruik van een zogenaamd accuraatheidsprofiel en gebruiken het
begrip totale fout (Hubert et al., 2004).
De beslissingsregels voorgesteld door het SFSTP worden beschreven als volgt. Een analytische
methode heeft als doel stalen te kwantificeren over een bereik van concentraties. Tijdens de validatie
worden er stalen gebruikt die dit bereik dekken en voor elk staal wordt er dan een βETI berekend. Het
zogenaamde accuraatheidsprofiel wordt bekomen door enerzijds de onderste limieten van de βETI’s met
elkaar te verbinden over de verschillende concentratieniveau’s en anderzijds door de bovenste limieten
te verbinden. Wat vervolgens gebeurt, is nagaan of dit profiel binnen de aanvaardbaarheidslimieten [-λ,
λ+ ligt. Wanneer dat het geval is, kan men besluiten dat de methode gevalideerd is over het beschouwde
concentratiebereik. Figuur 4.10. op de volgende pagina toont het accuraatheidsprofiel van een
“sandwich Enzyme-Linked Immunosorbent Assay” (ELISA) methode voor de bepaling van thyroïd
stimulerend hormoon (TSH) op gedroogde bloedvlekken van pasgeborenen. Deze methode zou dan
kunnen gebruikt worden bij de neonatale screening van congenitaal hypothyroïdisme (Boemer et al.,
2009).
In het voorbeeld van Figuur 4.10. worden de acceptatielimieten voor de totale fout gezet op [-
30%, 30%+. Het βETI, dat een regio beschrijft waar een proportie β van de toekomstige resultaten zal
vallen, werd op 80% gezet (β=80%). De methode wordt als gevalideerd beschouwd binnen het bereik
waarvoor het accuraatheidsprofiel binnen de aanvaardbaarheidslimieten voor de totale fout (±30%) valt.
De LLoQ is de kleinste concentratie die onder de experimentele omstandigheden met voldoende
nauwkeurigheid kan bepaald worden. Een analoge definitie kan gegeven worden voor de ULoQ. De LLoQ
en ULoQ worden bekomen door de kleinste en hoogste concentratie, waarvan de limieten voor het βETI
de acceptatielimieten overschrijden, te bepalen. In dit voorbeeld van Boemer et al. (2009) wordt de LLoQ
experimenteel bepaald op 17.48 mIU/L. De ULoQ bedraagt 250 mIU/L. Binnen dit bereik kan men zeker
zijn dat minstens 80% van de toekomstige resultaten een totale fout van hoogstens 30% zullen hebben
(Boemer et al., 2009). Dit is onmiddellijk gerelateerd aan het algemeen objectief van een analytische
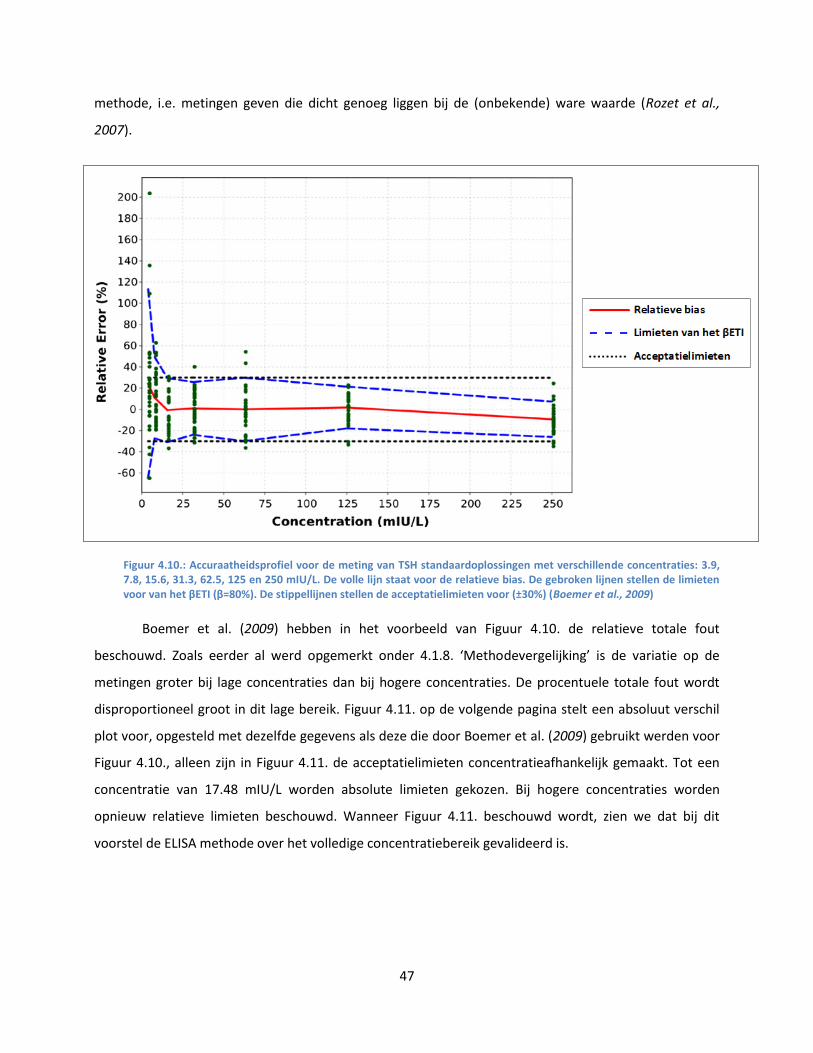
47
methode, i.e. metingen geven die dicht genoeg liggen bij de (onbekende) ware waarde (Rozet et al.,
2007).
Boemer et al. (2009) hebben in het voorbeeld van Figuur 4.10. de relatieve totale fout
beschouwd. Zoals eerder al werd opgemerkt onder 4.1.8. ‘Methodevergelijking’ is de variatie op de
metingen groter bij lage concentraties dan bij hogere concentraties. De procentuele totale fout wordt
disproportioneel groot in dit lage bereik. Figuur 4.11. op de volgende pagina stelt een absoluut verschil
plot voor, opgesteld met dezelfde gegevens als deze die door Boemer et al. (2009) gebruikt werden voor
Figuur 4.10., alleen zijn in Figuur 4.11. de acceptatielimieten concentratieafhankelijk gemaakt. Tot een
concentratie van 17.48 mIU/L worden absolute limieten gekozen. Bij hogere concentraties worden
opnieuw relatieve limieten beschouwd. Wanneer Figuur 4.11. beschouwd wordt, zien we dat bij dit
voorstel de ELISA methode over het volledige concentratiebereik gevalideerd is.
Figuur 4.10.: Accuraatheidsprofiel voor de meting van TSH standaardoplossingen met verschillende concentraties: 3.9, 7.8, 15.6, 31.3, 62.5, 125 en 250 mIU/L. De volle lijn staat voor de relatieve bias. De gebroken lijnen stellen de limieten voor van het βETI (β=80%). De stippellijnen stellen de acceptatielimieten voor (±30%) (Boemer et al., 2009)

48
Figuur 4.11.: Absoluut verschil plot opgesteld met dezelfde gegevens als gebruikt voor Figuur 4.10.
Hoffman et al. (2007) stellen een gelijkaardige procedure voor bij de methodevalidatie. Zoals
eerder vermeld, worden acceptatiecriteria voor analytische methoden vaak gekozen op basis van ad-hoc
regels. Ofschoon deze ad-hoc regels aan regulatoire eisen voldoen, houdt deze benadering toch
onbekende en ongecontroleerde risico’s in dat niet-geschikte analytische methoden weerhouden
worden (risico voor de patiënt) en geschikte methoden afgekeurd worden (risico voor de producent).
Hoffman et al. (2007) stellen eveneens een benadering voor op basis van de totale fout, gebaseerd op
het gebruik van een “β-content” tolerantie-interval. Het gebruik van de totale fout is een statistisch en
wetenschappelijk degelijke benadering. Ideale acceptatiecriteria zouden verzekeren dat minstens een
grote proportie (bijvoorbeeld β%) van de toekomstige observaties binnen acceptabele limieten zou
vallen, met een hoge graad van betrouwbaarheid (bijvoorbeeld γ%). Een “β-content” tolerantie-interval
lijkt in dit opzicht een voor de hand liggende keuze. Noteer dat deze applicatie van het TI de structuur
heeft van het statistisch testen van hypothesen. De nulhypothese (H0) is dat minder dan een proportie β
van de metingen binnen de aanvaardbaarheidslimieten [-λ, λ+ valt. De alternatieve hypothese (HA) is dat
minstens een proportie β binnen die grenzen valt. Bij de voorgestelde totale fout procedure wil men de

49
nulhypothese verwerpen ten voordele van het alternatief en op die manier de analytische methode
valideren (Hoffman et al., 2007).
Uit wat hierboven wordt beschreven, kan men opmerken dat het gebruik van het TI bij de
farmaceutische methodevalidatie meestal nog maar in een wetenschappelijke fase is. De benadering
wordt nog niet algemeen erkend en toegepast, al is er vandaag al software zoals e-noval
(http://www.arlenda.com/en/show-product/7/enoval), Agilent ChemStaion Plus Method Validation Pack
(http://www.chem.agilent.com/en-
US/Products/software/chromatography/chemstation/pages/default.aspx) en programma’s van VWR
(http://ru.vwr.com/app/catalog/Product?article_number=HPLC1.18988.0201) ter beschikking, die deze
nieuwe aanpak kunnen beschrijven. De vraag die vele wetenschappers zich stellen in deze fase, is of het
gebruik van het TI toelaat om zo’n belangrijke beslissing als het valideren van een analytische methode
te nemen. De minimale waarden voor β% die a priori moeten gekozen worden zijn meestal 80%, 90% of
95%, wat impliceert dat respectievelijk niet meer dan 20%, 10% of 5% van de metingen buiten de vooraf
gedefinieerde acceptatielimieten vallen. Op die manier wordt er via het gebruik van tolerantie-
intervallen een passend risicobeheer geïntroduceerd (Rozet et al., 2007). Daarbij komt nog dat deze
benadering via tolerantie-intervallen meer direct de prestatie van individuele metingen weerspiegelt.
Het gebruik van zo’n holistisch paradigma houdt een betere inschatting in van de prestatie van een
analytische methode (González & Herrador, 2007). Implementatie van deze benadering vereist echter
wel geschikte keuzes van de proportie β, het betrouwbaarheidsniveau en acceptatielimieten (Hoffman et
al., 2007).
Terwijl de verschillende statistische modellen en benaderingen die gebruikt kunnen worden bij
de farmaceutische methodevalidatie nog bediscussieerd worden, zijn er ondertussen al meer complexe
modellen op komst. Deze laatste zijn samen te vatten onder de noemers “Quality by Design” (QbD) en
“Process Analytical Technology” (PAT). Hierbij worden productieprocessen ontworpen, geanalyseerd en
gecontroleerd, waarbij men zich baseert op het begrijpen van de wetenschappelijke principes die met
het proces gepaard gaan en identificatie van de verschillende variabelen die de productkwaliteit kunnen
aantasten. Hierbij wordt getracht het proces in de realiteit te voorspellen met meer dan de pure
statistische retrospectieve analyse maar met technieken zoals “failure mode and effect analysis” (FMEA)
en andere risicobeoordelingen. (Rathore, 2009; Seely, 2003).

50
5. CONCLUSIE
Het meest robuuste en adequate kalibratiemodel voor de bepaling van de concentratie
vertrekkende van de piekoppervlakte bleek OLR geforceerd door nul te zijn. Uit de resultaten van de
validatie-experimenten kan besloten worden dat de methode voldoet aan de vooropgestelde
specificaties voor lineariteit en imprecisie. Zowel voor het Low IQC staal als voor het High IQC staal vielen
de CVwr en CVT binnen de specificaties voor een stabiel proces (voor CVwr is dat 2%, voor CVT 5%). De
detectielimiet werd als descriptieve meting beschouwd. De gemiddelde absolute hoeveelheid
ethylparabeen die wordt geïnjecteerd bij het bepalen van de LoD bedraagt 18.97 pg. Voor de
prestatiekarakteristiek juistheid kunnen we besluiten dat de methode voldoet aan de specificaties (5%)
op voorwaarde dat de metingen van juistheidsstaal 1 herhaald worden en binnen de limieten vallen.
Er stelden zich geen problemen bij het zelfstandig plannen en uitvoeren van de
methodevalidatie. Deze werd binnen de voorziene tijdspanne afgewerkt.
Bij de methodevergelijking wordt zowel een Bland & Altman benadering als een lineaire
regressieanalyse uitgevoerd. Deze twee benaderingen leveren verschillende conclusies op. Gebruik
makend van de Bland & Altman grafiek zou kunnen besloten worden dat de methodevergelijking voldoet
aan de vooropgestelde specificaties. Op basis van de resultaten die bekomen worden met de lineaire
regressieanalyse echter, komen we tot het besluit dat in het lage concentratiebereik zowel de
systematische als de totale fout niet voldoen aan de specificaties. Bij hogere concentraties voldoen de
systematische en totale fout wel aan de vooropgestelde limieten. Als algemeen besluit kunnen we dus
stellen dat de methodevergelijking niet voldoet aan de specificaties in het lage concentratiebereik. Bij
hogere concentraties wordt er wel aan de specificaties voldaan.
Bij de literatuurstudie werd aangeleerd dat de verschillende informatiebronnen met een kritisch
oog moeten gelezen worden. We kunnen besluiten dat het tolerantie-interval (TI) een aantal applicaties
vindt in het (bio)farmaceutisch veld, o.m. bij het bepalen van aanvaardbaarheidscriteria voor
farmaceutische productieprocessen, evaluatie van dosisuniformiteit en bepaling van de houdbaarheid
van geneesmiddelen. Het gebruik van het TI in de farmaceutische methodevalidatie is op enkele
uitzonderingen na nog in een wetenschappelijke fase, niet in een applicatiefase. Twee types van
tolerantie-intervallen hebben aandacht gekregen in de literatuur. Er wordt een onderscheid gemaakt
tussen het “β-expectation” TI (βETI) en het “β-content” TI (βCTI).

51
6. LITERATUURLIJST
Andersen, F.A. (2008). Final Amended Report on the Safety Assessment of Methylparaben, Ethylparaben, Propylparaben, Isopropylparaben, Butylparaben, Isobutylparaben, and Benzylparaben as Used in Cosmetic Products. International Journal of Toxicology, 27, 1-82. Bland, J.M., Altman, D.G. (1986). Statistical Methods for Assessing Agreement Between Two Methods of Clinical Measurement. The Lancet, 1, 307-310. Boemer, F., Bours, V., Schoos, R., Hubert, P., Rozet, E. (2009). Analytical Validation Based on Total Error Measurement and Cut-off Interpretation of a Neonatal Screening TSH-immunoassay. Journal of Chromatography B, 877, 2412-2417. Byford, J.R., Shaw, L.E., Drew, M.G.B., Pope, G.S., Sauer, M.J., Darbre P.D. (2002). Oestrogenic Activity of Parabens in MCF7 Human Breast Cancer Cells. J. Steroid Biochem. Mol. Biol., 80, 49-60. Chakraborti, S., Li J. (2007). Confidence interval estimation of a normal percentile. Amer. Statistician, 61, 331-6. Darbre, P.D., Aljarrah, A., Miller, W.R., Coldham, N.G., Sauer, M.J., Pope, G.S. (2004). Concentrations of Parabens in Human Breast Tumours. J. Appl. Toxicol., 24, 5-13. Darbre, P.D., Harvey P.W. (2008). Paraben Esters: Review of Recent Studies of Endocrine Toxicity, Absorption, Esterase and Human Exposure, and Discussion of Potential Human Health Risks. Journal of Applied Toxicology, 28(5), 561-578. Denyer, S.P. (1995). Mechanisms of Action of Antibacterial Biocides. International Biodeterioration & Biodegradation, 36(3-4), 227-245. El Hussein, S., Muret, P., Berard, M., Makki, S, Humbert, P. (2007). Assessment of principal parabens used in cosmetics after their passage through human epidermis–dermis layers (ex-vivo study). Experimental Dermatology, 16(10), 830-836. Golden, R., Gandy, J., Vollmer, G. (2005). A review of the endocrine activity of parabens and implications for potential risks to human health. Critical Reviews in Toxicology, 35(5): 435-458. González, A.G., Herrador, M.A. (2007). A Practical Guide to Analytical Method Validation, Including Measurement Uncertainty and Accuracy Profiles. Trends in Analytical Chemistry, 26(3), 227-238. Hahn, G.J., Meeker W.Q. (1991). Statistical Intervals: A Guide for Practitioners. John Wiley & Sons, Verenigde Staten. Harvey P.W. (2004). Discussion of Concentration of Parabens in Human Breast Tumours. J. Appl. Toxicol., 24(4), 307-310. Hauck, W.W., Shaikh, R. (2004). Modified Two-sided Normal Tolerance Intervals for Batch Acceptance of Dose Uniformity. Pharmaceut. Statist., 3, 89-97. Hoffman, D., Kringle, R. (2007). A Total Error Approach for the Validation of Quantitative Analytical Methods. Pharmaceutical Research, 24(6), 1157-1164. Hubert, P., Nguyen-Huu, J., Boulanger, B., Chapuzet, E., Chiap, P., Cohen, N., Compagnon, P., Dewé, W., Feinberg, M., Lallier, M., Laurentie, M., Mercier, N., Muzard, G., Nivet, C., Valat, L. (2004). Harmonization of Strategies for the Validation of Quantitative Analytical Procedures. A SFTP Proposal. J. Pharma. Biomed. Anal., 36(3), 579-586. http://ru.vwr.com/app/catalog/Product?article_number=HPLC1.18988.0201 (05/05/2011). http://www.arlenda.com/en/show-product/7/enoval (05/05/2011). http://www.chem.agilent.com/en-US/Products/software/chromatography/chemstation/pages/default.aspx (19/05/2011). Ishiwatari, S., Suzuki, T., Hitomi, T., Yoshino, T., Matsukuma, S., Tsuji T. (2007). Effects of Ethylparaben on Skin Keratinocytes. J. Appl. Toxicol., 27(1), 1-9.

52
Kiermeier, A., Jarett, R.G., Verbyla, A.P. (2004). A New Approach to Estimating Shelf-life, Pharmaceutical Statistics, 3, 3-11. Komka, K., Kemény, S., Bánfai, B. (2010). Novel Tolerance Interval Model for The Estimation of the Shelf Life of Pharmaceutical Products. J. Chemometrics, 24, 131-139. McDowall, B. (2010). Why System Suitability Tests Are Not a Substitute for Analytical Instrument Qualification or Calibration, Part 1. LCGC North America, 28(12), 1038-1041. Natrella, M. (2010). NIST/SEMATECH e-Handbook of Statistical Methods, http://www.itl.nist.gov/div898/handbook/ Mee, R.W. (1990). Simultaneous Tolerance Intervals for Normal Populations With Common Variance Technometrics, 32(1), 83-92. Novick, S., Christopher, D., Dey, M., Lyapustina, S., Golden, M., Leiner, S., Wyka, B., Delzeit, H., Novak, C., Larner, G. (2009). A Two One-sided Parametric Tolerance Interval Test Control of Delivered Dose Uniformity. AAPS PharmSciTech, 10(3), 841-849. Odeh, R.E., Owen, D.B. (1980). Tables for Normal Tolerance Limits, Sampling Plans and Screening. New York: Marcel Dekker Inc. Rathore, S.A. (2009). Roadmap for Implementation of Quality by Design (QbD) for Biotechnology Products. Trends in Biotechnology, 27(9), 546-553. Rozet, E., Hubert, C., Ceccato, A., Dewé, W., Ziemons, E., Moonen, F., Michail, K., Wintersteiger, R., Streel, B., Boulanger, B., Hubert, P. (2007). Using Tolerance Intervals in Pre-study Validation of Analytical Methods to Predict In-study Results. The Fit-for-purpose concept. Journal of Chromatography A, 1158, 126-137. SCCP/0874:2005. Extended Opinion on: Parabens, underarm cosmetics and breast cancer. Seely, R.J., Seely, J.E. (2003). A Rational, Step-Wise Approach to Process Characterization. BioPharm International. Shabir, G.A. (2007). Method Development and Validation of Preservatives Determination (Benzyl Alcohol, Ethylene Glycol Monophenyl Ether, Methyl Hydroxybenzoate, Ethyl Hydroxybenzoate, Propyl Hydroxybenzoate, and Butyl Hydroxybenzoate) Using HPLC. Journal of Liquid Chromatography & Related Technologies, 30(13-16), 1951-1962. Shabir, G.A., Lough, W.J., Shafique, A.A., Tony, K.B. (2007). Evaluation and Application of Best Practice in Analytical Method Validation. Journal of Liquid Chromatography & Related Technologies, 30, 311-333. Shao, J., Chow, S., (1994). Statistical Inferences in Stability Analysis. Biometrics, 50, 753-763. Soni, M.G., Carabin, I.G., Burdock, G.A. (2005). Safety Assessment of Esters of P-Hydroxybenzoic Acid (Parabens). Food and Chemical Toxicology, 43(7), 985-1015. Stöckl, D. (2007a). Laboratory Statistics & Graphics with EXCEL®. STT consulting, Horebeke, België, 141p. Stöckl, D. (2007b). Method validation With Confidence. STT consulting, Horebeke, België, 52 p. Technometrics (1990). American Statistical Association, Alexandria, VA, Verenigde Staten. The Merck Index, an encyclopedia of chemicals, drugs and biologicals, 13e editie (2001). Merck Research Laboratories, Whithouse Station, NJ, Verenigde Staten.
Vansteelandt, S. (2009). Statistiek en Farmaceutische Data Analyse.
Wald, A., Wolfowitz, J. (1946). Tolerance Limits for a Normal Distribution. Ann. Math. Statist., 17(2), 208-215.
Wang, X., Germansderfer, A., Harms, J., Rathore, A.S. (2007). Using Statistical Analysis for Setting Process Validation Acceptance Criteria for Biotech Products. Biotechnol. Prog., 23, 55-60.

APPENDIX
The manuscript titled Detection decisions defined by the standard deviation of the blank –
Questions form “analytical fresmen” (see below) has been submitted to Analytical Chemistry.

1
Detection decisions defined by the standard deviation of the blank – Questions from
“analytical freshmen”
By Arno Vermote and Manon Buyl
University of Gent, Faculty of Pharmaceutical Sciences, Laboratory for Analytical Chemistry
ABSTRACT
As pharmacy students in our 4th
academic year, we consider us “analytical freshmen”. Our master
thesises included a literature study about detection decisions based on the standard deviation of
the blank (sbi). The more the study progressed, the more we were puzzled. In the end, we were
left with more questions than answers: i) why is the literature on such a seemingly simple concept
(LC = μ + z1-α σ0) so divergent?; ii) different “k-values” are proposed for substituting “z”, which
one is correct?; iii) why was the blank dropped in the classical IUPAC paper?; iv) what is the
logic to substitute z * σ0 with t * sbi and what is the statistical meaning of the latter?; v) what is
the statistical meaning of imposing the confidence interval of a standard deviation upon a t-
statistic?; vi) is the confidence interval of LC adequately described by the confidence interval of
sbi, by the confidence interval of a percentile, or by another approach?; vii) why is there so little
communication between different analytical application fields for such fundamental issues as the
LC?; viii) with respect to LC, has Columbus's egg been found in a recent publication?
KEYWORDS
Limit of detection, Limit of the blank, prediction interval, confidence interval

2
MANUSCRIPT TEXT
To the Editor
We are two pharmacy students, just finishing our master thesis in our 4th academic year. The
general topic of our thesises was method validation, including a literature review about detection
decisions. The more the literature study progressed, the more we were puzzled about the wealth of
literature on such a seemingly simple concept and why there today, still, is no common understanding
about the subject (see Reference 1 and the literature cited therein).1 In the end, we were left with more
questions than answers. Here, we present these questions (see table 1) with a focus on the so-called
“critical value for detection decisions”.2

3
Table 1: Questions from “analytical freshmen”
Why is there so much literature on such a seemingly simple concept and why is there, still, no
common understanding about the subject?
There are different “k-values” proposed as substitue for “z”, which one is correct?
Why has xbarbi been dropped in the classical IUPAC paper?
In the classical IUPAC paper, what was the logic to substitue z * σ0 with t * sbi and what is the
statistical meaning of the latter?
What is the statistical meaning of imposing the confidence interval of a standard deviation upon a
t-statistic?
Is the confidence interval of LC adequately described by the confidence interval of sbi, by the
confidence interval of a percentile, or by another approach?
Why is there so little communication between the different analytical application fields for such
fundamental issues as the LC?
With respect to LC, has Columbus's egg being found?9

4
The critical value for detection decisions (LC) (also called limit of the blank, LoB) is part of the
general concept of the limit of detection.2 The definition of LC is common across analytical application
fields and given by the equation LC = µ + z1-α * σ0 (equation 1: see table 2), where μ is the population
mean of the blank measurements, z1-α is the one-sided z-value for a given probability (for example, 1.645
for 95% probability), and σ0 is the population standard deviation of the blank. A “generic” equation for an
experimentally estimated LC is given by the International Union of Pure and Applied Chemistry (IUPAC),
namely xL = xbarbi + k * sbi (equation 2; note, the IUPAC notation is retained here). In the equation, xL is
the smallest measure that can be detected with reasonable certainty for a given analytical procedure, xbarbi
is the mean of the blank measures, sbi is the standard deviation of the blank measures, and k is a numerical
factor chosen according to the confidence level desired.3
However, while the definition of LC is common across different application fields, the estimation
thereof shows striking differences between some, and the “k-factor” is one of the main reasons. For
example, Linnet4 used LC = xbarbi + Cn * z1-α * sbi (equation 3) while the classical IUPAC publication
2 uses
LC = t1-α,ν * sbi (equation 4). In equation 3, Cn is a bias correction for sbi.5 Calculation of LC from 5
measurements (example in Reference 2) yields LC = xbarbi + 1.75 * sbi with equation 3 and LC = 2.13 * sbi
with equation 4, a striking difference for us. So, which of the k-factors is correct and why is xbarbi dropped
in the classical IUPAC publication? Further, we were particularly intrigued to uncover the logic behind
the substitution of z * σ0 with t * sbi in the IUPAC publication2 and what the statistical meaning of the
latter was. From our basic statistical education, two equations came into our mind, the one of the
confidence interval of a mean µ = xbarbi ± t1-α,ν * sbi/SQRT[n] (equation 5; SQRT = square root) and the
other of the prediction interval xi = xbarbi ± t1-α,ν * sbi/SQRT[1 + 1/n] (equation 6). Substituting n in
equation 5 with 1 and infinite in equation 6 would lead to t * sbi in both cases. However, in the first case sbi
cannot be calculated anymore, and in the latter case it would lead to the z-statistic. Anyway, we were left
with the question what t * s really addresses in statistical terms.

5
Table 2: Equations used in the manuscript (consecutive order)
Number Equation Meaning
1 LC = µ + z1-α * σ0 Concept for LC
2 xL = xbarbi + k * sbi Generic experimental estimate for LC
3 LC = xbarbi + Cn * z1-α * sbi Tolerance interval concept for LC
4 LC = t1-α,ν * sbi IUPAC concept for LC
5 µ = xbarbi ± t1-α,ν * sbi/SQRT[n] Confidence interval for a mean
6 xi = xbarbi ± t1-α ,ν * sbi/SQRT[1 + 1/n] Prediction interval for the next result

6
There also arose questions in connection with the confidence intervals calculated in both concepts.
IUPAC2 used those of the standard deviation, while Linnet
4 used those of a centile. Unfortunately, Linnet
4
gives a simplified formula6 which underestimates the confidence interval of a centile for low n. Note, the
confidence interval of a centile is de facto a tolerance interval7, and indeed, tolerance intervals are used for
detection decisions in wastewater analysis.8 Because the above, we used a tabulated value for a one-sided
tolerance interval in the calculation below (k = 4.21, one-sided 95% confidence and coverage).8
Calculation of the upper limit (UL) of LC from 5 measurements gives ULLC = xbarbi + 4.21 * sbi with the
tolerance interval and ULLC = 2.13 * (2.37 * sbi) = 5.06 * sbi with the confidence interval of a standard
deviation. Again, we are left with more questions than answers. What is the statistical meaning of
imposing the confidence interval of a standard deviation upon a t-statistic? Why is the tolerance interval
concept not addressed as such in certain fields of analysis? Why is it not generally used? Why do different
application fields not cross-reference each other?
Last not least, it was stunning for us to read1 “Following promulgation (i.e., of Method 1631 B), a
lawsuit was filed challenging EPA on the validity of the method. The basis of the challenge included
several specific aspects of Method 1631 as well as the general procedures used to establish the MDL (i.e.,
Method Detection Limit) and minimum level of quantitation (ML) published in the method”. This
happened in 1999, and to the best of our knowledge, all efforts since have not resulted in an agreement
about the procedure to be used for the MDL in the Clean Water Act. We conclude with our last question:
has the scientific community found Columbus's egg?: “Many labored to find the key to accurately
estimating XD (i.e., “Currie Detection Limit”) but there were too many false leads, statistical errors,
notational confusions and completely untested assumptions. With hindsight, it is obvious that the non-
central t distribution was lurking at the heart of the whole matter and it should have been unmasked many
years sooner”.9

7
ACKNOWLEDGEMENT
We thank Prof. Dr. Linda Thienpont for encouraging us to write this letter and devoting senior
research scientist time for its guidance. We thank Dr. Dietmar Stöckl for providing guidance to
this letter and for his patience for answering our questions.

8
REFERENCES
1 Revised assessment of detection and quantitation approaches. U.S. Environmental Protection
Agency (EPA): Washington, 2004.
2 Currie L.A. Pure & Appl. Chem. 1995, 67, 1699-1723.
3 International Union of Pure and Applied Chemistry Goldbook Home Page.
http://goldbook.iupac.org/index.html (accessed May 25, 2011).
4 Linnet, K. Clin Chem Lab Med. 2005, 43, 394-399.
5 Gurland J.; Tripathi R.C. Amer. Stat. 1971, 25, 30-32.
6 Altman D.G. Practical statistics for medical research. Chapman & Hall: Boca Raton, 1997; pp 422.
7 Chakraborti S.; Li J. Amer. Stat. 2007, 61, 331-336.
8 Gibbons R.D. Statistical Methods for Groundwater Monitoring. John Wiley & Sons: New York,
1994.
9 Voigtman E.; Abraham K.T. Spectrochim. Acta B 2011, 66, 105-113.





















