
Total body glutathione depletion induces oxidative stress and disrupts the immune function in mice
15
PLEASE SCROLL DOWN FOR ARTICLE This article was downloaded by: [Abdalla, Maher] On: 16 July 2010 Access details: Access Details: [subscription number 924492018] Publisher Taylor & Francis Informa Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House, 37- 41 Mortimer Street, London W1T 3JH, UK Toxicological & Environmental Chemistry Publication details, including instructions for authors and subscription information: http://www.informaworld.com/smpp/title~content=t713653210 Total body glutathione depletion induces oxidative stress and disrupts the immune function in mice Maher Y. Abdalla ab ; Iman M. Hassan a ; Noor H. Mustafa a ; Lubna H. Tahtamouni a ; Iman M. Ahmad c a Department of Biological Sciences and Biotechnology, The Hashemite University, Zarqa, Jordan b Center for Cancer Research, National Cancer Institute, National Naval Medical Center, Bethesda, MD 20889, USA c Department of Radiography, The Hashemite University, Zarqa, Jordan First published on: 16 July 2010 To cite this Article Abdalla, Maher Y. , Hassan, Iman M. , Mustafa, Noor H. , Tahtamouni, Lubna H. and Ahmad, Iman M.(2010) 'Total body glutathione depletion induces oxidative stress and disrupts the immune function in mice', Toxicological & Environmental Chemistry,, First published on: 16 July 2010 (iFirst) To link to this Article: DOI: 10.1080/02772248.2010.494828 URL: http://dx.doi.org/10.1080/02772248.2010.494828 Full terms and conditions of use: http://www.informaworld.com/terms-and-conditions-of-access.pdf This article may be used for research, teaching and private study purposes. Any substantial or systematic reproduction, re-distribution, re-selling, loan or sub-licensing, systematic supply or distribution in any form to anyone is expressly forbidden. The publisher does not give any warranty express or implied or make any representation that the contents will be complete or accurate or up to date. The accuracy of any instructions, formulae and drug doses should be independently verified with primary sources. The publisher shall not be liable for any loss, actions, claims, proceedings, demand or costs or damages whatsoever or howsoever caused arising directly or indirectly in connection with or arising out of the use of this material.
Transcript of Total body glutathione depletion induces oxidative stress and disrupts the immune function in mice

PLEASE SCROLL DOWN FOR ARTICLE
This article was downloaded by: [Abdalla, Maher]On: 16 July 2010Access details: Access Details: [subscription number 924492018]Publisher Taylor & FrancisInforma Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House, 37-41 Mortimer Street, London W1T 3JH, UK
Toxicological & Environmental ChemistryPublication details, including instructions for authors and subscription information:http://www.informaworld.com/smpp/title~content=t713653210
Total body glutathione depletion induces oxidative stress and disrupts theimmune function in miceMaher Y. Abdallaab; Iman M. Hassana; Noor H. Mustafaa; Lubna H. Tahtamounia; Iman M. Ahmadc
a Department of Biological Sciences and Biotechnology, The Hashemite University, Zarqa, Jordan b
Center for Cancer Research, National Cancer Institute, National Naval Medical Center, Bethesda, MD20889, USA c Department of Radiography, The Hashemite University, Zarqa, Jordan
First published on: 16 July 2010
To cite this Article Abdalla, Maher Y. , Hassan, Iman M. , Mustafa, Noor H. , Tahtamouni, Lubna H. and Ahmad, ImanM.(2010) 'Total body glutathione depletion induces oxidative stress and disrupts the immune function in mice',Toxicological & Environmental Chemistry,, First published on: 16 July 2010 (iFirst)To link to this Article: DOI: 10.1080/02772248.2010.494828URL: http://dx.doi.org/10.1080/02772248.2010.494828
Full terms and conditions of use: http://www.informaworld.com/terms-and-conditions-of-access.pdf
This article may be used for research, teaching and private study purposes. Any substantial orsystematic reproduction, re-distribution, re-selling, loan or sub-licensing, systematic supply ordistribution in any form to anyone is expressly forbidden.
The publisher does not give any warranty express or implied or make any representation that the contentswill be complete or accurate or up to date. The accuracy of any instructions, formulae and drug dosesshould be independently verified with primary sources. The publisher shall not be liable for any loss,actions, claims, proceedings, demand or costs or damages whatsoever or howsoever caused arising directlyor indirectly in connection with or arising out of the use of this material.

Toxicological & Environmental Chemistry2010, 1–14, iFirst
Total body glutathione depletion induces oxidative stress and disrupts the
immune function in mice
Maher Y. Abdallaab*, Iman M. Hassana, Noor H. Mustafaa, Lubna H. Tahtamounia
and Iman M. Ahmadc
aDepartment of Biological Sciences and Biotechnology, The Hashemite University, Zarqa, Jordan;bCenter for Cancer Research, National Cancer Institute, National Naval Medical Center, Bethesda,MD 20889, USA; cDepartment of Radiography, The Hashemite University, Zarqa, Jordan
(Received 7 May 2010; final version received 9 June 2010)
The effect of total body glutathione (GSH) depletion induced by L-buthionine-(S,R)-sulfoximine (BSO) treatment on the murine immune system has not beenstudied previously. BSO was administered via drinking water for 14 days toinduce total body GSH depletion. A significant decrease in total GSH (TGSH) ofthe immune organs such as spleen, liver, peritoneal macrophages as well as bloodwas observed. The proliferative response of splenocytes against different types ofmitogens was significantly decreased in GSH-depleted mice compared to controls.The expression of several antioxidant enzymes was studied in both treated andcontrol mice. No significant change was seen in major antioxidant enzymes;manganese superoxide dismutase and catalase. However, the expression of hemeoxygenase-1 was increased in both liver and spleen in BSO-treated mice. Totalbody GSH depletion also increased lipid peroxidation as demonstrated by higherlevels of malondialdehyde in BSO-treated mice. All the above-mentioned changeswere reversed to near normal 14 days post termination of BSO treatment. Theseresults demonstrate that TGSH depletion induces oxidative stress and adverselyaffects the function of the murine immune system.
Keywords: glutathion; reactive oxygen species; immune system; hemeoxygenase-1
Introduction
Oxygen is the main oxidant in metabolic reactions designed to provide energy from theoxidation of a variety of organic molecules. Reactive oxygen and nitrogen species (ROSand RNS, respectively) are radicals that are constantly formed during normal aerobicmetabolism. ROS and RNS play a key role in many physiologic reactions, but aredeleterious when produced in excess or eliminated inadequately (Touyz 2003). Oxidativestress occurs when there is a disturbance in the equilibrium status of pro-oxidant/antioxidant systems in intact cells. When oxidative events occur; the pro-oxidant systemsoutweigh the antioxidant, leading to damage of lipids, proteins, carbohydrates, and nucleicacids. Oxidative stress alters the antioxidant systems by inducing or inhibiting proteinsthat participate in these systems, and by depleting cellular stores of antioxidant
*Corresponding author. Email: [email protected]
ISSN 0277–2248 print/ISSN 1029–0486 online
� 2010 Taylor & Francis
DOI: 10.1080/02772248.2010.494828
http://www.informaworld.com
Downloaded By: [Abdalla, Maher] At: 13:24 16 July 2010

compounds such as glutathione (GSH; Meister and Anderson 1983; Monk, Fagerstedt,
Crawford 1989; Blokhina, Virolainen, and Fagerstedt 2003).GSH is a tripeptide (L-�-glutamyl-L-cysteinylglycine) present in all mammalian tissues
and is especially concentrated in the liver (Lu 1999). GSH has a central role in sulfhydryl
homeostasis and serves as the major cytosolic antioxidant (Lu 1999). The L-buthionine-(S,R)-sulfoximine (BSO) is an irreversible inhibitor of �-glutamylcysteine synthetase; the
enzyme that controls the first step of GSH biosynthesis (Griffith 1982; Lorico et al.2005). Several studies reported the establishment of a murine mouse model in which
BSO continuously depletes intracellular GSH and results in oxidative damage (Watanabeet al. 2003; Reliene and Schiestl 2006). The effects of BSO on the murine immune system
are still poorly understood. The objective of this study was to evaluate the influence oftotal GSH (TGSH) depletion on the structure and functions of the murine immune
system.
Materials and methods
Reagents
BSO was obtained from Biochemika, Fluka, USA. Concanavalin A (Con A), lipopoly-saccharide (LPS), protease inhibitor cocktail, ethoxypropane 3-(4,5-dimethylthiazol-2-yl)-
2,5-diphenyltetrazolium bromide (MTT) solution, and N-1 naphthylenediaminedihydrochloride (NED) were purchased from Sigma, USA. Phytohemagglutinin (PHA)
was obtained from Remel Europe, England. 3-Aminopropyltriethoxysilane was obtainedfrom ACROS Organic, USA. Cell and tissue staining kit (HRP–DAB System); mouse
anti-manganese superoxide dismutase (MnSOD) polyclonal primary antibody werepurchased from R&D Systems, USA. Heme oxygenase-1 (HO-1) primary antibody was
from Stressgen Biotech Serologies, USA.
Mice and treatments
Male BALB/c mice, weighing between 23 and 32 g, were divided into two groups, controland experimental. Animals in the experimental group were treated with 20mM BSO via
drinking water for 14 days, while the control animals were treated with drinking wateronly. At the end of the experimental period, the animals were sacrificed by cervical
dislocation. The spleen and liver were excised and the blood was collected via heartpuncture.
Measurement of GSH levels
Liver and spleen samples were homogenized in 5% sulfosalicylic acid (SSA), then
centrifuged at 1200 g, 4�C for 5min, and the supernatants were collected for subsequentassays. For measurement of GSH in the blood, whole blood was collected from the heart
of control and treated animals using the method of Crowley, Gillham, and Thorn (1975)with modifications. Blood was prepared as mentioned above and the supernatant was
collected for assay. Blood was added to Drabkin’s solution and incubated for 15min atroom temperature to determine hemoglobin (HB) levels.
2 M.Y. Abdalla et al.
Downloaded By: [Abdalla, Maher] At: 13:24 16 July 2010

Assay procedure
Measurement of GSH levels was based on that originally described by Anderson (1985).
Fifty microliters of each sample prepared as described above was mixed with 100 mLdistilled water, 700 mL of 0.298mM nicotinamide adenine dinucleotide phosphate-oxidase
(NADPH) dissolved in sodium phosphate buffer at pH 7.5, and 100 mL of 6mM dithiobis-
nitrobenzoic acid (DTNB; Ellman’s reagent). The assay was started by the addition of
50 mL GSH reductase (GR; 266U/mL), and the rate of 5-thio-2-nitrobenzoate (TNB)
formation was monitored at 412 nm, every 10 s for 2.5min using a Jasco V-530
spectrophotometer. The oxidized GSH (GSSG) levels were determined using the same
DTNB assay when reduced GSH is masked by 2-vinylpyridine (2-VP; Griffith 1980). To
accomplish this, 12 mL of 1 : 1 mixture of 2-VP and absolute ethanol were added to 60 mLof sample for 2.5 h to block all reduced GSH and this was subjected to DTNB assay as
described for TGSH. TGSH and GSSG were determined from the standard curve and
normalized to protein mass.
Measurement of GSH level in mice peritoneal macrophages
Mouse peritonial macrophages were collected and washed with cold phosphate-buffered
saline (PBS) and centrifuged at 6500 g. DETAPAC buffer (Sigma, USA) was added and
then mixed with 5% SSA. TGSH and GSSG levels were determined as described above
(Griffith 1980; Anderson 1985). The results were normalized to the protein content.
Lymphocyte proliferation assay (MTT)
Serial dilutions of splenic lymphocytes were prepared in RPMI-1640 media containing 1%
penicillin/streptomycin, L-glutamine, and 10% fetal bovine serum. 1� 106 cells were
cultured into each well of a 96-well flat-bottomed tissue culture plate. Con A, LPS, and
PHA were added to a final concentration of 120, 5, and 20 mgmL�1, respectively. Media
alone were used as a negative control. Each assay was performed in triplicate. Plates were
incubated at 37�C for 48 h in a 5% CO2, 95% humidity incubator. The MTT assay was
performed as described previously (Mosmann 1983). Briefly, the MTT solution was
prepared by dissolving 5mg MTT/mL PBS and sterilized by filtering through a 0.45mmsyringe filter. Of the MTT solution 20 mL was added to each well, and the plates were
incubated at 37�C for 3 h in the CO2 incubator. The formation of formazan crystals was
checked using inverted microscope. Plates were then taken and 150 mL of each well was
discarded carefully using micropipette leaving the crystals only. These crystals were
dissolved with 200 mL dissolving solution (1 : 1 mixture of DMSO and isopropanol), and
the absorbance of the dark violet color solution developed in the wells was measured at
570 nm using a Jasco V-530 spectrophotometer.
Light microscopy
Liver and spleen tissues were fixed in 10% neutral formalin fixative and embedded in
paraffin. Tissues were prepared for routine hematoxylin and eosin staining. The
histological observation was carried out using a bright field microscope.
Toxicological & Environmental Chemistry 3
Downloaded By: [Abdalla, Maher] At: 13:24 16 July 2010

Immunohistochemistry
Liver and spleen tissue samples were fixed in 10% buffered formalin, embedded inparaffin, and cut into 6 mm thick sections. Representative sections were deparaffinized,rehydrated, and treated with antigen retrieval reagent in a microwave at full power (650W)for 5min, then washed with PBS. Streptavidin-biotin-peroxidase immunostaining wascarried out using cell and tissue staining kit HRP-DAB System (R&D, USA). Endogenousperoxidase activity was blocked in 3% H2O2 in methanol, followed by blockingnonspecific antibody binding with non-immune rabbit serum. Endogenous avidin andbiotin were blocked using the commercially available avidin and biotin blocking solutions.Sections were then incubated with HO-1 antibody (1 : 1000 dilution overnight inhumidity chamber), washed and then incubated with labeled secondary antibody.The primary/secondary antibody reaction was detected using high sensitivity streptavidinconjugated to horseradish peroxidase (HSS–HRP) and revealed by 3,3-diaminobenzidine(DAB) chromogen. Sections were counter stained with Mayer’s hematoxylin. Negativecontrols were processed as stated above except that the appropriate normal serum wassubstituted for the primary antibody.
Immunoblot analysis
Tissues samples were homogenized in lysis buffer (0.018M NaCl, 0.5M Na2–EDTA pH 8,1M Tris–base pH 7.4, 1% Triton� 100) to which 1X protease inhibitor cocktail solutionwas added. Tissue lysates were centrifuged and the supernatants were collected and theprotein concentration was determined using the Bradford Solution (Bio-Rad, USA). Ofthe cellular proteins 60 mg was mixed with one volume of 2X sample buffer containing 6%SDS and 10% �-mercaptoethanol, denatured by heating to 95�C for 5min. Proteins wereseparated on 5% acrylamide stacking gel and 12% resolving gels. Electrophoresedproteins were transferred (blotted) onto a 0.45mm nitrocellulose membrane (Bio-Rad,USA). To determine HO-1 and MnSOD protein levels in liver and spleen, the resultantmembranes were blocked with Tris-buffered saline (TBS) containing 5% (w/v) fat-free drymilk at 4�C overnight with continuous shaking. The membranes were then incubated withrabbit anti-HO-1 primary antibody and mouse anti-MnSOD primary antibody (1 : 2000and 1 : 4000, respectively in 2% fat-free dry milk in TBS), overnight at 4�C withcontinuous shaking. After washing with Tris-buffered saline with Tween (TBST),membranes were incubated with horseradish peroxidase-conjugated anti-rabbit immuno-globulin G and anti-mouse immunoglobulin G (IgG; 1 : 10,000 each in 2% fat-free drymilk in TBST) for 2 h at room temperature. Finally, the membranes were washed withTBST and bands were visualized using the 3,30,5,50 tetramethylbenzidine system (TMB;Sigma, USA).
Assessment of lipid peroxidation
Lipid peroxidation was determined by measuring thiobarbituric acid-reactive species(TBARS) as described by Ohkawa, Ohishi, and Yagi (1979) with modifications. Briefly,tissue samples from livers and spleens were homogenized in cold potassium chloride (KCl),mixed with 1% phosphoric acid, 0.8% TBA, and boiled in a water bath at 95�C for 1 huntil the detection of a pink color. The reaction was stopped using ice and the color wasextracted in n-butanol by centrifugation at 15,000 rpm. TBARS levels were measured at532 nm using a standard curve of MDA. The values are expressed in mM MDA.
4 M.Y. Abdalla et al.
Downloaded By: [Abdalla, Maher] At: 13:24 16 July 2010

Colorimetric determination of nitrite (NOZ2 ) concentration
Isolation of peritoneal macrophages was performed as described by Rouzer et al. (1982)with modifications. Briefly, control and treated mice were stimulated by 5mL intraper-itoneal (IP) injection of sucrose for 2 h, and then injected with 2mL RPMI completemedia. Macrophages were harvested and collected by centrifugation. The cell density wasadjusted to 1� 106 cells/mL and the viability of freshly isolated cells was determined usingtrypan blue exclusion test. Macrophages were cultured in triplicates with RPMI media in a96-well tissue culture plate. Cells were seeded with and without the simulation of 5 mg/mLLPS. Cultures were incubated for 48 h at 37�C, 5% CO2, and 95% relative humidity. Allwells were then treated with 1% sulfanilamide and 0.1% N-1-NED. Pink color wasmeasured after 10min using a plate reader. The nitric oxide (NO) concentration wasdetermined using a nitrite standard curve. The values are expressed in mM nitrite.
Statistical analysis
All results are expressed as means� SE. The statistical significance of bereaved differencesbetween means was determined using SPSS software (SPSS, version 14.00). The p valuesless than 0.05 were considered to be significant.
Results
BSO decreases body and organ weights
The toxicity of BSO was previously reported by Watanabe et al. (2003). In this study, theadministration of 20mM BSO did not produce deaths or signs of abnormalities throughthe period of administration. The body weight of treated animals was significantlydecreased compared to control animals by 20% as calculated by the ratio of body weightchange (Table 1). Organ weight (liver and spleen) also showed a significant decrease (10%and 27.41%, respectively) in mice treated with 20mM BSO as compared to controls(Table 1). However, no significant difference was found when percent organ weight tobody weight was compared within each group (Table 1).
BSO treatment causes total body GSH depletion
To verify whether BSO treatment induced TGSH depletion, GSH level was measured indifferent tissues and blood. GSH levels were significantly decreased in both spleen andliver. As shown in Figure 1, GSH was decreased in the spleen of treated mice more thanthreefold and by twofold in liver. Similarly, both peritoneal macrophages and whole blood
Table 1. Absolute and relative weights of spleen and liver.
Tissue Control Treated% Organ/bodyweight (control)
% Organ/bodyweight (treated)
Spleen (g) 0.2� 0.03 0.14� 0.01* 0.63� 0.03 0.48� 0.02Liver (g) 1.64� 0.1 1.5� 0.15* 5.1� 0.1 5.01� 0.18
Notes: Data are meanþ SEM of seven mice per group.*Significantly different from control (p5 0.05).
Toxicological & Environmental Chemistry 5
Downloaded By: [Abdalla, Maher] At: 13:24 16 July 2010

GSH levels were significantly reduced in BSO-treated mice compared to controls(Figure 1).
Total body GSH depletion disrupts murine immune function
In order to study the effect of TGSH depletion on immune functions, lymphocytes andmacrophages function was studied. Spleen and liver histological sections were alsoexamined.
B- and T-cell response to mitogen stimulation
The influence of GSH depletion on lymphocytes function was measured by stimulatingB-and T-cell with different mitogens (LPS, Con A, and PHA). When splenocytes fromBSO-treated mice were stimulated with Con A or PHA for T-cell stimulation; cellsexhibited significant reduction in proliferation (Figure 2). Similarly, in BSO-treated mice,B-cells stimulated with LPS showed significantly reduced proliferation compared tocontrols (Figure 2).
Effect of GSH depletion on macrophage function
The effect of GSH depletion on the function of macrophages was investigated bydetermining the nitrite levels after stimulation with LPS. Our results showed thatstimulated macrophages from BSO-treated mice exhibited higher nitrite concentrationthan stimulated control macrophages. However, the increase was only quantitative(3.31� 0.93 and 2.59� 0.4 mM nitrite, respectively; Figure 3).
Influence of GSH depletion on the morphology of spleen and liver tissues
The morphology of the spleen and liver of control and BSO-treated mice was examined.All organs were isolated after 14 days of treatment and processed for histologicalexamination. GSH depletion had no apparent effect on spleen ultrastructure
Spleen
Liver
Who
le blo
od
Mac
roph
ages
0.0
0.5
1.0
1.5 Control
Treated
*
***
GS
H+
GS
SG
(fo
ld c
hang
e)
Figure 1. Influence of BSO treatment on TGSH levels in various tissue organs and blood.Notes: Data represents mean� SEM, n¼ 7. *p5 0.05 vs. control group.
6 M.Y. Abdalla et al.
Downloaded By: [Abdalla, Maher] At: 13:24 16 July 2010

(Figure 4A and B). In contrast, GSH depletion exerted a marked effect on liver tissue ofBSO-treated mice. Multiple necrotic cells, especially in the central zone of hepatic lobule,were observed (Figure 4C and D).
Influence of GSH depletion on the expression of several antioxidant enzymes
To study the effect of TGSH depletion on oxidative status of immune organs, theexpression of major antioxidant enzymes such as MnSOD, CAT, and HO-1 were
examined. The protein levels of antioxidant enzymes were determined by immunoblotting.No marked differences were seen in the expression of MnSOD or CAT in the liver of BSO-treated mice (Figure 5). In contrast, HO-1 expression was significantly induced in liver of
Con A
PHALP
S0.00.51.01.52.02.5O
D 3.03.54.04.55.05.5
Control
Treated
* ***
Figure 2. Effect of BSO on B- and T-cell responses to mitogen stimulation in BSO-treated mice.Notes: 1� 106 of splenocytes from control and BSO-treated mice were cultured in the presence ofCon A, PHA, and LPS for 48 h. Proliferation was assessed by MTT assay. Graph represents themean� SEM, n¼ 6. *p5 0.05 vs. control, and **p5 0.01 vs. control.
Treat
ed
Contro
l0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
mM N
itrite
Figure 3. Influence of GSH depletion on the macrophage function.Notes: Macrophages were harvested from the peritoneal cavity of control and BSO-treated mice.Nitrite concentration was measured as indication of macrophage function. Graph represents themean� SEM, n¼ 6.
Toxicological & Environmental Chemistry 7
Downloaded By: [Abdalla, Maher] At: 13:24 16 July 2010

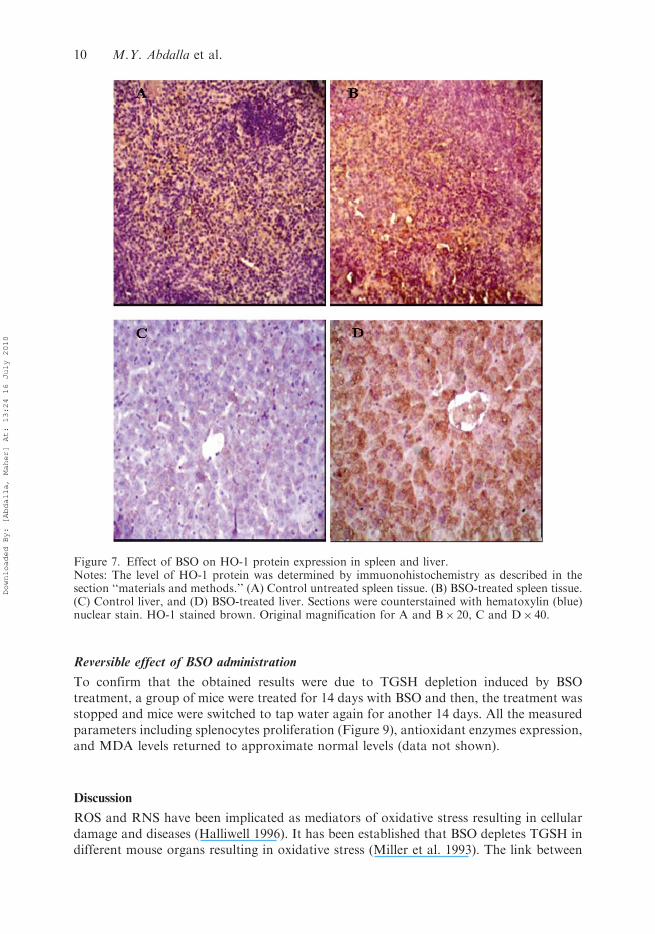
treated mice (Figure 5). Similar results were seen in spleen tissues where HO-1 was alsomarkedly induced by TGSH depletion (Figure 6). To confirm our results, HO-1immunohistochemistry was performed. As shown in Figure 7, an apparent increase inthe HO-1 protein expression was observed in liver and spleen of treated micewith higher HO-1 staining intensity in spleen tissues (Figure 7A and B). Similarobservations were seen in the liver tissues (Figure 7C and D). This enhanced stainingintensity indicates the induction of HO-1 enzyme in both liver and spleen tissues ofBSO-treated animals.
Influence of GSH depletion on malondialdehyde levels
To further explore the influence of oxidative stress induced by TGSH depletion onimmune organs, lipid peroxidation was measured using thiobarbituric acid reactive
Figure 4. Effect of GSH depletion on the morphology of spleen and liver.Notes: GSH depletion has no apparent effect on spleen tissue structure from BSO-treated andcontrol mice (A and B, respectively). In contrast, GSH depletion has a dramatic effect on livermorphology of BSO-treated and control mice (C and D, respectively). Arrows indicate necrotic cells.Original magnifications, A and B� 20, C and D� 40.
8 M.Y. Abdalla et al.
Downloaded By: [Abdalla, Maher] At: 13:24 16 July 2010

substances (TBARS) method by the measurement of malondialdehyde (MDA) (secondary
products of lipid peroxidation). Our results showed elevated MDA levels in BSO-treatedmice compared to controls, that reached an eightfold rise in spleen tissue, and fivefold
increase in liver tissues of treated mice (Figure 8).
Figure 5. (a) Immunoblot analysis of HO-1, MnSOD, and CAT antioxidant enzymes in livers ofcontrol and BSO-treated mice. and (b) Densitometric analysis of western blot results in (a)normalized to actin.Notes: Increased expression of HO-1 in liver of GSH depleted mice compared to controls. Nochange in the expression of MnSOD or CAT antioxidant enzymes. Lane 1 and 3 represent controluntreated liver tissue. Lanes 2 and 4 represent two different liver tissue samples from two differenttreated animals. Actin was used as loading control. *p5 0.05 vs. respective control.
Figure 6. HO-1 expression in spleen by immunoblotting.Notes: Increased HO-1 protein expression in GSH-depleted mice compared to controls. Theexpression of HO-1 protein was determined by western blotting as described in materials andmethods. Lane 1 represent control untreated tissue. Lanes 2 and 3 represents two different tissuesamples from two different treated animals. Actin was used as a loading control.
Toxicological & Environmental Chemistry 9
Downloaded By: [Abdalla, Maher] At: 13:24 16 July 2010
Reversible effect of BSO administration
To confirm that the obtained results were due to TGSH depletion induced by BSOtreatment, a group of mice were treated for 14 days with BSO and then, the treatment wasstopped and mice were switched to tap water again for another 14 days. All the measuredparameters including splenocytes proliferation (Figure 9), antioxidant enzymes expression,and MDA levels returned to approximate normal levels (data not shown).
Discussion
ROS and RNS have been implicated as mediators of oxidative stress resulting in cellulardamage and diseases (Halliwell 1996). It has been established that BSO depletes TGSH indifferent mouse organs resulting in oxidative stress (Miller et al. 1993). The link between
Figure 7. Effect of BSO on HO-1 protein expression in spleen and liver.Notes: The level of HO-1 protein was determined by immuonohistochemistry as described in thesection ‘‘materials and methods.’’ (A) Control untreated spleen tissue. (B) BSO-treated spleen tissue.(C) Control liver, and (D) BSO-treated liver. Sections were counterstained with hematoxylin (blue)nuclear stain. HO-1 stained brown. Original magnification for A and B� 20, C and D� 40.
10 M.Y. Abdalla et al.
Downloaded By: [Abdalla, Maher] At: 13:24 16 July 2010

GSH and immune function and immunological diseases has been established in variousstudies. Lehmann et al. (2007) showed that some chemical extracts such as fumaric acidesters mediate immunomodulatory effects by induction of the anti-inflammatory stressprotein HO-1 upon depletion of reduced GSH. Further, Collins, Unanue, and Harding(1991) suggested that several diseases such as cancer, allergic disorders, and acquiredimmune deficiency syndrome (AIDS) are characterized by GSH depletion in antigen-presenting cells (APC), which involve Th1 failure.
The role of cytokines and the critical switch between Th1 and Th2 is important indiseases such as cancer. In vitro and in vivo studies demonstrated that GSH depletion inmacrophages correlates with defective antigen processing and inhibits Th1-associatedcytokine production (Murata et al. 2002a; Murata, Shimamura, and Hamuro 2002b).In murine studies, GSH depletion in APC decreases the secretion of interleukin (IL)-12and leads to switch from the typical Th1 cytokine profile towards Th2 response patterns.This may be due to marked GSH depletion, which impairs the ability of APC to process
Spleen
Liver
0.0
2.5
5.0
7.5
10.0
ControlTreated
*
**
mM M
DA
Figure 8. Influence of BSO on lipid peroxidation.Notes: Spleen and liver from control and BSO-treated mice were processed for MDA measurementas described in the section ‘‘materials and methods’’. The concentration of MDA was extrapolatedfrom a standard curve of MDA (mM). Results shown are means� SEM, n¼ 7. *p5 0.05 vs.respective control, and **p5 0.01 vs. respective control.
ConA
LPS
PHA0
1
2
3
4
5
Post treatmentBSO-treatment
Control
OD
Figure 9. B- and T-cell response to mitogen stimulation 14 days after switching from BSO to water.Notes: Splenocytes from BSO-treated mice and from mice switched to water after 14 days of BSOtreatment were cultured in the presence of Con A, LPS, and PHA for 48 h. Proliferation was assessedby MTT assay. Results shown are means of the (optical density, OD) (570 nm)� SEM, n¼ 3.
Toxicological & Environmental Chemistry 11
Downloaded By: [Abdalla, Maher] At: 13:24 16 July 2010

and present the antigens through the major histocompatibility complex class II (Shortet al. 1996; Murata et al. 2002a; Murata, Shimamura, and Hamuro 2002b; Murata, Amao,and Junji Hamuro 2003). In addition, in human alveolar macrophages, Dobashi et al.(2001) found that GSH levels in APC play a central role in determining whether a Th1 orTh2 cytokine response will develop. It is possible to regulate the amount of IL-12 secretedby modulating the intracellular GSH/GSSG balance (Dobashi et al. 2001).
In this study, TGSH depletion was observed in treated mice compared to control.These findings confirmed an earlier study of GSH depletion induced by BSO in treatedmice (Miller et al. 1993; Watanabe et al. 2003). Our findings that GSH depletion adverselyaffected the ability of splenocytes to respond to different mitogen stimulation shows theimportant role of thiol compounds in normal immune functions (Figure 2). Data showedthat depleting GSH in T lymphocytes using BSO renders them unable to proliferate inresponse to different mitogen stimulation indicating a direct relationship between theproliferative response of lymphocytes and GSH availability (Hamilos and Wedner 1985;Droge et al. 1994; Kinscherf et al. 1994; Buchmuller-Rouiller et al. 1995; Forman andTorres 2002).
Oxidative stress induced by GSH depletion is likely to induce antioxidant enzymes suchas MnSOD and CAT. Monk, Fagerstedt, and Crawford (1989) reported that MnSODenzyme is increased during cellular oxidative stress. In contrast, our findings revealed thatliver and spleen tissues from BSO-treated mice showed no marked changes in theexpression of MnSOD or CAT (Figures 5 and 6). The finding that both enzymes were notinduced by GSH depletion implicates the involvement of other antioxidant enzymes suchas HO-1, which is a stress enzyme that is rapidly induced in response to cellular oxidativestress (Ewing and Maines, 1993; Immenschuh and Ramadori, 2000; Guo, Shin, and Cho2001). Our study showed by immunoblotting and immunohistochemistry that HO-1expression was significantly upregulated in both spleen and liver of BSO-treated mice(Figures 5–7).
Several studies reported that cellular oxidative stress affects the morphology of livertissue (Hayami et al. 1999; Cogger et al. 2004; Zhang et al. 2004). Our study found similarresults, and revealed that tissue sections of liver from BSO-treated mice showed numeroussigns of apoptosis and hepatic damage. The manifestations included the presence ofpyknotic nuclei around the central zone of hepatic lobule (Figure 4C and D).
Previous studies demonstrated that lipid peroxidation and lipid hydroperoxidesincrease during oxidative stress (Lykkesfeldt 2007). In the current study, measurement ofMDA levels demonstrated that lipid peroxidation was elevated in BSO-treated mice(Figure 8). This may be attributed to BSO treatment which induced a condition ofoxidative stress.
Finally, our finding that termination of BSO treatment reversed these effects may be ofclinical importance, since it indicates that oxidative stress and immune function disruptionassociated with GSH depletion could be reversed after restoring oxidant status. This mightbe of benefit for patients with diseases that produce TGSH depletion such as HIV,diabetes, cancer, neurodegenerative, and liver diseases (Franco et al. 2007). In thosepatients, restoring the level of GSH using drugs that contain N-acetylcysteine may restoreintracellular GSH levels and thus, normal immune system functions.
Acknowledgments
This study was supported by a grant from The Deanship of Research and Graduate Studies, TheHashemite University.
12 M.Y. Abdalla et al.
Downloaded By: [Abdalla, Maher] At: 13:24 16 July 2010

References
Anderson, M.E. 1985. Determination of glutathione and glutathione disulfide in biological samples.
Methods in Enzymology 113: 548–54.Blokhina, O., E. Virolainen, and K.V. Fagerstedt. 2003. Antioxidants, oxidative damage and oxygen
deprivation stress: a review. Annals of Botany 91: 179–94.Buchmuller-Rouiller, Y., S.B. Corradin, J. Smith, P. Schneider, A. Ransijn, C.V. Jongeneel, and
J. Mauel. 1995. Role of glutathione in macrophage activation: Effect of cellular glutathi-
one depletion on nitrite production and Leishmanicidal activity. Cellular Immunology 164:
73–80.Cogger, V.C., M. Muller, R. Fraser, A.J. McLean, J. Khan, and D.G. Le Couteur. 2004. The effects
of oxidative stress on the liver sieve. Journal of Hepatology 41: 370–6.
Collins, D.S., E.R. Unanue, and C.V. Harding. 1991. Reduction of disulfide bonds within lysosomes
is a key step in antigen processing. Journal of Immunology 147: 4054–9.
Crowley, C., B. Gillham, and M.B. Thorn. 1975. A direct enzymic method for the determination of
reduced glutathione in blood and other tissues. Biochemical Medicine 13: 287–92.Dobashi, K., M. Aihara, T. Araki, Y. Shimizu, M. Utsugi, K. Iizuka, Y. Murata, J. Hamuro,
T. Nakazawa, and M. Mori. 2001. Regulation of LPS induced IL-12 production by IFN-
gama and IL-4 through intracellular glutathione status in human alveolar macrophages.
Clinical and Experimental Immunology 124: 290–6.Droge, W., K. Schulze-Osthoff, S. Mihm, D. Galter, H. Schenk, H.P. Eck, S. Roth, and
H. Gmunder. 1994. Functions of glutathione and glutathione disulfide in immunology
and immunopathology. FASEB Journal 8: 1131–8.Ewing, J.F., and M.D. Maines. 1993. Glutathione depletion induces heme oxygenase-1 (HSP32)
mRNA and protein in rat brain. Journal of Neurochemistry 60: 1512–9.Forman, H.J., and M. Torres. 2002. Reactive oxygen species and cell signaling: Respiratory
burst in macrophage signaling. American Journal of Respiratory and Critical Care Medicine 166:
S4–8.Franco, R., O.J. Schoneveld, A. Pappa, and M.I. Panayiotidis. 2007. The central role of
glutathione in the pathophysiology of human diseases. Archives of Physiology and Biochemistry
113: 234–58.Griffith, O.W. 1980. Determination of glutathione and glutathione disulfide using glutathione
reductase and 2-vinylpyridine. Analytical Biochemistry 106: 207–12.Griffith, O.W. 1982. Mechanism of action, metabolism, and toxicity of buthionine sulfoximine and
its higher homologs, potent inhibitors of glutathione synthesis. Journal of Biological Chemistry
257: 13704–12.Guo, X., V.Y. Shin, and C.H. Cho. 2001. Modulation of heme oxygenase in tissue injury and its
implication in protection against gastrointestinal diseases. Life Sciences 69: 3113–9.
Halliwell, B. 1996. Antioxidants in human health and disease. Annual Review of Nutrition 16: 33–50.Hamilos, D.L., and H.J. Wedner. 1985. The role of glutathione in lymphocyte activation. I.
Comparison of inhibitory effects of buthionine sulfoximine and 2-cyclohexene-1-one by nuclear
size transformation. Journal of Immunology 135: 2740–7.Hayami, S., K. Ikeda, F. Sun, K. Tanaka, and S. Kojo. 1999. Increase of caspase-3 activity in rat
liver and plasma by thioacetamide. Biochemical Pharmacology 58: 1941–3.Immenschuh, S., and G. Ramadori. 2000. Gene regulation of heme oxygenase-1 as a therapeutic
target. Biochemical Pharmacology 60: 1121–8.Kinscherf, R., T. Fischbach, S. Mihm, S. Roth, E. Hohenhaus-Sievert, C. Weiss, L. Edler, P.
Bartsch, and W. Droge. 1994. Effect of glutathione depletion and oral N-acetyl-cysteine treatment
on CD4þ and CD8þ cells. FASEB Journal 8: 448–51.Lehmann, J.C.U., J.J. Listopad, C.U. Rentzsch, F.H. Igney, A. von Bonin, H.H. Hennekes, K.
Asadullah, and W.-D.F. Docke. 2007. Dimethylfumarate induces immunosuppression via
glutathione depletion and subsequent induction of heme oxygenase-1. Journal of Investigative
Dermatology 127: 835–45.
Toxicological & Environmental Chemistry 13
Downloaded By: [Abdalla, Maher] At: 13:24 16 July 2010

Lorico, A., D. Bratbak, J. Meyer, D. Kunke, S. Krauss, W.E. Plott, V. Solodushko, C. Baum, O.Fodstad, and G. Rappa. 2005. �-Glutamylcysteine synthetase and L-buthionine-(S,R)-sulfox-imine: A new selection strategy for gene-transduced neural and hematopoietic stem/progenitorcells. Human Gene Therapy 16: 711–24.
Lu, S.C. 1999. Regulation of hepatic glutathione synthesis: Current concepts and controversies.FASEB Journal 13: 1169–83.
Lykkesfeldt, J. 2007. Malondialdehyde as biomarker of oxidative damage to lipids caused by
smoking. Clinica Chimica Acta 380: 50–8.Meister, A., and M.E. Anderson. 1983. Glutathione. Annual Review of Biochemistry 52: 711–60.Miller, N.J., C. Rice-Evans, M.J. Davies, V. Gopinathan, and A. Milner. 1993. A novel method for
measuring antioxidant capacity and its application to monitoring the antioxidant status inpremature neonates. Clinical Science 84: 407–12.
Monk, L.S., K.V. Fagerstedt, and R.M.M. Crawford. 1989. Oxygen toxicity and superoxide
dismutase as an antioxidant in pyhysiological stress. Physiologia Planatarum 76: 456–9.Mosmann, T. 1983. Rapid colorimetric assay for cellular growth and survival: Application toproliferation and cytotoxicity assays. Journal of Immunological Methods 65: 55–63.
Murata, Y., M. Amao, and J. Junji Hamuro. 2003. Sequential conversion of the redox status of
macrophages dictates the pathological progression of autoimmune diabetes. European Journal ofImmunology 33: 1001–11.
Murata, Y., M. Amao, J. Yoneda, and J. Hamuro. 2002a. Intracellular thiol redox status of
macrophages directs the Th1 skewing in thioredoxin transgenic mice during aging. MolecularImmunology 38: 747–57.
Murata, Y., T. Shimamura, and J. Hamuro. 2002b. The polarization of Th1/Th2 balance is
dependent on the intracellular thiol redox status of macrophages due to the distinctive cytokineproduction. International Immunology 14: 201–12.
Ohkawa, H., N. Ohishi, and K. Yagi. 1979. Assay for lipid peroxides in animal tissues bythiobarbituric acid reaction. Analytical Biochemistry 95: 351–8.
Reliene, R., and R.H. Schiestl. 2006. Glutathione depletion by buthionine sulfoximine induces DNAdeletions in mice. Carcinogenesis 27: 240–4.
Rouzer, C.A., W.A. Scott, O.W. Griffith, A.L. Hamill, and Z.A. Cohn. 1982. Glutathione
metabolism in resting and phagocytizing peritoneal macrophages. Journal of Biological Chemistry257: 2002–8.
Short, S., B.J. Merkel, R. Caffrey, and K.L. McCoy. 1996. Defective antigen processing correlates
with a low level of intracellular glutathione. European Journal of Immunology 26: 3015–20.Touyz, R.M. 2003. Reactive oxygen species in vascular biology: Role in arterial hypertension. ExpertReview of Cardiovascular Therapy 1: 91–106.
Watanabe, T., H. Sagisaka, S. Arakawa, Y. Shibaya, M. Watanabe, I. Igarashi, K. Tanaka,S. Totsuka, W. Takasaki, and S. Sunao Manabe. 2003. A novel model of continuousdepletion of glutathione in mice treated with L-buthionine (S,R)-sulfoximine. Journal ofToxicological Sciences 28: 455–69.
Zhang, H.J., S.R. Doctrow, L. Xu, L.W. Oberley, B. Beecher, J. Morrison, T.D. Oberley, andK.C. Kregel. 2004. Redox modulation of the liver with chronic antioxidant enzyme mimetictreatment prevents age-related oxidative damage associated with environmental stress. FASEB
Journal 18: 1547–9.
14 M.Y. Abdalla et al.
Downloaded By: [Abdalla, Maher] At: 13:24 16 July 2010




















