
University of Groningen Experimental studies on signal transduction ...
154
University of Groningen Experimental studies on signal transduction pathways in rheumatoid arthritis Bijl-Westra, Johanna IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it. Please check the document version below. Document Version Publisher's PDF, also known as Version of record Publication date: 2005 Link to publication in University of Groningen/UMCG research database Citation for published version (APA): Bijl-Westra, J. (2005). Experimental studies on signal transduction pathways in rheumatoid arthritis. s.n. Copyright Other than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons). The publication may also be distributed here under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license. More information can be found on the University of Groningen website: https://www.rug.nl/library/open-access/self-archiving-pure/taverne- amendment. Take-down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons the number of authors shown on this cover page is limited to 10 maximum. Download date: 29-05-2022
-
Upload
khangminh22 -
Category
Documents
-
view
2 -
download
0
Transcript of University of Groningen Experimental studies on signal transduction ...

University of Groningen
Experimental studies on signal transduction pathways in rheumatoid arthritisBijl-Westra, Johanna
IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite fromit. Please check the document version below.
Document VersionPublisher's PDF, also known as Version of record
Publication date:2005
Link to publication in University of Groningen/UMCG research database
Citation for published version (APA):Bijl-Westra, J. (2005). Experimental studies on signal transduction pathways in rheumatoid arthritis. s.n.
CopyrightOther than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of theauthor(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons).
The publication may also be distributed here under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license.More information can be found on the University of Groningen website: https://www.rug.nl/library/open-access/self-archiving-pure/taverne-amendment.
Take-down policyIf you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediatelyand investigate your claim.
Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons thenumber of authors shown on this cover page is limited to 10 maximum.
Download date: 29-05-2022

Experimental studies on signal transduction pathways
in rheumatoid arthritis
Hannie Westra

Studies presented in this thesis were financially supported by Dutch Arthritis Association and Johnson and Johnson Pharmaceutical Research and Development, Raritan, New Jersey, USA The printing of this thesis was financially supported by University of Groningen, Dutch Arthritis Association, Groningen University Institute for Drug Exploration (Guide), Pharma-Bio Research (Zuidlaren), Tebu-Bio (Heerhugowaard) Copyright 2005 Johanna Westra ISBN-number: 90-367-22669-1 Printed by Drukkerij van Denderen, Groningen Cover illustration: Microscopic images of rheumatoid synovial tissue, stained for phospho-p38 MAPK (Santa Cruz antibodies). Upper: positive staining of synovial lining layer cells (brown); Lower: positive staining of synovial endothelial cells

RIJKSUNIVERSITEIT GRONINGEN
Experimental studies on signal transduction pathways in rheumatoid arthritis.
Proefschrift
ter verkrijging van het doctoraat in de Medische Wetenschappen
aan de Rijksuniversiteit Groningen op gezag van de
Rector Magnificus, dr. F. Zwarts, in het openbaar te verdedigen op
maandag 13 juni 2005 om 14.45 uur
door
Johanna Bijl-Westra
geboren op 10 december 1960
te Leeuwarden

Promotores: Prof. dr. P.C. Limburg Prof. dr. M.H. van Rijswijk Copromotor: Dr. M.A. van Leeuwen Beoordelingscommissie: Prof. dr. C.G.M. Kallenberg Prof. dr. P.P. Tak Prof. dr. E. Vellenga

Voor Heit en Mem

CONTENTS page
Chapter 1 General introduction and aim of the thesis 9
Chapter 2 Signal transduction pathways in rheumatoid arthritis with emphasis 13
on the p38 mitogen-activated protein kinase (MAPK) pathway.
Chapter 3 Effects of RWJ 67657, a p38 mitogen activated protein 29
kinase (MAPK) inhibitor, on the production of inflammatory
mediators by rheumatoid synovial fibroblasts.
Chapter 4 Strong inhibition of TNF-α production and inhibition 45
of IL-8 and COX-2 mRNA expression in monocyte-derived
macrophages by RWJ 67657, a p38 mitogen activated protein
kinase (MAPK) inhibitor.
Chapter 5 Monocytes and monocyte-derived macrophages differ 59
in regulation of signal transduction pathways.
Chapter 6 Chemokine production and E-selectin expression in 73
activated endothelial cells are inhibited by p38 MAPK
(mitogen-activated protein kinase) inhibitor RWJ 67657.
Chapter 7 Differential effects of NF-κB and p38 MAP kinase 89
inhibitors and combinations thereof on TNFα and IL-1β
induced pro-inflammatory status of endothelial cells in vitro.

Chapter 8 Differential influence of p38 mitogen-activated protein 107
kinase (MAPK) inhibition on acute phase synthesis in human
hepatoma cell lines and human liver slices.
Chapter 9 Summary, general conclusions and future perspectives. 125
Chapter 10 Nederlandse samenvatting 131
Dankwoord 138
List of abbreviations 140
List of publications 142

1
General introduction and aim of the thesis
Johanna Westra

Chapter 1
10
GENERAL INTRODUCTION AND AIM OF THE THESIS Rheumatoid arthritis is a chronic inflammatory disease, located in the synovial joints 1. This inflammatory process leads to degradation of cartilage and bone in the joints with consequent disability and reduced quality of life. Drugs that are intended to inhibit both the inflammatory and destructive processes in RA are the so-called DMARDS (disease modifying anti-rheumatic drugs). Even the gold standard of these drugs, methotrexate (MTX), has limited efficacy and its use may be hampered by side-effects 2. The breakthrough in treatment of RA in the last decade has been the development of TNF-blockers, thereby recognizing the key role of TNF-α in the inflammatory process 3. Although treatment efficacy of RA has dramatically improved with TNF-blockers, still not all patients do respond to this treatment. Moreover these drugs are expensive and severe side effects have been reported. A novel approach to treat inflammatory diseases might be the inhibition of intracellular signal transduction pathways. In short, signal transduction is the process by which a cell converts an extracellular signal into a response. A key role is played by proteins called kinases, which act as regulators of cell function by catalyzing (facilitating) the addition of a negatively charged phosphate group to proteins, resulting in the activation of the catalytic potential of the protein involved. One of the major signal transduction pathways in inflammation is the p38 mitogen-activated protein kinase (MAPK) pathway 4. Specific inhibitors for this pathway have been developed but until now only two of them have entered phase II clinical trials for RA 5;6. It still remains to be elucidated which pathways are important in inflammatory disease and especially in RA and what maybe the clinical importance of signal transduction inhibitors. The aim of the thesis was to investigate the potential of inhibition of signal transduction pathways in treatment of rheumatoid arthritis (RA) The study was performed in vitro using cells, which in some way are relevant players in the inflammatory process in RA. The main focus was on inhibition of the p38 MAPK pathway, but in addition the involvement of the NF-κB and the JAK/STAT pathways was investigated. In chapter 2 a general review is given on signal transduction pathways, which play a role in inflammatory processes in general and in RA more specifically. In chapter 3 the effects of p38 MAPK inhibition on inflammatory mediator production by rheumatoid synovial fibroblasts is investigated. These cells display aggressive invasive behaviour and are to a large extent responsible for the destructive process in the synovial joints in RA. In chapter 4 the effects of p38 MAPK inhibition on the products of monocyte-derived macrophages from healthy controls and RA patients was evaluated. In inflammation macrophages are responsible for the production of the major pro-inflammatory cytokines TNF-α and IL-1β, the key cytokines in RA. In chapter 5 the differences in reactivity towards p38 MAPK inhibition between monocytes and monocyte-derived macrophages were evaluated. In this study the use

Introduction and aim of the thesis
11
of a new technique, the kinase array is introduced. This technique allows the simultaneous determination of the activity of a large number of kinases in a cell lysate. In chapter 6 the effects of p38 MAPK inhibition on activated endothelial cells was investigated. These cells line the blood vessels and play an important role in recruitment of inflammatory cells into the inflamed synovium in RA. In chapter 7 the previous study was expanded by using both an NF-κB inhibitor as well as the p38 MAPK inhibitor in investigating the effects on endothelial cells. In chapter 8 the effects of p38 MAPK inhibition on the acute phase response (APR) was evaluated. Although the APR predominantly is regulated by the JAK/STAT pathway there is cross-talk between the pathways. In the case of p38 MAPK treatment in RA patients this could mean that the production of acute phase proteins may be influenced both directly as well as indirectly by the treatment. REFERENCES 1 Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis.
N.Engl.J.Med. 2001; 344: 907-16. 2 Whittle SL, Hughes RA. Folate supplementation and methotrexate treatment in rheumatoid
arthritis: a review. Rheumatology.(Oxford) 2004; 43: 267-71. 3 Feldmann M, Maini RN. Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned?
Annu.Rev.Immunol. 2001; 19: 163-96. 4 Kumar S, Boehm J, Lee JC. p38 MAP kinases: key signalling molecules as therapeutic targets
for inflammatory diseases. Nat.Rev.Drug Discov. 2003; 2: 717-26. 5 Haddad JJ. VX-745. Vertex Pharmaceuticals. Curr.Opin.Investig.Drugs 2001; 2: 1070-6. 6 Nikas SN, Drosos AA. SCIO-469 Scios Inc. Curr.Opin.Investig.Drugs 2004; 5: 1205-12.

2
Signal transduction pathways in rheumatoid arthritis with
emphasis on the p38 mitogen-activated protein kinase
(MAPK) pathway
Johanna Westra1
Pieter C Limburg1,2
From the Departments of 1 Rheumatology and 2 Pathology and Laboratory
Medicine, University Medical Center Groningen, The Netherlands

Chapter 2
14
CONTENTS
1. Rheumatoid arthritis 1.1 Pathogenesis 1.2 Therapy
2. Signal transduction pathways in RA 2.1 MAPK pathways
2.1.1 ERK 1/2 2.1.2 JNK 1,2,3 2.1.3 p38 MAPKα, β, γ, and δ 2.1.4 ERK5 / BMK1
2.2 NF-κB pathway 2.3 JAK-STAT pathway
3. p38 MAPK pathway 3.1 Identification 3.2 Downstream effects of p38
3.2.1 Gene expression 3.2.2 Post transcriptional regulation of mRNA stability
3.3 Inactivation by phosphatases 4 p38 MAPK inhibitors
4.1 development 4.2 p38 MAPK inhibitors in animal models for arthritis 4.3 p38 MAPK inhibitors in human studies
5 Conclusions

Signal transduction pathways in RA
15
1. RHEUMATOID ARTHRITIS Rheumatoid arthritis (RA) is a chronic inflammatory autoimmune disease, primarily located in the synovial joints, leading to destruction of the cartilage and bone as a result of the chronic disease activity 1. RA affects 0.5 - 1% of the population in the industrialized world is two to three times more frequent in women than men and can lead to disability and reduced quality of life. 1.1. Pathogenesis The cause of RA is still unknown, but both genetic and environmental factors appear to play a role. The association with certain human leukocyte antigen (HLA) DR4 subtypes and RA has been recognized for a long time 2, and now it is found that rather specific amino acid sequences, the so-called shared epitope (SE) confer the highest risk for developing RA 3. RA is characterized by the presence of autoantibodies, especially rheumatoid factors and antibodies to citrullinated proteins (anti-CCP) in the majority of the patients. Recently in a study in blood bank donors it was demonstrated that approximately half of patients with RA had specific serologic abnormalities (rheumatoid factor and anti-CCP antibodies) several years before the onset of symptoms 4. The earliest events in RA might involve activation of the innate immune system, which triggers a T-cell response possibly directed towards citrullinated proteins 5. Infiltrating T-cells in the synovial membrane may, by cell-cell contact, and activation by different cytokines, such as TNF-α, IFN-γ and IL-17, activate monocytes, macrophages and synovial fibroblasts. These cells than produce pro-inflammatory cytokines, mainly TNF-α, IL-1 and IL-6 6. As the disease progresses multiple cytokine networks enter a state of persistent activation, triggering the production of matrix metalloproteinases, ultimately resulting in irreversible damage of cartilage and bone 7. 1.2. Therapy DMARDS (disease modifying anti-rheumatic drugs) are drugs that are intended to inhibit both the inflammatory and destructive processes in RA. Of these DMARDS methotrexate (MTX) is the most commonly used and is regarded as the gold standard of DMARD therapy 8. At doses used in the treatment of RA, MTX is likely to act via a number of intracellular pathways. Upon transport into cells, MTX is converted to polyglutamated forms, which promote intracellular retention. This results eventually in induced release of adenosine, which has an anti-inflammatory effect on neutrophils and mononuclear cells 8;9. MTX modulates cytokine responses at a number of levels and may promote apoptosis of activated lymphocytes 10. Since five years a new DMARD has become available, Leflunomide, which is an active metabolite that inhibits dihydro-orotate-dehydrogenase, an enzyme involved in de novo pyrimidine synthesis 11. Inhibition of this enzyme affects various signal-transduction mechanisms, the generation of cytokines, and cell proliferation and migration. However, these DMARDS still have limited efficacy and may lead to toxicity problems. The pro-inflammatory role of cytokines and the involvement of different cell types led to the development of therapeutics to selectively target cytokines. The key role of TNF-α in the pathogenesis of RA was discovered both in experimental animal models and in RA patients by Feldmann, Maini and others 12. There are now

Chapter 2
16
three drugs clinically available for treatment that block the activity of TNF-α: Infliximab (chimaeric monoclonal antibody to human TNF), Adalumimab (human monoclonal antibody to TNF) and Etanercept (soluble TNF receptor construct) and one to block IL-1 activity: Anakinra (IL-1 receptor antagonist). The efficacy and possible toxic effects of these new drugs have been reviewed by Olsen and Stein 13. Although TNF blockade has been a major breakthrough in the therapy of RA in the last ten years, there are some drawbacks. About half of the patients in clinical trials do not achieve adequate clinical responses expressed as ACR50 responses, remission is rare and these drugs also have side effects, for instance increased risk of tuberculosis13. New drugs to inhibit the production and activity of inflammatory cytokines may be found in inhibitors of intracellular signal transduction pathways 14. These pathways are involved in the primary production of these cytokines, as well as in the responses generated by these cytokines. TNF-α and IL-1 induce a variety of signal transduction cascades that lead to the activation of transcription factors and next to the transcription and translation of genes, coding for inflammatory mediators. The cascades that are important in RA and the potential inhibitors will be discussed below. 2. SIGNAL TRANSDUCTION PATHWAYS IN RA Intracellular signal transduction pathways are intracellular mechanisms by which cells can react to extracellular stimuli such as stress and inflammatory cytokines. In short, intracellular signal transduction is the process by which a cell converts an extracellular signal into a response. A key role thereby is for proteins called kinases, which act as key regulators of cell function by catalyzing (facilitating) the addition of a negatively charged phosphate group to proteins. These signalling cascades ultimately lead to induction of gene transcription and translation into specific proteins. These inflammatory proteins, including cytokines, matrix metalloproteinases, and cyclo-oxygenase (COX-2) are involved in the pathogenesis of RA. The main intracellular signal transduction pathways implicated in RA include the mitogen-activated protein kinase (MAPK) pathways, nuclear factor-kappa B (NF-κB) and the Janus kinase (JAK-STAT) pathway. 2.1. Mitogen-activated protein kinase (MAPK) pathways (figure 1) There are four well-characterized families of MAPKs, acting by phosphorylation of specific serine (Ser), threonine (Thr) and tyrosine (Tyr) residues of target substrates thereby controlling important cellular functions, such as gene expression, mitosis, movement, metabolism and apoptosis. The MAPK isoforms themselves are phosphorylated by dual- specificity serine-threonine MAPK-kinases (MAPKK or MEK) which in turn are phosphorylated by upstream MAPK-kinase-kinases (MAPKKKs or MEKKs). The MAPK family include the extracellular signal-regulated kinases ERK1 and ERK2 (also known as the p42/p44 MAPK pathway), the c-jun NH2-terminal kinases JNK 1, JNK 2 and JNK 3, the four p38 enzymes, p38α, p38β, p38γ and p38δ, and the ERK5 or big MAP kinase 1 (BMK1) 15. All MAPKs share the amino-acid sequence Thr-Xxx-Tyr in which X differs: X is glutamic acid (Glu), proline (Pro) and glycine (Gly) for the ERK, JNK and p38 MAPK respectively 16.

Signal transduction pathways in RA
17
Figure 1. Mitogen-activated protein kinase (MAPK) pathways.
2.1.1. The ERK 1/2 signaling pathway is a major determinant in the control of cell growth, cell differentiation and cell survival. Growth factors, cytokines, viral infection, and carcinogens are the main activators of the proto-oncogene Ras, a small GTP-binding protein, which is the common upstream molecule of the MEKKs Raf (Raf1, A-Raf or B-Raf) 17. Activated Rafs phosphorylate MEK1/2, which in turn activate ERK1 and ERK2. ERKs can directly phosphorylate a set of transcription factors including Ets-1, c-Jun and c-myc, or activate RSK (ribosomal S6 kinase), which leads to the activation of the transcription factor CREB (cyclic AMP-responsive element-binding protein). MEK1 and MEK2 activity has been detected in a significant number of primary human tumor cells. Inhibitors of the ERK pathway (Ras-, Raf- and Src-inhibitors) are entering clinical trials as potential anti-cancer agents. PD98059 and U0126 are non-ATP competive MEK 1/2 inhibitors, which block phosphorylation and activation of ERK1 and 2 by MEK 18.
2.1.2. JNKs were discovered to bind and phosphorylate the DNA-binding protein c-Jun (a component of the AP-1 (activator protein) transcription complex) and to increase its transcriptional activity. Regulation of the JNK pathway is extremely complex and is influenced by many kinases. In general, stress or cytokines can initiate a series of events in which GTP-binding proteins such as Cdc42, Rac or Ras can activate protein kinases such as GCK (germinal centre kinase), which in turn can activate ASK (apoptosis stimulating kinase), TAK (TGFβ-activated kinase) and the MEKKs 1,2 and 3 19. Then MKK4 and MKK7 are activated, which are the direct activators of the JNK MAPKs. Targets of this pathway include c-Jun, ATF2 (activating transcription factor

Chapter 2
18
2) and ELK-1. JNK is one of the primary MAPKs required for expression of matrix metalloproteinases and joint destruction in models of inflammatory arthritis 20. The best known JNK2 inhibitor is SP600125, while another JNK pathway inhibitor CEP1347, has been reported to inhibit members of the MLK (mixed lineage kinase) family, which are upstream activators of the JNK pathway 18. 2.1.3. p38 MAPKs are, like the JNKs, stress-activated protein kinases, that mediate responses to cellular stress factors, such as UV light, osmotic shock and cytokines. The main activation route for p38 MAPK is through phosphorylation by MMK3 / MKK6 and possibly by MKK4, which in turn are activated by the MAPKKKs (figure 2). Members of the MAPKKK superfamily include MEKK 1-4, MLK, Tpl2 (tumor progression locus-2), TAK-1 and ASK-1 21. The MAPKKKs themselves are activated by small GTP-binding proteins, including Rac and Cdc42, partly involving p21-activated kinases (PAKs). In inflammation the most important route for activation is via TNF-α and IL-1 by ligation of their respective membrane receptors and recruitment of intracellular adaptor molecules. Activated p38 MAPKs can phosphorylate downstream kinases (MAPKAPK-2, MSK-1, PRAK and MNK) and transcription factors such as ATF-2, CHOP (C/EBP homologous protein) and ELK-1. Lipopolysaccharide (LPS) signal transduction is also one of the activation routes for
Figure 2. p38 mitogen-activated protein kinase (MAPK) pathway.

Signal transduction pathways in RA
19
p38 MAPK. LPS is a common constituent of Gram-negative bacterial outer membranes and is the principal initiator of septic shock. Signalling of LPS involves a receptor complex with Toll-like receptor 4 (TLR-4), CD14 and the adaptor molecules MyDD88 and IRAK (interleukin-receptor associated kinase). The route then resembles the IL-1 route: via TRAF6 and TAK1 p38 MAPK can be activated as well as the JNK and the NF-κB pathways 22. The p38 MAPK signal transduction pathway will be discussed in detail below.
2.1.4. The fourth and least studied MAP kinase pathway, BMK1 or ERK5, is activated in response to growth factors and stress. This pathway has not only been implicated in cell survival, proliferation and differentiation, but also in pathologic processes such as carcinogenesis, cardiac hypertrophy and atherosclerosis 23. MEK5 is the upstream kinase of BMK1, and can be inhibited by inhibitors of MEK 1/2 such as PD98059 and U0126. The MEK5 kinases identified thus far are MEKK2 and MEKK3, which are also known to regulate p38 MAPK and JNK activity through the activation of MKK3/6 and MKK4/7 respectively 23. 2.2. NF-κB pathway (figure 3) The transcription factor Nuclear Factor κ-B (NF-κB) is a key factor in the transcription of many inflammatory genes. NF-κB is a complex group of heterodimeric and homodimeric transcription factors, consisting of five members: c-Rel, RelA (p65 or NF-κB3), RelB, NF-κB1 (p50/p105) and NF-κB2 (p52/p100) 24. Normally these dimers bind to the specific inhibitors of NF-κB, known as IκB proteins. Upstream kinases, including members of the MAPKKK family and NF-κB- activating-kinase (NAK) can induce phosphorylation and degradation of IκB, thereby liberating the NF-κB dimers, which translocate to the nucleus and regulate gene transcription 24;25. The modification of the IκB proteins depends on the IKK (IκB kinase) complex, which is composed of three subunits: the catalytic subunits IKK-α(1) and IKK-β(2), and the regulatory subunit IKK-γ (also known as NEMO). These subunits have specific roles in the regulation of NF-κB activity. The activation of NF-κB has been implicated in the pathogenesis of RA. Translocation of nuclear p50 and p65 was demonstrated in RA synovial lining cells and in mononuclear cells of the sublining 26. It has been demonstrated that NF-κB suppression is beneficial in many models of inflammatory disease 27. Moreover, for many therapeutic agents it has been shown that at least some of their effects are due to NF-κB blockade. Efforts to block this pathway have led to the development of small-molecule inhibitors of various kinases and regulatory proteins and also to research in gene therapy. In arthritis IKK-β seems an attractive target for therapy, because it regulates cytokine production in many cell types including synovial fibroblasts 28;29. Although it is obvious that NF-κB plays a key role in inflammatory diseases, there are major safety concerns about inhibition of this transcription factor, because of the major role that NF-κB plays in host defence, homeostasis, cell survival and response to stress.

Chapter 2
20
Figure 3. NF-κB pathway. 2.3. JAK/STAT pathway (figure 4) The Janus kinase (JAK) family and the signal transducer and activator of transcription (STAT) transcription factors play an important role in cytokine signal transduction. Type I cytokine receptors (for colony stimulating factors, and several interleukins) and type II cytokine receptors (for interferons) lack intrinsic kinase activity and rely on Jak proteins to initiate signalling 30. In the case of IL-6 an association with the IL-6 receptor and gp130 subunits takes place that activates JAKs. This is followed by the phosphorylation of the tyrosine-based docking sites on the receptor and recruitment of STATs. They form homo-hetero-dimers and translocate to the nucleus, where they bind target sequences 31. Dimerization of the IL-6-type cytokine receptors does not only lead to activation of the JAK/STAT signalling pathway, but also of the MAPK cascade. Negative regulation of IL-6 signalling via the JAK/STAT pathway may occur in different ways 30. Suppressor of cytokine signalling (SOCS) proteins are induced in response to IL-6 binding and can bind directly to the JAKs. SH2-domain-containing tyrosine phosphatase-1 (SHP-1) either can dephosphorylate JAKs or activated receptor subunits. Protein inhibitors of activated STATs (PIAS) family members inactivate STAT dimers.

Signal transduction pathways in RA
21
Figure 4. JAK/STAT pathway.
THE p38 MAPK SIGNAL TRANSDUCTION PATHWAY 3.1. Identification The human p38α MAPK were first identified as the molecular target of pyridinylimidazole class of compounds, that were known to block TNF-α and IL-1 release from lipopolysaccharide (LPS)- stimulated human monocytes 32. Originally these proteins were designated cytokine-suppressive anti-inflammatory drug-binding proteins (CSBP) 33. Until now, five isoforms of p38 MAPK have been identified: p38β1 and p38β2 have more than 70% identity to p38α, whereas p38γ and p38δ have approximately 60% identity to p38α. Functional differences between the isoforms are related to their differential expression, activation, and substrate specificity 34. p38α, p38β and p38δ are widely produced in various tissues, while p38γ is expressed primarily in skeletal muscle. Inflammatory cells synthesize predominantly p38α and p38δ protein, but endothelial cells also produce p38β 35;36. ERK, JNK and p38 MAPK activation were almost exclusively found in synovial tissue from RA, but not osteoarthritis patients. p38 MAPK activation was observed in the synovial lining layer and in synovial endothelial cells 37. 3.2. Downstream effects of p38 MAPK activation 3.2.1. Gene expression One of the main downstream effects of the p38 MAPK pathway is regulation of gene expression, which can be at the transcriptional level, but also at the translational level,

Chapter 2
22
leading to protein synthesis. p38 MAPK has been implicated in the activation of various transcription factors including: ATF-2, MEF-2C, CHOP, and NF-κB 38-40. Other transcription factors are phosphorylated by downstream protein kinases that are themselves activated by phosphorylated p38 MAPK, such as MAPKAPK-2 (MAPK activated protein kinase). Furthermore MAPKAPK-2 deficient mice show diminished production of IL-6 and TNF-α 41. These effects are thought to be mediated by a mechanism involving mRNA turnover and protein translation. The p38 MAPK/MAPKAPK-2 pathway is crucial to inflammatory cytokine production. 3.2.2. Post transcriptional regulation of mRNA stability As will be discussed later, several p38 MAPK inhibitors have been developed which were shown to block the production of inflammatory cytokines. This inhibition seems to be a combined effect at the level of transcription and translation. Inducible cytokines usually have short lived mRNAs and contain an AU-rich element (ARE) in their 3’untranslated region responsible for their high turn-over rate 42. These AREs contain repeats of the motif AUUUA, and were discovered as instability elements. In the case of inflammatory response mRNAs, the instability is countered by signalling in the p38 MAPK pathway 43. Under normal conditions, these AU-rich elements are occupied by AU-binding proteins, thereby blocking translation or transcription. It has been demonstrated that upon stimulation these AU-binding proteins are phosphorylated in a p38 MAPK-dependent manner, resulting in their release, and allowing translation of these mRNAs 43;44. Inhibitors of p38 MAPK can target these events directly or via MAPKAPK-2. It has been demonstrated that both translation and stability of TNF-α mRNA are regulated by the p38MAPK pathway 45, whereas IL-6 and IL-8 mRNA stability is regulated by p38 MAPK, but the extent of inhibition of protein production varies with cell type 46;47. Activation of p38 MAPK has also been shown to enhance the mRNA stability of collagenase-1 (MMP-1) and stromelysin-1 (MMP-3 ) 48. Finally, Lasa et al demonstrated that the gluco-corticoid dexamethasone destabilizes COX-2 mRNA by inhibiting p38 MAPK. This effect was induced by expression of MAPK-phosphatase-1 (MKP-1) 49. 3.3. Inactivation by phosphatases For control of the MAPK signal transduction pathways dephosphorylation is necessary. Over the last decade a family of endogenous negative regulators of MAPK, the MAPK phosphatases (MKPs), also known as DUSPs (dual-specificity phosphatases) have been described 50. The MKPs dephosphorylate the tyrosine and threonine motifs of MAPK to deactivate MAPK-dependent signalling. At least 10 mammalian MKPs have been cloned and characterized, which have different subcellular distribution, substrate specificity, and expression patterns. MKP-1 was the first isolated MKP, and is a 39.5 kD protein that is preferentially localized in the cell nucleus. It is capable of dephosphorylating all MAPK families, although a preference for dephosphorylating p38 MAPK and JNK has been described 51. MKP-1 is an early response gene that is induced by the same stimuli that activate the MAPKs, such as cytokines, osmotic shock and UV radiation 52;53. Recently the expression of MKP-1 in

Signal transduction pathways in RA
23
rheumatoid synovial fibroblasts was demonstrated as well as a role for glucocorticoid dependent upregulation of MKP-152. 4. p38 MAPK INHIBITORS (figure 5) 4.1. Development Already in 1988 the inhibition of IL-1 production by the anti-inflammatory compound SK&F 86002 was reported 54, but it took until 1994 to discover that the molecular target of the pyridinyl imidazole class of compounds indeed proved to be p38 MAPK, originally known as CSBP 32. Since the original report of the efficacy of these compounds, they have become the most widely studied inhibitors of this kinase. The compounds have been used as framework for further synthetic work and have been utilized to elucidate the role of p38 MAPK in the immune system. Crystallographic and kinetic experiments have shown that the pyridinyl imidazole family of p38 MAPK inhibitors bind at the ATP binding site of p38 MAPK and compete with ATP for binding to active, phosphorylated p38 MAPK 55. When p38 MAPK is in the unactivated form, ATP is non-competitive with many p38 MAPK inhibitors. After the structure-activity relationship was established SB 203580 and other 2, 4, 5- triaryl imidazoles were prepared as pharmacological tools to regulate cytokine synthesis. A large number of preclinical studies have reported that specific and selective p38 MAPK inhibitors block the production of inflammatory cytokines in vitro and in vivo. Furthermore, the p38 MAPK pathway is involved in the induction of several other inflammatory molecules such as cyclo-oxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS).
Figure 5. p38 MAPK inhibitors.
The p38 MAPK inhibitor used in our studies, RWJ 67657, was developed by Johnson and Johnson Pharmaceutical Research and Development and was first described in 1999 56. RWJ 67657 (4-[4-(4-fluorophenyl)-1-(3-phenylpropyl)-5-(4-pyridinyl)-1H-imidazol-2-yl]-3-butyn-1-ol) has been shown to be effective in inhibiting the release of TNF-α from LPS-treated human peripheral blood mononuclear cells with an IC50 of 3 nM. In comparison to the literature standard SB 203580 this new compound proved to be approximately 10-fold more potent in all p38 MAPK dependent systems tested.

Chapter 2
24
Moreover the compound inhibited the enzymatic activity of p38α and β, but not of γ and δ, in vitro and had no significant activity against a variety of other enzymes. 4.2. p38 MAPK inhibitors in animal models for arthritis Several p38 MAPK inhibitors have been evaluated in animal arthritis models. Already in 1996 the pharmacological profile of SB 203580 (GlaxoSmithKline) was investigated in adjuvant-induced arthritis in Lewis rats 57, whereas in 1998 the pharmacologically effects of SB 220025 (GlaxoSmithKline) were investigated in an angiogenesis and chronic disease model 58. RPR-200765A (Aventis) reduced incidence and progression in the rat streptococcal cell wall (SCW) arthritis model at doses given orally 59, while prevention of the onset and progression of collagen-induced arthritis were reported for FR167653 (Fujisawa Pharmaceuticals) 60. Recently SB 242235 (GlaxoSmithKline) was evaluated in a new model of arthritis, pristane-induced arthritis, and demonstrated to significantly reduce all arthritis scores 61. 4.3. p38 MAPK inhibitors in human studies RWJ 67657 is one of the p38 MAPK inhibitors who until now have been used in human studies. The effects on clinical and cytokine response to endotoxaemia were studied in healthy human volunteers and reported by Fijen et al 62. Single oral doses of RWJ 67657 dose-dependently decreased symptoms and elevated cytokine levels, induced after administration of endotoxin. Furthermore single-dose pharmacokinetics and pharmacodynamics of RWJ 67657 were investigated in healthy male subjects 63. RWJ 67657 was rapidly absorbed (mean tmax = 0.6-2.5 h) and there were no significant adverse effects associated with single doses of this drug. This study demonstrates that RWJ 67657 has acceptable safety and pharmacokinetics to warrant further investigation in a repeat-dose setting. Other compounds that have been investigated in rheumatoid arthritis patients are VX-74564 and SCIO-469, which is now in phase II clinical trial 65. 5. CONCLUSIONS The discovery of p38 MAPK inhibitors have dramatically increased the understanding of signal transduction pathways involved in inflammation. It is widely expected that p38 MAPK inhibitors will have efficacy in arthritis and other inflammatory diseases. However, clinical trials have been stopped due to safety issues. One of the reasons for these undesirable effects might be the cross-reactivity against other kinases. Solutions for these problems might lie in the development of non-ATP competitive inhibitors or inhibitors, which target other molecules in the p38 MAPK pathway. REFERENCES 1 Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis.
N.Engl.J.Med. 2001; 344: 907-16. 2 Stastny P. Association of the B-cell alloantigen DRw4 with rheumatoid arthritis. N.Engl.J.Med.
1978; 298: 869-71. 3 McDonagh JE, Dunn A, Ollier WE, Walker DJ. Compound heterozygosity for the shared
epitope and the risk and severity of rheumatoid arthritis in extended pedigrees. Br.J.Rheumatol. 1997; 36: 322-7.
4 Nielen MM. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis and rheumatism 2004; 50: 380-6.

Signal transduction pathways in RA
25
5 Firestein GS, Zvaifler NJ. How important are T cells in chronic rheumatoid synovitis?: II. T cell-independent mechanisms from beginning to end. Arthritis Rheum. 2002; 46: 298-308.
6 Bombara MP, Webb DL, Conrad P et al. Cell contact between T cells and synovial fibroblasts causes induction of adhesion molecules and cytokines. J.Leukoc.Biol. 1993; 54: 399-406.
7 Dayer JM, Graham R, Russell G, Krane SM. Collagenase production by rheumatoid synovial cells: stimulation by a human lymphocyte factor. Science 1977; 195: 181-3.
8 Bathon JM, Martin RW, Fleischmann RM et al. A comparison of etanercept and methotrexate in patients with early rheumatoid arthritis. N.Engl.J.Med. 2000; 343: 1586-93.
9 Whittle SL, Hughes RA. Folate supplementation and methotrexate treatment in rheumatoid arthritis: a review. Rheumatology.(Oxford) 2004; 43: 267-71.
10 Genestier L, Paillot R, Quemeneur L, Izeradjene K, Revillard JP. Mechanisms of action of methotrexate. Immunopharmacology 2000; 47: 247-57.
11 Li EK, Tam LS, Tomlinson B. Leflunomide in the treatment of rheumatoid arthritis. Clin.Ther. 2004; 26: 447-59.
12 Feldmann M, Maini RN. Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned? Annu.Rev.Immunol. 2001; 19: 163-96.
13 Olsen NJ, Stein CM. New drugs for rheumatoid arthritis. N.Engl.J.Med. 2004; 350: 2167-79. 14 Smolen JS, Steiner G. Therapeutic strategies for rheumatoid arthritis. Nat.Rev.Drug Discov.
2003; 2: 473-88. 15 Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK,
and p38 protein kinases. Science 2002; 298: 1911-2. 16 Herlaar E, Brown Z. p38 MAPK signalling cascades in inflammatory disease. Mol.Med.Today
1999; 5: 439-47. 17 Chang F, Steelman LS, Lee JT et al. Signal transduction mediated by the Ras/Raf/MEK/ERK
pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia 2003; 17: 1263-93.
18 English JM, Cobb MH. Pharmacological inhibitors of MAPK pathways. Trends Pharmacol.Sci. 2002; 23: 40-5.
19 Barr RK, Bogoyevitch MA. The c-Jun N-terminal protein kinase family of mitogen-activated protein kinases (JNK MAPKs). Int.J.Biochem.Cell Biol. 2001; 33: 1047-63.
20 Han Z, Boyle DL, Chang L et al. c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J.Clin.Invest 2001; 108: 73-81.
21 Lee JC, Kumar S, Griswold DE, Underwood DC, Votta BJ, Adams JL. Inhibition of p38 MAP kinase as a therapeutic strategy. Immunopharmacology 2000; 47: 185-201.
22 Diks SH, Richel DJ, Peppelenbosch MP. LPS signal transduction: the picture is becoming more complex. Curr.Top.Med.Chem. 2004; 4: 1115-26.
23 Hayashi M, Lee JD. Role of the BMK1/ERK5 signaling pathway: lessons from knockout mice. J.Mol.Med. 2004; 82: 800-8.
24 Karin M, Yamamoto Y, Wang QM. The IKK NF-kappa B system: a treasure trove for drug development. Nat.Rev.Drug Discov. 2004; 3: 17-26.
25 Sweeney SE, Firestein GS. Signal transduction in rheumatoid arthritis. Curr.Opin.Rheumatol. 2004; 16: 231-7.
26 Handel ML, McMorrow LB, Gravallese EM. Nuclear factor-kappa B in rheumatoid synovium. Localization of p50 and p65. Arthritis Rheum. 1995; 38: 1762-70.
27 McIntyre KW, Shuster DJ, Gillooly KM et al. A highly selective inhibitor of I kappa B kinase, BMS-345541, blocks both joint inflammation and destruction in collagen-induced arthritis in mice. Arthritis Rheum. 2003; 48: 2652-9.
28 Andreakos E, Smith C, Kiriakidis S et al. Heterogeneous requirement of IkappaB kinase 2 for inflammatory cytokine and matrix metalloproteinase production in rheumatoid arthritis: implications for therapy. Arthritis Rheum. 2003; 48: 1901-12.
29 Tak PP, Gerlag DM, Aupperle KR et al. Inhibitor of nuclear factor kappaB kinase beta is a key regulator of synovial inflammation. Arthritis Rheum. 2001; 44: 1897-907.
30 Ortmann RA, Cheng T, Visconti R, Frucht DM, O'Shea JJ. Janus kinases and signal transducers and activators of transcription: their roles in cytokine signaling, development and immunoregulation. Arthritis Res. 2000; 2: 16-32.

Chapter 2
26
31 Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem.J. 2003; 374: 1-20.
32 Lee JC, Laydon JT, McDonnell PC et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 1994; 372: 739-46.
33 Obata T, Brown GE, Yaffe MB. MAP kinase pathways activated by stress: the p38 MAPK pathway. Crit Care Med. 2000; 28: N67-N77.
34 Kumar S, McDonnell PC, Gum RJ, Hand AT, Lee JC, Young PR. Novel homologues of CSBP/p38 MAP kinase: activation, substrate specificity and sensitivity to inhibition by pyridinyl imidazoles. Biochem.Biophys.Res.Commun. 1997; 235: 533-8.
35 Hale KK, Trollinger D, Rihanek M, Manthey CL. Differential expression and activation of p38 mitogen-activated protein kinase alpha, beta, gamma, and delta in inflammatory cell lineages. J.Immunol. 1999; 162: 4246-52.
36 Herlaar E, Brown Z. p38 MAPK signalling cascades in inflammatory disease. Mol.Med.Today 1999; 5: 439-47.
37 Schett G, Tohidast-Akrad M, Smolen JS et al. Activation, differential localization, and regulation of the stress- activated protein kinases, extracellular signal-regulated kinase, c-JUN N-terminal kinase, and p38 mitogen-activated protein kinase, in synovial tissue and cells in rheumatoid arthritis. Arthritis Rheum. 2000; 43: 2501-12.
38 Han J, Jiang Y, Li Z, Kravchenko VV, Ulevitch RJ. Activation of the transcription factor MEF2C by the MAP kinase p38 in inflammation. Nature 1997; 386: 296-9.
39 Zhu T, Lobie PE. Janus kinase 2-dependent activation of p38 mitogen-activated protein kinase by growth hormone. Resultant transcriptional activation of ATF-2 and CHOP, cytoskeletal re-organization and mitogenesis. J.Biol.Chem. 2000; 275: 2103-14.
40 Wesselborg S, Bauer MK, Vogt M, Schmitz ML, Schulze-Osthoff K. Activation of transcription factor NF-kappaB and p38 mitogen-activated protein kinase is mediated by distinct and separate stress effector pathways. J.Biol.Chem. 1997; 272: 12422-9.
41 Kotlyarov A, Neininger A, Schubert C et al. MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nat.Cell Biol. 1999; 1: 94-7.
42 Caput D, Beutler B, Hartog K, Thayer R, Brown-Shimer S, Cerami A. Identification of a common nucleotide sequence in the 3'-untranslated region of mRNA molecules specifying inflammatory mediators. Proc.Natl.Acad.Sci.U.S.A 1986; 83: 1670-4.
43 Frevel MA, Bakheet T, Silva AM, Hissong JG, Khabar KS, Williams BR. p38 Mitogen-activated protein kinase-dependent and -independent signaling of mRNA stability of AU-rich element-containing transcripts. Mol.Cell Biol. 2003; 23: 425-36.
44 Dean JL, Sully G, Clark AR, Saklatvala J. The involvement of AU-rich element-binding proteins in p38 mitogen-activated protein kinase pathway-mediated mRNA stabilisation. Cell Signal. 2004; 16: 1113-21.
45 Brook M, Sully G, Clark AR, Saklatvala J. Regulation of tumour necrosis factor alpha mRNA stability by the mitogen-activated protein kinase p38 signalling cascade. FEBS Lett. 2000; 483: 57-61.
46 Miyazawa K, Mori A, Miyata H, Akahane M, Ajisawa Y, Okudaira H. Regulation of interleukin-1beta-induced interleukin-6 gene expression in human fibroblast-like synoviocytes by p38 mitogen-activated protein kinase. J.Biol.Chem. 1998; 273: 24832-8.
47 Ridley SH, Sarsfield SJ, Lee JC et al. Actions of IL-1 are selectively controlled by p38 mitogen-activated protein kinase: regulation of prostaglandin H synthase-2, metalloproteinases, and IL-6 at different levels. J.Immunol. 1997; 158: 3165-73.
48 Reunanen N, Li SP, Ahonen M, Foschi M, Han J, Kahari VM. Activation of p38 alpha MAPK enhances collagenase-1 (matrix metalloproteinase (MMP)-1) and stromelysin-1 (MMP-3) expression by mRNA stabilization. J.Biol.Chem. 2002; 277: 32360-8.
49 Lasa M, Abraham SM, Boucheron C, Saklatvala J, Clark AR. Dexamethasone causes sustained expression of mitogen-activated protein kinase (MAPK) phosphatase 1 and phosphatase-mediated inhibition of MAPK p38. Mol.Cell Biol. 2002; 22: 7802-11.
50 Theodosiou A, Ashworth A. MAP kinase phosphatases. Genome Biol. 2002; 3: REVIEWS3009.

Signal transduction pathways in RA
27
51 Franklin CC, Kraft AS. Conditional expression of the mitogen-activated protein kinase (MAPK) phosphatase MKP-1 preferentially inhibits p38 MAPK and stress-activated protein kinase in U937 cells. J.Biol.Chem. 1997; 272: 16917-23.
52 Toh ML, Yang Y, Leech M, Santos L, Morand EF. Expression of mitogen-activated protein kinase phosphatase 1, a negative regulator of the mitogen-activated protein kinases, in rheumatoid arthritis: up-regulation by interleukin-1beta and glucocorticoids. Arthritis Rheum. 2004; 50: 3118-28.
53 Xu Q, Konta T, Nakayama K et al. Cellular defense against H2O2-induced apoptosis via MAP kinase-MKP-1 pathway. Free Radic.Biol.Med. 2004; 36: 985-93.
54 Lee JC, Griswold DE, Votta B, Hanna N. Inhibition of monocyte IL-1 production by the anti-inflammatory compound, SK&F 86002. Int.J.Immunopharmacol. 1988; 10: 835-43.
55 Young PR, McLaughlin MM, Kumar S et al. Pyridinyl imidazole inhibitors of p38 mitogen-activated protein kinase bind in the ATP site. J.Biol.Chem. 1997; 272: 12116-21.
56 Wadsworth SA, Cavender DE, Beers SA et al. RWJ 67657, a potent, orally active inhibitor of p38 mitogen-activated protein kinase. J.Pharmacol.Exp.Ther. 1999; 291: 680-7.
57 Badger AM, Bradbeer JN, Votta B, Lee JC, Adams JL, Griswold DE. Pharmacological profile of SB 203580, a selective inhibitor of cytokine suppressive binding protein/p38 kinase, in animal models of arthritis, bone resorption, endotoxin shock and immune function. J.Pharmacol.Exp.Ther. 1996; 279: 1453-61.
58 Jackson JR, Bolognese B, Hillegass L et al. Pharmacological effects of SB 220025, a selective inhibitor of P38 mitogen-activated protein kinase, in angiogenesis and chronic inflammatory disease models. J.Pharmacol.Exp.Ther. 1998; 284: 687-92.
59 Mclay LM, Halley F, Souness JE et al. The discovery of RPR 200765A, a p38 MAP kinase inhibitor displaying a good oral anti-arthritic efficacy. Bioorg.Med.Chem. 2001; 9: 537-54.
60 Nishikawa M, Myoui A, Tomita T, Takahi K, Nampei A, Yoshikawa H. Prevention of the onset and progression of collagen-induced arthritis in rats by the potent p38 mitogen-activated protein kinase inhibitor FR167653. Arthritis Rheum. 2003; 48: 2670-81.
61 Patten C, Bush K, Rioja I et al. Characterization of pristane-induced arthritis, a murine model of chronic disease: response to antirheumatic agents, expression of joint cytokines, and immunopathology. Arthritis Rheum. 2004; 50: 3334-45.
62 Fijen JW, Zijlstra JG, de Boer P et al. Suppression of the clinical and cytokine response to endotoxin by RWJ-67657, a p38 mitogen-activated protein-kinase inhibitor, in healthy human volunteers. Clin.Exp.Immunol. 2001; 124: 16-20.
63 Parasrampuria DA, de Boer P, Desai-Krieger D, Chow AT, Jones CR. Single-dose pharmacokinetics and pharmacodynamics of RWJ 67657, a specific p38 mitogen-activated protein kinase inhibitor: a first-in-human study. J.Clin.Pharmacol. 2003; 43: 406-13.
64 Haddad JJ. VX-745. Vertex Pharmaceuticals. Curr.Opin.Investig.Drugs 2001; 2: 1070-6. 65 Nikas SN, Drosos AA. SCIO-469 Scios Inc. Curr.Opin.Investig.Drugs 2004; 5: 1205-12.

3
Effects of RWJ 67657, a p38 mitogen activated protein
kinase (MAPK) inhibitor, on the production of
inflammatory mediators by rheumatoid synovial
fibroblasts
Johanna Westra1
Pieter C Limburg1,2
Peter de Boer3
Martin H van Rijswijk1
From the Departments of 1 Rheumatology, 2 Pathology and Laboratory Medicine,
University Medical Center Groningen, The Netherlands, and 3 Pharmaceutical
Research and Development, Johnson and Johnson, Saunderton, United Kingdom
Annals of the Rheumatic Diseases 2004; 63 (11): 1453 – 1459

Chapter 3
30
ABSTRACT Objective. To investigate the effect of the p38 mitogen activated protein kinase (MAPK) inhibitor RWJ 67657 on inflammatory mediator production by rheumatoid synovial fibroblasts (RSF). Methods. RSF were pretreated with RWJ 67657 and stimulated with TNF-α and/or IL-1β. Protein levels and mRNA expression of MMP-1, MMP-3, TIMP-1, IL-6 and IL-8 were determined, as was mRNA expression of COX-2 and ADAMTS-4. Results. MMP-3 production was significantly inhibited at 1 µM RWJ 67657, MMP-1 production at 10 µM, whereas TIMP-1 production was not inhibited. Significant inhibition of IL-6 and IL-8 protein production was already seen at 0.1 µM of RWJ 67657. mRNA expression profiles were in concordance with protein production. Significant inhibition of COX-2 mRNA expression already occurred at 0.01 µM. Conclusion. RWJ 67657 inhibits major proinflammatory mediator production in stimulated RSF at pharmacological relevant concentrations. These findings could have important relevance for treatment of rheumatoid arthritis. Key words. p38 MAPK inhibitor, synovial fibroblast, matrix metalloproteinase, cytokine, rheumatoid arthritis

p38 MAPK inhibition in RSF
31
INTRODUCTION The pathogenesis of rheumatoid arthritis (RA) involves complex interrelations between T cells, macrophages, fibroblasts and other immune cells. Growing evidence suggests that activated rheumatoid synovial fibroblasts (RSF) play a major role in both initiating and driving RA 1. Specially, RSF in the lining layer display numerous features of cellular activation that ultimately result in an aggressive, invasive behaviour. These cells can attach to the articular cartilage and invade the extra cellular matrix. Furthermore, RSF are important producers of inflammatory mediators such as cytokines and matrix-metalloproteinases (MMP). Many of these mediators are regulated by mitogen activated protein kinase (MAPK) pathways and downstream transcription factors 2. At least 3 subgroups of MAPK have been identified. These are the extra cellular signal regulated kinases (ERK), the c-Jun N-terminal or stress activated protein kinases (JNK/SAPK) and the p38 MAPK 3. In general ERK are activated by growth factors and hormones, whereas both JNK and p38 MAPK are activated by environmental stress and inflammatory cytokines 4. The involvement of p38 MAPK in the production of inflammatory mediators by fibroblasts has been reported in recent years. The role of p38 MAPK in relation to interleukin-6 (IL-6) and interleukin-8 (IL-8) production has been established in RSF 5. Also involvement of p38 MAPK in MMP-production was demonstrated in dermal fibroblasts 6 and gingival fibroblasts 7. An other matrix-degrading enzyme, aggrecanase-1 or ADAMTS-4 (a disintegrin and metalloproteinase with thrombospondin-1 motif), induced by cytokines in RSF, is important in cartilage degradation in RA 8. However its signal transduction pathways are not known at the moment. Prostaglandins have also been described as being under the influence of p38 MAPK 9. This has been confirmed in a study in which it was reported that glucocorticoids destabilize cyclo-oxygenase-2 (COX-2) mRNA by inhibiting the p38 MAPK route10. Interest in protein kinases as drug targets has increased in the recent years; in particular, p38 MAPK inhibitors have been developed 11-13, because p38 plays an important role as a major signal transducer responding to cellular stress stimuli such as cytokines. Because the production of interleukin-1 (IL-1) and tumor necrosis factor-α (TNF-α) is influenced by p38 MAPK, p38 MAPK inhibitors are expected to inhibit not only the production of these principal pro-inflammatory cytokines, but also their subsequent actions, leading to interruption of the vicious cycle that often occurs in inflammatory diseases. The use of p38 MAPK inhibitors therefore could provide an important advantage in therapy. The p38 MAPK inhibitor RWJ 67657 (4-[4-(4-fluorophenyl)-1-(3-phenylpropyl)-5-(4-pyridinyl)-1H-imidazol-2-yl]-3-butyn-1-ol) has been shown to inhibit the release of TNF-α from lipopolysaccharide-treated human peripheral blood mononuclear cells with an IC50 of 3 nM, as well as inhibiting the release of TNF-α from peripheral blood mononuclear cells treated with the super antigen staphylococcal enterotoxin B14. The compound was approximately 10-fold more potent than the reference standard p38 MAPK inhibitor SB 203580 in all p38 dependent in vitro systems tested. RWJ 67657 specifically inhibited the enzymatic activity of recombinant p38 α and β, but not of γ and δ in vitro, and had no significant activity against a variety of other kinases 14. Furthermore it was reported that this compound suppressed clinical and cytokine

Chapter 3
32
responses to endotoxin in healthy human volunteers 15. Recently a study was published on pharmacokinetics and pharmacodynamics of RWJ 67657 in humans 16. This study demonstrated acceptable safety and pharmacokinetic characteristics, warranting further investigation in a repeat-dose setting. In the present study we investigated the effects of RWJ 67657 on the release of pro-inflammatory mediators produced by RSF, after stimulation with IL-1β or TNF-α or both. Collagenase-1 (MMP-1), stromelysin-1 (MMP-3) and the tissue inhibitor of matrix-metalloproteinases (TIMP-1) were studied at both on protein- and on mRNA expression level. The same was done for IL-6 and IL-8. In addition we looked at the effects on mRNA expression levels of aggrecanase-1 (ADAMTS-4) and COX-2. MATERIALS AND METHODS Reagents RWJ 67657 was provided by Johnson and Johnson (R.W. Johnson Pharmaceutical Research Institute, Raritan, New Jersey, USA). Recombinant human IL-1β and recombinant human TNF-α were purchased from R&D Systems (Minneapolis, Minnesota, USA). Foetal calf serum (FCS) and Dulbecco’s Modified Eagle Medium (DMEM) were obtained from Biowhittaker (Verviers, Belgium). Anti-CD14 antibodies were from IQP (Groningen, The Netherlands) and anti-fibroblast antibodies (clone 5B5) were from Dako (Glostrup, Denmark). All reagents for RNA isolation and reverse transcriptase reaction were obtained from Invitrogen, Life Technologies (Gaithersburg, Maryland, USA). Reagents for real-time RT-PCR were obtained from Applied Biosystems (Foster City, California, USA). Specific antibodies to p38 MAPK, phospho-p38 MAPK and phospho-MAPKAPK-2 were purchased from Cell Signalling Technologies (Beverly, Massachusetts, USA). Isolation and culture of rheumatoid synovial fibroblasts (RSF) Synovial fibroblasts were isolated from synovium of 8 RA patients, who underwent total joint replacement. Synovium was minced and digested with 1 mg/ml collagenase (type 1A, Sigma, Zwijndrecht, The Netherlands) in DMEM (with L-glutamin and gentamycin) for two hours at 37ºC. The cell suspension was filtered through a cell strainer (70 µm) (Beckton Dickinson, Franklin Lakes, New Jersey, USA) and washed with phosphate buffered saline. Cells were cultured in a 5% CO2/37ºC incubator in DMEM with 10% FCS, and non-adherent cells were discarded after overnight incubation. At passage 3 the cell population consisted of CD14 neg/5B5 positive cells (fibroblast-like synoviocytes) and these cells were used for experiments until passage 8. For all experiments the cells were plated in 6-well or 48-well plates and serum starved for 24 hours in DMEM +1% FCS to synchronise cells in a non-activating and non-proliferating phase. Next they were pretreated with increasing concentrations (0.001 µM - 10 µM) of RWJ 67657 (stock solution 10 mM in DMSO) for 1 hour before stimulation with 1 ng/ml IL-1β and/or TNF-α.

p38 MAPK inhibition in RSF
33
Determination of MMP-1, MMP-3, TIMP-1, IL-6 and IL-8 levels in cell culture supernatants Confluent synovial fibroblasts (n=5) were plated in 48-well plates (10000 cells /ml per well) and treated as above. After 48 hours stimulation, supernatants were harvested and concentrations of MMP-1, MMP-3, TIMP-1, IL-6 17, and IL-8 18 were determined using enzyme linked immunosorbent assays (ELISAs) developed in our laboratory. The MMP-3 ELISA has been described previously 19. Briefly, 96-well plates (Greiner M129A) were precoated with F(ab)2 fragments of goat-anti-mouse IgG-Fc (Jackson, West Grove, Pennsylvania, USA) in 0.1 M carbonate buffer (pH=9.6) for at least 48 hours. Plates were subsequently coated with monoclonal antibody anti-MMP-3 (clone 10D6, R&D Systems) for 1 hour at 37°C. After sample incubation, bound MMP-3 was detected with rabbit-anti-human MMP-3 (Ab 810, Chemicon, Temecula, California, USA), and F(ab)2-goat-anti-rabbit IgG labelled with peroxidase (Zymed, San Francisco, California, USA). The colour-reaction was achieved with tetramethylbenzidin (TMB) (Roth, Karlsruhe, Germany). For the MMP-1 ELISA we used monoclonal anti-MMP-1 (clone 36665.111) and biotinylated goat-anti-human MMP-1 (both from R&D Systems). The TIMP-1 antibodies (R&D Systems) in the ELISA were monoclonal anti-TIMP-1 (clone 63515.111) and biotinylated goat-anti-human TIMP-1. The detection of the biotinylated antibodies was performed with streptavidin-poly-HRP (CLB, Amsterdam, The Netherlands) and TMB colour-reaction. RNA isolation Synovial fibroblasts (n=6) were plated in 6-well plates (0.5 x 106 cells/ well / 4 ml) and treated as mentioned before. After six or 24 hours of stimulation total RNA was isolated from the cells with TRIzol reagent according to the manufacturers instructions (Life Technologies). After DNase treatment (DNA-free, Ambion, Austin, Texas, USA) cDNA was synthesized from 2.0 µg of total RNA using M-MLV Reverse Transcriptase and oligo (dT)24. Real-time RT- PCR For quantitative detection of mRNA expression a fluorescence based real-time RT-PCR was performed, which allows relative quantification of steady-state mRNA. The amount of emitted fluorescence is proportional to the amount of PCR product and enables the monitoring of the PCR reaction 20. For the measurement of MMP-1, MMP-3, TIMP-1, ADAMTS-4, IL-6, IL-8, COX-2 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) 1µl of cDNA in duplicate was used for amplification by the real-time quantitative PCR system (ABI Prism 7900HT Sequence Detection System, Applied Biosystems) with specific Taqman primers/probes. The amount of target, normalized to an endogenous reference and relative to a calibrator, is given by: 2-∆∆CT in which CT is the threshold cycle. The results are expressed as fold induction relative to untreated samples. Western blotting to detect phosphorylation of p38 MAPK and MAPKAPK-2 Phosphorylation of p38 MAPK was analysed by western blotting. Synovial fibroblasts were plated in 6-well plates (0.5 x 106 cells/ well/4 ml) and treated as mentioned

Chapter 3
34
above. After stimulation with TNF-α and/or IL-1β up till 60 minutes, cell extracts were prepared by lysing the cells with 1x SDS sample buffer (containing 2% SDS, 10% glycerol, 50 mM dithiothreitol, 62.5 mM Tris-HCl (pH=6.8) and 0.01% brome-phenol blue). Cells were scraped off the wells and the lysates were subsequently sonicated for 5-10 seconds and boiled for five minutes. After centrifugation the samples were loaded onto a 10% SDS-PAGE gel and resolved by running at 200 V and 15 Watt constant. Semidry-blotting onto nitrocellulose membrane was followed by immunoblotting with specific antibodies to p38 MAPK, phospho-p38 MAPK and phospho-MAPKAPK-2. Enhanced chemi-luminescence (ECL) detection was done according to the manufacturers guidelines (Lumi-Lightplus, Roche Diagnostics, Mannheim, Germany). Effects of RWJ 67657 on phosphorylation of p38 MAPK and its downstream substrate MAPKAPK-2 were measured after incubation of the cells with the p38 MAPK inhibitor and stimulation with TNF-α and/or IL-1β for 30 minutes. STATISTICS One-way ANOVA with Dunnett’s post test or Bonferroni’s Multiple Comparison Test was employed using GraphPad Prism version 3.00 for Windows, GraphPad Software (San Diego, CA, USA). RESULTS Effect of RWJ-67657 on protein production by rheumatoid synovial fibroblasts Figure 1 shows the results of the production of MMP-1, MMP-3, TIMP-1, IL-6 and IL-8 by RSF (n=5) after stimulation with TNF-α and/or IL-1β, and also after pre-treatment with RWJ 67657 is depicted. Production of MMP-3 after stimulation with IL-1β without RWJ 67657 led to higher production compared to stimulation with TNF-α (15.0 fold), and the same was true to a lesser extent for MMP-1 (1.2 fold). TIMP-1 production was not induced when stimulated with either cytokine. Stimulation with both cytokines had a synergistic effect on MMP-3 production: 22.7 fold compared to TNF-α alone and 1.6 fold compared to IL-1β alone. Pre-incubation with RWJ 67657 resulted in a significant dose-dependent decrease in MMP-3 production when cells were stimulated with IL-1β alone or together with TNF-α. Only a high concentration (10 µM) of the p38 MAPK inhibitor had an effect on MMP-1 production, while for TIMP-1 there was no effect of p38 MAPK treatment. Stimulation with IL-1β led to higher productions of IL-6 and IL-8 than stimulation with TNF-α: 24.0 fold for IL-6 and 14.3 fold for IL-8. A dose-dependent decrease in IL-6 and IL-8 production was seen after pre-treatment with RWJ 67657. We calculated the average percentage inhibition caused by treatment with RWJ 67657 compared to stimulated cells is shown. More than 50% inhibition of MMP-3 and IL-8 production could be achieved at 1 µM, more than 50% inhibition of MMP-1 at 10 µM and more than 50% inhibition of IL-6 production at 0.1 µM. Control experiments were done by adding 0.1% DMSO (at a concentration of 10 µM RWJ 67657) to stimulated RSF (n=3). No significant inhibition of protein production could be detected as a result of the DMSO (data not shown).

p38 MAPK inhibition in RSF
35
Figure 1. Protein production of MMP-1, MMP-3, TIMP-1, IL-6 and IL-8 by rheumatoid synovial fibroblasts (n=5). Cells were stimulated with TNF-α and/or IL-1β for 48 hours and pre-treated with a concentration range of RWJ 67657 (t=-1h). Protein production was measured in supernatants by ELISA and expressed in ng/ml. Legends: unst = unstimulated, 0-10 = concentration RWJ 67657 added. Bars show mean and SEM. (* p< 0.05, ** p<0.001, Dunnett’s post test, tested against the stimulated control). IL, interleukin; MMP, matrix-metalloproteinase; TIMP-1, tissue inhibitor of matrix-metalloproteinases; TNF, tumor necrosis factor Effect of RWJ 67657 on mRNA expression A time-course study after stimulation with TNF-α and/or IL-1β was carried out to determine the time required for optimal mRNA expression. MMP-1 and MMP-3 mRNA expression was maximal after 24 hours, while IL-6, IL-8 and COX-2 mRNA had already reached maximum expression after six hours (data not shown).

Chapter 3
36
Figure 2. mRNA expression of MMP-1, MMP-3, TIMP-1 and ADAMTS-4 of rheumatoid synovial fibroblasts (n=3). Cells were stimulated with TNF-α and/or IL-1β for 6 and 24 hours and pre-treated with a concentration range of RWJ 67657. mRNA expression was determined with real-time RT-PCR (reverse transcriptase polymerase chain reaction) and results were expressed as -fold induction compared to unstimulated cells (fold induction=1). White bars represent values after TNF-α stimulation (scale on left vertical axis); grey bars represent values after IL-1β stimulation and black bars after IL-1β+TNF-α stimulation (scale on right vertical axis). Bars show means and SEM (* p<0.05, ** p<0.001, Bonferroni Multiple Comparison Test, tested against the stimulated control). ADAMTS, a disintegrin and metalloproteinase and metalloproteinase with thrombospondin-1 motif; MMP, matrix-metalloproteinase; TIMP-1, tissue inhibitor of matrix-metalloproteinases.

p38 MAPK inhibition in RSF
37
Figure 2 shows the mean levels (expressed as -fold induction compared with untreated cells =1) for MMP-1, MMP-3, TIMP-1 and ADAMTS-4 mRNA expression of three different RSF after six and 24-hour stimulation and after pre-treatment with RWJ 67657. As with protein production, mRNA expression of MMP-3 was much greater after stimulation with IL-1β than after stimulation with TNF-α: 40.1 fold after six hours of stimulation, and up to 149.4 fold after 24 hours. The equivalent values for MMP-1 mRNA expression were 3.4 fold after six hours, and 2.1 after 24 hours, although the absolute expression increased with time. TIMP-1 mRNA expression hardly increased after stimulation (maximum two- to threefold compared with unstimulated cells). ADAMTS-4 mRNA expression could be measured in synovial fibroblasts and increased with time. Again the expression after IL-1β stimulation was greater than after TNF-α stimulation (4.7-fold after six hours, 2.3-fold after 24 hours). Inhibition by RWJ 67657 could be detected for MMP-1 mRNA, MMP-3 mRNA and ADAMTS-4 mRNA at both time points and after treatment with all stimuli. However, significant inhibition was seen for MMP-1 mRNA expression after six hours of IL-1β stimulation at 1µM RWJ 67657 and more. Significant inhibition of MMP-3 mRNA expression also occurred after six hours of IL-1β stimulation at 0.1µM and more, and with both stimuli at 1 µM and more. TIMP-1 mRNA expression increased after 24 hours of stimulation with increasing concentrations of RWJ 67657. This effect was significant at 10 µM. ADAMTS-4 mRNA expression was significantly inhibited after six hours stimulation with both cytokines at 1 µM RWJ 67657 and more. Figure 3 shows mRNA levels, expressed as -fold induction compared with unstimulated cells (-fold induction =1) for IL-6, IL-8 and COX-2 after stimulation for six hours and after treatment of three different RSF with different concentrations of RWJ 67657. Again there was a difference in expression after stimulation with TNF-α or IL-1β. For IL-6, IL-8 and COX-2 respectively, stimulation with IL-1β gave 38.0-fold, 21.1-fold, and 18.3-fold higher expression than after stimulation with TNF-α. A significant inhibition of IL-6 mRNA expression was already seen at 0.01 µM RWJ 67657 when RSF were stimulated with IL-1β alone or together with TNFα. Inhibition of IL-8 mRNA expression was not significant, possibly because of a large interindividual response in IL-8 expression. COX-2 mRNA expression was significantly inhibited at 0.01µM RWJ 67657, when the cells were stimulated with IL-1β or IL-1β+TNF-α combination. To determine whether RWJ 67657 also affected cells that were already stimulated, 0.01 µM and 1 µM of p38 MAPK inhibitor was added before and one hour after TNF-α + IL-1β stimulation of two RSF cultures, and IL-6 and COX-2 mRNA expression was analysed. One hour after stimulation, phosphorylation of p38 MAPK had already reached maximum. When RWJ 67657 was added one hour before stimulation, the decrease in mRNA expression with RWJ 67657 concentrations of 0.01 µM and 1 µM was 59.8% and 97.9% respectively, for COX-2, and 38.4% and 71.5% for IL-6. When RWJ 67657 was added one hour after stimulation these values were 57.1% and 95.4% for COX-2 and 45.2 % and 81.0% for IL-6. This shows that the p38 MAPK inhibitor also inhibits inflammatory mediator production in previously activated rheumatoid synovial cells. Control experiments were carried out by adding 0.1% DMSO to stimulated RSF (RWJ 67657concentration 10 µM). Significant reduction by 0.1%

Chapter 3
38
Figure 3. mRNA expression of IL-6, IL-8 and COX-2 of rheumatoid synovial fibroblasts (n=3). Cells were stimulated with TNF-α and/or IL-1β for six hours and pretreated with a concentration range of RWJ 67657. mRNA expression was determined with real-time RT-PCR (reverse transcriptase polymerase chain reaction) and results were expressed as fold induction compared to unstimulated cells (-fold induction=1). White bars represent values after TNF-α stimulation (scale on left vertical axis); grey bars represent values after IL-1β stimulation and black bars after IL-1β+TNF-α stimulation (scale on right vertical axis). Bars show means and SEM (* p<0.05, ** p<0.001, Bonferroni Multiple Comparison Test, tested against the stimulated control). COX, cyclo-oxygenase; IL, interleukin; TNF, tumor necrosis factor. DMSO was seen only after IL-1β-induced IL-6 mRNA expression and TNF-α + IL-1β induced COX-2 mRNA expression. However, in both cases a significant reduction in mRNA expression was already found with 0.01 µM RWJ 67657 at non-inhibiting DMSO concentrations. Effect of RWJ 67657 on phosphorylation First we investigated the phosphorylation rate of p38 MAPK after stimulation with TNF-α and/or IL-1β in rheumatoid synovial fibroblasts. As shown in figure 4A, phosphorylation occurred rapidly and started after five minutes, reaching its maximum at 15 to 30 minutes for both stimuli. As expected, no inhibition of phosphorylation of p38 MAPK by RWJ 67657 was found (figure 4B). For MAPKAPK-2, which is a

p38 MAPK inhibition in RSF
39
Figure 4. (A) Representative presentation of phosphorylation of p38 MAPK in rheumatoid synovial fibroblasts after stimulation with TNF-α and/or IL-1β at different time points. Phosphorylation was measured by Western blot using specific antibodies. (B) Effect of RWJ 67657 on phosphorylation of the direct substrate of p38 MAPK, MAPKAPK-2, measured after 30 minutes of stimulation. IL, interleukin; MAPK, mitogen activated protein kinase; TNF, tumor necrosis factor.
direct downstream substrate of p38 MAPK, a strong inhibition of phosphorylation was demonstrated at concentrations down to 0.1 µM RWJ 67657. DISCUSSION In this study we showed significant inhibition by the p38 MAPK inhibitor RWJ 67657 of proinflammatory mediator and protease production in rheumatoid synovial fibroblasts. When inhibition was seen at the protein level, there was also inhibition at the level of mRNA expression, which means that this inhibition is at least at the level of RNA transcription. TNF-α and IL-1 are considered the most important cytokines in the process of inflammation in rheumatoid arthritis. Studies in experimental models have shown that TNF-α is indeed a pivotal cytokine in acute joint swelling, whereas IL-1β is the dominant cartilage destroying cytokine 21. Therefore we used both cytokines for activation of synovial fibroblasts to investigate the effects of a p38 MAPK inhibitor. Stimulation of RSF with IL-1β or TNF-α had different effects. Production of MMP-3 was greater after stimulation with IL-1β than with TNF-α, although there was a synergistic effect. Significant inhibition of induced production was seen when the cells were pretreated with 1 µM of the p38 MAPK inhibitor. MMP-1 protein production could be induced after stimulation (five- to sevenfold), but relevant inhibition was seen

Chapter 3
40
only at a concentration of 10 µM of RWJ 67657, which is too high for use in humans. The study by Parasrampuria et al 16 showed that a single oral dosage of 0.25 to 30 mg/kg resulted in plasma concentrations of RWJ 67657 between 0.01 µM and 6 µM. No up- or down regulation was seen for TIMP-1 production after stimulation and treatment with RWJ. TIMP-1 is constitutively expressed, and the expression is not influenced by TNF-α or IL-1β, and consequently not by p38 MAPK inhibition either. With quantitative real-time RT-PCR we measured mRNA levels of MMP-1, MMP-3, TIMP-1 and ADAMTS-4. IL-1β induced higher levels of mRNA expression for MMP-1, MMP-3 and ADAMTS-4 than TNF-α. Moreover, we showed inhibition of mRNA expression for these genes by the p38 MAPK inhibitor. Others have found that activation of p38 MAPK in human skin fibroblasts enhances MMP-1 and MMP-3 expression by mRNA stabilization 22. This is in agreement with our findings, indicating that inhibiting the p38 MAPK signal transduction route in RSF decreased expression of MMP-3 mRNA and to a lesser extent of MMP-1 mRNA. Work from our group has established the importance of MMP-3 as indicator for radiological progression in early RA, especially of joint space narrowing, which represents cartilage degradation 23. As others have shown that both aggrecanases and matrix-metalloproteinases degrade cartilage in human joints 24;25, the inhibition by RWJ 67657 could be of importance in the treatment of RA. The expression of TIMP-1 mRNA was only affected after 24 hours stimulation with increasing concentrations of RWJ 67657, which could have a protective effect by neutralising MMPs. As GAPDH levels were constant, possible adverse effects of RWJ 67657 did not induce this phenomenon. IL-6 and IL-8 are important cytokines in inflammation and both are present at high concentrations in synovial fluids of RA patients17. Strong induction of both cytokines in RSF could be demonstrated after IL-1β stimulation particularly. RWJ 67657 significantly inhibited this induced IL-6 and IL-8 production at 0.01 µM and 0.1 µM. Suzuki et al. 5 reported the decrease of IL-6 and IL-8 protein production after treatment with SB 203580, but no effect on mRNA expression, measured by traditional RT-PCR at a concentration of 30 µM. With quantitative real-time RT-PCR we detected inhibition of both IL-6 and IL-8 mRNA expression. Therefore this study showed that IL-6 and IL-8 are inhibited by RWJ 67657 both at the protein- and mRNA level. In 1998 Guan et al reported that the induction of COX-2 and production of PGE2 were directly linked to activation of MEKK1 and consequently to activation of p38 MAPK 26. Recently it was reported that COX-2 mRNA stability is under regulation of p38 MAPK10;27. In our study we demonstrated upregulation of COX-2 mRNA after stimulation with IL-1β or TNFα, and very strong inhibition by RWJ 675657 especially after IL-1β-induced expression. Effects of p38 MAPK inhibition are partly mediated through its downstream kinase MAPKAPK-2 and may involve phosphorylation of hsp2728. Our results showed that after 30 minutes p38 MAPK was already maximally phosphorylated and that MAPKAPK-2 phosphorylation was blocked at a concentration of 0.1 µM. Addition of RWJ 67657 to stimulated cells did not affect the inhibitory capacities of the compound, so inhibition of the p38 MAPK signal transduction route in activated cells is possible. p38 MAPK inhibitors have very potent effects on TNF-α production by LPS stimulated monocytes at low concentration. For

p38 MAPK inhibition in RSF
41
RWJ 67657 this was established at 3 nM 14. For monocyte-derived-macrophages 50% inhibition was reached at a concentration of 30 nM 29. Our study here clearly showed that IL-1β is a stronger inducer of expression of inflammatory mediators by synovial fibroblasts than TNF-α and may as such be the major cartilage destructive cytokine. p38 MAPK inhibitors such as RWJ 67657 inhibit both IL-1β and TNF-α, as well as their responses induced by these cytokines. This dual activity of p38 MAPK inhibitors may be of major importance in the treatment of RA and other inflammatory conditions. p38 MAPK inhibitor have effects on different cell types, which could enhance the therapeutic effects, but also increase the risk of side effects. In the past, clinical trials with other p38 MAPK inhibitors have been stopped due to safety issues. One of the reasons for undesirable effects might be the cross-reactivity against other kinases, which was not the case for RWJ 67657. Furthermore we excluded induction of apoptosis in RSF following incubation with RWJ 67657 by staining cells with Annexin V and propidium iodide as described previously 30 (data not shown). However, RWJ 67657 has been shown to have acceptable safety and acceptable pharmacokinetics characteristics, warranting further investigation 16. There were no adverse effects associated with single doses of this drug. While the preliminary pharmacokinetic data suggest a twice-daily dosing regimen, our data show significant effects at low concentrations. More research upon the effects of p38 MAPK inhibition on other cell types involved in inflammation will establish its applicability as drug in the near future. The results presented in this study are very promising. ACKNOWLEDGMENTS We gratefully acknowledge dr. Fred Breukelman and dr. Lex Boerboom for delivery of the synovial tissues. We thank dr. Marco Harmsen and dr. Sigga Asgeirsdottir for assistance with the PCR experiments and dr. Miek van Leeuwen for critically reading of the manuscript. We are grateful to mrs. Berber Doornbos-van der Meer for her excellent technical assistance. This work was supported by the Dutch Rheumatology Foundation and Johnson and Johnson Pharmaceutical Research and Development, Raritan, New Jersey, USA. REFERENCES 1 Pap T, Muller-Ladner U, Gay RE, Gay S. Fibroblast biology. Role of synovial fibroblasts in the
pathogenesis of rheumatoid arthritis. Arthritis Res. 2000; 2: 361-7. 2 Firestein GS, Manning AM. Signal transduction and transcription factors in rheumatic disease.
Arthritis Rheum. 1999; 42: 609-21. 3 Cobb MH, Goldsmith EJ. How MAP kinases are regulated. J.Biol.Chem. 1995; 270: 14843-6. 4 Dong C, Davis RJ, Flavell RA. MAP kinases in the immune response. Annu.Rev.Immunol.
2002; 20: 55-72. 5 Suzuki M, Tetsuka T, Yoshida S et al. The role of p38 mitogen-activated protein kinase in IL-6
and IL-8 production from the TNF-alpha- or IL-1beta-stimulated rheumatoid synovial fibroblasts. FEBS Lett. 2000; 465: 23-7.
6 Ridley SH, Sarsfield SJ, Lee JC et al. Actions of IL-1 are selectively controlled by p38 mitogen-activated protein kinase: regulation of prostaglandin H synthase-2, metalloproteinases, and IL-6 at different levels. J.Immunol. 1997; 158: 3165-73.
7 Ravanti L, Hakkinen L, Larjava H et al. Transforming growth factor-beta induces collagenase-3 expression by human gingival fibroblasts via p38 mitogen-activated protein kinase. J.Biol.Chem. 1999; 274: 37292-300.

Chapter 3
42
8 Yamanishi Y, Boyle DL, Clark M et al. Expression and regulation of aggrecanase in arthritis: the role of TGF- beta. J.Immunol. 2002; 168: 1405-12.
9 Guan Z, Baier LD, Morrison AR. p38 mitogen-activated protein kinase down-regulates nitric oxide and up- regulates prostaglandin E2 biosynthesis stimulated by interleukin-1beta. J.Biol.Chem. 1997; 272: 8083-9.
10 Lasa M, Brook M, Saklatvala J, Clark AR. Dexamethasone destabilizes cyclooxygenase 2 mRNA by inhibiting mitogen-activated protein kinase p38. Mol.Cell Biol. 2001; 21: 771-80.
11 Badger AM, Cook MN, Lark MW et al. SB 203580 inhibits p38 mitogen-activated protein kinase, nitric oxide production, and inducible nitric oxide synthase in bovine cartilage- derived chondrocytes. J.Immunol. 1998; 161: 467-73.
12 Branger J, van den BB, Weijer S et al. Anti-inflammatory effects of a p38 mitogen-activated protein kinase inhibitor during human endotoxemia. J.Immunol. 2002; 168: 4070-7.
13 Haddad JJ. VX-745. Vertex Pharmaceuticals. Curr.Opin.Investig.Drugs 2001; 2: 1070-6. 14 Wadsworth SA, Cavender DE, Beers SA et al. RWJ 67657, a potent, orally active inhibitor of
p38 mitogen-activated protein kinase. J.Pharmacol.Exp.Ther. 1999; 291: 680-7. 15 Fijen JW, Zijlstra JG, de Boer P et al. Suppression of the clinical and cytokine response to
endotoxin by RWJ-67657, a p38 mitogen-activated protein-kinase inhibitor, in healthy human volunteers. Clin.Exp.Immunol. 2001; 124: 16-20.
16 Parasrampuria DA, de Boer P, Desai-Krieger D, Chow AT, Jones CR. Single-dose pharmacokinetics and pharmacodynamics of RWJ 67657, a specific p38 mitogen-activated protein kinase inhibitor: a first-in-human study. J.Clin.Pharmacol. 2003; 43: 406-13.
17 van Leeuwen MA, Westra J, Limburg PC, van Riel PL, van Rijswijk MH. Interleukin-6 in relation to other proinflammatory cytokines, chemotactic activity and neutrophil activation in rheumatoid synovial fluid. Ann.Rheum.Dis. 1995; 54: 33-8.
18 de Bont ES, Vellenga E, Swaanenburg JC, Fidler V, Visser-van Brummen PJ, Kamps WA. Plasma IL-8 and IL-6 levels can be used to define a group with low risk of septicaemia among cancer patients with fever and neutropenia. Br.J.Haematol. 1999; 107: 375-80.
19 Posthumus MD, Limburg PC, Westra J, van Leeuwen MA, van Rijswijk MH. Serum matrix metalloproteinase 3 levels during treatment with sulfasalazine or combination of methotrexate and sulfasalazine in patients with early rheumatoid arthritis. J.Rheumatol. 2002; 29: 883-9.
20 Klein D. Quantification using real-time PCR technology: applications and limitations. Trends Mol.Med. 2002; 8: 257-60.
21 van den Berg WB, Bresnihan B. Pathogenesis of joint damage in rheumatoid arthritis: evidence of a dominant role for interleukin-I. Baillieres Best.Pract.Res.Clin.Rheumatol. 1999; 13: 577-97.
22 Reunanen N, Li SP, Ahonen M, Foschi M, Han J, Kahari VM. Activation of p38 alpha MAPK enhances collagenase-1 (matrix metalloproteinase (MMP)-1) and stromelysin-1 (MMP-3) expression by mRNA stabilization. J.Biol.Chem. 2002; 277: 32360-8.
23 Posthumus MD, Limburg PC, Westra J, van Leeuwen MA, van Rijswijk MH. Serum matrix metalloproteinase 3 in early rheumatoid arthritis is correlated with disease activity and radiological progression. J.Rheumatol. 2000; 27: 2761-8.
24 Lark MW, Bayne EK, Flanagan J et al. Aggrecan degradation in human cartilage. Evidence for both matrix metalloproteinase and aggrecanase activity in normal, osteoarthritic, and rheumatoid joints. J.Clin.Invest 1997; 100: 93-106.
25 Little CB, Flannery CR, Hughes CE et al. Aggrecanase versus matrix metalloproteinases in the catabolism of the interglobular domain of aggrecan in vitro. Biochem.J. 1999; 344 Pt 1: 61-8.
26 Guan Z, Buckman SY, Pentland AP, Templeton DJ, Morrison AR. Induction of cyclooxygenase-2 by the activated MEKK1 --> SEK1/MKK4 --> p38 mitogen-activated protein kinase pathway. J.Biol.Chem. 1998; 273: 12901-8.
27 Faour WH, He Y, He QW et al. Prostaglandin E(2) regulates the level and stability of cyclooxygenase-2 mRNA through activation of p38 mitogen-activated protein kinase in interleukin-1 beta-treated human synovial fibroblasts. J.Biol.Chem. 2001; 276: 31720-31.
28 Lasa M, Mahtani KR, Finch A, Brewer G, Saklatvala J, Clark AR. Regulation of cyclooxygenase 2 mRNA stability by the mitogen-activated protein kinase p38 signaling cascade. Mol.Cell Biol. 2000; 20: 4265-74.

p38 MAPK inhibition in RSF
43
29 Westra J, Doornbos-Van Der Meer B, de Boer P, van Leeuwen MA, van Rijswijk MH, Limburg PC. Strong inhibition of TNF-alpha production and inhibition of IL-8 and COX-2 mRNA expression in monocyte-derived macrophages by RWJ 67657, a p38 mitogen-activated protein kinase (MAPK) inhibitor. Arthritis Res.Ther. 2004; 6: R384-R392.
30 Bijl M, Horst G, Bijzet J, Bootsma H, Limburg PC, Kallenberg CG. Serum amyloid P component binds to late apoptotic cells and mediates their uptake by monocyte-derived macrophages. Arthritis Rheum. 2003; 48: 248-54.

4
Strong inhibition of TNF-α production and inhibition of
IL-8 and COX-2 mRNA expression in monocyte-derived
macrophages by RWJ 67657, a p38 mitogen activated
protein kinase (MAPK) inhibitor
Johanna Westra1
Berber Doornbos-van der Meer1
Peter de Boer3
Miek A van Leeuwen1
Martin H van Rijswijk1
Pieter C Limburg1,2
From the Departments of 1 Rheumatology, 2 Pathology and Laboratory Medicine,
University Medical Center Groningen, The Netherlands, and 3 Pharmaceutical Research and Development, Johnson and Johnson,
Saunderton, United Kingdom
Arthritis Research and Therapy 2004; 6 (4): R384 – R392

Chapter 4
46
ABSTRACT In inflammatory processes the p38 mitogen activated protein kinase (MAPK) signal transduction route regulates production and expression of cytokines and other inflammatory mediators. Tumor necrosis factor α (TNF-α) is a pivotal cytokine in rheumatoid arthritis (RA) and its production in macrophages is under control of the p38 MAPK route. Inhibition of the p38 MAPK route may inhibit production not only of TNF-α, but also of other inflammatory mediators produced by macrophages, and indirectly of inflammatory mediators by other cells induced by TNF-α stimulation. Here we investigate the effects of RWJ 67657, a p38 MAPK inhibitor, on mRNA expression and protein production of TNF-α and other inflammatory mediators, in monocyte-derived macrophages (MDM). A strong inhibition of TNF-α was seen at pharmacological relevant concentrations of RWJ 67657, but also inhibition of mRNA expression of IL-1β, IL-8 and COX-2 was shown. Furthermore, it was shown that monocyte-derived macrophages have a high constitutive production of MMP-9, which is not affected by p38 MAPK inhibition. The results presented here may have important implications for the treatment of rheumatoid arthritis. Key words. COX-2, matrix metalloproteinase, monocyte-derived macrophage, p38 MAPK inhibitor, TNF-α

p38 MAPK inhibition in MDM
47
INTRODUCTION Rheumatoid arthritis (RA) is characterized by chronic inflammation of synovial tissue and destruction of cartilage and bone in the joints 1. Macrophages play an important role in RA, as the rheumatoid synovium is intensively infiltrated by macrophages and their numbers correlate with clinical scores 2 and articular destruction in RA 3. RA patients with active disease display a faster generation of CD14+ myelomonocytic cells from the bone marrow and faster differentiation into HLA-DR+ cells than control individuals do 4. Activation of the monocytic lineage in inflammatory disease is not only restricted to synovial macrophages, but extends to circulating monocytes and other cells of the mononuclear phagocyte system 5. The activation state of monocytes /macrophages is characterized by increased expression and transcription of interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α), but also of other pro-inflammatory and regulatory cytokines and growth factors 6. Highly specific therapeutics have been developed to target these cytokines, like monoclonal antibodies, soluble receptors, binding proteins, and receptor antagonists. TNF-α blockade has been the major breakthrough in the therapy of RA during the past ten years. However more than half of the patients do not achieve adequate responses, remissions are rare and these drugs do have side effects 7;8. The importance of mitogen-activated protein kinases (MAPK) in cell biology has been reported in many studies concerning different inflammatory diseases. These MAPK belong to three families: the extracellular signal regulated kinases (ERK), the c-Jun N-terminal or stress activated protein kinases (JNK/SAPK) and the p38 MAPK. All three families have been shown to become activated in macrophages using a variety of stimuli both in primary cells and in cell lines 9. In the RA synovium p38 MAPK is predominantly activated in endothelial cells and in the lining layer 10. Inhibition of p38 MAPK therefore could provide an interesting target for intervention in inflammation as it occurs in the synovia in rheumatoid arthritis. In vitro stimulation of macrophages with LPS (lipopolysaccharide) leads to activation of MAPK cascades through the LPS receptor (CD14) or Toll-like receptors 9. The ability of bacterial toxins or super-antigens to induce pro-inflammatory responses leading to the production of TNF-α and IL-1 is relevant in view of the possible micro-organism aetiology in RA 11. Stimulation of monocytes with LPS induces a number of matrix metalloproteinases (MMPs), including two prominent monocytic MMPs: interstitial collagenase (MMP-1) and gelatinase-B (MMP-9). These enzymes are involved in the connective-tissue loss associated with chronic inflammatory diseases. In vivo a significant part of macrophage effector responses occur through cell-contact -dependent signalling with several inflammatory cells, mainly T-cells and fibroblasts. A few soluble stimuli are known to have a stimulatory effect on macrophages, like IL-15 and IL-17. It has also been reported that IL-17 induces the production of MMP-9 and cyclo-oxygenase-2 (COX-2, which is the rate-limiting enzyme in prostaglandin and leukotriene synthesis) in monocytes/macrophages 12. The p38 MAPK inhibitor RWJ 67657 (4-[4-(4-fluorophenyl)-1-(3-phenylpropyl)-5-(4-pyridinyl)-1H-imidazol-2-yl]-3-butyn-1-ol) has been shown to inhibit the release of TNFα from LPS-treated human peripheral blood mononuclear cells with an IC50 of 3 nM 13. Moreover this compound effectively inhibited endotoxin-induced clinical effects and cytokine release in normal healthy volunteers14. Furthermore a report was

Chapter 4
48
published in which pharmacokinetics and pharmacodynamics of RWJ 67657 were presented, showing that the compound has acceptable safety to warrant further investigation 15. Our group recently showed that RWJ 67657 significantly inhibited IL-6, IL-8, MMP-3 and COX-2 mRNA expressed by IL-1β and/or TNF-α stimulated rheumatoid synovial fibroblasts 16. It has been shown that differences occur in signalling pathways in myeloid cells in relation to the maturation stage of macrophages 17. Strong inhibition of TNF-α in monocytes has been reported due to p38 MAPK inhibition 13, but the effects in matured macrophages are not fully known. Therefore in this study we investigated the effects of the p38 MAPK inhibitor RWJ 67657 on the mRNA expression and production of inflammatory cytokines and MMPs by stimulated monocyte-derived macrophages (MDM). We investigated macrophages, differentiated in medium with pooled serum or by the method described by Plesner and co-workers 18 and compared MDMs from healthy controls and rheumatoid arthritis patients. Strong inhibition of mRNA expression and production of TNF-α by RWJ 67657 was found, as well as inhibition of IL-1β, IL-8 and COX-2 mRNA expression. MATERIALS AND METHODS Reagents RWJ 67657 was provided by Johnson and Johnson (R.W. Johnson Pharmaceutical Research Institute, Raritan, New Jersey, USA). Antibodies for flowcytometry were obtained from IQProducts, Groningen, The Netherlands. Lipopolysaccharide was purchased from Sigma-Aldrich (Zwijndrecht, The Netherlands). Recombinant human macrophage colony stimulating factor (M-CSF) and ELISA antibodies were from R&D Systems (Minneapolis, Minnesota, USA). Foetal calf serum (FCS) and RPMI 1640 culture medium were obtained from Biowhittaker (Verviers, Belgium). All reagents for RNA isolation and reverse transcriptase reaction were purchased from Invitrogen, Life Technologies (Gaithersburg, Maryland, USA). Reagents for real-time RT-PCR were obtained from Applied Biosystems (Foster City, California, USA). Specific antibodies to p38 MAPK, phospho-p38 MAPK and phospho-MAPKAPK-2 were purchased from Cell Signalling Technologies (Beverly, Massachusetts, USA) and detecting antibody peroxidase-swine-anti-rabbit was from DAKO (Glostrup, Denmark). Macrophage culture Blood was obtained from RA patients who had given informed consent, and from healthy laboratory workers. Peripheral mononuclear cells were isolated by Lymphoprep density gradient centrifugation from citrated blood. Cells were suspended in RPMI with gentamycin at 106 cells/ml and seeded in 6-well plates (Costar, Badhoevedorp, The Netherlands) in 3 ml or 0.5 ml in 24-well plates and cultured at 37°C in a 5% CO2 atmosphere. After 2 hours non-adherent cells were discarded and adherent monocytes were allowed to differentiate into macrophages in RPMI containing gentamycin, 50 ng/ml M-CSF + 1% FCS 18, while medium was refreshed at day 2. To compare methods monocytes were also differentiated in RPMI with gentamycin + 2% pooled human serum; on day 2 and 5 fresh medium was added to the wells. Cells were cultured for 5 or 7 days. We found no marked differences between

p38 MAPK inhibition in MDM
49
differentiation methods and decided to perform further experiments with macrophages differentiated with M-CSF and FCS. Flowcytometric analysis Expression of surface markers CD14 (LPS receptor), HLA-DR, CD18 (β2 integrin subunit), CD36 (GPIIIb, GPIV) and CD83 (dendritic cell marker) on monocytes in the PBMC-fraction and on differentiated macrophages was detected by flowcytometric analysis in an Epics-Elite Flowcytometer (Coulter Electronics, Mijdrecht, The Netherlands). Phosphorylation studies Phosphorylation of p38 MAPK in MDM was analyzed by western blotting. Cells were cultured in RPMI containing gentamycin, 50 ng/ml M-CSF + 1% FCS for 5 days and stimulated with 50 ng/ml LPS for various periods of time. Cell extracts were prepared by lysing the cells with 1x SDS sample buffer (containing 2% SDS, 10% glycerol, 50 mM dithiothreitol, 62.5 mM Tris-HCl (pH=6.8) and 0.01% brome-phenol blue). Cells were scraped off the wells and the lysates were subsequently sonicated for 5-10 seconds and boiled for 5 minutes. After centrifugation the samples were loaded onto a 10% SDS-PAGE gel and resolved by running at 200 V and 15 Watt constant. Semidry-blotting was performed onto nitrocellulose membrane and immunodetection was with anti-phospho-p38 MAPK and peroxidase-anti-rabbit-immnoglobulins. Enhanced chemiluminescence (ECL) detection was performed according to the manufacturers guidelines (Lumi-Lightplus, Roche Diagnostics, Mannheim, Germany). To determine the effect of RWJ 67657 on phosphorylation of p38 MAPK and its downstream substrate MAPK-activating protein kinase-2 (MAPKAPK-2) a concentration range of the p38 MAPK inhibitor (0, 0.01, 0.1, 1, 10 µM) was added 1 hour before stimulation with 50 ng/ml LPS for 30 minutes. Blotting experiments were performed with specific antibodies to p38 MAPK, phospho-p38 MAPK, and phospho-MAPKAPK-2. Determination of TNF-α, IL-1β, IL-6, IL-8, MMP-1, MMP-9 and TIMP-1 levels in cell culture supernatants MDM from 8 healthy controls and 9 RA patients, who were not treated with steroids, were cultured in 24 well-plates in RPMI containing gentamycin, 50 ng/ml M-CSF + 1% FCS for 5 days. The cells were subsequently pre-treated with increasing concentrations of RWJ 67657 (stocksolution 10 mM in dimethylsulfoxide [DMSO]), from 0. 01 µM -10 µM for 1 hour prior to stimulation with 50 ng/ml LPS for 24 hours. Cytokine (TNFα, IL-1β, IL-6 and IL-8), MMP-1, MMP-9 and tissue inhibitor of matrix metalloproteinases (TIMP)-1 levels were measured in cell supernatants by ELISA, using matched antibody pairs for ELISA and recombinant proteins as standards from R&D Systems. Detection limits for all cytokine ELISAs was 20 pg/ml. For optimal determination of MMP-1, MMP-9 and TIMP-1 96 well plates (Greiner M129A) were precoated with F(ab)2 fragments of goat-anti-mouse IgG-Fc (Jackson, West Grove, Pennsylvania, USA) in 0.1M carbonate buffer (pH=9.6) for at least 48 hours before coating of the capture antibody. After sample incubation and binding of the biotinylated detection antibodies, the color reaction was performed with

Chapter 4
50
streptavidin-poly-HRP (Sanquin, Amsterdam, The Netherlands) and tetramethyl-benzidin (TMB, Roth, Karlsruhe, Germany). The detection limit for MMP-1 and TIMP-1 was 1 ng/ml and for MMP-9, 0.1 ng/ml. RNA isolation and real-time RT-PCR Monocyte-derived macrophages from healthy controls were cultured in 6 well-plates in RPMI containing gentamycin, 50 ng/ml M-CSF + 1% FCS for 5 days. First macrophages were stimulated with 50 ng/ml LPS for different periods of time to determine optimal mRNA expression of TNF-α, IL-1β, IL-6, IL-8, MMP-9 and COX-2 genes. At the optimal time point the effect of p38 MAPK inhibition on LPS-stimulated MDM was determined by pre-treatment of the cells with increasing concentrations of RWJ 67657 for 1 hour. Total RNA was isolated from the cells with TRIzol reagent according to the manufacturers instructions (Life Technologies). After DNase treatment (Ambion DNA-free, Austin, Texas, USA) cDNA was synthesized from 1.0 µg of total RNA using M-MLV Reverse Transcriptase and oligo (dT)24 (Life Technologies). For detection of mRNA expression a fluorescence based real-time RT-PCR was performed, which allows relative quantification of steady-state mRNA. The amount of emitted fluorescence is proportional to the amount of PCR product and enables the monitoring of the PCR reaction. For the measurement of IL-1β, TNF-α, IL-6, IL-8, MMP-9, COX-2 and glyceraldehyde-3-phosphate dehydro-genase (GAPDH), 1µl of cDNA in triplicate was used for amplification by the real-time quantitative PCR system (ABI Prism 7900HT Sequence Detection System, Applied Biosystems) with specific Taqman primers/probes. The Assay-on-Demand numbers for the genes were: IL-1β: Hs00174097_m1, TNFα: Hs00174128_m1, IL-6: Hs00174131_m1, IL-8: Hs00174103_m1, MMP-9: Hs00234579_m1, COX-2: Hs00153133_m1, and GAPDH: Hs99999905_m1. The amount of target, normalized to an endogenous reference (GAPDH) and relative to a control sample, is given by: 2-∆∆CT in which CT is the threshold cycle. The results are expressed as fold induction relative to untreated samples. STATISTICS Paired T-tests were performed using GraphPad Prism version 3.00 for Windows, GraphPad Software (San Diego, CA, USA). RESULTS Macrophage differentiation Expression of surface markers on monocytes and macrophages was measured by flowcytometry. In figure 1 the mean fluorescence intensity (MFI) of CD14 and HLA-DR on monocytes (left panel) and macrophages differentiated in 50 ng/ml M-CSF and 1% FCS after 5 days (right panel) is shown, the number of cells measured in the flowcytometer. Macrophages differentiated in this way showed lower expression of CD14 compared to monocytes relative to the isotype control, whereas the HLA-DR expression did not differ between both cell types.

p38 MAPK inhibition in MDM
51
Figure 1. Expression of CD14 (bold grey line) and HLA-DR (bold black line) on monocytes (left graph) and macrophages, differentiated with 50 ng/ml macrophage-colony-stimulating factor and 1% FCS after 5 days (right panel), measured by flow cytometry and expressed in mean fluorescence intensity (MFI). The isotype control is an IgG2a antibody (dashed line), the blank is in solid fill. The mean fluorescence intensity (MFI) is shown on the x-axis, while on the y-axis the number of cells measured in the flowcytometer is expressed. PE = phycoerythrin.
Expression of CD18 increased with differentiation, while expression of CD36 decreased (data not shown). Expression of CD83 was not seen on matured macrophages, excluding differentiation into dendritic cells. For further experiments monocytes differentiated in medium with 50 ng/ml M-CSF and 1% FCS for 5 days were used. Effect of RWJ 67657 on phosphorylation of p38 MAPK and MAPKAPK-2 In figure 2A a representative example is shown of phosphorylation of p38 MAPK in MDMs after stimulation with 50 ng/ml LPS. Phosphorylation occurs already after 15 minutes, is maximal at 30-60 minutes and diminishes after 2 hours. p38 MAPK is constitutively expressed in the cells as is demonstrated in the control blot. The effect of RWJ 67657 on phosphorylation of p38 MAPK and its direct downstream substrate MAPKAPK-2 measured after 30 minutes of stimulation with 50 ng/ml LPS is shown in figure 2B. RWJ 67657 does not inhibit phosphorylation of p38 MAPK, but does inhibit its activity, as can be seen from the strong inhibition at 0.01 µM and the complete inhibition of MAPKAPK-2 phosphorylation at 0.1 µM RWJ 67657. The solvent, 0.1% DMSO, did not affect phosphorylation of either kinase. Effect of RWJ 67657 on cytokine and MMP production Stimulation of MDM with 50 ng/ml LPS for 24 hours resulted in increased production of TNF-α, IL-6, IL-8 and MMP-9 both in control macrophages and in rheumatoid arthritis macrophages (figure 3A). Production of IL-1β, MMP-1 and TIMP-1 was too low for detection. Before stimulation, TNF-α production was below detection limit, while after stimulation the mean production in control MDM was 2.204 (± 1.993) ng/ml and in RA-MDM 2.150 (± 1.816) ng/ml.

Chapter 4
52
Figure 2. Effect of RWJ 67657 on phosphorylation of p38 MAPK and MAPKAPK-2. (A) Representative presentation of phosphorylation of p38 mitogen-activated protein kinase (MAPK) in monocyte-derived macrophages after stimulation with lipopolysaccharide (LPS) at various time points. Phosphorylation was measured by western blotting using specific antibodies to p38 MAPK and phospho-p38 MAPK. (B) Effect of RWJ 67657 on phosphorylation of the direct substrate of p38 MAPK, MAPKAPK-2 (MAPK-activating protein kinase-2), measured after 30 minutes of stimulation. DMSO = dimethylsulfoxide. Pre-treatment of the macrophages with increasing concentrations of RWJ 67657 showed a dose-dependent decrease of protein production for TNF-α and IL-8. Inhibition of TNFα production was seen with an IC50 of 0.015 µM for control cells and 0.03 µM for RA cells (figure 3B). For IL-8 production the IC50 was 0.3 µM for control cells and 1.2 µM for RA cells. IL-6 and MMP-9 production was inhibited only at concentrations between 1 and 10 µM RWJ 67657. Pre-treatment of MDMs with 0.1% DMSO had no significant effect on protein production (data not shown). Effects of RWJ 67657 on mRNA expression To determine optimal stimulation time points for the genes involved in this study MDM of 2 healthy controls were stimulated during 0.5, 2, 4, 8, 12, and 24 hours with 50 ng/ml LPS. mRNA expression of TNF-α, IL-1β, IL-6, IL-8 and COX-2 was measured with real-time RT-PCR as depicted in figure 4. A significant increase of mRNA expression of all but MMP-9 was found after stimulation with LPS. After 4 and 8 hours of stimulation most genes were highly expressed, IL-8 mRNA expression was still increased after 12 and 24 hours. For measurement of the effects of the p38 MAPK inhibitor MDMs were stimulated for 4 hours.
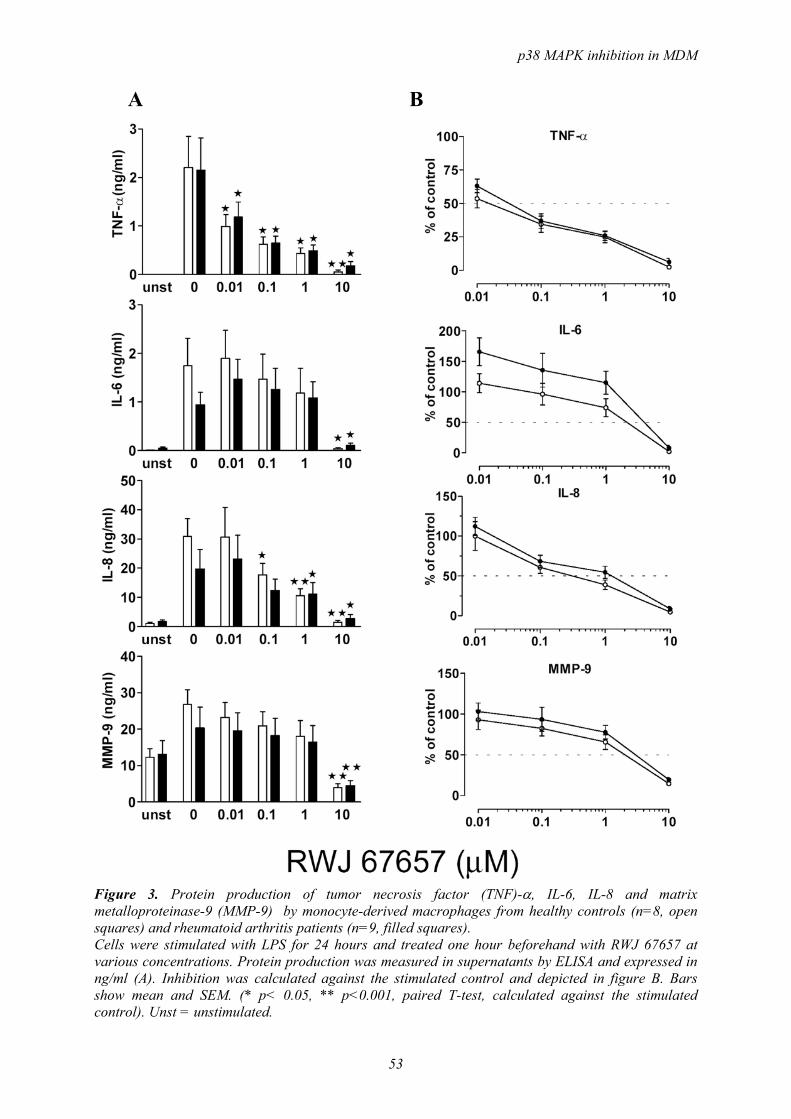
p38 MAPK inhibition in MDM
53
Figure 3. Protein production of tumor necrosis factor (TNF)-α, IL-6, IL-8 and matrix metalloproteinase-9 (MMP-9) by monocyte-derived macrophages from healthy controls (n=8, open squares) and rheumatoid arthritis patients (n=9, filled squares). Cells were stimulated with LPS for 24 hours and treated one hour beforehand with RWJ 67657 at various concentrations. Protein production was measured in supernatants by ELISA and expressed in ng/ml (A). Inhibition was calculated against the stimulated control and depicted in figure B. Bars show mean and SEM. (* p< 0.05, ** p<0.001, paired T-test, calculated against the stimulated control). Unst = unstimulated.
A B

Chapter 4
54
Figure 4. Time course of induction of mRNA expression of tumor necrosis factor (TNF)-α, IL-1β, IL-6, IL-8 and cyclo-oxygenase-2 (COX-2). Monocyte-derived macrophages from healthy controls (n=2) were stimulated for increasing periods of time with lipopolysaccharide (50 ng/ml). mRNA expression was determined with real-time RT-PCR and results were calculated as fold induction in comparison to unstimulated cells (fold induction=1).
As can been seen in figure 5 there is a dose-dependent decrease in mRNA expression with increasing RWJ 67657 concentration. TNF-α mRNA expression is inhibited by 48.4% at 0.01 µM and 65.8% at 0.1 µM p38 MAPK inhibitor. IL-1β, IL-8 and COX-2 mRNA expression was reduced by 40.2%, 56.6% and 65.0% respectively at 1 µM. MMP-9 mRNA expression is not induced and not inhibited and proved to be constitutively expressed at a high level. Control incubations with 0.1% DMSO had no significant effect on mRNA expression, as can been seen in figure 5. DISCUSSION In this study we have shown the significant inhibition of TNF-α production in MDMs by the p38 MAPK inhibitor RWJ 67657. The strong inhibition was seen both at the level of mRNA expression and protein production, so inhibition is already apparent at the level of transcription. p38 MAPK activity in RA is found predominantly in the synovial lining layer and in endothelial cells in the synovium 10. The lining layer consists mainly of fibroblasts and macrophages, both important players in the process of inflammation by the production of cytokines and degrading enzymes. Recently our group demonstrated strong inhibition of IL-6, IL-8, COX-2 and MMP-3 expression in rheumatoid synovial fibroblasts by the p38 MAPK inhibitor RWJ 67657 19. Studying macrophages in vitro raises some difficulties, because isolation and culture of macrophages from synovial tissue is disturbed by the overgrowth of fibroblasts. Therefore, macrophages differentiated from peripheral blood monocytes are widely used for in vitro studies. By
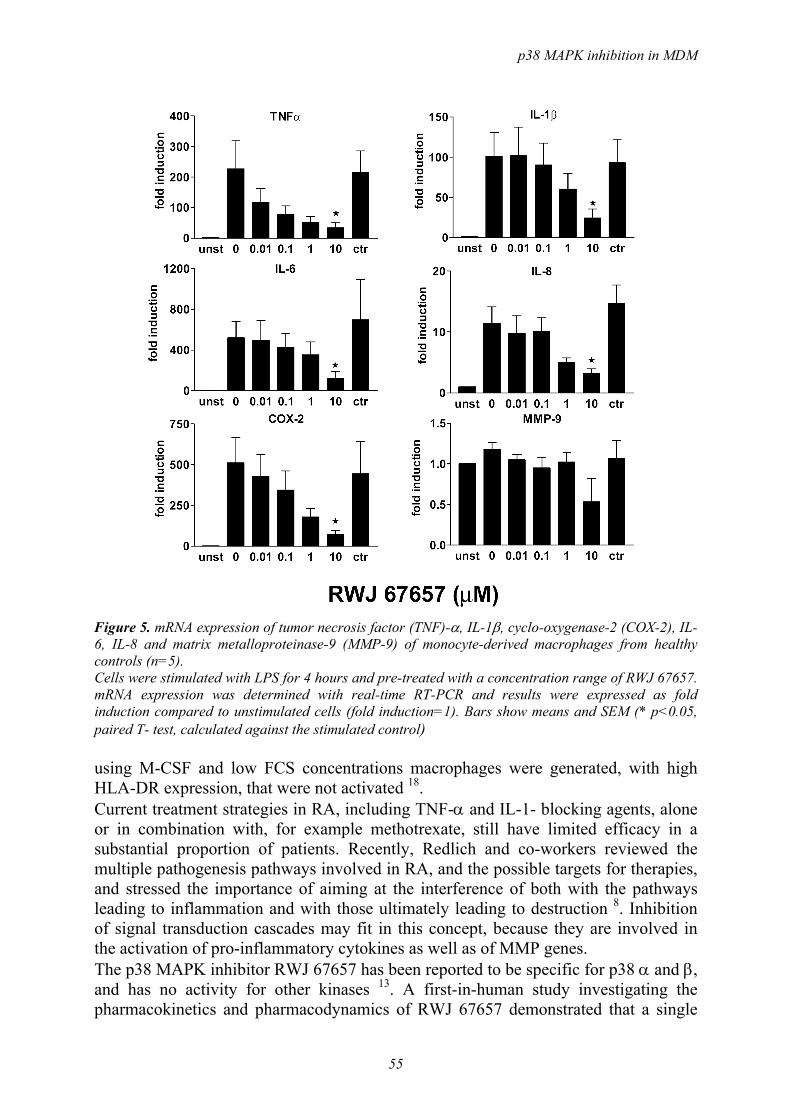
p38 MAPK inhibition in MDM
55
Figure 5. mRNA expression of tumor necrosis factor (TNF)-α, IL-1β, cyclo-oxygenase-2 (COX-2), IL-6, IL-8 and matrix metalloproteinase-9 (MMP-9) of monocyte-derived macrophages from healthy controls (n=5). Cells were stimulated with LPS for 4 hours and pre-treated with a concentration range of RWJ 67657. mRNA expression was determined with real-time RT-PCR and results were expressed as fold induction compared to unstimulated cells (fold induction=1). Bars show means and SEM (* p<0.05, paired T- test, calculated against the stimulated control) using M-CSF and low FCS concentrations macrophages were generated, with high HLA-DR expression, that were not activated 18. Current treatment strategies in RA, including TNF-α and IL-1- blocking agents, alone or in combination with, for example methotrexate, still have limited efficacy in a substantial proportion of patients. Recently, Redlich and co-workers reviewed the multiple pathogenesis pathways involved in RA, and the possible targets for therapies, and stressed the importance of aiming at the interference of both with the pathways leading to inflammation and with those ultimately leading to destruction 8. Inhibition of signal transduction cascades may fit in this concept, because they are involved in the activation of pro-inflammatory cytokines as well as of MMP genes. The p38 MAPK inhibitor RWJ 67657 has been reported to be specific for p38 α and β, and has no activity for other kinases 13. A first-in-human study investigating the pharmacokinetics and pharmacodynamics of RWJ 67657 demonstrated that a single

Chapter 4
56
oral intake of 0.25 ranging up to 30 mg/kg resulted in plasma levels of 0.01 µM to 6 µM 15. Furthermore the study showed that at the doses tested there were no significant adverse effects. p38 MAPK acts mainly through phosphorylation of its down-stream substrate MAPKAPK-2 and a variety of transcription factors 20. Mice that lack MAPKAPK-2 show increased stress resistance and survive LPS-induced endotoxic shock, due to a reduction of 90% in the production of TNF-α 21. The level and stability of TNF-α mRNA was not reduced in these mice, so the inhibition was at the post-transcriptional level. With western blotting we demonstrated complete inhibition of phosphorylation of MAPKAPK-2 at 0.1 µM RWJ 67657, and strong inhibition at 0.01 µM. In our study TNFα production was significantly inhibited at nanomolar concentrations RWJ 67657, and mRNA expression was also decreased, by nearly 50% at 10 nM. Inhibition of p38 MAPK activity leads to reduced TNF-α mRNA expression and therefore reduced TNFα production, while inhibition of MAPKAPK-2, as in MAPKAPK-2 knock-out mice, leads to reduced TNF-α production without affecting the mRNA levels. IL-6 production and mRNA expression were not inhibited by p38 MAPK inhibition, but IL-8 production and mRNA expression were inhibited for more than 50% at 1 µM RWJ 67657. Bhattacharyya and co-workers showed that LPS from Helicobacter pylori stimulates IL-8 release from cells of the monocytic lineage through activation of NF-κB and MAPK cascades 22, and we demonstrated that in MDM the p38 MAPK route plays an important role in this process. Already in 1992 Herzyk and co-workers published findings showing that macrophage and monocyte IL-1β regulation differs at multiple sites23. Macrophages did not differ from monocytes in LPS sensitivity but had limitations in IL-1β release. Our study shows mRNA expression of IL-1β in MDM, as well as a reduction of mRNA expression at 1 µM RWJ 67657, but protein production in MDMs was below the detection limit of the measurement. We found a reduction of 65% in COX-2 mRNA expression after treatment with 1 µM RWJ 67657. It was previously demonstrated that p38 MAPK plays a role in transcription and stabilisation of COX-2 mRNA 24. However, Caivano and Cohen showed that both p38 MAPK and ERK influence COX-2 mRNA expression through activation of mitogen- and stress-activated protein kinase-1 (MSK-1) 25, an activator of important transcription factors like ATF-2 and CREB. Our results indicate an important role for p38 MAPK in COX-2 mRNA expression in MDM. In synovial tissue, the presence of macrophages is often seen together with the expression of MMPs and TIMP-1 26. In our study MMP-1 and TIMP-1 could not be detected in the cell supernatants of MDMs after LPS stimulation, in contrast to high production of MMP-9. MMP-9 is associated with macrophages and peripheral blood mononuclear cells and has a broad substrate specificity and may contribute together with collagenases to the degradation of fibrillar collagens, basement membrane-components and stromal ECM molecules 27. Stimulation with LPS induced a two-fold induction of MMP-9 levels, which could not be inhibited by pretreatment with RWJ 67657 at low concentrations. Also, mRNA expression of MMP-9 could not be induced by LPS stimulation, and no inhibition by p38 MAPK inhibitor was observed. This latter finding is in accordance with the study by Lai and co-workers, which showed

p38 MAPK inhibition in MDM
57
that LPS induction of MMP-9 in monocytes is mainly regulated by the ERK1/2 pathway and not the p38 MAPK pathway 28. p38 MAPK inhibitors have effects on different cell types, thereby possibly enhancing the therapeutic effects, but increasing the risk of side effects. One of the reasons for undesirable effects might be the cross-reactivity against other kinases, which was not the case for RWJ 67657 13. The preliminary pharmacokinetic data suggest a twice-daily dosing regimen 15, while our data show significant effects at low concentrations. CONCLUSIONS A significant inhibition of TNF-α production and mRNA expression in LPS-stimulated monocyte-derived macrophages was observed after pre-treatment with RWJ 67657, a p38 MAPK inhibitor at pharmacological relevant concentrations. Inhibition of mRNA expression of IL-1β, IL-8 and COX-2 was also detected. MMP-9 was found to be constitutively produced at high levels and not inhibited by RWJ 67657. The results presented here could have important implications for the treatment of RA, since the drug used in this study has already proven to be safe in an in-human study 15. More research upon the effects of p38 MAPK inhibition on other cell types involved in inflammation will establish its applicability as a drug in the near future. ACKNOWLEDGEMENTS This work was supported by the Dutch Rheumatology Foundation and Johnson and Johnson Pharmaceutical Research and Development, Raritan, New Jersey, USA.
REFERENCES 1 Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis.
N.Engl.J.Med. 2001; 344: 907-16. 2 Tak PP, Smeets TJ, Daha MR et al. Analysis of the synovial cell infiltrate in early rheumatoid
synovial tissue in relation to local disease activity. Arthritis Rheum. 1997; 40: 217-25. 3 Mulherin D, Fitzgerald O, Bresnihan B. Synovial tissue macrophage populations and articular
damage in rheumatoid arthritis. Arthritis Rheum. 1996; 39: 115-24. 4 Hirohata S, Yanagida T, Itoh K et al. Accelerated generation of CD14+ monocyte-lineage cells
from the bone marrow of rheumatoid arthritis patients. Arthritis Rheum. 1996; 39: 836-43. 5 Burmester GR, Stuhlmuller B, Keyszer G, Kinne RW. Mononuclear phagocytes and rheumatoid
synovitis. Mastermind or workhorse in arthritis? Arthritis Rheum. 1997; 40: 5-18. 6 Kinne RW, Brauer R, Stuhlmuller B, Palombo-Kinne E, Burmester GR. Macrophages in
rheumatoid arthritis. Arthritis Res. 2000; 2: 189-202. 7 Smolen JS, Steiner G. Therapeutic strategies for rheumatoid arthritis. Nat.Rev.Drug Discov.
2003; 2: 473-88. 8 Redlich K, Schett G, Steiner G, Hayer S, Wagner EF, Smolen JS. Rheumatoid arthritis therapy
after tumor necrosis factor and interleukin-1 blockade. Arthritis Rheum. 2003; 48: 3308-19. 9 Rao KM. MAP kinase activation in macrophages. J.Leukoc.Biol. 2001; 69: 3-10. 10 Schett G, Tohidast-Akrad M, Smolen JS et al. Activation, differential localization, and
regulation of the stress- activated protein kinases, extracellular signal-regulated kinase, c-JUN N-terminal kinase, and p38 mitogen-activated protein kinase, in synovial tissue and cells in rheumatoid arthritis. Arthritis Rheum. 2000; 43: 2501-12.
11 Ebringer A, Wilson C. HLA molecules, bacteria and autoimmunity. J.Med.Microbiol. 2000; 49: 305-11.

Chapter 4
58
12 Jovanovic DV, Martel-Pelletier J, Di Battista JA et al. Stimulation of 92-kd gelatinase (matrix metalloproteinase 9) production by interleukin-17 in human monocyte/macrophages: a possible role in rheumatoid arthritis. Arthritis Rheum. 2000; 43: 1134-44.
13 Wadsworth SA, Cavender DE, Beers SA et al. RWJ 67657, a potent, orally active inhibitor of p38 mitogen-activated protein kinase. J.Pharmacol.Exp.Ther. 1999; 291: 680-7.
14 Fijen JW, Zijlstra JG, de Boer P et al. Suppression of the clinical and cytokine response to endotoxin by RWJ-67657, a p38 mitogen-activated protein-kinase inhibitor, in healthy human volunteers. Clin.Exp.Immunol. 2001; 124: 16-20.
15 Parasrampuria DA, de Boer P, Desai-Krieger D, Chow AT, Jones CR. Single-dose pharmacokinetics and pharmacodynamics of RWJ 67657, a specific p38 mitogen-activated protein kinase inhibitor: a first-in-human study. J.Clin.Pharmacol. 2003; 43: 406-13.
16 Westra J, Limburg PC, de Boer P, van Rijswijk MH. Effects of RWJ 67657, a p38 mitogen activated protein kinase (MAPK) inhibitor, on the production of inflammatory mediators by rheumatoid synovial fibroblasts. Ann.Rheum.Dis. 2004; 63: 1453-9.
17 Lucas DM, Lokuta MA, McDowell MA, Doan JE, Paulnock DM. Analysis of the IFN-gamma-signaling pathway in macrophages at different stages of maturation. J.Immunol. 1998; 160: 4337-42.
18 Plesner A, Greenbaum CJ, Lernmark A. Low serum conditions for in vitro generation of human macrophages with macrophage colony stimulating factor. J.Immunol.Methods 2001; 249: 53-61.
19 Lanyon P, Muir K, Doherty S, Doherty M. Influence of radiographic phenotype on risk of hip osteoarthritis within families. Ann.Rheum.Dis. 2004; 63: 259-63.
20 Kumar S, Boehm J, Lee JC. p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat.Rev.Drug Discov. 2003; 2: 717-26.
21 Kotlyarov A, Neininger A, Schubert C et al. MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nat.Cell Biol. 1999; 1: 94-7.
22 Bhattacharyya A, Pathak S, Datta S, Chattopadhyay S, Basu J, Kundu M. Mitogen-activated protein kinases and nuclear factor-kappaB regulate Helicobacter pylori-mediated interleukin-8 release from macrophages. Biochem.J. 2002; 368: 121-9.
23 Herzyk DJ, Allen JN, Marsh CB, Wewers MD. Macrophage and monocyte IL-1 beta regulation differs at multiple sites. Messenger RNA expression, translation, and post-translational processing. J.Immunol. 1992; 149: 3052-8.
24 Dean JL, Brook M, Clark AR, Saklatvala J. p38 mitogen-activated protein kinase regulates cyclooxygenase-2 mRNA stability and transcription in lipopolysaccharide-treated human monocytes. J.Biol.Chem. 1999; 274: 264-9.
25 Caivano M, Cohen P. Role of mitogen-activated protein kinase cascades in mediating lipopolysaccharide-stimulated induction of cyclooxygenase-2 and IL-1 beta in RAW264 macrophages. J.Immunol. 2000; 164: 3018-25.
26 Smeets TJ, Barg EC, Kraan MC, Smith MD, Breedveld FC, Tak PP. Analysis of the cell infiltrate and expression of proinflammatory cytokines and matrix metalloproteinases in arthroscopic synovial biopsies: comparison with synovial samples from patients with end stage, destructive rheumatoid arthritis. Ann.Rheum.Dis. 2003; 62: 635-8.
27 Murphy G, Knauper V, Atkinson S et al. Matrix metalloproteinases in arthritic disease. Arthritis Res. 2002; 4 Suppl 3: S39-S49.
28 Lai WC, Zhou M, Shankavaram U, Peng G, Wahl LM. Differential regulation of lipopolysaccharide-induced monocyte matrix metalloproteinase (MMP)-1 and MMP-9 by p38 and extracellular signal-regulated kinase 1/2 mitogen-activated protein kinases. J.Immunol. 2003; 170: 6244-9.

5
Monocytes and monocyte-derived macrophages differ
in regulation of signal transduction pathways
Johanna Westra1
Sander H Diks3
Berber Doornbos-van der Meer1
Karina Wessel1
Maikel P Peppelenbosch3
Martin H van Rijswijk1
Pieter C Limburg1,2
From the Departments of 1Rheumatology, 2Pathology and Laboratory Medicine, and 3Cell Biology, University Medical Center Groningen, The Netherlands
In preparation

Chapter 5
60
ABSTRACT Background. In rheumatoid arthritis (RA) macrophages play a key role in the inflammatory process. It has been reported that inhibitors of signal transduction pathways interfere with cell differentiation. As p38 MAPK (mitogen-activated protein kinase) inhibitors are now in phase II clinical trials in RA, it is relevant to know whether these inhibitors differ with respect to their effects on monocytes and macrophages, and whether they influence the differentiation process. Objective. Aim of the study was to investigate whether p38 MAPK inhibition has a different effect on mediators produced by LPS-stimulated monocytes or monocyte-derived macrophages (MDM). In addition we investigated whether p38 MAPK inhibition (by RWJ 67657) influences the process of differentiation of monocytes into macrophages. Methods. Monocytes and MDM were stimulated with 50 ng/ml LPS with or without pretreatment of 1 µM RWJ 67657. IL-1β, TNF-α, IL-8 and MMP-9 mRNA expression and protein production were measured with real-time RTPCR and ELISA. Furthermore, monocytes were left to differentiate in the presence of 0, 0.1 or 1 µM RWJ 67657. Cells were stimulated with LPS at day 1, 2, 3, 4, and 5, and cytokine and MMP-9 mRNA and protein levels were measured. Lysates of stimulated and unstimulated monocytes and MDM were also tested in a kinase array to identify which kinases were activated. Results. Monocytes produced more cytokines than MDM, but less MMP-9. Inhibition with the p38 MAPK inhibitor RWJ 67657 was more effective in monocytes than in MDM. Differentiation of monocytes into macrophages in the presence of the p38 MAPK inhibitor significantly reduced TNF-α, IL-8 and MMP-9 protein production by macrophages. Conclusions. It was demonstrated that monocytes respond better to p38 MAPK inhibition than monocyte-derived macrophages, and that differentiation is influenced by p38 MAPK inhibition. With kinome profiling we found that several kinases involved in the p38 MAPK pathway are activated both in stimulated monocytes and MDM. Differences in kinase activity were found between monocytes and MDM, that need further investigation.

Differences between monocyte and MDM signal tranduction
61
INTRODUCTION Macrophages are known to play an important role in inflammatory diseases such as rheumatoid arthritis. Rheumatoid synovium is intensively infiltrated by macrophages and their numbers correlate well with articular destruction 1 and clinical scores 2. Macrophages perform many functions including phagocytosis, killing of pathogens, and the release of cytokines, proteinases and other inflammatory mediators. In response to inflammatory conditions, monocytes migrate from the bonemarrow into the vasculature, and subsequently into extravascular tissues. Transmigration of monocytes is accompanied by changes in morphological, biochemical, and functional characteristics. Circulating monocytes posses a markedly different functional phenotype as compared to tissue macrophages. It has been demonstrated that the route of monocyte differentiation determines their cytokine production and phenotype expression. Two cytokines that have been indentified as promoters of monocyte differentiation are macrophage-colony stimulating factor (M-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF)3. In vitro maturation of monocytes into macrophages by M-CSF with only 1% FCS has been shown to yield a maximum of macrophages without inducing proliferation or activation 4. CSFs have been found to be present at elevated levels in the synovial fluid of RA patients. Also in collagen-induced arthritis models in mice, M-CSF has been shown to exacerbate the arthritis 5. Because of the crucial role synovial macrophages have in the inflammatory process in RA, they are target of therapy themselves, for instance by clodrate-containing liposomes 6 and recently by anti-FcγRI (CD64) antibody to which the plant toxin ricin A (RTA) was chemically linked (CD64-RTA) 7. In vitro stimulation of monocytes and macrophages by a variety of agents causes activation of signal transduction pathways and in particular activation of mitogen-activated protein kinases (MAPK) 8. Cytokine production by monocytes and MDM has been reported to be significantly reduced by treatment with specific p38 MAPK inhibitors 9;10. Stimulation with lipopolysaccharide (LPS) is not only regulated by CD14 but also involves Toll-like receptors, leading to activation of stress-induced signal transduction pathways. Differentiation of macrophages with serum is reported to be under influence of the p38 MAPK pathway 11. In monocytic cell lines differentiation was shown to be potentiated by p38 MAPK inhibitors, with concomitant up-regulation of the c-jun N-terminal kinase (JNK) pathway 12. Between 10-15% of the proteins encoded by the human genome are involved in intracellular signal transduction. Nearly a third of all the different types of proteins inside the cells are subject to phosphorylation, usally at multiple sites 13. Recently progress has been made with the preparation of arrays exhibiting specific consensus sequences for protein kinases. This kind of array would allow faster and more extensive analysis of activity of various intracellular signaling pathways. Usefullnes of peptide arrays was recently tested by Diks et al 14, who demonstrated that with this array the enzymatic activities of a large group of kinases could be determined in LPS-stimulated PBMC. In this study we investigated whether p38 MAPK inihibition had a different effect on mediators produced by LPS-stimulated monocytes or monocyte-derived macrophages. Furthermore we investigated whether a p38 MAPK inhibitor (RWJ 67657) did

Chapter 5
62
influence M-CSF-induced differentiation of monocytes into macrophages. Finally we tested stimulated and unstimulated monocytes and MDM in the kinase array. MATERIAL AND METHODS Reagents The p38 MAPK specific inhibitor RWJ 67657 was provided by Johnson and Johnson (R.W. Johnson Pharmaceutical Research Institute, Raritan, New Jersey, USA). Recombinant human macrophage colony stimulating factor (M-CSF) and ELISA antibodies were from R&D Systems (Minneapolis, Minnesota, USA). Foetal calf serum (FCS) and RPMI 1640 culture medium were obtained from Biowhittaker (Verviers, Belgium). All reagents for RNA isolation and reverse transcriptase reaction were purchased from Invitrogen, Life Technologies (Gaithersburg, Maryland, USA). Reagents for real-time RT-PCR were obtained from Applied Biosystems (Foster City, California, USA). Macrophage culture Blood was obtained from apparently healthy laboratory workers. Peripheral blood mononuclear cells (PBMCs) were isolated by Lymphoprep density gradient centrifugation from citrated blood. Cells were suspended in RPMI with gentamycin at 2.106 cells/ml and seeded in 3 ml in 6-well plates (Corning, Schiphol, The Netherlands) or 0.5 ml in 24-well plates and cultured at 37°C in a 5% CO2 atmosphere. The cells that adhered after two hours were used as monocytes or were allowed to differentiate into monocyte-derived macrophages (MDM) for five days in RPMI containing gentamycin, 50 ng/ml M-CSF + 1% FCS, while medium was refreshed at day two. Experimental set-up To investigate whether unstimulated or LPS stimulated monocytes and MDM responded differently to treatment with p38 MAPK inhibition, cells from four donors before and after differentiation were stimulated with 50 ng/ml LPS, with or without one hour pre-treatment with 1 µM RWJ 67657 during four hours for mRNA expression measurement or 24 hours for protein determination. After four hours stimulation cells were lysed in TRIzol reagent according to the manufacturers instructions (Life Technologies) and stored at -80°C until RNA isolation. For protein determination supernatants of treated cells were harvested and stored at -20°C until measurement. The effect of p38 MAPK inhibition on cell differentiation was investigated by incubating cells from four donors with 0, 0.1 or 1.0 µM RWJ 67657 during the entire differentiation period. At day 1, 2, 3, 4 and 5 cells were stimulated with 50 ng/ml LPS for four or 24 hours as described above. Before LPS stimulation RWJ 67657 was depleted from the medium by washing for 30 minutes, which experimentally has been shown to be sufficient (data not shown). RNA isolation and real-time RT-PCR RNA isolation was performed using TRIzol reagent according to the manufacturers instructions and mRNA expression of IL-1β, TNF-α, IL-8, MMP-9, and glyceralde-

Differences between monocyte and MDM signal tranduction
63
hyde-3-phosphate dehydrogenase (GAPDH), 1µl of cDNA in triplicate was used for amplification by the real-time quantitative PCR system (ABI Prism 7900HT Sequence Detection System, Applied Biosystems) with specific Taqman primers/probes as described earlier 10. In our experiments GAPDH and other genes of interest were always determined in the same RT-PCR run. Amplification was performed using standard conditions: denaturation at 95°C for 15 seconds, 40 cycles of amplification with annealing at 60°C for 1 minute, and extension at 50°C for 2 minutes. According to the comparative Ct (threshold cycle value) method described in the ABI manual, the resulting mRNA amount of the gene of interest was normalized to the housekeeping gene GAPDH, yielding the ∆Ct value. The ∆Ct value of unstimulated cells was subtracted from the average ∆Ct value of each sample, yielding the ∆∆Ct. The amount of target, normalized to an endogenous reference (GAPDH) and relative to the control sample, is given by: 2-∆∆CT. ELISA based determination of IL-1β, TNF-α, IL-8, and MMP-9 in cell culture supernatants Cytokine (TNF-α, IL-1β, and IL-8), and MMP-9 levels were measured in cell supernatants by ELISA, using matched antibody pairs for ELISA and recombinant proteins as standards from R&D Systems. Detection limits for all cytokine ELISAs was 50 pg/ml and for MMP-9 ELISA 100 pg/ml. Kinome profiling with kinase array To compare the activity of kinases between monocytes and MDM, kinase arrays were performed as described earlier 14. First it was investigated by western blotting whether monocytes were already activated by the isolation procedure and the adherence. Previously it was shown that unstimulated MDM had low phospho-p38 MAPK activity 10. PBMCs were isolated as described above and were left to adhere in 6-well plates at a concentration of 2.106 cells/ml (3 ml/well) in RPMI + 1% FCS. After 1, 2, 4, and 6 hours cells were either activated with 50 ng/ml LPS for 30 minutes and then lysed in 300 µl lysis buffer or were lysed unstimulated. Lysis buffer contained 2% SDS, 10% glycerol, 50 mM dithiothreitol, 62.5 mM Tris-HCl (pH=6.8) and 0.01% brome-phenol blue. Cells were scraped off the wells and the lysates were subsequently sonicated for 5-10 seconds and boiled for 5 minutes. After centrifugation the samples were loaded onto a 10% SDS-PAGE gel and resolved by running at 200 V and 15 Watt constant. Semidry blotting was performed onto nitrocellulose membrane and immunodetection with anti-phospho-p38 MAPK was performed, followed by incubation with peroxidase-anti-rabbit IgG. Enhanced chemi-luminescence (ECL) detection was performed according to the manufacturers guidelines (Lumi-Lightplus, Roche Diagnostics, Mannheim, Germany). Blots were exposed to HyperfilmTM (Amersham Biosciences, Roosendaal, The Netherlands) and developed. Subsequently, blots were stripped with RestoreTM Western Blot Stripping Buffer (Pierce, Rockford, IL, USA) and immunodetection with anti-p38 MAPK was performed.

Chapter 5
64
Peptide array analysis For kinase array 107 PBMC/well were plated in 4 ml RPMI + 1% FCS in 6-well plates, and were left to adhere or were differentiated to MDM with M-CSF for five days as described above. The following experiment was performed on both monocytes (after two hours of adherence) and MDM: one well was left unstimulated, one well was stimulated with 50 ng/ml LPS for 30 minutes. Stimulations were finalized by washing with ice-cold phosphate buffered saline. Next, cells were lysed in ice-cold kinase lysis buffer (100 µl/well): 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 mM NaF, 1 mM MgCl2, 1 µg/ml leupeptin, 1 µg/ml aprotin, 1 mM PMSF. The cell lysates were centrifuged at 10000 rpm for ten minutes. Peptide array incubation mix was produced by adding 10 µl of filter-cleared activation mix (50% glycerol, 50 µM [γ-33P]ATP, 0.05% v/v Brij-35, 0.25 mg/ml bovine serum albumin, [γ-33P]ATP (1000 kBq) to 50 µl of cell lysate. The lysate + peptide array mix was added onto the chips which consists of kinase substrate peptides on a glass slide (Pepscankinase, Pepscan Systems, Lelystad, The Netherlands) and the chips were kept at 37°C in a humidified stove for 75 minutes. The chips contain 1176 kinase substrate peptides in duplicate, and the consensus substrate peptides are approximately 9 peptides long. Subsequently the chips were washed twice with phosphate-buffered saline + 0.1% Triton, twice in 2 M NaCl, twice in demineralized H2O and then air dried. The chips were then exposed to a phosphor-imager plate for 72 hours, and the density of the spots was measured and analyzed with array software (ScanAlyze, Stanford University, Stanford, California, USA). STATISTICS Paired T-tests were performed using GraphPad Prism version 3.00 for Windows, GraphPad Software (San Diego, CA, USA). RESULTS Effects of p38 MAPK inhibition on mediator production by monocytes and monocyte-derived macrophages In table 1 the protein production (IL-1β, TNF-α, IL-8 and MMP-9) by monocytes and MDM before and after LPS stimulation and 1 µM RWJ 67657 pre-treatment is listed. The table shows that unstimulated monocytes did not produce TNF-α and IL-1β, and low IL-8 levels, whereas high levels were produced after LPS stimulation. Unstimulated MDM did not produce TNF-α and IL-1β, and low IL-8, but in contrast to unstimulated monocytes, produced unstimulated MDM high levels of MMP-9. In all situations there was inhibition due to pretreament with 1 µM RWJ 67657, but inhibition appeared to be more effective in monocytes than in MDM. Except for unstimulated monocytes MMP-9 production was less effectively inhibited compared to cytokines. mRNA expression was measured in stimulated monocytes and MDM, and mRNA expression in unstimulated cells was used as control (fold induction =1, table 2).

Differences between monocyte and MDM signal tranduction
65
In contrast to protein expression IL-1β mRNA was highly expressed in stimulated MDM. All cytokine genes were expressed at a higher level in MDM than in monocytes. MMP-9 mRNA was constitutively expressed in both monocytes and MDM and expression was not induced by stimulation (fold induction < 2). Again p38 MAPK inhibition was effective for cytokine mRNA expression, but not for MMP-9 expression.
Table 1. Protein production by monocytes and MDM with and without pre-treatment of 1 µM RWJ 67657. Cells were unstimulated or stimulated for 24 hours with 50 ng/ml LPS. Protein concentration of cytokines and MMP-9 was measured in supernatants by ELISA. BD = below detection, inh = % inhibition.
Monocytes, unstimulated
MDM, unstimulated
Monocytes, stimulated
MDM, stimulated
RWJ Mean (SEM) Inh Mean
(SEM) Inh Mean (SEM) Inh Mean
(SEM) Inh
- BD BD 2392(421) BD IL-1β pg/ml + BD BD 331 (63) 86% BD
- BD BD 2021(374) 1075(493) TNF-α pg/ml + BD BD 138 (39) 93% 587(305) 45%
- 1.7(0.6) 0.3(0.1) 116 (69) 11.7 (1.7) IL-8 ng/ml + 0.03(0.01) 98% 0.08(0.02) 74% 51.2(23.7) 56% 6.9(1.3) 41%
- 6.2(2.1) 79.8(19.2) 28.6(10.1) 97.0(13.5) MMP9 ng/ml + 1.1(0.5) 82% 52.3(13.0) 34% 22.7(9.1) 21% 61.4(11.0) 37%
Table 2. mRNA expression (fold induction) in stimulated monocytes and MDM with and without pre-treatment of 1 µM RWJ 67657. Cells were stimulated with 50 ng/ml LPS for 4 hours and mRNA expression of cytokines and MMP-9 was determined by real-time RT-PCR. Monocytes MDM
RWJ Mean (SEM) Inhibition Mean (SEM) Inhibition
- 341.6(140.9) 3434.0(1423.2) IL-1β + 84.9(42.9) 75% 1093.5(549.4) 68% - 175.7(28.0) 589.7(158. 6) TNF-α + 33.5(16.0) 81% 125.2(33.9) 79% - 67.8(30.2) 324.2(118.9) IL-8 + 15.3(10.0) 77% 207.9(89.6) 36% - 1.10(0.33) 1.50(0.12) MMP9 + 0.65(0.25) 42% 1.33(0.43) 11%

Chapter 5
66
Figure 1. Protein production by monocytes and MDM during differentiation in the presence of 0, 0.1, and 1.0 µM RWJ 67657. Cells were unstimulated or stimulated for 24 hours with 50 ng/ml LPS. Protein concentration of cytokines and MMP-9 was measured in supernatants by ELISA (∗ p<0.05, ∗∗ p<0.01, ∗∗∗ p<0.001, paired T-test, tested against MDM, untreated with RWJ 67657). Effects of p38 MAPK inhibition on differentiation As p38 MAPK inhibition might not only affect protein production and mRNA expression in cells but also might have an effect on the differentiation process, we left monocytes to differentiate in the presence of 0, 0.1 and 1.0 µM RWJ 67657. In figure 1 the protein production of cytokines and MMP-9 is shown in unstimulated and stimulated cells during the differentiation process. Figure 1 shows clearly that immediately after differentiation has started IL-1β production was decreased below detection limit of the assay, while TNF-α production was decreased by more than 70%. For all cytokines the presence of RWJ 67657 reduced the production during
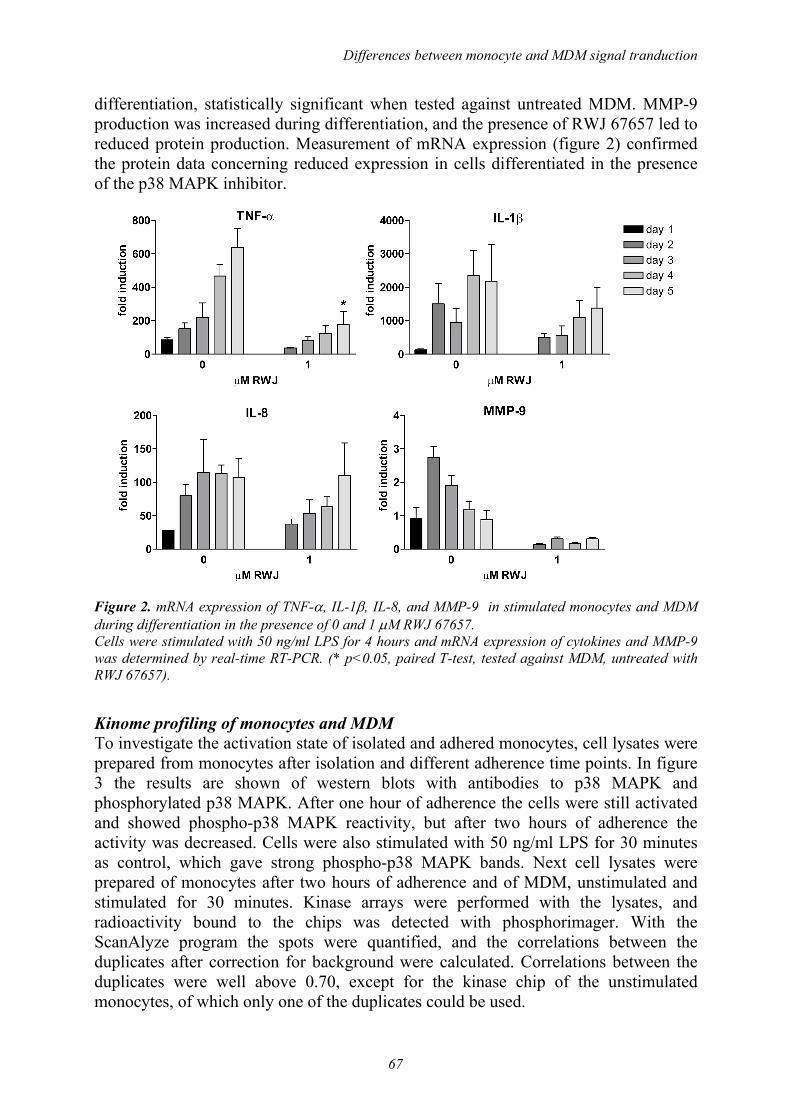
Differences between monocyte and MDM signal tranduction
67
differentiation, statistically significant when tested against untreated MDM. MMP-9 production was increased during differentiation, and the presence of RWJ 67657 led to reduced protein production. Measurement of mRNA expression (figure 2) confirmed the protein data concerning reduced expression in cells differentiated in the presence of the p38 MAPK inhibitor.
Figure 2. mRNA expression of TNF-α, IL-1β, IL-8, and MMP-9 in stimulated monocytes and MDM during differentiation in the presence of 0 and 1 µM RWJ 67657. Cells were stimulated with 50 ng/ml LPS for 4 hours and mRNA expression of cytokines and MMP-9 was determined by real-time RT-PCR. (* p<0.05, paired T-test, tested against MDM, untreated with RWJ 67657).
Kinome profiling of monocytes and MDM To investigate the activation state of isolated and adhered monocytes, cell lysates were prepared from monocytes after isolation and different adherence time points. In figure 3 the results are shown of western blots with antibodies to p38 MAPK and phosphorylated p38 MAPK. After one hour of adherence the cells were still activated and showed phospho-p38 MAPK reactivity, but after two hours of adherence the activity was decreased. Cells were also stimulated with 50 ng/ml LPS for 30 minutes as control, which gave strong phospho-p38 MAPK bands. Next cell lysates were prepared of monocytes after two hours of adherence and of MDM, unstimulated and stimulated for 30 minutes. Kinase arrays were performed with the lysates, and radioactivity bound to the chips was detected with phosphorimager. With the ScanAlyze program the spots were quantified, and the correlations between the duplicates after correction for background were calculated. Correlations between the duplicates were well above 0.70, except for the kinase chip of the unstimulated monocytes, of which only one of the duplicates could be used.

Chapter 5
68
Figure 3. Western blot of monocytes after different timepoints of adherence. Activation status of cells was measured with phospho-p38 MAPK, p38 MAPK was used as control. In table 3 results are shown of kinases involving in the p38 MAPK pathway. The results under control and LPS represent the radioactivity incorporated in the substrates (in Phosphorimager/Aida quantification units) and are the average of 2 measurements, except for the unstimulated monocytes results. The ratio of LPS stimulated samples and controls is given in the next column. From the results it appears that all substrates involved in the p38 MAPK pathway are phosphorylated after stimulation with LPS, although differences in intensity are seen between monocytes and MDM.
Table 3. Monocyte and MDM kinase activity. Depicted are kinases and substrate involved in the p38 MAPK pathway. Results represent the radioactivity in corporated in the substrates (in phosphorimager /Aida quantification units). (MK2-MAPKAPK2, cPLA2-cytosolic phospholipase A2, eIF4E-eukaryotic translation initiation factor 4E).
monocytes MDM
Kinase Substrate Sequence phos.
site
contr LPS LPS/
contr
contr LPS LPS/
contr
Raf1 MAPKK1 QLIDSMANS S-217 1823 3045 1.67 1255 3229 2.57
MKK3/6 p38MAPK DDEMTGYVA T-180 4783 8962 1.87 889 4209 4.73
MAPK MAPKAPK2 KVPQTPLHT T-300 6335 11398 1.80 5032 8679 1.72
MAPK cPLA2 SYPLSPLSD S-505 11649 14853 1.28 8729 12028 1.38
MK2 hsp27 LRGPSWDPF S-15 807 9021 11.18 2906 5643 1.94
MK2 hsp27 SRALSRQLS S-78 9149 12325 1.35 14072 14168 1.01
Mnk1 elF4E TKSGSTTKN S-209 2417 3729 1.54 2270 6602 2.91
STAT-1 LLPMSPEEF S-727 7324 10388 1.42 3436 8360 2.43

Differences between monocyte and MDM signal tranduction
69
DISCUSSION In the present study it was shown that monocytes respond better to p38 MAPK inhibition than monocyte-derived macrophages, and also that cell differentiation is influenced by p38 MAPK inhibition. The importance of synovial macrophages in inflammation has been established in several studies, and recently it has been reported that the number of synovial sublining macrophages does correlate well with clinical improvement after antirheumatic treatment (MTX, leflunomide, Infliximab, prednisone) 15. In previous work from our group it has been shown that TNF-α production by monocyte-derived macrophages was effectively inhibited by the p38 MAPK inhibitor RWJ 67657 10. As p38 MAPK inhibitors are in phase II clinical trials in RA 16 knowledge of the exact effects of such drugs is important. TNF-α production was higher and more effectively inhibited by RWJ 67657 (93%) in monocytes than in MDM (45%), whereas IL-1β production was inhibited in monocytes (93%), but was not excreted by MDM. This latter finding is in concordance with the study by Herzyk et al 17 and is related to the difficulty of release of IL-1β by macrophages. IL-8 production was tenfold higher in monocytes, but effects of p38 MAPK inhibitor treatment were comparable. In contrast to cytokine production was MMP-9 abundantly produced by MDM, even in unstimulated cells. Inhibition of MMP-9 by p38 MAPK inhibitor treatment in monocytes was only 21%, and in MDM 37%, indicating that pathways other than the p38 MAPK pathway must be involved in MMP-9 regulation. Lai et al demonstrated a dominant role for the ERK1/2 pathway in MMP-9 regulation in human monocytes 18. With real-time RT-PCR we found higher expression of TNF-α, IL-1β and IL-8 mRNA levels in MDM compared to monocytes. Although protein production in monocytes was higher, mRNA levels seem to accumulate in MDM. It has been known that activation of p38 MAPK has a stabilizing effect on mRNA of inflammatory genes. This effect has been described for TNF-α both for primary human monocytes 19 as well as for macrophage-like cell lines 20. The results from our study show that reduction of mRNA expression of TNF-α and IL-1β by p38 MAPK inhibition was approximately the same for monocytes and MDM (81% and 79% (TNF-α), and 75% and 68% (IL-1β) respectively), whereas IL-8 and MMP-9 mRNA was more reduced in monocytes than MDM due to RWJ 67657 treatment (77% and 36% (IL-8), 42% and 11% (MMP-9) respectively). From the differentiation experiments it could be demonstrated that alreay after one day of differerentiation with M-CSF, the properties of the monocytes changed: IL-1β was no longer produced, TNF-α production was very low, whereas MMP-9 production increased Differentiation in the presence of RWJ 67657 significantly reduced TNF-α protein and mRNA expression and IL-8 and MMP-9 protein production. Ayala et al reported that p38 MAPK inhibition at concentrations, which were reported to inhibit MAPKAPK-2 activation, blocked monocyte differentiation and chemotaxis 11. From our data we can not conclude that differentiation is blocked, but rather that protein production is decreased due to the p38 MAPK inhibition. The results from the kinase array showed that indeed the substrates involved in the p38 MAPK pathway were phosphorylated by activation both in monocytes and in MDM. From the literature it is known that in vitro MKK3, MKK4, and MKK6 all show a

Chapter 5
70
strong preference for phosphorylation of the tyrosine residue of the Thr-Gly-Tyr motif in the p38 MAPK substrate 21 and this is in concordance with our findings. Recently it was reported that oxidative stress of resident vascular cells and macrophages potently enhances eIF4E phosphorylation by the activation of Mnk-1 22. Phosphorylation of eukaryotic translation initiation factor 4E is associated with increased activity of the translational machinery. With this array the activation status of several kinases in a single experiment can be investigated, but more research is needed to understand the interactions of the intracellular signal transduction pathways. REFERENCES 1 Mulherin D, Fitzgerald O, Bresnihan B. Synovial tissue macrophage populations and articular
damage in rheumatoid arthritis. Arthritis Rheum. 1996; 39: 115-24. 2 Tak PP, Smeets TJ, Daha MR et al. Analysis of the synovial cell infiltrate in early rheumatoid
synovial tissue in relation to local disease activity. Arthritis Rheum. 1997; 40: 217-25. 3 Bender AT, Ostenson CL, Giordano D, Beavo JA. Differentiation of human monocytes in vitro
with granulocyte-macrophage colony-stimulating factor and macrophage colony-stimulating factor produces distinct changes in cGMP phosphodiesterase expression. Cell Signal. 2004; 16: 365-74.
4 Plesner A, Greenbaum CJ, Lernmark A. Low serum conditions for in vitro generation of human macrophages with macrophage colony stimulating factor. J.Immunol.Methods 2001; 249: 53-61.
5 Campbell IK, Rich MJ, Bischof RJ, Hamilton JA. The colony-stimulating factors and collagen-induced arthritis: exacerbation of disease by M-CSF and G-CSF and requirement for endogenous M-CSF. J.Leukoc.Biol. 2000; 68: 144-50.
6 Barrera P, Blom A, Van Lent PL et al. Synovial macrophage depletion with clodronate-containing liposomes in rheumatoid arthritis. Arthritis Rheum. 2000; 43: 1951-9.
7 van Roon JA, Bijlsma JW, van de Winkel JG, Lafeber FP. Depletion of synovial macrophages in rheumatoid arthritis by an anti-Fc{gamma}RI-Calicheamicin immunoconjugate. Ann.Rheum.Dis. 2004.
8 Rao KM. MAP kinase activation in macrophages. J.Leukoc.Biol. 2001; 69: 3-10. 9 Wadsworth SA, Cavender DE, Beers SA et al. RWJ 67657, a potent, orally active inhibitor of
p38 mitogen-activated protein kinase. J.Pharmacol.Exp.Ther. 1999; 291: 680-7. 10 Westra J, Doornbos-Van Der Meer B, de Boer P, van Leeuwen MA, van Rijswijk MH, Limburg
PC. Strong inhibition of TNF-alpha production and inhibition of IL-8 and COX-2 mRNA expression in monocyte-derived macrophages by RWJ 67657, a p38 mitogen-activated protein kinase (MAPK) inhibitor. Arthritis Res.Ther. 2004; 6: R384-R392.
11 Ayala JM, Goyal S, Liverton NJ, Claremon DA, O'Keefe SJ, Hanlon WA. Serum-induced monocyte differentiation and monocyte chemotaxis are regulated by the p38 MAP kinase signal transduction pathway. J.Leukoc.Biol. 2000; 67: 869-75.
12 Wang X, Studzinski GP. Inhibition of p38MAP kinase potentiates the JNK/SAPK pathway and AP-1 activity in monocytic but not in macrophage or granulocytic differentiation of HL60 cells. J.Cell Biochem. 2001; 82: 68-77.
13 Pelech S. Tracking cell signaling protein expression and phosphorylation by innovative proteomic solutions. Curr.Pharm.Biotechnol. 2004; 5: 69-77.
14 Diks SH, Kok K, O'Toole T et al. Kinome profiling for studying lipopolysaccharide signal transduction in human peripheral blood mononuclear cells. J.Biol.Chem. 2004; 279: 49206-13.
15 Haringman JJ, Gerlag DM, Zwinderman AH et al. Synovial tissue macrophages: highly sensitive biomarkers for response to treatment in rheumatoid arthritis patients. Ann.Rheum.Dis. 2004.
16 Nikas SN, Drosos AA. SCIO-469 Scios Inc. Curr.Opin.Investig.Drugs 2004; 5: 1205-12. 17 Herzyk DJ, Allen JN, Marsh CB, Wewers MD. Macrophage and monocyte IL-1 beta regulation
differs at multiple sites. Messenger RNA expression, translation, and post-translational processing. J.Immunol. 1992; 149: 3052-8.

Differences between monocyte and MDM signal tranduction
71
18 Lai WC, Zhou M, Shankavaram U, Peng G, Wahl LM. Differential regulation of lipopolysaccharide-induced monocyte matrix metalloproteinase (MMP)-1 and MMP-9 by p38 and extracellular signal-regulated kinase 1/2 mitogen-activated protein kinases. J.Immunol. 2003; 170: 6244-9.
19 Wang SW, Pawlowski J, Wathen ST, Kinney SD, Lichenstein HS, Manthey CL. Cytokine mRNA decay is accelerated by an inhibitor of p38-mitogen-activated protein kinase. Inflamm.Res. 1999; 48: 533-8.
20 Brook M, Sully G, Clark AR, Saklatvala J. Regulation of tumour necrosis factor alpha mRNA stability by the mitogen-activated protein kinase p38 signalling cascade. FEBS Lett. 2000; 483: 57-61.
21 Fleming Y, Armstrong CG, Morrice N, Paterson A, Goedert M, Cohen P. Synergistic activation of stress-activated protein kinase 1/c-Jun N-terminal kinase (SAPK1/JNK) isoforms by mitogen-activated protein kinase kinase 4 (MKK4) and MKK7. Biochem.J. 2000; 352 Pt 1: 145-54.
22 Duncan RF, Peterson H, Sevanian A. Signal transduction pathways leading to increased eIF4E phosphorylation caused by oxidative stress. Free Radic.Biol.Med. 2005; 38: 631-43.

6
Chemokine production and E-selectin expression in
activated endothelial cells are inhibited by p38 MAPK
(mitogen activated protein kinase) inhibitor RWJ 67657
Johanna Westra1
Joanna M Kułdo2
Martin H van Rijswijk1
Grietje Molema2
Pieter C Limburg1
From the Departments of 1 Rheumatology, and 2 Medical Biology section of Pathology
and Laboratory Medicine, University Medical Center Groningen, The Netherlands
International Immunopharmacology: in press

Chapter 6
74
ABSTRACT Endothelial cells play an important role in inflammatory diseases like rheumatoid arthritis by recruitment of inflammatory cells. The cytokines TNF-α and IL-1β are major inducers of endothelial cell activation and are stimulators of inflammatory signal transduction pathway involving p38 MAPK (mitogen-activated protein kinase). The present study investigated the effects of p38 MAPK inhibition on cell adhesion molecule (CAM) expression and chemokine production by endothelial cells both on mRNA and protein level. Pre-treatment of endothelial cells with the pharmacologically relevant concentration of 1 µM of the p38 MAPK inhibitor RWJ 67657 reduced TNF-α and IL-1β induced mRNA and membrane expression of E-selectin. Moderate inhibitory effects on ICAM-1 and VCAM-1 expression were found. Significant reduction of mRNA expression and protein production of the inflammatory cytokine IL-6 and the chemokines IL-8 and MCP-1 was demonstrated. Treatment with RWJ 67657 could lead to reduced leukocyte infiltration by the reduction of E-selectin expression and chemokine production. Key words. chemokine, endothelial cell, E-Selectin, MAPK inhibitor

p38 MAPK inhibition in EC
75
INTRODUCTION Rheumatoid arthritis (RA) is an inflammatory disease characterized by hyperplasia, increased vascularity, and infiltration of inflammatory cells into the synovial membrane 1. It is now known that the endothelial cells (EC), which line the lumen of the blood vessels, are not passive bystanders but are active responders to stimuli like activated leukocytes and cytokines 2. After stimulation EC can produce a number of inflammatory mediators and express cellular adhesion molecules (CAMs). Most CAMs involved in endothelial activation belong to three families, i.e., the integrins, selectins, and immunoglobulin superfamilies 3. The adhesion of leukocytes to EC takes place in four steps. The first step is a weak adhesion (tethering and rolling) mediated by E-, P- and L-selectins. The next step is leukocyte activation and is a consequence of interaction between chemokines and their receptors on leukocytes. Then firm adhesion takes place, which is mediated mostly by interaction between integrins on the leukocytes and vascular adhesion molecule (VCAM)-1 and intercellular adhesion molecule (ICAM)-1 on the EC. The final step is the transendothelial migration, which is directed by secreted chemokines bound to endothelial heparan sulphate glycosaminoglycans. Essential chemokines are interleukin (IL)-8 which attracts neutrophils, and monocyte chemo-attractant protein (MCP)-1, the main attractant for mononuclear cells 2;3. The cytokines TNF (tumour necrosis factor)-α and IL-1β, produced by activated macrophages in the synovium, have been identified as the major inducers of EC activation in RA 4. Blockade of these cytokines has proven to be effective in the treatment of RA, although Redlich et al recently concluded that targeting individual molecules such as TNF-α may not be sufficient in interfering with both inflammation and joint destruction 5. Successful drug treatment of RA is associated by a decrease in cytokine production, but also by a decrease in E-selectin and ICAM-1 expression 6. In vitro studies demonstrated that in TNF-α and/or lipopolysaccharide (LPS) stimulated human umbilical vein endothelial cells (HUVEC) the nuclear factor-κB (NF-κB), and p38 mitogen-activated protein kinase (MAPK) pathways are important in controlling adhesion molecule expression 7;8. Activation of all three stress- and mitogen-activated protein kinases (SAPK/MAPK) was found throughout the RA synovial tissue, whereas activated p38 MAPK predominantly was located in the synovial lining layer and in synovial endothelial cells 9. Interest in protein kinases and in particular in p38 MAPK as drug targets has increased in the last years as recently reviewed by Kumar et al 10. The p38 MAPK inhibitor RWJ 67657 (4-[4-(4-fluorophenyl)-1-(3-phenylpropyl)-5-(4-pyridinyl)-1H-imidazol-2-yl]-3-butyn-1-ol) has been shown to be effective in inhibiting the release of TNF-α from LPS-treated human peripheral blood mononuclear cells with an IC50 of 3 nM 11. This compound was approximately 10-fold more potent than the reference standard p38 MAPK inhibitor SB 203580 in all p38 dependent in vitro systems tested. RWJ 67657 specifically inhibited the enzymatic activity of recombinant p38 α and β, but not of γ and δ in vitro, and had no significant activity against a variety of other kinases 11. We recently demonstrated that RWJ 67657 significantly inhibited IL-6, IL-8, MMP-3 and COX-2 mRNA expression by IL-1β and TNF-α stimulated rheumatoid synovial fibroblasts 12. Furthermore Fijen et al showed that this compound induced a dose-dependent suppression of leukocyte and endothelial cell response after endotoxin challenge in

Chapter 6
76
humans 13. Recent pharmaco-kinetic and pharmaco-dynamic studies of RWJ 67657 in humans showed that the compound has an acceptable safety profile to warrant further investigations 14. In the present study the effects of p38 MAPK inhibition in IL-1β or TNF-α stimulated HUVEC concerning adhesion molecule expression and chemokine production were studied at the level of mRNA expression as well as protein production. Pre-treatment of activated endothelial cells with 1 µM RWJ 67657 reduced E-selectin mRNA and protein expression, and significantly reduced IL-8, MCP-1 and IL-6 production and mRNA expression. These results indicate that treatment with RWJ 67657 could lead to reduced leukocyte infiltration and therefore could have an important therapeutic benefit. MATERIALS AND METHODS Reagents RWJ 67657 was provided by Johnson and Johnson (R.W. Johnson Pharmaceutical Research Institute, Raritan, NJ). Recombinant TNF-α and IL-1β and ELISA antibodies for the detection of IL-8 and MCP-1 were obtained from R&D Systems (Minneapolis, MN). Antibodies for the detection of IL-6 were purchased from Sanquin (Amsterdam, The Netherlands). Specific antibodies to p38 MAPK, phospho-p38 MAPK and phospho-MAPK activated protein kinase (MAPKAPK)-2 were purchased from Cell Signalling Technologies (Beverly, MA) and detecting antibody peroxidase -swine-anti-rabbit was from DAKO (Glostrup, Denmark). Mouse anti-human E-selectin antibody (H18/7) was kindly provided by Dr. M. Gimbrone Jr., Harvard Medical School. Monoclonal anti-human VCAM-1 (CD106) and anti-human ICAM-1 (CD54), both labelled with phycoerytrin (PE) were from Beckton Dickinson (Bedford, MA). Monoclonal anti-human PECAM-1 (platelet endothelial cell adhesion molecule, CD31) was obtained from DAKO and PE-labeled goat-anti-mouse was from SBA (Southern Biotechnology Associates, Birmingham, AL). RNA isolation kit was from Stratagene (La Jolla, CA), quantitation of RNA was performed with Ribogreen RNA quantitation kit (Molecular Probes Europe BV, Leiden, The Netherlands). Other reagents for RNA isolation and reverse transcriptase reaction were purchased from Invitrogen (Breda, The Netherlands). Reagents, primers and probes for real-time RT-PCR were obtained from Applied Biosystems (Nieuwerkerk a/d IJssel, The Netherlands). Endothelial cell culture and stimulation HUVEC were obtained from the Endothelial Cell Facility at the UMCG (The Netherlands) as described previously 15. Primary isolates combined from 2 or 3 donors were cultured on 1% gelatin (Sigma-Aldrich, Zwijndrecht, The Netherlands) -precoated plastic tissue culture plates or flasks (Corning, Schiphol, The Netherlands) at 37º C under 5% CO2/95% air. The culture medium constisted of RPMI 1640 (Biowhittaker, Verviers, Belgium) supplemented with 20% heat-inactivated fetal calf serum (FCS), 2 mM L-glutamine, 5 U/ml heparin, 100 U/ml penicillin, 100 µg/ml streptomycin and 20 µg/ml endothelial cell growth factor, extracted from bovine brain according to the procedure described by Maciag 16. After attaining confluence, cells

p38 MAPK inhibition in EC
77
were detached from the surface by trypsin/EDTA (0.5/0.2 mg/ml) and split at a 1:3 ratio. In the experiments presented here HUVEC were used up to passage 4. In the experimental set-up HUVEC were grown to confluence in gelatin-coated 6-well plates. Fresh medium was added before cells were stimulated for 6 or 24 hours with 10 ng/ml TNF-α or IL-1β, with or 1 hour without pre-treatment with 1µM RWJ 67657 (stock solution 10 mM in DMSO = dimethylsulfoxide). Phosphorylation of p38 MAPK and MAPKAPK-2 in HUVEC HUVEC were pre-treated with 0, 0.1, 1, and 10 µM RWJ 67657 for 1 hour and stimulated for 30 minutes as mentioned above. Cell extracts were prepared by lysing the cells with 1X SDS sample buffer (containing 2% SDS, 10% glycerol, 50 mM dithiothreitol, 62.5 mM Tris-HCl (pH=6.8) and 0.01% brome-phenol blue). Cells were scraped off the wells and the lysates were subsequently sonicated for 5-10 seconds and boiled for 5 minutes. After centrifugation the samples were loaded onto a 10% SDS-PAGE gel and resolved by running at 200 V and 15 Watt constant. Semidry-blotting was performed onto a nitrocellulose membrane after which immunodetection with anti-p38 MAPK, anti-phospho-p38 MAPK, anti-phospho-MAPKAPK-2 and peroxidase labelled swine-anti-rabbit was performed. Enhanced chemi-luminescence (ECL) detection was performed according to the manufacturers guidelines (Lumi-Lightplus, Roche Diagnostics, Mannheim, Germany). RNA isolation and real-time RT-PCR HUVEC with or without 1 hour pre-treatment with 1µM RWJ 67657 were stimulated for 6 and 24 hours with 10 ng/ml TNF-α or IL-1β. Total RNA was isolated using the Absolutely RNA Microprep Kit according to the manufacturers guidelines. RNA was analysed qualitatively by gel electrophoresis and quantitatively by Ribogreen RNA Quantitation Kit. One microgram of total RNA was used for the synthesis of first strand cDNA using Superscript III RNase H- Transcriptase in 20 µl final volume containing 250 ng random hexamers and 40 units Rnase OUT inhibitor. For the measurement of mRNA for CD31, E-selectin, VCAM-1, ICAM-1, IL-8, MCP-1, IL-6 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) 1 µl of cDNA in triplicate was used for amplification by the Taqman real-time PCR system (ABI Prism 7900HT Sequence Detection System, Applied Biosystems) with specific Taqman primers/probes. In our experiments GAPDH and other genes of interest were always determined in the same RT-PCR run. Amplification was performed using standard conditions: denaturation at 95°C for 15 seconds, 40 cycles of amplification with annealing at 60°C for 1 minute, and extension at 50°C for 2 minutes. According to the comparative Ct (threshold cycle value) method described in the ABI manual, the resulting mRNA amount of the gene of interest was normalized to the housekeeping gene GAPDH, yielding the ∆Ct value. The ∆Ct value of unstimulated HUVEC was subtracted from the average ∆Ct value of each sample, yielding the ∆∆Ct. The amount of target, normalized to an endogenous reference (GAPDH) and relative to the control sample, is given by: 2-∆∆CT.

Chapter 6
78
Flow cytometric analysis HUVEC were stimulated for 6 and 24 hours as mentioned above and detached from the wells by short treatment with trypsin, and subsequently resuspend in FCS to neutralize the trypsin. After washing with PBS supplemented with 5% FCS, HUVEC were incubated with PE-labeled monoclonal antibodies against VCAM-1 (dilution 1:10) and ICAM-1 (1:25) for 45 minutes on ice or with monoclonal antibodies against CD31 (1:25) and E-selectin (undiluted) followed by incubation with PE-labeled goat-anti-mouse antibodies. Cells were fixed with 0.5% paraformaldehyde/PBS and adhesion molecule expression was detected by flow cytometric analysis in an Epics-Elite Flow cytometer (Coulter Electronics, Mijdrecht, The Netherlands). Non specific staining was assessed by staining with irrelevant isotype-matched monoclonal antibodies. The effects of RWJ 67657 on adhesion molecule expression was determined in 3 experiments with different HUVEC cultures. ELISA based determination of IL-8, MCP-1 and IL-6 in cell culture supernatants HUVEC were pre-treated with 1 µM RWJ 67657 for 6 and 24 hours and stimulated with 10 ng/ml TNF-α or IL-1β. IL-6 levels in cell supernatants were measured as described previously 17, IL-8 and MCP-1 were measured by ELISA, using matched antibody pairs for ELISA and recombinant proteins as standards from R&D Systems. In short, Corning high-binding ELISA plates were coated overnight with monoclonal antibodies in PBS. After blocking with 2% BSA (bovine serum albumin)/PBS diluted supernatants were added. Bound chemokines were detected with biotinylated polyclonal antibodies followed by incubation with peroxidase labelled Streptavidin (Sanquin, Amsterdam, The Netherlands). Colour reaction was performed with TMB (3’3’5’5’tetramethylbenzidin, Roth, Karlsruhe, Germany) and concentration of protein was determined with the SOFTmax PRO software (Molecular Devices, Sunnyvale, CA). Detection limits for ELISAs was 20 pg/ml for IL-6 and IL-8 and 50 pg/ml for MCP-1. STATISTICS Paired T-tests were performed using GraphPad Prism version 3.00 for Windows, GraphPad Software (San Diego, CA). RESULTS Effect of RWJ 67657 on phosphorylation of p38 MAPK and MAPKAPK-2 Upon stimulation with 10 ng/ml TNF-α and/or IL-1β p38 MAPK is rapidly phosphorylated within minutes, while after 60 minutes the level of phosphorylation is decreased (data not shown). In figure 1 the effect of RWJ 67657 on phosphorylation of p38 MAPK and its downstream substrate MAPKAPK-2 after 30 minutes of stimulation is shown. RWJ 67657 does not inhibit phosphorylation of p38 MAPK, but it does inhibit its activity as can be seen from the inhibition of MAPKAPK-2 phosphorylation at 0.1 µM. Complete inhibition was demonstrated at a concentration of 1 µM RWJ 67657. The solvent 0.01% DMSO did not affect phosphorylation of either kinase.

p38 MAPK inhibition in EC
79
Figure 1. Phosphorylation of p38 MAPK and MAPKAPK-2 in stimulated HUVEC after pre-treatment with 10, 1 and 0.1 µM RWJ 67657. HUVEC were treated with TNF-α and/or IL-1β for 30 minutes with or without 1 hour pre-treatment with RWJ 67657. Phosphorylation was measured by Western blot using specific antibodies against p38 MAPK, phospho-p38 MAPK and its direct downstream substrate MAPKAPK-2. Control incubations were performed with 0.01% dmso, the vehicle solvent for RWJ 67657.
Figure 2. Effect of RWJ 67657 pre-treatment on mRNA expression of CD31, E-selectin, VCAM-1 and ICAM-1 in HUVEC. Cells were stimulated with TNF-α and/or IL-1β for 6 or 24 hours and pre-treated with 1µM RWJ 67657 for 1 hour. mRNA expression was determined with real-time RT-PCR and results are expressed as fold induction compared to unstimulated cells (fold induction=1). Bars show means (n=4-6) and SEM. (* p< 0.05, paired T-test calculated against the stimulated control).
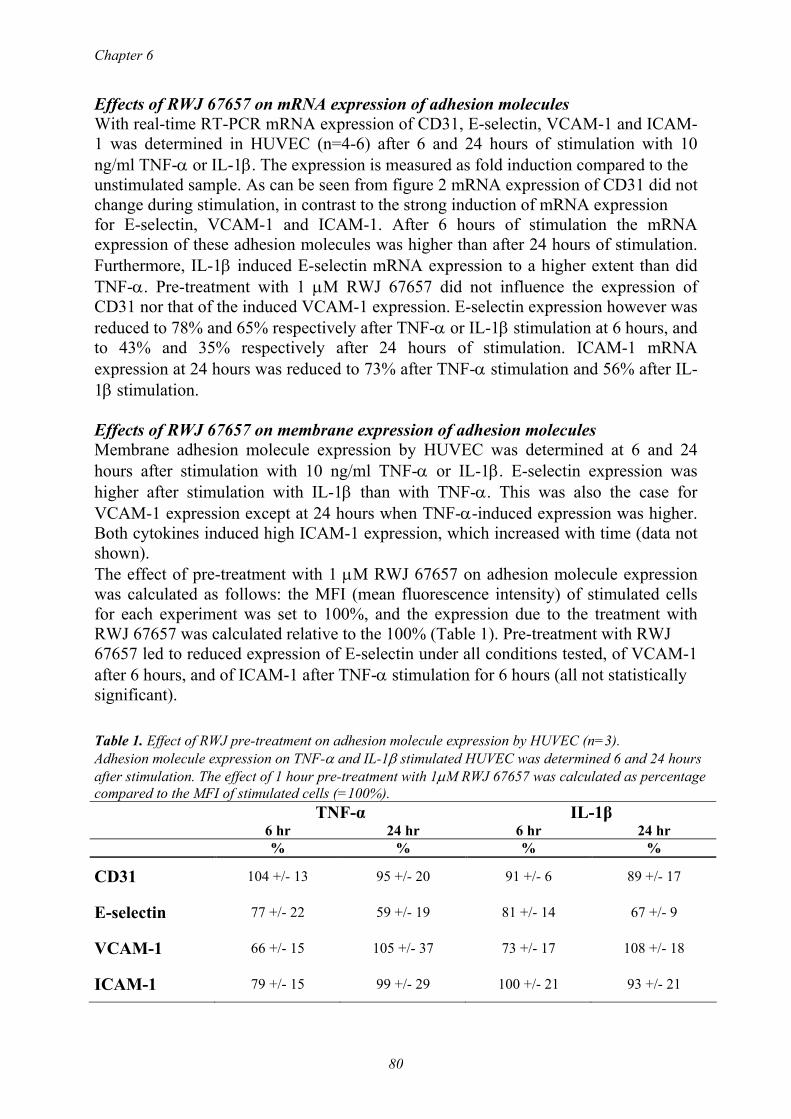
Chapter 6
80
Effects of RWJ 67657 on mRNA expression of adhesion molecules With real-time RT-PCR mRNA expression of CD31, E-selectin, VCAM-1 and ICAM-1 was determined in HUVEC (n=4-6) after 6 and 24 hours of stimulation with 10 ng/ml TNF-α or IL-1β. The expression is measured as fold induction compared to the unstimulated sample. As can be seen from figure 2 mRNA expression of CD31 did not change during stimulation, in contrast to the strong induction of mRNA expression for E-selectin, VCAM-1 and ICAM-1. After 6 hours of stimulation the mRNA expression of these adhesion molecules was higher than after 24 hours of stimulation. Furthermore, IL-1β induced E-selectin mRNA expression to a higher extent than did TNF-α. Pre-treatment with 1 µM RWJ 67657 did not influence the expression of CD31 nor that of the induced VCAM-1 expression. E-selectin expression however was reduced to 78% and 65% respectively after TNF-α or IL-1β stimulation at 6 hours, and to 43% and 35% respectively after 24 hours of stimulation. ICAM-1 mRNA expression at 24 hours was reduced to 73% after TNF-α stimulation and 56% after IL-1β stimulation. Effects of RWJ 67657 on membrane expression of adhesion molecules Membrane adhesion molecule expression by HUVEC was determined at 6 and 24 hours after stimulation with 10 ng/ml TNF-α or IL-1β. E-selectin expression was higher after stimulation with IL-1β than with TNF-α. This was also the case for VCAM-1 expression except at 24 hours when TNF-α-induced expression was higher. Both cytokines induced high ICAM-1 expression, which increased with time (data not shown). The effect of pre-treatment with 1 µM RWJ 67657 on adhesion molecule expression was calculated as follows: the MFI (mean fluorescence intensity) of stimulated cells for each experiment was set to 100%, and the expression due to the treatment with RWJ 67657 was calculated relative to the 100% (Table 1). Pre-treatment with RWJ 67657 led to reduced expression of E-selectin under all conditions tested, of VCAM-1 after 6 hours, and of ICAM-1 after TNF-α stimulation for 6 hours (all not statistically significant). Table 1. Effect of RWJ pre-treatment on adhesion molecule expression by HUVEC (n=3). Adhesion molecule expression on TNF-α and IL-1β stimulated HUVEC was determined 6 and 24 hours after stimulation. The effect of 1 hour pre-treatment with 1µM RWJ 67657 was calculated as percentage compared to the MFI of stimulated cells (=100%).
TNF-α IL-1β 6 hr 24 hr 6 hr 24 hr % % % %
CD31 104 +/- 13 95 +/- 20 91 +/- 6 89 +/- 17
E-selectin 77 +/- 22 59 +/- 19 81 +/- 14 67 +/- 9
VCAM-1 66 +/- 15 105 +/- 37 73 +/- 17 108 +/- 18
ICAM-1 79 +/- 15 99 +/- 29 100 +/- 21 93 +/- 21

p38 MAPK inhibition in EC
81
In figure 3 one of the three experiments is shown for adhesion molecule expression after TNF-α and IL-1β stimulation, and after RWJ 67657 pre-treatment followed by cytokine stimulation. The MFI values and percentage bright positive cells are indicated in the figures. As can be seen from the figure there was an overall reduced expression of E-selectin after treatment with the p38 MAPK inhibitor and also the number of positive cells was decreased. This reduction however was not statistically significant. VCAM-1 and ICAM-1 were moderately reduced. Control experiments with 0.01% DMSO showed no significant effects of the solvent on adhesion molecule mRNA or protein expression (data not shown).
Figure 3. Effect of RWJ 67657 pre-treatment on adhesion molecule expression on HUVEC after 6 and 24 hours. HUVEC were stimulated with TNF-α or IL-1β. E-selectin, ICAM-1 and VCAM-1 expression was measured by flowcytometry (thick black line) and after pre-treatment with 1µM RWJ 67657 (grey line). The mean fluorescence intensity (MFI) is depicted in the graph: first line cytokine stimulated expression, second line cytokine + RWJ 67657 treated expression. In brackets the percentage of bright positive cells is indicated. The thin black line represents the isotype control.
Effects of RWJ 67657 on chemokine and cytokine mRNA expression and production Quantitative mRNA expression of IL-8, MCP-1 and IL-6 was determined in HUVEC (n=4-6) after 6 and 24 hours of stimulation with 10 ng/ml TNF-α or IL-1β in the presence or absence of RWJ 67657. Stimulation with IL-1β induced higher mRNA expression of both IL-8 and IL-6 compared to stimulation with TNF-α (figure 4A). At both time points a marked inhibition of IL-8 and IL-6 mRNA expression was seen due to pre-treatment with 1 µM RWJ 67657. This inhibition was statistically significant in

Chapter 6
82
nearly all cases. MCP-1 mRNA expression was induced equally by TNF-α and IL-1β, and decreased in time. The inhibition seen by the p38 MAPK inhibitor was not statistically significant. The production of IL-8, MCP-1 and IL-6 protein by HUVEC after 24 hours of TNF-α or IL-1β stimulation is shown in figure 4B. Protein production was induced at high levels, especially IL-8 and MCP-1. Pre-treatment with 1 µM RWJ 67657 induced a significant reduction of IL-8, MCP-1 an IL-6 production. No effect of 0.01% DMSO on cytokine production was observed (data not shown).
Figure 4. (A). Effect of RWJ 67657 pre-treatment on mRNA expression of IL-8, MCP-1 and IL-6 in HUVEC. Cells were stimulated with TNF-α and IL-1β for 6 or 24 hours in the absence or presence of 1µM RWJ 67657 pre-treatment for 1 hour. mRNA expression was determined with real-time RT-PCR. Results are expressed as fold induction compared to unstimulated cells (fold induction=1). Bars show means (n=4-6) and SEM. (* p< 0.05, ** p<0.001, paired T-test, calculated against the stimulated control). (B). Effects of RWJ 67657 pre-treatment on protein production of IL-8, MCP-1 and IL-6 by HUVEC (n=5). Cells were stimulated with TNF-α or IL-1β for 24 hours and pre-treated with 1µM RWJ 67657 (t=-1hr). Protein production was measured in supernatants by ELISA and expressed in ng/ml. Bars show mean and SEM. (* p< 0.05, ** p<0.001, paired T-test, calculated against the stimulated control).
A B

p38 MAPK inhibition in EC
83
DISCUSSION The effects of RWJ 67657, a p38 MAPK inhibitior, on TNF-α and IL-1β stimulated endothelial cells were investigated in this study. Complete inhibition of MAPKAPK-2 phosphorylation was demonstrated at 1 µM. At this concentration we found inhibition of E-selectin expression, both at the level of mRNA and protein production. A significant inhibition of production of the chemokines IL-8 and MCP-1 was found, and also production of the pro-inflammatory cytokine IL-6 was significant inhibited. In inflammatory diseases the accumulation of leukocytes in a given tissue can lead to varying degrees of cell damage, extra cellular matrix disruption and organ dysfunction. Several attempts have been made to therapeutically block leukocyte adhesion to endothelium and thus control inflammation. This has been done for instance by the use of specific monoclonal antibodies to adhesion molecules in animal models 18, and also in RA patients 19. Also currently used anti-rheumatic agents, for example metho-trexate, glucocorticosteroids and leflunomide interfere to varying degree with the expression or function of different CAMs 20;21. Therapeutic strategies aimed at blocking chemokines and their receptors have also been studied. Recently Haringman et al reported relevant biological effects in RA patients treated with chemokine receptor 1 antagonist 22. In rheumatoid synovial tissue p38 MAPK is predominantly expressed in the lining layer and in endothelial cells 9 and therefore we wanted to investigate the effects of the p38 MAPK inhibitor on adhesion molecule expression and chemokine production. In our study we found a complete inhibition of phosphorylation of MAPKAPK-2, the direct downstream substrate of p38 MAPK at a concentration of 1µM RWJ 67567. The study by Parasrampuria demonstrated that after a single oral dose ranging from 0.25 to 30 mg/kg a plasma concentration of 0.01 to 6 µM of the p38 MAPK inhibitor could be reached in humans 14. We therefore decided to perform our study with 1 µM RWJ 67567, which equals a dose of 5-10 mg/kg. We did not compare our p38 MAPK inhibitor with the literature standard SB 203580 for two reasons: first, RWJ 67657 is already described to be 10-fold more potent than SB 203580 in all p38-dependent systems tested 11. Secondly, it has been demonstrated that SB 203580 can also block protein kinase B (PKB) activity at 3-5 µM 23, as well as JNK activities at 3-10 µM 24. Therefore SB 203580 is now considered not to be a specific p38 MAPK inhibitor. Different subsets of leukocytes use different (combinations of) cell adhesion molecules (CAMs) depending on the inflammatory stimulus and the site of inflammation. Furthermore, the functional consequences of downregulating cell adhesion molecule expression on the endothelial surface can be at the level of leukocyte rolling, adhesion, and transmigration. McCafferty 25 reported that in postcapillary venules (studied in the cremaster muscle venules, size ~30 µm in diameter) E-selectin knock out could completely abrogate antigen challenge induced leukocyte recruitment: leukocyte rolling as well as adhesion and transmigration were completely abolished in the E/P-selectin k.o. mice, while in the P-selectin k.o. mice only leukocyte rolling was affected. In contrast, leukocyte recruitment induced by local TNF-α administration was neither affected in P-selectin k.o. nor in P-/E-selectin k.o.. In both the antigen induced and TNF-α induced inflammation, the number of cells infiltrating and the cell types making up the infiltrate were similar. The main difference between the two models was the expression of VCAM-1 in the venular endothelium of the TNF-α

Chapter 6
84
induced inflammation, indicating that VCAM-1 is possibly able to control leukocyte recruitment to sites of inflammation, irrespective whether P- or E-selectin is present. Norman and colleagues recently reported on the role of CAMs during immune complex-dependent inflammation in the mouse cremaster muscle 26. While E-selectin inhibition by antibody infusion could abrogate leukocyte rolling on the venular endothelium (venules were 25-40 µm in diameter), its effects on leukocyte migration were limited. In contrast, treatment with anti-VCAM-1 antibody could inhibit leukocyte adhesion more than 70%, with subsequent functional consequence being a more than 80% inhibition of leukocyte migration. In the current study we showed that pre-treatment of HUVEC with RWJ 67657 inhibited E-selectin expression to 77%/59% (TNF-α induced, 6 hrs resp. 24 hrs) and 81%/67% (IL-1β induced, 6 hrs resp. 24 hrs). Moreover, we showed that VCAM-1 protein expression was inhibited at the 6 hours time point, 66 % (TNF-α induced) versus 73 % (IL-1β induced), while also ICAM-1 was inhibited at 6 hours after start TNF-α activation. Reports on p38 MAPK inhibition on ICAM-1 expression after TNF-α stimulation in literature are contradicting 27;28, whereas results after IL-1β stimulation have not been reported. Our results concerning VCAM-1 mRNA and protein expression corroborate the data by Pietersma et al 29, who stated that p38 MAPK regulates endothelial VCAM-1 expression at the post-transcriptional level. Extensive in vivo studies will be needed to answer the question whether the anti-inflammatory effects in the ranges reported here will affect leukocyte rolling, adhesion, and subsequent recruitment to the diseased tissue. The fact that at 1 µM RWJ 67657 phosphorylation of MAPKAPK-2 was completely blocked, while adhesion molecule expression was partly inhibited, indicates that other signal transduction routes are also important in regulating adhesion molecule expression in HUVEC. Activation through p38 MAPK and NF-κB have been described to act synergistically in inducing adhesion molecule expression in endothelial cells 7;8. Recently Viemann et al 30 reported that the induction of cell-surface receptor expression measured with oligonucleotide microarray was highly depended on IKK2/NF-κB activation , whereas there was additional modulation by p38 MAPK. With real-time RT-PCR they found that for instance VCAM-1 and IL-8 were dependent of both pathways, where as ICAM-1 expression was dependent of NF-κB activation alone. So both pathways play a role in endothelial activation, with CAM expression being more regulated by the NF-κB pathway, and chemokine production more by the p38 MAPK pathway. In our study we demonstrated that p38 MAPK inhibition affects E-selectin expression both at the mRNA and the protein level in HUVEC. The study by Fijen et al demonstrated that RWJ 67657 at a single dose of 5, 10 and 20 mg/kg prevented endotoxin-induced increase of circulating ICAM-1 and circulating E-selectin and also of integrins on neutrophils 13. They demonstrated that also in vivo neutrophil and endothelial activation could be prevented by RWJ 67657. With quantitative real-time RT-PCR we found that induction of IL-8 and IL-6 mRNA was higher after stimulation with IL-1β than with TNF-α, and a significant inhibition by RWJ 67657 was seen after both stimuli. In our study RWJ 67657 was added 1 hour before stimulation of the cells, but also administration of the compound up till 4 hours after stimulation of HUVEC induced marked reduction in IL-8 and IL-6 mRNA levels

p38 MAPK inhibition in EC
85
(unpublished results). MCP-1 mRNA and protein production was equally high after both stimuli. A significant inhibitory effect was demonstrated by RWJ 67657 treatment for IL-8, MCP-1 and IL-6 protein production. These observations corroborate data reported by others 31;32. Since chemokines play an essential role in maintaining the leukocyte-endothelial interactions after the initial interaction regulated by the selectins, the significant downregulation of IL-8 and MCP-1 could have an important effect on leukocyte infiltration in inflammatory disease. Previously we demonstrated significant inhibitory effects on inflammatory mediators produced by rheumatoid synovial fibroblasts 12 and we also showed significant inhibition of TNF-α production in monocyte-derived macrophages 33. p38 MAPK inhibitors have effects on different cell types, which could enhance the therapeutic effects, but also enlarge the risk of side effects. One of the reasons for undesirable effects might be the cross-reactivity against other kinases, which was not the case for RWJ 67657 11. Moreover, RWJ 67657 has been shown to have acceptable safety and acceptable pharmacokinetics to warrant further investigation 14. p38 MAPK inhibitors like RWJ 67657 have shown to effectively inhibit pro-inflammatory mediators in different cells in vitro, and also in vivo. Whether kinase inhibitors will have an important role in the treatment of RA will be dependent of safety issues, and will be investigated in the near future. ACKNOWLEDGEMENTS Annelie Rawee, Henk Moorlag and Berber Doornbos-van der Meer are gratefully acknowledged for excellent technical assistance. Supported by the Dutch Rheumatology Foundation, Johnson and Johnson Pharmaceutical Research and Development, Raritan, NJ, and the University Medical Center Groningen, The Netherlands.
REFERENCES
1 Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. N.Engl.J.Med. 2001; 344: 907-16.
2 Szekanecz Z, Koch AE. Cell-cell interactions in synovitis. Endothelial cells and immune cell migration. Arthritis Res. 2000; 2: 368-73.
3 von Andrian UH, Mackay CR. T-cell function and migration. Two sides of the same coin. N.Engl.J.Med. 2000; 343: 1020-34.
4 Gonzalez-Amaro R, Diaz-Gonzalez F, Sanchez-Madrid F. Adhesion molecules in inflammatory diseases. Drugs 1998; 56: 977-88.
5 Redlich K, Schett G, Steiner G, Hayer S, Wagner EF, Smolen JS. Rheumatoid arthritis therapy after tumor necrosis factor and interleukin-1 blockade. Arthritis Rheum. 2003; 48: 3308-19.
6 Smith MD, Slavotinek J, Au V et al. Successful treatment of rheumatoid arthritis is associated with a reduction in synovial membrane cytokines and cell adhesion molecule expression. Rheumatology.(Oxford) 2001; 40: 965-77.
7 Jersmann HP, Hii CS, Ferrante JV, Ferrante A. Bacterial lipopolysaccharide and tumor necrosis factor alpha synergistically increase expression of human endothelial adhesion molecules through activation of NF-kappaB and p38 mitogen-activated protein kinase signaling pathways. Infect.Immun. 2001; 69: 1273-9.

Chapter 6
86
8 Read MA, Whitley MZ, Gupta S et al. Tumor necrosis factor alpha-induced E-selectin expression is activated by the nuclear factor-kappaB and c-JUN N-terminal kinase/p38 mitogen-activated protein kinase pathways. J.Biol.Chem. 1997; 272: 2753-61.
9 Schett G, Tohidast-Akrad M, Smolen JS et al. Activation, differential localization, and regulation of the stress- activated protein kinases, extracellular signal-regulated kinase, c-JUN N-terminal kinase, and p38 mitogen-activated protein kinase, in synovial tissue and cells in rheumatoid arthritis. Arthritis Rheum. 2000; 43: 2501-12.
10 Kumar S, Boehm J, Lee JC. p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat.Rev.Drug Discov. 2003; 2: 717-26.
11 Wadsworth SA, Cavender DE, Beers SA et al. RWJ 67657, a potent, orally active inhibitor of p38 mitogen-activated protein kinase. J.Pharmacol.Exp.Ther. 1999; 291: 680-7.
12 Westra J, Limburg PC, de Boer P, van Rijswijk MH. Effects of RWJ 67657, a p38 mitogen activated protein kinase (MAPK) inhibitor, on the production of inflammatory mediators by rheumatoid synovial fibroblasts. Ann.Rheum.Dis. 2004; 63: 1453-9.
13 Fijen JW, Tulleken JE, Kobold AC et al. Inhibition of p38 mitogen-activated protein kinase: dose-dependent suppression of leukocyte and endothelial response after endotoxin challenge in humans. Crit Care Med. 2002; 30: 841-5.
14 Parasrampuria DA, de Boer P, Desai-Krieger D, Chow AT, Jones CR. Single-dose pharmacokinetics and pharmacodynamics of RWJ 67657, a specific p38 mitogen-activated protein kinase inhibitor: a first-in-human study. J.Clin.Pharmacol. 2003; 43: 406-13.
15 Mulder AB, Blom NR, Smit JW et al. Basal tissue factor expression in endothelial cell cultures is caused by contaminating smooth muscle cells. Reduction by using chymotrypsin instead of collagenase. Thromb.Res. 1995; 80: 399-411.
16 Maciag T, Hoover GA, Weinstein R. High and low molecular weight forms of endothelial cell growth factor. J.Biol.Chem. 1982; 257: 5333-6.
17 van Leeuwen MA, Westra J, Limburg PC, van Riel PL, van Rijswijk MH. Interleukin-6 in relation to other proinflammatory cytokines, chemotactic activity and neutrophil activation in rheumatoid synovial fluid. Ann.Rheum.Dis. 1995; 54: 33-8.
18 Issekutz AC, Ayer L, Miyasaka M, Issekutz TB. Treatment of established adjuvant arthritis in rats with monoclonal antibody to CD18 and very late activation antigen-4 integrins suppresses neutrophil and T-lymphocyte migration to the joints and improves clinical disease. Immunology 1996; 88: 569-76.
19 Kavanaugh AF, Schulze-Koops H, Davis LS, Lipsky PE. Repeat treatment of rheumatoid arthritis patients with a murine anti-intercellular adhesion molecule 1 monoclonal antibody. Arthritis Rheum. 1997; 40: 849-53.
20 Dimitrijevic M, Bartlett RR. Leflunomide, a novel immunomodulating drug, inhibits homotypic adhesion of mononuclear cells in rheumatoid arthritis. Transplant.Proc. 1996; 28: 3086-7.
21 Wheller SK, Perretti M. Dexamethasone inhibits cytokine-induced intercellular adhesion molecule-1 up-regulation on endothelial cell lines. Eur.J.Pharmacol. 1997; 331: 65-71.
22 Haringman JJ, Kraan MC, Smeets TJ, Zwinderman KH, Tak PP. Chemokine blockade and chronic inflammatory disease: proof of concept in patients with rheumatoid arthritis. Ann.Rheum.Dis. 2003; 62: 715-21.
23 Lali FV, Hunt AE, Turner SJ, Foxwell BM. The pyridinyl imidazole inhibitor SB203580 blocks phosphoinositide-dependent protein kinase activity, protein kinase B phosphorylation, and retinoblastoma hyperphosphorylation in interleukin-2-stimulated T cells independently of p38 mitogen-activated protein kinase. J.Biol.Chem. 2000; 275: 7395-402.
24 Clerk A, Sugden PH. The p38-MAPK inhibitor, SB203580, inhibits cardiac stress-activated protein kinases/c-Jun N-terminal kinases (SAPKs/JNKs). FEBS Lett. 1998; 426: 93-6.
25 McCafferty DM, Kanwar S, Granger DN, Kubes P. E/P-selectin-deficient mice: an optimal mutation for abrogating antigen but not tumor necrosis factor-alpha-induced immune responses. Eur.J.Immunol. 2000; 30: 2362-71.
26 Norman MU, Van De Velde NC, Timoshanko JR, Issekutz A, Hickey MJ. Overlapping roles of endothelial selectins and vascular cell adhesion molecule-1 in immune complex-induced leukocyte recruitment in the cremasteric microvasculature. Am.J.Pathol. 2003; 163: 1491-503.

p38 MAPK inhibition in EC
87
27 Fichtner F, Koslowski R, Augstein A, Hempel U, Rohlecke C, Kasper M. Bleomycin induces IL-8 and ICAM-1 expression in microvascular pulmonary endothelial cells. Exp.Toxicol.Pathol. 2004; 55: 497-503.
28 Ju H, Behm DJ, Nerurkar S et al. p38 MAPK inhibitors ameliorate target organ damage in hypertension: Part 1. p38 MAPK-dependent endothelial dysfunction and hypertension. J.Pharmacol.Exp.Ther. 2003; 307: 932-8.
29 Pietersma A, Tilly BC, Gaestel M et al. p38 mitogen activated protein kinase regulates endothelial VCAM-1 expression at the post-transcriptional level. Biochem.Biophys.Res.Commun. 1997; 230: 44-8.
30 Viemann D, Goebeler M, Schmid S et al. Transcriptional profiling of IKK2/NF-{kappa}B- and p38 MAP kinase-dependent gene expression in TNF-{alpha}-stimulated primary human endothelial cells. Blood 2004.
31 Goebeler M, Gillitzer R, Kilian K et al. Multiple signaling pathways regulate NF-kappaB-dependent transcription of the monocyte chemoattractant protein-1 gene in primary endothelial cells. Blood 2001; 97: 46-55.
32 Marin V, Farnarier C, Gres S et al. The p38 mitogen-activated protein kinase pathway plays a critical role in thrombin-induced endothelial chemokine production and leukocyte recruitment. Blood 2001; 98: 667-73.
33 Westra J, Doornbos-Van Der Meer B, de Boer P, van Leeuwen MA, van Rijswijk MH, Limburg PC. Strong inhibition of TNF-alpha production and inhibition of IL-8 and COX-2 mRNA expression in monocyte-derived macrophages by RWJ 67657, a p38 mitogen-activated protein kinase (MAPK) inhibitor. Arthritis Res.Ther. 2004; 6: R384-R392.

6
Chemokine production and E-selectin expression in
activated endothelial cells are inhibited by p38 MAPK
(mitogen activated protein kinase) inhibitor RWJ 67657
Johanna Westra1
Joanna M Kułdo2
Martin H van Rijswijk1
Grietje Molema2
Pieter C Limburg1
From the Departments of 1 Rheumatology, and 2 Medical Biology section of Pathology
and Laboratory Medicine, University Medical Center Groningen, The Netherlands
International Immunopharmacology: in press

Chapter 6
74
ABSTRACT Endothelial cells play an important role in inflammatory diseases like rheumatoid arthritis by recruitment of inflammatory cells. The cytokines TNF-α and IL-1β are major inducers of endothelial cell activation and are stimulators of inflammatory signal transduction pathway involving p38 MAPK (mitogen-activated protein kinase). The present study investigated the effects of p38 MAPK inhibition on cell adhesion molecule (CAM) expression and chemokine production by endothelial cells both on mRNA and protein level. Pre-treatment of endothelial cells with the pharmacologically relevant concentration of 1 µM of the p38 MAPK inhibitor RWJ 67657 reduced TNF-α and IL-1β induced mRNA and membrane expression of E-selectin. Moderate inhibitory effects on ICAM-1 and VCAM-1 expression were found. Significant reduction of mRNA expression and protein production of the inflammatory cytokine IL-6 and the chemokines IL-8 and MCP-1 was demonstrated. Treatment with RWJ 67657 could lead to reduced leukocyte infiltration by the reduction of E-selectin expression and chemokine production. Key words. chemokine, endothelial cell, E-Selectin, MAPK inhibitor

p38 MAPK inhibition in EC
75
INTRODUCTION Rheumatoid arthritis (RA) is an inflammatory disease characterized by hyperplasia, increased vascularity, and infiltration of inflammatory cells into the synovial membrane 1. It is now known that the endothelial cells (EC), which line the lumen of the blood vessels, are not passive bystanders but are active responders to stimuli like activated leukocytes and cytokines 2. After stimulation EC can produce a number of inflammatory mediators and express cellular adhesion molecules (CAMs). Most CAMs involved in endothelial activation belong to three families, i.e., the integrins, selectins, and immunoglobulin superfamilies 3. The adhesion of leukocytes to EC takes place in four steps. The first step is a weak adhesion (tethering and rolling) mediated by E-, P- and L-selectins. The next step is leukocyte activation and is a consequence of interaction between chemokines and their receptors on leukocytes. Then firm adhesion takes place, which is mediated mostly by interaction between integrins on the leukocytes and vascular adhesion molecule (VCAM)-1 and intercellular adhesion molecule (ICAM)-1 on the EC. The final step is the transendothelial migration, which is directed by secreted chemokines bound to endothelial heparan sulphate glycosaminoglycans. Essential chemokines are interleukin (IL)-8 which attracts neutrophils, and monocyte chemo-attractant protein (MCP)-1, the main attractant for mononuclear cells 2;3. The cytokines TNF (tumour necrosis factor)-α and IL-1β, produced by activated macrophages in the synovium, have been identified as the major inducers of EC activation in RA 4. Blockade of these cytokines has proven to be effective in the treatment of RA, although Redlich et al recently concluded that targeting individual molecules such as TNF-α may not be sufficient in interfering with both inflammation and joint destruction 5. Successful drug treatment of RA is associated by a decrease in cytokine production, but also by a decrease in E-selectin and ICAM-1 expression 6. In vitro studies demonstrated that in TNF-α and/or lipopolysaccharide (LPS) stimulated human umbilical vein endothelial cells (HUVEC) the nuclear factor-κB (NF-κB), and p38 mitogen-activated protein kinase (MAPK) pathways are important in controlling adhesion molecule expression 7;8. Activation of all three stress- and mitogen-activated protein kinases (SAPK/MAPK) was found throughout the RA synovial tissue, whereas activated p38 MAPK predominantly was located in the synovial lining layer and in synovial endothelial cells 9. Interest in protein kinases and in particular in p38 MAPK as drug targets has increased in the last years as recently reviewed by Kumar et al 10. The p38 MAPK inhibitor RWJ 67657 (4-[4-(4-fluorophenyl)-1-(3-phenylpropyl)-5-(4-pyridinyl)-1H-imidazol-2-yl]-3-butyn-1-ol) has been shown to be effective in inhibiting the release of TNF-α from LPS-treated human peripheral blood mononuclear cells with an IC50 of 3 nM 11. This compound was approximately 10-fold more potent than the reference standard p38 MAPK inhibitor SB 203580 in all p38 dependent in vitro systems tested. RWJ 67657 specifically inhibited the enzymatic activity of recombinant p38 α and β, but not of γ and δ in vitro, and had no significant activity against a variety of other kinases 11. We recently demonstrated that RWJ 67657 significantly inhibited IL-6, IL-8, MMP-3 and COX-2 mRNA expression by IL-1β and TNF-α stimulated rheumatoid synovial fibroblasts 12. Furthermore Fijen et al showed that this compound induced a dose-dependent suppression of leukocyte and endothelial cell response after endotoxin challenge in

Chapter 6
76
humans 13. Recent pharmaco-kinetic and pharmaco-dynamic studies of RWJ 67657 in humans showed that the compound has an acceptable safety profile to warrant further investigations 14. In the present study the effects of p38 MAPK inhibition in IL-1β or TNF-α stimulated HUVEC concerning adhesion molecule expression and chemokine production were studied at the level of mRNA expression as well as protein production. Pre-treatment of activated endothelial cells with 1 µM RWJ 67657 reduced E-selectin mRNA and protein expression, and significantly reduced IL-8, MCP-1 and IL-6 production and mRNA expression. These results indicate that treatment with RWJ 67657 could lead to reduced leukocyte infiltration and therefore could have an important therapeutic benefit. MATERIALS AND METHODS Reagents RWJ 67657 was provided by Johnson and Johnson (R.W. Johnson Pharmaceutical Research Institute, Raritan, NJ). Recombinant TNF-α and IL-1β and ELISA antibodies for the detection of IL-8 and MCP-1 were obtained from R&D Systems (Minneapolis, MN). Antibodies for the detection of IL-6 were purchased from Sanquin (Amsterdam, The Netherlands). Specific antibodies to p38 MAPK, phospho-p38 MAPK and phospho-MAPK activated protein kinase (MAPKAPK)-2 were purchased from Cell Signalling Technologies (Beverly, MA) and detecting antibody peroxidase -swine-anti-rabbit was from DAKO (Glostrup, Denmark). Mouse anti-human E-selectin antibody (H18/7) was kindly provided by Dr. M. Gimbrone Jr., Harvard Medical School. Monoclonal anti-human VCAM-1 (CD106) and anti-human ICAM-1 (CD54), both labelled with phycoerytrin (PE) were from Beckton Dickinson (Bedford, MA). Monoclonal anti-human PECAM-1 (platelet endothelial cell adhesion molecule, CD31) was obtained from DAKO and PE-labeled goat-anti-mouse was from SBA (Southern Biotechnology Associates, Birmingham, AL). RNA isolation kit was from Stratagene (La Jolla, CA), quantitation of RNA was performed with Ribogreen RNA quantitation kit (Molecular Probes Europe BV, Leiden, The Netherlands). Other reagents for RNA isolation and reverse transcriptase reaction were purchased from Invitrogen (Breda, The Netherlands). Reagents, primers and probes for real-time RT-PCR were obtained from Applied Biosystems (Nieuwerkerk a/d IJssel, The Netherlands). Endothelial cell culture and stimulation HUVEC were obtained from the Endothelial Cell Facility at the UMCG (The Netherlands) as described previously 15. Primary isolates combined from 2 or 3 donors were cultured on 1% gelatin (Sigma-Aldrich, Zwijndrecht, The Netherlands) -precoated plastic tissue culture plates or flasks (Corning, Schiphol, The Netherlands) at 37º C under 5% CO2/95% air. The culture medium constisted of RPMI 1640 (Biowhittaker, Verviers, Belgium) supplemented with 20% heat-inactivated fetal calf serum (FCS), 2 mM L-glutamine, 5 U/ml heparin, 100 U/ml penicillin, 100 µg/ml streptomycin and 20 µg/ml endothelial cell growth factor, extracted from bovine brain according to the procedure described by Maciag 16. After attaining confluence, cells

p38 MAPK inhibition in EC
77
were detached from the surface by trypsin/EDTA (0.5/0.2 mg/ml) and split at a 1:3 ratio. In the experiments presented here HUVEC were used up to passage 4. In the experimental set-up HUVEC were grown to confluence in gelatin-coated 6-well plates. Fresh medium was added before cells were stimulated for 6 or 24 hours with 10 ng/ml TNF-α or IL-1β, with or 1 hour without pre-treatment with 1µM RWJ 67657 (stock solution 10 mM in DMSO = dimethylsulfoxide). Phosphorylation of p38 MAPK and MAPKAPK-2 in HUVEC HUVEC were pre-treated with 0, 0.1, 1, and 10 µM RWJ 67657 for 1 hour and stimulated for 30 minutes as mentioned above. Cell extracts were prepared by lysing the cells with 1X SDS sample buffer (containing 2% SDS, 10% glycerol, 50 mM dithiothreitol, 62.5 mM Tris-HCl (pH=6.8) and 0.01% brome-phenol blue). Cells were scraped off the wells and the lysates were subsequently sonicated for 5-10 seconds and boiled for 5 minutes. After centrifugation the samples were loaded onto a 10% SDS-PAGE gel and resolved by running at 200 V and 15 Watt constant. Semidry-blotting was performed onto a nitrocellulose membrane after which immunodetection with anti-p38 MAPK, anti-phospho-p38 MAPK, anti-phospho-MAPKAPK-2 and peroxidase labelled swine-anti-rabbit was performed. Enhanced chemi-luminescence (ECL) detection was performed according to the manufacturers guidelines (Lumi-Lightplus, Roche Diagnostics, Mannheim, Germany). RNA isolation and real-time RT-PCR HUVEC with or without 1 hour pre-treatment with 1µM RWJ 67657 were stimulated for 6 and 24 hours with 10 ng/ml TNF-α or IL-1β. Total RNA was isolated using the Absolutely RNA Microprep Kit according to the manufacturers guidelines. RNA was analysed qualitatively by gel electrophoresis and quantitatively by Ribogreen RNA Quantitation Kit. One microgram of total RNA was used for the synthesis of first strand cDNA using Superscript III RNase H- Transcriptase in 20 µl final volume containing 250 ng random hexamers and 40 units Rnase OUT inhibitor. For the measurement of mRNA for CD31, E-selectin, VCAM-1, ICAM-1, IL-8, MCP-1, IL-6 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) 1 µl of cDNA in triplicate was used for amplification by the Taqman real-time PCR system (ABI Prism 7900HT Sequence Detection System, Applied Biosystems) with specific Taqman primers/probes. In our experiments GAPDH and other genes of interest were always determined in the same RT-PCR run. Amplification was performed using standard conditions: denaturation at 95°C for 15 seconds, 40 cycles of amplification with annealing at 60°C for 1 minute, and extension at 50°C for 2 minutes. According to the comparative Ct (threshold cycle value) method described in the ABI manual, the resulting mRNA amount of the gene of interest was normalized to the housekeeping gene GAPDH, yielding the ∆Ct value. The ∆Ct value of unstimulated HUVEC was subtracted from the average ∆Ct value of each sample, yielding the ∆∆Ct. The amount of target, normalized to an endogenous reference (GAPDH) and relative to the control sample, is given by: 2-∆∆CT.

Chapter 6
78
Flow cytometric analysis HUVEC were stimulated for 6 and 24 hours as mentioned above and detached from the wells by short treatment with trypsin, and subsequently resuspend in FCS to neutralize the trypsin. After washing with PBS supplemented with 5% FCS, HUVEC were incubated with PE-labeled monoclonal antibodies against VCAM-1 (dilution 1:10) and ICAM-1 (1:25) for 45 minutes on ice or with monoclonal antibodies against CD31 (1:25) and E-selectin (undiluted) followed by incubation with PE-labeled goat-anti-mouse antibodies. Cells were fixed with 0.5% paraformaldehyde/PBS and adhesion molecule expression was detected by flow cytometric analysis in an Epics-Elite Flow cytometer (Coulter Electronics, Mijdrecht, The Netherlands). Non specific staining was assessed by staining with irrelevant isotype-matched monoclonal antibodies. The effects of RWJ 67657 on adhesion molecule expression was determined in 3 experiments with different HUVEC cultures. ELISA based determination of IL-8, MCP-1 and IL-6 in cell culture supernatants HUVEC were pre-treated with 1 µM RWJ 67657 for 6 and 24 hours and stimulated with 10 ng/ml TNF-α or IL-1β. IL-6 levels in cell supernatants were measured as described previously 17, IL-8 and MCP-1 were measured by ELISA, using matched antibody pairs for ELISA and recombinant proteins as standards from R&D Systems. In short, Corning high-binding ELISA plates were coated overnight with monoclonal antibodies in PBS. After blocking with 2% BSA (bovine serum albumin)/PBS diluted supernatants were added. Bound chemokines were detected with biotinylated polyclonal antibodies followed by incubation with peroxidase labelled Streptavidin (Sanquin, Amsterdam, The Netherlands). Colour reaction was performed with TMB (3’3’5’5’tetramethylbenzidin, Roth, Karlsruhe, Germany) and concentration of protein was determined with the SOFTmax PRO software (Molecular Devices, Sunnyvale, CA). Detection limits for ELISAs was 20 pg/ml for IL-6 and IL-8 and 50 pg/ml for MCP-1. STATISTICS Paired T-tests were performed using GraphPad Prism version 3.00 for Windows, GraphPad Software (San Diego, CA). RESULTS Effect of RWJ 67657 on phosphorylation of p38 MAPK and MAPKAPK-2 Upon stimulation with 10 ng/ml TNF-α and/or IL-1β p38 MAPK is rapidly phosphorylated within minutes, while after 60 minutes the level of phosphorylation is decreased (data not shown). In figure 1 the effect of RWJ 67657 on phosphorylation of p38 MAPK and its downstream substrate MAPKAPK-2 after 30 minutes of stimulation is shown. RWJ 67657 does not inhibit phosphorylation of p38 MAPK, but it does inhibit its activity as can be seen from the inhibition of MAPKAPK-2 phosphorylation at 0.1 µM. Complete inhibition was demonstrated at a concentration of 1 µM RWJ 67657. The solvent 0.01% DMSO did not affect phosphorylation of either kinase.

p38 MAPK inhibition in EC
79
Figure 1. Phosphorylation of p38 MAPK and MAPKAPK-2 in stimulated HUVEC after pre-treatment with 10, 1 and 0.1 µM RWJ 67657. HUVEC were treated with TNF-α and/or IL-1β for 30 minutes with or without 1 hour pre-treatment with RWJ 67657. Phosphorylation was measured by Western blot using specific antibodies against p38 MAPK, phospho-p38 MAPK and its direct downstream substrate MAPKAPK-2. Control incubations were performed with 0.01% dmso, the vehicle solvent for RWJ 67657.
Figure 2. Effect of RWJ 67657 pre-treatment on mRNA expression of CD31, E-selectin, VCAM-1 and ICAM-1 in HUVEC. Cells were stimulated with TNF-α and/or IL-1β for 6 or 24 hours and pre-treated with 1µM RWJ 67657 for 1 hour. mRNA expression was determined with real-time RT-PCR and results are expressed as fold induction compared to unstimulated cells (fold induction=1). Bars show means (n=4-6) and SEM. (* p< 0.05, paired T-test calculated against the stimulated control).

Chapter 6
80
Effects of RWJ 67657 on mRNA expression of adhesion molecules With real-time RT-PCR mRNA expression of CD31, E-selectin, VCAM-1 and ICAM-1 was determined in HUVEC (n=4-6) after 6 and 24 hours of stimulation with 10 ng/ml TNF-α or IL-1β. The expression is measured as fold induction compared to the unstimulated sample. As can be seen from figure 2 mRNA expression of CD31 did not change during stimulation, in contrast to the strong induction of mRNA expression for E-selectin, VCAM-1 and ICAM-1. After 6 hours of stimulation the mRNA expression of these adhesion molecules was higher than after 24 hours of stimulation. Furthermore, IL-1β induced E-selectin mRNA expression to a higher extent than did TNF-α. Pre-treatment with 1 µM RWJ 67657 did not influence the expression of CD31 nor that of the induced VCAM-1 expression. E-selectin expression however was reduced to 78% and 65% respectively after TNF-α or IL-1β stimulation at 6 hours, and to 43% and 35% respectively after 24 hours of stimulation. ICAM-1 mRNA expression at 24 hours was reduced to 73% after TNF-α stimulation and 56% after IL-1β stimulation. Effects of RWJ 67657 on membrane expression of adhesion molecules Membrane adhesion molecule expression by HUVEC was determined at 6 and 24 hours after stimulation with 10 ng/ml TNF-α or IL-1β. E-selectin expression was higher after stimulation with IL-1β than with TNF-α. This was also the case for VCAM-1 expression except at 24 hours when TNF-α-induced expression was higher. Both cytokines induced high ICAM-1 expression, which increased with time (data not shown). The effect of pre-treatment with 1 µM RWJ 67657 on adhesion molecule expression was calculated as follows: the MFI (mean fluorescence intensity) of stimulated cells for each experiment was set to 100%, and the expression due to the treatment with RWJ 67657 was calculated relative to the 100% (Table 1). Pre-treatment with RWJ 67657 led to reduced expression of E-selectin under all conditions tested, of VCAM-1 after 6 hours, and of ICAM-1 after TNF-α stimulation for 6 hours (all not statistically significant). Table 1. Effect of RWJ pre-treatment on adhesion molecule expression by HUVEC (n=3). Adhesion molecule expression on TNF-α and IL-1β stimulated HUVEC was determined 6 and 24 hours after stimulation. The effect of 1 hour pre-treatment with 1µM RWJ 67657 was calculated as percentage compared to the MFI of stimulated cells (=100%).
TNF-α IL-1β 6 hr 24 hr 6 hr 24 hr % % % %
CD31 104 +/- 13 95 +/- 20 91 +/- 6 89 +/- 17
E-selectin 77 +/- 22 59 +/- 19 81 +/- 14 67 +/- 9
VCAM-1 66 +/- 15 105 +/- 37 73 +/- 17 108 +/- 18
ICAM-1 79 +/- 15 99 +/- 29 100 +/- 21 93 +/- 21
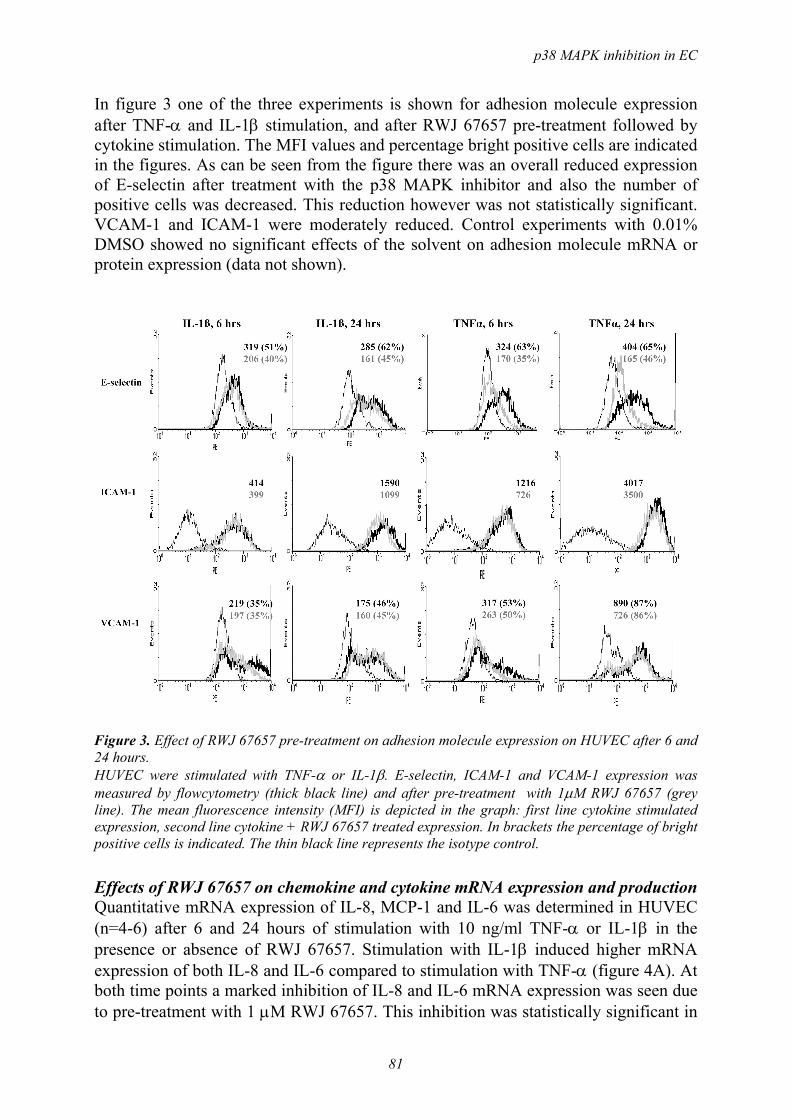
p38 MAPK inhibition in EC
81
In figure 3 one of the three experiments is shown for adhesion molecule expression after TNF-α and IL-1β stimulation, and after RWJ 67657 pre-treatment followed by cytokine stimulation. The MFI values and percentage bright positive cells are indicated in the figures. As can be seen from the figure there was an overall reduced expression of E-selectin after treatment with the p38 MAPK inhibitor and also the number of positive cells was decreased. This reduction however was not statistically significant. VCAM-1 and ICAM-1 were moderately reduced. Control experiments with 0.01% DMSO showed no significant effects of the solvent on adhesion molecule mRNA or protein expression (data not shown).
Figure 3. Effect of RWJ 67657 pre-treatment on adhesion molecule expression on HUVEC after 6 and 24 hours. HUVEC were stimulated with TNF-α or IL-1β. E-selectin, ICAM-1 and VCAM-1 expression was measured by flowcytometry (thick black line) and after pre-treatment with 1µM RWJ 67657 (grey line). The mean fluorescence intensity (MFI) is depicted in the graph: first line cytokine stimulated expression, second line cytokine + RWJ 67657 treated expression. In brackets the percentage of bright positive cells is indicated. The thin black line represents the isotype control.
Effects of RWJ 67657 on chemokine and cytokine mRNA expression and production Quantitative mRNA expression of IL-8, MCP-1 and IL-6 was determined in HUVEC (n=4-6) after 6 and 24 hours of stimulation with 10 ng/ml TNF-α or IL-1β in the presence or absence of RWJ 67657. Stimulation with IL-1β induced higher mRNA expression of both IL-8 and IL-6 compared to stimulation with TNF-α (figure 4A). At both time points a marked inhibition of IL-8 and IL-6 mRNA expression was seen due to pre-treatment with 1 µM RWJ 67657. This inhibition was statistically significant in

Chapter 6
82
nearly all cases. MCP-1 mRNA expression was induced equally by TNF-α and IL-1β, and decreased in time. The inhibition seen by the p38 MAPK inhibitor was not statistically significant. The production of IL-8, MCP-1 and IL-6 protein by HUVEC after 24 hours of TNF-α or IL-1β stimulation is shown in figure 4B. Protein production was induced at high levels, especially IL-8 and MCP-1. Pre-treatment with 1 µM RWJ 67657 induced a significant reduction of IL-8, MCP-1 an IL-6 production. No effect of 0.01% DMSO on cytokine production was observed (data not shown).
Figure 4. (A). Effect of RWJ 67657 pre-treatment on mRNA expression of IL-8, MCP-1 and IL-6 in HUVEC. Cells were stimulated with TNF-α and IL-1β for 6 or 24 hours in the absence or presence of 1µM RWJ 67657 pre-treatment for 1 hour. mRNA expression was determined with real-time RT-PCR. Results are expressed as fold induction compared to unstimulated cells (fold induction=1). Bars show means (n=4-6) and SEM. (* p< 0.05, ** p<0.001, paired T-test, calculated against the stimulated control). (B). Effects of RWJ 67657 pre-treatment on protein production of IL-8, MCP-1 and IL-6 by HUVEC (n=5). Cells were stimulated with TNF-α or IL-1β for 24 hours and pre-treated with 1µM RWJ 67657 (t=-1hr). Protein production was measured in supernatants by ELISA and expressed in ng/ml. Bars show mean and SEM. (* p< 0.05, ** p<0.001, paired T-test, calculated against the stimulated control).
A B

p38 MAPK inhibition in EC
83
DISCUSSION The effects of RWJ 67657, a p38 MAPK inhibitior, on TNF-α and IL-1β stimulated endothelial cells were investigated in this study. Complete inhibition of MAPKAPK-2 phosphorylation was demonstrated at 1 µM. At this concentration we found inhibition of E-selectin expression, both at the level of mRNA and protein production. A significant inhibition of production of the chemokines IL-8 and MCP-1 was found, and also production of the pro-inflammatory cytokine IL-6 was significant inhibited. In inflammatory diseases the accumulation of leukocytes in a given tissue can lead to varying degrees of cell damage, extra cellular matrix disruption and organ dysfunction. Several attempts have been made to therapeutically block leukocyte adhesion to endothelium and thus control inflammation. This has been done for instance by the use of specific monoclonal antibodies to adhesion molecules in animal models 18, and also in RA patients 19. Also currently used anti-rheumatic agents, for example metho-trexate, glucocorticosteroids and leflunomide interfere to varying degree with the expression or function of different CAMs 20;21. Therapeutic strategies aimed at blocking chemokines and their receptors have also been studied. Recently Haringman et al reported relevant biological effects in RA patients treated with chemokine receptor 1 antagonist 22. In rheumatoid synovial tissue p38 MAPK is predominantly expressed in the lining layer and in endothelial cells 9 and therefore we wanted to investigate the effects of the p38 MAPK inhibitor on adhesion molecule expression and chemokine production. In our study we found a complete inhibition of phosphorylation of MAPKAPK-2, the direct downstream substrate of p38 MAPK at a concentration of 1µM RWJ 67567. The study by Parasrampuria demonstrated that after a single oral dose ranging from 0.25 to 30 mg/kg a plasma concentration of 0.01 to 6 µM of the p38 MAPK inhibitor could be reached in humans 14. We therefore decided to perform our study with 1 µM RWJ 67567, which equals a dose of 5-10 mg/kg. We did not compare our p38 MAPK inhibitor with the literature standard SB 203580 for two reasons: first, RWJ 67657 is already described to be 10-fold more potent than SB 203580 in all p38-dependent systems tested 11. Secondly, it has been demonstrated that SB 203580 can also block protein kinase B (PKB) activity at 3-5 µM 23, as well as JNK activities at 3-10 µM 24. Therefore SB 203580 is now considered not to be a specific p38 MAPK inhibitor. Different subsets of leukocytes use different (combinations of) cell adhesion molecules (CAMs) depending on the inflammatory stimulus and the site of inflammation. Furthermore, the functional consequences of downregulating cell adhesion molecule expression on the endothelial surface can be at the level of leukocyte rolling, adhesion, and transmigration. McCafferty 25 reported that in postcapillary venules (studied in the cremaster muscle venules, size ~30 µm in diameter) E-selectin knock out could completely abrogate antigen challenge induced leukocyte recruitment: leukocyte rolling as well as adhesion and transmigration were completely abolished in the E/P-selectin k.o. mice, while in the P-selectin k.o. mice only leukocyte rolling was affected. In contrast, leukocyte recruitment induced by local TNF-α administration was neither affected in P-selectin k.o. nor in P-/E-selectin k.o.. In both the antigen induced and TNF-α induced inflammation, the number of cells infiltrating and the cell types making up the infiltrate were similar. The main difference between the two models was the expression of VCAM-1 in the venular endothelium of the TNF-α

Chapter 6
84
induced inflammation, indicating that VCAM-1 is possibly able to control leukocyte recruitment to sites of inflammation, irrespective whether P- or E-selectin is present. Norman and colleagues recently reported on the role of CAMs during immune complex-dependent inflammation in the mouse cremaster muscle 26. While E-selectin inhibition by antibody infusion could abrogate leukocyte rolling on the venular endothelium (venules were 25-40 µm in diameter), its effects on leukocyte migration were limited. In contrast, treatment with anti-VCAM-1 antibody could inhibit leukocyte adhesion more than 70%, with subsequent functional consequence being a more than 80% inhibition of leukocyte migration. In the current study we showed that pre-treatment of HUVEC with RWJ 67657 inhibited E-selectin expression to 77%/59% (TNF-α induced, 6 hrs resp. 24 hrs) and 81%/67% (IL-1β induced, 6 hrs resp. 24 hrs). Moreover, we showed that VCAM-1 protein expression was inhibited at the 6 hours time point, 66 % (TNF-α induced) versus 73 % (IL-1β induced), while also ICAM-1 was inhibited at 6 hours after start TNF-α activation. Reports on p38 MAPK inhibition on ICAM-1 expression after TNF-α stimulation in literature are contradicting 27;28, whereas results after IL-1β stimulation have not been reported. Our results concerning VCAM-1 mRNA and protein expression corroborate the data by Pietersma et al 29, who stated that p38 MAPK regulates endothelial VCAM-1 expression at the post-transcriptional level. Extensive in vivo studies will be needed to answer the question whether the anti-inflammatory effects in the ranges reported here will affect leukocyte rolling, adhesion, and subsequent recruitment to the diseased tissue. The fact that at 1 µM RWJ 67657 phosphorylation of MAPKAPK-2 was completely blocked, while adhesion molecule expression was partly inhibited, indicates that other signal transduction routes are also important in regulating adhesion molecule expression in HUVEC. Activation through p38 MAPK and NF-κB have been described to act synergistically in inducing adhesion molecule expression in endothelial cells 7;8. Recently Viemann et al 30 reported that the induction of cell-surface receptor expression measured with oligonucleotide microarray was highly depended on IKK2/NF-κB activation , whereas there was additional modulation by p38 MAPK. With real-time RT-PCR they found that for instance VCAM-1 and IL-8 were dependent of both pathways, where as ICAM-1 expression was dependent of NF-κB activation alone. So both pathways play a role in endothelial activation, with CAM expression being more regulated by the NF-κB pathway, and chemokine production more by the p38 MAPK pathway. In our study we demonstrated that p38 MAPK inhibition affects E-selectin expression both at the mRNA and the protein level in HUVEC. The study by Fijen et al demonstrated that RWJ 67657 at a single dose of 5, 10 and 20 mg/kg prevented endotoxin-induced increase of circulating ICAM-1 and circulating E-selectin and also of integrins on neutrophils 13. They demonstrated that also in vivo neutrophil and endothelial activation could be prevented by RWJ 67657. With quantitative real-time RT-PCR we found that induction of IL-8 and IL-6 mRNA was higher after stimulation with IL-1β than with TNF-α, and a significant inhibition by RWJ 67657 was seen after both stimuli. In our study RWJ 67657 was added 1 hour before stimulation of the cells, but also administration of the compound up till 4 hours after stimulation of HUVEC induced marked reduction in IL-8 and IL-6 mRNA levels

p38 MAPK inhibition in EC
85
(unpublished results). MCP-1 mRNA and protein production was equally high after both stimuli. A significant inhibitory effect was demonstrated by RWJ 67657 treatment for IL-8, MCP-1 and IL-6 protein production. These observations corroborate data reported by others 31;32. Since chemokines play an essential role in maintaining the leukocyte-endothelial interactions after the initial interaction regulated by the selectins, the significant downregulation of IL-8 and MCP-1 could have an important effect on leukocyte infiltration in inflammatory disease. Previously we demonstrated significant inhibitory effects on inflammatory mediators produced by rheumatoid synovial fibroblasts 12 and we also showed significant inhibition of TNF-α production in monocyte-derived macrophages 33. p38 MAPK inhibitors have effects on different cell types, which could enhance the therapeutic effects, but also enlarge the risk of side effects. One of the reasons for undesirable effects might be the cross-reactivity against other kinases, which was not the case for RWJ 67657 11. Moreover, RWJ 67657 has been shown to have acceptable safety and acceptable pharmacokinetics to warrant further investigation 14. p38 MAPK inhibitors like RWJ 67657 have shown to effectively inhibit pro-inflammatory mediators in different cells in vitro, and also in vivo. Whether kinase inhibitors will have an important role in the treatment of RA will be dependent of safety issues, and will be investigated in the near future. ACKNOWLEDGEMENTS Annelie Rawee, Henk Moorlag and Berber Doornbos-van der Meer are gratefully acknowledged for excellent technical assistance. Supported by the Dutch Rheumatology Foundation, Johnson and Johnson Pharmaceutical Research and Development, Raritan, NJ, and the University Medical Center Groningen, The Netherlands.
REFERENCES
1 Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. N.Engl.J.Med. 2001; 344: 907-16.
2 Szekanecz Z, Koch AE. Cell-cell interactions in synovitis. Endothelial cells and immune cell migration. Arthritis Res. 2000; 2: 368-73.
3 von Andrian UH, Mackay CR. T-cell function and migration. Two sides of the same coin. N.Engl.J.Med. 2000; 343: 1020-34.
4 Gonzalez-Amaro R, Diaz-Gonzalez F, Sanchez-Madrid F. Adhesion molecules in inflammatory diseases. Drugs 1998; 56: 977-88.
5 Redlich K, Schett G, Steiner G, Hayer S, Wagner EF, Smolen JS. Rheumatoid arthritis therapy after tumor necrosis factor and interleukin-1 blockade. Arthritis Rheum. 2003; 48: 3308-19.
6 Smith MD, Slavotinek J, Au V et al. Successful treatment of rheumatoid arthritis is associated with a reduction in synovial membrane cytokines and cell adhesion molecule expression. Rheumatology.(Oxford) 2001; 40: 965-77.
7 Jersmann HP, Hii CS, Ferrante JV, Ferrante A. Bacterial lipopolysaccharide and tumor necrosis factor alpha synergistically increase expression of human endothelial adhesion molecules through activation of NF-kappaB and p38 mitogen-activated protein kinase signaling pathways. Infect.Immun. 2001; 69: 1273-9.

Chapter 6
86
8 Read MA, Whitley MZ, Gupta S et al. Tumor necrosis factor alpha-induced E-selectin expression is activated by the nuclear factor-kappaB and c-JUN N-terminal kinase/p38 mitogen-activated protein kinase pathways. J.Biol.Chem. 1997; 272: 2753-61.
9 Schett G, Tohidast-Akrad M, Smolen JS et al. Activation, differential localization, and regulation of the stress- activated protein kinases, extracellular signal-regulated kinase, c-JUN N-terminal kinase, and p38 mitogen-activated protein kinase, in synovial tissue and cells in rheumatoid arthritis. Arthritis Rheum. 2000; 43: 2501-12.
10 Kumar S, Boehm J, Lee JC. p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat.Rev.Drug Discov. 2003; 2: 717-26.
11 Wadsworth SA, Cavender DE, Beers SA et al. RWJ 67657, a potent, orally active inhibitor of p38 mitogen-activated protein kinase. J.Pharmacol.Exp.Ther. 1999; 291: 680-7.
12 Westra J, Limburg PC, de Boer P, van Rijswijk MH. Effects of RWJ 67657, a p38 mitogen activated protein kinase (MAPK) inhibitor, on the production of inflammatory mediators by rheumatoid synovial fibroblasts. Ann.Rheum.Dis. 2004; 63: 1453-9.
13 Fijen JW, Tulleken JE, Kobold AC et al. Inhibition of p38 mitogen-activated protein kinase: dose-dependent suppression of leukocyte and endothelial response after endotoxin challenge in humans. Crit Care Med. 2002; 30: 841-5.
14 Parasrampuria DA, de Boer P, Desai-Krieger D, Chow AT, Jones CR. Single-dose pharmacokinetics and pharmacodynamics of RWJ 67657, a specific p38 mitogen-activated protein kinase inhibitor: a first-in-human study. J.Clin.Pharmacol. 2003; 43: 406-13.
15 Mulder AB, Blom NR, Smit JW et al. Basal tissue factor expression in endothelial cell cultures is caused by contaminating smooth muscle cells. Reduction by using chymotrypsin instead of collagenase. Thromb.Res. 1995; 80: 399-411.
16 Maciag T, Hoover GA, Weinstein R. High and low molecular weight forms of endothelial cell growth factor. J.Biol.Chem. 1982; 257: 5333-6.
17 van Leeuwen MA, Westra J, Limburg PC, van Riel PL, van Rijswijk MH. Interleukin-6 in relation to other proinflammatory cytokines, chemotactic activity and neutrophil activation in rheumatoid synovial fluid. Ann.Rheum.Dis. 1995; 54: 33-8.
18 Issekutz AC, Ayer L, Miyasaka M, Issekutz TB. Treatment of established adjuvant arthritis in rats with monoclonal antibody to CD18 and very late activation antigen-4 integrins suppresses neutrophil and T-lymphocyte migration to the joints and improves clinical disease. Immunology 1996; 88: 569-76.
19 Kavanaugh AF, Schulze-Koops H, Davis LS, Lipsky PE. Repeat treatment of rheumatoid arthritis patients with a murine anti-intercellular adhesion molecule 1 monoclonal antibody. Arthritis Rheum. 1997; 40: 849-53.
20 Dimitrijevic M, Bartlett RR. Leflunomide, a novel immunomodulating drug, inhibits homotypic adhesion of mononuclear cells in rheumatoid arthritis. Transplant.Proc. 1996; 28: 3086-7.
21 Wheller SK, Perretti M. Dexamethasone inhibits cytokine-induced intercellular adhesion molecule-1 up-regulation on endothelial cell lines. Eur.J.Pharmacol. 1997; 331: 65-71.
22 Haringman JJ, Kraan MC, Smeets TJ, Zwinderman KH, Tak PP. Chemokine blockade and chronic inflammatory disease: proof of concept in patients with rheumatoid arthritis. Ann.Rheum.Dis. 2003; 62: 715-21.
23 Lali FV, Hunt AE, Turner SJ, Foxwell BM. The pyridinyl imidazole inhibitor SB203580 blocks phosphoinositide-dependent protein kinase activity, protein kinase B phosphorylation, and retinoblastoma hyperphosphorylation in interleukin-2-stimulated T cells independently of p38 mitogen-activated protein kinase. J.Biol.Chem. 2000; 275: 7395-402.
24 Clerk A, Sugden PH. The p38-MAPK inhibitor, SB203580, inhibits cardiac stress-activated protein kinases/c-Jun N-terminal kinases (SAPKs/JNKs). FEBS Lett. 1998; 426: 93-6.
25 McCafferty DM, Kanwar S, Granger DN, Kubes P. E/P-selectin-deficient mice: an optimal mutation for abrogating antigen but not tumor necrosis factor-alpha-induced immune responses. Eur.J.Immunol. 2000; 30: 2362-71.
26 Norman MU, Van De Velde NC, Timoshanko JR, Issekutz A, Hickey MJ. Overlapping roles of endothelial selectins and vascular cell adhesion molecule-1 in immune complex-induced leukocyte recruitment in the cremasteric microvasculature. Am.J.Pathol. 2003; 163: 1491-503.

p38 MAPK inhibition in EC
87
27 Fichtner F, Koslowski R, Augstein A, Hempel U, Rohlecke C, Kasper M. Bleomycin induces IL-8 and ICAM-1 expression in microvascular pulmonary endothelial cells. Exp.Toxicol.Pathol. 2004; 55: 497-503.
28 Ju H, Behm DJ, Nerurkar S et al. p38 MAPK inhibitors ameliorate target organ damage in hypertension: Part 1. p38 MAPK-dependent endothelial dysfunction and hypertension. J.Pharmacol.Exp.Ther. 2003; 307: 932-8.
29 Pietersma A, Tilly BC, Gaestel M et al. p38 mitogen activated protein kinase regulates endothelial VCAM-1 expression at the post-transcriptional level. Biochem.Biophys.Res.Commun. 1997; 230: 44-8.
30 Viemann D, Goebeler M, Schmid S et al. Transcriptional profiling of IKK2/NF-{kappa}B- and p38 MAP kinase-dependent gene expression in TNF-{alpha}-stimulated primary human endothelial cells. Blood 2004.
31 Goebeler M, Gillitzer R, Kilian K et al. Multiple signaling pathways regulate NF-kappaB-dependent transcription of the monocyte chemoattractant protein-1 gene in primary endothelial cells. Blood 2001; 97: 46-55.
32 Marin V, Farnarier C, Gres S et al. The p38 mitogen-activated protein kinase pathway plays a critical role in thrombin-induced endothelial chemokine production and leukocyte recruitment. Blood 2001; 98: 667-73.
33 Westra J, Doornbos-Van Der Meer B, de Boer P, van Leeuwen MA, van Rijswijk MH, Limburg PC. Strong inhibition of TNF-alpha production and inhibition of IL-8 and COX-2 mRNA expression in monocyte-derived macrophages by RWJ 67657, a p38 mitogen-activated protein kinase (MAPK) inhibitor. Arthritis Res.Ther. 2004; 6: R384-R392.

7
Differential effects of NF-κB and p38 MAPK inhibitors
and combinations thereof on TNF-α and IL-1β induced
pro-inflammatory status of endothelial cells in vitro
Joanna M Kułdo1
Johanna Westra2
Sigridur A Ásgeirsdóttir1
Robbert J Kok3
Koen Oosterhuis4
Marianne G Rots4
Pieter C Limburg2
Grietje Molema1
From the Departments of 1 Pathology and Laboratory Medicine, Medical Biology
Section, Endothelial Cell and Vascular Drug Targeting Group, 2 Rheumatology, 3 Pharmacokinetics and Drug Delivery, and 4 Department of Therapeutic Gene
Modulation, University Medical Center Groningen, The Netherlands
Submitted

Chapter 7
90
ABSTRACT Endothelial cells actively participate in inflammatory events by regulating leukocyte recruitment via the expression of inflammatory genes such as E-selectin, VCAM-1, ICAM-1, IL-6, IL-8, and cyclo-oxygenase (COX)-2. In this study we showed by real time RT-PCR that activation of human umbilical vein endothelial cells (HUVEC) by TNF-α and IL-1β differentially affected the expression of these inflammatory genes. Combined treatment with TNF-α/IL-1β resulted in an exaggerated induction of IL-6, IL-8 and COX-2 gene expression. In contrast an attenuated increase in VCAM-1 and ICAM-1 gene expression was observed. Overexpression of dominant negative (dn)IκB protein blocking NF-κB signalling confirmed a major role of this pathway in controlling both TNF-α and IL-1β induced expression of most of the genes studied. While pyrrolidine dithiocarbamate (PDTC) and dexamethasone (DEX), both inhibitors of NF-κB controlled gene expression, exerted limited effects at 1 µM, the thioredoxin inhibitor MOL-294 that regulates the redox state of NF-κB mainly inhibited adhesion molecule expression. Its most pronounced effect was seen on VCAM-1 mRNA levels, especially in IL-1β activated endothelium. One µM RWJ 67657, an inhibitor of p38 MAPK activity, diminished TNF-α and IL-1β induced expression of IL-6, IL-8, and E-selectin, but had little effect on VCAM-1 and ICAM-1. Combined treatment of HUVEC with MOL-294 and RWJ 67657 resulted in significant blocking of the expression of the majority of genes studied. The inhibitory effects were much stronger than those observed by single drug treatment, implying the use of combinations of drugs affecting multiple targets in activated endothelial cells as a potential new therapeutic strategy. Key words. Endothelial cells, inflammatory gene expression, anti-inflammatory drugs, NF-κB, p38 MAPK

NF-κB and p38 MAPK inhibition in EC
91
INTRODUCTION Endothelial cells form the natural barrier between the blood and surrounding tissue. During inflammation they control leukocyte trafficking and actively participate in angiogenesis through differential expression of inflammation- and angiogenesis-associated genes, including cytokines, chemokines, growth factors, and adhesion molecules 1. Prevention of activation of endothelial cells has been suggested to be beneficial in the treatment of chronic inflammatory diseases like rheumatoid arthritis (RA) 2, and inflammatory bowel disease (IBD) 3. Furthermore, their position in the body makes them an easy accessible target for therapeutic intervention. The pro-inflammatory cytokines TNF-α and IL-1β, having a similar but not identical effect on gene expression, are often present simultaneously in chronic inflammatory diseases 4;5. They exert a prominent effect on the expression of pro-inflammatory genes in endothelial cells. This effect predominantly takes place through activation of intracellular signalling pathways involving NF-κB and p38 MAPK 6;7. The transcription factor NF-κB is present in the cytoplasm of unstimulated cells in an inactive form due to its association with the inhibitory protein IκB. Upon cytokine activation degradation of IκB and subsequent nuclear translocation of active NF-κB takes place 8. The p38 MAPK activation pathway engages diverse upstream kinases responsible for p38 MAPK activation, as well as downstream substrates 9. In endothelial cells both NF-κB and p38 MAPK are involved in the regulation of the expression of genes encoding E-selectin, VCAM-1, ICAM-1, IL-6, IL-8, and cyclo-oxygenase (COX)-2, among others 10-12. The regulation takes place on transcriptional and posttranscriptional levels 7;13. Both activated NF-κB and p38 MAPK have been shown to be present in RA and IBD lesions, and are therefore interesting targets for pharmacological intervention 14;15. However inhibition of NF-κB or p38 MAPK can have the serious drawback of undesired toxic effects on non-diseased cells 16. Incorporation of these drugs in endothelial cell specific drug delivery systems can theoretically overcome these undesired side-effects 17. The antioxidant and metal-chelating compound pyrrolidine dithiocarbamate (PDTC) 18, the glucocorticoid dexamethasone (DEX) 19, the thioredoxin inhibitor MOL-294 20, and the p38 MAPK inhibitor RWJ 67657 21 are potential candidates for incorporation in drug targeting constructs. Yet, limited data are available on quantitative comparison of the effects of these anti-inflammatory drugs on endothelial cell gene expression under pro-inflammatory conditions. In the current study we investigated the effects of TNF-α, IL-1β, and a combination of TNF-α/IL-1β activation on the kinetics and levels of expression of the pro-inflammatory genes E-selectin, VCAM-1, ICAM-1, IL-6, IL-8, and COX-2 by human umbilical vein endothelial cells (HUVEC). The importance of NF-κB signalling in TNF-α and IL-1β induced gene expression was investigated by overexpression of an IκB mutant inhibiting NF-κB signal transduction. Furthermore, we analysed the effects of the above-mentioned drugs on the expression levels of the pro-inflammatory genes chosen and their capacity to potentiate gene expression inhibition when added simultaneously. To quantitatively compare the effects of activators and drugs, both real time RT-PCR and, in distinct experiments, ELISA analyses of produced cytokines were performed.

Chapter 7
92
MATERIAL AND METHODS Endothelial Cells HUVEC obtained from the Endothelial Cell Facility (UMCG, Groningen, The Netherlands) were isolated from two umbilical cords to circumvent donor bias, and cultured as previously described 22. In short, the cells were cultured on 1% gelatin-precoated tissue culture flasks (Corning, Schiphol, The Netherlands) at 37°C under 5% CO2/95% air. The endothelial culture medium consisted of RPMI 1640 supplemented with 20% heat-inactivated FCS, 2 mM L-glutamine, 5 U/ml heparin, 100 IE/ml penicillin, 100 µg/ml streptomycin and 50 µg/ml endothelial cell growth factor supplement extracted from bovine brain. Upon confluence, cells were detached from the surface by trypsin/EDTA (0.5/0.2 mg/ml in PBS) and split at a 1:3 ratio into 12- or 6-well tissue culture plates (Corning). In the experiments performed, HUVEC were used up to passage 4. All experiments were performed with confluent HUVEC monolayers, except when using adenovirus encoded dominant negative (dn)IκB, for which confluency was 70%. The experiments were performed with at least two and in most cases with four different HUVEC isolates in independent experiments. Data shown are representative for the data from the different experiments. Activation of HUVEC Confluent HUVEC were activated with 1 and 10 ng/ml TNF-α (Boehringer, Ingelheim, Germany), and 1 and 10 ng/ml IL-1β (R&D Systems, Minneapolis MN, USA), added separately or in combination, for 6 hours (early gene expression) and 24 hours (late gene expression). After incubation cells were microscopically analyzed with regard to their morphology and consistently were found to be adherent and viable. MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxy-methoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, Promega Benelux BV, Leiden, The Netherlands) assays were occasionally executed according to the manufacturers protocol, to corroborate the light microscopy analysis. Dominant negative IκB Adenovirus Recombinant, replication-deficient adenovirus Ad5IκBAA, hereafter named dnIκB, was a gift from Prof. C. Trautwein from the Medical School of Hannover, Germany. Adenovirus contained an IκBα sequence, in which serine at positions 32 and 36 were substituted by alanine, and which was fused to influenza A virus hemagglutinin (HA)-tag. The expression was controlled by the cytomegalovirus promoter/enhancer 23. Virus was grown on HEK293 cells and purified from cell lysates by banding twice on CsCl gradients. Virus was desalted using a 10 kD slide-A-lyzer ® (Pierce Chemical Company, Rockford IL, USA) in Hepes/Sucrose buffer, pH 8.0 and stored at -80°C. Viral particles (vp) were determined by UV spectrophotometric analysis at 260 nm. Furthermore a standard limiting dilution assay was performed to determine the vp/plaque forming unit (pfu) ratio. As a control, adenovirus Ad5LacZ, containing the Escherischia coli β-galactosidase gene 24, was grown and purified as described above. Virus infection protocol For the transduction of HUVEC with dnIκB or the control virus Ad5LacZ, HUVEC were plated at 12,500 cells/cm2 in 6-wells tissue culture plates (Corning), and cultured

NF-κB and p38 MAPK inhibition in EC
93
overnight before actual transduction. The viral vectors diluted in DMEM (GibcoTM, Paisley, Scotland, UK) without FCS, were added at 500 pfu/seeded cell (corresponding to 7.5x103 vp/cell) and incubated for 90 min at 37°C. The incubation medium was then replaced by endothelial culture medium. Cells were subsequently incubated for 24 hours prior to activation to allow transgene expression. Western blot analysis of dnIκB expression in HUVEC After 24 hours of culturing, cells were detached from the surface by trypsin/EDTA treatment, lysed in the cell culture lysis reagent (Promega Corporation, Madison, WI, USA) and sonicated twice at 4°C for 5 sec. After centrifugation for 10 min at 10 000 g and at 4°C, cleared cell lysates were collected and the protein content was determined using the Bradford protein assay reagent (Bio-Rad Laboratories, Hercules, CA, USA), with BSA as the standard. Samples were then mixed with reducing SDS sample buffer, boiled for 5 min, and 30 µg of protein was loaded on SDS-PAGE 10% acrylamide gel. After separation proteins were electrophoretically transferred on a nitrocellulose membrane (Bio-Rad Laboratories). Blots were blocked in blocking buffer containing 5% non-fat dry milk in PBS/0.1% Tween for 2 hours. Next blots were incubated for 1 hr with rabbit anti-HA-probe antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA; dilution 1:200 in blocking buffer) for dnIκB detection, and rabbit anti-IκBα antibody (Santa Cruz Biotechnology; dilution 1:20 in blocking buffer) for both endogenous and dnIκB detection. Blots were washed with PBS/0.1% Tween and incubated for 1 hr with horseradish peroxidase-conjugated swine anti-rabbit antibody (DAKO, Glostrup, Denmark) diluted 1:2000 in blocking buffer. After washing as described above, detection was performed using enhanced chemoluminescence detection reagent (Amersham Corp., Arlington Heigths, IL, USA) according to the manufacturers protocol. Incubation of HUVEC with drugs The following drugs were used: PDTC (Sigma-Aldrich, Zwijndrecht, The Netherlands), DEX (9α-fluoro-16α-methyl-11β,17α,21-trihydroxy-1,4-pregnadiene-3,20-dione; Genfarma B.V., Maarssen, The Netherlands), MOL-294 (methyl (4R/S)-4-hydroxy-4-[((5S,8S)/(5R,8R))-8-methyl-1,3-dioxo-2-phenyl-2,3,5,8-tetra-hydro-1H-[1,2,4]triazolo [1,2-a]pyridazin-5-yl]-2-butynoate; kindly provided by Dr. M. Kahn from Pacific Northwest Research Institute, Seattle, Washington, USA), and RWJ 67657 (4-(4-(4-fluorophenyl)-1-(3-phenylpropyl)-5-(4-pyridinyl)-1H-imidazol-2-yl)-3-butyn-1-ol; kindly provided by Johnson & Johnson Pharmaceutical R&D, Raritan, New Jersey, USA). Stock solutions (10 mM) of PDTC, DEX, MOL-294, and RWJ 67657 were diluted in DMSO (Merck, Darmstadt, Germany). The stock solutions were diluted in endothelial culture medium to final concentrations as indicated in each experiment. Anti-inflammatory drugs were added to confluent HUVEC one hour before activation by TNF-α or IL-1β. After 6 and 24 hours of stimulation cells were analyzed microscopically with regard to their morphology and viability after which cells or supernatants were subjected to further analysis. By MTS assay the occurrence of toxic effects of drugs to the cells were excluded.

Chapter 7
94
RNA isolation and real time RT-PCR analysis Total RNA was isolated using the Absolutely RNA Microprep Kit (Stratagene, Amsterdam, The Netherlands) according to the protocol of the manufacturer. RNA was analyzed qualitatively by gel electrophoresis and quantitatively by RiboGreen RNA Quantitation Kit (Molecular Probes Europe B.V., Leiden, The Netherlands). One µg total cellular RNA was subsequently used for the synthesis of first strand cDNA using SuperScript III RNase H- Reverse Transcriptase (Invitrogen, Breda, The Netherlands) in 20 µl final volume containing 250 ng random hexamers (Promega) and 40 units RNase OUT inhibitor (Invitrogen). After RT-reaction cDNA was diluted with distilled water to 100 µl. Exons overlapping primers and Minor Groove Binder (MGB) probes used for real time RT-PCR were purchased as Assay-on-Demand from Applied Biosystems (Nieuwerkerk a/d IJssel, The Netherlands): housekeeping gene GAPDH (assay ID Hs99999905_m1), endothelial cell marker CD31 (PECAM-1, Platelet Endothelial Cell Adhesion Molecule 1, Hs00169777_m1), E-selectin (Hs00174057_m1), VCAM-1 (Hs00365486_m1), ICAM-1 (Hs00164932_m1), IL-6 (Hs00174131_m1), IL-8 (Hs00174103_m1), COX-2 (Hs00153133_m1). The final concentration of primers and MGB probes in TaqMan PCR MasterMix (Applied Biosystems, Foster City, CA, USA) for each gene was 900 nM and 250 nM respectively. As controls, RNA samples not subjected to reverse transcriptase were analyzed to exclude unspecific signals arising from genomic DNA. Those samples consistently showed no amplification signals. TaqMan real time RT-PCR was performed in an ABI PRISM 7900HT Sequence Detector (Applied Biosystems). Amplification was performed using cycling conditions: 2 min 50°C, 10 min 95°C, and 40 to 45 two-step cycles of 15 sec at 95°C and 60 sec at 60°C. Triplicate real time RT-PCR analyses were executed for each sample, and the obtained threshold cycle values (Ct) were averaged. According to the comparative Ct method described in the ABI manual (http://www.appliedbiosystems. com), gene expression was normalized to the expression of the housekeeping gene GAPDH, yielding the ∆Ct value. The average ∆Ct value obtained from resting, non-treated HUVEC was then subtracted from the average ∆Ct value of each sample subjected to the experimental conditions described, yielding the ∆∆Ct value. The gene expression level, normalized to the housekeeping gene and relative to the control sample, was calculated by 2 –∆∆Ct. IL-6 and IL-8 production measured by ELISA In designated drug combination treatment experiments, HUVEC medium was harvested, centrifuged and stored at -20°C prior to ELISA based cytokine quantification. Ninety-six-well plates (Corning) were pre-coated with MoAb.anti-IL-6, clone 6/16 (Sanquin, Amsterdam, The Netherlands) 1:1000 diluted in PBS for IL-6, and with MoAb.anti-IL-8 (R&D Systems) for IL-8 analysis. After blocking with 2% BSA/0.05% Tween in PBS, samples were incubated for 2 hours in incubation buffer containing 0.2% gelatine/0.05% Tween in PBS. After washing, bound IL-6 or IL-8 were detected with biotinylated polyclonal swine anti-human IL-6 (Sanquin) or polyclonal swine anti-human IL-8 (R&D Systems), respectively, in combination with streptavidin-E+ (Sanquin). Peroxidase activity was determined using tetramethylbenzidine (Roth, Karlsruhe, Germany) as substrate. IL-6 and IL-8 levels

NF-κB and p38 MAPK inhibition in EC
95
were calculated in the linear range of the assay from a standard curve (10-1000 pg/ml) using r-hIL-6 and r-hIL-8 (both from R&D Systems). STATISTICS Statistical significance of differences was studied by means of the two-sided Student’s t-test, assuming equal variances. Differences were considered to be significant when p<0.05. RESULTS In endothelial cells different gene expression patterns are induced by TNF-α and IL-1β To study gene expression profiles induced by TNF-α and IL-1β, HUVEC were activated with 1 and 10 ng/ml of both cytokines for 6 and 24 hours, and examined for the gene expression of E-selectin, VCAM-1, ICAM-1, IL-6, IL-8, and COX-2. The 6 hours time point reflects early gene expression profiles, while the 24 hours time point reflects late gene expression during prolonged activation, possibly comparable with prolonged exposure of the endothelium to activators in chronic inflammatory disorders.
Figure 1. Differences in endothelial gene expression profiles induced by TNF-α or IL-1β activation. HUVEC were stimulated with 1 or 10 ng/ml of TNF-α (black bars) or IL-1β (open bars) for 6 (A) or 24 (B) hours. mRNA levels of genes studied were determined with real time RT-PCR and adjusted to the expression of house keeping gene GAPDH. mRNA levels were normalized to untreated, control cells arbitrary set at 1. Note the different values on Y axis. The data present mean values ± s.d. (n=3). Expression of the endothelial cell marker CD31 remained constant under all conditions studied (figure 1), a result corroborating previously published studies 25. Patterns of pro-inflammatory gene expression, however, markedly differed with regard to the

Chapter 7
96
activator used and the incubation time studied. TNF-α induced the expression of adhesion molecules VCAM-1 and ICAM-1 to a higher extent than IL-1β, whereas IL-1β more profoundly affected IL-6, IL-8, and COX-2. For E-selectin a similar extent of expression was observed during activation by TNF-α and IL-1β with the exception of activation with 1 ng/ml of TNF-α or IL-1β at 24 hours. This apparent distinction was more pronounced at the early time point. At 6 hours, the level of gene expression induction was independent of the concentration of each activator, the only deviation being IL-1β induced IL-6 and ICAM-1 expression. Especially for TNF-α induced activation at 24 hours higher concentration of activator induced gene expression to the highest level. Stimulation of the expression of cell adhesion molecules was higher at 6 hours of activation compared to 24 hours of activation, while the opposite situation was observed for IL-8 and COX-2. IL-6 exhibited a mixed response, as its level of expression induced by TNF-α was highest at 24 hours, while IL-1β induced expression was higher at 6 hours. Adhesion molecule and cytokine gene expression are differently affected by TNF-α/IL-1β co-treatment Since the pro-inflammatory cytokines TNF-α and IL-1β can be present simultaneously at sites of inflammation, we investigated the gene expression profile of HUVEC when incubated with both TNF-α and IL-1β. A striking difference in gene expression was observed between cell adhesion molecules on one hand and cytokines and COX-2 on the other hand (figure 2). The observed level of gene expression of adhesion molecules upon co-treatment was lower than the expected level, which was calculated by summation of mRNA levels induced by separate cytokine treatment. This effect was most pronounced for VCAM-1 and ICAM-1 expression at 24 hours at 10 ng/ml concentration of both activators. In contrast, mRNA levels of cytokines and COX-2 were higher than expected from separate TNF-α or IL-1β treatment, with the strongest effects seen on IL-6 mRNA levels. Although the cytokine mediated effects on gene expression differed at the time points (as shown in figure 1), it is noteworthy that the pattern of deviation from the calculated values was similar at both time points. E-selectin was the only exception to this rule. Dominant-negative IκB mutant inhibits both TNF-α and IL-1β driven pro-inflammatory gene expression TNF-α and IL-1β induced signal transduction in endothelial cell relays mainly through the NF-κB and p38 MAPK routes 10;26. Yet the relative contributions of these pathways to the control of the expression of the genes under study are unknown. We investigated the functional relationship between NF-κB activity and pro-inflammatory gene expression in HUVEC using overexpression of dnIκB. HUVEC infected with LacZ adenovirus and HUVEC infected with dnIκB adenovirus were activated with 1 and 10 ng/ml of TNF-α or IL-1β for 6 and 24 hours. Approximately 70-80% of cells were infected by adenoviral infection as established with green fluorescent protein reporter protein (K.I. Ogawara et al, unpublished observations). Expression of dnIκB

NF-κB and p38 MAPK inhibition in EC
97
Figure 2. Effects of TNF-α and IL-1β co-treatment on HUVEC pro-inflammatory gene expression.
HUVEC were activated for 6 and 24 hours with mixtures of TNF-α and IL-1β in designated concentrations. Gene expression determined by quantitative RT-PCR, was adjusted to GAPDH and normalized to untreated, control HUVEC. The experimentally measured gene expression levels induced by TNF-α/IL-1β combination treatment were compared to the theoretical, expected gene expression levels calculated by summation of gene expression levels observed when cells were treated with TNF-α or IL-1β separately. This later value was arbitrary set at 1. Values above 1 represent situations where experimentally determined mRNA levels were higher than the expected additive value. Values below 1 indicate experimentally determined mRNA levels lower than the expected additive value. The gray area borders the lower through upper limit of the observed level of gene expression that is considered to be within the range of the expected additive level. The data present mean values ± s.d. (n=3).
transgene in HUVEC was confirmed by western blot analysis (figure 3A). In the conditions studied only a minor fraction of cells went into apoptosis upon TNF-α activation. Likely, NF-κB independent cell survival was controlled by serum derived growth factors as the cells were continuously cultured in medium containing FCS. An almost total inhibition of adhesion molecule expression induced by 10 ng/ml TNF-α or IL-1β was observed in dnIκB-expressing HUVEC compared to uninfected or LacZ infected cells, at both time points studied (figure 3B). A more limited inhibition of gene expression was observed for IL-6 and IL-8 at 6 hours, while at 24 hours the inhibition of these genes was also blocked. The limited COX-2 gene expression observed after 6 hours after start of activation was not NF-κB dependent. Possibly p38 MAPK and PKC control COX-2 expression here 27;28. However, after 24 hours a significant inhibition of COX-2 expression was observed in dnIκB expressing cells. Likely, during this longer time period cytokines expressed in an NF-κB independent manner induce, in an autocrine fashion, NF-κB dependent COX-2 expression 29;30. A similar pattern of inhibition of gene expression for all genes was seen with 1 ng/ml activator (data not shown).
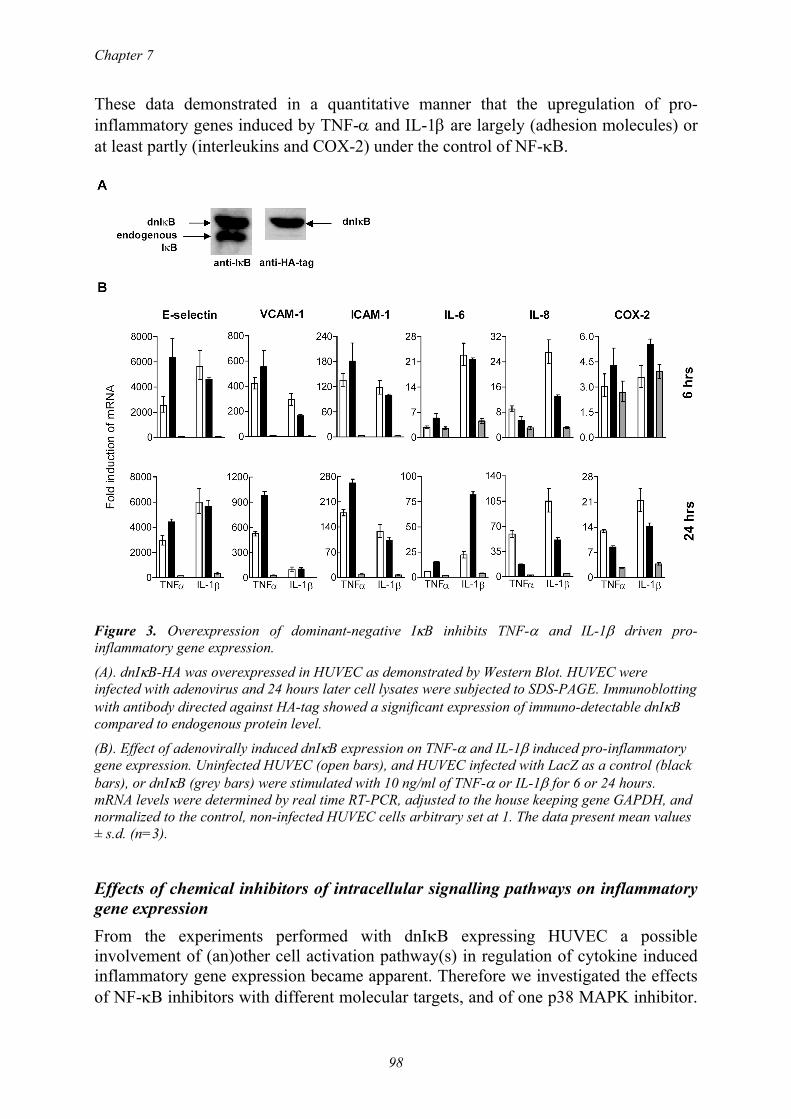
Chapter 7
98
These data demonstrated in a quantitative manner that the upregulation of pro-inflammatory genes induced by TNF-α and IL-1β are largely (adhesion molecules) or at least partly (interleukins and COX-2) under the control of NF-κB.
Figure 3. Overexpression of dominant-negative IκB inhibits TNF-α and IL-1β driven pro-inflammatory gene expression.
(A). dnIκB-HA was overexpressed in HUVEC as demonstrated by Western Blot. HUVEC were infected with adenovirus and 24 hours later cell lysates were subjected to SDS-PAGE. Immunoblotting with antibody directed against HA-tag showed a significant expression of immuno-detectable dnIκB compared to endogenous protein level.
(B). Effect of adenovirally induced dnIκB expression on TNF-α and IL-1β induced pro-inflammatory gene expression. Uninfected HUVEC (open bars), and HUVEC infected with LacZ as a control (black bars), or dnIκB (grey bars) were stimulated with 10 ng/ml of TNF-α or IL-1β for 6 or 24 hours. mRNA levels were determined by real time RT-PCR, adjusted to the house keeping gene GAPDH, and normalized to the control, non-infected HUVEC cells arbitrary set at 1. The data present mean values ± s.d. (n=3).
Effects of chemical inhibitors of intracellular signalling pathways on inflammatory gene expression From the experiments performed with dnIκB expressing HUVEC a possible involvement of (an)other cell activation pathway(s) in regulation of cytokine induced inflammatory gene expression became apparent. Therefore we investigated the effects of NF-κB inhibitors with different molecular targets, and of one p38 MAPK inhibitor.

NF-κB and p38 MAPK inhibition in EC
99
HUVEC were incubated with 1 µM of PDTC, DEX, MOL-294, and RWJ 67657 starting 1 hour before the addition of 10 ng/ml TNF-α or IL-1β. The choice of this fixed concentration was based on the experience that approximately 1 µM of drug can be delivered using drug targeting constructs. As shown in figure 4, the drugs affected inflammatory gene expression differently with respect to the activator used and the time interval studied. RWJ 67657 and MOL-294 were the most potent inhibitors showing downregulation of several inflammatory genes. Specific inhibition of p38 MAPK activity by RWJ 67657 resulted in blocking of gene expression of the interleukins at 6 hours and additionally of the adhesion molecules after 24 hours. MOL-294 treatment resulted in an inhibitory effect on both TNF-α and IL-1β induced
Figure 4. Effects of inhibitors of signal transduction on pro-inflammatory gene expression by HUVEC.
Drugs were added at 1 µM 1 hour before activation with 10 ng/ml of TNF-α or IL-1β. mRNA levels of designated genes at 6 or 24 hours were determined with real time RT-PCR, adjusted to the house keeping gene GAPDH, and normalized to untreated, non-activated control HUVEC arbitrary set at 1. The 100% value represents activation in the absence of drugs. 0% represents the mRNA level of control cells. Mean values ± s.d. (n=3); *, p < 0.05; **, p < 0.001 between cells activated with TNF-α or IL-1β in the absence of drugs, and cells treated with drugs.
adhesion molecule expression, with the most pronounced effect on VCAM-1 expression at both 6 hours and 24 hours. We consistently found significantly higher IL-8 mRNA levels at 6 hours after TNF-α treatment in combination with this drug, which is at present difficult to explain. DEX only downregulated IL-6 mRNA gene expression after 6 hours of TNF-α activation, and VCAM-1 mRNA levels after 24

Chapter 7
100
hours of IL-1β activation. Moreover, pretreatment of HUVEC with 1 µM DEX resulted in a significant increase in IL-6 mRNA level 24 hours after IL-1β stimulation compared to untreated cells. The minor modulatory effects of PDTC on gene expression was possibly due to the use of relatively low concentrations of this drug as compared to the concentrations used in other studies 18. Combination of drugs enhance inhibitory effects on gene and protein expression in HUVEC RWJ 67657 and MOL-294 affect different routes of the main pro-inflammatory activation pathways. Since they both showed pronounced inhibitory effects on TNF-α and IL-1β induced gene expression in HUVEC, we hypothesized that combination treatment employing these two drugs might result in enhanced inhibitory effects. Therefore the effects of simultaneous addition of these drugs in concentrations ranging from 0.1 to 10 µM on the expression levels of studied genes were investigated. These effects were measured at 6 hours after induction of activation by 10 ng/ml TNF-α or IL-1β. In all concentrations and combinations studied the drugs appeared not to be toxic to the HUVEC (data not shown). In the case of expression of the cell adhesion molecules an additive inhibitory effect of RWJ 67657/MOL-294 co-treatment was observed on TNF-α-mediated E-selectin gene expression (figure 5). One µM MOL-294 by itself downregulated E-selectin mRNA levels from 5725 (± 1583) fold increase to 4340 (± 614) fold increase (n=3 ± s.d.). Co-incubation with 0.1, 1, and 10 µM RWJ 67657 resulted in further statistically significant decrease of gene expression to respectively 3532 (± 485; p<0.05 vs. activated cells) fold, 2943 (± 283; p<0.05) fold, and 2117 (± 223; p<0.005) fold. Both TNF-α and IL-1β induced IL-6, IL-8, and COX-2 expression was inhibited in an additive way by drug combination treatment, with different concentration combinations being responsible for the enhanced inhibition of genes expression observed. To confirm this increase in the inhibitory potential of RWJ 67657 and MOL-294 combination treatment on the expression of IL-6 and IL-8, proteins were measured by ELISA in the supernatants of selected samples harvested at 6 hours (figure 6). The cytokine levels reflected the gene expression data, although the additive effects of drugs co-treatment were less pronounced. This may be due to the limit of detection of the ELISA assays used. A consistently found deviation from this pattern was IL-8 expression in samples treated with 10 µM MOL-294. Total blocking of the protein expression was paralleled by upregulated mRNA levels.

NF-κB and p38 MAPK inhibition in EC
101
Figure 5. Effect of combination treatment of HUVEC with RWJ 67657 and MOL-294 on pro-inflammatory gene expression. HUVEC were treated with RWJ 67657 and MOL-294, added alone or in combination in concentrations ranging from 0.1 – 10 µM. After 1 hour 10 ng/ml of TNF-α or IL-1β was added and cells were incubated for another 6 hours. mRNA levels were determined using real time RT-PCR. Data were adjusted to the house keeping gene GAPDH, and normalized to untreated, non-activated control HUVEC arbitrary set at 1. The data present mean values ± s.d. (n=3); *, p < 0.05 between cells treated by the combination of drugs and cells treated with each drug separately.

Chapter 7
102
Figure 6. Comparison of IL-6 and IL-8 gene and protein levels in HUVEC treated by RWJ 67657, MOL-294, or a combination. HUVEC were treated with designated concentrations of RWJ 67657 and MOL-294 for 1 hr, after which either TNF-α or IL-1β was added at 10 ng/ml to activate the cells. Fold induction of mRNA level (left axis) at 6 hours after start of activation was compared with the amount of secreted IL-6 or IL-8 protein determined by ELISA (right axis). Undetectable protein levels (ELISA detection limits are 20 pg/ml) are marked by asterisks. The data present mean values ± s.d. (n=3).
DISCUSSION In the current study we showed that TNF-α and IL-1β differentially activated inflammatory gene expression in endothelial cells in vitro. Of the four different inhibitors of intracellular signalling cascades studied, p38 MAPK inhibitor RWJ 67657 and the small redox active protein thioredoxin inhibitor MOL-294 were

NF-κB and p38 MAPK inhibition in EC
103
identified as the most potent drugs showing downregulation of a number of different inflammatory genes. Co-treatment with both drugs resulted in an enhancement of the inhibitory effect on pro-inflammatory gene expression. This observation can have important implications for (targeted) pharmacologic intervention to downregulate endothelial cell activation to reduce leukocyte infiltration in the diseased tissue. It can be envisioned that above mentioned drugs are either administered in a combination treatment protocol or both included in immunoliposomes harnessed with antibodies specifically recognizing activated endothelial cells 31. Immunoliposome based drug delivery systems can theoretically deliver low micromolar concentration of drugs. Besides the pharmacologic profile demonstrated here, 1 µM RWJ 67657 was shown to completely inhibit MAPKAPK-2 phosphorylation in HUVEC (Westra et al, Int Immunopharmacol 2005, in press). Low micromolar concentration of MOL-294 was furthermore able to block the DNA binding ability of NF-κB in HUVEC with IC50 value of VCAM-1 expression inhibition of 2.5 µM 20. Future studies on incorporation of these drugs in the carrier systems developed in our laboratory will allow us to investigate endothelial cell response to these drugs in vitro and in vivo. We found that the pattern of pro-inflammatory gene expression markedly differed depending on the activator used. IL-1β induced IL-6, IL-8, and COX-2 gene expression by HUVEC to a higher extent than TNF-α, whereas TNF-α more profoundly affected the expression of adhesion molecules. These data are in line with those reported by others, although these latter studies were not performed in a quantitative manner and did not compare all the genes in a direct manner 7;32. Both TNF-α and IL-1β are present simultaneously in pro-inflammatory diseases 4;5. Since limited data are available on endothelial cell pro-inflammatory gene expression after simultaneous cytokine treatment 33;34, we studied the effects of TNF-α/IL-1β co-treatment on gene expression by HUVEC. The most interesting observation arising from this experiment was the occurrence of diminished upregulation of adhesion molecule mRNA, while an additive or even increased induction of interleukins and COX-2 mRNA was found. A possible explanation is that TNF-α and IL-1β partly utilize the same cell activation pathways 35. A combinatory activation might saturate common cofactors or change downstream signaling specificity by utilization of other scaffold proteins leading to differences in response to both activators when compared to either activator alone 36. Since the molecular mechanism(s) of the observed attenuation /increases in gene expression induced by pro-inflammatory activator co-treatment is/are at present not known, we decided to perform our subsequent pharmacologic studies by using TNF-α and IL-1β separately. Still, this observation justifies further research, as it has important implications for the choice of the experimental conditions to study the pharmacologic potency of new chemical entities in the drug development pipeline. Both the dnIκB adenovirus and drug treatment experiments confirmed a major role of NF-κB and p38 MAPK pathways in regulation of gene expression in HUVEC as part of the inflammatory response induced by TNF-α or IL-1β 7;10-12. An almost total inhibition of adhesion molecule expression in dnIκB-expressing HUVECs was observed at both early and late time points of activation. The induction of IL-6 and IL-8 expression was also inhibited by dnIκB, but to a lower extent compared to the effects

Chapter 7
104
on the adhesion molecules, indicating considerable involvement of (an)other pathway(s) regulating IL-6 and IL-8. Ridley et al described that the expression of IL-6 and IL-8 induced by IL-1β in HUVEC was p38 MAPK dependent 12. In another study TNF-α induced IL-8 gene and protein expression was shown to be partly dependent on reactive oxygen species generation and activator protein 1 activation 37. Also from our pharmacological experiments, the conclusion seems justified that p38 MAPK is, at least partly, controlling expression of these genes. MOL-294 pretreatment of cells consistently resulted in IL-8 mRNA increases at the early time point compared to untreated controls. Analysis of protein levels, however, revealed a complete block of IL-8 production. This discrepancy may be a result of uncoupling of gene and protein expression, e.g., due to processing of proteins that affect the secretion and/or modifies their recognition sequence directing proteins for ubiquitination. Another explanation may be the fact that thioredoxin is required for efficient proteolysis catalysed by thiol-dependent Cys-proteases such as cathepsin 38. Cathepsins are known to be essential in processing of mature IL-8 protein at inflammatory sites 39. In summary, we performed a quantitative study on pro-inflammatory gene expression in HUVEC, in which we showed that TNF-α and IL-1β differentially induced cell adhesion molecule and cytokine gene expression when added alone. Combination treatment with both cytokines resulted in deviation from the expected induction of mRNA levels of the genes. This observation may be relevant for endothelial cell activation and its complex control mechanisms in inflammatory conditions in vivo. The demonstrated additive effects of combinations of anti-inflammatory drugs that inhibit NF-κB and p38 MAPK signal transduction forms the basis for further research to study whether co-administration can improve the efficacy of the (targeted) drugs to inhibit inflammatory disease activity. ACKNOWLEDGEMENTS We thank Henk Moorlag for technical support with endothelial cell culture and Naomi Werner for excellent technical assistance in RNA isolation.
REFERENCES
1 Cines DB, Pollak ES, Buck CA et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood 1998; 91: 3527-61.
2 Smolen JS, Steiner G. Therapeutic strategies for rheumatoid arthritis. Nat.Rev.Drug Discov. 2003; 2: 473-88.
3 Nielsen OH, Vainer B, Madsen SM, Seidelin JB, Heegaard NH. Established and emerging biological activity markers of inflammatory bowel disease. Am.J.Gastroenterol. 2000; 95: 359-67.
4 Redlich K, Schett G, Steiner G, Hayer S, Wagner EF, Smolen JS. Rheumatoid arthritis therapy after tumor necrosis factor and interleukin-1 blockade. Arthritis Rheum. 2003; 48: 3308-19.
5 Rogler G, Andus T. Cytokines in inflammatory bowel disease. World J.Surg. 1998; 22: 382-9. 6 Hoefen RJ, Berk BC. The role of MAP kinases in endothelial activation. Vascul.Pharmacol.
2002; 38: 271-3.

Chapter 7
106
to human umbilical vein and dermal microvascular endothelial cells. Microvasc.Res. 2001; 62: 383-91.
26 Surapisitchat J, Hoefen RJ, Pi X, Yoshizumi M, Yan C, Berk BC. Fluid shear stress inhibits TNF-alpha activation of JNK but not ERK1/2 or p38 in human umbilical vein endothelial cells: Inhibitory crosstalk among MAPK family members. Proc.Natl.Acad.Sci.U.S.A 2001; 98: 6476-81.
27 Billack B, Heck DE, Mariano TM et al. Induction of cyclooxygenase-2 by heat shock protein 60 in macrophages and endothelial cells. Am.J.Physiol Cell Physiol 2002; 283: C1267-C1277.
28 Schroer K, Zhu Y, Saunders MA et al. Obligatory role of cyclic adenosine monophosphate response element in cyclooxygenase-2 promoter induction and feedback regulation by inflammatory mediators. Circulation 2002; 105: 2760-5.
29 Schmedtje JF, Jr., Ji YS, Liu WL, DuBois RN, Runge MS. Hypoxia induces cyclooxygenase-2 via the NF-kappaB p65 transcription factor in human vascular endothelial cells. J.Biol.Chem. 1997; 272: 601-8.
30 Wu G, Mannam AP, Wu J et al. Hypoxia induces myocyte-dependent COX-2 regulation in endothelial cells: role of VEGF. Am.J.Physiol Heart Circ.Physiol 2003; 285: H2420-H2429.
31 Everts M, Koning GA, Kok RJ et al. In vitro cellular handling and in vivo targeting of E-selectin-directed immunoconjugates and immunoliposomes used for drug delivery to inflamed endothelium. Pharm.Res. 2003; 20: 64-72.
32 Zhao B, Stavchansky SA, Bowden RA, Bowman PD. Effect of interleukin-1beta and tumor necrosis factor-alpha on gene expression in human endothelial cells. Am.J.Physiol Cell Physiol 2003; 284: C1577-C1583.
33 Dagia NM, Goetz DJ. A proteasome inhibitor reduces concurrent, sequential, and long-term IL-1 beta- and TNF-alpha-induced ECAM expression and adhesion. Am.J.Physiol Cell Physiol 2003; 285: C813-C822.
34 Daxecker H, Raab M, Markovic S, Karimi A, Griesmacher A, Mueller MM. Endothelial adhesion molecule expression in an in vitro model of inflammation. Clin.Chim.Acta 2002; 325: 171-5.
35 Herlaar E, Brown Z. p38 MAPK signalling cascades in inflammatory disease. Mol.Med.Today 1999; 5: 439-47.
36 Chariot A, Meuwis MA, Bonif M et al. NF-kappaB activating scaffold proteins as signaling molecules and putative therapeutic targets. Curr.Med.Chem. 2003; 10: 593-602.
37 Yamagishi S, Inagaki Y, Nakamura K, Imaizumi T. Azelnidipine, a newly developed long-acting calcium antagonist, inhibits tumor necrosis factor-alpha-induced interleukin-8 expression in endothelial cells through its anti-oxidative properties. J.Cardiovasc.Pharmacol. 2004; 43: 724-30.
38 Kerblat I, Drouet C, Chesne S, Marche PN. Importance of thioredoxin in the proteolysis of an immunoglobulin G as antigen by lysosomal Cys-proteases. Immunology 1999; 97: 62-8.
39 Ohashi K, Naruto M, Nakaki T, Sano E. Identification of interleukin-8 converting enzyme as cathepsin L. Biochim.Biophys.Acta 2003; 1649: 30-9.

NF-κB and p38 MAPK inhibition in EC
105
7 Viemann D, Goebeler M, Schmid S et al. Transcriptional profiling of IKK2/NF-{kappa}B- and p38 MAP kinase-dependent gene expression in TNF-{alpha}-stimulated primary human endothelial cells. Blood 2004.
8 May MJ, Ghosh S. Signal transduction through NF-kappa B. Immunol.Today 1998; 19: 80-8. 9 Ono K, Han J. The p38 signal transduction pathway: activation and function. Cell Signal. 2000;
12: 1-13. 10 Denk A, Goebeler M, Schmid S et al. Activation of NF-kappa B via the Ikappa B kinase
complex is both essential and sufficient for proinflammatory gene expression in primary endothelial cells. J.Biol.Chem. 2001; 276: 28451-8.
11 Gustin JA, Pincheira R, Mayo LD et al. Tumor necrosis factor activates CRE-binding protein through a p38 MAPK/MSK1 signaling pathway in endothelial cells. Am.J.Physiol Cell Physiol 2004; 286: C547-C555.
12 Ridley SH, Sarsfield SJ, Lee JC et al. Actions of IL-1 are selectively controlled by p38 mitogen-activated protein kinase: regulation of prostaglandin H synthase-2, metalloproteinases, and IL-6 at different levels. J.Immunol. 1997; 158: 3165-73.
13 Pietersma A, Tilly BC, Gaestel M et al. p38 mitogen activated protein kinase regulates endothelial VCAM-1 expression at the post-transcriptional level. Biochem.Biophys.Res.Commun. 1997; 230: 44-8.
14 Schett G, Tohidast-Akrad M, Smolen JS et al. Activation, differential localization, and regulation of the stress- activated protein kinases, extracellular signal-regulated kinase, c-JUN N-terminal kinase, and p38 mitogen-activated protein kinase, in synovial tissue and cells in rheumatoid arthritis. Arthritis Rheum. 2000; 43: 2501-12.
15 Waetzig GH, Seegert D, Rosenstiel P, Nikolaus S, Schreiber S. p38 mitogen-activated protein kinase is activated and linked to TNF-alpha signaling in inflammatory bowel disease. J.Immunol. 2002; 168: 5342-51.
16 Chen LW, Egan L, Li ZW, Greten FR, Kagnoff MF, Karin M. The two faces of IKK and NF-kappaB inhibition: prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat.Med. 2003; 9: 575-81.
17 Everts M, Kok RJ, Asgeirsdottir SA et al. Selective intracellular delivery of dexamethasone into activated endothelial cells using an E-selectin-directed immunoconjugate. J.Immunol. 2002; 168: 883-9.
18 Munoz C, Pascual-Salcedo D, Castellanos MC et al. Pyrrolidine dithiocarbamate inhibits the production of interleukin-6, interleukin-8, and granulocyte-macrophage colony-stimulating factor by human endothelial cells in response to inflammatory mediators: modulation of NF-kappa B and AP-1 transcription factors activity. Blood 1996; 88: 3482-90.
19 Xu X, Otsuki M, Saito H et al. PPARalpha and GR differentially down-regulate the expression of nuclear factor-kappaB-responsive genes in vascular endothelial cells. Endocrinology 2001; 142: 3332-9.
20 Misra-Press A, Mcmillan M, Cudabaker E et al. Identification of novel inhibitor of the NF-κB pathway. Current Medicinal Chemistry- Anti-Inflammatory & Anti-Allergy Agents 2003; 1:29-39.
21 Parasrampuria DA, de Boer P, Desai-Krieger D, Chow AT, Jones CR. Single-dose pharmacokinetics and pharmacodynamics of RWJ 67657, a specific p38 mitogen-activated protein kinase inhibitor: a first-in-human study. J.Clin.Pharmacol. 2003; 43: 406-13.
22 Mulder AB, Blom NR, Smit JW et al. Basal tissue factor expression in endothelial cell cultures is caused by contaminating smooth muscle cells. Reduction by using chymotrypsin instead of collagenase. Thromb.Res. 1995; 80: 399-411.
23 Iimuro Y, Nishiura T, Hellerbrand C et al. NFkappaB prevents apoptosis and liver dysfunction during liver regeneration. J.Clin.Invest 1998; 101: 802-11.
24 Herz J, Gerard RD. Adenovirus-mediated transfer of low density lipoprotein receptor gene acutely accelerates cholesterol clearance in normal mice. Proc.Natl.Acad.Sci.U.S.A 1993; 90: 2812-6.
25 Murakami S, Morioka T, Nakagawa Y, Suzuki Y, Arakawa M, Oite T. Expression of adhesion molecules by cultured human glomerular endothelial cells in response to cytokines: comparison

8
Differential influence of p38 mitogen-activated protein
kinase (MAPK) inhibition on acute phase protein synthesis
in human hepatoma cell lines and human liver slices
Johanna Westra 1
Peter Olinga 3
Johan Bijzet 2
Berber Doornbos-van der Meer 1
Geny MM Groothuis 3
Martin H van Rijswijk 1
Pieter C Limburg 1,2
From the 1 Departments of Rheumatology, 2 Pathology and Laboratory Medicine,
University Medical Center Groningen, and 3 Department of Pharmacokinetics & Drug
Delivery, University of Groningen, The Netherlands
Submitted

Chapter 8
108
ABSTRACT Inhibition of intracellular signal transduction is considered to be an interesting target for therapy in inflammation. Especially p38 MAPK inhibitors have been developed and are now in phase II clinical trials for rheumatoid arthritis. This study was designed to investigate the influence of p38 MAPK inhibition on acute phase protein (APP) production, which is dependent on both JAK/STAT and p38 MAPK pathways. We investigated the effects of p38 MAPK inhibition on APP production and mRNA expression in four human hepatoma cell lines, after stimulation with IL-6 and/or IL-1β or TNF-α. These effects were also investigated in human liver slices, a model to mimic the liver in vivo. Two out of four cell lines produced C-reactive protein (CRP), especially after combined IL-6 and IL-1β stimulation. CRP production could be significantly inhibited by the p38 MAPK specific inhibitor RWJ 67657 at 1 µM, which is pharmacologically relevant. Fibrinogen production was also inhibited at 1 µM in all cell lines. Serum amyloid A (SAA) was produced in all four lines and was however not inhibited at 1 µM. In liver slices increased production of APP was detected after stimulation, but p38 MAPK inhibition reduced only fibrinogen production. Concluding we found that production and mRNA expression of CRP and fibrinogen, but not SAA production and expression, significantly were inhibited by p38 MAPK specific inhibitor in hepatoma cell lines. We think that in the case of p38 MAPK inhibitor therapy in rheumatoid arthritis SAA might be a better marker for disease activity than CRP and fibrinogen, because SAA is not directly affected by p38 MAPK inhibition. Key words. CRP, p38 MAPK, SAA, hepatoma cells

p38 MAPK inhibition in acute phase response
109
INTRODUCTION Rheumatoid arthritis (RA) is a chronic inflammatory disease, which leads to the destruction of cartilage and bone in the joints. Cytokines like interleukin (IL)-1β and tumour-necrosis factor (TNF)-α are major players in the pathogenesis of RA 1. During inflammation a number of physiological and metabolic changes, distant from the site of inflammation, are induced which collectively are called the acute phase response. This response is characterized by the change in concentration of many plasma proteins 2. The liver is a major target of systemic inflammatory mediators and is responsible for supplying many components for defence at the site of tissue damage, and is the producer of the acute phase plasma proteins (APP) after stimulation by cytokines3. Measurement of the APP serum CRP (C-reactive protein) is useful in managing disease, since the concentration reflects the inflammatory status of a patient. In RA, serial measurements of CRP are of prognostic value4. The most familiar indicator of the response of the acute phase proteins is the erythrocyte sedimentation rate (ESR), which largely depends on the concentration of fibrinogen. IL-6 is recognized as the principal regulator of most APP genes (the so-called type-2 or IL-6-specific APPs), including the three chains of fibrinogen, haptoglobin, and the protease inhibitors α1-antichymotrypsin, α1-antitrypsin and α2-macroglobulin. The IL-6 like cytokines IL-11, oncostatin M (OSM), leukaemia inhibitory factor (LIF) and ciliary neurothrophic factor (CNTF) induce type-2 APPs in a similar way. Type-1 APPs are regulated by the IL-1-like cytokines and include α1-acid glycoprotein (AGP), complement C3, serum amyloid A (SAA) and CRP. TNF- and IL-1-mediated stimulation of type-1 genes is synergistically enhanced by IL-6-like cytokines, while the production of IL-6-dependent (type-2) APPs usually is inhibited by IL-1-like cytokines 3. The IL-6-like cytokines bind to plasma membrane receptor complexes containing the common signal transducing receptor chain gp 130 (glycoprotein 130). Signal transduction involves the activation of JAK (Janus kinase) tyrosine kinase family members, leading to the activation of transcription factors of the STAT (signal transducers and activators of transcription) family 5. Upon activation, STAT proteins dimerize, translocate to the nucleus, and initiate transcription of the STAT-responsive genes. The importance of the JAK/STAT pathway in RA has not completely been established yet, but STAT1 and STAT3 seem to have both protective and pathogenic properties 6, with a regulating role for the suppressors of cytokine signalling (SOCS) 7. Dimerization of IL-6-type cytokine receptor not only activates the JAK/STAT pathway, but may also stimulate the mitogen-activated protein kinase (MAPK) cascade through activation of Ras, a GTP-binding protein 5. Simultaneous activation of the JAK/STAT and MAPK pathways in a rat hepatoma cell line has also been described for IL-22, an IL-10-related cytokine 8. A role for one of the MAPK homologues, p38 MAPK in IL-6 induced functions has been established in different studies 9;10. Furthermore p38 MAPK is involved in the apoptotic pathway in human hepatoma cell lines, because inhibition of p38 MAPK leads to reduced apoptosis in hepatocellular carcinoma 11. Activation of p38 MAPK in human hepatoma cells by TNF-α, leading to the production of RANTES, is correlated to alcoholic liver disease 12. p38 MAPK plays a dominant role in signal transduction pathways in inflammatory diseases and in the last years several specific inhibitors have been developed (for

Chapter 8
110
review see 13). The p38 MAPK inhibitor RWJ 67657 has been shown to significantly inhibit the release of TNF-α from lipopolysaccharide-treated human peripheral blood mononuclear cells 14, but also from macrophages 15. This compound is approximately 10-fold more potent than the reference standard p38 MAPK inhibitor SB 203580 in all p38 MAPK dependent in vitro systems tested. RWJ 67657 specifically inhibits the enzymatic activity of recombinant p38α and β, but not of γ and δ in vitro, and has no significant activity against a variety of other kinases 14. Since p38 MAPK inhibitors are now in phase II clinical trial for RA 16, it is important to know whether these inhibitors have a direct effect on the production of acute phase proteins, due to cross talk between the JAK/STAT and p38 MAPK cascades, in order to elucidate whether these APP are still a valuable marker for the disease activity during treatment with p38 MAPK inhibitors. In this study, we investigated the effect of p38 MAPK inhibition on IL-6, IL-1β and TNF-α induced acute phase protein production in four different hepatoma cell lines both at the level of mRNA expression and at the level of protein production. The effects on CRP, SAA, complement factor 3 (C3), fibrinogen, and albumin were studied. For mRNA analysis we studied the SAA-1 gene. Fibrinogen was studied by analyzing fibrinogen-β and fibrinogen-γ genes. Fibrinogen-β chain synthesis is considered to be the rate-limiting chain for assembly and secretion of mature fibrinogen. The promoter regions of both β- and γ-genes share the IL-6 responsive element, but the γ-gene lacks the C/EBP response element 17. To study the effects in a system that mimics the liver in vivo, we used human liver slices. The cellular architecture of the liver is retained in the human liver slices, making it the ideal in vitro preparation for multi-cellular processes. This model has been developed and validated for drug metabolism and toxicity studies 18. In addition, this system was used to detect the organ source of the acute phase protein procalcitonin, and it was shown that procalcitonin, in addition to SAA and CRP, in man originates from the liver 19. MATERIALS AND METHODS Reagents RWJ 67657 was provided by Johnson and Johnson (R.W. Johnson Pharmaceutical Research Institute, Raritan, NJ). The human hepatocellular carcinoma cell lines HepG2 and Hep3B, and the hepatoma cell line PLC/PRF/5 were purchased from the ATCC (American Type Culture Collection, Manassas, VA). The hepatoma cell line HuH7 was a kind gift from Dr. R. Kleemann (TNO, Leiden, The Netherlands). Recombinant human IL-1β, IL-6 and TNF-α were from R&D Systems (Minneapolis, MN). Foetal calf serum (FCS) and DMEM (Dullbeco’s-modified Eagle Medium) were obtained from Biowhittaker (Verviers, Belgium). Specific antibodies to p38 MAPK, phospho-p38 MAPK, STAT3 and phospho-STAT3 were purchased from Cell Signalling Technologies (Beverly, MA) and detecting antibody peroxidase-swine-anti-rabbit IgG was from DAKO (Glostrup, Denmark). Antibodies for CRP and fibrinogen ELISA were obtained from DAKO, capture-antibodies for C3 ELISA were from Calbiochem (San Diego, CA) and detecting antibodies were from ICN Biomedicals (Irvine, CA). Antibodies and ELISA for SAA were developed in our laboratory 20.

p38 MAPK inhibition in acute phase response
111
All reagents for RNA isolation and reverse transcriptase reaction were purchased from Invitrogen, Life Technologies (Gaithersburg, MD). Reagents for real-time RT-PCR were obtained from Applied Biosystems (Foster City, CA). Culture of hepatoma cell lines Cell lines were maintained in DMEM supplemented with 10% FCS and gentamycin in a humidified atmosphere of 5% CO2/95% air. Hep3B and PLC/PRF/5 were passaged twice a week in a 1:3 ratio, HuH7 in a 1:10 ratio. HepG2 was passaged weekly in a 1:3 ratio. For experiments, the hepatoma cells lines were grown to confluence in 12 wells (1 ml) or 24 wells (0.5 ml) tissue culture plates (Corning, Schiphol, The Netherlands). Activation of the cells with 50 ng/ml IL-6 and/or 10 ng/ml IL-1β or 10 ng/ml TNF-α was performed in DMEM with 1% FCS and 0.1 µM dexamethasone. Activation of p38 MAPK and STAT3 Phosphorylation of p38 MAPK and STAT3 in hepatoma cell lines was analyzed by western blotting. Confluent cells were stimulated with IL-6, IL-1β or TNF-α for various periods of time (15 and 30 min, 1, 2, 4, 6, and 24 hours) in DMEM with 1% FCS and 0.1 µM dexamethason. Cell extracts were prepared by lysing the cells with 200 µl 1x SDS sample buffer (containing 2% SDS, 10% glycerol, 50 mM dithiothreitol, 62.5 mM Tris-HCl (pH 6.8) and 0.01% brome-phenol blue). Cells were scraped off the wells and the lysates were subsequently sonicated for 5-10 seconds and boiled for 5 minutes. The samples were loaded onto a 10% SDS-PAGE gel and run at 200 V and 15 Watt maximum. Semidry blotting was performed onto nitrocellulose membrane and immunodetection with anti-phospho-p38 MAPK was performed, followed by incubation with peroxidase-anti-rabbit IgG. Enhanced chemi-luminescence (ECL) detection was performed according to the manufacturers guidelines (Lumi-Lightplus, Roche Diagnostics, Mannheim, Germany). Blots were exposed to HyperfilmTM (Amersham Biosciences, Roosendaal, The Netherlands) and developed. Subsequently, blots were stripped with RestoreTM Western Blot Stripping Buffer (Pierce, Rockford, IL) and immunodetection with anti-p38 MAPK was performed. The same procedure was followed for anti-phospho-STAT3 and anti-STAT3 immunodetection. Production of acute phase proteins by hepatoma cell lines Cells were stimulated with IL-6, IL-1β and TNF-α alone or in combination during 48 hours. The effect of p38 MAPK inhibition was investigated by pre-incubation for 1 hour with a concentration range of RWJ 67657 (0, 0.01, 0.1, 1, and 10 µM, diluted from stock solution of 10 mM in DMSO (dimethylsulfoxide)). Production of acute phase proteins was determined in the culture supernatants by ELISAs. For CRP and fibrinogen ELISA rabbit-anti-CRP (1:10000) or rabbit-anti-fibrinogen (1:4000) were coated in 96-well plates. Diluted supernatants were added with standards in a concentration range. After incubation conjugated rabbit-anti-CRP (1:2000) or -fibrinogen (1:4000) was added and detection was performed with the enzyme substrate tetramethyl-benzidin (TMB, Roth, Karlsruhe, Germany) and the reaction was stopped with 1M H2SO4. Plates were read in an ELISA reader at 450 nm and concentration of protein was determined with the SOFTmax PRO software (Molecular

Chapter 8
112
Devices, Sunnyvale, CA). The detection limits for the assays are 0.1 ng/ml and 40 ng/ml for CRP and fibrinogen respectively. For complement C3 ELISA, goat-anti-human C3 (1:2000) and peroxidase-conjugated goat-anti-human-C3 (1:2000) was used. Detection and calculations were done as described for the CRP and fibrinogen ELISAs. The detection limit for the assay is 80 ng/ ml. The SAA ELISA was performed as described before (15). Shortly, a capture monoclonal antibody (Reu.86.5, which reacts with all subtypes of SAA) was coated (1:1000), followed by incubation with diluted supernatants or standards. Detection was done with a peroxidase-conjugated monoclonal antibody (Reu.86.1, specific for SAA-1), followed by the substrate reaction. The detection limit of the assay is 2 ng/ ml. mRNA analysis of acute phase proteins Hepatoma cells (PLC/PRF/5 and Hep3B) were grown to confluence in 12 wells plates and stimulated for 24 hours as described above. One hour pre-treatment with 0, 0.1, 1, and 10 µM RWJ 67657 was done for all stimulations. Total RNA was isolated from the cells with TRIzol reagent according to the manufacturers instructions (Invitrogen, Life Technologies). DNase treatment (Ambion, Huntingdon, Cambridgeshire, UK) was performed and subsequently cDNA was synthesized from 2.0 µg of total RNA using M-MLV Reverse Transcriptase and oligo (dT)14-18 (Invitrogen, Life Technologies). For the measurement of mRNA for albumin, CRP, SAA, C3, fibrinogen-β, fibrinogen-γ and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) 1µl of cDNA in duplicate was used for amplification by the Taqman real-time PCR system (ABI Prism 7900HT Sequence Detection System, Applied Biosystems) with specific Taqman primers/probes. The Assay-on-Demand numbers for the genes were as follows: albumin: Hs00609411_m1, CRP: Hs00357041_m1, C3: Hs00163811_m1, fibrinogen-β: Hs00170586, fibrinogen-γ: Hs00241037_m1, and GAPDH: Hs99999905_m1. For SAA-1, suitable primers and probe were developed between the transit of exon 3 and 4 using the software program Primer Express 2.0 (Applied Biosystems) and the assay was ordered by Assay-by-Design. Amplification was performed using standard conditions: denaturation at 95°C for 15 seconds, 40 cycles of amplification with annealing at 60°C for 1 minute, and extension at 50°C for 2 minutes. According to the comparative Ct (threshold cycle value) method described in the ABI manual, the resulting mRNA amount of the gene of interest was normalized to the housekeeping gene GAPDH, yielding the ∆Ct value. The ∆Ct value of unstimulated cells was subtracted from the average ∆Ct value of each sample, yielding the ∆∆Ct. The amount of target, normalized to an endogenous reference (GAPDH) and relative to the control sample, is given by: 2-∆∆CT. Liver slices to mimic the liver in vivo Human liver tissue was obtained from livers procured from multi-organ donors. Consent from the legal authorities and from the families concerned was obtained from the explantation of organs for transplantation purposes. The human livers were handled as described before 21. Liver tissue cores (diameter 8 mm) were prepared, and stored in ice-cold University of Wisconsin organ preservation solution until slicing. Liver slices (200-300 µm thickness, wet weight 10-14 mg) were prepared with the Krumdieck slicer. The slices were incubated 24 hours in 3.2 ml Williams’ medium E

p38 MAPK inhibition in acute phase response
113
supplemented with 25 mM glucose, 50 µg/ml gentamycin and 0.1 µM dexamethasone. The 6-well tissue culture plates were continuously rocked back and forth (90/min) at 37°C under 5% CO2/95% O2. Liver slices were stimulated in triplicate with 50 ng/ml IL-6 or 10 ng/ml IL-1β during 24 hours with or without pre-treatment for 1 hour with 1 µM RWJ 67657. Supernatants were collected for measurement of acute phase proteins. The triplicate slices were collected and homogenized in TRIzol for isolation of mRNA according to manufacturers instructions. mRNA expression of acute phase proteins was performed as described above for the hepatoma cell lines.
STATISTICS One-way ANOVA with Dunn's Multiple Comparison Test was performed using GraphPad Prism version 3.00 for Windows, GraphPad Software (San Diego, CA).
Figure 1. Phosphorylation of p38 MAPK (A) and STAT3 (B) in hepatoma cell line PLC/PRF/5 after stimulation with cytokines. Hepatoma cells were stimulated with 10 ng/ml IL-1β or TNF-α or 50 ng/ml IL-6 in DMEM with 1% FCS and 0.1µM dexamethason for increasing periods of time. Phosphorylation was measured by Western blot using specific antibodies against p38 MAPK, phospho-p38 MAPK, STAT3 and phospho-STAT3.

Chapter 8
114
RESULTS
Activation of p38 MAPK and STAT3 phosphorylation of p38 MAPK and STAT3 in the cell lines Hep3B and PLC/PRF/5 was analysed after stimulation with IL-6, IL-1β and TNF-α after various periods of time. In figure 1 representative examples are shown of the Western blots. In figure 1A, a rapid phosphorylation of p38 MAPK is demonstrated after stimulation with IL-1β and TNF-α, but not after stimulation with IL-6. In figure 1B, STAT3 is strongly phosphorylated after IL-6 stimulation, whereas after IL-1β and TNF-α stimulation hardly any phosphorylation of STAT3 is detected.
Figure 2. Effect of RWJ 67657 pre-treatment on CRP (A) and SAA (B) production in hepatoma cell lines after stimulation with (combinations of) cytokines. Cell were unstimulated (○) or stimulated with IL-1β (▲), IL-6 (●), TNF-α (∆), IL-1β + IL-6 (■) or TNF-α + IL-6 (□) for 48 hours, with or without pre-treatment of 0.01, 0.1, 1, and 10 µM RWJ 67657. CRP and SAA levels were measured in cell supernatants by ELISA and expressed in ng/ml. In the graphs the mean and SEM is shown. (∗ p<0.05, ∗∗∗ p<0.001, One-way ANOVA with Dunn’s Multiple Comparison Test, tested against the stimulated control).

p38 MAPK inhibition in acute phase response
115
Production of acute phase proteins by hepatoma cell lines In the supernatants of the 4 different hepatoma cell lines the production of the acute phase proteins CRP, SAA, fibrinogen and C3 was measured by ELISA. CRP (type-1 APP) production could not be detected in HepG2 and HuH7 cell lines. In PLC/PRF/5, highest CRP production was seen after combined IL-1β and IL-6 stimulation, and to a lesser extent after TNF-α stimulation alone or together with IL-6. In Hep3B, CRP production was found exclusively after simultaneous stimulation with IL-1β and IL-6 (figure 2A). In both cell lines, CRP production was inhibited by RWJ 67657 both at 1 and 10 µM. High production of SAA was detected in all hepatoma cell lines after combined stimulation with IL-1β and IL-6 (figure 2B). Inhibition due to p38 MAPK inhibition was seen only at 10 µM in Hep3B and HuH7. No significant inhibition was seen in PLC/PRF/5 and HepG2.
Table 1. Effect of RWJ 67657 pre-treatment on fibrinogen production (µg/ml) in 4 hepatoma cell lines (mean of 6 experiments). Cells were unstimulated or stimulated with (combinations of) cytokines for 48 hours, with or without pre-treatment of 0.01, 0.1, 1, and 10 µM RWJ 67657. Fibrinogen levels were measured in cell supernatants by ELISA and expressed in µg/ml. The table shows the mean and SEM (∗ p<0.05, ∗∗ p<0.01, ∗∗∗ p<0.001, One-way ANOVA with Dunn’s Multiple Comparison Test, tested against the stimulated control). (BD = below detection limit, ND = not determined).
unstim IL-1β IL-6 IL-1β + IL-6
TNF-α IL-1β + TNF-α
µM RWJ PLC/PRF 0 2.77 2.13 10.33 5.92 2.35 8.77
0.01 2.47 1.77 10.42 5.57 2.05 8.00 0.1 2.68 1.65 8.67 5.12 1.87 7.20 1 2.35 1.45 8.23 4.57 1.48 5.58*
10 1.68 0.85 5.10* 2.48** 0.95 2.57***
µM RWJ Hep3B 0 BD BD 1.62 0.58 BD 0.35
0.01 1.25 0.62 0.30 0.1 0.80 0.43 0.20 1 0.60* 0.28 0.13
10 0.27*** 0.10** 0.10 µM RWJ HepG2
0 5.21 5.09 20.38 20.30 ND ND 0.01 4.82 5.45 18.08 17.61 0.1 4.30 5.40 17.97 16.58 1 3.93 4.41 15.15 11.44*
10 2.65 2.70 9.71* 5.73*** µM RWJ HuH7
0 2.88 2.90 10.93 5.77 ND ND 0.01 2.23 2.53 9.82 5.56 0.1 2.25 2.27 8.12 6.00 1 2.08 1.81 7.70* 4.47*
10 1.78 1.25 6.30*** 2.56**

Chapter 8
116
Results of fibrinogen production are shown in table 1. Fibrinogen is constitutively produced in the liver and in cultured hepatoma cell lines as well. Fibrinogen is a type-2 APP, and after stimulation with IL-6 a maximal fourfold induction of production is seen. In Hep3B cells fibrinogen production was not constitutive and could only bedetected after IL-6- or combined stimulation. Significant inhibitory effects of p38 MAPK at 10 µM were seen in all 4 cell lines after IL-6 stimulation alone or in combination of IL-6 and IL-1β, and in Hep3B, HepG2 and HuH7 at 1 µM, but not in PLC/PRF/5. In table 2, we show the results of C3 production in the hepatoma cell lines. Like fibrinogen, C3 is also constitutively produced, and production is induced after stimulation with all cytokines and combinations. Significant inhibition at 10 µM RWJ 67657 was seen in all 4 cell lines after IL-1β stimulation and in Hep3B and HepG2 after IL-1β + IL-6 stimulation. In HepG2, there was also significant inhibition of IL-6 induced C3 production at 1 µM. Pre-treatment of hepatoma cells with 0.1% DMSO had no significant effect on any acute phase protein production (data not shown).
Table 2. Effect of RWJ 67657 pre-treatment on C3 production (µg/ml) in 4 hepatoma cell lines (mean of 6 experiments). Cells were unstimulated or stimulated with (combinations of) cytokines for 48 hours, with or without pre-treatment of 0.01, 0.1, 1, and 10 µM RWJ 67657. C3 levels were measured in cell supernatants by ELISA and expressed in µg/ml. The table shows mean and SEM(∗ p<0.05, ∗∗ p<0.01, One-way ANOVA with Dunn’s Multiple Comparison Test, tested against the stimulated control). ND = not determined.
unstim IL-1β IL-6 IL-1β + IL-6
TNF-α IL-1β + TNF-α
µM RWJ PLC/PRF 0 0.72 1.57 2.03 1.62 1.22 1.47
0.01 0.73 1.28 1.37 1.37 1.25 1.52 0.1 0.65 1.20 1.65 1.33 1.12 1.45 1 0.60 1.07* 1.45 1.55 0.98 1.25
10 0.52 0.82** 1.55 1.12 0.72 0.88 µM RWJ Hep3B
0 0.82 3.13 3.85 5.27 1.80 3.23 0.01 0.67 3.72 5.27 6.00 2.05 3.17 0.1 0.65 3.42 4.37 6.00 1.82 2.95 1 0.65 2.57 2.67 4.97 1.48 2.48
10 0.55 1.72** 2.45 2.28* 1.32 1.95 µM RWJ HepG2
0 5.58 11.12 7.33 9.25 ND ND 0.01 4.70 12.95 6.77 7.63 0.1 5.22 11.53 8.30 7.45 1 4.70 8.70 5.77* 6.88
10 3.83 6.40* 4.97** 4.57* µM RWJ HuH7
0 0.48 2.67 1.42 2.83 ND ND 0.01 0.47 3.02 1.30 2.28 0.1 0.58 2.87 1.42 2.53 1 0.75 2.47 1.77 2.22
10 1.23 1.52* 0.92 2.07

p38 MAPK inhibition in acute phase response
117
mRNA analysis of acute phase proteins With quantitative RT-PCR mRNA expression of CRP, SAA-1, fibrinogen-β, fibrinogen-γ, complement C3, and albumin was measured in Hep3B and PLC/PRF/5. As previously mentioned CRP production by HepG2 and HuH7 cells could not be detected. In addition, CRP mRNA expression was not found in these 2 cell lines, and therefore the experiments were performed with the 2 other cell lines. Figure 3 shows the results of mRNA expression of 3 different experiments in Hep3B cells after 24-hour stimulation. Results of mRNA analysis in PLC/PRF/5 were comparable. CRP and SAA-1 mRNA expressions were abundantly induced, especially after combined stimulation with IL-6 and IL-1β. Pre-treatment with 0.1, 1 or 10 µM RWJ 67657 reduced CRP mRNA expression in a dose dependent way, but not SAA-1 mRNA expression. Reduction of
Figure 3. Effect of RWJ 67657 pre-treatment on mRNA expression of CRP, SAA-1, fibrinogen-β, fibrinogen-γ, complement C3 and albumin in Hep3B cells after stimulation with (combinations of) cytokines. Cells were stimulated with IL-1β, IL-6, IL-1β + IL-6, TNF-α, and TNF-α +IL-6 for 24 hours with or without pre-treatment with 0.1, 1, and 10 µM RWJ 67657. mRNA expression was determined with real-time RT-PCR, results are expressed as fold induction compared to unstimulated cells (fold induction = 1). Bars show mean (n=3) and SEM. (∗ p<0.05, One-way ANOVA with Dunn’s Multiple Comparison Test, tested against the stimulated control).

Chapter 8
118
CRP mRNA at 10 µM was significant both after IL-6 and combined IL-6/IL-1β stimulation. Fibrinogen and C3 mRNA are constitutively expressed, but can be induced by cytokine stimulation. Fold inductions after IL-6, combined IL-6/IL-1β and combined IL-6/TNF-α stimulation for fibrinogen-β and fibrinogen-γ were 45.0, 21.0, 7.7 and 13.8, 5.3, 2.5 respectively. mRNA induction for both fibrinogen-chains was significantly reduced by 10 µM p38 MAPK treatment after IL-6 stimulation. C3 mRNA expression was induced after all stimulations, but there was no inhibitory effect detectable due to pre-treatment with RWJ 67657. Albumin mRNA expression was reduced in all stimulated cells, and was further reduced with 10 µM p38 MAPK inhibitor in IL-6-stimulated cells. Effect of RWJ 67657 on protein production and mRNA expression of acute phase proteins in human liver slices The levels of the acute phase proteins CRP, SAA, fibrinogen and C3 were determined in the supernatants of liver slices which were stimulated with IL-6 or IL-1β, with or without pre-treatment with 1 µM RWJ 67657. In figure 4 the results are shown of 2 to 4 experiments, performed in triplicate. In unstimulated cultures, very high levels of CRP and SAA were present: 1090 +/- 247 ng/ml for CRP and 1346 +/- 193 ng/ml for SAA.
Figure 4. Acute phase protein production in human liver slices. CRP, SAA, fibrinogen and C3 levels were determined by ELISA in liver slice supernatants, after 24 hours stimulation with 50 ng/ml IL-6 or 10 ng/ml IL-1β with or without pre-treatment with 1µM RWJ 67657. Experiments (n=2-4) were performed in triplicate, bars show mean and SEM. (∗ p<0.05, One-way ANOVA with Dunn’s Multiple Comparison Test, tested against the unstimulated control). Stimulation raised the CRP and SAA levels to 2212 +/- 376 ng/ml and 2546 +/- 193 ng/ml after IL-6 stimulation, and 2101 +/- 220 ng/ml and 2199 +/- 274 ng/ml after IL-1β stimulation.

p38 MAPK inhibition in acute phase response
119
The induced levels differ significant (p<0.05) from the levels in unstimulated slices. Pre-treatment with 1 µM RWJ 67657 had no effect on the CRP and SAA levels. Fibrinogen was induced more than two-fold after stimulation (significant after IL-6 stimulation) and there was an inhibitory effect of p38 MAPK treatment, although not significant. Complement C3 levels were not induced and not inhibited in the liver slices. The expression of CRP, SAA-1, fibrinogen-β and -γ, and C3 mRNA in liver slices was conform the protein expression (data not shown). Unstimulated samples already had a high CRP and SAA-1 mRNA expression, which could be seen from the low number of cycli relative to GAPDH needed in the RT-PCR and expression was less induced by stimulation compared to the hepatoma cell lines. Albumin mRNA expression was reduced after cytokine treatment. No effects of p38 MAPK inhibition were seen for all genes tested. Statistical analysis was not possible due to the small number of experiments. DISCUSSION In this study, the effect of p38 MAPK inhibition on the acute phase response was investigated in hepatoma cell lines. We found significant inhibition of CRP and fibrinogen production at 1 µM RWJ 67657, which is considered a pharmacologically relevant concentration. The importance of the cytokines IL-1β and TNF-α in inflammation has been established, particularly by the success of cytokine blockade in the treatment of RA. Also the use of anti-IL-6 antibody intended to block the JAK/STAT pathway seems promising 22. The interest in the regulation of cytokine production by signal transduction pathways and transcription factors has led to new therapeutic targets 23. For RA several p38 MAPK inhibitors have been designed, which have been tested pre-clinically and in some cases even in clinical studies 13;16. In clinical trials, responses are measured by using the American College of Rheumatology (ACR) criteria and several laboratory markers, including acute phase proteins. It is important to know whether the levels of acute phase proteins are not directly blocked by therapeutic agents. Stimulation of hepatoma cells with IL-6 induced a weak p38 MAPK phosphorylation after 1 hour, in contrast to the rapid and strong phosphorylation by IL-1β and TNF-α stimulation. These cytokines could also induce a weak STAT3 phosphorylation in hepatoma cells, but only at a late time point (24 hours). In contrast to others 24;25 it was not possible for us to detect CRP production or mRNA expression by HepG2 and HuH7 cells making these cells less valuable as model to study acute phase reactions. Induction of CRP production and mRNA expression however was found in Hep3B and PLC/PRF/5 cells especially after combined IL-6 and IL-1β stimulation (in PLC/PRF/5 also after IL-6 and TNF-α stimulation). Pre-treatment with the p38 MAPK inhibitor at 1 µM did significantly reduce CRP production. The study by Parasrampuria 26 demonstrated that after a single oral dose of RWJ 67657 ranging from 0.25 to 30 mg/kg a plasma concentration of 0.01 to 6 µM of the p38 MAPK inhibitor could be reached in humans, so 1 µM is a pharmacologically relevant concentration. Recent studies investigating prognostic factors in RA 27, but

Chapter 8
120
also work from our group 4, have demonstrated that levels of CRP especially seem to be predictive for joint damage. All 4 cell lines showed high production of SAA, again especially after combined IL-6 and IL-1β stimulation, and inhibition by RWJ 67657 was seen only at 10 µM. SAA induction was also seen in PLC/PRF/5 after IL-6 and TNF-α stimulation and inhibition of SAA production at 1µM RWJ 67657 was seen in HuH7 cells after IL-1β stimulation. Strong induction of SAA-1 mRNA was demonstrated in PLC/PRF/5 and Hep3B after combined IL-6 and IL-1β stimulation. Our results confirmed the data reported by Hagihara et al 28, concerning the critical role of IL-6 in the synergistic induction of SAA gene expression in hepatoma lines. Moreover they found that anti- IL-6R Mab almost completely inhibited the synergistic increase of SAA-1 after triple cytokine stimulation, while IL-1ra and anti-TNF-α Mab only had moderate effects. In our study, the use of a p38 MAPK inhibitor had no effect on SAA production and expression, in contrast to the effects on CRP. Fibrinogen synthesis is mainly mediated by IL-6, and this was shown in our studies in the 4 hepatoma cell lines. Combined stimulation induces lower fibrinogen production, due to cross-talk between IL-6 and IL-1β signalling pathways leading to IL-1β dependent down-regulation of STAT1 phosphorylation 29. p38 MAPK treatment additionally reduced fibrinogen production independent of which stimulus was used. Fibrinogen plays an important role in the coagulation cascade, but is also an important acute phase protein, because the often-used ESR largely depends on the fibrinogen concentration. The fact that indeed fibrinogen production is reduced by p38 MAPK treatment could have important implications for the use of ESR as marker of disease activity. Finally, complement production was reduced in all cell lines at 10 µM RWJ 67657, but no effect on mRNA expression was seen. Albumin mRNA expression was reduced after stimulation with cytokines as expected, and no effect of p38 MAPK treatment was demonstrated. In human liver slices control incubations demonstrated a spontaneous release of acute phase proteins. This has been shown before 19 and may be related to the source of the human liver. The human liver slices are prepared from livers from brain-dead donors, and it is known that e.g. CRP is up regulated after brain-death, due to high levels of IL-6 30. We have shown increased levels of IL-6 in unstimulated slice culture medium after 24 hours, which were further increased after LPS stimulation 31. However, incubation of the human liver slices with IL-6 and IL-1β still led to an increase of the release of acute phase proteins. These results indicate there is induction of the acute phase proteins release possible as was seen in previously performed studies 19. However, in contrast to the results of the hepatoma cell lines, pre-incubation with RWJ 67657 did not lead to a significant reduction in acute phase proteins in human liver slices, although a tendency was seen in the case of fibrinogen. In humans a plasma concentration of 6 µM RWJ 67657 can be achieved after a single oral dose. In our study with liver slices we only used a concentration of 1 µM, which could be too low to inhibit production of for instance CRP. CRP and SAA are the major acute phase reactants in rheumatoid arthritis, but they have a different function and are differentially regulated 2, although both CRP and SAA are potently induced by combined IL-6 and IL-1β stimulation. The fact that CRP

p38 MAPK inhibition in acute phase response
121
production is inhibited at the pharmacological relevant concentration of 1 µM RWJ 67657, while SAA production is not, might be explained by differences in the acute phase responsive elements in the promoter regions of the two APP 32. Cunnane et al 33 demonstrated that SAA is the best marker for the assessment of inflammatory joint disease, while Rau et al demonstrated that in acute pancreatitis SAA had a wider dynamic range, but measurement of CRP provided an earlier differentiation between patients 34. The results presented in this study may implicate that SAA measurement should be included in clinical trials when p38 MAPK inhibitors are used. CONCLUSIONS In human hepatoma cell lines a significant inhibition of CRP and fibrinogen production and mRNA expression was found after treatment with a p38 MAPK specific inhibitor at pharmacologically relevant concentrations. SAA production however was not reduced in these cell lines. In human liver slices, which mimics an in vivo model, fibrinogen production was also reduced. As p38 MAPK inhibitors are now in phase II clinical trials, we encourage to include the use of SAA as marker for disease activity, because this acute phase protein does not seem to be affected directly by p38 MAPK treatment. ACKNOWLEDGEMENTS Supported by the Dutch Arthritis Association and Johnson and Johnson Pharmaceutical Research and Development, Raritan, New Jersey, USA. REFEFERENCES 1 Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis.
N.Engl.J.Med. 2001; 344: 907-16. 2 Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation.
N.Engl.J.Med. 1999; 340: 448-54. 3 Baumann H, Gauldie J. The acute phase response. Immunol.Today 1994; 15: 74-80. 4 van Leeuwen MA, van Rijswijk MH, Sluiter WJ et al. Individual relationship between
progression of radiological damage and the acute phase response in early rheumatoid arthritis. Towards development of a decision support system. J.Rheumatol. 1997; 24: 20-7.
5 Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem.J. 2003; 374: 1-20.
6 Ivashkiv LB, Hu X. The JAK/STAT pathway in rheumatoid arthritis: pathogenic or protective? Arthritis Rheum. 2003; 48: 2092-6.
7 Egan PJ, Lawlor KE, Alexander WS, Wicks IP. Suppressor of cytokine signaling-1 regulates acute inflammatory arthritis and T cell activation. J.Clin.Invest 2003; 111: 915-24.
8 Lejeune D, Dumoutier L, Constantinescu S, Kruijer W, Schuringa JJ, Renauld JC. Interleukin-22 (IL-22) activates the JAK/STAT, ERK, JNK, and p38 MAP kinase pathways in a rat hepatoma cell line. Pathways that are shared with and distinct from IL-10. J.Biol.Chem. 2002; 277: 33676-82.
9 Ahmed ST, Mayer A, Ji JD, Ivashkiv LB. Inhibition of IL-6 signaling by a p38-dependent pathway occurs in the absence of new protein synthesis. J.Leukoc.Biol. 2002; 72: 154-62.
10 Zauberman A, Zipori D, Krupsky M, Ben Levy R. Stress activated protein kinase p38 is involved in IL-6 induced transcriptional activation of STAT3. Oncogene 1999; 18: 3886-93.
11 Iyoda K, Sasaki Y, Horimoto M et al. Involvement of the p38 mitogen-activated protein kinase cascade in hepatocellular carcinoma. Cancer 2003; 97: 3017-26.

Chapter 8
122
12 Hirano F, Komura K, Fukawa E, Makino I. Tumor necrosis factor alpha (TNF-alpha)-induced RANTES chemokine expression via activation of NF-kappaB and p38 MAP kinase: roles of TNF-alpha in alcoholic liver diseases. J.Hepatol. 2003; 38: 483-9.
13 Kumar S, Boehm J, Lee JC. p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat.Rev.Drug Discov. 2003; 2: 717-26.
14 Wadsworth SA, Cavender DE, Beers SA et al. RWJ 67657, a potent, orally active inhibitor of p38 mitogen-activated protein kinase. J.Pharmacol.Exp.Ther. 1999; 291: 680-7.
15 Westra J, Doornbos-Van Der Meer B, de Boer P, van Leeuwen MA, van Rijswijk MH, Limburg PC. Strong inhibition of TNF-alpha production and inhibition of IL-8 and COX-2 mRNA expression in monocyte-derived macrophages by RWJ 67657, a p38 mitogen-activated protein kinase (MAPK) inhibitor. Arthritis Res.Ther. 2004; 6: R384-R392.
16 Nikas SN, Drosos AA. SCIO-469 Scios Inc. Curr.Opin.Investig.Drugs 2004; 5: 1205-12. 17 Gervois P, Vu-Dac N, Kleemann R et al. Negative regulation of human fibrinogen gene
expression by peroxisome proliferator-activated receptor alpha agonists via inhibition of CCAAT box/enhancer-binding protein beta. J.Biol.Chem. 2001; 276: 33471-7.
18 Olinga P, Hof IH, Merema MT et al. The applicability of rat and human liver slices to the study of mechanisms of hepatic drug uptake. J.Pharmacol.Toxicol.Methods 2001; 45: 55-63.
19 Nijsten MW, Olinga P, The TH et al. Procalcitonin behaves as a fast responding acute phase protein in vivo and in vitro. Crit Care Med. 2000; 28: 458-61.
20 Hazenberg BP, Limburg PC, Bijzet J, van Rijswijk MH. A quantitative method for detecting deposits of amyloid A protein in aspirated fat tissue of patients with arthritis. Ann.Rheum.Dis. 1999; 58: 96-102.
21 Olinga P, Merema M, Hof IH et al. Effect of human liver source on the functionality of isolated hepatocytes and liver slices. Drug Metab Dispos. 1998; 26: 5-11.
22 Nishimoto N, Yoshizaki K, Miyasaka N et al. Treatment of rheumatoid arthritis with humanized anti-interleukin-6 receptor antibody: a multicenter, double-blind, placebo-controlled trial. Arthritis Rheum. 2004; 50: 1761-9.
23 Smolen JS, Steiner G. Therapeutic strategies for rheumatoid arthritis. Nat.Rev.Drug Discov. 2003; 2: 473-88.
24 Kleemann R, Gervois PP, Verschuren L, Staels B, Princen HM, Kooistra T. Fibrates down-regulate IL-1-stimulated C-reactive protein gene expression in hepatocytes by reducing nuclear p50-NFkappa B-C/EBP-beta complex formation. Blood 2003; 101: 545-51.
25 Smith JW, McDonald TL. Production of serum amyloid A and C-reactive protein by HepG2 cells stimulated with combinations of cytokines or monocyte conditioned media: the effects of prednisolone. Clin.Exp.Immunol. 1992; 90: 293-9.
26 Parasrampuria DA, de Boer P, Desai-Krieger D, Chow AT, Jones CR. Single-dose pharmacokinetics and pharmacodynamics of RWJ 67657, a specific p38 mitogen-activated protein kinase inhibitor: a first-in-human study. J.Clin.Pharmacol. 2003; 43: 406-13.
27 Lindqvist E, Eberhardt K, Bendtzen K, Heinegard D, Saxne T. Prognostic laboratory markers of joint damage in rheumatoid arthritis. Ann.Rheum.Dis. 2004.
28 Hagihara K, Nishikawa T, Isobe T, Song J, Sugamata Y, Yoshizaki K. IL-6 plays a critical role in the synergistic induction of human serum amyloid A (SAA) gene when stimulated with proinflammatory cytokines as analyzed with an SAA isoform real-time quantitative RT-PCR assay system. Biochem.Biophys.Res.Commun. 2004; 314: 363-9.
29 Shen X, Tian Z, Holtzman MJ, Gao B. Cross-talk between interleukin 1beta (IL-1beta) and IL-6 signalling pathways: IL-1beta selectively inhibits IL-6-activated signal transducer and activator of transcription factor 1 (STAT1) by a proteasome-dependent mechanism. Biochem.J. 2000; 352 Pt 3: 913-9.
30 Amado JA, Lopez-Espadas F, Vazquez-Barquero A et al. Blood levels of cytokines in brain-dead patients: relationship with circulating hormones and acute-phase reactants. Metabolism 1995; 44: 812-6.
31 Elferink MG, Olinga P, Draaisma AL et al. LPS-induced downregulation of MRP2 and BSEP in human liver is due to a posttranscriptional process. Am.J.Physiol Gastrointest.Liver Physiol 2004; 287: G1008-G1016.

p38 MAPK inhibition in acute phase response
123
32 Steel DM, Whitehead AS. The major acute phase reactants: C-reactive protein, serum amyloid P component and serum amyloid A protein. Immunol.Today 1994; 15: 81-8.
33 Cunnane G, Grehan S, Geoghegan S et al. Serum amyloid A in the assessment of early inflammatory arthritis. J.Rheumatol. 2000; 27: 58-63.
34 Rau B, Steinbach G, Baumgart K, Gansauge F, Grunert A, Beger HG. Serum amyloid A versus C-reactive protein in acute pancreatitis: clinical value of an alternative acute-phase reactant. Crit Care Med. 2000; 28: 736-42.

9
Summary, general conclusions and future
perspectives
Johanna Westra

Chapter 9
126
SUMMARY, GENERAL CONCLUSIONS AND FUTURE PERSPECTIVES In this thesis the potential of signal transduction pathways as treatment target in rheumatoid arthritis (RA) was investigated. To explain the importance of intracellular signal transduction pathways in inflammation we first outlined the pathways involved in rheumatoid arthritis in chapter 2. In particular the p38 mitogen-activated protein kinase (MAPK) pathway has been found to be an interesting target for therapy in RA. The p38 MAPK is activated in different cells by inflammatory cytokines such as TNF-α and IL-1β, but also by other stress factors. The most important downstream effect of signal transduction pathways in inflammation is the induction of gene transcription and translation into specific proteins. These proteins, including cytokines, matrix-metalloproteinases, and cyclo-oxygenase-2 (COX-2) are involved in the pathogenesis of RA. Moreover the p38 MAPK pathway has been found to play a role in stabilizing mRNA of inflammatory genes, thereby influencing the efficiency of translation. The important advantage of inhibition of the p38 MAPK pathway is not only that the production of the major pro-inflammatory cytokines is inhibited, but also that the effects of these cytokines on other cells is inhibited. In chapter 3 the effects of the p38 MAPK specific inhibitor RWJ 67657 on the production of pro-inflammatory mediators by rheumatoid synovial fibroblasts (RSF) was investigated. There is growing evidence that activated RSF play a major role in both initiating and driving RA. These cells can attach to the articular cartilage and invade the extracellular matrix. Especially by the production of matrix metalloproteinases (MMPs) irreversible damage to cartilage and bone is induced. In this study synovial fibroblasts isolated from RA synovium were stimulated with TNF-α and/or IL-1β, and the effects of p38 MAPK inhibition on protein and mRNA levels of MMP-1 (collagenase 1), MMP-3 (stromelysin-1), tissue inhibitor of metalloproteinase -1 (TIMP-1), IL-6 and IL-8 were determined, as well as the mRNA expression of COX-2 and aggrecanase-1 (ADAMTS-4, a disintegrin and metalloproteinase with thrombospondin-1 motif). Although TNF-α is considered to be the most important pro-inflammatory cytokine in RA, stimulation with IL-1β induced higher levels of pro-inflammatory mediators in rheumatoid synovial fibroblasts. We found that MMP-3 production was significantly inhibited at 1 µM RWJ 67657, MMP-1 production at 10 µM, whereas TIMP-1 production was not inhibited. Significant inhibition of IL-6 and IL-8 protein production was already seen at 0.1 µM of RWJ 67657. The effects on mRNA expression profiles were in concordance with the inhibition of protein production. Significant inhibition of COX-2 mRNA expression already occurred at 0.01 µM, while inhibition of ADAMTS-4 mRNA was seen at 1 µM. Effects of RWJ 67657 below 5 µM are considered relevant, since these concentrations are achieved after a single oral dose in humans.

Summary
127
The inhibitory effects of RWJ 67657 on MMP-3 production are important since MMP-3 is present at very high levels in RA synovial fluid, and is not only a major degradative enzyme itself, but acts as activator of other MMPs as well. Another significant effect is the strong inhibition of COX-2 mRNA expression. COX-2 is selectively induced during inflammation and leads to the enhanced production of prostaglandins. Inhibition of prostaglandin production, either with general or selective cyclo-oxygenase inhibitors, is a major goal for treatment in RA. The results from this study are promising since production of inflammatory cytokines, MMPs, and COX-2 in rheumatoid synovial cells appears to be inhibited by p38 MAPK inhibition. The synovial lining layer consists mainly of synovial fibroblasts and macrophages. Monocytes/macrophages are the major producers of TNF-α and IL-1β, but also of other inflammatory cytokines and MMPs. The strong effects of p38 MAPK inhibitors was originally discovered by the inhibition of TNF-α production in LPS-stimulated monocytes, but the effects on macrophages were unknown. In chapter 4 we used monocyte-derived macrophages (MDM) from healthy controls and RA patients to investigate the effects of p38 MAPK inhibition on macrophages. Isolation and culture of macrophages from synovial tissue is disturbed by the overgrowth of fibroblasts, so for this study monocytes were differentiated with macrophage-colony stimulating factor (M-CSF) and low serum concentration, which generated macrophages with high HLA-DR expression, that were not activated. Treatment of MDM with increasing concentrations RWJ 67657 resulted in a highly significant inhibition of TNF-α production at 0.01 µM and significant inhibition of IL-8 at 0.1 µM. Effects on MMP production were not seen, since MMP-1 (collagenase-1) production by MDM was below detection limit and MMP-9 (gelatinase-2) production in these cells was constitutively high, and was only moderately induced by cytokines and not inhibited by p38 MAPK treatment. Inhibition at the level of mRNA expression was seen for TNF-α, IL-1β, IL-8 and COX-2. The effects on macrophages were not as strong as for the synovial fibroblasts, since higher concentrations of the p38 MAPK inhibitor were needed to reach significant inhibitory activity. From the previous chapter it was clear that there are differences between monocytes and monocyte-derived macrophages (MDM), for instance in regulation of IL-1β production, which is produced by monocytes but not excreted by macrophages. In chapter 5 therefore the reactivity of monocytes and MDM towards p38 MAPK inhibition was investigated, as well as the effects of p38 MAPK inhibition on differentiation of monocytes into macrophages. We found that monocytes produced much more cytokines (TNF-α, IL-1β and IL-8) than MDM, but that MMP-9 was more abundantly produced by MDM. Furthermore it was demonstrated that p38 MAPK inhibition of cytokine production was more effective in monocytes than in MDM. Differentiation of monocytes into MDM in the presence of the p38 MAPK inhibitor reduced both

Chapter 9
128
cytokine and MMP-9 production by MDM. To determine whether signal transduction pathways were altered due to cell differentiation the activation state of intracellular signal transduction pathways was assessed using a kinase array on both stimulated and unstimulated monocytes and MDM. Results showed that all kinases involved in the p38 MAPK pathway were activated in the stimulated cells. However there were a large number of kinases that were differentially activated in monocytes compared to MDM. Another group of cells involved in inflammation are the endothelial cells (EC), which line the bloodvessels. EC are not just passive bystanders but are active responders to stimuli like activated leukocytes and cytokines. Activated EC can produce a number of inflammatory mediators, such as chemokines and they express cellular adhesion molecules (CAMs), which play a major role both in recruitment as well as migration of leukocytes into the inflamed area. In chapter 6 the effects of p38 MAPK inhibition on expression of the adhesion molecules E-selectin, vascular cell adhesion molecule-1 (VCAM-1) and intracellular adhesion molecule-1 (ICAM-1) was investigated and the effects on the production of the chemokines IL-8 and monocyte chemo-attractant protein-1 (MCP-1). p38 MAPK inhibition at a relevant concentration (1µM) significantly reduced protein and mRNA levels of IL-8, MCP-1 and of IL-6. The effects on CAM expression were negligible. We could only demonstrate an insignificant reduction of E-selectin protein and mRNA expression. Since chemokines play an essential role in maintaining the leukocyte-endothelial interactions after the initial interaction regulated by the selectins, the significant downregulation of IL-8 and MCP-1 could have an important effect on leukocyte attraction and infiltration in inflammatory disease. Since the effects of p38 MAPK inhibition on CAM expression by endothelial cells were only modest the involvement of another inflammatory pathway was investigated in chapter 7. Overexpression of mutant IκB protein leading to continuous blocking of NF-κB mediated signalling confirmed a major role of this pathway in controlling both TNF-α and IL-1β induced expression of most of the genes studied. MOL-294, which inhibits thioredoxin involved in NF-κB signalling, inhibited adhesion molecule expression in contrast to pyrrolidine dithiocarbamate (PDTC) and dexamethasone (DEX), which both exerted limited effects at 1 µM. In the previous chapter we demonstrated that 1 µM RWJ 67657, an inhibitor of p38 MAPK activity, diminished TNF-α and IL-1β induced expression of IL-6, IL-8, and E-selectin, but had little effect on VCAM-1 and ICAM-1. Combined treatment of HUVEC with MOL-294 and RWJ 67657 resulted in significant blocking of the expression of the majority of genes studied. The inhibitory effects were much stronger than those observed by single drug treatment, indicating that the use of combinations of drugs affecting multiple targets in activated endothelial cells may offer new therapeutic possibilities.

Summary
129
Finally in chapter 8 a study was designed to investigate the influence of p38 MAPK inhibition on acute phase protein (APP) production, which is dependent on both JAK/STAT and p38 MAPK pathways. In diseases like RA acute phase proteins such as C-reactive protein (CRP) and the erythrocyte sedimentation rate (ESR), which is largely determined by the concentration of fibrinogen, are used as markers of disease activity. We investigated the effects of p38 MAPK inhibition on APP production and mRNA expression in four human hepatoma cell lines, after stimulation with IL-6 and/or IL-1β or TNF-α. These effects were also investigated in human liver slices, a model to mimic the liver in vivo. Concluding we found that production and mRNA expression of CRP and fibrinogen, but not SAA (serum amyloid A) were significantly inhibited by a p38 MAPK specific inhibitor in hepatoma cell lines. In liver slices increased production of APP was detected after stimulation, but p38 MAPK inhibition reduced only fibrinogen production. The consequences of a differential inhibition of the acute phase protein production may well be that in the case of p38 MAPK inhibitor therapy in rheumatoid arthritis SAA will be a better marker for disease activity than CRP and fibrinogen, because SAA is not directly affected by p38 MAPK inhibition. CONCLUSIONS In this thesis the potential of signal transduction pathways as treatment target in rheumatoid arthritis was investigated. We found that in cells, which are esential players in the inflammatory process in RA, p38 MAPK inhibition had important effects. In rheumatoid synovial fibroblasts MMP-3, IL-6, and IL-8 production and mRNA expression were significantly inhibited. The mRNA expression of COX-2 was dramatically reduced at very low p38 MAPK inhibitor concentrations. Previously it was shown that TNF-α production by LPS-stimulated monocytes was inhibited by p38 MAPK inhibition. We demonstrated that also TNF-α production by macrophages, is effectively reduced by p38 MAPK inhibition. Endothelial cells which are actively involved in recruitment of inflammatory leukocytes, have display a different response to p38 MAPK inhibitors. Chemokine expression is effectively inhibited by p38 MAPK inhibition, while cell adhesion molecule expression is regulated by the NF-κB pathway. Finally we showed that there is cross-talk between the p38 MAPK pathway and the JAK-STAT pathway in hepatoma cells, by demonstrating that C-reactive protein and fibrinogen are reduced in hepatoma cell lines by p38 MAPK inhibition. Summarizing we conclude that indeed p38 MAPK is one of the most important signal transduction pathways in RA and that inhibition has important effects on inflammatory cells. Because the NF-κB pathway also plays a role in this inflammatory process a combination of drugs that affect both pathways could even be more effective. Because however p38 MAPK inhibitors are not in clinical practice at the moment due to side effects, the search for new therapeutic targets is still going on.

Chapter 9
130
FUTURE PERSPECTIVES The research described in this thesis confirms the important role of the p38 MAPK pathway in pro-inflammatory responses. However p38 MAPK is not the only pathway involved in inflammation. There seem to be a large number of other kinases and transcription factors that might be promising therapeutic targets. Progression in this field has been hampered by the complexity of the different intracellular signal transduction pathways involved in chronic inflammatory diseases such as RA. Over the years the interest in studying synovial tissue has increased because, the synovium is recognized as the primary site of inflammation in RA. Synovial biopsies can be used to investigate the autoinflammatory response for diagnostic purposes and pathogenetic studies. Serial biopsies can be used to evaluate the effects of novel treatment modalities. A new approach to study the synovium would be the mapping of intracellular signaling systems and to investigate their linkage to normal and disease-related processes. A significant step forward has been made by the development of kinase arrays. Kinase arrays enable to study the activity of all (activated) kinases in whole tissue or cell lysates. One array may include over 1000 peptides, which are known substrate sequences for kinases. This new technique offers the opportunity to determine the intracellular signal transduction profile (IST) of the synovium of patients with RA at different stages of the disease and during different therapies. We recently started to investigate the IST routes (or profiles) in RA patients. We think it will be important to make such a profile from each RA patient, because the IST profile could be an important predictor for the individual response to the installed therapy. For this study the kinase array will be used, which makes it possible to investigate all involved IST routes at the same time. This kinase array already has been used in cultured cells, intestinal tissue and mice. We now want to extrapolate the findings towards synovial tissue. The findings as obtained by the kinase array will next be compared with findings on activationroutes in more conventional and well validated assays. We hope that by knowing the involved IST routes in RA, we will be able to treat a RA patient with the right therapy at the right time and make a basis for the development of new therapeutics. This could also mean that combinations of signal transduction inhibitors or combinations of signal transduction inhibitors with other drugs, for instance MTX, could be effective therapies in RA.

10
Nederlandse samenvatting
Johanna Westra

Chapter 10
132
SAMENVATTING (voor niet-ingewijden) Reumatoïde artritis (RA) is een chronische ontstekingsziekte, die met name gelocaliseerd is in de gewrichten. De ziekte komt voor bij 0.5 - 1% van de bevolking, en komt meer voor bij vrouwen dan bij mannen. Door de ontsteking in de gewrichten raken kraakbeen en bot beschadigd en dit kan leiden tot handicaps en verminderde kwaliteit van leven. De behandeling van RA is er ten eerste op gericht om de verschijnselen van ontsteking (pijn en koorts) te bestrijden en in de tweede plaats om de schade aan de gewrichten te beperken. Uit onderzoek is gebleken dat de zogenaamde cytokines tumor necrosis factor (TNF-α) en interleukine-1 (IL-1β) een centrale rol spelen in het ontstekingsproces. De laatste jaren is gebleken dat het blokkeren van deze cytokines met antilichamen een goede therapie is voor RA. Helaas reageren niet alle patiënten op deze therapie en komen ook incidenteel ernstige bijwerkingen van deze therapie voor. Een andere manier om de produktie en activiteit van deze cytokines te remmen is door het blokkeren van intracellulaire signaal transductieroutes. Deze routes zijn mechanismen waarbij een prikkel buiten de cel omgezet wordt in een reactie in de cel, meestal in de vorm van de produktie van ontstekingseiwitten. In deze signaal transductie routes spelen de zogenaamde kinases een belangrijke rol, doordat ze het signaal in de vorm van een fosfaatgroep aan elkaar doorgeven. In dit proefschrift is onderzocht of het remmen van een belangrijke intracellulaire signaal transductie route (de p38 mitogen-activated protein kinase (MAPK) route) een goede therapie zou kunnen zijn voor RA. In ons onderzoek is gekeken naar de gevolgen van remming met een specifieke p38 MAPK remmer (RWJ 67657) in cellen, die een rol spelen bij ontsteking. In hoofdstuk 2 is wordt een overzicht gegeven van de intracellulaire signaal transductie routes, die belangrijk zijn in ontsteking. Het is gebleken dat met name de zogenaamde p38 MAPK route van belang is bij RA. De p38 MAPK route wordt geactiveerd in verschillende cellen door cytokines, maar ook door andere stress factoren. Door de activatie van de p38 MAPK route gaan de cellen eiwitten produceren, die het ontstekingsproces in stand houden. Eiwitten die geproduceerd worden zijn bijvoorbeeld de eerdergenoemde TNF-α en IL-1β, maar ook andere cytokines en enzymen, die kraakbeen en bot kunnen beschadigen, de zogenaamde matrix-metalloproteinasen (MMPs). Verschillende p38 MAPK remmers zijn ontwikkeld, maar tot nu toe zijn nog maar twee remmers gebruikt in fase II klinisch onderzoek in RA. In dit hoofdstuk worden ook andere belangrijke routes, de NF-κB (nuclear factor -κB) en de JAK/STAT (Janus kinase/ signal transducer of activation and transcription) routes, besproken.

Nederlandse samenvatting
133
De ontsteking bij RA komt voor in de gewrichten en wel in de weefsel bekleding, het synovium. Daar zijn met name drie soorten cellen actief in het ontstekingsproces, de fibroblasten, macrofagen en endotheel cellen. Door de ontsteking gaan de synoviale fibroblasten zich sterk vermeerderen en produceren cytokines zoals IL-6 en IL-8, maar ook de eerder genoemde MMPs. In hoofdstuk 3 is gekeken wat het effect is van behandeling met de p38 MAPK remmer op de eiwitproduktie door synoviale fibroblasten, die geïsoleerd zijn uit synovium van RA patiënten. Wij vonden dat behandeling met RWJ 67657 de productie van IL-6, IL-8 en MMP-3 significant deed afnemen en in mindere mate de productie van MMP-1. Aangezien het MMP-3 een zeer belangrijk enzym is, dat niet alleen in grote hoeveelheden voorkomt in de synoviale vloeistof van RA patiënten, maar ook andere MMPs kan activeren, is deze bevinding van groot belang. In dit onderzoek is niet alleen gekeken naar de eiwit productie door de cel, maar ook naar de voorloper van de eiwitproductie, het zogenaamde mRNA in de cel. Gebleken is dat de p38 MAPK remmer het mRNA van IL-6, IL-8 en MMP-3 remt, als ook de mRNA expressie van cyclo-oxygenase-2 (COX-2). COX-2 is een belangrijke component in ontsteking en het gebruik van specifieke COX-2 remmers is een belangrijk onderdeel van therapie voor RA patiënten. In het bloed circuleren onder andere monocyten, die in weefsels uit kunnen rijpen tot macrofagen. Van deze cellen is bekend dat ze bij ontsteking TNF-α kunnen produceren. Om te onderzoeken wat het effect van p38 MAPK inhibitie is op deze cellen hebben we in hoofdstuk 4 monocyten uit het bloed van gezonde vrijwilligers en RA patiënten laten differentiëren tot macrofagen. Uit dit onderzoek bleek dat RWJ 67657 al bij lage concentraties effectief de productie van TNF-α kon remmen, terwijl ook IL-8 productie werd geremd. Het bleek ook dat de cellen wel veel MMP-9 konden produceren, maar dat deze productie niet geremd werd door p38 MAPK inhibitie. Op het gebied van mRNA expressie bleek dat ook het COX-2 mRNA in macrofagen sterk geremd werd door p38 MAPK inhibitie. In hoofdstuk 5 wordt nog wat dieper ingegaan op de verschillen tussen monocyten en macrofagen. Met name is gekeken naar de reactiviteit ten opzichte van p38 MAPK remming en ook naar de invloed van p38 MAPK inhibitie op differentiatie. We vonden dat monocyten in staat waren om meer cytokines (TNF-α, IL-1β en IL-8) te produceren, terwijl de macrofagen meer MMP-9 konden produceren. Ook bleek dat de remming van cytokine productie door p38 MAPK inhibitie in monocyten veel effectiever was dan in macrofagen. Bij differentiatie van monocyt naar macrofaag in aanwezigheid van RWJ 67657 werd de productie van zowel cytokines als MMP-9 door macrofagen sterk gereduceerd. Om te bepalen of signaal transductie routes veranderen tijdens differentiatie werd de intracellulaire activiteit van kinases als maat voor de activiteit van signaal transductie routes, meten met en kinase array, uitgevoerd op gestimuleerde en

Chapter 10
134
ongestimuleerde monocyten en macrofagen. Een kinase array is een methode waarmee de activiteit van meerdere kinases tegelijkertijd in een monster gemeten kan worden. Uit de array bleek dat alle kinases uit de p38 MAPK route geactiveerd waren in de gestimuleerde monocyten en macrofagen. Ook waren een groot aantal kinases differentieel geactiveerd in monocyten vergeleken met macrofagen. Een andere groep cellen die een rol spelen in ontsteking zijn de endotheelcellen (EC). Dit zijn cellen die bloedvaten bekleden en die een actieve rol spelen bij de migratie van witte bloedcellen (leukocyten) naar een ontstekingplaats. Geactiveerde EC produceren chemokines, die leukocyten aantrekken en ze dragen cel adhesie moleculen (CAM) op hun oppervlak die een interactie aan kunnen gaan met de CAMs op de leukocyten. In hoofdstuk 6 is gekeken naar het effect van p38 MAPK remming op de productie van chemokines en de expressie van CAMs (E-selectin, VCAM-1 en ICAM-1). Wij vonden dat behandeling van deze cellen met de p38 MAPK remmer de productie en mRNA expressie van de chemokines IL-8, MCP-1, maar ook van IL-6 sterk deed verminderen. Wat betreft de expressie van de CAMs bleek alleen E-selectin zwak te worden geremd door behandeling met RWJ 67657. Het feit dat de productie van de chemokines wel sterk wordt geremd is echter zeer belangrijk, omdat de chemokines een belangrijke rol spelen bij het in stand houden van de endotheel-leukocyte interactie. Omdat het effect van p38 MAPK remming op de CAM expressie in endotheelcellen zwak was, is de betrokkenheid van een andere signaal transductie route onderzocht in hoofdstuk 7. De stof MOL-294, waarvan bekend is dat het de NF-κB route remt, bleek de CAM expressie in endotheelcellen zeer sterk te verminderen. Gecombineerde behandeling van EC met zowel MOL-294 als RWJ 67657 bleek een sterker remmend effect te hebben op alle chemokines en CAMs die onderzocht waren dan wanneer ze apart gebruikt werden. Dit zou kunnen betekenen dat combinaties van geneesmiddelen, die verschillende routes als target hebben een beter effect hebben op ontsteking dan de geneesmiddelen apart. Tot nu toe zijn effecten van p38 MAPK inhibitie onderzocht op cellen die lokaal in het synovium voorkomen. In hoofdstuk 8 is onderzocht of p38 MAPK remming ook effecten heeft op een ander, systemisch, verschijnsel namelijk de acute fase reactie. Bij deze reactie worden onder invloed van cytokines (met name IL-6) die ontstaan door ontsteking in de lever, eiwitten geproduceerd die een maat zijn voor de ernst van de ontsteking. Deze acute fase eiwitten, met name CRP (C-reactive protein) en fibrinogeen, worden gebruikt om de ziekteactiviteit van RA patiënten te bepalen. De signaaltransductie route die IL-6 gebruikt bij het tot stand komen van de acute fase reactie is de JAK/STAT route en uit literatuur is gebleken dat er interactie is tussen de p38 MAPK route en de JAK/STAT route.

Nederlandse samenvatting
135
Het effect van p38 MAPK remming op de productie van diverse acute fase eiwitten door hepatoma cellijnen is bestudeerd en tevens de effecten op humane lever slices. De produktie en mRNA expressie van CRP en fibrinogeen bleek sterk te worden geremd, maar niet die van een ander acute fase eiwit, het serum amyloïd A (SAA). De conclusie uit deze laatste studie was dat SAA daarom een betere marker voor ziekteactiviteit van RA is in het geval van p38 MAPK inhibitor therapie. CONCLUSIES In dit proefschrift is onderzocht wat de gevolgen zijn van het remmen van signaal transductie routes, die belangrijk zijn in het ontstekingsproces, zoals zich dat afspeelt bij reumatoïde artritis. We vonden dat in cellen, die een cruciale rol hebben in de ontsteking, het remmen van de p38 MAPK route belangrijke gevolgen had. In synoviale fibroblasten werd een significante remming gevonden van de productie en mRNA expressie van IL-6, IL-8 en MMP-3. De mRNA expressie van COX-2 werd sterk geremd bij zeer lage concentraties van de p38 MAPK remmer. Ook werd de productie van TNF-α, het centrale cytokine in RA, significant geremd in macrofagen door RWJ 67657. Endotheelcellen die actief betrokken zijn bij de aantrekking van leukocyten bleken onder invloed van verschillende routes te staan. De productie van chemokines werd effectief geremd door p38 MAPK inhibitie, terwijl de expressie van adhesie moleculen gereguleerd bleek te zijn door de NF-κB signaal transductie route. Tenslotte werd aangetoond dat er interactie is tussen de p38 MAPK route en de JAK/STAT route, aangezien CRP en fibrinogeen productie gereduceerd werd in hepatoma cellijnen door p38 MAPK remming. Samenvattend concluderen we dat de p38 MAPK route een belangrijke rol speelt in signaal transductie in RA en dat remming van de route belangrijke effecten heeft op ontstekingscellen. Aangezien de NF-κB route ook een belangrijke rol speelt in het ontstekingsproces, zou een combinatie van geneesmiddelen die beide routes remmen heel erg effectief kunnen zijn. TOEKOMSTPLANNEN Het onderzoek beschreven in dit proefschrift bevestigt de belangrijke rol van de p38 MAPK route in het ontstekingsproces. De p38 MAPK route is evenwel niet de enige route. Sterker nog, het lijkt erop dat vele kinases en transcriptiefactoren betrokken zijn bij ontsteking. Vooruitgang in dit onderzoeksveld wordt bemoeilijkt door de complexiteit van de diverse signaal transductie routes. De afgelopen jaren is de interesse voor het bestuderen van synoviaal weefsel toegenomen, aangezien de ontstekingsreactie zich afspeelt in het synovium van RA patiënten. Door het nemen van synoviale biopten is men in staat om het synovium te bestuderen voor diagnostische redenen, maar ook voor onderzoek naar bijvoorbeeld de effectiviteit van therapie.

Chapter 10
136
Een nieuwe benadering om synovium te bestuderen zou zijn door het in kaart brengen van intracellulaire signaal transductie systemen en te onderzoeken of er een verschil is tussen normale en ziekte-gerelateerde processen. Een belangrijke stap in de goede richting is ontstaan door het ontwikkelen van kinase arrays. Door een kinase array is het mogelijk geworden om de activiteit van meerdere kinases in een cel- of weefsellysaat te bestuderen. Deze techniek maakt het mogelijk om het intracellulair signaal transductie (IST) profiel van het synovium van een RA-patiënt gedurende verschillende stadia van de ziekte en gedurende verschillende therapieën vast te stellen. Wij denken dat een dergelijk profiel een voorspellende waarde zou kunnen hebben voor individuele respons op een bepaalde therapie. De kinase array is al getest in cellen en diverse weefsels. Wij willen nu deze techniek toe gaan passen op synoviaal weefsel. Vervolgens zullen de bevindingen van de kinase array vergeleken worden met resultaten van reeds bestaande technieken. De verwachting is dat door het vaststellen van de betrokken IST routes in RA, we patiënten met de juiste therapie op het juiste moment kunnen gaan behandelen. Dit zou kunnen betekenen dat combinaties van signaal transductie remmers, maar ook combinaties van signaal transductie remmers met reeds bestaande geneesmiddelen effectieve therapieën zouden kunnen zijn voor RA.

Dankwoord
List of abbreviations
List of publications

Dankwoord
138
DANKWOORD Al in 1995/96 wist ik zeker dat ik zelfstandig onderzoek wilde gaan doen, omdat het me gewoon erg leuk leek en ik het gevoel had dat ik er geschikt voor was. Door persoonlijke omstandigheden duurde het echter nog tot 2000 voordat het idee echt structuur ging krijgen en voordat een geschikt onderwerp zich had aangediend. Signaal transductie inhibitie was een onderwerp dat bij niemand toen warme gevoelens opriep, maar wat mij in hoge mate intrigeerde. Aanvankelijk probeerde Piet (Limburg) me nog te weerhouden een promotietraject te starten, door me te vezekeren dat promoveren echt niet makkelijk was, maar dat mocht niet meer baten. Vanaf 1 maart 2001 ging het project echt van start en hebben Piet, Martin (van Rijswijk), mijn tweede promotor, en Miek (van Leeuwen), mijn co-promotor me altijd gesteund ook in tijden dat het op het werk of thuis niet zo goed ging. Bedankt voor het in mij gestelde vertrouwen. Veel mensen hebben in de afgelopen 4 jaren bijgedragen aan mijn onderzoek op een of andere manier en ik wil hen met name noemen en bedanken. De hoogleraren Cees Kallenberg, Paul-Peter Tak en Edo Vellenga, die mijn proefschrift hebben beoordeeld, wil ik hartelijk danken voor de genomen moeite. Verder natuurlijk eerst mijn paranimfen Annet Wilkens en André van Rossum, die het aandurven om op de grote dag samen met mij vooraan te staan. Direct daarna wil ik Johan Bijzet en Berber Doornbos bedanken, die op het lab een enorme bijdrage hebben geleverd, Johan in de vorm van allerlei hulp tot aan het lay-outen van het proefschrift toe en Berber voor haar uitstekende analytische werk. Alle mensen van de researchgroep Reumatologie en Klinische Immunologie, met name Marcel Posthumus, Bouke Hazenberg en Liesbeth Brouwer, bedankt voor de inhoudelijke bijdragen maar ook voor de inspirerende gesprekken. Van het lab wil ik Gerda Horst, Minke Huitema en de mensen van de diagnostiek bedanken voor alle hulp en gezelligheid. Niels Kouprie en Joan Vos verdienen een aparte vermelding voor alle geprikte buisjes bloed. Op iets grotere afstand hebben bijgedragen Fred Breukelman en Lex Boerboom door het leveren van synovium, Marco Harmsen en Sigga Asgeirsdottir door hulp bij het PCR-werk, Geert Mesander door te helpen bij het facsen en Jelleke Dokter door hulp bij het kweken. Allen dank! De mensen van de endotheelgroep: Ingrid Molema, Henk Moorlag en Robbert-Jan Kok wil ik hartelijk danken voor de prettige samenwerking. Dear Joanna, thanks for everything and let’s hope that chapter 7 will be accepted soon. Peter Olinga en Annelies Draaisma wil ik bedanken voor de samenwerking met het liverslice project. Met Sander Diks en Maikel Peppelenbosch is de samenwerking net gestart, ik hoop dat we nog een succesvolle tijd tegemoet kunnen zien. De studenten, die bij mij een keuzeproject of afstudeeropdracht hebben gedaan: Mirjan van Timmeren, Annelie Rawée, Hilde van der Schaar en Karina Wessel wil ik bedanken voor getoonde inzet en leuke samenwerking. Helaas zijn niet alle

Dankwoord
139
projecten een succes geworden, maar ik ben blij dat je toch nog een leuke werkplek hebt gevonden Hilde. Met 6 mensen op een kleine kamer zitten is niet altijd een pretje. Toch hebben we (in wisselende samenstelling) veel lol gehad en ook elkaar kunnen helpen bij computer- en andere problemen. Ik denk dat ik de eerste ben die de kamer gaat verlaten en ik zal jullie (Agnieszka, Alja, André, Edwin, Esther, Karina, Jan-Stephan en Wayel) dan vast nog gaan missen. Tenslotte wil ik de secretaresses Kiki Bugter en Janny Havinga bedanken voor hulp in allerlei vorm. Het klinkt als een cliché, maar toch is het waar. Dankzij onze fijne en veilige jeugd zijn mijn zusjes, broer en ik geworden tot wat we zijn en hebben we onderling en met onze ouders een heel erg goed contact. Ook het feit dat we mochten studeren wat we wilden en dat we in onze waarde werden gelaten is enorm belangrijk geweest. Met dit boekje wil ik jullie, heit en mem, hiervoor bedanken, maar ook mijn broer Arjen en zusjes Anneke, Foekje, Els, Siska, Irene en hun partners voor hulp en steun in moeilijke tijden. Lieve Marc, regelmatig verbazen wij ons erover dat twee mensen op alle fronten zo goed bij elkaar passen als wij. In jou heb ik mijn grote liefde gevonden en samen met onze jongens hebben we het heel erg goed. Bedankt voor je oprechte interesse, liefde, zorg en betrokkenheid tijdens mijn promotietraject. Ik ga niet beweren dat de computer nu weer vrij is voor de kinderen, want meestal moet ik tijd bevechten om even wat te kunnen doen. Maar toch bedankt Daan, Chris, Martijn, Steven en Fabian voor de broodnodige afleiding, drukte en gezelligheid. Dat ik bij de afdeling Reumatologie kan blijven is enorm fijn. Niet alleen is het moeilijk om als post-doc een baan te vinden, maar ik zou het heel erg jammer hebben gevonden als ik alle mensen waarmee ik zo prettig heb samenwerkt zou hebben moeten verlaten. Daarom: een toast op de toekomst !!! Hannie

List of abbreviations
140
LIST OF ABBREVIATIONS AP-1 activating protein kinase APP acute phase protein ASK apoptosis signal-regulating kinase ATF activating transcription factor BMK big MAP kinase CHOP C/EBP homologous protein C/EBP CCAAt/enhancer binding protein CREB cyclic AMP-responsive element-binding protein CRP C-reactive protein ERK extra-cellular signal-regulated kinase Hsp heat shock protein IκB inhibitor of NF-κB IKK IκB kinase IL interleukin IRAK interleukin related accociated kinase JAK janus kinase JNK c-Jun NH2 -terminal kinase MAPK mitogen activated-protein kinase MAPKK MAPK kinase kinase MAPKKK MAPK kinase kinase kinase MAPKAPK MAPK activated protein kinase MEF myocyte enhancer factor MEK MAPK or ERK kinase MEKK MEK kinase MKK MAPK kinase MLK mixed lineage kinase MNK MAPK interacting serine/threonine kinase MSK mitogen- and stress- activated protein kinase NAK NF-κB activating kinase NIK NF-κB inducing kinase NF-κB nuclear factor immunoglobulin κ chain enhancer B-cell PAK p21-activated kinase PRAK p38-regulated/activated protein kinase SAA serum amyloid A STAT signal transducer and activator of transcription SHP SH2-domain containing tyrosine phosphatase SOCS suppressor of cytokine signalling TAK TGFβ-activated protein kinase TGFβ transforming growth factor β TNF tumor necrosis factor

List of abbreviations
141
Tpl-2 tumor progession locus-2 TRADD TNF-receptor-1 associated death domain TRAF TNF-receptor associated factor

List of publications
142
LIST OF PUBLICATIONS Westra J, Kułdo JM, van Rijswijk MH, Molema G and Limburg PC. Chemokine production and E-selectin expression in activated endothelial cells are inhibited by p38 MAPK (mitogen activated protein kinase) inhibitor RWJ 67657. Int Immunopharmacol. 2005, in press. Westra J, Limburg PC, de Boer P, van Rijswijk MH. Effects of RWJ 67657, a p38 mitogen activated protein kinase (MAPK) inhibitor, on the production of inflammatory mediators by rheumatoid synovial fibroblasts. Ann Rheum Dis. 2004;63(11):1453-9. Westra J, Doornbos-van der Meer B, de Boer P, van Leeuwen MA, van Rijswijk MH, Limburg PC. Strong inhibition of TNF-alpha production and inhibition of IL-8 and COX-2 mRNA expression in monocyte-derived macrophages by RWJ 67657, a p38 mitogen-activated protein kinase (MAPK) inhibitor. Arthritis Res Ther. 2004;6(4):R384-92. Posthumus MD, Limburg PC, Westra J, van Leeuwen MA, van Rijswijk MH. Serum matrix metalloproteinase 3 levels in comparison to C-reactive protein in periods with and without progression of radiological damage in patients withearly rheumatoid arthritis. Clin Exp Rheumatol. 2003 Jul-Aug;21(4):465-72. Posthumus MD, Limburg PC, Westra J, van Leeuwen MA, van Rijswijk MH. Serum matrix metalloproteinase 3 levels during treatment with sulfasalazine or combination of methotrexate and sulfasalazine in patients with early rheumatoid arthritis. J Rheumatol. 2002 May;29(5):883-9. Posthumus MD, Limburg PC, Westra J, van Leeuwen MA, van Rijswijk MH. Serum matrix metalloproteinase 3 in early rheumatoid arthritis is correlated with disease activity and radiological progression. J Rheumatol. 2000 Dec;27(12):2761-8. Posthumus MD, Limburg PC, Westra J, Cats HA, Stewart RE, van Leeuwen MA, van Rijswijk MH. Serum levels of matrix metalloproteinase-3 in relation to the development of radiological damage in patients with early rheumatoid arthritis. Rheumatology (Oxford). 1999 Nov;38(11):1081-7. van Leeuwen MA, Westra J, Limburg PC, van Riel PL, van Rijswijk MH. Clinical significance of interleukin-6 measurement in early rheumatoid

List of publications
143
arthritis: relation with laboratory and clinical variables and radiological progression in a three year prospective study. Ann Rheum Dis. 1995 Aug;54(8):674-7. van Leeuwen MA, Westra J, Limburg PC, van Riel PL, van Rijswijk MH. Interleukin-6 in relation to other proinflammatory cytokines, chemotactic activity and neutrophil activation in rheumatoid synovial fluid. Ann Rheum Dis. 1995 Jan;54(1):33-8. van Leeuwen MA, Westra J, van Riel PL, Limburg PC, van Rijswijk MH. IgM, IgA, and IgG rheumatoid factors in early rheumatoid arthritis predictive of radiological progression? Scand J Rheumatol. 1995;24(3):146-53. van Leeuwen MA, Westra J, Limburg PC, de Jong HJ, Marrink J, van Rijswijk MH. Quantitation of IgM, IgA and IgG rheumatoid factors by ELISA in rheumatoid arthritis and other rheumatic disorders. Scand J Rheumatol Suppl. 1988;75:25-31. Kallenberg CG, Klaassen RJ, Westra J, Beelen JM, Ockhuizen T. Immunoglobulin genes, HLA-B8/DR3, and immune responsiveness to primary immunogen and mitogens in normal subjects. Clin Immunol Immunopathol. 1988 Jun;47(3):333-42. van Leeuwen MA, van Rijswijk MH, Westra J, de Jong HJ, Marrink J. [C-reactive protein; an expensive sedimentation?] Ned Tijdschr Geneeskd. 1986 Aug 2;130(31):1391-5. Dutch. Beukhof JR, Ockhuizen T, Halie LM, Westra J, Beelen JM, Donker AJ, Hoedemaeker PJ, van der Hem GK. Subentities within adult primary IgA-nephropathy. Clin Nephrol. 1984 Oct;22(4):195-9. Ockhuizen T, Beukhof JR, Westra J, Halie LM, Beelen JM, van der Hem GK. Immunogenetic differences in subpopulations of patients with IgA nephropathy (Berger's disease). Exp Clin Immunogenet. 1984;1(3):121-8. Ockhuizen T, Westra J, Aalders JG, de Bruijn HW. Immunoglobulin allotypes in patients with epithelial ovarian cancer. Exp Clin Immunogenet. 1984;1(4):189-92.

Stellingen behorende bij het proefschrift:
Experimental studies on signal transduction pathways in rheumatoid arthritis.
1. Remmers van p38 MAPK signaal transductie routes blokkeren niet alleen
de produktie van TNF-α, maar ook de effecten van TNF-α op andere cellen (dit proefschrift).
2. Bij p38 MAPK inhibitor therapie is het gebruik van specifieke COX-2
remmers niet nodig, aangezien de p38 MAPK remmers de inductie van COX-2 mRNA expressie in fibroblasten en macrofagen remmen (dit proefschrift).
3. Om geactiveerde endotheelcellen effectief te kunnen remmen zijn
tenminste 2 signaal transductie route remmers (NF-κB en p38 MAPK) nodig (dit proefschrift).
4. Serum amyloid A (SAA) is een betere indicator voor het meten van ziekte
activiteit dan C-reactive protein (CRP) en bloedbezinking bij patienten met reumatoïde artritis, die p38 MAPK inhibitor therapie krijgen (dit proefschrift).
5. Het niet specifiek zijn van de meest bestudeerde p38 MAPK remmer
SB203580 kan in plaats van “a-specific” beter “pharmacologically rich” genoemd worden. (Tim Perreira, JNJ, oktober 2004)
6. Wetenschappelijke tijdschriften, die niet gebruik maken van het “online
submission” systeem, prijzen zich vanzelf uit de markt.
7. Aangezien er 30.000 genen voor de mens zijn ontdekt en “slechts” ruim 500 kinases is het interpreteren van een kinase-array vergeleken met een gen-array een makkie.
8. Het afschaffen van het lekenpraatje voorafgaand aan de verdediging van het
proefschrift maakt het bijwonen van een promotieplechtigheid voor de geïnteresseerde leek een stuk minder aantrekkelijk.
9. Het is hoog tijd dat gedragsregels afgesproken worden over het publiekelijk
gebruik van mobiele telefoons.

10. Aangezien vrouwelijke sprinters de laatste jaren meer progressie hebben gemaakt dan mannelijke, zullen zij over 52 jaar sneller zijn (onderzoekers Universiteit Oxford).
11. Het is een voorrecht om het (bijna) uitzicht te hebben op het nieuwe FC-
Groningen stadion dat volgens www.euroborg.nl naast de Martinitoren, het Groninger Museum en de Gasunie het vierde beeldbepalende bouwwerk van de stad Groningen wordt.
12. Een gezin met een wisselende grootte variërend van 2 tot 7 personen houdt
een mens alert en bij de tijd. Hannie Westra Groningen, 13 juni 2005




















