
Ribosome association primes the stringent factor Rel for tRNA ...
Upload
independentCategory
view
2download
0
doi:10.1016/j.jmb.2008.04.005 J. Mol. Biol. (2008) 379, 579–588
Available online at www.sciencedirect.com
tRNA Integrity Is a Prerequisite for Rapid CCA Addition:Implication for Quality Control
Marcel Dupasquier†, Sangbumn Kim†, Konstantine Halkidis,Howard Gamper and Ya-Ming Hou⁎
Department of Biochemistryand Molecular Biology,Thomas Jefferson University,233 South 10th Street,Philadelphia, PA 19107, USA
Received 20 January 2008;received in revised form22 March 2008;accepted 2 April 2008Available online8 April 2008
*Corresponding author. E-mail [email protected].† M.D. and S.K. contributed equaAbbreviations used: AfCCA, Arch
CCA enzyme; ASL, anticodon stem–Escherichia coli CCA enzyme.
0022-2836/$ - see front matter © 2008 E
CCA addition to the 3′ end is an essential step in tRNA maturation. High-resolution crystal structures of the CCA enzymes reveal primary enzymecontact with the tRNA minihelix domain, consisting of the acceptor stemand T stem–loop. RNA and DNA minihelices are efficient substrates forCCA addition in steady-state kinetics. However, in contrast to structuralmodels and steady-state experiments, we show here by single-turnoverkinetics that minihelices are insufficient substrates for the Escherichia coliCCA enzyme and that only the full-length tRNA is kinetically competent.Even a nick in the full-length tRNA backbone in the T loop, or as far awayfrom the minihelix domain as in the anticodon loop, prevents efficient CCAaddition. These results suggest a kinetic quality control provided by theCCA enzyme to inspect the integrity of the tRNA molecule and to discri-minate against nicked or damaged species from further maturation.
© 2008 Elsevier Ltd. All rights reserved.
Keywords: kinetic quality control; tRNA surveillance; single-turnoverkinetics
Edited by J. DoudnaIntroduction
Many tRNA molecules must acquire the CCA se-quence at their 3′ end as an essential step in thematuration process.1 Also, tRNAs that are synthe-sized with the CCA sequence must be repaired inthe event of CCA degradation by nucleases. Theenzyme that catalyzes the synthesis and repair of theCCA sequence is ATP(CTP):tRNA nucleotidyl trans-ferase (the CCA enzyme), which is present in allthree domains of life and is essential for growth inorganisms that lack the CCA sequence in their tRNAgenes.2 According to sequence motifs in the catalyticdomains, the CCA enzymes are divided into twoclasses: the archaeal enzymes are assigned to class Iof the nucleotidyl transferase superfamily, while thebacterial and eukaryotic enzymes are assigned toclass II.3 Both classes of CCA enzymes use a singlecatalytic site to perform sequential nucleotide addi-tion without copying a nucleic acid template,4–11
ess:
lly to this work.aeoglobus fulgidusloop; EcCCA,
lsevier Ltd. All rights reserve
in contrast to template-dependent polynucleotidepolymerases. The generally accepted model of thetemplate-independent CCA addition assumes thatboth classes of CCA enzymes bind only to theacceptor stem–T stem–loop of tRNA (the minihelixdomain) for recognition.6 This model is based on co-crystal structures of the class I Archaeoglobus fulgidusCCA enzyme (AfCCA) bound to a full-length tRNA(Fig. 1a) and to minihelices.9,11 In these structures,the anticodon stem–loop (ASL) domain is projectedoutward, while the minihelix domains are in super-imposable positions among various substrates andare held in fixed locations by the enzyme. Consistentwith footprint and biochemical analyses,12–14 thesestructures reveal that the 3′ end of the minihelixdomain refolds at each step of nucleotide additionto place the terminus at the active site. The mini-helix domain is accommodated in an extended classI cleft that has a counterpart in class II enzymes withsimilar shape, charge, and dimensions.6,9 Indeed,tRNA minihelices are efficient substrates for bothclasses of CCA enzymes in steady-state kinetics.15–17
However, we show here by fast kinetics of the classII Escherichia coli CCA enzyme (EcCCA) that theminihelix domain is insufficient to account for thekinetics of CCA addition and that the full-lengthtRNA is necessary. Even a backbone break in ASL,distal from the acceptor end, causes a delay in CCA
d.

Fig. 1. (a) Crystal structure of the dimer ofAfCCA–tRNA complex (Protein Data Bank code 1SZ1). The tRNA backboneis highlighted in red and blue, whereas AfCCA subunits are represented by purple and green ribbons. (b) Schematicdrawing of the Val34dCr minihelix substrate for A76 addition, with ATP as the nucleotide donor. The minihelix is5′-labeled with 32P (50 nM), indicated by a red dot, and the time course of conversion to Val35dCr catalyzed by EcCCA(200 μM) is analyzed by 12% PAGE/7 M urea. The nomenclature of minihelices is established by the identity of the tRNA(Val), the length of the minihelices (from a 34-mer to a 35-mer), the DNA backbone (d), and the terminal ribose residue(from Cr to Ar). (c) Fitting the data of the time course in (b) to a single exponential equation to derive kapp=1.4±0.1 s
−1. (d)Representative time courses of A76 addition to Val34dCr by increasing concentrations of EcCCA (1–200 μM, as shown).Reactions were performed by preincubating EcCCA with the minihelix substrate, followed by rapid mixing with ATP(1 mM) over various periods, then terminated by mixing with 7 M urea as shown in (e). (e) Linear increase of kapp as afunction of EcCCA concentration based on data from (d), giving an estimated kon of 7×103 s−1 M−1.
580 Fast Kinetics of CCA Addition
addition. These results suggest the possibility thatthe CCA addition serves to kinetically discriminateagainst nicked or damaged tRNA in the qualitycontrol of tRNA maturation.Fast kinetics provides insights for understanding
enzyme mechanisms that are unattainable fromsteady-state kinetics. The steady-state kcat and Kmparameters are complex terms of all of the reactionsoccurring on the enzyme such that individual stepsare buried within these terms and are not resolvable.We addressed the limitations of steady-state kineticsby developing an assay for measuring transientkinetics of CCA addition, treating the enzyme as astoichiometric reactant relative to the substrate toallow for the isolation of specific rate and equilibriumconstants. Because both the enzyme and substratewere examined at micromolar concentrations, similarto those in cell physiology, the information obtainedby transient kinetics is biologically relevant.
Results
Kinetics of minihelices
Extensive steady-state kinetics studies of mini-helices were performed with class II EcCCA. Such
studies support the minihelix recognition model,showing that the dihydrouridine and ASL regionsare dispensable and that the only ribose requirementfor nucleotide addition is the last one in the mini-helix domain.15 For example, a 34-mer DNA mini-helix based on the sequence of E. coli tRNAVal thatterminates with C75 is an efficient substrate for A76addition, exhibiting a kcat of 1.3 min−1, similar to thekcat of 1.1 min−1 of a reconstituted full-length tRNA.16
This DNAminihelix, designated asVal34dCr (Fig. S1),was chemically synthesized with a deoxyribose back-bone up to the 1–72 base pairs and a ribose backboneat positions 73–75. Similarly, DNA minihelicesVal33dCr and Val32dAr, which terminate with C74andA73, respectively, are functional substrates for C75and C74 addition,15 although in these cases, oligoCsynthesis is observed when CTP is present (withoutATP) as the only nucleotide triphosphate.16–18
To extend steady-state studies of EcCCA, single-turnover kinetics was developed with minihelixsubstrates. The reaction of A76 addition to the DNAminihelix Val34dCr was performed on a rapid che-mical quench instrument and was monitored byfollowing conversion of the 5′-32P-labeled minihelixto the Val35dAr product. EcCCA was maintainedin molar excess of the substrate to ensure only oneturnover, while ATP was present at a saturatingconcentration (1 mM) relative to its Km (3 μM).17
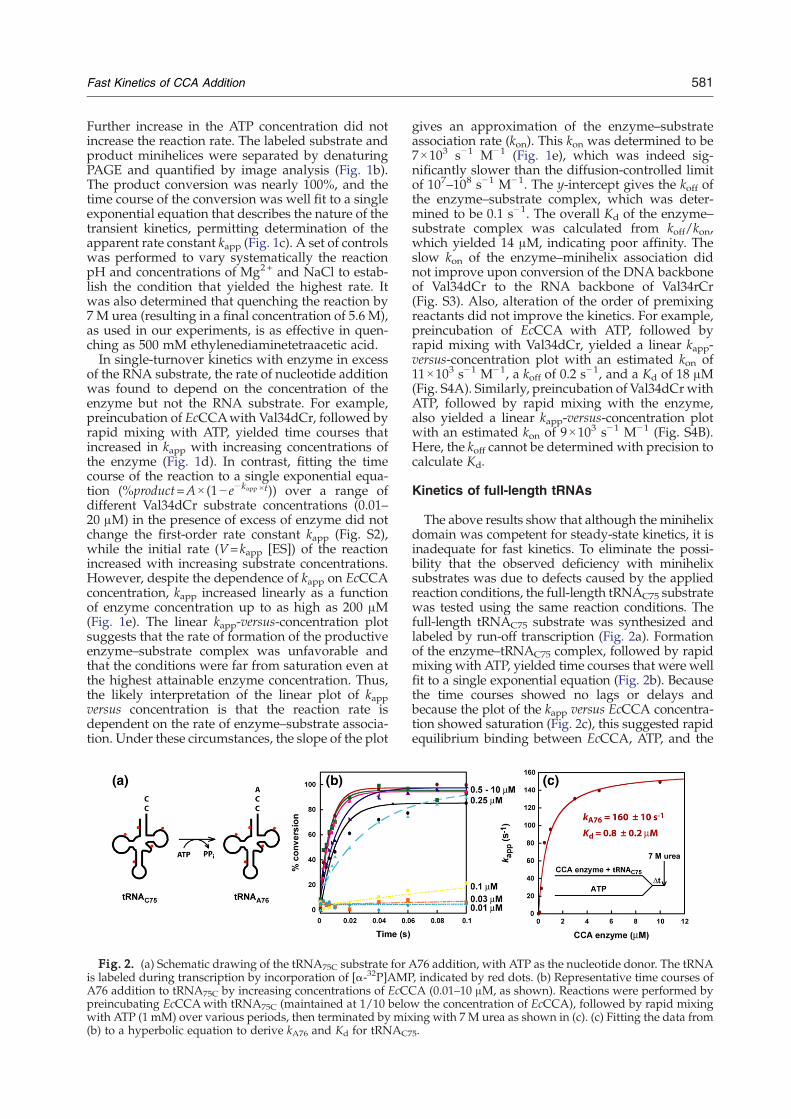
581Fast Kinetics of CCA Addition
Further increase in the ATP concentration did notincrease the reaction rate. The labeled substrate andproduct minihelices were separated by denaturingPAGE and quantified by image analysis (Fig. 1b).The product conversion was nearly 100%, and thetime course of the conversion was well fit to a singleexponential equation that describes the nature of thetransient kinetics, permitting determination of theapparent rate constant kapp (Fig. 1c). A set of controlswas performed to vary systematically the reactionpH and concentrations of Mg2+ and NaCl to estab-lish the condition that yielded the highest rate. Itwas also determined that quenching the reaction by7 M urea (resulting in a final concentration of 5.6 M),as used in our experiments, is as effective in quen-ching as 500 mM ethylenediaminetetraacetic acid.In single-turnover kinetics with enzyme in excess
of the RNA substrate, the rate of nucleotide additionwas found to depend on the concentration of theenzyme but not the RNA substrate. For example,preincubation of EcCCAwith Val34dCr, followed byrapid mixing with ATP, yielded time courses thatincreased in kapp with increasing concentrations ofthe enzyme (Fig. 1d). In contrast, fitting the timecourse of the reaction to a single exponential equa-tion (%product=A×(1− e− kapp ×t)) over a range ofdifferent Val34dCr substrate concentrations (0.01–20 μM) in the presence of excess of enzyme did notchange the first-order rate constant kapp (Fig. S2),while the initial rate (V=kapp [ES]) of the reactionincreased with increasing substrate concentrations.However, despite the dependence of kapp on EcCCAconcentration, kapp increased linearly as a functionof enzyme concentration up to as high as 200 μM(Fig. 1e). The linear kapp-versus-concentration plotsuggests that the rate of formation of the productiveenzyme–substrate complex was unfavorable andthat the conditions were far from saturation even atthe highest attainable enzyme concentration. Thus,the likely interpretation of the linear plot of kappversus concentration is that the reaction rate isdependent on the rate of enzyme–substrate associa-tion. Under these circumstances, the slope of the plot
Fig. 2. (a) Schematic drawing of the tRNA75C substrate for Ais labeled during transcription by incorporation of [α-32P]AMPA76 addition to tRNA75C by increasing concentrations of EcCpreincubating EcCCAwith tRNA75C (maintained at 1/10 belowith ATP (1 mM) over various periods, then terminated by mix(b) to a hyperbolic equation to derive kA76 and Kd for tRNAC7
gives an approximation of the enzyme–substrateassociation rate (kon). This kon was determined to be7×103 s−1 M−1 (Fig. 1e), which was indeed sig-nificantly slower than the diffusion-controlled limitof 107–108 s−1 M−1. The y-intercept gives the koff ofthe enzyme–substrate complex, which was deter-mined to be 0.1 s−1. The overall Kd of the enzyme–substrate complex was calculated from koff/kon,which yielded 14 μM, indicating poor affinity. Theslow kon of the enzyme–minihelix association didnot improve upon conversion of the DNA backboneof Val34dCr to the RNA backbone of Val34rCr(Fig. S3). Also, alteration of the order of premixingreactants did not improve the kinetics. For example,preincubation of EcCCA with ATP, followed byrapid mixing with Val34dCr, yielded a linear kapp-versus-concentration plot with an estimated kon of11×103 s−1 M−1, a koff of 0.2 s−1, and a Kd of 18 μM(Fig. S4A). Similarly, preincubation of Val34dCrwithATP, followed by rapid mixing with the enzyme,also yielded a linear kapp-versus-concentration plotwith an estimated kon of 9×103 s−1 M−1 (Fig. S4B).Here, the koff cannot be determined with precision tocalculate Kd.
Kinetics of full-length tRNAs
The above results show that although the minihelixdomain was competent for steady-state kinetics, it isinadequate for fast kinetics. To eliminate the possi-bility that the observed deficiency with minihelixsubstrates was due to defects caused by the appliedreaction conditions, the full-length tRNAC75 substratewas tested using the same reaction conditions. Thefull-length tRNAC75 substrate was synthesized andlabeled by run-off transcription (Fig. 2a). Formationof the enzyme–tRNAC75 complex, followed by rapidmixing with ATP, yielded time courses that were wellfit to a single exponential equation (Fig. 2b). Becausethe time courses showed no lags or delays andbecause the plot of the kapp versus EcCCA concentra-tion showed saturation (Fig. 2c), this suggested rapidequilibrium binding between EcCCA, ATP, and the
76 addition, with ATP as the nucleotide donor. The tRNA, indicated by red dots. (b) Representative time courses ofCA (0.01–10 μM, as shown). Reactions were performed byw the concentration of EcCCA), followed by rapid mixinging with 7 M urea as shown in (c). (c) Fitting the data from5.

582 Fast Kinetics of CCA Addition
tRNA substrate, which is in contrast with the kineticsof minihelices. Fitting the plot of kapp versus EcCCAconcentration to a hyperbolic equation revealed thesaturating kapp at 160±10 s
−1, which is defined as therate constant kA76 for A76 addition. This kA76 is acomposite term that includes all of the events up toand including nucleotidyl transfer. The curve fittingalso revealed the kinetic Kd at 0.8±0.2 μM for theaffinity of EcCCA with tRNAC75 (Fig. 2c). Here, thekinetic Kd is the Km of the single.-turnover reaction[Km(sto)]. In rapid equilibrium condition, the thermo-dynamics of binding controls the proportion of thetRNA substrate actually bound to the enzyme and,thus, the reaction rate kapp. The kinetic Kd [or Km(sto)]includes the possibility that a rearrangement of theinitial enzyme–NTP–tRNA complex may occur toform the productive enzyme–substrate interaction.Similar results were obtained by altering the premix-ing conditions. For example, premixing EcCCAwithATP, followed by rapid mixing with tRNAC75,showed saturation kinetics with kA76=170±10 s−1
and kinetic Kd=1.1±0.3 μM (Fig. S5A). PremixingtRNAC75 with ATP, followed by rapid mixing withEcCCA, showed saturation kinetics with kA76=130±10 s−1 and kinetic Kd=2.4±0.2 μM (Fig. S5B).The contrast between the full-length tRNA75C and
minihelices in fast kinetics emphasized that the tRNAwhole body is necessary for high-affinity and rapidproductive binding to the enzyme. Moreover, thekA76 value in the range of 130–170 s−1 for tRNAC75 iscomparable with the kpol value of 220 s−1 of thetemplate-dependent T7 RNA polymerase,19 indicat-ing that the rate-determining steps of these enzymestake place in similar time scales. The kineticKd of 0.8–2.4 μM for the enzyme affinity to tRNAC75 is in therange of the physiological concentration of tRNAprecursors, estimated at 2 μM.3
Characterization of nicked tRNAs
An earlier study showed that EcCCA has aphosphohydrolase activity that repairs aberranttRNA 3′ ends before nucleotide addition.20 Theability of EcCCA to discriminate against minihelicessuggested the further possibility that the enzymemight inspect the tRNAwhole body to reject nicks inthe tRNA phosphodiester backbone. To test thispossibility, two nicked substrates were prepared(Fig. S1). One was formed by hybridization of theG1–G57 fragment of E. coli tRNAVal with thechemically synthesized A58–C75 fragment to pro-duce a nick in the T loop of the minihelix domain.Such a T-nicked substrate has been shown to befunctional for recognition by EcCCA in steady-statekinetics.16 Also, a similar approach was used earlierto create a T-nicked substrate in the sequenceframework of E. coli tRNAPro, which was shown tobe functional for aminoacylation by E. coli prolyl-tRNA synthetase,21 an enzyme that recognizes thetRNA global structure.22 The other nicked substrateof E. coli tRNAVal was formed by hybridization ofthe G1–A37 fragment of the tRNAwith the G38–C75fragment to produce a nick in the ASL. In this case,
the G38–C75 fragment replaced the natural A38with G38 to allow efficient transcription in vitro. Asimilar ASL-nicked tRNA in the sequence frame-work of E. coli tRNAGln was previously constructed,where the backbone nick was readily sealed by T4RNA ligase to generate an intact tRNA that isrecognized by E. coli glutaminyl-tRNA synthetase,23
which also recognizes the tRNA global structure foraminoacylation.In the preparation of the two nicked substrates of
E. coli tRNAVal, the 3′ fragment was labeled with 32Pto allow analysis of hybridization by gel electro-phoresis in native conditions containing 10 mMMgCl2. Hybridization analysis revealed two pro-ducts in the generation of the T-nicked substrate(Fig. 3a, left) and one major product as well as oneminor product in the generation of the ASL-nickedsubstrate (Fig. 3a, right). In both cases, the majorproduct (indicated by red arrows) exhibited thesame gel mobility as that of the intact and full-lengthtRNA and was isolated from the native gel and usedas the nicked substrate, whereas the minor product(indicated by black arrows) exhibited aberrant gelmobility and was not used. The concentrations ofboth the gel-purified T- and ASL-nicked substrateswere determined by UV absorption.Because the ASL-nicked substrate has the A38G
substitution (Fig. 3b), which can induce an aberrantfolding (Fig. 3c), it was characterized further. Thefollowing three experiments support the notion thatthe ASL-nicked substrate, as isolated from Fig. 3a,existed in the proposed canonical structure (Fig. 3b).First, the ASL-nicked substrate is readily ligated byT4 RNA ligase 2 (Ref. 24) to 55% (data not shown).This high efficiency of ligation is not expected forthe putative aberrant structure, which is unfavor-able for ligation. Second, the ASL-nicked substrate isaminoacylated by E. coli ValRS with better efficiencycompared with the minor species, which might re-present the aberrant ASL-nicked tRNA (designatedas such in Fig. 3d). The rationale of this experimentwas based on extensive structural and biochemicalstudies of the tRNAVal–ValRS interaction, whichmust depend on direct enzyme recognition of thetRNA anticodon nucleotides.25 In the canonicalstructure, the anticodon nucleotides are accessiblefor recognition, although the presence of the nickadjacent to the anticodon triplet is expected to re-duce the aminoacylation activity relative to that ofthe intact tRNA. However, in the putative aberrantstructure, the anticodon nucleotides are inaccessible,preventing recognition by ValRS. More important,aminoacylation analysis also showed that the intactA38G mutant retained the activity of the wild-typetRNA, indicating that the A38G substitution did notsignificantly alter the tRNA structure. Third, theASL-nicked substrate was competent for lead cleav-age (Fig. 3e). The lead metal ion induces cleavagebetween nucleotide 17 and the conserved G18 in thecanonical tRNA tertiary core.26 This cleavage iscoordinated by precise positioning of lead in apocket formed by conserved residues in the D and Tloops.27 Substitutions in the tRNA tertiary core can

Fig. 3. Characterization of nicked tRNAs. (a) Formation of the T-nicked (left) and ASL-nicked (right) tRNAC75substrates by hybridization of an unlabeled 5′ fragment with a 32P-labeled 3′ fragment. Full-length and intact 32P-labeledtRNAC75 was run as a marker. The nicked substrates (indicated by the red arrows) that co-migrated with the intact tRNAmarker were isolated from gels and used for analysis of CCA addition, while the aberrant hybridization products thatmigrated slower than the intact tRNAwere not further analyzed for CCA addition and are marked by black arrows. (b)Sequence and cloverleaf structure of the A38G mutant of E. coli tRNAVal, where the A38G substitution is indicated by acircle. (c) Aberrant structure of the ASL-nicked A38G tRNAVal. (d) Aminoacylation of tRNAVal transcripts (2 μM) bypurified E. coli ValRS (4 μM). (e) Lead cleavage of intact A38G tRNA and the ASL-nicked A38G tRNA analyzed by 12%denaturing PAGE. The numbers on the sides of the gel indicate the lengths of tRNA or fragments.
583Fast Kinetics of CCA Addition
eliminate lead cleavage.27 Notably, the intact A38Gmutant of tRNAVal was a functional substrate for leadcleavage, generating a large fragment of 59-mer anda smaller fragment of 17-mer. These fragments werewell resolved by 12% denaturing PAGE (Fig. 3e). TheASL-nicked A38G mutant dissociated in the de-naturing PAGE into a mixture of two co-migratingfragments, a 39-mer and a 37-mer. Upon incubationof the ASL-nicked substrate with lead ions undernative conditions, one of the co-migrating fragments(the 37-mer) was cleaved into 17- and 20-mer frag-ments, while the 39-mer fragment remained intact.This cleavage pattern is consistent with the presenceof a prominent Pb2+ cleavage site in the D/T loopregion, suggesting that the ASL-nicked tRNA basi-cally adopts the canonical L-shaped tRNA fold.Together, the above results support the notion thatthe ASL-nicked tRNA adopts the normal tRNA
structure that is recognized for ligation, amino-acylation, and lead cleavage.
Kinetics of nicked tRNAs
Single-turnover kinetics of A76 addition to boththe T- and ASL-nicked substrates was measured bymixing a preformed EcCCA–substrate complex withATP. Both nicked substrates were competent forCCA addition, with more than 90% of substrateconverted to product at the end of the reaction.However, the T-nicked substrate exhibited slowkinetics, with kapp values ∼1000-fold lower thanthose of the intact tRNAC75 at equivalent enzymeconcentrations (Fig. 4a). A plot of kapp versus enzymeconcentration was linear up to 100 μM EcCCA,with aslow kon of 7×10
3 s−1 M−1 (Fig. 4a, inset) in a patternsimilar to that of minihelices. The ASL-nicked

Fig. 4. Kinetics of nicked tRNA. (a) Kinetics of A76addition to the intact (red), ASL-nicked (green), andT-nicked (blue) tRNAC75 as a function of EcCCA concen-tration. Inset: an expanded view of the kapp-versus-EcCCAconcentration plot for the T-nicked tRNAC75. (b) Kinetics ofC75 addition to the intact (red) and ASL-nicked (green)tRNAC74 as a function of EcCCA concentration. (c) Kineticsof C74 addition to the intact (red) and ASL-nicked (green)tRNAA73 as a function of EcCCA concentration. Single-turnover rates were monitored by incubating a tRNAsubstrate with EcCCA, followed by rapid mixing with thecorrect nucleotide donor.
584 Fast Kinetics of CCA Addition
substrate also exhibited a slower rate, but the kineticsof kapp versus enzyme concentration showed satura-tion, permitting the determination of kA76 (170±10 s−1), similar to that of the intact tRNA, but a10-fold higher kinetic Kd (8.6±1.3 μM) for enzyme–substrate association (Fig. 4a). Thus, EcCCA discri-minates against the ASL nick by reducing the affinityto the nicked substrate. Once stably bound to thenicked substrate, the enzyme catalyzes A76 additionat a rate identical with that of the intact tRNA.The kinetic discrimination of EcCCA against
minihelices and nicked tRNA substrates was fur-ther demonstrated for C75 and C74 addition. C75addition to the minihelix Val33dCr, performed inthe presence of a saturating concentration of CTP(5mM), produced oligoC (Fig. S6A and B), consistentwith previous observations.17,18 In this case, the rateof C75 addition based on summing all the productsof oligoC synthesis was the same as the rate based onsubstrate consumption. However, measurement ofkapp as a function of EcCCA concentration showed alinear increase of the rate with a rather slow kon of250 s−1 M−1 (Fig. S6C and D). In contrast, C75addition to the full-length tRNAC74 manifested twomajor improvements. First, the addition was pre-cisely one nucleotide without synthesis of oligoC(Fig. S7A and B), indicating that the presence of thetRNAwhole body strengthened the specificity of theenzyme. Second, the kinetics of C75 addition wassaturable (Fig. S7C and D), with kC75=170±20 s−1
and kinetic Kd=2.8±0.8 μM for the tRNAC74 sub-strate. Furthermore, introduction of a backbone nickbetween positions 37 and 38 in the ASL increased thekinetic Kd to 17.5±3.8 μM but had little effect on kC75(180±20 s−1) (Fig. 4b), indicating discrimination atthe binding step. These observations were recapitu-lated for C74 addition. In the presence of CTP only,the Val32dAr minihelix was a substrate for both C74addition and oligoC synthesis (Fig. S8A and B), andthe kinetics of nucleotide addition revealed a linearincrease of kapp as a function of enzyme concentra-tion, exhibiting an even slower kon of 25 s−1 M−1
(Fig. S8C and D) compared with that for C75addition. In contrast, the full-length tRNAA73 wasan efficient substrate for C74 addition, which wasrapidly followed by C75 addition (Fig. S9A and B).Kinetic analysis revealed a saturating kC74 of 170±20 s−1 and a kinetic Kd of 3.3±0.8 μM for thetRNAA73 substrate (Fig. S9C and D). Also, introduc-tion of a backbone nick at positions 37 and 38 raisedthe kinetic Kd to 16.9±4.2 μM but had only a smalleffect on kC74 (150±10 s−1) (Fig. 4c).
Discussion
The kinetic parameters of A76, C75, and C74addition to the full-length and ASL-nicked tRNAVal
substrates by EcCCA are summarized in Table 1.With the full-length tRNA, the enzyme catalyzesrapid nucleotide addition with a similar rate at allthree steps and has a physiologically relevant highaffinity. However, introduction of a backbone break

Table 1. Kinetic parameters of CCA addition
Intact tRNA ASL-nicked tRNADiscrimination[kN/Kd (intact)/kN/Kd (nicked)]kN (s−1) Kd (tRNA, μM)
kN/Kd(s−1 μM−1) kN (s−1)
Kd(tRNA, μM)
kN/Kd(s−1 μM−1)
A76 170±10 0.8±0.2 212 170±10 8.6±1.3 20 10.6C75 170±20 2.8±0.8 61 180±20 17.5±3.8 10 6.1C74 170±20 3.3±0.8 51 150±10 16.9±4.2 8.9 5.7
The reported kinetic Kd [or Km(sto)] values represent the monomer concentration of EcCCA. In crystal structures of the class I AfCCA, theenzyme exists as a dimer and binds two molecules of tRNA or the minihelix domain.6,7,9,11 In the crystal structure of the class II Bacillusstearothermophilus CCA, the enzyme also exists as a dimer and is modeled to bind two molecules of tRNA.5,6
Values reported are based on experiments performed by preincubating EcCCAwith a tRNA substrate in one syringe of the rapid quenchinstrument, followed by rapid mixing with NTP from the other syringe.
585Fast Kinetics of CCA Addition
to the ASL, the domain that is separate from the CCAend by the longest distance (~75 Å) in tRNA, reducesthe enzyme affinity to the nicked-substrate, suggest-ing that the proper activity of the minihelix domainfor CCA addition is dependent on the integrity of theASL. The overall discrimination against the nick foreach nucleotide addition is manifested by the ratio ofthe specificity factor kN/Kd for the intact tRNA to thefactor for the nicked substrate. This discriminationincreases from 5.7-fold for C74 addition to 6.1-fold forC75 addition to 10.6-fold for A76 addition, indicatinga progressively closer scrutiny of the tRNA substrateas the enzyme approaches completion of CCA syn-thesis. The discrimination against the T-nicked sub-strate is even stronger because reactions did not reachsaturation even at high enzyme concentrations(∼100 μM). This is rationalized by the fact that thenick is localized in the minihelix domain, which is indirect contact with the enzyme.The discrimination against backbone nicks by
EcCCA is likely shared in common by all class IICCA enzymes (including those of eukaryotes), giventhe similar properties of substrate recognition amongmembers of this class.28 In the lifetime of tRNAbiosynthesis, backbone nicks can be generated in anumber of ways. For example, misincorporation ofnucleotides, inappropriate processing and folding,and incomplete posttranscriptional modifications intRNA29 can lead to aberrant tertiary structures thatare prone to nuclease cleavage. Some bacterial andeukaryotic tRNAs are also subject to cleavage at theASL by specific toxins.30,31 In eukaryotes, the ASL isthe site that carries introns for certain tRNA precur-sors (usually between positions 37 and 38 as de-signated in Fig. S1). Splicing intermediates withoutproperly religated backbones that accumulate incells have been identified.32 Discrimination of thesenicked tRNAs from CCA addition offers a qualitycontrol mechanism that is manifested at the kineticlevel. Based on the estimated cellular concentra-tion of CCA enzyme at ~0.8 μM (calculated fromRef. 3), the kinetic quality control ensures that onlyfull-length and intact tRNAs are subject to rapidCCA addition at rates of ~30, 40, and 80 s−1 for theaddition of C74, C75, and A76, respectively (e.g.,kapp ¼ k76 � ½Enz�
Kd þ ½Enz�). As such, tRNAs with the CCA se-quence can quickly continue on in the maturationprocess or proceed to reactions for protein synthesis,
which in turn provide protein-binding partners (e.g.,aminoacyl-tRNA synthetases, elongation factors,and the ribosome) that protect the CCA sequence.In contrast, the backbone-nicked tRNAs are rejectedby the CCA enzyme and, without protein protection,are rapidly degraded by the various tRNA surveil-lance machineries that have been discovered in bothE. coli and yeast.29,33,34
The synthesis and maintenance of the CCAsequence confer “eligibility” to a tRNA for partici-pation in protein synthesis, even if the tRNA is stillat an early stage in the maturation process (e.g., inthe nucleus). Protein synthesis is a multistep processthat requires energy consumption for aminoacyla-tion of a matured tRNA, for accommodation of theaminoacylated tRNA at the ribosome, and for trans-location of the tRNA through the ribosome afterpeptide bond formation. Thus, in the overall matu-ration process of a tRNA, CCA addition representsan important checkpoint prior to becoming eligiblefor protein synthesis. The quality control of CCAaddition at the checkpoint can ensure that onlybackbone-intact tRNAs remain in the active proces-sing pathway as substrates for both the repair and denovo biosynthesis functions of the CCA enzyme. Forexample, in E. coli, where the CCA sequence is en-coded in all tRNA genes, EcCCA functions to rege-nerate missing nucleotides from the CCA sequencethat are removed by nucleases. In bacteria that do notencode the CCA sequence in some of their tRNAgenes (e.g., Bacillus subtilis), precursor tRNAsmust beprocessed by endonucleases to remove 3′ trailersequences,35 and the CCA sequence is added de novoas a prerequisite for efficient processing of the 5′end.36,37 A similar situation exists in the eukaryoticnucleus, where the CCA sequence is a determinantfor nuclear tRNA aminoacylation and for export oftRNAwith correctly processed 5′ and 3′ ends.38–40This study demonstrates that although tRNAmini-
helices are competent in steady-state kinetics andappear to embody all of the binding interactions withboth classes of CCA enzymes, they are not sufficientfor fast CCA addition. The minihelix domain isthought to have evolved earlier than the anticodondomain to serve as a tag for the replication of pri-mitive RNA genomes.41,42 Indeed, minihelices thatcarry major determinants for various tRNA functionshave been reported. For example, the minihelix

586 Fast Kinetics of CCA Addition
domain is recognized by RNase P for processing 5′leader sequences,43 and it provides the primary con-tact site for recognition of aminoacyl-tRNA by bac-terial elongation factor Tu.44,45 The minihelix oftRNAAla serves as the aminoacylation domain ofthe bacterial tmRNAmolecule that tags proteins syn-thesized from damaged mRNAs to degradation.46
More important, the minihelix domain of tRNAAla
contains the G3:U70 major determinant for amino-acylation with alanine.47,48 Although the domain byitself retains the specificity of aminoacylation, itscatalytic efficiency (kcat/Km) is reduced from that ofthe full-length tRNA.49 Upon analysis by single-turnover kinetics, the minihelix domain is likely to befurther reduced relative to that of the full-lengthtRNA. The inefficient interaction of the isolated mini-helix domain with EcCCA, and perhaps with otherenzymes as well, may arise from the fact thatalthough the isolated minihelix domain recapitulatesthe sequence of the coaxially stacked acceptor stemand T stem–loop, it lacks support from interactionswith the D loop. This could affect the conformation ofthe T loop and/or reduce the conformational rigidityof the acceptor stem–T stem module. Also, as seen inthe crystal structure of yeast tRNAPhe (Ref. 9), thecoaxial stacking of the acceptor stem and Tstem–loophas a small angle of 14° (Ref. 50), which may not beproperly maintained in the absence of the D and Tloop interactions.Our results here demonstrate that tRNA domains
distal from the minihelix domain (e.g., ASL) are det-erminants for rapid activity of CCA addition. Simi-larly, a recent study demonstrated that although theASL of a tRNAprovides all the information necessaryfor the anticodon–codon base pairing interaction onthe ribosome, the region of the tRNA distal from theASL, which involves the D stem of the tRNA tertiarycore, influences the fidelity of the anticodon–codonbase pairing.51 These studies, together with thosementioned above ofminihelices, emphasize the notionthat individual tRNA domains exhibit their biologi-cally relevant functions only in the context of theentire tRNAmolecule, even though these domains inisolation appear to possess the sequence and struc-tural information required for function. This notion islikely to hold in the interaction of tRNA with otherprotein-binding partners, such as those required fortRNA modification and processing. In the context ofthe tRNAwhole body, we further show that EcCCAeffectively discriminates against backbone nicks ordamage at each step of CCA addition, offering apreviously unrecognized surveillance step beforecommitting tRNA molecules to protein synthesis.
Materials and Methods
Preparation of EcCCA
A previous overexpression clone of wild-type E. coli ccagene was constructed by inserting the gene between theNdeI and BamH1 sites of pET-22b(+) (Novagen) and bychanging the TGA stop codon to the CTA leucine codon.18
Site-directedmutagenesis by QuikChange (Stratagene) wasperformed on this clone to remove most of the C-terminalextension sequence, leaving only a glycine codon betweenthe leucine codon of the gene and the His tag. The modifiedplasmid was transformed into E. coli BL21(DE3), andenzyme expression was induced for 4 h with 0.3 mM IPTGat 37 °C. The collected cells were sonicated and lysed in20 mM Tris–HCl, pH 8.0, 500 mM NaCl, 6 mM MgCl2,5 mM imidazole, 3 mM β-mercaptoethanol, and 5%glycerol. EcCCA was bound to the Co2+-chelated TALONresin (ClonTech) and eluted with 25–50 mM imidazole. Thefractions with the highest contents of EcCCAwere pooled,concentrated, and exchanged into a storage buffer of100 mM glycine, pH 9.0, 50 mM NaCl, 2 mM MgCl2,1 mM β-mercaptoethanol, and 40% glycerol. Enzymeconcentration was determined by Bradford assay.
Substrate preparation
DNA minihelices Val34dCr, Val33dCr, and Val32dAr,containing ribose moieties starting at position 73, andRNA minihelix Val34rCr were chemically synthesized byIDT (Coralville) and used directly. The full-length sub-strates of E. coli tRNAVal, tRNAC75, tRNAC74, and tRNAA73were prepared by run-off transcription by T7 RNApolymerase based on template sequences synthesizedfrom overlapping oligonucleotides. The full-length tran-scripts were separated from their templates and unin-corporated NTPs by 12% denaturing PAGE/7 M urea,localized by UV shadowing, and extracted from gels. TheT-nicked substrate was prepared by annealing the G1–G57fragment (transcribed by T7 RNA polymerase) with theA58–C75 RNA fragment (synthesized by IDT). The ASL-nicked substrate was prepared by annealing the G1–A37fragment (transcribed by T7 RNA polymerase) with theG38–C75 RNA fragment (which contained the A38Gsubstitution and was transcribed by T7 RNA polymerase).Both nicked substrates were formed by heating a mixtureof the 5′ fragment in twofold molar excess of the 3′fragment at 90 °C for 3 min, followed by slow cooling to37 °C for 10 min. The annealed mixtures were separatedby nondenaturing 12% PAGE/90 mM Tris, pH 8.0, 90 mMborate, and 10 mM MgCl2 (TBM) gel, and the annealedhybrids that co-migrated with a full-length tRNA stan-dard were extracted from gels and eluted in 10 mM Tris,pH 8.0. The nicked tRNA substrates were precipitated byethanol and stored in 10 mM Tris–HCl, pH 8.0, and 1 mMMgCl2. Such nicked substrates migrated to the sameelectrophoretic position as the intact full-length tRNA onnative gel analysis, indicating that the fragments in thenicked substrate did not dissociate during gel elution.Concentrations of full-length and nicked substrates weredetermined by UV absorption.Minihelices and the 3′ fragment of the T-nicked subs-
trate were labeled at the 5′ end with [γ-P]ATP (3000 Ci/mmol, Perkin Elmer) by T4 polynucleotide kinase (NEB)for 45 min at 37 °C. After heat inactivation of the kinase,the labeled substrates were separated from free ATP by aCentrispin-20 column (Princeton Separation). The full-length tRNAs and 3′ fragments of the ASL-nicked subs-trates were labeled internally by using [α-32P]ATP (3000Ci/mmol, Perkin Elmer) during the T7 transcription andwere purified by 12% PAGE/7 M urea.
Single-turnover kinetics
Single-turnover assays were performed using an RQF-3(KinTek). The reaction buffer for minihelices contained

587Fast Kinetics of CCA Addition
1 mM DTT, 50 or 100 mM glycine (pH 9.0), 5 or 10 mMMgCl2, 10% glycerol (including glycerol contributed bythe enzyme storage buffer), and the saturating concentra-tion of a specific nucleoside triphosphate (1 mM ATP or5 mM CTP). Increasing concentrations of EcCCA, as anno-tated in the figures, were mixed with 50 nM of a minihelixsubstrate. For the full-length tRNA substrate, both theenzyme and substrate concentrations increased but main-tained at a fixed 10:1 molar ratio. For each reaction, 10 μl ofeach syringe was loaded into the instrument. After diffe-rent reaction times (0.1–300 s for the minihelices and theT-nicked tRNA; 2–70 ms for the tRNAs), the reactionswere quenched with 80 μl of 7 M urea quench solution.The reaction samples were separated on 12% PAGE/7 Murea gel, exposed to a phosphor screen, scanned withStorm 820, and analyzed with ImageQuant 5.2 software(GE Healthcare).The fraction of product formation was calculated based
on the percentage of substrate that was converted toproduct. The fraction of substrate conversion over timewas fit to a single exponential equation: %product=A×(1− e−kapp×t), where A is the amplitude, t is the time, andkapp is the observed rate constant of the A76, C75, or C74incorporation. The data of kapp versus enzyme concentra-tion were fit to a hyperbolic equation: kapp ¼ k76 � ½Enz�
Kd þ ½Enz�, wherek76 is the catalytic rate constant and Kd is the dissocia-tion constant. Alternatively, a linear fit was obtained:kapp=koff+kon×[Enz], where koff is the intercept and kon isthe association rate constant.
Aminoacylation and lead cleavage analysis
Aminoacylation of tRNAVal transcripts (2 μM) was bypurified E. coli ValRS (4 μM) at 37 °C in 50 mM Tris–HCl,pH 7.5, 20 mMKCl, 4.0 mMDTT, 0.2 mg/ml bovine serumalbumin, 10 mM MgCl2, 2.0 mM ATP, and 20 μM [3H]valine (15,600 dpm/pmol). The activity of aminoacylationwas measured by monitoring the enzyme-catalyzedsynthesis of [3H]Val-tRNAVal collected on filter pads in5% trichloroacetic acid. Lead cleavage was performed byincubating lead acetate (1 mM) with intact (1.0 μg) or ASL-nicked (2.5 μg) A38G mutant of E. coli tRNAVal in 25 mMMops, pH 7.0, 1.5 mM spermine, and 15 mM MgCl2 for20 min at 42 °C. The reaction was terminated by thedenaturing loading dye, and the cleavage products wereresolved by 12% denaturing PAGE.
Acknowledgements
We thank Catherine Joyce for advice, Alan Weinerfor encouragement, and Eric Phizicky, MurrayDeutscher, Ken Johnson, and Jon Lorsch for com-ments on the manuscript. We also thank theanonymous reviewers for their very helpful sugges-tions. This work was supported by National Insti-tutes of Health grant GM068561 to Y.M.H.
Supplementary Data
Supplementary data associated with this articlecan be found, in the online version, at doi:10.1016/j.jmb.2008.04.005
References
1. Deutscher, M. P. (1982). tRNA nucleotidyltransferase.Enzymes, 15, 183–215.
2. Aebi, M., Kirchner, G., Chen, J. Y., Vijayraghavan, U.,Jacobson, A., Martin, N. C. & Abelson, J. (1990). Isola-tion of a temperature-sensitive mutant with an alteredtRNA nucleotidyltransferase and cloning of the geneencoding tRNA nucleotidyltransferase in the yeastSaccharomyces cerevisiae. J. Biol. Chem. 265, 16216–16220.
3. Yue, D., Maizels, N. & Weiner, A. M. (1996). CCA-adding enzymes and poly(A) polymerases are allmembers of the same nucleotidyltransferase super-family: characterization of the CCA-adding enzymefrom the archaeal hyperthermophile Sulfolobus shibatae.RNA, 2, 895–908.
4. Yue, D., Weiner, A. M. &Maizels, N. (1998). The CCA-adding enzyme has a single active site. J. Biol. Chem.273, 29693–29700.
5. Li, F., Xiong, Y., Wang, J., Cho, H. D., Tomita, K.,Weiner, A. M. & Steitz, T. A. (2002). Crystal struc-tures of the Bacillus stearothermophilus CCA-addingenzyme and its complexes with ATP or CTP. Cell,111, 815–824.
6. Xiong, Y., Li, F., Wang, J., Weiner, A. M. & Steitz, T. A.(2003). Crystal structures of an archaeal class I CCA-adding enzyme and its nucleotide complexes. Mol.Cell, 12, 1165–1172.
7. Okabe, M., Tomita, K., Ishitani, R., Ishii, R., Takeuchi,N., Arisaka, F. et al. (2003). Divergent evolutions oftrinucleotide polymerization revealed by an archaealCCA-adding enzyme structure. EMBO J. 22, 5918–5927.
8. Augustin, M. A., Reichert, A. S., Betat, H., Huber, R.,Morl, M. & Steegborn, C. (2003). Crystal structure ofthe human CCA-adding enzyme: insights into tem-plate-independent polymerization. J. Mol. Biol. 328,985–994.
9. Xiong, Y. & Steitz, T. A. (2004). Mechanism of transferRNA maturation by CCA-adding enzyme withoutusing an oligonucleotide template.Nature, 430, 640–645.
10. Tomita, K., Fukai, S., Ishitani, R., Ueda, T., Takeuchi,N., Vassylyev, D. G. & Nureki, O. (2004). Structuralbasis for template-independent RNA polymerization.Nature, 430, 700–704.
11. Tomita, K., Ishitani, R., Fukai, S. & Nureki, O. (2006).Complete crystallographic analysis of the dynamics ofCCA sequence addition. Nature, 443, 956–960.
12. Shi, P. Y., Maizels, N. & Weiner, A. M. (1998). CCAaddition by tRNA nucleotidyltransferase: polymer-ization without translocation? EMBO J. 17, 3197–3206.
13. Cho, H. D., Chen, Y., Varani, G. & Weiner, A. M.(2006). A model for C74 addition by CCA-adding en-zymes: C74 addition, like C75 and A76 addition, doesnot involve tRNA translocation. J. Biol. Chem. 281,9801–9811.
14. Cho, H. D., Verlinde, C. L. & Weiner, A. M. (2005).Archaeal CCA-adding enzymes: central role of a highlyconserved beta-turn motif in RNA polymerizationwithout translocation. J. Biol. Chem. 280, 9555–9566.
15. Shi, P. Y., Weiner, A. M. & Maizels, N. (1998). A top-half tDNA minihelix is a good substrate for theeubacterial CCA-adding enzyme. RNA, 4, 276–284.
16. Seth, M., Thurlow, D. L. & Hou, Y. M. (2002). Poly(C)synthesis by class I and class II CCA-adding enzymes.Biochemistry, 41, 4521–4532.
17. Hou, Y. M., Gu, S. Q., Zhou, H. & Ingerman, L. (2005).Metal-ion-dependent catalysis and specificity of CCA-adding enzymes: a comparison of two classes.Biochemistry, 44, 12849–12859.

588 Fast Kinetics of CCA Addition
18. Hou, Y. M. (2000). Unusual synthesis by the Escherichiacoli CCA-adding enzyme. RNA, 6, 1031–1043. [InProcess Citation].
19. Anand, V. S. & Patel, S. S. (2006). Transient statekinetics of transcription elongation by T7 RNApolymerase. J. Biol. Chem. 281, 35677–35685.
20. Yakunin, A. F., Proudfoot, M., Kuznetsova, E.,Savchenko, A., Brown, G., Arrowsmith, C. H. &Edwards, A. M. (2004). The HD domain of theEscherichia coli tRNA nucleotidyltransferase has 2′,3′-cyclic phosphodiesterase, 2′-nucleotidase, and phos-phatase activities. J. Biol. Chem. 279, 36819–36827.
21. Yap, L. P., Stehlin, C. & Musier-Forsyth, K. (1995). Useof semi-synthetic transfer RNAs to probe molecularrecognition by Escherichia coli proline-tRNA synthe-tase. Chem. Biol. 2, 661–666.
22. Liu, H., Peterson, R., Kessler, J. & Musier-Forsyth, K.(1995). Molecular recognition of tRNA(Pro) by Escher-ichia coli proline tRNA synthetase in vitro. NucleicAcids Res. 23, 165–169.
23. Sherlin, L. D., Bullock, T. L., Nissan, T. A., Perona, J. J.,Lariviere, F. J., Uhlenbeck,O.C.&Scaringe, S. A. (2001).Chemical and enzymatic synthesis of tRNAs for high-throughput crystallization. RNA, 7, 1671–1678.
24. Nandakumar, J. & Shuman, S. (2005). Dual mechan-isms whereby a broken RNA end assists the catalysisof its repair by T4 RNA ligase 2. J. Biol. Chem. 280,23484–23489.
25. Fukai, S., Nureki, O., Sekine, S., Shimada, A., Tao, J.,Vassylyev, D. G. & Yokoyama, S. (2000). Structuralbasis for double-sieve discrimination of L-valine fromL-isoleucine and L-threonine by the complex of tRNA(Val) and valyl-tRNA synthetase. Cell, 103, 793–803.
26. Krzyzosiak, W. J., Marciniec, T., Wiewiorowski, M.,Romby, P., Ebel, J. P. & Giege, R. (1988). Characteriza-tion of the lead(II)-induced cleavages in tRNAs insolution and effect of the Y-base removal in yeasttRNAPhe. Biochemistry, 27, 5771–5777.
27. Behlen, L. S., Sampson, J. R., DiRenzo, A. B. &Uhlenbeck, O. C. (1990). Lead-catalyzed cleavage ofyeast tRNAPhe mutants. Biochemistry, 29, 2515–2523.
28. Cho,H.D.,Oyelere,A.K., Strobel, S.A.&Weiner, A.M.(2003). Use of nucleotide analogs by class I and class IICCA-adding enzymes (tRNA nucleotidyltransferase):deciphering the basis for nucleotide selection. RNA, 9,970–981.
29. Alexandrov, A., Chernyakov, I., Gu, W., Hiley, S. L.,Hughes, T. R., Grayhack, E. J. & Phizicky, E. M. (2006).Rapid tRNA decay can result from lack of nonessen-tial modifications. Mol. Cell, 21, 87–96.
30. Ogawa, T., Tomita, K., Ueda, T., Watanabe, K.,Uozumi, T. & Masaki, H. (1999). A cytotoxic ribonu-clease targeting specific transfer RNA anticodons.Science, 283, 2097–2100.
31. Lu, J., Huang, B., Esberg, A., Johansson, M. J. &Bystrom, A. S. (2005). The Kluyveromyces lactis gamma-toxin targets tRNA anticodons. RNA, 11, 1648–1654.
32. Ho, C. K., Rauhut, R., Vijayraghavan, U. & Abelson, J.(1990). Accumulation of pre-tRNA splicing ‘2/3’intermediates in a Saccharomyces cerevisiae mutant.EMBO J. 9, 1245–1252.
33. Li, Z., Reimers, S., Pandit, S. & Deutscher, M. P. (2002).RNA quality control: degradation of defective transferRNA. EMBO J. 21, 1132–1138.
34. Kadaba, S., Krueger, A., Trice, T., Krecic, A. M.,Hinnebusch, A. G. & Anderson, J. (2004). Nuclearsurveillance and degradation of hypomodified initia-tor tRNAMet in S. cerevisiae. Genes Dev. 18, 1227–1240.
35. Deutscher, M. P. (2006). Degradation of RNA inbacteria: comparison of mRNA and stable RNA.Nucleic Acids Res. 34, 659–666.
36. Wegscheid, B. & Hartmann, R. K. (2006). Theprecursor tRNA 3′-CCA interaction with Escherichiacoli RNase P RNA is essential for catalysis by RNase Pin vivo. RNA, 12, 2135–2148.
37. Wegscheid, B. & Hartmann, R. K. (2007). In vivo and invitro investigation of bacterial type B RNase Pinteraction with tRNA 3′-CCA. Nucleic Acids Res. 35,2060–2073.
38. Lund, E. & Dahlberg, J. E. (1998). Proofreading andaminoacylation of tRNAs before export from thenucleus [see comments]. Science, 282, 2082–2085.
39. Sarkar, S., Azad, A. K. &Hopper, A. K. (1999). NucleartRNA aminoacylation and its role in nuclear export ofendogenous tRNAs in Saccharomyces cerevisiae. Proc.Natl Acad. Sci. USA, 96, 14366–14371.
40. Grosshans, H., Hurt, E. & Simos, G. (2000). Anaminoacylation-dependent nuclear tRNA exportpathway in yeast. Genes Dev. 14, 830–840.
41. de Duve, C. (1988). Transfer RNAs: the second geneticcode. Nature, 333, 117–118.
42. Weiner, A. M. & Maizels, N. (1987). tRNA-likestructures tag the 3′ ends of genomic RNA moleculesfor replication: implications for the origin of proteinsynthesis. Proc. Natl Acad. Sci. USA, 84, 7383–7387.
43. McClain, W. H., Guerrier-Takada, C. & Altman, S.(1987). Model substrates for an RNA enzyme. Science,238, 527–530.
44. Nissen, P., Kjeldgaard, M., Thirup, S., Polekhina, G.,Reshetnikova, L., Clark, B. F. & Nyborg, J. (1995).Crystal structure of the ternary complex of Phe-tRNAPhe, EF-Tu, and a GTP analog [see comments].Science, 270, 1464–1472.
45. Nissen, P., Thirup, S., Kjeldgaard, M. & Nyborg, J.(1999). The crystal structure of Cys-tRNACys–EF-Tu–GDPNP reveals general and specific featuresin the ternary complex and in tRNA. Structure, 7,143–156.
46. Keiler, K. C., Waller, P. R. & Sauer, R. T. (1996). Role ofa peptide tagging system in degradation of proteinssynthesized from damaged messenger RNA [seecomments]. Science, 271, 990–993.
47. Hou, Y. M. & Schimmel, P. (1988). A simple structuralfeature is a major determinant of the identity of atransfer RNA. Nature, 333, 140–145.
48. McClain, W. H. & Foss, K. (1988). Changing theidentity of a tRNA by introducing a G–U wobble pairnear the 3′ acceptor end. Science, 240, 793–796.
49. Francklyn, C. & Schimmel, P. (1989). Aminoacy-lation of RNA minihelices with alanine. Nature, 337,478–481.
50. Holbrook, S. R., Sussman, J. L., Warrant, R. W. & Kim,S. H. (1978). Crystal structure of yeast phenylalaninetransfer RNA: II. Structural features and functionalimplications. J. Mol. Biol. 123, 631–660.
51. Cochella, L. & Green, R. (2005). An active role fortRNA in decoding beyond codon:anticodon pairing.Science, 308, 1178–1180.
Copyright © 2022 FDOKUMEN




















