
tRNAHis guanylyltransferase catalyzes a 3'-5' polymerization reaction that is distinct from G-1...
6
tRNA His guanylyltransferase catalyzes a3-5 polymerization reaction that is distinct from G 1 addition Jane E. Jackman and Eric M. Phizicky † Department of Biochemistry and Biophysics, University of Rochester School of Medicine, Rochester, NY 14642 Communicated by Fred Sherman, University of Rochester Medical Center, Rochester, NY, April 15, 2006 (received for review April 1, 2006) Yeast tRNA His guanylyltransferase, Thg1, is an essential protein that adds a single guanine to the 5 end (G 1 ) of tRNA His . This G 1 residue is required for aminoacylation of tRNA His by histidyl-tRNA synthetase, both in vitro and in vivo. The guanine nucleotide addition reaction catalyzed by Thg1 extends the polynucleotide chain in the reverse (3-5) direction of other known polymerases, albeit by one nucleotide. Here, we show that alteration of the 3 end of the Thg1 substrate tRNA His unleashes an unexpected re- verse polymerase activity of wild-type Thg1, resulting in the 3-5 addition of multiple nucleotides to the tRNA, with efficiency comparable to the G 1 addition reaction. The addition of G 1 forms a mismatched GA base pair at the 5 end of tRNA His , and, with monophosphorylated tRNA substrates, it is absolutely specific for tRNA His . By contrast, reverse polymerization forms multiple GC or CG base pairs, and, with preactivated tRNA species, it can initiate at positions other than 1 and is not specific for tRNA His . Thus, wild-type Thg1 catalyzes a templated polymerization reaction acting in the reverse direction of that of canonical DNA and RNA polymerases. Surprisingly, Thg1 can also readily use dNTPs for nucleotide addition. These results suggest that 3-5 polymeriza- tion represents either an uncharacterized role for Thg1 in RNA or DNA repair or metabolism, or it may be a remnant of an earlier catalytic strategy used in nature. polymerase Saccharomyces cerevisiae THG1 tRNA modification A ll known polymerases catalyze the addition of nucleotides in the 5-3 direction, including classical DNA and RNA polymerases, as well as telomerase, reverse transcriptase, and a number of template-independent RNA polymerases (1–3). These polymerases universally catalyze the same reaction, in- volving attack by the polynucleotide 3-OH on the 5- triphosphate of an incoming NTP, to yield an extended polynu- cleotide and a pyrophosphate side product from the NTP (Fig. 1A). However, polymerization could just as easily occur chem- ically in the reverse (3-5) direction with the same functional groups, by attack of the 3-OH of the incoming nucleotide on a 5-triphosphorylated polynucleotide (Fig. 1B). We show here that tRNA His guanylyltransferase can act in this capacity as a reverse polymerase, catalyzing the 3-5 addition of multiple nucleotides to the polynucleotide chain in a reaction that is distinct from its known physiological role. tRNA His species are unique among tRNAs because they contain an additional universally conserved G residue at the 1 position (G 1 ), whereas only one other characterized tRNA has any nucleotide at that position (4, 5). In prokaryotes, G 1 is genome-encoded, forms a standard base pair with C 73 , and is retained during tRNA His maturation because RNase P cleaves pre-tRNA at the 1 position (6). By contrast, in eukaryotes, G 1 is added posttranscriptionally by tRNA His guanylyltransferase, which is encoded in yeast by the essential THG1 gene (7), and it catalyzes the addition of a guanine nucleotide to the 5 end of tRNA His across from residue A 73 of the tRNA, the nucleotide immediately preceding the CCA 3 end (7–9). G 1 addition activity is crucial for aminoacylation of tRNA His in vivo (10), consistent with the demonstration that G 1 is necessary and sufficient for histidyl-tRNA synthetase activity in vitro (11–13). Because G 1 modification is a strong determinant of histidyl- tRNA synthetase activity, Thg1 must be extraordinarily specific for tRNA His to prevent misacylation of other tRNAs. Indeed, Thg1 prefers its physiological substrate, monophosphorylated tRNA His (p-tRNA His ), which arises after cleavage by RNase P, by 10,000-fold over other p-tRNA species (14). As for amino- acyl-tRNA synthetases, where the anticodon is often a major determinant of tRNA specificity, the tRNA His GUG anticodon is the source of Thg1 specificity because it is both necessary and sufficient for Thg1 activity in vitro (14). The three-step enzymatic reaction catalyzed by Thg1 involves the activation of the 5 end of the tRNA by adenylylation, attack of the 3-OH of GTP on the activated intermediate, and removal of the 5-pyrophos- phate, yielding mature G 1 -containing tRNA His and AMP (Fig. 1B) (7, 8). These chemical steps are also strikingly similar to those catalyzed by aminoacyl-tRNA synthetases (14). However, despite these similarities between Thg1 and synthetases, as well as the proximity of their sites of action at the top of the tRNA aminoacyl-acceptor stem, there is no identifiable homology between Thg1 and the synthetases or even between Thg1 and any other enzyme family. Formally, the Thg1 G 1 addition reaction is the 3-5 extension of a polynucleotide chain, albeit by one nucleotide. Moreover, there is an alternative mode of Thg1 activity that more closely resembles a polymerase, in which triphosphorylated tRNA His (ppp-tRNA His ) is used directly for the addition of G 1 (Fig. 1B), bypassing the ATP requirement for activation (7). We report here that wild-type Thg1 can catalyze multiple rounds of reverse polymerization with certain substrates. While Conflict of interest statement: No conflicts declared. Abbreviations: CIP, calf intestinal alkaline phosphatase; PEI, polyethyleneimine; p-tRNA, monophosphorylated tRNA; ppp-tRNA, triphosphorylated tRNA. † To whom correspondence should be addressed. E-mail: eric[email protected]. © 2006 by The National Academy of Sciences of the USA Fig. 1. Nucleotide addition reactions catalyzed by 5-3 polymerases and tRNA His guanylyltransferase (Thg1). 8640 – 8645 PNAS June 6, 2006 vol. 103 no. 23 www.pnas.orgcgidoi10.1073pnas.0603068103
Transcript of tRNAHis guanylyltransferase catalyzes a 3'-5' polymerization reaction that is distinct from G-1...

tRNAHis guanylyltransferase catalyzesa 3�-5� polymerization reaction that isdistinct from G�1 additionJane E. Jackman and Eric M. Phizicky†
Department of Biochemistry and Biophysics, University of Rochester School of Medicine, Rochester, NY 14642
Communicated by Fred Sherman, University of Rochester Medical Center, Rochester, NY, April 15, 2006 (received for review April 1, 2006)
Yeast tRNAHis guanylyltransferase, Thg1, is an essential proteinthat adds a single guanine to the 5� end (G�1) of tRNAHis. This G�1
residue is required for aminoacylation of tRNAHis by histidyl-tRNAsynthetase, both in vitro and in vivo. The guanine nucleotideaddition reaction catalyzed by Thg1 extends the polynucleotidechain in the reverse (3�-5�) direction of other known polymerases,albeit by one nucleotide. Here, we show that alteration of the 3�
end of the Thg1 substrate tRNAHis unleashes an unexpected re-verse polymerase activity of wild-type Thg1, resulting in the 3�-5�
addition of multiple nucleotides to the tRNA, with efficiencycomparable to the G�1 addition reaction. The addition of G�1 formsa mismatched G�A base pair at the 5� end of tRNAHis, and, withmonophosphorylated tRNA substrates, it is absolutely specific fortRNAHis. By contrast, reverse polymerization forms multiple G�C orC�G base pairs, and, with preactivated tRNA species, it can initiateat positions other than �1 and is not specific for tRNAHis. Thus,wild-type Thg1 catalyzes a templated polymerization reactionacting in the reverse direction of that of canonical DNA and RNApolymerases. Surprisingly, Thg1 can also readily use dNTPs fornucleotide addition. These results suggest that 3�-5� polymeriza-tion represents either an uncharacterized role for Thg1 in RNA orDNA repair or metabolism, or it may be a remnant of an earliercatalytic strategy used in nature.
polymerase � Saccharomyces cerevisiae � THG1 � tRNA modification
A ll known polymerases catalyze the addition of nucleotides inthe 5�-3� direction, including classical DNA and RNA
polymerases, as well as telomerase, reverse transcriptase, and anumber of template-independent RNA polymerases (1–3).These polymerases universally catalyze the same reaction, in-volving attack by the polynucleotide 3�-OH on the 5�-triphosphate of an incoming NTP, to yield an extended polynu-cleotide and a pyrophosphate side product from the NTP (Fig.1A). However, polymerization could just as easily occur chem-ically in the reverse (3�-5�) direction with the same functionalgroups, by attack of the 3�-OH of the incoming nucleotide on a5�-triphosphorylated polynucleotide (Fig. 1B). We show herethat tRNAHis guanylyltransferase can act in this capacity as areverse polymerase, catalyzing the 3�-5� addition of multiplenucleotides to the polynucleotide chain in a reaction that isdistinct from its known physiological role.
tRNAHis species are unique among tRNAs because theycontain an additional universally conserved G residue at the �1position (G�1), whereas only one other characterized tRNA hasany nucleotide at that position (4, 5). In prokaryotes, G�1 isgenome-encoded, forms a standard base pair with C73, and isretained during tRNAHis maturation because RNase P cleavespre-tRNA at the �1 position (6). By contrast, in eukaryotes, G�1is added posttranscriptionally by tRNAHis guanylyltransferase,which is encoded in yeast by the essential THG1 gene (7), andit catalyzes the addition of a guanine nucleotide to the 5� end oftRNAHis across from residue A73 of the tRNA, the nucleotideimmediately preceding the CCA 3� end (7–9). G�1 additionactivity is crucial for aminoacylation of tRNAHis in vivo (10),
consistent with the demonstration that G�1 is necessary andsufficient for histidyl-tRNA synthetase activity in vitro (11–13).
Because G�1 modification is a strong determinant of histidyl-tRNA synthetase activity, Thg1 must be extraordinarily specificfor tRNAHis to prevent misacylation of other tRNAs. Indeed,Thg1 prefers its physiological substrate, monophosphorylatedtRNAHis (p-tRNAHis), which arises after cleavage by RNase P,by �10,000-fold over other p-tRNA species (14). As for amino-acyl-tRNA synthetases, where the anticodon is often a majordeterminant of tRNA specificity, the tRNAHis GUG anticodonis the source of Thg1 specificity because it is both necessary andsufficient for Thg1 activity in vitro (14). The three-step enzymaticreaction catalyzed by Thg1 involves the activation of the 5� endof the tRNA by adenylylation, attack of the 3�-OH of GTP onthe activated intermediate, and removal of the 5�-pyrophos-phate, yielding mature G�1-containing tRNAHis and AMP (Fig.1B) (7, 8). These chemical steps are also strikingly similar tothose catalyzed by aminoacyl-tRNA synthetases (14). However,despite these similarities between Thg1 and synthetases, as wellas the proximity of their sites of action at the top of the tRNAaminoacyl-acceptor stem, there is no identifiable homologybetween Thg1 and the synthetases or even between Thg1 and anyother enzyme family.
Formally, the Thg1 G�1 addition reaction is the 3�-5� extensionof a polynucleotide chain, albeit by one nucleotide. Moreover,there is an alternative mode of Thg1 activity that more closelyresembles a polymerase, in which triphosphorylated tRNAHis
(ppp-tRNAHis) is used directly for the addition of G�1 (Fig. 1B),bypassing the ATP requirement for activation (7).
We report here that wild-type Thg1 can catalyze multiplerounds of reverse polymerization with certain substrates. While
Conflict of interest statement: No conflicts declared.
Abbreviations: CIP, calf intestinal alkaline phosphatase; PEI, polyethyleneimine; p-tRNA,monophosphorylated tRNA; ppp-tRNA, triphosphorylated tRNA.
†To whom correspondence should be addressed. E-mail: eric�[email protected].
© 2006 by The National Academy of Sciences of the USA
Fig. 1. Nucleotide addition reactions catalyzed by 5�-3� polymerases andtRNAHis guanylyltransferase (Thg1).
8640–8645 � PNAS � June 6, 2006 � vol. 103 � no. 23 www.pnas.org�cgi�doi�10.1073�pnas.0603068103
investigating the tRNAHis determinants for Thg1 activity, wefound that alteration of the conserved nucleotide A73 of tRNAHis
unleashes a previously unexpected reverse polymerase functionof wild-type Thg1, resulting in the 3�-5� extension of the polynu-cleotide chain by multiple nucleotides. Characterization of Thg1reverse polymerization shows that this activity, unlike the addi-tion of G�1, is template-dependent, recognizing G�C Watson–Crick base pairs. Moreover, reverse polymerization is not spe-cific for tRNAHis or for starting at the �1 position of tRNA,provided that the activated (triphosphorylated) form of thetRNA substrate is used in the assays.
ResultsThg1 Adds Multiple Guanine Residues to the 5� End of A73C VarianttRNAHis Species. Although Thg1 normally adds a single guaninenucleotide to the 5� end of tRNAHis, Thg1 can add multipleguanine nucleotides to a variant tRNAHis that bears C73 insteadof the A73 that is universally conserved in eukaryotic tRNAHis
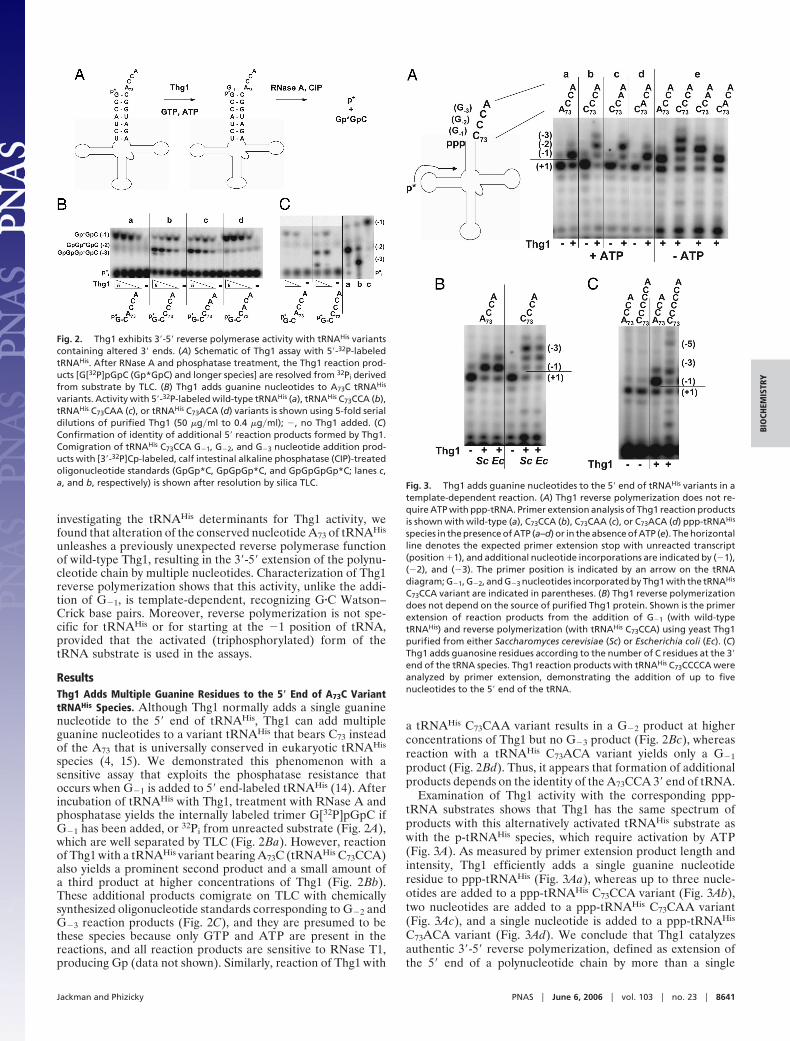
species (4, 15). We demonstrated this phenomenon with asensitive assay that exploits the phosphatase resistance thatoccurs when G�1 is added to 5� end-labeled tRNAHis (14). Afterincubation of tRNAHis with Thg1, treatment with RNase A andphosphatase yields the internally labeled trimer G[32P]pGpC ifG�1 has been added, or 32Pi from unreacted substrate (Fig. 2A),which are well separated by TLC (Fig. 2Ba). However, reactionof Thg1 with a tRNAHis variant bearing A73C (tRNAHis C73CCA)also yields a prominent second product and a small amount ofa third product at higher concentrations of Thg1 (Fig. 2Bb).These additional products comigrate on TLC with chemicallysynthesized oligonucleotide standards corresponding to G�2 andG�3 reaction products (Fig. 2C), and they are presumed to bethese species because only GTP and ATP are present in thereactions, and all reaction products are sensitive to RNase T1,producing Gp (data not shown). Similarly, reaction of Thg1 with
a tRNAHis C73CAA variant results in a G�2 product at higherconcentrations of Thg1 but no G�3 product (Fig. 2Bc), whereasreaction with a tRNAHis C73ACA variant yields only a G�1
product (Fig. 2Bd). Thus, it appears that formation of additionalproducts depends on the identity of the A73CCA 3� end of tRNA.
Examination of Thg1 activity with the corresponding ppp-tRNA substrates shows that Thg1 has the same spectrum ofproducts with this alternatively activated tRNAHis substrate aswith the p-tRNAHis species, which require activation by ATP(Fig. 3A). As measured by primer extension product length andintensity, Thg1 efficiently adds a single guanine nucleotideresidue to ppp-tRNAHis (Fig. 3Aa), whereas up to three nucle-otides are added to a ppp-tRNAHis C73CCA variant (Fig. 3Ab),two nucleotides are added to a ppp-tRNAHis C73CAA variant(Fig. 3Ac), and a single nucleotide is added to a ppp-tRNAHis
C73ACA variant (Fig. 3Ad). We conclude that Thg1 catalyzesauthentic 3�-5� reverse polymerization, defined as extension ofthe 5� end of a polynucleotide chain by more than a single
Fig. 2. Thg1 exhibits 3�-5� reverse polymerase activity with tRNAHis variantscontaining altered 3� ends. (A) Schematic of Thg1 assay with 5�-32P-labeledtRNAHis. After RNase A and phosphatase treatment, the Thg1 reaction prod-ucts [G[32P]pGpC (Gp*GpC) and longer species] are resolved from 32Pi derivedfrom substrate by TLC. (B) Thg1 adds guanine nucleotides to A73C tRNAHis
variants. Activity with 5�-32P-labeled wild-type tRNAHis (a), tRNAHis C73CCA (b),tRNAHis C73CAA (c), or tRNAHis C73ACA (d) variants is shown using 5-fold serialdilutions of purified Thg1 (50 �g�ml to 0.4 �g�ml); �, no Thg1 added. (C)Confirmation of identity of additional 5� reaction products formed by Thg1.Comigration of tRNAHis C73CCA G�1, G�2, and G�3 nucleotide addition prod-ucts with [3�-32P]Cp-labeled, calf intestinal alkaline phosphatase (CIP)-treatedoligonucleotide standards (GpGp*C, GpGpGp*C, and GpGpGpGp*C; lanes c,a, and b, respectively) is shown after resolution by silica TLC. Fig. 3. Thg1 adds guanine nucleotides to the 5� end of tRNAHis variants in a
template-dependent reaction. (A) Thg1 reverse polymerization does not re-quire ATP with ppp-tRNA. Primer extension analysis of Thg1 reaction productsis shown with wild-type (a), C73CCA (b), C73CAA (c), or C73ACA (d) ppp-tRNAHis
species in the presence of ATP (a–d) or in the absence of ATP (e). The horizontalline denotes the expected primer extension stop with unreacted transcript(position �1), and additional nucleotide incorporations are indicated by (�1),(�2), and (�3). The primer position is indicated by an arrow on the tRNAdiagram; G�1, G�2, and G�3 nucleotides incorporated by Thg1 with the tRNAHis
C73CCA variant are indicated in parentheses. (B) Thg1 reverse polymerizationdoes not depend on the source of purified Thg1 protein. Shown is the primerextension of reaction products from the addition of G�1 (with wild-typetRNAHis) and reverse polymerization (with tRNAHis C73CCA) using yeast Thg1purified from either Saccharomyces cerevisiae (Sc) or Escherichia coli (Ec). (C)Thg1 adds guanosine residues according to the number of C residues at the 3�end of the tRNA species. Thg1 reaction products with tRNAHis C73CCCCA wereanalyzed by primer extension, demonstrating the addition of up to fivenucleotides to the 5� end of the tRNA.
Jackman and Phizicky PNAS � June 6, 2006 � vol. 103 � no. 23 � 8641
BIO
CHEM
ISTR
Y

nucleotide. Because addition of multiple guanine nucleotides tothe ppp-tRNAHis variants is at least as efficient in the absence ofATP (Fig. 3Ae), we conclude that Thg1 does not require ATP toactivate the 5� end of the RNA for multiple-nucleotide additions;rather, we infer that the second and third guanine nucleotideadditions, like the first guanine nucleotide addition, are poweredby hydrolysis of pyrophosphate from the growing polynucleotidechain. Moreover, the slightly enhanced addition activity ob-served with the ppp-tRNAHis C73CCA variant in the absence ofATP suggests that ATP in fact inhibits the multiple-additionreaction. Because yeast Thg1 purified from E. coli exhibits thesame multiple-nucleotide addition activity, we conclude that thisactivity is intrinsic to Thg1 protein (Fig. 3B).
The addition of multiple guanine nucleotides by Thg1 is anefficient RNA reverse polymerization activity. Thg1 reversepolymerization with tRNAHis C73CCA and other variants occurswith similar efficiency (Figs. 2B and 3A) and on the same timescale (data not shown) as for the addition of G�1 to wild-typetRNAHis, whether the compared substrates are ppp-tRNAHis
species or p-tRNAHis species. Furthermore, there is an elementof template dependence in the reverse polymerization becausethe number of guanine nucleotides added to the 5� end of bothp-tRNAHis and ppp-tRNAHis variants appears to be directed bythe number of consecutive cytidine residues at the 3� end,starting at nucleotide 73 (Figs. 2B and 3). Indeed, up to fiveguanine residues are added across from the 3� end of tRNAHis
C73CCCCA (Fig. 3C).
Thg1 Reverse Polymerase Activity Is Template-Dependent BeyondPosition �1. Closer examination of the nucleotide dependence ofthe reverse polymerase activity of Thg1 demonstrates that it isstrictly template-dependent and recognizes G�C and C�G basepairs. Consistent with a templated nucleotide addition reaction,Thg1 can catalyze multiple rounds of reverse polymerization atthe 5� end of tRNAHis C73CCA only in the presence of GTP (Fig.4Ab), whereas with each of the other NTPs, Thg1 can catalyzeonly a single addition reaction. Furthermore, reverse polymer-ization is not restricted to the addition of guanine nucleotidebecause up to three cytidine nucleotides are added to the 5� endof tRNAHis G73GGA with CTP (Fig. 4Ac), but only one nucle-otide is added with each of the other NTPs. However, Thg1 doesnot appear to recognize U�A base pairs under these conditionsbecause tRNAHis U73UUA or A73AAA species are not sub-strates for reverse polymerization in the presence of ATP orUTP, respectively (Fig. 4 Ad and Ae). Thus, polymerizationbeyond the �1 position appears to require formation of acanonical G�1�C73 or C�1�G73 base pair and appears to extendfurther only if the next templated nucleotides at position 74 andbeyond are G or C residues. Moreover, Thg1 is not restricted tomultiple-nucleotide additions of a single NTP because additionto a tRNAHis G73CCA variant is somewhat more extensive in thepresence of both GTP and CTP than it is with GTP alone (Fig.4Af ). The relatively weaker extension observed with this sub-strate suggests that some nontemplated G�1 addition occursopposite G73, which would create a nonpaired and thereforenonextendable substrate. Indeed, as shown below, the additionof G is preferred at the �1 position.
Templated reverse polymerization by Thg1 beyond position�1 is in sharp contrast to the addition reaction that occurs at the�1 position because Thg1 can add any nucleotide at the �1position with each of the ppp-tRNA species examined for reversepolymerization (Fig. 4 Ab–Af ), as well as with wild-type tRNAHis
(Fig. 4Aa). However, consistent with the known role of Thg1,when all four NTPs are present, Thg1 adds exclusively G atposition �1 of wild-type p-tRNAHis opposite A73, as indicated byRNase T2 analysis of reaction products, which yields only Gp(Fig. 4B).
3�-5� Reverse Polymerization of ppp-tRNA Substrates by Thg1 Is NotSpecific for tRNAHis. Although our previous results demonstratethat the addition of Thg1 G�1 activity is at least 10,000-fold morespecific for tRNAHis than for other tRNAs when assayed in vitrowith the physiological p-tRNAHis substrate (14), Thg1 is notnearly as specific when assayed with ppp-tRNA substrates foreither G�1 addition activity (14) or reverse polymerizationactivity. Thus, Thg1 adds a single G residue to wild-typeppp-tRNAPhe, which normally contains an A73CCA 3� end (Fig.5A), and Thg1 efficiently adds up to three guanine residues to appp-tRNAPhe C73CCA variant (Fig. 5A). These results demon-strate that reverse polymerase activity does not inherentlydepend on recognition of tRNAHis and therefore could poten-tially occur with other substrates in vivo.
Reverse polymerization by Thg1 also does not need to initiateat the �1 position. Indeed, a G�1-containing tRNAHis C73CCAtranscript, which is the same molecule as the G�1 additionproduct that results from Thg1 activity on tRNAHis C73CCA, isa substrate for continued addition of up to two additionalnucleotides as demonstrated by primer extension (Fig. 5B).However, the corresponding G�1-containing wild-type tRNAHis
A73CCA 76-mer transcript cannot be extended further, presum-ably because it is the same molecule as mature tRNAHis, with anunpaired G�1 opposite A73 (Fig. 5B). Likewise, Thg1 adds twonucleotides to a tRNAHis transcript that initiates at residue �2;
Fig. 4. Nucleotide dependence of Thg1 reverse polymerization comparedwith the addition of G�1. (A) Nucleotide dependence of Thg1 activity withppp-tRNAHis species. Thg1 reaction products formed in the presence of indi-vidual NTPs and ppp-tRNAHis substrates (with 3� end sequence indicated beloweach panel) were analyzed by primer extension. Lanes G, A, U, and C show eachNTP added as indicated; �, no nucleotide; NP, no Thg1. (B) Thg1 is specific forthe addition of G at position �1 in the presence of all four NTPs. Thg1 reactionproducts with 5�-32P-labeled wild-type tRNAHis are shown in the presence ofGTP and ATP (a) or a mixture of all four NTPs (GTP, ATP, UTP, and CTP) (b–d),each at a final concentration of 1 mM (b), 0.4 mM (c), or 0.1 mM (d); �, noadded nucleotide; NP, no Thg1. (Left) RNase A and CIP treatment of reactionproducts, resolved by standard silica TLC. (Right) RNase T2 digestion of reac-tion products, to release 3�-labeled Np resulting from the addition of nucle-otide at position �1, resolved by polyethyleneimine (PEI)-cellulose TLC in 0.5M sodium formate (pH 3.5). Gp, Ap, Up, and Cp, migration of 3�-phosphory-lated standards as visualized by fluorescence quenching.
8642 � www.pnas.org�cgi�doi�10.1073�pnas.0603068103 Jackman and Phizicky
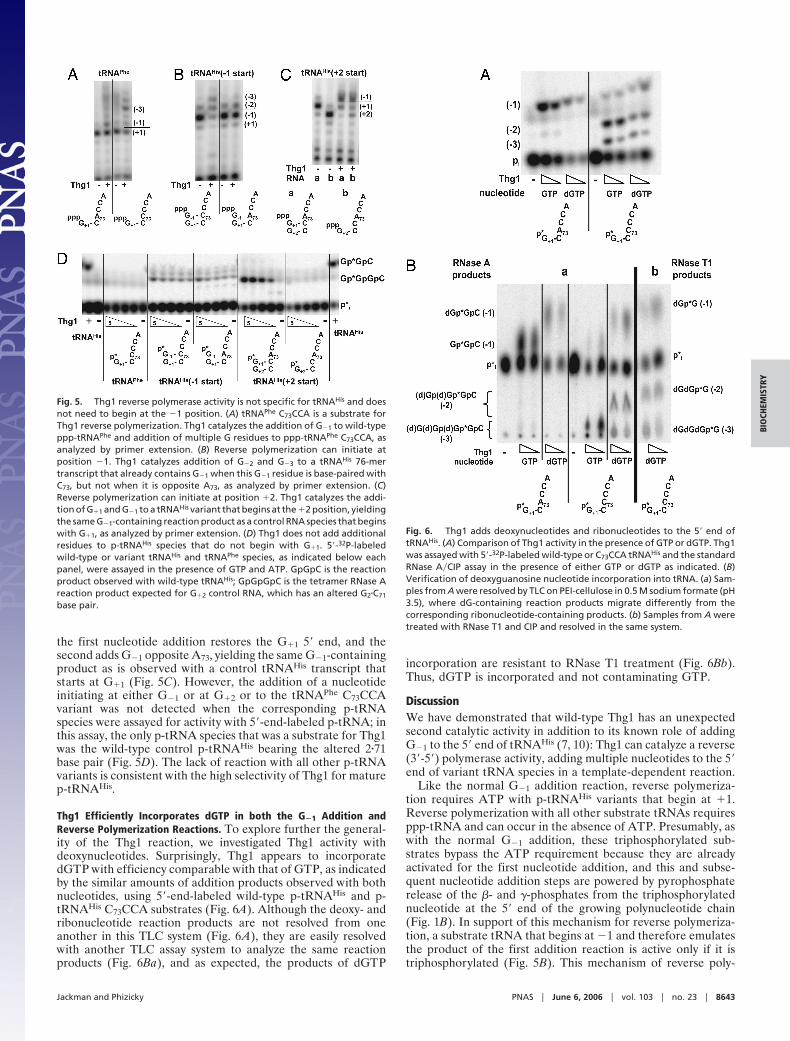
the first nucleotide addition restores the G�1 5� end, and thesecond adds G�1 opposite A73, yielding the same G�1-containingproduct as is observed with a control tRNAHis transcript thatstarts at G�1 (Fig. 5C). However, the addition of a nucleotideinitiating at either G�1 or at G�2 or to the tRNAPhe C73CCAvariant was not detected when the corresponding p-tRNAspecies were assayed for activity with 5�-end-labeled p-tRNA; inthis assay, the only p-tRNA species that was a substrate for Thg1was the wild-type control p-tRNAHis bearing the altered 2�71base pair (Fig. 5D). The lack of reaction with all other p-tRNAvariants is consistent with the high selectivity of Thg1 for maturep-tRNAHis.
Thg1 Efficiently Incorporates dGTP in both the G�1 Addition andReverse Polymerization Reactions. To explore further the general-ity of the Thg1 reaction, we investigated Thg1 activity withdeoxynucleotides. Surprisingly, Thg1 appears to incorporatedGTP with efficiency comparable with that of GTP, as indicatedby the similar amounts of addition products observed with bothnucleotides, using 5�-end-labeled wild-type p-tRNAHis and p-tRNAHis C73CCA substrates (Fig. 6A). Although the deoxy- andribonucleotide reaction products are not resolved from oneanother in this TLC system (Fig. 6A), they are easily resolvedwith another TLC assay system to analyze the same reactionproducts (Fig. 6Ba), and as expected, the products of dGTP
incorporation are resistant to RNase T1 treatment (Fig. 6Bb).Thus, dGTP is incorporated and not contaminating GTP.
DiscussionWe have demonstrated that wild-type Thg1 has an unexpectedsecond catalytic activity in addition to its known role of addingG�1 to the 5� end of tRNAHis (7, 10): Thg1 can catalyze a reverse(3�-5�) polymerase activity, adding multiple nucleotides to the 5�end of variant tRNA species in a template-dependent reaction.
Like the normal G�1 addition reaction, reverse polymeriza-tion requires ATP with p-tRNAHis variants that begin at �1.Reverse polymerization with all other substrate tRNAs requiresppp-tRNA and can occur in the absence of ATP. Presumably, aswith the normal G�1 addition, these triphosphorylated sub-strates bypass the ATP requirement because they are alreadyactivated for the first nucleotide addition, and this and subse-quent nucleotide addition steps are powered by pyrophosphaterelease of the �- and �-phosphates from the triphosphorylatednucleotide at the 5� end of the growing polynucleotide chain(Fig. 1B). In support of this mechanism for reverse polymeriza-tion, a substrate tRNA that begins at �1 and therefore emulatesthe product of the first addition reaction is active only if it istriphosphorylated (Fig. 5B). This mechanism of reverse poly-
Fig. 5. Thg1 reverse polymerase activity is not specific for tRNAHis and doesnot need to begin at the �1 position. (A) tRNAPhe C73CCA is a substrate forThg1 reverse polymerization. Thg1 catalyzes the addition of G�1 to wild-typeppp-tRNAPhe and addition of multiple G residues to ppp-tRNAPhe C73CCA, asanalyzed by primer extension. (B) Reverse polymerization can initiate atposition �1. Thg1 catalyzes addition of G�2 and G�3 to a tRNAHis 76-mertranscript that already contains G�1 when this G�1 residue is base-paired withC73, but not when it is opposite A73, as analyzed by primer extension. (C)Reverse polymerization can initiate at position �2. Thg1 catalyzes the addi-tion of G�1 and G�1 to a tRNAHis variant that begins at the �2 position, yieldingthe same G�1-containing reaction product as a control RNA species that beginswith G�1, as analyzed by primer extension. (D) Thg1 does not add additionalresidues to p-tRNAHis species that do not begin with G�1. 5�-32P-labeledwild-type or variant tRNAHis and tRNAPhe species, as indicated below eachpanel, were assayed in the presence of GTP and ATP. GpGpC is the reactionproduct observed with wild-type tRNAHis; GpGpGpC is the tetramer RNase Areaction product expected for G�2 control RNA, which has an altered G2�C71
base pair.
Fig. 6. Thg1 adds deoxynucleotides and ribonucleotides to the 5� end oftRNAHis. (A) Comparison of Thg1 activity in the presence of GTP or dGTP. Thg1was assayed with 5�-32P-labeled wild-type or C73CCA tRNAHis and the standardRNase A�CIP assay in the presence of either GTP or dGTP as indicated. (B)Verification of deoxyguanosine nucleotide incorporation into tRNA. (a) Sam-ples from A were resolved by TLC on PEI-cellulose in 0.5 M sodium formate (pH3.5), where dG-containing reaction products migrate differently from thecorresponding ribonucleotide-containing products. (b) Samples from A weretreated with RNase T1 and CIP and resolved in the same system.
Jackman and Phizicky PNAS � June 6, 2006 � vol. 103 � no. 23 � 8643
BIO
CHEM
ISTR
Y

merization is very similar to the pyrophosphate release thatpowers conventional 5�-3� polymerization, although in 5�-3�polymerization the pyrophosphate originates from the incomingNTP instead of the polynucleotide (Fig. 1B).
Under normal circumstances in the cell, tRNAs other thantRNAHis are presumably not substrates for reverse polymeriza-tion or G�1 addition by Thg1 because there is no ready sourceof activated 5� ends of the tRNAs. There is no naturally occurringmature ppp-tRNA that would be a substrate for Thg1, andactivation by adenylylation occurs only with 75-mer p-tRNAHis
through Thg1 recognition of the anticodon (14), consistent withthe crucial role of G�1-containing tRNAHis in charging tRNAHis
in vitro (11–13) and in vivo (10). Furthermore, tRNAHis nucle-otide addition is restricted to a single guanosine residue becauseThg1 adds only the single required G�1 opposite the discrimi-nator A73 (Figs. 2B and 3A). This feature emphasizes theimportance of the conserved A73 in eukaryotic tRNAHis becauseThg1 could add additional residues opposite the conservedprokaryotic discriminator, C73.
The incorporation of G�1 into tRNAHis in archaeal species ismore complicated. Despite the nearly universal conservation ofC73 in Archaea, there are at least some species that appear torequire posttranscriptional G�1 addition opposite C73 becausetheir tRNAHis genes lack a genomically encoded G�1, and thecorresponding organisms have a predicted Thg1 ortholog. Thus,archaebacterial Thg1 homologs may possess additional mecha-nisms to ensure addition of only a single guanine residue acrossfrom C73. Consistent with this possibility, the archaeal THG1orthologs are much less conserved than eukaryotic orthologs,and, although both human and Candida albicans THG1 or-thologs can complement a thg1 conditional yeast strain, thearchaeal ortholog from Methanobacterium thermoautotrophicumdoes not complement a thg1 mutant under the same expressionconditions (data not shown).
The results presented here, coupled with previous results, illus-trate a bewildering array of nucleotide recognition and use by Thg1protein. During the addition of G�1, Thg1 recognizes the 5� end ofATP for adenylylation, the 3� end of incoming NTPs for nucleotidyltransfer, and the 5�-pyrophosphate of the product tRNA forpyrophosphatase activity (Fig. 1). In addition, reverse polymeriza-tion of ppp-tRNAHis substrates by Thg1 at positions other than �1requires formation of a G�C or C�G base pair and the potential toform similar base pairs at subsequent addition sites, whereas anynucleotide can be added at position �1 whether or not a base paircan be formed (Fig. 4). Thus, Thg1 has the potential both torecognize and select for nucleotides that participate in Watson–Crick base pairing for polymerization and to recognize and selectguanosine in a non-Watson–Crick interaction opposite A73. Al-though Thg1 base pair recognition is limited to G�C but apparentlynot U�A base pairs (Fig. 4), it is not limited by the normal tRNA 3�end in the number of nucleotides that can be added because a tRNAwith five C residues at the 3� end (tRNAHis C73CCCCA) can beextended by up to five nucleotides (Fig. 3C). The multiple modes ofnucleotide selection by the relatively small (28 kDa) Thg1 suggestconformational changes during the course of the reaction, and theyare reminiscent of the multiple use of different nucleotides by theCCA-adding enzyme, which restructures its single active site foraccommodation of each new nucleotide substrate during nontem-plated addition to the 3� end of the tRNA (16–18).
We emphasize that the reverse polymerase activity describedhere is distinct from the addition of G�1 to tRNAHis that occursin vivo during tRNAHis maturation because the addition of G�1is not dependent on base pair formation, does not require prioractivation of the tRNAHis 5� end, and is absolutely specific forp-tRNAHis that arises after cleavage of pre-tRNA by RNase P.By contrast, reverse polymerization depends on base pair for-mation and is not specific for the correct 5� end of tRNAHis. Wealso emphasize that Thg1 catalyzes reverse polymerization on
tRNAHis and tRNAPhe C73CCA substrates at least as efficientlyas it catalyzes its known physiological G�1 addition reaction onwild-type tRNAHis (Figs. 2B, 3A, and 5A) and that both of thesereactions occur on the same time scale (data not shown). Thus,it is tempting to speculate that the reverse polymerase activityhas a function. It is possible that the reverse polymerizationactivity of Thg1 might simply be a remnant of an earlier 3�-5�polymerase activity of this family of proteins, which could havecompeted with or acted together with the activities of conven-tional 5�-3� polymerases for replication or repair functions.However, it is particularly difficult to reconcile the evolutionaryretention of a distinct Thg1 reverse polymerization activity thatforms G�C and C�G base pairs because such Watson–Crick basepair recognition and use would not be expected of an enzymewhose only role is to add G�1 opposite A73 of tRNAHis in vivo.Rather, the reverse polymerization described here may reflect anadditional Thg1 activity that is used in the cell. Indeed, there isan apparent 3�-5� polymerization activity that functions to editthe 5� end of mitochondrial tRNA in Acanthamoeba castellaniiand Spizellomyces punctatus although, unlike Thg1, this activitydoes not proceed beyond the normal 5� end of the tRNA(19–21). Because the gene product(s) that catalyze this reactionare unknown, the relationship between these activities is unclear.Thg1 might act in a similar or related capacity in 3�-5� polymer-ization of tRNA substrates, although this role would depend onsome additional mechanism to activate the 5� ends of the tRNAspecies.
The ability of Thg1 to use deoxynucleotides may also haveimplications for Thg1 function. Thg1 incorporates deox-yguanosine with an efficiency similar to its ribonucleotide coun-terpart for both reverse polymerization with tRNAHis C73CCAand the addition of G�1 with wild-type tRNAHis (Fig. 6). Thisproperty of Thg1 is in contrast to the highly selective preferencesexhibited by most 5�-3� RNA and DNA polymerases for theirnucleotide substrates (22). However, several members of thepolymerase X family of DNA damage repair polymerases alsoexhibit a lack of discrimination toward nucleotide sugar speciesin vitro, which may be important for their proposed functions invivo, including polymerization at points in the cell cycle whencellular dNTP levels are low and signaling the site of repair bymarking the chromosome with incorporated ribonucleotides (23,24). Thus, the ability of Thg1 to use deoxynucleotides suggeststhat any additional functions of Thg1 might employ eitherdNTPs or rNTPs. In this regard, Thg1 has recently been shownto associate with the origin recognition complex and to beimplicated in the G2�M transition in yeast (25), suggesting apotential role at this site.
Materials and MethodsSources of Thg1. Thg1 protein was purified from both S. cerevisiaeand E. coli as described in ref. 14. Unless otherwise indicated, theThg1 protein used for all assays was the yeast purified protein.
Variant tRNA Species. All tRNA variants with the exception ofC73CCCCA tRNAHis were cloned into a previously describedpUC13-based plasmid under control of the T7 RNA polymerasepromoter for purposes of in vitro transcription (7). tRNAHis andtRNAPhe variants were all created by mutagenic PCR, with theT7 RNA polymerase promoter encoded by the 5� PCR primerand subsequent ligation of the amplified PCR product into thePstI�BamHI sites of the previously described EMP835 (7). AlltRNA-containing plasmids were verified by sequencing. ThetRNAHis C73CCCCA RNA substrate was made by ligation of aphosphorylated synthetic RNA oligonucleotide (5�-ggagauggc-CCCCCA) to the 3� end of a truncated tRNAHis transcript,ending at A63, using 10 units��l T4 DNA ligase (USB Corp.) inthe presence of a complementary DNA oligonucleotide span-ning the ligation site.
8644 � www.pnas.org�cgi�doi�10.1073�pnas.0603068103 Jackman and Phizicky

Thg1 Activity Assay with [5�-32P]tRNA. [5�-32P]tRNA species wereprepared by phosphatase treatment of in vitro transcripts fol-lowed by labeling with [�-32P]ATP using T4 polynucleotidekinase (Roche Applied Biosciences) to a final specific activity of�7,000 Ci�mmol (1 Ci � 37 GBq). The [5�-32P]tRNA was usedas the substrate in a Thg1 assay containing 10 mM MgCl2, 3 mMDTT, 125 mM NaCl, 0.2 mg�ml BSA, and, unless otherwiseindicated, 1 mM each GTP and ATP. Enzyme titration assaysconsisted of 5-fold serial dilutions (�50 �g�ml to 0.4 �g�ml) ofpurified Thg1. Reactions were started by the addition of enzymeand carried out at room temperature. After 5–6 h, the reactionwas stopped by removal of an aliquot (4 �l) to a new tubecontaining 0.5 �l of 0.5 M EDTA and 0.5 �l of 10 mg�ml RNaseA followed by digestion for 10–30 min at 50°C and treatmentwith 0.5 unit of CIP in 1� phosphatase reaction buffer in a finalvolume of 6 �l at 37°C for 30 min. Reactions were spotted tosilica TLC plates (EM Science) and resolved in an n-propylalcohol�NH4OH�H2O (55:35:10) solvent system. The plateswere visualized and quantified with a PhosphorImager andIMAGEQUANT software.
5� End Analysis by Primer Extension. Thg1 reactions were carriedout for 6 h at room temperature in the same buffer describedabove with 1 �M unlabeled triphosphorylated tRNA in a 10-�lreaction volume containing 1 �M purified Thg1. Unless other-wise noted, each reaction contained a 1 mM concentration of theindicated NTP. Thg1-treated tRNAs were purified by extractionwith phenol�chloroform�isoamyl alcohol, concentrated by pre-cipitation with ethanol, and resuspended in 10 �l of buffercontaining 10 mM Tris�HCl (pH 7.5) and 1 mM EDTA. Twomicroliters of each purified tRNA was used as the template inprimer extension reactions employing a 5�-end-labeled primerspecific for tRNAHis (5�-ACTAACCACTATACTAAGA-3�) asshown in Fig. 2 A or a similar tRNAPhe-specific primer (5�-TGGCGCTCTCCCAACTG-3�). After the Thg1-treated tRNAtemplate was annealed to 1 pmol of primer in a final volume of5 �l, extension reactions were carried out with 8 units of avianmyeloblastosis virus reverse transcriptase (AMV-RT) (20 units��l; Promega) in the presence of a 0.4 mM concentration of eachdNTP in 1� AMV-RT reaction buffer (Promega) in a finalvolume of 10 �l at 37°C for 1–2 h. The resulting products wereresolved on 10% polyacrylamide�4 M urea gels, which weredried and visualized with the PhosphorImager.
Determination of 5� Nucleotide Identity by RNase T2 Digestion. 5�32P-labeled wild-type tRNAHis was used in Thg1 reactions in thebuffer described above with varied concentrations of all fourNTPs (1 mM, 0.4 mM, or 0.1 mM each) and 1 �M purified Thg1in a 10-�l reaction mixture for 6 h at room temperature. After6 h, a portion (4 �l) of the reaction was stopped, digested, andanalyzed as described above with EDTA and RNase A. Another4-�l aliquot was removed to a new tube containing 0.5 unit ofCIP in 1� alkaline phosphatase buffer in a final volume of 10 �lto remove the background from the remaining unreacted sub-strate tRNA. After 30 min at 37°C, the reaction products werepurified by extraction with phenol�chloroform�isoamyl alcoholand precipitation with ethanol. The resulting tRNA was resus-pended in a 10-�l reaction mixture containing 1 unit of RNaseT2 and 0.5 �g of RNase A in 20 mM sodium acetate (pH 5.2)�1mM EDTA and digested for 30 min at 50°C. Two microliters ofeach digestion reaction mixture was spotted to PEI-celluloseplates and developed in a 0.5 M sodium formate (pH 3.5) solventsystem. The positions of unlabeled nucleotide standards weredetermined by spotting 2 �l of 10 mM solutions of GMP, UMP,AMP, or CMP and visualizing the resultant spots by fluores-cence quenching.
Determination of Deoxyguanosine Incorporation by Resistance toRNase T1 Digestion. Thg1 reactions were carried out as describedabove with 5�-32P-labeled tRNAHis (wild-type and C73CCAspecies) in the presence of 0.1 mM ATP and either 1 mM GTPor 1 mM dGTP for 5 h at room temperature. After digestion withRNase A and CIP, as described above, a 2-�l portion of thereaction mixture was removed to a new tube containing 2 �g oftorula yeast tRNA and 0.5 unit of RNase T1 in 30 mM sodiumacetate (pH 5.2) buffer in a final volume of 5 �l. The RNase T1digestion was allowed to proceed for 30 min at 50°C, and then thereaction mixtures were treated with 0.5 unit of CIP in 1� alkalinephosphatase reaction buffer in a final volume of 6 �l. After 30min at 37°C, 2 �l of each reaction mixture (both the originalRNase A-treated and RNase A- � RNase T1-treated reactions)were spotted to PEI-cellulose TLC plates and resolved in the 0.5M sodium formate (pH 3.5) solvent system.
We thank S. Crary, S. Fields, M. Gorovsky, M. Gray, E. Grayhack, R.Green, and A. Hopper for advice and comments. This work wassupported by National Institutes of Health Grant GM52347.
1. Steitz, T. A. (1999) J. Biol. Chem. 274, 17395–17398.2. Aphasizhev, R., Sbicego, S., Peris, M., Jang, S. H., Aphasizheva, I., Simpson,
A. M., Rivlin, A. & Simpson, L. (2002) Cell 108, 637–648.3. Schurer, H., Schiffer, S., Marchfelder, A. & Morl, M. (2001) Biol. Chem. 382,
1147–1156.4. Sprinzl, M., Horn, C., Brown, M., Ioudovitch, A. & Steinberg, S. (1998) Nucleic
Acids Res. 26, 148–153.5. Schnare, M. N., Heinonen, T. Y., Young, P. G. & Gray, M. W. (1985) Curr.
Genet. 9, 389–393.6. Orellana, O., Cooley, L. & Soll, D. (1986) Mol. Cell. Biol. 6, 525–529.7. Gu, W., Jackman, J. E., Lohan, A. J., Gray, M. W. & Phizicky, E. M. (2003)
Genes Dev. 17, 2889–2901.8. Jahn, D. & Pande, S. (1991) J. Biol. Chem. 266, 22832–22836.9. Cooley, L., Appel, B. & Soll, D. (1982) Proc. Natl. Acad. Sci. USA 79,
6475–6479.10. Gu, W., Hurto, R. L., Hopper, A. K., Grayhack, E. J. & Phizicky, E. M. (2005)
Mol. Cell. Biol. 25, 8191–8201.11. Nameki, N., Asahara, H., Shimizu, M., Okada, N. & Himeno, H. (1995) Nucleic
Acids Res. 23, 389–394.
12. Rudinger, J., Florentz, C. & Giege, R. (1994) Nucleic Acids Res. 22, 5031–5037.13. Rudinger, J., Felden, B., Florentz, C. & Giege, R. (1997) Bioorg. Med. Chem.
5, 1001–1009.14. Jackman, J. E. & Phizicky, E. (April 2006) RNA, 10.1261/rna.54706.15. Marck, C. & Grosjean, H. (2002) RNA 8, 1189–1232.16. Xiong, Y., Li, F., Wang, J., Weiner, A. M. & Steitz, T. A. (2003) Mol. Cell 12,
1165–1172.17. Shi, P. Y., Maizels, N. & Weiner, A. M. (1998) EMBO J. 17, 3197–3206.18. Yue, D., Weiner, A. M. & Maizels, N. (1998) J. Biol. Chem. 273, 29693–29700.19. Lonergan, K. M. & Gray, M. W. (1993) Science 259, 812–816.20. Lonergan, K. M. & Gray, M. W. (1993) Nucleic Acids Res. 21, 4402.21. Bullerwell, C. E. & Gray, M. W. (2005) J. Biol. Chem. 280, 2463–2470.22. Rose, A. M., Joyce, P. B., Hopper, A. K. & Martin, N. C. (1992) Mol. Cell. Biol.
12, 5652–5658.23. Nick McElhinny, S. A. & Ramsden, D. A. (2003) Mol. Cell. Biol. 23, 2309–2315.24. Bebenek, K., Garcia-Diaz, M., Patishall, S. R. & Kunkel, T. A. (2005) J. Biol.
Chem. 280, 20051–20058.25. Rice, T. S., Ding, M., Pederson, D. S. & Heintz, N. H. (2005) Eukaryot. Cell
4, 832–835.
Jackman and Phizicky PNAS � June 6, 2006 � vol. 103 � no. 23 � 8645
BIO
CHEM
ISTR
Y




















