
Towards a consistent mechanism of emulsion polymerization-new experimental details
17
ORIGINAL CONTRIBUTION Towards a consistent mechanism of emulsion polymerization—new experimental details Klaus Tauer & Hugo Hernandez & Steffen Kozempel & Olga Lazareva & Pantea Nazaran Received: 11 September 2007 / Revised: 25 October 2007 / Accepted: 26 October 2007 / Published online: 3 December 2007 # Springer-Verlag 2007 Abstract The application of atypical experimental methods such as conductivity measurements, optical microscopy, and nonstirred polymerizations to investigations of the ‘classical’ batch ab initio emulsion polymerization of styrene revealed astonishing facts. The most important result is the discovery of spontaneous emulsification leading to monomer droplets even in the quiescent styrene in water system. These monomer droplets with a size between a few and some hundreds of nanometers, which are formed by spontaneous emulsification as soon as styrene and water are brought into contact, have a strong influence on the particle nucleation, the particle morphology, and the swelling of the particles. Experimental results confirm that micelles of low-molecular-weight surfactants are not a major locus of particle nucleation. Brownian dynamics simulations show that the capture of matter by the particles strongly depends on the polymer volume fraction and the size of the captured species (primary free radicals, oligomers, single monomer molecules, or clusters). Keywords Mechanism of emulsion polymerization . Particle nucleation . Radical capture . Swelling Introduction Aqueous radical emulsion polymerization is the technically most important synthetic route to polymer dispersions and is used to produce worldwide about 7.5 million metric tons of dry polymer [1]. From a more scientific point of classification, emulsion polymerization belongs to the class of heterophase polymerizations, which comprises further- more suspension, microsuspension, miniemulsion, micro- emulsion, dispersion, and precipitation polymerizations. All these techniques, which are leading to various kinds of polymer dispersions, are characterized by their heteroge- neous nature. Polymer dispersions are defined as colloidal systems where the polymer is finely distributed in a liquid- dispersion medium in the form of stable individual particles. It might be useful to define emulsion polymerization in a general way as polymerization or copolymerization in aqueous systems of any combinations of monomers, which lead to water-insoluble polymers or copolymers in the form of individual polymer particles with a size distribution of diameters in a range typically lower than 1 μm. The polymer or copolymer particles swell after nucleation with the monomers. These swollen particles represent the reaction loci where most of the monomer is polymerized. In many cases (especially in industrial systems), emulsifiers are present during the polymerization to stabilize the large interfacial area. Despite this characterization is quite rough, it reveals the two most important kinetic features of emulsion polymerization: particle nucleation and particle growth. The first can be considered as phase transition and the latter as sorption process, as matter — at least monomer and radicals — is captured by the particles. The mechanism of emulsion polymerization has been reviewed several times during the last decade [2–5]. In general, particle nucleation is considered to take place Colloid Polym Sci (2008) 286:499–515 DOI 10.1007/s00396-007-1797-3 K. Tauer (*) : H. Hernandez : S. Kozempel : O. Lazareva : P. Nazaran Max Planck Institute of Colloids and Interfaces, 14476 Golm, Germany e-mail: [email protected] O. Lazareva Department of Chemistry and Technology of High Molecular Weight Compounds, Lomonosov Moscow State Academy of Fine Chemical Technology, Prospect Vernadskogo 86, 119571 Moscow, Russia
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Towards a consistent mechanism of emulsion polymerization-new experimental details

ORIGINAL CONTRIBUTION
Towards a consistent mechanism of emulsionpolymerization—new experimental details
Klaus Tauer & Hugo Hernandez & Steffen Kozempel &Olga Lazareva & Pantea Nazaran
Received: 11 September 2007 /Revised: 25 October 2007 /Accepted: 26 October 2007 /Published online: 3 December 2007# Springer-Verlag 2007
Abstract The application of atypical experimental methodssuch as conductivity measurements, optical microscopy,and nonstirred polymerizations to investigations of the‘classical’ batch ab initio emulsion polymerization ofstyrene revealed astonishing facts. The most importantresult is the discovery of spontaneous emulsificationleading to monomer droplets even in the quiescent styrenein water system. These monomer droplets with a sizebetween a few and some hundreds of nanometers, whichare formed by spontaneous emulsification as soon asstyrene and water are brought into contact, have a stronginfluence on the particle nucleation, the particle morphology,and the swelling of the particles. Experimental resultsconfirm that micelles of low-molecular-weight surfactantsare not a major locus of particle nucleation. Browniandynamics simulations show that the capture of matter by theparticles strongly depends on the polymer volume fractionand the size of the captured species (primary free radicals,oligomers, single monomer molecules, or clusters).
Keywords Mechanism of emulsion polymerization .
Particle nucleation . Radical capture . Swelling
Introduction
Aqueous radical emulsion polymerization is the technicallymost important synthetic route to polymer dispersions andis used to produce worldwide about 7.5 million metric tonsof dry polymer [1]. From a more scientific point ofclassification, emulsion polymerization belongs to the classof heterophase polymerizations, which comprises further-more suspension, microsuspension, miniemulsion, micro-emulsion, dispersion, and precipitation polymerizations. Allthese techniques, which are leading to various kinds ofpolymer dispersions, are characterized by their heteroge-neous nature. Polymer dispersions are defined as colloidalsystems where the polymer is finely distributed in a liquid-dispersion medium in the form of stable individual particles.It might be useful to define emulsion polymerization in ageneral way as polymerization or copolymerization inaqueous systems of any combinations of monomers, whichlead to water-insoluble polymers or copolymers in the formof individual polymer particles with a size distribution ofdiameters in a range typically lower than 1 μm. Thepolymer or copolymer particles swell after nucleation withthe monomers. These swollen particles represent thereaction loci where most of the monomer is polymerized.In many cases (especially in industrial systems), emulsifiersare present during the polymerization to stabilize the largeinterfacial area. Despite this characterization is quite rough,it reveals the two most important kinetic features ofemulsion polymerization: particle nucleation and particlegrowth. The first can be considered as phase transition andthe latter as sorption process, as matter — at least monomerand radicals — is captured by the particles.
The mechanism of emulsion polymerization has beenreviewed several times during the last decade [2–5]. Ingeneral, particle nucleation is considered to take place
Colloid Polym Sci (2008) 286:499–515DOI 10.1007/s00396-007-1797-3
K. Tauer (*) :H. Hernandez : S. Kozempel :O. Lazareva :P. NazaranMax Planck Institute of Colloids and Interfaces,14476 Golm, Germanye-mail: [email protected]
O. LazarevaDepartment of Chemistry and Technology of High MolecularWeight Compounds, Lomonosov Moscow State Academy of FineChemical Technology,Prospect Vernadskogo 86,119571 Moscow, Russia

either via micellar or homogeneous nucleation in depen-dence on the hydrophilicity of the monomer and thesurfactant concentration. The micellar mechanism considersa smooth transition from a monomer swollen micelle to apolymer particle after entry of a free radical and is appliedfor hydrophobic monomers such as styrene. For morehydrophilic monomers such as methyl methacrylate, thehomogeneous nucleation mechanism assumes that a singlegrowing water-born oligomer radical precipitates when itbecomes insoluble in the aqueous phase. This single-chainprecipitation is rather unlikely for thermodynamic reasons[6, 7] and experimentally not proven. Growth of theparticles is mainly determined by both the monomer andthe radical concentration per particle. The thermodynamicsof swelling of the latex particles with monomer is mainlyhandled with the Morton–Kaizerman–Altier (MKA) equa-tion [8] despite the fact that it is not absolute and failedquantitatively to predict the monomer concentration perparticle [9, 10]. Since the early 1990s, radical entry ismodeled, assuming that only radicals with a certain criticalchain length are able to enter the latex particles from theaqueous phase [4, 11]. However, recently this quiterestrictive assumption was disproved experimentally [12].
This contribution describes new experimental resultsaiming to provide a better experimental base for theunderstanding of emulsion polymerization. Particularly,results will be presented regarding (1) particle nucleationin the absence and presence of both emulsifiers and seedparticles, (2) the state of the monomer in water, and (3) thesorption of matter by latex particles.
Experimental information
Materials and methods The water was taken from a Seralpurification system (PURELAB Plus) with a conductivity of0.06 μS cm−1 and degassed prior to use for the polymeri-zation. Styrene (Aldrich) was distilled under reducedpressure to remove inhibitors. Potassium peroxodisulfate(KPS from Sigma Aldrich), sodium dodecyl sulfate (SDS fromRoth), cetyl trimethyl ammonium bromide (CTAB fromFerak), and sodium perfluorooctanoate (SPFO from Lancaster)were used as received. Azobisisobutyronitrile (AIBN, Fluka)was recrystallized from methanol before use. Poly(ethyleneglycol)-azo-initiator (PEGA200) with an average molecularweight of 568 g/mol was synthesized as described elsewhere[13, 14]. 2,2′-Azobis(2-methyl-butyronitrile) (V-59) and 2,2′-azobis(2-methyl-N-(2-hydroxyethyl)propionamide) (VA-086)were purchased from Wako Pure Chemical Industries andused as received.
The latexes were characterized regarding solids contentwith a HR73 Halogen Moisture Analyzer (Mettler Toledo)
and average particle size (intensity-weighted diameter) witha Nicomp particle sizer (PSS Santa Barbara, USA) at afixed scattering angle of 90°. Two models were employeddiffering in the laser power: model 370 with a laser powerof 7 mW and model 380 with a laser power of 35 mW.
The zeta potential (ξ) and the z average particle size (DZ)were determined according to standard procedures with theZetasizer 4 (Malvern).
Analytical ultracentrifugation (AUC) was used to deter-mine the particle-size distribution of selected latex samples.The samples were measured with interference optics withwater as reference solvent in a Beckman Coulter OptimaXLI Analytical Ultracentrifuge (Palo Alto, CA, USA) at20,000 and 60,000 rpm and 25 °C. The sedimentationcoefficient distributions were evaluated by Sedfit 8.9 andthen transferred to the algorithm for the determination ofparticle size [4].
Molecular-weight distributions were determined by gelpermeation chromatography (GPC) and used to calculateweight and number average molecular weights (Mw, Mn).GPC was carried out by injecting 100 μl polymer solutions(solvent tetrahydrofuran) through a Teflon filter with amesh size of 450 nm into a thermo separation productssetup being equipped with ultra violet (UV; TSP UV1000)and refractive index (RI; Shodex RI-71) detectors intetrahydrofuran at 30 °C with a flow rate of 1 ml/min. Acolumn set was employed consisting of three 300×8 mmcolumns filled with a MZ-SDplus spherical polystyrene gel(average particle size 5 μm) having a pore size of 103, 105,and 106 Å, respectively. This column set allows a resolutiondown to molecular weights less than 500 g mol−1. Numberand weight average molecular weights (Mn, Mw) werecalculated based on polystyrene standards (between 500and 2×106 g mol−1 from PSS, Mainz, Germany).
Transmission electron microscopy (TEM) was per-formed with a Zeiss EM 912 Omega microscope operatingat 100 kV and the samples were placed on the grids viasuspension preparation.
Optical light microscopy was carried out with a KeyenceVH-X digital microscope (Keyence, Osaka, Japan) eitherwith an objective VH-Z100 or VH-Z500 allowing magni-fications up to 1,000 and 5,000-fold, respectively.
UV-fluorescence microscopy has been carried out withan Olympus BX51 (Hamburg, Germany) with an excitationwavelength of 330–385 nm. Emission was detected in thewavelength range between 400 and 420 nm.
Polymerizations The kinetic investigations on particlenucleation were carried out in an all-Teflon reactor with avolume of about 500 ml connected to a UV spectrometer(Spekol 11, Carl Zeiss Jena) and equipped with a variety ofprobes to characterize the changes taking place in theaqueous phase as described in detail elsewhere [5]. During
500 Colloid Polym Sci (2008) 286:499–515

these experiments, the stirring was adjusted so slowly thatonly the aqueous phase was homogenized without dispersingthe monomer which is confined on top of the water phase ina glass funnel. A fiber-optical quasi-elastic light-scatteringprobe (FOQELS) from Brookhaven Instruments, USA,wavelength 785 nm and scattering angle 135.93° was usedin some experiments as described in [6]. These nucleationexperiments were carried out at 70 °C according to thefollowing procedure. To avoid bubble nucleation during theinvestigations, the water was carefully degassed by vacuumtreatment and boiled. It was filled in the reactor at atemperature above 70 °C. In any case, the temperature, theoptical transmission (optical path length 9.8 cm), and theconductivity were recorded.
The polymer from the polymerizations in the presence ofsurfactant was isolated after freeze-drying the latex andwashing the solid repeatedly with water.
Surfactant-free emulsion polymerizations were carriedout in an all-glass 500 ml reactor at 70 °C. The experimentshave been carried out either under quiescent conditions orwith a stirrer speed of 300 rpm.
Results and discussions
Detection of particle nucleation
Particle nucleation in ab initio batch emulsion polymer-izations starts at extremely low solid contents typicallymuch below 1%. Thus, the experimental challenge to detectthe onset and follow particle nucleation is quite significant.It turned out that conductivity is an extremely good tool todetect the onset of nucleation in surfactant-free polymer-izations [7, 15–18]. This established experimental base isillustrated by the data summarized in Fig. 1. It is necessaryto point out that with all probes (conductivity, turbidity,dynamic light scattering, etc.), only properties of theaqueous phase are investigated. The monomer, resting ontop of the aqueous phase, is not dispersed by the low stirrerspeed that is just enough to homogenize the continuouswater phase.
After a certain time, allowing to equilibrate the monomerin water, polymerization was started by injecting theinitiator solution (here KPS dissolved in a few millilitersof water) at point 1. The conductivity rises sharply, andafterwards, it increases due to the decomposition of theperoxodisulfate linearly with time during a definite periodof time (tN). This behavior can be modeled nicely withperoxodisulfate decomposition kinetics [17]. Suddenly atpoint 2, within 1 s to the other, the slope of theconductivity–time curve changes to a smaller value. Thisbend is the point where massive particle nucleation takes
place, and tN denotes the nucleation time or the duration ofthe prenucleation period. The change of the slope takesplace as conducting species lose mobility due to incorpo-ration in the electrical double layer of the particles (mainlyprotons as outlined in [17]). Independent proof that particlenucleation takes place at the bend of the conductivity curvecomes from the fact that particles have only been detectedwith the FOQELS a few minutes after the bend at almostthe same time when transmission starts to decrease.
The smallest particles detected with the FOQELS have adiameter below 5 nm (between 2 and 3 nm) and arecomposed of oligomers with an average molecular weightof about 600 g/mol. These particles contain between 4 and15 oligomers, and hence, they prove the aggregativenucleation model which was derived based on the classicalnucleation theory [6, 7].
In accordance with the aggregative nucleation model, itis experimentally observed that the duration of theprenucleation period decreases with increasing initiatorconcentration or larger radical flux (cf. Fig. 2a). Theexperimental tN data converge to a value of about 6 minfor KPS concentrations greater than 1 mM. Higher initiatorconcentrations (KPS) do not shorten the nucleation periodthough the overall rate of polymerization after the particlenucleation period can be increased. This behavior duringthe prenucleation period illustrates the interaction of twoeffects: (1) the generation of nucleating oligomers (that arenucleable species) by reaction of the free primary radicals,stemming from the water soluble initiator, with themonomer molecules in water and (2) the nucleation stepafter a critical supersaturation of these oligomers has beenreached. The supersaturation — the ratio between concen-tration and solubility — is influenced by the initiatorconcentration on two counteracting ways within the frame
Fig. 1 Typical record of the initial period of surfactant-free emulsionpolymerization with on-line determination of transmission (T),conductivity (κ), and average particles size (D, FOQELS); experi-mental conditions: 70 °C, 400 g of water, 3.3 g of styrene, 0.067575 gof KPS (0.625 mM KPS in the reactor); monomer equilibration time,120 min; stirring, 70 rpm
Colloid Polym Sci (2008) 286:499–515 501

of radical polymerization kinetics. On the one hand, ahigher initiator concentration leads to higher supersatura-tion as the concentration of the nucleating species increases.On the other hand, a higher initiator concentration leads tolower supersaturation as the solubility of the oligomersincreases due to their lower molecular weight. At KPSconcentrations of 0.1 and 0.05 mM, no signs of particlenucleation were observed; that is, neither a bend in theconductivity curve has been observed nor particles havebeen detected by dynamic light scattering. Obviously, acritical KPS concentration exists, below which the concen-tration of oligomers is not high enough to reach a criticalsupersaturation within up to 3 h after KPS injection.
The lower the KPS concentration, the more scatter thedata of the nucleation time depicted in Fig. 2a. This result isalso in accordance with the classical nucleation theory as itpredicts a strong scatter on even small variation of theexperimental conditions due to the exponential dependenceof the rate of nucleation on the surface tension, thesupersaturation, and the temperature [19]. For high initiatorconcentrations, the high concentration of nucleable speciescauses a high supersaturation and reduces the influence ofthe always-present environmental scatter due to fluctuations.
In contrast to tN, the dependence of the average particlesize of the final latexes on the KPS concentration does notdisplay a simple dependence. The standard deviations ofthe experimental data points suggest that both the maxi-mum and the minimum are real. A quantitative interpreta-tion of this dependence is not possible, but qualitatively itcan be considered as the result of two effects running in theopposite direction with increasing amount of KPS. Theseeffects are, on the one hand, increasing stabilization due toa higher concentration of stabilizing end groups and, on theother hand, increasing destabilization due to the increasedionic strength in the continuous phase. Consequently, adependence with minima and maxima as depicted inFig. 2b seems reasonable. It is necessary to remark thatcomparing these data is somehow difficult as both themonomer conversion and the solids content are different.Nevertheless, the highest particle size at the lowest KPS
concentration is not to be explained at the moment. The muchhigher standard deviation might be understandable with theinherent scatter of the nucleation process as discussed alreadyfor the duration of the prenucleation period.
Surprisingly, if peroxodisulfate is substituted by nonionicazo-initiators such as AIBN, V-59, VA-086, or PEGA200, avery similar initial behavior of the conductivity is observedas illustrated by the data put together in Fig. 3. These azo-initiators possess a quite different solubility in water, but forall of them, a bend in the conductivity–time curve duringsurfactant-free emulsion polymerizations of styrene has beenobserved. Obviously, carbon radicals undergo in water sideoxidation reactions leading to ionic species that increase theconductivity in the aqueous phase and stabilize the particleseven in the absence of surfactants [20]. This is in accordancewith former results where, by means of nuclear magneticresonance spectroscopy, side oxidation reactions have beendetected during the decomposition of azo-initiators in water–benzene mixtures [21]. Note that V-59 has the lowestwater-solubility, and correspondingly, the change in theconductivity takes much longer (hours instead of minutes),but the extent of polymerization inside the monomer layer isextremely high.
In this context, it seems useful to remember the influenceof the hydrophobicity of the initiator during the microwave-induced miniemulsion polymerization of styrene [22]. Inthis study, it was found that under thermally pulsedconditions, an optimal hydrophobicity of the initiator isrequired to obtain high conversion and high molecularweight. KPS and V-59 were too hydrophilic and toohydrophobic, respectively, whereas AIBN and PEGA200turned out to be optimum for achieving high conversionand ultra-high molecular weights simultaneously.
All these results underline the important role of thereactions taking place in the continuous phase during theproduction of polymer dispersions via heterophase poly-merizations. The example of the styrene miniemulsionpolymerization shows that a restriction of the reaction to theoil phase by the application of extremely hydrophobicinitiators must not be an advantage per se.
Fig. 2 Dependence of thenucleation time (tN; a) and ofthe final average diameter(D from dynamic light scattering;b) on the potassium peroxodi-sulfate concentration (CKPS) forsurfactant-free emulsion poly-merization of styrene; otherexperimental conditions as men-tioned in the caption of Fig. 1;data points are average values offour repeats
502 Colloid Polym Sci (2008) 286:499–515

Although both the sharpness and the magnitude of thechange in the conductivity–time curve are, for PEGA200and V-59, not so large as for KPS or AIBN, it is observed inany case. Hence, the data of Fig. 3 prove that also fornonionic initiators, conductivity measurement of the aqueousphase is a valuable tool to detect the onset of particlenucleation in styrene emulsion polymerization. The TEMimages of Fig. 4 demonstrate that with AIBN, quite nicemonodisperse latexes can be obtained, however, at quite lowsolids content.
These data directly allow the conclusion that alsocarbon-centered radicals undergo some side reactions in
the continuous phase during aqueous emulsion polymeri-zation. Moreover, the side reactions must lead to hydro-philic groups covalently attached to the polymers and thusstabilizing the latex particles. The particles obtained bysurfactant-free emulsion polymerization of styrene initiatedwith AIBN are electrostatically stabilized and possess apH-dependent zeta potential [20]. Based on these results,one might also expect different decomposition mechanismof hydrophobic initiators inside the particles and in theaqueous phase. Indeed, this was experimentally provenwith a 13C-NMR study as described in [21].
Micellar nucleation — fact or fancy
Harkins considered in 1947 three loci where the polymer-ization of the monomer can initiate polymer particles: thesoap micelles, the aqueous phase, and the monomerdroplets [23]. The latter is only a very minor locus ofpolymerization but serves as storehouse from whichmonomer diffuses via the aqueous phase to the micellesor particles where it is polymerized. In dependence on thesoap concentration, the micelles or the aqueous phase aredominant during the initial reaction. Furthermore, heargued, based on X-ray scattering data [23–26], that themolecules grow from the soap micelle into the aqueousphase where they continue to propagate and eventuallyform polymer particles. At this point, micellar soapbecomes adsorbed soap and stabilizes the particles.
While referring to Harkins’ papers, Smith and Ewart donot consider that radicals can grow out of micelles. Theyassume that once a radical enters a micelle, it stays insideand polymerizes further, while more monomer diffuses intothis polymerizing region. This smooth transition from a
Fig. 4 TEM image of polystyrene latex particles obtained duringsurfactant-free emulsion polymerization initiated with AIBN after90 min; solids content of the latex phase below 1%; average particlesize 173 nm; polymerization conditions: all-glass reactor, 70 °C, 400 gwater, 20 g styrene, 0.08 g AIBN, stirrer speed 50 rpm
Fig. 3 Conductivity–time curves measured during the initial period ofsurfactant-free emulsion polymerization of styrene with different azo-initiators; the arrows indicate the bend in the curves; experimentalconditions: 70 °C, 400 g of water (for AIBN and V-59) or 410 g ofwater (for PEGA200 and VA-086), 3.75 g of styrene (for AIBN andV-59) or 3.3 g of styrene (for PEGA200 and VA-086), and thefollowing amounts of initiator added 120 min after placing the
monomer on top of the water phase: 0.072 g of VA-086, 0.112 g ofPEGA200, 0.075 g AIBN, and 0.075 g V-59 (VA-086 and PEGA200were added as aqueous solution to water, AIBN and V-59 were addedas solution in styrene to the monomer phase); stirring, 70 rpm; thedotted lines are just an extension of the initial slope for bettervisualization
Colloid Polym Sci (2008) 286:499–515 503
monomer-swollen micelle to a polymerizing monomer–polymer particle is the core of the micellar particle-formation mechanism [27]. With ongoing polymerization,more and more soap adsorbs to the particles until no soapremains in the micellar form, and the soap is no longereffective in particle formation. Smith and Ewart derived thefamous relation where the final number of particles (N)scales with the micellar soap (Smic) and the initiatorconcentration (I) as N / S3=5mic � I2=5. However, Roe [28]pointed out that this relation has only little to do with themechanism of particle formation but rather with thequestion when does particle formation cease. This relationis not helpful to learn about particle formation as it is in factindependent of the particle nucleation mechanism! More-over, there are all kinds of data published showing variousexponents for experimentally found N � Sα relations.Exemplarily, Bartholomé et al. [29] determined α=0.615for emulsion polymerizations of styrene with AmphoseifeC18 as emulsifier and 0.361 g/l KPS at 45 °C. Contrarily,Hansen and Ugelstad [30] obtained α=2.67 also for styreneemulsion polymerization but with SDS as emulsifier and0.6 g/l KPS at 60 °C.
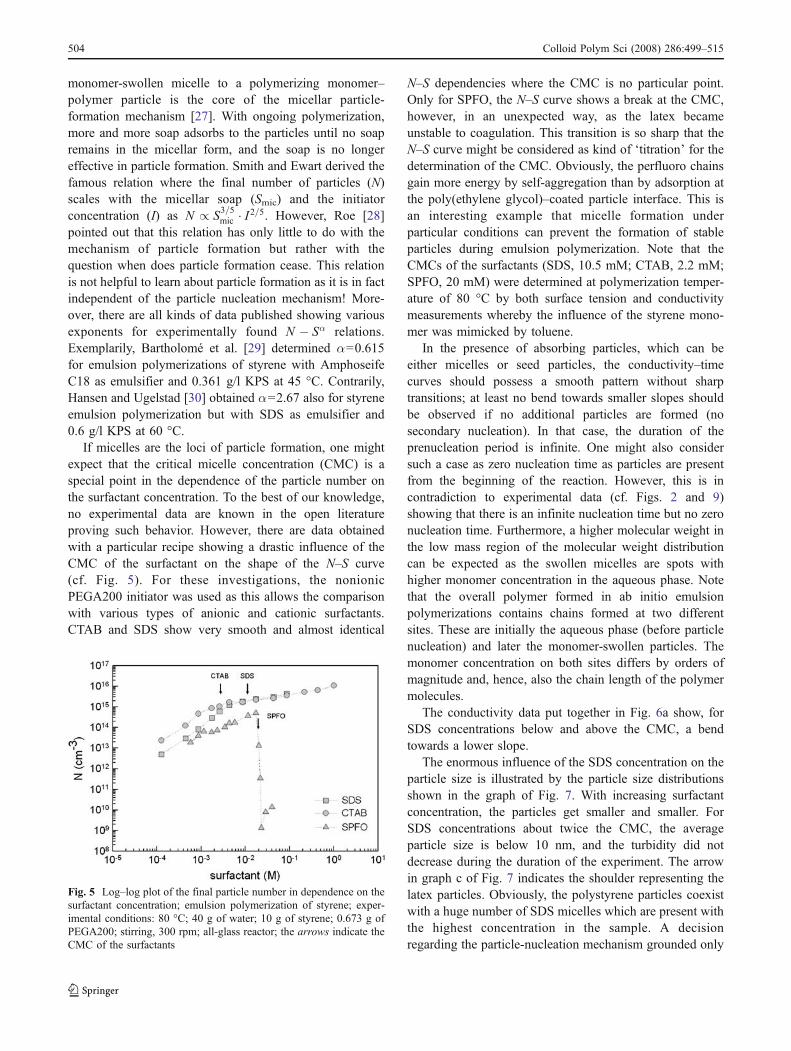
If micelles are the loci of particle formation, one mightexpect that the critical micelle concentration (CMC) is aspecial point in the dependence of the particle number onthe surfactant concentration. To the best of our knowledge,no experimental data are known in the open literatureproving such behavior. However, there are data obtainedwith a particular recipe showing a drastic influence of theCMC of the surfactant on the shape of the N–S curve(cf. Fig. 5). For these investigations, the nonionicPEGA200 initiator was used as this allows the comparisonwith various types of anionic and cationic surfactants.CTAB and SDS show very smooth and almost identical
N–S dependencies where the CMC is no particular point.Only for SPFO, the N–S curve shows a break at the CMC,however, in an unexpected way, as the latex becameunstable to coagulation. This transition is so sharp that theN–S curve might be considered as kind of ‘titration’ for thedetermination of the CMC. Obviously, the perfluoro chainsgain more energy by self-aggregation than by adsorption atthe poly(ethylene glycol)–coated particle interface. This isan interesting example that micelle formation underparticular conditions can prevent the formation of stableparticles during emulsion polymerization. Note that theCMCs of the surfactants (SDS, 10.5 mM; CTAB, 2.2 mM;SPFO, 20 mM) were determined at polymerization temper-ature of 80 °C by both surface tension and conductivitymeasurements whereby the influence of the styrene mono-mer was mimicked by toluene.
In the presence of absorbing particles, which can beeither micelles or seed particles, the conductivity–timecurves should possess a smooth pattern without sharptransitions; at least no bend towards smaller slopes shouldbe observed if no additional particles are formed (nosecondary nucleation). In that case, the duration of theprenucleation period is infinite. One might also considersuch a case as zero nucleation time as particles are presentfrom the beginning of the reaction. However, this is incontradiction to experimental data (cf. Figs. 2 and 9)showing that there is an infinite nucleation time but no zeronucleation time. Furthermore, a higher molecular weight inthe low mass region of the molecular weight distributioncan be expected as the swollen micelles are spots withhigher monomer concentration in the aqueous phase. Notethat the overall polymer formed in ab initio emulsionpolymerizations contains chains formed at two differentsites. These are initially the aqueous phase (before particlenucleation) and later the monomer-swollen particles. Themonomer concentration on both sites differs by orders ofmagnitude and, hence, also the chain length of the polymermolecules.
The conductivity data put together in Fig. 6a show, forSDS concentrations below and above the CMC, a bendtowards a lower slope.
The enormous influence of the SDS concentration on theparticle size is illustrated by the particle size distributionsshown in the graph of Fig. 7. With increasing surfactantconcentration, the particles get smaller and smaller. ForSDS concentrations about twice the CMC, the averageparticle size is below 10 nm, and the turbidity did notdecrease during the duration of the experiment. The arrowin graph c of Fig. 7 indicates the shoulder representing thelatex particles. Obviously, the polystyrene particles coexistwith a huge number of SDS micelles which are present withthe highest concentration in the sample. A decisionregarding the particle-nucleation mechanism grounded only
Fig. 5 Log–log plot of the final particle number in dependence on thesurfactant concentration; emulsion polymerization of styrene; exper-imental conditions: 80 °C; 40 g of water; 10 g of styrene; 0.673 g ofPEGA200; stirring, 300 rpm; all-glass reactor; the arrows indicate theCMC of the surfactants
504 Colloid Polym Sci (2008) 286:499–515

on the average particle size of the final latexes is notpossible. However, the conductivity data prove that SDSmicelles for styrene as monomer do not alter the nucleationbehavior. Additionally, the molecular weight data (cf. Table 1)give no hint that micelles are the loci where the initialpolymerization reaction takes place. The molecular weightdata in the low-molecular-weight region are independent ofthe SDS concentration. However, the average molecularweights in the high-mass portion of the molecular weightdistribution strongly depend on the surfactant concentration.This is an expected behavior, as the particles are smaller thehigher the SDS concentration, and smaller particles imbibeless monomer than larger particles [9, 10].
If instead of micelles seed particles are used as absorbingspecies, it is possible to find a critical volume fraction ofseed particles where the conductivity curves do not show
secondary nucleation (cf. Fig. 8b). For these particular seedparticles with an average size of 30 nm, the critical volumefraction is between 0.2 and 2%. At the lower seed-volumefraction, the bend in the conductivity curve clearly provessecondary nucleation (cf. Fig. 8a). However, for a seed-volume fraction of 2%, the conductivity curve is a straightline without any sign of bending towards lower slope. Thisbehavior was observed for both possibilities to start thepolymerization, that is, either by the addition of styrenemonomer or KPS. In the former case, the monomer wasadded 30 min after KPS, and in the latter case, the systemwas equilibrated with monomer for 2 h before starting thepolymerization. If the polymerization is started at low seed-volume fraction with the addition of monomer, theconductivity–time curve exhibits expectedly two bends(Fig. 8a). The first bend occurs immediately after monomeraddition and is due to the formation of less mobile speciesas free radicals react now with styrene molecules. Thesecond bend indicates the nucleation of new particles as inthis case the seed particles are unable to capture enoughnucleable species to keep their concentration below thecritical supersaturation.
Monomer concentration and particle morphology
Besides the flux of primary radicals, either adjusted by theinitiator concentration or the temperature [16], the durationof the prenucleation period (tN) can also be controlled bythe saturation of the aqueous phase with monomer. Varyingthe equilibration time of the aqueous phase with monomer(tequ) from zero to about 480 min reduces tN by almost afactor of 7 (cf. Fig. 9a). For monomer equilibration timeslonger than about 5 h, the duration of the prenucleationperiod is practically constant (6±0.6 min).
The comparison of the dependence of the duration of theprenucleation period on the monomer equilibration timeand the KPS concentration (Fig. 9b) reveals the commonnature of both dependencies. They possess similar shapeand lead practically to the same minimum value for tN of6.1±0.3 and 6.0±0.6 min for CKPS and tequ, respectively.The maximum must be different, as for lower KPSconcentrations (a concentration somewhere between 0.2
Fig. 7 Particle size distributions of the final latexes for emulsionpolymerizations of styrene in the presence of various amounts of SDS(curve a 0.11 mM; b 2.9 mM; c 15 mM) as determined with AUC;other experimental conditions: 70 °C, 410 g of water, 0.064 g of KPS,3.3 g of styrene; stirring, 70 rpm; the CMC of SDS under theseconditions is about 8 mM; the arrow indicates a shoulder in the sizedistribution of latex c representing the particles
Table 1 Average molecular weights of the high- and low-molecular-mass region of polystyrene particles obtained in the presence ofvarious amounts of SDS (the SDS was removed as described in the“Experimental information”)
CSDS
(mM)Mn / Mw (low molecularweight range; g/mol)
Mn / Mw (high molecularweight range; g/mol)
0.11 360 / 480 7.7×104 / 1.2×105
2.9 280 / 600 1.0×104 / 2.4×104
13 360 / 700 None
Fig. 6 Conductivity–time curves for emulsion polymerizations ofstyrene in the presence of various amounts of SDS (curve a 0.11 mM;b 2.9 mM; c 15 mM); other experimental conditions: 70 °C, 410 g ofwater, 0.064 g of KPS, 3.3 g of styrene with tequ=2 h; stirring, 70 rpm;the CMC of SDS under these conditions is about 8 mM
Colloid Polym Sci (2008) 286:499–515 505

and 0.1 mM), particle nucleation has not been detected (tNgoes to infinity). If the polymerization is started with themonomer addition (tequ=0), it takes 37.5 min untilnucleation at the particular KPS concentration.
Furthermore, for polymerizations in the presence ofabsorbent particles (either micelles or seed particles), theequilibration of the reaction system with monomer iscrucial (cf. Fig. 10). The experimental data of the durationof the nucleation period for polymerizations with seedparticles or surfactants are considered in dependence on thevolume fraction of the foreign material. For SDS concen-trations below the CMC, the resulting volume fraction isnegative, which has the physical meaning that no micellesbut different amounts of nonmicellar SDS are present.Besides the monomer equilibration, the nature of theabsorbent particles has a strong influence. Micelles,especially at higher volume fractions, show a completelydifferent dependence than polystyrene seed particles. Thesummarizing graph of Fig. 10 displays the ways how thepresence of absorbent objects influences the particlenucleation during emulsion polymerization. In relation tothe absorbent-free situations (characterized by tN-values in
regions A and B of Fig. 10), the following conclusions canbe drawn:
1. For monomer equilibration (tequ=2 h), the presence ofSDS is only of very minor influence on the duration ofthe prenucleation period as tN decreases almost linearlyfrom 10.5±1 to 8.1±1 min for surfactant-free conditionand for surfactant concentration five times the CMC,respectively.
2. If, for the situation with monomer equilibration SDSmolecules/micelles are replaced by seed particles withthe same volume fraction, tN remains practicallyunchanged up to a critical volume fraction. At highervolume fraction of seed particles, tN increases toinfinity, and particle nucleation ceases.
3. If the polymerization is started with the addition ofmonomer (tequ=0), the duration of the prenucleationperiod exhibits, at SDS concentrations below the CMCand at lower volume fractions of seed particles, aqualitatively similar pattern: tN displays a maximum independence on the volume fraction.
Fig. 8 Conductivity–time curvesfor emulsion polymerizations ofstyrene in the presence of seedparticles with a volume fractionof 0.2% (a) and 2% (b); thearrows indicate the start of thepolymerization by monomer andby KPS addition; experimentalconditions: 70 °C, 410 g ofwater, 0.064 g of KPS, 3.3 g ofstyrene; stirring, 70 rpm; thedotted lines are just an extensionof the initial slope for bettervisualization
Fig. 9 a Dependence of the duration of the prenucleation period (tN)on the equilibration time of the aqueous phase with styrene (tequ) andb comparison of tN in dependence on the reduced monomerequilibration time (tequ,r = tequ/tequ,max) and on the reduced KPS
concentration (CKPS, r = CKPS/CKPS, max); experimental conditions: 70 °C,400 g of water, 0.067575 g KPS for variable tequ, tequ=120 min forvariable amount of KPS, 3.3 g of styrene; stirring, 70 rpm
506 Colloid Polym Sci (2008) 286:499–515

4. For surfactant concentrations above the CMC and athigher volume fractions of seed particles, tN developsfor both systems oppositely as also observed under theconditions of monomer equilibration. The duration ofthe prenucleation period decreases until it reachesvalues also observed for monomer-swollen micelles,and it approaches infinity in the presence of seedparticles.
The data of Fig. 10 again reveal that the CMC isobviously no extraordinary point and exhibits no specialmeaning for particle nucleation. Contrarily, interestingfeatures are observed at SDS concentration far below theCMC but only under monomer-starved conditions (tequ=0).The explanation of the maxima in the tN–volume fractiondependences is neither straightforward nor possible withinthe frame of ordinary emulsion polymerization mechanisms[2–5]. Finding out the common features of surfactantmolecules at concentrations below the CMC and seedparticles might lead to a rationalization. A long prenucleationperiod (high tN) means that the rate of polymerization islow and vice versa. Obviously, the presence of already lowamounts of surfactants or seed particles influences the rateof polymerization in the aqueous phase considerably. Onereason might be an enhanced decomposition of theperoxodisulfate by the foreign material, although contra-dicting results have been published. Brooks et al. [31]observed an acceleration of the KPS decomposition rate in
the presence of SDS and/or polymer particles. Okubo andMori [32] reported that only free SDS molecules (neitheradsorbed nor bound in micelles) increased the KPSdecomposition rate but that this effect disappeared in thepresence of small amount of monomers. An enhanceddecomposition rate of KPS can be very likely excluded, asit cannot explain the occurrence of the maximum. Conse-quently, an influence on the monomer concentration inwater remains as possible explanation. As these polymer-izations were started with the addition of monomer, anycircumstances influencing the transfer of styrene from thereservoir to the water phase can affect the rate ofpolymerization and change the duration of the prenucleationperiod. Both the SDS molecules and the seed particlescontain hydrophobic portions, and hence, their presenceenhances the driving force for the monomer to enter theaqueous phase. This might be the reason for the initial dropof tN. However, the maximum points to the existence ofcounteracting influences. Seed particles can act as absorbentscapturing radicals and monomers, and thus, they can causealso a reduction in the rate of polymerization. Below theCMC, surfactant molecules are usually considered to bemolecularly dissolved, and consequently, they are not able toact likewise as absorbents. The situation is complicated, andunraveling requires changed paradigms especially regardingthe structure of the monomer-in-water system at surfactantconcentrations below the CMC.
All the results presented in Figs. 9 and 10 clearly revealtequ as new experimental parameter influencing the particlenucleation kinetics, likewise the initiator concentration.Moreover, the data raise immediately the question regardingthe state of the monomer-in-water solution. To get an idea, thetransmission measurements have been evaluated carefullyalready during the monomer equilibration period beforestarting the polymerization, and two kinds of experiments inother reactors have been carried out. It turned out that duringthe monomer equilibration period, the transmission in the all-Teflon reactor under the conditions of the standard experimentdecreases from 100 to about 91% continuously over about 3 hbefore reaching a constant value. With multi-angle laser-lightscattering in glass cuvettes, the development of a strongscattering signal in the aqueous phase was observed over aperiod of some hours after styrene was quiescently placed ontop of water [33]. Additionally, the styrene concentration inwater was determined by gas chromatography in samplestaken from the all-Teflon reactor [15]. The data sets for rG(radius of gyration from light scattering) and Csty (styreneconcentration in water from gas chromatography) show aninteresting correlation when plotted with a common time axis(cf. Fig. 11). The averaged time-dependent data sets showthat the average droplet size increases dramatically in thevicinity of the saturation concentration. Over the entireconcentration range d(2rG)/dCsty (not shown here) increases
Fig. 10 Nucleation time (tN) in dependence on the volume fraction ofabsorbent particles: SDS molecules (circles) or seed latex particles(squares); the open symbols represent data obtained with empty orunswollen particles (polymerizations started with monomer addition),and the grey symbols data points for swollen absorbent particles after2 h equilibration time with monomer (polymerizations started withKPS addition); the dotted lines mark the regions of tN=39±4 min andtN=10.5±1 min for absorbent-free polymerizations with zero (a) and2 h (b) monomer equilibration time, respectively; other experimentalconditions: 70 °C, 410 g of water, 0.064 g KPS, 3.3 g of styrene;stirring, 70 rpm; average data of four repeats for the polymerizationswith SDS
Colloid Polym Sci (2008) 286:499–515 507
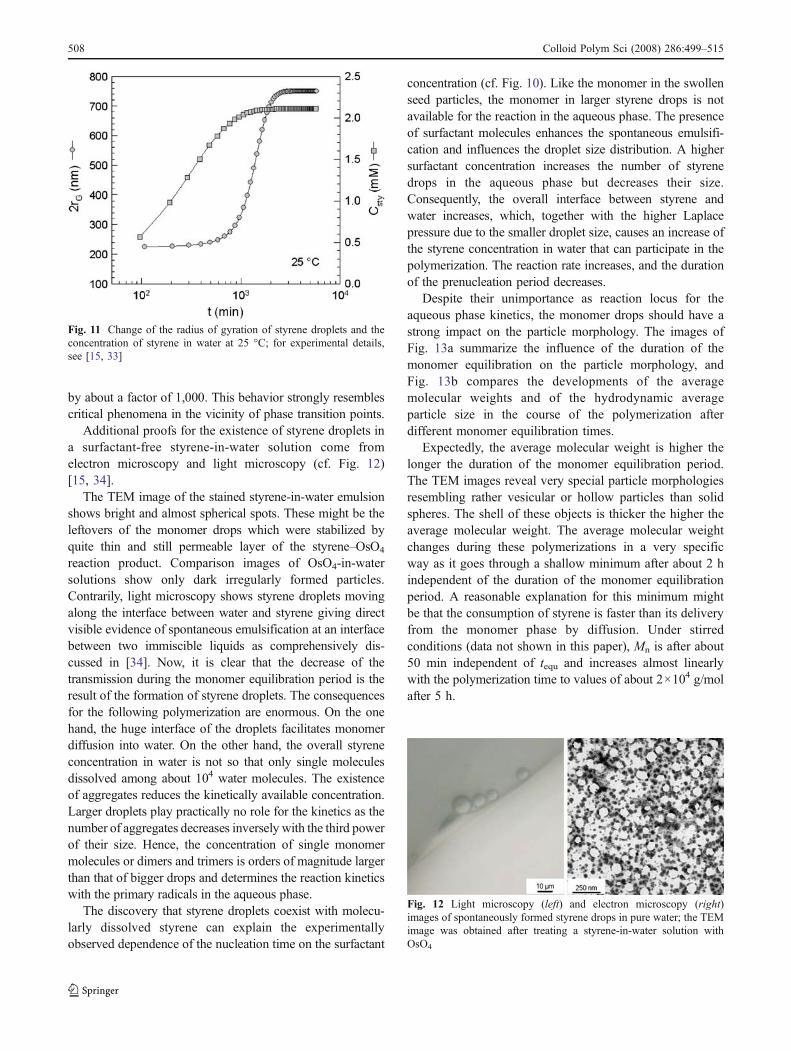
by about a factor of 1,000. This behavior strongly resemblescritical phenomena in the vicinity of phase transition points.
Additional proofs for the existence of styrene droplets ina surfactant-free styrene-in-water solution come fromelectron microscopy and light microscopy (cf. Fig. 12)[15, 34].
The TEM image of the stained styrene-in-water emulsionshows bright and almost spherical spots. These might be theleftovers of the monomer drops which were stabilized byquite thin and still permeable layer of the styrene–OsO4
reaction product. Comparison images of OsO4-in-watersolutions show only dark irregularly formed particles.Contrarily, light microscopy shows styrene droplets movingalong the interface between water and styrene giving directvisible evidence of spontaneous emulsification at an interfacebetween two immiscible liquids as comprehensively dis-cussed in [34]. Now, it is clear that the decrease of thetransmission during the monomer equilibration period is theresult of the formation of styrene droplets. The consequencesfor the following polymerization are enormous. On the onehand, the huge interface of the droplets facilitates monomerdiffusion into water. On the other hand, the overall styreneconcentration in water is not so that only single moleculesdissolved among about 104 water molecules. The existenceof aggregates reduces the kinetically available concentration.Larger droplets play practically no role for the kinetics as thenumber of aggregates decreases inversely with the third powerof their size. Hence, the concentration of single monomermolecules or dimers and trimers is orders of magnitude largerthan that of bigger drops and determines the reaction kineticswith the primary radicals in the aqueous phase.
The discovery that styrene droplets coexist with molecu-larly dissolved styrene can explain the experimentallyobserved dependence of the nucleation time on the surfactant
concentration (cf. Fig. 10). Like the monomer in the swollenseed particles, the monomer in larger styrene drops is notavailable for the reaction in the aqueous phase. The presenceof surfactant molecules enhances the spontaneous emulsifi-cation and influences the droplet size distribution. A highersurfactant concentration increases the number of styrenedrops in the aqueous phase but decreases their size.Consequently, the overall interface between styrene andwater increases, which, together with the higher Laplacepressure due to the smaller droplet size, causes an increase ofthe styrene concentration in water that can participate in thepolymerization. The reaction rate increases, and the durationof the prenucleation period decreases.
Despite their unimportance as reaction locus for theaqueous phase kinetics, the monomer drops should have astrong impact on the particle morphology. The images ofFig. 13a summarize the influence of the duration of themonomer equilibration on the particle morphology, andFig. 13b compares the developments of the averagemolecular weights and of the hydrodynamic averageparticle size in the course of the polymerization afterdifferent monomer equilibration times.
Expectedly, the average molecular weight is higher thelonger the duration of the monomer equilibration period.The TEM images reveal very special particle morphologiesresembling rather vesicular or hollow particles than solidspheres. The shell of these objects is thicker the higher theaverage molecular weight. The average molecular weightchanges during these polymerizations in a very specificway as it goes through a shallow minimum after about 2 hindependent of the duration of the monomer equilibrationperiod. A reasonable explanation for this minimum mightbe that the consumption of styrene is faster than its deliveryfrom the monomer phase by diffusion. Under stirredconditions (data not shown in this paper), Mn is after about50 min independent of tequ and increases almost linearlywith the polymerization time to values of about 2×104 g/molafter 5 h.
Fig. 11 Change of the radius of gyration of styrene droplets and theconcentration of styrene in water at 25 °C; for experimental details,see [15, 33]
Fig. 12 Light microscopy (left) and electron microscopy (right)images of spontaneously formed styrene drops in pure water; the TEMimage was obtained after treating a styrene-in-water solution withOsO4
508 Colloid Polym Sci (2008) 286:499–515

The change of the average hydrodynamic particle sizeduring the polymerization depends, especially within thefirst hour, strongly on the monomer equilibration time. Forshort equilibration times, DZ decreases drastically andremains almost constant thereafter. Basically, the TEMimages do not display this behavior. This discrepancy is astrong hint that the scattering structures in the dispersionsare no solid structures. During the stirred polymerizations,DZ increases with the polymerization time and is indepen-dent of the prenucleation period.
Besides the duration of the monomer equilibrationperiod, the monomer concentration can also be controlledby the hydrodynamics in the reactor. In the simplest case,this can be done by changing the stirrer speed, as shown bythe data of Fig. 14.
The principal particle morphology does not depend onthe hydrodynamic conditions. In either case — stirred andnonstirred — the hollow particle morphology is observed.However, the influence of the stirrer speed on the shellthickness of the hollow particles is enormous. The shellgrows thicker if the transportation of the monomer to thereaction loci is enhanced due to the stirring.
Both DZ and ξ reveal the strong influence of thehydrodynamics on the properties of the latex particles.Under quiescent conditions, the zeta potential is muchlower (much more negative), the shell around the vesicularor hollow particles is much thinner, and DZ decreasesduring the polymerization, whereas it increases under
Fig. 13 a TEM images of theparticle morphology after poly-merization time of 60 min forsurfactant-free emulsion poly-merizations of styrene withmonomer equilibration times of0 (A), 30 (B), and 180 min (C);experimental conditions: all-glass reactor, nonstirred, 70 °C,20 ml of styrene, 562.5 g ofwater, 1.25 g of KPS.b Development of the numberaverage molecular weight (Mn)and the average particle size(DZ, from ZetaSizer) during thesurfactant-free emulsion poly-merizations of styrene afterdifferent monomer equilibrationtimes of 0 (circles), 30(squares), and 180 min(triangles); experimental condi-tions: all-glass reactor, non-stirred, 70 °C, 20 ml of styrene,562.5 g of water, 1.25 g of KPS
Fig. 14 a TEM images showing particles after polymerization time of60 min for surfactant-free emulsion polymerizations of styrene withzero monomer equilibration time, nonstirred (A) and stirred with300 rpm (B); experimental conditions: all-glass reactor, 70 °C, 20 mlof styrene, 562.5 g of water, 1.25 g of KPS. b Development of theaverage particle size (DZ, from ZetaSizer) and the zeta potential (ξ)during the surfactant-free emulsion polymerizations of styrene withoutmonomer equilibration under unstirred (circles) and stirred (300 rpm;squares) conditions; further experimental details: all-glass reactor,nonstirred, 70 °C, 20 ml of styrene, 562.5 g of water, 1.25 g of KPS
Colloid Polym Sci (2008) 286:499–515 509

stirred conditions. In addition, stirring leads to higher solidscontent and to higher molecular weights. The latter is inaccordance with the less negative zeta potential of theparticles obtained under stirred conditions.
The data depicted in Figs. 10, 11, 12, 13, and 14 disclosethe importance of the ‘monomer-in-water state’ on theoutcome during the early stages of emulsion polymer-izations. Obviously, the monomer droplets formed aftercontacting styrene and water are templated or stabilized bysmall oligomeric particles formed by reaction of theprimary sulfate ion radicals with styrene molecules (ordimers or trimers). These oligomeric particles have a sizebelow 5 nm and are visible as small dark spots on the TEMimages (cf. Fig. 15) and consist of a few oligomers withhigher electron density due to the higher sulfur and oxygencontent than high molecular weight polymer. Note that thechemical structure of these oligomers must necessarily notbe regular as expected from radical polymerization kineticsas it is known that both �SO�
4 and also HO� radicals canreact with the phenyl ring to give the corresponding (vinyl)cyclohexadienyl radicals [35, 36]. After nucleation, theseoligomeric particles adsorb at the interface between thestyrene droplets and water as they are neither soluble inwater nor in styrene.
This scenario explains all the experimental observationsfrom the spontaneous emulsification to the formation ofvesicular or hollow particles. If, however, the polymeriza-tion conditions are changed in a way resulting in oligomerswith higher molecular weight which are soluble in thedroplets, solid particles should be formed. Indeed, this isproven by the data put together in Fig. 16a and b. Here, twopolymerizations with different KPS concentrations arecompared. The polymerization with the lower initiatorconcentration leads to polymers with higher averagemolecular weights and particles with lower averagediameters (cf. Fig. 16a). The former follows from theradical polymerization kinetics and the latter from thedependence of droplet and particle stability on the ionic
strength. The morphology of the particles is changed in theexpected way as the polymerization with the lower initiatorconcentration leads to solid particles.
Regarding the radical generation in the continuousaqueous phase, the polymerizations with AIBN correspondto polymerizations with low KPS concentrations. Conse-quently, the particle morphology is always solid asexemplarily shown in Fig. 4.
Absorption of matter by latex particles
Due to their large surface area, colloidal particles caneffectively absorb or imbibe all kinds of materials of eitherhydrophobic or hydrophilic nature [30]. Radicals andmonomers are for emulsion polymerizations of particularimportance. Despite the actual situation that the uptakemechanism of both monomers and radicals is considereddifferently and discussed contradictorily, there are generalprinciples involved which have to be considered.
It has been found that the Morton–Kaizerman–Altier(MKA) Eq. 1 [8] is not suited to describe quantitativelythe swelling of latex particles by organic solvents for thepolymer [9, 10]. In MKA Eq. 1, R is the gas constant, T thetemperature, v1 the molecular volume of the swelling agent,σ the interfacial tension between the particle and thecontinuous phase, χ the Flory–Huggins interaction parameterbetween polymer and swelling agent, φ2 the volume fractionof polymer in the swollen particles, and r the particle radius.To achieve a better agreement with experimental data, aswelling pressure was introduced that takes into account thework necessary for the disintegration of entangled polymermolecules. Additionally, the agreement with experimentaldata was improved if the concentration dependence of theFlory–Huggins interaction parameter and the dependence ofthe interfacial tension on the particle size were considered [9,10]. The investigation of swelling of large monodisperselatex particles by means of light microscopy revealed somenew experimental facts which, on the one hand, can improve
Fig. 15 TEM images illustrating the particle morphology as result ofthe deposition of oligomeric particles onto styrene droplets during theearly stage of unstirred emulsion polymerization; a tequ=5 min after40 min polymerization time, b tequ=120 min after 30 min polymer-
ization time, c tequ=180 min after 30 min polymerization time; furtherexperimental details: all-glass reactor, nonstirred, 70 °C, 20 ml ofstyrene, 562.5 g of water, 1.25 g of KPS
510 Colloid Polym Sci (2008) 286:499–515
the theoretical description and, on the other hand, offer novelpossibilities for the modification of latex particles.
2 � σr
� �� v1R � T ¼ � ln 1� φ2ð Þ þ 1� 1=j2ð Þ � φ2 þ χ � φ2
2
� �ð1Þ
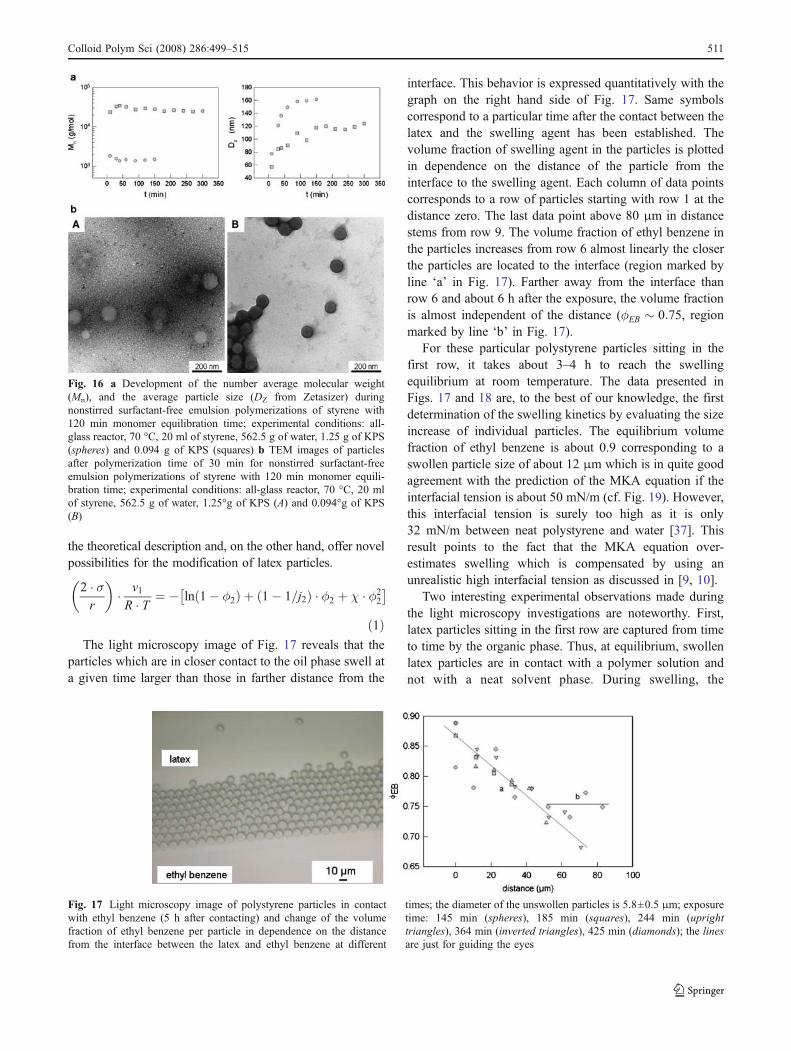
The light microscopy image of Fig. 17 reveals that theparticles which are in closer contact to the oil phase swell ata given time larger than those in farther distance from the
interface. This behavior is expressed quantitatively with thegraph on the right hand side of Fig. 17. Same symbolscorrespond to a particular time after the contact between thelatex and the swelling agent has been established. Thevolume fraction of swelling agent in the particles is plottedin dependence on the distance of the particle from theinterface to the swelling agent. Each column of data pointscorresponds to a row of particles starting with row 1 at thedistance zero. The last data point above 80 μm in distancestems from row 9. The volume fraction of ethyl benzene inthe particles increases from row 6 almost linearly the closerthe particles are located to the interface (region marked byline ‘a’ in Fig. 17). Farther away from the interface thanrow 6 and about 6 h after the exposure, the volume fractionis almost independent of the distance (φEB � 0:75, regionmarked by line ‘b’ in Fig. 17).
For these particular polystyrene particles sitting in thefirst row, it takes about 3–4 h to reach the swellingequilibrium at room temperature. The data presented inFigs. 17 and 18 are, to the best of our knowledge, the firstdetermination of the swelling kinetics by evaluating the sizeincrease of individual particles. The equilibrium volumefraction of ethyl benzene is about 0.9 corresponding to aswollen particle size of about 12 μm which is in quite goodagreement with the prediction of the MKA equation if theinterfacial tension is about 50 mN/m (cf. Fig. 19). However,this interfacial tension is surely too high as it is only32 mN/m between neat polystyrene and water [37]. Thisresult points to the fact that the MKA equation over-estimates swelling which is compensated by using anunrealistic high interfacial tension as discussed in [9, 10].
Two interesting experimental observations made duringthe light microscopy investigations are noteworthy. First,latex particles sitting in the first row are captured from timeto time by the organic phase. Thus, at equilibrium, swollenlatex particles are in contact with a polymer solution andnot with a neat solvent phase. During swelling, the
Fig. 16 a Development of the number average molecular weight(Mn), and the average particle size (DZ from Zetasizer) duringnonstirred surfactant-free emulsion polymerizations of styrene with120 min monomer equilibration time; experimental conditions: all-glass reactor, 70 °C, 20 ml of styrene, 562.5 g of water, 1.25 g of KPS(spheres) and 0.094 g of KPS (squares) b TEM images of particlesafter polymerization time of 30 min for nonstirred surfactant-freeemulsion polymerizations of styrene with 120 min monomer equili-bration time; experimental conditions: all-glass reactor, 70 °C, 20 mlof styrene, 562.5 g of water, 1.25°g of KPS (A) and 0.094°g of KPS(B)
Fig. 17 Light microscopy image of polystyrene particles in contactwith ethyl benzene (5 h after contacting) and change of the volumefraction of ethyl benzene per particle in dependence on the distancefrom the interface between the latex and ethyl benzene at different
times; the diameter of the unswollen particles is 5.8±0.5 μm; exposuretime: 145 min (spheres), 185 min (squares), 244 min (uprighttriangles), 364 min (inverted triangles), 425 min (diamonds); the linesare just for guiding the eyes
Colloid Polym Sci (2008) 286:499–515 511

chemical potential of the solvent in the particles increasesand in the liquid phase decreases until it is equilibrated.
Second, swelling of latex particles is cooperative innature. If a solution is in contact instead of a pure solventalso solute molecules are transferred into the latex particles.This happens even if the solute is a chemically differentpolymer as demonstrated by the images of Fig. 20 provingthe transfer of pyrene-labeled poly(methyl methacrylate)into polystyrene particles.
These results have important consequences for carryingout emulsion polymerization and for the modification ofpolymer latexes. There is a possibility to produce compositelatex particles simply by swelling with appropriate solu-tions. In monomer-flooded polymerizations, the monomerphase will always contain a certain amount of polymereither as a result of the capture of latex particle by
monomer droplets or as a result of direct polymerizationinside the monomer droplets because radicals can beabsorbed as well.
During emulsion polymerization of hydrophobic mono-mers, the main reaction loci for monomer conversion arethe latex particles. Besides swelling of the particles withmonomer, this requires also the capture of radicals from thecontinuous phase which then continue to grow inside theparticles. For the uptake of radicals by latex particles, fourmechanisms are discussed differing regarding the depen-dence of the capture rate coefficient (kc) on the particle size(kc / Dα). The collision model predicts α=2 [39, 40], thediffusion model α=1 [41, 42], the colloidal model alsoα=1 [43], and the propagational model α=0 [11]. Interest-ingly, experimental results have been published proving thevalidity of all models. Obviously, each of these models isonly valid for a certain range of experimental conditions.This is an insufficient situation showing that the generalbehavior regarding the absorption of matter by latexparticles has not yet been understood.
Recently, it has been shown that Brownian dynamics(BD) simulations [44] are quite useful to achieve a deeperunderstanding of the absorption of matter by latex particles.These numerical experiments allow effective simulation ofa variety of experimental conditions regarding the size andthe concentration of the particles. Note that models relyingon the Smoluchowski equation are insufficient, as it is validfor a single particle at infinite dilution [45]. However, inreal emulsion polymerizations, the volume fraction of thepolymer particles (φp) can be well above 50%.
With the Brownian dynamics simulations, the capturetime of a species by particles was numerically determined.The capture time (t) is the time between the introduction ofthe particular species in the system and its irreversiblecapture by the particles. The capture rate constant kc relateswith t according to Eq. 2 where N is the particle number inthe dispersion volume (that is the volume of the continuous
Fig. 19 Volume fraction of ethyl benzene in polystyrene particles independence of the particle size; the calculations have been carried outusing Eq. 1 with a Flory–Huggins interaction parameter of 0.5 and atemperature of 25 °C
Fig. 18 Swelling kinetics of polystyrene particles with ethyl benzeneat room temperature expressed as increase in the diameter (D, spheres)and volume fraction of ethyl benzene (φEB, squares); φEB values arecalculated with the average values of D for a given time
Fig. 20 Light microscopy images of swollen polystyrene latex particles;right, bright field image; left, fluorescence light image; the polystyreneparticles are in contact with a solution of poly((pyrenyl methacrylate)-co-(methyl methacrylate)) in ethyl benzene; images were taken about4 weeks after contacting the latex with the copolymer solution; thepyrene-labeled copolymer was prepared as described in [38]
512 Colloid Polym Sci (2008) 286:499–515

phase plus the overall particle volume) and NA isAvogadro’s number.
kc ¼ NA
N Ch i ð2Þ
The numerical simulations revealed that kc depends onthe particle size with a power which itself changeseffectively with the polymer volume fraction. For verylow polymer-volume fraction, kc / D (α=1) as predictedby the Smoluchowski Eq. 3 where
~Dr is the diffusion
coefficient of the absorbed species with a size much smallerthan the particles.
kc ¼ 2:~DrDNA ð3Þ
However, with increasing polymer volume fraction, kcincreases above the value given by Eq. 3. Thus, adimensionless number, Sm (Smoluchowski number), canbe defined by Eq. 4 relating the actual kc value to2:
~DrDNA.
Sm ¼ kc2π
~DrDNA
ð4Þ
The numerical results of the calculations (cf. Fig. 21) fora wide range of D and N values can be fitted in a satisfyingmanner by Eq. 5 where v=17.95 for the particular (mainlygeometrical) conditions of the simulation.
Sm ¼ 1þ vφp ð5ÞThe polymer volume fraction can be modified by the two
independent variables particle size and particle number. Forextremely low-volume fractions (φp < 10�4), the radicalcapture is determined by the Smoluchowski equation.These results of the numerical simulations, as given by
Eq. 5 and Fig. 21, are reasonable and fit with the variousexperimental data for radical capture by latex particles.
It is necessary to point out again that this behavior is notrestricted to radicals, but it should be valid for all kinds ofmatter with a size smaller than the absorbing particles.
Moreover, the absorption or capture rate depends on thesize of the absorbed species as expressed by their diffusioncoefficients (
~Dr). The smaller the species, the higher is the
capture rate by the larger particles as verified by numericalsimulations. Moreover, the simulations show that thecapture rate depends on the relative position of the absorbedspecies to the particle. The closer to the particle surface thespecies is generated or born or introduced, the higher thecapture rate. This is clearly in accordance with experimentaldata as shown exemplarily in Fig. 17.
The results of the numerical simulations based on theBrownian dynamics are useful to evaluate problems inrelation with the general mechanism of emulsion polymer-ization. A first example is related to the discussionregarding the micellar nucleation mechanism and concernsthe estimation of the number of micelles at which thecapture rate (rcm, Eq. 6) of a radical from the water phase[Raq] equals the propagation rate in water (rpaq, Eq. 7).From Eqs. 6 and Eq. 7 follows the quadratic Eq. 8 with thesolution 9 for the number of micelles (Nm).
rcm ¼ kcNm
NARaq
� � ¼ 2π~DrNA
υπNmD4m
6þ Dm
� �Nm
NARaq
� � ¼ 2π~Dr
υπNmD4m
6þ Dm
� �Nm Raq
� � ð6Þ
rpaq ¼ kp Maq
� �Raq
� � ð7Þ
N2m þ 6
υ:D3m
Nm � 3kp Maq
� �υ:2D4
m~Dr
¼ 0 ð8Þ
Nm ¼ 1
2
6
υ:D3m
þffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
6
υ:D3m
� �2
þ 12kp Maq
� �υ:2D4
m~Dr
s !ð9Þ
Considering styrene emulsion polymerization (50 °Cwith the propagation rate constant kp=237 l mol−1 s−1, themonomer concentration in water [Maq]=4.3∙10−3 M, thediameter of a swollen micelle Dm=10 nm, and~Dr ¼ 2� 10�9m2s�1) the propagation rate in the aqueousphase equals the rate of capture of radicals by micelles atNm=8.11×10
15 m−3 corresponding to a volume fraction ofmicelles as low as 4.25×10−9. This means that already atsurfactant concentrations just above the CMC, micelles cankinetically compete with the styrene molecules for the
Fig. 21 Dependence of the capture rate of small molecules byparticles in a polymer dispersion expressed by the Smoluchowskinumber (Sm) on the polymer volume fraction (φp ¼ π
�6 � D3 � N )
Colloid Polym Sci (2008) 286:499–515 513

radicals which is actually good for micellar nucleation. Thatmicellar nucleation is experimentally not observed mighthave several reasons. Either the radical grows out of themicelle into the water phase as Harkins [23–26] alreadysuspected, or the micelle is destroyed by the inside-growingradical (either by the heat of propagation or due to theincompatibility between the surfactant and the polymerchain), and the only millisecond lifetime of a low-molecular-weight surfactant micelle [46] is effectively still furtherreduced, or there exists an effective quite high energy barriercounteracting the entry of the radicals. BD simulations [47]with varying energy barrier clearly evidenced that the captureefficiency decays exponentially with the height of the energybarrier. Contrarily, the radical capture coefficient remainsalmost constant until certain “apparent” threshold energybefore it shows the expected exponential decay. This meansthat a radical hits the same particle several times before itgoes away again unless radical capture occurs. The thresholdenergy is larger than ten times kBT. The BD simulations donot contradict the experimental results showing that nucle-ation is practically unchanged in the presence of micelles butcan be prevented in the presence of seed particles with avolume fraction of about 2×10−2. The effective volumefraction for preventing nucleation must be much higher thanthe value estimated above for rcm=rpaq as the capture ratemust be so high that the accumulation of nucleable oligomersin the water phase can be prevented.
A second example deals with the question whether singlemolecules or nano-droplets contribute mainly to the swellingof latex particles. Applying Eq. 6 to the absorption ofclusters of monomers (C) by latex particles (NP) withdiameter D results in Eq. 10 for the absorption rate (rabc),where [C] is the cluster concentration relative to the volumeof water. For the diffusion coefficient of the clusterscomposed of nsm single monomer molecules,
~Dc, and the
cluster concentration hold the relations 11a and 11b,respectively. Dsm is the size of a single monomer moleculeand
~Dsm its diffusion coefficient.
rabc ¼ 2:~DcD υφp þ 1
� �NP C½ � 1� φp
� � ð10Þ
~Dc ¼ Dsm
D~Dsm ¼ n�1=3
sm~Dsm ð11aÞ
C½ � ¼ Maq
� �nsm
ð11bÞ
Equation 12, relating the absorption rate of clusters tothat of single monomer molecules (rabsm), shows that theabsorption rate for single molecules is higher than forclusters. Moreover, the absolute rate of monomer uptake(that is, the swelling rate of a particle) is also higher for the
absorption of single molecules, as only nsm molecules aretransferred per collision between a cluster and a particle.
rabc ¼ n�4=3sm rabsm ð12Þ
Conclusions
Emulsion polymerization is a quite complex process thatcomprises several steps proceeding on very different timescales. Particle growth takes place on time scales that are inthe order of the duration of the polymerization reaction, andhence, it is quite easy to study. However, some importantreactions such as particle nucleation or radical entry havemuch smaller characteristic time constants making on-lineinvestigation experimentally extremely challenging. Exper-imental results throwing light upon essential steps ofemulsion polymerizations such as particle nucleation andthe uptake of monomers and radicals by the particlesrequire particularly designed experiments rather than theanalysis of conversion–time curves of standard emulsionpolymerizations. Moreover, thinking out of the box ofestablishedmodels opens new possibilities to gain knowledge.
The analysis of particle nucleation requires slowingdown the reaction rate and a combination of variousmethods. Conductivity measurement is an extremely usefultool to identify the mechanism of particle nucleation. Theexperimental data obtained so far give no hint that micellesof low-molecular-weight surfactants are a major locus ofparticle formation. The data show that particle nucleationtakes place outside the micelles and can be understood asaggregative nucleation within the frame of the classicalnucleation theory [6]. Just after nucleation, the particleshave a size of a few nanometers (D<5 nm) and arecomposed of quite a few water-borne oligomers. Surfac-tants influence the particle nucleation kinetics in severalways. If the aqueous phase is not yet equilibrated withmonomer, the transfer of monomer to the aqueous phase isfacilitated, and subsequently, the duration of the prenuclea-tion period is shortened. The presence of surfactantmolecules in the aqueous phase causes the formation of ahigher concentration of smaller monomer drops due tospontaneous or forced emulsification. Moreover, surfactantmolecules lower the interfacial tension of the particle nucleiand increase thereby the rate of nucleation.
However, the presence of seed particles can effectivelyprevent the formation of new particles as, above a criticalvolume fraction, they act as absorbers for any kind ofsmaller-sized matter present in the dispersion.
Brownian dynamics simulations suggest that any kind ofspecies (radicals, monomers, and oligomers) can enter latexparticles in dependence on their size for a given polymervolume fraction. In this sense and in contradiction to the
514 Colloid Polym Sci (2008) 286:499–515

propagational entry model [11], primary radicals can enterlatex particles as it was proven also experimentally [12].Besides radical entry, also swelling of latex particles withsolvents or solutions can be treated as the simulations showthat particles can take up any object present in thedispersion where the capture rate is primarily controlledby their size (diffusion coefficient). Swelling of latexparticles with monomer must not necessarily take placevia the uptake of single molecules as also monomerdroplets are present in the aqueous phase. This spontaneousemulsification that takes place if two immiscible liquids arepresent [34] has enormous consequences for the mechanismof emulsion polymerization, not only for swelling but alsofor particle formation. Furthermore, it offers possibilities tomodify latex particles by post-polymerization treatment.
In industry, it is the state of the art to carry out semibatch-seeded emulsion polymerizations under monomer-starvedconditions [1]. Such production protocols are advantageousas they avoid all uncertainties connected with particlenucleation and the presence of a free monomer phase. Theresults presented here for batch ab initio polymerizations inthe presence of free monomer are at least relevant for theseed production where particle nucleation takes place. Underindustrially relevant conditions, that is, in the presence ofsurfactants and with stirring where emulsification is forced,the details of the nucleation process are not visible as thestate of the monomer in the continuous phase is fixed andcannot be varied in such width and detail as in the presentstudy.
Despite the fact that emulsion polymerization is one of theoldest industrial technologies for the production of polymers,new experimental approaches can still lead to the discoveryof new experimental facts and to better understanding.
Acknowledgements The authors gratefully acknowledge financialsupport from the Max Planck Society and technical assistance fromMrs. Ursula Lubahn, Mrs. Sylvia Pirok, Mrs. Rona Pitschke, Mrs.Heike Runge, Mrs. Antje Völkel, and Mrs. Eugenia Maximova (MPIof Plant Physiology). O. L. is thankful for a fellowship provided bythe German Academic Exchange Service (DAAD) within the MichailLomonosov Program.
References
1. Urban D, Distler D (2002) Introduction. In: Urban D, Takamura K(eds) Polymer dispersions and their industrial applications. Wiley-VCH Verlag GmbH, Weinheim, p 1
2. El-Aasser MS, Sudol ED (1997) Features of emulsion polymer-ization. In: Lovell PA, El-Aasser MS (eds) Emulsion polymeriza-tion and emulsion polymers. Wiley, New York, p 37
3. Fitch RM (1997) Polymer colloids. A comprehensive introduc-tion. Academic, San Diego
4. Gilbert RG (1995) Emulsion polymerization. Academic, London5. van Herk A (2005) Chemistry and technology of emulsion
polymerization. Blackwell, Oxford, UK6. Tauer K, Kühn I (1995) Macromolecules 28:22367. Tauer K, Kühn I (1997) Particle nucleation at the beginning of
emulsion polymerization. In: Asua JM (ed) NATO ASI Series,Series E: Applied Sciences, 335. p 49
8. Morton M, Kaizerman S, Altier MW (1954) J Coll Sci 9:3009. Antonietti M, Kaspar H, Tauer K (1996) Langmuir 12:6211
10. Tauer K, Kaspar H, Antonietti M (2000) Coll Polym Sci 278:81411. Maxwell IA, Morrison BR, Napper DH, Gilbert RG (1991)
Macromolecules 24:162912. Tauer K, Nozari S, Ali AMI (2005) Macromolecules 38:861113. Tauer K, Antonietti M, Rosengarten L, Müller H (1998) Macromol
Chem Phys 199:89714. Walz R, Bomer B, Heitz W (1977) Makromol Chem - Macromol
Chem Phys 178:252715. Kozempel S (2005) PhD thesis, Emulgatorfreie Emulsionspoly-
merisation—zonomerlösungszustand und Teilchenbildung, Uni-versity of Potsdam
16. Kühn I, Tauer K (1995) Macromolecules 28:812217. Tauer K, Deckwer R, Kuhn I, Schellenberg C (1999) Coll Polym
Sci 277:60718. Tauer K, Kühn I, Kaspar H (1996) Progr. Coll Polym Sci 101:3019. Laaksonen A, Talanquer V, Oxtoby DW (1995) Ann Rev Phys
Chem 46:48920. Tauer K, Mukhamedjanova M, Holtze C, Nazaran P, Lee J (2007)
Macromol Symp 248:22721. Tauer K (1995) Polym Adv Technol 6:43522. Holtze C, Antonietti M, Tauer K (2006) Macromolecules 39:572023. Harkins WD (1947) J Amer Chem Soc 69:142824. Harkins WD (1945) J Chem Phys 13:38125. Harkins WD (1946) J Chem Phys 14:4726. Harkins WD (1950) J Polym Sci 5:21727. Smith WV, Ewart RH (1948) J Chem Phys 16:59228. Roe CP (1968) Ind Eng Chem 60:2029. Bartholome E, Gerrens H, Herbeck R, Weitz HM (1956) Z
Elekrtochem 60:33430. Hansen FK, Ugelstad J (1979) J Polym Chem Part A Polym Chem
17:304731. Brooks BW, Makanjuola BO (1981) Makromol Chem-Rapid
Commun 2:6932. OkuboM, Mori T (1990) Makromol Chem-Macromol Symp 31:14333. Kozempel S, Tauer K, Rother G (2005) Polymer 46:116934. Tauer K, Kozempel S, Rother G (2007) J Coll Interf Sci 312:43235. McAskill NA, Sangster DF (1984) Aust J Chem 37:213736. Neta P, Madhavan V, Zemel H, Fessenden RW (1977) J Amer
Chem Soc 99:16337. Vijayendran BR (1979) J Appl Polym Sci 23:73338. Saito N, Kagari Y, Okubo M (2006) Langmuir 22:939739. Fitch RM, Tsai CH (1971) Polym Coll Proc Symp 7340. Gardon JL (1968) J Polym Sci Part A-1 Polym Chem 6:62341. Hansen FK, Ugelstad J (1978) J Polym Chem Part A Polym Chem
16:195342. Ugelstad J, Hansen FK (1976) Rub Chem Technol 49:53643. Penboss IA, Gilbert RG, Napper DH (1986) J Chem Soc-Farad
Trans I 82:224744. Hernandez HF, Tauer K (2007) Ind Eng Chem Res 46:448045. von Smoluchowski M (1906) Ann Phys 21:75646. Fuhrhop J-H, Köning J (1994) Membranes and Molecular
Assemblies. The Royal Society of Chemistry, Cambridge47. Hernandez HF, Tauer K (2007) Macromol Symp in press
Colloid Polym Sci (2008) 286:499–515 515




















